





























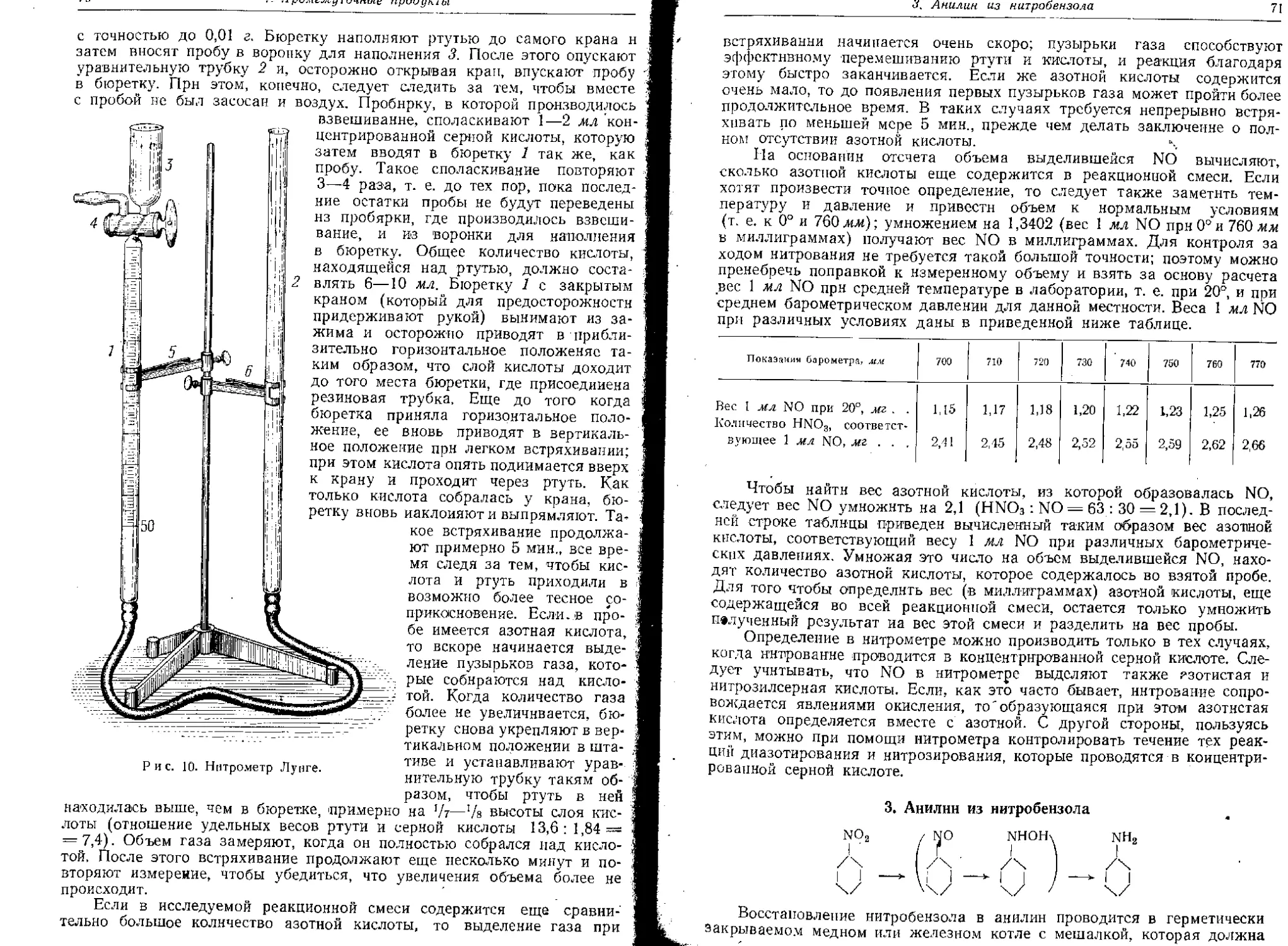





























































































































































Текст
GRUNDLEGENDE OPERATIONEN
DER FARBENCHEMIE
von
DR. HANS EDUARD FIERZ-DAVID
Professor an der eidgendssischen
technischen Hochscliule in Ziirich
und
DR. LOUIS BLANGEY
Professor an der eidgendssischen
technischen Hochscliule in Ziirich
Achte, zum Tell vermehrte Aaflage
mlt 57 Textabblldungen und 21 Tabellen
auf 24 Tafeln
Wien
Springer-Verlag
1952
Г. Э. ФИРЦ-ДАВИД, Л. БЛАНЖЕ
Основные
процессы
СИНТЕЗА
КРАСИТЕЛЕЙ
ПЕРЕВОД С НЕМЕЦКОГО
В. Р. СКВАРЧЕНКО, Ю. С. ШАБАРОВА
и Н. Я. ШУШЕРИНОЙ
ПОД РЕДАКЦИЕЙ
докт. хам., наук С. В. БОГДАНОВА
я канд. техн, наук И. В. ФО ДИ МАЯ А
1 3ДАТЕЛЬСfВ О ИНОСТРАННОЙ ЛИТЕРАТУРЫ
Москва — 1 957
I. ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ
Общие сведения о промежуточных продуктах
для производства красителей
Под промежуточными продуктами (или полупродуктами) для произ-
водства красителей понимают соединения, которые изготовляются из
исходных веществ, содержащихся в каменноугольной смоле, разнообраз-
ными химическими способами и ори помощи дальнейших сравнительно
простых превращений переводятся в искусственные красители (готовые
продукты).
В качестве примера можно назвать анилин, который получается
из бензола различными реакциями и затем может превращаться в много-
численные красители.
Реакции, служащие для получения промежуточных продуктов, яв-
ляются чаше всего простыми операциями, выполняемыми в различных
вариантах. Эти реакции во многих случаях протекают количе-
ственно по законам стехиометрии; однако часто наблю-
даются побочные реакции, которые неожиданно изменяют течение про-
цесса и сильно снижают ожидаемые выходы.
Поэтому одной из важнейших задач химика, работающего в области
красителей, является тщательное изучение именно этих нежелательных
побочных процессов, исследование их течения и, если это возможно,
подбор таких условий химической реакции, при которых получались бы
однородные химические продукты.
Эта цель не всегда достигается, так как часто ни при каких условиях
не удается направить реакцию только в одну строну. Но и тогда химик
должен иметь в виду, что при некоторых еще не известных условиях
можно было бы направить реакцию К одном желаемом направлении.
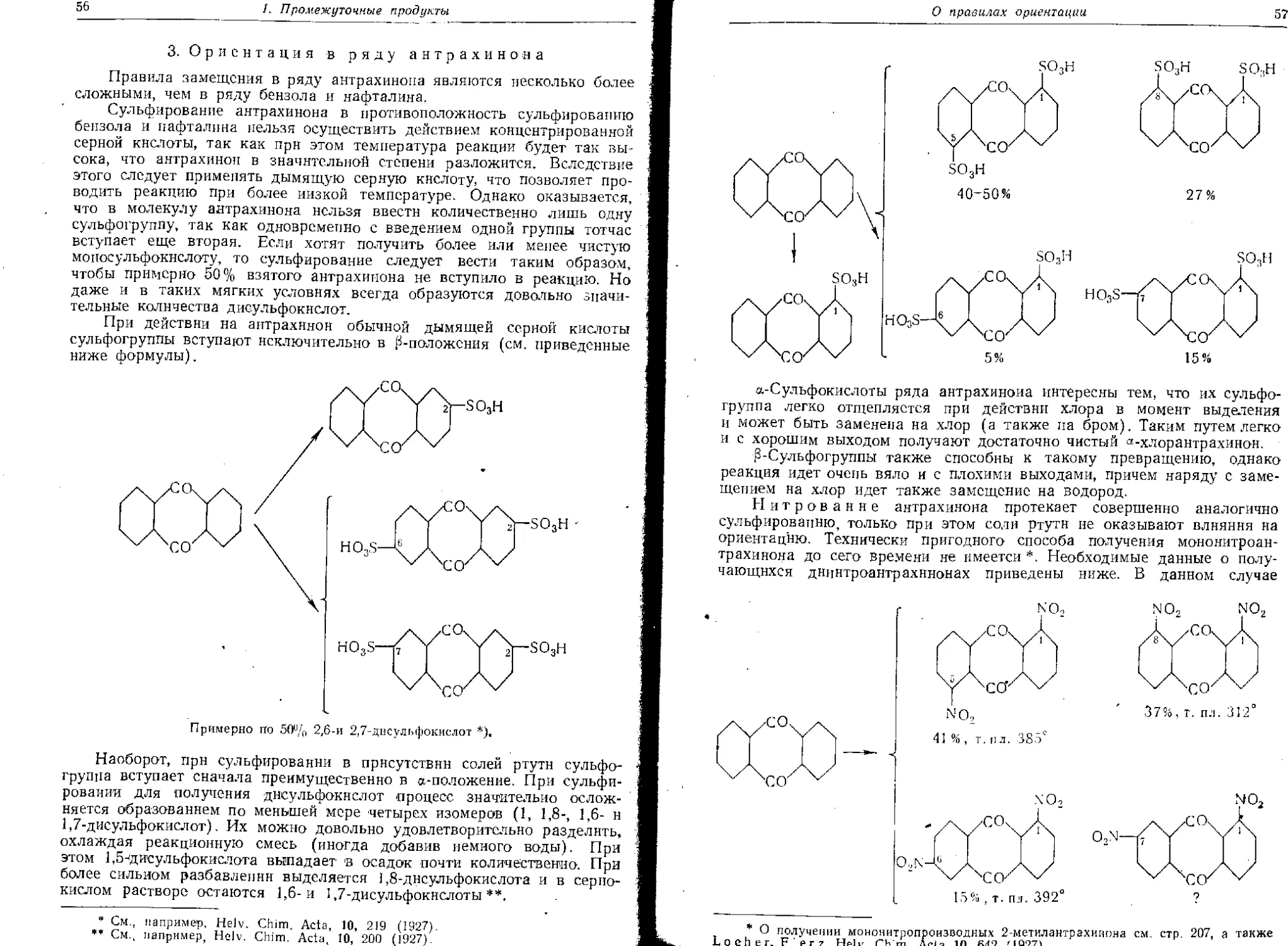
Важным примером является производство 1,8-а иинонафтол-3,6-дисуль-
фокислоты (Аш-кислота), которое, хотя и известно почти 60 лет, все
еще тщательно изучается во многих лабораториях; однако до последнего
времени пе удалось еще достичь удовлетворительных выходов.
В некоторых случаях достигаются количественные выходы, ио обра-
зующиеся продукты неоднородны, т. е. хотя продукт и получают в рас-
считанных количествах, но он представляет собой смесь, состоящую
частью из изомеров, частью из аналогов, которые иногда приходится г
разделять физическими методами. В качестве примера можно назвать
изомерные нитротолуолы (орто, мета и пара), каждый из которых ни-
когда не получается в чистом вндэ, так что для их разделения пришлось
выработать специальные надежные и в то же время дешевые методы.
Иногда индивидуальные промежуточные продукты удается получить
обходным путем, вводя, например, заместители (чаще всего сульфо-
группу), которые затем снова отщепляют (см. получение о-хлортолуола,
«П'Т'ГЪ ЧТГ».Т ттл Л л л Л Z-. л ъ „
12 /. Промежуточные продукты
тельные изомеры не могли образоваться. Примером может служить так
называемая «кислота Тобиаса» (2-нафтиламип-1-сульфокислота), кото-
рую не удается получить непосредственно из Р-нафтиламнна; ее получают
при строго определенных условиях из fS-цафтола (см. стр. 176 и сл.).
Основные процессы химии красителей базируются, как уже упоми-
налось, па простых химических реакциях. Часто можно один и тот же
промежуточный продукт получить совершенно различными путями;
в этом случае точный производственный расчет (калькуляция) должен
показать, какой способ является наиболее выгодным. Не всегда способ,
кажущийся самым дешевым, оказывается лучшим: часто должны быть
приняты во внимание побочные обстоятельства. Они могут определяться
стоимостью аппаратуры, если калькуляция покажет, что приобретение
дорогого аппарата, необходимого для производства, невыгодно вследствие
небольших количеств производимого продукта. С другой стороны, сле-
дует принимать во внимание использование получаемых при производ-
стве побочных продуктов, которые иногда вообще не находят применения
(например, примулни), а иногда являются желательными или необходи-
мыми для другого производства (например, сульфат хрома при произ-
водстве антрахинона). При обсуждении способа производства следует
всегда иметь в виду аппараты, в которых проводится процесс. На произ-
водстве в противоположность научно-исследовательской лаборатории
невозможно или возможно лишь в исключительных случаях работать
со стеклянной аппаратурой. Нужно также иметь в виду, что применяемые
химикалии часто сильно действуют па материал аппаратов, 'так что амор-
тизация последних становится существенным фактором.
Промежуточные Продукты, которые применяются для изготовления
искусственных органических красителей, принадлежат большей частью
к ароматическому ряду. В иих, кроме метильных групп, наиболее часто
встречаются в качестве заместителей прежде всего галоиды (особенно
хлор), нитро-, амино-, окси-, алкокси-, сульфо- и карбоксильные
группы, а также альдегидные и кетонные группы, причем последние
могут быть в хиноидной форме. Все эти и многие другие реже приме-
няемые заместители могут быть введены в молекулу поодиночке или
в сочетании с нескЪлькими другими. Замещения могут происходить в раз-
личной последовательности и самым различным образом, так что воз-
можности здесь безграничны. На практике обычно1 применяются анало-
гичные процессы, так что химик, знающий основные положения этих
процессов и овладевший методами, может сразу определить, как проще
всего получить требуемое соединение. Всегда надо учитывать, чтопочти
во всех случаях используются Стехиометрические количества хнмика-
лиев и что лишь в редких случаях оказывается необходимым применять
количества, большие или меныпие, чем те. которые соответствуют хими-
ческому уравнению. Для достижения надлежащего перемешивания
во время реакции часто приходится использовать растворители. Следует
также отметить, что интенсивное перемешивание часто является необхо-
димым, чтобы реакция прошла удовлетворительно. Часто решающую
роль играет соблюдение определенной температуры реакции во избежа-
ние образования изомеров, нежелательных в данном случае.
В виду изложенного часто совершенно необязательно давать
для получения какого-либо продукта точные «рецепты»; очень часто
достаточно указать химику, по какому принципу вещество получается,
и он иа основании собственного опыта сможет тотчас составить удовлет-
ворительный рецепт.
Поэтому в прилагаемых к настоящей книге таблицах соединения
Различные процессы химии промежуточных продуктов 13
ство рассматривается как исходное, из которого получаются его произ-
водные. Такая форма изображения имеет то преимущество, что начи-
нающий химик получает ясное представление об уравнениях реакций,
а специалист непосредственно на основании этих таблиц сможет уста-
новить, как получить нужное соединение из имеющегося исходного.
Чтобы сделать эти таблицы более удобными для пользования, перед
.большинством соединений даются ссылки на приведенные в книге мето-
дики их получения или на те места книги, где рассматриваются спе-
циальная литература, патенты, «научные данные и др. Конечно, н е-
возможно в сравнительно небольшом лабораторном
руководстве предусмотреть все возможност я. Есте-
ственно поэтому, что таблицы не являются полными
и характеризуют лишь общие направления. Настоящая
книга рассчитана на лиц, не имеющих опыта, и предназначается в каче-
стве учебного пособия в химико-технической лаборатории. Таблицами
может воспользоваться также и специалист, если вопрос касается обла-
сти, не являющейся его специальностью.
В тексте материал также расположен в генетической последователь-
ности. Сначала рассматриваются важнейшие процессы: хлорирование,
нитрование и восстановление, сульфирование н щелочное плавление
в применении к простейшему исходному веществу—-бензолу. Затем из
синтезированных таким Путем промежуточных продуктов с помощью
цикла реакций, которые располагаются в определенной последователь-
ности и могут считаться типичными, получаются .все более сложйые
производные. Аналогично рассматриваются производные гомологов бен-
зола, затем — нафталина и, наконец, антрахинона. Это помогает изу-
чающему ознакомиться, с одной стороны, со всем ходом получения
основных веществ и, с другой стороны, с возможностью дальнейшей
переработки этих веществ в более сложные промежуточные продукты;
начинающий химик таким образом сможет рассматривать каждый про-
цесс не только сам по себе, но в связи с целым комплексом методов
синтеза. .
Различные процессы химии промежуточных продуктов
В настоящем разделе рассматриваются в общем виде некоторые
важнейшие процессы, причем для отдельных соединений приведены осо-
бые примечания. Касаться всех вариантов нельзя, так как это завело бы
слишком далеко,
1. Сульфирование
Под сульфированием (сульфурация, сульфурирование, сульфонация)
понимают введение в молекулу группы SO3H. Этот процесс применяется
потому, что продукты в виде свободных сульфокислот или, что очень часто
бывает, в виде их солей обладают большой растворимостью в воде.
"Обычно имеют дело с дешевыми натриевыми солями.
Сульфирование производится:
1) обычной концентрированной серной кислотой (уд. .вес 1,84);
2) 100%-ной серной кислотой (моногидратом);
3) дымящей серной кислотой (олеумом), с содержанием от 5 до
70% SO3;
4) хлор сульфо новой кислотой в растворителе нлн без него;
5) запеканием (сухой нагрев) кислого сульфата амина, иногда в ва-
кууме.
Реже сульфог уппа вво ится косвенными путями, а именно:
14
I. Промежуточные продукты
6) заменой атома галоида на сульфогруппу действием сульфита
натрия;
7) действием бисульфита иа нитросоединение, на хинон или на ок-
сим хиноиа (нитрозофенол);
8) окислением сульфиновой кислоты, меркаптана или дисульфида.
Иногда можно также
9) с помощью бисульфитного производного формальдегида вводить
CH2SO3H-rpynny.
2. Нитрование
Под нитрованием (нитрация) понимают введение в молекулу нитро-
группы — NOs. Нитрование проводят;
1) разбавленной нли концентрированной азотной кислотой;
2) нитрующей смесью, т. е. смесью азотной и серной кислот, которая
иногда содержит небольшое количество воды;
3) вещество, подлежащее нитрованию, можно сначала сульфировать
и затем нитровать сульфокислоту; при этом сульфогруппа отщепляется
и заменяется на ннтрогруппу (пикриновая кислота, см. стр. 136);
4) окисляют разбавленной азотной кислотой полученное вначале
нитрозосоединение (тропеолин, см. стр. 245);
5) обрабатывают диазосоединение горячей разбавленной азотной
кислотой; при этом одновременно с нитрогруппой вводится и оксигруппа
(например, получение нитрокрезола из «-толуидина).
При введении двух нитрогрупп нитрогруппы вступают >в основном
в метя-положение друг к другу. Однако реакция никогда не протекает
лишь в одном направлении (см., например, м-динитробензол, стр. ЛОЗ).
3. Восстановление
Из реакций восстановления химик, работающий в области краси-
телей, чаще всего имеет дело с переведением нитросоединения в амин.
Однако большую роль в химии красителей играют также и многие дру-
гие реакции восстановления.
Восстанавливают:
1) водой и железом с добавлением незначительных количеств кис-
лоты (соляной, серной или уксусной, а иногда и смеси их). При этом
«нейтральном» способе восстановления Вешана — Бриммейера можно
применять лишь определенный вид железа— серый чугун; его всегда
следует испытывать на пригодность в качестве восстанавливающего
средства. Другие сорта железа в большинстве случаев непригодны
(многие примеры такого рода приводятся ниже);
2) железом и таким количеством кислоты, что все используемое для
восстановления железо переходит ,в раствор в виде закисной соли,
В этом случае можно применять всякое малоуглеродистое железо, так
как оно не дает загрязнений в виде мелких кусочков графита; подходят,
например, железные гвозди, жесть, сталь и т. д. (см. получение Аш-кис-
лоты, стр. 189);
3) сероводородом и его солями (в настоящей книге приведено1 много
Примеров);
4) цинковой пылью и кислотой или щелочью; реже — оловом;
5) гидросульфитом (см., например, получение п-аминосалициловой
кислоты из феннлазоСалициловой, стр. 142);
6) электролитическим путем, например получение гидразобензола из
нитробензола, н-аминофенола из нитробензола (с одновременно идущей
перегруппировкой промежуточно образующегося фенил гидрокси ламииа);
Различные процессы химии промежуточных продуктов 1;
7) с помощью Fe(OH)2 (проводится редко);
8) порошком алюминии, например получение бензантрона, хиииза-
рина из пурпурина (применяется редко);
9) водородом в присутствии катализаторов;
10) действием SO2 (при этом часто идет и сульфирование).
4. Введение оксигруппы
Оксигруппа вводится в молекулу различными путями:
1) .'плавлением сульфокислоты с едким натром. Этот метод часто
называют «кали-плавление»; это название объясняется тем, что прежде
применяли почти исключительно едкое кали. В настоящее время в ог-
gOMHOM большинстве случаев используется более дешевый едкий натр,
зависимости от обстоятельств применяется разбавленная илн концен-
трированная гидроокись. При работе с разбавленным NaOH (30—60%),
когда температура реакции выше температуры кипения, применяют авто-
клав. Давление совершенно не влияет на такие процессы, а является
лишь нежелательным следствием температуры.
Имеются два в-ида щелочного плавления:
а) с добавкой окислителя, когда наряду с заменой сульфогруппы
ОН-группой в молекулу вступает еще одна оксигруппа (см. Ализ'арин,
стр. 281);
б) с добавкой гидроокиси щелочноземельного металла; при этом
образующийся во время реакции нерастворимый сульфит щелочно-
земельного металла выпадает в осадок и не восстанавливает конечного
продукта реакции (ряд антрахинона, см. 1,5- и 1,8-диоксиантрахиноны,
стр. 213);
2) заменой на ОН-группу «подвижного» галонда, например получе-
ние динитрофепола из динитрохлорбензола и Др. (см. в разделе «Серни-
стый черный», стр. 301);
3) кипячением соли диазония, иногда в присутствии соли меди (гид-
рохинон из п-аминофенола, гваякол из о-анизидина);
4) обработкой амина бисульфитом и омылением образующегося
промежуточного соединения (метод Бухерера, стр. 162);
* 5) нагреванием амина с кислотами или щелочами под давлением;
6) заменой находящихся в орто-положении в солях диазония суль-
фогруппы, атома хлора или нитрогруппы на ОН-группу при действии
слабой щелочи.
5. Введение амино- и алкоксигрупп
Во многих случаях аминогруппу и алкоксигруппу удается ввести
в ароматическую молекулу методами, аналогичными применяемым при
введении окснгруппы.
Так, например, нитрохлорбензол и соединения антрахинонового ряда
можно во многих случаях перевести в амино- и алкокси производные..
Амино- и алкоксисоединения можно также получить заменой сульфо-
группы и оксигруппы; последняя замещается при нагревании с аммиаком
илн, лучше, по методу, предложенному Бухерером — нагреванием
с сульфитом аммония. »
Примером замены сульфогруппы на аминогруппу может служить
получение аминоантрахинона. Однако 2-аминоантрахииои в последнее
время обычно, синтезируют из 2-хлор антрахинона, так как при этом
промежуточные продукты
получается более чистый продукт. С другой стороны, о- и п-нитроапилины
теперь получают почти исключительно из соответствующих хлориитро-
производных (СМ. Стр. 86).
Алкильные эфиры фенола, нафтола и оксиантрахинона в очень мно-
гих случаях можно получить, обрабатывая галоидопроизводные алкоголя-
том при высокой температуре (под давлением; см. получение анизолов,
стр. 91). Можно, конечно, также и алкилировать фенолы (см. стр. 134),
В каждом отдельном случае применяют наиболее дешевый метод. В ка-
честве энергичных алкилирующих средств в известных случаях приме-
няют диалкйлсульфаты, например в случае ди метоксид ибенз а итрона
(кубовый ярко-зеленый С).
6. Методы окисления
Разработано много методов окисления органических соединений.
Окисляют:
1) кислородом воздуха, очень часто с применением катализатора.
В качестве примера может служить получение фталевого ангидрида из
нафталина с применением воздуха и окислов ванадия (метод Воля—
Данцига; см. стр. 153), а также аналогичное производство антрахинона
из антрацена. При этом можно исходить из ие совсем чистого антрацена;
2) хромовой кислотой. Этот метод применяют при получении многих
красителей гетероциклического ряда (метиленовый голубой). Окисление
проводят в присутствии щавелевой кислоты;
3) перекисью марганца или так называемым «марганцовым шла-
мом», который представляет собой закись-окись марганца (см. Ксиле-
иовый голубой Фау, кислотный ярко-голубой 3, стр. 269); ' .
4) гипохлоритом натрия (см. Дииитростильбендисульфокислота,
стр. 150);
5) азотиой кислотой (редко);
6) двуокисью свинца (в случае трифенил метановых красителей);
7) питрозилсерпой кислотой (аурин по Заидмейеру);
8) хлориым железом (в случае некоторых трифенилметановых кра-
сителей, иапрнмер гельве.ция голубого);
9) путем хлорировании в боковой цели (в случае толуола и ксилола)
и последующим омылением полученного хлор производного. Например;
толуол —> хлористый бензил —бсизи.тоиый спирт
или
толуол —> хлористый бензилиден —> бензальдегид;
10) действием избыточного количества одного из реагирующих ве-
ществ (см. Галламииовый синий). Аналогичных примеров много (Мель-
дола голубой).
7. Введение галоида
Галоид, обычно хлор или бром (редко йод или фтор*), вводят
в большинстве случаев, действуя, например, элементарным хлором на
вещество, в котором хотят произвести замещение. Часто оказывается
необходимым применять катализатор, так как иначе происходит присо-
единение .хлора, а ие замещение водорода. В качестве катализатора чаще
всего применяют железо (хлориое железо), иногда железо с добавкой
йода, реже — соединения сурьмы, серы или фосфора.
* См„ например, герм., пат. 551882 (Frdl., XIX, 1625), и герм. пат. 575593 (Frdl..
XX, 475).
О практической работе в органической технической лаборатории
17
Вместо элементарного мора (брома) в некоторых случаях приме-
няют гипохлорит натрия в присутствии минеральных кислот (хлорирова-
ние ацет-о-толуидина). Хлор в момент образования реагирует в таких
случаях очень энергично и без нежелательных побочных реакций.
Если введение галоида по какой-либо причине не удается осуще-
ствить, действуя хлором, или если хлор не вступает в то место молекулы,
где хотят произвести замещение, то прибегают (редко!) к реакции Занд-
мейера (см. пример иа стр. 145).
Иногда, пользуясь специальным приемом, можно избежать прове-
дения реакции Запдмейера (которая довольно дорога и сложна для
выполнения). Примером является получение о-хлортолуола из п-толуол-
сульфокислоты (см. стр. 147 и патент*).
Фенолы можно также галоидировать иным способом. При этом из
фенолята и гипохлорита получается главным образом (75—80%) о-хлор-
фенол, а нз фенола и хлористого сульфурила (SO2CI2) п-хло.рфенол
(около 78%) **.
Особым случаем является хлорирование с заменой сульфогруплы на
галоид. Эта реакция особенно важна для химии антрахинона, ио из-
вестна также и в ряду бензола (см. стр. 215).
Область, в которой работает химик по промежуточным продуктам,
кои е чно, ие исчершывается всем и перечиел енными реакциями; эти реакции
являются лишь главнейшими. В настоящей книге будут рассмотрены еще
некоторые другие, менее важные реакции, как например алкилиро-
вание аминов, введение альдегидной группы (—СНО)
по Зандмейеру, фепилирование. Хотя и эти реакции иногда при-
меняются в широком масштабе (например, получение алкил- и бензил-
анилинов и толуидинов), оии все же значительно менее важны, чем упо-
мянутые выше процессы.
При проведении любого технического процесса
химик должен добиваться наибольших выходов с наи-
меньшими затратами. Методы, с успехом применяемые в науч-
ном исследовании, часто оказываются негодными в технике. Следует
также Припять во внимание то обстоятельство, что производимые про-
межуточные продукты должны быть возможно более чистыми, так как
незначительные отклонения в чистоте очень часто вызывают большие
потери конечного продукта в производстве красителей. Поэтому посту-
пающие в продажу так называемые промежуточные продукты часто
являются химически чистыми, и требования, предъявляемые к ним за
последние годы, значительно выросли,
О практической работе в органической химико-технической
лаборатории
Общие замечания
Особое внимание начинающего! работать в органической химико-
технической лаборатории -следует обращать иа то, что не только в ана-
литической и чисто научной, ио точно так же и в технической лаборато-
рии первым залогом успеха является абсолютная чистота в работе.
Это касается как используемых аппаратов и посуды, так и применяемых
веществ. Содержать посуду в чистоте здесь часто труднее, чем в анали-
тической лаборатории, во-первые, потому, что в этом случае значительно
чаще имеют дело с сильно окрашенными и, к сожалению, нередко
* Герм. пат. 294638 (Frdl., ХП, 908).
** См. стр. 132 (Frdl., VII, 90).
л fipuuyKTW
смолистыми веществами, и потому, во-вторых, что часто используется
непрозрачная реакционная посуда, чистоту которой нельзя так легко
установить; следовательно, се необходимо особенно тщательно контроли-
ровать. Именно поэтому из металлических аппаратов отдают, если
возможно, предпочтение тем, которые открываются, разбираются и во
всех своих частях легко доступны. В тех случаях, когда это невозможно,
особое внимание следует обращать па то, чтобы посуда основатель по очи-
щалась тотчас после ее использования. Как можно более быструю
очистку использованной посуды и аппаратов следует рекомендовать и
в других случаях. Так, очень часто удастся достигнуть безупречной чи-
стоты, применяя горячую воду, иногда с добавлением незначительных ко-
личеств кислоты или щелочи. Остатки же, которые высохли и покрылись
коркой, удаляются значительно' труднее, часто лишь с применением силь-
нодействующих средств, таких, например, как концентрированная серная
кислота и хромовая кислота. Во многих случаях весьма полезны приме-
няемые в домашнем обиходе порошки для очистки, которые удаляют пре-
имущественно маслянистые и смолообразные загрязнения.
Если химик очищает свои аппараты не сам, а предоставляет их очи-
щать под своим непосредственным наблюдением, он должен обращать
особое внимание на то, чтобы для очистки не давали посуду, содержащую
остатки легковоспламеняющихся, взрывчатых или сильнодействующих
ядовитых веществ (например, эфир, щелочные металлы, амид натрия, ди-
метилсульфат, фосген и т. д.). При несоблюдении этого правила часто
происходили несчастные случаи, так как персонал, производящий очистку,
может не предвидеть опасности. Для такого рода остатков также нельзя
использовать лабораторные раковины. Остатки надо обезвреживать по-
возможности иа открытом воздухе.
Рабочий стол должен быть как можно более свободным и также
содержаться в чистоте. При соблюдении этих условий можно, напри-
мер, вновь собрать рассыпанные вещества и таким образом спасти опыт,
который иначе надо было бы проводить заново. Для сохранения покры-
тия стола при опытах, которые требуют продолжительного сильного на-
гревания, следует ставить горелку не прямо на стол, а па огнеупорную
и изолирующую подкладку (асбестовые или этернитовые пластинки).
Не находящиеся в работе аппараты нельзя оставлять на рабочем месте.
Там, где Это возможно, целесообразно более громоздкую аппаратуру не
устанавливать на рабочем месте, а освободить его для опытов, проводи-
мых в стеклянной посуде; сюда же помещают приспособления для титро-
вании, приборы для определения температуры плавления и т. п. Все
необходимое для взятия контрольных проб (чистая и су-
хая стеклянная посуда, пробирки, стеклянные палочки и трубки, ма-
ленькие воронки и фильтры, наиболее употребительные реактивы и раз-
личная индикаторпаи бумага) всегда должно быть готовым
к ра боте и стоять на рабочем месте.
Так же важно, как и содержание в чистоте посуды, применение ч и-
сты х материалов. В аналитической и исследовательских лаборато-
риях, где, как правило, имеют дело только с небольшими количествами
веществ и вопрос о цепе ие играет никакой роли, применение химически
чистых химикалиев является чем-то само собой разумеющимся. Однако
применение последних в технике часто оказывается невозможным из-за
дороговизны. Необходимо особо указать, что в технической лаборатории
в целях приближения к условиям производства необходимо применять
технические исходные материалы. Однако следовало бы взять за правило
при получении нового продукта и при разработке нового спо-
соба синтеза сначала применять возможно более чистые вещества, не-
О практической работе в органической технической лаборатории 1У
взирая на их стоимость и техническую доступность. Только после этого
следует проверить, можно ли достигнуть того же результата, используя
технические материалы.
Кроме, того, нужно сделать следующие отдельные замечания. Приме-
няемые в органической промышленности неорганические хими-
калии в большинстве случаев пригодны в том виде, .в каком их выпу-
скает промышленность. Имеются, однако, важные исключения. Такг
азотная кислота, применяемая при нитровании первичных аминов в рас-
творе серной кислоты, нс должна содержать окислор азота (см. стр. 149);
проведению некоторых реакций диазотирования может мешать содержа-
щаяся в технической соляной кислоте серная кислота, если последняя
образует с применяемым амином труднорастворимый сульфат; при ще-
лочном плавлении наличие хлората в вводимой в реакцию едкой щелочи
может привести в нежелательному окислению и даже взрыву и т. д. Во*
всех подобных случаях необходимо обязательно применять продукты, ко-
торые свободны от соответствующих вредных примесей. Надо также
иметь в виду, что сильно гигроскопичные (например, олеум, хлор сульфо-
новая кислота), легко окисляемые (например, сульфит и бисульфит) или
неустойчивые (например, растворы гипохлорита) продукты часто после
длительного хранения не содержат начального количества активного ве-
щества; такие продукты перед употреблением надо непременно титровать,
В лаборатории целесообразно применять д и с т и л л и р ов а и и у ю
воду также и для технических работ, так как это позволит избежать
образования мути вследствие выпадения в осадок кальциевых солей. Но
прежде чем какой-нибудь способ перенести в производство, следует про-
верить, нс вредит ли применение обычной воды. С другой стороны, нали-
чие воды в качестве загрязнения в органических жидкостях может ока-
заться очень вредным (см., .например, хлорирование нитробензола,
стр. 107). В таких случаях требуется тщательное удаление воды, которое
для высококилящих жидкостей целесообразнее осуществлять путем пере-
гонки с отбрасыванием первого погона.
Загрязнения в органических исходных продуктах яв-
ляются, в общем, гораздо более вредными, чем в случае неорганических
химика лиев. Здесь часто наличие совсем незначительных количеств со-
провождающих веществ оказывается достаточным, чтобы заметно ухуд-
шить выходы и степень чистоты конечного продукта. Поэтому органиче-
скаи промышленность стремится изготовлять промежуточные продукты
в возможно более чистом виде, ив самое последнее времи в этой области
достигнуты уже значительные успехи, так что сейчас многие органические
промежуточные продукты можно, иметь в почти химически чистом виде.
Это касается особенно тех веществ, которые очищают перегонкой (в част-
ности, в вакууме). Напротив, все продукты, которые нужно выделять вы-
саливанием, неизбежно содержат неорганические соли. Однако содер-
жание с о л е й не вредит в подавляющем 'большинстве случаев. Нужно,
конечно, только принимать во внимание наличие этих солей при расчете
необходимого количества вещества. Во всех исходных материалах, содер-
жащих соли, необходимо определять содержание чистого вещества.
Содержание первичных аминов можно определить титрованием раство-
ром нитрита, а веществ, способных сочетаться с диазосоединеииямн, —
титрованием диазососдинением. При определении содержания других
продуктов должны использоваться методы, подходящие для каждого от-
дельного случая (см. аналитическую часть).
Начинающего химика часто смущает более или менее сильная окра-
ска тех технических продуктов, которые в совершенно чистом виде
должны быть бесцветными. Эта ок аска, как п авило, не имеет значения
20 I. Промежуточные продукты
для технического применения. Очистка может понадобиться только тогда,
когда окраска очень интенсивна и свидетельствует о наступившем разло-
жении или окислении. Но гораздо .вреднее наличие изомерных или род-
ственных соединений, которых нельзя обнаружить по -внешнему виду
(например, присутствие моносульфокислоты в дисульфокислотах или на-
оборот). Так, например, jw-фенилендиамин, который содержит лишь не-
значительный процент орто- и пара-изомеров, при применении в каче-
стве компонента в синтезе азокрасителей дает значительно худшие вы-
ходы -и более загрязненные красители, чем чистое .мега-соединение.
л<-Фенилецдиамии, загрязненный примесью изомеров, кроме того, меиее
стоек при хранении (см. стр. 104 и 107). Методы испытания органических
исходных материалов на чистоту и их очистка проводятся, как описано
ниже в разделе об очистке и проверке чистоты конечных продуктов
(см. стр. 43).
Количественные соотношения, взвешивания
В органической технической лаборатории почти всегда работают
с молярными количествами, что весьма упрощает все расчеты.
Наиболее употребительной загрузкой является ’/ю, а в случае большого
молекулярного веса */ео граммолекулы. Эти количества достаточны, на-
пример, для предварительного испытания красителей и а красящие свой-
ства и позволяют с удовлетворительной точностью определить выходы
(взвешивание следует производить с точностью до 0,1 г иа обычных тех-
нических весах). В качественных предварительных опытах можно исполь-
зовать и меньшие количества (0,01—0,02 моля). При синтезе исходных
веществ или промежуточных продуктов, которые будут затем применять-
ся во многих опытах, обычно исходят из 1 или 2 молей. В этом случае
достаточно взвешивать с точностью до 1 а. Химик, работающий в техни-
ческой лаборатории, должен привыкнуть с самого начала взвешивать или
измерять все применяемые вещества (например, при нейтрализациях и
др. реакциях), а ие просто приливать их из сосуда, в котором они хра-
нятся. Количества веществ, введенных в реакцию, следует записывать
в лабораторном журнале; позднее, когда понадобится повторить опыт,
эти данные очень пригодятся.
Течению многих реакций благоприятствует наличие избытка одного
из реагентов. Но весьма часто обязательно требуется применение точно
стехиометрических количеств. В таких случаях удобно для проведения
серийных опытов иметь необходимые реагенты в виде растворов, содер-
жащих точно 1 моль вещества,_ например, в 100, 200, 500 или 1000 мл.
Если для опытов требуется особая аппаратура, имеющаяся в наличии
лишь одного размера, то количества используемых реагентов должны,
конечно, соответствовать ее емкости.
При применении исходных веществ, содержащих кристаллизационную
воду, соли, влагу или какие-либо загрязнения, -следует определять в них
содержание чистого вещества. Некоторые из применяемых для этой цели
методов описаны в аналитической части. Результаты таких определений
целесообразно выражать ие в процентах, а количеством (Л1) сырого про-
дукта, в котором содержится 1 моль чистого вещества. Если, например,
для технической Аш-кислоты М равно 382, то это значит, что 382 г этого
технического продукта содержат 1 моль Аш-кнслоты. Таким образом, без
особого расчета для опыта, требующего ’/ю моля, надо взять 38,2 г техни-
ческого продукта, независимо от того, является ли ои свободной кислотой,
какой-нибудь солью, содержит или ие содержит кристаллизационную
воду.
О практической работе в органической технической лаборатории
21
Лабораторный журнал, записи
Химик, работающий в технической лаборатории, должен обязательно
тсчг[О описывать все проводимые им опыты. Поэтому студент должен при-
учить себя с самого начала вносить в свою лабораторную тетрадь
результаты всех выполненных работ и датировать свои записи. При этом
не следует буквально переписывать применяемые методики, но нужно
описывать работу так, как ода действительно производилась, чтобы
по этим записям можно было в любое время точно воспроизвести опыт,
не вводя отдельных новых условий. Сюда относится прежде всего точное
указание примененных количеств веществ, температуры и продолжитель-
ности реакции, а также размеров и характера использованной аппара-
туры. Надо также записывать все наблюдения, сделанные при проведении
реакции, например возникновение или исчезновение окраски или осадка,
выделение газа, самопроизвольное повышение температуры и т. д. Осо-
бенное внимание при описании опыта следует обращать иа то, как конт-
ролировалось течение реакции, как определялся ее конец, как устанавли-
валась чистота продукта, и т. д.
После окончания работы составляется отчет, в котором кратко сум-
мируются полученные результаты и описывается способ, оказавшийся
оптимальным. Это' описание должно быть настолько подробным, чтобы,
пользуясь им, каждый химик смог повторить опыт точно таким же обра- -
зом.
Аппаратура
Раньше в учебных химико-технических лабораториях стремились
приучить студентов к пользованию возможно более примитивными, со-
стоящими из простейших составных частей аппаратами. Теперь считают,
что надо пользоваться ие простейшими аппаратами, а наиболее целесооб-
разными, дающими возможность экономить время и труд. При одинако-
вой производительности предпочитают, конечно, простейшую аппаратуру.
Следует избегать ненужных усложнений, преимущества которых ие оправ-
дывают необходимых издержек. Но, как и прежде, весьма желательно,
чтобы химик, работающий в технической лаборатории, мог пользоваться
в случае необходимости также и простейшими средствами. Например,
исключительно ценно, если ои обладает некоторыми навыками в стекло-
дувном деле и может сам изготовить для себя простой прибор из несколь-
ких стеклянных трубок,
Мешалки
Во многих реакциях технической органической химии для достижения
хороших результатов обязательно требуется постоянное энергичное пере-
мешивание реакционной смеси. Оно особенно необходимо тогда, когда
одно или -несколько реагирующих веществ нерастворимо в реакционной
жидкости и суспендировано в ней в жидком или твердом виде, Даже
когда во время реакции образуется осадок, перемешивание также даето
преимущества, так как способствует кристаллизации и позволяет избе-г
жать образования сильно пересыщенных растворов, которые затем сразу
затвердевают. Хорошее перемешивание гомогенных жидкостей необхо-
димо в тех случаях, когда один из реагентов прибавляется медленно, для
того чтобы он распределился равномерно и не смог оказаться местами
в избытке. Это особенно важно при сильно экзотермических реакциях, на-
пример при нитрованиях, когда местный избыток реагента может при-
вести к слишком эне гвчпой реакции и даже взрыву.
22 I- Промежуточные продукты
Поскольку органические реакции обычно протекают во времени, пе-
ремешивание от руки так, как оно часто применяется в аналитической ла-
боратории, как правило, непригодно. Следует пользоваться механиче-
скими прибора ми для перемешивания, которые могут при-
водиться в движение с помощью небольшой водяной турбины (давление
воды должно составлять ие меиее 2 атм). Если требуется значительно
большее усилие, например для быстро вращающихся мешалок в аппара-
тах для восстановления или для вращающихся автоклавов, то применяют
электромотор; выгоднее всего использовать электромотор с трансмиссией,
от которого можно было бы приводить в движение одновременно различ-
ные аппараты. В последние годы появились небольшие электромоторы,
которые можно прикреплять непосредственно к мешалке.
Тип самой мешалки зависит прежде всего от характера реакционной
смеси, а также от формы и материала сосуда, в котором мешалка должна
работать.
Конечно, удобнее всего применять открытые сосуды, например хими-
ческие стаканы, эмалированные сосуды, чашки, открытые котлы и т. д.
Но их следует употреблять только тогда, когда можно ие бояться значи-
тельных потерь вещества или растворителя в результате испарения и
когда ие предвидится выделения паров или газов, которые вследствие
ядовитости, горючести, дурного запаха и т. д. требуется поглощать или
отводить в тягу. При этом надо учитывать, что очень многие из промежу-
точных продуктов, применяемых в органической промышленности, как
например нитробензол, анилин, динитробеизол, дицитрохлорбензол, иитро-
аиилин, фенол и т. д., являются сильными ядами, которые могут действо-
вать на организм ие только при попадании в желудок, но также и через
кожу и в виде паров или пыли —через легкие. Открытые сосуды не
подходят также тогда, когда вследствие вредного действия кислорода
воздуха или содержащихся в ием влаги или углекислоты следует избегать
доступа воздуха.
Когда по одной из приведенных выше причин открытые сосуды при-
менять нельзя, обычно пользуются котлами, имеющими крышки, или
круглодониыми колбами (чаще всего трех- или п яти гордыми), снабжен-
ными обратным холодильником или же вертикально поставленной трубкой
для конденсации паров нли связанными с поглощающим сосудом для за-
держания ядовитых н неприятно пахнущих газов. Если выделяются лишь
незначительные количества газов, то достаточно хорошо действующей
тяги. Кроме тех случаев, когда приходится работать под давлением, за-
крытая аппаратура должна, конечно, иметь отверстие для выравнивания
давления. Это отверстие можно в случае необходимости снабжать труб-
ками с хлористым кальцием или натронной известью для поглощения
влаги и углекислоты воздуха. Часто оказывается достаточным лишь за-
труднить доступ воздуха при помощи вытянутого из стекла капилляра или
тампона из ваты.
Необходимо сделать несколько замечаний относительно мате-
риала, из которого изготовляются реакционные со-
суды. Для проведения реакций восстановления железом в ’слабокислом
растворе по Бешаиу, для плавления с едкими щелочами и полисульфи-
дами и т. п., а также для работы при высоком давлении, когда имеют
дело не со слишком малыми количествами вещества (в последнем случае
пользуются запаянными трубками), применяют только металлические со-
суды. Кроме того, следует применять металлическую аппаратуру в тех
случаях, когда ее емкость превышает 6—8 л. Во всех других случаях
можно, как правило, использовать сосуды из стекла; это стало возмож-
ным особенно с тех пор, как появилось стекло пи екс и подобное еми
О практической работе в органической технической лаборатории 23
сорта, устойчивые к колебаниям температуры, а также достаточно проч-
ные в механическом отношении и стойкие — в химическом. Из такого ма-
териала можно изготовлять, например, достаточно прочные трех- и пяти-
горлые колбы и сложные приборы.
Применение прозрачной стеклянной аппаратуры имеет в лаборатории
большое преимущество, состоящее в том, что можно точно наблюдать все
течение реакции. Именно поэтому рекомендуется применять по возмож-
ности стеклянные сосуды, когда реакция проводится впервые. С другой
стороны, для химика, работающего в технической лаборатории, очень
важно привыкнуть иметь дело также и с непрозрачными аппаратами, мо-
делирующими производственные, и контролировать течение реакции при
помощи отбора проб, как это бывает иа производстве.
Р и с, 1. Фарфоровые мешалки, приспособленные -для подвешивания на загнутую
стеклянную палочку.
В лабораториях высших школ принято использовать в качестве м е-
ш а л к и несколько раз согнутую стеклянную палочку
’(см,, например, рис, 19 на стр, 94 и рис. 20 па стр. 98). Такие мешалки
можно легко изготовить самому; их удобно применять в случае легкопод-
вижных жидкостей. Однако оии оказываются слишком малоэффектив-
ными, когда имеют дело с кашицеобразными смесями. Для перемешива-
ния таких смесей следует применять плоские мешалки.
В технической лаборатории для открытых сосудов часто применяют
прямоугольные пластинки длиной около 40 см и шириной
около 5 см, изготовленные из толстого или, лучше, ребристого стекла,
которые укрепляют в держателе, имеющем форму вилки, при помощи
двух зажимпых винтов. Их легко содержать в чистоте; они, кроме того,
обеспечивают очень энергичное перемешивание и позволяют регулировать
в широких пределах его интенсивность при постоянном числе оборотов
путем изменения наклона пластинки. Недостатком таких мешалок яв-
ляется их ломкость.
Менее хрупкими и очень ^удобными являются поступившие в послед-
нее время в продажу лопастнцр мешалки из фарфора, которые
могут просто навешиваться ца загнутую стеклянную палочку и имеют
различную величину и форму (см. рис. 1, а — в).
Если требуется поднять со дна тяжелый осадок, например железный
ПППйШаК И ПЫ 1 UUbTimm Лг.гТТГ. mrs v^tmr iv П U'ji г ТТ l-I.O т-г.п .,r,w r,
24 Z. Промежуточные продукты
меняя мешалку, имеющую форму пропеллера (см, рис. 17,
стр. 88); при этом мешалка должна доходить до самого дна сосуда и по
возможности целиком его касаться. Для этих же целей могут с успехом
применяться мешалки в форме якоря или лопатки.
В закрытых котлах, как правило, применяется подходящая мешалка,
которая обычно имеет форму якоря и касается дна и стенок котла (см.
рис. 36, стр. 298).
Труднее всего осуществлять энергичное перемешивание в стеклянных
колбах (круглодонных, трехгорлых и т, п.), так как они имеют сравни-
тельно узкое горло, которое мешает вводить достаточно широкие мешалки.
Для таких случаев сконструированы специальные мешалки, которые
позволяют осуществлять интенсивное перемешивание также и при неболь-
шом диаметре; к ним относятся пропеллерные мешалки
(рис. 6, а, стр. 66) .центробежные мешалки с выдвигающимися
при вращении лопастями, мешалка Витта в виде колокола и т, д.
Все указанные приспособления при достаточном числе оборотов переме-
шивают очень интенсивно легкоподвижные гомогенные жидкости. Они,
одиако, не работают в случае очень вязких и особенно кашицеобразных
смесей. Для перемешивания последних лучше всего применять простую
стеклянную лопатку (рис. 6, б, стр. 66), которая бы доходила
почти до стенки колбы. Такую лопатку можно приготовить самому, скру-
тив плотной спиралью конец стеклянной палочки, а потом расплющив ее.
Надо учитывать, что действительно интенсивное перемешивание воз-
можно лишь, когда сосуд не слишком переполнен. В открытых сосудах
большого диаметра, именно в чашках и плоских котлах, жидкость при пе-
ремешивании легко приходит в волнообразное движение и может выплес-
нуться через край сосуда. В этих случаях надо устанавливать «вол по-
рез», в качестве которого в лабораторных условиях можно применять
толстую стеклянную палочку, установленную вертикально недалеко от
края сосуда.
Эффективное перемешивание даже и кашицеобразных реакционных
смесей может быть достигнуто не только вращением, ио также и очень
быстрым коротким движением вперед и назад (вибрацией) мешалкн,
имеющей форму шайбы и приводящейся в движение электромагнитом.
Такое приспособление, имеющееся в продаже под названием «виброме-
шалка», может быть установлено герметически без сальника и поэтому
особенно подходит для реакций, которые ведутся при повышенном или по-
ниженном давлении (см. рис. 31, а, стр. 170).
О конструкциях и применении автоклавов см. специальный раз-
дел, стр. 312 и сл.
Фильтрование
В аналитической лаборатории, как правило, приходится отфильтро-
вывать практически нерастворимые осадки, которые можно промывать
любыми количествами жидкости; органические же продукты в большин-
стве случаев более или менее легко растворимы в применяемых раство-
рителях. Поэтому необходимо добиваться возможно более 'полного уда-
ления маточного раствора с использованием минимального
количества промывной жидкости. Для этой цели мало под-
ходит фильтрование под влиянием одной лишь силы тяжести. Вследствие
этого в органической , технологии пользуются преимущественно такими
методами фильтрования, при которых между верхней и иижнсй сторо-
нами фильтра имеется значительная разность давлений. Эта разность
давлений может быть получена созданием избыточного давления иад
фильтром или созданием разрежения под фильтром (отсасывание .
О практической работе в органической технической лаборатории 25’
На производстве предпочитают первый метод, осуществляемый чаще
всего в фильтр-прессах, где избыточное давление создается с по-
мощью сжатого воздуха (см. соответствующий раздел, стр, 310 и сл,).
Однако фильтр-пресс нельзя применять при работе в лабораторных
условиях.
Напротив, центрифуга для фильтрования, в которой
избыточное давление создается за счет центробежной силы, может при-
меняться также и для таких количеств вещества, с которыми обычно-
имеют дело в технической лаборатории. Центрифуга для фильтрования
состоит из цилиндрического закрытого внизу и открытого сверху металли-
ческого или фарфорового решетчатого сосуда («корзины»), поверхность
которого имеет отверстия; этот цилиндрический сосуд может приводиться
в быстрое вращение; стекающая из [[его при этом жидкость собирается
в кожухе, охватывающем цилнидр. Для фильтрования внутренняя сто-
рона цилиндрического сосуда выкладывается -соответствующей фильтрую-
щей тканью. При достаточно большом числе оборотов жидкость, содер-
жащаяся в осадке, удаляется из него настолько полно, что он становится
почти сухим. Но центрифуга для фильтрования годится лишь для крупно-
зернистых веществ. Тонкие осадки проходят через фильтр или спрессовы-
ваются в плотную массу, которая больше не пропускает жидкость.
Последнее особенно часто бывает, когда кристаллы осадка имеют форму
листочков. Поэтому число органических продуктов, которые могут с успе-
хом фильтроваться на центрифуге, пе слишком велико. Наиболее полез-
ной оказывается центрифуга при разделении изомеров, один из которых
застывает при охлаждении жидкой смеси (см,, например, о- и н-нитро-
хлорбепзол, стр. 84) *.
Другой метод создания разности давлений — отсасывание — приме-
няется на-производстве только в случае хорошо выкристаллизовываю-
щихся и достаточно грубых осадков. При той высоте слоя вещества иа
фильтре, которая встречается при работе в больших масштабах, тонкие
осадки слишком бы замедляли фильтрование. В лаборатории, где на
фильтре образуются слои осадка толщиной не более нескольких санти-
метров, фильтр для отсасывания, называемый иутч-филь-
т р о м (см. рис. 13, стр. 77), применяется гораздо чаще. Однако он непри-
годен также и в лабораторном масштабе, если имеют дело с вязкими,
смолистыми или студнеобразными продуктами, а также с мелкокристал-
лическими осадками, которые сначала проходят сквозь фильтр, а затем
забивают его. Затруднения вызывают также осадки с кристаллами
в форме листочков, которые легко образуют на фильтре непроницаемый
слой. В таких случаях можно облегчить фильтрование, применяя нутч-
фильтр таких размеров, чтобы слой вещества на нем был очень тонким;
при этом полезно также во'время выливания суспензии на фильтр совсем
слабо ее отсасывать и постепенно перемешивать, чтобы предотвратить по
возможности образование плотного осадка.
В качестве фильтрующего материала применяют для крупнозерни-
стых осадков фильтровальную ткань, а для мелкозернистых — фильтро-
вальную бумагу. В случае очень мелких осадков, способных проходить
сквозь фильтр, можно применять фильтровальную бумагу, сложенную
вдвое или втрое; иногда хорошие результаты дает плотная фильтроваль-
ная бумага. В больших воронках для отсасывания рекомендуется иа
” Центрифугу для фильтрования* не следует смешивать с часто применяемой
в биохимической лаборатории центрифугой для осаждения, при помощи которой тон-
чайшие осадки не фильтруются, а прижимаются к стенке сосуда и таким путем от-
деляются от жидкости.
26 I. Промежуточные продукты
фильтровальную бумагу класть еще фильтр из ткани, который предохра-
няет бумагу от разрывов и облегчает удаление осадка с фильтра. Бу-
мажные и тканевые фильтры должны быть точно такого размера, как дно
воронки для отсасывания, и ни в коем случае не загибаться па стенки
воронки, иначе легко образуются складки, по которым в фильтрат может
попасть часть осадка. При работе с сильнокислыми жидкостями, кото-
рые разъедают бумагу и хлопчатобумажную ткань, можно применять
шерстяной войлок. Более удобными и применимыми даже при работе
с концентрированной серной кислотой являются поступившие в последнее
время в продажу пористые стеклянные фильтры с различным разме-
ром пор.
Для того чтобы осуществить эффективное промывание на иутч-
фнльтре с затратой минимального количества промывной жидкости, очень
важно перед каждым приливанием ее возможно более полно удалять
маточный раствор из осадка на фильтре. Это достигается постоянным
дроблением и перемешиванием осадка (после того, как стечет большая
часть фильтрата) для более плотного сцепления отдельных его частиц,
а также систематическим приглаживанием шпателем, которое не дает
образовываться трещинам и разрывам в находящемся на фильтре слое
вещества. Наконец, отсасывающее действие можно усилить, сильно при-
жимая осадок па фильтре пестиком или перевернутой стеклянной проб-
кой. Только когда, несмотря на все эти меры, жидкость перестает сте-
кать, приливают при выключенном вакууме промывную жидкость и дают
ей получше всосаться в осадок, который впитывает сс, как губка. После
этого снова включают вакуум и отсасывают жидкость возможно более
полно, постоянно разглаживая и выжнмая осадок. Чередующиеся прили-
вание и отсасывание небольших количеств промывной жидкости с «пере-
крыванием» осадка повторяются аналогичным образом до тех пор, пока
загрязнения не будут удалены в желательном размере. Достижение
успеха в этой операции облегчается правильным выбором величины
воронки для отсасывания. Сильно отсосанный осадок не доджей запол-
нять воронку до самого верха, так как в этом случае не останется места
для приливания промывной жидкости. Кроме того, осадок не должен
образовывать очень топкого слоя, поскольку при этом потребуется слиш-
ком большое количество промывной жидкости и будет труднее избежать
образования разрывов и отверстий в осадке.
Мелкокристаллические осадки, проходящие, несмотря ца все предо-
сторожности, через нутч-фильтр или закупоривающие его, лучше всего
фильтровать через большой складчатый фильтр. Поскольку
в этом случае в осадке остается очень много жидкости, фильтр по окон-
чании промывания вместе с содержимым распластывают на пористой под-
кладке, например на толстом слое дешевой фильтровальной бумаги (рас-
тительная бумага),-н распределяют осадок по возможности равномерно.
Если отсосано такое количество жидкости, что остаток образовал пасту,
то последняя без большого труда отделяется от фильтра. В таком виде
осадок еше содержит много маточного раствора, который удаляют, отжи-
мая осадок на винтовом прессе (см. рис. 21, стр- 98). Для этой
цели осадок завертывают в обычную фильтровальную ткань, а затем
в плотную фильтрпрсссную салфетку, которая может выдержать давле-
ние пресса. Сдавливание производится очень медленно, вначале без при-
менения сколько-нибудь значительного усилия. Жидкость должна иметь
время пройти между частицами осадка, а также через фильтровальную
ткань и фильтрпресспую салфетку; в противном случае возникает такое
.высокое гидростатическое давление, что ткань рвется или оса ок п охо-
О практической работе в органической технической лаборатории
27
дйт через пес. Только в конце, когда остается лишь совсем немного
жидкости, пресс затягивают со всей силой.
Наибольшие затруднения возникают при фильтровании аморфных
хлопьевидных и студнеобразных осадков. В этих случаях больше всего
подходит сложенная в несколько раз фильтровальная ткань, которую
помещают в обычную стеклянную вороику таким же образом, как и глад-
кую фильтровальную бумагу. Осадок можно затем отжать на прессе
прямо в такой ткани, причем это следует делать особенно медленно и
осторожно.
Фильтрование происходит всегда значительно скорее при нагревании,
чем иа холоду, поскольку вязкость с повышением температуры умень-
шается. Поэтому в тех случаях, когда вещество устойчиво и обладает
малой растворимостью, его фильтруют в горячем виде.
Вообще фильтруемость в большой степени' зависит от физических
свойств осадка, а последние в свою очередь зависят от условий осажде-
ния или кристаллизации. Очень часто полезно проводить осажден не или
кристаллизацию при повышенной температуре и перемешивании. Часто
дает хорошие результаты добавление солей и поддержание определенной
кислотности или щелочности. Иногда осаждение проходит лучше всего
в точно нейтральном растворе. Однако условия в разных случаях могут
быть столь различными, что общих правил установить нельзя.
Перегонка
Перегонка, в тех случаях, когда она применима, является самым де-
шевым способом выделения и очистки продуктов реакции. Основные цеди
перегонки:
а) уда'лсиие растворителя,
б) очистка приблизительно однородного продукта реакции (ректифи-
кация) ,
в) разделение нескольких продуктов реакции, имеющих различные
температуры кипения (дробная перегонка).
По характеру выполнения различают:
1. Перегонку при атмосферном давлении,
2. Перегонку при уменьшенном давлении (перегонка в вакууме).
3, Перегонку с водяным паром.
1. Перегонка при атмосферном давлении
а) Отгонка растворителя. Растворитель, как правило, кипит при зна-
чительно более низкой температуре, чем продукт, который из него выде-
ляется. Поэтому обычно не требуется специальной установки для фрак-
ционирования. Однако продукты, летучие с водяным паром, могут захва-
тываться низко кипящим и растворителями. В таких случаях рекомен-
дуется применять простой дефлегматор, например дефлегматор Гсмпеля,
наполненный стеклянными бусами. Форма перегонного сосуда значения
не имеет. Часто растворитель Можно отгонять прямо из того сосуда,
в котором проводится реакция. Может оказаться целесообразным отго-
нять растворитель из колбы, которая затем будет использована для пере-
гонки основного продукта. В этом случае, чтобы не пользоваться сравни-
тельно большим сосудом, в колбу не приливают сразу весь раствор, а
добавляют его из капельной воронки по мере отгонки растворителя. Если
остаток после отгонки растворителя застывает при обычной температуре,
то рекомендуется использовать широкогорлую колбу, чтобы облегчить его
извлечение.
28 Л Промежуточные продукты
В качестве источника тепла при перегонке низкокипящих и легко-
воспламеняющихся растворителей (например, спирта, бензола и осо-
бенно эфира) применяют паровую баню с электрическим или паровым
обогревом. Большие количества подобных жидкостей всегда следует пере-
гонять в особой «эфириой комнате», в которой нет открытого пламени и
запрещено курение. Там, где обстоятельства вынуждают отгонять эфир
или подобные вещества, нагревая водяную баню на открытом пламени
газовой горелки, необходимо приемник с одной стороны герметично
соединять с холодильником, а с другой стороны —с резиновой трубкой,
которая доходит до пола; таким образом избегают попадания нескондеи-
енровавшихся паров эфира на рабочий стбл невдалеке от пламени.
Растворители, кипящие при температурах выше 100°, отгоняются, как
правило, па масляной баие, которая нагревается иа 20—30° выше тем-
пературы кипения растворителя. Очень высококипящие растворители,
например нитробензол, целесообразно отгонять в . вакууме или
с водяным паром.
Растворы, которые могут поглотить воду при предварительной обра-
ботке, целесообразно высушивать до отгонки раствори-
теля; высушивание осуществляется выдерживанием в течение несколь-
ких часов, лучше всего1 в теченйе ночи, над одним нз известных осушите-
лей (CaCls, ZnCls, Na2SO4, КгСОз, NaOH, КОН, СаО и т. д.). Осушитель
следует выбирать таким образом, чтобы он не растворялся в раствори-
теле и ие реагировал ни с одним из имеющихся продуктов. Так, напри-
мер, для высушивания аминов нельзя применять СаСБ и ZnCls, а для
высушивания кислот — КзСОз, КОН и т. д. Индифферентный Na2SO4
может применяться почти всегда, но, для того чтобы быть активным, оп
должен обезвоживаться перед употреблением. Перед отгонкой раствори-
теля раствор следует отфильтровать от осушителя, так как иначе осуши-
тель может при нагревании отдать поглощенную воду.
При нагревании жидкостей в гладкостенных, особенно в стеклянных
сосудах легко наступает перегрев, что вызывает внезапное очень бурное
вскипание («толчки»). При этом может быть легко выбита пробка или
жидкость выброшена через холодильник, что в свою очередь может при-
вести не только к потере вещества и времени, но при работе с легковос-
пламеняющимися жидкостями — и к пожарам. Поэтому при перегонке из
стеклянных сосудов, а также при кипячении с обратным холодильником
никогда не следует забывать помещать в колбу кипятильные ка-
мешки' (глиняные черепки или кусочки пемзы величиной с горошину)
или кипятильные палочки (палочки толщиной со спичку из по-
ристого дерева, которые ставятся в колбе вертикально н доходят до се
горла). Деревянные палочки имеют то Преимущество, что по окончании
перегонки даже в случае кристаллизации остатка их можно легко вынуть;
конечно, такие палочки можно применять лишь для жидкостей, которые
ие действуют на древесину.
В качестве холодильника для низкокипящих растворителей
лучше всего применять змеевиковый холодильник, который энергично
охлаждает и занимает мало места. За неимением змеевикового холодиль-
ника можно пользоваться достаточно длинным наклоненным вниз обрат-
ным холодильником. При более высоких температурах кипения подходит
обычный холодильник Либиха, а для еще более высококипящих раство-
рителей — простой воздушный холодильник.
Удобнее всего соединять холодильник с перегонным сосудом при
помощи резиновых пробок. Однако следует иметь в виду, что
некоторые растворители (например, бензол, лигроин и т. д.) действуют
на резину, при этом может ие только повреждаться пробка, но также
О практической работе в органической технической лаборатории 29
загрязняться вещество. В таких случаях пользуются корковыми
пробками, которые до сверления следует тщательно обжать. При
наличии паров, разъедающих резиновые и корковые пробки, целесооб-
разно применять холодильник со шлифом.
Вода как растворитель, если она не содержит веществ, перегоняю-
щихся с водяными парами, обычно не отгоняется, а выпаривается из
открытых сосудов. Выпаривание всегда производится при перемешива-
нии. Оно значительно ускоряется, если пары быстро удаляются током
воздуха. Это достигается вращением деревянного пропеллера над фарфо-
ровой чашкой или подобным сосудом; если испарение производится из
стакана, то применяется отсасывание или продувание воздуха через
стеклянную трубку, находящуюся непосредственно над поверхностью.
Таким образом удается осуществить быстрое испарение при температурах
80—90°, не прибегая к кипячению. Такой способ, конечно, нельзя приме-
нять при наличии легкоокисляемых веществ. В таких случаях лучше
всего применять испарение в вакууме (см. соответствующий раздел).
б) Ректификация? При ректификации обычно работают с веще-
ствами, имеющими высокие температуры кипения, так что интенсивного
охлаждения не требуется. Чтобы по возможности избежать соприкосно-
вения паров с резиновыми или корковыми пробками, целесообразно
использовать в качестве сосуда для перегонки колбу с длинной
отводной трубкой, которой часто оказывается достаточно для
охлаждения. В случае надобности можно присоединять холодильник
Либиха или воздушный холодильник, а также надевать на отводную
трубку муфту для водяного охлаждения. При работе с застывающими
веществами для избежания закупоривания лучше охлаждать приемник и
применять не очень длинную, но широкую отводную трубку. Чтобы пары
нс перегревались слишком сильно, при перегонке высококипящих веществ
отводная трубка должна быть расположена в нижней части горла колбы.
При ректификации всегда применяют термометр. Его надо уста-
навливать таким образом, чтобы верхний край шарика находился как раз
против отводной трубки. Только в этом случае будет правильно показана
температура переходящих паров. О температурных поправках см. ниже,
в разделе «Испытание продуктов на чистоту» (стр. 43).
То, что отгоняется ниже температуры кипения данного вещества
(остатки растворителя, неизменное исходное вещество и т. п.), собирается
отдельно как «первый Погон». Содержащуюся в небольшом количестве
воду должно, если это только возможно, удалять тщательным высушива-
нием еще до1 отгоики растворителя Jcm. выше), так как ее присутствие
часто затрудняет быстрое и четкое установление температуры кипения.
Основная фракция должна перегоняться примерно в пределах одного гра-
дуса. То, что перегоняется выше этого предела, является «последним
погоном» и собирается отдельно. Часто из этого погона повторной пере-
гонкой можно выделить еще некоторое количество чистого продукта.
Нагревание чаще всего производится на масляной бане. Ее рекомен-
дуется применять особенно в тех случаях, когда в конце перегонки
остается значительное количество остатка, который нельзя сильно пере-
гревать. В других случаях можно с некоторыми предосторожностями*
производить перегонку на голом пламени, иногда с применением во-
ронки Бабо.
в) Дробная перегонка. Когда требуется разделить несколько про-
дуктов, температуры кипений которых различаются не очень значительно,
то простая перегонка из колбыЪказывается уже недостаточ'ной. Для этой
цели следует применять особую насадку для фракционирования, называе-
мую ф р акциони ров оч н ой колонной. Действие подобных
установок основано па том, что поднимающиеся из перегонного сосуда
пары все время приходят в соприкосновение с жидкими компонентами
смеси, благодаря чему более высококнпящее вещество конденсируется и
удерживается, а в холодильник попадает лишь более ннзкокипящее, Такое
действие проявляется в известной степени уже тогда, когда лары подни-
маются по длинной пустой трубке, иа стенках которой происходит частич-
ная конденсация. Оно значительно усиливается, когда трубка имеет
расширения и сужения и особенно, когда на стенках трубки сделаны вы-
дающиеся внутрь выступы или острия, которые способствуют частичной
конденсации и увеличивают площадь соприкосновения пара и жидкости.
Насадки такого рода применяются очень часто. Они удобны, для разделе-
ния веществ, температуры кипения которых не слишком близки. Но они
не подходят, когда разница в температурах кипения составляет лишь
несколько градусов, как это обычно бывает у изомеров. В таких случаях
площадь соприкосновения пара и жидкости следует увеличить еще
больше. Это достигается заполнением колонки соответствующими напол-
нителями, лучше всего кольцами Рашига, т. е, Цилиндрическими коль-
цами из какого-нибудь материала (для лабораторных целей чаще всего
из стекла), высота которых равна их диаметру и которые расположены
в колонке совершенно беспорядочно. Кроме того, требуется, чтобы коли-
чество стекающей обратно жидкости можно было регулировать; для этого
на верхнем конце колонки устанавливается дефлегматор, т. е. холо-
дильник с регулируемым охлаждающим действием, а другая часть ко-
лонки изолируется от потерь тепла вследствие излучения. Дефлегматор
в зависимости от температуры кипения вещества можно охлаждать водой
или другой жидкостью, а также воздухом (см, рис. 37, стр. 305). Чем
большая часть паров конденсируется в дефлегматоре, тем более четко
происходит разделение; перегонка при этом продолжается, конечно, соот-
ветственно дольше. Поскольку при дробной перегонке имеет значение
также и равномерность нагрева, его следует осуществлять при помощи
бани — водяной, масляной или графитовой — в зависимости от темпера-
туры кипения.
При помощи хорошо действующей ректификационной колонки можно
также и в лабораторном масштабе с одного раза практически полностью-
разделить, например, бензол, моно- и дихлорбеизолы (см. стр, 63).
Изомерные же дихлорбеизолы, температуры кипения которых различаются'
лишь на 4—5°, не удается полностью разделить даже при работе в боль-
ших масштабах. Однако жидкие смеси изомеров, отличающихся незначи-
тельной разницей в температурах кипения и являющихся в чистом виде-
твердыми веществами, можно при тщательном фракционировании разде-
лить на отдельные фракции, настолько обогащенные разными изомерами,
что они выкристаллизовываются при охлаждении. Кристаллы отделяются
центрифугированием от жидкой части, которая вновь поступает на про-
изводство (см. разделение 2,4- и 2,6-нитрохлортолуолов, стр, 144).
2. Перегонка в вакууме
О перегонке в вакууме см. специальный раздел «О перегонке в ва-
кууме в лаборатории и на производстве», стр. 304.
3. Перегонка с водяным паром
Перегонка с водяным паром является на производстве сравнительно-
дорогой операцией, так как она требует много пара н очень много воды
для охлаждения. Однако для лабораторных условий она очень удобна,.
JUUUUJt? u i ^AIMWLMJU jlUV<J}Jt2'i OftUll
01
так как с ее помощью можно гладко отделить летучие с водяным паром
органические соединения от нелетучих неорганических веществ и от вы-
сокомолекулярных смолистых побочных продуктов, В некоторых случаях
возможно также разделение изомеров, если только один из них перего-
няется с водяным паром (например, о- и п-литрофепол, см. стр. 134),
Если перегонять попеременно то из кислого, то из щелочного раствора,
то можно отделить летучие с паром вещества кислого и основного харак-
тера друг от друга, а также от нейтральных продуктов.
Если пар берут из паропровода, то между ним и перегонным сосудом
рекомендуется помещать предохранительный сосуд (например, колбу для
отсасывания) с поднимающейся вверх трубкой; это позволяет с одной
стороны, задерживать увлеченную парами сконденсированную воду и,
с другой стороны, регулировать давление (см. водоуловитель 2 на
рис. 23, а, стр. 128). Если пар получают в лаборатории, то парообразова-
тель снабжают трубкой, поднимающейся вертикально вверх.
В качестве перегонного сосуда лучше всего подходит к р у г л о д о н-
н а я колба с длинным горлом, которая ставится косо таким
образом, чтобы жидкость, разбрызгивающаяся при вводе пара, попадала
на стенку колбы, по не в ее горло *. Трубка для ввода пара должна дохо-
дить почти до самого дна колбы. Отводная трубка должна быть широкой
и косо срезанной вттизу; благодаря этому увлеченные капли легко сте-
кают обратно в колбу и не попадают в холодильник. Для избежания
переброса и вспенивания колба должна быть заполнена не. более чем на
одну треть. Если в силу каких-либо особых причин перегонная колба
ставится вертикально или перегонка ведется из котла, то необходимо
отводную трубку снабжать каплеуловителем, чтобы холодильник.
был предохранен от забрызгивания. В перегонном сосуде не должно соби-
раться слишком много воды; для этого его следует нагревать во время
перегонки. Нагревание кругл од он ных колб целесообразно проводить на
воронке Бабо.
Поскольку теплота испарения воды очень велика, охлаждение должно
быть интенсивным, если хотят вести перегонку быстро. При перегонке
веществ, которые при обычной температуре остаются жидкими, лучше -
всего присоединять одни за другим два нисходящих обратных холодиль-
ника. Для застывающих веществ берут только один обратный холодиль-
ник, к которому присоединяют один прямой. Последний должен иметь
тонкостенный, широкий и ие сужающийся у нижнего конца фор-
штос. Застывший дистиллят можно тогда удалять, расплавляя его путем
периодического выключения воды в холодильнике. Может также ока-
заться целесообразным держать холодильник настолько теплым, чтобы
нс наступала кристаллизация, и использовать в качестве приемника
круглодоиную колбу, охлаждаемую струей воды. При перегонке высоко-
плавящихся веществ, которые обязательно застывают в холодильнике,
можно применять только один прямой и возможно более широкий холо-
дильник,. снабженный прнсдособленисм для выталкивания в виде спе-
циально изогнутой толстой проволоки или стеклянной палочки (см. при-
способление 6 на рис. 23,а и 5 на рис. 23,6, стр. 128, 129).
Если в перегонном сосуде остаются большие количества смолистых
веществ, то они могут включать и удерживать значительные количества:
летучего с водяным паром вещества. В таких случаях рекомендуется
* Особенно удобны приемники е гтубусом, которые раньше применялись при.
перегонке из реторт. Тубус здесь служит для ввода пара, а горло — для отвода ди-
стиллята.
J2 l. Промежуточные продукты.
извлечь смолу, тщательно измельчить се после охлаждения и затем вновь
поместить в перегонную колбу.
Разделение различных веществ дробной перегонкой с водяным паром
возможно (лишь до некоторой степени) только тогда, когда разница
в летучести с паром очень велнка. Иногда можно произвести разделение
соединений, обладающих различными основными или кислыми свой-
ствами; для этой цели можно, например, отгонять сначала из довольно ;
снльнокнслого раствора амин с наиболее слабыми основными свой- j
ствами, затем нз несколько менее кислого раствора — более сильное осно- ?
вание и, наконец, нз нейтрального или щелочного раствора — амнн с наи- ’
более сильными основными свойствами, j
Перегонку веществ, лишь трудно отгоняемых с обычным водяным
паром, можно значительно ускорить, если пар перегревать. Но, j
конечно, не имеет смысла вводить перегретый пар в водный раствор или z
суспензию, так как последние нельзя нагреть выше температуры их кипе- ;
ния. Напротив, перегоняемое вещество следует брать сухим илн предвари- )
тельно испарить содержащуюся в нем воду. После этого перегонную |
колбу нагревают до температуры пара, что лучше делать на масляной )
бане. Пароперегреватель и вводную трубку прн сильном перегреве
нельзя соединять резиновой трубкой или резиновой пробкой; для этой 3
пели применяют металлическую трубку с уплотнениями нз асбеста илн g
корковую пробку, J
Выделение продуктов реакции >
Если в результате реакции выделяется органическое соединение, ко-
торое обладает кислыми или основными свойствами и, следова-
тельно, может образовывать с о л н, то начинающий химик часто сомне- 3
вается, является ли полученный продукт свободным соединением илн его )
солью; в литературе достаточных сведений часто не имеется. Этот вопрос j
будет поэтому обсужден здесь несколько более подробно; кроме того, ]
будут рассмотрены наиболее целесообразные способы выделения соедиие-
ний такого рода, ?
а) Вещества основного характера
Алифатические амины — обычно легко летучие и легко раство- J
римые в воде жидкости — являются сильными основаниями; в сво-
бодцом состоянии они окрашивают лакмус в синий цвет и дают с мине- i
ральными кислотами нейтральные соли. Следовательно, реакция па лак- J
Мус сразу показывает (независимо от физического состояния), является 1
ли данное соединение основанием или солью. Во многих случаях для ]
выделения и идентификации пригодны солн пикриновой илн других по- 1
добньгх ей кислот, которые хорошо кристаллизуются н имеют характер- 1
ные температуры плавления. I
Совсем по-другому ведут себя технически более важные а р о м а т и- 1
ч е с к и е амины, В качестве простейшего примера возьмем анилин, 1
Свободный анилин в воде растворим мало (растворимость его составляет 1
примерно 3%|), Водный раствор не вызывает посинения лакмуса; следо- |
вательно, анилин является слабым основанием. С сильными минералы |
нымн кислотами: соляной, серной и азотной — он дает соли, которые
легко растворимы в воде. Такие растворы имеют сильнокнслую реакцию т
по лакмусу; это говорит о том, что при растворении имеет место гидроли- ;
тическоё расщепление, которое, однако, нс доходит до выделения свобод- с
кого анилина, Концентрация ионов водорода в таком растворе оказы-
вается недостаточной, чтобы вызвать посинение конго бумажки. Поэтому
Выделение продуктов реакции
33
с помощью конго можно установить, находится ли минеральная кислота
в количестве, избыточном по сравнению с необходимым для образования
соли. Соли анилина выделяются в неизмененном виде при упаривании их
водных растворов. Из этого следует, что если в растворе анилина, кис-
лом по конго, выпадает осадок илн наступает кристаллизация, то выде-
лившийся продукт может быть только солью анилнпа, но нс свободным
основанием. Такие осадки могут, например, образовываться при обра-
ботке концентрированного раствора солянокислого анилина сульфатом
патрня; это объясняется тем, что сернокислый анилин растворим значи-
тельно хуже, чем солянокислый. Но осадок образуется также и при при-
бавлении концентрированной соляной кислоты, так как солянокислая
соль растворима гораздо меньше в концентрированной соляной кислоте,
чем в чистой воде. Особенно легко образуются осадки с солями ще-
лочных металлов органических сульфокислот (напри-
мер, с натриевой солью цафталнпсульфокислоты), которые часто дают
с ароматическими аминами груднорастворимые соли. В случае анилина
можно, конечно, сразу установить, является лн осадок свободным осно-
ванием или солью, поскольку основание при обычной температуре пред-
ставляет собой жидкость, а соль является твердым веществом. В случае
твердых оснований один внешний внд осадка не позволяет сделать каких-
либо заключений.
Со слабыми кислотами, например уксусной, аннлнн не дает
солей, устойчивых по отношению к воде. Отдельные аро-
матические амины дают с ледяной уксусной кислотой кристаллические
ацетаты; последние, однако, разлагаются водой с выделением свободного
основания. Это свойство попользуется в технике, например для выделе-
ния л-ксилидина из смеси его с изомерами. Следовательно, если в вод-
ном растворе соли анилина заменить анион минеральной кислоты на
анион уксусной кислоты (этого можно достигнуть, например, прибавле-
нием ацетата натрия), то при соответствующей концентрации выделится
свободный аннлнн.
Гомологи анилина, а также нафтнламнна и многоядерные амнны,
содержащие не более одной аминогруппы в каждом ядре, как например
бензидин, днамнноднфеиилметан, ди- и тр нами потри фенил мет ан н т, д.,
ведут себя аналогичным образом. Если в одном ядре находятся две
аминогруппы или более, то основность несколько увеличивается, а раство-
римость в воде увеличивается значительно, однако характер соединения
при этом существенно не меняется. При введении заместителя в ядра все
'названные амины образуют вещества, которые обладают такими же свой-
ствами, если, конечно, заместители не меняют в значительной степени
основности. Такого рода заместители можно назвать почти индиффе-
рентными; к ним относятся алкоксигруппы (ОСНз, ОСаНз и т. д.) и ацил-
амипогруппы (NHCOCH3, NIICOCeEU и т. д.), Аннзндин н фенетндин,
моноацетил- и монобензонл-га-фенилеидиамин в отношении образования
солей ведут себя так же, как аннлнн. Введение алкилов в аминогруппу
по вызвшает существенных изменений; моно- и ди мстил анилин, моно- н
диэтиланнлнн, этнлбензиланилин и т, д. ведут себя подобно анилину.
Введение второго ароматического остатка в аминогруппу полно-
стью подавляет основные свойства; дифениламин, феннлнафтнламин и
подобные им вещества, а также карбазол не образуют уже солей с вод-
ными растворами кислот. Основность может быть также снижена или
полностью подавлена при введении в ядро определенных заместителей,
называемых отрицательными или усиливающими кислотные свойства.
Наиболее сильно действует в этом отношении ннтрогруппа, несколько
слабее — галоиды. Еще более слабым является влияние карбонильной
группы в альдегидах, кетонах, эфирах и амидах карбоновых кислот н т. д.
Сульфоны ведут себя подобно кетонам. Азогруппа также оказывает отри-
цательное влияние. Большое значение имеет не только характер замести-
теля, по и его положение относительно аминогруппы. Наиболее сильное
влияние-оказывают заместители в орто-положении, немного слабее —
в парс-положении и намного слабее — в м ета-положе ни и. Так, л«-нитро-
анилии еще образует солянокислую соль, которая растворяется в воде без
разложения. В случае л-иитроапилина для предотвращения диссоциации
необходима по крайней мере 10%-иая соляная кислота; о-нитроапилин
образует солянокислую соль лишь с концентрнровэнной соляной кисло-
той; в этом случае при добавлении даже незначительных количеств воды
выпадает свободное основание. Диннтроанилины вообще ие дают солей
с водными растворами кислот. То же самое наблюдается для трихлор-
анилина и дихлорнитроаиилииа. У амииоантрахиноиов под влиянием
обеих СО-групп основные свойства также настолько ослаблены, что они
образуют соли, например, Только с 75—80 %-ной серной кислотой, а из
разбавленных кислот выделяется свободное основание.
Все амины с сильно отрицательными заместителями спо-
собны выделяться в виде свободных оснований даже из сильнокислых по
конго растворов. В случае окрашенных соединений соль и свободное осно-
вание можно обычно различить по цвету. Так, нитроамииы — ярко-жел-
того цвета, а их соли бесцветны. Соли амииоантрахиноиов также бес-
цветны, в то время как свободные основания имеют окраску от оранже-
вой до красной. Аминоазосоедипения при солеобразоваиии меняют цвет
с желтого па красный или с оранжевого на фиолетовый. Нитрозирован-
ные амины (например, нитрозодиметил а нилин) окрашены в зеленый
цвет, когда они являются свободными основаниями, а их соли желтого'
цвета. Для бесцветных веществ этн методы идентификации, конечно,
неприменимы. В таких случаях характер осадка легко определить по его
поведению в отношении инертных растворителей — эфира, бензола и т. п_
Свободные основания в них обычно растворяются более или менее легко,
а соли нерастворимы.
Следует также отметить, что диазососдииеиия являются го-
раздо более сильными основаниями, чем амины, из которых онн полу-
чены. Так, раствор диазотированного л-нитроанилииа можно как угодно-
разбавлять водой или даже нейтрализовать ацетатом натрия и прн этом
основание дна зон и я не выделится в свободном состоянии. Легкость,. .
с какой идет разрушение дназосоедииения, оказывается в этом случае
значительно большей, чем в сильнокислом растворе.
б) Вещества- кислого характера
Среди веществ кислого характера, которые способны образовывать
соли с основаниями, следует различать три главные группы — фенолы,
карбоновые кислоты Н сульфокислоты.
Фенолы можно сравнить с ароматическими аминами. Они обра-
зуют с едкими щелочами соли, которые, как правило, растворяются в воде
без видимого разложения и имеют в растворах сильнощелочную реакцию
по лакмусу. Однако из этих растворов фенолы легко выделяются даже
при действии самых слабых кислот, например угольной или сернистой.
Онн имеют нейтральную реакцию по тиазоловой и аурамииовой индика-
торным бумажкам; избыток щелочи, сверх необходимой для солеобразо-
вания, можно определить по покраснению тиазоловой бумажки или
соответственно по обесцвечиванию аурамииовой. Из раствора, имеющего^
щелочную реакцию по этим индикаторам, можно выделить путем выпа-
иыоеление продуктов реакции
35
ривания или высаливания только соли фенолов. Как ароматические амины
с уксусной кислотой, так и фенолы с аммиаком уже не дают, как правило,
устойчивых в воде солей. Поэтому в водных растворах щелочные соли
фенолов можно перевести в свободные фенолы добавлением солей аммо-
ния и при подходящей концентрации свободные фенолы можно выделить
из растворов.
Отрицательные заместители действуют на фенолы точно так же, как
и на ароматические амины, ио, конечно, в обратном смысле: их действие
заключается в усилении кислотных свойств оксигруппы.
о- и /2-Хлорфеиолы обладают значительно более сильными кислот-
ными свойствами, чем фенол; это имеет место в еще большей степени
у о- и д-иитрофеиолов. Трииитрофенол, или пикриновая кислота, является
настоящей кислотой. Ее соли нейтральны и не разлагаются ни углекис-
лотой, ии солями аммония. Из растворов солей пикриновой кислоты,
имеющих нейтральную реакцию, можно хлористым Натрием или хлори-
стым калием высолить соответствующие пикраты. Следовательно, фенолы,
содержащие отрицательные заместители, можно выделить в виде фено-
лятов из нейтральных или слабощелочных по лакмусу растворов; чтобы
решить в сомнительных случаях, состоит ли осадок из свободного фенола
или из соли, его надо специально исследовать, так же как в случае ами-
нов. Для питрофенолов показателен цвет осадка. Свободные нитрофенолы
имеют бледно-желтый цвет или бесцветны, а их щелочные соли ярко-жел-
того цвета. Если соединения не окрашены, можно, как правило, исследо-
вать раствор в инертном растворителе. Из растворов, им'еющих кислую
реакцию, Могут, конечно, выпадать только свободные фенолы.
У а мин о фенолов свойства амино- и оксигруппы сохраняются и
заметного влияния этих групп друг иа друга нс наблюдается. Так, в неко-
торых случаях можно, например, высолить поваренной солью из раствора
амипофенола в избыточном количестве щелочи его натриевую соль, а из
раствора. в минеральной кислоте — солянокислую; из раствора же ам-
миака, бикарбоната, уксусной кислоты или из нейтрального раствора вы-
деляется свободный аминофенол.
Карбоновые кислоты обладают значительно более сильными
кислотными свойствами, чем фенолы. Они окрашивают лакмусовую бу-
магу в красный цвет н дают со щелочами соли, которые имеют нейтраль-
ную реакцию по лакмусу. Посинения конго они не вызывают; лишь
в отдельных случаях наблюдается фиолетовое окрашивание. В противопо-
ложность фенолам онн растворяются в растворах бикарбонатов и осо-
бенно карбонатов щелочных металлов, а также в водном аммиаке. Но,
с другой стороны, при действии на их-соли сильных минеральных кислот
они выделяются в свободном состоянии.
Если в молекуле имеются одновременно а м и ио- и карбоксиль-
ная групп ы, то они влияют друг на друга лишь в незначительной сте-
пени. Антраниловая кислота растворяется в содовом растворе так же
легко, как и бензойная, а в соляной кислоте так же легко, как анилин.
Поэтому при выделении аминокарбоиовой кислоты в свободном состоя-
нии следует тщательно избегать даже незначительного избытка минераль-
ной кислоты или щелочи. Избыток уксусной кислоты не мешает выде-
лению.
К третьей группе соединений кислого характера относятся сульфо-
кислоты, которые играют очень важную роль в технической химии,
хотя с теоретической точки зрения оин менее важны и поэтому в учебни-
ках и лекциях по органической химии упоминаются лишь кратко. Сульфо-
кислоты по своему поведению резко отличаются от карбоновых кислот
Они являются такими же сильными, как обычные минеральные кислоты,
например соляная или серная. Последние поэтому нельзя приме-
нять для выделения свободных сульфокислот. Если, например, раз-
бавленный водный раствор натриевой соли бензол сульфо кислоты
обработать эквивалентным количеством соляной кислоты, то обра-
зуется одинаковое количество ионов Н+, Na+, С1~ и С6Н55Оз". Если же
разбавление недостаточно для полной ионизации, то устанавливается рав-
новесие между НС1, NaCI, СбН55О3Н и C6H5SO3Na и указанными че-
тырьмя нонами, т, е. образуются все четыре возможные комбинации этих
четырех ионой.
Только когда концентрация (например, при упаривании) повышается
настолько, что превышается предел растворимости одного нз четырех
продуктов, последний начинает выделяться; дальнейшее упаривание мо-
жет в конце концов привести к преимущественному или исключительному
образованию этого одного продукта, или, вернее, пары продуктов (напри-
мер, натриевой соли бензолсульфокислоты и НС1 или бензолсульфокис-
лоты и NaCI). Возможность выделения зависит'исключительно от соот-
ношений растворимостей, которые очень различны у разных сульфокислот
и не могут быть предсказаны теоретически. Опытным путем, однако, уста-
новлено, что свободные сульфокислоты, как правило, очень легко раство-
римы в воде, В концентрированных растворах соляной и серной кислот
растворимость часто бывает значительно меньшей. Например, при сульфи-
ровании нафталина обычной серной кислотой при повышенной темпера-
туре в условиях, когда образуется в качестве главного продукта нафта-
лин-₽-сульфокислота (см. стр. 167), при последующем разбавлении реак-
ционной смеси небольшим количеством воды (примерно 3 частн воды иа
4 частн примененной серной кислоты) и охлаждении нафталин-p-сульфо-
кислота выделяется почти количественно и в свободном от примесей со-
стоянии*. Для дальнейшей очистки ее можно растворить в небольшом ко-
личестве воды и осадить концентрированной соляной кислотой, так как
в примерно 10%-иой соляной кислоте она очень мало растворима. Все
изложенное относится лишь к тому случаю, когда в растворе отсутствует
ион металла. Когда же исходят из соли сульфокислоты нли вносят ноны
металла, добавляя в раствор хлористый натрий или глауберову соль, то
обычно в осадок выпадает соль сульфокислоты, поскольку соответствую-
щая соль большей частью растворима труднее, чем свободная сульфокис-
лота. Если, например, упомянутую выше реакционную смесь, Полученную
при сульфировании нафталина, ие разбавлять небольшим количеством
воды, а влить в раствор поваренной соли, то, несмотря иа большой избы-
ток в растворе свободной серной кислоты, образуется большое количе-
ство осадка натриевой солн и а фтал и и-\6-сульфокислоты. Щелочную соль
сульфокислоты, как правило, це удается перевести в свободную суль-
фокислоту добавлением сильной минеральной кислоты. В случае необхо-
димости такое превращение можно, например, осуществить, приготовив
бариевую соль сульфокислоты и затем обработав ее рассчитанным коли-
чеством серной кислоты. Превращение в этом случае происходит количе-
ственно, поскольку барий удаляется в виде нерастворимого BaSO4. Можно
также приготовить свинцовую соль и разложить ее .сероводородом; при
этом, как и в предыдущем случае, металл удаляется в виде нераствори-
мого соединения.
Д и - и полисульфокислоты способны при насыщении лишь
части сульфогрупп образовывать кислую соль, которая, можно полагать,
будет менее растворима и может быть выделена. Но это, однако, яв-
ляется исключением; в 'Случае полисульф окис л от осадки, получающиеся
* Witt, Вег., 48. 750 (1915).
Выделение продуктов реакции
37
из содержащих соли растворов, а особенно осадки, получающиеся в ре-
зультате высаливания, как правило, также состоят из нейтральной соли.
Все. что было сказано выше о сульфокислотах, справедливо только
тогда, когда в молекуле наряду с сульфогруппой нет аминогруппы или
другой группы основного характера. Амииосульфокнслоты зна-
чительно отличаются по своим свойствам от обычных сульфокислот. Выше
уже упоминалось, что при сливании водных растворов солянокислого ани-
лина и натриевой соли нафталипсульфокяслоты выпадает труднораство-
римая соль анилина и нафталинсульфоки,слоты. Аналогичный процесс
происходит также, когда амнно- и сульфогруппа находятся в одной и той
же молекуле. При этом полярности внутримолекулярно уравновешиваются
и образуется внутренняя соль, которая обычно трудно растворима. Если,
например, подкислять минеральной кислотой водный раствор сульфаиило-
вокислого или нафтионовокислого натрия, то соответствующая внутренняя
соль выпадает почти количественно. Одиако эти соединения обычно ие
изображают как внутренние солни называют их не солями, а сво-
бодными сульфаниловой и нафтионовой кислотами. В этих так называе-
мых свободных аминокислотах кислотный характер сульфогруппы еще
сохраняется. Они дают нейтральную реакцию по конго, а по лакмусу —
кислую; в растворах щелочей, а также карбонатов и бикарбонатов ще-
лочных металлов они ведут себя так, как будто аминогруппа отсутствует,
и растворяются с образованием соответствующих солей. Амнносульфокис-
лоты образуют соли также н с органическими основаниями.' Так, при сли-
вании водных растворов натриевой соли сульфаниловой кнслоты и соляно-
кислого аннлнна выпадает осадок сульфаниловокислого анилина. Основ-
ные свойства аминогруппы в аминосульфокислотах полностью подавлены.
Они не образуют солей даже при действии большого избытка минераль-
ной кйсЛоты. Н е су Щ е с’т И V е г со л ин окисло й ~и сер н ок и ал о й*
ГТГ.ТТ! 1Г"'(! у льфа л иловой кислоты. Это имеет место также н
в случае групп, обладающих сильными основными свойствами, например
для диазогруппы. Выпадающее при диазотировании сульфаниловой кис-
лоты труднорастворИ'Мое дназосоединение, несмотря на избыток соляной
кислоты, ие является хлоридом, а представляет собой внутреннюю соль,
в которой диазониевая группа и сульфогруппа -нейтрализуют друг друга.
Для амитгосульфокисл'от, содержащих несколько амнно- или
сульфогрупп, действует правило, согласно которому каждая группа
основного характера нейтрализует одну сульфогруппу, и наоборот. Если
в соединении имеется одинаковое количество амнно- и сульфогрупп, то его
поведение аналогично поведению моно а ми но моносульфо кислоты. Так,
Л£-фениленднаминодисульфокнслота или беизиднндисульфокислота ведут
себя так же, как сульфаниловая кислота. Если же количество аминогрупп
превышает количество сульфогрупп, то избыточные аминогруппы со-
храняют свою способность образовывать соли с кислотами. .м-ФеиилеиДН-
аминомоносульфокнслота растворяется в соляной кислоте, как анилин.
Следовательно, При выделении таких сульфокислот, которые содержат
аминогрупп больше, чем сульфогрупп, следует, как н в случае амииокар-4
боновых кислот, обращать внимание на то, чтобы избежать избытка мине-
ральной кислоты, так как иначе вещество может снова раствориться.
Когда, наоборот, сульфогрупп в соединении больше, чем аминогрупп, то
при подкислении щелочных солей образуется столько свободных сульфо-
групп, сколько имеется в соединении аминогрупп. От остальных же суль-
фогрупп металл не отщепляется^ Эти сульфогруппы ведут себя так же,
как сульфогруппы в бензол- н нафталиисульфокнслотах, т. е. свободная
сульфокислота образуется только в исключительных случаях, когда со-
3$
/. Промежуточные продукты
мостей. Осадок представляет собой, как правило, кислую соль, в ко-
торой каждой аминогруппе соответствует свободная сульфогруппа, а
остальные сульфогруппы связаны со щелочным металлом. На это обстоя-
тельство следует обратить особенное виимаиие, так как такого рода соеди-
нения в технике обычно носят названия свободных кислот. Так, С-кислота,
амино-Г-кислота, Ц-кислота и т. д. в действительности являются моно-
натриевымн солями 1,4,8- 2,6,8- и 2,4,8-пафтиламииодисульфокислот; так
называемая кислота Коха представляет собой дипатриевую соль 1-иа-
фтиламин-3,6,8-трисульфокислоты, а Аш-кислота — моиоиатриевую соль
1,8-амиионафтол-3,6-дисульфокислоты. Наличие оксигруппы в таких со-
единениях имеет значение лишь постольку, поскольку в сильнощелочных
растворах она может реагировать со щелочью. Образующиеся при этом
соли имеют щелочную реакцию и обычно значительно легче растворяются,
чем нейтральные соли. Поэтому, если хотят выделить фено л- или
и а ф т о л с у л ь ф о к и с л о т ы, выбирают такие условия, которые исклю-
чают образование соли по оксигруппе.
Очистка продуктов
Как уже упоминалось в разделе, посвященном перегонке, эта опе-
рация в том или ином оформлении является обычно самым дешевым мето-
дом очистки. Но она, конечно, может применяться только для очистки ве-
ществ, перегоняющихся без разложения. Но и в этом случае перегонка
сама по себе может только тогда привести к цели, когда температуры
кипения подлежащих удалению загрязнений или побочных продуктов зна-
чительно отличаются от температуры кипения очищаемого вещества.
Вещества, температуры кипения которых лежат очень близко друг к другу,
могут разделяться тщательной дробной перегонкой с достаточной для
технических целей полнотой, хотя и не абсолютно полно. Так, например,
не удается освободить одной только перегонкой технический бензол
(т. кип. 80°) от всегда содержащегося в нем тиофеиа (т. кип. 84°), а тех-
нический о-питротолуол (т. кип. 220е) всегда содержит небольшие коли-
чества изомеров (т. кип. 228 и 238°); содержание этих изомеров, правда,
так невелико, что не мешает их техническому применению. Если требуется
высокая степень чистоты, то может оказаться необходимым сочетание пе-
регонки с каким-нибудь другим методом очистки.
Наряду с перегонкой важнейшим методом очистки, особенно в лабо-
раторных условиях, является перекристаллизация. При перекри-
сталлизации вещество, подлежащее очистке, растворяют при нагревании,
чаще всего при кипячении с обратным холодильником, в подходящем рас-
творителе и отфильтровывают в горячем состоянии от нерастворившихся
загрязнений. Прозрачный фильтрат оставляют медленно охлаждаться;
при этом должна выкристаллизоваться большая часть растворенного ве-
щества, а растворимые загрязнения остаться по возможности в растворе.
Если кристаллизация происходит медленно, то ее можно ускорить пере-
вешиванием. При работе с веществами, которые очень склонны к образо-
ванию пересыщенных растворов, может оказаться необходимым вызвать
кристаллизацию, царапая стеклянной палочкой по стейке сосуда или
внося в раствор для затравки кристаллик вещества. Можно также приме-
нять охлаждение во льду или выдержку в холодильном шкафу. Количе-
ство растворителя следует выбирать таким образом, чтобы оно было до-
статочным для полного растворения вещества при нагревании (при этом,
конечно, не следует принимать во внимание нерастворимых загрязнений)
и чтобы растворимые загрязнения па холоду оставались по возможности
в растворе. Сильно загрязненные nnoavKTbi пекомеид ется пепел пепекпи-
Очистка продуктов
39
сталлизацией промыть на холоду растворителем, который бы легко рас-
творял загрязнения, но трудно — основной продукт. Для удаления окра-
шенных или коллоидных загрязнений бывает полезно добавлять живот-
ный, кровяной или активированный уголь или другие сильные адсор-
бенты. Адсорбция идет лучше при нагревании, чем па холоду, и требует
некоторого времени: например, раствор кипятят примерно в течение
>/2 часа с обесцвечивающим средством, применяя обратный холодильник,
и затем фильтруют в горячем состоянии *.
Для успешного проведения перекристаллизации решающую роль
играет, конечно, выбор растворителя. Когда в литературе на этот
счет нет достаточных сведений, то подходящий растворитель находится
с помощью пробирочных' опытов. Первое условие, которому должен удов-
летворять растворитель, состоит, конечно, в том, чтобы он растворял при
нагревании намного лучше, чем на холоду; это необходимо для того,
чтобы при охлаждении вещество выпадало как можно полнее. Кроме того,
растворитель должен или совсем ие растворять загрязнений (и тогда их
можно удалять фильтрованием в горячем состоииии) или растворять их
так хорошо, чтобы они при охлаждении оставались в растворе и не выпа-
дали в осадок вместе с основным продуктом. Не всегда можно найти рас-
творитель, который бы удовлетворял полностью этим условиям. В этом
случае необходимо повторять перекристаллизацию, сменив при этом
растворитель. Если имеется в распоряжении несколько подходящих
растворителей, то предпочтение следует отдать тому, который растворяет
нс слишком легко, но и не слишком трудно. При этом следует также учи-
тывать количество очищаемого вещества. Прн перекристаллизации боль-
ших количеств растворитель надо выбирать таким образом, чтобы веще-
ство в нем хорошо растворялось и чтобы для растворения при нагревании
достаточно было, например, 1 л растворителя на 200 г вещества. При ис-
следовательской же работе с 0,1 а того же вещества пришлось бы рабо-
тать с 0,5 мл такого растворителя, что уже затруднительно. В этом случае
следует отдать предпочтение тому растворителю, который растворяет труд-
нее в 10—20 раз и позволит работать с 5—10 мд. Если одни растворители
растворяют слишком легко, а другие — слишком трудно, то лучше всего
применять смесь растворителей, например разбавленный спирт,
разбавленную уксусную кислоту, смесь бензола и лигроина, эфира и хло-
роформа и т. д. Однако кристаллизация при этом часто проходит ие так
хорошо, как при применении однородных растворителей, и переработка
маточников также встречает затруднения. Этим приемом пользуются по-
этому только в случае необходимости,.
Простое растворение вещества в растворителе и его выделение путем
полного испарения растворителя ие являются перекристаллизацией и, за
исключением возможного удаления нерастворимых примесей путем филь-
трования, ие способствуют очистке. Иногда, правда, для достижения
обильного образования кристаллов бывает необходимо отогнать после
фильтрования часть растворителя. Чтобы растворимые загрязнения
остались в растворе, нельзя отгонять слишком много растворителя, В слу-
чае очень трудно растворимых веществ такое растворение и частичную ,
отгонку можиб производить непрерывно. Для этого применяют экстра-
гирование неочищенного продукта. Когда работают с иизкокипящими
растворителями, пользуются аппаратом Сокслета, а при работе с высоко-
кипящими растворителями применяют экстракционный тигель Нолле, ко-
торый подвешивают в горле экстракционной колбы; он действует так
же, как аппарат Сокслета.
'♦ Г1Т*л nnnuLTii nd/.FrnnnTi unrrinn лбалл ootutl TJZ U П A-T U M Tit unu VFJTPM
40
7. Промежуточные продукты
В маточных растворах, полученных после перекри-
сталлизации, наряду с растворимыми загрязнениями остается также
бо^ее или менее значительная часть очищаемого вещества. Чтобы полу-
чить дополнительные количества этого вещества, часть растворителя отго-
няют, образовавшиеся после охлаждения кристаллы отфильтровывают,
полученный фильтрат упаривают и т, д. до тех пор, пока не перестанут
образовываться кристаллы, пригодные для дальнейшего использования.
Эти продукты, выделенные из маточных растворов, обычно не являются
такими чистыми, как первая фракция, но они могут быть очищены 'по-
вторной перекристаллизацией.
Следует также отметить, что растворимость в растворителях одного
и того же типа тем выше, чем выше их температура кипения. Так, веще-
ство, которое очень трудно растворяется в кипящем бензоле (т, кнп. 80°),
будет несколько лучше растворяться в кипящем толуоле (т. кип, 111е) и
еще лучше в кипящем ксилоле (т. кнп. 140°) или кипящем хлорбензоле
(т, кип. 132°). Поэтому для перекристаллизации очень трудно раствори-
мых веществ (например, производных антрахинона) бывает необходимо
использовать такие вы с ококн пящ и е растворители, как. три-
хлорбензол, нитробензол, тетралин, анилин, фенол и т- п. Эти растворители
имеют то преимущество, что растворимость в них при температуре их ки-
пения значительна, а на холоду — очень невелика. Вследствие этого веще-
ство из них выкристаллизовывается почти полностью. Их недостаток со-
стоит в том, что то же самое наблюдается и дли загрязнений, которые при
нагревании также растворяются, а при охлаждении выпадают вместе
с очищаемым веществом. В таких случаях удовлетворительной очистки ие
происходит. Но все же правильно проведенная перекристаллизация
является надежным способом очистки. Но в промышленных масштабах
она довольно дорога, особенно когда применяются органические
растворители, поскольку регенерация последних невозможна без потерь.
Поэтому в технике очень часто отдают предпочтение так называемому
пер еос а ж дев и ю, которое прн правильном проведении также может
оказаться очень эффективным. Под переосаждением понимают растворе-
ние продукта в подходящем растворителе иа холоду нли при нагревании
и последующее осаждение добавлением какого-нибудь вещества, умень-
шающего растворимость или полностью осаждающего. Сюда относится
растворение в спирте и осаждение добавлением другого растворителя, на-
пример воды, эфира и т, д, В технике из соображений экономии предпо-
читают, конечно, такие методы переосаждення, по которым обходятся без
органических растворителей, т. е, работают только в чисто водной среде.
В качестве примера можно привести очень часто применяемое для сульфо-
кислот и красителей растворение в воде и осаждение поваренной илн ка-
кой-либо другой подходящей солью (сульфатом натрия, хлоридом калия,
сульфатом аммония и т. д.). При этом, чтобы получить более грубый и
легче фильтруемый осадок, осаждение, если возможно, производят при
нагревании. Если при нагревании осаждение происходит достаточно
полно, можно также и фильтровать в горячем состоянии; однако чаще для
завершения осаждения раствору дают предварительно охладиться. Пере-
мешивание во время охлаждения ускоряет кристаллизацию и делает ее
более равномерной. Если раствор непрозрачный, то его перед добавлением
осадителя фильтруют. Для того чтобы вместе с осадителем Не вводить
новых загрязнений/ последний, если это возможно, употребляют в виде
прозрачного раствора (например, насыщенный отфильтрованный раствор
поваренной соли). Количество осадителя следует выбирать таким обра-
зом, чтобы продукт выпадал возможно более полно, а загрязнения не вы-
пйлалн. осле осаж ения осалок отсасывают и промывают иа путч-
Очистка продуктов
41
фильтре раствором солн, концентрация которого соответствует концентра-
ции соли в фильтруемой смеси. Так, если для осаждения к водному
раствору прибавлялся равный объем насыщенного раствора поварен-
ной соли, то для промывания применяют смесь равных объемов
насыщенного раствора соли и воды. Для того чтобы осадок не содержал
слишком большого количества соли, его в конце можно промыть неболь-
шим количеством более разбавленного солевого раствора или, если про-
дукт не слишком хорошо растворим, очень небольшим количеством хо-
лодной воды. При переосаждении сульфокислот или их солен в лабора-
торных условиях можно в целях дальнейшей очистки промыть под
конец осадок спиртом и затем эфиром. Этим путем часто удается удалить
окрашенные или смолистые загрязнения. Кроме того, осадок после этого
высыхает гораздо быстрее и при этом ие склеивается и ие изменяет
окраски с поверхности; обычно таким путем получают сразу рассыпчатый
светлый порошок. Промывание спиртом, однако, никогда не следует пред-
принимать, не убедившись предварительно (взяв отдельную пробу), что
вещество в нем заметно не растворяется. Это относится к большинству
солей сульфокислот, ио ии в коем случае ие ко всем; некоторые солн рас-
творимы в спирте очень хорошо.
Другой употребительный метод переосаждеиия применим к соедине-
ниям, которые сами по себе в воде трудно растворимы, ио образуют легко*
растворимые в воде соли. К таким соединениям могут относиться основа-
ния, которые растворяются в разбавленных кислотах и могут быть оса-
ждены из этих растворов щелочами, а также вещества кислого харак-
тера — карбоновые кислоты, сульфокислоты и фенолы, которые раство-
ряются в щелочах и вновь выпадают при добавлении кислот. Такого рода
переосаждение способствует в основном удалсияю только тех загрязнений,
которые не обладают основными или кислыми свойствами н которые, сле-
довательно, остаются иераствореиными при действии кислот или щелочей
и могут быть отфильтрованы. Загрязнения, имеющие тот же химический
характер, что н основной продукт, удалить таким путем едва ли воз-
можно. Однако эффективность этого метода значительно повышается при-
менением двух модификаций.
Первая модификация — дробное о с а ж де и н е. Если, иапримерг
неочищенный продукт, обладающий основными свойствами, растворить
в строго необходимом количестве соляной кислоты и добавлять требую-
щееся для нейтрализации количество щелочи не сразу, а вначале только
небольшую часть —около 5—10%, то примеси, обладающие более сла-
быми основными свойствами, если только онн имеются, выпадут в первую
очередь. После фильтрования и дальнейшей нейтрализации фильтрата бу-
дет выпадать значительно более чистое основание. Если же, наоборот,
загрязнения являются более сильными основаниями, чем основной про-
дукт, то они выпадут только в конце нейтрализации. В этом случае более
чистое основание получается при прибавлении к раствору лишь 80 или
90% соляной кислоты, необходимой для нейтрализации. Этот метод
можно применять и для очистки соединений кислого характера. Хороших
результатов можно достигнуть только при разделении соединении, сила
кислотности или основности которых неодинакова. Это часто имеет место
в случае смесей изомеров, В предыдущей главе упоминалось, что атом
галоида, иапрнмер, сильно уменьшает основные свойства аминогруппы,
находящейся в ароматическом 'ядре, если он находится по отношению
к ней в орто- или пара-положении* если же он находится в .мета-пол оже-
ним, то влияние его значительно меньше. Если при реакции в качестве
главного продукта образуется, иапрнмер, амнн, содержащий хлор в мета-
42
I. Промежуточные продукты
орто- или «ара-изомера, то последний можно удалить дробным осажде-
нием.
Более общее применение, однако, находит другой метод, который со-
стоит в том, что растворенную соль не переводят опить в свободное осно-
вание или кислоту, а сначала осаждают как таковую, В случае щелочных
солей кислот это чаще всего осуществляется с помощью высалива-
ния, как это описано выше. Дли высаливании солей аминов можно
вместо поваренной соли применять избыток кислоты, использованной дли
растворения. Уже было упомянуто, что, например, солинокислый анилни
очень легко раствориетси в воде, но плохо в концентрированной соляной
кислоте. Дли многих аминов такое различие в растворимости еще более
велико. Так, например, для почти полного осаждении солянокислого
а-нафтил амина из раствора оказывается достаточным добавление сравни-
тельно небольшого избытка соляной кислоты. Можно также не осаждать
легко растворимую соль как таковую, а переводить ее в более
труднорастворимую соль, например солянокислую в серпокис-
лую или щелочную соль в бариевую или свинцовую, если последние
трудно растворимы. Можно напомнить, что обычный способ очистки, на-
пример бензидина, состоит в растворении неочищенного основания в раз-
бавленной соляной кислоте и последующем осаждении его в виде серно-
кислой соли добавлением сульфата натрия; сернокислый бензидин
практически нерастворим и полностью выпадает в осадок, в то времи как
сернокислая соль образующегося в качестве.побочного продукта изомер-
ного основания остается в растворе. Осажденная тем или другим путем
соль до переведения в свободное соединение (основание или кислоту)
должна быть, конечно, отсосана и промыта. Во многих случаях примене-
ние находит и сама соль, В качестве примера следует привести антранило-'
вуго кислоту, которую можно очищать как растворением в соляной кислоте
и осаждением в виде солянокислой солн, так и растворением в растворе
соды и высаливанием образовавшейся натриевой солн. Продукты, очищен-
ные таким образом, бывают обычно намного чище, чем вещества, очи-’
щенные растворением в кислоте с последующим осаждением действием
щелочи, или наоборот. Дли технических целей они бывают обычно доста-
точно чистыми. Когда к чистоте вещества предъявляются особенно высо-
кие требования, можно достигнуть и более совершенной очистки. Для
этого выделенную соль перекристаллизовывают или персосаждают до пре-
вращения в исходное свободное соединение.
Аналогичный описанному способ переосаждения, который особенно
полезен дли очистки трудно растворимых производных антрахинона, со-
стоит в том, что неочищенный продукт, обладающий слабыми основными
свойствами, растворяют в концентрирован ной серной кис-
лоте; раствор затем осторожно разбавлиют водой настолько, чтобы вы-
пала сернокислая соль, но не происходило гидролиза. Обычно дли этой
цели подходит разбавление до содержании серной кислоты в растворе
70—80%. Выделенную таким образом сернокислую соль отсасывают на
стеклянном фильтре и промывают серной кислотой соответствующей кон-
центрации, После этого свободное основание выделяется перемешиванием
сернокислой солн с большим количеством воды.
Органические соединения можно очищать многими другими мето-
дами, кроме описанных выше. Примером может служить возгонка, хрома-
тография, переведение в более легко кристаллизующиеся производные
(дли аминов, например, в ацетильные, бензоильные или другие ацильные
производные, для кислот—в хлорапгидриды, амиды, сложные эфиры
и т. д.). Но все эти методы применяются главным образом в исследова-
тельских лабораториях. Они конечно, находят применение также и в тех-
Испытание продуктов на чистоту
43
нических лабораториях и ими должен владеть химик таких лабораторий,
но подробное рассмотрение их не должно входить в задачу технического
учебника.
Испытание продуктов на чистоту
Когда получают в первый раз неизвестное вещество, то критерием его
чистоты и однородности служит постоииство его свойств при повторных
очистках. Такое вещество подвергают очистке различными методами (пе-
регонке, перекристаллизации по -возможности из различных растворите-
лей, очистке через соли, эфиры, амиды и т. д.) и если его свойства
после этих обработок не изменяются, то его считают чистым н одно-
родным. Этот метод, однако, не является абсолютно надежным.
Иногда случается, что вещества, считавшиеся чистыми, при повторном
исследовании более тонкими методами оказывались смесями различных
соединений.
Но в технической лаборатории гораздо чаще имеют дело с извест-
ными, описанными в литературе соединениями, В этом случае для кон-
троля чистоты, как правило, довольствуются тем, что свойства получен-
ного препарата согласуются с литературными данными. Физические
константы являются свойствами, которые могут быть очень легко вы-
ражены в числах; из физических констант наиболее легко определяются
температура плавления и температура кипения.
Об определении температуры кип сии я было уже кратко
упомянуто в разделе, посвященном перегонке. Здесь следует еще указать
на некоторые необходимые поправки. Температура кипения зависит,
.как известно, от давления; поэтому при более точных определениях сле-
дует учитывать атмосферное давление. Поправки, вызванные этим, правда,
нс особенно велики. При показаниях барометра свыше 700 мм они не пре-
вышают 2°, Более значительные ошибки могут иногда быть обусловлены
тем, что парами кипящего вещества окружен не весь термометр, а только
его шарик. Для части столбика ртути, которая ие нагрета до температуры
паров и называется обычно «выступающим столбиком ртути», вводится
поправка, которая пропорциональна длине (/) выступающего столбика и
разности между температурой кипения и температурой помещения (t),
«Эта поправка равна 0,00015° 1-1 и при высоких температурах кипения
может составлять даже несколько градусов, Прн указании температуры
кипения следует всегда отмечать, к какому давлению она относится и вво-
дилась ли поправка на выступающий столбик ртути.
Иногда соответствующая температура кипения не может служить до-
казательством чистоты вещества. Это относится к тем случаям, когда
имеется смесь веществ, кипящих при одинаковой или почти одинаковой
температуре; при этом иа основании температуры кипения нельзя опреде-
лить, имеется ли примесь. Но когда речь идет о веществах, полученных
собственноручно, то, как правило, уже знают, может ли присутствовать
примесь, кипящая примерно при той же температуре. Если такая возмож-
ность имеется, то определение температуры кипения не может, конечно,
быть достаточным для контроля чистоты вещества. Когда же присутствие
близко кипящих примесей исключено, то на основании правильной и чет-
кой температуры кипения можно считать, что вещество не содержит зна-
чительных количеств примесей и что оно достаточно чисто для техниче-
ских целей. Но все это не является еще доказательством того, что
вещество полностью очищено: с нарами ниже кипящего продукта могут
переходить небольшие количества даже значительно более высоко кипя-
щего соединения и температура кипения прн этом заметно не изме-
ЛИТРЯ t""1 ГТ(Ч Q ТЛ ГТГ_ ur-i iznrnn т- i ur/xiT.zxi-T'.n пп НоОггОС п Ча т. ггп п пт/лт'Л1 кх
44
/. Промежуточные продукты
высокие требования, нельзя довольствоваться только определением темпе-
ратуры кипения.
Температура плавления значительно более чувствительна,
к загрязнениям, чем температура кипения. Известно, что растворение ка-
кого-либо постороннего вещества в жидкости понижает температуру ее
застывания; точно так же понижается и температура плавления твердого
вещества, когда оно содержит примесь, растворяющуюся в нем при плав-
лении. Слишком низкая температура плавления является, таким образом,
неопровержимым доказательством наличия загрязнения. Однако правиль-
ная температура плавления ие всегда является надежным доказатель-
ством чистоты соединения; загрязнения, которые не растворяются в рас-
плавленном основном веществе, не оказывают влияния на его температуру
плавления. Такие загрязнения можно обнаружить, поскольку, когда они
присутствуют, не образуется совершенно прозрачного расплава. На это
обстоятельство следует обращать внимание при определении температур
плавления. Кроме того, бывает, что два изомера или гомолога или два <
сходных между собой вещества, которые' как раз и могут получаться
одновременно, оказываются изоморфными и дают смешанные кристаллы "
или образуют молекулярное соединение, которое имеет соб-
ственную температуру плавления и ведет себя как индивидуальное соеди-
нение. Характерным примером могут служить ацетил- и «-бутирил-^-ами-
нофенолы. Первый из них плавится при 145°, второй — при 138° а экви-
молекулярная смесь их — при 154—155°. Молекулярные соединения
такого рода, как правило, нельзя разделить на составные части с по-
мощью перекристаллизации. В таких случаях доказать неоднородность
обычно удается только получением производных. Подобные случаи ветре-
чаются не так часто, однако и не столь редко, как это иногда считают; их
нельзя упускать из виду во избежание ошибок.
Другим затруднением при определениях температур плавления яв- ,
ляется наличие Кристал л”и зад ион но связанного раство-
рителя. Известно., что многие вещества кристаллизуются с кристалли-
зационной водой, кристаллизационным спиртом, кристаллизационным бен-
золом и т. д. Часто этот кристаллизационио связанный растворитель уле-
тучивается при определении температуры плавления еще до плавления'
вещества и не влияет на температуру плавления. Но бывает также, что»
намного ниже действительной температуры плавления наступает кажу-
щееся плавление, которое представляет собой растворение в кристаллиза-
циоино связанном растворителе. Таким примером является в неорганиче-
ской химии кристаллическая сода, которая легко растворяется в собствен-
ной кристаллизационной воде, В органической химия такие примеры
можно найти главным образом среди высокомолекулярных соединений, <
особенно среди соединений ряда трифенилметана, которые очень прочно
удерживают кристаллнзациоино связанный растворитель. Соединения та-
кого рода плавятся при совершенно различных температурах в зависи- J
мости от того, с каким растворителем они кристаллизационио связаны,
т. е. в зависимости от того, из какого растворителя они перекристаллизо-
вывались; если это обстоятельство не учитывать, можно власть в боль-
шие ошибки. Поэтому температуру плавления следует определять только. J
для тех образцов вещества, которые высушены до полного удаления
кристаллизационио связанного растворителя.
Затруднения в определении температуры плавления встречаются
также для тех веществ, которые начинают разлагаться еще до того,,
как вещество расплавится. Примером может служить фталевая кислота,
которая плавится при 206—208°, но уже значительно ниже этой темпера-
туры начинает отщеплять воду н поевпашаться во (йтлярвый яигиптп
Испытание продуктов на чистоту
45
температура плавления которого 131°, Если прибор для определения тем-
пературы плавления нагревать медленно и осторожно, так чтобы проба
вещества долгое время находилась при высокой температуре, то фталевая
кислота еще задолго до достижения температуры плавления превратятся
полностью в ангидрид, который и расплавится. Установленная таким
образом температура плавления окажется слишком низкой. В таких слу-
чаях перед введением пробы прибор следует предварительно нагреть до
температуры, которая лишь немного ниже температуры плавления фтале-
вой кислоты, и дальнейшее нагревание производить как можно быстрее,
чтобы фталевая кислота не успевала терять воду и плавилась в неизме-
ненном виде. Только при соблюдении этих условий температура плавления
будет определена довольно правильно. Часто (особенно в случае высоко-
плавящихся веществ) плавление происходит с разложением, В каждом
из таких случаев может оказаться, что наблюдалась ие температура
плавления иензмеиившегося вещества, а температура, при которой веще-
ство разлагается, образуя жидкий продукт распада. Точки разложения за-
висят не только от температуры, как обычные точки плавления, но также
и от продолжительности нагревания, поэтому кажущаяся тем-
пература плавления будет тем выше, чем быстрее велось нагревание.
Рассмотренные выше трудности реже являются причиной того, что
в литературных данных о температуре плавления одного и того же веще-
ства часто встречаются расхождения в несколько градусов. Гораздо чаще
это объясняется тем, что недостаточно очищенное вещество .было принято
за чистое, н главным образом тем, что по-разному выполнялось само
определение температуры плавления. Прежде всего следует напомнить,
что здесь так же, как и при определении температуры кипения, необходима
поправка на выступающий столбик ртути, которая далеко не всегда учи-
тывается. Эту поправку можно сделать менее необходимой, если пользо-
ваться введенными Пинке укороченными термометрами,
каждый из которых предназначен для измерения только ограниченной
области температур, например от 0 до 50°, от 50 до 100°, От 100 до 150°
и т. д., так что выступающий столбик охватывает не более 50°. Вследствие
этого поправки становятся столь незначительными, что ими можно пре-
небречь. Следует, конечно, иметь полный набор таких термометров и
знать заранее приблизительное значение температуры плавления, чтобы
Правильно выбрать нужный термометр. Поэтому укороченные термо-
метры используют прежде всего для окончательного установления темпе-
ратур плавления, например перед их опубликованием.
Другим источником ошибок при определении температур плавления
является то, что температура капилляра, где находится вещество, и тем-
пература шарика ртути термометра не всегда одинаковы. Наиболее на-
дежным способом достижения равенства температур капилляра н шарика
термометра является использование в качестве банн для нагревания
открытого стаканчика с мешалкой; перемешивание обеспечивает равно-
мерное нагревание всей находящейся в стаканчике жидкости. Для того
чтобы можно было осуществлять перемешивание без особых затруднений,
аппарат должен быть открытым. Жидкость же, служащая для нагрева, не г
должна быть ни летучей, ни сильно гигроскопичной. Можно рекомендо-
вать парафиновое масло, удобное для применения при умеренных темпе-
ратурах; при более высоких температурах оно быстро темнеет. Поэтому
при определении температур плавления, превышающих 200°, парафино-
вое масло приходится очень час*го менять. По этой причине более удобно
пользоваться закрытыми приборами, которые позволяют применять в ка-
честве жидкости для нагревания концентрированную серную кислоту.
Сериая кислота остается бесцветной вплоть до температуры ее кипения
ное
Р ис, 3, Прибор Тиле для опре-
деления температуры плавле-
ния.
/--канал для выравнивания давления;
2—пламя для нагрева,
определении
для
температуры пла-
вления.
/ _ канал ддя выравци.
палия давления.
(около 280°); если же она окрасится вследствие попадания какого-л ибо*
органического вещества, то ее легко вновь обесцветить добавлением не-
большого кристаллика азотнокислого калня. Простейший прибор такого'
рода состоит из круглодониой колбочки с длинным узким горлом, в ко-
тором с помощью пробки укреплен термометр (см, рис. 2); в пробке вы-
резан узкий каиал для выравнивания давления. Применяя такой прибор,
можно получить удовлетворительные результаты, если обеспечить медлен-
и равномерное нагревание. Этого можно достигнуть, или нагревая
совсем маленьким пламе-
нем самую середину кол-
бочки, нли применяя ис-
сколько большее пЛамя, I
ио производи обогрев кол-
бочки постоянными круго- *
выми движениями. Следу- J
ет также следить за тем,. 5
чтобы капилляры с веще- ]
ством всегда устаиавлива- 4
лись на одинаковой высоте
н притом так, чтобы ве- 1
щество находилось рядом >
с серединой ртутного ша- ч
рика термометра. Иногда; ]
равномерного нагревания
можно достигнуть, иагре- i
вая не самый сосуд непо- 1
средственно под термоме- Л
тром, а отходящее в сто- 1
рону колено, вследствие I
чего жидкость, служащая 1
для нагревания, циркули- 1
руст. По этому принципу ‘ 1
устроен прибор,сконструи- 1
роваиный Тиле (см. рис. ]
3). Одиако при более тща- 1
тельцом исследовании ока- 1
залось, что возникающая |
циркуляция жидкости не я
является равномерной: сто- 1
рона, обращенная к нагреваемому колену, нагревается заметно сильнее, 1
чем противоположная сторона. Ввиду этого надо обращать внимание на; .1
то, чтобы капилляр с веществом устанавливался всегда одинаково, ;|
а именно между наиболее и наименее нагретой сторонами. При Сравне-
нии двух проб обе должны устанавливаться на одинаковом расстоянии ' 1
от нагреваемого колена. -|
В последнее время стали предпочитать приборы, в которых вместо- 1
жидкости для обогрева применяется металлический блок, еде- 1
данный обычно из меди (см. рис. 4). Капилляр с веществом входит I
в узкую щель, через которую можно наблюдать процесс плавления,. 1
термометр вставляется в находящееся в непосредственной близости от- 1
верстие. Большое преимущество таких приборов состоит в том, что их. 1
можно применять.для температур выше 280°, т. е. превышающих темпе- |
ратуру кипения серной кислоты. При пользовании такими приборами еле- 1
дует особенно тщательно следить, чтобы нагревание было медленным »’ I
равномерным. Этого можно достигнуть, применяя электрообогрёв с по- 1
мощью тока, сила которого имеет постоянное значение, а также с по-
мощью очень маленького пламени газовой горелкн (микрогорелка), кото-
рая устанавливается всегда в одном месте. Если, кроме того, проверить
термометр путем определения с его помощью в тех же условиях темпе-
ратуры плавления ряда абсолютно чистых соединений с точно известными
точками плавления, то с этим прибором
можно производить действительно падежные
определения.
Независимо от конструкции применяемо-
го прибора наиболее надежные результаты
получают при одновременном определении
температур плавления исследуемого веще-
ства и абсолютно чистой пробы того же
вещества; определение должно производить-
ся на том же приборе и в точно
тех же условиях. Для этой цели сле-
дует выбирать капилляры по возможности
одного диаметра, одинаково наполнять их
весьма топко измельченным веществом и сле-
дить за тем, чтобы наполнение было как
можно более плотным, что достигается лег-
ким постукиванием зажатого между двумя
пальцами капилляра по столу; можно также
заставлять его падать иа стол через ие слиш-
ком короткую стеклянную трубку. После
этого оба капилляра прикрепляют иа равной
высоте к термометру и помещают в прибор
таким образом, чтобы они были одинаково
удалены от источника тепла.
Такие же предосторожности следует со-
блюдать при проверке идентичности двух
веществ на'основании определения так назы-
ваемой температуры плавления
смешанной пробы. В этом случае
Рис. 4. Прибор Изели с ме-
таллическим блоком и освеще-
нием с трех сторон для опре-
деления температур плавле-
ния. Пока вещество не спечет-
ся, лучше включать только
боковое освещение. При на-
ступлении спекания включают
также и заднее освещение, что
помогает установить момент,
когда плав станет прозрачным.
Прибор изготовляется также н
с электрическим обогревом.
два капилляра наполняют исследуемыми
веществами, а третий — их смесью; темпера-
'fypy плавления всех трех проб определяют
одновременно на том же самом приборе.
Если смесь плавится ниже, чем каждое из
веществ в отдельности, то это является на-
дежным доказательством того, что эти веще-
ства отличаются друг от друга. Если же
смесь плавится при той же темпер а-
туре, то вероятно, что вещества идентичны,
однако абсолютной уверенности это еще ие
дает, так как может случиться, что они
илй взаимно нерастворимы, или образуют
смешанные кристаллы, или дают молеку-
лярное соединение, которое имеет ту же
7 — блок; 2 —приспособление для
укрепления блока; 3—капилляры для
определения температуры плавления;
4 -термометр; 5 — заднее освещение;
б —боковое освещение; 7 —лупа;
S — микрогорелка; 9 —цилиндр из
слюды; 10 — регулятор полами воз-
духа; 11 — штатив; 72—включение
освещенин.
точку плавления *. Поэтому довольство-
ваться определением только температуры плавления смешанной пробы не
рекомендуется; в целях дальнейшего контроля следует определять также
и другие свойства.
КоИег L,, Brandstatter М., Вег., 75, 496 (1942).
► j t jjuAicjTijg j ичпрц' ttpuMynivi
Неопытные химики часто сомневаются, какую фазу процесса плавле-
ния считать точкой плавления. Можно дать следующее определение:
плавление начинается, когда вещество будет стекать вниз по стенкам ка-
пилляра; оно заканчивается, когда расплавленное вещество становится
совершенно прозрачным. Интервал температур между началом и концом
плавления называется температурой плавления. Когда плавление нача-
лось, следует прекратить дальнейшее нагревание и поддерживать темпе-
ратуру по возможности постоянной; только убедившись, что плавление
при этой температуре не заканчивается, можно ее осторожно немного
повысить. Чистые однородные вещества, плавящиеся без разложения, =
имеют обычно резкую температуру плавления, т. е. между началом и
концом их плавления лежит интервал не более }/°. Для загрязненных
или плавящихся с разложением веществ этот интервал может быть зна-
чительно большим. Часто перед плавлением наступает размягчение и
слипание вещества, ио это еще ие является плавлением, а лишь спека-
нием. Точные данные относительно температуры плавления можно запи- '
сать, например, следующим образом: т. пл. 124—125°, спекается, начи- ;г
иая со 118°. . 1
О мало распространенном в настоящее время микроопределеиии )
температуры плавления по Кофлеру* здесь можно только упомянуть, *
Определение температуры плавлении просто в выполнении, требует '
небольшой затраты времени и минимальных количеств вещества, поэтому, ;;
несмотря на все неточности, оно является наиболее употребительным
методом контроля. Ойо, конечно, неприменимо для веществ, жидких при
обычной температуре, а также для твердых веществ, которые при нагре- !-
вании обугливаются или разлагаются не плавясь. К последней группе":
относится прежде всего большинство сульфокислот, а также их соли
с металлами. Из жидких веществ очень многие перегоняются без разло- S
жения. В этом случае критерием чистоты должна служить температура
кипения. Очень полезным для проверки чистоты является, кроме того, j
определение плотности, которое легко осуществляется, если только;?
в распоряжении имеетси достаточное количество вещества. Сложнее опре-
деляются, но более чувствительны к загрязнению показатель пре- ;
л о м л е н 'И я н для оптически активных веществ — о п т и ч е с к о е в р а-
щ е и и е. Эти свойства очень ценны дли идентификации и проверки чи-
стоты, например в ряду терпенов.
Из оптических свойств как жидких, так и твердых веществ следует :
упомянуть цвет и флуоресценцию. Большинство органических ;
соединений в чистом состоянии бесцветно. Неочищенные продукты почти
всегда в большей или меньшей степени окрашены. При обычных опера- ;
циях очистки (перегонка, переосаждеиие, перекристаллизация, перекри- j
сталлизация с добавлением животного угли н т. и.) большая часть окра- 4
шнвающих загрязнений обычно удаляется. Слабая окраска, однако, 4
очень часто удерживается весьма прочно и не может быть удалена без ,
сравнительно больших потерь. Конечно, даже совсем слабая окраска '
бесцветного в чистом состоянии вещества является доказательством того,::
что оно не совсем чисто. Но количество окрашивающего загрязнения:
бывает почти всегда столь незначительно, что ие мешает применению ,
продукта даже в чисто научных целях и не имеет значения при техниче-
ском использовании. Трудно, конечно, установить, какая степень окраски
допустима. Можно, пожалуй, сказать, что для технических целей вполне
пригодно вещество, раствор которого ие имеет отчетливой окраски в тон-
* Kotler, Mikroskopische Methoden z.ur Identifizierung organischer Substanzen.
Beiheft zur Zlscbr. d. VDCh. Nr. 36 (1940).
rj wrruc i fttt tf* L’f/it/f/i V
49
ком слое или в виде отдельных капель. Во многих случаях окраска мо-
жет быть значительно более сильной; допустимая степень окраски зави-
сит от того, для каких целей вещество применяется, и должна устанавли-
ваться в каждом отдельном случае.
Для идентификации продуктов, которые сами по себе окрашены,
и особенно для идентификации красителей очень ценным может оказаться
определение спектра поглощения. Этот метод реже приме-
няется для проверки чистоты вещества, так как наличие загрязнений
часто не вызывает сдвига полос поглощения- Для определения чистоты
красителя в технике применяется прежде всего пробное крашение,
если имеется для сравнения чистый препарат.
Цепным индикатором чистоты часто является флуоресценция.
В нафталиновом ряду можно различить и распознать одновременно обра-
зующиеся изомеры или другие близкие соединения на основании наличия
флуоресценции. Например, щелочные растворы !{3-нафтола и почти всех
его сульфокислот обладают сильной флуоресценцией; ие флуоресцируют
только сульфокислоты, содержащие сульфогруппу в положении 1. Следо--
вательио. если при получении 2-нафтол-1-сульфокислоты из Р-цафтола
щелочной раствор полученного продукта ие обладает флуоресценцией, то
это указывает на отсутствие леизмснившегося ^-нафтола и изомерной
сульфокислоты (см. стр. 176). Кроме того, натриевая соль 2-иафтол-б-
сульфокислоты (соль Шеффера) дает в водном растворе лишь совсем
слабую фиолетово-синюю флуоресценцию. Соли обеих образующихся
одновременно 2,3,6- и 2,6.8-дисульфокислот (Р- н Г-соль) флуоресцируют
значительно сильнее, и цвет флуоресценции с ни е-зеленый. При прибавле-
нии соды флуоресценция всех трех растворов значительно усиливается,
ио характер ее ие изменяется. Чтобы фиолетово-синяя флуоресценция
соли Шеффера стала совсем незаметной, достаточно присутствии незна-
чительных количеств Р- или Г-соли. Наличие хорошо различимой фио-
летово-синей флуоресценции доказывает, что соль Шеффера практически
не содержит дисульфокислот (см. стр. 174).
У всех кристаллических веществ важным признаком чистоты является,
конечно, форма кристаллов. Наличие загрязнений, имеющих
иную кристаллическую форму или находящихся в аморфном состоянии,
может быть установлено иногда даже при простом рассмотрении, ио чаще
под микроскопом. Иногда также бывает, что загрязнение кристаллизуется
совместно с основным продуктом и не может быть непосредственно обна-
ружено. Однако такие смешанные кристаллы часто имеют другую
форму, чем чистое соединение; в этом случае присутствие загрязнения
можно установить, если имеется для сравнения чистый препарат. Про-
верка чистоты сульфокислот,. затрудненная отсутствием определимых
температур плавления, облегчается, с другой стороны, тем, что из них
можно получать многочисленные соли, часто обладающие хорошо разли-
чимой характерной формой кристаллов.
Проведение всех испытаний иа чистоту значительно облегчается,
когда для сравнения в наличии имеется препарат,
чистота которого не вызывает сомнений. Если такого препарата иет, то
его можно приготовить тщательной очисткой продукта собственного,
получения. Довольно кропотливый, но очень широко применяемый метод
испытания состоит в том, что пробу испытуемого вещества перекристал-
лизовывают и полученные кристаллы сравнивают с остатком, находя-
щимся в маточном растворе. ДЛя этого маточный раствор подвергают
фракционному упариванию и отфильтровывают кристаллы, образующиеся
из каждой фракции; последние маточники, как правило, настолько обо-
гащаются загрязнениями, что их легко распознать.
Прн проверке чистоты следует, конечно, основываться не только на
упомянутых выше физических свойствах, но также н и а химическом
поведении 'исследуемого вещества. Однако здесь чрезвычайно
трудно дать общие положения. Метод испытания должен меняться в раз-
личных случаях в соответствии с химическими свойствами испытуемого
вещества н предполагаемыми загрязнениями; при этом очеяь важно
общее знакомство с органнческимя реакциями, которые в настоящей
работе яе могут быть подробно рассмотрены. Следует только указать на
одну реакцию, которой в научной литературе уделяется обычно мало
внимания, но которую в технических лабораториях применяют очень ча-1
сто и с успехом. Это — реакция образования азокрасителей. Ояа ямеет
значение для всех соединений, которые можно диазотировать или соче-
тать с дяазосоедннениями. Красителя, которые образуются при этом из
могущих присутствовать побочных продуктов, яапример изомеров, при
правильном выборе компонентов оказываются достаточно отлячцымя от
красителя, полученного из основного вещества, и могут быть легко обна-
ружены. Подходящие компоненты в каждом отдельном случае должны
подбираться экспериментально, для чего требуется некоторый навык.’
В качестве прямера можно привести упоминавшуюся выше соль Шеф-
фера. Уже отмечалось, как можно установить наличие примеси Р-соли
в соли Шеффера. Рассмотрим теперь, как, наоборот, доказать присут-
ствие соли Шеффера в Р-соли. Совершенно чистая Р-соль при сочета-
нии ее содового раствора с дн азо с о единенном, полученным из моно-,
ацетил-п-фенялендяамяна, дает почти нерастворимый в содержащей
соль реакционной жидкости сине-красный краситель. После фильтрова-
ния получают бледно-розовый фильтрат. Если обработать таким же обра-
зом соль Шеффера, то образуется желто-красный, гораздо легче раство-
римый краситель, который в условиях опыта в осадок не выпадает. Если
в Р-соля содержится даже незначительное количество соля Шеффера, то
после отфильтровывания красителя получается более желтый я более
интенсивно окрашенный фильтрат, чем в случае чистой Р-соли. По интен-
сивности окраски можно даже судить о количестве присутствующей
соля Шеффера.
Начинающие химики, работающие в технической лаборатории, часто
задают вопрос: «Когда не вполне чистый продукт можно считать
пригодным для технических целей?» На этот вопрос можно от-
ветить: «Когда этот продукт подходят для намеченной целя, т, е. когда
имеющиеся загрязнения яе помешают его применению». Справедливости
этого зависят, конечно, от того, каково будет применение. Один и тот же
продукт может быть достаточно чистым для одной целя и оказаться со-
вершенно непригодным для другой. Продукт, использовавшийся прежде?'
без всяких затруднений, может оказаться непригодным даже для той же
самой цели, если будут изменены методы переработки. Во всяком случае,,
пригодность для технических целей меняется в различных случаях и мо-
жет быть установлена только экспериментально. ;
Чтобы помочь начинающим, можно, пожалуй, сказать, что вещества^
плавящиеся ниже на 2—3°, чем нужно, обычно еще применимы для TeXJ
ничеекях целей. О том, что не слишком сильная окраска в большинстве
случаев не мешает, уже упоминалось. Наконец, следует еще указать на?
то, что неорганические солн, которые, конечно, содержатся во всех про-
дуктах, выделенных высаливанием, почти никогда яе влияют иа возмож-
ность применения этих продуктов и обычно даже ие считаются загрязне-
ниями. Наличие ясоргаяических солей надо принимать во внимание лишь
при расчете количеств.
О правилах ориентации
51
О правилах ориентации
Химику, работающему в области красителей, часто бывает необхо-
димо синтезировать новый промежуточный продукт определенного строе-
ния я с определенным положением заместителей или найти новый способ
получения уже известного продукта. Решение этих задач значительно
облегчается, когда хороню известны все закономерности, касающиеся
места вступления заместителей, и которые наблюдаются при замещениях
в различных ароматических системах. Поэтому эти «правила ориентации»
следует обсудить несколько более подробно.
1. Ориентация в риду бензола
Монозамещеняые производные бензола существуют только в одной
форме. По прн вступления в молекулу второго заместители возможно
образование трех изомеров — орто, мета я пара. Какое из трех соедине-
ний в каждом отдельном случае действительно получается, зависит
в меньшей степени от характера вновь вступающего заместителя и от
особых условий реакции; главную роль здесь яграет природа уже имею-
щегося заместителя. По направляющему, иля «ориентирующему», дей-
ствию содержащихся в соединения заместителей последние можно раз-
делить на два ряда. Заместители первого ряда, к которым относятся
алкилы и арялы (дифенильная связь), галоиды —ОН, —OR*, <—О-ацил,
—NHe, —NHR*, —NR*, —NH-ацнл, —NR^-ацил, —N=N— и другие,
направляют вступающий заместитель исключительно иля почти исключи-
тельно в орто- я нпрп-положения. К заместителям второго ряда относятся
группы - NO2, —SO3H, —SO2C1, — SO2R*, - СООН. — COOR*.
—CONHR*, —COR*, —СНО, —CN н т. д.; этя группы направляют пре-
имущественно — я ляшь в редких случаях исключительно — в л/ега-поло-
жение.
Преимущественное образование орто- или ппрп-соединеняя при на-
личия заместителя первого рода завясит яе только от уже имеющегося,
но я в значительной степени также от вновь вступающего заместителя;
часто также оказывают влияние условия реакции, особенно температура.
Сульфогруппа вступает чаще всего н лара-положение, причем в случае
хлорбензола и толуола лишь в одно положение; то же самое имеет
место и в случае фенола, если сульфировать его пря нагревания; если
сульфирование вести на холоду, то образуется значительное количество
фенол-о-‘сульфокислоты. Пря галоидировании и нитрования обычно обра-
зуются оба изомера одновременно; кроме того, при иитрованяи толуола
образуется также несколько процентов м-нитротолуола.
Ориентирующее влияние первичной, вторичной я третичной амино-
групп сильно ослабляется в присутствии больших количеств концентри-
рованной серной Кислоты; поэтому пря нитрования в растворе концентри-
рованной серной кислоты, а также при сульфярованяя концентриро-
ванной серной кислотой иля олеумом иногда образуются значительные
количества жега-соединений. г
Направляющая сила заместителей, ориентирующих в era-положе-
ние, всегда гораздо меньше, чем заместителей первого рода. Это находит
свое выражение в том, что, как правило, наряду с л^ета-производным '
в качестве побочных продуктов образуются также оба других изомера.
Если в бензольном ядре уже имеются два заместителя, то их ориен-
тирующее вляяияе на третий вновь вступающий заместитель может быть
* Н>алкил ИЛИ апил
л J t/J ftpuuyfii bb
совпадающим; в случае же несовпадающей ориентации ориентир у ющи1
влиянии могут конкурировать между собой. Первое происходит в тон
случае, когда два заместителя, принадлежащих к одному роду, иахо
дятся в .мега-положении друг к другу или когда заместители, относящиеся
к различным родам, занимают друг относительно друга орто- или пара
положения. Например:
Вступление нового заместителя происходит в этом случае исключнтельн
в места, определяемые совместным влиянием обоих уже имеющихся за
местителей. Направления замещений указаны в формулах стрелками.
Если же оба заместителя одного и того же рода находятся в орте
или пара-положен ни или если они принадлежат к разным родам и нахе
дятся в лгета-положеиин друг к другу, как это показано в приведении»
ниже примерах, то они направляют в разные места н их влияние в из
вестной степени взаимно уничтожается.
Следствием этого является образование всех возможных изомеров
Только когда одним из заместителей является окенгруппа, его влияний
остается преобладающим. Так, например, в случае и-крезола, п-хлорфе
нола, л-ацетаминофеиола каждый новый заместитель вступает исключи
тельио в орго-положеиие по отношению к оксигруппе. Алкилирование ил]
ацилирование заметно ослабляют силу ориентирующего действия окси
группы. Все же оно остается таким же по силе, как действие свободней
или ацилированной аминогруппы, и значительно более сильным, чем дей
ствне галоидов и алкильных групп. Два последних типа заместителей
очень сходны по своему ориентирующему действию; если они располо
жены таким образом, что их ориентирующие действия не совпадают, т<
всегда получаются смеси возможных изомеров в приблизительно равны:
количествах (например, при нитровании п-хлортолуола).
Ориентирующее действие аминогруппы, сильное само по себе, ка1
уже упоминалось, значительно ослабляется в присутствии больших коли
честв концентрированной серной кислоты. Это особенно относится к сво
бодным аминам и в меньшей степени к их ацильным производным. Так
ацето-л-толуидид нитруется водным раствором-азотной кислоты исключи1
тельио в орго-положеини к ацетаминогруппе; в растворе же коицеитри1
рованной серной кислоты наряду с 3-иитроизомером образуются значи1
тельные количества 2-нитро-4-ацетаминотолуола. Сам л-толундин дает
при нитровании в присутствии очень большого количества концентриро-
ванной серной кислоты только 2-нитро-4-аминотолуол (см. стр. 149).
Совершенно аналогично ведут себя л-хлораиилип и его ацетильное про-
изводное. При сульфировании аминов концентрированной серной кисло-
той или олеумом влияние других заместителей также может значительно
усилиться или даже стать преобладающим. Напротив, прн сульфирова-
нии методом запекания (см. стр. 116) сульфогрулпа вступает всегда
только в орто- или лара-поЛоженне по отношению к аминогруппе; при
этом заместители, направляющие в другие положения, влияния не ока-
зывают. При нитровании можно также полностью избежать образования
производных лгиитроанилииа, если нитровать арилсульфо- (особенно
п-толуол сульфо-) производные аминов* азотной кислотой в водном рас-
творе или в органическом растворителе **.
При дальнейшем замещении соединений (уже имеющих два замести-
теля) из различных изомеров, которые могут образоваться в соответствии
с приведенными выше правилами, обычно получаются главным образом
несимметричные (замещенные в 1,2,4-положениях) производные. Сим-
метричные (1,3,5-) тризамещениые продукты образуютси лишь с трудом,
а смежные (1,2,3-) производные получаются чаще всего только в качестве
побочных продуктов в совсем незначительных количествах (исключение
составляет хлорирование p-нитротолуола, см. стр. 144).
Если ориентирующие влияния заместителей первого и второго родов
не совпадают, то определяющим или даже преобладающим будет вообще
влияние заместителя первого рода.
Сказанное о введении третьего заместителя в двузамещенные про-
изводные ряда бензола относится также и к дальнейшему введению чет-
вертого заместителя в трнзамещеиные производные. Прн этом следует
только иметь в виду большую склонность к образованию симметричных
(1,2,4,5-замещеииых) форм; это часто приводит к тому, что такие сим-
метричные формы получаютси в качестве главного продукта даже в тех
случаях, когда в соответствии с действием имеющихся трех заместителей
можно было бы ожидать преобладающего образования другого изомера.
Так, при нитровании 2-хлортолуол-4-сульфокислоты образуется главным
образом 5-иитро-2-хлортолуол-4-сульфокислота, хотя в положение 6 на-
правляют одинаково и метильная группа и сульфогруппа, а в положение
5 — только хлор. То же самое наблюдается п-ри нитровании 1,2-днхлор-
бензол-4-сульфокислоты, 4-иитро-2-хлортолуола и т. д.
2. Ориентация в ряду нафталина
В ряду нафталина уже при введении первого заместителя возможно
образование двух изомеров а и ?. Прн нитровании и при галоидирова-
нии получается практически только а-соедииенне. Напротив, при суль-
фировании концентрированной серной кислотой образуется уже смесь
обоих изомеров ***, состав этой смеси зависит от температуры сульфиро-
вания: на холоду в качестве главного продукта реакции образуется
«-сульфокислота, а прн температурах выше 120—130°—,3-сульфокис-
лота. Следует также отметить, что образующиеся нафталннсульфо-
кислоты прн нагревании с" концентрированной серной кислотой Win
* п-Толуолсульфа ни лиды также и в других отношениях ведут себя очень сходно
с фенолами: они растворяются в едких щелочах, некоторые из них сочетаются с ди-
азосоединениями и т. д.
.** Герм. пат. 157859, 164130, 163516 (Frdl.. VIII, 104—НО).
' Нафталин-а -сульфокислота без примеси изомера может быть получена суль-
фированием хлорсульфоновой кислотой в органическом растворителе.
с олеумом часто претерпевают перегруппировки, причем между различи
нымн изомерами устанавливается равновесие, зависящее от темпера*.;
туры.
При вступлении второго заместителя, а также при дальнейшем.:
замещении наблюдаются закономерности, значительно отличающиеся от?
действующих в бензольном ряду. Их можно обобщить следующим обра*^
зом: сильно ориентирующие заместители первого рода (окси-, алкокси-/
амино- и ацил аминогруппы в отсутствие концентрированной серной кис*-;
лоты), находящиеся в положении 1 нафталинового ядра, направляют,
вновь вступающий заместитель, как и в ряду бензола, в орто- и лара-
места, т, е. в положения 2 и 4. Прн наличии заместителей первого рода/
обладающих более слабым ориентирующим действием (галоиды, амино-;;
группа в присутствии концентрированной серной кислоты), наряду с заФ
мещеннем в положение 4 идет замещение не в 2-, а в 5-положеиие. •
Заместители первого рода, находящиеся в положении 2, напра-?
вляют частью в положение 1 (такое влияние проявляется тем больше,,
чем сильнее ориентирующее действие заместителя), частью в другое?
ядро, преимущественно в положение 8, и в меиьшей степени в положе*’
ние 6; -нафтиламии дает при сульфировании все четыре замещенные 1
в другом ядре моносульфокислоты одновременно. Положение 3 заме-1
щается лишь в редких случаях и то лишь тогда, когда по меиьшей мере -
один заместитель уже находится в другом ядре. Исключение составляет ?
действие двуокиси углерода на (3-иафтолят натрия, которое приводит при j
более низкой температуре к 2,1-оксинафтойной кислоте, а прн более вы-S
сокой — 2,3-нзомеру. ;
Заместители второго рода направляют вновь вступающий замести-
тель преимущественно в другое ядро; мета-положение того же ядра за- J
мешается обычно лишь тогда, когда другое ядро уже замещено, (
Прн введении второго и следующих заместителей действует в об-
щем то же правило, по которому галонды и нитрогруппа вступают j
преимущественно в «-положение, в то время как сульфогруппы при бо- j
лее низких температурах замещают главным образом a-место, а при
более высоких — преимущественно р-место.
Наряду с упомянутыми правилами для процесса сульфирования
действует правило Армстронга и Винна, по которому сульфогруппы при
прямом сульфировании никогда не вступают в орто-, пара- или пери- ?
положения друг к другу *, Это сильно ограничивает число возможных J
изомеров, которое иначе было бы чрезвычайно велико. Сульфирование
нафталина представлено приведенной ниже схемой (днсульфокислота, >
заключенная в скобки, получается лишь в незначительных количествах). -
ственно к 1,5- н 1,8-дниитронафталину. 3
При нитровании нафталинсульфо кислот замещаются главным о бра-
зом «-положения несульфированного ядра. Если сульфогруппы имеются |
в обоих ядрах, то нитрогруппа вступает в то «-место, которое ие нахо- 3
днтся в орто- или дара-положенин к сульфогруппе. Если такого места
не имеется, то нитрогруппа вступает как в дара-положенне к сульфо- 1
группе, так и в вместо, находящееся в лшта-псложении к сульфогруппе; :
в результате образуются смесн. Нафт ал ин-1,3,5,7-тетрасульфокислота -1
не нитруется.
Сульфирование нафтолов и иафтиламииов рассматривается ниже :>
при описании соответствующих соединений.
* Редкие, и ле имеющие промышленного значения исключения из этого правила
иногда встречаются при наличии других сильных ориентирующих заместителей, но не
при замещениях одними только сульфогруппами.
Дальнейшее нитрование «-нитрона фтал ина приводит пренмуще-
SO;1H
56
А Промежуточные продукты
3. Ориентация в ряду антрахинона
Правила замещения в ряду антрахинона являются несколько более
сложными, чем в ряду бензола и нафталина.
Сульфирование антрахинона в противоположность сульфированию
бензола и нафталина нельзя осуществить действием концентрированной
серной кислоты, так как при этом температура реакции будет так вы-
сока, что антрахинон в значительной степени разложится. Вследствие
этого следует применять дымящую серную кислоту, что позволяет про-
водить реакцию при более низкой температуре. Однако оказывается,
что в молекулу антрахинона нельзя ввести количественно лишь одну
сульфогру пну, так как одновременно с введением одной группы тотчас
вступает еще вторая. Если хотят получить более или менее чистую
моносульфокнслоту, то сульфирование следует вести таким образом,
чтобы примерно 50% взятого антрахинона не вступило в реакцию. Но
даже и в таких мягких условиях всегда образуются довольно значи-
тельные количества дисульфокнслот.
При действии на антрахинон обычной дымящей серной кислоты
сульфогруппы вступают исключительно в ^-положения (см. приведенные
ниже формулы).
Примерно по 50% 2,6-и 2,7-ди сульфокислот *).
Наоборот, прн сульфировании в присутствии солей ртути сульфо-
группа вступает сначала преимущественно в a-положение. При сульфи-
ровании для получения дисульфокнслот процесс значительно ослож-
няется образованием по меньшей мере четырех изомеров (1, 1,8-, 1,6- н
1,7-дисульфокислот). Их можно довольно удовлетворительно разделить,
охлаждая реакционную смесь (иногда добавив немного воды). При
этом 1,5-'ди1сульфокислота выпадает в осадок почти количественно. При
более сильном разбавления выделяется 1,8-днсульфокислота и в серно-
кислом растворе остаются 1,6-и 1,7-дисульфокяслоты **,
* См., например, Helv. Chim. Acta, 10, 219 (1927).
** См., например, Helv. Chim. Acta, 10, 200 (1927).
О правилах ориентации
57
а-Сульфокислоты ряда антрахинона интересны тем, что их сульфо-
группа легко отщепляется при действии хлора в момент выделения
и может быть заменена на хлор (а также па бром). Таким путем легко
и с хорошим выходом получают достаточно чистый «-хлорантрахинон,
Р-Сульфогруппы также способны к такому превращению, однако
реакция идет очень вяло и с плохими выходами, причем наряду с заме-
щением на хлор идет также замещение на водород.
Нитрование антрахинона протекает совершенно аналогично
сульфированию, только при этом соли ртути не оказывают влияния на
ориентацию. Технически пригодного способа получения мононитроан-
трахинона до сего времени не нмеетси *, Необходимые данные о полу-
чающихся днлнтроантрахннонах приведены ниже. В данном случае
* О получении мононитропроизводных 2-метилантрахинона см. стр, 207, а также
Looher. F'nr? Иь1г ГК™ bio in RJO /10071
os
i. Промежуточные продукты
также оказалось, что 1,5-производное является наименее растворимым
и может быть очень легко получено в чистом виде *.
Для реакции нитрования антрахинонсульфокислот наблюдаются те
же закономерности, что и в ряду нафталина. Всегда образуются смеси
различных изомеров, которые легко разделяются в случае 1,5-производ-
ного и трудно — во всех других случаях.
Аминирование антрахинона можно проводить различными спосо-
бами. Для этой цели используют замену сульфогруппы на — NHj,
а также и восстановление соответствующих питросоединений. Восста-
новление нитросоединений идет здесь очень легко прн кипячении
с раствором сернистого натрия; при этом всегда промежуточно обра-
зуется зеленое, легко растворимое в сернистом натрии производное
гидроксиламина, после чего чистый амин выпадает в виде красного
кристаллического осадка, который достаточно только промыть. При за-
мене сульфогруппы, происходящей под действием аммиака прн повы-
шенных температурах (следовательно, под давлением, см. стр. 208
и 210), необходимо образующийся сульфит аммония удалять в про-
цессе реакции, так как в противном случае производное антрахинона
подвергнется глубоко идущему восстановительному расщеплению. В ка-
честве добавок, делающих сульфит безвредным, используют вещества,
окисляющие его, например пиролюзит, мышьяковую кислоту, .^-нитро-
бензол сульфокислоту; применяют также осаждение сульфита добавле-
нием солей щелочноземельных металлов или солей магния (см. стр. 209).
Такого рода аминирование идет не количественно, так что при получе-
нии, например, ^-аминоантрахинона в настоящее время чаще всего
исходят нз 2-хлор антрахинон а, который легко получается из фталевого
ангидрида и хлорбензола **.
Введение оксигруппы в молекулу антрахинона производится нагре-
ванием соответствующей сульфокислоты с гидроокисью щелочноземель-
ного металла, при этом осаждается вредно действующий сульфит. Прн
применении едкой щелочи наблюдаются нежелательные побочные реак-
ции, которые иногда, а особенно в присутствии окислителей, приводят
к образованию производных антрахинона с более высокой степенью
окисления (см. при описании ализарина на стр. 281). Хянизарин,
т, е. 1,4-диоксиантрахниоц, в настоящее время получают из фталевого
ангидрида и 1,4-хлорфенола конденсацией под действием коицентриро-
+ NH4CI
* См. Не f t ц Helv. Chim. Acta, 14, 1404 (19311.
Герм. пат. 75288 (FrdL, HI, 260).
Герм. пат. 75288 (FrdL, HI, 260); пат. США 1746736 (Chi. 1930, J, 2798),
О правилах, ориентации
59
ОН О—ВО ОН
’ 1 г
3.
\/\С0/ Y 1 V/Xco/'4/ ^/Xco/Y
он о—во он
ХиниЗарин **
ванной серной кислоты в присутствии борной кислоты, которая пере-
водит образующийся хинизарин в эфир борной кислоты и предохраняет
его, таким образом, от дальнейших 'изменений ***.
При дальнейшей переработке упомянутого выше производного ан-
трахинона иногда возникают значительные осложнения, что здесь
можно лишь кратко отметить.
а-Аминоантрахцнон может быть сравнительно легко просульфиро-
ван в положении 2 дымящей серной кислотой в присутствии бисуль-
фата. 1-Амино-2-сульфокислота антрахинона гладко получается также
запеканием кислого сульфата 1-амииоаитрахинона при 200°***'*, она яв-
ляется очень важным исходным веществом для получения ценных
кислотных красителей ряда антрахинона *****.
Вместо аминогруппы могут быть введены также и другие аминные
остатки. Особенно важными являются антримиды, то есть диантрахн-
нониламины, которые конденсацией могут быть переведены в коричне-
вые кубовые карбазоловые красители ряда антрахинона. Такая
конденсация осуществляется чаще всего с помощью хлористого алюми-
ния и лишь в редких случаях действием двойной солянокислой соли
алюминия и натрия или хлористого алюминия и пиридина. Подробнее
об этом можно узнать в монографии Г. Кренцлейна ******. в этой книге
описано много синтезов, часть из которых приобрела большое промыш-
ленное значение, как например синтез дибензпиренхинона (нндантрен
золотисто-желтый ГК или кубовый золотисто-желтый ЖХ), который по-
лучается из бензантрона и хлористого бензоила в присутствии хлористого
алюминия и кислорода.
Галоидирование антрахинона идет лишь с трудом. Амино- и окси-
производные, напротив, галоидируются легко; лучше всего реакция
идет с бромом. Так, из а-амииоаитрахинона в ледяной уксусной кислоте
«гладко образуется 1-амиио-2,4-дибромантрахинон, являющийся важ-
ным "исходным продуктом для получения ценных кислотных красите-
лей для шерсти (см. стр. 211).
Для полноты здесь следует упомянуть и так называемую реакцию
Бона — Шмидта, состоящую в окислейин антрахинона дымящей серной
кислотой (иногда в случае нитросоедииений применяется добавка серы
и борной кислоты). В настоящее время эта реакция в технике приме-
няется мало*******.
Вследствие повышенной реакционной способности антрахинона
в отношении различных агентов в ряду его производных могут быть
проведены такие синтезы, которые неизвестны для соединений ряда
бензола и нафталина. Можно упомянуть индантреновый плав, прн ко-
Или п-хлорфенол; см. герм. пат. 255031 (FrdL, XI, 588).
** Губен, Антрацен и антрахинон (1929).
*** См. стр. 216, а также герм. пат. 2&503I (Frdl., XI, 588).
Huber. Диссертация, Zurich, 1931, S. 49.
***** Fierz-David, Kunstliche Organische Farbstoffe, S. 77.
p Креццлейн, Хлористый алюминий в органической химии, Гл. ред. хим.
лит, Москва, 1935.
' Fierz-David. Kiinstliche Organische Farbstoffe, S. 538.
i. промежуточные продукты
тором две. молекулы антрахинона связываются двумя NH-группами,
и дибензантроновый плав, когда две молекулы бензантрона образуют
ценные кубовые красители. Повышенная реакционная способность про-
является часто в еще большей степени у производных антрагидрохи-
нона. Примером может служить синтез основания хиинзаринового
зеленого — кислотного зеленого атрахнпонового (см. стр. 284).
4. Ор и е нтац н я в ряду карбазола
Карбазол в реакциях замещении ведет себя как производное ди-
фениламина и образует в первую очередь производные, замещенные
СМ NO.
/. Хлорбензол 61
в орто- и яара-положеннях к аминогруппе. Ввести заместитель
в м ета-положе пне к азоту удастся только косвенным путем. Для этого
известны два способа: 1) предварительное замещение одного нлн обоих
лпрд-положсний. сильными ориентирующими заместителями первого
родя, которые направляют вновь вступающий заместитель в жета-поло-
жепие по отношению к азоту и которые затем удаляются; 2) введение
нужных заместителей и, кроме того, двух аминогрупп (в положении 2
н 2Z) во взятый в качестве исходного вещества дифенил и последующее
замыкание полученного производного с образованием ядра карбазола»
например см. формулу на стр. 60.
А. СОЕДИНЕНИЯ РЯДА БЕНЗОЛА
1, Хлорбензол
Хлор вступает в идро бензола только в присутствии переносчиков
галоида. В их отсутствии идет лишь реакция присоединения с образо-
ванием гексахлорцнклогексаиа. В качестве переносчика в технике при-
меняется только железо — целесообразно в виде тонкого порошка.
Хлорирование лучше всего проводить приблизительно прн 30°. Прн
более низких температурах реакция идет плохо, а более высокие
благоприятствуют образованию побочных продуктов (ди- и полнхлор-
бензолы; часто и СбНеС16, особенно на солнечном свету). Хлор посту-
пает нз баллона через редукционный вентиль, который позволяет очень
точно регулировать скорость подачи газа. Здесь, как и во всех аиало-
* гичных реакциях хлорирования, важно полное отсутствие влаги;
присутствие даже следов ее сильно замедляет хлорирование и способ-
ствует образованию побочных продуктов. Поэтому перед реакционным
сосудом должны быть установлены по меньшей мере две обычные про-
мывные склянки с концентрированной серной кислотой или, лучше,
одна так называемая «спнральнаи промывная скляика» достаточных
размеров. В последней пузырьки газа проходят по спирали длинный
путь через серную кислоту. За промывными склянками для задержа-
ния увлеченных газом капелек серной кислоты ставится осушительная
колонка, наполненная пемзой, нлн включенная «наоборот» промывная
склянка, содержащая немного стеклянной ваты (см. рис. 5).
В качестве реакционного сосуда при хлорировании используют пре-
имущественно довольно высокий и узкий стеклянный цилиндр, в котором
хлор должен пройти через высокий столб жидкости. Этот сосуд соеди-
няется (лучше всего посредством шлифа) с возможно более длинным
и эффективным обратным холодильником; если пришлифованный холо-
дильник отсутствует, то соединение производится с помощью обрабо-
танной ацетил целлюлозным лаком корковой пробки. Сосуд, где произ-
водится хлорирование, помещается в водяную баню, через которую
может циркулировать холодная вода. Выделяющийся нз холодильника
i. промежуточные продукты
Хлор
Раствор
щелочи
Водя-
ная
Ваня
Р и с. о, Лабораторный прибор для хлорирования.
хлористый водород отводится в круглодонную колбу с раствором едкого
натра илн с холодной водой; эта колба не должна быть плотно закрыта,
чтобы воздух мог выходить из прибора. Отводная трубка устанавли-
вается непосредственно над поверхностью поглощающей жидкости,
при этом, чтобы избежать засасывания, ее нельзя погружать в жид-
кость.
Отдельные части прибора должны быть собраны по возможности
так, чтобы концы стеклянных трубок находились встык друг к другу и
соединялись короткими кусками толстостенной вакуумной резиновой
трубки. Там, где неизбежны более длинные соеди-
нения, резиновую трубку применять не следует,
так как она быстро разрушается хлором; в таких
случаях используются стеклянные или свинцовые
трубки; последние перед употреблением надо тща-
тельно очищать и высушивать продуванием.
В сосуд для хлорирования емкостью 500 мл по-
мещают 312 г * (4 моля) сухого бензола и 5 г
железного порошка н затем пропускают
довольно быстрый ток хлора.
Реакции начинается только после того, как
в бензоле растворится значительное количество хло-
ра; начало реакции определяется по внезапному
повышению температуры. После этого скорость про-
пускания хлора временно уменьшают н включают
водяное охлаждение. За-
тем ток хлора можно сно-
ва увеличить и отрегули-
ровать его так, чтобы тем-
пература реакционной
смесн была постоянной и
равнялась приблизительно
30°. Прн очень быстром
пропускании хлора он так-
же поглощается почти це-
ликом. Хорошая работа
обратного холодильника
должна обеспечить воз-
можно более полное воз-
вращение бензола, захва-
ченного выделяющимся
хлористым водородом.
Образующийся при реакции хлористый водород отводится в двух-
литровую колбу, в которой находится вода или смесь 300 мл раствора
едкого натра (уд. вес 1,26) и 700 мл воды. Ход реакции нужно время
от времени контролировать взвешиванием. Можно определять убыль
в весе баллона с хлором или увеличение веса сосуда, в котором произ-
водится хлорирование, нли увеличение веса поглотительного сосуда.
В технике обычно взвешивают баллон с хлором. В лаборатории часто
не имеется подходящих весов, которые при такой большой нагрузке
обладали бы чувствительностью до 1 г. Удобнее всего взвешивать
поглотительный сосуд; это, конечно, можно делать только тогда, когда,
* Количество, которое можно прохлорировать в лабораторных условиях'за один
день.
1. ллорйензол
как в данном случае, хлор потребляется целиком. Все же для контроля
рекомендуется взвешивать (по крайней мерс в конце реакции) также
п сосуд, в котором проводилось хлорирование (определяется увеличение
в весе). При этом следует иметь в виду, что поглощение 1 г-атома хлора
обусловит увеличение веса реакционного сосуда на 34,5 г (С1—И),
увеличение веса поглотительного сосуда па 36,5 г (Cl + Н) и умень-
шение'веса баллона с хлором на 71 г (СЬ).
Реакцию прекращают, когда поглотится 80% того количества хлора,
которое необходимо по расчету для образования монохлорбензола, т. е.
когда вес поглотительного сосуда увеличится на 117 г. При дальнейшем
хлорировании количеетво более высокохлорированных побочных про-
дуктов быстро увеличивается, а выход монохлорбензола почти не уве-
личивается. Железу дают осесть, жидкость декантируют в делительную
воронку и тщательно промывают разбавленной соляной кислотой, затем
раствором соды и, наконец, водой. После этого продукт реакции, вес
которого составляет примерно 410—420 г, подвергают дробной перегонке,
которую целесообразно заканчивать в вакууме. Лучше всего приме-
нять колонку высотой не менее 60 см, наполненную стеклянными коль
цами *и снабженную дефлегматором, как описано на стр. 304. Такая
колонка позволяет достичь разделения в результате одной перегонки.
Получают приблизительно следующие фракции. При атмосферном дав-
лении-.
1) 70—100° — бензол с небольшой примесью хлорбензола ~ 70 г;
2) 120—130° — почти чистый хлорбензол—295 г.
При 9 мм остаточного давления:
3) до 55° — моно- и дихлорбеизол ~ 10 а;
4) 55,5° — о- и п-дихлорбензол ~ 40 а;
5) очень небольшое количество вышекипящих продуктов и немного
непсрегоняющего остатка.
Из фракции 4 при охлаждении приблизительно до 10° и центри-
фугировании получают около 12 г химически чистого л-дихлорбен-
зола.
Выход мопохлорбензола 295 г, т. е. 65%, считая на взятый в реак-
цию бензол, и 84%, считая па вступивший в реакцию.
Технические примечания, Хлорбензол стал важным промежуточным
* продуктом для получения целого ряда других соединений (см. раздел 6 и табл. 1—5).
Он производится в количествах 2000 иг и более в один прием; в производстве исполь-
зуются чугунные котлы с мешалкой и обратным холодильником.
Вводят только около 00% теоретического количества хлора, вследствие чего
количество образующихся более высокохлормроваиных продуктов сильно снижается,
а выход монохлорбензола, считая на вступивший в реакцию бензол, значительно по-
вышается. Бензол, не вступивший в реакцию, всегда возвращается в цикл и снова
хлорируется. Ректификация проводится очень точно (о дробной перегонке см. также
стр. 304 и сл.).
Соляная кислота, образующаяся при подобных процессах хлорирования, соби-
рается в бутылях. Незначительная примесь хлора нейтрализуется добавлением неболь-
шого количества бисульфита натрия. Такая соляная кислота находит большое при-
менение на заводах красителей, так как она является дешевой и очень чистой.
Дихлорбепзол, всегда образующийся в незначительных количествах наряду
с монохлорбензолом, представляет собой смесь орто- и пара-изомеров. Последний из
них можно легко получить в чистом виде, так как он при обычной температуре пред-
ставляет собой твердое вещество; он служит для приготовления целого ряда проме-
жуточных продуктов (см. раздел 6 и табл. 1) и под названием «глобол» применяется
как средство против моли. Жидкий о-дихлорбензол лишь с трудом освобождается
от примеси ziapa-изомора. Поэтому его дальнейшая переработка затруднительна и он
чаще всего используется как высококипящий и довольно инертный раство-
ритель.
Интересный метод хлорирования бензола состоит в том, что раствор хлора в бен-
золе пропускают через контактную колонку, заполненную железными стружками.
64
i. промежуточные n poo унты
Количество образующихся ди- и полихлорбензолов можно значительно снизить.
Для этого хлор непрерывно приводят н соприкосновение с большим избытком стекаю-
щего вниз бензола (это производится в трубке) и смесь пропускают через катализа-
тор в сосуд, в котором производится кипячение; не вступивший н реакцию бензол
отгоняется из этого сосуда через эффективную ректификационную колонку снова
в реактор (где он хлорируется), а образовавшийся хлорбензол остается в сосуде.
Таким образом удается превратить в моцохлорбепзол до 95% бензола.
Общие сведения о галоидировании
В технике хлорирование в огромном большинстве случаев осуществляется пря-
мым действием газообразного хлора. Вещества, которые при температуре реакции на-
ходятся в жидком состоянии, обычно обрабатываются газообразным хлором без до-
бавления растворителя, как это, например, описано выше для бензола; твердые же
вещества должны, как правило, растворяться в подходящем растворителе или суспен-
дироваться. Конечно, следует применять только такие растворители, которые в данных
условиях не взаимодействуют с хлором или взаимодействуют с ним лишь с трудом.
Из неорганических жидкостей в качестве растворителей применяются вода и кон-
центрированная серная кислота, а из органических — главным образом четыреххло-
ристый углерод, тетрахлорэтан, нитробензол, о-дихлорбензол, трихлорбензол и ледя-
ная уксусная кислота. Всегда следует следить за тщательностью перемешивания, так
как оно обеспечивает равномерность действия галоида и более полное его исполь-
зование. Лучше вводить хлор в возможно более тонко распределенном состоянии,
пропуская его предварительно через цилиндр из пористого материала. Кроме того,
рекомендуется, особенно когда хлор трудно поглощается, применять высокий и срав-
нительно узкий сосуд, чтобы газ проходил через возможно более высокий столб жид-
кости.
Многие реакции хлорирования протекают гладко лищь в отсутствие даже следов
воды, В таких случаях хлорируемое вещество, а также растворитель и хлор следует
тщательнейшим образом высушивать. Но даже н тогда, когда не требуется применять
сухой хлор, его все равно пропускают через промывную склянку с концентрирован-
ной серной кислотой. Это делается для того, чтобы можно было наблюдать скорость
пропускания газа. Ход хлорирования контролируют, взвешивая или баллон с хлором,
или реакционный сосуд; можно также взвешивать сосуд для поглощения хлористого
водорода, если в последний не попадает не вступивший в реакцию хлор. При этом
следует иметь в виду, что в молекулу вступает только половина вошедшего в реак-
цию хлора, а другая половина выделяется в виде хлористого водорода. Надо также
помнить, что как хлор, так и хлористый водород иногда хорошо растворяются в ре-
акционной смеси, вследствие чего может показаться, что хлора поглотилось слиш-
ком много. Кроме того, выделяющийся хлористый водород может увлекать легколету-
чие вещества или растворитель. Этого стараются избежать, применяя достаточна
большой и эффективный обратный холодильник.
Хлор очень ядовит, поэтому весь прибор для хлорирования всегда следует уста-
навливать под хорошо действующей тягой и тщательно проверять его герметичность;
следует также по возможности не соединять отдельные части прибора длинными
кусками резиновой трубки, так как последняя быстро разрушается хлором; герме-
тичность всех соединений обеспечивается их перевязыванием.
Условия, в которых следует проводить различные реакции хлорирования, зави-
сят от природы исходного вещества и могут колебаться в широких пределах. Арома-
тические углеводороды, а также ‘ их галоидо- и нитроироизводиьге хлорируются
в ядро чаще всего только в присутствии переносчиков галоида *. В технике в ка-
честве переносчика используется преимущественно железо как таковое или в виде
хлорного железа; иногда также применяются соединения йода с железом, реже — сам
йод, сурьма или хлориды последней. Реакцию обычно проводят при комнатной или
слегка повышенной температуре, Для введения хлора в боковые цепи гомологов аро-
матических углеводородов следует работать при полном отсутствии названных выше
переносчиков, ирп высокой температуре и желательно при освещении; в таких слу-
чаях часто бывает полезной добавка фосфора или его хлоридов.
Вследствие сильного окисляющего действия хлора на амины последние редко
хлорируются в свободной форме. Таким исключением является, например, н-нитроани-
лип, который в присутствии концентрированной соляной кислоты довольно гладко
хлорируется и образует при этом 2-хлор- и затем 2,б-дихлор-4-нитроанилин;
* В отсутствие переносчиков хлор только присоединяется или вступает в боко-
вую цепь, если опа имеется.
/, Хлорбензол
N03, N02 NO,
Аминогруппа чаще всего защищается ацетилированием или введением других кислот-
ных остатков; кроме того, во избежание побочных реакций следует работать при
охлаждении, С такими соединениями хлор реагирует очень легко даже в отсутствие
переносчиков.
Еще легче пропсхюдмт галоидирование фенолов; поэтому при прямом действии
галоидов реакцию трудно остановить на стадии образования однозамещенного про-
изводного. Для получение монохло'рироваиных фенолов применяются более мягкие
хлорирующие агенты. К таким агентам относятся, с одной стороны, гипохлорит
натрия (в эквимолекулярном количестве); который в щелочном растворе'гладко хло-
рирует фенол с образованием мо н о хл о размещенного (хлор вступает преимущественно
в орто-положепне к оксигруппе), и, с другой стороны, хлористый сульфурил (SOgCla),
который при действии на фенол образует главным образом «ара-соединение (см.
стр. 132). Хлористый сульфурил применяется для хлорирования не только фенолов,
но многих других соединений, в особенности ряда антрахинона, так как он действует
очень равномерно и легко дозируется.
В некоторых случаях хлорирование проходит особенно энергично и гладко,
когда действуют хлором в момент образования, Хлор получают -в самой реакционной
смеем, окисляя соляную кислоту такими окислителями, как например гипохлорит
(хлорирование ацет-о-толуидида), хлорат или азотная кислота (получение хлоранила
с применением царской водки, см. стр. 133)'.
В некоторых случаях хлор может замещать не только атомы водорода, но также
и некоторые заместители, например сульфогруппу, карбоксил, нитрогруппу и т. п.
Именно такие реакции часто применяются в ряду антрахинона (ом. стр. 215). Они
также известны и в ряду бензола. Так, например п-толуолсульфохлорид при обра-
ботке хлором при высокой температуре в отсутствие железа и сурьмы, но в присут-
ствии пятихлориедбго фосфора дает л-хлорбензотрихлорид; при этом происходит за-
мена ЗО2С1-груипы на хлор®.
cio2s—6 9-сн3 —— С1^А СС13
Почти псе сказанное выше о хлорировании относится также и к бромиро-
ванию. Только обычно бром вводится в реакционную смесь в жидком виде (до-
бавляется по Каплям). В Отдельных случаях лучшие результаты получаются, когда
действуют парами брома. Бромирование часто протекает более гладко, чем хлори-
рование (особенно в случае чувствительных .веществ), и дает более однородные и
лучше кристаллизующиеся продукты, так что иногда высокая цена брома вполне
оправдывается лучшими выходами, Если при бромировании в качестве растворителя
применяют концентрированную серную кислоту, то образующийся бромистый водород
в большем или меньшем количестве окисляется серной кислотой опять до брома.
Поэтому в таких случаях брома берется меньше, чем требуется по расчету (иногда
только половинное количество или немного больше).
Иодирование и фторирование применяются в химии красителей
лишь в редких случаях. Трифторметцлбепзолы легко получаются при действии кон-
центрированной фтористоводородной кислоты на соответствующие бензотрихлориды.
При прямом галоидировании не всегда удается вообще ввести атом хлора вили
брома в желаемое место или только в него. Так, дихлорбензол нельзя получить
дальнейшим хлорированием мопохлорбензола; о-хлортолуол образуется при хлориро-
вании толуола всегда только вместе4 с «ара-изомером, от которого его отделить прак-
тически пс удастся. В таких случаях обычно прибегают к реакции Зандмсйера (за-
мена аминогруппы па галоид через диазосоединения), хотя в производственных ус-
ловиях она довольно дорога, сложна в выполнении и редко протекает гладко. При-
бегают также к искусственному приему; он состоит в том, что место, в которое
* Герм, пат, 98433 (Frdl., V, 41)
нежелательно введение галоида, замещают сульфогруппой; последнюю после проведе-
ния реакции галоидирования снова отщепляют (см. получение о-хлортолуола из
и-толуолсульфокнслоты *, стр. 147).
2. Нитробензол
В трехгорлую колбу, снабженную хорошо действующей мешалкой,
термометром и капельной воронкой (см. рис. 6, а и б), помещают 100 а
бензола. Затем при энергичном перемешивания в течение '/з часа
туда добавляют по каплям
a б
Р и с. б. а — трехгорлая колба со снаб-
женной затвором пропеллерной мешалкой,
термометром и воронкой для' загрузки;
б-- трехгорлая колба со снабженной затво-
ром лопастной мешалкой, обратным холо-
дильником и капельной воронкой.
охлажденную смесь 11U г
азотной кислоты (уд. вес
1,44; 75% HNO3) н 170 а кон-
центрированной серной кис-
лоты (уд, вес 1,84). Темпера-
туру реакционной смеси внеш-
ним охлаждением поддержива-
ют при 50°. Вместо стеклян-
ной колбы можно также при-
менять фарфоровый стакан с
хорошо закрывающейся крыш-
кой или закрывающийся эмали-
рованный котел. Следует всег-
да следить за тем, чтобы тер-
мометр и мешалка с самого на-
чала были погружены в жид-
кость. Чтобы быть совершенно
уверенным в том, что нитрова-
ние пройдет гладко, можно так-
же применять азотную кислоту
уд. веса 1,46 (80% НЫОз); на
производстве же вполне обхо-
дятся кислотой уд. веса 1,44.
Поме прибавления всей нит-
рующей смесн перемешивание
при 50° продолжается еще 2*ча-
са; под конец температура под-
нимается до 60°. Конец нитро-
вания можно определить ана-
лизом пробы при помощи нит-
рометра: количество азотной
кислоты, определенное на осно-
вании анализа пробы, должно
соответствовать тому неболь-
шому ее избытку, который вво-
дился в реакцию. Нитробензол,
плавающий на поверхности кислоты, отделяют иа делительной воронке,
промывают сначала небольшим количеством воды, затем раствором соды
и опять водой. Нитробензол, имеющий после этого нейтральную реак-
цию (проба на лакмус!), перегоняется (см. рис. 7) **. Сначала отго-
няется немного воды н совсем немного оеизола, а затем переходит чи-
стый нитробензол.
* Герм. пат. 294638 (Frdl., XII, 908).
** С помощью этого простого прибора перегоняют без холодильника Либиха
высококипящце жидкости; лишь иногда применяют орошение приемника водой,
2. Нитробензол
67
Выход чистого нитробензола примерно 150 г, т. е. около 95% от
теории; т. кии. 205°.
Технические примечания. Нитробензол является одним из важнейших
продуктов для производства красителей. Он служит для получения анилина и бен-
зидина, а также ряда других промежуточных продуктов, особенно лета-зам еще иных
(см. табл. 6); кроме того, из него получаются имеющие большое значение нигрозины.
Нитробензол часто применяется также в качестве растворителя и мягкого окислителя
(плав фуксина, синтез хинолина). На производстве для получения нитробензола
берут сразу до 1500 аг бензола н получают выходы около 98%. При работе с такими
большими количествами процесс продолжается до 12 час. и используется до 97%
азотной кислоты. Течение процесса нитрования контролируется количественным опре-
делением остаточной азотной кислоты в смеси кислот; определение производится
с помощью нитрометра Лунге. Отработанная кислота в конце процесса должна содер-
жать лишь 1% азотной кислоты. В большинстве случаев нитробензол применяется без
дальнейшей очистки; для получения в чистом виде его всегда перегоняют в вакууме.
На рис. 8 и 9 показан нитратор с внутренним охлаждением, применяемый для
нитрования ароматических углеводородов, а также делительная воронка со смотро-
вым стеклом и с расположенным внизу краном из сплава свинца с сурьмой или из
керамики. Аппараты, применяемые для нитрования бензола, должны быть гомогенно
освинцованы, так как остающаяся после нитрования кислота вследствие большого
разбавления разъедает железо.
Общие сведения о нитровании
Нитрование ароматических соединений осуществляется на производстве в огром-
ном большинстве случаев с помощью смеси азотной и концентрированной серной
кислот (нитрующая смесь, нитрующая кислота). Серная кислота связывает воду,
образующуюся при нитровании, а также содержащуюся в азотной кислоте; она под-
держивает необходимую концентрацию последней и тем самым делает возможным
почти полное ее использование. Серная кислота является также отличным раствори-
телем для очень многих веществ, благодаря ее сравнительно большой теплоемкости
.она исключительно удобна для поглощения тепла реакции и поэтому способствует
спокойному и равномерному течению реакции. Большой избыток серной кислоты
Может даже защитить свободную аминогруппу от действия азотной кислоты
(см. 2-Нитро-4-толуидин, стр. 149)'.
На заводах красителей всегда ищется целый ряд нитрующих смесей, приготов-
ленных смешиванием при охлаждении азотной кислоты различных концентраций'
о концентрированной серной кислотой или с олеумом. В таких смесях содержание
воды и соотношение азотной и серной кислот подобраны так, чтобы можно было
проводить любые реакции нитрования. Чем труднее нитруется вещество, тем меньше
воды должна содержать применяемая нитрующая смесь; в некоторых случаях исполь-
зуют даже такие нитрующие смеси, которые содержат свободный серный ангидрид
(см. стр. 136).
68
/. промежуточные продукты.
Нитрование является сильно экзотермическим процессом, поэтому при его про-
ведении, как правила, требуется энергичное охлаждение, тем более что во многих
случаях реакция протекает гладко только при низких температурах; при повышен-
ных же температурах происходит главным образом окисление или идут другие по-
бочные реакции. Если при нитровании одновременно образуется несколько изомеров,
то температура имеет известное, однако обычно не очень большое влияние на соот-
ношение получающихся продуктов. Чтобы избежать вредного повышения темпера-
туры в том месте, куда прибавляется азотная кислота или нитрующая смесь, при
нитровании совершенно необходимо применять постоянное и
сильное перемешивание. Такое перемешивание особенно необходимо тогда,
когда нитруемое вещество не растворяется в применяемой для нитрования кислоте,
Рис. 8. Нитратор с винтообраз-
ной мешалкой и с наружным н
внутренним охлаждением.
7 — труба, подводящая воду для йиутрен-
него охлаждений; 2 — рубашка ^ля обо-
грева р охлаждения.
Р и с. 9. Делительная воронка и
экстракционным аппарат с про-
пеллерной мешалкой и смотровым
стеклом.
как это, например, имеет место в случае бензола. Если такие вещества не переме-
шивать, то образуются два слоя, и когда на их границе начинается реакция, то
вследствис_псрсгрсвания могут произойти взрывы. Однажды на анилиновом заводе
в Руммельсбурге при получении нитробензола произошел сильнейший взрыв. При-
чина заключалась в том, что при прибавлении нитрующей смеси мешалка сначала
не работала; она была включена с опозданием. С тех пор на производстве введено
приспособление, позволяющее производить прибавление кислоты только в том случае,
когда включены мешалка и водяное охлаждение. В лаборатории несчастные случаи
происходят даже при включенной мещалкс, когда последняя вначале приводит в
движение лцгпь поверхность жидкости; в тот момент, когда мешалка начинает по-
гружаться в жидкость, происходит реакция, идущая подобно взрыву. Поэтому и в
лаборатории надострого следить за тем (особенно прн работе в непрозрачных реак-
ционных сосудах), чтобы мешалка и термометр с самого начала были правильно
погружены в жидкость.
Приведенный и описанном выше примере способ работы — приливание нитрую-
щей смеси к нитруемому веществу — применяется при сравнительно немногих, но тех-
ничсски важных реакциях нитрования. Гораздо чаще нитруемое вещество растворяют
в концентрированной серной кислоте и затем в полученный раствор при персме-
шпвании и охлаждении прибавляют по каплям азотную кислоту или нитрующую
смесь. Использование нитрующей смеси имеет в данном случае то преимущество, что
тепло, выделяющееся при смешивании азотной it серной кислот, может быть отве-
дено заранее и ие прибавляется к теплоте реакции нитрования. Если нитрование
следует за сульфированием, то его, как правило, можно проводить, непосредственно
используя сульфурационную массу (см. 2-Нитрохлорбензол-4-сульфокислота, стр. 97)'.
При некоторых реакциях нитрования, например при нитровании первичных
аминов, требуется полное отсутствие азотистой кислоты в кислоте, применяемой для
нитрования; в таких случаях азотную кислоту перед употреблением (или перед
смешиванием с серной кислотой)’ следует освободить продуванием воздуха от окислов
азота (см. нитрование га-толуиднна, стр. 149). Можно также вместо азотной кислоты
вносить в сернокислый раствор селитру; этот способ, однако, менее удобен, так как
селитра плохо растворяется в серной кислоте при низких температурах, что мешает
достигнуть равномерного течения реакции.
Слишком продолжительное воздействие нитрующей смеси часто неблагоприятно
влияет на выход и чистоту продукта, поэтому рекомендуется реакционную смесь под-
вергать дальнейшей переработке тотчас по окончании нитрования. Контроль за рас-
ходом азотной кислоты, а также установление конца реакции легко осуществляются
с помощью нитрометра Лунге.
Следует также указать на то, что в некоторых случаях присутствие концентри-
рованной серной кислоты оказывает влияние на направление замещения при нитро-
вании. В присутствии серной кислоты ослабляется, например, ориентирующее влия-
ние ацетилированной или фармилирогганзой аминогруппы и в еще большей степени
свободной аминогруппы; ввиду этого влияние других наличных заместителей па ме-
сто вступления нитрогруппы становится заметным, а иногда и решающим. В неко-
торых случаях, когда такое влияние является нежелательным, приходится отказы-
ваться от применения нитрующей смеси.
Вещества, которые под действием концентрированной серной кислоты очень
легко сульфируются, омыляются или подвергаются каким-либо другим превращениям,
также не следует нитровать в растворе серной кислоты. Во всех этих случаях при-
ходится или применять водный раствор азотной кислоты соответствующей концен-
трации или нитровать концентрированной азотной кислотой вещество, растворенное
или суспендированное в органическом растворителе; последний со своей стороны,
должен быть возможно более инертным в условиях реакции по отношению к азотной
кислоте (например, ледяная уксусная кислота, нитробензол, о-дихлорбензол).
Непрямое введение нитрогрупп в ароматическое ядро в технике почти не при-
меняется.
Нитрометр Лунге
Этот простой прибор очень удобен для контроля за течением реак-
ции, нитрования, проводимых в концентрированной серной кислоте.
Применение этого прибора для определения кислот, получаемых из
окислов азота, в особенности азотной и азотистой кислот, основано на
их свойстве (которое присуще также нх солям и эфирам) быстро и
количественно восстанавливаться уже прн комнатной температуре ме-
таллической ртутью в присутствии концентрированной серной кислоты
до ХО; последняя выделяется в газообразном состоянии, н объем ее из-
меряется.
2HNO3 4- 6Hg 4- SHaSOi 2NO 4- 3HgaSO4 + 4НаО.
Нйтросоединеиия в противоположность эфирам азотной кислоты
при этом не реагируют.
Нитрометр (рис. 10) состоит из бюретки с делениями до Vio ли „ем-
костью 50 ли, которая одновременно является н реакционным сосудом /;
эта бюретка снабжена специальным краном с двумя косыми отвер-
стиями, позволяющими соединять ее или с воронкой для наполнения'3
или с выпускным капилляром 4 и уравнительной трубкой 2, соединен-
ной с ней толстостенной резиновой трубкой.
Для определения взвешивают в маленькой пробирочке 1—2 г (при-
близительно 1 мл) реакционной смеси, полученной при нитровании
г Л I и “«flute; rtjU'MUJ/A. i hl
с точностью до 0,01 г. Бюретку наполняют ртутью до самого крана н
затем вносят пробу в воронку для наполнения 3. После этого опускают
уравнительную трубку 2 и, осторожно открывая крап, впускают пробу
в бюретку. Прн этом, конечно, следует следить за тем, чтобы вместе
с пробой не был засосан и
Р и с. 10. Нитрометр Лунге.
воздух. Пробирку, в которой производилось
взвешивание, споласкивают 1—2 мл кон-
центрированной серной кислоты, которую
затем вводят в бюретку 7 так же, как
пробу. Такое споласкивание повторяют
3—4 раза, т. е. до тех пор, пока послед-
ние остатки пробы не будут переведены
нз пробярки, где производилось взвеши-
вание, и из 'воронки для наполнения
в бюретку. Общее количество кислоты,
находящейся над ртутью, должно соста-
влять 6—10 мл. Бюретку 1 с закрытым
краном (который для предосторожности
придерживают рукой) вынимают из за-
жима и осторожно приводят в прибли-
зительно горизонтальное положение та-
ким образом, что слой кислоты доходит
до того места бюретки, где присоединена
резиновая трубка, Еще до того когда
бюретка приняла горизонтальное поло-
жение, ее вновь приводят в вертикаль-
ное положение прн легком встряхивании;
при этом кислота опять поднимается вверх
к крану и проходит через ртуть. Как
только кислота собралась у крана, бю-
ретку вновь наклоняют и выпрямляют. Та-
кое встряхивание продолжа-
ют примерно 5 мин., все вре-
мя следя за тем, чтобы кис-
лота и ртуть приходили в
возможно более тесное со-
прикосновение. Если.® про-
бе имеется азотная кислота,
то вскоре начинается выде-
ление пузырьков газа, кото-
. рые собираются над кисло-
7 той. Когда количество газа
более не увеличивается, бю-
ретку снова укрепляют в вер-
тикальном положении в шта-
тиве и устанавливают урав-
нительную трубку такям об-
разом, чтобы ртуть в ней
находилась выше, чем в бюретке, примерно на 7т—Vs высоты слоя кис-
лоты (отношение удельных весов ртути и серной кислоты 13,6: 1,84 =
= 7,4). Объем газа замеряют, когда он полностью собрался над кисло-
той, После этого встряхивание продолжают еще несколько минут и по-
вторяют измерение, чтобы убедиться, что увеличения объема более не
происходит.
Если в исследуемой реакционной смеси содержится еще сравни-
тельно большое количество азотной кислоты, то выделение газа при
J. Анилин из нитробензола
71
встряхивании начинается очень скоро; пузырьки газа способствуют
эффективному перемешиванию ртути и кислоты, и реакция благодаря
этому быстро заканчивается. Если же азотной кислоты содержится
очень мало, то до появления первых пузырьков газа может пройти более
продолжительное время. В таких случаях требуется непрерывно встря-
хивать по меньшей мере 5 мин., прежде чем делать заключение о пол-
ном отсутствии азотной кислоты.
Па основании отсчета объема выделившейся NO вычисляют,
сколько азотной кислоты еще содержится в реакционной смеси. Если
хотят произвести точное определение, то следует также заметить тем-
пературу и давление и привести объем к нормальным условиям
(т. е. к 0° и 760мм); умножением на 1,3402 (вес 1 мл NO прн0°и 760 мм
в миллиграммах) получают вес NO в миллиграммах. Для контроля за
ходом нитрования не требуется такой большой точности; поэтому можно
пренебречь поправкой к намеренному объему и взять за основу расчета
вес 1 мл NO прн средней температуре в лаборатории, т. е. при 20°, и при
среднем барометрическом давлении для данной местности. Веса 1 мл NO
при различных условиях даны в приведенной ниже таблице.
Показания барометра, .«.и 700 710 720 730 740 750 760 770
Бес I мл NO при 20°, мг . . 1,15 1,17 1,18 1,20 1,22 1,23 1,25 1,26
Количество HNO3, соответст- вующее 1 мл NO, мг . . . 2,41 2,45 2,48 2,52 2,55 2,59 2,62 2,66
Чтобы найтн вес азотной кислоты, из которой образовалась NO,
следует вес NO умножить на 2,1 (HNO3: NO — 63 : 30 — 2,1). В послед-
ней строке таблицы приведен вычисленный таким образом вес азотной
кислоты, соответствующий весу 1 мл NO при различных барометриче-
ских давлениях. Умножая это число на объем выделившейся NO, нахо-
дят количество азотной кислоты, которое содержалось во взятой пробе.
Для того чтобы определить вес (>в миллиграммах) азотной кислоты, еще
содержащейся во всей реакционной смеси, остается только умножить
полученный результат иа вес этой смеси и разделить на вес пробы.
Определение в нитрометре можно производить только в тех случаях,
когда нитрование проводится в концентрированной серной кислоте. Сле-
дует учитывать, что NO в нитрометре выделяют также азотистая и
нитрозилсерная кислоты. Если, как это часто бывает, нитрование сопро-
вождается явлениями окисления, то'образующаяся при этом азотистая
кислота определяется вместе с азотной. С другой стороны, пользуясь
этим, можно при помощи нитрометра контролировать течение тех реак-
ций диазотирования и нитрозирования, которые проводятся в концентри-
рованной серной кислоте.
3. Анилин из нитробензола
Восстановление нитробензола в анилин проводится в герметически
закрываемом медном или железном котле с мешалкой, которая должна
каплям в течение Л/4 часа 123 г (1
зол восстанавливается в анилин с
Рис. 11. Медный закрытый редуктор
С мешалкой, капельной воронкой и
обратным холодильником для веществ,
летучих с водяным паром.
доходить до его дна. В лаборатории обычно применяют котелок, пока-
занный па рис. 11. В прибор, снабженный обратным холодильником и
капельной воронкой, помещают 200 г измельченных в топкий порошок
чугунных стружек, 300 мл воды и 20 мл соляной кислоты
(30%-ной). Чтобы железо «протравить», смесь кипятят в течение 10 мин.
Затем при постоянном быстром перемешивании (железо должно нахо-
диться в вихреобразном движении) в кипящую смесь добавляют по
моль) нитробензола, Нитробсн-
енльпым выделением тепла (внешний
обогрев приходится уменьшить или
совсем выключить); железо при
этом окисляется главным образом
в FesO-t, который выпадает в виде
плотного хорошо фильтрующегося
осадка. Непрерывное кипячение с
обратным холодильником продол-
жают до тех пор, пока с холодиль-
ника не станет стекать бесцветный
дистиллят. Затем к реакционной
смеси прибавляют осторожно (вспе-
нивание!) 15 г соды и перегоняют
анилин с водяным паром. Пар вво-
дят через дополнительный штуцер,
холодильник присоединяют при по-
мощи согнутой стеклянной трубки
(которая должна иметь такой же
диаметр, как и трубка для ввода
пара) к главному отверстию/ третий
штуцер остается закрытым.
Анилин растворим в воде (Зав
100 г воды). Поэтому к его водной
суспензии добавляют такое количе-
ство поваренной соли, чтобы образо-
вался 20%-ный раствор соли, в кото-
ром анилин совершенно нерастворим.
После многочасового стояния в де-
лительной воронке аннлнн можно от-
делить и перегнать, нагревая открытым пламенем (см. рис. 7, стр. 67).
Первая фракция содержит следы бензола и немного воды; 99% основ-
ной фракции перегоняется при 182°. Из 123 а нитробензола получается
примерно 85 г анилина (т. е. около 91 % от теории).
Технические примечания. В производственном масштабе анилин пе-
регоняется с водяным паром, уже насыщенным анилином; для этого паровой котел
питают водой, полученной при этой перегонке. На заводе Вейлер-Тер Меера просто
экстрагируют анилиновую воду нитробензолом, причем основание извлекается нз
жидкости полностью, смесь нитробензола с анилином затем восстанавливается, как
указано выше. Благодаря этому становится ненужным применение парового котла
с анилиновой водой, которое всегда связано с некоторыми неудобствами.
На производстве железо добавляется понемногу и берут меньше воды *. Выходы,
получаемые в технике, являются практически количественными. Так, из 100 кг бен-
зола получают примерно 110 кг чистого анилина. Последний перегоняется в вакууме
в количествах от 10 000 до 30 000 кг. Нагревание осуществляется с-помощью системы
змеевиков, обогреваемых паром и помешенных внутри котла.
* Рисунки аппаратуры, применяемой в технике, имеются, например, в книге U 11-
гп а и п В., Enzyklopiidie der technischen Chemie, 2 Aufl., Bd. 1, S. 465 467, Berlin
1928.
В последнее время в технике нашло применение восстановление нитробензола
в анилин водородом или водяным газом в присутствии соответствующих катализа-
торов; преимущество этого способа по сравнению с восстановлением при помощи же-
леза состоит в том, что он позволяет вести процесс непрерывно, вследствие чего при
тоц же производительности требуется значительно меньшая аппаратура *.
Можно также упомянуть, что иногда в качестве восстановителей применяются
сульфиды щелочных металлов (когда они особенно дешевы).
Рис, 12. Гидравлический пресс с автоматически вы-
ключающимся насосом.
7—цилиндр из литой стали; 2 —плита пресса: 5 — труба, подводя-
щая нагнетаемую воду (250 ат}; 4 — насос, выключающийся при
240 атм. (давление можно регулироилть, передвигая груз; один
насос может легко обслужить 4—6 прессов); ,5—головная плита
из литой стали.
Следует еще указать на принципиально новый способ получения анилина, впер-
вые примененный американской фирмой «Дау» и получивший большое техническое
значение. Упомянутая фирма получает большие количества анилина (а также и фе-
нола, см, стр. 82) из хлорбензола (и аммиака; реакция ведется при очень высоких
Давлениях и температурах (до 340 атм и около’340°). При этом образуются анилин и
хлористый аммоний,
С началом промышленного получения анилина, наеденного впервые в Англин,
связаны первые успехи промышленности красителей. Анилин до сего времени остался
* См., например, герм. пат. 436820 (Frdl., XV, 391. I. GJ.
iiримежуточные продукты
одним из важнейших продуктов этой промышленности. Значительная часть его и
в настоящее время расходуется для получения анилинового черного на волокне.
Большие количества анилина идут как непосредственно, так п после превращения
в многочисленные другие промежуточные продукты (см. табл. 7) для получения
красителей различных классов. Анилин является важным исходным веществом также
для производства фармацевтических препаратов. Однако самым крупным потребите-
лем производных анилида в настоящее время является, вероятно, резиновая промыш-
ленность, которая расходует в качестве ускорителей вулканизации громадные коли-
чества дифеннлгуавидина, тиокарбанцлида и других серусодержащих производных
анилина.
Анилин, как и нитробензол, а также многие их гомологи и продукты замещения
являются сильными ядами. Они вызывают так называемое «анилиновое отрав-
ление», представляющее собой отравление крови, сопровождающееся цианозом (т. е.
связанным со слишком большим содержанием в крови углекислого газа посинением
кожи, которое особенно отчетливо проявляется на губах и на ногтях), и являются
тем более опасными, что могут попадать в организм не только через желудок, но
также и через дыхательные пути и даже через кожу. Поэтому на всех производствах,
где получают или перерабатывают такие продукты, следует заботиться о хорошей
вентиляции, интенсивном отсасывании пыли и паров, а также о самом тщательном
соблюдении чистоты (мытье рук перед каждым приемом пищи, купание и смена
одежды после окончания работы). Еще более неприятным, чем острое, сопровождаю-
щееся цианозом отравление, является часто встречающийся при многолетней работе
на производстве анилина рак мочевого пузыря*, который часто распознают, слишком
поздно, когда больному нельзя уже оказать радикальной помощи. Помимо упомяну-
тых мер предосторожности, рабочие таких производств должны находиться под по-
стоянным врачебным контролем и переводиться на другое, безвредное производство
при появлении первых симптомов опасности.
Общие сведения о восстановлении
Описанный выше метод восстановления железом в присутствии очень небольших
количеств кислоты, предложенный Бешаном и Бриммейером, применяется в технике
наиболее часто. Такое иосстаиовление протекает в общем очень гладко в тех случаях,
когда соблюдаются приводимые ниже условия.
1. Сосуд, в котором производится восстановление, должен быть изготовлен из
металла. В производственных условиях применяются только железные аппараты, в ла-
боратории могут с успехом использоваться также и медные. Сосуды из стекла, фар-
фора и эмали неприменимы **.
2. Для восстановления должен применяться чугун в виде стружек,
получаемых при сверлильных и токарных работах. Стружка должна быть измельчена
в тонкий порошок на шаровой мельнице и предварительно обработана органическим '
растворителем, чтобы удалить жир. Для применения в чувствительных реакциях вое- '
становления чугунный порошок рекомендуется просеивать через сито для удаления ।
грубых частиц. Отходы ковкого железа, гвозди и т. п. непригодны. !
3. Мешалка должна доходить почти до самого дна сосуда н вращаться так
быстро, чтобы чугунный порошок, несмотря па свою тяжесть, находился в сильном
впхреобразном движении,
4, Следует также принимать меры к тому, чтобы промежуточная стадия восста- >
новления — образование фепилгидроксиламина (см. приведенную выше схему) была ;
пройдена возможно более быстро. Для этой цели рекомендуется, по крайней мере при (
работе в лабораторном масштабе, медленно вводить нитросоединения в восстанавли-
вающую смесь так, чтобы каждая порция тотчас после введения восстанавливалась
до амина. На производстве, где приходится считаться со значительным удлинением
времени реакции, иногда поступают как раз наоборот.
Восстановление нитробензола в анилин протекает, как показывает приведенная
выше схема, через стадии образования нитрозобензола и фенилгидроксиламинз, В то .!
время как первая из названных стадий протекает настолько быстро, что се едва
можно уловить, февилгидроксиламин в подходящих условиях может быть получен
с хорошим выходом в качестве основного продукта реакции. Это соединение также
* Согласно новым данным, рак мочевого пузыря вызывает прежде всего р-наф-
тиламин.
** Если все-таки в лаборатории работают с такими сосудами, то необходимо до-
бавлять небольшое'количество растворимой медной соли; тогда на поверхности же-
леза выделится металлическая медь, что значительно повысит реакционную способ-
ность железа. В последнее время в продажу поступили особенно активные сорта
железа «для восстановления», которые можно применять без добавок ‘при работе
в стеклянных сосудах.
3. Анилин из нитробензола
71
легко восстанавливается дальше до амина, но оно легко подвергается н другим пре
вращениям. Так, оно конденсируется с большой легкостью, особенно в присутствие
щелочен, с промежуточно образующимся нитробензолом и дает при этом азоксибен-
зол (см. получение бензидина, стр. 114); может происходить и конденсация двух мо-
лекул феннлгидроксиламина с образованием азобензола (желтое окрашивание!).
Хотя и эти соединения прн дальнейшем восстановлении переходят через гидразобен-
зол в конце концов в анилин, но такое восстановление часто проходит значительно
более медленно и менее гладко, чем прямое восстановление феннлгидроксиламина
в амин, Для избежания этой побочной реакции реакционная смесь в продолжение
всего восстановления должна оставаться отчетливо слабокислой. С другой стороны,
фенилгидроксиламин под действием разбавленной серной кислоты может перегруппи-
роваться в п-амидофенол, а также превращаться в галоидозамещенные анилины при
реакции с галоидоводородными кислотами. Поэтому, для того чтобы реакция Проте-
кала гладко, при 'восстановлении применяют уксусную кислоту. 'Однако во многих
случаях такие же результаты получают и при работе с достаточно разбавленными
соляной или серной кислотами, Вопрос о том, какую кислоту и в каком количестве
выгоднее всего Применять, решается в каждом отдельном случае экспериментально *.
Восстановление по Бешану — Бриммейеру обычно проводится при температуре
кипения, а При работе с особенно чувствительными веществами (например с п-нитро-
эодиметлланилином) —примерно п!ри 80°. Поскольку при реакции выделяется тепло,
обогрев регулируется таким образом, чтобы избежать чрезмерно сильного кипения.
Некоторые реакции восстановления сопровождаются сильным вспениванием. В таких
случаях реакционный сосуд заполняется не больше, чем наполовину, и нитросоедине-
ние вносится очень медленно и осторожно; если пена поднимается все же слишком
высоко, то ее сбивают, впрыскивая несколько капель холодной воды.
Характер дальнейшей обработки зависит от природы полученного при восстано-
влении продукта. Если последний представляет собой основание, летучее с водяным
ларом, то его после подщелачивания реакционной смеси перегоняют с водяным ларом.
Иногда после подщелачивания реакционную смесь отсасывают от Хелезного шлама.
Амнносоединения, растворимые в воде или щелочах — особенно сульфокислоты и кар-
боновые кислоты, — остаются при этом в фильтрате, откуда они выделяются путем
подкисления, высаливания или упаривания; нерастворимые в щелочах основания
остаются в железном шламе и извлекаются оттуда с помощью подходящих органиче-
ских растворителей. Часто можно не выделять продукт восстановления, а пускать
непосредственно в дальнейшую переработку раствор, полученный после удаления
остатков железа.
Описанный выше метод применим для восстановления в амины не только нитро-
соединений, но также и нитрозосоединений, азосоединений и т. п.
Восстановление нитросоединений можно, конечно, проводить и с большим коли-
чеством кислоты, чтобы все железо переходило в раствор. Такой метод применяется
на Производстве в тех случаях, когда нитросоединения, полученные в концентрирован-
ной серной кислоте, не удается отделить от последней простым путем.
Метод, однако, целесообразно Применять только тогда, когда продукт восста-
новления может быть выделен прямо из кислого реакционного раствора без осажде-
ния железа (см. получение Аш-кислоты, стр. 189). При таком способе восстановления
в противоположность описанному выше методу лучшие результаты получаются при
применении отходов ковкого железа.
Наконец, железо может восстанавливающим образом действовать также и в при-
сутствии щелочи: при этом нитробензол переходит последовательно в азокси-, азо- и
гидразобензол. В этом случае следует применять особенно тонко измельченный чугун-
ный порошок, который должен быть перед употреблением протравлен (отдельная
операция).
Железо оказывается непригодным в качестве восстановителя, если в соединении
имеется несколько питрогрупп, а восстановить требуется лишь одну или если наряду
с нитрогруппой имеется азогруппа, которая должна остаться незатронутой. Такого
рода «частичное восстановление» в технике обычно осуществляется при помощи серо-
водорода; в качестве источника сероводорода чаще всего используют сернистый нат-
рий (NaaS) или сульфгидрат натрия (NaSH). Этот метод служит не только для ча-
стичного восстановления; очень часто он применяется для восстановления иитррсоеди-
иений ряда антрахинона, а также других нитросоединений, которые не содержат
других способных восстанавливаться групп. Реакция проводится ие только в водном,
* В лабораторных условиях часто бывает целесообразно применять кислоты
больше, чем рекомендуется в прописях, предназначенных для производства (прибли-
зительно до Vs эквивалента на ( моль нитросоединения). Нельзя, однако, заходить
слишком далеко в этом отношении, так как если в раствор перейдет слишком много
железа, то при подщелачивании выпадет объемистый осадок гидроокиси железа,
который очень затруднит фильтрование.
/. Промежуточные продукты
но и в спиртовом растворе, поэтому применяется для восстановления веществ, которые
трудно реагируют вследствие полной нерастворимости в водной среде. Подробности
приведены при описании синтеза м-нитро анилин а (см. стр. 104).
Из большого числа восстанавливающих агентов, используемых- в лабораторной
практике, многие находят применение также и в производственных условиях. Так,
прежде всего следует назвать цинк н ццпкопую пыль, которые можно применять для
самых разнообразных восстановлений в кислом и в щелочном растворах при нагрева-
нии и ня холоду; однако эти восстановители вследствие дороговизны употребляются
в технике только в тех случаях, когда восстановление с. помощью более дешевых
средств — железа и сернистого натрия — не удается или дает худшие результаты
(подробнее см, при описании получения бензидина стр, 114), Сказанное выше ка-
сается еще в большей степени также и значительно более дорогих олова и хлористого
олова, которые в технике применяются лишь в исключительных случаях.
В качестве восстановителя очень часто употребляется гидросульфит натрия
NaaSsO4 (см. стр, 142); по и он также не может широко Применяться на производ-
стве пз-за высокой цены. В специальных случаях, особенно при получении и приме-
нении кубовых красителей, гидросульфит играет важную роль (см. стр. 287). .Можно
попутно упомянуть о применении гидросульфитов и сульфоксилатов для разрушения
окрасок при вытравном печатании, для обесцвечивания окрашенного волокна, а также
для беления.
Сернистая кислота н ее соли являются дешевыми восстановителями, но они
могут использоваться только в некоторых особых случаях. При нх применении наряду
с восстановлением часто происходит и сульфирование (см. получение 1-нафтиламин-
2,4-дисульфокислоты из 1-нитронафталина и 1,2,4-амцнонафтолсульфокислоты из ни-
трозо- р-партола стр. 158 и 178) при восстановлении диазобензола до фенилглдразина
вначале также образуется N-сульфокислота. которая затем расщепляется при энергич-
ной обработке соляной кислотой 1см. стр. 89 и 117).
Восстановление с помощью глюкозы и щелочи применяется в особых случаях,
в частности для переведения нитросоединений в азокси- и азосоединения.
Следует также указать, что в промышленности красителей иногда находит при-
менение электролитическое восстановление, а также восстановление молекулярным во-
дородом в присутствии подх’одящих катализаторов.
4. Бензол сульфокислот а
CftCO;!
или
SO3Na
NagCO3
или NasSO4
В железном, фарфоровом или эмалированном сосуде, снабженном
мешалкой и обратным холодильником, осторожно смешивают в течение
часа 200 г бензола н 450 г о л е у м а, содержащего 10% SO3; сле-
дят за тем, чтобы температура не поднималась выше 75°. По окончании
смешивания (но не раньше!) температуру повышают до ИО13. Примене-
ние более высокой температуры не рекомендуется, так как при этом
легко образуется днеульфокнелота *, Приблизительно через 1,5—2 ча-
са бензол полностью исчезает. Реакционную смесь выливают в 1 л воды
и нейтрализуют, кипятя при хорошем перемешивании с 450 а порошко-
образного мела; вместо мела можно также применять соответствую-
щее количество гашеной извести**. Нейтральный раствор каль-
* Бензолдисульфокислота получается гораздо легче, чем это можно предполо-
жить ца основании литературных данных (см. получение бензол-.«-дисульфокислоты
стр. 131).
** В лаборатории лучше применять углекислый кальций, так как вследствие не-
растворимости небольшой избыток его ие мешает, а выделение углекислого газа спо-
собствует хорошему перемешиванию. Слишком сильного псиенивания и переброса
пены можно легко избежать црц применении достаточно больших сосудов и осторож-
ной работе. Гашеная известь, напротив, очень часто образует вязкие комки', которые
обычными лабораторными мешалками измельчаются медленно и поэтому медленно
4. Ьензолсульфокислота
77
Рис. 13. Лабораторный
нутч-фильтр.
«левой соли отфильтровывают на большом нутч-фильтре (см. рис. 13)
от гипса; последний тщательно промывают 'водой (с растиранием) и от-
жимают. Общее количество раствора составляет примерно 1,5 л. Затем
кальциевые соли переводят в натриевые. Для этого к раствору при
нагревании добавляют соды до щелочной реакции по фенолфталеину
(пли сульфат натрия). Продукт, содержащий около 99% бензолсульфо-
ната в виде Na-соли, пригоден дли дальнейшей переработки в фенол. На
нейтрализацию расходуется около НО г каль-
цинированной соды. Раствор натриевой соли
отфильтровывают в горячем состоинии от вы-
павшего СаСО3. Прозрачный фильтрат упари-
вают на голом огие до тех пор, пока не начнут
выделяться кристаллы бензолсульфокислого
натрия. При охлаждении об разуете и паста из
солей и воды, которая содержит приблизи-
тельно 50% сухого вещества; последнее со-
стоит примерно из 90% бензолсульфокислого
натрия, 7% сульфата натрия н соды (считая
на сухое вещество) и небольшого количества
кальциевых солей. Приготовленная таким об-
разом паста может для получения фенола не-
посредственно сплавляться со щелочью (см.
получение фенола, стр. 80).
Если требуется выделить бензолсульфо-
кислый натрий, то пасту выпаривают иа водя-
ной бане досуха.
Для получения (препарата, не содержащего
неорганических солей, можно отсосать полу-
ченную пасту и высушить осадок. Прн этом,
конечно, неизбежны значительные потери.
Технические примечания. На производ-
стве сульфируют до 1200 кг бензола за один прием.
Типе обычно отделяют от жидкости на фильтрах (та-
ких же, какие применяются в производстве соды).
Удельный вес раствора натриевой соли составляет
приблизительно 1,06—1,08, раствор концентрируется при-
мфно до уд. веса 1,21 (плотность измеряется прн 100°).
Бензолсульфокислоту можно также получить совершенно другим путем. Вместо
того чтобы сульфировать бензол в жидком состоянии, можно пропускать его пары
через серную кислоту (уд. вес. 1,84), нагретую до 100—140°. Образующаяся вода
непрерывно отгоняется при этом вместе о непрореагировавшим бензолом. Таким
образом получают в конце концов раствор бензолсульфокислоты и совсем незначитель-
ное количество серной кислоты. Этот раствор можно не обрабатывать известью,
а прямо после нейтрализации содой сплавлять с едким натром. Таким же образом
гладко сульфируется и толуол. Этот новый способ был разработан американской
компанией «Бакелит».
Общие сведения о сульфировании
Сульфирование, так же как галоидирование и нитрование, играет важнейшую
роль в производстве красителей тем более, что в огромном большинстве «случаев
реагируют; это обстоятельство может обусловить добавление слишком большого коли-
чества извести, которую потом придется нейтрализовать.
В технике дело обстоит по-иному. Здесь сильное пенообразовапие мешает значи-
тельно больше, чем в лаборатории, а измельчение извести нс представляет трудностей
вследствие применения значительно более эффективных приспособлений для переме-
шивания. Поэтому на производстве для нейтрализации по крайней мере основного ко-
личества кислоты чаще всего применяют гашеную известь и только в случаях, когда
следует избегать щелочной реакции, остаток кислоты нейтрализуют мелом.
/а к Промежуточные продукты
растворимость последних в воде обусловлена наличием сульфогрупп. Кроме того,
сульфокислоты являются исключительно важными промежуточными продуктами для
получения фенолов, особенно в ряду нафталина.
Сульфирование осуществляется чаще всего при помощи серной кислоты. При-
меняется обычная концентрированная серная кислота, моногидрат, а также дымящая
кислота (олеум) с различным содержанием SO3, Температуры, при которых проводится
сульфирование, также колеблются в широких пределах (от 0 до 200°) в зависимости
от реакционной способности исходного вещества. Сульфирование протекает легче
в ряду нафталина, чем в ряду бензола; антрахинон сульфируется значительно труднее.
Реакция сильно облегчается при наличии оксигрупп фенольного характера; таким же
образом, но несколько слабее, влияют алкокси-, амино- и ациламиногруппы; самое
слабое влияние оказывают алкилы. Сульфирование затрудняется в присутствии галои-
дов, карбонильных и карбоксильных групп, сульфогрупп и особенно нитрогрупп.
Углеводороды, содержащие две нитрогруппы, обычно не сульфируются. Для дальней-
шего сульфирования сульфокислот часто бывает целесообразно применять не свобод-
ные кислоты, а соли их со щелочными металлами; при этом в некоторых случаях
добавка сульфата натрия способствует более гладкому течению реакции. Температура,
при которой ведется сульфирование, часто оказывает влияние на место вступления
сульфогруппы. Это наблюдается в случае фенолов и в ряду нафталина (подробнее об
этом сказано в главе о правилах ориентации). В таких случаях уже образовавшиеся
сульфокислоты могут при нагревании с серной кислотой изомеризоваться в той или
иной степени. Процесс изомеризации останавливается по достижении равновесного
состояния, характерного для данной температуры. Это явление имеет особенн-о боль-
шое практическое значение в ряду нафталина *. В ряду антрахинона сульфированию
в а-положение способствует присутствие солей ртути (см, главу о правилах ориен-
тации).
При сульфировании образуется вода, которая разбавляет серную кислоту, поэтому,
Чтобы последняя сохранила нужную концентрацию до конца реакции, необходим зна-
чительный ее избыток. При сульфировании олеумом в реакцию обычно вступает только
свободный серный ангидрид, так что и в этом случае по окончании сульфирования
остается большое количество избыточной серной кислоты. Отделение этого избытка
серной кислоты от образовавшихся сульфокислот легче всего осуществляется в случае
аминосульфокислот, которые содержат одинаковое число сульфо- и аминогрупп **;
такие сульфокислоты настолько трудно растворимы в воде, и особенно в разбавлен-
ной серной кислоте, что при разбавлении реакционной массы практически полностью
выпадают в осадок, который достаточно отфильтровать и промыть. Выделение других
сульфокислот производится в технике двумя способами. Первый способ состоит
в том, что растворенную в воде реакционную массу нейтрализуют гидратом окиси
кальция (гашеная известь, известковое молоко^ или углекислым кальцием (мел или
тонко измельченный известняк), и отфильтровывают выпавший в осадок гипс. Каль-
циевую соль полученной сульфокислоты, содержащуюся в фильтрате, переводят с по-
мощью соды или сульфата натрия в натриевую соль. После этого снова отфильтровы-
нают. от СаСОз (а также от гипса) и упаривают до начала кристаллизации или,
в случае надобности, досуха. К реакционной массе можно также сразу прибавить не-
обходимое для образования натриевой соли количество сульфата натрия; после
отфильтровывания гипса непосредственно получают раствор натриевой соли сульфо-
* Эти перегруппировки обычно объясняют, исходя из предположения, что при
повышенной температуре сульфогруппы снова отщепляются и устанавливается со-
стояние равновесия, например
. SOyH
Lso,,h
нафталина отщепление сульфогрупп (как
Экспериментально установлено, что в ряду
замещение на водород под действием горячей содержащей воду серной кислоты,
так и на оксигруппу при щелочном плавлении) идет значительно легче, если они
находятся в а-, а ие в ^-положении. В соответствии с этим кажущаяся миграция
идет преимущественно, но обычно не полностью, нз а- в p-положение. Следует осо-
бенно подчеркнуть, что наблюдаются миграции также из одного а-положения в дру-
гое а-положение (надример, нафтиламин сульфокислоты 1,8—»-1,4 —*-1,5)’ или из одного
(3-положения в другое p-положение (например, иафталиндисульфокислоты 2,7 -> 2,6)Г,
** Полисульфокислоты моноаминов ведут себя, как сульфокислоты углеводородов;
их кислые соли, в которых одна сульфогруппа свободна, а другие связаны со
щелочным металлом, обычно легко высаливаются. Моносульфокислоты диаминов, так
же как свободные амины, растворяются в избытке минеральной кислоты (см. стр. 37);
4, Бензолсульфокислота
79
кислоты. Этот способ, называемый в технике «известкование», применим для
любых сульфокислот, кроме тех немногих случаев, когда образуются трудно раство-
римые кальциевые соли; он, однако, неудобен и довольно дорог из-за необходимости
работать с большими количествами жидкости, которую потом приходится снова упа-
ривать. Поэтому часто предпочитают к разбавленной водой реакционной массе добав-
лять соль щелочного металла (обычно NaCi или NasSO^, реже — КС1 илн
(NH^aSO^ и, таким образом, высаливать соответствующую соль образовав-
шейся сульфокислоты. Этот метод является обычным, но не всегда применим, так как
не псе сульфокислоты можно осадить таким нутом достаточно полно. Следует также
иметь в виду, что выделенные этим методом соли сульфокислот загрязнены, как
правило, неорганической солью, применявшейся для осаждения; это, правда, в боль-
шинстве случаев не мешает дальнейшему их применению.
Ароматические амины часто можно с успехом сульфировать особым, применимым
только для веществ этой группы способом, который состоит в нагревании в сухом
состоянии их кислых сульфатов (содержащих точно 1 моль серной кислоты на каждую
аминогруппу) до 170—220°; нагревание лучше производить в вакууме. При применении
этого способа, который называется «запекание», сульфогруппа вступает всегда только
в орто- или пара-положение к аминогруппе (это относится также и к многоядерным
соединениям). Таким путем можно получить однородный продукт также и в тех слу-
чаях, когда другие методы приводят к образованию смеси изомеров (например,
д-нафтиламнн при запекании дает исключительно 1,4-нафтиламиносульфокислоту, в то
время как при применении других методов образуются смеси 1,4- и 1,5-нафтиламино-
сульфокиюлот) или вообще не позволяют получить изомер желаемого строения (как
например имеет место в случае ^-ксилидина, который при запекании сульфируется
в орто-положение, а при сульфировании другими методами дает вследствие ориенти-
рующего влияния обеих метильных групп сульфокислоту с сульфогруппой в мета-
положении по отношению к аминогруппе).
Метод запекания более подробно рассмотрен при описании получения сульфани-
ловой ’(стр. 116) и нафтионовой (стр. 161) кислот.
Избыток серной кислоты, который, как это упоминалось выше, является неизбеж-
ным при сульфировании серной кислотой или олеумом, вызывает иногда неже-
лательные побочные реакции, как например дальнейшее сульфирование или перегруп-
пировку образовавшейся вначале сульфокислоты. Этих побочных процессов можно
избежать, если сульфирование проводить рассчитанным количеством хлорсульфоновой
кислоты в ис взаимодействующем с ней органическом растворителе (для этой дели
обычно используется нитробензол). Более подробно такой метод сульфирования рас-
сматривается при описании синтеза 2.1-нафтол-сульфокислоты (см. стр. 176).
Если для сульфирования применяется большой избыток хлорсульфоновой кислоты
без растворителя, то, как правило, образуется не свободная сульфокислота, а соот-
ветствующий сульфохлорид. Так, например, из толуола получают большие количества
смеси о- и п-толуолсульфохлоридов:
+ H2SO4 -Ь- НС1
SOgCI
Й0
11 (JUMtMLyTUHttDlt! m
о-Толуолсульфохлорид используется в производстве сахарина, а мара-изомер,
который был раньше обременительным побочным продуктом, находит в последнее
время разнообразное применение в производстве красителей и в фармацевтической
промышленности.
В последней цз названных реакций, как и при сульфировании концентрированной
серной кислотой пли олеумом, в качестве побочных продуктов образуются небольшие
количества сульфонов. Их образование объясняется взаимодействием уже полученной
сульфокислоты с пеизменившимся исходным веществом при водоотннмающем действии
сульфирующего агента:
\ SOsH -L / \ = / \ SO2- / \
нйо
По сравнению с прямым сульфированием непрямое введение сульфо групп играет ;
в химии красителей сравнительно ограниченную роль; оно все же применяется для !
получения некоторых технически важных соединений. На производстве для этой цели .
применяются следующие способы: ;
1. Замена «подвижного» атома галоида на сульфогруппу под действием сульфита •;
натрия. «Подвижный» атом галоида может быть связан с алифатической частью моле- i
кулы, как например в хлористом бензиле, или находится в ароматическом ядре; в пог
следнем случае он должен быть активирован отрицательными заместителями, в осо- [
бенностц нитрогруппами [см. получение 2,4-дмннтробензолсульфокиСлоты из 2,4-дини- .;
трохлорбензола (стр. 94) и нитробензол-2,5-дисульфокислоты из о-нитрохлорбензол-п- <
сульфокислоты (стр. 98)]. \
2. Действие бисульфита на нитросоединение, на хинон или на хиноноксим :
(нитрозофенол). При этом одновременно происходит восстановление нитро-, а также J
и нитрозогруппы до аминогруппы. Примерами могут служить получение 1-нафтнламин- '
2,4-дисульфокислоты из а -нитронафталина и бисульфита (побочным продуктом при
этой реакции является нафтионовая кислота) и производство 1,2,4-аминонафтолсульфо- -
кислоты из нитрозо-£-нафтола и бисульфита (см. стр. 179).
3. Окисление сульфиновой кислоты, меркаптана или дисульфида, В о-нитрохлор- I
бензоле, например, хлор недостаточно подвижен, чтобы прямо заменяться на сульфо- )
группу, как это описано в п. 1; однако при действии Na2S2 он довольно гладко пере- ;
ходит в о.о'-днннтродифепилдисульфид, который прн окислении дает о-нитробепзол-
сульфокислоту*.
Cl -Н Na—S—S—Na+С1—/ \
з
о2м
:-SO3H [
no2 o2n
no2
4. Действие бисульфитного производного формальдегида на амин или фенол, при
котором группа СНгЗОзН вводится в первом случае в аминогруппу, а во втором слу-
чае в ядро в орто- и нара-положение к оксигруппе. Полученные таким путем про-
изводные фенолов исключительно устойчивы, а СН250зН-группа, связанная с азотом,
легко опять отщепляется при действии омыляющих агентов. Использование этого
способа для синтеза азокрасителей рассмотрено в специальном разделе (см. стр. 226).
5. Фенол
О Na
2NaOH =
Na2SO;i + Н2О
В качестве исходного материала применяют неочищенную примерно
50%-пую пасту бепзолсульфокислого натрия, полуденную, как описано-
в разделе 4, стр. 77. В отдельной пробе определяют содержание в пасте
сухого вещества и условно принимают, что все оно является чистым
бензолсульфокислым натрием.
Helv. Chim, Acta, 12, 663.
Теоретически возможно с 1 весовой частью едкого патра сплавить
2,25 части бе изол сульф о кисло го натрия; однако практически в плав
вводится только 1,65 части.
Аппарат для проведения плавления (см. рис. 14а).
Лучшим материалом для изготовления лабораторного аппарата, в ко-
тором проводится плавление, является медь *, которая вследствие хоро-
Р ii с. 14а. Котел для щелочного плавления.
Рис. 146. Котел для плавления с цен-
тральной трубкой для термометра
Z — приклепанная полоса для укрепления
котла; 2 —отверстие для ввода газов, напри-
мер при индиговом плаве; 5 — загрузочное
отверстие.
Шей теплопроводности позволяет экопо-'
мить много газа. Ввиду высокой тем-
пературы плавления необходимо, чтобы
мешалка касалась всей поверхности
плавильного котла (см. рисунок!). Термометр помещается в медную
трубочку, которая снизу запаяна твердым припаем и наполнена сухим
цилиндровым маслом таким образом, что термометр покрыт им не
меньше чем на 10 см. Целесообразно также устанавливать термометр
просто в полой оси мешалки (см. рис. 146).
Аппарат, в котором проводится плавление, устанавливается прямо
на маленькой печи Флетчера. В( него помещается 200 г твердого ед-
кого натра (большими кусками), не содержащего хлората, и 100'лы
* Важно, чтобы плав пс соприкасался с двумя различными металлами, по-
скольку п .этом случае образуется гальваническая пень, что иызынает нежелательные
явления окисления и восстановления.
воды (плавление с едким натром, содержащим хлорат, снижает выход
и очень опасно: взрывы!). Едкий натр расплавляют на сильном пла-
мени, и он становится совершенно прозрачным. Повышение темпера-
туры до 270° сопровождается вспениванием; затем образование пены
прекращается. Нагревание продолжают до 29(Г и, хорошо перемешивая
плав, добавляют небольшими порциями такое количество предвари-
тельно нагретой приблизительно до 100° пасты бензоле ул ьфо-
к и слога натрия (при этой температуре паста находится в жидком
состоянии), которое соответствует 330 г сухого вещества (осторожно,
защитные очки!). Ла прибавление соли, которое регулируется таким
образом, чтобы температура плава в котле все время была от 290 до
300°, требуется около 45 мин.
По окончании введения соли температуру в течение 72 часа подни-
мают до 325° и продолжают перемешивание еще 40 мин. После этого
плав еще в горячем состоянии выливают на железный противень
с низкими бортами. Застывшую массу после охлаждения измельчают,
помещают снова в котел и обрабатывают 500 мл вод ы. Большая часть
твердого вещества при осторожном нагревании растворяется; нераство-
ренной остается корка сульфита натрия. Полученный раствор сливают
и добавляют свежую воду до тех пор, пока нс растворится весь плав.
Для этой цели требуется не более 2 л воды. Затем вес количество
раствора помещают в фарфоровую чашку, нагревают на открытой печи
Флетчера до кипения и обрабатывают 50% -ной серной нли концент-
рированной соляной кислотой почти до полного исчезнове-
ния реакции по тиазоловой бумажке. Раствору дают немного охладиться
и отсасывают через большой лабораторный путч-фильтр в предвари-
тельно подогретую колбу. К еще теплому прозрачному фильтрату до-
бавляется прн перемешивании концентрированная соляная кис-
лота; прибавление ведется до тех пор, пока фильтрат не начнет
отчетливо и устойчиво окрашивать конго бумажку в синий цвет. После
охлаждения фенол трижды экстрагируют бензолом (порциями по
400 мл). Бензол отгоняют и перегоняют фенол при обычном давлении
илн, лучше, в вакууме. Выход обычно составляет 85% от теоретически
возможного, т. с. из 100 весовых частей бензола получают 90—100 ве-
совых частей фенола с т, пл. около 38°,
Технические примечания. Плавление проводится в котлах емкостью
до 2400 л, которые обогреваются голым огнем. Мощность мотора мешалки'составляет
5 л. с. Плавление бензолсульфокислого натрия в количестве, которое соответствует
1000 кг бензола, прод»лжается на производстве в среднем 6 час. (включая смешива-
ние реагентов).
Фенол не экстрагируется, а выделяется добавлением серной кислоты (уд. вес
1,38), отделяется от маточного раствора, содержащего соли и менее 0,4 г сырого фе-
нола в 1 л, и перегоняется с водяным паром п высокой ректификационной колонне.
На практике особенно эффективными оказались колонны Рашига (см. соответ-
ствующий раздел). Таким образом из 75%-ного фенола получают 95%-нь1Й; остаток
представляет собой воду и «соль».
Полученный таким образом неочищенный фенол всегда перегоняют в вакууме.
Первый погон и вода, выделяющаяся при перегонке, содержат довольно много фенола,
который используется при следующей операции плавления. Остаток также снова воз-
вращается в производство; это позволяет избежать сжигания пека. Температура кипе-
ния фенола около 186е при 760 мм атмосферного давления.
Для измерения температур часто пользуются регистрирующими термометрами,
при помощи которых* очень удобно контролировать процесс.
В технике фенол можно также получать совершенно 'Другим путем; для этого
хлорбензол обрабатывают едким натром при высокой температуре (около 340°) и вы-
соком давлении (около 320 атм) в присутствии небольшого количества меди. При
5. Фенол
83
этом образуются фенолят и поваренная соль. Этот способ был введен в производ-
ственную практику американской фирмой «Дау» *. О получении таким же путем
анилина (из аммиака и хлорбензола) было сказано выше (см. стр. 73).
Общие сведения о щелочном плавлении
Щелочное плавление позволяет осуществлять замену связанной с ароматическим
ядром сульфогрупны на оксигруппу и таким путем синтезировать многочисленные
важные в техническом отношении фенолы и их производные. Щелочное плавление,
особенно в ряду нафталина, является наряду с сульфированием, нитрованием и
восстановлением наиболее часто используемой операцией.
Применяются два существенно отличающихся друг от друга способа щелочного
плавления. Можно, во-первых, как в описанном выше случае, работать с действи-
тельно расплавленной безводной или почти безводной едкой щелочью в
Рис. 15. Кривые зависимости давления паров водных
растворов едкого натра от температуры.
открытом сосуде или, во.вторых, использовать водный раствор щелочи, концентра-
ция которого может колебаться в широких пределах, и применять автоклавы для
достижения необходимой высокой температуры (см, температурную кривую на
рис. 15). Преимущество последнего способа состоит в том, что он позволяет лучше
регулировать реакцию и путем выбора подходящей концентрации и температуры
поддерживать условия, благоприятные для каждой отдельной сульфокислоты.
К тому же при работе в автоклаве исключается влияние кислорода воздуха,
которое при высоких температурах часто бывает вредным. По этим причинам
при работе в автоклаве, особенно с недостаточно устойчивыми веществами, часто»
получают более чистые продукты и лучшие выходы, чем при плавлении в открытом
котле. Последнее, конечно, является более, дешевым, и его всегда предпочитают в тех
случаях, когда оно дает такие же результаты, как и автоклавный метод.
В качестве щелочи чаще всего употребляют как наиболее дешевую едкий натр.
Однако в некоторых случаях успеха >южцо достигнуть только при применении более
легко плавящегося и более энергично действующего едкого кали. Иногда применяются
также смеем едкого натра и едкого кали, которые обладают более низкими темпера-
турами плавления.
См. Hale W, J„ Britton Е. С., Ind. engjn, Chem., 1928, 114—124.
81 /. Промежуточные продукты
В случае ди- и полисульфокислот реакцию часто можно вести так, чтобы замена
сульфогрупп па оксигруппу происходила ступенчато (частичное щелочное плавление).
Этим путем из бензол-ч-д и сульфокислоты можно в мягких условиях получить фенол-
,и-сульфокислоту, а и более жестких условияхрезорцин (см. стр, 131); точно так
же нафталин-1,5-дисульфокислота превращается сначала в 1-нафтол-б-сульфокислоту,
а затем в 1,5-диоксин<тфталип (оба эти продукта являются ценными дзосоставляющнми
в синтезе азокрасителей). В ряду нафталина при частичном щелочном плавлении на
оксигрупиу н первую очередь всегда заменяются схльфогруппы, находящиеся в .г-поло-
жениях. Имеющие очень важное промышленное 'значение 2,5,7-,2,8,6-, 1 Д4-аминонаф-
толмоносульфокислоты и 1 8,3,6-, 1,8/1,6-, 1,8,2,4-аминонафтолдисульфокислоты легко по-
лучаются таким путем из соответствующих нафтилами иди- и трисульфокпслот.
Следует иметь в виду, что при щелочном плавлении часто идут следующие
побочные реакции;
а) замена сульфогруппы на Н, а по на ОН, с одновременным образованием
сульфата вместо сульфита. Этот побочный Процесс обычно не имеет существенного
значения; однако в некоторых случаях он может стать основным (например, 2,3-ди- *
оксипафталии-6-сульфокислота даст при щелочном плавлении не 2,3,6-триоксицафтаЛин,
а 2,3-диоксищтфталин);
б) образующийся сульфит может оказывать восстанавливающее действие, если
имеются легко восстанавливающиеся группы. По этой причине, например, щелочное
плавление сульфокислот, содержащих иитрогруипы, протекает, как правило, не
гладко. Иногда, особенно и ряду антрахинона, такого восстанавливающего действия
можно избежать, добавляя окислители или гидроокись щелочноземельного металла;
последняя осаждает сульфит в виде нерастворимой соли и таким образом делает его /
безвредным;
в) расплавленная щелочь может оказывать и обратное окисляющее действие, J
Так, например, известно, что из антрахинон-(3-сульфокислоты образуется нс р-окси- ;
антрахинон, а ализарин (1,2-дноксиантрахинон). Добавлением окислителя можно па- |
править реакцию только по этому пути; 1
г) кроме сульфогруппы, на оксигруппу могут обмениваться также и другие за- |
местителщ особенно аминогруппы. Если эта реакция является нежелательной, ее еле- j
дует по возможности ограничить, работая в мягких условиях (не следует применять 1
излишне высоких температур). |
Как уже упоминалось выше, галоид (практически это касается только хлора), |
так же как и сульфогруппу, можно замещать на оксигруппу с помощью щелочного |
плавления. Эта реакция идет легче и присутствии меди. Как лучше получать данный 1
фенол из сульфокислоты пли из хлорпроизводпого, зависит от того, какой из этих а
продуктов дешевле и доступнее,
Шелочппо плавление применяется не только для получения фенолов, но также |
и для проведения разнообразных реакций конденсации, например для замыкания цик- J
лов. Здесь следует только упомянуть о синтезе индиго из фенилглицина пли из Я
фенилглицин-о-карбоновой кислоты, а также получении индантреиа из р-аминоантрахи- |
нона и дибепзантрона из бензантрона. Для успешного проведения реакций конден- |
сации такого типа, осуществляемых с помощью щелочного плавления, часто является j
весьма существенным отсутствие даже следов воды и немедленное связывание воды, j
образующейся в процессе реакции. Для достижения этой цели в плав добавляют амид Я
натрия или окись кальция. В последнем случае имеет место собственно не плавление,'Я
а только спекание реакционной смеси. Я
Удаление образующейся при реакции воды может быть ускорено при приме- j
нении вакуума или продувании инертного газа. 3
6. Производные хлорбензола 1
о- и п- Нитрохлорбензолы 1
\ /~С1 I
/—\ rt l Ь°2 '
\ / ° 1
o2N—/ ч>—Cl I
I J \__/ Л
Ннтрола'ние хлорбензола всегда приводит к образованию смеси, состоящей при- Я
б т из и толь но из '/я (’ П ‘7з л-нитрохлорбензола. Вол юная чисть последнего вы кристал- Л
лйзнвииается при охлаждении и может быть отделена простым центрифугированием,
и- и п-питрохлороензолы 8 Л
Остающаяся смесь вследствие слишком малой разницы в температурах кипения не
может быть разделена на составные части с помощью одной только дробной пере-
гонки. Однако тщательным фракционированием с применением эффективной ректи-
фикационной колонны удается получить фракции, настолько обогащенные пара-изо-
мером (первые фракции) и орто-изомером (последние фракции), что охлаждением и
центрифугированием нз них могут быть выделены, соответственно, пара и орто-соеди-
нения *,
На производстве некрист.тллцзующисся промежуточные фракции и отделенная
центрифугироваТщсм эвтектика всегда пускаются снова в работу, так что в конце кон-
цов достигают полного разделения. В лаборатории следует удовлетворяться получе-
нием основного количества п- и части о-нитрохлорбензола.
112 а хлорбензола (1 моль) помещают в эмалированный
нитратор или в круглодоппую колбу и нагревают иа водяной бане до
40°. По достижении этой температуры при сильном перемешивании до-
бавляют по каплям нитрующую смесь, приготовленную из 60 г H2SO(
(уд, вес 1,84) и 68 a HNO3 (уд. вес 1,52); добавление ведут таким об-
разом, чтобы температура реакции была 40—50°. При сильном превы-
шении этой температуры образуется некоторое количество динитросое-
динения. Все количество нитрующей смеси прибавляют приблизительно
за 10 мин. Перемешивание продолжается еще около 2 час. Реакцион-
ную смесь медленно охлаждают и -выливают и а лед при периодическом
сильном перемешивании стеклянной палочкой. Добавлением льда под-
держивают температуру около 0°, Образовавшуюся кашицу после
продолжительного стояния фильтруют, промывают холодной водой и
тщательно отсасывают. Осадок помещают в центрифугу и центри-
фугируют при 15° до тех пор, пока не перестанет отделяться жид-
кость. Таким путем получают около 80 г практически чистого пара-про-
дукта.
Отделенная на центрифуге жидкость состоит нз небольшого коли- -
честна воды и эвтектики (примерно 30% п- и 70% о-нитрохлорбензола).
Она соединяется с фильтратом и помещается в делительную воронку,
где эвтектика отделяется от кислоты и промывается до нейтральной
реакции.
Эвтектику подвергают затем тщательной дробной перегонке в ва-
кууме с ’применением ректификационной колонки, описанной на стр. 304.
После отделения небольшого первого погона, состоящего из воды и сле-
дом хлорбензола, собирают первую главную фракцию (начиная со 105°
при 10 мм остаточного давления). Перегонку продолжают до тех пор,
пока не отгонится 3/г от общего количества жидкости. Из полученной
таким образом первой фракции после охлаждения и центрифугирования
при 15° выделяется еще несколько гра'ммов л-ннтрохлорбепзола. Остав-
шиеся а/э жидкости лучше перегонять в вакууме без колонки. После
охлаждения и центрифугирования при 15° из этой фракции выделяется
около 10 з о-нитрохлор бензол а.
Эвтектику, выделенную прн центрифугировании обеих фракций,
соединяют и снова подвергают дробной перегонке и кристаллизации.
Такую обработку повторяют, пока это будет целесообразно.
Пол ученные таким образом продукты уже достаточно чисты. Если
хотят получить их в химически чистом виде, то во время центрифугиро-
вания в центрифугу вдувают 1—2 мл распыленного бензола. В чистом
виде соединения обладают следующими температурами кипении и плав-
ления: 1
* Герм. пат. 97013 (Frdl., V, 47).
ло
/. Промежуточные продукты.
Соединение Т. пл., °C Т. кип., °C при 10 мм (испр.) Т, кнп„ вС при 727 (искр.)
n-Нитрохлорбензол 82,5 106 234
о-Нитрохлорбепзол 32,5 109,5 241
Точная крива и температур плавления смесей о- и n-нитро хлорбен-
зола, с помощью которой можно непосредственно определить содержа-
ние орто- и пара-продукта в смеси, опубликована *.
л-Нитроаиилин н о-нитроанилин из л- и а-нитрохлор бензола
+ 2МНЙ
NH3
|/Xj +NH.JC1:
I
no2
+ 2NH3 = |
-j- NH4C1
1 моль ti- иитрохл орбензол а (или о-ннтрохлорбензола) на-
гревают в автоклаве в течение 10 час. с 10 молями аммиака до 170°.
В реакцию берется 25%-ный аммиак, давление повышается до 35 атм
Рис. 16. Кривые зависимости давления водных раство-
ров аммиака от температуры.
(см, рис. 16). После охлаждения содержимого автоклава давление спу-
скается. Нитроанилин выделяется в кристаллической форме, Он обычно
еще содержит следы ие вступившего в реакцию иитрохлорбензола. По-
этому нитро'аинлин перекристаллизовывают из кипящей воды. При
Holleman, de Bruyn, Rec. trav. chim., 19, 192.
п-Нитрофенилоксаминовая и п-аминофенилоксаминовая кислоты
87
этом следует учитывать, что иитроанилин немного летуч с водяным па-
ром и сильно ядовит. Кипячение поэтому следует вести с обратным холо-
дильником. Выходы составляют в среднем 95% от теории.
Технические примечания. На производстве поступают несколько иначе.
Как только реакция окончилась, избыточный аммиак выпускают в конденсационную
установку (вода). Затем содержимое автоклава, в котором при 150° все количество
нитроанилина находится в растворенном состоянии, продавливается через железный
фильтр-пресс. Нитроанилину не дают выкристаллизоваться; раствор сразу стекает
в холодную воду, вследствие чего выпадает мелкокристаллический продукт, что
является желательным, так как в таком виде он может гладко диазотироваться.
Таким же путем получают соответствующие нитроанилины из 1,4-дихлор-5-
нитробедзола и аналогичных соединений. Чем больше «отрицательных» заместителей
имеется в молекуле вещества, тем легче проходит эта реакция.
В качестве примера можно привести получение л-нитроанилин-о-сульфокислоты
(см. стр. 92).
Описанный способ в значительной степени вытеснил более старый метод полу-
чения п-нитроанилина (нитрование ацет- или форманилида, см. Стр. 120), хотя себе-
стоимость при получении его обоими способами примерно одинакова. По новому
способу легче получать продукт, совершенно не содержащий изомеров; кроме тоги,
способ дает очень желательную часто возможность использования п-нитрохлорбен-
зола, который образуется при нитровании хлорбензола в значительно больших коли-
чествах, чем о-изомер (см. стр. 84). Большая потребность в последнем для получе-
ния о-нитро ан изола (из которого готовятся о-анизидин и дианизидин, см. стр. 91)'
приводит к тому, что на производстве накапливается такое количество п-нитрохлор-
бензола, которое не может быть полностью использовано для иных целей, чем полу-
чение гс-нитроанилина.
п-Нитроанилин применяется прежде всего для производства многочисленных азо-
красителей и для образования их на волокне (пара-красный). Из него получают
также n-фенилендиамин и его производные (см. описание следующего синтеза).
«Нитроанилин также находит применение в синтезе азокрасителей, однако в значи-
тельно меньших масштабах. Путем конденсации с бензальдегидом и сульфирования
получается ценный желтый краситель, применяемый для крашения шерсти *.
n-Ннтрофенилоксаминовая и п-аминофенилоксамииовая кислоты **
NH3
соон
I I + I
\/ соон
NOa
NH—СО—СООН
NH—СО—СООН
nh2
п- А м чмоф ен и л о -
ксйминсвая
кислота
138 г (1 моль) л-питроаиилииа и 225 г (2,5 моля) сухой
(безводной!) щавелевой кислоты нагревают, хорошо перемеши-
вая, в реакционном котле (см. рис. 36, а н 36,6) при 110°. п-Ннтроаин-
лин и щавелевая кислота должны быть предварительно тонко измельчены
и тщательно смешаны. Чтобы достигнуть хорошего смешивания, в котел
помещают 6 стальных шаров диаметром 25 мм, которые служат для
обеспечения тонкого измельчения. Сначала температуру медленно под-
нимают до 100° (в течение двух часов); при этом образуется щавелево-
кислый л-ннтроанилин. Энергичное перемешивание смеси прн 110° про-
должают в течение 12 час. Образующуюся воду отсасывают с помощью
водоструйного насоса. Прн ‘этом увлекается некоторое количество
* Герм. пат. 289111 (Frdl., ХИ, 308).
** Описан также синтез 1-антрахинсчюксаминовой кислоты и индантрена красного
5 ГК (Curtis, Диссертация, Zurich — Weida, 1929).
оо
л промежуточные продукты
щавелевой кислоты. По окончании реакции (когда реакционная масса,
находящаяся в котле, станет нерастворимой в воде) сырой продукт обра-
батывают l’/г л холодной воды, а затем отфильтровывают от раствора
щавелевой кислоты. Осадок промывают на иутч-фильтре тремя порциями
(по 250 мл) холодной воды.
Выход технически чистой rt-питрофсиилоксамиповой кислоты со-
ставляет 95% от теории, т. пл. 205—207°, после перекристаллизации из
воды т. пл. 210°.
Восстановление. Для восстановления берется четвертая часть <
полученного продукта (1/4 моля), п - Н и т р о ф е н и л о к с а м и н о в а я )
кислота прибавляется в '<
течение 40 мии. в нагретую
до кипения смесь 200 г ч у- ’
гуиного порошка (см.
Рис. 17. Редуктор с пропеллерной мешалкой
Для восстановления по Бешану — Бриммейеру.
Рис. 18. Редуктор с винтообраз-
ной мешалкой.
/ — загрузочное огиерстие с крышкой,
закрепляющейся с пом о шью <к.1бы;
2 — патрубок для трубы, служащей для
разгрузки.
стр. 72 и 74), Г л воды н 50 мл концентрированной соля-,
иой кислоты. Реакционная смесь должна все время кипеть н
сильно перемешиваться (см. рис. 17 и 18). Восстановление считается ,)
законченным, когда вытек реакционной жидкости на фильтровальной )
бумаге будет бесцветным. Затем при температуре кипения прибавляют ‘
30 г осажденного мела и кипятят еще ‘/г часа (переведение
в кальциевую соль). После этого для полного осаждения железа по- ;
степенно вносят 45 г кальцинированной соды и раствор, имеющий
отчетливую щелочную реакцию, отфильтровывают от же-
лезпого шлама. Остаток в горячем состоянии промывают 200 мл 2 %-кого J
раствора соды. К прозрачному фильтрату добавляют концентрирован-
ную соляную кислоту до кислой реакции по конго. Выпавшие
белые кристаллы «-амипофеиилоксаминовой кислоты после охлаждения
п-Нитрофенол гидразин
89
отфильтровываются и промываются небольшим количеством холодной
воды. После высушивания обычно получают около 42 г п-аминофенмл-
оксамшювой кислоты, т. с. примерно 93% от теории.
гг-Аминофенилоксами новая кислота является важным промежуточным продук-
том для получения полиазокрасителей. Остаток щавелевой кислоты в противополож-
ность ацетильной группе легко отщепляется при омылении, благодаря чему амино-
группа становится опять свободной и может диазотироваться.
п-Нитрофеиилгидразин *
/—X /—X + С1
O2N—р;—NH2+NaN°2-f-2HCl = O2N— / N= N -|- NaCl 4- 2НаО
у—, + С]
ф—N=N 'I’ NaaSOs + NaHSOs =
O2V-/ \_N-NH SO3Na 4- NaCl
SO3Na
O2N—N—NH SO3K 4- HC14- 2H,0 =
>—/ [
SOsK
~ O3N—/ NH—NH2 HC1 4- 2KHSO4
34,5 г (’/4 моля) n - нитро а и и л и и а диазотируются по методике,
описанной па стр. 223. В раствор диазосоединения (который можно не
фильтровать), охлаждаемый снаружи смесью льда и поваренной соли,
при постоянном механическом перемешивании медленно добавляют каль-
цинированную соду. Добавление ведут до тех пор, пока раствор не
станет вызывать лишь слабого посинения конго бумажки (для этого тре-
буется 18—20 а). При этом следует тщательно следить за тем, чтобы сода
тотчас распределялась в растворе и чтобы реакция в отдельных- частях
раствора ие становилась щелочной; температура должна цо возможности
поддерживаться около 0° и ни в коем случае не должна превышать 5°.
Последний остаток свободной кислоты нейтрализуют небольшим количе-
ством бикарбоната натрия. Раствор отфильтровывают через
складчатый фильтр от отдельных коричневых хлопьев; полученную про-
зрачную светло-желтую жидкость при постоянном перемешивании при-
ливают к охлаждаемой извне до температуры ниже 10° смеси 155 г
(0,55 моля) раствора бисульфита натрия (уд. вес. 1,36) и 21 мл
(l/t моля) едкого иатра (уд. вес 1,38). При этом образуется про-
зрачный нейтральный по конго раствор интенсивного зеленовато-желтого
цвета. К этому раствору прибавляют 120 г хлористого калия (прн
этом выпадает в осадок трудно растворимая калиевая соль нитрофеиил-
гидразиидисульфокислоты) и перемешивают до тех пор, пока хлористый
калий не перейдет полностью в раствор. Чтобы осаждение быле более
полным, реакционную смесь оставляют иа ночь в холодном месте. Густую
желтую кашицеобразную массу тщательно отсасывают (с растиранием
и отжиманием) и промывают (охлажденным 10%-иым раствором хлори-
стого калия. Осадок, который при правильном отсасывании должен
* Bamberger Е., Kraus, Вег., 29, 1834.
весить не более 170 г, помещают в фарфоровую чашку и обрабатывают
140 мл концентрированной соляной кислоты (уд. вес. 1Д7); при-
бавление кислоты производят постепенно при размешивании пестиком.
Размешивание продолжают до тех пор, пока не образуется совершенно
гомогенная кашица; последнюю оставлиют стоять 4 часа при комнатной
температуре. Затем реакционную смесь нагревают на водяной бане,
снова тщательно размешивая пестиком, до превращения мелкокристал-
лической желтой кашицеобразной массы в крупнокристаллическую
бледно-красную суспензию солянокислой соли п-нитрофенилгидразнна.
Нагревание на водяной бане продолжают еще ’Д часа; при этом не дол-
жен обнаруживаться запах нитробензола, а также происходить осмоле-
нне. После стояния в течение ночи (желательно в холодильном шкафу)
осадок отсасывают и промывают на фильтре охлажденной смесью равных
объемов концентрированной соляной кислоты и воды. Осадок, получен-
ный после отсасывания, растворяют в 300 мл тепловатой воды, отфиль-
тровывают от незначительного остатка (коричневые хлопья) н нейтра-
лизуют основную часть соляной кислоты добавлением 25 мл 25%-ного
раствора аммиака (лакмус должен еще окрашиваться
ный цвет; в противном случае следует добавить уксусной
чтобы реакция стала снова кислой); осаждение заканчивают
нием раствора 15 г кристаллического ацетата натрия
в
кислоты,
добавле-
в 40 мл
воды; после полного охлаждения кристаллическую оранжево-желтую
массу отсасывают, к фильтрату добавляют еще некоторое количество
ацетата, чтобы убедиться в полноте осаждения; осадок промывают хо-
лодной водой и сушат в сушильном шкафу с паровым обогревом при-
мерно прн 80°.
Выход около 30 г, т. е. 75—80% от теории, т. пл. 156—157° (спе-
кается за несколько градусов до плавления). Полученный таким образом
неочищенный сырой продукт пригоден в большинстве случаев без даль-
нейшей очистки. Прн перекристаллизации из 20-кратного количества
спирта получают совершенно чистый п-нитрофеннлгндразин в виде корич-
невых с синеватым отливом узких листочков,
при 157—158°.
которые
четко
плавятся
Совершенно аналогичным образом из о-иитроа пилила* полу-
чают о-нитрофенилгндразнн; только прн этом для выделения
калиевой соли сульфокислоты раствор после прибавления хлористого ка-
лия следует еще сильно подкислить концентрированной соляной кис-
лотой **.
Технические примечания, Нитрофенилгидразины применяются для
получения нитрофснилпиразолонод ***, из которых после восстановления нитро-
группы в аминогруппу получают ценные конечные азосоставляюшие для прямых азо-
красителей, которые могут на волокне подвергаться дальнейшему диазотированию и
сочетанию или обрабатываться формальдегидом ***♦.
Нитрофенилгидразины (п- и о-} являются, кроме того, ценными реактивами,
которые применяются в исследовательских лабораториях для выделения и идентифи-
кации альдегидов и кетонов. Особенно важную роль в этом отношении они играют
в химии углеводов.
* Диазотирование в этом случае целесообразно проводить по методике, опи-
санной для 3-нитро-4-тол ундина (стр. 223), с применением 100 мл концентрированной
соляной кислоты и 120 г льда на каждые 0,25 моля о-нитроанилина.
* * См. Miiljer Н., Monti gel С,, Reich stein Т., Helv. China. Acta, 20,
1472.
**'• См. стр, 118. , ,
*«•* См., например, герм. пат. 278871/2, 287071, 289350 (Frdl., XU,. 334—339);
герм. пат. 296141 (Frdl., Xlll, 527).
о-Нитроанизол из о-нитрохлорбензола
С1
I
O-NO^
CIT.ONa
128« "
OCHg
,/Xi-NO2
В автоклав без мешалки емкостью 1 л, снабженный масляной баней,
помещают 600 мл сухого метилового спирта, в котором предва-
рительно растворяют 23,0 г металлического натрия (обратный холо-
дильник!) и 158 г чистого о-н и т р о х л о р б е и з о л а с т. пл. ~ 32° и
т. кип. 243°. Смесь нагревают в плотно закрытом автоклаве в течение
1 часа до 120°, оставляют при этой температуре на 3 часа и затем в те-
чение еще одного часа поднимают температуру до 128°. Давление при
этом составляет 8—40 атм. По окончании реакции метиловый спирт вы-
пускают через вентиль в хорошо действующий холодильник. Полученный
таким образом метиловый спирт может без дальнейшей очистки снопа
вводиться в реакцию. Продукт реакции извлекается из автоклава, кото-
рый после этого споласкивается горячей водой для растворения и удале-
ния выпавшей поваренной соли. Неочищенный продукт дважды промы-
вается пятикратным объемом горячей воды, отделяется от водного слоя
и перегоняется в вакууме.
Выход составляет 136 г, т. с. 88% от теории; т. кип. 141° (15 лш).
Технические примечания. Аналогичным путем можно получить целую
серию алкильных и фенильных эфиров ряда бензола (а также ряда нафталина и
антрахинона). В то время как в случае нитроанизола удовлетворительные результаты
получаются при применении безводного метилата, часто оказывается выгоднее ис-
пользовать щелочь (например, при получении динитроаниэола из динитрохлорбен-
зола) ; прн этом наличие небольшого количества влаги не мешает реакции. Кроме
того, во многих случаях полезно применять только 90% от теоретически рассчитан-
ного количества щелочи.
В качестве интересного в техническом отношении примера можно привести по-
лучение I-амино-2,4-днметокси-5-хлорбензола. Процесс можно изобразить следующей
схемой:
1-Нитро-2,4,5- 1-Ни тро-2,4-диме-
трихлорбензид токси-5—хлорбензол
1-Амино-2,4-ди-
метокси-5-хлорбензол
Нафтол АС ИТР (азотол О)
1-Амино-2,4-диметокси-5-хлорбснзол является исходным веществом для полу- '
чсния ценного нафтола АС ИТР (азотола О), который применяется для образования *
на хлопчатобумажном волокне пунцово-красных окрасок. Упомянутые выше реакции ;
протекают так же, как п при синтезе о-ннтроаиизола, поэтому не имеет смысла рас-
сматривать их подробнее. i
о-Нитроанизол служит для получения обоих особенно важных для производства ’
азокрасителей оснований — о-аннзидина и о-дианцзидина. Первое из этих оснований j
получают восстановлением по Бсшану—Бриммсйеру, как это описано при рассмотре- (
иии синтеза анилина (стр. 71). о-Дианизидин образуется аналогично бензидину
(см. стр. 114): на о-нитроанизол действуют спиртовым раствором щелочи и цинковой \
пылью или раствор о-нитроанизола в сользент-нафте обрабатывают едким натром и ч
цинкоек й пылью; полученный таким образом гидразоанизол подвергают «бензиди- ;
новой перегруппировке». 7
Получение о-питроанпзола метилированием о-нитрофенола описано на стр. 134. <
й-Нитрохлорбензол-о-сульфокислота и я-нитроанилин-о-
сульфокислота из /2-нитрохлорбензола
100 г л-нитрохлорбензола смешивают при 50° со 100 г моногидрата *
и прн перемешивании добавляют 280 г олеума, содержащего 25% SOa. Нагревают j
при 100—110° до тех нор, пока нитрохлорбензол полностью не вступит в реакцию.
Затем реакционную смесь выливают в ледяную воду (300 г льда н 300 г в о д ы);
и высаливают 200 г .поваренной соли. Через 24 часа фильтруют и отжимают. (
Выход невысушенного Продукта составляет в среднем 280 г. Для получения ч
л-нитроанилинсульфокислоты измельченный осадок нагревают в течение 8 час. в авто- •(
клаве до 150° с равным по весу количеством концентрированного аммиака *
(20% NHs). Давление достигает примерно 6 атм (стальной манометр!). При охлажде- 3
нии аммонийная соль л-нитроанилин-о-сульфокислотъг выпадает в виде крупных,
твердых кубических кристаллов янтарного цвета. Кристаллы отделяют; вес их состав1
ляет около 100 г. На производстве маточный раствор аммонийной соли обрабатывают *
известью для выделения аммиака.
л-Нитроанилин-о-сульфокислота применяется главным образом для произвол- (
ства азокрасителей; например, с ^-нафтолом она дает ценный лак красный П (лак
красный Б). Продукт его восстановления, л-фенилендиаминсульфокислота, исполь- >
зуется для получения азиновых красителей, а в виде монобензоильного произвол- 1
ного—для синтеза азокрасителей. :
л-Нитрохлорбензолсульфокислота находит широкое применение для получения ;
диаминодифениламинсульфокислоты и амииодифениламинсульфокислоты. Схемы по-
лучения последних приводятся ни^ке. )
1. Диаминодифениламинсульфокислота J
и MgO (или
СаСО3) в вод-
ном растворе;
кипячение в
деревянном
чане
п-Фенилсн-
диамив
U (4 fl-nut риипилич-о-сулырокислотл
Основная
реакция
км
Восстаю) вление
по Бешану
Легко раствори-
мая натриевая
соль
(О
N03
кн3
Диамнноли-
ф ин илам ин-
сульфокислота
(И)
Натриевая соль
трудно растворимая;
легко отделяется
Промежуточный продукт (1) даст при сочетании с аминонафтолсульфокислотами
ценные азокрасители, применяющиеся для крашения хлопчатобумажных тканей
в темные цвета.
2, Аминодифениламинсульфокислота (III)
и аминофенилтолиламинсульфокислота
+‘АНИЛИГ! (или о-толуиаин)
н автоклаве при 140°;
перемешивание; MgO -|-пода^
Восстановление
по Бешану
Амнноли-
фениламик
сульфЛислота
Эта составляющая (III) дает и с р о л и — дисазокра<ители, получающиеся путем
диазотирования и сочетания с а-иафтиламином с последующим повторным диазотиро-
ванием и сочетанием с солью Шеффера или другими азосоставляющими.
Следует также отметить, что на фенил амино- или ариламиногруппу
могут замещаться не только атомы галоидов, но также н нитро- и сульфо-
группы. Легкая подвижность нитрогрупп особенно ярко выражена в тех
случаях, когда они связаны с ядром антрахинона; это используется для
получения некоторых важных соединений ряда антрахинона; рассмотреть
их в настоящей работе не представляется возможным. Многие производ-
ные бензола, содержащие несколько питрогруцп, также дают интересные
продукты конденсации.
2Л-Динитрохлорбензол из хлорбензола
С1 С1
I ;
+ 2HNO3=f/Xi-N°3 + 2ЩО
I
no2
В железный сосуд для нитрования (см. рис. 19) вливают 280 г ни-
трующей смеси, содержащей 50% азотной кислоты*, и при очень
хорошем перемешивании добав-
ляют по каплям 113 г (1 моль)
хлорбензола; при этом
поддерживается температура
ниже 5°. После того как при-
бавлено все количество хлор-
бензола, перемешивание при
5—10° продолжают еще час.
Рис. 19. Сосуд для сульфирования
н нитрования.
Стакан из листового железа пли лучше из
чугуна (фарф ра> стоила) диаметром около
11 с..1 и высотой 20 с.и. Стакан закрывается
точно пригнанным свинцовым кружком
с отиерстпямл для мешалки, термометра и
вводной трубки, Очень удобной по форме
явл!|стс11 мети а лк а, показанная на рисунке,
Во всех случаях, когда не требуется осо-
бой механической прочности, мешадка мо-
жет инготсн.ипвся из стеклянной палочки.
Вообще же следует предпочесть железные
мешалки. I Оправляющие т р у 0 Р11 it и лучше
всего изгото ниять из меди, п ^сксльку и же-
лезо, и стекли очень хорош,) скользят по
меди и при этом не происходит неприятных
случаен заедания мешалки. Шкив целесо-
образно додать из бр 1изы, тик как бронза
также очень хорошо скользит _ по меди;
(смазки -- вазелин}- Термометр с верхней'
[пк ал ай доджей быть погружен возможно
глубже. Кроме того, слеаусс отмстит,., что
во" всех случаях, когда требуется тщательно
следить за цнеденцем жшдкостн, необходим»
пользоваться капельной норочкой с капель-
ным наконечником. Стакан иимешают па
[|[|Очи).|н треножник, а цапр:|цля[ошую мед-
ную трубку прочло укрепляют с лимитно
надежи,,го зажим|1. Тик >кй укрепляют тер-
MO.VCTJI и капельную BopoHiry, причем ме-
шалкЗ не должна их задеиап,.
Затем температуру медленно поднимают до 50° и выдерживают реакцион-
ную смесь при этой температуре еще в течение одного часа; после этого к
хорошо перемешиваемой смеси очень осторожно добавляют по каплям еще
Смесь 140 г моногидрата и 140 г азотной кислоты УД- веса 1,52.
350 г концентрированной серной кислоты ц нагревают в течение
*/> часа при 115 3 Реакционной смеси дают охладиться и выливают се в
2 л ноды; ири этом она тотчас застывает в иветло-окслтую массу. По-
следнюю отделяют от маточного раствора и расплавляют под слоем воды
для удаления кислоты. Полученное таким образом вещество является
почти химически чистым.
Выход йз 113 г хлорбензола составляет 200 г, т. пл. 51°.
Технические примечания. Динитрохлорбенэол производится в больших
количествах. Он служит для ролунения сернистого первого и других ценных кра-
сителей. Он является также исходным материалом для получения целого ряда про-
дуктов конденсации, синтезируемых заменой подвижного атома хлора на остатки
основного или другого характера. Из динптрохлорбензола получаются динитроацн-
лци, дпнитрофевол и Пикриновая кислота, дцнитроанизол в т, д. В приведенной
схеме представлена только небольшая часть всех практически используемых утревра-
Щенпй (см. также табл. 5) * * ***.
Выше было отмечено, что в лаборатории, для того чтобы нитрование проходило
более гладко, применяют 10%-пый избыток азопюн кислоты. В технике удастся
обходиться, применяя значительно меньший избыток; последний к тому же почти
полностью регенерируется (так как отработанная кислота разделяется на серную и
Дзотную кислоты при обработке водяным паром в децитрациочных башнях).
* См. стр. 106.
44 Carter, Zlschr. gcs. Spreng- n. SchicBw-esen, 8, 205 и 251 (1913).
*** Следует помнить, что дннитрохлорбензол оказывает очень неприятное дей-
ствие. Он, а особенно его растворы, вызывают экзему и невыносимый зуд.
95
/, Промежуточные продукты
.«-Фенилендиаминсульфокислота из 2,4-динитрохлорбензола *
202 г (1 моль) 2,4 - д и п и т р о х л о р б е н з о л а (см. стр. 94) сме-
шивают с 500 г с и и р т а тех и и ч., недепатурировацного добавкой пи-
ридиновых оснований. При этом следует соблюдать осторожность вслед-
ствие неприятного действия дииитрохлорбензола (см. примечание на
стр. 95). К полученному раствору добавляют 64 а (1 моль) SO2 в виде
возможно более концентрированного раствора сульфита натрия.
Последний готовят, добавляя к точно рассчитанному количеству раствора
технического бисульфита натрия концентрированный раствор . едкого
натра. 40%-ный раствор едкого натра добавляют до тех пор, пока не
исчезнет желтоватая окраска раствора и фенолфталеиновая бумажка не
станет очень слабо окрашиваться в красный цвет. Часть сульфита выпа-
дает в осадок даже при повышенной температуре, но это не имеет зна-
чения. Полученную смесь диншрохлорбензола, сульфита, воды и спирта
нагревают на водяной бане до кипения в течение 5 час. при хорошем
перемешивании. Затем смесь охлаждают до возможно более низкой
температуры погружением в холодную воду. Натриевую сачь динитро-
бензол сульфокислоты, выделившуюся в виде красивых, блестящих жел-
тых листочков, отсасывают на нутч-фнльтре и сильно отжимают па вин-
товом прессе. Восстановление проводят точно так же, как и в случае
л-динитробепзо.ча (см. стр. 106), Полученный таким образом раствор
м-фенил он диаминсульфокислоты не является достаточно чистым и не
может еще применяться для производства азокрасителей. Поэтому рас-
твор следует упарить до 400 мл и обработать 100 г поваренной
соли. При подкислении соляной кислотой свободная сульфо-
кислота выпадает в виде красивых кристаллов. Важно взять правильное
количество соляной кислоты, так как в избытке последней сульфокислота
опять растворится. Конго бумажка должна окрашиваться не в голубой,
а в отчетливый слабо-фиолетовый цвет. Осадок отфильтровывают через
2 дня и промывают очень небольшим количеством воды.
Выход чистого вещества составляет 125 а, или около 66% от
теории.
Технические примечания. Такого рода реакции с сульфитом лучше
всего проводить н гомогенно-освинцованных котлах (см, стр. 323.). Важно, чтобы
в жидкость не попало следов других металлов. Присутствие нескольких миллиграм-
мов меди или железа приводит к тому, что не образуется даже следов нужного
соединения. Как динитробензолсульфокислоту, так ц полученную л-фецилендиамин-
сульфокислоту лучше нс фильтровать на фильтр-прессе, а отделять центрифугиро-
ванием. Остаток спирта удаляют отжиманием на гидравлическом прессе; полученный
спирт после ректификации на колонке может снова применяться. При тщательной
работе на одной операции теряют не более 5% спирта.
Описанный способ является примером непрямого введения сульфогруппы; он
основан на использовании подвижности хлора .. ароматических соединениях, содер-
жащцх наряду с хлором отрицательные заместители (см, также описание синтеза
следующего препарата).
* Герм, пат G5240 (Г'гсП., ill, 41),
Анилин-2 ^-дисульфокислота из хлорбензола
97
лг-Фенилеиднаминсульфокпслоту можно также легко синтезировать, сульфируя
олеумом прн 160я растворенный в моногидрате чистый -и-фенилендиамин. л-Феннлспди-
ампнеульфокнелота используется как составляющая прн получении азокрасителей.
Анилии-2,5-дисульфокислота из хлорбензола
а) 2 - Н и т р о х л орбензол -4 - су ль фо кислота
из хлорбензола*
Сульфуратор или круглодонпую колбу с хорошо действующей ме-
шалкой, термометром и капельной воронкой (см. рнс. 6 и 20) помещают
в сосуд большего размера, который может наполняться водой, служащей
для охлаждения или обогрева. В котел (или колбу) вносят 220 а моно-
гидрата н 125 а 20%-ного олеума. Затем при перемешивании в те-
чение примерно 1/э часа добавляют по каплям 112,5 г (1 моль) хлор-
бензола. При этом температура поднимается от 70 до 80°. Когда
после окончания добавления температура начнет падать, внешний сосуд
заполняют горячен водой и нагревают ее до кипения. Перемешивание
на кипящей водяной бане продолжают до тех пор, пока при разбавлении
пробы водой не прекратится выделение масла и пока разбавленная проба
при кипячении не перестанет пахнуть хлорбензолом (на это обычно тре-
буется около двух часов). При этом, как правило, совершенно прозрач-
ного раствора пе получается, так как при реакции образуются незначи-
тельные количества дихлорднфепилсульфона, который выделяется уже
не в виде масла, а в виде кристаллических хлопьев.
Реакционную смесь охлаждают и затем к ней в течение 1’/2—2 час,
добавляют <ю каплям при охлаждении льдом и псремешиваини 65 г
азот пой кислоты (уд. вес 1,52); добавление ведут таким образом,
чтобы температура оставалась в пределах от 15 до 20°, После этого пре-
кращают охлаждение и для окончания реакции перемешивают еще 3—
4 часа при комнатной температуре. Часть образовавшейся нитрохлор-
бенз од сульфокислоты выпадает; благодаря этому вся реакционная смесь
застывает в довольно твердую массу, которую, чтобы сделать возможным
дальнейшее перемешивание, можно измельчить стеклянной палочкой или
шпателем. Убедившись (в результате определения содержания в пробе
азотной кислоты с помощью нитрометра), что-все количество азотной
кислоты, за исключением введенного в реакцию небольшого ее избытка
(около 2 г), израсходовано, реакционную смесь выливают в 1 л насы-
щенного раствора поваренной соли, к которому добавлено '/%л
вод|х. Полученной смеси дают охладиться при перемешивании; после
этого отсасывают, промывают 20%-пым раствором повареннойсоли
и отжимают па винтовом прессе (рис. 21).
Вес влажного отжатого осадка около 370 г; удержание сухого ве-
щества около 72%.
Перекристаллизацией из неболвйюго количества горячей воды из разбавленного
раствора поваренной соли из разбавленного спирта можно получить натриевую
е('ль 2-нитрохлорбен?,ол-4-сульфокислоты в чистом состоянии.
* Герм. пат. 116759 (Frdl., V, 931).
98
Л Иромежуточные продукты
б) Нитробензол - 2,5 • д ис ул ь фоки с л о т а *
2-нитрохлорбензол-4-сульфокислоты
400 мл кипящей воды
и
ряют
3
Полученный по описанной выше методике влажный осадок натрие-
вой соли 2 - нитр ох л о р б е изо л - 4 - су л ь ф о кисл оты раство-
отфильтровывают от небольшого коли-
чества остатка, В фильтрате раство-
ряют 300 г кристаллического с у л fa-
фита натрия (Na2SO3 • 7I IaO);
полученный раствор не должен вы-
зывать покраснения фенолфталеино-
вой бумажки; если это имеет место,
то осторожно добавляют несколько
капель соляной кислоты до исчезно-
вения щелочной реакции. Нейтраль-
ный раствор кипятят в течение 2 час.
с обратным холодильником. Раствор,
А
и с. 21, Винтовой пресс с хомутом из
ковкого железа.
Плита пресса покрыта листовой медыо; продукты
помешаются между дощечками из древесины
твердой породы.
Рис. 20, Сульфуратор, автогеиио сва-
ренный, особенно пригоден для работы
с олеумом.
принимающий при этом ярко-жел-
тую окраску в горячем состоянии, об-.-
рабатывают 100 г твердой пова-‘
рениой соли. После того как;
последняя полностью растворится,^
раствор оставляют для охлаждения^
(желательно в холодильном шкафу) 1
на ночь. Выпавший осадок (густаяЗ
кристаллическая масса) отсасывают^
и тщательно при измельчении про-’
мывают холодным насыщенным раствором поваренной солн до тех пор/
пока стекающий фильтрат не будет иметь лишь очень слабую желтую'
окраску. Натриевую соль нитробензол-2,5-дисульфокислоты можно по--
* Герм, пат, 77192 (Frdl,, IV, 37),
лучить в чистом состоянии перекристаллизацией из разбавленного спирта.
Вещество, полученное после отсасывания, можно восстанавливать, не
подвергая дальнейшей очистке.
в) Анилин- 2,5 - д и су л ь ф о'к и слота
Восстановление осуществляется по методике, которая более подробно
приводится при описании получения анилина (см. стр. 71). Влажный
осадок нитро бензол-2,5-дисульф окис лоты, полученной, как это описано
в разделе «б», вносится небольшими порциями приблизительно в течение
1 часа в кипящую и энергично перемешиваемую смесь 200 г желез-
ного пор о ш к а, 1л воды и 20 мл ледяной уксусной кис-
лоты (см. рис. 17). После этого реакционную смесь кипятят еще около
7г часа при перемешивании, добавляя воду (вместо испарившейся), до-
водят добавлением кальцинированной соды до отчетливо щелочной
реакции (па это требуется около 30 г соды) и отсасывают в горячем
состоянии от железного шлама. Последний еще раз кипятят с водой,
опять отсасывают и промывают горячей водой до тех пор, пока проба
фильтрата после диазотирования не перестанет давать заметного окра-
шивания со щелочным раствором ^-нафтола или Р-соли. Соединенные
фильтраты подкисляют небольшим количеством концентрированной с о-
ляиой кислоты до появления кислой реакции по лакмусу и при
перемешивании упаривают приблизительно до 800 мл, К полученному
раствору добавляют еще 60 в поваренной соли, сильно подкис-
ляют, добавляя 200 мл концентрированной соляной кислоты, и
оставляют на ночь для охлаждения, Бесцветную густую кашицу отсасы-
вают, промывают насыщенным раствором поваренной соли, отжимаю?,"
высушивают и измельчают в порошок. Получают 210—220 г мононатрие-
вой соли анилин-2,5-дисульфокислоты в виде белого порошка, иа 1 г
которого расходуется 3 мл 1 и. раствора нитрита. Таким образом, выход
составляет 45 е по нитриту, т..е. около 65% от теории (по хлорбензолу).
2-Нлтрохлорбензол-4-сульфокислоту можно получать не только нитрованием
хлорбензол-«-сульфокислоты, но также и сульфированием о-нитрохлорбензола
(см, табл. 3), Последний способ связан с необходимостью выделения о-нитрохлорбен-
зойа и отделения его от образующегося одновременно с ним пара-изомера; поэтому
он сложнее и дороже, чем описанный выше метод. Однако и он может оказаться
полезным, если потребность в п-нитрохлорбснзолс настолько велика, что является
желательным использование побочно образующегося о-питрохлорбензола, Однако
и настоящее время дело обстоит иначе; для производства о-нитроанизола потреб-
ляется такое большое количество о-нитрохлорбензола, что часть побочно образую-
щегося пара-изомера необходимо перерабатывать в п-нитроанилин.
2-Нитрохлорбснзол-4-сульфокислота дает в результате восстановления о-хлорме-
тащзловую кислоту, которая находит применение для синтеза азокрасителей. Однако
значение нитрохлорбецзолсульфокислоты определяется ее способностью обменивать
атом хлора па различные группы атомов. Например, при обработке раствором едкого
натра получают о-нитрофенол-п-сульфокислоту, а из нее восстановлением — о-амино-
фснол-п-сульфокислоту; аналогично при действии раствора едкого натра в метиловом
спирте получают о-нитроанизол-п-сульфокислоту, а из нее — о-анизидин-п-сульфо-
киелоту, При реакции с аммиаком нитрохлорбензолсульфокислота превращается
в о-интроанилин-п-сульфокпелоту, а при реакции с анилином и его производными —
в о-нитродифениламин-п-сульфокислоту или ее производны^ (см, табл. 2).
К обменным реакциям такого рода относится также замена хлора на сульфо-
группу, происходящая под действие^ сульфита натрия; эта реакция приводит к по-
лучению нитробензол-2,5-дисульфокислоты, которую нельзя получить прямым суль-
фированием. Получающаяся при восстановлении последней апплип-2,5-дисульфо-
кислота применяется для производства азокрасителей, особенно вторичных бис-
и трисазокраситслей типа Сириуса светопрочно-голубого (см. стр. 250).
100
/. промежуточные продукты
а) Н и т р о -п-дихл ор б е изо л из n-дих л о р бе изо л а
С1 С1
I ।
-j- hno3 = |/X|“’NOa Н м3о
V Y
Cl с 1
В трехгорлую колбу, снабженную мешалкой, термометром и капель-
ной воронкой, помещают 147 г ([ моль) грубоизмельченного п - д и х л о р-
бензола (побочный продукт, получающийся при хлорировании бен-
зола, см, стр. 63) и приливают 300 г м о н о г шдр а та. Затем при хоро-
шем перемешивании в течение часа или полутора часов добавляют по
каплям смесь 68 г азотной кислоты (уд. вес [ ,52) и 68 г м о но-
ги д р а т а; смесь азотной кислоты н моногидрата готовится медленным
приливанием последнего в азотную кислоту при охлаждении. Как только
температура достигнет 30°, начинают слегка охлаждать водой и регули-
руют добавление по каплям таким образом, чтобы температура остава-
лась в пределах от 30 до 35°. Кристаллы дихлорбензола постепенно рас-
плавляются, и после прибавления примерно половины всего количества
нитрующей смеси образуется почти прозрачный слой масла (эвтектика
n-дихлорбензола и его ннтросоединсния). Следует следить за тем, чтобы ‘
энергичная работа мешалки обеспечила достаточное перемешивание.
Дальнейшее течение реакции сопровождается выделением кристалличе-
ского нитро-н-дихлорбензола. Иногда, особенно при плохом перемеши-
вании, питро-я-дихлорбензол начинает выпадать довольно неожиданно и
со значительным выделением тепла. Поэтому на этой стадии реакции i
опыт ни в коем случае нельзя оставлять без присмотра. Как только за-
мечают, что температура начинает слишком сильно повышаться, добавле-
ние нитрующей смеси временно прекращают и усиливают в случае
необходимости охлаждение, чтобы предотвратить разогревание более чем
до 50°. По окончании добавления смесн получают густую кристалличе-
скую массу светло-желтого цвета, Перемешивание продолжают еще 3— i
4 часа и затем, взяв пробу, устанавливают с помощью нитрометра, из-
расходовапа ли вся азотная кислота, за исключением взятого избытка.
Коион нитрования можно также установить по температуре плавления
(56—57°) образца, который получают после разбавления пробы водой и j
отсасывания. Убедившись, что нитрование закончено, реакционную смесь (
выливают в смесь 700 г льда и 300 воды, отсасывают крупно-
кристаллический осадок и промывают его холодной водой. Для удаления
последних остаткой кислоты продукт расплавляют приблизительно в ‘/а л
горячей воды, хорошо перемешивают и оставляют стоять, пока, он не
застынет с образованием лепешки. После охлаждения воду сливают
и повторяют эту операцию еще раз. Освобожденное таким образом
4-\Хлор-2-а.чинофенал из п-дихлорбензола
ю:
полностью от следов кислоты светло-желтое кристаллическое веществе
высушивается при комнатной температуре па фильтровальной
бумаге.
Выход [86—188 г, т. е. 97—98% от теории, т. пл. 56—57°.
Пр и меча ни я. Нитро-ч-дихлорбснзол может быть восстановлен обычным пу-
тем (с применением железа и небольшого количества кислоты) в л-дихлорани-
л н н. С другой стороны, хлор, находящийся в орто-положениц относительно нитро-
группы, «подвижен». Он может замещаться (не так легко, как -в 2,4-дицитрохлорбен-
аоле, но легче, чем в о-нцтрохлорбензоле) на самые различные заместители. Замена
хлора на оксигруппу при действии водного раствора щелочи описана в следующем
разделе. При действии раствора натрия в метилоном спирте образуется 4-хлор-2-
н в т ро а н и з о л. который прн восстановлении переходит в 4-х л о р-2-а н и з и д и н
(азоамин красный К, основание для прочно красного Р). При обработке аммиаком
под давлением получается 4-х лЪ р-2-н и т р о а н и л и н (основание для прочно крас-
ного ЗГЛ). Вес названные основания являются важными исходными веществами для
получения азокрасителей, особенно пигментов, лаковых красителей и ледяных краси-
телей на волокне.
б) 4-Х лор-2-нитрофенол нз нитро-я-дихлорбензола
Cl ONa
I 1
2Na0H №и j_ NaCt + Hp .
Cl Cl
48 г (*/ч моля) нитро-п-дихлорбензола нагревают в желез-
ном автоклаве с, мешалкой при хорошем перемешивании с 86 а (3/4 моля;
избыток 50%) раствора едкого натра (уд. вес 1,38) и 600 мл
воды. Нагревание (на масляной бане) продолжается [5 час; при'ЭтОм
температура внутри автоклава должна быть 145° (температура масляной
бани 186—190°). Давление ис превышает. 3 атм. В автоклаве после
охлаждения образуется кашицеобразная масса из хорошо образованных
красных кристаллов, представляющих собой трудно растворимую натрие-
вую, соль n-хлор-о-нитрофенола. Их отсасывают, промывают разбавлен-
ным раствором соляной кислоты, осадок растворяют примерно в ’/е л
кипящей воды, отфильтровывают от небольшого количества остатка
и сильно подкисляют еще горячий раствор концентрированной соляной
к и сут отоПосле охлаждения крупнокристаллический осадок свобод-
ного Хлорйитрофенола отсасывают, промывают холодной водой и сушат
при комнатной температуре на фильтровальной бумаге. Полученный про-
дукт при правильном проведении реакции имеет светло-желтый цвет, пла-
вится при 87—88° и может без дальнейшей очистки-применяться для вос-
становления. Из полученного вначале щслочнсй-о фильтрата при
подкислении выделяется еще несколько граммов менее чистого 4-хлор-2-
нитрофенола, который можно получить в совершенно чистом виде путем
перегонки с водяным паром.
Общий выход составляет около 37 а, т. е. 85% от ^теории.
Для достижения хороших результатов совершенно необходимо: 1) перемеши-
вать с возможно большей скоростью, какую только допускает работа в автоклаве;
прн этом мешалка должна идти почти по самому дну (описанный на стр. 319 вра-
щающийся автоклав в этом случае непригоден); 2) точно выдерживать нужную тем-
пературу; 3) применять раствор щелочи с концентрацией, не превышающей указан-
ную в приведенной выше методике. Если раствор щелочи слишком концентрирован
или температура слишком высока, то происходит осмолеиие, сопровождающееся от-
щеплением аммиака; выход в этом случае значительно снижается.
102 /. П ромежуточные продукты
в) 4 - X л о р - 2 - а ми н о ф с н о л из 4-хлор-2-нитрофенола
Поскольку /г-хлор-о-нитрофснол легко отгоняется е водяным паром, аппарат,
в котором производится восстановление, должен быть снабжен хорошо закрываю-
щейся крышкой с обратным холодильником (см. рис. ]], стр. 72). Последний реко-
мендуется держать под небольшим вакуумом, что предотвратит возможность выхода
паров из отверстия, через которое вводятся реагенты; и этом случае через отвер-
стие будет всасываться небольшое количество воздуха. Несмотря на эту меру,
хлориитрофенол легко расплавляется уже на ложке, если его вносить в порошко-
образном состоянии. Поэтому удобнее вносить его кусками величиной с горошину
или с лесной орех. Такие куски готовят, грубо измельчая кусок хлорнитрофенола,
полученный при расплавлении последнего в фарфоровой чашке и последующем
охлаждении. На производстве нитросоединение вводится в жидком виде из обогре-
ваемого сосуда.
Аппарат, где производится восстановление, нагревают на кипящей
водяной бане, предварительно загрузив в него 50 г тонко нзмельченных
чугунных стружек (см. стр. 74), 200 мл в о д ы и 25 мл 2 и. с о-
ляной кислоты. Затем прн энергичном перемешивании в течение
1 —1,5 часа вносят 34,7 г (2/10 моля) 4-хлор-2-нитрофсиола.
после чего смывают хлориитрофенол с холодильника, с крьипки или ,с ме-
шалки и продолжают перемешивание прн нагревании па кипящей водя-
ной бане еще не менее ’А часа. Восстановление считается законченным,
когда хлориитрофенол не отгоняется более в холодильник, а также когда
полностью пропадает его запах и капля реакционной смеси даст иа филь-
тровальной бумаге почти бесцветный вытек, который не желтеет при сма-
чивании раствором едкого натра. После этого котсл открывают и, про-
должая перемешивание, добавляют сначала очень осторожно 25 мл 2 и.
раствора соды, чтобы осадить растворенное железо, и затем еще
25 мл раствора едкого натра (уд. вес 1,38), чтобы растворить
хлораминофепол. Железный иглам отсасывают в горячем состоянии и
тщательно промывают горячей водой при растирании и отжимании»
Промывание продолжают до тех пор, пока подкисленная проба фильтрата
при добавлении раствора нитрита ие будет окрашиваться лишь в блед-
но-желтый цвет. К еще теплому фильтрату добавляют концентрирован-
ную соляную кислоту до появления кислой реакции на лакмус
(на это требуется 25—28 мл кислоты); небольшой избыток кислоты ней-
трализуют несколькими каплями концентрированного раствора уксусно-
кислого натрия. Уже в теплом растворе образуется значительный осадок
(блестящие листочки), который при охлаждении делается еще обиль-
нее. Добавлением гюварегпгой соли можно добиться более полного оса-
ждепия. Осадок оставляют стоять гга ггочь в закрытом сосуде; после
этого отсасывают, промывают сначала 15%-ным раствором поваренной
соли, затем холодной водой й сушат в сушильном шкафу с паровым
обогревом.
В ы х од составляет окол 26 г, т. е. 90% от теории.
При правильном проведении восстановления и использовании чистого нитросоеди-
нения получается продукт, окрашенный в слегка серый цвет; температура его плавле-
ния лишь немного ниже, чем в случае чистого 4-хдор-2-амипофенола (140—141°). Его
можно без дальнейшей очистки применять для получения азокрасителей. В случае
необходимости 4-хлор-2-амипофенол можно получить в, совершенно чистом виде пере-
кристаллизацией из горячей воды или растворением в горячей разбавленной соляной
кислоте с последующим фильтрованием и добавлением раствора соды до точно ней-
тральной реакции.
Примечания, Восстановление может проводиться сернистым натрием точно
по той же методике, какая вринедшга при описании синтеза аминофенолдцсульфокис-
лоты (стр. 139). Выделение проводится путем централизации.содой кислого раствора,
отфильтрованного предварительно от серы.
4-Хлор-2-амнпофсиол применяется главным образом для получения синих хро-
мирующихся азокрасителей. Продукты сочетания его с хромотроповой кислотой и
7, Производные нитробензола
103
с ацетилированной Аш-кнелепой были лучшими из продажных синих хромирующихся
красителей до открытия хромирующихся трифенилметановых красителей (эриохром-
азурол Б).
Последние превосходят азокрасителе по яркости оттенков, но уступают им по
светопрочное™.
7. Производные нитробензола
jn-Динитробензол из нитробензола
В сосуд для нитрования из стекла или железа с мешалкой н термо-
метром помещают 500 г серной к и сл оти (моногидрат) и за-
тем при хорошем внешнем охлаждении приливают 70 г безводной
(уд. вес 1,52) или эквивалентное количество не менее чем 85%-иой
азотной кислоты. Вместо этого можно также применять соответ-
ствующее количество 30%-ной нитрующей смеси. К хорошо размешивае-
мой кислотной смесн добавляют по каплям в течение получаса 123 г с у-
хого, чистого нитробензола и охлаждают снаружи льдом,
чтобы температура не поднималась' выше 15—20°. После прибавления
всего количества нитробензола перемешивание при комнатной темпера-
туре продолжают еще в течение 1 часа. Нагреванием приблизительно до
35° выпавший дннитробензол переводят снова в раствор и выливают при
хорошем перемешивании на 1 кг льда. Выделившийся динитробензол
отфильтровывают й промывают холодной водой. Сырой продукт дважды
нагревают, каждый раз приблизительно с 400 мл воды до расплавления,
причем первый раз добавляют соду до ясно щелочной реакции по лак-
мусу.
Полученный’ таким образом динитробензол содержит еще значитель-
ные количества 1,2- и 1,4-изомеров, от которых можно легко освободиться.
Как Г,2-, так и 1,4-изомер, будучи эмульгированы в воде, легко реагируют
с- сульфитом иатрня н переходят прн этом в легко растворимые ннтро-
сульфокислоты; 1,3-Изомер в этих условиях в реакцию практически не
вступает. \
NO., -ЫОЁ
I J 1
^—NOa Na2SO3 —> SOuNa.’H- NaNOa
NO2
I
Na2SO3 *" | | ф- haNO2
I _
SOaNa *
Для очистки сырого продукта его нагревают в 500 мл воды до
расплавления (примерно до 80°), при этом добавляют 5г смачиваю-
щих веществ (ядровое мыло, ализариновое масло, игапон Т, некаль
ЬИкс, смачиватель ИБ и т. д.). При очень хорошем перемешивании
к маслянистой суспензии прибавляют в течение получаса 20 г кристал-
104
i. Промежуточные продукты
лического сульфита натрия и перемешивают после этого еще 2 часа при .
90—95°. Водный раствор становится при этом темно-коричневым, и оба .
образовавшихся нежелательных изомера переходят в раствор. Реакциои-
ной смеси дают охладиться до комнатной температуры при постоянном ;
перемешивании и отфильтровывают твердый осадок от маточного рас-
твора. Продукт реакции еще раз расплавляют, нагревая с 500 мл чистой
воды, и оставляют охлаждаться, продолжая перемешивать. Полученные-
белые мелкие кристаллы it-ди нитро бензол а сушат при температуре, не .
превышающей 90°.
Выход чистого к-динитробепзола с т. пл, 90,7—91,4° (температура’
застывания 90.5—91°) составляет 140—150 а т. е. 83—90% от теории. ?
Примечания. Дннитробензол очень ядовит. Поэтому при плавлении его;
с водой и при очистке сосуды, в которых выполняется работа, следует тщательно?
закрывать. Особенно опасны спиртовые растворы, которые могут вызывать 1
цианоз и долго не заживающие экземы. Все три изомерных динитробензола заметно
растворяются в горячей воде. !
При дальнейшем нитровании нитробензола наряду с лг-ди Нцтро бензолом всегда'?
образуется некоторое количество (5—15%) о- и «-соединений, притом в увеличенном !
количестве с повышением температуры нитрования *. Чтобы в результате последую- Т
щего восстановления получить л*-фенилендиамин, устойчивый при хранении и дающий?
хорошие выходы красителей, необходимо обязательно освободить At-ди нитробензол от?
о- и «-изомеров. Готовый л<-фенилендиамин можно освободить от примеси изомеров)
лишь с трудом, однако достаточно даже весьма незначительной примеси о- и п-фени-J
лендиамина, чтобы значительно снизить стойкость продукта. Кроме того, Эти изомеры :
вследствие сильного восстанавливающего действия разрушают диазосоединения. Ввиду?
этого если в качестве составляющей для получения азокрасителей применяют загряз-,:
нениый Л4-фенилендиамин, то при сочетании наблюдается сильное вспенивание вслед-!
ствие выделения азота; полученный краситель загрязнен продуктами разложения?
диазосоединения, л выход его снижается (см. стр. 258 и 262).
Ai-Динитробензол применяется главным образом для синтеза лг-ннтроанилина ич
лг-фенилендиамина.
и-Ннтроанилин из и-динитро бензола
К 1 Ю г кристаллического сернистого натрия (Na2S 9112О)
прибавляют 80 мл воды и пропускают сероводород до полного?
насыщения. При этом образуется прозрачный раствор сульфгид-)
рата натрия (NaSH). Одновременно в двухлитровом стакдпе из?
железа или из стекла растворяют 4 г некаля БИкс (смачивателя НБ;
или другого аналогично действующего эмульгатора) и 10 г х л о р ис
сто го аммония в 420 мл горячей воды; к полученному раствору)
прибавляют при 90° 84 г (1/2 моля) чистого к-дннитробензола; при?
этом перемешивают с такой скоростью, чтобы образовывалась возможно?
более тонкая эмульсия (соблюдать осторожность, так как пары очень
ядовиты!). Затем температуру снижают до 85° н добавляют по каплям
в течение т/ч часа при непрерывном сильном перемешивании получен-
ный по описанной выше методике раствор сульфгидрата натрия, следя,
чтобы температура была в пределах от 80 до 85°. Поскольку при реак-
ции выделяется тепло, нагревание во время добавления раствора сле-
См, Wyler О,, Helv. Chim. Acla, 15, 23 (1932).
м-Нитроанилин из м-динитробензола
105
дует уменьшить или временно прекратить. После того как весь раствор
сульфгидрата прибавлен, перемешивание (без нагревания) продолжают
еще в течение 5 мин. После этого прибавлением льда температуру сни-
жают до 20°. перемешивают еще около 1 часа при комнатной темпера-
туре, отсасывают выпавшие желтые кристаллы л-нитро анилина и про-
мывают их холодной водой. Осадок не высушивая кипятят со смесью
250 мл воды и 80 мл концентрированной технической соляной
кислоты до тех пор, пока все количество ж-иитроанилина нс перей-
дет в раствор. Затем ему снова дают охладиться (чтобы выпал в осадок
не вступивший в реакцию м-ди нитро бензол, который, заметно растворим
в горячей воде), отфильтровывают от осадка, состоящего главным об-
разом из динитроазоксибензола, нагревают и к фильтрату в горячем
состоянии приливают аммиак до сильнощелочной реакции. Когда
реакционная смесь совсем остынет, отсасывают очищенный л-нитро-
ацилин, промывают, его водой и сушат. Выход 62 г, т. пл. 110—112°.
Полученный таким образом продукт достаточно чист для многих целей.
Химически чистый ди нитро а пилин получают перекристаллизацией при-
мерно из 4 л кипящей воды (при этом остается незначительный смоли-
стый остаток). Он представляет собой золотисто-желтые иглы, плавя-
шисся прн 114°.
Сырой продукт можно перекристаллизовать прямо из воды и без предваритель-
ного переосажденпя; однако в этом случае часть невосстановленного динитробензола
(если он имеется) выкристаллизовывается вместе с нитроанилином. *
Общие замечания о реакциях частичного
восстановления
Если из многих имеющихся в молекуле соединения щгтрогрупп нужно восстано-
вить только одну, то обычно применяющийся‘метод восстановления с помощью железа
(как в сильнокислой, так и в почти нейтральной среде) оказывается непригодным. То
же самое имеет место, когда в молекуле наряду с нитрогрущюй имеются другие легко
иог.станавливаемые группы, например азогруппа *. Во всех этих случаях при примене-
нии железа восстановлению подвергаются все способные восстанавливаться группы.
Для такого рода «частичных в ос ста но эле пи н» в технике обычно используют сернистый
натрий или другие сульфиды. Однако во многих случаях, чтобы избежать слишком
далеко,идущего восстановления, следует работать осторожно также и при применении
этих восстановителей: прибавлять их только постепенно, избегая избытка, и поддер-
живать температуру не выше той, которая обязательно требуется для проведения
реакции, Теммература, наиболее благоприятная для проведения реакции, бывает раз-
личной от Случая к случаю; она может изменяться в пределах от 0 до 100° и должна,
как и другие условия проведения реакции, устанавливаться на основании специальных
опытов для каждого отдельЕкй'о вещества. Восстановление проводят обычно в водном,
а иногда и в водно-спиртовом растворе.
•Если и соединении, которое, подвергается частичному восстановлению, обе нитро-
группы занимают 2- и Ч-цоложениЧПТо^отношению к алкильной, окси- и алкоксильной
группам или к аминогруппе, то восстанавливается, кщс правило, питрогруппа, находя-
щаяся в положении 2.
Сернистый натрий в процессе реакции переходит главным образом в тио-
сульфат натрия в соответствии с уравнением
4\ - - NO2 + GNaaS 4- 7Н3О ~^4\ — NHa [- GNaOH 3Na2S2O3.
Поэтому для восстановления каждой нитрогруппы расходуется .приблизительно
полтора моля сернистого натрия **. Образующийся при реакции едкий натр часто ока-
зывает очень вредное действие. В таких случаях лучше применять сульфгидрат натрия
* См. стр. 259. "
Восстановление -идет не всегда точно по приведенному выше уравнению, по-
этому оптимальные, количества сернистого натрия могут несколько отличаться от те-
оретически рассчитанных и их следует для каждого отдельного случая определять
экспериментально.
106
/. Промежуточные продукты
(NaSH), т. е. насыщенный сероводородом раствор сернистого натрия. В технике такие
растворы получают При поглощении сероводорода, выделяющегося из сернистых пла-
вов. Восстановление без образования едкой щелочи происходит по следующему
уравнению:
-1Х' — КО3 + GNaSH ф- Н2О = -1Х — ХН, + 3Na2S2O;!.
Если восстанавливаемое вещество содержит кислую группу, то последняя может
нейтрализовать щелочь (см., например, получение пикраминовой кислоты, стр. 138).
Можно также к раствору восстанавливаемого вещества заранее прибавить сульфат
магния, с тем чтобы образующийся NaOH тотчас превращался в плохо растворимый
осадок гидрата окиси магния; последний, вследствие очень незначительной раствори-
мости, может придать раствору лишь слабощелочную реакцию.
Некоторые нитросоединения следует восстанавливать в аммиачном растворе
точно рассчитанным количеством сероводорода. Часто, однако, недостаточно точное
определение веса сероводорода, и приходится прибегать к специальным приемам. Так,
например, динитрофенол только тогда удовлетворительно восстанавливается в нитро-
аминофснол, когда он применяется в виде мелкокристаллической натриевой соли, ко-
торая непосредственно получается при омылении динитрохлорбензола (см. сернистый
черный, стр. 301); восстановление проводится приблизительно при 60° в аммиачном
растворе точно рассчитанным количеством сероводорода. Полученный нитроамино-
фенол лучше всего очищать перекристаллизацией из кипящей воды.
При сочетании трудно растворимого диазосоединения с описанной на стр. 96
34-фен и ленд и аминсульфокислотой образуется дешевый хромирующийся коричневый
краситель для шерсти. Сочетание проводят при 28° и течение 2—3 дней в возможно
более концентрированных и полностью нейтральных растворах.
(кислотный ХрОМ
коричневый 2К)
-к-Феиилендиамии из -и-дииитробеизола
В редуктор (см. стр. 71 и рис. 17) помещают 1,5 л воды и 400 г тон-
ких железных стружек (см. стр. 72). Железо протравляют 20 мл
концентрированной соляной кислоты и смесь кипятят в течение
5 мин. Затем при непрерывном перемешивании прибавляют 168 г чи-
стого дниитр обензол а порциями ие больше чем по 2 г. Жидкость
сначала становится желтой, что указывает па образование .и-питроаии-
лина. Прибавление каждой • порции динитробензола сопровождается
настолько сильным вспениванием, что поверхность приходится обрыз-
гивать водой. Для правильного течения восстановления необходимо,
чтобы реакционная смесь все время кипела. Следующую порцию ди-
литро бе изол а прибавляют лишь после того, как проба на фильтроваль-
ной бумаге будет бесцветной. При слишком быстром прибавлении
жидкость становится коричневой, что связано с образованием
азоксисоедиисш|й. Об азовавшисся азоксисоединсния п епятствуют
м-Нитрохлорбензол из нитробензола
107
дальнейшему восстановлению, и реакцию восстановления следует пре-
кратить. Это явление одно из самых неприятных в технике восстанов-
ления. Оно может быть вызвано также плохим качеством железа,
поэтому всегда необходимо предварительно проверить реакционную
способность применяемого образца железа. Прн некотором навыке без
труда можно восстановить динитробензол в течение 40 мин. К концу
реакции образуется светло-коричневый, а иногда почти бесцветный
раствор, который быстро темнеет. Кипятят еще по крайней мере в те-
чение 5 мин. и прибавляют вместо испарившейся столько воды, чтобы
объем жидкости был равен приблизительно 2 л, что соответствует со-
держанию 45 а диамина в 1 л раствора.
К кипящему раствору очень осторожно прибавляют небольшими
порциями твердую кальцинированную соду (около 10 а) до отчетливой
щелочной реакции на лакмус и кипятят еще в течение 5 мин. для пол-
ного разложения растворимых железных соединений гидроксил а мнн а,
всегда образующихся в небольшом количество. После того как проба
раствора на фильтровальной бумаге не дает больше темного пятна
с раствором сернистого натрия (1:10), ее можно фильтровать. Пробу
всегда следует проводить и при работе с большими количествами *, так
как это предохраняет от многих неприятностей. Жидкость фильтруют
в предварительно нагретую колбу и прозрачный раствор подкисляют
соляной кислотой до слабокислой реакции на лакмус. Такой
раствор лг-фенилендиамииа вполне устойчив. Выход 100%-ного про-
дукта около 95 г. Аналитическое определение проводят с помощью
диазотированного анилина в очень разбавленном растворе при 0° в тех
же условиях, как в случае Аш-кислоты, только без прибавления соды
(см. аналитическую часть).
Этот технический раствор достаточно чист н его можно использо-
вать для многих целей, однако в большинстве случаев для получения
более высоких выходов диамин следует иметь в более чистом виде.
Для этого пеподкисленНый водный раствор упаривают сначала на го-
лом пламени, -а затем желательно в вакууме до тех пор, пока содержа-
ние основания не повысится до 40%.' После этого можно либо полностью
отогнать воду в вакууме и перегнать остаток, либо, что лучше, просто
выморозить диамин при 0°. Чтобы раствор закристаллизовался, в этом
случае необходимо внести для затравки кристаллик фенилендиамина.
Очищенный диамин кристаллизуется с 0,5 молями воды в красивые бе-
лые призмы и вполне устойчив.
Аналогично ла-фениленДнамину полурот также его гомолог
1,2,4-тол у ил ендиамин. В этих'чже условиях восстанавливают 2,4-диннтро-
хлорбензол, 2,4-дшштроа11Изол?'7мгитроанилин и другие нерастворимые
нитрососдинения.
jw-Нитрохлорбеизол из нитробензола
Хлорирование нитробензола в лабораторных условиях требует большой тщатель-
ности и удается лишь в том случае, если реакция проводится при полном отсутствии
Елаги. Даже следы воды препятствуют реакции хлорирования или чрезвычайно сильно
* Если реакция па железо после длительного кипячения не прекращается, то
Уследи осаждают небольшим количестиом сернистого аммония.
1U8
/. 11 ромежуточные продукты
ее замедляют. Поэтому предварительным условием успешного проведения реакци]
является тщательное высушивание как аппаратуры, так и исходных веществ. В каче
стве переносчика хлора лучше всего применять безводное хлорное железо. Продажно!
хлорное железо рекомендуется нагреть в круглодонной колбочке в вакууме, просасы
пая слабый ток высушенного хлора или хлористого водорода до начинающейся вод
гонки, и тотчас же перенести в склянку с хорошо притертой пробкой.
Исходный нитробензол может быть высушен следующим образом: в круглодоя
ной колбе в течение нескольких часов нагревают при 80—100’ нитробензол, просасы;
вая в это же время с помощью слабого вакуума через не слишком узкий капилля
сильный ток высушенного воздуха.
Для хлорирования 246 г (2 моля) нитробензола может быть нспользова!
аппаратура, указанная при описании методики получения хлорбензола (см. рис.
стр. 62). В этом случае можно работать без обратного холодильника, так как натр,
бензол кипит при значительно более высокой температуре, чем бензол; для начала реа:
нои колбу слегка нагревают на водяной бане, которую в случае необходимости исиол:
зуют и для охлаждения. Выделяющийся хлористый водород сначала пропускают чер<
колонку, наполненную хлористым кальцием, и только после этого поглощают, ка
описано в методике получения хлорбензола. В качестве переносчика достаточно испоя
зовать 5 г заранее приготовленного хлорного железа. Температура реакционнс
смеси должна быть 40—45°, так как при хлорировании нитробензола с повышение
температуры также увеличивается количество образующихся побочных продукте:
Интенсивность поглощения хлора нитробензолом значительно меньше, чем в случа
хлорирования бензола. Пойлощается только слабый ток хлора. Реакция продолжаете
при данной загрузке около 6 час. Течение реакции контролируется взвешивание!
колбы. Хлорирование ведется до достижения привеса 85 г. Ввиду значительно;
растворимости хлора и хлористого водорода в реакционной смеси этот привес соответ
ствуст приблизительно двум молям хлора, фактически вошедшим в реакцию.
После того как будет достигнут указанный привес, прекращают пропускать хло{
реакционную смесь оставляют стоять на некоторое время, затем жидкость декантирую
в делительную воронку и тщательно промывают соляной кислотой, раствором сод!
и водой. Если тотчас же не образуется белая кристаллическая масса, то смесь быстр'
переливают в стакан, где она через некоторое время частично застывает; фильтруют
охлаждают до 10°, центрифугируют и получают 95 г практически чистого л^-нитра
хлорбензола. Незастывшую часть соединяют с отцентрифугированной жидкости
и подвергают фракционированной перегонке в вакууме. Перегонку лучше всего вест!
на колонке высотой 80 с.и, снабженной дефлегматором (см. стр. 304).
Выделяют следующие фракции:
1. 85—106° (9 лмг) около 40 а, жидкая.
2. Ю6—108° (9 мм) » 130 г, застывает.
3. Выше 108°жидкий остаток.
Фракция 1 состоит цз нитробензола с примесью нитрохлорбензола.
Из фракции 2 после охлаждения и центрифугирования подучают около 1001
чистого лг-н и т р ох л о р б е н з о л а. Из жидкой части, полученной при центрифугиро
вании, повторной перегонкой можно выделить лишь следы нитрохлорбензола;эта част,
в основном состоит из Побочных продуктов реакции, растворимых Друг в друг
(3,6-дихлорнитробензол, п- и о-ннтрохлорбензолы и др.).
Фракция Зи остаток состоят также из этих побочных продуктов. Если остата
обогащен гексахлорбензолом, то последний при длительном стоянии частично выпадаеэ
Выход л(-нитрохлорбензола составляет около 60%, или, считая на во шедши]
в реакцию нитробензол, 75% от теории.
Чистый м-нитрохлорбензол кипит при 107° (9 хм); плавится при 44.5°.
сн3 1 С Ну 1 1 2-Хлор-4-диметиламииобеизальдегйд снз СН3 1 ! CH3
1 1 —> |/ХГ -SOyll _z^. /X SO3H -SO3H SO3Na
NOg no2 Y NHOH CH2O ЛС1 XaOH l"eSU4 * Il > C]I 1 NHa CHO 1
ZVCI -> -C! !/4|-C1 1 O~CI Q-ci
NO3 1 N‘H2 N(CH;i)a 1 N’ (СНВ)Й N (CH3)2
2-Хлор-4-диметиламинобензальдегид
109
а) я - То ли л гидр окси л а м и п сульфокислота
47,8 г (0,2 моли) 100 % -ного я-н и т р ото л уо л i,o - с у л ь ф о к и с-
л.о го ri атрия * и 8 г х л ор ис то г о аммония растворяют в 200 мл
горячей воды и точно нейтрализуют аммиаком. Раствор помещают
в железный редуктор и охлаждают до 25° при перемешивании. Вытес-
няют воздух углекислотой и прибавляют через широкое сопло в течение
3—4 мип. 40 г цинковой пыли. Цинковая пыль должна попадать
на реакционную массу в топко распыленном виде. Во время восста-
новления необходимо быстрое механическое перемешивание и наружное
охлаждение льдом. Реакция начинается внезапно, и температура быстро
повышается приблизительно следующим образом:
Время (мин.)........ 0 I 2 3 4 5 6 7 8 9.
Температура (°C) ... .25 26 38 46 40 32 28 22 18 1 5
Тотчас же отфильтровывают, отжимают и промывают осадок окиси цинка
четыре раза порциями воды по 20 мл. Фильтрат и промывные воды
переносят в мерный цилиндр; общий объем состав л яетм240 мл.
Из этого раствора берут 2 мл пробы для определения содержания
л-тол и л гидроксил амиисульфокислоты С помощью фол и и Г о в о й
жидкости.
Пробу разбавляют до 50 мл и смешивают с 30 мл фелинговой жид-
кости **. Нагревают на горелке Теклу с шумящим пламенем до кипения
и кипятят точно две минуты (по секундомеру). Затем охлаждают по-
гружением в холодную воду и фильтруют через тигель- со стеклянным
фильтрующим дном. Закись меди два раза промывают порциями воды
по 25 мл. Фильтрат выливают н колбу для отсасывания хорошо опола-
скивают. Содержимое тигля растворяют в избытке смеси серной кислоты
и сульфата железа (III)***. Закись меди окисляется при этом по урав-
нению
* Си2О + Нй5О4 Fea(SO4)s ->.2CiiSO4 + 2F'eSO4 Л- Н2О.
Отсасывают, промывают водой н титруют получающуюся соль же-
леза (II) 0,1-н. раствором перманганата калия.
. Фелингов а жидкость расходуется' не в стехиометрических количе-
ствах. Приводимая криваи (рис. 22) выражает зависимость между
количеством перманганата, пошедшим на титрование, и содержанием
производного гидроксила мнна\ Концентрация выход я-тол нл гид рокси л-
амиисульфокислоты вычисляете^ при помощи этой кривой.
Например, израсходовано 7,9')югО;1 и. раствора перманганата калия.
7,9 мл 0,1 и. раствора перманганата калия соответствуют 11,1 мл 0,1 н.
раствора гидроксиламиисульфокислоты.
'120.11,1
—10 qqq = 0,133 моля гидрокси^^минсульфокисдоты.
I
—— ----------------- I
* Получают из п-нитротолуола так жф как д-нитробензолсульфокислоту из
нитробензола, см. стр. 111. \
** Получают смешиванием равных частей «ади «б».
а. 69,278 г химически’ чистого кристаллического ’медного купороса растворяют
в воде и доводят объем до 1 л.
б. 346 г сегнетовой соли и 130 г едкого натра растворяют в воде и также до-
водят объем раствора до 1 л.
*** 50 г сульфата железа (111). и 200 г концентрированной серной кислоты рас-
творяют в воде и доводят объем раствора до 1 л.
j. промежуточные продукты
Выход составляет, таким образом, 66,5%. 0,125 моля л-толил
гидроксиламнпсульфокпслоты содержится в 225 мл полученного рас
твора.
Рис. 22, Кривая для определения количества п-толилгидро-
ксиламинсульфокьслотн.
б) 2-Хлор-4-диметиламинобеизальдегид
15,6 г м-х л о р д и м е т и л а и и л и н а * (0,1 моля) растворяю’
в 40 мл концентрированной соляной кислоты и после охлаждена
раствор смешивают с 7,7 мл 39%-него раствора формальдегиде
Смесь прибавляют при хорошем перемешивании к раствору 0,125 мол:
n-т о л и л г и д р о к с и л а м и п с у л ь ф о к н с л о т ы, т. е. к 225 М.
указанного выше раствора. Сразу же после этого прибавляют 70 м.
20%-ного раствора железного купороса. Реакционная масс:
разогревается до 40°, и через несколько минут начинают выделятьс:
* Приготовляют из .и-хлораиилииа по методике получения диметлланплииа, опи
санной на стр. 122, лг-Хлоранилин получают из .и-иитрохлорбензола (стр, 107) точн<
так же, как получают анилин из нитробензола (стр. 71); он очень ядовит.
М-Питробензолсульфокислота и метаниловая кислота из нитробензола
111
кристаллы. Раствор вскоре сгущается в вязкую кашицеобразную массу,
перемешивание которой облегчает ее фильтрование в дальнейшем. Бен-
зилиденовое производное отсасывают через 12 час. и промывают
20%-ным раствором поваренной соли. Маточный раствор подщелачи-
вают едким натром и перегонкой с водяным пйро.м регенерируют из него
3,6 с хлордимстиланилина. I
Бензилиденовое производное растворяют! для омыления в 300 мл
теплого 2%-ного раствора соды, отфильтровывают от остатков карбо-
ната железа и размешивают в течение часа на водяной баие с 5 мл кон-
центрированного раствора едкого натра (уд. вес 1,38). Раствор
становится мутным, и альдегид выделяется в виде маслянистых капель,
застывающих при охлаждении. Его растворяют в соляной кислоте
(1:1) и осаждают 2 н. раствором соды. Получают 13,2 г сухого аль-
дегида ст. пл, 81°. Выход составляет, таким образом, 72%, считая на за-
груженное основание, или 94% с учетом регенерированного основания.
Альдегид можно перекристаллизовать из лигроина. Его применяют, на-
пример, для получения красителя синего для шерсти 5Б (см. стр. 272).
г--"'”" \
м-Нитробензолсульфокислота и метаииловая кислота
из нитробензола
К трехкратному количеству олеума (25% SO3), помещенному в
чугунный котсл и нагретому до 70°, осторожно приливают 123 г (1 моль)
нитробензола. Смесь быстро нагревают до 100—110°; следует из-
бегать перегрева, так как в этом случа’е может произойти внезапное об-
угливание. После прибавления всего количества нитробензола смесь про-
должают нагревать прн ПО—115° до тех пбр, пока проба, вылитая в воду,
совершенно не перестанет пахнуть нитробензолом. Если через полчаса
после смешения реагентов сульфирование не закончено, то это указывает
на недостаток SO3. В этом случае по каплям прибавляют еще 50 а
олеума, и если необходимо, то через прлчаса снова прибавляют его
в таком же количество. Если же оАеум/действительно содержал 25% SO3,
то нет необходимости прибавлять его больше, чем трехкратное количество
но отношению к нитробензолу. Массе дают охладиться, и при хорошем
механическом перемешивании ее выливают на 500 г льда. Нитробензол-
сульфокислога растворяется, а небольшое количество сульфона остается
в осадке. \
Кислота может быть выделена из раствора различными способами,
например так, как это описано на -стр. 76, для бензол сульфокислоты.
Предпочитают, однако, высаливать сульфокислоту, так как ее натриевая
соль практически нерастворима в насыщенно^ растворе поваренной соли.
При непрерывном механическом. перемешивании всыпают небольшими
порциями 200 г поваренной соли. Натриевая соль цитробепзол-
сульфокислоты выделяется в виде густой кашицеобразной массы, и для
того чтобы реакционная масса снова стала подвижной, ее надо длитель-
ное время перемешивать. Примерно через 10 час. ее фильтруют на боль-
шом иутч-фнльтре через бумажный фильтр и сильно и долго отжимают
/. Промежуточные продукты
на винтовом прессе через хлопчатобумажную ткань. Натриевая соль мо-
жет применяться без дальнейшей очистки для технических целей, пере-
кристаллизацией из воды ее получают в чистом виде.
Восстановление проводят так же, как это было описано для получения
анилина (см. стр, 71), только железо не протравляют, так как количество свободной.,
минеральной кислоты, содержащейся в отжатом осадке, достаточно для начала реак-
Нин. В двухлитровый железный или медный редуктор помещают 250 г мелко измсль-,,
ченного серого чугуна и 1 л воды. Нагревают на голом пламени до кипения’
и вносят при интенсивном перемешивании и постоянном кипении маленькими порциями
в течение часа измельченный отжатый осадок натриевой соли нитробензосульфокис-л1
лоты. Время от времени прибавляют взамен испаряющейся воду так, чтобы объем.
жидкости все время был равен приблизительно литру. После прибавления всего коли-:
чества соли кипятят еще в течение 20 мин. и капельной пробой на фильтровальной:
бумаге проверяют, обесцветился ли раствор; он должен быть лишь свстло-коричне- ‘
ным, по ни в косм случае не темно-коричневым или темно-желтым. После обесцвечи-
вания раствора очень осторожно, чтобы не происходило сильного вспенивания, при-1
бавляют кальцинированную соду до сильнощелочпоп реакции на лакмус до тех пор,)
пока капельная проба раствора на фильтровальной бумаге не будет больше чернеть»
при прибавлении раствора сернистого натрия. Отсасывают на нутч-фнльтре железный,
шлам и хорошо его Промывают горячей водой. После упаривания фильтрата до
600 мл и поДкислепИЯ соляной кислотой до кислой реакции па конго выпадает
метандловая кислота в виде мелкокристаллического осадка. Многие заводы предпо-
читают прямо перерабатывать концентрированный раствор метаниловой кислоты, так,)
как она хорошо растворима и при ее выделении всегда .теряется 10—]5% вещества.
Эти потери компенсируются, однако, высокими выходами красителей. Выход кис-)
лоты определяют простым титрованием минеральнокислого раствора нитритом
натрия; он составляет около 90%. т. е. примерно 155 а ]00%-ной кислоты.
Примечания. Для осаждения л-нитробензолсульфокпслоты вместо поварен-
ной соли можно с успехом в атом случае применять также железный купорос. Же-
лезная соль л:-нитробензолсульфокислоты довольно плохо растворима в 20%-пой сер-
ной кислоте и легко может быть отфильтрована. В остальном восстановление проте-
кает точно так же, как описано выше. Если сульфировать бензол моцогидратом и
нитровать бенэолсульфокислоту при 100°, то, согласно, данным Обермиллера *, наряду':
с .м-нитросульфокислотой образуется значительное количество о-и п-нитросульфокислот, /
которые могут быть относительно легко отделены в виде магниевых солей; таким 1
образом можно получить цепную о-аминобснзолсульфокислоту. Получение ортанило- j
вон кислоты из о-н итр ох лорбензола подробно описано **. I
Следует отметить, что и другие л-нитросульфокислоты дают довольно трудно/
растворимые соли закиси железа. Например, 2-нитропафталин-4,8-дисульфокислоту"
можно получить этим методом в чистом виде*** ****. Этот метод разделения, впрочем, ,
давно был известен в технике. 2-Нафт[1лаМ[1п-4,8-дисульфокислота описана на стр. ]97.:
Аналогичные методы сульфирования. Точно так же сульфируют д-нитрохлорбен- <
зол, и-нитротолуол, о-нитрохлорбензол, хлорбензол и др. Этим методом часто не^
удается просульфировать ди нитросоединения. Динитрохлорбензол при обработке i
дымящей серной кислотой разлагается со взрывом; так же ведет себя динцтр0толуол.ч
Для получения, например, динитрохлорбецзолсульфокислоты исходят из /г-нитрохлорД
бензола. Его сульфируют по методике, описанной выше, и сульфокислоту затем деи-т
ствием нитрующей смеси (50% ПаЗСД + бО^/о HNOs) при низкой температуре превра--*
щают в д ин нт рохл о р бенэолсульфокислоту. Заменой хлора на оксигруипу и частичным-;
восстановлением эту крслоту Превращают далее в 4-иитро-2-аминофенол-6-сульфокис-\
лоту (нитрокнелота Ш). которая применяется для получения хромирующихся кра-’
сигелей. . J
Динитронафталцны при действии олеума превращаются в нафтазарин **м, ;
Технические примечания. При сульфировании больших количеств реакД
цию проводят в котлах с паровой рубашкой, в которую по желанию можно пропу-.
скать пар или холодную воду. Реакционная смесь часто очень сильно разогревается,,
и поэтому приходится работать с большой осторожностью, так как в противном случае’’
может произойти опасный перегрев вли даже взрыв. Высаливание производят в дере-
вянных чанах, осадок отжимают сначала на фильтр прессах, а затем на гидравличе-.;
ских прессах с салфетками из волосяной ткани при 250 ат, Восстановление, выпарива-1
ние и выделение проводят так, как было уже описано.
* См. герм, пат. 281176 (Frill., XII, 125).
** Helv. chim. Acta, 12, 663.
*** франц. пат 7346]6 (I. G).
**** Stockar W„ Диссертация, Zurich, 1942.
2,2'-Бензидиидисульфокислота из нитробензола
NHOH
Гидроксиламии- Аяойензол-
бензолсульфо- ' дисульфо-
кислота кислота
NH---NH
Ги др а з р б е к а ол ди сул ьф о -
/ кислота
Азоксибензол-
днсульфонислота
2,2Г-Беизидинли-
сульфокислотя
Получение нитробензолсульфокислоты подробно изложено прн опи-
сании методики получения метаниловой кислоты. Метод восстановления
отличается от применяемого в подобных реакциях только тем, что в дан-
пом случае работают намеренно с разбавленным водным раствором и вос-
становление проводят в три стадии. Получение беизидиидисульфокислоты
удается осуществить при применении минимальных количеств едкого
натра и цинковой пыли.
При применении не совсем чистой натриевой соли отжатый осадок
нитробеизолсульфокислого натрия, соответствующий 100 г нитробен-
з о л а, растворяют в воде и прибавляют.около 30 г соды так, чтобы рас-
твор был строго нейтральным иа лакмус. Доводят объем до 1,5 л, охла-
ждают до 10°, затем прибавляют Юг рористого аммония и
энергично перемешивают жидкость с помощью пропеллерной мешалки.
Затем в течение двух минут Прибавляют порциями по чайной ложке
120 г цйнковой пыли. Прибавлением толченого льда поддержи-
вают температуру около 20”» и перемешивают в течение 20 мин. Затем
прибавляют в один прием 120 г 30% -ного-раствора едкого натра и
нагревают без перемешивания дК. 70°. Бесцветный раствор тотчас же
окрашивается в оранжево-желтый ^вст; вследствие образования азо- и
азоксибепзолдисульфокислот. Оставляют стоять не мепее чем на 3 часа;
лучше всего, однако, оставить па ночь.
На Другой день раствор осторожно нейтрализуют/прибавлением по
каплям примерно 90 г копцептрироващюй-щр_л_.я,.иб й кислоты до
исчезновения реакции па тиазоловую бумагу. Нагревают при перемеши-
вании до 80° и прибавляют еще 40 г цинковой пыли. Если через
5 мин. раствор не обесцветится, то медленно по цаплям при 75—80° при-
бавляют еще соляную кислоту. Когда раствор станет нейтральным, то
стП) обесцвечивание, т. с, переход от грязного коричневого цвета к чи-
стому светло-серому, происходит менее чем за 5 сек. Раствор содержит
теперь гидр азосульфокислоту; его объем составляет около 1,8 л. Тотчас
Же отфильтровывают, чтобы не произошло дальнейшее восстановление
в мстаниловую кислоту, и цинковую пыль хорошо промывают; охлаж-
дают и при 20° прибавляют 120 г концентрированной соляной
кислоты. Через несколько минут образуется блестящий бесцветный
осадок твердых кристаллов 2,2'-бензидиндисульфокислоты, а раствор ста-
новится желтым, так как происходит частичное диспропорционирование на
азобензолсульфокислоту и мстаниловую кислоту. Для обесцвечивания рас-
твора прибавляют несколько граммов цинковой пыли. Хотя бензидинди-,
сульфокислота чрезвычайно трудно растворяется в воде (1 л воды рас-’
творяет менее одного грамма кислоты), она очень медленно выделяется из
раствора. Поэтому ее необходимо оставить стоять на 2 дня для того,
чтобы кислоту можно было отфильтровать и промыть холодной водой^
Выход около 65 г.
Технические примечания. Кристаллизация 2,2^бецзндиндисульфокис'
лоты в больших количествах (из объемов 4000—5000 л) продолжается не менее треа
суток. Для более быстрого охлаждения в деревянный чан помещают свинцовый змсевид
через который пропускают холодную роду. Вследствие трудной растворимости сульфсу
кислоты ее нельзя непосредственно диазотировать. Для этого кислоту растворяю!
в необходимом количестве соды и воды, смешивают нейтральный раствор с нитритом
натрия и вливают смесь тонкой струей в соляную или серную кислоту.
Как все производные бензидина, замешенные в о-положенци по отношению к ди
фенильной связи, бензидин -2,2'-дисульфокислота дает не прямые к р а си т ел 1
для хлопка, а кислотные красители для шерсти, исключительно прочные к валю
и к стирке (например, кислотный антраценовый красный Г, см. стр- 263). Продукт со.
четапия диазотированной кислоты с двумя молями салициловой кислоты применяете*
как краситель для печатания ца хлопчатобумажных тканях (хромоцитронин, протрав'
ной чисто желтый, см. стр, 264)',
Бензидин из нитробензола
Zti-NaOU
"I- я „О
NH---NH
КН3( НС1)
Промежуточ-
ные про-
дукты — азо-
кенбгнзол и
азобензол
Бензидин,
около 850/0
125 г нитробензола смешивают с 250 мл о-д и х л о р б е и?
зола (можно также применять сольвентнафту с т. кип. 170°) и поме;
гцают в стеклянную колбу с эффективной мешалкой и обратным холо
дильником.
К смеси прибавляют попеременно цинковую пыль, размешай
ную с о-д и хлорбензолом до консистенции подвижной кашицеоб
разной массы, и 50%-ный раствор едкого натра. Смесь нитробей
зола и о-дпхлорбензола нагревают до 115—125° и прибавляют сначал.
5 мл 50%-ного раствора едкого натра, а затем суспензию, содержащут
10 г цинковой пыли. Восстановление должно начаться быстро, в протш
иом случае смесь нагревают до 130°. После того как начнется реакция
попеременно прибавляют указанные реагенты так, чтобы температура вс
время была 115—120°. Необходимо следить за тем, чтобы на дне колб1
не скапливалась не вступившая в реакцию цинковая пыль, так как эт
может вызвать вдруг бурную реакцию. Прибавление раствора едкоп
натра и цинковой' пыли следует проводить таким образом, чтобы ео
закончить за 3 часа. Всего прибавляют 250 г 50 %-ного раствора щелоч;
и 260 г цинковой, пыли. Раствор сначала окрашен в красный -цвет, а за
Бензидин из нитробензола 115
тем постепенно становится бесцветным или вследствие наличия цинката
натрия — белым.
Размешивают до обесцвечивания раствора, что может продолжаться
в зависимости от условий работы и качества цинковой ныли 4—10. час.
Если долгое время не происходит обесцвечивания, то осторожно прибав-
ляют несколько миллилитров воды, и если восстановление в гидразобен-
зол опять-таки пройдет не полностью, то прибавляют еще немного цин-
ковой пыли и раствора едкого натра.
После окончания восстановления прибавляют небольшими порциями
воду до тех пор, пока цинковый шлам не отделится резко от раствора
гидразобензола, и сливают о-дихлорбстрольный раствор с осадка. Цин-
ковый шлам дважды промываютзд^болыпими порциями о-дихлорбен-
зола,
Перегруппировка гидразобензола в бензидин. Смешивают о-дихлор-
бензольный раствор г и д р а з о б/е и з о л а с равным количеством тонко
измельченного льда и прибавляют ЗОО-дгллсонцентрированной соля-
ной кислоты. Солянокислый бензидин (вмеЬте с образовавшимся
дифенилином) переходит в водный слой. Через 3 часа нагревают до 80°,
прибавляют еще 500 мл горячей воды и отделяют о-дихлорбе.нзол.
о-Дихлорбензол дважды экстрагируют небольшим количеством воды дня
того, чтобы быть уверенным в полном извлечении бензидина. Если рас-
твор мутный, то его фильтруют горячим.
К раствору солянокислого бензидина прн энергичном- перемешива-
нии прибавляют раствор 100 г безводного сульфата натрия (или
соответствующее количество кристаллической соли), Нерастворимый
сернокислый бензидин тотчас же выпадает, а побочные продукты
остаются в растворе.
Через час отфильтровывают сернокислый бензидин и хорошо промы-
вают его водой [примечание; если раствор при перегруппировке
окрашивается в красный цвет (образование азобензола), то для полного
восстановления прибавляют несколько граммов железных стружек].
Получение свободного основания. Еще влажный сернокислый бензи-
дин смешивают с пятикратным количеством горячей воды н прибавляют
твердую кальцинированную соду (около 45 г) до отчетливой и неисче-
зающей щелочной рейкцнн на лакмус. Пойле Охлаждения свободное осно-
вание отфильтровывают н промывают небольшим количеством воды.
Основание получают в виде серых зерен, которые сушат при 100°. Если
хотят получить совершенно чистое основание, то его перегоняют в хоро-
шем вакууме нз саблевидной колбь/ Т.,пл. 128°, Вы^од около 75%
от теории, в остатке содержался дифенили-н н побочные^продукты.
Технические примечания. Производство бензвдтша является одним из
важнейших процессов и химии красителей, т^кцеак-ИЗ”этого основания получают мно-
гие ценные, хотя часто и непрочные прямые красители. Аналогично можно получать
из о- н .н-нитротолуолов оба толидина (орто- и .мега). Красители, получаемые из
я-толндяна, плохо выбираются хлопчатобумажной тканью, но являются хо-
рошими красителями для шерсти. Из о-нитроанизола точно так же получают о-ди-
анизидищ
Отходы цинковой пыли и окиси цинка, ^сли они сухие, не следует просто вы-
брасывать в мусорный ящик, так как они способны к самовозгоранию.
Помимо описанного широко применяемого метода, С)<1ествует также другой ме-
тод восстановления железными стружками; однако он трудно выполним. Разработан
Также электролитический метод, который состоит в том, что восстанавливают на ка-
тоде ц гидразобензол суспензию нитробензола в растворе едкого натра. Насколько
известно, этот метод применяется только фирмой С1ВА (Общество химической про-
мышленности в БазелсЦ Этот метод не связан с применением цинковой пыли и дает
хорошие выходы. Лучше всего восстановление проходит в том случае, если нитробен-
зол и раствор едкого натра имеют одинаковый удельный вес.
Перегнанный бензидин дает наилучшне результаты, но он немного intinwji
116
I. Промежуточные продукты
реГйанного. При перегонке теряют лишь очень немного вещества; остаток представляет
собой загрязнение. Все же в настоящее время в технике по возможности из-
бегают получать свободное основание бензидина, так как оно является канцеро-
генным веществом, н применяют непосредственно невысушенный сернокислый бен-
зидин,
В заводских условиях можно восстанавливать сразу 200—500 кг нитроПроизвод-
ного, что требует аппаратуры больших размеров. При проведении восстановления
в больших масштабах берут меньше растворителя, так как в этих условиях можно
осуществить более эффективное перемешивание, чем в лабораторных условиях. Вос-
становление продолжается до 24 час,
В лабораторных условиях восстановление по описанной методике удается
успешно провести только при хорошем перемешивании реакционной смеси. При работе
с небольшими количествами, когда невозможно Энергичное перемешивание всей массы,,
о-дихлорбензол лучше заменить растворителем, смешивающимся с водой, например
спиртом,
8. Производные анилина
Сульфаниловая кислота из анилина (метод запекания)
(У
В железной чашке смешивают 1 моль (105 г) серной кислоты;
д. вес
1,84)
с 1 молем
(93 г)
анилина.
Анилин помещают в
чашку:
и к нему при хорошем перемешивании тонкой струей приливают сернук»1
кислоту. В производственных условиях реакцию проводят в железном.
котле и перемешивают от руки железной кочергой. Образовавшуюся гуе
стую массу еще в горячем состоянии тотчас же намазывают на железный
лист (15 X 15 см) с загнутыми краями (2 см). Слой должен быть толщи-i
пой приблизительно 1 см, при работе с большими количествами — 8 см
Массу помещают в сушильный шкаф (причем расстояние листа от обо
греваемой поверхности должно быть не меиее 5 сл1) и нагревают в тече
ние 8 час. при температуре воздуха 190° при помощи бунзеновской го
релки с насадкой в виде грибка. Лист вынимают из шкафа и выколачи
в а ют из пего готовую сульфаниловую кислоту. После этого получаете
примерно 90%-ный продукт светло-серого цвета; в нем еще содержите
около 3% исходного анилина и немного угля. Эту неочищенную сульфа
ниловую кислоту можно непосредственно применять для многих целет
для чего се растворяют в таком количестве соды, чтобы раствор нме
сильнощелочную реакцию па лакмус. В описываемом случае требуете
около 60 а с о,д ы и 500 мл воды. Затем раствор кипятят, возмещая ш
паряющуюся воду, до тех пор, пока легко летучий анилин не будет увд
чей водяным паром. После этого фильтруют через ватный фильтр и пол
чают раствор сульфаниловой кислоты, который в большинстве случае
удовлетворяет требованиям техники. Для выделения чистой сульфанил!
вой кислоты раствор подкисляют серной кислотой до сидыюкислой peai
ции на конго. -Выпадает чистая сульфаниловая кислота, которая, о ди а к
не может еще применяться для аналитических целей (см. аиадитическу:
часть),
Выход сырого вещества около 175 г и около 140 г 1ререосажденцог
ггпппскта.
J "( /f - tpi -<J-гПС-'j ИЛ-ц/’Ла/УиЛЛЛМ/Ч U»J иул<м|/илилипиа AWL.' UIUL
Общие сведения о методе запекания
Описанный выше так называемый метод запекания может быть применен
с целью получения соответствующих сульфокислот * не только из анилина, но также
из ряда других ароматических оснований (например, толуидинов, ксилидинов, хлпр-
анплпнов, а-нафтиламипа, бензидина, аминоантрахинонов, дегидротиотолуидина
и т. д.), В отличие от обычного сульфирования в присутствии избытка серной кис-
лоты или олеума при сульфировании методом запекания применяют серную кислоту
в количестве, точно соответствующем теоретическому, и избегают, таким образом, об-
разования продуктов дальнейшего сульфирования. Кроме того, как правило, при запе-
кании не образуются изомерные соединения. Напротив, сульфогруппа вступает почти
всегда только в пара-положение или, если пора-положение занято, в орто-положение
по отношению к аминогруппе, и никогда не происходит замещения в тиета-положение
пли в другое кольцо. Обычно это объясняют тем, что при нагревании кислого сульфата
с отщеплением воды сначала образуется сульфаминовая кислота, которая затем пере-
группировывается в амипосульфокислоту. Длим не объясняется, однако, почему можно
сульфировать методом запекания -'та^йе третичные основания, такие, как, на-
пример, диметилапилин, из которых невозможно образование сульфаминовой
кислоты.
Для гладкого течения реакции очень важноу-чтобы кислый сульфат был совер-
шенно однороден, т. е. чтобы в отдельных частях не было мало серной кислоты,
а в Других слишком много, Па практике этого не всегда легко достигнуть, так как
при смешении основания с серной кислотой легко образуются комки, которые препят-
ствуют равномерному перемешиванию. Поэтому часто Применяют водную серную кис-
лоту, а также предварительно разжижают растворителем твердое основание (см.
получение нафтионовой кислоты, стр. J6I); в этих случаях, однако, необходимо
очень основательно опять удалить воду при температуре ниже температуры реакции.
Для более равномерного высушивания реакционной массы ее распределяют тонким
слоем.
Наиболее благоприятная температура реакции находится в области 170—220°;
в разных случаях она может быть различной и устанавливается для каждого осно-
вания опытным путем.
Целесообразно проводить реакцию в вакууме: в этих условиях уменьшается воз-
можность обугливания и сульфирование протекает не только более гладко, но и ско-
рее. Поэтому в настоящее время в технике применяют для запекания исключительно
вакуумные печи, которые нагревают либо непосредственно на огне, либо, что лучше,
обогревают перегретым паром. Желаемой температуры можно достигнуть также при
помощи электрообогрева, который легче регулировать, при этом оказываются ненуж-
ными паровые плиты, выдерживающие высокое давление. Применяемый в лаборато-
рии аппарат для запекания под вакуумом изображен на стр. 162 (см. рис. 30),
/
1 ‘(«‘Сульфафен илХ‘-^"м^тил‘^‘пиРазолс,н
< из Сульфаниловой, кислоты
- а) Феи и лгидразип-п -сульфокислота
NH—NHSO8Na
SO3Na
вся
NH—N‘H3
^О8Н
N
fil
NH2 N+ N—N— SO3Na
J ? I
। Y UNOa Ma2SO, /\ SO2
I ! — -> । | -------► [ |
r - । Y
SO..H SO7 SO,,Na I
Техническую сульфанилову кХк и с л о т у в количестве, соответ-
ствующем 104 а (0,6 моля) 100 %-иойд, растворяют в 400 мл горячей
.воды и 33 г кальцинированной соды, фальцуют, охлаждают и прн
перемешивании медленно прибавляют 70 г концентрированной серной
кислоты. Охлаждают льдом и прибавляют по каплям при энергичном
перемешивании в течение 1/ч часа раствор 42 г нитрита натрия в
* Huber, Диссертация^ Zurich, 1931.
100 мл воды; при этом температура не должна подниматься выше 12°.
Перемешивают еще в течение */< часа и проверяют, имеет ли раствор
сильнокислую реакцию иа конго и вызывает ли он лишь слабое посине-
ние йодокрахмальной бумажки. В случае отрицательной реакции на йодо-
крахмальиую бумажку следует прибавить еще несколько капель раствора
нитрита. Выделившееся кристаллическое диазосоединение основательно
отсасывают на путч-фильтре и промывают для удаления избытка серной
кислоты небольшим количеством охлажденной воды.
Влажное* диазосоедипение медленно' вносят при постоянном пере-
мешивании в охлаждаемый снаружи льдом раствор 340 г кристалличе-
ского сульфита натрия (Na2SO3 - 7Н2О) в 500 мл воды так,
чтобы температура раствора все время была ниже 5°. Раствор становится
оранжевым, по ои должен оставаться прозрачным и давать лишь слабо-
щелочную реакцию на лакмус. Перемешивают еще в течение часа при
охлаждении льдом, затем нагревают до кипения и при постоянном кипе-
нии и перемешивании в течение часа прибавляют 400 мл концентри-
рованной соляной кислоты (уд, вес 1,17). Раствор светлеет и под
конец становится светло-желтым. Для полного обесцвечивания при-
бавляют еще немного цинковой пыли. Ф енил-гидр азинсульфо -
кислота в значительном количестве выделяется уже из горячего раствора
в виде белых блестящих чешуек. Оставляют охлаждаться на ночь,
отсасывают, промывают холодной водой и сушат в паровом сушильном
шкафу.
Выход 106 г (около 94% от теории)'.
Маточный раствор упаривают наполовину и получают еще несколько
граммов фенил гидразинсульфокислоты.
б) л-Сульфофени л - 3- мети л - 5- пиразол он.
СНг СООС2Н5
| 5 NH
сн=~со +нХ
сн—СОЧ / \
Г, 5 2 >-ЗОйН
СН—C==N ---------7
+Н2О-гС2Н5ОН
Готовят суспензию ]8,8 г (0,1 моля) ф е и и л г и д р а з и н с у л ь ф о к и с л о т ы ,
в 50 мл воды, прибавляют 6 г кальцинированной соды и нейтрализуют возможный <
избыток соды разбавленной соляной кислотой до нейтральной реакции на лакмус.
Затем медленно, по каплям, при перемешивании прибавляют 13 а ацетоуксусного :
эфира, нагревают до ]00° и перемешивают в течение часа при этой температуре.’:
Раствору при перемешивании дают охладиться, подкисляют 18 мл концентрированной
соляной кислоты, отсасывают выпавшую пцразолонсульфокислоту, промывают’
ее холодной водой и сушат. Получают 24 г кислоты в виде слегка желтого порошка,,.,
выход около 94% от теории.
Технические примечания. Фенилметилпиразолонсульфокислота Приме-,
няетец, например, для получения светопрочного желтого Г [кислотного желтого свето-
прочного (см. стр. 238)]. Сам фенилметилпиразолон (из фепилгидразина и ацетоуксус-
ного эфира), так же как и его многочисленные производные, является цепной азо-:
составляющей при получении главным образом желтых азокрасителей, отличающихся
в большинстве слуАаев.очснь хорошей прочностью.
* Сухая дназосульфаниловая кислота может сильно взрывать.
п-АминосщетаниАид из анилина
119
Фенилметилпиразолон является, кроме того, чрезвычайно важным исходным ве-
ществом при производстве фармацевтических препаратов (антипирин, пирамидон и др.).
п-Аминоацетанилид из анилина
/ ^-NIL —> / \_\НСОСНУ —> NHCOCH3 —>
—> H3N—>-NHCOCH3
а) Ацетанилид из анилина .
< /-КНа -}- СНдСООН NHCQCH3 + Н3О
Установка; полулитровая кругл од он пая колба, помещенная в масляную баню
и соединенная через хорошо действующую' фрдкщщнировочную колонку с нисходящим
холодильником. Целесообразно применять колонку>, описанную на стр. 304, заполнен-
ную стеклянными кольцами с высотой слоя не мейее 20 с,м; нижняя часть колонки
изолирована, а верхняя — снабжена дефлегматором, который может охлаждаться про-
сасыванием или продуванием тока воздуха.
В реакционной колбе смешивают 186 г (2 моля) анилина н 180 г
(3 моля) ледяной уксусной кислоты, вносят кипятильные ка-
мешки — смесь закипает с трудом — и нагревают до слабого кипения на
масляной бане (температура масляной бани 150—160°). Как только ко-
лонка прогреется и пары достигнут дефлегматора, через него начинают
пропускать довольно' быстрый ток воздуха, чтобы большая часть паров
конденсировалась и снова стекала в колонку; при этом происходит лишь
совсем медленная перегонка прн температуре, самое большее и а 2° пре-
вышающей температуру кипения воды. В этих условиях перегонка почти
полностью прекращается примерно через час. Затем медленно в течение
часа повышают температуру масляной баии до 190° и нагревают прн
'этой температуре до тех пор, пока практически ие прекратится перегонка
при температуре не выше 102°. При этом отгоняется около 45—50 г ди-
стиллята, состоящего из 30—40%-иой уксусной кислоты. В колбу прибав-
ляют еще 60 г (1 моль) ледяной уксусной кислоты и снова
нагревают масляную бапю до Г^Р—190°, медленно отгоняя в течение часа
дистиллят с т. кип- 100—102°, После этого ацетилирование в основном за-
копчено. Для того чтобы оно прошло до конца, медленно повышают тем-
пературу масляной бани ДО' 220°; при этом температура отгоняющихся па-
ров под конец возрастает, до 110°. Нагревание продолжают до тех пор,
пока в пробе реакционной шаосы _нельзя будет обнаружить сле-
дов анилина. Для испытания каплю реакционной смеси при помощи
стеклянной палочки помещают в пробирку, смешивают с кусочком
льда н несколькими каплями разбавленной соляной кислоты и при-
бавляют несколько капель разбавленного раствора нитрита. Если
ешс имеется свободный анилин, то образуется диазобепзол, который прн
выливании в содовый раствор Р-соли дает интенсивное, оранжево-крас-
ное окрашивание. Посл0 того как эта проба перестает давать окрашива-
,иас, масляную баню охлаждают примерно до 180°, удаляют фракциони-
ровочную колонку и остйдок уксусной кислоты пряностью отгоняют в ва-
кууме па масляной бане, нагретой до 180°; целесообразно при этом проса-
сывать через капилляр слабый ток воздуха. После перегонки еще горячий
остаток выливают в фарфоровую чашку или на медный лист, где он
застывает при охлаждении в твердую кристаллическую массу, бесцвет-
izu 1. Промежуточные продукты
ную, — если исходный анилин также был бесцветным,—с т. пл. 114—
115°. Выход не менее 265 г (98% от теории).
Для нитрования можно прямо применять неочищенный продукт. Пере-
кристаллизацией из кипящей воды получают совершенно бесцветные бле-
стоящие, без запаха кристаллы ацетанилида. Такой ацетанилид может
быть использован для фармацевтических целей, где требуются особенно
чистые препараты.
б) «-Нитроацетанилид из ацетанилида
/ NHCOCH3 4- HNO;i — > O3N - / NH—СОСН3 Д- Н3О
+ nhcochA
J
135 a (1 моль) сухого тонко растертого ацетанилида вносят
при перемешивании в 540 г концентрированной серной кислоты
(уд. вес 1,84), находящейся в чугунном стакане с хорошо закрывающейся’
крышкой, термометром и капельной воронкой (см. рис. 19) либо в трех-
горлой колбе. Температура не должна повышаться выше 25°, так как
иначе может произойти омыление. Ацетанилид полностью растворяется
через ]-—2 часа. После этого из капельной воронки прибавляют в тече-
ние часа при энергичном перемешивании и очень хорошем охлаждении
смесью льда с солью * 105 а 62 % -ной азотной кислоты (уд. вес
1,38), смешанной со 105 г серной кислоты (уд. вес 1,84). При
нитровании температура не должна превышать 2—3°; целесообразно под-
держивать более низкую температуру (около —5°), так как чем ниже"
температура, тем меньше образуется о-питрососдинения. При правиль-
ном проведении нитрования обходятся, как это часто бывает в производ-
ственных условиях, теоретическим количеством азотной кислоты. Н е-
большой избыток кислоты, однако, не вредит, так как вторая нитро-
группа относительно трудно вступает в молекулу. После прибавления
всей нитрующей смеси раствор перемешивают еще 3 часа прн 0° и опре-..
деляют при помощи нитрометра (см. стр. 70), вступило ли в реакцию
все количество азотной кислоты, за исключением взятого небольшого из^-
бытка. Далее проверяют, не пахнет ли анилином вылитая в воду пробам
смеси после кипячения с едким натром. -
После этого реакционную жидкость выливают при хорошем перемер
шиванин в смесь 350 г в-оды и 350 г льда. Тотчас же выпадает^
нитроацетанилид, который можно отфильтровать уже через час, не опа-;;
саясь потерь. Осадок основательно промывают водой, затем взмучивают]
в 700 мл воды, прибавляют соды до отчетливой щелочной реакции-;
па лакмус и кипятят.
При этой обработке омыляется только о-иитр о ацет анилид. Фильтруют)
при 50° и хорошо промывают водой. ' j
Выход обработанного таким образом продукта составляет околей
90% от теории. 1
Ацетильное производное всегда омыляют раствором едкого натра^
Нитроацетацил ид, полученный в виде отжатой влажной лепешки, взбал-)
* Для получения хорошей охладительной смеси необходимо отвешивать со-;
ставные части смеси (3 части измельченного льда и I часть поваренной соли) И;
хорошо их перемешивать. ;
п-Аминоацетанилид из анилина
12
тывают с равным по весу количеством воды и образовавшуюся суспен-
зию кипятят с 200 г 35%-ного раствора едкого натра. Реакция
должна быть все время отчетливо щелочной. Примерно через 3 часа от-
бирают пробу; если она полностью растворяется в 15 % -ной соляной кис-
лоте с образованием прозрачного раствора, то омыление закопчено.
Раствору дают охладиться до 40° и отфильтровывают. Продукт тщатель-
но промывают хм одной водой и получают химически чистым.
Из 93 г анилина получают около 100 г п-нитроанилина.
в) л-А м и н о а ц е т а н и л и д из «-н н т р о а ц е т а н и л и д а
Эту азосоставляющую получают восстановлением нитроацетанилида
в нейтральной среде почти так же, как было уже несколько раз описано.
В железном стакане с пропеллерной мешалкой (см. рис. 17, стр. 88)
сильно кипятят в течение небкольких минут смесь 250 г измельченных
чугунных стружек / 15 мл 40 %-ной уксусной кислоты и
500 мл воды. Затем при'1 постоянном перемешивании и кипячении при-
бавляют небольшими порциями влажный ннтроацетаннлид, по-
лученный нз 1 моля ацетанилида, с такой скоростью, чтобы проба рас-
твора на фильтровальной бумаге все время оставалась бесцветной. После
прибавления всего количества нитроацетанилида кипятят еще 10 мин.,
все время добавляя воду взамен испарившейся. Затем прн 70° очень
осторожно прибавляют соду до слабощелочной реакции. За 20 мин.
можно легко восстановить все количество нитроацетапилида, полученное
из 93 г аннлнна. Если при нейтрализации раствор кипятить илн добавить
слишком много соды, то аминоацетанилид легко омылится. При 70°, од-
нако, не удается полностью осадить железо, перешедшее в раствор, а это
совершенно необходимо. Поэтому остаток металла осаждают прибавле-
нием минимального количества сернистого аммония до тех пор,
пока капля раствора на фильтровальной бумаге прн прибавлении серни-
стой щелочи пе будет оставаться бесцветной. Лишь после этого можно
фильтровать раствор.
Раствор, отделенный от железа и окиси железа, выпаривают на го-
лом огне до объема 400 мл. При охлаждении выделяется амнноацетани-
лид в виде красивых длинных игл. /В ыхо д из 93 г анилина составляет
около 75 г чистого основания. Маточный раствор, который всегда содер-
жит около 15% загрязненного* продукта, после однодневного стояния
упаривают и снова оставляют кристаллизоваться.
Полученный таким образом продукт является для технических целей
достаточно чистым. Для ркончательной очистки его можно перекристал-
лизовать еще раз из шестикратного количества воды, причем полезно
прибавлять небольшое количество животного угля. В производственных
условиях при упаривании раствора в вакууме и без перекристаллизации
получается чистый аминоацетаннлид.
При омылеини амино ацетанилида, которое проводят точно так же,
как в случае иитроанилина, получается «-фенилендиамии, имеющий так-
же широкое примененне/Он легко окисляется, н омыление поэтому про-
водят либо в отсутствий воздуха, либо при кипячении с разбавленной
серной кислотой; п-фенйдендиамнн "получают также восстановлением
л-нитроанилина и примеиягбт-главным образом для получения коричне-
вых окрасок окислением на волокне. •
Технические примечания. В технике для получения п-нитроанилина
имеете ацетанилида применяют более дешевый форманилид, который нитруют при
очень низкой температуре (—20°), чтобы по возможности избежать образования
о-изомера.'Получающийся в качестве побочного продукта о-нитроанилин, естественно,
fJMjnvjruy J UJJUlSyKl Ot,
также используют; он находит ограниченное применение в производстве азокрасителей,
главным образом для получения пигмента (ганза желтый 5Г, пигмент желтый свето-
прочный 23, пигмент оранжевый прочный, литоль прочно-желтый Р), Омыление ннтро-
форманилида проводят в присутствии рассчитанного количества едкого натра; из обра-
зующегося при этом муравьинокислого натрия регенерируют муравьиную кислоту, ?
Этот способ раньше был единственным для получения л-нитроанилина. В настоящее
время он почти полностью вытеснен методом получения из п- нитрохлор бензола и
аммиака (см, стр. 86), Выгодность того нли другого из указанных способов опреде- ;
ляется соотношением цен,
ф
Диметилаиилии (диэтил- и этилбензилаиилии) *
Для получения диметил анилина применяют железный автоклав на з
60 ат рабочего давления с чугунным вкладышем, масляной баней, мано- 1
метром и т, д. Применяемый для алкилирования метиловый спирт не J
должен содержать следов ацетона или этилового спирта, так как эти за- |
грязнеиия вызывают несоразмерное повышение давления. Чистоту спирта j
надо поэтому проверить с помощью йодоформной реакции, I
Дйэтидапидип
93 а чистого анилина смешивают со 105 г чистого метило-
в о г о спирта и 9,4 г 94%-ной серной кислоты (уд, вес 1,84). j
Закрывают автоклав и нагревают масляную баню до 200°. Давление |
поднимается примерно до 30 ат; нагревают в течение 6 час, при 215°, j
Затем дают остыть и к смеси добавляют 25 г 30%-ного едкого ч
натра. Для расщепления частично образовавшейся сернокислой солн
четвертичного аммониевого основания, которая разлагается лишь при
высокой температуре на серную кислоту, спирт и третичный амин, реак-
ционную смесь необходимо нагревать в автоклаве еще в течение 5 час. 3
при 170°*. Затем содержимое автоклава перегоняют с водяным паром, д
диметилаиилии полностью выделяют поваренной солью из вод- |
него раствора, отделяют в делительной воронке и перегоняют через не- j
большую колонку с шариковым дефлегматором. Получают почти химиче- d
ски чистый диметилаиилии в виде бесцветной жидкости, содержащей, 4
однако, всегда немного мопо.метил анилин а, |
Выход составляет 117 г; т, кнп, 192°. J
Чистоту полученного вещества определяют, смешивая 4 мл диметил- ’
анилина с 2 мл уксусного ангидрида, Прн этом температура должна
подняться максимум на 1° (проба с уксусным ангидридом),
Дизтиланилин. В лаборатории диэтиланилин получают также очень просто, 'й
Реакцию следует проводить лишь в эмалированном автоклаве, так как в этом случае .1
* Образование четвертичных аммониевых оснований наблюдается особенно при
получении этилбензиланилина и метилбензиланилина.
вместо серной кислоты применяют соляную. Серная кислота расщепляет этиловый
спирт па воду, этилен и уголь.
130 г сухого солянокислого анилина нагревают в эмалированном авто-
клаве со 140 г 95%-ного спирта в течение 8 час. при 180°. Давление повышается
до 30 ат. Если автоклав достаточно прочен, то можно нагревать до 200°; давление
в этом случае поднимается до 55 ат. Потом содержимое автоклава переносят в стек-
лянную колбу, отгоняют спирт и этиловый эфир, а оставшуюся смесь моно- и диэтил-
анилина смешивают со ПО а 30%-ного раствора едкого натра. Затем эту массу
энергично размешивают при комнатной температуре с 40 г n-т олуолсульфохл fl-
ри д а. При этом моноэтиланилин превращается в толуолсульфоэтиланилид, который
не перегоняется с водяным паром. Таким образом, можно отогнать совершенно чистый
диэтиланилин. Чистоту полученного соединения проверяют пробой с уксусным анги-
дридом и в случаё необходимости повторяют обработку п-толуолсульфохлоридом.
Выход около 120 г.
Оставшееся толуожульфопронэводное можно омылить концентрированной
серной кислотой и йыделдть, таким образом, моноэтиланилин.
Приведенный выше метрд/ получения диметил- и диэтиланилина не является
вполне удовлетворительным, нр его можно рекомендовать для небольшого производ-
ства. Более дешевый и более" рациональный метод получения заключается в том, что
берут меньшее количествц.ейирта "й кислот^ и полученную смесь сразу омыляют едким
натром. Моноалкилпроизводное, которое/предварительио не отделяют от диалкилпро-
пзводного, превращают взанмодействц/м с хлористым бензилом в алкилбенэилпро-
изводное. Последнее кипит при_зндийтельно более высокой температуре, чем диалкнл-
производное, и может быть легко от него отделено простой дробной перегонкой в ва-
кууме. Можно применять, например, следующую методику.
Этилбензиланилан. 260 г сухого солянокислого анилина нагревают со
150 мл спирта в течение 12 час. в эмалированном автоклаве при 200°. Давление
21 ат. Дальнейшая обработка такая же, как в случае диэтиланилина (однако без
обработки n-толуолсульфохлоридом). Полученная смесь оснований весит 230 г\ практи-
чески она не содержит анилина и состоит примерно из 38% диэтил- и 62% моно-
этиланилина.
В полулитровой трехгорлой колбе с мешалкой, обратным холодильником, термо-
метром и капельной воронкой смешивают 100 г этой смеси с 60 г кальцинированной
соды, увлажненной несколькими каплями воды. Нагревают прн энергичном переме-
шивании до 50а и медленно прибавляют при этой температуре из капельной воронки
'68 г хлористого бензила, примерно 5% избытка, считая на содержащееся
в смеси количество моноэтиланнЛина; последнее определяют по плотности смеси (см.
табл, на стр. 124). После прибавления всего количества хлористого бензила темпера-
туру повышают до 100° и перемешивают до тех пор, пока не прекратится выделение
газа (около 3 час.), Затем выливают в воду для удаления неорганических солей,
отделяют маслянистый слой и фракционируют в вакууме. Вследствие большой раз-
ницы в температурах кипения легко удается, достичь разделения. Почти количественно
выделяют 38 г диэтиланилина, содержавшегося в исходной смеси, и, кроме того, около
100 а .этилбензиланилина, \
Разделение моно- и дяэтиланилинов мож'но также проводить, действуя на смесь
оснований концентрированной соляной кислотой- Хлористоводородная соль моноэтил-
анилииа выкристаллизовывается, и ее центрифугируют; из пее получают основное ко-
личество моноэтиланилина как такопого. Оставшаяся смесь может быть превращена
в диэтиланилин и этидбепзиланилин действием хлористого бензила и соды по мето-
дике, описанной выше. * > . '”~_
Технические примечания. В заводских условиях разогревание боль-
шого автоклава, продолжающееся 4—6 час., надо проводить очень осторожно, Как
только температура достигнет примерно 190°, давление само собой быстро повышается
от 10 до 30 ат. После окончания реакции избыточный метиловый спирт вместе с эфи-
ром выпускаются из автоклава и конденсируются. Расщепление четвертичного аммо-
ниевого основания проводят в очень больших котлах, в количестве от 3000 до 5000 кг
дчметиланилина, \
Существуют различные методы определения содержания моноэтил- и диэтилани-
лина в данной смеси. Один из методов приведен в кйиге Лунте *. Этот метод основан
на определении повышения температуры при смейивании определенного количества
смеси оснований с точно измеренным количеством чистого уксусного ангидрида. Более
Удобным и более точным методам является определение плотности смеси исследуемых
оснований. Приведенная ниже таблица цозврляет вполне точно определить количе-
ственный состав смеси по плотности -(пфи Пш) - Необходимые условием, естественно,
является полное отсутствие примеси исходного анилина, %ак это имеет место при
применении методики, описанной на стр. 122.
* Lunge, Untersuchungsinethoden, 6 AuFI., В. IV, S. 625.
124
/. промежуточные продукты.
Разделение даже в лабораторных условиях больших количеств (от I кг и выше)
удается дробной перегонкой. Для этого применяют, например, колонку высотой 1 м
с внутренним диаметром 5 см, наполненную стеклянными кольцами (по Рашиту)
шириной и высотой по I см. Кольца лежат на проволочной сетке с крупными отвер-
стиями, чтобы жидкость могла не задерживаясь стекать, Колонка изолирована асбе-
стом, за исключением верхних 10 см. В качестве перегонного сосуда применяют мед-
ную колбу, так как это обеспечивает равномерное кипение,.
Удельные веса моно- и дпэтиланилинов:
моноэтиланнлИП при 15° С 0,9643, т. кип. 204°,
ди^тпланилин при 15° С 0,9389, т. кип. 214°.
Числа и таблице немного отличаются от приведенных в литературе (Г. Видмер);
они основаны на собственных опытах.
Содержание диэтиланилина (%) в зависимости от плотности смеси
моно- и диэтиланилина при 15°С
Плотность Диэтнланнлин, | Плотность Ди этила НИЛИН, Плотность Диэтиланилнн
0,9646 0 0,9565 33,2 0,9470 70,0
0,9643 1 0,9560 35,2 0,9465 71,9
0,9640 2,3 0,9555 37,2 0,9460 73,8
0,9637 3,7 0,9550 39,1 0,9455 75,6
0,9634 4,8 0,9545 41,0 0,9450 77,5
0.9630 6,4 0,9510 43,0 0,9445 79,4
0,9626 8,2 0,9535 45,0 0,9440 81,3
0,9622 9,8 0,9530 47,0 0,9435 83,2
0,9618 11,6 0,9525 48,9 0,9430 85,0
0.9614 13,3 0,9520 50,8 0,9425 86,7
0,9610 15,0 0,9515 52,7 0,9420 88,4
0,9605 17,0 0,9510 54,7 0,9415 90,2
0,9600 19,1 0,9505 56,7 0,9410 92,1
0,9595 21,2 0,9500 58,6 0,9405 93,9
0,9590 23,3 0,9495 60,4 0,9400 95,6
0,9585 25,3 0,9490 62,3 0,9395 97,4
0,9580 27,3 0,9485 64,2 0,9-390 99,2
0,0575 29,2 0,9480 66,1 0,93875 100,0
0,9570 31,2 0,9475 68,0
Этилбензиланилнисульфокислота *
В стеклянную колбу с 'мешалкой помещают 150 г моногидрата и
осторожно в течение 15 мин. прибавляют по каплям 150 г этилбен-
знл апилииЬ; при этом с помощью внешнего охлаждения поддержи-
вают температуру" не выше 50°. Затем прибавляют по каплям 150 5
В I а п g е у, F i е г х, Slamm. Helv. Chim. Acta, 25, J162.
Тетраметил-п.п'-диаминодифенилметан из диметиланилина и формальдегида 12;
с л е у м а, содержащего 60 % SOg, и нагревают при 60° до тех пор,
пока небольшая проба, разбавленная водой, не перестает мутнеть при
прибавлении разбавленного раствора соды. Нагревание продолжают
около трех часов. Затем реакционную массу выливают в 1 л воды.
Свободная сульфокислота выпадает через 12 час. Ее отфильтровывают и
промывают водой. Таким образом получают чистое вещество с выходом
примерно 72% от теории. Маточный раствор, содержащий серную кис-
лоту, нейтрализуют известью, превращают в натриевую соль (до-
бавление глауберовой соли) и, после того как отфильтруют гипс,
упаривают до объема 400 мл. При подкислении получают вторую фрак-
цию сульфо кнелотк (сульфокислота часто выпадает лишь после трення
о стенки сосуда стеклянной палочкой или внесения затравки), выход ко-
торой составляет окаЛо 5/% от теории. Из остающегося маточного рас-
твора поваренной сольф/высаливают натриевую соль изомерной «-кис-
лоты (около 15% от тесфии).
Тетраметил-/гщ'-диамииодифеии л метан из диметиланилина
и формальдегида
<CH3)gN-/~\ + СИ2О + /~\-К(СН;;)й —*
—> (CH3)3N-/~^>- СН3--/_____>-N(CHJa + НаО
242 г (2 моля) диметиланилина, 90 г 40%-пого водного рас-
твора формальдегида (1 моль -ф- 20% избытка) и примерно I а
чистой сульфациловой кислоты кипятят в течение 8 час. с
обратным холодильником при сильном перемешивании. Прн этом не про-
исходит видимых изменений реакцио'йярй смеси. фЧерез 8 час. отбирают
пипеткой пробу светло-желтой эм ул ьснй'-и -охлаждают. Масло должно
полностью застыть и при кипячении лишь очень слабо пахнуть диметил-
анилином. В противном случае надо еще продолжить кипячение с обрат-
ным холодильником.
После окончания реакции реакционную смесь перегоняют с водяным
паром 'до тех пор, пока не отгонятся полностью димстиланилин и фор-
мальдегид. Диметиланилина должно отогнаться, однако, всего несколько
капель. Как только-в дистилляте исчезает диметилапилип, остаток выли-
вают в большое количество холодной воды, при этом основание мо-
ментально застывает. Воду декантируют и кристаллическую массу много
раз основательно промывают водой для освобождения от формальдегида.
Затем расплавляют под водой и оставляют кристаллизоваться.
Так получают основание в виде твердой желтовато-белой кристалли-
ческой лепешки. Температура плавления продукта 80—90°. Перекристал-
лизацией из 500 мл спирта подучают чифтое основание в виде почти
белых блестящих кристалликов ф правильней т. пл. 91 °?
Выход неочитецпого продукта количественный, перекристаллизо-
ванного основания — свЪипе-90%. ।
Тетраметилдилминодифеиилметан (называемый в технике «метановое основание»),
является исходным соединением для получения аура мина (см. об аурамине, стр. 266).
Окислением перекисью свинца он превращается в тетраметилдиамипобеизгидрол
(сокращенно «гидрол»), см. следующую методику.
I ZD
/. Промежуточные продукты
Тетраметил-щп'-диаминобензгидрол (гндрол Михлера)
(СНя).\-<^ / <;Н \ N (СН;1), -у РЬО2 —
Н
7-х 1 /—X
—> (CH!t)aN—/ J>—>-N (CHJ2 + PbO
OH
Тетраметилдиамиподифенилметан окисляют в
точно рассчитанным количеством двуокиси свинца
разбавленном кислом растворе
в виде пасты. Высушенную дву-
окись свинца уже нельзя достаточно тонко измельчить и ее не следует применять для
реакции окисления. Окислительная способность пасты двуокиси свинца должна обяза- ,,
телыю определяться титрованием (см. аналитическую часть, стр. 350). Для приме- '
нения в лабораторных условиях можно, однако, и без анализа приготовить пасту .
двуокиси свинца с точно известным содержанием; для этого растворяют в воде
отвешенное количество азотнокислого свинца и при непрерывном кипячении нрибав-
ляют полученный на холоду и отфильтрованный раствор хлорной извести * до полного ;
осаждения снинца. Свинец полностью осажден, если бесцветный вытек капельной -
пробы па фильтровальной бумаге больше не чернеет при прибавлении раствора сер-
нисгого натрия или аммония. Смесь кипятят еще некоторой время, чтобы она пере-
стала пахнуть хлором, отфильтровывают выпавшую днуокись свинца на нутч-фильтреР
хорошо промывают водой и не высушивая растирают с небольшим количеством воды ;
в однородную пасту.
25,4г (0,1 моля) тетраметилдламинодифеннлметана
растворяют в смеси 1400 мл воды и 60 мл азотной кислоты (уд.
вес 1,38). Раствор смешивают с 600 г льда и сразу прибавляют при
очень сильном перемешивании двуокись свинца в виде
пасты, полученной из точно 0,1 моля = 33,1 г азотнокислого свинца.
Перемешивают 'примерно —3/л часа н получают совсем прозрачный
желтый раствор, Прибавляют 20 г безводного или 40 г кристаллического
сульфата натрия и осаждают таким образом свинец в виде не-
растворимого сульфата. Его отфильтровывают и промывают водой. Про-
зрачный фильтрат медленно прибавляют по каплям при перемешивании'
к раствору 100 г кальцинированной соды в 800 мл воды, При прибав-
лении первых же капель выделяется в большинстве случаев маслянистый
слой, который закристаллизовывается при трен ни стеклянной палочкой
о стенки сосуда или при внесении .затравки твердого гидрола. Прн даль-
нейшем прибавлении гидрол выделяется в виде сероватых кристалличе-
ских хлопьев. После осаждения их тотчас же отфильтровывают, тщатель-
но промывают холодной водой и сушат при комнатной температуре.
Выход 26 г, т. с, около 96% от теории.
Полученный таким образом светло-серый неочищенный продукт с
т. пл. 90—92° в большинстве случаев непосредственно применяется для
получения красителя **. Ои полностью очищается перекристаллизацией из
двадцатикратного количества низкокипящего лигроина и образует почти
* Хлорная известь, как и хлористый кальций, очень хорошо растворима п воде;
нерастворимый остаток, полученный при смешении с водой, представляет собой угле-
кислый кальций и гидрат окиси кальция.
ws Содержащийся в большинстве случаев в неочищенном продукте дигидриловый
эфир
(СНЯ)2М-СВН4 /CJU-N (СНЯ)В
)СН—О—СЦ<
(CH!;)aN-Cf:H./ V’f;lI,-N (СНЛ
(бессчетные призмы с т. пл. 200—201°. почти нерастворимые в холодном эфире) кон-
денсируется так же, как свободный гидрол, и поэтому реакции не мешает.
бесцветные иглы или призмы, плавящиеся при 101 —103°, Растворяется
в уксусной кислоте с образованием глубокой силен окраски, в избытке
минеральной кислоты окраски не дает.
Гидрол может быть также получен менее удобным способом — восстановле-
нием кетона Михлера в щелочной среде. Гидроксильная группа гидрола чрезвы-
чайно реакционноспособна; так, например, она этилируется уже при кипячении со
спиртом. Техническое значение гидрола определяется его способностью легко кон-
денсироваться с различными ароматическими соединениями с образованием лейко-
соедпнепий трифенилметановых красителей (см. получение зеленого С для шерсти,
стр. 274).
Тетр амети л - д нами иобензоф еиои
(кетон Михлера)
N(CH3)2 N(b^)a N(CH3)2 N(CH3)2
4 + COCL —Г4)
4// V’'//' ' • r. CO—-
COC1 + Изомер
N(CH3)3HC1
2
Конденсация диметиланилина с фосгеном не дает однородного про-
дукта и, помимо щн'-соединсния, приводит к образованию также значи-
тельных количеств о,о'- н о,«'-соединений. Фосген, необходимый для
этой реакции, берут из баллона *.
Чтобы полностью связать всю образующуюся соляную кислоту, при-
меняют небольшой избыток диметил анилина,
В 520 г (4,3 моля) чистого сухого дн мети л а нил и на, поме-
щенного в стеклянную колбу, пропускают при хорошем перемешивании
99 г (1 моль) фосгена, при этом \смпература нс должна превышать
24°. Количество поглотившегося фосген^ определяют, периодически взве-
шивая реакционную колбу. После окончания пропускания фосгена пере-
мешивают еще 24 часа при 24—28° и затем, проверяют, исчез ли запах
фосгена. Если фосген остался, то прибавляют еще 10 г диметиланилина
и оставляют на несколько часов. Затем прибавляют 40 г мелкозернистого
хлористого цинка и перемешивают 2—3 часа для того, чтобы
хлористый цинк полностью перешел в раствор. Осторожно повышают
температуру в течение двух часов до 55° и оставляют стоять в течение
12 час. при этой температуре. Под конец перемешивают еще 2 часа
при 80°.
После этого конденсация окоцчеца. Реакционную массу разбавляют
5 л воды и осторожно йрнбавляют концентрированную с ол я ну ю кис-
лоту (около 40 г) до исчезновения запаха диметиланилина. Не вошед-
ший в реакцию диметиланилин и небольшое количество побочно об-
разовавшегося продукта переходят в раствор. Так как кетон Михлера
значительно менее сильное основание, чей диметиланилин, то он ие
растворяется. Кристаллический осадок отфильтровывают и промывают.
Затем его кипятят с пятикратным количеством воды ив том случае,
если опять появится запах диметиЛанилмна, прибавляет еще немного
концентрированной соляной кнелотф]. Кетон Михлера отфильтровывают
и высушивают при 100°, /
Выход 177 г, т. с. 6крло^з6% QT теории.
Технически чистый кетон, еми нужно, можно* перекристаллизовать
пз высококипящсго бензина (т. кип. 180^). При перекристаллизации
* См., например, Ней mann, Die AnilfnfarbstoIIe.
больших количеств его нагревают до 140° и дают охладиться. Темпера-
тура плавления полученного таким образом кетона Михлера 170—172°.
Аналогичным образом получают тетраэтилдиаминобензофенон. Этот
кетон очищают через кислую сернокислую соль, которая прекрасно, кри-
сталлизуется.
Технические пр и'м с ч а н и я. Кетон Мцхлсра и его гомологи являются
важными исходными веществами для получения ценных трпфевилметановых красите-
лей — виктория голубого (основного синего) и виктория голубого БО (основного
синего 3).
Дифеииламии из анилина и анилиновой соли
93 а аннлннанЭЗа солянокислого анилина (анили-
новой солн) нагревают в течение 20 час. при 230° в эмалированном авто-
клаве с эмалированной гильзой для термометра. Давление поднимается
до 6 агм. Если не имеется эмалированной гильзы для термометра, то
Рис.- 23а. Лабораторная 5гстановка для перегонки с перегретым воднггым паром.
7 — парообразователь; 2 — водоотделитель; 3 — плроперегрепате/п,; ^ — перегонный сосуд н масляной бане;
5 —холодильник с широкой трубкой; в — приспособление для проталкивания твердого вешества, застыв-
шего н холодильнике; 7 —приемник для дистиллята; 8 — газовая горелка; 9— водяное охлаждение.
можно просто нагревать при указанном давлении, следя за температурой
масляной бани. Она‘примерно на 25° выше, чем действительная темпе-
ратура внутри автоклава. Через два часа осторожно спускают через
вентиль воду, так как даже следы ес вредно сказываются на течение
реакции. Продувание повторяют три раза в течение чаеа, при этом выде-
ляются также немного амидина и аммиак. Нагревать более 20 час. не
имеет смысла, так как при этом выход может только уменьшиться. После
Рис. 236. Лабораторный Сбсуд'для перегонки с перегретым
водяным паром.
Z—медный куб; масляная баня е термометром; 3— соединение с паро-
перегревателем; 4 — хо.т.1.(1 л(.ннк; > — просиос.’блениг для проталкивания,
твердого ыещесгна, настывшего в холодильнике.
охлаждения содержимое, автоклава переносят в фарфоровую чашку н
прибавляют 1 л воды. Затем нагревают до подкисляют 30%-иой со-
ляной кислотой до кислой реакции ,йа конго и оставляют опять
охлаждаться на ночь. Неочищенный дифениламин выделяется в виде
Рис. 24. Производственная установка дли /перегонки с перегретым водяным паром
а-нафтиламипа, дифенила и пн а и т. д,
7 - инод перегретого пара; 2 — пере гони 1лй\сост'л с лаелленно нрашаюшейся мешалкой; 3 — змеевиковый
' Х0ЛТ)/1И,П,1|ИК. . ' ’
/
твердой лепешки, которую можно легко отделить от маточного раствора,
так как дифениламин не образует соли с разбавленной соляной кисло-
той. Отфильтрованный дифениламин расплавляют еще раз с некото-
рым количеством воды, экстрагируют небольшим количеством соляной
кислоты и промывают разбавленным раствором соды. По,ту ценный таким
образом дифениламин очень загрязнен.
Поэтому его следует перегнать с перегретым водяным паром. Для
этого собирают аппаратуру, как показано па рис. 23а и 236; перегонный"
сосуд берут емкостью примерно 500 мл. Масляную баню .нагревают до>
25(Р п затем приводят в действие перегреватель на обычной печи Флет-
чера. Пар должен быть тщательно высушен п перегрет до 300° При хо- ;
рошен перегонке легко удается с одной частью воды отогнать ’/а части;
основания. Дифениламин получают в виде почти бесцветной жидкости, )
застывающей в бледно-желтую лепешку. После сливания воды получают :
около 100 с совершенно чистого дифениламина с т. пл. 51"'.
Из кислого маточного раствора можно регенерировать около 35 г
анилина.
Технические примечания. Должны быть эмалированы не только ниж-:
няя часть, но и крышка антоклаи;!. Следы железа или меди снижают выход дифе-
ниламина на 30—35% и вызывают осмоление. Экстракцию соляной кислотой проводят,
в деревянных чанах; перегонку с перегретым водяным паром осуществляют в при-;
боре, изображением на рис. 24. Для перегревания водяпого пара применяют новые!
аппараты, например так называемый пароперегреватель Гейцмаиа и др. С од-j
ной частью воды можно перегонять при 230" до одной части дифениламина.
Анилид ацетоуксусной кислоты
nh -со—аъ—со—сн:1
+ СН3—СО—(ДЬ—COOCvH- —> I I
— С2Г1,рЦ
Агги Л ид
{шетоуксу (•-
ЕТОЙ КИСЛО-
ТЫ
Анилиз реагирует с NaOH, давая
При сочетании с диазо составляю ши мн 11* замещается на— К — X —It.
В колбе с обратным холодильником нагревают до кипения 156 г
(1,2 моля) свсже.перегпаиного ацетоуксусного эфира (т. кип.'
69° при 12 мм), 500 мл сухого толуола и 0,5 мл пиридина. К сла-л
бокипящей жидкости добавляют по каплям в продолжение 5 час. 93 г
(1 моль) свсжеперегпаипого анилина, смешанного с 250 мл то-1
луола и 0,5 мл пиридина с такой скоростью, чтобы постоянно;
отгонялось приблизительно стблько же толуола, „сколько его прибавляют.;
После прибавления всего количества смеси кипятят еще 2 часа с обрат- ;
ным холодильником. Раствор должен быть окрашен не в красный, а ;
светло-желтый цвет. Еще горячую жидкость охлаждают в стакане; при
этом' выделяется иногда лишь после внесения затравки чистый анилид|
ацетоуксусной кислоты. Фильтруют через 12 час., промывают 50 мл то-;
луола и сушат при 60% Выход около 150 г, т. с. около 85% от теории?
Помимо желаемого анилида, может получиться также несколько процен-
тов побочных продуктов, В этом случае анилид ацетоуксусной кислоты;
не полностью растворяется в разбавленном растворе едкого натра. При
осаждении прозрачного фильтрата разбавленной соляной кислотой полу-
чают химически чистый анилид с г. пл. 85°.
Тсхнпч ech’ire прим сч ани я. Анилид ацетоуксусной кислоты применяется
для нолуиеипя ценных нерастворимых нигментов желтого инета — таила желтых
(см. стр. 239) я.
9. Производные бенаолсульфокнслоты
Бензол-ju-дисульфокислота
SO„H
\ I
О + 2s°;i —>
X'/4SO;iH
К 78 г (1 моль) б с н зю л а ори хорошем перемешивании в течение
2 час, по каплям прибавляю-/ 250 г 20%шого о л е ум а с такой скоростью,
чтобы температура по превышала 40—45°. Затем за следующее 2 часа
таким же образом прибавляют 200 г 66%-ного олеума, при этом темпе-
ратура повышается примерно до 75°. Под копен, нагревают еще 1 час
при 90% Реакционную массу выливают в 2 л воды и горячий раствор
нейтрализуют при перемешивании 400 г мела. Отфильтровывают осадок
гипса на путч-фильтре и хорошо промывают водой, В горячий фильтрат
прибавляют кальцинированную соду (около 100 г Na2COs) до слабоще-
лочной реакции на фенолфталеиновую бумажку. Фильтру/от, промывают
осадок карбоната кальция, фильтрат упаривают досуха и, наконец, оса-
док сушат при 130—140°.
Выход около 250 г, или 90% от теории.
Бензол-ллдпсульфокислота применяется Для получения фенол-.и-сульфокислоты и
резорцина. Дальнейшее сульфирование бензод-,ч-ДИ сульфокислоты в трисульфокислоту
удается осуществить при нагревании до 25рС| в течение нескольких часов ГСа-соли
бензол-л-дисудьфокислоты с 66%-ным олеумом в присутствии ртути.
\
Резорцин4
SOsNa ONa
1 ‘ I
| । -ф- 4\аОН —> । | З- SNa^SCXj -ф- 2НИО
'X/'4SOBNa 4'/'X-ONa
Во вкладыше аппарата для запекания под вакуумом (см. рис. 30,
стр. 162) растворяют при нагрева пин до 200° 32 г (0,8 'моля) твердого
едкого натра в 20 мл воды, затем вносят 28,2 г (0,1 моля) на-
триевой соли бе пт одш м-дисульфокисдоты (полученной,
как указано выше) и /йереме’шивают до получения однородной массы.
Вкладыш помещают в’ аппаратная запекания. Сначала в вакууме при
200° отгоняют воду, затем повышают температуру до 320° и выдержи-
вают 6 час. при этой температуре и давлении 12—15лгж.яПосле охлажде-
ния застывшую массу растворяют в псбодьшо.м количестве горячей воды,
подкисляют концентрированной с о л я ц о й кислоте й до сильнокислой
реакции на конго и кипячением удаляет SC%. Профильтрованный, если
это необходимо, раствор подвергают исчерпывающему экстрагированию
" Z i с g I с г Е., Диссертация, Zurich, 1928; Ficrz -Davrd nnd В 1 a n g e у,
Naditmg zu den Kimstljchen Organischcn Farbsloffrii, S. 58.
J. Промежуточные продукты.
эфиром в экстракторе (Кучер-Штейделя) до тех пор, пока проба
с ГеС13 не будет давать больше окрашивания, Эфирную вытяжку
сушат обезвоженным сульфатом натрия и после отгонки раство-
рителя остаток перегоняют в вакууме. При 80° (12 мм) отгоняется около
0,5 г фенола, который собирают отдельно, а затем при 156—159° пе-
регоняется около 8,5 г чисто белого резорцина ст. пл. 109—110°.
Выход около 75—80% от теории.
Щелочная плавка бензол-лг-дисульфокислоты приводит к получению резорцина
только в отсутствие воды. С водным раствором едкого натра под давлением получают
не резорцин, а фснол-,и-сульфокислоту или при более высокой температуре — фенол
и продукты разложения. Полностью избежать образования фенола в качестве побоч-
ного продукта не удастся *,
Плавку лучше всего проводить в вакууме; для этого целесообразно применять
аппарат для сульфирования аминов методом запекания под вакуумом. При упари-
вании плава, содержащего воду, масса вспенивается; для того чтобы це забивалась
отводная трубка, ее делают достаточно широкой, и вкладыш заполняют не больше,
чем наполовину. Для получения высоких выходов резорцина необходимо применять
примерно в два раза больше едкого натра, чем требуется по теории, т. е. 8 молей на
1 моль бензолднеульфокислого натрия.
10. Производные фенола
о- н /г-Хлорфенол нз фенола
он
он
•% SO3+HC1
Cl
Смесь обоих изомеров получают по методу Дюбуа (1866 г.) дей-
ствием избытка хлористого сульфурила на фепол. Эту смесь разделяют
фракционированной перегонкой. Берут 380 г (4 моля) фенола и 610г
(4,5 моля) хлористого сульфурила.
В трехгорлую литровую колбу, снабженную мешалкой, термометром,
капельной воронкой я трубкой для пропускания газа, помещают расплав-
ленный фенат и добавляют по каплям в течение 12 час. хлористый суль-
фурил. Температура должна быть 20—25°. Перемешивают еще в течение
5 час. прн 10° и затем в течение 12 час. пропускают через реакционную г
смесь сжатый воздух.
Смесь перегоняют в вакууме с дефлегматором. о-Хлорфснол перего- .
няется при 75—90° (20 жж), а пара-изомер — при НО—115° (20 жл).,;
Получают приблизительно 130 г о-хлорфенола (25%) и 320 г н-хлор-\
фенола (около 62%),
Примечания. Свободный хлор действует на фенол так энергично, что тотчас ]
же образуется трихлорфенол (наряду с неизмененным фенолом), Монохлорфенол;
можно также получить, если прибавлять по каплям при охлаждении к раствору фе- i
ноля в едком натре рассчитанное количество раствора гипохлорита натрия и потом :
подкислить; одпако при этом образуется главным образом о-хлорфенол.
Хлорфенол имеет стойкий отвратительный запах, поэтому с ним
необходимо обращаться осторожно.
* См. F i е г z Stamm, Helv. Chrm, Acta, 25, 364,
Хлоранил *
В двухлитровую трехгорлую колбу, снабженную герметически уста-
новленной мешалкой (в качестве запорной жидкости применяют концен-
трированную серную .кислоту или парафиновое масло), трубкой для про-
пускания газа и обрат’вдм холодильником, помещают 47 г (0,5 моля)
фенола или 65 г о- или л,-х ло рфен о л а и 1 л концентрированной тех-
нической соляной кнефоты (уд. вес 1,17). Обратный холодильник
снабжается стеклянной трубкой, которая выводится прямо в тягу. При
энергичном перемешиваний,'-А результате которого исходные вещества
образуют тонкую эмульсию, пропускают довольно сильный ток хлора.
Хлор можно особенно пе сушить, хотя его и пропускают через склянку
с концентрированной серной кислотой для того, чтобы можно было сле-
дить за скоростью его пропускания. Происходит саморазогревание при-
мерно до 40° Через 4 часа реакционную колбу нагревают на водяной
бане до 70° и пропускают далее прн этой температуре хлор до полного
насыщения, для чего требуется около 20 час, Постепенно па стенках
колбы и в холодильнике образуются блестящие кристаллы, После насы-
щения реакционной смеси хлором трубку, через которую пропускают
хлор, заменяют капельной воронкой и прибавляют пр. Каплям при посто-
янном перемешивании и 80—85° в течение 3 чао.,,2-50' мл азотной кис-
лоты (уд. вес 1,38). Сначала реакция идет/довольно бурно, но скоро
замедляется, Жидкость становится красной,/и выделившиеся ранее кри-
сталлы расплавляются. После прибавления] азотной кислоты перемеши-
вают еще в течение 20 час. при 85° (температура водяной бани). Посте-
пенно выделяются тяжелые желтые кристаллы в виде листочков. После
охлаждения кристаллы отсасывают, промывают сначала 2 л воды, а затеи
250 лы спирта для удаления красноватой маслянистой примеси, Продукт-
сушат прн 80° и получают практически чистый хлоранил с т. пл, 285—286° _
Выход 70—75 а, т. е. около 60% от теории.
Примечания. По вышеописанной методике все хлорпроизводные фенола пре-
вращаются jb хлоранил. Метод дает возможность, таким образом, использовать по-
бочные продукты хлорирования фенола (см. стр. 132), которые вследствие сильного
запаха и ядовитости часто являются трудно удаляемыми отходами производства.
В хлораниле имеются два, находящихся в ларя-исложении один к другому, очень
реакционноспособных атома Хлора, которые могут замещаться различными другими
группами. Например, с аннлййом легкб получают 2г5-дианилин-3,б-дихлорхинон, кото-
О
II ___
~~ II
рый уже сам обладает свойствами кубов^гр красителя для шерсти, а при осернении
может быть Превращен в интенсивные прочные кубовые красители* **, С сернистыми
соединениями, такими, как сернистый натрий? тиосульфат, роданистый калии и т. д.
* Kempf R., Moehrke Н., герм. пат. 256034 (Frdl., Ж, 193).
** Герм, пат, 263382, 265195/6, 270401 (Frdl., XI, 257); germ. pat. 236074 (Frdl.
X, 282).
и уили’м ytvvttwi ripwyiv i ы
хлоранил реагирует очень легко, и получающиеся при этом серусодержашпе соединения
могут конденсироваться, например, с тиосульфокиелотой диметил-л-фенилен дням ина
(см. Метиленовый голубой, стр. 278) с образованием синих- сернистых красителей *.
Эта реакции особенно интересна потому, что она протекает уже на холоду при стехио-
метрических соотношениях реагирующих веществ, т. е. в условиях, аналогичных обра-
зованию азокрасителей.
Следует упомянуть также, что хлоранил может с успехом применяться в лабо-
ратории в качестве мягкого окислителя, например для превращения лейкосоединений
трифеннлметанового ряда в соответствующие красители, так как он, даже будучи
взятым в избытке, не разрушает их. Однако хлоранил для применения с этой целью
в технике очень дорог (см. с гр. 353) .
о- и n-Нитрофенолы и их алкильные эфиры
О-Алк.
О-Алк.
।
no2
94 г ф е н о л а расплавляют в 20 мл воды и жидкую смесь добавляют
по каплям при 20° и сильном перемешивании в 150аазотнокислого
натрия, растворенного в 400 .мл воды и 250 г концентрирован-
ной серной кислоты. После прибавления перемешивают еще
2 часа. Затем сливают маточный раствор со смолистой смеси нитросоеди-
нений, расплавляют смолу в 500 мл воды н прибавляют мел до нейтраль-
ной реакции на лакмус. Промывные воды сливают и промывание повто-
ряют. Сырой, отмытый от азотной кислоты нитрофенол перегоняют с во-
дяным паром (с широким холодильником). Отгоняют около 40 г чистого .
□-нитрофенола. Остаток в колбе охлаждают и через 24 часа отфильтровы-
вают от маточного раствора. Затем этот остаток кипятят с 1 л 2 %-пой :
соляной кислоты и фильтруют через складчатый фильтр. Из горя-
чего раствора выкристаллизовывается чистый п-нитрофенол в виде почти .
белых длинных игл. Если надо, то остаток экстрагируют еще раз. .
Выход составляет около 40 г орто- и 40 а пара-изомера. Обра-
ботка неочищенного нитрофенола раствором едкого натра, рекомендуемая '
различными методиками, очень вредна, так как щелочь тотчас же осмо-
ля ст.
Технические примечания. При перегонке больших количеств приме-
няют змеевиковый холодильник, который помещают в теплую воду для того, чтобы
вешество не застывало, или прямой холодильник, через который пропускают теплую
воду. Для уменьшения расхода пара его целесообразно перегревать до 110а.
о- н л-1 Гитрофеполы являются исходными веществами Для получения о- и л-феие-
тидинов и о- и п-анизидшгов. Из о-гштроанизола получают дианизидиц, который дает
наиболее яркие прямые голубые красители (прямой чисто голубой, диамин чисто го-
лубой, чдкаго голубой и др.).
А 4 к и л н р о а а п и е н и т р о ф с н о л о в
Эфиры нитрофенолов получают следующим образом: 1 моль нитро-
фенола растворяют , в 400 мл в о д ы, растворе 1 моля едкого
натра и 80 а соды. В этот раствор прибавляют 500 мл 90%шого
* .1 гг 1 i ц s, Р., Munch Е., герм. цат. KJ70J2 (ETclL, V | И, 752).
спирта (метилового или соответственно этилового) и охлаждают до 10°.
Затем вносят 1,75 моля хлористого метила или хлористого
этила*. Смесь нагревают в автоклаве 8 час, при 100°, давлении 4—5 ат
и перемешивании или вращении. Алкилирование после этого считают
оконченным. Содержимое автоклава выливают в воду, отделяют алкил-
эфир и спирт ректифицируют. Алкильное производное, промытое неболь-
шим количеством щелочи и водой, не должно больше содержать нитро-
фенола,
Так как работать с хлористым метилом и этилом нелегко, ниже при-
водится наиболее удобная методика. Смесь нитрофенола, соды и щелочи
рис. 25а. Загрузка хлористого
алкила в лабораторной автоклав.
Рис. 256. Вспомогательный баллон
из газовой трубы для загрузки хло-
ристого алкила.
с водным спиртом помещают в автоклав, и последний закрывают. Не
обязательно добиваться того, чтобы вещества полностью растворились.
Автоклав эвакуируют с помощью водоструйного пасоса и закрывают вен-
тиль. Небольшой баллон для хлористого алкила 1, изготовленный из га-
зовой трубы диаметром 50 мм с винтовым затвором 3, присоединяют
к автоклаву при помощи медной трубки 4 и так называемого раккорда 2
(рис. 256). Хлористый алкил заранее отмеряют, в охлажденном мерном
цилиндре и переносят в баллон, который д.^'я этой цели помещают
к ледяную воду**. Баллон / присоединяют к автоклаву* 3 (рис. 25а)
и поворачивают таким образом (1а), чтобы хлористый алкил стекал
* Можно также, особенно для нитрокрезолов, с успехом применять следующую
загрузку: I моль «фенола», 600 мл спирта, 1,8 моля NaOH и 1,75 моля хлористого
алкила, без воды и соды. ’ 9
” При применении хлористого метила его растворяют в равном количестве
охлажденного метилового спирта и наполняют этим раствором балле#!.
к
136
/. Промежуточные продукты
в соединительную трубку. Баллон нагревают очень горячей мокрой тряп- ,
кой до тех пор, пока до него можно еще дотронуться, и лишь после этого г
открывают вентиль 2 автоклава (рис. 25а). Теплый хлористый алкил
стремительно направляется в разреженное пространство автоклава, и че-
рез несколько секунд вентиль автоклава закрывают. Как показал опыт,
таким путем можно в автоклав перевести по крайней мере 98% всего
количества хлористого алкила. Баллон, в котором устанавливается при
этом нормальное давление, можно отъединить и начать нагревать авто-
клав на масляной бане, как было указано выше.
В технике алкилирование проводят в больших горизонтальных или
вертикальных котлах (см. о хризофенипе, стр. 254). Хлористый алкнл !
получают из соляной кислоты и спиртового раствора хлористого цинка *. г:
Его перевозят в больших железных цилиндрах и храпят в резервуарах. А
Для употребления продукт разливают во взвешенные стальные баллоны/
нз которых затем его перекачивают или передавливают в реакционный-
сосуд нагреванием через сифонную трубку. '
Алкильные эфиры о- и п-нитрофеиолов также могут быть получены из о- или,,
соответственно, гг-нитрохлорбензолов со спиртовой щелочью (см. стр. 91).
Триннтрофенол (пикриновая кислота)
Фенол-
дисул ьфокислота
Тринитрофенол
В сульфуратор (стеклянный, железный илн фарфоровый) помещают j
94 а фенола первого сорта. Нагревают прн перемешивании до 100° и
вносят 300 г моногидрата с такой скоростью, чтобы температура
все время была ниже 110°. Затем нагревают I час при 100—110° для :
завершения сульфирования всего количества фенола. Большая часть '
фенола в этих условиях превращается в дисульфокислоту. Сульфуратор
охлаждают снаружи льдом н солью до тех пор, пока реакционная масса
не охладится до 0°, и затем при этой же температуре приливают по кап-
лям в течение 3 час. 3,5 моля азотной кислоты в виде 50%-иой
нитрующей смесн.
Продажная нитрующая смесь готовится из очень концентрнройанной азотной
кислоты и олеума также высшей концентрации. Обычно в больших железных котлах
с водяным охлаждением смешивают азотную кислоту уд. веса 1,48 с 40%-ным олеу-Д
мом. В лаборатории этот способ не следует применять, так как он очень опасен. Для
лабораторных целей достаточно смешать азотную кислоту уд. веса 1,50—1,52 с рав-
ным по весу количеством моногидрата.
После прибавления всего количества азотиой кислоты к фенол- i
сульфокислоте реакционную массу оставляют на ночь при комнатной тем- а
пературе и на следующий день очень медленно нагревают на водяной
бане до 30° в течение часа при перемешивании. Затем нагревают до 45% j
* Вместо хлористого цинка можно также применять хлорное железо.
Тринитрофенол (пикриновая кислота)
137
но не выше, так как иначе может произойти внезапное саморазогревание
массы, и если даже взрыва не произойдет, смесь может выбросить нз
котла. Однако при 45° реакция не проходит до конца. Поэтому небольшую
часть реакционной массы (около 50 мл) переносят в полуторалитровый
фарфоровый стакан и нагревают при перемешивании на песчаной бане.
Р и с. 26. Керамический путч-фильтр для сильнокислых осадйов.
1 — трубка к иакууму; 2 — трубка дли пере.тавликасия фильтрата; 3 — натрубпк для вцуска воздуха;
4 — трубка для спуска промывных вол или для соединения с канализацией; 5 —стопорный болт.
Температура быстро поднимается до 110—125°. К нагретой части при
постоянном перемешивании медленно добавляют по каплям остаток реак-
ционной смеси, следя за тем, чтобы не происходило вспенивания. Нагре-
вают еще полчаса при 110—120° и затем при 120° прибавляют по каплям
воду, пока концентрация серной кислоты не достигнет 40%. Как показы-
вает простое вычисление, для этого необходимо около 700 мл воды. Вы-
деляются окислы азота; количество их невелико, если нитрование прово-
дилось правильно. При охлаждении количественно выпадает пикриновая
Рис. 27. Переносный фильтр на козлах для крупнокристаллических осадков.
кислота, не растворяющаяся в 40%-ной серной кислоте. В маточном рас-
творе находится немного смолы и других продуктов разложения. Филь-
труют па холоду через хлопчатобумажный фильтр и промывают холодной
водой. Промытая пикриновая кислота является химически чистой. Полу-
чается около 210 а чистого продукта.
Аналогичным путем получают желтый Марциуса (2И-диннтро-1-наф-
тол) и нафтоловый желтый (2*4щинитро-1-нафтол-7-сульфокислота),
а также д-инитрокрезол и другие полниитросое^инения/П и достаточной
1.3.S
/. П ро.чеж уточные продукты
концентрации нитрующей смеси можно применять почти теоретическое
количество азотной кислоты.
Технические примечания. Все описанные выше жидкости являются
спльиокнслыми, и поэтому их следует фильтровать через кислотоупорные материалы;
рекомендуется применять для этой цели только керамические путч-фильтры (рис. 26)
или фильтровальные салфетки из нитроцеллюлозы (нитрофильтры). Окислы азота,
выделяющиеся при нитровании, конденсируются в башнях с кольцами Рашига в азот-
ную кислоту.
Пикраминовая кислота
он он
I
NO3 NO2
В стеклянном или железном сосуде емкостью не мсиее 2,5 л раство-
ряют 10 г пикриновой кислоты и 10 г 35%чного едкого
натра в 600 мл воды. Нагревают до 55°, сильно перемешивают и
прибавляют тонкой струей в течение 10 мин. раствор 40 г кристалли-
ческого сернистого н а т р и я в 100 лтл воды. Затем вносят пор-
циями (по чайной ложке) 127,5 г растертой в порошок пикриновой
кислоты и одновременно вводят в течение 10 мин. еще 220 г серни-
стого натрия в 400 мл воды. Все количество пикриновой кислоты
должно быть прибавлено за то же время, что и сернистый натрий. Если
температура поднимается выше 65°, то надо прибавить немного льда.
После добавления пикриновой кислоты и сернистого натрия перемеши-
вают еще 10 мии. Затем добавляют сразу 400 г льда, н тотчас же пол-
ностью выпадает пикрамииовокислый натрий. Его отфильтровывают после
десятичасового стояния и промывают 10%-ным раствором соли. Свобод-
ную п икрам и новую кислоту пат уча ют нагреванием до 80° при перемеши-
вании ее натриевой соли с 500 мл воды и подкислением разбавленной
серной кислотой. Раствор должен иметь чуть кислую реакцию на конго.
Охлаждают, фильтруют через 10 час. и получают выход около 100 г
100%-ной кислоты.
Вариант. Частичное восстановление пикриновой кислоты, естественно,
можно проводить различными способами. Вместо того чтобы постепенно
вводить пикриновую кислоту и, таким образом, нейтрализовать ею обра- ;
зующуюся щелочь *, можно также восстанавливать натриевую соль пи-
криновой кислоты и одновременно приливать необходимое количество
соляной кислоты.
Так, например, растворяют 137,5 г (0,6 моля) пикриловой кис- ,
лоты в 1200 мл воды и 36 г соды при 50°. При этом не происходит :
полно'го растворения. После полного выделения углекислоты прибавляют ,•
при хорошем перемешивании в течение получаса 1 моль (240 г) кристал- ;
личсского сернистого натрия, растворенного в 450 мл воды. Одно- j
временно приливают раствор 108 г 30%-ной соляной кислоты ..
в 300 мл воды так, чтобы продолжительность приливания кислоты была "
примерно на 1 мин. больше, чем сернистого натрия. После того как все
прибавлено, размешивают еще в течение получаса без нагревания и через
12 час. фильтруют. 'Осадок промывают 100 мл насыщенного раствора по-
варенной соли. Затем растворяют сырой пикрамииовокислый пат-
* Уравнение реакции: 1Х — NO? 7IisO •= 4Х—NH;>-J-6Nr;iOH-| ЗМацЗаОз.
2-Аминофенол-4.6-дисульфокислота из фенола
139
рий в 2 л воды и профильтрованный раствор выливают в нагретую до
90° смесь 70 мл 30% -ной соляной к и с л о т ы и 400 мл воды. Чистая
пикраминовая кислота полностью выпадает через 24 часа. Ее отфильтро-
вывают и промывают небольшим количеством воды. Отжимают и сушат
при 80°; выход около 100 г, т. е. 83% от теории.
Из двух описанных способов первый более пригоден при работе с большими
количествами, так как он дает возможность легче регулировать постепенное приба-
вление веществ. .
Пикраминовая кислота в последнее время приобрела большое значение в качестве
дназосоетавляющей. Ее диазосоединение при сочетании с несульфированными азо-
составляющими дает очень прочные хромирующиеся красители для шерсти, которые
отличаются тем. что они могут окрашивать при одновременном прибавлении
в красильную ванну хромовой кислоты (однохромовые красители, например метахро-
мовый коричневый, кислотный однохром коричневый 3).
ОН NH.>
( I—'
osn-1/^-n2-/ >-NH,
I Cl
no2
Эти красители в большинстве случаев очень трудно растворимы и в сухом со-
стоянии взрывают, поэтому их измельчают с большим количеством глауберовой соли
или же выпускают в продажу в виде водных паст.
Диазосоединение пикраминоной кислоты является вообще первым соединением
такого рода. Его получение послужило началом важнейших работ П. Грисса.
2-Аминофенол-4,6-дисульфокнслота из фенола
1. В железном сульфураторе расплавляют при 50° 94 г чистого фе-
нола и прибавляют при хорошем перемешивании в течение 10 мин. 300 г
моногидрат а. Нагревают 1 час при 100°, затем охлаждают до 50° и
прибавляют еще 300 г олеума, содержащего 60% SOj *. После этого
нагревают еще 1 час при 100°, охлаждают льдом с солью до —5° и при
температуре от —5° до 0° добавляют по каплям смесь 100 г 65 %-ной
азотной к ис лоты и 100 а моногидрата. Нитрование продол-
жается около 3 час. Процесс\не следует вести быстрее, так как иначе
образуется пикриновая кислота:, Через 10 час. смесь выливают на 500 г
льда и 1 л воды и высаливают фитро фенол ди сульфо кислоту 300 г пова-
ренной соли. Общий объем составляет примерно 2 л. Сульфокислота или
ее натриевая соль выпадают в виде красивого светло-желтого осадка;
через 10 час. ее фильтруют, промывают на фильтре 200 мл насыщенного
раствора поваренной соли и отжимают. Выход около 380—400 г.
2. Восстановление. Влажную лепешку нитрофенолдпсульфокислоты
измельчают на кусочки величиной с горошину н вводят в течение полу-
часа в 480 г кристаллического сернистого натрия (Na2S + 9Н2О),
расплавленного при 100° с 50 мл воды. Нагревают до 108° и приливают
ио мере испарения воду так, чтобы объем оставался все время постоян-
ным. Масса становится сначала коричневой, потом красно-коричневой, и
затем образуется красная кристаллическая корка. Примерно через час
раствор немного светлеет. В общей сложности нагревают 2,5 часа при
105°, добавляя все время воду но мере ее испарения. Подкисляют
соляной кислотой до отчетливо кислой реакции па конго и оставляют на
* Можно-брать также лишь пол овин} этого количества олеума, как это делают
в производственных условиях. В лаборатории же лучше исходить из указа иных коли-
честв. ' —
ночь. Серу и аминосульфокислоту отфильтровывают; осадок растворяют
при кипячении в 700 мл воды и фильтруют горячим. Бесцветный рас-
твор целесообразно смешать со 120 г хлористого калия и оставить охла-
ждаться. Уже при 90° прекрасно выкристаллизовывается бесцветная
кислая калиевая соль амино фенол дисульф окис лоты, через 10 час. ее
фильтруют и отжимают.
Выход составляет около 230 г сухого вещества.
Салициловая кислота из фенола
В настоящее время салициловую кислоту получают исключительно
по методу Коль бе-Шмита взаимодействием абсолютно сухого фенолята
натрия с сухой углекислотой сначала при обычной температуре, затем
при 125° и давлении 4—7 ат. Салициловая кислота получается, почти
с количественным выходом, если исходная соль хорошо высушена и тонко
измельчена. Топко измельченную соль получают высушиванием и раз-
малыванием в вакууме.
В автоклав с мешалкой н вентилем для введения углекислоты поме-
щают 94 г чистого фенола и смешивают его с 1 молем (40,1 г)
100%-иого не содержащего углекислоты* едкого натра, растворен-
ного в 100 мл воды. Раствор упаривают при 100°, уменьшенном давлении,
и постоянном перемешивании до тех пор, пока не оттопится вся вода.
Сухой фенолят извлекают из автоклава и возможно скорее измельчают:
в предварительно нагретой фарфоровой ступке. Для того чтобы защитить
фенолят от влаги, его тотчас снова помещают в автоклав вместе
с 5—10 железными или каменными шариками диаметром около 14
которые при перемешивании в вакууме производят дальнейшее измельче-'
иие вещества. Снова нагревают фенолят в вакууме при 1-65° до тех порф
пока масса не станет абсолютно сухой. На это требуется 5—6 час.; затем;
охлаждают до 30° и пропускают при постоянном перемешивании углекис-.®
лоту из баллона, регулируя редукционным вентилем давление так, чтобы;
оно было ие выше 1 ат. Через 2 часа медленно поднимают давление^
до 5 пт и температуру до 125°. Затем, спустя несколько часов отключают.]
баллон с углекислотой и спускают давление. По охлаждении желтоватыйл
порошкообразный салицилат растворяют в 400 мл воды и осаждают^
125 г 30 % -ной соляной кислоты. Выпадает почти чистая салици-|
лов а я кислота; ее фильтруют при 30° и промывают небольшим количе-Уз
ством воды от оставшегося фенола. Для очистки ее можно перегнать^
с перегретым до 140° водяным паром или перекристаллизовать из горя-1
чей воды, предварительно осадив примеси с помощью хлористого оловаЛ
взятого в количестве 5% от веса кислоты **. J
Выход чистой перегнанной салициловой кислоты из 94 г фенолаг|
достигает 125 г.
Технические примечания. В технике применяется та же аппаратура, |
что и в лаборатории, с тем различием, что с самого начала пользуются очень сильнойа
мешалкой и шарами для дробления, для того чтобы соль для измельчения нс приходи-]
* Едкий натр, совершенно не содержащий углекислоты, получают, растворяя егсщ
в равном объеме воды ц оставляя на день при 50°. Профильтрованный через асбест я
раствор титруют ио фенолфталеину. ' 4
** Герм.‘'пат. 65131 (1892) (Frdl., Ill, 826). 1
п-Аминосалициловая кислота
141
лось извлекать из автоклава. Для этой цели применяют также особую мешалку с вхо-
дящими одна в другую лопастями; в этом случае отпадает необходимость в шарах. Для
очистки салициловую кислоту можно также сублимировать и токе горячего воздуха;
при этом получают довольно красивый, однако не совсем чистый продукт. Выход кра-
сителей из перегнанной кислоты всегда выше. Реакция протекает почти количественно,
н из 94 кг фенола получают до 137 кг салициловой кислоты.
Аналогичным образом получают о-крезотичовую кислоту из о-крезола. В этом
случае, однако, операцию всегда надо доводить сразу до конца, так как натриевая
соль о-крезола самовозгорается. Около 20% о-крезола возвращается в неизмененном
виде, и крезотиновую кислоту необходимо переосаждать нз воды *. Тем не менее она
обходится не дороже салициловой кислоты, потому что более низкие выходы крезо-
тнновой кислоты окупаются более дешевой ценой крезола.
Салициловая п крезотицовля кислоты используются для получения азокрасите-
лей и трифенилметановых красителей типа аурича (эриохромазурол). Эти кислоты
обусловливают способность красителя соединяться с металлическими протравами,
в частности хромовой, а также образовывать комплексные соединения с солями соот-
ветствующих металлов в субстанции.
Как известно, салициловая кислота находит также широкое применение в фар-
мацевтической промышленности.
По методу Кольбе — Шмита прн температурах выше 2203 получают из ^-нафтола
технически очень важную 2-окси-З-иафтойную кислоту.
n-Аминосалициловая кислота
Салициловая кислота при нитровании не образует однородного продукта и не
гладко нитрозируется. Поэтому я-амнносалициловую кислоту в большинстве случаев
получают восстановлением азокрасителя, полученного из диазотированного анилина
и салициловой кислоты:
КН3 -|- 2НС1 + NaNO3 — > / N = N р’ -4- NaCI ф- 2Н2О
/ У ~N=Np! ф- / "у—ONa —> / N=N—/ ОН ф- NaCI
4COONa XCOONa
/ X_N=N—ОН 4- 2NaBS2O4 ф- 4NaOH —>
XCOONa
—> NHa ф- H3N-/ >-OH 4Na2SO3
XCOONa
а) Получение а з\о красителя
18,6 г (0,2 моля) -анилина растворяют в смеси 45 мл концен-
трированной соляной кислоты (уд. вес 1,17) и 45 мл воды.
Охлаждают смесью льда с солью до 0° и при энергичном перемешивании
медленно, по каплям, прибавляют раствор 14 г нитрита натрия
в 40 мл воды, следя за тем, чтобы температура ие поднималась выше 2°.
После окончания прибавления перемешивают еще в течение 10 мин., за-
тем, убедившись, что раствор имеет сильнокислую реакцию на конго
и дает слабую нитритную реакцию на йодокрахмальную бумажку **, осто-
рожно при перемешивании внбсяТ'4чг кальцинированной соды для ней-
трализации избытка соляной кислотйц Раствор после этого должен еще
иметь слегка слабокислую реакцию конго. Его приливают по каплям
* Растворяют в соде, осаждают пря кипении с разбавленной соляной кислотой и
фильтруют горячей. у •
** Если' этого нет, то надо пря/бавить еще несколько капель раствора нитрита
натрия.
Промежуточные
проаукты
лрп перемешивании в охлажденный до 0° раствор 28 г (0,2 моля0,4 г
избытка) с а л нцн л о в о и к п с л о т ы в 33 мл едкого натра (уд.
вес. 1,38) н 67 мл воды, к которому прибавлено еще 2 а кальцинирован-
ной соды. Внешним охлаждением поддерживают температуру не выше 5°.
Сочетание тотчас же начинается, раствор вначале окрашивается в интен-
сивный 'желтый цвет и ди азосоед иной и е мгновенно исчезает. Когда при-
бавлена примерно 'половина раствора, начинает выделяться краситель, и
вскоре образуется густая коричнево-желтая масса. Сочетание протекает
теперь медленно, и после приливания всего дпазораствора следует пере-
мешивать еще в течение 5—6 час. при 0 -5° до полного исчезновения
диазосоединения. Краситель почти полностью осаждается в виде мелких
кристаллов; его при желании легко можно получить в чистом .виде после
отсасывания и промывки кристаллов раствором поваренной соли. Для
проведения реакции 'восстановления такое выделение, однако, ие обя-
зательно.
Вос с т а новление
Краситель, полученный в виде густой массы, смешивают с 80 мл
едкого натра (уд. вес 1,38) и нагревают до 80°. При этом полу-
чается прозрачный раствор, в который вносят при 80—90° и перемеши-
вании в течение часа 80 а порошкообразного гидросульфита
натрия*. Раствор светлеет н через несколько минут после прибавле-
ния всего количества гидросульфита становится бесцветным. Если,
однако, через L/4 часа раствор не обесцветится, то это значит, что в реак-
цию был взят гидросульфит низкого качества **. В этом случае следует
прибавить его еще немного до тех пор, пока раствор не обесцветится.
Если необходимо, то прибавляют, кроме того, еще немного раствора
едкого натра до сильцощслочцой реакции на фенолфталеиновую бумажку.
Как только раствор станет бесцветным, что означает конец восстановле-
ния, отгоняют образовавшийся анилин с водяным паром; при этом пере-
гонную колбу нагревают так, чтобы объем жидкости оставался прибли-
зительно постоянным (около 0,5 л). После полной отгонки анилина
оставшуюся в колбе жидкость фильтруют через складчатый фильтр и
медленно, хорошо перемешивая, прибавляют в еще горячий фильтрат
концентрированную соля н у ю к и с л о т у до слабокислой реакции па
лакмус; если исходить из указанных количеств, то для этого требуется
около 80 мл соляной кислоты (уд. вес 1,17), Лминосалиццдовая кислота
выделяется в виде отчетливо кристаллического сероватого осадка. Сле-
дует избегать избытка соляной кислоты, так как аминосалициловая кис-
лота в ней вновь растворится. Чтобы избежать этого, соляной кислотой
нейтрализуют только большую часть щелочи и подкисляют под конец
уксусной кислотой. На следующий день густую массу отсасывают па )
нутч-фидьтре, хорошо промывают холодной водой и сушат при 80°.
Выход 26—28 а, т. е. около 85—90% от теории. Продукт'растворяется
как в сильно разбавленной соляной кислоте, так и в щелочи с образо-
ванием прозрачных растворов. ;
Примечания. Салициловая кислота относится к соединениям, которые
трудно сочетаются. Для того чтобы сочетание протекало относительно быстро и .
гладко, необходимо работать с возможно более концентрированмымп растворами (см. ;
* Вследствие ядовитости выделяющихся паров анилина реакцию проводят в хо-
рошо действующем нытяжиоМ шкафу,
** безводный пидроаульфит натрия, цока оц абсолютно сухой, вполне устойчив
даже в присутствии кислорода; при доступе влаги о и быстро разлагается, выделяя
SO2 ц сцекаясь. Хороший гидросульфит должен быть рыхлым порошком без запаха.
стр. 227). Поэтому при диазотировании. так же как и при сочетании, не следует при-
бавлять лед в реакционную смесь, как это обычно делают, а надо охлаждать ее
только снаружи.
Азокраситель можно восстанавливать также другими восстановителями, напри-
мер железом в присутствии небольшого количества кислоты, или цинковой пылью
н щелочной среде. Гидросульфит дает, однако, лучшие результаты. Вместо иродаж-
погс) сухого гидросульфита можно также применять сырую реакционную смесь, полу-
чаемую ДСЙСТ1И1СМ сернистой кпслоть| на смесь цинковой пыли и едкого натра.
Лмлпосалициловая кислота, подобно салициловой кислоте, применяется для по-
лучения азокрасителей, которым она сообщает протравные свойства.
11. Производные» толуола
Беизальхлорид и бензальдегид из толуола
а) Беизальхлорид
В приборе для хлорирования, изображенном на рис. 5, стр. 62, нагре-
вают до кипения 460 с (5 молен) сухого толуола и 10 г пятнхло-
р исто го фосфора и пропускают ток сухого хлора до тех пор,
пока не будет достигнут привес 345 а *, что происходит примерно через
6 час. Образовавшуюся смесь неизмененного толуола, бензил- бензаль- и
бензотрнхлорнда перегоняют па колонке со стеклянными бусами, от-
дельно отбирая фракцию с т. кип. 160—225°, Она содержит в основном
беизальхлорид с т. кип. 204э, а также немного бензил- и бензотрнхлорида.
Точной перегонкой, особенно в технике, удается удовлетворительно раз-
делить эти вещества.
С НО
СООИ
б) Б е н з. а л вщ е г и д
Беизальхлорид, применяемый для получения бензальдегида, не дол-
жен содержать бензилхлорида, поэтом? Сначала его тщательно перего-
няют, отбрасывая фракцию, кипящую ниже 180°.
161 г (1 моль) бепзальхлорида, содержащего немного бензо-
трихлорида, нагревают при 30° и перемешивании в течение получаса
в маленькой стеклянной колбе с 0,5 г желез по го порошка. Затем
прибавляют 25 мл воды и осторожно нагревают, пока примерно при 100°
не начнет выделяться соляная кислота. Некоторое время реакция про-
текает сама собой; ее доводят до конца, слегка подогревая. Затем при-
бавляют соды до отчетливой-щелочной реакции па лакмус и отгоняют
бензальдегид с водяным паром,^Остаток в колбе фильтруют и подкис-
ляют соляной кислотой до постоянной кислой реакции; по охлаждении
выпадает образовавшаяся чисто /белая бензойная кислота. Дистиллят,
-—— • ---------.. /
* Солнечный илн ультрафиолетовый свет чрезвычайно облегчает хлорирование
в боковую цс[|Ь, особсчпю в случае/хлор толуол о и (см. стр. !*>) В лаборатории
с успехом применяют лампу мощностью 600--1000 вт.
1-?4 I. Промежуточные продукты
перегнанный с водяным паром, содержит, помимо бензальдегида, также
некоторое другие вещества, которые нельзя полностью удалить при пере-
гонке. Поэтому весь дистиллят растворяют в растворе технического би- ;
сульфита натрия и после длительного стояния отделяют маслянистый слой.
В зависимости от количества воды берут от 230 до 350 г б ц с у л ь ф и т а .
натрия (с содержанием 25% SOj). К осветленной жидкости цриба- 5
вляют соду цлн раствор едкого и а т р а до ясно щелочной реакции, , !
снова отделяют в делительной воронке и перегоняют, наконец, прн атмо- j
сферном давлении. 1
Выход бензойной кислоты около 12 г; бензальдегида—до 80 г]
(т. кип. 178—179°).
Технические примечания. Толуол в отличие от бензола нельзя хлори- J
ропать в железных сосудах, так как в присутствии железа хлор вступает в бензольное з
ядро. Поэтому при хлорировании толуола необходимо применять эмалированную, J
фарфоровую или стеклянную аппаратуру (см. 2,6-Дихлорбепзальхлорид, стр. 146). ч
Часто реакцию проводят, не прибавляя пдтихлорпстого фосфора, так как оп лишь 1
ускоряет процесс и не является, таким образом, всегда обязательным. Разложение |
бензальхлорида проводят в медной аппаратуре, а бензальдегид отделяют в больших и
освинцованных делительных воронках со смотровыми стеклами. Вышеописанный ме- |
тод * совершенно вытеснил старый метод получения бензальдегида из хлористого бен- 1
зила действием воды и нитрата свинца. j
В проиэнодствс распространен также другой способ, условия которого весьма |
благоприятствуют образованию бензойной кислоты, являющейся цепным продуктом. 1
Он состоит в окислении толуола двуокисью марганца или манганитом в концентри-
рованной серной кислоте (см. ксиленовый голубой ФауС — кислотный ярко-голубой 3, |
стр. 269). |
Другой метод состоит н том, что толуол превращают в бензилхлорид, который 1
омыляют водным раствором соды (при 120°) в бензиловый спирт, а последний пере- |
водят в бензальдегид бихроматом в 80%-ной серной кислоте. |
Бензальдегид является не только промежуточным продуктом для получения |
различных трифенилметановых красителей, по оег применяется также в еще больших а
количествах в качестве отдушки для так называемого миндального мыла. Но дешевые I
сорта этого мыла фальсифицируются нитробензолом (мирбановое масло); фальенфи- я
кацию узнают по пожелтению мыла. 1
2,6- Дихлорбензальдегид1 из о-нитротолуола |
сн3
а) Н и т р о х
Хлорирование о-нитротолуола приводит к образованию приблизительно г/з 2,6-
и ’А 2,4-нтпрохлортолуолов. Получающийся из 2,4-нитрохлортолуола 2,4-дихлорбен-
.зальдегид не дает пенных красителей, и поэтому целесообразно изомерные нитрохлор-
толуолы разделять тщательной фракциоищщ перегонкой в вакууме.
* Герм. пат. 85493 (Гг(Ц., IV. 115).
G i п d г р ц х L., Hclv. Cpim, Acla, XII, 927.
Хлорирование с успехом проводят в сравнительно высоком и узком
стеклянном цилиндре (см. рис. 5, стр. 62), снабженном трубкой для про-
пускания хлора, термометром и обратным холодильником. В этот сосуд
помещают 5 г железного порошка и 0,5 г иода и затем про-
пускают хлор, чтобы образовалось иодохлорное железо. Через не-
сколько минут прибавляют 548 г (4 моля) абсолютно сухого о-нитро-
толуола и пропускают сильный ток сухого хлора. Иодохлорное железо
вскоре растворяется, образуя темно-коричневый раствор, н темпера-
тура очень быстро поднимается. Во избежание слишком сильного разо-
гревания реакционный сосуд помещают в воду, нагретую до 50—60°. Хлор
прекращают пропускать, когда поглотится 95% рассчитанного количе-
ства (131 а), что происходит примерно через 4 часа. В реакционной смеси
остаются растворенными в значительных количествах хлор и хлористый
водород, которые необходимо удалить перед взвешиванием путем отсасы-
вания. Полученный тем неокрашенный продукт хлорирования тщательно
промывают в делительной воронке разбавленной соляной кислотой, затем
водой и под конец отмывают кислоту щелочью. Фракционируют в ва-
кууме на хорошо действующей колонке, снабженной дефлегматором, как
это описано па стр. 305; охлаждение дефлегматора регулируют таким
образом, чтобы на одну отгоняющуюся каплю в колонку стекало
15—20 капель флегмы. Сначала отгоняется иепрореагировавший о-нитро-
толуол (около 50—60 г), затем температура быстро повышается до 114,6°
(прн 11 мм). Первая фракция (около 250 г) является химически чистым
2,6-нитрохлортолуолом (т. пл. 37е). Из последующих фракций центри-
фугированием в фарфоровой центрифуге выделяют также часть 2,4-иитро-
хлортолуола в чистом виде (около 90 а; т. пл. также 37°). Отцентрифуги-
рованную эвтектику (около 260 г) можно снова подвергнуть фракционной
перегонке. На производстве ее перегоняют~вместе со следующей партией.
б) 2,6 - X л о р т о л у и д и и
Нитрохлортолуол восстанавливают по методу Бешапа. К кипящей
смеси 100 г мельчайшего железного порошка, 200 мл воды и
20 г технической соляной кислоты прибавляют по каплям в тече-
ние 2 час. 100 г нитро производного. Реакционный сосуд поме-
щают в масляную баню, прибавляют 20 г соды, нагревают с нисходя-
щим холодильником до 200е в баие и отгоняют хлортолуидин с перегре-
тым до 140° водяным паром. Основание целиком легко удается отогнать
с тремя частями воды. Его отделяют в делительной воронке и для
очистки перегоняют в вакууме: т. кип. 105—110° (10 мм), 240° при атмо-
сфериом давлении.
Перегонка не является абсолютно необходимой; ее рекомендуют
только для полного удаления железа.
Выход составляет около 94 % от теории.
в) 2,6 - Д н х л о р т о л у о л
Растворяют 141,5 г (1 моль) 2,6-х л о р т о л у н д и и а в 1 л воды и
450 г 30%-ной соляной кислоты при 80° и раствору дают при пере-
мешивании охладиться до 30°. Затем прибавляют лсд до тех пор, пока
температура ис понизится до 5е. Часть солянокислого хлортолуидина вы-
падает. Диазотируют, прибавляя 70 г 100%-ного нитрита натрия,
растворенного в 200 мл воды (о диазотировании см. стр, 218 и далее).
Температура может повыситься до 16°; объем должен составлять около
1,4 л. Диазотирование можно считать оконченным, ес,-й реакция иа йодо-
крахмальную бумажку не исчезает через 10 мин.
Раствор соли диазония в течение получаса прибавляют к раствору
хлористой меди, который готовят, пропуская сернистый газ в р ас-
твор 200 г медного купороса н 200 г поваренной соли ,
в 800 ли воды. Избыток сернистого газа из раствора хлористой .
меди надо предварительно удалить кипячением *,
Раствор хлористой меди лучше всего нагреть в керамиковом сосуде, 1
пропуская пар, и прибавить к нему прн механическом перемешивании рас- J
твор солн диазония. Для того чтобы не происходило потерь дихлор- J
толуола, сосуд должен быть хорошо закрыт и температура ие должна |
превышать 95°. Раствор по окончании реакции переносят в четырехлитро- 1
вую стеклянную колбу и отгоняют дихлортолуол с водяным паром. Отго- J
ияется около 141 г, т. е. 88% от теории.
Полученный таким образом продукт еще недостаточно чист. Поэтому
его встряхивают в делительной воронке сначала с 5%-ами серной кисло-i
той (уд. вес 1,84), затем промывают водой и, наконец, очищают два раза J
40%-иым раствором едкого иатра. Перегоняют, собирая фракцию |
185—192°. |
г) 2,6 - Д и х л о р б е и з а л ь х л о р и д |
Хлорирование дихлортолуола в дихлорбензальхлорид в лаборатории 1
осуществляется очень просто. В сухой кипящий дихлортолуол про- |
пускают, по возможности иа солнечном свету, хлор до привеса в 69 г-1
иа 161 г дихлортолуола, что достигается обычно через 2 часа. Сосуд дол- j
жен быть сиабжеи очень хорошим обратным холодильником, чтобы выде- J
ляющнйся хлористый водород ие увлекал с собой бензальхлорид. Про- 1
дукт перегоняют в вакууме. Прн 116—119° (16 мм) перегоняется Я
около 1%, при 120—130° около 95% д и х л о р б е и з а л ь х л о р и д а; .3
остаток представляет собой полихлориды и смолу. При атмосферном |
давлении 2,6-днхлорбеизальхлорид кипит при 250°. |
д) Омыление 2,6-дихлорбеизальхлорида
2,6-Дихлор б сиз альхлорид омыляется значительно труднее бензаль- -я
хлорида. Так, он не омыляется (ни водой н железом, ин известью или '1
едким кали даже под давлением при 150°. Альдегид удастся получить 1
лишь омылением концентрированиой серной кислотой. Однако при этом 1
значительная часть осмоляется. я
100 г 2,6-д ихлорбеизальхлорида и 200 г с е р и о й к и с- 1
лоты (уд. вес 1,84) перемешивают в течение 12 час. при 55°. Раствор я
выливают в 1 л воды,' сливают с разбавленной серной кислоты и пер его- Я
цяют с водяным паром. Получают, таким образом, около 30 г чистого
2,6-д и х л о р б е и з а л ь д е г и д а с т. пл. 7 Г. Я
Технические примечания. За последние годы 2,6-дихлор бензальдегид 'Я
стал довольно важным Промежуточным продуктом, так как он является исходным :-Я
веществом для получения многих красителей ряда аурина (эриохромаэурол и т. д.)..<Я
Он интересен с технической точки зрения, так как его получают с помощью трех -Ж
* Раствор хлористой меди можно также получать следующим образом.- 100 а-.-Я
медного купороса растворяют в 0,5 л воды, прибавляют 50 е цинковой пыли до пол- Ж
ного осаждения меди н жидкость сливают. Осадок нагревают с разбавленной соля- Я
ной кислотой до тех пор, пока цинй полностью не растворится, прибавляют 100 г по- Я
варенной соли и еще „раз раствор 100 г медного купороса и нагревают четверть часа Я
при 80°. Я
По Другому способу кипятят с обратным холодильником солянокислый раствор Я
хлорной меди с медными 'стружками до обесцвечивания раствора и сливают его Я
с оставшейся меди. Я
Бензидин-3,3'-дикарбоновая кислота
147
методов хлорирования. Осуществление двух первых методов хлорирования даже в про-
изводственном масштабе не вызывает трудностей, однако третий не легко осущест-
вить, так как большие стеклянные сосуды часто оказываются недолговечными, а же-
лезную, медную, оловянную или другую подобную аппаратуру нельзя применять;
эмаль же при такой высокой температуре растрескивается. Поэтому применяют
фарфоровые сосуды или сосуды из стекла пирекс, которые нагревают на песчаной
бане перегретым водяным паром. Можно также обогревать газом. Паровой обогрев
обеспечивает, р случае если лопается стекло, лучшую защиту от пожара, однако
в стальных трубках, обогреваемых паром, возникает столь высокое давление (200 ат),
что не исключена опасность взрыва. Поэтому предпочитают нагревать просто на пес-
чаной бане.
Следы железа (заводская пыль) или сурьмы (из резиновых пробок или трубок)
препятствуют хлорированию в боковую цепь.
Получение 2,6-дихлортолуола является редким примером применения реакции
Зандмейера в технике. Ранее в промышленности получали этим методом также
о-хлортолуол (который затем Превращают в о-хлорбензальдегид)1 из о-толуидина. Од-
нако в этом случае лучше использовать метод *, осуществляемый по следующей схеме:
и5о
----.—:------>
конц. EaSQ4
SO.C1
Пар
дрн
сн3
Если имеется дешевый n-толуолсульфохлорид, то его сначала омыляют концен-
трированной еервой кислотой, после чего можно гладко хлорировать в сернокислом
растворе в присутствии железа как катализатора в орто-положенйе по отношению
к СНз-группе, В заключение продувкой водяным паром отщепляют сульфогрулпу
и получают .с отличным выходом о-хлортолуол. Этот метод, малопригодный в лабо-
ратории, дает в Производственных условиях превосходные результаты.
В о-хлорзамешенном бензальдегиде атом хлора легко может замещаться сульфо-
группой при нагревании хлорбензальдегида с сульфитом натрия при 150°. Сульфирован-
ные в орто-положение бензальдегиды дают устойчивые к щелочам трифеннлметановые
красители (патентованный голубой, кислотный ярко-голубой 3, эриоглауцин, ксилено-
вый голубой).
Прн работе з большом масштабе продукты, полученные в промежуточных ста-
диях процесса, не перегоняют. Необходимо перегонять лишь дихлортолуол, чтобы:
получить его совершенно сухим.
Другие соединения достаточна лишь отделить от маточного раствора в гомо-
генно освинцованных делительных воронках. Медные соли в растворе при помощи
цинковой пыли или железа снова превращают в хлористую медь, причем потери пр»
этом редко превышают 2%.
2-Хлор-6-ниТротолуол и 2 хлор-6-т\)луидин имеют также техническое значение.
Первый является исходным веществом при получении 4,4'-дихлориндиго, которое
дальнейшим хлорированием или бромированием превращают в 4,5,4/,5'-тетрагалоиди-
рованное индиго (например, очень зеленого оттенка яркое индиго 4Г), 2-Хлор-6-толу-
идии под названием основание Дл я прочно алого Т Р находит примене-
ние для получения азокрасителей на волокне (нафтолы АС — азотолы). Изомерный
4-хлор-2-толуидин, получающийся восстановлением побочно образующегося 4-хлор-
2-ннтротолуола, применяется под названием основание для прочного крас-
ного КБ (азоамина красного С) с той же целью.
Бензид ии-3,3'-дикарбоновая кислота из о-иитробензойной кислоты
НС!
Герм. пат. 294638 (Frdl., XII, 9D8J.
118
1. Промежуточные продукты
33,4 г (0,2 моля) чистой о-и итр обеизойной кислоты* рас-
творяют при нагревании до 100° в 120 мл раствора едкого иатра
(уд. вес 1,38) и 40 мл воды, а затем при энергичном перемешивании
очень небольшими порциями вносят цинковую пыль таким образом,
чтобы температура реакционной смеси без дальнейшего нагревания оста-
валась между 100 и 105°. Цинковую пыль прибавляют до тех пор, пока
раствор, который сначала окрашен в темно-коричнево-желтый цвет,
опять не обесцветится. Обычно требуется -в зависимости от качества
40—50 г цинковой пыли, которую прибавляют примерно в течение чет-
верти часа. После того как раствор обесцветится, его разбавляют 600 мл
горячей воды, отфильтровывают и а иутч-фильтре избыточную цинковую
пыль и промывают ее горячей водой. К еще теплому фильтрату прибав-
ляют 20 мл 2 и. уксусной кислоты и несколько капель раствора
бисульфита (чтобы предохранить раствор от окисления), а затем по
каплям при хорошем перемешивании 140 мл соляной кислоты
(уд. вес 1,17). Лакмусовая бумажка должна сильно краснеть, однако
конго не должно еще синеть, для того чтобы преждевременно не происхо-
дила перегруппировка (предварительное прибавление уксусной кислоты
позволяет легче достичь нужной кислотности раствора). После того как
профильтрованная проба пе дает осадка при прибавлении уксусной кис-
лоты — в противном случае прибавляют еще немного кислоты,— раствору
дают охладиться; выделившуюся в кристаллической, хорошо фильтрую-
щейся форме гидразобензойную кислоту отсасывают и тщательно про-
мывают холодной водой. Хорошо отжатый светло-коричневый осадок
(около 50 а) растирают в однородную пасту с 80 мл воды, смешивают
с 60 мл соляной кислоты (уд. вес 1,17) и нагревают при перемешивая ни
полчаса прн 95—100°, добавляя воду взамен испарившейся. При этом
все переходит в раствор, кроме незначительного остатка темного’ цвета,
который отфильтровывают на нутч-фильтре и промывают горячей смесью
15 мл концентрированной соляной кислоты и 30 мл воды. Окрашенный
в фиолетовый цвет фильтрат снова нагревают до 70—80°, нейтрализуют
при перемешивании аммиаком (около 50—60 мл 25 %-ного раствора)
до исчезновения кислой реакции иа конго и окончательно осаждают, при-
бавляя раствор 30 г кристаллического уксуснокислого натрия
в 70 мл воды. Профильтрованная проба опять-таки ие должна давать
осадка пи с уксусной кислотой, ни с раствором уксуснокислого натрия.
Выделившуюся бензидинднкарбоновую кислоту отфильтровывают еще
в теплом состоянии, хорошо промывают теплой водой и сушат в паровом
сушильном шкафу.
Выход бенз идин дикар бонов ой кислоты, получающейся в виде зеле-
новато-серого порошка, около 26 а, т. е. 95% от теории.
Можно получить очень чистый желтовато-зелеиоватый продукт, если после пере-
группировки солянокислый фильтрат охладить, отсосать выкристаллизовавшуюся хло- ,
ристоводородиую соль, снова размешать се с горячей водой и перевести в свобод-
ную бензидиндикарбоновую кислоту прибавлением уксуснокислого натрия. Из ма- ;
точного раствора, полученного после отсасывания хлористоводородной соли, остаток >
продукта можно осадить, как это было описано выше. •;
Технические примечания. бис-Диазотированная бензидиндикарбоновая
кислота дает с двумя молями 1,8-а минон а ф тол-2,4-ди сульфокислоты (чикаго-кислота, J
см. стр. 194) дисаэокраситсль, медный комплекс которого применяется как яркий си- 1
ний, чрезвычайно светопрочный краситель для вискозного щелка. *
* Полученной окислением о-нитротолуола двуокисью марганца в серной кислоте.
См. герм. пат. 179689 (Frdl., VIII, 151).
2-Нитро-4-толуидин из /г-толуидина
си.
CH,
NH3
NH-,
53,5 г (0,5 моля) л- то л у и ди иа вносят при перемешивании в 1000 г
концентрированной серной кис л о ты (уд. вес 1,84) с такой
скоростью, чтобы без внешнего охлаждения температура реакционной
смеси не превышала 40°. В этих условиях п-толуидин быстро раство-
ряется. Раствор охлаждают смесью льда с солью * до —5° и при пере-
мешивании медленно по каплям прибавляют смесь 34 г 93 %-ной азотной
кислоты, не содержащей о кис лов азота, и 66 г концентри-
рован ной серной кислоты (уд. вес 1,84), следя за тем, чтобы
температура смеси не превышала 0°. Если нет готовой нитрующей смеси
илн азотной кислоты, не содержащей окислов азота, то ее получают про-
пусканием сухого тока воздуха при нагревании до 40—50° в возможно
более 'концентрированную азотную кислоту до ее полного обесцвечивания.
Затем кислоту охлаждают в токе воздуха, определяют процентное содер-
жание титрованием пробы нормальным раствором щелочи, отвешивают
количество, соответствующее 31,5 г HNO3, и медленно при охлаждении
прибавляют примерно двойное количество концентрированной серной
кислоты. Серная кислота, в которой растворяют толуидин н из которой
готовят нитрующую смесь, естественно, ие должна содержать SOs, в про-
тивном случае будет образовываться опять азотистая кислота или нитро-
знл-серная кислота.
После полного прибавления нитрующей смеси перемешивают еще
в течение 5—6 час., причем температура не должна превышать 0° до тех
пор, пока анализ пробы нитрометром не покажет, что израсходовано все
количество азотной кислоты. После этого реакционную массу выливают
прн перемешивании на лед £2500 г). Большая часть трудно растворимой
сернокислой соли 2-иитро-4-аминотолуола выделяется в виде красновато-
бсльгх кристаллов. Через несколько часов осадок хорошо отсасывают на
иутч-фильтре и заливают холодной водой для удаления следов кислоты.
Затем осадок снова растворяют^ кипящей воде, отфильтровывают от не-
большого смолистого остатка и еще теплому раствору прибавляют ам-
миак до щелочной реакции. Пос^е охлаждения оранжево-желтый кристал-
лический осадок свободного орйовання отфильтровывают на путч-фильтре,
хорошо промывают холодной''водой и сушат при комнатной температуре
Выход 60—62 а;т. е. около 80% от теории. Т. пл. 76—77°.
Можно выделить еще некоторое количество вещества, нейтрализуя аммиаков
сернокислый маточный раствор. Сначала раствор следует нейтрализовать так, чтобы
на конго бумажке оставалась еще слабая синяя окраска. При охлаждении разогрев-
шегося раствора выделяется немного сернокислого нитротолуидина, загрязненного
смолой. Осадок отфильтровывают и переводят, как и основную часть, в свободно^
основание. Фильтрат обрабатывают затем аммиаком до щелочной реакции, причем
выпадает еще несколько граммов основания. Вторая и третья фракции плавятся лиш^
на 1° ниже, чем основная фракция. Вместе они составляют 7—8 г, т. е. 10%, так чтс
общий выход достигает 90%.
Общие замечания. Первичные ароматические амины, основные свойству
которых не слишком сильно ослаблены отрицательными заместителями, часто можне
довольно гладко нитровать, не ацетилируя аминогруппу и не защищая ее другиь
подобным способом; в этом случае амины растворяют в очень большом количеству
* CM. сноску на стр. 120.
150
Промежуточные продукты
концентрированной серной кислоты (берут по крайней мере десятикратное количе-
ство) и нитруют при возможно низкой температуре, во всяком случае ниже 0°, ни-
трующей смесью, не содержащей следов азотистой кислоты.
В этих условиях ориентирующее влияние аминогруппы настолько мало, что поло-
жение, которое занимает нитрогруппа, определяется другими находящимися в моле-
куле заместителями. У соединений, содержащих в пара-положении к аминогруппе
атом галоида или алкильную, арильную или алкоксильную группы, реакция протекает
практически лишь в одном направлении и нитрогруппа становится в орто-положение
к этим заместителям и, следовательно, в л/sra-положение относительно аминогруппы.
Так происходит в случае «-толуидина, л-анизидина, «-фенетиднна, п-хлоранилина,
а также диаминов, например бензидин, 4,4'-диаминодифепилметан и др. Если один
из упомянутых заместителей находится в орто-положении к аминогруппе, то нитро-
группа входит преимущественно в лпрд-положсние к нему, но также в некоторой сте-
пени и в свободное орто-положение. Так, из о-толуидина получается 4-нитро-2-амино-
толуол и немного б-ннтро-2-амннотолуода; о-анизидин превращается главным образом
в 4-нитро-2-аминоанизол и т. д. Указанное выше ориентирующее влияние наблю-
дается также при нитровании вторичных и третичных оснований в присутствии очень
больших количеств концентрированной серной кислоты.
Сернокислая соль пронитрованного основания лишь редко является настолько
трудно растворимой, что она сразу же выделяется при разбавлении реакционной
массы, как это описано, например, в предыдущем случае. Иногда поваренной солью
осаждают хлористоводородную соль. Однако чаще всего для выделения продукта
следует нейтрализовать все имеющееся большое количество серной кислоты. Для ней-
трализации не следует брать известь, хотя она и дешева, потому что вместе с основа-
нием будет выпадать также образующийся гипс. В технике применяют окись магния
(из-за очень малого эквивалентного веса ее требуется относительно небольшое коли-
чество): либо нейтрализуют аммиаком, который затем удаляют из фильтрата известью
и-применяют вновь. Рекомендуется проводить нейтрализацию ступенчато, при этом сна-
чала в большинстве случаев выпадают примеси, а затем осаждается чистое вещество.
Дииитростильбеидисульфокислота и диагми костил ьбеидисульфокислота
из «нитротолуола (совместное окисление двух молекул)
а) Дииитростнльб’ендисульфокислота
100 г п-н итротолуола сульфируют так же, как нитробензол
(см. стр. Ш), и сульфокислоту выделяют в виде натриевой соли. Отжа-
тый осадок растворяют в соде и 1 л воды .при 60°. Соды требуется около
50 г, если ее идет больше, то это указывает на то, что соль была плохо
отжата. Раствор отфильтровывают от окиси железа, которая почти
всегда присутствует, разбавляют до 2 л и нагревают до 50°.
Нагревание ведут на водяной бане, причем реакционный сосуд помещают в во.
дяиую баню на подставку, для того чтобы обеспечить более равномерное нагревание,
К раствору при перемешивании в течение получаса прибавляют по каплям 160 г
35%-ного едкого патрй; при этом не должен выпадать нерастворимый осадок.
Затем в течение 3 час., также по каплям прибавляют смесь 300 г 35%-ного едкого
натра и 1700 г 5%-ного раствора гипохлорита натрия. Содержание гипохло-
рита натрия точно определяют титрованием Ае2Оз и берут количество раствора, соот-
ветствующее 85 г NaOCl. Не следует забывать, что устойчив только такой раствор
б-Хлор-З-аминобензойная кислота 151
гипохлорита, который содержит в избытке не менее 5% NaOH; это особенно надо
помнить пр» получении раствора гипохлорита. Температура не должна превышать 60°,
в противном случае образуются желтые красители стильбенового ряда. Рас-
твор оставляют стоять по крайней мере на 4 часа при 55°; в продолжение этого вре-
мени раствор при пробе на подокрахмальную бумажку должен давать отчетливую
реакцию на активный хлор (гипохлорит). Затем раствор охлаждают до 15°, прибавляют
400 гповареннной соли и оставляют стоять в течение дня. Ди нитростильбенди-
сульфокислый натрий, выделяющийся в виде желтого кристаллического осадка, отфиль-
тровывают и промывают небольшим количеством раствора поваренной соли.
Выход неочищенной соли около 100 г.
б) Восстановление
в диамипостильбендисульфокисл.оту
Трудно растворимую натриевую соль динитростилыбендисульфокис-
лоты растворяют в 300 мл горячей воды и одновременно нейтрализуют сво-
бодную щелочь небольшим количеством разбавленной соляной кис-
лоты. Этот раствор в течение получаса приливают к 200 г железных
стружек, протравленных 20 мл 40 % -ной уксус ной к и слоты. Вос-
становление протекает по известной уже схеме (см,, например, стр. 71).
Нейтрализованный содой и отфильтрованный от . железного шлама
раствор подкисляют соляной кислотой до сильнокислой реакции на конго,
при этом выпадает ди аминостиль бенди су ль фоки слота в виде желтоватого
мелкокристаллического осадка. Ее через 10 час. отфильтровывают и хо-
рошо промывают.
Выход из 100 г «-нитротолуола около 70 г 100%-ной сульфо-
кислоты. В противоположность аналогично построенной 2,2'-бензидии-
дисуль фо кислоте она плохо диазотируется.
Технические примечания. Описанный метод получения диаминостиль-
бендисульфокислоты впервые был предложен Грином и с падением цены на хлор пол-
ностью вытеснил метод Леонга рда. Старый метод состоял в восстановлении прямого
желтого К, который получается действием концентрированного раствора едкого натра
на n-нитротолуолсульфокислоту. По этому методу ди аминокислота может быть полу-
чена с выходом в наилучшем случае только 48% от теории, и для восстановления
требуются большие количества цинковой пыли или сернистого аммония. Кроме того,
цо методу Грина продукт получается гораздо более чистый; если не применять слиш-
ком концентрированные растворы, то образующаяся диаминостильбендисульфокислота
не содержит примеси диаминодибензилднеульфокислоты, в присутствии которой обра-
зуется значительно менее интенсивный хризофенин. Наличие дибензильного производ-
ного легко можно установить двумя реакциями. Во-первых, краситель нз Аш-кислоты
и дибензильного производного гораздо краснее, чем полученный из стильбенового
производного, и, во-вторых, «хризофенин» из дибензильного производного при дей-
ствии минеральной кислоты становится не циста синим, а почти красно-фиолетовым.
Относительно малое содержание диаминрдйбензилдисульфокислоты можно тотчас же
определить, сравнивая ,полученные- -образцы с чистым красителем. Окисление в ди-
нитрокислоту проводят в бетонных чанах. Необходимо помнить, что очень малые ко-
личества железа или даже меди, тотчас же разлагают раствор гипохлорита; по этой
же причине не следует применять и дерево.
6-хлор-З-иминобеизоиная кислота
С1 С1 С1
NO3
\ Т. ггл. 165°
Для этой реакции можно применять,
кислоту, образующуюся в качестве побочм
NHa
например, о-хлорбензойную
ГО ПО дукта ппи no.nvu.pwww
152
Л Промежуточные продукты
о-хлорбензойного альдегида. Технический продукт целесообразно перед 1
обработкой предварительно растворить в разбавленном растворе едкого |
натра, профильтровать и снова осадить. J
32 г чистой о-хлорбензойной кислоты растворяют при 1
перемешивании в фарфоровом или железном сосуде в 160 г моногнд- |
рата н охлаждают ниже 0° смесью льда с солью. Затем по каплям |
прибавляют в течение часа смесь 16 а 80%-ной азотной кислоты |
и 40 г 100 %-ной серной кислоты. Температура должна быть ниже 1
0°, в противном случае образуются нежелательные побочные продукты. - 3
К концу реакции частично выделяется иитросоединепие, и реакционную j
массу оставляют на 10—12 час. при комнатной температуре. На следую- ;а
шнй день медленно напревают до 60° и выливают массу на 400 г льда. £
Ннтрохлор бензо иную кислоту отфильтровывают и, лучше всего, дважды J
перекристаллизовывают из 1 л кипящей воды, прн этом удаляется не- J
большая примесь неизмененной хлорбензойной кислоты. В технике от 1
этой операции отказываются, но в лаборатории ее следует проводить. 1
Таким образом получают продукт с т. пл. 164—165°. 1
Выход совсем чистого продукта достигает 92 % от теории, 1
т. е. около 37,5 г. 1
Восстановление. 20,2 г (0,1 моля) нитрохлорбеизойиой 1
кислоты растворяют в 5,5 г соды и 70 мл воды и подкисляют 10 мл 1
40 %-ной у кс у с ной кислоты. Этот раствор прибавляют по каплям .3
к суспензии 100 г ц и н к о в о й п ы л и в 250 мл кипящей воды, к которой- i
прибавлено 4 мл 40 % -пой уксусной кислоты. Непрерывно кипя- |
тят при постоянном перемешивании, причем масса часто сильно велению 1
вается, так что необходимо вестн реакцию в большом сосуде (эмалиро- |
ванный котел емкостью 3 л). 1
Реакцию ведут медленно, так что восстановление продолжается около 2 час. Я
Затем прибавляют в раствор 5 г соды и фильтруют. Упаривают до 200 мл и получают Я
раствор, который непосредственно можно применять для получения азокрасителей, не, .1
выделяя хлораминобензойную кислоту. Выход титруемого вещества составляет т 1
около 92% от теории. „ Я
Если хотят выделить хлораминобензойную кислоту как таковую, то раствор под- 'I
кисляют соляной кислотой, которую не следует брать в избытке. я
12. Галламид и галловая кислота из таннина
Таннин или другое
дубильное веще-
ство па основе гал-
ловой кислоты
(NHt)2SO;i + NHs
он
СООН
Галловая кислота
но-
conh2
Галламид
Важнейшими исходными материалами, из которых получают, галла- |
мид и галловую кислоту, являются чернильные орешкн н сумах (rhus. |
13. Производные бензола из нафталина 153-'
coriaria), дубильные вещества которых либо расщепляют раствором
едкого натра на сахар и талловую кислоту, либо превращают действием
сульфита аммония в сахар, галловую кислоту и галл амид. При этом по-
лучаются приблизительно равные количества амида н кислоты.
200 г т а н н и н а, 200 мл воды, 400 г 20 % -ного аммиака и 100 г-
раствора бисульфита натрия (25% SO2) нагревают в течение'
12 час. на водяной бане при 50° в бутылке из-под минеральной воды
с каучуковым затвором. Для полного растворения содержимое бутылки
несколько раз встряхивают. Затем раствор упаривают до 400 мл в боль-
шой колбе под уменьшенным давлением.
По охлаждении очень осторожно прибавляют соляную кислоту
до точно кислой реакции и а лакмус. Через 24 часа галла мид полностью*
выпадает из раствора. В лаборатории часто бывает необходимо сначала:
сильно охладить небольшую порцию и растереть ее стеклянной палочкой:
для того, чтобы вызвать начало кристаллизации.
Трудно растворимый галЛамид отфильтровывают и хорошо промы-
вают. К маточному раствору прибавляют 100 г 30%-ного раствора ед-
кого натра н аммиак отгоняют в вакууме. Затем раствор снова упа-
ривают до 300 мл и подкисляют концентрированной соляной кислотой до*
чуть кислой реакции иа конго. Натриевая соль галловой кислоты выде-
ляется в течение нескольких суток в виде мелкого кристаллического-
осадка, который отфильтровывают и, не промывая, отжимают. Соль
растворяют в 100 мл воды и осаждают кислоту из растрора соляной,-
кислотой.
Выход галламида и галловой кислоты — по 60 г.
Технические примечания. При работе в больших масштабах применяют
растворы дубильных веществ, которые получают, экстрагируя, по принципу противо-
тока горячей умягченной водой материалы, содержащие дубильные вещества. Эти'
растворы упаривают в вакууме до уд, веса 1,26. Реакцию проводят в больших бетон-
ных чанах, рассчитанных на уменьшенное и повышенное давление. Кристаллизация
галламида длится 10—14 суток, а соли еще дольше. Растворы дубильных веществ,
очень склонны к брожению, и поэтому их следует, особенно летом, быстро перераба-
тывать. Чистоту галламида определяют отговкоя аммиака из отвешенной пробы
с едким натром, поглощением его нормальной соЛяной кислотой и обратным титрова-
нием. Хороший галламид является 92%-ным.
Гйлловая кПслота и ее амид применяются в больших количествах для получениях
оксазинов (см. гаАдаминовый синий).
)
13. Производные бензола из нафталина
Получение фталевого ангидрида каталитическим
। окислением нафталина *
Теплота горе-
ния нафталина
4- Ы31 ккал’,яоль
Теплота горе-
ния фталевого
ангндрина
+ 450 ккал[.ноль
Каталитическое окисление нафталина можно легко осуществить
в лаборатории, однако количества фталевого ангидрида, которые можно-
~~ —7'----------
* Герм. .пат. 347610, 379822 Wohl-Danzig (Frdl., 14, 450); JTlnd. Chem. Soc,, 11,.,
185—196 (1934); 14, 638—643 (1&37).
154
/. Промежуточные продукты
получить с одной операции, весьма незначительны. Важным является
прежде всего поддержание оптимальной температуры и применение под- '
ходящих катализаторов. Если окисление проводят в лаборатории, то ?
аппаратуре следует уделять особое внимание. Необходимо также отме-
тить, что для достижения более высоких выходов продукта реакции реко- ;
мендуется применять пылеуловитель Коттреля, хорошо задерживающий ,;
тонкоизмслъчениые частицы. Без этой аппаратуры всегда теряется часть
продукта реакции.
Получение катализатора
Пемзу измельчают таким образом, чтобы получились кусочки вели-Ц
чиной 3 мм, и просеивают через сито с отверстиими диаметром 3 мм.
Кусочки должны быть по возможности одинакового размера. Неочи-
щенные кусочки пемзы (200 г) вносят в -горячий наиболее концентри-
рованный раствор ванадиевокислого аммония. После того как кусочки
пемзы полностью пропитываются раствором, их вынимают и сушат
в фарфоровой чашке на водяной бане. Должно быть -нанесено при-
близительно 3 г ванадия па 200 г пемзы. Высушенную массу нагре-
вают при 400° до- тех пор, пока -зерна не примут красно-коричневую
окраску. Очень важно, чтобы катализатор не нагревалси выше той тем-
пературы, при которой в дальнейшем будут проводить реакцию. Это
правило относится ко многим каталитическим реакциям: катализа-
тор не следует перегревать выше температуры
реакции.
Проведение реакции. Аппаратура
Схема процесса лучше всего поясняется рис. 28. Нафталин поме-
щают в -сосуд 2, который нагревают до 110°, Через трубку 1 пропускается
Рис. 28, Схема аппаратуры для получения фталевого ангидрида каталитическим
окислением.
/ — подвод воздуха; 2—расплавленный нафталин,- 3 — графитовая баня; 4 — электрообогрев; 5 — заземление;
6 — искровой индуктор; 7—стеклянная вата; 8—- катализатор; 3—шаровой приемник; 10 — алюминиевая
заземленная фольга; 11 — отходящие газы; 12 — термоэлемент для измерения температуры; 13 — металличе-
ская очень Жесткая проволока; один полюс искрового индуктора соединен с проволокой, другой зазем-
лен; 14 — термометр.
ток влажного* воздуха. При окислений очень важно присутствие па-
ров воды. Необходимый ток воздуха -нагнетается проще всего при
помощи обычного водоструйного насоса. Количество- проходящего через
Фталимид из фталевого ангидрида
155.
установку воздуха измеряется известными лабораторными приборами и
потому здесь не описываемыми. Воздух должен пропускаться с такой
скоростью, чтобы через систему за 1 час проходило 200 л воздуха и
5 а нафталина. Важно, чтобы соотношение между воздухом и наф-
талином было таково, чтобы не образовывалось взрывчатой смеси,
обусловливающей потерю фталевого ангидрида. Газообразную смесь
(воздуха с парами нафталина) пропускают через каталитическую
трубку с внутренним диаметром приблизительно 5 см, длина слоя ката-
лизатора — 10 см. Кусочки пемзы удерживаются стеклянной ватой.
Трубка снабжена электрообогревом, обычно применяемым в технической
лаборатории.
Газы, выходящие из каталитической трубки, поступают сначала
в шаровой приемник 9, а затем проходят через пылеуловитель, представ-
ляющий собой стеклянную трубку с внутренним диаметром 8 см. Внут-
ренние стенки трубки обложены гладкой алюминиевой фольгой, которая
легко вынимается. Фольга тщательно заземлена металлической проволо-
кой (водопровод). Через трубку проходит металлическая проволока или
посеребренная стеклянная палочка, по которой подается напряжение
10 000 в. Это напряжение, которое в данном случае может быть напря-
жением (перемени ого. тока, получают любым способом: проще всего с по-
мощью искрового индуктора'ШШ трансформатора с прерывателем. Важно,
чтобы искровое поле индуктора составляло по крайней мере 25 мм. Необ-
ходимо также, чтобы/внутренний диаметр трубки, в которой происходит
улавливание пыли, был настолько велик, чтобы исключалась всякая воз-
можность проскакивания искры между металлической проволокой и ме-
таллической фольгой, так как искра легко может вызвать взрыв. Взрыв
нс опасен, однако прн этом сгорает часть продукта реакции.
После того1 как вся аппаратура собрана и тщательно проверена,
можно* начинать окисление. Для этого нагревают каталитическую трубку
до 450° (термопара) и начинают пропускать смесь воздуха с парами
нафталина. Окисление происходит по мере того, как газы проходят
через катализатор; часть фталевого ангидрида оседает в шаровом прием-
нике, ио основное erb количество задерживается в пылеуловителе Кот-
трелл. Фталевый ангидрид оседает на небольшом участке в самом начале
электрического пылеуловителя в виде красивых игл длиной до 5 см. При
желании процесс можи^ продолжать, и если применять чистый нафталин,
то катализатор в течете нескольких недель не меняет своей активности.
Выход еЮставлйеттоКоло 90—95% от теории. Если поддерживаете и пра-
вильная температура реакции и если скорость пропускания не очень
велика, то получаемый фталевый ангидрид является практически хими-
чески чистым. Прн слишком быстром пропускании нафталина полу-
чается продукт желтоватого или желтого цвета нз-за примеси 1,4-нафто-
хинопа. Время от времени продукт реакции извлекают, вынимая
алюминиевую фольгу, и затем продолжают процесс. В лаборатории за
5 час. можно легко получить 20 г чистого фталевого ангидрида.
Фталимид из фталевого ангидрида
( 1 О + NHs-> i I NH -ь Н2О
В трехгорлую колбу, снабженную термометром, трубкой для подвода
таза н широкой вертикальной отводной трубкой, йВмещают 148 г
{I моль) чистого фталевого ангидрид а.^Ангидрид расплавляют
156 /. Промежуточные продукты
и нагревают далее до 170°. При этой температуре в расплавленную;,
массу начинают пропускать сильный ток аммиака. Аммиак полностью
поглощается; выделяющиеся пары воды увлекают некоторое количество $
возгоняющегося фталевого ангидрида. Время от времени расплавляют
осевший на горле колбы фталевый ангидрид. Пропуская аммиак, темпе-
ратуру медленно повышают до 240° для того, чтобы не застывал обра-J
зующийся фталимид, т. пл. которого около 230°. После того как начнет 1
выделяться аммиак, реакция в основном закончена. Из предосторожности I
аммиак пропускают еще в течение 10 мии. и тотчас же выливают пла^д
в фарфоровую чашку, где он застывает. Получают 130—135 г фталнмиддЦ
с т. пл. 223 (пеислр.), содержащего в виде примеси самое большее следы!
неизмененного фталевого ангидрида. |
.'--1
Антраниловая кислота из фталимида
| | NH -у NaOCI -j- 3NaOH-> | | ф Na С] + Na3CO8 + Н2О’ 1
\/'xCOON’a 1
73,5 г (0,5 моля) тонко измельченного фталимида смешивают;!
с 250 мл (воды и 125 г льда и прибавляют 55 мл раствора едкого натра я
(уд. вес 1,38). После того как все растворится, прибавляют еще 250 г!
льда и затем сразу приливают раствор гипохлорита натрия, не содержа-!
щего хлората, в количестве, соответствующем содержанию 37 г активного'!
хлора; раствор гипохлорита должен содержать 80—90 а активного хлора!
на литр раствора и иа 1 моль NaOCl 2 моля NaOH *, к раствору пред-!
варительно прибавлено еще 20 мл раствора едкого иа т ра!
(уд. вес 1,38). Капля реакционной смеси должна окрашивать несколько!
капель анилиновой воды в слегка фиолетовый цвет. Раствор хорошо!
перемешивают и оставляют стоять во льду иа 3—4 часа, затем прибав^!
ляют несколько1 капель раствора бисульфита натрия, чтобы разложить-!
небольшой избыток гипохлорита, и нагревают до 80°. Прн этой темпе-Я
ратуре осторожно (вспенивание) нейтрализуют концентрированной соля-!
ной кислотой до тех пор, пока лакмусовая бумажка не перестанет синетб'Я
(требуется около 130 мл), отфильтровывают в .горячем состоянии <хгд
возможных неорганических примесей и еще горячий фильтрат подкис-И
ляют 40 мл концентрированной соляной кислоты и 12 мл ледя-^И
ной уксусной кислоты. Следует избегать избытка соляной. кнс«
лоты, так как антраниловая кислота в ней снова может раствориться^
Из теплого еще раствора начинает выделяться кристаллический светлотИ
коричневый осадок, количество которого значительно увеличивается прНД
охлаждении. Оставляют на ночь, затем отсасывают, промывают холодной™
водой и сушат в паровом сушильном шкафу. Я
В ыход 57—58 г, т. е. около 84% от теории; т. пл. 144—145°. Л
Оставшаяся в растворе антраниловая кислота может быть в’ыделенаМ
при помощи уксуснокислой меди в виде почти нерастворимой медноЙД
соли, которую отфильтровывают, затем опять суспендируют в воде и раз-Д
лагают, пропуская сероводород; отфильтровывают сернистую меДйЯ
и фильтрат выпаривают до небольшого объема. При охлаждении выкри-Ж
сталлизовывается еще несколько^ граммов антраниловой кислоты. Я
* Такой раствор можно легко приготовить, пропуская при охлаждении льдомИ
71 г хлора в смесь 400 мл раствора едкого натра (уд, пес 1,38) и 400 г льда. Содер-ЗМ
жание активного хлора определяют йодометрически. Продажный раствор гииохлоритЯц^И
если он долго стоял, содержит значительные количества хлората и для данной селя4И
не пригоден.
a -j j ыд punmp j или re и -444-пх;рг «л-
Б. СОЕДИНЕНИЯ РЯДА НАФТАЛИНА
Для получения всех производных нафталина с хорошими выходами
необходимо в качестве исходного материала применять очень чистый
нафталин. Если нет под рукой чистого нафталина, то его можно очистить
либо перегонкой, либо нагреванием с 5 % -иым (по весу) количеством
концентрированной серной кислоты. Однако в настоящее время можно
получать чистый нафталин от коксохимической промышленности.
14. а-Нитронафталин и а-нафтиламин *
NO., NHo
I
Нафталин в общем значительно более реакционноспособен, чем бен-
зол. Так, например, нитрование нафталина протекает очень энергично
и легко может привести к, образованию полип ит рос о единен ни. С другой
стороны, так как нафталин приходится нитровать при температуре ниже
его температуры пл а в лепир, то азотная кислота в этих условиях не дей-
ствует на большие куски | нафталина. Поэтому необходимо1 нафталин
тонко растирать так, чтоб^ он проходил через сито с 400 отверстиями
па 1 см2 **. ч
В смесь ЮЗ г 62 %-пой азотной кислоты (уд. вес 1,38) и 300 г
80%-иой серной кислоты вносят 128 г нафталина. Непре-
рывно размешивают в течение 6 час. при 50° и повышают, наконец, тем-
пературу в течение 1 часа до 60°, затем охлаждают. Нитронафталии
всплывает иа поверхности кислоты в виде пористой лепешки, которая со-
держит примерно 90—92% а-нитронафталина, около 4—5% Р-нитроиаф-
талина, 2—3% Лрнитронафталина и около 0,5% 2,4-динитро-1-нафтола
(желтый Марциубр).
Сырой продукт расплавляют несколько раз в кипящей воде для
полного удалений кислоты, и одновременно нафталин отгоняется с па-
рами воды._3атбм расплавленный продукт выливают при хорошем пере-
мешивании в холодную воду, где ря и застывает в виде небольших
шариков.
Если хотят получить совершенно чистый нитронафталин, его сушат плавлением
при 120° в воздушной печи. Затем прибавляют ]0%-ное (по весу) количество лигро-
ина с т. кип. 150° (можно также применять технический ксилол или кумол); филь-
труют теплым через фильтр и оставляют па некоторое время в закрытом сосуде.
Образующуюся кристаллическую лепешку переносят на хлопчатобумажную ткань и
хорошо отжимают- Эту операцию очистки следует повторить несколько раз, до тех
пор пока нитронафталин не будет плавиться при 61°. Получается он в виде желтых
блестящих кристаллов. Значительная часть нитронафталина всегда остается в маточ-
ном растворе и может быть выделена оттуда при отгонке растворителя.
Сырой нитрон афта ли.н восстанавливают по Беш ан у железом в при-
сутствии небольшого количества соляной кислоты.
* См. также ДУ f 11 О. N., Chem. Ind., 215 (1887); Paul L., Ztschr. angew.
Chem., 10, 145 (1897). >
** Нафталин, кристаллизующийся листочками/ очень трудно превратить в поро-
шок. Поэтому рекомендуется сначала его расплавить и дать застыть в кристалличе-
скую лепешку, которую гораздо легчо измельчить в порошок, нафталин в виде
слоистых кристаллов.
J. J J J
J Oi
В железный редуктор с якорной мешалкой (см. рис. 11, стр. 72) по- J
мещают 200 г железных стружек, 100 г воды и 10 мл 30 %-ной |
концентрированной соляной кислоты. Нагревают на кипя-а
щей водяной бане и вносят небольшими порциями нитронафталнн, лучше.!
всего так, как это описано на стр. 102 для восстановления 4-хлор-2-нитро- |
фенола. При постоянном перемешивании в течение 4 час, восстанав-ли-J
вают 1 моль (173 г) н и т р о н а ф т а л и и а, считая ла воздушно-сухое"!
вещество. При более быстромЦ
Рис. 29. Чугунный реакционный котел на
1 ати с мешалкой, весом 12 кг, масляная
баня из меди.
проведении процесса может про-я
исходить образование иежела«|
тельных азосоединеиий. К смеси!
прибавляют соду до отчетливой!
щелочной реакции; затем содер-|
жимое реакционного сосуда!
м<тж ио пер енести в чаш ку. |
В лаборатории образовавшийся!
а-иафтиламни лучше всего вы-!
делять перегонкой с перегретым!
водяным паром. Для этого про-j
дукт реакции вместе с водой*!
железом и окисью железа пере- J
носят в котел с масляной баней,J
изображенный иа рис. 29. Воду!
полностью отгоняют, нагревая,^
масляную баию до 200° при по-|
стоя-нном перемешивании, и за-. J
тем пропускают перегретый дф|
250° водя ио й пар (см. рнс. J
23а) *. При быстрой перегонке!
легко удается отогнать с одной!
частью воды ’А—1 часть иа-1
фтиламииа. С основанием все-J
гда перегоняется небольшое ко-1
личество очень тонкого желеэ-J
ного порошка, графита из чугуна н окиси железа. Перегонку кончают,
если с перегретым точно до 260° паром больше ие отгоняется вещества!
или же отгоняется окрашенный продукт. В зависимости о? способа иа-я
греваиия перегонка продолжается 1—1,5 часа. В котле остается черная!
очень тонкая масса, обладающая пирофорными свойствами, поэтому ее|
нельзя просто выбрасывать. Нафтиламии после охлаждения отделяют от|
маточного раствора, расплавляют и сушат при 110° в воздушной печи*1
Лишь после этого приступают к перегоике в вакууме. Получают бесцвет-J
иое кристаллическое основание.
Выход чистого а-нафтиламина (т. пл. 50°) около 110 г из 1 мол;
нафталина.
Продукт можно выделять также следующим образом? после окончания реакций®
восстановления смесь нейтрализуют содой и отгоняют полностью воду в вакууме*!
Остаток трижды экстрагируют бензолом, бензол отгоняют при атмосферном давле^З
нии и затем в вакууме перегоняют a-нафтиламин. J
\Я
Технические примечания 1
а) Нитронафталин. Отработанная при нитровании кислота частично снова воз-Л
вращается в производство. К ней прибавляют концентрированную серную кислоту я:|
* На рисунке схематически изображена аппаратура без мешалки. Для более лег-^
кого разделения окиси железа и нафтиламина смесь рекомендуется перемешивать. 1
повышают, таким образом, ее концентрацию до 80%, Оставшаяся часть отработанной
кислоты применяется для подкисления щелочных плавов и для других целей. При
достаточном измельчении (дезинтеграция при 60°) нафталина нитрование его удается
провести почти количественно. Нитронафталин применяется также для получения
1,5- н 1,8-динитроиафталипов. Баденский анилиновый и содовый завод применял нитро-
нафталин для получения диазосоединения 1-амино-2-нафтол-4-сульфокислоты. Нитро-
нафталин при нагревании с бисульфитом натрия дает 1-нафтиламин-2,4-дисульфокис-
лоту и немного нафтионовой кислоты. Нафтиламиндисульфокислота может диазотиро-
ваться и превращаться далее при действии бикарбоната натрия и гипохлорита натрия,
в диаэосоединение аминонафтолсульфокислоты. Ниже дана схема этой замечательной,,
реакции *.
Этот довольно хороший метод был вытеснен более дешевым способом Занд--
мейера (см. стр. 178).
б) а-Нафтилалшн, Восстановление проводится в аппаратах, неоднократно ранее -
описанных. Так как реакционная масса имеет кашицеобразную консистенцию, то.
нельзя применять пропеллерную или лопастную мешалку, а следует работать с якор-
ной мешалкой, изображенной, например, на рис. 31. Перегонку с водяным паром про-
водят в аппарате, схема которого представлена на рис. 24. Пропускаемый пар почти,
всегда дополнительно нагревают в специальном перегревателе.
При работе в большом масштабе значительную часть я-нафтиламина можно -
просто слить в виде жидкости; остаток железа, окиси железа и я-нафтиламнна сме-
шивают с древесными опилками и несколько раз экстрагируют бензолом. Из смеси
Железа, окиси железа н опилок бензол отгонйЩт с водяным паром; остаток приме-
няется в литейных цехах в качестве связующего при брикетировании железных стру-
жек, используемых в вагранках.
а-Нафтиламин применяется в качестве начальной, конечной и, особенно, сред-
ней азосоставляю14ей для получения многих важных азокрасителей; он используется,
также для получения красителей других классов. а-Нафтиламин является также
исходным соединением и при получении ряда промежуточных продуктов, из которых"
некоторые будут опцсаны в дальнейшем. Подобно анилину, он нашел также новое
интересное применение в другой области. Его применяют, особенно в Северной Аме-
рике и Австралии, в качестве флотационного агента при обогащении бедных руд; про-
цесс заключается в том, что тонко измельченную породу с 0,5%-ным (по несу) коли-
чеством нафтиламина и неочищенного ксилола энергично перемешивают (эмульгируют),
с большим количеством воды. При этом тяжелая руда, несмотря на большой удельный;
вес, поднимается вместе с пеной на поверхность и может быть легко отделена.
* Герм/ лат. 160536, 157325, 156440 (Frdl V11I, 656—657),
160
1. li fjujnejtcyj<j4nmc иуииупля
Феннл- а-нафтиламин
В колбу Клайзена на 3Д литра помещают: 143 г (1 моль) а-на-
фтила мин а, 175 г анилина н 3 г сульфаниловой кислоты.
Колба снабжена термометром, погруженным в реакционную массу,
я трубкой, соединенной со склянкой для поглощения выделяющегося ам-
миака. Отводную трубку колбы закрывают и колбу закрепляют косо так,
чтобы отводная трубка была немного направлена вверх. Смесь нагревают' ;
на голом огне при энергичном кипении в течение 40—48 час.; необходимо
следить за тем, чтобы пары анилина не достигали резиновой пробки,,
которая в противном случае сильно бы разрушалась. Выделяется аммиак;
температура кипения смеси во время реакции повышается от 195 до
215°. После окончания реакции колб)7 закрепляют в вертикальном поло-
жении и содержимое перегоняют в вакууме. Основное количество непро-
реагировавшего анилина (около 80—85 г) перегоняется при 70° (12 мм).
Затем при неравномерном кипении, значительном вспениваини и толчках
перегоняется в незначительном количестве промежуточная фракция
(8—10 а), содержащая, помимо анилина и небольших количеств а-на- <
фтиламина, в основном фенил-а-нафтиламин; последний можно отделить
от первичных аминов обработкой разбавленной соляной кислотой. При
дальнейшем нагревании жидкость начинает кипеть опять равномерно .1
и спокойно; отгоняется желтоватое масло, основная часть которого кипит -;
при 212—216°, а остаток перегоняется при 225°; масло при охлажде- *
нин застывает в бесцветную кристаллическую массу (190—195 a), i
т. пл. 55—60°. Общий выход составляет около 200 г, т. е. 91 % от теории. 4
Продукт перекристаллизовывают из небольшого количества спирта 4
и получают в виде бесцветных игл с т. пл. 62°. /
Фенил-а-нафтнламин применяется так же, как и алкилпроизводные а-нафтил-,;
амина, главным образом для получения основных дифенилнафтилметановых красите- ;
лей (виктория голубого, основного синего). J
а-Нафтол из а-нафтиламииа
ч*Нафтол
143 г л -на фтиламина, 110г серной кис лоты (уд. вес-1,84)
и 1 л воды нагревают-под давлением 14 ат до 200°. Нафтилами» пред-4
варительно расплавляют в горячей воде и затем при энергичном пере-
л кислига из ct-к аерти ламина
101
мешивании приливают тонкой струей кислоту. Автоклав должен быть
освинцован или эмалирован. Крышка может быть железной, так как сер-
ная кислота нелетуча. Нагревать автоклав необходимо .на масляной бане,
иначе, особенно в производстве, трудно избежать перегрева, от которого
свинец легко может расплавиться.
После восьмичасового нагревания охлаждают и отделяют нафтол от
маточного раствора, из которого выделяют сульфат а.ммония. а-Нафтол
расплавляют в небольшом количестве воды и, когда он застынет, отде-
ляют от жидкости. а-Нафтол получается почти химически чистым. Для
полной очистки его можно перегнать в вакууме.
Выход 94—95% от теории; т. пл. 94°.
Технические примечания. Описанный выше способ является самым луч-
шим и наиболее дешевым. Имеется, однако, и другой способ, аналогичный методу по-
лучения ^-нафтола, а-Нафталиисульфокислын натрин плавят с едким натром при
290—300° (не выше). Сульфируют при 80—90° и высаливают из возможно более кон-
центрированного раствора. В этом случае можно также избыток кислоты удалить из-
вестковым молоком или мелом, обработать содой и упаренную натриевую соль пла-
вить. Полученный таким образом з-нафтол, однако, не является чистым.
Нафтионовая кислота из а-нафтиламина
j Нафтионовую кислоту получают из кислого сернокислого нафтиламина так на-
зываемым методом запекания, т, е. длительным нагреванием сухого соединения пре-
имущественно в вакууме.
SO3H
Получение кислого сернокислого нафтиламина
В трехгорлой колбе с капельной воронкой, мешалкой и нисходящим
холодильником нагревают до 120—125° 73,5 г 70%-пой серной кис-
ло ты. и при хорошем перемешивании в течение получаса прибавляют
по каплям нагретый до 50° раствор 75 г 1-нафтиламина в 15 г
бензола. В этих условиях полностью исключается возможность обра-
зования комков нсрастворившегося основания. Бензол при этом непре-
рывно отгоняется. Прозрачная, слегка красноватая жидкость быстро
застывает. Осадок сушат в вакууме при-120° в течение 18 час. При при-
менении 70%-ной серной кислоты • н чистого 1-иафтиламина получают
очень чистый кислый сернокислый нафтиламии, что имеет очень важное
значение для процесса запекания.
Запекание
Превращение кислого сернокислого а-нафтиламипа в нафтионовую
кислоту проводят в аппарате для запекания под вакуумом, представлен-
ном ца рис. 30. В аппарат помещают 75 г тонко растертого кислого
сернокислого нафтиламина и нагревают в течение 8 час. до
180° при 10—-15 мл остаточного давления. После охлаждения получается
светло-серая лепешка, которую растворяют в 500 мл в о д ы с Добавкой
20 г безводной с од ы. Кипятят, фильтруют и раствор экстрагируют
бензолом для удаления не вошедшего в реакцию нафтиламин4, нагре-
бают еще раз до кипения, прибавляют немного соляной кислоты
до начинающегося помутнения, затем прибавляют животный уголь и
фильтруют горячим. Охлажденный раствор подкисляют соляной кисло-
топ. Отфильтровывают выделившуюся бесцветную нафтионовую кислоту
и сушат при 100°. Выход 60—65 г, т. с. 89—95% от теории.
О методе запекания см. стр. 117,
Рис. 30, Аппарат для запекания иод вакуумом,
1 — термометр для масляной бани! 2—медная прокладка; —термометр для
измерения температуры реакционной млеем; 4 — вакуум; .5 — масло; б — свилцочая
прокладка; 7 — вкладыш; 8 — реакционная масса.
1-Нафтол-4-сульфокислота (кислота Невнль-Вннтера)
нз нафтионовой кислоты (реакция Бухерера)
кислота Винтера (1-нафтол-
- 4-сулъфокислота)
100 s 100 %-него нафтионата растворяют в 200 мл воды, при-'-’
бавляют 600 г раствора бисульфита на 'Гр и я (25 % SOa) и кипятят у
в течение одного дня с обратным холодильником. Затем добавляют •
30%-ный едкий натр до покраснения тиазоловой бумажки и кипятят ?
до тех пор, пока не прекратится выделение аммиака. Подкисляют соля-
ной кислотой до постоянной минеральнокислой реакции; по охла-
ждении выделяется кристаллическая кислота Невиль-Винтера. Ее раство-
ряют в воде и отфильтровывают от оставшейся нафтионовой кяслоты.
Выход до 80% от теории.
j<j. ^улофикислоты. нафталина 16;
Нафтолы, так же как и нафтнламины, прн действии сульфита превращаются
в нестойкие промежуточные соединенна, которым Бухсрер, открывший эту реакцию
приписывает строение серии сток в слых эфиров нафтолов (формула I), а Ворожцог
рассматривает цх как продукт присоединения бисульфита к кето-форме нафтола (фор.
мула II). Эти промежуточные соединения при действии аммиака превращаются в со.
отвстствующпе нафтиламцры, а едкие щелочи омыляют их в нафтолы. Поэтому
можно, смотря по обстоятельствам, превращать нафтолы в нафтнламины (см. стр. 176
178) либо, наоборот, нафтнламины прекращать в нафтолы *.
Далее, вместо аммиака можно Применять первичные или вторичные, алифатиче-
ские, а для ^-замещенных также и ароматические амины; таким образом можно иолу,
чать также алкилированные и арилированные нафтнламины,. исходя из первичны^
производных нафтиламина либо из соответствующих нафтолов**.
Реакция Бухерера применима также для некоторых соединений ряда бензола ц
ряда антрацена ***. Однако практическое значение она имеет только в ряду нафта-
лина. Реакция Бухерера применима для производных я- и ^-нафтолов и нафтилами-
нов, но неприменима для а -производных, содержащих в орто- или .мета-положении,
и для 3-производных, содержащих в .чети-положении к окси- или аминогруппе сульфо-
группу. Поэтому, например, у И- или Т-кислоты (2-амино-5-нафтол-7-сульфокислота,
соответственно 2,8,6) вступает в реакцию только аминогруппа, а не оксигруппа, «за-
щищенная» сульфсгруппой, стоящей в лета-положении (см. фенил-у-кислота, Стр, 185},.
В 2,8-диоксинафталин-6-сульфокислоте по . этой же причине реагирует только окси-
группа, находящаяся в положении 2 (см. стр. 185).
\1-Нафтод-4-сульфок11слота может быть также получена кипячением диазотиро-
ванной нафтйоновой кислоты.. Она находит применение главным образом как азосо-
ставляющая! для азокрасителей.
15. Сульфокислоты нафталина
Сульфокислоты нафталина непосредственно находят л’ишь ограничен-
ное применение в производстве красителей. Однако они имеют большое
значение в технике как промежуточные вещества при производстве дру-
гих' важнейших полупродуктов (Р-нафтола и Р-нафтил амина, диоксина-
фталинов и аминонафтолов, большого числа нафтнламии-, нафтол- и
а м пиона ф тал с ул ь ф окис лот).
В главе о правилах ориентации уже указывалось, что положение суль- .
фогруппы, входящей в молекулу нафталина, зависит в основном от тем-
пературы реакции сульфирования: на холоду сульфогруппа вступает
преимущественно в а-, а при нагревании в ^-положение. Поэтому прн по-
лучении чх-сульфо кислоты нафталин сульфируют при возможно более
низкой температуре, во всяком едучае при температуре ниже темпера-
туры плавления нафталина, который поэтому должен применяться в виде
тонкого порошка (подробнее см. при описании синтеза 1-нафтИламин-5-
н -8-сульфокислот, стр. 191). При получении р-сульфокислоты реакцию
проводят, наоборот, при наиболее высокой температуре, при которой,
однако, не может еще иметь место разложение, а именно прн 160—170°.
Одиако и в этих условиях образуется также всегда некоторое количество
а.-нафталинсульфокислоты, причем, по согласующимся данным различ-
ных исследователей, самое меньшее 15%. Исследования О. Витта****
в некоторой степени объясняют сложные соотношения в реакции.
г
Сульфирование проводят по-разному, в зависимости от того, будет ли получаемая
нафталинсульфокислота без выделения далее непосредственно нитроваться или
сульфироваться либо она будет выделена как таковая перед дальнейшей переработ-
кой. В первом случае, при котором все равно нужно применять избыток кислоты, вы-
годно пользоваться методом’Витта (в технике, однако, известным и раньше), приме-
няя избыток серной кислоты с целью перевода а-сульфокислоты, которая сульфируется
* Герм. пат. 109102 (Frdl. V, 164); герм, пат: 115335, 117471, 126136 (Frdl, VI,
187—190).
** Герм. пат. 121683 и 122570 (Frdl., VI, 192—194).
*** Герм. пат. 550707 (Frdl. XIX, 1899); Helv. Chim. Acta, 29, 1756 (1946).
**** Witt О,, Вег, 48, 743 (1915).
легче, чем fi-нзомер, в дисульфокпслоту (ем. получение кислот Клене). Во втором слу-
чае, напротив, нз экономических соображений необходимо обходиться возможно мень-
шими количествами серной кислоты (см. ]3-Нафталп1[сульфок1!слота н ]3-Нафтол,
стр. 167j.
1-Нафтиламин-6- и -7-сульфокислоты (кислоты Клеве)
1 -Нафтиламин-6- н -7-сульфокислоты давно приобрели важное зна-
чение в производстве полназокраентелей прежде всего в качестве сред-
них азосоставляющих. Эти кислоты применяются при производстве чер-
ных красителей для хлопка типа прямого черного (колумбия чер-
ного ФФ), а также для получения ..ряда диазотирующихся красителей,
капримср, пафтогси синий, замбезху черный, прямой диазосиний, прямой
диазочерный К и др. Затем сульфокислоты этого типа часто применяются
при получении красителей типа Прямого голубого светопрочного (см.
стр. 250)". X..,
В данном случае сульфирование лучше всего проводить по методике
О. Витта.
128 г нафталина лучшего качества нагревают при постоянном
перемешивании до 165° на голом пламени в сосуде для сульфирования,
или нитрования, описанном на стр, 94 (рис. 19), Затем медленно при
этой же температуре вносят 206 г 94%-пой серной кислоты (уд. вес
1,84). Кислоту следует прибавлять в продолжение по крайней мере полу-
часа, так как иначе получается слишком много а-сульфокислоты, которая
для процесса теряется. Нагревают еще в течение получаса при 165° для
того, чтобы а-иульфокнелоту по возможности полностью превратить в ди-
сульфокислоту и получить в дальнейшем смесь 1,6 и 1,7-кислот, практи-
чески не содержащую изомерных а-кислот. Постепенно смеси дают остыть
при перемешивании до 60°; ее не следует специально охлаждать, так как
иначе на стенках сосуда может образоваться прочносидящий. осадок.
Если, однако, такой осадок все же образовался, то его надо сиять со
стенок н мешалки с помощью острого железного шпателя и измельчить.
После того как температура понизится до 60°, массу разбавляют 300 г
90%-нон с с р н о й кислоты. (В технике па этой стадии моносульфо-
кислоту сжатым воздухом передавливают через выводную трубу в нн-
тратор, в котором находится серная кислота, необходимая для разбавле-
ния. Не следует охлаждать слишком сильно, так как 8-сульфокислота
может застыть в трубопроводах и закупорить нх.) Затем массу при по-
стоянном перемешивании охлаждают холодной водой до 25° и очень
медленно начинают прйбавлять по каплям 103 г (1 моль) 62%-ной
азотной кислоты (уд. вес 1,38). Получающееся нитросоединение
1 -Нафтиламин-6- и -7-сульфокислоты
165
гораздо легче растворяется в серной кислоте, чем пафталинсульфокис-
лота, поэтому после цапала нитрования ие следует опасаться того, что
реакционная масса может затвердеть. После прибавления нескольких
граммов азотной кислоты реакционную массу охлаждают льдом до 1(Р
и далее проводя!' нитрование при этой температуре. После того как будет
прибавлено около половины всего количества азотной кислоты, необхо-
димо убедиться, не образовался ли твердый осадок на стенках сосуда
и на мешалке и нет ли больших комков в реакционной массе; осадок
следует соскоблить шпателем и комки размельчить, так как твердые
корки и плотные комки закристаллизовавшейся нафталинсульфокислоты
даже при очень длительном стоянии не поддаются нитрованию. При ра-
боте с большими количествами для разбавления из экономии берут мень-
шее количество серной кислоты и опасность затвердевания здесь больше;
поэтому во избежание трудностей необходимо особенно внимательно сле-
дить за тем, чтобы масса ие затвердела, и, если надо, разламывать твер-
дые частицы. Все количество азотной кислоты прибавляют, примерно в
течение 2,5 чае., оставляют стоять не меньше чем па 12 час. и затем, если
в пробе (анализ при пСвдещи нитрометра) содержится не более 2% взя-
той в реакцию азотной кислоты, вязкий, но прозрачный коричневатый
раствор выливают в 2 л воды. При этом почти не происходит выделения
азотистой кислоты.
Кислый раствор нагревают до 75° н прибавляют 20%-ный раствор
сульфата железа (II) до тех пор, пока нс прекратится выделение
окислов азота. Затем продувают или просасывают через жидкость воздух
до исчезновения мгновенного посинения йодокрахмальпой бумажки
от капли раствора, сильно разбавленного водой *, что необходимо во из-
бежание образования побочных продуктов при последующем восстанов-
лении.
К смеси прибавляют 50 а углекислого магния и затем, пока
свободная серная кислота полностью ire будет нейтрализована, тонко
растертый в порошок мел. Для этого требуется около 320 г мела (можно
также нейтрализовать гашеной известью, но прн этом необходимо сле-
дить, чтобы реакционная смесь не имела сил ьнощ ел очной реакции, так
как свободная щелочь или гидраты окиси щелочноземельных металлов
разрушают нитросульфокислоты). Гипс отфильтровывают на большом
путч-фильтре, хорошо отжимают н тщательно' промывают горячей водой
до тех пор, пока промывные воды не будут окрашены лишь в бледно-
желтый цвет. Отмывать дальше не имеет смысла, иначе, будет слишком
много промывных вод. Общий объем отфильтрованной жидкости доджей
составлять примерно 2,8 л.
Восстановление. Раствор нитросульфокислот подкисляют уксусной
кислотой до слабой, но отчетливой кислой реакции. В редуктор емкостью
2,5 л (лучше всего медный) помещают 250 г мелкого порошка серого
чугуна (см. об анилине, стр. 71 и 74) и 250 мл воды. Нагревают до
кипения на голом огне и протравляют железный порошок 10 мл ледяной
уксусной кислоты. Кипятят в течение некоторого времени, прибавляют
* Образующаяся из сульфата железа III) соль окиси железа также дает реак-
цию на йодокрахмальную бумажку, но лишь через 1—2 сек., причем иосинение начи-
нается но краям пробы, При известном навыке обе реакции легко отличить друг
от друга. Однако для начинающего вместо йодокрахм'альной бумажки вернее приме-
нять Г^-диаминодифенилметац-З^'-сульфон л(так называемый «сульфон»), который не
реагирует с солями окиси железа; при этом также не мешает и сильная минераль-
ная кислота. Подробнее об этом см. главу об азокрасителях, раздел о диазотировании
анилина (стр. 220, сноска 2).
](55
i. Промежуточные продукты
20 г кристаллического уксуснокислого натрия и при очень
хорошем перемешивании (железо также должно быть в вихревом дви-
жении) и сильном кипении прибавляют в течение часа по каплям слабо-
кислый раствор нитросульфокислот, Значительная часть раствора испа-
ряется, и общий объем жидкости составляет около 1 л. После прибавле-
ния всего раствора нитросульфокислот кипятят еще в течение 20 мин.;
капельная проба ла фильтровальной бумажке должна быть почти бес-
цветной. Полного обесцвечивания, однако, не происходит, но, во всяком
случае, раствор ие должен быть интенсивного коричневого или желтого
цвета. Кипящий еще раствор нейтрализуют окисью маги и я (при-
мерно 20 г), до тех пор пока капля на фильтровальной бумажке ие
станет давать хорошо фильтрующийся осадок окиси железа, а реакцию
по лакмусу — слабую, ио ясно щелочную.
Затем капельной пробой на фильтровальной бумаге с сернистым на-
трием или сернистым аммонием определяют полноту осаждения железа.
В случае неполноты осаждения остаток железа осаждают сернистым
аммонием. Безусловно, необходимо осадить последние остатки солей
железа, иначе кислоты Клеве при дальнейших операциях быстро окис-
ляются.
Раствор магниевых солей кислот Клеве фильтруют на иутч-фильтре
и, если необходимо, упаривают до 1 л (в фарфоровой чашке на голом
пламени: над чашкой вращается деревянный пропеллер).
Концентрированный раствор подкисляют примерно 100 лм концен-
трированной соляной кислоты ДО1 сильнокислой реакции на
коиго и при постоянном перемешидатпт-оставляют кристаллизоваться на
день в стеклянном стакане. Обе/ кислоты Клеве выпадают медленно, по
раз они выделились, то в воде больше не растворяются. Осадок отфиль-
тровывают через 24 часа и хорошо промывают большим количеством
воды.
q Разделение неочищенных кислот. Осадок растворяют в
800 мл горячей воды и в таком количестве аммиака (нли раствора
соды), чтобы раствор имел отчетливую щелочную реакцию. К раствору
прибавляют поваренную соль до получения 10%-ного раствора пова-
ренной соли. Трудно растворимая натриевая соль 1,7-кислоты выде-
ляется в течение дин в виде блестящих желтоватых листочков; ее отфиль-
тровывают и промывают сначала 10%-ным раствором поваренной соли, .
а затем совсем небольшим количеством холодной воды. Фильтрат под-
кисляют с О' л я и о й кис л отой до отчетливой реакции на конго и
оставляют на два дня, время от времени перемешивая. 1,6-Кнслоту от-
фильтровывают, промывают водой и сушат при 100°.
Выход свободной 1,6-кислоты Клеве (мол. вес 223) около 80 г;
натриевой сод и 1,7-кислоты Клеве (мол, вес 245) — около 75 а.
Маточный раствор содержит еще зачнтслызые количества загрязненного веще-
ства; его отбрасывают, либо при работе с большими количествами, выпаривают и
выделяют таким образом дополнительно некоторое количество кислот.
Технические примечания. Чистые 1,6- и 1,7-нафтиламиносульфокислоты
практически дают идентичные красители. Часто бывает выгодно применять смесь этих
кислот, особенно при получении черных полиазокрасителей, которые в этом случае
дают более интенсивные окраски, например колумбия черный, прямой диазочерный К,
замбези черный Фау (см. об этом в литературе).
1,7-Кислота Клеве обычно получается более чистой, чем 1,6-кислота Клеве, по-
этому для получения сложных красителей чаще применяют 1,7-кислоту. Содержание
1-нафтиламиго8-сульфокислоты в 1,7жислоте Клеве можно легко определить диазо-
тированием и кипячением полученного раствора диазоеоединений — 1-иафтиламин-в-
сульфокислота дает при этом нафтосультои в виде нерастворимого осадка,
который можно отфильтровать и взвесить (см, стр. 193).
^Нафталинмоносульфокислота и ^-нафтол
167
Р-Нафталиимоносульфокислота и р-иафтол
Если Р-нафтали^сульфокислоту получают для того, чтобы превратить
ос далее в р-нафтол, то необходимо возможно полнее использовать сер-
ную кислоту, так как Р-нафтол настолько дешев, что для его получения
необходимо применять лишь наиболее экономичный метод.
В сосуде, описанном иа стр. 94, нагревают на голом огие до 165°
прн постоянном перемешивании 256 г (2 моля) нафталина. Затем в
течение получаса по каплям прибавляют 280 г 94%-вой серной кис-
лоты (уд. в,ес 1,84)-.-Пламя регулируют, таким образом, чтобы темпе-
ратура реакционной массы была 163—168°. Капельную воронку заменяют
согнутой стеклянной трубкой, которую плотно вставляют в крышку со-
суда (на пробке нли асбестовом картоне). Через трубку в процессе реак-
ции сульфирования отгоняется вода и нафталин. Смесь нафталина и сер-
ной кислоты нагревают при 165° и непрерывном перемешивании в течение
1 часа, затем 1 -час при 167°, 1 час при 170° и, наконец, еще 1 час при
173°. За это время в приемнике собираются 30 г воды и около 25 г на-
фталина, Значительное количество нафталина осаждается также.с тече-
нием времени, на крышке сосуда; этот нафталин, однако, не используется.
Нагревание прекращают и установку разбирают. Образовавшаяся масса
содержит, помимо нафталинсульфокислоты, также некоторое количество
сульфонов, немного свободной серной кислоты, дисульфокислоту и смолу.
Смесь должна быть почти бесцветной. Ее выливают еще горячей в 1,8 л
воды.
Раствор свободной сульфокислоты частично нейтрализуют осторож-
ным прибавлением 60 г сод ы при хорошем перемешивании. Затем посте-
пенно прибавляют 360 а поваренной соли. Вскоре раствор засты-
вает в большие комки, затрудняющие дальнейшее перемешивание. Не-
смотря иа это, необходимо продолжать перемешивать до тех пор, пока
жидкость снова не станет однородной, так как только в этом случае про-
исходит полное растворение соли н удае*гся получить хорошо фильтрую-
щийся осадок. Продолжительность перемешивания зависит от быстроты
размешивания, однако оно должно продолжатьси ие менее 6 час., так •
как в противном случае ие происходит полного выделения. Осадок от-
фильтровывают на большом иутч-фильтре с хлопчатобумажным филь-
тром и хорошо отжимают. Затем его переносят на влажную плотную
хлопчатобумажную салфетку и сначала осторожно, а затем возможно
сильнее отжимают на винтовом прессе. Отжимать следует не менее 2 час.,
иначе в осадке останется сдишком много маточного раствора. Твердую
как камень массу измельчают и окончательно сушат при 100—120°.
Выход «Р-соли» около 165%, считая на нафталин, т. е. 400—420 з.
Маточный раствор, содержащий немного а-кислоты, смолу и следы
р-кислоты, можно переработать на глауберову соль.
Плавление нафталинсульфокислого натрия является одной из
важнейших технических операций органической технологии. Не удиви-
тельно, что при низкой цене нафтола лишь немногие заводы занимаются
168
/. Промежуточные продукты
Растворимость ₽-нафталннсульфокислого натрия в зависимости
от количества NaCl н Na-jSCU
(по американским данным)
[5-Соль н NaCl (г/100 г раствора)
25“ С 30° с 40° С 50° С, 65° С
[ЬСоль NaCl Й -Со .11'1 NaCl [ТОоль NaCl 3-Соль NaCl (ЬСоль NaCl
5,58 1 0 6,24 0 7,98 0 9,75 0 14,6 0
3,46 2,38 1,21 4,84 1,46 5,62 ; 4,15 2,9 8.47 2,93
0,31 । 9,19 0,16 13,08 0,65 8,47 ; 2,17 5,42 ' 6,12 3,81
0,15 0 0 1 13,16 16,81 26,43 0 26,5 [5-Соль и 0 Na.SO 26,70 4 (г/100 г 1.05 0 раствс 8,39 26,8 >ра) 1,96 1,26 0 7,19 10,83 27,2
3,42 1,97 1,97 1,81 4,3 2,85 5,72 ' 2,87 11,75 1,68
2,41 3,06 0,26 13,23 2,18 5,83 3,49 5,35 7,37 5.28
1,78 4,34 0 29,1 1.2 8,48 1,93 8,24 6,7 5.45
0,93 0,62 0,52 0,10 0 7,4 9,25 10,52 13,15 21,9 1 0,77 0 f 10,92 32,5 Х\ 1,42 0 10,01 31,9 1,90 3,14 0,25 0 12,0 10,86 26,96 31,0
его производством, необходимым условием которого является дешевизна
таких материалов, как уголь, сода и серная кислота.
Для того чтобы натриевая соль хорошо плавилась с едким натром,
ее надо хорошо измельчить. В лаборатории для этого соль проще всего’
размолоть иа мощной кофейной мельница.
В установленный непосредственно на небольшой печи Флетчера коте-
лок для плавления, описанный иа стр. 81 (см. рис. 14), загружают 200 г’
твердого едкого натра в кусках, не содержащего хлората, и 60 мл-
воды (плавление с едким натром, содержащим хлорат, снижает выходу
и, кроме того, очень опасно; может произойти взрыв!). Едкий натр рась ’
плавляют иа большом пламени, плав получается прозрачным как вода;;
вспенивающуюся жидкость постепенно нагревают до 270°, после чего
вспенивание прекращается. После этого при постоянном перемешивании
порциями по чайной ложке вносят порошкообразную натриевую солы :
р-Нафталинмоносудьфок.ислота и $-нафтол
169
Температуру' медленно повышают до 290°. Сухая натриевая соль посте-
пенно исчезает и появляется темный жидкий блестящий пафтолят натрия.
Ввиду того что нафтоляг—жидкий, оказывается возможным вводить го-
раздо больше натриевой соли, чем это указывается в большинстве пропи-
сей. В лаборатории легко удается ввести на 1 часть щелочи 1,5 части соли,
т. е. в нашем случае 300 г натриевой соли (в технике при хорошей аппа-
ратуре и нагреве генераторным газом удается ввести на 1 часть едкой
щелочи даже 2,8 частей соли и избежать прн этом подгорания и образо-
вания густой массы). К тому времени, когда температура поднимется до
290°, должна быть внесена приблизительно половина (150 г) всего коли-
чества натриевой соли. Затем очень осторожно нагревают до 300°, а к
моменту прибавления 3/4 всего количества соли (225 а) —до 305°; под
конец, когда уже прибавлено все количество соли, нагревают до 318°.
Нельзя перегребать выше этой температуры. Вследствие выделения суль-
фита натрия пЛдв становится крупичатым и нафтолят постепенно заме-
няет натриевую <оль, которая медленно исчезает. Плав выдерживают
ровно 15 мин. прн'ЗДв^и тщательно следят за тем, чтобы нс происходило
перегрева. Все время необходимо размешивать. Весь процесс плавления
с момента прибавления соли продолжается около часа; при слишком
быстром введении соли масса пригорает и, следовательно, уменьшается
выход.
Содержимое плавильного котла выливают иа противень с низкими
бортами. По охлаждении плав измельчают, снова загружают в котел и
прибавляют 0,5 л воды. При осторожном нагревании легко удается
растворить большую часть плава; ио всегда остается н ер а с творен ной
корка сульфита натрия. Сливают первую часть раствора и прибавляют
еще свежей воды до тех пор, пока все не растворится, Не следует рас-
ходовать более 2_л воды. Соединенные растворы нагревают до кипения
в фарфоровой чашке на открытой печи Флетчера и прибавляют 50%-ную
серную кислоту до тех пор, пока почти ие исчезнет реакция иа
тиазоловую бумажку. Слегка остывший раствор отсасывают через боль-
шой иутч-фильтр в предварительно нагретую колбу. Нейтрализованный
и профильтрованный раствор бесцветен или слегка желтоват, его объем
около 3 л.
Прозрачный раствор нагревают до кипения и при перемешивании при-
бавляют 50%-ную серную кислоту до сильнокислой реакции иа
лакмус *. При этом не должно быть запаха сернистой кислоты; р-иафтол
не растворяется в сульфите натрия в присутствии небольшого количе-
ства бисульфита. Сначала нафтол выделяется в виде масла, которое
тотчас же застывает. Его можно отфиДьтро-вать уже через час, теряются
лишь следы нафтола. Осадок отфильтровывают иа иутч-фильтре через
фильтр из хлопчатобумажной ткани и хорошо промывают холодной во-
дой. Перед перегонкой нафтол сушат при низкой температуре в вакуум-
ном сушильном шкафу или в теплой камере. При слишком высокой тем-
пературе нафтол плавится и возгоняется.
Выход высушенного неочищенного продукта из 300 г Р-соли около
150 г (93%-иого), перегнанного— 135 г: т. пл. 122°.
Во многих случаях может быть использован неочищенный продукт,
однако к нафтолу, выпускаемому в продажу, предъявляются особые тре-
бования н поэтому его подвергают основательной очистке. В настоящее
время применяется только перегонка в вакууме.
* Если нет возможности отфильтровать сырой fi-иафтол в течение часа, Лучше
применять концентрированную соляную кислоту, так как иначе выпадает сульфат
натрия.
170
Л Промежуточные продукты
л -нафталин-
Технические примечания. Сульфирование нафталина проводят всегда
в больших чугунных котлах (на IOOO-—3000 л). Нагрев производится либо непосред-
ственно генераторным газом, либо с помощью паровой рубанки (рис. 31а), рассчитан-
ной по меньшей мере на давление
в 6 от, для того чтобы можно было
достигнуть требуемой температуры
174°,
Вместо того чтобы
сульфокислоту превращать при дли- )
тельном нагревании и р-кислоту, в
технике применяют специальный -
прием: в сульфомассу после много- ;
часового нагревания продувают во- j
дяной пар, при этом «-кислота рас-
щепляется на нафталин и серную
кислоту, а З-кислота остается неиз- (
мененной. Таким образом получают
смесь серной кислоты с /3-кислотой,
не содержащую изомеров. *
В технике сульфирование про- ?
водят в условиях существенно отли-
чающихся от лабораторных. Так, ft- I
нафталинсульфокислый натрий ие-—i
осаждают поваренной солью, потому ‘ J
что это было бы слишком дорого, а
прибегают к следующему интерес- 3
ному техническому приему. К раз-
бавленной сульфомассе вместо пова- •;
<ренной соли прибавляют фильтрат, д
полученный в качестве отхода прн "
производстве нафтола и содержащий з
сульфит натрия. При этом выде* )
ляется сернистая кислота, которая i
в свою очередь непосредственно про- i
пускается в разбавленный водой плав
ft-нафтола (деревянный вентилятор). )
При этом осаждается ^-нафтол, в то
время как маточный раствор служит j
для выделения солн /3-нафталнн- .
сульфокислоты. Только этим путем э
вообще возможно дешево получать ’
/3-нафтол. Далее, осторожно экстрагируя сырой Р-нафтол разбавленным раствором
щелочи, можно извлечь содержащийся в небольшом количестве более легко рэство- а
рммый в щелочи а-нафтол. Об определении ^-нафтола в присутствии а-нафтола j
см. в аналитической части. На некоторых заводах промытый сырой нафтол расплав-
ляют в закрытом железном котле (водный маточный раствор отбрасывают) м . сушат
его сначала в вакууме. Затем фильтруют горячим на небольшом фильтрпрессе. Все
соли, которые содержатся в сыром нафтоле, вместе с неорганическими примесями, ;
остаются на фильтр-прессе; после этого нафтол очень гладко, без сильного осмоле- Д
пня, перегоняют в вакууме. Технический ^-нафтол 99,9%-ной чистоты. За последние
годы он выпускается в продажу в ферйе чешуек; с таким нафтолом удобнее ра-
ботать и он не пылит. ;
Рис. 31а.' Котел для сульфирования и нитро-
нания с паровой рубашкой (с двойными
стенками).
Сульфирование £-нафтола !
Сульфирование ^-нафтола в зависимости от концентрации примеияе- р
мой серной кислоты и от температуры реакции приводит к ряду моно- и
полисульфокислот. Здесь подробнее будут описаны лишь некоторые ти- \
пичные случаи, дающие возможность начинающему химику составить <
представление об этой отрасли производства промежуточных продуктов. 5
Первым продуктом взаимодействия концентрированной серной кис- i
лоты и ^-нафтола является 2-цафтол-1-сульфокислота (см. также
стр. 176). Од [га ко эта кислота очень неустойчива и в присутствии избытка ;
серной кислоты уже на холоду перегруппировывается в 2-нафтол-8-
сульфокислоту (кислота Байера, кроцеиновая кислота), которая в свою
Сульфирование ^-нафтола
171
очередь, далее, на холоду частично, а при нагревании полностью превра-
щается в 2,6-кислоту (кислота Шеффера).
При избытке кислоты, кроме того, всегда образуются в небольших
количествах 2-нафтол-3,6- и 2-нафтоЛ-6,8-дисульфокислоты, которые
вследствие своей способности давать с солями диазония красители
красноватого или соответственно желтоватого цвета более известны под
названием Р- и Г-кислот. В зависимости от того, сульфируют при нагре-
вании или на холоду, получается больше Р- или больше Г-кислоты. При
достаточном избытке серной кислоты эти «сульфокислоты оказываются
основными продуктами реакции. При сульфировании в очень жестких
условиях (олеумом при нагревании) обе кислоты окончательно превра-
щаются в 2-иафтол-3,6,8-трисульфокислоту. Все названные кислоты
являются важными исходными соединениями прежде всего для произ-
водства азокрасителей. Большинство из них практически нельзя не-
посредственно получить в индивидуальном виде; поэтому, как правило,
приходится тщательно выделять каждую кислоту из смеси изомеров:
При сульфировании иа холоду или при умеренной температуре реак-
ционная смесь, помимо названных кислот, часто содержит также значи-
тельные количества 2-нафтол-1,6-дисульфокислоты или 2-нафтол-1-,
3,6-трисульфокислоты. Эти соединения ие представляют технической цен-
ности, поэтому у них отщепляют сульфогруппу, стоящую в положении 1:
Для этого сульфомассу разбавляют водой и перед дальнейшей обработкой
нагревают в течение 0,5—1 часа при 90—100°.
Для разделения сульфокислот используют различную растворимость
их солей со щелочными металлами. Из трех технически наиболее важных
и ь большинстве случаев сдидвремеино образующихся кислот — моносуль-
фокяслоты-2,6- (кислота Шеффера) и дисульфокислот-2,3,6 (Р-кислота)
и -2,6,8 (Г-кислота) — первая дает натриевую соль, наиболее трудно рас-
творимую в холодной воде. Относительно небольшая растворимость этой
соли не очень сильно уменьшается в присутствии поваренной соли. На-
триевая соль Р-кислоты, напротив, значительно легче растворима в чи-
стой воде, но даже относительно небольшое количество пава вен нп» т™
172
1. Промежуточные продукты
резко уменьшает ее растворимость. Натриевая сачь Г-кислоты легко
растворима даже в растворе, содержащем большое количество поварен-?
нои соли; напротив, калиевая соль этой кислоты лишь умеренно раство-л
рима в холодной воде и еще менее растворима в растворе хлористого
калия. Приведенные выше дацщые относятся к средним солям, у которых
все имеющиеся сульфогруппы связаны со щелочными металлами, а окси-
группа остается свободной. Из нейтрального или из кислого раствора —
вопреки противоположным литературным данным — всегда выделяются'-
эти сади, а не кислые соли, у которых из двух сульфогрунн связана^
с металлом лишь одна, В грелочных растворах, напротив, образуются^
основные соли, у которых водород оксигруппы также связан со щелочным1^
металлом. Этн соли во всех аду чаях значительно легче растворимы, чему
средние солн, и неудобны для разделения. Поэтому разделение проводят?
всегда в нейтральном или кислом растворе, ’•
Практически поступают 'Следующим образом; нейтральный или слабо-1
кислый раствор натриевой соли, нс содержащий по возможности поварен-;
ной соли, упаривают до определенного, зависящего' от состава реакцнон-?
ной смеси объема, чтобы при охлаждении выкристаллизовывался^
возможно полнее лишь один 2-нафтол-б-сульфокиадьгй «атрий. После от- ;
деления осадка фильтрат, сади нужно, упаривают и прибавляют столько;
поваренной соли, чтобы в растворе ее содержалось 18—20%. При охла-?
ведении выделяется в ос.иовп1ом/ЛЗбль'4<-кислоты. Ее отфильтровывают и?
к маточному раствору прибавляют хлорйутый/Калий для выделения ка-б
лиевой солн Г-кислоты, ' '
Для получения действительно чистых продуктов надо ис только точно,
соблюдать оптимальные в каждом отдельном случае концентрацию и:;
количество соли, иа надо также особенно тщательно проводить отсасываб
ние и промывание отдельных фракций при кристаллизации с тем, чтобьГ
по возможности полностью отделять маточный раствор, применяя МИНН-
мальное количество жидкости для промывок.
Следует подчеркнуть, что исходный ^-нафтол должен применяться
виде тонкого порошка. Если пренебречь этим элементарным условием,
то просульфируется лишь часть нафтола; причем грубые комки нс вегу-;
па ют в реакцию и плавают в реакционной массе, вследствие чего нё‘
всегда достигаются одинаковые результаты. То же имеет место- и в том
случае если реакционную смесь просто оставить стоять, а не перемеши-
вать се все время. Аппаратура такая же, как описана при получеии)
^-нафталинсульфокислоты (см, стр. 94, рис, 19),
2-Нафтол-6-сульфокислота (кислота Шеффера),
2 нафтол-3,6- и 2-нафтол-6,8-дисульфокислоты (Р-н Г-кислоты)
В сосуде с 250 г ко и ц е п тр н р о в а н и ой серной кислоть1
(уд. вес 1,84) вносят при перемешивании 144 г (1 моль) чистого р-н а
фтола в виде тонкого порошка. Температура при этом повышается са:
мое большее до 30°. Затем смесь нагревают в течение 8 час. при перемс
шивании иа кипящей водяной бане. Реакционную смесь, лучше всег
еще теплую, выливают в 1 л воды и перемешивают в течение часа пр;
95—100°,' если необходимо, то прибавляют воду, следя за тем, чтоб!
объем оставался ие менее 1 л. Затем прибавляют еще 0,5л воды н 100 .
кальцинированной ,(илн соответствующее количество кристаллической
глауберовой соли и при 90° и перемешивании нейтрализуют ме
лом (требуется около, 190 а). Мел ладо вносить очень осторожно не
большими порциями, особенно- под конец, чтобы избежать слишком силь
ного вспенивания. Нагревают До кипения, отфильтровывают из горяче;
2-Нафтол-б-сульфокислота и 2-нафтол-3.6- а 2-нафтол-6,8-дисульфокислоты 173
раствора выделившийся гипс и промывают горячей водой, тщательно пе-
ремешивая, до тех пор, пока проба фильтрата, к которой прибавлена
сода, не будет давать с диазобензолом лишь очень слабую окраску. Филь-
трат, соединенный е промывными водами, снова нагревают до кипения
и для удаления оставшихся еше соединений кальция * прибавляют каль-
цин ированмую соду (окало 30 г) до слабого покраснения фенолфта-
леиновой бумажки и до тех пор, пока проба фильтрата не будет больше
давать осадка при дальнейшем 'Прибавлении соды. Осадок карбоната
кальция отсасывают и промывают горячей водой, фильтрат подкисляют
концентрированной соляной кислотой (около 40 жл) и упаривают
до 1 л. Охлаждают при перемешивании; при этом объем ввиду продол-
жающегося испарения заметно уменьшается; по охлаждении он должен
составлять 800 мл. Оставляют па ночь; затем густую кашицеобразную
массу возможно лучше отсасывают на путч-фильтре, тщательно пригла-
живают шпапелсм и отжимают пестиком или широкой стороной стеклян-
ной пробки, следя за тем, чтобы в отсасываемом осадке тге образовались
трещины и отверстия и чтобы в нем по возможности не оставалось ма-
точного раствора. Если при отжимании из осадка больше ие стекает
жидкость, то вакуум отключают и приливают 50 мл холодной воды. Тща-
тельно следят за тем, чтобы ,не образовалось каналов, через которые
стекала бы промывная вода, вместо того чтобы равномерно промыть весь
осадок. Затем снова присоединяют сначала слабый вакуум, который под
конец увеличивают как возможно, причем отжимом и замазыванием
трещин способствуют отсасыванию. Таким образом промывают водой
порциями в 50 мл еще два раза. При правильном проведении этой опе-
рации получают слегка влажный осадок который можно окончательно
отжать па винтовом прессе; этот метод рекомендуется при работе с боль-
шими количествами. После высушивания в паровом сушильном шкафу
получают 150—155 г технически чистого 2 - н а ф т о л - 6 - с у л ь ф о-
кяслого натрия, на 1 г которого расходуется 36,5—37 мл 0,1 и,
раствора соли диазония.
Выход около 56% от теории. Продукт в большинстве случаев может
быть использован без дальнейшей очистки. При желании можно изба-
виться от следов дисульфокислот перекристаллизацией из трех — четы-
рехкратного количества горячей воды.
Фильтрат, соединенный с промывными водами, снова упаривают до
300 мл и к еще горячему раствору прибавляют 40 г твердой поварен-
п о: й со л и. После стояния в течение ночи отсасывают образовавшийся
осадок и трн раза промывают порциями по 20 мл полунасыщенным рас-
твором поваренной соли (смесь равных объемов насыщенного раствора
'и воды), при этом поступают так же, как в случае первого осадка.
После высушивания в паровом сушильном шкафу получают 50—-55 г
сырой Р-соли, на 1 г которой расходуется 23—26 мл 0,1 л. раствора соли
диазония.
Выход 12,5—13%,Об очистке см. ниже.
Фильтрат снова кипятят и прибавляют 60 г хлористого калия.
При охлаждении и стоянии в течение ночи происходит обильное образо-
вание кристаллов. Осадок отсасывают^ как было описано выше, и промы-
вают 10 % -ным раствором хлористого калия, Высушенный в паровом
сушильном шкафу продукт весит 40—45 г. Он в основном состоит из
1-соли, и на 1 а его расходуется 23—25 мл 0,1 п. раствора соли /г-нитро-
Лказобензола.
* Полное удаление кальция необходимо лишь в том случае, если требуется по-
лучить прозрачный раствор вещества даже н присутствии елды.
174 1. Промежуточные продукты
Вых од около 10% от теории.
Оставшийся маточный раствор содержит еще около 12% исходного
вещества в виде сульфокислот, титруемых раствором соли п-тштродиазо-
бензола, которые, однако, не удается выделить. Помимо небольших ко-
личеств соли Шеффера и Р- и Г-солей, в растворе содержатся также’
другие подробно ие изученные изомеры.
Неочищенная Р-соль, полученная по способу, описанному выше, всегда содержит',
немного соли Шеффера. При точном соблюдении методики эта примесь составляет?
не более 5%. Такой продукт применим для многих целей. Совершенно чистую Р-соль?
можно получить следующим образом: )
50 а неочищенного вещества растворяют в 250 мл кипящей вод
прибавляют 25 е поваренной соли и раствор отфильтровывают горячим от;
небольшой мути. Оставляют на ночь в холодильном шкафу, затем отсасывают и про-
мывают описанным выше способом в три приема 50 мл смеси одной части насыщен-”;
кого раствора поваренной соли и трех частей воды. Таким образом получают 65—(
70% взятого неочищенного продукта в виде совсем чистой Р-соли в том случае, если4'
исходная соль содержала не более 5% других сульфокислот. В противном случае?
операцию следует повторить.
Фильтрат упаривают до половины первоначального объема (т. е. до 125 мл).?
При охлаждении выкристаллизовывается 25% взятой смеси. Эта вторая фракция^
настолько обогащена солью Шеффера, что основное количество ее можно выделить’-
в чистом виде перекристаллизаций из четырехкратного количества воды. Из маточ<
ноте раствора осаждают поваренной солью смесь соли Шеффера и Р-соли; эту смесь 1
можно разделить далее на ее составные даепг-яоцтореиием уже описанных операций!1;
Фракция, полученная осаждением х/ористым калием, состоит из Г-соли, загряз-":
ненной не более чем 5% Р-соли. В лаборатории ее м^жидяЬчистить перекристаллиза--.
дней из воды. В технике чаще всего д^я очистки используют свойство Г-соли значн-(
тельно труднее вступать в реакцию азоЬочетания с диазососдинениями, чем Р-соль jjJ)
соль Шеффера. В пробе титрованием диазобензолом или диазоксилолом в сильно раз-1'!
бавленно.м растворе определяют содержание сульфокислот, легко вступающих в реак- Й
цшо сочетания. Затем на нечищенную Г-соль действуют менее энергично сочетающимся^
диазосоедииением, .чаше всего диазоксилолом, в количестве, соответствующем количе-а
ству сульфокислот, определенному титрованием. В этих условиях в реакцию сочетания^
вступают только примеси; образующийся Краситель отделяют — он продается под на-.й
званием «пунцовый» (кислотный алый)—и получают раствор чистой Г-соли, который^
непосредственно может быть использован, например, для получения азокраОтелей. $
О чистоте полученных продуктов можно судить по флуоресценции н по отноше-3
нню к диазотированному ацетил-п-фенилендиамину. Чистая соль Шеффера в нейтраль-и
ном растворе не обнаруживает заметной флуоресценции; а щелочном растворе оиава
дает не очень интенсивную фиолетово-синюю флуоресценцию. Р-соль и Г-сол&я
обнаруживают зеленовато-синюю флуоресценцию, слабую уже в нейтральной^
и очень сильную в щелочном растворах. Незначительной примеси одной из этих соЛеИ
к соли Шеффера достаточно, чтобы погасить флуоресценцию последней. В случала
неокрашенных продуктов таким путем можно установить наличие очень незначителЬ-||
пых примесей соли Шеффера и сравнением со смесями известного состава можнда
Приблизительно определить количество этих примесей. Этот метод не является точными
и надежным в случае сильно окрашенных неочищенных продуктов.
С диазотированным ацетил-п-фецилецдиамином (л-а.миноацетанилидом) солД
Шеффера дает хорошо растворимый Оранжево-Красный краситель, который осаждаетсЯЯ
лишь очень большим количеством поваренной соли. Р-соль с указанным диазосоеди-йй
нением, наоборот, дает краситель, практически полностью осаждающийся уже соасеМЙИ
небольшим количеством поваренной соли в виде блестящих с бронзовым блескомй|
кажущихся в проходящем свете синевато-красными кристалликов; фильтрат при этоэет
лишь очень слабо окрашен в чуть голубовато-розовый цвет. Более интенсивная нЯ
желтоватая окраска фильтрата указывает на присутствие соли Шеффера. Г-Сол1Я
с названным диазосоединением в разбавленном слабом содово-щелочном растворяя
вообще не сочетается либо сочетается очень медленно. Немедленное появление интен-Я
сивной красной окраски свидетельствует о наличии примесей (чаще всего соли Шеф*М
фера или Р-соли). Наличие Г-соли в продуктах, содержащих главным образом дв«И
другие сульфокислоты, устанавливают на основании того, что фильтрат, полученныЙЯ
после высаливания н фильтрования красителя и не вступающий более в реакцию аэо-Я
сочетания, показывает^ интенсивную флоуресценцию, характерную для Г-соли, и обра*Я
зует красители е энергично сочетающимися дпазосоединениями, например с диазо-'Я
соединением rt-нитроаннлина. ?Я
Для титрования соли Шеффера и Р-соли с успехом применяют диазоти'рованныйи
ацетил-/г-фенилендиамин в слабом содово-щелочном растворе, а для определения^
2-Нафтол-6,8-дисульфокислота и 2-нафтол-3,6-дисульфокислота
175
Г-соли — диазотированный л-нитроанилин в присутствии бикарбоната. Разность между
обоими титрованиями в случае смесей дает возможность определить количество нахо-
дящейся в смеси Г-соли.
Об определении соли Шеффера и Р-солн в случае их совместного присутствия
см, аналитическую часть.
2-Нафтол-6,8-дисульфокислота (Г-кислота)
и 2-нафтол-3,6-дисульфокислота (Р-кислота)
Если желают получить в качестве основного продукта реакции Г-соль,
то следует увеличить количество, и повысить концентрацию серной кис-
лоты и сульфирование проводить при умеренной температуре, во всяком
случае не выше 60°.
В 200 г моногидрата при хорошем перемешивании и охлажде-
нии проточной водой вносят 72 г (0,5 моля) чистого порошкообразного
й>-и а ф то л а; температура при этом не должна превышать 20°. После
перемешивания в течение часа при комнатной температуре проба реак-
ционной смеси должна при прибавлении воды дать прозрачный раствор
и ие мутнеть при кипячении. Затем, опять-таки при охлаждении водой,
прибавляют по каплям 100 г 20%-ного олеума с такой скоростью,
чтобы температура оставалась около 20°. Затем нагревают иа водяной
бане до 55—60° и перемешивают при этой температуре в продолжение
40 час. до .тех пор, пока разбавленная водой и слегка подщелоченная
содой проба ие начнет при действии диазотированного ацетил-я-фенилен-
диамина.образовывать легко высаливаемый синевато-красный краситель
на основе Р-ооли и нс перестанет давать трудно осаждаемый оранжево-
красный краситель иа основе соли Шеффера (см. стр. 174). К концу
реакции сульфирования выделяется большей частью кристаллический
осадоас и образуется плотная, однако еще хорошо размешиваемая масса.
Сульфомассу выливают в 1 л воды, прибавляют 75 г кальцинированной
(или соответствующее количество кристаллической) глауберовой
соли и перемешивают в течение 1 часа при 95°, приливая воду взамен
испарившейся. Пр^ этом образовавшиеся в небольшом количестве
2-нафтол-1,6-ди- и -1,3,6-трисульфокислоты превращаются соответственно
в кислоту Шеффера или Р-кислоту. -Затем горячую реакционную смесь
нейтрализуют при перемешивании 300 г мела; при этом ие происходит
сильного вспенивания и мел можно прибавлять довольно' быстро. Отса-
сывание н промывание осадка гипса, удаление остаточных количеств
кальция путем осаждения содой (около 25 г), повторное подкнелеине
фильтрата концентрированной соляной кислотой (около 30 мл)
проводят точно так же, как и при получении кислоты Шеффера. Под к&
цец упаривают до 400 мл и к горячей смеси прибавляют 60 г твердой
поваренной соли. Охлаждают при перемешивании; выделяется в основ-
ном образовавшаяся Р-ооль и немного соли Шеффера. Оставляют на
ночь, затем отсасывают и промывают полунасыщениым раствором пова-
ренной соли, как это уже было подробно описано при выделении солн
Шеффера (стр. 173). Вес высушенного продукта около 65—70 г, что соот-
ветствует, по данным титрования, около 120 мл 1 н. раствора диазотиро-
ванного ацетил-п-фенилендиамииа и около 150 мл 1 н. раствора диазоти-
рованного п-нитроанилина; таким образом выход составляет около 30%,
считая на взятый '|3-нафтол, и продукт состоит примерно на 4/5 из
Р-соли с незначительной примесью соли Шеффера и на 1/5 нз Г-соли. Из
продукта можно получить чистую Р-соль по методу, приведенному иа
стр. 174. '
Фильтра? снова нагревают до кипения, прибавляют 100 г хлори-
стого калия и охлаждают прн, перемешивании. После стояния
176
1. Промежуточные продукты
в течение ночи отсасывают, трижды промывают 10%-иым раствором хло-
ристого калия порциями по 50 мл, каждый раз хорошо отжимают, но воз-
можности тщательно отсасывают и сушат н паровом сушильном шкафу.
Выход 140—150 г, что соответствует по титрованию п-нитродиазо-
бензолом примерно 300 мл 1 н. дназораствора, или около 60% от теории.
При тщательном проведении реакции получают совершению бесцветную
чистую Г-содь; эта соль с диазобензолом в слабощелочном разбавленном
растворе немедленно красителя не образует.
Маточный раствор содержит еще около 10%, считая на взятый р-нафтол, слож-
ной смеси различных сульфокислот, которую нельзя разделить способом, применимым
в технике,
2- Нафтиламин-1-сульфокислота (кислота
из ^-нафтола
Тобиаса)
ОН
SO3H
/I /ОН
8О3Н
A /NH2
Н1Ц
ci so л
а) 2-Нафтол -1-сульфокис лдэ та из ₽ - н а ф т о л а
В литровую пятигорлую колбу, снабженную лопатообразной мешал-
кой со ртутным затвором, капельной (воронкой, термометром, отводной
трубкой и трубкой для пропускания раза, помещают 72 г (0,5 моля)
сухого 6-нафтола н заливают егф 280 г совершенно безводного
нитробензола*, нагретого1 примерно до 100°. Затем реакционной
смеси дают охладиться при перемешивания; часть растворившегося иа- .(
фтола при этом снова выпадает. При непрерывном перемешивании и ох-
лаждении льдом медленно прибавляют по каплям 35 мд (62 г) х л о р- ’
сульфоновой кислоты, нс содержащей серной киедоты **; ’
температура не должна подниматься выше 5°. Выделяющийся хлористый >
водород поглощают в специальном поглотителе, соединенном через хлор-
кальциевую трубку; выделившийся (3-нафтол быстро переходит в раствор
и к концу реакции образовавшаяся нафтолсульфокислота начинает ча- (
стично выпадать. После окончания прибавления хлорсульфоновой j
кислоты оставшийся хлористый водород вытесняют пропусканием тока
сухого воздуха,
Питробепзольный раствор вместе с суспендированным в нем осадком г
встряхивают сначала со смесью. 300 мл воды и 100 г льда, а затем еще \
два раза с холодной водой порциями по 100 мл. Соединенные водные-
вытяжки фильтруют через смоченный фильтр и насыщают при перемеши- '
вании 200 г поваренной солн; при этом выделяется в виде бесцвет- 1
пых листочков 2-нафтол-1-сульфокислый натрий. Перемешивают до тех
пор, пока поваренная соль полностью не растворится, оставляют, лучше г
всего, на ночь и затем отсасывают, дважды заливая насыщенным раство-
ром поваренной соли. Полученный осадок непосредственно превращают;
* Нитробензол сушат простой перегонкой, отбрасывая первую фракцию, содер- '
жашую воду,
** Хлорсульфоновая кислота жадно поглощает воду, разлагаясь на серную кис-«
лоту и хлористый водород. Поэтому ее следует хранить в герметически закрываю-
щихся сосудах; обычные стеклянные сосуды с притертыми пробками для этод5 цели ;
редко бывают пригодными^ Если имеющаяся в наличии хлорсульфоновая кислота ,•
не является безупречной, то ее следует предварительно перегнать в приборе, совер- ;
шенно изолированном от влапн воздуха.
2-Нафтиламин-!-сульфокислота из ft-нафтола
177
в 2-нафтиламип-1-сульфокислоту; следует только предварительно убе-
диться в том, что он не флуоресцирует в содово-щелочном растворе н не
дает с диазобензолом растворимого в воде оранжево-красного азокра-
сителя, т, е. не содержит изомерных пафтолсульфокнслот.
Если хотят получить 2-нафтол- 1-сульфокислый натрий в чистом виде, то отфиль-
трованный осадок снова растворяют в небольшое количестве холодной воды, нейтра-
лизуют, если надо, содой, дополнительно встряхивают с бейзолом для удаления
нитробензола, увлеченного осадком, фильтруют и снова осаждают поваренной солью;
отсасывают, промывают насыщенным раствором поваренной солн, отжимают и сушат.
Продукт получается чистым, если не считать, что он содержит поваренную соль.
Для очистки от неорганических солей его можно перекристаллизовывать из спирта.
Для определения содержания в полученном веществе взвешенную пробу кипятят
два часа с десятикратным количеством 20%-ной сернрй кислоты в колбе с обратным
холодильником; при этом сульфогруппа опять отщепляется и выделяется ^-нафтол,
который растворяют в разбавленной щелочи и титруют 0,1 и. раствором диазобензола.
б) 2 - Н а ф тн л a ji и н 1- сульфокислота
1 из 2-иафтол-1-сульфокислоты
Полученный отжатый осадок 2-нафтол-1-сульф окислого
натрия помещают в эмалированный автоклав емкостью 1 приливают
250 мл раствора 20%-ного аммиака, прибавляют раствор бисуль-
фита аммония, полученный насыщением прн охлаждении 60 мл
20%-ного раствора аммиака сернистым газом, и нагревают в течение
8. час. при 145—150° (температура реакционной массы). .При работе
с бодьцщми количествами аммиак отгоняют и используют при проведении
повойхреакции. В лабораторных условиях удобнее охладить содержимое
автоклава и при перемешивании насытить твердой поваренной
солью. При этом образовавшаяся натриевая соль 2-пафтнламин-
1-сульфокислоты практически полностью выделяется. После стояния
в течение ночи соль отсасывают, хорошо промывают насыщенным
раствором поваренной соли для удаления аммиака и солей аммония,
отжатый осадок снова растворяют в горячей воде, фильтруют и подкис-
ляют еще горячий фильтрат концентрированной соляной кислотой
до отчетливой кисдрй реакции на конго. После полного охлаждения от-
сасывают выделившуюся в виде иголочек свободную 2-нафтиламии-
1 -сульфокислоту, промывают холодйой водой и сушат.
Выход 93 г, т. е. около 83% от теории (считая на Р-иафтол),
Как упоминалось на стр. 170, 2-нафтол-1-сульфокислота является первым про-
дуктом взаимодействия р-нафтола с концентрированной серной кислотой. Однако она
не может быть получена таким путем, так как, сразу же перегруппировывается в при-
сутствии избытка серной кислоты, которого не-удается избежать.
Только что описанный метод, позволяющий сульфировать при низкой температуре
в отсутствие свободной серной кислоты и значительного избытка сульфирующего
агента, дает возможность получать вообще лишь одну единственную сульфокислоту.
Последняя либо вовсе не может быть получена другим путем, либо не может быть
получена без примеси других кислот, так как концентрированная серная кислота
легко ее- изомеризует или сульфирует дальше. Описанным методом можно также
гладко получать моносульфокислоты соединений, которые иначе тотчас же превра-
щаются в дисульфокислоты, как например карбазола, 4,4'-диоксидифенилметана и др.
2-Нафтол-1-сульфокислота при обычных условиях не вступает в реакцию азосо-
четания с диазосоединениями, так как у нее замещено единственное место, способное
к сочетанию. Однако при нагревании и в кислой среде сульфогруппа замещается на азо-
группу; в Этом случае образуются те же красители, какие получаются из ^-нафтола.
Это свойство кислоты было использовано для получения на волокне паракрасного *.
2-Нафтол-1-сульфокислота находит применение главным образом в качестве
промежуточного продукта для получения 2-нафтиламин-1 -сульфокислоты, которая
после диазотирования и сочетания с -нафтолом дает применяемый в больших коли-
чествах ценный лито ль красный Р (лаковый красный С}.
Герм. пат. 204702 (Frdl., IX, 408).
j. промежуточные продукты
Превращение 2-нафтол-1-сульфокислоты в соответствующую нафтиламинсульфо-
кислоту является еще одним примером реакции Бухсрсра, описанной ранее подробно
для 1-нафтол-4-сульфок[1слоты (стр. 162).
Получение аминоиафтолсульфокислоты
из оксийитрозосоединеиия (хинонмойооксима)
1-Амиио-2-нафтол-4-сульфокислота из ^-нафтола
1. Нитрозо-^-нафтол. В трехлитровом стеклянном стакане раствор
ряют при 50° 72 г (0,5 моля) * ^-нафтола в 65 а 35%-ного раствора^
едкого иатра и 750 мл воды. К этому раствору, который должен,;
давать слабую, ио отчетливую реакцию иа тиазоловую бумажку, при-)
бавляют 36 г 100%-ного нитрита натрия и охлаждают до 0° при-1
бавлением воды и льда до- объема 1,5 л. При хорошем перемешивании 3
в течение 3 час. прибавляют около 160 а 40%-пой серной кислоты, j
К концу прибавления раствор должен иметь отчетливую кислую реакцию (
на конго и давать реакцию на йодокрахмальную бумажку; Не менее чем J:
через час иитрозонафтол отфильтровывают иа большом иутч-фильтре
и тщательно промывают. Он получается достаточно чистым, если был_Д
чистым исходный ^-нафтол.
2. Восстановление и превращение в аминонафтолсульфокислоту. !
Влажный нитрозонафтол (его не следует сушить) смешивают
в стеклянном стакане с небольшим количеством воды и охлаждают льдом
до 5°, К однородной массе сразу прибавляют 320 г раствора б и с у л ь- ;
фита натрия (содержащего примерно 25 %, SO2 *=^-Нито оз о нафтол
вскоре переходит в раствор; иногда осторожно прибавляют ещф немного
разбавленной щелочи. / '*
Раствор содержит смолистые вещества и поэтому его следует про-
фильтровать (при высаливании образовавшегося юисульфитного ооеди-’
нения получают краситель, применяющийся в ситцепечатании, — эльзас- ;
ский зеленый Н или диоксин Н — протравной зеленый Бс); железный
лак очень светопрочен. » .
-j-NsHSO3
„Диоксин" 1~Амино-2-иафтол-4-сульфокислот|
* Применяется 0,5 моля, так как иначе пришлось бы работать со слищком боль^
шими объемами.
** Содержание SO2 в растворе бисульфита необходимо определять титрованием*;
На 1 моль ^-нафтола применяют 2,5 моля SOa. •;
$-Нафтиламин из ^-нафтола
179
Объем профильтрованного раствора около 1 л. Его переносят в ста-
кан и при 25° смешивают со 100 г серной кислоты (уд. вес. 1,84), раз-
бавленной 200 мл воды. После этого раствор должен иметь сильную
мннеральнокислую реакцию (см. стр. 191). Через час его осторожно
нагревают до 50° и оставляют на ночь. Содержимое стакана застывает
в плотную массу свободной аминоиафтолсульфокислоты. Ее отфильтро-
вывают н хорошо промывают водой.
Выход составляет, считая на взятый ^-нафтол, около 90%.
О диазотировании 1-амино-2-нафтол-4-сульфокислоты см. Азокрасители, стр. 219
и 225; о получении этого диазосоединения другим путем см. стр. 159. Сочетанием
с jj-нафтолом сначала Баденский анилиновый и содовый завод, а также одновременно
и независимо фирмы Гейги (Зандмейер) и Калле (Эльбель) получили очень
прочный сине-черный хромирующийся краситель, известный под названием палатин
хромовый черный 6Б*, эриохром црне-черный Р или салициновый черный У (кислотный
хром сине-черный А). Сочетанием с д-нафтолом фирма «Гейги» получила также
очень прочный краситель — эриохром сине-черный Б**. Интересно, что сочетание
с а-нафтолом в сильнощелочной среде идет только в одго-положение относительно
оксигруппы.
Диазосоединение настолько устойчиво, что его можно без опасений сушить и ни-
тровать нитрующей смесью в концентрированной серной кислоте (Зандмейер-Гаген-
бах **•). Пронитрованное диазосоединение с а- и (З-нафтолами дает наиболее дешевые
и чуть ли не самые прочные хромирующиеся черные красители для шерсти (эриохром
черный Т и А. кислотный хром черный С и кислотный хром черный специальный) *♦**.
Описанный выше метод сульфирования сернистой кислотой находит, далее, при-
менение для получения n-амииофенолсульфокислоты из нитрозодиметиланилина и би-
сульфата натрия. При образовании дисульфокислоты одновременно происходит от-
щепление диметиламиногруппы с образованием производного п-аминофенола.
Таким образом, при этом образуется также чистый диметиламин, являющийся
товарным продуктом.
^-Нафтиламии из ₽-нафтола
В автоклаве с мешалкой и масляной баней нагревают 144 г
(1 моль) 100%-ного р-иафтола
и 600 г раствора сульфита
* Герм. пат. 156440 (Frdl,, VIII, 656).
** Герм. пат. 181326 (Frdl,, VIII, 668),
'*** **•* Герм. пат. 164655 (Frdl,, VIII, 647).
**•* Герм. пат. 169683 (Frdl., VIII, 673L
80
I. Промежуточные продукты
аммония*. К смеси прибавляют еще 125 г 20%-кого аммиака.
Смесь нагревают в течение 8 час, при 150° (температура реакционной
смеси) и около 6 ат давления (стальной манометр). Дают автоклаву
остыть, и измельчают полученную лепешку [3-нафтиламина в ступке. Из-
мельченную лепешку тщательно промывают па путч-фильтре водой. Рас-
твор сульфита аммония может быть использован несколько раз. Промытое
основание растворяют в 1,5 л воды и 110 г соляной кислоты, не
содержащей серной кислоты, и фильтруют теплым от оставшегося в не-
большом количестве нафтола. Фильтрат смешивают с раствором 100 г
безводного сульфата натрия, растворенного в 250 мл воды; выде-
ляется серпокислый нафтил а мин. Оставляют иа ночь, отфильтровывают
осадок и хорошо промывают его холодной водой. Для многих целей можно
прямо применять высушенный сернокислый иафтиламин.
Для получения свободного основания влажную сернокислую соль
смешивают с 1 л воды и прибавляют 60 г кальцинированной соды, ко-
торую растворяют в небольшом количестве воды. Вследствие того, что
сернокислая соль трудно растворима, выделение продолжается несколько
часов, но его можно ускорить постоянным перемешиванием и нагреванием
до 80°. Затем отфильтровывают, промывают, сушат при 80° и получают
около 130 г сухого основания или 85—95% от теории.
Технические примечания. Для такого рода реакций ..совершенно необ-
ходимо применять автоклавы с масляной баней или паровой рубашкой. Нафтил амин
выделяется на дне реакционного сосуда и виде маслянистого слоя; поэтомуДесли не
применять масляной бани, несмотря на перемешивание, тотчас Же произойдет перегрев
и большая часть нафтиламина превратится в динафтиламин и продукты разложения.
Эти же замечания относятся и к получению а-нафтола (см. стр. 1G0). -
fi-Нафтиламин в технике чаще всего перегоняют в вакууме. Необходимо рабо-
тать очень осторожно, так как он легко разлагается. Если нс выделяют свободное
основание, то для сульфирования хорошо высушенный тонкий порошок сернокислого
нафтиламина смешивают с 1% соды (см. также примулин, стр. 2.97) и вносят в сер-
ную кислоту или олеум.
Метод Бухерера совершенно вытеснил старый метод, который заключался в на-
гревании нафтола с аммиаком; этот метод давал выход лишь 70% и требовах при-
менения давления в 50—60 ат.
Подробнее о реакции Бухерера см. стр. 1G2.
2-Амиио-8-иафтол-6-сульфокислота и 2 амиио-5-
иафтол-7-сульфокислота (у-кислота и И-кислота)
из р-иафтиламина
Эти важные сульфокислоты можно получить различными путями,
а именно у-кислоту -получают тремя методами, И-кислоту—двумя. Ис-
ходным веществом во всех случаях является Р-иафтол, Его можно пре-
вратить в обе аминоиафтолсульфокислоты по схеме, приведенной чиже.
Здесь будет описан лишь метод получения из р-нафтиламина, В техни-
ческих примечаниях даны некоторые сведения о методах, в которых исхо-
дят из В-цафтола через его сульфокислоты и которые предпочитают в на-
стоящее время, так как они позволяют избежать получения 8-пафтил-
амина, являющегося, как известно, наиболее опасным канцерогенным
веществом (так же, как и основание бензидина).
* Раствор сульфита аммония получают, насыщая сернистым газом 250 а
20%-ного аммиака и прибавляя к полученному бисульфиту аммония еще '250 г
аммиака.
2-Амино-8-пафгол-6-сульфокислота
18
2-А мино-5-нэфтол-
-7-сульфок ис лота
(«зо-Т-кислота,
Й~кислота)
2-Нафт о л-1-
суль фокислота
2-Наф тиламн н-1~
сульфокислота
а) 2-Нафтиламин- 6,8 - д и сульфокислота
и 2 - н а ф тил амин- 5,7 дисульфокислота
из ^-иафтиламииа
В 800 г 15%-ного олеума вносят в течение 10 мни, 1 моль (192 г)
измельченного в тонкий порошок сернокислого ^-нафтил-
ами на, хорошо растертого с 1 г «кальи ини подл и ~ ” гт~.
182
/. Промежуточные продукты
температура не должна подниматься выше 50°. Небольшую пробу выли-
вают в небольшое количество воды и проверяют, как она растворяется
в соде. Моносульфокислота обычно образуется в течение первых 15 мни.
Охлаждают до 40° и прибавляют при непрерывном перемешивании в те-
чение 15 мин, 350 г олеума, содержащего 66% SQ3. В первый день суль-
фируют при 55° и на второй день при 85°, причем образующиеся сначала
2-нафтиламин-5,7- и -1,5-дисульфокислоты гладко превращаются в 2-наф-
тила мии-1,5,7-трисульф окис лоту:
По окончании сульфирования (которое в промышленности проводят
с различной скоростью) дают охладиться до 50° и выливают при постоян-
ном перемешивании в смесь 800 а ледяной воды и 1100 г льда, после
чего температура должна быть 60°. Выпадает чистая 2-нафтиламии-6,8-ди-
сульфокислота; ее через 6 час. отфильтровывают при 20° и отжимают.
Осадок весит около 210 г (соответствует 30 г NaNOa *).
* Это представляет собой принятое в технике обозначение. Так как на граммоль
какого-либо амина всегда расходуется при диазотировании 69 г NaNOz (= 1 молю
азотистой кислоты), то в общем говорят, что выход по нитриту соответствует Х%,
или X г. Так, если ла данное количество иафтиламинсульфокислоты расходуется 17 а
NaNO2, то говорят: получается 17 г нафтиламиисульфокислоты по нитриту, В техни-
ческих рецептах пишут также; «берут 7 г анилина по нитриту», т. е, берут
такое количество анилина, на которое расходуется 7 г нитрита, или десятая часть грам-
молекулы. Другими словами, в технических указаниях почти всегда рекомендуется брать
граммоль или килограммоль какого-нибудь амина, так как тогда для получения ка-'
кого-нибудь азокрасителя следует брать ранные количества нитрита, кислоты и щелочи.
В технике такое выражение; «выход соответствует 35 г по нитриту» — означает,
что истинны выход составляет 50%.
2-Амано*8*нафтал-6*сульфокислдта
183
Фильтрат, объем которого около 2300 мл, нагревают до кипения
в чашке; при этом температура повышается до 125°. Через 4 часа нагре-
вания при 125° (следует доливать воду взамен испаряющейся), отщеп-
ляется сульфогруппа в положении Г* и в течение 2 суток при 0° выде-
SO3H SO3H
ляется 2-иафтиламии-5,7-дисульфокислота, После фильтрования
и отжима оиа весит около 170 г; оставшаяся сер пая
кислота имеет примерно 40%-иую концентрацию и со-
держит в литре около 22 г сульфокислоты, которая те-
ряется, Количество 2 - и а ф ти л а м и н - 5,7 - ди с ул ь ф о кис-
лоты соответствует примерно 26 a NaNOa.
Отжатую 2-иафтиламии-6,8-дисульфокислоту растворяют в 700 мл
кипящей воды и прибавляют 70 г поваренной соли, Моиоиатриевая соль
чистой 2-иафтиламии-6,8-дисульфокислоты выделяется настолько обильно,
что содержимое сосуда превращается в твердую массу. Лепешку раздав-
ливаюФ,. фильтруют через 12 час, и сильно отжимают. С другой стороны,
отжатую 2-нафтил а ми и-5,7-ди сульфокислоту растворяют также в 850 мл
горячен воды и аналогичным образом осаждают 85 г поварен-
ной соли.
Чистые сульфокислоты отличаются очень характерной флуоресценцией — синей для
2-нафтила мин-6,8-дисульфокислоты и зеленой для 2-нафтиламин-5,7-дисульфокислоты.
В случае последней кислоты флуоресценция бывает отчетливой только у очень чистого
вещества н ее маскирует уже небольшая примесь 2-нафтиламин-б,8-дисульфокислоты.
Далее, обе кислоты различно ведут себя по отношению к уксуснокислому раствору
диазотированного п-нмтроанилина, 2-Нафтиламин-6,8-дисульфокислота дает с послед-
ним в разбавленном растворе лишь совсем слабую желтую окраску, обусловленную
образованием диазоаминосоединения; 2-нафтил а мин-5,7-дисульфокислота, напротив,
тотчас же образует настоящий красный краситель. Таким образом, возможно на осно-
вании интенсивности окраски сделать заключение о чистоте продукта. Кроме того,
диазотированная 2-цафтиламин-6,8-дисульфокислота сочетается с Р-соЛью и дает
очень трудно растворимый красный азокраситель, который даже при очень большом
разбавлении тотчас же выпадает в осадок, растворяющийся при кипячении с образо-
ванием красного раствора, 2-Нафтиламин-5,7-дисульфокислота дает очень легко рас-
творимый оранжево-красный краситель.
По более новому методу получения 2-нафтиламин-5 7-дисульфокислоты, позво-
ляющему избежать применения J3-нафтил а мин а, исходят из 2-нафтиламин-1-сульфокис-
лоты, которая в свою очередь получается из Р-нафтола через 2-нафтол-1-сульфокис-
лоту (см, стр, 176, 177), 2-Нафтиламин-1-сульфокислоту сульфируют и получают одну
2-нафтиламин-1,5,7-трисульфокислоту, от которой отщепляют сульфогруппу в положе-
нии 1, как это было описано выше.
б) 2 • Ами но - 8 • и а фтол - 6 - сул ьф оки сл ота
(у-кмслота). Мол. вес 239
ОН
* Можно кипятить также с обратным холодильником.
]«4
/. Промежуточные продукты
Плавление чистых нафтиламиндисульфокислот ие представляет труд- |
иостей; необходимо только, чтобы эти кислоты по возможности были сво- ]
бодными от солей. В автоклаве с мешалкой в течение 7 час. нагревают 1
при 205—210° 35 г по нитриту чистой сухой 2 - н а ф т и л а м и и - 6,8 - 1
д и с у л ь ф о к и с л о т ы (или соответствующее количество влажного 1
продукта) с 220 г едкого натра (не содержащего хлората) и 120 мл
воды. Давление поднимается до 14 ат. После охлаждения спускают дав- 1
лен не, содержимое автоклава разбавляют водой до 1 л (плав не должен . З
сильно пахнуть аммиаком) и подкисляют коп цеитриров а нно^ ]
серкой кислотой до отчетливой минерально кислой реакщтщ Tpjgi
буется около 250 г концентрированной серной кислоты. Через'”несколько |
часов отфильтровывают и хорошо промывают холодной водой. у-Кислота 'I
очень трудно растворима в воде. Отжимают и сушат при 100°. Ц
Выход нз 35 г нитрита около 105 г (95 г 100%-иой) чистой y-khcl 1
лоты, т. с. 80% от теории. \ - J
Y-Кислота определяется путем сочетания разбавленного сильноще- -я
лочного раствора с 1 н. раствором диазобеизола; параллельно у-кислоту я
диазотируют в очень разбавленном мииеральиокислом растворе (общие .1
данные о такого рода определениях см, в аналитической части). Числа,, 3
получающиеся при этих реакциях с анилином н нитритом, должны совпа- ..1
дать с точностью До одного процента, как и в случае Лш-кислоты. Если .1
плавление проводилось при слишком низкой, температуре, то нитритное 'Ji
число будет выше, чем число азосочетаиия. у-Кислота должна быть не д|
менее чем 91 %-ной. -я
в) 2-Амиио-5-нафтол-7-сульфокислота
(И - к и с л о т а, и з о - у - к и с л о т а) я
Мол. вес 239 Д
ОН
Плавление проводят точно так же, как и в случае, у-кислоты. ре-т
комендуется только брать немного больше воды, а именно вместо 120 мл j
160 мл. Нагревают в течение 7 час. при более низкой температуре (200—|
205°). Выход 2-амино-5-иафтол-7-сульфокислоты 95 г J
100%-нон (105 а 92%-ной). 1
Щелочное плавление обеих кислот дает однородные продукты, так^и
как a-стоящая сульфогруппа гораздо более реакционноспособна, чем |
£-стоящая.
И-кислота, полученная По вышеописанному способу, в общем доста-Я
точно чиста для технических целей.' Ее можно очистить еще дальше|
высаливанием ее натриевой соли и особенно переводом в труднораство-J
римую, хорошо кристаллизующуюся цинковую соль. 1
Технические примечания. К сульфируемому веществу прибавляют содуу
только для того, чтобы препятствовать образованию комков при смешивании с серной |
кислотой. Выделяющаяся углекислота разрыхляет смесь. Для этого достаточны очень?*
небольшие количества соды. В технике часто поступают несколько иначе, чем. было^
описано выше. ОбычнЪ сернокислый 0-нафт ила мин или свободное основание прямо
вносят в 40%-ный олеум. Отдельные сульфокислоты при правильном проведении
процесса разделить относительно легко, так как в большом масштабе они выделяются
лучше, чем в лаборатории. Чаще всего фильтруют через деревянные фильтр-лрессы,
с нитрофильтром (пронитрованная ткань) (см. стр, 32G). Очищенные кислоты или их
Фенил-Ч-кислота
185
кислые соли можно с успехом центрифугировать. Различные маточные растворы, кото-
рые в лабораторных условиях представляют собой неразделимые смеси, в зависимости
от степени их чистоты разделяют или перерабатывают вместе. С этой целью их пол-
ностью нейтрализуют содой и перерабатывают на соль, т. е. упаривают в многокор-
пусном выпарном аппарате в вакууме до тех пор, пока не начнет выпадать трудно-
растворимая поваренная соль. Ее отделяют центрифугированием, а маточный раствор
снова поступает в производство, Оказывается, что при дисульфнрованни всегда исче-
зает известная часть вещества, определяемого нитритом, Частично это объясняется
прямым сгоранием вещества, а частично образованием очень легко растворимых суль-
фонов или также сульфамидов; эти соединения легко обнаружить по их желтой окраске.
У-Кислота и Й-кислота, так же как и Аш-кислота являются важнейшими произ-
водными аминонафтолов в промышленности азокрасителей. Они применяются в боль-
ших количествах для получения красителей для шерсти и хлопка, у -Кислота приме-
няется в настоящее время еще в больших количествах, поэтому ее получают как нз
р-нафтиламнна, так и из нафтол-Г-кислоты. И-кислота в настоящее время приобре-
тает все более важное значение и вскоре достигнет значения у-кислоты, так как азо-
красители, получаемые из нее и ее многочисленных производных (см. табл. 13 и 14),
отличаются особенно хорошим сродством к хлопку, обладают чистыми тонами и ча-
стично высокой светопрочностью (см. бензо-светопрочный голубой 2 ГЛ, стр. 250).
Два других метода получения у-кислоты будут описаны очень коротко.
/. О 2-нафтол-6,8-дисулъфокислоте
Нагреванием с 25%-ным аммиаком и сульфитом аммония при 140° (давление
20 ат) нафтол-Г-соль превращают в амино-Г-кислоту. Метод выделения не предста-
; вляет особого интереса (NHa отдувают в конденсационную установку). Удается легко
; достигнуть превращения на 92% от теоретического, после чего всю реакционную массу
' плавят с едким натром.
\2. О нафтолдисулъфокислоте Г и 2,8-диоксинафталин-6-сульфокислоте
SO3H
/вк/А
г Т 2
ho3s—1 J
ОН
А NH3
ПГ^У-ОЙ 135°
он
I Т
НОзЗ-^Д^
У—кислота
Щелочное плавление
с NaOH и небольшим
ОН количеством воды при
200°, без давления HO3S
2,8-Диоксинафталин-б-сульфокислота.
Ее не выделяют, а смешивают с суль-
фатом аммония и часть свободной
шелочн нейтрализуют серной кислотой
NH?
Так получают вполне удовлетворительную У-кислоту: этот метод имеет то пре-
имущество, что давление в процессе реакция не превышает 15 ат. Интересно, что
амидируется лишь (3-гидроксильная группа, а а-гидроксильная группа не затрагивается
(см. стр. 162).
Таким образом, как это ни поразительно, можно изменить последовательность
описанных выше реакций, т. ё. сначала провести щелочное плавление и после этого
амидировать по Бухсреру. При этом даже не применяют водный аммиак, а исполь-
зуют более дешевый сернокислый аммоний, который едкой щелочью плава разлагается
на сульфат и аммиак. Далее, для того чтобы шла реакция Бухерера, оказывается
достаточным то количество сульфита, которое образуется при щелочном плавлении.
Фенил-у-кислота
ОН
186
1. Промежуточные продукты
В ко,лбе с обратным холодильником нагревают в течение 24 час.
239 г 100 % -ной у - к и с л о т ы, 750 г раствора бисульфита натрия,
содержащего 25% SO2, 750 мл воды и 200 г анилина. Затем прибав-
ляют концентрированный раствор со д ы до отчетливой щелочной реакции
и анилин отгоняют с водяным паром. При подкислении соляной кислоты
выделяется чистая феиил-у-кислота.
Выход 270 г 90 %-ной кислоты, т. е. 75—80%. ,
. 1-Амино-2-этоксннафталии-б-сульфокислот^
(этоксикислота Клеве)
NO3
/\/\/ОН /\/\/ОСзН5 /\/\/ОС2Нз
111 ~> I I I —> i I I
\/\/ \/\/ \/\/
а) 2 - Э то к с н и а ф та л и и
В пятигорлой колбе, снабженной мешалкой, термометром, капельной
воронкой и обратным холодильником, при нагревании растворяют 144 г
(1 моль) ^-нафтола в 58,5 мл 96%-иого спирта и прн сильном
перемешивании прибавляют по каплям 20 мл 78%-ной сер пой кис-
лоты с такой скоростью, чтобы температура ие превышала 80°. После
прибавления всего количества кислоты реакционную смесь медленно на-
гревают на масляной бане до 120° и выдерживают при этой температуре
и постоянном перемешивании 2 часа. Горячую еще смесь выливают
в предварительно нагретую делительную воронку, ннжиий водный слой
отбрасывают, а маслянистый слой несколько раз промывают теплой
водой (60°) до исчезновения кислой реакции. Для удаления непрореаги-
ровавшего ^-нафтола красновато-коричневое масло перемешивают долгое
время при 60° со смесью 15 мл 30%-иого едкогоиатран55юг воды,
снова отделяют в делительной воронке и промывают до нейтральной
реакции водой, нагретой до 60°. Перегонкой с перегретым до 150° водя-
ным паром получают почти бесцветное масло, которое при охлаждении
застывает с образованием серебристых белых кристаллов. Выход 120—
130 а, т. е. около 70—75% от теории. После одной перекристаллизации
из спирта продукт имеет т- пл. 36—37°.
Остающийся в щелочной вытяжке (3-нафтол в технике выделяют и снова вводят
в реакцию; таким образом можно повысить выход до 95%.
б) 1 - Н и т р о - 2 - э т о к с и н а ф т а л и н
В приборе, описанном для получения 2-этокси нафталин а,
растворяют 86 г (0,5 моля) 2-этокси нафта л ина в 74 г о-ди-
хлорбензола и нагревают до 40° иа водяной бане. Затем при хоро-
шем перемешивания* в течение 2 час. прибавляют по каплям 192 г
38 % -ной азотной кислоты так, чтобы температура не поднималась
выше 43°, После дальнейшего 6-часового перемешивания при 43° кислот-
ный слой отделяют в делительной воронке и остающееся красно-корич-
1в. 1-Нафтиламин-8,6-дисульфокислота
187
левое масло четыре раза промывают нагретой до 60° водой порциями по
70 мл, затем в течение получаса перемешивают при 60° с 500 мл 10%-ного
раствора соды, снова отделяют в делительной воронке, промывают теп-
лой водой до нейтральной реакции и отсасывают выделившееся при охла-
ждении основное количество нитропродукта. Из фильтрата частичной от-
гонкой о-дихлорбензола в вакууме можно выделить еще некоторое коли-
чество кристаллического вещества. Неочищенный продукт, полученный
в виде оранжевых кристаллов (82—85 г, 75—77%), содержит еще не-
значительное количество изомерных 6- и 8-нитро-2-этоксииафталицов, ко-
торые можно удалить повторной перекристаллизацией из спирта. Очи-
щенный таким образом продукт получается в виде желтых кристалличе-
ских игл с т. пл. 102,5°. Выход около 67 г, т, е. 62% от теории.
в) 1 Ам и и о - 2 это ксин афта л ни
Нитроэтоксииафталии восстанавливают по Бешану железом и соля-
ной или уксусной кислотой аналогично тому, как это было описано при
получении а-нафтнламииа (см. стр. 157). Продукт восстановления также
можно отогнать с перегретым водяным паром и очистить перегонкой
в вакууме. Образуются бесцветные призмы с т. пл. 51°, которые иа свету
Постепенно окрашиваются в фиолетовый цвет.
\ г) 1-Амино-2-этоксииафталии-6-сульфокислота
В ..трехгорлой колбе, снабженной мешалкой и термометром, нагре-
вают иа водяной бане до 50° 200 г 92,5%-иой серной кислоты и
виоеят при постоянном перемешивании 47 г измельченного в тонкий по-
рошок 1- амино -2- этокс и нафтали я а с такой скоростью, чтобы
температура ие поднималась выше 58°. Образующийся прозрачный рас-
твор перемешивают еще 2 часа при 60°, затем медленно, по каплям, при
перемешивании приливают к 500 мл нагретой до 60° воды. Сульфокис-
лота тотчас же выделяется в виде белого осадка, который по охлаждении
отсасывают и промывают водой, Выход почти количественный.
Технические примечания. 2-Этоксннафталин под названием «новый
н е р о л и н» применяется как душистое вещество.
1-А'мино-2-этоксинафталин-6-сульфокислота применяется прежде всего как про-
межуточное соединение для получения прямых вторичных полиазокрасителей, как на-
пример сириус голубой 6Г; анилин-2,5-днсульфоки слота -► а-нафтиламин ->1-амияо-
2-этоксинафталин-6-сульфокислота-»-фенил-И-кислота. Этоксигруппа обусловливает
сдвиг оттенка в зеленовато-синий. Сочетание, с конечной составляющей целесооб-
разно проводить в присутствии пиридина (см. атр. 230).
16, 1- Нафтил амин-3,6-дисульфокислота
(кислота Фрейнда)
В сосуде, описанном на стр. 94, нагревают до 165° прн постоянном
перемешивании 128 г чистого нафталина н прибавляют к нему по
каплям в течение 15 мин. 400 г 100%-ной серной кислоты (моно-
гидрата). Нагревают в .течение 1 часа при 165°; при этом днеульфнрова-
иие заканчивается *. Нагревать выше 165° не рекомендуется, так как
* Можно довольно легко убедиться в том, насколько далеко прошло сульфиро-
вание. Для этого сульфомассу нейтрализуют карбонатом бария и действием необхо-
димого количества соды получдют натриевую соль; по количеству затраченной соды
рассчитывают, сколько имеется сульфогрушн 53 г NaaCOg =!SOgH.
Не следует применять мел, так как гипс растворим в сульфокисло-
тах ряда нафталина. ,
188
1. Промежуточные продукты
Кислота Фрейнда
(1,3,6 = 5,2, 7)
иначе образуется до 30% 2,6-дисульфокислогы, которая, однако, дает
совершенно аналогичный продукт, как и 2,7-изомер,
Охлаждают до 15° и нитруют, прибавляя по каплям в течение часа
103 г 62%-пой азотной кислоты (уд. вес 1,38) так, чтобы темпе-
ратура ие поднималась выше 30°, иначе легко могут образоваться ди-
иитросоедииения. Массу оставляют стоять при комнатной температуре ие
менее чем иа 10 час, и затем выливают при перемешивании в 1 л холод-
ной воды. Нагревают до 70° и пропускают воздух до прекращения выде-
ления окислов азота; избыток азотной кислоты разлагают сульфатом
железа (II), как это было описано при получении кислот Клеве
(стр. 165). Для получения натриевой соли прибавляют 150 г безводного
(или соответствующее количество кристаллического) сульфата
натрия. Избыток серной кислоты удаляют при помощи мела (около
400 а) и после отфильтроваиия гипса раствор восстанавливают так же,
как и в случае кислот Клеве (стр. 165), с той лишь разницей, что железо
протравляют не уксусной, а соляной кислотой (20 мл коицен-,
трироваииой кислоты).
Раствор, полученный после восстановления, нейтрализуют содой
(проба с сернистым аммонием иа полноту осаждения железа!), после
фильтрования упаривают до 800 мл и к кипящему раствору прибавляют
ЮОгхлористого калия. После того как хлористый калий раство-
рится, подкисляют при 100° 100 мл 30%-иой СОЛЯНОЙ КИСЛОТЫ И:
дают охладиться. Через 12 час. обильно выделяется кислая калиевая^
соль и вся масса застывает. Фильтруют на большом иутч-фильтре с двойд’
иым бумажным фильтром и остаток споласкивают маточным раствором.’’
Под конец заливают осадок 200 мл насыщенного раствора поваренной.,;
соли и, как только возможно, сильно отжимают на винтовом прессе.
Влажный продукт весит 300—320 г, сухой-— около 270 а?
и с о отв етств ует примерло 35 а нитрита натрия*. ''
Маточный раствор, особенно нри неправильном провидении восстановления;•;
темного цвета и соответствует 22 г нитрита натрия. Он содержит аминонафтол--
сульфокислоты, которые при диазотировании дают фиолетовый раствор. Вместо калие-’
вой соли можно также получить натриевую соль; тогда для осаждения вместо 100
.хлористого калия применяют 100 г поваренной соли. Однако осадок фильтруется эна-,
чительно хуже и конечный продукт поэтому не получается таким чистым, как в слу-
чае калиевой соли. Растворением осадка в четырехкратном по весу количестве воды
при 100" и кристаллизацией получают очень чистую кислоту Фрейнда, особенно при-
годную для получения сложных полиазокрасителей.
* См. сноску на стр. 182.
17.
1-Амино-8-нафтол-3,6-дисульфокислота
189
17. 1-Амино-8-нафтол-3,6-дисульфокислота (Аш-кислота)
а) 1-Нафт илам ин - 3,6,8 - трисульфокислота
Jk и с л о т а Коха)
В литровом железном сосуде (или стеклянной колбе, или фарфоровом
стакане с хорошо закрывающейся свинцовой крышкой) нагревают до 150°
128 г чистого нафталина и при хорошем перемешивании прибавляют
по каплям 140 а моногидрата так, чтобы температура ие поднима-
лась выше 160—165°. Это количество моногидрата прибавляют приблизи-
тельно 33 Ю—13 мни., затем перемешивают еще полчаса при 160—165°,
охлаждают сульфомассу до 100° и снова прибавляют, по возможности
быстрее, 260 г моногидрата. Охлаждают до 30° и при этой темпе-
ратуре медленно в течение 30 мин. прибавляют по каплям 400 а олеума,
содержащего 60% SO3. Перемешивают еще 3 часа при 25° и нагревают
7 час. при 165°. Трисульфирование (кроме неизбежного тетрасульфирова-
ния) после этого заканчивается. Реакционный сосуд охлаждают ледяной
водой до 10°, прибавляют внутрь 15 г льда и по каплям 103 г 62%-ной
азотной кислоты (уд. вес 1,38), Температура ие должна превышать
10°, так как иначе будет происходить сильное окисление. Азотную кислоту
следует прибавлять в течение часа. После прибавления всего количества
азотной кислоты смесь оставляют стоять иа ночь. Затем массу выливают
в 2 л Голодной воды. Выделяются окислы азота н температура может
подняться до 70°. Окислы азота удаляют продуванием воздуха в течение
приблизительно часа при 70° Прибавляют при 75—80° 20%-ный раствор
сульфата железа (Ц) до уех прр, пока проба не будет больше
давать реакцию на окислы азота (пробу, разбавленную водой, испыты-
вают иа йодокрахмальную бумагу или реактивом «сульфон» (ср. стр, 165,
сноска).
Восстановление проводят следующим образом: сернокислый раствор
охлаждают до 30° и небольшими порциями постепенно прибавляют 180 а
железных гвоздей при непрерывном перемешивании. Темпера-
тура в течение 2 час, поднимается примерно до 60°, Перемешивают в те-
чение 4 час, и нагревают раствор в продолжение часа до 70°. Железо
частично переходит в раствор, и реакционную массу оставляют иа ночь.
Выделяется железная соль 1-нафтиламин-3,6,8-трисульфокнслоты (кис-
лоты Коха), Затем нагревают до 90°, при этом соль переходит в раствор.
Раствор сливают с непрореагировавшего железа и при хорошем пере-
мешивании прибавлением,, твердой поваренной соли осаждают кислую
натриевую соль кислоты Коха. Поваренной с о Л и берут столько,
Чтобы получился ее 18%-иый раствор. Сульфат железа (II) в этих усло-
виях не выпадает. Массу охлаждают при постоянном перемешивании до
199
1. Промежуточные продукты
25° и вскоре (при долгом стоянии может выделиться железный купорос)
отфильтровывают через тройной фильтр на большом путч-фильтре. Оса-
док хорошо промывают 20%-ным раствором поваренной соли^для удале-
ния соли железа и сорной кислоты; затем осадок растворяют при нагре-
вании в 1 л воды и необходимом количестве соды^ снова фильтруют
па путч-фильтре. Раствор нагревают до 80° и осадУдают кислоту Коха
18% поваренной соли (считая на объем) и соляной кислотой до сильно-,
кислой реакции на конго. 1-Нафтиламии-3,6,8-трисульфо.кислота выпадает )
в виде белоснежной кислой натриевой соли. Ее отфильтровывают через
12 час. и хорошо отжимают иа винтовом прессе. Получают1 около 350 г
влажного продукта, соответствующего приблизительно 38—42 г нитрита,
т. е. с выходом около 55—60% от теории.
Технические примечания. Производство кислоты Коха является одним?
из важнейших процессов в химии красителей, потому что получающаяся из нее?
1-амино-8-нафтол-3,6-дисульфокнслота, а также аналогичная ей хромотроповая кислота
(1,8-диоксинафталин-3,6-дисульфокислота) принадлежат к промежуточным продуктам,.!,;
которые наиболее часто используются при промышленном получении красителей.?
В производстве обычно сульфируют 2 моля (256 кг) нафталина и для облегчения про-’
цссса сульфирования прибавляют сульфат натрия. Необходимую температуру поддеру
живать довольно трудно. Чаще всего нагревают с помощью водяного пара в аппара-!
тах с двойными стенками (см. рис. 31а, стр. 170), что позволяет быстро нагревать иJ
охлаждать. Особенно трудно проводить нитрование потому, что при работе с боль-;
щими количествами образовавшаяся нафталин-1,3,6-трисульфокислоТа легко выпадает?
в осадок. Вследствие этого находящийся в растворе остаток будет подвергаться да-<
леко идущему окислению азотной кислотой. Поэтому часто начинают нитровать уже;;
при 80°; эта температура, правда, слишком высока, но имеет то преимущество, чтоС
в этих условиях удается избежать кристаллизации. После прибавления небольшого?;
количества азотной кислоты можно легко охладить реакционную массу до 20°, уж$£
нс опасаясь выделения трисульфокислоты. Остальные операции проводятся почти^
так же, как в лаборатории, и потому они не требуют дальнейших пояснений. 3
Эти операции исследовались во всех вариантах, из которых упомянем следую-’’
щие; вместо того чтобы восстанавливать кислую нитромассу, ее можно известковать)
(нейтрализовать известью или мелом) и затем восстанавливать с совсем небольшим;
количеством кислоты, как описано (см. стр. 165) в случае кислот Клеве (1.7-и 1,6-наф-?
тиламинсульфокислот). Далее, нитротрисульфокислоту можно легко высолить и от-)
фильтровать. Однако это связано с техническими трудностями, так как переработка'
таких сильнокислых масс неприятна. Сульфирование можно также проводить и так,;
чтобы в качестве промежуточного соединения образовывалась главным образом нафо
талин-2,7-дисульфокислота (вместо 1,6-дисульфокислоты, как это имеет место при!
проведении реакции по вышеописанной методике), * ?
Исходя из 2,7-нафталиндисульфокислоты, Аш-кислоту можно1 получать принци*
пиально другим путем, как это видно ИЗ схемы, приведенной ниже. !)
NOaNOa
io. i *пафтиламин-5-сульфокислота й 1-нафтиламин~8~сульфокислота
191
Выходы Аш-кислоты при этом почти такие же. как и по описанному методу, однако
она получается нс такой чистой.
б) Щелочное плавление 1 - и а ф т и л а м и и - 3,6,8 -
трису ль фокнслотьт до 1 - а м и но - 8 - нафтол - 3,6-
дисуль'фокислоты (Аш-кислоты)
При щелочном плавлении описанной аминокислоты образуется так
называемая Аш-кислота (1-амиио-8-нафтол - 3,6 - д и с у л ь-
фо кислота), являющаяся важнейшим промежуточным продуктом
в промышленности красителей, получение которой представляет собой ти-
пичный пример такого рода реакций.
Для получения Аш-кислоты с хорошим выходом надо, чтобы темпе-
ратура ие поднималась выше 190° и чтобы едкий натр был по меньшей
мере 30%-иым. При проведении плавления в лаборатории рекомендуется
брать следующие количества: ияфтиламиитрисульфокислоты
по нитриту 28, т. е.«около 280 г влажного продукта, 130 г едкого
натра, 130 мл воды.
Плавление проводят в автоклаве. Этим аппаратам, играющим столь
важную роль как в лаборатории, так и в технике, посвящена специаль-
ная глава (см. ниже). В аппарат загружают указанное количество ве-
1 ществ и плавление проводят при постоянном перемешивании и иагрева-
\ иии до 178—180° в течение 8 час. при давлении 7 ат. По охлаждении
спускают избыточное давление через вентиль и открывают автоклав.
Если., плавление было проведено правильно, то плав Аш-кислоты имеет
грязноватую тупую желтую окраску. Если масса слишком светлая, то
плавление ие закончено; если она коричневая и очень сильно пахнет
аммиаком, то плавление проводилось слишком долго. Одиако даже прь/
осторожном плавлении всегда отщепляется немного аммиака.
Получают густую сиропообразную массу, смешанную с зернами кри-
сталлов безводного сульфита натрия. Эту массу переносят в керамиковый
сосуд емкостью 2 л, разбавляют 1 л воды и подкисляют 50%-ной серной
кислотой до постоянной сильиокислой реакции иа конго. Надо иметь в
виду, что бумажка коиго может синеть от действия серцистой кислоты,
которая, однако, быстро улетучивается (тяга!). Амиионафтолдисульфо-
кислбта выделяется в виде мелких белых кристаллов кислой натриевой
соли; Ьиа очень трудно растворима в концентрированном растворе суль-
фита натрия *. Все же для полноты выделения ее оставляют стоять на
. несколько часов. Затем отфильтровывают и осадок хорошо промывают
10%-иым раствором соли, к которому, прибавлен 1% соляной кислоты.
Не следует промывать слишком долго, так как это ведет к потере Аш-кис-
лоты. Отжимают на виитовбм прессе и сушат прн 100°.
Выход составляет около 100 г 100%-ной Аш-кислоты, или ПО г
86%-ной кислоты.
Аналитическое определение см. в аналитической части, стр. 344.
18. 1-Нафтиламии-5-сульфокислота
и 1-иафтнламнн-8-сульфокнслота
Получение 1-нафтил амии-5- и 1 -нафтил амии-8-сульфокнслот очень
сходно с получением кислот Клеве. Они принадлежат к наиболее употре-
бительным промежуточным продуктам.
* Растворимость Аш-кислоты: при 18’ в воде 0,93%, при 18° в 10%-ном NaCI
г 0,053%, при 18е в 10%-лэм NaCl + 0,8% НС1 0,023%, при 100° в воде 8%.
Нафтосультон
IV
SO3H SO3H
1-Амино-8~нафтол-
-4-сульфокиСлотз
(С-кисл^та или чй-
каго-С-кислота)
1-Амино-8-нафтол-
~2,4~дису льфокис лота
(2С-кислота, Чикаго—
-СС-кислота или
СС-кислота)
1-Нафтилами«-8-сульфокислота диазотируется азотистой кислотой (нитритом
натрия) в минеральнокислом растворе при 25 , При нагревании этого диазосоедине-
ния в водном растворе до 55° образуется с количественным выходом нафтосуль-
тон (1). По количеству полученного сультона можно судить о чистоте исходной
кислоты. Нафтосультон в свою очередь почти всегда превращается в нафтосультои-
сульфокислоту, которая дает с диазосоединениями очень чистые и светопрочные кра-
сители. В последние годы эти красители в значительной мере утратили свое значение.
Далее, 1-нафтила^ ин-8-сульфокислоту можно превращать в армированные
соединения. Нагревая свободную 1 -нафтиламин-8-сульфокислоту с анилином и.соля-
нокислым анилином, получают технически важную N-ф е н и л-1-н а ф т и л а м и н-8-
19. На фталин-1,5-дисульфоки слота
19
Реакция осуществляется при нагревании до 160° в эмалированном котле на мае
ляной бане одной части чистой свободной сульфокислоты с трехкратным количествен
анилина (n-толуидина) в присутствии солянокислого анилина или соответствен^
солянокислого п-толуидина. Воду, которая всегда содержится в веществах, отгоняю-
в вакууме; после этого нагревают в течение 24 час. при постоянном перемешивании
Затем избыток анилина осторожно отгоняют в вакууме, разлагают анилиновую сож
образовавшейся фенилированной кислоты точно рассчитанным количеством раствор;
едкого натра; выделившийся анилин отгоняют с водяным паром и получают, таким
образом, раствор желаемой феннлнафтиламинсульфокислоты; этот раствор можно
прямо вводить в реакцию сочетания с раствором диазотированной Аш-кислоты
в уксуснокислой среде.
При правильном проведении процесса нет необходимости выделять кислоту.
Так, получают светопрочный кислотный синий 2 К (сульфон кислотный синий Р),
При сульфировании в соответствующих условиях нафтил аминсульфокислоты
олеумом в зависимости от степени воздействия, получают ди- или трисульфо-
кислоты нафтиламина (илн ангидрид последней, — нафтсультамдисулъфо-
кислоту) *. Оба соединения дают прн щелочном плавлении соответствующие амино-
пафтолсульфокислоты Ш и IV. Эти сульфокислоты являются исходными соедине-
ниями для получения красителей для шерсти и хлопка.
1-Н а ф т и л а м и н-5-с у л ь ф о к и с л о т а—менее важное соединение, и о иеЙ
упомянем лишь вкратце. Ее либо диазотируют и сочетают с аминами или нафтолами»
либо превращают в 1-е м и н о-5-н а ф т о л-7-с у л ь ф о к и с л о т у (М-кислоту).
SO3H SO3H $О3Н ОН
Как видно из формул, 1-нафтиламин-5-сульфокислоту в отличие от 1.8-кислоты
сначала ацетилируют, так как иначе она разрушается серным ангидридом. Такое
ацетилирование в производстве красителей имеет довольно важное значение
(см. амидонафтоловый красный Г, кислотный ярко-красный).
19. Нафтални-1,5-дисульфокислота
В пятигорлой полулитровой колбе с мешалкой (гидравлический за-
твор;— концентрированная серная кислота), термометром, хлоркальцие-
вой трубкой и капельной воронкой емкостью 200 мл, снабженной хлор-
кальциевой трубкой, помещают 300 г моногидрата н охлаждают льдом до
4-5°. Затем при этой температуре н перемешивании вносят в течение
15 мин. небольшими порциями 128'а (1 моль) чистого порошкообразного
нафталина. Перемешивают еще полчаса и после этого, продолжая охла-
ждать льдом и следя за тем, чтобы температура ие превышала 30°, .мед-
ленно прибавляют По каплям 300 г олеума, содержащего 66% SO3. После
прибавления всего количества олеума, что продолжается около 1,5 часа,
смесь медленно нагревают до 40° и перемешивают в течение 8 час, при
этой же температуре.
Очень густую, но еще легко перемешиваемую массу выливают при
размешивании в смесь 1200 мл насыщенного рдствора поваренной соли
и 600 мл воды; при этом происходит саморазогревание до 60°. Нагревают
четверть часа при 90°, в продолжение этого времени мелкий сначала оса-
док становится крупнозернистым, и смесь оставляют охлаждаться при
перемешивании. На следующий день кристаллический осадок отсасывают
без сильного отжима и, не допуская образования трещин, промывают
800 мл насыщенного раствора поваренной соли; бумажка коиго и е должна
* Wielopolski, Диссертация, Zurich — Linz, 1936.
196 1, Промежуточные продукты
больше синеть. Для того чтобы вытеснить основное количество поварен-
ной соли, удерживаемой осадком, его промывают с начал /смесью 100 мл
насыщенного раствора поваренной соли и 200 мл воды, /атом еще 100 мл
чистой воды и под конец хорошо отжимают. Полученном таким образом
нафталин-1,5-дисульфокислый натрий после сушки в паровом сушильном
шкафу содержит еще 1 моль кристаллизационной водьР ц весит около
220—230 г (что соответствует выход}' около 65%); если промывка про-
водилась правильно, оп практически свободен от изомеров и лишь содер-
жит немного поваренной соли, и может применяться непосредственно для
многих целей.
Совершенно чистый продукт, не содержащий поваренной соли, легко
получают перекристаллизацией из небольшого количества кипящей воды.
Его можно очистить с меньшими потерями, если растворить ранее полу-
ченный неочищенный продукт в 800 мл кипящей воды, смешать с равным
объемом насыщенного раствора поваренной соли, перемешивать неко-
торое время при нагревании и оставить охлаждаться иа ночь. Красивый
кристаллический осадок надо сильно отсосать, промыть для удаления
поваренной соли 200—300 мл холодной воды, спиртом и эфиром и сушить
в паровом сушильном шкафу. Так получают около 180 г совсем чистого
продукта, не содержащего поваренной соли, что соответствует 51%-иому
выходу.
Из промывных вод, полученных при промывании сырого и перекри-
сталлизованного продукта разбавленным раствором соли и водой, при
упаривании до половины объема можно получить еще вторую фракцию,
которую легко очистить, как это только что было описано.
1-Нафтол-5-сульфокислота
52,5 г (0,15 моля) нафталин-1,5-дисульфокислого натрия (перекри-
сталлизованного из воды и высушенного в паровом сушильном шкафу)
помещают во вращающийся автоклав, заливают 200 мл 16%-пого рас-
твора едкого натра и нагревают при постоянном перемешивании в тече-
ние часа до 260—265°, затем выдерживают при этой температуре
5 час. Охлаждают до 90°, открывают автоклав и выливают содержи-
мое в стакан, а автоклав ополаскивают возможно малым количеством
горячей воды. К еще горячему раствору при перемешивании прибав-
ляют концентрированную соляную кислоту до нейтральной реакции на
лакмус и фильтруют. Фильтрат снова кипятят, опять осторожно под-
кисляют концентрированной соляной кислотой (выделяется SO2!) до
сильного и неисчезаюшего посинения коцго-бумажки и прибавляют,
наконец, 50 г поваренной соли. По охлаждении выпавшую мононатрие-
вую соль 1-нафтол-5-сульфокислоты хорошо отсасывают на нутч-фильтре,
промывают раствором поваренной Соли и сушат в паровом сушильном
шкафут
Чистоту полученной соли целесообразно определять титрованием 0,1 и. <
раствором диазотированного ацетил-я-фепнлендиамнпа (методику диазо-.
тирования см стр. 222, титрование см. стр. 3-14, 345). -
В ыход составляет около 80—85%.
Продукт можно очистить от соли перекристаллизацией из неболь- 1
шого количества кипящей воды. Для очистки может быть также при- ч
менен перевод’в хорошо кристаллизующуюся цинковую соль. (
Технические примечания. 1 -Нафтол-5-сульфокислотэ является _ важной ;
азосостаилягощей .тля получения лаковых азокрасителей, как например гелно- (
бордо, (лак бордо СК.) (см. стр. 241). 4
Нафтцдаминдисульфокислоты
197
При плавлении на фталин-1.5-дисульфокнслоты с концентрированной щелочью вто-
рая сульфогруппа также замещается на окспгруппу. Таким образом получают
1,5-диоксниафталнн, который также является важной азосостапляющей для получения
азокрасителей, в особенности хромирующихся, например диамант черный II фау.
1- Нафтил амин-4,8-дисульфокнслота, 2-иафтил амин-4,8-
дисульфокислота н 1-нафтнламин-3,8-дисульфокислота
900 г моногидрата охлаждают в котле с мешалкой до 5° и при
хорошем перемешивании вносят небольшими порциями в течение четвер-
ти часа 384 г чистого порошкообразного нафталина. После приба-
вления всего количества нафталина перемешивают еще в течение 30 мин.
и затем прибавляют по каплям 900 г олеум а, содержащего 64% SO3,
с такой скоростью, чтобы температура ие превышала 30°. Затем массу
медленно нагревают до 40° и выдерживают прн этой температуре в те-
чение 8 час.
Приливают 10 мл воды и нитруют, прибавляя по каплям 305 г
62%-иой азотной .кисло ты (уд. вес 1,38) при 15—20°. Оставляют
стоять на ночь и затем выливают в 3,5 л воды.
е-кислота
Переработку осуществляют двумя способами: известкуют и восстанавливают по
Бешану (см. стр. 71 и 72) или иитрокислоты высаливают 1250 г поваренной
соли, Второй метод состоит в том. что 2-нитронафталинД,8-дисульфокислоту оса-
ждают 200 г железного купороса, отфильтровывают и маточный раствор из-
весткуют.
Дальнейшая обработка такая же, как в случае кислот Клеве, но только без при-
бавления окиси магния.
Если 2-нитронафталин-4,8-дисульфокнслота осаждена железным купоросом, то
как ее, так и маточный раствор после известкования восстанавливают по Бошану.
Восстановлеи.ные растворы упаривают так. чтобы они содержали примерно до
г 20% твердого веществами перерабатывают дальше.
1-Нафтиламин-4,8-д «сульфокислоту осаждают в виде средней натриевой соли
таким количеством поваренной соли, чтобы получился 20%-ный раствор поваренной
соли (уд. вес 1,26), и выделившуюся соль отфильтровывают. 1-Нафтиламин-3,8-ди-
сульфокислоту осаждают соляной кислотой в виде кислой натриевой соли. Если из-
вестковалась вся масса, то маточный раствор от 1-нафтиламии-3,8- дисульфо-
кислоты нейтрализуют, упаривают до тех пор, пока выделится поваренная соль,
сливают с нее раствор и оставляют кристаллизоваться при 20 . Выделяется средняя
натриевая соль 2-нафтиламин-4,8-дисульфокислоты. Ее растворяют в воде (раствор
должен иметь уд. вес 1,20) и подкисляют концентрированной соляной кислотой до
кислой реакции на конго, дают охладиться и отфильтровывают.
В зависимости от методики работы получают приблизительно следующие вы-
ходы: общий выход продуктов реакции составляет 55—68% от теории. На долю
1-нафтиламин-4,8-дисульфокислоты приходится около 70%, 1-нафтиламин-3,8-дисуЛЬ-
фикослоты — 16% н 2-нафтиламин-4,8-диеульфокислоты — 14%. Кроме того, обра-
зуются еще изомерные кислоты—1-иафтиламин-4,7- и 2-нафтиламин-4,7-дисульфокис-
лоты, которые, однако, трудно отделяются от 2-нафтилам ин-4,8- дисульфокислоты *.
Поразительно, что даже при очень тщательной работе никогда не удается по-
лучить больше 70% вещества, титруемого нитритом, хотя азотная кислота и израсхо-
довалась (нитрометрическое определение). Причина в том, что при восстановлении,
как и в других аналогичных случаях, амин разрушается. Необходимо всегда, как и
в случае Аш-кислоты и других нафтиламинсульфокислот, перед восстановлением
удалить и разрушить азотную кислоту (см. стр. 165).
нафт о л-4-сульфокислота
so3H
III
Нафтсультамднсульфо-
кислота (дает желтую соль
1 “Амино —8—нафтол-
-2,4-д и сульф ок и с-
Аммонийная соль
1-иафт о л-3,8-Яи сульфо-
кислоты, Е-кислота
Edelmann R. F., Диссертация, Zurich, 1925.
1-Амино-8-нафтол~4,6-дисульфокислота
19!
Описанные выше сульфокислоты являются важными исходными соединениями
для получения азокрасителей и других красителей, например светопрочного синего
для шерсти Б Л *. р
1-Нафтиламин-4,8-дисульфокислота щелочным плавлением может быть превра-
щена в 1-амино-8-нафтол-4-сульфокиелоту (С-кнелоту). Эта кислота может быть
также просульфирована дальше и полученный сультам превращен 'в очень ценную
чикаго-кислоту. Далее 1-нафтиламин-3,8-дисульфокислота нагреванием ее кислой
натриевой соли с водой. при 180—200° может быть превращена в I-нафтол-3,8-ди-
сульфокислоту. Наконец, 2-нафтиламин-4,8-дисульфокислота применяется для получе-
ния прямых красителей, как например нафтоген синий РР (прямой дназосиний 1Q **,
Ход реакций пояснен на схеме, а также табл. II.
1 -Амино-8-нафтол-4,6-д и сульфокислота ?
( К-кислота)
МО2 SOgNa
Na
SO3Na
NH£ SO3Na
AA
I I I
X/X-/4SOf;Na
SO3H
' NHa OH
I I
, SO3H
a) 1 - H а ф т и л а м и н - 4,6,8 - т р н с у л ь ф о к н с л о т а
В пятигорлой литровой колбе нагревают 800 г моногидрата до 80®.
Прн этой температуре вносят небольшими порциями при перемешивании
332 г (1 моль) тонко измельченной дннатрневой соли нафталин-1,5-
дисульфокислоты (получение см. стр. 195, 196), предварительно пере-
кристаллизованной из воды и высушенной при 120°, и одновременно при-
бавляют по каплям 470 г 66%-ного олеума. Помутневшую вследствие
выделения ангидрида 1,5-днсульфокислоты *** смесь нагревают 6,5 часа
при 90-—95°, затем еще полчаса при 99°, при этом раствор становится про-
зрачным. Сульфирование окончено, если вылитая на лед\и смешанная
с поваренной солью проба остается. прозрачной при стоянии. Сульфо-
массу медленно смешивают с 250 мл воды, причем температура не должна
подниматься выше 90°, и перемешивают еще час при 90°.
Охлаждают до —5° и прибавляют по каплям при сильном перемеши-
вании смесь 68 г азотной кислоты (уд. вес 1,52) и 40 мл, концентриро-
ванной, серной кислоты с такой скоростью, чтобы температура не под-
нималась выше 2°. Перемешивают еще в течение часа и “контролируют
расход азотной кислоты при помощи нитрометра. Густой прозрачный рас-
твор, окрашенный в оранжевый цвет, выливают при перемешивании
в смесь 4 л воды н 2 кг льда и пропускают в течение получаса воздух
для удаления нитрозных газов. После этого нейтрализуют, прибавляя
около 1,5 кг мела, отсасывают выделившийся гипс и три раза промывают
водой. Соединенные фильтраты (около 9 л) упаривают в вакууме на
* Fierz- David, Kiinsfliche organische Farbstoffe, S. 332.
** Fierz-David, Kiinsfliche organische Farbstoffe, S. 160.
*** Helv. Qnim. Acta, 32, 631.
водяной бане с температурой 50—55° до 4 л и отфильтровывают допол-
нительно выделившийся гипс. J
Восстановление можно осуществлять при помощи сульфгидрата
аммония или водородом в присутствии никелевого катализатора.
у
а) Восстановление сульфгидратом амМония.
Приготовление сульфгндрата аммония: в 600 г 23%-ного аммиака,
содержащего 138 г NHs, пропускают прн охлаждении льдом сероводород
до привеса в 276 г.
Половину полученного ранее раствора ннтрокислот (2 л) смешивают
с 50 мл 23%-ного раствора аммиака н нагревают до 60°. Прн этой тем-
пературе и перемешивании прибавляют по каплям 320 мл сульфгндрата
аммония н нагревают затем до кипения. После получасового кипячения
подкисляют соляной кислотой до кислой реакции на конго н снова кипя-
тят в течение часа. После двухчасового стояния отфильтровывают вы-
павшую серу. Общий выход амипосоединеннй определяют титрованием
нитритом натрия аликвотной части раствора. Оставшийся раствор упа-
ривают в вакууме до 400 мл-, при этом снова выделяется немного гипса,
который отфильтровывают. Фильтрат нагревают до 80°, прибавляют 50 а
поваренной соли и сильно подкисляют соляной кислотой. По охлаждении
отфильтровывают выделившуюся в виде динатрневой солн 1-нафтнламнн-
4,6,8-трнсульфокислоту н определяют ее выход титрованием аликвотной
части осадка нитритом натрия. Для дальнейшей очистки сырой продукт
можно растворить в рассчитанном количестве раствора соды н снова оса-
дить соляной кислотой,
Общий выход амнносоедннепнй в растворе (по титрованию) 68% от
теории, выход выделенной нафтиламинтрнсульфокислоты 45% от теории,
считая на взятую нафталин-1,5-дисульфокнелоту.
Восстановление водородом-в присутствии
никелевого катализатора
Приготовление катализатора. В круглодонной колбе с широким воз-
душным холодильником нагревают в течение часа в вакууме до 180° 100 г
формиата никеля (двухводного), смешанного со 100 г парафина и 20 г
парафинового масла, и затем быстро повышают температуру до 240°. При
этой температуре формнат разлагается н вакуум снижается вследствие
выделения газов. В продолжение 3 час. температуру повышают далее до
260°. Разложение окончено, когда давление снова понизится до 18 мм.
Массу выливают на железный противень, дают застыть и как можно тща-
тельнее удаляют верхний слон парафина. Оставшуюся мерную массу
непосредственно перед употреблением промывают на путч-фильтре боль-
шим количеством горячей воды, пока не выплавится основное количество
парафина. Оставшийся порошок промывают чистым спиртом для удале-
ния воды, затем несколько раз замешивают с петролейпым эфиром н от-
сасывают, пока порошок не будет тотчас же сухнм и совершенно рыхлым.
Только полное удаление парафина обеспечивает хорошую смачиваемость
катализатора водой.
Восстановление. Аппаратура: автоклав на 60 ат рабочего давления,
снабженный масляной баней, эмалированным вкладышем н вибромешал-
кой. Вибрация передается в радиальном направлении при помощи вала
нз стали V2A, на конце которого внутри автоклава находится наклонно
установленная тонкая пластинка. Эта пластинка, с одной стороны, за-
ставляет катализатор циркулировать в жидкости, с другой стороны, спо-
собствует тонкому распределению газа в жидкости; таким образом про-
исходит очень тесное соприкосновение между тремя фазами.
500 мл полученного ранее раствора нитросульф окне л от помещают
в автоклав и при помощи уксусной кислоты н ацетата натрия доводят pH
раствора примерно до 5, после чего прибавляют 4 г катализатора. Авто-
клав закрывают н трижды на-
полняют водородом до давле-
ния 20 ат н снова выпускают
его (благодаря этому содержа-
ние кислорода воздуха в авто-
клаве снижается до 0,001 %);
затем автоклав наполняют прн
комнатной температуре водоро-
дом до давления 50 ат, прове-
ряют герметичность и нагре-
вают до 50°. Восстановление
начинается прн включении виб-
ромешалкн; за ходом восста-
новления можно следить по
уменьшению давления;
Расчет поглощенного коли-
чества водорода. Определяют:
объем водорода, который равен
разности между объемом авто-
клава и объемом жидкости в
автоклаве,
Давление водорода прн 0°
„О__(Роб Рв) ' 273
b ~~ t -j- 273 1
где Рвб — общее давление прн
t° С, измеренное ма4-
нометром,
— давление паров воды
при Р С (из таб-
лицы) .
Рис. 316. Вибромешалка.
1 — вибратор; 2 — кнопка регулятора; 5— винтовой за-
жим; 4 — врэшаюшийся стержень; 5 — фланец; 6 —мем-
брана (резиновая, неопреновая или силиконовая):
7 — стеклянная горловина; S — пробка; 9 — пластинка
мешалки; 10— токоподводяшнй кабель. Мешалка мо-
жет быть соединена с вибратором также таким обра-
зом, чтобы вибрация распространялась в радиальной
направлении, а не по оси.
Объем поглощенного водорода при 0° равен произведению уменьше-
ния давления водорода прн 0° на объем водорода.
Обработка. Спускают избыточное давление через вентиль, открывают
автоклав, содержимое взмучивают и переносят в стакан. Катализатор
отфильтровывают и фильтрат упаривают в вакууме примерно до 100 мл.
Упаренный раствор нагревают до 80° и прибавляют 10 г поваренной
соли. По охлаждении выкристаллизовывается динатрневая соль кислоты.
Выход приблизительно такой же, как прн восстановлении сульф-
гндратом аммония.
б) 1 - А м н и о - 8 • н а ф то л - 4,6 - днсульфокислота
Во вращающемся автоклаве плавят при 180° в течение 7 час. 0,5 моля
дннатрневой соли 1-нафталнн-4,6,8-трисульфокислоты со 170 мл воды и:
170 г едкого натра. По охлаждении плав переносят в стакан,
точно нейтрализуют концентрированной соляной кислотой, быстра
фильтруют. Фильтрат нагревают до 80°, подкисляют соляной кислотой
до снльнокисрой реакции на конго н охлаждают. Мононатриевая соль
'202
I. Промежуточные продукты
1-амино-8-нафтол-4,6-дисульфокислоты кристаллизуется в виде тонких
игл и хорошо перекристаллизовывается.
Выход 123 г 94%-ного продукта, т. е. около 68% от теории.
Технические примечания. Бензоильное и хлорбензоильное производ-
ные 1-амино-8-нафтол-4,6-ди сульфокислоты применяются для получения моноазокраси-
телей различных цветов, начиная от красного и кончая фиолетовым; эти красители,
вследствие чрезвычайно чистых оттенков являются особенно ценными лаковыми кра-
сителями (антозины фирмы И. Г.) *.
1-Нафтиламин-4,б,8-трисульфокислота не имеет большого значения в производстве
красителей, но она является исходным веществом в фармацевтической промышленности i
для получения германина (лекарства против трипанозом). Неудовлетворительный .?
выход при получении 1-нафтиламин-4,б,8-трисульфокислоты объясняется побочными
реакциями, избежать которые до сих пор не удается (частичное образование нафта-
лин-1-3,5,7-тетрасульфокислоты, которая не нитруется, частичное вхождение нитро- 7
группы в положение 2 вместо положения 1 и особенно окислительное действие '
.азотиой кислоты, Приводящее к образованию нафтолсульфокислот, которые затем -
очень легко превращаются в производные динитронафтола).
20. Нафталиитетракарбововая-1,4,5,8-кислота
нз пирена
О
ос хсо
чо
Диангидрид
нафталин- ;
тетракарбоно-j
вой кислоты
а) Т е т р а х л о р и и р е н
В трехгорлой колбе, защищенной от доступа влаги, снабженной Мб:;
шалкой, термометром и обратным холодильником, растворяют прн натре-;
вании до 130° 101 г (0,5 моля) пирена в 1500 г сухого т р и х л о
бензола (нли о-дихлорбензола). При энергичном перемешивании прей?
пускают ток сухого хлора до прнвеса 65 г. Под конец реакции выдёЧ
ляется тетрахлор пирен и жидкость превращается в густую массу. Конй$
хлорирования можно также определить по температуре плавления виде-;
лившегося хлорпирена; хлорирование окончено, если продукт плавится^
прн 355° (неправд.). Реакционную смесь охлаждают, отсасывают на путч-
фильтре, выделившиеся кристаллы промывают небольшим количество^
бензола н сушат в паровом сушильном шкафу.
Выход неочищенного хлорпирена 130 г. Чистый 3,5,8,10-тетрахлор-З
пирен можно получить перекристаллизацией из большого количества?,
нитробензола в виде бесцветных игл с т. пл. 368°. J
* Герм. пат. 272062/4; (Frdl., IX, 460/2).
20. Нафталинтетракарбоновая-1,4,5,8-кислота из пирена
203
б) Нафталинтетракарбоновая кислота
В трехгорлой колбе, снабженной капельной воронкой, термометром
и хлоркальциевой трубкой, размешивают 130 г полученного продукта
хлорирования с 2400 г 20%-ного о л е у м а н нагревают в течение часа до
100°. Охлаждают до 70° н при этой температуре прибавляют 1300 а кон-
центрированной серной кислоты. Затем нагревают до 120° и пол-
часа выдерживают при этой температуре. Красно-коричневую жидкость
снова охлаждают до 70° и в течение часа прибавляют по каплям 260 г
азотной кислоты (уд. вес 1,52). Температура прн этом повышается
от 70° до 130° и выделяются окислы азота. По окончании реакции еще
недолго нагревают до 160°, причем раствор заметно светлеет. Охлаждают,
выливают в 10 л воды (сильное выделение окислов азота!) и оставляют
на ночь. На следующий день отфильтровывают выделившийся желтова-
тый продукт, промывают водой и затем вносят в 1 л 2 н. раствора соды;
почти весь осадок растворяется. Коричневато-желтый раствор непродол-
жительно кипятят с животным углем и фильтруют. При подкислении
фильтрата концентрированной соляной кислотой выделяется чистая бе-
лая нафталинтетракарбоновая кислота, которая после трехчасовой сушки
при 110° превращается в диангндрид.
Выход 60—70 г, т. е. 45—52 % от теории (считая на пирен).
Технические примечания. Нафталинтетракарбоновую кислоту получают
проще всего аналогично фталевой кислоте — окислением соответствующего много-
ядерного углеводорода — пирена. Вместо того чтобы сначала хлорировать, пирен
можно окислить бихроматом и серной кислотой в пиренхинон (смесь 3,8- и 3,10-хино-
нов) и полученный хинон окислить дальше хлорной известью в присутствии гидрата
окиси кальция в нафталинтетракарбоновую кислоту по схеме
3,10- 3,8-
-Пиренхинон
ноос соон
ноос соон
Этот способ удобнее при работе с большими количествами; выход примерно такой же.
Нафталиитетракарбоновая кислота и ее ангидрид легко реагируют, подобно фта-
левому ангидриду, с о-диаминами с образованием интенсивно окрашенных бмс-имид-
азолов, которые в отличие от соответствующих производных фталевой кислоты
обладают свойствами кубовых красителей. Из о-фенилендиамина при этом обра-
зуется смесь двух изомеров 1 (цис), и П (транс), известная под названием индан-
трен алый 2Г (кубовый алый 2Ж).
ZU I
/. yi U4HDIV lipU'JtJMDl
I ('{"C)
Il (транс)
Иил(|нтрен ярко-оранжевый ГР (кубооый ярко-
оранжеЕзый)
Индан треп алый 2Г (кубовый алый 2 Ж)
Выделенная из смеси rpawc-форма (11) является ценным красителем — нндантрен
ярко-оражневый ГР (кубовый ярко-оранжевый).
Пирен принадлежит к высокомолекулярным углеводородам, которые лишь срав-
нительно недавно стали выделять в технических масштабах из высококиггящих фрак-
ций каменноугольной смолы (а также из продуктов гидрирования каменного угля)’*.
Это новое исходное вещество очень быстро подверглось широким испытаниям ** и
оказалось пригодным в особенности для синтеза кубовых красителей. Так, например»
3,8-дибснзоилпирен при запекании с хлористым алюминием*** дает пнраптрон, ранее
получавшийся из [З-метилантрахинона:
С„НПСОС!
+ XI ‘
-гКаС|
Бензпиренхиноны, являющиеся ценными кубовыми красителями, и также содер-
жащие пиреновое кольцо, получаются, однако, не из пирена, а из бензантрона или из
нафталина.
В. СОЕДИНЕНИЯ РЯДА АНТРАХИНОНА
21, Аитрахннон
1-й метод. Окисление антрацена****
Антрацен
Антрахинон
Для получении антрахинона нельзя применять очень грязный антра-
цен, так как иначе потребуется слишком много хромовой кислоты. В на-
стоящее время коксохимические заводы поставлиют 80—92%-ный антра-
* Kruber О.; Вег., 64, 84 (1931); герм, пат. 639240 и 640580, >
** Vollmann. Н„ Becker Н., Correll М Streeck Н., Ann., 531г<
1—159 (1937), _ у
*** Kriinzlein Р., Fortschritte der Friedel-Craftsschen Reakiion und ihre'
lechnische Verwertung, Zt. Angew, Cheui., 51, 373 (1938).
**** Gnehm R., Die Anthracenfarbstoffe, Braunschweig, 1897.
2!. Антрахинон 205
цен, анализ которого производится с помощью известных методов *. Про-
дажный продукт всегда кристаллизуют из пиридиновых оснований.
Антрацен перед окислением надо возогнать с перегретым примерно
до 200° водяным паром, так как только таким путем его можно получить
достаточно тонко раздробленным.
300 г сублимированного, еще влажного антрацена (считая на
100%-ный продукт) размешивают в большом освинцованном железном
сосуде в 6 л во ды и одновременно растворяют в ней 600 г б нхр о м эта
натрия. Нагревают на печи Флетчера до 80° н прибавляют по каплям
в течение 10 час. 1800 г 50 %-ной серной кислоты. В смеси все
время должна присутствовать хромовая кислота; необходимо перемеши-
вать деревянной или стеклянной мешалкой. Под конец кипятят в тече-
ние 2 час., приливая воду взамен испарившейси. Отфильтровывают н
основательно промывают. Из маточного раствора можно получить хро-
мовые квасцы или сульфат хрома.
Хорошо высушенный неочищенный антрахинон содержит, помимо
других примесей, еще немного неизмененного антрацена. Перед даль-
нейшей переработкой его тщательно очищают. Сначала большинство
-примесей удаляют частичным сульфированием н, наконец, перегоняют
еще раз с перегретым водяным паром.
Измельченный в порошок сухой нсочищеный антрахинон нагревают
до 120° с 2,5-кратцым (по весу) количеством серной кислоты
(уд. вес 1,84) до тех пор, пока не прекратится выделение сернистой кис-
лоты. После прекращения выделения сернистой кислоты, что происходит
примерно через 3 часа, охлаждают до 80° и прн этой температуре прибав-
ляют по каплям в течение часа такое количество воды, чтобы получилась
30%-ная серная кислота. В этих условиях антрахинон осаждается в легко
фильтрующейся форме; если же выливать реакционную массу в воду, то
получается слизистый осадок, который отфильтровать невозможно. По
охлаждении отфильтровывают и тщательно промывают водой. Очищен-
ный антрахинон возгоняют с водяным паром, перегретым до 240—260°,
либо еще раз повторяют очистку, причем серная кислота должна лишь
слабо окрашиваться. Очищенный антрахинон получается в виде тонкого
порошка, слегка окрашенного в желто-зеленый цвет.
Выход может составлять до 106 частей 100%-ного антрахинона
из 100 г чистого антрацена (аппаратура изображена па рис. 23 и 24).
Технические примечания. В технике окисление антрацена приводят
в очень больших освинцованных деревянных или гомогенно освинцованных железных
сосудах. Нередко применяют чаны емкостью 15 000—25 000 л. Сульфат хрома, полу-
чаемый в качестве побочного продукта, имеет большое значение в калькуляции, так
как он применяется в больших количествах для дубления кожи и не может быть
получен так дешево другим путем. Этим обстоятельством и объясняется, почему еще
до сих пор проводят окисление хромовой кислотой в больших масштабах, хотя ан-
трацен может быть окислен и антрахинон также и другими технически выгодными'
методами, например окислами азота и воздухом или просто воздухом в присут-
ствии соединений ванадия п качестве катализатора. По последнему методу в настоя-
щее время получается значительное количество антрахинона. Если синтетические ду-
бители вытеснят хромовый метод дубления кожи, то можно не сомневаться, что
хороший метод окисления хромовой кислотой со временем исчезнет.
Перегонка антрацена и антрахинона осуществляется в аппаратах, очень похожих
па те, какие применяются при перегонке дифениламина (см. стр. 129). Только в этом
случае пар конденсируется в больших камерах размерами примерно 3X5X5 jk,
орошаемых холодной водой из разбрызгивателя. Дно камеры затянуто миткалем,
пропускающим воду и задерживающим сублимат.
* Lunge, UtilersuchunKsmeihoden.
2-й метод. Получение из фталевого ангидрида и бензола
СО
co.
/\/СО „
! I I I + НЙО
а) о - Бензоил бензойная кис лота
148 г (1 моль) фталевого ангидрида, не содержащего еле- ,
дов фталевой кислоты *, смешивают в трехгорлой колбе, снабженной ‘
мешалкой и термометром, с 520 г совершенно сухого бензола, Затемг (
в один прием вносят 267 г (2 моля) ** гранулированного безводного- -
хлористого алюминия (куски размером с горошину) н к колбе-л
присоединяют обратный холодильник с хлоркальцневой трубкой на конце,
Медленно нагревают прн постоянном перемешивании до 70° и выдержи-
в а ют прн этой температуре до тех пор, пока не прекратится выделение- )
хлористого водорода. Реакционную смесь осторожно (вспенивание!) н
выливают прн перемешивании в раствор 400 г кальцинированной соды -
в 2,5 л воды н избыток бензола отгоняют с водяным паром. После от- j
гонки бензола убеждаются в том, что смесь имеет щелочную реакцию,
в противном случае прибавляют еще немного соды. После этого фнль-
труют горячий раствор от гидрата окиси алюминия и осадок промывают-j
горячей водой, кипятят в литре воды, снова отсасывают горячим и про-
мывают до тех пор, пока проба фильтрата при подкислении не будет- -
больше давать осадка. Еще горячие соединенные фильтраты подкисляют- j
концентрированной соляной кислотой до сильнокнслой 1
реакции па конго. По охлаждении отсасывают выпавшую бензоилбензой- <
ную кислоту, промывают водой н сушат, 5
Выход 215—220 г, т. е. 95—97% от теории. J
б) Антрахинон ;;
Полученную, как выше описано, сухую беизоилбензойну к>;(
кислоту растворяют в шестикратном (по весу) количестве серной
кислоты (уд. вес 1,84) и нагревают в течение 1—2 часа прн 150°, По--
охлаждении выливают в воду, отфильтровывают выделившийся антрахн-J
нои и тщательно промывают горячей водой. Для удаления следов беи1--*
зоилбензойной кислоты осадок можно прокипятить в воде, содержащей^
немного соды, снова профильтровать, промыть н высушить, S
Выход почти количественный.
Аналогичным путем, если вместо бензола применять толуол или соответственно^'
хлорбензол, получают fi-метил- и jj-хлорантрахиноны, (См, описание синтез»!..;
следующего препарата, а также стр, 58,) При применении хлорбензола рекомен-
дуется хлористый алюминий постепенно вносить при 40—50° и перемешивании ЙЙ
затем нагревать при 80° до окончания выделения хлористого водорода ***,
* Проба при нагревании в пробирке должна тотчас же давать прозрачный плав-.;
и при этом не должно происходить выделения газов. В противном случае фталевый;!
ангидрид следует обезводить осторожным нагреванием в фарфоровой чашке до npe—j
кращения вспенивания’прозрачного плава и охладить в эксикаторе,
** Такое количество хлористого алюминия необходима брать для того, чтобььс
не образовался дифенилфталид,
Пат, США 1746736,
ж, i-лмино-^-метилантрахинон
207
22. 1-Амино-2-метилантрахинон *
а) 2-Метнлантрахннон
Конденсация толуола с фталевым ангидридом проте-
кает в присутствии хлористого алюминия уже при комнатной
температуре и полностью заканчивается в течение 12-—15-часового пере-
мешивания. Циклизацию целесообразно проводить в присутствии 5%-ного-
олеума (10 частей на 1 часть толуилбензойной кислоты) при нагрева-
нии на водяной бане в течение 2 час. В остальном поступают точно так.
же, как прн получении антрахинона нз фталевого ангидрида и бензола
(см. стр. 206). Выход 2-метнлантрахннона с т. пл. 170—174° составляет
85—88% от теории, считая на фталевый ангидрид.
б) 1- Нитро-2 мети л антрахинон
В круглодонной колбе с мешалкой. растворяют 100 г 2-метнлан-
трахннона в 900 г серной кислоты (уд. вес 1,84). Раствор
охлаждают охлаждающей смесью до- 0° н медленно вносят прн хорошем
перемешивании 50 г тонко растертого азотнокислого калия.
Температура реакционной смеси при этом по возможности должна быть,
около 0° и во всяком случае не должна подниматься выше 5°; иначе
образуются в значительном количестве полинитроооединення и продукты
окисления. Ннтропро-изводное постепенно выделяется в виде микроско-
пических иголочек. После 15-часового перемешивания прн 0° смесь вы-
ливают на 5 кг льда, отсасывают выделившийся слегка желтоватый
продукт и промывают водой до нейтральной реакции.
С целью очистки полученный таким образом сырой продукт без предваритель-
ной сушки кипятят в течение 6 час. (с обратным холодильником) с раствором 100 а1
сульфита натрия в 1 л воды. При этом 1-нитро-2-метилантрахкнон не изме-
няется, а его изомеры при обмене нитрогрупп на сульфогруппы переходят в раствор.
Продукт отсасывают горячим и промывают горячей водой до тех пор, пока промыв-
ные воды, которые сначала окрашены в темно-коричневый цвет, не станут бесцвет-
ными. После сущки получают около 98 г, т, е. 82% от теории 1-нитро-2-метилантрахи-
нона в виде тонких сероватых кристалликов с т. пл. 265—267°, свободного от изоме-
ров и достаточно чистого для применения в большинстве случаев. Он содержит лишь
незначительное количество неизмененного метилантрахйнона, который может быть,
легко удален экстрагированием спиртом. Обработанный таким образом препарат
является химически чистым и плавится при 269—270°.
* Locher, Fierz, Helv. Chitn. Acta, 10, 642.
ZUD
в) 1 Амино - 2 м ети л а нтр ахи но н
Еше влажный нитро мет и л антрахинон, очищенный нагреванием с суль-
фитом, восста на вливают кипячением в течение 2 час. (с обратным холодильником))
с раствором 200 г кристаллического сернистого натрия в 2 л воды. Отфиль-
тровывают горячим на нутч-фильтре и промывают горячей водой до бесцветных про-
мывных вод. Затем отсасывают возможно лучше и сушат при не слишком высокой
температуре (около 80°), чтобы избежать потерь вследствие возгонки.
Выход около 80 г, т. е. 75% от теории; т. пл. 198—200°. Однократной перекри-
сталлизацией из толуола можно получить химически чистый 1-амцно-2-метилантрахи-
нон с т. пл. 201—202°.
23. 2-Амнноантрахннои
' /wC°Y>
/\/СО\/\/5ОзН NH3 /\/C0\/\/NI13 ,
1 I II * I II — -j- H3AsO3
а) Аитрахннон-2-сульфокислый натрий
(«серебристая соль»)
100 г возможно более чистого антрахинона вносит при переме-
шивании в 125 г олеума, содержащего 18% SOa- После полного сме-
шения постепенно нагревают прн медленном перемешивании до 135°
и в течение 3 час. выдерживают при этой температуре. Дают охладиться
до 50° и при этой температуре в течение 5 мин. прибавляют по каплям
еще 80 г 66%-ного олеума и снова нагревают в течение 4 час. при 110°.
По охлаждении выливают в 1,5 л воды, нагревают до 80°, отфиль-
тровывают не вошедший в реакцию антрахинон и промывают неболь-
шим количеством горячей воды. К фильтрату при нагревании прибав-
ляют 150 г поваренной соли и оставляют охлаждаться при
перемешивании. После стояния по меньшей мере в течение 10 час. выде-
лившийся в виде листочков антрахиноп-2-сульфокислый натрий отфиль-
тровывают, промывают небольшим количеством 10%-ного раствора пова-
ренной соли, отжимают и сушат. Вых од составляет приблизительно
73 г; 37 г антрахинона выделяются в неизмененном виде.
Ант ра хинон-2-е ул ьфокислый натрий, который благодаря своему внешнему виду
носит в технике название «серебристой солц». может быть перекристаллизован из
воды; полученный таким образом продукт является почти химически чистым и, бу-
дучи высушен на воздухе, содержит 1 молекулу кристаллизационной воды.
. б) 2-Амипоантрахиион
Во вращающийся автоклав, описанный на стр. 318, помещают 50 а
а н т р а х и п он-2-с у л ь ф о к и с л о го натрия, 120 г 25 %-него а м-
м и а к а, 25 г х л о р и с т о г о а м эд о1 н и я и 36 г м ы ш ь я к о в о к и с-
л ого на тр и я. Автоклав медленно нагревают до 120'; затем в течений
часа до 200° и выдерживают при этой температуре в течение 12 час<
Давление поднимается приблизительно до 40 ат. После охлаждения,),
в случае необходимости, спускают давление через спускной вентиль авто*;
клав а, Открывают автоклав и отфильтровывают выделившийся 2-аминов
антрахинон. После, промывания водой осадок кипятят в 200 мл воды^
к которой добавлено 3—4 мл раствора едкого натра (уд. вес 1,38)^
снова отсасывают и промывают водой. Для дальнейшей очистки осадоК^
некоторое время кипятят с 200-—300 лтл 5%-ной соляной к и с л о т ыл
24. 1-Алшноантрахинон. и 1,4-диаминоантрахи.нон
209
охлаждают до 50—60°, отфильтровывают на путч-фильтре и тщательно
промывают горячей водой. После высушивания продукт весит около
30 г и плавится при 300—301° Он почти химически чистый, однако его
целесообразно еще перекристаллизовать из кипящего хлорбензола или
переосадить из концентрированной серной кислоты *. Установлено, что
совершенно чистый 2-аминоаптрахинон дает более чистый и более
устойчивый к хлору индантрен синий (кубовый синий О) (см,
стр. 287).
Технические примечания. Раствор сульфита аммония, образующийся
при взаимодействии антрахнконсульфокислоты с аммиаком, оказывает неблагоприят-
ное действие па конечный продукт. Поэтому эту соль удаляют, окисляя ее в сульфат.
Окисление проводят различными способами. Раньше с этой целью применяли, на-
пример, пиролюзит; в настоящее время используют мышьяковую кислоту (в случае
2-аминоантрахинона) и л:-нитро бензо лсульфокислый натрий (в случае 1-аминоантра-
хинона). Можно также сульфит обезвредить осаждением его по мере образования
с помощью хлорида щелочноземельного металла, например хлористым барием. Опи-
санный здесь способ имеет то преимущество, что выделенный аминоантрахинон прак-
тически не содержит золы. Прибавление хлористого аммония имеет целью нейтрали-
зацию освобождающейся при реакции щелочи **, оказывающей вредное действие.
Маточный раствор, получающийся при мышьяковом способе, содержит мышьяковистую
кислоту и является, естественно, ядовитым, поэтому с ним надо обращаться осто-
рожно. В технике растворы часто обезвреживают известковым молоком. Ядовитость
таких отходов, однако, часто преувеличивают; если их, например, спускать в большие
реки, то они вряд ли могут вызвать отравление рыбы. Избыточный аммиак в технике
отгоняется в холодную воду и может быть снова непосредственно введен в реак-
цию ***.
Общие сведения о'сульфировании антрахинона см. па стр. 54.
24. 1-Аминоантрахиион и 1,4-Д.иамйиоантрахнион
Получение а-аминоантрахинона очень похоже на получение /J-изомера,- только
сульфирование проводят в присутствии соли ртути, которая способствует вхождению
сульфогруппы на 98% в а-положение.
NH.
т-иитрнбензод
с j 3 ь ф ок полый
натрий
’ См. герм. пат. 421205 (Frdl., 14, 1495)!. Растворяют, например, 30 г полученного
продукта в 240 г концентрированной серной кислоты при температуре не выше 35° и1
прибавляют 135 г 52,5%-ной серной кислоты (уд. вес 1,53), Выпадающий в' виде почти
бесцветных кристалликов сульфат по охлаждении отсасывают на стеклянном нутч-
фильтре н промывают 82—83% -ной серной кислотой и размешиванием с водой снова
переводят в свободное основание.
* * Получающаяся мышьяковистая соль действует как свободная Щелочь.
* ** Герм. цат. 347683 (Frdl., X1II, 398) (фирма «Гейги»);.
210
I. Промежуточные продукты
а) Антрахинон-1-сульфокислота
208 г сухого чистого антрахинона тщательно растирают с 4 г
осажденной о к и с н р ту т и или с у л ь ф а т а р т ут н (II) и при перемеши-
вании вносят в 200 мл олеума, содержащего 20% SO3. Перемешивают ;
при 50° до полного растворения, затем нагревают в течение часа до 130—
135° и далее в течение 2 час. прибавляют по каплим 50 а о л е у м а, со-
держащего 60% SQ3 (или соответствующее количество олеума другой.;
концентрации), после этого перемешивают еще час при 135°. После охла- 4
ждеиня выливают в 2 л ледяной воды, нагревают до кипения и *
отфильтровывают не вошедший в реакцию антрахинон (около 60 а). ?’
Фильтрат снова нагревают до кипения и прибавляют при перемешивании )
также горячий раствор 80ахлорнстого калия в небольшом коли-
честве воды. Калиевая соль анатрахинон-а-сульфокислоты выкристалли- ;
зовывается очень хорошо. Охлаждают при перемешивании до 60° и
выдерживают прн этой температуре еще около 6 час. Затем отсасывают 4
прн 60° на предварительно нагретом нутч-фильтре, промывают насыщен-
ным раствором хлористого калия и под конец небольшим количеством 4
воды и сушат. -6
Выход около 190 г, т. е. 58% от теории.
Из маточного раствора при охлаждении выкристаллизовывается еще 15—20 е
менее чистой калиевой соли антрахинонсульфокислоты с небольшой примесью антрй- /
хинон-1,5-дисульфокислого калия. Калиевая соль днсульфокислоты еще очень хорошо
растворима при 60° и довольно трудно растворима на холоду. *
б) 1 - А м и н о а и т р а х и н о и
60 г калиевой солн а и т р а х и н о н-а-с у л ь ф о к и с л о т ы,4
120 г 24 %-ного аммиака и 21 г и ит р о б е и з о л с у л: ь ф о к и с л о г о
натрия нагревают во вращающемся автоклаве в течение 4 час. доJ
170—175° и выдерживают 12 час. при этой температуре. Давление по- i
вышается до 24-—27 ат. После охлаждении содержимое автоклава xo-"j!
рошо отсасывают и промывают небольшим количеством горичей воды,4
Затем осадок кипятят с 300 мл воды, слегка подкисленной соляной{
кислотой, отсасывают горячим на нутч-фильтре и тщательно пр'омы-^
вают горячей водой. Сушат при 90° н получают около 39 г (т. е. 95% от;;
теории) технически чистого 1-амииоантрахинона с т. пл. 238°. После
перекристаллизации из ксилола амин получают в виде черно -красных,^
маленьких призм с металлическим блеском, плавящихся при 241°. J
Выход около 75% от теории. j
в) АнтрахинонилП-оксамнновая кислота*
В снабженном несколькими стальными шарами котле с мешалкой^
(котел подобного типа изображен на стр. 298) нагревают на масляной;
бане прн 115—120° (температура реакционной смеси) прн перемешиваний
44 г 1-а м и н о а и т р а х и н о и а и 132 г кристаллической щавеле^
вой кислоты до тех пор, пока плав, первоначально имевший крас*^
ный цвет, ие станет коричнево-желтым и не застынет, что происходят^
примерно через полтора часа. Реакции окончена, если проба после ки^
пячения с водой плавится при 224—226°. В этом случае прибавляют
350 мл горячей воды, кипятят полчаса, отсасывают горичим, промываюй
горячей водой и сушат. Получают около 57 г (т. е. 98% от теории) зад
мещенной оксамниовон кислоты, плавящейся прн 224 -226°. й
* Noelting, Wortmann, Вег,, 39, 642' Curtis Диссертация, Zurich-]
Weida. 1929. t
24. 4-Аминоантрахинон и 1,4-диаминоантрахинон
211
г) 4-Нитроайтрахиноиил-1-оксаминовая кислота
57 г полученной по описанной выше Методике замещенной окса-
ми новой кислоты растворяют в 570 г серной кислоты (уд.
вес 1,84), охлаждают до 0° н вносят прн 0—2° и хорошем перемешивании
21,7 г высушенного при 120° и хорошо измельченного азотнокислого
калия. Перемешивают еще в течение 6 час. прн 0° и оставляют на ночь
в холодильном шкафу. Затем выливают на 2 кг льда, отсасывают, остаток
снова размешивают в 2 л воды, отсасывают и основательно промывают
холодной водой.
д) 1,4-Диаминоантрахинон
Полученный по пункту «г» влажный осадок заливают 600 мл воды.
Нагревают До кипения и осторожно прибавляют соды до отчетливой
щелочной реакции. Коричневый хлопьевидный осадок превращаетси при
этом в смолообразную массу. Прибавляют 200 г кристаллического сер-
нистого натрия и кипятят 2 часа (с обратным холодильником)
при перемешивании; прн этом нитрогруппа восстанавливается н одно-
временно отщепляется остаток щавелевой кислоты. Отфильтровывают
горячим и промывают горячей водой до тех пор, пока промывные воды
не будут почти бесцветными.
В противоположность обычной методике работы прн промывании
осадка не дают жидкости стечь полностью и сильно отсасывают только
под конец промывания. Получают около 45 г (95% от теор'ин, считан
на 1-аминоантрахнион) сухого 1,4-диамииоантрахннона с т. пл. 260—265°.
Технические примечания. Из 1,4-диаминоантрахинона бензоилирова-
нием получают индантрен красный 5ГК, а из 1,5-диаминоантрахинона — индантрен
желтый ГК (кубовый желтый ЖХ) (см. стр. 288).
а-Аминоантрахинон применяется не только для получения 1,4-днаминоантрахи-
нона; он является также исходным соединением для получения многих важных
антрахиноновых красителей, из которых опишем в общих чертах получение ализари-
нового чисто голубого Б (кислотного чисто голубого антрахинонового).
Ё 10 частях ледяной уксус-
NP1 ной кислоты при 50е абра-
2 бзтывают в течение 2 дней
2,5 молями брома. Затем
выливают в воду с небо----—
льшнм количеством бисуль-
фита для нейтрализации
избытка Галойда
Вт
1-Амино-2,4-диброма нтрд-
хинон, т.пл. 222° (из ледя-
ной уксусной кислоты)
NH
1 моль 1-а мино-2,4-дибр ом антрахинон а раство-
ряют в 7 частях сухого ft-толуидина и нагревают
до 200® точно с 1 молем уксуснокислого натрия
(безводного). Для полного удаления воды отго-
няют немного n-толуидина. Когда проба в хло-
роформе будет иметь такой же оттенок, как и
«тип» (эталон), то все количество разбавляют
равным объемом спирта, фильтруют и промы-
вают спиртом. Выход 82% от теории.
212
/, Промежуточные продукты
Затем сульфируют 6 частями моногидрата. Основание растворяют в кислоте
при 25° и осторожно прибавляют по каплям в продолжение часа около 1,5 части
олеума, содержащего 66% БОз; температура не должна подниматься выше 45°, так
как иначе краситель разрушится. Если проба растворяется в растворе соды, то реак-
ционную смесь выливают в большое количество воды, прибавляют 10% от объема
поваренной солн и отфильтровывают. Осадок промывают 10%-ным раствором соли и
•сушат при 90°. Выход количественный.
Ализариновый чисто голубой Б \
(кислотный чисто голубой аитра- <
хиноновый) 3
Технические примечания. Ализариновый чисто голубой Б (кислотный 4
чисто голубой антрахиноновый) красит в чистый синий цвет, приближающийся по
тону к окраске анилинового голубого (трифенилрозанилин). Он дает более пнтен-
сцвную окраску очень хорошей светопрочцости. Краситель особенно ценен для утя- 1
желейного фосфатом олова шелка, для крашения которого он часто и применяется. л
Если на антрахинон-а-сульфокислоту действуют не аммиаком, а амином, то |
получают соответствующие производные 1-амипоаптрахцпоца; так, например, при 1
действии монометиламина получают 1-метиламиноантрахинон, который, как и пеме- 1
тилированное основание, является важным исходным соединением для получения |
ценных антрахиноновых красителей. 1
Сульфогруппа в а -сульфоантрахиноие может замещаться не только на амино- 3
или замещенную аминогруппу; еще более легко происходит замещение на атом галоида, j
Так, из калиевой соли а-антрахинонсульфокислоты при кипячении в 15%-пой соляной 1
кислоте и постепенном прибавлении хлората натрия получается с количественным вы-;. Я
ходом а-хлораптрахипон. Полученный таким образом а-хлорантрахинон совершенно Я
чист. Я
р-Сульфокислота также способна к такому замещению, но реакция протекает
значительно медленнее. Я
Т. пл. 1бГ
Прн действии алкоголятов или фенолятов образуются
хинона (а-оксиантрахинона).
эфиры эритрооксиантра-1
25. 1,5 и 1,8-Диоксиантрахинон, 1,5-дихлорантрахинон 213
25, 1,5 и 1,8-Дноксиаитрахннон, 1,5-днхлор антрахинон
Хризазин
а) А н т р а х и н о н -1,5 и 1,8 - д и с у л ь ф о к и с л о т ы *
200 г очищенного н высушенного при 140° антрахииоиа хорошо
растирают с 4 а осажденной окиси ртути н прн перемешивании вно-
сят в течение получаса в 400 г 18%-пого олеума, причем одновре-
менно температуру медленно поднимают до 75—80°. После прибавления
всего количества антрахинона нагревают до 120°, перемешивают час при
этой температуре и затем в продолжение 3—4 час. прибавляют по кап-
лям 140 г 66%-ного олеума. Следят за тем, чтобы па стенках со-
суда и на мешалке ие оставалось шевступившего в реакцию антрахинона.
Сульфирование заканчивается при дальнейшем перемешивании при 120°
в течение 10—12 час. Конец реакции определяют по пробе, которая дол-
жна легко растворяться в воде и давать прозрачный раствор. Охлаж-
дают до 50° и медленно при перемешивании'разбавляют 100 г кон-
центрированной серной кислоты. После стояния в течение
ночи выделяется с почти количественным выходом чистая свободная
антрахинон-1,5-дисульфокислота. Отфильтровывают иа стеклянном нутч-
фильтре, промывают 50 мл концентрированной серной кислоты н как
можно лучше отсасывают. Осадок снова растворяют в 2 л горячей
* Fierz, Krebser, Hclv, Chim, Acta, 10, 200.
воды, фильтруют горячим и смешивают с также горячим раствором
100 г хлористого калия в 300 мл воды; при этом выделяется I
трудно растворимая калиевая соль сульфокислоты. Убеждаются в пол- J
ноте осаждения, отфильтровывая пробу и прибавляя в фильтрат еще ;
немного хлористого калия—осадок не должен больше выпадать. Дают )
охладиться, затем отсасывают, промывают 200 мл холодной воды н
сушат. I
Выход 192—196 г антрахинон-1,5-дисульфокис л ого калия, т. е, J
45—46% от теории. |
Концентрированный сернокислый раствор, отфильтрованный от 1,5-дисульфо- а
кислоты, медленно смешивают с равным объемом воды при хорошем перемешивании во я
избежание местного перегрева, который может вызвать отщепление сульфогрупп. J
Оставляют стоять на 2—3 часа; в течение этого времени выделяется большая часть -я
1,8-дисульфокислоты. Ее отсасывают на стеклянном нутч-фильтре и промывают I
44%-ной серной кислотой. Свободную кислоту переводят, как в случае 1,5-изомера, J
в калиевую соль; для этого осадок растворяют в 1,5 л горячей воды и смешивают
с раствором 80 г хлористого калия в 200 мл воды.
Выход 98—100 г антрахинон-1,8-дисульфокислого калия, т. е. около 23% от -1
теории,
Маточный раствор, полученный после выделения 1,8-дисульфокислоты, содержит^
еще около 30% введенного в реакцию антрахинона в виде сложной смеси дисульфо-' л
кислот (главным образом 1,7-кислоты, наряду с меньшим количеством 1,6-кислоты •• а
незначительные количества 1,5- и 1,8-кислот и следы 2,6- и 2,7-кислот), которые не Я
удается разделить простым путем. В технике эту смесь перерабатывают на «сере- Я
бристую соль» продолжительным нагреванием разбавленного сернокислого раствора а
при 180—200°; сульфогруппы, стоящие в a-положении, отщепляются, и получаю-pj
Щуюся антрахинои-2-сульфокислоту наряду с небольшой примесью образовавшегося 1
снова антрахинона перерабатывают так же, как это было указано при описании полу- Я
чения алтрахинон-2-сульфокислого натрия (см, стр. 208), Ц
Вместо того чтобы свободные 1,5- и 1,8-дисульфокислоты переводить в калие- Я
вые соли, можно также нейтрализацией содой получать менее трудно растворимые
натриевые соли, ' Jj
В технике часто не разделяют 1,5- и 1,8-дисульфокислоты, а выделяют их вместе, Я
разбавляя сульфомассу сначала небольшим количеством концентрированной серной Я
кислоты, а затем водой до получения раствора серной кислоты примерно 66%-ной Я
концентрации. •Я
Я
б) 1,5- и 1,8-Диоксиаитрахиноиы Я
44,4 г хорошо высушенного и тонко измельченного а итрахн нои-1
1,5- или 1,8-д нс ул ь ф о к и с л о г о калия (или их -смесь), 700 мл Я
воды, 46 г гидрата окиси кальция и водный раствор 5,5
хлористого кальция нагревают в течение 20 час. в автоклав^Я
с мешалкой при 195—200° при. хорошем перемешивании. Давление со--Я
ставляет 14—16 ат. По охлаждении содержимое автоклава переносятЯ
в стакан, нагревают прн перемешивании до кипения и осторожно под-Я
кисляют концеитрироваииой соляной кислотой до сильИО'Я
кислой реакции на коиго. Кипятят до исчезновения запаха SO3, отсасы*Я
вают зеленовато-желтый осадок и промывают горячей водой до ией'Я
тральной реакции промывных вод. Я
Выход 20—21 г, т. е. около 85% от теории. Я
1,5- Диоксиантрахинон (антраруфин)'! плавится при 280°, 1,8-изомер (хризазин)) —;Я
при 191°. Если исходят из смеси 1,5- и 1,8-дисульфокислот, то получают смесь диокси.--Я
соединений, которая рщдезко плавится при 235—255°. Я
Оба диоксиантрахинона являются важными исходными соединениями для полу*Я
чения ряда ализариновых кубовых красителей. Во многих случаях оба изомера дают Я
очень похожие продукты, так что можно непосредственно использовать смесь этих-Я
оксисоедииений (см. ализаринсафироль 13 и СЕ, кислотный синий антрахиноновый) <Я
2b. i,ь-динитроантрахинон и tд-аиамияоантрахинон
в) 1,5-Дихлорантрахииои
82 г а н т р а х и н о н-1,5-д и с у л ь ф о к и с л о г о калия взмучивают в 1 л
воды н 140 г концентрированной соляной кислоты, нагревают до
98—99° и при этой температуре и перемешивании прибавляют по каплям в течение
3 час, раствор 172 г хлората калия в 1500 мл воды. Перемешивают прн этой
температуре до тех пор, пока из отфильтрованной горячей пробы при охлаждении не
перестанет выделяться хлорантрахинонсульфокислота. Для этого требуется около
3 час. Отсасывают горячим, промывают горячей водой до исчезновения кислой реак-
ции ппомывных вод и сушат.
Выход 45 г, т, е. около 88%; т, пл, 243—244°.
Оба атома хлора легко замещаются на остатки аминов, феноксигруппу и др,,
с образованием новых технически важных промежуточных продуктов,
26. 1,5 Дииитроаитрахннон и 1,5-диаминоантрахинон
Изомерные днннтроантрэхиноны
а) Ьб-ДииитроанФрахииои
100 г чистого антрахинона с т. пл. 278—279° растворяют в
2000 а моногидрата. При 25° и хорошем перемешивании прибав-
ляют в продолжение 30 мин. по каплям 460 г нитрующей смеси (230 г
азотной кислоты (уд. вес 1,52) и 230 г Моногидрата). Масса
разогревается приблизительно до- 80° и уже во время прибавления ни-
трующей смеси начинает выделяться 1,5-дниитроаитрахинои, Массу на-
гревают еще 2 часа при 125° н оставляют охлаждаться. Чисто желтый
осадок отсасывают на стеклянном иутч-фильтре и промывают 100 мл
моногидрата. Затем промывают водой до нейтральной реакции
промывных вод. После сушки осадок весит 56 г, т. е. выход составляет
около 40% от теории. В концентрированной серной кислоте находятся
1,8-, 1,6- и 1,7-динитроантрахиноны, не имеющие технического значения.
б) Восстановление 1,5-динитроантрахнноиа
до 1,5 - д и а м и и о а и т р а х и и о и а
Если 1,5-д инитроантрахииои далее подвергают восстановле-
нию, то его лучше не сушить. Влажный осадок суспендируют в 500 мл
воды и прибавляют при 80° концентрированный раствор 350 г серни-
стого натрия (содержащего кристаллизационную воду), Раствор на-
гревают до 100° при хорошем перемешивании. 1,5-Дниитроаитрахиион
сначала переходит в раствор с зеленой окраской (образование раство-
римого в щелочах производного гндроксиламииа), н вскоре выделяется
красный кристаллический 1,5-диамниоаитрахинои. Через час осадок от-
фильтровывают и промывают водой до тех пор, пока промывные воды
не станут бесцветными. После сушки получают около 40 г практически
химически чистого 1,5-днаминоаитрахинома.
216
1. Промежуточные продукты
27. Хнннзарнн из л-хлорфенола
Это соединение получают в технике конденсацией прн 160—210е*
фталевого ангидрида с /г-хлорфенолом в концентрированной серной
кислоте в присутствии борной кислоты. В этих условиях образуется бор-
ный эфир хнннзарина, который можно выделить, прибавив лед, и омы-
лить в хнннзарнн нагреванием с водой нлн раствором соды. Выпавший
сырой хннизарин следует очистить переюсажденнем, перекристаллизацией
или возгонкой.
270 мл концентрироваиной серной кислоты;
25 г борной кислоты;
96 г фталевого ангидрида;
26 г л-хлор фенол а.
Смесь борной и серной кислот в трехгор лой колбе с мешалкой и
термометром нагревают на масляной бане при 50° до полного растворе-
ния борной кислоты. Прн этой температуре и хорошем перемешиваний
вносят в течение часа, друг за другом, фталевый ангидрид и п-хлюрфе-
нол. Температуру медленно поднимают до 160& и нагревают при этой
температуре в течение 3 час. В продолжение четвертого часа нагревают
до 210° и выдерживают прн этой температуре 4 часа. Надо следить за
тем, чтобы, безусловно, не происходило перегревания.
Реакционную массу разбавляют до достижения уд. веса 1,71, прибав-
ляя медленно по каплям при 180° смесь 150 г концентрирован^
ной серной кислоты и 130 мл воды. После полного охлаждения
выливают на лед, отсасывают выделившийся сырой продукт (эфир бор-
ной кислоты), кипятят его в течение 10 мин. с 1 л воды при хорошем
перемешивании, фильтруют горячим н промывают 1 л горячей воды.
Маточный раствор и промывные воды собирают отдельно, так как из
первого выкристаллизовывается избыточная фталевая кислота (около
40% от взятого ангидрида).
Для очнеткн сырого хнпизарина отфильтрованный осадок затирают
с 50 мл раствора едкого натра (уд. вес 1,53), разбавляют до 1,5 л
горячей водой н нагревают до кипения, пропуская острый пар. При
сильном перемешивании и медленном прибавлении концентрирован пой
соляной кислоты хиннзарин выделяется в виде мелких кристаллов-
С целью дальнейшей очистки его кипятят в 1 л воды, содержащей соля-
ную кислоту, фильтруют горячим, сушат прн 80—90° и перекристаллизо-
вывают из хлорбензола. Выход 35 г, т. е. около 70% от теория; т. пл. 194°.
Примечание. Обладающий сильным и «прилипчивым» запахом п-хлорфенол
в лаборатории можно заменить эквивалентным количеством гидрохинона.
II. КРАСИТЕЛИ
Г. АЗОКРАСИТЕЛИ
В настоящее время группа азокрасителей включает самый большой
н самый разнообразный ассортимент искусственных органических краси-
телей. Поэтому они будут рассмотрены несколько более подробно. Сна-
чала будут изложены общие принципы получения азокрасителей и дан
ряд прописей для проведения диазотирования, особенно оправдавшихся
в лабораторных условиях. Затем приведены примеры диазотирования н
получения простых и сложных азокрасителей применительно' к производ-
ственным условиям.
Методы определения даны в аналитической части.
О ПОЛУЧЕНИИ АЗОКРАСИТЕЛЕЙ в лаборатории
а) Основные условия
1. Применение чистых исходных веществ.
2. Точное соблюдение относительных количеств реагирующих ве-
ществ.
3. Энергичное и постоянное перемешивание нлн встряхивание при
проведении реакций диазотирования и сочетания.
4. Защита диазосоединенин от нагревания и света. Применение диа-
зосоединенни непосредственно после их получения.
Примечание к пункту /. Следует особенно следить за тем, чтобы применяемые
исходные соединения не содержали изомеров или других соединений сходного строе-
ния (например, дисульфокислот в моносульфокислотах и наоборот), которые могли
бы принимать участие в образовании красителей; наоборот, ие слишком сильная
коричневатая или серая окраска многих технических продуктов в большинстве слу-
чаев нс имеет значения. Необычно темная окраска или осмоление указывают на на-
ступившее окисление или разложение; в этом случае вещества необходимо очистить
перегонкой или перекристаллизацией. Наличие поваренной сати или других неорга-
нических солей щелочных металлов в общем не мешает, но такие примеси следует
учитывать при расчетах взятых в реакцию количеств (ср. 2).
Примечание к пункту 2. Количество нитрита, взятое для диазотирования, должно
точно соответствовать теоретическому, если проводят сочетание в кислом'растворе,
а также если сочетание проводят в щелочной среде, но краситель выделяют при под-
кислении. Избыток нитрита в этих условиях способствовал бы диазотированию или
питрозировапию составляющих, участвующих в сочетании, а также, возможно, изме-
нил бы готовый краситель. Если сочетание и выделение красителя проводят в щелоч-
ном растворе, то небольшой избыток нитрита не оказывает вредного действия. Не-
достаток нитрита приводит к образованию диазоаминосоединения или к сочетанию,
диазотируемого основания с самим собою что во всяком случае вредно.
Обычно берут небольшой избыток (3—5%} конечной азосоставляющей, что
ускоряет сочетание. При получении дне- и полиазокрасителей среднюю азосостав-
ляющую, наоборот, надо брать в точно теоретическом количестве во избежание обра-
зования побочных красителей.
Для того чтобы можно было применять точные количественные соотношения
ZJO
//. красители.
выделяемые в технике перегонкой (например, анилин и его гомологи, хлоранилин,
анизидин и фенетидин, нафтиламин, фенол, крезол, резорцин, нафтол, дифениламин
и т. Д.), если получены свежими, в общем можно считать практически чистыми и
100%-ными. Если вещества потемнели при длительном хранении или даже осмоли-
лись, то перед употреблением их следует перегнать еще раз, в случае необходимости
в вакууме. Соединения, которые нельзя выделить перегонкой, редко бывают 100%-
ными. Так, сульфокислоты и их соли содержат в большинстве случаев примеси неор-
ганических солей, а также часто кристаллизационную воду. Если данные о содержа-
нии чистого продукта в используемом ненадежны, их следует проверить. Определение
проводят для диазотируемых аминов с помощью растворов нитрита, а для азосостав-
ляющих — диазосоединеиия (см. аналитическую часть). Найденное содержание чи-
стого вещества в техническом продукте проще всего выражать кажущимся молеку-
лярным весом (Л/), т. е. числом граммов технического продукта, содержащим 1 моль
чистого вещества. Например, если на 1 г технической Аш-кислоты при титровании
израсходовано 26,8 мл 0,1 н. раствора диазосоединения, то М = 10 000 : 26,8 — 373.
Для получения 0,1 моля красителя берут, таким образом, 37,3 г этой Аш-кислоты.
Технический нитрит также не бывает 100%-ным, и его следует перед употребле-
нием титровать. В лабораториях, где диазотирование проводят часто, целесообразно
иметь в запасе нитрит в трех формах:
а) нормальный раствор нитрита (1 моль NaNOa в литре); приготовление (см.
стр. 340);
б) 5 н. раствор нитрита (1 моль NaNOs в 200 мл); готовится аналогично (а);
в) твердый нитрит. Технический нигрит сушат в паровом сушильном шкафу,
тонко измельчают и хранят в хорошо закрытой банке (так как иначе он снова при-
тянет влагу). Содержание чистого нитрита определяют титрованием пробы.
Примечание к пункту 4. Разложение диазосоединений ускоряется при нагревании
и часто также на свету; оно протекает тем быстрее, чем более шелочную реакцию
имеет раствор, если только щелочь не вызывает перегруппировку в антидиазотаты.
Последние, Правда, устойчивы, но либо вовсе не способны к сочетанию, либо соче-
таются лишь частично. Избыток кисдоты способствует устойчивости диазосоединений.
Признаком начавшегося разложения является выделение азота, которого нужно,
безусловно, избегать хорошим охлаждением, защитой от яркого света и, насколько
возможно, поддерживанием кислой реакции. В обшем диазосоединения разлагаются
тем легче, чем сильнее основность аминов, лежащих в их основе; отрицательные заме-
стители (галоиды, нитро-, сульфогруппы и другие аналогичные заместители) повы-
шают устойчивость диазосоединений. Особое место занимают ввиду отличающегося
строения о-оксидиазосоединения, которые являются чрезвычайно устойчивыми, по
крайней мерс в отсутствие щелочи.
Помимо обычного разложения с выделением азота и превращением в соответ-
ствующий фенол, некоторые диазосоединения претерпевают еще и другие превраще-
ния, а именно замещение группы, стоящей в орто-положении к диазогруппе, на окси-
группу. К таким заместителям относятся галоиды, алкокси-, ннтро- и сульфогруппы.
Замещение происходит тем быстрее, чем выше температура и чем щелочнее раствор.
Замещению благоприятствует наличие отрицательных заместителей (как, например,
галоидов, нитро-, сульфо-, карбонильных и др. групп); замещение протекает особенно
легко, если две диазогруппы, стоящие в лшга-положении друг к другу, действуют
совместно. Эта реакция используется в технике для получения о-оксидиазосоединений.
В тех случаях, когда она не желательна, реакцию следует проводить при низкой тем-
пературе и в возможно более кислой среде.
Необходимо, чтобы сочетание всегда проводилось сразу
же после диазотирования. Ни в коем случае дназосоеднне-
ния нельзя оставлять на длительное время или даже на ночь.
При выделении диазосоединеиия в твердом виде надо всегда внимательно следить за
тем, чтобы оно оставалось влажным. Даже относительно устойчивые диазосоединення
могут в сухом виде сильно взрывать (исключение: о-оксидиазосоедннения ряда нафта-
лина).
б) Диазотирование
Диазотирование протекает по уравнению:
RNHa ф NaNOa ф 2 НС!* —> R—N^N | С1 ф NaCI ф 2 Н2О
* В большинстве случаев при диазотировании в водных растворах применяют
соляную кислоту, так как она с ароматическими аминами вообще дает наиболее легко-
растворимые соли. При применении серной кислоты, естественно, берут 0,5 моля ее
вместо 1 моля соляной кислоты.
6) Диазотирование
219
Одна свободная сульфогруппа замещает моль соляной кислоты,
например.
/NH2
R< + NaNO3 + НС1 —>
Х5О.,Н
/N=N
RXSo3 +NaCl + 2HaO
В то время как количество нитрита должно точно соответствовать
теоретическому, кислоту всегда берут в избытке, а именно в лабораторных
условиях, как правило, сверх 2 эквивалентов кислоты, требуемых по
уравнению реакции, берут еще избыток 0,5 эквивалента. Для аминов,
являющихся слабыми основаниями, соли которых сильно гидролизуются
в воде (например, для хлор- и нитро анилинов), избыток кислоты повы-
шают до 1—3 эквивалентов. В тех случаях, когда основание не раство-
ряется даже в таком избытке кислоты, например ннтрохлоранилин, диа-
зотирование проводят в суспензии, если основание имеется в виде тонко-
дисперсной пасты; если же оно является сухим, то сто можно перевести
н пасту растворением в концентрированной серной кислоте, вылива-
нием этого раствора в ледяную воду, отсасыванием и промывкой. Диин-
троанилины и нитродихлора ни л нны н другие аналогичные соединения
не удается диазотировать н этим методом; такие основания можно
гладко диазотировать лишь в концентрированной серной кислоте.
Сульфокислоты н карбоновые кислоты могут диазотироваться, как
обычные амины, если они хорошо растворяются в воде. в присутствии
кислот. Во многих случаях, когда это условие не выполняется, рекомен-
дуется подлежащие диазотированию кислоты в виде их солей со щелоч-
ными металлами растворять в воде, смешивать этн нейтральные рас-
творы с нужным количеством раствора нитрита и смесь выливать при
хорошем перемешивании в разбавленную соляную кислоту (обратное
лиазотированне); .сульфокислота и азотистая кислота при этом одно-
временно переходят в .свободное состояние и, как правило, успевают
прореагировать друг с другом до того, как произойдет осаждение трудно
растворимой кислоты. Однако если свободная сульфокислота очень
трудно растворима — это имеет место именно н случае некоторых амино-
азокрасителей, которые должны быть продиазотированы дальше с целью
получения полиазокрасителей, — то оиа выпадает раньше, чем может
произойти ее диазотирование. Такую кислоту трудно продиазотировать
до конца, особенно если и диазосоединение практически нерастворимо.
В таких особых случаях может оказать благоприятное действие примене-
ние значительного избытка нитрита и прибавление с самого начала не-
большого количества нитрита к соляной кислоте. Кроме того, раствор
сульфокислоты довольно сильно разбавляют и медленно прнбавляютего
при хорошем перемешивании в соляную кислоту, содержащую нитрит.
По окончании диазотирования, диазосоединение отфильтровывают, бла-
годаря чему удаляется избыток нитрита.
1,2- и 2,1-аминонафтолы, их сульфокислоты и другие производные не
могут быть гладко продиазотированы ни по одному из описанных ме-
тодов, так как они в кислом растворе окисляются азотистой кислотой
в соответствующие хиноны. Однако диазотирование амннонафтолов
в виде их солей с минеральными кислотами или сульфокислот амино-
нафтолов, содержащих одну свободную сульфогруппу, проходит безуко-
ризненно, 'если обрабатывать эти соединения нитритом в отсутствие до-
полнительного количества кислоты и в присутствии либо эквивалентного
количества цинковой соли, либо небольшого количества медной солн;
в последнем 'Случае надо по окончании диазотирования удалять медь,
цинк же, в общем, не мешает.
Особое место занимают также те основания, которые, помимо пер-
вичной аминогруппы, содержат в молекуле также вторичную амино-
группу, как например моноэтил-л-фенилепдиамин и особенно п-амнио-
дифениламин и его производные. Эти основания можно довольно гладко
прсдиазотировать так, чтобы имипогруппа осталась незатронутой; од-
нако образующиеся при этом диазосоедиисния очень медленно соче-
таются и, кроме того, очень легко разлагаются. Поэтому поступают та-
ким образом, что берут' на 1 моль основания 2 моля нитрита. В этих
условиях диазотируется первичная аминогруппа, а вторичная одновре-
менно при этом ни третируете я. Эти нитрозированные диазосоед и нения
гораздо легче вступают в реакцию азосочетания и обладают намного
большей устойчивостью. После сочетания из готового красителя нитрозо-
группу удаляют нагреванием с кислотой или с основанием либо слабыми
восстановителями такими, как бисульфит (красители — вариамины).
Следует еще отметить, что кои центр и ров энная соляная
кислота в присутствии нитрита выделяет хлор; это, естественно, может
повлечь за собой образование побочных продуктов. Поэтому диазоти-
руемая смесь не должна содержать более чем 20%-иуЮ свободную НС1.
При диазотировании в разбавленных кислых растворах нет надобно-
сти, как это часто рекомендуют, очень медленно прибавлять раствор
нитрита. Для оснований, которые особенно склонны к образованию диа-
зоаминосоединений или к сочетанию с самими собой, с целью избежания
таких побочных реакций даже лучше все количество нитрита приба-
вить сразу. Необходимо только следить за тем, чтобы было1 достаточное
охлаждение, которое достигается лучше всего прибавлением к реаги-
рующему раствору льда.
Примеры
1, А Н и Л Н и
9,3 г* (0.1 моля) анилина (который в случае необходимости
очищают перегонкой) растворяют в стакане в 125 мл 2 н. соляной
кислоты, раствор охлаждают снаружи льдом при перемешивании
мешалкой, действующей от турбинки или электромотора. По охлажде-
нии раствора в него вносят несколько кусочков льда и прибавляют из
капельной воронки по каплям 100 мл 1 н. раствора нитрита. Не сле-
дует прибавлять нитрит очень медленно', но необходимо следить за тем,
чтобы температура не поднималась выше 0° и чтобы каждая прибав-
ляемая капля тотчас же смешивалась с жидкостью. Если надо, то при-
бавляют еще немного льда. 11с должно иметь места выделение газа, по-
мутнение или окрашивание раствора. Перемешивают еще в течение
10 мин. и проверяют, окрашивает ли капля раствора, с одной стороны,
в синий цвет бумажку конго и вызывает ли она, с другой стороны,
слабое посинение йодокрахмальной бумажки или реактива «суль-
фона» **. В противном случае прибавляют по каплям раствор нитрита
* Указанные во всех примерах количества рассчитаны для 100%-ных веществ;
в случае загрязненных веществ, естественно, надо брать соответственно большие ко-
личества (см. стр. 218).
4,4/-Диаминодифе11НЛметан-2,2''-сульфон (Вег,, 27, 2806),
НЙМ— / СН2--/ у -МН2
\ ЗОу/
Применяют, примерно, 10%-ный раствор основания в разбавленной соляной кис-
лоте, наносят несколько капель этого раствора па фильтровальную бумагу и прибав-
Примеры реакций диазотирования
221
до положительной реакции на нитрит и также оставляют на несколько
минут. Если же, наоборот, реакция на нитрит слишком сильная, то
вносят несколько капель очень разбавленного раствора солянокислого
апплица до тех пор, пока реакция почти не исчезнет. Для этой поправки,
во всяком случае должно потребоваться лишь несколько капель раство-
ров нитрита или анилина. Если же этого количества недостаточно; то
значит при отвешивании или отмеривании реагентов была допущена гру-
бая ошибка, либо был взят раствор нитрита не той концентрации. В та-
ком случае этот опыт отбрасывают и ставят новый опыт, избегая по-
вторения сделанных ошибок. Так же поступают, в особенности в том
случае, если получается мутный пли сильно окрашенный раствор.
При работе с не очень большими количествами вместо стакана и механического
перемешивания можно использовать коническую илн круглодонную колбу и встряхи-
вать ее рукой. В таком случае отказываются от внешнего охлаждения и к раствору
прибавляют больше льда (при указанных загрузках около 100 г). Колба должна
быть достаточно большой, чтобы не происходило разбрызгивания при сильном встря-
хивании. Одной рукой колбу все время встряхивают, перемешивая в пей жидкость,
а другой льют тонкой струей раствор нитрита прямо иа мерного цилиндра. Капли
раствора, оставшиеся на стенках сосуда, тотчас же смывают при помощи пррмывалкн.
Затем встряхивают еще несколько минут и проверяют, как описано выше.
Если падо получить возможно более концентрированный дпазораствор, то вместо
нормального раствора нитрита берут 5 н. раствор или твердый нитрит. В последнем
случае надо раствор перемешивать особенно хорошо. Вместо 2 н. соляной кислоты
можно также применять соответственно меньшее количество более концентрированной
кислоты. Во всяком случае воды следует брать столько, чтобы солянокислая соль
применяемого основания растворилась хотя бы при нагревании, Если при охлаждении
наступает кристаллизация, то надо следить за тем, чтобы выделялись возможно более
мелкие кристаллы, поэтому при охлаждении следует перемешивать или встряхивать.
При соблюдении этих условий выделившаяся солянокислая соль вновь легко раство-
ряется при прибавлении нитрита, В случае трудно растворимой солянокислой соли
следует сначала растворить основание в горячей воде при прибавлении лишь эквива-
лентного количества соляной кислоты; остаток соляной кислоты (которая оказывает
высаливающее действие) прибавляют только после растворения,
Подобным образом диазотируют гомологи и алкоксипроизводные
анилины, а также производные всех этих соединений, содержащие атом
галоида в жега-положении к аминогруппе, многоядериые основания,
такие, как бензидин, ди а ми но ди- и тр и феи ил метан, ди аминодифеи иловый
эфир и другие аналогичные соединения и их производные, в общем те
основания бензольного ряда, солянокислые соли которых не гидроли-
зуются при растворении в воде. Количество воды определяется по дан-
ным предыдущего абзаца.
Сульфо- и карбоновые кислоты, если они достаточно хорошо раство-
ряются в разбавленной соляной кислоте, также можно диазотировать по
описанной методике.
2. я-Хлоранилин
12,75 г (0,1 моля) д-х л о р а н и л и и а растворяют при нагревании
в 100 мл воды и 30 мл концентрированной солянойкиСлоты, охлаж-
дают при перемешивании или встряхивании, смешивают со льдом и
поступают далее, как описано, в примере 1.
Также можно диазотировать о-хлоранилин, jit-нитроанилин и дру-
гие гомологи и аналоги этих аминов, обладающие близкой основностью,
причем количество' воды берут в зависимости от растворимости вводи-
мого в реакцию амина.
ляют каплю испытуемого раствора. В присутствии нитрита получается синее йсустой-
чивое окрашивание. Этот реагент несколько менее чувствителен, чем йодокрахмальная
бумажка, но оц может применяться также в сильнокислой среде.
II. Красители
3. 2,5 Д ихл о р а ни л и н
В литровой круглодонной колбе 16,2 г (0,1 моля) 2,5-дихлор-
ан и л и н а обливают 50 мл концентрированной соляной кислоты
и 200 мл воды и кипятят до полного растворения (за исключением воз-
можных примесей в дихлор анилине). Охлаждают под водяным краном
при сильном встряхивании, чтобы солянокислая соль выкристаллизо-
валась в виде мелких кристаллов. Прибавляют около 100 г льда и при по-
стоянном встряхивании медленно приливают 100 мл ] и. раствора и н-
трита. Встряхивают до полного растворения осадка и проверяют, как
обычно, реакцию иа конго и йодокрахмальную бумажку и, если надо,,
фильтруют. Так же диазотируют другие дигалоидозамещенные основания.
4. Ацет-п-феиилендиамии
Солянокислая соль этого основания трудно растворяется на холоду; нагревание
с кислотами исключено из-за возможности омыления. Поэтому не добиваются полного
растворения, а довольствуются лишь получением солянокислой соли в возможно более
мелком виде, для чего поступают следующим образом.
15,0 г (0,1 моля) тонко растертого м о и о а ц ет-п-ф е й и леи-
диамина постепенно растирают в ступне с 50 мл 2 и. соляной кислоты,
прибавляя для охлаждения кусочки льда. Массу переносят в стакан,
ступку ополаскивают холодной водой, стакан охлаждают снаружи льдом.
В массу также добавляют немного льда и еще 75 мл 2 и. соляной
кислоты и при перемешивании прибавляют по каплям 100 мл 1 н.
раствора нитрита. Перемешивают при охлаждении льдом до тех пор,
пока все ие растворится. Небольшое количество иерастворившихся гру-
бых комков можно1 удалить фильтрованием.
5. Аминоазобензол
Амииоазобеизол и многие другие я-амииоазосоедииения мало рас-
творимы даже в кипищей разбавленной соляной кислоте. В этом случае
также не добиваются полного растворения, а поступают, как описано
в примере 4, с той лишь разницей, что особого охлаждения при расти-
рании с соляной кислотой не требуется.
6. (х-Нафтиламии
В литровой круглодонной колбе смешивают 14,3 г (0,1 моля)
os-и а ф т и л а м и и а, 120 мл горячей воды и 10 мл концентрированной
соляной кислоты, не содержащей примеси серной кислоты. Ки-
пятят до полного растворения, прибавляют еще 12 мл концентрирован-
ной соляной кислоты и охлаждают под -водяным краном до ком-
натной температуры при сильном встряхивании. Полученную густую
массу смешивают с 200 г измельченного льда, хорошо перемешивают и
в одни прием прибавляют при встряхивании высушенный и измель-
ченный нитрит в количестве, соответствующем 6,9 г чистого NaNOa-
Колбу тотчас же закрывают резиновой пробкой и энергично встряхивают
до получения почти прозрачного раствора. Отдельные комочки соляно-
кислого нафтиламина, не растворившиеся при встряхивании, измельчают
стеклянной палочкой 'и снова встряхивают до растворения. Через
10—15 мии. диазотирование должно быть окончено. Убеждаются в том,
что раствор имеет сильнокислую реакцию на конго и дает слабую
реакцию на нитрит с йодокрахмальиой бумажкой. Диазотирование про-
Примеры реакций диазотирования
223
ходит тем лучше, чем быстрее был прибавлен нитрит и чем скорее
удалось добиться основательного смешения. Хотя ие удается полностью
воспрепятствовать образованию амииоаз ©нафталина, одиако последнее
должно проявиться в выделении лишь незначительного количества тем-
ных хлопьев, которые отфильтровывают по1 окончании диазотирования.
Остаток на фильтре должен быть почти невесомым, фильтрат должен
быть светло-желтым и совершенно прозрачным. Ни в коем случае филь-
трат ие должен мутнеть после фильтрования, что означало бы недоста-
ток кислоты или нитрита. Если при диазотировании образовался обиль-
ный фиолетово-коричневый осадок, то это значит, что опыт ие удался;
возможность последующих исправлений исключается.
Гладкое диазотирование достигается легче, если количество соляной кислоты
увеличить, а также если применять небольшой избыток нитрита при условии, что
избытки реактивов не мешают дальнейшей переработке диазораствора.
₽-На фтил а мин диазотируют по этой же методике.'
7. п-Нитроаиилин
13.8 г (0,1 моля) n-иитроаиилина растворяют при нагревании
в смеси 30 мл воды и 30 мл кои центрированной соляной кислоты
и охлаждают при встряхиваини под водяным краном до комнатной тем-
пературы. Прибавляют 80 г льда и при постоянном встряхивании в
одни прием вносят высушенный и измельченный нитрит в количе-
стве, соответствующем 6,9 г чистого NaNCh- Встряхивают до тех пор,
пока осадок, который в большинстве случаев перед этим выпадает, снова
не растворится. Охлаждают еще в течение 10 мии. во льду, проверяют
реакцию на конго и реактив «сульфон» и отфильтровывают от небольшого
количества иерастворившихся хлопьев. Осадка должно быть немного,
а фильтрат должен быть прозрачным и почти бесцветным; во всяком слу-
чае, ои не должен опять мутнеть. Дназораствор довольно устойчив в тем-
ноте, ио очень чувствителен к свету.
8. о-Ннтроаннлин
13,8 г (0,1 моля) о-и нтроаиилина раствориют, нагревая почта
до кипения, в 50 мл коицеитрироваииой соляной кислоты. Охла-
ждают при перемешивании до 30°; при этом образуется плотная почта
бесцветная кашица солянокислой соли о-иитроанилнна, лишь после этого
разбавляют 100 мл воды, вносят 100 г льда и при постоянном перемеши-
вании прибавляют по каплям 20 мл 5 н. раствора нитрита. Пере-
мешивают, пока почти все ие растворится, а нитрит останется лишь
в следах; в процессе перемешивания, если надо, прибавляют еще лед.
После фильтрования получают почти бесцветный, прозрачный н немут-
иеющий диазораствор.
9. 3 - Нитр о - 4 - то л у и д и н
15,2 г (0,1 моля) 3-ии тро-4 -толуидина возможно тоньше из-
мельчают и хорошо растирают в ступке с 50 мл концентрированной
соляной кислоты. Оставляют стоять до тех пор, пока оранжево-
желтое основание полностью ие превратится в бесцветную солянокислую
соль и при растирании ие будут обнаруживаться желтые точки. Затем
прибавляют 100 г льДа и постепенно при переметив а и ин пестиком 20 мл
5 н. раствора нитр нт а, Почти все основание переходит при этом в рас-
твор; оставшиеся комочки растирают пестиком. Наконец, фильтруют от
незначительного остатка и получают почти бесцветный раствор.
Если интротолупдин имеется в виде пасты, то его можно диазотировать, как
описано в примере 10.
10, 2-Нитро-4-хлораиилин
172,5 г 10 % -ной пасты 2 - н н т р о - 4 - х л о р а н и л и н а смешивают
в стакане с 30 мл концентрированной соляной кислоты и 100 г
льда и при перемешивании прибавляют по каплям 100 мл 1 п, рас-
твора нитрита. Перемешивают при охлаждении льдом почти до пол-
ного растворения и до тех пор, пока не останутся лишь следы нитрита, и
отфильтровывают от возможного незначительного осадка.
Изомерные нитрохлоранилины в виде пасты можно диазотировать подобным же Э
образом, Если же основания имеются в сухом виде, то их диазотируют, как описано
в примере 11, либо превращают в пасту. Для этого основания растворяют в десяти-
кратном количестве концентрированной серной кислоты, раствор выливают при пере-
мешивании на лед. отсасывают выпавший осадок и промывают холодной водой до '
удаления серной кислоты, 1
11.2,4-Дииитроанилии
В маленькую круглодонную колбу помещают 80 г концентрирован-
пой серной кислоты; колбу охлаждают льдом или лучше охла-
ждающей смесью и медленно вносят при перемешивании высушенный ;
порошкообразный н и т р и т в количестве, соответствующем 6,9 г чистого
NaNO2, так, чтобы температура не подымалась выше +10°; следят за J
тем, чтобы нитрит тотчас же распределялся в жидкости и чтобы в кис- 1
лоту не попадала вода. Не должны выделяться бурые пары, и жидкость j
должна оставаться бесцветной. После внесения всего количества нитрита
перемешивают еще в течение 10 мин., затем вынимают колбу из охла-
ждающей смссн и помещают ее в холодную воду. После того как со- |
держимое колбы будет иметь ту же температуру, что и вода, последнюю J
начинают постепенно нагревать до 70° при постоянном перемеши- j
вании реакционной смеси до полного' растворения нитрита, Температура 1
реакционной массы не должна быть выше температуры воды, и при этом уз
также пе должны выделяться газы и раствор не должен окрашиваться.
Полученный прозрачный раствор охлаждают холодной водой илн льдом а
'(при этом часто выкристаллизовывается бисульфат) и постепенно при 20° <1
вносят 18,3 г (0,1 моля) порошкообразного 2,4 - д н н н т р о а и и л и н a.
Перемешивают при комнатной температуре до тех пор, пока смешанная $1
со льдом проба не будет давать лишь очень слабую реакцию на нитрит. :Я
Выливают на 240 г льда (не больше!), отфильтровывают от не- Ц
значительного осадка и полученный раствор используют немедленно'. Ц
При работе с небольшими количествами целесообразнее брать немного больше 1
серной кислоты, чем рекомендуется выше, если это не вызывает осложнений при /3
дальнейшей работе. При разбавлении берут столько льда, чтобы получилась 25%-ная Я
серная кислота. Чем больше разбавлена кислота, тем быстрее происходит замена ,Л
нитрогруппы на оксигруппу.
Так же могут 6й1ть продиазоггиро<ваны все первичные амины, основ-
пость которых настолько мала, что они не растворяются в водных рас- 1
творах кислот, как например три- и тетрагалоиданилицы, дигалоидни- Л
троанилины, дииитроацилииы, амицоацтрахинопы и др. Если это
необходимо, то для полного растворения амина увеличивают количество
серной кислоты. Сернокислая соль диазоантрахиноиа очень трудно рас-
творима в разбавленной серной кислоте. Поэтому после выливания кон-
це нтрнр о в ан ного сернокислого раствора на лед, соль можно отфильтровать
и освободить таким образом от значительного количества избыточной
серной кцслоты,
12. Сульфаниловая кислота
17,3 г (0,1 моля) сульфаниловой кислоты растворяют
в 50 мл 2 и. раствора соды и 50 мл воды, смешивают со 100 мл
1 п, раствора нитрита и выливают прн перемешивании в смесь 125мл
2 в. соляной кислоты и 100 г льда. Большая часть трудно рас-
творимой соли диазония выпадает в осадок. Если хотят получить очень
чистую соль диазония н ие боятся при этом потерь, то соль отсасывают
й для сочетания размешивают в небольшом количестве воды.
Этот метод пригоден для всех сульфо- и карбоновых кислот, трудно раствори-
мых в разбавленной соляной кислоте, В случае особо трудно растворимых соединений
раствор следует очень медленно вводить при сильном перемешивании в разбавлен-
ную соляную кислоту. Для тех нафтиламинсульфокислот, которые склонны вступать
сами с собой в реакцию сочетания, рекомендуется к соляной кислоте прибавлять не-
много нитрита. В таких случаях целесообразно применять избыток нитрита, если он.
ис помешает в дальнейшей работе или может быть удален фильтрованием диазосо-
ед имения.
13. 1 - Амино-2-нафто л -4-сульфокислота*
24 г (0,1 моля) топко измельченной 1 -амицо-2-нафтол-4-
сульфокислоты перемешивают со 100 мл воды и небольшим
количеством льда. Прибавляют концентрированный водный раствор
2 & сернокислой меди и затем по каплям при перемешивании и
охлаждении льдом 20 мл 5 н. раствора нитрита. Амиионафтолсульфо-
кислота постепенно растворяется, образуя раствор желтого цвета. Нерас-
творившнеся комочки отфильтровывают. К фильтрату прибавляют 30 мл
концентрированной соляной кислоты, в результате чего почти все
диазосоединенне выпадает в осадок. Его отсасывают н промывают
разбавленной соляной кислотой. Это диазосоединение очень устойчиво,
и его можно, не опасаясь сушить и освободить таким образом от соляной
кислоты.
Этим Методом можно диазотировать все другие сульфокислоты 1,2- и 2,1-амино-
Нафтолов, а также и сами аминонафтолы в виде их солянокислых солей. В зависимо-
сти от растворимости диазосоединений от случая к случаю изменяют лишь метод их
выделения, которое необходимо проводить для того, чтобы удалить медь. Очень легко
растворимые свободные кислоты можно выделить в виде бариевых солей.
Диазосоединеиия можно не выделясь, если проводить диазотирование не в при-
сутствии сернокислой меди, а в присутствии сернокислого цинка, как описано в ли-
тературе **,
14. п - А м и и о д и ф е н и л а м и н ***
23,3 г (0,1 моля) сернокислого д-а м и иод и фе н и л а ми н а тща-
тельно растирают в ступке с 30 мл концентрированной соляной кислоты
и переносят в стакан, споласкивая холодной водой. Смешивают со льдом,
* Герм. пат. 171024 и 172446 (Frdl. УЩ, 640, 646).
** Герм. пат. 175593 и 176618 (Frdl.. VIII, 648, 651).
*** Герм, пат. 508585 (Frdl.. XVII, 967).
In Зак, 1905, Г Э. Фипи-Лавия, Л. Влапжп
при хорошем перемешивании прибавляют 40 мл 5 н. раствора н’итр ит а
и продолжают перемешивание при охлаждении льдом до тех пор, пока
не прореагирует азотистая кислота. При этом первичная аминогруппа
диазотируется, а вторичная одновременно питрозируется. После соче-
тания из полученного красителя можно отщепить иитрозогруппу, лучше
всего при помощи мягкого восстановителя, как например бисульфит
натрия.
в) Сочетание
1. Теоретическая часть
В реакцию сочетания с диазосоединсниями вступают:
а) Феполы.
б) Кетосоединения алифатического характера, склонные к еноли-
зации, имеющие «реакционноспособную метиленовую группу», находя-
щуюся в открытой цепи или в изо- или гетероциклическом кольце, т. е.
соединения общей формулы
X—СНЙ— СО—Y5-yX—СН= С (ОН)—Y,
где X — отрицательная группа (например, COR, COOR, CN, SC^R
И т. д.), к этим соединениям относятся также пиразолоны.
в) Первичные, вторичные и третичные амины, у которых амино-
группа непосредственно связана с ароматическим ядром.
д) Пиррол, индол и подобные им циклические системы.
Сложные и простые эфиры фенолов ие способны в нормальных усло-
виях к реакциям сочетания; аналогично ведут себя ацилированные
амины (например, ацетилированные или бензоилированные). Исключе-
ние представляют производные сульфокислот первичных аминов, напри-
мер n-толуолсульфанилид; эти соединения растворимы в щелочах и ведут
себя по отношению к диазосоединениям, подобно фенолам.
Ароматические соединения, содержащие в о- или я-положеииях друг
к другу две амино- или две оксигруппы или одну амино- и одну окси-
группы, как азосоставляющие не подходят. Они восстанавливают диазо-
соединения, превращаясь сами в хиноны *.
Первичные мопоамины ряда бензола при сочетании с диазоооедине-
н ия ми дают наряду с аминоазо со единения ми (а в некоторых случаях и
исключительно) изомерные диазоаминопронзводные; образование по-
следних протекает тем легче, чем меньше кислотность среды, в которой '
происходит азосочeraиие. Метильные или алкоксильные группы, находя- ;
щиеся в орто- и особенно в лета-положениях к аминогруппе, благо-
приятствуют образованию аминоазокрасителей; особенно гладко соче-
таются основания, в которых одновременно замещены таким образом
орто- и лета-положения по отношению к аминогруппе, иапрнмер
ОСН3
осн3
* Во многих случаях можно провести довольно гладко реакцию сочетания путем j
добавления тиосульфата” пли роданидов. Образующийся при этом краситель можно ;
диазотировать дальше и сочетать с подходящей конечной азосоставляющей для по-
лучения хромирующегося о-оксиазокрасителя— эриохромвердона А и С. Герм. пат. <
224024 и 221025; Geigy (Frdl., X, §44/5).
«7
ичетание
227
Для получения аминоазокрасителей из оснований, склонных к обра-
зованию диазоамипосоединений, можно основания действием формаль-
дегида и бисульфита превратить в метил-ю-сульфокислоты;
RNH. -I- СН3О 4- NaHSO. —> RNHCH3SO.,Na + Н2О.
Последние при сочетании с диазосоедннениями образуют азосоединения,
при омылении которых получаются аминоазокрасители *.
Вообще сочетание легче протекает с фенолами, чем с аминами, и
значительно легче с соединениями нафталинового ряда, чем с соедине-
ниями бензольного ряда. Бета-соединения ведут себя подобно фенолам
бензольного ряда. Отрицательные заместители (галоиды, иитро-, сульфо-,
карбоксильная, карбонильная группы и т. д.) ускоряют сочетание, если
они находятся в диазососдипенни, и замедляют его, если находятся
в азосоставляющей; положительные заместители оказывают обратное
влияние. Вторая окси- или аминогруппа в азосоставляющей сильно благо-
приятствует сочетанию, если находится в лега-положении к первой,
а в случае производных нафталина, также если она находится в другом
ядре.
Сочетание может быть ускорено нагреванием, увеличением щелоч-
ности (соответственно уменьшением кислотности) среды, повышением
концентрации, а также добавлением связывающих воду веществ.
Первые два из названных средств благоприятствуют как реакции
сочетания, так и разложению диазосоединений. Поэтому, если! сочетание
протекает медленно, нужно в каждом отдельном случае проверить,
является ли выгодным повышение температуры илн щелочности. В боль-
шинстве случаев, однако, не всегда при медленно протекающих сочета-
ниях наилучший результат получают, если ведут реакцию иа холоду и
в возможно' более нейтральной среде; при этом приходится мириться
с большой продолжительностью сочетания. В противоположность этому
повышение концентрации реагирующих веществ не вносит осложнений.
Поэтому при вяло идущем сочетании всегда стремятся работать при
возможно более высокой концентрации. Когда диазосоединение плохо
растворимо (как, иапрнмер, ди азосоед и пения многих сульфокислот), его
следует отфильтровать и таким образом удалить главную массу жид-
кости. Подобно повышению концентрации действует добавление индиф-
ферентных солей (особенно поваренной соли) или смешивающихся
с водой органических растворителей, таких, как спирт илн пиридин.
По-видимому, эти добавки связывают воду и таким образом повышают
концентрацию; пиридин при этом одновременно связывает и кислоту. ;|
При сочетании диазосоединений с аминами или фенолами ряда бен-
зола азогруппа всегда вступает в дара-положение (если оно свободно)
по отношению к амино- или оксигруппе, в других же случаях — в одно
из орто-положений и никогда не вступает в ткета-псложение. Феиолк,
имеющие свободные орто- и пара-положения, могут также реагировать
с двумя и даже с тремя молекулами диазосоедииёнйй с образованием
бис- и соответственно трис-азокрасителей. В некоторых случаях заме-
стители, стоящие в орто- или пара-положениях (например, SO3H или
СООН-группы), могут быть вытеснены азогруппой,
Бета-иафтол, р-нафтил амин и их производные всегда сочетаются
только в соседнее a-положение; если последнее занято, то сочетание,
либо не происходит, либо стоящий в этом месте заместитель вытесняется
азогруппой (например, у 1,2-нафтолсульфокислоты). Сульфогруппа,
* Герм. пат. 131860 (Agfa) (Frdl.. VI, 872).
стоящая в положении 8, затрудняет или вовсе препятствует вступлению j
азогруппы в положение 1.
Альфа-нафтол, а-нафтиламин и их производные сочетаются вообще ;
в положение 4; однако азогруппа легко вступает частично (а у а-нафтол а •
в 'исключительных случаях даже целиком) в положение 2. Альфа-нафтол }
склонен также к образованию дне азокрасителей путем сочетания в поло-,:
жения 2 и 4. Производные п-нафтола и а-нафтиламина, у которых ?
положение 4 замещено, сочетаются исключительно в положение 2; еелн^
заняты 2 и 4 положения, то сочетание вообще не происходит. Сульфо- j
группа, стоящая в положении 3 или 5, сильно препятствует вступлению
азогруппы в положение 4. Поэтому 1,3- и 1,5-нафтолсульфокислоты и со-)
ответственно нафтил аминсульфокислоты и их производные сочетаются;^
с большинством диа'зосюед|И1нений только в положение 2. В противопо-1
ложность этому очень энергично сочетающиеся ди аз ©соединения (прежде/
всего пронитрованные в орто- или дара-положения, или, многократно-
галоидированные дназосоединеиия, а в меньшей степени также диазо-;
производные д-амииоазобензола и т. п.) дают смесь обоих изомеров. j
I У азосоставляющих, содержащих одновременно ароматическую^
I аминогруппу и фенольную окенгруппу, решающее значение для .места-)
| вступления азогруппы имеет аминогруппа, если сочетание ведется?
у в кислой среде, и‘оке игрун Па, если сочетание ведется в щелочной среде. ’
Если сначала сочетание ведут в кислой среде (со стороны аминогруппы),;
’ то можно затем просочетать со второй молекулой диазосоединения’
г в щелочной среде (со стороны оксигруппы); для обратной последователь*)
: ностн отсутствует по крайней мере технически приемлемый способ. 1
i Способность дназосоединепий к сочетанию сильно понижается при;
наличии оксигруппы, стоящей и орго-положении по отношению к азо-;
группе. Во многих случаях вообще невозможно провести гладко реакцию!
образования красителя. Кроме того, многие о-оксидиазосоедииения полу-1
чаются ^акже с трудом. Во всех таких случаях можно использовать:
в качестве 'исходных веществ вместо о-оксиамииосоединений соответ-
ствующие о-алкоксиаминосоединения н удалять алкильный остаток из-
готового красителя, например путем обработки соединениями меди или?
хрома *.
2. Практическая часть
Целесообразность условий сочетания определяется в первую очеред
природой азосоставляющей и во вторую очередь также природой диазо
соединения.
а) Фенолы и способные к енолнзацни кетосоедн
йены я, как правило, сочетаются в содово-щелочных растворах. ЕсЛ1
азосоставляющая содержит сульфо- или карбоксильную группу или во
обще является соединением, растворимым и воде (например, резорцин):
то ее прямо растворяют в необходимом количестве раствора соды. Во все’
других случаях азосоставляющую растворяют по возможности в экви'
валентном количестве разбавленного раствора едкого натра и затем д£
бавляют необходимое количество соды.
Для полного растворения многих азосоставляющих, наприме
нафтол AS (азотолы) **, требуется избыток раствора едкого натра; в
*Ге,рм. пат. 47^997 545624, 575645, 590872 (I. G.) (Frdl., XVI, 969, XV11I, Ю2:
XIX, 1965, 1967.
** Добавление спирта очень облегчает растворение эриламидов 2,3-оксинафт01
ной кислоты; во многих случаях это является единственно возможным способом ра
творения азосоставляющей.
наприМ!
в) Сочетание
229
всяком случае берут его пе больше чем безусловно необходимо. За
отдельными исключениями следует избегать ненужного большого избытка
едкой щелочи, так как он почти всегда быстро разрушает диазосоедн-
нения. Соду целесообразно взять в таком количестве, чтобы она была
переведена кислотой диазораствора (свободной и связанной с диазогруп-
пой) в бикарбонат, однако без выделения свободной углекислоты. Таким
образом устраняют возможность образования пены. Расчет, при котором,
конечно, следует принять во внимание, хотя бы приблизительно, и коли-
чество примененной щелочи, основан на следующих уравнениях реакции,
например:
Диазотирование:
(I)
,Х—NHa + 2,5 НС1 + NaNO2 —> X—№=N | Cl 4. NaCl + 0,5 HCI -j-2HB0.
Сочетание с фенолом, растворенным в эквивалентном количестве едкого
натра;
- (Н)
X—N=N I Cl -J-0,5 MCI 4- H—Y—ONa 4- 0,5 NaBCO3 —> X—N-N—Y—OH 4-
-I- 1,5 NaCl 4- 0,5 NaHCOa-
Сочетание с солью фенолсульфокнелоты: (Ш)
+ — /ОН /ОН
X--N^N [ Cl + 0,5 НС1+ Н—4-l,5Na.,CO4—X-N=N—4-
\sO3Na L XSO3Na
-p 1,5 NaCl -p 1,5 NaHCO3.
Таким образом определяют необходимый минимум соды; избыток в об-
щем безвреден.
Кислый диазораствор медленно и при хорошем перемеши-
вании вводят нз капельной воронки в щелочной раствор азосостав-
ляющей. Сочетание протекает часто почти моментально, в таком случае
можно работать при обыкновенной температуре. Однако если реакция
Требует длительного времени, то обычно выгодно проводить сочетание
при охлаждении льдом в отсутствие яркого солнечного света. Однако
бывают также случаи, когда сочетание должно быть, наоборот, активи-
ровано слабым нагреванием, В этом отношении установить какие-либо
правила неиозможио.
Наиболее благоприятные условия нужно определять в каждом от-
дельном случае опытным путем, поскольку их нельзя почерпнуть нз
патентной литературы.
Для установления конца реакции сочетания каплю реакционной
смеси наносят на фильтровальную бумагу, и иа образоваишийся вокруг
пятпа красителя бесцветный вытек наносят раствор легко сочетающейся .
азосоставляющей, обычно водный раствор р-иафтолята натрия, Р-соли
или резорцина и, смотря по обстоятельствам, содовый раствор или раз-
бавленный раствор едкого натра. Если еще имеется диазосоединение,
не вступившее в реакцию, то на этом месте появляется соответствующая
окраска. В случае легко растворимых красителей, которые не дают бес-
цветного вытека, отбирают сначала небольшую пробу реакционной жид-
кости в пробирку, добавляют поваренной соли для осаждения красителя
и проводят испытание с обработанной таким образом смесью. Подобным
же образом можно установить наличие не вступившей в реакцию азо- ;
составляющей, нанося диазораствор па бесцветный вытек. ;
При применении диазосоедииеций с нитрогруппой в орто- или пора- !
положении, которые при действии соды перегруппировываются в ,
щелоч-
обмену,
случае
темпе-
изодиазотаты, далее нс сочетающиеся, целесообразно избегать присут-
ствия соды и заменять ее уксуснокислым натрием или, в особых случаях,
веществами, связывающими кислоту, не сообщающими раствору
ной реакции, например углекислым кальцием нли окисью магния.
Аналогично поступают, когда диазосоединение склонно к
о-стоящего заместителя на оксигруппу (см. стр. 218). В этом
рекомендуется проводить сочетание прн возможно более низкой
ратурс и избегать избытка щелочи при растворении азосоставляющей^
в отдельных случаях нейтрализуя его добавлением нескольких капел^
уксусной кислоты. При особенно чувствительных диазосоедннениих, па^
I пример в случае 2,4-динитро диазобензола, сочетание должно протекать
в мипералыгокислой среде, при возможно более низкой температуре ц
^возможно быстрее. Если применяемая азосоставляютцая растворима:
только в едких щелочах, то ее растворяют сначала как обычно, но до
прибавления диазосоедииення опять осаждают в возможно более мелко?
раздробленном состоянии добавлением разбавленной соляной или серной?
кислот (иногда в присутствии диспергирующих средств). !
Многие реакции сочетания .диазосоединений, очень чувствительных
к щелочам, протекают наиболее гладко, если применять пиридин вкаче-1
стве вещества, связывающего кислоту. Это имеет значение особенно при
получении вторичных полиазокрасителей с алкильными эфирами 1-ами-
но-2-нафтола или их сульфокислотами, как средними составляющими
(3) Амииы, как правило, сочетают в слабокислых или нейтральных’
растворах. В зависимости от соотношений растворимостей аминов посту-
пают' при этом различно.
Амины, которые растворимы как в воде, так
в разбавленных минеральных кислотах (например, ж-фе-
иилендиамин, многие нафтил а мин по л и сульфокислоты и т. д.), растворяют'
в воде. Если оин способны к многократному сочетанию, то, чтобы его
избежать, диазораствор следует добавлять к раствору азосоставляюте$
медленно при перемешивании. В других случаях проще приливать рас,
твор азосоставляющей в диазораствор. У энергично сочетающихся диазо|
соединений сочетание начинается тотчас и оканчивается через короткое
время. В случае трудно реагирующих дназосоединений снижают кислот^
ность добавлением уксуснокислого натрия и, если и после этого сочетай^
идет медленно, нейтрализуют содой и поддерживают возможно более ней-
тральную среду до окончания реакции). При большой продолжительности
сочетания рекомендуется работать прн охлаждении льдом.
Амины, которые плохо нли совсем нерастворим^
в воде, но растворимы в разбавленных минеральны^
кислотах (таковыми в большинстве случаев являются несульфиро*
ванные моноамины), растворяют в разбавленной соляной или серий
кислоте н смешивают с диазораствором, как указывалось выше. Если Ж
при этих условиях сочетание протекает медленно, то и в этом случа
снижают кислотность раствором уксуснокислого натрия или содой
однако лишь настолько, чтобы раствор еще окрашивал бумажку конП
в совсем ' “
как при
реакции
слабо синий цвет во избежание осаждения составляющих. Tai
сочетании всегда выделяется кислота, то по мере протеканий
X-\^N|C1-PH-Y-NH3C1 —► X-N=N—Y-NM3C14-НС1 4
нужно добавлять уксуснокислый натрий или соду. Чтобы остаток диазо^
соединения ввести в реакцию сочетания, в большинстве случаев необхбт
* Герм. пат. 45D998. 453133. 476080, 478045 (Frdl., XV, 521/2 XVI, 996/7).
unur
димо в заключение полностью нейтрализовать раствор; это возможно
только тогда, когда остается так мало азосоставляющей, что она вся
находится в растворе, несмотря на плохую растворимость.
Если сочетание протекает очень вяло, рекомендуется добавление
спирта нлн другого индифферентного смешивающегося с водой органи-
ческого растворителя, который хорошо растворяет азосоставляющую
с самого начала, и тогда можно работать в уксуснокислой илн нейтраль-
ной среде, Одиако когда азосоставляющая склонна к образованию диа-
зоаминосоединения (например, ^-толуидин), поддерживают при всех
обстоятельствах такую сильную кислотность раствора, какую только воз-
можно.
А м и и ы, которые не растворимы ни в воде, нн в раз-
бавленных кислотах (дифениламин, фениляафтиламин и т. п,),
в лабораторных условиях лучше всего сочетать в органических раствори-
телях (спирт, метиловый спирт, ацетон, пиридин и т. пД В технике такую
азосоставляюшую эмульгируют при помощи раствора мыла или другого
диспергирующего средства; этот способ особенно хорошо применим
тогда, когда диазосоединение сульфировано, и поэтому образующийся
краситель растворим в воде.
Сульфированные амины в виде их щелочных солей рас-
творяют в воде и прибавляют по меньшей мере столько раствора
уксуснокислого натрия, сколько соответствует количеству минеральной
кислоты, содержащейся в диазорастворе и выделяющейся при сочетании.
В приготовленный таким образом раствор прибавляют при перемешива-
нии диазосоединеиие. В случае необходимости раствор частично или пол-
ностью нейтрализуют содой, чтобы ускорить сочетание.
у) Аминофенолы, особенно амниоиафголсульфо-
кислоты. Здесь место сочетания зависит от условий сочетания, причем
в кислых растворах решающее влияние оказывает амино-, а в щелоч-
ных — оксигруппа. Поэтому, чтобы получить индивидуальный краситель,
в зависимости от желаемых результатов сочетаКУГптибо'в ясно выражен-
ной щелочной, либо в отчетливой минерально кислой среде. Для получе-
ния _пер;вичнш днеа зо крас и тел ей всегда следует сочетать первое диазо-
соёдинение в кислой а второе — в щелочной среде.
Для сочетания в кислой среде растворяют азосоставляющую в виде
ее соли со щелочным металлом в воде и добавляют соляной кислоты до
кислой реакции на конго. Образующуюся в большинстве случаев суспен-
зию добавляют к диазораствору н перемешивают прн охлаждении до
окончания образования красителя. Если реакция протекает очень вяло,
то ее можно ускорить, нейтрализуя избыточную минеральную кислоту
уксуснокислым натрием настолько, чтобы еще оставалась слабокислая
реакция на конго.
Для сочетания в щелочной среде, как и у фенолов (см, п. а), до-
бавляют соду. Если диазосоеднненше чувствительно к щелочи, то реко-
мендуется сначала добавить лишь небольшую часть соды только для
растворения а зос оставляющей, а остаток прибавлять одновременно
с диазосоединением таким образом, чтобы раствор все время оставался
слабощелочным. Добавление диазораствора в этом случае должно про-
исходить по каплям довольно медленно и прн энергичном перемеши-
вании.
3) Бензидиновые красители. При получении смешанных
дне азокрасителей из бензидина и подобных ему оснований, кроме выше-
указанного, следует еще иметь в виду, что при сочетании с первой азо-
составляющей следует избегать условий, которые могут привести
к образованию симметричного дисазокрасителя. Поэтому сначала всегда
l\jJUCUJEAU
сочетают с более трудно вступающей в реакцию азосоставляющей, а за-
тем уже—с более легко вступающей. Если обе азосоставляющие мало
отлич'аются друг от друга в своей способности к азосочетанию, то прн
первом сочетании выбирают возможно более мягкие условия (слабая
щелочность или сильная кислотность). Далее, в противоположность
обычным приемам при хорошем перемешивании вводят раствор первой
азосоставляющей в раствор бцс-диазосоедииения, а не наоборот, чтобы
избежать временного присутствия избытка азосоставляющей. Это легко
осуществить, так как бис-диазосоедипеиия бензидина и его1 производных
под действием соды при низкой температуре не изменяются. Со второй
азосоставляющей сочетание начинают, само собой поиитно, только тогда*
когда первое полностью закончилось и бцс-диазососдинение совершенно
исчезло; второе сочетание проводят тогда в обычных для моноазокраси-
телей условиях.
г) Дальнейшая переработка
1. Красителя без сульфо- или карбоксильной
групп
а) Ам й н о аз окр а с ите л и. Красители, которые содержат не-
сколько аминогрупп (например, комбинации с л - фенил енди амином)
имеют вообще ярко выраженный основной характер (хризоидин, бнемарк
коричневый). Они образуют с минеральными кислотами устойчивые
в воде растворимые соли и, как правило, применяются в виде этих солей.
Такие соли получают либо подкисляя реакционную смесь после сочета-
ния соответствующей кислотой (например, соляной) и высаливая краси-
тель солью этой кислоты (например, поваренной солью), либо осаждая
основание красителя щелочью, последующим фильтрованием, вторичным
растворением в желаемой кислоте и кристаллизацией соли.
Аминоазокрасители с одной только аминогруппой (типа амипоазо-
беизола) являются слабыми основаниями, В большинстве случаев оии
дают трудно растворимые и легко гидролизующиеся водой соли. Если
хотят выделить краситель в виде такой соли, например солянокислую
соль, то ее осаждают из реакционной массы после сочетания путем до-
бавления значительного избытка соляной кислоты и промывают после
фильтрования разбавленной соляной кислотой, но ие водой. Однако .
если такие красители должны применяться для окрашивания масел, жи-
, ров и т. п., то онн должны быть выделены в виде свободных оснований
н не должны содержать солей. В этом случае по окончании сочетания
подщелачивают, отсасывают и тщательно промывают водой.
Краситель может быть очищен перекристаллизацией из органиче-
ского растворителя.
б) Оксиазокраснтели. За малыми исключениями, здесь речь
идет об о-оксиазокрасителях, нерастворимых в воде и растворе соды,
которые поэтому прн их образовании легко выпадают из раствора. Та-
кие красители служат либо для окрашивания спиртовых н нитролаков,
масел, жиров и т. п., либо для изготовления пигментов. В первом случае
физическая форма красителя безразлична. По^яе окончания образования
красителя осадок отфильтровывают, тщательно промывают водой и су-
шат. В случае необходимости краситель можно очистить перекристалли-
зацией из органического растворителя. Напротив, пигменты только от-
фильтровываются и* промываются и, как правило, применяются в виде
паст. Поэтому всякая последующая очистка или иная дополнительная
обработка исключается и, следовательно, при сочетании необходимо уже
заботиться о том, чтобы краситель выпадал сразу в совершенно чистом
г) Дальнейшая переработка
233
виде и в желаемой степени дисперсности. Последнего достигают доба-
влением перед сочетанием в раствор азосоставляющсй небольшого коли-
чества ализаринового масла, если это не удается без применения добавок,
2. Красители с сульфо- или карбоксильными
группами
Онн, как правило, выделяются высаливанием в виде солей щелочных
металлов, растворимых в воде. При этом следует обращать внимание иа
то, чтобы краситель, с одной стороны, выпадал возможно полнее, но,
с другой стороны, чтобы могущие присутствовать примеси оставались по
возможности растворенными и чтобы не было излишне много неоргани-
ческой солн в готовом красителе. Поэтому соли берут не больше, чем
требуется для осаждения. Во многих случаях бывает уже достаточно соли,
образовавшейся совместно с красителем. Однако обычно приходится до-
бавлять еще соли. Если возможно, применяют для высаливания раствор
соли, так как при этом можно считать гарантированным, что осажденный
краситель не содержит иерастворенной соли, и, кроме того, удается из-
бежать загрязнения красителя нерастворимыми примесями, всегда содер-
жащимися н небольших количествах в поваренной соли. Только в том
случае, когда краситель так легко растворим, что длн его выделения
необходимо полное насыщение раствора поваренной солью, используют
твердую соль. В отдельных случаях более полного осаждения красителя
можно достичь добавлением не поваренной соли, а хлористого калия или
сернокислого аммония. Если средние щелочные соли хорошо растворимы
даже в насыщенных растворах соли, то, если это необходимо, краситель
можно выделить в виде свободной кислоты или кислой соли его пу-
тем добавления кислоты вместе с солью. Если такие красители в виде
свободной кислоты вследствие плохой растворимости не применимы в кра-
шении, их можно опять перевести в соли щелочных металлов растворе-
нием в минимальном количестве воды с добавлением необходимого
количества щелочи и упариванием досуха. Можно также после высуши-
вания просто перемешать с точно необходимым количеством соды; тогда
при добавлении воды одновременно происходит образование солн н рас-
творение.
Часто бывает очень трудно выделить краситель в хорошо фильтруе-
мой и промываемой форме. В большинстве случаев, ио не всегда, вы-
годно проводить осаждение при 70—80°, реже при температуре кипения,
и еще некоторое время размешивать при этой температуре. Благодаря
этой операции часто удается слизистый вначале осадок перевести в кри-
сталлическую или зернистую форму. Но может быть и наоборот, что
краситель, выпавший на холоду в кристаллической форме, при нагрева-
нии разжижается. Какого-либо правила здесь установить иельзи. Наилуч-
шие условия устанавливают в каждом отдельном случае опытным путем.
Кроме температуры осаждения н концентрации соли структуру осадка
иногда определяет также щелочность раствора. Так, например, хорошо
фильтрующийся осадок иногда получают только из нейтральных рас-
творов.
Если осаждение проводят при нагревании, то выгодно проводить
фильтрование или отсасывание горячего продукта; фильтрование идет
тогда значительно быстрее и возможные примеси остаются в растворе.
Только в том случае, если при нагревании ие удается провести полного
осаждения, может возникнуть необходимость перед фильтрованием опять
охлаждать. Для промывания применяют раствор соли, который по темпе-
ратуре и концентрации соответствует примерно первоначальному раствору.
Если при сочетании образуются побочные красители, то для лучшего
удаления их полезно добавлять в промывную жидкость немного содового
раствора. Если краситель таким путем полностью не освобождается от
побочных продуктов, то его нужно далее очистить путем переосаждення.
Для этой цели его растворяют в горячей воде, отфильтровывают, если i
необходимо, и опять высаливают приблизительно при такой же темпера- •
туре и тем же количеством соли, как прн первом осаждении. ;
Готовый промытый краситель сильно отжимают для возможно более |
полного удаления содержащегося в нем солевого раствора, затем сушат
и измельчают в порошок. Однако очень плохо растворимые красители
лучше сохранять в виде пасты и только в пробе определять содержание
сухого вещества.
Красители, содержащие несколько ннтрогрупп, во избежание взрыва
нли совсем ие сушат, или сушат с осторожностью при умеренной темпе-
ратуре.
ТЕХНИЧЕСКИЕ ПРОПИСИ ПРИМЕРОВ ДИАЗОТИРОВАНИЯ
Анилин (толуидины, ксилидины, лг-нитроаннлИн)
Размешивают стеклянной палочкой 9,3 г (0,1 моля) анилина в 30 мл
горячей воды и тонкой струей приливают 25 мл концентрированной со-
ляной кислоты. Раствору дают несколько охладиться и примерно при 40°
добавляют столько льда, чтобы температура понизилась до 0° и в рас-
творе оставалось еще немного льда. Затем быстро при очень хорошем
перемешивании добавляют 7 г 100%-ного нитрита натрия в виде
20%-ного раствора *. Концентрацию этого раствора лучше всего уста-
навливать ио чистой сульфаниловой кислоте и готовить его заранее**. •;
Диазотирование считается законченным, когда капля раствора соли -
диазония немедленно реагируете йодкрахмальной и конго-бумажкой. '
Каждую операцию диазотирования следует проверять прн помощи обоих у
реагентов. Продолжительность диазотирования — 2 мин., в больших
масштабах — 30 мин., коиечиан температура должна быть около 7°, 5
общий объем — 250 мл.
Иногда при работе с «-толуидином и хлораиилниом во время охла-
ждення льдом выделяется немного солянокислой соли, которая, однако, 1
при диазотировании быстро исчезает. ,5!
п Нитроанилин '•?
Так как я-ннтроанилин не образует устойчивых в воде солей, то эточ
основание следует вводить в реакцию в очень тонко раздробленном внде.Р
13,8 г (0,1 моля) технического нитроанилииа растворяют прн 80-—*
90° в 30 мл концентрированной соляной кислоты и 30 мл воды; прозрач- ;
ный раствор выливают тонкой струей прн хорошем перемешивании -
в 50 мл воды и 50 а тонко измельченного льда. К концу температура^
около 8°, Затем при энергичном перемешивании в один прием др-)
бавляют 7 г нитрита натрия в виде 20%-иого раствора. Температуру
поднимается до 15°, и за несколько секунд раствор становится прозрач-)
ным. Испытывают на конто- и нитритную бумажки. При работе с боль-?
шимн количествами нитрит также следует приливать очень быстро под^
поверхность жидкости, в противном случае образуются большие колнц^-?
ства диазоамииосоединения. , ;
* Объемные проценты: I л — 200 г 100%-ного нитрита натрия.
** См, аналитическую часть, :
ьензиаин [о-толиаин, о-оианизидим)
23.
а-Нафтиламин
14,3 г (0,1 моля) а-иафтиламина растворяют в 22 а 30 %-ной соляной
кислоты н 100 мл горячей воды и раствор охлаждают до 0° добавлением
200 а льда. Затем прибавляют 60 г солн и, когда температура упаде!
примерно до —5°, 20 г 20 % - ной серной кислоты и быстро добавляют 7 г
100%-ного нитрита натрия в виде 20%-ного раствора. Диазотирование
заканчивается за несколько минут; при этом трудно растворимый серно-
кислый иафтнламнп переходит в раствор. Конечный объем составляет
около 800 мл, а конечная температура — несколько ниже 0°.
Сульфаниловая кислота (метанилован кислота, нафтионован кис-
лота, ннтроаннлнисульфокнслоты, хлораинлинсульфокислоты, диамнно-
стильбендисульфокислота, сульфокислота примулина н т. и.)
17,3 г (0,1 моля) 100%-ной сульфаниловой кислоты растворяют
в 5,5 а соды н 100 мл воды1* **. Затем добавляют 25 мл концентрирован-
ной соляной кнслоты и диазотируют прн хорошем перемешнваини 35 мл
20%-ного раствора нитрита натрия. Диазотирование длится примерно
10 мни., температура может подниматься до 15°.
Дназосоедннення, содержащие сульфогруппу, в' большинстве случаев
плохо растворимы н выпадают в виде внутренних солей в форме белых
нлн желтых кристаллических осадков. Так как многие амнносульфокнс-
лоты также очень» трудно растворимы, то приходятся их' диазотировать
обходным путем? Та к, вместе с натриевой солью сульфокислоты раство-
ряют необходимое количество нитрита натрия и выливают эту смесь
в кислоту.
Дальнейшие затруднения возникают также вследствие того, что опре-
деленные амины, например кнслоты Клеве, сочетаются сами с собой,
поэтому для ннх требуется применение примерно 5% избытка нитрита
натрия,
Беизидии (о-толидин, о-диаиизидии)
18,4 г (0,1 моля) технически чистого бензидина растворяют при 70°
в 23 мл 30%-иой соляной кнслоты и 150 мл воды*'*. Этот раствор охла-
ждают до 30—40° н прибавляют примерно 50 г льда, после чего выде-
ляется часть солянокислой соли. Затем прн хорошем перемешивании
добавляют еще раз 23 мл соляной кислоты, которую предварительно раз-
бавляют небольшим количеством воды. Прн этом выделяется еще больше
солн, после этого быстро, в течение 10 сек. вливают 70 мл 20 %-ного рас-
твора нитрита натрия. Температура достигает 10—12° и раствор должен
за 1 мнн. стать прозрачным. При более низкой температуре последние
следы сернокислого бензидина часто исчезают лишь через 8—10 Мии.
Испытывают на коиго- и ннтрнтную бумажки. Раствор почти нейтралей,
по в этом случае не образуется дназоаминосоеднненнн, как это происхо-
дит при диазотировании анилина.
Толндии н днаиизиднн нельзя кипятить; лучше всего их растворять
при температуре ниже 40° нли тонко измельчить и суспендировать.
В производстве растворы с половинным количеством соляной кнслоты
оставляют на ночь и лишь иа следующий день прибавляют лед и остаток
соляной кислоты.
* Сульфокислоты в виде солей растворяют, естественно, только в воде.
** Для получения совершенно прозрачного раствора следует применять соляную
кислоту, не содержащую серной.
zoo
//. Красители
ПРИМЕРЫ АЗОСОЧЕТАНИИ
Простые сочетания с окснсоеднненнем
Кислотный оранжевый А, илн оранжевый II
(кислотный оранжевый)
он
но3з-/
лоты в
удаления
ной кислоты
Затем добавляя
Р и с. 32. СТакан с делениями
для сочетания.
Растворяют 17,3 г (0,1 моля) 100%-ной с у л ь ф а н и л о в о й кис-
6 г с о д ы и 200 мл воды н кипятят с водяным паром для
прнмесн анилина. После фильтрования прибавляют 30 мл кон-
центрированной С О Л Я " ~
н охлаждают до 20°.
небольшое количество льда, понижают -
температуру до 10° н диазотируют прн £
температуре ниже 15° 7 г 100%-ного
нитрита натрия до появления неис-
чезающен реакции на нитритную н конго- i
бумажки. :
Одновременно растворяют 14,4 г ;
(0,1 моля) ^-нафтола в смеси 15 г
30 %-ного раствора едкого натра,.
25 г сод ы и 200 мл в о д ы; раствор ₽-иа-
фтола должен быть прозрачным. Затем ]
раствор охлаждают льдом до 3° и тонкой
струей добавляют к нему суспензию диа- (
зотированной сульфаниловой кислоты;
при этом температура ие должна превы- ;
шать 8°. Через час суспензию образовав- j
шегося красителя нагревают до кипения
в фарфоровой чашке па голом пламени и 31
к кипящему раствору добавляют пор- <5
циями 100 г поваренной соли. Кра- |
ситель полностью выделяется и может |
быть легко отфильтрован на большом
нутч-фильтре при 50°. После отжима при J
помощи винтового пресса вещество сушат при 100°. 3
Выход составляет около 50 г, но точно его определить можно j
только путем сравнительного крашения. На рис. 32 изображен лабора- |
торный прибор для сочетания, 1
Технические примечания. Кислотный оранжевый А вследствие деше-
визны и живости оттенка является одним из наиболее часто применяющихся моноазо-
красителей. В технике сочетание проводят в очень больших чанах из пичпайна (белой
американской сосны) объемом от 15 000 и более литров или даже в бетонных чанах„
выложенных керамическими плитками, объемом до 40 м3. На рис. 56, стр. 334, пока- ..
зана общая заводская установка с чанами для диазотирования и сочетания, а также
монтежю и фильтр-прессы. Отфильтрованный краситель не отжимают на гидравли- f
ческих прессах, а продувают на фильтр-upeccc сжатым воздухом в течение 1—3 час. т
и затем непосредственно сушат па медном противне, Для этого применяют обычно' '
вакуумные сушильные шкафы, которые не только делают возможной быструю сушку„ '
но также обеспечивают большую сохранность красителя. Расчет Ci он мости этого-
красителя подробно излагается ниже (см. стр. 335 и сл,).
лцетил-АШ-кислота и амиоонафтол красный Г 23;
В этом особом случае азосочетание можно также проводить и без применение
соды, р-Нафтол обычно растворяют в необходимом количестве едкого натра и сме
шиваюг этот раствор с почти нейтральным раствором диазотированной сульфаниловой
кислоты. ]3-Нафтол выпадает в очень тонко раздробленном виде. После этого всю масс^
основательно перемешивают и при 0° прибавляют такое количество едкого натра, какое
необходимо для образования нейтральной натриевой соли кнелотноге
оранжевого. Сочетание протекает мгновенно, и краситель отфильтровывают на холоду
без предварительного подогревания. Таким путем получают очень концентрированный
краситель, который плавится на сушильном противне. Этот простой метод здесь не
выбран, потому что большинство красителей подобным образом получить нельзя.
Однако приведенный метод применим почти во всех случаях сочетания с ^-нафтолом
(рокселлин, яркий оранжевый и т. д.)*.
Ацетил-А ш-кислота и амндонафтол красный Г
(кислотный ярко-красный)
СН3СО—NH ОН
HO3S—
35 г 100%-ной Аш-кислоты (0,1 моля) растворяют прн 50° в 6 г
соды и 200 мл воды. При энергичном перемешивании в течение
’А часа добавляют 17 а уксусного ангидрида, при этом амино-
группа Аш-кнслоты ацетилируется полностью, одновременно происходит
также частичное ацетилирование оксигруппы. Для того чтобы убедиться
в .полноте ацетилирования, небольшую пробу раствора подкисляют со-
ляной кислотой, прибавляют несколько капель раствора нитрита натрия,
а затем раствор соды до щелочной реакции. Если содержится еще много
Аш-кислоты, то появляется синяя окраска (самосочетание диазотирован-
ной Аш-кислоты), по мере того как ацетилирование протекает дальше,
окраска становится слабее и более красной (сочетание диазотированной
Аш-кислоты с ацетил-Аш-кислотой); если больше иет иепрореагировав-
шей Аш-кислоты, то появляется только желтая окраска, вследствие
нигрозироваиня (эту реакцию удобно проводить на фильтровальной бу-
маге) . Когда с помощью этой реакции убеждаются, что ацетилирование
прошло полностью, прибавляют 25 г кальцинированной соды и нагре-
вают прн перемешивании 1 час при 90—95°, все время добавляя воду
взамен испарившейся. Прн этом ацетильная группа, стоящая у кисло-
рода, отщепляется (эго можно контролировать с помощью титрования
раствором диазобензола), а ацетильный остаток в аминогруппе ие за-
трагивается **.
Полученный содово-щелочный раствор ацетил-Аш-кислоты исполь-
зуется непосредственно для сочетания с различными диазосоедннениями;
образующиеся прн этом азокрасители дают окраски, отличающиеся
большой светопрочностью, красотой и исключительной ровнотой. Напри-
мер, с диазотированным анилином получают важный краситель амидо-
нафтол красный Г (кислотный ярко-красный).
Диазотируют 9,3 г анилина (0,1 моля), как указано на стр. 234,
и смешивают раствор соли диазония с охлажденным льдом раствором
а цетил-Аш-кислоты, содержащим соду. Спустя 12 час. высаливают
на холоду поваренной солью (20% от объема жидкости).
* St hul't z, Farbstof ftabellen.
** Если це производить обработку содой, то теряется около 30% исходного ве-
щества и краситель оказывается загрязненным продуктами разложения диазососди-
нений.
а. /\ раса гели
Отфильтрованный краситель хорошо отжимают на винтовом прессе и су-
шат при 50°.
Выход около 50 г.
Если вместо анилина применяют аминоацетанилид, описанный на
стр. 71, то получают краситель синеватого оттенка амидонафтол
красный 6 Б (кислотный красный ЗС), который является еще более
светопрочным, чем марка Г.
Технические примечания. Описанные красители в значительной сте-
пени вытеснили аналогичные красители из хромотроповой кислоты (1,8-диоксина-
фталин-3,6-дисульфокислота). Они дешевле последних и более светопрочны.
Интересно, что ацетилирование нельзя проводить на производстве в деревянной
аппаратуре. Аппаратура нз содержащего смолу пичиайиа почти всегда придает
красителю мутный оттенок, Поэтому ацетилируют в эмалированных сосудах. Далее
в технике применяют несколько меньшие количества уксусного ангидрида. Чтобы из-
бежать омыления, краситель отжимают на гидравлическом прессе и быстро сушат
в вакуумном сушильном шкафу при 60°. В таких случаях благоприятно влияют не
только низкая температура, но также, возможно, быстрая сушка.
Желтый светопрочный Г (фирмы Байера)
Желтый светопрочный Г является простейшим представителем пирй-
золоиовых красителей. Последние получают двумя различным способами:
из диоксивиииой кислоты и фенилгидразииов (тартразин) и сочетанием
фепилметилпиразолонов с диазосоединеииями. Второй путь проще и
поэтому вытеснил старые способы получения. Из производного феиил-
гидразипа, иапрнмер из феиилгидразинсульфокислоты (описана на
стр. 117) и ацетоуксусного эфира, получают пиразолои (см. стр. 118)
и сочетают его с диазотированным анилином.
/СЬЧ --N2—/
у-ОН —сн3-С "с-ОИ
N N----<Z/~S0»H N—N-/2)>SO,H
1-Сульф офени л-3-.митил-5-пи разили и
Желтый светопрочный Г
В формуле метилфеиилпиразолонсульфокислоты атом водорода,
который при сочетании замещается на азогруппу, обозначен звездочкой.
Этот атом водорода находится в орто-пол ожени и к оксигруппе +, которая
обусловливает возможность сочетания. Опа действует точно так же, как
оксигруппа в фенолах и нафтолах и в азокрасителях из о-амииофеиолов
или о-аминонафтолов н имеет лакообразующие свойства. Образуются кра-
сители типа
ОН‘
но
Так, например, из 1,2,4-а ми ио нафтол сульфокислоты (описана на стр. 178)
и феиилметилпнразолона (из фенилгидразина и ацетоуксусного эфира)
образуется эриохром красный Б (фирмы Гейги) (кислотный хром крас-
ный), очень прочный хромирующийся краситель для шерсти.
с Н3— С ЧС—N,- / SOyH
II I
Й—N-QH;
- Эриохром красный К (Xarcj|6‘jx)
(КПГ .ЧОТИЫИ Хрии Б par Jlliltj 1
26 г (0,1 моля) 100%-ного сульфофеиилметилпиразо-
л о п а растворяют в 6 г с од ы и 120 мл в од ы и добавляют
к раствору 30 а уксуснокислого натрия. При 0° смешивают
с раствором фенилдиазония, приготовленным из 9,3 г анилина, и переме-
шивают до тех пор, пока небольшая проба после высаливания ие переста-
нет давать красную окраску со щелочным раствором резорцина, что
обычно наступает через 4—6 час. Затем нагревают до кипения и выса-
ливают 100 г поваренной соли.
Выход концентрированного красителя составляет около 40 г.
Эти пиразолоновые красители, особенно * более сложные, прочнее, чем Прочный
желтый (см. стр. 243). Красители из о-сульфоаминов, как например из л-толуидин-
.w-сульфокислоты или л-хлоранилин-о-сульфокислоты, относятся к наиболее свето-
прочным желтым красителям, которые вообще известны. Но светопрочность может
быть-еще больше повышена, если для получения азокрасителя применять вместо суль-
фофенилпиразолонов хлорзамещенные фенилпиразолоны. Ксиленовый желтый
(фирмы Сандоз) является хлорпроизводным такого типа; несмотря на довольно вы-
сокую цену, краситель вследствие своей исключительной светопрочности находит все
большее применение. Здесь имеются почти неограниченные возможности различных
комбинаций; одну из них представляет собой поляр желтый 5Г (фирмы Гейги),
который имеет следующее строение:
СНЯ
S°sH N = C
i
Cl—Q N—Cz
Остаток 4-хлор- Хт,
апнлик-З-сульфо-
кислоты Остаток
ацето-
уксус-
ного
• эфира
Остаток Остаток
я-амино- л-толуол-
фепола сульфо-
кислоты
Поляр желтый 5Г (Рихард), желтый д.чя швейцарской кавалерии.
н.-Хлор-щсуЛьфофснилгидразнн конденсируют с ацетоуксусным эфиром и обра-
зующийся пиразолон сочетают с диазо-гс-аминофенолом в уксуснокислом растворе.
Образующийся чувствительный к щелочи азокраситель обрабатывают при 70°
и-толуол сульфохлорид ом в присутствии соды и моля едкого натра, благодаря чему
оксигруппа этерифицирустся. Этерифицированный краситель совершенно нечувстви-
телен к щелочи и дает прочные к валке окраски на шерсти.
Технические примечания. Получение пиразолоновых красителей осу-
ществляется просто. Арилгидразины конденсируют' в большинстве случаев в эмали-
рованных сосудах, чтобы возможно меньше потерять дорогостоящих веществ. Про-
цессы диазотирования и сочетания ничем не отличаются от описанных ранее,
Ганза желтый Г (пигмент желтый светопрочный)
15,2 г чистого З-иитро-4-толуидииа диазотируют по про-
писи, приведенной на стр. 223. Отфильтрованный’ прозрачный диазорас-
твор охлаждают льдом до 2—5° и для понижения кислотности добавляют
такое количество уксуснокислого натрия, чтобы бумажка коиго еще окра-
шивалась в фиолетовый цвет,
.Готовят при 50° раствор 17,7 г чистого анилида ацетоуксус-
ной кислоты в 300 мл воды и примерно 12 мл 35%-ного раствора
едкого натра (последнего должно быть взято ровно столько, чтобы
получился прозрачный раствор), Охлаждают и добавляют 25 г кристал-
лического уксуснокислого натрия; затем осторожно приба-
вляют столько разбавленной уксусной кислоты, чтобы появилась
кислая реакция на лакмус, но не выпадал осадок. Затем при очень хо-
рошем перемешивании добавляют по каплям прозрачный раствор
диазосоединеиия и перемешивают еще в течение 12 час. Выделившийся
полностью краситель отфильтровывают и-тщательно промывают горнчей
дистиллированной водой. Остаток на фильтре замешивают в ступке
в однородную пасту, которую для изготовления пигментов лучше всего
в таком же виде смешать с субстратом. Для определения содержания
сухого красителя сушат отвешенную небольшую пробу.
Выход количественный.
Технические примечания. Ганза желтый Г представляет собой чисто
желтый краситель. Более зеленоватые марки получаются, если заменить нитротолу-
идин на о-нитроанилин или о-нитро-н-хлоранилин, а также иногда анилид ацетоук-
сусной кислоты на о- или n-хлоранилид ацетоуксусной кислоты. Все эти красители
являются исключительно светопрочными и удовлетворяют всем требованиям, предъ-
являемым к пигментам в отношении прочности к маслу и спирту*. Они являются
наиболее ценными пигментами.
Перманент красный ГГ (пигмент оранжевый прочный) **
18,3г (0,1 моля) чистого 2,4 - д и н и т р о а н и л и и а» диазотируют л
по прописи, приведенной на стр. 224, и при перемешивании и охлажде-
нии льдом медленно добавляют по каплям раствор диазосоединення-
в концентрированной серной кислоте (ие разбавленной льдом) в рас-
твор 15,5гР-нафтола в 400 м.л спирта с такой скоростью, чтобы
температура ие превышала 5°. Сочетание начинается тотчас и спустя
несколько минут после добавления диазосоединеиия заканчивается. Пере-
мешивают еще 1 час, полностью выделившийся краситель отсасывают на
нутч-фильтре и заливают три раза небольшими порциями спирта до тех
пор, пока не появятся бесцветные капли фильтрата. Затем основательно
промывают горячей водой до полной нейтральности фильтрата, сушат
в паровом сушильном шкафу и возможно тонко измельчают.
Выход 32—33 г (около 95—98% от теории). Краситель предста-
вляет собой яркий ораижево-красный порошок, совсем нерастворимый
в воде и очень мало растворимый в обычных органических растворителях.
Технические примечания. Перманент красный ГГ (или литоль оранжевый
прочный Р) является, как и пигмент желтый светопрочный, очень ценным пигментом
с выдающейся светопрочлостью и очень хорошей прочностью к маслу и спирту. '*
Описанный выше метод сочетания в спиртовом растворе является наиболее j
простым и надежным лабораторным методом получения совершенно чистого красителя, •.
чистота оттенка которого отвечает современным очень высоким требованиям. Приме-
няемый избыток jJ-нафтола значительно ускоряет сочетание и благодаря этому пред- 7
отвращается разложение диазосоединеиия.
Для получения красителя в больших масштабах чаще всего применяют водную ,’.'
эмульсию р-нафтола, , которую получают при медленном добавлении соляной или
* Герм. пат. 257488 (Frdl1,, XI, 452) и герм. пат. заявка F33J90 (Frdl., X], 455).
** Laudi R., герм. пат. 217266 (Frdl., fx, 418).
Гелиобордо Б Л
241
серной кислот к раствору р-нафтола в едком натре в присутствии диспергирующего
средства, как например некаль БИкс (смачиватель НБ, натриевая срль изопропилна-
фталинсульфокислоты). Во всяком случае, сочетание должно проводиться в среде мине-
ральной кнслоты, иначе образуется коричневатый, тупой, совершенно непригодный
краситель.
Гелнобордо Б Л (лак бордо СК)*
он
О 4/Y
SOsNa
а) Получение натриевой соли
14,3 г (0,1 моля) а-н афтиламина растворяют прн кипячении
в 280 мл воды и 10 мл концентрированной соляной кислоты.
Охлаждают при встряхивании водой под краном, чтобы выкристаллизо-
вавшийся солянокислый нафтил амии получить в возможно более'дис-
персном состоянии, затем смешивают с 75 мл концентрированной соля-
ной кислоты и опять охлаждают. К полученному раствору добав-
ляют 250 г мелко истолченного льда и при сильном встряхивании
сразу добавляют 7,1 г сухого и измельченного нитрита н,а три я.
Затем сильно встряхивают до тех пор, пока солянокислый' нафтнламин
полностью не перейдет в раствор, что происходит через короткое время.
После 15-минутного стонния отфильтровывают от небольшого остатка
прозрачный светло-корнчневый свободный от пеиы раствор, который еще
должен давать слабую реакцию на нитрит, и фильтрат приливают при
перемешивание в охлажденный льдом раствор 27 г натриевой соли
(96 %-нон) 1,5-иафтолсульфокислоты (о получении ее см.
стр. 196) в 200 мл воды. Перемешивают в течение получаса прн
охлаждении льдом, отсасывают желто-коричневый кристаллический оса-
док, промывают его ледяной водой и смешивают с 500 мл ледяной воды.
В полученную суспензию прибавляют по каплям прн охлаждении льдом
и сильном перемешивании 80 мл 2 н. раствора соды таким обра-
зом, чтобы каждая капля тотчас распределилась в жидкости. Как
только весь раствор соды добавлен, сочетание заканчивается и капля
реакционной жидкости должна давать отчетливо синее окрашивание лак-
мусовой бумажки, ио не давать красного окрашивании фенолфталеино-
вой бумажки. ’ После этого прекращают охлаждение и -медленно на-
гревают при перемешивании до 80—90°. При этом темно-коричневый
желатинообразный вначале краситель быстро превращается в крнсталлн-.
ческии. Прибавляют в количестве 0,1 от объем’а жидкости насыщен-
ный р а с т в ор не содержащей кальция поваренной солн;
раствор отсасывают горячим, тщательно промывают горячим, не содержа-
щим кальция 5%-ным раствором поваренной соли, последнюю вытес-
няют холодной водой и сушат в паровом сушильном шкафу.
Выход около 37 а, т. е. 92% от теории.
б) Получение кальциевого лака
10 г полученного, как указано выше, красителя кипятят с обратным
холодильником в 250 мл воды и 25 мл 2 н. раствора едкого
и а т р а до полного растворения. Затем охлаждают, фильтруют через
* Герм, пат, заявка F 28279 (1910) (FfdL, X, 946).
складчатый фильтр и фильтрат медленно прибавляют по каплям в
сильно размешиваемый раствор 8 а кристаллического уксуснокис-
лого кальция н 30 мл 2 и, уксусной кислоты в 500 мл
воды при 25—30°. Каждая падающая капля должна тотчас вызывать
образование очень тонкокристаллического ярко-красного осадка, По
окончании приливания смесь нагревают до 80—90°, осадок отсасывают
горячим, тщательно промывают его горячей водой и сушат в паровом
сушильном шкафу.
Выход количественный.
Примечание. Гелиобор до БЛ (лак бордо СК), который даже в виде своей
натриевой соли довольно трудно растворим, дает совершенно нерастворимую кальцие-
вую соль яркого сине-красного цвета, которая полностью отвечает требованиям,
предъявляемым к красочным лакам, и является особенно ценной для изготовления
полиграфических красок. Однако самые незначительные загрязнения сказываются на
чистоте оттенка. Чтобы помешать образованию таких нежелательных побочных продук-
тов. необходимо проводить диазотирование о-нафтиламина так, чтобы це образова-
лось следов аминоазонафталина или его диазосоединения; этого удается достичь,
применением необычно большого избытка соляной кислоты. Кроме того, сочетание
необходимо проводить не как обычно в щелочной среде, а таким образом, чтобы
кислая смесь диазосоединения и азосоставляющсй постепенно нейтрализовалась. Это
даст возможность избежать частичного сочетания в положении 4. Так как при сме-
шении диазосоединення и азосоставляющей в кислом растворе выделяется в хоро-
шей кристаллической форме плохо растворимая а-нафтилдиазониевая соль 1,5-нафтол-
сульфокислоты, то рекомендуется эту соль отфильтровать, вследствие чего удаляется
избыток соляной кислоты. На результаты лакообразования существенное влияние
оказывают также условия переведения натриевой соли в кальциевую.
1,5- Нафтолсульфокислоту получают щелочным плавлением нафталин-1,5-дисуль-
фокислоты (ср, стр. 196)*, а также из 1-нафтиламип-5-сульфокислоты (ср. стр. 191)
во Бухсрсру.
Простые сочетания с аминами
тг-Амнноазобензол нз аннлнна
28 г (0,3 моля) анилина растворяют в 350 мл 2 и. соляной ;
кислоты, охлаждают снаружи холодной водой до 20° и затем коло- ,
тым льдом до 0°, Полученный раствор дназотируют 22 г технического, 4
нитрита н а т р н я, растворенного в 150 мл ледяной воды. Ди- ..
азотирование может быть проведено менее чем за 1 мии. Раствор дол- '.
жен оставаться совершенно прозрачным, появление мути указывает на л
недостаток соляной кислоты, Спустя 10 мин. диазотирование заканчн- j
васдея (испытание н^ конго- и йодокрахмальную бумажки), j
К полученному* раствору соли диазония добавляют 31г анилина,.
растворенного в 200 мл воды, 100 г льда, 40 мл 2 н. уксусной. ’<
* Ewer, Pick герм. пат. 41934 (Frdl., 1, 398).
п-Аминоазобензоя из анилина
243
кислоты и 120 мл 2 н, соляной кислоты, Спустя 10 мин, при
постоянном перемешивании добавляют по каплям смесь 50 мл 25 %-ного
аммиака и 400 мл вод ы, следя за тем, чтобы все время оставалась
отчетливо кислая реакция на лакмус. Аммиак приливают под уровень
жидкости, Продолжительность приливания 2—3 часа. К этому времени
реакция на диазотированный анилин (проба с раствором Аш-кислоты)
исчезает. Желтое диазоаминосоединение выделяется в виде легко филь-
трующегося осадка. Его отфильтровывают и хорошо промывают дистил-
лированной водой, Осадок, отжатый .на пути-фильтре, смотря по продол-
жительности отсасывания, весит 120—170 г.
Перегруппировка диазоаминосоединения, Осадок смешивают в со-
суде с мешалкой со 140 г анилина, при этом ои быстро почти полностью
растворяется. Вода, которая содержалась в диазоамииосоединеиии, вы-
деляется в основном иа поверхности масла и может быть легко удалена
при помощи маленькой пипетки. После этого добавляют 10 г соляно-
кисл ого анилина (анилиновой соли) и смесь медленно нагревают
при постоянном перемешивании на водяной бане до 30°, Спустя 2 часа
осторожно повышают температуру максимум до 40° и оставляют при
этой температуре на ночь, На следующее утро отбирают небольшую
пробу реакционной смеси в пробирку и нагревают до 100°. Если пере-
группировка закончилась, азот ие выделяется и ие чувствуется запаха
фенола. В этом случае можно нагревать смесь еще 1 час при 50°, ио это
в большинстве случаев ие обязательно,
Маслянистую смесь выливают при хорошем перемешивании в 200 мл
концентрированной соляной кислоты и 300 мл воды. Выпавший
графитоподобный осадок солянокислого аминоазобеизола после охлаж-
дения дважды промывают на нутч-фильтре водой, подкисленной соля-
ной кислотой (4 мл НС1 иа 100 мл. воды), Полученный таким образом
сырой продуют является уже достаточно чистым. Он может быть пере-
кристаллизован из большого количества воды с добавлением соляной
кислоты, причем маточник применяется опять для кристаллизации; вслед-
ствие этого отсутствуют потери, Применяют 1%-ную соляную кислоту.
Для перекристаллизации указанных выше количеств требуется около 3 л
воды, которая используется в виде маточных растворов около четырех
раз. Часто солянокислый амииоазобензол выделяется сначала красного
цвета, а затем быстро переходит в графитоподобную форму.
. Чистую соль сушат при 50—55°.
Выход 49—50 г, т. с, 70 % от теории.
Свободное основание получают при длительной обработке соляно-
кислой соли разбавленным раствором соды или аммиака.'Но чаще всего
для дальнейшей переработки применяют сол .шокислую соль. *
Получение по топ же схеме аминоазотолуола протекает с еше лучшим выходом.
Прочный желтый. Прочный желтый является дисульфокислотой амино-
азобензола. Первая сульфогруппа вступает главным образом в пари-положение к азо-
группе, при этом образуется желтый краситель для шерсти умеренной прочности.
Лишь при введении в молекулу Красителя второй сульфогруппы в орто-положение
к. аминогруппе (или азогруппе) удается значительно повысить све,топрочность.
Это сульфирование осуществляется очень просто, Сухой солянокислый амино-
азобензол вносят в трехкратное количество 25 %-ного олеума и перемешивают при 25°
до тех пор, пока проба не будет гладко растворяться л соде. Затем при постоянном
хорошем перемешивании повышают температуру до 40° и нагревают при этой темпе-
ратуре до тех пор. пока проба полностью не растворится в большом количестве воды,
что происходит приблизительно через 5 час. Сульфомассу выливают на шестикратное
количество льда и высаливают мопоиатриевую соль дисульфокислоты поваренной
солью, взятой из расчета 200 г на литр, Осадок цвета мяса отфильтровывают, тща-
тельно промывают 15%-ным раствором поваренной соли, отсасывают и обрабатывают
небольшим количеством воды, а затем прн 5СГ таким количеством воды, чтобы окраска
244
II. Красители
стала чисто желтой; количество соды зависит от тщательности промывки осадка.
Прочный желтый можно выделять из раствора не высаливанием, а выпа-
риванием при 90° досуха.
Выход составляет около 200%, считая на исходное вещество.
Аминоазобензол в технике получают в больших эмалированных котлах емкостью
300—400 л. Выделение кислотой проводят в обыкновенных деревянных чанах. Маточ-
ный раствор, содержащий анилин, обрабатывают известью н анилин отгоняют с во-
дяным паром, потери составляют около 15%,
Вопреки противоречивым утверждениям, прочный желтый является не таким
светопрочным красителем, как тартразин, и значительно менее прочен, чем пиразоло-
новые красители, имеющие сульфогруппу в орто-положении к азогруппе. Аминоазобен-
зол является важным исходным веществом для получения многих дисазокрасителей.
Сочетание диазотированного аминоазоберзола с фенолами и другими азосоставляю-
щими приводит к получению вторичных дисазокрасителей, первым из которых был
так называемый бибрихский алый (комбинация; аминоазобензолдисульфокислота 4~
+ (3-нафтол). Поэтому красители этого класса вторичных дисазокрасителей называют
вообще красителями типа бибрихского алого. Диазотирование аминоазобензола продол-
жается несколько часов. Свежеприготовленный солянокислый аминоазобензол суспенди-
руют в пятикратном количестве воды и добавляют на 1 моль солянокислой соли еще
150 г соляной кислоты, Перед диазотированием надо установить необходимое количе-
ство нитрита натрия путем диазотирования небольшой пробы при большом разбавлении.
Диазотирование аминоазобенэола проводится при 10—14° и на производстве про-
должается обычно целый день. Готовая диазотированная масса должна быть либо
тотчас переработана, либо охлаждена льдом до 0°.
Подобно анилину аминоазобензол можно конденсировать с динитрохлорбелзо-
лом. При этом образуется фенилазодинитродифениламнн, почти нерастворимое
Красное кристаллическое вещество. Действием моногидрата его легко превращают
в моносульфокислоту, причем получается нитроазокраситель такого же состава, как
описанный на стр. 247 азожелтый Г (фирмы «Вейлер-тер-Меср»), Но он отличается
от последнего своей совершенной однородностью и не отщепляет при кипячении азо-
тистой кислоты.
Поэтому, несмотря на сравнительно высокую цену, некоторые красильщики для
крашенин шелка предпочитают его обыкновенному азожелтому.
Азофлавин Ф Ф (фирмы БАСФ)
динитроди- у!
фениламнн Азофлавнн ФФ j
а) Конденсация аминоазабензола с динитрохлорбензолом. Нагревают 100 г
100%-ного еще влажного солянокислого аминоазобензола, 100 г д И- у.
Тропеолин. или оранжевый /V
245
н итро х л ор бензо л а, 250 г кристаллического уксуснокислого натрия и
600 г —' 90%-ного спирта при перемешивании с обратным холодильником л тече-
ние 6 час. Продукт конденсации выделяется в виде красно-коричневых блестящих
кристаллов, которые отфильтровывают от горячего раствора и промывают не-
большим количеством спирта. После сушки при 100° вес осадка око-
ло 115 г.
б) Сульфирование, Одну часть продукта конденсации вносят в 3 части моно-
гидрата и перемешивают при 30° в течение 1 часа, после чего осторожно повышают
температуру до 45°. Через 1—2 часа проба полностью растворяется в разбавленном
растворе соды. Затем смесь выливают в шестикратное количество воды, высаливают
краситель, отфильтровывают, промывают 15%-ным раствором поваренной соли и
затем растворяют в небольшом количестве горячей воды с добавлением необходимого
количества соды для получения прозрачного раствора. Высаливают 15% (по
объему) поваренной соли, причем сначала получается желатинообразный осадок
натриевой соли, который быстро превращается в красивые легко фильтрующиеся
кристаллы.
Из 100 г продукта конденсации получают около 125 г концентрированного краси-
теля. Аэофлавин Фф имеет оттенок слабонитрованного тропеолнна и обладает боль-
шой устойчивостью к кислотам, свойственной высоконитрованному азожелтому.
Тропеолин, или оранжевый IV (кислотный желтый метаниловый К)
н азожелтый нз сульфаниловой кнслоты н дифениламина
Сочетание сульфаниловой кислоты с дифениламином является инте-
ресным примером сочетания в мииеральнокислой среде. В этом случае
невозможно работать с нейтральными или щелочными растворами, так
как ди азосульф а ии левая кислота тотчас разлагается содой, и хотя
это и неожиданно, ие реагирует в нейтральном растворе. Затем дифенил-
амин в воде совершенно нерастворим, поэтому сочетание нужно
вести в водпо-спиртовом растворе. При известных условиях можно рабо-
тать без спирта, но в этом случае остается так много дифениламина, что
затрудняется нитрование красителя. Между тем кроме оранжевого IV
очень ценными красителями являются его иитропроизводные. Поэтому
сочетание лучше проводить в спиртовом растворе, тем более что полу-
чающийся при этом более высокий выход красителя компенсирует неболь-
шие потери спирта.
Уже упоминалось, что дифениламин препятствует нитрованию; в еще
большей степени препятствуют также и другие примеси, которые
имеются в оранжевом IV. Если тропеолнн перерабатывают в азожелтый,
то кислота тропеолииа должна быть абсолютно чистой; незначительные
примеси понижают выход на 30—50%. Ес-ли имеется чистая кислота тро-
пеолина, то нитрование осуществить довольно легко. Эта реакция инте-
ресна тем, что нитро соединение удается получить через нитрозамин и
нитрамин. Тропеолин нитрозируют азотистой кислотой и окисляют затем
очень слабой азотиой кислотой. В виде промежуточного соединения об-
разуется нитрамин, который под влиянием минеральной кислоты сразу
перегруппировывается в питросоединеиие, подобно тому как феиилнитра-
мин Бамбергера перегруппировывается в о-нитроанилии. Аналогичное яв-
ление наблюдается у метиленового зеленого (см. стр. 280),
Тропеолин часто применяется для крашения шерсти, так как дает
окраски чистого оттенка и достаточно прочные к свету и стирке. При при-
менении в смесях со миегими красителями ои значительно повышает
интенсивность последних. Это свойство тропеолина особенно важно для
многотоинажного красителя кислотного черного 4В, который представ-
ляет собой смесь, состоящую из 45% нафтолового сиие-черного Б, 45%
нафтиламинового черного Д, 5% тропеолииа и 5% прочного- красного А
Фау (кислотного красного Ж) Опыты замены в этой смеси тропеолина
другими желтыми красителями показали, что лишь метаниловый желтый,
который получается из метан иловой кислоты и дифениламина кислот-
И. Красители
ный желтый метаииловый), обладает такими же свойствами. Повышение
интенсивности достигает 30%.
Устойчивый к кислоте азожелтый хорошо окрашивает шелк, утяже-
ленный фосфатом олова, Азожелтый незаменим для получения прочных
к воде желтых и коричневых окрасок.
а) Т роп ео л ин, или оранжевый IV
(кислотный желтый метаниловый К)
Дифенил-
амин
Диазпсуль-
фапилоиая
кислота
Оранжевый IV, или тропеолнн (кислотный желтый
метанилопый Kj
52 г (0,3 моля) 100%-иой сульфаниловой кислоты и 16г
соды растворяют в 300 мл воды и кипятят для удаления прнмеси
анилина с водяным паром. Затем отфильтровывают от примесей и под-
кисляют ,35 г концентрированной серной кислот ы. Внешним ох-
лаждением понижают температуру до 12° и диазотируют 22 г н и т р и т а
натрия, который растворяют в небольшом количестве воды. Спустя
1 час отфильтровывают плохо растворимую диазосульфаниловую кис-
лоту *, споласкивают маточным раствором иа иутч-фильтре и замеши-
вают кристаллы с 250 мл 90%-ного спирта**. Охлаждают до 12° и
смешивают с 38 г тонко измельченного дифениламина, прн этом
краситель не образуется. Затем закрывают сосуд крышкой из картона
или листового свинца и добавляют при хорошем перемешивании 12 г
концентрированной соляной кислоты. В течение часа поддержи-
вают температуру при 12°, затем 2 часа при 14° и 2 часа при 18°, в за-
ключение повышают ео н нагревают на водяной бане до 35°. Краситель,
который разбрызгался по стенкам стакана, нужно с.мыть небольшим ко-
личеством спирта. Во время реакции не должно происходить выделения
газа, Если возможно, перемешивают еще 6 час., и на другой день раз-
бавляют 1 л воды, нагретой до 50°. Нерастворимую кислоту тропеолина
отфильтровывают и очень тщательно промывают водой до тех пор, пока
цвет промывных вод не сделается чисто желтым. Затем снимают кислоту
с путч-фильтра, при этом наблюдается интересное явление: масса, ка-
жущаяся твердой после такой обработки, делается совершенно жидкой.
В технике это явление служит верным критерием для определения сте-
пени чистоты полученной кислоты: чем более жидкая кашица образуется
из твердого отпрессованного осадка, тем чище тропеолии. Блестящее
серовато-голубого цвета тесто замешивают с 200 мл воды, кипятят и до-
бавляют 30 г поташа. Прекрасные кристаллы калиевой соли краси-
теля выделяются в течение 24 час., их отфильтровывают и сушат
при 100°. Получают около 75 г концентрированного
* Влажная диазосульфаниловая кислота безопасна, однако в совсем сухом со-
стоянии она чрезвычайно взрывчата.
** Спирт не должен быть денатурирован пиридиновыми основаниями; бензол же,
напротив, безвреден
Тропеолин, или оранжевый /V
247
красителя (натриевая соль плохо растворима и неприглядна на вид,
поэтому ее неохотно применяют в качестве красителя).
б) Азожелтый
(индийский желтый, гели нити нит. п,)
HO3S-
NaNOg
NO
/ \^HNO3 рэзб.
X---/
Натриевая соль нитрозотронсо.иша
Перемешивают свежую очень хорошо отмытую кислоту тропеолина
с 300 лы воды и прибавляют при 5° 16 г 100 % -ного нитрита нат-
рия. При этом нужно очень медленно перемешивать, чтобы ие образо-
валось пены, которая затрудняет последующее нитрованне. Спустя два
часа, после того как выпадет нитроз амин светло-желтого цвета, добав-
ляют 40 а 60%-ной азотной кислоты, после чего опять переме-
шивают в течение 2 час., затем осторожно повышают температуру до 68°.
Масса начинает пениться, постепенно темнеет и в течение 25 мин, пол-
ностью переходит в раствор. Нагревают еще в течение 10 мни. до 71°,
разбавляют 500 мл воды, нейтрализуют 25 а соды и осаждают кра-
ситель 200 г поваренной соли. Он выделяется в течение дня в виде
ораижево-красного рыхлого осадка, который спустя 24 часа отфильтро-
вывают, отжимают н сушат при 60°, так как при более высокой темпе-
ратуре наступает разложение.
Выход составляет около 100 г.
Маточный раствор всегда сильно окрашен, так как нитрованне ни-
когда не протекает гладко и под действием азотной кислоты часть тро-
псолипа превращается в питроднфсниламии и диазосульфаииловую кис-
лоту. Образование диазосоедипения можно легко установить, если каплю
иитромассы вначале нанести на фильтровальную бумагу и иа светло-
желтый вытек нанести каплю щелочного раствора Аш-кнслогы; при этом
тотчас образуется красный азокраситель нз сульфаниловой кислоты и
Аш-кнслоты.
Полученный таким образом азожслтый нечувствителен к разбавлен-
ным минеральным кислотам, по не удовлетворяет всем требованиям,
предъявляемым к красителям для шелка. Путем энергичной обработки
еще большим количеством азотной кислоты получают более зеленоватые,
совершенно устойчивые к кислоте марки.
При получении марки Г (кислотного желтого) поступают несколько
иначе, чем при получений обыкновенного азожелтого. Вместо 40 г
60 % -ной азотной кислоты берут 90 г и начинают нитрование при
40°. В продолжение 2 час. повышают температуру до 70° и выдерживают
при 70° 2 часа. При этом происходит дальнейшее вступление нитрогрупп,
и полученный краситель более устойчив к кислоте; он требует дальнейшей
обработки, так как выпадает в виде липкого и нефнльтрующегося осадка.
Поэтому в иитромассу добавляют 100 г поваренной солн, раз-
бавляют до 1 л и перемешивают прн 70° до тех пор, пока выпавшие
хдопья не превратятся в светло-оранжевый порошок, что происходит в те-
чение 1—2 час. Только после этого разбавляют 500 л/л воды и посту-
пают так же, как в случае азожслтого.
Выход эзожелтого Г (кислотного желтого) составляет около 95 г.
Нитрованный тропеолин при растворении в горячей воде отщепляет азотистую
кислоту, вследствие него медная красильная аппаратура корродирует. Поэтому в тех
случаях, когда требуется не содержащий азотистой кислоты азожелтый, его получают
следующим способом. Свежеотфилътровапный азожелтый нагревают до 90° с четырех-
кратным по весу количеством воды, благодаря чему большая часть азотистой и азотной
кислот отщепляется. Спустя 3 часа добавляют еще 5% бисульфита натрия для удале-
ния последних следов азотной кислоты. Масса выделяет бурые газы и довольно сильно
пенится, поэтому для этой операции нужно применять большие чаны. При такой
обработке всегда теряется 15—20'% красителя (см. также Азофлавин ФФ, стр. 244).
Технические примечания. Диазотирование и сочетание сульфаниловой
кислоты проводят в больших эмалированных сосудах. В качестве мешалки часто при-
меняют толстые стеклянные палки, укрепленные в деревянных брусках, не соприка-
сающихся с жидкостью. Диазосульфаниловая кислота отсасывается на путч-фильтре
(см. рис. 26, стр. 137). Вместо эмалированной гильзы для термометра применяется
бамбуковая, которая служит очень долго. Хорошая кислота тропеолииа должна быть
совсем жидкой и легко передавливаться из сосуда для сочетания. После промывки и
сильного продувания на фильтр прессе влажная кислота тропеолииа, полученная нз
38 кг дифениламина, весит (довольно точно) 200 кг. Отклонения в ту или другую
сторону иа 10 кг указывают на наличие загрязнений. Спирт собирают' и после ней-
трализации содой перегоняют. При этой операции теряется около 15%.
Нитрование проводится в чанах из американской сосцы (пичпайн) емкостью
около 2500 л. Они снабжены хорошим вытяжным устройством (см. рис, 56, стр. 334)
и служат свыше года. Место, куда вдувают пар для нагревания, нужно защитить от
струи пара доской, укрепленной деревянным болтом.
Вторичные дне- и полназокрасители
Нафтиламнновый черный Д (Вейнберг, Касселла, 1888)
Кислота Фрейнда Моноазокраситель (практи-
ческой ценности не имеет)
0,1 моля кислоты Фрейнда (кислая натриевая соль
СюНвОе SBNNa, мол. вес 325) растворяют в 300 мл теплой воды, н
S = SOsH.
Вторичные дис~ и полиазокрасители
249*
5,0 г соды. Обычно кислота Фрейнда бывает 75—80%-ная, так как
вследствие легкой растворимости ее приходится выделять большим коли-
чеством поваренной соли. Дают раствору остыть до 30°, добавляют
столько льда, чтобы температура упала до 0°, и смешивают с 30 мл кон-
центрированной соляной кислоты. Обычно часть сульфокислоты
выпадает в осадок. Добавляют в течение 5—10 мин. прн хорошем пере-
мешивании раствор 7 г технического нитрита натрия; прн этом
температура не должна подниматься выше 6°,
Если в течение 5 мни. сохраняется слабая, но отчетливая реакция
па свободную минеральную кислоту (по конго-бумажке) и азо--
тистую кислоту (по йодокрахмальной бумажке), то диазотирование за-
канчивают *.
14,3 г чистого a-и афт и ламина растворяют в 12 мл концентри-
рованной соляной кислоты н 200 мл кипящей воды и при по-
стоянном перемешивании оставляют охлаждаться до 50°. При этом
обычно выделяется немного солянокислой соли. Этот раствор амииа тон-
кой струей выливают в хорошо перемешиваемый раствор соли диазония,.
предварительно добавляя мелконзмельченный л ед, чтобы температура
оставалась ниже 5°. После трехчасового перемешивания прибавляют в
реакционную смесь по каплям в продолжение 2 час. раствор 10 а-
соды и оставляют стоять иа ночь. На другой день добавляют 30 г кон-
центрированной соляной кислоты н, понизив температуру до 0°"
добавлением 300 г льда, диазотируют моноазокраситель раствором
6,8 г нитрита натрия в 50 мл воды. Продолжительность диазо-
тирования прн 0—5° 15 мин.
Проверяют на полноту диазотирования **. Затем добавляют опять,
как оцнсаио выше, 14,3 г нафтил амина и спустя 3 часа—10 г-
соды. Через дальнейшие 5 час. вводят еще в продолжение получаса рас-
твор 35 г соды в 100 мл холодной воды; спустя час, медленно нагре-
вают до 80° и добавляют поваренной солн в количестве 18% от
объема. Выпавшяй в виде хорошо фильтрующегося осадка иафтилами-
новый черный затем отжимают на прессе. Сухой краситель
весит около 70 а. 4%-ное крашение шерсти из слабо сернокислого
раствора дает глубокую черную окраску с незначительным коричневым
оттенком ***.
Технические примечания. Нафтиламиновый черный Д можно полу-
чать различными способами. Многие заводские химики предпочитают высаливать.-
диазосоединение моноазокрасителя и очищать его фильтрованием. Этот способ имеет
то преимущество, что таким путем удаляют цепрореагировавший а-нафтиламнн.
* Кислота Фрейнда подобно многим другим аминам с отрицательными группами
(NOs, SO3H, Cl) дает через некоторое время реакцию на йод и незаконченном
диазотировании, поэтому диазотирование следует считать законченным лишь тогда,
когда реакция наступает в течение 0,1 сек. Для начинающего легче проводить испы-
тание с реактивом «сульфон» (ср, стр. 220, сноска 2).
** Такие темноокрашенные диавосоединелия часто бывает невозможно испытать,
непосредственно ца азотистую кислоту с помощью йодокрахмальной бумажки. По-
этому наносят каплю исследуемого раствора или суспензии на небольшую щепотку
соли, которую помещают на тонкую фильтровальную бумагу. Соль осаждает окрашен-
ное вещество и если теперь приложить реактивную бумагу к обратной стороне
фильтра, то можно легко определить, имеется ли избыток минеральной и азотистой.,
кислот.
*** Gnelun, Taschenbuch.
2 oil
//. красители
вследствие чего устраняется возможность присутствия в конечном продукте нераство-
римого непрочного к трению жирорастворимого красителя.
—N4I2
При этом способе работы получается краситель значительно более чистого и
интенсивного оттенка. Особенно важным является отделение диазосоединеиия, если
из него хотят получить краситель для хлопка, так как любое загрязнение мешает
дальнейшей переработке. Так, например, из этого диазосоединеиия сочетанием с эфиром
ж-амино-н-крезола и последующим сочетанием с Й-кислотой получают очень прочный
к свету краситель для хлопка типа сирруса голубого.
Фрейнда „ крезндина11
Если вместо крезидина (эфира аминокрезола) применяют я-тол ундин, то по-
. лучают прямой прочно фиолетовый РР.
Нафтнламиновый черный принадлежит к важнейшим азокрасителям, так как
он обладает большой Красящей способностью, а также относительно хорошей свето-
прочностью, как и большинство красителей тина бибрихского алого. Он не
совсем прочен к кипячению, поэтому растворы его, особенно если они щелочные, не
следует напрасно кипятить. Наряду с этим красителем существует еще большое число
подобных продуктов, о которых приведены данные в учебниках, а также в таблицах
Шульца, Относительно метода получения следует заметить, что для растворения
а-нафтиламина нельзя применять соляную кислоту, содержащую в значительном ко-
личестве и серную, так как серпокислый нафтнламин очень трудно растворим. Вместо
соды для нейтрализации свободной минеральной кислоты при сочетании целесооб-
разно применять муравьинокислый натрий (он дешевле уксуснокислого).
Бензосветопрочио-голубой 2 ГЛ. Сириус светопрочно-голубой Г*
(прямой голубой светопрочный К)
SO3H
а) А н и л и п - 2,5 - д и с ул ь ф о к и с л о т а -> а-нафтил амии
А н и л и н-2,5-д и с у л ь ф о к и с л о т у ** (см. получение мононат-
риевой соли, стр, 97), взятую в количестве, 'соответствующем 50 мл
* Светопрочные 'Красители выпускаются в Германии под названием «сириус—-(
светопрочные красители».
** Необходимо определить концентрацию исходных материалов, аиилипдисульфо-
кислоты и нафтил аминасульфо кислоты титрованием нитритом, фелнл-И-кислоты титро-
запнем ил азобензолом (см, аналитическую часть, стр. 3-11—345),
Вторичные due- и полиазокрасители 251
1 н. раствора нитрита, растворяют в 150 ли горячей воды, в случае не-
обходимости фильтруют п при перемешивании добавляют 8 мл 30%-ной
с о л я и о й кислоты. Охлаждают до 0° и быстро вливают 17,5 мл
20% -иог'о раствора нитрита натрии. Дизотирование продол-
жается около 15 мин.
Вследствие плохой растворимости солянокислого а-пафтнламнна со-
четание в лабораторных условиях целесообразно проводить в водно-спир-
товой среде *. Поэтому раствор соли диазония разбавляют равным объе-
мом 96 %-ного спирта ив хорошо перемешиваемый раствор очень
медленно добавляют по каплям раствор 7,15 г свежсперегнанного а-н а ф-
т и л а м и п а в 100 мл спирта; при слишком быстром добавлении значи-
тельная часть азо-составляющей может быть увлечена выпадающим кра-
сителем и ле вступит в реакцию. Очень густую кашицу оставляют на ночь
и затем отфильтровывают полностью выделившийся краситель. Он пред-
ставляет собой коричнево-красную бронзирующую пасту, которую для
дальнейшей переработки не сушат, Для получения чистого конечного кра-
сителя продукты реакция освобождают от невступившего в реакцию
<х-пафтнламина. Для этой цели краситель растворяют в 300 мл теплой
воды с добавлением вычисленного количества соды, охлаждают до 0°.
оставляют на час при этой температуре н отфильтровывают от выделив-
шегося кристаллического а-нафтиламина. Фильтрат непосредственно при-
меняется для дальнейшего диазотирования.
б) А и илип- 2,5 .дисульфокислота ->а - нафтила мии -+
1-нафт и л амин- 7- сульфокис лота
Профильтрованный раствор мо ио аз о красителя смешивают с 22 мл
20%-ного раствора нитрита натрия (30%-иый избыток **) и
из капелыюй воронки прибавляют по каплям к хорошо размешиваемой
охлажденной до 10° (но не ниже) смеси 30 мл концентрированной соля-
ной кнслоты и 150 мл воды. Целесообразно сначала за один прием до-
бавить около десятой части раствора красителя и дожидаться, пока реак-
ция диазотирования полностью не закончится, что определяют по изме-
нению окраски от фиолетовой до коричневой. Если реакция начата таким
образом, то при дальнейшем добавлении раствора красителя она проте-
кает легко и быстро и заканчивается приблизительно через 2 часа. Проба
полностью и правильно продиазотированного моноазокрасителя после со-
четания с (3-нафтолом в содовом растворе дает глубокую и чистую фио-
летовую окраску. Если еще имеется испродиазотированный краситель, то
получается грязный сильно красноватый оттеиок. В этом случае Добав-
ляют к раствору соли диазопия еще 2—3 мл 20 %-ного раствора нитрита
натрия и в течение некоторого времени перемешивают. После окончания
реакции диазосоедииеиие высаливают поваренной солью (15% от объема
жидкости), добавляют несколько кусочков льда, фильтруют и осадок
трижды промывают 15%-иым раствором поваренной соли, чтобы пол-
ностью удалить избыточную азотистую кислоту. Коричиево^селтую пасту
соли диазония следует защитить от тепла и света, такж'йк иначе оиа
* При тщательной работе удается получить хорошие результаты и без примене-
ния спирта. Для этого растворяют «-нафтиламиц при кипячении в 100 мл воды и
28 мл 2 н. соляной кислоты, отфильтровывают в случае необходимости, дают
охладиться до 50° и при этой температуре, прежде чем начнет выкристаллизовы-
ваться солянокислая соль, довольно быстро вводят в диазораствор, в который добав-
лены кусочки льда; после этого медленно прибавляют по каплям раствор 7 г кри-
стадличнекого уксуснокислого ратрия в 20 мл воды,
** Наличие избытка способствует полноте диазотирования.
2,52 /7. Красители
быстро темнеет. Маточный раствор окрашен в темный цвет и не соче-
тается больше с ^-нафтолом.
Для получения дисазокрасителя 1-нафтилами н-7-с у л ь ф о к и с-
лоту (кислота Клеве) в количестве, соответствующем 50 мл 1 н. рас-
твора нитрита натрия, растворяют в 150 мл воды при добавлении не-
обходимого количества соды и снова подкисляют разбавленной соля-
ной кислотой до слабокнслой реакции на конго. В полученную
суспензию при размешивании медленно приливают диазосоедннение, раз-
мешанное в 300 мл ледяной воды. Одновременно в смесь добавляют нз
капельной воронки концентрированный раствор уксуснокислого,
и а т р и я в таком количестве, чтобы реакционная смесь все время остава-
лась слабокислой на конго. Тотчас происходит изменение цвета в интен-.
сивно фиолетовый. После 5—7-часового перемешивания полностью ней-i
трализуют концентрированным раствором уксуснокислого н а-.
тр и я и оставляют на ночь. Образующийся краситель ввиду его хорошей;
растворимости следует высаливать в кислой среде. Нагревают до 50°, до-)
бавляют поваренной соли (20% от объема жидкости), а затем 15% -иой4
соляной кислоты до сильно-кислой реакции на конго. Краситель выпа-)
дает в виде синего порошка; его отфильтровывают, для очистки еще раз ?
растворяют в 500 мл воды с добавлением соды и осаждают добавлением')
100 г поваренной соли и 15%-ной соляной кислоты. 1
в) Получение трисазокрасителя j
Переосаждеипый дисазокраситель замешивают с 75 мл 2 н. раствора^
соды и 150 мл воды, добавляют такое количество нитрита на-)
т р и я, которое соответствует 3,5 г чистого NaNOa, и нагревают до 80° да|
полного растворения. Затем охлаждают до 60° и прозрачный раствору
вводят при перемешивании в течение 0.5 часа в смесь 30 мл концентри*|
роваиной соляной кислоты и 200 мл воды, к которой постепенно)
добавляют 300 г льда так, чтобы температура все время оставалась
около 8°. Образуется совершенно прозрачный раствор диазосоединения||
Его в продолжение часа прибавляют по каплям в постоянно охлаждае-я
мый льдом до 0° раствор 15 г ф е н и л-И-к и с л о т ы (в переЯ
счете на 100%-иую) в 300 мл воды и 25 г соды (большое количествам
соды обусловливает образование при сочетании бикарбоната, вследствие!
чего не происходит вспенивания, вызываемого выделением свободной угЦ
лскислоты). По окончании прибавления перемешивают еще в течения
часа н оставляют на ночь. На другой день нагревают до кипения, добавя
ляют поваренную соль (15—20% от объема жидкости) и фильтруют прз|
80°. Наконец, промывают 5%-ным раствором поваренной соли и с ушата
блестящую бронзирующую пасту при 100°. Маточный раствор очеим
сильно окрашен и всегда содержит некоторое количество фенил-И-киС^
лоты, ее избыток необходим для достижения хорошего выхода. .’.та
В ы х о д 40 г. Я
Краситель окрашивает в голубой цвет и при правильно проведенном
получении окрашивает и из остаточных ванн в такой же, только болеЯ
слабый цвет. «Я
Технические примечания. Красители этого класса производят на обЫЧЦ
них для азокрасителей установках, одна из которых схематически изображена ТЦД
рис. 56, стр. 334. Вследствие неустойчивости диазосоедииения необходимо применятаИ
фильтр-пресс такой величины, чтобы можно было нею загрузку целиком поместить Rin
нем. В таком случае диазосоединение можно переработать тотчас после выгрузки МЯя
фильтр-пресса. Краситель целесообразнее производить в холодное время года и
ручать работу только опытным работникам. Рекомендуется, если это возможно. ПРЗм
Дис~ и полиазокрасители из диаминов
253
верять в лаборатории с малым количеством промежуточного вещества (например,
1/1000) каждую последующую стадию процесса до того, как сс проводить в произ-
водстве, Благодаря этому удастся избежать многих ошибок, Следует иметь также
в качестве образцов отдельные чистые промежуточные вещества и путем тщательного
сравнения с ними тотчас устанавливать, нормально ли протекает производственный
процесс.
Дне- и полиазокрасители из диаминов
Бисмарк коричневый Г и Р
(основной коричневый)
Бисмарк коричневый Г
NH3
I I
СН8 NH3
Бисмарк коричневый Р
Эти красители представляют собой смесь различных высокомолеку-
лярных соединений, в которой преобладают, однако, продукты, отвечаю-
щие вышеприведенным формулам. Рецепты, имеющиеся в литературе,
мало удовлетворительны, так как в них всегда рекомендуется смешивать
кислый раствор диамина с нитритом натрия. Гораздо лучше осторожно
подкислять нейтральный раствор диамина и нитрита или же вводить
нейтральную смесь в продолжение 12 мин. в необходимое количество
соляной кислоты. Необходимо применять нитрита несколько больше, чем
следует по уравнению реакции
3 моля диамина -|-2NaNO2 ф- 4НС1-> I моль красителя -j- (2НС1) -j-2NaCl -ф 4Н2О.
Избыток составляет для ^-феинлендиамина ~24%, для толуилеидй-
амина ~20%, т. е. на 3 моля диамина применяют 2 моля нитрита плюс
24 и соответственно 20% от этого количества. В обоих случаях, после
того как образуется краситель, полностью исчезает диамин, в чем можно
убедиться путем высаливания пробы.
Бисмарк коричневый Р (везувин Р н т. д.)
(основной коричневый)
36,6 г чистого толуилен диамина растворяют в 1 л воды
при 40° и после охлаждения раствор смешивают с 16,5 г 100%-ного н и т-
рита натрия. Затем путем добавления льда до^^дят объем до
1600 мл и при постоянном перемешивании в течение 20 мин. добавляют
иод уровень жидкости 60 4 концентрированной соляной к н с-
л оты, разбавленной 40 мл воды. Раствор тотчас становится темно-ко-
ричневым и выделяется довольно много азота. Конечная температура
~ 10°. Спустя 8 час. высаливают 300 г соли, через 3 часа отфильтровы-
вают и чрезвычайно легко растворимый краситель споласкивают на
фильтр его маточным раствором. После сушки при низкой температуре
(на производстве в вакууме) вес сухого красителя состав-
ляет около 40 а.
Окраски по хлопку, протравленному танинном и рвотным камнем,
прочны к стирке, дешевы и интенсивны, но непрочны к свету, Несмотря
па эго, обе коричневые марки широко применяются для хлопка, шелка,
бумаги и особенно кожи. Краситель в смеси с азожелтым дает прочные
к свету и трению коричневые окраски гга мебельной коже. Марка Г по-
лучается таким же образом, только в большинстве случаев краситель не
выпадает в виде хороших кристаллов и поэтому неудобен для фильтро-
вания. Этот недостаток можно частично устранить, применяя больший
избыток нитрита.
Хризофенин ГОО (хрнзофенин)
37 г (0,1 моля) 100%-ион диа миности л ьбеид и сульфо-
кис лоты растворяют в И г соды и 200 мл воды и после охлаж-
дения осаждают кислоту добавлением 50 мл (около 60 г) 30 %-ной со-
ляной кислоты. Добавлением льда понижают температуру до 50,,
и диазотируют в течение 2 час. 14 г 100%-ного нитрита натрия.
К концу диазотирования должен оставаться небольшой, но вполне замет- ’
ный избыток азотистой кислоты. Добавляют столько льда, чтобы темпе-
ратура понизилась до 0°, а затем 20 г фенола, расплавленного с не-
большим количеством воды. В хорошо перемешиваемую суспензию бис-.
дназосоедипения и фонола добавляют сразу 50 г соды, растворенной
в 200 г воды*, и такое количество льда, чтобы температура после сме- *
шения растворов равнялась 8°. Сначала все переходит в раствор и через
некоторое время выпадает часть яркого желтого. Спустя 2 часа нагре- "
вают до 70° и добавляют 100 г поваренной соли и столько с о- ’
ляной кислоты, чтобы краситель полностью выпал, но без из-
менсния окраски от желтой до синен. После охлаждения фильтруют н
отсасывают осадок при помощи насоса, по возможности, досуха. Он ве-
сит 180 г. У
Этилирование. Влажный осадок разбавляют водой до веса 200 г и «
перемешивают с 50 г твердой кальцинированной соды и 30 г 35 % -ного
* В противоположность многим предположениям фенол нс так легко сочетается
с циазосоставляющнмц. Он часто образует дназоэфир, который и принимают за азо-
соединение, Но можно получить краситель с гораздо лучшим выходом, если посту-
пать так, кдк указано пргг гюлученпп яркого желтого, а именно сначала перемешать
минеральнокислый раствор соли диазония с фенолом и затем добавить соды (но
не едкого натра).
раствора едкого натра. Тестообразную смесь помещают во вра-
щающийся автоклав или в автоклав с мешалкой н добавляют еще 250 г-
90%-ного спирта. В автоклав загружают 40 г хлористого
этила, как описано на стр, 134, и затем нагревают смесь при постоян-
ном перемешивании в течение 10 час. до 100ъ (максимальное давление
6 атм). После охлаждения открывают автоклав, разбавляют содержимое
двойным объемом 10%-ного раствора поваренной соли и от-
фильтровывают прекрасно выкристаллизовавшийся краситель. Если ди-
аминостидьбен дисульфокислота не содержит днамннодибензи л дисульфо-
кислоты, то полученный краситель приблизительно и а 20% крепче самого
концентрированного товарного продукта.
Выход сухого концентрированного красителя составляет около 70 а.
Технические примечания. Хрнаофенин является важнейшим прямым
желтым красителем. Вследствие светопрочностн окрасок на шерсти, шелке и хлопке,
а также дешевизны он вне конкуренции. Этилирование можно проводить ие только*
в спиртовой среде, но и в водном растворе в присутствии извести. В обоих случаях
существенно, чтобы образующееся алкильное производное выделялось тотчас. Какой
нз обоих способов более целесообразен, определяется ценой на спирт. Мы предпочитаем
спиртовый метод, так как он непосредственно дает готовый полностью растворимый
продукт высокой концентрации; кроме того, давление в этом случае не поднимается,
выше 6 атм, в то время как при этилировании в присутствии извести развивается
давление до 25 и более атмосфер.
Хризофенпн дает с минеральными кислотами характерную синюю окраску. Заме-
чательным с научной точки зрения является то, что он является исключительно ин-
тенсивным красителем, хотя и не содержит ауксохромных г^унп, как этого
требует теория цветности Витта, Конец алкилирования яркого желтого опре-
деляют следующим образом; небольшую пробу растворяют в воде и смешивают с не-
сколькими каплями уксусной кислоты, Каплю слабо уксуснокислого раствора наносят-
на фильтровальную бумагу л смачивают желтое пятно 10%-ным раствором соды.
Если алкилирование закончено, то нс должно происходить изменения окраски в крас-
но-желтую или почти красную, В производстве отбирают время от времени пробы,
при помощи крана для отбора проб. Не следует никогда перекачивать в котел все
количество хлористого этила в один прием, а подавать его порциями по 10—15 кг.
Нагревание производится при помощи паровой рубашки; применяют горизонтальные -
цилиндрические котлы с горизонтальной мешалкой, сальник которой хорошо охлаж-
дается, так как в противном случае спирт тотчас растворяет смазку. Расход хлори-
стого этила составляет 180% от теории.
Бензидиновые красители
Бензидин можно комбинировать со всеми фенолами и аминами, ко-
торые применяют для получения азокрасителей. При этом оказывается,,
что только одна из двух диазогрупп б«с-диазобснзидина реагирует энер-
гично, другая же, напротив, реагирует довольно вяло. Это дает возмож-
ность получать не только бензидиновые красители, содержащие две оди-
наковые составляющие (фенолы или амины), но во многих случаях
также так называемые «смешанные» бензидиновые красители. Послед-
ние образуются только в том случае, если первая азосоставляющая кра-
сителя реагирует не слишком легко, так как в противном случае наряду
с промежуточным соединением у?
IЮ — X - X’ / /—\ N—N—X
тотчас образуется дисазокраснтель. Одиако перечисление лишь важней-
ших вариантов этих промежуточных соединений увело бы слишком
далеко, поэтому ограничимся немногими примерами. Особенно важным
является промежуточное соединение, которое образуется при однократном
сочетании салициловой кислоты с бензидином. Это соединение получается
очень однородным, так как вторая молекула салициловой кислоты мо-
жет быть введена лишь с большим трудом после сочетания в содовом рас-
творе при избытке едкого натра. Также легко удается просочстать одну
дпазогруппу бис-диазобензидина с моноазокрасителем из п-нитроанн-
.лина и Аш-кислоты, а также с Аш-кислотой в минеральнокислом рас-
творе. Оба случая рассматриваются далее.
Промежуточное соединение бензцднна с салициловой кислотой
(о-толиднн о-крезотниовая кислота *)
18,4 г (0,1 моля) технического бензидина бис-ди азотируют, как
описано на стр. 235. Прозрачный бис-диазораствор. быстро выливают ;
в раствор 15 г чистой салициловой кислоты и 40 г кальцици- ii
ровапной соды в 300 мл воды при 5°. Выделяется оранжево-желтое
промежуточное соединение; реакция считается законченной, когда бес-
цветный вытек от капли реакционной смеси на фильтровальной бумаге
не образует синего ореола со щелочным раствором Аш-кислоты. Мед-
ленно перемешивают до полного исчезновения реакции на бис-диазобен-
зидин, на что обычно требуется при 12° 1 час.
Диазосоединсние
/СООН
НО—V- N--/ /—<С_ /—0Н
сочетается со многими аминами и фенолами, образуя некоторые важные
красители. Если, напркмер, к промежуточному соединению прибавить со-
дово-щелочной раствор у-кислоты, то образуется диамииовый коричне-
вый М (прямой коричневый КХ). Интересно, что расходуется только 4
85% теоретического количества у-кислоты. С другой стороны, если про^ 5
межуточное соединение подкислить уксусной кислотой и затем смешать х
с раствором у-кислоты, который имеет еще отчетливо-кислую реакцию
на лакмус, то при 12—28° образуется в течение 12 час. важный краси-
тель — диаминовый прочно-красный Ф (прямой красный X); он вслед- ,
ствие наличия в молекуле остатка салициловой кислоты дает с хромовой
протравой прочные к валке окраски па ше.рсти.
Диаминовый прочно-красный Ф
(прямой красный X)
Диаминовын коричневый М
(прямой коричневый КХ)
Прн щелочном сочетании дназогруппа вступает в орто-положенне гЦ
оксигруппе, при уксуснокислом сочетании — в орто-положение к NH3-
группе. ;
* Соединение о-толидина с о-крезотиновой кислотой производят в больших коли-!
чествах. Салициловая кислота малопригодна для сочетания с диазотированным толи-
дином, так как красители из о-толиднна и салициловой кислоты очень плохо рас- '
творяютея.
Дианил коричневый ЗГИ
257
Днанил коричневый ЗГН (прямой
коричневый ЖХ)
применяемым прямым аао-
Этот краситель принадлежит к наиболее
красителям, так как он обладает исключительной красящей способностью.
Ои непрочен к свету н кислотам. Сначала получают моноазокраснтель,
сульфохризондин Г
NHa n2-/—s)^SOsH
NH3
Его готовят следующим образом: 17,3 а (0,1 моля) сульфанило-
вой кислоты диазотируют, как указано на стр. 235, и суспензию
диазосоединеиия, которая должна иметь слабую минеральнокислую реак-
цию, прибавляют по каплям в раствор 10,8 г чистейшего л-феннлен-
д нам нна. Целесообразно диамнн подкислять 5 мл концентрированной
соляной кнслоты и устанавливать раствор на 10%-ное содержа-
ние диамина. За ходом сочетания следят по вытеку на фильтровальной
бумаге, смоченной щелочным раствором Аш-кислоты, и прибавляют ди-
азосоедннение до тех пор, пока вытек начнет давать совсем слабое по-
краснение. Диамин полностью исчезает, однако образование красителя
еще не произошло. Спустя 2 часа при 5° осторожно прибавляют по кап-
лям при постоянном перемешивании 10%-ный раствор соды, пока
полностью не будет нейтрализована минеральная кислота. Для этого тре-
буется около 6 г соды. Через дальнейшие 3 часа прибавляю? при 5° еще
5,5 г соды в продолжение часа и оставляют стоять иа ночь. Утром до-
бавляют 10 г соды, растворенной в небольшом количестве воды, и
опять оставляют стоять на 3 часа. Безусловно необходимо вести сочета-
ние сульфаниловой кислоты и диамина с этнкЭ предосторожностями, так
как в противном случае образуется неконцентрнрованный конечный кра-
ситель. Натриевая соль сульфохрнзоидина большей частью выделяется
в виде светлого красно-коричневого прекрасно кристаллизующегося
осадка. Объем всего раствора составляет около 500 мл, и его не следует
увеличивать. Эту суспензию смешивают при 10° с промежуточным
соединением бензидниа с салициловой кислотой (см.
стр. 256) и медленно перемешивают в течение 5 час. Затем осторожно на-
гревают до 30° и оставляют стоять 12 час. После этого кипятят и высали-
вают краситель 200 г поваренной солн. Он выпадает чистого ко-
ричнево-красного цвета и очень хорошо фильтруется; маточный раствор
содержит немного сульфохрпзондипа. Выход сухого красителя состав-
ляет около 95 г. Окрашивает ровно хлопок только тогда, когда он сме-
шан с кальцинированной содой (10% от веса красителя). Избыток или
недостаток соды действует неблагоприятно, точно так же, как в случае
прямого глубоко черного ЕВ пли прямого черного К (см. стр. 261).
Технические примечания. Если вместо лгфенилендиампва применять
1,2,4-толувлендиамин, то получают совершенно аналогичный краситель, который, од-
нако, несколько более прочен к кислоте. Так как в цен пара-положение к аминогруппе-
замешено, то из этого следует, что приведенная выше формула красителя тгз лг-фени-
лсидиамппа правильна, т. е. вторая азогруппа находится между обеими аминогруп-
пами, а не в гсара-положеннп к одной из них * *.
Напротив, если диамин сочетать сначала с промежуточным соединением из бен-
зидина и салициловой кислоты, а затем с диазотированной сульфаниловой кислотой, то-
получают изомерный краситель, который имеет следующую формулу;
СООН
Интересно, что этот краситель не представляет никакой практической ценности..
Очень важно применять совершенно чистый диамин, так как следы о- илн «-фе-
нилендиамина разрушают большую часть дНазосульфапиловой кислоты, а также про-
межуточное соединение из бензидина и салициловой кислоты. Раствор вспенивается,
и краситель делается тупым и слабым. При применении чистейших материалов вместо-
неочищенного технического раствора диамина выход повышается приблизительно*
на 40%,
Оба красителя как из фснплсп-, так и из толуилендвамина применяются в боль-
ших количествах для изготовления смесовых красителей.
Днамнновый зеленый Б (фирмы Касселла) — (прямой зеленый)
ОН NH,
14,5 г ** чистого п-нитроанилина диазотируют (стр, 234) и в
течение я/4 часа к охлажденному льдом прозрачному раствору соли ди-
азоиия прибавляют по каплям 34,1 г Аш-ки слоты (100%-ной), кото-
рая растворена па холоду в 100 мл воды и 5,5 г сод ы. Чтобы не о бра-
зовывалось комков, необходимо хорошее механическое перемешивание.
Диазотированный нитроаиили-н сочетается в течение 4—5 час, с Аш-кисло-
* См., напротив, Schmidt, HagcnbScker, Ber„ 54, 2201.
*,f' Вместо 13,8 а, требующихся по теории; этот небольшой избыток необходим,
так как при длительном сочетании лиазосоедииепис частично разлагается.
тол, при этом освобождается эквивалентное количество соляной кислоты.
Смесь оставляют стоять по меньшей мере па 12 час, и на другой день,
предварительно нагрев до 50°, смешивают с 20 г 30%-го раствора МаОН
и 40 с соды. Моноазокраситель
ОН NH.,
HO.S
образует раствор красивого синего цвета;
200 г пов аренпо й с о л и. Спустя
в легко фильтрующейся форме блестящая
сасывают на нутч-фильтрс и отжимают. Маточный раствор окрашен в
интенсивно синий цвет, ио даже при насыщении поваренной солью не
дает больше пригодного красителя и поэтому может быть удален.
Если этот краситель из лг-нитроаиг'лина. не выделяя, просочетать при 5° в содово-
щелочном растворе с вычисленным количеством диазотированного анилина, то полу-
чают важный краситель — нафтоловый сине-черный Б (кислотный сине-черный).
ОН NH.
его высаливают добавлением
несколько часов выделяется
натриевая соль, которую от-
— Na—?
HC^S-i
-MX
SO:iH
В этом случае моноазокраситель ие выделяют. Избыток диазотированного ани-
лина вреден. Высаливанием поваренной солью (15%) при 90° полуЧают нафтоловый
сине-черпый Б (кислотный сине-черный) -краситель с красивым бронзовым блеском,
Попутно следует упомянуть, что восстановлением нафтолового енне-че.рпого Б серни-
стым натрием при 25:' получают ценный темно-зеленый азокраситель — а з о-т е м но-
ле л с н ы й. *
ОН NHa
__ II"
\ / — \ /- ХН2
~ HO,S-^/x/(-SO,H “
Спустя 3 часа краситель осаждают при 50л добавлением поваренной соли (15%)
и небольшого количества серной кислоты. Он плохо растворим в бикарбонате; маточ-
ный раствор сильно окрашен.
Следует заметить, что красители из /г-нитроадилинз могут быть восстановлены
вычисленным количеством сернистого натрия до азокрасителей из п-фепилепдиамина
количественно. Таким образом получают новые амнноазокрасители, которые можно
диазотировать еще раз п сочетать с другими азосоставляющрми. Подобные амино-
азокрасители получают также путем омыления красителей из ацетил-я-фенилендиа-
мина *.
IX-N3 / >-NO^-7H3O+6Na2S
—i./fX-.-No - / 'N—\H3-j-('iNa0H-j-3Na3S3O3
Натриевую соль растворяют в 500 мл воды и 40 г соды при 80’
н при постоянном перемешивании охлаждают до 20°. Затем добавляют
столько льда, чтобы температура упала до 4°. Часть красителя вы-
падает в тонко измельченном состоянии. К этой суспензии добавляют по
каплям раствор б«с-диазобензидина, который получают, как указано на
стр. 235; раствор бензидина добавляют до тех пор, пока вытек пробы на
фильтровальной 'бумаге будет давать со щелочным раствором Аш-ки-
слоты слабую, по отчетливую синюю окраску. Эта окраска сначала
быстро исчезает, так что нужно добавлять еще бис-д и азораствор.
* Применяют также формил- и оксалил-/г-фепнлендиамины (ср. стр. 88),
1 7'-
Расходуют всего около 18,6 г бепзндииа; образование промежуточ-
ного продукта продолжается 0,5 часа.
Промежуточное соединение имеет следующую формулу:
ОН NHa
II _ '
/Му-—-/'У')— N3—C_/_NOa
/ HOgS-yy^SOgH
Бензидин^
ОН
К этому соединению добавляют 12 г фенола, расплавленного
с небольшим количеством воды, и оставляют на 3 часа прн 10°, затем
медленно повышают температуру до 30° н оставляют па ночь. После
этого подогревают до 60° и добавляют столько 30%-ного раствора ед-
кого натра (около 40 г), чтобы все растворилось*. Добавляют
150 г поваренной соли и приливают по каплям разбавленную
серную кислоту до тех пор, пока ,не выпадет весь краситель (проба
на вытек на фильтровальной бумаге). Отфильтровывают, отжимают и су-
шат при 90°. Выход концентрированного красителя ~ 110 г. Можно также
выделить краситель без применения едкого натра, нагревая до' 90° н
высаливая нз горячего раствора 300 г поваренной солн, но в этом
случае краситель не получается такям концентрированным.
Технические примечания. Диаминовый зеленый Б (прямой зеленый),
несмотря на свою умеренную светопрочность, является одним из наиболее применяе-
мых зеленых красителей для хлопка. Он служит для крашения изоляционной обмотки
для телефонных и других медных проводов, а также для изготовления смесовых
красителей. Если вместо фенола применить салициловую кислоту, то ее нужно про-
сочетать в первую очередь, так как в качестве второй азосоставляющей она плохо
сочетается с бензидином. При этом образуется диаминовый зеленый Г, который,
однако, применяется значительно реже, так как образование красителя протекает не
так гладко и вследствие этого цена его значительно выше. В технике нагревание
всегдапзедут путем пропускания пара и избегают отжимать такие красители, так как
они проходят через салфетки.
Прямой глубоко-черный ЕВ (Байера)
(прямой черный 3)
Nib ОН
Получение днаминового зеленого Б (прямой зеленый) является при-
мером сочетания Аш-кнслоты с n-ннтроанилином в минералънокислой
среде; эти соединения очень легко реагируют с образованием моноазо-
красителя, причем диазогруппа вступает в орто-положение к амино-
группе. С бензидином сочетание идет значительно менее энергично, и
необходимо постоянно нейтрализовать выделяющуюся при реакции1 ми-
* Нитроазокрасители нельзя обрабатывать щелочью в присутствии древесины
или восстановителей.
неральную кислоту. Вопреки данным патентной литературы эту реакцию
нельзя проводить в уксуснокислом растворе, так как в присутствии
уксуснокислого натрия Аш-кпслота тотчас сочетается в орто-положение
к оксигруппе. Этот факт дал повод ко многим патентным спорам, ко-
торые, однако, были окончены в пользу владельца патента, основанного
на сочетании в минеральнокислой среде.
а) Промежуточное соединение
N;
NH3OH
I I
Диазотируют 19,2 г 100%-ного бензидила (стр. 235), темпера-
туру доводят до 10—12°. В этот бис-дназораствор прибавляют по каплям
в течение часа профильтрованный раствор 34,1 г Аш-кислоты
и 5,5 г 'Соды в 300 мл воды. Раствор Аш-кнслоты должен давать отчетливо
кислую реакцию на лакмус. Перемешивают при 12° в течение 3 час. и по
каплям очень осторожно добавляют в продолжение 2 час. раствор 5,5 г
соды в 60 мл воды; при этом раствор должен все время сохранять
минеральнокнелую реакцию. Спустя 3 часа при 12° добавляют по кап-
лям, если необходимо, разбавленный раствор соды до слабой, но
отчетливой реакции на конго-бумажку н оставляют на ночь в холодном
месте. Спустя 12 час. пробы па вы^ек не дают реакцию на бне-дназобен-
зидин (с раствором Аш-кнслоты) и реакцию на Аш-кислоту (с диазоти-
рованным нитроа нил ином). Промежуточное соединение выделяется
в виде порошкообразного осадка.
б) Пр омежуточное соединение
Диазотируют 8,8 г чистого анилина, как описано на стр. 234, и
раствор соли диазония добавляют прн 5° к первому промежуточному
соединению. Если необходимо, добавляют немного льда. К хорошо пере-
мешиваемой смеси быстро прибавляют раствор 26 г соды в 120 г хо-
лодной воды. Тотчас все переходит в раствор, а затем полностью вы-
падает новое промежуточное соединение. Нельзя применять слишком
много соды, так как иначе часть промежуточного соединения просоче-
тается с Аш-кислотой в орто-поло жен не к оксигруппе. 'Сочетание можно
легко контролировать пробой на вытек на фильтровальной бумаге.
Реакция на диазобензол исчезает иногда не полностью, поэтому к полу-
чению красителя приступают спустя 15 мин.
Ко второму промежуточному соединению добавляют 11 г чистей-
шего, растворенного в небольшом количестве воды ж-ф енилендиа-
м и н а, который очень быстро сочетается с дназосоединеннем. При этом
всегда часть образующегося красителя переходит в раствор. Выдержи-
вают в течение часа прн 14°, а затем осторожно нагревают до 50° и
добавляют еще 10 г соды. После этого добавляют 120г поваренной
соли и прн постоянном перемешивании подкисляют приблизительно
20 мл концентрированной соляной кислоты, нежа краситель пол-
62
II. Красители
постью не выделится. При 50° он не растворим в 10 %-ном растворе по-
варенном соли и бикарбонате, однако только в том случае, если он пред-
варительно не был прокипячен. Продукт реакции отлично фильтруется н
после отжатия сушится при 100°, Выход концентрированного красителя
составляет около 100 г. Для того чтобы ои хорошо выбирался хлоп-
чатобумажным волокном, его нужно смешать с содой (6% от веса).
Если вместо феиилендиамнпа применить лгт о л у и -
л сн ди амин, получают глубоко1 черный Фау (прямой черный К),
который обладает несколько1 более красным оттенком, В этом случае
также необходимо после- нагревания добавить немного соды, чтобы полу-
чился хорошо фильтрующийся продукт.
Технические примечания. Описанный черный краситель для хлопка
является наиболее важным прямым черным красителем. Он служит прежде всего для
крашения таких материалов, как хлопок, полушерсть, кожа и др. Его получают на
больших установках, применяя только чистейшие исходные вещества. Исполь-
зуя перекристаллизованный из воды л-фепилендиамин, достигают наиболее
высокого выхода красителя, который даже при крашении из остаточных дани дает
чистый черный цвет.
Конго красный
Диазотируют 18,4 г технического бензндииа, как описано иа
стр. 235, и смешивают этот раствор с раствором 50 г 100%-ного ,н аф т и-
оиатан 50 г уксуснокислого натрия в 200 мл вод ы. В те-
чение часа температуру поддерживают при 5°, затем медленно повышают
до 20° н оставляют стоять при этой температуре в течение 5 час,; затем
температуру поднимают до 30°, перемешивают в течение 24 час, и иа тре-
тий день нагревают до 55°, Через 2,5 суток после начала сочетания смесь
кипятят и добавляют 40 г окиси магния. Выпадает плохо раствори-
мая магниевая соль конго красного. Ее фильтруют и тщательным промы-
ванием полностью удаляют загрязнения, Промытую магниевую соль за-
мешивают с 500 мл кипящей воды и добавляют 15 г соды, при этом
осаждается карбонат .магния, а краситель переходит в раствор в виде
натриевой соли. Горячий раствор фильтруют, углекислый магний
промывают водой н из фильтрата осаждают конго красный добавлением
поваренной соли (15% от объема). Выпавший краситель имеет светло-
красный цвет и после сушки весит 70 г.
HO3S
Техн и ч е с к и с прим с и а в и я. Конго красный является первым бензиди-
новым красителем; несмотря па его большую чувствительность к кислотам, он приме-
i\Ul.’/U/rtOTU tfrt I V/nfJ£U WT<J< U
пястся в больших количествах, так как является самым красивым прямым краси-
телем.
В технике проводят сочетание часто несколько иначе, чем в лаборатории. Соче-
тание можно весьма ускорить, если нафтионат смешивать с бпс-диазораствором бен-
зидина при 85е при очень хорошем перемешивании. В этом случае работают с неболь-
шими количествами и проводят по 8 — 10 операций в день.
Избыток нафтионата часто регенерируют.
Наряду с конго красным большую роль играет бензо пурпурин 4Б из
о-т о л и д и н а и нафтионовой кислоты. В этом случае нельзя проводить сочетание
в горячем растворе, так как бис-диазососдинспие о-толцдина очень легко разлагается.
Этот краситель несколько менее чувствителен к кислотам, чем конго красный, и так
же, как и последний, находит широкое применение, особенно на Востоке. Установлено,
что в странах района Средиземного моря, с мало развитой промышленностью, где
атмосфера не содержат сернистой и серной кислот, красители такого типа оказы-
ваются более прочными, чем у пас.
Кислотный антраценовый красный Г и хромоцитроннн
(протравной чисто-желтый)
ОН
ОН
Кислотный антраценовый
красный Г
Беизидинднсульфокислоту вследствие плохой растворимости нельзя
диазотировать обыч’ным способом. Ее. растворяют в соде или едком
натре, полученную натриевую соль смешивают с нитритом н выливают
в кислоту.
Кислотный, антраценовый красный Г. 34,4 г б е н з и д н н-2,2'-д н-
сульфокислоты (100%-ной) растворяют в 300 мл воды и 11 г
соды прн нагревании и прн 20° смешивают с 14 г нитрита натрия
(100%-ного). Полученный раствор вливают в смесь 60 мл 30%-ной со-
ляной кнслоты, 200 мл воды и 100г льда. При этом темпера-
тура может подняться до 25°. Диазотирование заканчивается в течение
нескольких минут. бцоДназоооедниенне смешивают с 30 а Р-н а ф то л а,
который растворяют точно в таких же количествах воды, едкого натра,
соды и льда, как указано для кислотного оранжевого А. Аналогично ведут
и дальнейшую обработку. Нужно иметь, однако, в виду, что бке-дназо-
б енз иди ид исуль'ф окис лота может выделиться в виде плохо растворимого
грубого кристаллического осадка, который ие реагирует со щелочным
раствором р-нафтола. В этом случае необходимо обработать охлажден-
ный льдом бис- ди азораствор таким количеством щелочи, пока не обра-
зуется растворимая натриевая соль диазот ат а, которая мгновенно соче-
тается с 0-нафтолом. Кислотный антраценовый красный Г дает без
протравы устойчивую к валке окраску на шерсти.
Хромоцитронин (протравной чисто-желтый). Смешивают бис-диазо-
раствор бензидннднсульфокислотьт с 32 г чистой салициловой
кислоты, растворенной прн 5° в 80 а соды и 200 лы воды. Через
12 час. образовавшийся краситель при охлаждении высаливают пова-
ренной солью (20%), сильно отжимают и сушат прн 60°.
В этом случае нет необходимости растворять выделившееся бие-диазосоедине-
ние, так как хромоцитронин сам тотчас переходит в раствор. Напротив, готовый краси-
тель следует, особенно в производстве, перед высаливанием фильтровать, чтобы ом
при ситцепечатании, в котором он находит большое применение, не забивал печатных
валов. Из деревянных чанов всегда отщепляется довольно много древесины, которая
мешает в набивке тканей. Хромоцитронин — красивый желтый краситель, хромовый
лак которого отличается прочностью к свету, воде и хлору. Благодаря своей большой
растворимости он глубоко проникает в хлопчатобумажную ткань, так что тонка®
ткань отпечатывается с обеих сторон.
Особые методы
Эриохромфлавин А*
С N=N—Z ОН ~^Н О—/ N=N—/ \-О К
ноос соон соон соон
Т. пл, 288°. с разложением Т. пл. 277°, с разложением
Раствор 17г 100% -ной х л о р а м и >н о б е н з о й и о й кислот ы, ко-
торая может быть получена, как описано на стр. 151, подкисляют 30 мл
30%-ной соляной кислоты н охлаждают льдом приблизительно до
—2°. К раствору пшрбавляют в течение нескольких минут 7 г нитрита
натрия (35 мл 20%-ново раствора). При правильном получении ис-
ходного вещества раствор соли диазония остается бесцветным и про-
зрачным. Его смешивают с раствором 15 г салициловой кислоты
(10% избытка) в 15 ли 30%-ного едкого натра, 40 г соды и
300 мл воды. Спустя 2 часа сочетание заканчивается, после чего на-
гревают до 80° и добавляют поваренной соли (15% по объему),
и столько соляной кислоты, чтобы выпал краситель. Краситель
отфильтровывают н отжимают па винтовом прессе.
Полученный таким образом краситель не имеет практической цен-
ности и непосредственно перерабатывается дальше. Измельченный оса-
док помещают во вращающийся автоклав (см. стр. 318), добавляют
50 мл 30% -ного ед кого натра н 200 мл вод ы, а также 1 г с е р и о-
кнелой меди. Затем закрытый автоклав нагревают при постоянном
вращении в течение 10 час. прн 140° (давление 4 ат).
В течение этого времени хлор заменяется иа оксигруппу. Чистый
красно-коричневый раствор отфильтровывают от окиси меди и при 80°
осаждают азосалициловую кислоту добавлением 15%-ной соляной
кислоты, Она в воде нерастворима. Выход составляет около 90—
95% от. теории, т. е. около 28 г азосалициловой кислоты. Из 156 г чистой ,
хлорбензолы ой кислоты легко можно получить 230 г чистой азосалици-
ловой кислоты; в случае загрязненных исходных веществ н неправиль-
ном проведении реакции восстановления нитросоедипения выход может
упасть больше чем вдвое. Ввиду нерастворимости красителя в воде для
крашения шерсти 1 г его растворяют в аммиаке н затем подкисляют
уксусной кислотой ДО' отчетливо кислой реакции. При последующем хро-
мировании бихроматом натрия получают более полные окраски и совер-
шенно устойчивые к кипячению.
Кегп А., Диссертация Ziirich, 1921; герм. пат. 278613 (Frdl,, XII, 323).
солнечный желтый Г из п-нитротолуола
265
Технические примечания. Азосалициловая кислота была предложена
вследствие затруднений с использованием о-хлорбензойной кислоты, которая
получается в качестве отхода при изготовлении о-сульфобензойного альде-
гида. Фирма «Гейги» имела на складе около 30 000 кг о-хлорбензойной кислоты,
прежде чем С. Метлеру удалось разрешить проблему ее использования. Азосалицило-
вая кислота является наиболее прочным желтым хромирующимся азокрасителем, про-
изводным салициловой кислоты. Она может конкурировать с описанным ранее хромо-
цитронином, который на шерсти даст неровные окраски. Краситель из о-крезоти-
новой кислоты дает на шерсти па 25% более интенсивные окраски, чем краситель
из салициловой кислоты.
Замещение атома хлора на оксигруппу протекает при каталитическом действии
окиси меди. Вместо нее можно с тем же успехом применять осажденную («цемент-
ную») медь или тонко измельченную «листовую медь», так называемую «натуркупфер»..
Солнечный желтый Г нз л-нитротолуола
100 г л-н н т р от о л у о л а вносят в 280 а 25 %-ного олеума н
нагревают до 100° прн постоянном перемешивании прн помощи железной
или стеклянной мешалки. Сульфирование должно быть закончено в те-
чение 20 мнн., если необходимо, добавляют еще немного дымящей сер-
ной кислоты. Проба, вылитая в воду, должна дать вполне прозрачный
раствор без запаха «-нитротолуола.
Как только сульфирование закониено, дают охладиться до 40° н
выливают массу в 300 мл воды и 300 г льда. Затем сульфокислоту"
высаливают в виде ее натриевой соли добавлением 250 г поваренной-
с ол и, фильтруют после охлаждения и хорошо отжимают. Рекомен-
дуется во время выделения беспрерывно механически перемешивать и:
осадок залить на нутч-фильтре 15%-ным раствором поваренной соли,,
чтобы удалить большую часть серной кислоты. Отжатый осадок весит
около 220 г.
Конденсация. Полученный осадок растворяют приблизительно
в 400 мл воды н в таком количестве соды, чтобы реакция раствора была
нейтральной, после чего объем доводят до 1000 мл и температуру до
65°. Затем в течение 15 мин. при постоянном перемешивании нитрото-
луол сульфокислоту смешивают с 200 г 35%-ного раствора едкого-
натра и осторожно повышают температуру до 69°. Не следует при-
бавлять раствор щелочи слишком быстро, так как образуются крупные-
комки нерастворимой натриевой соли, которые не вступают в реакцию.
Затем непрерывно перемешивают сначала 1 час при 70°, затем 1 час
при 74° и, наконец, 1 час прн 76°; прн этом следует избегать местного
повышения температуры выше указанной. Этого лучше всего удастся
достичь нагреванием на водяной бане.
За это время конденсация заканчивается и суспензию красителя
нейтрализуют '>-220 а концентрированной соляной кислоты таким:
образом, чтобы цвет еще не переходил в коричневато-черный. После
полного охлаждения фильтруют и сушат при 80°.
Выход чистого красителя составляет ~ 170 г.
Примечания. Конденсация сульфопроизводного «-нитротолуола впервые слу-
чайно наблюдалась Вальтером; в ходе исследований было затем найдено, что в зави-
симости от условий реакции образуются различные красители с более желтым или
более красным оттенком. Если проводить конденсацию несколько более энергично,
например, с большим количеством щелочи и при более высокой температуре, то от-
тенки становятся несколько более зеленоватыми. Путем осторожного окисления с по-
мощью хлорноватистокислого натрия можно получить красители с еще более зеленым
оттенком (солнечный желтый ЗГ, полифенил желтый ЗГ).
Восстановлением этих красителей получают значительно более красные оттенки,
которые могут доходить до красновато-оранжевого. Если, например, указанный выше
краситель нейтрализовать только частично и после конденсации непосредственно вое-
266 ,
II. Красители
становить 40 г кристаллического сернистого натрия (например, 1 час
прн 75s) п затем подкислить, как указано выше, то получают микадо оран-
жевый Р, довольно светопрочный краситель для хлопка.
Красители такого рода в противоположность хрнзофенпну обладают общим свой-
ством, а именно они совсем не окрашивают из нейтральной ванны
шерсть и шелк, а на растительных волокнах обладают высокой прочностью
к свету и даже к стирке. Они применяются также для окраски бумаги, Строение этих
красителей с достоверностью неизвестно; при энергичном восстановлении, например
цинковой пылью и щелочью или аммиачным раствором сернистого аммония, все они
дают смесь днаминостильбен- и днаминодцбензнлдисульфокнслот. В производстве эти
красители получают в небольших железобетонных чанах с железобетонными мешал-
ками, которые служат в течение многих лет. Медь также является очень подходящим
материалом, но дорога.
Д. ДИ- И ТРИФЕНИЛМЕТАНОВЫЕ
КРАСИТЕЛИ
Аурамнн 00 (аурамин) (по Т. Зандмейеру *) 1
Смесь 127 г (0,5 моля) 4,4'-т етр а м е ти л д и а м и в о д и ф е н и л- .4
метана (см. стр, 125), 32 ? серы, 70 г хлористого аммония и J
1000 г чистой поваренной соли нагревают до 110° в плавильном 1
котле с мешалкой (см. рис. 36 а и 36 б). Необходимо; чтобы исходные |
вещества были очень тонко измельчены и не содержали влаги. В течё- 1
ние 2 час. температур^повышают до 130° **, причем пропускают в anna- j
рат из баллона быстрый ток сухого аммиака. Аммиак уносит последние 1
следы влаги; примерно при 140° начинается интенсивное выделение серо- 1
водорода, которое продолжается в зависимости от скорости пропускания 'J
аммиака в течение 10—15 час. За 5 час. при постоянном перемешивании
температуру повышают до 175°, выделяющийся сероводород поглощают 1
концентрированным раствором едкого натра. Полезно также несколько
прикрыть вентиль для отходящего газа и этим создавать небольшое из- J
бы точное давление, примерно 0,2 атм (манометр!). Аммиак пропускается Я
со скоростью 5 пузырьков в секунду и проходит через промывную склянку Я
с 50 %-дым раствором едкого кали и через две колонки с кусками ед- Я
кого натра ***. ’ Я
После прекращения выделения сероводорода котел открывают н пе- Я
репосят порошкообразную коричнево-желтую массу в большую фарфо- Я
ровую ступку. Для растворения поваренной соли добавляют 3 л воды. Я
После этого краситель отфильтровывают, а затем растворяют при 60° Я
примерно в 1,5 л воды; температуру не следует поднимать выше, так.Я
как аурамин легко разлагается, К прозрачному раствору (остаток со- Я
стоит из небольшого количества серы и кетона А^ихлера) прибавляют Я
1 л раствора поваренной соли, полученного выщелачиванием; при этомШ
аурамин выпадает в виде красивых блестищик золотистых листочков. Я
ЕГыход чистого, сухого красителя может достигачъ 155 а. АурамннЯ
окрашивает хлопок, протравленный таннИном и рвотным камнем, в чн- 'Я
стый желтый цвет. Я
примечания, Аурамин является важнейшим желтым ос- а
он очень ценится вследствие сто чрезвычайно чистого |
в котлах, обогреваемых при помощи масляцой бани; темпер^-
Технические
ионным красителем,
оттенка. Его получают
туру следует выдерживать очень точно, так как прн малейшем отклонении снижается
* Приведенный метод получения аурамина носит в патентной литературе имя '|
Феера. Действительным автором этого метода является Зандмейер (герм, пат, 53614;д
.Frdl'., II, 60). 1
** Температура масляной бани примерно ria 25° гчяшс. А
*** Аммиак нельзя сушин, хлористым кальцием: нее амины реагируют с ним, j
Малахитовый зеленый 267
выход. Иногда применяются л котлы Фредсркцнга, которые дают возможность точно
регулировать температуру обогрева. Очень большую роль играет чистота поваренной
соли. Следы хлористого кальиия или хлористого магния, которые присутствуют в вы-
варочной солн, оказывают вредное действие, Наилучшсп является галицийская камен-
ная соль, которая представляет собой практически химически чистый продукт.
Аммиак сушат в колонках, заполненных твердым едким натром. Вводят лишь такое
количество аммиака, чтобы создавалось избыточное давление в 0,5 атм, а затем при
помощи циркуляционного насоса заставляют аммиак циркулировать над размеши-
ваемой смесью солей. Сероводород поглотают и в виде сернистого натрия применяют
для восстановления. На хорошо налаженном производстве выход достигает 132%; это
значит, что из каждых 100 кг тетрамстилднампнодифекцлметана получают до 132 кг
чистого 100%-ного аурамина. Определение выхода в этом случае чрезвычайно затруд-
нительно, так как только очень немногие могут точно оценить концентрацию выкраски
иа протравленном таннином хлопке. Поэтому теперь не производят пробных выкра-
сок этим красителем, а пробу красителя гидролизуют разбавленной соляной кислотой
па водяной бане; после прибавления едкого натра отгоняют выделившийся аммиак,
поглощают его нормальной соляной кислотой и избыток последней оттитровывают.
На многих заводах выкраску аурамином производят имеете с точно известным коли-
чеством синего основного красителя; получающийся зеленый цвет гораздо легче
оцепить.
Наряду с аурамином 00 производят еще аурамин Г, который получают из
монометил-о-толуидлпа. Он еще чище и зеленее, чем марка 00, Краситель из диэтил-
апилина не производят, потому что при высаливании он выпадает таким смолистым,
что его дальнейшая обработка невозможна.
Аурамин идет в больших количествах для крашения хлопка и прежде всего.для
крашения бумаги. Одни лишь шведские спичечные фабрики расходуют в год 8 ваго-
нов аурамина для окраски спичечных коробок в желтый цвет.
Малахитовый зеленый *
N(CH3)a _
а) Лейкооснованне малахитового зеленого
Смесь из 36,3 г (0,3 моля) диметиланилина, 24 г (0,2 моля)
30% -ной соляггой кислоты н 10,6 г (0,1 моля) бенз альдегида
нагревают в круглодонной колбе емкостью 300 мл с обратным холодиль-
ником в течение 12 час. Чтобы не окислилось слишком много альдегида,
холодильник закрывают ватой. Во время проведения реакции необходимо
энергичное перемешивание. Через 12 час. бензальдегид исчезает почти
полностью; добавляют 12 г кальцинированной соды и отгоняют избы-
ток диметиланилина. Его можно легко регенерировать. Лейкооснование
малахитового зеленого по охлаждении отделяют от воды, измельчают и
промывают еще раз.
Выход сухого продукта составляет примерно 24 г.
* Формула малахитового зеленого дана схематично, соответственно предложен-
ной в Uelv. С1рш. Acta, 1, 210 (1918).
б) Окисление л е й ко о снования в краситель
N(CH3)a N(CH3)3
I I
N (СН3)2 N(CH3)3
16,5 г (0,05 моля) чистого лейкоосновання растворяют
в 300 мл в о д ы и 20 г концентрированной соляной кнслоты, раз-
бавляют раствор льдом и устанавливают на 400 мл и 0°. К раствору,
который энергично перемешивают, добавляют в один прием пасту
перекиси свинца (приготовлена точно из 0,05 моля азотнокислого
свница = 16,5 г) *. Через-2 часа добавляют раствор 25 г глауберо-
вой солн, причем свитец выпадает в виде нерастворимого сульфата,
н фильтруют. Основание красителя осаждают примерно 15 г кальциниро-
ванной соды и отфильтровывают. Большей частью оно выпадает в виде
смолистой массы.
Выход сухого продукта составляет примерно 16 а, или около 100%
от теории.
Кристаллизация малахитового 'зеленого
Проведение кристаллизации в лаборатории связано с трудностями,
так как хорошие кристаллы получаются лишь при работе с большими
количествами. 120 г основания (лучше в несколько раз больше) рас-
творяют в 72 г кристаллической гцавслевой к и с л о т ы и 300 г ди-
стиллированной воды и кипящий раствор отфильтровывают от приме-;
сей. После этого к еще горячему раствору добавляют возможно более
концентрированный раствор 7 г щавелевокислого аммонии и
оставляют стоять в месте, защищенном от воздушных потоков. Лучше
всего, если жидкость для кристаллизации ставят в большой сосуд с го-
рячей водой, для того чтобы замедлить охлаждение, В течение дня дают'
температуре снизиться до 70° и отфильтровывают красивые кристаллы,
Прн дальнейшем охлаждении маточный раствор выделяет еще некоторое
количество менее чистого красителя — так называемого малахитового,
зеленого II.
Выход из 1 частн основания составляет 1,45 частн щавелево-
кислой соли, или 145% исходного материала.
Технические примечания. Малахитовый зеленый получается и в на-
стоящее время в больших количествах' он служит для крашения шелка, утяжелен-
ного оловом, шерсти и бумаги. В смеси с другими красителями дает чистые дешевые
смешанные окраски средней прочности. Употребляется также для печатания по шелку,
и хлопку, однако его прочность не удовлетворяет современным требованиям, так что
его потребление сокращается.
Конденсацию проводят только в присутствии минеральных кислот; старый способ
с хлористым цинком уже давно не применяют. Не применяется также метод получе-
ния из бензотрихлорида по Дббнеру. Конденсацию проводят в присутствии соляной
или серной кислот. Конденсация в присутствии соляной кислоты протекает быстрее,
’ О получении см. стр. 126.
однако в этом случае требуются эмалированные аппараты; конденсация в присутствии
серной кислоты может быть осуществлена в гомогенно освинцованных сосудах. Далее,
важно применять не слишком большие количества кислоты, так как в этом случае
конденсация частично может пойти по иному направлению, В качестве побочного
продукта образуется бензгидрол
который естественно цс может дать краситель. В технике всегда получают различ-
ные фракции, так как существуют различные требования к внешнему апду красителя.
Щавелевокислая соль малахитового зеленого имеет следующую формулу: -j-
+ ЗСЕН2О^. Кристаллизация больших количеств (I—6 л/3) раствора длится несколько
суток, при этом часто получаются очень красивые кристаллы. Добавка щавелевокис-
лого аммония как ускорителя кристаллизации напоминает сходные приемы в химии
алкалоидов, и необходимость ее установлена чисто эмпирически. Дальнейшие данные
см. при описании ксилспового голубого.
Конденсацией бензальдегида или о-хлорбензальдсгида с этилбензцланцлниом,
сульфированием полученного лейкоосноваиия олеумом и окислением лсйкосульфо-
кцелоты перекисью свинца получают светло-зеленый СФ— желтоватый (кислот-
ный зеленый для кожи) или эрионнридип Б. Оба красителя имеют малую свето-
ирочно'сть, но, несмотря на это, благодаря своим живым оттенкам они находят еще
широкое применение.
Ксиленовый голубой Фау С (кислотный ярко-голубой 3)
(фирмы «С а ндо з»)
Ксиленовый голубой относится к группе так называемых патенто-
ванных голубых красителей, которые являются сульфированными трн-
фе и ил мет ано вы ми красителями, устойчивыми к щелочам, Для всех этих
соединений характерно наличие сульфогруппы в орто-положении к цен-
тральному атому углерода. Общая формула их
Na
Заидмейер первый выяснил связь между строением и устойчивостью
по отношению к щелочам; его эриоглауцин, имеющий приведенную ниже
формулу, был первым красителем, при изучении которого был установ-
лен этот важный факт,
Сйщ
I " \ /—ч
SO3—С6Н4—СН2—N—CeH4 / =С—< >
। J
О-—SO 2
Na3
Эриоглауцин А цз этилбензиланилицсульфокислоты
и 1,2- беггзальлегггдсульфокислоты
Вероятно, между гидроксилом карбинола н сульфогруппой обра
зуется внутренний ангидрид, который обусловливает большую устойчи
вость к соде и едкому натру. Это предположение подтверждается тем
что красители, имеющие приведенную ниже формулу, совершенно нерас
творимы.
сн/
_ ^Ог11
so3H
^З^-Толуолди-
сульфок^слота
II
СНО
SO3H
1,2,4-Бенвальдегид-
дисульфокислота
III
(с2нрг1ч
Л ей кооснова ние ксиленов ого
голубого Фау С
IV
(C2H5)2N
Ксиленовый голубой Фау С
V
а) Толуолдисульфокислота (II)
46 г (0,5 моля) чистого толуола смешивают с 80 z моногид-
рата, приливая последний по каплям к кипящему толуолу в течение
1/4 часа. Затем нагревают 1 час прн 125°, при этом толуол полностью ис-
чезает. Смесь охлаждают до 30° и в течение 7г часа при очень хорошем
перемешивании добавляют по каплям 220 г олеума, содержащего 66%
SO3. Нагревают^ 4 часа при 125°, толуол полностью прн этом превра-
щается в дисульфокислоту. Затем смесь разбавляют 400 г сер ной ки с-
лоты (уд. вес 1,84) и выливают жидкость в фарфоровый стакан с хо-
рошей железной мешалкой.
б) Бенза льдегиддисульфокнслота (III)
1\ полученному раствору ди сульфокислоты прибавляют при постоян-
ном перемешивании небольшими порциями 125 г 80%-ного марта нцо
во го шлама*. Прибавление продолжается 2—3 часа, температура
смеси около 25°, После того как добавлено все количество марганцового
шлама, перемешивают еще 3 часа при 30°, а затем медленно нагревают
до 120°. При этой температуре масса загустевает н дальнейшее переме-
шивание часто становится невозможным. Темная окраска двуокиси мар-
ганца постепенно переходит в светлосерую, В лабораторных условиях
редко удается довести окисление полностью до исчезновения двуокиси,
и приходится прерывать процесс. Смесь оставляют на 12 час., затем до-
бавляют 2 л воды и гашено й и з вест и до полного исчезновения
мин ера льнокислой реакции. Реакция не должна быть слишком щелоч-
ной (по лакмусу), так как избыток щелочи разлагает бензальдеглд-
дпсульфокислоту. К гипсовой кашице добавляют концентрированный
раствор соды до тех пор, пока отфильтрованная проба нс будет давать,
осадка прн дальнейшем прибавлении соды. После этого отфильтровы-
вают гипс и окись .марганца,* хорошо промывают осадок н, если воз-
можно', замешивают его еще раз с водой н фильтруют второй раз. Свет-
лый, слабощелочной раствор упаривают в вакууме в токе СО? до объема
250 мл и отфильтровывают в случае необходимости от выделившегося
гипса и окнеи марганца. Выход можно определять следующим образом:
к отмеренной пробе прибавляют (в присутствии уксуснокислого натрия)
уксуснокислый раствор фенилгидразипа известной концентрации до тех.
пор, пока высоленная проба не перестанет реагировать прн дальнейшем,
прибавлении гидразина. С этим реактивом тотчас же образуется интен-
сивная желтая окраска. Метод определения не очень точен.
в) Конденсация в лейкосоедипенне (IV)
Полученный раствор кипятят в течение двух дней в колбе с обрат-
ным холодильником с 45 г серной кислоты (уд, вес. 1,84) н 100 г
чистого диэтиланилина, затем прибавляют около 100 г 30%-ного
раствора едкого иатрадо енльношелочной реакции и отгоняют из-
быток амнна с водяным паром. В случае надобности щелочной раствор
фильтруют и затем подкисляют примерно 50 г концентрированной
серной кислоты до ясно кислой реакции. В течение 24 час. выпа-
дает внутренняя соль лейкосоедииения в виде тонких белых игл; ее от-
фильтровывают и тщательно промывают водой. После сушки прн 80°
полученный продукт весит около 70 г.
г) Окисление в краситель (V)
Это окисление сходно' с окислением в случае малахитового зеленого,
50 г лейкосоедииения растворяют прн кипячении в воде, в кото-
рую добавлено 8 г кальцинированной соды; трудно растворимое лей-
косоединение почти совсем нерастворимо в холодном растворе соды.
Среда должна быть точно нейтральной (по лакмусу). Раствор разбавляют
до объема 1000 мл, охлаждают до 0° и при очень энергичном механиче-
ском перемешивании добавляют в одни прием смесь 15 г концентриро-
ванной серной кислоты и точно 22 г 100%-ной пасты дву-
окиси свинца (см. стр. 126). Оставляют стоять на час при 0—5°,
* Количество марганцового шлама рассчитывают по содержанию МпОа и при-
меняют ровно 100 г МпОг. Марганцовый шлам является отходом сахаринового произ-
водства и соответствует приблизительно формуле МщСЦ,
затем нагревают до 80° п отфильтровывают от сернокислого свинца.
Раствор упаривают (лучше в вакууме) до объема 600 мл и добавляют
50 г поваренной соли. В течение дня краситель выкристаллизо-
вывается, его отфильтровывают и заливают небольшим количеством на-
сыщенного раствора поваренной соли. К кристаллам добавляют
несколько капель концентрированного раствора аммиака для нейтрали- -
зацнн остатка минеральной кислоты и затем их сушат в небольшой -
.фарфоровой чашке. ’
Выход концентрированного красителя около 32 г.
Если объем жидкости после отфильтровывания сернокислого свинца не превы- ;
шает примерно 1 л, то краситель можно высолить из голубого раствора добавлением
20% поваренной соли (по отношению к количеству жидкости) без предварительного J
упаривания. (Вначале следует на небольшой пробе в пробирке убедиться в том, что 1
это возможно,) При наличии избытка кислоты раствор становится зеленым; если 1
это имеет место, то перед высаливанием следует добавить соды до тех пор, пока 1
раствор не станет голубым. 1
Технические примечания. Беизальдеглддисульфокислота так хорошо а
растворима, что ее невозможно выделить. Окисление проводят в больших рычажных
тестомесильных аппаратах (впервые их построила фирма «Вернер и Пфлейдерер»).л|
Аппараты обогреваются при помощи паровой рубашки; вследствие прочности кон- 1
струкции в них можно хорошо пеумешивать реакционную массу. Для разбавления J
можно обойтись несколько меньшим количеством серной кислоты. Нейтрализация суль-1
фомассы известью и упаривание производят известными методами, однако в данном- |
случае имеется следующее неудобство: нагревательные трубы очень быстро покрыва- 1
ются гипсом, для полного осаждения которого нельзя применять избыток соды вслед-
стене большой чувствительности бензальдегиддисульфокислоты. Конденсацию проводят
в гомогенно освинцованных котлах с мешалкой; окисление—в деревянных чанахJ
< пропеллерной мешалкой из ясеневого дерева. Упаривание производят в вакууме, J
л отделение хорошо выкристаллизовавшегося красителя — центрифугированием. Из на- J
точного раствора при добавлении анилина выпадает загрязненный краситель, который J
выпускается в продажу как марка II. J
В последнее время 1,2,4-бензальдегиддисульфокислоту получают не из толуола, ^
а из «-толуол сульфо хлорид а. Вследствие применения этого метода, а также расшире- J
ння других областей применения n-толуолсульфохлорида цена на него значительно Я
возросла.
SO2CI SO3H Я
Омыление® конц. НаЧО;
4-SO.j бО’.'о-ный
с Н3
л-Толуолсуль-
фохлорид
сн3
Л-ТО-1 уолсуль-
фо кислота
SOgH
SO3H
Lso,
'3Н
Мп,
—SO3H
вд
СН3
1, 2, 4-Талуол дисуль-
фо кислота
сно
I. 2, ЪБенэальдегид-
дисульфокислота
Синий для шерсти 5Б
9,2 г (0,05 моля) о-х л о р-л-д иметила ми нобенз а л ьде г и дй-Н
(ом. стр. 108) кипятят с обратным холодильником при перемешивании«
* Вместо того чтобы омылять п-толуолусульфо хлорид, его можно вначале суль-1
фировать. Омыление происходит с образованием хлорсульфоновой кислоты; при до-1
бавленпи после сульфирования небольшого количества воды происходит почти мгно-I
венное удаление соляной кислоты. j
с 32 г эт и л б е и з и л а п и л и н с у л ь ф о ки сл о ты и 225 г 12,5 % -ной
серной кислоты в течение 24 час. При осторожной нейтрализации
едким натром натриевая соль лейкосоединен и я выпадает в виде
вязкой массы. Маточный раствор декантируют, остаток растворяют в
0,5 л воды и добавляют равный объем насыщенного раствора пова-
ренной солн., Лейкосоедннение, выпавшее в виде белых хлопьев,
отфильтровывают и промывают 10%-ным раствором поваренной соли.
Сушат его в вакууме при 100°. Получают 36 г лейкосоединення, что со-
ставляет 91 % от теории.
7,9 г (0,01 моля) лейкосоединення растворяют при интен-
сивном перемешивании в 100 мл 50%-ной уксусной кислоты и
одновременно добавляют 30 мл щавелевой кислоты 1:10 и
12,5 мл раствора двухромовокислого натрия 1:10. Смесь
тотчас становится темно-енней. Окисление заканчивается за 10 мии. Раз-
бавляют равным объемом насыщенного раствора поваренной
соли и нейтрализуют уксусную кислоту аммиаком. Краситель вы-
падает в виде красного бронзирующего шлама, который быстро застывает
в стекловидную массу. Ее растворяют в горячей воде, фильтруют и
высаливают прибавлением равного объема насыщенного раствора п о-
варенной соли. Таким путем получают 7,8 г очищенного красителя.
Он окрашивает шерсть в яркие синие тона. Краситель выбирается шер-
стью из нейтральных и слабокислых растворов. Светопрочность окрасок
невысока.
Окисление
1R Зак. lOCki. Г_ Й tbunrr.Лооиг,
Виктория голубой Б (основной синий)
КетонМихлера (стр. 127) конденсируют с ф ей и л-а-п а ф тил^
амином (стр. 160) таким образом, что кетон смешивают с 26%-ным’
от его веса количеством толуола н точно с одной молекулой фенил-
а-нафтиламина. Затем прибавляю^ точно 1 моль РОС1з и перемешивают
реакционную массу до загустевания. Температура должна повыситься,
до 75—80°. Примерно через 45 мин. реакционная масса становится та-
кой густой, что дальнейшее перемешивание невозможно. Содержимое,-
реакционного сосуда выливают в десятикратное количество вод ы,
кипятят для разложения продукта присоединения фосфорной кислотыj
и добавляют едкого натра, пока вся масса не приобретет блестящую
зеленоватую окраску. Толуол отгоняют с водяным паром, сливают щелочь;
н сушат прн 80—90°-
Выход количественный.
Аналогичным образом получают из тетраэтилкетоиа и этил-а-наф-)
тиламина виктория чисто голубой БО (основной синий 3) )
Зеленый для шерсти С
N(CH3)2
Галламиновый синий из амида галловой кислоты
275
13,5 г (0,05 моля) тетра метил n,n'-jn и а м е н о б е н з г и д р о л а
(стр. 126) вносят при перемешивании в 120 г серной кислоты
(уд. вес. 1,84) таким образом, чтобы температура не превышала 40°.
После полного растворения охлаждают льдом примерно до 5° и прн
5—10° при перемешивании вносят такое количество технической Р-сол и,
которое соответствует 21,8 г (0,0625 моля) чистого 2-н а ф т о л-3,6-д н-
сульфокнслого натрия. Перемешивают в течение 2 час. при
обычной температуре, затем в течение часа нагревают до 60° н выдер-
живают при этой температуре примерно 2 часа до тех пор, пока раз-
бавленная водой проба при прибавлении уксуснокислого натрия будет
давать еще совсем слабую, не усиливающуюся прн нагревании синюю
окраску. Смесь выливают в 700 мл холодной воды и вызывают кри-
сталлизацию, протекающую чрезвычайно медленно, внесением затравки
илн трением стеклянной палочки о стенки сосуда. Перемешивают в те-
чение нескольких часов и оставляют стоять на ночь. Осадок, похожий
на песок, отсасывают, промывают холодной водой до исчезновения кис-
лой реакции на конго, затем промывают два раза спиртом и два раза
эфиром и сушат в вакуум-эксикаторе.
Выход 24 г рыхлого, серовато-белого порошка, т. е. около 86%
от теории.
Полученное таким образом лейкосоедннение растворяют
в 480 мл холодной воды, в которую добавлено 15 г кальцинированной
соды, вносят 160 а льда н прн энергичном перемешивании прибав-
ляют в одни прием пасту двуокиси свинца, приготовленную
точно нз 14.3 г азотнокислого свинца (стр. 126). Смесь тотчас окраши-
вается в темно-синий . цвет; в течение ’/а часа ее перемешивают прн
комнатной температуре, нагревают в течение часа до 80°, отфильтровы-
вают горячей от углекислого свинца н промывают последний горячей
водой. После охлаждения фильтрат подкисляют 20 мл концентрирован-
ной соляной кислоты* и при перемешивании к нему добавляют
200 г поваренной солн. Краситель, который выпадает в виде брон-
зирующих кристалликов, оставляют на ночь, а затем отсасывают на
нутч-фильтре, промывают раствором поваренной соли и сушат (целе-
сообразно в вакууме) прн 50—60°. Получают 24 г красителя, содер-
жащего соль.
Зеленый для шерсти С окрашивает шерсть из кислой ванны в интенсив-
ные сине-зеленые тона; он является одним из наиболее употребляемых зеленых краси-
телей для шерсти наряду с нафталиновым зеленым Фау (кислотным зеле-
ным Ж), который получают аналогичным методом из тетраэтилдиаминобензгидрола
и 2,7-нафталиндисульфокислоты.
Е. ОКСАЗИНОВЫЕ И ТИАЗИНОВЫЕ КРАСИТЕЛИ
Галламииовый синий из амнда галловой кислоты
Нагреванием нитрозодиалкиланилинов с галловой кислотой или ее
амидом получают хорошо характеризуемые соединения—оксазины.
Галловая кислота получается исключительно нз природных дубильных
веществ.
* Окраска раствора не должна при этом переходить нз синей в зеленую; подоб-
ная перемена окраски свидетельствует об избытке соляной кислоты, которая препят-
ствует полноте осаждения и в случае необходимости должна быть нейтрализована
уксуснокислым натрием.
276
/Л Красители
а) Нитрозодимстилапилин
100гдиметил анилина смешивают с 200 г 30% -ной соляной'
кислоты, после охлаждения добавляют 300 г льда. Затем в течение
5 час. по каплям прибавляют возможно более концентрированный рас-
твор 60 г нитрита натрия (100%-ного), причем стакан помещают
в ледяную воду. Иодокрахмальной бумажкой определить наличие сво-:
бодной азотистой кислоты нельзя, так как солянокислый нитрозодиме-
тиланилин сам с ней реагирует. Избыток азотистой кислоты определяют;
только по запаху. Попятно, что реакционная смесь все время должна
быть кислой (по кои го). Через 6 час. отфильтровывают, смывают осадок
па нутч-фильтр маточным раствором и отсасывают возможно полнее.
Под конец хорошо отжимают на винтовом прессе и влажный соляно- :
кислый нитрозодиметил анилин измельчают насколько возможно. Его не'
следует сушить, а применять свежий и влажный. В производстве для
получения в достаточно сухом виде только центрифугируют.
n-Нитрозодиэтиланилин получают сходным методом, однако вследствие огром- £
ной растворимости солянокислого
нитрозодиэтиланилина
при
нитрозировании
нельзя^
применять воду; употребляют только концентрированную соляную кислоту и насы*.;
щенный раствор нитрита натрия; работают прн внешнем охлаждении. В технике. таК^<
же как в случае получения тропеолииа, работают в эмалированных сосудах.
б) Г а л л а м и и о в ы й с и н и й J
В стеклянной колбе с обратным холодильником и мешалкой (см<
рис, 6, стр, 66) растворяют 20 г (см, стр. 152) примерно 92%-ногф
амида галловой кислоты** в 500 мл 90%-ного спирта и до*
бавляют при постоянном кипении в три приема солянокислы®
н и т р о з о д и 'М е т и л а п и л и н, полученный из 75 г диметиланилина,:
Лучше всего добавлять каждую порцию с интервалом в 15 мин. и всЩ
операцию закончить за 45 мнн. Затем кипятят с обратным холодильнич-
ком еще 4 часа и оставляют стоять на 12 час. Галлам инов ый сини®
выпадает в виде красивого, блестящего бронзирующего осадка, кото-
рый отфильтровывают и промывают водой. Спирт перегоняют. ВыхоД
чистого галл аминового синего составляет около 40 г. Из спиртовогй
маточного раствора получают серый, похожий на нигрозин краситель^
который на хлопке с уксуснокислым хромом дает очень прочные серЫЧ
окраски; этот продукт называют «метиленовый серый». J
Галлам и новый синий нерастворим в воде и поэтому нй
может применяться как таковой. При помощи различных реакций ers
можно, однако, Превратить в легко растворимую форму.
* Одна молекула нитрозодиметиланилина является окислителем.
** Анализ амида галловой кислоты производят отгонкой аммиака с едким нат*
ром и титрованием его.
На водяной бане нагревают при 50° 1 часть галламинового
синего с 6 частями раствора бисульфита натрия, содер-
жащего 25% SOs, до прекращения выделения сернистой кислоты. Это
происходит примерно через 1 час, затем нагревают прн 85° в течение
1—3 дней до тех пор, пока смесь не станет серо-зеленой. Полученный
таким образом краситель является сульфокислотой лейкосо ед ипения (ве-
роятно, комплексной сульфосолью?); он окрашивает шерсть с уксусно-
кислым хромом в красивые и пройные темно-синие (флотские синие)
цвета. Он может использоваться и в ситцепечатании, однако другие кра-
сители этой группы его превосходят. Так, восстановлением галламино
вого синего сероводородом получают л ей косо единение — модерн
фиолетовый (фирмы «Дюран и Гюгенен»), образующий на хлопке
чрезвычайно чистый и прочный хромовый лак.
СО— .чли
N(CHs)a °
НС1
Модерн фиолетовый
.50 г галламинового синего растворяют примерно в 40 а
30%-ного едкого натра ш 400 мл воды, к прозрачному'раствору до-
бавляют 50 г кристаллического сернистого натрия. К нагретому
до 60° раствору в течение часа добавляют около 100 г концентриро-
ванной соляной кислоты до неисчезающей окраски конго-бумажки.
Синяя окраска раствора исчезает и он становится почти бесцветным,
его отфильтровывают от выпавшей серы; солянокислую соль лейкосо-
единения осаждают добавлением 150 г поваренной соли, отфиль-
тровывают, промывают небольшим количеством насыщенного раствора
поваренной соли и хорошо отжимают. Краситель сушат при 60° в ва-
кууме, так как он быстро окисляется в исходное соединение.
Вых од около 55 г.
Технические примечания. Техническая аппаратура состоит из эмали-
рованного чугунного котла и обратного холодильника из свинцовой трубки. Процесс
переработки 40 <кг амида галловой кислоты длится около 12 час.
Вследствие легкой окисляемости дезинтеграция модерн фиолетового должна про-
водиться на холоду. В противном случае может произойти самовоспламенение. По-
видимому, причина этого явления заключается в наличии чрезвычайно мелкодисперс-
ной серы.
Оксазины являются по преимуществу красителями для печати. Кроме производ-
ных диметиланилина, получают также и производные диэтиланилнна, которые отли-
чаются очень чистым зеленоватым оттенком (целестин голубой). Если
применять вместо амида галловой кислоты галловую кислоту, то получают г а л л о;Ц и а-
нины, случайно открытые Г. Кёхлином. Он пробовал при помощи таннина и рвот-
ного камня фиксировать нитрозо диметил анилин на хлопке и получил при этом синие
красители, которые он признал как оксазины. Галлоцианины плохо получаются в рас-
творах этилового спирта, поэтому работают с метиловым спиртом, который вследствие
своей ядовитости и летучести менее удобен. Наряду с простыми оксазинами имеется
еще очень большое количество сложных продуктов конденсации, описывать которые
здесь мы не имеем возможности.
Попутно можно отметить, что первым оксазином, нашедшим техническое при-
менение, был синий Мел дола, нафтоловый синий или бенгальский
синий (основной темно-синий 2К); его получают из солянокислого нитрозодиметил-
анилина и [3-нафтола. Он очень прочен, но нс дает, однако, красивых оттенков. Пыль
этого красителя так сильно раздражает слизистые оболочки, что многие совсем нс мо-
гут с ним работать. Несмотря на это, он и сейчас еще довольно распространен.
Метиленовый голубой нз диметнланилина
Реакция образования метиленового голубого интересна как с на-
учной, так и с технической точки зрения, и ее следует несколько пояс-
нить, прежде чем приступить к описанию самой методики получения
красителя.
Диметиланилин обрабатывают в солянокислом растворе нитритом
натрия; восстановлением образовавшегося нитрозодиметиланилина по-
лучают п-аминодиметил анилин.
а) я-Аминодиметиламилин
(CHft)3N
(СН3)3 N
Диметиланилин к-НитрозодимС’
тнланнлин
NO
б) n-Амииодиметиланилин в кислом растворе окисляют вместе с мо-
лекулой диметнланилина, одновременно в молекулу вводят остаток тио-
серной кислоты. Это происходит благодаря тому, что окисление проводят
в присутствии тиосерной кислоты в момент ее образования
(in statu nascendi).
(CHs)aNS
1
so3H
Тиосульфи кислота
П'амин&диметил-
анилина
(CH3)2NS N(CH3)2
I
so3..,.
Тиг1суи|ьфокислата
зеленого Бнидшед.тера
в) Эта тиосульфокислота при дальнейшем окислении в результате ;
замыкания цикла превращается в метиленовый голубой.
Мети,ген оный голубой
а) л-Аминоднметиланилин ',1
24,2 г (0,2 мюля) чистого диметнлаи илина растворяют в 75
концентрированной (30%-ной) соляной кислоты, раствор охлаж- £
дают, добавляют 150 а льда и приливают по каплям в течение одного ••
часа 14,7 г 100 %-ново нитрита натрия в виде 20%-ного раствора-
Нитрозирование заканчивается полностью за 4 часа. Затем добавляют
еще 110 г 30%-ndfi соляной кислоты, 200 г льда и в течение)
’А часа при механическом перемешивании прибавляют 35 г хорошей )
цинковой пыли, Температура без вреда может подниматься до 25°-
Раствор становится бесцветным ib нейтральным в отношении минераль-
ной кислотности; его фильтруют и цинковую пыль промывают неболь-
шим количеством воды.
б) Тиосульфокислота зеленого Биндщедлера
Окисление на этой стадии следует вести в присутствии раствора
хлористого цинка, который ие должен оказывать никакого восстанав-
ливающего действия. Необходимый раствор приготовляют, растворяя
листовой цинк в концентрированной соляной кислоте *. Тиосерную кис-
лоту применяют в виде тиосернокислого алюминия, который столь сильно
диссоциирован, что реагирует как свободная тиосернан кислота.
Прежде чем приступить к получению метиленового голубого, готовят
растворы необходимых реагентов. Необходимость предварительного при-
готовления растворов обусловлена тем, что вещества должны приба-
вляться быстро и прн надлежащих температурах.
Раствор 1; 38 г чистого сернокислого алюминия в 60 мл
воды'
» II: 52,5 г кристаллического тиосернокислого натрия
в 50 МЛ воды
» III: 57 г д в у х р о м о в о к и с л о г о натрия до объема
90 мл воды
» IV: 20 г диметил апи л ина в 27 г концентрированной с о-
ляцой кислоты
г V : 25 г тонко измельченного пиролюзита (МпО^), замешан-
ного с 30 мл воды в однородную пасту
Прозрачный нейтральный раствор л-ам и под и метил анилина подкис-
ляют 4 г концентрированной серной кислоты до минер ал ьнокис-
лой реакции и добавляют 100 г 50%-ного, раствора хлористого
цинка, не обладающего восстанавливающим действием.
Стакан ставят на войлочную подкладку и вставляют трубку для
пропускания пара. Прн хорошем перемешивании при комнатной темпера-
туре добавляют раствор I, затем раствор II и через 2 сек. 7з раствора
III (что соответствует 19 г двухромовокислого натрия). Пропусканием
сухого пара за одну минуту повышают температуру до 40°, добавляют
раствор IV и остаток раствора III и быстро нагревают до 70°. Вследствие
образования тиосульфокислоты зеленого Биндшедлера раствор стано-
вится темным зелено-синим. Как только температура достигает 70°, до-
бавляют пасту V и повышают температуру до 85°.
Добавление пиролюзита имеет целью превращение освобождающейся при замы*
кании цикла сернистой кислоты в безвредный дитионат. С таким же успехом вместо
пиролюзита можно применять сернокислую медь (40 г), при этом окисная соль меди
превращается в нерастворимую CtijO.
Прн 85° раствор приобретает красивый бронзовый блеск и образо-
вавшийся краситель выпадает из концентрированного раствора хлористого
цинка. Через 7г часа раствору дают охладиться до 50° и добав-
ляют 70 г концентрированной серной кислоты; последняя раство-
ряет соль марганца, гидроокись алюминия н окись хрома. Отфильтро-
* В технике поступают следующим образом; к продажному раствору хлори-
стого ниика добавляют двухромовокислый натрий до тех пор, пока не получится рас-
твор, не оказывающий восстанавливающего действия. Обычно на 100 кг цинкового
раствора требуется 100 -250 г бихромата.
вывают при 20° и промывают небольшим количеством 10%-ного рас-
твора поваренной соли. Неочищенный краситель растворяют в 1 л
воды прн 100°, отфильтровывают от нерастворившейся частн; из про-
зрачного фильтрата краситель осаждают добавлением 50 г обычного
50%-ного раствора хлористого цинка и 150 г поваренной
соли. Через 24 часа выпадает двойная соль с хлористым цинком в виде
великолепного красного с бронзовым блеском осадка, который отфиль-
тровывают и промывают небольшим количеством 10%-ного раствора
поваренной соли. Сушат при температуре не выше 50° и получают при-
мерно 44 а чистого концентрированию го красителя.
/
Технические примечания. Описанный выше метод принадлежит Бернт-
сену и Ульриху; Ульрих рекомендовал применение тиосернокислого алюминия. Доба-
вление пиролюзита или сернокислой меди получило общее употребление. Следует одно-
временно получать не очень большие количества красителя, так как очень важно
Рис. 33. Центрифуга с нижней выгрузкой.
быстрое подогревание. В производстве готовый краситель фильтруют обычно только
на рамных фильтр-прессах или на фильтрах на козлах (см. рис. 27); после того как
жидкость стечет, краситель перекладывают в небольшие мешки и подвергают центри-
фугированию (см, рис. 33).
Метиленовый голубой очень ценится благодаря чистоте оттенка и дешевизне
получения; он употребляется в больших количествах для крашения таинированногО'
хлопка. В печати по шелку для получения вытравных тканей применяют метиле-,
новый голубой, не содержащий цинка. Его получают следующим обра-’-
зом: обыкновенный краситель растворяют в воде, цинк осаждают содой и отфильтро-
вывают легко растворимое основание метиленового голубого. При добавлении пова-
ренной соли и соляной кислоты выпадает не содержащий металла метиленовый голу-
бой в виде хороших кристаллов. В производстве кристаллизация продолжается не-
сколько суток и может быть ускорена охлаждением холодной водой, циркулирующей
в свинцовых трубах.
Интересным является нитропроизводное — метиленовый з ел е-
н ы и. Нитрование проводят аналогично нитрованию тропеолииа, при-
чем можно нитровать неочищенную двойную соль с хлористым цинком.
а) Протравные красители
281
Влажный неочищенный метиленовый голубой (в таком
виде, в каком он был получен) замешивают с 50 лл воды и 20 г 62%-ной
(уд. вес 1,38) аз от ио и кислоты и добавляют при 25° 5 г ни-
трита натрия в минимальном количестве воды. Затем осторожно,
при хорошем перемешивании, поднимают температуру до 50° н оставляют
при такой температуре иа 2 часа.
Затем смесь разбавляют 200 г насыщенного раствора пова-
ренной соли и через 12 час. фильтруют. Сырой продукт растворяют
в 1 л воды при температуре не выше 60°, отфильтровывают и высали-
вают добавлением 150 г поваренной солн и 50 г 50%-ного рас-
твора хлористого цинка. Через 12 час. отфильтровывают и су-
шат при 45° до тех пор, пока не станет возможным краситель измель-
чить в порошок. Такой краситель содержит еще окосю 20% воды. Если
его высушить полностью, то концентрация красителя резко падает и
часть его становится нерастворимой.
Выход из приведенных выше количеств составляет около 38 г
концентрированного продукта.
Метиленовые голубой и зеленый разбавляются декстрином, так как прибавление
соли очень сильно уменьшает растворимость. Метиленовый зеленый применяется глав-
ным образом в смеси с кампешем для крашения шелка в черный цвет по железной
протраве, значительное применение находит он и для крашения шелка оловянно-
фосфатного утяжеления. Полученные таким образом окраски являются наиболее кра-
сивыми и прочными черными окрасками на шелке.
Если вместо диметиланилина применять диэтиланилин или моночетил-о-толуидин,
то получаются чистые зеленоватые тиазиновые голубые (например, тионин
голубой и др. — основной голубой), которые служат для получения наиболее чистых
голубых окрасок на шелке. Однако вследствие конкуренции более прочных ализа-
риновых красителей они несколько утратили свое былое значение. Неалкилнрованный
метилено1шй голубой — хлорид диамцнофеназтиония или фиолетовый Лаута —
употребляется в ограниченных количествах для получения чистых фиолетовых тонов.
Его и сейчас еще получают старым методом: совместным окислением п-фенилен-
диамина и сероводорода хлорным железом.
Ж- АНТРАХИНОНОВЫЕ КРАСИТЕЛИ
2-Антрахинонсуль-
фокислота
а) ПРОТРАВНЫЕ КРАСИТЕЛИ
Ализарин
1,2-Оксиантрахинон-
сульфокислота
Ализарин (1,2-диоксиантрахинон) образуется при щелочном плав-
лении 2-антрахинбнсульфокнслого натрия (серебристой соли); при этом
не только сульфогруппа замещается па ОН, но одновременно вводится
еще вторая оксигруппа. Поэтому прибавление окислителя влияет поло-
жительным образом на течение реакции.
Получение ализарина щелочным плавлением было впервые введено
в технику Каро; добавление окислителя, а именно селитры, было впер-
вые применено' Биндшедлером и Бушем в Базеле (предшествен-
никами 'фирмы Циба). В начале семидесятых годов, по предложению
Коха, перешли к применению хлорноватокислых солей; в настоящее
время большей частью употребляют дешевый электролитический хлор-
новатокислый натрий*
Смесь 100 г 100%-ной серебристой солн (см, стр. 208), 260г
100%-hgdo едкого натра, 28 г хлорноватокислого натрия
и такого количества воды, чтобы общий объем смеси составлял 670 л<4,
нагревают при постоянном перемешивании в автоклаве с мешалкой до
185°, Давление поднимается до 5—6 ат. Через 48 час, охлаждают и про-
веряют, готов ли плав. Для этого отбирают 2 мл плава, осаждают али-
зарин необходимым количеством концентрированной соляной кислоты
Рис. 34, Котел с паровым или водяным обогре-
вом системы Фредеркинга.
и дважды экстрагируют фильтрат небольшим количеством эфира. Рас-
твор, в котором полностью отсутствует ализарин, разбавляют до 15 мл (
н наблюдают флуоресценцию, которая обусловлена наличием непрореа- ;
тировавшей серебристой соли, нли моиоокснантрахннонсульфокислоты.
Флуоресценция должна отсутствовать илн быть совсем слабой. Если
нужно, то нагревают еще 24 часа при 190°. Затем плав разбавляют ’
двумя литрами воды и из кипящего раствора осаждают ализарин г
50%-ной серной кислотой. Фильтруют При 50° и промывают алн- !
зарин до тех пор, пока фильтрат не будет содержать солей. Ализарин '
не сушат, так как высушенный краситель плохо окрашивает. Выход опре-;
деляют по содержанию сухого остатка н пробной выкраской, Пасту
всегда устанавливают на 20%-ное содержание ализарина,
Из 100 г чистой серебристой солн получают при-
мерно 60 г чистого ализарина.
Технические примечания. Ализарин явился первым натуральным кра-
сителем, который с успехом производился искусственным путем. Этот синтез был
триумфом молодой еще тогда промышленности синтетических красителей; долгое
время ализарин оставался ее важнейшим продуктом. Мировое производство 100%-ного
ализарина составляло ежегодно около 2800 000 кг, из которых около 2000 000 кг
производил Баденский анилиновый и содовый завод. В последнее время, однако,
потребление ализарина резко упало вследствие конкуренции более легко применяе-
мых красных азокрасителей, в особенности также столь же прочных, получаемых при
помощи нафтолов АС (азотолов),
В настоящее время плавление для получения ализарина все больше проводят
в аппаратах Фредеркйцга (см. рис. 34) или в действующих аналогичным образом более
новых котлах системы Замесрейтера (см. рис. 35, а и б). Так как применяемые в плаве
хлорноватокислые соли * сильно портят аппаратуру, то всегда работают с вклады-
шами из щелочеупорного чугуна, которые можно легко заменить. Имеется много
Рис. 35 (а и б), Реакционный котел системы Замес-
рейтера,
Котел а нагревается нли охлаждается снаружи змеевиком. Змеевик
соединен с котлом при помощи медных теплопроводяших коль-
цевых держателей (б), находящихся между отдельными витками
змеевика. В отличие от аппаратов, которые обогреваются по
системе Фредеркинга (см. рис. 34), в этом случае стенки котла не
испытывает давления. После того как котел перестает быть
годным, обогревательное устройство можно перенести на другой
котел. Подобные котльт особенно пригодны, например, для суль-
фирования, когда попеременно нужно охлаждать и нагревать (см.
Аш-кислота, стр. 18У и ся.)<
вариантов аппаратуры подобного рода. В технике применяются очень большие за^
грузки; иа больших заводах из одного котла сразу получают 2000—2500 кг 100%-ного
ализарина, из которого готовят 20- или 16%-ную пасту. Концентрацию устанавли-
вают на основе определения содержания сухого красителя и пробной выкраски. Далее,
в технике возможно применение значительно меньших количеств едкого натра,
а именно только 110% от количества, необходимого теоретически (в нашем примере
лишь около 40 г вместо 260 г|). Высушенный ализарин снова можно перевести в легко
красящую пасту растворением в буре и осаждением из раствора уксусной или серной
кислотой. Красители типа ализарина вследствие их трудной растворимости следует
осаждать из кипящих, растворов, так как только в этом случае они получаются
в тонкодисперсной форме. Для экспорта в восточные страны готовят и твердые пре-
параты введением в краситель крахмала до образования твердых кусков; последние
при кипячении в воде клейстеризуются и легко окрашивают.
* На многих заводах хлорноватокислым солям предпочитают селитру; последняя
меньше разъедает железо и восстанавливается прямб до аммиака, что представляет
преимущество. С другой стороны, хлорноватокислые соли оказывают своего рода очи-
щающее действие.
б) КИСЛОТНЫЕ КРАСИТЕЛИ
Хииизарин зеленый, ализарнициаиин зеленый Г
(кислотный зеленый антрахиноновый)
В 1,5—2-литровой круглодонной колбе, снабженной мешалкой, термо-
метром и нисходящим холодильником, сплавляют 500 г n-тол ундина
с 60а (0,25 моля) хинизарина (см, стр, 216) и примерно при 80°при
перемешивании вносят смесь из 18 г б о р н о й к и с л о т ы, 30 г хл о ри-
стого олова и 16г мела, Нагревают массу до 110°, выдерживают час
при этой температуре, затем час при 120° и, наконец, еще 2 часа при 130°.
При этом отгоняется образующаяся при реакции вода и некоторое коли-,:
чество n-толуидина, Реакционную массу охлаждают до 70°, разбавляют
350 мл спирта и переливают после полного охлаждения в достаточно',
большой, хорошо закрывающийся сосуд, Колбу и мешалку смывают?
350 мл с п и р т а. Реакционную массу оставляют стоять закрытой на ночь;.
затем отсасывают образовавшийся осадок и размешивают его еще два<
раза со с п и р т о м (каждый раз примерно по 300 мл спирта). Осадок
отсасывают и промывают спиртом на нутч-фильтре до почти бесцветного
фильтрата.
Для очистки полученного таким образом сырого основания его кипя-
тят примерно с 1600 мл воды н 70 мл концентрированной соляной
кислоты, отсасывают горячим и промывают горячей водой до нейтраль-
ной реакции. Остаток обрабатывают таким же образом примерно 1600 мл-
воды и 40 мл едкого натра (уд. вес 1,38), После сушки получают,
85—90 г о с п овання красителя,
Для сульфирования 25 г основания красителя вносят при перемеши-1
вании в 250 г 10 %-ного олеума, Температура при этом поднимается до .
, 40—45и н для быстрейшего растворения в течение 2—3 час. ее поддержи-
вают на этом уровне. Оставляют стоять на сутки и затем смесь выливают
в 1 л воды, реакционный сосуд и мешалку также смывают 1 л воды,-
К разбавленному кислому раствору, нагретому до 50—60°, добавляют
250 г поваренной соли; при охлаждении краситель выкристаллизо-
вывается в хорошо фильтруемой форме. Выпавший краситель отсасывают,
отжимают и вновь растворяют примерно в 1,5 л горячей воды, нейтрали-
зуют примерно 15 г кальцинированной соды и фильтруют при 70—80°
через складчатый фильтр, который затем промывают горячей водой. В еще
горячий фильтрат добавляют 500 г поваренной соли. Образовав-
шийся краситель отсасывают, отжимают и сушат.
Выход около 50 а.
Ализарин-сафнрол Б н СЕ
(кислотный сниий антрахиноновый)
Ализа; ии-иафирол СЕ
Сульфирование и нитрование. 50 а хорошо высушенного
и тонко измельченного 1,5 -диоксиантрахинона или его смеси
<с 1,8-диоксиантрахиноном (см. стр. 213) вносят при перемешивании при
комнатной температуре в 188 а 20 %-ного олеума. Затем медленно в те-
чение часа температуру поднимают до 100° 'и выдерживают 2 часа при
100°, следующие 2 часа примерно при 105° и, наконец, столько же при
110°, пока проба ие будет полностью растворяться в холодной воде. Как
только это достигнуто, охлаждают до 25—30° и при этой температуре
добавляют 272 г моногидрата. Затем в течение 1 часа прибавляют
по каплям смесь 36 г азотной кислоты (уд. вес 1,50) . н 109 г
олеума (20% SO3) * причем реакционный сосуд охлаждают водой так,
чтобы температура не превышала 30°. После окончания прибавления тем-
пературу медленно повышают до 35° и выдерживают 2 часа при 35°, нагре-
вают следующие 2 часа до 55° и, наконец, еще 2 часа до 80°. Реакционную
* Смешивание следует производить осторожно: олеум вливают медленно при
перемешивании в сильно охлажденную азотную кислоту.
И. Красители
смесь охлаждают до 30° и при перемешивании так быстро, как только
позволяет ледообразование, выливают в 200 мл холодной воды.
Температура при этом поднимается до ИО—115° и образуется примерно
75%-ная серная кислота. Дают охладиться и после двухдневного стояния
отсасывают возможно сильнее па керамическом или стеклянном нутч-
фильтре (в случае его отсутствия отсасывают через асбест). Кислый оса-
док растворяют снова примерно в 1 л в о д ы и отфильтровывают от не-
большого остатка. Фильтрат должен быть совершенно прозрачным и не
должен мутнеть при стоянии.
Восстановление. Восстановление проводят концентрированным:
раствором сульфгндрата натрия NaSH (о получении его в лаборатории см.
стр. 106). Количество сульфгидрата, необходимое для реакции, опреде-
ляют следующим образом: пипеткой отбирают 25 мл отфильтрованного
раствора нитросоедииения в коническую колбу емкостью 750 мл, разбав-
ляют 350 мл горячей воды и затем нейтрализуют раствором соды до
появления неисчезающей красной окраски. При 60—70° из бюрсткн
прибавляют разбавленный раствор сульфгидрата натрия (10 Мл. концен-
трированного раствора NaSH разбавляют до 100 мл) до появления чистой
синей окраски раствора. Затем добавляют по 1 мл разбавленного рас-
твора сульфгндрата, пока бесцветный вытек высоленной пробы иа филь-
тровальной бумаге не будет давать ясного почернения (FeS) от раствора
сернокислого железа. По количеству израсходованного сульфгндрата
рассчитывают количество концентрированного раствора NaSH, необходи-
мое для восстановления всего количества нитрораствора.
Основное количество нитрораствора нейтрализуют при перемешива-
нии кальцинированной содой до иеисчезающей красной окраски, нагре-
вают раствор до 60—65° и при этой температуре при перемешивании мед-
ленно вливают концентрированный раствор NaSH; количество
концентрированного раствора рассчитывается иа основании данных пред-
варительного опыта. Еще 3 часа выдерживают при 60—65°, затем посте-
пенно добавляют поваренную соль (10% от объема жидкости) н
дают раствору остыть при перемешивании. Полностью выделившийся кра-
ситель отсасывают н промывают 15%-иым раствором поваренной соли до
бесцветного фильтрата, отжимают и сушат при 90°,
Выход около 105 г ализарин-сафирола Б.
Отщепление одной сульфогруппы. 25 г полученного,
как указано выше, красителя заливают 800 мл горячей воды, при 90°
приливают 46 г едкого натра (уд. вес 1,38) и нагревают до пол-
ного растворения красителя. Затем медленно прибавляют раствор 10 а
Na2S в 50 мл воды и перемешивают при 95—100° (общий объем
все время должен составлять около 1 л) до тех пор, пока отобранная
проба при разбавлении и подкислении серной кислотой будет давать уже
не синий, а виино-красный фильтрат. Затем тотчас прибавляют 200 г
поваренной соли и при перемешивании дают раствору охладиться.
Когда температура упадет до 30—40°, отсасывают выделившийся краси-
тель, промывают его 10%-иым раствором поваренной соли, отжимают
и сушат.
Выход около 22—24 а ализарин-сафирола СЕ (кислотного синего
антрахинонового).
Технические примечания. 1,5- и 1,8-Диоксиантрахиноны дают при при-
менении описанного метода синтеза столь сходные красители, что часто можно при-
менять их смесь. Для получения ализарин-сафирола СЕ не является необходимым
выделение ализарин-сафирола Б. Раствор, восстановленный еульфгидратим натрия,
можно прямо обрабатывать далее сернистым натрием.
в) Кубовые красители
2,87
в) КУБОВЫЕ КРАСИТЕЛИ
Индантрен синий PC (кубовый синий О)
из [3-аминоантрахинона
N. N’ -дигидро-1,2,1 2-аитрахиноназина
индантрен синий PC (кубовый синий -О)
50 г чистого 3 - а м и и, о а н*гр а х и н о н а (см, стр. 207) тщательно
перемешивают с 25 г уксуснокислого калия и 6 а азотно-
кислого калия. Небольшими порциями эту смесь вносят в нагретый
до 210° плав 150 г едкого кали и 20 мл воды. Плавление прово-
дят в никелевом тигле или в тигле из стали V2A. Мешалка должна быть
сделана из такого же материала. Железо нс следует применять. Смесь
вносят с такой скоростью, чтобы за 20 мин. прибавление было закончено.
После этого плав при постоянном хорошем перемешивании нагревают еще
5 мии. при 215—220° (но не выше). По окончании плавления немедленио
выливают плав в 1000 г льда, остаток плава в реакционном сосуде также
смывают небольшим количеством воды. Весь плав растворяют в воде; для
высаливания красителя добавляют 40 мл концентрированной серной
кислоты. Это имеет целью высаливание красителя. Затем водно-щелоч-
ной раствор нагревают до 60° и добавляют 30 г гидросульфита
натрия. Образуется синее лейкооснованис индантрона синего PC, кото-
рое нерастворимо в растворе имеющихся солей. После охлаждения рас-
твора осадок отфильтровывают иа нутч-фильтре через хлопчатобумажный
фильтр и промывают 2%-ным раствором едкого натра (па каждый литр
раствора добавляют 5 а гидросульфита натрия). Промывают дополучения
прозрачного светло-синего фильтрата. Осадок замешивают с 500 мл
воды при 60° и продувают воздух для окисления лейкосоедипения.
Окисление считается закопченным, если небольшая проба вещества не
растворяется в большом количестве воды. Краситель отфильтровывают и
отмывают водой. После сушки получают 22 г индантрена синего
PC. Краситель из синего гидросульфита ого куба окрашивает хлопокв ин-
тенсивный синий цвет; окраски отличаются исключительной с вето проч-
ностью. Прочность по отношению к хлору умеренная, опа может быть
улучшена хлорированием, например хлорированием сернокислого рас-
твора в присутствии небольшого количества нитрита натрия (марка
ГЦД — кубовый голубой О). Можно также хлорировать хлористым суль-
фурилом в нитробензоле (марка БЦС — кубовый голубой К) или хло-
ром в хлорсульфоиовой кислоте, иногда в присутствии переносчика, на-
пример хлорного железа или пятихлористой сурьмы.
Несмотря па многочисленные опыты, до сих пор не удалось повысить выход
кубового синего О: он составляет всего 45% от теории. Всегда образуются побочные
продукты, кик например ализарин и др. Во время плзидеиия часть красителя непре-
рывно разрушается.
Предложенныи недавно метод получения красителя* нагреванием 1-хлор-2-амнно-
антрахинона с йодистой медью (реакция Ульмана — Гольдберга) дает еще менее удо- -
влетворптельные результаты. ?
Чем чище применяемый амнноантрахннон, тем краситель прочнее к действию е
хлора. Можно также дополнительно очищать готовый краситель: для этого сгообра- '
батывают в концентрированной серной кислоте окислителем (например, двуокисью
марганца); все примеси при этом разрушаются. Очищенные подобным образом марки
поступают в продажу под названием индаптрена ярко-синего. Они немного ярче и )
прочнее по отношению к хлору, чем исходный краситель. Применение уксуснокислого
калия было впервые предложено в германском патенте**. В этом же патенте указы-
валось, что, кроме ацетата (или формиата), целесообразно прибавлять окислитель. :
Важно также вести плавление не слишком долго и прн не слишком высокой темпера-Г
туре. На производстве поэтому плавят небольшие количества, например одновременно ;
по 20 яг. Реакционный сосуд сделан цз чистого никеля или нержавеющей стали.
Индаитреи желтый ГК (кубовый желтый ЖХ)
1,5-Диаминоаитра- Ия да нт ре н желтый ГК
хинон (кубовый желтый ЖХ)
растворяют в.
1,5-Диамииоантрахинон (см, стр. 215) растворяют вл
20-кратном количестве 1,2-дихлорбензола (можно применять также j
нитробензол) и добавляют медленно при 140° рассчитанное количество!
хлористого бензоила. Выделяется хлористый водород (тяга!).^
После прекращения выделения хлористого водорода, которое продол<
жается примерно один час, дают смеси остыть и отфильтровывают)
краситель. Выход количественный. j
1,5-Дибевзоилами.но антрахинон окрашивает из слабощелочного гидросульфитного;
куба при 45° в желтый цвет с отличными свойствами [буква К (X) при названий!
красителя означает, что красителем следует красить не в очень горячем растворе^
в противном случае он разрушается. Холодное кубовое крашение!]***.
Аналогичным образом при бензилировании 1,4-диаминоантрахинона (стр. 209),]
получают индантрен красный 5 ГК, а из 1-амипоантрахицона (стр. 210)—;
алгольжелтыйВГ.
3. ИНДИГОИДНЫЕ КРАСИТЕЛИ
Индиго
1. Индиго по Гейману ****
а) Феннлглнцин-о-карбоновая кислота *****
/x/COONa /Х/СООН
] I + CfCHBCOON,i = ( 4- NaCI
'Ч'/'\М112 Х'/Х'ХН — CIL, — СООМа
137 г (1 моль) антраниловой кислоты (стр. 156) замеши-
вают с небольшим количеством воды и точно нейтрализуют примерна
* Герм, пат. 656944; Frdl., 24, 868- Dll Pont de Nemours.
** Герм. пат. 382178 (Pope W. J. и Scottish Dyes Ltd); Frdl., XlV, 871.
*** Hefti, Hefv.’ Chim. Acta, XlV, 1404; Schmidt R. E., герм. пат. 225232
(Frdl., IX, 1197).
**** Герм, пат. 56273 [Frdl., Ш, 281 (В. A. S. F.)]_
***** Герм. пат. 127178 jFrdl., VI, 538 (В. A. S. F.)].
120 г едкого натра (уд. вес 1,38), 94,5 г (1 моль) монохлор у кт
с у с н о и кнслоты, растворяют в 200 мл в о д ы н осторожно при пере-
мешивании точно нейтрализуют примерно 55 г кальцинированной соды.
Смешивают оба раствора и оставляют на 4 суток при 40°. Выкристалли-
зовавшуюся моноиатриевую соль фенилглицин-о-карбоновой кислоты от-
сасывают на иутч-фидьтре, промывают небольшим количеством холодной
воды и сушат до постоянного веса в паровом сушильном шкафу.
Выход примерно 75% от теории.
б) Индокснлкарбоновая кислота
/х/СООн / С(\^
! \ 4- СаО = | | CH —COONa + Са(ОН)3
СН2 — COONa X/XNH/
В шаровой мельнице, в условиях, исключающих проникновение влаги,
перемалывают 25 г едкого натра, полностью обезвоженного плавле-
нием в железном тигле, 25 г едкого кали, обезвоженного таким же
способом, 7,5 г едкой извести, обезвоженной прокаливанием в фар-
форовом тнгле (следует избегать спекания) и 25 а хорошо высушенной
мононатриевой соли фепилглицин-о-карбоновой кис-
лоты. Полученную таким образом однородную смесь помещают в аппа-
рат для запекания под вакуумом (см. рис. 30, стр. 162) и нагревают
в графитовой бане в вакууме сначала 2 часа при 150°, затем 6 час, при
230^235°. Образуется твердая масса, равномерно окрашенная в желто-
кориДцевый цвет.
' в) И п л н г о
/\/СО\ /Х/С°Х /NHX/^
‘2 \ \ CH —COONa + G2= | | С = С I i — 2NaHCO3
X/XNh/ Х/Xnh/ Xqo/Х/
Плав, получение которого описано выше (б), растворяют в 2 л в о д ы
и при 80° пропускают сильную струю воздуха до тех пор, пока от-
фильтрованная проба при встряхивании с воздухом не выделяет больше
индиго. Выпавший краситель отсасывают, промывают водой, кипятят
с разбавленной соляной кислотой для полного удаления извести, снова
отфильтровывают, тщательно промывают водой и сушат в паровом су-
шильном шкафу.
Выход 12—12,5 а, или 80—82% от теории, считая на фенилглицин-
о-карбоповую кислоту.
Приведенный выше метод получения индиго, открытый ГеГ|Маном и разработан-
ный Баденским анилиновым и содовым заводом, явился первым методом, применен-
ным с успехом в больших масштабах. Долгое время Этот метод применялся наряду
с другим методом фирмы «Deutsche Gold- und Silherscheideanstalf», согласно кото-
рому фенилглицин сплавляют с едким кали и едким натром с добавкой амида натрия,
и лишь в последнее время был вытеснен последним,
2. Индиго по 7. Зандмейеру *
Несмотря на то, что в настоящее время синтез индиго по Занд-
мейеру не применяется, этот синтез является столь интересным примером
совместной работы ученых и техников, что он должен найти отражение
в этой книге.
* Sandmever Т., Zeitschr, Farben- u. Textilchem, 1903, Heft 7, S. 129; Helv.
Chjm. Acta, 2, 2,34 (1919).
19 Зак. 1905. Г. Э. Фирц-Давид Л. Блаиже
Прежде чем описывать отдельные операции, следует разобрать хи
мизм процессов.
а)
Нагреванием анилина с CS? получают тиокарбапплид А.
Гофмана
б)
Тиокарбанилид обессеривается
при
помощи
основного
глекислого
свинца и одновременно присоединяется синильная кислота, в
чего образуется гидроциан карбодифенил имид Лаубенгепмера
резу
льтате
НО’-Н3ч
-------->
+1ES
У
Г идрлцианкарбодифепмиипц
Т. пл. т =
в) Гидроцианкарбодифепилимид при действии желтого сернистого
аммония превращается в тиооксамин дифенил амидин, или «тиоамид»
S = С
I
NHm
г) «Тиоамид» под действием концентрированной серной кислоты
гладко превращается в а-изатина пил ид.
а- Изатнгта^г'-ИгД
Т. пл. 124”
д) а-Изатиналилид может быть превращен в индиго различными'
способами. Его восстанавливают в спиртовой среде при помощи разбав-1
лепного раствора сернистого аммония или превращают в а-тиоизатий,;
который при действии щелочи тотчас дает индиго *. В технике приме-!
иялся последний способ. \
/Х| -—-NH
I । -j- Анилин
II С =S
кЛ'|1<Я|ЗП711ГГ
* Формулы индиго и его производных па стр. 290, 292—295 приведены в соответ- '
ствин с. оригиналом, в ^uc-форме (см. Котай И. М,, Химия красителей, М. 1956,
стр. 450 сл.).— Прим. ред. >
a) 1 иокарба пилил
186 г чистого а н и л ина в 100 г чистого сероуглерода кипятят
с обратным холодильником до прекращения выделения сероводорода (это
продолжается около 2 диен). Затем температуру масляной бани повы-
шают до 160° и отгоняют избыток сероуглерода. Расплавленный тиокарб-
анилид выливают на жестяным лист и после охлаждения измельчают
в порошок. Неочищенный продукт реакции целесообразно кристаллизо-
вать из спирта, при этом получают блестящие кристаллы с т. пл. 151°.
Выход составляет около 200 г очищенного продукта. (В технике боль-
шей частью отказываются от перекристаллизации тлокарбапилида, однако
в этом случае при дальнейших операциях всегда образуются небольшие
количества маслообразного побочного продукта, получение которого
можно избежать путем предварительной очистки.)
б) Гпдроцианкарбодифенилимид
350 г азотнокислого свинца растворяют в 1 л горячей
воды и при 95° осторожно осаждают 120 г кальцинированной соды;
выпавший осадок тщательно промывают водой. Влажный основной
углекислый свинец смешивают с 600 а 90%-ного спирта в двух-
литровой колбе с мешалйой и обратным холодильником (см. рис. 6).
Перемешивание продолжают до тех пор, цока не образуется совершенно
однородная паста. Затем быстро добавляют 228 г (1 моль) тонко измель-
ченного ти о к а р б а п и л и да и при 25° добавляют около 60 г техниче-
ского цианистого натрия (1,3 моля) *. При энергичном перемеши-
вании в течение часа повышают температуру до 77° и отфильтровывают
небольшую пробу. Бесцветный раствор не должен окрашивать основной
углекислый свинец (небольшая проба на кончике ножа) в черный цвет.
Если, одпако, появляется черная окраска, то нагревают еще час и снова
отбирают пробу. Если и в этом случае не произойдет полного обессерива-
ния, то добавляют еще немного углекислого свинца и цианистого натрия.
Если, однако, количества необходимых реагентов были правильно рассчи-
таны, то дополнительного прибавления не потребуется.
После того как исчезнет реакция на серу, смесь нагревают до кипения
и отфильтровывают горячий раствор, Остаток еще дважды экстрагируют
!/j л спирта и дают гидроцианкарбо дифенил имиду выкристаллизо-
ваться. Первая фракция совершенно чиста; опа весит примерно 160 а.
После упаривания м а точного раствора выделяется еще около 40 а почти
чистого продукта. Выход составляет около 98%. Гидроцианкарбодифе-
нилимид кристаллизуется в виде красивых желтоватых призм, т. пл. 137°.
Маточные растворы следует обрабатывать очень осторожно, так как они
содержат синильную кислоту.
в) «Т и о а м и д»
Присоединение сероводорода к гидроцианкарбодифенил имиду проис-
ходит очень легко, если последний топко измельчен, поэтому на вальцовой
мельнице или просеиванием веществу придают необходимую степень дис-
персности. 200 г г и др о ци а нк ар бо ди ф е п и я и м и д а эмульгируют
при 35° и энергичном перемешивании с 500 г желтого раствора сер-
нистого аммония**. Раствор сернистого аммония готовят сле-
дующим образом: в смесь 25 г измельченной серы и 460 г 20%-ного
* Следует определять содержание в пианистом натрии HCN.
'* Добавлением равного объема спирта реакция чрезвычайно ускоряется.
19*-
раствора аммиака пропускают 35 а сероводорода. Если гидро-
цианкарбодифенилимид был достаточно тонко измельчен, то сероводород
присоединяется количественно в течение 12 час. Реакция считается окон*,
чеиной, если промытая проба растворяется в разбавленной соляной?
кислоте. Вещество отфильтровывают и тщательно промывают водой^
Продукт достаточно чист дли дальнейшей переработки. 1
Выход около 220 а. Продукт кристаллизуется из спирта в желтых^
призмах с т. пл. 162°.
г)а-Изатинаиилид V
Замыкание цикла с образованием производных изатина происходи!
в строго определенных условиях. Необходимо работать с горячей серной
кислотой.
В течение ][/4 часа вносят 200 г чистого, тонко измельченного сухог^
«тиоамида» в 800 г 94%-ной серной кислоты (уд. вес 1,84)|
температура должна быть точно 94°. Смесь разогревается довольно сильа^
и ее следует охлаждать. После того как прибавлено все количество тиба
амида, нагревают в течение 1 часа прн 106—108°; за это время Д>екрд£
щается выделение SO2. Раствор охлаждают до 20° и соединение непосреш
ственно переводят в солянокислый .а-изатинанилнд, для чего раствор пр|
непрерывном перемешивании выливают тонкой струей в смесь 1 л во дь|
2 да л ь д а и 500 гповаренной соли. Солянокислая соль изатинашй
лида выделяется в виде светлого, красно-коричневого осадка, смешанной!
с тоикоизмельчепной серой. .
Если желательно получить чистый анилид, то осадок фильтруют Я
тщательно промывают 20%-ным раствором поваренной соли. Затем с о ля
освобожденную от кислоты, перемешивают с водой и с раЦ
бавлеиным раствором соды до тех пор, пока реакции ие станет ели
бощелочиой. Осадок, состоящий из анилида и серы, отфильтровывают!
хорошо промывают и экстрагируют сухую смесь холодным сероугл.и
родом. В заключение анилид перекристаллизовывают из горячем
спирта. Он получается в виде темных игол с т. пл. 126°. ВыхсДЯ
200 г тиоамида составляет до 150 г чистого вещества. При кипячен™
с небольшим избытком разбавленной серной кислоты анилидная групий
отщепляется в виде анилина н прямо выпадает чистый изатин, т. пл. 200-Л
201°. Он хорошо растворим в горячей воде и перекристаллизовывается фМ
нее. Изатин применяют для получения ценных кубовых красителей.
Из ос-изатипанилида ДЯ
Из изатина
II
/С\
СО S
СО(р) СО
Трибромпроизводяое = циба
фиолетовый Б
Дибромдроиз водное циба
фиолетовый 3 Б
Монобромпроизводное = циба
серый Г
Тиоиндиго алый Р
циба краопый Г =
дцбромдроизводное
Еще более важными являются
кубовые красители, получаемые непосредствен^
Конденсацией а-изатинанилида с [J-окситионафтеном. Г. Энги впервые заметил, ЧЯ
изатин и его анилид при конденсации дают совершенно различные красители. П₽Я
Индигозоль 0,4 Б (броминдигозоль)
293
конденсациях изатина обменивается кислород, находящийся в ^-положении, в то время
как аннлид странным образом отщепляет анилин, а затем образует красители, пред-
ставляющие собой продукты конденсации в а-положении. Продукты а-конденсации
являются значительно более пенными, чем их изомеры (см. формулу на стр, 292).
д),а- Тиоизатин и индиго
Для получения иидиго из сернокислого раствора изатинанилида нет
необходимости выделять чистый изатин ан ил ид или его солянокислую соль
и можно получить тиоизатин непосредственно из раствора. Пропусканием
сероводорода в раствор 45 г едкого натра в 150 мл воды
готовят раствор сульфгидрата натрия. Этот раствор смешивают с серно-
кислым раствором нзатниаиилида, полученного из 200 г тиоамида. Сме-
шивание производят одновременным выливанием обоих растворов в 6 л
ледяной воды. При этом все время должен быть небольшой, но до-
статочно отчетливый избыток сероводорода. Восстановление продолжается
около 7э часа, тиоизатин выделиется в виде объемистого коричневого
осадка. В растворе остаемся сернокислый анилин. Тиоизатии отфиль-
тровывают лишь после того, как небольшая проба отфильтрованного рас-
твора не дает осадка с сернистым натрием; обычно это имеет место при-
мерно через час. Затем промывают до тех пор, пока удельный вес маточ-
ного раствора не станет равным 1,007. Промытый таким образом осадок
размешивают с 3 л воды и добавляют концентрированный раствор соды
до тех пор, пока не будет достигнута не исчезающая силыющел очная реак-
ция. Требуется около 30 г соды. Образование иидиго происходит очень
быстро; целесообразно нагревать при 60° в течение еще 1 часа и оставить
па ночь при перемешивании. На следующий день индиго и серу отфиль-
тровывают, хорошо промывают и сушат при 80°. Сухой краситель экстра-
гируют двойным по весу количеством сероуглерода и полу-
чают 80 г чистого индиго.
Технические примечания. Синтез индиго по Зандмейеру протекает не-
обычайно гладко. В расчете на анилин красителя получают около 80% от теорети-
чески возможного количества, В начале столетия непродолжительное время по этому
способу работал завод Гейги; себестоимость 100%-ного товара составляла 10,80 фран-
ков за 1 кг. Индиго, получаемое по методу Зандмейера, переводится в куб легче, чем
какой-либо другой синтетический продукт, и оно тотчас нашло применение в краше-
нии, Весь процесс получения индиго удается проводить без расхода спирта, так как
оказалось, что все вещества при достаточно тонком измельчении хорошо реагируют
в водной среде. Основное затруднение представляет не синильная кислота, а серо-
водород, который является опасным промышленным ядом, так как уже через корот-
кое время притупляется чувствительность к его запаху. Сернистый свинец разлагают
концентрированной соляной кислотой на PbCls + H3S, которые возвращаются в произ-
водство. Метод, предложенный в Германии фирмой «Deutsche Gold- und Silberscheidean-
stalt», вскоре вытеснил этот многообещавший метод. По последнему методу, в резуль-
тате новейших улучшений выход повысился до 85%, так что с ним не представлялось
возможным конкурировать.
Индигозоль 04 Б* (броминдигозоль)
I NH NH I
Вг Вг
Циба голубой 2Б (бромитгдиго)
* Герм. пат. 563958 (1925), Scottish D es Ltd (Frdl,, 18, 1513).
И, Красители
В литровую пятигорлую колбу со стеклянной мешалкой (гидра вл и*
веский затвор с концентрированной серной кислотой) термометром, ка-
пельной воронкой и обратным холодильником (воронка и холодильник
с поглощающими влагу трубками) помещают 325 мл чистого совершенно
сухого пиридина и при энергичном перемешивании и интенсивном охла-:
ждсиии добавляют 47,5 мл хлорсудьфоповой кислоты с такой скоростью;
чтобы температура ие превышала 25°. Затем реакционную смесь охла-
ждают до 0° и при этой температуре вносят хорошо перемешанную смесь-
57,5 г циба голубого 2Б (броминдиго) и 80 г восстановленного железа/.
После окончания внесения смеси прекращают охлаждение и перемети-.,
вают в течение ’Л часа, а затем осторожно нагревают до 40°, Теплоты^
реакции достаточно, чтобы поддержать температуру некоторое время на?
этом уровне; когда оиа начнет падать, нагревают до 55° и выдерживают?
при этой температуре 20 мин. Затем при перемешивании охлаждают при*)
мерно до 20° и выливают смесь в 1,5 л воды. Оставляют па ночь стоять
при 0°, затем отсасывают пиридиновую соль лейкосернокислого эфира, ксн
торая выделяется почти полностью, на стеклянном нутч-фильтре и промы^
вают холодной водой.
Остаток растворяют при нагревании в 1 л воды с прибавлением’
125 мл едкого иатря (уд, вес 1,38), железо отфильтровывают и про-;
мывают два раза по 175 мл горячей воды- Коричневый фильтра^
оставляют на ночь при 0°, затем отсасывают выкристаллизовавшийся ин-
дигозоль и сушат его при 60° в сушильном шкафу. Получают около 65 г-
примерно 90 %-ного нндигозоля. Из маточного раствора после пасыще;’
ния поваренной солью .можно выделить небольшое количество менее чиу
стого продукта. -
Тел-ми ческ ие примеч ани я. Как это впервые установил М. Бадер, лейко-:
соединенна индиго ц многочисленных других кубовых красителей могут быть превра-
щены действием хлорсульфоновой кислоты в присутствии пиридина или других тре-
тичных оснований в серпокислые эфиры, которые в отличие от самих лейкосоединенкй
устойчивы на воздухе в виде щелочных солей; в продажу они поступают под наава-,
пнем п ют пг о и о л е н (и кубозодей), Ими можно красить, или печатать и на волокне'
омылением и огип’ЛСггнем снова превратить в краситель. Для их применения не тре-
Тиоиндиго
295
буется приготовления куба, что при печатании является существенным упроще-
нием. Метод получения индпгозолей в деталях может подвергаться многочисленным
изменениям и для каждого кубового красителя должен быть найден наиболее прием-
лемый.
Известный интерес представляет и более новый метод, который позволяет полу-
чать ипдигозоль в воднощелочном кубе. Согласно американским патентам*, кубовые
красители вначале переводятся в куб-действием гидросульфита натрия и едкого натра
в отсутствие воздуха, затем лейкосоединення превращают в эфиры действием про-
дуктов присоединения серного ангидрида к третичным аминам.
R'ONn -l R3N — SOs = R' •• OSO3Na -p Ra\.
В качестве таких продуктов присоединения могут применяться
(CH3)3N-SO3
Из тг нал |< илам tiua
СН.,-СН2х ,С2Н-
о/ >n+<
хсн3—сн/ xso,;
zCH3-CHSx ,С3НВ
CH /
ХСН2— СИ/ 4SOf
Из
Нз
К-МК>1.тп>тп»1:ид1[ па
Тиоиндиго
S S
;/Х/ ЧСН —> ХСН2
< - N 1 ! !
I у1___СОН 1 ।--со
X / х / --------1 ----
-СН„СООН
I I
^/^СООН
/\/SH
I i
\/\соон
Тиося.ч ициловая
кислота
Получение о-к а р б о к с и-ф с и и л т и о г л и ко лево й
кислоты (О-к и слоты)
Диазотируют антраниловую кислоту; соль диазония действием рас-
твора полисульфида натрия превращают в смесь тиосалицилоЛй и ди тис-
салициловой кислот. Для этого вначале готовят три следующих рас-
твора.
1. 68,5 г (0,5 моля) антраниловой кнслоты, 500 мл воды,
78,5 г соляной кислоты (уд. нес 1.17).
П. 34,5 г 100%-ного нигрита натрия,
75 мл воды,
* Пат. США 2396582 и 2403226.
а. красители
III. 85 г сернистого натрия кристаллического (NaaS * 9Н3О),
11,1 г серного цвета,
100 мл воды.
Раствор I получают внесением антраниловой кислоты в водный рас
твор соляной кислоты, раствор III получают кипячением серы в раствррг
сернистого натрия. После того как раствор III станет прозрачным, добав
ляют в пего еще 12аедкогонатра (уд. вес 1,38). Раствор I охлаждаю1
в литровом стакане до 0° и при энергичном механическом перемешивани:
медленно приливают раствор II, температура при этом поднимается до 5*
Йодокрахмальиая бумажка слегка окрашивается в синий цвет. Раствор II
помещают в четырехлитровый стакан, в котором находятся 500 г л, ьда 1
при энергичном перемешивании по порциям в течение 10 мин. в щелочно!
раствор полисульфида прибавляют раствор соли диазония. Смесь сг^ьи
пенится, и образуется желтый осадок. Размешивают еще 2 часа, а зате!
подкисляют 90 а соляной кислоты (уд. вес 1,17) до кислой реакцц
по конго. Выпавшие тио- и дитиосалициловые кислоты отсасывают н
иутч-фильтре и отмывают от минеральной кислоты. Для дальнейше
обработки осадка необходимо вначале восстановить дитиосалицнловуя
кислоту до тиосалициловой. К смеси кислот, помещенной в двухлитровый
фарфоровый стакан, содержащий 500 мл воды, постепенно при перем^
шивании добавляют 25,8 г со ды и нагревают смесь до кипения. Реакций
должна быть кислой иа лакмус. Затем восстанавливают 100 г ж е л е з i
(применяемого для восстановления по Бешану, см. стр. 75) в тече
иие 2 час. при 95°. По мере испарения время от времени добавляю,
воду. Для определения конца восстановлении отбирают небольшую
пробу, сильно ее подщелачивают, кипятят в течение короткого временй
фильтруют и фильтрат подкисляют соляной кислотой. Выделившуюся
кислоту отсасывают, промывают и слегка подсушивают па глиняной та
релке. Кислота должна быть хорошо растворима в холодном спирте (ие
восстановленная ди тиосалицилов а я кислота очень плохо растворима Я
спирте). ?
Если проба дает положительный результат, то всю реакционную
смесь подщелачивают добавлением 60 г раствора едкого натр;
(уд. вес 1,38) и затем при 95° приливают раствор 52 г моиохлор
у ксу си ой к ис л оты и 29г соды в 150 мл в о д ы. Жидкость должн?
все время иметь щелочную реакцию; ее выдерживают еще ’Д часа npi
90°, затем дают охладиться и отстояться. После отсасывания остатк!
железа его промывают сильно разбавленным раствором едкого натра 1
при хорошем охлаждении осаждают образовавшуюся О-кислоту 150 г с о
ля и ой кислоты (уд. вес .1,17). Чем холоднее раствор, тем красив©
кристаллы выделяющейся кислоты. Рекомендуется также проводить оса-
ждеиие возможно медленнее и при энергичном механическом перемешива*
иии. О-кислоту отсасывают на иутч-фильтре, отмывают от кислоты и су*
шат при 80°.
Выход около 85 г (80% от теории).
Получение 3-окситиоиафтеиа
50 г хорошо высушенной О-кислоты и 100 а твердого технического
едкого иатра* перемалывают возможно тонко в течение 24 час, в ша-
ровой мельнице. Порошкообразную смесь помещают в аппарат для запе-
кания под вакуумом (см, рис. 30, стр. 162); после эвакуирования масля-
ную баню аппарата в течение 2—3 час. возможно равномерно нагревают
Примулин (по Грану)
297
до 205°, При этой температуре реакционную массу выдерживают 24 часа.
По охлаждении продукт запекания вынимают. Он равномерно окрашен
в желтый цвет и весит примерно 135 г.
Окисление плава в тиоиндиго
Плав растворяют при перемешивании в 1 л в о д ы и приливают 50 г
концентрированной серной кислоты, которая предварительно не-
сколько разбавляется водой. Затем добавляют 40 г серного цвета и
перемешивают 3 часа при 95°, причем время от времени опять добавляют
иемиого воды. Образовавшемуся красителю дают осесть, отсасывают на
иутч-фильтре и промывают холодной водой. Сушат при температуре
около 80°.
Выход 25 г (70%, считая иа О-кислоту); продукт содержит еще
серу; для очистки он может быть вновь выделен из куба.
I
И. СЕРНИСТЫЕ ПЛАВЫ
Примулин (по Грииу)
Хлораминовый желтый ФФ (нафтамин желтый НН)
и тиазоловый желтый
!
,,Бис“-дегидротио-л -толуидин или
основание примулина
При реакции с ароматическими аминами сера действует большей
частью замещающим образом. При этом два ароматических ядра атомом
серы связываются в тиосоединеиия. Однако всегда образуются раз-
личные вещества; получить только один определенный продукт реакции
совершенно невозможно. Точно так же при действии серы на «-толуидин
вопреки различным противоречивым данным образуются четыре легко
распознаваемых продукта реакции: небольшое количество иепрореагиро-
вавшего n-тюлуидина, затем тиотолуидин, дегидротиотолуиДЙи и бис-де-
гидротиотолуидин. Приведенные выше формулы разъясняют сказанное.
В котелке с мешалкой и обратным холодильником (рис. 36 а и б)
нагревают до 180° 214 г (2 моля) «-толуидина, 140 г порошко-
образной серы (ие серного цвета!) н 2 з кальцинированной
соды1*. Выделяющийся сероводород поглощают раствором едкого натра
* Соду добавляют для нейтрализации кислых примесей, всегда содержащихся
в сере. Если не добавлять соду, то плав примулина получается почти всегда темным,
а иногда даже черным,
и, красители
или кусками влажного едкого натра, находящимися в колонке. При-
мерно через 8 час. выделение сероводорода ослабевает, температуру мед- 3
ленпо поднимают до 220° и выдерживают при этой температуре в течение
5 час, Сероводород больше почти не
выделяется, и плав выливают на
плоский жестяной противень, где он
застывает в светло-желтую кристал-
лическую лепешку.
Выход 325 г.
медной масляной баней для получе-
Разделение плава
Метод 1, Охладившийся твер-
дый плав измельчают в топкий по-
рошок и тщательно смешивают с
кальцинированной содой (3 % отве-
са плава), чтобы избежать образова-
ния комков при сульфировании. 100 г
плава вносят в 300 г м о н о г и д р а-
т а, причем температура может под-
ниматься как угодно. После того как
все растворено, что происходит при-
мерно через 1 час, раствор охлаж-
дают до 25° и при постоянном пере-
Р и с. 33. Железный плавильный котелок
иля иримулипа, иидиго, шелочпыл планов и т. д. Емкость около 450 мл,
мешивании и очень хорошем охлаждении прибавляют по каплям в течение :
часа 200 г олеума (66% SO3); температура должна быть ниже 30°. j
После пятичасового перемешивания прн 30° температуру повышают до 40®'
и нагревают до полного растворения небольшой пробы сульфомассы в раз- ;
бавленпом растворе аммиака. Полное растворение наступает большей .<
частью через 10 час., однако рекомендуется операцию продолжить, так
как для облегчения последующего фильтрования масса должна быть у
полностью просульфироваца. Сульфомассу выливают в смесь 500 г льда
П 500 мл водь: и через 12 час. отфильтровывают. Сульфокислота очень
хорошо промывается холодной водой; при этом большая часть толуидин-
сульфокислоты и тиотолундинсульфокислоты удаляется. После того как
промывные воды будут давать лишь слабо минсралыюкислую реакцию,
осадок растворяют примерно в смеси 50 г аммиака (20% NH3) и
800 .ял воды; раствор доводят при. температуре 80° до объема 1200 лм.
Трудно растворимая аммонийная соль дегидротиотолуидинсульфокислоты
полностью выделяется в течение двух суток, ее отфильтровывают и про-
мывают небольшим количеством 5%-пого раствора аммиака. Примулин,
содержащийся в маточном растворе, осаждается 15%-ным раствором по-
варенной соли при кипячении,
Выход сухой аммоний пой соли составляет примерно 25 г приму-
лина — около 80 г концентрированного продукта.
Метод 2, Измельченный в тонкий порошок плав экстрагируют спиртом (не
слабее чем 90%-ным), При этом растворяются только толуидин, тиотолуидин и соб-
ственно дегидротио толуидин, *в то время как чистое основание примулина остается
нерастворенным. Спиртовый экстракт упаривают досуха н нагреванием сухого остатка
прн 250° отгоняют толуидин и часть тиотолуидина. Полученные продукты сульфируют
25%-ным олеумом.
Метод 3. Сульфирование проводят точно так, как приведено выше, Отмытую
сульфокислоту растворяют при 80° в двадцатикратном объеме воды, содержащей
необходимое количество раствора едкого натра, затем добавляют поваренную соль
до образования 8%-ного .раствора соли и при 75s отфильтровывают примулин. Де-
Гндротиотолуидин в виде легко растворимой натриевой солн сульфокислоты остается
в растворе, из которого он выделяется высаливанием.
\ Примулин, найденный Грином, был первым синтетическим желтым
прямым красителем, который после диазотирования на волокне и проявле-
ния\фенолами и аминами давал прочные к стирке окраски. С 3-иафтолом
оп дает примулин красный — краситель, который раньше применялся
в огромных количествах. Прочность к стирке хороша, по светопрочность
неудовлетворительна; он ие дает также белой вытравки, а только жел-
тую, так как основание примулина устойчиво к любому вытравляющему
средству.
. Нафтамин желтый НН (также ФФ) и тиазоловый желтый
Дсгидротнотолуидин сначала являлся побочным продуктом, и когда
началось производство примулина, его сплавлением с серой превращали
в основание примулина. Теперь положение совершенно изменилось — не
имевший ранее значения дегидротиотолуидин стал основным продуктом.
Его можно выделить из сырого глава перегонкой в высоком вакууме *,
Однако невозможно вести цлав таким образом, чтобы образовывался
только од Ии дегидротнотолуидип; противоположные утверждения яв-
ляются неправильными. В настоящее времи примулин превратился в по-
бочный продукт. Из дегидротиотолуидина получают разнообразные краси-
тели. Свободное основание или его сульфокислоту диазотируют и диазосо-
единение сочетают с различными нафтолсульфокислотами, нМгример с так
называемой а-кнелотой, 1,3,8-нафтолдисульфокислотой; полученный кра-
ситель отличается большой чистотой оттенка и при вытравке дает безукориз-
ненную бель. Подобные прямые красные красители поступают в продажу
под различными названиями и большей частью обозначаются как краси-
тели типа эрика красного* **. Наряду с настоящими азокрасителями
Brunner W., Диссертация, Ziirich, 1943 г.
** Эрика Ц (AG1A) является комби нацией дегндротиоксилидина и е-кислоты.
С 1,3,6-нафто л ди сульфокислотой получают сходный краситель.
duo
/Л Красители
из дегидротиотолуидина получают еще два важных желтых красителя;.)
Один из этих красителей — нафтамии желтый НН (также хлор*й
аминовый желтый) фирмы «Калле» — получают окислением дегидротното*т
луидинсульфокислоты хлорноватистокислым натрием, другой краситель —)
тиазоловый желтый, или желтый Клейтона, — образуется
при взаимодействии диазосоедииения дстидротиотолуидиисульфокислоты1;
со второй молекулой того же соединения. При этом образуется описывае-J
мое ниже диазоамииосоедннсние.
Следует, кроме того, отметить, что прн алкилировании «примулинов»'
образуются красивые желтые основные, а также кислотные красители,
которые, однако, не имеют большого значения (тиофлавии Т и С).
Нафтамии желтый НН
Дегидротиотолуидинсульфокислота —N^N-дегияротиотолуидинсульфокнслота
67,4 а (0,2 моля), что соответствует 14а нитрита натрия чистой
100 % -ной аммонийной соли дегндротиотолуидинсульУ
фоки с лоты молекулярного веса 337, растворяют точно в 8,2
100%-ного едкого натра и 300 мл воды; раствор кипятят для удален
ния аммиака. Следы аммиака мешают окислению. Примерно через ча$
запах аммиака исчезает, раствор доводят до объема 500 мл и темпера?)
туры 20° и смешивают его с 10,5 a HOCI (в виде примерно 5%-иого рас^
твора хлорноватистокислого иатрин). Содержание аммоний*]
ной соли и гипохлорита должно быть точно определено титрованием;)
Температура поднимается примерно на 4°. Через час небольшую пробу^
нагревают в пробирке и высаливают. Осадок должен быть ораижевог^
цвета и йодокрахмальиая бумажка должна давать четкую реакцию иа^
хлорноватистую кислоту. В противном случае добавлнют еще немного пК
похлорита. Кипятят через 5 час., осаждают 15% поваренной солЯ
и фильтруют, у
Выход составляет примерно 75 г концентрированного красителя. <
Нафтамин желтый НН является наиболее светопрочным желтым красителем для
хлопка; он вполне устойчив к действию хлора. Поэтому он применяется в большн®!
количествах, особенно в США, где при стирке всегда употребляют отбеливающий
вещества. Он не такой чистый, как хризофенин, и уступает последнему по интенсив^
ности окраски. Л
Тиазоловый желтый, или желтый Клейтона
Дегмдротиотолуидинсульфокислота —N2—NH-дегидротиотолуидинсульфокислота.
Д егид р оти от о л у и д и нс у л ьф о к и с л от у в количестве, со;
ответствующем 14 г нитрита натрия, растворяют в 25 а 30%-ноге
едкого натра, разделяют раствор пополам и одну половину под
кисляют 25 мл концентрированной соляной кислоты. Этот раствор
диазотируют прн 10° в течение 2 час. 7 г 100%-ного иитрнта натри?
Оранжево-желтое дназосоединенне смешивают затем со второй поло
виной раствора сульфокислоты, к которой предварительно добавляй!
25 г кальцинированной соды в небольшом количестве воды й
25 мл концентрированного раствора аммиака. Сочетание,
проводит при 4—5°; целесообразно, чтобы растворы были по возможно-;
сти более концентрированными. Через 2 часа нагревают до 30° и остав*;
ляют стоять иа ночь. На другой день нагревают до 80° и высаливают
20% поваренной соли.
Выход концентрированного красителя около 85 г.
Тиазоловый желтый (желтый Клейтона, мимоза и т. д.) в противо-
положность хлораминовому желтому является наиболее непрочным из
всех желтых красителей, и удивительно, что такой продукт мог вообще
найти применение.
При действии едкого натра тиазоловый желтый меняет окраску —
нз чисто желтого он превращается в ярко-красный, поэтому его приме-
няют как реактив иа щелочь (см. о тиазоловой бумажке стр. 349). Как
уже упоминалось, он чрезвычайно непрочен, однако обладает очень чи-
стым оттенком и большой красящей способностью и поэтому все же,
к сожалению, применяется для крашенин дешевых текстильных товаров,
При сульфировании оснований красителей типа примулина «методом запекания»
образуются сульфокислоты, дающие более светопрочные азокрасители, чем азокраси-
тели, полученные из обычных сульфокислот. Предполагают, что при «запекании»
сульфогруппа вступает в орто-положение к аминогруппе, благодаря чему значительно
повышается светопрочность окрасок. На эту зависимость было уже указано при опи-
сании пиразолоновых красителей.
Технические примечания. Прпмулиновый плав проводят в котлах, снаб-
женных обратным холодильником, нагреваемых на масляной бане. В рубашку обрат-
ного холодильника пропускается теплая вода, чтобы возгоняющийся н-толуидин не
забивал трубки. Сероводород поглощается раствором едкого натра и используется
для восстановления. В самом начале производства сероводород сжигали в топке
котла, но это является нерациональным и неприятным для окружающего населения.
Экстракция спиртом проводится в железных котлах с ситчатым дном и фильтром,
причем испаряющийся спирт все время возвращается обратно, подобно тому, как это
происходит в аппарате Сокслета. После отгонки спирта из экстракта массу вновь
нагревают на масляной бане до 240°, до полной отгонки толуидина.
Аналогичные продукты можно получить из самого примулина, (хлораминовый
Желтый, тиазоловый желтый и т. д.), но все эти красители не находят применения,
так как они намного тупее по оттенку, краснее и слабее по концентрации только что
описанных желтых.
Сернистый черный Т из динитрохлорбеизола
С1 он
Краситель
неизвестного
строения
ли,
В стеклянном или железном сосуде * при перемешивании нагревают
До 90°- смесь 120 мл в од ы и 70 г 2,4-д и и и т р ох л орбензол а и при-
бавляют по каплям в течение 2 час. 80 г 35%-ного раствора едкого
натра, причем реакция ие должна быть сильнощелочиой. Нагревание
продолжают до полного растворения пробы в воде, в случае необходи-
мости добавляют еще едкого натра. Суспензию дииитрофенолята
натрия охлаждают до 45° и добавляют раствор 50 г серы в 125 мл
воды и 125 г кристаллического сернистого' натрия. Затем темпе-
ратуру повышают до 60°, общий объем должен составлять 600 мл. На-
гревают осторожно на водяной бане до 80°, а затем в течение 27а час,
на масляной бане до 105°. Без перемешивания кипятят с обратным холо-
дильником в течение 30 час. и разбавляют затем 600 мл воды. При 60°
продувают в жидкость воздух до выпадения красителя, который от-
фильтровывают и сушат при 70°. Вых о д составляет примерно 70 г.
Например, в котле, изображенном па рис. 36а и б.
Красителем окрашивают хлопок при кипячении в присутствии взятого
в четырехкратном от веса красителя количестве кристаллического сер- я
нистого натрия и добавляя поваренную соль (100% от веса хлопка). У
Технические примечания. Сернистый черный Т является наиболее рас- J
пространенным из всех применяемых черных сернистых красителей и отличается света- д
прочностью и прочностью к стирке. В производстве на загрузку берут 500—1500 ка- .я
диннтрохлорбензола. Котел для плавления вмещает 12 000 л, котел для окисления—: ,J|
около 30 000 л. При таких больших загрузках нагревание оказывается излишним, так м
как достаточно теплоты реакции. Котлы делаются из чугуна и обычно быстро разъ- я
сдаются. Из маточных растворов выделяют тиосернокислый натрий, который находит -Я
применение в фотографии и в текстильной промышленности. Часть его используют на '“я
заводах красителей для получения метиленового голубого. Цена на сернистый черный Я
Т чрезвычайно низка, поэтому его могут успешно производить только те заводы, на 1И
которых полностью выделяются побочные продукты, Тс заводы, которые не произ-‘ ДЯ
водят хлорбензол и динитрохлорбензол, не могут выдержать конкуренции. Динитро- ’Дя
х-лорбензол не должен содержать 2,6-изомера. ’ jl
К. ФТАЛОЦИАНИНЫ Я
Моиастраль прочио-голубой Б С — гелиоген голубой Б
(пигмент голубой фталоцианиновый) ,-Я
В трехгорлую колбу емкостью 500 мл, снабженную термометром,. 1
мешалкой и трубкой, помещают 37 г (0,25 моля) фталевого анги-. |
д р и д а, 40 г мочевин ы, 0,2 г м с.> л и б д е и о во к и ел о го а м м о- |
ния и 260 г 1,2,4-тр и х л о р бензол а. При интенсивном перемешива* |
нии нагревают на масляной бане в течение часа до 195° в колбе (тем-.-Я
пература масляной бани около 22(F). Затем 4 часа при постоянном пере-. :|
мешииании возможно точно поддерживают указанную температуру и
в это время постепенно маленькими порциями добавляют 10 г х л о р и-. -J
с г о й мед и. Следят за тем, чтобы трубка не забивалась возгоняющимся Д
фталевым ангидридом, для чего время от времени возогиавшсеся веще-/ Д
ство расплавляют. После охлаждения отсасывают густую, зеленовато- I
синюю кашицеобразную массу на стеклянном нутч-фильтре. Красно*’ |
фиолетовый кристаллический остаток промывают последовательно горя-» 1
ним спиртом, горячей 2 п. соляной кислотой, горячим 2 п. едким натром /1
и, наконец, горячей водой. Каждым из этих растворителей промывают 4
до получения бесцветного фильтрата. После сушки получают мелкокри-
сталлический фиолетово-синий порошок.
Выход 27—28 г (75—78% от теории).
Примечания. Фталоцианин меди был впервые получен Г. Дпзбахом * при
попытке получить фталоннтрпл нагреванием о-дибрпмбеизола с Сп2(СК)2. Спустя
некоторе время независимо от него на заводах фирмы «Scottish Dyes» 'при получе-
нии фталимида в железных сосудах было обнаружено образование синего красителя.
Детальные исследования, проведенные Липстедом ** с сотрудниками, показали, что
в этом случае образуется железное соединение вещества, для которого Лицстед пред-
ложил название фталоцианин и формулу I. По строению это соединение чрезвычайно
похоже на порфин (II)—вещество, лежащее в основе гемоглобина ц хлорофилла.
Фталоцианин
\Ли нет ед установил, что фталоцианины легко образуются при нагревании до высо-
ких Температур в присутствии подходящих металлов или соединений металлов фтало-
нитрила или других о-динитрилов или каких-либо соединений или смесей, которые при
нагревании могут превращаться в о-днпитрилы. Фталоцианины склонны давать ком-
плексные соединения с многочисленными металлами. Особенной устойчивостью отли-
чается медное соединение. Его можно возгонять в вакууме примерно при 600', кипя-
тить с разбавленными кислотами или щелочами; даже в холодной концентрированной
серной кислоте он растворяется без изменения, В воде и обычных органических рас-
творителях он совершенно нерастворим, он отличается также совершенно исключи-
тельной светопрочпостью н удовлетворяет наивысшим требованиям, предъявляемым
к пигментам.
При энергичном хлорировании (содержание хлора в хлорированном фталоциа-
нине должно быть минимум 14%) образуется яркий зеленый пигмент, который отли-
чается выдающейся прочностью. Фирма «Imperial Chemical Industries» обладает
многочисленными патентами в области фталоцианинов. Сульфокислоты подобных со-
единений являются прямыми красителями, которые, одна ко, не отличаются очень
высокой прочностью.
* Dieshach Н. de, von dcr Weid Е. Helv. Chim. Acta, 16. 886 (1937).
** Вег., 72. 93 (1939).
III. ТЕХНИЧЕСКИЕ УКАЗАНИЯ
л. перегонка в вакууме в лаборатории
И НА ПРОИЗВОДСТВЕ
Перегонка под уменьшенным давлением, или проще—перегонка
в вакууме, относится к важнейшим операциям в производстве красите*
лей. Некоторые продукты перегоняют под уменьшенным давлением, по-?
тому что они при обычном атмосферном давлении разлагаются при тем»]
пературе кипения. Кроме того, перегонка в вакууме имеет и другие’
преимущества. При понижении температуры кипения уменьшаются поу
тер и от лучеиспускания и появляется возможность применять для на-й
гревапия вместо огня пар. Таким образом устраняется опасность пожара*
и аппаратура может быть установлена в любом производственном помеу
щеиии. Очень важным и определяющим значение этого метода является’
также и то обстоятельство, что под уменьшенным давлением легче раз-4
деляются смеси, состоящие из веществ с близкими температурам^:
кипения. Так, только с помощью вакуумной перегонки удается удовле-j
творительно разделить три изомерных нитротолуола; алкилбензнлани^
лины разделяются гладко на отдельные составные части также только
в вакууме. ;/
Аппараты, применяемые в лаборатории для дробных перегонок уже
в течение ряда лет, оказываются во многих случаях иеудовлетворитель*
иыми, так как не могут обеспечить правильной дефлегмации смеси. Чау
сто слишком большое значение придают форме перегонной колонны и
полностью игнорируют то, что наиболее важно, — проведение перегонкИ:
таким образом, чтобы была обеспечена правильная дефлегмация?
Под дефлегмацией понимают систематическое вымывание вышекипящих
составных частей смеси в колонне, осуществляемое определенной частью’
дистиллята, непрерывно стекающей в колонне обратно. Для этой цели
сверху колонии устанавливается холодильник (дефлегматор), сконструи*
роваиный таким образом, что имеется возможность отводить обратно;
в колонну любое количество 'дистиллята. i
На рисунках 37, а и б показаны колонки из стекла, которые с успе-
хом применяются в лаборатории. Колонки наполняются небольшими-
стеклянными кольцами типа колец Рашита (т. е. такими, у которых выу
сота равна диаметру). Эти кольца расположены в колонке совершенно;
беспорядочно и обусловливают превосходное раздробление стекающей;
обратно жидкости. '
Дефлегматор охлаждается в зависимости от требующейся^
эффективности воздухом, водой, спиртом и т. д. Дефлегмацию можно, иа-
пример, вести точно при 100°, если непрерывно пропускать по каплям
в охлаждающий сосуд дистиллированную воду. При работе в больших’
масштабах можно* снабдить дефлегматор холодильником, с режимом, при
котором охлаждающаяся жидкость будет находиться при температуре
кипени н.
Очень важно, чтобы при тщательной дефлегмации происходило рав-
номерное обратное стекание дистиллята. Поэтому необходимо хорошо
изолировать всю колонну, что, например, можно осуществить с помощью
рубашки, находящейся под вакуумом (рис. 37,6), и охранять колонку от
потоков воздуха. Источник тепла (например, масляная баия) должен
обеспечивать постоянную температуру. Так как перегонке может мешать
даже простое движение людей мимо аппарата, то целесообразно операции
Рис. 37. Ректификационная колонна с дефлегматором,
такого рода проводить не в общем рабочем помещении, а в отдельной
лаборатории.
Далее, вакуум должен быть совершенно постоянным. По-
этому при применении водоструйных насосов необходимо, чтобы напор
воды был достаточно большим и исключал возможность засоса воды
в систему. Если напор воды недостаточен, то можно выйти из положения,
снабдив водоструйный насос дес яти метров ой и достаточно широкой
нисходящей трубкой, которая действует как барометрическая
трубка и обеспечивает постоянное разрежение, зависящее от упругости
паров воды при данных условиях.
Для работы с большими количествами можно конструировать аппа-
раты из железа. Так, в лаборатории авторов в течение ряда лет
Зак Г. -♦ Фиггг-Папин. JT. Плат же
применялась колонна высотой 2,5 м с перегонным сосудом ем*
костью 4 л. ;
С помощью таких колонн можно очень легко разделить изомерны^
нитротолуолы, изомерные нитрохлорбензолы и т. д. Можно даже фрак^
ционировать смеси, содержащие очень близко кипящие компоненты, на*
пример нитрохлортолуолы.
Конечно, необходимое условие для отделения чистого
ства от эвтектики центрифугированием
веще*
то mJ
заключается в
Р и с. 38.
Колонии а и б — ко.чонны по К‘у бер гик и; в — колонка по Рашигу. Диаметр
колонны 50—120 см. Высота 8—16 .и. Верхняя пасть (1— 2 л) ггри ректификации
охлаждается снаружи; остальная1 часть колонны (7—15 м) хорошо изолируется,
причем верхнее отверстие закрывается.
чтобы вещества кристаллизовались (см., например,
чтобы вещества кристаллизовались (см., например, нитро
хлорбензол на стр. 85). Например, в лаборатории не удается разделит?
1,2- и 1,4-хлортолуолы. Возможность разделения должен показать опьй
с большим количеством вещества. \
Фракционные колонны, применяемые в технике, построены т^кнЙ
же образом; кроме того, для достижения хорошей дефлегмации скош
струированы особые колонны. Три колонны такого типа показаны нЗ
рис. 38 а, б и в. При работе в больших масштабах, когда Не^
требуется особой -дефлегмации (прн перегонке анилина, нафтола нл»(
дифениламина), для обогрева применяют сжатый пар, масло илий
открытое пламя. На рис. 40 (см. стр, 306) показана совсем простая^
установка для перегонки в вакууме, которая очень хорошо работает,
несмотря на свою кажущуюся простоту. Чтобы сублимат (вещества, за-
стывающего при комнатной температуре) не попадал в насос, нужно
установить между вакуумным насосом и приемником так называемую
воздушную камеру.
Вакуум получают с помощью насосов, которые позволяют дости-
гать остаточного давления примерно до 50 мм ртутного столба. Более
низкие давления, примерно до 8 мм, получают, включая последовательно
два насоса. Но и в этом отношении наблюдаются перемены. В послед-
ние годы все более входят в употребление новые ротационные насосы,
например насос, представляющий собой компрессор и вакуумный на-
сос (системы Витте), показанный на рис. 39. Механизм его действия
виден из эскиза. Посредством подвижных пластин захватывается опре-
деленный объем воздуха, который сжимается при движении к выходному
Р и с. 39. Ротационный компрессор и вакуумный насос.
отверстию. Машина эта двойного действия, т. е. может служить как ком-
прессором, так и вакуумным насосом. Она может создавать до 4 атм
избыточного давления и до 12 мм разрежения. Аппарат необходимо
охлаждать, как это следует из эскиза. Он присоединяется непосред-
ственно к мотору трехфазиопо тока, который делает 1500—2000 об/мин;
потери на передачу силы при этом минимальны. Часто также включают
два ротационных насоса Друг за другом.
При перегонке в вакууме жидких веществ применяют простые котлы,
часто очень больших размеров. Так, например, в вакууме перегоняют
до 20 000 кг анилина, используя котлы с целым рядом паровых змееви-
ков. 8-Нафтол и подобные ему соединения нельзя перегонять в столь
больших количествах. В этом случае перегоняются 2000 кг и более, н
для этого требуется уже другая аппаратура. Высокая температура пере-
гонки! 3-нафтол а не позволяет применять для нагревания пар, хотя в этой
области уже имеются многообещающие опыты с аппаратами Фредер -
книга. Наиболее удобно нагревать газовым пламенем, так как оно осо-
бенно легко поддается регулированию. Очень часто работают без масля-
ной бани, но это связано с некоторым риском, так как остающаяся
смола может пригореть, вследствие чего ее трудно извлечь из котла
и она теряет ценность *. В производстве иет необходимости пропускать
воздух во время перегонки под вакуумом, так как при работе в большом
масштабе не происходит толчков. Напротив, вся аппаратура должна
Нафтольиая смола является многотоннажным товарным продуктом. Она пред-
ставляет собой черную, блестящую, как стекло, хрупкую массу, которая применяется
как изоляционный материал для соединительных муфт электрических кабелей.
308
ZZZ. Технические указания
быть хорошо изолирована и вес трубы, которые могут закупориться, J
должны быть легко доступны и иметь приспособления для обогрева, 1
Приемник оборудован рубашкой для обогрева и охлаждения. По окоича- |
ним перегонки масса через выводную трубу выдавливается из котла в за- |
крытые формы, что позволяет избежать неприятного действия выделяю- Ц
щихся паров, Между насосом и приемником устанавливают большую*!
воздушную камеру для улавливания воды и сублимата. Особенно много !
сублимата в виде мельчайших, снежинок, которые могли бы засорить на-
сос, переходит при перегонке р-нафтола. J
Р и с, 40. Установка для перегонки в вакууме легко застывающих веществ (нафтол!
фенилеидиамнн и т. д.).
Аппараты для работы с болыгщми количествами (1000—3000 кг) енабчеепы мегиадкой для предсхранен|
от сгригорличя,
/—перегонный сосуд; 2 —приемник е пар; in ой или охлаждающей рубашкой; 3 — обогреваемые парс
трубки, дли предохранения от зас’г|яваиия; ^ — резервуар для воды и сублимат?; о — к насосу. Т
В крупных установках пользуются несколькими термометрами. Одй
из них доходит до диа перегонного куба и показывает температуру сЫ
рюй смеси. По показаниям этого термометра определяется начало 1
конец перегонки. Перегонку следует заканчивать, когда разность межД,
температурой переходящих паров и остатком в кубе превысит 50°; в про
тивпом случае пёк может пригореть.
На рис. 40 изображен аппарат для перегонки ^-нафтола, Он обогрв
вается газом (3 кольцевые газовые горелки) и рассчитан примерно Ki
1000 кг. Перегонка такой загрузки продолжается около 4 час. Количв
ство пока составляет в среднем 5%, считая иа сырой нафтол*. Нафт$
выливают в формы, напоминающие сахарные готовы, и после раздр0
бления застывшей массы выгружают.
Для жидкостей применяют змеевиковые или .Трубчатые холодильник^
Последние содержат пучки труб по 20—30 штук. Такие холодильник!
можно подогревать, так что их применяют - при переработку
* Блльщиистпо аппаратов для перегонки нафтола снабжено мешалкой, котора!
предохраняет массу от пригорания ла дне.
дифениламина или других легко плавящихся веществ, если почему-
либо перегонка их с водяным паром не представляется удобной
(см. стр. 128, 129).
Часто иа нисходящей части холодильника устраивают смотровое
стекло для наблюдения за стекающей жидкостью.
В лаборатории очень хорошо зарекомендовал себя прибор, изобра-
женный на рис. 41. Перегонная колба ив хорошего стекла снабжена
двумя горлами — одним для капилляров, другим —для термометра.
Такое устройство препятствует перебросу брызг жидкости. Чаще всего
Рис. 41. Схематическое изображение вакуумной перегонки в лаборатории.
7 —винтовой зажим; 2 — колба Кзайзена с капилляром; 3~ водяное охлаждение; 4 -приемник; 5 — мано-
метр; б —предохранительный кран; 7 — предохранительная сцляниа; 8 — ц вакуум-насосу.
нагревают ие на Голом огне, а на масляной бане, температуру которой
держат иа 30—40° выше температуры перегонки. Капилляр вытяги-
вается из обыкновенной тонкой стеклянной трубки и должен быть по-
движным, как шелковая нить. Он должен касаться дна колбы, сверху он
закрыт винтовым зажимом, находящимся на резиновой трубке. Воздуха
пускают ровно столько, сколько необходимо для спокойной перегонки.
В качестве приемника не применяют сложных сосудов, а просто
берут перегонную колбу с длинным горлом. Для собирания разных
фракций прерывают перегонку и сменяют приемник, для чего требуется
всего несколько секунд. Способ охлаждения показан на рисунке. Мано-
метр включают не иа пути к насосу, а присоединяют его при помощи
особой трубки, благодаря чему устраняется возможность его загряз-
нения перегоняемой жидкостью. Большое значение имеет так называе-
мый предохранительный край, позволяющий немедленно впускать не-
много воздуха, как только появится опасность переброса перегоняемой
жидкости. При помощи этих приспособлений можно- очень быстро1 про-
вести любую. перегонку. Насос должен быть отделен от прибора боль-
шой предохранительной склянкой. Можно также рекомендовать
присоединять водоструйный насос к главной линии водопровода, чтобы
работа иасоса ие зависела от колебания давления. В очень многих слу-
чаях можно в качестве приемника вместо перегоииой колбы применять
так называемую «вурст-колбу». Применение этого устройства особенно
рекомендуется для начинающих.
Выполнение перегонки. Пускают насос и нагревают масляную баню
до нужной температуры. В большинство случаев сперва отгоняется вода.
или растворитель, так что разрежение следует уменьшить, приоткрывая:
предохранительный край. После того как плав начинает спокойно ки-
петь, регулируют капилляр, через который должен проходить быстрый ток
мельчайших пузырьков воздуха. Через некоторое время начинается пере-
гонка, которая в случае жидких веществ не представляет никаких затруд-
нений; затвердевающие же вещества вроде р-нафтола, нафтиламина и;
т. д. легко закупоривают отводную трубку. Поэтому еще до начала пе-
регонки нагревают горло колбы, чтобы перегреть первые капли. Иногда,
чтобы заставить дистиллят пройти через трубку, следует подогреть бун-
зеновской горелкой даже место, закрытое пробкой. При хорошем стекле
почти исключается опасность взрыва; несмотря на это, рекомен-
дуется всегда работать с защитными очками. Перепонку1
следует вести по возможности быстро. 200 г -нафтола перегоняют, иа-*
пример, за.. 15—20 мин., слегка охлаждая приемник. Манометр отклю-!
чают от прибора и только время от времени проверяют разрежение.!
Очень часто начинающие пытаютси сделать свои приборы герметшй
ними, замазывая пробки парафином, коллодием и другими веществами,;
что является совершенно недопустимым. Гораздо проще предварительно
тщательно пропитать хорошую пробку горячим расплавленным парафин
ном, после чего уже не потребуется добиваться дальнейшей герметич-1
иости. Только при перегонках в высоком вакууме следует применять ре-!?
зииовые пробки или, лучше, использовать спаянные приборы *. 1
Перегнанное вещество расплавляют нагреванием на голом огне и?
выливают жидкость в небольшую фарфоровую чашку. Застывшая масса?!
достаточно чиста и не нуждается в перекристаллизации. 3
М. ФИЛЬТР ПРЕССЫ j
Химик, работающий в области красителей, для отделения твердого.’
вещества от жидкостей охотно использует в лаборатории воронки для!
отсасывания. На производстве же они применимы только для крупнозер-^
иистых веществ, которые даже в толстом слое ие создают слишком боль-^
шого сопротивления для прохождения жидкости. Тонкие осадки, которые!
образует огромное большинство красителей, на производстве приходится:
фильтровать на фильтр-прессах (см. рис. 56, стр. 334 и схемы на !
рис. 42, а—з).
Суспензия, подлежащая фильтрованию, передавливается с помощью!
сжатого воздуха из монтежю (см. монтежю на рис. 56, стр. 334) через.-
трубопровод (5) (рнс. 42, а) в’фильтр-пресс. По сплошному питающему!
каналу (6) и через вертикальные отверстия (7), соединяющие полости рам :
фильтр-пресса с каналом (рис. 42,6, в, г), суспензии стекает в камеры-
рам (8) и их заполняет. Твердая фаза остается в камерах; маточный рас-!
твор проходит через ткань (9) и по желобам плит стекает через краны !
в корыто (10) и в случае, если он далее ие перерабатывается, напра-
вляется в канализацию (см. рис. 42, б—г: фильтрование). Вначале, пока .
* Так приходится поступать на производстве прн получении гваякола, при пере-
гонке которого применяется ртутный насос.
Фильтр-прессы
л i
ткань, особенно новая, не покрылась слоем твердого вещества, фильтруют,
применяя слабое давление, затем устанавливают полное давление
(обычно несколько атмосфер). Когда пресс заполнен, то маточный рас-
твор стекает только по каплям; после заполнения фильтр-пресса можно
приступать к продувке. В случае необходимости осадок иа фильтре про-
Р и с. 42. а — фильтр-пресс; б, в-—рамы; г — фильтрация;
Д е — промывные плить;; ж — неподвижные плиты для прес-
сования; з — промывание.
мывается. Для промывания можно использовать опорожненный мон-
тежю или, если имеют дело с трудно растворимыми осадками, то исполь-
зуют воду непосредственно из водопровода; промывную воду пропускают
через два боковых канала (//), находящихся в головной плите фильтр-
пресса и ведущих к желобкам в промывных плитах (см. рнс. 42, д—з-,
промывание). При промывании краны промывной камеры IF должны
быть закрыты. Когда маточный раствор вытеснен и промывная (вода
стекает чистой, отфильтрованную массу через те же каналы (//) про-
!ехнические указания
дувают досуха сжатым воздухом. Затем пресс открывают (отвинчива-
нием) и наполненные рамы разгружаются при помощи деревянных ло-
паток в ящик {12) и ткаць очищают соскабливанием*. После этого пресс
закрывают н пускают снова в работу.
Если при фильтровании стекает мутный фильтрат, то его собирают
и возвращают обратно. Но, это, допустимо только1 в самом начале
фильтрования. Причиной того, что в течение продолжительного времени
стекает мутный фильтрат, могут быть перекос фильтровальной ткани или
ее разрыв во время фильтрования. Если мутная жидкость стекает из
крана какой-либо плиты, то его следует закрыть; нельзя, однако, за-
крывать кран у промывной плиты, так как при этом мутная жидкость
пойдет по пути, обратному движению промывной воды {11), и попадет
во все промыв'ные камеры. В этом случае плохо отфильтрованная жид-
кость будет задержана четным краном и возвращена обратно.
Подробные описания многочисленных специальных видов фильтр-
прессов и других аппаратов для фильтрования, применяемых в технике,
имеются в специальных руководствах **.
Н. О КОНСТРУКЦИИ И ПРИМЕНЕНИИ АВТОКЛАВОВ
Автоклавы, или сосуды для работы под давлением, применяются'
всегда в тех случаях, когда температура реакции превышает темпера-
туру кипения вещества, или когда при нагревании выделяются газы,
имеющие значение для хода реакции. В производстве красителей чаще
всего приходится иметь дело, с водными растворами и смесями, но су-
щественное значение имеет также работа со спиртами и хлористыми алки-
лами. Применяют давления до 60 атм н нагревание приблизительно до
300°. Эти пределы относятся к описываемому ниже типу аппаратуры.
Существуют вертикальные или горизонтальные автоклавы с мешал--
ками или без иих. Когда смесь однородна, перемешивание обычно ие<
применяется; если же образуются различные слои или если лерераба<
тывается смесь твердых и жидких веществ, то необходимо непрерывное;
перемешивание. В качестве примера реакции, идущей под давлением,;
и не требующей перемешивания, можно назвать получение диметилани-;
лина; примером же реакций, где необходимо постоянное перемешивание^
являются все реакции щелочного плавления.
Автоклавы представляют собой цилиндрические сосуды емкостью;;
100—10 000 л с фланцем, к которому болтами и гайками прикрепляется.)
крышка. Дли большей прочности днишу автоклавов обычно придается-,
сферическая форма. Автоклавы изготовляются почти всегда из чугуна ;
или литой стали. Последняя Обеспечивает наибольшую безопасность при;
высоких давлениях (о стали и железе см. также в разделе о конструк-,
ционных материалах). Наряду с чугуном и литой сталью применяют'
также и листовую сталь. Автоклавы из листовой стали изготовляются'
с толщиной стенок до 40 мм. Они илн склепываются или свариваются;
автогенным способом. Предубеждение некоторых заводов против свар-".
ных автоклавов совершенно неосновательно и объясняется только тем,?
что раньше техника автогенной сварки была несовершенной.
* На все эти операции затрачивается довольно много ручного труда. Вследствие
этого на производстве пользование фильтр-прессом стоит дороже, чем иутч-фнльтром'
для отсасывания иля центрифугой. Поэтому, когда это позволяет характер осадка,
последним отдается предпочтение.
** См,, например, U 1 1 m a n n, Еnzyklopadie der tcclin. Chemie, 2. Auff. Bd. V,
S. 358,
О конструкции, и применении автокланов
313.
Слабым местом каждого автоклава являются болты, которые по-
этому всегда изготовляются из лучшего литого железа ручной ковки.
На крышке имеется несколько прилитых штуцеров, которые отчетливо-
видны на рис. 43 и 44. Они служат для укрепления арматуры и
сальника мешалки. Опора мешалки, на которой укреплен привод, дол-
жна иметь такую высоту, которая позволяет в любое время осматривать,
н сменять набивку. Сам сальник, через который проходит ось мешалки,
Рис. 43. Большой автоклав из литой
стали.
Емкость 1500 л; максимальное давление 40 ат
(сальник расположен близко к опоре для
мешалки).
Рис. 44. Большой автоклав из чугуна
с паровым обогревом.
Емкость 1400 л\ максимальное давление 25 от.
Охлаждаемый водой сальник легко доступен
благодаря тому, что опора для мешалки доста-
точно высока. Справа наверху виден кран,
с пимошыо которого можно отбирать пробы.
должен быть сконструирован возможно более простым и допускать охла-
ждение водой. Отливаются также полые сальники, через которые цирку-
лирует вода. Охлаждение циркулирующим маслом или лабиринтные-
уплотнения, с успехом применяющиеся в паровых турбинах, здесь не
нужны, так как условия работы совершенно другие. На штуцерах,
имеющихся на крышке, наряду с двумя манометрами и двумя термо-
метрами имеется также два спускных вентиля со стальным конусом.
В последние годы в некоторых случаях отказываются от применения;
предохранительных клапанов, так как последние, часто плохо выполняют
свои функции и являются источником недоразумений. Двух термометров
и двух манометров достаточно для того, чтобы точно следить за ходом
процесса. На крышках больших автоклавов устраивается особое:
u, 2 rArtwutrcziuv указания
отверстие для загрузок (люк); причем только это отверстие регулярно от-
крывается. Для герметичности крепления крышки во фланец корпуса авто-
клава закладывается специальная кольцевая прокладка. В качестве про-
кладочного материала применяется медь, свинец, освинцованное железо
и асбестовый картон. Кольца должны быть тщательно обточены и имел»
ширину 20—50 мм, а толщину 1—6 мм. Свинец легко выжимается дав-
лением болтов, ио зато он очень устойчив к действию аммиака. Идеаль-
ным прокладочным материалом является медь, но она разъедается ам-
миакам. Прн низких давлениях очень часто можно применять асбесто-
вую прокладку, но оиа неудобна тем, что почти всегда разрывается,
при открывании: автоклава. При завинчивании крышки сперва слабо,
затягивают болты, расположенные на противоположных сторонах, затей;
подтягивают сильнее все болты по кругу и, наконец, окончательно за*,
тягивают, ударяя тяжелым свинцовым молотком по длинному ключу;;
При таком способе работы совершенно исключается возможность с<К
рвать болт при затягивании.
Автоклав не должен входить в непосредственное соприкосновений
с веществом, так как каждое плавление разъедает стенки, вследствие
чего прочность аппарата с течением времени может значительно умеий
шиться, и его надо будет перевести, как говорят, в низший разряд. Пой
этому в автоклав почти всегда вставляется вкладыш. Последний укреЩ
ляют, заливая сплавом из свинца и олова зазор между ним и стенкам®
автоклава. Вкладыш нельзя вставлять в автоклав просто без этой лро|
кладки, так как вследствие недостаточной теплопроводности стенки авто|
клана могут сильно перегреться и даже раскалиться. При устаиовям
вкладыша его сначала укрепляют в автоклаве с помощью поперечной
балки и затем через железную воронку вливают металл. Если вкладыЙ
покрыт эмалью или свинцом, то покрытие защищают, обкладывая вну,^
треинюю сторону вкладыша мокрой тканью или наполняя его водож|
воду в этом случае следует сперва подогреть, а затем охлаждать при
помощи змеевика. Нагревание предупреждает застывание сплава. Ииата
он может не дойти до дна и застыть на полпути. Если же вода не буд^Ц
охлаждаться, то она нагреется до кипения и полностью испарится им
вкладыша. Недостаточная теплопроводность может быть следствием НЯ
только наличия воздуха между стенками автоклава 'и вкладышем. В еЩИ
большей степени понижают теплопроводность коркн соли внутри CO*S
суда. Поэтому в тех случаях, когда выделяются солн, всегда следуем
применять перемешивание; прн этом мешалка должна как можно блиайН
подходить к стенкам н очищать их. Но когда выделяются большие колИЙ
чества соли, то оказывается бесполезным и самое лучшее перемешивая
ние. Известен случай, когда слон соли толщиной всего в 4 см вызвала
перегрев автоклава до красного каления. При внутренней температур®
240° и давлении 48 атм автоклав раздулся, как стеклянный баллон, й
дал трещину на дне. Вырвавшиеся пары настолько охладили сталь, чтздЗ
опасность миновала. Однако если автоклав был бы из серого чугуи^И
то, без сомнения, произошел бы взрыв *.
Из изложенного следует, что автоклав,
помещать в баню. —
если это возможно, иаД01
Применяют масляные бани или бани из сплава^
свинца н олова. Даже тогда, когда в реакционной массе не образуется^
корок, мешающих теплопередаче, при высоких температурах, имеют м^|
сто явления, которые обусловливают желательность применения свиН*|
* К такого рода процессам относится, например, плавление ^-нафтола, которое,^
если его проводить не на металлической бане, безусловно, приведет к разрушению^
любого сосуда, работающего под давлением. j
wrtu руъции u применении автоклавов
did
ново-оловянной бани. Если автоклав подвергается непосредственному
нагреванию, то сплав, которым залит вкладыш, становится жидким;
вкладыш при этом всплывает, упирается в крышку автоклава и пре-
пятствует сжатию болтов при охлаждении. Вследствие этого с течением
времени возникает такое напряжение, йто автоклав становится негерме-
тичным; к тому же растягивание болтов представляет большую опас-
ность. При неправильном нагреве страдает не только автоклав, но и
перерабатываемое вещество1. Такие случаи подробно обсуждались при
описании получении а-нафтола и ^-нафтиламина.
Автоклав загружают либо через хорошо закрывающийся люк, либо,
если это возможно, вещество засасывают в автоклав через штуцер для
спуска давления, создавая в
нем разрежение; при этом ме-
тоде загрузки отсутствуют усло-
вия для негерметичности авто-
клава. Вода снльио расширяет-
ся при нагревании (по Менде-
лееву, при нагревании от 0 до
250° оиа расширяется на 20%
своего первоначального объе-
ма), поэтому автоклав следует
заполнять не более чем на 80%
его общего объема. Если весь
сосуд заполнен, то возникает
настолько высокое давление,
что он разрывается. При каж-
дом автоклаве должна нахо-
диться таблица с точными дан-
ными о его общем объеме, ма-
ксимальном давлении, макси-
мальном заполнении, а также
о роде вкладыша.
Расчет автоклава произво-
дится инженером на основании
официальных норм. Однако
рекомендуется сделать прове-
рочный расчет каждого аппа-
рата даже первоклассного ма-
шиностроительного завода и
после этого передавать проект
станций.
Автоклав для эксплуатации
Рис. 45. Автоклав в разрезе.
на утверждение официальных ии-
уста па вливается в готовой кладке
с помощью общего мостового крана. Кладка должна быть укреплена
стальными тягами (стержнями), расположенными на расстоянии 30 см
один от другого, а выступающие концы тяг должны соединяться
болтами, под которыми подложены стальные подкладки размером
25 X 25 ем, находящиеся на обмуровке. Автоклав илн баня поме-
щаются на кольце, вмазанном в кладку, как изображено на рнс. 43;
заполненный автоклав нагревают хорошим углем. Если крепление вы-
полнено правильно, то даже первое нагревание можно проводить без
особой предосторожности, хотя лучше начинать нагревать на малом
пламени. Колосниковая решетка должна быть чистой; в случае плохой
тяги вынимают один или два колосника. Рекомендуется устраивать об-
щую топку для ‘каждых двух больших автоклавов. Это дает возмож-
ность работать независимо от других соседних аппаратов. На производстве
нагревание автоклава всегда продолжается несколько часов, ио если
кладка еще не остыла, \го требуется очень мало дров или угля для под»;
держания нужной температуры. При температурах выше 200° излучение
тепла настолько сильное, что часть автоклава, выступающую из топки^
необходимо изолировать железиым кожухом с асбестовой футеровкой^
Для охлаждения снимают кожух, состоящий из отдельных частей, я
одновременно открывают топоч-
ную дверцу и заслонку. Мож-
но зиачнтельно ускорить охла-
ждение автоклава, если выпу-
скать часть газов в воздух ли-
бо в конденсационную уста-
новку (в случае спирта или
аммиака); обмуровка при этом
не слишком охлаждается, что
оказывается выгодным для
Р и с. 16. Лабораторный автоклав
с мешалкой.
Литая сталь; рабочее дянленис- 60 ат- объем
1 .1; пес 33 ъе; вес масляной бани (из мели?
11 к?..
проведения следующей операции. Во время плавления надо особенна
тщательно следить за автоклавом. Температура масляной баи»
должна быть примерно на 30° выше внутренней температуры, и пока|
зания обоих термометров и манометров должны совпадать в предела»
двух делений. Если наблюдаются большие расхождения, то термси
метры надо проверить; ииогда приходится прервать процесс. Следит»
за производством можио даже из лаборатории с помощью телетермОя
метра; также вге шире применяют самопишущие манометры, который
позволяют осуществлять последующий контроль. Все происшествие!
подробно записывают в журнал, чтобы имелись документальные'данные!
в том случае, если произойдет авария пли несчастный случай. Если,
несмотря на все предосторожности, происходит существенное нарушение
режима, как например быстрое неожиданное повышение давления или
температуры, то немедленно прекращают обогрев, открывают вентиль и
заслонки и оставляют автоклавное и соседнее с ним помещение. Взрыв
автоклава, изображенного, например, иа рис. 43 и 44, может разрушить
целый завод. Однако, так как все аппараты для работы под давлением
изготовляются с восьмикратным запасом
при надлежащем обслуживании не пред-
ставляет опасности. Каждый аппарат для
работы под давлением ежегодно прове-
рястси инспектором котлонадзора, для
чего эти апараты хорошо очищаются и
охлаждаются. Инспектор может начать
обследование автоклава, лишь убедив-
шись в том, что свеча, помещенная вну-
три автоклава, горит спокойно. В боль-
шинстве случаев открывают только люк
и через него внутрь накачивают воздух.
Эту работу должны проводить два ра-
ботника. О результатах обследования
составляется официальный отчет. Часто
изготовляют шаблон, с помощью кото-
рого можно точно измерить каждое изме-
нение стенок. При измереииях вынимают
вкладыш, для чего разжигают внутри
огонь, который поддерживают с помощью
сжатого воздуха. Когда свииец распла-
виться, вкладыш немного поднимают
кверху и передвигают при помощи’крана.
Свииец вычерпывают железным ковшом
в формы.
Автоклав устанавливают в высоком
И светлом помещении, оборудованном
мостовым краном. На рис. 56 (см. стр. 334)
дай разрез заводского помещения произ-
водства красителей с примыкающим
автоклавным помещением. На рисунке
видио, как материал поступает в авто-
клав и как готовая масса выдавливается
по трубе непосредственно в другое поме-
прочности, то работа с ними
Рис. 48. Вертикальней автоклав
из литой стали.
Рабочее давление 60 ат; объем 1 л;
пес 30 лгу.
щение для переработки.
Лабораторные автоклавы по конструкции подобны про-
мышленным. На рис. 46—48 показаны общие виды автоклавов из сталь-
ного литья с мешалкой" и без нес (на рис. 47 первый автоклав в раз-
резе). Как уже отмечалось, вкладыш тщательно заливается. Если для
нагревания вместо масла используют сплав свинца с олово-м, то надо
применять железную баню, так как медь разрушается свиппом. В лабо-
ратории часто отказываются от охлаждения сальника, так как через
пего возможны лишь оч'снь незначительные потери. Сальник следует
охлаждать только при очень высоких давлениях и температурах, ио
в этих случаях лучше применять вращающиеся автоклавы, которые опи-
саны ниже. Следует еще указать, что мешалка должна вращаться вправо,
чтобы ие развинчивался виит сальника. В лаборатории, так же как и
в производстве, гайки надо затягивать осторожно и равно-мерно, при
этом нельзя j
тить нарезку,
ключом, установив автоклав в подставку, чтобы ои не вращался при з
винчиванин гаек,
Крышка автоклава может быть выпуклой или плоской, как изобр,
жено в разрезе на рис. 47 н иа рис. 46 и 48. В общем автоклавы с пл<
Ской крышкой предпочтительней, так как на ней лучше закрепляется cti
нина привода мешалкн н легче уплотнить штуцеры. На рнс. 48 изобр;
жен вертикальный автоклав с выпуклой крышкой со штуцерами дл
арматуры.
,арять по дайкам молотком, так как легко можно испор!
Поэтому достаточно подтянуть' гайки длинным гаечньй
Р и с. 49. Вращающийся автоклав нз ковкого железа с чер-
вячным приводом.
Вес II кг\ объем 400 .«.г; рабочее давление 100 ат.
Автоклав нагревают на печи Флетчера, а затем на хорошей бунг
новской горелке, которую помещают прямо под серединой автоклав}
ие следует нагревать несколькими горелками с разных сторон. Abti
клав должен быть защищен от потоков воздуха и изолирован кожухб
из жести, вследствие чего. экономится около 70 % газа. Дл
охлаждения весь аппарат вынимают нз бапи, помещают на железн!
треугольник и дают маслу стечь в бапю. Нагревание и охлажденИ
продолжается примерно I час. В непредвиденных случаях поступай
по правилам, принятым на производстве, с соответствующими иомен*
ниями. Пока в автоклаве имеется давление, нельзя подтягивать гайВ
сальник же подтягивать можно без всякого onaci
на крышке;
В остальном следует руководствоваться правилами, приведенными нИЖ
(стр. 320). Д
Вместо того чтобы применять эмалированный вкладыш, можно пОу
крыть эмалью крышку и внутреннюю поверхность автоклава, однако эту-;-
операцию удовлетворительно проводят лишь немногие заводы. -fl
В лабораторных условиях при работе под давлением около 20 ат
с перемешиванием встречаются значительные трудности, так как прихо-
дится сильно подтягивать и охлаждать сальник, Поэтому авторы в тече- .
ние- ряда лет применяют аппарат известной конструкции с некоторыми
изменениями. На рис. 49 и 50 представлен такой вращающийся авто-
клав в 0|бщем виде и в разрезе. Отверстие этого автоклава сужено,
чтобы было меньше болтов, а весь корпус изготовлен нз целого куска
вала ковкой стали. Для того чтобы можно было измерять давление и
температуру, автоклав устанавливают в наклонном положении, а угол
наклона может быть изменен по желанию; во всяком случае, угол на-
клона подбирается таким образом, чтобы содержимое' автоклава ие со-
прикасалось с прокладкой. Наверху имеется отверстие для манометра,.
под ним — отверстие для термометра, который закрепляется с помощью*
асбеста. Автоклав опирается ие на ось с «черенком», а иа бронзовую,
втулку, проходящую через опорные колонки (см. рис. 51). Поэтому для
вращения требуется очень малая сила.
Автоклав вмещает 400 мл, веснт 11 кг и рассчитан на давление до
100 ат'. Столько же веснт под ставка к автоклаву. Прн нагревании авто-
клав закрывают железным колпаком; тогда расход газа на 80%
меньше, чем у автоклавов другой конструкции. Попытки заменить доро-
гостоящие болты простым винтовым затвором ие дали результатов, так
как последний всегда начинает пропускать примерно при 180°. Прн за-
винчивании прокладка просто зажимается иа крышке, и нельзя добиться
герметичности. Горелка должна, как показано иа рисунке, помещаться
под нижней частью автоклава, заполненной веществом, для того чтобы
прокладка не перегревалась.
Вращающийся автоклав особенно пригоден при работе с кашице-
образными массами нли суспендированными твердыми веществами...
j елниысш указания
Из серого чугуна изготовляют, кроме того, опоры для мешалок,
чанов, автоклавов и других сосудов. Зубчатые колеса для более легкого
хода должны быть фрезерованьи желательно также все крупные при-
воды снабжать шарикоподшипниками, что экономит энергию и сма-
зочные материалы. Станина и неподвижные (головные) плиты фильтр-
прессов отливаются из чугуиа, но струны не делают из чугуна, так как"
чугуи не обладает достаточной прочностью к растяжению. Автоклавы;*
рассчитанные на рабочее давление до 40 ат, изготовляются из серого
чугуна. Для работы при более высоких давлениях применяют стальио
литье, так как чугунное литье при отливке деталей слишком больши:
размеров пузырится и, кроме того, пришлось бы делать слишком тал
стые стенкн. Стальной автоклав, изображенный на рис. 43, имеет, иа
пример, толщину стеики 80 и весит 10 т. Автоклав диаметро*
1200 мм, рассчитанный на рабочее давление до 40 ат, изготовленный и
серого чугуна, должен иметь стенки толщиной около 400 мм и веси
около 60 т. Такие чудовищные аппараты технически неприемлемы, хо
• бы из-за огромных напряжений, возникающих при нагревании. Из с
рого чугуна изготовляют также котлы для плавления в произведет
нафтолов; добавка 1—3% никеля чрезвычайно повышает устойчивое
чугуна к щелочам. Расплавленные щелочи, особенно ед ко е к а л
вызывают сильную коррозию железа. Чугун, легированный 12% к pel
и и я и 4—6% алюминия, полностью или частично устойчив к кислф
там. Этот сплав железа—кремния—алюминия довольно сильно разру
шается только соляной кислотой, которая вообще является кислотой
наиболее сильной по своему корродирующему действию. Этот спл?
. впервые был применен в Англии под названием «а й р о и э к» и «тэ i
т а й р о :•;». ф
Аналогичны сплавы «кизельгус», «ацидур» и «клюзайрон», в
они очень легко поддаются отливке, однако они, к сожалению, хруп:
и обладают твердостью стекла, так что нх приходится об раб ат:
вать па шлифовальных кругах. Эти сплавы оказались особенно пригс
ными для изготовления аппаратуры для перегонки азотной кислот
Они также находят применение н для других специальных целей. Э
сплавы ввиду их хрупкости не применяют для изготовления вкладыш
автоклавов.
В тех случаях, когда требуется большая прочность, следует прим
нятъ кузнечное железо, литое железо и сталь. Из эт
сортов железа изготовляют струны для фильтр-прессов и гндравлическ!
прессов. Головные плиты для этих прессов следует отливать из стал
так как чугун недостаточна прочей. В настоящее время прнмеия!
также швейцарскую электросталь, Из стали изготовляют также спирал
ные трубки пружинных манометров, применяющихся при работе с аммн
ком. В качестве материала для изготовления обручей деревянных чаи
применяют пойосовое железо, а для армированных железобетонных ч
нов — арматурное железо.
В последнее время на рынке появились особенно устойчив!
сплавы железа, получившие быстрое расп ростр а пенне. В особенное'
сплавы иикель—хром—железо, содержащие в большинстве случае
очень мало углерода. Эти сплавы довольна устойчивы к щелочам и кисл
там и применяются там, где необходимы материалы, устойчивые к хим
ческим агентам. Назовем различные VA (Фау А)-стали, соответствуют
английским маркам S-80(C-80), которые в зависимости от своего на1!
значения имеют различный состав. Эти металлы оказываются, например^,
особенна устойчивыми к концентрированной азотной кислоте; другй^
кислоты, за исключением соляной кислоты, на них также мало дей-»
1. Металлы
323
ствуют. Особенно большую роль эти стали играют в аппаратуре для син-
тезов под высоким давлением, которая в производстве красителей при-
меняется мало. Едкое кали, даже расплавленное, едва действует иа эти
сплавы, так что оии могут применяться при индантреиовом плавлении
(для этого случая также вполне подходит никель).
Медь раньше применялась более широко; она часто бывает необхо-
дима и в настоящее время. Из нее делают черпаки, корзинки для центри-
фуг, трубопроводы и главным образом противни для сушки, которые
почти всегда изготовляют из меди. Медь Неустойчива к аммиаку в при-
сутствии воздуха, поэтому противни часто лудят. Аппаратуру для пере-
гонки спирта обычно делают из меди.
Олово в чистом виде применяется мало, его используют,' однако,,
в сплавах, например бронзе*, сплаве для баиь ** и в особенности для
лужения железной и медной аппаратуры (гомогенное оевннцование, см,
ниже).
Цинк в большинстве случаев применяется не в чистом виде, а в
виде латуни и сплава для подшипников и для поверхностного гальва-
нического покрытия жести.
Алюминий, напротив, все больше используется вследствие боль-
шой устойчивости к разбавленной, а также концентрированной азотной
кислоте. Из него часто делают трубопроводы для азотной кислоты
и нитраторы, однако он ие устойчив к заводским атмосферным
газам.
Никель применяется лишь для получения специальных сплавов.
Среди цветных металлов наиболее употребляемым и часто незаменимым
является свинец. Почти все фильтр-прессы снабжены свинцовыми,
трубами, из свинца также делают и специальные трубопроводы' для
кислых |ц щелочных жидкостей. Головные плиты и входные штуцеры
фильтр-прессов также покрывают листовым свинцом, При нагревании
металлов, покрытых свинцом, часто наблюдается повреждение свин-
цовой оболочки; она отстает, вспучивается и разрывается. Поэтому свииец.
не просто накладывают на металл, а его наплавляют на защищаемую,
поверхность (легируют). Аппараты, поверхность которых тесно связана
со свинцовой оболочкой, называют гомогенно освинцован-
ными; гомогенное освинцовывание играет все большую роль в произ-
водстве красителей. Цилиндрические аппараты, например вкладыши Для
автоклавов и т. д., по предложению Кюнле, Коппа и Кауша освинцовы-
вают следующим образом: сосуд быстро вращают и в него вливают
расплавленный свинец. Таким образом полностью закрываются все поры
металла; этим методом можно освинцовывать емкости до 6000 л весом
Ю г. Железо или медь перед гомогенны^ освиицовываиием всегда
следует лудить, так как иначе свинцовое покрытие мало прочно. Это
покрытие часто бывает довольно толстым — 2 мм и уолее, так что
на покрытие очень больших аппаратов расходуются тысячи килограммов
свинца.
Этими краткими сведениями, естественно, не исчерпываются всевоз-
можные случаи применения металлов в промышленности красителей^
одиако они достаточны для того, чтобы показать, какую большую
роль в этой области играют материалы, из которых изготовляется аппа-
ратура.
* Входной штуцер фильтр-прессов, а также краны чанов почти всегда делаются
из лучшей бронзы.
** Сплав равных частей свинца и олова при нагревании почти ие расширяется.
32-1 ///, Технические указания
2. Неметаллические материалы
К неорганическим материалам b первую очередь принадлежат це-1
менты и различные сорта керамики.
В тех случаях, когда требуется абсолютная устойчивость' к кисло- .
там, керамика оказывается единственным годным материалом.
Правда, в известных случаях для этой цели можно использовать также :
свинец, однако каждый из химиков, работающих на производстве, ср-2
временем приходит к выводу, что рано или поздно даже наиболее тац- ;
дельно освинцованная аппаратура потребует дорогостоящего ремонт^.
Если установка должна работать неограниченное время без перерыва, -<
то в качестве материала для нее следует применять только керамику и $
в редких случаях природные кислотоупорные камни. J
Краны небольших размеров очень часто изготовляют из керамики, и -х
при надлежащем обращении они служат неограниченное время. В слу- j
чае горячих жидкостей применение керамиковых кранов не рекомендуется, |
так как они лопаются. Для того чтобы краны не заедало, их надо пра-
вильно смазывать. Так называемые армированные керамические
краны более устойчивы к удару и нагреванию. Защитный кожух дё-Т|
лается из освинцованного железа и в большинстве случаев укреп-
ляется нарезным болтом, легким вращением которого от руки можно
освободить втулку крана, Эти армированные крапы полностью вытеснили
старые краны из гартблея (сплав сурьма — свинец). Керамику приме- 1
ня ют также в качестве материала для изготовлений трубопроводов, вен- J
тилей и центрифуг. Корзинки керамических центрифуг заключают в ко-
жух из листовой стали во избежание разрыва под действием центробеж- ч|
ной силы. Существуют очень сложные конструкции, на которых мы ие21
можем здесь останавливаться. 1
Широкое применение находят керамические резервуары, которые из- а
тотовляются либо цельнолитые, либо составляются из отдельных частей. Ц
Цельные резервуары можно изготавливать емкостью до 5000 л, но они Я
очень дороги и чувствительны к незначительным колебаниям температурЫгЦ
На заводе можно самим изготовить кислотоупорный резервуа р,.Я
если имеется хороший каменщик. Железный котел обмазывают цементом'/Я
и на затвердевшем слое при помощи обычного же цемента укрепляютД
кислотоупорный клинкер или глазурованные керамические плитки. Отдель- д
ныс плитки должны быть разделены швом шириной 6 мм. Эти швы за-Я
называют кислотоупорным цементом. Тонким деревянным шпателем сиа-Я
чала заполняют швы наполовину н цемент сушат, нагревая весь аппаратЯ
паровым змеевиком, что продолжается около 14 суток. Лишь после того, 19
как совсем высохнет первый слой кислотоупорного цемента, шов запол-Дд
няют целиком и снова сушат. Изготовление котла емкостью 5000 л про-Я
должается около 2 месяцев. Когда цемент схватится, котел наполняйте
2%-пой серной кислотой и оставляют на 3 суток. При этом кислотоупор-Я
ный цемент окончательно затвердевает и можно не опасаться того, что* Я
швы станут неплотными. Такие сосуды выдерживают, если они тща^Я
тельио изготовлены, даже 80%-ную горячую серную кислоту, а такжеЯ
давление и вакуум. Изготовляют также котлы с двумя слоями кислото-Я
упорных плиток, которые накладывают так, чтобы швы первого рядалЯ
покрывались плитками второго ряда. Но они значительно дороже и не-Я
многим устойчивее хорошо сделанных котлов с одним слоем. Я
Щелочные и Нейтральные жидкости можно хранить в цементных Я
резервуарах, которые часто армированы железом. Так как при нагрева- з
нии возникает огромное напряжение, то армировку следует тщательно
рассчитывать. В производстве красителей применяют также цементные 4g
з. материалы органического приинимиспи.ч
чаны, которые, однако, рекомендуется выложить кислотоупорными
плитками, так как даже очень слабые кислоты быстро разъедают це-
мент. Из железобетона можно также изготовлять мешалки и с успехом
применять их в особых случаях.
Пол в заводских помещениях выкладывают кислотоупорными плит-
ками, швы между надми заливают серой. Она очень прочно держится на
камне и не смывается, как асфальт, горячей водой. В сухих помещениях
делают просто хороший цементный пол.
Стекло из-за хрупкости находит ограниченное применение, но все
же им иногда приходится пользоваться. Оно незаменимо при операциях
хлорирования при высокой температуре (см. также ди хлор бенз альдегид).
Из стекла часто делают трубопроводы для хлора, а также стеклянные
мешалки, состоящие из железной или деревянной по-перечной балки с за-
жатым в ней стеклянным стержнем. Плавленый кварц етце мало при-
меняется, кварцевые ла^тпы для хлорирования находят напротив, все
большее распространение (см, стр. 143).
Фарфор находит применение только в лаборатории и в колористи-
чсской лаборатории. Считающаяся прочной посуда из закаленного стекла
слишком часто лопается, так что ее не следует рекомендовать для про-
изводства.
Специальным видом стекла является эмаль, которой особенно
часто покрывают чугун. Получение хорошей, кислотоупорной эмали
трудно, и поэтому часто заводские аппараты эмалируются дважды. Эта
эмаль не столь «красива», как эмаль па предметах домашнего обихода,
зато она значительно прочнее последней. Так как эмалированный ап-
парат, имеющий повреждение хотя бы в одном месте, становится непри-
годным для работы, то при обслуживании аппаратов следует соблюдать
чрезвычайную осторожность. До эмалированных сосудов никогда не
следует дотрагиваться металлическими предметами, а только деревян-
ными. Изготовляются очень сложные эмалированные аппараты, они рас-
цениваются по весу и являются чрезвычайно дорогими. Очень распро-
странены эмалированные черпаки.
3. Материалы органического происхождения
Среди природных материалов первое место занимает дерево. Его
применяют для изготовления чанов для получения красителей, мешалок,
различных площадок и в особенности для строительства заводских по-
мещений, В последнее время деревянные постройки уступили место
железобетонным. Дерево, как это ни удивительно, чрезвычайно устой-
чиво ко всем реагентам: оно разрушается только с поверхности, и по-
врежденный верхний слой предохраняет от дальнейшего разрушения.
В первую очередь применяют американскую белую сосну (иичпайн)
наряду с лиственницей и обыкновенной сосной. Бук не употребляется
вследствие того, что он легко растрескивается; дубовые чаны, наоборот,
встречаются часто: они дороги, но очень прочны. Другие древесные по-
роды, кроме ясеня, вследствие своей дороговизны не применяются.
Чаны нз готов л я юте я емкостью до 20 000 л. Мешалки делают из
ясеневого дерева, они прикрепляются к приводам при помощи манжет
(см. также о чугуне, стр, 321). Такие большие чаны редко (как это
показано на рис. 56) устанавливаются на помостах, большей частью их
устанавливают непосредственно на полу. Монтежю ставят под полом
или на одном уровне -и жидкость всасывают при помощи вакуума. В этом
случае монтежю должны быть' внутри снабжены распорным стержнем;
для того чтобы опи не разлетелись при 2—3 ат давления, их охватывают
326
7/Л, Технические указания
сильными стальными обручами. Наряду с выводной сифоинон трубкой
вставляют небольшую воздушную трубку для перемешивания сжатым
воздухом суспензии выпавшего красителя. При недостаточном перемеши-
вании может случиться, что большая часть красителя останется на дне
монтежю. Все железные обручи должны быть тщательно окрашены су-
риком, иногда окрашивают даже весь чаи. Если жидкость в чане нагре-
вают до кипения, то для предохранения помещения от выделяющихся
паров и дтя безопасности чаны снабжают крышками, также необходима^
хорошо действующая вытяжная труба, Это устройство ясно показано на
рис. 56 (см, стр, 334). Для усиления тяги в вытяжную трубу вставляется^-
воздушная нли паровая трубка. /
Часто из дерева делают плиты и рамы фильтр-прессов; в тех слу-
чаях, когда обработке подвергаются спиртовые жидкости, применяют
вместо смолистой сосны лиственницу или лучше дуб. На фильтр-прессах
применяются небольшие бочечные краны иа грушевого дерева.
Кожа идет для изготовления приводных ремней трансмиссий, кожа-
ных манжет гидравлических прессов и для других менее важных
целей.
Из искусственных органических материалов следует прежде всего
упомянуть каучук. Он употребляется в различных видах, из иего делают
шланги, покрытия центрифуг (эбонит), черпаки, а также эбонитовые
краны. Гуммированные центрифуги оказались весьма пригодными, однако
они редко применяются в промышленности красителей. В таких аппаратах
центрифугируют галловую кислоту, однако чаще работают с медными кор-
зинами или корзинами, освинцованными шоопированием. Хлорироваииый
каучук и синтетический хлоркаучук (неопрен) употребляется для.
кислотоупорной футеровки трубопроводов, сосудов и т. д.
Пластмассы, как например бакелит, поллопас, цибанит н родственные
им вещества, применяются только в специальных случаях, так как их:
механическая прочность большей частью недостаточна. В этих случаях
лучше применять эбонит, который может служить также в каче-.
стве материала для трубопровода для концентрированной соляной
кислоты.
Фильтровальные ткани, которые делаются нз х л о п ч а т о-;
бумажного волоки а, джута, пеньки н шерсти, обрабаты-
ваются органическими веществами, В фильтр-прессах применяется хлоп- '
чатобумажная ткань, редко шерстяная. Сильнокислые осадки отжимают
через ткань из верблюжьей шерсти; одно время были в продаже;
волосяные ткаии, по прочности они превосходили все известные ткани,
В качестве фильтровальной ткани, но не для фильтр-ирсссов часто приме-
няют так называемые ннтрофильтры*. Их делают всегда из гото-
вой специальной фильтровальной материи с одинаковой основой и утком,
так как обычная хлопчатобумажная ткань при нитровании садится. Кис-
лотоупорные фильтры изготовляются следующим образом: сухой суровый
хлопчатобумажный фильтр слабо натягивают на алюминиевую раму и
окунают па 1 час в 85—88%-ную азотную кислоту при 15—20°, после?
этого иа 20 мин. в серную кислоту (уд. вес 1,84), затем его тщательно,
промывают. Такие фильтры устойчивы даже по отношению к 60%-ной
серной кислоте при 100°, но быстро разрушаются кислыми растворами
солей железа. В последнее время применяются фильтры нз п о л и винил-
хлорида, которые, отличаются большой устойчивостью и прочностью.
Они очень устойчивы к кислотам. Применяются также стеклянные
фильтровальные ткани, так как они также чрезвычайно
* Ннтрофильтры имеют очень умеренную механическую прочность.
Техническая характеристика производства
327
устойчивы к кислотам. Новые материалы типа естественных и искус-
ственных хлоркаучуков (неопрен) также показали себя с луч-
шей стороны,
П. ТЕХНИЧЕСКАЯ ХАРАКТЕРИСТИКА ПРОИЗВОДСТВА
Общая стоимость мировой продукции красителей чрезвычайно мала,
она достигает 500 миллионов франков в год (1913), что составляет
’/ы часть стоимости годового сбора шерсти, не составляет даже 7s части
стоимости урожая хлопка и составляет лишь 7s часть стоимости добычи
каучука. Красители производятся в условиях сильной конкуренции и
представляют собой высококачественные продукты. Производство ии
одного из товаров, обращающихся на мировом рынке, пе требует столько
энергии, знаний и настойчивости, как производство красителей.
Развитие промышленности красителей привело к тому, что многие
секреты, которые ранее строго охранялись, стали всеобщим достоянием.
Как видно из данных, приведенных в энциклопедии технической химии
Ульмана, многие способы производства уже длительное время известны
большинству предприятий. Возможность перемены рабочими мест работы
также привела к тому, что в относительно короткое время каждое значи-
тельное улучшение производства становится известным конкуренту. Успех
деятельности больших заводов красителей определяется поэтому не секре-
тами производства, а многолетними традициями, отличной, организацией
и выпуском оригинальных красителей, производство которых защищено
патентами.
Было бы ошибочным предполагать, что только выпуск оригинальных
красителей может на длительное время обеспечить прочное положение
заводу красителей, однако в эту ошибку впадают не только молодые, не-
опытные химики; часто подобное мнение разделяют и опытные техники
и коммерсанты. Оригинальные красители являются своего рода краси-
выми цветами иа фоне обычных фабрикатов, и наряду с весьма доход-
ными оригинальными марками следует производить по возможности пол-
ный ассортимент рядовых красителей. Завод красителей может существо-
вать как крупное производство, лишь если ои вырабатывает в большом
масштабе массовые продукты. Такими массовыми, или ведущими, про-
дуктами в первую очередь являются черные красители, например прямой
черный 3, хромирующиеся черные красители различного строения (диа-
мант черный П Фау, эрнохром черный Ти т. д.). Наряду с черными краси-
телями, потребность в которых составляет более 50% общей потребности,
следует отметить синие красители, в первую очередь индиго, кубовый
синий О, прямой чисто-голубой и сернистый сииий; затем следуют красные
красители и, наконец, желтые, такие, как хризофенин и нафтамии жел-
тый НН.’
Только наличие этих массовых марок дает предприятию возможность
сбывать свои оригинальные марки и сводить до минимума общие из-
держки производства. В специальной части книги неоднократно указыва-
лось, насколько важным является полное использование всех побочных
соединений, получающихся в производстве промежуточных продуктов.
Здесь следует добавить лишь следующее? различные заводы красителей
вступают между собой в соглашения, в соответствии с которыми они снаб-
жают друг друга важнейшими промежуточными продуктами по себестои-
мости и обмениваются опытом по их производству. Благодаря подобным
соглашениям становится возможным производить каждый промежуточный
продукт в очень больших масштабах и рационально использовать побочные
продукты, как например азотистую и сернистую кислоты, сероводород,
32«
Ill. Технические указания
тиосульфат и глауберову соль, ПонДгно, что такие объединения
заводов красителей должны производить Также и неорганические про-
дукты для того, чтобы быть независимыми в отношении едкого натра,
серной и соляной кислот, а также соды и хлора, а если возможно, то и
поваренной соли и угля,
Оборудование завода красителей должно быть наиболее современ-
ным; самой большой ошибкой, которая, к сожалению, встречается часто, if
является применение старой, плохо работающей аппаратуры. Часто пред-t’
ставляется необходимым в течение ночи переделывать установку для':
производства нового красителя; делом руководителя является обеспеченней
наиболее рациональной аппаратурой. Единовременная основательная ре/
конструкция аппаратуры почти всегда является более дешевой, чем ис-
пользование нерационального оборудования, требующего большого числа
рабочих и занимающего много места’, Почти всегда оказывается, что ко-
ренная, хотя и дорогая, реконструкция является в конечном счете наибо-
лее дешевой. Расчетами занимается специальное расчетное бюро, которое;
получает данные от инженера и производственного химика.
Для правильного функционирования такого сложного производства,-
как производство красителей, необходимо строгое разделение труда, хоро-
шая организация. Высшее руководство находится всегда в руках коммер-
сантов и химиков, которые ведают каждый своей областью, но всегда ,
работают в полном контакте по всем вопросам, Коммерческий директор,
занимается снабжением и сбытом продукции, в то время как дирекция пб S
химической части руководит производством, научно-исследовательской и
колористической лабораториями. Промежуточным звеном является ипфор- ;
мациошю-реклампое так называемое пропагандное бюро, в .задачу, кото- j
рого входит ведение текущих дел, включая рассмотрение рекламаций,.;
исследование новых своих и чужих красителей, составление проспектов.;
с выкрасками н т. д,
Поэтому работа химика на заводе красителей может быть весьма раз- /
личной в зависимости от того, где он работает: в колористической лабора- J
тории, в научной лаборатории, иа производстве, в патентном бюро или^
в каком-либо другом отделе. Задачей химика, работающего в научно-ис- /
следовательской лаборатории, является разработка новых научных про- !
блсм при постоянном изучении специальной литературы. Следует подчерк- ;
путь, что не имеет смысла приступать к решению какой-либо проблемы:^
до тщательного изучения вопроса по всем доступным источникам, Поэтомуд
на всех хорошо организованных заводах красителей имеется специальный^
информационный отдел, который по требованию па основании тщательно
ведущихся записей составляет справку, дающую возможность быстро иФ
полностью ориентироваться в литературе вопроса, Часто бывает необхо-ъ
димым распространить определенную реакцию на различные области и
систематически синтезировать, может быть, сотни красителей и лрепара- J
тов, так как оказывается, что в большинство случаев лишь очень немногие 5
из испытываемых соединений являются ценными (Эрлих — Гата 606)-
Если дирекция по согласованию с различными отделами, например коло- ;
ристической лабораторией, фармацевтической лабораторией или каким-/
либо другим отделом, находит какое-либо новое соединение или какой--;
либо новый метод достаточно интересным, то опыты производятся .
в несколько больших масштабах.
Для этой цели,имеется опытная установка, являющаяся про-
межуточным звеном, связывающим лабораторию и производство. На этой
установке находятся аппараты больших размеров, чем в лаборатории, но
не такой величины, как производственные аппараты. Предварительные
опыты дают представление о том, как пройдет реакция в больших мае- л
Техническая характеристика производства
329
штабах; часто такие опыты дают возможность сэкономить большие
суммы.
Далее на этой стадии ставится вопрос о том, следует лн запатенто-
вать новое вещество или метод, Патентное бюро должно решить, будет ли
действенной защита изобретения патентом или же, если сделанное наблю-
дение является ценным, лучше держать его в тайне до тех пор, пока не
будет разработана вся область и тем самым будет исключена опасность
обхода патента, Только в очень редких случаях отказываются от получе-
ния патента и пытаются держать изобретение в тайне; подобный метод
является ненадежным, и к нему прибегают только в случае крайней не-
обходимости.
Через определенные пром ежу тки времени химики отчитываются
о своей деятельности перед дирекцией для того, чтобы последняя всегда
была в курсе дела. Подобные отчеты сдаются ежемесячно или через боль-
шие, но также равномерные промежутки времени; они обрабатываются
иод наблюдением дирекции.
Прежде чем какой-либо продукт пускают в производство, в каль-
куляционном бюро подсчитывают его стоимость. Необходимые
данные это бюро получает от дирекции и заводского инже-
нера, В разделе «Р» приводится пример расчета для того, чтобы можно
было составить приблизительное представление о том, как определяется
стоимость красителя.
Производство, Все производство делится на три чдети: химико-
техническую. а нал ити ко-колористическую и инженерную,
Аппаратура обычно быстро портится вследствие разрушающего дей-
ствия реагентов, часто бывают нужны большие изменения в аппаратуре,
поэтому соотношение рабочих-химиков к ремонтным рабочим (слесари,
трубопроводчики, столяры, маляры, каменщики) составляет 2 ; 1, Мастер-
ские, которыми руководит производственный инженер, являются в первую
очередь ремонтными мастерскими. Если необходим ремонт или изменение
существующей установки, то химик, ведающий производством, обращается
к инженеру; если речь идет о значительных изменениях, то необходимо
предварительное разрешение дирекции. Все работы выполняются в соот-
ветствии (Якартой заказа; после окончания работы последняя поступает
в калькуляционное бюро для расчета.
Большие заводы красителей имеют собственные мастерские для изго-
товления аппаратуры, однако и они сдают заказы на крупную аппаратуру
на сторону машиностроительным заводам, с которыми они связаны до-
говорами, что обеспечивает быстроту и дешевизну поставок. Рекомен-
дуется не применять слишком разнообразную аппаратуру, для того- чтобы
замена какого-либо котла или какой-нибудь мешалки могла быть произве-
дена немедленно из запасов, имеющихся на складе. Часто достаточно
иметь небольшое количество запасных частей для многих аппаратов, если
эти запасные части взаимно заменяемы.
Расходы по производству складываются из расходов по ремонту
и по амортизации аппаратуры, а также производственных рас-
ходов различных видов. Часть из них вычисляется очень точно, часть же
определяется общей суммой и называется общими расходами, К расходам,
которые могуч быть определены сравнительно легко, относится заработ-
ная плата, которая рассчитывается на основании рабочих журналов
мастеров и производственных химиков. Далее подсчитывается расход пара
па основании показаний паромера, а также расход сжатого воздуха и
вакуума.
Расход пара. На заводах красителей расходуется значительное
количество лара; оно определяется количеством воды, которое необходимо
330 III. Технические указания
нагреть, и количеством выпаренной водыЛОсобенно большое количество |
пара расходуется на выпаривание жидкостей, полученных в результате >1
процессов восстановления. В настоящее время больше применяются мио- |
гокорпусные выпарные аппараты (двойного и тройного действия). В этих J
аппаратах пар используется два или три раза, причем отходящий пар из 1
первого корпуса отводится во второй корпус, в котором испаряется жид- I
кость, находящаяся под вакуумом. Эти аппараты устроены или подобно .л|
многокорпусным выпарным аппаратам, применяемым в сахарной п ромы ш-ЗЯ
леиности, или же они состоят из нагревательного сосуда, который на- -я
ходится рядом с резервуаром для жидкости. Жидкость заставляют прохо-.^а
дить через трубчатый испаритель, благодаря чему достигается энергичная^»
циркуляция, кроме того, иакипь (большей частью гипс) оседает толыда'Ц
в вспомогательных котлах, трубки которых могут быть сменены за''ие-..Я
сколько часов. Благодаря подобному многократному использованию парой
удается понизить расход угля пе менее чем на 25%; большие заводхой
красителей применяют почти исключительно трехкорпусные выпарин^д
аппараты, отличающиеся большой производительностью. Значительным
лучше можно использовать пар, если нагревать его ие до 5 ат, а додИ
60—100 ат. Прежде чем поступить в заводскую паровую сеть, пар высо-$а1
кого давления приводит в действие паровую турбину или поршиевухб^Д
паровую машину, работающую на перегретом паре. Перепад давления озЦЯ
60—100 ат до 5 ат дает такое ’Количество энергии, что завод можй^И
отдавать на сторону избыточный электрический ток. Было предложенная
также понижать давление пара до 2 ат, однако в этом случае приходнтйКЯ
применять слишком большие трубопроводы, и потери от лучеиспусканиями
особенно зимой, переходят допустимые пределы, В последнее время вве-ЗЯ
ден новый способ наиболее экономичного использования пара; пРиицйЗД®
этого метода, однако, известен давио. Пары выпариваемой жидкости отсаЯЯ
сывают при помощи турбовоздуходувки из герметически закржЯИ
того выпарного аппарата и под давлением около 3/4 ат проводят в систеймИ
труб, находящихся в таком же аппарате. При сжатии отходящих паро^И
выделяется значительное количество тепла; таким образом экономят
80% топлива. Подобные конструкции быстро входят в употребление. Этот®
способ использования пара заслуживает самого серьезного внимания.
Сжатый воздух и вакуум, Наряду с паром расходуется®
также сжатый воздух. В большинстве случаев работают с избьггочшя|^Н
давлением в 2—3 ат, которое создают при помощи поршневого или
тационного насоса. Количество потребляемого сжатого воздуха зависш^И
в первую очередь от числа имеющихся фильтр-прессов, так как боЛ(^Н
шая часть воздуха расходуется ими. Перед разгрузкой пресса каждадМИ
осадок долго продувается, т. е. через осадок пропускают сжатый в°зду»Н
до тех пор, пока не будет вытеснена основная масса маточного раствори®
Пресс в 40 рам при продувке потребляет в час 100 м3 сжатого воздум^И
(избыточное давление 2 атм).
Поэтому стоимость сжатого воздуха является довольно значительной»®
должна быть точно подсчитана, Вполне оправдали себя изображенные Ня®
рис, 39 (см. стр. 307) компрессор и вакуумный насос системы ВиттМ^Я
Стоимость воды также должна быть определена (согласно точным
мерениям инженера), так как она расходуется в больших количества?^®
особенно для охлаждения конденсаторов,
Задачи заводского химика, Работа химика иа завоД||®
является одной из наиболее интересных, так как химическими реакциявда®
не так прост» управлять, за ними следует тщательно следить и направляТ^Я
в желательную сторону. Химик всегда должен быть в курсе дела и в дета«'*Д
лях знать кажд ю стадию процесса. ДяН
Техническая характеристика производства
331
Организационные производственные меропри я-
ти я. Необходимое для производства сырье заказывается по специальным
картам, которые за день отправляются на склады и в редких случаях дру-
гому заводу. Химикалии накануне вечером доставляются в производствен-
ное помещение для того, чтобы к началу производства все было готово.
Химик отвечает за готовые продукты до момента их вывоза в сухом виде
из производственного помещеяия. Так как многие красители чувстви-
тельны к повышенной температуре и требуют поэтому осторожной обра-
ботки, то сушилка всегда должна находиться под наблюдением химика,
чтобы ои мог следить за влиянием сушки на концентрацию и оттенок кра-
сителя. О подобных случаях упоминалось при описании получения мети-
леиового зеленого и азожелтого.
Рис, 52. Вакуумный сушильный шкаф для красителей системы Пассбурга.
К о jfo ристическая лаборатория. Г отовый краситель по-
ступает из сушильного отделения в колористическую лабораторию, где
производят крашение небольшой средней пробы параллельно с типом
(эталоном). Полученные данные немедленно сообщаются в дирекцию,
в калькуляционное бюро и химику, дли того чтобы все эти инстанции
были в курсе дела. Очень часто небольшая проба красителя отбирается
д,пя крашения сейчас же после разгрузки фильтр-прессов, для того чтобы
установить, не произошло ли ошибки в процессе производства кра-
сителя.
Сушка. В последнее время сушку все чаще производят в вакуум-
сушильиых шкафах, так как оказалось, что они расходуют меньше пара
и продукт получается более концентрированным. На рис. 52 показан со-
временный вакуум-сушильный шкаф, применяемый на многих заводах.
Стойкие промежуточные продукты, как например (3-иафталинсульфокислый
натрий я простые азокрасители, сушат на обычных паровых плитах или
даже в канальных печах, работающих по принципу противотока. Но и
в этих случаях все больше применяют вакуум-су шильные шкафы, которые
обеспечивают экономию времени и места. Для сушки 1 кг красители
в вакуум-су шильном шкафу расходуется 6—6,5 кг пара, а в канальной
печи без вакуума около 9 кг.
332 III- Технические указания
Рис. 53. Эскиз дезинтегра-
тора „перплекс’1.
/ — загрузка; 2 — неподвижнее
пальцы; 3 — пальцы, враша|ОШнеся
со скоростью J-00—2000 об/мин;
4 — сито.
Во время сушки с целью ее ускорения продукт следует хотя бы один ’
раз измельчить. Так как при измельчении фильтр-прессных лепешек обра- j
зуется много пыли, то из сушильных помещений проводится отсос
пн л и. \
В последнее время пары из сушильных шкафов конденсируют в ба- 1
рометрических'конденсаторах, благодаря чему насосы значительно меиьше
портятся от кислых или щелочных ларов. J
Установка товарного продукта на тип. После того как |
несколько партий (загрузок) высушено, их измельчают и устанавливают 1
на определенную типовую концентрацию. Измельчение и смешение лроиз-а
водят обычно в смесовых отделениях, которые работают под руководством JI
колористической лаборатории (эта лаборатория ие имеет ничего общего^!
с коммерческой лабораторией информационно -реклам н.ош&З
бюро, так как она служит совершенно ииннЦ
целям).
Измельчение. В настоящее времяЦ
красители измельчают иа дезинтеграторах, схе-.я
магически изображенных на рис, 53 и 54. Про-л|
изводительиость этих машин превосходит вН
10—15 раз производительность старых дробцЛЬ*|Я
ных мельниц с бегунами и даже барабанов
железными шарами; в то же время красителЬ|Я
получается значительно более тоикоизмельченуЯ
ным. Много рекламаций по поводу недостачи
точной растворимости продукта следует пиипидД
сать неправильному измельчению, так как пршИ
применении старой аппаратуры красителвИ
спрессовывается в очень твердые слоистые та(3|Я
летки, которые с трудом растворяются. -Л|
Если возможно, для сокращения временяИ
перемешивания в мельницу одновременно с нраЖИ
сителем загружают необходимое колнчестВяИ
наполнителя, Концентрированный красит&ч^И
перелопачивают с наполнителем (глаубероВяМ
соль, поваренная соль, сода, декстрин) и смесЯМ
эту загружают в мельницы. Мельница,
браженная иа рисунке, снабжсиа автоматтаМ
ческим ситом и магнитом для удаленпМ
железных частиц, всегда присутствующих в красителе. Краситель ИЯ^В
мельчается в потоке ударными пальцами особой формы и до тех п0яМ
находится в вихреобразиом Движении, пока не достигнет такой топинЗД^И
что будет проходить через сито (см. рис. 53). При этом вследствяИ
действия центробежной силы захватывается много воздуха, которьг^И
необходимо выводить из аппарата. Особые фильтрующие мешки продси^Ж
говатой формы 7 (см. рис. 54) дают возможность воздуху выходиТН^И
одновременно они задерживают пыль. Основная масса летучей пы<®|М
оседает в камере 6t причем воздушный поток поступает тан ген цн а льВЯМ
к стенкам камеры. Если измельчают очень мягкие вещества, как НмН
пример нафтол или нафталин, то сито удаляют, так как оно очей|г^И
легко засоряется. Измельченный продукт послушает по короткому чер^И
вяку непосредственно в смесевой барабан, в котором он тщательйжЯ
перемешивается в течение нескольких часов. На рис. 55 изображали
такой современный смесевой барабан, снабженный переключайте
щимся червяком, который дает возможность производить загрузк^^Д
и разгрузку автоматически. Эти смесевые аппараты вмеш,г.ЮЯИ|
Рис. 54. Дезинтегратор для красителей.
I—загрузочная воронка; 2 — вибрирующий лоток, подающий измельчаемый материал с магнитными
уловителями; 3— дезинтегратор; 4 — мотор; 5 — пылецровод; 6 — пыльная камера; 7 —фильтрующие мешки.
Рис, 55. Аппарат для смешения красителей с автоматической загрузкой и выгрузкой
‘ (системы Хехст).
Рис. 56. Производственное помещение на заводе красителей,
гидравлический _пресс; 2— фильтр-пресс с деревянными рамами; 3—монтежю с железными распорками; 4 и 5 —деревянные чаны с механическими мешалками;
тяга с пароструйным вентилятором; 7 — трансмиссия; 8^ материальный трубопровод от автоклава; 9~ подвижной кран, (10 mi; 1Q — автоклав емкостью 1.о лгв;
71 и 12 — грузоподъемники (3 m). ,
Пример калькуляции простого красителя
335
до 4000 кг, и со временем они полностью вытеснят старые неэкономич-
ные смесевые барабаны, особенно при выпуске мяоготоннажных продук-
тов. Часто применяют и более простые смесители, которые работают при
помощи сжатого воздуха и вакуума, наподобие смесителей для силосова-
ния зерна. Некоторые красители следует измельчать вне производствен-
ного помещения (см. рис. 56), так как они либо огнеопасны (красители
из пикрамииовой кислоты), либо дают неприятную пыль при измельчении
(бенгальский синий или нафтоловый синий, см. стр. 277). Если в колори-
стической лаборатории установлено, что краситель удовлетворяет по от-
тенку и концентрации, то он поступает в у п а к о в о ч н о е отдел еН’Ие,
откуда направляется в продажу. Дирекция, калькуляционное бюро и про-
изводственный химик извещаются о плохом или хорошем выходе и
оттенке красителя. Г1а сдаче продукта, будь то краситель или промежу-
точный продукт, деятельность заводского химика заканчивается.
Р. ПРИМЕР КАЛЬКУЛЯЦИИ ПРОСТОГО КРАСИТЕЛЯ*
Оранжевый Н-кислотный оранжевый А
(сульфаниловая кислота — р-нафтол, см. стр. 236)
Расчет стоимости продукции промышленности красителей всегда производится,
калькуляционным бюро. Последнее ежедневно, еженедельно или ежемесячно получает
от различных производств необходимые данные, на основании которых стоимость.
может быть установлена самым точным образом. Должность калькулятора должно.,
занимать лицо, пользующееся доверием; калькулятор наряду с дирекцией является
важнейшим лицом на современном заводе красителей.
Цена продукта складывается из стоимости материалов и заработ-
ной платы. Каждая сумма, определяющая цену продукта, должна быть обоснована.,
точными справками.
Рассчитываем отдельные компоненты.
^-Нафтол
а) Натриевая соль $-нафталинсульфокислоты
Франки
260 кг нафч^лина по 11 фр. за 100 кг ........... 28,60
280 кг серной кислоты по 2,70 фр. за 100 кг. 7.56
60 кг соды по 9 фр. за 100 кг .............. 5,40
60 кг угля ло 2 фр. за 100 кг............ 1,20
350 кг соли по 1,4 фр. за 100 кг......... 4,90
Выход соли р-нафталинсульфокислоты 165% = 429 кг............. 47,66
ЮО кг стоят 11,10 фр.
Эта цена является заготовительной, или, как ее обозначают, ценой 1..
Она включает только стоимость материалов, покупаемых или получаемых заводом от.
других производств (серная кислота с завода кислот и т. д.). К этой цене добавляется,,
целый ряд так называемых издержек производства самого различного типа. Онн скла-
дываются из расходов на заработную плату, стоимости ремонта и амортизации аппа-
ратуры, стоимости салфеток для отжатия отфильтрованного осадка, сушки соли
сульфокислоты, транспортировку ее, измельчения, энергии, лара и воды. Все эти.
затраты должны быть точно определены для того, чтобы получить правильную кар-
тину в целом. Совершенно очевидно, что эта большая работа может быть выполнена,
на заводе только обученным персоналом.
* Цены и издержки производства, принятые в этих расчетах, относятся к 1913—
1914 гг. применительно к большому государству с собственным углем. Пример должен
лишь показать начинающему химику, как из многих отдельных элементов складывается
окончательная цена сравнительно очень>простого красителя. В Швейцарии, однако,,
следовало уже и в 1913 г, считать по более высоким ценам.
Till
Ш. 1 ехнические указания
Расчет заработной платы проводят на основании ежедневных записей в рабочих
журналах, которые ведутся мастерами и содержатся в строгом порядке.
Заводский химик должен возможно меньше заниматься такого рода администра-
тивной деятельностью, так как это мешает его непосредственно химической работе.
Но он регулярно, не реже чем раз в неделю, должен контролировать рабочий журнал
и журнал химикатов; проверенные числа сообщаются в калькуляционное бюро лишь
с его визой.
Таким же образом подсчитывают а другие элементы общей стоимости на осно-
вании проверки складов, ремонтных мастерских, а также данных завод-
ских ин Жеиерм. Время от времени производятся испытания, и на основании
точных измерений определяют расход пара и воды на данный продукт.
Вес эти издержки производства можно распределить на отдельные продукты
различным способом. Для простоты мы расходы относим на 100 кг сухого товара.
Предположим, что установлены следующие издержки на получение сухой соли
^-нафталинсульфокнслоты;
Франки
Заработная плата—-2 часа по 0,8 фр. на 100 кг (учитывай
расходы па страхование и социальные учреждения) .... 1,60
Энергия — 4 квт-ч по 1 сайт, (прессование, перемешивание
сжатый воздух)............................................. 0,16
Сушка и измельчение — 20 сайт, иа 100 кг .................... 0,20:‘
Издержки производства на 100 кг р-нафталинсульфокислой .соли 1,96
Стоимость сырья (цеца I) на 100 кг соли
^-нафтали нсульфокислотм ............................... 11,10
13,06
б) Плавление натриевой соли
Для облегчения расчетов предположим, что плавится 100 кг, хотя
обычно плавят большие количества, например 400—2000 кг.
Франки
100 кг «нафталиновой соли».............. 13,06
45 кг NaOH ио 17 фр. за 100 кг .............. 7,65
15 кг угля по 2 фр. за 100 кг ............... 0,30
20 кг серной кислоты по 2,7 фр. за 100 кг ......... 0,54
Зарплата по операциям плавления, растворения и перегонки 5,00
Топливо для плавления и перегонки, ,вода для охлаждения
и сжатый воздух . . . , .............. 2,00
Амортизация (на 100 кг 5 фр., выход 45 кг) ....... . 2,25
Всего. . 30,80
Вычитается стоимость полученного сульфита и глауберовой
соли................................................... 2,00
45 кг полученного чистого 9-иафтола стоят................... 28,80
Следовательно, 1 кг чистого нафтола стоит.................... 64 сайт.
К этому следует добавить общие расходы, которые составляют 5%,
поэтому себестоимвсть [3-нафтола составит округленно 67 сайт, за 1 кг.
Сушат н канальных печах отходящим теплом котлов для плавления.
Пример калькуляции простого красителя 337
Сульфаниловая кислота
а) Нитробензол
Франки
100 кг бензола по 32 сайт......................... 32,00
110 кг HNO3 75%-пой по 40 сайт, ............. 44,00
170 кг H3SO4 по 2,70 фр. на 100 кг................. 4,60
80,60
Выход 154 кг
Ремонт и амортизация 50 сайт, на 100 кг ........... 0,77
Зарплата 35 сапт. на 100 кг......................... 0,54
Всего па 154 кг нитробензола . . . 81,91
Вычитается 3 фр. за стоимость отработанных кислот, воз-
вращаемых в производство..................... 78,91
1 кг нитробензола с общими расходами стоит около 55 сайт.
б) Восстановление нитробензола
Предполагаем, что восстанавливают только 154 кг нитробензола, хотг
обычно сразу восстанавливают до 2000 кг.
Франки
Нитробензол 154 кг (округленна)................... 85,00
Железо 200 кг цо 3 сайт........................... 6,00
Соляная кислота 20 кг по 4 сапт.................... 0,80
Известь 8 кг по 1,25 фр. за 100 кг ............ . 0,10
Пар, ремонт, амортизация, перегонка, энергия и т. д, на
154 кг нитробензола.............................. 5,00
** Всего... 96,90
Выход: 110 кг. Себестоимость, включая общие расходы, составляет около 95 сайт,
за 1 кг,
в) Сульфирование анилина
ПО кг серной кислоты по 2,7 фр. за 100 кг.......... 2,97
93 кг анилина по 0,95 фр.......................... 88,35
91,32
Зарплата: 5 час. по 80 сайт........................ 4,00
Пар (печи для запекания!), уголь................' . 2,50
Амортизация па 100 кг сырого товара по 10 сайт..... 0,17
Сумма . . 97,99
В ы х о д: 163 кг 100°/о-ной сульфаниловой кислоты.
Себестоимость, включая общие расходы, около 65 сайт, за 1 кг.
Таким образом мы подсчитали стоимость обоих промежуточных про-
дуктов. Опа, конечно, колеблется в зависимости от условий, н это пока-
зывает, как трудно получить действительно точные числа.
Мы принимаем, что сульфаниловая кислота стоит 65 сайт, и р-наф-
тол — 67 cam;
338
Ш. Технические указания
Получение красителя нз сульфаниловой кислоты и ^-нафтола
Исходим из килограмм-моля и умножаем загрузки для получения
кислотного оранжевого, которые приведены на стр. 236, на 10 000.
Франки
176 кг сульфаниловой, кислоты по 65 сант. за 1 кг.............. 112,45
60 кг соды по 7 сант.'............................................ 4,20
144 кг р-нафтола по 67 сапт. за 1 кг.............................. 96,48
144 кг едкого натра уд. веса 0,88 по 6 фр. за 100 кг.............. 8,64
110 кг серной кислоты 2,70 фр. за 100 кг........................... 2,97
70 кг нитрита натрия 51 фр. за 100 кг.......................... 35,70
250 кг соды по 7 сант. за 1 тег.............................. . . 17,50
800 кг льда по 80 сант. за 100 кг................................ 6,40
200 кг соли по 1,4 фр. за 100 кг .................................. 2,80
Всего. . . 287,14 -г
Выход концентрированного товара, загризпенпого солью и содой, составляет 4,
~ 400 кг.
Франки
9,60
32,00
16,00
16,00
Зарплата 12 час. по 80 сант,..............................
расходы на 100 кг сухого концентрированного товара:
Сушка 400 кг по 8 фр. за 100 кг...........................
Перемешивание и измельчение по 4 фр. за 100 кг............
Воздух, пар, кода, энергия цо 4 фр. за 100 кг.............
Всего. . . 73,60 , -г |
Расходы по крашению относят либо к производственным, либо к общим расхо- л
дам, К производственным расходам следует, однако, относить только расходы по Крй-
шеиию производственными образцами после смесевого отделения, стоимость же работы -^
в информационно-рекламной лаборатории следует считать расходами по рекламирО- .-я
ванию. Эти последние значительно выше расходов по крашению заводскими образцами;.^
Обе эти статьи расходов нельзя объединять. Поэтому здесь мы учитываем лишь®
расходы ц о заводскому крашению, которые составляют по 1,8 фр.
00 кг изготовленного товара—итого 7,20 фр.
Франки
Расходы по Крашению........................................... 7,20
Прочие расходы.............................................. 73,60
Сумма расходов без общих производственных расходов.............. 80,80
Себестоимость кислотного оранжевого без общих производствен- .
них расходов за 400 кг........................................ 287,14 ='7|
Сумма . . 367,94 Ц
К этой себестоимости должна быть прибавлена определенная сумма, котораяЦ
слагается из так называемых общих производственных расходов. Она складывается И#-и
следующих величин: стоимость перевозки по железной дороге, содержание дворовыз^
рабочих, склада, охраны завода (вахтер, ночные сторожа и т. д). Сюда же следуетМ|
отнести расходы по аналитической и производственной лабораториям, за исключение!^
заработной платы химикам-исследователям. Эти суммы, смотря по размерам сбываемоя^$
продукции, могут сильно колебаться. Однако можно считать, что эти общие произвоДуй!
ственные расходы составляют 5—7% стоимости полученного красителя. Для некотОуул
рых продуктов вследствие сильной конкуренции отдельные расходы не учитываются,’:«
но эти расчеты зависят от особого решения высшей дирекции и находятся в компе-^
тецпии коммерсантов.* З
Таким образом, если принять размер общих расходов в 6% и прибавить к укЯ;
запцой выше сумме еще 6% от- 367,94 фр., что составляет 22.08 фр., то действитель-(-т
ная себестоимость (цена 11) составит 390,02 фр. за 400 кг чистого товара, или- |
около 0,98 фр. за 1 кг. Краситель доводится, как это указано на стр. 332, до желае- м
мой концентрации прибавлением соли. Д
IV. АНАЛИТИЧЕСКАЯ ЧАСТЬ
Б производстве красителей необходимо точное аналитическое опре-
деление исходных материалов. Для этого применяются как физические,
так н химические методы. Для многих продуктов определяются только
физические константы — температура плавления, температура застывания
и температура кипения. Так, например, анилин, толуидин, нитросоеди-
нения и др. характеризуются только этими константами. Б некоторых
случаях определяют еще удельный вес (плотность) и даже показатель
преломления монохроматического света. В большом труде Лунгс *
можно найти все необходимые указания. Часто этн константы точно
фиксируются в договорах и имн руководствуются прн возникающих недо-
разумениях. Промежуточные продукты поступают теперь на рынрк
в таком чистом состоянии, что ни одно обоснованное желание не остается
не выполненным.
Из закупленных материалов отбирается небольшая средняя проба,
которая немедленно исследуется в аналитической лаборатории. Способ
отбора пробы часто устанавливается в договоре. Нагревание при опре-
делении темпергугуры плавления и застывания также производится боль-
шей частью по заранее обусловленным методам, На производстве очень
часто определяют содержание чистого вещества в техническом растворе
и лишь в конце процесса производства определяют общий выход. На
больших заводах постепенно все больше переходят к взвешиванию про-
изводственных растворов в заводской аппаратуре, для чего применяются
весы с предельной нагрузкой до 40 000 кг с чувствительностью до
100 г.
В производстве красителей принято в качестве характеристики для
каждого продукта приводить его молекулярный вес, который
просто вычисляется из химической формулы. Так как многие вещества
применяются в различной форме, например бензидин в виде сернокислой
соли и в виде основания, кислоты Клеве в виде свободных кислот и в виде
натриевых солей и т. д„ то принято каждое вещество всегда рассчиты-
вать по одному иг тому же молекулярному весу. Тогда естественно, что
соль содержит меньше чистого вещества, чем свободная кислота, По-
этому при покупках следует обращать внимание не только на цепу за
килограмм чистого товара, по также на молекулярный вес вещества.
Если, например, 1 кг бензидина (мол. вес 181) стоит 3 фр., а 1 кг серно-
кислого бензидина (мол, вес 282) 2 фр., то 1 кг чистого основания в сер-
нокислом бензидине стоит 2 • ~ = 3,02 фр., т. е. цена и в том и в дру-
гом случаях одинакова. Часто указывают содержание чистого вещества
Lunge, Uniersuchungsmeihodcn.
ллилц; «чеслия 4UCTb
в процентах. В приведенном выше примере сульфат является 65,2 %-иым J
(мол. вес 184); это значит, что если требуется 184 кг основания бензн- .)
дина (мол. вес 184), то следует взять 282 кг сульфата. J
Приготовление исходных титрованных растворов I
В торговой практике содержание нитрита натрия всегда определяют |
известным методом окисления перманганатом калия, одиако этот метод 1
дает всегда несколько завышенные результаты. Небольшая ошибка обу-J
словлена тем, что перманганатом калия, кроме азотистой кислоты, окис-J
ляются все другие окисляющиеся вещества. Несмотря иа это, данный 1
метод определения применяется на ряде заводов. Для химиков, работаю--1
щих иа заводах красителей, имеет значение лишь один метод определе-я
ния — при помощи сульфаниловой кислоты, по которому при некотором
навыке можно получить точные и более надежные результаты.
Получение чистой су л ьфаниловой кислоты J
250 г технической сульфаниловой кислоты растворяют в таком коли-1|
честве соды, чтобы образовался сильнощелочной раствор, и кипятят ДОЯ
полного удаления анилина. Объем составляет примерно 1 л. РастворЛ
фильтруют и сильно подкисляют концентрированной соляной кислотой.Я
Через 12 час. осадок отфильтровывают, промывают небольшим количе-Я
ством воды и растворяют кристаллы в 400 мл воды и таком количестве^
соды (примерно 60 г), чтобы образовался нейтральный раствор. ГорячийЯ
раствор при непрерывном перемешивании охлаждают до 0° и отфильтро-Я
вывают сульфаииловокислый натрий- Если имеется небольшая центри-Я
фуга, то маточный раствор полностью отделяют на ней. КристалЛыЯ
растворяют в 500 мл дистиллированной воды, раствор фильтруют и поД’Я
кисляют чистой концентрированной соляной кислотой. Перемешиванием
обеспечивает выпадение только небольших кристаллов, через день ихЯ
отфильтровывают, тщательно промывают осадок дистиллированной водойМ
до полного вымывания поваренной соли. Очищенные таким образомН
кристаллы перекристаллизовывают еще раз из кипящей дистиллирован-Я
ной воды и затем сушат в сушильном шкафу прн 120° до постоянногаЯ
веса. Кристаллы сохраняют в хорошо закрывающейся склянке с. прнтер-Я
той пробкой. Полученная таким образом сульфаниловая кислота почтаМ
белого цвета и содержит менее 0,01% примесей. Точно 173 г веществам
растворяют в 100 мл чистого аммиака (20% NH3) и при 17,5° растворе
разбавляют до 1 л, Такой раствор при хранении в темном месте остаетсвдИ
в течение многих месяцев без изменения, одиако через каждые 3 месяцяМ
его следует проверять. Этот раствор применяется для установки титрам
нормального раствора NaNO>. М
Приготовление 1 н. раств'Ора нитрита (и. NaNOs) М
75 г технического нитрита натрия растворяют в небольшом количе-Л
стве воды, раствор фильтруют и разбавляют при 17,5° точно до 1 л. ЭтиМЯ
раствором титруют 50 мл нормального раствора сульфаниловой кислотЫ-'Я
следующим образом. М
Раствор отмеряют пипеткой и выливают в стакан емкостью 0,5
разбавляют 200 Лл ледяной воды и подкисляют 25 мл концентрированнойЛ
соляпой кислоты. Из бюретки прибавляют под уровень жидкости раствор я
нитрита; после того как прибавлено 45 мл, остаток прибавляют по каП-;(Я
лям до тех пор, пока небольшая капля при легком саприкосиове-||
Определение аминов
341
нии (не следует размазывать!) с йодокрахмальной бумажкой не даст
немедленно (!) совсем слабую, но иеисчезающую синюю окраску. Диазо-
тирование длится примерно 10 мин. По количеству израсходованного
нитрита можно легко вычислить, сколько воды следует прибавить, чтобы
раствор был точно нормальным. Лучше раствор установить точно и ра-
ботать без поправки. Небольшой излишний труд прн точной установке
раствора вознаграждается в дальнейшей работе.
После того как приготовлены растворы сульфаниловой кислоты н
нитрита, приготовляют еще 1 н. раствор анилина. 200 мл чистого
анилина перегоняют из небольшой колбы (см. рис. 7 стр. 67), перегонку
ведут с такой скоростью, чтобы она была закончена в течение 3/4 часа,
Для приготовления раствора применяют анилин, перегоняющийся в пре-
делах 0,5° (между 184 и 185°). Следует отметить, что продажный анилин
химически чист. Удельный вес при 17,5° должен находиться в пределах
1,0260—1,0265. *
Точно 93 г чистого анилина растворяют в 150 мл чистой 30%-иой
соляной кислоты и при 17,5° разбавляют раствор до 1 л.
Если растворы нитрита натрия н сульфаниловой кислоты пригото-
влены правильно, то иа титрование 100 мл раствора сульфаниловой кис-
лоты или 100 мл 1 н. раствора апнлниа расходуется, ровно 100 мл ни-
трита.
Приготовление 0,1 и. раствора ф ен и л д’и азоиия
К 50 мл раствора анилина, помещенным в 0,5-литровую мерную колбу,
прибавляют 50 мл концентрированной соляной кислоты и раствор охла-
ждают, помещая его в ледяную воду. Затем при взбалтывании при-
бавляют 50 мл 1 и. раствора нитрита и оставляют стоять в ледяной
воде в течение 20 мии. По истечении этого времени реакция иа азоти-
стую кислоту совершенно исчезает, раствор разбавляют ледяной водой до
метки в 500 мм и раствор готов для анализа. Раствор следует готовить
не менее чем 20 мин., так как в указанных условиях для диазотирова-
ния требуется столько времени. Такой раствор остается без изменения
в темноте при 0° примерно в течение 4 час.; всегда следует употреблять
свежий раствор.
Определение аминов
а) Прямое определение
Амииы титруют в очень разбавленных солянокислых растворах ни-
тритом натрия, а затем проводят сочетание полученной соли диазония
с точно определенным количеством фенола, большей частью с солью
Шеффера. Этим контролируется процесс диазотирования. Некоторые
вещества, как например Аш-кислоту, амино-Р-кислоту ,и т. д., в одной
пробе диазотируют, а в другой сочетают с диазотированным анилином
или с иным диазосоединеиием. Иногда удается определить два вещества
в смеси; это возможно в том случае, если одно из них реагирует намного
быстрее, чем другое. Имея некоторый навык, можно довольно точно
определять содержание Г- и Р-соли в их смеси; Р-соль сочетается очень
быстро и дает с диазотированным анилином красный краситель; Г-соль
сочетается позже и дает желтый краситель. Наряду с подобными мето-
дами имеется еще целый ряд специальных методов, дающих возможность
определять отдельные составные частн смеси.
Для определения числа сочетания наряду с хлористым феиилдиазо-
нием употребляются лишь немногие диазосоставляющие. Многие заводы.
342
IV. Аналитическая часть
например, употребляют вместо анилина ж-ксилидин, однако в этом нет
никаких преимуществ, так как этот раствор менее устойчив, чем раствор
анилина. Применение в некоторых случаях /таминоацетанилида имеет
то преимущество, что ои несколько энергичнее сочетается и довольно
устойчив (см. о хромотроповой кислоте, стр. 190, 238); о- и л-нитроани-
липы применяются реже.
На 1 а нитрита берут 5 г соды или, если сочетание проводят в уксус-
нокислой среде, минимум 15 г уксуснокислого натрия; для нитроанилинов
требуется двойное количество. Если вещество содержит сульфогруппы,
то берут еще больше соды или ацетата. Температура сочетания ие должна
превышать 5°, и раствор должен быть очень разбавленным (примерно
1%-ным).
Избыток соли диазоиия определяют пробой па вытек, при этом легко
растворимые красители предварительно высаливают. В качестве реаген-
тов употребляют амины и фенолы, легко вступающие в реакцию соче-
тания, например резорцин, Р-соль, раствор Аш-кислоты. На некоторых
заводах применяют свежие растворы сип ильной кислоты, которые дают
желтую окраску. Избыток определяемого фенола или амина открывают,
простой пробой па вытек с раствором диазопия. Потери при этом так.
малы, что ими можно пренебречь.
б) Косвенное определение
Многие амины нельзя определять прямым диазотированием. Они
либо дают диазоамииосоединения, либо образуют соли диазония, кото-
рые окрашивают йодокрахмальную бумажку в черный цвет, подобно сво-
бодной азотистой кислоте. Поэтому такие амииы, например питроаиилин,
дихлоранилиц и т. д., определяют косвенным путем. Растворяют, напри-
мер, 0,01 моля соответствующего амина в концентрированной или<
несколько разбавленной соляной кислоте, раствор выливают в воду сод
льдом и диазотируют значительным избытком нитрита натрия. В мерной*)
колбе доводят прозрачный раствор диазоиия до точно определенного^
объема, а затем из бюретки или из мерного цилиндра прибавляют ЭТОТ^
раствор диазоиия к сильно содовощелочному раствору р-нафтола (содеру^
жапие последнего в растворе точно известно) до тех пор, пока в пробё^
красителя на вытек с раствором диазония не будет больше обнаруЖи-у"
ваться присутствие -нафтола. Обычно выбирают такие количественны^
соотношения, чтобы от израсходованного количества миллилитров легкв|
было перейти к процентному содержанию исследуемого амина. -у
Например, 3,45 г я-нитроанилина (— моля) растворяют в 10
30%-иой соляной кислоты и 10 мл воды. Прозрачный раствор выливают^
в смесь 50 г льда и 50 мл воды и добавляют 2 г 100%-ного нитрита-1
натрия в виде 20%-ного раствора. Вследствие наличия избытка азотиу|
стой кислоты (примерно 0,2 г NaNCh) образуется прозрачный раствору
в мерной колбе его доводят до объема 250 мл.' Й
100 мл этого раствора, отмеренные мерным цилиндром, прибавляю!^
порциями при хорошем перемешивании к раствору 1,44 г 100%-Hordhj
^-нафтола, 2 мл 30%-кого едкого натра и 20 г соды в 300 мл лединой'у
воды. Пробой и а вытек на фильтровальной бумаге с раствором диазония^
определяют момент исчезновения реакции на %нафтол. Если иитроанн-г
лип 100%-ный, то* его расходуется ровно 100 мл. Большей частью рас-
ходуется 101 —102 мл.
100
°', содержание амида =± —- --- 100.
количество мл
ииреиеление насртолов
313
Определение нафтолов
3-Нафто л
1,44 г (0,01 моля) нафтола растворяют в 2 мл 30%-ного раствора
едкого натра, разбавляют до 400 мл и прибавляют 25 мл 10%-ного
раствора соды. Из мерного цилиндра или из охлаждаемой льдом бюретки
добавляют холодный 0,1 и. раствор фенилдиазоиия до тех пор, пока проба
на вытек с раствором того же фенилдиазопия не будет больше давать
орапжево-красиой окраски. Очень часто через несколько секунд примеси
также дают цветное пятно. Однако окраска всегда мутноватая, и при
некотором навыке ее легко отличить от окраски, образуемой нафтолом.
Израсходованное количество миллилитров даст непосредственно процент-
ное содержание ^-нафтола. Чистота хорошего товара не менее 99,5%.
а - Н а ф т о л
а-Нафтол сочетается значительно легче, чем -нафтол, и в щелочном
растворе он дал бы завышенные результаты. Поэтому сочетание произ-
водят в уксуснокислой среде следующим образом.
а-Нафтол растворяют так же, как и нафтол, разбавляют, как ука-
зано выше для ^-нафтола, осаждают разбавленной уксусной кислотой
в присутствии 25 мл 25 %-кого раствора уксуснокислого натрия. После
этого проводят сочетание так, как это описано для ^-нафтола; после того
как исчезнет реакция на а-иафтол, растворяют, добавляв едкий натр,
снова осаждают уксусной кислотой и прибавляют «анилиновый раствор»
до исчезновения реакции иа а-иафтол. Часто приходится добавлять до
30% раствора диазоиия, так как краситель адсорбирует нафтол.
Подобным методам определяют только а-иафтол, так как й-нафтол
не сочетается в уксуснокислой среде. Если после определения а-нафтола
желают определить содержание Й-иафтола, то добавляют до тех пор
0,1 п. раствор /г-нитрофенилдиазония, пока весь |3-нафтол не вступит в со-
четание. Таким образом довольно легко в нечистом а-нафтоле опреде-
лить содержание обоих нафтолов.
Диоксинафта л ины (мол. вес 160)
Диоксииафталины определяют точно так же, как а-иафтол. Они
очень быстро сочетаются, а окраска от вторичного сочетания чаще всего
сильна и очень нечиста, так что определение конца сочетания не пред-
ставляет затруднений.
Определение а ми иосульфокислот
0,01 моля кислоты растворяют в необходимом количестве раствора
соды, разбавляют примерно до 250 мл, добавляют 25 мл концентрирован-
ной соляной кислоты и титруют 1 н. раствором нитрита. Количество по-
шедших иа титрование миллилитров, умноженное на 10, дает процентное
содержание. Всегда следует наносить каплю на нитритную бумагу при
легком соприкосновении; при размазывании нельзя получить точных
результатов.
Следует иметь в виду, что многие очень энергично действующие соли
диазония (особенно если они имеют сильнокислую реакцию вследствие
наличия минеральной кислоты) быстро вызывают синюю окраску иа
йодокрахмалыюй бумажке; следует также точно знать чувствительность
бумажки. Сульфаниловую, метаниловую и нафтиламинсульфокислоты
диазотируют при 15°. Кислоты Клеве нельзя определять таким простым
способом, так как они тотчас сочетаются сами с собой, В этом случае
лучше всего добавлять основное количество нитрита к нейтральному рас-
твору, а затем подкислять при хорошем перемешивании. Однако можно
и непосредственно диазотировать при 0°, добавляя нитрит до тех пор, *
пока начальная фиолетовая окраска не перейдет в чисто коричневую. 1
Предпочтительней, однако, косвенный метод, так как он является более. *
быстрым.
жестко; если нитритное число меньше, чем число сочета
Определение аминонафтол сульфокислот
Всегда производят два определения различными методами. Прежде
всего определяют нитритное число, т. е. количество нитрита, пошедшего
на титрование. После этого определяют число сочетания, т. е. количество,
израсходованного раствора диазония. Если оба числа совпадают, то
можно считать, что плавление аминонафтолсульфокислоты шло пра-:
вильно. Завышенное нитритное число указывает, что плавление прово-
дилось
ния, то это указывает, что плавление проводилось при слишком высокой
температуре. У правильно приготовленной аминонафтолсульфокислотай
нитритное число и число сочетания должны совпадать в пределах одного^
процента.
Едва лн есть необходимость подчеркивать, что все определения, ка^
и всякие определения в аналитической химии, следует производить
раза.
6
соль,
МОД,-
раз*
соды,
Аминонафтолдисульфокислота-1, 8, 3,
(Аш - к и с л от а)
а) Нитритное число (вычисляется на кислую натриевую
вес 341).
3,41 г Аш-кнслоты растворяют в 5 мл 10%-ного раствора
бавляют до 250 мл, осаждают 25 jo концентрированной соляной кислотй
и диазотируют прн 5° 1 н. раствором нитрита натрия. Аш-кислота дае^
красивое яичио-желтое ди аз осо единение, которое выпадает при высали-*
вании в виде прекрасных кристаллов. Количество миллилитров, умно-
женное на 10, дает процентное содержание,
б) Число сочетания. 3,41 г Аш-кислоты растворяют в 50 мл 10%-ногр,
раствора соды, разбавляют до 300 мл и при 0° добавляют 0,1 н. раствор
диазотированного анилина до минимального избытка. Избыток опредед
ляют следующим образом: на фильтровальную бумагу насыпают немног^
соли и на нее наносят несколько капель красного раствора продукта со-'
четаиия. Через 5 мин. бесцветный вытек перечеркивают ди азотированными
анилином. Если имеется еще Аш-кислота, то немедленно образуется?
красное окрашивание. Таким же образом определяется избыток диазо-^
тированного апилнна при помощи .Аш-кислоты. Последние количеств^
Аш-кислоты лишь с трудом отделяются от красителя, так что при пО-э
следнем испытании следует ждать четверть часа. В конце реакции имеет^
место более или менее сильное вторичное сочетание. Чем чище Аш-кнс<|
лота, тем слабее это вторичное окрашивание. Нитритное число хорошей^
Аш-кнслоты примерно на 0,3% выше, чем число сочетания. Израсходуй
ванное количество раствора диазотированного анилина в миллилитрах^
равно процентному содержанию.
Все а мин опафтол дисульфокислоты н моносульфокислоты опреде-
ляются подобным образом. Раствор диазония помещают в мерный ци-
линдр емкостью 100 мл и прямо по делениям отсчитывают проценты. На
многих заводах имеются бюретки с ледяным охлаждением; такие бюретки
изящны, ио сложны. Для перемешивания употребляют стеклянную па-
лочку, конец которой загнут в большую петлю. Сочетание проводят в чи-
стой фарфоровой чашке.
Определение моно- и ди сульфокислот нафтола,
моно- и дисульфокислот диоксинафталина
П ря мер: кислота Невиль-Винтера
(1,4- нафто л сульфокислот а) (мол. вес 224)
Сочетание с 0,1 н. раствором диазотированного анилина проводят
точно так же, как это указано в случае Аш-кнслоты, под конец краситель
высаливают в чашке, вследствие чего легко определить остаток нафтол-
сульфокислоты. Если исходить из 2,24 г нафтол сульфокислоты, то израс-
ходованное количество миллилитров раствора анилина сразу дает про-
центное содержание. Температура сочетания 0°.
Точно так же анализируют соль Шеффера, Р-соль и другие нафтол -
суль фоки слоты. Сультоны же следует предварительно расщепить неболь-
шим количеством горячего раствора едкого натра.
Для некоторых нафтол- и аминонафтолсульфокислот и родственных
соединений выгоднее применять вместо раствора фенилдназония гораздо
более устойчивый раствор диазотированного п-амнноацетаиилида, кото-
рый во многих случаях дает более трудно растворимые или .более легко
высаливаемые красители/так что конечную точку титрования легче рас-
познать. Чистый п-аминоацетанилид легко получить перекристаллизацией
технического продукта из горячей воды. Диазотирование проводят по
методуг указанному на стр. 222.
Моно- и ди сульф оки слоты диоксинафталина сочетаются очень быстро
и притом дважды; поэтому их сочетают с анилином или несколько более
энергичным и-аминоацетанилидом в уксуснокислых растворах в присут-
ствии уксуснокислого натрия. У многих кислот сочетание продолжается
несколько часов, например у диоксина фталин дисульфокис лоты-1,8,3,6
(хромотроповой кислоты). Образующийся при этом краситель хорошо
растворим и вследствие этого чрезвычайно медленно отделяется от непро-
реагировавшей хромотроповой кислоты, так что в этом случае следует
работать очень осторожно. 1
В некоторых случаях можно анализировать различные сульфокис-
лоты в смесях; полученные данные, однако, редко являются точными.
Так, например, удается довольно точно определить холь Шеффера
(нафтолсульфркислый натрий-2,6) наряду с Р-солью (иа фтол дисул ь-
фокислый натрий-2,3,6). Производят это следующим образом: сна-
чала в отдельной пробе при помощи реакции с «анилиновым раствором»
определяют общее содержание веществ, вступающих в сочетание. Дру-
гую пробу растворяют в минимальном количестве воды и обрабатывают
20-кратным объемом 96 %-ного спирта. Р-соль выпадает, можно опре-
делить в осадке ее содержание, а в экстракте — моиосуль фоки слоту. Не-
обходимо после прибавления спирта встряхивать осадокв течение */г часа,
так как в противном случае осадок адсорбирует слишком много соли
Шеффера.
Другой метод заключается в том, что вначале определяют общее
содержание в смеси при помощи «анилинового раствора», а после этого
во второй пробе формальдегидом удаляют соль Шеффера. Например,
растворяют 5 г смесц в 100 мл воды, добавляют 5 мл 30%-ной чистой
соляной кислоты .и 2,5 мл раствора формальдегида и нагревают
1 час на водяной бане. Разность между двумя определениями дает содер-
жание моносульфокислоты. 3
Третий метод — йодометрический—-основан на том, что йод реаги- ?
рует с Р-солью и с солью Шеффера и притом лучше всего в присутствии J
бикарбоната натрия. Вначале титруют 0,] н. раствором йода, применяя
избыток йода, который затем оттитровывают обратно. Другую пробу раз- |
деляют при помощи спирта, как это указывалось выше, и вновь тнтруют й
йодом. Этот метод является наилучшим, так как метод сочетания дает 'I
завышенные результаты.
Определение иафтиламиндисульфокислоть1-2,5,7 в смеси .'••а
с нафтиламиндисульфокислотой-2,6,8 йя
Определение обеих этих кислот в смеси очень просто. Вначале опре^лА
деляют общее содержание веществ, титруемых NaNOa. Затем титруют ж
2-нафтил амин-5,7-дисульфокислоту гинобромитом натрия.
2,5,7-кислота в солянокислом растворе гладко присоединяет
атома брома, в то время как 2,6,8-кислота к брому совершенно илдиффе-ЗЯ
рентпа. Имеется поразительная аналогия с солью Шеффера и с Р-солью^д
Для выполнения определения известное количество исследуемых киС^И
лот растворяют в 300-кратном количестве воды и прибавляют на каждыми
2 г сульфокислоты 20 мл концентрированной соляной кислоты. ДобавлеЦИ
иием льда понижают температуру до 0° и прибавляют 0,2 н. раетжшИ
NaOBr до явного посинения йодокрахмальной бумажки. 0,2 н. растваюМ
гипобромита готовят следующим образом: 14 г чистого КОН растворявж|И
в 500 мл воды и примерно 200 г чистого льда, при хорошем перемешиваем
нии добавляют 16 г брома. Затем разбавляют раствор до 1 л и устанавлмН|
вают титр по одному из известных методов (например, по мышья ков нстойОИ
кислоте). ч
Чистая 2,6,8-кислота даже в самых разбавленных растворах в пр£бяИ
сутствии соляной кислоты уже с одной каплей раствора гипобромита^И
мгновенно дает реакцию на свободный галоид. щИ
Объемное определение красителей методом ьЦИ
восстановления по Кнехту* , 'ЯИ
В некоторых случаях вместо определения концентрации и оттенкя^М
красителя методом крашения определяют содержание восстанавливаемыми^
веществ при помощи треххлористого титана (T1CI3). Этот изящный
год применим лишь в тех случаях, когда исследуемый продукт явЩИ
ляется однородным; его нельзя' применять, если исследуемый красителЦИ
состоит из различных изомеров или если в нем содержатся примеси, кЙ"||М
торыс также восстанавливаются. Поэтому этот метод мало распростраие^И|
па старых производствах красителей, имеющих хорошо оборудоваину^И
колористическую лабораторию, однако он применяется на небольших ЗД-^И
водах в Англии и Америке, так как гораздо проще произвести проетоб^М
титрование, чем деликатное крашение, результаты которого, конечИбОЯ
являются решающими. Оценка окраски, естественно, всегда сильно за,*Дд
висит от индивидуальных факторов и она может быть верно произведении
только обученным и совершенно надежным персоналом. . -уД
* Knecht Е., Hibbert Е,, New Reduction Methods in Volumetric Analysis^
with additions, new edition, Longmans, Green and Co., London. 1918.
Принцип метода Кнехта
Треххлористый титан (TiCl3) является чрезвычайно легко окисляе-
мым веществом, с водой он дает растворы красивой фиолетовой окраски;
при присоединении кислорода он гладко переходит в бесцветную
Ti(OH)4, или титановую кислоту. Раствором треххлористого титана из-
вестной концентрации удается количественно восстановить очень многие
красители различных классов. Конец восстановления определяют по
исчезновению окраски раствора исследуемого красителя.
При помощи треххлористого титана можно с большой степенью точ-
ности анализировать все азокрасители, далее тиазины, сафранины, три-
фенилметановые красители, индиго и многие другие. Приводимые крат-
кие. данные разъясняют этот метод; дальнейшие подробности можно
найти в работах Кнехта.
Приготовление раствора треххлористого титана
Принцип. Точно известное количество соли двухвалентного же-
леза окисляют до трехвалентного железа и восстанавливают последнее
треххлористым титаном; в качестве индикатора применяют роданистый
калий.
В качестве соли двухвалентного железа Кнехт применял двойную
соль сернокислого аммония и железа — так называемую соль Мора; он
окислял ее 0,02 и. рартйором перманганата калия в серной кислоте до
окисного соединения,/после чего оттитровывал обратно раствором трех-
хлористого титана. Другой такой же простой метод заключается в том,
что так называемую «цветочную проволоку» растворяют в чистой соляной
кислоте и образовавшийся раствор хлористого железа окисляют несколь-
кими каплями брома, избыток брома удаляют кипячением и затем ти-
труют раствором треххлористого титана *.
1. Продажный примерно 15%-ный раствор треххлористого титана
разбавляют, смешивая 100 мл раствора со 100 мл чистой примерно
30 %-иой HCI, и нагревают до кипения, после этого разбавляют дистилли-
рованной водой при 18° до 1 л. При хранении в атмосфере углекислого
газа нли водорода раствор в течение нескольких суток остается без из-
менения; через определенные промежутка времени его следует, однако,
проверять.
2. Для установки титра раствора треххлористого титана при помощи
железа употребляют раствор хлорного железа, который содержит
в литре точно 2 г железа. Реакция этого раствора должна быть миие-
ральнокислой, должно содержаться примерно 5%: НС1 или серной кис-
лоты.
Пример: найдено, что 6,85 мл треххлористого титана соответствуют
25 мл хлорного железа. Индикатором является, как указывалось, рода-
нистый калий (проба иа вытек). Так как раствор треххлористого железа
содержит 2 г железа в литре, то 1 мл TiCla соответствует 0,05 6,85 =
= 0,007299 г железа.
Пример 1. Метилоновый голубой (мол. вес 319,6).
Для восстановления одного моля метиленового голубого необходимы
2 эквивалента железа или 112 г Fe, поэтому 1 мл раствора треххлори-
стого титана соответствует:
0,007299-319,6 пАоло-з х гл мег,
-----— = 0,02083 г метиленового голубого CjgHjgNgSCl.
* См., например Тред в ел, Аналитическая химия, часть ТТ.
348
1 И. Аналитическая часть
Титрование метиленового голубого
Растворяют точно 0,2 г метиленового голубого (не содержащего:
цинка) в круглодонной колбе емкостью 250 мл примерно в 100 мл водЦ?
пропускают из аппарата Киппа углекислоту и нагревают до кипений)
В кипящий раствор добавляют из бюретки раствор треххлористого т#Р
тана до обесцвечивания.
При титровании метиленового голубого, не содержащего цинка (т
повой образец фирмы Гейги), на 0,2 г пошло 7,86 мл TiCb (средн
значение трех титрований). Содержание чистого красителя составляв
следовательно: 0,02083X7,86=0,1637 г, или 0,1637X100:0,2=81,8%.
Пример 2. Метиленовый зеленый (мол. вес 364,6).
Метиленовый зеленый является иитропроизводпым i
голубого; для его восстановления требуется 6 эквивалентов
Отсюда следует:
, 0,0 07299 V364.6
1 мл раствора треххлористого титана = —-------------
красителя.
Для титрования 0,2 г метиленового зеленого расходуется
хлористого титана; таким образом,
0,00809X20,00=1,618 г метиленового зеленого, или 80,9%.
Переход цвета ие очень четкий.
метиленов*
( железа.
—.0,00809
20 мл тр<
Азокрасители
Определение азокрасителей лучше всего производить в присутств
тартрата натрия, который препятствует осаждению трудно растворил®
красителей кислого характера (например, бензопурпурин*). Желт}
красители трудно титровать, так как тартрат титана имеет иитеисив
желтую окраску. Д
Восстановление проходит количественно по следующему уравиенн;
R—N = N—R' + 4TiCf_, 4- 4HCl=4TiCl4 X RNHg f- R'NHS. /
Следовательно, для определения нужно знать только молекулярно,
вес и число восстанавливаемых азогрупп; титрование проводится точЙ
так же, как указано выше.
Трифенилметаиовые красители
Восстановление происходит согласно следующему уравнению;
(Х)3ОН X 2TiCl3 + 2HCl=2TiCl4 + (Х)3Н Н3О.
Титруют до обесцвечивания; переход цвета большей частью оче$
резкий. Кнехт титровал таким образом фуксин, эозин, малахитовый зел^
ный, кислотный зеленый, анилиновый голубой, родамин и другие кр^
сители. 3
Сафранин ведет себя точно так же, как метиленовый голубой.
Имеющиеся в нашем распоряжении данные показывают, что мето;
Кнехта во многих случаях является более быстрым и таким же иа
дежь'ым, как и метод крашения, и поэтому его всячески следует ре
комендовать.
Knecht, loc. cit, S. 31—32
Наиболее употребительные реактивные бумажки
1. Красная и синяя лакмусовая бумажка. Чувстви-
тельна ко всем сильным н слабым кислотам и основаниям. Краснеет от
кислот, синеет от оснований.
Приготовление. Применять только лучший лакмус. Кубики, которые
содержат 50—90% гипса, измельчают и один раз экстрагируют спиртом.
После этого 4—5 а красителя растворяют в ] л воды и пропитывают рас-
твором чистую фильтровальную бумагу. Ее высушивают затем на бечев-
ках и разрезают на узкие полоски. Для получения красной лакмусовой
бумажки к раствору добавляют несколько капель уксусной кислоты, для
получения синей лакмусовой бумажки — несколько капель раствора
аммиака. Чем меиее резкой является окраска бумаги, тем она чувстви-
тельнее.
2. Бумажка конго. Реактив на сильные кислоты. Минераль-
ные кислоты дают синюю окраску, сильные органические кислоты — фио-
летовую.
Приготовление. 0,5 г концентрированного конго красного раство-
ряют в 1 л воды и добавляют 5 капель- уксусной кислоты. Фильтроваль-
ную бумагу пропитывают теплым раствором и сушат в чистом месте.
3. Тиазоловая бумажка (бумага мимоза). Реактив на сво-
бодную щелочь. Щелочами окрашивается в красный цвет; ее следует
предпочесть куркумовойДГеактивной бумаге. Готовят так же, как и бу-
мажку конго, только не Прибавляют уксусной кислоты. Аммиак действует
иа эту бумагу только в больших концентрациях.
4. Фенолфталеиновая бумажка. При действии щелочей
становится красной. Реагирует на аммиак и раствор соды, от действия
бикарбоната не изменяется. С успехом применяется для более точного
определения реакции.
Приготовление. 1 г фенолфталеина растворяют в 1 л горячей воды
и пропитывают горячим раствором фильтровальную бумагу.
5. Йодокрахмальная' бумажка (нитритная бумажка). Ре-
актив на азотистую и хлорноватистые кислоты (соли). От небольшого
количества окислителя она становится сине-фиолетовой, от большого —
коричневой. К ней следует слегка прикасаться, а не размазывать!
Приготовление. 10 г чистого крахмала растирают с небольшим коли-
чеством воды и при хорошем перемешивании прибавляют 1 л кипящей
воды. После охлаждения прибавляют 2 г йодистого калия и пропитывают
этим раствором чистые листы фильтровальной бумаги; высушивают в чи-
стом месте. Приготовленная таким образом бумага дает четкую реакцию
с раствором, в котором в лнтре 1%-ной соляной кислоты содержится
1 капля 1 и. раствора нитрита, и, следовательно, очень чувствительна.
6. Свинцовая бумага. Реактив на сероводород.
Приготовление. Фильтровальную бумагу пропитывают раствором,
содержащим 5 г азотнокислого свинца в литре воды, и высушивают ее на
воздухе, не содержащем сероводорода. Вместо этой бумаги можно при-
менять также влажную фильтровальную бумагу, пропитанную раствором
железного купороса или раствором свинцового сахара.
Растворы реактивов для пробы иа вытек
1. Р а с т в о р А ш - к н с л о т ы, 1 % -ный с 5 % соды. Этот раствор
дает возможность определять пробой иа вытек легко сочетающиеся диазо-
соедипения. Вместо Аш-кислоты применяют также Р-соль, р-нафтол, си-
нильную кислоту и т. Д.
2. Раствор резорцина. 1%-ный с 5% соды. Этот растворЯ^И
реагирует со всеми диазосоединениями, даже с такими, которые с Ащ.
кислотой не реагируют (аминоиафтосульфокислоты-1,2,4), 'Ян
3. Диазотированный п - нитроанилин. Реагирует coJ^Hh
всеми фенолами и аминами. Его следует хранить в темном месте, через
1—2 дня он с содой дает желтую окраску, поэтому его следует употреб^^^Ви
лять с осторожностью. С таким же успехом вместо n-нитроанилииа можноЛмр!
употреблять о-хлоранилин. 'ЯМКУ
4. Реактив «сульфон». 10%-ный раствор 4.4^-диамииоднФе*1иИ^
нилметан-2,2/-сульфона в 2 н. соляной кислоте (ср. стр. 220, сноска
При стоянии раствор окрашивается в зеленый цвет и из него выделяет^|ияВ|
темный осадок; несмотря на это, он длительное время годен для употреб#З^Вн|
ленйя. Нитрит дает интенсивную, ио нестойкую синюю окраску; реякп
не мешают сильные кислоты и слабые окислители (например, соли трех^ЙяИи
валентного железа). <
5. Раствор сернистых щелочей для определения тяжелых
металлов в растворе (железо, медь, олово и т. д.). '
ЙвмВВ
Определение качества цинковой пыли
1 г цинковой пыли растворяют вместе с 4 г бнхромата натрия в i
воды и добавляют при хорошем встряхивании 20 мл 20%-ной ссрноЙйн^^^
кислоты.
Из этого раствора отбирают 250 мл и разбавляют 900 мл воды.
тем добавляют 150 мл 20%-иого раствора серной кислоты и 100 мл 0,1
раствора йодистого калия. Раствор оставляют стоять в темном
в течение ’Д часа и затем оттитровывают избыток йода 0,1 и. растворо||В^И1?у
тиосульфата.
Для определения концентрации бихромата взвешивают точно 0,80$|м^
и анализируют, как указано выше.
Расчет: В — миллилитры раствора тиосульфата иа 0,800 а бихромат^дИКр^
А — миллилитры на 4 г бихромата -% цинковая пыль,
(В X 1,25 — А) X 1,308 = содержание циика в %.
Определение качества двуокиси свинца
Между двумя часовыми стеклами отвешивают хорошую среднюя|^^и|^
пробу 3—5 г пасты. Затем добавляют точно 5 г соли Мора и <жваДМк%
все это в колбу емкостью 200 мл. Нагревают уг часа на кипящей
бане и добавляют 25 мл концентрированной сериой кислоты.
Нагревают до кипения и по охлаждении оттитровывают избыток
Мора перманганатом калия. .
Йз
V. ОБ АНАЛИЗЕ ТОВАРНЫХ КРАСИТЕЛЕЙ
Определение строения неизвестного товарного красителя относится
к наиболее интересным и в то же время к наиболее трудным задачам
химика, работающего в области красителей. В прежние времена, когда
имелись только природные красители, задача идентификации была от-
носительно простой. Во многих случаях по внешнему виду продукта
можно было определить его происхождение. Кошениль, индиго,, ализарин
(крапп) и многие другие красители имеют сталь характерный вид, что
даже ие имеющий осдбаго опыта может без труда .установить, с чем
он имеет дело. Единственный вопрос, который следовало выяснить —
пригоден ли продукт для крашения, ие является ли он. испорчен-
ным или фальсифицированным. Многочисленные применявшиеся не-
органические краски—берлинская лазурь, окись хрома, окись железа,
киноварь и т. д.— могли быть определены непосредственно' при помощи
чисто качественных реакций. С появлением искусственных органических
красителей задачи химика-красочника стали совсем иными. Если еще
в начале возникновения промышленности красителей было относительно
легко отличить друг от друга, например, фуксин, анилиновый голубой,
метиловый фиолетовый или весьма простые азокрасители, то с^ появ-
лением на рынке все большего количества неизвестных красителей дело
значительно изменилось. Вначале пытались выйти из этого трудного
положения путем создания таблиц, в которых каждый известный кра-
ситель был точно описан. Но со временем появилось такое количество
новых красителей, что стало невозможным проследить за их появлением,
и известные таблицы красителей Шульца или Грина стали все больше
и больше устаревать. Вопрос теперь уже не заключался в том, «с. ка-
ким из описанных в таблице красителей идентичен имеющийся про-
дукт», а в том, «что представляет собой краситель, который еще нигде
в научной литературе точно ие упоминался».
Поэтому широко использовавшиеся ранее труды ие могли стать
основой для современного анализа красителей: необходимо было разра-
ботать новые методы взамен упоминаемых в устаревших трудах.
Подробное изложение в рамках данной книги всех известных фак-
тов завело бы нас слишком далеко. Однако и для начинающего было бы
интересно, хотя бы в общих чертах, ознакомиться с принципами новых
методов анализа красителей.
Поэтому в конце главы приведена литература по анализу красите-
лей и иа трех примерах описано, как следует поступить, чтобы узнать
строение неизвестного красителя. Автор изложил это подробно в док-
ладе, с которым рекомендуется ознакомиться *.
* J. Soc. Dyers and Colour, 45, ТЗЗ (1929).
• wu анализе товарных красителей
Так как отсутствуют точные основы, придерживаясь которых можно
было бы действовать аналогично тому, как это имеет место в иаучио-
обосиоваиной аналитической химии, следует удовлетвориться методами/
которые частично не 'имеют ничего общего с научным исследованием;
Однако по существу методы современного анализа красителей осно-
ваны на тех же принципах, что и классические приемы анализа. Прежде:’
всего для обеспечения правильной работы необходимо ясное представ^
ление о всех имеющихся литературных источниках. Литературы мождВ
не оказаться или она имеется в виде исключения в научных публика-
циях, которые затем собираются в справочниках Бейльштейиа и Гме??
лин-Краута. Имеются, одпако, и иные источники, которые во многих^
случаях являются полезными. yd
Этими литературными источниками являются, с одной стороны, паЛ
тенты, которые публикуются, с другой стороны, специальны®
журналы по химии красителей и, наконец, сообщения технй
ков, которые нам часто могут дать указания, к какому разделу отнсШ
сится изучаемый продукт.
В патентах часто находят упоминания об исследуемом продукте
часто на упаковке красителя стоит маркировка «запатентовано». Следсяжи
тельно, можно предположить, что патент опубликован. Далее, большем
частью этот же краситель можно найти в специальных журналах под РУ^И
рикой «Выпущены». Можно, следовательно, предположить, что дата вэяш
тия патента близка ко времени появления красителя иа рынке. Дал®Д
большей частью известно, кто является производителем красителя; тМЯ
ним образом, уже вначале имеются три рода данных: во-первых, ф£|Д
наличия патента, во-вторых, имеется определенная дата продазЯД
красителя, в-трстьих, большей частью известен производитель. Из этоиИ
можно заключить, что краситель был запатентован производителем, ’ИД
должен существовать патент иа его имя или на его имя переведен и щИ
до начала продажи патент был выдан или по крайней мере была сделаздД
заявка. Подобный род исследований, попятно, ие имеет ничего обш^Д
с настоящей наукой; ои является, как уже раз упоминалось, своего роДД
видом детективной деятельности ;|Д
Кроме факта существования патентов, имеется еще ряд важных (ЯД
стоятельсгв. Согласно патентным законам большинства стран, требу&пЯД
чтобы общий метод пояснялся лишь отдельными характерными приЯнД
рами, и не обязательно прямое упоминание того продукта, который буДЙД
производиться. Напротив, согласно швейцарскому патентному праву, ПяД
теитной защите подлежат лишь те вещества, которые точно описаны в пяД
тенте. Кроме того, каждый патент должен относиться к какому-лИмД
одному продукту. Итак, если известно, что краситель запатеитовай-ЦД
Швейцарии, то можно предположить, что он точно опнсаи в определённой
патенте. Поэтому швейцарские патенты могут быть с большой пользЙИ
привлечены для выяснения строения красителя. Однако известны мнбгаМ
численные случаи, когда эти методы расшифровки полностью отказывавд^И
а именно тогда, когда патент ие опубликован или когда краситель прояИ
водится после прекращения действия патента другой фирмы или е(ЗДН
произошло изменение названий. В этом случае часто можно устаиовияИ
строение путем сравнения свойств красителя со свойствами извести одД
продукта. -’Д
Первый источник, который давал возможность справиться о характерЗД
красителя, — известные таблицы Артура Грииа (Arthur Green)- ’./Jl
От tn на сегодня так устарели, что едва ли ими можно пользоваться^
Простые цветные реакции теперь не приводит к цели. Поэтому
обходимо искать новые пути, которые очень часто являются затяжными и
по которым ие всегда достигают цели.
Прежде всего необходимо установить, к какой группе краситель отно-
сится. Это сравнительно легко сделать, так как различные классы краси-
телей дают различные реакции. Если краситель является кубовым, то его
ищут в классе кубовых красителей н наблюдают цвет куба. Индигоидные
и тиоиндигоидиые красители дают бесцветные или желтые кубы. В кон-
центрированной серной кислоте они обычно образуют зеленовато-желтый
раствор и выпадают из щелочных восстановленных растворов при окисле-
нии воздухом в неизмененном виде. Кубовые антрахиноновые красители
дают обычно интенсивно окрашенные кубы, так что краситель типа иидиго
легко можно отличить от антрахиноновых кубовых красителей. Далее,
многие антрахиноновые кубовые красители дают при перегонке с цинковой
пылью антрацен или его производные.
Многие гетероциклические красители аналогично обычному индиго
при обработке восстановителями обесцвечиваются и при обратном окисле-
нии переходят в прежнее состояние. Сюда относятся азины, тиазины, оке*
азииы, акридины и другие им подобные красители. В противоположность
им трифенилметановые красители хотя и обесцвечиваются при восстанов-
лении, ио продукты их восстановления окисляются обратно воздухом зна-
чительно труднее, т. е. лейкооснования этого класса являются относи-
тельно стойкими. Напротив, хлоранилом (тетрахлорбензохнноном-1,4) оии
окисляются обратно о.чёиь легко и количественно (см. стр. 132).
В известных случаях удается получить аналитически чистые краси-
тели. Лучше всего это достигается в случае кубовых красителей, которые
очень часто могут быть перекристаллизованы из высококипяших раствори-
телей — хлорбензола, нитробензола, а также из ледяной уксусной кислоты
или пиридина. Например, тетрабромиидиго (циба голубой 2Б) можно
легко получить в аналитически чистом виде из дихлорбензола; аналогично
можно очистить и другие кубовые красители типа индаитреиа синего (ку-
бового синего О). В этом случае очень часто ценные, заключения может
дать количественный химический анализ, проведенный научной лабора-
торией, Для установления содержания алкоксильиых групп применяют
специальные методы анализа (Цейзсля и т. д.).
Следует иметь я виду и спектроскопический метод Формаиека, кото-
рый основан и а установлении кц»ксимума поглощения. Так как очень мно-
гие группы красителей, согласно исследованиям Формаиека, дают харак-
терные спектры поглощения, то применение этого метода часто является
полезным. Однако таблицы Формаиека устарели, они помогают только
в тех случаях, когда хотят справится о красителе, который в них описан,
ио оин ие дают ответа,так же как и таблицы Грина и таблицы красителей
Шульца, если речь идет о неописанных красителях.
Для азокрасителей существуют методы, которые во многих слу-
чаях дают хорошие результаты. Азокрасители в большинстве случаев
могут быть подвергнуты «восстановительному расщеплению», т. е, можно
молекулу расщепить по—N =N-rpynne так, что получаются два амииа,
которые могут быть исследованы раздельно. Одновременно при этом вос-
станавливаются, конечно, и иитрогруппы. В качестве восстановителей
могут быть использованы различные вещества: гидросульфит, хлористое
олово и соляная кислота, цинковая пыль и многие другие. Универсальных
восстановителей не существует. В известных случаях при применении гид-
росульфита может произойти сульфирование «продукта расщепления».
В случае применения хлористого олова во время восстановления иногда
имеет место перегруппировка первоначально образующегося гидразосо-
единения по типу * бензидиновой илн семидииовой перегруппировки
23 Зак 1905. Г. Э. Фирц-Давид+ Л. Кланке г
354
V. Об анализе товарных красителей
й
(например, оранжевый 1, т. е. продукт сочетания диазосульфаниловой
кислоты с 1-нафтолом, дает прн восстановлении солянокислым раствором
хлористого олова нс только 1,4-аминонафтол и сульфаниловую кислоту,
а вследствие внутримолекулярной перегруппировки частично и сем и дин)’.
С другой стороны, восстановление гидросульфитом идет лишь до гидразо-
соедипения, так что и здесь расщепление не удается. Однако такого рода
случаи представляют исключение.
Не вдаваясь в излишние подробности, в рамках настоящего обзора 3
необходимо рассмотреть лишь общие принципы анализа азокрасителей.
Прежде всего (как и при всех анализах красителей) нужно твердо
установить, является ли продукт однородным. Это производят путем рас-*.'^
пыливаиия небольшой пробы по смоченной фильтровальной бумаге. Если
имеют дело со смесью, то часто это узнают по образованию различно;Ц
окрашенных пятен *. Затем пробуют очистить краситель путем переоса-
ждення до получения однородного продукта. Поэтому во всех случая^- О
стараются получить возможно более концентрированный и чистый аиа^Я
литический материал. Это имеет значение для всех крас1^£||
телей вообще. .—
Восстановительное расщепление проводят лишь после того, как путем^Ц
предварительных опытов установят иаилучший вид восстановления. Полупи
ченный раствор исследуют, причем часто уже из теплого раствора выэд*^Я|
ляется более или менее чистая часть продукта восстановления. Ее отфилб^В
тровывают, перекристаллизовывают и по возможности количественнй^Я
анализируют. Во многих случаях продукт восстановления бывает уже||да
описан и может быть идентифицирован с помощью необходимых реакцнвдЯ
Для этой цели может оказаться полезной работа А. Бруннера (см. список!1И
литературы в конце этой главы). ЛОИ
При восстановлении хлористым оловом олово из раствора удалтйнН
электролитически. Этот метод удаления олова легко осуществляется Йя
имеет то большое преимущество перед осаждением его сероводородом, чтОН
в раствор не вводится дополнительных веществ. / 'ЖИ
Смесь различных веществ можно разделить как в этом, так и в иексйд|
торых других случаях с помощью метода хроматографии
с ко й адсорбции Цвета, Одним из вариантов его является хроматсНЯИ
графия на бумаге, которая во многих случаях дает отличные результатшяИ
Основателем этого метода является Гоппельсредер **, хотя ему и не УдЗ&аН
лось достичь практического успеха. Этот метод был позднее детальйм|Я|
разработан и улучшен ***. ' -рйМ
Если удается получить чистые продукты восстановления, их загежоЯ
количественно анализируют и пытаются по патентной литературе усгайО^Я
вить связь между данным продуктом и примером какого-либо патента. ЗДииВ
многих случаях, одиако, нет необходимости проводить количественный
мический анализ, так как полученные продукты могут быть хорошо из$яМ
вестными веществами (анилин, Аш-кислота, сульфаниловая кислотШШ
и т, п.), а также потому, что имеющиеся таблицы позволяют их идентгоЯи
фицировать. Например, 1 -амино-7 -кислота, 7-амино-Аш-кислота
быть идентифицированы непосредственно при помощи точно описаинЗДМ|
цветных реакций. Цветные реакции проводят следующим образом: разба^Яя
лениым раствором исследуемого вещества смачивают фильтрОвальиуЮМЗ|
* Смесь нерастворимых в воде красителей можно иногда распознать, распыляяуЯ
маленькую пробу на концентрированную серную кислоту, которую обычно помещаются
в небольшое углубление на белой пористой пластинке, ' ~,.Нд
** G о р р ь 1s г о с d е г F„ Capjllaranalyse, 1901.
*~* Schaeffer A., H.'indbueh der 1'arberci, Bd. IV, 1950, S, 52, Chimia, 5, 256
(1951); Textil-Praxjs, 6, 127 -130 (1951). Л
V. Об анализе товарных красителей. 355
бумагу п на нее наносят капли различных веществ, которыми могут быть
растворы солей металлов, кислоты, щелочи и такие окислители, как хлор-
ное железо, перекись водорода и т. п. По наблюдаемым окраскам в очень
многих случаях тотчас можно заключить о строении «продукта расщепле-
ния»; вместе с этим становится известной часть красителя. Если «про-
дукты расщепления» исследованы достаточно, то можно затем либо на
основании патентов, либо на основа пип полученных результатов перейти
к синтезу красителя. Когда это -удается сделать, задача решена.
Далее будут описаны анализы трех красителей, которые при своем
появлении б&ли неизвестны. Три выбранных примера должны показать,
как в данных случаях следует подходить, чтобы достигнуть цели.
1. Поляр ярко-красный ЗБ (фирмы «Гейги»), Он не был найден
в немецких патентах.
2. Беп зосветопроч по-с еры й БЛ (фирмы «Байер»), Для пего
может быть в литературе на идея патент,
3. С у ль фофлавин яркий (фирмы И. Г.). В данном случае, в до-
ступном каждому сообщении, была названа фамилия изобретателя и
тем самым было дано указание на немецкий патент, вследствие чего
анализ оказался излишним, .
/П о л яр ярко-красный ЗБ
Этот краситель только в последнее время был включен в карты
образцов красителей, прочных к кислотам и к валке, фирмы «Гейги»
в Базеле. Так как названная фирма в течение нескольких лет не заявляла
к патентованию получение прочных к валке азокрасителей, то следует
предположить, что поляр ярко-красный либо описан в старых патентах,
либо вообще не может патентоваться.
Восстановление гидросульфитом: растворяют 30 г псреосаждснного
красителя в 250 мл воды и в таком количестве соды, чтобы раствор имел
отчетливую щелочную реакцию па лакмус. В кипящий раствор всыпают
небольшими порциями гидросульфит до тех пор, пока раствор нс сде-
лается бесцветным. Избытка гидросульфита следует по возможности
избегать, так как легко может выделиться сера.
Всплывающее на поверхность желтого раствора продукта восстано-
вления желтовато-коричневое масло при охлаждении кристаллизуется.
После двукратной перекристаллизации из водного спирта с добавлением
животного угля основание плавится при 68°, Раствор продукта восстано-
вления после фильтрования подкисляют и добавляют поваренной соли;
при этом через короткое время образуется объемистый белый осадок,
который отфильтровывают и промывают раствором поваренной соли. Из
5() а продажного продукта получают 9—10 г очищенного основания и 15 а
сульфокислоты.
Основание имеет следующие свойства: растворимо в разбавленной
соляной кислоте только при нагревании, при охлаждении выпадает со-
лянокислая соль с т, пл.* 168—170°, которая гидролизуется водой. Если
нагревать солянокислый раствор до 80°, то происходит частичное разло-
жение с образованием масла, летучего с водяным паром. Основание
содержит галоид, ио не содержит серы. Определение азота и галоида
дает соотношение хлора к азоту 1:1, молекулярный вес 219. Темпера-
тура плавления ацетильного производного 166°.
Полученная при высаливании сульфокислота диазотируется и соче-
тается, во всем остальном является очень устойчивой. Ее свойства иден-
тичны свойствам N-аиил-Атп- или К-кисл ты
Зэо
i'. ud анализе товарных красителей
Восстановление хлористым оловом. Раствор 20 а очищенного краси-
теля в 250—300 мл воды нагревают в круглодонной колбе с обратный
холодильником и мешалкой до кипения. К кипящей смеси добавляют рас-
твор 40 г хлористого олова в 100 мл концентрированной соляной кислоту
(уд. вес 1,19) и кипятят 3 часа. Если в течение этого времени ие про-
исходит полного обесцвечивании раствора, то добавляют еще некоторой
количество раствора хлористого олова в соляной кислоте.
Олово из раствора удаляют электролизом. Для этого жидкость иат
,’1ивают в кислотоупорную глиняную ячейку емкостью 350 мл и помещают
ее в стакан из фарфора или стекла пнрекс, который наполняют 10%-Hofc
серной кислотой таким образом, что у анода и катода устанавливаете^
одинаковый уровень. Олово выделяется прн 80—90° иа электроде, изгое
товлеппом из медпой сетки, В качестве анода служит пластина из элек*-
тродного угля. Все количество олова, которое применялось для BoccTft-j
н овл е ни я 20 г красителя, удаетси таким образом выделить в течение 4-rs
5 час. при э. д. с. 8 в и токе 6—8 а. Электролиз продолжают до выделу
иия водорода. Раствор, нз которого по удалении олова ничего ие вы-
кристаллизовывается, упаривают в вакууме до половины объема. Прй!
охлаждении выделяетси светло-серый порошок, который дает реакции;
7-амиио-Аш-кислоты. Спектр окисленного раствора имеет следующей:
полосы. .
Х'= 530, 490 да (из 7-амино-Аш-кислоты X = 528, 491 да).
Фильтрат, который имеет сильный запах толуол сульфохлор ид а; вь$«
паривают досуха. Серый остаток обрабатывают содой и встряхиваю^
с эфиром. Из эфирного раствора кристаллизуется вещество с т. пл. ]4$?*
которое растворяется как в сап я ной кислоте, так и в растворе едког
натра, по ие растворяетси в соде. Хлорное железо вызывает появленн
красной окраски солянокислого раствора. Содержащее галонд веществ
которое диазотируется и сочетается с Р-солью с образованием красител
является, по-видимому, хлораминофенолом.
1 г сульфокислоты, полученной при восстановлении гидросульфиту
кипятят с обратным холодильником с 20 мл 10%-иой соляной кислота
Уже через короткое время раствор окисляется воздухом или окислит
лями, приобретая характерную красную окраску, которую дает 7-амин<
1,8-аминонафтолсульфокислота. Так как известно, что красители —щ
ляры содержат толуолсульфогруппу, наиболее вероятным является пре;
положение, что связанный с азотом ацильный остаток представлиет собо
я-толуол сульф окис лоту.
Идентификацию основания проводили следующим образом. Так кв
при восстановлении хлористым оловом образуется амииофенол, мож^
считать, что основание представляет собой сложный или простой эфи
Кипятят 8*г основания с 8 мл 20 %-ной соляной кислоты в колбе с ииех<
дящим холодильником и пропускают водяной пар. Отгониется слезотощ
вая жидкость, тяжелее воды, с т. кип. 175°. С азотнокислым серебро
при нагревании образуется хлористое серебро. Окисление перманганата
калия в нейтральном растворе протекает очень быстро и приводит к Ш
лучению бензойной кислоты, т. пл. 121°. Итак, в дистилляте содержите
хлористый бензил. ' 4
Жидкость в колбе подщелачивают, фильтруют, подкисляют и обрв*;
батывают раствором соды. В течение дни выкристаллизовываются белые1
листочки с т. пл. 139°, которые идентичны с хлораминофенолом, полу*
ченным при восстановлении красителя в кислой среде. Они идентичны
‘также с хлорамннофенолом, который соответствует 2-нитро-4-хлорфеиолу.
Поэтому основание явлиется бензиловым эфиром 4-хлор-2-амииофенола,
Он может быть получен синтетически следующим образом. 1,4-Дихлор-2-
v. ut> анализе товарных красителей
нитробензол нагревают с 2,5 моля 10%-ного раствора едкого натра в авто-
клаве в течение 10 час. при 150—160°*. Образовавшуюся натриевую соль
нитрохлорфенола кипятят с хлористым бензилом в спиртовом растворе
по Фрише ** в течение 5 час. и восстанавливают полученный нитроэфир.
Полученное таким образом основание плавнтси при 68° смешаииаи проба
с основанием из красителя не дает депрессии.
Итак, поляр ярко-красный ЗБ фирмы «Гейги», согласно данным ана-
лиза, имеет следующее строение.
Поляр ярко-красный ЗБ
Синтез. 0,1 ъ0ля Аш-кислоты растворяют в 0,2 моля соды и 200 мл
воды. При 60—70° при перемешивании добавлиют небольшими порциями
толуол сульфохлор ид до тех пор, пока раствор не перестанет реагировать
с нитритом, Для этого применяют 2—3-кратный избыток, так как окси-
группа реагирует также с толуолсульфохлоридом. Затем добавляют
столько соды, чтобы образовался 10%-иый раствор, и кипятят в течение
0,5 часа для отщепления остатка толуолсульфокислоты, связанного с гид-
роксильной группой. К охлажденному льдом раствору добавляют раствор
соли диазоиия, полученной из бензилового эфира 4-хлор-2-аминофенола,
После многочасового перемешивания нагревают до 60° н высаливают не-
большим количеством поваренной соли. Краситель очищают псреосажде-
инем.
Как растворы, так и выкраски не показывают разницы между типом
и синтетически полученным красителем.
Тип Синтезированный краситель
к НаО = 564,5; 522 р,р, Совершенно идентичны; полосы
резче, чем у типа, особенно при 565 р-р.
A CyijOH — 561; 522,5; 490 р-р- Идентичны
X H2SO4 =616; 581 р,р. Идентичны
Обе полосы не очень отчетливы
Моноазокрасители, содержащие в качестве азосоставляющей N-to-
луолсульфо-Аш-кислоту, не являются сами по себе новыми, так как они
упоминаются уже в патенте ***.
Применение для получения азокрасителей бензилового эфира амино-
фенола также описано в другом патенте ****, так что понятно, почему
поляр ярко-красный не может быть запатентован.
* Ср. стр. 101.
** Ann., 224, 141 (1884)'г
*** Герм. пат. 1Э0О81' (Ciba) [Frdl., VI, 866 (1900)].
**** Герм, пат, 142899 (М. L. В.) [Frdl., VII, 466 (1902)1
С научной точки зрения интересен факт легкого расщепления бензи-
лового эфира хлораминофенола с образованием хлористою бензила при
простом кипячении с разбавленной соляной кислотой *,
Б е из ос вет оп ро ч по с ер ы й БЛ (фирмыБайер)
Бензосветопрочно-серьш БЛ в основном состоит из синсвато-серогсГ*
красящего вещества с небольшой примесью оранжево-красно-коричневого
красителя и иногда немного прямого глубоко черного, При распылнваинн '
сухого порошка красителя на влажную фильтровальную бумагу легко Т-
можно распознать смесь.
При переосаждении (растворение в горячей воде и высаливание) по- у
лучают почти чистый синевато-серый краситель, маточный раствор окра-
шивает в тупой коричнево-серьгй цвет,
Восстановление, При восстановлении переосажденного краси-
теля хлористым оловом и соляной кислотой образуется коричневый Д
раствор. Из горячего раствора выпадает очень мало осадка, поэтому его У
непосредственно подвергают электролизу и немного упаривают, При охлщ пщ
ждепии раствора получают светло-коричневое, довольно чувствительное .
к воздуху вещество, которое при конденсации с фенаптренхиноном дает .у
соединение, характеризующееся в растворе концентрированной серной ; \ \
кислоты полосой поглощения при Л 611,0 уу, Полученный фенантразнн
идентичен с фенантразином из 1-амино-у-кнслоты, Итак, в красителе со-
держится у-кислота (сочетание в кислой среде) в качестве конечной
азосоставляющей, так как у-кнелота сочетается только один раз. По уу.../
другим реакциям соединение представляет собой также 1-амипо-у-кнс- "У'у:
лоту, УУ-
При дальнейшем выпаривании полученного при восстановлении рас- уд
твора выпадает втораи фракция, свойства которой не очень характерны, уУ..
так как продукт неоднороден, Вещество не дает характерной реакции дЛ
с хлорным железом, следовательно, не содержит оксигруппы (т. е, фе-У ,уХу
вольного гидроксила). Поэтому речь может идти только о нафтила мин- - д
сульфокислотах или об их амннопроизводных; продукта конденсации / У.
с фенантренхиноном не образуется,
В качестве третьей фракции получается легко растворимое вещество^..
которое дает слабую голубую флуоресценцию в водном илн слабощелоч-
ном растворе, подобно нафтиламинсульфокислотам типа кислоты ,у_;У,
Фрейнда нлн Лорана или кислоты IV, С нитритом и соляной кислотой об- у. К.
разуется диазосоединение, продукт сочетания которого с Р-солью после :-у'
подкисления характеризуется двумя не очень резкими линиями поглоще- У
нии при ).— 515,8 н 490,8 уу (-кислота IV), -У
Восстановление гидросульфитом и последующая перегонка щелочного . У/у :у
раствора с водяным паром не приводят к получению в дистилляте основа- , •••'
ния; следовательно, в красителе пе содержится простейшего ароматнче- ууу'д
ского амина бензольного ряда, а только сульфокислоты либо нафтил- -
амин, у у-
Так как такие светопрочные прямые красители являются обычно _
трисазокрасителями, то один нз них был синтезирован из 2,4,8-нафтнл-
аминдисульфокислоты, а-нафтнламипа, кислоты Клеве и -f-кислоты (со-
четание в кислой среде)
Scliort W, Г., Si wart М. L.. J. Cbem. Soc. (London), 1929, 553.
SO3U NH.J
SO3H I I
SO,;H SOsH
]3ензосветопрочно-с^[>1.гй БЛ*
Краситель окрашивает хлопок из слабощелочной ванны в синевато-
серый цвет, оттенок которого практически идентичен с оттенком, полу-
ченным при крашении очищенным беизосветопрочно-серым БЛ,
Получение красителя, 0,1 моля кислоты IV диазотируют
(как сульфаниловую кислоту) и к диазосоединению добавляют муравьи-
нокислый раствор нафтиламина; сочетание продолжается 24 часа. Нагре-
тый до кипения раствор высаливают и осаждают щелочью. Моноазокра-
ситель диазотируют обратным способом, диазосоединение отфильтровы-
вают и сочетают с кислотой Клеве в муравьинокислой среде, через 24 часа
подогревают (медленное сочетание), подщелачивают, высаливают и 4 раза
переосаждают, пока маточный раствор не приобретет красно-фиолетовый
цвет, Ди саз окра сите ль в щелочном растворе фиолетового цвета, а в кис-
лом — голубого, окрашивает хлопок. Его диазотируют, как указано выше,
выделяют диазосоединение н сочетают с Т -кислотой в уксуснокислой среде
(Y-кислоту растворяют в соде и подкисляют уксусной кислотой). После
сочетания медленно подогревают до 90°, слабо подщелачивают аммиаком,
высаливают и фильтруют горячим.
Сульфофлавин яркий
Этот красивый желтый краситель появился па рынке около 1930 года.
Он представляет собой желтый порошок, который легко растворяется
в воде с образованием чисто желтой окраски. Полученные с этим краси-
телем выкраски отличаются исключительно чистым почти ослепляющим
желтым тоном, который даже приблизительно не может быть достигнут
ни одним из известных красителей. Окраски интенсивно флуоресцируют
в свете ртутной лампы. Краситель содержит азот, но ие расщепляется при
восстановлении, следовательно, в нем отсутствует азогруппа. Итак, кра-
ситель представляет собой новое неизвестное соединение, которое, ве-
роятно, было запатентовано. Действительно, в сборнике Фридлендера * **
приведен патент***, а в следующих томах —4 дополнительных па-
тента ****, в которых описаны новые соединения следующей общей фор-
мулы, представляющие собой очень яркие желтые красители:
R
i
N
СО СО
I I '
00
NH2
R = H, алкил, арш или циклоалкил
* Герм, пат, 293184 (Frdl., ХТП, 515)',
** Forfschrftte der Teerfarbenfabrfkation, Bd. 16. S. 850.
*** Герм, пат. 494446,
**** Герм, чат. 49906&, 502570, 50Ц291, 554586,
В данном случае не было необходимости делать анализ красителя,^
так как вскоре после появления он был упомянут в книге Г. Креиц- ^
лейна. * На стр, 59 книги Кренцлейна мы читаем; f
«Большой прогресс в отношении чистоты оттенка представляет собой j
по сравнению с до сих пор наиболее чистым хинолиновым желтым кра-i
ситель сульфофлавин яркий (Эккерт, Хёхст, 1929), не отио-;
сящийся к классу трифепилметана», j
Действительно, в упомянутом выше патенте в качестве изобретателя
назван Эккерт, Так как во всех других патентах, в которых приводится^
имя этого же изобретателя, описываются продукты с совершенно другими^
свойствами, можно прямо принять, что в вышеназванном патенте упоми-^
нается и сульфофлавин яркий, соответствующий указанному выше строрЧ?
нию. Итак, в этом особом случае изучение легко доступной лнтерату^ш
освобождает химика от необходимости анализировать краситель.
Литература: Friedlaendcr Р„ Fortschritte der Teerfarbenfndustrie und ventrafi-^
dter Industriezwelge, Werden, Sein und Vergehen der kiinstlichen organise!^
Farbstoffe, Vortrag, gehalten von Dr. Georg Kranzlein, Verlag von Ferdinfifii
Enkc, Stuttgart, 1935.
Литература no анализу красителей Д
1. F r i e d I a n d e r P., Fierz-David H, E., Fortschritte der Teerfarbenfabrikatltj®
Springer, Berlin, 1877—1940.
2. Winther Adolf, Patente der organischen Chemie, 1877—1905, 5
3. Green Arthur, The Analysis of Dyestuffs., 3 ed,r London, 1920,
4. Schultz G., Farbstofftabellen, 7. Aufl, mit ErgSnzungsband, Leipzig, 1939. Д
5, Colour Index и дополнительный том, London, 1924. 1
6. Form a nek J., Untcrsuchung und Nachwefs organischer Farbstoffe atif spektr
kopischem Wege, Berlin, Springer, 1908—1927.
7. Brunner A,, Analyse der Azofarbstoffe, Berlin, Springer, 1929.
8. F i e г z - D a v i d H. E„ The Analysis of Dyestuffs, Yesterday and To-day, Socb
of Dyers and Colourists, 45, 1929, p, 133.
9. Fierz-David, Matter M., Azo- and An thr a quinonoid Dyes containing 1
Cyanuric Ring, Society of Dyers and Colourists, 53, 424 (1937).
10. Дальнейшая литература имеется в вышеназванной книге A. Brunner
стр. 117, Так как очень многие из получаемых продуктов расщепления ни:
точно не описаны, то большие справочники, такие, как Beilstejns Handbuch '
organischen Chemie и др., помогают очень мало. Напротив, безусловно необ
димо подробно изучить имеющиеся патенты и количественно анализиров:
полученные соединения. См. введение в раздел об анализе,
И, В этой связи следует еще указать на список литературы у Fierz-Matt
(цнт. 9), особенно на Forster, Напсоп Society of Dyers and Colourists, 42, J
(1926).
12. Schaeffer A., Handbuch der Farberei, Bd, 1V, 1950.
* Крен ц л eiim Г,, Прошлое, настоящее и будущее искусственных оргаяи^
ческих красителей, М,, ОНТИ, 1935 стр, 59,
VI. ОПРЕДЕЛЕНИЕ СВЕТОПРОЧНОСТИ *
Под действием дневного нли солнечного света окраски претерпевают
изменения: оии выцветают и бледнеют. В зависимости от характера кра-
сителя и глубины окраски обесцвечивание происходит с различной ско-
ростью, Существуют окраски, которые обесцвечиваются в течение одного
солнечного дня или нескольких часов, У других заметное вьщветание
наблюдается лишь спустя несколько дней, педель и даже месяцев. Таким
образом, окраски обладают совершенно различной светопрочиостью.
Быстрое увеличение числа синтетических красителей заставило уже
давно классифицировать красители по степени их прочности. Сначала
для этой цели отдельные заводы красителей применяли сравнительные
йлкраски повышающейся светопроч пости. Эти сравнительные эталоны,
применявшиеся лишь на данном заводе, выбирались произвольно, и от-
сутствовала какая-либо связь между эталонами различных заводов кра-
сителей. Поэтому понятны все более настойчивые требования потреби-
телей об установлении единообразной системы определения светопроч-
ности.
База для этого появилась лишь после основания в 1911 году «Гер-
манской комиссии по вопросам определения прочности». Для нормиро-
вания светопрочности были предложены 8 степеней, из них первая озна-
чала наинизшую, а восьмая высшую светопрочиость.
В результате этих работ были впервые изданы в 1914 и 1916 гг.
сравнительные эталоны для хлопка (1) и для шерсти (2), и затем после
длительного перерыва между 1926 и 1928 гг. появились 5-степенные эта-
лоны для неутяжелениого (3) и утяжеленного шелка (4), а также для
вискозы (5) и ацетатного искусственного шелка (6). Только после этого
представилась впервые возможность классифицировать красители по
светопрочности по единой системе.
Однако нскоре оказалось, что этот метод имеет серьезные недостатки.
Прежде всего наличие шести серий эталонов, для которых требуется
33 красителя, осложняло работу; кроме того, вследствие разнооттеноч-
иости окрасок затруднялась надежная оценка светопрочности.
С другой стороны, что оказалось особенно важным, отсутствовала
связь между отстройкой эталонов иа различных волокнах; поэтому пока-
затели светопрочности, полученные на различных волокнах, нельзя было
сравнивать между собой. Этот недостаток стал особенно чувствительным
вследствие мощного развития промышленности искусственного шелка и
особенно применении смешанной пряжи и смешанных тканей- Красиль-
щику было совершенно невозможно выбрать иа основании таблиц
По г. Рису.,
прочности красители с одинаковой светопрочностью для окрашивания сме-
шанной пряжи топ в топ. Поэтому нужно было найтн какие-то вспомога-
тельные средства.
На основании многолетних работ фирма «И. Г. Фарбениндустри» пы-
талась составить типовые шкалы для отдельных текстильных волокон и
привести их в соответствие друг с другом, но убедилась, что этим путем
нельзя достичь единообразия, так как работами было совершенно отчет-
ливо показано, что на степень выцветания, кроме света, сильное влияний*
оказывают н такие факторы, как температура, 'интенсивность света, в осо-
бенности влажность воздуха, а также характер субстрата. Окраски иа
хлопке и искусственном шелке оказались особенно чувствительными “
к влажности, они выцветают гораздо быстрее во влажном воздухе, чем
в сухом. Наоборот, в случае шерсти наблюдаются незначительные коле-
бания, что и было установлено путем сравнительного облучения окрасок
по шерсти и хлопку в различных климатических условиях в 18 точках
земного шара.
Эти факты были положены в основу вновь созданной шкалы нз
8 эталонных окрасок на шерсти. Одновременно были найдены способы,
позволяющие избавиться от разнооттеночности, которая мешает работе; -A-yf
для этого было предложено ограничиться определенным рядом окрасок,
обладающих сходными оттенками. Особенно подходящей для этого ока-
залась серая шкала, так как с пей легче всего сравнивать окраски раз-
личных цветов. К сожалению, имеется недостаточное количество красите- '
лей серого цвета. В ряду синих оттенков дело обстоит гораздо благо-. :
приятнее. Путем искусного выбора красителей и интенсивности окраску
удалось составить шкалу из 8 эталонов, которые характеризуются уве-
личением степени светопрочности приблизительно в геометрической про- —
грессии; для достижения следующей, более высокой, степени прочности '’.У
требуется действие двойного количества света по сравнению с предше- . <<
ствующей степенью. Следует особенно подчеркнуть эту прогрессивность,
так как из наличия показателей светопрочности 1—8 нельзя сделать вы-
вода о таком построении шкалы. Лишь исходя из этих данных, можно. у. У,
правильно понять порядок величин показателей светопрочности и пра- г У-',
вильно их интерпретировать. Лучше всего величина этой прогрессии У
иллюстрируется практическим примером. Если 1-й эталон отчетливо вы- ' ,г'
цвел после одного-двух дней облучения, то продолжительность облучения '
следующих эталонов должна выражаться следующими числами: * <У
Эталон...................... 1 2 3 4 ,5 б 7 8 • "
Продолжительность облучения . . 2 4 8 16 32 (14 128 256 дней У У Л У
В зависимости от яркости дневного света и продолжительности соЛ- /
печного освещения имеет место большее или меньшее отклонение от этих ы1';-;.
чисел, поэтому их^нужно рассматривать как приблизительные средние уу У
величины. .. ?&*'
Эти числа показывают, что баллы от 1 до 3 соответствуют низкой У'т--
светопрочности, в то время как 4-й является средним, а 5-й более высо- <•>/'-
ким баллом. Для 6-го балда время экснозиции быстро возрастает и для ,
8-го достигает указанной выше продолжительности. Этн показатели
прочности удовлетворяют самым высоким требованиям; практически при .
столь длительном облучении большинство текстильных тканей заметно А
повреждается или полностью разрушается. На этом основании показа- ус-
тели светопрочности-для первых четырех баллов выражают целыми чис-
лами, а последующие — с половинным числом.
Эта новая синяя шкала была принята немецкой Комиссией по во-
просам определения прочности и впервые была опубликована в 1932 г.
в шестом издании «Verfahren, Normen und Туреп». В последующее время
введены еще некоторое улучшения шкалы, поэтому в восьмом издании
были введены новые эталоны.
Степень светопрочности 1 :0,8% ярко-голубой длч шерсти ФФР экстра
2:1,0% ярко-голубой для шерсти ФФБ
3 :1,2% яркий индоцианин 613
4:1,2% супрамин голубой ЕГ
5 : 1,О°/о цнанантрол РИкс
6:3,0% ализариновый непосредственно голу-
бой ЗГЛ
7:3,0% ни ди го золь 06Б
8 :3,0%, нндигозоль голубой АГГ
Хотя методики крашения точно разработаны, однако каждому пред-
приятию трудно самому изготовить эту синюю шкалу. Чтобы с -самого
начала устранить все причины ошибок, шкала изготовляется централи-
зованным путем и распространяется бесплатно немецкой комиссией по
вопросам определения прочности, а также немецкими нли швейцарскими
заводами красителей.
Введение в практику этой синей шкалы значительно упростило опре-
деление. Сдало возможным проводить сравнительное определение проч-
ности окрйсок при любой интенсивности и при любом цветовом топе и,
что особенно важно, на различных текстильных материалах. Благодаря
этому синяя шкала сделалась абсолютным масштабом для
определения светопрочности. Это простое вспомогательное средство дало
возможность каждому красильщику выражать светопрочность окрасок
наиболее изящным способом в баллах.
Определение светопрочности проводится очень просто. Испытуемый
образец и сипюю шкалу закрывают наполовину картоном, и открытую
часть облучают дневным светом (см. рис. 57).
Облучаемые образцы находятся на расстоянии
по меньшей мере 2 см от стекла, проницаемого
для ультрафиолетовых лучей, в хорошо вентили-
руемом ящике при наклоне образца в 45° к югу.
Как только на испытуемом образце заметно о т-
ч е т л и в о е выцветание, закрывают картоном
узкую полосу облучаемой части шкалы и образ-
ца и продолжают облучение до появления сле-
дующего отчетливого изменения, таким же об-
разом закрывают еще раз и облучают. Если рис> г)7 Ступенчатое
хотят сопоставить любое количество окрасок оснещение.
с различной светопрочностью с синен шкалой,
то нужно несколько изменить методику. В этом случае, не принимая во
внимание промежуточных изменений окрасок, облучают еннюю шкалу до
тех пор, пока не будет заметного отчетливого выцветания 4-го эталона.
Затем снова перекрывают полоску на шкале и одновременно на всех
испытуемых образцах и облучают до тех пор, пока ие обнаружится изме-
нение 6-го эталона. Затем, снова перекрывают полоску н освещают до
отчетливого изменения 7-го эталона. Таким образом устанавливают три
степени выцветания испытуемого образца н шкалы. Для уста-
новления светопрочности следует сопоставить характер выцветания испы-
туемого образца с характером выцветания синей шкалы; отдельные эта-
лоны шкалы характеризуют степень светопрочности. Степени сеетопроч-
пости от 1 до 4 выражают в целых числах и от 4 до 8 с точностью до
364
VI. fjnpeuejtenue caeiufipu^nuviu
половины единицы; эти числа отвечают следующим степеням прочностям
1 — незначительная, 3—умеренная, 5 — хорошая, 6—^очень хороша^
7 — превосходная, 8 — отличная, io
Таким образом проблема определения светопрочности окрас ожу
нашла простое н удобное разрешение. л
Далее необходимо было установить одинаковые методики оценка?
красителя. Известно, что светлые окраски выгорают скорее, чему"
темные. Поэтому один и тот же краситель имеет различную светопроч*
ность в зависимости от интенсивности окраски, В качестве первого важ4:
нейшего шага необходимо было точно установить интенсивность'
окраски, для которой проводилось определение прочности. Для это&
цели были разработаны так называемые вспомогательные тип
т. е, стандартные окраски одинаковой интенсивности в ваяк
иейшие цвета — желтый, оранжевый, красный, фиолетовый, голубой, зГ
левый, коричневый, серый. Исключение представляет ф л о т с к
синий и черный. В соответствии с практикой для этих цветов бь$
приняты более насыщенные окраски. ?
Второй важный фактор, который надлежит учитывать, это за О
с и м о с т ь светопрочиости от характера текстиль нот о fcij
л о к н а. Например, окраски на хлопке прямыми красителями отличают^
от светопрочиости окрасок на блестящей вискозе или матовом нску$
ствениом шелке. В еще большей степени отличаются по светопрочной
окраски на утяжеленном и неутяжелеином натуральном шелке, прич
на первом они гораздо менее устойчивы, чем на втором. Исходя из это:
было необходимо разработать вспомогательные типы * окрасок иа вЙ
нейших видах текстиля: на хлопке, искусственном шелке, ацетата)
искусственном шелке, утяжеленном натуральном шелке, неу тяжелен»
натуральном шелке, шерсти. у
Благодаря этим двум мерам удается достичь и 1
обходимого единообразия. Эти вспомогательна
типы соответствуют по интенсивности окраски Й'
всех цветах и на всех текстиль пых волокнах, что п о з в I
л нет сравнивать показатели светопрочиости и а в С Й
волокнах и для всех ассортиментов красителей.
Флотский синий и черный занимают, как уже упоминалось, особ1
положение. Вследствие того что интенсивность их окраски отличаем
от интенсивности по другим цветам, темио-синие и черные окраски ti
гут сравниваться только друг с другом.
Одиако такой способ сравнения ие всегда удовлетворяет. КаждСи
красильщику необходимо дать сведения о светопрочиости не ТоЛЬ
окрасок среднего тоиа, но и окрасок светлого и темного тоне
Поэтому было установлено все красители оценивать в окрасках тф
интенсивностей. Для эт^рго светлые и темные окраски готовят в совё
шеино определенном отношении к вспомогательным ТВ
п а м, а именно */з • 1 2. Это значит, что светлые тона получают при гф#
меиении трети количества красителя, а темные тона — двойного количФ
ства красителя по сравнению с тем, какое необходимо для достиженМ
интенсивности вспомогательного типа (среднего тоиа), В особых сЛУ*.
чаях делают исключения нз этого правила; так, светло-розовые иЛ4Ы
светло-серые тоиа можно оценивать в интенсивности */6 или среднВГфд
тона, а насыщенный коричневый в четырех- или шестикратной интсисйЯ >
* Эти вспомогательные типы окрасок можно заказать германской илн швейцар-
ской Комиссии по вопросам определения прочности, а также швейцарским или гер-
майским заводам красителей.
ности. Это соотношение должно быть каждый раз отмечено и во всяком
случае должны быть приведены данные для интенсивности среднего
топа. В случае флотского синего производят оценку окраски двойной
интенсивности, а черного — глубоко черного цвета. Вследствие разной
красящей способности красителей окраски в */з и двойную против сред-
него тона ие являются одинаковыми по интенсивности и поэтому не
сравнимы между собой. Поэтому в проспектах жирным шрифтом отме-
чаются сравнимые данные в интенсивности среднего тона.
Этот новый метод лежит в основе оценок светопрочиости, произво-
димых германскими и швейцарскими заводами красителей, которые
в своих таблицах приводят данные о светопрочиости всех красителей на
соответствующих видах волокна и в трех интенсивностях.
. Таким образом, была впервые построена система оценки красителей,
полностью удовлетворяющая требованиям практики. Таблицы прочности
дают возможность красильщику отбирать одинаковые по светопрочностн
красители для смешанных пряжи и тканей и одновременно получать
данные о поведении красителя в светлых или темных тонах.
Подобный способ используется в Англии и Америке. В результате
многолетних самостоятельных работ английского Общества красилыци.-.
ков и колористов были разработаны красные и синие эталоны на шерсти,
Красные эталоны вскоре уступили место синим эталонам, так что и
в Англии обдучают с применением синей шкалы. С английской н опи-
санной выше синей шкалой получаются очень сходные результаты. Тем
самым созданы предпосылки для установления международных норм
определения светопрочиости, тем более что английская синяя шкала
была принята Американской ассоциацией текстильных химиков и коло-
ристов. Близкие к завершению переговоры швейцарской и германской
комиссий с Англией и Америкой по вопросам установления светопроч-
ное™ были прерваны второй мировой войной. Следует надеяться, что
ми ого обещавшая работа в последующее мирное время будет закопчена *.
Литература: Verfahren, Typen und Normen fur die Priifung und Beurteilung der
Echthcitseigenschatten usw., Berlin, Verlag Chemie, 1939. Там же находятся ме-
тодики определения других видов прочности, например к стирке, хлору, щело-
чам, кислотам и т. д.
* Международной организацией по стандартизации (International Organization
for Standardization, сокращенно I.S.O.), в результате многолетних согласований между
представителями отдельных стран в августе 1956 г, рекомендован для принятия стра-
нами — участниками этой организации проект методик испытания прочности окрасок
к разным воздействиям, в частности, к свету. Принципы методики и эталоны в основ-
ном соответствуют изложенным на стр. 363. — Прим, ред,
предметный указатель
Автоклавы 312—322
- общие указания ко работе 320
Азиновые красители, см. Красители
Азоамин красный К 101
— красный С 147
Азобензол 75, 115
Азобензолднсульфокислота ИЗ
Азожелтый 245, 247
— Г 247, 248
Азокрасители, общая часть 217—234
— восстановительное расщепление 354
-- практическая часть 228—234
— титрование по Кнехту 348
Азоксибенаол 75
АзоксибензолДисульфокислота 113
Азосалициловая кислота 265
Азотёмно-зеленый 259
Азотная кислота в качестве окислителя 14
Азотол О 91 и сл,
Азотолы 228
Азофлавин ФФ 244
Айронак 322
Алголь желтый ВГ 288
Ализарин, см. 1,2-Диоксиантрахинон
Ализариновый чисто голубой Б 211 и сл.
Ализарин-сафирол Б и СЕ 214, 285 и сл.
Ализарин-цианин зеленый Г 281
Алкилбензиланилины, накуум-псрегоцка
304
АААлкилморфолипы 295
Л’-Алкилпиперидины 295
Алкилхлорид, см. Хлористый алкил
о 'А лкоксиаминосоединения для оксиазо-
красителей 228
А л кок си-г руппа, введение 15 *
Альдегидная группа, введение 17
Алюминий 15, 323
— хлористый 59, 204, 206, 207
----- двойная соль с пиридином 59
— — — — с хлористым натрием 59
Амидокафтоловый красный 6Б 238
----Г 195, 237
Аминоазобензолдисульфокислота 243, 244
п-Аминобепзол 242
— диазотирование 222
Аминоазокраситель 232
]-Аминоантрзхинон 59, 209, 210, 288
— производные 212 *
* Указатель составлен М. И. .Михай-
ловой.
2-Амипоантрахинон 15, 58, 84, 208, 287
- - получение, технические примечания'
208 < ?•
1-Аминоантрахщюн-2-сульфокислота 59
п-Амипоацетанилид 119, 121, 222, 238, 345
7-Амнно-Аш-кислота 354, 356 ___\
о- Аминобснзолсульфокислота 112
1-Амино-гамма-кислотэ 354, 358
Аминогидрохинондиметиловый эфир, см4
2,5- Диметоксиани.'1Ин - С
Амипо-Г-кнслота, см. 2-Нафтиламин-6,8-i
дисульфокнслота
Аминогруппа, введение 15 >
— защита ацилированием 65
— ориентирующее действие 52
1-Амино-2,4-дибромантрахинон 59, 211 -й
п-Аминодиметнланилин 278 ,
п-Аминодиметиланнтин-тиосульфокислотй
278 " J
1-Амнно-2,4-диметокси-5-хлорбензол 91, 926
л-Аминодифениламцн, диазотирование 220^
Амннодифениламинсульфокислота 92, 93щ^
4- Амино-1,3-ксилол-6-е у л ьфокнслота 79 ЛЙ
Амино-2-метилантрахинон 207 и с л.
[,4-Аминоиафтол 354
1-Амино-8-нафтол-2,4-днсульфокислота' 84^.j
148, 193, 194, 198, 199 V.
1-Амино-8-нафтол-3 6-дисульфокислота 34S;r'
10, П, 14, 184, 189. 190, 191, 349, 354,;^
151, 185, 237^
— ацетилирование 237
- красители из нее
258 • 261, 357
— определение 344
— растворимость 191
1-Амяно-8-пафтол-4,6-дисульфокислота
199, 201, 202
— бензоильные производные 202
— хлорбепаоильные производные
1- Амино-2-нафтол-4-сУльфокислота
159, 178
— диазотирование 225
1-Амино-5-нафтол-7-сульфокислота
1 - Амино-8-нафтол-4-сульфокислота
194, 197
2-Ам1що-5-пафтол-7-сульфокислота
180, 184, 185, 250
202 . ?
76, ДО».
195 .
193. ,
84,
ИЗО, 184, 100, ZOU дд;
2-Амино-8-нафтол-6-сульфокиСЛОта 84, 1W,
180, 183, 184, 185, 256 '
Аминонафтолсудьфокислота 103, 219, 2о
Амипопафтолы 163
— диазотирование 219
Предметный указатель
367
п-Амипосалицплоная кислота 1-1, 111
Амппосульфокислоты, выделение 37
— определение 343, 344
— получение методом запекания 117
п-Амннофепилоксаминовал кислота 68, 89
Аминофеннлтолиламписул ьфокнелота 93
л-Аминофенол 14, 15, 7о, 239
2-Лмццофенол-4,6-дпсульфоккслота 139
4-Амщюфенол-3,5-дисульфокнс лота 179
о-Амннофеиол-л-сульфокислота 99
п- Аминофенолсульфокнслота 179
Амгшофенолы, выделение 35
— сочетание 231
1-Амино-2-этоксинафталин 187
1-Амино-2-этоксгшафталнн-6-сульфокис-
лота 18?
Амины алифатические 32
— ароматические 32
— ароматические пс-рвичные, нитрование
149, 150
— выделение 32 и сл.
— определение 341 и сл.
— сочетание 230
Аммиак, нодные растворы, кривые зави-
симости давления оТ температуры 86
Аммониевые основания четвертичные 122
Аммоний гядросульфид 200
— ванадиево-кислый 154
— молибденовокислый 302
— сернистый 292
— сульфгидрат, приготовление 200
Анализ красителей объемный по Кнехту
346
Аналитическая часть 339 и сл.
о-Анизидин 15, 87, 92, 134
о -А низидин-л-сульфокислота 99
л-Анизиднн 134, 150
Анизолы 16, 91
Анилин 354
— диазотирование 220, 234, 341
— из нитробензола 71 и сл.
— из хлорбензола 73
каталитическим методом 73
— перегонка 306
— применение 116, 119, 122, 128, 130,
141, 160, 186, 237, 238, 242, 260, 261,
290, 291
— растворимость в воде 72
— свойства 32
— сульфирование 337
технические примечания 72
— токсичность 22, 74
Анилин-2,5-дисульфокислота 97, 99, 187,
250, 251
Анилиновый голубой 212
— черный 74
Анилин-.и-сульфокислота, см. Метаниловая
кислота
Лнилин-о*сульфокислота 112
Анилип-тг-сульфокислота, см. Сульфанило-
вая кислота
Антипирин 119
Аитозины 202
Антраниловая кислота 156, 289, 295
Аитраруфип, см. 1,5-Диоксиантрахинон
Антрахинон 16, 204—206, 208
— алкильные и фенильные эфиры 91
— аминирование '.58
Антрахинон, нведсние оксягрунпы 58
— применение 208—210, 213, 215
— сульфирование об
— титрование 57
Автрахипон-гпдроксиламнн 58
Антрахинон-1,5-дисульфокислота 56, 57
213, 214
Литр,1хинои-1,5-дисульфокнслотз калиевая
соль 210, 214, 215
Антрахинон-1,6-днсульфокислота 56, 57
214
Антрахинон-1,7-дисульфокнслота 56 57
214
Антрахинон-1,8-днсульфокислота 56 57
213, 214
Антрахинои-2, 6-днсульфокислота 56, 214
Антрахинон-2, 7-дисульфокнслога 56, 214
Антрахинонил- l-оксаминовая кислота
210
Антрахинон-моносульфокислота 56
Антрахинононые красители, см. Красители
1-Антрахинон-оксаминовая кислота 87
Антрахинон, оксипроизводное 58
— ориентация в ряду 56
— сульфирование об, 208, 209, 213
Антрахинон-1-сульфокислота 56, 57, 209
210, 212
Антрахинон-1-сульфокислота, калиевая
соль 212
Аптрахиион-2-сульфокислота 56—57, 84,
208, 212, 214, 281
Антрахннон-судьфокислоты 56 и сл.
Антрахиноны, галоидирование 59
Антрацен 10, 16, 204 и сл.
Аптримиды 59
Аппаратура лабораторная 21
— материалы для изготовления 321
Аридсульфамиды 53
Армстронга и Винца правило ориента-
ции 54
Аурамин 266
Аурамин Г 267
— ОО 266 и сл.
Аурины 16, 14]
Аценафтен 10
1-А|1етамино-8-нафтол-3,6-дисульфокислота
103, 237
п-Ацетаминофеиол 52
Ацетанилид 87, 119, 120, 121
Ацетил-Аш-кислота, см. 1-Ацетамино-8-
н а фто л-3,6-лису ль фоки с лота
Ацетоуксусная кислота, анилид 130, 239,
240
--- о- и ц-хлоранилиды 240
Ацетоуксусный эфир 118, 130, 238
Ацет-п-толуидин 52
Ацет-о-толуидин 17, 65
Ацет-п-фенилендиамин ]21, 259
— диазотирование 222, 345
Ацидур 322
Аш-кислота, см. 1-Амино-8-нафтол-3,6-ди-
сульфокислота
Балер 295
Байера кислота, см. 2-Нафтол-8-сульфо-
кислога
Бакелит 326
Бенгальский синий 277
368
Предметный указатель
Бензальдегид 143, 144, 267, 269
Бензальдегидднсульфокцслота 271, 272
Бензальдсгид-2-сульфокислота 147, 269
Бензальхлорид, получение 143 и сл.
- технические примечания 144
Бензантрон 15, 59, 60, 84, 204
Бензгидрол 269
Бензидин 114 и ел., 258, 260—262
— (7ас-дназотирование 235
— канцерогенность 116
— получение, технические примечания
115
—. — электролитическим восстановле-
нием 115
Бензг1Дин-3.3/'-днкарбоновая кислота 147
Бецзидин-2,г'-дисульфокислота 114. 118,
263, 264
— получение, технические примечания
114
Бензидиновая перегруппировка 92
Бензидиновые красители, см. Красители
Бензидин с салициловой кислотой, про-
межуточное соединение 256, 258
Бензиловый спирт 144
Бензилхлорид, см. Хлористый бензил
о-Бенэоилбензойная кислота 206
Бензоилхлорид, см. Хлористый бензоил
Бензойная кислота 144
Бензол 9, 30, 66, 76, 112, 131, 206
— алкильные и фенильные эфиры 91
— ориентация в ряду 51
Бензол-л/-дисульфокислота 76, 84, 131,
132
Бснзолсульфокислота 76, 80—82, 112
— получение по способу компании „Ба-
келит" 77
Бензольного ряда соединения 61 и сл.
Бензопурпурин 4Б 263
Бензосветопрочно-голубой 2ГЛ 185, 250
Бепзоснетопрочно-серый БЛ, анализ 358
Бензотрихлорид 65, 143, 268
Бешана — Бриммейера .метод 14, 22, 74,
75, 92, 93, 145, 157, 197
Бибрихскип алый 244
ОмоДегидротио-л-толуадин 297
(7нс-Диазобен<чидин 259
Сшс-Диаэобепзндиндисульфокислота 264
Бисмарк коричневый Г и Р 253
Бона - Шмидта реакция 59
Борная кислота 59, 216, 284
------ эфир 59 *
Бромнндиго 293, 294
Броминдигозоль 293 и сл.
Бромирование 65
Бромистый водород 65
Бронза 323
Бгхерера реакция 15, 162, 178, 180, 185,
“242
Вакуум-насосы 307, 330
Вакуум-фильтр 24
Ван тмя окись 16, 164
Варитмины 220
Везувии Р и Г 2 53—254
Виб зомешчлка 24, 200
Виктория голубой 160
------Б 128, 274
— чисто голубой БО 128, 274
Водород, применение при каталитическом
восстановлении 15, 200
— хлористый, хюрирующий агент 64
Воздух в качестве окислителя 16
— сжатый 326, 330
Волнорез 21
Воля—Данцига метод 16
Воронка Бабо 29, 31
Восстановление 14, 74, 76
— каталитическое 15, 76, 200
по Бешану -Брпммейеру, см. Бе-
шана Бриммейера метод
— сернистым натрием, уравнение реак-
ции 105
— частичное 75, 105
— электролитическое 14, 76
Вулканизации ускоритель 74
Выделение продуктов реакции 32 и с л.
Выпарные аппараты 362
Высаливание 40 и сл., 79
Выходы 17
Галламид, см. Галловой кислоты амид
Галламиновый синий 16 153, 276
Галловая кислота 152, 153, 275, 277, 326
Галловой кислоты амид 152, 163, 27з
Галлоциаиины 277
Галоида переносчики 64
Галоидирование, об дне сведения 16, 64
Галоид подвижной 80, 96
I лмма-кнелота, см. 2-Амино-8-нафтол-6-
сульфокислота
Ганза желтые 131
-----Г 239
-----5Г 122
Гваякол 15, 310
Гвозди железные, применение для восста-
новления 74, 189
Гексанитродифепнламин 95
Гексахлорциклогексан 61
Гелиэнтин 247
Гелиобордо Л Б 196, 241, 242
— Са-лак 241
Гелиоген голубой Б 302
Гельвеция голубой 16
Германии 202
Гндрлзоанизод 92
Гидразобензол 14, 75, 115
Гидразобензолдисульфокислота 113
Гидроксиламипбензолсульфокислота 113
Гидрол Михлера, см. Михлера гидрол
Гидрохинон 15
Гидроцианкарбодифенилимид 290, 291
Г-кислота, см. 2-нафтол-6,8-дисульфокйС?.
лота
Глобол 63
Глубоко-черный Фау 262
Глюкоза в качестве восстановителя 76
Грина метод 151
Грисс 139
Губен 59
„Дау" — фирма, методы получения ани-
лина и фенола 73, 83
Лебпсра реакция 268
Лсгидротиоксилидин 299
Дсгидротио-п-толуидин 297, 299
Предметный указатель
369
Дегндротио-п-толуидинсульфокислота 299
к сл.
Дерево 325
Дефлегматор 27, 30, 304
Дефлегмация 304 и сл.
Диазоаминобензол 243
Диазобензол 76
Диазосульфаниловая кислота 118
Диазотирование 218 и сл.
— технические прописи 234
Дналкилсульфат 16
Диамант черный П Фау ]97, 327
1,4-Диаминоантрахиион 211, 288
1,5-Диаминоантрахинон 211, 215, 288
Диаминовый зеленый Б 258 —260
— зеленый Г 260
— коричневый М 256
прочно-красный Ф 256
— чисто голубой 134
Диаминодибензилдисульфокислота 151, 255
Диаминодифениламинсульфокислота 92, 93
ФД'-Диаминодифенилметан Д50
1,7-Диамино-8-нафтод-3,6-дисульфюкислота
354, 356 Д/
1 2-Диамино-8-нафтол-6-сульфокислота 354,
358
Диаминостильбендисульфокислота
150—151, 254, 255 >
О-Дианизидии 78, 92, 115, 134
— бис-диазотирование 235
2,5-Дианилино-3,6-дихлорхинон 133
Дианил коричневый ЗГН 257
Диантрахинониламины 59
Дибензантрон 84
1,5-Ди-(бенэоиламино)-антрахинон 288
Дибензоилпирен 204
Дибензпиренхинон 59, 204
о-Дибромбензол 303
Дигидриловый эфир 126
Диметиламин 179
Диметиланилин 122, 125, 127, 226, 267, 276,
277, 278
2,5-Диметоксианилин 226
Диметоксидибензантрон 16
Динафтиламин 180
о-Динитрилы 303
2,4-Динитроанизол 91, 94, 107
2,4-Динитроанилин 95, 240
— диазотирование 224
1,5- Дииитроаптрахинон 57, 58, 215
1,6- Динитроантрахинон 57
1,7- Динитроантрахинон 57
1,8- Динитроантрахинон 57, 215
1,6- и 1,7-Динитроантрахиноны 215
м-Динитробензол 14, 103, 104, 106,
107
— очистка 103
— токсичность 22, 104
2,4-Динитробензолсульфокислота 80,.96
о- и п-Динитробенэолы 103 и сл.
Динитродифениламин 95
о, о'-Динитродифенилдисульфид 80
Динитрокрезол 137
1,5-Динитрона фталин 54, 159
1,8-Диннтронафталин 54, 159
Динитронафталины 112, 157
2, 4-Динитро-1-нафтол, 137, 157
Динитронафтол, производные 202
2,4-Динитронафтол-7-сульфокислота 137
Динитростильбендисульфокислота 151 152
Динитрофенол 15, 95, 301
2,4-Динитрохлорбензол 15. 80 91. 94 95
96, 101, 107, 112, 245, 301 ’ ' '
— токсичность 95
Динитрохлорбекзолсульфокислота 112
1,2- Диоксиантрахинон 15, 58, 84, 281 и сл.,
287
1,4- Ди оксиантрахинон 58, 216
1,5' Диоксиантрахинон 15, 213—214, 285,
286
1,8-Диоксиантрахинон 15, 213—214, 286
Диоксивинная кислота 238 t
4,4<-Диоксидифенилметан-моносульфо-
кислота 177
Диоксин Н 178
1,5-Диоксинафталин 84, 197
2,З-Д иоксина фталин 84
1,8-Диоксинафталнн-3,6-дисульфокислота
190, 238
2,3- Диоксинафталин-6-сульфокислота 84.
2,8-Диоксинафталин-6-сульфокислота 163,
181, 185
Диоксинафталины 163
— определение 343
Диперинафтиндандион 202
Дисбах Г 303
Дитиосалицнловая кислота 295 и сл.
Дифенил 60
Дифениламин 128 и сл„ 205, 231 245, 246
306, 309
— получение, технические примечания
130
Дифенилгуанидин 74
Дифенилин 114
Дифенилметановые красители, см. Краси-
тели
Дифенилфталид 206
2,5-Дихлоранилин 101
— диазотирование 222
1,5-Дихлорантрахинон 213, 215
2,4- Дихлорбензальдегид 144
2,6-Дихлорбензальдегид 144
— получение, технические примечания
146
2,6-Дихлорбензальхлорид 144, 146
.и-Дихлорбензол 65
о-Дихлорбензол 61, 63, 64, 69, 114, 202
л-Дихлорбензол 61, 63, 64, 100
1,2-Дихлорбензол-4-сульфокислота 53
Дихлорбензолы 30
Дихлордифенилсульфон 97
4,4'-ДихлОрИНДИГО 147
2,6-Дихлор-4-нитроанилин 64
1,4-Дйхлор-5-нитробензол 87, 100
2,6- Дихлортолуол 145 и сл.
Диз гиланилин 122, 271, 277, 281
— физические константы 124
Дубильные вещества 153
Дюбуа, метод получения хлорфенолов 132
Железо 14, 16, 64, 74, 75, 76, 143/-321,
323
— гидрат закиси 15
-окиси 72
— для восстановления 74
— ковкое, см. Ковкого железа отходы
Г*
370
Предметный указатель
Железо хлорное 16, 108
Желтый Клейтона 300
— Марциуса 157
Замбези черный 164
— — Фау 166
Заместители второго рода 51
— первого рода 51
Зандмсйера реакция ]6, 17, 65, 145, 147,
293
Запекания метод 53, 79, 116, 117, 161
--- аппаратура 162
Зеленого Бпндшедлера тиосульфокислота
278, 279
Зеленый Бнндшедлера 281
— для шерсти С 274 и сл.
а-Иза тина ни лид 290, 292, 293
Известкование 76, 78 и сл.
Измельчение 332
И зо-гамм а-к нс лота, см. И-Кислота
И-Кислота, см. 2-Ами[Ю-5-нафтол-7-суль-
фокнелота
Индаитрен алый ГГ 203
— желтый ГК 211, 288
— золотисто-желтый ГК 60
— красный 5ГК 87, 211
— синий 209
— _ ГЦД. БИС 287
----PC 287
— ярко-ораиженый ГР 204
— ярко-синий 288
Инден 10
Индиго 84, 288 и ел., 327
— лейкосоединение 295
— получение но Гейману 288
------- по Зандмейеру 289 и сл.
— 4, 5, 4', 5/-тетрагалоидированцое 147
Индигозо.ти 295
Индигозоль О4Б 293
Индийский желтый 247
Индоксилкарбоноиая кислота 289
Йод 145
Йодирование 65
Йодометрическое определение сульфокис-
лот 346
Калькуляционное бюро 329, 331
Канцерогенные вещества 74, П6, 180
Карбазол 10
— моносудьфокислоты 177
— ориентация в ряду 60
Карбазоловые кубовые красители, см. Кра-
сители
Карбазольное ядро 6]
о-Карбоксифенилтиогдиколевая кислота
295
Катализатор ванадиевый, получение 154
— восстановления 200
— никелевый, приготовление 200
— окисления 154 1
— хлорирования 16
Каучук' 326, 327
хлорированный 326
Кварц 325
Kt><vac 9
Керамика 70, 324
Кетон Мцхлера,
Кетосоединения
228
см. Мидлера
способные к
кетон
енолнзацни
О
I
Кбхлин 277
Кпзельгус 322
-ирс^ше, no-
Кипятильные камешки 28
— палочки 28
K СМ- 2-^Ф"-«мин-4,8.дисуль.
Дошыота С“' ада*п,’ам»"-в.8-дисуль-
5Ж^и^:це1ам™°-8-“‘*-
фокмлота 1-Ам""°-8-«а»™''-а.6-лисуаь-
Байера см. 2-Нафтол-8-сульфокислота
, см. 2-Нафтол-6,8-днсульфокислота
— гамма, см. 2-Ампно-8-нафтол-6-сульфо-
кислота J v
”о^а СМ' 2'^М111г0'5’н3ФТ0Л-7-сульфокис-
— индоксилкарбоновая 289
К, см. 1-амино-8-нафтол-4,б-дисульфо-
кислота v
о-карбоксифенилтиогликолевая 295
— Клеве, см. Ы/афтиламии-б- и 7-суль-
фокислоты
Коха, см. 1-Нафтиламин-З, 6, 8-три-
сульфокислота
— кргщеиновая, см. 2-Нафтол-8-с¥льфо-
кислота ' .
М, см. 1-Амицо-5-нафтОл-7-сульфокис- ।
лота
— нафтионовая,,см. 1-Ыафтиламин-4-суль-
фокислота
— Невиля — Винтера, см. 1-Нафтол-4-
сульфокислота
— О, см. О-Карбоксифенилтиогликолсвая
кислота ,
— отработанная 67
Р, см. 2-Нафтол-3,6-дисульфокислота
— С, см. 1-Амиио-8-нафтол*4-сульфокис-
СС, см. I-Амино-8-нафтол-2, 4-дисуль-
фЪкислота
— Фрейнда, см. I-Нафтиламин-З, 6-дисуль-
фокислота
— хромотроповая, см. I, 8-Диоксинаф-
та лин-3, 6-дисульфокислота
— Ц, см. 2-Нафтиламин-4,8-дисульфокис-
лота
— чикаго-С, см. 1-Амино-8-нафтол-4-суль*
фокислота ,
— чикаго-СС, см. 1 -Амино-8-нафтол-2, 4-
дису льфокислота
— Шеффера, см. 2-Нафтол-6-сульфокис-
лота
— эпсилон (s), см. I-Нафтиламин-3,8-ди-
сульфокислота
Кислотные антрахиноновые [красители, см.
Красители
Кислотный алый 174
антраценовый красный Г Ц4. 263
-- желтый 247, 248
--- метанилоцый К 245, 246
> '$
X
Al
Кислотный зеленый антрахиноновый 60, 284
---Ж 275
— красный ЗС 238
— одпохром коричневый 3 139
- - оранжевый 236
---А 236, 335
— - сине-черный 259
- - синий антрахиноновый 2]4, 285 и сл,
— — 2К 195
— хром коричневый 2К 106
--- красный 238
---сине-черный А 179
--- черный С 179
— — — специальный 179
— чисто голубой антрахиноновый 211, 212
— ярко-голубой’ 3 16, 147, 269
— ярко-красный 195, 237
Кислотоупорное литье 321
К-кислота, см. 1-Амино-8-нафтол-4,6-дн-
сульфокислота
Клеве-кислоты, (Ai. I-Нлфтиламин-6- и
7-сульфокислоты
Клузаирон 322
Кнехт, методы анализа 3'17, 348
Ковкого железа отходы 74, 75, 189
Кожа 326
Колбы круглодонные 22. 31
Колонна перегонная 324 и сл. '
— Рашига, см. Ряшггга колонна
— ректификационная 39
— фракциомировочная, см, Фракциониро-
вочная колонна
Колумбия черный 166
------- фф 164
Кольбе — Шмидта метод 149, 141
Компрессор ротационный 307, 330
Конго красный 262
Коха кислота, см. 1-Иафтиламин-З,6,8-три-
сульфокислота
Краны керамиковые 324
Красители азиновые 147
— азо 217—234
— анализ по Кнехту 346 н сл.
— антрахиноновые 21] и сл., 281 и сл.
— бензидиновые 231, 255 и сл.
— дифеиилметановые 266 и сл.
— для холодного крашения Ю1
— из о-амипонафтолов 238
— из о-аминофецолов 238
— калькуляция 335 и сл.
— кислотные антрахиноновые 59
— кубовые 133, 204
— кубовые карбазоловые 61
— кубовые, лейкосоедииения 295
— лаковые 101, 177, 339—-342
— оксазинов ые 275 и с л.
— оксиазо 232
— пиразолоновыс 119, 239
— полиазо 80. 248 и с л,
— сернистые синие 134, 327
стильбеновые желтые 151
— таблицы Грина 351 и сл.
— таблицы Шульца 351
— товарные, анализ, установка ыа тип
332, 351 и сл,
— трифеинлме та новые 147, 266 и сл,
— хромирующиеся сицие 1()2
— хромирующиеся черные ,327
Краситель желтый для шерсти 87
— сицнй для вискозы 148
Крашение пробное 49, 331
Крсзидин 226, 250
о-Крезол III
л-К резол 52
о-Крезотнповая кислота 141
о-Крезогнновая кислота —|- <э-толидин со-
единение 256 ’
Кренцлепи 59, 360
Кристаллы, форма 49
Кроцсиповая кислота, см. 2-Нафтол-8-еуль-
фокислота
Ксилеиовын голубой 147
--Фау с 16, 269
— желтый 239
л/-Кснлидн1[ 79
л-Ксилидин 226
Щ-Кснлидинсульфокнслоты 79
Ксилидины, диазотирование 234
Ксилол 10, 16
Кубовое крашение холодное 288
Кубовые красители, см. Красители
Кубовый алый 2Ж 203, 204
— голубой О 287
— желтый ЖХ 211. 288
— золотисто-желтым ЖХ 60
— СИНИЙ О 201), 287. 327
— ярко-зеленый С 16
— ярко-оранжевый 204
Кубозоли 295
Лаборатория информационно-рекламного
бюро 332
- колористическая 331
— практическая работа 17
Лабораторная аппаратура 21, 42
Лабораторные записи 21
Лабораторный журнал 21
— отчет 21
Лак бордо СК 196, 241—242
— красный Б 92
----П 92
Лаковые красители, см. Красители
Лаковый красный С 177
Латунь 323
Леонгарда метод получения диаминостиль-
бендиеульфокислоты 151
Лицстед 303
Литоль красный Р 177
— оранжевый прочный Р 240
— прочно-желтый Р 122
Магний, соли 58
Малахитовый зеленый 267 и сл.
+— — лейкооснование 267
Марганец, окись—закись 16
7— перекись 16, 144, 148, 279
Марганцевый шлам 16, 271
Мастерские ремонтные 329
Медь 81, 323
— в качестве катализатора 82, 84,
265
— листовая 265
— хлористая 146
Метациловая кислота 111, 113
Метановое основание 125
Мета хромовый коричневый 139
372
Предметный указатель
1-Метиламиноантрахинон 212
2-Метилантра хинон 57, 204 206, 207
Метиленовый голубой 16, 278, 280, 281
— — титрование по. Кнехту 347 и сл.
Метиленовый зеленый 280—281
— серый 276
Метиловый спирт 122
Метилхлорид, см. Хлористый метил
Метод Вешана — Бриммейера, см. Ве-
шана — Бриммейера метод
— Бона — Шмидта, см. Бона — Шмидта
реакция
— Бухерера, см. Бухерера реакция
— Воля —Данцига 16
— выделения продуктов 32
— Грина 151
—- Дебнера, см. Дебнера реакция
— Дюбуа 132
— Зандмейера, см. Зандмейера реакция
запекания, см. Запекания метод
— Кнехта 347, 348
— Кольбе — Шмидта 140, 141
— Леонгарда ]51
— хроматографической адсорбции 354
Методы окисления, см. Окисление
1-Метоксиантрахинон 212
Мешалки 2] и сл.
— в виде колокола 24
— вибрационные, см. Вибромешалка
— лопастные 23
— пропеллерные 24
— стеклянные 23, 325
— фарфоровые 23
— центробежные 24
Микадо оранжевый 266
Мимоза 301
Мирбановое масло 144
Михлера гидрол 125, 126, 275
— кетон 127, 274
М-кислота, см. 1-Амино-5-нафтол-7-суль-
фокислота
Модерн фиолетовый 277
Монастраль прочно-голубой БС 302
Монобензоил-п-фенилендиаминсульфокис-
лога 92
Монометил-о-толуидин 267, 281
Моноэтиланилин 123
Монтежю 325 и сл,
Мочевина 302
Мыло миндальное 144
Мышьяковая кислота 58, 209
Натр едкий, водные растворы, кривые за-
висимости давления паров от темпера-
туры 83
Натрий амид 84, 289
— гидросульфит 14, 76, 143, 287, 353,
355
— гипохлорит 16, 17, 65, 132, 150, 300
— двухромовокислый в качестве' окис-
лителя 16, 273, 279
— ^-нафталинсульфокислый, раствори-
мость 167 и сл.
— нитрит, раствор 340
— нитрит твердый 218
- сернистый, см. Сернистый натрий
— сульфгидрат 75, 105, 286
— хлорат 65, 282
Натрий цианид 291
Нафтазарип 112
Нафталин 9, 16, 53, 55, 59, 84, 157, 164,
167, 187, 189, 192, 195, 197
— алкильные я фенильные эфиры 91
— галоидирование 53
— нитрование 53
— окисление 153 и сл.
— ориентация в ряду 53
— сульфирование 53 ШЯк
Нафталин-1, 3-дисульфокислота 55 .••ЗгаИг
Нафталин-1, 5-дисульфокислота 55,84 195, '
196, 197, 199, 200, 242
— ангидрид 199
— нитрование 197 мш
Нафталин-1, 6-дисульфокислота 55, 190
Нафталин-1, 7-дисульфокислота 55 '. Ж
Нафталин-2, 6-дисульфокислота 55, 188
Нафталин-2, 7 (-3, 6)-днсульфокислота 55, ’4mL
188, 275 ... -jjef"
— нитрование 188
Нафталиновый зеленый Фау 275
Нафталин-а-сульфокислота 53, 55-,163. 170г
— нитрование 192
Нафталин-0-сульфокислота 53, 55, 163, 165, /ж
167, 170 ' '
— нитрование 165
Нафталинсульфокислоты 53, 55, 163, 164 '
и сл,
— нитрование 53
— хлорирование 53
^-Нафталинсульфокислый натрий, раство- .
римость 167 и сл.
I, 4,5,8-Нафталинтетракарбоновая кислота
202, 203
— — диангидрид 202, 203
Нафталин-1, 3, 5, 7-тетрасульфокислота. 54,
55, 189, 202
Нафталин-1, 3, 5-трисульфокислота
Нафталин-1, 3, 6-трисульфокислота
190
— нитрование 189
Нафталин-1, 3, 7-трисульфокислота
Нафталин-4, 6, 8-трисульфокислота,
триевая соль 201
Нафтамин желтый НН (ФФ) 297, 299, 300,
327
а-Нафт»*амип 79, 93, 157—161, 187, 241,
249, 250, 251, 358
— диазотирование 222, 235, 249
— получение, технические примечания 153
— сочетание 228
^-Нафтиламин 12, 54, 74, 163, 179, 180, 18Ъ
184,185,315
— диазотирование 223
— сочетание 227
— токсичность 74, 180
I-Нафтиламин-2, 4-дисульфокислота
159
1 -Нафтиламин-3, 6-дисульфокислота
и сл., 248, 249, 250
f-Нафгиламин-З, 8-дисульфокислота
и сл.
1-Нафтиламин-4,7-дисульфокислота
1-Нафтиламин-4, 8-дисульфокислота
197 и сл.
2-Нафтнламин-1,5-дисульфокислота
182
55
55, 189,
55
дина-t.
76, 80,
187 Ш
197 *
198
112,
Предметный указатель
373
2-Нафтиламин-4, 7-дисульфокислота 198
2-Нафтиламин-4,8-дисульфокислота 113,
197 и сл., 358
2-Нафтиламин-5,7-дисульфокислота 181,
182, 183
— определение в смеси с 2-нафтнламик-
6,8-дисульфокислогой 346
2-Нафтиламин-6,8-дисульфокислота 181,
183, 184, 185
— оцределение в смеси с 2-нафтиламиН-
5, 7-дисульфокислотОй 346
Нафтиламиновый черный Д 248-—250
1-Ндфтиламин-4-сульфокислота 79, 80, 159,
161, 162, 262, 263
ЬНафтиламин-5-сульфокислота 79, 191
и сл., 195, 242
1-Нафтиламин-6-сульфокислота 164—166,
190
1-Нафтиламин-7-сульфокислота 164—166.
190, 251, 252
1-Нафтиламия-8*сульфокислота 166, 191
и сл.---------
2-Нафтиламин-1-сульфокислота 12, 176, 183
Нафтиламинсульфокислоты 163
1-Нафтиламин-2, 4, 8-трисульфокислота 195
1-Нафтиламин-З, 6,8-трисульфокислотл 189
1'На^тиламин-4, 6, 8-трисульфокислота 199
2-Нафтиламнн-1,5* 7-трисульфокислота 182,
183
Нафтиламины алкилированные 163
— арилированные 163
Нафтионовая кислота, см. 1-Нафтиламин-
4- суль фокислота
Нафтоген синий 164
----- РР 199
Нафтол, алкильные эфиры 16
1-Нафтол 160, 161, 179, 180, 315, 354
— определение 343
— сочетание 228
2-Нафтол 12, 163, 167, 178—180, 236, 240
263, 277, 306, 307, 335, 349
— определение 343
— перегонка 307, 308
— сочетание 227
— сульфирование 170—177
—; технические примечания 170
этиловый эфир, см. 2-Этокс ин афта лин
Нафтол АС, растворение 228
— АС-ИТР 92
Нафтол-Г-кислота 185
I-Нафтол-З, 6-дисульфокислота 299
1-Нафтол-3, 8-дисульфокислота 198, 299
2-Нафтол-1, 6-дисульфокислота 171, 175
2-Нафтол-3,6-дисульфокислота 49, 50 17]—
176, 275, 345, 346, 349
— определение в смеси 174
— флуоресценция растворов Г74
2-Нафтол-6,8-дисульфокислота 49 171—
176, 183, 185
— определение в смеси 174
" флуоресценция растворов 174
Нафтоловый желтый С 137
— сине-черный Б 259
— синий 277
1-Нафтол-З-сульфокислота, сочетание 228
1-Нафгол-4-сульфокислота 162, 178 345
— опре еление 345
I - Нафто л-5- сульфокислота 84, 196 242
— сочетание 228 ’
2-Нафтол- 1-сульфокислота 49 79 170
176—178, 183
2-Нафтол-6-сульфокислота 49, 50, 93, 171—
175, 345, 346
— определение 174, 345, 346
— флуоресценция растворов 174
2-Нафтол-8-сульфокислота 170
Нафтолсульфокислоты 163
- 2-Нафтол-1, 3, 6-трисульфокислота 171, 175
2-Нафтол-3, 6, 8-трисульфокислота 171
р-Нафтольная смола 307
^-Нафтолят натрия 54
Нафтеультам-2, 4-дисульфокислота 195, 198
/ Нафтсультон 166, 193, 194 V
Нафтсультоисульфокислота 194
Невиля — Винтера кислота, см. 1-Нафтол-
-4-сульфокислота
Неопрен 326, 327
Нероли 93
Нерелин новый 187 . ...
Никель 322, 323
Нитратор 67, 68, 94
4-Ннгро-2-амнноанизол 150
2-Нитро-4-аминотолуол 53, 149
6-Нитро-2-аминотолуол 150
4-Нитро-2-амицофенол 95, 106
4-Нит ро-2-аминофенол-б-суАьфокислота 112
о-Нитроанизол 8/, 91, 92, 99, 115, 134
и-Ннтроаннзол 134
о-Нитроанизол-л-сульфокислота 99
Нитроанилин, токсичность 22
.и-Нитроанилин 76, 104
— диазотирование 221, 234
— производные 53
о-Нитроанилин 16, 86, 90, 121, 240
— диазотирование 89, 223
— применение 87, 89
л-Нитроанилин 16, 86, 99, 107, 121, 122,
258, 259, 260
— диазотирование 223, 234
— диазотированный 350
— получение, технические примечания 87
— применение 87, 89
— хлорирование 64
о-Нитроанилин-л-сульфокислота 99
л-Нитроамилин-о-сульфокислота 87, 92
I-Нитроантрахинон 57
4- Н итроантрахинон- 1-оксаминовая кислота
211
2-Нитро-4-ацетаминотолуол 52
З-Нитро-4-ацетаминотолуОл 52
л-Нитроацетанилид 120, 121
'о-Нитробензойная кислота 147
Нитробензол 14, 28, 64, 66 и сл., 69, 71, 72,
74, 103, 107, 108, 111, ИЗ, 114
— восстановление 115
— восстановление каталитическое 73
— получение, технические примечания 67
— токсичность 22, 74
Нитробензол-2, 5-дисульфокислота 80, 99
л<-Нитробензолсульфокислота 80, 112
— натриевая соль 209
— получение, технические примечания 112
о-Нитробензолсульфокислота 80, 112
Нитрование, общие сведения 14, 67
1-Нитро-2,4-диме»окси-5-хлорбенэол 91
374
Предметный указатель
о-Нитроднфенилямин-п-сульфокислота 99
Нитро-я-дпхлорбензод 100, 101
Нитрозилссриэя кислота 16
Нитрозобензол 71
Нитрозодиалкплапилпн 273
Нитрозодимстиляпилин 179
и-Нитрозоднметиланилпи 276, 277, 278
— восстановление 75, 278
п-Нитрозодиэтиланнлин 276
Нитрозо-З-иафтол 76, 89, 178
я-Нитроз‘офенол 80
Нитрокислота Ill 112
Нитрокрезол 14
Нитрокрезоли 13т
]-Нитро-2-метилантрахинон 207, 203
Нитрометр Лунге 67, 69 71
1-Нитронафталии 51, 76, 80, 157, 158
— полтчение, технические примечания 158
2-Нитрона фталиц 157
1 -Нитронафталин-3,6-дисульфокис дота 188
1 -Нитронафталин-3, 8-дисульфокислота 197
1 - Ни трона фтал ин-4,8-дисульфокислота 197
2-Нитронафталин-4, 8-дцсульфокислота 112,
197
1-Нитронафталиц-5-сульфокислота 192
1-Нитронафталнн-6-сульфокцслота 164
1-Нитропафталин-7-сульфокиСлота 164, 192
I-Нптроиафталин-8-сульфокислота 192
1-Нитронафтали н-3, 6, 8-трисульфокислота
189
1-Нитронафталин-4, 6,8-трисульфокислота
199
2-Нитро-4-толуидин 67, 149
З-Нитро-4-толуидин, диазотирование 223,
239
о-Нитротолуол 11,53, 115, 144, 145, 148
^-Нитротолуол 11, 51, 115
л-Нитротолуол II, 108, 112, 150, 265
л-Нитротолуол-о-сульфокислота 109, 265
и-Нитротолуолсульфокислота 151
Нитротолуолы 11, 304, 306
1-Нитро-2, 4,5-трихдорбензол 91
О-Нитрофепилгидразин 90
л-Нитрофенилгидразин 89, 99
л-Нитрофенилоксаминовая кислота 87, 88
Нитрофенилпиразолоны 90
о-Нитрофенол 31, 92, 134
— алкильные эфиры I34, 136
л-Нитрофенол 31, 134
— алкильные эфиры 134, 136
о-Нитрофепол-л-сульфокислота 99
Нитрофенолы, алкилирование 134 и сл.
Нитрофильтры 138, 326
о-и л-Нитроформа нилиды 121
2-Нитро-4-хлоранилин, см. 4-Хлор-2-иитро-
анилин
л<-Нитрохлорбелзол 108
й-НитрохлОрбснзол 80, 84-и сл,, 86 91, 99,
101, 112, 136
л-Нитрохлорбеизол 84 и сл., 86, 92, 112,
136
о-и л-Нитрохдорбензолы, физические кон-
станты 86 ,
— разделение 25, 85, 306
Нитрохлортолуолы 144
— разделение 306
I- Нитро-2-этоксипафталин 186
б-Нитро-2-этоксинафталин 187
8-Нитро-2-эгоксипа(фтал11н 187
Нитрующая кислота 67
— смесь 67, 68, 136
Нутч-фильтры 25
— керамические 137, 138
— лабораторные 25
— стеклянные 26
Окисление, методы 16 'Ж
О-кислота, см. о-Карбокснфенилтиоглико- ж
левая кислота W
Оксазиновые красители, см. Красители Ж
Оксалил-л-фенилепдиамин 259
Оксиазокрасптелп 232
Оксиаминосоединения, сочетание 228
2-Оксиантрахщтон 84
Оксиантрахинон, алкильные эфиры. 16
- эфиры 212
Оксигруипы, введение 15
Оксинафталин, см, Нафтол
2, 1-Окси нафтой на я кислота 54
2, 3-Оксинафтойпая кислота 54, 141 _________
З-Окситиоиафтен 293, 296 и сл.
Олеум 67, 78
Олово 14,’ 76, 323, 356
— хлористое 76, 284, 353, 354, 356
Оранжевый И 236 335
— IV 245—247 ’
Орешки чернильные 152
Ориентации правила 51
Ориентация в ряду антрахинона 56
— антрацена 53
— ----- бензола 51 л
----- — карбазола 60
Ориентирующее действие 52 ' - 1
Ортаниловая кислота 112
Осадки, промывание, „перекрывание" 24, 26
Осаждение дробное 41 г
Освинцовывание гомогенное 323
Основание для прочно-алого ТР 147
-----прочно-красного ЗГЛ 101
- — прочно-красного КБ 147
- — прочно-красного Р 101
Основной голубой 281 *
— коричневый 253 - .\ >
: - синий 128, 160, 274 -
—синий 3 128, 274
— темно-синий 277, 281
Палатин хром черный 6Б 179
Пара-красный 87, 177
Пар, расход 329
Пароперегреватель 32
— Гсйцмапд 130
Патентованный голубой 147
Перегонка 27, 29, 304 и сл.
-в вакууме 30, 145, 304 и сл,
— в лаборатории 27—32
— дробная 29, 30, 32, 124, 304, 332
— при атмосферном давлении 27
— с водяным паром 30 и сл..14б
— с перегретым паром 32, 128, 129
Перегонная колонна 304 и сл.
Перекристаллизация 38
„Перекрывание" осадка 2б
Перемешивание 2[ и сл.
-_ механические приборы 22
Переосаждение 40
11 реометныи ун.и.аи.тчло
0/0
Перманент красный ГГ 240
Печи канальные 331
Пигмент голубой фталоцианиновый 302
— желтый светопрочный 239
— - — 23 122
— оранжевый прочный 122, 240
Пигменты 101, 122, 239—242
Пикраминовая кислота 138
Пикриновая кислота 95, 136, 138
Пиразолоновые красители, см. Красители
Пирамидон 119
Пирантрон 204
Пирен 9, 202—204
Пир^нхинон 203
Пиридин 9, 130, 187, 230, 231; 294, 295
Пиролюзит, см. Марганца перекись
^Пичпайн 238, 248 , 325
“Плав дибензангроновый 60
— индантреновый 59
— сернистый 29? и сл.
— фуксииовыц'67
— щелочнойлВ!
------ аппаратура 81
------- общие сведения 83 и сл.
— — побочные реакции 84
Плавления температуры определение 44
и сл., 46 ,
Пластмассы 326
Плитки кислотоупорные 325
Плиты паровые .сушильные 331
Полиазокрасители, см. Красители
Поливинилхлорид 326
Полифеиил желтый ЗГ 265
Полихлорбензолы 61, 64
Поллопас 326
Поляр ярко-красный ЗБ 355
— — анализ 355
----спектры поглощения 357
— желтый 5Г 239
Порфин 303
Посуда, чистка 17 и сл.
Правила ориентации 51
Правило Армстронга и Винна, см. Арм-
стронга и Винна правило
Пресс гидравлический 73
— винтовой 26
Прессы 25
Прибор Тиле 46
'Примулин 12, 297
— основание 297
— плав, разделение 298 и сл.
Пробки корковые 29
— резиновые 28
Продукты органические, чистота 18, 19, 43
- — испытание на чистоту 43
— - очистка 38 н сл., 43 у
-----промежуточные ] 1
---- цвет 48
Производство,техническая характеристика
327—335
Промывная склянка спиральная 61
Протравной зеленый Бс 178
— чисто желтый 114, 263 и сл.
Прочно-желтый 243
Пунцовый 174
Пурпурин 15
Прямой глубоко-черный ЕВ 260
— голубой светопрочный К 250
Прямой дназосиний 164
— -- К 199
- диазочерный К 164, 166
- желтый К 151
— зеленый 258 и сл.
— коричневый Ж 257
--КХ 256
— красный X 256
- - прочно-фиолетовый РР 250
— черный 164
— — 3 260 и сл.
--К 262
— чисто голубой 134, 32у
Растворители 39 н сл.
Растворитель, отгонка 27
— кристаллизационно связанный 44
Расходы производственные 329
Рашпга колонна 30, 82
— кольца 30, 124, 138, 304
Реагенты, количественные соотношения 20
Реактивные бумажки 349
Реактивы для пробы на вытек 349
Реакции, см. Методы
Реакция Дебнера 268
Реакционные сосуды, см. Сосуды
Реакционный котел системы Замссрейтера
Редуктор 72 , 74, 88
Резервуары керамические 324
—- кислотоупорные 324
— цементные 324
резорцин 84, 131, 350
Ректификационная колонна 30
Ректификация, см. Перегонка
Р-кислота, см. 2-Нафтол-З, 6-дисульфоки-
слота
Роданиды 226
Ртуть в качестве катализатора сульфиро-
вания 131, 210, 213
— окись 131, 210, 213
— соли 56, 57
Салициловая кислота 140, 142, 256 , 258,"
260, 264, 265
----— получение, технические примечания
141
Салицин черный У 179
Сахарин 80
Светло-зеленый СФ желтоватый 269
Светопрочно-желтый Г 118, 238
Свинец 323
— перекись (двуокись), паста 350
— — определение 350
— — получение 126
---- применение 116, 125
— углекислый 291
Серебристая соль 208, 214, 281, 282
Серная кислота, теплоемкость 67
Сернистые щелочи в качестве восстанови-
теля 73, 75, 76, 104, .138, 208, 211, 215,
277, 287
Сернистый ангидрид в качестве восстано-
вителя 15'
— натрий 75, 76, 104, 138, 208, 211, 215,
, 277, 286, 296
Сернистый синий 134, 327
Сернистый черный 95, 301
Сероводород и его соли в качестве вос-
становителя 14
Сероуглерод 290
Синий для шерсти 5Б 11, 272
--------БЛ 199
— Me ль дола 16, 277
Сириус голубой 6Г 187
— светопрочно-голуб ой Г 250
— светопрочно-синий 99
С-кислота, см, 1-Амино-8-нафтол-4-сульфо-
кислота
Смешение 322
Смола каменноугольная 9
— Р'нафтольная 327
Соединения ряда бензола 61 и сл.
Сокслета аппарат 39
Солнечный желтый Г 265
--- ЗГ 265
Сосна американская белая, см. Пичпайн
Сосуды металлические 22
— реакционные, материал для изготовле-
ния 22
— стеклянные 22
Сочетание, общие сведения 226 и сл.
Спектры поглощения 49, 353, 356, 357, 358
Сплав для бань 323
— для подшипников 323
— железа — кремния—алюминия 322
— никель — хром — железо 322
— сурьма — свинец (гартблей) 67, 324
СС-кислота, см. 1-Амино-8-нафтол-2, 4-ди-
сульфокислота
Сталь 322, 353
— С-80 (S-80) 322
— Фау A (VA) 200, 322
Стекло 325
— закаленное 325
— пирекс 22, 147, 356
Стильбеновые желтые красители 151
Стол лабораторный рабочий, содержание
18
Стружки чугунные 72, 74, 121
Сульфаниловая кислота 79, 116 117, 125,
160, 236, 245, 246, 257. 258, 335, 337,
338, 354
— диазотирование 225, 235
- чистая для анализа 340, 341
Сульфиновая кислота 80
Сульфирование, общие сведения 13, 77
Э-Сульфобензойный альдегид 265
Сульфокислоты, выделение 35, 36
— йодометрическое определение 364
— перегруппировка 78
Зульфоксилаты 76
2ульфонация, см. Сульфирование
Зульфон кислотный синим Р 195
Сульфон “-реактив 165, 189, 220, 350
Сульфоны 80
-(п-Сульфофеннл)-3-метил-5-пиразолои
117—119, 238, 239
^ульфофенилпиразолоны 239
]ульфофлавин яркий 355, 359, 360
^ульфохризоидин Г 257
!у льфураторы 94 *
Вульфу рация, см. Сульфирование
Сульфурил хлорид, см. Хлористый сульфу-
рил
Сумах 153
Сушильные шкафы вакуумные 331
Сушка 331
Таннин 152
Тартразин 238
Температура кипения, определение 43
— плавления смешанной пробы 47
— — определение 44 и сл., 46
Термометр регистрирующий’ 82
— укороченный 45
— Цинке 45
Тетраметил-л, л'-диаминобензгидрол, см.
Михлера гидрол
Тетраметил-л, п'-диаминобензофенон, см.
Михлера кетон
Тетраметил-л, л<диаминодифенилметан
125. 126, 266, 267 *
Тетрахдорпирен-3,5,8,10 202
Тетрахлорэтаи 64
Тетраэтилдиаминобензофенон 128, 274
Тетраэтилкетон 274
Тиазиновые голубые 281 __L
Тиазоловый желтый 297, 299, 300
Тиоамид 290, 291, 292
а-Тиоизатин 290, 293
Тиоиндиго 295
— алый 292
Тиокарбанилид 74, 290
Тионин голубой 281 .
Тиоокса миндифениламидин, см. Тиоамид
Тносалициловая кислота 295
Тиосерная кислота 278
Тио-л-толу идин 297 . .
Тиофлавин Т и С 300
Тип (эталон) 331
Титан хлористый, раствор 347
Титрование по Кнехту, см. Метод Кнехта
Титрованные растворы исходные 340
Ткани фильтровальные, стеклянные 326
-------- хлопчатобумажные 26, 326
Тобиаса кислота, см. 2-Нафтиламин-1-суль-
фокислота
X-Толидин 115
о-Толидин 115, 235, 263
— диазотирование 235
о-Толидин о-крезотиновая кислота, сое-
динение 256
л-Толилгидроксиламинсульфокислота 109^
лг-Толуидин 250 *
й-Толуидин 147, 150
л-Толуидин 14, 52, 150, 211, 264, 297
— нитрование 69, 149
л-Толуидин-.«-сульфокислота 239
Толуидины, диазотирование 234
1,2,4-Толуилендиамин 107, 253, 258, 262
Толуол 9, 16, 51. 65, 77, 79, 130, 143, 144,
207, 270
Толуолдисульфокислота 270
л-Толуолсульфанилид 53, 226
л-ТолуОлсульфо-Аш-кислота 357 й
О-Толуолсульфохлорид 79, 80 J
л-Толу олсульфохлорид 65, 79, 80, 123, 147*
239; 272, 357
л-Толуолсульфоэтиланилид 122, 123
Трансмиссии 22
Триалкиламины 295
предметный указатель
377
Трииитрофенол 136, 138, 139
Трифенилметановые красители, см. Краси-
тели
----титрование по Кнехту, см. Метод
Кнехта
Трифенилрозанилнн 212
Трифторметилбензолы 65
1,2,4- Трихлорбензол 64, 91, 202, 302
Трихлорфенол 132
Тропеолин 14, 245—247
Турбовоздуходувки 330
„Тэйтайрон" 322
Углеводороды алифатические 10
Углерод четыреххдористый 64
Уксусная кислота ледяная 64, 69, 99, 119
Уксусный ангидрид 237
Ультрафиолетовый свет 143
Упаковочное отделение 335
Упаривание 29
Фарфор 325
Фенантрен 9
о-Фенетидин 134
л-Фенетидин 134, 150
Фенил-азо-динитродифениламин 244 '>
Фенилазосалициловая кислота 14
Фенил-гамма-кислота 185—186
Фенилгидразин 76, 118, 238
Фенилгидразнн-п-сульфокислота 117, 118,
238
Феннлгидроксиламин 14, 74
— перегруппировка 75
Фенилглицин 84, 289
Фенилглицин-о-карбоновая кислота 84,
288
Фенилдиазоний, раствор 0,1 н. 341
л-Фенилендиамин 20, 104, 106-107, 253,
257, 258, 261
— сульфирование 97
о-Фенилен диамин 2оЗ
п-Фенилендиамин 87, 121, 259, 281
лг-Фенилендиаминсульфокислота 96, 97,106
л-Фенилендиаминсульфокислота 92
Фенил- И-кислота 187. 252
Фенилметилпиразолон 118, 119, 238
Фенилметилпиразолонсульфокислота 118,
238
Фенил-я-нафтиламин 160, 231, 274
N-Фенил-!-нафтиламин-8-сульфокислота
193, 194
Фенол 22. 51, 80—84, 132—134, 136, 139,
140, 228, 254, 260
— алкильные эфиры 16
Фенол-2,4-дисульфокислота 136
Фенол-м-сульфокислота 84
Фенол-о-сульфокислота 51
Фенолы, галоидирование 65
— получение,технические примечания 82
— разделение 34, 35
Физические константы 43
Фильтровальная бумага 25
— ткань 25
Фильтрование 310
—• в лаборатории 24
Фильтр-пресс 25, 184, 310 и сл., 322, 323,
326, 330
Фильтрующий материал 25, 138, 326
Фильтры кислотоупорные 326
Фиолетовый Лаута 281
Флотация 159
Флуоресценция 48, 49
Формальдегид 90, ц0, 125
Форм а пек, спектроскопические таблицы 353
Форманилид 87, 121
Формил-л-фенилендиамин 259
Фосген 127
Фосфор 64
— пятихлористый 65, 143
— хлориды 64, 65
Фракционировочная колонна 29, 30, 124,
304 и сл.
Фредеркинга котел 267, 282, 283, 307
Фрейнда кислота, см. 1-Нафтиламин-3,6-
дисульфокислота
Фталевая кислота 203
Фталевый ангидрид 16, 58, 59, 153, 155,
206, 207, 216, 302
Фталимид 155, 156, 302
Фталонитрил 303
Фталоцианин 303
— сульфокислоты 303
— хлорирование 303
Фторирование 65
Химикалии неорганические 19
Хинизарин 15, 58, 216, 284
Хннизариновый зеленый 284
Хинолин 9, 67
Хиноноксим 8, 178
Хиионы, действие бисульфита 80
Хлор в момент образования 17, 65
— токсичность 64
6-Хлор-З-аминобензойная кислота 151, 264
Хлораминовый жёлтый 297
4-Хлор-2-аминофенол 102, 356
2-Хлор-4-аминофенолбензиловый эфир 356
4-Хлор-2-анизидин 101
Хлоранизидин П, см. 4-Хлор-2-анизндин
Хлоранил 65, 133
лг-Хлоранилин ПО
о-Хлоранилин, диазотирование 221
— диазотированный 350
л-Хлоранилин 150
— ацетильное производное 53
— диазотирование 221
л-Хлоранилин'-И-сульфокислота 239
1-Хлорантрахинои 57, 212
2-Хлорантрахинон 15, 58, 206
о-Хлорбензальдегид 147, 269
о-Хлорбензойная кислота 151, 152, 265
Хлорбензол 51, 58 61 и сл., 84, 85, 94, 97,
11*
—Получение, новейшие методы 63 и сл.
— технические примечания 63 и сл.
Хлорбензол-л-сульфокислота 97, 99
л-Хлорбензотрихлорид 65
2-Хлор-4-диметиламинобензальдегид 108,
ПО, 272
-и-Хлордиметиланилин 110
5-Хлор-2, 4-диметоксианилин 92
Хлорирование 61—65, 133
Хлористое олово, см. Олово хлористое
Хлористый алкил 135, 136
— бензил 123, 144
— бензоил 59, 288
378
Предметный указатель
Хлористый водород, Хлорирующий агент
64
— мстил 135
— сульфурил 65, 132
— этил 135, 255
Хлоркаучук 326, 327
о-Хлормс'таниловая кислота 99
л-Хлорметаниловая кислота, см, п-Хлор-
анил и п-ль сульфокислота
4-Хлор-2-[ШТроанизол 101
2-Хлор-4-нитроанилин 64
4-Х лор-2-нитроанилин 101, 240
— диазотирование 224
2-Хлор-5-нитробензойная кислота 151—152
Хлорнитробензолы, см. Нитрохлорбепзолы
1 -Хлор-2-нитробепзол-4-сульфокислота 69,
80, 97, 98, 99
1-Хлор-4-11итробецзол-2-сульфокислота 92
2-Хлор-4-нитротолуол 53
4-Хлор-2-иитротолуол 53, 144, 145, 147
6-Хлор-2-цитротолуол 53, 144, 145, 147
2-Х.тор-5-нитротолуол-4-сульфокислота ,53
4-Хлор-2-нитрофенол 101, 102, 356
Хлорсульфонопая кислота 79, 176, 294
п-Хлор-Д(-сульфофенилгидразин 239
4-Хлор-2-толуцдин 147
6-Хлор-2-толуидии 145, 147
о-Хлорголуол 11, 17, 52, 65, 147
л-Хлортолуол 52, 65
2-Хдортолуол-4-сульфокислота 53, 147
1,2- и 1,4-хлортолуолы, разделение 306
Хлорфеннлниразолоны 239
о-Хлорфенол 17, 65, 132
л-Хлорфенол 17, 52, 58, 65, 132, 133, 216
Хлоруксуспая кислота 289, 296
Холодильник 22, 28, 29, 30, 31, 38, 39, 102,
202, 304, 308 , 309
Холодное крашение, красители, см. Кра-
сители
Хризазин, см. 1,8-Диоксиантрахинон
Хризен 9
Хризофенин 136, 151, 254, 255, 327
— ГО О 254
Хроматографической адсорбции метод, см.
Метод
Хромирующиеся красители синие 102
— — черные 327
Хромовая кислота, см. Натрин двухромо-
вокислый
Хромовый коричневый Р 106
Хромотроповая кислота, см. 1,8-Диокси-
нафталин-3,6-дисульфокислота
Хромоцнтронин 114, 263, 264
Хромсульфат 12
Царская водка 65
Целестиновый голубой 277
Цементы 324
Центрифуги 25
- керамические 324
Циацоз 74, 104
Циба голубой 2В 293, 294
— красный Г 292
— серый Г 292
— фиолетовый Б и ЗБ 292
Цибанит 326
Цинк 76, 323
Цинк, окись 115
— хлористый 127, 268, 279, 280
Ципковая пыль 14, 76, 92, 109, 113, 114,
115, Ц8, 127, 143, 148, 152,266,278, 353
— — определение качественное 350
Ц-кислота, см. 2-Нафти,тамин-4, 8-дисуль-
фокислота
Чапы 325, 326
— деревянные 325
- — цементные 324
Чернильные орешки 152
Чикаго голубой 134
Чикаго С-кислота, см. 1-Амнно-8-нафтол-
4-сульфокислота
Чикаго СС-кислота, см. 1-Амнно-8-нафтол-
2,4-дисульфокислота
Чугун 321, 322
Чугун серый 14, 112, 314, 321, 322
Шерсть 26, 326, 327
Шеффера соль, см. 2-Цафтол-6-сульфоки-
слота
Щавелевая кислота 16, 87, 210, 268, 273
Щелочи сернистые, см. Сернистые щелочи
Щелочное плавление 281
— — аппаратура 81
— — общие сведения 83 ,
— — побочные реакции 84
Эбонит 326
Эвтектика 85, 145
Экземы 95, 104
Экстрагирование 39
Экстракционный тигель Нолле 39
Электромоторы 22
Эльзасский зеленый Н 178
Эмаль 325
Эпсилон-кислота, см. 1-Нафтиламин-3,8-
дисульфокислота
Эрика красный 299
Эрика Ц 299
Эриовиридин 269
Эриоглауцин 147
Эритрооксиаптрахинон, эфиры 212
Эриохромазурол Б ЮЗ, 141, 146
Эриохромвердон А и С 226
Эриохром красный Б 238
— сине-черный Б и Р 179
Эриохромфлавин A 264
Эриохром черный А 179
---Т 179, 327
Этиланилип 122, 123
Этилбснзиланилин 122—124, 269
Этилбензиланилинсульфокнслота 124, 269,
273 ' * \
Этил-а-нафтиламин 274
Этил-п-фенилендиамин 220 z
Этилхлорид, см. Хлористый этил
Этокси-Клеве-кислота 186
2-Этоцси11афталИ([ 186, 187
Яркий желтый 254
— сульфофлавин, см. Сульфофлавин яр-
кий
Яркое индиго 4Г 147
СОДЕ РЖАН И Е
Предисловие................................................... 5
Предисловие, авторов.......................................... 7
Об основах производства красителей........................... 9
1. ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ........ ................................. 11
Общие сведений о промежуточных продуктах для производства красителей 11
Различные процессы химии промежуточных продуктов................... ]3
О практической работе в органической химико-технической лаборатории 17
Выделение продуктом реакции ................................... , 32
Очистка продуктов.................................................. '38
Испытание продуктов на чистоту ................................... 43
О правилах ориентации.........'..................................... 5]
А. Соединения ряда бензола.............................................. 61
1. Хлорбензол.........................................................
2. Нитробензол- . . . -..............................................
3. Анилин из нитробензола............................................
4. Бензолсульфокислота...............................................
5. Фенол ............................................................
б. Производные хлорбензола...........................................
о* п л-Ннтрохлорбеизолы...............................................
п-Нитроацилин и о-нитроапилин из п- и о-нитрохлорбеизола..............
п-Нитрофенилоксаминовая и л-амцнофенилоксаминовая кислоты ............
л-Ннтрофенилгидразип .................................................
о-Нитроанпзол из о-нитрохлорбензола ..................................
л-Нитрохлорбензол-о-сульфокиСлота п л-иитроаиилин-о-сульфокислота из
п-цитрохлорбензола ................................................
2,4- Дииитрохлорбензол из хлорбензола................................
лт-Фенилепдиаминсульфокислота из 2,4-динитрохлорбензола..............
Анилин-2,5-дисульфокислота из хлорбензола............................
4-Хлор-2-аминофеиол из л-дихлорбензола ..............................
7. Производные нитробензола ,........................................
.и-Динитробензол из нитробензола ....................................
^-Нитроанцлин из jf-динитробецзола...................................
.и-Фе ни леи ди амии из хрдииитробензола.............................
.«-Нитрохлорбензол из нитробензола...................................
2-Хлор-4-диметиламйнобензальдегид....................................
л(-Нитробензолсульфокислота и метениловая кислота из нитробензола
2,2/-Бензидипднсульфокислота из нитробензола..........................
Бензидин из нитробензола ........................................ . .
8. Производные анилина...............................................
Сульфаниловая кислота из анилина (метод.Дапекания) . .................
1-(л-Сульфофепил)-3-метил-5-пиразолО[1 из сульфаниловой кислоты ....
л-Аминоацетанцлид из анилина .........................................
Диметиланилин (диэтил- и этнлбензиланилип) ...........................
Этилбензнланилинсульфокислота.........................................
Тетра мети л* л, л'-диаминодифеиилметач из ди.метияанилнпа и формальде-
гида ................................................................
Тетраметцл-л, л'-диаминобенэгидрод (гидрол Мйхлера) .................
Тетраметил-л, л7-диаминобензофенон (кетон Мйхлера) .................-
Дифениламин из анилина и ацилиновой соли ..............
Анилид ацетоуксусной кислоты .........................................
9. Производные бензолсульфокислоты....................................
Бензол-.и-дисул'ьфокислота............................................
61
66
71
76
80
84
84
86
87
89
91
92
94
96
97
100
103
103
104
106
107
108
111
из
114
116
116
117
119
122
124
125
126
127
128
130
131
131
380
Содержание
Резорцин .............................................................
10. Производные фенола...............................................
о- и п-Хлорфенол из фенола............................................
Хлоранил .............................................................
о- и п-Нитрофенолы и их алкильные эфиры...............................
Тринитрофенол (пикриновая кислота) ...................................
Пикраминовая кислота ....................... .........................
2-Аминофенол-4,6-дисульфокислота из фенола............................
Салициловая кислота из фенола ........................................
л-Аминосалициловая кислота............................................
11. Производные толуола................................................
Беизальхлорид и бензальдегид из толуола ..............................
2,6-Д ихлорбензальдегид из о-нитротолуола ...........................
Бензидин-3, З'-дикарбоповая кислота из о-нитробензойной кислота . . . .
2-Нитро-4-толуидин из л-толуидина.....................................
Динитростильбендисульфокислота и диаминостильбендисульфокислота нз
л-нитротолуола (совместное окисление двух молекул)..................
6-Хлор-З-аминобензойная кислота . ...................................
12. Галламид и галловая кислота из таннина ..........................
13. Производные бензола из нафталина............................ .
Получение фталевого ангидрида каталитическим окислением нафталина . .
Фталимид из фталевого ангидрида........................................
Антраниловая кислота из фталимида ....................................
Б. Соединения ряда нафталина..............................................
14. «-Нитронафталин и а-нафтиламин...................................
Фенил-а-нафтиламин....................................................
а-Нафтол из «-нафтиламина......................................... ; .
Нафтионовая кислота из «-нафтиламина..................................
1-Нафтол-4-сульфокислота (кислота Невиль-Винтера) из нафтионовой кислоты
(реакция Бухерера) .................................................
15. Сульфокислоты нафталина..........................................
1-Нафтилам ин-6- и -7-сульфокислоты (кислоты Клеве)................
^-Нафталинмоносульфокислота и ^-нафтол................................
Сульфирование ^-нафтола ..............................................
2-Нафтол-6-сульфокислота (кислота Шеффера), 2-нафтол-3,6- и 2-нафтол-
6,8-ДИСуЛЬфОКИСЛОТЫ (Р- и Г-КиСЛОТЫ)................................
2-Нафтол-6,8-дисульфокислота (Г-кислота) и 2-нафтол-3,6-дисульфокислота
(Р-кислота).........................................................
2-Нафтиламнн-1-сульфокислота (кислота Тобиаса) из ^-нафтола...........
Получение аминопафтолсульфокислоты из оксинитрозосоединения (хинон-
монооксим) ..........................................................
1-Амино-2-нафтол-4-сульфокислота из ^-нафтола.........................
Е-Нафтиламин из ^-нафтола.............................................
2-Амино-8-нафтол-6-сульфокислота и 2-амино-5-нафтол-7-сульфокислота
(у-кислота и И-кислота) из ^-нафтиламина............................
Фенил-7-кислота.......................................................
i 1-Амино-2-этоксинафталин-6-сульфокислота (этоксикислота Клеве)........
J 16. 1-Нафтиламин-3,6-дисульфокислота (кислота Фрейнда)................
17. 1-Амино-8-нафтол-3,6-дисульфокислота (Аш-кислота) .
18. 1-Нафтиламин-5-сульфокислота и 1-нафтиламнн-8-сульфокислот5 ....
19. Нафталин-1,5-дисульфокислота......................................
1-Нафтол-5-сульфокислота..............................................
1-Нафтцламин-4.8-Дисульфокислота, 2-нафтиламин-4.8-ДИсульфокислота и
1-нафтиламин-3,8*ДиСульфокислота....................................
1-А мино-8-нафтол-4,6-дисульфокислота (К-кислота).....................
20. Нафталинтетракарбоновая-1, 4, 5, 8-кислота нз пирена..............
В. Соединения ряда антрахинона ...........................................
21. Антрахинон........................................................
22, 1-Амино-2-метиланхрахинон.........................................
23. 2-Аминоантрахинон.................................................
24. 1-Аминоантрахинон и 1,4-диаминоантрахинон.........................
25. 1,5 и 1,8-Диоксиантрахинон, 1,5-дихлорантрахинои..................
26. 1,5-Динитроантрахинон и 1,5-диаминоаптрахиион.....................
27. Хннизарин из л-хлорфенола , . ....................................
131;^
132^
132 >
133I
134 J
13^^
133^
13Й
14O&I
14111
143®
143Ж
14Ш
147Й
149.Я
' 15«
15Й1
15$Ж
ШЭД
1«Н
15ГШ
Mi
16МЯ
16в|
здаМ
1бЙЯ
ж!®
«||
17йМВ
17«^К
!W
isMS?
187«Ж
19&
1!
1!
20Ы
2(Ю
2W
2151'
216^
и
Содержание
381
Н. КРАСИТЕЛИ..................................................... 217
Г. Азокрасители........................................................... 217
О получении азокрасителей в лаборатории................................. 217
а) Основные условия................................................... 217
б) Диазотирование..................................................... 218
Примеры................................................................ 220
в) Сочетание.......................................................... 226
г) Дальнейшая переработка.............................................. 232
Технические прописи примеров диазотирования 234
Анилин (толуидины, ксилидины, л/-нитроднилин) . . . .................. 234
л-Нитдоанилин.......................................................... 234
и-Нафтиламин........................................................... 235
Сульфаниловая’кислота (метаниловая кислота, нафтионовая кислота, нитро-
анилинсульфокислоты, хлоранилинсульфокислоты, диаминостильбенди-
сульфокислота, сульфокислота примулина и т. п.)..................... 235
Бензидин (о-толидин, о-дианнзидин)..................................... 235
Примеры азосочетаний..................................................... 236
Простые сочетания с оксисоединением...................................... 236
Кислотный оранжевый А, или оранжевый 11 (кислотный оранжевый) . . . 236
Ацетил-Аш-кислота и амидонафтол красный Г (кислотный ярко-красный) . . 237
Желтый светопрочный Г.................................................. 238
. Ганза желтый Г (пигмент желтый светопрочный) ......................... 239
Перманент красный ГГ (пигмент оранжевый прочный) . .................... 240
Гелнобордо БД (лак бордо СК) . . ».................................. 241
Простые сочетания с аминами.............................................. 242
л-Аминоазобензол из анилина......................................... 242
Тропеолин, или оранжевый IV (кислотный желтый метениловый К) и азо-
желтый из сульфаниловой кислоты и дифениламина.................... 245
Вторичные дис- и полиазокрасители .................................... 248
Нафтиламиновый черный Д (Вейнберг. Касселла, 1888)..................248
Бензосветопрочно-голубой 2 ГЛ. Сириус светопрочно-голубой Г (прямой
голубой светопрочный К).............................................. 250
Дис- и поли азокрасители из диаминов.................................. 253
Бисмарк коричневый Г и Р (основной коричневый)....................... 253
Хризофенин ГОО (хризофеннн)............................................ 254
Бензидиновые красители................................................... 255
Промежуточное соединение бензидина с салициловой кислотой (о-толидин -j-
-|-о-крезотиновая кислота).......................................... 256
Дианил коричневый ЗГН (Прямой коричневый ЖХ) . . ’................... 257
Диаминовый зеленый Б (фирмы Касселла) (прямой зеленый).................. 258
Прямой глубоко-черный ЕВ (Байера) (прямой черный 3).................. 280
Конго красный.......................................................... 262
Кислотный антраценовый красный Г и хромоцитронин........................ 263
Особые методы............................................................. 264
1 Эриохром флавин А...................................................... 264
Солнечный желтый Г из п-нитротолуола.................................... 265
Д. Ди- и трифенилметановые красители...................................... 266
Аурамин 00 (аурамин) (по Т. Зандмейеру).............................. 288
Малахитовый зеленый..........................• j ,................... 267
Ксиленовый голубой Фау С (кислотный ярко-голубой 3) (фирмы «Сандоз") 269
Синий для шерсти 5Б.................................................. 272
Виктория голубой Б (основной синий) ................................. 274
Зеленый для шерсти С................................................ 274
Е. Оксазиновые и тиазиновые красители.................................. 275
Галламиновый синий из амида галловой кислоты ..... .................. 275
Метиленовый голубой из диметнланилина................................ 278
Ж. Антрахиноновые красители............................................. 281
а) Протравные красители .............................................. 281
Ализарин............................................................. 281
б) Кислотные красители............................................... 284
Хинизарин зеленый, ализаринцианин зеленый Г (кислотный зеленый антра-
хиноновый) ........................................................ 284
Ализарин-сафирол Б-и СЕ (кислотный синий антрахиноновый)............ 285
в) Кубовые красители ................................................. 287
382
Содержание
Индантрен ciiheiei PC (кубовый синий О) из р-аминоантрахинона......... 287
Индантрен желтый ГК (кубоный желтый ЖХ)............................... 288
3. Индигоидные красители........................ . ....................... 288
Индиго.............................................................. 288
Иидпгоюль 04 Б (броминднгозоль) ...................................... 293
Тиоиндиго ............................................................. 295 '
И. Сернистые плавы........................................................ 297
Примулцн (по Грину)................................................. 297
Сернистый черный Т из диннтрохдгорбензолз......................... 301
К. Фталоцианины ....................................................... 302
Монастраль прочно-голубой БС — гелпоген голубой Б (пигмент голубой
фталоцианиновый).................................................. 302
IIJ. ТЕХНИЧЕСКИЕ УКАЗАНИЯ .............- -............................ 304
Л. Перегонка в вакууме в лаборатории и на производстве.................... 304
М. Фцльтр-прессы........................................................ 310___
Н. О конструкции и применении автоклавов ............................... 312
Общие указания по работе с автоклавами................................ 320
О. Материалы для изготовления аппаратуры, применяемой в производстве кра-
сителей . ........................................................ 321
1. Металлы.................................................... . . . ; 321
2. Неметаллические материалы...................................... 324
3. Материалы органического происхождения.............................. 325
П. Техническая характеристика производства ............................... 327
Р, Пример калькуляции простого красителя ................................. 335
Оранжевый 11 — кислотный оранжевый А (Сульфаниловая кислота — ^-нафтол) 335
^-Нафтол........................................................ . 335
Сульфаниловая кислота ................................................ 337
Получение красителя из сульфациловой кислоты и 3-нафтола.............. 338
IV. АНАЛИТИЧЕСКАЯ ЧАСТЬ................................................ 339 .
Приготовление исходных титрованных растворов.......................... 349
Определение аминов................................................ 341
Определение нафтолов .... ............................................ 343
Определение аминосульфокислот......................................... 343
Определение амипонафтолсульфокислот .................................. 344
Определение моно- и дисульфокислот нафтода, моно- и дисульфокислот
диоксинафталинэ..................................................... 345
Определение нафтиламипдисуЛ[.фокислоты-2, 5, 7 в смеси с нафтиламин-
дисульфокислотой-2, 6, 8.............................................. 346
Объемное определение красителей методом восстановления по Кнехту 346
Принцип метода Кнехта............................................... 347
Приготовление раствора треххлористого титана.......................... 347
Титрование .метиленового голубого..................................... 348
Азокрасителя .......................................................... 348 (
Трифенилметановые красители .......................................... 348
Наиболее употребительные реактивные бумажки . ........................ 349
Растворы реактивов для пробы на вытек................................. 349
Определение качества цинковой пыли.................................. 359
Определение качества двуокиси свинца.............................. . 359
I. ОВ АНАЛИЗЕ ТОВАРНЫХ КРАСИТЕЛЕЙ.........................................351
о. ОПРЕДЕЛЕНИЕ СВЕТОПРОЧНОСТИ............................................. ^61
Чредметный указатель.................................................... 366 t
'хе.мы I—2!.............................................................. вкл-
Г. Э, Ф и р и. - Д а з и д, Л. Г, л а п ж г
ОСНОВНЫЕ ПРОЦЕССЫ СИНТЕЗА
КРАСИТЕЛЕЙ
Pe.iar.-гор В. А. ЗАХАРЬЕВСКИЙ
Технический редактор В. С. Герасимова
Художник Б. Н. Глазков
Сдано в производство 19/IF 195? г.
Поягпкдпо к печати 12/VII 1957 г.
Бумага 70xl08’/l(i=:I3,S бум. л.
37 печт л. в т/'i 12 дел.
Уч.-изд. л, 37,5. Изд. Я: 3/3429.
Цена 28 р. 25 к. Зак. 1905.
ИЗДАТЕЛЬСТВО
ИНОСТРАННОЙ ЛИТЕРАТУРЫ
Москва, Ново-Алексеевскаи, 32.
Министерство культуры СССР
Главное управление полиграфической
п р омышл ен н ост и
4-е тип. им. Еэг. Соколовой
Ленинград, Измайловский пр,, 29.