









































































































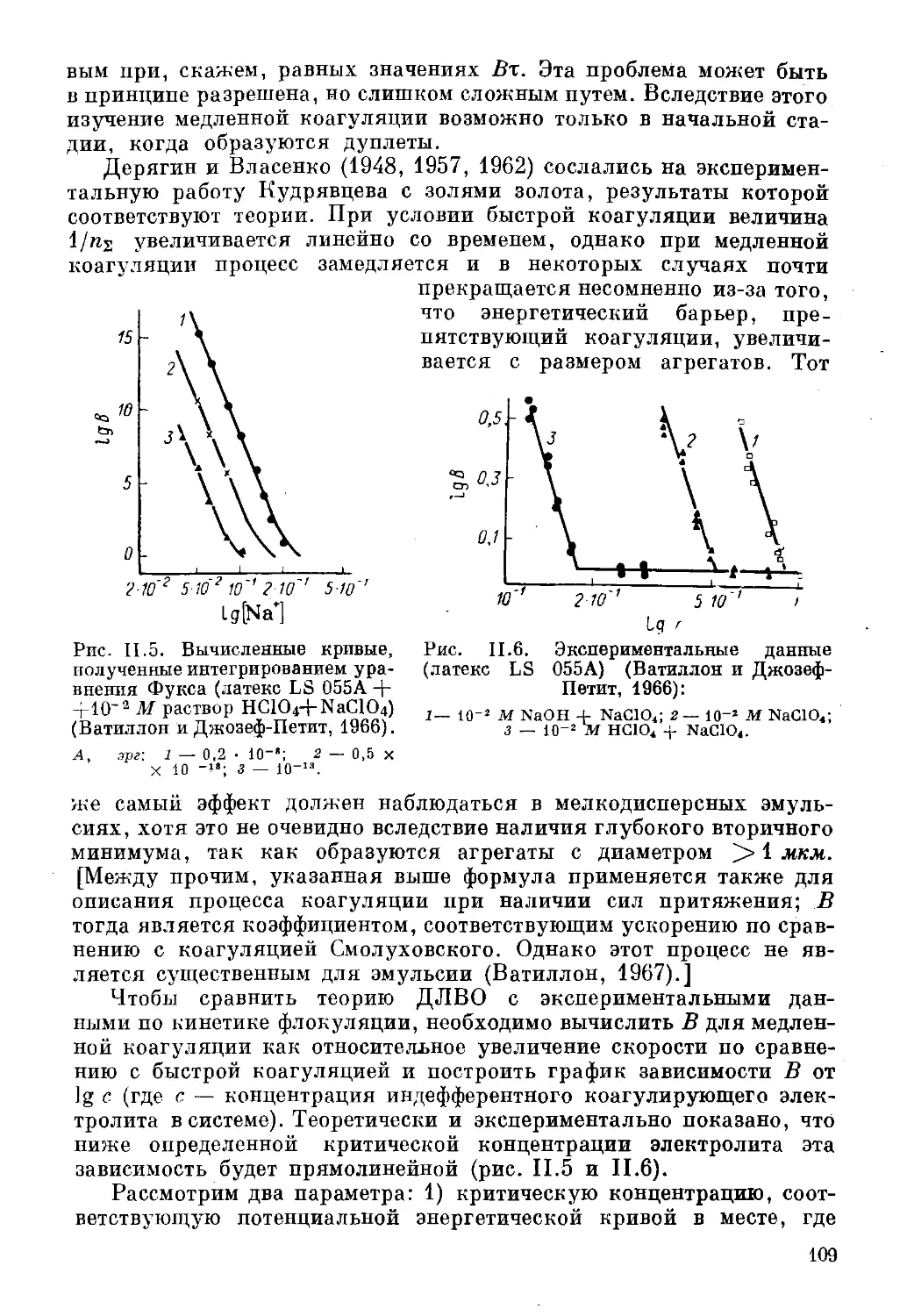








































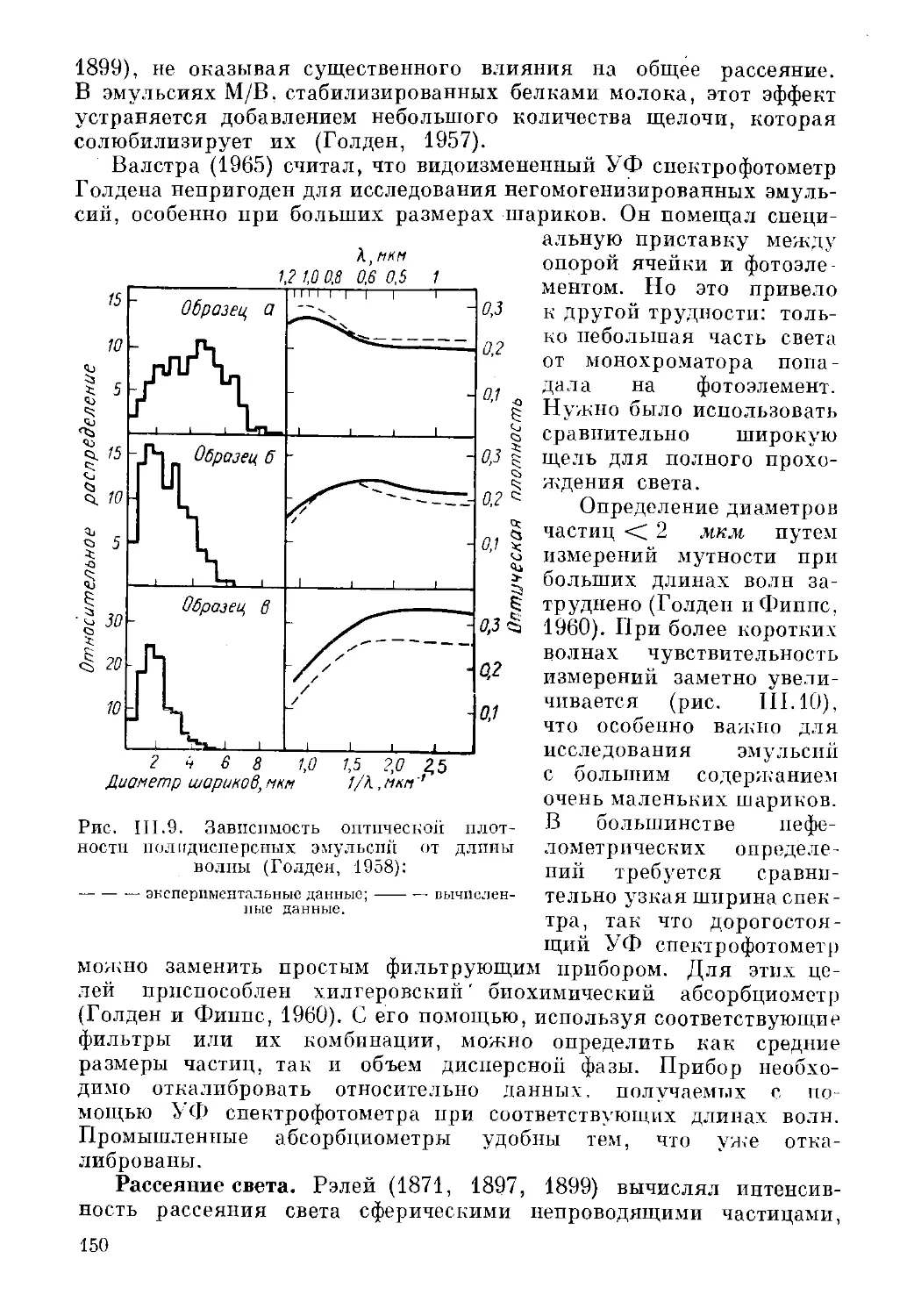
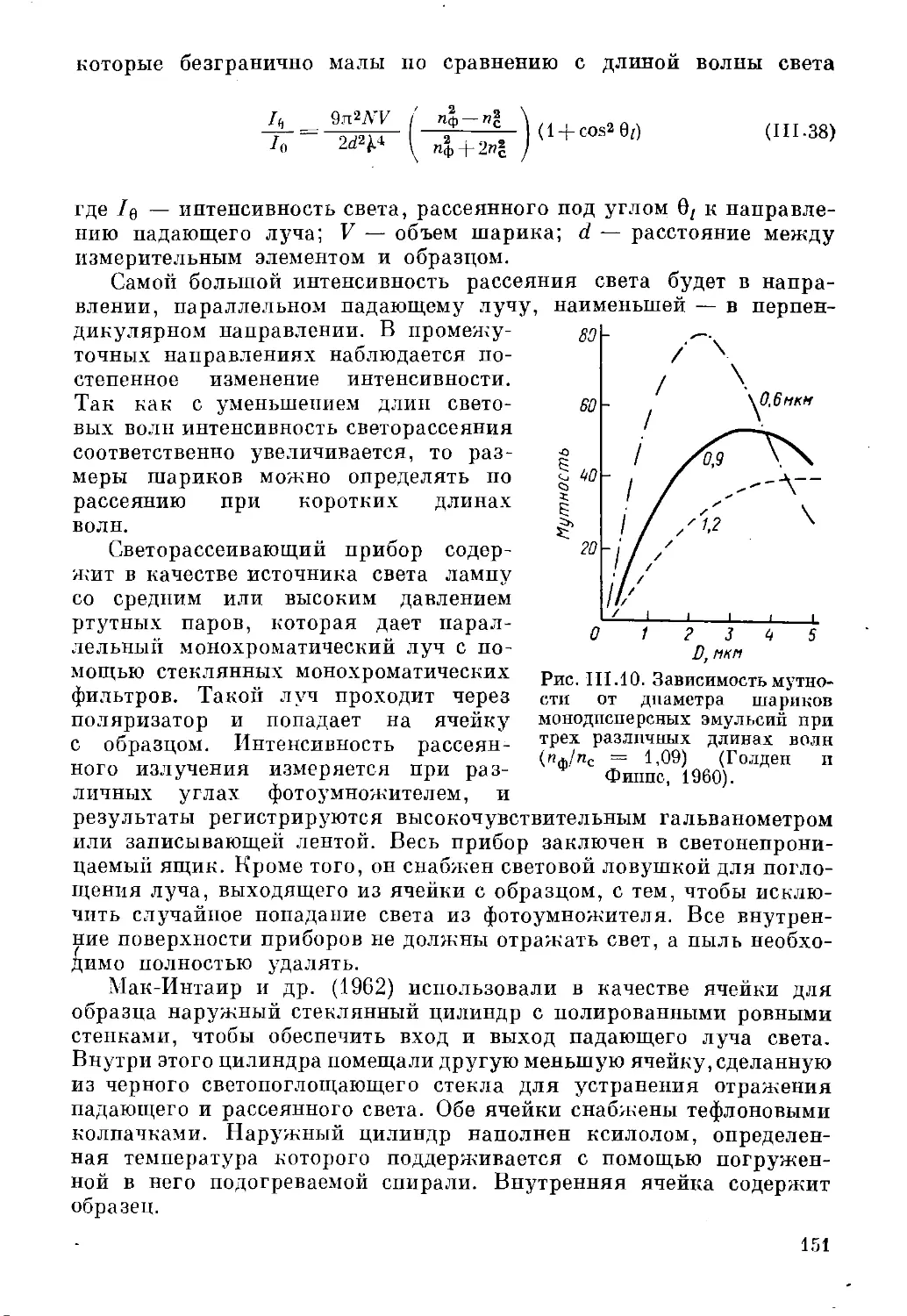



















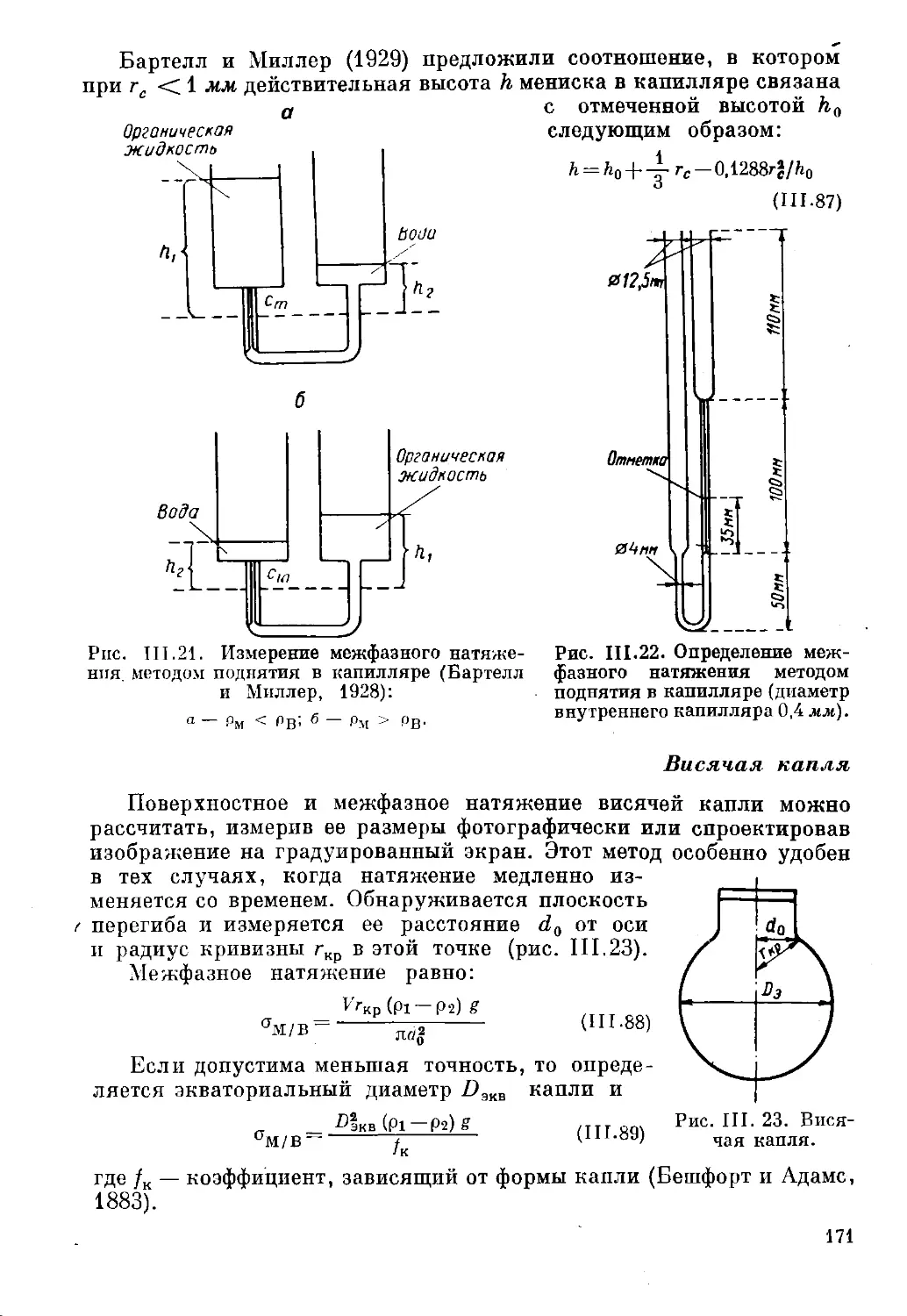
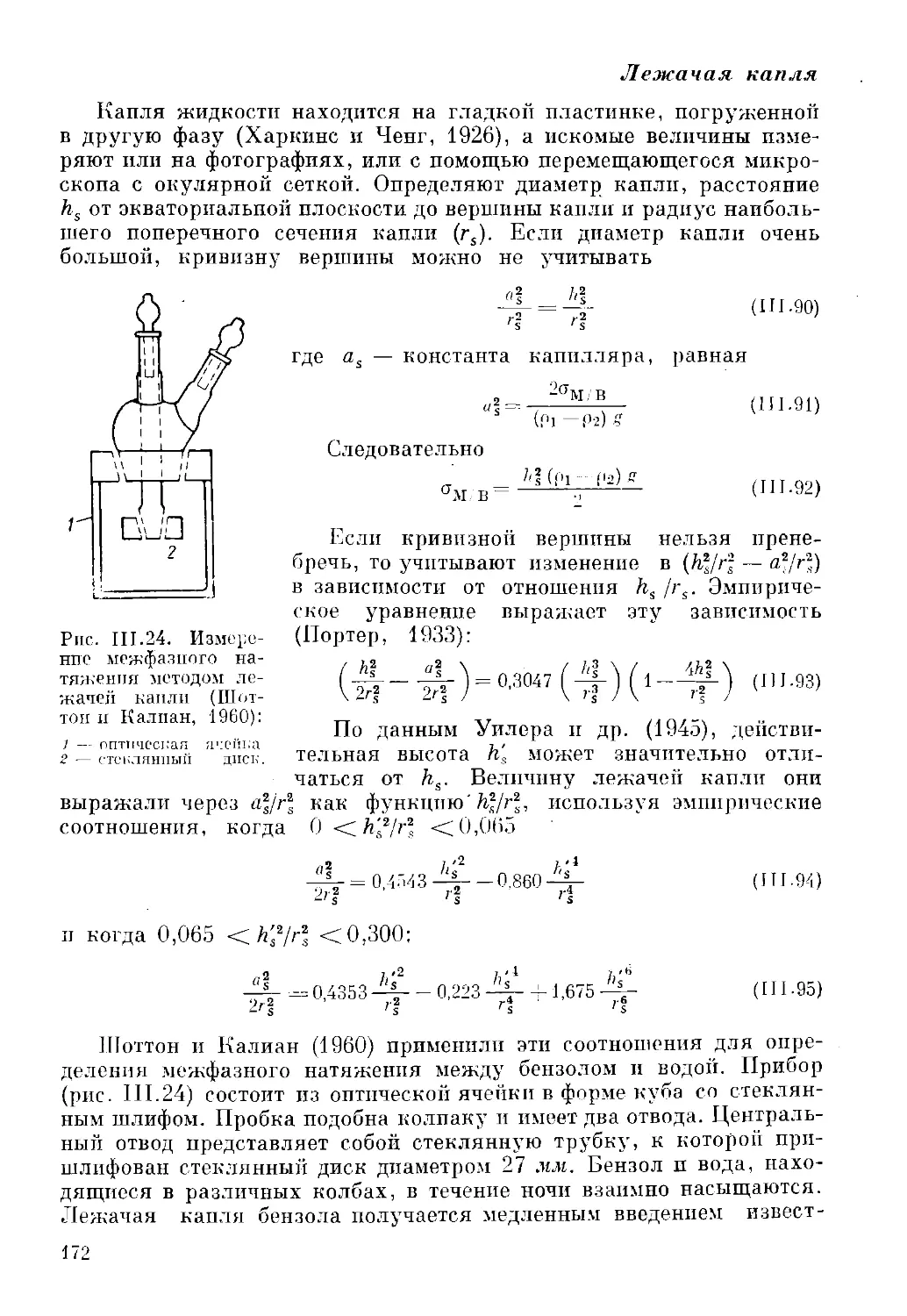













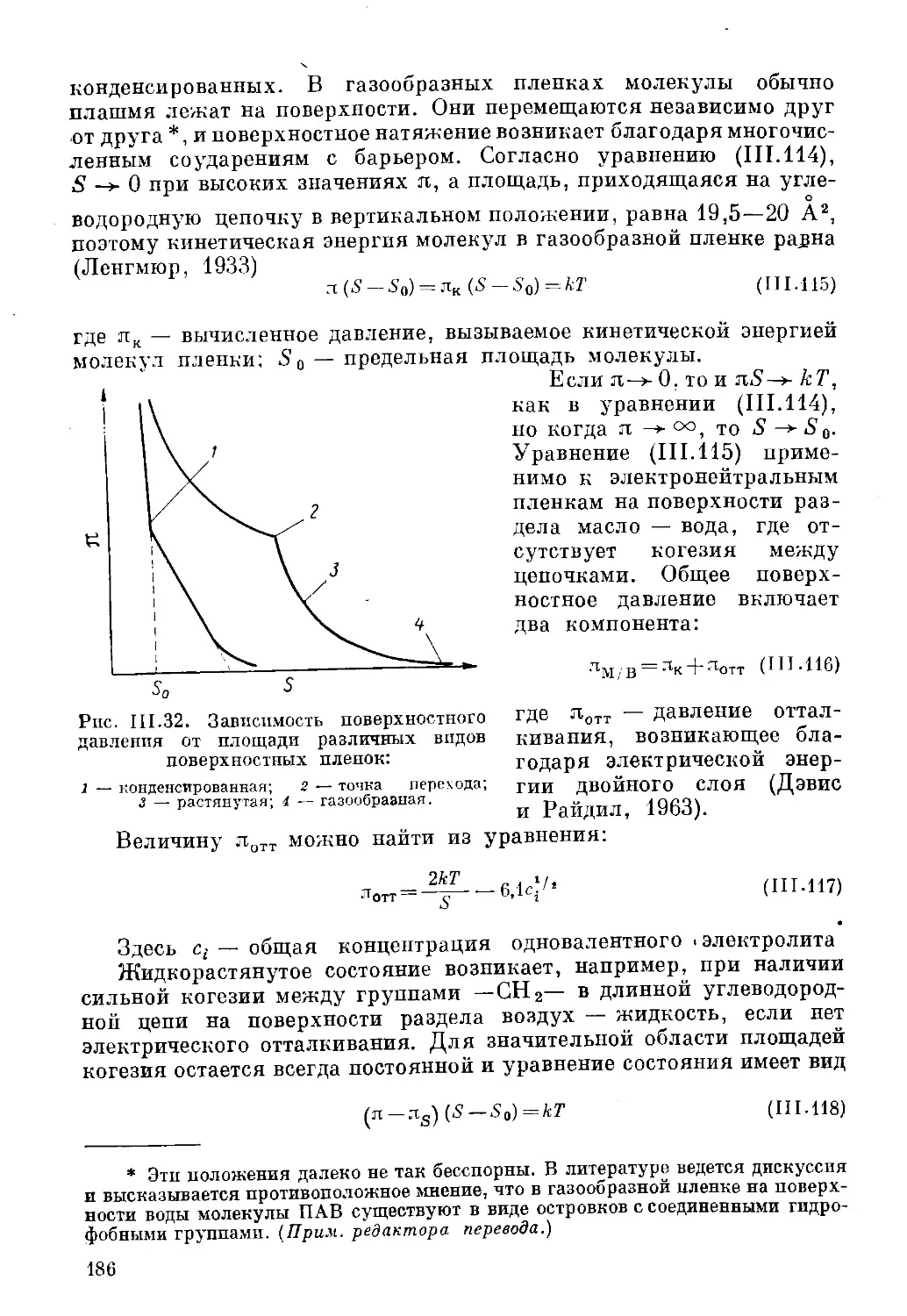


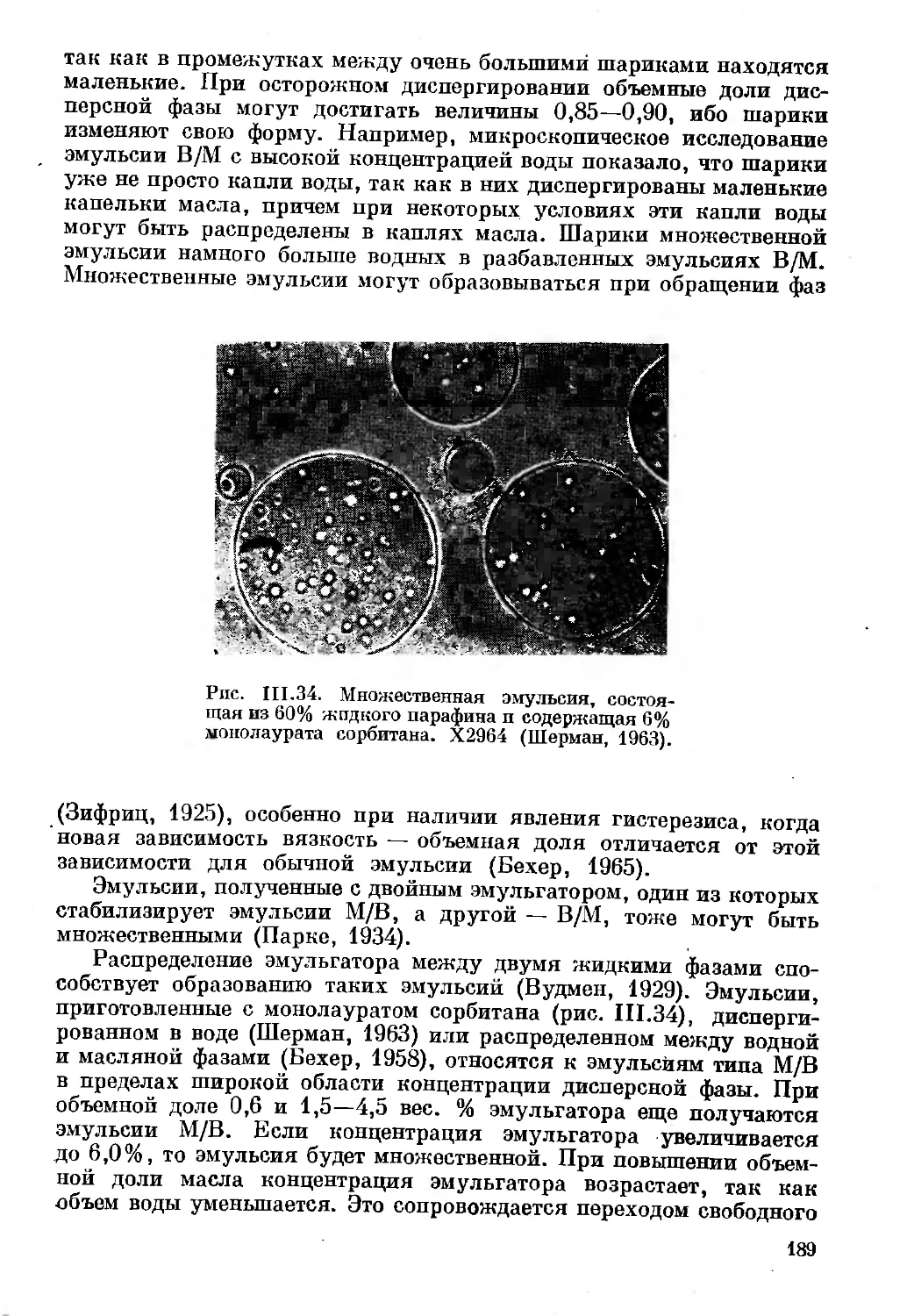




























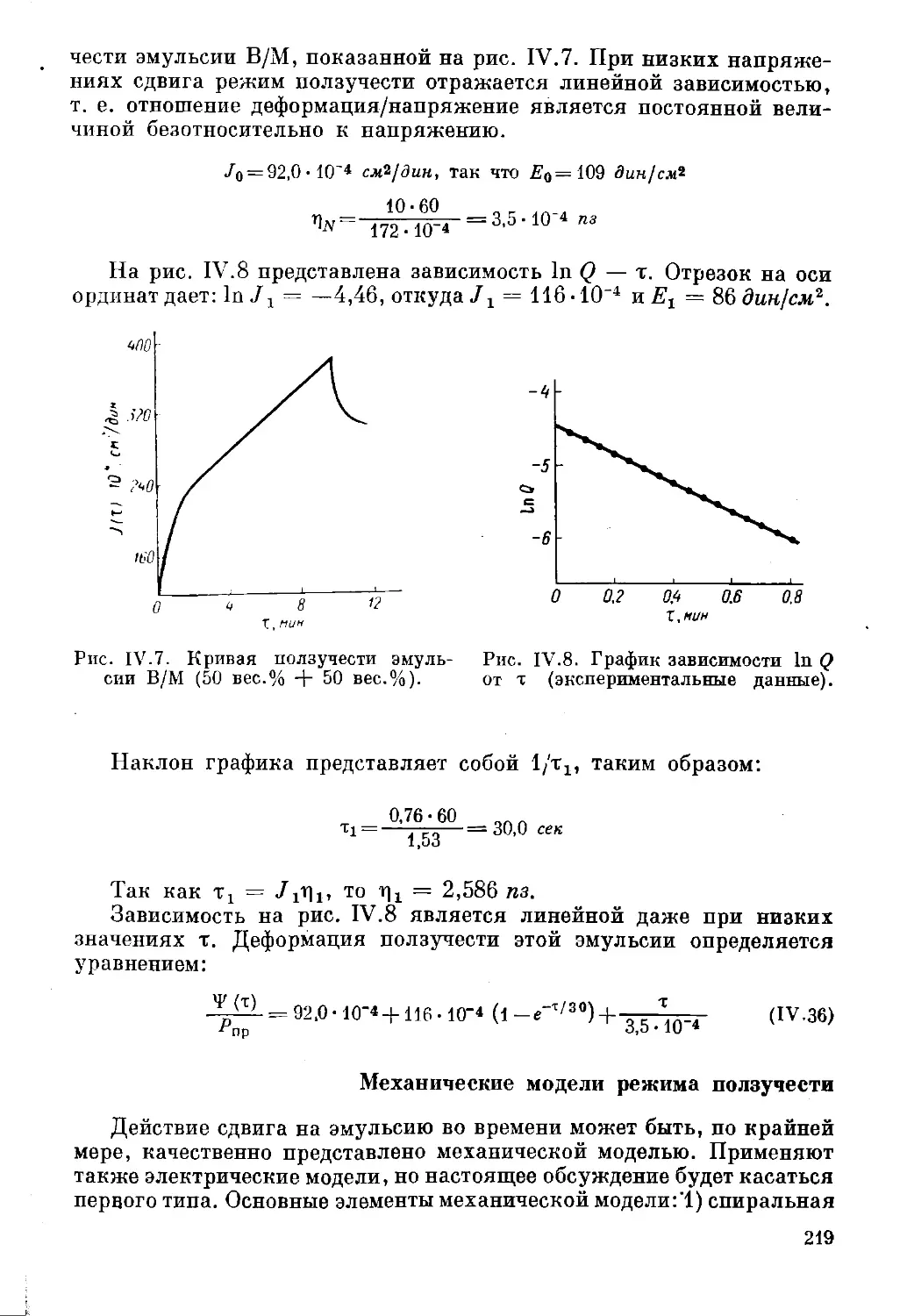






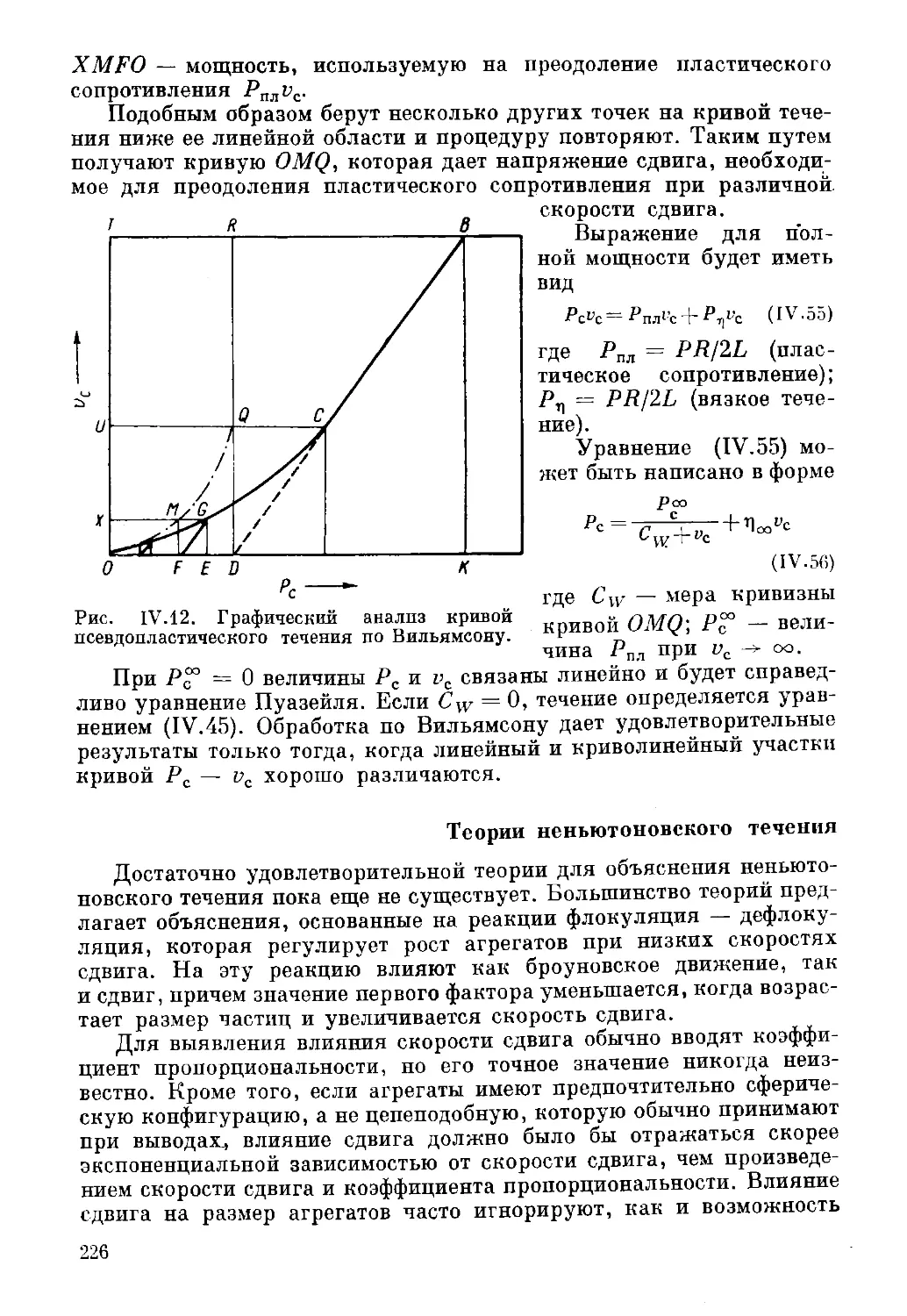























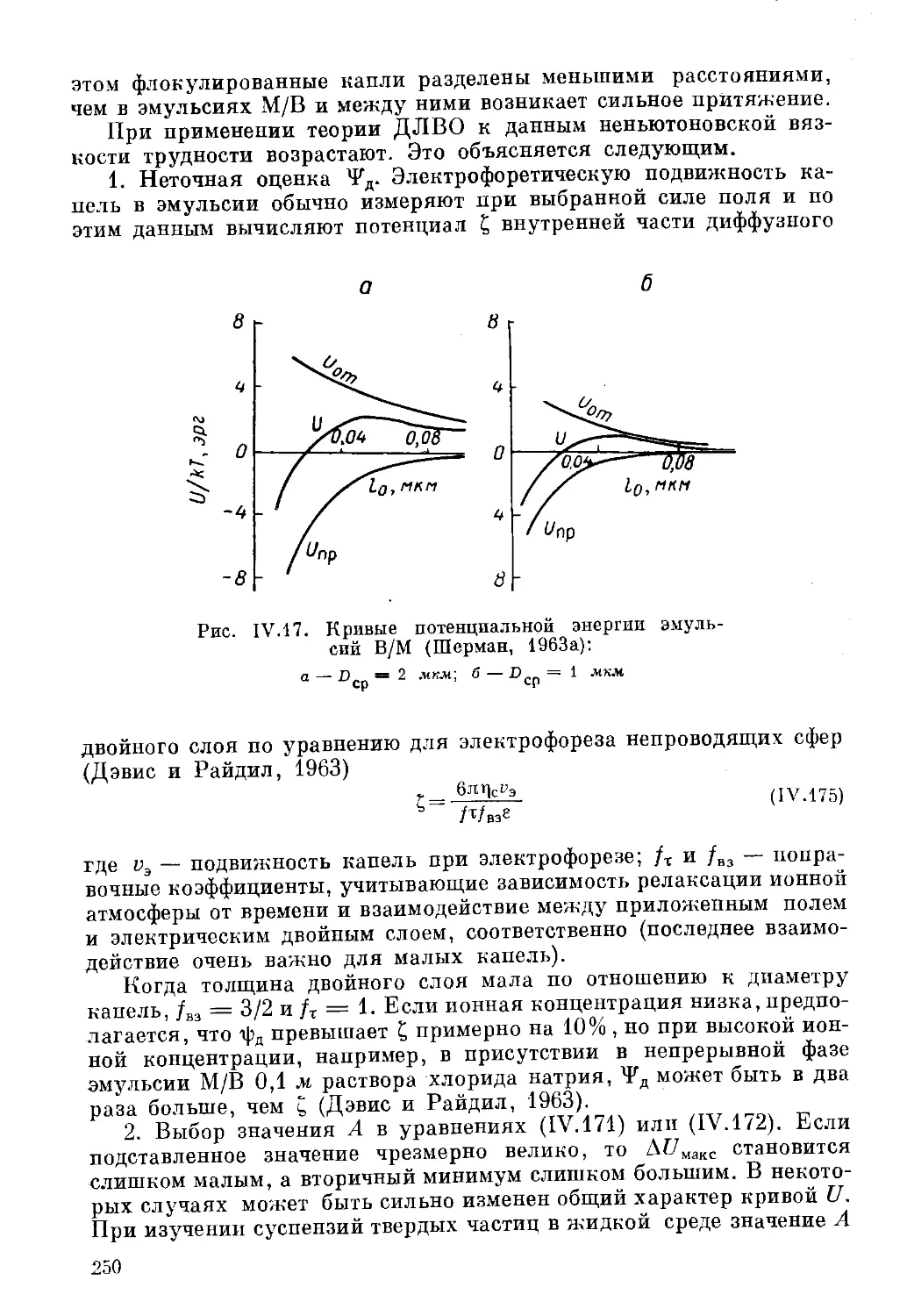















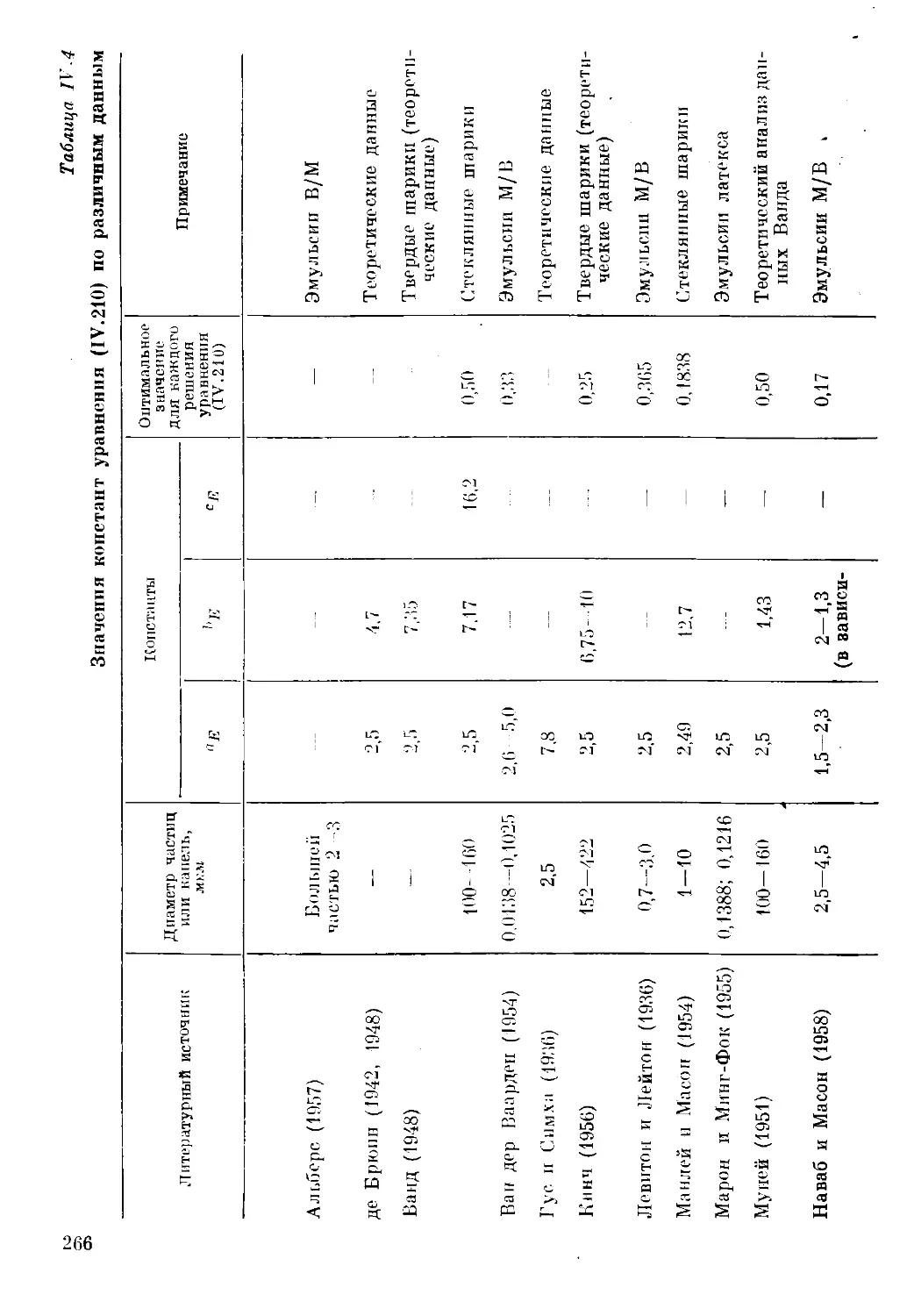










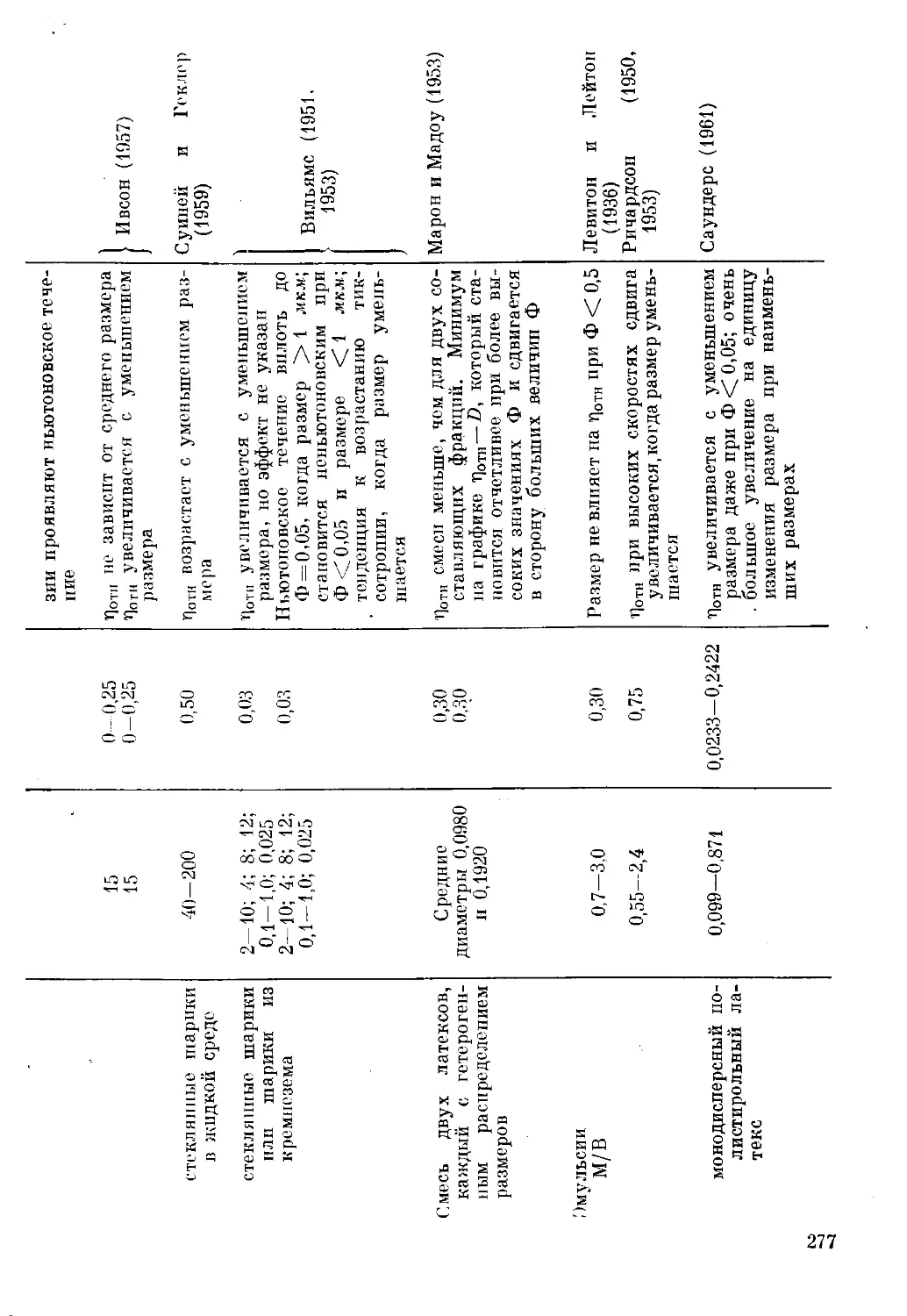


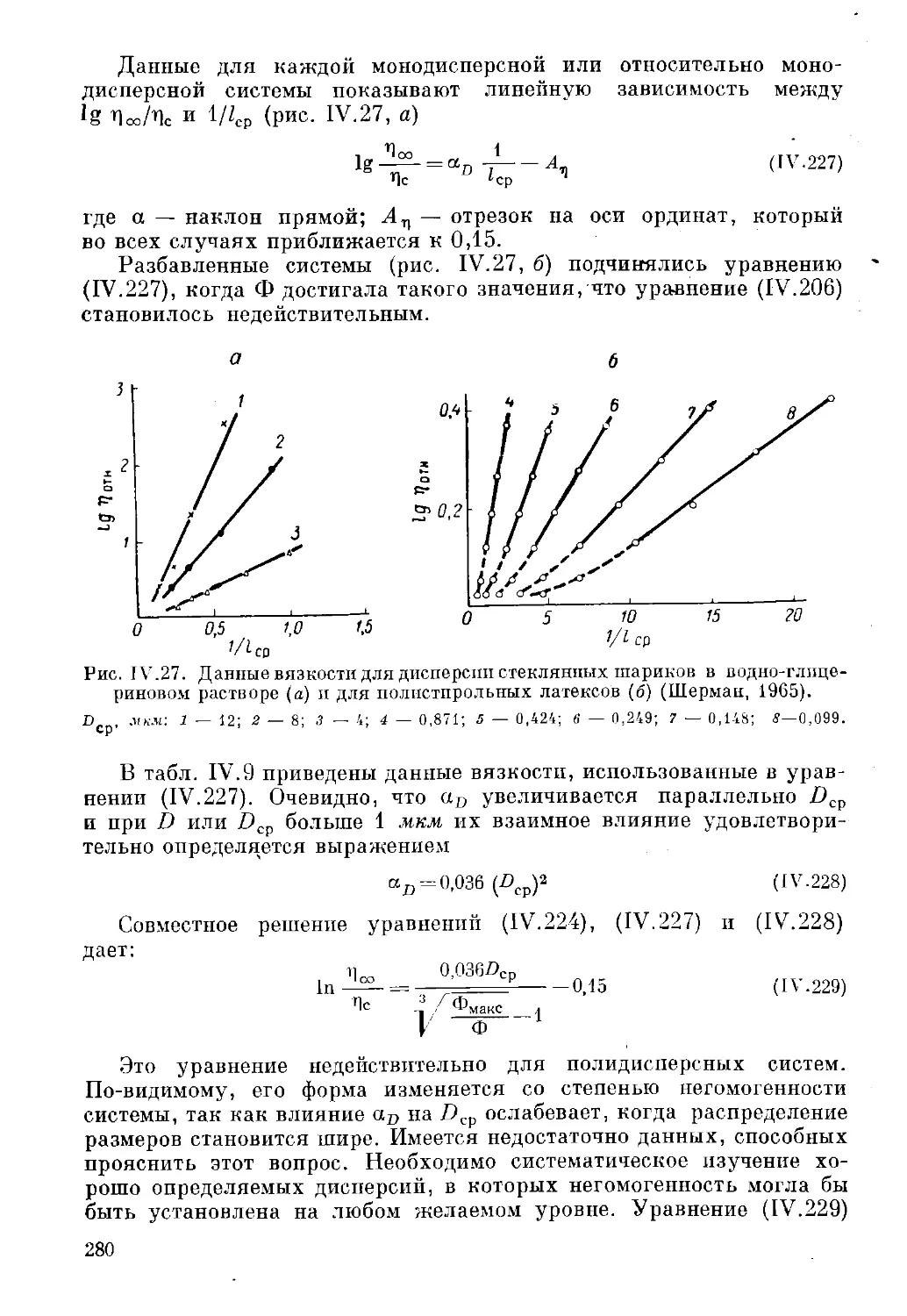


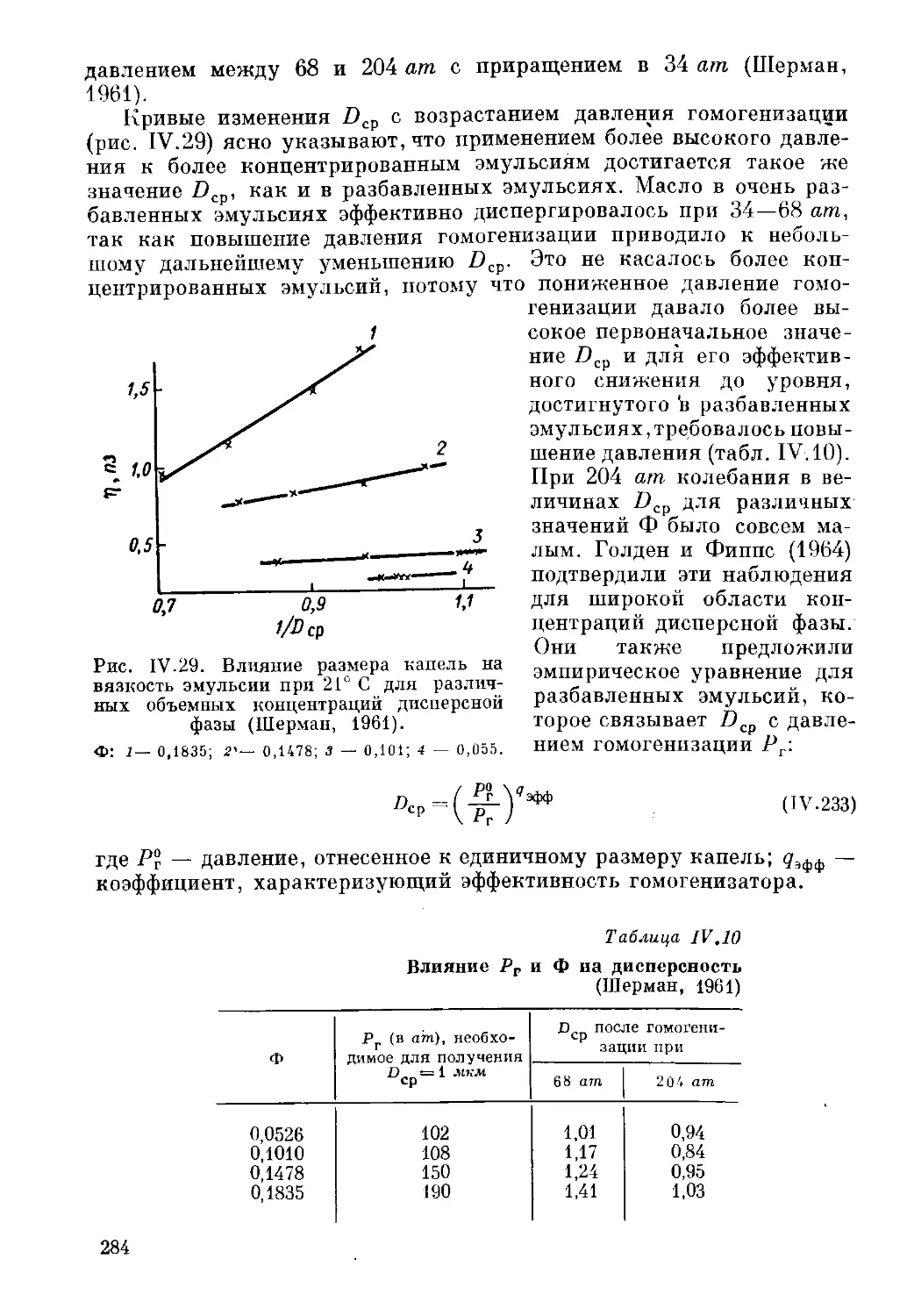











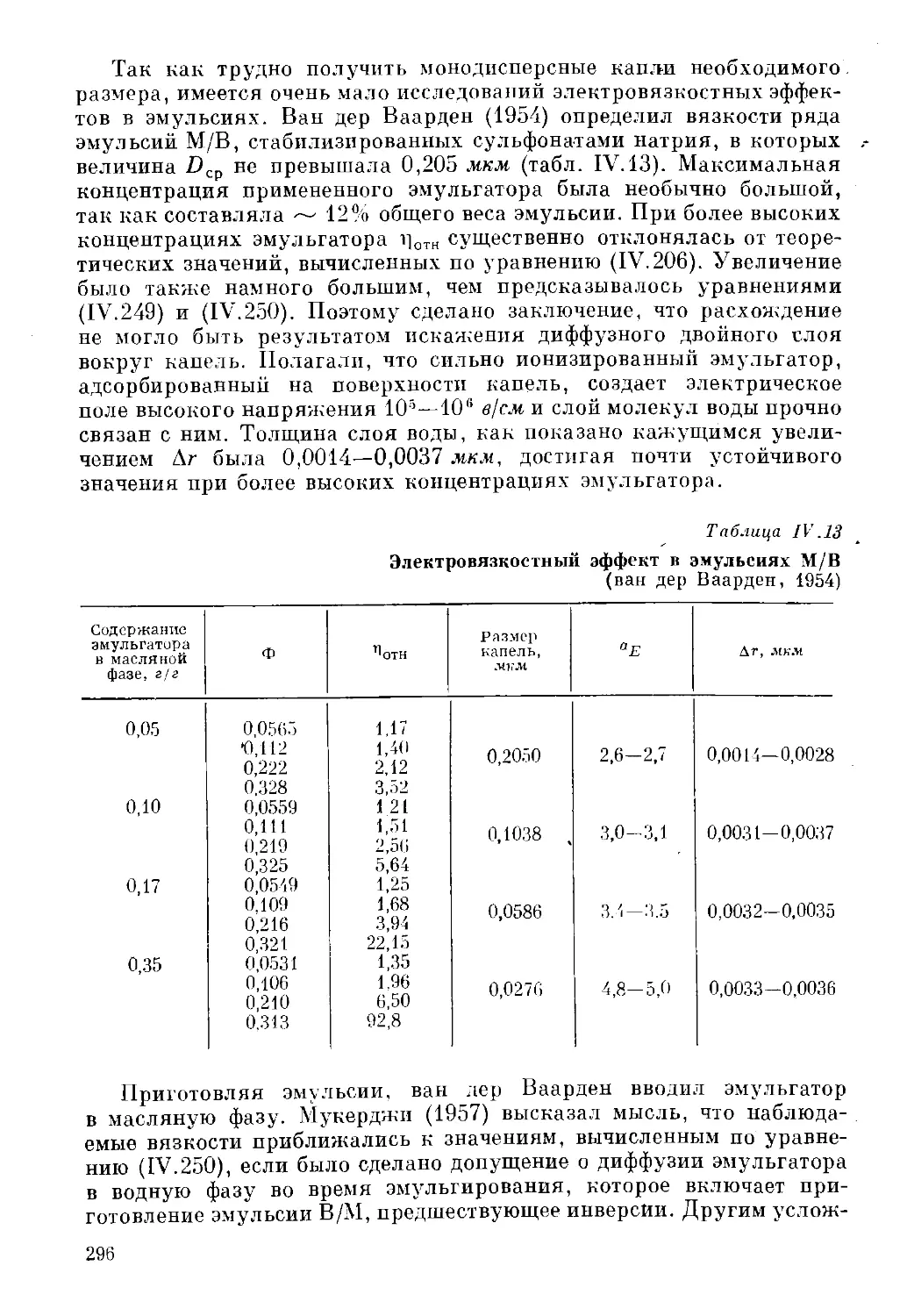




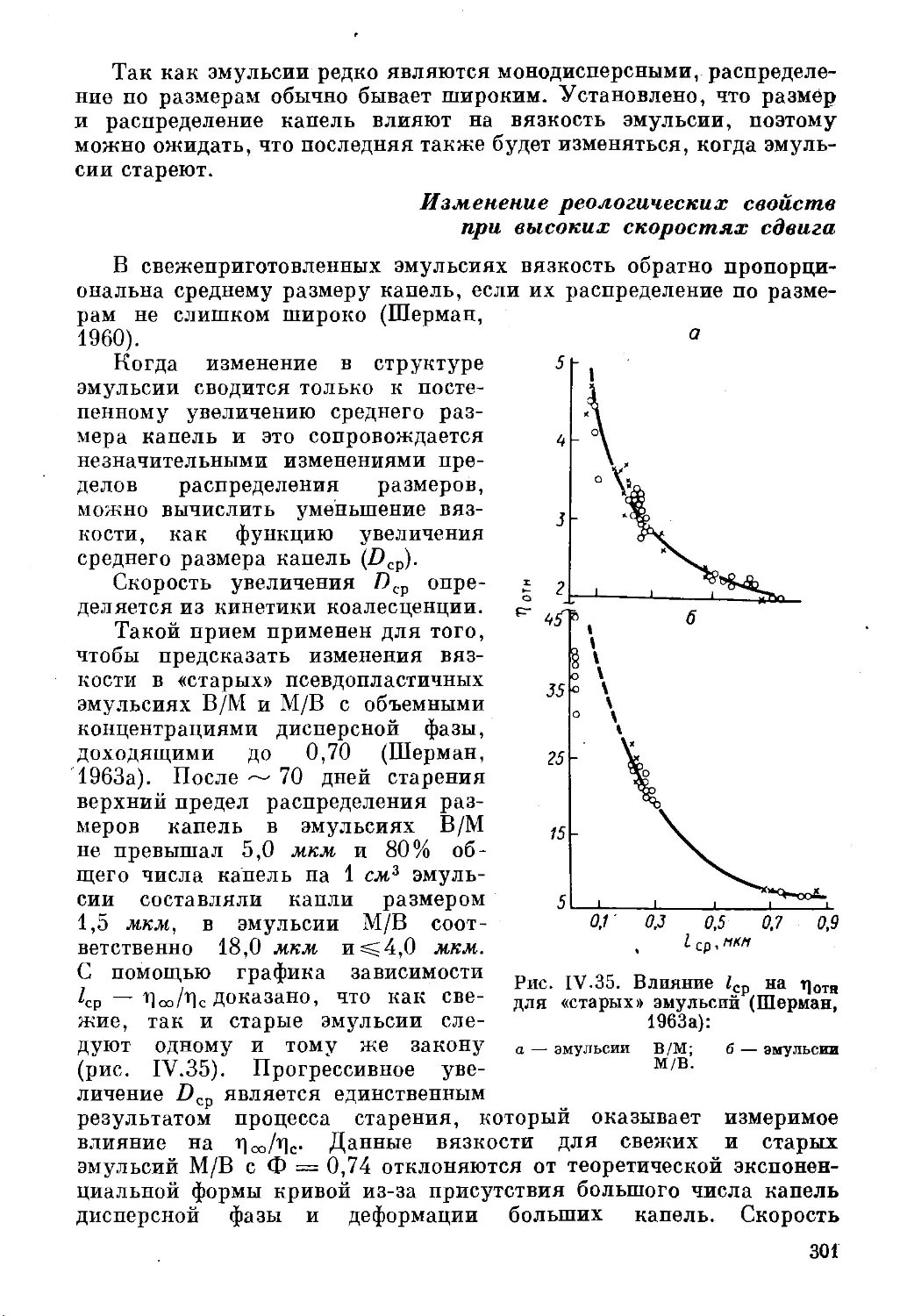
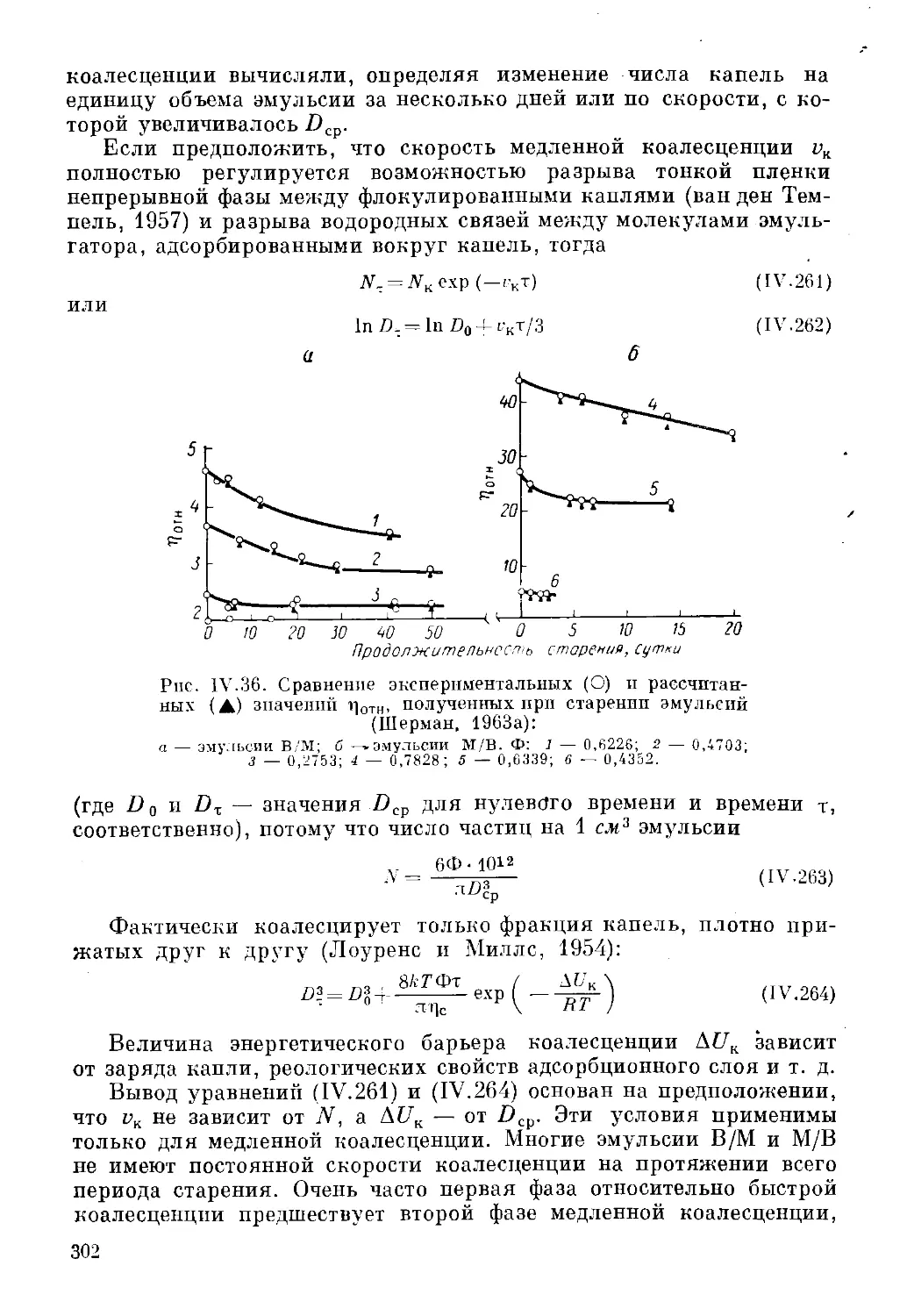
































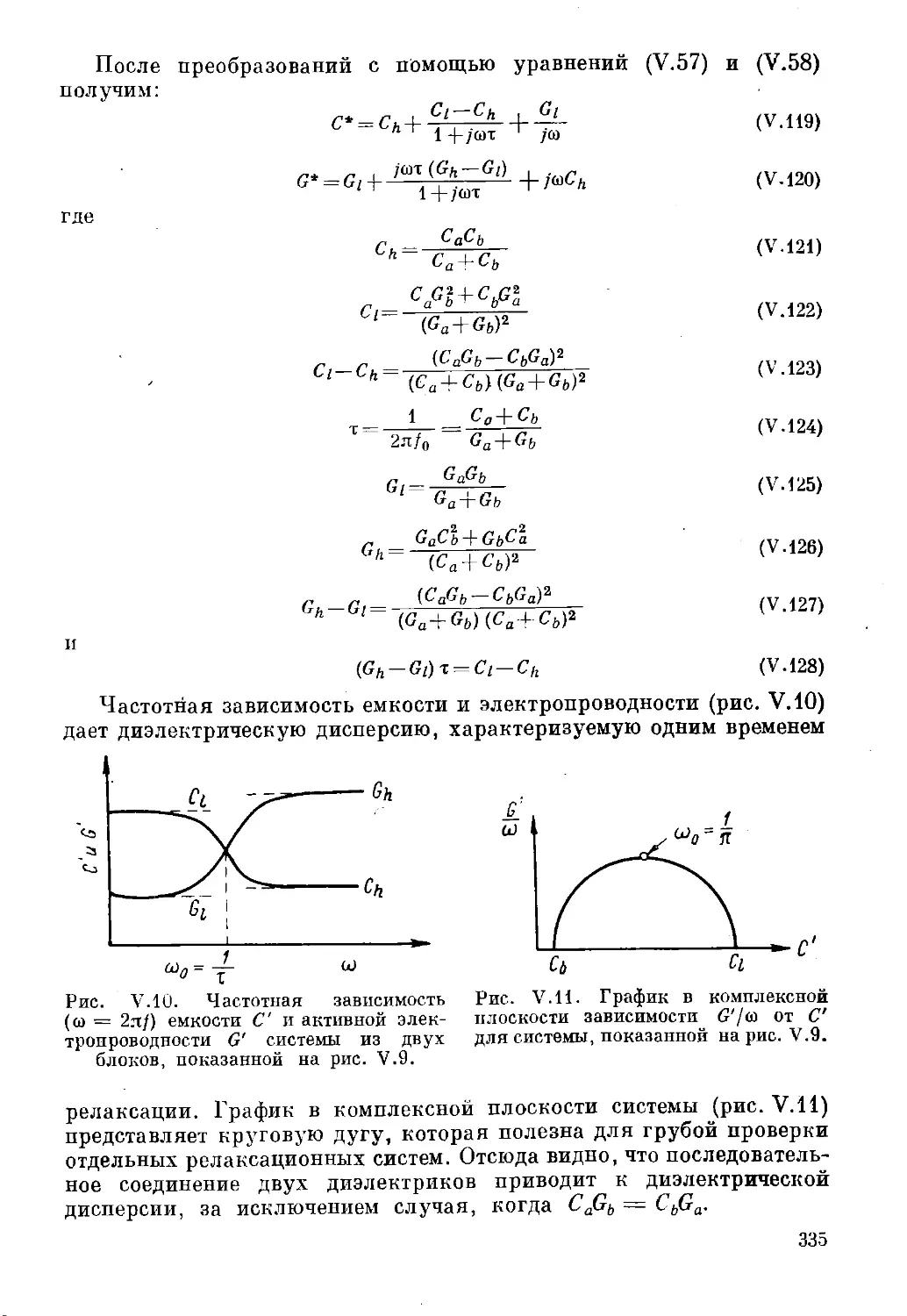















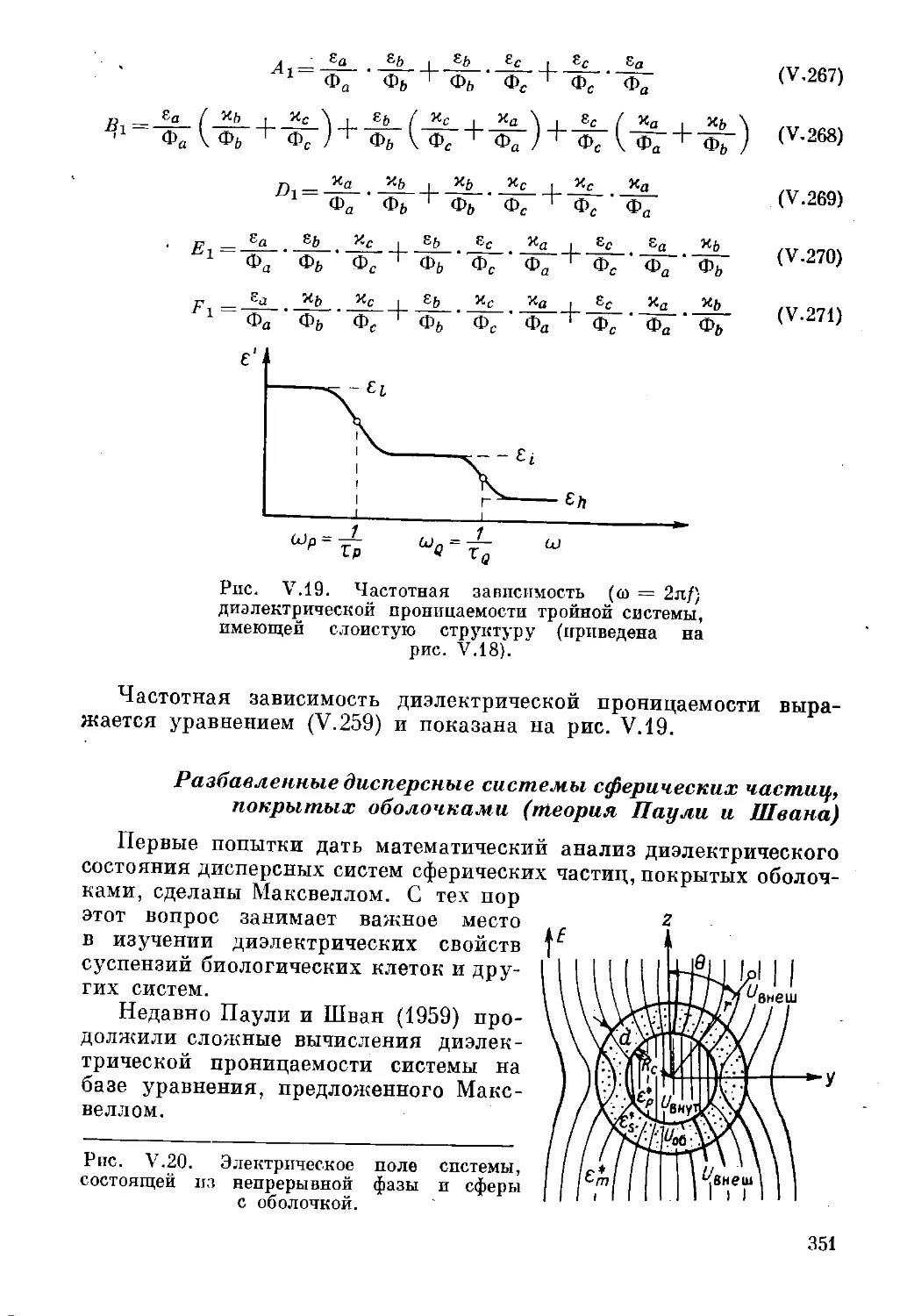











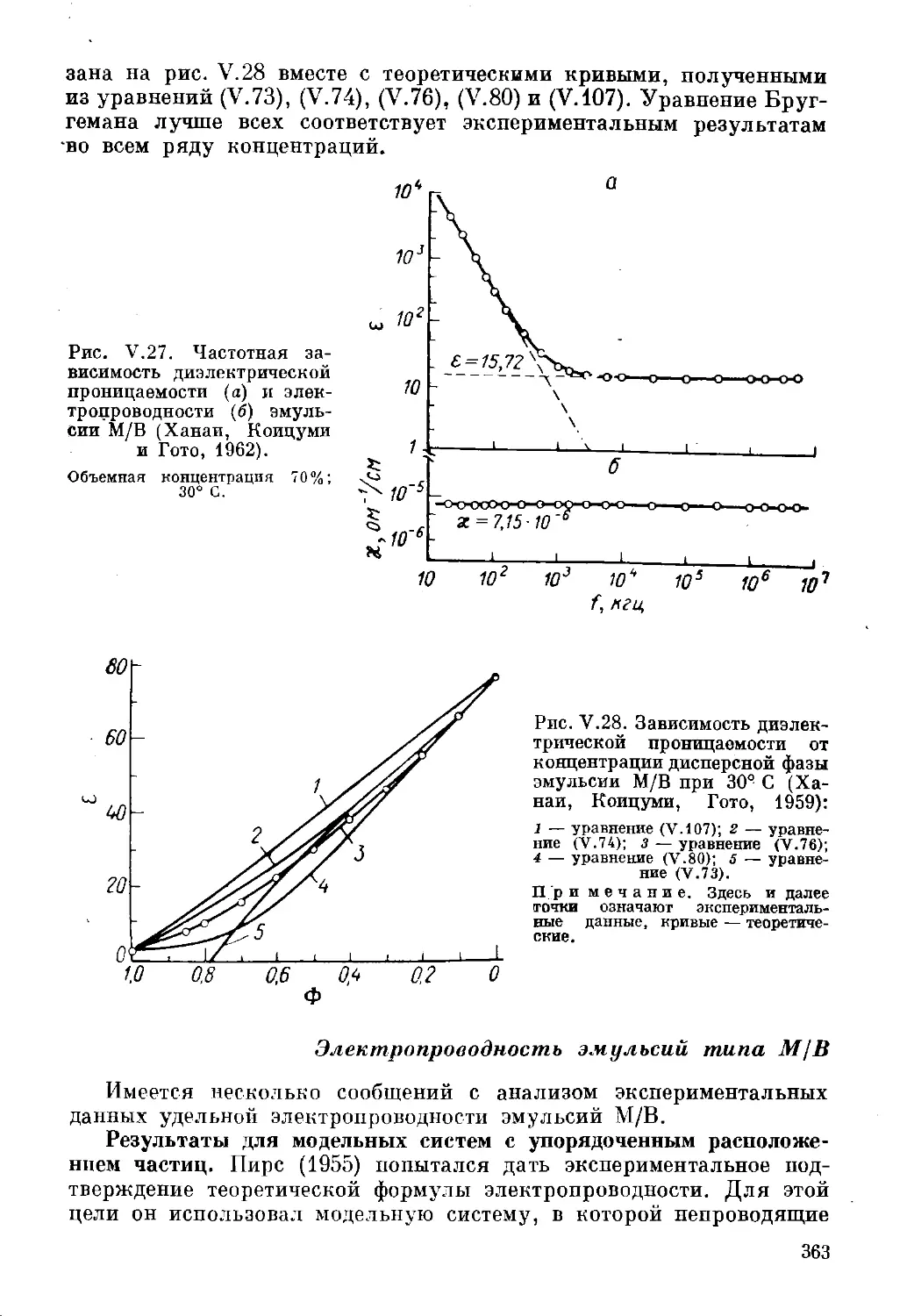





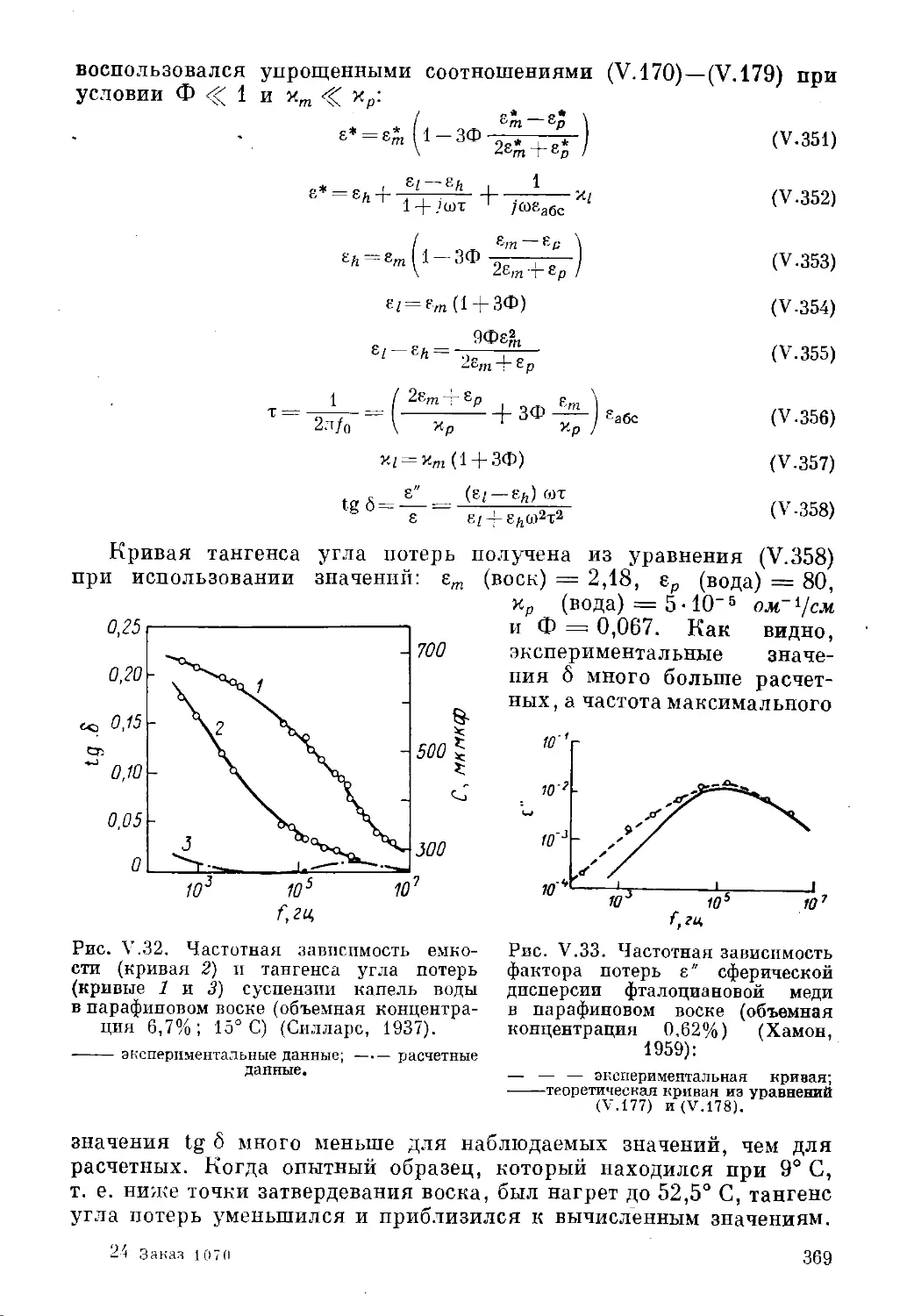





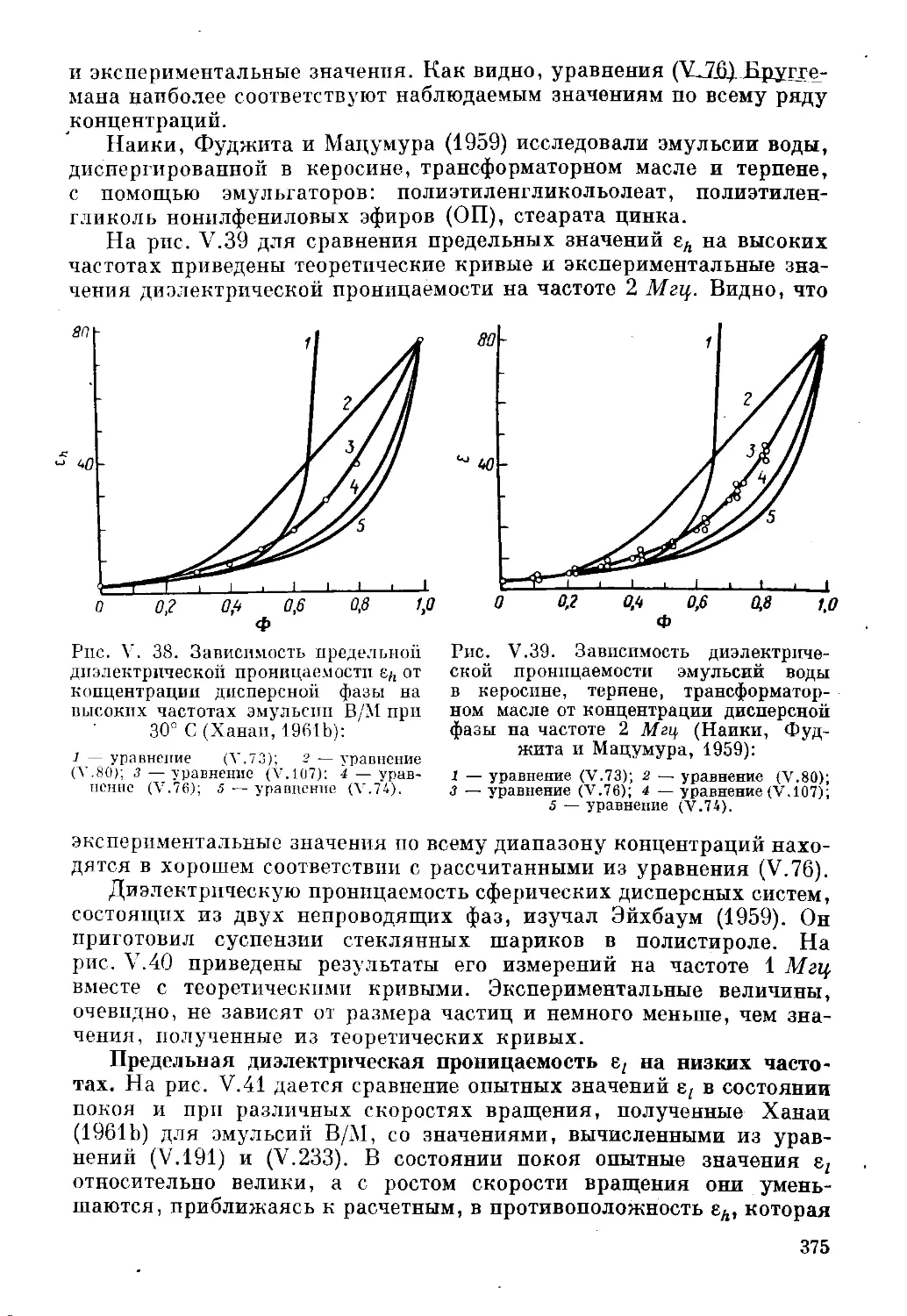





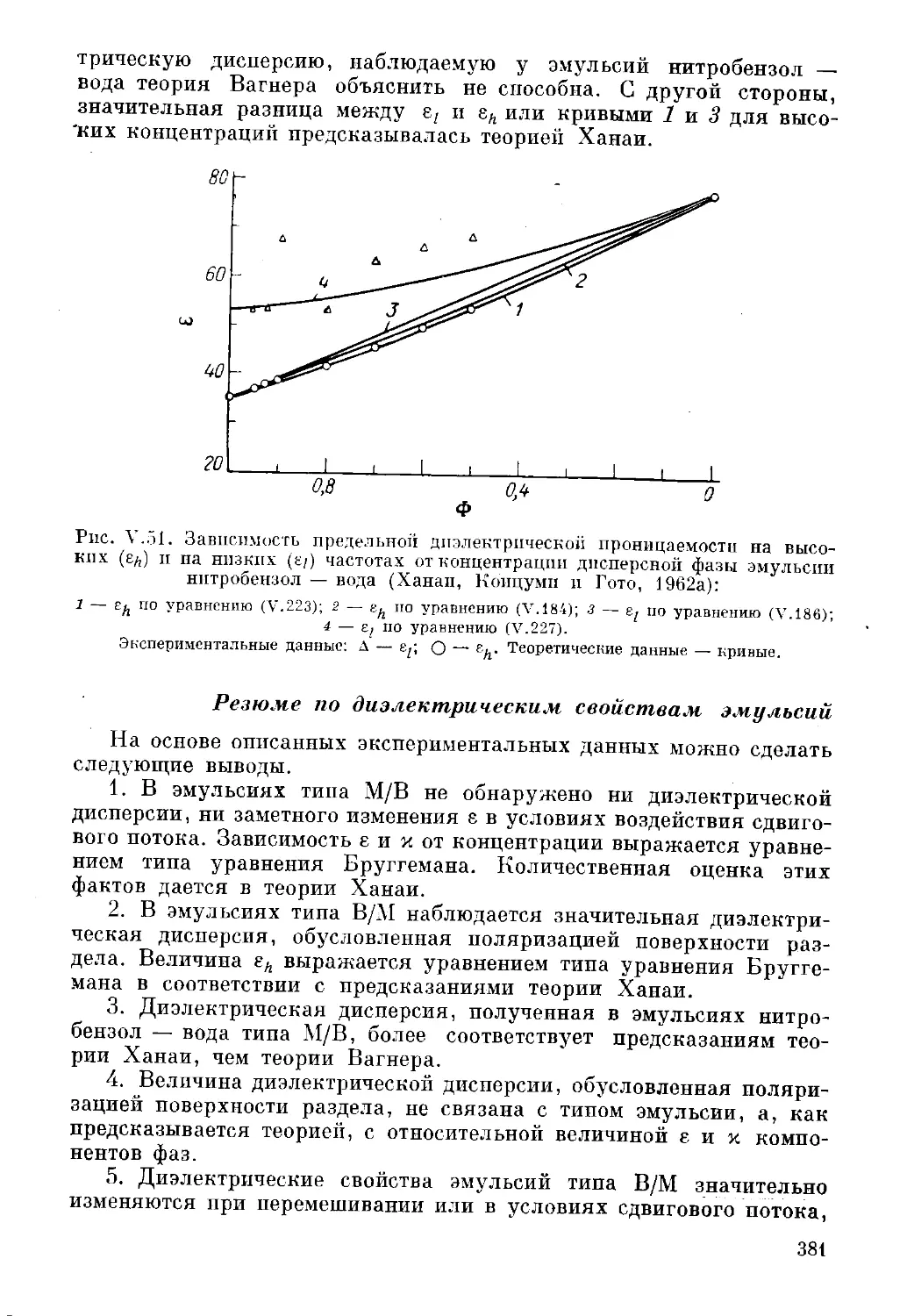

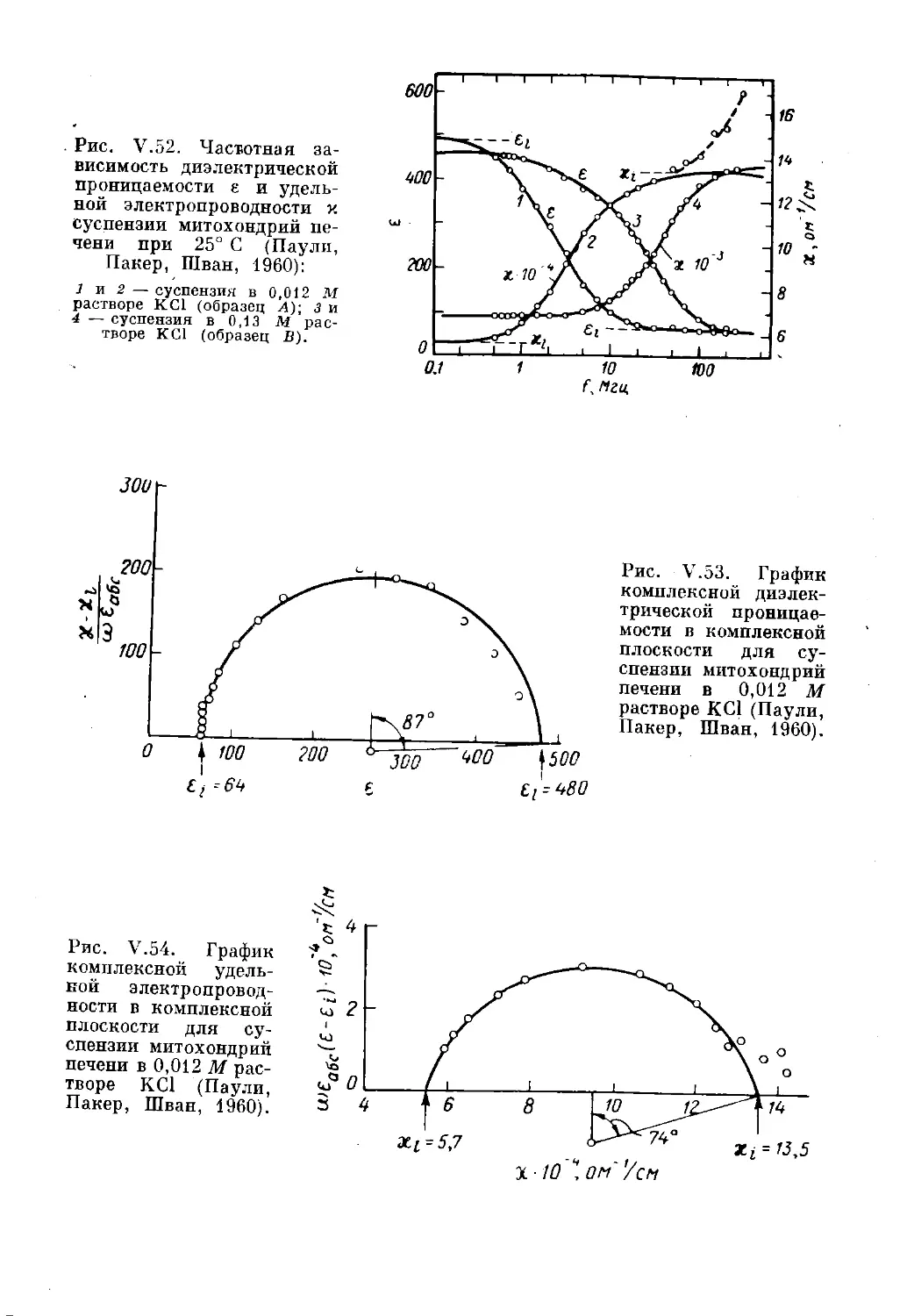



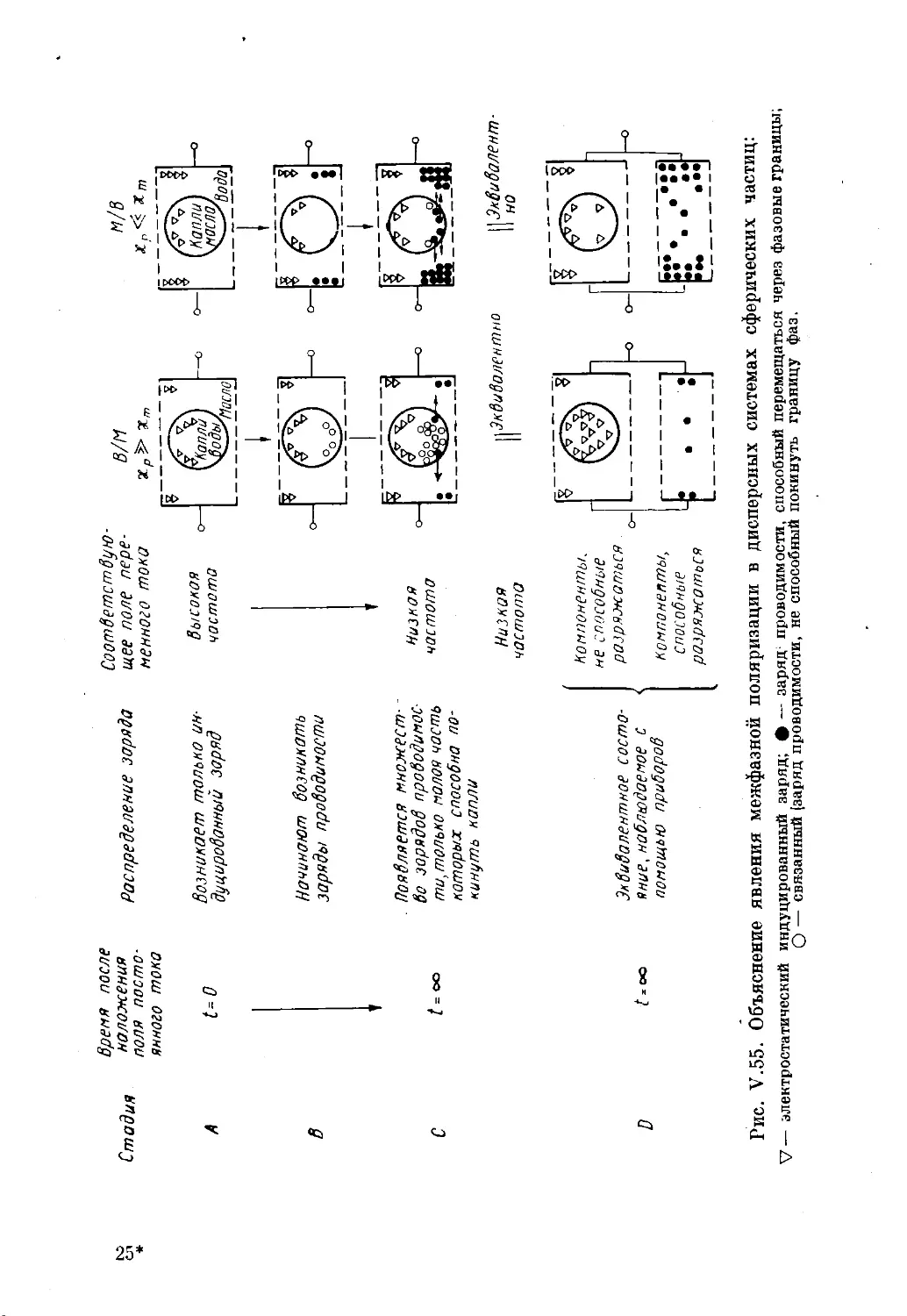









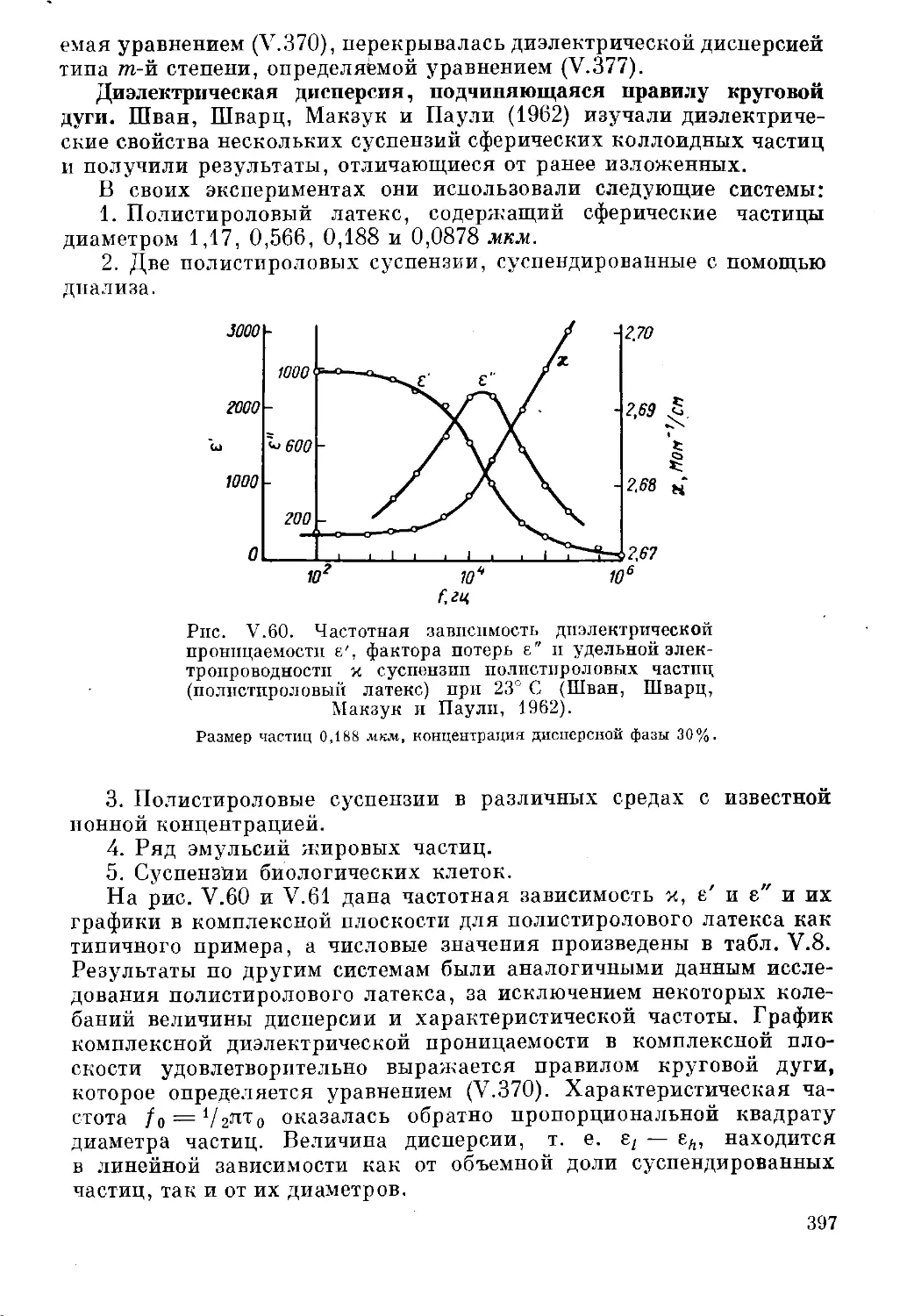






































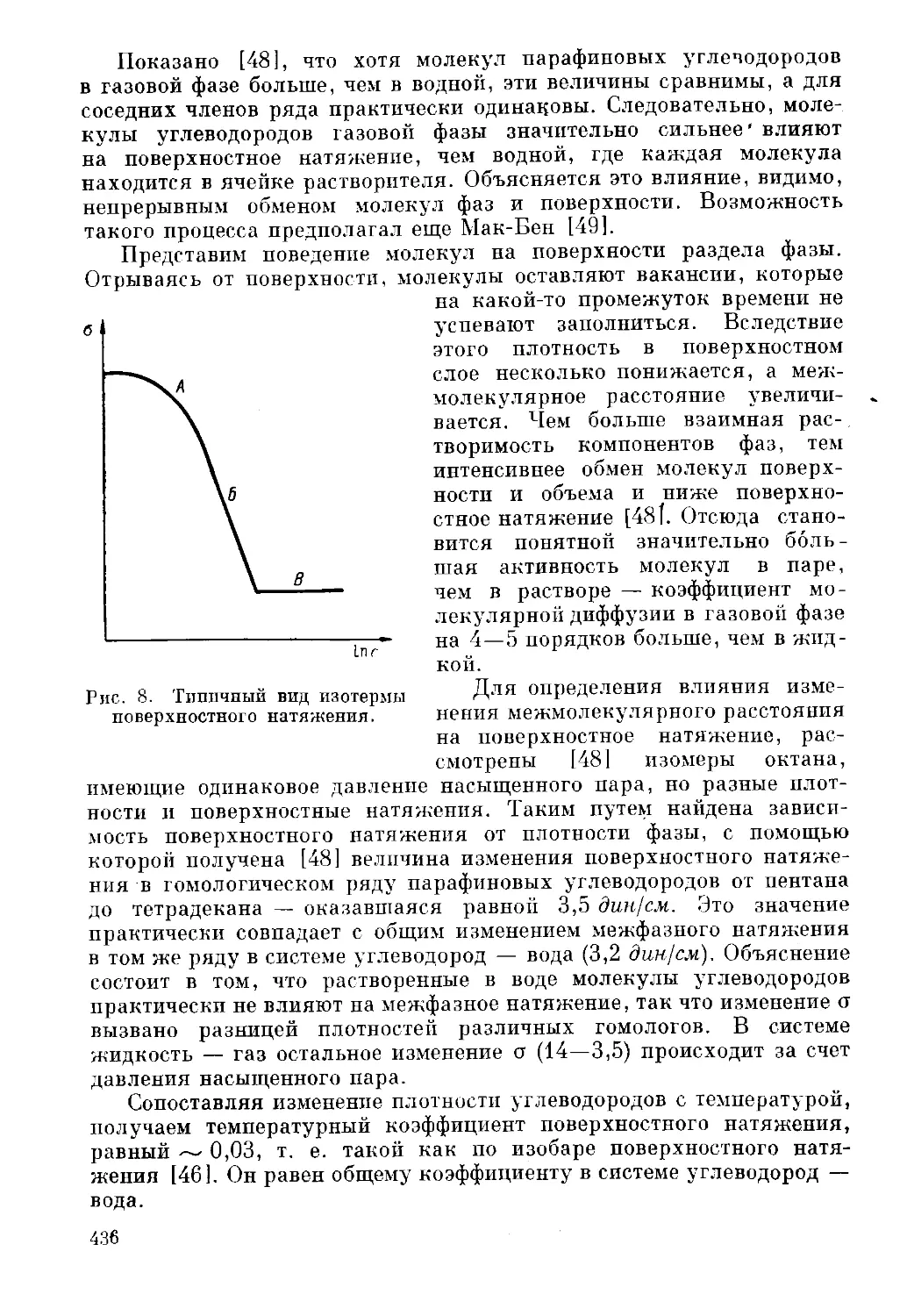

















Текст
ш
Е
MULSION SCIENCE
Edited by
PHILIP SHERMAN
Academic Press.
London and New York
1968
Б1! I
') СО
Э
М У Л Ь С И И
Перевод с английского
под редакцией
докт. техн. наук А. А. АВРАМЗОНА
Издательство «X и м и я»
Ленинградское отделение
1972
УДК 541.182
Эмульсии.
Ф. Шермана.
ред. А- А.
«Химия», Л
табл. 24, рис. 146.
Под редакцией
Пер. с англ. под
Абрамзона. Изд-во
,, 1972, стр. 448.
В книге на современном уровне
рассмотрены важнейшие вопросы теории и
практики эмульсионных систем: теория
стабильности, свойства п реология
эмульсий. Впервые и достаточно обстоятельно
публикуется материал об электрических
свойствах эмульсий, который должен
способствовать развитию метрологии этой
системы.
Освещены вопросы влияния строения
молекул поверхностно-активных веществ
(ПАВ) на получение и свойства
эмульсий, а также методы оценки
эмульгирующей способности и выбора ПАВ в
качестве стабилизатора дисперсионной системы.
Книга может быть полезна для
специалистов самых различных областей, в
которых находят применение эмульсин:
химиков и биологов, фармацевтов и
металлургов, пищевиков, нефтяников,
строителей, текстильщиков ii т. д.
367037
Перевод с английского
Канд. техн. паук Ю- М.
Канд. хм. наук 3. Н.
Н. В. Д45
Канд. /х
Л- И. Доро,
Сокольского
С лавиной,
Малаховой,
2-5-
9—72
В настоящее время трудно
назвать область пауки и техники,
в которой не применяются
эмульсии.
Несмотря на большую
практическую важность эмульсий,
единственная обстоятельная книга на
русском языке, посвященная им, —
монография Клейтона со статьей акад.
Ребиндера, вышедшая в 1950 г.
За последние 20 лет теория и
практика эмульсий значительно продви-
ПРЕДИСЛОВИЕ нулась вперед. Так, заграницей была
РЕДАКТОРА переиздана книга Клейтона (1954 г.),
ПЕРЕВОДА вышла несколькими изданиями книга
Бехера «Эмульсии, их теория и
практическое применение» и, наконец, в
1968 г. опубликована представляемая
вниманию читателей монография,
освещающая основные проблемы
теории, получения и свойств эмульсий.
Данную монографию нельзя
рассматривать как энциклопедию в
области эмульсий, какой была в свое
время известная книга Клейтона.
Несмотря на значительный объем,
книгу можно рассматривать лишь
как краткий обзор основных
теоретических положений и
библиографию, настолько обширна в настоящее
время литература об эмульсиях.
Не традиционной является
последняя глава книги, посвященная
электрическим свойствам эмульсий.
До настоящего времени метрология
эмульсий находится в зачаточном
состоянии. Знание электрических
свойств может способствовать
развитию количественных методов
исследования эмульсий.
5
Каждая глава написана специалистом по данному вопросу,
внесшим собственный вклад в науку об эмульсиях, что делает книгу
особенно ценной.
Мной написана дополнительная глава и сделан ряд замечаний
по тексту. Основная цель этого материала — осветить вновь
вышедшие работы, обратить внимание на многие нерешенные и
дискуссионные моменты, отметив различие существующих подходов и мнений.
Кроме того, по темам глав составлен дополнительный список работ,
опубликованных за период с момента выхода книги до сдачи перевода
в редакцию.
Все переводчики книги являются исследователями, работающими
в области изучения эмульсий. Главу I перевел канд. техн. наук
Сокольский, главу II — канд. хим. наук Славина, главу III —
ипж. Абрамова, главу IV — канд. хим. наук Малахова и главу V —
инж. Дорофеева.
А. Абрамаон
ПРЕДИСЛОВИЕ
РЕДАКТОРА
КНИГИ
В течение последних лет
опубликован ряд книг по коллоидной
химии и теории поверхностных
явлений. В них, однако, удивительно
мало внимания уделено эмульсиям, и
изложение этого важного предмета
ведется элементарно, если не
архаично. Например, многие детали
процесса приготовления эмульсий в
лабораторных масштабах оказываются
невоспроизводимыми при
производстве эмульсий, стабильных в
течение сколько-нибудь длительного
времени. Кроме того, обработка
данных по вязкости эмульсий дает
основания полагать, что после
классической работы Эйнштейна о
дисперсии сферических частиц в
разбавленных растворах достижения в этой
области весьма скромные, хотя
прошло уже более полувека.
В настоящее время возрос
интерес к свойствам эмульсий. Появились
многочисленные публикации в
журналах, которые способствуют
лучшему пониманию эмульсий.
Данная книга, являющаяся
достаточно полной и критической
монографией — результат
содружества коллектива ученых.
Каждая глава этой книги
планировалась так, чтобы дать
исчерпывающую характеристику положению
теории в свете современных
представлений, а не отсылать читателя к
оригинальной литературе. Там, где
для более глубокой оценки
обсуждаемого предмета требуется знание
других теорий, они описаны более
подробно.
7
Весь материал разделен на пять глав: принципы получения
эмульсий, стабильность эмульсий, общие свойства, реология,
электрические и диэлектрические свойства. Последние две главы отчасти
перекрывают друг друга в том смысле, что электрические и
диэлектрические свойства могут быть использованы для изучения
структуры коагулированных эмульсий. Новые достижения, описанные
в последней главе, могут быть использованы для изучения мембран
на поверхности раздела фаз. В главе о стабильности эмульсий
рассмотрены вопросы, связанные с изменениями при хранении их в
нормальных условиях, а также описаны теории тонких жидких пленок,
поверхностной вязкости и т. д. Стабильность при низких или
высоких температурах и при центрифугировании обсуждается в главе III,
так как установлено, что механизмы коалесценции капель иные.
В книге изложены лишь общие принципы диспергирования, без
подробного описания промышленных диспергаторов. Наконец,
медленные процессы объяснены на основе структуры эмульсий вместо
чисто феноменологических описаний, часто применяемых в реологии.
ф . Шерман
Глава I
ПРИНЦИПЫ
ПОЛУЧЕНИЯ
ЭМУЛЬСИЙ
Е.С.Р. Гопал
Эмульсии в настоящее время
применяют во многих
отраслях промышленности,
сельского хозяйства, а также для научных
исследований. Поэтому
неудивительно, что техника эмульгирования
быстро развивается, правда, методом
«проб и ошибок». Теория
эмульгирования до сих пор явно отстает
от практики. Приготовление
эмульсий пока еще остается эмпирической
областью.
Появление значительного
количества доступной информации в области
физики и химии эмульсий
способствовало прогрессу в их
производстве. Нужно отметить, что основы
знаний об эмульсиях относятся чаще
всего к идеализированным моделям
или к простым системам. Если эти
теории попробовать применять к
производству, то придется ввести так
много различных параметров, что
успех будет очень ограниченным.
С другой стороны, в
производстве эмульсий в промышленных
масштабах приходится сталкиваться
с противоречивыми требованиями,
что заставляет принимать
компромиссные решения. Так возникла
дилемма: для промышленности лучше,
когда эмульсия сложная, для
изучения лучше, когда эмульсия
простая.
Диспергирование и коалесценция
В значительной степени эта
дилемма возникла вследствие того, что
получение эмульсий есть результат
двух конкурирующих процессов, а
именно: диспергирования всего
объема жидкости с образованием
отдельных капель и коалесценции этих
капель с образованием большого
объема жидкости.
Эмульсии термодинамически
неустойчивы. Более целесообразным
9
оказывается рассмотрение системы в метастабильном состоянии
в противоположность изучению взаимодействия капель. При
экспериментальном изучении эмульвий следует учитывать баланс
между процессами образования и коалесценции капель. Для
теоретических анализов, очевидно, удобнее рассматривать эти
процессы как два различных. Поэтому в производстве эмульсий
выделяют две самостоятельные проблемы: а) образование новых
капель; б) стабилизация этих капель по мере их образования.
Такой подход, предложенный Штаммом и Крамером много лет
назад (1926), имеет много преимуществ.
Полагают, что капли удерживаются в метастабильном равновесии,
не сливаясь, под действием электрических сил. Этот вопрос подробно
будет обсужден далее (глава II). Здесь же рассмотрены
исключительно процессы диспергирования большого объема жидкости на
малые капли.
Быстрые и медленные процессы
Имеется, вероятно, одна причина, по которой процесс
диспергирования не изучался так хорошо, как кинетика коалесценции. Внут-
рифазовое диспергирование — «быстрый» процесс, протекающий в
течение секунды или менее. Коалесценция — процесс сравнительно
медленный. Обычно коалесценция длится минуты, часы и даже
месяцы и, следовательно, может быть детально изучена. Исследование
быстрых процессов требует значительной модернизации
оборудования даже в случае простых систем.
Естественно ожидать, что в быстрых процессах динамические
свойства будут иметь большее значение, чем равновесные. В качестве
примера рассмотрим мощность, необходимую для образования
эмульсии. Допустим, что масло (межфазное натяжение а = 1 дин/см)
должно быть заэмульгировано со скоростью —'500 л/ч. Если капли
имеют радиус порядка 1 мкм, то мощность, которая требуется для
образования новой поверхности, составит 5-Ю"4 л. с. В более ранней
литературе такие расчеты нередки, хотя в действительности
требуется мощность порядка 2 л. с. Расхождение обусловлено
пренебрежением работой, затрачиваемой на приведение жидкости в
движение во время эмульгирования. Используя некоторые простые модели
для описания процесса образования эмульсий, можно вычислить
потери мощности на преодоление вязкости (Монк, 1952; Субрама-
ньям, 1966). Эта величина оценивается от 0,1 до 10 л. с, что
соответствует опытным данным. Таким образом, в большинстве случаев
процесс разрыва поверхности, по-видимому, вызван явлениями,
происходящими в жидкой фазе, с учетом электрических и
диффузионных факторов. Объяснение механизма действия облегчается при
использовании термодинамических параметров, таких как
поверхностная энергия. Природа и концентрация компонентов оказывают
косвенное влияние, как и природа поверхности и вязко-эластичные
свойства.
10
ОСНОВНЫЕ МЕТОДЫ ПРИГОТОВЛЕНИЯ ЭМУЛЬСИЙ
Термодинамически устойчивое состояние двух несмачивающихся
жидкостей отвечает минимуму свободной поверхности, причем более
тяжелая жидкость располагается под более легкой. Требуется
проявить немалую изобретательность, чтобы получить метастабиль-
ную эмульсию с большим числом капель одной из жидкостей,
диспергированных в другой. Капли требуемых размеров могут быть
получены двумя различными методами. Один заключается в
выращивании капель из малых центров каплеобразования, другой состоит
в дроблении больших капель. Второй метод часто применяют как
в лабораторной, так и в производственной практике. Коммерческие
фирмы выпускают широкий ассортимент установок для
промышленного приготовления эмульсий. Для понимания процессов, которые
будут рассмотрены ниже, ограничимся ознакомлением с общими
принципами, лежащими в основе работы различной аппаратуры.
Контроль
давлений и температуры
Конденсационный метод
Для пересыщенного пара любой жидкости характерно осаждение
капель в центрах конденсации, которые могут быть в системе. Такие
центры бывают естественные (например, частички пыли или ионы)
и искусственные (специально вводимые
частицы). При отсутствии посторонних
частиц центры конденсации могут
образовываться самопроизвольно (при
относительно высоком перенасыщении)
посредством объединения молекул в очень
маленькие капли порядка 10"6 см в диаметре.
Пересыщенный пар конденсируется в этих
центрах, вследствие чего капли
увеличиваются в размерах. Конденсационным
методом обычно получают аэрозоли
Синклер и Ламер, 1949; Фукс и Суту-
гин, 1965).
Такой же метод может быть
использован для приготовления эмульсий, и это
лучше всего иллюстрирует рис. 1.1, на
котором изображено устройство,
разработанное Зумнером (1933). Пар одной
жидкости (дисперсная фаза) инжектируется под поверхность другой,
образующей внешнюю фазу (дисперсионную среду). В таких условиях
пар становится пересыщенным и конденсируется в виде капель
размерами порядка 1 мкм. Эти капли стабилизируются в жидкости,
содержащей соответствующий эмульгатор. Жидкость, которая должна
быть диспергирована, нагревают в отдельном сосуде. Для
контроля нагрева температуру и давление пара поддерживают
постоянными при определенных значениях. На размер образующихся капель
Рис. 1.1. Принципиальная
схема конденсационного
метода.
И
существенным образом влияют давление инжектируемого пара,
диаметр впускного сопла и эмульгатор. Чтобы создать неизменные
условия, в течение всего периода нагревания под соплом держат
какую-либо жидкость, выполняющую вспомогательную роль. Когда
температура и давление достигнут требуемых значений, сосуд,
содержащий эту вспомогательную жидкость, быстро заменяют сосудом
с той жидкостью, которая должна быть дисперсионной средой в
приготавливаемой эмульсии. Жидкость выдерживают при постоянной
температуре, а также осторожно перемешивают, чтобы обеспечить
равномерность распределения центров каплеобразования. Даже при
не очень высоких концентрациях эмульсий этим методом легко
получают капли с размерами до 20 мкм.
Как было указано выше, конденсационный метод широко
применяют для получения аэрозолей. Если в слегка пересыщенный пар
ввести очень маленькие (с размерами до 10"6 см) частички и позволить
всем этим центрам каплеобразования одинаково расти в течение
некоторого времени, то, как было установлено, все капли будут иметь
практически одинаковые размеры. Если такие аэрозоли станут
оседать в жидкой среде, то тем самым образуется эмульсия. Применение
этого метода будет рассмотрено далее (стр. 58).
Дисперсионные методы
Эмульсию легко приготовить, прикладывая внешнюю силу.
Существует три метода: смешения, гомогенизации и коллоидной
мельницы. Обычно аппаратура для приготовления эмульсий этими
методами характеризуется широким интервалом
производительности — от малых лабораторных до больших промышленных
установок. Нецелесообразно их перечислять, ниже будут описаны в общих
чертах лишь три основные типа. Однако перед тем, как выбрать ту
или иную установку, следует решить, будет ли ее применение
действительно выгоднее, чем использование простых методов
эмульгирования.
Метод прерывистого встряхивания
Легко продемонстрировать образование эмульсии, если пробирку,
в которую налиты две жидкости, энергично встряхивать. Бриге
(1920) установил, что прерывистое встряхивание с постоянными
интервалами между толчками гораздо более эффективно, чем
непрерывное. Например, для приготовления эмульсии 60% бензола
в 1% растворе олеата натрия необходимо непрерывное встряхивание
в течение 7 мин (за это время машина-трясучка сообщает ~3000
толчков). Такая же эмульсия может быть приготовлена посредством
только пяти встряхиваний вручную в течение 2 мин, если интервалы
между каждыми двумя толчками составят 20—30 сек. Если
изобразить графически зависимость между числом толчков, сообщаемых
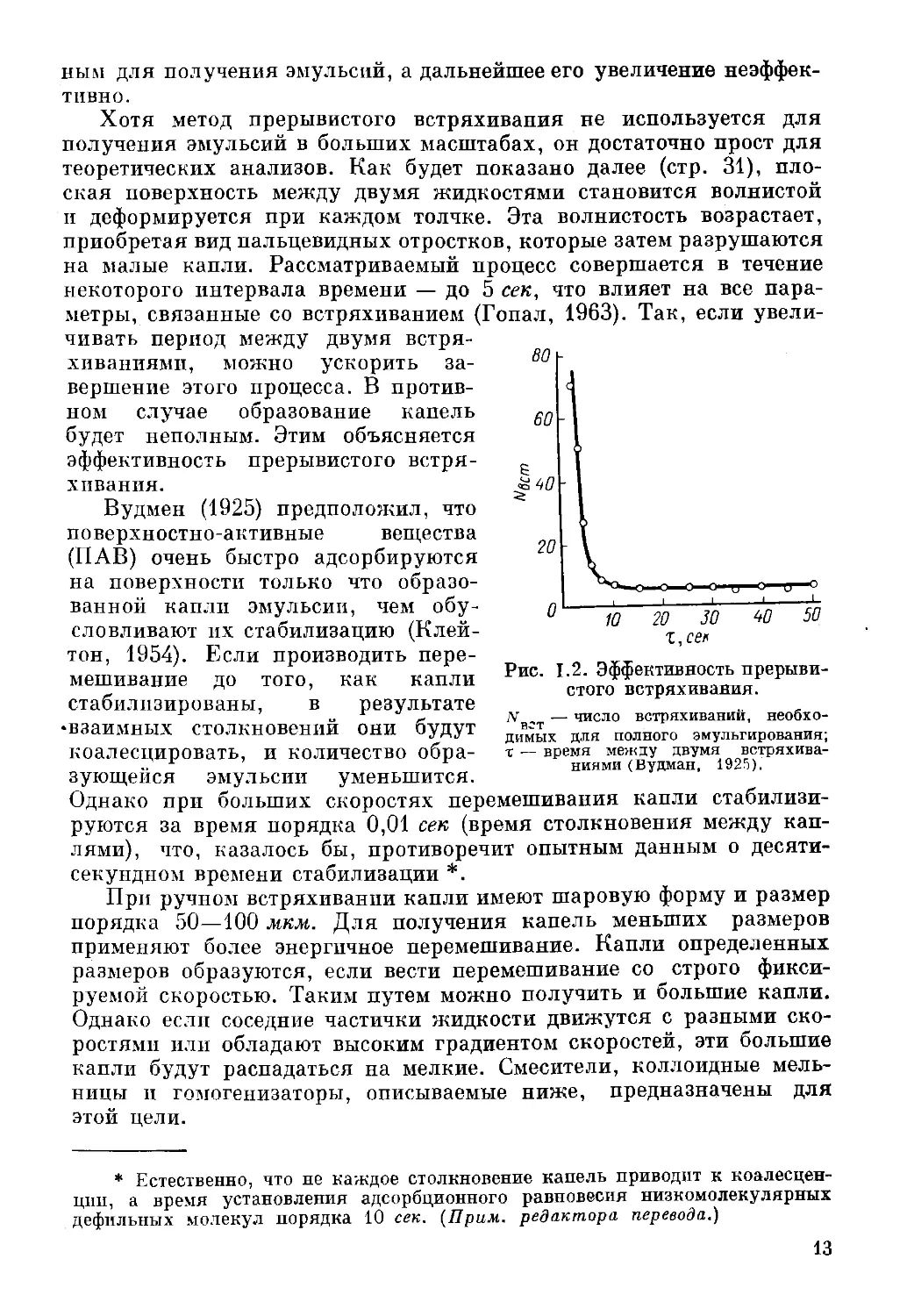
эмульсии, и интервалом между толчками (рис. 1.2), то из этого
графика будет видно, что интервал в 10 сек является достаточ-
12
во
60
\ьо
20
ным для получения эмульсий, а дальнейшее его увеличение
неэффективно.
Хотя метод прерывистого встряхивания не используется для
получения эмульсий в больших масштабах, он достаточно прост для
теоретических анализов. Как будет показано далее (стр. 31),
плоская поверхность между двумя жидкостями становится волнистой
и деформируется при каждом толчке. Эта волнистость возрастает,
приобретая вид пальцевидных отростков, которые затем разрушаются
на малые капли. Рассматриваемый процесс совершается в течение
некоторого интервала времени — до 5 сек, что влияет на все
параметры, связанные со встряхиванием (Гопал, 1963). Так, если
увеличивать период между двумя
встряхиваниями, можно ускорить
завершение этого процесса. В
противном случае образование капель
будет неполным. Этим объясняется
эффективность прерывистого
встряхивания.
Вудмен (1925) предположил, что
поверхностно-активные вещества
(ПАВ) очень быстро адсорбируются
на поверхности только что
образованной капли эмульсии, чем
обусловливают их стабилизацию
(Клейтон, 1954). Если производить
перемешивание до того, как капли
стабилизированы, в результате
•взаимных столкновений они будут
коалесцировать, и количество
образующейся эмульсии уменьшится.
Однако при больших скоростях перемешивания капли
стабилизируются за время порядка 0,01 сек (время столкновения между
каплями), что, казалось бы, противоречит опытным данным о десяти-
секундном времени стабилизации *.
При ручном встряхивании капли имеют шаровую форму и размер
порядка 50—100 мкм. Для получения капель меньших размеров
применяют более энергичное перемешивание. Капли определенных
размеров образуются, если вести перемешивание со строго
фиксируемой скоростью. Таким путем можно получить и большие капли.
Однако если соседние частички жидкости движутся с разными
скоростями или обладают высоким градиентом скоростей, эти большие
капли будут распадаться на мелкие. Смесители, коллоидные
мельницы и гомогенизаторы, описываемые ниже, предназначены для
этой цели.
О1
10
20 30
г, сен
чо 50
Рис. 1.2. Эффективность
прерывистого встряхивания.
N — число встряхиваний, необхо-
в-т
димых для полного эмульгирования;
х — время между двумя
встряхиваниями (Вудман, 1925).
* Естественно, что не каждое столкновение капель приводит к коалесцен-
цип, а время установления адсорбционного равновесия низкомолекулярных
дефильных молекул порядка 10 сек. (Прим. редактора перевода.)
13
Смесители
Смешение жидкостей — хорошо известная операция в химической
технологии. Промышленность выпускает смесители разнообразных
размеров — от малых, с рабочей емкостью <1 л, до больших, с
емкостью до нескольких кубических метров. Принцип образования
смесей можно пояснить с помощью рис. 1.3. Предположим, что
обыкновенное весло вращается в большом цилиндрическом сосуде
и приводит жидкость в движение, причем ее поверхность принимает
форму, близкую к параболической. Частицы жидкости приобретают
круговое движение по горизонтали и лишь в незначительной
степени — вертикальное перемещение. Равномерное распределение
\/
\
^_
\
_>' п
^7
Рис. 1.3. Схема движения жидкости в сосуде с отбойными
перегородками:
а — с пропеллерной мешалкой; 6 — с турбинной мешалкой; 1 —
мешалка; 2 — отбойные перегородки.
частиц но всему объему сосуда получается лишь когда имеются
боковые вертикальные течения.
Равномерного смешения можно достичь, установив вблизи стенок
сосуда вертикальные отбойные перегородки, которые отклоняют
жидкость вверх. Далее, при использовании мешалки пропеллерного
типа частицы жидкости приобретают импульсы как в
горизонтальном, так и в вертикальном направлениях (см. рис. 1.3, а), что
способствует смешению. Перемешивание эффективно, когда течение
становится турбулентным во всем объеме аппарата. Применение
турбинной мешалки (см. рис. 1.3, б) позволяет значительно
увеличить скорость кругового движения. Центробежные силы
разбрасывают частицы жидкости по всему объему смесителя, чем достигается
большая эффективность перемешивания.
Возможны различные решения при проектировании смесителей.
Для усиления боковых течений могут быть использованы две
турбинные мешалки, нижняя создаст движение вверх, верхняя —
вниз. Турбинные мешалки могут устанавливаться эксцентрично
или даже сбоку. Чтобы получить смешение требуемого качества,
применяют смесители периодического действия. В смесителях
14
непрерывного действия равномерное смешение в больших объемах
достигается так быстро, что не требуется дополнительной выдержки
смеси.
В большинстве смесителей обычно происходит незначительный
нагрев из-за преодоления вязкости жидкости и других потерь. Хотя
это и допустимо для малых смесителей (например, в маслобойных
аппаратах), в смесителях большого размера обычно
предусматривают охлаждающие устройства. Смесители с пропеллерными
мешалками применяют для приготовления эмульсий с малой или средней
вязкостью, а смесители с турбинными мешалками — для эмульсий
с большой вязкостью. Диаметр капель в таких эмульсиях — порядка
5 мкм.
Коллоидные мельницы
В коллоидной мельнице эмульгирование происходит при
выдавливании жидкости в узкий зазор между ротором, вращающимся с
большой скоростью, и неподвижным статором. Схема коллоидной
мельницы представлена на рис. 1.4. Смешивающиеся жидкости поступают
тиОкиспю
Жидкость под
давлением
л вращающемуся
мотору
Рис. 1.4. Схема вертикальной
коллоидной мельницы:
1 — статор; 2 — ротор; 3 — обмотка
и подшипник вала ротора.
Эмульсия
Рис. 1.5. Устройство клапана
одностадийного генератора:
1 — станина гомогенизатора; 2 — подшипник
стержня; 3 — црюкимная пружина; 4 —
клапан; 5 — конический стержень; 6 — винт
с рукояткой.
сверху через соответствующую трубу в полость статора, протекают
через узкий зазор между поверхностями статора и ротора и, наконец,
вытекают из мельницы. Ротор отцентрирован и может развивать
скорость 1000—20 000 об/мин. Зазор между поверхностями ротора
и статора может быть уменьшен до 0,0025 см. Вследствие большой
скорости и малого зазора возникают большие касательные
напряжения, которые в сочетании с центробежными силами обусловливают
15
почти мгновенный разрыв жидкой струи на капли. Жидкость течет
в зазор либо иод действием собственного веса, либо под небольшим
избыточным давлением.
Изготавливают различные варианты коллоидных мельниц.
Например, они бывают вертикальными (как на рис. 1.4) или
горизонтальными. Поверхности ротора и статора могут быть как ровными,
так и неровными — с зубцами и прорезями. Эти прорези делают
радиальными, спиральными или концентрическими, что, как
полагают, увеличивает турбулентность и улучшает смешение. Обычно
в конструкции предусматривают возврат эмульсии п повторное
пропускание через мельницу, что дает более тонкое измельчение.
В настоящее время коллоидные мельницы чаще всего изготавливают
из дюралюминия или из нержавеющей стали, но иногда природа
смешиваемых жидкостей или экономические соображения диктуют
выбор иного конструкционного материала. Регулировкой скорости
вращения ротора и зазора между ротором и статором можно
приспособить коллоидную мельницу для жидкостей с различными вязко-
стями или иными характеристиками. Выпускаемые
промышленностью мельницы в большинстве случаев имеют производительность
10—20 000 л/ч. Вследствие больших касательных напряжений и
потерь на трение температура в них быстро возрастает. В мельницах
больших размеров всегда применяют охлаждение.
Пастообразную массу в коллоидную мельницу подают под
небольшим давлением. Чем выше вязкость образующейся эмульсии, тем
меньше количество выходящей из мельницы смеси. Диаметр капель
в эмульсиях, получаемых посредством коллоидных мельниц,
порядка 2 мкм.
Гомогенизаторы
Гомогенизаторы — это устройства, в которых диспергирование
жидкости достигается пропусканием ее через малые отверстия под
высоким давлением. Гомогенизаторы используют для получения
эмульсий с размерами капель 1 мкм и менее. Общеизвестно
применение этих устройств для гомогенизации молока — уменьшения
размеров капель жира в молоке.
В типичных гомогенизаторах жидкость продавливается через
отверстие под давлением до 3,5-107 н/м2, при этом насосы имеют
различную производительность. Поперечное сечение отверстий —
порядка 10"4 см2. Различные модели гомогенизаторов отличаются
конструкцией отверстий и способом регулирования их размеров.
Детали клапана показаны на рис. 1.5. Жидкость под большим
давлением продавливается через кольцеобразную полость между
неподвижным отверстием и подвижным коническим стержнем. Стержень
перемещается с помощью винтового механизма. Так, при подаче
стержня внутрь кольцевого отверстия площадь его сечения
уменьшается. И конический стержень, и отверстия изготавливают из
прочных материалов, например из закаленной нержавеющей стали,
чтобы не было эрозии под воздействием высокоскоростных струй.
16
Возникающие в кольцевом пространстве высокие напряжения и
большой градиент скоростей вызывают разрыв жидкости на капли.
Крепление подвижного стержня может быть осуществлено посредством
винта, но, по-видимому, прижимное пружинное устройство,
показанное на рис. 1.5, более удобно. Стержень такой конструкции можно
подвергать высокочастотным колебаниям, которые способствуют
диспергированию жидкости (это используют при звуковых и
ультразвуковых методах получения эмульсий, рассматриваемых далее —
стр. 45). В некоторых моделях клапанов отверстие закрывают
горизонтальной пластиной или втулкой. Известны и другие варианты
клапанов, нередко остроумные по конструкции.
Промышленность выпускает гомогенизаторы различных типов
и емкостей. В большинстве аппаратов предусмотрена возможность
повторного диспергирования. Некоторые модели имеют две стадии
гомогенизации, и два выпускных отверстия. Было найдено,
что иногда в гомогенизаторе попадаются достаточно крупные капли.
Если же составить своеобразный тандем из двух аппаратов, имея,
например, в первом давление 300 am, а во втором — 30 am, то
крупные частицы будут образовываться в первом и разрушаться во втором
гомогенизаторе.
В гомогенизаторах можно диспергировать как жидкости, так
и пастообразные материалы. Вследствие использования высокого
давления вязкость среды мало сказывается на производительности
аппарата. Возрастание температуры в работающих гомогенизаторах
незначительное, поэтому обычно не требуется охлаждающее
устройство.
Если пропустить через гомогенизатор предварительно смешанные
•жидкости, то образовываемая эмульсия может иметь размер частиц
до 1 мкм даже при однократной обработке. В случае раздельно
взятых жидкостей требуется многократное прохождение их через
гомогенизатор для получения столь мелких капель. Гомогенизаторы —
экономически наиболее выгодные аппараты для производства
высокодисперсных эмульсий.
Энергия, требуемая для эмульгирования
Как уже отмечалось, энергия необходима не только для
образования новых поверхностей, но и для преодоления внутреннего трения
жидкости и приведения ее в движение. Потребляемая установкой
для эмульгирования мощность будет зависеть от целого ряда
факторов: скорости прохождения жидкости через гомогенизатор, ее
вязкости, поверхностного натяжения, использованного эмульгатора,
размера частиц, концентрации эмульсий, подъема температуры,
а также размера и типа самого а/ппаддта. Все эти разнообразные
данные учитываются в соответствталцив^моделях, их классификация
дана Гриффином (1950). На РиэдВЙ^ЧеЛтом из его работы,
ориентировочно показаны области потрвюш»мо\яЮюгии смесителя,
коллоидной мельницы и гомогенизатор*./ \ \\
3267007 ff££\
; Мяч. «нем. *Л,
Как видно из рис. 1.6, в простых смесителях затрачивается
значительно меньше мощности, чем в коллоидных мельницах или
гомогенизаторах при той же
производительности.
Поэтому на химических заводах
предпочитают применять
смесители. Однако следует
отметить, что смесители
производят эмульсии с более
крупными частицами, чем
аппараты других типов.
Гомогенизаторы потребляют
меньше мощности, по сравнению
с коллоидными мельницами,
но они дороже в
эксплуатации из-за высоких
давлений, приводящих к
преждевременному износу и поломке деталей. Таким образом, все
три типа установок дополняют друг друга.
/ v'. чэО 4500 45000
Производительность. л/ч
Рис. 1.6. Расход мощности па
эмульгирование в различных аппаратах:
1 — коллоидная мельница; 2 — гомогенизатор,
3 — смеситель.
Другие методы эмульгирования
Хотя получение эмульсий в смесителях, коллоидных мельницах
и гомогенизаторах сейчас является обычным для промышленного
производства, за последнее время появились и другие методы, по
крайней мере для специальных целей. Это, прежде всего, звуковые
н ультразвуковые методы, которые постепенно внедряются в
промышленность. Ввиду интенсивного развития современной
ультразвуковой техники, этим методом посвящен специальный раздел.
Электрические методы получения эмульсий в настоящее время
используют лишь в лабораторных масштабах.
Часто приходится слышать о «самоэмульгирующихся» системах.
Это неправильное употребление термина, так как эти системы
все же требуют небольшого количества эмульгатора, хотя и в
меньшей степени, чем обычные вещества. Изредка встречается истинно
самопроизвольное эмульгирование (см. ниже).
ФИЗИКО-ХИМИЧЕСКИЕ ФАКТОРЫ ,
ВЛИЯЮЩИЕ НА ОБРАЗОВАНИЕ ЭМУЛЬСИИ
В предыдущем разделе о методиках приготовления эмульсий
упомянуто было кратко. В общих чертах все методики сводятся
к разбиванию жидкостей на малые капли. Однако для получения
действительно хороших эмульсий необходимо знать многие детали
и тонкости. Так, применяя первоначально довольно большие
внешние силы, нужно затем прилагать весьма незначительные усилия
для получения эмульсии с требуемыми свойствами. Тонкости при
образовании эмульсий, бчевндно, зависят от свойств конкретных
18
эмульсий и проявляются в виде разнообразных хитроумных
приемов. В этом разделе детально рассмотрено влияние различных'
факторов на качество эмульсий.
При получении эмульсии желательно контролировать некоторые
ее характеристики, а именно: а) концентрацию; б) стабильность;
в) размер частиц; г) вязкость. Можно было бы определять и другие
свойства, такие как диэлектрическая проницаемость,
электропроводность или цвет, но они относительно специфичны и, кроме того,
изменяются незначительно. Поэтому только первые четыре
представляют наибольший практический интерес. Эти свойства не являются
независимыми друг от друга. Так, чем меньше размер частиц, тем
больше вязкость и стабильность эмульсии. Концентрированные
эмульсии обычно очень вязкие. Для достижения высокой
стабильности или высокой концентрации требуются совершенно
противоположные условия. Все эти взаимосвязи более подробно рассмотрены
в главе IV. В настоящей главе ограничимся вопросом о влиянии
этих свойств на способы образования эмульсий.
Выбор эмульгатора
Первые исследователи свойств эмульсии считали, что
поверхностное натяжение а является очень важным фактором, определяющим
стабильность и размер частиц. Приводились доводы, что большая
величина а означает и большую энергию, затрачиваемую на
образование новой поверхности и, следовательно, это
неблагоприятствует образованию эмульсии. Поэтому стремились к уменьшению а
тем или иным путем. Как установлено в настоящее время, работа,
"затрачиваемая на образование новой поверхности, представляет
собой лишь часть общей энергии, потребляемой в процессе
приготовления эмульсии. Несомненно, низкое значение поверхностного
натяжения способствует диспергированию, но более важны те
изменения, которые происходят в двойных электрических слоях *,
образующихся возле этих поверхностей. Двойной электрический
слой обеспечивает устойчивость эмульсии, препятствуя коагуляции
частиц, и показывает, будут ли образовываться эмульсии типа вода
в масле (В/М) либо масло в воде (М/В). Изменение поверхностного
натяжения — проявление тех изменений, которые происходят в
природе самой поверхности.
Для промышленного производства наиболее важной
характеристикой эмульсии является ее устойчивость. Основные требования
к эмульгаторам сформулированы Коббом (1946). Эмульгаторы должны:
1) уменьшать поверхностное натяжение до 5 дин/см для
эмульсий, которые приготавливают перемешиванием, и до 0,5 дин/см для
эмульсий, не требующих интенсивного перемешивания; 2) достаточно
* Автор явно преувеличивает влияние двойного электрического слоя на
свойства эмульсий. Ниже этот вопрос будет подвергнут более подробному
обсуждению. (Прим. редактора перевода.)
2*
19
быстро адсорбироваться на каплях, создавая тонкий слой, не
изменяющийся при столкновениях капель и препятствующий
коагуляции и коалесценщш; 3) иметь специфическую молекулярную
структуру с полярными и неполярными группами; 4) хорошо растворяться
в дисперсионной среде; 5) придавать эмульсии определенный
электрокинетический потенциал; 6) влиять на вязкость эмульсии; 7)
обладать эмульгирующими свойствами даже при малых количествах;
8) быть дешевыми; 9) быть безопасными в обращении и нетоксичными *.
Для удовлетворения разнообразных требований различных
систем было получено множество эмульгаторов. Бехер (1965) описал
свыше 800 выпускаемых
промышленностью ПАВ, однако этот
список далеко не полный.
Рационально используемые эмульгаторы
стабилизируют капли, как только
последние образуются. В зависи-
-ц - - мости от типа стабилизируемых
капель получаются эмульсии В/М
~WoiO№0,005*ioia,05D,OSOj3 o'i7 или М/В. Эмульгатор может быть
Концентрация ростдора опеотанатрия,н. охарактеризован специальным
числом — гидрофильно-липофильным
Рис. 1.7. Зависимость радиуса ча- балансом (ГЛБ). Если число ГЛБ
спщ от концентрации ПАВ в заключено в пределах 3-6, об-
эмульсиях, приготовленных в высо- v " „ .,, '
коскоростном смесителе. разуется эмульсия В/М.
Эмульгаторы с числом ГЛБ, равным 8 —
13, дают эмульсию М/В. Изменяя
природу эмульгатора и его концентрацию, можно добиться
обращения фаз эмульсии. Вопросы устойчивости эмульсин подробно
рассмотрены в главе III.
Эмульгаторы влияют па процесс разрыва поверхности и
образование капель и, следовательно, па поверхностное натяжение о" и,
в меньшей степени, па вязкость. При введении небольших количеств
эмульгатора поверхностное натяжение быстро уменьшается, а но
мере увеличения его концентрации о стремится к определенному,
достаточно малому по величине, значению. Дальнейший рост
концентрации эмульгатора незначительно влияет на о". Итак, малая
величина о способствует эмульгированию. Поэтому по мере
увеличения концентрации эмульгатора процесс образования эмульсии
происходит все легче, возрастает ее стабильность, а размер капель
уменьшается. При этом возрастание концентрации эмульгатора
свыше оптимальной величины уже не улучшает стабильность
эмульсии и не дает более мелких капель. Типичными в этом отношении
являются результаты, полученные Юрген-Лохманн (1951) с эмуль-
* Условия 4—6 не обязательны, так как в ряде случаев ПАВ, не
растворимые в непрерывной фазе, стабилизируют эмульсии. Например, олеат натрия
в присутствии солей (NaCl, Na,SC>4 и др.) стабилизирует обратные эмульсии,
в то время как в органической фазе после расслоения не обнаруживаются даже
следы олеата натрия. (Прим. редактора перевода.)
1:0
сией 15 объемн. % нефти в воде с олеатом натрия. Эмульсии
приготавливали в высокоскоростном смесителе в течение- 30 мин. При
возрастании концентрации олеата натрия от 0 до 0,005 н. радиус
капель уменьшался от нескольких микрометров до 0,12 мкм, но
дальнейшее увеличение концентрации эмульгатора вплоть до 0,16 н.
вызывало незначительное уменьшение их размеров (рис. 1.7).
Аналогичные результаты получены и в других работах.
В промышленности количество используемого эмульгатора
ограничено его стоимостью. Для образования эмульсий употребляют
эмульгаторы в минимальных количествах, которые дают
приемлемые для практических целей результаты. Приблизительная величина
этой оптимальной концентрации эмульгатора может быть вычислена,
исходя из площади поверхности эмульсии, толщины поверхностного
слоя и критической концентрации молекул.
Способ введения эмульгатора
Наиболее распространенными являются четыре способа
введения эмульгатора в эмульсию: 1) растворение в воде; 2) растворение
в масле; 3) образование мыла; 4) прерывистое введение.
По первому способу эмульгатор растворяют в водной фазе,
затем при перемешивании добавляют масло. Обычно таким путем
образуется эмульсия М/В. Если же требуется получить эмульсию
В/М, то масло следует добавлять до тех пор, пока не произойдет
обращения фаз, или же вводить водный раствор в масло.
Второй способ является обратным первому. Эмульгатор
растворяют в масляной фазе, куда затем добавляют воду. Обычно так
образуется эмульсия В/М. Если требуется получить эмульсию М/В,
добавление воды должно быть продолжено вплоть до обращения
фаз, либо масло надо вводить малыми количествами в воду.
Казалось бы, оба способа симметричны, и поэтому при
неизменных условиях способ растворения в воде будет давать эмульсии
М/В, в способ растворения в масле — эмульсии В/М. Это
справедливо, если условия действительно неизменны, а число ГЛБ
эмульгаторов одинаково. Обычно эмульсии М/В требуются чаще. Было
найдено (Кобб, 1946), что в большинстве случаев способ растворения
в масле дает лучшие эмульсии, чем способ растворения в воде,
причем капли оказываются достаточно малыми и одинаковыми по
размеру.
Способ образования мыла *, пригодный для эмульсий,
стабилизируемых мылами, рассчитан на протекание реакции образования
мыла на поверхности раздела фаз. Одна составная часть
эмульгатора — жирная кислота — растворяется в масле, а другая часть —
* Описания данного способа получения эмульсий, называемого также in
sity, можно найти в недавно вышедшей книге Бернштейна, 1969. (Прим.
редактора перевода.)
21
щелочь — в воде. Когда обе фазы соединяются, на поверхности их
раздела образуется мыло, создавая устойчивую эмульсию. В
зависимости от природы мыла могут быть получены эмульсии М/В или В/М.
По способу прерывистого введения воду и масло добавляют
к эмульгатору периодически, малыми порциями. Этот способ
получил широкое распространение в парфюмерной и пищевой
промышленности.
Во всех случаях, где мыла могут служить эмульгаторами, способ
образования мыла предпочтителен. Он дает устойчивые эмульсии
с очень малыми размерами капель. В качестве примера приведем
результат, полученный Доре (1946) с эмульсиями оливкового масла
в водном растворе олеата натрия, приготовленными в
высокоскоростном смесителе. Когда эмульсии получали по способу растворения
в воде, -—-48% капель имели размеры <1 мкм, в то время как
некоторые капли достигали 12 мкм в диаметре. В эмульсиях,
приготовленных по способу образования мыла, 69% всех капель имели
размеры <1 мкм и не было пи одной капли крупнее 8 мкм.
Возможное объяснение этому состоит в том, что па поверхности капли мыло
образуется очень быстро, а при иных способах распределение
эмульгатора вблизи поверхности происходит вследствие диффузии, т. е.
медленпо. В результате капли стабилизируются и быстрее, и лучше.
Способ введения второй фазы
Так же, как существуют различные пути введения эмульгатора,
могут быть разными и способы слияния различных жидкостей.
Например, масло можно вводить в воду, воду — в масло или оба
компонента одновременно — в установку, в которой происходит
образование эмульсии. Очевидно, эмульсия М/В будет легче получаться
при добавлении масла в массу воды, а эмульсия В/М — при
добавлении воды в масло. Это используют в технике, когда добиваются
обращения фаз. Такое обращение зависит от относительных
количеств обеих фаз и природы эмульгатора. Этот вопрос подробнее
рассмотрен ниже (стр. 66).
Известно, что при смешении с помощью пропеллерных или
турбинных мешалок в гомогенизаторах или коллоидных мельницах
получают высокодисперсные и устойчивые эмульсии. Например
(Доре, 1946), в эмульсиях, приготовленных способом растворения
в воде, 72% капель имели размеры < 1 мкм после однократного
прохождения через гомогенизатор и не было капель крупнее 6 мкм.
•Аналогичные результаты получены и с другими эмульсиями.
Время перемешивания
Влияние времени перемешивания на размеры капель в
эмульсиях исследовано в некоторых работах (Гопал, 1959 с). Установлено,
что продолжение перемешивания сверх оптимального времени мало
улучшает качество эмульсий. При нормальных условиях эмульгиро-
•■>•>
вания средние размеры капель уменьшаются очень быстро в течение
первых нескольких секунд и постепенно достигают предельного
значения за 1—5 мин. Стабильность и вязкость эмульсий изменяются
аналогично. Таким образом, если время перемешивания больше
оптимального, то затраты мощности оказываются невыгодными.
Это и понятно. При образовании эмульсии, как было указано
выше, происходят два процесса — диспергирование и коалесцен-
ция. В течение первых нескольких секунд перемешивания
преобладает первый процесс — диспергирование, а коалесценция
распространяется лишь на малое число капель. Чем больше в процессе
перемешивания образуется отдельных капель, тем более частыми
будут и соударения между ними. После нескольких минут
перемешивания коалесценция будет происходить столь же часто, как
и диспергирование, т. е. оба процесса станут равновесными. Именно
условиями равновесия определяются величина концентрации
эмульгатора, размер капель и другие характеристики эмульсии *.
Хотя все эти соображения верны, трудно оценить количественно
скорость возрастания концентрации эмульсии и, следовательно,
определить оптимальное время перемешивания. При несколько
идеализированных условиях эмульгирования ультразвуком найдено,
что скорость диспергирования приблизительно пропорциональна
поверхности S между объемами жидкостей и скорости коагуляции
Vc2 (где V и с — объем и концентрация эмульсии, соответственно).
Таким образом, скорость изменения концентрации эмульсии может
быть дана соотношением (Гопал, 1961)
1|^ = а5-ргея (1.1)
•где т — время; аир — константы.
Это связано с двумя упрощающими предположениями (которые,
однако, не всегда справедливы), а именно: в процессе
диспергирования основная роль отводится разрыву объема жидкости на
отдельные капли, в процессе коалесценции — столкновениям капель **.
Уравнение (1.1) дает:
«-«„** '„-(тт)"": Ч-^У" (,'2>
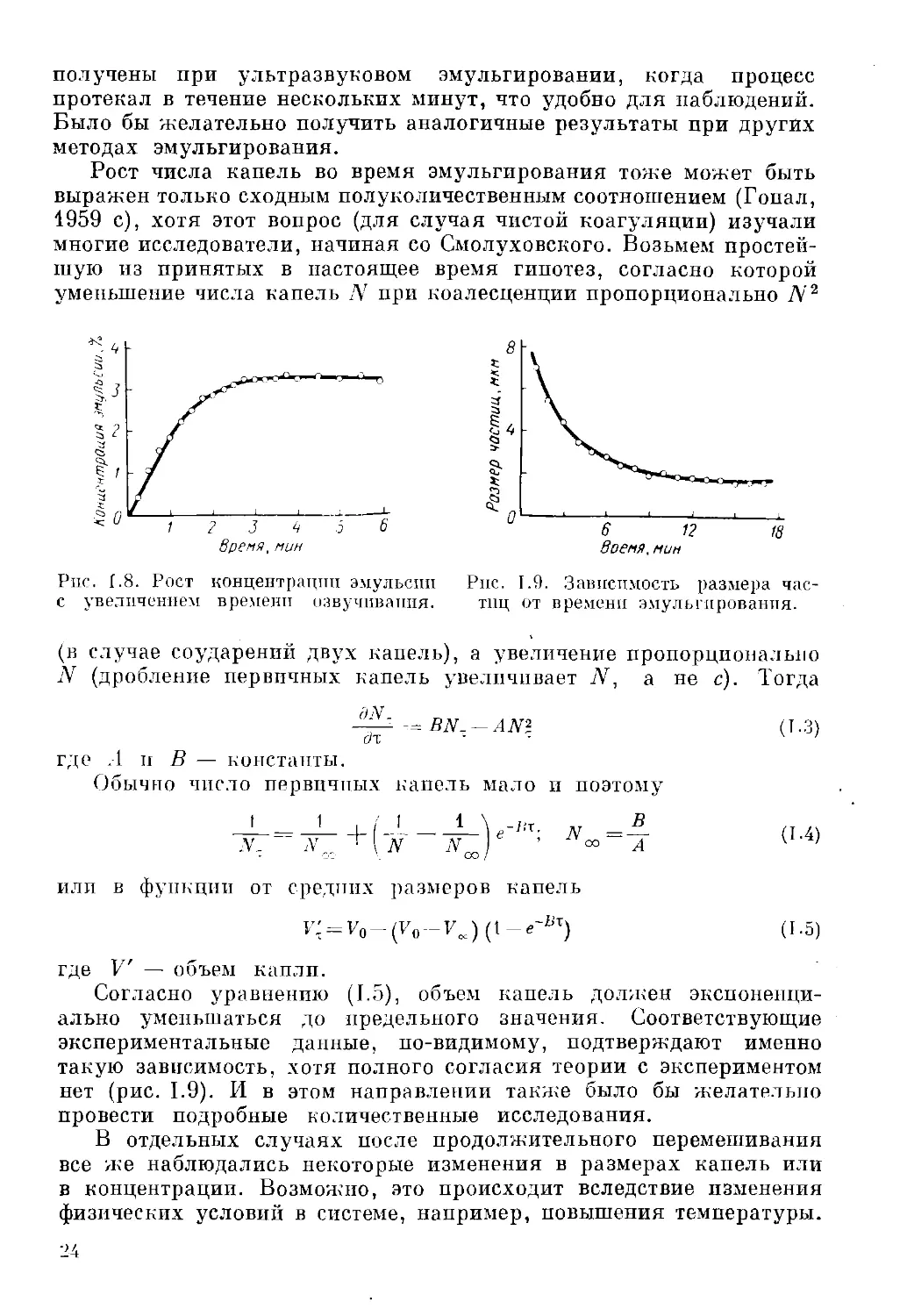
Концентрация эмульсии сначала быстро возрастает и вскоре
достигает предельного значения с (рис. 1.8). Эти результаты были
* Гораздо более вероятно другое объяснение. С уменьшением размера *
капель их жесткость возрастает, и диспергируются они хуже. При
определенной интенсивности перемешивания капли дробятся только до какого-то предела.
Подтверждается такое объяснение следующими опытами. При перемешивании
окрашенной п неокрашенной эмульсий, полученных в одинаковых условиях,
дальнейшего дробления капель не наблюдается, и процент окрашенных капель
не изменяется, т. е. коалесценция не происходит. (Прим. редактора перевода.)
** Это условие может быть допустимо только для случаев нестабилизирован-
ных диспергированных систем типа углеводород — вода. (Прим. редактора
перевода.)
23
получены при ультразвуковом эмульгировании, когда процесс
протекал в течение нескольких минут, что удобно для наблюдений.
Было бы желательно получить аналогичные результаты при других
методах эмульгирования.
Рост числа капель во время эмульгирования тоже может быть
выражен только сходным полуколичественным соотношением (Гопал,
1959 с), хотя этот вопрос (для случая чистой коагуляции) изучали
многие исследователи, начиная со Смолуховского. Возьмем
простейшую из принятых в настоящее время гипотез, согласно которой
уменьшение числа капель N при коалесценции пропорционально Л'2
§<
6 12 18
воемя. мин
Рпс. [.8. Рост концентрации эмульсии Рнс. 1.9. Зависимость размера час-
с увеличением времени озвучивания. тиц от времени эмульгирования.
(в случае соударений двух капель), а увеличение пропорционально
N (дробление первичных капель увеличивает N, а не с). Тогда
(IN.
с/т
BN- — ANI
где Л п В — константы.
Обычно число первичных капель мало и поэтому
I __ 1
N- ~~ Л'.
1
~N'
"оо=4
или в функции от средних размеров капель
VI = V,
-Ь'Тч
о-(Го-Г,)(1-«-*т)
(Т.З)
(1.4)
(1-5)
где V — объем капли.
Согласно уравнению (1.5), объем капель должен
экспоненциально уменьшаться до предельного значения. Соответствующие
экспериментальные данные, по-видимому, подтверждают именно
такую зависимость, хотя полного согласия теории с экспериментом
нет (рис. 1.9). И в этом направлении также было бы желательно
провести подробные количественные исследования.
В отдельных случаях после продолжительного перемешивания
все же наблюдались некоторые изменения в размерах капель или
в концентрации. Возможно, это происходит вследствие изменения
физических условий в системе, например, повышения температуры.
Эти изменения влияют на константы в расчетных уравнениях, что
и вызывает отмеченное расхождение в значениях величин
предельных размеров капель и концентраций. Конечно, это имеет лишь
сугубо академический интерес. Что касается практики
эмульгирования, то надо отметить, что оптимальным временем перемешивания
является интервал 1—5 мин, и дальнейшее перемешивание
заметного улучшения свойств эмульсий не вызывает.
Интенсивность перемешивания
При неизменных компонентах эмульсии, способах их введения
и той же самой аппаратуре имеется еще один параметр, изменение
которого оказывает существенное влияние на свойства эмульсии —
это интенсивность перемешивания. Обычно более быстрое
перемешивание дает лучшие эмульсии. В последние годы делаются попытки
найти количественные соотношения между параметрами,
описывающими перемешивание, и размерами частиц или удельной
поверхностью капель.
Образование эмульсий в простых смесителях подробно изучали
многие исследователи (Вермелен, Вильяме и Ланглуа, 1955; Роджер,
Трайс и Руштон, 1956; Хютиг и Штадлер, 1957; Павлушенко и
Янишевский, 1959; Родригес, Грос и Энгл, 1961; Ландау и Про-
хазка, 1964). Было установлено, что поверхность капель возрастает
при увеличении скорости вращения п и диаметра DK мешалки и
при уменьшении диаметра DCM смесителя. Все это соответствует
более длительному перемешиванию жидкости. Удельная
поверхность 5уд возрастает пропорционально разности плотностей
дисперсионной среды и дисперсной фазы рс — рф. Чем больше отлича-
.ются жидкости по плотности, тем больше отличие в скоростях,
которые приобретают капли этих жидкостей при прочих равных
условиях. Но чем больше градиент скорости, тем больше
касательные напряжения, которые вызывают диспергирование. Величина
Sya также возрастает при уменьшении поверхностного натяжения а,
ибо требуется совершить меньшую работу для образования новых
поверхностей.
Помимо этого, вязкости дисперсной фазы (т]ф) и дисперсионной
среды (пс) также играют определенную роль. Теоретические
расчеты показывают, что значение SyK несколько уменьшается при
увеличении отношения г|ф/г|с. На это указали Павлушенко с
сотрудниками, но Роджер, а также Салливан и Линдсей (1962) нашли
противоположную зависимость. Такое увеличение может
происходить, если г)ф возрастает настолько, что препятствует коалесценции
и, следовательно, сдвигает равновесие в сторону образования
большего числа капель и большего значения 5уд. Однако имеются
определенные трудности в объяснении экспериментальных результатов.
Вязкость дисперсионной среды может отличаться от вязкости
чистого растворителя из-за присутствия эмульгатора. Роджер, Трайс
и Раштон (1956) нашли приближенное количественное соотношение
25
(без учета влияния вязкости) для смешения пропеллерными
мешалками в смесителях с отбойными перегородками:
D*npcVl. !
Dc
ехр
З,6(рс-Рф)
Рс
' (1-6)
Это соотношение используют при выборе аппаратуры для
экспериментов. Авторы попытались согласовать величину Sya с
критерием Вебера D^ n2pja, представляющим собой отношение
сдвиговых сил к силам поверхностного натяжения в системе. Теория
смешения, основанная на статистической теории турбулентности, была
дана Шинаром (см. стр. 42).
Эмульгирование ннжекцией одной жидкости в другую изучалось
рядом авторов (Хайворф и Трейбал, 1950; Симес и Кауфман, 1957;
Скот, Хайес и Холланд, 1958; Макдоно, Томм и Холланд, 1960).
Течение струй здесь сходно с теми процессами, которые происходят
в гомогенизаторе. И в этом случае также найдено, что удельная
поверхность SyR увеличивается по мере возрастания разности
плотностей рс — р. или уменьшения поверхностного натяжения а.
Капли становятся крупнее, если сопло, через которое пропускают
жидкость или вязкость, увеличивается. Макдоно с сотрудниками
нашли, что
■"уд
• (АР) /*а-
/бЧф/е
(1.7)
где АР — изменение давления при прохождении через сопло.
Авторы также попытались получить корреляцию между
размерами капель, с одной стороны, и кинетической энергией текущей
струи и энергией, требуемой для преодоления внутреннего трения, —
с другой. Зависимость 5уд от АР объясняет, почему гомогенизаторы
(с их большим значением АР) столь эффективны при изготовлении
эмульсий.
Вообще эксплуатационные характеристики смесителей изучены
в достаточной степени, чего нельзя сказать о коллоидных мельницах
и гомогенизаторах. Такие исследования имели бы большую
ценность, так как позволили бы создать новые модели аппаратов с
оптимальными характеристиками. Кроме того, результаты этих
исследований должны стать проверкой теоретическим представлениям и
тем самым способствовать большему пониманию сущности явления.
Температура
Обычно изменение температуры оказывает на эмульсии лишь
косвенное воздействие: изменяется поверхностное натяжение,
вязкость, адсорбция эмульгатора и др. Поскольку при возрастании
температуры и вязкость, и поверхностное натяжение уменьшаются,
то и эмульгирование обычно происходит легче. Нередко температура
повышается вследствие энергичного перемешивания жидкости.
Классическим примером может служить производство маргарина и
майонеза, когда готовые эмульсии охлаждают после того, как они были
26
приготовлены при несколько повышенной температуре. В ряде
случаев нельзя допускать увеличения температуры, например для
предотвращения роста бактерий. По некоторым данным, значительное
повышение или понижение температуры ведет к коагуляции частиц—
ухудшению качества эмульсий. Изменение температуры влияет на
адсорбцию эмульгатора, что иногда является нежелательным.
Поэтому в химическом машиностроении предусматривают охлаждающие
устройства, чтобы предотвратить сильный разогрев при
образовании эмульсий.
Как упоминалось выше, иногда нужно контролировать цвет
или проводимость эмульсий. Это достигается введением подходящих
добавок, которые не влияют на другие свойства эмульсий. Такие
вещества рассмотрены в книгах Клейтона и Беркмана. Для контроля
свойств эмульсий обычно имеется несколько способов. Например,
вязкость можно изменять как введением добавок в дисперсионную
среду, так и изменением размеров частиц дисперсной фазы, причем
второй способ более эффективен.
ИЗУЧЕНИЕ РАЗЛИЧНЫХ МОДЕЛЕЙ
ЭМУЛЬГИРОВАНИЯ
Нестабильность течения жидкости
Процесс эмульгирования, описанный в предыдущих разделах,
полезно рассматривать с различных позиций, например с точки
зрения гидродинамической нестабильности. При свободном течении
смесь двух жидкостей стремится остаться в виде двух отдельных
термодинамически устойчивых фаз. И только при сообщении системе
энергии течение становится нестабильным, образуется взвесь одной
жидкости в другой. Вопросы устойчивости движения жидкостей и
эмульсий описаныв монографиях Лина (1955)и Чандрасекхара(1961).
Основной принцип, положенный в основу изучения устойчивости,
очень прост. На первоначально заданное течение накладывается
небольшое по величине возмущение и определяется, будет ли со
временем амплитуда возмущения уменьшаться или увеличиваться.
Если возмущение затухает, система возвращается к
первоначальному состоянию — устойчивому течению. Если же, напротив,
амплитуда возмущения возрастает, то это соответствует неустойчивому
течению, когда первоначальный поток разбивается на несколько
отдельных потоков.
Несжимаемая жидкость подчиняется уравнению неразрывности
уг; = 0 (1.8)
и уравнению движения Навье — Стокса щ.,
l^ + ^^f-ivP+JLvP+iLv^ (I.9)
где v — скорость; F — внешняя сила.
27
Если коэффициент вязкости г\ равен нулю, то уравнение (1.9)
сводится к уравнению Эйлера. К этому уравнению задаются
граничные условия. Для вязких жидкостей тангенциальные и нормальные
составляющие скорости должны быть продолжены через внешнюю
поверхность. Для невязких жидкостей остается только одна
нормальная составляющая скорости, так как жидкости могут
скользить относительно друг друга. Кроме того, тангенциальная
составляющая напряжения (в вязких жидкостях) должна быть
продолжена через границы. Давление на обе стороны внешней поверхности
соответствует уравнению Лапласа
*-*i = ° (-£-+-£-) ' (Ы0)
где ri и Го — радиусы кривизны границ поверхности.
Допустим теперь, что задана начальная скорость жидкости v0,
удовлетворяющая всем необходимым уравнениям. Чтобы
определить, будет ли течение устойчиво (т. е. может ли существовать
неопределенно долго) или неустойчиво (скачкообразно станет
переходить к другому, стабильному течению), зададим скорость малого
возмущения v. Тогда скорость течения станет v0 -г v. Так как и
новая скорость должна удовлетворять всем необходимым
уравнениям, то для v выбирают группу уравнений, например, из
следующих:
v^o = o; v ("0+^ = 0;, v^ = o
Уравнения Навье — Стокса и Эйлера нелинейны из-за члена
(уу) v, а так как уравнения для v выводятся из них, то это
усложняет искомое решение. Однако, рассматривая физическую сущность
явления, можно в указанных уравнениях пренебречь определенными
членами или величинами второго порядка малости и получить
приближенное линейное уравнение для v. Так же можно получить и
зависимость v от времени, которая во многих случаях имеет вид:
j/'0expivx (v — обычно комплексное число).
Допустим, что "V — действительное число и равно со. Значит малое
возмущение v накладывается на колебания скорости течения v0
в виде некоторой функции времени с периодом 2я/со. Эти колебания
будут, естественно, затухать вследствие внутреннего трения
(вязкости) системы. Начальное течение жидкости v0, даже если оно
каким-либо образом выведено из состояния равновесия,
возвращается в исходное положение. Это происходит при стабильном
течении.
Допустим теперь, что v — комплексное число, равное со — iQ.
Тогда зависимость v от времени учитывается множителем
ехр От ехр тх. Это значит, что возмущение экспоненциально
возрастает с течением времени, амплитуда возмущения удваивается за
время 0,693/Q. Какими бы ни были сначала величины и0и v,
вследствие экспоненциального роста v вскоре превзойдет и0, и
результирующее движение уже не вернется в исходное состояние. Течение vg
28
становится нестабильным. Таким образом, распространение малого
возмущения полностью изменяет форму течения.
В качестве примера рассмотрим плоскую поверхность между
двумя жидкостями, которые подвержены малым возмущениям.
Если система стабильна, на поверхности возникает рябь и
образуются поверхностные волны. Если система нестабильна, амплитуда
возмущения возрастает, и через некоторое время поверхность
приобретает очертания, напоминающие скрещенные пальцы, — так
слои жидкости проникают один в другой. Эта поверхность уже не
имеет никакого сходства с первоначальной плоской.
Последняя стадия нестабильности, а именно проникновение
слоев одной жидкости в другую, не учитывается уравнениями,
написанными выше. Из этих уравнений получаются лишь линейные
приближенные решения, основанные на допущении о малости v
в сравнении с v0. Это, действительно, так в самом начале
нестабильного течения, но вскоре амплитуда возмущения становится
достаточно большой. Линейное приближение теперь уже неприемлемо,
и требуется применить полностью нелинейное уравнение. Такая
задача вызывает значительные математические трудности, и для
растворов обычно не решается. Лучшее, что можно сделать, это дать
приближенные решения точных уравнений. Нередко ограничиваются
тем, что указывают на природу нестабильности.
Для инженерных расчетов нужны гидродинамические уравнения,
учитывающие все статистические соотношения и измеренные
величины. Такие данные, например, потребовались бы для
проектирования заводов, производящих эмульсии. Неважно, если бы при этом
не вскрывалась физическая сущность процесса. Динамика
жидкостей также не в состоянии разрешить задачу, она лишь позволяет
более или менее глубоко проникнуть в происходящий процесс,
но не дает формул, по которым можно было бы рассчитывать эмуль-
. гирующие машины. Прогресс здесь может быть достигнут только
в результате использования приближений.
Виды гидродинамической нестабильности
Некоторые виды нестабильности изучены детально. Многие из
них встречаются в различных процессах, другие — только при
эмульгировании. Перед тем, как перейти к обсуждению
эмульгирования с точки зрения гидродинамической нестабильности, следует
остановиться на характерных чертах каждого вида нестабильности.
Ради краткости опустим все вычисления, которые можно найти
в специальной литературе (Чандрасекхар, 1961).
Переход от ламинарного течения к турбулентному — один из
рассматриваемых видов нестабильности. Он широко известен и имеет
важное практическое значение. Эксперименты показывают: когда
число Рейнольдса pdvfi] (где d — характеристический размер)
достигает критического значения порядка 1000, течение
параллельными струями (ламинарное) становится нестабильным (турбулентным);
29
силы внутреннего трения, которые препятствовали развитию
небольших возмущений, уступают место инерционным силам. Этот
вид нестабильности, называемый некоторыми авторами
нестабильностью Толмина — Шлихтинга (Острах и Костель, 1965),
может встречаться в газах и жидкостях, в гомогенных и
гетерогенных системах.
Другой вид нестабильности — Кельвина — Гельмгольца,
наблюдается, когда две жидкости движутся с разными тангенциальными
скоростями относительно поверхности раздела. Кинетическая
энергия движения обусловливает некоторое волнообразное возмущение
поверхности, возрастающее по амплитуде, и это ведет к смещению
жидкостей. Разрыв поверхности раздела происходит в этом случае
даже при малых сдвиговых скоростях, когда течение ламинарное.
По мере возрастания нестабильности внутреннее трение (вязкость)
и поверхностное натяжение уменьшаются.
Следующий вид нестабильности — Рэлея — Тейлора.
Поверхность между двумя жидкостями будет нестабильной, если она
испытывает ускорение от более легкой жидкости к более тяжелой, и
устойчивой, если ускорение направлено противоположно. В качестве
примера рассмотрим стакан с водой, стоящий на столе. Вода
испытывает ускорение, равное/равитационному, но направленное вверх,
из воды в воздух. Следовательно, поверхность раздела этих двух
фаз стабильна. Если стакан перевернуть, жидкость не станет падать
как единое целое; сначала поверхность начнет вытягиваться,
принимая пальцеобразную форму, и далее вода будет вытекать
аналогично падению капель с края плоской пластины (Гопал,
1959).
Еще один вид нестабильности — Бенарда — происходит
вследствие флуктуации плотности. Она может возникнуть и в гомогенных
системах, подобно нестабильности Толмина — Шлихтинга, тогда
как нестабильность Кельвина — Гельмгольца и Релея — Тейлора
характерны для гетерогенных систем. Флуктуации плотности
состоят в том, что под влиянием тех или иных причин (например,
градиентов температуры, концентрации) более тяжелые слои
оказываются над более легкими. Тогда под действием гравитационных сил
начнется перераспределение слоев жидкости, чему, однако, будут
препятствовать силы внутреннего трения.
Имеется несколько других видов нестабильного течения. Так,
течение Куэтта: устойчивое течение жидкости между двумя
вращающимися коаксиальными цилиндрами становится неустойчивым,
если скорость вращения внутреннего цилиндра превосходит
теоретическое значение. Если же внутренний цилиндр неподвижен, то
течение будет стабильным при любых скоростях вращения внешнего
цилиндра. Устойчивость жидких цилиндров и струй будет
рассмотрена далее (стр. 34). Для астрономии представляет интерес вопрос
об устойчивости жидких систем в поле действия гравитационных
и центробежных сил. Соответствующие ссылки можно найти в
монографиях, уже цитированных выше.
30
В следующих разделах будет показано, как представления о
гидродинамической стабильности можно использовать при изучении
различных стадий образования эмульсий.
Разрушение поверхностей
Пусть в пробирку налиты две жидкости с плотностями pj и р2
(Рг >Pi)- Пробирку энергично встряхивают в течение некоторого
времени. Образуется эмульсия, которая в зависимости от
обстоятельств может быть стабильной или нестабильной. Вопрос состоит
в том, почему и как большой объем жидкости распадается на
отдельные капли. Ответ заключается в анализе устойчивости данного
движения. Очевидно, в этом случае скорости течения будут не очень
большими (в отличие от нестабильности Толмина — Шлихтинга),
отсутствуют сколько-нибудь значительные тангенциальные
составляющие скорости (в отличие от нестабильности Кельвина — Гельм-
гольца), нет неблагоприятных градиентов плотности (в отличие от
нестабильности Бенарда). Преобладающим видом течения будет
колебательное движение вверх и вниз, что соответствует
нестабильности Рэлея — Тейлора. Если ручным встряхиванием удастся
достичь движения, близкого к синусоидальному с частотой
~3 кол/сек и амплитудой ~ 10 см, то максимальное ускорение составит
3,6-103 см/сек2. В определенные моменты движения алгебраическая
сумма этого переменного ускорения и ускорения силы тяжести
(0,98-103 см/сек2) может достичь величины, являющейся критической
для нестабильности Рэлея — Тейлора. Более подробно этот вопрос
рассмотрен в работе Гопала (1963). Здесь ограничимся анализом
принципа расчета.
Примем в качестве плоскости XY поверхность раздела между
двумя жидкостями; ось Z направлена вертикально вверх. В момент
•времени т = 0 жидкость испытывает ускорение g, направленное
вверх. Это эквивалентно положению, когда жидкость свободна,
а внешние тела испытывают ускорение g, направленное вниз.
Обычное ускорение силы тяжести производит такое же действие, как
ускорение более легкой жидкости от более тяжелой, когда это
ускорение направлено вверх. Уравновешенность этих двух
противоположно направленных ускорений определяет стабильность
жидкостей. Если бы g было отрицательным, т. е. поверхность раздела
жидкостей испытывала бы ускорение вверх от более легкой жидкости
к более тяжелой, то это привело бы к нестабильности и к
образованию волн возмущения. Из анализа размерности следует, что
амплитуда волны возмущения пропорциональна cos kx (где к = 2п/к).
(Известно, что волны любых форм, согласно теореме Фурье, могут
быть представлены в виде суммы синусоидальных или косинусол-
дальных волн.) Линеанизированное уравнение для этого случая:
dvr , dv,
31
Уравнения движения:
dvx , 1 дР dvx l dp
—а— j— = 0 — з—Ь# = 0 (1-12)
дх ' р дх dz ' р дх '
Величина давления
Р = Ро-РВ' + Р-^- (1-13)
Потенциал скорости Ф находят с помощью выражений:
дФ _ ЭФ
°х ~~ ~ ~дх~ ' "2 ~~ ЫГ
Значение z для новерхности при т = 0 было равно 0, а в другие
моменты z = £ (х, т). Чтобы получить непрерывную нормальную
скорость через поверхность (vz)i = {v2)2 при £ —0 и нулевую
скорость для волн возмущения вдали от поверхности, следует взять:
ф1 = Л/ (т) cos кх ехр (—kz);' ф2 = — Af (т) cos kx exp (fez)
Это дает скорость поверхности
Чг (0) = -7Г = И-
г v ' дх dz
= kAf (x) cos /ca:
-о
и тогда
Z, = kAcoskx^f(x)dx (I.14)
Поверхность, таким образом, изменяется синусоидально с
течением времени, период этих изменений должен быть определен.
Уравнение давления на поверхности:
Дифференцируя по т и используя выражения (1.13) и (1.14),
получим:
(p2 + Pi)-^- + (p2-p1)«*/(T)+o*a/(T) = 0 (I.15)
Так как скорость жидкости равна нулю при т = 0, решение
этого уравнения будет иметь вид
/(T)=shQx
где
Qa=-(P2-Pi)gfc-afc»
Р2 + Р1
Если g положительно (например, ускорение силы тяжести),
то Q мнимое, и поверхность совершает синусоидальные колебания,
частота которых может быть найдена из известного уравнения
Кельвина для волн (Лемб, 1945):
na^ (P2~Pi)ek + ak3 n 17ч
Р2 + Р1
32
Благодаря вязкости, которой часто пренебрегают, колебания
будут постепенно затухать, и поверхность в конце концов станет
ровной. Таким образом, это — стабильное течение.
Если g отрицательно (например, при ускорении от более легкой
жидкости к более тяжелой), величина Q в уравнении (1.16) станет
положительной, когда к будет меньше, чем
Для частного случая
w-l—35—J
ускорение будет максимальным и определится выражением:
о-. _ 2 [-(P2-Pi)g]3/*
макс
3 (pa + PiH3a)'''
Амплитуда волны возмущения экспоненциально увеличивается
с течением времени. Поэтому через некоторое время одна из волн —
компонент разложения Фурье — с данной величиной к станет
преобладающей среди всех остальных компонент. Такие возросшие до
максимума колебания слоев жидкости происходят повсюду в
рассматриваемом объеме, и первоначально ровная поверхность
жидкости полностью разрушается.
Можно уточнить вычисления несколькими способами. Все они
дают одинаковый результат, так как рассматривают волны
возмущения, описываемые уравнением z = £, (х, у, т). Внутреннее трение
уменьшает действие этих волн, причем тем значительней, чем меньше
колебания. На первых стадиях волны возмущения малы и
выражаются чистой синусоидой В cos kx. Затем амплитуда начинает
возрастать (практически с момента т = 0), и форма волны
соответствует выражению В cos kx ch Qt. Подробное исследование этого
нелинейного уравнения показывает, что более тяжелая жидкость
проникает в виде длинного узкого клина в более легкую жидкость,
а последняя — в виде короткого тупого клина в более тяжелую.
Тяжелая жидкость от сообщаемого ей ускорения имеет амплитуду
/ Рг Pi g \ T2 Легкая жидкость сохраняет постоянную скорость
1
( Ps~Pi f,^ )2. Из уравнения непрерывности следует, что слой тяже-
\Р2+ Pi б /
лой жидкости должен быть узким. В дальнейшем ее поверхность
претерпевает разрывы и приобретает форму пальцев или витков
резьбы, как отмечалось выше.
Утверждения о движении струй на последних стадиях
рассматриваемого процесса основаны на предположении, что в системе не
происходит в это время никаких других изменений. В
действительности же положение усложняется двумя обстоятельствами:
неустойчивостью самой струи и возможностью изменения ускорения. [Вопрос
о неустойчивости струй будет рассмотрен ниже (стр. 34).]
3 Заказ 1070
33
Стабильная
поверхность
в
нестабильно?,
поверхность
"I
Распад струй на капли схематически изображен на рис. 1.10, а~г.
Изменения ускорения, очевидно, воздействуют на рост скорости.
В частности, если дестабилизирующее ускорение действует в
течение конечного промежутка времени, поверхность возмущения будет
расти в течение этого периода, и
а ■ ■ ->— именно с этого момента начнет
колебаться с конечной амплитудой.
Так как здесь возникают два
потока жидкости, движущиеся в
противоположных направлениях, то
рассматриваемый случай относится
к нестабильности Кельвина — Гельм-
гольца (рис. 1.10, а, д, е).
Другой важный случай имеет место,
когда ускорение — периодическое.
(Подробнее см. на стр. 49.)
Различные аспекты
нестабильности Рэлея — Тейлора были
экспериментально изучены многими
исследователями. Многократно проверены
в различных условиях
нелинейность волн возмущения,
стабильность при ускорении в одном
направлении и нестабильность при
ускорении в противоположном
направлении. Однако обстоятельных работ
по приложению этой теории к
проблеме образования эмульсий не
проведено. О достигнутых в этом
направлении результатах сообщено
в обзоре Гопала (1963). Приведенные
выше теоретические расчеты не могут
быть использованы непосредственно
для промышленного производства
эмульсий, так как во всех случаях
необходимо учитывать рекомбинацию
частиц. Кроме того, ускорения
изменяются от места к месту и с
течением времени, так что обязательно
будут образовываться капли различных размеров. Поэтому нужен,
такой расчет, где были бы использованы законы статистики.
Рпс. 1.10. Схема разрушения
плоской поверхности жидкости:
а — малое начальное возмущение
поверхности; б — возмущение растет
(сплошнап линия) — ускорение
направлено к более тяжелой жидкости,
поверхность только колеблется (пунктир-
пая линия) — ускорение направлено
к более легкой жидкости; в — тяжелая
жидкость узкими гребнями
выбрасывается вверх, а легкая жидкость
тупыми клиньями устремляется вниз —
ускорение, создающее нестабильность,
продолжает действовать; г — столбики
жидкости распадаются на капли; S —
возмущение принимает форму
колебания с большой амплитудой — ускорение,
создающее нестабильность, действует
короткий промежуток времени; е —
типы капель, образующихся в
случае д.
Распад струи
Распад струй жидкости на капли — это явление, которое
наблюдается при гомогенизации и на последних стадиях разрыва плоских
поверхностей. Этот вопрос впервые изучал Рэлей (1878, 1892).
Он показал, что вытекающая из круглого отверстия цилиндриче-
34
екая струя может стать нестабильной по двум причинам. Во-первых,
в результате поперечных деформаций расширения, когда
синусоидальные волны возмущения направлены вдоль радиуса цилиндра
(рис. 1.11, а). Ось струи остается прямой, но струя изменяет форму,
образуются сужения вдоль оси. Когда длина волны возмущения
превысит размеры цилиндра, струя станет неустойчивой. Во-вторых,
из-за синусоидальных деформаций (рис. 1.11, 6). Здесь ось струи
становится синусоидальной, но сечение остается неизменным по
всей длине струи.
После классических работ Рэлея Вебер (1931) применял
гидродинамический анализ для решения различных задач. Томотика
Рис.
в о о о>
1.11. Нестабильная деформа- Рис. 1.12. Стадии разрушения цилин-
цня струи: дрической струи на капли:
а — ось струи — прямая линия, радиус
изменяется синусоидально; б — ось струи —
синусоидальная, радиус не изменяется.
а — поперечные деформации, вызывающие
колебание на поверхности струи; б —
усиление деформации; в — разрыв струи
на капли.
(1935) и сравнительно недавно Кремнев и Равдель (1953, 1954)
изучали устойчивость цилиндрических струй, в которых большое
значение имеют силы внутреннего трения и поверхностное
натяжение, а инерциальные силы отсутствуют. Во многих работах
приведены разнообразные эмпирические корреляционные соотношения,
основанные на анализе размерностей или на приближенных моделях.
Эти соотношения полезны для практических целей, они
систематизированы в работе Мюссе (1955).
Используя метод анализа, описанный выше, можно указать
условия нестабильности для случая, когда длина волны в
результате деформации расширения становится больше, чем периметр
струи. Чтобы избежать ненужных осложнений, допустим, что на
струю не действуют никакие посторонние силы. Потенциал скорости
для тела цилиндрической формы описывается функцией Бесселя
/0 (кг) (Лэмб, 1945) и должен быть взят в виде:
ф = Л/ (т) /0 (кг) cos/cz
На свободной поверхности струи г = а + £,. Тогда
dt дф
-^ = — — | = —кAf (г) /о (ка) co.s kz
дт
дг
Отсюда
£ = — кА/д (ка) с )s kz j / (т) dx
что аналогично выражению (1.14).
(Ы8)
3*
35
Давление волны возмущения на поверхность
Р =р —= p4/0(/ca)cosi-z-^- (I.19)
Остается вычислить давление а (1/г4 + 1/г2), которее можно
получить из граничных условий.
Кривизна определяется выражениями \\а — Z,/'а2 и —d^L/dz2.
Отсюда давление возмущения
P'==kAcI (ка) cos kz (-^--kA ^ f (т) dx (1.20)
Приравняв эти два выражения для давления, получим
дифференциальное уравнение для / (т) решение которого будет иметь вид:
/(T) = sh Qt
и
ckl'(ka)(l-k-2gi) ч
f>a4B(ka) (1-г])
Величина Q2 отрицательна при ка^>\, это соответствует
колебаниям на поверхности струп. Если ка < 1, т. е. длина волны
возмущения больше периметра струи, амплитуда возрастает
пропорционально sh fit, и возникает нестабильность. На поверхности
струи образуются округлые впадины и выступы, радиус которых
непрерывно растет до тех пор, пока струя не разбивается на
отдельные капли. Это схематично изображено на рис. 1.12. Фотографии
различных стадий процесса даны Румшейдтом и Мэзоном (1962).
Физический смысл нестабильности вытекает из уравнения (1.18)
и (1.20). При ка < 1 силы поверхностного натяжения вызывают
возрастание давления во впадинах и уменьшение его в выступах.
Возникающий перепад давлений гонит жидкость из областей сжатия
в области расширения. Неравномерность сечения струи возрастает
еще больше, что соответствует росту амплитуды волны возмущения.
Наибольший рост возмущения — при максимальном значении
отношения
каГа (ka)(i—kW)
h (ка)
Это происходит при к2а2 = 0,4858 или 2п/к = 4,508-2а. Именно
такое значение длины волны расширения соответствует наибольшей
нестабильности.
Учитывая силы внутреннего трения, Вебер получил выражение
для условия максимальной нестабильности 2л/к = 2ло 1/2 (Зи -f- 1),
где у = r|/|/2pao". Отсюда 2п/к = 4,4-2а. Для жидкостей с большой
вязкостью эта величина является неопределенной. Приведенные
условия согласуются с предельными случаями, которые
рассматривал Рэлей. Подобным же образом к нестабильности ведет и анализ
синусоидальных деформаций. Большей частью она возникает при
больших скоростях струи и проявляется в турбулентности.
*36д „ .
Рассмотренные теоретические выкладки относятся только к
сформировавшейся нестабильности. Нелинейные уравнения,
получающиеся для тел цилиндрической формы, не могут быть решены
строго математически. Так как проблема распада струй имеет
большое прикладное значение, накоплено значительное количество
экспериментальных данных, позволяющих связать некоторые параметры
процесса. К таким параметрам относятся вязкость, плотность
и поверхностное натяжение обеих жидкостей (той, которую
инжектируют, и той, в которую вводят струю), а также скорость и диаметр
струи. Из теоретического анализа следует, что разрыв поверхности
жидкости происходит, главным образом, при больших скоростях,
при турбулентном режиме течения.
При больших скоростях капли измельчаются до очень малых
размеров. Эти явления изучены многими исследователями, и
установлены различные количественные соотношения, связывающие
перечисленные выше параметры с размерами капель, степенью
измельчения и т. д. Полученные данные наилучшим образом
согласовывались с уравнениями, основанными на анализе размерностей
или на приближенных моделях явления. Мюссе (1955)
проанализировал все эти работы и тем дал повод для более критического
теоретического и экспериментального изучения.
Проблема распада струй аналогична вопросу об образовании
капель из тонких слоев жидкости. В обоих случаях рассматривается
гидродинамическая модель, выводятся статистические соотношения,
которые должны соответствовать экспериментальным фактам. Все
эти теоретические и опытные работы обобщены Фразером, Эйзенкла-
мом, Домбровским и Хассоном (1962).
Диспергирование капель
Распад больших капель жидкости на малые происходит на
последних стадиях эмульгирования. Очевидно, если жидкая сфера
должна быть деформирована и разрушена, к ней нужно приложить
6 в г
Рис. 1.13. Типы деформаций жидких сфер:
а — первоначальная сфера; б — вытянутый сфероид,
растяжение вдоль оси X; в — сплющенный сфероид,
сжатие вдоль оси Z; г — деформация произвольной
формы.
достаточно большие усилия. Величина таких усилий
обусловливается динамическим или вязкостным сопротивлением. Хотя капли
могут быть раздроблены различными способами, были изучены лишь
основные типы деформаций сферических капель. Возникающие при
этом виды течения в настоящее время хорошо известны (Гинце, 1955).
Начальные стадии трех основных типов деформаций капель
показаны на рис. 1.13. Сферическая капля (рис. 1.13, а) может
37
быть растянута в виде продолговатого сфероида (рис. 1.13, б),
причем сфера получается вращением эллипса вокруг главной оси.
Канля вытягивается и далее, образуя нитевидный цилиндр,, который
разрывается на отдельные капли (см. стр. 31). Второй тип
деформации капли — расплющивание до сфероида (рис. 1.13, в); сфера
получается вращением эллипса вокруг малой оси. По мере действия
а 6 в ^у
-*■ х
</♦
г*
W*
ж
I
О
ч —
Рис. 1.14. Схематическое изображение течений, деформирующих жидкую сферу:
а — течение параллельными слоями (в = const); б — течение Куэтта (v = By); в —
вращение жидкости (и = ту; v = —ах); г — плоское гиперболическое течение (их = Gxl2;
t>„ = —Gy/2); д — асимметричное гиперболическое течение, направленное против оси
<г, = dr/2; v — —Gi2/2); е — асимметричное гиперболическое течение, направленное
вдоль оси (г = —G2r/2; и = Gjz/2); ж — течение произвольной формы.
сил расплющивания капля превращается в узкую полосу жидкости
и разрушается на более мелкие капли. Третий тип деформации —
полностью неопределенная форма капли (рис. 1.13, г). В отдельных
участках, где имеются впадины и выпуклости, из данной капли
могут отделяться новые капли — меньшего размера.
На рис. 1.14 представлены возможные схемы движения жидкости
вокруг капли для каждого из рассмотренных типов деформаций.
Для первого случая — деформации капли вдоль оси х — возникают
течения сдвига Куэтта (рис. 1.14, б), плоское гиперболическое
38
(рис. 1.14, г) или асимметричное гиперболическое (рис. 1.14, е) по
направлению оси вращения. Для второго случая — деформации
капли вдоль оси z — характерны течения параллельными слоями
(рис. 1.14, а), вихревое (рис. 1.14, в) или асимметричное
гиперболическое (рис. 1.14, д) в направлении, противоположном оси вращения.
В третьем случае — деформации произвольной формы — возникает
турбулентное течение (рис. 1.14, ж), где векторы скорости в любой
точке пространства изменяются произвольно как по величине, так
и по направлению. Силы, вызывающие деформации капель, могут
возникать от сил либо динамического, либо вязкостного
сопротивлений, причем обычно преобладает, в зависимости от условий, тот
или другой вид сопротивления. Величина деформации капли и
способность ее к разрыву зависят не только от формы течения, но
и от физических свойств обеих фаз — от их плотности, вязкости
и поверхностного натяжения.
Первый тип деформации капли (вытянутый сфероид),
обусловленный действием вязких сил в плоском гиперболическом и в
сдвиговом течениях, изучал впервые Тейлор (1934), позднее Томотика
(1936) и другие. Для плоского гиперболического течения: как
полагают,
где G — модуль сдвига.
Если капля радиусом г находится в жидкости, то в последней
возникает определенное возмущение, следовательно, на каплю
действуют силы. Тангенциальные составляющие этих сил создают внутри
капли градиент скорости, нормальные составляющие — давление,
которое для малых деформаций имеет величину
/19г)ф+ 1611С \
АР — ^с(16г);+16т1с)соЯ2ф (1.22)
где ф — угол, отсчитываемый от оси Y.
Приравняв выражение (1.22) капиллярному давлению АР =
= а (1/^+ 1/г2), можно рассчитать радиус кривизны капли.
Уравнение (1.22) показывает, что вдоль оси Y (ср = 0) сфера будет
выравниваться, а вдоль оси X — удлиняться, так что в результате
капля примет вид сфероида, вытянутого вдоль оси X как главной.
Если Ьг — длина главной оси, L2 — длина короткой оси, то
Lx — L2 __ Grr\c 19т)ф+16т1с
Li+Li <Г~' 16г)ф + 16Чс
Для малых деформаций теория превосходно согласуется с опытом.
В случае больших деформаций, как показывают эксперименты,
существенным оказывается отношение *1ф/т1с. Если это отношение мало,
то новые (очень мелкие) капли образуются за счет вытягивания
концов первоначальной большой капли (рис. 1.15, а). Если отношение
г1ф/г1с велико, то вся капля постепенно вытягивается в длинную нить,
39
которая далее дробится на отдельные капли, по-видимому, по
механизму, рассмотренному ранее (см. стр. 34). Такое дробление
усиливается, если имеется какое-либо возмущение. Это объясняется
возрастанием общей нестабильности системы. Тщательная опытная
проверка поведения капель в условиях сдвигового течения была
выполнена недавно Румштейдтом и Мэзоном (1961).
Течение Куэтта во многом сходно с гиперболическим течением
при условии ф = л/4. Поэтому капля под действием малых
деформаций в этом случае примет форму вытянутого сфероида с главной осью,
направленной под углом ф = л/4 (рис. 1.16). При больших
деформациях положение зависит от отношения вязкостей Лф/Лс- Когда
дисперсная фаза имеет малую
вязкость (Пф/Лс "С 1). капля
принимает 2 -образную
форму. ; Из заостренных
концов этой капли
отделяются частички
жидкости в виде мелких новых
капель (рис. 1.16, а). Когда
центральная
часть капли вытягивается
Рпс. 1.15. Схема деформации капли при в цилиндр, причем в сере-
плоском гиперболическом течении, когда дине этого цилиндра об-
непрерЫГ„/ГГ^Гй пСЛпГ™. СДЮГа Разуется утоньшение, из
О вязкость (.Пф/Лс <Ss 1) > капля
СТГ") <=^=> о о ° принимает 2-образную
^— ° форму. ; Из заостренных
концов этс
ляются ча
: сти в виде
Os^jr-^. капель (рис
от нуля до большой величины;
которого возникают новые
°-(VV<i; s-oyV^1-. мелкие капли (рис. 1.16,6).
В некоторых случаях вся
капля вытягивается в длинную цилиндрическую нить,разрушающуюся
на капли, если имеется какое-либо возмущение. Механизмы каплеоб-
разования при Цф/цс ^ 1 весьма разнообразны. Вязкая капля
достигает максимальной деформации и ориентируется при ср = я/2
(рис. 1.16, в). Эти эксперименты также описаны в работе Румштейдта
и Мэзона (1961).
О поведении капли в асимметричном гиперболическом течении
вдоль оси X (см. рис. 1.14, е) можно судить по уравнению (1.22),
в котором надо заменить G на — G2. Капля будет вытянута вдоль оси
Z (ф = 0), а так как капля остается симметричной, это означает, что
она примет форму сфероида, вытянутого вдоль оси. Если в уравнении
(1.22) G заменить на Gv, можно получить представление о поведении
капли в симметричном гиперболическом течении. Капля будет
сплющиваться у полюсов и примет форму обжатого сфероида. На практике
очень трудно получить такие сечения, и экспериментальных
доказательств существования именно этих форм капель нет. Деформации
капель, приводящие к образованию формы расплющенного сфероида,
возникают лишь при вращениях жидкости с очень большой скоростью
и поэтому представляют ограниченный интерес. Аналогичные
деформации, происходящие в течении параллельными слоями, вовсе не
наблюдаются в процессе эмульгирования, но с ними сталкиваются
40
в некоторых вопросах метеорологии (разрушение водяных капель
потоком воздуха), при проектировании двигателей внутреннего
сгорания (инжекция жидкого топлива) и др. В этих случаях силы
внутреннего трения играют меньшую роль, чем силы инерции. Особенности
образования капель зависят от скорости воздушной струи.
Результаты хорошо коррелируются с числом Вебера We = pv2r/a,
которое представляет собой безразмерное отношение динамической
энергии р v2r3 к поверхностной энергии от2 капли радиусом г. В то
время, как величина числа Вебера изменяется от 1 до 20,оказывается,
изменяется и характер разрыва жидкости на капли. Подробно
об этом можно прочесть , „
в обзоре Гинце (1955). ° — ^5-
С деформациями треть- ( Л 7VH /<? /у?~
) типа — с произвольной \ J \S (^y aoo~/^
его типа
формой капель —
встречаются в турбулентном
течении. Флуктуации да- б '45°,
вления вызывают появле- /"~\ /£ЯГ /О 0 (^~>
ние в отдельных участках I J (^у ^^у^ --, °"
сначала небольших вы- ^^
пуклостей, которые
постепенно разрастаются и g t it$-/
становятся очагами воз- s "ч /Н>3
никновения новых капель. ( J / jr /^/ ^Z__^>
Такой путь каплеобразо- —' ^
вания, очевидно, трудно Рис_ 1Л6- Схема деформации капли при
зафиксировать. Наилуч- течении Куэтта, когда скорость сдвига непре-
шее, что здесь можно пред- рывно возрастает от нуля до большой вели-
принять, это использо- чины:
вать какие-либо корреля- a-(\l\)<i; б - <VV " 1; "-'Ус1»1'
ции. Например, если число
Вебера достигнет какого-то определенного значения, можно
сказать, что динамическое давление превысило силы когезии, и это
вызывает разрыв поверхности жидкости и образование капли.
Такой подход бывает продуктивен и будет использован далее
(см. стр. 42).
В процессе эмульгирования, очевидно, происходит распад
больших капель на малые под действием деформаций рассмотренных выше
типов. Под влиянием полей сдвиговых и других сил жидкость
перемешивается, и первоначально крупные капли становятся все мельче
и мельче. По мере того как перемешивание продолжается, средние
размеры частиц будут постепенно уменьшаться, если нет
рекомбинации. В частности, как было показано ранее (стр. 22), размеры частиц
достигают предельного минимального значения, если приняты меры
по предотвращению коалесценции.
Мы рассмотрели лишь модели нестабильных капель. Чтобы
применить эти идеи, например, для предсказания размеров частиц в
реальных эмульсиях, нужно иметь полную информацию о конечных
41
стадиях дробления капель. Пока что этого нет. Для таких сложных
систем нелинейные уравнения гидродинамики не могут быть решены,
а корреляционные зависимости имеют ограниченное применение.
Даже при наличии «всей» математической информации проблему
эмульгирования нельзя решить. Течение в разных аппаратах не
одинаково, и нестабильность устанавливается на различных участках
в разные интервалы времени. Внутренние связи в жидкости
распространяются на слои различной толщины, и образующиеся капли
имеют разный диаметр. Таким образом, монодисперсность при
образовании капель не достижима. Наиболее перспективным для
изучения является статистический метод. Ниже рассмотрены основные
идеи этого метода и некоторые результаты его применения.
Статистические эффекты турбулентности
Очевидно, интенсивное перемешивание, которое производится
с целью получения эмульсий, создает турбулентное течение
в жидкости. Действительно, числа Рейнольдса подтверждают это;
например, для смесителя с размером лопасти 10 см, скоростью ее
вращения 1000 об/мин, вязкостью жидкости 0,01 пз число Рейнольдса
достигает миллиона. Для столь выраженного турбулентного течения
очень велики флуктуации скоростей, и можно использовать
статистическое рассмотрение, впервые предложенное Колмогоровым (1941а-).
В последние годы метод Колмогорова был применен к проблеме
эмульгирования рядом исследователей (Колмогоров, 1949; Гинце, 1955;
Гопал, 1959а; Шиннар и Чарч, 1960; Штейдл, 1960). Рассмотрим
кратко основные положения этих расчетов.
При турбулентном течении движение частиц вызывается, главным
образом, большими вихрями, в то время как основные потери на
внутреннее трение происходят в малых вихрях. Кинетическая энергия
больших вихрей передается малым вихрям, где и расходуется на
преодоление сил внутреннего трения. Этот переход идет разными путями
с различной скоростью, так что можно говорить о независимости
малых вихрей друг от друга и от главного течения в жидкости.
Важно лишь общее количество кинетической энергии, полученное
ими. Потери этой энергии в единице массы малого вихря w, вязкость
и, плотность р — таковы основные характеристики малого вихря,
принимаемого как независимое целое в статистическом рассмотрении.
По Колмогорову, в вихре на единицу длины поглощается энергия:
'-(£)"■
Пусть энергия всего течения 2w/l. (В типичных случаях это
соответствует 1,25 л. с. на 450 л жидкости и тогда б приблизительно
равно 25 мкм). Это очень малая величина по сравнению с длиной I
большого вихря, который часто имеет размер всего сосуда (например,
10 см). В то время как течение всей жидкости является анизотроп-
42
ным, в ближайшем окружении малого вихря оно изотропно. Это
называется местной изотропной турбулентностью. в
Из предположения о местной изотропии следует, что флуктуации
скорости на расстоянии, большем г, должны быть меньше I и могут
зависеть только от w, г, г\ и р. Возможны два случая: I ^> r ^> б
и I 3> б ^> г. Когда г 3> б, на этом расстоянии укладываются
несколько независимых вихрей, динамические силы будут играть
большую роль, чем силы внутреннего трения. Пусть и2 (г) — флуктуация
среднеквадратичной скорости за пределами г. Колмогоров показал,
что
J^) = b1(ur)'h; Z»r»S; 6i=const (I.23)
Если г-f 8, силы внутреннего трения преобладают и
b%pwr2
У2(г) = _«; . Zx>6»r; Ь2 = const (I.24)
Эти положения можно применить для вычисления диаметра DMaKC
наибольшей капли, которая остается неразрушенной в
турбулентном течении. Было найдено, что в обычных аппаратах без
специальных устройств для гомогенизации Омакс > б. Согласно Тейлору,
вязкие деформации капель происходят при условии, что .течение остается
однородным по крайней мере на расстоянии размера капли.
Следовательно, это условие невыполнимо при DMaKC — б. Поэтому можно
ожидать, что возникающие в турбулентном режиме давления способны
разрушить капли в таких аппаратах. Капля разрывается под
действием динамических сил, возникающих вследствие градиента
скоростей, который образуется на расстоянии, равном диаметру капли.
Поэтому число Вебера как критерий разрушения капли можно
представить в виде
We=^^=b1pz^>WeKpHT (I25)
ИЛИ
/ п \ э /.
,/s = const; / > £>макс 3> б
(*)
Это соотношение получено Колмогоровым (1949) и Гинце (1955).
В разбавленных эмульсиях, где рекомбинация мала, соотношение
(1.25) хорошо согласуется с опытными данными о зависимости
размера частиц от сообщаемой энергии, плотности и поверхностного
натяжения. Такие исследования были проведены несколькими
авторами (Гинце, 1955; Шиннар и Чарч, 1960; Шиннар, 1961). Величина
DMaKL — диаметр капель, которые уже разрушаются в турбулентном
течении — будет несколько меньше, чем это следует из соотношения
(25). По данным многих авторов, опытная величина составляет ~95%
от расчетной. Ряд авторов трактуют эту величину как средний
диаметр или наиболее вероятный. Обусловленная этим ошибка,
по-видимому, будет меньше той неопределенности, которая вводится при
теоретических расчетах. Некоторые авторы придерживаются иных
позиций, считая Омшс определенным параметром, имеющим физический
43
смысл верхнего предела частицы, которая остается устойчивой
в эмульсиях. Эти авторы (Мугеле, 1960; Шлейчер, 1962) в
подтверждение своей точки зрения подвергают сомнению отправные
предпосылки для вывода уравнения (1.25). Это, пожалуй, сомнительный
аргумент, ибо теории турбулентности — статистические, и значения
v2 (г) в уравнениях (1.23) и (1.24) характеризуют лишь наиболее
вероятные величины. Далее, разрушение капли тоже трактуется с
позиций теории вероятности, так что и DuaK(. и другие величины в
соотношениях (1.26) — (1.28) будут иметь статистический характер.
Конечно, такие статистические теории показывают, что капли будут
двигаться согласно логарифмическому закону. Теоретически
логарифмический закон распространяется на капли лйбых размеров, от
нуля до бесконечности, хотя крайние значения очень редки и обычно
не наблюдаются.
Возвращаясь к критическому числу Вебера как критерию
разрушения капли, мы можем получить соотношение D 4C б для случая,
когда вязкость системы большая или когда каЧгли очень малые.
Колмогоров (1949), используя уравнение (1.24), получил
\U'= ■■=- — ^\\екрит (l-^Ь)
или
— const; 1л 6 „. Z)M
f)2U>
■no
Но Шиннар (1961) указал, что в предельном случае б > D вязкие
напряжения превосходят силы инерции в момент разрушения капли.
Он предложил использовать метод Тейлора и получил
B^c(i9L )'''' = const (Г. 27)
Оба соотношения — (1.26) — (1.27) — еще недостаточно проверены
экспериментально.
С таких же позиций Шиннар и Чарч (1960) рассматривали действие.,
турбулентности при коалесценции. Если кинетическая энергия
относительного движения между двумя каплями р£>?шн v2 (D) будет больше
энергии адгезии между ними, то рекомбинация не происходит. Энер-
гию адгезии можно рассчитать из известных условий стабильности
коллоидных растворов и из формулы WaDMKl{ (где Wa — энергия
адгезии между двумя сферами одинакового диаметра). Поэтому
Ошш(р*ы2)1!'= const (I.28)
Это соотношение справедливо для относительно
концентрированных эмульсии, где деструкция и рекомбинация находятся в
динамическом равновесии.
Колмогоров (19416) первым указал и на другое важное
применение статистической теории турбулентности — к процессу
образования капель. Он показал, что разрушение капель можно рассматривать
как процесс Маркова, т. е. как результат проявления большого числа
случайных событий. Используя основную теорему теории
вероятности, он получил распределение капель по размерам согласно
логарифмическому закону. Позднее Эпштейн (1947, 1948) и другие дали
несколько более простой анализ вероятностного процесса
измельчения.
Проблема эмульгирования более сложная, чем измельчение из-за
явления рекомбинации частиц и изменения их размеров с течением
времени. Это требует внесения определенных добавлений в
математический аппарат теории. Подробнее об этом сказано в работе Гопал
(1959а). Здесь следует отметить лишь, что логарифмический закон
есть следствие статистической природы турбулентности.
В последние годы показано, что логарифмический закон имеет
солидную теоретическую базу. Наблюдающиеся иногда отклонения
от этого закона происходят вследствие неисправного оборудования
или других посторонних причин (Гопал, 19596; Ирани, 1959; Смит
и Иордан, 1964). Было бы желательно согласовать параметры
логарифмического закона и турбулентного течения, но до сих пор это
не сделано.
ЭМУЛЬГИРОВАНИЕ ЗВУКОМ И УЛЬТРАЗВУКОМ
Образование аэрозолей и эмульсий при интенсивном
ультразвуковом воздействии впервые наблюдали Вуд и Лумис (1927), которые
работали с кварцевым генератором большой мощности частотой
200 кгц. По мере развития ультразвуковой техники открывали все
новые и новые явления, что привело к целому потоку исследований
в этом направлении. Однако большинство работ было качественным,
и лишь Солнер с соавторами попытались установить
полуколичественные закономерности. Обзор Солнера (1944) посвящен ранним работам
по эмульгированию ультразвуком — исследованиям, не выходящим
за рамки лабораторных испытаний. После изобретения Полманом
жидкостного свистка (Жановский и Полман, 1948) началась вторая
фаза — попытки применения ультразвуковых методов
эмульгирования в промышленности. Использование звуковой энергии для полу-
'чения эмульсий, казалось, сулило значительный экономический
эффект. Обзоры Кроуфорда (1955) и Бехера (1965) относятся именно
к этому времени. Третья фаза началась с трезвого анализа всех
достоинств и недостатков звуковых методов эмульгирования по
сравнению с другими известными методами. Дальнейшее развитие техники
эксперимента к этому времени позволило установить, наконец, и
количественные закономерности процесса. Обзор Недужего (1962)
дает описание положения дел на 1960 г.
Генераторы звуковых и ультразвуковых волн
Ультразвуковая область частот лежит выше предела слышимости
человека О 15 кгц) и распространяется вплоть до 109 гц. Для
эмульгирования и других технологических целей должен применяться
ультразвук большой мощности, что возможно лишь на частотах
45
< 5 Мгц. Далее, поскольку речь идет об эмульсиях, ограничимся
рассмотрением возбуждения звуковых волн в жидкостях. Это
достигается превращением электрической энергии в механические
высокочастотные колебания. Источники таких колебаний (их называют
также трансдуцерами) бывают четырех типов: пьезоэлектрические,
магнитострикционные, электромагнитные и жидкоструйные. Пере-
зцислим здесь кратко основные особенности этих трансдуцеров,
^Щрлее подробное описание генераторов звуковых и ультразвуковых
вблн имеется в литературе (Гюнтер и Болт, 1955; Кроуфорд, 1955;
Фредерик, 1965; Браун и Гудман, 1965).
Кристаллы кварца (и некоторые другие) обладают
пьезоэлектрическими свойствами, они образуют электрические заряды на своих
поверхностях при механических деформациях. В последние годы было
установлено, что пьезоэлектрическими свойствами обладают в
заметной степени титанат барпя, цирконат свинца, метаниобат свинца.
Эти керамические материалы весьма перспективны, ибо из них можно
изготовить трансдуцеры любой формы. После искусственной
поляризации они служат генераторами ультразвука. Когда пластина пьезо-
электрика находится в переменном электрическом поле, она излучает
механические колебания, амплитуда которых зависит как от
приложенного напряжения, так и от свойств самой пластины. Если
приложенная частота совпадает с частотой собственных колебаний
пластины, то амплитуда колебаний будет резонансной, т. е. наибольшей.
В этом случае в энергию звуковых волн переходит значительная
часть электрической энергии. Резонансная частота пластины обратно
пропорциональна ее толщине. Пластина кварца толщиной 1 см
имеет частоту ~300 кгц. Таким образом, для частот > 100 кгц
обычно используют пьезоэлектрические трансдуцеры. Ультразвук
столь высокой частоты распространяется прямолинейно. Это является
достоинством при лабораторных исследованиях, ибо дает возможность
точно контролировать энергию ультразвука. Следовательно,
эмульгирование ультразвуком может быть проведено при вполне
определенных условиях.
Типичное оборудование, используемое в лабораторной практике,
показано на рис. 1.17. К кварцевой или керамической пластине
подводят электрод, через который ей сообщают напряжение до 5000 в
высокой частоты. Пластина погружена в трансформаторное масло,
выполняющее роль электрического изолятора высокого напряжения
и передающего механические колебания среде. Пластину крепят
с помощью обода таким образом, чтобы рабочие поверхности имели
наибольшую полезную площадь. Частота электрического поля
регулируется для достижения резонанса с пластиной. Фонтан масла над
работающей пластиной представляет собой ультразвуковое поле.
Жидкость, которую собираются эмульгировать, помещают в этот
фонтан в специальном сосуде с акустическим окном. В качестве
последнего обычно используют тонкий металлический или
пластмассовый лист, который служит основанием (дном) сосуда. Звуковые
колебания проходят сквозь акустическое окно и вызывают эмульгирова-
40
ние жидкости в сосуде. Иногда под пластиной-трансдуцером создают
воздушную полость. Вследствие больших потерь на границе воздух —
твердое тело почти вся звуковая энергия отражается от этой полости,
что усиливает волны, распространяющиеся вверх. С
пьезоэлектрическими генераторами ультразвука проводили свои первые опыты Вуд,
Солнер и др. В настоящее время ультразвуковая техника достигла
высокой степени совершенства, и процесс эмульгирования
ультразвуком изучен детально (Бил и Скоэн, 1955; Кришман, Венкатасубра-
мангам и Гопал, 1961).
Обычно для получения
эмульсий требуется
акустическая энергия порядка десятков
ватт на квадратный сантиметр
поверхности *, что достигается
о-К
Т1
т
-f-
J-
Х- -.
J-
LJ
Рис. 1.17. Схема устройства для
эмульгирования ультразвуком:
1 — сосуд для эмульгирования; 2 —
акустическое окно; 3— передающая среда
(трансформаторное масло); 4 — пластина трансдуцера;
5 — воздушная «подушка».
Рис. 1.18. Магнитострикционный
трансдуцер (а) и вид отдельной
пластины (б).
I — высота, равная полуволне звука
(звук распространяется в направлении I).
применением стандартных кварцевых или керамических трансду-
церов. И керамические, и особенно кварцевые пластины весьма
хрупки. Генерирование радиочастот в диапазоне мегагерц
обходится сравнительно дорого, а эксплуатация таких установок
требует квалифицированного обслуживания. Кроме того,
производительность самого процесса эмульгирования на этих частотах мала.
Поэтому пьезоэлектрические трансдуцеры обычно не применяют
в промышленности, тогда как в лабораторных исследованиях их все
еще используют достаточно широко.
Магнитострикционный эффект — это изменение размеров
ферромагнитного материала, помещенного в переменное магнитное поле.
У большинства ферромагнетиков относительные деформации малы,
но у никеля, пермендюра и ферритов они достаточно большие.
Трансдуцер сделан из листов соответствующего ферромагнитного материала,
листы выштампованы по определенной форме и собраны в пакет.
На рис. 1.18 показана типичная форма пакета и отдельного листа.
* Эта величина называется интенсивностью и характеризует скорость
распространения энергии в единице объема вещества. (Прим. переводчика).
47
7
Провода с электрическим током укладывают вокруг пластин, причем
для повышения к. п. д. по этим проводам пропускают и переменный,
и постоянный ток одновременно. Магнитострикционные генераторы
ультразвука дают большую амплитуду колебаний. Мощность 100 —
5000 вт может быть получена на приборах, рассчитанных на частоты
20 — 100 кгц, что соответствует резонансной длине I порядка 10 см.
К магнитострикционным трансдуцерам подводят не высокое,"
а обычное напряжение, поэтому их можно погружать непосредственно
в жидкость, что облегчает воздействие звука. При этом сам прибор
охлаждается. Тем не менее эти трансдуцеры не находят широкого
распространения для эмульгирования в промышленных масштабах.
Вероятно, это связано с тем, что низшую частоту 20 кгц можно
получить и без применения
электронной аппаратуры — бо-
\ Выпуск лее простыми методами,
которые мы сейчас рассмотрим.
Трансдуцеры электромагнит-
Голобо Вибрирующая ного типа обычно представляют
струи \ Упластина собой вибрирующие пластинки,
-=»-^ "zt^t которые колеблются под воздей-
ствием вспомогательных магни-
Рис. 1.19. Схема работы жидкостного тов. Они имеют ограниченное
свистка: применение и поэтому не будут
а — вибрирующая пластина закреплена здесь .ООСуждаТЬСЯ.
в центре; б — пластина закреплена с краю. \j_ „
' Ультразвуковые струйные
генераторы могут быть
предназначены для работы с газами или с жидкостями. Жидкоструйные
генераторы, работающие на звуковых частотах, оказались очень удобными
для промышленного эмульгирования. Их потенциальные возможности
использованы далеко не полностью, и несомненно, что в будущем
их роль будет все более возрастать. Схема работы жидкостного
свистка показана на рис. 1.19. Когда струя жидкости ударяется
об острое ребро, в струе возникает нестабильность в виде волны
возмущения, которую в акустике называют волной основного тона.-
В жидкостном свистке жидкость подают во внутреннюю область под
давлением 100 am, а в струе скорость составляет 50 м/сек. Прошедшая
через входное сопло жидкость приобретает форму тонкой струи
и направляется на острое ребро пластины, особым образом
укрепленной в своей центральной части. В пластине возбуждаются колебания,
и при резонансе акустическая энергия в струе резко возрастает.
Вся система регулируется так, чтобы резонансная частота пластины
(15—30 кгц) достигалась при оптимальных давлениях и скорости
жидкости, а также расстоянии между соплом и краем пластины.
Этот тип струйного генератора, впервые предложенный Жанов-
ским и Полманом (1948), усовершенствован в результате работ
многих исследователей. В настоящее время его широко применяют для
эмульгирования и диспергирования. Вибрирующую пластину можно
крепить либо в центральных точках (рис. 1.19, а), либо на одном
48
конце (рис. 1.19, б); в последнем случае получается как бы
консольный вибраторов камеру, где установлена пластина, подают несколько
струй, что обеспечивает резонанс пластины, так что акустическая
энергия сохраняется неизменной и максимальной. Насос, который
подает жидкость в генератор, служит также и для циркуляции
жидкости.
Струйный генератор используют для различных целей. Очевидно,
он просто может работать как обыкновенный источник непрерывного
течения жидкости или как смеситель. Основное его применение —
в качестве аппарата для эмульгирования, так как в малом объеме
у края вибрирующей пластины концентрируется большая
акустическая энергия и возникает кавитация. Согласно уравнению (25), такая
большая плотность энергии обусловливает малый размер
образующихся капель эмульсии. Поэтому звуковые генераторы
оказываются весьма эффективными. Например, в гомогенизаторах для
получения частиц размером 1 мкм при производительности 5000 л/ч
требуется мощность 40—50 л. с, а в струйных генераторах при этих
же условиях достаточно 5—7 л. с. В гомогенизаторах давление 500 —
2000 am, а в струйных генераторах — 75—100 am. Конструкция
аппаратов довольно простая. Единственный элемент, который
требует повышенного внимания, — это вибрирующая пластина. При
работе в жестких условиях она должна быть заменена уже через
несколько месяцев. Наконец, следует указать, что струйные
генераторы легко могут быть перестроены на диспергирование твердых тел.
Механизм эмульгирования звуком
В первых опытах по эмульгированию звуков обычно погружали
пробирку, содержащую жидкости, в трансформаторное масло и
подвергали ее озвучиванию. Акустическая энергия передавалась через
стеклянные стенки пробирки так, что казалось, будто вибрируют
сами стенки, особенно вблизи поверхности, и именно они вызывают
эмульгирование. Более поздние исследования показали, что
основной процесс — иной. Например, эмульсия легко образуется в
устройстве, изображенном на рис. 1.17, где звуковые волны входят в
акустическое окно не вызывая в пробирке колебаний с заметной
амплитудой. В настоящее время известно два механизма эмульгирования
звуком: один основан на явлении кавитации, второй — на
представлениях о поверхностных волнах. Главные работы в этом
направлении проведены давно. Мы рассмотрим лишь итоги и укажем на
некоторые нерешенные проблемы.
Для объяснения механизма эмульгирования ультразвуком
используют представления о поверхностных волнах, которые имеют место
в случае нестабильности Рэлея — Тейлора (см. стр. 30). Вопрос
о стабильности поверхности жидкости при периодическом ускорении
впервые был подробно изучен Бенджаменом и Урселлом (1954),
однако безотносительно к проблеме эмульгирования. Независимо от
них Сорокин (1957) и другие исследователи, в том числе Экнадиосянц
4 Заказ 1070
49
(1963), установили, что процесс образования аэрозолей под
воздействием ультразвука несомненно протекает с участием
поверхностных волн. Как установлено с помощью киносъемок, капли тумана
мгновенно образуются в отдельных точках вдоль стоячей волны,
когда амплитуда колебаний пластины становится достаточно большой."
Ланг (1962) и Штамм (1966) показали, что диаметр капель тумана
колеблется от 1/4 до 1/10 длины стоячей волны. Все эти наблюдения
подытожили Пешкин и Рако (1963).
Маринеско (1946) впервые предположил, что эмульгирование под
действием ультразвука возникает вследствие нестабильности
капиллярных волн. Туман можно рассматривать в гидродинамической
модели как своеобразную «эмульсию», в которой плотность
дисперсной среды очень мала. Тогда к туману применимы общие положения
о неустойчивости поверхностных волн, изложенные выше (см. стр. 31).
Для двух жидкостей неопределенных размеров при условии Z ~ О
уравнение (1.15) принимает вид:
(p2 + Pi)-Q-+(P2-pi)A7(T)ftcoso)T-i-aA-3/(T)=0 (I.29)
Его можно представить и в другой форме, известной как
уравнение Матью:
_-|_ (*.;-(? cos т)Ф = 0 (1.30)
Решая уравнение (1.30), получаем Ф =' г|з (т) ехр (2т[где г|з 00—
периодическая функция времени; Q (а, 6) — характеристическая
экспонента].
Для определенных областей значений а и В величина Q — мнимо©
число; это соответствует колебанию всей системы в целом. Для
других значений а и В (т. е. для иных р, к, gQ, со и a) Q — действительное
число, что соответствует нестабильности. Амплитуда поверхностной
волны экспоненциально возрастает, и это приводит к разрушению
жидкости на капли. Пешкин и Рако нашли, что наибольшая неста—
бнльность возникает при условии:
^TP,)/°? + f Ш\ ,~2,5 - (1.31)
(Р2 + Р1)Ш2 (P2 + Pl)^2
Здесь g'o/w2 равно амплитуде звуковой волны на поверхности
жидкости. Поэтому, когда интенсивность звуковой волны достигает
некоторой критической величины, пульсация поверхности
перерастает в нестабильность, и жидкость разбивается на капли. Анализ
уравнения Матью показывает,что колебания поверхности будут
устойчивыми, пока частота вынужденных колебаний вдвое меньше частоты
источника звуковых волн. Согласно Кельвину, для ряби с длиной
волны 2л/к частота определяется выражением [аР/(р3 + pj)l "
З.г-.
и длина устойчивой поверхности волны равна 2л у 4а/j (p2 + рх) со2.
Отсюда можно определить и размеры капель, однако иногда такие
расчеты оказываются недостаточными.
5(1
Можно поставить очень простой опыт для выявления
нестабильности Рэлея — Тейлора в процессе образования эмульсии. Для этого
используют устройство, изображенное на рис. 1.17. Жидкость, которую
требуется заэмульгировать, помещают в оболочку из тонкой фольги.
Сначала звуковые волны проходят сквозь акустическое окно
нормально к поверхности жидкости, и образуется эмульсия. Затем
пьезоэлектрический кристалл переставляют так, чтобы звуковые волны
распространялись тангенциально к поверхности жидкости. Если
при этом на поверхности не возникает нестабильность Рэлея —
Тейлора, которую считают ответственной за образование эмульсии, то
эмульсия не образуется. Следовало бы проверить эту идею с
системой масло — вода, где, как полагают, некоторое значение имеет
кавитация, и с системой ртуть — вода, где, очевидно, кавитации
не будет. Насколько нам известно, такие опыты еще не проведены.
Бонди и Солнер (1935) предположили, что кавитация является
причиной образования эмульсий. Это предположение принято как
в общем верное еще задолго до введения понятий о нестабильности
поверхностных волн. Кавитация — это образование в жидкости
полостей с последующим их быстрым захлопыванием. Это явление
известно в гидравлике уже более 60 лет. Всякий раз, когда
мгновенное давление в какой-либо точке жидкости становится
отрицательным, жидкость разрывается в этой точке, образуя пустоту, сразу же
заполняющуюся паром самой жидкости или газами, растворенными
в ней. Разрушение этих полостей порождает ударные волны,
действие которых в масштабах рассматриваемых точек весьма
значительно. Кавитация иногда нежелательна (в частности, она вызывает
износ гребных винтов кораблей, лопастей турбин), однако, в
некоторых случаях ее стараются получить (например, для целей очистки).
Обзор о действии кавитации в поле большой интенсивности звука
•был сделан недавно Сиротюком (1963), а некоторые общие вопросы
можно найти в упомянутой выше литературе.
Для понимания процесса кавитации необходимо проанализировать
поведение пузыря воздуха, находящегося в акустическом поле с
переменным давлением Р0 — Ра sin cot, где Ра — амплитуда давления
(Нолтинг и Непира, 1950, 1951). Существует несколько механизмов,
посредством которых в жидкости образуются такие пузыри-зародыши
кавитации (Сиротюк, 1963). Вот основные из них: а) флуктуации
температуры, что дает избыточный пар жидкости; б) очень мелкие
твердые частицы примесей, нарушающие структуру жидкости;
в) уже существующие газовые пузыри — примеси растворенных
газов; г) ионы, возникающие под действием космических лучей или
естественной радиации. Когда такой пузырь находится в поле с
переменным звуковым давлением, характер явления зависит от
отношения частоты вынужденных колебаний со к частоте собственных
колебаний пузыря со 0, причем
1 ГЗх /„ . 2о\-]Ч*
4*
51
где р — плотность жидкости; х = Ср/Су для газа, находящегося
в пузыре радиуса г0.
При со ^> со о пузырь совершает колебания, более или менее близ- .
кие к гармоническим. При ш Сйово время полупериода с
отрицательным мгновенным давлением пузырь резко увеличивается в
размере, а в следующий полупериод, когда мгновенное давление
становится положительным, пузырь лопается, особенно если Р3 достигает
критической величины. Сдвиг фаз, который при этом происходит,
зависит от Ря, Р0 и г0. Таким образом, разные пузыри начнут
разрушаться в различные промежутки времени и с различной скоростью.
В момент захлопывания кавитационного пузыря мгновенные скорости
достигают сверхзвуковых величин, и образуются ударные
микроволны. Если, например, радиус пузыря составляет 1 мкм, частота
4 Мгц, амплитуда давления 4 am, то, как показывает расчет, в
ударных микроволнах давление может достигать нескольких тысяч
атмосфер за период в 1/4 мксек. В действительности давление будет меньше,
ибо вследствие вязкости и сжимаемости ударные волны затухают
и, кроме того, пузыри могут начать разрушаться в результате
нестабильности Рэлея — Тейлора.
Необходимо отметить также и зависимость порога интенсивности;
от частоты звука. Этот порог возрастает от 0,1 вт/см2 при 1 кгц до
10 вт/см2 при 10 мгц. Другими словами, кавитация легче возникает
на низких частотах.
На то, что ультразвуковое эмульгирование связано с кавитацией,
по-видимому, указывают сходные зависимости этих двух процессов
от таких параметров, как плотность, частота, содержание
растворенного газа и примесей (Недужий, 1965). Приведем здесь основные
аргументы.
1. Для начала процессов кавитации и эмульгирования требуется
превзойти одинаковый порог интенсивности звука.
2. Для обоих процессов наблюдается сходная зависимость порога
интенсивности звука от частоты, а именно: с возрастанием частоты
пороговое значение интенсивности увеличивается.
3. Как показали измерения Недужего, с увеличением
интенсивности звука число очагов кавитации возрастает и размеры таких
пузырей-зародышей определенным образом зависят от изменения
интенсивности. Аналогичная зависимость размеров капель
эмульсии от интенсивности предусматривается теорией, в которой
механизм образования эмульсий связан с действием поверхностных
волн.
4. Между размерами очагов кавитации в момент их образования
и длиной поверхностной волны (согласно Недужему) не наблюдается
корреляции.
5. В некоторых системах установлено наличие двух порогов
интенсивности звука, один из них соответствует эмульсии М/В,
второй — В/М. Это можно сравнить с различными порогами кавитации
для двух жидкостей, в то время как нестабильность поверхностных
воли для обеих жидкостей одинакова.
52
6. Солнер и другие исследователи установили, что образование
эмульсий в легких жидкостях, таких как вода, спирты, масла,
зависит от содержания растворенного газа и величины внешнего давления
(Солнер, 1944). От этих же самых факторов зависит и возникновение
кавитации. В системе ртуть — вода такая зависимость не
наблюдалась, что было объяснено действием поверхностных волн. Эти
эксперименты следовало бы сейчас, спустя четверть века, повторить,
тщательно замерив характеристики звукового поля и содержание газов
и других примесей.
Можно визуально наблюдать, как образуются при кавитации
эмульсии М/В и В/М (рис. 1.20). Кавитационные пузыри
увеличиваются в размерах в дисперсионной среде и захлопываются в
дисперсной фазе. Жидкость в окрестности пузыря и часть нижележащей
Рис. 1.20. Схема образования капель жидкости, когда в
расположенной над ней другой жидкости происходит
разрушение кавитационных пузырей.
жидкости перемещаются по направлению к центру захлопывающегося
пузыря. Образующиеся струи дробят капли дисперсной фазы. Наблю-
'дая это явление качественно, мы, однако, не можем его оценить
количественно, т. е. определить размеры и число капель, которые при этом
образуются.
Из всего сказанного можно сделать вывод, что характер
ультразвукового эмульгирования, как мы его сегодня понимаем,
заключается одновременно и в кавитации, и в нестабильности
поверхностных волн. Именно этим дуализмом можно объяснить разнообразные
явления, наблюдающиеся при эмульгировании. По-видимому, в
различных условиях эксперимента проявляются либо кавитация, либо
нестабильность. Оба эти механизма дополняют друг друга.
Можно указать, при каких условиях преобладает тот или иной
механизм. Например, нестабильность Рэлея — Тейлора тем больше,
чем больше разность плотностей двух рассматриваемых жидкостей.
Очевидно, в системе ртуть — вода, где разность плотностей велика,
будет проявляться, в основном, механизм нестабильности
поверхностных волн. В системах масло — вода с малой разностью
плотностей большее значение будет иметь кавитация. При низких интенсив-
ностях звука преобладает механизм нестабильности, при больших —
доминирует кавитация (Розенберг и Экнадиосянц, 1961; Гершензон
53
и Экнадиосянц, 1964; Фоглер и Тиммерхауз, 1966). Было бы жела--
тельно более четко разграничить сферы действия каждого
механизма.
Общая характеристика эмульсий,
полученных звуковыми методами
Эмульсии, образованные звуковыми и ультразвуковыми методами,
вообще говоря, не отличаются от эмульсий, полученных в
гомогенизаторах, коллоидных мельницах или смесителях. Размеры частиц,
устойчивость и другие свойства зависят от характеристик
использованной при эмульгировании акустической аппаратуры, а также от
времени озвучивания. Можно сказать, что для практически любой
системы двух жидкостей акустические методы позволяют получить
столь же хорошие эмульсии, как и любые другие методы
эмульгирования.
Ультразвуковое эмульгирование дает возможность приготовлять
достаточно концентрированные (до 30%) высокодисперсные эмульсии
без использования поверхностно-активных веществ. Это может
быть особенно ценно там, где присутствие посторонних веществ
допустимо лишь в малых количествах или вовсе исключено.
Основными параметрами, которые характеризуют озвучивание,
являются интенсивность и частота звука. Интенсивность может быть
постоянной внутри всей рассматриваемой системы и непостоянной *.
Для выяснения роли этих параметров было проведено много работ
по ультразвуковому эмульгированию, но большинство таких
исследований велось на установках лабораторного типа; обзор сделан
Недужим (1962). Сравнительно мало имеется данных о работе
промышленных установок. Следует надеяться, что этот пробел вскоре
будет восполнен.
Ранее было упомянуто, что для производства эмульсий
ультразвуковым методом требуется интенсивность порядка 0,2 вт/см2
и частота 20 кгц. Когда система из двух жидкостей озвучивается,
концентрация образующейся эмульсии вначале быстро возрастает
и вскоре достигает предельного значения (см. стр. 22). Если
интенсивность звуковой волны возрастает, эмульгирование завершается
в более короткий срок.
Изменение частоты влияет на производительность
эмульгирования, но не вызывает обращения эмульсий. Более низкие частоты,
как правило, оказываются более выгодными для эмульгирования.
При 5 Мгц эмульгирование происходит лишь при очень больших
интенсивностях. Чем ниже частота, тем меньшая интенсивность звука
требуется. Однако при частоте < 20 кгц звук становится слышимым,
поэтому ниже этой частоты звуковые колебания большой интенсив-
* Частота, в отлпчие от интенсивности, зависит только от характеристики
трансдуцера. Для данного излучателя звука частота и, следовательно, длина
волны — постоянны, так как дисперсией здесь можно пренебречь. (Прим.
переводчика.)
54
ности не применяют в промышленности. Итак, наиболее эффективной
для эмульгирования является область частот 20—50 кгц.
Кроме параметров звуковой волны, на эмульгирование влияют
и свойства самих жидкостей, а именно: плотность, поверхностное
натяжение, вязкость, а также свойства и количество эмульгатора.
Влияние всех этих факторов в большинстве случаев такое же, как
и. при обычных методах эмульгирования.
Ультразвуковым методам эмульгирования присущи особенности.
Так, в установке с пьезоэлектрическим трансдуцером, изображенной
на рис. 1.17, можно одновременно получить эмульсии В/М и М/В.
Поэтому используют специальные стабилизаторы, аналогично тому,
как это делается при методе прерывистого встряхивания (см. стр. 12).
Если в сосуде для эмульгирования, который озвучивается в
непрерывном режиме, нет принудительного течения жидкости, то в
областях вблизи узлов стоячей волны, где амплитуда наименьшая, будет
наблюдаться коагуляция капель. Если такое озвучивание
происходит достаточно долго, то эта коагуляция может привести к
расслаиванию эмульсии. Расслаивания не происходит, если обеспечено
принудительное течение жидкости.
Уже в первых опытах по ультразвуковому эмульгированию
отмечалось влияние на процесс внешнего давления и наличия в жидкости
растворенных газов. Легкие жидкости, такие как вода, спирты,
масла, не образуют эмульсии, если внешнее давление <4ат или
жидкости полностью дегазированы. Возможно, это связано с тем,
что при таких условиях не возникает и кавитация. Однако в случае
ртути и других тяжелых жидкостей при тех же условиях эмульсия
образуется. По-видимому, здесь проявляется иной механизм эмуль-
тирования *.
Следует отметить, что эмульгирование звуком и ультразвуком
весьма перспективно, хотя в настоящее время и не имеет широкого
применения в промышленности.
ПОЛУЧЕНИЕ ЭМУЛЬСИЙ ЭЛЕКТРИЧЕСКИМИ МЕТОДАМИ
Все методы эмульгирования, рассмотренные выше, имеют
механическую природу, они заключаются в разбивании большого объема
жидкости на капли малых размеров с помощью механических,
гидродинамических процессов. Оказывается, такой же эффект может быть
достигнут за счет действия сил электрического поля. Этот метод
электрического «дробления» известен давно, но стал привлекать
к себе внимание лишь в последние годы, главным образом после
работ Фонегута и Небауэра (1952, 1953). Обзор более ранних работ
дан Дрозпном (1955).
* Для легких жидкостей, эмульгирование которых связано с кавитацией,
характерно оптимальное внешнее давление 2—4 am: при меньших давлениях
захлопывание воздушных пузырей затруднено, при больших давлениях полости
не образуются вовсе. Для тяжелых жидкостей такой зависимости не
наблюдается, так как эмульгирование связано с нестабильностью Рэлея — Тейлора.
(Прим. переводчика.)
55
Аппаратурное оформление
Предположим, что жидкость, которую нужно диспергировать,
помещена в сосуд, оканчивающийся тонким капилляром (с
внутренним диаметром — 1 мм). Пусть жидкости сообщен большой
положительный потенциал. Если этот потенциал медленно увеличивать,
то капли, которые выходят из капилляра, постепенно вытягиваются
и образуют устойчивые нити (рис. 1.21, а). При дальнейшем
увеличении потенциала нити становятся тоньше (рис. 1.21, б) и затем
разбиваются на капли (рис. 1.21, б). Наконец, при весьма высоком
напряжении облако очень мелких капель со значительной скоростью
±
Т
±
Т
Рис. 1.21. Стадии образования аэрозоля электрическим
диспергированием:
а — 2 кв\ б — 4 кй; в — о кв; г — 8 кв.
выделяется из капилляра (рис. 1.21, г). Радиус капель порядка
1 мкм, капли заряжены. Указанные величины потенциалов соответ7
ствуют лишь порядку действительных значений, которые зависят
от ряда условий. Найдено, что напряжение может быть уменьшено,
если нижнюю заземленную пластину держать достаточно близко
от капилляра. При этом возрастает градиент потенциала, что
увеличивает скорость и давление истечения капли, а также уменьшается
экранирующее действие поверхностного заряда. Напряжение можно
уменьшить и специальной конструкцией металлических капилляров,
используя их в случае малопроводящих и легких жидкостей: воды,
масел, спиртов и т. п.
Механизм образования капель
Чтобы разобраться в довольно сложном механизме
рассматриваемого явления, представим себе сначала сферическую каплю
жидкости, находящуюся в электрическом поле. Согласно законам
классической электростатики, сила, действующая на поверхность
жидкости, т. е. давление, определяется выражением
1
ДР:
(е-
1 dp
(1-32)
56
где Ет и Ен — тангенциальная и нормальная составляющие вектора
напряженности электростатического поля Е (Ен обусловлен электро-
стрикцией); е — диэлектрическая проницаемость жидкости.
Равенство (Г.32) можно преобразовать к виду:
АР = ±(е.~1)Ц2Е1-Е*) (1.33)
Если поверхность капли нормальна к Е, то вблизи этой
поверхности величина АР положительна, если тангенциальна к Е, АР —
отрицательна. Это электростатическое давление уравновешено
поверхностным натяжением, и, следовательно, капля принимает форму
сфероида, вытянутого вдоль вектора Е. О степени вытянутости капли
можно судить, измеряя ее эксцентриситет, который связан с
остальными параметрами следующим образом (О'Конски и Тачер, 1953;
Гопал, 1958; Найяр и Мурфи, 1959; Аллан и Мэзон, 1962)
9е' (е-е')гЕо
4(т(е —2е')2
(1-34)
где Е0 — напряженность внешнего поля; е' — диэлектрическая
проницаемость внешней среды.
Соотношение (1.34) обосновано для малых деформаций и
подтверждено экспериментально (О' Конски и Гюнтер, 1955; Аллан
и Мэзон, 1962). Экстраполяция к большим величинам Е0 показывает,
что капля становится все более вытянутой в нить и разрывается на
более мелкие капли, так как поверхностное натяжение уже не может
уравновесить электростатические силы.
По-видимому, в таких или приблизительно таких условиях
оказываются капли жидкости вблизи конца капилляра. Очевидно, здесь
надо учитывать, что вновь образуемые капли оказываются
заряженными и, следовательно, взаимодействуют друг с другом. Это влияет
и на стабильность всей системы капель.
Рэлей (1882) показал, что если заряд Q малой капли превосходит
величину
<? = (16nr3(j),/J (I.35)
то волна возмущения на ее поверхности станет неустойчивой, ампли-
туда этой волны начнет увеличиваться. Анализ этой задачи
существенно упрощается, если использовать выражение для энергии
заряженной частицы:
£/ = 4пг2а + -|^ (1.36)
Капля стремится уменьшить свою энергию путем распада на более
мелкие капли. Пусть число капель N, радиус каждой капли г' =
= rN'1/3, заряд q = QN'1. Тогда:
U'=Anr*oN,h+ Q\, (I.37)
57
Из условия OU'/dN = 0 получим:
N--9L-. г, / 4яг«а V/..
g = (4.V3a)'/s (Г.38)
Конечно, кинематика распада достаточно сложная, и
образующиеся вновь капли имеют разные размеры — в пределах некоторого
интервала. Порядок средней величины капель, как показали
эксперименты (Гендрикс, 1962; Рице, 1964; Гинее, 1966), согласуется
с уравнением (1.38).
Указанным путем может быть дано количественное объяснение
электрического «дробления» жидкостей. Однако это явление более
сложное. Используя стробоскопическое свечение, Джонсон (1963)
нашел, что нити жидкости (рис. 21, б, в) действительно содержат
множество капелек, которые вытекают из конца капилляра с
определенной периодичностью. В настоящее время эта последняя стадия
процесса — вытягивание жидкости в нити и их последующий распад
на капли — не поддается теоретическим расчетам.
Приготовление монодисперсных эмульсий
Производство аэрозолен методом электрического «дробления»
представляет немалый интерес в том отношении, что размеры
образующихся частиц весьма близки друг к другу, точнее, интервал
размеров достаточно узок. Если через полученный таким образом
аэрозоль пропустить световой пучок, то свечение рассеянного света
(эффект Тиндаля) будет очень ярким, что и указывает на
монодисперсность коллоидной системы. Типичное распределение частиц
по размерам представлено на рис. 1.22. Используя это свойство,
Наваб и Мэзон (1958) получили эмульсию, близкую к
монодисперсной.
Идея их метода схематично изображена на рис. 1.23. Жидкость,
которая должна быть диспергирована, помещена в сосуд,
оканчивающийся капиллярной воронкой. Последняя соединена с
положительным полюсом источника высокого напряжения. Сосуд
вставлен в большую круглодонную колбу, на дне которой уложен
заземленный электрод. В колбу налита жидкость, которая должна
служить в эмульсии дисперсионной средой. Образующиеся при
истечении из капилляра мелкие капли дисперсной фазы, погружаясь
в жидкость, дают эмульсию. Установлено, что при непосредственном
погружении капилляра в жидкость получается 1%-ная эмульсия.
Для получения эмульсий с большими концентрациями
устанавливают зазор в 2—3 мм между концом капилляра и поверхностью
жидкости в колбе. Изменяя величину приложенного напряжения
и регулируя зазор, можно получить эмульсии с определенным
размером частиц. Капли эмульсии получаются с размерами в интервале
1 —10 мкм, а распределение частиц по размерам — узкое (см.
58
рис. 1.22). Для улучшения свойств эмульсий жидкость в колбе
перемешивают и вводят эмульгатор. Таким путем получают устойчивые
эмульсии типа М/В и В/М с концентрацией до 30%.
Следует упомянуть о работе Вачтела и ла Мера (1962). Они
получали монодисперсные аэрозоли методом испарения и конденсации,
причем капельки приобретали электрический заряд во время
коронного разряда. Пропуская эти заряженные капельки через жидкость
с эмульгатором, получали стабильные монодисперсные эмульсии.
2 4 б
Диаметр частиц, мкм
Рис. 1.22. Распределение частиц по
размерам при электрическом
диспергировании.
Рис. 1.23. Схема установки для
получения эмульсий методом
электрического диспергирования.
В некоторых эмульсиях почти все частицы имели размер 0,5—
2,0 мкм. Очевидно, таким путем можно получить эмульсии из
любых аэрозолей.
Недостатки
электрических методов эмульгирования
Электрические методы эмульгирования в настоящее время
находятся в стадии развития и совершенствования. Они имеют ряд
очевидных преимуществ, из которых главное — высокая
монодисперсность получаемых эмульсий. Известно, что многие
физико-химические свойства эмульсий зависят от размеров частиц и степени
дисперсности. Невозможность контроля этих свойств в процессе
эмульгирования серьезно тормозит развитие в этой области. Поэтому
электрические методы заслуживают серьезного внимания, особенно
для исследовательских целей. Эти методы позволяют также получать
эмульсии обоих типов с меньшей концентрацией эмульгатора, чем
посредством других методов. Наконец, здесь может быть точно
определена концентрация дисперсной фазы.
59
Однако электрические методы имеют и недостатки. Так, если
жидкости обладают заметной вязкостью, то эмульгирование
затруднительно или вообще невозможно. Наличие заряда у капель
искажает измерения. Если будут найдены способы для нейтрализации
зарядов капель, то электрические методы станут идеальными для
многих исследовательских работ.
САМОПРОИЗВОЛЬНОЕ ЭМУЛЬГИРОВАНИЕ
Самопроизвольным называют такое эмульгирование, которое
происходит без внешнего воздействия. Иногда эмульсии образуются
в результате очень слабого встряхивания двух жидкостей или
перемешивания их с очень малой интенсивностью. Такие случаи
эмульгирования в промышленной практике часто называют
самопроизвольным эмульгированием. Известны также случаи, когда при
соприкосновении двух жидкостей и принятии специальных мер
по предотвращению внешних воздействий, все же получают
эмульсии. Следовательно, такое самопроизвольное (спонтанное)
эмульгирование действительно может происходить, и в зависимости от
взятых компонентов оно завершается в течение различных интервалов
времени — от нескольких часов до нескольких дней.
Хотя внешнее воздействие полностью отсутствует, в самой
системе вблизи поверхности происходят внутренние
физико-химические процессы, что приводит к разрушению этой поверхности. В
процессе участвуют гравитационные силы, которые преодолеваются
внутренними силами. В результате капельки более тяжелой жидкости
оказываются распыленными («самопроизвольно») в более легкой.
Так, если чистый толуол осторожно привести в соприкосновение
с водой, то не образуется никакой эмульсии. Однако при
использовании раствора толуола с 10% метанола начнут проявляться
внутренние процессы. Через некоторое время органическая фаза станет
мутнеть из-за образования эмульсии, тогда как слой воды останется
прозрачным. Если же взять раствор толуола с 40% метилового
спирта, то, наоборот, помутнение начнется в воде, тогда как
органические вещества сохранят прозрачность.
После открытия спонтанного эмульгирования Гэдом в 1878 г.
найдены многочисленные системы жидкостей, которым свойственно
самопроизвольное эмульгирование. Обзор ранних работ дан Мак-
Беном и By (1937). Несмотря на большое число работ, посвященных
этому вопросу, механизм явления остается дискуссионным.
Прогресс здесь следует ожидать в раскрытии природы процессов,
происходящих на поверхности.
Механизмы самопроизвольного эмульгирования
Обычно указывают на существование трех различных
механизмов. Рассмотрим их.
1. В большинстве случаев, когда каплю более легкой жидкости
помещают на поверхность более тяжелой жидкости, самопроизволь-
60
ное эмульгирование начинается с неустойчивого движения
поверхности. Тонкие струи более тяжелой жидкости неожиданно
отрываются от поверхности (рис. 1.24, а), проникают в более легкую
жидкость и оставляют в ней малые капли. Как полагают, в этом явлении
проявляется одна из форм поверхностной нестабильности.
Действительно, как было установлено Квинке еще в 1888 г., в месте
соприкосновения двух жидкостей возникают неравновесные условия, и
поверхностное натяжение уменьшается. Неравновесные условия могут
возникать вследствие образования химических соединений в отдельных
точках или неоднородного поглощения молекул раствора на
.поверхности. От этих точек начинает быстро распространяться
турбулентность. Нити одной жидкости проникают в другую, где они распадаются
на капли, которые перемещаются внутри каждой фазы. Стабилизация
Рис. 1.24. Схема основных механизмов при спонтанном
эмульгировании:
а — поверхностная турбулентность; б — диффузионный механизм; в —
механизм, обусловленный отрицательным поверхностным натяжением.
капель дает эмульсию. Таков в общих чертах механизм
поверхностной турбулентности при самопроизвольном эмульгировании.
Например, когда толуол или ксилол осторожно вводят в
соприкосновение с водным раствором додециламина, образуется система,
в которой происходит самопроизвольное эмульгирование. Эту
систему изучали многие авторы (Каминский и Мак-Бен, 1949; Гартинг
и Райе, 1955; Дэвис и Хейдон, 1957). При концентрации
эмульгатора >0,1 М самопроизвольное эмульгирование происходит в
водной фазе. Капельки масла стабилизируются вследствие адсорбции
на своей поверхности активного вещества. Равновесное
поверхностное натяжение остается постоянным (порядка 1 дин/см при
изменении концентрации эмульгатора от 0,4 до 0,03М), и нет тенденции
к тому, чтобы оно стало отрицательным. В этой системе происходит
значительное перемешивание в поверхностных слоях. Под
микроскопом видно, что отдельные капли или нити ксилола прорывают
поверхность раздела двух жидкостей, некоторое время движутся в воде,
а затем возвращаются обратно. Во время такого «путешествия»
от них отделяются более мелкие капли, которые остаются в воде
и стабилизируются.
2. В некоторых случаях движение поверхности жидкости очень
мало или незаметно. Вероятный механизм, теперь называемый
61
диффузионно-неподвижным, предложен Гурвичем (1913) и Рашевским
(1928). Эту систему изучали многие авторы (Мак-Бен и By, 1937;
Дэвис и Хейдон, 1957).
Подобный механизм можно наблюдать, когда раствор метанола
или этанола в толуоле осторожно вводят в соприкосновение с водой.
Предполагается, что спирт диффундирует в водную фазу, неся с собой
некоторое количество толуола. Смесь вода — спирт с толуолом
образует трехфазную систему, в которой толуол менее растворим,
чем спирт. Поэтому на некотором расстоянии от поверхности воды,
когда растворение зайдет уже достаточно далеко, выделяется толуол
в виде малых капелек *. Капельки толуола перемещаются в водной
фазе и со временем там стабилизируются.
Здесь также наблюдается поверхностная турбулентность, но,
вероятно, она не является основным механизмом процесса. Так,
если вместо воды взять разбавленный (0,001 М) раствор лаурил-
сульфата натрия, поверхностная турбулентность подавляется, а
самопроизвольное эмульгирование продолжается. Кроме того, не
наблюдается корреляции с величиной поверхностного натяжения, и имеется
только связь с коэффициентом распределения.
3. Если поверхностное натяжение стремится стать
отрицательным, то обычно это ведет к самопроизвольному возрастанию
внутренней поверхности (поверхность раздела обеих жидкостей). Во всех
подобных системах наблюдается наличие определенных предельных
концентраций, ниже которых не происходит самопроизвольного
эмульгирования. Для выяснения причины этого явления
потребуются точные измерения поверхностного натяжения при различных
концентрациях.
В качестве примера укажем на систему, которая образуется,
когда раствор цетилового спирта в толуоле вводят в соприкосновение
с водным раствором додецилсульфата натрия. Найдено, что
самопроизвольное эмульгирование происходит, когда концентрации
спирта и соли превосходят четко определенные пределы. Измеренное
при малых концентрациях поверхностное натяжение (Дэвис и Хейдон,
1957) в обоих указанных растворах было экстраполировано к нулю.
При более высоких концентрациях поверхностное натяжение,
очевидно, отрицательное, и это является причиной самопроизвольного
возрастания внутренней поверхности. Некоторые авторы подвергали
сомнению результат экстраполяции отрицательного поверхностного
натяжения. Основанием для этого был широко известный случай,
когда отрицательное поверхностное натяжение, полученное
экстраполяцией (Штекельберг, Клокнер, Морхауэр, 1949), при тщательной
проверке оказалось хотя и небольшой (~ 0,005 дин/см), но
положительной величиной (Менсфилд, 1952). Однако при самопроизвольном
* В работе [М. В. Островский, Р. X. Бареибаум,
А. А. А б р а м з о в, Коллоиди. ж., 32, № 4, 565 (1970)] экспериментально
показан п количественно описан такой механизм самопроизвольного
эмульгирования, наблюдаемого в процессе массопереноса. Даны условия его появления
и методы расчета. (Прим. редактора перевода.)
02
эмульгировании поверхностное натяжение, как и некоторые другие
характеристики, является динамическим, и если даже в целом оно
положительно, в отдельных участках может быть отрицательным.
Обычно считают, что эти три механизма будто бы взаимно
исключают друг друга. Однако более подробное изучение приводит
к выводу, что все они взаимосвязаны. Так, поверхность жидкости
принимает пальцеобразную форму вследствие поверхностной
турбулентности. Этот процесс достаточно продолжителен: он протекает
в течение минут и даже часов. Возможно, что такая форма жидкости
образуется также и в результате взаимной диффузии молекул обеих
жидкостей через поверхность раздела. С другой стороны,
неравномерная диффузия сама может послужить причиной поверхностной
нестабильности, как это будет рассматриваться далее. Отрицательное
поверхностное натяжение * обусловливает термодинамическую
неустойчивость и как следствие этого — движение жидкости и разрушение
поверхности. Таким образом, на кинематику течения жидкости
отрицательное о влияет так же, как и уменьшение а, но более интенсивно.
Взаимосвязанность указанных трех механизмов затрудняет
описание процесса самопроизвольного эмульгирования. Дискуссионным
является вопрос, какой из этих механизмов первичный, а какой —
вторичный. По-видимому, можно утверждать, что в отдельных
частных случаях доминирующими являются разные механизмы (Дэвис
и Райдил, 1961).
Примеры расчетов для систем
с самопроизвольным эмульгированием.
Рассмотренные выше модели не дают возможности произвести
какие-либо количественные расчеты процесса. Существующие методы
расчета самопроизвольного эмульгирования разрабатываются и
основаны на грубо приближенных моделях, так что в этом направлении
многое еще предстоит сделать. Ниже дан краткий обзор сделанному.
Хейдон (1958) вычислил величину работы неоднородной
адсорбции. Наблюдения за каплей воды, взвешенной в толуоле с 4%
ацетона показывают, что эта капля испытывает беспорядочные
пульсации. Частота этой пульсации уменьшается с течением времени,
и движение исчезает, когда ацетон распределяется между толуолом
и водой в соответствии с коэффициентами распределения.
Количество энергии, затраченной в процессе пульсации, может быть
определено по амплитуде или иной характеристике пульсации.
Поглощенная энергия может быть определена по изменению физико-
химических свойств системы. Обе эти величины достаточно хорошо
согласуются друг с другом, что свидетельствует в пользу
справедливости исходных посылок, а именно: движение капли по
поверхности жидкости обусловлено неоднородным перемещением третьего
* Само существование отрицательного поверхностного натяжения глубоко
дискуссионно и может рассматриваться лишь как рабочая гипотеза, но не как
установленный факт, на котором могут базироваться теории. (Прим. редактора
перевода.)
63
компонента — ацетона через поверхность раздела двух жидкостей
(толуол — вода).
Как показали Стернлинг и Скривен (1959), при изменении
поверхностного натяжения возрастает вероятность возникновения
поверхностной турбулентности. В этом, собственно говоря, заключается
эффект Марангони, пнтерес к которому вновь возрос в последнее
время (Скривен и Стернлинг, 1960, 1964; Смит, 1966).
С помощью гидродинамических уравнений, составленных из
условий движения жидкости в диффузионных ячейках вблизи
плоской поверхности, рассчитывали поле скоростей. Из уравнений
диффузии вычисляли градиенты концентрации растворенных
веществ, которые пропорциональны изменению поверхностного
натяжения. На поверхности раздела происходят одновременно
гидродинамический и диффузионный процессы, которые могут
контролировать механизм массопереноса. В ряде случаев оба процесса идут
в одном направлении, скорости движения частиц складываются,
и результирующая скорость значительно возрастает. Такое
состояние аналогично нестабильности Бенарда (см. стр. 30), что приводит
к турбулентности.
Возникновение поверхностной турбулентности облегчается,
если растворенное вещество перемещается из фазы с более высокой
вязкостью или с меньшим коэффициентом диффузии.
Таким образом, если вещество диффундирует в одном
направлении, система может быть стабильной, если в другом — нестабильной.
Неустойчивость также возрастает вследствие чрезмерного градиента
концентрации вблизи поверхности.
Авторы подчеркивают, что использование для теоретического
анализа простой модели не позволяет согласовать теорию с опытом.
Полученные анализом результаты подтверждаются экспериментом
лишь качественно (Линде и Зерт, 1965). В то же время теоретические
анализы важны, ибо необходимо твердо выяснить механизм, который
вызывает турбулентность при диффузии через поверхность.
Рассмотрим расчеты (Субрамангам и Гопал, 1967) по
поверхностной нестабильности от синусоидальных перемещений *. Пбложим,
что поверхность раздела между двумя жидкостями с плотностями рй
и р,2 задана в момент времени т = 0, и перемещение ее совершается
по гармоническому закону a cos kx. Тогда, если поверхностное
натяжение о0 постоянно, поверхность колеблется но закону:
* В настоящее время предложен н разрабатывается механизм
возникновения межфазной турбулентности, основанный на предположении, что
турбулентность возникает благодаря несоответствию поверхностного сопротивленпя
движущей силе массопереноса. Поток диффундирующего вещества производит
удары по границе, вызывая колебания последней. Движение поверхности
раздела передается пограничным слоям фаз. С помощью такого механизма удалось
количественно объяснить ряд имеющихся в настоящее время
экспериментальных факторов по возникновению и протеканию процесса межфазной
турбулентности [М. В. Островский и др., ЖПХ, 40. 1319 (1967)]. (Прим.
редактора перевода.)
64
a cos kx cos сот где со2 описывается хорошо известным уравнением
Кельвина (1.17).
Предположим далее, что поверхностное натяжение уменьшается
на некотором участке до величины ст0 — о. Тогда, как показывает
анализ, в этом месте происходит увеличение амплитуды колебаний
с течением времени, а на остальных участках колебания поверхности
продолжаются почти с той же частотой Кельвина. Этого следует
ожидать, так как поверхность должна растягиваться там, где
поверхностная энергия уменьшилась. При о~х < сг0 возникает
нестабильность, несколько менее выраженная чем та, которую рассматривали
ранее (см. стр. 30), ибо в данном случае увеличение амплитуды
линейное, а не экспоненциальное, т. е. более медленное. Но когда
о1 > а0 и поверхностное натяжение становится отрицательным
при х ~ 0, нестабильность растет экспоненциально во времени.
В этом случае образование капель было бы практически мгновенным.
Так, этот расчет дает определенную, хотя и идеализированную модель
для внутриповерхностной турбулентности *.
ПРОБЛЕМА ДЕЭМУЛЬГИРОВАНИЯ
Выше рассмотрены вопросы, связанные с процессом получения
эмульсий, — диспергирование больших масс жидкостей и
образование капель. При эмульгировании одновременно протекает и
противоположный процесс — малае капли рекомбинируют, образуя капли
больших размеров. Обычно такую рекомбинацию предотвращают,
используя вещества, которые создают энергетический барьер,
препятствующий сближению частиц. Этот вопрос подробнее рассмотрен
во второй главе.
Иногда необходимо выделить из эмульсии ее составные части.
В качестве примера, имеющего важное практическое значение,
укажем на нефтяную эмульсию. Сырая нефть в момент ее выхода
на поверхность земли часто содержит воду в виде эмульсии В/М.
Наличие в нефти воды затрудняет ее очистку, образуется пена,
выделяется битум, трубопроводы и емкости-танки подвергаются
усиленной коррозии. Возрастает также потребляемая мощность, и скорее
изнашивается оборудование, так как увеличивается вязкость и объем
продукта (Монзол и Стензл, 1946). Итак, нефть должна быть
высушена (до концентрации воды — 1% вместо первоначальных 10—
60%). Другими важными примерами деэмульгирования являются
такие известные процессы, как выделение сливок из молока, каучука
из латекса. Экономическое значение этих проблем рассмотрено
в литературе (Беркман и Эглоф, 1941; Клейтон, 1954; Бехер, 1965).
* В данном разделе не нашел отражения подробно разработанный меха-
низы самопроизвольного эмульгирования, возникающего при очень низких,
но положительных значениях межфазного натяжения, которые сопоставимы
с энергией теплового движения. При этом получаются термодинамически
устойчивые дисперсные системы. Эта теория разрабатывается школой акад. П. А, Ре-
биндера и освещена в многочисленных публикациях — см., например,
дополнительную литературу к этой главе. (Прим. редактора перевода.)
5 Заказ 1070
65
Седиментация, обращение и коалесценция ■
Разрушение эмульсий может быть трех видов: седиментация,
обращение (фаз) и коалесценция. Такая классификация удобна
для практических целей.
Седиментация наблюдается, например, при отделении сливок
от молока. При этом не происходит полного разрушения эмульсии,
а образуются две эмульсии, одна из которых богаче дисперсной
фазой. Так, в обычном молоке содержится 8 — 10% жира, а в
сливках — 30—35%. Известно, что капля радиусом г и плотности рф
будет всплывать в более тяжелой жидкости с плотностью рс и
вязкостью Г| со скоростью, которая определяется уравнением Стокса:
, WP'-РФ) (Г.39)
9ti
Если рф > рс, то капля станет опускаться на дно под действием
силы тяжести. Таким образом, осаждение капель в эмульсии —
седиментация — есть следствие образования больших капель и
большого различия в плотностях обеих жидкостей. Для типичных
эмульсий г я^ 1 мкм, рс «^ рф ^ 0,2 г/см3, ц ^ 0,01 пз и г;имеет порядок
нескольких сантиметров в сутки. Чтобы ускорить процесс, например
для получения масла, обычно применяют центрифугирование, где
ускорение (центробежное) более чем в сто раз превышает ускорение
силы тяжести. При экстракции каучука из латекса используют
специальные вещества, которые способствуют слипанию частиц,
увеличивая эффективный радиус г.
Обращение фаз — нестабильное состояние эмульсии, когда
неожиданно происходит изменение типа эмульсии от В/М к М/В
или наоборот. На обращение фаз влияют объемная концентрация
компонентов, природа и количество эмульгатора. При изменении
концентрации и благоприятном сочетании всех остальных факторов
обращение фаз происходит, когда концентрация достигает ~ 75%.
Эта величина близка к теоретическому значению 74%, что
соответствует плотной упаковке жестких шаров одинакового размера.
Но совпадению этих величин не следует придавать большого
значения, так как в концентрированных эмульсиях капли могут не иметь
сферической формы, а кроме того, обращение фаз происходит и при
иных концентрациях. Согласно схеме, предложенной Шульманом
и Кокбейном (1940), при возрастании концентрации масла в
эмульсии М/В капли масла сталкиваются друг с другом и соединяются
таким образом, что теперь уже вода оказывается в виде капли
(рис. 1.25). Эта упрощенная схема является, вероятно, правильной.
Следует добавить, что на обращение фаз влияют также температура
и динамика процесса эмульгирования.
~J Коалесценция — полное разрушение эмульсии, когда
выделяются в чистом виде отдельные компоненты эмульсии.
Коалесценция — это переход каждого составляющего компонента из мета-
стабильного состояния в термодинамически устойчивое. При раз-
66
о
L Mac no)
Вода
Г7^
L-Маслоч
рушении эмульсии можно выделить две стадии. В первой, называемой
флокуляцией, капли дисперсной фазы образуют агрегаты-клюстеры,
которые легко распадаются при слабом перемешивании. Во второй
стадии, собственно коалесценции, капли в клюстерах соединяются
в одну большую каплю.
Этот процесс необратим в том смысле, что для разрушения столь
больших капель на малые и воссоздание эмульсии требуется очень
сильное перемешивание. При
коалесценции происходит
видимое невооруженным глазом
разделение фаз.
В последние годы было
проведено много работ по изучению
кинетики флокуляции и
коалесценции. Установлено, что фло-
куляция происходит, когда две
капли приближаются друг к другу
на расстояние двойного
молекулярного слоя и далее остаются
рядом, что соответствует
минимуму их потенциальной энергии. Это
явление, как было отмечено
Смолуховским около 50 лет
назад, во многом сходно с
явлением столкновения молекул газа
при условии, что обе частицы
прекращают движение после
столкновения. Используя
математический аппарат этой теории,
можно определить число клюсте-
ров, состоящих из 1, 2, 3, . . .
капель, как функцию времени.
Вычислив общее число клюстеров
и их распределение по размерам
по кинетике флокуляции.
Что касается коалесценции, то здесь возникает новая стадия —
разрушение устойчивого поверхностного слоя. Исследование
поведения, например, капель масла, образующихся в воде при
коалесценции в эмульсии М/В, позволяет получить нужную информацию
о различных аспектах проблемы. Так как интерпретация полученных
данных связана с вопросом о стабильности эмульсий, то
соответствующее обсуждение будет дано в главе II.
Рис. 1.25.
обращения
Механизм
фаз.
можно решить любую задачу
Техника разрушения эмульсий
В промышленных масштабах эмульсии разрушают химическими
методами, нагреванием, осаждением под действием силы тяжести
или центробежных сил, электрическими методами и др. Обычно
5*
67
используют одновременно несколько способов. Ниже будут описаны
в основных чертах применяемые в технике установки.
Самыми распространенными, по-видимому, являются химические
методы разрушения эмульсий. Их действие заключается в удалении
барьеров, препятствующих коалесценции. Химические вещества —
деэмульгаторы нейтрализуют действие защитного слоя. Например,
сероуглерод н четыреххлористый углерод растворяют эти защитные
пленки. Частичное разрушение этих пленок происходит и при
обращении эмульсий из В/М в М/В и наоборот. Многовалентные кислоты
и соли нейтрализуют действие электрических сил двойного защитного
слоя. Итак, для каждой эмульсин имеется специальный «свой» де-
эмульгатор, который оказывает оптимальное действие; выбрать такое
Легкая
жидкость С
*Э
(V+-1- +
+■ +■ + t
txxx
1- + + 4
-T-I-I
\
X.
r
t
у
i
t
л- ■+-
-л- +-
+- +
3=
Тяжелая_
жидкость"
Рис. 1.26. Схема отстойника для
эмульсий.
Эмульсия
разделения грубых
вещество можно только после тщательного изучения свойств
эмульсии. Беркман и Эглоф (1941) приводят перечень деэмульгаторов
п области их применения. В установках для разрушения эмульсии
деэмульгатор лучше всего вводить вначале работы, он ускоряет
также и разделение фаз.
Грубые эмульсии, например нефтяные, содержат капли довольно
больших размеров. Поэтому целесообразно для разделения
жидкостей просто выдержать их в отстойнике. Такое разделение, однако,
является частичным — существует градиент концентрации, а очень
малые капли остаются во взвешенном состоянии. Механизм
сепарации простым отстаиванием заключается в действии на каждую каплю
гравитационных и выталкивающих сил. Этот вопрос рассмотрен
Аткинсоном н Фришвотером (1958).
Схема простого отстойника дана на рис. 1.26. Чтобы жидкости
не смешивались, подбирают опытным путем скорость течения, я вблизи
впуска эмульсин устанавливают перегородки. Обычно время
разделения составляет ~1 ч. Более эффективно оборудование, где
используют центробежные силы. Установка такого типа схематически
изображена на рис. 1.27. Конические диски расположены внутри
центрифуги. Более тяжелая жидкость выталкивается к периферии
68
Эмульсия
\
Легкая
j~~~~жидкость
"7 Тяжелая
~$^>С1/Зкость
и через обод отводится. Более легкая жидкость собирается вблизи
центра. На такой установке время сепарации составляет несколько
минут. Обзор современного состояния техники сепарации дал Ландус
(1965).
Для электрических методов разрушения эмульсии характерны
два случая: первый — когда капли заряжены, второй — когда они
электронейтральны, но приобретают дипольный момент,
индуцируемый в постоянном или переменном электрическом поле. Таким
образом, в эмульсиях, где частицы не заряжены, происходит коалесцен-
ция диполей. Это можно наблюдать визуально, если две капли
поместить рядом друг с другом в электрическое поле с
напряженностью Е: капли вскоре начнут
притягиваться друг к другу. Для двух
жидких сфер одинакового радиуса г
с диэлектрической проницаемостью е,
расстоянием между ними в масле I
и диэлектрической проницаемостью
масла е' силы притяжения составят:
Величина ('/О3 пропорциональна
концентрации дисперсной фазы и почти
не зависит от размеров частиц, так что
для многих эмульсий фактор (r/l)i
является почти константой в течение
времени сепарации. Коалесцеиция
пропорциональна Е2г2.
Однако, когда капли очень крупные,
электрическое поле стремится их
деформировать, и в конце концов капли
претерпевают разрыв, аналогичный
описанному ранее (стр. 56) Поэтому, если
размеры капель становятся большими, следует уменьшить величину
напряженности электрического поля, а также градиент скорости.
В типичном аппарате, показанном на рис. 1.28, это осуществляют
прикладыванием напряжения (от постоянного тока или чаще от
выпрямленного переменного) к нижнему из двух электродов,
смонтированных внутри сосуда с эмульсией. Саму эмульсию подают по
патрубку в область между этими двумя электродами — туда, где
величина напряженности электрического поля наибольшая. Так как
вода, содержащая примеси, проводит электрический ток, то между
нижним электродом и дном сосуда возникает электрическое поле,
правда слабое, и в этой области скапливаются большие капли воды.
Течение жидкости устанавливают так, чтобы в случае, если капли
не достигнут достаточного размера, эмульсия вновь возвращалась
бы в межэлектродное пространство, где протекает коалесцеиция.
В хорошо спроектированных аппаратах концентрация капель может
Рис. 1.27. Сепаратор для
разделения эмульсий.
69
быть уменьшена до 0,1%. Поэтому электрические методы разрушения
эмульсии становятся распространенными. Обзор этих методов дал
Уотерман (1965).
Нагревание также используют для разрушения эмульсий. Многие
эмульсии можно разделить на составляющие их компоненты простым
нагреванием до высокой температуры с последующим отстаиванием.
Вероятно, нагревание ускоряет химические реакций, которые могут
протекать в эмульсиях, изменяет природу поверхностного слоя,
уменьшает вязкость. Таким образом, возникают условия,
благоприятные для протекания процесса распада эмульсии.
Масло
Коалесцениия
мелких капель
-^-*-4££5g
Коалесценция
крупных капель'
t- +■ +
+ + -h -Ь + +-
+ t- -+-
++>:
+ +
szi
Энульсия —r^_
VI
+-
-I
*4LI>t——]—•
Ж+У-
. Вода
Рис. 1.28. Схема аппарата для разделения эмульсий
электрическим методом.
Иногда для сепарации фаз используют фильтрацию, если капли
имеют большую вязкость и ведут себя как жесткие тела. Кроме того,
как полагают, взвешенные в эмульсии капли взаимодействуют с
веществом фильтра, вследствие этого изменяется поверхностный слой
и протекает коалесценция. Для этого метода характерна частая
смена фильтров, что является его недостатком.
В промышленности применяют все эти методы в различных
сочетаниях.
ЛИТЕРАТУРА
Allan R. S., Mason S. G., Proc. Roy Soc, A267, 45 (1962).
Atkinson E., Freshwater D. C, Br. Chem. Eng., 3, 554 (1958).
Beal H. M., Skauen D. M-, J. Am. Pharm. Assoc, 44, 487 (1955).
В е с h e r P. Emulsions: Theory and Practice, New York, 1965.
Benjamin Т. В., U г s e 1 1 F.. Proc. Roy. Soc, A225, 505 (1954).
В е г k m a n S., E g 1 о f f G., Emulsions and Foams, New York, 1941.
В о n d у С., S 6 1 1 n e г К., Trans. Faraday Soc, 31, 835 (1935).
Briggs T. R., J. Phys. Chem., 24, 120 (1920).
Brown В., Goodman J. E., High Intensity Ultrasonics, London, 1965.
Chadrasekhar S., Hidrodynamic and Hidromagnetic Stability, Oxford,
1961.
70
Christiansen R. M., Hixson A. N., Ind. Chem. Eng., 49, 1017
(1957).
С 1 ay ton W.,_ Theory of Emulsions and their Technical Treatment, ed. by
* С (j. Sumner, London, 1954.
Cobb R. M. K., in «Emulsion Technology», ed. by H. Bennett, New York,
1946, p. 7.
Crawford A. E., Ultrasonic Engineering, London, 1955.
D a v i e s J. Т., Н а у d о n D. A., in «Proceedings of the Second
International Congress of Surface Activiti», ed. by J. H. Schulman, vol. 1, London,
1957, p. 417.
Da vies J. Т., R ideal E. K., Interfacial Phenomena, London — New
York, 1961.
D orey R., in «Emulsion Technology», ed. by H. Bennett, New York, 1946,
p. 119.
D г о z i n V. G., J. Colloid Sci., 10, 158 (1955).
Epstein В., J. Franklin Inst., 244, 471 (1947).
Epstein В., Ind. Eng. Chem., 40, 2289 (1948).
F о g 1 e r H. S., T i m m e r h a u s K. D., J. Acoust. Soc. Am., 39, 515 (1966).
Fraser R. P. et al., Am. Inst. Chem. Eng. J., 8, 672 (1962).
Frederick J. R., Ultrasonic Engineering, New York, 1965.
G о p a 1 E. S. R., J. Indian Inst. Sci., 40A, 152 (1958).
G о p a 1 E. S. R., Kolloid Z., 162, 85 (1959 a).
G о p a 1 E. S. R., Kolloid. Z., 164, 1 (1959 b).
Gopal E. S. R., Kolloid. Z., 167, 17 (1959 c).
G о p a 1 E. S. R., Curr. Sci., 28, 392 (1959 d).
Gopal E. S. R., Kolloid. Z., 175, 126 (1961).
Gopal E. S. R., in «Rheology of Emulsions», ed. by P. Sherman, Oxford,
1963, p. 15.
Griffin W. C. in «Encyclopedia of Chemical Technology», ed. by R. E. Kirk
and D. F. Ofhmer, vol. 5, New York, 1950, p. 706.
Gurwitsch L., Wissenschaftliche Grundlagen der Erdolbearbeitung,
Berlin, 1913.
Hartung H. A., Rice С. К., J. Colloid Sci., 10, 436 (1955).
H а у d о n D. A., Proc. Roy Soc, A243, 483 (1958).
Hay worth С. В., Treybal R. E., Ind. Eng. Chem., 42, 1174 (1950).
Hendricks С D., J. Colloid Sci., 17, 249 (1962).
H i n e s R. L., J. Appl. Phys., 37, 2730 (1966).
H i n z e J. O., Am. Inst. Chem. Eng. J., 1, 289 (1955).
H u e t e г Т. F., Bolt R. H., Sonics, New York, 1955.
H u t t i g G. F., S t a d 1 e r H., Monatsh., 88, 150 (1957).
Irani R. R.. J. Phys. Chem., 63, 1603 (1959).
J a n о v s к v W., P о h 1 m a n R., Z. angew. Phys., 1, 222 (1948).
Jehnson С Nature, 197, 1092 (1963).
Jiirgen-Lohmann L., Kolloid Z., 124, 77 (1951).
К a m i n s к i A., M с В a i n J. W., Proc. Roy Soc, A198, 447 (1949).
KrishnanR. S., V e n к a t a s u b r a m a n i a n V.-S., GopalE.S.R.,
J. Colloid Sci., 16, 41 (1961).
Lamb H., Hydrodynamics, New York, 1945.
Landau J., Prochazka J., Coll. Czech. Chem. Comm., 29, 1866 (1964).
L a n d i s D. M., Chem. Eng. Progress, 61, (10), 58 (1965).
Lang R. L., J. Acoust. Soc. Am., 34, 6 (1962).
L i n С. С, Theory of Hydrodynamic Stability, Cambridge, 1955.
L i n d e H., S e h г t В., Uber. dt. Akad. Wiss. Berl., 7, 341 (1965).
Manfield W. W., Austr. J. Sci. Res., 5A, 331 (1952).
Marinesco N., Chim. Ind., 55, 87 (1946).
M с В a i n J. W., Woo Т., Proc. Roy Soc, A163, 182 (1937).
M с D о n о u g h J. A., T о m m e W. J., H о 1 1 CD., Am. Inst. Chem. Eng.
J.. 6, 615 (1960).
Miesse С. С, Ind. Eng. Chem., 47, 1690 (1955).
Monk G. W., J. Appl. Phys., 23, 288 (1952).
71
М о n s о n L. Т., S t e n z e 1 R. W.. in. «Colloid Chemistry», ed. by J.
Alexander, vol. 6, New York, 1946, p. 535.
Mugele R. A., Am. Inst. Chem. Eng. J., 6, 3 (1960).
N a w a b M. A.. Mason S. G., J. Colloid Sci., 13. 179 (1958).
N а у у а г N. К., Murthy G. S., Proc. Natl Inst. Sci. India, 25A, 373 (1959).
Noltigk B. E., Neppiras E. A.. Proc. Phys. Soc. (London), 63B.
674 (1950).
Noltigk B. E.. Neppiras E. A., Proc. Phvs. Soc. (London), 64B, 1032
(1951).
O- Rons к v С. Т.. Gmitber R. L., J. Colloid Sci., 10. 563 (1955).
O'Ronski СТ.. T h а с h e r H. C, J. Phys. Chem., 57. 955 (1953).
Ostiach S.. К о с s t e 1 A., Am. Inst. Chem. Eng. J.. 11. 294 (1965).
P e s к i n R. L., R а с о R. J., J. Acoust. Soc. Am.. 35, 1378 (1963).
Raschevskv N. V., Z. Phys., 46, 568; 48, 513 (1928).
Ravleigh, Proc. Lond. Math. Soc, 10, 4 (1878).
Ravleigh, Phil. Mag., 14, 184 (1882).
Ravleigh, Phil. Mag.. 34. 145 (1892).
Rodger W. A., Trice V. G., Rush ton J. H., Chem. Eng. Progr.,
52, 515 (1956).
Rodriguez F., G г о t z L. C, E n g 1 e D. L., Am. Inst. Chem. Eng.
J., 7. 663 (1961).
Rumscheidt F. D.. Mason S. G., J. Colloid Sci., 16, 238 (1961).
Hiimscheidt F. D., Mason S. G., J. Colloid Sci.. 17, 260 (1962).
Rusliton J. H.. J. Am. Oil Chem. Soc. 33, 598 (1956).
R у с e S. A.. J. Colloid Sci., 19, 490 (1964).
Schulman J. H., Cockbain E. G-, Trans. Faraday Soc, 36, 661
(1940).
Scott L. S.. Hayes W. В., Holland С D., Am. Inst. Chem. Eng..
4. 346 (1958). '
S с riven L. E., S torn ling С V., Nature (London), 187. 186 (1960).
Scriven L. E., Stcrnling С V.. J. Fluid Mech., 19. 321 (1964).
Shinnar R., J. Fluid Mech., 10, 259 (1961).
Shinnar R., Church J. M., Ind. Eng. Chem., 52, 253 (1960).
S i e m e s W., К a u i I m a n n J. F., Chem. Ing. Techn., 29, 32 (1957).
Sinclair D.. La Мог V. К., Chem. Rev., 44, 245 (1949).
Slciclitr С A., Am. Inst. Eng. J., 8, 471 (1962).
Smith K. A.. J. Fluid Mech., 24, 401 (1966).
Smith J. E., Jordan M. L., J. Colloid Sci., 19, 549 (1964).
Sol 1 n e r K... in «Colloid Chemistry», cd. by J. Alexander, vol. 5, New. York,
1944, p. 337.
S t а с к с 1 b e r g M. V., Klockner E., Mohrhaucr P., Kolloid Z.,
115, 53 (1949).
Stamm A. I.. К г а о m e r E. O., J. Phys. Chem., 30, 992 (1926).
Stamm K.. Chem. Abstr., 64, 15017b (1966).
Stei dl H., Coll. Czech. Chem. Comm., 25, 515 (1960).
Sterling С V., Scriven L. E., Am. Inst. Chem. Eng. I., 5, 514
(1959).
Subramanvam S. V.. G о p a 1 E. S. R., Kolloid Z. u. Z. Poly-
mere, 210, 80 (1966).
Sullivan D. M., Lindsev E. E., Ind. Eng. Chem. Fundamentals,
1, 87 (1962).
Sumner С G., J. Phys. Chem., 37, 279 (1933).
Taylor G. I., Proc Roy. Soc, A146, 501 (1934).
T о m о t i к a S., Proc Roy. Soc. A150. 322 (1935).
T о m о t i к a S., Proc Roy. Soc, A153, 302 (1936).
V e г m e u 1 e n Т., Williams G. M., L a n g 1 о i s G. F., Chem. Eng.
Progr., 51, 85F (1955).
Vonnegut В., Neubauer R. L., J. Coll. Sci., 7, 616 (1952).
V о n n e g u t В.. Neubauer R. L.. J. Coll. Sci., 8, 551 (1953).
W а с h t e 1 R. E., La M e г V. К., J. Coll. Sci., 17, 531 (1962).
72
Waterman L. С, Chem. Eng. Progr., 61 (10), 51 (1965).
Weber C, Z. angew. Math. Mech., 11, 136 (1931).
Wood R. W., LoomisA.L, Phil. Mag., 4, 417 (1927).
Woodman R. M., J. Pomol., 4, 95 (1925).
Гершензон Э. Л., Экнадпосянц О. К., Акустич. ж., 10, вып. 2,
156 (1965).
Колмогоров А. Н., ДАН СССР, 30, 301; 31, 538; 32, 16 (1941 а).
Колмогоров А. Н., ДАН СССР, 31, 99 (1941 б).
Колмогоров А. Н., ДАН СССР, 66, 825 (1949).
К ремне в Л. Я., Равдель А. А., ДАН СССР, 90, 405 (1953).
К р е м н е в Л. Я., Равдель А. А., Коллоидн. ж., 16, № 1, 17 (1954).
Н е д у ж и и С. А., Акустич. ж., 8, вып. 4, 481 (1962).
Недужий С. А., Акустич. ж., 10, вып. 4, 456 (1964).
Розенберг Л. Д., Экнадпосянц О. К., Акустич. ж., 6, вып. 3,
370 (1961).
С и р о т ю к М. Г., Акустич. ж.. 8, вып. 2, 216 (1963).
Сорокин В. И., Акустич. ж., 3, вып. 3, 262 (1957).
П а в л ушен ко И. С, ЯнпшевскийА. В-,ЖПХ,31, № 8, 1215 (1959).
Фукс Н. А., Сутугин А. Г., Усп. хим., 34, 116 (1965).
Экнадпосянц О. К., Акустич. ж., 9, вып. 2, 247 (1963).
Дополнительная литература
Bernard P., Fr. pat., 1498294, 1967. Процесс приготовления стабильных
эмульсий или дисперсий.
В е с h е г P., J. Coll. Interf. Sci., 24, № 1, 91 (1967). Влияние перемешивания
на распределение капель эмульсии по размерам.
В е с h e г P., Nonionic Surfactants, 1967. Эмульгирование (неионными ПАВ).
J a i n S. P.. Sri v ast av a S. N., Bull. Chem. Soc Japan, 42, №5,1171 (1969).
Механизм эмульгирования твердыми эмульгаторами (гидроокисью железа).
LangeJ., J ochinke Н., Fette, Seifen, Anstrichm., 69, № 10, 718 (1967).
Влияние условий эмульгирования на качество анионных эмульсий парафина.
L i e t г G., Parfum Kosmet., 48, № 2, 29 (1967). Условия образования и
существования эмульсий В/М, особенно в косметических композициях.
Mukerjee L. N., Shukla S. D., Indian J. Appl. Chem., 30, № 1—2.
17 (1967). Роль поверхностного натяжения в приготовлении и
устойчивости эмульсий, стабилизированных гидрофильными коллоидами.
Mueller R., Canad. Text. J., 84, № 16, 34 (1967). Основные принципы
диспергирования систем.
R e h f е 1 d S. J., J. Coll. Interf. Sci., 24, № 3, 358 (1967). Влияние
начальной концентрации ПАВ и времени эмульгирования на величину капель
и распределение эмульспп бензола в воде.
Rohbins M. L., Schulman J. H., Chem. Abstr., 68, №20, 90210
(1968). Микроэмульспи в жидких углеводородах.
Романова 3. Т., Б а р а м б о й м Н. К. Коллоидн. ж., 31, № 4, 573
С1969). Изменение поверхностного натяжения в процессе образования
и смешивания эмульсий п суспензий.
Saphir J., S a phi г Н., Seifen-Ole-Fette-Wachse, 95, № 5, 143 (1965).
Проблемы эмульгирования и гомогенизации.
Schonauer G., Koll. Z. u. Z. Polymer., 233, №1—2, 908 (1969).
Электронномпкроскопнческое определение размера капелек в молоке
и других эмульсиях методом отпечатков.
Shotton Е., Davis S. S., J. Pharm. Pharmacol., 20, № 6, 430 (1968).
Нспользоваппо счетчика Коултера для анализа эмульсионных систем.
Soichi Hayashi, Noriko Sasaki, J. Japan Oil Chem. Soc, 16,
№ 8, 467 (1967). Соотношение между межфазной вязкостью п
способностью к эмульгированию.
Soichi Hayashi, Noriko Sasaki, J. Japan Oil Chem. Soc, 16,
№ 10, 554 (1967). Требование ГЛБ, межфазной вязкости и параметров
растворимости для эмульгирования.
73
Snbramangan S. V., G о p a 1 E. S. R., Indian .1. Technol., 5, Л» 5,
139 (1967). Самоэмулынрованпе, обусловленное локальным изменением
межфазного натяжения.
Т о k i w a F., Imamura Т., J. Am. Oil Chem. Soc, 47, № 4, 117 (1970).
Эмульгирующее действие различных типов неорганических электролитов.
Wachtel E., Diss. Abstr. В., № 10, 3424; Chem. Abstr., 67, № 18, 85248
(1967). Приготовление монодисперсных эмульсии.
W а 1 s t г а Р., О о г t a r j u H., J. Coll. Interf. Sci., 29, Л» 3, 424 (1969).
Определение дисперсионного состава с помощью счетчика Коултера.
А в е р б у х Ю. И., II а в л у ш е н к о И. С, Костин Н. М., ЖПХ,
42, № 9, 1085 (1969). Определение межфазной поверхности при
механическом перемешивании несмачпвающихся жидкостей.
А в е р б у х Ю. П., П а в л у ш е н к о И. С, Костин Н. М., ЖПХ,
43, № 9, 2005 (1970). Изменение дисперсионного состава неустойчивых
эмульсий после перемешивания в аппаратах с мешалкой.
А н и с и м о в Б. Ф., Е м е л ь я н ч е н к о В. Г., М а д а н е н к о -В. П.,
Труды Института химии нефти и природных солей, Л» 2, 1970, стр. 13.
Деформация и диспергирование частиц дисперсной фазы обратимых
эмульсий в однородном электрическом поле.
Бернштейн А. В.. Самопроизвольное эмульгирование битумов, Изд.
«Наукова думка», Киев, 1969.
Бернштейн А. В., К уйма М. II., Зельдин И. М., Коллопдн.
ж.. 31, Л; 3, 330 (1969). Влияние высших жирных кислот на
эмульгирование битумов.
Залкнп В. М., Коллопдн. ж., 32, № 4, 521 (1970). Жидкие эвтектики—
самопроизвольно образующиеся двухфазные дисперсные системы.
Каган С. 3., Ковалев Ю. Н., Молочко в а М. И., ТОХТ, 3,
•N» 5, 728 (1969). Исследование дробления капель в аппарате с мешалкой
в отсутствие коалесцепцпп.
Кокорев Д. Т., Ю д а е в В. Ф., сб. «Проблемы тепло-массопереноса»,
Изд. «Энергия», 1970, стр. 136. К частотной зависимости дисперсности
эмульсий, получаемой с помощью ультразвуковых аппаратов.
К о р е ц к и й А. Ф.', Глаголев Г. М., Коллопдн. ж., 32, № 4, 625 (1970).
К механизму образования природных нефтяных эмульсий.
М а в л ю т о в а М. 3., А с ф а г а н И. И., Нефтепром. дело, № 4, 35 (1969).
Эффективность использования деэмульгаторов как функция температуры
и минерализации водной фазы.
М о ч а л о в а О. С, Никитина С. А., Коллопдн. ж., 30, Л"» 2, 264
(1968). Квазнспонтанное эмульгирование на поверхности двух жидкостей
в присутствии ПАВ.
Островский М. В.. Бареибаум P. X-, Абрам зон А. А.,
Коллопдн. ж., 32, ЛЬ 4, 565 (1970) Условие возникновения эмульсин при
массопереносе.
П е б а л к В. JI-, ЗапковскаяБ. П., 3 а и к о в с к и й А. В., ТОХТ,
5, Л° 1, 69 (1971). Полпдпсперсность эмульсий и размеры капель.
Русанов А. И., Щ у к п н Е Д., Р е б п н д е р П. А., Коллоидн. ж.,
30, № 4, 573 (1968). К теории диспергирования (сообщение I).
Термодинамика монодпеперсных систем.
Симакова Г. А., Чалых А. Е., Никитина С. А., Коллоидн.
ж., 32, № 6, 899 (1970). Исследование массопереноса в системе вода —
ксилол.
Т е л е с м и н А. Б., Труды Таганрогского радиотехнического пн-та, вып. 21,
1969, стр. 250. Принципы эмульгирования на гидродинамических
излучателях.
Шелудко А. Д., Годпшн. Софийск. ун-та, хим. фак., Л° 62, 93 (1967).
О механизме эмульгирования.
Щукин Е. Д., Федосеева Н. П.. Кочанова JI. А., Ребпн-
д е р П. А.. ДАН СССР, 189, Л» 1, 123 (1969). Об условиях образования
стабпльных эмульсий в системе парафин — оксихпнолпн вблизи
критической области смешения.
Г л а в"а [II
ТЕОРИЯ
СТАБИЛЬНОСТИ
ЭМУЛЬСИЙ
Дж. А. Китченер
и П. Р. Масселъвайт
ПОВЕРХНОСТЬ РАЗДЕЛА ФАЗ
И СТАБИЛЬНОСТЬ
ЭМУЛЬСИИ
С термодинамической точки
зрения эмульсия есть двухфазная
система с дисперсной фазой,
содержащей микроскопические капли
диаметром 0,1—100 мкм. Такие
дисперсии никогда не являются
полностью устойчивыми из-за того,
что поверхность раздела между
фазами обладает свободной энергией;
при соединении двух капель
происходит уменьшение межфазной
поверхности. Следовательно, коалес-
ценция капель — это
самопроизвольный процесс, в то время как
эмульгирование требует затраты работы.
Самопроизвольное эмульгирование
наблюдается только в определенных
системах, где две фазы
предварительно взаимно ненасыщенны.
Работа, необходимая для увеличения
межфазной поверхности, черпается
из свободной энергии смешения за
счет массопереноса (см. гл. I).
Истинно стабильные растворы,
содержащие коллоидные мицеллы, не
должны классифицироваться как
эмульсии, так как они не имеют
термодинамической фазы, которая может
существовать отдельно.
При рассмотрении
неустойчивости дисперсных систем необходимо
делать различие между коалесцен-
цией, седиментацией и флокуляцией
(коагуляцией) (см. стр. 66).
Известно, что для приготовления
стабильной эмульсии с
определенной концентрацией дисперсной
фазы необходимо добавить третий
компонент — стабилизатор. Можно
различить четыре класса
стабилизирующих агентов. Наименее
эффективными являются простые
неорганические электролиты.
Например, при добавлении тиоцианата
75
калия (KCNS) к воде в небольшой концентрации можно получить
временную разбавленную эмульсию М/В, называемую гидрозолем
масла. Ее высокая устойчивость по сравнению с чистой взвесью М/В
моя;ет быть объяснена возникновением двойных электрических слоев
на водной стороне межфазной поверхности, которые образуются
вследствие предпочтительной адсорбции одного типа ионов. Например,
SCN~ дает малый отрицательный потенциал на межфазной
поверхности (Ч измен и Кинг, 1940). Действительная плотность
поверхностного заряда, вероятно, мала, так как адсорбция слабая. Такая
стабилизация может быть объяснена действием сил отталкивания
между электрическими двойными слоями, приводящим к уменьшению
скорости столкновения капель (см. ниже). Однако этот тип
стабилизатора слишком слаб для получения эмульсии нужной
концентрации и с достаточным временем «жизни».
Следующий класс — высоко поверхностно-активные соединения
(мыла и детергенты). Это соединения, содержащие одну или более
гидрофильную группу (анионную, катионную или неионную) и одну
или более гидрофобную группу (алкильнып или арильный
углеводород, фторзамещенный углеводород и т. д.). Такие дифильные
молекулы сильно адсорбируются на межфазной поверхности;
с их помощью могут быть приготовлены довольно устойчивые
эмульсии.
Еще большая стабильность может быть получена при
использовании высокомолекулярных соединений: протеинов, каучука, смолы,
резины, крахмала и других полисахаридов (например, декстрин,
метнлцеллюлоза, лигносульфонат) и также синтетических
полимеров (поливиниловый спирт и т. д.). Из-за большого количества
гидрофильных и гидрофобных групп каждая молекула
адсорбируется па поверхности во многих точках и поэтому прочно
удерживается.
Четвертый класс эмульгирующих агентов составляют тонкоиз-
мельченные нерастворимые порошки. Под микроскопом можно
увидеть, что частицы образуют монослой, который обволакивает каплю.
Условия для получения таких оболочек очень строгие и определяются
химической природой частиц, а не их химическим составом.
Невозможно охарактеризовать устойчивость эмульсии простым
методом, применимым при всех обстоятельствах. Известно, что
скорости расслоения разных типов эмульсий сильно отличаются.
Например, нестабилизированные масляные гидрозоли расслаиваются
в течение секунд или минут. Процесс разрушения происходит в
результате столкновения капель под действием броуновского движения,
седиментации, случайных конвективных токов. Все это приводит
к коалесцепции. При слабом перемешивании процесс ускоряется,
так как малые и большие капли двигаются с различными скоростями
и поэтому чаще сталкиваются. Этот процесс называется ортокинети-
ческой флокуляцией. При интенсивном перемешивании большие
капли растягиваются и делятся. Если вести перемешивание в
течение достаточно долгого времени с умеренной интенсивностью, то
7(1
можно достичь динамического равновесного состояния
распределения капель по размеру.
Аналогичные явления наблюдаются при облучении эмульсий
ультразвуком: волны сжатия заставляют капли сталкиваться, что
способствует их разрыву.
Таким образом, на устойчивость масляного гидрозоля будет
влиять его концентрация, распределение капель по размеру и
условия разрушения.
Стабилизированные эмульсии устойчивы в течение часов, дней
или месяцев. Эмульгирующий агент образует в этом случае
молекулярный барьер между жидкостями. Этот барьер способен
противостоять давлению или динамического рода (когда концентрированная
эмульсия перемешивается, капли моментально соединяются вместе
вследствие инерционных эффектов) или статического (как при
образовании сливок).
Центрифугирование эмульсии или частичное испарение
дисперсионной среды способствует уплотнению капель; в результате
образуются полиэдры. Так как полиэдр имеет большую площадь
поверхности, чем сфера объема, этот процесс сопровождается растяжением
слоя эмульгирующего агента и может привести к его разрыву (Волд
и Грут, 1962, 1963, 1964; Рефельд, 1962; Гаррет, 1962). Последнее
вызывает коалесценцию капель у некоторых эмульсий, которые,
в общем, расслаиваются медленно.
При длительном хранении на устойчивость эмульсий могут влиять
и другие факторы, например, переход мелких капель в большие
посредством диффузии *. Различие химического потенциала между
жидкостью внутри большой и малой капель возникает в связи с тем,
что лапласовское давление внутри капли обратно пропорционально
радиусу (Ар = 2а/г). В результате влияние радиуса на
растворимость выражается аналогично уравнению Кельвина
, Lx ^ aV i 1 1_\
П La RT \п' г2 )
где Li и L2 — растворимость частиц радиуса гх иг2;о — межфазное
натяжение; V ' — молярный объем жидкости внутри капли.
Процесс сокращения и окончательного исчезновения наименьших
пузырьков наблюдается в пенах. В эмульсиях он менее очевиден,
во-первых, потому, что растворимость дисперсной фазы очень низка
(например, 0,01% для парафина в воде) и, следовательно, скорость
диффузии от малых капель к большим незначительна; во-вторых,
экспериментально трудно наблюдать очень малые капли. (Между
прочим, информация об образовании, распределении по размерам
* Этот вопрос требует дальнейшего изучения, так как в ряде случаев
исследователи наблюдали, что при разрушении эмульсий остаются как раз мелкие
капли [А. В. Бромберг, Коллоид, ж., 8, 117 (1946)]. (Прим. редактора
перевода.)
77
и исчезновении субмикронных капель в эмульсиях недостаточна
в основном из-за отсутствия совершенной измерительной
аппаратуры).
Другие факторы, ограничивающие продолжительность «жизни»
стабилизированных эмульсий — бактериальное действие и
замораживание. В процессе замораживания зарождаются кристаллы льда
и затем растут, захватывая воду. Масляные капли (если эмульсия
М/В) сжимаются. Кроме того, любая растворенная соль в отдельных
участках эмульсии становится высококонцентрированной и, вероятно,
кристаллизуется. Не удивительно, что оболочки, которые
предотвращают коалесценцию капель, разрываются. Противостоят
замораживанию только эмульсии, имеющие жесткую оболочку вокруг капель,
например, молочные сливки, но даже и они являются неустойчивыми
при продолжительном хранении в условиях низкой температуры.
Каждый тип эмульсии должен иметь свои особенности, зависящие
от присутствия солей, коллоидов и неэлектролитов (например,
сахар), от их взаимодействия при кристаллизации независимо от того,
было ли охлаждение медленным или быстрым и было ли
кристаллизовано само дисперсное масло. Подробные обзоры по устойчивости
эмульсий даны Лангом (1965), Люкассен — Рейндерс (1964), Ван
Вурст Вадером (1964) и Дэвисом (1964).
Поверхностные силы в жидких пленках
Когда в жидкой среде две капли приближаются друг к другу
(например, под действием сил тяжести), поведение системы
определяется взаимодействием гидродинамических и поверхностных сил.
Гидродинамические силы вызывают вязкую текучесть жидкой среды
между каплями и искажение формы капель вследствие давления,
возникающего между ними. Радиальное движение жидкости между
каплями способствует циркуляции жидкости внутри каждой капли.
Искажение формы капель сдерживается поверхностным натяжением,
так как любое отклонение от сферической формы приводит к
увеличению поверхности капли (Чэпелир, 1961).
Механизм сближения двух капель в жидкой среде или подхода
капли к межфазной поверхности жидкость — жидкость был
рассмотрен многими исследователями (Чэпелир, 1961; Чарльз и Масон,
1960а, 1960b; Аллен и Масон, 1961, 1962; Барток и Масон, 1959;
Румшепдт и Масон, 1961; Манлей и Масон, 1952; Принсен и Масон,
1965; Маккей и Масон, 1963).
Для относительно больших капель, когда центробежные и
поверхностные силы являются сравнимыми, наблюдается растекание
капли. При этом жидкость временно попадает в «ловушку» около
линии сближения капель, образованную впадинами (рис. П.1, а).
(Гидродинамические аспекты образования «впадины» рассмотрены
теоретически Линдбладом, 1964; Платикановым, 1964; Френкелем
и Майзельсом, 1962). Если капли не будут защищены эмульгирующим
агентом, будет происходить коалесценция.
78
В малых каплях (или больших, сближающихся очень медленно)
впадины не образуются. Поверхности капель неизбежно сближаются
по мере того, как среда между ними вытесняется, но остаются
выпуклыми (рис. II.1, б). Таким образом, место ближайшего подхода
поверхностей двух капель лежит на линии центров и в той точке,
где находящаяся между каплями жидкость вытечет, в незащищенной
системе будет происходить коалесценция.
Проведены исследования по определению времени коалесценции
двух соприкасающихся капель или капли со слоем жидкости (Джил-
леспи и Райдил, 1956; Кокбейн и Мак Роберте, 1953; Зоннтаг и Кларе,
1963). Данные, полученные с
обычными «чистыми» жидкостями,
вызывают сомнение, так как очень
вероятно, что присутствие следов
примесей ПАВ будет иметь
некоторое влияние на время
коалесценции. Кроме того, время
последующих коалесценции капель сильно
различалось. Это говорит о том,
что слияние капель в значитель- Рис. II.1. Схема сближения капель:
НОЙ Степени определялось неКО- а. — образование впадины; б — искажение
Фбез впадины; в — образование плоской
а КТО- пленки.
ром (возможно, случайными
вибрациями), а не регулярным утончением жидкой пленки под
действием силы тяжести.
Введение ПАВ сильно увеличивает время коалесценции.
Между каплями образуется тонкая жидкая пленка (см. рис. 11,1, в).
Такие пленки можно наблюдать визуально, если большие капли
масла осторожно соединить в концентрированном растворе мыла;
капли, заключенные в почти невидимые пленки раствора мыла,
становятся полиэдрическими, как и в пене.
Образование пленок между масляными каплями показывает,
что действие поверхностных сил, препятствующих слиянию капель,
для параллельного слоя жидкости никогда не может возникнуть
просто из гидродинамических сил и инвариантного поверхностного
натяжения. По аналогии с подобной системой газ — жидкость, для
которой имеются более полные данные, можно уверенно
предположить, что следует различать два типа жидких пленок,
соответствующих неустойчивой и стойкой пенам (Китченер и Купер, 1959).
Неустойчивая пленка — это такая, в которой поверхностные силы
достаточны, чтобы образовать толстую пленку в динамическом
состоянии, но она не способна выдержать равновесное давление в
статическом состоянии.
Пленка может быть образована, если она обладает упругостью.
Так, для растворов ПАВ характерна поверхностная упругость Гиббса
и Марангони. Однако она не обеспечивает сохранения равновесной
толщины жидкой пленки в случае преобладания гидростатических сил,
которые всегда стремятся сделать пленку тоньше. Гиббс указывал
79
на существование другой, еще неизвестной, силы, действием
которой можно объяснить, например, высокую устойчивость мыльных
пленок.
Мыльные пленки могут быть сохранены в течение многих месяцев
при условии, что они являются горизонтально расположенными
и тщательно защищены от пыли и внешних помех. Большую-жидкую
пленку типа М/В/М получить трудно преимущественно из-за того,
что помехи значительно сильнее передаются через жидкость, чем
через пузырьки газа. Пленки с малой поверхностью легко образуются
и ведут себя аналогично мыльным пленкам между воздушными
пузырьками — становясь все тоньше, они дают интерференцию света
и окончательно превращаются в «черные» пленки. То же самое
применимо к пленкам типа В/М/В, толщина которых может достигать
нескольких миллиметров. В этом случае применяют маслораствори-
мые ПАВ. Тонкая пленка, стабилизированная лицитином, изучена
рядом исследователей в качестве модели основной оболочки
в биологических ячейках (Ханг и Томпсон, 1965; Ханг и др., 1964;
Ханаи и др., 1964; ван ден Берг, 1965; Хайдон и Тейлор, 1966).
При рассмотрении сил взаимодействия между коллоидными
частицами в иенах или эмульсиях удобно использовать элементарную
модель, введенную Дерягиным. Согласно модели, взаимодействие
возникает при наличии дополнительной силы или расклинивающего
давления, направленного под прямым углом к плоскости жидкой
пленки. Эта сила является поверхностной силой второго рода в
отличии от поверхностного натяжения, которое действует вдоль плоскости
раздела фаз и называется поверхностной силой первого рода. Такая
трактовка не единственная, но она удобна по отношению к удельной
поверхностной энергии как переменной величине, зависящей от
свойств системы (Дерягин и Щербаков, 1961).
Таким образом, выражение для общей удельной поверхностной
энергии тонкой плоско-параллельной пленки жидкости будет иметь
вид
(п+ \ Л dh J
\ со
где л — расклинивающее давление; h — толщина пленки.
Работа соединения поверхностей — малая величина, сравнимая
с работой образования поверхности раздела.
Хотя понятие расклинивающего давления введено для объяснения
устойчивости коллоидных систем, последние работы показали, что
должна учитываться возможность появления как сил притяжения,
так и сил отталкивания — другими словами я может быть
отрицательной величиной. Дэвиси Райдил (1961) утверждали, что имеется мало
данных о существовании сил притяжения между каплями эмульсии.
Однако, тогда универсальную тенденцию к флокуляции следует
приписать адгезии при контактировании. Но коллоидные свойства
эмульсий зависят от тех же факторов, что и свойства золей и тонких
80
жидких пленок, а для последних имеется множество данных о
наличии сил притяжения *.
Например:
1. Кинетические и равновесные измерения на свободных жидких
пленках (Китченер, 1964 и Шелудко, 1966).
2. Существование тактоидов и слоев Шиллера, а также обратимо
набухающих монтмориллонитовых глин. В работах Хачису и Фуру-
сава (1962, 1963, 1966) доказано наличие сил притяжения между
микроскопическими пластинками трехокиси вольфрама. Получена
цветовая интерференция между временно ассоциированными парами
пластинок. При этом цвета изменялись с содержанием электролита
в растворе именно в том направлении, как ожидалось согласно
современной теории, по которой существует равновесие между силами
притяжения и отталкивания.
3. Несколько исследователей прямо измерили дальнедейству-
ющие силы Ван-дер-Ваальса между отшлифованными пластинками.
Их результаты согласуются в пределах ошибки опыта с теоретически
вычисленными. Это показывает, что теория Лондона является
надежной для очень больших расстояний между макроскопическими телами
(0,1 — 1 мкм). При очень малых расстояниях величина энергии коге-
зии неполярных жидкостей хорошо согласуется со значением,
вычисленным на основе теории Лондона для случая притяжения между
молекулами, находящимися в тесном контакте. Поэтому нельзя
считать, что эта теория, справедливая для больших и малых
расстояний, не будет применима для промежуточных расстояний (например,
100 —1000 А), используемых в теории коллоидной стабильности, где
прямые измерения невозможны.
4. Исследователи наблюдали под микроскопом самопроизвольное
сжатие капель с выдавливанием дисперсионной среды и образованием
гексагональных поверхностей капель **. Так как внешняя сила не
применялась и коалесценция не происходила, флокулированные
осадки очень легко перестраивались при малейшем сотрясении
образца. Это были относительно большие капли (40 мкм в диаметре)
с низким межфазпым натяжением (Nujol, диспергированный с
неионным ПАВ, Lissapol N), что способствовало наблюдению за
расплющиванием поверхностей взаимнопритягивающихся капель.
Доказательством наличия сил притяжения между
макроскопическими телами служит неустойчивость тонкой жидкой пленки. Она
подвергается действию отрицательного расклинивающего давления,
* Механический перенос результатов с систем Ж — Т и Т — Г на систему
Ж—Ж далеко не всегда правомочен и требует подтверждения. В эмульсионных
системах капли жидкости разделены жидкой же прослойкой, часто имеющей
р.андерваальсовы силы более значительные, чем в дисперсной фазе. Вопрос
о дальнедействующих силах в эмульсионных системах является дискуссионным
и требует экспериментальной и теоретической разработки. Так что скорее правы
Дэвис и Ридел. (Прим. редактора перевода.)
** Эти опыты ие являются доказательными, так как расплющивание
крупных капель происходит под действием сил тяжести. (Прим. редактора
перевода.)
б Закна 1 (iTii
81
которое увеличивается по мере того, как пленка становится тоньше.
Это применимо к свободным пленкам (в пенах) и к жидкости между
малыми каплями. Значение этого фактора для эмульсий впервые
рассмотрено де Врисом (1957, 1958, I960).
Таким образом, при отсутствии защитного механизма утончение
пленки при постоянном внешнем давлении резко ускоряется, когда
толщина пленки достигает радиуса действия сил притяжения.
Разрушение чистых жидких пленок так внезапно, что очень трудно
уловить этот момент даже с помощью высокоскоростной фотографии
(Чарльз н Масон, 1960b). Лишь Шелудко (1962) исследовал толщину,
при которой мгновенно разрушаются пленки пены. Микроскопические
пленки существуют более продолжительное время, чем толстые,
так как последние сильнее подвержены действию внешних помех.
Врий (1966) предложил новое обоснование механизма разрыва
мыльных пленок.
Превосходное экспериментальное исследование динамики
утончения пленок выполнено Зоннтагом, Нетзель и Кларе (1966).
Используя усовершенствованную методику определения микроотра-
ження (для пленки М/В/М) и емкости (для пленки В/М/В), они
успешно измерили отрицательное и положительное расклинивающее
давление для систем вода — циклогексан и вода — октан с толщиной
пленки 10U —1000 А. Для неионных ПАВ с ростом концентрации
КС1 от 10" 3 до 2 н. равновесная толщина водной пленки колебалась
от 870 до 85 А, а устойчивой масляной пленки — от 23 до 48 А. Как
можно было видеть под микроскопом, утончающаяся прослойка
внезапно образует черные пятна (очень тонкие поверхности). Это
явление возникает в связи с тем, что отрицательное давление резко
увеличивается с уменьшением толщины пленки только в случае,
если последняя имеет строго определенный размер. Положительные
расклинивающие силы начинают действовать в очень тонких черных
пленках. Не пспользуя термодинамику расклинивающего давления
для определения источника молекулярных сил, перечислим лишь
силы, действующие в коллоидных системах:
1. Дальнедействующие поверхностные силы
вандерваальсовы силы притяжения между частицами;
силы электрического двойного слоя (отталкивания между
поверхностями одинакового знака, притяжения — для
противоположных знаков).
2. Блпжнедействующие поверхностные силы
химическая связь молекул с поверхностными группами
посредством ионной, ковалентной и водородной
связей;
физическая связь молекул посредством дипольного
взаимодействия и т. д.;
гидрофобная связь — ассоциация двух негидратирован-
ных частиц в водной среде;
борновские силы отталкивания между атомами (в
действительности, жесткость атомов).
82
Тщательное исследование коллоидных систем (включая эмульсии)
нужно начинать с рассмотрения природы химических соединений
на поверхности частиц, так как они оказывают основное влияние
на взаимодействие частиц. Из-за отсутствия аппаратуры, пригодной
для прямого исследования, данные о структуре и составе
поверхностного слоя должны быть получены при изучении адсорбции, £-потен-
циала и т. д. Правда последние работы по ядерно-магнитному
резонансу и спектроскопии дисперсных систем, вероятно, позволят
получить информацию о структуре воды около поверхностей раздела фаз
(Клиффорд и др., 1965; Клиффорд и Петика, 1964, 1965а, 1965b).
Так как большинство эмульсий содержит водную фазу,
специфические структурные свойства воды — ее тенденция к построению
"открытых каркасов с помощью многократных водородных связей —
являются существенным фактором в теории эмульсий. К сожалению,
количественных данных о структуре воды все еще не имеется, Немети
и Шерага (1962а, 1962Ь, 1962с) развили полуэмпирическую теорию,
которую они назвали «гидрофобная связь». «Связь» является,
возможно, неудачным термином, потому что выражение «гидрофобная
связь» означает особый вид взаимодействия между соприкасающимися
группами, тогда как ассоциация вызывается специфическим
связыванием воды с водой. На это обстоятельство давно указал Хартлей
(1936) и в действительности оно было доказано количественным
путем.
Выражение «гидрофобная ассоциация» не содержит этого
неправильного истолкования. Гидрофобная молекулярная группа ведет
себя как постороннее тело в водной среде; ее присутствие
предохраняет молекулы воды, образующие наиболее благоприятные (пусть
даже временные) ассоциационные структуры. Разрушение
структуры, вероятно, начинается на расстоянии нескольких молекулярных
диаметров от гидрофобной группы. Когда две такие группы подходят
друг к другу, происходит значительное структурирование воды.
То же самое наблюдается при сближении двух гидрофобных
поверхностей (например, парафиновые капли). Явление, обратное
гидрофобной ассоциации, называется гидрофильным отталкиванием (сильно
гидратированные молекулярные группы будут отталкиваться).
Одно время химики-коллоидники объясняли большинство
явлений существованием сольватационных оболочек. Однако с
современной точки зрения влияние гидратации не так велико. Даже наиболее
сильные гидрофильные группы, а именно ионы, связывают только
один или два молекулярных слоя воды, в то время как умеренно
гидрофильные группы (такие как —ОН, —СООН, —NH2) просто
соединяются в воде водородными связями. Поэтому гидратированная
поверхность (например, целлюлозы) не оказывает значительного
действия на расстоянии нескольких ангстрем *.
* Известный химик-коллопдник Дерягнн считает, что гидрофильные
поверхности влияют на структуру воды на значительном расстоянии — возможно
несколько сотен ангстрем от межфазной поверхности (Дерягин, 1964).
6*
83
Силы отталкивания обычно объясняют действием электрического
двойного слоя или простым молекулярным стерическим
препятствием при наличии оболочек макромолекулярных веществ.
Поэтому состав поверхностных слоев определяется, по-видимому,
ближнедействующими поверхностными силами. Они, однако, не
должны прямо влиять на устойчивость золей или эмульсий против
коагуляции, так как, если частицы приблизятся на расстояние
нескольких молекулярных диаметров, вандерваальсовы силы
притяжения станут такими большими, что частицы останутся соединенными
независимо от того, слипнутся они в действительности или коалесци-
руют. Обратное явление наблюдается для самопроизвольно
диспергируемых коллоидов, например, глобулярных протеинов; для этих
веществ константа Гамакера (см. стр. 93) очень близка к константе
воды, так что даже тонкий гидратационный слой достаточен, чтобы
удержать молекулы на расстоянии, где энергия притяжения Ван-дер-
Ваальса мала по сравнению с тепловой энергией.
Поверхностная нестабильность и динамика
устойчивости эмульсий,
стабилизированных низкомолекулярнымн ПАВ
Слабое эмульгирование может быть получено с любым ПАВ,
т. е. с любым соединением, которое понижает поверхностное
натяжение между двумя жидкостями. Последнее связано с адсорбцией ПАВ
на межфазной поверхности и влияет как на легкость
диспергирования при получении эмульсии, так и на скорость разрушения жидкой
пленки между каплями. Согласно некоторым взглядам, существенным
фактором стабилизации является эластичность пленки. Ниже
изложена хорошо известная теория этого явления Марангони и Гиббса *.
Рассмотрим сначала равновесие между поверхностью и раствором
ПАВ. Изотерма адсорбции Гиббса дает понижение межфазного
натяжения (— da) в результате добавления ПАВ
— с1а = 1\ПТ dln(rti)
где Г; — величина адсорбции компонента г на единицу площади
поверхности (по отношению к растворителю как неадсорбирующемуся
компоненту); я,- — термодинамическая активность компонента.
* Применение эффекта Марангони — Гиббса к объяснению устойчивости
эмульсий не является плодотворным, так как эффект Марангони возникает
в неравновесных системах, а эмульсии, как правило, системы долгоживущие
и равновесие в них устанавливается в начальные моменты образования.
Эластичность же Гиббса наиболее сильно проявляется при таких концентрациях
ПАВ, при которых оно не является стабилизатором эмульсии. Эффектом
Марангони — Гиббса может быть объяснена устойчивость «толстых» пленок, в то время
как устойчивость эмульсий обусловлена свойствами «тонких» пленок.
Эластичность Гиббса и устойчивость «толстых» пленок исследуется в
последнее время в работах Кротова и Русанова [сб. «Вопросы термодинамики
гетерогенных систем, вып. 1, Изд. ЛГУ им. А. А. Жданова, стр. 157]. (Прим.
редактора перевода.)
84
Имеются три особых случая, когда aL может быть просто связана
с концентрацией. Если ПАВ — неионное соединение в
разбавленном растворе, то at ^q (где с,- — объемная концентрация). Когда
ПАВ является электролитом, молекулы которого диссоциируют
на два иона, а примеси других электролитов содержатся в
незначительном количестве *, d(ln а,) = 2d (In v±c,), где у+ —
коэффициент активности иона электролита при высокой
концентрации.
Наконец, для слабого раствора ПАВ, добавленного к относительно
более концентрированному раствору поверхностно-неактивного
электролита (например, NaCl), с? (In a,) = d (In y±ct), где ум —
коэффициент активности ПАВ в этой среде.
Если растворы ПАВ разбавленные, коэффициенты активности
могут быть вычислены по теории Дебая — Хюккеля. При
концентрации ПАВ выше ККМ (критическая концентрация мицелообразова-
ния) практически невозможно применить теорию Гиббса и обычно
предполагают, что адсорбция в данном случае равна адсорбции ПАВ
при концентрации ниже ККМ. Однако это не всегда правильно
(Салиб и Китченер, 1964).
Правильность теории Гиббса проверена путем прямого
определения величины Г; и сравнением полученных результатов со значением,
вычисленным при измерении поверхностного натяжения (Дэвис
и Райдил, 1961).
Уравнение Гиббса часто применяют для вычисления адсорбции
на межфазных поверхностях эмульсий М/В. Благодаря значительной
межфазной поверхности, эмульсии являются удобными системами
для определения адсорбции посредством измерения падения
концентрации эмульгирующего агента. Кокбейн (1954) успешно измерил
поверхностные концентрации додецилсульфата натрия на межфазной
поверхности эмульсии типа М/В и показал применимость уравнения
Гиббса. Трудности возникают, когда замедляется достижение
постоянного значения поверхностного или межфазного натяжения,
например, в случае сильно разбавленных растворов, следов высоко
поверхностно-активных примесей или при наличии макромолекул.
Во-первых, все методы, связанные с увеличением межфазной
поверхности — например, метод счета капель или метод дю Нуи — дают
завышенные результаты (Педдэй и Расселл, 1960). Во-вторых,
применение равновесной формулы к системе, поверхностное натяжение
которой все еще медленно уменьшается (например, протеины),
является сомнительным, так как скорость понижения о может быть
* Это уравнение противоречит экспериментальным фактам, так как, со"
гласно уравнению, при диссоциацгш поверхностная активность (da/dlnc) должна
была бы увеличиться в два раза, а на самом деле наблюдается обратное явление
[J. Raidles, D. Schiffrin, Trans. Faraday, 62, 2403 (1966);
E. E. Малахова, Н. А. А б р а м з о н, ДАН СССР, 180,1157(1968)].
Физическое объяснение этого явления заключается в том, что из двух ионов
алкплсульфоната, например, поверхностно-активным может быть только один.
(Прим. редактора перевода.)
85
0,03
разной при различных исследуемых концентрациях. Действительно,
адсорбция протеинов, видимо, является необратимой в любой момент
времени. Следовательно, для таких систем уравнение Гиббса не
может быть использовано.
Поверхностная эластичность возникает вследствие
неоднородности поверхностного или межфазного натяжения. Она отсутствует
в чистых жидкостях, но существует в растворах при наличии
адсорбции. Изменение а происходит по двум причинам. Если, например,
образуется свежая поверхность (как в струе жидкости при истечении
из трубы) или возрастает существующая (когда возникают пузырьки),
то увеличение поверхности раздела фаз
опережает процесс адсорбции
(подведение ПАВ к поверхности раздела).
Следовательно, среднее местное
значение Г; становится ниже равновесного
и поверхностное натяжение
увеличивается по сравнению с таковым для
чистого растворителя. Например, при
определении поверхностного натяжения
методом осциллирующей струи для
очень разбавленных растворов мыла
установлено, что для первой 0,001 сек
после образования новой поверхности
ее натяжение практически равно
натяжению чистой воды и -—"0,05 сек
требуется для понижения с до
равновесного значения (—40 дин/см) (рис. II. 2).
Наоборот, если поверхность
уменьшается, то локальное натяжение
понижается по сравнению с
равновесным, так как требуется определенное
время для десорбции и диффузии IIАВ. Это различие между
динамическим и статическим натяжениями известно как эффект
Марангонн. Была предпринята попытка количественного
обоснования этого эффекта на основе уравнения Шишковского (1908). Эта
проблема трудна из-за сложностей конвективного переноса,
потенциальных энергетических барьеров адсорбции и стерических
ограничений к проникновению молекул в адсорбционный слой, уже
частично занятый молекулами ПАВ. Качественно ясно, что этот эффект
является наибольшим в системах с очень разбавленными растворами
высоко поверхностно-активных соединений, включающих
высокомолекулярные поверхностно-активные вещества.
Недавно разработаны методы определения влияния изменения
межфазной поверхности на поверхностное натяжение раствора ПАВ
(Майзельс и др., 1961; ван ден Темпель и др., 1964). Например, когда
поверхность раствора додекандиола-1,2 увеличивали посредством
передвижения барьера, поверхностное натяжение падало на 4 дин/см.
Гарнером и Мина (1958, 1959) использован метод сжатия струи для
0,01 0,0?
Время, сек
Рис. П. 2. Зависимость
поверхностного натяжения от
времени при различных
температурах для и,015н. раствора
лаурата натрия.
86
систем М— В. Этот полуэмпирический метод определения
мгновенного натяжения подвергался критике.
Второй причиной изменения поверхностного натяжения является
эффект Гиббса. Он проявляется в расширяющейся пленке жидкости
или в жидкой прослойке между двумя соприкасающимися каплями
масла и заключается в том, что пленка не может быть утончена до
предела, обедняясь ПАВ. Результирующий коэффициент
эластичности дается Гиббсом в следующем виде:
. d In a
°-"№)-«<$-)-Г7%-
dc
где S — площадь, занимаемая определенным количеством раствора
ПАВ (концентрации с).
Коэффициенты эластичности Гиббса можно легко вычислить
по кривой зависимости поверхностного натяжения от концентрации.
Китченер и Кумпер (1959) приводят некоторые данные для растворов
додецилсульфата натрия, из которых видно, что слабая эластичность
проявляется с 0,0001 М растворами при толщине пленки 0,01 —
0,001 мм. Можно ожидать, что с более концентрированными
растворами при образовании очень тонкой пленки коэффициенты
эластичности будут выше. (Например, 0,1 М растворы при толщине пленки
0,1 мкм имеют коэффициент эластичности Гиббса —100 дин/см.
Таким образом расширение на 1% будет увеличивать местное
натяжение на 1 дин/см для каждой стороны пленки.)
Как и эффект Марангони, эластичность Гиббса возрастет с ростом
поверхностной активности вещества, т. е. с увеличением da/d In a.
Измерить эластичность жидкой пленки (например, пленки мыла)
посредством ее статического растягивания на рамке не представляется
возможным. Причина состоит в том, что пленка растягивается лишь
по периметру («кольцо Гиббса» или «плоская рамка») и натяжение
изменяется чуть заметно. Однако Майзельс и Кокс (1961, 1962)
разработали уникальный метод измерения изменения толщины пленки
с натяжением и получили значение —10 дин/см для подвижных
пленок дегергентов и 100 дин/см для жестких смешанных пленок.
По-видимому, сравнимые результаты можно ожидать в случае
пленки М/В.
В динамической системе эффекты Марангони и Гиббса
способствуют временной стабилизации жидкой пленки, так как в любой
точке, где за счет внешних сил пленка утончается до предела,
возникает местное увеличение поверхностного натяжения,
противодействующее утончению. Градиент поверхностного натяжения
проявляется не только в поверхностном монослое, но и в части
близлежащей жидкости вследствие действия сил вязкости. Согласно этому
механизму, названному поверхностным переходом, возможна
стабилизация любых потенциальных точек разрыва. Наоборот, на
утолщенной поверхности происходит падение местного натяжения, что
87
вызывает утончение пленки. В результате образуется сравнительно
однородная но толщине пленка. Пленка утончается до тех пор, пока
не возникнут силы отталкивания, уравновешивающие внешнее
давление, или, если пленка переходного типа не достигнет
критической толщины.
Трудно в настоящее время определить относительный вклад
эффектов Гиббса и Марапгони в реальных системах. Пленочный
эффект Гиббса можно вычислить, но проблематичным остается
наличие градиента поверхностного натяжения. Эластичность Гиббса
практически должна быть равна нулю для концентрированных
растворов ПАВ, так как do/dc >=& 0 при концентрации ПАВ выше ККМ.
Однако такие растворы являются сильно эластичными. Исследования
по затуханию волн позволят, вероятно, разъяснить эту проблему.
Когда волна проходит вдоль жидкой поверхности раздела,
наблюдается некоторое затухание: амплитуда колебаний уменьшается
из-за разности значений вязкости по объему жидкости. Затухание
значительно усиливается, если жидкость является раствором ПАВ
или имеется нерастворимый монослой. В этом случае волны
расширяют и сжимают поверхность, вызывая противосилы, которые
отсутствуют в чистых жидкостях.
Теоретические и экспериментальные работы по затуханию волн
были опубликованы Левичем (1941), Хансеном и Манном (196.3),
Люкассеном и Хансеном (1966), ван ден Темпелем и ван ден Ритом
(1966), Девисом и Восе (1965) и Шелудко и Тиссеи (1966).
Большинство авторов нашли, что для данного ПАВ имеется максимум
затухания, величина которого превышает предсказанную Левичем для
твердых нерастворимых мопослоев. Основная часть энергии
затухания рассеивается микроциркуляционными токами, возникающими
в жидкости ниже поверхности раздела под действием градиента
поверхностного натяжения. Ван ден Темпель и ван ден Рит успешно
разрешили эту проблему для случая, когда влиянием вязкости
поверхностного слоя можно пренебречь. Измерения коэффициента
затухания на межфазной поверхности масло — вода не были
проведены, но можно с уверенностью предположить, что они
основаны на тех же принципах, что и для поверхности раздела
воздух — вода.
Имеется значительный, но не полный параллелизм между
затуханием воли и устойчивостью пен. Затухание определяется
влиянием ПАВ на объемное состояние жидкости, в то время как высокая
устойчивость пены (или устойчивость эмульсии) в основном
определяется взаимодействием слоев. Шелудко и Чернов (1964) указали,
что некоторые соотношения между ценообразованием и
эластичностью Марангопи — Гиббса могут быть ошибочными. Например,
максимальное затухание в растворах изовалериановои кислоты
происходит примерно при той же самой концентрации, что и
максимальное ценообразование. Однако растворы пропионовой и масляной
кислот пенятся сильнее, чем растворы изовалериановои кислоты,
несмотря на то, что для последних характерно более сильное затуха-
88
ние волн. Каприловая кислота дает затухание, сравнимое по
величине с затуханием ПАВ Удепон Г и ОП-20, с помощью которых можно
получить более устойчивые пены. Кроме того, максимальное пено-
образование с этими детергентами происходит при концентрации в два
или три раза большей, чем концентрация, соответствующая
максимальному затуханию волн.
Видимо, эффект Марангони характеризует первоначальное
состояние пены, когда пленки еще толстые, но устойчивость определяется
другими эффектами. Шелудко (1962) показал, что стойкие пены
образуются в системах, которые дают «черные» пленки. Как было
упомянуто ранее, следует принять во внимание поверхностные силы
второго рода, чтобы объяснить метастабильную пленку, и поэтому
классическая химическая термодинамика поверхности не является
адекватной. Дерягин, Мартынов и Гутоп (1965) разработали
термодинамику свободных от жидкости пленок (например, для пленок
тоньше, чем толщина двух адсорбционных слоев Гиббса).
Смешанные поверхностно-активные вещества. Имеется много
данных о наличии примесей, сильно влияющих на поверхностное
натяжение растворов. Например, длинноцепочные спирты, кислоты,
неноныые дпфильные ПАВ дают хорошо известный минимум на
кривой а — с солей с парафиновыми цепями (в частности, лауриловый
спирт в натрий-лаурилсульфате). В действительности неионные
примеси солюбилизируются ионным монослоем, так как гидрофобные
цепи могут проникать между молекулами первичного ПАВ, которые
уже ориентированы на поверхности. (При концентрации выше ККМ
неионные молекулы солюбилизируются мицеллами обратно в
основной объем фазы.) Таким образом, присутствие основного ПАВ
усиливает поверхностную активность примеси, и общее понижение
поверхностного натяжения а будет больше, чем сумма понижений а,
полученная этими веществами в отдельности.
Ван ден Темпель и Люкассен — Рейндерс (1965) разработали
теорию эластичности Гиббса для растворов с малой и высокой
концентрацией активных компонентов, а Принс и ван ден Темпель (1964)
применили эту теорию к экспериментальным данным для системы
лаурат натрия — лауриповая кислота. Чтобы учесть взаимодействие,
необходимо иметь данные о поверхностном натяжении ряда тройных
систем. Указанные выше авторы нашли, что в присутствии ионов
лаурата лауриновая кислота имеет более высокую поверхностную
активность. Из этого следует, что при некоторых условиях малая
концентрация примесей вызывает большую эластичность Гиббса,
чем высокая концентрация.
Принс и ван ден Темпель получили коэффициент эластичности
Гиббса ^=d03 дин/см. Это, по-видимому, объясняет высокие
результаты, сообщенные Майзельсом и др. для пленок «нечистых»
детергентов. Авторы показали также поразительную связь между
наблюдаемой устойчивостью пены растворов лаурата натрия и
эластичностью Гиббса, вычисленной с поправкой концентрации и рН на
степень гидролиза мыла в растворе. Они заключили, что устойчивость
89
пены обусловливается в основном эластичностью пленки, а не
поверхностной вязкостью (Майзельс и др., 1961).
Ван ден Темпель нашел, что пены и эмульсии не слишком
чувствительны к незначительным примесям (это замечено при коалес-
ценции). Единственное различие состоит в том, что маслорастворимые
ПАВ экстрагируются в масляную фазу, из которой они адсорбируются
слабее, чем из воды. Однако нет данных о межфазном натяжении
на границе раздела масло — вода для тройных растворов.
Динамическая эластичность межфазных слоев должна объяснять влияние
незначительных количеств примесей на распределение капель по
размерам при эмульгировании.
Взаимодействие двух (и более) эмульгирующих веществ (одного —
маслорастворимого и другого — водорастворимого) изучено Алек-
сапдером и Шульманом (1940). Определенные комбинации таких
веществ дают высокую стабильность, обычно связанную с очень
низким межфазным натяжением (например, 0,1 дин/см) и
сопровождающуюся совместной адсорбцией. Однако отсутствуют
количественные данные, из которых можно было бы вычислить адсорбцию
двух ПАВ по уравнению Гиббса. Следует также ожидать обратимость
совместной поверхпостпоп активности по аналогии с антипенными
или пеноразрушающими агентами. Чтобы ликвидировать поверх-
постную эластичность, нужно или высолить эмульгирующий агент
химическими способами, или вытеснить его подавляющим избытком
агента на поверхности (даже если он обладает более низкой
поверхностной энергией). Возможно, силиконы или фторуглеводороды
будут действовать как эмульгирующие агенты при использовании,
например, в процессах экстракции яеидкость — жидкость.
Роль межфазной вязкости. Со времени Плато (1873) некоторые
авторы предполагают, что высокая поверхностная вязкость может
способствовать стабильности жидких пленок (обзор этих работ дан
Китченером, 1964). Если вязкость и не является причиной
статической метастабильиости, то она может способствовать динамической
стабилизации, которую обеспечивает объемная вязкость в
результате замедления экструзии жидкости между двумя поверхностями.
Имеется много данных о вязких адсорбционных слоях на
межфазной поверхности воздух — вода, и они часто связываются с высокой
поверхностной эластичностью Марангони — Гиббса.
Принс и ван ден Темпель (1967) аргументировали против теории
вязкости следующим образом: «Были сделаны попытки (Майлс,
Росс, Шедловский, 1950) объяспить стабилизирующий эффект
поверхностно-активных веществ медленным растеканием,
обусловленным поверхностной вязкостью сдвига. Для медленно растекающейся
пленки требуется больше времени до образования областей меньшей
толщины и разрыв происходит в области достаточно низкой
(критической) толщины. С другой стороны, такие медленнорастекающиеся
пленки становятся быстрорастекающимися при несколько
увеличенной температуре (Джонс, Дюргам, Эвене и Кэмп, 1957), но это не
влияет па стабильность пены. Более того, при небольшом увеличе-
90
нии концентрации основного компонента поверхностная вязкость
сдвига мгновенно падает до значения, сравнимого с поверхностной
вязкостью сдвига чистой воды или разбавленного раствора чистого
поверхностно-активного вещества (Лоуренс и Блеки, 1954). Пленки,
полученные из таких растворов, являются быстрорастекающимися,
но имеют более высокую стабильность, чем пленки без примеси.
Следовательно, стабилизирующий эффект примеси не может быть
объяснен увеличением поверхностной вязкости сдвига или медленным
растеканием».
Возвращаясь к эмульсиям, отметим два факта, противоречащих
мнению об определяющей роли межфазной вязкости как фактора,
влияющего на стабильность. Во-первых, высокая межфазная вязкость
редко встречается (кроме высокомолекулярных веществ). Лоуренс
и Блеки (1954) показали, что дифильные молекулы, дающие высокую
поверхностную вязкость с детергентами на поверхности воздух —
вода, показывают незначительную межфазную вязкость на
поверхности масло — вода. [Может быть имеет смысл проверить эту работу
с чувствительным межфазным вискозиметром Девиса и Майерса
(I960).] Тем не менее, согласно Шульману и Райдилу (1937) и Шуль-
ману и Кокбейну (1940), эмульсионные системы, для которых
характерны жесткие пленки (например, цетилсульфат натрия и
холестерин), способны образовать эмульсии типа В/М.
Во-вторых, в любой эмульсии, приготовленной с ПАВ,
адсорбционный слой делает поверхность жесткой; капли, как правило,
таких размеров, что любое тангенциальное давление сдвига, которому
они могут быть подвержены, непосредственно противодействует
градиенту поверхностного натяжения, возникающему при
бесконечно малом изменении а. Хорошо известно, что капли с диаметром
>1 мм имеют нешарообразную форму при перемещении в
низкоконцентрированных водных растворах ПАВ, так как они подчиняются
закону Стокса, а не Гадамарда (1911). Разные участки капель могут
одновременно иметь несколько различное натяжение. Установлено,
что в данном случае происходит запаздывание процесса адсорбции —
десорбции, т. е. наблюдается эффект Марангони. Поэтому, когда
соприкасаются две такие капли эмульсии, они коалесцируют
медленно *.
ПРИМЕНЕНИЕ ТЕОРИИ УСТОЙЧИВОСТИ
ЛИОФОВНЫХ КОЛЛОИДОВ К ЭМУЛЬСИЯМ
Значительная часть современных фундаментальных работ па
теории стабильности эмульсий посвящена вопросу, в какой степени
теория устойчивости коллоидов (сейчас классическая), развитая
независимо друг от друга Дерягиным и Ландау (1941) и Вервеем
* В последнее время точка зрения об определяющем влиянии вязкости
непрерывной фазы поддерживается в работах Фриберга [S. F г i b e г g,
J. Wilton, Am. Parfum. a. Cosmet., 85, № 12, 27 (1970)]. Этот фактор,
видимо, играет существенную роль в обратных эмульсиях п прямых,
стабилизированных высокомолекулярными ПАВ. (Прим. редактора перевода.)
91
и Овербеком (1948), объясняет свойства эмульсий. Следует отметить,
что «теорию ДЛВО» (название которой состоит из инициалов
основных авторов теории) первоначально применяли для систем с
классическими неорганическими солями. Устойчивость содержащихся
в них субмикроскопических твердых частиц объяснялась
электростатическим зарядом последних. Нужно подойти с осторожностью в
применении этой теории к микроскопическим каплям масла,
стабилизированным адсорбируемыми эмульгирующими агентами. Она может
довольно хорошо описывать стабилизацию эмульсий двойным
электрическим слоем (например, ионными ПАВ) против коагуляции.
Однако ее нельзя использовать для определения скорости коалесцен-
ции капель эмульсии, ибо этот процесс зависит от вытеснения или
разрушения адсорбированной пленки. Кроме того, она не применима
к эмульсиям, стабилизированным твердыми частицами или
гидрофильными коллоидами *.
В дополнение к первоначальным публикациям теории ДЛВО
имеется несколько хороших кратких ее изложений, например, Овер-
бек (1952а), ван Ольфен (1963) и Шелудко (1966а). Здесь дано только
краткое описание сущности этой теории. Некоторые статьи,
предлагающие модификации и усовершенствование этой теории, будут
рассмотрены ниже.
Краткое изложение теории ДЛВО
Суть теории ДЛВО заключается в том, что дисперсные
частицы подвергаются действию двух родов дальнедействующих сил:
1) вандерваальсовы силы притяжения;
2) электростатическое отталкивание между двойными
электрическими слоями одинакового знака.
Они могут проявляться, когда две частицы сближаются в
результате броуновского движения. Эти силы определяют, будут ли
частицы коагулировать.
Так как указанные силы действуют независимо друг от .друга
и имеют различные источники, они могут быть вычислены отдельно.
Общее взаимодействие между частицами определяется суммирова-
* Имеются еще доводы против приложения теории ДЛВО к
эмульсионным системам. В прямых эмульсиях, стабилизированных ионными ПАВ.
со значительным двойным электрическим слоем капельки органической фазы
разделены водной прослойкой, а величины дисперсионных сил на единицу
объема жидкости в полярных и неполярных жидкостях близки. Таким
образом, на расстоянии > 10 А капельки органической жидкости не притягиваются
друг к другу вандерваальсовымп силами. В случае обратных эмульсий в
непрерывной фазе отсутствует двойной электрический слой (серьезных доказательств
существования двойного слоя в этих эмульсиях не имеется). Не наблюдается
параллелизма между стабильностью эмульсий по отпогпештю к коалесценцпи
и наличием двойного электрпческого слоя. Вопрос о силах притяжения и
отталкивания капель эмульсий можно считать открытым и требующим как
экспериментального, так и теоретического решения. При стабилизации эмульсий
следует учитывать не только силы отталкивания — притяжения между каплями,
но п адсорбцию ПАВ. Иначе трудно объяснить, почему только определенные
ПАВ являются стабилизаторами эмульсий. (Прим. редактора перевода.)
92
нием сил с учетом расстояния между частицами. Если вандервааль-
совы силы превышают силы отталкивания при всех расстояниях,
дисперсия является неустойчивой и будет коагулировать с такой же
скоростью, с какой сталкиваются частицы. В противном случае
энергетический барьер противодействует столкновению частиц. Когда
высота энергетического барьера сравнима с изменением
кинетической энергии коллоидных частиц (тепловой энергии — кТ),
коагуляция будет замедляться, но не прекращаться. Если энергетический
барьер значительно выше чем кТ, скорость коагуляции практически
равна нулю. Ввиду того, что высота энергетического барьера —
интересующий нас параметр, обычно строят кривые энергий
взаимодействия, а не сил.
Вандерваальсова энергия
взаимодействия двух дисперсных частиц
Энергия взаимодействия (отрицательная при притяжении) двух
бесконечно малых элементов объема dV\ и dV2 вещества 1,
расположенных на расстоянии Н в вакууме, дается теорией Лондона
Величина р может быть определена из экспериментальных данных
и имеет значения, лежащие в пределах 10~59—10"57 эрг-смв для
малых молекул и зависящие от их электронных свойств. Чтобы
вычислить энергию взаимодействия между определенными частицами,
нужно проинтегрировать уравнение (П.1) по всем элементам объема.
Это выполнено для пары одинаковых сфер Гамакером, который
получил следующее выражение
^=iLir4i^+4r+ln-V-] (II-2)
где г — радиус сферы; Нп — расстояние между центрами сфер.
Следуя Гамакеру, выражение л2тг2|3 обычно заменяют на А и
называют константой Гамакера — Лондона. Теоретические расчеты А
показывают, что ее значение для жидкостей лежит в пределах от
0,5-Ю"12 эрг для воды до 4-10-12 эрг для четыреххлористого
углерода. Эксперименты, основанные на явлениях смачивания, привели
к подобным результатам (Фоукес, 1964). Шелудко и Платиканов
(1965) получили значение А = 1,5 ■ 10"13 дин для бензола и А =
— 3,1 -10"13 дин для хлорбензола при изучении микроскопических
тонких пленок. Но. к сожалению, ни теория, ни эксперимент в
настоящее время не могут дать точных значений.
Энергия взаимодействия для двух сферических частиц вещества 1,
помещенных в непрерывную среду 2 ниже, чем энергия,
получаемая по уравнению (П.2), из-за взаимодействия между молекулами
среды и между частицами и средой. Гамакер показал, что общее
93
взаимодействие имеет ту же самую форму множителя [квадратные
скобки уравнения (2)], но А тогда составит
А = Аг 1 + А-22 — 2Axi
где А 2 2= лаге2р2для вещества 2; А ^ 2 = n^ra^jPi 2 Для
взаимодействия между молекулами вещества 1 и 2.
Из-за недостатка данных обычно принимают f>4 2 = Vfii if>2 г-
Выражение для энергии взаимодействия по теории Лондона
применяют для двух электронных осцилляторов, имеющих
одинаковую характеристическую частоту, но различные поляризуемости.
В случае различных частот взаимодействие будет слабее. Из этого
следует, что общее значение А всегда положительно, независимо
от того, имеет ли А± ^ или А2 i большую энергию когезии.
Следовательно, капли эмульсии будут всегда притягиваться вандервааль-
совыми силами, имеющими равное значение для капель
определенного размера независимо от типа эмульсии, так как
a^IVaTi-Va^?
Величина .4 является разностью между двумя величинами,
отличающимися не более чем в два раза, значение которых, к сожалению,
не может быть вычислено с точностью более ±25%; значения А
такие большие, что даже порядок ее величины может быть
сомнительным. Следует отметить, что для различных пар жидкостей
возможна широкая область значений А, колеблющихся от нуля до
—2-КГггэрг (для СС14 — Н20). Для системы парафин — вода
А <=& Ю- 13 эрг. Эти колебания энергии притяжения будут отражаться
на стабилизации против флокуляции (Джонсон, Гольдфарб и Пе-
тика. 1965). В настоящее время необходимо определить значения А
различными методами.
Анализ цифровых значений формулы показывает, что вандер-
ваальсово взаимодействие капель эмульсии становится
значительным только при совсем малых расстояниях между каплями. Например,
для капель tr = 0,5 мкм и А = 10~13 эрг, находящихся на
расстоянии 0,1 мкм, энергия взаимодействия примерно равна тепловой
энергии kT (величина которой при комнатной температуре равна
4,1-10~14 эрг).
Поэтому в практике достаточно взять простую форму уравнения
Гамакера, полученную из уравнения (П.2) путем замены Нц — 1г =
= Н (где Н <^ г). Приблизительная формула:
Эта энергия пропорциональна размеру капель и не понижается
резко с увеличением расстояния между ними. Результаты Гамакера,
изложенные выше, должны быть усовершенствованы с учетом
влияния диэлектрической проницаемости среды на лондоновские силы.
За исключением областей аномальных дисперсий эта величина лежит,
9',
вероятно, в пределах 1,5—2 для различных жидкостей (ср. квадрат
показателя преломления для длины волны видимой области спектра,
в которой жидкости являются прозрачными). Вопрос трансмиссии
лондоновских сил через среду до сих пор дискутируется (Макла-
члан, 1963, 1965).
Второе усовершенствование, особенно важное для эмульсионных
систем — учитывает влияние замедления сил Лондона между
атомами, находящимися на относительно больших расстояниях,
сравнимых с характеристическими длинами волн электронов Лондона
(—10~5 см, т. е. 0,1 мкм для прозрачных диэлектриков). Согласно
теории Казимира и Польдера (1946, 1948), величина энергии
взаимодействия Лондона делится на коэффициент F, равный
f = 1.01 — 0,14? Для 0<р<3
r 2,45 2,04 _ . .
F = -5- Для 3<р<оо
P P2
где p = 2nvHJc; v — лондоновская1 частота; На — расстояние
между атомами; с — скорость света.
Чтобы учесть это уменьшение, необходимо выполнить
интегрирование уравнения Гамакера с включением коэффициента F в
уравнение (11.1). Некоторые примеры таких вычислений даны Овербеком
(1952) в графической форме. Лондоновская энергия притяжения
капель эмульсии (диаметром 1 мкм), которые находятся на
расстоянии 0,1 мкм, вычисленная с учетом коэффициента F, уменьшается
на —0,1 от величины энергии, полученной по уравнению (II.2).
Чтобы облегчить вычисление Шенкель и Китченер (1960) вывели
приблизительную, но достаточно точную для практических целей
формулу:
Аг Г __ 2,45 , 2,17 0,59 "1
~ #0 L 60р + 180р2 420р» J
где Н0 — расстояние между поверхностями сфер; р = 2nvH0/c.
Следует отметить, что русские ученые разработали
альтернативный путь для вычисления взаимодействия диэлектриков (Лившиц,
1955, 1956; Дзиазлошинский и др., 1960). Использование этого метода
для вычисления энергий взаимодействия коллоидных частиц требует
знаний диэлектрических свойств в пределах широкой области
частот — данных, которые отсутствуют в настоящее время для многих
веществ. Поэтому химики-коллоидники вынуждены прибегать к
грубым приближениям, предлагаемым теорией Лондона. Однако эта
теория разработана довольно хорошо в применении к дальнедей-
ствующим силам между отшлифованными поверхностями,
поверхностной энергии неполярных жидкостей и энергии адсорбции
простых неполярных молекул на твердых телах — например, бензол
на графите (Киселев, 1965). Можно с уверенностью предположить,
что эта теория дает правильный порядок величины энергии
взаимодействия коллоидных частиц.
95
При вычислении вандерваальсового притяжения между каплями
необходимо рассмотреть влияние оболочки эмульгатора.
Адсорбционный слой будет иметь константу Гамакера, отличную от
константы дисперсной фазы, которую он защищает. Пренебрежение
указанным фактором дает незначительную ошибку для
низкомолекулярных эмульгаторов (например, мыло), но оказывает
существенное влияние в случае эмульсий, стабилизированных разветвленными
цепями оксиэтилированных веществ. Такие системы как бы
окружены толстым слоем концентрированного раствора гидрофильного
коллоида с константой Гамакера, сильно отличающейся от
константы масляной или водной фазы. Волд (1961) вычислил влияние
таких оболочек на вандерваальсово притяжение капель. Если кон^
станта Гамакера оболочки близка к константе дисперсионной среды,
оболочка будет барьером, препятствующим сближению капель на
расстояние действия вандерваальсовых сил. Это учитывается в
вычислениях. [Например, стабилизация воды в масляной эмульсии
поверхностно-активными веществами с очень длинной маслораство-
римой частью молекулы (Люкассен — Рейндерс, 1964).] Однако
Волд не принимал во внимание эффект «замедления», которым нельзя
пренебречь для частиц такого размера, как капли эмульсии.
Электростатическое отталкивание
Согласно теории ДЛВО, стабилизация происходит в
результате действия сил отталкивания между коллоидными частицами,
которые несут двойные электрические слои. Двойные слои могут
быть описаны классической теорией Гун — Чэпмена или ее
модификацией. Частицы сами несут электрический заряд и окружаются
диффузным слоем ионов равного и противоположного по знаку
заряда; отталкивание происходит при перекрытии диффузных слоев.
Так как теория взаимодействия перекрывающихся диффузных слоев
непроста, здесь будет приведена единственная приемлемая рабочая
формула.
Полное описание теории дано Вервеем и Овербеком (1948).
Предполагается, что двойной слой возникает благодаря равновесию между
ионами в растворе и ионами на поверхности частиц. Хотя система
является электрически нейтральной, катионы и анионы не одинаково
распределяются между межфазной поверхностью и раствором. В
дополнение к ионам, возникающим из растворителя (Н+ и ОН-),
в растворе могут быть ионы эмульгатора (например, Н+ из
карбоксильной кислотной группы) и ионы, полученные в результате
диссоциации растворенных электролитов. Природа поверхностного
заряда будет зависеть от адсорбции и концентрации присутствующих
ионов. Обычно преобладает один вид ионов, который становится
потенциалопределяющим ионом для системы, в то время как другие
присутствующие электролиты не оказывают особого влияния на
поверхность и могут считаться индифферентными.
96
В таких системах электрический двойной слой, как установлено,
характеризуется тремя параметрами: Q — поверхностная плотность
заряда; г|з — поверхностный потенциал; б = 1/А; — толщина
диффузной части двойного слоя. Здесь к — параметр Дебая,
определяющийся ионной силой раствора:
к = \ гкТ )
Связь между зарядом и потенциалом описывается теорией Гуи —
Чэпмена. Например, для электролита, имеющего п+==п_ = га и
2+ — Z_ = 2
<?=(-^-)'/! [ехр(мф/2А7')-ехр(-м1|)/2М')] (II.5)
Если двойной слой образуется вследствие обратимой адсорбции
из относительно большого объема раствора, то потенциал
определяется концентрацией потенциалопределяющих ионов, в то время
как индифферентные ионы в основном влияют на толщину диффузного
слоя. Метод вычисления для капель эмульсии рассмотрен ниже.
Типичные значения if) лежат в области 25 н- 100 мв, а значения б,
которые могут быть рассмотрены как расстояния между
поверхностью и центром заряда противоионов, колеблются от 1000 А (для
дистиллированной воды) до 10 А [для 0,1 н. раствора (1 : 1)
электролита]. Обычно считают, что если две коллоидные частицы, несущие
подобные двойные слои, соприкасаются (например, в результате
броуновского движения), поверхностный потенциал при их
взаимодействии остается постоянным; это означает, что адсорбционное
равновесие устанавливается очень быстро. Альтернативно можно
постулировать, что поверхностный заряд остается постоянным в
результате медленной адсорбции. Видимо, истина находится между
указанными двумя предположениями, которые, к счастью, не
приводят к сильно отличающимся оценкам энергии взаимодействия.
Расчеты энергии взаимодействия двойных слоев в растворах были
опубликованы Дерягиным и Ландау в 1941 г. и независимо от них
более подробно Вервеем и Овербеком в 1948 г. Ранее Дерягин (1934)
показал, что, если рассчитать расклинивающее давление между
параллельными пластинами, взаимодействие между телами в форме,
например, сфер, цилиндров можно вычислить, по крайней мере,
приблизительно. Оказывается, что только для взаимодействующих
параллельных слоев имеется довольно простая и ясная формула,
справедливая при определенных условиях. Уравнения для
параллельных слоев в настоящее время решены с высокой точностью
с помощью ЭВМ, и результаты опубликованы в форме таблиц (Деверёз
и де Брюн, 1963).
Так как соответствующие таблицы для взаимодействия
сферических двойных слоев являются не совсем точными *, необходимо
* Ф. Г. С у и т о н, Научное сообщение ORNL-2791, UC-4, Мин. торговли
США.
7 Заказ 1ПТ0
97
иснользовать приблизительную формулу, справедливую при
определенных условиях. Эта формула в применении к эмульсиям строго
справедлива только для низких потенциалов (гг)з < 25 мв), сфер
с большим радиусом г, сравнимым с толщиной двойного слоя (т. е.
г ^ 1), и для слабого взаимодействия между двойными слоями
(т. е. расстояние Н0 между поверхностями сфер сравнимо с величиной
1/к). Таким образом, потенциальную энергию пары одинаковых
сфер можно выразить формулой:
(7л = -|-еп|;2 1п[1 + ехр(-А:#0)] (II.fi)
Формула (II.6) может быть использована как приближение для
большинства практически важных систем. Вервей и Овербек дали
таблицы для высоких потенциалов и систем, частицы которых малы
по сравнению с толщиной двойного слоя.
Использование теории Гуи — Чэпмена в ее первоначальной
форме пренебрегает такими моментами, как дискретность заряда
иона, конечный радиус иона, местная диэлектрическая поляризация
среды и т. д. Ясность по этому вопросу внесена Хейдоном (1964)
и Спарнейем (1962). Наиболее важное уточнение учитывает
специфическую адсорбцию противоинов по теории Штерна; последующее
уточнение проведено Вервеем и Овербеком (1948). Однако с точки
зрения стабильности коллоидов адсорбция Штерна способствует
уменьшению эффективного поверхностного потенциала,
применяемого для вычисления энергии взаимодействия, которое в любом
случае ограничено довольно малыми значениями.
В случае эмульсий (в отличие от золей) следует рассматривать
два фактора, прежде чем решить, применима ли указанная выше
формула, а именно: возможность искажения капель при их
взаимодействии и наличия диффузных слоев внутри самих капель. Если
капли стабилизируются вследствие отталкивания двойных слоев,
то сильное сближение способствует расплющиванию поверхностей;
потенциальный энергетический барьер, противодействующий
соприкосновению капель, будет больше, чем вычисленный при
предположении недеформированных сфер, при этом эффективный радиус
кривизны увеличивается. Математического истолкования этого
эффекта еще не существует. Влияние внутреннего диффузного слоя
в масляной и водной фазах было рассмотрено Вервеем и Овербеком.
Так как некоторая часть поверхностного заряда нейтрализуется
внутренними противоионами, то поверхностный потенциал
уменьшается; но па отталкивание между каплями, благодаря
взаимодействию их внешних двойных слоев, не влияет наличие внутренних
двойных слоев.
Кривые суммарного взаимодействия
Так как отталкивание двойного слоя и вандерваальсово
притяжение действуют независимо, то общее взаимодействие определяется
путем их суммирования. Вычисления показывают, что общая кривая
98
взаимодействия сферических частиц имеет потенциальный
энергетический барьер AU (рис. Н.З), значительно превышающий величину
средней тепловой энергии коллоидных частиц (кТ). Это говорит
о высокой устойчивости капель к флокуляции, но не к седиментации.
Нет фактора, который нарушал бы порядок величины барьера.
Эксперимент подтверждает, что такие системы действительно
являются стабильными.
Если поверхностный потенциал уменьшается или ионные силы
увеличиваются (одновременно), то энергетический барьер
понижается до значения, сравнимого с величиной кТ, показывая, что
система будет подвергаться медленной флокуляции. Переход от
высокой стабильности через
медленную флокуляцию к
быстрой (т. е. к исчезновению
потенциального энергетического
барьера) является
непрерывным, без резкой флокуляцион-
ной точки. Поэтому важно
рассмотреть зависимость между
кривой потенциальной энергии
и скоростью флокуляции. При
этом надо учитывать, что
величина общей энергии является
разностью между двумя
большими (почти равными)
значениями. Следовательно,
вычисленная кривая очень
чувствительна к игнорированию
различных факторов.
Сопоставление теоретических и экспериментальных данных при медленной
коагуляции связано с большими трудностями. Тем не менее, это
единственное средство проверки теории стабильности, так как
пределы высокой стабильности или быстрой флокуляции являются
независимыми переменными.
Вторичный минимум
В своей монографии Вервей и Овербек обратили внимание на
важное различие между эмульсиями (или другими грубыми дисперсиями)
и классическими субмикроскопическими золями: для первых
характерно наличие вторичного минимума значительной глубины О кТ).
Теория ДЛВО предсказывает вторичный минимум для всех
лиофобных коллоидов при условии, что отталкивание двойного слоя
будет достаточным для преодоления вандерваальсова притяжения
на некотором расстоянии — другими словами, при наличии барьера
отталкивания всегда должен существовать вторичный минимум
на больших расстояниях. Он возникает вследствие того, что
притяжение уменьшается с расстоянием более медленно, чем отталкивание.
Однако, чтобы оказывать заметное влияние на свойства дисперсий,
60
ЬО
-= 20
-
1
50
100^
150
i
200
—""""\Г>
о
И, А
О
-20
Рис. П.З. Вычисленная кривая
энергии взаимодействия для капель
парафинового масла, стабилизированных
бычьим сывороточным альбумином
(радиус капель 1,24 мкм, 0,01 М
раствор КС1, £=20 мв, 4 = 1,72-10-13 эрг)
(Срнвастава и Хейдон, 1964).
7*
99
глубина вторичного минимума должна быть больше кТ в несколько
раз. Ввиду того, что величины притяжения и отталкивания примерно
пропорциональны радиусу частиц, вторичный минимум становится
больше для относительно крупных частиц. Он также должен быть
более заметен для параллельных пластинок, чем для сферических
частиц.
Вычисленные кривые потенциальной энергии показывают, что
вторичный минимум будет значительным для сфер с радиусом 10 мкм
и незначительным — с радиусом 0,1 мкм; важно, что при радиусе
сферы 1 мкм он будет зависеть от величины константы Гамакера А
и от поправки на замедление. Оба эти фактора являются
неопределенными, следовательно, значение вторичного минимума для типичных
эмульсий теорией устанавливается неопределенно.
Вероятно, наиболее надежные вычисления проведены Хейдоном
и Сриваставой (1964), результаты которых для парафиновых капель
радиусом 1,24 мкм в растворе протеина и 0,01М КС1 представлены
на рис. II.3. Согласно их вычислениям, эта система будет показывать
обратимое образование парных частиц (см. стр. 115). В практике
ассоциация во вторичных минимумах не будет останавливаться
на паре частиц, так как тройные и высшие группы удерживаются
сильнее, чем нары. Следовательно, положение равновесия в
ассоциированных системах будет смещаться в сторону тесно упакованных
мультиплетных групп. Равновесные константы увеличиваются со
сложностью структуры (К( < К2<. Ко и т. д.). Схематически это
можно изобразить так
к, к, О О к, 00
О -0;z_* 00 : 00-0 ^12 00 : 00-0 ^zl 00
где О — представляет каплю.
Глубина минимума для данной частицы будет пропорциональна
числу ее ближайших соседей, а константа равновесия для
ассоциации — диссоциации выражается формулой: exp AU/kT. Отсюда
следует, что такая система, очевидно, будет образовывать большие
флокулированные осадки.
Специфическая особенность вторичного минимума заключается
в том, что частицы остаются разделенными пленкой раствора, и
следовательно, могут быть снова разделены при малейшем механическом
воздействии. Обычно принимается, что структурная вязкость
концентрированных эмульсий возникает при флокуляции во вторичном
минимуме п исчезает при сдвиге (см. гл. II).
Экспериментальные данные
Чтобы установить, в какой степени стабильность эмульсии
(против флокуляции) может быть объяснена отталкиванием двойного
слоя, необходимо определить параметры г|з, к, А и г, а также связать
стабильность с кривыми потенциальной энергии.
100
Роль электрического двойного слоя
Для межфазной поверхности масло — вода не существует строгого
метода определения г|з. Сильные электрические двойные слои
возникают вследствие адсорбции анионного или катионного поверхностно-
активного вещества, что доказывается явлением электрофореза или
«поверхностного потенциала». Однако последний плохо определим
экспериментально и теоретически для поверхности М/В и, конечно,
не равен г|). Из-за отсутствия лучшего метода обычно предполагают,
что ^-потенциал равен г|). Качественно известно, что для
стабильности эмульсий требуется ^-потенциал (любого знака), больший
•— 30 мв (Повис, 1914), но количественное его значение точно не
установлено. Поэтому необходимо рассмотреть эту проблему детально.
Строго определенный ^-потенциал — гипотетическая величина,
вычисляемая из скорости электрофореза на основе более или менее
усовершенствованной теории этого явления. Ранее использовали
уравнение Гельмгольца — Смолуховского:
где г] — вязкость; и — электрофоретическая скорбеть частицы
в поле силы Е.
Это уравнение основывается на модели, по которой подвижная
часть двойного слоя может иметь любое распределение (как слой
Гуи), по предполагается движение в среде со средним отношением
вязкости л к диэлектрической постоянной е. Большинство авторов
принимают значения этих параметров, равными параметрам воды.
Однако другие считают, что вода в области диффузного двойного
слоя имеет аномальные свойства вследствие высокой локальной силы
поля. Ликлема и Овербек (1961) заключили, что е, вероятно, не
изменяется, а г) может увеличиваться, но надежные значения «вязко-
электрической константы» для воды отсутствуют.
Состояние воды у поверхности полностью еще не установлено.
Дерягиным и другими исследователями показано, что значительные
слои воды в действительности являются неподвижными. Имеется
множество данных, согласующихся с этой теорией, но они не
являются абсолютными. Большинство исследователей
предполагают существование одного или двух молекулярных слоев вокруг
ионов, связь которых ослабевает при увеличении расстояния.
Имеются некоторые данные против наличия толстых вязких слоев,
полученные из кинетики утончения пленки пены. Ликлема, Шолтен
и Майзедьс (1965) нашли, что утончение описывается
гидродинамическим уравнением, основанном на предположении о нормальной
вязкости; они установили, что любые вязкие слои не могут достигать
толщины 10 А. Тем не менее, эффективная вязкость внутри слоя Гуи
остается неопределенной в теории электрофореза.
При изучении электрофореза помимо вязкости необходимо
учитывать два фактора. Один — поправка Генри на искажение поля,
101
вызванное присутствием частицы. Эта поправка зависит от отношения
толщины двойного слоя (1/А) к радиусу частицы (г) и легко
определяется. Второй фактор — поправка на искажение поля,
возникающее вследствие миграции противоионов (релаксационный эффект).
Ее важно учитывать при относительно высоких потенциалах,
связанных с умеренно толстыми диффузными слоями (0,1 < кг < 100).
Оказалось, что для потенциалов вплоть до 100 мв можно применять
теорию Овербека, а для систем с потенциалом > 100 мв большую
точность дают расчеты Вирсема (1967). Частицы, имеющие такие
высокие потенциалы, находятся обычно в высокостабилизированном
состоянии. Переход же от быстрой к медленной флокуляции обычно
происходит с потенциалом — 25 мв или менее. Поэтому можно
пользоваться лишь теорией Овербека.
Тем не менее этой дилеммы не избежать даже при улучшении
методов вычисления ^-потенциала, так как не ясен вопрос, является
ли ^-потенциал реальной величиной, применимой в теории
ДЛВО. С теоретической точки зрения ^-потенциал — это
разность потенциалов, возникающих в объеме раствора и на
электрокинетической плоскости скольжения. Если двойной слой частично
находится в твердой или вязкой среде (как утверждают некоторые
русские исследователи), то только его подвижная часть должна
учитываться при измерении ^-потенциала.
Одно время предполагали, что плотность Q заряда двойного слоя
можно получить из ^-потенциалов посредством применения теории
Гуи — Чэпмена [см. уравнение (5)], что отождествляет £-потен-
циал и \|l Если бы это было справедливо, то можно было бы
определять ^-потенциал по адсорбционной плотности. Однако
экспериментально доказано, что значения Q, вычисленные по ^-потенциалу,
прогрессивно уменьшаются по сравнению с ионными,
адсорбционными плотностями при увеличении плотности заряда, и при высоких
плотностях заряда расхождение является десятикратным (Хейдон,
1960; Салиб и Китченер, 1964). Другими словами при этих условиях
большая часть противоионов, присутствующая в адсорбированном
слое, является электрокинетически инактивной. Наиболее вероятное
объяснение заключается в том, что противоионы, сильно связанные
со слоем Гельмгольца (т. е. противоионы, находящиеся в слое
Штерна), являются действительно неподвижными. Конечно,
альтернативно можно доказать, что высокий потенциал может привести
к толстому слою неподвижного растворителя. Салиб и Китченер
показали как, в принципе, этот вопрос может быть решен путем
точных измерений отрицательной адсорбции ко-ионов; но попытка
выполнить такие измерения с порошком графита, имеющим
физически адсорбированный слой ионного поверхностно-активного
вещества, оказалась неудачной по техническим причинам. Простейшая
гипотеза заключается в том, что ^-потенциал — потенциал в жидкости
за слоем Штерна. Это выполнимо, если плотность заряда низка
и противоионы слабо ассоциированы. Для высокой плотности заряда
можно предположить, что ^-потенциал — потенциал диффузной части
102
двойного слоя, и поскольку эта диффузная часть способствует дальне-
действующему отталкиванию, приближение является
справедливым.
Определение скорости флокуляции (коагуляции)
Ввиду отсутствия резкого перехода от устойчивого состояния
систем к неустойчивому, необходимо измерить скорость их
флокуляции и сопоставить ее с теоретическими данными. Имеются два хорошо
известных экспериментальных метода измерения скорости
флокуляции эмульсии — на основе оптических свойств (мутность или
рассеивание света) и метод счета частиц.
Оптический метод
В случае эмульсий без адсорбционного слоя (белых) необходимо
рассмотреть зависимость между светорассеянием и распределением
частиц по размеру. Теория строго справедлива только для очень
разбавленных сферических дисперсий, поскольку оптическая
интерференция между частицами усложняет исследование. Если размер
частиц превышает длину волны света (< 0,1 Я), светорассеяние
описывается теорией Релея, согласно которой рассеяние
пропорционально квадрату объема частиц. Поэтому флокуляция будет
сопровождаться увеличением светорассеяния или мутности.
Троелстра (см. Овербек, 1952) вывел зависимость между
светорассеянием и временем для случая, когда кинетика флокуляции
соответствует теории Смолуховского (см. ниже), а флокулированные
осадки имеют такую же степень светорассеивания, что и сферы,
равные по массе. По этой теории светорассеивание прямо
пропорционально времени; после времени полурасслаивания светорассеяние
должно увеличиться в три раза. Однако эксперименты с
классическими золями установили, что светорассеяние увеличивается в
меньшей степени, чем предсказывалось теорией. Эти отклонения, по
сравнению с линейной зависимостью (кривая вогнута к оси времени)
показали, что агрегаты рассеивают меньше света, чем сферы равной
массы из-за деструктивной интерференции. В самом деле, при
определенных условиях, чем быстрее флокуляция золя, тем более
открытой является структура флокулированного осадка и тем меньше
рассеивание света.
Ватиллон и Джозеф-Петит (1966) показали, что линейное
увеличение коэффициента затухания со временем и незначительное
отклонение, предсказанное теорией Троелстра. наблюдается в
разбавленных монодисперсных латексах с очень мелкими частицами (60 нм)
в ранней стадии флокуляции, но в общем, это несправедливо для
частиц с радиусом 1 мкм. Для капель, сравнимых по размеру с X,
рассеивание света отдельной частицей может быть рассмотрено по
теории Ми (см. ван де Хулст, 1964); даже для сфер оно является
сложной функцией длины волны, показателя преломления и радиуса.
В настоящее время используют таблицы функций рассеивания
света для сферических частиц (Пангонис, Геллер и Якобсон, 1957),
103
и экспериментаторы предпочитают проводить измерения с длиной
волны света, дающей максимум затухания, соответствующий размеру
п показателю преломления частицы относительно среды.
Ватиллон и Джозеф-Петит обнаружили, что при коагуляции латек-
сов происходило уменьшение затухания при длинах волн, дающих
максимальное затухание и увеличение — при больших длинах волн.
Для любых длин волн можно па основании таблицы рассеяния света
и теории Смолуховского предсказать ход кривой рассеяние — время.
Кривые, построенные Ватиллоном и Джозеф-Петитом, показали, что
при условии измерения с длиной волны максимальной адсорбции
соответствующий график должен быть линейным вплоть до 0,2т
(где т — время). Предполагают, что этот график имеет одинаковую
форму как для медленной, так и для быстрой коагуляции. Начальный
наклон кривой можно принять как меру соответствующей скорости
коагуляции.
Подобная методика использована Оттевиллом и Шоу (1966).
Они вычислили мутность, которая ожидалась в момент 5%-ного
уменьшения общего числа частиц и сравнили время достижения
мутности для латексов, обработанных различными электролитами. Эту
методику можно применять для эмпирического сравнения скоростей
даже для гетеродисперсных коллоидов при использовании
предположения, что закономерности роста агрегатов не изменяются во
времени. Это предположение реально, если ограничиться малыми
степенями флокуляции, при которых образуются дуплеты, триплеты
и т. д., и несправедливо, если образуются мультиплетные агрегаты
частиц, так как форма флокулированного осадка (и, следовательно,
степень светорассеяния) зависит от скорости флокуляции.
Теория, применяемая указанными выше авторами, ограничивается
описанием монодисперсных золей. Она основывается па теории
коагуляции (которая рассматривается ниже) и на предположении, что
дуплеты, триплеты и т. д. рассеивают свет, как сферы равного объема,
которое не совсем правильно. Оттевилл и Шоу смогли сравнить метод
светорассеяния с методом счета, используя монодисперсный латекс
с частицами диаметром 0,4 мкм и получили хорошее согласие между
обоими методами.
Метод счета частиц
Простой, но очень трудоемкий метод изучения флокуляции
заключается в разбавлении образца эмульсии и подсчете числа частиц
в единице объема под микроскопом. При этом смешение должно быть
осторожным; разбавляющая среда может быть защитным
гидрофильным коллоидом (таким как желатин) или неионным детергентом.
Кинг и Мукерджи (1939, 1940) использовали этот метод при изучении
скорости коалесценции, они определяли распределение частиц по
размеру для получения межфазной поверхности эмульсий как
функции времени. Для облегчения измерения и подсчета капли
фиксировали в слабом геле желатина и увеличенное оптическое изображение
проектировали на экран.
104
По другому методу известный объем разбавленной эмульсии
помещали в тонкую микроскопическую ячейку (используемую для
подсчета кровяных телец) и подсчитывали число капель, находящихся
в измерительной сетке. Сривастава и Хейдон (1964) применяли этот
метод для определения кривых распределения по размеру. Они
наблюдали как за флокуляцией (уменьшение числа отдельных капель),
так и за слабой коалесценцией (уменьшение общего числа капель),
так как дуплеты, триплеты и т. д. можно ясно различить при
увеличении в 1425 раз для эмульсий с распределением по размеру 1—5 мкм.
Для предотвращения отстаивания дисперсной фазы при длительном
эксперименте трубки, содержащие
эмульсию, осторожно
перевертывались; как было показано, это не
вызывало значительных изменений в
коагуляции.
При более совершенном методе
применяют ультрамикроскоп.
Достаточно разбавленный коллоидный
раствор пропускают через сильно
освещенную сбоку ячейку. Когда
дисперсные частицы проносятся
через освещенную зону в микроскопе
видны вспышки света. Подсчитав
число таких вспышек в
определенный промежуток времени, можно
определить концентрацию частиц.
Эта методика с визуальным
подсчетом использована Ватиллоном и ван
виллом и Вилькинсом (1960).
Описано несколько струйных ультрамикроскопов с
автоматическим фотоэлектрическим счетчиком. Вероятно, наилучшим из них
является ультрамикроскоп Дерягина и Власенко (1948, 1957, 1962)
(рис. II.4). Кроме того, посредством добавления электронного
импульсного усилителя и импульсного высотного анализатора (в
качестве обычных сцинтилляционных радиационных счетчиков) можно
получить распределение частиц по размеру. Однако данных о
применении этого метода к эмульсиям не имеется.
Отчасти подобный струйный принцип использован в
промышленном счетчике Коултера, который первоначально был разработан для
подсчета кровяных телец, а сейчас нашел широкое распространение
при измерении размера частиц дисперсных порошков и т. д.
Принцип его действия следующий. Дисперсию пропускают через
микроотверстие и замеряют электропроводность. Каждая проходящая
через отверстие частица дает электрический импульс, величина
которого приблизительно пропорциональна объему частицы. Хорошие
результаты получены для систем с диаметром частиц вплоть до 0,3 мкм.
Грегори (1963) с помощью счетчика Коултера проследил изменение
распределения по размеру частиц синтетического латекса. В принципе
ш
2L
6
«вСЕВДЕ
Рис. П.4. Схема автоматического
ультрамикроскопа:
1 — источник света; 2— объектив; з —
кювета; 4 — фотоумножитель; 5 —
усилитель; 6 — механический счетчик
импульсов.
Гриндербеком (1954) и Отте-
105
прибор можно использовать для измерения скорости флокуляции
эмульсий (включая мелкие и крупные гетеродисперсные эмульсии).
Однако здесь возможно разрушение крупных агрегатов из-за высокой
скорости сдвига в отверстии. (Скорости сдвига в струйном микроскопе
значительно ниже.) Вахтель и ла Мер (1962) успешно применили
счетчик Коултера для стабильных эмульсий с диаметром капель
вплоть до 0,6 мкм, а Хигучи с сотрудниками (1963) — для
фармацевтических эмульсий.
Скорость флокуляции
и теория энергетического барьера
Теория кинетики флокуляции подробно изложена в литературе,
часть материала обобщена Овербеком (1952). Здесь будут рассмотрены
только основные моменты этой теории, так как вся проблема
представляет большие теоретические и математические трудности.
Механизм быстрой коагуляции. Классическая теория быстрой
коагуляции первоначально монодиснерсных золей дана Смолухов-
ским (1916). Предполагалось, что частицы (радиуса г) сталкиваются
друг с другом только под действием броуновского движения и
слипаются при соприкосновении. Таким образом, из отдельных частиц
получаются дуплеты, триплеты и т. д. Обычно число агрегатов,
содержащих х частиц ко времени т, дается выражением:
_ пп (t/tk)*-i
* <1-гт/тк)*«
где п0 — первоначальное число частиц; тк — время коагуляции
(время, необходимое для уменьшения общего числа частиц до
половины первоначального). Согласно этой теории, общее число частиц
«о 3ii 2-10-11 . .
и - - — — [сек]
2 1-т-т/тк к АкТп0 п0
Отсюда видно, что величина l/n^ будет линейно увеличиваться
со временем.
Прямая проверка теории быстрой коагуляции Смолуховского
была проведена с различными золями, к которым добавляли избыток
коагулирующего электролита, Ван-Аркелем и Кройтом (1920),
Турнлло (1926), Туркевичем (1959), Хигучи и др. (1963). Обычно
получалась линейная зависимость 1/сп от времени и порядок
величины наклона прямых имел предполагаемое значение. Оттевилл и
Растоги (1960) определили константу скорости коагуляции для золя
иодида серебра — К0 = 1/г-п0 = 3,2-10-12, а Оттевилл и Вилькинс
(1962) нашли К о = 2,1 -Ю-11 для арахиновокислых золей.
Трудность при исследовании коагуляции золей заключается в
полидисперсности, приводящей к повышенным скоростям, а также в
несферичности форм частиц (Бусф, 1954), которая способствует
возрастанию вероятности столкновения. Единственными системами,
действительно близкими к теоретически рассмотренной модели, являются
монодисперсные синтетические полимерные латексы. Но даже и в этих
106
системах возможны осложнения из-за сил притяжения, действующих
на малых расстояниях.
Шоу и Вервеем при исследовании монодисперсных и полистироль-
ных латексов установлено (частное сообщение), что константа
скорости для наиболее быстрой коагуляции значительно изменяется
при изменении концентрации частиц (уменьшается при увеличении
концентрации) и размера частиц (уменьшается при увеличении
радиуса). Значения К0, найденные для указанных латексов,
колеблются от 3-Ю-12 и ниже, являясь, таким образом, меньшими, чем
теоретическое значение 5-Ю-12. Причина этого отклонения не совсем
ясна, однако можно предполагать, что экспериментальные значения
будут приближаться к теоретическим при экстраполировании к
бесконечному разбавлению.
Другая трудность в применении теории Смолуховского к
обычным эмульсиям — влияние ортокинетической коагуляции. Она
проявляется в том, что в высокополидисперсных системах, подвергающихся
коагуляции, мелкие частицы исчезают значительно быстрее, чем
крупные — эффект Вернера (1932). Ортокинетическая коагуляция
заключается в увеличении скорости столкновения частиц сверх
скоростей, обусловленных броуновским движением, возникающим
из-за различных скоростей движения больших и малых частиц
в гравитационном поле или при конвекции. Этот эффект ясно
демонстрируется, например, в дисперсиях угольной сажи, к которым
добавляют определенное количество соли, чтобы вызвать медленную
коагуляцию. В некоторых случаях золи, медленно коагулирующие
при стоянии, мгновенно коагулируют при интенсивном встряхивании.
Такой эффект является авто каталитическим, так как при росте
агрегатов неравенство скоростей увеличивается. В типичных эмульсиях
с размером капель 0,1 —10 мкм и более ортокинетическая
коагуляция может быть более важной, чем обычная коагуляция. Поэтому ни
теория Смолуховского, ни любое ее усовершенствование не
применимы к процессам быстрой и медленной коагуляции.
Изучение ортокинетической коагуляции проведено Свифтом
и Френдлендером (1964) и Гиди (1965). Процесс описывается
полностью, если известна функция распределения частиц по размеру.
Свифт и Френдлендер (1965) вывели эту функцию, на основании чего
получили уравнение, приводимое к классической формуле
Смолуховского. Их решение не зависит от особенностей кинетики.
Функции распределения являются «самосохраняющимися», так
как при графическом выражении в безразмерном виде они стремятся
сохранить свою форму. Для проверки нескольких функций
использованы эмульсии М/В без эмульгатора; кривые оказались примерно
самосохраняющимися. Эта работа продолжена Гиди (1965) и Гиди
и Лилли (1965). Предложенные ими уравнения предсказывают, что
скорость коагуляции для гетерогенных золей больше, чем для
первоначально гомогенных. Кроме того, они считают, что уравнение
Смолуховского для броуновского движения согласуется с подобными
уравнениями для гетерогенных золей, когда отношение среднего
107
свободного пробега к радиусу частицы равно нулю и значение
функции пх = Ь Ус„/Ф находится между 1 и 4, где Ъ — избранный размер
частиц; V — объем частицы; сп —концентрация частиц; Ф —
объемная доля. Этот вопрос требует дальнейшего изучения.
Джовановиком (1965) проведено исследование ортокинетической
флокуляции на разбавленных суспензиях порошков,
диспергированных в органической жидкости. Его метод вычисления среднего
диаметра частиц и удельной поверхности для седиментирующих систем,
в которых происходит ортокинетическая флокуляция, разработан
теоретически и подтвержден экспериментально. Этот метод может
быть полезным при изучении полидисперсных эмульсий.
Механизм медленной коагуляции. При наличии энергетического
барьера между частицами уменьшается возможность их столкновения.
Смолуховский рассмотрел этот случай путем формального введения
параметра а — доли броуновских столкновений, вызывающих
слипание частиц. В результате время коагуляции тк увеличивается в 1/а раз.
Однако этот формализм не раскрывает связь а с энергией
взаимодействия частиц. Следует отметить, что эта зависимость не выражается,
как в химической кинетике, простым коэффициентом Максвелла —
Больцмана а =ехр (—AU/kT), где Л£/—потенциальный
энергетический барьер (Лоуренс и Майлс, 1954), так как концентрация частиц
в активированном состоянии является также функцией потока
частиц. Другими словами, это есть случай диффузии через относительно
высокий потенциальный энергетический барьер.
Рассматриваемая проблема впервые решена Фуксом в 1934 г.
для аэрозолей н применена к гидрозолям Дерягиным в 1940 г.
Теория имеет определенные допущения, включая применение
формулы Стокса — Эйнштейна для коэффициента диффузии частиц.
[Дерягин (1956, 1966) предложил заменить эту теорию теорией
взаимного приближения сфер.]
Теория Фукса приводит к следующей рабочей формуле: по
сравнению с быстрой коагуляцией скорость медленной коагуляции
уменьшается па коэффициент В, который называется коэффициентом
стабильности и дается выражением:
со
B = 2r j ехр (f/'/A-Г) -^-
По данной кривой потенциальной энергии (например, из теории
ДЛВО) коэффициент стабильности можно вычислить графическим
или расчетным интегрированием и затем сравнить его с
экспериментальными значениями.
Не следует ожидать, что В будет иметь такое же значение для
отдельной сферической частицы, приближающейся к мультиплетной
частице, как для двух равных сфер, так как U — функция размера
и структуры. Поэтому кинетика медленной коагуляции не является
просто кинетикой быстрой коагуляции, умноженной на постоянный
фактор. Соотношение дуплетов, триплетов и т. д. не будет одинако-
108
вым при, скажем, равных значениях Вт. Эта проблема может быть
в принципе разрешена, но слишком сложным путем. Вследствие этого
изучение медленной коагуляции возможно только в начальной
стадии, когда образуются дуплеты.
Дерягин и Власенко (1948, 1957, 1962) сослались на
экспериментальную работу Кудрявцева с золями золота, результаты которой
соответствуют теории. При условии быстрой коагуляции величина
1/res увеличивается линейно со временем, однако при медленной
коагуляции процесс замедляется и в некоторых случаях почти
прекращается несомненно из-за того,
;\ что энергетический барьер,
препятствующий коагуляции,
увеличивается с размером агрегатов. Тот
г W
Lg[Na*]
Рис. II.5. Вычисленные кривые,
полученные интегрированием
уравнения Фукса (латекс LS 055A +
+10-2 М раствор HC104+NaC104)
(Ватиллон и Джозеф-Петит, 1966).
0,2 ■ 10-'; 2 — 0,5 х
-1"; 3 — Ю-18.
10 ' 2 10 ' 5 10'
Lg r
Рис. II.6. Экспериментальные данные
(латекс LS 055A) (Ватиллон и Джозеф-
Петит, 1966):
1— 10"2 М NaOH 4- NaC104; 2— 10"2 М NaC104;
3 — 10"2 M HC104 4- NaC104.
эрг: 1 —
X 10
же самый эффект должен наблюдаться в мелкодисперсных
эмульсиях, хотя это не очевидно вследствие наличия глубокого вторичного
минимума, так как образуются агрегаты с диаметром > 1 мкм.
[Между прочим, указанная выше формула применяется также для
описания процесса коагуляции при наличии сил притяжения; В
тогда является коэффициентом, соответствующим ускорению по
сравнению с коагуляцией Смолуховского. Однако этот процесс не
является существенным для эмульсии (Ватиллон, 1967).]
Чтобы сравнить теорию ДЛВО с экспериментальными
данными по кинетике флокуляции, необходимо вычислить В для
медленной коагуляции как относительное увеличение скорости по
сравнению с быстрой коагуляцией и построить график зависимости В от
]g с (где с — концентрация индефферентного коагулирующего
электролита в системе). Теоретически и экспериментально показано, что
ниже определенной критической концентрации электролита эта
зависимость будет прямолинейной (рис. II.5 и П.6).
Рассмотрим два параметра: 1) критическую концентрацию,
соответствующую потенциальной энергетической кривой в месте, где
109
барьер исчезает; 2) угол наклона линейной части графика, который
может быть связан с семейством определенных кривых
энергетического барьера по методу Фукса. Первый параметр обычно оказывается
правильным, второй — плохо согласуется с имеющимися данными
из-за использования ^-потенциала и вычисления константы Гамакера
(^4). Так, согласно последним работам Оттевилла и Шоу (1966),
связь этого параметра с размером частиц для латексов не
установлена, как предполагалось ранее; угол наклона должен был быть
пропорционален размеру частиц, но в действительности оказалось,
что он почти не зависит от размера частиц. Причина этих
разногласий не совсем ясна.
УСТОЙЧИВОСТЬ К КОАЛЕСЦЕНЦИП,
ОБУСЛОВЛЕННАЯ МЕХАНИЧЕСКИМИ СВОЙСТВАМИ
МЕЖФАЗНЫХ СЛОЕВ
Высокомолекулярные стабилизаторы
Прочные оболочки, образуемые на межфазной поверхности
высокомолекулярными веществами, такими как резина, каучук или
протеины, известны уже несколько столетий. Качественная
характеристика результатов ранних исследований опубликована в работе
Серралаха и Джонса (1931, 1933). Эти авторы и другие (Таубман
и Корецкий, 1958; Холмес и Камерон, 1922; Ребиндер и Венстрем,
1930; Мартинез Морено и др., 1960, 1961; Шоттон и др., 1964)
наблюдали под микроскопом образование довольно толстых оболочек
на различных поверхностях раздела. Большинство исследований,
однако, проведено с маслами, которые могли содержать ПАВ. Кроме
того, методы получения высокомолекулярных веществ и
определения их физико-химических свойств были несовершенны.
К сожалению, большинству исследователей не удалось выяснить,
имели ли эти системы адсорбционный слой или микроскопический
осадок на межфазной поверхности. Для высокомолекулярных ПАВ
типично, что их адсорбция является очень медленной и практически
необратима. Некоторые протеины после адсорбции становятся
нерастворимыми в воде. Если такие монослои сжимать, происходит их
разрушение с образованием микроскопических осадков. Последние
остаются на межфазпой поверхности в виде прочной эластичной
оболочки, снять которую можно с помощью металлической сетки.
Гетерогенность оболочки обнаруживается посредством неоднородного
рассеяния света при исследовании под микроскопом в темном поле.
Априорно ясно, почему капли в такой «капсюле» неограниченно
устойчивы против коалесценции, однако количественные
закономерности этого явления неизвестны.
Наиболее удобная система для изучения — дисперсия чистого
масла в разбавленном растворе чистого протеина, например,
сывороточный альбумин (в противоположность растворам желатина,
неустойчивым при комнатной температуре). Доказано, что адсорбция
сывороточного альбумина ограничивается монослоем, содержащим
110
1—2 мг адсорбента на 1 м2 (Кокбейн, 1956; Бисвас и Хейдон, 1963).
Наблюдаемые расхождения в результатах можно объяснить
различными процессами гомогенизации. Согласно данным, большинство
молекул не переплетаются и расположены в виде свободных
спиралей. Стабильность таких эмульсий к коалесценции в основном была
приписана «механическим свойствам» межфазных пленок (Кумпер
и Александр, 1950; Лоуренс, 1952; Дерягин и др., 1954; Никитина,
1966; Кокбейн и Мак-Роберте, 1953; Бисвас и Хейдон, 1960, 1962а,
1962b; Сривастава и Хейдон, 1964; Нильсен и др., 1958; Эль Шими
и Измайлова, 1966). Только сравнительно недавно была
систематически обоснована корреляция в работах Хейдона и др. между
стабильностью эмульсий и прочностью монослоев.
Исследованы механические свойства монослоев протеинов
(Тачибана и др., 1957; Тачибана и Инокучи, 1953; Мотомура, 1964;
Мотомура и Матуура, 1963; Мотомура и Соботка, 1964). Однако следует
быть осторожным в интерпретации результатов, так как
органические разветвленные молекулы могут видоизменять структуру.
Например, Тачибана и др. (1957) показали, что изопропиловый спирт
может полностью и мгновенно удалить эластичный компонент из
монослоев яичного альбумина (как установлено при использовании
реологической модели Максвелла—Фойгта). Большинство авторов
применяли метод Криддла и Медера (1955). Мотомура (1964) было указано,
что в этом случае возможны ошибочные результаты при осцилля-
ционных экспериментах. Однако, если показать, что структура
монослоя не разрушается при вибрации, то этим методом можно получить
результаты с помощью простой формулы.
Наиболее тщательное изучение данного вопроса проведено Бисва-
сом и Хейдоном (1962а, Ь) на эмульсии петролейного эфира,
стабилизированной растворами бычьего сывороточного альбумина (БСА).
Результаты показывают прямую связь между стабильностью
эмульсий и вязкоэластичными параметрами пленок стабилизатора,
образуемых на межфазной поверхности петролейный эфир — вода. Эти
данные сопоставлены со стабильностью капель масла, находящихся
на межфазной поверхности масло — раствор протеина.
Бисвас и Хейдон получили двумерные релаксационные кривые
предела текучести пленки методом Тачибана и Инокучи (1953) и
выразили их в форме реологической модели Максвелла — Войгта,
определив таким образом цифровые данные для коэффициентов
эластичного сдвига и вязкости. В действительности они нашли, что эти две
величины тесно связаны. Это объясняется образованием молекулами
протеина сетчатых структур. Каждый из двух параметров может быть
рассмотрен при анализе связи стабильности с коалесценцией
(табл. И.1).
Нильсен и др. (1958) связали механические свойства различных
поверхностей раздела, стабилизированных высокомолекулярными
ПАВ, со стабильностью капель, хотя в качестве параметра для
исследования была выбрана поверхностная жесткость, а не модуль
упругости. Жесткость была измерена с помощью колебательного торзионного
111
Таблица 11-1
Зависимость стабильности эмульсий от поверхностной вязкости
(Бисвас и Хейдон, 1962а, Ь)
Стабилизатор
БСА (рН 5,0^
БСА (рН 5,0) 0,01 М
раствор КС1
Пепсин (рН 2,5)
G',
дин/см
19
19
40
Поверхностная вязкость
при сдвиге
па
7,6 • 103
2,1-103
4,5-103
дин/см
0,116
0,165
0,116
Стабильность xi / ,
/ 2
сек
М/В
4500
4440
6000
В/м
5
5
маятника, вследствие чего структура некоторых веществ могла
быть разрушена. Лоуренс (1952) подверг сомнению выбор жесткости
в качестве стабилизирующего фактора на том основании, что жесткая
пленка не способна «залечить» малейшие повреждения адсорбционного
слоя. Наблюдаемая зависимость стабильности от жесткости может,
таким образом, быть проявлением другого параметра, определяющего
стабильность. Только полный анализ вязко-эластичных свойств
адсорбционных пленок внесет полную ясность.
Упомянутые выше соотношения получены для вязко-эластичных
параметров сдвига. Можно предположить, что более пригодными для
обоснования стабильности являются свойства адсорбционной пленки
при расширении. Двумерная сжимаемость и эластичность пленки
при расширении выражаются кривыми сила — площадь, однако
данных о вязкости при расширении для макромолекул в литературе
не содержится. В практике для жесткой макромолекулярной пленки
модуль упругости при расширении сопоставляется с модулем сдвига
(Бисвас и Хейдон, 1960, 1962а, 1962b). Другие параметры (например,
^-потенциал и межфазное натяжение), по-видимому, должны иметь
малое влияние на стабильность к коалесценции.
Значительную часть изученных высокомолекулярных
эмульгаторов составляют водорастворимые соединения, дающие эмульсии
М/В; получить эмульсию типа В/М с такими веществами не удалось.
Это показывает, что межфазная эластичность является
несущественной (ван дер Ваарден, 1958). Вероятно, устойчивость будет зависеть
от плотно упакованной пленки, которая противодействует утончению
жидкой прослойки между каплями. Можно считать эффективным
высокомолекулярный эмульгатор, образующий эластичный гель:
он разбухает в непрерывной фазе, а попытке к сжатию этого геля
препятствуют большие осмотические силы (давление набухания).
Если эта точка зрения правильна, можно сразу определить масло-
растворимые поверхностно-активные полимеры, дающие эмульсию
типа В/М. Блочный сополимер с короткими гидрофильными-
звеньями и длинными гидрофобными цепями может соответство-
112
вать этому требованию, но необходима некоторая корреляция на
«доброкачественность» растворителя. Исследования на полиоксиэти-
леновом гликоле и блочных сополимерах окиси этилена, которые
растворимы в обеих фазах (например, ксилол и вода), привели к
постулированию «надмолекулярных» стабилизирующих слоев (Никитина
и Таубман, 1963; Зотова и Трапезников, 1964). Водный 5%-ный
раствор неионного эмульгатора стабилизирует межфазную
поверхность ксилол — вода, при этом получается чрезвычайно устойчивая
система. Несомненно, некоторая стабильность в этом случае
обусловлена отсутствием распределения стабилизатора, однако величина
этого эффекта и времени, необходимого для получения эмульсии,
говорит о наличии другого стабилизирующего фактора. В статье
Таубмана (1965) сообщается, что тонкие пленки неионного
эмульгатора оказываются серыми в белом свете, и, таким образом, прослойка
между поверхностями составляет порядка 500 А.
В заключение отметим, что величина поверхностных
реологических параметров не является мерой полной стабильности (ван дер
Ваарден, 1958). Ребиндер (1946) подчеркнул важность структурно-
механических свойств эмульсий для практики.
Стабилизация тонко измельченными порошками
Давно известно, что эффективно стабилизируют эмульсии против
коалесценции определенные высокодисперсные порошки.
Химическая природа этих частиц является менее важной, чем их
поверхностные свойства. Основные требования к ним *: 1) размер частиц
должен быть очень малым по сравнению с размером капли; 2) частицы
должны иметь определенный угол смачивания в системе масло —
вода — твердое. Твердые, сильно гидрофильные частицы (например,
двуокись кремния в среде с рН = 10) легко переходят в водную фазу;
наоборот, сильно гидрофобные частицы, в частности, твердые частицы
с очень длинными углеводородными цепями) переходят в масло.
Эмульгирование происходит частицами с соответствующим балансом
гидрофильное™ и гидрофобности, причем непрерывная фаза образует
с поверхностью раздела острый угол. Например, окись алюминия
(глинозем) способствует образованию эмульсий М/В, а газовая
сажа — В/М. Такая зависимость от смачивания изучена Шульманом
и Леем (1954) и Такакува и Такамори (1963).
Механизм стабилизации порошками должен быть изучен
количественно. Их действие преимущественно заключается в
предотвращении утончения жидкой прослойки между каплями. Необходима
непрерывная оболочка; вероятно, важен гистерезис угла контакта
для предотвращения смещения мениска. Гладкие сферические
частицы непригодны; хорошие результаты получаются с пластинчатыми
по форме частиц порошками (такими как бентонитовая глина).
Подобные частицы, находящиеся в равновесии на поверхности жидкости,
* Эти условия впервые подробно обсуждены Ребиндером (19-46). (Прим.
редактора перевода.)
'8 Заказ 1070
ИЗ
притягиваются одна к другой благодаря капиллярности. Чем меньше
радиус кривизны мениска между ними, тем сильнее притяжение
и, следовательно, значительнее когезия слоя порошка на
поверхности. Действительно, капля жидкости, деформирующаяся в
присутствии избытка порошка, способна сохранить искаженную форму при
снятии давления, так как плавающий слой частиц настолько плотно
сжат, что может выдержать отрицательное внутреннее давление.
Все сказанное выше, вероятно, объясняет явление «ограниченной
коалесценции», наблюдаемое не только с твердыми эмульгаторами,
а также с протеинами и т. д. Необходима определенная плотность
упаковки частиц или молекул эмульгатора для наибольшей
прочности пленки; первоначально образованные слои могут не иметь
времени или вещества для достижения этого состояния (Люкассен —
Рейндерс и ван ден Темпе ль, 1963). Общее уменьшение площади
поверхности, сопровождающее коалесценцию, сжимает слой до
необходимой формы).
НЕКОТОРЫЕ ВЫВОДЫ
ИЗ ИССЛЕДОВАНИЯ СТАБИЛЬНОСТИ ЭМУЛЬСИЙ
В ранней работе Кинга и Мукерджи (1939, 1940) показана разница
в кинетике скоростей коалесценции эмульсий с различными типами
эмульгаторов. Однако первой действительной проверкой
современной теории для эмульсий были работы вад ден Темпеля (1953а, Ь, с).
Им впервые вычислена сила электрического двойного слоя на каплях
масла для мыл и детергентов из данных о поверхностном натяжении
с помощью уравнения Гиббса. Он также показал, что высокий
энергетический барьер между каплями будет обеспечен при значениях
гр вплоть до 125 мв. Более низкие значения получены для систем,
содержащих NaCl, и еще ниже — MgCl2. Эти расчеты подтвердились
измерением ^-потенциалов, величина которых, как оказалось, имела
одинаковый порядок с величиной •§. Исключение составляли системы
с поливалентными солями, где величина ^-потенциалов была намного
ниже, вероятно, из-за адсорбции иротивоионов в слое Штерна.
Во второй статье ван ден Темпель (1953b) развил упрощенную
кинетическую модель флокуляции и коалесценции. Для флокуляции
в разбавленной суспензии закон изменения числа п капель имеет
следующий вид:
Согласно теории Смолуховского, константа флокуляции К0
должна быть примерно равна 1011 см3/сек. С другой стороны, если
скорость флокуляции выше скорости коалесценции, то число капель
к моменту времени т будет равна *
п = п0 [1 — ехр (\—К-с)]/Кт
где К — константа скорости коалесценции, сек'1.
* Ван ден Темпель утверждает, что коалесценцпя описывается уравнением
первого порядка. Однако он исследовал эмульсии различной дисперсности,
а дисперсность резко влияет на стабильность эмульсий. По нашему мнению
114
Для систем, в которых флокуляция и коалесценция протекают
со сравнительными скоростями, было выведено кинетическое
уравнение, включающее как константу Ка, так и К.
В третьей статье ван ден Темпель (1953с) приводит
экспериментальные данные, полученные путем микроскопического анализа
эмульсий масла с плотностью 1,01 г/см3 в растворах ПАВ. Оказалось,
что эти эмульсии являются умеренно стабильными. Кинетика чистой
флокуляции в начальной стадии следовала приблизительно
ожидаемой зависимости: 1/с„ линейна от времени т. При добавлении солей
двухвалентных катионов увеличение скоростей более эффективно,
чем при добавлении одновалентных катионов, анионы же не
оказывают влияния. Хотя этот вывод и ожидается из теории двойного слоя,
абсолютное значение К0 найдено высоким: 10"12—30-Ю"11 см3/сек,
т. е. несмотря на присутствие стабилизатора оно иногда превышало
теоретическое значение для быстрой коагуляции.
Ван ден Темпель указал несколько вероятных причин высокого
значения К0.
1. Хотя соприкосновению капель препятствует высокий
электрический барьер двойного слоя, микроскопические капли
притягиваются на больших расстояниях и должны флокулировать во
вторичном минимуме. Следовательно, из-за этого значение К0 должно
превышать 10" и сек~*.
2. Эффект Мюллера для полидисперсных золей предсказывает
большие скорости, чем для монодисперсных.
3. В эмульсиях с довольно широкой областью распределения
частиц по размеру любое гравитационное движение (оседание)
увеличивает скорость столкновения (ортокинетическая флокуляция).
4. Ортокинетическая флокуляция может быть вызвана
перемешиванием при смешении эмульсий с растворами солей.
В последующей статье (1957) ван ден Темпель приводит
результаты исследований высококонцентрированных эмульсий (например,
80% медицинского парафина в растворе додецилсульфата натрия,
аэрозоль ОТ). Эти эмульсии можно рассматривать как полностью
флокулированные, так как частицы соприкасаются со многими
соседними; существует только устойчивость к коалесценции. Значение
константы скорости коалесценции К колеблется от 10"3 сек~1 для
нестабильных эмульсий до 10"7 сек'1 для стабильных.
(Высокостабильные эмульсии, такие как эмульсии, защищенные протеиновыми
пленками, имеют меньшие скорости коалесценции, но являются
неудобными для экспериментального изучения). Некоторые эмульсии
показали ограниченную коалесценцию, причем значение К
уменьшалось до нуля после достижения определенной стадии.
Лоуренс и Миллс (1954) провели тщательное исследование
кинетики коалесценции разбавленных эмульсий (1%-ных) типа М/В,
процесс коалесценции капель должен соответствовать уравнению второго
порядка, так как капли практически всегда коалесцируют попарно. Это
подтверждается эксперимента льпг,*-" ::п;гтты.мп ряда авторов (см. стр. 417). (Прим.
редактора перевода.)
115
используя фотомикрографическип анализ размера частиц. Эмульсии
были гомогенизированы до размера частиц '--2 мкм. (Для таких работ
рекомендован 3,3-дитолил, так как он имеет плотность, близкую
к плотности воды, и высокий показатель преломления.) Исследование
показало, что средний объем частицы (пропорциональный 1/с„)
увеличивался линейно, согласно теории Смолуховского, как для
нестабилпзированных эмульсий (гидрозоли масла), так и для
эмульсий, стабилизированных олеатом натрия. Однако абсолютное
значение скорости коалесценции составляло только 0,001 скорости
коагуляции Смолуховского даже для нестабилпзированных дисперсий.
Авторы указали, что капли парафина в воде имели отрицательный
^-потенциал, который мог объяснить степень стабилизации. В
присутствии мыла скорость коалесценции уменьшилась до порядка
10"5 скорости Смолуховского.
Сривастава и Хейдон (1964), исследуя высокостабильные к
коалесценции эмульсии (1%-ный петролейный эфир, диспергированный
в 0,1%-ном бычьем сывороточном альбумине + 0,01 М раствор КС1),
смогли измерить скорость флокуляции. Оказалось, что около изо-
электрической точки протеина флокуляция нроисходила в течение
нескольких часов; при других значениях эмульсии становились
полностью диспергированными или некоторое время слабо
агрегировали.
Особенно интересные наблюдения проведены на эмульсиях со
значениями и-потенциала 20 .мвили —20 мв. Обнаружено почти
постоянное соотношение одиночных и двойных капель, что указывает на
обратимое равновесие между флокуляцией п диспергированием.
Авторы считают, что эти результаты соответствуют теории ДЛВО.
Для капель диаметром 2 мкм и -ф «^ 20 мв энергетический барьер
должен быть таким высоким, чтобы предотвратить соприкосновение,
а вторичный минимум — неглубоким (—8 к Г), чтобы вызвать
обратимую агрегацию (см. рис. II.3). Кроме того, установлено, что
соотношение дуплетов увеличивалось примерно на вычисленную
величину при изменении ^-потенциала от —20 до —23 мв.
Для точной проверки систем по теории ДЛВО монодисперсные
полимерные латексы почти идеальны. Хотя строго они и не
являются эмульсиями, но абсолютно эквивалентны им, когда речь
идет о первичной ступени флокуляции (за исключением отсутствия
вторичного минимума, по крайней мере, для латекса с размером
частиц < 0,1 мкм). Однако Оттевилл и Шоу (1966) и Ватиллон и
Джозеф-Петит (1966) в своих работах показали, что эксперимент хорошо
согласуется с теорией ДЛВО только качественно. Причина этих
расхождений до сих пор не установлена.
Ряд очень интересных экспериментов выполнен с целью прямого
измерения сил, действующих между каплями. Первую значительную
работу провел ван ден Темпель (1958). Он использовал метод Деря-
гина и Титиевской (1957) для измерения толщины водной прослойки
между каплями масла, находящимися под известным
гидростатическим давлением. Для парафиновых капель в 0,01 М растворе доде-
110
цилсульфата натрия наблюдались стабильные равновесные нленки
о
толщиной в пределах от —220 до 100 А (последняя в присутствии
0,2 М раствора КС1). При такой толщине прослойки вандерваальсово-
давление (намного превышающее гидростатическое)
уравновешивается отталкиванием двойного слоя. Другими словами,
расплющенные капли находятся во вторичном минимуме.
Эти результаты подтверждены Девисом (1964) на смесях додецил-
сульфата натрия и неионного детергента НД15 с использованием
циклогексана в качестве масляной фазы. Наличие вторичного мини-
. мума подтверждается только на разбавленных растворах. Толщина же-
прослойки при большой ионной силе зависит от стерических
затруднений предельного монослоя, как в тончайших черных мыльных
пленках.
Для систем В/М исследования на липидных мембранах (Хейдовт
и Тейлор, 1966; Зонтаг, Нетзель и Кларе, 1966) показали, что даль-
недействующее отталкивание отсутствует, но маслорастворимое ПАВ
обусловливает эластичность пленки В/М/В, которая затем быстро
утончается до бимолекулярной структуры. Этот бимолекулярный
слой удерживает некоторое количество растворителя и является
жидким; вандерваальсово сжатие уравновешивается осмотическими
силами.
Новая кинетическая теория коалесценции предложена Гилли
и Книгтом (1965) на основе динамики столкновения сфер, способных
к слабой деформации. Они предположили (отчасти произвольно),
что возможность коалесценции двух сталкивающихся капель должна
быть пропорциональна j (SP) dx (где S —"поверхность; Р —
давление) независимо от стадии процесса коалесценции и что дальнедей-
ствующими силами притяжения или отталкивания можно пренебречь.
Установлено, что эмульсии, имеющие первоначально гауссовское
распределение частиц по размеру, должны сохранять такое
распределение, и обратная величина общей площади поверхности должна
линейно увеличиваться со временем. Полученные данные должны
быть в соответствии с формой зависимости Смолуховского.
В своей работе Гилли и Книгт использовали газо-кинетическую»
формулу для частоты столкновений, а не классическую диффузную-
модель. Они отметили, что только для очень неустойчивых эмульсий
скорость диффузии является определяющей; при медленной
коалесценции определяющими будут частота и продолжительность столкно-
. вений. Это интересный подход, но он не учитывает некоторые
фундаментальные трудности, такие как поверхностная эластичность при
столкновениях, образование дуплетов, триплетов и т. д. в области
вторичного минимума.
ЛИТЕРАТУРА
Al exander A. E., Schulman J. H., Trans. Faraday Soc, 36, 960
(1940).
Allan R. S., Mason S. G., Trans. Faraday Soc, 57, 2027 (1961).
Allan R. S., Mason S. G-, J. Colloid Sci., 17, 383 (1962).
В art ok W., Mason S. G., J. Colloid Sci., 14, 13 (1959).
117
Biswas В., Haydon D. A., Proceedings of the Third International
Congress on Surface Activity, Cologne, 1960.
Biswas В., Haydon D. A., Kolloid. Z., 185, 31 (1962 a).
Biswas В., Haydon D. A., Kolloid Z., 186, 57 (1962 b).
Biswas В., Haydon D. A., Proc. Roy Soc, A271, 296 (1963).
Booth F., Discuss. Faraday Soc, 18, 104 (1954).
С a s i m i г Н. В. G., Polder D., Nature (London), 158, 787 (1946).
С a s i m i г Н. В. G., Polder D., Phys. Rev., 73, 360 (1948).
Chapelear D. C, J. Colloid Sci., 16, 186 (1961).
Charles G. E., Mason S. G., J. Colloid Sci., 15, 105 (1960 a).
Charles G. E.. Mason S. G., J. Colloid Sci., 15, 236 (I960 b).
Charles G. E., Mason S. G-, J. Colloid Sci., 16, 150 (1961).
Cheesman D. F., King A., Trans. Faraday Soc, 36, 241 (1940).
Clifford J.. Pethica B. A., Trans. Faradav Soc, 60, 1483 (1964).
Clifford J., Pethica B. A., Trans. Faraday Soc, 61, 182 (1965 a).
Clifford J., Pethica B. A., Trans. Faraday Soc, 61, 1276 (1965 h).
Clifford J., Pethica B. A., Senior W. A., Ann. N. Y. Acad. Sci.,
125, 458 (1965).
Cockbain E. G., Trans. Faraday Soc, 50, 874 (1954).
С о с к b a i n E. G., J. Golloid Sci., 11, 575 (1956).
Cockbain E. G., McRoberts T. S., J. Colloid Sci., 8, 440 (1953).
Criddle D. W., Meander A. L., Jr., J. Appl. Phys., 26, 838 (1955).
Cum per С W. N., Alexander A. E.. Trans. Faraday Soc, 46, 235
(1950).
D a v i e s J. Т., in «Recent Progress in Surface Science», ed. by J. F. Danielli,
K. G. A. Pankhurt and A. C. Riddifort, vol. 2, New York — London,
p. 129.
D a v i e s J. Т., Mayers G. R. A., Trans. Faraday Soc, 56, 690 (1960).
D a v i e s J. Т., R i d e a 1 E. K., Interfacial Phenomena New York —
London. 1961.
Da vies J. Т., Voso R. W., Proc. Roy. Soc.-. A286, 218 (1965).
Derjaguin B. V., Kolloid Z., 69, 155 (1934).
Derjaguin B. V., Trans. Faraday Soc, 36, 203 (1940).
Darjaguin B. V., Nineteenth Symposium of the Society of
Experimental Biology, Swansea 1964, p. 55.
Derjaguin B. V., Discuss. Faraday Soc, 1966.
Derjaguin B. V., Landau L., Acta. phys. chim. URSS, 14, 633 (1941).
Derjaguin B. V., Ti tie vskaya A. S., Proceedings of the 2nd
International Congress on Surface Activity, vol. 1, London. 1957, p. 536.
Derjaguin B. V., Vlasenko G. J., J. Colioid Sci., 17, 605 (1962).
Derjaguin B. V. et al., Discuss. Faraday Soc, 18, 32 (1954).
Derjaguin B. V., M a r t i n о v G. A., Gutop Yu. V., Kolloid Z.,
27, 298 (1965a).
Derjaguin B. V., M а г t i n о v G. A., Gutop Yu. V., Kolloid Z., 27,
257 (1965b).
DeVries A. J., Foam Stability, Delft, Holland, 1957.
DeViies A. J., Rec Trav. chim., 77, 81, 209, 283, 383 (1958).
DeVries A. J., Third International Congress on Surface Activity, vol. 2,
London, 1960, p. 566.
D e V e r e u x O. F., de В г u у n P. L., Interaction of Plane Parallel Double
Layers, Cambridge, Massachusetts, 1963.
D u у v i's E. M., Tesis Utrecht University, 1964.
D z у a z 1 о s h i n s к i i I.E., Soviet. Phys. JETB, 37, 161 (1960).
El S h i m i A. F., I z m a i 1 о v a V. N., Tenside, 3, 23 (1966).
Ewers W. E., Sutherland K. L., Aust. J. Sci. Res., A5, 697 (1952).
F о w к e s F. M., Ind. Eng. Chem., 56, 40 (1964).
Frankel S. P., Mysels K. J., J. Phys. Chem., 66, 190 (1962).
Garner F. H., M i n a P., Trans. Faraday Soc, 54, 1607, 1616 (1958).
Garner F. H., Mina P., Trans. Faraday Soc, 55, 1627 (1959).
Garrett E. R., J. Am. Pharm. Assoc, 51, 35 (1962).
118
Gillespie Т., R i d e a 1 E. K., Trans. Faraday Soc., 52, 173 (1956).
Gregory J., Ph. D. Thesis, University of London, 1963.
Hachisu S., F u г u s a w a K., Sci. Light (Tokyo), 11, 157 (1962).
Hachisu S., Furusawa K., Sci. Light (Tokyo), 12, 1, 157 (1963).
Hachisu S., Furusawa K., Sci. Light (Tokyo), 15, 115 (1966).
H a d a m a r d J. S., С R., 122, 1735 (1911).
Hanai Т., Haydon D.A., Taylor J. Proc. Roy. Soc, A281, 377 (1964).
H a n a i Т., Н а у d о n D. A., J. Theoret. Biol., 11, 539 (1966).
Hansen R. S. Mann J. A., J. Colloid Sci., 18, 805 (1963).
Hartley G. S., Aqueous Solutions of Parafin Chain Salts, Paris, 1936.
H a u d о n D. A., in «Recent Progress in Surface Sciense», ed. by J. F. Dani-
clli, K. G. A. Pankhurst, A. C. Liddiford, New York — London, 1964.
Haydon D. A., Third International Congress on Surface Activity, Collogne
B, 1960, p. 341.
Haydon D. A., Hanai Т., Taylor J., J. Theoret. Biol., 9, 278
(1965 a).
Haydon D. A., Hanai Т., Taylor J., J. Theoret. Biol., 9, 424 (1965 b).
Haydon D. A., Hanai Т., Taylor J., J. Theoret. Biol., 9, 433,
(1965 c).
Haydon D. A., Hanai Т., Taylor J., J. gen Physiol., 48, 59 (1965 d).
Haydon D. A., Hanai Т., Redwood W. R., Ann. N. Y., Acad.
Sci., 137, 731 (1966).
Haydon D. A., Srivastava S. N., Proceedings of the Fourth
International Congress of Surface Activity, Brussel, 1964.
Haydon D. A., Taylor I., Discuss. Faraday Soc, 42, 51 (1966).
H i d у G. M., J. Colloid Sci., 20, 123 (1965).
H i d у G. M., L i 1 1 у D. K., J. Colloid Sci., 20, 863 (1965).
H i g u с h i W. I., О к a d a R., Stelter G. A., L e m b u r g e r A. P.,
J. Am. Pharm. Assoc, 52, 49 (1963).
Hill R. A. W., Knight J. Т., Trans. Faraday Soc, 61, 170 (1965).
Holmes H. N., Cameron D. N., J. Am. Chem. Soc, 44, 66 (1922).
Huang C, Thompson Т. Е., J. Molec. Biol., 13, 183 (1965).
Huang C, Thompson Т. Е., Ann. N. Y., Acad. Sci., 137, 740 (1966).
Huang C, Thompson T. E., W h e e 1 d о n L., J. Molec. Biol., 8,
148 (1964).
Johnson G. A., Goldfarb J., Pethica B. A., Trans. Faraday
Soc, 61, 2321 (1965).
Jones T. G., Durham K., Evans W. P., Camp M., Second
International Congress on Surface Activity, vol. 1, London, 1957, p. 225.
King A., Murkherjee L. N., J. Soc. Chem. Ind. (London), 58, 243
(1939).
King A., Murkherjee L. N., J. Soc. Chem. Ind. (London), 59, 185
(1940).
Kiselev A. V., Discuss. Faraday Soc, 40, 195, 203 (1965).
Kitchener L. A., in «Recent Progress in Surface Science», ed. by
J. F. Danielli, K. G. A. Panklhorst, A. C. Liddiford, vol. 1, New York —
London, 1964 a, p. 57.
Kitchener J. A., in «Recent Progress in Surface Science» ed. by J. F.
Danielli, K. G. A. Pankhurst, A. C. Liddiford, vol. 1, New York — London,
1964, b, p. 51.
kitchener J. A., Cooper C.F., Quart. Rev. chem. Soc, 13, 71 (1959).
L a n g e H., J. Soc. Cosmet. Chem., 16, 697 (1965).
Lawrence A. S. C, Nature (London), 170, 232 (1952).
Lawrence A. S. С, В 1 а к е у В. С, Discuss. Faraday Soc, 18, 268 (1954).
Lawrence A. S. V., Mills O. S., Discuss. Faraday Soc, 18, 98 (1954).
L e v i oh V. G., Acta physicochim. URSS, 14, 307, 321 (1941).
L i f s h i t z E. M., Soviet Phys. JENB 2, 73 (1956).
L i n d b 1 a d N., J. Colloid Sci., 19, 729 (1964).
L u с a s s e n J., H a n s e n R. S., J. Colloid Sci., 22, 32 (1966).
Lucassen-Reynders E. H., Kolloid Z., 197, 137 (1964).
119
Lucassen-Reynders E. H., van den Tempel M., J. phys.
Chem., 61, 731 (1963).
L у klem a J., 0 v er b e e к J. Th. G., J. Colloid Sci., 16, 501 (1961).
Lyklema J., Scholten P. C, Mysels K. J., J. Phys. Chem.,
69, 116 (1965).
Mackay G. D. M.. Mason S. G., Can. J. Chem. Eng., 41,203 (1963).
Maclachlan A. D., Proc. Roy. Soc, A271, 387 (1963).
Maclachlan A. D., Discuss. Faraday Soc, 40, 239 (1965).
Mauley R. St. J., Mason S. G., J. Colloid Sci., 7, 354 (1952).
Martinez Moreno J. M. et al., J. Am. Oil Chem. Soc. 37, 582
(1960).
Martinez Moreno J. M., Catalina D. F., Gomez Her-
г е г a C, Fette, Seifen, Anstrichm., 63, 915 (1961).
Miles G. D., Ross J., Shedlovski L., J. Am. Oil Chem. Soc,
27, 268(1950).
Motomura K., J. Phys. Chem., 68, 2826 (1964).
Motomura K., Matuura J., J. Colloid. Sci.. 18, 295 (1963).
Motomura K., S о b о t к а Н., Proceedings of the Fourth International
Congress on Surface Activity, Cologne, 1964.
Mysels K. J., Cox M. C, J. Phys. Chem., 65, 1107 (1961).
Mysels K. J., Cox M. C, J. Colloid Sci., 17, 136 (1962).
Mysels K. J., Cox M. C, Skewis J. D., J. Phys. Chem., 65, 1107
(1961).
Nemethv G., S с he rag a H. A., J. Chem. Phys., 36, 3382 (1962 a).
Nemethy G., Scheraga H. A., J. Chem. Phys., 36, 3401 (1962 b).
Nemethy G., Scheraga H. A., J. Chem. Phys., 66, 1773 (1962 c).
Nielsen L. E., Wall R.. Adams G., J. Colloid Sci., 13, 441 (1958).
Ni kit in a S. A., Tenside, 3, 25 (1966).
Ottewil R. H., Rastogi M. L., Trans. Faraday Soc, 56, 866 (1960).
Ottevill R. H., Shaw J. N., Discuss. Faraday Soc, 42, 154 (1966).
Ottewill R. H., Wilkins D. J., J. Colloid. Sci., 15, 512 (I960).
Ottewill R. H., Wilkins D. J., Trans. Faraday Soc, 58, 698 (1962).
Overbeek I. Th. G., in «Colloid Science», ed. by H. R. Kruyt, Amsterdam,
1952 a.
Overbeek I. Th. G., in «Colloid Science», ed. by H. R. Kruvt. Elsevier —
Amsterdam, 1952 b, p. 278.
Overbeek J. Th. G., in «Colloid Science», ed. by H. R. Kruyt, Elsevier —
Amsterdam, 1952 c, p. 286.
Padday J. F., Russell D. R., J. Colloid Sci., 15, 503 (1960).
Pangonis W. J., Heller W., Jacobson A., Tables of Light
Scattering Functions for Spherical Particles, Detroit — Michigan, 1957.
Platikanov D., J. Phys. Chem., 68, 619 (1964).
Powis F., Z. phys. Chem., 89, 186 (1914).
Princen H. M., Mason S. G., J. Colloid. Sci., 20, 156 (1965).
P r i n s A., van den Tempel M., The IV International Congres on
Surface Activity, Brussels, 1967.
Re h binder P. A., Kolloid Z., 12, 157 (1946).
Rehbinder P. A., Wenstrom E. K., Kolloid Z., 53, 145 (1930).
Rehfield S. J., J. Phys. Chem., 66, 1969 (1962).
Rumscheldt E. D., Mason S. G., J. Colloid Sci., 16, 238 (1961).
Saleeb F. Z., Kitchener J. A., The IV International Congress on
Surface Activity, Brussels, 1964.
Schulman J. H., Cock bain E. G., Trans. Faradav. Soc, 36, 651,
661 (1940).
Schulman J. H., Leja J., Trans. Faraday Soc, 50, 598 (1954).
Serralach J. A., Jones G., Ind. Eng. Chem., 23. 1016 (1931).
Serralach J. A., Jones G., Ind. Eng. Chem., 25, 816 (1933).
Schenkel J. H., Kitchener J. A., Trans. Faraday Soc, 56", 161
(196ii).
S h e 1 u d к о A., Proc. K. ned. Akad. Wet., B65, 76 (1962 a).
120
S h e 1 u d к о А., Ргос. K. ned. Akad. Wet., B65, 91 (1962 b).
Sheludko A., Colloid Chemistry, Elsevier — Amsterdam, 1966 a, p. 198.
Sheludko A., Colloid Chemistry, Elsevier — Amsterdam, 1966 b, p. 173.
Sheludko A., P 1 a t i к a n о v D., Kolloid Z., 175, 150 (1965).
Sheludko A., Tchernev R., The IV International Congress on Surface
Activity, Brussels, 1964.
Sheludko A., T h i e s s e n D., Proceedings of the III International
Congress on Surface Activity, Berlin, 1966; Tenside, 3, 23 (1966).
Sheludko A., Platkanov D., Manev E., Disc. Faraday Soc,
40,253 (1965).
Sherman P., J. Soc. Cosmet. Chem., Symposium on Emulsions, Harrogate,
March, 16,591, 1965.
Shotton E. et. al., Proceedings of the IV International Congress on Surface
Activity, Cologne, 1964.
S о n n t a g H., Z. phys. Chem., 221, 365 (1962 a).
Sonntag H., Z. phys. Chem., 221, 373 (1962b).
Sonntag H., Klare H., Z. phys. Chem., 223, 8 (1963).
Sonntag H., Klare H., Tenside, 2, 365 (1965).
Sonntag H., Netzel J., Tenside, 3, 45, 46 (1966 a).
Sonntag H., Netzel J., Proceedings of the III International Conference
on Surface Active Substances, Berlin, 1966b; Tenside, 3, 25 (1966).
Sonntag H., Netzel J., Kolloid Z., 211, 122 (1966 c).
Sonntag H.. Netzel J., Tenside, 8, 296 (1966 d),
Sonntag H., Netzel J., Klare H., Kolloid Z., 211, 121 (1966).
S p a a r n a v M. M. J., in «Rheology of Emulsions», ed. by P. Sherman, Oxford,
1962.
Srivastava S. N., H а у d о n D. A., Proceedings of the IV
International Congress on Surface Activity, Brussels, 1961.
Swift D. L., Fried lander S. K., J. Colloid. Sci., 19, 621 (1964),
T а с h i b a n а Т., I n о к u с h i K., J. Colloid Sci., 8, 341 (1953).
Tachibana Т., Inokuchi K., Inokuchi Т., Biochem. biophys.
acta, 24, 174(1957).
Takakuwa Т., Takamori Т., Proceedings of the VI International
Congress on Processes of Mineralogy, Cannes — Oxford, 1963.
T u о г i 1 1 a P., Kolloidchem. Beih., 22, 191 (1926).
Turkevich J., Am. Sci., 47, 97 (1959).
van Ark el A. E., Kruyt H. R. Rec. trav. chim., 39, 656 (1920).
Van den Hulst H. C, Light Scattering by Small Particles, New York,
1964.
Van den Berg, J. Molec. Biol., 12, 290 (1965).
Van den Tempel M., Rec. trav. chim., 72, 419 (1953 a).
Van den Tempel M., Rec. trav. chim., 72, 433 (1953 b).
Van den Tempel M., Rec. trav. chim., 72, 442 (1953 c).
Van den Tempel M., Proceedings of the 2 nd International Congress
on Surface Activity, vol. 1, 1957, p. 439.
Van den Tempel M., J. Colloid Sci., 13, 125 (1958).
van den Tempel M., Erkens Th. F., van Voorst Vader F., Trans.
Faraday Soc, 60, 1171 (1964).
van den Tempel M., Lucassen J., Lucassen-Reyn-
ders E. H., J. Phys. Chem., 69, 1798(1965).
Van der Waarden M., Kolloid Z., 156, 116 (1958).
Van Olphen H., An Introduction to Clay Colloid Chemistry, New York,
1963.
Van Voorst V a d и г F., Fette, Seifen, Anstrichm., 66, 44 (1964).
Verwey E. J. W., О v e r b e e к J. Th. G., Theory of Stability of Lyop-
hobic Colloids, Elsevier — Amsterdam, 1948, p. 128.
Void M. J., J. Colloid Sci., 16, 1 (1961).
Void R. D., Groot R. C, J. Phys. Chem. (Ithaca), 66, 1969 (1962).
Void R. D., Groot R. C, J. Soc. Cosmet. Chem., 14, 233 (1963).
Void R. D., Groot R. C, J. Colloid. Sci., 19, 384 (1964).
121
V г i j A., Discuss. Faraday Soc, 42, 23 (1966).
W а с h t e 1 R. E., La M e г V. К., J. Colloid Sci., 17, 531 (1962).
Wang С S., Fried lander S. K., J. Colloid Sci., 22, 128 (1966).
W a t i 1 1 о n A., Van Grunderbeck F., Bull. Soc. chim. Belg., 63,
115 (1954).
Watillon A., Joseph-Petit A. M., Discuss. Faraday Soc, 42, 143
(1966).
Wigner G., Kolloid Z., 58, 167 (1932).
Wiersema P. H., Thesis of the University of Utrecht, 1964.
Дерягин Б. В., ДАН СССР, 109,967 (1956).
Дерягпн Б. В., Власенко Г. К., ДАН СССР, 2,155 (1948).
Дерягин Б. В., Власенко Г. Я., Коллопдн. ж., 4, 249 (1957).
Дерягин Б. В., Щербаков Л. М., Коллопдн. ж., 20, 33 (1961).
Д ж о в а н о в и к Д. С, Коллопдн. ж., 203, 48 (1965).
Зотова К. В., Трапезников А. А., Коллоидн. ж., 26, 263 (1964).
Лпфшпц Б. М., ЖЭТФ, 29, 94 (1955).
Никитина С. А., Таубман А. В., ДАН СССР, 149, 905 (1963).
Таубман А. Б., Коллопдн. ж., 27, 238 (1965).
Таубман А. Б., Корецкпн А. Ф., Коллоидн. ж., 20, 631 (1958).
Дополнительная литература
A d a m s о n A. W., J. Coll. Interf. Sci., 29, № 2, 261 (1969). Модель мицел-
лярных эмульсий.
Anioloncska M., Leszczynskia H., Z. Eisner Farm. Pol.. 23,
№ 5—6, 403 (1967). Влияние температуры на стабильность эмульсионных
мазевых основ.
В imlinger G., Am. Perfum. a Cosmet., 82, №.12, 31 (1967). Стабильность
эмульсий.
Brown A. H., Brit. Chem. Eng., 13, №12, 1719(1968). Механизм коалес-
ценцпи жидкостей.
Brown A. H., Chem. a. Ind., Л? 30, 990 (1968). Коалесценция жидкостей.
D a v i e s J. Т. at al., Recent Progress in Surface Science, 2 ed., New York,
1964. Обзор.
Ford R. E., Furmidge С G. L., J. Coll. Interf. Sci., 22, №4,331
(1966). Исследование межфазных поверхностей эмульсий.
Gagnare В., Double Liaison, №143, 823(1967). Поверхностно-активные
вещества.
Н а г t 1 a n d S., Trans. Inst. Chem. Eng., 46, № 10, 275 (1968). Коалесценция
жидких капель на поверхности жидкость — жидкость. Влияние ПАВ.
Hartland S., Trans. Inst. Chem. Eng., 45, № Зт, 97 (1967). Коалесценция
жидких капель на поверхности жидкость — жидкость.
Hodgson Т. D., Lee J. С, J . Coll. Interf. Sci., 30, № 1, 94 (1969).
Влияние ПАВ на коалесценцию капли на поверхности раздела.
I s h i d a S., S о п о d а Т., Y о s h i d а Т., Jukagaku, 17, № 10, 562 (1968).
Стабильность эмульсий. I. Стабильность капель на поверхности
раздела М/В.
Jain S. P., Sri vast a va S. N.. Kolloid. Z. u. Z. Polym., 235, № 1,
1230 (1969). Роль частично флокулированных золей в стабильности
эмульсий.
К aye R. С, S eager H., Instr. Pract., 20, 733 (1966). Измерение
стабильности эмульсий.
L а с h a m p t F., Villa R. M., Am. Perfum. a Cosmet., 82, № 1, 29 (1967).
Новое в изучении эмульсий.
Lachampt F., Villa R. M., Am. Perfum a. Cosmet., 85, № 1, 27 (1970).
Эмульсии.
L у k 1 e m a J., Adv. Coll. Interf. Sci., 2, № 2, 65 (1968). Основы устойчивости
лиофобных коллоидных систем.
122
M a h г о u s H., Diss. Abstr., 28, № 8, 3360 (1968). Влияние некоторых
полимерных веществ на стабильность эмульсий гексан — вода.
Marrucci G., Chem. Eng. Sci., 24, № 6, 975 (1969). Теория коалес-
ценции.
Mukerjee L. N., Gupta О. P., Indian J. Appl. Chem., 31, № 3, 90
(1968). Использование твердых эмульгаторов (гидроокисей Сг, Al, Fe).
Mukerjee L. N., Gupta O. F., Indian J. Appl. Chem., 32, № 4, 215
(1969). Использование твердых веществ для стабилизации эмульсий.
Часть III. Влияние физического состояния порошка гидроокиси алюминия
на стабильность эмульсий.
Mussel 1 white P. R., Kitchener J. A., J. Coll. Interi. Sci., 24,
K« 1, 80 (1967). Предельная толщина протеиновых пленок.
N i k i t i n a S. A., Abh. Deutsch. Akad. Wiss., Berlin 1966, (6), 609 (1967).
Образование структуры в адсорбционных пленках в эмульсиях и их
стабильность.
Petersen R. V., Umschau, 69, JV« 3, 85 (1969). Неводные эмульсионные
системы.
Prakash С, Srivastava S. N., Bull. Chem. Soc. Japan, 40, № 8,
1756 (1967). Роль макромолекул в стабильности эмульсий и пепсин как
эмульгатор.
Prince L. M., J. Coll. Interf. Sci., 23, № 2, 165(1967). Теория водных
эмульсий. Отрицательное поверхностное натяжение на поверхности
раздела В/М.
Prince L. M., J. Coll. Interf. Sci., 29, № 2, 216 (1969). Теория водных
эмульсий. II. Механизм искривления пленок на поверхности раздела М/В.
Rao U. N., Indian J. Pharm., 29, № 6, 182 (1967). Влияние концентрации
мыла на стабильность эмульсий.
Rimlinger С, Parfum., Cosmet., Savons, 9, № 12, 553 (1966).
Стабильность эмульсий.
Rimlinger С, Parfum., Cosmet., Savons, 11', № 1, 22 (1968). Основные
проблемы эмульсий.
Rimlinger G., Parfum., Cosmet., Savons, 13, № 8, 605 (1970). Обобщение
«смешанной» теории эмульгирования.
R i t с hi F. Mc, Nature, 215, № 5106, 1159 (1967). Барьер для коалесценцпи
в стабильных эмульсиях.
Robinson J. R., Am. Perfum. Cosmet., 83, № 8, 27 (1968). Эмульсии.
Rosa no H. L., Peiser R. C, Eydt A., Rev. Fr. Corps Gras., 16,
JN» 4, 249 (1969). Микроэмульсии.
S с h m о 1 k a J. R., Fed. Proc, 29, № 5, 1717 (1970). Теории эмульсий
(обзор).
S о n n t a g H., Abh. Deutsch. Akad. Wiss., Berlin, 1966, (6), 551 (1967).
Свойства жидких пленок между каплями эмульсии.
Sonntag Н., К 1 а г е Н., Tenside, 4, № 4, 104 (1967). Влияние
водорастворимых ПАВ, внешних электрических полей и температуры на
стабильность к коалесценции эмульсии В/М.
Sumner С. G., J. Appl. Chem., 7, №9, 504 (1967). Фазовые отношения
и стабильность эмульсий.
Torza S., Ma sop S. G., Sci., 163, № 3869, 813 (1969). Коалесценция двух
капель, состоящих из несмешнвающихся жидкостей.
Wiese G. R., Heal у Т. W., Trans. Faraday Soc, 66, № 2, 490 (1970).
ч Влияние размера частиц на стабильность коллоидов.
Zlochower I. A., Schulman J. H., J. Coll. Interf. Sci., 24, № lr
115 (1967). Изучение молекулярного взаимодействия и подвижности на
поверхности раздела ж — ж посредством ЯМР. •"•
Бабанян Г. А., Ахмадеев М. Х-, ДАН СССР, 179, № 1, 123 (1968). ^
Исследование процесса коалесценцпи капель в эмульсиях методом ско- -
ростной киносъемки.
Барбой В. М., Глазман Ю. М., Ребиндер П. А., Ф у к с Г. И.,
Щукин Е. Д., Коллоидн. ж-, 32, № 4, 480 (1970). О термодинамически
равновесных двухфазных дисперсных системах.
123
Боброва Л. Е-. Измайлова В. Н., X о л л е р В. А., Коллоидн.
ж.. 32, № 6, 662 (1970). Тепловой эффект ирп гелеобразованин желатины.
Бовкун О. П., Маркина 3. Н., Цпкурина Н. Н., Коллоидн.
ж., 32, Л« 6, 831 (1970). Исследование эмульгирующей способности хелата
натрия.
Воларович М. П., Авдеев Н. Я-., Коллоидн. ж., 32, Лг 1, 38 (1970).
Характеристика кинетики коалесценции каиель на поверхности раздела
ж — ж.
Иванова Н. Н. и др.. Коллоидн. ж., 31, Х« 1, 63 (1969). Влияние
электролитов и температуры на устойчивость латекса, стабилизованного
неионным эмульгатором.
И з м а й лова В. Н. и др., ДАН СССР, 191, Л» 5, 1081 (1970). Исследование
межфазного п объемного структурообразованпя в концентрированных
эмульсиях бензола, стабилизованных желатиной.
Никитина С. А., Симакова Г. А., Коллоидн. ж., 31. Л» 5, 73и
(1969). Эмульгирующее действие оксиэтплпрованных жирных спиртов
(спнтанолов).
Пригородов В. Н., Никитина С. А., Таубман Л. В.,
Коллоидн. ж-. 30, № 4, 569 (1968). Стабилизация концентрированных
эмульсий в условиях переноса поверхностно-активного эмульгатора через
поверхность раздела фаз.
Романова 3. Т., Баренбойм Н. К., Труды Московск. технол. нн-та
легк. пром., вып. 36, 1969, стр. 89. Определение устойчивости дисперсных
систем.
Т а н а к и М., Н а к а д а К., О я м а И., Кагаку кагаку, 34, № 8, 893
(1970). Коалесценция диспергированных капель в реакторе с
перемешиванием .
Эль- Ш и м и А. Ф., И з м а й л о в а В. Н., Коллоидн. ж., 33, Л! 2, 287
(1971). Влияние прочности межфазных адсорбционных слоев яичного
альбумина на коалосцснцшо углеводородных капель.
СТАБИЛЬНОСТЬ ЭМУЛЬСИИ
ПРИ ЗАМЕРЗАНИИ
И ОТТАИВАНИИ
Глава III
ОСНОВНЫЕ
СВОЙСТВА
♦ ЭМУЛЬСИЙ
И ИХ СТРОЕНИЕ
Ф. Шерман
Хотя структурные изменения в
эмульсиях, подвергающихся
действию низких температур,
изучались подробно, механизм этих
процессов известен недостаточно.
Эмульсии для исследования
температурных влияний приготавливают
двумя способами: 1) замораживается
весь объем эмульсии; 2)
замораживается тонкая плёнка эмульсии,
нанесенная на предметное стекло.
При замерзании водной фазы
эмульсии типа М/В появляются
кристаллы льда, которые выталкивают
шарики масла в сужающиеся каналы
незамерзшей жидкости (Янг, 1934).
При этом концентрация
электролитов в еще незамерзшей воде
увеличивается, вода все более
переохлаждается, электрический заряд эмульсии
уменьшается (Боросихино и др., 1961).
В результате роста кристаллов льда
шарики масла сжимаются,
вытягиваются в нити и соединяются.
Согласно Лебедеву и др. (1962),
последующие процессы зависят от условия
контакта поверхности шарика и
адсорбционного слоя эмульгатора.
Когда вязкость поверхности шарика
достигает вязкости твердого вещества,
гидрофобная часть адсорбированных
молекул эмульгатора теряет свою
подвижность. Это предотвращает
деформированные шарики масла,
находящиеся под давлением кристаллов
льда, от восстановления. В той части
поверхности шарика, которая не
защищена эмульгатором, начинается ко-
алесценция, зависящая от природы
эмульгатора (Поспелова и др., 1962),
его концентрации, степени покрытия
эмульгатором поверхности шарика
и от природы дисперсной фазы (Кист-
лер, 1936). Длина гидрофобной
125
цепочки молекулы эмульгатора влияет на степень коалесценции, а
природа гидрофильной группы—на степень дегидратации при
замерзании эмульсии. Оптимальный баланс между этими группами
уменьшает коалесценцию при замерзании.
Поспелова и др. (1962) предположили, что размер пустот между
кристаллами льда влияет на стабильность при замерзании, так как
шарики диаметром меньше, чем ширина пустот, не должны
подвергаться давлению. Это замечание может помочь объяснить зависимость
стабильности от скорости замерзания, которая влияет на размер
кристаллов льда и, следовательно, на размер пустот. Практически как
скорость замерзания, так и скорость таяния, влияют на стабильность
(Синглетон и др., 1960). Замерзание разбавленных эмульсий типа М/В
при двух различных скоростях показало, что более быстрое
замерзание и умеренная скорость оттаивания приводят к большей
неустойчивости. Медленное оттаивание после замерзания при любой скорости
не дает шариков диаметром > 7 мкм, хотя, очевидно, после быстрого
замерзания размер частиц должен быть больше, чем после
медленного, при всех скоростях оттаивания.
Найдено, что на степень коалесценции влияет также температура
плавления масляной фазы. Дисперсность эмульсий типа М/В,
Приготовленных с фракционированным хлопковым маслом (температура
плавления —5° С) или сафлоровым (температура плавлеЕЙш —12° С),
не изменялась при различных скоростях замерзания и оттаивания.
Очевидно, природа масляной фазы влияет на растворимость в ней
эмульгатора благодаря равновесию твердое вещество — жидкость,
существующему в масляной фазе при различных температурах. Если
это предположение верно, тогда для увеличения стабильности при
низких температурах должны быть использованы масла с низкими
температурами плавления.
Коле и др. (1959) определили, что при быстром замерзании
эмульсии масла в воде распределение шариков по размерам не
увеличивалось до значения, полученного при медленном замерзании. Если
эмульсии замерзали в интервале температур от —2 до —10° С,
растущие на стенках сосуда кристаллы льда выталкивали шарики масла
в центр. Однако при очень низкой температуре (—79° С) таких
миграций масляных шариков не наблюдалось.
Механизмы замерзания всего объема эмульсии и ее тонкой пленки
оказались подобными. Рохов и Масон (1936) нашли, что
устойчивость эмульсий типа четыреххлористый углерод — бензол и вода,
стабилизированных 0,5% раствором олеата натрия, зависит от
температуры замерзания. При —6° С кристаллы льда толкали шарики
друг к другу; последние сплющивались, но не коалесцировали при
оттаивании. Выдерживание эмульсии в течение 10 мин при —11,8° С
приводило к разрушению слоя олеата натрия вокруг шариков и к
коалесценции.
Поведение других эмульсий аналогично, поэтому важным
фактором стабильности при замораживании является наличие
адсорбционного слоя вокруг шариков.
126
Подобным же образом вели себя эмульсии типа М/В,
стабилизированные 2,5—10% раствором сухого молока, при медленном
уменьшении температуры тонкой пленки до 7° С (Шерман, 1966). При слабом
давлении на пленку во время замораживания белковая оболочка
вокруг шариков разрушалась. Смежные шарики соединялись
посредством узких «протоков» масла, и иногда внутри агрегата шариков
образовывалось несколько таких цепочек. Возможно, что при
перемешивании эмульсии во время замораживания шарики из-за частого
столкновения коалесцируют еще до замерзания. Заканчивается коале-
сценция из-за разрыва оболочек.
При низких температурах эмульгирующие свойства многих
протеинов уменьшались вследствие необратимой агрегации (Боросихино
и др., 1961). Кроме того, плотность адсорбционного протеинового
слоя вокруг шариков будет снижаться (в результате увеличения
поверхности) при превращении в лед воды, связанной водородными
связями с адсорбционным слоем.
Степень переохлаждения воды в эмульсиях типа В/М зависит от
природы эмульгатора (табл. III.1). При использовании ряда неионо-
генных эмульгаторов переохлаждение достигалось при 12—15° С
(Фокс, 1959). Шарики воды среднего диаметра (2—4 мкм)
диспергировались в «нуйоле», содержащем 5% эмульгатора. При
диспергировании водной фазы таким образом содержание любой примеси,
способной катализировать образование центров кристаллизации, снижалось
до минимума. Можно предположить, что кристаллы льда образуют
сферу на внутренней поверхности каждого водного шарика и
дальнейший рост их катализируется этой поверхностью.
Таблица III.1
Влияние эмульгатора на переохлаждение воды в эмульсиях В/М
(Фокс, 1959)
Эмульгатор
Моноолеат сорбитана
Полутораолеат сорбитана
Триолеат сорбитана
Окисленное соевое масло
'Ланолин
Переохлаждение, °С
12,0
13,0
13,5
13,5
15,5
Краевой угол
между центрами
кристаллизации
льда и
поверхностью
шариков воды
68
72
74
74
84
Число молекул
в центре
кристаллизации льда
635
585
590
590
480
Аналогичные явления переохлаждения наблюдались в эмульсиях
М/В, когда дисперсная фаза являлась смесью триглицерида и
керосина (Шкода и ван ден Темпель, 1963) или только триглицеридов
(Фиппс, 1964). В эмульгированной форме триглицерид
кристаллизуется при более низкой температуре. Степень переохлаждения
127
зависит от природы эмульгатора. Кристаллизацию ускоряют
преимущественно те эмульгаторы, чья гидрофобная цепь подобна цепи три-
глицерида (табл. III.2). Такие эмульгаторы ускоряют процесс
образования центров кристаллизации ориентацией молекул триглицерида,
лежащих у поверхности шарика. Ниже ККМ влияет и концентрация
эмульгатора. С увеличением среднего размера шарика в пределах
2 — 6 мкм возрастает количество жира, кристаллизующегося при 38СС.
Найдена логарифмическая зависимость между температурой
замерзания и диаметром шарика.
Таблица Щ.2
Влияние эмульгатора на переохлаждение жировых шариков
тристеарата глицерина в эмульсиях М/В
(ван ден Темпель, 1963)
Объемное содержание масляной фазы: 44%.
Состав масляной фазы: 30% тристеарата глицерина в 70% керосина.
Концентрация,
и
0,03
0,06
0,03
2%
Эмульгатор
Пальмптат натрия (рН 10,5)
Капрпнпл пзоцпанат
Лаурпл пзоцпанат
Миристил пзоцпанат
Стеарил изоцианат
Лаурпл саркозпнат (рН 7.6)
Пальмптпл саркозпнат (рН 6,0)
Децп.исульфопат натрия
Додецплсульфонат натрия
Октадецплсульфонат натрия
Додецплсульфат натрпя
Лаурилдскагликодевый эфир
Нонилфснолдекагликолевып афпр
Аэрозоль ОТ
Эмульфор О
Казспнат натрия (рН 8,5)
лаждение °С *,
41,3
50,9
48,3
42,8
36.5
54,3
47,4
50,5
49,3
47,3
47.8
55,9
5в,3
55,3
51,4
48,2
* Температура плавления тристеарата глицерина принята равной 72,3° С.
Эти выводы указывают на влияние гетерогенности на первом этапе
кристаллизации триглицерида, так как вероятность появления
катализируемых центров замерзания в шарике понижается с уменьшением
числа шариков в единице объема эмульсии. Действительно,
профильтрованный или подогретый триглицерид кристаллизуется медленнее.
Кристаллизация триглицерида в шариках керосина уменьшает
стабильность эмульсий М/В. Эмульсии будут разрушаться, если их
подогреть после замораживания; исключение составляют эмульсии,
стабилизированные казеинатом натрия.
СТАБИЛЬНОСТЬ ЭМУЛЬСИЙ
ПРИ ПОВЫШЕННЫХ ТЕМПЕРАТУРАХ
Об изменениях, происходящих в эмульсиях при повышенных
температурах, известно гораздо меньше, чем при условиях
замораживание — оттаивание. Выдержка эмульсий при температуре выше
128
нормальной часто используется для ускорения процесса старения
и определения длительности их устойчивости. Льюис и Дроммонд
(1953) показали, что интервал температур 30—40° С является
оптимальным для разбавленных эмульсий М/В, приготовленных с
эмульгаторами: полиэтиленгликоль-40-моностеарат, камедь акации, лау-
рилсульфат натрия, марсельское масло и полисорбитан-80.
В эмульсиях, содержащих заэмульгированный твердый жир, не
наблюдается определенного характера устойчивости в пределах
температур 5—40° С (Вуд и Катакалос, 1963) при выдержке в течение
100 дней и более. При высоких температурах плавится жировая фаза,
изменяется растворимость компонентов в ней и может произойти
обращение фаз.
Эмульсии В/М, стабилизированные стеаратом натрия, обращаются
в эмульсии М/В, если нагревание сопровождается легким
встряхиванием (Вельман и Тартар, 1930). При кратковременном
выдерживании на холоду происходит восстановление первоначальных эмульсий.
Эмульсии В/М с различным содержанием стеарата натрия обращаются
при разных температурах. Очевидно, существует равновесие между
противоположными тенденциями эмульгатора, которое смещается
температурой в сторону одного типа эмульсии. Другими факторами,
влияющими на температуру фазового превращения, являются
химические составные части масляной фазы и в меньшей степени длина
гидрофильной цепи эмульгатора (Шинода и Араи, 1964).
Температура обращения фаз зависит от природы масляной фазы.
Если в качестве масляной фазы для эмульсий, стабилизированных
нонилфенил-9,6-полиоксиэтиленовым эфиром (ОП-10), используются
бензол, гептан, гексадекан, толуол, четыреххлористый углерод
и жидкий парафин, то бензол дает самую низкую температуру
обращения фаз (—20° С), а гексадекан — самую высокую (~110° С).
Эмульгатор хорошо растворяется в бензоле в отличие от гексадекана.
Следовательно, чем лучше эмульгатор растворим в масляной
фазе, тем ниже температура обращения фаз. Этот вывод подтверждает
значение растворимости эмульгатора в непрерывной фазе для
устойчивости эмульсий при высоких температурах.
УСТОЙЧИВОСТЬ К УЛЬТРАЦЕНТРИФУГИРОВАНИЮ
Один из методов оценки стабильности эмульсий заключается
в том, что последние подвергаются центрифугированию при больших
скоростях с целью определения скорости отделения дисперсной фазы
(Мерилл, 1943; Лацкар и Маклей, 1943). Метод непригоден для
устойчивых или вязких эмульсий. Для измерения стабильности он, в
сущности, неприменим, так как дает информацию только о конечной
стадии коагуляции. Волд и Грут (1962, 1963, 1964) подробно
изучили устойчивость к ультрацентрифугированию при скоростях
^ 25 000 об/мин; скорость отделения дисперсной фазы определяли
фотографически. Ультрацентрифугирование имеет то преимущество,
что оно способствует агрегации шариков задолго до коалесценции.
9 Заказ 1070
129
Получается высококонцентрированная эмульсия, скорость коалес-
ценции которой можно подробно изучать (Рефельд, 1962).
Ультрацентрифугированием образец эмульсии М/В разделяется
на три слоя: верхний слой — отслоившаяся масляная фаза,
средний — эмульсия, шарики которой находятся в плотной упаковке,
нижний — водный слой. После пятиминутного
ультрацентрифугирования граница между слоем эмульсии и воды более не изменяется,
так как почти вся дисперсная фаза из эмульсии удалена. В этом
случае шарики имеют полиэдрическую форму и отделены друг от друга
небольшими прослойками. Единственное уменьшение объема этой
высококонцентрированной системы может происходить в результате
коалесценции. В течение первых нескольких минут
ультрацентрифугирования масло отслаивается быстро. Вероятно, изменение
сферической формы шариков в полиэдрическую увеличивает площадь
поверхности масляной фазы, и эмульгатор, первоначально имеющийся
в достаточном количестве, не может адсорбироваться на межфазной
поверхности. Следовательно, масло отслаивается с установившейся
низкой скоростью.
На устойчивость при ультрацентрифугировании влияет чистота
эмульгатора, его концентрация, если она ниже ККМ, время,
объемная концентрация дисперсной фазы, площадь поверхности шариков
и концентрация добавляемых солей.
В эмульсиях нуйол/вода с концентрацией дисперсной фазы 0,5,
содержащих 0,2% технического додецилсульфата натрия, понижалась
скорость отслаивания масла при уменьшении скорости
ультрацентрифугирования от 56 100 до 10 589 об/мин. При последней скорости
отслаивание масла не наблюдалось в течение 2,5 ч. Волд и Грут
(1962) пришли к заключению, что коалесценция может не начинаться
до тех пор, пока сила поля ультрацентрифугирования лишь едва
превышает силу разрыва адсорбционных слоев эмульгатора. Ниже
ККМ с увеличением концентрации додецилсульфата натрия
стабильность возрастала, при ККМ —оставалась постоянной (Рефельд, 1962).
Стабильность также увеличивается по мере того, как площадь
масляной фазы возрастает. Высота слоя некоагулированной эмульсии
изменяется симбатно с объемной долей Ф дисперсной фазы. Ее
увеличение вызывает повышение гидростатического давления в
межфазном пространстве, через которое вода удаляется с участка
коалесценции (Грут и Волд, 1964).
Влияние Ф на стабильность зависит от количества эмульгатора,
адсорбировавшегося на поверхности раздела М/В. В разбавленных
эмульсиях, полученных добавлением водной фазы к готовой эмульсии
с Ф = 0,5, при содержании 0,2% додецилсульфата натрия
наблюдалось уменьшение установившейся скорости отслаивания масла с
увеличением Ф. Наоборот, увеличение концентрации масла в эмульсиях,
содержащих 0,4% додецилсульфата натрия, приводило к
возрастанию отделения масла.
В образцах с низким содержанием эмульгатора коалесценция
способствует выделению додецилсульфата натрия, который затем опять
130
адсорбируется. Такая адсорбция на оставшейся межфазной
поверхности приводит к уменьшению скорости отслаивания масла. Этим
компенсируется противоположная тенденция, вызванная
уменьшением высоты слоя сливок. В эмульсиях с большим содержанием
эмульгатора реадсорбция намного меньше, так как поверхность
раздела М/В первоначально более насыщена.
При равновесной концентрации додецилсульфата натрия в
водной фазе независимо от того, является ли она ниже или выше ККМ,
скорость отслаивания масла изменяется обратно пропорционально
логарифму общей концентрации иона натрия, находящегося в
эмульгаторе и добавленном количестве хлорида натрия.
Так как скорость отслаивания масла остается постоянной после
нескольких минут центрифугирования, то коалесценция не
происходит ни в слое эмульсии, ни на стенках сосуда. Поверхность
раздела эмульсия — вода не может быть местом коалесценции, ибо
масло должно было бы подниматься вверх через оставшийся слой
высококонцентрированной эмульсии противоположно потоку
водной фазы.
Волд и Грут (1964) показали аналогию между устойчивостью
эмульсий к ультрацентрифугированию и стабильностью пен. В пенах
тонкие пленки водной фазы разделяют полиэдрические газовые
ячейки. Чем тоньше пленка, тем больше вероятность того, что
газовые ячейки будут разрушаться. Поэтому верхние слои пен менее
стабильны вследствие вытекания жидкости.
В эмульсиях, подвергнутых ультрацентрифугированию,
существует аналогичная ситуация, но полиэдрические газовые ячейки
замещены здесь искаженными шариками масла, а водные пленки,
разделяющие их, более тонки в верхнем слое эмульсии.
Перед коалесценцией масляных шариков водные пленки
становятся настолько тонкими, что разрываются, вызывая разрушения
адсорбционного слоя эмульгатора. Скорость утончения водных
пленок можно контролировать по вытеканию жидкости или по вязкости
потока внутри пленок. Однако эти экспериментальные методы не
всегда точны. Известно (см. гл. II), что толщина пленки определяется
расклинивающим давлением при взаимодействии электрических
двойных слоев, а сжатие происходит благодаря центробежному полю
и силам притяжения Ван-дер-Ваальса в тонких пленках. Поэтому,
если электростатическое отталкивание уравновешивается
центробежным давлением, толщина пленки должна составить —8,4 А.
Если скорость вытекания определяется вязкостью потока в
пленках при условии, что последние представляют собой параллельные
пластины, средняя скорость для гравитационного потока выражается
формулой (Адамсон, 1960).
где рс — плотность водной фазы; б — расстояние между пластинами.
9*
131
При добавлении соли к эмульсии величина б изменяется почти
экспоненциально. Следовательно, скорости вытекания водной фазы
и отслаивания масла должны также приблизительно экспоненциально
зависеть от концентрации соли. Но скорость отслаивания масла, как
отмечено выше, уменьшается с увеличением логарифма общей
концентрации иона натрия и поэтому не может влиять на скорость коа-
лесценции.
Существует другая возможность. Вытекание происходит в
большей степени через большие каналы в тех границах, где соприкасаются
плоские водные пленки (Майзельс и др., 1959). Скорость вытекания
зависит от ширины этих каналов и контролируется установившимся
равновесием между давлением ультрацентрифугирования и
давлением, обусловленным изгибами каналов. Вычисление показывает, что
в этом случае каналы должны быть на порядок меньше, чем
предполагается.
Грут и Волд (1964) пришли к выводу, что повышение стабильности
происходит по мере увеличения адсорбции додецилсульфата натрия
на поверхности раздела М/В. После коалесценции шариков
освободившийся эмульгатор переносится водной фазой в нижнюю часть
оставшегося слоя эмульсии. Во время переноса часть эмульгатора вновь
адсорбируется на оставшейся поверхности раздела М/В, причем
степень адсорбции зависит от ее первоначальных значений и от высоты
слоя эмульсии. Эта дальнейшая адсорбция может изменить
реологические свойства граничной пленки.
Центрифугирование, создавая определенные физические
изменения в слое эмульсии, все же может быть использовано как быстрый
метод определения старения эмульсий при нормальных условиях.
При центрифугировании основным процессом является коалесценция
в эмульсиях, а при нормальных условиях — флокуляция.
ВЛИЯНИЕ ГЛБ НА ОБРАЗОВАНИЕ ЭМУЛЬСИЙ
Гриффин (1949,1954) ввел полуэмпирический метод подбора одного
или смеси нескольких эмульгаторов для получения эмульсий.
Основой этого метода является гидрофильно-липофильный баланс (ГЛБ),
заключающийся в том, что молекула любого поверхностно-активного
вещества (ПАВ) состоит как из гидрофильных, так и гидрофобных
групп, отношение содержания которых влияет на характер
эмульгирования.
Эмульгатор В/М имеет низкий ГЛБ,солюбилизирующее вещество —
высокий и эмульгатор М/В — промежуточное значение (табл. III.3).
Относительное значение гидрофильных и гидрофобных групп было
впервые определено при исследовании эмульсий со смесью неион-
нных эмульгаторов типа В/М и М/В, содержащих одну и ту же
масляную и водную фазы. С изменением соотношений эмульгаторов
противоположного характера эффективность любой комбинации
определяли визуально по отслаиванию дисперсной фазы, имеющему
максимальное значение при определенной концентрации гидрофильного
132
Таблица Ш.З
Влияние ГЛБ на характер диспергируемости в воде
(Гриффин, 1956)
Область
ГЛБ
1—4
3—fi
6-8
8-10
10-13
13
Способность диспергироваться в воде
Отсутствует
Плохая
Молокоподобная дисперсия при
встряхивании
Устойчивая молокоподобная
дисперсия
Почти прозрачная дисперсия
Прозрачный раствор
Соответствующее применение
Эмульгатор В/М
Смачивающий реагент
Эмульгатор М/В
»
Эмульгатор М/В, солюбили-
знрующий реагент
вещества в смеси. Следует заметить, что с помощью системы ГЛБ
можно определить только тип образующейся эмульсии, но не
эффективность эмульгирования.
Обычно выбор соответствующих эмульгаторов или их смесей
производят методом «проб и ошибок». Недавно предложено несколько
определенных значений ГЛБ неионных ПАВ п масел для подбора
смеси эмульгаторов.
Значения ГЛБ для эфиров многоосновных жирных кислот
приблизительно могут быть вычислены по формуле, полученной из
эмпирических данных
*глб = 20(1—^) (Ш.2)*
где Ъ — число омыления эфира; Ак — кислотное число жирной
кислоты.
В тех случаях, когда трудно определить точное значение чисел
омыления (например, эфиры таллового масла, ланолин, смола)
используют другое выражение:
где сэт и ссп — соответственно содержание оксиэтилена и
многоатомного спирта, вес. %.
Для производных окиси этилена и веществ, в которых
гидрофильной группой является окись этилена, это выражение сокращается:
Nrw=^f- (III.4)
Диссоциация ионной группы ПАВ способствует увеличению их
гидрофильности. Поэтому значение ГЛБ нельзя определить лишь по
весовому содержанию.
* В настоящем издании в формулах и уравнениях число ГЛБ, определяющее
гидрофильно-липофильнып баланс, обозначено -^ГЛБ. (Прим. редакции.)
133
Растворимость многих ПАВ в воде приблизительно можно
сопоставить с их значениями ГЛБ (табл. III.3). Если эмульгатор
растворим в воде, то его ГЛБ вычисляют по температуре помутнения, т. е.
той температуре, при которой 5% водный раствор дает резкое
изменение мутности (рис. III.1). Температура помутнения для
производных оксиэтилена в большей степени зависит от длины полиоксиэтиле-
новой цепи и, следовательно, от ГЛБ, чем от их концентрации
(Гриффин, 1956).
Предложены и другие методы определения значений ГЛБ. Грин-
вальд и др. (1956) разработали метод титрования, заключающийся
в том, что 1,0 г эмульгатора растворяют в смеси из 4% бензола и 96%
100
I
80
ВО -
W
18
4
12
К
16
глв
? 6 10 М
Водное иисло
Рис. III. 1. Зависимость
температуры помутнения от ГЛБ (Грпффин,
1956).
Рис. III.2. Зависимость ГЛБ от
водного числа для эфпров многоатомных
сппртов (Гринвальд и др., 1956):
1 — с окисью этилена; 2 — без окиси этилена.
диоксана и титруют дистиллированной водой при осторожном
помешивании до неисчезающего помутнения.
Выбранная смесь растворителей в отличие от других позволила
отчетливее определить конец титрования и получить лучшую
воспроизводимость результатов, особенно для ионных эмульгаторов.
Содержание диоксана в воде может изменяться, что сказывается на
титровании. Для предотвращения этого влияния объем бензола
подбирают таким, чтобы значение водного числа самой смеси было
21,6—21,8 мл. При титровании следует наблюдать за температурой,
ибо водное число изменяется приблизительно на 0,08 мл/град.
Природа изменения, совпадающего с возникновением неисчезающего
помутнения, неизвестна, однако это может соответствовать инверсии
эмульсий.
В этой точке размеры шариков дисперсной фазы становятся
намного меньше. Значения водного числа и ГЛБ, вычисленные по
приближенной формуле, дают линейную зависимость: значение водного
134
числа повышается с увеличением ГЛБ (рис. III.2), хотя градиент
зависит от классификации включенного вещества.
При растворении производных окиси этилена в воде образуются
водородные связи между атомами кислорода и молекулами воды:
—СН2—О—СН.2—
i
I
н
I
о-н
Выделяющуюся при этом энергию можно измерить
калориметрически (Ракз и Орбан, 1965) с помощью запаянного сосуда Дьюара,
снабженного мешалкой,
используя термопары медь-константан.
Для жидких гидрофильных
ПАВ справедливо эмпирическое
соотношение:
ЛГГЛБ = 0,42(3+ 7,5 (ПГ.5)
где Q — теплота гидратации.
Для твердых ПАВ требуется
более сложное соотношение, так
как превращения всплывающей
фазы вызывают соответствующие
изменения дополнительно к
энергии, выделяющейся при
гидратации. В бинарных смесях ПАВ
наблюдаются слабые
положительные отклонения от значений ГЛБ,
вычисленных при условии, что
вещества не взаимодействуют, а значения ГЛБ двух компонентов
суммируются.
Значение ГЛБ можно определить, не прибегая к исследованию
поверхностных свойств системы. Например, Бехер и Беркмейер
(1964) указывали, что хотя эффективность отделения из смеси в
газожидкостной хроматографии зависит от полярности субстрата, можно
определить ГЛБ непосредственно, используя ПАВ и пропуская
масляную фазу через колонку.
При пропускании 3,0 мл смеси этанола и гексана (50 : 50) в
колонку (с неионным адсорбентом) получены два хорошо выраженные
максимума на хроматограмме. Один из них, обычно появляющийся
раньше, принадлежал гексану. При построении зависимости ГЛБ от
отношения времени удерживания этанола ту и гексана ту получены
прямые, причем с ростом ГЛБ увеличивалось ту (рис. III.3).
Статистические исследования ГЛБ в области 2—18 показали, что
ЛГГЛБ = 8,55ту—6,36 (II 1.6)
где ту -= Ту/Ту1.
ГЛБ
Рис. III.3. Зависимость отношения
времени удерживания от ГЛБ для
эфиров полиоксиэтилированного сор-
битана и жирных кислот и для
эфиров многоатомного спирта сор-
бптана и жирных кислот (Беркмейер
п др., 1964).
135
Время удерживания чувствительно к изменениям температуры
и уменьшается с ее увеличением. Значения ГЛБ для смесей
эмульгаторов, полученные из уравнения (II 1.6), незначительно отличаются от
теоретических, рассчитанных из предположения, что значения для
двух компонентов смеси суммировались. В этом случае (Ракз и Орбан,
1965) отклонение отрицательно.
Стабильность эмульсий зависит от химической природы
эмульгатора и фаз. Оба фактора влияют на значение ГЛБ. Можно сопоставить
стабильность со свойствами растекания дисперсной фазы (Росс и др.,
1959) следующим образом. Когда масляный шарик поднимается на
-20 -16 -12 -8 -4 0 4
Коэффициент
растекания
-30 -38 -46 -54 -62
Коэффициент растекания
Рпс. III.4. Зависимость коэффи- Рис. III.5. Зависимость коэффициента
циента растекания касторового растекания дистиллированной воды по
масла по поверхности 1% водных
растворов ПАВ
п др.,
от ГЛБ
1959).
(Росс
поверхности 1% растворов касторового
масла от ГЛБ (Росс и др., 1959).
поверхность столба эмульсии М/В при расслаивании может
наблюдаться два случая: шарик возвращается в объем эмульсии при
перемешивании, стабильность не изменяется или же он растекается по
поверхности эмульсии, что приводит к отделению фазы. Для
обеспечения стабильности необходимо, чтобы коэффициент растекания был
отрицательным. Визуальное определение отделения фазы указывает,
что оптимальная стабильность достигается в эмульсиях М/В, когда
коэффициент растекания не слишком мал, а именно от 0 до —5, что
соответствует низкому поверхностному натяжению.
Отслаивание фазы, коэффициент растекания и значение ГЛБ
сравнивались для различных масел и смесей двух эмульгаторов
(спен-80 и твин-80) в разных соотношениях и для эмульсий,
приготовленных из них. Коэффициенты растекания, полученные из данных
межфазного натяжения, являются характеристикой способности
масла растекаться по 1% водному раствору смеси эмульгаторов.
В области значений ГЛБ 4—15 коэффициент растекания линейно
увеличивается с повышением ГЛБ (рис. III.4). Выше значения ГЛБ,
равного 8, зависимость линейна и отклонение от нее в нижней
части графика объясняется неполной растворимостью эмульгатора
в воде.
136
Для подтверждения этих результатов сделано подобное
сопоставление с использованием коэффициента растекания дистиллированной
воды на маслах, содержащих два эмульгатора в различных
пропорциях и охватывающих область ГЛБ приблизительно от 4 до 10.
На рис. III.5 показано, что коэффициент растекания линейно
уменьшается с увеличением ГЛБ по всей области. Отклонения появлялись
при значениях ГЛБ выше 10, так как эмульгаторы в этих случаях
меньше растворялись в масле.
Сравнительно устойчивые данные для эмульсий В/М не были
получены. Однако, используя значение ГЛБ как показатель
стабильности, можно заключить, что устойчивость эмульсий В/М зависит от
величины коэффициента растекания. Так как каждый эмульгатор
имеет определенное значение ГЛБ, то можно обеспечить оптимальную
стабильность приготовленных из них эмульсий М/В или В/М
(табл. III.4).
Таблица III-4
Значение ГЛБ, необходимое для эмульгирования или солюбилизации
(Гриффин, 1954)
Вещества
Воск
микроциста л лин
парафин
пчелиный
Ланолин, безводный
Масло
витамин (свободный жир)
витамин (с жирами или маслами)
минеральное, легкое
минеральное, тяжелое
хлопковое
эфирное
Нефть
Стеариновая кислота
Стеариловый спирт
Эфиры
Эмульсия
В/М
4
5
8
4
4
4
Эмульсия
М/В
9,5
9
10-16
15
10-12
10,5
7,5
10,5
17
13
Солюбилиза-
ция в воде
16.5
15,0
15.5
16,5
14
По-видимому, стабильность должна зависеть от взаимодействия
между гидрофобными группами эмульгатора и масляной фазой
(Гриффин, 1954). Например, в случае масляной дисперсной фазы и смеси
эмульгаторов спен-80 (моноолеат сорбитана) — твин-80 (полиокси-
этилен моноолеат сорбитана) или спен-80 — твин-20 (полиоксиэтилен
монолаурат сорбитана) для оптимальной эмульсионной стабильности
необходимо значение ГЛБ в области 10, концентрация смеси
эмульгаторов от 0,2 до 30% (Гриффин, 1956).
С другой стороны, концентрация не влияет на стабильность
эмульсий В/М, полученных посредством инверсии эмульсий М/В. Так как
концентрация смеси эмульгаторов возрастает, то ГЛБ, при котором
137
эмульсия В/М превращается в эмульсию М/В, тоже увеличивается.
При слишком большой концентрации смеси эмульгаторов происходит
резкое повышение ГЛБ, необходимое для инверсии, потому что часть
смеси уже составляет дисперсную фазу.
Дэвис (1957) предположил, что тип образующейся эмульсии и,
следовательно, понятие ГЛБ можно объяснить через относительные
скорости коалесценции эмульсий М/В и В/М и распределение
эмульгатора между масляной и водной фазами *.
Если шарики имеют потенциал поверхности ijj, то коэффициент
отталкивания пропорционален г)Д а скорость коалесценции масляных
шариков в эмульсиях М/В выражается уравнением
vM/B=Kiexv(-B^/RT) (III.7)
где К1 — гидродинамический фактор столкновения, равный
4ФА'г/Зг]с; г|с — вязкость водной фазы; В — постоянная, равная
0,24, которая выведена эмпирически из теории кинетики
коалесценции (Лоуренс и Миллс, 1954).
Если капли эмульсии не заряжены, стабилизация осуществляется
связыванием молекул воды в гидратный слой на поверхности. Это
ведет к повышению энергетического барьера, который контролируется
количеством молекул воды, присоединенных к каждой молекуле
адсорбированного эмульгатора.
Для большинства эмульсий действуют оба фактора стабилизации.
Уравнение (III.7) в этом случае принимает вид
гм/в = АГ1РХР(ч 77г J (III-8)
где IИ — общий энергетический барьер; Qf— доля поверхности
частиц, покрытой адсорбировавшимися молекулами эмульгатора.
В эмульсиях В/М коалесценции водных шариков, между которыми
содержатся тонкие пленки масла, происходит, когда шарики воды
находятся очень близко друг от друга. Скорость коалесценции v
зависит от гидродинамического фактора К2 и от числа п групп
—СИ,— в каждой углеводородной цепи (каждая группа —СН2 —
* Теория Дэвиса весьма логично объясняет эмпирическую систему ГЛБ,
однако в ее основе лежит предположение, что при диспергировании двух
жидкостей образуется множественная эмульсия п выживают только капли более
устойчивой эмульсии. Это предположение не подтверждается при цпспергированип
многих систем (эмульсия одного типа образуется за доли секунды; время жизни
капель даже без стабилизатора порядка нескольких секунд). Такие наблюдения
легко осуществить, помещая небольшой аппарат с мешалкой в поле зрения
микроскопа или скоростной мпкрокпноустяновки.
К тем же результатам, что и Дэвпс, пришел Осипов, исходивший пз
предположения, что стабилизация эмульсий лимитируется не отталкиванием
адсорбционных слоев эмульгатора, как предполагал Дэвис, а удерживанием молекул
стабилизатора на поверхности раздела фаз.
Видимо, при рассмотрении стабилизации эмульсий следует учитывать оба
фактора. (Прим. редактора перевода.)
138
вносит приблизительно 300 кал в энергетический барьер, так как
водный мостик будет установлен через группы —СН2—
vB/m = Kr,exv(-2nQrm/RT) (III.9)
Объединяя уравнения (III.8) и (III.9), получим:
-^м * ехр 1+о.2Ау+е,2*-™*Л
VU/B Л1 I НТ )
Когда ^в,м/ум/вЗ> li образуется эмульсия М/В, если Ум/в/ув/м <С
•С 1 — эмульсия В/М.
Если вязкости водной и масляной фаз одинаковы и Ф = 0,5,
уравнение сокращается:
1>р/м 0,24г|з2 + е, Ytf-600"8;
1п_в/м_ = /^j r_ (nL11)
"М/В rti
Уравнение (III.11) можно переписать следующим образом:
*r-lni^«=-^. + !* я (Ш.12)
600В; им/в 6009; г 600
В большинстве случаев условия уравнения (III.10) и (III.11) не
выполняются, и взаимосвязь должна быть более сложной.
Значения ГЛБ для ПАВ могут быть вычислены при отнесении
групповых чисел (табл. II 1.5) к каждой составной части радикала.
Используется формула:
Л'гЛБ = 2^г„д-«ЛгСНг + 7 (II 1.13)
где Лггид— гидрофильное групповое число; NCn2 — групповое число
группы —СНп —.
В табл. III.6 приведены экспериментальные и вычисленные
значения ГЛБ.
Из уравнений (III.12) и (III.13), принимая групповое число
равным 0,475 для группы —СН2—, имеем:
^глб-? _ 1>гид _„ (П114)
0,47.5 0,475
1п(7ГШ-)==2-2в/(лгглБ-7) (III.15)
\ М/В /
Гидрофильное групповое число для гидратированной группы
равно Н/1260; гидрофильное групповое число для группы, имеющей
заряд, составляет 1,9 ■ 10_ 4 ар2/Э^.
Уравнение (III.15) предполагает, что ни эмульсия В/М, ни М/В
не образуются, когда ГЛБ равно 7 или 6;- малая величина.
139
Взаимосвязью между ГЛБ и степенью гидратации объясняют
наблюдаемое соотношение между ГЛБ и температурой помутнения, так
как обе величины являются функцией энергии дегидратации ПАВ.
Дэвис (1957) указывает, что величина ГЛБ групп, имеющих заряд,
зависит и от г|з, и от 6;-.
Таблица III .5
Групповые числа ГЛБ
(Дэвис, 1957)
Группа
-S04Na+
-соо-к*
—COCTNa+
N (третичный амин)
Эфир (сорбитановое кольцо)
Эфир (свободный)
-соон
Гидроксил (свободный)
—О—
Гидроксил (сорбитановое кольцо)
Липофилыше
-СН-
— СН-2 —
-СН3-
= СН-
С ложные
-(СНг-<
-(СН2-(
:н2-о) о
]Н2СН2—О)-
Групповос
число
38,7
21,1
19,1
9,4
6,8
2,4
2,1
1,9
1,3
0,5
-0,475
0,3,4
-0,15
Число ГЛБ группы не является постоянным, ибо г|з
контролируется общей концентрацией и 0,.
Распределение эмульгатора между водной и масляной фазами
определяется его свободной энергией переноса Д<7В->м из воды
в масляную фазу
ЬСъ^^ПТЫ^/Съ) (II 1.16)
где см и св — концентрации эмульгатора в масляной и водной
фазах, соответственно.
Гидрофильные и гидрофобные части молекул эмульгатора связаны
с AG„ ,, соотношением
В->-М
дсв+м = 2 дсв^м-8°0" (III. 17)
где 2 ^в->-м — свободная энергия переноса гидрофильной группы;
800 — разность свободных энергий группы —СН2— в воде и в масляной
фазе, кал/моль (для группы —ОН эта разность равна 3200 кал/моль).
140
Таблица Ш-6
Сравнение значений ГЛБ, полученных экспериментально
и из групповых чисел
(Дэвис, 1957)
ОЭ— оксиэтиленовые группы.
ПАВ
Лаурилсульфат натрия
Олеат калия
Олеат натрия
Твин-80 (моноолеат сорбитана с 20-к
Моноолеат сорбитана с 10-ю ОЭ
Твин-81 (моноолеат сорбитана с 5-ю
СмНзтЖСНа-СНаОН) (СН2-СН2-0-
Монолаурат сорбитана
Метанол
Этанол
Пронанол
Моноолеат сорбитана с 2-я ОЭ
Бутанол
Моноолеат сорбитана
Монопальмитат сорбитана
Моностеарат сорбитана
Спен-80 (моноолеат сорбитана)
Монолаурат пропиленгликоля
Дистеарат сорбитана
Моностеарат глицерина
Моностеарат пропиленгликоля
Тристеарат сорбитана
Цетиловый спирт
Олеиновая кислота
Тетрастеарат сорбитана
ОЭ)
ОЭ)
-СН2-СН2ОН)
Значение ГЛБ
экспериментальное
40
20
18
15
—13,5
10
10
8,6
—7
7,0
—7
6,7
5,9
4,3?
4,5
—3,5
3,8
3,4?
из групповых
чисел,
приведенных в
табл. III.5
40
20
18
15,8
12,5
10,9
10
8,5
8,4
7,9
7,4
7,0
7,0
7,2
6,6
5,7
5,0
4,6
3,9
3,7
1,8
(может содержать мыло)
2,1
1,0
1
—0,5
2,1
1,3
1
0,3
При сопоставлении уравнений (III.16) и (III.17) получаем:
RT
800
1п-^- =
2Afii
в-*м
ьм
800
(III.18)
Тип эмульсии и распределение эмульгатора между масляной
и водной фазами связаны друг с другом уравнением (III.12), в
котором
■'в / м
Св ^0,75^
(III.19)
"м/в \ °-м
# = 0,75Д(?В_^М (для незаряженной гидрофильной группы) (III.20)
0,324i])2/8/= Л^в-^м (для заряженной гидрофильной группы) (III.21)
141
Из уравнений (III.9) и (III.14) получаем:
лгглб-7 = °.361пЫсм) (111.22)
гидрофильное число неионной группы равно
AG^M (II 1.23)
1680
гидрофильное число ионной группы равно
А(?в-^м (III.24)
1680
где AGg_>M и AGg_^ — свободная энергия переноса неполярной
и полярной гидрофильных групп, соответственно.
Уравнение (III.22) дает зависимость между ГЛБ и типом эмульсий.
По правилу Банкрофта (1913), непрерывной фазой будет та, в которой
лучше растворим эмульгатор. Выражение (III.23) справедливо при
некоторых условиях, а именно: масло не должно растворяться в
водной фазе и наоборот. Во многих эмульсионных системах используется
достаточно высокая концентрация эмульгатора, поэтому эти
условия нарушаются, и теорию нельзя использовать для предсказания
типа образующейся эмульсии.
Дэвис (1960) создал эмульгирующую установку для изучения
влияния некоторых факторов, включая и ГЛБ, на тип эмульсии.
Основными ее частями являются две плоские пластины, верхняя из
которых образует ротор, а нижняя — статор. Масляную и водную
фазы из отдельных емкостей подают в отверстия статорной пластины.
При вращении верхней пластины происходит процесс
эмульгирования. Вытекает эмульсия через специальное сопло, и тип эмульсии
зависит от того, чем смачивается сопло. Эмульсии В/М покрывают
его непрерывной пленкой, эмульсии М/В только отдельными каплями.
Изучалось влияние ГЛБ на концентрацию дисперсной фазы (Фи),
при которой происходит инверсия.
При скорости ротора 1850 обIмин, петролейном эфире и воде
в качестве жидких фаз и использовании гладких пластин, образующих
ротор и статор, значение Фи линейно увеличивалось с повышением
ГЛБ в пределах от 6,7 до —16. При более низких значениях ГЛБ
величина Фи возрастала с уменьшением ГЛБ
Влияние ГЛБ на Фи зависит от скорости вращения ротора, от
масла и его вязкости, концентрации эмульгатора, относительного
смачивания поверхностей водной и масляной фазами и от отношения
скоростей вытекания масляной и водной фаз из емкости.
Наблюдаемая взаимосвязь между Фи и ГЛБ согласуется с тем, что
относительные скорости коалесценции влияют на тип эмульсии. В
эмульгирующем аппарате два возможных типа эмульсии не образуется при
равных скоростях, очевидно, потому, что эмульгатор изменяет
относительные энергии и относительные скорости смачивания ротора
и статора водной и масляной фазами.
142
Так как сам метод эмульгирования влияет на Фи, то выводы из
данных, полученных с помощью эмульгирующего аппарата,
неприемлемы для всех случаев. Например, найдено, что при добавлении
водной фазы, содержащей эмульгатор, и полном ее диспергировании
с помощью энергичного ручного встряхивания величина Фи для
превращения эмульсии В/М в М/В была больше, чем в случае
механического перемешивания при непрерывном медленном добавлении
водной фазы к масляной. В последнем случае предыдущая порция
водной фазы полностью не диспергировалась к моменту подачи
следующей.
Уравнения (III.7) — (III.24) основываются на предположениях
о механизмах коалесценции, которые расходятся с существующими
взглядами о преобладающем значении утончения жидкой пленки
между шариками и о важности свойств сдвига и расширения
адсорбционной пленки эмульгатора вокруг шариков. Последний фактор
влияет на скорость разрыва пленки эмульгатора до коалесценции
шариков.
Ригельман и Пихон (1962) пересмотрели опубликованные данные
н нашли, что устойчивые эмульсии М/В можно получить при
значениях ГЛБ от 2 и выше 17. При низком ГЛБ эмульсия В/М не
образуется. Исследования Петерсена и др. (1964) подтвердили это
предположение. Они нашли, что при эмульгировании глицерина и
олеиновой кислоты не было корреляции между ГЛБ нескольких классов
неионных эмульгаторов и типом образующейся эмульсии.
Приведем еще пример несоответствия экспериментов с ГЛБ. Число
ГЛБ монолаурата сорбитана равно 8,6 (см. табл. III.6), и он должен
стабилизировать эмульсии керосин — вода (см. табл. III.3). Это
верно для низких концентраций эмульгатора, добавленного к водной
фазе, но при концентрациях 5 — 6% эмульсия М/В превращается
в эмульсию В/М, когда объемная концентрация масла высока (Шер-
ман, 1963). Монолаурат сорбитана в зависимости от условий
стабилизирует как эмульсии М/В, так и В/М. Подобное поведение
наблюдалось с производными полиоксиэтилена.
Это двойственное поведение нельзя объяснить с помощью
значений ГЛБ. Тем не менее, несмотря на такие недостатки, понятие
ГЛБ оказывается пригодным при получении большого числа
эмульсий, особенно, если используется смесь эмульгаторов
противоположного характера.
Если ГЛБ для смеси эмульгаторов нельзя определить с помощью
указанной выше формулы из-за того, что неизвестны величины ГЛБ
двух жидких фаз, нужно приготовить ряд эмульсий с
соответствующими масляной и водной фазами. Смеси двух эмульгаторов со
значениями ГЛБ, расположенными в противоположных концах его шкалы,
например твин-80 и снен-80 (см. табл. III.6), используются в
различных соотношениях. Все эмульсии необходимо готовить одним
методом, с несколькими процентами эмульгирующей смеси. Стабильность
эмульсий оценивается по скорости коалесценции, и ГЛБ,
соответствующий оптимальной стабильности, является наиболее подходящим.
143
РАСПРЕДЕЛЕНИЕ ШАРИКОВ ПО РАЗМЕРАМ
В большинстве теорий об эмульсиях требуется определение
размера шариков (если эмульсии монодисперсные) или распределение
их по размерам (в случае полидисперсных эмульсий). Эти данные
нужны для объяснения, например, реологических свойств, для оценки
стабильности эмульсии и эффективности процесса гомогенизации.
Оптические методы
Микроскопия
Распределение частиц по размерам наиболее просто определяют
микроскопически, хотя это и очень трудоемко. Образцы можно
исследовать двумя путями. В первом случае эмульсию разбавляют в 20 —
30 раз непрерывной фазой, затем образец помещают на предметное
стекло с небольшой впадиной в центре. Значения размеров шариков
определяют сопоставлением их с градуированной сеткой,
находящейся в окуляре микроскопа. Увеличение при этом должно быть
^ 700—800 раз. Особенно важно большое увеличение при высокой
дисперсности. Таким способом определяют размеры 500—2000
шариков.
Можно использовать для подсчета капель и фотографии. Для
этого призму присоединяют к окуляру, а изображение проектируют
на градуированный экран, получая большие увеличения. Эмульсию
разбавляют обычно в 1000 раз. Каплю такого образца помещают
в ячейку для измерения, число шариков определяют при различных
уровнях. Число шариков в 1 см3
6Ф
N=-W (III.25)
Di = Y ~Ш (Ш.26)
где Di — средний диаметр шариков.
Данные этих методов обычно хорошо согласуются, хотя во
втором случае труднее подсчитать все шарики. Небольшие ошибки при
подсчете дают большие неточности в значении Dt, чем
непосредственное измерение его при большом числе шариков.
При низкой вязкости дисперсной фазы броуновское движение
шариков можно уменьшить, смешан образец с глицерином или
другими вязкими многоатомными спиртами.
Предложено несколько механических методов с тем, чтобы
облегчить или совсем исключить утомительный визуальный подсчет.
Один из них основан на принципе двойного изображения шарика
в микроскопе (Барнет и Тимбрелл, 1962). Луч в микроскопе,
расщепленный между окуляром и объективом, позволяет наблюдать два
144
идентичных изображения каждого шарика. С помощью
вращающегося предметного столика изображения могут сливаться или
сдвигаться относительно друг друга, а градуированная шкала
показывает степень сдвига. Вначале два изображения совпадают
(рис. III.6, а). При небольшом сдвиге появляются серые изображения,
а область их пересечения будет темней (рис. 6, б). С увеличением
сдвига изображения отделяются друг от друга, и между ними
появляется светлый зазор (рис. 6, г). Изображения могут
соприкасаться (рис. 6, в) при соответствующей степени сдвига. Одновременно
градуированная шкала электрического счетчика указывает размер
шарика. Ручное управление позволяет увеличивать степень сдвига
постепенно с тем, чтобы соответствовать десяти областям размера,
которые заранее отобраны. Каждый шарик относится к области
соответствующего размера.
Рис. III.6. Двойное изображение при микроскопировании
(Барнет п Тимбрелл, 1962).
Регистрацию такой сортировки производят на одном из десяти
электромагнитных счетчиков. Опытный исследователь может
рассортировать 600 шариков за 10 мин с погрешностью < 0,25 мкм. Хотя
область применения такого прибора ограничивается увеличениями,
даваемыми микроскопом, все же точность здесь больше, чем при
использовании обычного микроскопа, и данные различных
исследователей достаточно хорошо согласуются.
Можно исследовать фотографии и шарики классифицировать
в одну из десяти областей, используя вращающийся шаблон с десятью
дырками различных размеров (Бехер, 1964). Ось шаблона
присоединена к выключателю, имеющему одиннадцать положений, так что
число частиц в каждой области размера регистрируется. Острие
пробивает дырку в каждом изображении шарика для
предотвращения повторного исследования одной частицы.
Более усовершенствованный метод исследования
микрофотографий производится с помощью фазовоконтрастного микроскопа,
предложенного Нарасимхамом и др. (1965). Фотографии
проектируют на экран, разделенный как вертикально, так и горизонтально.
Каждый шарик сортируется, это записывается на соответствующем
блоке серии счетчиков. Практически за 15 мин можно
классифицировать 1100—1250 шариков.
10 Закаэ 1070
145
Измерение отражательной способности
Ллойд (1959) наблюдал, что интенсивность окрашивания
эмульсии М/В, приготовленной из кукурузного масла, снижается при
уменьшении размера шариков. Исходя из этого, приготавливали
эмульсии М/В, в которые добавляли красный краситель в
концентрации, достаточной для достижения адсорбции 0,710 при 450 нм.
Адсорбцию света определяли по формуле
a = lgrB/rn (ПГ-27)
где Тв и Тш — коэффициент пропускания для воды и масляного
раствора красного красителя, соответственно.
Для достижения нужной адсорбции неокрашенных масел,
например керосина, требуется больше красителя, чем для кукурузного
масла.
Отражательную способность таких эмульсий определяли при
450 нм в спектрофотометре, снабженном отражающей приставкой,
и данные сопоставляли с размерами частиц, определенными
микроскопически. Найдена линейная зависимость между этими
параметрами для размеров шариков 1 мкм ]> D > 30 мкм. Отражательная
способность (в %) увеличивалась с уменьшением диаметра шариков
lg(p = algZ)s-|-lgP (III.28)
где аир — постоянные характеристики эмульсионной системы;
Ds — средний диаметр поверхности шарика.
Это соотношение верно и при других длинах волн, но с
различными значениями двух постоянных (табл. III.7).
Так как
6Ф
Z)s = — (III.29) .
луд
(где 5уд — удельная межфазная поверхность) и
Ф=7ё# (ш-30>
то отражательная способность пропорциональна удельной
межфазной поверхности.
Приборы поглощения
При прохождении луча света через слой эмульсии, в которой
коэффициенты отражения двух жидких фаз различны, часть света
может поглощаться, а часть рассеиваться на поверхности шариков.
На оба явления расходуется энергия луча, уменьшая проходящий
свет. О размере частиц можно судить из данных о пропускании и
рассеивании света. Теория одна и та же в любом случае.
Нефелометрия. При пропускании параллельного пучка света
с длиной волны Я через разбавленную монодисперсную систему,
1-40
все шарики которой имеют диаметр D и коэффициент преломления
Пф, а коэффициент преломления непрерывной среды — пс, рассеяние
света равно 4/4 nD2L (здесь L — коэффициент рассеяния света,
являющийся функцией отношения Пф/пс и nDnJX). Выражение
оптической плотности слоя эмульсии толщиной Z, находящейся
в ячейке, основано на законе Ламберта — Вера (Лотиан и Чэппел,
1965)
Oi = \g(-^-\ = ~lgeND^Ll (III.31)
где 10 и I — интенсивности падающего и проходящего света,
соответственно.
Таблица III. 7
Влияние дисперсной фазы
на постоянные уравнения (III.28)
(Ллойд, 1959)
Дисперсная фаза
Керосин
Кукурузное масло
Парафин
а.
0,558
0,500
0,548
Р
55,8
64,0
76,4
Таким образом, оптическая плотность пропорциональна D2L.
Для полидисперсных эмульсий оптическая плотность составит
Oi = -£- Hg eN{ ^ LiPiBi (III.32)
где Li— коэффициент светорассеяния для шариков диаметром D t;
рс — доля частиц диаметром Dt в эмульсии.
Если эмульсия содержит с (в г) дисперсной фазы на 100 г
эмульсии, каждый кубический сантиметр которой разбавлен до / (в см3).
то количество дисперсной фазы в единице объема разбавленного
образца выражается уравнением
-jr WiXi ^ р^ = °.01 ("J ) Рэ (Ш .33)
где р,- и рэ — соответственно, плотности шариков и неразбавленной
эмульсии.
Подстановка Ni в уравнение (III.32) дает:
_ Q,0lblcp3lgeJ^LiPiD2i
fpi 2 л'д'?
i
10*
(II 1.34)
147
Мутность разбавленной эмульсии определяется как ее
оптическая плотность в ячейке длиной 1 см, содержащей 1% дисперсной
фазы:
£з = -^(|) (HI-35)
На коэффициент рассеяния света L, значение которого не
превышает 5, влияют следующие факторы (Голден, 1958).
1. Отношение размера частиц к длине волны проходящего света:
2. Отношение Пф/пс.
3. Поглощение света шариками. (В видимой части спектра
и вблизи инфракрасной поглощение обычно незначительно.)
4. Теоретическое значение L важно лишь в случае, когда углы,
противолежащие освещаемой поверхности образца при источнике
(Q„CT) и детекторе (£2дет), близки нулю. При отсутствии этого
условия L имеет низкие значения, особенно при более коротких длинах
волн, так как v увеличивается (Голден, 1958).
5. Шарики могут рассеивать свет, предварительно рассеянный
другими шариками, тем самым увеличивая общий результат (Вал-
стра, 1965). Интенсивность светового луча уменьшается поэтому
согласно ехр( —sn), но не (1 — s„), где sn — общее рассеивающее
поперечное сечение. Многократное рассеяние не происходит при
ехр ( — sn) *=« (1 — s„), для чего на практике требуется, чтобы
О, < 0,05.
6. Если шарики плотно упакованы, то при рассеянии световых
волн наблюдается их интерференция (Валстра, 1965). Это присуще
концентрированным эмульсиям или тем, в которых до измерения
произошла флокуляция.
Значение коэффициента рассеяния света для сфер, не
поглощающих его, вычисляют по уравнению
/. = 2-4-sin 1 + 4" О-™^) (III.37)
где S = 2v^—--—J -
Уравнение (III.37) справедливо, если гаф/ис -+1 и D/2 > X,
и применяется для значения v вплоть до 7. При более низких
значениях необходимо использовать уточненное соотношение (Геллер
и др., 1967). На рис. III.7 (Голден, 1958) показаны значения L
в зависимости от v для эмульсии М/В с отношением коэффициентов
преломления 1,09 (гаф= 1,452 и ис=1,33). Голден (1957) изучал
пропускание света разбавленными эмульсиями М/В с помощью УФ
спектрофотометра с широкой шкалой длин волн. Для длин волн
до 1,3 применялся стандартный фотоэлемент, и проходящий свет
измерялся в ячейке длиной 1 см. Для поддержания наименьших
148
Рпс. 111.7. Значения коэффициента
светорассеяния в зависимости от v
(гф/пс = 1,09) (Голден, 1958).
значений (QnCT-f- Одет) коробка фотоэлемента монтировалась за 0,5
диаметра неподвижной аппаратуры.
При этих условиях значения Q„CT в вертикальной и
горизонтальной плоскостях были приблизительно равны 7,5° и 3,5°,
соответственно, а углы Q1ICT и QaeT —9°
и 5°. Средний диаметр
шариков в этих эмульсиях 3 мкм,
и при Ф <=» 0,04 общее
поперечное сечение площади
составило ~ 190 см2/мл.
Результаты многократного рассеяния
можно подсчитывать при Ф >
> 0,005. Поэтому эмульсии
перед исследованием
разбавляли до Ф = 0,002. На рис. III.8
показана зависимость
коэффициента светорассеяния
монодисперсных эмульсий от 1/Х.
Уэлс (1962) рассчитывал
величины мутности
монодисперсных латексов с
диаметрами частиц 0,05—5,4 мкм для длин волн 0,65, 0,80, 0,95
и 1,10 мкм.
На рис. III.9 приведены данные оптической плотности для
полидисперсных эмульсий при различных значениях 1/А,. Кривые здесь
более пологие, чем в случае монодисперсных эмульсий.
А, пкн
/,J 1,11,0 0,8 0,7 0,6 0,5
Рис. III.8. Зависимость
коэффициента
светорассеяния монодиснерсных
эмульсий от длины
волны (Голден, 1958).
D, мкм: 1 — 5; 2 — 3;
3—2; 4 — 1; 5 — 0,5.
//А, мкп'
Если в непрерывной фазе содержатся мицеллы, они, как и
шарики, будут рассеивать свет. При значительных длинах волн
рассеяние света мицеллами небольших диаметров будет уменьшаться
в соответствии с выражением (1Д)4 формулы Рэлея (1871, 1897,
149
К, пни
1,2 1,0 0,8 0,6 0,5
Образец а
10
I 5
г>
<s
1)
<Ъ
^ 15
§. 10
-I 1 L
Образец 6
ъ
■3 зо
<| 20
Образец в
10
Л
I—i—I г
1899), не оказывая существенного влияния на общее рассеяние.
В эмульсиях М/В, стабилизированных белками молока, этот эффект
устраняется добавлением небольшого количества щелочи, которая
солюбилизирует их (Голден, 1957).
Валстра (1965) считал, что видоизмененный УФ спектрофотометр
Голдена непригоден для исследования негомогенизированных
эмульсий, особенно при больших размерах шариков. Он помещал
специальную приставку между
опорой ячейки и
фотоэлементом. Но это привело
к другой трудности:
только небольшая часть света
от монохроматора
попадала на фотоэлемент.
Нужно было использовать
сравнительно широкую
щель для полного
прохождения света.
Определение диаметров
частиц <С 2 мкм путем
измерений мутности при
больших длинах волн за-
ё труднено (Голден и Фиппс,
£ Л Г\ПГ\\ ТТ ^ ._ .
2 н 6 8 1,0
Диаметр шариков, ним
-I 1 1 L.
0,3 |
0,2 <=
о;
0.1 §
"о
Hi
0,2
0,1
1,5 2,0 2,5
J/K.mtf'
Рис. III.9. Зависимость оптической
плотности полидисперсных эмульсий от длины
волны (Голден, 1958):
- экспериментальные данные;
ные данные.
вычпслен-
0,3 § 1960). При более коротких
волнах чувствительность
измерений заметно
увеличивается (рис. ШЛО),
что особенно важно для
исследования эмульсий
с большим содержанием
очень маленьких шариков.
В большинстве пефе-
лометрических
определений требуется
сравнительно узкая ширина
спектра, так что
дорогостоящий УФ спектрофотометр
можно заменить простым фильтрующим прибором. Для этих
целей приспособлен хилгеровский' биохимический абсорбциометр
(Голден и Фиппс, 1960). С его помощью, используя соответствующие
фильтры или их комбинации, можно определить как средние
размеры частиц, так и объем дисперсной фазы. Прибор
необходимо откалибровать относительно данных, получаемых г,
помощью УФ спектрофотометра при соответствующих длинах волн.
Промышленные абсорбциометры удобны тем, что уже отка-
либрованы.
Рассеяние света. Рэлей (1871, 1897, 1899) вычислял
интенсивность рассеяния света сферическими непроводящими частицами,
150
которые безгранично малы по сравнению с длиной волны света
h
9л2ЛТ
/о
2^4,
2
Пф-
2
Пф-
(1 + COS2 9;)
(111-38)
0,6 нкн
где /е — интенсивность света, рассеянного под углом 0; к
направлению падающего луча; V — объем шарика; d — расстояние между
измерительным элементом и образцом.
Самой большой интенсивность рассеяния света будет в
направлении, параллельном падающему лучу, наименьшей — в
перпендикулярном направлении. В
промежуточных направлениях наблюдается
постепенное изменение интенсивности.
Так как с уменьшением длин
световых волн интенсивность светорассеяния
соответственно увеличивается, то
размеры шариков можно определять но
рассеянию при коротких длинах
волн.
Светорассеивающий прибор
содержит в качестве источника света лампу
со средним или высоким давлением
ртутных паров, которая дает
параллельный монохроматический луч с
помощью стеклянных монохроматических
фильтров. Такой луч проходит через
поляризатор и попадает на ячейку
с образцом. Интенсивность
рассеянного излучения измеряется при
различных углах фотоумножителем, и
результаты регистрируются высокочувствительным гальванометром
или записывающей лентой. Весь прибор заключен в
светонепроницаемый ящик. Кроме того, он снабжен световой ловушкой для
поглощения луча, выходящего из ячейки с образцом, с тем, чтобы
исключить случайное попадание света из фотоумножителя. Все
внутренние поверхности приборов не должны отражать свет, а пыль
необходимо полностью удалять.
Мак-Интаир и др. (1962) использовали в качестве ячейки для
образца наружный стеклянный цилиндр с полированными ровными
стенками, чтобы обеспечить вход и выход падающего луча света.
Внутри этого цилиндра помещали другую меньшую ячейку, сделанную
из черного светопоглощающего стекла для устранения отражения
падающего и рассеянного света. Обе ячейки снабжены тефлоновыми
колпачками. Наружный цилиндр наполнен ксилолом,
определенная температура которого поддерживается с помощью
погруженной в него подогреваемой спирали. Внутренняя ячейка содержит
образец.
г 3 4 S
D, мкп
Рис. III. 10. Зависимость
мутности от диаметра шариков
монодисперсных эмульсий при
трех различных длинах волн
{njnc = 1,09) (Голден и
Фиппс, 1960).
151
Ван дер Ваарден (1954) определял средние диаметры шариков
в эмульсиях М/В по интенсивности света, рассеянного в
направлении, перпендикулярном падающему лучу, который испускался
а
ртутной лампой (4358 А). Средние диаметры колебались от 138 до
1025 А; для эмульсий с диаметрами капель, большими ОАк,
применима поправка для отклонений от закона рассеяния Рэлея (Доти
и Штейнер, 1950). Диаметры вычисляли и по интенсивности
рассеянного света, когда 8, = 0 в уравнении (III.38). Такую
интенсивность можно получить экстраполяцией данных рассеяния при
различных значениях 8;. Достаточно хорошо согласовывались данные,
вычисленные с помощью других методов, но так как в
полидисперсных эмульсиях в отличие от монодисперсных наблюдалось более
сильное рассеяние, значения диаметров колебались от малых до
больших.
Если капли велики по сравнению с К, то волны, рассеянные ими,
будут интерферировать. Это уменьшает интенсивность рассеянного
света согласно
V4 V sin к Ada
pm=ZZ^Ji— (IIU9)
где проведено суммирование по всем парам рассеивающих элементов
г и у; к = 2я/А/; к' — длина волны света в образце; А = 2sin 8:/2;
д.ц — расстояние между элементами i и ;' (Дебай, 1927).
Уравнение (III.39) можно упростить
2
Р (6,) = Г~ (sin х-cos х)Л (II 1.40)
где х = kAD/2.
Доти и Штейнер (1950) на основе этого выражения представили
таблицы поправочных коэффициентов.
Поточный ультрамикроскоп
Концентрацию частиц в стационарном объеме можно определить
с помощью ультрамикроскопа (Зидентопф и Жигмонди, 1903),
однако это длительный процесс. Дерягин и Власенко (1962)
предложили прибор, в котором число частиц подсчитывают по числу
световых вспышек. Стеклянная ячейка состоит из двух коаксиальных
трубок. Образец при контролируемой скорости протекает в одном
направлении через внутреннюю трубку и возвращается через
наружную. На конце ячейки есть окошко, через которое образец
просматривается с помощью микроскопа. Когда частица пересекает
наблюдаемое поле, появляется световая вспышка. Вспышки
подсчитывают или непосредственно, или автоматическим фотоумножителем,
электрические импульсы из которого попадают на усилитель
постоянного тока и затем регистрируются автоматическим счетчиком
152
с помощью микрореле. Число частиц на 1 см3 вычисляют по
уравнению
N = -p^ (III.41)
' всп
гггр т/ Уобт и 7 =: ^52
Д м Z а Д»,р (2-Л5ар/Л8н)
Здесь ивсп — число вспышек; FBCn — объем образца, проходящего
через ячейку в течение времени регистрации пвсп; Vo6ui — общий
объем образца, пропускаемого через ячейку; Dнар и DBU — диаметры
наружной и внутренней трубок ячейки, соответственно.
Диаметр частиц можно вычислить, если известна объемная
концентрация дисперсной фазы. Недавно высказано предположение, что
с высокоимпульсным анализатором и измерением скорости можно
вывести функцию, относящуюся к дисперсности.
Поточный ультрамикроскоп, применяющийся для определения
размеров твердых частиц в широком диапазоне (0,05—10 мкм),
не нашел применения при исследовании эмульсий. Преимущество
метода в том, что можно проводить измерения в широкой области
концентраций частиц (1 —1010 частиц на 1 см3) без большого
увеличения, так как не требуется одновременная дифференциация
нескольких частиц в отличие от обычного микроскопа.
Другие методы
Цен трифугирование
Седиментация под влиянием центрифугирования происходит
при скорости, зависящей от размера капель эмульсии. Пинтер и
Зильвесмит (1962) фракционировали эмульсии М/В по размерам
частиц седиментацией в колонке с сахарозой. Они получали слои
с различными плотностями. Градиент плотности создавали при
смешивании 50 и 30% растворов сахарозы. Смесь помещали в пробирку
емкостью 100 мл и далее вводили 1 мл эмульсии М/В в 50% растворе
сахарозы. В нижнюю часть пробирки вводили 5 мл 60%
раствора сахарозы. Центрифугирование проводили при скорости до
2800 об/мин.
Плотность сахарозы в любой точке пробирки можно выразить
Уравнением
Рсах = "с [De-Dp) + p, (HI.42)
где ас — постоянная градиента; De и Dp — расстояния от центра
вращения начального слоя эмульсии до каждого последующего
слоя; р{ — плотность слоя, непосредственно находящегося выше
места введения эмульсии.
Аналогично можно выразить вязкость сахарозы в любой точке
пробирки
1\cn = $c(De-Dp) + r\t (111.43)
где рс — постоянная градиента; r\t — вязкость слоя, прилегающего
к месту введения эмульсии.
153
Закон Стокса для седиментации с помощью центрифугирования
выражается следующим образом:
dDp О2С02£>р(рф-рсах)
dt
18т)с
(III.44)
где а» — скорость вращения; рф — плотность дисперсной фазы;
т — время.
После интегрирования получаем:
D =
18
I С02Т
4i—$cDe
I Рф —('/-
- arD«
ln-
«с(Ч/ —РсДе) -(Рф-Р/
— Ol^r-arD.
МРф— РГ
-а,Д,
In
-р/-г"е {De — Dp)
Рф —Р/
(III-45)
Так как все величины известны, то можно определить Z>.
При вычислении (о2т нужно
учитывать как время ускорения, так
и время торможения. Это удобно
выполнять графически.
Строится зависимость со2 от т.
Площади под кривой во время
ускорения и торможения определяются
планиметрически. При скорости
центрифугирования, превышающей
2000 об/мин, эти площади
одинаковые, величина со2т равна
произведению квадрата максимальной угловой
скорости к моменту начала
центрифугирования и времени начала
торможения. При низких скоростях
центрифугирования значение г
получается делением разности между
площадями под кривыми ускорения
и торможения на со2, добавлением полученного к интервалу
времени, соответствующего наибольшей скорости. Для получения со2т
результат умножается на максимальную угловую скорость.
На рис. III.11 представлены значения миграции шариков
плотностью 0,939 г/см3 и диаметром 0,1—0,5 мкм. При диаметре 0,2 —
0,45 мкм наблюдается прямолинейная миграция, при <; 0,2 мкм
шарики мигрируют быстрее. Точность этого метода проверяли
центрифугированием монодисперсных латексов с известным
диаметром частиц. Значения диаметров частиц 0.53. 0,74 и 1,08 мкм.
определенных с помощью электронного микроскопа, соответствовали
0,556, 0,74 и 1,17 мкм при центробежной седиментации. Таким
образом, для частиц с указанными диаметрами метод оказался вполне
удовлетворительным, но при диаметре 0,26 мкм полученная величина
равнялась 0,19 мкм. Причина такого расхождения неизвестна,
1? м
Рис. 111.11. Миграция шариков.
плотностью 0,939 г/см3 в растворе
сахарозы (Ппнтер и Зильве] смит,
1962).
I — расстояние после
центрифугирования и течение 4 ч при 2800 об/мин.
154
возможно, что передвижение этих меньших частиц из раствора
сахарозы высокой вязкости в тот, куда они первоначально
вводились, может замедляться.
Счетчик частиц (счетчик Коултера)
С помощью этого прибора удается очень быстро исследовать
большое число частиц. К сожалению, его применение для эмульсий
М/В ограничено тем, что эмульсии необходимо разбавлять
раствором электролита.
Небольшой объем эмульсии (—20 мл) разбавляют 400 мл деиони-
зированной дистиллированной воды, после чего 1 мл этой дисперсии
Рис. II 1.12. Схема счетчика частиц:
1 — главный усилитель; 2 — пороговое устройство; 3 — импульсный
усилитель; 4 — счетная машина; 5 — цифровой регистр; 6 — счетчик
«пуск — стоп»; 7 — горизонтальная развертка; S — осциллоскоп.
смешивают с 200 мл 0,5% раствора хлорида натрия и определяют
однородность распределения масляных шариков в непрерывной
фазе.
Стеклянную трубку специальной конструкции (рис. III.12),
содержащую электрод, вводят в сосуд, в котором размещен второй
электрод. Верхняя часть трубки присоединена через краник к
вакуумному насосу, а отвод — к ртутному манометру. У основания
трубки имеется небольшое отверстие, через которое проходят
шарики. Если краник открыт, то через отверстие поступает поток
жидкости, контролируемый вакуумом. Одновременно ртуть движется
вверх в правую часть лимба манометра. Если же краник сразу
закрывается после того, как ртуть принимает свое первоначальное
положение, то скорость движения потока через отверстие
приблизительно равна 30 см3/сек. Начальное движение ртути при закрытом
кранике передается счетному механизму, регистрирующему число
частиц в данном объеме дисперсии, и одинаковые по размерам
шарики сразу же подсчитываются.
155
Проходя через отверстие, каждый шарик дает информацию
об объеме электролита, эквивалентном его собственному объему.
Электропроводность масляных шариков намного меньше
электропроводности водной фазы, поэтому сопротивление между
электродами мгновенно изменяется. Такое изменение сопротивления равно
АЛ=-^ =— (111.46)
Луд1у
Q1
'-'от
1-
1
1
-Луд l/ЯуД 2
S
200т
где ЯуД1 и Луд2 — удельное сопротивление электролита и шарика;
SQT — площадь отверстия, перпендикулярная оси; 5 — площадь
шарика, перпендикулярная оси отверстия; х — отношение длины
шарика, параллельной оси отверстия, к диаметру эквивалентной
сферы.
Изменение сопротивления создает короткий импульс
напряжения, величина которого зависит от объема шарика. Каждый импульс
усиливается и подается на один из ряда порогов контура с
регулируемым уровнем напряжения. Как только достигаются уровни
заранее выбранного напряжения или превышают его, импульсы подсчи-
тываются. Счетчик подсчитывает количество частиц с диаметрами,
не превышающими более чем на 30% диаметр отверстия. Возможно
применение отверстий с диаметрами 30—2000 мкм. Вместе с 70-ю
цифровой выборкой можно подсчитать 5000 импульсов в 1 сек,
измеряя диаметры шариков 5= 1 мкм. Вахтель и ла Мер (1962) не смогли
определить диаметры < 0,6 мкм, так как этот предел достигает
уровня электронного шума прибора.
Исследование данных распределения по размерам
Средний размер шарика можно вычислить несколькими путями
из данных распределения, полученных с помощью любого прибора,
описанного выше. Основные методы вычисления (Далла Балле,
1948) диаметра:
среднегеометрическое значение диаметра
ог--| -Pi^A).- ■ • Ac (III.47)"
2 «is л
среднеарифметическое значение диаметра
_1inD __ n1D1^n2Di + n3Da+. . -+пхРх пп.48}
средний диаметр
^^Г (.11.49,
156
средний поверхностный диаметр
средний объемно-поверхностный диаметр
2jnD
средний объемный диаметр
»-№
где пъ щ, п3, . . ., пх —число капель с диаметрами Dl:D2, D3, . . ., Dx.
При расчете среднего диаметра необходимо по свойствам
изучаемой эмульсии подобрать наиболее подходящий метод. Например,
если требуется общая площадь поверхности для данной объемной
концентрации дисперсной фазы, то средний диаметр можно
подсчитать по уравнению (III.51). Когда вязкость при высокой скорости
сдвига нужно сопоставить со средним диаметром, необходимо
применить уравнение (III.52).
Может возникнуть вопрос о применимости уравнений (III.51)
и (III.52) к эмульсиям с большим числом шариков диаметром
<Г 1 мкм. Этими шариками создается большая общая площадь
поверхности, но на вязкость они влияют в меньшей степени, чем
шарики с диаметром > 1 мкм. Мелкие шарики оказывают влияние
и'на величину среднего диаметра, так как в указанные выше
уравнения диаметр шарика входит во второй или третьей степени. Поэтому
в таких случаях требуется другой метод вычисления. Для
определения среднего объемного диаметра можно пользоваться
уравнением:
DV f "1 + ге2 + "3 + • ■ • + пХ
з/ v? Г~
У ^F
При изучении реологических и других свойств важно определять
распределение по размерам вблизи среднего значения. Две эмульсии
с одним и тем же средним размером шарика могут проявлять
различные свойства, потому что распределение по размерам вблизи
среднего значения неидентичны. Джеллинек (1950) ввел коэффициент
(111.50)
(111.51)
(III-52)
157
неоднородности, определяемый как «среднеквадратичное отклонение
кривой коэффициента распределения от диаметра»
F = -
f (D-Dap)vndl
оо
(III.54)
где vn — скорость изменения числа шариков с определенным
диаметром.
Для монодисперсных эмульсий F = 0. Интегрируя уравнение
(III.54), получим
F = — -.D§p (III.55)
8 12
Диаметр капель, пкм
16
20
Рис. III.13. Гистограмма распределения капель по размерам
в эмульсии М/В (Шварц и Беземер, 1956).
где среднее значение площади выражается формулой:
Yns
Самым простым методом представления данных частоты
распределения по размерам является гистограмма, по оси ординат которой
откладывается содержание шариков (в %), а по оси абсцисс — их
размер. Каждый прямоугольник гистограммы показывает
содержание шариков в пределах данного размера. Сумма площадей всех
прямоугольников должна быть равна 100%. Распределение по
объему и по площади поверхности представляется аналогично.
Если плавная кривая, проведенная через вершины прямоугольников,
симметрична относительно вертикальной оси, то она называется
нормальной вероятностпой кривой. Однако чаще кривая
асимметрична (рис. III. 13), так как верхний предел распределения
растянут намного дальше от размера оптимальной частоты, чем
нижний.
158
Симметричная кривая частотного распределения по размерам
подчиняется обычному вероятностному закону (Далла Балле, 1948)
2»
G(D)= *• ехр
Д 1 2л
■(^—Dap)2
2Д2
где G (D) — частота наблюдения диаметра D;
отклонение, равное:
(Ш.56)
нормальное
■V
2>(Я-Аф)а]
Асимметричное распределение превращается в симметричное
при замене размеров шариков на графике на их логарифмы. Такой
кривой соответствует уравнение
У; » Г — (lnD-lnDr)2
G(D) = -
ln Дг J 2л
эхр [-
21п2 Дг
(III.57)
где
пДг=1/
2 [« (In О—ln/Jr)2
2 "
Теоретический средний объемный
диаметр для эмульсий можно вычислить,
используя уравнение (III.57):
Диаметр капель
Рис. 111.14. Кривая
распределения капель по
размерам.
°у-
V ~Ъ
2иД8
2»
\ I "^ — т=г- С DS охр Г -
V У п Ig Дг I' 2л 3 Р L
(In О— 1пОг)2
2 1п2 Дг
Iх
'x'dln D = /ехр (3 In Д.+ 4,5 In2 Дг)
(111.58)
Согласно другому методу, строится график зависимости
содержания капель, которые меньше данного размера, от размера капель.
Он представляет собой S-образную кривую (рис. III. 14). По ней
можно определить средний диаметр, т. е. размер более чем 50%
йапель. Математически ^кривая выражается следующим образом
G(D) = .
dN^
dD
(Ш.59)
где ND — число шариков, больших или меньших определенного
размера D.
Если ND (D) является функцией, определяющей результативную
кривую, то для двух шариков с диаметрами D, и D2 имеем:
Dt
!fD(D1)-ND{D2)-
dND
dD
■dD,
G (D) dD
(111.60)
159
Шварц и Беземер (1956) для определения распределения по
размерам предложили простое линейное уравнение с двумя
параметрами
lnVB = ln<D + -j-^ jS- (Ш.61)
■"макс JJ
где Vj) — общий объем дисперсной фазы с диаметром шариков
ниже выбранного диаметра D', Dx — характеристический диаметр;
^макс — максимальный диаметр.
График зависимости In VD от D является прямой, по которой
можно вычислить Dx и й41акс. Исследование опубликованных данных
показало, что для эмульсий, приготовленных механическим
перемешиванием, уравнение (III.61) применимо в отличие от эмульсий,
полученных инверсией фаз. Дальнейшее ограничение этой
зависимости состоит в резком отклонении от линейной зависимости при
D < 2 мкм (Шерман, неопубликованные данные).
ЭЛЕКТРОФОРЕЗ
Электрические свойства межфазной поверхности масло — вода
влияют на устойчивость и реологические свойства эмульсий.
Данные об этих свойствах можно получить из электрофоретических
исследований, проводимых одним из двух методов. В первом случае,
когда применяется метод движущейся границы, изучается объемное
движение частиц, а во втором — микроскопически определяется
скорость отдельных частиц.
Прибор, используемый в исследованиях, которые проводят с
помощью первого метода, состоит из стеклянной U-образной трубки,
в открытые концы которой помещены платиновые электроды. Трубку
заполняют эмульсией М/В ниже края каждого электрода, а затем
вводят достаточное количество дистиллированной воды с тем, чтобы
покрыть их. К электродам подводят постоянный ток и измеряют
скорость, с которой поверхность раздела вода — эмульсия движется
в верхнюю часть одного из лимбов U-образноп трубки. Поток можно
направить в противоположную сторону и изучать скорость
движения в другом лимбе. Измерения проводят при различных
напряжениях, подтверждая, что скорость не зависит от подаваемого
потенциала.
Второй метод заключается в изучении скорости движения
шариков в стеклянном капилляре под влиянием градиента потенциала.
Предложено несколько конструкций ячейки. В обычной закрытой
трубке возникает два эффекта:
а) движение шариков относительно непрерывной фазы — электро-
форетический эффект;
б) движение непрерывной фазы относительно заряженных
стенок стеклянной трубки — электроосмотический эффект.
Эти эффекты приводят к параболическому распределению
скоростей частиц на расстоянии от стенок трубки. Действительную
160
электрофоретическую скорость получают измерением движения
частиц на различных глубинах. К сожалению, градиент скорости на
этих глубинах значителен, поэтому и скорость шариков существенно
изменяется в зависимости от глубины, что и определяет погрешности.
Смит и Лиссе (1936) предложили ячейку (рис. III.15), состоящую
из двух стеклянных капилляров различных диаметров. Концы
капилляров помещают в две широкие стеклянные трубки,
содержащие платиновые электроды и крышки для введения образца. В такой
ячейке скорость шариков вдоль оси более узкого капилляра
соответствует электрофоретической скорости при выполнении условия
h
R-i
2 Г
L
Д-2
0<
III.62)
Вид сбоку
\m
дид сверху л
А -«-I /
А А ^
где Zj, Z2 и R i, R2— длины
п радиусы соответственно узкого
и широкого капилляров.
Обычно li = 10 см; 12 =
= 14,34 см; R = 0,03 см и Д2 =
= 0,048 см. В таком случае
поток возвращается через капилляр
большего диаметра. Широкие
трубки настолько больше
капилляров, что падение потенциала
в первых невелико. В более узком
капилляре градиент потенциала
равен U/l{ (где U — падение
потенциала между электродами).
Скорость непрерывной среды в обычной закрытой трубке (Ламб,
1888) равна
Рис. III.15. Система из двух ячеек
для микроэлектрофореза (Смит и
Лиссе, 1936):
1 — объектив микроскопа; 2 —
конденсатор; 3 — электрод; 4 — широкий
капилляр; 5 — узкий капилляр.
Ph
4NcZ
R%-fR\
(Ш.63)
где Ph — градиент гидростатического давления, параллельный оси
капилляра радиусом R; R0 — радиус произвольного
прямолинейного потока.
Скорость на поверхности капиллярной трубки можно выразить
уравнением
поэтому
и --^
s~ 8r)cl
I 2Rl а\
(Ш.64)
(111.65)
Если приложен градиент потенциала, то скорость шариков
находят из выражения:
и = "ф + Ис = "ф + "а(-дГ !} (III -66)
где Цф — действительная электрофоретическая скорость шариков.
11 Заказ 1070 161
Из уравнения следует, что ис = 0, если 2Щ= R2, т. е. R0 =
= ± R 1/2. Неподвижные уровни с нулевым электростатическим
потоком будут при условии, что i?0 — 0,707/?. Это и есть глубина,
на которой нужно определять электрофоретическую скорость.
Конструкция из двух капилляров имеет следующие
преимущества по сравнению с одним капилляром:
а) изменение скорости с глубиной незначительно вблизи
стационарного уровня, поэтому небольшие погрешности в фокусировке
микроскопа не вызывают серьезных ошибок в определяемой скорости;
б) глубина поля зрения не так важна;
Вид сверху *
Эмульсия
Рис. III.16. Микроприбор для электрофореза (Дуглас,
1947):
1 — медный электрод; 2 — пробка из фильтровальной бумаги;
з — электродные ответвления; 4 — скрепляющее устройство.
в) градиент скорости вблизи стационарных уровней мал, так
что вращение шариков наименьшее;
г) дает более согласующиеся результаты.
Дуглас (1947) применял ячейку из предметных стекол (рис. III.16),
скрепленных парафином. Отводы для электродов, впускную и
выпускную трубки тоже прикрепляли к ячейке с помощью парафина.
Разность потенциалов источника составляла 120 в, постоянный ток
от потенциометра подавали на электрод через миллиамперметр
и выключатель. Использовали медные электроды, погруженные
в сульфат меди. Электродная жидкость, отделяемая от образца
пробками или скатанной фильтровальной бумагой, просачивалась в
соответствующий раствор электролита. В качестве буфера для эмульсий
М/В применяли 0,001 н. раствор хлорида натрия. Ячейку помещали
на столик микроскопа. Стационарные уровни рассчитывали из
уравнения:
h =
±*Y~-
128z
(Ш.67)
162
где z — половина глубины ячейки; у — половина ее
ширины.
Бенгем и др. (1958) вместо обратимых электродов серебро —
хлорид серебра или медь — сульфат меди, которые загрязняют
образец солями серебра или меди, предложили применять
платиновые электроды. Они состоят из платинового цилиндра с впаянной
платиновой проволокой и помещены с помощью пробки из
натриевого стекла или полиэтилена в емкость со ртутью.
При фокусировке микроскопа на уровень нулевого электро-
осматического потока в ячейке с круговым поперечным сечением
могут появляться погрешности (Генри, 1938), потому что луч света,
входящий при таком уровне, загибается вверх. Точечный фокус
объектива относится к точечному фокусу в воздухе, как
_ "_£^ + („м_„обр) (Ш.б8)
т—"воз1Гпм Ч
где пвоз, км, иибр — показатели преломления воздуха, материала
ячейки и образца, соответственно; /i?0T — толщина стеклянной
электрофоретической ячейки кругового сечения; mROT и qR0T —
расстояния от поверхности ячейки до фокусной точки в воздухе и
до действительной фокусной точки в образце, соответственно; R0T —
радиус отверстия ячейки.
Из уравнения (III.65) получаем:
Следовательно :
-1 (111.69)
•4ut
№)
d{R0/R) -**S'
Ошибка в 5% при вычислении R/R0 приводит к ошибке 0,20as
в измеряемой скорости. В табл. III.8 приведены значения поправок
регулировки хорошо настроенного микроскопа, если внутренний
диаметр стеклянной ячейки равен 2 мм, наименьшая толщина стенок
0,25 мм, j = 0,25 мм; пвоз = 1,000, пм = 1,515 (стекло) и побр =
= 1,342. Поправки, полученные при 0 С и фокусировке на
верхней внешней поверхности трубки, сведены в графу 2.
< Электрофоретическая скорость эмульсии при градиенте
потенциала определяется не менее чем из 20—30 показаний для двух
направлений потока. Перед исследованием каждый образец
эмульсии необходимо разбавлять непрерывной фазой, а среднюю скорость
определять при нескольких разностях потенциалов, убеждаясь, что
прилагаемое напряжение не влияет на результаты. Обычно поток
бывает небольшим, даже в хорошо проводящей системе, так что
любое повышение температуры за счет него незначительно.
Электрофоретическая скорость выражается уравнением (Генри,
1931):
и=Ее^ (111.70)
6лт]с
И*
163
Таблица III.8
Точность настройки при использовании электрофоретической ячейки
кругового поперечного сечения
(Генри, 1938)
Глубина действительной фокальной
точки от вершины отверстия, мм
0 (вершина отверстия)
0,2
0 293 (нулевой уровень)
0,4
0,6
0,8
1,0
1,2
1,4
1,6
1,707 (пулевой уровень)
1,8
2,0
Исправленные
значения
величины
регулировки, ММ
0,165
0,310
0,375
0,448
0,580
0,705
0,825
0,939
1,049
1,153
1,207
1,254
1,350
Значения
величины
регулировки,
вычисленные линейной
ин тери оляцней,
мм
0,165
0,283
0,339
0,402
0,520
0,639
0,757
0,876
0,995
1,113
1,176
1,231
1,350
Л, мм
0
0,027
0,036
0,046
0,060
0,066
0,068
0,063
0,054
0,040
0,031
0,023
0
где Е — прилагаемое напряжение; ес — диэлектрическая
постоянная непрерывной среды; пс — вязкость непрерывной среды; £ —
электрокинетический потенциал; к — функция Дебая — Хюккеля.
Если kR > 1, то функция Генри / (kR) = 1,5, когда же kR <"< 1,
то f{kR) = 1,0.
Представлены значения подвижности шариков нуйола,
диспергированных в воде, равные 3,65 мкм/(сек-в-см) (Лимбург, 1926; Гинн
и др., 1952), 4,0 мкм/(сек-в-см) (Раувелл, Александер, 1952) и
4,45 мкм/'(сек■ в■ см) (Паули и Вуд, 1940). Олеат или лаурат натрия
увеличивают подвижность, в то время как хлорид натрия
уменьшает ее.
Описанные методы измерения электрофоретической скорости
пригодны для эмульсий М/В, диэлектрическая постоянная водной
фазы которых велика. Для большинства эмульсий В/М величина е
не превышает 5, и испытания в таких ячейках приводят к
беспорядочному движению шариков, которые могут притягиваться к
электродам, а затем отталкиваться (ван дер Минне и Германи, 1952).
Это явление называется диэлектрической поляризацией и
наблюдается в сильных полях.
Ячейка, наиболее пригодная для измерений в эмульсиях В/М,-
состоит из стеклянного капилляра, окруженного стеклянным
кожухом, в котором можно создавать вакуум (рис. III.17). Образец
вводят через отвод капилляра. Для устранения проводимости
поверхности капилляра его обрабатывают кремнием при 35° С
в течение 0,5 ч, после чего помещают в кожух и закрепляют песочно-
164
тальковым цементом. Однородность электрического поля достигается
при устранении проводимости поверхности капилляра, увеличении
сопротивления стенок за счет более тонкого стекла, создании
вакуума в кожухе. Ван дер Минне и Германи (1952) пользовались
капилляром с размерами: длина —
13,0 см, внутренний диаметр —
0,2 см, толщина стенок —
0,22 см. Расстояние между
концами спиралевидных
платиновых электродов равнялось 4 см.
Альберс (1957) подобную
ячейку использовал для
изучения подвижности шариков
в эмульсии В/М,
стабилизированной олеатами различных
металлов. Прилагаемое
напряжение не превышало 100 в/см
с тем, чтобы предотвратить
явление электростатической
индукции. Электрофоретическая
подвижность изменялась в
области (0 — 140)-Ю-3 мкм/(сек-в
емого олеата. При олеате бария подвижность равна нулю, при
олеате магния — наибольшая.
ПОВЕРХНОСТНОЕ И МЕЖФАЗНОЕ НАТЯЖЕНИЕ
Данные межфазного натяжения характеризуют эффективность
диспергирования в процессе образования эмульсии. Эти данные
используют для вычисления коэффициентов растекания, краевых
углов, вандерваальсовых сил притяжения между шариками и
взаимодействия между поверхностями раздела (Фоукес, 1964).
Динамические методы
,, Метод отрыва кольца (метод дю Нуи)
Силу отрыва кольца от поверхности раздела масло — вода
измеряют тензиометром. Кольцо подвешивают к коромыслу весов,
соединенному с закручивающейся нитью (рис. III.18). С помощью винта
медленно опускают столик, где находится стеклянная чашка с
жидкостью, поверхностное натяжение которой измеряют. Для
предотвращения движения кольца вместе с поверхностью раздела
к нити кручения прикладывают вращающий момент. Это достигают
с помощью коромысла с прикрепленными зеркалами. Если сила,
передаваемая вращающим моментом, направлена вверх и превышает
направленную вниз силу удержания кольца межфазной
поверхностью, то кольцо отрывается от последней. Величина вращающего
2 3
,>irrhr^
0
Рис. II 1.17. Ячейка для электрофореза
эмульсий В/М (ван дер Минне и
Германи, 1952):
1 — электрод; 2 — стеклянная трубка; 3 —
плоская пластинка; 4 — стеклянный кожух;
5 — плоская отшлифованная поверхность.
см) в зависимости от использу-
165
момента регистрируется на градуированном диске прибора, который
после калибровки показывает измеряемое межфазное натяжение
в динах на сантиметр. Большую чувствительность дает оптический
метод, при котором луч света проектируется на зеркало, укрепленное
на нити кручения и, отражаясь, попадает на градуированную шкалу.
Перед наслоением масла на водную фазу кольцо должно быть
хорошо смочено водой. Если масляная фаза тяжелее водной, кольцо
покрывают кремнием для лучшего
смачивания маслом.
Благодаря методу
уравновешивания действующей на кольцо
силы, граничная пленка
подвергается минимальному искажению
до отрыва кольца. Динамические
изменения межфазного натяжения
при старении межфазной
поверхности возникают при
использовании упора для■коромысла.
Сила, отрывающая кольцо от
межфазной поверхности, равна сумме
веса кольца и жидкости,
поднимаемой с ним. Ее можно легко
подсчитать. Максимальное
усилие Рмякс равно произведению
межфазного натяжения оМ/В на общий
периметр кольца
Ри
jm/ -
<Ь
/в-
2л#к
(1П.71)
Рис. III. 18. Схема тензиометра для
измерения межфазного натяжения
(Александер, 1949):
1, 3 — зеркало; 2 — лампа; 4 —
противовес и демпфер; 5 — пластинка
закручивания; 6 — платиновое кольцо.
где DK — средний диаметр кольца.
По данным Харкинса и Иордана (1930), X. Фрейда и Б. Фрейда
(1930), максимальное усилие на кольце зависит от DJSVK и DJDK
(здесь DH — диаметр нити кольца; VK — объем жидкости,
поднимаемой с кольцом). Уравнение (III.71) можно переписать, введя
поправочный коэффициент Fc:
FcPvwc (I II. 72)
JM/B"
2nDK
Значение Fc изменяется от 0,75 до 1,45 в зависимости от формы
и размера кольца. Если не учитывать эту поправку, ошибка может
достигать 30 %.
Зюидема и У отер (1941) считают, что поправочный коэффициент
важен только в случаях, когда плотности двух фаз существенно
отличаются или же показания прибора низки. Проанализировав
таблицу поправок, составленную Харкинсом и Иорданом, которые
оказались достаточно сложными, они с помощью экстраполяции
нашли поправочный коэффициент
Dr
Pi —P2
3
(III.73)
166
где pi и р2 — плотности двух жидких фаз; В1 и В2 — константы*
со значениями 0,7250 и 0,0009075 независимо от используемого
кольца; В3 — константа, зависящая от отношения DJDK, так как
В3 = 0,4534 — i,679D„/DK (III-74)
Вычисления F'c как функции Py^Jp^ — p2 при различных
значениях D показывают, что чем больше диаметр кольца, тем меньше
поправочный коэффициент.
Для точности проводимых измерений кольцо должно лежать
строго на межфазной поверхности. Тонкие платиновые кольца легко
деформируются, поэтому применяют толстые кольца.
Пластинка Вильгельми
Тонкую платиновую или стеклянную пластинку подвешивают
к коромыслу тензиометра вместо платинового кольца. Можно
пластинку подвешивать к коромыслу весов. Максимальное усилие при
отрыве определяют осторожным уравновешиванием чашки весов
небольшими гирьками. Если стеклянная пластинка полностью
смачивается нижней водной фазой, ее необходимо до погружения
промыть нагретой хромовой кислотой или заменить шероховатой
слюдяной пластинкой. Слюдяная пластинка, покрытая слоем сажи (Чис-
мен, 1946), смачивается масляной фазой. Согласно элементарной
теории
PMaKC="lg = 2(lim + dnA)aM/B (III.75)
где 1пл и <1ПЛ — длина и толщина пластинки; т — вес
уравновешивающих гирек.
Если dnn <1ПЛ, то
%/в=Ф^ (Ш-76)
■"пл
Лучшие результаты получают, когда пластинка не отрывается
от межфазной поверхности, а остается частично погруженной в
водную фазу. В этом случае учитывают поправку на плавучесть. Рус-
сен (1946) обосновал применение этого метода при условии, когда
плотность масляной фазы меньше или больше плотности водной
фазы, т. е. когда мениск вогнутый или выпуклый.
Если масляная фаза легче водной, то рассматриваемые силы
направлены вниз в результате межфазного натяжения [в
соответствии с уравнением (75)], плавучести Ву пластинки, ее веса М и
усилия /, вызванного поверхностным натяжением масляной фазы.
Компенсирующее усилие Р'ылкс i уравновешивающее эти силы, равно:
^макс = 2(гпл-Нпл)<Тм/В + М-,В4,+/ (111.77)
Для совместного определения Ву a J измеряют усилие Р",
необходимое для отрыва пластинки от межфазной поверхности таким
образом, чтобы она была погружена только в масляную фазу
P" = M-^By-Ji^-dep2gj+J (II 1.78)
167
где de — расстояние между краем пластинки и поверхностью раз-
га масло — вода
отовления пласт
Таким образом:
дела масло — вода; гп — радиус проволоки, используемой для
изготовления пластинки Вильгельми.
-2
пр
°м'"=—2(1па+^)— (Ш-79)
Если масляная фаза тяжелее водной и в нее погружена пластинка,
то действующие силы направлены вверх в результате межфазного
натяжения [в соответствии с уравнением (75)1, веса пластинки,
архимедовой силы погруженной пластинки и усилия /', возникающего
благодаря верхней водной фазе. Компенсирующее усилие равно:
Ршкс = -2(1пл-тЛпл)оВ1М^М-Ву + Г (ИГ.80)
Величины Ву и / определяют совместно при отрыве пластинки
от поверхности раздела и перемещении ее вверх на небольшое
расстояние (de). Плавучесть при этих условиях увеличивается на А5у:
АВу= xrlvdcpig + 2nr%p (pi — p2) deg (II 1.81)
В этом случае
Ри=Р»шс-(.Ву + АВу)-±-1 (II 1.82)
Поэтому
п *макс лпу /ТП йо.
стм/в— п77 4-rf \ Uii-b.j)
^ 1'пл т^"пл;
Паддей (1957) усовершенствовал метод Вильгельми в
соответствии с законом Абрибата — Догнона (1939), заключающегося в том,
что сила, действующая на пластинку, частично уравновешивается
грузом, а частично силой, возникающей в подвижной электрической
катушке при пропускании тока через нее. В методе Паддея груз
замещался дополнительной катушкой и прибор давал показания
(в дин/см), включая концевую поправку на длину пластинки при
калибровке. Поверхностные натяжения непрерывно образующихся
новых поверхностей можно определить, нагнетая жидкость в
верхнюю часть вертикальной открытой трубки и давая затем ей
возможность опускаться вниз по межтрубному пространству.
Измерения проводились в верхней части трубки. Можно получать также
сравнимые данные динамического поверхностного натяжения.
Статические методы
Объем капли
Один из наиболее широко используемых статических методов
заключается в определении веса или объема капли, которая
медленно отрывается от кончика вертикально расположенного
капилляра. При этом, если определяется поверхностное натяжение
жидкости, капля падает в воздух, при измерении межфазного
натяжения— в жидкость, представляющую другую фазу.
168
Капли отрываются, когда гравитационное усилие
уравновешивает силу поверхностного или межфазного натяжения. В этом случае
mg = V (Р1-Р2)г=2я/-Кам/В (III.84)
или
mg V (pi— рг) g
Um/b 2ягк 2лгк
где гк — радиус кончика капилляра; V — объем капли.
Однако этим уравнением не пользуются при расчетах, потому
что не вся капля отрывается от капилляра (рис. III.19). Иногда
объем капли на 40% меньше вычисленных значений. Необходимо
1.0 -
\
0.9 Д
0.8- \
^ \
\
0.7- V
о,- ^-^\ ■
ОМ i .
0 01 0.8 1.2 ■ 1,6
Рис. 111.20. Зависимость
поправочного коэффициента от rK[V '*
при определении объема канлн
(Харкинс и Браун, 1919).
На рис. III.20 представлена зависимость fv от rJV /з. Очевидно,
что кончики капилляров должны быть так подобраны, чтобы
величина rJV lz понижалась в пределах 0,75—0,95, в которых fv
изменяется минимально.
Шприц-микрометр с острым, как иголка, кончиком пригоден
для измерений межфазного натяжения этим методом, особенно
в случаях, когда имеются небольшие объемы двух жидкостей.
Наружная сторона кончика должна хорошо смачиваться жидкостью,
в которую опускаются капли, в противном случае величина гк не
будет соответствовать радиусу кончика. Кончики из нержавеющей
стали после обработки кремнием становятся гидрофобными. Если
в шприце содержится жидкость с большей плотностью, чем в
пробирке, то шприц ставят в вертикальное положение и капли медленно
опускаются в пробирку. Если же плотность жидкости в шприце
ниже, то его переворачивают и капли поднимаются в более плотную
жидкость. Всю установку необходимо термостатировать.
Аналогично можно применять микробюретку, градуированную
по 0,02 мл. При этом кончик заменяется равной трубочкой узкого
использовать поправочный
коэффициент /у как функцию rJV "
(Харкинс и Браун, 1919, 1926). Поэтому
уравнение (III.84) принимает вид:
а У(Р1-Р-2) (111.85)
Ы/В"2лг11/У(гж/Г''-)
Рпс. II 1.19. Изменяющаяся форма капли,
падающей из капилляра (Адамсон, 1960).
169
диаметра при условии, что плотность опускаемых Капель больше,
если же последняя меньше, то его заменяют U-образным
капилляром.
Бюретки необходимо тщательно обезжиривать и хранить
открытыми. Между сериями измерений их промывают хромовой кислотой
и споласкивают. Средний объем капли определяют по изменению
объема в микробюретке или в шприце-микрометре после отрыва
нескольких капель. Средний вес капли устанавливают по
увеличению веса сосуда.
Независимо от способа определения объема или веса капель
необходимо создать их медленный отрыв с тем, чтобы они не были
слишком большими. Харкинс и Браун (1919) достигли этого с
помощью наконечника. Капли образовывались в закрытой склянке,
соединенной с изогнутой трубкой, которую можно подключить
к всасывающей системе. Жидкость подавали сифоном из емкости,
в которой поддерживался соответствующий уровень ее для
обеспечения медленного вытекания под действием силы тяжести. В первые
моменты получение капель ускорялось всасыванием, а затем уже
регулировалось только силой тяжести.
Высота поднятия в капилляре
Бартелл и Миллер (1929) использовали прибор, показанный
на рис. III.21.
Широкие трубки, выполняющие роль емкостей для двух
жидкостей, соединены узкой U-образной стеклянной трубкой. Один отвод
U-образной трубки является капилляром, в котором создается
поверхность раздела масло — вода. Если органическая жидкость
легче водного раствора, то ее вводят в широкую трубку над
капилляром, в противном же случае эту трубку заполняют водным
раствором. Поверхность раздела образуется в верхней части капилляра,
затем введением соответствующей жидкости в широкую трубку
над капилляром ее уровень опускается до градуированной метки ст.
Для расчета межфазного натяжения измеряют высоту поднятия
жидкостей в двух широких трубках над градуированной меткой —
h1 и h2.
Джел линек и Ансон (1949) применили подобный прибор
(рис. III.22), введя небольшое изменение в его конструкцию для
определения поверхностного натяжения.
Межфазное натяжение равно
%/B=(AlPl"^P2)^ <Ш.8в>
где гс — радиус капилляра у градуированной метки.
Для очень точных определений межфазного натяжения
необходимо делать поправку на высоту мениска.
170
Бартелл и Миллер (1929) предложили соотношение, в котором
при тс < 1 мм действительная высота h мениска в капилляре связана
а с отмеченной высотой hQ
следующим образом:
Органическая
жидкость
Л,
_L.
h = h0 + ^-rc-0,i288r*/h0
(Ш.87)
bouu
ъ.
01?jm
Вода
Органическая
жидкость
Отметка
04wr \) " I -
Рис. III.21. Измерение межфазного
натяжения методом поднятия в капилляре (Бартелл
и Миллер, 1928):
а — рм < рв; б — рм > рв.
Рис. III.22. Определение
межфазного натяжения методом
поднятия в капилляре(диаметр
внутреннего капилляра 0,4 мм).
Висячая капля
Поверхностное и межфазное натяжение висячей капли можно
рассчитать, измерив ее размеры фотографически или спроектировав
изображение на градуированный экран. Этот метод особенно удобен
в тех случаях, когда натяжение медленно
изменяется со временем. Обнаруживается плоскость
! перегиба и измеряется ее расстояние d0 от оси
и радиус кривизны гкр в этой точке (рис. III.23).
Межфазное натяжение равно:
°м/в= Ш1 ("1-88)
Если допустима меньшая точность, то
определяется экваториальный диаметр D3KB капли и
-D!kb(pi-
-P2)g
(Ш.89)
Рис. III. 23.
Висячая капля.
где /к -
1883).
им/в~ /
'к
коэффициент, зависящий от формы капли (Бешфорт и Адаме,
171
Лежачая капля
Капля жидкости находится на гладкой пластинке, погруженной
в другую фазу (Харкинс и Ченг, 1926), а искомые величины
измеряют или на фотографиях, или с помощью перемещающегося
микроскопа с окулярной сеткой. Определяют диаметр капли, расстояние
hs от экваториальной плоскости до вершины капли и радиус
наибольшего поперечного сечения капли (rs). Если диаметр капли очень
большой, кривизну вершины можно не учитывать
где as
константа
Следовательно
JM В
= 3
капилляра,
2(Тм;в
(Ci — Р-2) S
'<! (in (ч) ff
(111.90)
равная
(Ш.91)
(111.92)
Если кривизной вершины
бречь, то учитывают изменение
в зависимости от отношения h.
нельзя
в (Л»/г|
/г
Рис. III.24.
Измерение межфаяного
натяжения методом
лежачей капли (Шот-
топ и Калпан, 1960):
ское уравнение
(Портер, 1933):
j4 а\_
выражает эту
0,3047
прене-
- я2/г»)
. Эмпириче-
зависимость
т
(1)1.93)
оптическая
стеклянный
чеш.'а
диск.
выражали через а*/г
соотношения, когда
По данным Уилера и др. (1945),
действительная высота h's может значительно
отличаться от hs. Величину лежачей капли они
! как функцию' Щг\, используя эмпирические
0 <Л;'2/'1 < 0,065
= 0,4543-
К
и когда 0,065 <h'*lr\ < 0,300;
2г!
= 0,4353
hf
0,223-
-0,860
h'*
'й1
1,675-
7 '*>
(III.94)
(III.95)
Шоттон и Калиан (1960) применили эти соотношения для
определения межфазного натяжения между бензолом и водой. Прибор
(рис. 111.24) состоит из оптической ячейки в форме куба со
стеклянным шлифом. Пробка подобна колпаку и имеет два отвода.
Центральный отвод представляет собой стеклянную трубку, к которой
пришлифован стеклянный диск диаметром 27 мм. Бензол п вода,
находящиеся в различных колбах, в течение ночи взаимно насыщаются.
Лежачая капля бензола получается медленным введением извест-
172
ного объема через боковой отвод пробки в ячейку, предварительно
заполненную водой. Через определенные промежутки времени
капля фотографируется, затем определяется экваториальный
диаметр и высота от него до вершины капли.
Дисменкес (1959) рассмотрел влияние ошибки в определении
величины hs на размер капли. В основном ошибки связаны с
трудностью точного определения D3KB. Другая, менее важная причина
ошибки вызвана неточным определением вершины капли [см.
уравнение (III.90)]. Вычисления показали, что для капли с us/hs = 1,03
неправильное нахождение вершины вызывает появление ошибки
в величине hs, не превышающей 0,001 см, в случае, если ошибка
в h., связанная с неверным нахождением D3KB, равна 0,025. С
увеличением капли ошибка в величине hs, получаемая от неверного
определения расположения вершины, будет меньше, чем при
неточном определении D3KB. Поэтому наибольшая точность достигается
при исследовании больших капель.
Адам (1941) полагал, что методу лежачей капли присущи
значительные ошибки даже при оптимальных условиях. Из-за недостатков
оптической системы изображение капли не вполне совершенно, что
и вызывает новые экспериментальные значительные ошибки. Он
приводит примеры различных значений поверхностного натяжения
ртути.
СМАЧИВАЕМОСТЬ
Когезия и адгезия
К силам, действующим в эмульсиях, относятся: 1) когезия между
молекулами непрерывной фазы; 2) когезия между шариками
дисперсной фазы, подвергнутых изменению адсорбировавшимся
эмульгатором; 3) взаимодействие между шариками и дисперсионной
средой. Когезию можно определить следующим образом. Если столбик
жидкости с площадью поперечного сечения 1 см2 разделить на две
части, образуются две новые поверхности с той же площадью
поперечных сечений; работа, совершенная при этом, т. е. работа коге-
зип WK, равна 2а.
Представим себе столбик из двух несмешивающихся жидкостей
разных объемов с поверхностными натяжениями ам и ав- Если
жидкости разделить по межфазной поверхности, межфазное
натяжение которой в каждой ее точке равно ом/в, то возникают новые
поверхности с поверхностными натяжениями ам и ав. Затраченная
при этом работа называется работой адгезии Wa и равна (Дюпре,
1869):
Wa = oM + oB-om/B (Ш.96)
Растекание одной жидкости
по поверхности другой
Понятия когезии и адгезии можно распространить на свойства
растекания капли одной жидкости по поверхности другой, не
смешивающейся с первой. Если капля масла растекается на воде, то
173
межфазная поверхность воздух — вода заменяется двумя новыми;
первая образуется между двумя жидкостями, а вторая, равная по
площади первой, между маслом и воздухом. Коэффициент
растекания /р определяется как
/р = сВ-(ам+аМ/в) (Ш.97)
Капля масла растекается, т. е. значение /р положительно, при
условии, что (сгм + о-м/в) <ов. Если же (сгм + стМ/в) > 0"в,
величина /р отрицательна и капля не растекается. Присутствующие
примеси могут влиять на коэффициент растекания. Так как WK =
= 2ам, то
fP=Wa-WK (III.98)
При медленном растекании масла по поверхности воды жидкости
взаимно насыщаются. Величина /р будет уменьшаться вследствие
снижения ав. Краевой угол равен нулю при условии, что значение
/ положительно. Растекание прекращается, когда о"в<м) — °м(в) +
~г о"М(В) (где В (М) и М (В) означает, что вода насыщена маслом и
масло насыщено водой, соответственно). В этой точке краевой угол
все еще равен нулю.
Когда масло быстро растекается по поверхности воды и жидкости
не насыщены, очевидно, после взаимного насыщения краевой угол
будет равен нулю. Поэтому из уравнения (III.96) следует, что
^. = 2ffM(B) ("1-99)
так как работа адгезии масла в данном случае равна работе когезии,
а ам после насыщения водой уменьшилось.
Согласно теории адсорбции Гиббса, небольшое увеличение а
одной жидкости происходит при ее насыщении другой с большим
значением а (Девис и Райдил, 1963). Например, а бензола равно
28.2 дин/см, а при насыщении водой оно увеличивается только до
29.3 дин/см. Жидкость с большим значением а, однако, легко
адсорбирует монослой менее полярной жидкости.
Если уравнение (III.99) верно при всех условиях, то растекание
будет всегда на одной из двух жидкостей до момента их взаимного
насыщения, когда коэффициент растекания равен нулю. Это в
основном справедливо для малополярных масел, таких как хлороформ и
бензол, в отличие от более полярных, например гептиловып спирт,
и не растекающихся масел — сероуглерод и йодистый метилен.
Незначительное уменьшение коэффициента растекания, что
происходит при взаимном насыщении жидкостей, приводит к увеличению
краевого угла. Тогда ав(М) < о-м(В) ~г сгм/в.
Поверхностные натяжения двух жидкостей можно связать с их
межфазным натяжением (Джирифалко и Гуд, 1957)
ам/в = стм + ов-2Лу((Тм(1в),/2 (III.100)
где AY — постоянная, вычисляемая из молярных объемов (Ум, Ув)
или из свободной энергии адгезии (AF|I/B) для межфазной поверх-
174
ности между двумя жидкостями и их соответствующих свободных
энергий когезии (Л^м, AF%):
Значения функции Av для органических жидкостей, не
образующих сильные водородные связи с водой, находятся в пределах
0,51—0,78, для жидкостей, имеющих такие связи, — в области
1,00—1,15 или чаще выше 0,84. Это подтверждает предположение,
что в основном когезионные силы в этих жидкостях возникают
в результате водородных связей. В уравнении (III.100) взаимная
растворимость жидкостей не учитывается, хотя последняя
предполагает увеличение величины Av, и следовательно, уменьшение
значений 0"м/В-
Можно считать, что поверхностное натяжение возникает в
результате действия двух независимых видов межмолекулярных сил,
которые суммируются (Фоукес, 1962). Например, в воде основными
молекулярными силами являются силы, обусловливающие
водородную связь (Fh) и дисперсионные силы (Fv), поэтому ее
поверхностное натяжение можно выразить как
'в
v (III.102)
Для насыщенных углеводородов преобладающими являются
дисперсионные силы:
ам = 4У (III.103)
Межфазное натяжение на границе раздела насыщенный
углеводород — вода определяется, главным образом, дисперсионными
силами между молекулами углеводорода и воды:
в-21/ав*<# (II1-104)
Уравнения (III.100) и (III.104) справедливы для взаимодействий
между маслом и водой в отсутствие любых ПАВ или в случае, если
на поверхности адсорбированы неионные производные окиси
этилена (Бехер, 1963). В изучаемых системах концентрации веществ,
растворимых в воде, превышали ККМ, а межфазные натяжения
водных растворов определены для большого числа масляных фаз.
Выявлены два основных вида закономерностей. График зависимости
межфазного натяжения от содержания окиси этилена в ПАВ
представляет кривую с явно выраженным минимумом (рис. III.25) при
условии, что масляная фаза — алифатический углеводород, эфир
жирной кислоты, силиконовое или хлопковое масло. Если масляной
фазой являются ароматические углеводороды, спирты, четырех-
хлористый углерод, олеиновая кислота, касторовое или льняное
масло, то высокое межфазное натяжение наблюдается при более
175
низком содержании окиси этилена (рис. III.26), чем в первом
случае. В обоих случаях межфазное натяжение уменьшается с
удлинением полиоксиэтиленовой цепи. Первая зависимость получена при
А\- <0,6, а вторая — когда Av >0,6, кроме случаев с эфирами
жирных кислот. Очевидно, вторая зависимость преобладает, когда
масляная фаза имеет водородные связи. Бехер (1963) указывал, что в
ароматических углеводородах я-орбитали могут давать результаты,
подобные водородным связям.
20
Ю
ю
го
ю-
ю го зо
Содержание окиси этилена,
мол. dpn-i
Рис. III.25. Зависимость межфазного
натяжения растворов алифатических
углеводородов от содержания окиси
этилена (Бехер, 1963):
а —ПОЭ (полиоксиптилен) нонилфекол/октан;
б — ПОЭ понплфенол/толуол.
10
ю го зо
Содержание окиси
этилена, мол. дол я
Рис. III.26. Зависимость межфазного
натяжения растворов ароматических
углеводородов от содержания окиси
этилена (Бехер, 1963):
а —ПОЭ лауриловый спирт/октан; б —
ПОЭ лауриловый спирт/толуол.
Значения Av, пропорциональные о"М/В и вычисленные по
данным поверхностного и межфазного натяжений, достаточно хорошо
согласуются с теорией (табл. II 1.9). Величины овг и aBv
рассчитаны для водных растворов ПАВ при условии, что aMv можно
вычислить по уравнению (III.103). Рассмотрим эти данные:
а) доля дисперсионных сил в поверхностном натяжении водных
растворов ПАВ увеличивается в гомологических рядах с
возрастанием содержания окиси этилена;
б) доля дисперсионных сил для данного ПАВ зависит от
природы масляной фазы, особенно при высоких содержанпях окиси
этилена;
в) величина силы, обусловленной водородными связями, очень
близка к межфазному натяжению, но остается всегда немного ниже;
176
Нес
„ «5
3 2
1 '.
2 °<
се
9
И
О
К
к
с
о
я
о
и
S
ь. я
о
я"
ь. я
е
>
к
^
1
х
3
о
j
3
2
-j
^
о
5
s
р4
1
м
3
о
с
о
3
п
■а
-
сг
*~г
2
1
к
§
в
5
з
?.
Е
о
Ь-
■Ч
Ь, Я
о
д
ь, и
D
3!
О
я
о
^>
•*,
^
ь, я
о
2
b,'ffl
о
я
й
о
а
о
Ч
о
Я
<;
И
о
о_
см
о"
СП
щ"
см
со
о"
с
СО
X
о'
СД_
[>-"
СМ
х"
О
ст.
СМ
я
—
сс
см
Е-
г~
О
:3
§
о
о_
СО
о"
LO
ОС
см
v-H
О
ю
=0
о"
=1
t^
о
с"
см
V-
х"
X
х"
см
ь
о_
^
о
:3
3
со
о
о"
г~
СМ
о.
со
о
со"
■X
с.
о"
СО
см
V-
^-Г
ю
см"
г~-
СО"
СО
S;
«
С
:'Я
2
-гн
О
о"
V-
о"
о
см"
СО
~_
^Г
г^
СЛ
о"
°°
*г*
со
со-
со
со
см"
v-<
00"
со
н
г~
и
:3
1
чч
та
о
о
1^
CD
LO"
СО
о
00
г~
CD
о"
^
см"
LO
о"
vc
см_
со"
«с;
W
V-
н
~.
°
=4
|
^*
CD
о"
X
-
~»
СО
см
CD
V?
СМ
со
о"
с.
Ю
о
см
"Ч.
см
cd_
t~-"
см
ч
2
со
ел
о"
СМ
ЧГ.
со
lo
см
см
~*
г~
со
о"
см_
со"
СО
со"
VrH
LO
cd"
CM
ч
X
CD
о"
CM
CM
X
CM
Nf
LO
c~
o"
,_,
со"
CO
со"
r~
4f
vr>
CD"
CM
14
о
!>■
cd
о"
CD
X
CM
CM
,_,
X
o"
LO
o"
CO
cd"
CO
*—
o"
CO
4
о
LO
CD
o"
о
CO
CO
о
CO
CD
CO
о
CD
o"
о
о"
CM
*<r"
CM
X
о
CM_
vc
CO
14
о
en
X
o"
LO
X
LO_
CO
CD
X
CO
CD
o"
LO_
sp
LO_
LCj"
CO
CD_
vc"
o_
o"
VP
4
о
о
^s|s|l|l|sls|s|l|l|ll
oooooooooo
1- Заказ 1070
177
г) основная доля в межфазном натяжении масло-водного
раствора ПАВ принадлежит Овн, так как сгмн очень мало, если оно
вообще существует.
Влияние водородных связей на межфазное натяжение зависит
от ориентации молекул ПАВ на поверхности раздела.
Вторую зависимость (см. рис. III.26) для веществ с меньшей
длиной оксиэтпленовых цепей можно объяснить действием водородных
связей. С увеличением же длины оксиэтпленовых цепей молекулы
ПАВ ориентированы таким образом, что все располагаются в водной
части межфазной поверхности. Труднее объяснить первую
зависимость (см. рис. III.25). Возможно, в этом случае молекулы,
ориентированные лшюфильными цепями к воде, переходят в
положение, когда часть гидрофильной оксиэтиленовой цепи удерживается
маслом.
Растекание жидкости
по поверхности твердых веществ
Для стабилизации эмульсий применяют достаточно
высокодисперсные порошки (Пикеринг, 1907). Получаемый тип эмульсий
зависит от избирательной смачиваемости твердых частиц одной из
фаз (Скарлетт и др., 1927). Если частицы лучше смачиваются
масляной фазой, образуется эмульсия типа В/М, в случае лучшего
смачивания водой — эмульсия типа М/В. Исследования, проведенные
Шулъманом и Леем (1954), подтвердили эти выводы. Кроме того,
они показали, что в присутствии ПАВ и твердых эмульгаторов
образуется эмульсия М/В, если краевой угол, измеренный со
стороны водной фазы, почти равен 90°.
Для поверхности раздела твердое тело — жидкость, когда
жидкость растекается по твердой поверхности, можно применить
уравнение (III.96)
И'а (Т: Ж) = аТ -Г °Ж - СТТ / Ж ( П Г • 105>
где Т, Ж и Т/Ж — поверхности раздела твердое тело — воздух,
жидкость — воздух, твердое тело — жидкость.
К сожалению, ни а , ни о" непосредственно нельзя определить.
Жидкость может не растекаться по твердой поверхности, а только
образовывать с ней угол 6, называемый краевым углом (рис. III.27).
Подобное явление наблюдается с жидкостями, не смешивающимися
друг с другом. Различные натяжения можно вычислить по
уравнению
ат=стТ/Ж + аж cos 9 (III.106)
которое справедливо при всех значениях 6.
При сопоставлении уравнений (III.105) и (III.106) получаем:
^а(Т/Ж) = стж(14-созе) (Ш.107)
Поэтому, если известны краевой угол и поверхностное натяжение
жидкости, можно вычислить работу адгезии.
178
Согласно Адаму и Ливингстону (1958), Wa( — это работа,
необходимая для удаления жидкости с поверхности твердого
вещества, хотя сгт — межфазное натяжение на границе твердое
вещество — насыщенный пар, но не поверхностное натяжение твердого
вещества (Харкинс, 1952).
В эмульсии воздух (см. рис. 27) замещен другой жидкой фазой,
поэтому уравнение (III. 106) принимает вид
Ят/ж.^т/ж.+^ж./ж,0089» (III.108)
где Жх и Ж2 — две жидкости; 0 —краевой угол между жидкостями.
Шероховатость поверхности твердого вещества влияет на
краевой угол. Например, если поверхность ровная и краевой угол > 90°,
Воздух
Воздух
б г/ж Твердое Вещество бг бт/ж Твердое вещество бТ
Рис. III.27. Краевой угол смачивания между
жидкостью и твердым веществом:
а — 9 < 90°; б — 9 > 90°.
его значения возрастают для неровных поверхностей. Когда
краевой угол <С 90° для ровных поверхностей, то с увеличением
шероховатости поверхности он будет еще больше уменьшаться. При
наличии небольшой шероховатости As ее можно определить по
выражению (Вензель, 1936)
As = ^- (Ш.Ю9).
Ь COS 62
где 0j и 02 — краевые углы для шероховатой и гладкой
поверхностей, соответственно.
При экспериментальном определении краевого угла большая
трудность возникает в связи с созданием гладкой поверхности
(Китченер, 1960). Если твердое вещество можно получить в виде
гладкой ровной пластинки, то самым простым методом измерения
краевого угла для одной жидкости, предложенным Адамом и Джессо-
ном (1925), является метод наклонной пластинки Харкинса и Фоу-
кеса (1940).
Пластинку с помощью специального устройства можно наклонять
под любым углом или опускать и поднимать относительно
поверхности жидкости. Таким образом, краевой угол можно измерять
оптически при условиях погружения или удаления пластинки из
воды. Угол наклона подбирают таким, чтобы поверхность жидкости
12*
179
не искажалась в месте соприкосновения с пластинкой. В случае
пзмеренпя краевого угла при погружении пластинку опускают так,
чтобы покрыть жидкостью ее сухую часть, и через 1—2 мин
определяют угол. Угол, под которым пластинку вынимают, измеряют при
легком поднятии ее через тот же интервал времени. Чашка или
кювета с жидкостью должна иметь парафинированные стеклянные,
нейлоновые или тефлоновые барьеры для удаления любых
загрязнений с поверхности, ибо они могут влиять на поверхностное
натяжение .
Краевой угол между твердым веществом и двумя жидкими
фазами можно измерить подобным образом. В этом случае возможна
существенная разница (гистерезис)
между углами погружения и поднятия,
указывающая, что если твердая поверхность
покрыта пленкой одной жидкости, то
замещение ее другой жидкостью
происходит очень медленно. Обычно
имеющиеся небольшие количества твердого
вещества не позволяют сделать его гладким.
Несколько видоизмененный метод
наклонной пластинки состоит в том, что
твердое вещество частично погружают
Рис. III.28. Измерение в жидкость, находящуюся в плоской
краевого угла смачивания кювете (рис. II 1.28). При этом угол между
с помощью наклонной пла- твердым веществом и поверхностью жид-
стпнкп. кости подбирают таким, чтобы мениск
оставался горизонтальным.
Перед исследованием образцы твердых веществ должны быть
специально обработаны. Вырезанные' образцы шлифуют
наждачными материалами, затем кожаным ремнем и, наконец,
высокосортной фильтровальной бумагой (Фоукес и Харкинс, 1940). После
этого их промывают дважды перегнанным этанолом и высушивают
под вакуумом в эксикаторе.
Шульман и Лея (1954) получали высокоактивные и легко изгота- <
вливающиеся поверхности раскалыванием кристаллов сульфата
бария. Один образец вводили в оптическую кювету, содержащую
15 мл водного раствора ПАВ. Капли масла наносили на
кристаллическую поверхность хорошо оттянутой стеклянной палочкой,
а количество контролировалось шприц-микрометром. Краевой угол
на межфазной поверхности твердое вещество — масло — вода
измеряли с помощью передвижного микроскопа, соединенного с
угломером. Краевые углы на межфазной поверхности воздух — вода —
твердое вещество определяли при замещении масляных капель
пузырьками воздуха.
На краевой угол влияют длина цепочки ПАВ и рН, если его
концентрация выражена в молях. Иначе говоря, влияние
оказывает и концентрация ПАВ. Если ПАВ имеют длинные
углеводородные цепочки, то краевой угол возрастает почти до 180°, так что
180
поверхность твердого тела почти полностью смачивается маслом.
Порошки, смачиваемые таким образом, не удерживаются в
эмульсиях на межфазной поверхности масло — вода; они диспергируются
в масляной фазе, образуя неустойчивые эмульсии.
Если твердое вещество доступно только в виде высокодисперсного
порошка, то краевой угол можно измерить видоизмененным методом
Волкова (1934). Резиновую пробку (рис. III.29) вводят на
определенный уровень в нижний конец тонкостенной стеклянной трубки
постоянного диаметра, отградуированной в миллиметрах
(Студебекер и Сноу, 1955). Затем на поверхность пробки помещают кружок
фильтровальной бумаги. Хорошо высушен- Л
ный порошок насыпают высотой в 0,5 см,
затем плотно спрессовывают плунжером
и покрывают кружком фильтровальной
бумаги. Операцию введения и спрессовывания
порошка повторяют трижды, каждый раз
удаляя верхний кружочек фильтровальной
бумаги. Затем сверху на поверхность
порошка помещают платиновый диск и стеклянную
с выемкой трубочку, диаметр которой
меньше, чем у основной трубочки. Далее верхний
конец основной трубки закрывают
резиновой пробкой с вставленной в нее
трубочкой, а из нижнего конца трубки удаляют
пробку п фильтровальную бумагу. Жидкость
вводят через стеклянную трубочку в
верхней пробке и устанавливают время,
необходимое для ее протекания через колонку,
заполненную порошком.
Скорость, с которой протекает жидкость, определяется на
световом фоне. Зависимость времени протекания от квадрата расстояния
является прямолинейной. Отклонения от прямой указывают, что
порошок в колонке спрессован не одинаково.
Считают, что колонка из порошка подобна ряду капилляров,
поэтому принимают режим Вашбурна — проникновение жидкости
в капилляры. Если при смачивании твердого вещества краевой угол
равен нулю, то
Рис. III.29. Схема
прибора для измерений
краевых углов смачивания
порошков (Студебекер и
Сноу, 1955):
1 — резиновая пробка; 2 —
фильтровальная бумага; 3—
порошок; 4—
перфорированный диск; S — стеклянная
трубочка.
/2 —-
1 а —
2тц
(III.110)
а при определенном значении его
0"2 COS 0ГТТ2
'1 =
2Т)2
(111.111)
где 1а и 1Ъ — длина колонки, пересекаемая жидкостью.в моменты
времени i1 и т2; ох и а2 — поверхностные натяжения двух
жидкостей; У]г и 1"!2 — соответственно их вязкости; гт — радиус трубки.
181
Если 1а = lb, т. е. 0',5 см, то
C0Sq = ^L.2±=K±L (III.112)
Таким образом, при определении краевого угла, образуемого
любой жидкостью и порошкообразным твердым веществом,
сравнивается время прохождения жидкостями с разным смачиванием
определенного расстояния в колонке.
Педдей (1959, 1960) предположил, что более важным параметром,
чем краевой угол, является коэффициент растекания. Его легче
измерить, особенно когда мениск капли искажен. Из уравнений
(III.90), (III.97) и (III.106) получаем
/р = ат-(ат/ж + аж) = ож(со8е-1) = -^- (III.113)
для находящейся на твердой поверхности неподвижной капли,
которая с последующим увеличением объема достигает максимальной
постоянной высоты hs (при условии, что система выдерживалась
для установления равновесия). Этот принцип можно применить
для поверхностей раздела жидкость — твердое вещество — жидкость.
Рзй и Бартелл (1953) и Педдей (1960) показали, что краевые углы
при погружении и поднятии подобны на гладких поверхностях
в отличие от шероховатых, когда среднее значение угла близко к
значению его для гладкой поверхности. На последней сильно искажен
только мениск поднятия, в то время как на шероховатой
поверхности искажены оба мениска. Если, кроме шероховатости,
поверхность еще и загрязнена, то среднее значение коэффициента
растекания изменяется со степенью шероховатости.
СВОЙСТВА ПЛЕНОК ЭМУЛЬГАТОРА
НА ПОВЕРХНОСТЯХ РАЗДЕЛА МАСЛО — ВОДА
Физические свойства слоя эмульгатора, адсорбированного на <
поверхности раздела масло — вода, влияют на реологические
свойства эмульсии, ее стабильность. Эти проблемы обсуждаются в
других разделах книги. Сведения об адсорбции или ориентации
молекул эмульгатора получают при изучении модели плоской
поверхности раздела масло — вода, которую можно рассматривать
как поверхность шарика с бесконечно большим диаметром.
Основной принцип таких методов — определение площади, занимаемой
каждой адсорбированной молекулой, при изменении давления
на поверхности пленки.
Возможно, самый простой способ заключается в использовании
метода Впльгельми для определения межфазного натяжения (см.
стр. 167). Если масляная фаза легче водной, то в чистую
стеклянную чашку вводят сначала водную фазу; примеси удаляют
с помощью воздушной струи, создаваемой капиллярами, которые
присоединены к водяному насосу (Александер и Теорелл, 1939).
182
Затем несколько капель масла осторожно опускают на поверхность
воды. Примеси, остающиеся еще на ней, перемещаются к краям
чашки, откуда удаляются воздушной струей. После этого добавляют
остальное масло. Эмульгатор, растворенный в растекающемся по
поверхности веществе, осторожно вводят на поверхность раздела
с помощью шприца-микрометра. Когда образуется пленка,
растекающееся на поверхности вещество, например, петролейный эфир
или изопропиловый спирт, должно быстро переходить в смежную
масляную или водную фазу.
Межфазные натяжения измеряют как перед, так и после введения
эмульгатора, и разность поверхностных натяжений представляет
поверхностное давление я. Эмульгатор добавляют несколько раз
п в каждом случае определяют межфазное натяжение. При введении
эмульгатора площадь, занимаемая отдельной молекулой,
уменьшается. Соответствующие значения площади вычисляют из размеров
чашки и мольной концентрации используемого эмульгатора.
Недостаток методов, в которых используют кольцо или пластинку
для получения взаимосвязей сила — площадь, заключается в
определении краевого угла при высоких давлениях на поверхности, когда
пленки жесткие (Ван де Зеггерен и др., 1959, Педдей, 1960). Кроме
того, при увеличении давления пленка растекается (Брукс и Пе-
тика, 1964).
Метод Ленгмюра — Адама лишен этих недостатков. Он применим
для поверхностей раздела воздух — жидкость. Длинную узкую
кювету с плоскими парафинированными краями наполняют до краев
маслом или водой. Эмульгатор наносят на поверхность с помощью
шприца-микрометра, и образующийся мономолекулярный слой
сжимают подвижным парафинированным предметным стеклом (рис. III.30).
При уменьшении площади пленки сила действует на легкий
дюралюминиевый поплавок. Чтобы молекулы пленки не проникали за
поплавок, к нему одним концом присоединяют шелковую нить,
другой конец которой лежит на поверхности кюветы. Сила, действующая
на поплавок, слегка его смещает. Это передвижение определяют
с помощью устройства лампа — зеркало — шкала. Смещение
поплавка на 1 мм дает регистрирующееся отклонение зайчика на 20 см.
Одна из горизонтальных шкал, расположенная вдоль длины
кюветы и указывающая положение подвижного барьера, может
быть использована для регистрации смещения поплавка.
Передвижение поплавка регулируют пружиной, калибровку прибора
выполняют небольшими гирьками, а также с помощью масел (например,
касторового) или олеиновой кислоты с известным распределением
давления по поверхности. Кривые сила — площадь можно записывать
автоматически с помощью механизма, который одновременно
приводит в движение предметное стекло и барабан фотоаппарата,
регистрирующего отклонение (Александер, 1948). Поверхность жидкости
перед измерениями очищают несколькими передвижениями
предметного парафинированного стекла и удалением примесей
воздушной струей.
183
Эскью и Даниэль (1940) приспособили эту кювету для пленок,
растекающихся по поверхности раздела масло — вода, однако
оказалось, что она пригодна только для масел, которые тяжелее воды.
Просачивание пленки и вытекание жидкости из кюветы можно
устранить, заключив ее в подвижное кольцо, которое можно сжимать
между подвижными стеклянными барьерами (Брукс и Мак Ритчи,
1961).
Кольцо может быть изготовлено из слюды. Недостаток этого
метода — ограниченное сжатие кольца.
Рпс. 111.30. Схема весов для измерения поверх-
постного давления нерастворимых монослоев на
поверхности раздела воздух — вода (Алексан-
дер, 1949):
1 — зеркало; 2 — лампа; 3 — поплавок; 4 — жолоЛ;
S — подвижно» барьер; в — пленка; 7 — шкала.
Трудности, возникающие при использовании описанной кюветы
для поверхности масло — вода, связаны с краевым углом. Для
сохранения определенной площади пленки надо, чтобы вода лишь едва
смачивала кювету. Это достигают с помощью кюветы, сделанной
из специальных сортов нейлона или фторопласта (Джонс и др.,
1963) и вставленной в стеклянную кювету; поплавок и барьер
изготовляют из этого же материала (рис. III.31). Для устранения
перетекания за барьер можно применить нити из нейлона или терилена.
Определение зависимости сила — площадь при низких давлениях
затруднено из-за тяжести нити. Поэтому в таких случаях уместно
использовать кювету, состоящую из двух пластинок стекла пирекс,
скрепленных параллельно с помощью двух стеклянных палочек
(Брукс и Петика, 1964). Такую рамку вставляют в стеклянную
кювету. Кювета и барьеры гидрофобны, что достигается обработ-
184
кой 2% раствором диметилдихлорозила в четыреххлористом
углероде. Если масляная фаза легче водной, то внутренние стороны
стеклянной рамы и нижнюю барьеров обрабатывают абразивной
бумагой, после чего они становятся гидрофильными: Когда масляная
фаза тяжелее водной, края стеклянной рамки и барьеров должны
быть гидрофобными для предотвращения переливаний через края.
Давление пленок измеряют с помощью гидрофобной пластины
Вильгельми из слюды, покрытой слоем газовой сажи, краевой угол
которой на границе с масляной фазой равен нулю.
Рпс. III.31. Весы для измерения поверхностного давления
нерастворимых монослоев на поверхности раздела масло — вода.
Соотношение сила — площадь иногда называют поверхностным
уравнением состояния аналогично соотношению давление — объем
для газа
7iS = kT (III.114)
где л. — вычисленное давление пленки, которое учитывает только
двумерное кинетическое движение в пленке с кинетической
энергией в каждом направлении; S — площадь, занимаемая отдельной
молекулой.
Предполагается, что нет ни притяжения, ни отталкивания между
молекулами, поэтому уравнение (III.114) справедливо, когда л -*■ О
или S —>- °°. Давление пленки при любом значении S обычно больше
на поверхности раздела масло — вода, чем в случае воздух — вода,
потому что молекулы масла проникают в промежутки
углеводородных цепей эмульгатора, уменьшая силы притяжения между ними.
Кривые сила — площадь (рис. III.32) характерны для трех
основных видов пленок: газообразных, жидких растянутых и
185
конденсированных. В газообразных пленках молекулы обычно
плашмя лежат на поверхности. Они перемещаются независимо друг
от друга *, и поверхностное натяжение возникает благодаря
многочисленным соударениям с барьером. Согласно уравнению (III.114),
£ -»- 0 при высоких значениях я, а площадь, приходящаяся на угле-
о
водородную цепочку в вертикальном положении, равна 19,5—20 А2,
поэтому кинетическая энергия молекул в газообразной пленке равна
(Ленгмюр, 1933)
x(S-S0) = nK(S-SQ)=kT (III. Ц5)
где лк — вычисленное давление, вызываемое кинетической энергией
молекул пленки; S0 — предельная площадь молекулы.
Если я-»- 0, то и nS-*- kT,
как в уравнении (III.114),
но когда я -»- °о, то S -*■ S 0.
Уравнение (III.115)
применимо к электронейтральным
пленкам на поверхности
раздела масло — вода, где
отсутствует когезия между
цепочками. Общее
поверхностное давление включает
два компонента:
лМ/В = лк + Лотт (П1.116)
Рис. III.32. Зависимость поверхностного
давления от площади различных видов
поверхностных пленок:
где яотт — давление
отталкивания, возникающее
благодаря электрической
энергии двойного слоя (Дэвис
и Райдил, 1963).
Величину яотт можно найти из уравнения:
1 — конденсированная; 2 — точка перехода;
з — растянутая; 4 — газообразная.
2кТ
S
■6,lcV»
(III.117)
Здесь ci — общая концентрация одновалентного . электролита
Жидкорастянутое состояние возникает, например, при наличии
сильной когезии между группами — СН2— в длинной
углеводородной цепи на поверхности раздела воздух — жидкость, если нет
электрического отталкивания. Для значительной области площадей
когезия остается всегда постоянной и уравнение состояния имеет вид
(л — ns)(5 — S0)=kT
(III.118)
* Эти положения далеко не так бесспорны. В литературе ведется дискуссия
и высказывается противоположное мнение, что в газообразной пленке на
поверхности воды молекулы ПАВ существуют в виде островков с соединенными
гидрофобными группами. (Прим. редактора перевода.)
186
где л8— поверхностное давление, возникающее в результате
действия сил когезии.
Для цепи с Сщ величина ns «=° 10 дин/см, а для цепи с С22 —
приблизительно 22 дин/см. Конфигурация молекул с
ненасыщенными углеводородными цепями препятствует плотной упаковке
и уменьшает долю л5. При увеличении площадей или при высоких
температурах жидкорастянутые пленки превращаются в
газообразные.
В конденсированных пленках молекулы плотно прижаты и
углеводородные цепи ориентируются параллельно друг другу и
перпендикулярно поверхности раздела фаз. Такая конфигурация
напоминает кристаллическое состояние. В этом случае значение л3 очень
высоко и находится примерно между 50 и 100 дин/см.
Для двухкомпонентной системы, состоящей из растворенного
вещества 1 и растворителя 2, уравнение адсорбции Гиббса можно
представить следующим образом
Лт + /Чф1 + Аф2 = 0 (III.119)
где Г — избыток адсорбированного вещества в поверхностном слое;
ц — химический потенциал.
Если геометрическая поверхность определена таким образом,
что избыток растворителя на поверхности равен нулю, то
й(т + Л^1=0 (III.120)
Для разбавленных растворов эмульгатора
И1 = М-1 + лг1пс№ (III.121)
поэтому
da= — ^RTdlncy! (III.122)
где R — газовая постоянная; ct и Yi — концентрация и коэффициент
активности растворенного вещества.
Следовательно
Г=—^т-'-тт^ (III.123)
HI a In ciYi
При использовании разбавленных растворов эмульгатора
концентрация его на поверхности значительно превышает
концентрацию в объеме фазы, поэтому rt равно концентрации вещества на
1 см2 в адсорбированном монослое. Ее величину определяют из
графика а — In CjYi или о" — In c4 проведением касательной к кривой
в соответствующей точке и подстановкой полученного значения
do/dlnc^i или da/d In ct в уравнения (III.122) или (III.123).
Так как J^iVS-lO"16 = 1 (где iV—число Авогадро, равное 6,023 х
X 1023), то можно вычислить площадь, занимаемую молекулой
в монослое, и построить кривую. Если поверхностная концентрация
187
ионного эмульгатора вычислена для условия, когда нет
электролита, то RT в уравнении (III.123) можно заменить на 2RT (Хайзоп
и Филлипс, 1958) *.
МИКРОЭМУЛЬСИИ
При добавлении водных растворов мыла к равному объему масла,
например к бензолу, образуется непрозрачная гетерогенная
система, которая становится прозрачной при введении небольшого
количества спирта (Шульман и др., 1959; Стокениус и др., 1960).
Если от;м < °"т,в (рис. III.33), то образуется микроэмульсия В/М,
при а > о — микроэмульсия М/В. Диаметр шариков в таких
системах равен 80—800 А в зависимости от длины цепи спирта (Бау-
кот и Шульман, 1955; Кук
и Шульман, 1965), так что
нижний предел этой области
сравним с диаметром сферических
анионных мицелл (■—50 А)
и неионных мицелл (~100 А).
Самопроизвольное
эмульгирование наблюдается в случае,
когда межфазное натяжение
практически равно нулю **.
Так как для образования
микроэмульсий требуются
растворы мыла и спирта, то
возможно, что метастабильная
отрицательная свободная
энергия разрушает поверхность
раздела, способствуя
образованию шариков масла или воды. Стабильность микроэмульсий
зависит от этой отрицательной свободной энергии, потому что при
коалесценции существует сила, которая опять увеличивает
межфазную площадь. На распределение спирта между различными фазами
влияет природа масла (Кук и Шульман, 1965). Масло и эмульгатор,
используемые для получения микроэмульсий, должны быть
одинаковы по своей структуре ***.
МНОЖЕСТВЕННЫЕ ЭМУЛЬСИИ
Теоретическая предельная объемная доля дисперсной фазы
в эмульсии равна 0,74 при условии, что все шарики одинакового
диаметра. Практически же эмульсия редко бывает монодисперсной,
* Это положение экспериментально не доказано п требует проверки.
(При.и. редактора перевода.)
** Самопроизвольное эмульгирование данного типа подробно исследуется
школой акад. П. А. Ребиндера — см. дополнительную литературу к главе I.
(Прим. редактора перевода.)
*** В последнее время в Институте физической химии АН СССР (Таубман,
Никитина и др.) получены новые данные о существовании микроэмульсии
на поверхности обычных эмульсий. (Прим. редактора перевода.)
Бензол
Спирт
Мыло
Спирт
Вода
Спирт
Непрерывная
масляная
фаза
Поверхность
раздела
Дисперсная
фаза
водная
фаза
б„у^
Orryft
Рис. III.33. Предлагаемая структура
микроэмульспй (Кук и Шульман, 196.5).
188
так как в промежутках между очень большими шариками находятся
маленькие. При осторожном диспергировании объемные доли
дисперсной фазы могут достигать величины 0,85—0,90, ибо шарики
изменяют свою форму. Например, микроскопическое исследование
эмульсии В/М с высокой концентрацией воды показало, что шарики
уже не просто капли воды, так как в них диспергированы маленькие
капельки масла, причем при некоторых условиях эти капли воды
могут быть распределены в каплях масла. Шарики множественной
эмульсии намного больше водных в разбавленных эмульсиях В/М.
Множественные эмульсии могут образовываться при обращении фаз
Рис. III.34. Множественная эмульсия,
состоящая из 60% жидкого парафина п содержащая 6%
монолаурата сорбитана. Х2964 (Шерман, 1963).
(Зифриц, 1925), особенно при наличии явления гистерезиса, когда
новая зависимость вязкость — объемная доля отличается от этой
зависимости для обычной эмульсии (Бехер, 1965).
Эмульсии, полученные с двойным эмульгатором, один из которых
стабилизирует эмульсии М/В, а другой — В/М, тоже могут быть
множественными (Парке, 1934).
Распределение эмульгатора между двумя жидкими фазами
способствует образованию таких эмульсий (Вудмен, 1929). Эмульсии,
приготовленные с монолауратом сорбитана (рис. III.34),
диспергированном в воде (Шерман, 1963) или распределенном между водной
и масляной фазами (Бехер, 1958), относятся к эмульсиям типа М/В
в пределах широкой области концентрации дисперсной фазы. При
объемной доле 0,6 и 1,5—4,5 вес. % эмульгатора еще получаются
эмульсии М/В. Если концентрация эмульгатора увеличивается
до 6,0%, то эмульсия будет множественной. При повышении
объемной доли масла концентрация эмульгатора возрастает, так как
объем воды уменьшается. Это сопровождается переходом свободного
189
монолаурата сорбитана из водной фазы в масляную, в которой
он хорошо растворим, причем при высоких концентрациях масла
и эмульгатора в воде последнего остается очень мало.
<
ОПРЕДЕЛЕНИЕ ТИПА ЭМУЛЬСИЙ
Существует несколько методов определения типа эмульсий (М/В
или В/М), но ни один из них не является вполне совершенным.
Метод разбавления. В пробирку с водой добавляют каплю
эмульсии (Бриггс, 1914), которая при осторожном встряхивании
равномерно распределяется в объеме воды, если эта эмульсия типа М/В.
Если же эмульсия типа В/М, то капля не диспергируется. Эта проба
дает лучшие результаты с разбавленными эмульсиями.
Окрашивание непрерывной фазы. Каплю эмульсии помещают
на предметное стекло микроскопа рядом с несколькими кристаллами
растворимого в воде красителя (Ньюмен, 1914). Пластинку
наклоняют так, чтобы капля и краситель соприкасались. Если окажется,
что непрерывная фаза окрасилась, это эмульсия М/В. В противном
случае опыт повторяют с жирорастворимым красителем,
подтверждая, что это эмульсия типа В/М. Водорастворимыми красителями
являются метилоранж и бриллиантовый синий, а маслораствори-
мыми — судан III и фуксин.
Эту пробу можно проводить с объемом эмульсии в пробирке.
Например, если при добавлении нескольких кристалликов
водорастворимого красителя и встряхивании происходит равномерное
окрашивание образца по всему объему, то это эмульсия типа М/В.
В концентрированных эмульсиях труднее определять окрашенную
площадь.
Троннер и Бассьюс (1960) развили этот метод. На кружки
фильтровальной бумаги, смоченные 20% водным раствором хлорида
кобальта и затем высушенные, они помещали каплю эмульсии.
Эмульсия М/В вызывает быстрое появление розового окрашивания,
с эмульсией В/М никаких цветовых изменений не происходит.
Множественные эмульсии или смесь эмульсий М/В и В/М вызывают
медленное появление слабо-розового окрашивания.
Электропроводность. Определение типа эмульсии по
электропроводности предложено много лет назад. Водная непрерывная
фаза обладает намного большей электропроводностью, чем масляная'.
В процессе измерения в эмульсию помещают два электрода, и если
неоновая лампа загорается, это эмульсия типа М/В. Эмульсии М/В,
стабилизированные неионными эмульгаторами, могут давать низкую
электропроводность, а эмульсии В/М будут повышать
электропроводность с увеличением концентрации дисперсной фазы.
Множественные эмульсии могут обладать электропроводностью даже в
случае, если они диспергированы в масляной непрерывной фазе.
190
ЛИТЕРАТУРА
А Ь г i b a t M., Dognon А. С. R., С. г., 208,1881 (1939).
Adam N. К., in «Physical Chemistry of Surface», London, 1941.
Adam N. K., Jessop G., J. Chem. Soc, 122, 1865 (1925).
Adam N. K., Livingston H. K., Nature (London), 182, 128(1958).
Adamson A. W., in «Physical Chemistry of Surfaces», New York, 1960.
A 1 b e г s W., Doctoral Dissertation, University of Utrecht, 1957.
Alexander A. E., in «Surface Chemistry», London, 1949, p. 123.
Alexander A. E., Те ore 11 Т., Trans. Faraday Soc, 35, 727 (1939)
Antonoff G., J. chim. phys., 5,364, 372 (1907).
Askew F. A., Danielli J. F., Trans. Faraday Soc, 36, 785 (1940).
Bancroft W. D., 17,54(1913).
Bangham A. D., He and D. H., Seaman G. F., Nature (London),
182, 642 (1958).
В a r n e t t M., T i m b r e 1 1 V., Pharm. J., 189, 379 (1962).
Bart ell F. E., Miller F., J. Am. Chem. Soc, 50, 1961 (1928).
Bashforth F., Adams J. C., in «An Attempt to Test the Theories of
Capillary Action», Cambridge, 1883.
Becher P., J. Soc. Cosmet. Chem., 9, 141 (1958).
В е с h e r P., J. Colloid. Sci., 18, 665 (1963).
Becher P., J. Colloid Sci., 19, 468 (1964).
Becher P., in «Emulsions. Theory and Practice», New York, 1965, p. 169.
Becher P., Birkmeier R. C, J. Am. Oil Chem. Soc, 41, 169 (1964).
В h a t n a g a r S. S., J. Chem. Soc, 117, 542 (1920).
Боросикхино В. И., Скрилев Л. Д., Мокрешин С. Г.,
Коллоидн. ж., 23, 563 (1961).
Bowcott J. E., Schulman J. H., Z. Elektrochem., 59, 283 (1955)
Briggs Т. L., J. Phys. Chem., 18,34 (1914).
Brooks J. H., MacRitchie F., J. Colloid Sci., 16, 442 (1961).
Brooks J. H., Pethica B. A., Trans. Faraday Soc, 60, 208 (1964).
С h e e s m a n D. F., Ark. Kemi, 22B, I (1946).
Cole L. J. N., Kluepfel D., Lusena С V., Canad. J. Biochem.
Physiol., 37, 821 (1959).
Cooke C. E., SchulmanJ. H., in «Surface Chemistry», ed. by P. Ekwall,
K. Groth and V. Runnstrom-Reio, Munksgard — Copenhagen, 1965, p. 231.
Dalla Valle J. M., in «Micromeritics», New York, 1948.
D a v i e s J. Т., Proceedings of the 2nd International Congress on Surface
Activity, vol. I, 1957, p. 426.
D a v i e s J. Т., Proceedings of the 3rd International Congress on Surface
Activity, vol. 2, 1960, p. 585.
Davies J. Т., Rideal E. K., in «Interfacial Phenomena», New York —
London, 1963.
Debye P., Phys. Z., 28, 135 (1927).
Derjaguin B. V., Vlasenko G. Ja., J. Colloid Sci., 17, 605 (1962).
Dismuskes E. В., J. Phys. Chem., 63, 312 (1959).
Doty P., Steiner R. F., J. Chem. Phys., 18, 211 (1950).
Douglas H. W., J. Sci. Instrum., 24, 103 (1947).
D u p г ё A., in «Theorie Mechanique de la Chaleur», 1869, p. 393.
Fowkes F. M., J. Phys. Chem., 66, 382 (1962).
Fowkes F. M., Ind. Eng. Chem., 56, 40 (1964).
Fowkes F. M., Hark ins W. D., J. Am. Chem. Soc, 62, 3377 (1940).
F о x P. G., Nature (London), 184, 546 (1959).
Freud В. В., Freud H. Z., J. Am. Chem. Soc, 52, 1772 (1930).
G i b b s J. W., in «Collected Works», vol. I, New Haven, Connecticut, 1928,
p. 230.
G i n n M. E., Anderson R. M., Harris J. C, J. Am. Oil Chem.
Soc, 41, 112(1964).
Girifalco L. A., Good R. J., J. Phys. Chem., 61, 904 (1957).
G о u 1 d e n J. D. S., Spectrovision, (4), 3 (1957).
191
Goulden J. D. S., Trans. Faraday Soc, 54. 94 (1958).
Goulden J. D. S.. Phipps L. W.. Proceedings of the III International
Congress on Surface Activity, vol. 3. 1960. p. 190.
Goulden J. D. S., Sherman P., J. Dairy Res., 29, 47 (1962).
G r e e n w a 1 d H. L., Brown G. L., F i n e m a n M. W., Analvt. Chem.,
28,1963(1956).
Griffin W. C, J. Soc. Cosmet. Chem., 1. 311 (1949).
Griffin W. C. J. Soc. Cosmet. Chem., 5, 249 (1954).
Griffin W. C. Off. Dig. Fed. Paint Yarn. Prod. Clubs, 28, 466 (195G).
G г о о t R. C, Void R. D., Paper presented at the IV International
Congress on Surface Activy, Brussels, 1964.
Наг к ins \V. D., Brown F. E., J. Am. Chem. Soc, 41, 499 (1919).
Harkins W. D., Cheng Y. C, J. Am. Chem. Soc, 43, 35 (1921).
Hark ins W. D., Brown F. C, Int. Crit. Tabl., 4, 435 (1926).
Harkins W. D., Jordan H- F.. J. Am. Chem. Soc, 52. 1751 (1930).
Harkins W. D., in «The Physical Chemistry of Surface Films», New York,
1952.
Haugaard G., Pettinati J. D., I. Dairy Sci.. 42, 1255 (1959).
H a user E. A., in «Advances in Colloid Science», vol. I, New York, 1942,
p. 319.
На у don D. A., Philips J. N.. Trans. Faraday Soc, 54, 698 (1958).
Heller W., J. Chem. Phys., 26, 920. 1258 (1957).
Henry D. C, Proc Roy. Soc, A133, 106 (1931).
Henrv D. C, J. Chem. Soc, 1938, 997.
lellinek H. H. G., J. Soc Chem. Ind. (London), 69, 225 (1950).
J el line к H. H. G., Anson H. A., J. Soc. Chem. Ind. (London), 68,
108 (1949).
Jones T. G., P ethic a B. A., Walker D. A., J. Colloid Sci., 18,
485 (1963).
К i s t 1 e г S. S., J. Am. Chem. Soc, 58. 901 (1936).
Kitchener J. H-, Proceedings of the III International Congress on Surface
Activity, vol. 2, 1960, p. 426.
Lamb H., Phil. Mag., 25, 52 (1888).
Langmuir I., J. Chem. Phys.. I, 756 (1933).
Lawrence A. S. C, Mills O. S., Discuss. Faraday Soc, 18, 98 (1954).
Лебедев А. В. и др., Коллоида, ж., 24, 482, 487 (1962).
Levi us H. P., Drommond F. G., J. Pharm. Pharmac, 5, 743 (1953).
Limburg H., Rec trav. chim., 45, 854 (1926).
Llothian C. F., Chappel F. P., J. Appl. Chem., I, 475 (1951).
Lloyd N. E., J. Colloid Sci., 14, 441 (1959).
Latzkar H., Ma clay W. D., Ind. Eng. Chem., 35, 1294 (1943).
M с I n t у г e D. a. oth., J. Phys. Chem., 66, 1932 (1962).
Merrill R. C, Ind. Eng. Chem. Analyt. Ed., 15,743 (1943).
van d e г M i n n e J. L.. Hermanie P. H. J., J. Colloid Sci., 7, .
600 (1952).
Mukerjee L. M., Barthwal J. P., Z. phys. Chem., 209, 80 (1958).
M у s e 1 s K. J., S h i n о d a K., F г a n к e 1 J., in «Soap Films», London,
1959.
Narasimham P. a. oth., J. Colloid Sci., 20, 473 (1965).
Newmann F., J. Phys. Chem., 18, 34 (1914).
Northrup J. H., Kunitz M-, J. Gen. Physiol., 7, 729 (1925).
Padday J. F., Proceedings of the 2nd International Congress on Surface
Activity, vol. 3, 1957, p. 136.
Padday J. F., J. Sci. lustrum., 36, 256 (1959).
Padday J. F., Proceedings of the III International Congress on Surface
Activity, vol. 2, 1960, p. 457.
Parke, J. Chem. Soc, 1934,1112.
Petersen R. V., Ham ill R. D., McMahon J. D., J.Am. Pharm.
Assoc, 53,651 (1964).
Phipps L. W., Trans. Faraday Soc, 60, 1878 (1964).
192
Pickering S. U., J. Chem. Soc, 91, 2002 (1907).
Pinter G. G., Zilversmit D. В., Biochem. biophys. acta, 59, 116
(1962).
Plant Protection Ltd., in «The Physics Exhibition Handbook», Institute of Physics,
London, 1965, p. 148.
Porter A. W., Phil. Mag., 15, 163 (1933).
Поспелова К. А., Воробьева Т. А., Зубов П. И., Коллоидн.
ж., 24,511 (1962).
Powell В. D., Alexander A. E., Canad. J. Chem., 30, 1004 (1952).
Powney J., Addison С. С, Trans. Faraday Soc, 33, 1243 (1937).
Powney J., Wood L. J., Trans. Faraday Soc, 36, 57 (1940).
Racz I., Or ban E., J. Colloid Sci., 20, 99 (1965).
Rav B. R., Bartell F. E., J. Colloid Sci., 8, 214 (1953).
Rayleigh D., Phil. Mag., 41, 107; 274; 447 (1871).
Rayleigh D., Phil. Mag., 44, 28 (1897).
Rayleigh D., Phil. Mag., 48. 375 (1899).
Rehfeld S. J., J. Phys. Chem., 66, 1966 (1962).
Riegelmann S., Pichon G., Am. Perfumer, 77, 31 (1962).
R о chow T. G., Mason С W., Ind. Eng. Chem., 28, 1296 (1936).
R о s s S. a oth., J. Phys. Chem., 63, 1681 (1959).
Rumscheidt F. D., Mason S. G., J. Colloid Sci., 16, 238 (1961).
Ruyssen R., Rec trav. chim., 65, 580 (1946).
Scarlett A. J., Morgan W. L., Hilde brand J. H., J. Phys.
Chem., 31, 1166 (1927).
Schulman J. H., Leja J., Trans. Faraday Soc, 50, 598 (1954).
Schulman J. H., Stoeckening W., Prince L., J. Phys. Chem.,
63,1677 (1959).
Schwarz N., Bezemer C, Kolloid Z., 146, 139, 145 (1956).
Seifriz W., J. Phys. Chem., 29, 738(1925).
Sherman P., in «Rheology of Emulsions», ed. by P. Shermann, London,
1963, p. 73.
Sherman P., J. Food Sci., 31, 699 (1966).
Shi no da K., Arai H., J. Phys. Chem., 68, 3485 (1964).
Shotton E., Kalyan K., J. Pharm. Pharmacol., 17, 109 (1960).
S i e d e n t о p f M., Z s i g m о n d у R., Ann. Phys., 10, I (1903).
Singleton W. S., Benerito R. R., White J. L., J. Am. Oil
Chem. Soc, 37,88(1960).
Skoda W., van den Tempel M., J. Colloid Sci., 18, 568 (1963).
Smith M. E., Lisse M. W., J. Phys. Chem., 40, 399 (1936).
Stoeckenius W., Schulman J. H., Prince L., Kolloid Z.,
169, 170 (1960).
Studebaker M. I., Snow С W., J. Phys. Chem., 59, 973 (1955).
Tronnier H., Bussius H., Seifen-Ole-Fette-Wachse, 86, 747
(1960).
Void R. D., Groot R. C, J. Phys. Chem., 65 (1962).
Void R. D., Groot R. C, J. Soc. Cosmet. Chem., 14, 233 (1963).
Void R. D., Groot R. C, J. Colloid Sci., 19, 384 (1964a).
Void R. D., Groot R. C, J. Phys. Chem., 68, 3477 (1964b).
Van de Hulst H. C, in «Light Scattering by Small Particles», New York,
1957, p. 176.
Van den Waarden M., J. Colloid Sci., 9, 215 (1954).
Van de Zeggeren F., Courval C, Goddard H. D., Canad.,
J. Chem., 37,1937 (1959).
Wachtel R. E.. La M e r V. K., J. Colloid Sci., 17, 53 (1962).
Wales M., J. Phys. Chem., 66, 1768 (1962).
Walstra P., Br. J. Appl. Phys., 16, 1187 (1965a).
Walstra P., Spectrovision, 13, 7 (1965b).
Washburn E. W., Phys. Rev., 17, 273 (1921).
We 11m an V. E., Tartar H. E., J. Phys. Chem., 34, 379 (1930).
W e n z e 1 R. N., Ind. Eng. Chem., 28, 98 (1936).
13 Заказ 1070
193
Wheeler 0. L., Tartar H. V., Livingstone H. C, J. Am-
Chem. Soc, 67, 2115(1945). ч
Wolkowa Z. E., Kolloid Z., 67, 280 (1934).
Wood J. H., Catacalos G., J. Soc. Cosmet. Chem., 14, 147 (1963).
Woodman R. M., J. Phys. Chem., 33, 88 (1929).
Woodman R. M., J. Soc. Chem. Ind. (London), 54 70T (1935).
Young P. A., PI. Physiol., 9, 795 (1934).
Zuidema H. H., Water G. W., Ind. Eng. Chem. Anal. Ed., 13, 312
(1941).
Дополнительная литература
А о к i M., Matsuzaki Т., Jakugaku Lasshi, 87, № 7, 761 (1967).
Температура инверсии фаз в эмульсиях с неионогеннымп ПАВ.
А г a i H., Shinoda К., J. Coll. Interf. Sci., 25, № 3, 396 (1967). Влияние
смесей масел и неионных ПАВ на инверсию фаз в эмульсиях.
В а г с 1 е t L. et. al., Trav. Soc. Pharm. Montpellies, 29, № 2, 101 (1969). Физико-
химическое изучение эмульсий и неионпых ПАВ.
В erg we in К., Fette, Seifen, Anstrichm, 69, № 5, 353 (1967). Технология
эмульсий и значение ГЛБ.
Blonchard P., Parfum., Cosmet., Savons, 12, № 2, 82 (1969). Измерение
ГЛБ эмульгаторов методом, основанном на инверсии фаз.
В о о i j H. L., J. Coll. Interf. Sci., 29, № 2, 865 (1969). Экстракция масла из
эмульсий.
Brawn A. H., Chem. Ind., № 30, 990 (1968). Жидкая коалесценция.
FribergS., MandellL., LarssonM., J. Coll. Interf. Sci., 29, № 1, 155
(1969). Мезаморфные фазы как фактор, определяющий свойства
эмульсий.
Gillbery G., Lehtinen H., Friberg S., J. Coll. Interf. Sci., 33,
№ 1, 40 (1970). Исследование условий, определяющих стабильность
микроэмульсий с помощью ЯМР и ИК-спектрофотометрии.
Griffin W., С, Ranauto H. J., Adams A. D. Sperialities, 2, № 11,
15 (1966). Дальнейшее изучение эмульсионных систем.
Groot R. С, Void R.D., Proceedings of the IV International Congress
on Surface, 1964, vol. 2, 1967, p. 1233. Ультрацентрифугирование
эмульсий с различными количествами масляной фазы.
Н ackett W., Deterg. Spec, 6, № 11, 28 (1960). Как выбирать эмульгатор.
Н е i t 1 a n d V., Р е t г е L., Seifen — Ole — Fette — Wachse, 95, № 10,
362 (1969). Проницаемость эмульсий В/М парами воды.
Jombik J., Gruntova Z., Schiller P., Cesk. Farm., 17, № 2, 419 (1968).
Использование электронов отдачи для определения стабильности
эмульсий М/В.
К lare H., Sonntag H., Abh. Deutsch. Akad. Wiss., Berlin, 1966, (6), "
^ 597 (1967). Химическое и электрическое деэмульгирование эмуль-
V сий В/М.
Lin J.T., Lambrechts J. С, J. Soc. Cosmet. Chem., 20, № 3, 185 (1969).
Влияние начальной концентрации ПАВ на инверсию фаз в
эмульсиях.
Hank D., М a t i j е v i с F., К е г к е г М., J. Coll. Interf. Sci., 29, № 1,
164 (1969). Определение величины частиц светорассеянием.
Маг А., М a s о n S., Koll. Z. u. Z. Polym., 224, № 2, 161 (1968).
Коалесценция в трехфазных системах.
Narazaki H., Seechaku, 11, Л» 1, 34 (1967). Успехи в технологии
эмульсий М/В.
N a wre у J. E. et al., J. Chem. Eng. Data, 13, № 3, 297 (1968). Тепловая
проводимость ппщевых эмульсий М/В.
Netzel J., Sonntag H., Abh. Deutsch. Akad. Wiss., Berlin, Kl. Chem.,
1966, № 6, 589 (1967). Стабильность эмульсий М/В.
Parsons W. H., Doadero D. L., J. Pharm. Sci., 58, Д° 12, 1449 (1969).
1£4
Изучение градиента плотности в некоторых эмульсиях М/В с помощью
Y-лучевого анализа.
Prakash С, Srivastava S. N., Bull. Chem. Soc. Japan, 42, № 7,
1787 (1969). Влияние некоторых катионных ПАВ на эмульсии,
стабилизированные макромолекулами эмульгатора — альгинатом натрия и
гуммиарабиком.
R a h m H., S о 1 i v a M., S p e i s е г P., Pharm. acta Helv., 44, № 6, 356
(1969). Общее приложение системы ГЛБ к эмульсиям.
R a n n у М., Prum. Potravin., 20, № 9, 271 (1969). Определение свойств
детергентов и эмульгаторов.
R i d d i с k Т. M., Amer. Perfum. a. Cosmet., 85, № 7, 31 (1940). Контроль
устойчивости эмульсий с помощью дзета-потенциала.
S a i t о Н., Nippon Kagaku Zasshi, 90, № 10, 1020 (1969). Влияние
температуры и длины гидрофильной цепи неионных ПАВ на свойства
эмульсий.
Saito H., S h i n о d а К., J. Coll. Interf. Sci., 32, № 4, 647 (1970).
Стабильность эмульсий типа В/М как функция температуры и длины
гидрофильной цепи эмульгатора.
S a s t г у G., Srivastava S., Indian J. Chem., 6, № 12, 728 (1968). Роль
биологических веществ в стабилизации эмульсий.
S a s t г у G. Т., S г i v a s t a v a S. N., J. Coll. Interf. Sci., 33, № 3, 468 (1970).
Изучение межфазного натяжения в водно-масляной эмульсии,
стабилизированной смесью лецитина-холестерина.
S с h о t t H., J. Pharm. Sci., 58, № 12, 1443 (1969). ГЛБ и точка мутности
неионных ПАВ.
S e i 1 1 е г М., Legras Т., Puisenex A., Ann. Pharm. France, 25, № 11,
723 (1967). Характеристики эмульсий парафинового масла в воде при
критических значениях ГЛБ.
Sherman P., Proceedings of the IV International Congress on Surface Active
Substances, 1964, vol. 2, 1967. Коагуляция эмульсий М/В, содержащих
малые глобулы.
Shi nod а К., J. Coll. Interf. Sci., 24, № 1, 4 (1967). Корреляция между
растворимостью неионного ПАВ и типом стабилизированной
дисперсии.
Shinoda К., А г a i H., I. Coll. Interf. Sci., 25, № 3, 429 (1967). Влияние
объемов фаз на температуру инверсии эмульсий с неионными ПАВ.
Shinoda К., Nippon Kagaku Zasshi, 89, № 51, 435 (1968). Использование
температуры инверсии и ГЛБ для выбора эмульгатора.
Shinoda К., S a i t о Н., J. Coll. Interf. Sci., 30, № 2, 258 (1969).
Стабильность эмульсий М/В как функция температуры и ГЛБ. Эмульгирование
по методу инверсии фаз.
Shinoda К., Takeda H., J. Coll. Interf. Sci., 32, № 4, 642 (1970). Влияние
добавления солей в воду на ГЛБ неионного ПАВ (на температуру
инверсии фаз).
Srivastava S. N., Н а у d о n D. A., Proceedings of the 4th International
Congress, vol. 2, 1964, p. 1221. Эмульсии М/В, стабилизированные
протеинами.
Suzuki A., H о N., H i g u с h i W., J. Coll. Interf. Sci., 29, № 3, 552 (1969).
Предсказание изменения дисперсности эмульсий и суспензий посредством
digital счетчика.
Walstra P., J. Coll. Interf. Sci., 27, №3, 493 (1968). Определение спек-
тротурбодиметрическим способом распределения частиц в
эмульсиях М/В.
Walstra Р., О о г t u г j и Н., J. Coll. Interf. Sci., 29, № 3, 424 (1969).
Определение с помощью счетчика Коултера распределения частиц по
размерам в эмульсиях М/В.
Авдеев Н. Я., Коллоидн. ж., 32, №5, 635 (1970). Характеристика
распределения дисперсной фазы эмульсий.
Бовкун О. П., Маркина 3. Н., Цикурина Н.И., Коллоидн. ж.,
32, № 6, 831 (1970).
13*
195
ч
Г о л ы ш е в а Г. П., К и р и ч е н к о Г. С, М а к о л е ц Б. И., ЖПХ, 43,
Л» 10, 2188 (1970). Исследование устойчивости эмульсий переменного
состава методами математической статистики.
Е н а л ь е в В. Д. и др., Коллоидн. ж., 31, № 1, 53 (1969). Влияние добавок
регулятора гидрофильно-гидрофобного баланса на свойства суспензий
бентонита и эмульсий, стабилизированных ими.
Колпаков Л. В., Н и к и т и н а С. А., Т а у б м а н А. В. и др.,
Коллоидн. ж., 32, № 2, 229 (1970). Электронно-микроскопический метод
исследования дисперсных систем с жидкими фазами.
Ш в а р о в Е. П., Никитина С. А., Чернышева Н. М., Коллоидн.
ж., 32, Л» 6, 916 (1970). Структурообразование в межфазных слоях
и объеме водных растворов сополимера с малеиновой кислотой и
устойчивость стабилизированных эмульсий М/В.
X а р и н С. Е., А ф и н о г е н о в а И. Н., Изв. вузов. Хим. и хим. технол.,
12, № 6, 762 (1969). Некоторые свойства эмульсий коллоидной степени
дисперсности.
ОСНОВНЫЕ ТИПЫ
ТЕЧЕНИЯ
Глава IV
РЕОЛОГИЯ
ЭМУЛЬСИЙ
Ф. Шерман
Реологию обычно определяют как
науку о деформации и
течении материалов. Согласно Фред-
риксону (1964), «целью реологии
является предсказание системы сил,
необходимой для того, чтобы вызвать
деформацию или течение тела, или,
наоборот, предсказание деформации
или течения, возникающих от прило- .
жения данной системы сил к телу».
Ее методы могут быть использованы
для изучения структуры эмульсий,
консистенция которых колеблется в
пределах от жидкостей до твердых
тел. Приложение силы к жидкости
вызывает течение. Если эту силу
удалить, жидкость не возвращается
в свое первоначальное состояние —
она претерпевает необратимую
деформацию. Ответная реакция твердого
тела на приложенную силу зависит
от того, является ли оно эластичным
или пластичным. Эластичное твердое
тело подвергается деформации, но не
течет. После удаления силы оно
возвращается в свое первоначальное
состояние и, следовательно,
проявляет обратимую деформацию.
Пластичное или вязкоэластичное твердое
тело ведет себя таким же образом,
если приложенная сила не превышает
критической величины. В противном
случае оно течет, как жидкость.
При удалении приложенной силы
пластичное твердое тело не
возвращается полностью в исходное
состояние.
Очень разбавленные эмульсии
ведут себя подобно простым жидкостям
и проявляют ньютоновское течение.
В более концентрированных системах
шарики взаимодействуют друг с
другом и флокулируют, образуя
агрегаты, которые обладают вязкоэла-
стичными свойствами. Замороженные
197
эмульсии или эмульсии, содержащие большую долю твердого
вещества в непрерывной фазе, могут вести себя как твердое тело.
Ньютоновское течение может быть проиллюстрировано простой
моделью: жидкость заключена между двумя параллельными
пластинами; площадь каждой составляет S (см2), расстояние между
ними 1ПЛ (в см) (рис. IV. 1). Сила F, приложенная к верхней пластине,
заставляет ее двигаться со скоростью v относительно нижней
пластины. Не вся жидкость, заключенная между двумя пластинами,
движется с одинаковой скоростью. Скорость изменяется с
увеличением расстояния от верхней пластины, имея максимальную величину
в слое, прилегающем к этой пластине, и приближаясь к нулю в слое,
соприкасающемся с нижней пластиной. Степень изменения скорости
*~ имакс Подвижное
пластина
Рпс.
Неподвижная
пластина
IV. 1. Мидель, иллюстрирующая ньютоновское
течение.
жидкости с расстоянием от верхней пластины, т. е. средняя
скорость сдвига vc дается как и/1пл. В общем случае, для некоторой
точки, лежащей в слое, который расположен на расстоянии х1 от
верхней пластины, vc = dv/dxt. Сила, приложенная на единицу
площади верхней пластины (F/S), представляет напряжение сдвига
Рс. Вязкость г\ жидкости дается отношением PJvc и, так как ц
не зависит от приложенной силы, она может быть определена из
единичных измерений Рс и vc.
Большинство эмульсий проявляет при течении более сложное
(неньютоновское) поведение (рис. IV.2). В некоторых случаях
увеличение vc прогрессивно растет на единицу приращения Рс вплоть
до предельного значения Рс; таким образом, ч\ уменьшается. При
более высоких значениях Рс отношение PJvc и, следовательно, ц
остаются неизменными. Такой характер течения наблюдается, если
во время сдвига происходит разрушение структуры.
В пределах этой категории систем те эмульсии, которые текут, как
только приложено усилие (давление), проявляют псевдопластичные
свойства, в то время как эмульсии, требующие определенного
минимального напряжения сдвига или предельного его значения (Р°с)
для того, чтобы установилось течение, обладают пластичностью.
В растекающемся потоке vc уменьшается при возрастании Ps, что
приводит к увеличению т|. На рис. IV.2 такое поведение предста-
198
вляет собой зеркальное отображение нсевдопластического течения
при отсутствии предельного напряжения сдвига и пластического
течения, если предельное напряжение сдвига имеется.
Многие порошки и уплотненные дисперсные материалы
проявляют способность к растеканию. При сдвиге, прежде чем
отдельные частицы смогут двигаться относительно друг друга, геометрия
упаковки становится более рыхлой благодаря первоначальному
увеличению в объеме. В сообщениях, содержащих данные по
пластическому течению, часто приводят
два разных значения напряжения (Хау-
винк, 1958). Это экстраполированное
предельное напряжение сдвига
(отрезок ОБ на рис. IV.2) и верхнее
предельное напряжение сдвига (отрезок ОА).
Чтобы установить, действительно
ли достигнуто предельное напряжение
сдвига, необходимо экспериментально
найти зависимость Рс — vc вплоть до
малых значений vc, а не
экстраполировать линейный участок, который в
действительности может быть кривой
псевдопластического течения.
Так как вязкость
псевдопластичных, пластичных и текучих систем
изменяется со скоростью сдвига,
определения вязкости, сделанные при
отдельных случайных скоростях сдвига,
не имеют большого значения. В
частности, когда сравниваются режимы
течения двух разных эмульсий, их вязкости должны быть измерены
в широкой области скоростей сдвига. Поэтому, если одна из
эмульсий имеет большую вязкость, чем другая при каком-либо одном
значении скорости сдвига, из этого не следует с неизбежностью, что
такой порядок относится к другим скоростям сдвига. Определения
«одной точки» могут привести к неточным выводам. Когда имеем
дело с неньютоновским течением, должны быть указаны скорости
сдвига, при которых определялась вязкость.
Градиент PJvu линейного участка кривой неньютоновского
течения часто рассматривают как «кажущуюся вязкость»*. Если
слабое сдвиговое усилие стационарно прикладывают к
концентрированным эмульсиям, часто оказывается, что равновесное
напряжение не устанавливается мгновенно. Вместо этого Рс понижается
в течение периода времени, обусловленного структурными
изменениями, до тех пор, пока не будет достигнуто равновесное значение.
Необходимый интервал времени уменьшается, если скорость сдвига
увеличивается. Когда сдвиговое усилие устраняют, структура вновь
* Эффективная вязкость.
Градиент скорости
Рис. IV.2. Типы режимов
течения:
1 — пластическое; 2 —
псевдопластическое; 3 — ньютоновское; 4—
растекающееся, без предельного
напряжения сдвига. Предельное
напряжение сдвига: А — верхнее
(Хаувинк); В —
экстраполированное (Бингам); С — нижнее
(Хаувинк).
199
восстанавливается за определенное время. Это явление иногда
рассматривают как тиксотропию, хотя термин первоначально был
введен для определения изотермического перехода золь ^± гель.
В настоящее время его часто применяют в более широком смысле
для описания изотермических обратимых переходов между
состояниями высокой и низкой вязкости. Умштеттер (1935) подчеркивал
роль влияния времени на вязкость структурированных систем и
определял ее для некоторого времени т
П^Чравн-Ичо — Чрапн)« "' "С (IV. 1)
ГД° Лравн — равновесная вязкость после бесконечного времени;
г] о — начальная вязкость; тр — время релаксации.
Напряжение сдвига
Рис. IV.3. Трехмерная диаграмма Рис. IV.4. Гистерезисные эффекты
(Хаувинк, 1952). в тпксотропной эмульсин:
а — предельное значение постоянно; б —
предельное значение переменно.
Кривая зависимости скорости сдвига от напряжения не
определяет полностью поведения таких систем, так как должна быть
введена третья ось — времени (рис. IV.3).
Если скорость сдвига не остается постоянной и вначале
возрастает, а затем понижается без учета времени на установление
равновесной структуры при некоторой определенной скорости сдвига,
идущая вверх кривая напряжения не совпадает с идущей вниз
кривой, образуя гистерезисную петлю (рис. IV.4). Таким образом,
предельное напряжение сдвига может зависеть или не зависеть
от «истории» сдвига.
МЕТОДИКА ИЗУЧЕНИЯ РЕОЛОГИЧЕСКИХ СВОЙСТВ
Общие замечания
Для исследования реологических свойств жидких и
полутвердых эмульсий пригодны вискозиметры многих конструкций. Здесь
рассмотрены лишь вискозиметры общего назначения. К ним отно-
200
сятся: 1) капиллярные; 2) с коаксиальными цилиндрами; 3) конус-
пластина; 4) с падающим шариком. В табл. IV. 1 приведены
уравнения, с помощью которых получены значения vc, Рс, Р°с и т) путем
измерений с этими вискозиметрами и с учетом их геометрии.
Указаны также поправки для случаев, связанных с различным течением
образцов. Кроме этих вискозиметров, существуют и другие, которые
применяют реже, например, пьезоэлектрический и вибрационный
вискозиметры, а также пенетрометры.
Большинство вискозиметров (за исключением вискозиметров
с падающим шариком и ультразвукового) можно применять для
измерений как ньютоновских, так и неньютоновских жидкостей.
Вискозиметр типа конус-пластина является единственным, который
при малом угле конуса 9К обеспечивает постоянные условия сдвига
для всего образца. В капиллярном вискозиметре скорость сдвига
изменяется от нуля на оси капилляра до максимума у стенки
капилляра. Когда измерения производят в вискозиметре с коаксиальными
цилиндрами, один из цилиндров вращается. Если вращается
внешний цилиндр, измеряется скручивающее усилие, передаваемое
внутреннему цилиндру. В этом случае скорость сдвига изменяется
от максимума у поверхности вращающегося цилиндра до минимума
у поверхности внутреннего цилиндра. Однако при соответствующей
конструкции, когда зазор между двумя цилиндрами мал, этот
градиент сдвига минимален. Если внешний цилиндр неподвижен,
а внутренний цилиндр вращается, вязкость вычисляют из
вязкостного сопротивления, оказываемого последнему образцом.
Капиллярные вискозиметры не могут быть использованы для
изучения влияния времени сдвига на вязкость при какой-либо
заданной скорости, так как образец в капилляре непрерывно изменяется.
Далее будет показано, что информация об изменениях такого рода
полезна для объяснения структуры концентрированных эмульсий.
Этот недостаток может быть использован также, когда интересует
мгновенная вязкость, предшествующая изменениям в эмульсиях,
испытывающих зависимые от времени разрушение и восстановление
структуры.
Ошибки капиллярной вискозиметрии
При интерпретации данных, полученных с помощью
капиллярной вискозиметрии, необходимы поправки для различных явлений,
происходящих во время течения.
1. Так как опытный образец переходит из широкого резервуара,
в котором он содержится, в капилляр с диаметром, намного более
узким, образец деформируется вокруг буртика резервуара. Этот
эффект можно рассматривать как увеличение эффективной длины
капилляра от L до L + AL (где AL = nRK). Эффективное
напряжение сдвига у стенки капилляра в этом случае составит:
PRK _ PRK
с~ 2(L+AL) 2(L±nRK) K *>
где Р — приложенное давление; RK — радиус капилляра.
201
IS5
О
ts5
Таблица JY.1
Уравнения для вычисления параметров,
относящихся к четырем основным классам вискозиметров
Обозначения: (7i — экстраполированное значение скручивающего усилия для Q = 0; p—плотность ртути; ft —константа для перевод;)
Схема виекознмотра
Ньютоновская пяг>-
кость т]
Капилляр
\?к,(сн3сечение)/сек
1\ = пРП*к/Н1А'к
Коаксиальные
цилиндры
Конус-плаетияа
Пг С) G,дин/С»
о
а, рад/се к
1 1
V /?2 /?2
Ti = G АЛ1 ill
1 >.тгЬ Г)
бк./Х^
3G / Q *
2я«а/ Вк
Скатывающийся
или падающий шарик
/^^CSk
-ф'
^v
v^,en/сек
= _2_ / Рш —Робр\
1 9 V * У
Неньютоновская
вязкость Т)
Скорость сдвига
Напряжение сднига
Предельное
напряжение сдвига Р%
Необходимые
ципиальные
правки
прин-
по-
.яД^Р,
'■оо-
dVK
dp
+
&Lk
1
Р
4
d*VK
dP*
"o = 0
Pc =
РПК
2L
Pl=p0gpnK
Po = Pvc„n
Входной эффект;
кинетическая энергия,
сообщаемая
образцу; пристеночный
эффект; турбулентное
течение; течение в
виде «пробки»
Для малых значений 9
Чсо = -
,с° 4я/гц0
ч-Х
2Q
1{* (Л L
J<1 \ щ щ
2Q
Щ \~R\~~Ri)
J_ 1_\
Я1~ Щ)
4п/гц
Рс1^2
In (Л2/Л1)
Краевой эффект;
турбулентное течение
в области концов
е,-
Рг =
3G
2п№
P°C = PCG2
Математическая теория
но разработана
3i
W = ^!(e< = 90o)
im.ii = 0 (9, = 0°)
Краевой эффект;
пристеночный эффект
Для п предложены значения в пределах 0 —1,2. Поправка для
этого «краевого эффекта», или более правильно «входного эффекта»,
является минимальной, когда длина капилляра намного
превосходит его радиус, например, при L/RK = 200/1. Следовательно, эффект
сравним с перепадом давления, вызванным течением в капилляре.
2. Когда в узкий капилляр поступает эмульсия, может
возникнуть осевая миграция дисперсной фазы. Это вызвано флуктуациями
концентрации вдоль капилляра, главным образом уменьшением
концентрации, происходящим в слоях, которые прилегают к стенке
капилляра. В этом случае жидкость непрерывной фазы вблизи
стенки капилляра действует при малом сдвиге как смазывающий
слой и дисперсная фаза «скользит» вдоль него. Ванд (1948) высказал
мысль, что чистый эффект заключается в увеличении объемной
концентрации дисперсной фазы на [1/(1 — г) RK]2 (где г — радиус
частиц). Однако это выражение может описать изменение
концентрации только в центральной части капилляра с радиусом RK — г
и не может дать изменения концентрации для всего капилляра (Хиг-
гинботем, Оливер и Уорд, 1958).
Истинная относительная вязкость т)0Т11 для эмульсий дается
выражением
-i—i=«x(~—Л (iv.3)
tjoth V Цх I
где Цх — кажущаяся относительная вязкость эмульсии, измеренная
в капилляре х; Нх — поправочный коэффициент для слоя
непрерывной фазы вблизи стенки капилляра х.
Величину Н х определяют из уравнения
Я* = (1-в/Л,Г* (1V.4)
где б — толщина гипотетического слоя чистой жидкости
непрерывной фазы; Rx — радиус капилляра х.
Значение б можно вычислить, если измерить вязкость в двух
разных капиллярных вискозиметрах (.г и у), так как
нх *-****
(Нх\_ 1-6/л?
V Ну ) \-bjRx
а значения Rx, Ry и (HJHy) известны.
3. Не все прилагаемое давление Р используется для сдвига
образца; некоторая часть его расходуется на сообщение кинетической
энергии образцу, когда он заполняет капилляр. Этот феномен
оказывает большее влияние, чем «входной эффект». Давление,
фактически используемое для сдвига образца, дается выражением
Яф-Я-робрЧ/алЭДа (IV. 7)
201
(IV.5)
(1V.6)
где робр — плотность образца; VK — объем жидкости, протекающий
через капилляр в 1 сек; а — коэффициент кинетической энергии.
Когда течение является ньютоновским, а = 1 при условии
параболического распределения скоростей. Опубликованные значения а
колеблются в пределах 0,7—2,0. Ван Вазер, Лайонс, Ким и Колвелл
(1963) считают, что в этом случае происходили отклонения в
распределении скоростей, вызванные непостоянством просвета капилляров
и краевыми эффектами.
Для бингамовского пластического течения
а= (2-h/Pc) (IV-8)
где Р° = PR 0/2L; Pc = PRJ2L; R0 — расстояние от некоторой
точки капилляра до его оси, по которой вытянут столбик потока.
При неньютоновском течении а может быть определена по
уравнению
а= (4» + 2) (5.+3)
[3(3«+1)2] (1V'9)
где п —■ константа.
4. Напряжение Р°с, необходимое для того, чтобы вызвать течение
в некоторой точке капилляра на расстоянии R0 от оси, дается
выражением: Р° = PRJ2L. Вблизи оси R0 очень мало, так что Р должно*
было бы быть бесконечно большим для того, чтобы PRJ2L
превысило Р°с. Следовательно, у оси всегда существует тонкий слой
образца, который двигается через капилляр как твердая пробка.
Букингем (1921) и Рейнер (1926) при обработке данных
неньютоновского течения через капилляр ввели коэффициент для этого явления
(1 -Ч™- SLVK V 3 Зрз ) <IV-10)
.где т]пл — пластическая вязкость; р — давление, соответствующее Р°.
Когда Р ^> р, членом pi/3p3 можно пренебречь и таким образом
v-SH'-i")
5. В выведенном Пуазейлем (1840) уравнении для ньютоновских
жидкостей (см. табл. IV. 1) предполагается ламинарное течение
по всему капилляру. Рейнольде (1883) показал, что отклонения,
которые проявляются при высоких скоростях течения, вызваны
изменением характера течения от ламинарного до турбулентного.
Переход наблюдается, когда число Рейнольдса превышает 2000
2vcpRKp
Re = (IV. 12)
где vcp — средняя скорость образца (vcp = VJnR%).
Ламинарное течение превалирует, когда Re < 1000, а также
в промежуточной области, где Re = 1000—2000, при условии, что
образец не претерпевает турбулизации при вхождении в капилляр.
205
При неньютоновском течении переход часто происходит при том
же значении Re, что и для ньютоновского течения.
6. С капиллярами сильно различающихся размеров один
образец может дать совершенно разные кривые напряжение — скорость
сдвига. Варьирование длины капилляра, по-видимому, имеет
небольшой эффект; основное влияние оказывает изменение радиуса
(Скотт Блэйр, 1958). Если радиус капилляра уменьшить,
экспериментально определяемая вязкость также снизится. Одно из
объяснений, предложенных для этого феномена, состоит в том, что
уравнения Пуазейля, Букингема — Рейнера и другие выведены путем
интегрирования, основанного на предположении, что сдвигающиеся
слои имеют бесконечно малую толщину. Это предположение не
обосновано, когда частицы в суспензии или капли в эмульсии
относительно велики в сравнении с радиусом капилляра (Дин и Скотт
Блэйр, 1940).
7. В табл. IV. 1 скорость сдвига у стенки капилляра дается
выражением iVJnR^. В действительности это относится только
к ньютоновскому течению, а истинная скорость сдвига выводится
путем умножения AVJnRl на поправочный коэффициент (3 + £>)/4,
где Ъ — угол наклона кривой lg ^VJnr\ — lg PRJ2L (стенка
капилляра) (Рабинович, 1929). Если Ъ остается постоянным в
широкой области напряжений сдвига, достаточно отдельного его
вычисления, но если он меняется с изменением напряжения сдвига,
градиент должен быть определен для нескольких значений
последнего.
Ошибки вискозиметрии
с коаксиальными цилиндрами
При вискозиметрии с коаксиальными цилиндрами следует
учитывать следующие моменты.
1. Исследование усилий, оказываемых образцом на
искривленную поверхность цилиндра, приводит к уравнению для течения
в вискозиметре с коаксиальными цилиндрами (см. табл. IV. 1).
Если внутренний цилиндр полностью погружен, силы действуют
на оба его конца. Когда верхний конец внутреннего цилиндра не
покрыт образцом, эффект, вызываемый действием сил на нижний
конец, может быть оценен путем погружения внутреннего цилиндра
в образец до различных уровней и определения отношения круговая
скорость/угловая скорость. График зависимости этого отношения
от глубины погружения /г.ц является линейным, причем на оси йц
отсекается отрицательный отрезок. Он представляет собой поправку
A/ju, которая должна быть прибавлена к ha в уравнении для
ньютоновского течения (см. табл. IV. 1). При неньютоновском течении
краевой эффект может иметь большее значение. Скорость сдвига
у концов ниже, чем в зазоре между двумя цилиндрами, поэтому
вязкость выше в этих слоях. В этом случае поправка на краевой
эффект должна быть определена для каждого образца при каждом
использованном значении Q.
2^6
2. Краевой эффект увеличится, если в области концов цилиндра
будет создано турбулентное течение. Для ньютоновских вязкостей
больших, чем 1 пз, поправка почти постоянна, но когда вязкость
<С 1 пз, эффект возрастает (Линдсей и Фишер, 1947).
Ошибки вискозиметрии с падающим шариком
В случае вискозиметрии с падающим шариком необходимы
поправки для различных явлений.
1. Для вычисления ньютоновской вязкости по данным,
полученным для шарика, падающего через образец с постоянной
скоростью иш (в см/сек), используется закон Стокса (см. табл. IV. 1).
Если радиус шарика велик в сравнении с радиусом трубки, через
которую он падает, т. е. г ^> R, должно быть применено более
сложное уравнение вследствие торможения, производимого стенкой
трубки. Ладенбург (1907) считает, что в первом приближении
поправка должна относиться к иш, так что
ьш = »ш( 1 + 2,4-^-) (IV.13)
Факсен (1922) указал, что постоянная должна быть равна. 2,1
вместо 2,4. Он видоизменил уравнение (IV. 13) в форму, которая
давала значение вязкости для ньютоновских жидкостей с точностью
в пределах 1% от полученной в капиллярном вискозиметре
% = ЧЯ* [l-2,104 -^- + 2,09 (-^У-0,95 (-£-)'] (IV-14)
где r\st — вязкость, полученная по закону Стокса.
Бэкон (1936) нашел, что уравнение (IV.14) справедливо для
отношений r/R >0,32.
Скотт Блэйр и Остьюзен (1960) показали, что на отдельных
образцах могут быть проведены опыты при использовании очень
маленьких шариков. Материал, не поддающийся сдвигу, становится
пригодным для опытов, если между измерениями осторожно вращать
трубку. Для исследования в широкой области вязкостей применены
нейлоновые шарики. Когда разница плотностей между образцом
и нейлоновым шариком оказывалась незначительной и, таким
образом, возрастала возможность серьезных ошибок, применяли
металлические шарики.
Системы, лишь слегка отклоняющиеся от ньютоновского режима,
могут быть исследованы путем применения шариков различных
радиусов и плотностей (Вильяме и Фулмер, 1938).
2. Одно из предположений при выводе закона Стокса состоит
в том, что трубка, содержащая образец, имеет бесконечную длину.
На практике это, конечно, неосуществимо, поэтому должно быть
учтено влияние, вызываемое двумя концами трубки. Этот эффект
описывается выражением r/hx, где hx — расстояние от шарика до
основания трубки (Ладенбург, 1907; Альтрихтер и Люстиг, 1937).
207
3. Имеется криволинейная зависимость между скоростью
падения шарика (иш) и углом наклона 6В трубки. Флауерс (1914) показал,
что сила сопротивления FB связана с иш уравнением
FB = AFr]vul + BFpo6pil1 (IV.15)
где AF и Вр — константы с размерностями, соответствующими L и L2.
Величина BF приближенно равна пг2/2.
Вязкостное сопротивление Fr\ = AFi\vm дается как разность FB
и инерционного члена BFpulv%1
/■т=4/3пгЗ(рш-робр) g sin^-Bpp^l, (IV.16)
где рш — плотность шарика.
Пьезоэлектрический вискозиметр
Принцип действия ультразвукового вискозиметра иной, чем
у описанных выше. Вязкость связана с коэффициентом затухания
колебаний для ньютоновских материалов соотношением (Рич и Рот,
1953)
4 (РРрбп.,)»
Л = - (IV. 17)
л^рр0бр
где р — коэффициент затухания; рр — плотность металлического
резонатора; бпл — ширина (0,3 см) боковых поверхностей
металлической вибрирующей пластинки; vp — частота резонанса (28 X
А 103 циклов!сек).
Пластинка удерживается умышленно слабо, так что образец
подвергается сдвигу только на ее боковых поверхностях и
некоторый эффект, обусловленный ее краями, незначителен. Кроме того,
когда волны распространяются вдоль длины пластинки, сводится
к минимуму противодействие, вызываемое слишком сильным
отклонением ее от среднего положения.
Эффективная скорость сдвига является функцией частоты
резонанса, амплитуды и глубины сдвигаемого слоя образца,
примыкающего к датчику. На практике достигаются очень высокие скорости
сдвига — порядка 104—103 ceK'i.
Если образец обладает высокой упругостью при сдвиге (Gc),
реакция торможения, данная уравнением (IV. 17), будет
видоизменена в соответствии с выражением
(Робр)
'/2
2РР
(GS + оп!2)
,ai'/«
2
(IV.18)
где со — угловая скорость.
При г\ — 10 пз величина со — 1,8-105, так что Gc должно
превысить — 106 дин/см2, прежде чем окажет влияние на торможение.
208
Пенетрометры
Конус, иглу или шарик, прикрепленные к основанию
металлического стержня, вначале удерживают в неподвижном состоянии
в слабом контакте с поверхностью образца. Когда приводят в
действие выключающий механизм, конус, игла или шарик проникают
в образец с заданной силой в течение заранее определенного времени,
после чего измеряют глубину проникновения. Основные недостатки
этой методики: 1) площадь контакта между проникающим телом и
образцом не остается постоянной в течение опыта; 2) образец
перемещается в направлении, противоположном движению
пенетрометра.
Предельное напряжение сдвига для конического пенетрометра
вычисляют по глубине проникновения с помощью соотношения
(Ребиндер и Семененко, 1949)
Pl=If!L ■ (iv.19)
где Кп — константа, значение которой зависит от свойств образца,
взятого для опыта — обычно приближается к 2; т — вес конуса
и других движущихся частей "пенетрометра; hn — глубина
погружения конического пенетрометра.
Константу пенетрометра определяют из выражения
Кп = — cos2¥nch9n (IV.20)
я
где фп — угол конуса пенетрометра.
Институт нефтяных смазочных средств включил в испытания
модели ASTM5-25 конус малого угла (30°), наложенный на конус
широкого угла (90°). При таком замысле геометрия системы
становится сложной, когда глубина погружения превышает высоту конуса
в 30° и уравнения (IV. 19) и (IV.20) более не применимы. Этот
недостаток преодолевают, используя полированный прямоугольный
конус (Хейтон, 1959). Для длительной службы он должен быть
сделан из алюминия с наконечником из твердой стали.
Исследования с конусами, имеющими различные углы (Самбук
и Найдет, 1959), показали, что
hn\h(cfJ2) = Ks (IV.21)
где Ks — константа.
Воспроизводимость более удовлетворительна, когда hn
ограничена пределами 7,5—20 мм.
Одна из моделей стержневого пенетрометра работает на тех же
принципах, что и конический пенетрометр. Другая модель состоит
из металлического стержня, увенчанного небольшой площадкой.
Основание стержня приводят в контакт с поверхностью образца
и глубину или скорость проникновения определяют при различных
нагрузках площадки. Если нагрузка преобразована в напряжение
14 Заказ 1070
209
Таблица IV .2
Промышленные вискозиметры
Тип вискозиметра
Принцип действия
Применения и ограничения
1. Ротационные
а. Коаксиальные
цилиндры
Стормера
Haake
Rotovicko
Epprecht
(Drage)
Rheomat
Portable
Ferranti
Неподвижный внешний стакан, внутренний
ротор. Возможны другие геометрические
конструкции последнего. Ротор приводится в
движение с помощью грузов и блока. Усилие
изменяется путем применения различных грузов
Фиксированный внешний стакан и
внутренний ротор. Возможны некоторые комбинации
стаканов и роторов различных размеров. Ротор
приводится в движение крутящим усилием
динамометра. Десять основных скоростей — от 3,6
до 582 об/мин с помощью редукционного
механизма могут быть понижены до 1/10 или 1/100
этих значений. Отсутствие других мер
предосторожности против краевых эффектов, кроме
переливания из кольца на прилегающую к
стакану крышку ротора. Некоторые роторы
изготовляют рифлеными для предотвращения
скольжения, другие делают из пластмассы для
исследований при высоких температурах
Фиксированный внешний стакан и внутренний
вращающийся поплавок цилиндрической формы,
но с коническими верхом и низом. Возможны
отдельные комбинации стаканов и роторов с
различными размерами. Вязкое торможение
поплавка измеряется через вращение мотора,
свободно подвешенного на проволоке.
Регистрируется угловое отклонение пружины. Имеются
пятнадцать скоростей
Наружный вращающийся цилиндр приводится
в движение синхронным мотором. Вязкое
торможение внутреннего цилиндра измеряется с
помощью комбинированной пружины со стрел-
Имеются краевые эффекты. Искривляются
линии обтекания потока. Прибор калибруют
по жидкости с известной вязкостью.
Полученные константы прибора включают
коэффициенты для указанных эффектов
В узкой области вязкости доступно
снижение скоростей сдвига до 10~2 сек~1. Можно
измерять ньютоновскую и неньютоновскую
вязкости. Структура восстанавливается при
низкой скорости сдвига после воздействия
высокой. Контроль за температурой
недостаточно удовлетворительный
Имеются краевые эффекты. Конические
концы снижают турбулентные эффекты. Для
многих комбинаций градиент скорости
настолько велик, насколько велика ширина
зазора между поплавком и стаканом.
Поскольку взвешенные системы имеют большой
момент инерции, трудно измерять зависимые
от времени эффекты или предельное значение
Концевой эффект почти исключен.
Торможение у основания внутреннего цилиндра
незначительно. Наблюдается расхождение
между данными вязкости, полученными с различ-
б. Вращающийся вал
Brookfield
Synchro-Lect-
ric
Конус-пластина
Ferranti-
Shirley
реогопиометр
Вейзенберга
Haake Roto-
visko
кой. Имеются девять комбинаций цилиндров
различных размеров. Коробка скоростей с пятью
передачами дает скорости от 1 до 300 об/мин.
Торможение внутреннего цилиндра сведено к
минимуму, так как концы обоих цилиндров
находятся в одной и той же плоскости
Измеряют вязкую силу сцепления на валу,
вращающемся в образце. Вал (имеется до
7 моделей) приводится в движение
посредством бериллиево-медной пружины синхронным
мотором
Вращающийся конус с малым углом и
неподвижная нижняя плоская тарелка. Имеются три
конуса — большой, средний и малый. Вершина
конуса почти касается пластины и образец
срезается в зазоре между ними. Вязкое
торможение иа конусе вызывает вращательный момент
на электромеханическом динамометре.
Требуется меньше, чем 0,5 мл образца
Конус жестко фиксирован, плоский нижний
стол вращается. Предоставляется выбор углов
конуса и диаметров столов. Измеряется
скручивание (т. е. тангенциальное напряжение),
передаваемое конусу, и нормальная сила,
действующая на стол. Два синхронных мотора приводят
в движение коробку передач с 60 скоростями.
Необходим очень малый объем образца. В
вибрационных опытах стол вибрирует около своей
оси и колебательное движение передается
конусу через образец
Вращающийся конус и стационарная плита.
Имеются три конуса из нержавеющей стали.
Скручивающее усилие, развиваемое при
вращении конуса, измеряется динамометром
ными комбинациями цилиндров, когда
исследуются дисперсии с малой вязкостью при
одной и той же скорости сдвига. Не очень
прочный прибор
Форма вала приводит к тому, что
чрезвычайно трудно рассчитать скорость сдвига.
Не пригоден для абсолютных измерений
вязкости. Имеются краевые эффекты,
искривление линий обтекания потока
Конус малого угла обеспечивает
постоянную скорость сдвига по всему образцу.
Пригоден для измерений ньютоновских и
неньютоновских потоков при скоростях сдвига,
превышающих несколько сек_1. Особенно
подходит для измерений при высокой
скорости сдвига. Практически отсутствует
концевой эффект; образец удерживается в зазоре
поверхностным натяжением
Необходима постоянная скорость сдвига
для всего образца. Пригоден для измерений
ньютоновских и неньютоновских течений.
Широкая область скоростей сдвига: 10"* —
18 • 103 сек~1. Для определения упругих и
вязких компонентов может быть использован
колебательный сдвиг
Пригоден для измерений ньютоновских и
неньютоновских течений, в частности, при
очень высоких скоростях сдвига. При этих
условиях возникает опасность фрикционного
теплового эффекта
Продолжение
Тип вискозиметра
Принцип действия
Применения и ограничения
2. Капиллярные
а. Стеклянные
капилляры
U-трубка
Оствальда
Caniion-Fcnskc
Уббедоде
б. С переменным
давлением
lnstron
Bheomelor
Tecline
Printing
Запасное расширение, из которого
фиксированный объем образца течет вниз через
капилляр в приемное расширение, расположенное на
более низком уровне
Резервуар и приемный шарик расположены
па одной и той же вертикальной оси.
Фиксированный объем образца вытекает из резервуара
через капилляр в приемный шарик. Имеется
ряд капилляров разной длины п диаметров
Визкозиметр в виде U-образной трубки с
третьим отводом. Принципы истечения аналогичны
описанным выше. Имеется ряд капилляров
разной длины и диаметров
Образец выдавливается из приемной камеры
через капилляр плунжером, прикрепленным к
подвижному ползуну (крейцкопфу), который
может двигаться с разными скоростями.
Имеется серия капилляров с различными
диаметрами и длиной. Сила, потребная для движения
плунжера при каждой скорости, измеряется с
помощью грузовой ячейки, расположенной на
крейцкопфе
Образец продавливается сжатым воздухом
через горизонтальную капиллярную трубку.
Воздух, вытесняемый при этом, двигает капли
воды вдоль другой горизонтальной трубки и
отмечается время, необходимое для перехода
Малая движущая сила (сила тяжести).
Низкое напряжение сдвига. Пригоден для
измерений ньютоновской вязкости.
Охватывается широкая область вязкостен благодаря
капиллярам различных размеров
Такая конструкция позволяет снизить
ошибки. Пригоден для измерений
ньютоновской вязкости, а ц соединении с внешним
источником давления—для непьютоповской
вязкости
Жидкость, поступающая из капилляра,
течет только по стенкам нижнего шарика,
образуя «взвешенный слой». В результате
пет необходимости использовать для опыта
фиксированный объем образца. Пригоден для
измерений ньютоновской вязкости
Доступна широкая область скоростей сдвига
и напряжений сдвига. Пригоден для
измерений ньютоновской и иепьютоновской вязко-
стей и исследований при высоких скоростях
сдвига
Пригоден для измерений ньютоновских
вязкостен в пределах 0,003—2000 па.
Требуется лишь небольшой объем образца.
Попеременное использование давления и всасывания
воздуха позволяет проделать повторные
опыты с одним и тем же образцом
Бнпгпма
3. С соплом
Рсдвуда, Сэйболта
4. С падающим
шариком Гопплера
5. Пьезоэлектрический
Ultraviscoson
Пенетрометры
игла или конус
Хатчинсона
между двумя делениями. В автоматизированной
модели фотоэлектрические элементы
контролируют электрические приборы для определения
времени операции и записи
Образец выдавливается через капилляр в
приемник сжатым воздухом или другим
источником давления. Скорость течения измеряется
по вытеснению воздуха из приемника, которое
регистрируется с помощью потокомерн.
Скорости течения записываются в широкой области
давлений
Состоят из резервуара, отверстия и приемника.
Измеряется время протекания фиксированного
объема образца через отверстие
Цилиндрическая стеклянная трубка содержит
образец. Учитывается время, необходимое для
того, чтобы стальной шарикоподшипник
прокатился через среднюю градуированную секцию
трубки. Таким образом гарантируется
установление определенной скорости шарика до
момента достижения им тест-секции. Имеется
комплект шести шарикоподшипников разных
диаметров. Если жидкий образец непрозрачен, время
падения шарика может быть определено
электрически
Тонкий листик легированной стали на конце
дитчика вибрирует с ультразвуковой частотой.
В образце вокруг листка создаются волны
ультразвукового сдвига. Счетчик переводит
энергию, потребную для создания этого движения,
в вязкость. Имеется автоматический
компенсатор температуры
Стержень с иглой или конусом падает
вертикально в образец после освобождения из
стационарного положения. Градуированная шкала
регистрирует глубину проникновения. Для
конического пенетрометра рекомендуются гладко-
угольные конусы
Особенно пригоден для исследований при
высоких сдвигах
Время истечения принимается как
произвольная мера вязкостп, хотя они связаны не
простой зависимостью. Не может быть
применен для абсолютных измерений
Затруднительно определение напряжения
сдвига. Пригоден для измерений только
ньютоновской вязкости
Наличие одной скорости сдвига; пригоден
только для ньютоновских вязкостей или для
обнаружения отклонений от фиксированной
вязкости. Снабжен многоаондовым датчиком
для получения нескольких измерений при
повороте переключателя
Форма проникающего тела сложна. Не
предпринимаются попытки теоретической
обработки определений даже ньютоновской
вязкости
сдвига, а проникновение — в градиент скорости, тогда для
неньютоновских материалов кривая будет получаться похожей на
кривую псевдопластической или пластической деформации, как
показано на рис. IV.2. Подобная кривая может быть получена
с помощью конического пенетрометра при использовании конусов
различного веса.
Общий обзор вискозиметров
В табл. IV.2 приведены характеристики вискозиметров основных
типов, в общих чертах описаны их главные рабочие части,
способ действия и недостатки.
Вискозиметры с коаксиальными цилиндрами и типа конус-
пластина особенно пригодны для изучения жидких и
полутвердых эмульсий, так как они дают возможность изменять в широкой
области скорости сдвига вплоть до очень малых значений.
Капиллярные вискозиметры более подходят для изучения высоких
скоростей сдвига. Они не чувствительны к краевым эффектам
и температурным флуктуациям, вызванным распространением
тепла, не наблюдается в них и эффект Вейзенберга.
В вискозиметре с коаксиальными цилиндрами вязкоэластичный
материал может подниматься по внутреннему вращающемуся
цилиндру благодаря тенденции течь в направлении, нормальном к
направлению напряжения сдвига. Когда это происходит, объем образца
в зазоре между двумя цилиндрами снижается ниже требуемого
уровня и в вычисления вязкости вносятся ошибки. Возможно,
основным преимуществом капиллярного прибора является то, что
путем подбора капилляров с подходящими размерами может быть
исследована более широкая область консистенций, чем в
вискозиметре с коаксиальными цилиндрами. С последними можно изучать
концентрированные эмульсии с густой консистенцией только тогда,
когда имеется широкий зазор между двумя цилиндрами. Это
нарушает постоянство условий сдвига.
Для многих практических целей наиболее пригоден прибор
Хааке и Ротовиско, так как он включает два типа вискозиметров —
коаксиальные цилиндры и конус-пластина. Реогониометр
Вейзенберга удобен для измерения как вращательного, так и
колебательного сдвига. Колебательный сдвиг представляет особый интерес
при изучении структуры эмульсий, ибо малые амплитуды вызывают
меньшие повреждения структуры, чем вращение.
Исследование стационарной структуры эмульсий
по режиму их ползучести
Реологические исследования не ограничиваются измерениями
вязкости при слабом сдвиге. Большинство эмульсий, кроме очень
разбавленных, проявляет вязкоэластичные свойства, обусловленные
214
флокуляцией капель. В начальной стадии сдвига они ведут себя
подобно твердому телу, а затем обнаруживают свойства
жидкостей «в том смысле, что работа деформации сдвига не сохраняется
полностью, как в твердых телах, и не рассеивается полностью,
как в жидкостях» (Фредриксон, 1964).
Исследования при этих условиях могут дать другие
реологические параметры, помимо вязкости, которые можно использовать
для характеристики эмульсии.
Для такого рода исследований
пригоден вискозиметр с коаксиальными
цилиндрами. В модели прибора,
примененной в лаборатории автора
(рис. IV.5), поверхность обоих
цилиндров снабжена мелким рифлением для
предохранения образца от скольжения
вдоль стенок. Наружный цилиндр
удерживается неподвижно, в то время
как внутренний, подвешенный за
крутильную головку на тонкой
скручивающейся проволоке, медленно
вращается с помощью устройства блок-
груз *.
Угловое вращение внутреннего
цилиндра измеряют оптически по
отражению луча света, который
отбрасывается лампой через зеркало,
прикрепленное к торзионной проволоке,
на градуированную шкалу. Отсчеты
берут через короткие интервалы
времени.
Напряжение, возникающее в
зазоре между цилиндрами, определяют
как
Рис. IV.5. Вискозиметр с
коаксиальными цилиндрами
для исследований ползучести
эмульсий.
Т„
R<,—Ri
(IV.22)
где R1 и R2 — радиусы внутреннего и внешнего цилиндров,
соответственно; соц — угловое вращение внутреннего цилиндра.
Приложенное сдвигающее усилие
пр n(R! + R2)h0
(IV.23)
* В более поздних моделях смещение во времени регистрируется
автоматически с использованием преобразовательных датчика, измерителя и X — Y-pe-
гистратора.
215
где тг — вес груза, действующего через блок диаметра D6n;
h0 — высота, до которой доходит образец в вискозиметре.
Деформация ползучести
/(т) =
(n2 — Ri)mrgD6n
-К
(IV. 24)
так как только ооц и тг являются переменными
. Ползучесть
I Восстановление
В , Lj.
"Jo
Е
Рис. IV.6- Модель кривой ползучести.
Детальный реологический анализ может быть проведен способом,
при котором деформация ползучести изменяется со временем,
рис. IV.6 иллюстрирует вид кривой деформации ползучести для
вязкоэластичного режима. Она может быть разделена на три
характерных области.
1. Область мгновенной упругой деформации /0 (область А В
кривой на рис. IV.6), в которой связи между флокулированными
каплями в эмульсии упруго растягиваются. Если напряжение сдвига
216
снимается, наблюдается полное восстановление связей. Величина J0
дается как
J* = -jr = lTL (IV.25)
где Е0 — модуль мгновенной упругости; W0 — напряжение в области
АВ кривой ползучести.
2. Область запаздывающей упругой деформации Jx (область ВС
кривой на рис. IV.6), в которой связи разрушаются и вновь
формируются
/, = ^- = ^1 = ^(1-^4 (IV.26)
т
" Пр
где Ех — модуль запаздывающей упругости; W^ — напряжение в
области ВС кривой ползучести; /ср — средняя деформация всех
включенных элементов; тср — время среднего запаздывания, равное
^срЛср (здесь Чср — средняя вязкость, обусловленная
запаздывающей упругостью).
Не все связи между каплями разрушаются и формируются
вновь с одной п той же скоростью, поэтому, если желательна более
детальная информация, величина /ср должна быть заменена спектром
i деформаций:
л=2'/(1—-т/т')
(IV.27)
3. Область ньютоновской деформации JN (область CD кривой
на рис. IV.6). Когда связи разорваны, т. е. время, необходимое для
их восстановления, больше, чем период опыта, отдельные капли
свободно перемещаются одна относительно другой. Величина JN
пропорциональна времени нагрузки:
'Ijv *пр
где r],v — ньютоновская вязкость; Ws — напряжение в области CD
кривой ползучести.
Полная деформация при ползучести
/(т) = /0 + /т + / =/0+V/,. (l-<r^')+-f- <IV-29>
г
или в ее более простой форме
/(т) = /0 + /ср (l ~е"'~ч) +~ (IV-30)
'Ijv
217
Когда т -> О
J(r) = J0 (IV.31)
Когда х -> оо
^(T) = /0 + 2/' + /iv (IV-32)
г
Уравнение (IV.30) может быть представлено в виде:
т т , /—-МтО + ^о + ^ср + т/чср \
-~ = -:г^=1п т— (IV-33)
lcp 'срЧср \ Jcp /
На графике зависимости правой части уравнения (IV.33) от х
наклон линии представляет собой — 1/^Срт1ср' откуда могут быть
вычислены г)ср и тср, поскольку Jx известно.
Когда требуется более детальное описание механизма
запаздывающей упругости, уравнение (IV.27) преобразуется следующим
образом (Инокучи, 1955).
Пусть
■2*-
-о— UV.34)
^пр
а также
Q = '%Jie ''''' плп In Q = In Jt -т/тг (IV.35)
I
Таким образом, Q представляет собой отрезок между
экстраполированной линейной частью кривой ползучести и криволинейным
участком в некоторое время т.
Зависимость In Q — т дает прямую линию при больших
значениях т, из которой могут быть вычислены время отдельного
запаздывания (х^) и деформация ползучести /1.
Эти значения подставляют в уравнение (IV.27). Если оно не точно
описывает форму экспериментальной кривой, следует построить
зависимость In (Q — J\e~%IXi) — т, чтобы определить значение времени
второго запаздывания (т2) и второй деформации ползучести (/2).
Этот график также должен быть линейным при больших значениях т
и условии, что хг 1>т2. Подобно этому, если оказывается
необходимым, может быть определено время третьего запаздывания (т3) путем
построения зависимости In (Q — /1e~x/'Cl — J2e T/T2) от т.
Этот прием повторяют до тех пор, пока не будет получено
достаточно значений периодов запаздывания и деформации, чтобы с
помощью уравнения (IV.27) адекватно представить вид последней.
Проиллюстрировать применение этого метода для анализа
запаздывающей упругости можно на примере исследования кривой ползу-
218
чести эмульсии В/М, показанной на рис. IV.7. При низких
напряжениях сдвига режим ползучести отражается линейной зависимостью,
т. е. отношение деформация/напряжение является постоянной
величиной безотносительно к напряжению.
/0 = 92,0-иГ* см^/дин, так что E0=i№ дин/см*
10-60
% = "
172-10"*
= 3,5-10-4 пз
На рис. IV.8 представлена зависимость In Q — т. Отрезок на оси
ординат дает: In Jх = —4,46, откуда J x = 116 • 10~4 и Ег = 86 дин/см2.
Рис. IV.7. Кривая ползучести эмуль- Рис. IV.8. График зависимости In Q
сии В/М (50 вес.% + 50 вес.%). от т (экспериментальные данные).
Наклон графика представляет собой 1/Tlt таким образом:
0,76 • 60
1,53
-=30,0 сек
Так как тх = /imi» to 4i = 2,586 пз.
Зависимость на рис. IV.8 является линейной даже при низких
значениях т. Деформация ползучести этой эмульсии определяется
уравнением:
ЧЧт)
= 92,0-10-4+116-10-* (1-е~'с/30)-
3,5 • 10_4
(IV.36)
Механические модели режима ползучести
Действие сдвига на эмульсию во времени может быть, по крайней
мере, качественно представлено механической моделью. Применяют
также электрические модели, но настоящее обсуждение будет касаться
первого типа. Основные элементы механической модели:'!) спиральная
219
to
2'.
JL
пружина, действие которой подчиняется закону Гука; 2)
амортизатор, состоящий из цилиндрического контейнера с ньютоновской
жидкостью, смонтированного с плунжером; последний имитирует
ньютоновское течение. Растяжение пружины (модуль упругости Е0)
играет роль сдвига и пропорционально приложенной силе,
символизирующей напряжение сдвига. Скорость растяжения в амортизаторе
пропорциональна приложенной силе.
Фрикционная константа
(коэффициент трения) амортизатора
соответствует ньютоновской
вязкости Г|у.
Пружина вместе с амортизатором
(рис. IV.9, а) составляет максвеллов-
ское тело. Когда приложено
напряжение Рпр, напряжение в области
мгновенной упругой деформации
в амортизаторе (Ч1*,-) и в пружине
идентичны и аддитивны. Под
действием напряжения Pnv/E0 пружина
растягивается мгновенно, в то время
как амортизатор удлиняется
постепенно со скоростью -Рпр/г|л- = dWi/dx.
Пружина и амортизатор при
параллельном соединении (рис.
IV.9, б) образуют элемент
Кельвина — Войгта, в котором они
испытывают одинаковые деформации.
Амортизатор оказывает тормозящее
сопротивление упругой деформации
пружины.
В этом случае модуль упругости
пружины Ех, вязкость
амортизатора — т]ср. Реакция запаздывающей
упругости при моделировании ее
в виде тела Кельвина — Войгта
дается уравнением
-Из
Рис. IV.9.
Части механической
модели:
а — элемент Максвелла; б — элемент
Кельвина — Войгта; в — модель
режима ползучести, составленная из
четырех элементов; 1,2 — пружины; 3, 4 —
амортизаторы.
ИЛИ
*т =
мер —J-- -f-^V-^cp— Рпр
^ср (1-*-Т^М =Рпр/сР (1 -е"Т/Ч
(IV. 37)
(IV. 38)
Уравнение (IV.36) представлено моделью (рис. IV.9, б), состоящей
из элемента Максвелла и элемента Кельвина — Войгта, соединенных
последовательно. Выражение, включающее полную деформацию,
имеет вид:
¥(т) =Pnp/0 + Pnp/cp(l-eT/Tcp)-f-^- (IV.39)
%
220
Если на механическую модель внезапно накладывается
напряжение сдвига, пружина 1 мгновенно растягивается. Это сопровождается
растяжением пружины 2 под действием запаздывающей упругости
и постепенным вытягиванием амортизатора 4. Когда напряжение
сдвига устраняется, пружина 1 мгновенно сжимается до своей
первоначальной длины, но пружина 2 делает это медленнее из-за эффекта
—щ_ торможения, вызываемого
амортизатором 3. На амортизаторе 4
ovvH I 1—I-° удаление напряжения сдвига
I—V4AH
Г
Мгновенно) упруго»
деформация
o-vv/W—
oVWnA-
«WVW4—
9
-^Н
Деформация
запаздывающей упругости
9
-^-с
■^h*
9
Ч
Ньютоновское
течение
L....
Рис. IV.10. Простая механическая Рис. IV.И. Детализованная меха-
модель, воспроизводящая зависимость ническая модель вязкоэластичного
деформации от времени. режима эмульсии В/М для
линейного участка кривой ползучести.
не сказывается. Реакция этой модели на приложение напряжения
и его последующее устранение показана в виде диаграммы на
рис. IV.10.
Для полной механической аналогии упруго-вязкого режима,
представленного линейным уравнением (IV. 29), модель должна
была бы содержать бесконечное число последовательных элементов
Кельвина — Войгта, имитирующих деформацию запаздывающей
упругости (рис. IV.И).
Измерение динамических реологических свойств
Методики динамических измерений особенно полезны при
изучении упруго-вязких и других структурированных систем. Когда
применяют колебания малых амплитуд, структура изменяется в меньшей
221
степени, чем при использовании слабых вращательных сдвигов,
и полученная информация приближается к таковой в состоянии
нулевого сдвига. Если амплитуду увеличивают, структура разрушается
в большей степени, чем при увеличении вращательного сдвига.
Многие структурированные системы проявляют линейную вязко-
эластичную реакцию, т. е. режим, который при очень малом
напряжении характеризуется линейным дифференциальным уравнением
с постоянными коэффициентами между деформациями сдвига и
напряжениями сдвига и их производными по времени (Вейзенберг, 1964).
Для таких систем динамические измерения имеют особые
преимущества.
Модифицировав соответствующим образом вискозиметр с
коаксиальными цилиндрами, можно дополнительно (или вместо) к
вращательному движению добиться колебательного движения внутреннего
цилиндра. Для этого наружному цилиндру сообщают
синусоидальные колебания посредством металлического стержня,
присоединенного к его основанию, а также к мотору переменной скорости с
помощью узла эксцентрика (Ван Вазер и др., 1963). Внутренний и
наружный цилиндры колеблются с одинаковой частотой, однако имеется
разница фаз, так как первый подвергается скручивающему усилию,
являющемуся результатом вязкого торможения образца между
цилиндрами.
Движение внутреннего цилиндра определяют на основе принципа
торзионного маятника (Моррисон и др., 1955; Шульберг и др.,
1959)
<A'+y+/>^+(ir+4-^+hr+H6«=G<- (IV-40)
где / — момент инерции маятника; X, Y — инструментальные
постоянные; Н — фактор геометрической формы; Кт — коэффициент
торможения прибора; Кж — коэффициент жесткости прибора; т]д —
динамическая вязкость; Ел — динамический модуль; 6Ц — угловое
отклонение; Gt — вращающее усилие на внутреннем цилиндре,
вызываемое единицей тока, проходящего через вращающий мотор;
i — ток, идущий через мотор.
Уравнение (IV.40) справедливо, когда длина поперечных волн
в образце велика в сравнении с размерами образца. Когда величина /
очень мала в сравнении с (X + Y) и i изменяется синусоидально
Чд = Я{[г (/,//„) sin 9]/шо-Ят} (IV-41>
£Д = Я IZ (/,//„) cos 9--Яж + (Х +У) eoj] (IV .42)
где /г и /е — амплитуды тока i и углового отклонения 0 ,
соответственно; Z — постоянная прибора; q — отставание фаз между
отклонением маятника и током; со о — угловая скорость наружного
цилиндра.
Угловое отклонение внутреннего цилиндра изменяется путем
изменения амплитуды активирующего тока.
Фактор формы Н и запаздывание фазы q даются выражениями:
9 = cos-i [{fl + fl-f*)/2fif<A (IV.44)
где fq — амплитуда разницы фаз между током и отклонением
маятника.
Такано (1964) сравнил реологические данные, полученные при
простом и колебательном сдвигах на одних и тех же суспензиях.
Он нашел, что для псевдопластичных систем кажущаяся вязкость
при низких скоростях сдвига подобна динамической вязкости,
измеряемой при низких частотах. Для пластичных систем, однако,
наблюдались расхождения между двумя рядами данных, причем
кажущаяся вязкость при низких скоростях сдвига иногда была выше,
чем динамическая вязкость при низких частотах. Эти расхождения
приписывались различным путям, которыми разрушались и
восстанавливались сетчатые структуры флокулированных частиц под
влиянием простого и колебательного сдвига: «. . .зависимость
кажущейся вязкости от скорости сдвига связана со структурными
изменениями сетчатой системы, вызванными сдвигающими силами, в то
время как частотная зависимость динамической вязкости проистекает
главным образом от релаксации сетчатых структур, образованных
частицами в среде».
В этих опытах вместо обычной торзионной проволоки для
подвешивания внутреннего цилиндра вискозиметра использован .мотор,
создающий момент вращения. Таким образом торзионную константу
этого цилиндра можно было изменять в широкой области путем
изменения тока, питающего вращающий мотор (Комбе и Такано, 1963).
Угловое отклонение внутреннего цилиндра измерялось специальным
детекторным узлом и записывалось электрическим регистратором.
Подобные опыты с колебанием системы могут быть проведены
с помощью модифицированного вискозиметра конус-пластина или
реогониометра Вейзенберга.
При изучении свойств эмульсий, в которых капли флокулированы,
угловое отклонение должно оставаться небольшим, чтобы
коагулированная структура радикально не изменялась.
АНАЛИЗ НЕНЬЮТОНОВСКОЙ ВЯЗКОСТИ
Общее математическое рассмотрение
Делалось много попыток математического определения вида
кривой Рс — vc для пластического течения (см. рис. IV.2). Бингам
(1922) предложил уравнение для идеализированной системы, в
которой зависимость Рс — vc линейна на всем протяжении, т. е. если
течение не начинается до тех пор, пока не будет превзойдено Р°с
Vc=A-(Pc-P°c) (IV.45)
223
гДе Лот ~ вязкость при такой скорости сдвига, дальнейшее
увеличение которой не приводит к изменению вязкости.
Оствальд (1925) и де Вель (1925) независимо друг от друга
модифицировали уравнение Пуазейля с тем, чтобы распространить его
применимость на пластическое течение. Оба пришли к одному результату
или в более детальной форме
где ns — характеристика структуры, развитой внутри образца,
равная единице для ньютоновской жидкости; yv — некоторая
предельная величина; г\' — вязкость.
Против уравнения (IV.46) выдвинуто много возражений. Для
некоторых систем г)' не является постоянной — наблюдаются
флуктуации с колебаниями в ns, которые могут происходить от изменений
Рс. Кроме того, г)' не имеет строгой размерности вязкости.
Гершель и Балкли (1926) и Скотт (1931) вывели степенные
уравнения, напоминающие уравнение (IV.46):
Vc^^—{Pc-Vv)"s (IV. 47)
С/геиенное уравнение «. . .не является единым законом для
различных веществ, т. е. различные значения ns дают различные
численные результаты: перед нами столько же различных правил, как и
значений п, или для каждого вещества имеется свой закон. Их можно
назвать «индивидуальными законами», если подобные существуют».
Тем не менее степенные уравнения являются, по-видимому,
полезными эмпирическими формулами. Любой график Рс — vc с
небольшой криволинейностью даст приблизительно прямую линию
при нанесении на двойную логарифмическую шкалу. Для таких
систем не оправдывается предположение, что уравнение степенного
закона должно иметь физический смысл.
Другие системы, например концентрированные эмульсии М/В,
дают прямую линию в весьма широкой области условий и, возможно,
что для них степенное уравнение имеет не только эмпирическое
значение (Скотт Блэйр, 1965). Доказательством служит следующее.
Если j — доля связей, разрывающихся при данном напряжении Рс,
тогда
d) bs
(IV. 48)
dPc Рс
где bs — константа.
Это выражение отрицательно, потому что i/bs увеличивается
с возрастанием Рс. После интегрирования получим:
l = K„-bslnPc (IV.49)
где Ки — константа интегрирования.
224
При In Pc = О, т. е. Рс = 1 дин/см2, полагаем / = jlt тогда
Ки = ь и
bslaPc=j1-~j (IV.50)
Теперь рассмотрим влияние скорости сдвига на разрушение
связей
drc vc
что после интегрирования дает
/01п!>с = /а-/ (IV.52)
где 70 — доля связей, разрушающихся при некоторой скорости сдвига;
/ 2 — значение / при vc = 1 сек'1.
Из уравнений (IV.49) и (IV.52) имеем
is In Рс — /01 n ус = /i — /2
откуда
pbS/!v
Uc= e(h-i,)/iv
Уравнения (IV.48) и (IV.51) не могут выполняться при низких
значениях Рс и vc. При этих условиях флокуляция преобладает над
дефлокуляцией, т. е. связи между частицами не разрываются,
а вместо этого или происходит дополнительная флокуляция и
образуются новые связи, или флокулированные частицы сдвигаются ближе
друг к другу и образуют более компактные структуры. При
критической верхней скорости сдвига все связи будут разрушены, так что
уравнение (IV.54) не будет справедливо за этой точкой. Тем не менее
оно должно иметь силу в широкой области условий.
Вильямсон (1929) ограничил свое внимание изучением
псевдопластического течения. Он разработал графический прием для
вычисления по кривой Рс — vc полной мощности Pcvc, необходимой для
линейного течения. Приводится детальное описание обработки данных,
так как идеи Вильямсона были впоследствии развиты другими
исследователями.
При псевдопластическом течении часть сдвигающего усилия
необходима для разрыва агрегатов частиц, другая часть используется
на то, чтобы вызвать вязкое течение при более высоких скоростях
сдвига.
На рис. IV. 12 приведен графический анализ кривой
псевдопластического течения по Вильямсону. Кривая OGCB является кривой
псевдопластического течения. Область RDOT определяет мощность,
необходимую для преодоления пластического сопротивления при
ис = В, а область BRDK — мощность, расходуемую на вязкую
деформацию. Из точки G, ниже которой кривая прямолинейна,
проводят линию GF параллельно BD. Затем опускают перпендикуляры
из точек G и F. Область GMFE представляет собой мощность,
потребляемую для преодоления вязкостного сопротивления Pnvc, а область
(IV.53)
(IV.54)
15 Заказ 107U
225
XMFO — мощность, используемую на преодоление пластического
сопротивления Рпл vc.
Подобным образом берут несколько других точек на кривой
течения ниже ее линейной области и процедуру повторяют. Таким путем
получают кривую OMQ, которая дает напряжение сдвига,
необходимое для преодоления пластического сопротивления при различной.
скорости сдвига.
Выражение для
полной мощности будет иметь
вид
PcVc^Pn^c + P^c (IV.55)
где Рпл = PR/2L
(пластическое сопротивление);
Рл = PR/2L (вязкое
течение).
Уравнение (IV.55)
может быть написано в форме
и
X
т
R
1
1
/
M/G
0 С
у/
/ /
/
/
/
/
/
в
Реп
с
Cw + i
-+ЧС
О F E D К (IV.50)
с где С\у — мера кривизны
Рис. IV.12. Графический анализ кривой коивои ОМО; Р? — вели-
псевдопластического течения по Вильямсону. ' ™f ^
чина Рпл при vc -> оо.
При Р£° = 0 величины Рс и ус связаны линейно и будет
справедливо уравнение Пуазейля. Если Cw = 0, течение определяется
уравнением (IV.45). Обработка по Вильямсону дает удовлетворительные
результаты только тогда, когда линейный и криволинейный участки
кривой Рс — vc хорошо различаются.
Теории неньготоновского течения
Достаточно удовлетворительной теории для объяснения
неньютоновского течения пока еще не существует. Большинство теорий
предлагает объяснения, основанные на реакции флокуляция — дефлоку-
ляция, которая регулирует рост агрегатов при низких скоростях
сдвига. На эту реакцию влияют как броуновское движение, так
и сдвиг, причем значение первого фактора уменьшается, когда
возрастает размер частиц и увеличивается скорость сдвига.
Для выявления влияния скорости сдвига обычно вводят
коэффициент пропорциональности, но его точное значение никогда
неизвестно. Кроме того, если агрегаты имеют предпочтительно
сферическую конфигурацию, а не цепеподобную, которую обычно принимают
при выводах^ влияние сдвига должно было бы отражаться скорее
экспоненциальной зависимостью от скорости сдвига, чем
произведением скорости сдвига и коэффициента пропорциональности. Влияние
сдвига на размер агрегатов часто игнорируют, как и возможность
226
влияния размера агрегатов на константы скорости флокуляции
и дефлокуляции.
Только две теории предлагают альтернативные подходы. Одна
из них, основанная на теории абсолютных скоростей процессов,
предполагает наличие структур, составленных из определенных элементов
потока. Другая теория, основанная на кинетике реакций, не делает
предположений, касающихся структуры агрегатов, кроме тех, что
структурные изменения происходят при наложении сдвига. Ни одна
из теорий не может быть легко применена к экспериментальным
данным.
«Импульсная» теория псевдопластичности
Гудив распространил на псевдопластичность концепцию о том,
что складываются два независимых эффекта. Он полагал, что на
режим течения концентрированных эмульсий и дисперсий влияют
ньютоновский эффект, при котором сдвигающая сила
пропорциональна скорости сдвига, и тиксотропный эффект, при котором
сдвигающая сила постоянна независимо от скорости сдвига. Между
частицами, находящимися в контакте, устанавливаются связи; во время
сдвига эти связи растягиваются, искривляются, рвутся и
восстанавливаются. Этот процесс сопровождается переносом количества
движения (кинетической энергии) от движущегося слоя к соседнему
более медленно движущемуся слою
^- = r]OCTi>c + pT (iv.57)
или после деления всех членов на vc
Л00 = г1ост-г-РтК (IV.58)
где F — сила; S — площадь; т|ост — остаточная вязкость; р\ —
коэффициент тиксотропии, который равен кинетической энергии,
передаваемой от одного слоя другому взаимодействием частиц.
Величины т)ост и £>г являются константами.
Ньютоновский и тиксотропный эффекты, выраженные
уравнениями (IV.57) и (IV.58), выведены следующим образом. Сила FT,
противодействующая искажению связей в тиксотропной составляющей,
дается в виде
FT = E0x (IV.59)
где Eq — модуль упругости; х — растяжение связей (х «« Ifvct);
If — расстояние, на котором действует сила F.
Каждая тиксотропная связь имеет среднее время жизни
кр
(IV.60)
E0lFvc
где FKp — критическое значение FT, при котором связь разрушается.
15* 227
Импульс, передаваемый при сдвиге от одного подвижного слоя
к соседнему более медленному слою, обратно пропорционален vc:
F -
S*P fT E0lFvcx% F^p
Frdx=\j E0lvcxdx = 2~~= 2E0l vc (IV-61)
о о
Полная сила между двумя плоскостями является произведением
среднего импульса FKp/2 и числа импульсов в секунду NCBlF/xKp
FKpNCBlF
2ткр
(IV. 62)
где iVCB — число связей на 1 ом3; ткр — критическое время,
необходимое для разрушения связей.
Предполагается, что продолжительность импульса в тиксотроп-
ной составляющей течения контролируется скоростью сдвига. Однако,
если энергия, необходимая для разрыва связи, много меньше кТ,
тогда связи разрушаются главным образом под влиянием тепловой
энергии и превалирует ньютоновское течение. При этих условиях
вероятность разрыва связей в секунду выражается как
-L. = Ve-E'hT (TV.63)
где t.y — среднее время жизни ньютоновской связи; v — частота,
с которой происходит перераспределение энергии между связью
и окружающей средой; Е — энергия, необходимая для разрыва связи.
Когда сдвиг действует на связь только в течение времени тЛ-,
импульс 1СЛ становится равным
I^^^L (IV.64)
а полная сила FN вычисляется как в уравнении (IV.62):
NcbE0Ifvc ,TV й-.
Fn. N = ^ ( 0)
Таким образом Fn_N прямо пропорциональна скорости сдвига.
Уравнение (IV. 65) для ньютоновской составляющей течения
определяется твердо установленной экспоненциальной зависимостью между
вязкостью и температурой.
Общая действующая сила F в уравнении (IV.57) является суммой
^п. т И г п д-.
Образование и разрушение связей происходит под влиянием как
сдвига, так и броуновского движения. Гудив (1939) полагал, что
эффектом, вызванным последним фактором, можно пренебречь в
простых тиксотропных системах, в системах со слабым броуновским
движением и при высоких скоростях сдвига. Джиллеспи (1960а, Ь) обобщил
эти выводы, рассмотрев эффекты, вызываемые броуновским
движением. Однако его теория применима лишь для не очень
концентрированных эмульсий (с объемной долей Ф <0,3), так как основана на
228
лимитирующих предположениях. Скорость образования каплей
связи определялась как
,,ТК- = я1Лгк_я2Лгсв ср (IV.66)
dN_
где NK — число капель в свежей эмульсии; ЛГСВ_ ср — среднее число
связей на каплю; Кг и К2 — константы скорости образования и
разрушения связи, соответственно.
Так как связи образуются под влиянием и сдвига, и броуновского
движения, то
K1 = (Ks + Kivc)N^e (IV.67)
где А'3 и Ktvc — константы скорости флокуляции, вызванной
броуновским движением и сдвигом, соответственно; 7VcB — число связей,
установившихся между двумя контактирующими каплями.
Аналогично
K2 = K& + K6vc (IV.68)
где К& и K6vc — константы скорости дефлокуляции от броуновского
движения и сдвига, соответственно.
При равновесии dNCB cp/dx = 0 и
Л^св. сР = -%^ (IV.69)
В каждой связи участвуют две капли, поэтому 7VCB в уравнении
(IV.62) может быть вычислено:
K,Ni
NCB^^X (1V.70)
Из уравнений (IV.62) и (IV.67), полагая ткр = 1/К2, получаем:
^•т=—щ— <IV-71>
Возвращаясь к уравнению (IV.67), отметим, что для разбавленных
систем, в которых любые две капли имеют контакт не более чем
в одной точке, используется определение К3, данное Смолу ховским
(1916а, Ь, 1917)
K3=8nMrmW (iv.72)
где гэфф — эффективный радиус контакта; Д — коэффициент
диффузии для двух капель; W — вероятность флокуляции, обоснованная
рассмотрением сил взаимодействия между каплями:
КА=—f?- (IV.73)
Можно показать, что Кв в уравнении (IV.68) дается выражением
#6 = -^ <IV-74)
'кр
где /ср — среднее значение 1р; 1кр — критическое разделяющее
расстояние, при котором происходит разрушение в отсутствие теплового
влияния.
229
Решая совместно уравнения (IV.57), (IV.67), (IV.71) и (IV.74),
получим
где полная энергия связи между двумя каплями равна 0,5Е01^РАТ'СВ.
При высоких скоростях сдвига эффект, вызываемый броуновским
движением, отсутствует:
F EollpN'^NlKiVz
Т=^1'^ iKb/Ke + 2vc <IV-76>
Значения некоторых параметров можно теперь определить
графически. Уравнение (IV.76), которое очень напоминает уравнение (IV.56)
Вильямсона, после деления на ис будет иметь вид:
EoI^N'^NlKt
ЧЭФФ-ЧОО+ AK5/Ke + 2vc (П-77)
где 1^фф — эффективная вязкость.
При высоких значениях vc график зависимости г)Эфф — l/vc
должен быть линейным. Отрезок, отсекаемый на оси ординат, равен
Лэфф- Уравнение (IV.76) можно представить как
1 iKb/Ke + 2vc
Мэфф-Моо E0llpN'cbNlKi
(IV. 78)
и график зависимости 1/(т]Эфф — ^оо) °т ис выражается прямой
линией, наклон которой дает EollpNcsN^Ki, а отсекаемый отрезок —
„ ,„ .,, £„,. . Константы Къ и К6 нельзя определить непосредственно
£0'крЛ СЕЛ'кЛ4
по отсекаемому отрезку и приходится обращаться к указанным выше
приемам. Для частиц латекса с объемными долями в пределах
0,13—0,28 полное притяжение между двумя частицами наблюдалось
при —40 кТ.
Имеются сообщения о немногих экспериментальных проверках
уравнения (IV.56). Шенграу и др. (1961) модифицировали его с тем,
чтобы получить более удовлетворительное совпадение с
экспериментальными данными:
или „
1 = 1 . / cw \ / 1 N
Pc-r]VQ Р£° \ Р™ )\ L'c -
Зависимость 1/(Рс — Лоо^с) от 1/ус должна быть линейна.
Практически, однако, такого случая не наблюдалось ни при низких, ни при
высоких скоростях сдвига, поскольку экспериментальные данные
были ниже теоретических значений.
Для характеристики этой области графика введено
дополнительное уравнение для пластического течения:
Pc»=P? + r\co»c (IV.80)
230
Предполагая, что вязкоэластичное сопротивление, обратно
пропорциональное скорости сдвига, уменьшается пропорционально
остаточному сопротивлению, получаем
где Ъ1 — коэффициент пропорциональности.
Интегрирование дает
Рс1 = Ьсопре"Ь'°с "" (IV.82)
где Ьсопр — коэффициент вязкоэластичного сопротивления.
Решение уравнений (IV.80) и (IV.82) приводит к выражению:
ркорр = рс 2 _рс 1 = рсо + ^ _Ьсопре-Ь,ас (Iv.83)
Предельное значение дается как
Р*°П=р?-Ьеоир
так как е~й,0с -> 1 при vc ->■ 0.
Уравнение (IV.83) имеет ограниченное применение, потому что Ьх
неизвестно. Шенграу и др. (1961) выбрали произвольное значение
0,001, так как е~ *"с «. . .должно иметь удобную величину».
Теория, основанная на кинетике агрегации
Ван ден Темпель (1963) и де Врис (1963) также рассматривали
влияние процесса флокуляция — дефлокуляция на вязкость
эмульсий. Однако в отличие от Гудива, Джиллеспи и других
исследователей они связывали это влияние с кинетикой агрегации капель, а не
со скоростью образования связей. Согласно Смолу ховскому, частота
столкновений между агрегатами различного размера, вызванных
броуновским движением, выражается как
К и = AnMijRijNiNj (IV.84)
где Rt: — радиус взаимодействия между агрегатами, содержащими
i капель, и агрегатами с числом капель j; Nt и N- — число агрегатов
типа i и j на 1 см3 эмульсии, соответственно.
Когда агрегаты типа i и j сталкиваются, они образуют агрегаты
типа к; столкновение последних с агрегатами других размеров
вызывает дефлокуляцию. Таким образом, скорость образования агрегатов
типа к можно представить выражением:
=* Т 2 in^4RHNiNi -Nk 2 ^iRiNi (IV.85)
dNk
di _
231
Здесь R{- примерно равно диаметру капель/), если эмульсия
монодисперсна, или среднему их диаметру Z)cp, если распределение не
является широким и
и Злт|с/)
где Пс — вязкость непрерывной среды.
Таким образом:
dNk
2кТ
dx
Зл11с
^ Л-,Л^-2Л^2Л'«
(IV.86)
\i+j=k
К этому необходимо добавить влияние градиента скорости на оба
процесса:
12 !-(й'/)з,'свд-^ 2 4 (адз''сЛ''
(IV.87)
i+j=k
Ван ден Темпель (1963) сделал дальнейший шаг по сравнению
с де Врисом (1963) и ввел два дополнительных члена, учитывающих
разрушение агрегатов под влиянием градиента скорости. Он полагал,
что агрегаты принимают форму длинных цепочек, но считал, что это
предположение справедливо только для агрегатов, не содержащих
большого количества капель. Согласно ван ден Темпелю, два эффекта,
характеризуемые добавочными членами, прямо пропорциональны
скорости сдвига. Дополнительными членами являются
2 2Nm (Л'пр'-'с) -Л^ (Л"пргс) (к - 1)
(IV..
m=k+l
где ^пр — коэффициент пропорциональности.
При этом предполагается, что агрегат типа m в форме длинной
цепочки содержит (пг — 1) связей между каплями. Для эмульсий
В/М, где капли флокулируют намного быстрее, чем в эмульсиях
М/В, и образуют более сложные агрегаты, предположение,
касающееся структуры агрегатов, не является справедливым.
В условиях равновесия уравнения (IV.86) —(IV.88) могут быть
объединены:
dNi
dx
= 0 =
2кТ
Зт)с
2*
N,-2N
\i+i=k
+ (Л"пр!'с)
СО
2
+ TV°D*\
2 ЗД-2Л-* 2ЛГ<'
i=l
Nm~Nk{k~\)
m=»fc+l
(IV.89)
Из уравнения (IV.89) можно найти число одиночных и сдвоенных
капель, а также число агрегатов больших размеров путем введения
232
соответствующих значений для к.
капель (N г) дает
Решение для числа одиночных
1кТ 9
(IV.90)
где TV = 2 Nk.
*=i
Для числа сдвоенных капель (N2)'-
2кТ 9
0 = -z—(2NNs-Nl) + ±vcD*(2NN2-Nl) + Knpvc{2N-2N1-
-3JV2) (IV.91)
Рис. IV. 13. Зависимость отношения
числа агрегатов типа к к общему
числу капель от скорости сдвига
в эмульсии М/В, содержащей 1010
первичных капель на 1 см3 (Ф =
3Чс v ' -1' ' 3
Коэффициент пропорциональности устанавливают с помощью
зависимости скорость сдвига — вязкость при установившемся состоянии.
Рис. IV. 13 иллюстрирует
влияние скорости сдвига на размер
агрегатов капель в эмульсиях М/В
с Ф = 0,50, NK = 1010 частиц
на 1 см3 эмульсии и Кпр = 20.
При совместном использовании
уравнений (IV.269) и (IV.91)
построена кривая влияния скорости
сдвига на вязкость. Коэффициент
набухания /н принят равным 1,60
и предполагается постоянным для
всей области скоростей сдвига.
При очень низких скоростях
сдвига обнаружены расхождения
между теоретической кривой и
экспериментальными данными, так
как агрегаты оказались крупнее,
чем предполагалось при разработке
теории. Кроме того, предположение о том, что значение /н не зависит
от скорости сдвига, не является строгим. Когда скорость сдвига
увеличивается, размер агрегатов уменьшается и, следовательно, /н
должен снижаться. Если Nx сравнимо с NT (где Nx — число капель на
1 см3 эмульсии ко времени т от момента приготовления) в уравнении
(IV.269) точное значение /н не очень заметно влияет на результат,
но когда Nt < Nx, как в эмульсиях В/М, предположение об
устойчивом значении /н вносит большое расхождение.
Другие теории,
включающие процесс флокуляции — дефлокуляции,
вызванный сдвигом и броуновским движением
Кросс (1965) предположил, что флокулированные частицы
расположены в виде беспорядочно перепутанных цепочек, размеры которых
зависят от скорости сдвига. Как и Джиллеспи (1960а, Ь), он считал,
что на процесс разрушения связей влияют и сдвиг, и броуновское
движение. С другой стороны, он утверждал, что образование связей
зависит только от броуновского движения. Совместно с Денни
0,50; Рс
20) (ван ден Темпель,
1963).
233
и Бродки (1962) Кросс сделал допущение, что дефлокуляция,
вызванная сдвигом, зависит в некоторой степени от vc.
Если Nv — среднее число связей при некоторой скорости сдвига
vc, тогда при равновесии
^ = 0 = K3N,-(Kb + Kivcc')Xv (IV.92)
где vcl — постулируется как монотонная функция.
Следовательно
Nv= KsN* (IV.93)
*5
+Некогда ve = о, nv = n°0
K& + Kevcc< 1 + (К,/Кл)4*
(IV.94)
Nv
N%
BXN% =
%
11-
:Ч-Л,
-Ч со
-Лоо
Если отношение К6/Ка велико, т. е. дефлокуляция зависит
главным образом от сдвига, тогда она происходит при низком значении vc.
Выведена аналогия между вязкостью беспорядочных цепочек
из шариков и объемной вязкостью линейного полимера. Последняя
зависит от молекулярного веса (числа сегментов на цепь), так что
tt = Tloo+BlJV«' <IV-95>
где г\- — вязкость при средних значениях vc; Bx — константа.
Когда N- = Nv, % = ц.
В(.-0
Таким образом
(IV.96)
(IV.97)
Совместное решение уравнений (IV.94) и (IV.97) дает:
4-4oo = Ci--4oo)(i+4;^) <IV-98)
Это основное уравнение, устанавливающее связь между вязкостью
и скоростью сдвига.
Для определения значений KJKb и с1 требуется
утомительная графическая процедура. При больших скоростях сдвига,
is
когда -гЛ f^1 >1, уравнение (IV. 98) приводится к виду
Т| — Т]„
ti--ti = — (IV. 99)
И
lg (l--^oo) = ]g (^jKv)^Cl]g l'C (IV'100)
234
Когда строится зависимость lg (г\^ — т]^) — lg vc, градиент дает
значение Cj, а отсекаемый на оси ординат отрезок есть lg ( °° ) •
Чтобы найти отношение K6fKb, уравнение (IV.99) переписывают:
(чо-Чоо)-§7 # = 4-400 (IV.101)
Таким образом, график т^- — (г\- — т^) t>£> должен быть
линейным с отсекаемым отрезком, равным г\, и градиентом,
соответствующим Кв/Къ.
С другой стороны, если уравнение (IV.98) записать в ином виде
—!— = ( *+<*./*.)* \ (IV.102)
то получим линейную зависимость 1/(Лз — Tloo) — y£s гДе
1/(т] — rico) — отсекаемый отрезок и —-^— градиент.
"1 "loo
Когда Ка/Кь мало, допущение, что -^- и§<>1, не может быть
точным даже при vc-> оо. Тогда график % — у£> будет
криволинейным и определить т^ путем экстраполяции не представляется
возможным. В этом случае уравнение (IV.98) может быть переписано
в другой форме:
При очень низких скоростях сдвига
или
X^X+^.J* (IV.104)
Г)- Т| Я5 Т)
Таким образом, величина 1/г|^ должна давать линейную зависи-
К IК
мость от у£< с отсекаемым отрезком 1/т] и градиентом ———-. С другой
стороны, уравнение (IV.98) представляет собой выражение
ч~ч-
ч--Чоо = ——*-г (IV.105)
а зависимость п- от (т) — г)-) i>{j« — прямую линию с отрезком т]^,
1
и градиентом .
При средних значениях К6/Кь уравнение (IV.98) применяют
в форме: „ „
Vc'Tr + ^+T?1^' (IV-106)
235
Величину т]- определяют при трех различных значениях vc,
причем ц исключают путем вычитания и таким образом решают два
уравнения для Кв/Къ и (К6/Къ) л со-
При всех значениях КЬ\КЪ остается неизвестной только константа
сг. Она определена Кроссом (1965) чисто эмпирически. В соответствии
с уравнением (IV.102) строилась зависимость 1/(т)й — Лоо) от vll
для различных произвольных значений сх. При с1 = 2/3 получался
линейный график. Это значение оказалось пригодным для многих
других систем.
Кэссон (1959) также допускал, что агрегаты частиц принимают
форму длинных цепей. На основе статистики он пришел к выводу
что цепь имеет «. . .большее число возможных конфигураций, чем
более компактное устройство. Большие объемы осадков, полученных
для флокулированных суспензий. . . могут быть объяснены только
путем постулирования некоей открытой структуры, которая
заставляет частицы формировать группы подобные цепям». Хотя он
признавал, что агрегаты не будут образовывать жесткие комочки, для
упрощения математического анализа была принята конфигурация
жестких цилиндрических палочек с большим отношением длины к
ширине. Начав работать с очень разбавленными системами, Кэссон
рассмотрел скорость рассеяния энергии, вызываемого всеми
палочками во время течения.
Эффектами, вызываемыми броуновским движением, пренебрегали,
так как они уменьшаются с увеличением скорости сдвига, а
модельные системы, для которых была развита эта трактовка, имеют
непрерывную фазу высокой вязкости. Влияние сдвига на разрушение
и восстановление агрегатов определяется уравнением
— = 1 + (аса1 —1) Ф+Чс/2ас<*2Ф/Ус/! (IV.107)
Чс
где ах и а2 — константы, при которых отношение осей равно:
1 (th 9^ cos Qy)
^^Iw^' a°~ (th 9^008 9^)2+1
Qx и 6^ — ориентация палочек при сдвиге по отношению к осям х, у, z.
Уравнение (IV. 107) распространено на более высокие
концентрации путем учета влияния увеличения объемной концентрации Ф на
величину ДФ. Обозначая новую объемную концентрацию как Ф *,
можно написать:
ф* = Ф(1 —ДФ) + ДФ (IV. 108)
Тогда увеличение объемной концентрации
<2Ф = Ф*-Ф (IV. 199)
И
дф=т^=1г <IV-110)
Для вычисления вязкости т] * при увеличении объемной
концентрации Ф* считают, что непрерывная фаза имеет вязкость ц, а объем-
230
ная концентрация диспергированной в ней фазы составляет АФ.
Тогда из уравнения (IV.107) следует, что
;1+ (acai — i) АФ + Ч,гаса2 ^ф/ис'
и из уравнения (IV. 110)
dr| = T]* — т)= T)(acai —1) +
ч'/гаса-2
„У.
Т^Ф~
(IV.111)
(IV.112)
Подставляя ^4С вместо (асах — 1) и 5С вместо аса2/ус/2 имеем:
dt] <2Ф
(у>сг, + 5сг,'/.) ~Т^Ф <IV113>
Интегрирование с граничными условиями г) = г\0 при Ф = 0
дает:
Для получения зависимости между напряжением сдвига Pq
и скоростью сдвига vc обе части уравнения умножают на Ус/г
1/2 , £civ/z
V*"/»
:Р'>' =
(1-ф)
АС
[(тЬО с -1] <IV-115>
или
где
Рси = кХ'' + к0
Кг
(1-Ф)
А„/2
Чс
(1-Ф)
(IV. 116)
(IV.117)
-«с^с-г/ 1 \лс'' Л аса2 г/ 1 \
ас(Х2
"с"1"1
аса1 — 1
C'J
]-
- — 1
(IV.118)
Из уравнения (IV.116) следует, что график Рсг — Vq должен
быть линейным, с градиентом Кс и отсекаемым отрезком К0.
Уравнения (IV.И7) и (IV. 118) тогда дают а1 и а2 при условии, что известно
ас. К сожалению, это не так и для систем, изученных Кэссоном, ас
принималось равным 0,5I/z.
Все теории течения, описанные выше, предполагают для
структуры агрегатов конфигурацию длинных цепей. Справедливость этого
предположения исследовал недавно Вейманн (1965). Он отметил, что,
хотя статистика указывает на цепи как весьма возможную
конфигурацию, наиболее вероятной конфигурацией в действительности
является спираль, в которой поперечные связи снижают
потенциальную энергию.
237
Образование временных пар капель
во время течения
Во всех ранее обсужденных теориях, относящихся к процессу
флокуляции — дефлокуляции, свойства потока рассмотрены в
зависимости от образования связей и явлений разрушения или агрегации.
Кригер и Догерти (1959) основывали свою интерпретацию на
допущении, что в результате концентрационных флуктуации, вызванных
броуновским движением, в какой-то промежуток времени некоторые
капли будут разделены расстоянием, меньшим чем диаметр одной
капли. Связи между каплями не устанавливаются. Вместо этого
в
О
ч>
р <р
сР
Рис. IV.14. Схематическое
изображение типов потока:
а — сближенные сферы, вращаются
независимо; б — сферы вращаются как жесткая
гантель; « — действительный тип потоков
(скорость движения жидкости между сферами
замедлена) (Кригер и Догерти, 1959).
пары капель вращаются около
общего центра тяжести, потому
что иначе скорость сдвига,
который возник бы в пленке
жидкости, разделяющей капли, была
бы очень высокой (рис. IV.14).
Вращение пары капель
продолжается до того момента,
когда сдвигающее усилие
разделит их.
При vc = 0 распределение
одиночных сфер (Лг1) и пар (N2)
на единицу объема дается
выражением
2iV,
кобр
дисс
(IV.119)
где кобр и А:дисс соответственно константы скорости образования
и диссоциации пар под влиянием броуновского движения.
Если vc ф 0, вводится добавочная реакция диссоциации пар
с константой скорости к'ЯИСС:
*дисс
N*
2N,
(IV-120)
В этой теории не рассмотрена возможность образования пар под
влиянием сдвига. Полную скорость образования пар получают путем
объединения уравнений (IV.119) и (IV.120):
При равновесии
""jT-= ^'обр-^i ~ (^дисс + ^дисс) Лг2
dN2
d%
= 0
И
или
kobpN\ = (/Сднсс + /сдНСС) Лг2
Ni __ fc06p
(IV. 121)
(IV.122)
Л'}
:+*Д
дисс
238
Деление на к^^ дает:
лг„ 4-:..._-/*■
(IV.123)
-** 2 "ДИСС/^ДИСС
"1 1 Т "-дисс/ Ядисс
Когда ус = О, А;'дисс = 0 и
|f=-r^ <IV-124>
JVf «дисс
где N i п N 2 — число одиночных сфер и пар на единицу объема при
vc = 0.
Полагаем, что
Ч-
— = hN1±hN2 = fxN^ + {h-2fi)Ni (IV.125)
Лс
где Nx — iVx + 2iV2; Л и /а — константы.
При гс -> °°
-^Г- = Л^ (IV. 126)
Лс
Когда vc -> О
J- = /iiVt + (/j-2/i)JV2 (IV.127)
Заменяя ДЛ^ в уравнениях (IV.125) и (IV.127) на TiooAlc и поделив
первое на второе, имеем:
Ч--1га ^
— = ^=- (IV.128)
Л-Чсо iV2
Из уравнений (IV. 123) и (IV. 124):
Л Лео + ™диссМдисс
(IV.129)
Теперь предположим, что N и N { не очень отличаются от 7VtB
(это будет зависеть от природы эмульсии и вряд ли применимо
к эмульсиям с масляной непрерывной средой):
Лт.-Лсо !
(IV. 130)
Л — 1 со 1 ~Ь ^ДиссМдисс
Если это уравнение записать в виде
1 __ ^ + Лгдисс Ддисс (IV 131)
1—4 со П-Чоо
оно напоминает уравнение (IV. 102) с той лишь разницей, что в
последнем введена степенная функция vc.
Так как АдПСС и АДиСС относятся к реакциям первого порядка
&дисс~ 1/тс и Адисс ~ 1/т6р (где те и тбр — среднее время жизни пар
капель, диссоциирующих под влиянием сдвига и броуновского
239
движения, соответственно). Уравнение (IV. 128) тогда может быть
записано так:
^--Чос !
= V"T7 /—Г (IV.132)
Величина тбр — это время, необходимое для того, чтобы две
сферы, образующие пару, разошлись на расстояние Z', при котором
они больше не взаимодействуют:
(О2
Тб?=2Д (IV.133)
где Д — коэффициент диффузии, равный kT/3nDr[c; D — диаметр
капли.
Считается, что расстояние V пропорционально D:
Д2
(2')2 = а,— (IV.134)
Здесь aL — константа, приблизительно равная 1.
Подставляя значения (Г)2 и Д в уравнение (IV.133), получаем:
?бР= £т1 (IV.135)
Время, необходимое для того, чтобы капли данной пары
передвинулись в положение, при котором они могут разделиться, равно
а,, я/2 oiiTf
т^-1^г=^- <1V-I36>
где я/2 — угол вращения; vJ2 — угловая скорость пары капель;
ат — константа, которая включена по той причине, что выражение
для угла вращения является приблизительным.
Из уравнений (IV. 135) и (IV. 136) имеем
тбр = За/ДЗ^^ ЗРС03"/ __ Рс ,.„ . .
тр " Ъа.кТ 8cczkT КСК (iv-i.1t)
где Кск — константа скольжения.
Тогда уравнение (IV. 132) принимает вид:
Чт-Чс
Ч-Чоо 1 + (За^ячс"с/8«,АГ) 1-(^с/«ск)
(IV.138)
Неизвестными являются только численные значения ат и а;;
их получают с помощью графической процедуры методом «проб
и ошибок».
Строят зависимость т|- от КСК/(КСК -+- ^с) Для выбранных значений
Хск и определяют постоянные уравнения (IV. 138).
240
Теория абсолютных скоростей процессов
Ри и Эйринг (1955) и Кис и др. (1960) рассмотрели
неньютоновское течение с точки зрения теории абсолютных скоростей процессов
(Глесстон и др., 1941). Для этого они предположили, что во время
течения частица не может двигаться мимо своих соседей до тех пор,
пока не преодолеет потенциальный энергетический барьер. Они
полагали, как и Вильямсон (1929), Г удив (1938) и Джиллеспи (1960а, Ь),
что существует два основных типа течения: один — ньютоновский
и другой — неньютоновский с различными характеристиками.
Поверхность каждого элемента потока разделяется на локализованные
площади sx, s2, ■ ■ -i sn> которым соответствует напряжение сдвига
Р Р Р
iCll-ic2l'',l-£c/l'
Все элементы в одной и той же плоскости сдвига двигаются
с одинаковой скоростью vc. Ньютоновские элементы не соединяются
с подобными им элементами на противоположной стороне
поверхности сдвига, поэтому они сдвигаются легко. Неньютоновские
элементы соединяются с подобными себе, так что они могут сдвигаться
только с трудом.
Скорость сдвига определяется уравнением
тр п
где
1
{KlU)n2k'n =TP"
к'п — константа скорости течения га-й группы элементов [для
преодоления потенциального энергетического барьера AU = (кХ2К3)п/2кТ];
К — расстояние, которое проходит элемент между равновесными
положениями; A,t — расстояние между плоскостями течения
элементов данного вида;Я2^3—площадь поперечного сечения данного
элемента потока; тр „ — среднее время релаксации, AFn — свободная
энергия активации для течения га-го элемента; h — постоянная
Планка.
Напряжение дается уравнением:
п
Рс = ^хпРсп (IV.141)
где snPcn—сила, действующая на п-ю группу элементов.
Делая подстановку для Рс й в уравнении (IV. 141) с помощью
уравнения (IV. 139), получаем:
//
Pc=2"I^ sh~lTP«v' (IV. 142)
/1=1
(IV.139)
(IV. 140)
16 Заказ 1070
241
Изменение вязкости со скоростью сдвига определяется как:
Рс -<ГЧ «лтр л
lp nvc
sh Д£7РС
(IV.143)
л=1
Заменяя sh AUPcn на т „ vc [см. уравнение (IV. 139)], имеем:
i snxp'n sh-i тр„и с
Когда тр „ ь>с -> 0
Когда тр „ vc -*■ оо
н^-^ Д£//^трп,с
п = 1
i-mSh"iTpni-c 1
.. sh-iTpBic
hm ■ = 0
Тр nVc
(IV.144)
(IV. 145)
(IV. 146)
Предполагается, что вся область vc удовлетворяет условиям:
I. тр1г;с < 1
II. таис5* 1
III. Tp3i>c » 1
IV. трпус (где rJj 4).
Группа I представляет ньютоновские элементы, группы II — IV —
неньютоновские. Элементы группы' II отличаются от элементов
групп III и IV тем, что они проявляют ньютоновское поведение при
низких скоростях сдвига и становятся неньютоновскими только при
более высоких скоростях сдвига.
На практике найдено, что п обычно не превышает 3. Это значение
применяют для частиц латекса при низких скоростях сдвига (Марон
и Пирс, 1956), причем уравнение (IV. 144) принимает вид
Ч(1'с) =
slTpl
s2tp
Sll ! Тр 2l-c
s3Tp3
sh"1 т
рЗ^С
AUi
Mii
rp2t'c
Д#я
Дрз'-'с
где s1rpl/AU1 — вклад ньютоновской составляющей.
Дифференцирование уравнения (IV. 144) дает:
dt) (vc)
d In vc
ЛЧ (vz)
ь/г^р п
UJn
sh i (tp nvc)
(тряГс)2+1
lp л"с
(IV.147)
(IV. 148)
ьр П
d (In i;c)2 \Un v*
sh"i (tp nvc) ■
(tp n^c)2
v7
Tp fl^c
[(Tpni'c)a+l]V
[(t„„vc)2-M]
(IV. 149)
Для определения значения различных параметров строят
зависимость п от lg vc (рис. IV.15). Необходимо иметь достаточно данных,
чтобы на графике площадка у нижнего конца шкалы скорости сдвига
была видна так же хорошо, как у верхнего конца. Точка перегиба
242
на кривой наблюдается, когда d2^ {vc)/d (In yc)a = 0, при этом из
уравнения (IV. 149) имеем:
Tpi;,- = 2,90
где vt — значение vc в точке перегиба.
Тогда из уравнений (IV. 148) и (IV. 150) следует, что
-\^й 1 = 0.290-2,303-^7-
L d lg vc J AU
Совместное решение уравнений (IV. 150) и (IV. 151) дает:
S
то
(IV. 150)
(IV.151)
(IV.152)
* 4
*^L
т?^ ^s. Точка перегиба
г j
lqvc
•
Ч. Точка перегиба
4
Рис. IV.15. Дналпз неньютоновской вязкости (Ким
н др., 1960).
Таким образом могут быть определены тр1 и sJAU^, вязкость т)1
основного элемента потока затем вычисляют из уравнения (IV. 144).
Процедуру повторяют с графиком зависимости (rj — г) t) от lg vc.
При этом кривая является горизонтальной в широкой области
скоростей сдвига, но при высоких значениях vc может наблюдаться
другая точка перегиба. Если это так, вычисления повторяют до
определения Т] 2.
Если строят зависимость [ц — (п 4 + т]2)] от lg vc, получают
прямую линию для всех значений vc (см. рис. IV. 15). Эта линия
представляет ньютоновский элемент вязкости.
К сожалению, точки перегиба на графиках т) — lg vc очень трудно
установить, так что описанная выше графическая процедура обычно
не является легкой для интерпретации данных течения эмульсий.
Когда имеются только два типа элементов течения, параметр тр
определяют из измерений вязкости ца, т]р, t)v при трех различных
скоростях сдвига va. yp, Vf (Габриш и др., 1963). Можно показать, что
Tp^lg-iXc/2»«
(IV. 153)
16*
243
где XG — константа, равная:
XG =
, / и3
v&
b-^-K'^b-t-irb-f: k'hr—iri"1+-
Va V.,
Марон и Пирс (1956) и Марон и Сиско (1957) применили теорию
неньютоновского течения Ри — Эйринга к эмульсиям латекса с
объемной концентрацией дисперсной фазы 0,25—0,60. Для объемных
концентраций вплоть до 0,43 в уравнении (IV.144) п равнялось 2,
но при более высоких концентрациях п составило 3, как в уравнении
(IV.147), в пределах области сдвига 1 — 15 сек-1. Вклад, вносимый
третьим элементом течения, приобретает возрастающее значение
при нижнем пределе этой области сдвига. Эмульсии латекса не были
монодисперсными и течение при низких скоростях сдвига «. . .могло
зависеть не только от релаксационной реакции более мелких частиц,
которые будут иметь меньшее тр, но и более крупных частиц, для
которых тр будет больше. При более высоких скоростях сдвига
влияние последнего будет прогрессивно уменьшаться и система станет
зависимой только от одного элемента течения латекса».
Марон и Сиско (1957) отметили, что теория Ри — Эйринга не
предсказывает экспериментально наблюдаемую зависимость
концентрации от различных параметров. Для определения разных элементов
течения они применили довольно утомительную графическую
процедуру. Уравнение (IV. 147) может быть написано так:
slTpl 52Tn2 s3Tp3
где
sh-iTn2iv sli-iTn3i;c
Трг^'с трз1'с
При трус -> 0 из уравнения (IV. 154) следует:
slTnl , s2Tp2 , s3Tp3
Зависимости r| (uc) — A 2 экстраполировались к vc = 0. Строили
графики зависимости для нескольких значений тр 2 и т) от А 2. При
подходящих величинах тр2 графики были линейными при высоких
значениях vc, но проявляли криволинейность при низких значениях vc.
Через линейные участки кривых проводили прямые линии и
определяли наклон — s2Tp2/A£/2 и отсекаемый на оси отрезок — SitpJAUt.
Затем из уравнения (IV. 155) вычисляли SjTpo/AC/з- В конечном счете
Д3 определяли для каждого значения vc из уравнения (IV. 154),
представленного в виде:
Д3 = — (1\ . 156)
s3Tp3
ДЕ/3
Через А3 и vc выражали параметр тр3.
244
Кинетическая теория неньютоновского течения
При неньютоновском течении с увеличением скорости сдвига
происходят некоторые структурные изменения, вызванные
разрушением агрегатов. Объяснение этого явления, данное Денни и Бродки
(1962), основывается на кинетике реакции. При рассмотрении
механизма изменения структуры не было сделано каких-либо
специфических допущений и не предполагалось наличие нескольких элементов
течения, как в теории Ри — Эйринга. Авторы ввели допущение
только о том, что разрушение невозможно при нулевой скорости
сдвига, и таким образом игнорировали разрушение под действием
броуновского движения.
Изменение структуры может быть представлено следующим
образом :
(IV.157)
АСТ
неразрушенная
структура
>■ R
-Ч -°ст
k2
разрушенная
структура
Уравнение (IV. 157) решается с помощью кинетического
уравнения:
-^~=К (Лет)"' ~h (Яст)"2 (ГУ.158)
где /с4 или A'j и к2 — константы скорости прямой и обратной реакций,
соответственно; пу и п2 — порядок реакции.
При некоторой скорости сдвига частичное разрушение структуры
и неразрушенная фракция описывается выражениями:
ч| — Чу
ВС,= ^—^Г (IV. 159)
ц-
■п-
V
~Пп
-Псо
"'loo
А"= n-n (IV160>
1 'со
где т), т)а и y]^ — вязкости при vc -> 0, промежуточных равновесных
значениях vc и при vc -v о°, соответственно.
Решая совместно уравнения (IV.158) — (IV.160), получим:
п~т,0° ;-ч(^Г-*.(-^Г (iv.161)
dT l^-^oo/ 1*1-11
Эффект, производимый при сдвиге дисперсной системы,
рассматривается как аналогия катализу, так как т)от достигается только
при высоких скоростях сдвига, поэтому
т1 —^00 \ dx J \ т| — т|
*-Ч^Г <-62>
245
где член, отражающий разрушение, включает степень С{ скорости
сдвига.
При равновесии для некоторой скорости сдвига dr\Jdx = О,
следовательно:
К I ~— I ^с = к1
А" =
*1
Л —По \п'
(л-л0)"
А'2 (Л^-Лсо)"'^^-^»)'"""1
Уравнение (IV. 164) может быть записано иначе:
lg (Л-чо)" =gi lg b.c + K lg(T,_tloo)n,-«.
(Л„-Лм) '
(IV. 103)
(IV. 164)
(IV. 165)
Зависимость левой части уравнения (IV. 165) от lg vc должна быть
линейной при условии, что применяются точные значения iti и п2,
с градиентом с4. По отсекаемому на оси отрезку может быть вычислено
значение К. Наибольшей трудностью при такой трактовке является
получение значений п4 и п2 методом «проб и ошибок». Для эмульсии
латекса линейный график получен с п2=2 и rat = 1 и дал К =
= 3,60-10" 3.
Интерпретация зависимого от времени изменения структуры
включает интегральную форму уравнения (IV. 162); это ведет к более
сложным зависимостям. Для постоянной скорости сдвига, п2=2
и nt = 1 уравнение (IV. 162) может быть записано следующим образом
^-а^'К-О
_А. (л-л,)2
2 (Л-Лео)
так как kt = к2К
где
d\\v
■■ i k2 dx
J C-2 —C3T]„-t-C4T]J
^-Л'-с'Лсо+^/Л-Лсо)
^з = -[А'^' + (2л/Л-ЛСо)]
1
(IV. 166)
(IV.107)
Интегрирование уравнения (IV. 167) и определение константы
интегрирования для т = 0 и вязкости п дают:
th-i
2с4Л + сз
,(с§ —4с2с4)
'/=
-th-
2с4Ли_гС3
(с§— 4с2с4)
= А:2т (IV.168)
где с| — 4с2с4 = Я i£ (#i# + 4).
246
Подобным образом может быть выведена константа ujj в этом
случае величинами, которые должны быть введены в уравнение (IV.167),
являются:
^ = -{[211/^(4-11^)]}+4'
Из уравнения (IV. 168) видно, что график зависимости
th"1 —с4Чо + са от т дает Градиент Д;2(с2 _ 4с2с4)'/»/2, откуда может
L(c§ —4c2e4) "J
быть определена константа к2-
Таким же путем получают и константу к\.
В зависимости от времени разрушения структуры для К найдено
значение —10~и.
С точки зрения этой теории различие между независимым и
зависимым от времени изменением структуры заключается в более низком
значении ftj для последнего. Применение теории к вязкости эмульсий
дало различные значения kt ш к2 для разных концентраций
дисперсной фазы. Пока нет опубликованных данных, показывающих
возможность проинтегрировать эти константы скорости так, чтобы
подтвердить справедливость кинетической теории. Согласно Денни и Бродки,
«наибольшим недостатком метода является то, что обратное
вычисление для восстановления основной диаграммы сдвига по известным
значениям констант потребовало бы больше информации, чем значения
констант; последующее развитие должно рассматриваться скорее как
способ связать течение с фундаментальными свойствами материалов,
чем изображение течения жидкости».
Теория Дерягина—Ландау—Вервея—Овербека (ДЛВО)
Взаимодействие между каплями в эмульсии, особенно внутри
агрегатов, может быть интерпретировано как проявление сил
отталкивания и притяжения (Дерягин, 1934, 1939, 1940; Вервей и Овербек,
1948). В эмульсии М/В силы отталкивания обычно являются
электростатическими, их величина зависит от электрического заряда,
концентрации электролита, размера капель и расстояния 10 между ними.
Если все капли являются сферическими и идентичными по размеру
D, то при условии, что толщина диффузного слоя (1/^) много меньше,
чем диаметр капель, т. е. %DI2 > 300, выражение для энергии
отталкивания будет иметь вид
eD4>*
иот = -—■ {In [1 + ехр (-х/0)]} (IV. 169)
где ^д — потенциал наиболее удаленной части двойного слоя; е —
диэлектрическая постоянная непрерывной фазы.
247
Более точное выражение выведено Вервеем и Овербеком:
Uo, = ~ (IV.170)
азе;
где 2 — валентность ионов, присутствующих в непрерывной
<р — функция от yj0 и z.
Овербек (1952) дает таблицу, иллюстрирующую эту взаимозави
симость.
% = 40мв
0
-60\-
Рпс. IV.16. Кривые потенциальной энергии эмульсий М/В
(Шерман, 1963а):
0 — D = 3 мкм; X — D = 6 мкм.
Энергия притяжения Unp, которая действует на большем
расстоянии, чем энергия отталкивания, не подвержена влиянию полярной
природы непрерывной фазы
A j £>2 t D2 , „ , Г . D2
12 {W'
U
пр-
где А — константа Лондона
Когда l0 < D
'^+2Ь[
Ван-дер-Ваальса.
'^Л (ПМ71)
'пр-
AD
Ж"
(IV.172)
Вычисленные значения А должны быть откорректированы на
эффекты релаксации (Шенкель и Китченер, 1960). Эмульсии редко
являются монодисперсными, но, если распределение размеров не
слишком широкое, D в уравнениях (IV.169), (IV. 171) и (IV.172),
по-видимому, может быть заменено на Dcp.
Общая потенциальная энергия взаимодействия U между каплями
масла в эмульсиях М/В является суммой Unp -f- U0T. На рис. IV. 16
приведены характерные типы кривых энергии притяжения — оттал-
248
кивания и общей потенциальной энергии взаимодействия для эмульсии
М/В, стабилизированной неионным эмульгирующим агентом моно-
лауратом сорбитана, содержащим—0,5 вес. % мыла (Шерман, 1963а).
При определенном расстоянии между каплями кривая U имеет
максимальное значение (АС/макс). Если капли при движении
оказываются ближе друг к другу, чем эта критическая дистанция, то
потенциальный энергетический барьер (ДС/макс) будет преодолен. Когда
Д£/макс не превышает нескольких кТ, некоторые капли в состоянии
перескочить барьер. При таком очень малом расстоянии капли в
первичном минимуме взаимно удерживаются большими силами
притяжения, если только слой эмульгирующего агента вокруг каждой капли
препятствует быстрой коалесценции. Напротив, если AUuaKC более
15—20 кТ, капли не могут преодолеть энергетический барьер во
вторичном минимуме (где силы притяжения слабы и обычно не
превышают нескольких кТ) и флокулируют.
В эмульсиях В/М толщина диффузного двойного слоя (1//)
составляет несколько микрометров, так что здесь при увеличении
расстояния между каплями энергия отталкивания убывает намного
медленнее, чем в эмульсиях М/В. Вследствие этого при вычислении Uor
для эмульсий В/М необходимо скорее вводить поправку,
учитывающую эффект взаимодействия между многими частицами при их
сближении (Альберс и Овербек, 1960), чем принимать во внимание
отталкивание между двумя изолированными каплями, как это
обычно делается для эмульсий М/В:
РДМ«*" j • ихЯсф+1 -у,ХЙд|Ь /сф-е-*'сФ
1<"- 4 1 8'88 -/.зд»ф ' Ц +
+-~;(Дсф"сф) -—« [^^^-.-x/^'v-/-^) (IV,173)
где ЛСф — радиус гипотетической сферы, на которой расположены
двенадцать капель при наибольшей возможной близости к
центральной капле; /сф — среднее расстояние от центра сферы, на
котором может быть найдена любая из двенадцати капель
[пределами его являются 0 и (Дсф — D)].
Радиус сферы находят из выражения:
0.905Д
лсФ=-^=- (IV.174)
В эмульсиях В/М с объемной концентрацией Ф = 0,40—0,74
величина АС/макс в зависимости от значения % понижается на 60—100%.
Для большинства эмульсий наибольшее влияние оказывает член
е-*(Дсф-'сф)
■#сф — гсф
На рис. IV. 17 изображены кривые потенциальной энергии
взаимодействия для эмульсий В/М с частицами двух различных
диаметров. Значение U0T значительно ниже, чем в эмульсиях М/В, поэтому
величина ДЕ/макс очень мала и вторичный минимум отсутствует. При
249
этом флокулированные капли разделены меньшими расстояниями,
чем в эмульсиях М/В и между ними возникает сильное притяжение.
При применении теории ДЛВО к данным неньютоновскои
вязкости трудности возрастают. Это объясняется следующим.
1. Неточная оценка Уд. Электрофоретическую подвижность
капе ть в эмульсии обычно измеряют при выбранной силе поля и по
этим данным вычисляют потенциал £ внутренней части диффузного
§
-*
^
Рис IV.17. Кривые потенциальной энергии
эмульсий В/М (Шерман, 1963а):
а — D
ср
2 мкм; б — D = 1 мкм
двойного слоя по уравнению для электрофореза непроводящих сфер
(Дэвис и Райдил, 1963)
у_ блчс^э (IV.175)
где „э _ подвижность капель при электрофорезе; U и /вз —
поправочные коэффициенты, учитывающие зависимость релаксации ионной
атмосферы от времени и взаимодействие между приложенным полем
и электрическим двойным слоем, соответственно (последнее
взаимодействие очень важно для малых капель).
Когда толщина двойного слоя мала по отношению к диаметру
капель / = 3/2 и /т = 1. Если ионная концентрация низка,
предполагается "что г|зд превышает I примерно на 10% , но при высокой
ионной концентрации, например, в присутствии в непрерывной фазе
эмульсии М/В 0,1 м раствора хлорида натрия, WA может быть в два
раза больше, чем £ (Дэвис и Райдил, 1963)
2. Выбор значения А в уравнениях (IV.171) или (IV.1/2). Если
то
Д£Л
становится
подставленное значение чрезмерно велико,
сташком малым, а вторичный минимум слишком большим. В
некоторых случаях может быть сильно изменен общий характер кривой U
При изучении суспензий твердых частиц в жидкой среде значение А
250
может быть рассчитано из скорости флокуляции частиц или из
физических данных, к которым применима, например, модификация Мел-
вин-Хьюза для формулы Слейтера — Кирквуда (Мелвин-Хьюз,
1959). В эмульсиях капли вслед за флокуляцией коалесцируют,
поэтому очень трудно изучать скорость флокуляции изолировано от
последующего процесса коалесценции. Кроме того, обычно не
доступны физические данные, необходимые для другого метода
вычисления А. Овербек (Волд, 1961) и Фоукс (1964) оценивали А по
величине вклада дисперсионных сил в поверхностную свободную
энергию. Константа Лондона — Ван-дер-Ваальса для капель масла,
диспергированных в водной среде, дается следующим выражением
amib = (Vamim-V^WbY - (IV.176)
где 4м/м и АВ/ъ — соответственно, константы взаимодействия
между молекулами масла и между молекулами воды при отсутствии
другой жидкости.
Аналогично:
ab,m = (Va~^b-Vam/mY (IV.177)
Согласно Овербеку
^м/м = 24лгмам (IV. 178)
и
кв/в = 24лгв(Тв (IV.179)
где ам и ав — поверхностное натяжение масла и воды,
соответственно; /м и ^в — расстояния, отделяющие центры атомов соседних
молекул масла- и воды, соответственно.
Для дисперсий жидкого парафина в воде А = 5,4-10"12 эрг, для
воды в жидком парафине А = 3,0-10"12 эрг. Уравнения (IV.176)
и (IV. 177) не учитывают поправку на ослабление притяжения между
каплями, когда они покрыты слоем эмульгирующего агента. В
зависимости от толщины слоя эмульгатора и диаметра капли коэффициент
снижения мог быть между 5—50 (Волд, 1961). Так, для упомянутых
примеров А должно было бы снизиться до 10"12 и до 10"13 эрг.
С другой стороны, величина А может быть оценена по данным;
вязкости (Альберс и Овербек, 1960). Когда эмульсия подвергается
сдвигу, агрегаты капель разрушаются при увеличении скорости
сдвига до тех пор, пока не будет достигнута полная дефлокуляция.
Последнюю стадию дефлокуляции при сдвиге можно
рассматривать как разрыв сил притяжения между остаточными парами капель.
В этих парах капли разделяются, когда силы притяжения
превзойдены гидростатической силой, создаваемой непрерывной фазой при
сдвиге. Для монодисперсной эмульсии при отсутствии деформации
сдвига критическая скорость сдвига, которая является причиной
разрушения, дается выражением
^"=18n^sin(2aKFl) <IV-180>
25t
где акр — критический угол между линией, соединяющей центры двух
капель, и направлением сдвига в момент разрушения (а —- 30°).
График зависимости вязкости от скорости сдвига дает значение
умин1 выше которого вязкость остается постоянной.
Альберс и Овербек предполагают, что для эмульсий В/М величина
10 равна удвоенной толщине слоя эмульгатора вокруг капель. Так,
для моноолеата сорбитана, стабилизирующего эмульсии В/М, 10 =
= 40 А, если углеводородная цепь эмульгатора полностью вытянута,
и несколько меньше (—25 А), если она в какой-то степени
искривлена. С учетом этих двух возможностей по данным вязкости (при
Dcp= 1,1— 2,0мкм) вычислено значение ЛВ/м = (0,5—2,0) • 10~12эрг
(Шерман, 1936а), которое дает правильный порядок величины.
В каждой эмульсии наблюдалось узкое распределение капель по
размерам, что позволяло заменять D в уравнении (IV. 180) на Dcp.
Вычисленные подобным путем Альберсом и Овербеком (1960) значения
Лв/м оказались равными 10~14—10~15 эрг. Однако тщательное
исследование их кривых Рс — vc показало, что значения i>MHII слишком
низки, так как эти кривые никогда не были точно линейными, т. е.
дефлокуляция никогда не была полной. Авторы признавали, что
неожиданно низкие значения ЛВ/м могли быть вызваны отчасти
неполной редисперсией, и считали, что должен быть включен
коэффициент не превышающий 2 или 3. В действительности же эти значения
необоснованы и поэтому рассчитанные и экспериментальные
значения иыт дают довольно большие расхождения.
Тем же путем могут быть обработаны данные вязкости для
эмульсий М/В, если принять, что 10 будет теперь отвечать флокуляции во
вторичном минимуме. Например, эмульсии М/В, стабилизированные
монолауратом сорбитана, с Dcp = 3,5—6,0 мкм и /0 = 60—80 А
дали Лм в ^0,5-10" 12 эрг (Шерман, 1963а).
Преимуществом получения Л в/и или ЛМ;в из данных вязкости
является то, что влияние слоя эмульгатора учтено в выражении для
общей силы, противодействующей распаду во время сдвига, и не
требует самостоятельного рассмотрения. При использовании этой
методики, помимо чрезмерного упрощения модели для последней стадии
дефлокуляции, ограничением является лишь то, что концентрация
дисперсной фазы не должна быть слишком высокой, иначе капли
из разрушенных пар могут быть немедленно притянуты в
расположенные вблизи агрегаты.
Другой путь для определения ЛВ/м по данным вязкости
разработан на основе дополнения Джиллеспи к методике обработки данных
непьютоновского течения Гудива (Джиллеспи и Вайли, 1962).
Величину Лв/М вычисляют из выражения
Лв/м-9Сл1* (ам+ов-2амв) (IV. 181)
Комбинируя это уравнение с уравнением (IV. 172) получают:
° пр
7 == ^ (]\т 1Я91
0 4яА»(ам+ств-2омв)
252
и
А=*(«м + 4-*нв)(-^)' (rV'183)
Когда вязкость не зависит от скорости сдвига, уравнение (IV.75)
приводит к выражению для общей энергии притяжения между двумя
частицами, которая приближается к С/Пр, если U0T очень мало
Р°с=—^-f [ЛГк(ЛГк-^кр)] - (IV.184)
где 7VKp — число частиц на 1 см3 при критической объемной
концентрации дисперсной фазы, при которой впервые наблюдается режим
неньютоновского течения; К± дается уравнением (IV.73).
Значения Р° определяют по данным вязкости для неньютоновских
эмульсий с различными объемными концентрациями дисперсной
фазы. Затем вычисляют значение Unp с помощью уравнений (IV.73)
и (IV. 184) и вводят в уравнения (IV. 182) и (IV. 183), получая величины
10 и А. Таким путем Джиллеспи и Вайли нашли для эмульсий В/М,
стабилизированных монолауратом сорбитана, значения Лв/М =
= 3,9-Ю-12 эрг и /0= 2,3 А. Они пришли к заключению, что
каждая капля вместе с окружающим ее слоем эмульгатора действует как
единая система, так что реально 10 — это расстояние между
наружными поверхностями слоя эмульгатора, покрывающего соседние
капли, а не удвоенная толщина слоя эмульгатора, как полагали
Альберс и Овербек (1960). Вероятно, это правильно, когда химические
составы дисперсной фазы и слоя эмульгатора подобны. Однако во всех
других случаях подразумевается, что вычисленные значения А
относятся к взаимодействию между соседними слоями эмульгатора, а не
к взаимодействию между каплями, видоизмененными
адсорбированным эмульгатором.
При флокуляции капель во вторичном минимуме в эмульсиях
М/В силы притяжения слабы, поэтому вязкость в стационарном
состоянии не слишком высока. Если прикладывать сдвигающее усилие,
вязкость не будет падать очень быстро с увеличением скорости
сдвига, потому что кривые потенциальной энергии взаимодействия
показывают, что U изменяется медленнее, чем увеличивается 10,
так как вторичный минимум представляет собой широкую плоскую
выемку.
С другой стороны, эмульсии В/М имеют высокие вязкости в
стационарном состоянии при очень низких скоростях сдвига, вызванные
флокуляцией при малых разделяющих расстояниях. Например,
в опытах по ползучести в вискозиметре с коаксиальными цилиндрами
эмульсия В/М (50% + 50%) с Dcp = 1,4—1,8 мкм имела вязкость
в пределах (2,0—2,5) -105 пз и модуль упругости 1200—2300 дин/см2
(Шерман, 1965).
Растительные эмульсии М/В, стабилизированные протеинами
молока, проявляют более сложное поведение. Так как максимум
на кривой потенциальной энергии взаимодействия не превышал
253
10 кТ при.Оср =1,5 мкм, капли с диаметром < 0,5 мкм, для которых
£/макс много ниже, флокулировали в первичном максимуме, а капли
с диаметром 1,5 мкм и больше — во вторичном. Анализ
характера ползучести этих эмульсий указал на существование двух
различных сил связи. В этом случае модуль упругости для частиц,
флокулирующих в первичном минимуме, составил только
~200 дин/см2, а ньютоновская вязкость 104—105 па.
Таким образом, данные неньютоновской вязкости могут быть
качественно интерпретированы на основе теории ДЛВО,
количественные же соотношения между величиной сил взаимодействия и
реологическими параметрами все еще не установлены.
Гидродинамическое взаимодействие
в дефлокулированных системах
Гидродинамическое взаимодействие между каплями в
концентрированных системах рассмотрено Симха (1952), однако безотносительно
к возможности агрегации или влиянию сдвига. Его наблюдения
относятся к ньютоновскому компоненту неньютоновского течения. Капли
имеют конечный размер, и это становится особенно существенным
в концентрированных эмульсиях, где расстояние между дефлокули-
рованными каплями мало и часто меньше, чем их диаметр.
Взаимодействие с каплями, расположенными на некотором
расстоянии от центральной эталонной капли, иное, чем с ближайшими
к ней каплями — оно будет снижаться из-за эффекта экранирования.
Дистанция, на которой происходит это взаимодействие, определяется
экранирующим расстоянием Zs.
Предложена клеточная модель, где вокруг эталонной капли
выделяется концентрическое сферическое пространство диаметром Dc$,
определяющее границу, за которой отсутствует взаимодействие
между эталонной каплей и всеми другими. Значение Dc$ зависит от
концентрации дисперсной фазы. Оно будет зависеть также от D или
Dcp, хотя это положение математически не подтверждено.
Уравнение вязкости для очень разбавленных эмульсий (Эйнштейн,
1906) преобразовано из
-7-=11отн=1 + 2,5Ф (IV. 185)
В
Чотн=1--2,5Явз (IV.186)
с тем, чтобы учесть поправку на взаимодействие в присутствии
экранирующего эффекта. Здесь Квз (функция у = D/Dci>) — коэффициент
взаимодействия между эталонной каплей и всеми другими:
v ** ' * У' (TV Щ1\
вз 4(1 + г/)10-25г/3(1 + г/)4 + 42!/5 v '
При у г£ 0,5 коэффициент Квз возрастает медленно, а 'при у =
= 0,5—1,0 — намного быстрее.
254
В случае низких концентраций дисперсной фазы D^
пропорционально расстоянию между каплями, при более высоких
концентрациях — пропорционально разности этого расстояния и диаметра
капель. Отсюда следует предположение, что в разбавленных
эмульсиях
J/ = V- (IV.188)
в концентрированных эмульсиях
У (IV.189)
где Кф константа, равная у 8Фмакс.
При этом максимальная концентрация дисперсной фазы Фмакс
будет зависеть от геометрии упаковки. Для гексагональной и простой
кубической плотной упаковки Кф = 1,81 и 1,61 соответственно.
Введение этих значений для Кф в уравнение (IV. 186) приводит к двум
взаимоисключающим уравнениям" вязкости:
Чотн=1+2,5Ф
т]0ТН=1 + 2,5Ф
Когда Ф -> Фмакс
1 4- 25 Ф — Ф'Л -I 625 ф2-ь . • -1 (IV.190)
1 + 4Кф Ф 2Кф W + 16ЛГ5Ф * + J llV 1Щ
+ 4Кф Ф+ 4К% Ф + Кф^ + 16ЛГф "г" ■ ■ ■ J
(IV.191)
54 Г Ф2 П
5Кф Kl-Ф/Фмакс)
От Ф = 0 до ФмаКс величина Кф изменяется только от —1,3
ДО <2.
РЕОЛОГИЯ МОДЕЛЬНЫХ ДИСПЕРСИЙ
Течение в вискозиметре
с коаксиальными цилиндрами
Вращательное и смещающее движения капель в момент сдвига
оказывают большое влияние на явления, вносящие вклад в вязкость
эмульсий: столкновение между каплями; флокуляция, приводящая
к образованию агрегатов; циркуляция жидкости внутри агрегатов
и т. д. Некоторые из этих явлений уже обсуждены в предыдущих
разделах, другие обсуждаются ниже.
Масон и сотрудники с помощью оригинальных методик провели
широкое изучение реологии более чем десятка модельных систем.
Для изучения течения Куэтта они использовали вискозиметр
с коаксиальными цилиндрами, изготовленными из тюбингов стекла
пирекс. Вискозиметр действовал не на обычном принципе. Цилиндры
могли вращаться в противоположных направлениях при 0—2 об/мин,
255
так как каждый из них независимо нриводился в действие мотором
с переменной скоростью. Максимальный градиент скорости от
неподвижного слоя до середины зазора составлял 2 сек"1.
Зазор между цилиндрами заполняли жидкостью, образующей
непрерывную фазу, затем с помощью шприца вводили капли
дисперсной фазы. Через микроскоп наблюдали их поведение относительно
оси Y.
Период вращения твр стеклянных шариков диаметром 150 или
290 мкм, которые были суспендированы в высоко вязкой кукурузной
патоке, хорошо согласуется с теорией, развитой на основе
рассмотрения Джеффри (1922) вращательного движения твердых эллипсоидов
(Тривелиан и Масон, 1951)
твр = ^ (IV.Ш)
и
сос = 4- (IV. 194}
где о)с — постоянная угловая скорость.
Период вращения был пропорционален градиенту скорости и не
зависел от размера шариков. Постоянная угловая скорость играет
существенную роль при математической обработке связанных с
вязкостью зависимостей для дисперсий и эмульсий.
Барток и Масон (1957, 1958), используя теорию Тэйлора (1932),
вывели уравнения для линий обтекания потока как внутри, так и
снаружи жидких шариков, суспендированных в другой жидкости.
Циркуляция жидкости внутри капель вызывается вязким торможением,
порождаемым непрерывной фазой. Константа линий обтекания для
течения внутри капель в ламинарном сдвигающем потоке дается
выражением
К
Обт ~
4~М-£+1
т 1
(IV. 195)
где гв — радиус-вектор, измеряемый от начала координат системы
®y — угол полярных координат, измеряемый от оси Y или У;
Лф — вязкость дисперсной фазы.
Величина Кобт будет максимальной, когда отношение Лф/Лс < ilг-
Это имеет место при условии:
^L = A \Jk I (Ivi96)
D"- 5 5cos26y ^v.iyt,;
Когда 0У = 0, уравнение (IV.196) приобретает вид:
' Мер
+ 1|
(IV.197)
4j _ 3 ^Vt)c +1j
Д2 5 5 cos 26 у
На рис. IV. 18 и IV. 19 показаны типы внутренней циркуляции
в каплях, когда il<j/nc равно 1 и £Д соответственно. В первом случае
линии обтекания имеют овальную форму, причем их эллиптичность
256
уменьшается при смещении от начальных линии, во втором — четкие
зоны циркуляции развиваются вокруг каждой из точек застоя,
близких к центру капель.
Период циркуляции жидкости (ту) вдоль линии обтекания внутри
сферы дается выражением:
tvvc 2(г)ф/т|с+1)
-7-^ = —- — (IV.198)
4л /(2V,lc+5)(2V,lc-1)
Когда т]ф/т1с ->■ °°, как для жестких шариков, TvvJ4n = 1.
Период циркуляции (ts) на
поверхности капли:
rSVc W'Hc + l
4л ^■Пф/чсСлф/чс+г)
(IV. 199)
-1 -
-2 -1 О
Рис. IV.18. Циркуляция внутри
жидкой капли в поле действия сдвига
ПРИ Лф/Лс = 1 (Барток и Масон, 1958).
-1
1
0,5; г-
,0; а - 7,5.
Рис. IV.19. Циркуляция внутри
жидкой каплп в поле действия сдвига
при r]Jr\c = 1/4 (Барток и Масон,
1958).
Сс :1 — 1,952; 2 — 1,9; 3 — 1,8; 4 —
0,9; 5 ~ 0,1.
Из уравнений (IV. 198) и (IV. 199) следует:
(т1ф/Лс)2 + 2т1ф/т]с
(IV.200)
Когда Vic >т> ху >х£
4(1ф/11с)2 + 8г1ф/г]с — 5
Это видно из значений отношения
Ty/Ts, вычисленных для широкой области значений т)ф/т)с.
На вязкость разбавленных эмульсий Т|ф/т1с влияет, если
циркуляция жидкости внутри капель не подавлена (Наваб и Масон, 1958)
и данные согласуются с уравнением Тэйлора [см. уравнение (IV.220)]
при условии, что деформация капель при сдвиге мала. Когда жидкость
не может циркулировать внутри капель, например, из-за присутствия
жесткой пленки эмульгатора, уравнение Тэйлора неприменимо.
При высоких скоростях сдвига капли жидкости деформируются
и могут даже разрываться в зависимости от величины т|ф/т]с (Румшейдт
и Масон, 1961). Искривление капель с диаметрами 500—2000 мкм
изучалось при градиентах скорости 0,003—40 сек-1 и отношении
1/ Заназ 1070
257
т]ф/т]с, изменяющемся между 0,0004—29, с помощью
модифицированной формы вискозиметра с коаксиальными цилиндрами. При более
низких градиентах скорости все капли деформировались в
продолговатые сфероиды с главной осью, расположенной под углом л/4
независимо от т]ф/1]с. При более высоких градиентах скорости
наблюдались четыре различных типа поведения капель при увеличении
деформации (рис. IV.20), существенно зависящей от т]ф/,пс, но независимой
от размера капли (табл. IV.3).
0^ ^...^'
(Х)0^г<=^=:>0-0
12 3 4
§0^
1 2 3
Ф 0 0<Е>
7 2 3 4
Рис. IV.20. Изменение
степени деформации с
увеличением скорости сдвига
(Румшейдт и Масон, 1961):
а—деформация типа I, 'U/^c —
-- 2. Ю-4; б — деформация
типа II, '1ф/Лс = 1,0;
в—деформация типа III
типа III, Г1ф/Т1с =0,7;
деформация типа IV, ЛЛ/ПГ =
0,0.
Таблица IV.3
Влияние т)ф/г|с на периоды циркуляции
11ф 1с
1,0
1,5
2,0
Ti-t'c/',n
1,51
1,25
1,15
TWTs
1,31
1,15
1,08
V^c
5,0
10,0
100,0
xvvc;in
1,0.3
1,01
1,000
Tv.'Ts
1.02
1,005
1,000
I. т]ф/т]с = 1,3- Ю-4 —^0,19. Капли принимали форму сигмы
с заостренными концами, вторичные капли с диаметром 50 мкм
разъединены.
II. т)фЛ]с = 0,03 -т- 1,0. При градиенте скорости, достаточном для
разрушения капли, центральная часть ее вытягивалась в цилиндр,
в середине появлялся узкий перешеек. Перешеек утоньшался до тех
пор, пока капля не разделялась на две меньшие капли одинаковых
размеров; это сопровождалось отделением нескольких очень малых
капель.
258
III. т)ф/т]с = 0,7 -=- 2,2. Это вариант деформации типа II для
случая, когда градиент скорости достигал критической величины для
разрушения капли. Капля вытягивалась в длинную тонкую нить
и в этот момент разрывалась на несколько меньших капель.
IV. Лф/Лс — 3,8 н- 29. Эллипсоидальная форма, принятая при
низких градиентах скорости, все сильнее деформировалась при более
высоких градиентах скорости, но разрушения или разделения не
происходило.
О
о
С),
о
Ч:
61
<А
9
а
Рис. IV.21. Микрофотография столкновения и разделения капель
(Барток и Масон, 1957):
1 — сближение капель; 2 — кажущийся контакт; з — перед моментом
столкновения; 4 — после момента столкновения; 5 — зеркально отображенное
разделение; б — удаление капель.
Резкое разграничение в значениях т]ф/т]с для типов деформации
I, II или III отсутствовало, однако деформация типа IV проявлялась
только при высоких отношениях т)ф/т]с. Низкие концентрации
эмульгирующего агента, растворенного в каплях, не изменяли их поведения
при деформации. Например, капли воды в силиконовом масле
с ^ф/Лс — 2 • 10" 4 показали деформацию типа I. Когда к каплям было
добавлено 0,005 или 0,5% твин-20, т]ф/т]с не изменилось, как не
изменился и характер деформации, хотя межфазное натяжение упало
с 38 до 20 и 6,6 дин/см соответственно. Точно также капли
окисленного касторового масла, диспергированные в кукурузной (маисовой)
патоке, с г\ф/цс = 0,7 проявили деформацию типа II как с добавкой
0,5% спен-85, так и без нее. В присутствии эмульгирующих агентов
при постоянном градиенте скорости деформация капель
увеличивалась со временем. Диффузия ПАВ к межфазной поверхности раздела
вызывала снижение внутренней циркуляции.
17*
259
Дополнительно к изучению поведения при сдвиге отдельных сфер
и капель изучено влияние сдвига (*£ 3 сек'1) на сближение,
столкновение и разделение твердых сфер и жидких капель (Барток и Масон,
1957). При использовании вискозиметра, в котором коаксиальные
цилиндры изготовлены из нержавеющей стали, и при рассмотрении
вдоль оси Z найдено, что траектории сближения и разъединения
сталкивающихся твердых сфер диаметром 107 мкм или жидких сфер
с диаметром —-100 мкм криволинейны. Когда две сферы подходили
близко друг к другу (рис. IV.21), они никогда фактически не имели
контакта, но тем не менее образовывали дуплет, который вращался
как жесткая гантель. Эта модель впоследствии использована Криге-
ром и Догерти (1959) при выводе уравнения течения. Вращение
дуплета согласовывалось с уравнениями Джеффри (1922) для
продолговатых сфероидов и это подтверждало, что между двумя сферами,
образующими дуплет, жидкость иммобилизована.
Экспериментальные данные также подтверждали, что траектории сближения и
разъединения были зеркальным отражением одна другой. Так как период
вращения твердых сфер, подвергавшихся повторным столкновениям,
не изменялся, следует, что дуплеты вращались с той же угловой
скоростью и/2, что и единичные сферы.
Если при вращении дуплет повернется на угол 20 0 прежде чем
разъединится, время жизни дуплета тд составит:
T«=^[th_1(i"th90)] (IV-201)
Для прямолинейного сближения двух сфер
5л
6vc
Максимальное время жизни дуплета равно:
5л
2 ус
(IV.202)
(IV. 203)
Жидкие сферы диаметром > 500 мкм вели себя при сдвиге иначе,
чем твердые и более мелкие жидкие сферы. Смещение между центрами
капель в дуплете вдоль оси У увеличивалось при столкновениях.
Кроме того, период вращения дуплета уменьшался при вращательно-
поступательном движении. ч
Течение в круглых трубках
Течение, подчиняющееся закону Пуазейля в горизонтальных
и вертикальных круглых трубках, впоследствии изучали (Гольдсмит
и Масон, 1962) путем рассмотрения по оси Z поля движения с помощью
подвижного микроскопа. В дополнение к жидким сферам диаметром
75—300 мкм применены полистирольные шарики с диаметром 450 —
600 мкм и высоковязкими маслами в качестве непрерывной среды.
Угловое вращение в большей части трубок находилось в соответствии
с теорией Джеффри (1922), но период вращения вблизи стенок трубки
260
оказался больше ожидаемого. Во всех системах жидких сфер во время
течения происходила миграция в направлении оси трубки, причем
скорость ее возрастала с радиусом капли, скоростью течения и
радиальным расстоянием. В других отношениях жидкие капли вели себя
точно так же, как в специальном вискозиметре с коаксиальными
цилиндрами. Так, при низких градиентах скорости, капли
деформировались в вытянутые сфероиды с главной осью, расположенной под
углом я/4; одновременно осуществлялась циркуляция внутренней
жидкости введением в капли малых количеств непрерывной фазы.
Твердые шарики проявляли незначительную осевую миграцию.
Разница в поведении жидких и твердых сфер объяснялась различием
между общими силами (Fp), действующими на сферы в той и другой
системах:
5я» (4У/ЛД*) Д'ЛфТк [у, , 2чф/г|сУ мф , 3\ J_ ( Пф 6 \1
Р 8(V*lc+l) IA + 5 /Uc + 5/ 4Ue+TJJ
(IV.204)
где /Сф — деформация жидких сфер.
Сила FP действует таким образом, что капля передвигается в зону
более низкого градиента скорости. Для твердых сфер D = О, так что
FP = 0.
ФАКТОРЫ,
ВЛИЯЮЩИЕ НА РЕОЛОГИЧЕСКИЕ СВОЙСТВА ЭМУЛЬСИЙ
При интерпретации реологических данных для эмульсий должно
быть принято во внимание, что много других факторов, помимо
феноменологических эффектов, перечисленных ранее, могут оказывать
некоторое влияние. Все рассмотренные факторы находятся в
зависимости от химической природы и других свойств ингредиентов,
использованных при получении эмульсий.
Ниже перечислены основные составные части эмульсии и указаны
факторы, обычно связанные с ними, которые могут влиять на
реологическое поведение.
1. Дисперсная фаза:
а) объемная доля Ф; гидродинамическое взаимодействие между
каплями; флокуляция;
б) вязкость (Чф); деформация капель при сдвиге;
в) размер и характер распределения капель, методика
приготовления эмульсии; межфазное натяжение между двумя жидкими
фазами; поведение капель при сдвиге; взаимодействие с непрерывной
фазой; взаимодействие капель;
г) химический состав.
2. Непрерывная фаза:
а) вязкость (т]с) и другие реологические свойства;
б) химический состав, полярность, рН; потенциальная энергия
взаимодействия между каплями;
в) концентрация электролита, если среда полярна.
261
3. Эмульгирующий агент:
а) химический состав; потенциальная энергия взаимодействия
между каплями;
б) концентрация и растворимость во внутренней и наружной
фазах; тип эмульсии; инверсия эмульсии; солюбилизация жидких
фаз в мицеллах;
в) толщина пленки, адсорбированной на каплях, и ее
реологические свойства, деформация капель при сдвиге; циркуляция жидкости
внутри капель;
г) электровязкостный эффект. ч
4. Дополнительные стабилизирующие агенты: пигменты,
гидроколлоиды, водные окислы; влияние на реологические свойства жидких
фаз и межфазной пограничной области.
Такая форма представления удобна, но не слишком
удовлетворительна, так как предполагает, что каждый фактор действует
независимо от других. На практике два или более факторов действуют
одновременно и общий эффект отличается по величине от суммы
индивидуальных вкладов.
При интерпретации экспериментальных данных фактор
взаимодействия часто игнорируют, что приводит к необоснованным
заключениям. Иллюстрацией этого служит простой пример. Две эмульсии
с различными объемными концентрациями Ф дисперсной фазы
приготавливают из одинаковых ингредиентов с применением одного
и того же метода предварительного смешения и гомогенизации. Затем
сравнивают их вязкости т] в широкой области скоростей сдвига.
Непосредственные заключения, касающиеся влияния Ф на г\ могут быть
сделаны только в том случае, если будет показано, что средний
размер капель и распределение размеров около среднего значения
являются одними и теми же для обеих эмульсий. Однако, возможно,
что более концентрированная эмульсия будет иметь больший средний
размер капель и более широкое распределение размеров. В этом
случае эффекты, связанные с Ф и размером капель, действуют
одновременно. Поэтому, если не будут сделаны некоторые поправки, наиболее
интересующий фактор не может быть изучен. В общем, действующие
факторы оказывают больший эффект, когда Ф увеличивается, т. е.
когда капли расположены ближе друг к другу и создается ,болыпе
точек контакта.
Реология эмульсий изучена значительно меньше, чем реология
коллоидных дисперсий, главным образом потому, что эмульсии
являются системами намного более трудными для исследования.
Дисперсную фазу составляет способная к деформации жидкость,
а эмульгирующий агент создает третью фазу в форме слоя,
адсорбированного вокруг капель, который видоизменяет силы когезии между
каплями, а также силы между каплями и непрерывной фазой. Если
при сдвиге капли лишь слегка искривлены, деформацию можно
вычислить (Тэйлор, 1934) из выражения:
_L-B_ __ i^Lc (19/16Чф + Т1с\
262
где Ьи В — размеры наибольшей и наименьшей осей; а — межфазное
натяжение.
Малые капли, не превышающие нескольких микрометров в
диаметре, обычно испытывают меньшую деформацию даже при высоких
скоростях сдвига.
Информация, касающаяся поведения таких капель при течении,
часто может быть получена путем аналогии по данным для
коллоидных дисперсий сравнимого размера. Для значительных деформаций
интерпретация данных более затруднительна, если капли не
принимают хорошо определяемую форму, например, эллипсоидальную.
Реология коллоидных систем разработана в деталях.
Дисперсная фаза
Объемная доля
Вязкость т] разбавленной эмульсии (Ф <0,05), содержащей не
деформируемые сферические кадли, дается (Эйнштейн, 1906, 1911)
уравнением
т)отн=-^-=1 + а£Ф (IV.206)
при условии, что отсутствует взаимодействие между каплями,
разделяющее их расстояние значительно превосходит их диаметр,
отсутствует скольжение у межфазной поверхности раздела и т) возрастает
при рассеянии энергии или вязком торможении, возникающем при
видоизменении движения жидкости вблизи поверхности капель.
Константа аЕ имеет значение 2,5.
Уравнение (IV. 206) может быть записано как
-^-1 = \я = аЕф <IV-2°7)
гДе Луд — удельная вязкость (относительное увеличение вязкости
эмульсии).
Отсюда
or^-=1W=aE <IV-208>
Ф-э-0
где Tixap — характеристическая вязкость.
Когда строят график г)уд — Ф, получают прямую линию с
градиентом, равным аЕ. Поскольку уравнение (IV.208) действительно только
при Ф <0,05, для очень разбавленных эмульсий требуются данные
вязкости большой точности. Недавно было сделано предположение,
что эту зависимость нельзя применять, если Ф > 0,01 (Кинч, 1954).
Далее будет показано, что лимитирующее значение для Ф зависит от
размера капель, т. е. фактора, который не фигурирует в уравнении
(IV.208).
Большинство эмульсий, представляющих практический интерес,
превышает оптимальную концентрацию, для которой действительно
263
уравнение (IV.208). При увеличении Ф взаимодействие между
каплями возрастает, благодаря более тесному сближению
принадлежащих им линий течения в непрерывной фазе, которые в конце концов
частично совпадают. На этой стадии общая вязкость не является
больше суммой эффектов, вызванных индивидуальными каплями.
Первая количественная обработка (Гут и Симха, 1936) этого
увеличенного гидродинамического взаимодействия привела к выражениям:
т1от„=1 + аЕф+14-1*2 (IV.209)
или
При такой обработке игнорировали возможность включения
других факторов, например, столкновений между каплями, слабой
агрегации (Джиллеспи, 1963а) и эффективного объема, занимаемого
каплями или скоплениями их. Последующие попытки теоретического
подхода к проблеме, в которых принимали во внимание один или
более дополнительных факторов, а также экспериментальные
исследования привели к выводу, что аЕ < 14,1 связана с Ф2.
Все относящиеся сюда уравнения более точно выражаются
многочленом с Ф, чем квадратичной функцией, так как последняя форма
приводит к существенным ошибкам при более высоких значениях Ф
11уд = аЕФ+ЬЕф2 + СЕф3 + ^ф4 ' ' ' (IV.210)
где bE, сЕ, dE — дополнительные константы.
В табл. IV.4 представлены значения для аЕ, ЬЕ и сЕ, приведенные
в опубликованной литературе. Ввиду недостатка информации,
относящейся конкретно к эмульсиям, включены данные для суспензий
твердых шариков в жидкой среде. Существенно, что, несмотря на
большую разницу в размере частиц или капель и распределении по
размерам между различными исследованными системами, значение
аЕ обычно остается 2—5, в то время как значение ЬЕ изменяется в
широких пределах. Небольшое число значений приведено для сЕ %или
констант, связанных с еще более высокими степенями Ф.
В высококонцентрированных эмульсиях (Ф >0,4—0,5 или более
низкие значения Ф для очень малых размеров капель) т]отн
увеличивается в большей степени с возрастанием Ф, чем это предсказывается
уравнением (IV.209). Течение становится псевдопластическим и
незначительное приращение Ф вызывает резкий рост т)отн. В конечном
счете эмульсии могут проявить пластическое течение.
При этих условиях резко возрастает агрегация капель, поскольку
с увеличением Ф растет число капель, находящихся в тесной близости
в каждый данный момент времени. При низких скоростях сдвига,
не вызывающих серьезных изменений в структуре агрегатов, каждый
агрегат ведет себя как отдельная сфера с объемом, большим, чем сумма
объемов составляющих его капель, потому что внутри структуры
удерживается некоторое количество непрерывной фазы (Муней, 1946).
Это изменяет соотношение эффективных объемов дисперсной и непре-
264
рывной фаз. В условиях оптимальной упаковки агрегаты связываются
в непрерывную сетку.
Внутри агрегатов капли удерживаются вместе силами притяжения
Лондона — Ван-дер-Ваальса, так что структура агрегатов, очевидно,
будет очень сильно ощущать влияние сдвига. Если скорость сдвига
возрастает, вязкость будет падать, потому что вслед за
первоначальным сжатием структуры при очень малом сдвиге агрегаты
разрушаются и жидкость непрерывной фазы высвобождается. Таким
образом, отношение эффективных объемов постепенно уменьшается до
тех пор, пока не будет соответствовать теоретическому отношению,
если только не будут затронуты эффекты растворения.
Уравнение (IV.20) может быть модифицировано путем введения
экспоненциального члена, учитывающего поправку на возможность
переноса капель из одной плоскости сдвига в другую (Томас, 1965)
при бесконечно большом сдвиге, когда агрегация отсутствует
т1уД='г£ф + ьяф2+ ' ' ' +АтехР(ВтФ) (IV.211)
где Ат и Вт — коэффициенты.
Для их значений не имеется объяснения и, более того, они
фактически не являются константами, так как их величина будет
изменяться с размером частиц и другими факторами, рассмотренными
ниже.
«Свободный» объем непрерывной фазы, в котором в
концентрированных эмульсиях двигаются друг относительно друга капли при
высоких скоростях сдвига, определяется как 1 — Уфр (где Уфр —
фракционный объем непрерывной фазы, связанный с каплями). На
основе этого выведены уравнения вязкости:
W^+T^ (IV.212)
или
. 1п^^ф (IV.213)
Константа аЕ имеет несколько интерпретаций. При значении 2,5
она удовлетворительно отвечает экспериментальным данным в том
случае, если капли не слипаются и не взаимодействуют с непрерывной
фазой. Робинсон (1949, 1957) рассматривал аЕ как коэффициент
трения, поскольку для твердых дисперсий ее точное значение зависело
от формы частиц и неровностей поверхности. Согласно Мунею (1951)
и Марону с сотрудниками (1951, 1953), V^v определяет эффект
уплотнения, который возрастает, когда вместе располагаются частицы
более чем одного размера. В простейшем примере, когда имеются
частицы только двух размеров, V$p является функцией отношения их
размеров. По мнению Ванда (1948), V$p представляет собой константу
гидродинамического взаимодействия. Свини и Геклер (1954) нашли,
что Уфр изменяется от 1,00 до 1,47, причем с уменьшением размера
частиц Уфр возрастает. Саундерс (1961) также наблюдал, что в
265
Таблица IV .4
Значения констант уравнения (IV.210) по различным данным
Литературный источник
Алг.бсрс (1957)
де Брюпн (1942, 1948)
Ванд (1948)
Ван дрр Ваардрн (1954)
Туе. и Симха (1936)
Кннч (1956)
Левитом и Лейтон (1936)
Маилей и Масон (1954)
Марон и Минг-Фок (1955)
My ней (1951)
Наваб и Масон (1958)
Диаметр частиц
или капель,
JVtKJU
Большой
частью 2 -3
—
—
100--100
0.0138—0,1025
2,5
152—422
0,7—3,0
1-10
0,1388; 0,1216
100—160
2,5-4,5
"Е
—
2,5
2,5
2,5
2,0 5,0
7,8
2,5
2,5
2,49
2,5
2,5
1,5-2,3
Константы
"к
—
4,7
7,35
7,17
6,75- -10
-
12,7
-■-
1,43
2-1,3
(в вависи-
ск
-
16,2
—
—
-
—
—
—
Оптимальное
значение
для каждого
решения
уравнения
(IV. 210)
—
-
0,50
0,33
■-
0,25
0,365
0,1838
0,50
0,17
Примечание
Эмульсии В/М
Теоретические данные
Твердые шарики
(теоретические данные)
Стеклянные шарики
Эмульсии М/В
Теоретические данные
Твердые шарики
(теоретические данные)
Эмульсии М/В
Стеклянные шарики
Эмульсии латекса
Теоретический анализ
данных Ванда
Эмульсии М/В i
Роско (1952)
Сайто (1950)
Саундерс (1961)
Симха (1952)
Томас (1965)
Хиггинботем и др. (1958)
Ченг и Шахман (1955)
Шерман (1950а)
Эйлере (1941)
Эйрих и др. (1936а)
Эйрих и др. (1936Ь)
to
0,0990—0,8710
Бесконечно
большой
53-178
0,2640
1,0-3,0
Большей
частью 2,7
125—205
4-5,5
мости от
значения
2.5
6,29-7,64
2,64-6,4
10,05
—10
0-9,7
4,94
7,8
7,8
26,9—36.3
^50
8,78
0,05
Очень
мелкие шарики
испытывают сильное
броуновское
движение
0,24
0,09
<0,25
0,325
0,02-0,08
0,47
0,797
0,20
0,08
Монодисперсные твердые
шарики (теоретические
данные)
Твердые шарики
(теоретические данные)
Эмульсии латекса'
Монодисперсные твердые
шарики (теоретические
данные)
Обзор опубликованных
данных
Метилметакрилатные
шарики
Полистирольный латекс
Эмульсии В/М
Эмульсии асфальт/вода
Стеклянные шарики
Споры
монодисперсных латексах с размером капель < 1 мкм Уфр зависят
от размера, колеблясь от 1,118 до 1,357.
Значение 2,504 для аЕ не зависит от размера капель, если при
вычислении принимается во внимание толщина слоя эмульгатора
вокруг капель.
При низких значениях Ф уравнения (IV.212) и (IV. 213) сводятся
к уравнению (IV.206). При других значениях Ф получен степенной
ряд, подобный уравнению (IV.210) с подобной же зависимостью от
размера капель.
Бринкман (1952) записал уравнение (IV.210) как
Чот„ = J— (IV.214)
(1-Ф) Е
Здесь аЕ = 2,5, если капли двигаются независимо одна от другой.
В тех случаях, когда даже при высоких скоростях сдвига
наблюдается частичная агрегация (некоторые эмульсии латексов), величина
аЕ >2,5 (Джиллеспи, 1963 b). Гатчек (1911) изучал течение
неньютоновских эмульсий и нашел, что при Ф > 0,05 линейную часть кривой
напряжение сдвига — скорость сдвига можно представить в
следующем виде:
Лотн= з^Г (IV.215)
Однако Сибри (1930, 1931) наблюдал, что уравнение (IV.215)
давало значения относительной вязкости ниже, чем
экспериментальные. Расхождение приписывалось увеличению эффективного объема
капель (АУЭфф), вызванному гидратацией слоя эмульгатора. Таким
образом
Чотн= , * (IV.216)
1-УДУэффФ
Значение AV.^ для всех эмульсий составило 1,3. Так как
эмульсии приготовлены с одной и той же непрерывной фазой, Зибрее
допускал, что АУЭфф могло зависеть от химической природы эмульгатора,
его концентрации и химической природы непрерывной фазы.
Уравнение (IV.216) имеет еще более ограниченное применение,
чем уравнения (IV.212) и (IV.213), по той причине, что АУэфф иногда
уменьшается с увеличением Ф и его значения внезапно падают ниже
единицы (Бротон и Сквайре, 1938). Его величина также изменяется
для различных дисперсных фаз эмульсий М/В (Томе, 1941) и
эмульгаторов в концентрированных эмульсиях В/М, имеющих одинаковые
жидкие фазы и одну и ту же объемную концентрацию дисперсной
фазы (Шерман, 1955а).
Ричардсон (1933) вычислил «сжимаемость» эмульсии, для которой
Ф увеличивается на АФ по аналогии с влиянием переменного
давления на материал, подчиняющийся закону Гука. Из этой зависимости
268
он вывел выражение относительной вязкости для концентрированных
эмульсий. При любой скорости сдвига
1пт1отн=*яф (IV.217)
где Kr — константа.
Позднее это уравнение было эмпирически улучшено Бротоном
и Сквайрсом (1938) для достижения более близкого согласования
с их экспериментальными данными
1п%т„ = *яф + вн (IV.218)
где Вп — константа.
Уравнения (IV.217) и (IV.218) не нашли широкого применения,
помимо тех эмульсий, для которых они первоначально выведены.
Например, в серии эмульсий В/М (Шерман, 1950а) значения как KR
так и Br изменялись с Ф и концентрацией эмульгатора. При Ф > 0,5
расхождения были чрезвычайно большими.
Вязкость
Тэйлор (1932) распространил выводы Эйнштейна для
гидродинамики разбавленных суспензий твердых сфер на разбавленные
суспензии капель в жидкой среде. Он исходил из допущений: 1) пленка
любого эмульгатора вокруг капель не препятствует передаче
тангенциального и нормального напряжений от непрерывной фазы к
дисперсной; 2) отсутствует скольжение на межфазной границе раздела
масло — вода. Эти напряжения вызывают циркуляцию жидкости
внутри капель, которая ослабляет искажение линий обтекания
вокруг капель. Окончательный эффект зависит от отношения т]ф/т)с:
^=i + aEm^-)o (IV.219)
„ Чф-Г2/5Чс
Выражение — отвечает внутренним потокам в пределах
Чф + 'Пс
капель. Когда щ > цс, уравнение (IV.219) сводится к уравнению
(IV.206); при всех других условиях величина tioth ниже, чем в случае
дисперсии твердых сфер с идентичным значением Ф. Левитон и
Лейтон (1936) видоизменили уравнение (IV.219) с тем, чтобы расширить
область его применения на более концентрированные эмульсии
сФ^ 0,4:
1пТ1оТн-^(:^^)(Ф + Ф"/3 + ф,,/3) (IV-220)
При низких значениях Ф уравнения (IV.219) и (IV.220)
идентичны. Член Ф6'» введен в соответствии с мнением Смолуховского
(1916b) о том, что он аналогичен следующему члену Ф для раскрытия
ряда уравнения (IV.210); член Ф11'3 — для достижения лучшего
согласия между теоретическими и экспериментальными
величинами т)отн и не имеет теоретического объяснения.
Справедливость уравнения (IV. 219) подтверждена изучением
целого ряда разбавленных эмульсий М/В (Наваб и Масон, 1958),
в которых капли слишком малы (2,5—4,5 мкм), чтобы испытывать
269
Таблица IV.5
Данные вязкости разбавленных эмульсий М/В при 25° С, использованные
в уравнении Тейлора (Наваб и Масон, 1958)
Состав эмульсий
СП
Т'ф + Чс~
экспериментальное
.шачение
теоретп,т '-
си ■
Данные, согласующиеся с теорией Тейлора
Бутилбснзоат в 25% водном растворе саха-
'ра + 1% тшш-20
Бутилбензоат/вода-(-1% твин-20
Бутилбензоат/вода +0,5% твин-20
Раствор касторового масла в бутилбензоате/
вода -f-1 % твин-20
2,67
2,67
2,67
4,89
2,08
0 934
0,908
0,934
1,28
2,86
2,94
5,23
0,06-0,16
0,04—0,16
0,04-0,16
0,07-0,16
1,82
2,11
2,13
2,23
J.84
2,11
2,12
2,26
2,7
4,5
4,6
6,8
Бутилбензоатный раствор CClj в 45% водном
растворе сахара + 1% твии-20
Бутилбензоатный раствор CCI4 в 45% водном
растворе сахара + 0,2 твин-20
Бутилбензоатный раствор ССЦ в 33% водном
растворе глицерина-)-1% твин-20
Данные, отклоняющиеся от теории Т з й л о р а
3,79
1,99
1,91
2,32
2,98
0,53
0,51
0,78
0,06-0,16
0,06-0,16
0,06—0,17
2,01
1,99
1,83
1,52
1,50
1,66
7,5
6,3
8,5
значительную деформацию при скоростях сдвига вплоть до 900 сек'1.
В табл. IV.5 приведены некоторые типичные данные, включая
значения для ЬЕ, вычисленные по уравнению (IV.210). Когда слой
эмульгатора вокруг капель масла не подавляет внутреннюю
циркуляцию масла, экспериментальные и теоретические значения
/ Т)(Ь+2/бт1с\ -п
аЕ I—-*— 1 близко согласуются. В противном случае теоретические
значения всегда ниже экспериментальных. Для эмульсий, подчиняв
ющихся уравнению (IV.219), аЕ I — 1, а также ЪЕ
увеличивались при возрастании т]ф/т]с. Внутренняя циркуляция всегда
понижается, если ПАВ адсорбируются на каплях. Течение жидкости
вокруг капель заставляет адсорбированное вещество перемещаться
с фронта капли в ее тыл, и это создает градиент поверхностного
давления (Линтон и Сусерленд, 1957). Градиент в несколько дин
на квадратный сантиметр достаточен для предотвращения движения
внутри капель, и они двигаются как капли жидкости высокой
вязкости или даже как твердые сферы.
Во многих эмульсиях капли окружены слоем эмульгатора,
который проявляет при сдвиге вязкоэластичные свойства. Если эта
пленка противодействует возрастанию равновесного межфазного
натяжения при увеличении площади поверхности, капли ведут себя
как твердые сферы и отношение 'Пф/'ПС не влияет на tioth (Олдройд,
1953, 1955). С другой стороны, вязкая межфазная пленка не влияет
на тип реологического поведения, проявляемого эмульсией, хотя
значения параметров могут быть переменными. Влияние вязкости,
проявляющейся при сдвиге межфазной пленки (tip"), и ее
поверхностной вязкости' (rfs1), которая является двумерным эквивалентом
объемной вязкости, на Лотт как показали измерения в опытах с
медленным достижением устойчивого состояния, дается выражением:
„ i ,^[> + 2/^lc + 2/5(2riW + 3T1^)/r]0
%ТН + Чф + т1с + 2/5(2г1?.л+Зг1п3л)/г ( ^
Таким образом, вязкая межфазная пленка оказывает влияние,
которое эквивалентно увеличению Т1ф на 2/5 (2т]Рл -f- Зт|дл)/г.
Величина (2т)Рл + Зт]"л) вычислена для нескольких разбавленных
эмульсий М/В (Наваб и Масон, 1958) и найдено, что она лежит в
пределах 0,95-10" 4—0,014-Ю-4 сек'1. Эти данные подобны (но не строго
сравнимы) значениям г\рл для пленок ПАВ на границе раздела
воздух — вода (Джоли, 1956).
При исследовании влияния т]ф на ti0TH отношение Лф/Лс надо
вычислять осмотрительно, ибо оно не обязательно должно быть
таким же, как до эмульгирования. Обычно эмульгатор растворен
или эмульгирован в жидкости, которая служит непрерывной фазой.
В некоторых случаях, например, когда приготовляют эмульсии М/В
с монолауратом сорбитана (спе"н-20), эмульгатор диспергируется
перемешиванием в водной фазе, хотя он растворим в масляной фазе. После
добавления масла эмульсия гомогенизируется; это сопровождается
271
переходом заметной части эмульгатора в масляную фазу, в
результате чего tic уменьшается, а г|ф увеличивается (Шерман, 1963b).
Для таких систем т|ф и т]с следует определять путем исследования
коагулированных эмульсий.
Любой эффект, вызываемый т]ф, в концентрированных эмульсиях
анализировать труднее из-за наложения эффектов, связанных с
взаимодействием капель, агрегацией и, вероятно, также деформацией.
Томе (1941) исследовал ряд эмульсий М/В, приготовленных с
одиннадцатью различными органическими жидкостями в качестве
дисперсной фазы и с лауратом натрия, лауратом калия, мнристатом
калия, как эмульгаторами. При этом не только отсутствовала
корреляция между т)ф и лотн, хотя отношение Цф/цс и изменялось
примерно от 0,3 до 4, но и имелись некоторые формы взаимодействия
между эмульгатором и масляной фазой. Например, в ряду эмульсий
масло/0,002Л/ раствор лаурата калия наименьшее значение п0ТН
наблюдалось с анилином как дисперсной фазой, в то время как в ряду
эмульсий масло/0,002М раствор олеата калия анилин дал наиболее
высокое значение Т|отн.
Подобные результаты получены Шоттоном и Уайтом (1960),
которые нашли, что эмульсия М/В с наиболее высоким значением ц011{
приготовлена из масла с наименьшим значением г]фпри Ф = 0,1—0,6.
Ни в одной из публикаций не приведены данные о размере капель.
Однако, судя по принятой методике повторной гомогенизации, вряд
ли он будет таким, чтобы капли испытывали значительную
деформацию при сдвиге.
Отсутствие корреляции между Цф и лотн обнаружено также
в высококонцентрированных неньютоновских эмульсиях (Шерман,
1955b). Большие изменения в отношении г|ф/т)с достигнуты при
использовании 0—80% глицерина в дисперсной фазе, однако,
несмотря на это, ни вязкость при высокой скорости сдвига (л, о-,), ни
экстраполированное предельное значение не показали какого-либо
заметного изменения (табл. IV.6).
Таблица IV.6
Влияние т)ф на вязкость эмульсий В/М
Состав масляной фазы, вес. %: 89,02 минерального масла (г) = 0,136 пз), 8,29 глицерил
монорицинолеата, 2,69 лецитина сои; Ф = 0,717; 25° С.
Содержание
глицерина в
водной фапе.
вес. %
0
6,5
13.0
19,5
25,0
70,0
80,0
Иф, пз
0,0114
0.0140
0,0170
0,0232
0,0295
0,2167
0,5588
V пз
0,2475
0.2475
0,2475
0,2475
0,2475
0,2475
0,2475
Vic
0,046
0,057
0,069
0,094
0,119
0,875
2,257
Ноо' пз
3,10
3.10
3,16
3,10
3,14
3,06
3,00
Экстраполированное
предельное
значение
2623
2523
2432
2450
2623
2926
2600
272
При содержании в водной фазе 36,30% пропиленгликоля,
глицерина или сорбита, изменения в т)ф/г)с были намного меньше и на т]отн
не оказывалось значительного влияния (табл. IV.7). Однако, когда
к масляной фазе добавляли небольшое количество газовой сажи
в качестве дополнительного стабилизирующего агента, три эмульсии
показали отчетливую разницу в реологических свойствах.
Таблица IV.7
Влияние т)ф на вязкость эмульсий В/М,
содержащих газовую сажу в масляной фазе
(Шерман, 1955b)
Состав масляной фазы, вес. %: 89,52 минерального масла (т) = 0,126 пз); 1,86
окисленного рапсового масла (т)= 104,4 пз); 8,62 лецитина сои. Ф = 0,661; 25°С.
Добавка 36,3 вес. %
компонента к водной
фазе, вес. %
Экстраполированное предельное
значение
Пропиленглнколь
Глицерин
Сироп сорбита
Без добавки газовой сажи
0,323
0,0275
0,0200
0,1437
0,1437
0,1437
Добавка 5,3 вес. %
Пропиленглнколь 0,0323
Глицерин 0,0275
Сироп сорбита 0,0200
2,10
2,17
2,18
газовой сажи
6,0
8,9
10,6
1615
1514
1640
3632
5449
6659
Эмульсия, содержащая в водной фазе пропиленгликоль, т. е.
с наиболее высоким значением т]ф, дала наименьшую величину ц^
и низкое экстраполированное предельное значение. Экстраполяция,
вероятно, связана с данными удельной адсорбции для газовой сажи
- в различных водных средах, так как состав масляной фазы был одним
и тем же для трех эмульсий. Удельная адсорбция, которая в этом
случае отражает степень, до которой газовая сажа смочена водной
фазой, была наименьшей в водной фазе, содержащей пропилен-
гликоль.
Таким образом, химический состав дисперсной фазы может влиять
на конфигурацию поверхности раздела масло — вода даже при
ОТСУТСТВИИ ВЛИЯНИЯ Г|ф на Цсо-
Распределение капель по размерам,
Уравнение (IV.206) не содержит члена, включающего размер
капель, и, так как многие зависимости вязкости более
концентрированных систем являются результатом распространения этого
уравнения, любой эффект, связанный с размером капель, игнорируется.
В большом количестве опубликованной литературы не содержится
сведений о степени дисперсности изучаемых систем. Немногим лучше
положение, когда эти данные имеются: средний размер капель может
18 Заказ 1(17 0
273
быть в пределах от <0,1 до > 100 мкм. Для примера несколькими
сотрудниками проверена справедливость уравнения (IV. 210) на
предварительно опубликованных данных, где размеры для различных
систем избирательно варьировались от одного крайнего предела -
до другого. Влияние эффекта этих вариаций на значения
выведенных констант (см. табл. IV.4) не было оценено.
Уравнения (IV.215) —(IV.218), которые определяют в разных
формах вязкость концентрированных эмульсий, также не содержат
выражений, отражающих влияние размера капель. Поэтому не
удивительно, что для гипотетических констант в этих уравнениях,
например коэффициента гидратации, сообщались различные
величины: в одном случае они относились к эмульсиям, в которых размер
капель был иногда < 10 мкм, в другом — к эмульсиям такого
широкого распределения размеров, что включались капли диаметром
20-340 мкм. (Сибри, 1930, 1931).
До последнего времени данные о размере капель можно было
найти лишь в работах Левитона и Лейтона (1936), а также
Ричардсона (1950, 1953). Левитон и Лейтон наблюдали, что вязкость
разбавленных жидких эмульсий М/В не изменялась, когда диаметр
капель уменьшался от ~ 3,0 до — 0,7 мкм. Они допускали, что
размер капель оказывает небольшое влияние на вязкость
разбавленных эмульсий, если капли неплотно упакованы. Исследований с более
концентрированными эмульсиями для подтверждения этого взгляда
не было проведено.
Ричардсон исследовал концентрированные эмульсии М/В с Ф =
= 0,75, очень вязкие, со склонностью к неньютоновскому течению.
Он нашел, что г^ пропорциональна обратной дроби среднего
размера капель (-Dcp), а произведение r|00.DCp не изменяется при
небольшом разбросе размеров около среднего значения.
Единственными уравнениями, которые включают выражение для
размера капель, являются уравнение (IV.221) и второе,
предложенное Гопалом (1960):
2 2т)оЛф / 1
25
о
'ср /J
ф
Когда т]ф -> со
11отн=1 + [2.5 + |^(^)]ф (IV-223)
Уравнения (IV.221) и (IV.222) применимы только к очень
разбавленным эмульсиям. Несколько уравнений описывают дисперсию
твердых сферических частиц в жидкой среде, но даже в них не
включено специфическое выражение для размера частиц. Вместо этого
предложены взаимоисключающие формы, зависящие от того, будет ли
суспензия иметь гомогенное или гетерогенное распределение по
размерам (Роско, 1952; Мари и Отатаке, 1956; Пинг и Любберс, 1957).
274
В табл. IV.8 приведены данные о влиянии размера капель или
частиц на вязкость эмульсий и дисперсий твердых частиц.
Размер капель оказывает сильное влияние на вязкость
псевдопластических эмульсий В/М и М/В, стабилизированных неионными
эмульгаторами, в широкой области Ф (Шерман, 1960). При Dcp <
< 2 мкм в эмульсиях В/М величина г\т значительно увеличивалась
с уменьшением размера капель при всех значениях Ф (рис. IV.22).
Эффект менее выражен в эмульсиях М/В и проявляется только при
Ф >0,5 (рис. IV.22, б).
6
— з
\
?
Ofi
0.3
С:
%0;2
Р-
0,1
~
N.
'-^
8
— 1 1_
\tf
N
Orn. ""■
J
Dcp.tKM
Рис. IV.22. Влияние Дср на т)^ для эмульсий, стабилизированных полутора-
олеатом сорбитана:
а — эмульсии В/М; б — эмульсии М/В. Ф: 1 — 0,7280; 2 — 0,6739; 3 — 0,5704; 4 — 0,3707;
J — 0,1809; 6 — 0,6775; 7 — 0,5802; 8 — 0,4805; 3 — 0,3260.
Это различие связано с реологическими свойствами
соответствующих пленок ПАВ вокруг капель, причем эмульгаторы типа В/М
дают пленку с высокой поверхностной вязкостью, а эмульгаторы
типа М/В — с низкой вязкостью. Если эти данные нанести в форме
кривых т]отн — Ф для выбранных значений среднего диаметра капель
(рис. IV.23), становится ясно, что любое уравнение, связывающее г\0ТН
с Ф, должно включать выражение для D или Dcp.
При высоких скоростях сдвига капли в эмульсии полностью
деформированы, структура разрушена и капли находятся на равных
расстояниях одна от другой (являются эквидистантными). Если
йапли ведут себя как твердые сферы, среднее расстояние между
ними может быть вычислено из уравнения:
ср-
D,
ср
V
ф
-1
(IV.224)
где Фм
максимальная объемная доля дисперсной фазы.
18*
275
Таблица IV .8
Влияние размера частиц или капель на вязкость (но данным различных авторов)
Система
.Чо.чи fie кто пита
Синтетический латекс-
iii.iii гель
Суспензии
кремнезем
полимстплметакри-
латные шарики
Размер,
МИМ
0,010-0,200
0,052-0,325
0,0075—0,0457
53-76; 76-89;
76-178; 152-178
152-177
2-15; 2-30:
15-30; 30-42;
30-54; 40—54;
150-240;
345-435
Ф
SS 0,015
0,193—0,600
0 0,03
0,025-0,325
>
0,30
0—0,225
Наблюдения
чотн увеличивается с уменьшением
размера; очень большое
инкрементальное увеличение при
наименьших размерах
Г|отн увеличивается, когда размер
уменьшается и распределение по
размерам становится более
однородным
т)отн увеличивается с уменьшением
размера; прогрессивно
возрастающее инкрементальное увеличение
при уменьшении размера
г]отн не проявляет зависимости от
размера или распределения по
размерам
"Чотн не зависит от абсолютного
размера, по является функцией
распределения размеров, уменьшаясь
с увеличением диапазона размеров
до постоянной величины
чотн ПРИ любом значении Ф меньше,
чем т)оти для суспензии,
содержащей только один из входящих
размеров. Уменьшение очень, резкое,
когда Ф>0,175 и размеры в смеси
различаются настолько широко,
насколько возможно. Все суспен-
Литература
Хаузер и Ле Бью
(1939)
Фрилинг (1963)
Гринберг и др. (1965)
Хиггинботем и др.
(1958)
Уорд и Витмор (1950)
Ивсон (1959)
стеклянные шарики
в жидкой среде
стеклянные шарики
или шарики из
кремнезема
Смесь двух латексов,
каждый с
гетерогенным распределением
размеров
,')мульсии
М/В
монодисперсныи по-
листирольный
латекс
15
15
40-200
2-10; 4; 8; 12;
0.1—1,0; 0,025
2-10; 4; 8; 12;
0,1-1,0; 0,025
Средние
диаметры 0,0980
н 0,1920
0,7—3,0
0,55-2,4
0,099—0,871
0,0233-0,2422
зии проявляют ньютоновское
течение
не зависит от среднего разме
т)отг( увеличивается
размера
с уменьшением
ера |
шм \ И
Г)оти возрастает с уменьшением
размера
тЬтп увеличивается с уменьшением
размера, по эффект не указан
Ньютоновское течение вплоть до
Ф = 0,05, когда размер > 1 мкм;
становится неньютоновским при
Ф<С0,05 и размере <; 1 мкм.;
тенденция к возрастанию тик-
сотропии, когда размер
уменьшается
т)отн смеси меньше, чем для двух
составляющих фракций. Минимум
па графике т)отн — D, который
становится отчетливее при более
высоких значениях Ф и сдвигается
в сторону больших величин Ф
Размер не влияет на г]отн при Ф <; 0,5
т)отн при высоких скоростях сдвига
увеличивается,когда размер
уменьшается
"По™ увеличивается с уменьшением
размера даже при Ф < 0,05; очень
. большое увеличение на единицу
изменения размера при
наименьших размерах
всон (1957)
Суиней
(1959)
и Геклор
Вильяме (1951
1953)
Марон и Мадоу (1953)
Левитон и Лейтон
(1936)
Ричардсон (1950,
1953)
Саундерс (1961)
Во многих случаях Фмакс «» 0,74 при узком распределении
размеров. Микроскопическое исследование эмульсий В/М обнаружило,
что при Ф > 0,75 появлялись множественные эмульсии. Зависимость
т1оо/'Пс ~ 4р Для всех эмульсий В/М, приготовленных с одним
эмульгатором, давала экспоненциальную кривую, охватывающую данные
вязкости для всех значений Ф (рис. IV.24). Этот метод представления
данных вязкости имеет очевидные преимущества. Более того, он
дает объяснение влиянию Dcp на п^/г^ с точки зрения взаимодействия
между каплями. При Zcp <0,5 мкм "Ц^/Цс прогрессивно
увеличивалось с уменьшением
очень большим.
Zcp, при Zcp <0,1 мкм возрастание цт/цс было "
Рис. IV. 23. Данные вязкости для
эмульсий В/М, стабилизированных полутора-
алеатом сорбитана (Шерман, 1965):
1,4; 2 — 2,0; 3 — 3,3; 4 — 0.
Рис. IV.24. Влияние /
ср
нат|с
для
эмульсий, стабилизированных сеск-
виолеатом сорбитана.
D , мкм: 1
Уравнение (IV.224) показывает, что для малых капель
критическая величина Zcp достигается при более низких концентрациях
дисперсной фазы, чем для больших (рис. IV.25). Если Dcp =s; 2 мкм,
значение 1ср понижается до 0,5 мкм при Ф <^ 0,5.
Более крупные капли в эмульсиях М/В, достигающие «=< 16 мкм
в диаметре, по-видимому, претерпевают деформацию при высоких
скоростях сдвига. В этом случае данные не могут быть точно
интерпретированы тем же методом, что для эмульсий В/М. Кроме того,
более концентрированные эмульсии М/В оказались множественными
(Шерман, 1936b). Представление данных, согласно уравнению
(IV.224), давало экспоненциальную зависимость между Лоо/Лс и ^ср
при Фг^0,634 (рис. IV.26). При еще более высоких
концентрациях дисперсной фазы экспериментально полученное
значение 1ср было ниже теоретического, ожидаемого в случае
деформации капель.
278
Данные для эмульсий М/В согласуются с уравнением
Чсо = *
ч Д
'Р
(IV.225)
где Н^ — градиент; Сп — отрезок на оси х\са-
Когда Ф = 0,678, константа Ся = 0, так что произведение
Лоэ^ср = const. Это же наблюдали Ричардсон (1950, 1953) и Лоуренс
и Ротвелл (1957) для эмульсий М/В, стабилизированных олеатом
натрия, при Ф = 0,65. Значение этих фактов не следует
переоценивать, так как они не
подтверждаются для других
концентраций дисперсной фазы.
0.3
ф
0,5
0,7
Рис. IV.25. Влпянпе Всп на концент- Рис. IV.26. Влияние I,
JT 11*-/. IV.-О. и.ШЛППС Lf Ср
рацию Ф, при которой ZCD снижается
до 0,5 мкм.
FP,,
для эмульсий М/В.
Лоз/Лс
Соответствующее уравнение вязкости может быть выведено из
рис. IV.24 (Шерман, 1965). Так как In (Цсо/цс) = 0, если 1/1ср очень
мало,
1-/ZC
"ср
In Лоо/Лс достигает своего максимального
э, уравнение принимает форму
ср
значения при
In-
1
■Пс
'л 7
ср
(IV.226)
где aD — константа, зависящая от Dcp (даже при 1ср-+0 значение
Лоэ/Лс зависит от Z>cp).
Уравнение (IV.226) применено к данным вязкости (как
ньютоновской, так и неньютоновской) ряда систем, для которых известны
размеры частиц и их распределение. Сюда относились эмульсии В/М
и М/В, стабилизированные неионными эмульгаторами, синтетические
латексы и дисперсии твердых шариков. Несколько систем были
монодисперсными. Многие имели узкое распределение по размерам,
поэтому в качестве характерного размера была принята величина Dcp.
Для дисперсий твердых сфер использовали значение Dcp,
установленное ранее, или за среднее бралась половина между
минимальным и максимальным значениями (Робинсон, 1949, 1957).
279
Данные для каждой монодисперсной или относительно
монодисперсной системы показывают линейную зависимость между
lS Псо/Пс и 1/Zcp (рис. IV.27, а)
\g^- = an-^--A. (JV.227)
Чс
'П
ср
где а — наклон прямой; А^ — отрезок на оси ординат, который
во всех случаях приближается к 0,15.
Разбавленные системы (рис. IV.27, б) подчинялись уравнению
(IV.227), когда Ф достигала такого значения, что уравнение (IV.206)
становилось недействительным.
о/*
2 0,2
//>-
О
10
'/'ср
15
го
О 0,5 1,0 1,5
'A CD
Рис. IV.27. Данные вязкости для дисперсии стеклянных шариков в
водно-глицериновом растворе (а) и для полистпрольных латексов (б) (Шерман, 1965).
D„ , мкм: J — 12; 2 — 8; 3 — 4; 4 — 0,871; 5 — 0,424; 6 — 0,249; 7 — 0,148; ^—0,099.
В табл. IV.9 приведены данные вязкости, использованные в
уравнении (IV.227). Очевидно, что aD увеличивается параллельно Dcp
и при D или Dcp больше 1 мкм их взаимное влияние
удовлетворительно определяется выражением
(IV. 228)
(IV.228)
Ид = 0,036 (Dcp)2
Совместное решение уравнений (IV.224), (IV. 227)
дает:
In = -
0,036Д
<=Р
Пс
i Лфм
-0,15
(IV. 229)
У
ф
-1
Это уравнение недействительно для полидисперсных систем.
По-видимому, его форма изменяется со степенью негомогенности
системы, так как влияние aD на Dcp ослабевает, когда распределение
размеров становится шире. Имеется недостаточно данных, способных
прояснить этот вопрос. Необходимо систематическое изучение
хорошо определяемых дисперсий, в которых негомогенность могла бы
быть установлена на любом желаемом уровне. Уравнение (IV.229)
280
Система
Размер частиц,
мкм
Эмульсии В/М:
моноолеат сорбитана
полутораолеат сорбитана
триолеат сорбитана
моноолеат полиоксиэтилен
сорбитана
Эмульсии М/В:
асфальт
латекс
монолаурат сорбитана
Дисперсии твердых сферических
частиц:
метилметакрилатные шарики
стеклянные шарики
1,3-2,8
1,0-2.5
1,0-3,3
2,7-3,2
Моно дисперсные
3,4-5,7
2-15
15-30
3-4
10—20
20-30
Моно дисперсные
Таблица IV.9
Данные, использованные в уравнении (IV.227)
(Шерман, 1965)
D , мкм
2,0
1,75
2,5
3.0
2,7
0,195
0,1388
0,11
4,5
9,4
21,9
3,5
15
25
130
4
8
ап
0.10
0,15
0,21
0,38
0,32
0,05
0,02
0,03
0,52
2,2
5,2
1,0
2,5
4,5
30
1
2,3
Оптимальное
значение Ф
0.50
0.57
0.50
0,40
0,57
0.60
0.60
0,52
0,64
0,13
0,20
0,37
0,50
0.45
0.50
0,40
0,50
Литературный источник
■
Шерман, 1965
Эйлере, 1941
Марон и др., 1951
Марон и Минч-Фок,
1955
Марон и Леви-Паскаль,
1955
Шерман, 1965
1 Ивсон, 1959
Робинсон, 1951
Робинсон, 1949
Робинсон, 1957
Ванд, 1948
1
Вильяме, 1953
не применяется при Dcp <С 1 мкм: на графике, связывающем aD
и Dcp, криволинейность увеличивается, когда Dcp уменьшается
(рис. IV.28). Это указывает на заметное усиление влияния,
вызванного Dcp.
Расстояние 1ср в уравнении (IV.224) связано зависимостью с
расстоянием Ls, на котором капли конечного размера, находясь на
противоположных сторонах гипотетического пространства,
ограниченного сферой, взаимодействуют в присутствии .лежащих между
ними капель (Симха, 1952; Кинч, 1954):
■А
ср
+ ДсР/2
(IV. 230)
Коэффициент гидродинамического
взаимодействия определяется как:
Kv
D,
р-\
2Lsj
DcJ2LS
(IV. 231)
Рпс
IV. 28.
Для Dcp/2LS = 0,5—1 Кт
соответствует 2lcp/Dcp. Коэффициент Квз
значительно увеличивается, когда Dcp/2LS >
> 0,5. Поэтому, согласно этой
теории, чоо/^с будет также возрастать
для £>ср, равного 1 или 2 мкм при 1ср
менее 0,5 и 1,0 мкм, соответственно.
Дальнейшее развитие теории Симха (1952) выявляет ее
недостатки, так как уравнение течения для концентрированных
дисперсий выводится путем обобщения уравнения (IV.206)
(< 1 .iiK.it) на aD (Саундерс,
1961).
Мое
Мс
= 1-
-а£Кв
,Ф
(IV.232)
при этом аЕКвз~^>2,Ъ.
Данные табл. IV.5 показывают, что гидродинамическое
взаимодействие между твердыми шариками влияет только на более высокие
степени Ф при раскрытии рядов, даваемых уравнением (IV.210).
Кроме того, совместное решение уравнений (IV.224) и (IV.231)
показывает, что Квз не испытывает влияния Dcp. К такому же заключению
можно прийти, если в уравнение (IV.231) ввести выражение Симха
для Ls. Это противоречит экспериментальным данным.
Томас (1965) проанализировал опубликованные данные по
вязкости в широкой области и нашел, что в случае зависимости
Т1отн _ ф разброс данных от средней величины tioth составлял ±20%
при Ф = 0,02 и ±75%, когда Ф = 0,5. Приводимые размеры частиц
колебались от 0,099 до 435 мкм. Томас предположил, что влияние Dcp
и vc могло быть сведено до минимума путем экстраполяции
данных к D -v оо и vc -> оа. При использовании метода
наименьших квадратов получен степенной ряд Ф, напоминающий урав-
282
нение (IV.210), который дал результаты, подобные предсказанным
теорией Симха (1952). Это подтверждает, что последняя теория не
принимает во внимание влияние D или Dcp на вязкость.
Суини и Геклер (1954) и Саундерс (1961), по-видимому, впервые
показали в полуколичественной форме, что размер частиц или капель
влияет на свойства потока. Для размеров 0,099—0,871 мкм
некоторое влияние имелось уже при Ф <0,05. При еще меньших размерах
(0,0075—0,0457 мкм) это влияние проявляется при более низких
значениях Ф (Гринберг и др., 1965). Поэтому уравнение (IV.206)
не применимо для указанных размеров даже в пределах ограниченной
области концентраций, для которой оно первоначально
формулировалось. При диаметре частиц 0,264 мкм отклонения от уравнения
(IV.205) наблюдались, когда Ф превышало 0,02 (Ченг и Шахман,
1955), в то время как при 0,088 мкм величина т]отн проявляла
зависимость от скорости сдвига при неожиданно низком значении Ф
(Коллинз и Вейленд, 1963):
Зависимость 1ср как от Ф, так и от Dcp помогает объяснить
противоречивые взгляды, отраженные в табл. IV.8. Когда Dcp > 10—
20 мкм, он будет вызывать значительное влияние на tioth или Т)оо/т1с
только при умеренно высоких значениях Ф. С другой стороны, когда
Dcp <z 1 мкм, этот эффект выявляется при Ф = 0,05.
Усиление влияния размера частиц на т]отн с уменьшением
размера (особенно <1—2 мкм) ставит вопрос, на который в настоящее
время не имеется простого ответа. Все практически важные эмульсии
имеют какое-то распределение по размерам, так как они никогда
не бывают истинно монодисперсными, но Dcp все же представляет
некоторое среднее значение этого распределения. Когда Dcp меньше
нескольких микрометров, эффект, вызываемый присутствием капель,
диаметр которых ниже Dcp, будет больше эффекта, связанного с
каплями размером более Dcp.
Все методы вычисления среднего размера, не считая простого
среднего арифметического, включают степени соответствующих групп
размеров. Для размеров <с 1 мкм, которые фактически оказывают
наибольшее влияние на вязкость, эти степени не делают
значительного вклада в средний размер. Это значит, что график т]отн — Dcp
не отражает полного влияния капель размером менее Dcp. Если эти
размеры в основном < 1 мкм и составляют умеренную часть общего
числа капель на кубический сантиметр эмульсии, график этого типа
может быть ненадежным. Точно так же, когда сравниваются данные
вязкости для эмульсий с одинаковыми значениями Dcp, необходимо
определить, будет ли одним и тем же характер распределения.
Дисперсионный анализ должен проводиться в установленном
порядке, если из измерений вязкости не следует, что различные
эмульсии будут иметь идентичные средние размеры просто потому,
что они приготовляются по одной и той же методике. Это касается
случаев, когда сравниваются эмульсии с различными
концентрациями дисперсной фазы. Например, эмульсии М/В с величиной Ф,
колеблющейся от 0,0526 до 0,1835, были гомогенизированы под
283
давлением между 68 и 204 am с приращением в 34 am (Шерман,
1961).
Кривые изменения Dcp с возрастанием давления гомогенизации
(рис. IV.29) ясно указывают, что применением более высокого
давления к более концентрированным эмульсиям достигается такое же
значение Dcp, как и в разбавленных эмульсиях. Масло в очень
разбавленных эмульсиях эффективно диспергировалось при 34—68 am,
так как повышение давления гомогенизации приводило к
небольшому дальнейшему уменьшению Dcp. Это не касалось более
концентрированных эмульсий, потому что пониженное давление
гомогенизации давало более
высокое первоначальное
значение Dcp и для его
эффективного снижения до уровня,
достигнутого в разбавленных
эмульсиях, требовалось
повышение давления (табл. IV.10).
При 204 am колебания в
величинах Dcp для различных
значений Ф было совсем
малым. Голден и Фиппс (1964)
подтвердили эти наблюдения
для широкой области
концентраций дисперсной фазы.
Они также предложили
Рис. IV.29. Влияние размера капель на ЭМПИрИЧеское уравнение для
вязкость эмульсии при 21 С для различ- ,r J r „ "
ных объемных концентраций дисперсной разоавленных эмульсии, ко-
фазы (Шерман, 1961). торое связывает Dcp с давле-
ф: 2- 0,1835; г-- 0,1478; з - o.ioi; 4 - о,оо5. нием гомогенизации Рг:
д
ср
-т
эфф
(IV. 233)
где Рг — давление, отнесенное к единичному размеру капель; дэфф
коэффициент, характеризующий эффективность гомогенизатора.
Таблица IV.10
Влияние PF и Ф на дисперсность
(Шерман, 1961)
ф
0,0526
0,1010
0,1478
0,1835
Р (в am),
необходимое для получения
D =1 мкм
ср
102
108
150
190
D после
гомогенизации при
68 am
1,01
1,17
1,24
1,41
2 0 4 am
0,94
0,84
0,95
1,03
284
Как </Эфф, так и Р°г уменьшались с увеличением Ф. Для более
концентрированных эмульсий с Ф > 0,15 график lg D — lg Pr
не был больше строго линейным; кроме того, скорость, при которой
эмульсия текла через гомогенизатор, также влияла на Dcp.
Непрерывная фаза
Вязкость эмульсий часто выражают как разность вязкостей
эмульсии и непрерывной фазы. Необходимо отметить, что tic — это вязкость
чистой непрерывной фазы, а не вязкость той жидкости, к которой
добавлены другие вещества. Так, обычно в этой фазе растворяются
или диспергируются эмульгатор, гидроколлоиды, тонко
раздробленные твердые вещества и т. д. Каждый компонент вносит свой вклад
в т]с. Если в результате этих добавок непрерывная фаза приобретает
свойства неньютоновской жидкости, эмульсия также будет
проявлять такие свойства, даже если она очень разбавлена.
Массоперенос эмульгатора, вызванный адсорбцией на
поверхности капель, снижает г\с, но убыль концентрации является
незначительной до тех пор, пока эмульгатор проявляет тенденцию
к диффузии через межфазную границу раздела.
В свежеприготовленных эмульсиях капли под действием диффузии
сталкиваются друг с другом, что приводит к флокуляции. Когда
скорость коалесценции мала по сравнению со скоростью флокуляции,
капли образуют агрегаты прогрессивно возрастающего размера.
Далее будет показано более детально, что скорость флокуляции
определяется общим уравнением (Смолуховский, 1916, 1917)
ЯК (Т/Т,, )*"!
Nk= 1 u\ (IV.234)
(l+T/T1/f)*+l
где ti/, — время, принятое для уменьшения NK до NJ2.
Для неполярной непрерывной фазы Ti/2 находят по формуле:
Ч = Ш7 <IV-235>
Зг),
лт.
Для полярной непрерывной фазы (Дэвис и Райдил, 1963)
'■/.-WW •"•"'"
где АС/фл — потенциальный энергетический барьер флокуляции
в первичном минимуме.
Таким образом, NK увеличивается с повышением г|с. Кроме того,
расстояние, разделяющее капли в пределах агрегатов, зависит от
полярности непрерывной фазы. Оно будет меньше в неполярной
среде, чем в полярной, потому что диффузная природа двойного слоя
в первой (Альберс и Овербек, 1960) такова, что силы притяжения
будут больше. Эти факторы отражаются на поведении обоих типов
эмульсии при низких скоростях сдвига; при этом удобно изучать
285
структуру агрегатов. Агрегаты в эмульсиях с неполярной
непрерывной средой будут разрушаться медленнее с увеличением
скорости сдвига, чем агрегаты в полярной среде. Точно так же, чем
больше NK, тем выше т)отн.
Недавние исследования на тонких пленках, не превышающих
100—250 А толщиной, показывают, что их вязкость значительно
больше, чем вязкость той же жидкости в объеме. Для водных пленок
эти расхождения приписывали влиянию электрического заряда
(Дерягин и Самыгин, 1951; Дерягин и Гитиевская, 1959). Подобные
явления потом наблюдали для пленок неводнон природы (Фукс,
1958; Карасева и Дерягин, 1959). В высококонцентрированных
эмульсиях капли разделены очень тонкими пленками непрерывной
фазы даже при высоких скоростях сдвига. Если результаты изучения
вязкости тонких пленок могут быть распространены на концентри--
рованные эмульсии, возможно, что высокая г|отн последних отчасти
вызвана необычно большим значением т|с при этих условиях. С другой
стороны, первоначальное быстрое снижение т|отн, наблюдающееся
при применении сдвига, может быть вызвано некоторым падением Г|с,
так как при частичной дефлокуляции увеличивается расстояние
между каплями.
Эмульгирующий агент
Химическое строение и концентрация
Выше обсуждалось влияние эмульгатора на внутреннюю
циркуляцию жидкости в каплях при высоких скоростях сдвига и его
воздействие на вязкость. Химическая природа эмульгатора также
Таблица IV.11
Влияние строения молекулы эмульгирующего агента
на вязкость эмульсий
(Шерман, 1955а)
Эмульгирующий агент
Лецитин сои
Моноолеат сорбптана
Полутораолеат сорбптана
Триолеат сорбитана
Моноолеат нолиатиленгликоля
(200)*
Моноолеат пропиленгликоля
Моыолаурат диглпколя
Продутое соевое масло
Полприцинолеат глицерина
Полприцпнолеат полнэтиленгли-
коля
ф
0,688
0,687
0,687
0,686
0,686
0,686
0,686
0,686
0,686
0,686
Чс
0,168
0,176
0,176
0,169
0,151
0,149
0,144
0,184
0,358
0,93
^оо
1,10
2,67
2,60
3,22
2,10
2,20
2,14
2,72
4,21
3,90
Лес
■"с
6,55
15,14
14,77
19,03
13,91
14,76
14,86
14,78
11,76
4,15
полированное
предельное
значение
807
1456
1766
1922
1214
1614
1221
2339
2825
2371
* Относится к молекулярному весу полиэтиленгликолевой цепи.
286
оказывает влияние на т]отн (Вильсон и Паркес, 1936; Бротон
и Сквайре, 1938; Самнер, 1940). Концентрированные эмульсии с
одинаковыми концентрациями дисперсной фазы, приготовленные из
одних и тех же масляной и водной фаз, но с разными эмульгаторами,
показали заметную разницу в MooAle (табл. IV.11).
Так как размер капель также оказывает влияние на Цо^/Цс
действие, вызываемое составом эмульгатора, может исследоваться
изолированно только в случае, если данные вязкости представлены
графически как функция от 1ср. Это показано для некоторых
эмульсий на рис. IV.30. Эмульсии М/В, приготовленные с монолауратом
сорбитана, при малых значениях /ср проявляют аномальные эффекты
из-за деформации кацель.
Поэтому экстраполяция данных
(пунктирная линия) дает
только теоретические значения
для "Цос/ц,. при 1ср <0,5 мкм.
Эмульсии В/М,
стабилизированные моноолеатом полиокси-
этилен сорбитана, обращаются,
когда /ср <0,5 мкм.
Химическая природа
эмульгатора важна также для
исследований при малых сдвигах.
В обоих типах эмульсий она
влияет на расстояние,
разделяющее флокулированные капли:
в эмульсиях В/М капли
разделены расстоянием,
приблизительно соответствующим
двойной длине углеводородной цепи (Альберс и Овербек, 1960),
в эмульсиях М/В эмульгатор влияет на величину потенциального
энергетического барьера Д£/Макс, препятствующего тесному
сближению капель.
Концентрация эмульгатора влияет на т)отн и тем сильнее, чем
выше концентрация дисперсной фазы. Анализ некоторых публикаций
показал, что эффект мог бы быть удовлетворительно описан
эмпирическим уравнением, которое имеет близкое сходство с уравнением
(IV.218) при замене KR концентрацией эмульгатора, умноженной
на константы (Шерман, 1959).
Повышенную вязкость, вызванную высокой концентрацией
эмульгатора, часто приписывают увеличенной адсорбции
эмульгатора вокруг капель. Это может быть верно в отдельных случаях
(например, с протеинами), но для многих эмульгаторов
маловероятно, чтобы адсорбционный слой был больше толщины одной
молекулы. Как только формируется плотный мономолекулярный слой
вокруг капель в эмульсиях, приготовленных с этими эмульгаторами,
все избыточные молекулы эмульгатора ассоциируются, образуя
мицеллы в непрерывной фазе. Эти мицеллы лишают подвижности
287
Зг
»*'
0,1
0,3
0,5
Lcp,«
0,7
0,9
Рис. IV.30. Влияние химической
природы эмульгирующего агента на
вязкость эмульсии:
1 — монолаурат сорбитана (В/М); 2 — моно-
олеат сорбитана (В/М); з — смесь
мороженого (М/В); 4 — триолеат сорбитана (В/М);
. S — ПОЭ моноолеат сорбитана (В/М).
жидкость непрерывной фазы внутри себя так же, как это происходит
с мицеллами масла в водной среде. Для растворов многих ПАВ это
выражается в нелинейности графика вязкость — концентрация,
но в эмульсиях проявляется суммарный эффект. Потеря подвижности
непрерывной фазы изменяет отношение объема дисперсной фазы
к «свободной» непрерывной фазе, приводя к соответствующему
увеличению чоо/Лс- Чем больше избыток эмульгатора, тем больше объем
иммобилизованной непрерывной фазы.
На рис. IV.31 показано, как увеличивается Лоо/Лс для эмульсий
В/М в пределах области Ф = 0,37—0,68, если концентрация моно-
олеата сорбитана возрастает от 1,5 до 6,0% (Шерман, 1963b). Чтобы
*\- 1 1 1_
0,1 0,3 0,5
I ср, мкп
Рис. IV.31. Влияние концентрации эмульгатора на
вязкость эмульсии (Шерман, 1963b).
Концентрация моноолеата сорбитана, %: 1 — 1,5; 2 —
3,0; 3 — 4,5; 4 — 6,0.
исключить эффект, вызванный переменным значением Dcp, данные
выражают как зависимость г|га/г|с от 1ср.
С помощью такого графика вычисляют чистое влияние
иммобилизации жидкости на 1Qоо/Лс Для трех значений 1ср. При этом Z)cp берут
равным 1,50 мкм в каждом примере (табл. IV.12). Концентрацию
эмульгатора выражают как число, кратное ККМ. В приведенных
примерах Лот/^с увеличивается линейно и приблизительно с той же
скоростью, с какой возрастает концентрация эмульгатора.
Экстраполяция данных дает >1оо/чс ПРИ ККМ. Каждая избыточная молекула
эмульгатора иммобилизует 28-Ю-23 мл масляной фазы.
Концентрация эмульгатора оказывает влияние на концентрацию
дисперсной фазы, при которой эмульсия обращается (Шерман, 1950а;
Бехер, 1958), а также на вязкость, непосредственно предшествующую
обращению. В эмульсиях В/М т)ет приблизительно линейно
увеличивается с ростом Ф до— 0,5 при Dcp = 2 мкм. При Ф >0,5 для
эмульсий, содержащих 2,0 — 5,0% эмульгатора, или Ф я« 0,6 в при-
288
Таблица IV .12
Влияние концентрации эмульгатора
на вязкость эмульсий В/М (/>ср = 1,504 мкм)
Концентрация моноолеата
сорбитана
вес. %
1,5
з.о
4,5
6,0
1,5
3,0
4,5
6,0
1,5
3,0
4,5
6,0
кратное от ККМ
1
88,2
.182,8
284,6
294,4
1
72,9
150,1
232,1
319,7
1
64,1
131,8
203,0
277,5
t„, JltKJU
ср'
1
} 0,15
1
1
0,25
>
\
\ 0,35
)
Tlog/tlg
2,99
3,07
3,16
3,25
3,36
2,48
2,55
2,63
2,71
2,80
2,20
2,25
2,31
2,38
2,45
сутствии 1,0% эмульгатора величина г)^ возрастает быстрее и скорость
возрастания прогрессивно увеличивается (рис. IV.32). При всех
концентрациях эмульгатора, кроме наименьшей, ц^ достигает
Рис. IV.32.
Зависимость вязкости
при 21° С от
концентрации
дисперсной фазы (Шер-
ман, 1950а).
Концентрация
эмульгатора, %: 1 — 1,0;
г — 2,0; 3 — 3,5;
4 — 5,0.
го w во во
Концентрация дисперсной фазы, вес. %
100
19 Заказ 107 0
289
максимального значения при Ф = 0,75—0,80. Это почти соответствует
теоретической величине для плотно упакованных сферических
частиц. С 1,0% эмульгатора максимум т]от будет при Ф = 0,625.
Дальнейшее слабое увеличение Ф вызывает инверсию в эмульсии М/В,
сопровождаемую быстрым падением г^ до величины, немного
большей, чем вязкость водной фазы.
Растворимость ё двух фазах;
концентрация ионов водорода
Многие полиоксиэтиленовые (ПОЭ) производные сорбитана
растворимы в масле, а в воде лишь диспергируются. Тип эмульсии,
полученной с этими эмульгаторами, зависит от фазы, в которую
8
/ О о
2 о- о—
J*—*—
1
-О сг-
ь " —**"
1
О О ~
— г—*
1
i
1
\
}
L
6
рЫ
10
Рис. IV.33. Влияние рН на вязкость (Шерман,
1950b).
1 — моноолсат маннита; 2 —■ моноолеат маннитана;
3 — полутораолсат сорбитана.
они внесены, их концентрации и способа приготовления эмульсии.
При определенных концентрациях дисперсной фазы эмульсия
обращается и это сопровождается быстрым изменением вязкости.
Подобным образом ведет себя монолаурат сорбитана. Если
эмульсии М/В приготовлены с 1,5—6,0% монолаурата сорбитана,
диспергированного в водной фазе, капли дисперсной фазы появляются
при более высокой концентрации последней; их число, размер
и структурная сложность возрастают, когда концентрация
эмульгатора увеличивается. При Ф = 0,727 и 6,0% эмульгатора эмульсия
обращается при более низкой концентрации дисперсной фазы, чем
в случае применения меньших концентраций эмульгатора. Это сопро-
290
вождается падением iioo/r)c от 78,6 до 1,46 (Шерман, 1963b) и
объясняется распределением монолаурата сорбитана между двумя фазами,
даже если он сначала диспергирован в водной фазе.Степень переноса
в масляную фазу увеличивается, когда концентрация эмульгатора,
первоначально присутствующего в водной фазе, повышается. Подобно
этому, если монолаурат сорбитана распределен между двумя фазами
перед тем, как была приготовлена эмульсия, инверсия
контролируется общим количеством эмульгатора (Бехер, 1958).
Стабилизированные 2,5% полутораолеата сорбитана, моноолеата
маннитана или моноолеата маннида эмульсии, в которых водной
фазой является один из буферных растворов, охватывающих область
рН от 3 до 10, не показывают изменений т^ вплоть до рН 9,0
(Шерман, 1950b). При немного большем рН эмульсии обращаются в
разбавленные эмульсии М/В и это сопровождается быстрым
понижением rico (рис. IV.33). Эмульгаторы нерастворимы в буферных
растворах при рН 7,0, но их растворимость возрастает, когда рН
повышается до 9,0.
Реологические свойства
адсорбционной пленки эмульгатора
К сожалению, еще не доказана возможность изучения
реологических свойств непосредственно в эмульсиях. Поэтому применяют
модельные системы в виде пленок, адсорбированных на плоских
стационарных поверхностях раздела масло — вода (Криддл, 1960).
Имеются некоторые сомнения в возможности корреляции результатов
таких исследований с поведением пленок эмульгатора, подвергаемых
сдвигу в эмульсиях.
Когда сдвиг применяют к межфазной пленке, составляющие ее
молекулы, а также молекулы масляной и водной фаз,
расположенные в непосредственной близости, смещаются со своих равновесных
положений. Развиваемое напряжение влияет на ассоциативные
молекулярные перегруппировки. Это явление теоретически
рассмотрено Джоли (1954, 1956) и Олдройдом (1953, 1955). Джоли (1954,
1956) применил теорию абсолютных скоростей реакций Эйринга,
которая дает следующее уравнение (Эвелло и Эйринг, 1937; Мур
и Эйринг, 1938):
tiff Aw _ [Avn„hexV(AF/kT)] ,-,„,.
2кТ 2кТ (iv.aj/j
где s — площадь молекулы; Аупл — градиент скорости в пленке;
AF — свободная энергия на молекулу.
Когда
Чрл Aun„s < 2кТ (IV.238)
уравнение (IV.237) приводится к виду:
"? = (-гМтг) ^.2г»
10*
291
Межфазная пленка проявляет способность к ньютоновскому
течению, когда действительно уравнение (IV.238). Молекулы в монослое
занимают ограниченное число устойчивых состояний равновесия
1,2,..., г — 1, г с молекулярными площадями Sj, s2,. . ., Si_t, st.
Экспериментально найденная величина представляет собой среднее
значение для смеси молекул в двух смежных устойчивых состояниях
« = (1-аи) Ч +а«-1««-1 (IV.240)
где а;_4 — часть общего числа молекул на 1 см2 в состоянии i — 1.
Подобно этому AF определяется из выражения
AF = (l~ai.1)AFi + ai_lAFi-1 (IV.241)
где Af,-_i и AF[ — энергия активации для состояний i — 1 и г,
соответственно.
Величина AF t включает два члена: один — для энергии,
потребной для того, чтобы проделать дырку в пленке, через которую могут
проходить близлежащие активированные молекулы; другой — для
энергии, необходимой молекуле для перехода из равновесного
положения в ближайшее свободное положение.
Величина поверхностной вязкости (г\%) включает вклад молекул
масляной и водной фаз, ближайших к пленке эмульгатора, которые
двигаются с ней во время сдвига, если отсутствует скольжение.
Применение метода Джоли к поверхности раздела масло — вода ,
потребовало ввода выражения для молекул масляной фазы
AF = AF3K + AFB + AFM (IV.242)
где А^эм. Д^в и Д^м — свободные энергии активации межфазного
слоя молекул эмульгатора, воды и масла, соответственно.
В результате совместного решения уравнений (IV.239) и (IV.242)
получим:
т]| = т]"л ехр
кТ "+" кТ
(IV.243)
Уравнение (IV.243) показывает, что пленки с высокой вязкостью
должны проявлять свойства неньютоновских систем. Перемещение
молекулы из состояния i в ближайшее свободное положение в ряду
молекул, параллельном направлению потока, сопровождается
временным смещением молекул из соседних рядов. Если более
удаленные молекулы смещены из состояния i прежде, чем другие молекулы
возвратились в свои первоначальные положения, энергия активации,
связанная с начальным смещением, несколько меньше AF{ и дается
как AFI
\F = AFi-wi(AFi-\F'i) (IV.244)
где
u>, = exp(--—J ) (IV.245)
\ Ji/ruilp. пл /
T,n^0,^y(^yexp[i^g^l] (IV.246)
292
u>i — вероятность того, что смещенная молекула i не займет свое
первоначальное положение; тп — масса отдельной молекулы; U{_k —
энергия взаимодействия между соседними молекулами в состоянии
i — к; тр пл — время релаксации для молекул межфазной пленки,
которое приближается к среднему времени, принимаемому для
возвращения смещенной молекулы в ее первоначальное положение;
0,94 — коэффициент, учитывающий уменьшение расстояния между
молекулами во время сдвига, вызванное течением от lt до минимум
0,94/,.
Олдройд (1955) отмечал, что когда молекулы эмульгатора в
межфазной пленке относительно далеко отстоят друг от друга
(например, при действии сил отталкивания), работа, необходимая для того,
чтобы сдвинуть пленку, много меньше, чем для изменения ее
площади. В этом случае межфазное натяжение в любой точке является
простой функцией локального поверхностного расширения или
скорости, с которой оно изменяется. Если молекулы в межфазной пленке
упакованы настолько тесно, что они связаны, например,
водородными связями, тогда, чтобы сдвинуть пленку с постоянной площадью,
требуется произвести значительно большую работу.
Метод возмущения, согласно Фрелиху и Заку (1946), привел
к уравнению (IV.221) для очень разбавленных эмульсий, в которых
капли окружены жидкой межфазной пленкой, и к уравнению
(IV.206) для эластичной пленки. Для мелких капель оба уравнения
дают одинаковые результаты. Это следует также из уравнения
(IV.205), показывающего, что деформация капель незначительна,
когда Dcp мало. На реологических свойствах эмульсий не будет
серьезно отражаться реология межфазной пленки, если капли малы
и жидкость не циркулирует внутри них. Когда капли велики и
окружены вязкой пленкой, они деформируются под влиянием сдвига
таким же образом, как и нестабилизированные капли. Вязкость
эмульсии будет тогда зависеть от реологических свойств межфазной
пленки.
В концентрированных эмульсиях деформация может происходить
из-за плотной упаковки капель. Вязкость эмульсий, в которых
капли деформируются при сдвиге, возрастает быстрее с
концентрацией дисперсной фазы, чем для эмульсий с каплями идентичного
размера, окруженными эластичной пленкой.
Если эмульсии перемешивать с большой скоростью,
центрифугировать или очень сильно охлаждать, реологические свойства
межфазной пленки будут влиять на скорость разрыва пленки
и коалесценции капель.
Присутствие пленки эмульгатора вокруг капель приводит к
возможности скольжения у границы с непрерывной фазой. Когда
скольжение имеется, оно вызывает разрыв в тангенциальной скорости,
которая пропорциональна напряжению сдвига, передаваемому через
межфазную границу. Уравнение (IV.221) выведено в предположении,
что на межфазной границе скольжение отсутствовало. Принимая
это во внимание Гопал (1960, 1961) модифицировал уравнение (IV.219)
293
для разбавленных эмульсий. В результате получены уравнения
(IV.222) и (IV.223). Для нулевого скольжения Кск ->- °о, а для
условия свободного скольжения Кск ->■ 0. Олройд (1953) ранее вывел
уравнение подобного вида
и показал, что как время релаксации, так и время запаздывания
должны увеличиваться при скольжении.
Когда #ск -»- 0
Лотн = 1 т- Ф (IV. 248)
Таким образом, при свободном скольжении т]ф не влияет на
вязкость эмульсии. Однако она оказывает влияние на время релаксации
и запаздывания.
Электровязкостный эффект
Когда подвергают сдвигу очень разбавленные эмульсии,
содержащие электрически заряженные капли, нарушается симметрия
двойного электрического слоя вокруг каждой капли. Оказывается
затронутым взаимодействие между ионами двойного электрического
слоя и электрическим зарядом на поверхности капель, что приводит
к дополнительному рассеянию энергии и повышенной вязкости
(Конвэй и Добри-Дюкло, 1960).
Смолуховский (1916) внес поправку в уравнение (IV.206) для
сферических жестких частиц с тем, чтобы учесть первый
электровязкостный эффект. Для эмульсий оно принимает вид:
4yA = "**[l-r-!j^T (-§)'] (IV.249)
где £ — электрокинетический потенциал заряженных капель, х —
удельная электропроводность эмульсии.
Таким образом, увеличение г]Уд, вызванное электрическим
зарядом, запишется как
аЕф f eg у
т]схг2 \ 2п /
Уравнение (IV.249) показывает, что первый электровязкостный
эффект будет оказывать измеримое влияние на г\ул только тогда,
когда г не превышает нескольких сот ангстрем. Это противоречит
предположению Смолуховского, принятому при выводе уравнения,
о том, что толщина двойного слоя мала в сравнении с г. Противоречие
преодолевается только тогда, когда ионная сила превосходит — 0,01.
Бус (1950) вывел более сложное уравнение, развернув уравнение
(IV.206) в виде степенного ряда заряда на каплях. Он ввел выражение
Ф
294
для толщины диффузного слоя 1/у,концентрации ионов с,- и
валентности иона z,-\
Чуд = аяФ [* + ?< (-jffr) (1 + У/2)^ (у/)] (IV.250)
где
еА"г ^ 1ф2»/тр
1
" = i
1
<? — элементарный заряд; Z (уг) — сложная функция от уг, в которой
(1 + yr2) Z (уг) уменьшается, когда %г возрастает; f —
коэффициент трения иона i в непрерывной фазе.
Когда уг мало, т. е. толщина двойного слоя велика
так что электровязкостный эффект заметно способствует
увеличению г\уд и его вклад составляет — 1Д"у.
Когда yj велико, т. е. толщина двойного слоя мала
Z('fr) = -42n(ir)i (IV.252)
Электростатический вклад в г|уд является тогда чрезвычайно
малым, потому что двойной слой испытывает небольшие искажения
во время течения.
Эти заключения противоречат классической концепции, которая
предсказывает измеримый электровязкостный эффект только тогда,
когда двойной слой тонок в сравнении с г. Разногласие возникло
из-за ограниченности основного предположения, на котором развита
теория Смолуховского (1916). Работа Буса также предсказывает
более низкий вклад электровязкостного эффекта в т]уд. Это
согласуется с экспериментально найденными данными.
Стрит (1958) вывел более простую форму уравнения (IV.250),
принимая во внимание относительное движение между непрерывной
и дисперсной фазами во время течения:
^ = V> Ol^b (■§) « + *&] <IV-253>
В более концентрированных эмульсиях расстояние между
каплями таково, что их двойные слои могут перекрываться и т|уд
увеличивается благодаря их взаимному отталкиванию. Этот эффект,
известный как второй электровязкостный эффект, впервые наблюдали
Хармсен, Ван Шотен и Овербек (1953).
Величина этого эффекта прямо пропорциональна Ф. При
постоянном Ф он возрастает с уменьшением ионной силы, потому что
двойной электрический слой при этом увеличивается, повышая тем самым
вероятность взаимодействия двойных слоев.
295
Так как трудно получить монодисперсные капли необходимого
размера, имеется очень мало исследований электровязкостных
эффектов в эмульсиях. Ван дер Ваарден (1954) определил вязкости ряда
эмульсий М/В, стабилизированных сульфонатами натрия, в которых
величина Z)cp не превышала 0,205 мкм (табл. IV. 13). Максимальная
концентрация примененного эмульгатора была необычно большой,
так как составляла — 12% общего веса эмульсии. При более высоких
концентрациях эмульгатора нотн существенно отклонялась от
теоретических значений, вычисленных по уравнению (IV.206). Увеличение
было также намного большим, чем предсказывалось уравнениями
(IV.249) и (IV.250). Поэтому сделано заключение, что расхождение
не могло быть результатом искажения диффузного двойного слоя
вокруг капель. Полагали, что сильно ионизированный эмульгатор,
адсорбированный на поверхности капель, создает электрическое
поле высокого напряжения 105—106 в/см и слой молекул воды прочно
связан с ним. Толщина слоя воды, как показано кажущимся
увеличением Лг была 0,0014—0,0037 мкм, достигая почти устойчивого
значения при более высоких концентрациях эмульгатора.
Таблица IV .13
Электровязкостный эффект в эмульсиях М/В
(ван дер Ваарден, 1954)
Содержание
эмульгатора
в масляной
фазе, г/г
0,05
0,10
0,17
0,35
Ф
0,0565
•0,112
0,222
0.328
0,0559
0,111
0,219
0,325
0,0549
0.109
0,216
0,321
0,0531
0,106
0,210
0.313
^ОТН
1.17
1,40
2,12
3,52
121
1,51
2,56
5,64
1,25
1,68
3,94
22,15
1,35
1.96
6,50
92,8
Размер
напелъ,
Л1}ГЛ1
0,2050
0,10.38 ,
0,0586
0,0276
аЕ
2,6-2,7
3,0-3,1
3.4 -.4.5
4,8-5,0
Дг, мкм
0,0014—0,0028
0,0031-0,0037
0,0032-0,0035
0,0033-0,0036
Приготовляя эмульсии, ван лер Ваарден вводил эмульгатор
в масляную фазу. Мукерджи (1957) высказал мысль, что
наблюдаемые вязкости приближались к значениям, вычисленным по
уравнению (IV.250), если было сделано допущение о диффузии эмульгатора
в водную фазу во время эмульгирования, которое включает
приготовление эмульсии В/М, предшествующее инверсии. Другим услож-
296
няющим фактором могло быть то, что мицеллы в непрерывной
фазе при избытке эмульгатора изменяли его ионную
концентрацию.
Наблюдения ван дер Ваардена можно объяснить также
следующим образом. Во-первых, из-за очень малого размера капель в его
эмульсиях уравнение (IV. 206) не было пригодно для большинства
концентраций дисперсной фазы, которые он использовал.
Следовательно, не удивительно, что он нашел расхождение между
рассчитанными и экспериментальными значениями т]отн. Во-вторых, хотя
каждая эмульсия была достаточно монодисперсной, не все они имели
капли одного и того же размера.
При возрастании концентрации эмульгатора размер капель
уменьшался от 0,205 до 0,0276 мкм, однако этот факт не был принят
во внимание. В указанной
области размеров капель
уменьшение этой величины сильно
влияет на вязкость. Если
построить график зависимости
■Потн — 1/^cpi получается серия
прямых линий. При этом их
градиенты aD зависят от
размера капель. Сравнение этих
результатов с
соответствующими данными для
монодисперсных латексных систем с
подобным размером частиц,
подчиняющихся закону Ньютона
(Саундерс, 1961) и не
обладающих электровязкостным
эффектом, показывает, что две
серии данных согласуются
весьма хорошо (рис. IV.34).
Поэтому возможно, что эмульсии ван дер Ваардена не проявляли
электровязкостного эффекта.
В некоторых: эмульсиях В/М капли имеют ^-потенциал, равный
100 ме, так что мог ожидаться первый электровяэкостный эффект.
Однако эмульсии (Ф = 0,03—0,33), содержащие различные
эмульгаторы и имеющие ^-потенциалы от 15 до 100 мв, при применении
уравнения (IV.206) к данным вязкости (Альберс, 1957) дали примерно
одно и то же значение аЕ. Величина первого электровязкостного
эффекта, полученная по уравнению (IV.250), равна ~ 1%. Таким
образом, эффект был мал в системах с низкой диэлектрической
постоянной. В эмульсиях В/М толщина двойного электрического слоя
составляет несколько микрометров, так что в более
концентрированных эмульсиях мог ожидаться второй электровязкостный эффект.
Но так как двойной слой является очень диффузным, увеличение
вязкости, вызванное последним эффектом, должно было бы быть
также малым.
Рис. IV.34. Корректировка данных
ван дер Ваардена для капель
различных размеров, предшествующая оценке
влияния электровязкостного эффекта
(Шерман, 1965):
х — данные ван дер Ваардена; о — данные
Саундера.
297
Когда следы электролита удаляли из латекса путем диализа,
вязкость при низких скоростях сдвига ощутимо увеличивалась
и становилась неньютоновской (Бродинан и Келли, 1965).
Удаление электролита увеличивало толщину диффузного
двойного слоя, в результате чего в стационарном состоянии внутри
агрегатов удерживалось значительно больше непрерывной фазы. Это
увеличивало эффективную объемную концентрацию дисперсной фазы,
так как при низких скоростях сдвига агрегаты перемещались как
отдельные единицы. Добавка электролита к диализованному латексу
изменяла зависимость, и вязкость уменьшалась при увеличении
концентрации электролита до тех пор, пока не достигала
минимального значения. Это сопровождалось изменением режима от
неньютоновского до ньютоновского. Лаурилсульфат натрия был гораздо
менее эффективным, чем хлорид натрия. Например, 1,71 • 10~5 моль
лаурилсульфата натрия на 1 г латекса снижали вязкость при 1 сек'1
от 505 до 425 пз, а та же концентрация хлорида натрия снизила
вязкость до 0,367 пз.
Гндроколлоиды и пигменты
Гидроколлонды, растворенные в водной непрерывной среде,
увеличивают вязкость непрерывной фазы пропорционально
концентрации; вязкость в этом случае может быть неньютоновской.
Если концентрация гидроколлоида высока, эмульсия может принять
гелеподобную консистенцию. Должно быть проведено различие
между неньютоновским течением, обусловленным дисперсной фазой,
и неньютоновским течением вследствие образования структуры
в непрерывной фазе. Некоторые гидроколлоиды, например камедь
акации (Шоттон, и Уайт, 1963), стабилизируют эмульсии в отсутствие
эмульгатора, адсорбируясь на поверхности раздела масло — вода.
При этом образуется жесткая гидратированная пленка.
Тонко раздробленные пигменты также мигрируют к границе
раздела масло — вода и образуют защитный слой вокруг капель.
Все водные окислы (например, гидратированные формы пятиокиси
ванадия, окиси железа и алюминия) поверхностно активны.
Поэтому, помимо некоторого увеличения вязкости
свежеприготовленной эмульсии, происходящего в процессе их применения, может
наблюдаться дальнейший ее рост во время хранения, вызванный
прогрессирующей гидратацией окислов. В конце концов, вокруг
каждой канли образуется слой геля. Примером могут служить
концентрированные эмульсии В/М, в которых окись алюминия
(глинозем) размешана в водной фазе (Шерман, 1955с). Когда к водной фазе
добавляют пропиленгликоль до концентрации 20%, эти изменения
замедляются в зависимости от концентрации пропиленгликоля.
При более высоких концентрациях прониленгликоля образование
слоя геля полностью подавляется. Другие полиспирты оказывают
тот же эффект.
Применение пигментов, гидроколлоидов и других ПАВ в
эмульсиях выдвигает интересную проблему математической интерпретации
298
их вязкости, так как они являются, по существу, системами с двумя
дисперсными фазами. Тем не менее, эта проблема привлекает мало
внимания. Непрерывная фаза — жидкость и эмульгатор — образует
начальную коллоидную дисперсию; структура становится более
сложной, когда в ней диспергируется другая жидкая фаза. В
простейшем случае очень разбавленная дисперсия сферических твердых
частиц в непрерывной фазе подчиняется уравнению (IV.206):
11° = %(1 + «ЕФа) (1V.254)
где т)а — вязкость суспензии, содержащей объемную
концентрацию Фа твердых частиц.
Когда внутри этой дисперсии эмульгируется небольшой объем
(Фь) другой жидкой фазы, результирующая вязкость может быть
выражена одним из двух путей. С одной стороны, если т)"
представляет вязкость новой непрерывной фазы, то
Т)««-ь = if (1 +аЕФь) = т)с [(1 + а£Фа) (1 + аЕфЬ)] (1V.255)
С другой стороны, т)с — вязкость непрерывной среды, в которой
диспергированы как твердая, так и жидкая фазы:
11в+ь = т)с [1 + а£(Фа + Ф»)] (1V.256)
Разница между уравнениями (IV.255) и (IV.256) заключается
в выражении а% ФаФь, которое не является значительным, если
Ф" и Ф6 малы. При более высоких концентрациях дисперсной фазы
уравнение (IV.210) заменяет уравнение (IV.206) и два метода
интерпретации приводят к большим расхождениям.
Можно провести аналогию с исследованием изменений вязкости,
проведенным Ивсоном (1959). В этом случае дисперсию
приготавливали с твердыми шариками более чем одного размера. Ивсон
предположил, что дисперсию шариков только с двумя различными
размерами можно рассматривать как суспензию больших шариков
в непрерывной фазе, состоящей из малых шариков в жидкой среде.
Для очень разбавленных дисперсий его выражение для вязкости
этой смеси идентично уравнению (IV.255). Дальнейшее изучение
проводили на дисперсиях, содержащих шарики трех различных
размеров. Предполагали, что вязкость конечной суспензии может
быть описана двумя способами. Во-первых
«« = т)°тнт)2тнч£тн (IV.257)
что для очень разбавленных систем могло быть сведено к
выражению:
где г|оТН, т]оТН и т)отн — относительные вязкости суспензий,
содержащих шарики увеличивающегося размера в общей жидкой фазе,
с объемными концентрациями Ф", Ф6 и Фс, соответственно.
299
Другой способ рассматривает конечную систему как суспензию
наиболее крупных шариков в дисперсионной среде, содержащей "
шарики двух меньших размеров. Тогда
ггё;г"=»1§Й1§тя (IV. 258)
где г|отя — относительная вязкость суспензии, содержащей шарики
с концентрацией Фа -f- Ф".
Если полная объемная концентрация шариков < 0,1,
экспериментально полученные значения чой"1^ хорошо согласовывались
со значениями, вычисленными по уравнению (IV.257) или (IV.258).
При более высоких концентрациях проявлялось расхождение,
которое возрастало, когда объемная концентрация увеличивалась. Однако
уравнение (IV.258) всегда давало значения, более близкие к
эксперименту, чем уравнение (IV.257).
Коллинз и Вейленд (1963) приняли другое приближение. Они
предположили, что увеличение удельной вязкости (т|Уд), когда
суспензия палочкообразных частиц смешана с суспензией сферических
частиц, давалось как
м$ - ч$* + иудл "г- д%д (l v •259)
где т|5$ и г)"дл — увеличение удельной вязкости суспензий шариков \
и палочек, соответственно; Аиуд — увеличение вязкости,
возникающее из-за взаимодействия между двумя типами частиц и зависящее
от концентраций их обоих.
Непрерывная фаза для обоих типов частиц была одной и той же;
объемные концентрации для шариков Фс* и палочек фпал были очень
низки
Д11уД = /(г)ФсфФпал (iv.260)
гДе / (g) — функция геометрических параметров двух типов частиц.
Экспериментально найдено, что т|уд возрастает линейно с
произведением Фс* и Фпал. Это можно объяснить тем, что шарики
препятствуют ориентации палочек при течении под влиянием сдвига. Если
обе твердые фазы — шарики, то, по-видимому, выражение для Ат|уд
в уравнении (IV.259) должно быть приведено к уравнению (IV. 256).
ИЗМЕНЕНИЕ РЕОЛОГИЧЕСКИХ СВОЙСТВ
ЭМУЛЬСИЙ ПРИ СТАРЕНИИ
Старение эмульсий приводит к ухудшению их реологических
свойств. Вначале капли флокулируют и создают агрегаты, которые
внутри своих структур иммобилизуют жидкость непрерывной фазы.
При высокой скорости флокуляции наблюдается заметное
возрастание вязкости в случае очень низких скоростей сдвига. Этот процесс
не может быть изучен при высоких скоростях сдвига из-за
разрушения агрегатов, происходящего в результате обратимой природы
флокуляции. Коалесценция приводит к уменьшению числа капель
на единицу объема эмульсии и к увеличению размеров
индивидуальных капель.
3(0
Так как эмульсии редко являются монодисперсными,
распределение по размерам обычно бывает широким. Установлено, что размер
и распределение капель влияют на вязкость эмульсии, поэтому
можно ожидать, что последняя также будет изменяться, когда
эмульсии стареют.
Изменение реологических свойств
при высоких скоростях сдвига
В свежеприготовленных эмульсиях вязкость обратно
пропорциональна среднему размеру капель, если их распределение по
размерам не слишком широко (Шерман,
1960). а
Когда изменение в структуре
эмульсии сводится только к
постепенному увеличению среднего
размера капель и это сопровождается
незначительными изменениями
пределов распределения размеров,
можно вычислить уменьшение
вязкости, как функцию увеличения
среднего размера капель (Dcp).
Скорость увеличения /Зср
определяется из кинетики коалесценции.
Такой прием применен для того,
чтобы предсказать изменения
вязкости в «старых» псевдопластичных
эмульсиях В/М и М/В с объемными
концентрациями дисперсной фазы,
доходящими до 0,70 (Шерман,
1963а). После ~ 70 дней старения
верхний предел распределения
размеров капель в эмульсиях В/М
не превышал 5,0 мкм и 80%
общего числа капель на 1 см3
эмульсии составляли капли размером
1,5 мкм, в эмульсии М/В
соответственно 18,0 мкм и =^4,0 мкм.
С помощью графика зависимости
4Р — ЛооЛк доказано, что как
свежие, так и старые эмульсии
следуют одному и тому же закону
(рис. IV.35). Прогрессивное
увеличение Dcp является единственным
результатом процесса старения, который оказывает
влияние на ilooAlc- Данные вязкости для свежих
0.3 0,5 0.7
I ср, мкн
0,9
Рис. IV.35. Влияние 1ср на т)отн
для «старых» эмульсий (Шерман,
1963а):
а — эмульсии
В/М;
М/В.
б — эмульсии
измеримое
и старых
эмульсий М/В сФ = 0,74 отклоняются от теоретической
экспоненциальной формы кривой из-за присутствия большого числа капель
дисперсной фазы и деформации больших капель. Скорость
301
коалесценции вычисляли, определяя изменение числа капель на
единицу объема эмульсии за несколько дней или по скорости, с
которой увеличивалось Dcp.
Если предположить, что скорость медленной коалесценции vK
полностью регулируется возможностью разрыва тонкой пленки
непрерывной фазы между флокулированными каплями (ван ден Тем-
пель, 1957) и разрыва водородных связей между молекулами
эмульгатора, адсорбированными вокруг капель, тогда
или
N, = NKcxp(—rKT)
In D. = In D0 -L- l-kt/3
(IV.261)
(IV.262)
10 40 50 ' 0 5 10 15 20
Продолжительность старение, сутки
Рис. IV.36. Сравнение экспериментальных (О) п
рассчитанных (А) значений г|отн. полученных при старении эмульсий
(Шерман, 1963а):
а — эмульсии В/М; б —. эмульсии М/В. Ф: 1 — 0,6226; 2 — 0,4703'
3 — 0,2753; 4 — 0,7828; 5 — 0,0339; в — 0,4352.
(где D0 и Dx — значения Dcp для нулевого времени и времени т,
соответственно), потому что число частиц на 1 см3 эмульсии
6Ф ■ Ю12
Л1уСр
(IV. 263)
Фактически коалесцирует только фракция капель, плотно
прижатых Друг к другу (Лоуренс и Миллс, 1954):
Dl = DI -г ехр
RT
(IV. 264)
Величина энергетического барьера коалесценции Af/K зависит
от заряда капли, реологических свойств адсорбционного слоя и т. д.
Вывод уравнений (IV.261) и (IV.264) основан на предположении,
что fK не зависит от N, a AUK — от DCf. Эти условия применимы
только для медленной коалесценции. Многие эмульсии В/М и М/В
не имеют постоянной скорости коалесценции на протяжении всего
периода старения. Очень часто первая фаза относительно быстрой
коалесценции предшествует второй фазе медленной коалесценции,
302
В общем, первая фаза обнаружена только в тех эмульсиях, которые
содержат, по крайней мере, 10% капель с диаметром г£ 0,5 мкм.
Следовательно, зависимость D%p — т или In D — х должна быть
исследована в течение длительного времени, пока не будет достигнута
линейность, соответствующая медленной коалесценции. Затем
определяют значения vK и AUK для медленной коалесценции. Значение
vK для эмульсий В/М составило 17,7 - Ю-8 сект1 и для эмульсий М/В
31,9• 10"8 сек-1; AUK для обоих типов эмульсий—6—7 ккал-молъ'1.
Анализ этих значений и кривых r\oo/r]c — lcp для свежих эмульсий
позволил предсказать скорости снижения TlooAlc на всем протяжении
периодов старения. Теоретические и экспериментальные данные
удовлетворительно совпали (рис. IV.36).
Изменение реологических свойств
при низких скоростях сдвига
При высоких скоростях сдвига вязкость в основном определяется
взаимодействием между индивидуальными каплями; при низких
скоростях — силами притяжения между флокулированными каплями
Рис. IV.37. Агрегат капель в эмульсии В/М. Х2964.
и кажущимся увеличением Ф до /НФ (где /н — коэффициент
набухания). Точное значение /„ зависит не только от структуры
агрегатов, на которую влияет, например, число капель (рис. IV.37),
толщина электрического двойного слоя, время старения, но также
и от скорости сдвига.
Эмульсии В/М более пригодны для изучения структуры агрегатов,
чем эмульсии М/В. Электрический двойной слой диффузен в эмульсиях
303
В/М, так что капли флокулируют быстро и влияние
агрегирования на вязкость проявляется в начале периода старения (Шерман,
1966). Если предположить, что применима теория быстрой флок'уля-
ции Смолуховского (1916, 1917), то среднее время (ti/2) для неассо-
циированных капель на 1 см3 эмульсии, необходимое для того, чтобы
уменьшилось наполовину первоначальное их число при Dcp =■
= 1,25 мкм, Ф = 0,47 и iVK = 4,6-10й, составит 64 сек. За время т
160
120
8U
W
I
' \S"~~~-
С ~~*_
fill
i
' »
„ i
□ 4
* 5
»6
- i
0 10 20 30 W 50 60
Время действия сдвигающего усилия, сек
Рис. IV.38. Восстановление структуры при скорости
1,2
сек'1 после приложения сдвигающего усилия
в течение 5 мин при скорости 215,5 сек'1.
Продолжительность старения, ч: 1 — свежие эмульсии; 2 — 24
3 — 44; 4 — 08; 5 — 189; б — 450.
между приготовлением этой эмульсии и измерением ее
вязкости (~ 1000 сек) уменьшение в Nк было незначительным (Шерман,
1964).
Число неассоциированных капель (iVj), агрегатов, содержащих
две капли (N2), агрегатов, содержащих к капель (Nk), дается
уравнениями:
ЛГ,=
NK
(1-г-т/т
Так как т ^> Ti
и Л\ = 1.66-109.
304
Л', ^
.\k =
■No (т/т,,
\k-l
(l+T/T,,,)**!
(IV. 265)
(T/TV,)S
■ =• iV-2 ■ ■ ■ =- Л'*
(tV.266)
Таким образом, эмульсия содержала намного больше агрегатов,
чем неассоциированных капель, когда ее вязкость измеряли в
первый раз.
Когда эту эмульсию подвергали сдвигу в вискозиметре с
коаксиальными цилиндрами в течение 5 мин при скорости 215,46 сек'1,
вязкость снизилась до 10,5 пз. При уменьшении скорости сдвига
до 1,33 сек'1 вязкость повысилась до равновесного значения 139 пз
примерно за 30 сек (рис. IV.38), причем наибольший подъем
наблюдался в течение первых 10 сек. Что касается свежей эмульсии, то
в дальнейших опытах во время старе- ,
ния подобный минимум вязкости
достигался при 215,46 сев-1, но равновесная
вязкость снижалась при 1,33 сек'1 со
временем старения. Наибольшее
снижение равновесной вязкости
приходилось на первые 50 ч старения, и это
вызвано главным образом быстрой
коагуляцией капель диаметром =^ 0,5 мкм.
По этим данным можно вычислить
кажущееся увеличение объема фракции
дисперсной фазы, вызванное
изменением агрегатов, когда устранены
условия сильного сдвига. Таким же путем
Муней (1946) интерпретировал тиксо-
тропное восстановление сложных латек-
сов, использовав эмпирическое
уравнение tlcoAlc — Ф) которое он вывел,
анализируя ряд данных вязкости Эй-
лерса (1941) для эмульсии с
большинством капель диаметром 2,7 мкм:
Ю 20 30
Время действия сдвиеиюшего
уеипия, сен
Рис. IV.39. Кажущаяся
объемная доля дисперсной фазы при
скорости 1,33 сек'1 через
различные интервалы после
приложения сдвигающего усилия
в течение 5 мин при
скорости 215,5 сек"1:
уравнение (IV.267); 2
нение (IV.269).
урав-
\ 1-^0,5Ф
Ф
ехр
1.25Ф
1-Ф
(IV.267)
Хотя это уравнение удовлетворительно соответствует данным
Эйлерса, оно не может быть применено к эмульсиям, имеющим сильно
отличающиеся размеры капель. Уравнение (IV.229) выражает более
характерную зависимость для истолкования кривой г|ет/г]с — ф.
Кажущееся значение Ф (Фк) определено через различные интервалы
времени (при восстановлении структуры) путем построения
зависимости экспериментально полученных данных "*1 со/Лс от Ф« найденного
отсчетом соответствующих значений. Вычисленные таким образом
данные для свежей эмульсии приведены на рис. IV.39 вместе с
соответствующими данными, определенными по уравнению (IV.267).
Истинное равновесное значение Ф и степень восстановления
структуры были в действительности меньше полученных по
уравнению (IV.267). Кроме того, из уравнения (IV.229) следует, что
непосредственно после сдвига при 215,46 сек'1 Фк = 0,55, а его
теоретическое значение — 0,47, т. е. такая скорость сдвига не
20 зяказ 1070
305
полностью разрушает флокулированную структуру. Это
подтверждается кривой вязкость — скорость сдвига, показывающей, что
вязкость не понижается до устойчивого минимального значения,
пока скорость сдвига превышает 750 сек'1.
Согласно Мунею (1946)
ли
-/„(#,-#!)]
(IV. 268)
Так как N{ относительно мало по сравнению с Лгт для
эмульсий В/М
6ФК Фк
/н=„пз v =~^- (IV.269)
nDlpN.
Ф
Значения /н при 1,33 сек-1, полученные для «старой» эмульсии
при использовании уравнения (IV.269) совместно с
уравнением (IV.229), оставались постоянными независимо от изменений Dcp.
Однако при замене уравнения (IV.229) уравнением (IV.267) этот
прием становился неприменимым, так каг. /н тогда уменьшалась
со временем старения. Единственным геометрическим
расположением капель, при котором Dcp не влияет на /„при /н = 1,35, является
додекаэдральная упаковка, где капли составляют 74,03% от объема
агрегатов. Полагая, что все агрегаты при сдвиге разрушаются
приблизительно до одного и того же размера, можно вычислить среднее
число капель в агрегате как при высоких, так и при низких скоростях
сдвига (табл. IV. 14) путем вычисления кажущегося диаметра
каждого агрегата по уравнению (IV.229). Более высокие значения /н
получены при скоростях сдвига более низких, чем 1,33 сек~1. При
0,133 сек~1 /н = 1,4, а при 10~4 —10"5 сек"1 /н > 1,6, так что капли
располагались внутри агрегатов более свободно. Беспорядочная
плотная упаковка монодисперсных сфер дает Ф = 0,637 при /„ =
= 1,56, а эквипропорциональная смесь сфер, имеющих отношение
размеров 1/3,8, —Ф = 0,68 при /,, = 1,47 (Еразанис, Бартлет и
Ниссан, 1962).
Таблица IV.14
Изменение структуры агрегатов в эмульсиях В/М при старении
(Шерман, 1966, неопубликованные данные)
Время
старения,
ч
0
24
44
08
189
405
мкм
1,25
2,09
2,29
2,46
2.97
3 27
<ji
уравнение
(IV.267)
0,71
0,70
0,68
0,67
0.66
0,64
к
уравнение
(IV.229)
0,64
0,63
0,63
0.63
0,66
0 63
уравнение
(IV.267)
1,51
1,49
1,45
1,43
1,40
1.3(5
t
н
уравнение
(IV.229)
1,36
1,34
1,34
1,34
1.34
10
Равновесное среднее
число капель на
агрегат при скорости
сдвига
215,45 се>~1
31.8
5,3
3,8
2,8
1,6
1,0
1,33 сек-
231.0
49,3
33,4
24,6
12.Г.
8,2
306
Вязкость т] при низкой скорости сдвига эмульсий В/М можно
представить аналогично уравнению (IV.229)
lg
цу*
/лФ
(IV.270)
.с /н = ехр (0,28гг°-15) в области сдвига Ю-4—103 секгК
Когда на один и тот же график зависимости вязкости от среднего
размера капель наносились данные для нескольких эмульсий В/М
200 400 600
Продолжительность старения, ч
Рис. IV.40. Влияние Dcp на г|/г|с для Рис. IV.41. Сравнение
экспериментальна™ образцов эмульсий. ных (О) и вычисленных (х) аначений
_,. „,„. , вязкости, полученных при старении
Скорость сдвига, сек ': а — 0,133 о — • J r ' г
1,20; в - 10,77. ЭМУЛЬСИИ.
Скорость сдвига, сек-1: а— 0,133; б — 1,20;
в — 10,77.
с различными исходными средними размерами капель, расхождения
в вязкости наблюдались при любом размере. Чем короче период
старения, тем выше вязкость. Аналогично этому свежая эмульсия
с данным средним размером капель всегда имела более высокую
вязкость, чем эмульсия, достигшая того же размера после старения.
Все свежие эмульсии содержали капли диаметром < 0,5 мкм,
которые исчезали в течение первых 50—60 ч старения. Наблюдаемые
расхождения в вязкости при любом выбранном среднем размере
капель вызваны различиями в концентрации мелких капель.
Эмульсия, достигшая узкого распределения размеров капель путем
старения, содержала меньшее число частиц с диаметром •< 0,5 мкм, чем
свежеприготовленная эмульсия с тем же средним размером. Когда
20*
307
обе эмульсии старели в течение нескольких дней, эти различия
сводились к минимуму, потому что субмикроскопические капли
исчезали из обеих эмульсий.
В реологических исследованиях средний размер капель обычно
вычисляют как диаметр сферы среднего объема. Этот параметр не
объясняет того существенного вклада, который вносится в вязкость
каплями с диаметром < 1 мкм. Вычисление требует суммирования
кубических степеней размеров разных фракций и кубическая степень
диаметров < 1 мкм ничтожно мала.
Более показательная оценка среднего диаметра выведена из
предположения, что каждая эмульсия представляет собой бинарную
смесь капель диаметрами < 0,5 и >0,5 мкм. Диаметр для фракции
капель большего размера получен обычным путем, для области
меньших размеров принята величина 0,25 мкм. Затем вычислено среднее
арифметическое для обеих областей. На графике зависимости
вязкости от исправленных значений среднего размера капель данные
и свежей и старой эмульсий для трех низких скоростей сдвига
ложились на одну и ту же кривую (рис. IV.40). Если средний размер
капель уменьшался, то вязкость увеличивалась, но увеличение на
единицу измерения размера капель было намного больше, чем
наблюдалось при высоких скоростях сдвига.
При совместном решении уравнений (IV.262) и (IV.265) можно
получить величину уменьшения вязкости для любого периода
старения. Значения вязкости, вычисленные этим путем, хорошо
согласуются с величинами, определенными экспериментально
(рис. IV.41).
ЛИТЕРАТУРА
А 1 b е г s W., Doctoral dissertation, University of Utrecht, 1957.
A 1 b e r s W., Overbeds, I. Th. G., J. Colloid Sci., 15, 489, 510 (1960).
A 1 t г i с h t e r F., Lustig A., Phys. Z., 38, 786 (1937). American Society
for Testing Materials ASTM 5-25; D217-52T.
Bacon L.R., J.Franklin, Inst., 221, 251 (1936).
BartokW., Mason S. G., J. Colloid Sci., 12, 243 (1957).
В art ok W., Mason S. G., J. Colloid. Sci., 13, 293 (1958).
Be cher P., J. Soc. Cosmet. Chem., 9, 141 (1958).
Bingham E., Fluidity and Plasticity, New York, 1922.
Booth F., Proc. Rov Soc, A203, 533 (1950).
В г in km an H. C, J. Chem. Phys., 20, 571 (1952).
Brodynan J. G., К e 1 1 у E. L., J. Colloid. Sci., 20, 7 (1965).
Broughton J., Squires L., J. Phys. Chem., 42, 253 (1938).
de Bruijn H., Rec. trav. chim., 61, 263 (1942).
de В r u i j n H. Proceedings of the I International Congress on Rheology, vol. 2,
1948. p. 95.
Buckinghan E., Proc. Am. Soc. Testing Materials, 21, 1154 (1921).
С a s s о n N., in «Rheology of Disperse Systems» ed. bv С. С Mill, London,
1959. p. 84.
Cheng P. Y., S с h а с h m a n H. K., J. Polymer Sci., 16, 19 (1955).
Collins D.J., Wayland H., Trans. Soc. Rheol., 7, 275 (1963).
Conway B.E., Dobry-Duclaux A., in «Rheology, Theory and
Applications», ed. by F. Eirich, vol. 3, New York — London, 196(i, p. 83.
С г i d d 1 e D. W., in «Rheology Theory and Applications», ed. by F. Eirich,
vol. 3, New York — London, 1960, p. 429.
308
Cross M. M., J. Colloid Sci., 20, 417 (1965).
Davies J.T., Rideal E. K., Interfacial Phenomena, New York — London,
1963, p. 129.
Denny D. А., В г о d к е у R. S., J. Appl. Phys., 33, 2269 (1962).
Derjaguin B. V., Kolloid Z., 69, 155 (1934).
Derjaguin B. V., Trans. Faraday Soc, 36, 203 (1939).
Derjaguin B. V., Acta physicochim., 10, 333 (1940).
Derjaguin B. V., Samygin M. H., Discuss. Faraday Soc, 18, 24 (1951).
Derjaguin B. V., Titijevskaya A. S., Proceedings of the II
International Congress on Surface Activity, vol. 1, 1957, p. 211.
Din F. J., S с о t t Blair G. W., J. Appl. Phys., 11, 574 (1940).
E i 1 e г s H., Kolloid Z., 97, 313 (1941).
Einstein A., Ann. Phys., 19, 289 (1906).
Einstein A., Ann. Phys., 24, 591(1911).
Eiiich F. R., В u n z 1 M., M a r g а г e t h a H., Kolloid Z.,.74, 275 (1936a).
Eiiith F. R., В u n z 1 M., M a r g a r e t h a H., Kolloid Z., 75, 20 (1936b).
E v e s о n G. F., J. Oil Colour Chem. Assoc, 40, 456 (1957).
E v e s о n G. F., in «Rheology of Disperse Systems», ed. by С. С Mill. London,
1959 p. 61.
Ewe 11 R. H., Eyring H., J. Chem. Phys., 5, 726 (1937).
F a x e n H., Ark. Mat. Astr. Fys., 17, 1 (1922).
Flowers A. G., Proc Am. Soc. Test. Mater., 14, 565 (1914).
F о w к e s F. M., Ind. Eng. Chem., 56, 40 (1964).
Fredrickson A. G., Principles and Applications of Rheology, New Jersey,
1964.
Fro hiiсh H., Sack R., Proc Roy. Soc, A185, 415 (1946).
Fryling C.F, J. Colloid. Sci., 18, 713 (1963).
Фу kc H., Коллоида, ж., 20, 705 (1958).
Gabrysh A. F., Eyring Н., S h i m i г u M., A say J., J. Appl.
Phys., 34, 261 (1963).
Gillespie Т., J. Cooloid. Sci., 15, 219 (1960a).
Gillespie Т., J. Polymer. Sci., 46, 383 (1960b).
Gillespie Т., W i 1 e у R., J. Phys. Chem., 66, 1077 (1962).
Gillespie Т., J. Colloid Sci., 18, 32 (1963 a).
G i 1 1 e s p i e Т., in «Rheology of Emulsions», ed. by P. Sherman, London, 1963 b,
p. 115.
Glasston S., Laidler K. J., Eyring H., The Theory of Rate Pro-
cgssgs New York 194:1.
Goldsmi'th H.L.,'Mason S. G., J. Colloid. Sci., 17, 448(1962).
G о о d e v e C.F, Trans. Faraday Soc, 35, 342 (1939).
G о u 1 d e n J.D.S., Phipps L. W., J. Dairy Res., 31, 195 (1964).
Greenberg S. S., Jarnutowski R., Chang T. N., J. Colloid Sci.,
20, 20 (1965).
Guth E., Simha R., Kolloid Z., 74, 266 (1936).
Haighton A. J., J. Am. Oil Chem. Soc, 36, 345 (1959).
Harmsen G. J., van Schooten J., Overbeek J. Th. G., J.
Colloid Sci., 8, 64 (19S3).
Hatschek E., Kolloid Z., 8, 34 (1911).
Ha user E. A., Le В e a u D. S., Kolloid Z., 86, 105 (1939).
H e r s с h e 1 W. H., В u 1 к 1 e у R., Kolloid Z., 39, 291 (1926).
H i g g i n b о t h a m G.H., Oliver D. R., Ward S. G., Brit. J. Appl.
Phys., 9, 372 (1958).
H о u w i n к R., Elasticity, Plasticity and Structure of Matter, New York, 1958.
I n о к u с h i K., Bull. Chem. Soc. Japan, 28, 453 (1955).
J e f f e г у G. В., Proc Roy. Soc, A102, 161 (1922).
J о 1 у М., Proceedings of the II International Congress on Rheology, London,
1954, p. 365.
Joly M., J. Colloid Sci., 11, 519 (1956).
Kambe H., Takano M., Report No. 377, Aeronautical Research
Institute, University of Tokyo, 1963.
309
К а р а с е в а В. В., Д е р я г и н Б. В., ЖФХ, 33, 100 (1959).
Kim W. К. a. oth., J. Appl. Phys., 31, 358 (1960).
Krieger I. M., Dougherty T. J., Trans. Soc, Rheol., 3, 137 (1959).
К у n с h G. J., Br. J. Appl. Phys., Suppl. № 3, 85 (1954).
Kynch G. J., Proc. Roy. Soc, A237, 90 (1956).
Ladenburg R., Ann. Phys., 23, 447 (1907).
Lawrence A. S. С, М i 1 1 s O. S., Discuss. Faraday Soc, 18, 98 (1954).
Lawrence A. S. C, R о t h w e 11 E., Proceedings of the II International
Congress on Surface Activity, vol. 1, 1957, p. 499.
Leviton A., Leighton A., J. Phys. Chem., 40, 71 (1936).
L i n d s 1 e у С.Н., Fischer E. R., J. Appl. Phys., 18, 988 (1947).
Linton M-, Sutherland K., Proceedings of the II International
Congress on Surface Activity, vol. 1, 1957, p. 494.
Mauley R. St. J., M a s о n S. G., Canad. J. Chem., 32, 763 (1954).
M a r i Y., О t a t a k e N., Chem. Eng. Tokyo, 20, 488 (1956).
Maron S.H., Belner R. J., J. Colloid. Sci., 10, 523 (1955).
Maron S. H., Elder E. H., J. Colloid. Sci., 9, 263 (1954).
Maron S. H., L e v у - P a s с a 1 A., J. Colloid. Sci., 10, 494 (1955).
Maron S.H., Madow B. P., J. Colloid. Sci., 8, 130 (1953).
Maron S. H. Madow В. Р., К г i e g e г I. M., J. Colloid. Sci., 6, 584
(1951).
Maron S.H., Ming Fok S., J. Colloid. Sci., 10, 482 (1955).
Maron S. H, Pierce P. E., J. Colloid. Sci., 11, 80 (1956).
Maron S. H., Sisko A. W., J. Colloid. Sci., 12, 99 (1957).
Moelwyn Hughes E.A., Physical Chemistry, London, 1959.
Mooney M., J. Colloid. Sci., 1, 195 (1946).
Mooney M., J. Colloid. Sci., 6, 162 (1951).
Moore W. J., Eyring И., J. Chem., Phys., 6, 391(1938).
Morrison Т.Е., Zapas L. J., De Witt T. W., Rev. Sci., Instrum.,
26, 357 (1955).
Mukerjee P., J. colloid Sci., 12, 267 (1957).
N a w a b M. A., M a s о n S. G., Trans. Faraday Soc, 54, 1712 (1958).
Oldroyd J. G., Proc. Roy Soc, A218, 122 (1953).
Oldroyd J. G., Proc Pov Soc, A232, 567 (1955).
Ostwald W., Kolloid Z., 36, 99, 157, 248 (1925).
Overbeek J. Th. G., in «Colloid Science», ed. by H. R. Krugt, vol. 1,
Amsterdam, 1952 p. 258.
Ping A. P., L e u b b e г s R. H., Am. Inst. Chem. Eng. J., 3, 111 (1957).
Poiseuille J. L. M., С. г., 11, 961 (1840).
Rabinowitsch R., Z. phys. Chem., A145, 1 (1929).
Raja Go pal E. S., Z. phys. Chem., 23, 342 (1960).
Raja Gopal E. S., Rheol. acta, 1, 584 (1961).
Ребпндер П. А., Се мен ен ко Н. Н., ДАН СССР, 64,835(1949).
Ree Т., Eyring H., J.Appl. Phys., 26, 793 (1955).
Reiner M., Kolloid Z., 39, 80 (1926).
Reiner M., Deformation, Strain and Flow. London, 1960.
Reynolds O., Phil. Trans. Roy Soc, 174, 935 (1883).
Rich S.R., Roth W., J. Appl. Phys., 24, 940 (1953).
Richardson E.G., Kolloid Z., 65, 32 (1933).
Richardson E. G., J. Colloid Sci., 5, 404 (1950).
Richardson E. G., J. Colloid Sci., 8, 367 (1953).
Robinson J., J. Phys. Colloid Chem., 53, 1042 (1949).
Robinson J., J. Phys. Colloid Chem., 55, 455 (1951).
Robinson J., Trans. Soc. Rheol., 1, 15 (1957).
Roscoe R., Brit. J. Appl. Phys., 3, 267 (1952).
R u m s с h e i d t F. D., M a s о n S. G., I. Colloid Sci., 16, 238 (1961).
Saito H., J. Phys. Soc. Japan, 5, 4 (1950).
S a m b u с E., Naudet M., Rev. fr. Cps gras, 6, 10 (1959).
Saunders F. L., J. Colloid. Sci., 16, 13 (1961).
Schenkel J.H., Kitchener J., Trans. Faraday Soc, 56, 161 (I960).
310
Scott J. R., I.R.I. Trans.. 7, 2 (1931).
Scott Blair G. W., Rheol. acta, 23, 123 (1958).
Scott Blair G. W., О о s t h u i s e n J. C. Brit. J. Appl. Phys., 11,
332 (1960).
Scott Blair G. W., Paper Presented at Annual Meeting, French Rheology
• Society, Paris, 1965.
Shangraw R., Grim W., Mattocks A. M., Trans. Soc. Rheol., 5, 247
(1961).
Sherman P., J. Soc. Chem. Ind. (London), 69, Suppl. Issue № 2, 70 (1950 a).
Sherman P., J. Soc. Chem. Ind. (London), 69, Suppl. Issue № 2, 74 (1950 b).
Sherman P., J. Colloid Sci., 10, 63 (1955a).
Sherman P., Kolloid Z., 141, 6 (1955b).
Sherman P., Mfg. Chem., 26, 306 (1955 c).
Sherman P., Kolloid Z., 165, 156 (1959).
Sherman P., Proceedings of the III International Congress on Surface
Activity, vol. 2. 1960, p. 596.
Sherman P., Fd. Technol. (London), 15, 394 (1961).
Sherman P., J. Phys., Chem., 67, 2531 (1963a).
Sherman P., in «Reology of Emulsions», ed. by P. Sherman, London, 1963 b,
p. 77.
Sherman P., Paper Presented at the IV International Congress on Surface
Activity, Brussels, 1964.
Sherman P., Proceedings of the IV International Congress on Rheology,
New York, 1965, p. 605.
S h о t t о n E., White R. F., J. Pharm. Pharmacol., 12, 108T (1960).
S h о t t о li E., White R. F., in «Rheology of Emulsions», ed. by P. Sherman,
London, 1963, p. 59.
Shoulberg R. H., Zimmerli F. H., Kohler О. С, Trans. Soc.
Rheol.. 3, 27 (1959).
Sibree J.O., Trans. Faraday Soc, 26, 26 (1930).
Sibree J.O., Trans. Faraday Soc, 27, 161 (1931).
S i m.li a R., Somcynsky Т., J. Colloid Sci., 20, 278 (1965).
von Smoluchowski M., Phys. Z., 17, 557, 585 (1916 a).
von Smoluchowski M., Phys. Z., 18, 190 (1916 b).
von Smoluchowski M., Z. phys. Chem., 92, 129 (1917).
Street N., J. Colloid Sci., 13, 288 (1958). .
Sumner С G., Trans. Faraday Soc, 36, 372 (1940).
Sweeney R. H., G e с к 1 e г R. D., J. Appl. Phys., 25, 1135 (1954).
Takano M., Bull. Chem. Soc. Japan., 37, 78 (1964).
Taylor G. I., Proc. Roy. Soc, A138. 41 (1932).
Taylor G. I., Proc. Roy. Soc, A146, 501 (1934).
van den T e m pe 1 M., Proceedings of the II International Congress on Surface
Activity, vol. 1, 1957, p. 439.
van den T e m p e 1 M., in «Rheology of Emulsions», ed. by P. Sherman, Lon-
■ don, 1963. p. 1.
Thomas D. G., J. Colloid Sci., 20, 267 (1965).
T г e v e 1 у a n B. J.. M a s о n S. G., J. Colloid Sci., 6, 354 (1951).
Toms B. A., J. Chem. Soc, 1941, 542.
Umstatter H., Kolloid Z., 70, 174 (1935).
Vand V., J. Phys. Colloid. Chem., 52, 277, 300 (1948).
V e г w e у E. J. W., О verbeek J. Th. G., Theory of the Stability of Lyo-
phobic Colloid, Amsterdam, 1948.
de Vries A. J., in «Rheology of Emulsions», ed. by P.Sherman. London,
1963, p. 43.
Void M. J., J. Colloid. Sci., 16, 1 (1961).
van der Waarden M., J. Colloid Sci., 9, 215 (1954).
de Waele A., Kolloid Z., 36, 332 (1925).
Ward S. G., W h i t m or e R. L., Brit. J. Appl. Phys., 1, 286(1950).
Van W a z e г J. R., Lyons I. W., К i m K. Y., Col well R. E.,
Viscosity and Flow Measurement, New York, 1963, p. 202.
311
Weissenberg K., in «The Testing of Materials by means of the Rheogo-
niometer», Farol Research Engineers, Bognor, 1964.
Weymann A. D., Proceedings of the IV International Congress on Rheology,
vol. 3, 1965, p. 573.
Williams J.C., Fulmer E. J., J. Appl. Phys., 9, 760 (1938).
Williams P. S., Discuss. Faraday Soc, 11, 47 (1951).
Williams P. S., J. Appl. Chem., 3, 120 (1953).
Williamson R. V., lnd. Eng. Chem., 21, 1108 (1929).
Wilson С L., Parke s J., Quart. J. Pharm. Pharmac, 9, 188(1936).
Yerazanis S., Bartlett J. W., Nissan A. H., Nature (London),
195, 33 (1962).
Дополнительная литература
Barry В. W., J. Coll. Interf. Sci., 32, № 3, 551 (1970). Реология эмульсии,
стабилизированных додецилсульфатом натрия и высшими спиртами.
С а г 1 е s s J. Е., Н а 1 1 w о г t h G. W., J. Coll. Sut. Sci., 26, № 1. 75 (1968).
Вязкость эмульгирующих агентов на поверхности масло—вода.
Doroszkowki A., Lambourne R., J. Coll. Interf. Sci., 26, № 1,
128 (1968). Определение константы Ван-дер-Ваальса по реологическим
свойствам эмульсий.
L in J. Т., J. Soc. Cosmet. Chem., 19, № 10, 683 (1968). Влияние начальной
концентрации ПАВ на вязкость эмульсий.
М u k е г j с е L., S h u к 1 a S., Indian J. Appl. Chem., 32, Л» 4, 247 (1969).
Значение вязкости и гидратации гидрофильных коллоидов для
образования эмульсий и пх стабильности.
Matsumoto S., Sherman Р. В., J. Coll. Interf. Sci., 30, Л. 4, 525 (1969).
Вязкость мпкроэмульспй.
Mukerjee L., S h e г m a R. G., Indian i. Appl. Chem., 31, N° 5—6, 164
(1968). Зависимость вязкости от величины капель. (Morosowa К.,
Mimata К., М i t s u i Т., Nippon Kagaku Zasshi, 90, № 4, 352 (1969).
Реологические зависимости эмульсий М/В. I. Явление загустевают при
сдвиге, наблюдаемое в эмульсиях, представляющих собой тройные
системы — жидкий углеводород — поверхностно-активное вещество — вода.
Rimlinger G., Bull. Tech. Guttenfosse SFPA, N° 64, 65 (1968).
Взаимосвязь между составом эмульсии и ее вязкостью.
Sherman P.. J. Coll. Interf. Sci., 24, N° 1, 97 (1964). Изменение
реологических свойств эмульсий при перемешивании.
Sherman P., J. Coll. Interf. Sci., 27, N° 2, 282 (1968). Реологические
изменения эмульсий при перемешивании.
Shotton Е.. D а г е i s S. S., J. Pharm. Pharmacol., 20, N° 6, 439 (1968).
Влияние концентрации эмульгатора на реологические свойства эмульсий
М/В, стабилизированных анионными мылами.
Suzuki К. a. oth., Bull. Cbem. Soc. Japan, 42, № 10, 2773 (1969).
Неньютоновское поведение эмульсий В/М.
Т а 1 m a n F. A., D a vies P. J., Powan E. M., J. Pharm. Pharmacol.,
19, N° 7, 417 (1967). Реология некоторых эмульсий MB,
стабилизированных конденсированными пленками.
Савосонкова М. Ф., Р у б и к о в а Ю. С. Л у к ь я н о в А. Б., Масло-
жир, пром., 35, № 7. 28 (1969). Тпксотроппческие свойства эмульсий,
стабилизированных фосфатами крахмала.
Глава V
ЭЛЕКТРИЧЕСКИЕ
СВОЙСТВА
ЭМУЛЬСИИ
Т. Ханаи
Изучение диэлектрических свойств
дисперсных систем имеет
практическое и теоретическое
значение, так как информация о их
внутренней структуре и свойствах
может быть получена без
нарушений системы в процессе измерений.
В начале прошлого века Максвелл,
создав теорию электромагнетизма,
начал изучать диэлектрические
свойства веществ, обусловленные
их гетерогенностью. Примерно в то
же время коллоидные дисперсии
рассматривались как один из видов
гетерогенных систем. Позднее Дебай
предложил теорию полярных
молекул, рассматривая их как частный
случай диэлектриков. Такая
трактовка вызвала большой интерес среди
исследователей, в результате чего
теория полярных молекул получила
широкое применение и была
распространена на область коллоидного
состояния вещества. Это влияние
можно проследить на примере
исследований диэлектрических свойств
макромолекулярных и
протеиновых растворов, адсорбции молекул
на порошках твердого вещества
и т. д. По этому вопросу имеется
значительное число работ как
обзорного, так и оригинального
характера.
С другой стороны, за последние
годы стало известно много
диэлектрических явлений, характерных для
коллоидных дисперсий, которые не
могут быть объяснены с помощью
теории полярных молекул. Поэтому
нужно искать какой-либо
аналитический метод для их толкования.
Несмотря на значительный интерес
к этой области найдено небольшое
число публикаций по
диэлектрическим свойствам коллоидных
дисперсий, которые изложены в форме,
доступной для химика-коллоид-
ника.
313
В данной главе изложены в общих чертах проблемы, которые
встречаются при изучении диэлектрических свойств дисперсных
систем. Объяснены некоторые основные положения теории
диэлектриков и описаны методы измерений диэлектрической
проницаемости и электропроводности. Вместе с тем, проблемы, касающиеся
применения теории полярных молекул, рассмотрены здесь в тесной
связи с теориями молекулярных диполей и гетерогенных дисперсных
систем.
Электропроводность эмульсий представляет интерес как метод
определения дисперсности.
Как показано ниже (стр. 334), электропроводность является
неотъемлемой частью диэлектрических свойств дисперсных систем
и должна рассматриваться как теоретически, так и экспериментально
вместе с диэлектрической проницаемостью. Термин «электрические
свойства эмульсий» включает вопросы возникновения заряда,
поляризации и двойного слоя на поверхности раздела и т. д. Эти явления
выходят за пределы данной главы.
ОСНОВНЫЕ СВОЙСТВА ДИЭЛЕКТРИКОВ
Явления в диэлектриках
Если эбонитовую палочку потереть о мех, то она заряжается
отрицательно, а мех положительно. Это явление хорошо известно,
как электризация трением или трибоэлектричество.
Если заряженную палочку поднести к кусочкам бумаги, они
притянутся. Объясняется это следующим образом: на поверхности
бумаги, близкой к палочке, индуцируется положительный заряд,
на противоположной части — отрицательный. Сила притяжения
между палочкой и прилежащей частью листка, будет больше, чем
отталкивающая сила, возникающая между палочкой и его
противоположной частью. В результате бумажный листок притягивается
палочкой. Другими словами, заряды на эбонитовой палочке создают
вокруг нее электрическое поле и любой предмет, внесенный в это
поле, заряжается вследствие электростатической индукции. Это
самое распространенное электрическое явление.
Величина индуцированного заряда определяется
диэлектрической проницаемостью.
Диэлектрическая проницаемость,
электрическое смещение,
напряженность поля и потенциал
Для количественной оценки электростатической индукции
рассмотрим конденсатор (рис. V.1, а), состоящий из двух параллельных
пластин с площадью поверхности 5 (в см*) и расстоянием d (в см)
между ними. Когда пластинам конденсатора сообщается заряд QS
(где Q — плотность электрического заряда), то между ними
возникает однородное электрическое поле. Силовая характеристика поля
314
называется электрическим смещением D. Можно предположить,
что D пропорционально QS. В соответствии с электростатической
теорией (теорема Гаусса)
D = knQ (V.1)
Если в это поле поместить диэлектрик (рис. V.1, б), то на
поверхностях, прилежащих к пластинам конденсатора, будет
индуцироваться заряд PS (P — плотность заряда) противоположного знака.
Вектор Р называется вектором поляризации диэлектрика. Так как
индуцированный заряд PS расположен близко к заряду на
пластине QS, имеющему противоположный знак, то PS и часть QS
взаимно компенсируются. В результате реальный заряд каждой
пластины станет равным QS — PS, а напряженность поля будет Е
в
QS-PS
\U
Рис. V.I. Модельный конденсатор для объяснения явлений
в диэлектриках:
а— свободный или общий заряд QS; б —связанный или
поверхностный заряд PS; в — нескомпенсированный заряд QS—PS.
(рис. V.1, в). Заряд QS — PS называется нескомпенсированный
или несвязанным зарядом, a^QS и PS — свободным или общим и
связанным пли поверхностным зарядом, соответственно. Напряженность
электрического поля Е, как мы предположили, пропорциональна
QS — PS и выражается в соответствии с теоремой Гаусса следующим
образом
E = in-9-iO^(Q — P) (V.2)
где Е — в в/см; Р — в к/см2.
Ясно, что при постоянной величине заряда QS значение вектора
смещения постоянно. Отношение P/Q должно быть мерой индукции.
В соответствии с электромагнитной теорией, диэлектрическая
проницаемость е, как мера электростатической индукции, определяется
отношением свободного заряда QS к нескомпенсированному
заряду QS — PS:
Qs 1 _n.<niij^-^< /у з)
QS-PS 1-
— q. ю11 —
■P/Q * Ш Е
1
В вакууме индуцированного заряда не возникает, т. е. Р = О
и, следовательно, е = 1.
В обычных случаях PS не может превышать QS. В крайнем случае
имеем PS = @5, который даст е = +оо. Как правило, е находится
в пределах 1 ^ е < оо.
315
В примере, иллюстрированном на рис. V.1, где QS = 5, и PS = 3,
е можно вычислить так:
QS 5
QS-PS 5-3
= 2,5
Потенциал между двумя пластинами определяется как работа,
совершенная против сил электрического поля при перемещении
единицы электрического заряда от одной пластины к другой
d
U = J \Edx = EdK (V.4)
о
где U —■ в в; d — в см.
Свойства заряда Р,
обусловленного электростатической индукцией
Заряд находится на поверхности диэлектрика и не способен
покинуть ее. Можно отметить следующие его свойства.
Заряд PS появляется в тот момент, когда к электродам приложен
заряд QS, и немедленно исчезает, как только заряд QS будет снят.
Индуцированный (или поверхностный) заряд, возникающий на
фазовых границах, может быть найден из уравнения (V.3)
/>=(1_¥)<3- (v-5)
и не зависит от времени t. Строго говоря, уравнение (V.5) является
не совсем точным. Например, известно, что полярные жидкости
проявляют замедленную поляризацию. Такая диэлектрическая
дисперсия обусловлена замедленной ориентацией молекулярных
диполей на очень высоких частотах, порядка нескольких сотен мегагерц.
Тогда временную зависимость исключить нельзя, и характер ее
необходимо учитывать. Тем не менее, уравнение (V.5) полезно для
качественного понимания физического смысла поляризации
поверхности раздела, которая обычно наблюдается на более низких
частотах.
Соотношение между емкостью
и диэлектрической проницаемостью
Диэлектрическую проницаемость рассчитывают, исходя из
электростатической емкости конденсатора, а не по величине
индуцированного заряда Р.
Емкость (в ф) конденсатора с параллельными пластинами
определяют из уравнения:
С = ^ (V.6)
Из уравнений (V.6), (V.4), (V.1) и (V.3) следует, что
C=4K-9-10UEir <V-7>
316
или
С=габсе^- (V.8)
где еабс — абсолютная диэлектрическая проницаемость среды,
равная:
еабс = 4я,, д, 1QU = 8,8541 • 10-14 ф/см (V.9)
Из уравнения (V.8) величина емкости С0 в вакууме (т. е. е = 1)
определяется как еабс
С0 = ^бс4- (V.10)
"к
Из уравнений (V.8) и (V.10) получим:
4- = г (V.11)
С помощью этого соотношения можно экспериментально
определить диэлектрическую проницаемость.
Электрический момент
и вектор электрической поляризации
Рассмотрим систему, состоящую из двух зарядов +е и —е,
находящихся на расстоянии I друг от друга (рис. V.2). Электрический
момент всей системы определяется произведением el. Когда
расстояние I мало, электрический момент
называется дипольным моментом. +Д ~^
Рассмотрим более общий случай ^^^-^ I """^-^
(см. рис. V.1): две пластины с пло- ~ "
щадью поверхности S расположены на Рис- V-2' Электрический мо-
расстоянии d друг от друга. Тогда
электрический момент диэлектрика (см. рис. V.1, б) определяется
как произведение
PS-dK (V.12)
Так как объем материала, введенного в пространство между
пластинами конденсатора, равен SdK, то электрический момент
единицы объема составит
-т£-=р (V13>
Отсюда следует, что вектор поляризации Р равен электрическому
моменту единицы объема. Выражение (V.13) является очень важным
в развитии диэлектрической теории гомогенных систем, таких как
молекулярные растворы. При теоретическом анализе молекулярных
растворов электрический момент Р для единицы объема,
определяемый уравнением (V.13), вычисляется как сумма дипольных моментов
молекул, находящихся в единице объема. Благодаря аддитивности
вектора поляризации, вычисление электрического момента
упрощается .
317
Активная и удельная электропроводность
и сопротивление
Представим параллельные пластины конденсатора (см.
рис. V.1, б), пространство между которыми заполнено
электропроводным материалом. Активная электропроводность G — мера
проводящих свойств — определяется как постоянная величина,
пропорциональная отношению силы тока / к потенциалу U между
двумя пластинами (закон Ома):
UG = I (V.14)
где / — в a; G — в ом'1.
В этом случае полное количество электричества, проходящее
через материал, определяет величину силы тока
/ = ^-(<ЗпРов5) . (V.15)
где t — в сек.
Другими словами, заряд проводимости <2ПрОВ5\ протекающий
в единицу времени, в соответствии с уравнением (V.15), должен
постоянно поддерживаться на пластинах, чтобы обеспечить
непрерывный ток /.
Удельная электропроводность х (в ом'1 /см), характеризующая
электропроводность материала, независимо' от его геометрии дается
уравнением
б=х-£- (V.16)
Сопротивление (в ом) конденсатора определяется как
л=4" (v-17)
Свойства зарядов проводимости
Заряды проводимости обладают следующими свойствами.
1. Достигнув границы с диэлектриком, заряды могут перейти
в более проводящую фазу, смежную с границей, или на электроды»
2. Будем считать время t — временем движения зарядов.
Интегрируя уравнение (V.15) и используя уравнение (V.14),
получим
Qnv0BS=GUt (V.18)
при условии, что U поддерживается постоянным. Величина Qnp0B
возрастает пропорционально времени t.
3. Величина х пропорциональна величине тепла, выделяющегося
в единицу времени.
318
Соотношение явлений электропроводности и смещения.
Комплексные величины поля переменного тока
В соответствии с теорией поля переменного тока
± = 1* (V.19)
где ;* = У— 1; со — угловая частота;со = 2л./; / — частота, гц.
Реальный конденсатор (см. рис. V.1, б) характеризуется двумя
свойствами: смещением и электропроводностью. В этом случае общий
заряд, находящийся на пластинах, равен сумме двух видов зарядов,
а именно: смещения (QS) и проводимости (Q„PObS)- Используя
уравнения (V.6), (V.14), (V.15) и (V.19), можно получить выражения
для общего заряда:
TIP
Qo64 = UC + — (V.20)
G
0общ = (/(С+_)
Уравнение (V.20) обычно записывается в виде
<?общ= UC* (V.21)
где С* — комплексная емкость
С*=С+— (V.22)
/со
Или уравнение (V.22) запишется как
C* = C-jC" (V.23)
где С" — мнимая часть комплексной емкости.
С"=— (V.24)
со
Уравнение (V.21) является частным случаем уравнения (V.6)
для полной электропроводности.
Подстановка уравнений (V.8) и (V.16) в уравнение (V.22) дает:
С* = еабс(е + —^— )-£- (V.25)
Это уравнение можно записать в виде
C* = Ea6cE*-f- (V.26)
dK
где е* — комплексная диэлектрическая проницаемость.
г* = е+~— (V.27)
.'шеабс
319
Уравнение (V.27) можно записать иначе
е* = е — /р."
(У. 28)
где е" — мнимая часть комплексной диэлектрической проницаемости
или фактор потерь
* *■• 1,7975-1012 (V.29)
соеабс
/
Уравнение (V.26) представляет частный случай уравнения (V.8)
для полного значения емкости.
Когда Сие зависят от частоты, они обозначаются С'ие'и
означают реальную часть комплексной величины.
Как показывает уравнение (V.29), е" утрачивает свой физический
смысл при постоянном токе (/ = 0), тогда как у, является всегда
измеряемой величиной.
Тепло, которое выделяется при прохождении тока за время одного
колебания, дается выражением: ^iJjfe", где U0 — амплитуда
потенциала U. Таким образом, е" пропорционально количеству тепла,
выделенного за одно колебание.
Тангенс потерь tg б или диссипация энергии является мерой
потери энергии и находится из выражения:
Угол определяется как
= tg-i (J
(V.30)
(V.31)
Энергетический фактор cos cp, применяемый на практике, дается
уравнением:
cos cp = ^in 6
l/p2
Е2 + е"
(V.32)
Некоторые примеры диэлектрической проницаемости
и электропроводности
В табл. V.1 приведены значения е и соответствующие значения
PS/QS и и для некоторых типичных материалов. i
Таблица V.1
Несколько примеров значений г и х
Вещество
Вакуум
Гексан
Диэтиловый эфир
Нитробензол
Вода
е
1,0
2,0
4,4
35,0
80,0
PS
QS
0,0
0,50
0,77
0,97
0,99
1
ом ■ см
0
Ю-12
10-9
ю-'
10-4—10-7
320
Если конденсатору (см. рис. V.1) сообщается заряд, равный 100
(QS = 100), то индуцированный или поверхностный заряд PS в
вакууме равен нулю, а в воде — 99. В любом случае выполняется
условие 0 г^ P/Q <1. В действительности, величина е для чистых
веществ с низким молекулярным весом колеблется между 2 и 100
в противоположность х, имеющему широкий диапазон значений.
ЭКСПЕРИМЕНТАЛЬНЫЕ МЕТОДЫ ИЗУЧЕНИЯ
ДИЭЛЕКТРИЧЕСКИХ СВОЙСТВ
Основные положения
Существует целый ряд методов для измерения диэлектрических
свойств материалов. Применение их требует необходимых знаний
и технических навыков, рассмотрение которых выходит за пределы
настоящей главы. Поэтому по ходу изложения будут даваться ссылки
на соответствующие труды по экспериментальной технике, так
например, Паулс п Смит (1960). Чтобы понять связь между
диэлектрической проницаемостью и удельной электропроводностью,
рассмотрим некоторые принципы и методы измерений на частотах ниже
нескольких мегагерц.
Общими принципами различных экспериментальных методов
является измерение емкости С0, сопротивления R0 или
электропроводности G0 пустого конденсатора-ячейки и тех же параметров кон-
денсатора-ячейкп, заполненного диэлектрическим материалом.
Из уравнений (V.22), (V.25) и (V.26) комплексная емкость образца
выражается как
С*=С + —= С + -Лг (V.33)
и
C* = e.fce'A = Coe-=Ce(e + _J^) (v..y,)
Величины С и R в уравнении (V.33), рассматриваемые вместе,
определяют импеданс 1/(G + /и С) конденсатора, наполненного
образцом.
Таким образом, имеем [подобно уравнению (V.11)]:
& = ~ (V.35)
„ еабс г, _ еабс ,,. „„.
*—сГ "с^Г ( 36)
х __ G
й)еабс шСо
(V-37)
Ниже рассмотрены два принципиальных метода измерения С
и G или R.
21 Заказ 1070
321
Мостовой метод
На рис. V.3 показан измерительный мост. Сопротивление плеч
(Rl и R2) известно. Переменный конденсатор с емкостью С5 и
сопротивлением Rs подстраивается таким образом, что в точках А и В
может быть некоторое напряжение, а стрелка индикатора показывает
нуль. При условии равновесия моста
получаем следующие отношения:
R-
Jh
R2
(V.38)
(V.39)
Мостовой метод применяют на
частотах от 200 гц до 100 кгц.
Мост, плечи которого имеют
сопротивления R1 и R2, не дает достаточной
чувствительности и точности при
измерении импеданса в широком диапазоне
частот. Для измерений в диапазоне от
30 гц до 5 мгц общепринятой является мостовая схема Коле
и Гросс (1949), плечи которой содержат индуктивности.
Рис. V.3. Схема измеритель
ного моста:
1 — генератор; 2 — образец
3 — индикатор.
Резонансный метод
Измерительное устройство, используемое Хартшорном и Бардом
(1936), показано на рис. V.4. Генератор с постоянной частотой / =
= со/2л индуктивно связан со вторичной резонансной цепью так,
■00
Рпс. V.4. Резонансный метод:
а — резонансная схема; б — эквивалентная схема; 1 — генератор; 2 — ячейка.
чтобы сообщать ей постоянное напряжение U0. Резонансная цепь
(рис. V.4, а) состоит из катушки с индуктивностью L и
сопротивлением RL, переменного конденсатора с емкостью Сь, конденсатора-
ячейки с емкостью С и электропроводностью G. Ее упрощенная
эквивалентная схема дана на рис. V.4, б, где
Ct = C + Cv (V.40)
322
Потенциал между точками А я В определяется как
ТГ~ (G + j<aCt)-i
U 0 RL + )(oL
(G + fwCt)-
RL—j<oL
Z2
где
Тогда
U0
U
Z=/?£^ffl2£2
RL + j(oL
\GZ^ + RL + j(ii(CtZ2~L)]
(V.44)
(V.41)
(V.42)
(V.43)
Z2
[ (GZ2 -
+ flL)2 + ffi2(C(Z2-L)2] (V.45) 2
Условие для резонанса
напряжения
dCt \ U
= 0
(V.46)
дает величину емкости Ct как
Сь Cf-L/Z Ca Ct
Рис. V.5. Заряд в потенциальном
попе между точками А и В (U2A_B\
при емкости Ct-
[С,
t\P =
'Р 22 ДЯ +Ш2£2
(V.47)
где Ср — резонансная емкость.
Из уравнений (V.45) и (V.47) получаем отношение резонансного
напряжения Up к напряжению U при любой величине емкости Ct
' t/p I2
|i/P
\Uo/U\2 . |
f/o/f/pla "h
0)2 (C<Z2-L)2
(GZ2+
flr^2
(V-4i)
Это уравнение показывает, что | С/ |2 изменяется симметрично С,;
что касается емкости, то Ct = Ср = LIZ*, как вытекает из рис. V.5.
Два значения емкости Са и Сь при условии
ип
■-Uv 0,7071
IT P
которое соответствует уравнению (V.48), определяются как
с GZ*+RT
toZ2
21*
(V.49)
(V.50)
323
Эти соотношения приводят к
Са + Сь = 2Ср • (V.51)
И
АС = Са-Сь= м22 L' (V.52)
ИЛИ
0 = —2 2? О'.оЗ)
Значения емкости С и электропроводности G- конденсатора-ячейки
получают следующим образом:
1) Cv p при резонансе и АС (приращение между Са и Сй) измеряют
в цепи с конденсатором-яче11кой; 2) в цени без конденсатора-ячейки
измеряют соответствующие величины C'vp и ДС".
Принимая во внимание уравнения (V.51) п (V.53) имеем:
C = C'Vp— Cvp (V-54)"
с= СЬа + Clb _ Сеа + Сд (у55)
с=со(ДС_ДГ) (V56)
Резонансный метод применяют на частотах порядка от 10 кгц
до 100 мгц, за исключением высоконроводящих систем.
Электрические величины и их измерения
Известны несколько методов диэлектрических измерений,
отличные от описанных выше. Существенной особенностью всех методов
является измерение одной из следующих пар: е и х, е и г", г и tg б.
Одновременное измерение 8 и и в широком диапазоне частот
является существенным как теоретически, так и экспериментально,
так как приводит к более точно сбалансированному состоянию моста
и более точным измерениям. Кроме того, общая теория диэлектриков
учитывает явления электропроводности и предполагает частотную
зависимость диэлектрических характеристик.
В соответствии со свойствами зарядов (см. главу II), измерение
двух электрических величин независимо друг от друга может
интерпретироваться, как различие между двумя видами зарядов: смещения
и проводимости.
Некоторые аспекты диэлектрических свойств
в зависимости от размеров частиц
При изучении диэлектрических свойств коллоидных дисперсных
систем измеряют такие макроскопические величины, как
диэлектрическая проницаемость и электропроводность. Их рассматривают
в связи с внутренней структурой этих систем.
324
Анализ диэлектрических свойств различных систем, приведенных
в табл. V.2, связанный с размерами частиц, можно проводить двумя
методами. Один метод состоит в том, чтобы распространить
макроскопическую обработку, пригодную к крупнодисперсным системам
(диаметр частиц > 10 мкм), на мелкодисперсные системы. Другой
метод, основанный на молекулярной теории гомогенных растворов,
использует методику, применяемую к молекулярным дисперсиям
(— Ю-7 см), к довольно круинодисиерсным системам.
Таблица V.2
Различные системы и размеры их частнц
Микроскоп
Оптический
"
Электронный
Вид
дисперсности
Грубые
Коллоидные
Молекулярные
Атомы
Диаметр частиц,
см
ю-1
10-2
10-з
10"*
(длина волны
видимого света)
Ю"5
ю-6
Ю--
10"8
Примеры
Яйца лягушки; мелкий
песок
Картофельный крахмал;
крупные эмульсии
Красные кровяные
шарики; жировые
шарики молока
Средние бактерии
Мелкие эмульсии;
частицы коллоидного
золота
Высокополимерные
молекулы; коллоидный
угольный порошок;
мицеллы солюбилизи-
рованных систем
Молекулы протеина; ле-
цитиновые
бимолекулярные пленки
Молекулы
органического растворителя;
молекулы воды
С помощью первого метода исследуют некоторые
крупнодисперсные системы, такие как эмульсии и суспензии, вторым — растворы
несложных молекул и некоторых полимеров в свете диэлектрической
теории полярных молекул. Однако для систем, являющихся
промежуточными (со средним размером частиц), таких как
микроэмульсии, солюбилизированные системы воды в растворителях,
коллоидные дисперсные системы и растворы полимеров, до сих пор не имеется
общепринятых методов определения диэлектрических свойств.
ТЕОРИЯ ДИЭЛЕКТРИЧЕСКОЙ ПРОНИЦАЕМОСТИ
И ЭЛЕКТРОПРОВОДНОСТИ ГЕТЕРОГЕННЫХ СИСТЕМ
Сделано много попыток выразить диэлектрическую Проницаемость
сложных систем через диэлектрическую проницаемость фаз и их
компонентов применительно к сферическим дисперсным системам.
325
Ниже приведено несколько типичных случаев, псследованных как
теоретически, так и полуэмпирически.
Основные обозначения, использованные в уравнениях:
Ф — объемная доля фазы;
т — время релаксации диэлектрической дисперсии;
тр и Тд — время релаксации для тройной системы;
Ах — число сфер в единице объема.
Индексы
а, Ь, с — составные элементы конденсатора или составные фазы системы;
h, I — предельные значения на высоких и низких частотах, соответственно;
i — промежуточная величина;
* — комплексная величина;
' — реальная часть комплексной величины;
" — мнимая часть комплексной величины;
т — непрерывная среда;
р — дисперсная фаза;
s — оболочки частиц.
Рассмотрим прежде соотношения между величинами. Принимая
во внимание уравнения (V.22) —(V.24) и (V.27) —(V.29), получим:
С*--
Ga q , Ga _ „* Gt, Gb
/си a ^ /to ' - b /со h ' /со '•
c*— -^- —c ' -^-
c /со cT~ /со
c* = c+~=c — ic"
;co
-*— v*a -•■ i yia — с ic"
;сиеабс /СОЕабс
* Хл . Щ ■ ■■
/ШЕабс /COSa6c
r* X° г ' Xc r if"
^шеабс /ШЕабс
,* xm i "m _ - ,v."
;cufia6c УШЁабс
* "XP ( *p
p /coEa6c ^ ' /coea6c v p
;'coea6c ;coEa6c
e* = = в ч = e — /£
(V-57)
(V.58)
(V.59)
(V.60)
t
(V.fil)
(V.62)
(V.63)
(V.64)
(V.65)
/coea6c ;wea6c
Полуэмпирическая формула
На первой стадии полуэмпирического подхода к гетерогенным
смесям, диэлектрическая проницаемость е смеси, состоящей из двух
фаз, рассматривалась без достаточно строгой теоретической аргу-
326
ментации. Это позволило показать аддитивное соотношение
относительно некоторой функции / (е) в зависимости от объемной доли Ф
фракции:
/(е) = (1-Фй)/(еа) + Фь/(е6) (V.66)
Функциональная зависимость / (е) предполагалась следующей:
Винер (1912)
/(в) = в (V.67)
/(e) = 4" (V-68)
Беер (Лихтенекер, 1926)
f(*) = Vbte (V-69)
Лорентц — Лоренц (Лихтенекер, 1926)
/(s)=^i (V.70)
Лихтенекер (1926)
/(e)=lge (V.71)
Винер (1912)
/Н = -Ц^- (V-72)
В уравнении (V.72) и является параметром, который
рассматривается как функция еь/еа, Ф, формы частиц, структуры дисперсной
системы и т. д.
Фактически эта формула не всегда хорошо описывает
экспериментальные значения е в противоположность предложенной
полуэмпирической формуле.
Уравнение Релея
Первая попытка вычислить диэлектрическую проницаемость
сферических частиц дисперсной системы с помощью математического
анализа электростатического поля сделана Релеем в 1892 г. Он
вывел уравнение для дисперсных систем, в которых сферические
частицы с одинаковыми радиусами упорядочено расположены в узлах
простой кубической решетки, находящейся в непрерывной фазе
(рис. V.6). Это уравнение имеет вид:
■ , ЗФ
1-
£р + 2е„
Ер—Ь;п л'» U
-Ф-1,65——, ф '
Ер т~о" e"i
(V.73)
Уравнение Винера
Для разбавленных дисперсий, где Ф <^ 1, членом,
содержащим Ф10/', в уравнении (V.73) можно пренебречь. Винер (1912)
получил следующее уравнение:
г-гт = вр-8т ф (у74)
Е i 2ё/п ер-г2бт
Г- 327
Как будет показано ниже, уравнение (V.74) выведено также
Вагнером (1914) для статистического распределения сферических
частиц.
Уравнение Бруггемана
Бруггеман (1935) применил уравнение (V.74) к
концентрированным дисперсным системам на основе следующих предположений:
с>Ф-р^-фХ--&
I I ) u ( I I t-1 1 ' I i-t 1 i ) T^
бЩ&Шъ
^-.O
Pnc. V.6. Модельная система сфер с одинаковым
радиусом, расположенных в узлах кубической решетки, для
уравнения Релея.
1) уравнение (V.74) справедливо для процесса с бесконечно малым'
приращением концентрации дисперсной фазы; 2) наибольшая
концентрация дисперсной си«емы будет достигнута в условиях
непрерывного процесса.
При допущении первого предположения из уравнения (V.74)
можно получить соотношение для бесконечно малого
приращения Ае и АФ':
2е + ер —Дф'
Де = -
38 (8—Ер)
1 —Ф'
(V.75)
Второе предположение позволяет интегрирование по е (е„,, е)
и Ф' (О, Ф), приводящее к уравнению:
е —е
—ер \ s /
Ф
(V.76)
328
Используемые математические выкладки аналогичны
преобразованиям в теории Ханаи, которая подробно будет рассмотрена
далее (стр. 341).
Уравнение Бётчера
Бётчер (1952) рассмотрел дисперсные системы, плотно агрегати-
рованные двумя видами сферических частиц тир, при условии,
что дисперсионная среда и дисперсная фаза не отличаются друг
от ДРУга и развил следующую теорию.
Он предположил, что электрическое поле Fp внутри сферы
компонента р, определяемое негомогенным окружением рис. V.7, а,
Эквивалент
Р[[с. V.7. Эквивалентная схема, не учитывающая
распределение частиц по размерам, для уравнения
Бётчера
эквивалентно полю сферы, помещенной в диэлектрик,
диэлектрическая проницаемость которого е (рис. V.7, б).
'В соответствии с электростатической теорией
F 38
р 2е + ер
Е
(V.77)
где Е — напряженность электрического поля в точке, бесконечно
удаленной от сферы.
Аналогично имеем соотношение для сферы с диэлектрической
проницаемостью ет:
38 " (V.78)
Fm =
2е + е„
Вектор поляризации Р единицы объема всей системы равен сумме
векторов поляризации обоих видов сфер т и р без учета
распределения по размерам, т. е.
Р = еЛс(в-1)£ = (1-Ф)ЕЛ(ет-1)^ + Феавс(вр-1)/?р (V.79)
Подставив уравнения (V.77) и (V.78) в уравнение (V.79), получим:
Зе
Ер+28
■Ф
(V.80)
323
Уравнение (V.80) Бётчера не показывает изменений е обратной
эмульсии, так как по условию непрерывная фаза т не отличается
от дисперсной.
Уравнение Кубо— Накамура
Введение дисперсной фазы в виде сфер в непрерывную фазу
приводит к некоторому возрастанию е дисперсной системы в результате
увеличения электрического момента. Наивысшая концентрация
может быть достигнута постепенным добавлением. Основываясь
на этом, Кубо и Накамура (1953) разработали диэлектрическую
теорию сферических систем.
В соответствии с
электростатической теорией, Fp внутри
сферы с диэлектрической
проницаемостью ер и радиусом Rc,
помещенной в гомогенную среду
с диэлектрической
проницаемостью е (рис. V.8), находится из
уравнения
38 Е (V..81)
F„
2s-
а потенциал ^ виеш
определяется как
вне
Рис. V.8. Сфера, помещенная
в однородное электрическое поле Е.
U»
--Ez+E ■
сферы
■ cos 9
(V.82)
Величины г, В и ср являются переменными в общепринятой
сферической системе координат, см. рис. V.8. Приращение потенциала £/внеш,
обусловленное введением сферы, равно:
Л^ вне
-Е№
cos 9
(V.83)
Приращение Л£/Впеш по оси z, связанное с изменением внешнего
потенциала на AU, составит:
Д^„
д
~dz~
AF
^^ внеш — 1
COS 9
дг
-^—^ ) ДГв„еш (V.84)
3COS2 9 —1
(V.85)
Приращения вектора поляризации АРпнеш 2 (в к/см2) по оси и
электрического момента А МвнеШ2 по всему пространству вокруг сферы,
соответствующие приращению AFBHeui 2 (в в/см), равны
А^вкешг — fa6c (е Ч Ы" вне
(V.86)
330
AAfBHelU2 = jj'f APBBtal2r* sin Q drdQdy (V.87)
г=со 9=тс cd=«2tc
A*fBHem2 = ea6c(e-l)£7?3-iL— J J J ~r sine drdQdi? (V.88)
Если г, 6 и ф не зависят друг от друга, приращение АМвнеш г
дается выражением:
СО и 21
АЛ/внеш2 = 8абс (8-1) ER% 1"+1г \ Ц- \ (3cos2 6-1) sin 6 d9 f ЙФ (V.89)
а О О
Интегралы каждого из этих переменных дают:
2-я:
j йф = 2я (V.90)
о
j"(3cos29 —l)sined6 = 0 (V.91)
о
со
У^=11пг]»_юо (V.92)
яс
Уравнение (V.89) примет определенный вид благодаря
уравнениям (V.91) и (V.92).
Чтобы избежать трудностей, Кубо и Накамура использовали
довольно оригинальный метод вычисления. Они предположили,
что если электрическое поле меньше некоторой величины Fc, то им
можно пренебречь, так как влияние объемных элементов, далеких
от центра сферы, незначительно.
Электрическое поле вне сферы определяется как
,„._[(_iSg~.y+(-f.ibr)T" (V-93)
Пнеш= eEpP~2Ss • -ff- E (3 cos2 e+ DV 2 (V.94)
Интегрирование по г дает:
En —s Я3 \ Ч
-Цр-Е (3cos2e+l) /g (V.95)
ер+2е Fc ,
Вместо уравнения (V.92) можно записать
i-f-"a)-T,»(vS-f)+Tb""—+« <»•*>
331
Используя выражения (V.90) и (V.96), уравнение (V.88) можно
привести к виду:
(е-1)(ер-Е)
АЛ/„нсш г = еабс ,с , 0<Л Е2лЩ
3 nsp + 28 " Fe
S<3
COS2 9 — 1) X
x sin 8 сШ + -|- Г (3 cos2 6— 1) sin 8 In (3 cos2 6+1)
dQ
(V.97)
При использовании уравнения (V.91) первый член уравнения
(V.97), содержащий Fc, исчезнет. Интегрируя по частям, получим:
Мв
где
(е-1)(ер-е) 4л „,_
;£"* ^+2i с —Л^
4л
9^3
-=0,1Ц387
(V.98)
(V.99)
Приращение электрического момента внутри сферы АМвауг равно
разности моментов после введения сферы гр и до введения ее в поле:
АМвнут = [еа6с (8Р—1) /'р—Еабс (6—1) £]
AM
4л
тут— сабс
(г„-в).(2г+1) 4
2е-
3
(V.100)
(V.101)
Если число сфер в единице объема обозначить Av, приращение
вектора поляризации АР, которое равно приращению
электрического момента в единице объема, станет равным:
ЛР=еавс
АР = (АМвиеш z + AAfBHyT) Av
(fp-£)[(2 + c)e + (l-c)]
4л
ьрт
2е
Л3£ А у
Используя уравнения (V.5), (V.2) и (V.103), получим:
Де =
АР
(8р-8)[(2Ч-с)е-Ь(1-с)] 4л
Д1 Av
(V.102)
(Л -103)
(V-104)
еабс-£ 2е -f- Ер
Так как множитель (4л/3) i?3 Av) определяет объем сфер,
введенных в единицу объема дисперсной системы в процессе
постепенного добавления, то можно записать, что
3 с 1 —Ф
(Производная величины Ф' описывается далее (стр. 341).
Из уравнений (V.104) и (V.105) получим:
2s + 8r
(8р-е)[(2 + С)8 + (1-с)]
Де^
ДФ'
1 —Ф'
(V.105)
(V.106)
Интегрирование уравнения (V.106) по всем значениям е (ет, е)
и Ф' (О, Ф) дает:
Зе„
(2 + с)ер + (1-с)
lg-
(2-с)Ер-2(1-с)
lg
(2+с)е + (1-с)
[(2 + с)ер + (1-с)](2+с) ь (2 + e)e„+(i-e)
lg(l-O) (V-107)
Теоретическая формула для удельной электропроводности
сферических дисперсных систем
В соответствии с теориями электростатических и
квазиэлектростатических полей, удельная электропроводность играет роль,
аналогичную диэлектрической проницаемости. Поэтому уравнения,
выражающие функциональную зависимость для диэлектрической
проницаемости, также пригодны для удельной электропроводности.
Уравнение типа Релея. Рунге (1925) и Мередит и Тобиас (1960)
привели следующее уравнение для удельной электропроводности:
1-
ЗФ
' 2х,?
-Ф —0,523-
*р + 4/з*т
ф'°/о
(V.108)
tp лт лр -
В отличие от уравнения (V.73) числовой коэффициент здесь равен
0,523 (или л/6), а не 1,65 (или 0,523л). Мередит и Тобиас согласились
с исправлением, внесенным Рунге, и применили позднее значение
0,525.
Для эмульсий М/В (хт ^> яр) уравнение (V. 108) имеет вид
ЗФ
2 + Ф-0,523-з/4ф,°/3
а для эмульсий В/М (и,„ < хр)
ЗФ
1 + -
1 —Ф —0.523Ф
(V.109)
(V.110)
Уравнение типа Винера. Вагнер (1914) показал, что для
хаотического распределения сферических частиц удельная
электропроводность разбавленных дисперсных систем определяется уравнением:
Ф
(V.111)
Для эмульсий М/В (кт > Хр) уравнение (V. 111) сводится к виду
я 2(1—Ф)
хт 2+Ф
а для эмульсий В/М (кт < кр)
v. 1+2Ф
у.т ~~ 1 —Ф
(V.112)
(V.113)
333
Уравнение типа Бруггемана. Де ла Рю и Тобиас (1959) получили
следующее выражение для к:
*--*/» ( xm \V.
Хт Хо
' = 1 —Ф
(V.114)
Для эмульсий М/В (хт > кр) это уравнение примет вид
_^_ = (1_ф)'/' (V.115)
а для эмульсий В/М (%т ^ у,р)
1
(1_ф)3
(V.116)
ТЕОРИЯ КОМПЛЕКСНОЙ ДИЭЛЕКТРИЧЕСКОЙ ЛРОНИЦАЕМОСТИ
ГЕТЕРОГЕННЫХ ДИСПЕРСНЫХ СИСТЕМ.
ПОЛЯРИЗАЦИЯ ПОВЕРХНОСТИ РАЗДЕЛА,
ОБУСЛОВЛЕННАЯ ГЕТЕРОГЕННОЙ СТРУКТУРОЙ ,
Выше диэлектрическая проницаемость и удельная
электропроводность дисперсных систем рассмотрены независимо друг от друга.
Этот теоретический подход является правильным, поскольку е не
является величиной того н;е порядка, что и х/соеабс. Однако в
практических системах обе величины следует рассматривать одновременно
в виде комплексной диэлектрической проницаемости. Результаты
характеризуются диэлектрической дисперсией, обусловленной так
называемой поляризацией поверхности раздела.
Бинарная система,
обладающая сосредоточенной емкостью и сопротивлением
Общая формула. В качестве простого примера бинарной смеси
рассмотрим диэлектрические характеристики системы, показанной
на рис. V.9, которая представляет последовательное соединение двух
Рис. V.9. Последовательное соединение двух
блоков, обладающих емкостью и
электропроводностью.
блоков, обладающих емкостью и сопротивлением.В этом случае имеем
следующие соотношения для С* и G*:
С
J__ = J_ , 1
J_.=J_+-
с* с*а '
GI
(V.117)
(V.118)
После преобразований с помощью уравнений (V.57) и (V.58)
получим:
Cl — Ch i Gl
где
c* = c*+TW + 7^ (V-119)
S' = g|+/OT/+;~g,)+/«>C* (V-120)
с>=^Ык (V121)
CaGb + ClPl
C'= (S. + ft)' <V'«*>
г _г _ (CaGb — CbGa)2
' h~ (Ca^-CbMGa + Gb)2 ( 6)
Т=^г=-^х^ <v-124>
Gl = 4^TZ <VM25>
_ GgCb + GbGa ,v .oc.
C*-G'-<Ga+Cft)«7e+C6)» (V'127>
и
{Gh-Gi)i = Ci-Ch (V.128)
Частотная зависимость емкости и электропроводности (рис. V.10)
дает диэлектрическую дисперсию, характеризуемую одним временем
Рис. V.10. Частотная зависимость Рис. V.11. График в комплексной
(о = 2я/) емкости С и активной элек- плоскости зависимости С/<в от С
тропроводности G' системы из двух для системы, показанной на рис. V.9.
блоков, показанной на рис. V.9.
релаксации. График в комплексной плоскости системы (рис. V.11)
представляет круговую дугу, которая полезна для грубой проверки
отдельных релаксационных систем. Отсюда видно, что
последовательное соединение двух диэлектриков приводит к диэлектрической
дисперсии, за исключением случая, когда CaGb = CbGa.
335
Аппроксимация для частного случая. При условии
Ca<i Cb (V.129)
и
£а»Сй (V.130)
уравнения (V.121) и (V.127) упрощаются:
Ch=Ca (V.131)
Ci = Cb (V.132)
Ci-Ch = Cb (V.133)
2л/о "a
G, = G6 ' (V.135)
Gh = Ga (V.136)
Gh~Gi = Ga (V.137)»
Бинарная система, имеющая слоистую структуру
(теория Максвелла)
Общая формула. Максвелл (1892) объяснил диэлектрическую
дисперсию гетерогенных систем с помощью слоистой модели,
показанной на рис. V.12, а.
п 6
'Л
е»\
ч
■*■+•• *-
щ
iff
У/ хау/
• е*:
Рис. V.12. Слоистая модель в случае бинарной
гетерогенной системы.
G точки зрения теории импеданса, рис. V.12,a рассматривается
аналогично рис. V.12,6. Последний является эквивалентной схемой
системы, показанной на рис. V.9, со следующими изменениями
бабе
е-а
1 —ф '
с* —>
*а
1-ф
с* —
Сь-
еабсЁ
Ь'абс
У-!>
Ф
ф
(V.138)
(V-139)
336
С учетом соотношений (V.138) и (V.139) уравнения (V.117)—(V.128)
преобразуются к следующему виду:
-^- = 4-<1-Ф) + 4гФ (V.140)
^ = ^(1_Ф) + _^ф (V.141)
(V.142)
(V.143)
(V.144)
(V.145)
(V.146)
(V.147)
(V.148)
g,_E._ ^"~° чг-аг-*- \> ^ч fV.149)
' " [*Ь-гФ(£а-ЧтУ-Ь+Ф(Ха-Хь)}2 К '
1 _ еь+Ф(га-гь) ^ (улщ
„* ..* 8ь
" ~^а е; + Ф(е;
'■ -*" х* + Ф(х
,* v, I №(Y.h-Y.l)
с - Е"
Св ^ ей+Ф(еа
с еахЛ-Ф(ейха-
' [ИЬ+Ф(У.а-
„ х, . (efcxa-
-е!)
/С08абс
а-О
+ /шеабсеА
— е/,)
—eaxg)
*ь)]2
еахь) хаФ
"' "" "-a ' [хь + Ф(хв-Х6)]Я
(еахь —ейха)2
ф(1 — ф)
2 л/0 И& + Ф(*а — ИЬ)
х, = ха —-^ r (V.151)
"Хб + Ф(ха —х.,)
"Л= [е» + Ф(еп-е6)]» (УЛ52)
е/г | (ще-а — xfls&) еаФ
Хй=Ха-^-+ :ь,°^7г „°». (v.153)
х* х,- (ВаХь-еьха)»Ф(1-Ф) ,
(y.h — х/)т=(е/ — Eft) еабс (V-155)
Как видно из уравнений (V.142) и (V. 143), диэлектрическая
дисперсия, характеризуемая одним временем релаксации, возникает когда
еах^^е0иа. Уравнение (V. 146) для предельной диэлектрической
проницаемости на высоких частотах является идентичным предельной
формуле Винера (1912).
Аппроксимация для частных случаев. К эмульсиям М/В, которые
представляют практический интерес, можно применить теорию
22 Заказ 1070 337
Максвелла. Так как х водной фазы значительно выше х масла, то
уравнения (V.147), (V.151) и (V.152) можно упростить следующим образом.
а) ха > щ (эмульсия М/В).
Уравнения (V.146), (V.147), (V.151) и (V.152) сводятся к
выражениям:
£* = е« ч+о%а-ч) • (V"156)
E/ = £b-7F - (V.157)
f = i (V.158)
б) Щ 5> * а (ЭМУЛЬСИЯ В/М).
Уравнения (V.146), (V.147), (V.151) и (V.152) сводятся к
выражениям:
Е/, = ЕЦ „ , **, -у (V-160)
Eft—* (^а — tb)
"'-^т=ф- <v-16*>
Х/ 1 (V.162)
У-п ~ 1-Ф
Г Г75 гТФ (V.163)
Разбавленные дисперсные системы
сферических частиц (теория Вагнера)
Вывод основных выражений. Вагнер (1914) предложил теорию
поляризации поверхности раздела для дисперсной системы, в которой
сферические частицы равномерно распределены в дисперсной среде
(система разбавленная).
Рассмотрим в среде с комплексной диэлектрической
проницаемостью е„* некоторую сферическую область R0, содержащую N
маленьких сфер с радиусом Rc и комплексной диэлектрической
проницаемостью е* (рис. V.13, а). Подобно уравнению (V.82) потенциал,
обусловленный потенциалом маленькой сферы в точке Р, которая
находится на расстоянии г от сферы, определяется так:
UBHem = -Ez+E я.^9р. я cose (V.164)
fcp i -ътГ
Второй член уравнения (V.164) выражает влияние каждой
маленькой сферы. Поэтому потенциал, обусловленный влиянием N сфер,
338
заключенных внутри сферической области радиусом Л0, определяется
уравнением
U
N внеш"
= — Ez + E-
-г* Я*
«5 + 2^
NcosQ
(V.165)
при условии, что взаимодействием между маленькими сферами
пренебрегаем, как это предполагается для разбавленных дисперсных
систем.
^с* внеш
/9^у
Сферическая
■ область
Рис. V.13. Модель Вагнера разбавленных дисперсных систем
сферических частиц:
а — в сферической области радиусом Rc заключено iV сфер; б — сфера й0
с диэлектрической проницаемостью s* эквивалентна сферической области й0,
содержащей Л' маленьких сфер.
Если считать, что эффективная диэлектрическая проницаемость
сферической области радиусом R0 определяется е*, то потенциал
точки Р, обусловленный сферой R0 (рис. V.13, 6), будет равен:
U.
£* ввеш
= -Ez+E
8*+28j,
-^-cose
(V.166)
Вагнер предположил, что уравнение (V.166) должно быть подобно
уравнению (V.165). Объемная доля Ф маленькой сферы равна
частному от деления общего объема маленьких сфер на объем области i?0:
Ф =
Используя это соотношение, получим
N
£о Рп
Ф
е ~i~26m ep + 2em
или для комплексных удельных электропроводностей
хт
Х-п
У-п
-У-п
Л ~Т~ ZXm
,9у*
Ф
22*
(V.167)
(V.168)
(V.169)
339
Уравнение Винера (V.74) соответствует случаю только реальных
чисел в уравнении (V.168).
Общее уравнение. После преобразований с помощью выражений
(V.62) и (V.63), уравнения (V.168) и (V.169) записываются так
_ « 2г*т + е*-2Ф(г*т-г*)
г*~ет 2г*т + е*р + Ф(г*т-ер) ( <V"170)
e* = E/i + _^Z^ + ^<_ (V.t71)
2х?п^у.*-2Ф(у.*т-у.*)
2у.*т^х*^Ф(х*т-У.*р)
(V.172)
где
** = "<+ ^'ffi^ +i°**6c*h (V.173)
2еш. + ер-2Ф(8т-Е0)
^ = гт 9£ *^ ф( \ (V.174)
ет (2хш + хр)2 + [(9вр -2е,д) х»г - 8s„,xmxp + ?т*|] Ф -2&т (хт-хр)2 Ф2
[2хт + хр^Ф(х„1-Хр)]2
х, 9(ерхт — £тх.р)хтФ
£г = е„
S;—е/г
(V.
(V
(V
(V
.175)
.176)
.177)
.178)
v.m [2хт + хр+Ф(хт — ир)]2
9(етКР-ерхт)2ф(1-Ф)
= [2ет + Ер + Ф(8т-8р)] [2ит + Хр+Ф(хт-Хр)]2
_ 1 _ 2ет + 8р+Ф(ет —ер)
%~ 2я/0 "~~ 2хт. + хр+Ф(хт-хр) Еабс
2х,„+хр — 2Ф (хт—Хр)
х' = х" 2^ + хр + Ф(хн,-хр) (УЛ79)
_ х„, (28m + ep)2+[(9xp-2xm)ea,-8xmemep-f хтеР1 Ф-2хт(ет-ер)2ф2
*" [2ет^ер+Ф(8т-ер)Р
(А'.180)
8А , 9 (хрет—хтер) етФ
r.k = *m-^-+ -..,. , р ^Ф/Ё rw (V.181)
8т 12еш —£p-j- Ф (8т — fcp;j^
_ 9(хтЕр~Хр8т)2Ф(1-Ф)
Х"_>{/~[2хт-гХр-Ф (хт-хр)] [2ет-|-8р+Ф(ет-ер)]2 (Х -182)
(хА— х,)т' = (е/— еА)еабс (V.183)
Из уравнений (V.171) u (V.177) видно, что диэлектрическая
дисперсия характеризуется одним временем релаксации, когда гт у.р -^ еру.ш.
Графическое изображение уравнения (V.171) в комплексной плоскости
представляет полукруглую дугу. Уравнение (V.174) для ей на
высоких частотах является таким же, как уравнение (V.74) Винера для 8
сферических дисперсных систем, а уравнение (V.179) для v.t анало-
З'.О
гично уравнению (V.111) для х дисперсных систем, используемых
Фрике (1924).
Аппроксимация для частных случаев, а) хт > хр (эмульсии М/В).
Уравнения (V.174), (V.176), (V.179), (V.181) сведутся к следующим:
2ет + ер —2Ф (ет —ер)
ЬЛ-
е;
х/,
Хт
2бт + еР —
— cm
6/ =
= £«1
8/г
£т
-2Ф I
2ет + еР4
-е Х/' +
лт
2(1 —Ф)
2+Ф
х, 2(1
х,п 2
[2еш + е
(ет —sD)
-Ф(ет-ер)
9ерФ
(2 + Ф)2
9ерФ
' (2+Ф)2
-Ф)
+ Ф
9е„,ерФ
о+Ф(ет-ер)]2
9етерФ
У-т ." 2ет + 8р + Ф(ет-ер) [2ет + ер + Ф (sm-sp)]2
(V.184)
(V.185)
(V.186)
(V.187)
(V.188)
(V.189)
б) хр > хт (эмульсии В/М).
Уравнения (V.174), (V.176), (V.179) и (V.181) сведутся к
следующим:
2ет + ер —2Ф (ет — ер)
^ = е'" 2Ет + ер+Ф(ет + ер) ^19°>
х, 1—2Ф
х,„ "" 1-Ф
_^L = Г Зе^ I2 ф
хр L 2ет + ер + Ф (?т — ер) J
(V.192)
(V.193)
Концентрированные дисперсные системы
сферических частиц (теория Ханаи)
Вывод основных выражений. Теория Вагнера получена для
разбавленных систем. Ханаи (1960, 1961а, 1961b) разработал теорию для
концентрированных дисперсных систем на основе теории Вагнера.
Процесс получения концентрированных дисперсий
рассматривался как последовательное добавление бесконечно малого количества
дисперсной фазы AFP в дисперсионную среду Vm. На промежуточной
стадии i (рис. V. 14), когда общий объем равен V, концентрация
дисперсной фазы — Ф', а диэлектрическая проницаемость — е*,
добавление бесконечно малого количества дисперсной фазы AVp приводит
341
к следующей стадии i -\- 1. Тогда объем станет равным V + AFp,
концентрация — Ф' + АФ', а диэлектрическая проницаемость —
е* + Ае*. Ханаи предположил, что соотношение (V.168) Вагнера
справедливо для этого процесса с бесконечно малым изменением
концентрации. Другими словами, значения е„ие*в уравнении (V.168)
должны быть заменены следующим образом:
Стадия
Общий объем
Концентрация
Количество
дисперсной фазы
Диэлектрическая
проницаемость
1
*т
0
0
Cm
о* v р*
?
Vrn'Wp
АФ'
4 Up
£m*4t'
£* -->
е* -f
1
V
ф'
1/ф'
с'
- Ае*
i« I
1/*ДУ0
i .1
Ф ♦ ДФ
\,4>'*UVp
с'+&с'
(V.194)
Окончательно
*т - VB
Фкснечн
Уо
£ качечн
I
Рис. V.14. Получение концентрированной дисперсной системы в процессе
постепенного добавления дисперсной фазы.
Как видно из рис. V.14, изменение концентрации между стадиями
i -\- 1 и i составит:
УФ'+АУ
Р
ДФ' = Ф' =
V + AVp
V + AVp
(1-Ф') (V.195)
С другой стороны, величина Ф в уравнении (V.168) определяется
выражением:
AVP
ф = у + Ш-р <v-196>
Таким образом, имеем:
АФ'
Ф = ТЗоГ <V'-197)
Окончательное уравнение (V.168) приводит к виду:
2е* + е* _дф'
Ав*= . rtt, (V.198)
3e*(g*_6*) 1_ф
Эти математические преобразования проще, чем у Бруггемана
(1935), который вывел уравнение для диэлектрической проницаемости
342
концентрированных дисперсных систем, состоящих из непроводящих
материалов.
Последовательным добавлением бесконечно малых порций
дисперсной фазы [этот процесс описывается уравнением (V.198)] будет
достигнута конечная концентрация Ф и диэлектрическая
проницаемость е*.
Исходя из этого, получим основное уравнение:
, (С) г*
ф
2е* + е;
Зе* (е*—е
d£* =
1 —Ф'
= 1д(1-Ф)
(V.199)
*(е'Х)
§р (£р, £р) |
О £' €р е'т
Реальная часть
%
51
*-£
Т e*(e\z")
£/77 £р £
Реальная часть
Рис. V.15. График комплексной диэлектрической проницаемости
в комплексной плоскости:
а — эмульсии M/B; б — эмульсии В/М.
На рис. V.15 дан график е* в комплексной плоскости. Сложный
интеграл в левой части уравнения (V.199) интегрируется вдоль
некоторого контура С — от г*т до е*.
Контур С определяется соотношением между е и г" на каждой
стадии в процессе постепенного добавления. Следовательно, мы не
можем заранее иметь аналитическое выражение для контура С.
Вычисление сложного интеграла. Подынтегральная функция
в уравнении (V.199)
2е* + £р 1 1
/ (S*) = 38*(8*-е£) = ~ Зе*" + е*-е* (V.200)
имеет два полюса: е* = 0 и е* = е£. Один полюс (е* = 0) является
началом комплексной плоскости (см. рис. V.15), положение другого
полюса заранее не определено.
В эмульсиях может быть так, что х дисперсной системы окажется
выше у. масляной фазы и ниже и водной фазы. Тогда физический смысл
частично выражается следующими соотношениями:
343
для эмульсий типа М/В (см. рис. V.15, а)
*>хр>0 и s">ej>0, когда ит>ир и гщ>ер (V.201)
для эмульсий типа В/М (см. рис. V. 15, б)
ир>х>0 и 8p>s">0, когда хр>хт и ер>е^ (V.202)
Из этого вытекает, что не существует полюса функции / (е*)
определяемой уравнением (V.200), в некоторой области, которая
ограничена контуром С и прямой е^е*, так как в этих пределах
функция имеет постоянное значение. С помощью теоремы Коши
доказывается, что интеграл / (е*) вдоль контура С равен интегралу вдоль
контура г*пг*.
Точку Z* на прямой е,*„е* находят из уравнения:
2* = e,1i4-*(e*-8m) при я [0,1] (V.203)
Отсюда получают:
dZ* = (e*~eZl)dx (Y.204)
Сложный интеграл уравнения (V. 199) вычисляют следующим
образом:
| f(e*)ds* = J_ /(s*)d8*= j f(Z*)dZ*
(V.205)
(С)-,*
J /(8*)^-|[-3^+ТО|-_
dZ*
, (С) =*
(V.206)
/ (e*) As*
£* (С);*
da:
(V.207)
\ / (s*) Аз*
£* (C)E*
3 g V s*
+ lg
Em 8p
(V.208)
Таким образом, получено следующее уравнение, которое можно
рассматривать, как уравнение Бруггемана, распространяющееся
на комплексные числа:
' = 1-Ф
(V.209)
344
Каждый член уравнения (V.209) преобразуется:
e*-s£=[(e-Ep)2 + (e."-R-)2],/'exp(_/th-i
eS, -ej =[(em-Ep)a +(е--«;)»]'/• cxp/-/th-i.
С = (е*, + ЕЙ)'/' exp (-^Ь_1-Ц-)
8* = (e2-bs"2),/2 exp(—/th-i-^-Л (V.213)
Подставляя уравнения (V.210)—(V.213) в уравнение (V.209)
ii разделив реальную и мнимую части, получим:
[<е-е,).+ (е.^,Ч(е»1 + ей)'/1 =(1_ф), (у ш)
Ё-£р /
р" о" ,
sm ер /
(V.210)
(V.211)
(V.212)
[(вщ- ер)2 + (е'т- Ер)2] (е2 + в"2)'/
и
1-р" —г р" (г — eD) {г"т — е"„) — (вт — г0) (е" — в")
th-i bSm ,ЕГ! =3th-i-) ^ P f '" Pi (v.215)
6Em + eem (e —ep) (Em —6p) + (s — Ep) (em—8P)
Уравнения (V.214) и (V.215) являются общими выражениями для
определения е и е" или ■/..
Уравнение (V.215) может быть переписано в следующем
упрощенном виде
г"т _ е"
ет ё Х(3-Х2)
1 +ik.il. ^-з^2
6/77. 6
(V.216)
где
*= i^^F 8"-8" (V-217)
1 -f- '
Ет Ер Е Бр
Уравнение (V.216) можно преобразовать:
(esm— Ems") [(в — 8Р) (em —Sp) + (e" —Ер) (е" — ер)] X
X {[(Е-Ер) (Ет-Ёр) + (Е"-Ер) (Ет-8р)]2 —
— 3[(Е-Ер) (Е^-Ер)-(Е"-Ер) (Em-6p)]2j =
= (sem+8'£m) [(Е — Ер) {г"т — Ер) — (ё"— Ер) (Ет—Ер)] (3[(б — Ёр) (ет — Ер) +
+ (Ё"-8р)(Е^-Е;)]2-[(е-Ер)(Е^-8р)-(8"-ер)(8т-Ер)]2} (V-218)
Аппроксимация для частного случая, а) Предельные
значения е и х на высоких частотах.
Из уравнений (V.214) и (V.216) получим следующие соотношения,
включающие еЛ, %h, &t и v.t независимо от типа эмульсии — М/В
или В/М.
345
На высоких частотах (со ->- ©о, ет > е"т, ер > е* и е ^> е"):
-г^Ч-^У"-1-*- ^-^
fcm ьр \ hft /
/ 3 1 \ [V-m—V-p . У-р \ у.т
ХП7Г-Г--77 )=3ll Г + 1Г=Н~Т^ (v'220)
V Eft —Ep Eft У \ Em—Ep Eft—Ер / Em
На низких частотах (со -> 0, е^, > em, е", > ер, е" > е):
/3 1 \ / ят—ер Ер \ гт
е/( L)=3 ~-\ — — . (V.221)
\У.1—У.р У.1 1 \У-т—У-р У-1 — y-pl У-т '
*'-*" f*»^h=l-0 (V.222)
Ут —У.р\ У.1
Уравнение (V.219) является таким же, как уравнение (V.76)
Бруггемана для диэлектрической проницаемости сферических
дисперсий.
б) У-т л> "лр (эмульсии М/В).
Уравнения (V. 219)—(V.222) сводятся к следующим:
(V.223)
(V.224)
(V.225)
(V-226)
Если допустить, что щ > Хр, уравнения (V.225) и (V.226)
запишутся так:
2е/—Зе„ ,
--(1-Ф)'/2 (V.227)
Eft-
Em-
Xft
У-т
£, = •
У-1-
V;
"Ер V Eft
Eft (Eft"
Е/71 (E/n "
У1 [о£рУ.т
У-т
~"ЛР / У-т
,п \ У-1
.)"-..
- Ер) (2Ет
— Ф
+ Ер)
-ер) (2ей + ер)
+ (2ет-
(2хг-|-хР
\7.
ЗЕр) Щ\
)
-_Ф
2Ет-ЗгР
у-1
(1-Ф)"'2 (V.228)
У.т
в) у.р > хт (эмульсии В/М).
Уравнения (V.219)—(V.222) примут вид:
SP &h t Em v _ , ф (V.229)
_Xft __ 3Eft (Eft — Em)
Xp (Ep-b26ft) (8p —Em)
(V.230)
щ Ып—yi)
Xm(xp + 2x,)
Xp V X( /
(V.232)
346
Когда хр > Я[, уравнения (V.231) и (V.232) можно записать таким
образом:
*'=*>* (1-ф)3 (V-233)
^t-ji^w (v-234>
Оценка величины диэлектрической дисперсии, обусловленной
поляризацией поверхности раздела. Для эмульсий неполярного
масла в воде гр является величиной бесконечно малой по сравнению
с ет. Уравнение (V.184) и (V.186) из теории Вагнера в данном случае
упростятся
8ft ~ гт 2 (2j ~^) «* ы (V. 235)
и уравнения (V.223) и (V.227) из теории Ханаи сведутся к виду
ел«=<ет(1-Ф),/2^е/ (V.236)
Числовая оценка этих соотношений подтверждает их
приблизительное равенство. Теории Ханаи и Вагнера предсказывают, что
величина диэлектрической дисперсии слишком мала, и не может быть
обнаружена в эмульсиях М/В.
Наоборот, можно предположить, что диэлектрическая дисперсия,
обусловленная поляризацией поверхности раздела, может
наблюдаться даже в эмульсиях М/В при условии, что масло будет иметь
высокую диэлектрическую проницаемость (например, нитробензол).
Это предположение подтверждается экспериментально (см. стр. 360).
Нужно отметить, что выражения для к или е, вывод которых дан
выше (см. стр. 319), включены в теорию поляризации
поверхности раздела, рассматриваемую в данной главе. Следовательно, для
полного описания этих величин необходимо их одновременно
измерить.
Тройные системы,
обладающие сосредоточенной емкостью и сопротивлением.
Упрощенная модель тройной системы (как и бинарной) может быть
представлена в виде цепи с последовательным соединением блоков,
обладающих сосредоточенной емкостью и сопротивлением (рис. V.16).
Комплексная емкость С* всей системы определяется как
(V.237)
Последующие вычисления и преобразования формулы не сложны,
но довольно громоздки. Ханаи, Хейдон и Тейлор (1965) сделали эти
вычисления, чтобы вывести аналитическую формулу, показывающую
частотную зависимость комплексной емкости.
347
Переписав уравнение (V.237) и сделав подстановку уравнений
(V.57) п (V.58), получим:
с*=-
С* p-k р-к
а^ ь^ с
с*
с*ас*ь+с*с*с + с*с*а
(.Ga + mCa) {Gb + i<uCb) (Gc + mCc)
с:* =
i
;со '1С~а^}шСа) (Gb + тСь) + (Gb~mCb)(Gc + juC~cy
-r(Gc + iaCc)(Ga^ja>Ca)
Ob
A
,Cb
Pnc. V.16. Последовательное соединение трех блоков,
обладающих емкостью и электропроводностью.
(V.238)
(V.239)
(V.2',0)
Знаменатель уравнения (V.240) представляет квадратичное
уравнение и приводится к следующему виду методом разложения на
множители:
г*_ 1 (Ga + i^Ca) (Gb + mCb) (Gc + jaCc)
(V.241)
/со (1 -г /штр) (l-j-/'coTQ)
Методом частных дробей уравнение (V.241) можно преобразовать:
Ci-Ci . С{-Ск , 1
где
3'i8
c* = ch +
Gt
С i-С г-
Ci =
1
1—/штр 1-р/шТд ' jco
Г Г, Г
Ch= —
С a {GbGc)i f Cfc (g,Ga)^ + Cc (GaGfc)2
(V.242)
(V.243)
(V.244)
C,- — Ch —
1
^(ТР-Т<?)
1
■ £ -г ^р + — CaCftC, - %l GaGbGc (V -245)
Tp
£ — FTq — CaCbCc -j- t-qGcPjjGc I (V.246)
Л£»2
[CQGS(C6Gc-CcGu)4-C^(C,<?a-CaGc)2--
-|-CcG?(CaG6-CftGa)2]
(V.247)
Gr-
1
GaGbGc
D
1
ЧГ
ВА-УB^—AAD
2D
2A
2nfQ ~ B+VBi — bAD
2n/p
1
(всегда тр > rQ, т. е. cop < aQ и /р < /Q)
Л = CaCft -f CftCc + CcCa
■ B = Ca(Gb^Gc) + Cb(Gc + Ga) + Cc(Ga + Gb)
D = GaGb + GbGc-yGcGa
E = CaCj,Gc -f- CbCcGa -\- CcCaGb
F = CaGbGc -f- CbGcGa 4- CcGaGh
Сi
XL
■ Cn
Tp
Рис. V.17. Частотная зависимость (со = 2л/) емкости
для системы, состоящей из трех сосредоточенных им-
пендансов (показана на рис. V.16).
(V.248)
(V.249)
(V-250)
(V.251)
(V-2521
(V.253)
(V.254)
(V.255)
B2-iAD--(CaGb-CbGcyi-<-(CbGc-CcGb)2+(CcGa-CaGc)2-
-2 (CaGb - CbGa) (CbGc - СcGb) - 2 (C,GC - CcGb) (CcGa -CaGc) -
~2(CcGa-CaGc)(CaGb-CbGa) (V.256)
Из уравнения (V.242) видно, что система, содержащая три
сосредоточенных импеданса, имеет две дисперсные области,
характеризующиеся двумя отдельными временами релаксации тр и Тд(рис. V.17),
за исключением случая, когда
Са
Gn
Сь_
Gb
Тройная система, имеющая слоистую структуру
Тройная система, имеющая слоистую структуру (рис. V.18, а),
может рассматриваться как эквивалентная системе, состоящей из
последовательно соединенных конденсаторов (рис. V.18, б).
349
Если в уравнениях (V.242) —(V.255) произвести следующую замену
^а ~~*" ^а
Ф„
Сь -* f'a6c
ч
Фн
С£-еавс-^-, С*-еа6с8*
Ф
У-с
Фг
(V.257)
(V.258)
////} ■ ■ •' -"^
U
Фг
\щ
УЛа/
/'■'. ,
' ■ ■ //
'*а/,
^12.
Фа
■* и»
:£ь:.
:Хь[
4>ь
■* ^
1
'€с
'//У/
уу
'Н
Ш
фс
Рис. V.18. Слоистая модель тройной гетерогенной системы.
получим следующие уравнения, выраженные через е и у.
£;—£,- i £, —Eh | 1_
= ffc-
Ea Eft
1 -i- /штг
ДОЕабс
-у.г
1
£h~-
Фа Фь Фс
(V.259)
(V-260)
е; =
Г Efl / *Ь У-с у, Ч (Л^.^о\2 + ±-[^-.^-\2Л-7Ц-
Ы— Ej
^1(ТР-Т«)
, 2 1 Ед Eft Ее
-Ea6c£l-rflTpT"Ea6c т " фа " фь фс
£j—Ей =
^Ср-^)
1 2 Я2 ЯЬ *с
Еабс ХР Фа' Фь' Фс
Еабс£1 — FlxQ 8абс т
' Фа " Фб " Фс
1 , У-.
_>^_ _*с_
Еабс '° Фа ' ФЬ " ФС
Уа J^b_ J_L_ _1_
Х/_ Фа" ФЬ ' Фс Dl
TiJ = 2Д1 абС
2^1
Si-1 fiJ-^iZ)!
еабс
(V.261)
(V.262)
(V.263)
(V.264)
(V.265)
(V.2G6)
Аг =
Ф„
Фь
ч
Еа
+ Фй ' Фс ^ Фс ' Фа
' Фа
~ф7ч"фГ + "ф7,/'",~ фДфЛ фй /"'" фД ф« ^ фJ
Д1 =
Ф„
Фь "^ Фь
"фТ
+ ■
фе
Яа
Ф„
Е-,
Fi =
(V.267)
(V-268)
(V.269)
(V.270)
(V.271)
Рис. V.19. Частотная зависимость (ев = 2л/)
диэлектрической проницаемости тройной системы,
имеющей слоистую структуру (приведена на
рис. V.18).
Частотная зависимость диэлектрической проницаемости
выражается уравнением (V.259) и показана на рис. V.19.
Разбавленные дисперсные системы сферических частиц,
покрытых оболочками (теория Паули и Швана)
Первые попытки дать математический анализ диэлектрического
состояния дисперсных систем сферических частиц, покрытых
оболочками, сделаны Максвеллом. С тех нор
этот вопрос занимает важное место
в изучении диэлектрических свойств
суспензий биологических клеток и
других систем.
Недавно Паули и Шван (1959)
продолжили сложные вычисления
диэлектрической проницаемости системы на
базе уравнения, предложенного
Максвеллом.
Рпс. V.20. Электрическое поле системы,
состоящей из непрерывной фазы и сферы
с оболочкой.
351
Далее рассмотрены общие теории поляризации поверхности
раздела сферических частиц суспензий с оболочкой и применение этих
теорий.
Вывод основного уравнения. Рассмотрим электрическое поле
в случае, когда сфера радиусом Rc с диэлектрической проницаемостью
ер, имеющая оболочку толщиной d и диэлектрическую
проницаемость е*, помещена в дисперсную среду с диэлектрической
проницаемостью еД и находится в однородном электрическом поле Е
(рис. V.20).
В соответствии с потенциальной теорией электрического поля,
потенциал С/анут внутри сферы, потенциал оболочки Uo6 и потенциал
^внеш в дисперсной среде можно найти таким образом:
— 9е,„ es
U™rr= (2em+81)(2es-bep) + 2(ein-8,)(8s-eF)Ul Er C°S 9 (V'272)
_ — 3em(2?s — sp)
06~~ (2sm + ss) (2ss-j-ep)-2(em-es) (es-ep) vx X
X Er cos 9 -f -ъ i—Г7^—i r-nr, Г7 ; £/?2-^-cos8 (V.273)
(2em-)-es)(28s4-ep) + 2(Em —es)(bs — 8P) v1 cr* v '
и
(8m —es) (2es-{-ep)-r(em-r2es)(8s—ep) У!
Ьтеш=-Ег соя 0- (2em + es)(2e^-ep) + 2(8m-e,)(es-8p)L-1 X
X£№cf d)3-Lcos6 (V.274)
где
С другой стороны, рассмотрим систему, в которой частицы
радиусом Rc с оболочкой толщиной d диспергированы в некоторой
сферической области радиусом R0, являющейся дисперсной средой с
диэлектрической проницаемостью е^.
Так же как в теории Вагнера, потенциал UNBueui, обусловленный
наличием N частиц с оболочками, определяется уравнением
_ (sm —es) (2es4^8p) + (em + 2es) (es —ер) rj
L v внеш = ~ Er C°S G (2e,n + es) (28s + e.p) + 2(em-ss) (e,-ep) v X
X£7V(/?c + d)3-i-cos9 (V.276)
с учетом уравнения (V.275).
На рис. V.21, б изображена сфера радиусом R0 с диэлектрической
проницаемостью е*, которая является эквивалентной сферической
области, показанной на рис. 21, а. Потенциал вне сферы R0 может
быть записан как
^е* ВНеш = -Er cos 6- Е ^-^ Л» ± cos 9 (V.277)
352
Приравняв уравнения (V.276) и (V.277), получим окончательное
выражение для е*
где
Ет —е _ (em — es)(2es + ep)4-(6m + 2es)(es — ър) vx
2ет + е — (2em+es) (2es + ер) + 2 (sm — es) (es — ep) Vl
-<
Rc + d V
Ra
N
(V.278)
(V.279)
Таким образом, выражение для е* можно записать так:
(l + 2v2) га[(1-}-2у1) ер + 2(1-У!) es]+2(l~v2) em[(l-Vl) ep + (2+v1)es]
~~S,n (l-^)es[fl + 2i'i)Bp+2(l-i'i)8s] + (2 + I;2)Fm[(l-!;1)ep + (2 + yi)8s]
(V.280)
6
n, внеш
C.BHflL'
Сферическая
область
Рис. V.21. Разбавленная дисперсия сферических частиц:
а — п сферической области радиусом Я0 заключено N сфер с оболочкой,
толщина которой d\ 6 — сфера радиусом Но с диэлектрической
проницаемостью е* эквивалентна сферической области Д0 с тем же радиусом,
содержащей Л' маленьких сфер.
Применение уравнения (V.280) к комплексным величинам (со
звездочками) и преобразования с помощью уравнений (V.62) и (V.65)
дает
Е* = х* = А2 + тЕ2+(т)^В2
е*т у*т C2 + iu>F2+{ju>)*D2
„» _ КтА2 + /ю (Xm-g2 + ea6cSm^2) + (/to)2 (И/пД2 + еабс6т#2) + (/т)3 еабсет.В2
(V.281)
где
АоЕабсСа [1 +;ш (^а/Са) +{Щ2 {D2JC2)\
Л2=(1 + 2у2)х8я2 + 2 (1 — Уг) *тй2
■82 = [(1 +2у2) esc2 + 2 (1 - v2) г,„ d2] е*бс
Ci. = (1 — v2) х,а2 + (2 + р2) и„,62
D2 = [(l—v2) ssc2 + (2 + р2) e;n d2] е|бс
£,2=[(l+2y2)(ssa2 + >«sc2) + 2(l — y2) (em&2 + xmd2)] еабс
•in Заказ 1070
(V.282)
(V.283)
(V.284)
(V.285)
(V.286)
(V.287)
353
^2= [(1 —г-г) (esa.2-j-xsc2)-L(2 + i;2) (emb2 + xm <22)] еабс (V.288)
a2 = (l + 2yi)xp + 2(l-i'i)xs (V.289)
b-2 = (1 — »i) Xp 4- (2 - i-!) xs (V.290)
c2.--=(l 4.2i-i) ep + 2 (l-^i) es '(V.291)
d2 = (1 — i-'i) ep-r (2+ i-j) es (V.292)
Вывод общих уравнений. Так как в знаменателе уравнения (V.282)
выражение в скобках имеет квадратичную форму относительно /со,
уравнение можно переписать
у.тА-2 --/со (у.тЕ-г — гябсЪтА*) -f- (/(•>)2 (х,„Я2 -МабсЕт^г) -1-
. 4- (/(о)3 еабсе/и^5
(V.293)
~~ /С08абсС2(1+/(0Тр) (1— /COTQ)
Величины Тр и Тд определяются как
tp + tQ = F2/C2 (V.294)
rpXXQ = Di/C2 (V.295)
и приводят к следующим выражениям:
(V.296)
(V.297)
- 1L. 2iV2
(всегда тР > xQ, т. е. соР < coQ и /> < fQ).
Методом частных дробей уравнение (V.293) можно преобразовать
е« = еЛ4- ,6(Т£' + ."Т6* +—— щ (V.298)
или
/ШТр (х,- — щ) /ШТп (хЛ — X,-)
** = *' + l^/опр + l^xQ ^^ <V"299>
ХР
Ц
1
СОр
1 __
1
2л/р
1
2 л/Q
/•■2 V
^2+1
Fl-
2Сг
2D.,
' F* -
Г 2
ACiD-i
- 4С202
где
В2
^h = ^m~jy (V..300)
e'~e' = F г /т ГТт- (^^я-^тр + 50 (V.301)
Еабс£т ХтТл
Ьабс^2 (тр~tq) ^q \ ч ч /
^ 2 5-2 \ Я„ / A2F2 Е2
! А2 В2 \ хш / A2F2 Е2 \
(«абсбт —XmTQ) (£2 — Л2Тд) —
£Г
еабс С2(хр — Тд)
354
Е/ — Е„
С-г бабс
A2F2 E2
С\ С2
(еабс-^—х„Л52
2С2 —(A2F2-C2E2)
еабс
(х/ — х/)тр=(8; — Sj)ea6c
(У-h—Kl) TQ = (<=i — 6ft) еабс
#2^2
«1
х< =
С 2 (Тр— Тд)
/ В2 А2 \ (
y-h — Kl = Km I "д — )— 8абс?т (
/ gjjf 2 £2 \
£2_\
«2 /
е!,бс-~—Хт') (^~- Е-А+(£абс£т -HmTQ) Л2
В2
(V.304)
(V.305)
(V.306)
(V.307)
(V.308)
(V.309)
(V.310)
(V.311)
н
си
-fc»
со
со
•
Xi
(€1-ен)л-—Т-
р 2л; ?лтР
J ' Ч—*-
f„=
tOQ 7
в 2тг глт.
1дЛ
Рис. V.22. Частотная зависимость диэлектрической
проницаемости е и удельной электропроводности х
суспензии сферических частиц с оболочками.
Из этих выражений видно, что дисперсная система сферических
частиц с оболочками имеет две области диэлектрической дисиерсии,
которые характеризуются двумя временами релаксации: тР — на
высоких частотах и xQ — на низких частотах, как показано на
рис. V.22, на котором раскрывается смысл применяемых символов.
Из уравнений (V.283) — (V.292) получают функциональную
зависимость диэлектрической проницаемости и электропроводности.
Например, Сигиура, Кога и Акабори (1964) рассчитали е; как
&1 = ~С*~ [^ет + Эри^ (apEp + as8s)]
(V.312)
2i*
355
Здесь параметры а„„ ар и as соответственно равны:
am=2(2 + i-j)(l-r4)(l-i.-i)!1xJxS, + 4(2 + i-i)(l-«2)(2 + i-i)(l-i.-i)x.
X xsJV<m-M (1 -i'.2)2 (1 + 2«i) (1 -i'i) я5к|хт + 2 (2 + i--2) (1 -у2) X
X(2-l'1)2xsl<21 + 4(l-i'2)2(4 + ^ + 4il)xlXpKm+(l + 2(.-.2)(l-i-.2)x
X (1 -f- 2i>i)2 x|xp + 8 (1 - i>2)2 (2 + vi) (1 - li) xfx„, -f 4 (1 + 2v2) (d - y2) x
X (i-vt) (1 + 2i>i) x»xp + 4 (1 + 2у2) (1-i-s) (1 -i'i)2 x| (V.313)
«p = 9vix| (V.314).
as = (l+2i'i)(l-i-i)xJ + 4(l-wi)2t,Kp + 2(2 + i;i)(l-i-i)K! (V.315)
В результате уравнения (V.313) и (V.315) незначительно
отличаются от данных Паули и Швана (1959).
Аппроксимация для частного случая с низкой
электропроводностью оболочек. Суспензия сфер с оболочками интересна, как
упрощенная модель различных систем, таких как суспензия
биологических клеток или частиц, обладающих поверхностной удельной
электропроводностью. Необходимые приближения сложных
уравнений, выведенных выше, можно продолжить в соответствии с
истинными свойствами каждой системы.
Рассмотрим случай, когда xs оболочек бесконечно мала по
сравнению с у вне и внутри частиц, т. е.
xj«xm, xs«xp (V.316)
а толщина пленки является бесконечно малой по сравнению с
радиусом частиц
d<ZRc (V.317)
Это условие справедливо для суспензий некоторых биологических
клеток (см. стр. 382).
Для практического применения можно вывести приближенное
уравнение при условиях (V.316) и (V.317) без какого-либо
ограничения относительно v2; с тем чтобы включить результаты Паули
и Швана (1959), которые были получены при условии v2 <C 1.
Если предположить, что
x=d/Rc (V.318)
неравенство (V.317) можно записать так:
i< 1 (V.319)
Тогда получим приближенную формулу для уравнения (V.275):
i>!««l— Ъх (V.320)
При условиях (V.316), (V.319) и (V.320) уравнения (V.283)-(V.292)
могут быть приведены к следующему упрощенному виду:
А3 = 3 [ [(1 + 2v2) xp + 2 (1 -i>2) xm] >с3 + 2 (1 - v2) xpxmz } (V.321)
Д3 = е!бс [(1 + 2f2) ep + 2 (i-v2) em) es (V.322)
Сз=3 [ l(i-v2)xp + (2+vi)xm\ys + (2 + vi)Hpxmx} (V.323)
356
Da=3el6c[(l-vi)?.p + (2 + v2)em]et (V.324)
Ea=3ea6c[(l->r2i-2)y.p-{-2(i-v2)y.m]£s (V.325)
^3=36360 [(1- i-z)«p+(2 + i'2)xm]es (V.326)
a3 = 3x„ (V.327)
b3 = 3(xs + Xp*) (V.328)
c3 = 3ep (V.329)
^3 = 3es (V.330)
Удельная емкость Cs (в ф/см2) и активная электропроводность
Gs (в ом~*/см2) для оболочек толщиной d (в см) определяется
аналогично уравнениям (V.8) и (V. 16), которые характеризуют конденсатор
с параллельными пластинами:
Cs = ^ea6c (V.331)
Gs=~ (V.332)
Объединив эти уравнения с уравнением (V.318), получим
£^ = ЛСС, ' (V.333)
и
J = ficCs (V.334)
С помощью уравнений (V.321) —(V.330) и (V.333), (V.334) при
условиях (V.316) и (V.319) без каких-либо ограничений относительно
и.2 из уравнений (V.296) —(V.311) после нескольких громоздких
вычислений можно получить следующие уравнения:
р ^р (l4-2f2)eP4-2(l-i;2)em
E/I Pm "71—^i7— гр-и» 1 p (v.335)
(1 — v2) ep -r (Z -f- y2J em
= Ет(2у.т + *р)2— [(9ер — 2ет)х^-8вотхтир + ирЕт] i;2 — 2em (xm —xp)2 к»
' [(l-i'!)XpT(2t4)g"
(V.336)
. _fu = 9(Ep>tm-E,nxp)a^(-l-^) ,
' [(l-"i)ep+(2 + ^)em][(i-B2)4p + (2 + i'2)xm]« (V"W/')
T«- 2n/Q-F3 ^ (l-r2)xp+(2+,2)xm Ea6c (V'338>
(1 + 2^)^ + 2(1-^) xm
*•-*» (1-,2)хр + (2 + ,2)хт <V'339>
_«m (2«w+ ep)a+ [(йкр — 2ит) s%, -8xmsmsp +xmsp] u2 — 2xm (ew-ep)2 pjj
Л [(l-»g)ep + (2+Bj)el„]»
(V.340)
X/k_x.= 9(xmep-xpem)ai>a(l-»>i)
* ' [(l-«'j)Xp+(2 + i;i!)xm][(l-i'2)ep + (2 + i;2)em]i lv"*">
357
•р-
1 , l-f-2 _ 1
?з ,, г *р 2тг-2 ' х„
2л/р 6'
3
"="<А ". , ' : ;т , . (V.342)
Vv-p ' 2 -г- у2 хт У »
L v *р ' 2+^2 хт л
1 — BCGS
«,„«„,111^11 V'°, 2|'-:-1 ,"■;- (V.344)
8'""-ii^w-^ ■ ' •-■ - ^ <"45>
v 9u2xpx,
(2 + u2)[(l—у2)хр + (2 + У2)'<т] i ' jig ( 1 1 l~Vi -L
(V.346)
_v = 9i'-2 [(2 + !■->) r^xp -~ (1 - p) epx,„] /
ИЛ *' (2 + ^2)[(1-^)8р^(2-И'2)еш]2х
1л_л с (1 —'-'a) (gmxp —gPxm)2
w c ' хтхр[(2Ч-"2)^хрП-(1 — i>-2)gpxm]
x —-j j-— — (v.d^/;
e^
p 2 + 1--2 X„
9v2ncCs
Fa6c(2 + l>2)2
In-2^1 |~2 + 1'2 _ 1 — t-'2 1
Одинаковые функциональные зависимости выражений,
полученных для е, — еЛ и е, — е;, являются результатом допущенных
приближений и предположения, что е,- — еЛ — величина бесконечно
малая по сравнению с е( — е,-.
Некоторые свойства систем с низкой электропроводностью
оболочек. Из полученных выше приближенных результатов можно
сделать некоторые выводы, представляющие интерес для
диэлектрической дисперсии с двумя отдельными временами релаксации — хР
и xQ.
Диэлектрическая дисперсия с временем
релаксации xq (высокие частоты). Можно легко
увидеть, что функциональная зависимость уравнений (V.335)—(V.341)
хорошо согласуется с уравнениями (V. 174) —(V. 183) теории Вагнера
для простых сферических дисперсных систем. Это означает, что ди-
358
электрическая дисперсия, возникающая на высоких частотах, с
временем релаксации Xq характеризуется диэлектрической дисперсией,
являющейся результатом сферической дисперсии суспендированных
частиц (ер, хр) без оболочек в дисперсионной среде (ет, хт).
Таким образом, величина диэлектрической дисперсии считается
малой для случая, когда непрерывная среда имеет высокую
диэлектрическую проницаемость (подробно см. стр. 347). Следовательно,
диэлектрическая дисперсия, характеризуемая временем релаксации
Тд, может перекрываться диэлектрической дисперсией с хР.
Диэлектрическая дисперсия с временем
релаксации хР (низкие частоты). Диэлектрическая
дисперсия с хР характеризуется уравнениями (V.342)—(V.348) как
совершенно независимая от ет и ер. В этом случае существование
оболочек играет важную роль в диэлектрической дисперсии, которая
иногда может быть большой величиной для тонких оболочек. Поэтому
соответствующий анализ данных диэлектрической дисперсий дает
возможность получить подробную информацию о Cs, Gs, как и о хр,
которые не измеряли непосредственно. (Пример приведен на стр. 382)
Несферические дисперсии
и общие случаи смесей
Математические теории дисперсий несферических частиц очень
сложны и выходят за пределы данной работы. Их выводы не всегда
удобно применять для объяснения экспериментальных результатов,
так что здесь даются только ссылки на соответствующую литературу.
Несферические дисперсии. Дисперсные системы эллипсоидных
и сфероидальных частиц рассмотрены во многих работах, так как их
форма хорошо определена. Перрин (1934) исследовал процесс
диэлектрической релаксации. Польдер и Ван Сантен (1946) описали
эллипсоидные дисперсии, которые впоследствии исследовались Альт-
шуллером (1954).
Математическая теория удельной электропроводности или
емкости эллипсоидных суспензий разработана Фрике (1924, 1925а,
1925b). Принимая во внимание обе величины — е и х, Фрике
рассмотрел диэлектрическую релаксацию эллипсоидных суспензий (1953)
и слоистых частиц сферической, эллипсоидной и цилиндрической
формы (1953b, 1955).
Мендел (1961) дал теорию поляризации поверхности раздела для
суспензий сфероидов.
Диэлектрическое поведение дисперсных систем, состоящих из
сфероидальных капель, искаженных приложенным электрическим
полем, теоретически рассмотрели Течер (1952) О'Конски и Течер
(1953) и Гопал (1958) и экспериментально О'Конски и Гунтер (1955).
Недавно Шварц, Санто, Шван (1965) рассмотрели теоретически
ориентацию частиц под действием внешнего электрического поля.
После ранних работ Релея (1892) и Максвелла (1892) многие
исследователи пытались вывести теоретически формулу диэлектрической
359
проницаемости гетерогенных смесей, состоящих из диспергированных
частиц различной формы. Уравнения, полученные Винером (1912),
Рунге (1925) и Бруггеманом (1935), до сих пор являются полезными
для вычисления диэлектрической проницаемости дисперсий
сферических, сфероидных, цилиндрических, иглоподобных и пластино-
подобных частиц.
Общие случаи смесей. Общая оценка уравнений, полученных
для диэлектрической проницаемости дисперсных систем, дается
в статьях Нисель (1952), (1953), Брауна (1955), де Лур (1956) и Рей-
нольдса и Хок (1957). Иран (1956) предложил теорию для комплексной
диэлектрической проницаемости бинарных смесей.
Хигучи (1958) применил новое уравнение для бинарных смесей
к экспериментальным данным в суспензиях.
ЭКСПЕРИМЕНТАЛЬНЫЕ ИССЛЕДОВАНИЯ
ДИЭЛЕКТРИЧЕСКОЙ ПРОНИЦАЕМОСТИ
И ЭЛЕКТРОПРОВОДНОСТИ ЭМУЛЬСИЙ.
ПОДТВЕРЖДЕНИЕ ПОЛЯРИЗАЦИИ
ПОВЕРХНОСТИ РАЗДЕЛА
Поляризация поверхности раздела и результирующая
диэлектрическая дисперсия обнаруживаются в гетерогенных системах, таких
как эмульсии, суспензии и в некоторых изоляционных материалах
мозаичной структуры. Ниже приведено несколько примеров
диэлектрической дисперсии, связанной с видом гетерогенной структуры.
Пленки углеводородов в водной фазе;
пример бинарной системы,
обладающей сосредоточенной емкостью и сопротивлением
С целью моделирования мембран некоторых биологических клеток
с упрощенным составом Ханаи, Хейдон и Тейлор (1964) пробовали
создать бимолекулярную пленку яичного лецитина в водном растворе.
Их прибор показан на рис. V.23.
Тонкая пленка лецитина образуется на
отверстии диаметром 1,4лш с одной из
сторон тефлонового стакана,
опущенного в 0,1 н. раствор NaCl.
Результирующая комплексная
емкость измеряется с помощью
платиновых электродов, соединенных с
мостом переменного тока. Частотная
к мосту переменного тока
Рис. V.23. Прибор для получения лецитино-
вых пленок и измерения их электрических
свойств:
1 — платиновые электроды; 2 — тонкая пленна;
3 — 0,1 н. раствор NaCl; 4 — тефлоновый стакан.
3G0
зависимость емкости С, активной электропроводности и
мнимой части С" = G/co комплексной емкости и их графики в
комплексной плоскости показаны на рис. V.24 и V.25.
4500
3000
%
1500
С^ШОпф
10
10
10^
Г,гц
Рис. V.24. Частотная зависимость емкости С,
электропроводности G и мнимой части С' = G/ш комплексной емкости
(Ханаи, Хейцон и Тейлор, 1964).
На графике С; = 4040 пф и Ga = 1,015 Мом~1 приводят к /„ =
= Ga/2nC, = 40,0 кгц, которую можно сравнить с величиной /о =
= 36 кгц, полученной по максимальной точке С".
3000
Сг^ОМпф
Рис. V.25. График комплексной емкости в
комплексной плоскости системы, состоящей из леци-
тиновой пленки и 0,1 н. раствора NaCl (Ханаи,
Хейдон и Тейлор, 1964).
Данные те же, что на рис. V.24.
Воспользуемся обобщенной моделью, изображенной на рис. V.26.
Кроме того, так как для приведенной системы допускаются
соотношения
Са (~ 0,7 пф) <г Сь (~ 4000 пф)
Ga (~ Ю-з 0M-i) - Gb (—10-ю 0M-i)
361
то для анализа результатов могут быть использованы упрощенные
уравнения (V. 131)— (V. 137). Учитывая полученные величины
площади пленки (S = 1,05 ммг) и емкости {Сь = Сt «* 4040 пф), емкость
пленки на единицу площади вычисляют как Cb/S = Ct/S =
= 0,385 мкф/смг. Предполагается, что данная пленка является би-
а
0,1Н NaCl
Пленка
Ga
Gb
.Са
Чг
Сь
Рис. V.26. Схема последовательного
соединения растьоиа ГчаС1 и лецптпно-
вой пленки (а) п эквивалентная
электрическая цепь для этой схемы (б).
молекулярным слоем лецитина
с полярными группами,
обращенными в сторону водной
фазы, и что емкость
представляет конденсатор с
параллельными пластинами, состоящими
из гидрокарбонатных групп,
диэлектрическая проницаемость
которых оценивается в 2,05.
Из уравнения (V.8) следует,
что толщина пленки d = '"У =
ь
= 47,2 Л. Эти результаты
хорошо согласуются с данными,
полученными для расстояния,
которое соответствует двойной
длине цепочки с 18 атомами
углерода молекулы
лецитина — 46 А.
Нужно отметить, что
макроскопическая теория
поляризации поверхности раздела все еще является справедливой даже
для таких маленьких величин, как несколько десятков ангстрем.
Диэлектрическая проницаемость эмульсий типа М/В
Диэлектрическая проницаемость эмульсий типа М/В плохо
изучена из-за их значительных проводящих свойств по сравнению
с эмульсиями В/М. Систематические данные для количественного
анализа сообщены Ханаи, Копцумн и Гото (1959). Они приготовили
эмульсии нуйол/четыреххлористый углерод в воде с
эмульгирующими агентами твин-20 и спен-20 н измеряли е в широком диапазоне
концентраций (0 —85?-о) и частот (от 20 гц до 5 Мгц).
Рис. V.27 иллюстрирует зависимость е и у. 70%-ных эмульсий
М/В от частоты.
На низких частотах е быстро растет с уменьшением частоты
вследствие поляризации электродов, тогда как у. остается постоянной
независимо от частоты. В этом диапазоне частот диэлектрическая
дисперсия, обусловленная поляризацией поверхности раздела, не
обнаружена.
Предельное значение диэлектрической проницаемости на частоте
более 5 кгц считается истинной диэлектрической проницаемостью
эмульсий. Зависимость ее от концентрации Ф масла графически пока-
362
зана на рис. V.28 вместе с теоретическими кривыми, полученными
из уравнений (V.73), (V.74), (V.76), (V.80) и (V.107). Уравнение Бруг-
гемана лучше всех соответствует экспериментальным результатам
•во всем ряду концентраций.
Рис. V.27. Частотная
зависимость диэлектрической
проницаемости (а) и
электропроводности (б)
эмульсии М/В (Ханаи, Коицуми
и Гото, 1962).
Объемная концентрация 70%;
30° С.
sot
Рис. V.28. Зависимость
диэлектрической проницаемости от
концентрации дисперсной фазы
эмульсии М/В при 309 С
(Ханаи, Коицуми, Гото, 1959):
1 — уравнение (V.107); г —
уравнение (V.74); 3 — уравнение (V.76);
4 — уравнение (V.80); S —
уравнение (V.73).
П.'р и мечание. Здесь и далее
точки означают
экспериментальные данные, кривые —
теоретические.
Электропроводность эмульсий типа М/В
Имеется несколько сообщений с анализом экспериментальных
данных удельной электропроводности эмульсий М/В.
Результаты для модельных систем с упорядоченным
расположением частиц. Пирс (1955) попытался дать экспериментальное
подтверждение теоретической формулы электропроводности. Для этой
цели он использовал модельную систему, в которой непроводящие
363
1,0 0,8 0.6 QM 0,2 О
1,0
0,8
0,6
ом
0,2
0
-
-
-
>^
г
\У/
/У^2
i i i i , i , i
1,0
0.8
0.6
Of>
о.г
0
- 1
УМ
-
3
6 /
/
2
t . I . L
ОМ 0,2
Ф
1,0 0,8 0,6 ОМ 02 О
Ф
1.0Т
1.0 0,8 0.6 ОМ 0,2 О
бакелитовые полусферы диаметром —25 мм, правильно
расположенные в узлах кубической решетки, были опущены в воду. Эта система
аналогична эмульсии М/В с упорядоченно расположенными
сферическими частицами, что соответствует предположению, принятому при
выводе уравнений (V.73), (V.74) и (V.111).
На рис. V.29, а показана зависимость х/хт — Ф, полученная
Пирсом на частоте 400 гц для концентраций вплоть до 45%, и
приведены теоретические кривые. Данные Пирса (точки на рисунке)
лучше согласуются с уравнением (V.112), чем с уравнением (V.115).
Отсюда можно сделать вывод, что уравнение (V.112) справедливо
для систем с правильно упорядоченными частицами. Пирс не провел
экспериментов для исследования электропроводности произвольных
систем на модельных средах.
Мередит и Тобиас (1960) измеряли электропроводность модельных
систем небольших элементарных ячеек кубической решетки из
непроводящих луцитовых сфер, опущенных в воду. Результаты
исследований представлены на рис. V.29, б вместе с теоретическими кривыми.
Как видно, экспериментальные значения при концентрации 51%
не согласуются с уравнением (V.112). Авторы считали, что такое
расхождение может быть вызвано пренебрежением высшими членами
ряда, опущенными при вычислении Релеем потенциала
электрического ноля. Они вывели модифицированное уравнение Релея:
Ко У-т ^ ю < ,
ЗФ-1,227 —, Ф >*
Ир i q У-т
ф_0,409 ———, Ф/з-0,906 »___?_ ф'°/.
_4
3 "'" "" ' 3
хт хр + ~о" К/п
(V.349)
Соответствие уравнения (V.349) с экспериментальными данными
достаточно удовлетворительно, как видно из рис. V.29, б.
Тщательный анализ теоретических величин подтверждает, что
модифицированное уравнение очень близко к уравнению (V.115)
при концентрациях < 60%. Отсюда следует, что значения,
наблюдаемые авторами, хорошо согласуются с уравнением (V.115), которое
выведено для произвольных дисперсных систем с высокой
концентрацией.
Рис. V.29. Зависимость удельной электропроводности от концентрации
различных дисперсных фаз:
а — правильно упорядоченные непроводящие бакелитовые полусферы (Пирс, 1955); б —
кубически расположенные сферы луцита (Мередит и Тобиас, 1960); в — суспензия песка
(Окер-Блум, 1960); г — суспензия стеклянного порошка [О — фракция III (175—210 мкм),
V — смесь фракций I и VI (49—6400 мкм)\ (Де ла Рю и Тобиас, 1959); д — суспензия
песка (О) и полистироловых частиц (V) (Де ла Рю и Тобиас, 1959); е — кровь собаки (Фрике,
1924); 1 — уравнение (V.112); 2 — уравнение (V.115); 3 — уравнение (V.110); 4 —
уравнение (V.349); S — уравнение Фрике для эллипсоидов.
365
Результаты, полученные в случае практических эмульсий и
суспензий. Рассмотрим примеры произвольных дисперсных систем,
таких как реальные эмульсии и суспензии.
Данные о зависимости v.jy.m — Ф для суспензии песка сообщены
Окер-Блюмом (1900). Он приготовил суспензию добавлением песка
к горячему раствору соли, содержащему 3% желатина, затем
охладил при непрерывном перемешивании. На рис. V.29, в его данные
сравниваются с двумя теоретическими кривыми. Как видно,
экспериментальные значения ближе к уравнению (V.115), чем к уравнению
(V.112). Строго говоря, эти результаты не пригодны для точного
анализа теоретических выражений, выведенных для сферических
дисперсий, поскольку частички песка только приблизительно имели
сферическую форму.
Де ла Рю и Тобиас (1959) изучали удельную электропроводность
суспензий стеклянного бисера, песка, полистероловых цилиндров
в электролитическом растворе, а также влияние размеров и форм
суспендированных частиц на электропроводность.
Значения диаметров суспендированных частиц и используемая
непрерывная фаза приведены в табл. V.3. На рис. V.29, г показаны
результаты измерений для фракции III и смеси фракций I и VI,
а также представлены теоретические кривые. Как видно, зависимость
х/х,„ — Ф хорошо отражается уравнением (V.115) для широкого
распределения частиц по размерам. Данные же в узкой области
распределения частиц по размерам попадают между значениями,
предсказываемыми уравнениями (V.112) и (V.115).
Таблица V.3
Вид суспендированных частиц и значения п уравнения (V.350)
Суспендированные
частицы
Стеклянный
бисер
Песок Долины
смерти
Морской песок
Полистирол
Форма
Сферы
Неправильная
То же
Цилиндры
Фракция
I
II
III
IV
V
VI
Размеры
частиц,
мкм
49-77
107—175
175-210
500—840
3100-3400
6100-6400
150-175
150-175
Диаметр
0,3 мм; длина
3,0 мм
Непрерывная
фаза
Раствор
ZnBr2,
близкий к
насыщению
Раствор
ZnBr.2
0-1 н. раствор
КС1
Эмпирическая
постоянная
1,43
1,52
1,45
1,47
1,48'
1,50
1,58
1,55
1,53
Влияние формы суспендированных частиц рассмотрено на
суспензиях полистироловых цилиндров и двух видов песка (рис. V.29, д).
Экспериментальные данные отклоняются от уравнения (V.112)
и близко приближаются к кривой, полученной из уравнения (V.115).
366
Кроме того, Де ла Рю и Тобиас рассмотрели применимость
уравнения (V.115) при условии, чтобы эмпирическая формула
~ = ({-Ф)п (V.350)
аналогичная уравнению (V.115), будет удовлетворительно выражать
наблюдаемые величины при соответствующем выборе значений га.
Численные величины п, полученные из экспериментальных данных
методом наименьших квадратов, представлены в табл. V.3. Все
эти значения очень близки к 1,5, имеющемуся в
уравнении (V.115).
Фрике (1924) сравнил теоретические кривые с данными,
полученными Стюартом (1899) при изучении удельной электропроводности
крови собаки в растворе соли (рис. V.29, е) и установил, что
уравнение.^. 112) не соответствует наблюдаемым величинам. Он объяснил
это несоответствие несферической формой частиц и вывел новое
уравнение для дисперсий эллипсоидных частиц на базе уравнения
Винера. Из рис. V.29, е видно, что кривая, предсказанная уравнением
Фрике, хорошо согласовывается с экспериментальными данными.
Все исследования, упомянутые выше, проведены на
фиксированных частотах. Однако, так как гетерогенная структура эмульсий
может давать диэлектрическую дисперсию, обусловленную
поляризацией поверхности раздела, то необходимо принять во внимание
зависимость удельной электропроводности эмульсий от частоты, как
и от концентрации. Результаты, приведенные на рис. V.27, не
достаточны для количественного анализа электропроводности вследствие
неустойчивости величины электропроводности водной фазы,
использованной в опытах. Вместе с тем, диэлектрическая проницаемость
является величиной устойчивой и достаточно воспроизводимой,
поэтому ее можно использовать для количественного анализа.
Ханаи, Коицуми, Сугано и Гото (1960) измеряли х и С эмульсий
нуйол/четыреххлористый углерод в 0,5 н. растворе хлорида натрия
с неионным эмульгатором спен-20, твин-20 и цетиловым эфиром
полиоксиэтиленгликоля. На рис. V.30 приведен пример частотных
характеристик Сии. Как видно, величина С значительно возрастает
с уменьшением частоты вследствие электродной поляризации, тогда
как у. остается постоянной независимо от частоты. Эти результаты
показали, что диэлектрическая дисперсия, обусловленная
поляризацией поверхности раздела, не обнаруживается в диапазоне частот
20 гц — 5 Мгц. На рис. V.31 представлены теоретические кривые
и график зависимости наблюдаемых значений х/хт от Ф.
Выводы о диэлектрической проницаемости эмульсий типа М/В.
На основе анализа экспериментальных данных для эмульсий М/В
и родственных суспензий можно сделать вывод, что уравнения (V.74)
и (V. 111) справедливы для систем с упорядоченным расположением
сфер одинаковых размеров, тогда как уравнения (V.76) и (V.114)
приемлемы для произвольных дисперсий вплоть до высоких
концентраций.
367
Связь полученных результатов с поляризацией поверхности
раздела будет отмечена далее. Величины еии для эмульсий М/В не
показали диэлектрической дисперсии, обусловленной поляризацией
поверхности раздела, и хорошо
согласуются с уравнениями
(V.223) и (V.228).
Следовательно, теория поляризации по-
Рпс. V.30. Частотная зависимость
емкости, диэлектрической проницаемости (а)
и удельной электропроводности (б)
эмульсин М/В (Ханан, Коицуми, Сига-
но, Гото, 1960).
Объемная концентрация 70%; 25° С.
Рис. V.31. Зависимость удельной
электропроводности эмульсии М/В
от концентрации дисперсной фазы
при 25° С (Ханаи, Коицуми, Спгано
и Гото, 1960):
1 — уравнение (V.112); i — уравнение
(V.115).
верхности раздела Ханаи является пригодной для
количественного описания диэлектрических свойств эмульсий
типа М/В.
Диэлектрические свойства эмульсий типа В/М
В противоположность эмульсиям М/В, диэлектрические свойства
гетерогенных дисперсных систем типа В/М весьма чувствительны
к изменению концентрации, частоты и условий потока. Со времени
теоретических работ Максвелла (1892) и Вагнера (1914)
диэлектрическую дисперсию, обусловленную поляризацией поверхности
раздела, изучали многие исследователи. Первая попытка сделана
Силларсом (1937), который измерял емкость и тангенс потерь
капель воды в парафине (рис. V.32). Для количественной оценки он
368
воспользовался упрощенными соотношениями (V.170)—(V.179) при
условии Ф < 1 и хт < хр:
= еА-
1-ЗФ
ег — ей
1 + ;шт
28*
1
£а =
1-ЗФ
/<й8абс
я/
с
2е„
£/ =
ez — £/,=
:(1 + ЗФ)
9Фе;>
2л/0
2£т
2sm ~г Ер
ЗФ
е„
tg6
х/ = у.т(1-г-ЗФ)
__ Е" (g;— Eft) OJT
е е; — 8/jW2t2
ьабс
(V.351)
(V.352)
(V.353)
(V.354)
(V.355)
(V.356)
(V.357)
(V.358)
Кривая тангенса угла потерь получена из уравнения (V.358)
" е. '
при использовании значении:
0,25
(воск) = 2,18, ер (вода) = 80,
кр (вода) = 5-Ю-5 ом~ 1/см
и Ф = 0,067. Как видно,
экспериментальные
значения б много больше
расчетных, а частота максимального
f,2U,
Рис. V.32. Частотная зависимость
емкости (кривая 2) п тангенса угла потерь
(кривые 1 w. 3) суспензии капель воды
в парафиновом воске (объемная
концентрация 6,7%; 15° С) (Силларс, 1937).
экспериментальные данные; —
данные.
расчетные
Рис. V.33. Частотная зависимость
фактора потерь е" сферической
дисперсии фталоциановой меди
в парафиновом воске (объемная
концентрация 0,62%) (Хамон,
1959):
— — — экспериментальная кривая;
теоретическая кривая из уравнений
(V.177) и(У.178).
значения tg б много меньше для наблюдаемых значений, чем для
расчетных. Когда опытный образец, который находился при 9° С,
т. е. ниже точки затвердевания воска, был нагрет до 52,5° С, тангенс
угла потерь уменьшился и приблизился к вычисленным значениям.
24 Заказ 107(1
369
На основании этого Силларс сделал вывод, что значительное
расхождение между наблюдаемыми и расчетными значениями может быть
обусловлено наличием мелких трещин в результате сжатия воска
при низких температурах.
Хамон (1953) приготовил дисперсию фталоцианина меди, который
является органическим полупроводниковым материалом, в
парафиновом воске; объемная концентрация составила 0,62%. Зависимость
е" — / для образца, имеющего форму диска, показана на рис. V.33.
Там же приведены теоретические кривые, вычисленные из уравнений
(V.171), (V.177) и (V.178) при условии хт < кр и Ф < 1. Так как у.р
и е;) исследуемого образца зависели от частоты, Хамон при решении
4
J
юг ю3 юч
Рис. V.34. Частотная зависимость е/е,„ и lg 6
модельной системы с прямоугольным расположением нолн-
стиролннтробензоловых сфер в полптене (объемная
концентрация 46,5%; 17° С):
1 п 2 — экспериментальные лначения; 3 н 4 — по уравнениям
(Л".170) — (Y.17H); 3 п в — по уравнениям (Л".351) —(Y.358)
при Ф ,: 1.
уравнения Вагнера использовал числовые значения, найденные
различными способами на каждой частоте.
Соответствие между наблюдаемыми и расчетными значениями
является удовлетворительным, за исключением частот ниже 5-Ю4 гц,
когда наблюдаемые значения становятся значительно больше
вычисленных. Хамон объяснил это расхождение незначительной
неоднородностью в свойствах образца. Однако он не сообщил данных
по реальной части диэлектрической проницаемости, которые
необходимы для дальнейших обсуждений.
Карадли и Джексон (1953) подтвердили диэлектрическую
дисперсию, обусловленную поляризацией поверхности раздела, на
модельной системе, представляющей собой кубическое расположение
сферических элементов плохо проводящего материала в изоляционной
среде. Суспендированные частицы приготовлены из сильно вязкой
смеси иолистирола и нитробензола в соотношении: 5 г первого на
15 см3 второго, для которых ер = 22 и у.р = 3,25 ■ 10"4 ом~[/см при
370
170° С. Исследуемые образцы получены механической обработкой
квадратной решетки полусферических углублений на двух плоских
листах полиэтилена (гт = 2,30). Углубления заполняли вязкой
смесью полистирол — нитробензол и точно располагали их один
под другим. Затем образцы соединяли вместе и запаивали
погружением в расплавленный парафиновый воск. Образец содержал один
слой кубического ряда сферических элементов. Результаты
исследований представлены на рис. V.34.
Для кривых 3 и 4 получено лучшее соответствие с
экспериментальными значениями, чем для кривых 5 и 6, имевших слишком
Рис. V.35. Частотная зависимость фактора потерь е"
дисперсной системы капель воды в шерстяном воске
при 20° С (Драйден и Мекинс, 1957).
Концентрации воды, %: 1 — 0; 2 — 9,3; 3 — 18,7; 4 — 28 З-
л — 38,1. ' '
грубое приближение. К сожалению, теория Ханаи не может
рассматривать данные рис. V.34, потому что частотная зависимость
диэлектрической проницаемости в ней освещена недостаточно.
Кроме того, Карадли и Джексон провели теоретический анализ
диэлектрического поведения систем с кубическим расположением
сфер. Принимая во внимание высокую степень взаимодействия между
сферами в концентрированных системах, они вывели уравнение,
подобное уравнениям (V.170) —(V.178), а для разбавленных систем
получили такие же выражения, как уравнения (V.351)—(V.356).
В процессе исследований диэлектрических свойств шерстяного
воска Драйден и Мекинс (1957) получили различные пики частотной
зависимости фактора потерь (рис. V.35). Они объяснили это
межфазной поляризацией. Шерстяной воск, являющийся сложной смесью
органических эфиров, обладает способностью образовывать
эмульсии В/М. Авторы приготовили сферические дисперсии сильным
24*
371
перемешиванием воска и капель воды в стакане с помощью маленькой
проволочной лопаточки. Данные исследования образцов с
содержанием воды <10% не пригодны для количественного теоретического
анализа, потому что величина диэлектрических потерь в самом
шерстяном воске довольно значительна по сравнению с небольшим
влиянием низких концентраций воды.
Остальные результаты можно рассматривать в свете теорий
релаксационной частоты и величины диэлектрической дисперсии.
Используя данные: у.р = 1,2-Ю"6 ом~1/см (для дистиллированной
воды), у.т <^ хр, &Р— 80, sm = 3 и Ф = 0,381, из приближенного
уравнения (V.356) получаем /0 = 24,1 кгц, а из уравнения (V.178) —
/0 — 23,6 кгц. Из рис. V.35 видно, что экспериментальное значение
частоты (220 кгц) больше расчетного в ~9 раз. Такое расхождение
можно объяснить увеличением эффективного значения у, ,
обусловленным ионными примесями, которые введены в воду в процессе
перемешивания. Это предположение подтвердилось возросшей
электропроводностью водной фазы, выделенной из исследуемого образца
после измерений.
В табл. V.4 приведено сравнение величин диэлектрической
дисперсии е/ — еЛ, полученных из уравнений (V.190) и (V.191) теории
Вагнера и уравнений (V.229) и (V.233) теории Ханаи. Так как
реальная часть величины диэлектрической проницаемости не была
измерена, значения диэлектрической дисперсии et — ей получены
интегрированием площадей, описываемых кривыми на рис. V.35, с
помощью соотношения:
+ со
4-th = ~ j z"(f)d(\gf) (V.359)
-со
Как видно из табл. V.4, измеренные значения е1 — ед распола-'
гаются между расчетными.
Таблица V.4
Сравнение величин диэлектрической дисперсии
для систем капель воды в шерстяном воске
ф
Н,0
0,093
0,187
0,283
0,381
£/
ментальные
данные *
4,2
5,2
6,9
11,1
расчетные
уравнение
(V.191)
4,3
5,5
7,0
9,1
уравнение
(V.233)
4,5
6,0
8.7
13,4
Ч~г1г
ментальные
данные -
1,13
1,56
2,03
3,85
расчетные
уравнения
(V.191)
и (V.190)
1,08
1,46
2,05
2,86
уравнения
(V.233)
п (V.229)
1,2
1,7
2,8
4,7
* Экспериментальные значения взяты из работ Дрмйдсна и Мекннса (1957).
372
Чтобы понизить диэлектрическое поглощение самой дисперсной
фазы, Драйдеи и Мекинс в дальнейших экспериментах использовали
в качестве непрерывной фазы смесь нефтяного желе с 3—10%
раствором шерстяного воска. График зависимости е" — lg / для этих
систем, в противоположность рис. V.35, имеет симметричную форму
относительно частоты максимальных потерь, а площади,
описываемые кривыми е", т. е. et — еЛ, больше полученных расчетным путем
из теории Вагнера. Например, при содержании воды 10 и 20%
площадь, полученная по экспериментальным значениям, на 25 и 70% ,
соответственно, больше площади, вычисленной по теории Вагнера.
При использовании теории Ханаи эти величины становятся еще
больше. Такое расхождение объяснено широким распределением
частиц воды по размерам (0,5—5,5 мкм) в этих системах. Кроме того,
значения е; — е/г могут быть больше вследствие эффекта агломерации,
как в экспериментах Ханаи (см. стр. 375), когда эти значения
уменьшались с ростом сдвигового потока.
Анализ межфазной поляризации
в эмульсиях типа В/М
Систематические данные по эмульсиям В/М сообщены Ханаи
(11)61 Ь), который исследовал диэлектрические свойства
приготовленных им эмульсий воды в смеси нуйол/четыреххлористый углерод
с эмульгаторами арлацен-83, спен-20 и спен-60. Он обнаружил
значительное влияние перемешивания и сдвигового потока на
диэлектрические свойства эмульсий. Измерения он проводил с помощью
двойного цилиндрического вискозиметра типа Грина с фиксированным
отвесом и вращающейся чашкой.
На рис. V.36, а, б приведены графики частотной зависимости е,
е" и х для эмульсий В/М с 80%-ной объемной концентрацией в
состоянии покоя и при различных скоростях вращения чашки
вискозиметра. На рис. V.37 дан график результатов этих исследований.
Значительная диэлектрическая дисперсия на частоте выше 30 кгц
может быть объяснена межфазной поляризацией, которая в эмульсиях
М/В не была обнаружена. Постепенный рост е с понижением частоты
ниже 1 кгц можно отнести за счет поляризации электродов.
Значительное понижение е под действием сдвигового напряжения, как
видно из рис. V.36, а может быть результатом изменения состояния
агломерации дисперсных частиц. Подробно это будет рассмотрено
ниже.
Предельные значения диэлектрической проницаемости на
высоких частотах. Предельную диэлектрическую проницаемость ей
на высоких частотах (см. рис. V.36, а и V.37) можно рассматривать
в некотором смысле, как диэлектрическую проницаемость
сферических дисперсных систем, состоящих из двух непроводящих фаз.
Поэтому наблюдаемые значения еА следует рассматривать в свете
уравнений (V.73), (V.74), (V.76), (V.80) и (V.107). На рис. V.38 для
сравнения даны теоретические кривые, полученные из уравнений,
373
800 v-
Г,гц
f.zn
Рис. V.36. Частотная зависимость диэлектрической проницаемости е (а),
фактора потерь s" (б) и электропроводности у. (в) эмульсии В/М (объемная
концентрация 80%; 30? С) (Ханай, 1961).'
Скорость вращения чашки вискозиметра, об/мин: 1 — 0; 2 — 100; 3 — 200; 4 — 300.
Ш
~" ?00 -
е, 800
Рис. V.37. Частотная зависимость е п е" эмульсии В/М (объемная
концентрация 80%; 30° С) (Ханап, 1961b)
Скорость вращения чашки вискозиметра, об/мгт: 1 — 0; 2 — 100; 3 — 200!
4 — 300.
и экспериментальные значения. Как видно, уравнения (VJZfi).Б-ругге-
мана наиболее соответствуют наблюдаемым значениям по всему ряду
концентраций.
Наики, Фуджита и Мацумура (1959) исследовали эмульсии воды,
диспергированной в керосине, трансформаторном масле и терпене,
с помощью эмульгаторов: полиэтиленгликольолеат, полиэтилен-
гликоль нонилфениловых эфиров (ОП), стеарата цинка.
На рис. V.39 для сравнения предельных значений ел на высоких
частотах приведены теоретические кривые и экспериментальные
значения диэлектрической проницаемости на частоте 2 Мгц. Видно, что
80-
Рпс. V. 38. Зависимость предельной
диэлектрической проницаемости e/j от
концентрации дисперсной фазы на
высоких частотах эмульсии В/М прп
30° С(Ханаи, 1961b):
1 — уравнение (Л'.73); 2 — уравнение
(Y.80); 3— уравнение (Л".107): 4 —
уравнение (V.76); 5 — уравнение (V.74).
Рис. V.39. Зависимость
диэлектрической проницаемости эмульсий воды
в керосине, терпене,
трансформаторном масле от концентрации дисперсной
фазы на частоте 2 Мгц (Наики,
Фуджита и Мацумура, 1959):
1 — уравнение (V.73); 2 — уравнение (V.80);
3 — уравнение (V.76); 4 — уравнение (V.107);
5 — уравнение (V.74).
экспериментальные значения по всему диапазону концентраций
находятся в хорошем соответствии с рассчитанными из уравнения (V.76).
Диэлектрическую проницаемость сферических дисперсных систем,
состоящих из двух непроводящих фаз, изучал Эйхбаум (1959). Он
приготовил суспензии стеклянных шариков в полистироле. На
рис. V.40 приведены результаты его измерений на частоте 1 Мгц
вместе с теоретическими кривыми. Экспериментальные величины,
очевидно, не зависят от размера частиц и немного меньше, чем
значения, полученные из теоретических кривых.
Предельная диэлектрическая проницаемость ez на низких
частотах. На рис. V.41 дается сравнение опытных значений ег в состоянии
покоя и прп различных скоростях вращения, полученные Ханаи
(1961b) для эмульсий В/М, со значениями, вычисленными из
уравнений (V.191) и (V.233). В состоянии покоя опытные значения гг
относительно велики, а с ростом скорости вращения они
уменьшаются, приближаясь к расчетным, в противоположность ед, которая
375
не реагирует на сдвиговое напряжение. Следовательно, влияние
сдвига на е становится заметным на частотах, когда
электропроводность фаз влияет на диэлектрическую проницаемость дисперсной
системы. По результатам, представленным на рис. V.41, еще не ясно,
будут ли совпадать экспериментальные и расчетные значения е,
при большой скорости сдвига.
Некоторыми исследователями дано экспериментальное
подтверждение теоретической формулы для et. Так, Жульен (1941) измерял е^
эмульсий ртути в касторовом
масле при концентрациях <; 40%
(рис. V.42). Он установил
хорошее соответствие между
опытными значениями е, и полученными
по уравнению (V.233).
1000
<? юо -
Рис. Л*.40. Зависимость
диэлектрической проницаемости от концентрации
дисперсной фазы суспензии порошка
оконного стекла в полистироле на
частоте 1 Мгц (Эйхбаум, 1959):
1— уравнение (V.76); 2 — уравнение (Л".74);
О — 200 ыеш; ч* — 100 меш.
Рис. V.41. Зависимость предельных
значений диэлектрической
проницаемости t:i от концентрации
дисперсной фазы эмульсии типа В/М
на низких частотах (Ханан, 1961b):
1 — уравнение (V.191); 2 — уравнение
(V.233). Скорость вращения чашки
вискозиметра, об/мин: О — 0; X) — 100;
О 200; -О — 300.
Пирс (1955) исследовал диэлектрическую проницаемость эмульсий
при концентрациях вплоть до 63% на частоте 1 кгц. Результаты
измерений для частиц, размеры которых находились в пределах 2—40 мкм,
приведены на рис. V.43. Поскольку морская вода, используемая как
дисперсная фаза, имеет высокую электропроводность, эмульсии на
очень высоких частотах показали диэлектрическую дисперсию,
обусловленную межфазной поляризацией. Значения диэлектрической
проницаемости, наблюдаемые Пирсом, соответствовали предельным
zt на низких частотах. Пирс сделал вывод, что уравнение (V.233)
справедливо для дисперсных систем с беспорядочным распределением
сферических частиц, например, для эмульсий.
Наики, Фуджита и Мацумура (1959) изучали диэлектрическую
проницаемость систем типа ртуть — вазелин на частоте 2 Мгц.
Данные их измерений показаны на рис. V.44. Опытные значения
немного выше полученных из уравнения (V.233).
376
0.8 г
О 195 1.90 1,85 1.80 1.75
1д(1-Ф1
Рис. V.42. Зависимость
диэлектрической проницаемости эмульсии ртути
в масле от концентрации дисперсной
фазы (Жульен, 1941):
1 — уравнение (V.233); 2—уравнение (V.191);
О — эмульсия ртути в машинном масле;
ф — эмульсия ртути в касторовом масле.
1.8 1.7 1,6
lglt-Ф!
Рис. V.43. Зависимость
диэлектрической проницаемости эмульсии
морской воды в мазуте от
концентрации дисперсной фазы на частоте
1 кгц (Пирс, 1955):
1 — уравнение (V.233); 2 — уравнение
(V.191).
<о 4
Рис. V.44. Зависимость
диэлектрической проницаемости эмульспп
ртути в вазелине от концентрации
дисперсной фазы на частоте 2 Мгц
(Напкп, ФуджитаиМацумура, 1959):
1 — уравнение (V.233); 2 — уравнение
(V.191); 3 — уравнение (V.107).
«Г7-
^'
10'
10
,-И
—г
-
-
-
у*
о
'
it
о
i ■
о
J
1—1—г-
1/
j<S
> -8- °
О
—I i
' V I
2J
О
I I
О 0,2
0.4 0,6
Ф
1000
100
ю
-1
5
*
0)
0,8 1.0
Рис. V.45. Зависимость предельной х/
и относительной и//хт удельной
электропроводности эмульсии В/М от
концентрации дисперсной фазы на низких
частотах (Ханаи, 1961b):
1 — уравнение (V.234); 2 — уравнение
(V.192). Скорость вращения чашки
вискозиметра, об/мин (при 100 гц): О — 0;
О — 100; О- — 200; -О — 300.
Предельная удельная электропроводность v4 на низких частотах.
Согласно данным Ханаи по эмульсиям типа В/М (1961b),
приведенным на рис. V.36, б, на частотах <1 кгц не обнаружено
определенной зависимости от сдвигового напряжения и не получены предельные
значения х по всему диапазону низких частот.
На рис. V.45 значения хг на частоте 100 гц, взятые из рис. V.36, в
сравниваются со значениями, вычисленными из уравнений (V.192)
и (V.234). Оказалось, что опытные значения х, лежат ниже
вычисленных.
10 г
Рис. V.46. Зависимость удельной
электропроводности от концентрации
дисперсной фазы модельной системы
кубически расположенных
латунных полусфер в водопроводной воде
на частоте 400 гц (Пирс, 1955):
1 — уравнение (V.234); 2 — уравнение
(V.192).
Рис. V.47. Зависимость удельной
электропроводности от концентрации
дисперсной фазы модельной системы
кубически расположенных латунных сфер
в водопроводной воде на частоте
>50 кгц (Мередит и Тобиас, 1960):
1 — уравнение (V.234); 2 — уравнение
(V.349); 3 — уравнение (V.110); 4 —
уравнение (Y. 192); .5 —плотная упаковка.
Экспериментальное подтверждение теоретической формулы для х,
в случае сферических дисперсных систем дано уже многими
исследователями.
На рис. V.46 дан график зависимости х/хт — Ф, наблюдаемой
Пирсом (1955) для электролитической модельной системы с
кубическим расположением латунных полусфер в водопроводной воде.
Опытные значения хорошо согласуются с полученными из уравнения
(V.192).
Мередит (1959) измерял х эмульсий В/М с объемной
концентрацией <; 50% на частотах 1 и 10 кгц. Найденные им значения можно
рассматривать как предельные для х на низких частотах, хотя в его
сообщении диэлектрическая дисперсия, обусловленная межфазной
поляризацией, не была отмечена. Экспериментальные значения х
находятся между величинами, полученными из уравнений (V.192)
и (V.234).
378
Мередит и Тобиас (1960) изучали удельную электропроводность
модельной системы, представляющей кубически расположенные
латунные сферы в водопроводной воде. Результаты этих измерений
в сравнении с теоретическими кривыми показаны на рис. V.47.
Экспериментальные данные, за исключением высоких концентраций,
лежат ближе к значениям,
полученным из уравнения (V.349).
Предельная удельная
электропроводность хЛ на высоких
частотах. Вопрос о предельной
электропроводности v.h на высоких
частотах вне диэлектрической
дисперсии, обусловленной межфаз-
ной поляризацией, в литературе
совершенно не обсуждался.
Единственная попытка в этом
направлении сделана Ханаи (1961b)
(см. рис. V.36, в). Даже в его
экспериментах не получено в
диапазоне использованных частот
определенных значений nh.
Следовательно, установить
справедливость уравнений (V.103) и
(V.230) оказалось невозможным.
Значения хЛ, полученные на
частоте 3 Мгц, приведены на
рис. V.48 вместе с теоретическими
кривыми, вычисленными из
уравнений (V.193) и (V.230). Видна
общая тенденция
экспериментальных значений хЛ: с ростом
скорости вращения они уменьшаются,
приближаясь к теоретическим.
Предполагаемое объяснение состоит в том, что под влиянием
сдвигового напряжения агломераты частиц могут быть разрушены на
отдельные частицы.
Диэлектрические свойства
эмульсий нитробензола в воде
Выше теоретически предсказывается, что в эмульсиях М/В может
наблюдаться диэлектрическая дисперсия при условии, если масляная
фаза имеет высокую диэлектрическую проницаемость. Чтобы
обнаружить это явление, Ханаи, Коицуми и Гото (1962а) исследовали
диэлектрические свойства эмульсий нитробензола в воде,
приготовленные с помощью эмульгатора твин-20. На рис. V.49 показана
частотная зависимость е и к этих эмульсий при 70%-ной объемной
концентрации. Быстрый рост е на частотах < 30 кгц происходит в результате
электродной поляризации. С увеличением частоты О 100 кгц) можно
С1
- 0.01
0,001
О 0.? 0.4 0.6 08 W
ф
Рис. V. 48. Зависимость предельной
удельной электропроводности X/, от
концентрации дисперсной фазы
эмульсии В/М на высоких частотах
(Ханаи, 1961Ь):
1 — уравнение (V.193); 2 — уравнение
(V.230). Скорость вращения чашки
вискозиметра (при 3 Мгц), об/мин: О — оЧ
Ъ — 100; О 200; -О — 300.
379
обнаружить диэлектрическую дисперсию, сопровождающуюся
значительным увеличением электропроводности. График в комплексной
плоскости представляет собой дугу
(рис. V.50). При средних скоростях
сдвига (вплоть до 40 сек'1)
заметного изменения е и х эмульсий
нитробензола в воде не
наблюдалось. Диэлектрическая дисперсия
объяснена поляризацией
поверхности раздела.
Предельная диэлектрическая
проницаемость гн на высоких
частотах. На рис. V.51 даны
экспериментальные значения еЛ при
различных дисперсных фазовых
концентрациях в эмульсиях
нитробензол — вода. Экспериментальные
значения еЛ находятся в хорошем
согласии с теоретическими
кривыми. В случае эмульсии
нитробензол — вода, кривые расположены
очень близко друг к другу, поэтому
трудно сделать вывод, какая из них
лучше соответствует наблюдаемым
значениям.
Предельная диэлектрическая
проницаемость е1 на низких
частотах. Экспериментальные значения
е, показаны на рис. V.51.
Согласно теории Вагнера, величина
диэлектрической дисперсии, т. е.
разница между ег и eft или между
кривыми 2 и 4, является очень малой
величиной как при низких, так
и при высоких концентрациях, хотя при средних концентрациях
эти значения довольно велики. Следовательно, большую диэлек-
Г,гц
Рис. V.49. Частотная зависимость
диэлектрической проницаемости
(а) п удельной
электропроводности (б) эмульсин нитробензол —
вода (Ханап, Коицуми п Гото,
1962а).
Объемная концентрация 70%; 20° С.
Рпс. V.50. Зависимость
г" от 8 в комплексной
плоскости для эмульсии
нитробензола в воде
(Ханаи. Коицуми и
Гото, 1962).
Объемная концентрация
70%; 20° С.
380
трическую дисперсию, наблюдаемую у эмульсий нитробензол —
вода теория Вагнера объяснить не способна. С другой стороны,
значительная разница между 8; и еЛ или кривыми 1 и 3 для высо-
'ких концентраций предсказывалась теорией Ханаи.
лм 1 1 i I i 1 i i i I
0,8 0,Ь О
Ф
Рис. V.51. Зависимость предельной диэлектрической проницаемости на
высоких (е/,) п на низких (8/) частотах от концентрации дисперсной фазы эмульсии
нитробензол — вода (Ханап, Концумп и Гото, 1962а):
1 — £/j по уравнению (V.223); 2 — еЛ по уравнению (V.184); -3 — е1 по уравнению (V.186);
4 — е/ по уравнению (V.227).
Экспериментальные данные: Д — е^; О — £/,• Теоретические данные — кривые.
Резюме по диэлектрическим свойствам эмульсий
На основе описанных экспериментальных данных можно сделать
следующие выводы.
1. В эмульсиях типа М/В не обнаружено ни диэлектрической
дисперсии, ни заметного изменения е в условиях воздействия
сдвигового потока. Зависимость е и х. от концентрации выражается
уравнением типа уравнения Бруггемана. Количественная оценка этих
фактов дается в теории Ханаи.
2. В эмульсиях типа В/М наблюдается значительная
диэлектрическая дисперсия, обусловленная поляризацией поверхности
раздела. Величина zh выражается уравнением типа уравнения
Бруггемана в соответствии с предсказаниями теории Ханаи.
3. Диэлектрическая дисперсия, полученная в эмульсиях
нитробензол — вода типа М/В, более соответствует предсказаниям
теории Ханаи, чем теории Вагнера.
4. Величина диэлектрической дисперсии, обусловленная
поляризацией поверхности раздела, не связана с типом эмульсии, а, как
предсказывается теорией, с относительной величиной е и х
компонентов фаз.
5. Диэлектрические свойства эмульсий типа В/М значительно
изменяются при перемешивании или в условиях сдвигового потока,
381
тогда как в эмульсиях типа М/В и нитробензола в воде влияние сдвига
не наблюдается. Подробно это изложено далее (стр. 405).
6. Влияние размеров частиц на диэлектрические свойства слишком
мало и сделать определенных выводов по этому поводу невозможно.
7. Как видно из рис. V.31 и V.45, по теоретическим и
экспериментальным данным х эмульсии — величина того же порядка, что и х
непрерывной фазы, которая электропроводна у эмульсий М/В и не-
проводяща у эмульсий В/М. Эти характеристики очень полезны для
определения типа эмульсий с помощью грубого, но простого
измерения сопротивления постоянному току (Бехер, 1957).
Диэлектрические свойства суспензий
биологических клеток
Живые клетки, такие как эритроциты или дрожжи, содержат
проводящее ядро, окруженное мембранной оболочкой. Последняя
состоит из липидов белков и полисахаридов и имеет относительно
высокое или очень высокое сопротивление, которое соответствует
высокому сопротивлению лецитиновых пленок. Следовательно,
суспензии этих клеток в электролитическом растворе представляют
пример суспензий сфер, покрытых непроводящей оболочкой. Теория
таких систем описана выше (см. стр. 341).
С тех пор как Паули и Шван (1959) предложили строгий метод
анализа результатов диэлектрических измерений таких систем,
появилось достаточно много сообщений об экспериментальных
исследованиях суспензий биологических клеток. Так, суспензии печеночных
митохондрий исследовали Паули, Пакер и Шван (1960),
пневмококков — Паули и Шван (1906), интактных дрожжей — Кога и Акабори
(1964), неплотных дрожжей — Сугиура и Кога (1965а) и
разрушенных дрожжей — Сугиура и Кога (1965b).
Ряд других исследований по диэлектрическим свойствам суспензий
вплоть до 1957 г. собраны в замечательном обзоре Швана (1957).
Здесь рассмотрены результаты исследований Паули, Покера
и Швана (1960) с тем, чтобы показать, как анализировать данные
измерений. Они изучали диэлектрическую проницаемость и
электропроводность суспензии печеночных митохондрий в растворе хлорида
калия.
На рис. V.52 дан график частотной зависимости е и х для
суспензий двух образцов А ж В: первый содержит 0,012 М раствор КС1,
второй — 0,13 М раствор КС1. На рис.У.53 и V.54 приведены
зависимости этих величин в комплексной плоскости для образца А.
Как видно, диэлектрическая дисперсия в этих образцах много больше
величины, предсказываемой теорией Вагнера для структуры, не
имеющей оболочек. Таким образом, эту диэлектрическую дисперсию
можно рассматривать как эквивалентную описанной выше (см.
стр. 351) и обозначенной тР. Другими словами, результаты
приблизительно можно выразить уравнениями-
г* = е<+ Л"Ё'' Ь-Г-^—*i (V.360)
382
Рис. V.52. Частотная
зависимость диэлектрической
проницаемости е и
удельной электропроводности и
суспензии митохондрий
печени при 25° С (Паули,
Пакер, Шван, 1960):
1 и 2 — суспензия в 0,012 М
растворе КС1 (образец А); 3 и
4 — суспензия в 0,13 М
растворе КС1 (образец В).
600
ш
■
гоо
0
1—1 1—1—г-
^-^^--fii
\
*ю У
1 1 Г 'i
—1—
>£
J2
е,
т—г-
«О:
Y
-Т?а«<
1 i
- i i—i i
fb
/"г Ю~3
э п очДЦЭо— _
'
0.1
10
f,Mzn
100
-«•<:
- ю
JOOt-
Ct -64
Ш \500
Рис. V.53. График
комплексной
диэлектрической
проницаемости в комплексной
плоскости для
суспензии митохондрий
печени в 0,012 М
растворе КС1 (Паули,
Пакер, Шван, 1960).
Рис. V.54. График
комплексной
удельной
электропроводности в комплексной
плоскости для
суспензии митохондрий
печени в 0,012 М
растворе К.С1 (Паули,
Пакер, Шван, 1960).
Xt- 5,7
Х-10 , ом'/см
или
/ШТр(»С,-— Щ) .
14 * = *« + ГГТТ^ И"*^ (V.361)
которые вытекают из уравнений (V.298) и (V.299) при условии
coTq <С !• Что касается кривых 1 и 2 на рис. V.52, то рост величины
электропроводности с возрастанием частоты выше 50 Мгц
предполагается началом высокочастотной дисперсии, обозначенной в
уравнении (V.298) буквой Xq.
Чтобы оценить хр, t>2, Cs по наблюдаемым значениям е;, е;, хг,
у.,-, у.т и /Р, делают дальнейшее предположение
RCGS «хри RCGS € У-т (V -362)
которое эквивалентно неравенствам:
Хр Лс Лт «с
Тогда уравнения (V.342)—(V.344) приводятся к виду:
*t = xm 2(п~"'2) (V-364)
8/— 6j = 77Г-; гт- (\ -36o)
еабс(2 + у2)2
^Р = -Л- = лсС8(' —+ 4=ZL-—") (v-366)
Затем производят следующие вычисления:
а) с помощью значений хг и ~/.т из уравнения (V.339)
получают хр;
б) так как прямое измерение v2 путем определения объема
упакованной ячейки не всегда успешно из-за неполноты упаковки, то
для его оценки используют уравнение (V.364);
в) величину Cs можно оценить как с помощью уравнения (V.365),
так и уравнения (V.366), используя полученные выше значения v2
и кр.
Результаты вычислений вместе с экспериментальными
значениями, полученными из рис. V.52, V.53 и V.54, приведены
в табл. V.5.
Так как характеристическая частота /р, 6 для е-дисперсии не
идентична частоте /PiK для х-дисперсии, как видно из рис. V.53
и V.54, то для вычисления С, берется среднее геометрическое между
двумя характеристическими частотами. Для вычисления Cs
используют упрощенные формы уравнений (V.365) и (V.366), которые
получаются при условии t>2 С 1-
Как видно из табл. V.5, менаду величинами Cs, полученными из
разных уравнений, установлено довольно хорошее соответствие.
Таким образом, можно сделать вывод, что величина Cs для мембран
384
Таблица V.5
Сравнение теоретических и экспериментальных данных
для суспензий печеночных митохондрий
(Паули, Пакер и Шван, I960)
Параметры
Концентрация раствора КС1
Средний радиус
Единица
измерения
ммолъ
мкм
Образец
А
12,0
0,75
Образец
В
130,0
0,75
Экспериментальные значения
У.р
v2
cs
cs
■*pl
fp
[из
[из
[из
[из
ит
'^■т
Щ
Щ
е;
е;
= V 'Р, е 'Р, у-
Вычисленные
уравнения (V.339)]
уравнения (V.364)]
уравнения (V.365)]
уравнения (V.366)]
Мо.ч~1/см
»
»
Мгц
1,84
0,57
1,35
480,0
64,0
2,8
значения
Мом~1/см
мкф/см2
»
1,07
0,60
0,50
0,61
0,58
18,7
6,8
13,7
440,0
65,0
27,0
10,0
0,53
0,49
0,62
0,54
печеночных митохондрий (0,5—0,6 мкф/см2) почти не зависит от
концентраций ионов окружающей среды.
Кроме того, установлено, что хр внутренней части клетки
приблизительно в два раза меньше хт внешней среды для обоих
образцов. Это означает, что клетки митохондрий способны регулировать
свою внутреннюю концентрацию ионов пропорционально их
содержанию во внешней среде.
Интересно отметить, что значения Cs, полученные в этих
исследованиях, лежат в диапазоне 0,5—2 мкф/см2, который, как
установлено, характерен для других биологических мембран. Если та часть
мембраны, которая оказывает влияние на Cs, состоит только из липи-
дов, то ее диэлектрическая проницаемость должна быть величиной
довольно малой, а именно: 2—5.
С помощью уравнения (V.331) можно вычислить соответствующую
толщину d, которая лежит в пределах 30—80 А. Этот диапазон
значений d соответствует толщине двойного слоя молекул линида,
а их диэлектрические свойства аналогичны таковым лецитиновых
пленок, описанных ранее (см. стр. 360).
ОБЪЯСНЕНИЕ ДИЭЛЕКТРИЧЕСКОЙ ДИСПЕРСИИ,
ОБУСЛОВЛЕННОЙ ПОЛЯРИЗАЦИЕЙ ПОВЕРХНОСТИ РАЗДЕЛА
Одной из важных особенностей поляризации поверхности раздела
является тот факт, что в эмульсиях типа В/М наблюдается
диэлектрическая дисперсия, которая не была обнаружена у эмульсий
25 Заказ 1070
385
типа М/В. В настоящем разделе эти особенности будут объяснены
в упрощенной форме, нестрого теоретически.
На рис. V.55 показаны некоторые стадии распределения зарядов.
Они соответствуют дисперсии сферических частиц, помещенных
между параллельными пластинами конденсатора, к которым
приложено напряжение переменного тока. Заряд, вызванный
электростатической индукцией (v), накапливается вблизи электродов и
фазовых границ. На границах фаз имеется два вида зарядов:
связанный (О) и несвязанный (•). Первый заряд — электростатический,
связанный с фазовой границей, не может разрядиться, второй —
способен перемещаться через фазовые границы диэлектриков и
быстро разряжаться на электродах. Эти особенности не свойственны
каждому заряду, а лишь в среднем являются функцией
электропроводности и диэлектрической проницаемости двух фаз, образующих
границу.
В начальный момент, когда на электроды подается напряжение
постоянного тока, в пространстве между пластинами появляется
некоторое количество индуцированного заряда v (рис. V.55,
стадия А). Следовательно, распределение заряда наглядно показывает
поведение бинарной смеси, составные фазы которой обладают
собственными диэлектрическими проницаемостями.
Спустя немного времени появляются заряды проводимости,
приводящие к совершенно различным состояниям распределения заряда
в эмульсиях В/М и М/В.
В эмульсиях В/М, где электропроводность дисперсной фазы
выше, чем непрерывной среды, заряды о возникают внутри частиц
(рис. V.55, стадия В). Так как сферические частицы сохраняются
изолированными благодаря окружению масляной фазы, которая
вообще является эффективным изолятором, заряды о не способны
перейти из сфер в масляную фазу.
Через определенный промежуток времени (рис. V.55, стадия С)
внутри сферических частиц накапливается заряд о. Тогда только
небольшая часть заряда (показанная на рисунке двумя темными
кружками) может разрядиться вследствие незначительной
электропроводности масляной фазы. Кроме того, в окружающей масляной
фазе появляется небольшой заряд проводимости, который быстро
разряжается иа электродах (на рисунке показано четырьмя темными
кружками).
С другой стороны, прибор, используемый для измерения
диэлектрических свойств, обладает способностью различать только два
вида зарядов: одни — способны разряжаться на электродах,
другие — не способны.
Все заряды о, находящиеся внутри сферических частиц (рис.У.55,
стадия С) регистрируются с помощью прибора как индуцированные
заряды v. Следовательно, результат, наблюдаемый с помощью
прибора за время t ->- °° (рис. V.55, стадия D) определяет только
два вида зарядов: v — приводящий к наблюдаемой диэлектрической
дисперсии, и • — к наблюдаемой проводимости.
386
Стадия
Время после
наложения
поля
постоянного тока
t=0
N00
Распределение заряда
Возникает только
индуцированный заряд
Начинают возникать
заряды проводимости
Появляется множест- -
во зарядов
проводимости, только малая часть
которых способна
покинуть капли
Соответствующее поле
переменного тока
высокая
частота
Низкая
частота
Низкая
частота
В/П
Пасло
Эквивалентно
М/В
jc„« хг
Води
О!
Эквивалент-
I но
Эквивалентное состо-
£-оо вние, наблюдаемое с
помощью приборов
Компоненты,
не способные
разряжаться
Компоненты,
Способные
разряжаться
О
>• •
•• •
>м
.••I
• ••
• •«
• ••
Рис. V.55. Объяснение явления межфазной поляризации в дисперсных системах сферических частиц:
— электростатический индуцированный заряд; ф — заряд; проводимости, способный перемещаться через фазовые границы;
О — связанный (заряд проводимости, не способный покинуть границу фаз.
При сравнении стадий А и D легко обнаружить, что величина
заряда V является относительно малой за время t = 0 и значительно
возрастает за время t —>- °°. Это означает, что величина
диэлектрической проницаемости мала в момент времени 1 = 0и велика ири
t ->- °о.
Теперь рассмотрим соотношение между временем действия
постоянного и переменного тока. В момент приложения напряжения
постоянного тока к электродам, т. е. когда t = 0, система находится
в переходном неуравновешенном состоянии. После
продолжительного времени (при t ->- °°) система приходит в состояние равновесия.
Если приложено напряжение переменного тока низкой частоты,
один полупериод является таким продолжительным, что система
способна перейти в следующее равновесное состояние в каждый
момент изменения направления переменного поля. Отсюда следует,
что характеристики состояния в поле неременного тока низкой
частоты такие же, как и за время t —>- °° в поле постоянного тока.
С другой стороны, очень высокая частота напряжения
переменного тока, периоды которой очень короткие, изменяет поле так
быстро, что в течение полупериода система остается только в
переходном состоянии, далеко от состояния равновесия. Это означает,
что характеристика состояния в поле переменного тока высокой
частоты соответствует начальной стадии переходного процесса (т. е.
t = 0), в поле постоянного тока.
Дело в том, что различные значения диэлектрической
проницаемости в моменты времени ( = 0и t = °° связаны с изменением ее
в зависимости от частоты приложенного переменного тока.- Эта
частотная зависимость диэлектрической проницаемости определяется
поляризацией поверхности раздела, потому что заряды,
накапливающиеся на поверхности раздела или границе между двумя фазами,
приводят к возрастанию наблюдаемой поляризации диэлектрика.
С помощью уравнений (V.15), (V.18) и (V.62) можно сделать
количественную оценку частотной зависимости удельной
электропроводимости в эмульсиях В/М. На начальной стадии действия
напряжения постоянного тока, что равносильно действию переменного
поля высокой частоты, величина заряда проводимости,
возникающего, в основном, внутри капель воды (рис. V.55, стадия А,
система В/М), увеличивается ео временем довольно быстро. Это
приводит к большому значению наблюдаемой электропроводности, как
видно из уравнений (V.18) и (V.15). При t -»- оо (рис. V.55, стадия С,
система В/М), т. е. при низких частотах, распределение заряда о
внутри сферических частиц достигает состояния равновесия и уже
дальнейшего возрастания у. со временем не наблюдается. В
результате вся система на стадии С имеет низкие значения у. при условии,
что окружающая непрерывная фаза имеет низкую удельную
электропроводность.
В системах М/В, где электропроводность непрерывной фазы
выше, чем у диспергированных частиц, во внешней фазе появляется
большое число зарядов проводимости, которые разряжаются на
388
электродах (обозначено шестью кружками • на рис. V.55, стадия В).
После продолжительного периода времени внутри масляных
сферических капель появляется небольшой по величине заряд
проводимости (обозначено четырьмя кружками • и двумя о на рис. V.55,
стадия С). Так как непрерывная среда является хорошо проводящей,
то большая часть зарядов проводимости капель может перейти через
границу раздела и только немногие из них задерживаются внутри
капель. Однако измерительные приборы зарегистрировали заряд о,
как заряд v и выдали информацию относительно части зарядов v
и о (рис. V.55, стадия D, система М/В).
Сравнение стадий А и D в случае систем М/В показывает, что
количество зарядов v в момент времени t = 0 почти такое же, как
и при t = оо. Другими словами, диэлектрическая проницаемость
на высоких частотах приблизительно такая же, как и на низких.
Из этого следует, что в системах..М/В диэлвктрлческая
проницаемость почти не зависит от частоты. Что касается наблюдаемой
электропроводности систем М/В, то на всех стадиях (А, В и С) можно
увидеть незначительный рост заряда О со временем. Следовательно,
электропроводность систем в этом случае фактически не изменяется
с частотой.
Изложенное объяснение представляет собой грубый
количественный анализ.
НЕКОТОРЫЕ ВИДЫ ЧАСТОТНОЙ ЗАВИСИМОСТИ
ДИЭЛЕКТРИЧЕСКОЙ ПРОНИЦАЕМОСТИ
Имеется несколько типов диэлектрической дисперсии, т. е.
частотной зависимости е и х.
Ниже дано краткое описание соответствующих математических
моделей для облегчения оценки диэлектрических характеристик
различных видов коллоидных систем.
Правило полукруглой дуги
для релаксационной системы
Имеется несколько примеров диэлектрического поведения систем,
графическое изображение которых в комплексной плоскости дает
полукруглую дугу. Комплексная диэлектрическая проницаемость
определяется уравнением
K«=e'-je"=s/,+ f ,~".S/t (V.367)
или же
e'-=e*+Tfw (V-368)
и
(Y1T
e' = (e/-eft) l + [m)t ; «о = 2я/ (V.369)
Первоначально функциональная зависимость уравнения (V.367)
выведена в теории поляризации поверхности раздела, предложенной
Максвеллом (1892). Дебай (1929), принимая во внимание ориентацию
389
молекулярных диполей, подверженных влиянию внешнего
электрического поля, вывел теоретическое выражение для аномальных
диэлектрических дисперсий полярных жидкостей и твердых частиц,
соответствующее уравнению (V.367). Система, характеризующаяся
диэлектрическими свойствами, определяемыми уравнением (V.367),
называется простой релаксациоршой системой в приближении
Дебая.
Этот вид диэлектрических дисперсий, обнаруженный в целом
ряде жидких диэлектриков (например, вода, спирты и некоторые
протеиновые растворы), зависит от свойств полярных частей
молекул.
Подробно с этим можно ознакомиться в монографиях Дебая
(1929), Смита (1955), Гиипеля (1954а. 1954b) и Бетчера (1952).
Хотя поляризация поверхности раздела дает диэлектрическую
дисперсию, идентичную по форме уравнению (V.367), ее механизм
не соответствует теории полярных молекул Дебая.
Правило круговой дуги
для распределения времени релаксации
Для многих диэлектриков графики е* в комплексной плоскости.
т. е. диаграммы зависимости мнимой части е" диэлектрической
проницаемости от ее реальной части е', представляют круговую
дугу. Чтобы выразить математически круговую дугу в комплексной
плоскости, Коле и Коле (1961) предложили следующую
эмпирическую формулу
f,= f'^'f" = f" + TW ">P>«> (V-370,
(^-ел)Г1 + (шт0Гсоз(-^-р)1
е'-«?А = 4 . (V.371)
1 + 2(сот0)3соз(^-Н + (0)т0)23
или
^г —рЛ) (coT0)3sin (~ (5
1 + 2(шт0)"соз -^-р +(<от„)
(V.372)
где р — эмпирическая постоянная; т0 — вероятное время
релаксации.
Согласно теории диэлектриков, поведение, отражаемое правилом
круговор! дуги, является результатом стохастического распределения
времени релаксации при данных исследоваР1Р1ях. Это распределение
в каждом случае характеризуется коэффициентом р и связано с
величиной т0 для данного диэлектрика.
390
Правило скошенной дуги
для распределения времен релаксации
В некоторых случаях график е* в комплексной плоскости
представляет скошенную дугу, аналитическое выражение которой
следующее
где
-f=A=-
8/ —eft
(1>Р>0)
(l+/«)To)'J
s'— еЛ = (е/ — eA)cos3cDcos ((5Ф)
е" = (е/ — eft) cos9 Ф sin (РФ)
tg Ф = о)т0
(V.373)
(V.374)
(V.375)
(V-376)
Правило т-й степени
для распределения времен релаксации
График е* в комплексной плоскости для некоторых гетерогенных
систем представляет прямую линию. ей пересекает ось г', а частотная
зависимость е*, е', е" и х определяется следующими выражениями:
8* — еЛ=Л (т)'т
е' — Fft= Л cos ( —- m J
(V.377)
(V.378)
(V.379)
Таблица V.6
Значения показателя степени т в правиле m-й степени (е', е", а, ш~т),
полученные из уравнений (V.378), (V.379) и (V.381)
для различных систем
s" = A sin
(*-)
OJ
Система *
Суспензии в водной фазе:
калин
VjOi (свежий)
V2Ot (старый)
А1263
картофельный крахмал
стеклянный порошок**
т
0,814
0,235
0,286
0,529
0,348
1,15
Система
Суспензии в масле:
TiO (рутил)
TiO (анатаз)
Высокодисперсные
системы:
желатин (<47%)
желатин (>47%)
т
0,644
0,530
0,34
-0,2
* Частотная зависимость диэлектрической проницаемости этих систем приведена на
рис. V.56 —V.58 и V.62.
** Величина т больше единицы, что с точки зрении диэлектрической теории,
включающей уравнения (V.377) — (V.381), является невероятным.
391
Таблица V.7
Виды диэлектрической релаксации
Вид
Частотная аависимость
График п комплексной
плоскости
Аналитическое выражение
Пример
Правило
полукруглой дуги для
простых
релаксационных систем
- Ч --
в/ — iy,
1 + ДОТ
Иода; спирты
Правило круглой
дуги для
распределении времени
релаксации
]-■!-(/шт„)
1 > р > О
Большинство
полярных жидкостей и
твердых тел
Правило скошенной
дуги для
распределения времени
релаксации
Правило т-к степени,
Клиновой тин
с,- Е'~е"
1>р>о
еЛ £■
: — e/j = А (до)-
1 > т > О
Гликоль, пропилен-
1'ЛИКОЛЬ
Желатиновый гель;
суспензии
Поляризация
поверхности раздела
(Максвелл —
Вагнер)
со
CD
СО
-6/,=
+ 7
в/ — ел
1 + /сот
;соеабс
Эмульсии
v. = fa6c^ sin(y '") ы^т (V..381)
где Л и т — эмпирические константы.
Этот тип частотной зависимости сформулирован Швайдлером
(1907), Фрике (1932) и др. и проявляется в гидрозолях, суспензиях,
твердых диэлектриках и т. д. Несколько примеров дано далее
(стр. 394). Числовые значения показателя степени т приведены
в табл. V.6 (см. стр. 391).
Этот тип диэлектрической дисперсии иногда называют правилом
иг-й степени, которое вытекает из ее функциональной
зависимости.
В реологии распределения времени релаксации для такого случая
иногда называют клиновым видом распределения.
Типы диэлектрических дисперсий, описанных выше,
представлены в табл. V.7 (см. стр. 392).
ДИЭЛЕКТРИЧЕСКИЕ СВОЙСТВА
НЕКОТОРЫХ ДИСПЕРСНЫХ СИСТЕМ
Существует большое число работ по исследованиям
диэлектрических свойств дисперсных систем, полное описание которых выходит
за пределы данной главы.
Здесь рассмотрены лишь типовые системы, хорошо изученные
или разработанные в последние годы.
С точки зрения состояния дисперсности, гетерогенные системы
можно разделить на три вида: грубодисперсные (эмульсии и
суспензии), высокодисперсные (микроэмульсии, гидрозоли и солюбилизи-
рованные системы В — М) и молекулярные (взаимно смешивающиеся
растворы и растворы высокомолекулярных соединений).
Суспензии
Диэлектрические дисперсии, подчиняющиеся правилу m-ii
степени. Фрике и Куртис (1935а, Ь, 1936, 1937) исследовали
диэлектрические свойства различных суспензий. Они измеряли е суспензий
каолина, целлюлозы, Fe203, V203, A1203, растворимого
картофельного крахмала и стеклянного порошка, диспергированных в воде
или в растворе неорганических солей, органических кислот, мыл
и желатина.
Некоторые данные пэказаны на рис. V.56. Как видно,
диэлектрическая дисперсия характеризуется правилом т-ii степени,
аналитическое выражение которого определяется уравнениями (V.378)
и (V.381).
Результаты исследования зависимости е от размеров частиц
в суспензиях стеклянного порошка представлены на рис. V.57.
394
График зависимости lg (e — ел) — lg / дает семейство прямых линий
с одинаковой величиной угла наклона т независимо от размеров
частиц, а величина е — еА на фиксированной частоте обратно
пропорциональна диаметру частиц.
О
Ю
10е 10' 10й 10ь Ю2 'О3
f,zu
-
— д/
— З'
I
1 1 '
1^
, \А
2У
2/
Зр
V
—
—
1
10"
10
Рис. V.56. Частотная зависимость диэлектрической проницаемости е — е/,
(а) и удельной электропроводности и (б) различных суспензий (Фрике и Куртис,
1937):
1 — 45% каолина (диаметр 0,5 мкм) в 0,5% растворе опеата натрия; 2 —
свежеприготовленный 1% раствор ViOj; 2' — 1% раствор VsOs 3-месячной давности; 3 —■ 5% раствор
растворимого картофельного крахмала; 4 — 1% раствор А1г03.
Анализ этих результатов приводит к выводу, что поверхность
раздела между частицами и дисперсной средой, возможно, обладает
особыми диэлектрическими свойствами.
Фрике и Куртис разработали теорию
для объяснения характеристик
межфазной удельной электропроводности,
являющейся результатом комплексной
электропроводности, которая действует
в направлении, параллельном границе
раздела фаз. Эта теория объясняла
экспериментальные результаты, хотя <.
происхождение особых свойств меж- I
фазной электропроводности все еще
остается непонятным.
Рис. V.57. Частотная зависимость
диэлектрической проницаемости 8 — в^ суспензии
стеклянного порошка в 0,005 М
растворе КС1 (Фрике и Куртис, 1937).
Размер частиц, мкм: 1 — 80; 2 — 20; Л — 2,5.
395
В опытах Фрике и Куртиса дисперсионной средой была вода.
Они утверждают, что диэлектрические характеристики определяются
молекулами воды на поверхности суспендированных частиц, а объем
водной фазы не вносит существенного вклада.
Чтобы сделать эти выводы более понятными, Гото, Ханаи и Кои-
цуми (1958) провели ряд исследований, измеряя е и е" суспензий
порошка окиси титана в
жидком парафине, в
котором вода не растворяется
Рис. V.58. Частотная зависимость f'. e' — е/г,
е" суспензии окиси титана (рутил) в
жидком парафине (размер частиц 0,4 мкм,
содержание влажности 0,7%, объемная
концентрация порошка 13°о, т — 0,644, е/г=-
= 3,78):
нижняя кривая — 20° С; верхняя кривая — 50° С;
А — сухой порошок.
Рис. V.59. График е* = ё'— is"
в комплексной плоскости для
суспензии окиси титана при
20° С:
О-
20° С;
5(1° С.
(рис. V.58 и V.59). Как видно, при воздушной сушке на
поверхности суспендированных частиц адсорбировалось некоторое
количество воды, приводящее к значительной диэлектрической дисперсии,
подчиняющейся правилу т-й степени. Аналитическое выражение,
согласно результатам Фрике, дается уравнениями (V.378) — (V.380).
Когда порошок высушивали при 150° С в течение 3 ч, частотная
зависимость полностью исчезала, величина е оставалась в пределах
значения еЛ но всему диапазону частот, а е" была величиной
слишком малой для регистрации. Таким образом, получено
экспериментальное подтверждение тому, что адсорбированная вода влияет па
диэлектрические характеристики, хотя точный механизм этого
явления до сих пор остается непонятным.
При использовании в опытах более гигроскопичного порошка
(например, крахмал и бентонит) зависимость е' и е" от частоты
усложнялась: диэлектрическая дисперсия типа круговой дуги, определя-
306
емая уравнением (V.370), перекрывалась диэлектрической дисперсией
типа то-й степени, определяемой уравнением (V.377).
Диэлектрическая дисперсия, подчиняющаяся правилу круговой
дуги. Шван, Шварц, Макзук и Паули (1962) изучали
диэлектрические свойства нескольких суспензий сферических коллоидных частиц
и получили результаты, отличающиеся от ранее изложенных.
В своих экспериментах они использовали следующие системы:
1. Полистироловый латекс, содержащий сферические частицы
диаметром 1,17, 0,566, 0,188 и 0,0878 мкм.
2. Две полистироловых суспензии, суспендированные с помощью
диализа.
270
2,69 ^
Z68 й
3000
гооо
1000
0
-
1000
"^600
200
B~°~~a^°^>t
I I I 1 L.
с"
1 ■
/
/х
\ .
\
1 Г""-^
V
/0*
Г.гц
10
?,67
в
Рис. V.60. Частотная зависимость диэлектрической
проницаемости е', фактора потерь е" п удельной
электропроводности у. суспензии полистироловых частиц
(полистироловый латекс) при 23° С (Шван, Шварц,
Макзук и Паули, 1962).
Размер частиц 0,188 jiiujh, концентрация дисперсной фазы 30%.
3. Полистироловые суспензии в различных средах с известной
ионной концентрацией.
4. Ряд эмульсий жировых частиц.
5. Суспензии биологических клеток.
На рис. V.60 и V.61 дана частотная зависимость х, е' и е" и их
графики в комплексной плоскости для полистиролового латекса как
типичного примера, а числовые значения произведены в табл. V.8.
Результаты по другим системам были аналогичными данным
исследования полистиролового латекса, за исключением некоторых
колебаний величины дисперсии и характеристической частоты. График
комплексной диэлектрической проницаемости в комплексной
плоскости удовлетворительно выражается правилом круговой дуги,
которое определяется уравнением (V.370). Характеристическая
частота /о = 1/2лто оказалась обратно пропорциональной квадрату
диаметра частиц. Величина дисперсии, т. е. е/ — ел, находится
в линейной зависимости как от объемной доли суспендированных
частиц, так и от их диаметров.
397
Согласно сообщению Швана и др., некоторые физические
процессы (такие как релаксация вследствие поляризации поверхности
раздела, электрофоретического движения суспендированных частиц
или отсутствия частотной зависимости поверхностной
электропроводности) не достаточны для объяснения экспериментальных
данных.
Они предположили, что величина поверхностной
электропроводности складывается из двух частей, одна из которых зависит, а другая
не зависит от частоты. Хотя они и не дали удовлетворительного
объяснения физического механизма, приводящего к таким
характерным свойствам поверхностной электропроводности, введение частот-
Рпс. V.61. График комплексной диэлектрической проницаемости
в комплексной плоскости для суспензии полпстироловых частиц
(полистпроловый латекс).
Данные те же, что на рис. V.60.
ной зависимости электропроводности поверхности раздела достаточно
хорошо объясняет высокую диэлектрическую проницаемость
коллоидных суспензий на низких частотах.
Шварц (1962) предложил диэлектрическую теорию, исходя из
явления поляризации противоионного облака вокруг
суспендированных частиц. Согласно его теории, смещение противоионов вокруг
Таблица \'.8
Характерные величины диэлектрической дисперсии,
полученные для полистироловой суспензии (полистпроловый латекс)
Диаметр
частиц,
МЬ'М
1.17
0,566
0,188
0,088
г1
8500
2500
2480
525
ek
—
44
46
Мом~1 /см
2,2814
2,8177
2,6735
6,4803
у.
(200 кгц),
Мам-1 /см
2,2872
2,8254
2,7030
6,4896
/о
(максимальные
потери), кгц
0,48
2,0
14 0
80
398
суспендированных частиц эквивалентно емкости поверхности,
проявляющейся в релаксации, регулируемой диффузией носителей.
Этот результат можно выразить через дополнительную
диэлектрическую проницаемость суспендированных частиц, величина которой
превышает истинную диэлектрическую проницаемость на низких
частотах. С помощью вероятных значений плотности и подвижности
концентраций на поверхности частиц, теория дает удачное
объяснение статической диэлектрической проницаемости и
характеристической частоты, наблюдаемых Шваном и др. (1962) на
полистироловых суспензиях.
Джонсон и Нил (1962) изучали свойства других видов суспензий.
Они измеряли фактор потерь в диапазоне частот от 30 кгц до 5 Мгц
в дисперсных системах порошка алюминия, порошка карбида
кремния, фибры в воде, этиленгликоле, водном ацетоне и водном
глицерине. Ими замечено два вида поглощения; первое в диапазоне
частот 10—30 кгц, которое соответствовало правилу т-й степени,
другое — в области нескольких мегагерц с простым видом
релаксации диэлектрической дисперсии. К сожалению, рассмотрение
механизма диэлектрического поглощения нельзя продолжить из-за
недостатка данных по реальной части диэлектрической проницаемости.
Желатиновые гели
Исследования диэлектрических свойств желатиновых гелей
проводили Фрике и Якобсон (1939), Фрике и Паркер (1940). Они
измеряли е и и желатиновых
гелей различных
концентраций в диапазоне частот от
2 кгц до 65 Мгц. Один из
результатов измерений
Фрике (1939) показан на
рис. V.62. Как видно,
частотная зависимость е и х
желатиновых гелей
подчиняется правилу т-ж степени,
аналитическое выражение
которого дается уравнениями
(V.378), (V.379), (V.381).
Величина показателя
степени т, которая для
концентраций < 47% равна ~ 0,34,
снизилась до 0,2 при
высоких концентрациях.
Фрике, Якобсон, Паркер
свойства этих систем зависят от межфазного слоя
ориентированных молекул воды, однако убедительного объяснения они не
дали.
Рис. V.62. Частотная зависимость
диэлектрической проницаемости е' и удельной
электропроводности х45%-ного
желатинового геля (Фрике и Якобсон, 1939).
Показатель степени т = 0,232 (е' :: со-т).
предположили, что диэлектрические
399
Гидрозоли
На ранней стадии исследований диэлектрических свойств
коллоидных дисперсных систем внимание и усилия исследователей были
сконцентрированы, главным образом, на измерениях в гидрозолях.
Различные результаты этих исследований собраны в обзоре Хей-
мана (1934). Клейтон (1943) в своей монографии дает широкий обзор
ранних исследований диэлектрических свойств золей.
Диэлектрические измерения, как правило, являются
трудоемкими и дают низкую точность из-за относительно высокого
значения х, свойственного дисперсным средам. В указанных работах
показана довольно высокая диэлектрическая проницаемость
гидрозолей. Например, Эрера (1922, 1923, 1930) для золя V205 получил
е = 1280.
На основании имеющихся результатов можно сделать вывод, что
такие большие значения е являются результатом адсорбции молекул
воды на поверхности суспендированной фазы (как и в суспензиях
и в желатиновых гелях). Экспериментальные результаты,
полученные в ранних работах, следовало бы проверить с помощью более
совершенной техники в широком диапазоне частот.
Солюбилизированные систем ы
Солюбилизированные системы представляют значительный
интерес, так как дисперсности данных систем, грубодисперсных и
молекулярных различаются.
В литературе имеется несколько примеров систематических
исследований диэлектрических свойств солюбилизированных систем.
Ввиду трудностей измерений, связанных с требуемой точностью,
системы солюбилизированной воды в неполярном растворителе более
целесообразны, чем обратные системы.
Кратин и Робертсон (1965) измеряли е воды, солюбилизированной ■
в смеси четыреххлористого углерода и циклогексапа с 1,3% про-
пилендиаминдиолеата. Степень солюбилизации воды в масляной
фазе была < 0,62 объемн. %. Это недостаточно для дальнейшего
рассмотрения. Коицуми и Ханаи (1967) изучали диэлектрические
свойства воды, солюбилизированной в керосиновом растворе
аэрозоля ОТ (диоктилсульфосукцинат натрия). Исследуемые ими образцы
приготовлены следующим образом: известное количество воды
вводили в керосиновый раствор (140—220° С), содержащий 1,5.г
аэрозоля ОТ на 10 мл раствора.
Диэлектрическая проницаемость и удельная электропроводность
образцов, полученных таким образом, не зависела от частоты
в области частот < 100 кгц.
На рис. V.63 дан график зависимости ez и ■а1 от температуры.
У безводного образца (кривая 1) ег уменьшается, a xz растет с
повышением температуры, что характерно для большинства полярных
жидкостей. Для образцов с высокой концентрацией воды (кривые 4
и 5) характерно своеобразное поведение: увеличение температуры
400
100
10 -
10'
10'
10
го
зо чо
Температура, °С
50
60
Рис. V.63. Зависимость диэлектрической проницаемости £;(а) и удельной
электропроводности щ(6) системы воды, солюбилизпрованнои в керосиновом растворе
аэрозоля ОТ, от температуры.
Керосиновый раствор содержит 1,5 г аэрозоля ОТ на 10 мл раствора. Объемная
концентрация воды всей системы, %:
1 — 0; 2 — 5; 3 — 10; 4 — 15; .5 — 20.
Рис. V.64. Сравнение значений
диэлектрической
проницаемости различных дисперсных
систем:
1 — уравнение (V.393); 2 —
уравнение (V. 190); 3 — уравнение (V. 223);
4 — уравнение (V.229); 5 —
уравнение (V.184); в — уравнение (Y.386).
Значения е, наблюдаемые при 20° С:
Q — эмульсия М/В (Ханаи, Кои-
цуми и Гото, 1959); © —
предельные значения е эмульсий В/М на
высоких частотах (Ханаи, 1961j;
О — смесь диоксан — вода (Акер-
лоф и Шорт, 1936); V — солюбили-
эированная вода в керосиновом
растворе аэрозоля ОТ.
н,о
26 Заказ 1070
401
приводит к слишком высоким значениям et и y.L, измерение которых
оказалось невозможным.
На рис. V.64 дан график зависимости значений е от концентрации
воды при 20° С. Установлено, что эта зависимость не является моно-
100 \—i 1 1 1 1 1 1 т—i i 1 14
to
«о
f,2U,
Рис. V.65. Частотная зависимость диэлектрической проницаемости е', фактора
потерь е" и удельной электропроводности х системы воды, солюбилизированной
в керосиновом растворе аэрозоля ОТ при 5° С.
Керосиновый раствор содержит 5 г аэрозоля ОТ на 10 мл раствора. Объемная концентрация
воды всей системы 15 %.
тонно возрастающей и по своим значениям отлична от таковой для
молекулярных растворов и эмульсий типа В/М.
На частотах >> 100 кгц можно обнаружить уменьшение
диэлектрической проницаемости, обусловленное, вероятно, диэлектрической
Рис. V.66. График комплексной
диэлектрической
проницаемости в комплексной плоскости
для системы воды,
солюбилизированной в керосиновом
растворе аэрозоля ОТ.
Данные те же, что на рис. V.64.
дисперсией. В системах с высокой концентрацией аэрозоля ОТ
диэлектрическая дисперсия сдвигается в сторону низких частот, как
показано на рис. V.65 и V.66. Очевидно состояние растворенной воды
в солюбилизироваином растворе значительно отличается от
состояния ее в молекулярных растворах, однако это требует
дальнейшего подтверждения.
ДИЭЛЕКТРИЧЕСКАЯ ТЕОРИЯ ГОМОГЕННЫХ СИСТЕМ;
МОЛЕКУЛЯРНЫЕ РАСТВОРЫ
Диэлектрические теории чистых жидкостей до сих пор широко
обсуждаются. Ниже даны некоторые математические модели для
молекулярных растворов, т. е. гомогенных систем, с тем, чтобы
сравнить результаты этих теорий с экспериментальными данными.
402
Уравнение Дебая
Согласно теории Дебая (1929), е смеси, состоящей из нескольких
различных жидкостей, определяется уравнением:
е—1
(V.382)
где а,- и р., — поляризация и дипольныи момент г'-го вида молекул;
к — постоянная Больцмана; Т — абсолютная температура; JV,- —
число молекул г'-го вида в единице объема смеси.
В случае чистой жидкости уравнение (V.382) примет вид:
е,-+ 2
<=^N,
<*(a<+w)
(V.383)
где е,- и Nip — диэлектрическая проницаемость и число молекул
в единице объема чистой жидкости, состоящей из г'-го вида молекул.
Совместное решение уравнений (V.383) и (V.382) дает
е + 2
1 _У е<-1 Nt _V ef-l
2 Z* е, + 2 'Nip Z e, + 2 '
(V.384)
где Ф,- — объемная доля г'-го вида молекул.
Отношение NJNip = Ф,- допускается вследствие аддитивности
объемов. Уравнение (V.384), идентичное уравнению (V.70), можно
преобразовать:
2^тф'=0 (v-385)
г
Для бинарных смесей, состоящих из фаз а и Ь, уравнение (V.385)
сводится к виду:
~=ГТфа + ^=^фь = 0 (V.386)
86-
Уравнение Онзагера
По теории Онзагера (1936), е многокомпонентной смеси, исходя
из предположения аддитивности объемов, определяется уравнением
^ Зе(е
2е + и?
■Ф,=
^ 2Ni
е("?+2)
2e + nf
(V.387)
где и,— показатель преломления чистой жидкости г'-го вида молекул.
В случае чистой жидкости уравнение (V.387) примет вид:
26*
Зе((е,— л?)
2г* + «?
- inNi
2е,- + п\
2 l*f
ЗА:Г
(V.388)
403
Чтобы исключить \ih решим совместно уравнения (V.388)
ц (V.387). Используя соотношение NJN' 1р = Ф,, получим:
х V у-^-Ф; = 0 (V.389)
£± гг(2е4-п?
i
Для бинарной смеси уравнение (V.389) сводится к виду
(г-еа)(2еае^) (г-Ч) (2^--^) ф
еь (2е-ь п«)2
или
(F-
еа(2е+ !.»)«
(е — efl) (2fae + nJ)
Fe(2e-r"S)a
-еа)(2еае + «4а) Ма
(e-e(,)(2ebe + nj) ,„ _ Q {у ш)
Eb(2e + ng)2 Р(,
(е —еь) (2Efes + nt) ЛГь
e0(2e + nS)2 f'a eft(2c + n*)« Рь
mft = 0 (V.392)
где с и иг — весовая и мольная доли фаз, соответственно М и р —
молекулярный вес и плотность чистой жидкости соответственно.
Если жидкость является неполярной, уравнение (V.390) в случае
еа «= Па упрощается:
е-£а п. , (е — еь) (2гьь + пь) ... n ,v эдч,
l^+i7 °ai Bb(2e+n«)2 Фь = ° {Л -393)
Существует несколько теорий, в которых рассматривается
диэлектрическая проницаемость чистых жидкостей, например, теории
Кирвуда (1939) и Фройлиха (1949). Полученные ими теоретические
выражения содержат параметры, которые отражают взаимодействие
между молекулами и влияние молекул, препятствующее ориентации
соседних молекул. Поэтому без подробных данных о структуре
жидкостей в смеси эти выражения использовать нельзя.
СРАВНЕНИЕ ДИЭЛЕКТРИЧЕСКИХ ПРОНИЦАЕМОСТЕЙ
РАЗЛИЧНЫХ ДИСПЕРСНЫХ СИСТЕМ
Как отмечено выше (см. стр. 334), диэлектрические свойства
эмульсий зависят не только от объемных долей компонентов фаз,
но и от типа эмульсий, в противоположность бинарным
молекулярным растворам (см. стр. 402). На рис. V.64 приведены значения
диэлектрических проницаемостеп бинарных систем воды и других
жидкостей се ^2.
Согласно полученным выводам (см. стр. 360), экспериментальные
значения диэлектрической проницаемости эмульсий (строго говоря,
предельные значения на высоких частотах) хорошо согласуются
с теоретическими уравнениями (V.223) и (V.229) (см. рис. V.64).
Если жидкие компоненты растворяются один в другом, то их
смесь будет находиться в состоянии молекулярного раствора. Для
оценки е таких растворов применяют уравнения (V.390) и (V.393),
поскольку более строгой теории еще нет. Величина е для смеси диок-
404
сан—вода, экспериментально полученная Акерлофом и Шортом
(1936), сравнивается с теоретическими кривыми на рис. V.64, хотя
теория Онзагера не может строго применяться к водным системам.
Из рис. V.64 видно, что теоретические и экспериментальные
значения е молекулярных растворов характерны и для дисперсных
состояний. Интересно отметить, что этот рисунок представляет
иллюстрацию двух методов оценки диэлектрических свойств систем,
показанных в табл. V.2 (см. стр. 325). Очевидно, что
крупнодисперсные системы (размеры частиц > 10"4 см) можно рассматривать
в свете диэлектрической теории гетерогенных систем, а гомогенные
системы (размеры частиц •< 10"6 см) — в свете молекулярной теории
диэлектриков.
Диэлектрические свойства солюбилизированных систем
представляют особый интерес, поскольку размеры их частиц являются
промежуточными. Подробнее об этом сказано выше (см. стр. 400).
ДИЭЛЕКТРИЧЕСКИЕ СВОЙСТВА СИСТЕМ
В ДИНАМИЧЕСКОМ СОСТОЯНИИ
Реологические свойства гетерогенных систем, таких как эмульсии
и суспензии, в динамическом состоянии представляют значительный
интерес.
Результаты экспериментальных исследований суспензий
Работы Партса. Значение информации о реологии при изучении
диэлектрических свойств дисперсных систем впервые отмечено Пар-
тсом (1945). При исследовании суспензий угольного порошка в лаках
он заметил, что емкость системы растет со временем после
прекращения движения в ней. На рис. V.67 сравниваются результаты,
полученные для двух образцов, один из которых был более тиксо-
тропен, чем другой. Парте предположил, что диэлектрические
свойства связаны с реологическими и образование агломератов является
причиной наблюдаемых характеристик.
Эмпирический анализ Воета. Влияние движущегося потока на
диэлектрическую проницаемость широко исследовал Воет (1947).
Он наблюдал диэлектрическое поведение нескольких видов
суспензий при поперечном потоке, вращающемся в вискозиметре
коаксиального типа.
На рис. V.68 показана зависимость е от концентрации, в
состоянии покоя и при сдвиге для суспензий угольного порошка в
различных органических средах.
При низких концентрациях минерального и льняного масел
изменение е после сдвига не обнаружено. При концентрациях >5%
минерального масла и >7% льняного величина е значительно
уменьшилась. В случае касторового масла понижение е наблюдалось
при любой его концентрации.
405
Уменьшение е при сдвиге Воет объяснил разрушением
агломератов суспендированных частиц, так как при низких концентрациях
в условиях ньютоновского потока, когда агломераты отсутствовали,
подобного изменения не наблюдалось. При высоких же
концентрациях в суспензиях обнаружено пластическое течение, указывающее
на наличие агломератов частиц. Воет проанализировал полученные
результаты, основываясь па уравнении (V.76).
20 г
50 100
Х.мин
Рис. V.67. Изменение
диэлектрической проницаемости
суспензии угольного4 порошка
в лаках со временем после
прекращения перемешивания
при частоте 1 кгц (Парте, 1945):
1 — концентрация угольного
порошка 14,2%, вязкость лака
562 сп.э; 2 — концентрация
угольного порошка 12,4%, вязкость лака
886 спв.
О 2 к 6 8 W
Объепная концентрация дисперсных частиц,%
Рис. V.68. Зависимость диэлектрической
проницаемости от концентрации дисперсной
фазы суспензии угольного порошка в кастё-
ровом масле (кривые 1, 1'), в льняном масле
(кривые 2, 2') и минеральном масле
(кривые 3, 3') при частоте 5 кгц (Воет, 1947):
Для суспендированных частиц с высоким значением е или к при
условии ет < ер и е < ер уравнение (V.76) упрощается:
(1 —Ф)3
Для низких концентраций (Ф < 1) оно примет вид:
е = ет(1 + ЗФ)
(Y.394)
(V.395)
Для несферических суспензий Воет ввел коэффициент а,
учитывающий несферическую форму:
е = Ет(1 + ЗаФ)
(V..396)
Очевидно, что для сферических дисперсных систем а=1, для
несферических — а > 1. В крайнем случае а < 1 например в
системах, содержащих пластино- или нитеподобные частицы,
перпендикулярно ориентированные к направлению электрического поля.
(Теория этого вопроса дана далее — стр. 409.)
406
Воет предположил, что уравнение (V.396) можно применять
к предельно низким значениям е при наивысшем сдвиговом
напряжении.
При статическом состоянии агломератов, уравнение (V.396)
преобразуется
e=sm (1 + ЗбаФ) (V.397)
где б — коэффициент агломерации.
Из примеров, показанных на рис. V.68, видно, что уравнение
(V.396) достаточно справедливо для трех систем в случае сдвига
при а = 3,9 независимо от среды. Отклонение значения а от единицы
означает, что частицы угольного порошка имеют несферическую
форму.
Величина 8, соответствующая разности значений е в состоянии
покоя и при сдвиге, зависит от концентрации и вида среды. Так,
значение 8 в касторовом масле оказалось довольно большим даже
при низких концентрациях, что указывает на значительный рост
агломератов частиц. Фактически в условиях сдвигового потока в этих
системах обнаружена большая подвижность значений б.
Измеряя е суспензий порошков цинка и двуокиси титана в
касторовом, минеральном и льняном маслах, Воет получил те же
результаты, что и для суспензий угольного порошка: для суспензий цинка
а = 1,4, для суспензий двуокиси титана а = 1,5.
При исследованиях суспензий алюминиевого и медного порошков,
форма которых представляла пластиноподобные листки, Воет
получил значения а значительно больше единицы: для суспензии
алюминия а. = 6,2, для суспензии меди а = 4,5.
Воет и Суриани (1952) изучали диэлектрические свойства
нескольких дисперсий пигментов в средах, диэлектрическая
проницаемость которых одного порядка с величиной диэлектрической
проницаемости частиц, например, милори синий, бензидиновый желтый,
хромовый желтый, монастраль синий в лаках, льняном и
минеральном масле. Эти исследования не дали значительной информации
о внутренней структуре дисперсий из-за недостаточности уравнения
(V.397) и довольно слабого влияния сдвигового течения в изучаемых
системах.
Дальнейшие исследования особенностей удельной
электропроводности и тангенса угла потерь суспензий угольного порошка
в условиях сдвига проведены Бонди и Пентером (1953) и Воетом
(1957).
Результаты экспериментальных исследований
эмульсий
Значительное влияние сдвига на диэлектрическую проницаемость
эмульсий обнаружил Воет (1947). На рис. V.69 показана
зависимость е от концентрации дисперсной фазы эмульсий 0,5 н. водного
раствора едкого натра в минеральном масле в случае покоя системы
и при сдвиге. Полученные данные хорошо согласуются с уравнением
407
(V.396) при а = 2,0. Крайне высокий коэффициент агломерации
установлен в случае статического состояния системы. На рис. V.70
дан график изменения б, полученного с помощью уравнения (V.397),
во времени сразу же после прекращения действия сдвиговой силы.
Диэлектрическая проницаемость продолжала изменяться в течение
5 мин.
Чтобы получить минимальное значение е, соответствующее
полному разрушению агломератов, понадобилась довольно высокая
скорость перемешивания:
> 100 об/мин.
Подобные результаты для
других эмульсий типа В/М полу-
0 5 10
Объемная концентрация
дисперсных частиц, %
Рис. V.69. Зависимость
диэлектрической проницаемости от концентрации
дисперсной фазы эмульсии 0,5 н.
раствора NaOH в минеральном масле
(Воет, 1947):
1 — покой; 2 — сдвпг.
Рис. V.70. Изменение коэффициента
агломерации 6 во времени после
прекращения перемешивания
эмульсии 0,5 н. раствора NaOH в
минеральном масле.
Объемная концентрация эмульспп 10 %
(Воет, 1947).
чены Ханаи (1961b), часть из которых приведена на рис. V. 36 и V. 37
(см. стр. 374). Согласно данным Ханаи, представленным на рис. V.41,
экспериментальные значения диэлектрической проницаемости 8/экс
Таблица V.9
Величина констант (5 и к в уравнении'* (V. 398)
при различных концентрациях Ф в эмульсиях В/М
(Ханаи, Коицуми, Гото, 1963)
ф
0,1
0,2
0,3
0,4
Р
0,43
5,0
15,7
38.5
k, об/мин
108
311
341
270
Ф
0,5
0,6
0,7
0,8
Р
104,0
135,0
253,0
441,0
к у об/мин
190
175
138
85
408
для эмульсий уменьшаются с ростом скорости перемешивания и
определяются следующей эмпирической формулой
8i3KC = 8/Teop+P«"iV/'' (V.398)
где £теор — значения диэлектрической проницаемости, полученные
из уравнения (V.233) для сферических дисперсных систем,
идентичные значениям, найденным из уравнения (V.394); р и к —
эмпирические константы, значения которых приведены в табл. V.9.
Величина (3 показывает влияние агломератов на диэлектрическую
проницаемость. Согласно результатам Воета, обычно к > 100 об/мин.
В экспериментах Ханаи величина е( экс была выше расчетных
значений 6; теор вплоть до 300 об/мин, что соответствует 39 сек'1.
Теоретический анализ
несферических дисперсных систем
Многими исследователями сделаны попытки дать теоретический
анализ диэлектрической проницаемости несферических дисперсных
систем (ссылки на их работы указаны выше — см. стр. 359).
Альтшуллер (1954) рассмотрел диэлектрическую проницаемость
разбавленных суспензий сфероидальных частиц (ер),
диспергированных в непрерывной среде с диэлектрической проницаемостью гт,
и рассчитал теоретический линейный коэффициент р
е = ет(1 + рФ) (V.399)
Воет предположил, что коэффициенты (3 и а относятся как
Р = 3а (V.400)
Установлено, что расчетные значения р зависят не только от
осевого соотношения суспендированных сфероидов и гр/гт, но и от
направления и степени ориентации относительно приложенного
электрического поля.
Выводы, сделанные Альтшуллером в результате теоретического
анализа, можно выразить через малую и большую оси эллипсоида.
Случай, когда гр > гт. 1. Для частиц, большая ось которых
ориентирована параллельно электрическому полю, значения
диэлектрической проницаемости (р > 3, а ]> 1 при ер ^> вт) выше,
чем в случае беспорядочной ориентации, и максимальны (р >3,
а >1), когда ер > ет.
2. При беспорядочной ориентации диэлектрическая
проницаемость принимает средние значения ф >3, а >■ 1 при ер 3> ет),
которые больше рассчитанных из уравнений (V.76) и (V.74) для
сферических частиц.
3. Для частиц с ориентацией большой оси перпендикулярно
электрическому полю значения диэлектрической проницаемости (Р <
<3; а <1) ниже, чем при беспорядочном распределении и в случае
сферических частиц, и минимальны (Р = 0, а = 0), когда ер = гт.
409
4. Значения диэлектрической проницаемости (р1 ]>3, а >> 1 при
гр У> £т) ПРИ беспорядочной ориентации выше, чем у сферических
дисперсий.
Случай, когда гр = гт. Диэлектрическая проницаемость всей
системы равна ет независимо от объемной доли, формы и ориентации
частиц.
Случай, когда ер < гт. 1. Для частиц с ориентацией большой
оси параллельно электрическому полю значения диэлектрической
проницаемости выше, чем при беспорядочной ориентации и в случае
сферических дисперсий, и максимальны, когда ер = гт.
2. При беспорядочной ориентации частиц, диэлектрическая
проницаемость принимает промежуточные значения, которые ниже
значений для сферических дисперсий.
3. Для частиц, большая ось которых перпендикулярна
электрическому полю, значения диэлектрической проницаемости меньше,
чем в случае беспорядочной ориентации и сферических дисперсий,
и минимальны, когда гр <^ ет.
4. При произвольной ориентации частиц значения
диэлектрической проницаемости ниже, чем у сферических дисперсий.
Комментарии по суспензиям
Влияние агломератов, формы, ориентации и деформации
дисперсных частиц на диэлектрические свойства систем можно рассматривать
в свете уже приведенных теоретических выводов.
В случае суспензий должны быть приняты во внимание влияние
агломерации, формы и ориентации частиц, в то время как в случае
эмульсий, ввиду сферической формы их частиц, существует влияние
только агломерации и деформации.
Поскольку влияние движущегося потока, т. е. уменьшение
диэлектрической проницаемости при сдвиге, наблюдается в системах
при условии ер ]> гт, ограничимся замечаниями только
относительно этого случая.
Обычно при проведении электрических измерений вискозиметр
вращающегося типа вызывает сдвиговый поток в направлении,
перпендикулярном электрическому полю, которое приложено между
чашкой и подвесом вискозиметра.
При сдвиге большие оси несферических частиц будут
ориентироваться в направлении сдвигового потока, перпендикулярном
направлению электрического тока, что соответствует условию
минимального сопротивления (см. случай А, п. 3 — стр. 409).
Влияние агломератов. Если агломераты несферических
суспендированных частиц находятся в статическом состоянии с
произвольной ориентацией, этот случай соответствует описанному выше (см.
случай А, п. 2).
При перемешивании агломераты распадаются на множество
отдельных частиц. Из теоретических предположений при сдвиге
естественно ожидать падение величины диэлектрической проница-
410
емости при условии, что отдельные частицы имеют более сферическую
форму по сравнению с их агломератами.
Форма и ориентация суспендированных частиц. Если при сдвиге
отдельные частицы ориентируются в направлении,
перпендикулярном электрическому полю, то ожидается, что значения
диэлектрической проницаемости будут ниже, чем у сферических дисперсий
(см. случай А, п. 3). Однако этого не наблюдается в случае,
показанном на рис. V.68 (а = 3,9), несмотря на близкую к сферической
форму угольного порошка по сравнению с алюминиевым и медным.
Таким образом, анализ а, сделанный Воетом, указывает на то,
что агломераты суспензий распадаются на отдельные частицы,
которые остаются неполностью ориентированными перпендикулярно
полю.
Комментарии по эмульсиям
Влияние агломератов. Как видно из рис. V.41, V.69 и V.70,
предельные значения диэлектрической проницаемости на низких
частотах сильно уменьшаются под действием сдвигового напряжения
вследствие разрушения агломератов, что и предсказывается
теоретически в случае (п. 2 и 4). Согласно результатам Ханаи (рис. V.37),
изменения диэлектрической проницаемости на высоких частотах
при перемешивании не обнаружено, когда условие ер ^> ет не
соблюдается.
Воет и Суриани (1952) и Альтшуллер (1954) отметили, что влияние
агломератов значительно уменьшается в системах, где е
суспендированных частиц одного порядка с е среды. По-видимому, влияние
агломератов на величину диэлектрической проницаемости ел
слишком мало (см. рис. V.37), чтобы быть заметным.
Деформация частиц. Деформация частиц эмульсий при высокой
скорости перемешивания подтверждена несколькими
исследованиями, такими как Зилберберг и Кун (1954), Барток и Масон (1959).
Если для измерений используют вращающийся вискозиметр,
то деформированные частицы в нем ориентируются таким образом,
что больший диаметр располагается перпендикулярно
электрическому полю. Согласно теоретическим предположениям (см. случай А,
п. 3), а <1, что противоречит полученному из уравнения (V.76)
значению а = 2. Возможно, положительное влияние агломератов
на величину а является достаточно большим, чтобы перекрыть
отрицательное влияние деформации на а. Фактически при исследованиях
эмульсий Воетом найдены большие значения 8 (см. рис. V.70),
полученные без учета влияния деформации.
Экспериментальные значения ez в исследованиях Ханаи
(рис. Л7.41) выше теоретических, что является следствием влияния
агломератов даже при скорости вращения 300 об/мин.
Выводы, сделанные по результатам разных исследований
дисперсных систем, можно свести к следующему:
411
1. Значительное уменьшение диэлектрической проницаемости
обусловлено, главным образом, наличием агломератов
диспергированных частиц, хотя убедительного теоретического объяснения
этому до сих пор не сделано.
2. Влияния, обусловленные наличием агломератов и
ориентацией частиц, все еще полностью не разделены.
3. Влияние деформации частиц на диэлектрическую
проницаемость предполагается весьма незначительным.
4. Чтобы объяснить влияние ориентации частиц необходимо
одновременное измерение двух диэлектрических прошщаемостей:
одной в направлении, параллельном электрическому полю, другой —
перпендикулярном ему (Якобсон, 1953).
* * *
При объяснении диэлектрических свойств некоторых коллоидных
систем удачноц оказалась только теория поляризации поверхности
раздела. Возможно, другие теории дают разумные объяснения
результатов, но большинство из них остаются на стадии
феноменологических объяснений или гипотез. Как отмечено (см. стр. 404),
соотношение между диэлектрическими свойствами жидкостей или растворов,
состоящих из полярных молекул, и суспензий сферических частиц
все еще ни теоретичЪски, ни экспериментально не освещено.
Дальнейшие исследования необходимы для определения критериев границ
применения электрических методов в коллоидных системах.
Большинство коллоидных систем, представляющих практический
интерес, имеют гидрофильную структуру. "Это затрудняет измерения
диэлектрической проницаемости и электропроводности и снижает
их точность. За последние двадцать лет измерительная техника
значительно улучшилась. Поэтому некоторые ранее изученные
системы нужно исследовать еще с помощью имеющейся более
совершенной техники.
Вопросы изменения диэлектрической проницаемости при чпере-
мешивании остаются нерешенными. В этой связи должны быть
ценными исследования молекулярных слоев, образующихся на
поверхности раздела.
Диэлектрический метод должен получить широкое применение
в области фундаментальных и практических коллоидных
исследований.
После подготовки этой монографии автор получил от профессора
Тобиаса следующие данные, опубликованные в работах: R. E.
Meredith, С. W. Tobias. J. Electrochem. Soc, 108, 286 (1961); R. E.
Meredith, С W. Tobias, Advances in Electrochemistry and
Electrochemical Engineering, Ed. by P. Delahay and С W. Tobias, vol 2, New
York, 1962, ch. 2, p. 15. В первой работе новое уравнение,
выведенное Мередитом и Тобиасом, определяет удельную
электропроводность эмульсии воды в пропиленкарбонате с достаточной точностью,
412
но соответствие между уравнением (V.115) и экспериментальными
значениями в эмульсиях М/В лучше.
В следующей обзорной работе рассмотрены теоретические
соотношения и проведено сравнение с экспериментальными данными
по электропроводности гетерогенных систем. Сделано также
заключение, что экспериментальные значения и в случае сферических
дисперсных систем соответствуют уравнениям Бруггемана
[уравнениям (V.114) — (V.116)] особенно для широкого распределения
частиц по размерам. Экспериментальные данные для кубически
упорядоченного распределения сфер хорошо выражаются
уравнением (V.349) Мередита и Тобиаса вплоть до 0,5 объемн.%.
ЛИТЕРАТУРА
А к е г 1 о f G., S h о г t О. A., J. Am. Chem. Soc, 58, 1241 (1936).
Altshuller A. P., J. Phys. Chem., 58, 544 (1954).
В а г t о к W, Mason S. G., J. Colloid Sci., 14, 13 (1959).
В е с h e г Р., Emulsions. Theory and Practice, New York, 1957, p. 77, 327.
Bondi A., Pent her С J., J. Phys. Chem., 57, 72 (1953).
В б t t с h e г С. J. F., Theory of Electric Polarization, New York, 1952, p. 419.
Brown W. F. В., Jr., J. Chem. Phys.. 23, 1514 (1955).
Bruggeman D. A. G., Ann. Phys., 24, 636 (1935).
Cole K.S., Cole R. H., J. Chem. Phys., 9, 314 (1941).
Cole R.H., Gross P. M., Jr., Rev. sci. Instrum., 20, 252 (1949).
С г a t i n P. D., R о b e г t s о п В. К., J. Phys. Chem., 69, 1087, (1965).
Curtis H.J., Fricke H., Phys. Rev., 48, 775 (1935b).
D e b у е P., Polare Molekeln, Кар. 3, Leipzig, 1929.
De L о о г G. P., Thesis, University of Leiden, 1956.
D г у d e n J. S., M e а к i n s R. J., Proc. Phys., Soc, 70B, 427 (1957).
Eichbaum B. R., J. Electrochem. Soc, 106, 804 (1959).
Errera J., Kolloid. Z., 31, 59 (1922).
Error a J., Kolloid. Z., 32, 157, 240, 373 (1923).
Errera J., Kolloid Z., 51, 104 (1930).
Fricke H., Phys. Rev., 24, 575 (1924).
Fricke H., Phys.. Rev., 26. 678 (1925a).
Fricke H., Phys. Rev., 26, 682 (1925b).
Fricke H., Phil. Mag., 14, 310 (1932).
Fricke H., J. Phys. Chem., 57, 934 (1953a.).
Fricke H., J. Appl. Phys., 24, 644 (1953b).
Fricke H., J. Phys. .Chem., 59, 168 (1955).
Fricke H., Curtis H. J., Phys. Rev., 47, 974 (1935 a).
Fricke H., Curtis H. J., J. Phys. Chem., 40, 715 (1936).
Fricke H., Curtis H. J., J. Phys. Chem., 41, 729 (1937).
Fricke H., J а с о b s о n L. E., J. Phys. Chem., 43, 781 (1939).
Fricke H., Parker E., J. Phys. Chem., 44, 716 (1940).
Frohlich H., Theorv of Dielectrics, Ch. 2, Oxford, 1949.
G о t о h R., H a n a i i'., Koizumi N., Nature (London), 181, 406 (1958).
Guillien R., Ann. phys., 16, 205 (1941).
Hamon B. V., Austr. J. Phys., 6, 304 (1953).
Hanai Т., Kolloid Z., 171, 23 (1960).
H a n a i Т., Kolloid Z., 175, 61 (1961 a).
Hanai Т., Kolloid Z., 177, 57 (1961b).
Hanai Т., Bull. Inst. Chem. Res. Kyoto Univ., 39, 341 (1961c).
Hanai Т., H а у d о n D. A., T а у 1 о r J., Proc Roy. Soc, A281, 377 (1964).
Hanai Т., Haydon D. A., TaylorJ., J. Theoret. Biol., 9, 278 (1965).
Hanai Т., К о i z u m i N., Gotoh R., Kolloid Z., 167, 41 (1959).
413
Hanai T. u. a., Kolloid Z.. 171, 20 (1960).
Hanai Т., К о i г u m i N., Gotoli R., Kolloid Z., 184, 143 (1962 a).
Hanai Т., Koizumi N., G о t о h R.. Bull. Inst. Chem., Res. Kyoto
Univ., 40, 240 (1962 b).
Hanai Т., Koizumi N., G о t о h R., in «Emulsion Rheology». ed. by
P.Sherman, Oxford, 1963, p. 91.
Hartshorn L., Ward W. H., Proc. Inst. Elect. Engrs, 79, 597 (1936).
Hey man E., Kolloid Z., 66. 229. 358 (1934).
Higuchi W. I., J. Phys. Chem., 62, 649 (1958).
von H i p p e 1 A. R., Dielectrics and Waves. New York — London, 1954 a.
von H i p p e 1 A. R., Dielectric Materials and Applications, New York —
London, 1954 b.
Irie F., Ann. Phys. [61, 19, 31 (1956).
Jacobson В., Rev. sci. Instrum., 24, 949 (1953).
Johnson G. A., Neale S. M., Kolloid Z., 185, 130 (1962).
Khar adlyM.M.Z., J ackson W.. Proc.Inst. Elec. Engrs, 100(111), 199(1953),
К irk wood J. G., J. Chem. Phvs.. 7. 911 (1939).
К u b о М.. Nakamura S.. Bull. Chem. Soc. Japan. 26, 318 (1953).
Lichtenccker K.. Phys. Z.. 27. 115 (1926).
Mandel M., Physica. 'sGrav.. 27, 827 (1961).
Maxwell J. C, Electricity and Magnetism, vol. 1, Oxford, 1892.
Meredith R. E., Thesis. University of California, Contract W-7405-eng-48,
1959.
Meredith R. E., T о b i a s С \Y.. J. Appl. Phys.. 31, 1270 (1960).
N a i к i Т.. F u j i t a K.. M a t s u m u г a S.. Mem. Fac. ind Arts Kyoto
tech. Univ., 8, 1 (1959).
Niesel \Y., Ann. Phys.. [6], 10. 336 (1952).
Niesel \V., Ann. Phys. [6], 12, 410 (1953).
Окег-Blom M., Arch. Ges. Physiol.. 79, 510 (1900).
О' К о n s к i С. Т., Gun the r R. L.. J. Colloid. Sci., 10, 563 (1955).
О' К on ski С. Т., Thacher H. С J. Phys. Chem., 57, 955 (1953).
Onsager L., J. Am. Chem. Soc, 58. 1486 (1936).
Parts A., Nature (London), 155. 236 (1945).
P а и I v H., Biophys.. 1, 113(1963).
Paul у H., Packer L., J. Biophys. Biochem. Cytol., 7, 603 (1960).
Paul v H., Рас k-e r L.. S с h w a n H. P.. J. Biophys. Biochem. Cytol.,
f, 589 (1960).
P a u 1 у H, Schwan H. P.. Z. Naturforsch., 14b, 125 (1959).
Реагсе С A. R., Br. J. Appl. Phys., 6, 113 (1955).
P err in F., J. Phys. Radium, 5, 497(1934).
Polder D., Van Santen J.H., Physica, 'sGrav.. 12, 257 (1946).
P о w 1 e s J. G., Smyth С. Р., in «Physical Methods of Organic Chemistry»,
- ed. bv A. Weissberger, Pt 3, Ch. 18, 19, New York — London, p. 2553,
2599.
Go pal E. S. R., J. Indian Inst. Sci., 40. 152 (1958).
Rayleigh, Phil. Mag., 34. 481 (1892).
Revnolds I.A., Hough J. M.. Proc. Phys. Soc, B70. 769 (1957).
d e 1 a R u e R. E., T о b i a s С W., J. Electrochem. Soc, 106, 827 (1959).
Runge I., Z. tech. Phys., 6, 61 (1925).
Schwan H. P., in «Advances in Biological and Medical Physics» ed by
J. H. Lawrence and С A. Tobias, vol. 5. New York — London, 1957,
p. 147.
Schwan H. P., M о г о w i t z H. J., Biophys J., 2, 395 (1962).
Schwan H. P. a. oth., J. Phys. Chem.. 66. 2626 (1962).
Schwa rz G., J. Phys. Chem., 66, 2636 (1962).
S с h w a r z G., S a i t о M., S с h w an H. P., J. Chem. Phys., 43, 3562 (1965).
von Schweidler E. R., Ann. Phys., 24, 711 (1907).
S i 1 b e r b e r g A, Kulm W., J. Polymer Sci., 13, 21 (1954).
Sillars R. W., Proc Inst. Elec. Engrs (London), 80, 378 (1937).
Smyth СР., Dielectric Behavior and Structure, New York, 1955.
414
Stewart G. N., J. Physiol., 24, 356 (1899).
S u g i u г a Y., К о g a S., J. Gen. Appl. Microbiol. (Tokyo), 11, 175 (1965 a).
Sugiura Y., Koga S., Biophys. J., 5, 439 (1965b).
S u g i u г a Y., К о g a S., Akabori H., J. Gen. Appl. Microbiol. (Tokyo)
10, 163 (1964).
Sumner С G., Clayton's The Theory of Emulsions and Their Technical
Treatment, London, 1954, p. 236, 241.
S u t h e i m G. M., Introduction to Emulsions, Brooklyn — New York, 1947
p. 15.
Thacher H. C, Jr., J. Phys. Chem., 56, 795 (1952).
Voet A., J. Phys. Colloid. Chem., 51, 1037 (1947).
Voet A., J. Phys. Chem., 61, 301 (1957).
Voet A., S u r i a n i L. R., J. Colloid Sci., 7, 1 (1952).
Wagner K. W., Arch. Electrotech., 2, 371 (1914).
Wiener O., Abh. sachs. Akad. Wiss., Math.-Phys. Kl., 32, 509 (1912).
Дополнительная литература
Chapman J.D., J. Phys. Chem., 72, № 1, 33 (1968). Влияние температуры
на диэлектрические свойства эмульсий воды в масле.
Dubois-Violette E., Parodi О., J. Phys. Colloq. (Paris), 4, 57
(1969). Нематические эмульсии. Магнитные поля и пьезоэлектрический
эффект.
Н о 1 z n е г G. W., Perfum. Essent. Oil Rec, 58, № 10, 714 (1967). Удельная
электропроводность и электролиз эмульсий.
К а у е R. С, S e a g e r H., J. Pharm. Pharmacol., 19, № 2, 78 (1967). Новый
метод определения при помощи диэлектрических измерений
распределения капель в эмульсии по размерам.
Беньковскии В.Г. пдр. Труды Института химии нефти и природных
соединений, Алма-Ата, 1970. Коалесценция и поведение эмульсий типа
М/В в однородном электрическом поле.
Григоров О. Н., Свердлова Н. С, Башенова А. В., Вестн.
ЛГУ. Сер. физ.-хим., 23, № 10, 94 (1968). Электрохимическая активность
концентрированных эмульсий, стабилизированных твердыми
эмульгаторами.
Панченков Г. М. и др., Хим. и технол. топлив и масел, № 2, 34 (1970).
Исследование движения заряженных водяных капелек в однородном
электрическом поле.
Панченков Г. М., Ц а б е к Л. К., ЖФХ, 42, № 8, 2027 (1968).
Колебания сферической капли эмульсии, помещенной во внешнее однородное
электрическое поле (коагуляция эмульсий).
Панченков Г. М., Ц а б е к Л. К., Поведение эмульсий во внешнем
электрическом поле, Изд. «Химия», 1969.
Панченков Г. М., Ц а б е к Л. К., Коллоида, ж., 31, № 6, 887 (1969).
Тепловая (броуновская) коагуляция эмульсии, возмущенная
гравитационным и электрическим полями.
Свердлова Н. С, Григоров О. Н., Вестн.- ЛГУ. Сер. физ.-хим., 23,
№ 16, 133 (1968). Электрохимическое поведение концентрированных
эмульсий керосина в воде, стабилизированных желатиной.
Дополнение
ЭМУЛЬГИРУЮЩИЕ
СВОЙСТВА
ПАВ
А. А. Абрамзон
В представленной книге
недостаточно полно отражено влияние
строения молекул ПАВ и фаз
на эмульгирующие свойства, а эти
проблемы являются центральными
в теории эмульсий. Цель данной
главы — восполнить, насколько это
возможно, указанный пробел и
рассмотреть ряд свойств эмульсий в
связи со строением ПАВ, опираясь,
в основном, на вновь вышедшие
работы.
Важнейшим свойством эмульсий
является их устойчивость. При
анализе устойчивости дисперсных
систем обычно исходят из
предположения лимитирования процесса
стабилизации или адсорбцией молекул
(частиц) стабилизаторов, или.
отталкиванием адсорбционных слоев.
Рассмотрим факторы,
определяющие стабильность эмульсий, исходя
из обеих предпосылок. Чтобы не
подпустить капли на расстояние,
при котором происходит коалесцен-
ция, молекулы или частицы
стабилизатора должны иметь определенное
строение, а также прочно и с плотной
упаковкой располагаться на
поверхности. При этом устойчивость
эмульсий обусловливается^ тремя
факторами [1]:
1. Геометрическим —
минимальные отрезки омолекул эмульгатора
в фазах (— 6 А) и определенное
соотношение размеров полярной и
неполярной групп.
2. Энергетическим—минимальная
прочность удержания ПАВ в
адсорбционном слое (работа адсорбции
— 4 ккал/молъ *).
* Следует отметить, что при 25° С
вещества существуют в жидком
состоянии, когда их изобарно-иаотермический
потенциал испарения достигает 3,5—
4 ккал/моль, т. е. это значение, когда
межмолекулярные силы обеспечивают
устойчивость жидкости.
4И.
3. Концентрационным — наличие насыщенного адсорбционного
слоя, предельная адсорбция, т. е. Г/Гмакс-+ 1 (здесь Г и Гызкс —
адсорбция и предельная адсорбция, соответственно).
Часто считают фактором устойчивости эмульсии двойной
электрический слой. Однако с этим согласиться нельзя, так как нет
даже качественной корреляции между существованием двойного
электрического слоя и свойствами эмульсий (см. стр. 92). Двойной
электрический слой определяет лишь толщину прослойки
непрерывной фазы между капельками эмульсии, как бы увеличивая длину
отрезка молекулы в водной фазе.
Естественной характеристикой стабильности эмульсий должна
быть константа скорости разрушения системы. Поскольку капли
коагулируют и коалесцируют попарно, константа должна
рассчитываться по уравнению второго порядка.
Для коагуляции это положение не подвергалось сомнению после
работ Смолуховского, но для коалесценции единого мнения нет.
Дэвис [2] принимает второй порядок. Ван ден Темпель утверждает
правильность уравнения первого порядка [3] для коалесценции.
Однако при проверке на строго монодисперсных эмульсиях (величина
элементарной капли экспоненциально влияет на стабильность
эмульсии [4]) показано [5], что коалесценция эмульсий описывается
уравнением второго порядка. Еще ранее второй порядок
коалесценции показал Грищенко [6] на жировых шариках молока. В наших
работах [5, 7] получена зависимость константы скорости
коалесценции К от объема фаз, что указывает на более высокий порядок
скорости, чем первый, так как Кх — l/сек, а К2 — л/(сек-число
частиц).
Таким образом
Тп0(п0 — х)
где х — число коалесценции ко времени т; п0 — число капель в
единице объема эмульсии в начальный момент.
СТАБИЛИЗАЦИЯ ЭМУЛЬСИЙ
НИЗКОМОЛЕКУЛЯРНЫМИ ПАВ
В соответствии с изложенными взглядами величина К должна
быть функцией прочности удержания ПАВ на поверхности, т. е.
работы адсорбции, плотности адсорбционного слоя, возрастающей
параллельно с концентрацией ПАВ, размера элементарной капли,
температуры. Если предположить, что процесс коалесценции
аналогичен реакции, описываемой теорией соударений, то зависимость К
от перечисленных параметров должна быть экспоненциальной.
Предэкспонента в этом случае является функцией физических свойств
фаз (таких как вязкость, плотность), а также соотношения объемов
фаз и величины капелек эмульсии.
27 Закяз 10 70
417
Явный вид данной функции найден экспериментально [5]. Так как
это имеет большое значение для возможности предсказания
стабильности эмульсий, обсудим подробнее анализируемую зависимость.
Определяли зависимости К по формуле (1) от указанных
параметров для двух типов эмульсий: искусственной монодисперсной,
состоящей из 50 строго одинаковых капель, и предельной
высококонцентрированной с узким дисперсионным составом.
Преимущество искусственной эмульсии — возможность строго
контролировать равновесную концентрацию ПАВ в фазах и иметь
определенный размер капель.
Однако такую эмульсию
можно получить только с
небольшим числом капель.
Предельная
высококонцентрированная эмульсия
является реальной системой
и в то же время с узким
дисперсионным составом.
На таких эмульсиях
получены [5] линейные
зависимости К от 1/VK и i/T
во всем исследованном
интервале (где VK — средний
объем капли эмульсии; Т —
абсолютная температура).
Зависимости К от работы
адсорбции W и
концентрации с имеют максимум
(рис. 1 и 2). На участке
до максимума зависимость
прямолинейна в координатах lg К — И7' , , где а — постоянная,
которая при равновесном значении концентрации равна постоянной
уравнения Лэнгмюра [5]: г
р 1 максс
~~ с-\-<х
Величина W изменялась в гомологическом ряду ПАВ, так что
зависимость на рис. 2 отражает также влияние длины углеводородной
цепи на стабильность эмульсии.
На участке до максимума эмульгирующей способности (так
называемого максимума Донана) формула для константы скорости
коалесценцип имеет вид
w
Рис. 1. Зависимость К от с эмульсий
гептана в воде, стабилизированных калиевы
ми солями:
1 — капринат;
— миристат;
пальмитат.
лаурат;
K=Ze к (2)
где Z и а — постоянные.
Следует обратить внимание на факт, замеченный Шерманом [8]:
механизм коалесценцип капель изменяется при диаметре капли
порядка микрона.
418
Остановимся на причинах появления максимумов
эмульгирующей способности. Интересно, что для объяснения этого явления
использованы те же физические предпосылки, что и при установлении
типа эмульсий. В настоящее время нет единой точки зрения ни по
тому, ни по другому вопросу. Все теории, посвященные данной
проблеме, можно разделить на два класса: 1) геометрические,
рассматривающие тип образующейся эмульсии как функцию геометрии
молекулы ПАВ; 2) энергетические, связывающие эти явления со
взаимодействием молекул ПАВ с жидкостями фаз (растворимость
и т. д.).
S 6
5 4
" стеорото = 630 ■ 10 '
I Кстеорота~550 10
В W,Kxon/tionb
Рис. 2.'Зависимость К от W эмульсий гептана
(кривая 1) и толуола (кривая 2) в воде,
стабилизированных калиевыми солями карбоновых кислот (сггав=
= 0,5 моль/'л).
Одна из первых теорий — теория ориентированных клиньев —
предусматривала искривление поверхности в сторону одной из фаз
вследствие различных диаметров «головы» и «хвоста» дифильной
молекулы ПАВ. Базировалась эта теория на фактах: соли
одновалентных металлов карбоновых кислот образуют прямые эмульсии,
а многовалентных — обратные.
В противовес этой теории приводили следующие данные [91:
1. Соли одновалентного серебра не стабилизируют прямые
эмульсии. Отметим, что довод этот не основателен, так как соли серебра
и карбоновых кислот не диссоциируют в водных растворах и слабо
гидратированы. Следовательно, они должны рассматриваться не
как «клин», а как «цилиндр» и, естественно, не могут стабилизировать
эмульсии.
27*
419
2. Радиус кривизны обычных эмульсионных капель в ~ 1000 раз
больше длины молекулы ПАВ. Поэтому эффект будто бы может
сказываться только на очень маленьких капельках [9]. Если
эмульсию стабилизирует насыщенный адсорбционный слой, то достаточно
отношение диаметра головы к диаметру хвоста, равное 1,005, чтобы
создать тенденцию к искривлению поверхности в сторону дисперсной
фазы. Величина (1,005—1) того же порядка, что и отношение радиуса
микронной капли к длине молекулы ПАВ — 20 А/5000 Л. В
последних работах [5, 10] показано, что только насыщенный адсорбционный
слой стабилизирует эмульсии против коалесценции.
Подтверждается теория клиньев п данными по влиянию изомерии
молекул ПАВ на тип эмульсии: нормальные изомеры образуют
прямые эмульсии, а разветвленные — обратные.
Другие объяснения базируются на влиянии растворимости ПАВ
на тип эмульсии и максимум Донана. Действительно, получило
широкое распространение правило Банкрофта. Высшие члены
гомологических рядов ПАВ плохо растворимы в воде и слабо
стабилизируют эмульсии (например, стеарат натрия). При повышении
температуры растворимость их увеличивается параллельно с
повышением эмульгирующей способности. Однако, например, в ряду
четвертичных аммониевых оснований при определенных
концентрациях все гомологи растваримы в водной фазе, но высшие
стабилизируют эмульсии хуже [11].
В настоящее время широкое распространение получила так
называемая система ГЛБ. Согласно этой системе, тип образующейся
эмульсии объясняется не из геометрии молекулы ПАВ, а с помощью
энергии взаимодействия ее с фазами в зависимости от соотношения
растворимости в разных фазах. Но система ГЛБ базируется на
положениях, которые не подтверждаются для многих систем, и не
учитывает влияния геометрических особенностей молекул ПАВ,
влияющих на тип эмульсии.
Таким образом, причины образования того или иного типа
эмульсии и максимума Донана до сих пор окончательно не установлены.
Требуются дальнейшие поиски, чтобы выявить справедливость
геометрического или энергетического объяснения, а возможно, и
совместить их. Последнее кажется весьма вероятным.
Приведем одно из возможных объяснений, базирующееся на
совмещении геометрической и энергетической концепций.
Поверхность раздела фаз должна искривляться в какую-либо
сторону молекулами ПАВ, но адсорбционный слой нельзя
рассматривать как статический. Молекулы ПАВ поверхности и объема
непрерывно обмениваются, причем преимущественно с той фазой, в которой
растворимо данное ПАВ. Ориентировочный расчет скорости
обновления поверхности дает среднее время нахождения молекулы на
границе раздела фаз, составляющее—5 • 10'7 сек [12, 13].
Естественно, что при этом адсорбированные молекулы несколько втянуты
в фазу, в которой они растворимы, и это оказывает влияние на
тенденцию к изгибу адсорбционного слоя в сторону непрерывной фазы.
420
Среднестатистическая сдвинутость адсорбционного слоя внутрь фазы
по сравнению с поверхностью раздела чистых жидкостей
подтверждается исследованием энергетики процесса адсорбции в системах
жидкость — жидкость [14]. Приведенное объяснение показывает
влияние и растворимости, и геометрии молекул ПАВ на тип эмульсии.
Аналогично объясняется [11] и максимум Донана. При удлинении
углеводородной цепи растворимость ПАВ понижается, молекулы
углубляются во вторую фазу, и при каком-то числе углеродных
атомов в цепи отрезки молекул в фазах сравниваются, поверхностный
слой не искривляется и эмульгирующая способность понижается.
Стабильность является одним из основных свойств эмульсий,
однако недостаточным для полной характеристики, так как
необходимо знать геометрические и концентрационные параметры системы,
т. е. размер капель и концентрацию их. Эти параметры зависят
от метода получения и физических свойств гетерогенной системы
(поверхностного натяжения, вязкости, плотности фаз и т. д.).
Результаты дисперсного анализа и соотношение объемов непрерывной
и дисперсной фаз наиболее полно характеризуют эти параметры.
Зная объем дисперсной фазы V^ и общее число капель эмульсии п
легко получить средний объем капли, входящий в уравнение (2):
Для характеристики ПАВ в качестве эмульгатора удобно
использовать значения введенной Бромбергом [15] и Кремневым [16]
площади поверхности предельной эмульсии iSM, которую можно
рассчитать по формуле [7]
с .... соус .„.
^ сю — ^ (*)
1 макс
где с0 — начальная концентрация ПАВ в непрерывной фазе; Vc —
объем непрерывной среды.
Эта формула верна во всех концентрационных пределах.
Указывалось [7, 17], что зависимость Sco от концентрации эмульгатора
ограничена в области, высоких концентраций, однако этот вывод
сделан из-за ошибочной методики проведения дисперсионного
анализа, когда все мелкие капли принимались равными микронным.
При высоких концентрациях ПАВ в некоторых системах имеются
капли меньше микрона в диаметре.
Таким образом, величина Soz является показателем того, какую
межфазную поверхность может стабилизировать данное ПАВ
в эмульсии.
Для характеристики ПАВ весьма показательна концентрация,
при которой ПАВ начинает стабилизировать эмульсии — се [7].
Эта концентрация оказалась равной той, при которой достигается
предельная адсорбция [1, 10]. Следовательно, се можно с
достаточным приближением рассчитать по уравнению адсорбции Лэнгмюра:
с* = 77 ГГ\ Г ПР" Г *" ^"акс (4)
\' макс/-' ) 1
421
Итак, для выбора ПАВ в качестве эмульгатора необходимо и
достаточно знать три показателя [71:
1) характеристику стабильности эмульсии — константа
скорости коалесценции; 2) показатель величины межфазной
поверхности эмульсии, которую может стабилизовать данное ПАВ —
поверхность предельной высококонцентрированной эмульсии;
3) предельную концентрацию, выше которой ПАВ является
стабилизатором эмульсии.
Для характеристики получаемой эмульсии необходимо иметь
еще результаты дисперсионного анализа и данные о концентрации
капель эмульсии, от которых в свою очередь зависит стабильность
эмульсии, как видно из уравнения (2).
СТАБИЛИЗАЦИЯ ЭМУЛЬСИЙ
ВЫСОКОМОЛЕКУЛЯРНЫМИ ПАВ
Высокомолекулярные поверхностно-активные вещества (ВМ ПАВ)
следует выделить в отдельную группу стабилизаторов эмульсий.
Обусловлено это тем, что структура их на поверхности иная, чем
дифильных низкомолекулярных эмульгаторов. Отличают их и
внешние показатели. Так, для стабилизации единицы"межфазной
поверхности эмульсий с помощью высокомолекулярных соединений (ВМС)
требуется значительно больше ПАВ, зато эмульсия в этом случае,
как правило, значительно стабильнее эмульсий на дифильных
эмульгаторах типа мыл. Диспергируются же фазы с помощью последних
лучше вследствие более значительного понижения поверхностного i
натяжения.
Природа создала совмещенный эмульгатор — белково-фосфо-
липидный комплекс [181, включающий оболочечный белок и лецитин
или кефалин и имеющий преимущества обоих классов эмульгаторов.
Такими оболочками стабилизированы жировые шарики молока,
каучуковые частицы млечного сока (латекса), оболочки
эритроцитов и т. д.
ВМ ПАВ широко распространены как стабилизаторы в природных
и синтетических эмульсиях. Такие стабилизаторы как желатина,
агар-агар, сапонин известны давно, однако теория ВМ ПАВ развита
слабо. Наибольшее число фактов собрано об оболочках жировых
шариков молока [181.
В последнее время особое значение приобрели оболочечные
структуры в связи с развитием техники получения микрокапсюль, которые
представляют собой защитные оболочки из ВМС различной степени
плотности и проницаемости.
Характерным отличием многих ВМ ПАВ является необратимая
адсорбция их на межфазной поверхности. Сливки, например, можно
промывать водой, а жир экстрагировать эфиром. Оболочка будет
сохранять свою форму [18]. Прямые эмульсии, стабилизированные
поливиниловым спиртом (ПВС), можно разбавить водой, при этом
422
не наблюдается коалесценции. Кульман [19] на индивидуальных
каплях показал необратимый характер адсорбции ЛВС.
В других отношениях адсорбция ВМ ПАВ также отличается
от адсорбции обычных ПАВ. Если равновесие на поверхности в
случае низкомолекулярных ПАВ устанавливается, как правило, за
несколько минут, то структура ВМС — за несколько часов [20, 21],
что объясняется сложностью последней.
В зависимости от условий молекулы ВМ ПАВ в предельных
случаях могут располагаться как вертикально, так и горизонтально
на поверхности. Однако при высоких концентрациях наиболее
устойчиво промежуточное состояние и для переведения молекул
ВМ ПАВ в вертикальное положение необходимо длительное боковое
поджатие [22]. Классические методы определения ориентации на
межфазной поверхности не применимы к ВМ ПАВ. Предложены
другие методики [22], основанные на том, что после определенного
предела молекулярный вес ВМ ПАВ не влияет на их поверхностно-
активные и эмульгирующие свойства [4, 22, 23].
Структура ВМС на поверхности раздела фаз эмульсий. Если
на свободных поверхностях жидкость — жидкость и жидкость — газ
возможна любая ориентация в зависимости от условий, то на
поверхности стабильных эмульсий молекулы ВМ ПАВ образуют
трехмерную сетку определенных параметров [4, 24]. При исследовании
жировых шариков молока разработаны [25, 26] методики выделения
оболочечных защитных структур, состоящих из белковых молекул
и имеющих под микроскопом вид сот размером в несколько микрон.
Такие структуры представляют собой беспорядочное
переплетение цепей полимеров [27]; участки цепей образуют сетку с ячейками
порядка десятков ангстрем.
Получены химические доказательства сетчатой структуры ВМ
ПАВ на поверхности раздела стабильных высококонцентрированных
и концентрированных эмульсий [4], а также определены
геометрические показатели этих структурных образований. Методика работы
была следующей. Из 1 мл водного раствора частично ацетилирован-
ного поливинилового спирта определенной концентрации получали
высококонцентрированную предельную эмульсию по методике
Кремнева [28]. Микроскопически определяли площадь поверхности
всех капель эмульсии (Sea). Эта величина по мере возрастания
концентрации ВМ ПАВ проходит через максимум, лежащий в области
— 5% ПВС в воде. До этого значения концентрации поверхность
эмульсии, отнесенная к общему числу звеньев полимера в 1 мл
раствора, остается постоянной, а следовательно, и толщина
адсорбционного слоя (б) сохраняет постоянное значение. При
концентрациях более высоких, чем ~ 5% ПВС величина Sот понижается,
а б резко возрастает.
Подтверждают полученные данные исследования толщин водных
черных пленок, стабилизированных ПВС [29]. Определенное
значение порядка 1000 А хорошо совпадает с полученным при
исследовании эмульсий значением [4].
423
Ланкфельд и Ликлема [23] при исследовании эмульсий,
стабилизированных ПВС, определили площадь, приходящуюся на одну
молекулу, которая оказалась близкой к найденной нами [4].
Таким образом, минимальная толщина прослойки между каплями
эмульсии и площадь поверхности, приходящаяся на молекулу ВМ
ПАВ, найденные различными методами и авторами, совпадают
(табл. 1).
Таблица 1
Сравнение толщины адсорбционного слоя
и площади эмульсии на молекулу ПВС
по данным различных авторов
Авторы
Площадь эмульсии
на молекулу, А2
Толщина
адсорбционного слоя, А
Абрамзон и Громов [4]
Измайлова п Зонтаг [29]
Лпклема п Лапкфельд [23]
300-2000
900-3000
700-1900
750-1500
Молекулы ВМС в адсорбционном слое не могут иметь ни
вертикальной, ни горизонтально-!! ориентации по чисто геометрическим
параметрам, так как толщина прослойки между каплями меньше
длины молекулы ВМ ПАВ, а на площади, приходящейся на одну
молекулу ПАВ, может разместиться меньше 10% всех звеньев.
Следовательно, возможна или сетчатая структура, построенная
из беспорядочно переплетенных цепей полимерных молекул, или '
структура из свернутых в клубок полимерных молекул. Последнее
исключается, так как показано [4] на разных образцах полимера,
что эмульгирующие свойства ВМС не зависят от молекулярного
веса в случае одинаковой дисперсности эмульсий.
Имеется и другое доказательство в пользу сетчатой
структуры [24]. В защитном слое непредельной эмульсии иногда
присутствуют молекулы ВМС, не соприкасающиеся с поверхностью раздела
фаз. Доказывается это опытами по адсорбции на капельках
разбавленной эмульсии, которую определяли по понижению межфазного
натяжения в системе жидкость — жидкость до и после
эмульгирования. В ряде случаев адсорбировалось такое количество ВМ ПАВ,
которое не могло разместиться на поверхности даже при насыщенном
мономолекулярном слое.
Ланкфельд и Ликлема [23] полагают, что ВМС образуют на
границе раздела фаз переплетающиеся петли, удерживающиеся
на поверхности небольшим числом звеньев.
Стабилизирующие свойства структур ВМ ПАВ. Зависимость
стабилизирующих свойств частично ацетилированных
поливиниловых спиртов от молекулярного веса и числа гидрофобных групп
изучена в достаточно широких пределах. При этом все исследования
проведены на эмульсиях с одинаковым дисперсионным составом [4,
24], так как стабильность экспоненциально зависит от среднего
42!
размера капель [5]. Следовательно, по этим данным можно судить
о сравнительной стабилизирующей способности исследованных ПАВ.
Если же синтезировать эмульсии в одинаковых условиях
перемешивания, то с образцами разной поверхностной активности будут
получаться эмульсии различной дисперсности.
Исследования долгоживущих эмульсий толуола в воде
одинаковой дисперсности показали [24], что устойчивость их не зависит
от молекулярного веса ПВС и процентного содержания ацетатных
групп. В условиях же перемешивания стабильность зависит от
скорости адсорбции и поверхностной активности, та в свою очередь —
от процентного содержания ацетатных групп, а скорость
адсорбции — от молекулярного веса.
Сопоставление эмульгирующих свойств различных ВМ ПАВ
позволяет сделать вывод [27], что во всех случаях ВМС
стабилизируют эмульсии, образуя трехмерную сетчатую структуру с очень
близкими геометрическими свойствами. Существование этой
структуры подтверждается независимыми методами и результатами
различных исследователей.
Ниже приведены данные о количестве высокомолекулярного
эмульгатора, приходящегося на 100 см2 поверхности эмульсии
в.оптимальном защитном слое:
Количество
эмульгатора
Эмульгатор на 100 см*
межфазной
поверхности, мг
Агар-агар . . . . 0,4
Желатин 0,19
Казеин 0,08
Натриевая соль метилфталил целлюлозы 0,2
Оболочечные белки жировых шариков молока .... 0,04
Поливинилбутираль 0,24
Поливиниловый спирт 0,09
Поливинилформаль 0,21
ПоливинилциКлогексанон 0,15
Сапонин 0,03
Частично ацетилированпый ПВС 0,03—0,07
Интересно, что ВМС во всех случаях подчиняются правилу Банк-
рофта в противоположность низкомолекулярным ПАВ. Это
объясняется преимущественным образованием структуры с одной стороны
поверхности раздела фаз.
Априорным требованием к ВМ ПАВ является наличие в цепи
чередующихся гидрофильных и гидрофобных групп, которые
предопределяют как адсорбцию ВМС на границе раздела, так и
образование сетчатых структур у поверхности.
Свойства структуры ВМ ПАВ. Прочность адсорбционных слоев
сапонина исследована в работе Трапезникова и Зотовой [30]. В серии
работ Ребиндера и Измайловой с сотрудниками [29] изучены
реологические свойства поверхностных слоев белков и поливиниловых
спиртов, а также показан параллелизм между прочностными
425
свойствами межфазных слоев макромолекул и устойчивостью капель
углеводородов, стабилизированных этими слоями. '
Однако для создания теории недостаточно знания геометрии
защитных слоев и их прочностных свойств. Цель теории ВМ ПАВ —
предсказать, какие ВМС и как нрочно стабилизируют эмульсии.
Необходимо определить количественную связь между строением
молекулы ВМ ПАВ и ее стабилизирующими свойствами. Эти вопросы
ждут своего разрешения.
ЗАВИСИМОСТЬ АДСОРБЦИИ
В СИСТЕМЕ ЖИДКОСТЬ — ЖИДКОСТЬ
ОТ СТРОЕНИЯ МОЛЕКУЛЫ ПАВ
В формуле (2) для константы скорости коалесценции свойства
фаз и структура ПАВ влияют на W и а. Значение последней само
является функцией W, так как известно [31], что
' -"'
Другие величины в формуле (2) связаны следующим образом:
Z — с концентрацией дисиерсной фазы, видом эмульсии и вязкостью 1
фаз [5], Ук — с поверхностным натяжением и интенсивностью
диспергирования, а также с вязкостью и плотностью фаз.
При адсорбции ПАВ на границе раздела жидкость — жидкость
закономерности несколько изменяются по сравнению с адсорбцией
в системе жидкость — газ. Характерной особенностью
рассматриваемых систем является возможность адсорбции с обеих сторон границы
раздела. В связи с этим понятие поверхностной активности
перестает быть однозначным и возникает вопрос об оценке ее по
отношению к каждой из граничащих жидкостей. В работах Ребиндера [32]
и Таубмана и Бурштейн [33] указано на резкое различие
поверхностной активности разных фаз. Показано, что адсорбция со стороны
полярной и неполярной фаз осуществляется в результате перехода
различных частей дифильной молекулы. Это предопределяет разную
энергетическую основу процесса адсорбции из каждой фазы.
Величину работы адсорбции можно получить как из
экспериментальных данных, так и расчетным путем пз значений энергии
взаимодействия различных частей адсорбирующихся молекул с полярной
и неполярной фазами.
В общем случае работу адсорбции определяют как разность
стандартных химических потенциалов ПАВ (|х°) на поверхности
и в объеме фазы. Выразив концентрации ПАВ в фазах через
мольные доли N и приравнивая поочередно потенциал вещества в
межфазном слое и в каждой из фаз, получим
N.
W* = И°в - V-% = Л Т In —f- -f- Лаб макс (5)
426
где а — поверхностное натяжение; 5макс = 1//\,акс — площадь моля
ПАВ в предельном поверхностном слое; индексы в, о, S относятся
к воде, органической фазе и поверхности.
Ns
W0 = h°-vl% = RT In-jf--AaSM3KC (6)
Произведение Дст5макс, согласно Жуховицкому [34], отражает
изменение свободной поверхностной энергии при адсорбции.
Очевидно, при с —>- О, величина AoSUiKC —>- 0 и выражения (5) и (6)
примут вид уравнений работы адсорбции Лэнгмюра
WB=RTln^L = RTlliJ_ (7)
W0 = RT In -J^ = RT In -L- (8)
Ребиндером [32] показано, что в случае распределения ПАВ
между фазами может применяться уравнение Гиббса, причем расчет
адсорбции может вестись по любой из граничащих фаз. Он нашел,
что при равновесном распределении ПАВ
да _ да
Следовательно, расчет адсорбции можно вести по любой фазе.
В то же время из этого выражения следует, что при св Ф с0
поверхностная активность, а, следовательно, и работа адсорбции будут
различными для разных фаз, несмотря на то, что а имеет одно и то же
значение.
В формулу работы адсорбции входит толщина поверхностного
слоя б. Показано [35], что толщина слоя жидкости, определяющего
поверхностное натяжение, составляет 6 А. На этом основании для
толщины межфазного слоя на границе раздела двух жидкостей
принята [38] величина 12 А, так как адсорбция определяется по
изменению поверхностного натяжения, а все то, что лежит за
границей этих 12 А, не влияет на поверхностное натяжение.
Уравнения (7) и (8) получены для идеальных бесконечно
разбавленных .растворов. Поэтому при их использовании необходима
экстраполяция к нулевой концентрации ПАВ.
При адсорбции молекулы ПАВ на поверхности раздела фаз будет
изменяться энергия взаимодействия той части молекулы, которая
перешла из одной фазы в другую. Изменением поверхностного
натяжения в результате адсорбции можно пренебречь, так как при с —>■ О
и До -> 0. Таким образом, для работы адсорбции дифильной
молекулы из водной фазы имеем
WB = Gl-05
где G% — G" — разность свободных энергий неполярной части
молекулы в водной и органической фазах.
427
Из органической фазы
W'Q=Gg-Gg
где GJS — GS — разность свободных энергий полярной части
молекулы, перешедшей из одной фазы в другую.
Таблица 2
Значения свободной энергии испарения (AG„)
и взаимодействия (AG0) различных групп с растворителями
Группа
-СН
—I
-Вг
-С1
СвЩ—
-он
-N0-2
Чс = о
-О-
—с=с-
AG„,
и1
ккал/малъ
0,3
3,0
2,4
1,8
4,3
3,8
4,1
2,3
0,7
0,3
вода
—0,1
0,7
0,6
0,6
0,4
5,1
3,5
3,8
2,0
—
AGo, '>кя-1 'моль
парафины
0,3
2,6
, 2,1
1,6
4,0
20
2,0
1.5
0.4
0,5
бензол
0,28
3.0
2,4
1,8
4,3
2.3
3,5
2,2
0.7
0,i
•
сец
0.3
2,9
2,1
1,7
4.3
2,2
2,9
1,9
0,6
—
cs2
3,0
2,3
1,5
4.1
2,5
3,2
1,3
0,3
Значения свободной энергии различных групп органических
соединений в разных растворителях приведены в табл. 2 [36].
В качестве примера
рассмотрим закономерности
взаимодействия гомологического
ряда спиртов (ПАВ) с
органическими растворителями и водой
[36]. На рис. 3 приведены
зависимости коэффициентов
активности у спиртов и
растворителей от мольной доли Лг
компонентов в системе.
Коэффициент активности связан со
свободной энергией
растворения уравнением [37]:
AG=— ПТ\пу
Величину AG определяли
при бесконечном разбавлении
данного вещества в растворе,
Рис. 3. Зависимость lg Y1/V2 от состава т е по отрезку, отсекаемому
раствора для систем: на оси ординат Полученные
О - зТаноЛл__пентан;л. - этанол - гексан: ЗНаченИЯ Приведены В табл. 3.
428
Таблица 3
Изменение свободной анергии спиртов
при растворении (ДСр) в органических жидкостях
Спирт
Метанол
Этанол
Пропанол
Бутанол
Октанол
Додеканол
Среднее
циклогексан
-1,84
-1,90
-1,80
-1,85
AG_. ккал/молъ
парафиновые
углеводороды
-1,65
-1.68
-1,72
-1,60
-1,50
-1,65
четырех-
хлор истый
.углерод
-1,'б0
—1,50
-1,50
-1,45
-1,51
бензол
-1,50
-1.46
-1.50
—1,37
—1,46
Из дачных рис. 3 видно, что при растворении спирта в различных
углеводородах зависимость одна и та же.
На рис.4 представлена зависимость lg7i/72 0T мольной доли
различных спиртов в определенном алифатическом углеводороде.
Из данных рисунка и табл. 3
видно, что значение AG для всех
спиртов очень близко, т. е.
величина взаимодействия гид-
роксильной группы различных
спиртов в данном растворителе
практически одна и та же.
Кроме того, lg у растворителя
в различных спиртах регулярно
уменьшается от метанола к
октанолу. Следовательно,
энергия взаимодействия
растворителя со спиртами повышается
с увеличением числа метилено-
вых групп в спирте, т. е.
уменьшением влияния
полярной группы.
На рис. 5 приведена
аналогичная зависимость для систем
алифатический спирт — вода,
из которой видно, что спирты
взаимодействуют с водой
по-разному. Различие рис.4 и 5
объясняется одинаковым
взаимодействием углеводорода и
алифатической группы спирта.
Рис. 4. Зависимость lg yjy2 от состава
раствора для систем:
О — метанол — парафиновые углеводороды;
Д — этанол — парафиновые углеводороды;
X — пропанол — парафиновые
углеводороды; а — бутанол — парафиновые
углеводороды; 9 — октанол — парафиновые
углеводороды.
429
Приведенные зависимости являются общими для разных
классов ПАВ [36].
Проанализируем процесс адсорбции, в котором алифатическая
цепь молекулы ПАВ переходит из водной фазы в органическую, а
полярная группа остается в воде. Изменение энергетического состояния
при этом можно оценить на основе данных о растворимости
углеводородов в воде. Свободная энергия растворения .
углеводородов аддитивна и равна на атом водорода 0,42 ккал [371.
Следовательно, работа адсорбции из водной фазы в системе вода —
насыщенный углеводород
может быть определена
расчетным путем с помощью
выражения [38]
WB = 0,42«H (9)
где ?гн — число атомов
водорода в углеводородной цепи,
перешедшей при адсорбции
в органическую фазу.
Очевидно, что
наибольшее значение работы
адсорбции для каждого соединения
независимо от типа
полярной группы будет
соответствовать переходу в
органическую фазу всей
углеводородной цепи. Показано [38],
что в системе алифатический
углеводород — вода
экспериментальная и рассчитанная
по уравнению (9) величина W
для органических кислот
и спиртов нормального
ряда, совпадают. При расчете
принимали, что вся угле-
органической фазе, а полярная
Рис. 5. Зависимость lg V1/Y2 от состава
раствора для систем:
О — метанол — вода; Д — этанол — вода; X —
пррпанол — вода; с — бутанол — вода.
водородная цепь находится в
группа — в водной.
Иные результаты по сравнению с кислотами и спиртами полу1
чены при изучении адсорбции солей карбоновых кислот, алкил-
сульфонатов- и сульфатов натрия. Для этих соединений
экспериментальные значения работы адсорбции значительно ниже
полученных по формуле (9). Возможными причинами расхождения могут
являться следующие. В случае адсорбции солей вследствие их
хорошей растворимости в воде часть алифатической цепи может быть
втянута в водную фазу. С увеличением общей длины цепи значения
работы адсорбции солей возрастают, но в меньшей степени, чем
у кислот и спиртов, т. е. увеличивается число метиленовых групп
в обеих фазах.
430
С другой стороны, более низкие значения W могли быть
результатом некоторого повышения межфазного натяжения в результате
влияния отрицательно поверхностно-активных ионов. Однако
дополнительное введение солей в систему с ионогенными ПАВ
значительно понижает поверхностное натяжение [14]. Таким образом,
это предположение не позволяет удовлетворительно объяснить
полученные данные. Предпочтительной является, очевидно, точка зрения
о частичной втянутости молекул ПАВ в водную фазу.
Изменение природы катиона в пределах I группы периодической
системы, несмотря на известное различие степени их гидратации,
на поверхностной активности солей карбоновых кислот заметно
не отражается, в то время как различные соли фторзамещенных
карбоновых кислот отличаются по своей поверхностной
активности [39].
Сопоставление экспериментальных (И^ЭКСп) и расчетных (WpaC4)
значений работы адсорбции солей позволяет установить, какая
часть углеводородной цепи погружена в водную или органическую
фазы [14]:
^расч " эксп
042
(Ю)
Значения эти для нескольких классов ПАВ приведены в табл. 4.
Таблица 4
Показатели адсорбции ПАВ
ПАВ
Алкплсульфаты
Алкплсульфонаты
Окспэтплцро ванные
алкплфеполы [41]
Соли четвертичных
аммониевых
оснований
Щелочные соли
карбоновых кислот
И' в системе
"м
парафиновый
углеводород— •
вода, ккал/моль
0.24
0,29
Из гептана в
воду на оксп-
этпльную
группу 0,44
0,26
0,24
пмна
максимуме
Донана
12
14
—
16
16
пм последнего
эмульгирующего члена
ряда
14
16
—
18
18
Отметим, что введение высаливающих реагентов практически
не влияет на адсорбцию кислот и спиртов, что может служить
подтверждением перехода в органическую фазу всех звеньев их цепей.
Адсорбцию из органической фазы следует рассматривать как
результат перехода полярной группы ПАВ из органической в водную
фазу. Таубман [33], Позднышев и Петров [40, 41] указывают, что
431
при этом работа адсорбции будет предопределяться изменением
энергетического состояния полярных групп (WQ = G% — G") и не
должна зависеть от длины алифатической цепи. Иными словами
в пределах гомологического ряда работа адсорбции должна быть
практически постоянной, что в действительности наблюдается для
карбоновых кислот и спиртов [42, 43]. Типичный вид изотермы
поверхностного натяжения по обеим фазам приведен на рис. 6,
который показывает разницу в гомологическом ряду поверхностной
активности по водной фазе и отсутствие ее по органической [38].
Ч,,морь/л 0,05 ° °-05 сt.моль/л
Рис. 6. Изотерма поверхностного натяжения в системе
гептан — вода — жирная кислота как функция концентрации
кислот в водной (св) и органической (с0) фазах.
Кислота: Н —пропионовая; 0 —масляная; х —валериановая;
* — капроновая; # — онантовая; Д — лауриновая.
Рассмотрим зависимость работы адсорбции от разности
полярностей граничащих фаз. Показано [14], что работа адсорбции связана
с межфазным натяжением линейной зависимостью:
WB = 0,42«н - (5о - о) 5макс (11)
И7о-^о-(50-а)6'макс (12)
где W0 — работа адсорбции в системе парафин —вода;
о—межфазное натяжение в отсутствие ПАВ.
Таким образом, в ряде случаев можно теоретически рассчитать
работу адсорбции или, зная ее величину для одной системы,
предсказать ее значение для другой.
Уравнения (7), (8) и (11), (12) получены для бесконечно
разбавленных растворов. Позже показано [44], что значение работы
432
id 40
адсорбции для различных классов ПАВ сохраняется постоянным
в широком концентрационном интервале в случае расчета по
уравнениям (5) и (6). Эти значения равны полученным по уравнениям (7)
и (8).
ЗАВИСИМОСТЬ ПОВЕРХНОСТНОГО НАТЯЖЕНИЯ
ОТ СТРОЕНИЯ МОЛЕКУЛ ФАЗ
Известно, что процесс эмульгирования и свойства эмульсий
зависят от поверхностного натяжения фаз и межфазного натяжения
гетерогенной системы. Поверхностное натяжение а является одним
из основных параметров, определяющих диспергируемость системы.
Чем ниже а, тем мельче капли эмульсии при определенном
перемешивании и тем стабильнее система.
Другой параметр, определяющий
стабильность эмульсий, — работа
адсорбции W. Из уравнения (11)
видна прямолинейная
зависимость W от а.
Рассмотрим взаимосвязь
поверхностного натяжения со
структурой молекул. В простейшем
случае, когда силовые поля
молекул жидкости «симметричны»,
химических связей (типа
водородных) между молекулами
граничащих фаз не образуется,
жидкости практически не
смешиваются (низкое давление
насыщенного пара в системе
жидкость — газ), а определяется
лишь разностью полярностей
фаз ЛЯ, которая является функцией диэлектрической
проницаемости е. Ребиндер [45] показал параллелизм между е и а жидкостей.
Зависимость о — е криволинейна, но выпрямляется в координатах
(рис. 7) а — ЛЯ, где Я = ^~--
Другой характеристикой энергии взаимодействия является
теплота испарения ЛЯИ. Поскольку величина о отнесена к единице
площади (1 см2), то и теплоту испарения следует отнести к единице
объема (1 см3), а не к 1 г-мол.
На рис. 7 приведена зависимость с — ДЯ для систем
органическая жидкость — пар и вода. Используемые жидкости не
смешиваются со второй фазой; молекулы их можно считать
симметричными. Зависимости о = f (ЛЯ) и о = / (ЛЯН) имеют
аналогичный характер и совпадают для систем жидкость — газ
и жидкость — жидкость. Обе зависимости описываются линейной
функцией [35]
а = Л-|-ВДЯ(ДНи) (13)
где А и В — постоянные.
Рис. 7. Зависимость поверхностного
натяжения а от разности
полярностей фаз ЛЯ в системах
органическая жидкость — газ (X) и
органическая жидкость — вода (•).
28 Заказ I 070
433
Для систем жидкость — пар П газовой фазы можно принять
равной нулю, так как для газов и паров П = 1 и АП = П1
(конденсированной фазы). Величина G соответствует поверхностному
натяжению при разности полярностей, равной нулю, и имеет значение
24 дин/см. Меньшее значение а свидетельствует или о значительной
взаимной растворимости фаз, или об ориентации молекул на
поверхности, или о химических связях между молекулами граничащих фаз.
Выигрыш энергии, полученный в результате действия
перечисленных факторов, может быть рассчитан по формуле [35]
&F = (^теор —аsKcn) ^макс (14)
где атеор и аэксп — теоретические и экспериментальные значения
поверхностного натяжения.
Молекулы, содержащие полярные и неполярные группы
(несимметричные), с низким давлением насыщенного пара (]> 1 мм рт. ст.),
дают значения а, соответствующие неполярной части молекулы.
Так, все алифатические кислоты, эфиры, спирты, кетоны, альдегиды
и амины, несмотря на резкую разницу в длине цепи и значениях П
и АЯ„, имеют практически то же значение о, что и парафины. Этот
эффект начинает проявляться уже у соединений, имеющих
углеводородную цепочку в два-три углеродных атома (5—6 А) и с
очевидностью показывает, что в чистой жидкости молекулы на границе
раздела фаз ориентированы своей полярной частью в жидкость,
а неполярной — в газовую фазу. Аналогичная картина наблюдается
в ряду ароматических соединений. В данном случае только следует
учитывать, что электронные эффекты могут передаваться по
бензольному кольцу, изменяя тем самым в нешироких пределах
поверхностное натяжение.
Используя факт ориентации молекул некоторых веществ, можно
найти значение толщины слоя, определяющего поверхностное
натяжение. Так как о для всех веществ, имеющих парафиновую цепочку,
независимо от их полярности равно а парафинов, то межфазное
натяжение определяет только та часть ориентированной молекулы,
которая непосредственно граничит с другой фазой. Толщину слоя,
определяющего поверхностное натяжение, можно считать равной
длине углеводородной цепи в два-три углеродных атома. Например,
у диметилформамида, уксусного ангидрида, ацетонитрила, в которых
ориентируется лишь метальная или этильная группы, значение а
выше, чем у соединений, содержащих более длинную парафиновую
цепочку, однако ниже, чем следовало бы иметь по уравнению (13).
Таким образом, хотя алкильная группа их и ориентирована в
газовую фазу, полярная группа оказывает влияние на поверхностное
натяжение.
Часть дезориентированных молекул на поверхности раздела фаз
можно найти по уравнению
т„
— =е в* (15)
т
434
где тд. — число дезориентированных молекул; т — общее число
молекул; AFA — свободная энергия дезориентации.
Ниже приведены отношения mjm при различных значениях AFA:
AF , ккал/моль
0
1
2
3
4
5
6
7
8
9
Д
1,0
0,2
3,6 • 10-2
7,0 • 10-з
1,3 ■ Ю-з
2,0-10-4
4,5 • 10-5
9,2 • 10-«
1,7-Ю-6
3,2-10-'
Как видно, при 25" С ощутимо дезориентируются лишь молекулы
жидкостей, у которых AFfl = 2 ккал/моль. Примерный расчет [46]
показывает, что AFA этого процесса достигает ~ 5 ккал/моль.
Поэтому о асимметричных молекул с алифатической группой
практически равно а парафиновых углеводородов и дезориентация не
обнаруживается современными методами исследования.
Зависимость поверхностного натяжения от взаимосмешиваемости
фаз, т. е. присутствия молекул, составляющих основу одной фазы,
во второй. В случае систем жидкость — газ это давление
насыщенного пара (Р), а систем жидкость — жидкость — взаимная
растворимость жидкостей смакс.
Поверхностное натяжение и взаимосмешиваемость фаз —
функции межмолекулярных сил. Оба эти свойства не являются
параллельными функциями, так как поверхностное натяжение зависит
от ориентации молекул на поверхности, химических связей и
присутствия молекул одной фазы во второй. Да и выражаются они в
разных размерностях: о — в эрг/см2, а взаимосмешиваемость —
в моль/л.
Для выяснения зависимости о — Р и а — смакс найдены [361
функции а = f (Р или сыакс) для одного класса веществ с
одинаковыми значениями АД, отличающиеся лишь величинами Р и смакс.
Обе зависимости выражаются уравнением
амаке-а = ДГА In Р(сыаве) (16)
где огнакс и h — постоянные.
Первая зависимость проявляется при более низких
концентрациях. Так, в гомологическом ряду парафиновых углеводородов
(от пентана до гексадекана) о изменяется на ~ 14 дин/см, а на
границе с водой — всего на 3 дин/см [47]. Объяснить эти данные можно
с помощью двух факторов [48]: 1) изменение растворимости в воде
и давления насыщенного пара ряда углеводородов; 2) увеличение
в гомологическом ряду плотности, т. е. уменьшение
межмолекулярного расстояния.
28*
435
б
Показано [48], что хотя молекул парафиновых углечодородов
в газовой фазе больше, чем в водной, эти величины сравнимы, а для
соседних членов ряда практически одинаковы. Следовательно,
молекулы углеводородов газовой фазы значительно сильнее' влияют
на поверхностное натяжение, чем водной, где каждая молекула
находится в ячейке растворителя. Объясняется это влияние, видимо,
непрерывным обменом молекул фаз и поверхности. Возможность
такого процесса предполагал еще Мак-Бен [49].
Представим поведение молекул на поверхности раздела фазы.
Отрываясь от поверхности, молекулы оставляют вакансии, которые
на какой-то промежуток времени не
успевают заполниться. Вследствие
этого плотность в поверхностном
слое несколько понижается, а
межмолекулярное расстояние
увеличивается. Чем больше взаимная
растворимость компонентов фаз, тем
интенсивнее обмен молекул
поверхности и объема и ниже
поверхностное натяжение [481. Отсюда
становится понятной значительно боль -
шая активность молекул в паре,
чем в растворе — коэффициент
молекулярной диффузии в газовой фазе
на 4—5 порядков больше, чем в
жидкой.
Для определения влияния
изменения межмолекулярного расстояния
на поверхностное натяжение,
рассмотрены [48] изомеры октана,
имеющие одинаковое давление насыщенного пара, но разные
плотности и поверхностные натяжения. Таким путем найдена
зависимость поверхностного натяжения от плотности фазы, с помощью
которой получена [48] величина изменения поверхностного
натяжения в гомологическом ряду парафиновых углеводородов от пентана
до тетрадекана — оказавшаяся равной 3,5 дин/см. Это значение
практически совпадает с общим изменением межфазного натяжения
в том же ряду в системе углеводород — вода (3,2 дин/'см). Объяснение
состоит в том, что растворенные в воде молекулы углеводородов
практически не влияют на межфазное натяжение, так что изменение о
вызвано разницей плотностей различных гомологов. В системе
жидкость — газ остальное изменение а (14—3,5) происходит за счет
давления насыщенного пара.
Сопоставляя изменение плотности углеводородов с температурой,
получаем температурный коэффициент поверхностного натяжения,
равный —' 0,03, т. е. такой как по изобаре поверхностного
натяжения [46]. Он равен общему коэффициенту в системе углеводород —
вода.
In r
Рис. 8. Типичный вид изотермы
поверхностного натяжения.
436
Уравнение (16) зависимости поверхностного натяжения от
взаимосмешиваемости компонентов фаз не следует идентифицировать
с уравнением зависимости давления насыщенного пара от
поверхностного натяжения [50, 51], в которых а выступает как
характеристика межмолекулярных сил. Уравнение (16) по физическому
смыслу является аналогом уравнения Гиббса, Г = — дг" *~яьГ~ и поп"
ностью совпадает с ним на прямолинейном участке изотермы
поверхностного натяжения, типичный вид которой приведен на рис. 8.
Участок А, как известно [52], соответствует адсорбции до
достижения ГЫЙКС, наклонный прямолинейный участок Б при предельной
адсорбции описывается уравнением о"макс — а = Я7Тмакс In с,
идентичным уравнению (16). Понижение поверхностного натяжения на
этом участке можно объяснить непрерывным обменом молекул ПАВ
поверхности и объема [44]. Участок В характерен для мицелло-
образующих систем.
Для практических целей большое значение имеет установление
взаимосвязи межфазного и поверхностных натяжений. Так, правило
Антонова «межфазное натяжение в системе жидкость — жидкость
равно разности поверхностных натяжений взаимнонасыщенных
жидкостей» используется уже более полувека, несмотря на
постоянную критику и действительно значительное число отклонений.
Анализ правила Антонова дан нами ранее [48]. Здесь остановимся
только на основных его положениях.
Если заменяются поверхности между жидкостью и паром одной
поверхностью между жидкостями, ликвидируется зависимость от
давления насыщенного пара и возникает зависимость от взаимной
растворимости компонентов фаз, возможно изменение ориентации
молекул на поверхности и в ряде случаев образование химических
связей (водородных) между молекулами граничащих фаз. Из
сказанного ясно, что само поверхностное натяжение не может являться
характеристической величиной в правиле Антонова. Когда
используются поверхностные натяжения взаимнонасыщенных жидкостей,
в определенной степени компенсируется разность от влияния
давления насыщенного пара и взаимосмешиваемости жидкостей. Если обе
жидкости обладают низким давлением насыщенного пара,
практически нерастворимы и молекулы их симметричны, а также не
взаимодействуют химически, то такие системы должны достаточно хорошо
подчиняться правилу Антонова без условия взаимного насыщения
растворителей. С учетом изменения всех указанных факторов
получим уравнение в общем случае, связывающее межфазное и
поверхностное натяжение [48]:
ац-^-с+ЛПв-^ —^ (П)
где Д^н
— свободная энергия образования водородной связи.
437
В настоящее время для описания температурной зависимости
поверхностного натяжения предложен целый ряд эмпирических
уравнений, из которых наибольшее распространение получило
уравнение Этвеша. Подробно этот вопрос изложен в книге Партинг-
тона [53]. Термодинамические уравнения поверхностного натяжения
обстоятельно проанализированы в монографиях Оно и Кондо [54]
и Русанова [55]. Поэтому остановимся лишь на некоторых новых
фактах, относящихся к теме обзора.
Интересно отметить, что почти все органические жидкости имеют
близкий температурный коэффициент поверхностного натяжения.
Для неполярных жидкостей вблизи нормальной температуры его
значения равны 0,08—0,11 дин/'(см-град), для полярных — 0,1—0,13
и для воды — 0,15. Температурный коэффициент межфазного
натяжения органических жидкостей на границе с водой ниже, чем каждой _
из жидкостей отдельно, и составляет-—0,03 дин/(см-град). Кроме
того, межфазное натяжение в ряде систем с повышением температуры
возрастает.
Для объяснения этих фактов следует выяснить, какой вклад
вносит в температурный коэффициент каждый из факторов,
определяющих поверхностное натяжение. С этой целью построены [46]
зависимости поверхностного натяжения гомологического ряда
нормальных парафиновых углеводородов от температуры при
постоянных давлениях насыщенного пара — изобары поверхностного
натяжения. По углу наклона этих прямых найдены температурные
коэффициенты только за счет теплового расширения жидкостей.
Значения их оказались равными ~ 0,03 дин/(см-град), т. е. общему
температурному коэффициенту в системе жидкость — жидкость,
так как изменения, происходящие в результате растворимости
парафиновых углеводородов, при повышении температуры практически
не сказываются на поверхностном натяжении. Общий температурный
коэффициент поверхностного натяжения парафиновых
углеводородов равен ~ 0,1 динI(см-град). Таким образом, за счет изменения
давления насыщенного пара dajdT = 0,07.
Такое соотношение коэффициентов подтверждается данными о
температурных коэффициентах систем жидкость — жидкость, в которых
взаимная растворимость жидкостей с повышением температуры
уменьшается. При этом поверхностное натяжение во всех случаях
возрастает, т. е. эффект изменения о вследствие взаимной
растворимости компонентов фаз превалирует над эффектом уменьшения о" из-
за повышения интенсивности теплового движения молекул жидкости.
Исследованы [46] температурные зависимости поверхностного
натяжения веществ, молекулы которых ориентируются на поверхности
раздела фаз, например, нормального ряда спиртов. Результаты
аналогичны полученным с парафиновыми углеводородами. Общий
температурный коэффициент равен— 0,1 дин/(см-град), а по
изобарам ~ 0,03. Следовательно, при изменении температуры
происходит незначительная дезориентация поверхностного слоя, что
объясняется с помощью формулы (15) в случае Д/^д "}р RT.
138
ВЫБОР ПАВ
ДЛЯ СТАБИЛИЗАЦИИ ЭМУЛЬСИИ [56J
Как видно из материала книги, единственной возможностью
для выбора ПАВ, кроме «метода проб и ошибок» и интуиции
исследователя, является система ГЛБ, но эта система, а также ее
модификации и попытки теоретического объяснения [2, 57, 58] позволяют
предсказать лишь тип получаемой эмульсии, да и то только с
энергетических позиций, т. е. не учитывают особенности строения ПАВ.
Однако известно, что изомеры ПАВ с разветвленными
алифатическими цепями стабилизируют обратные эмульсии, а с нормальными —
прямые.
Для практических целей необходимо выбрать ПАВ,
предопределяющее нужную устойчивость эмульсии и стабилизирующее
определенное количество дисперсной фазы. Следует также определить,
в каких концентрационных пределах данное ПАВ является
стабилизатором. Кроме того, существует ряд показателей, которыми
необходимо задаться, учитывая целевое назначение эмульсии. К ним
относятся: время жизни; дисперсность; тип эмульсии; химические
свойства ПАВ.
Следовательно, ПАВ в качестве эмульгатора должно отвечать
следующим основным требованиям:
1. Физические и химические свойства ПАВ должны
соответствовать свойствам фаз, методам получения и цели применения эмульсии
с учетом дефицитности эмульгатора и экономики производства.
2. Предопределять нужную стабильность системы.
3. Стабилизировать требуемое количество дисперсной фазы в
дисперсионной среде.
Синтезируя эмульсию, исследователь может задаться временем
ее жизни или периодом полураспада (г или т./2), а также
дисперсностью. Последнее свойство можно регулировать в широких
пределах с помощью используемого метода и интенсивности
диспергирования. Кроме того, необходимо выбрать класс ПАВ, который
определяется природой полярной группы, так как гидрофобная
часть, как правило, отличается лишь числом метиленовых групп
и наличием ароматических колец.
Имеется достаточное количество ПАВ самых различных классов
с примерно одинаковой эмульгирующей способностью. Однако при
выборе их следует учитывать физико-химические свойства всей
системы и область применения эмульсии. Так, в кислой среде должны
применяться катионоактивные эмульгаторы, а в щелочной — ани-
оноактивные. Если в полярной фазе присутствует значительное
количество солей, то лучше использовать неионогенные эмульгаторы,
как и при колебаниях рН среды. Нужно принимать во внимание
возможное химическое взаимодействие между ПАВ и компонентами
фаз (например, гидролиз, комплексообразование), а также
биологическую активность ПАВ *.
* Перечень промышленных ПАВ содержится в книгах [59 — 61].
439
При выборе конкретного ПАВ прежде всего необходимо учесть
тип эмульсии (М/В или В/М), который определяется и геометрией
молекулы, и энергетикой ее взаимодействия с фазами.-Однако еще
нет достаточно удовлетворительного математического описания этих
явлений. Поэтому будем учитывать оба фактора как
«арифметическую сумму».
Таким образом, необходимо:
1. Часть молекулы, которая находится в непрерывной фазе,
должна иметь больший диаметр, чем в дисперсной.
2. ПАВ должно лучше растворяться в непрерывной фазе.
Геометрические особенности ПАВ можно выявить, построив или
начертив модель молекулы по данным межмолекулярных и ван-
дерваальсовых радиусов и валентных углов.
На предпочтительную растворимость ПАВ в той или иной фазе
указывает коэффициент распределения (^р), который можно
рассчитать по данным свободной энергии взаимодействия растворителей
с различными группами, входящими в молекулу ПАВ (см. табл. 2).
Расчет ведется по формуле
Sag0-Sagb
Кр = е~ Ят (18)
где 2 AG0 и 21 AGB — суммы свободных энергий взаимодействия
групп в фазах.
Оба показателя должны указывать на один и тот же тип
эмульсии. В противном случае эмульсии мало стабильны и легко
оборачиваются.
Расчет стабильности эмульсии. Задавшись xi 2 по формуле:
%i/2 = \/К-п0, рассчитывают константу скорости коалесценции.
Время полного распада эмульсии связано с К более сложной
зависимостью, так как последние капли значительно крупнее начальных.
Можно еще задаться определенным числом коалесценции ко
времени х, тогда расчет следует вести по формуле (1).
Получив К, можно определить свойства ПАВ по формуле (2).
Значения Z и а рассчитать в настоящее время не представляется
возможным. Поэтому воспользуемся табл. 5, взятой из работы [561,
в которой приведены зависимости Z и а от среднего диаметра
элементарной капли с точностью, приемлемой для инженерных решений.
Член с/с ~г а в формуле (2) можно принять равным единице
для стабильных эмульсий, т. е. когда достигнута предельная
адсорбция на межфазной поверхности: Г/Гмакс-+ 1.
Выбрав значения Z ш а, зная К, VK и Т, рассчитывается W.
Поскольку класс ПАВ уже выбран, остается найти число метиленовых
групп в алифатической цепи «
W
где Wnu — работа адсорбции, приходящаяся на метиленовую группу
(определяется по методикам, указанным выше—стр. 431 и табл. 4).
440
При использовании класса ПАВ, не указанного в таблице и
растворимого только в водной фазе, можно с достаточным приближением
принять [56]:
1
Wnu
W.
теор
Величина WTeop рассчитывается по формулам (11) и (12).
В табл. 4 приведены числа углеродных атомов в алифатической
цепи, которые соответствуют максимуму Донана. ПАВ данного
класса с большим числом атомов углерода в качестве стабилизаторов
эмульсий применять нельзя.
Расчет количества ПАВ, Таблица 5
необходимого для стабили- Значения Z и а концентрированных
зации эмульсии. Соотноше- эмульсий
ние между количеством
дисперсной фазы и
дисперсионной среды, как
правило, задается целевым
назначением эмульсии. Отсюда
количество ПАВ
определяется по формуле
материального баланса:
Средний
капли, м
< 1,35 • Ю-»
> 1,35-10-5
(1-3) -Ю-з
Z,
(сек«число частиц)
0,5-10-21
1,0-10-18
1,0-Ю-10
а
1,8-
1,2-
2,0-
м'
10»
1015
109
M = c0Vc = rMSKCS +ceVc + CeK?V<b
(19)
Предельную концентрацию эмульгирования можно
ориентировочно рассчитать по формуле:
сй=Ю0а (20)
Если же необходимо знать, сколько заэмульгируется дисперсной
фазы данным раствором ПАВ, можно воспользоваться формулой (3).
9.
10.
11.
12.
13.
14.
15.
ЛИТЕРАТУРА
А. А. А б р а м з о н, З.Н.Славина, Коллоидн. ж., 31, 635 (1969).
J. Т. D a v i s, Proceedings of the II International Congress on Surface
Activity, vol.1, London, 1957, p. 426.
M. van den T e m p e 1, Proceedings of the II International Congress on
Surface Activity, vol. 1, London, 1957, p. 439.
А. А. А б р а м з о н, Е.В.Громов, Коллоидн. ж., 31, 795 (1969).
З.Н.Славина, Автореф. канд. дисс, ЛХФИ, 1972.
А. Д. Г р и щ е н ко, Молочн. пром., №4, 12 (1966).
А. А. А б р амз он, З.Н.Славина, ДАН СССР, 186, 116 (1969).
Ph. Sherman, Proceedings of the IV International Congress on Surface
Activity, vol. 2, Brussels, 1964; J. Phys. Chem., 67, 2531 (1963).
В.Клейтон, Эмульсии, ИЛ, 1950.
A. D.Sheludko, Proceedings of the IV International Congress on Surface
Activity, vol. 2, Brussels, 1964.
А. А. Абрамзон и др., Коллоидн. ж., 34, 156 (1972).
J.I.Bikerman, Physical Surfaces, New York, 1970, p. 112.
Я. д е Бур, Динамический характер адсорбции, ИЛ, 1962.
Е.Е.Малахова, А. А. А б р а м з о н, ДАН СССР, 180, 1157 (1967).
А. В. Б р о м б е р г, Коллоидн. ж., 8, 117 (1946).
441
16. Л. Я. К р е м н е в, С. А. С о с к и н, Коллопдн. ж., 9, 269 (1947).
17. Л. Я. К ремне в, Автореф. докт. дпсс, ЛТИ, им. Ленсовета, 1955.
18. П.Кинг, Оболочки жировых шариков молока, Пищепромпздат, 1956.
19. R. A. Kuhlman. Nature, 207, Л» 5003, 1289 (1965).
20. К. Fukawa, Т. A s a k u г a, H.Daimon, Kobunshi Kagaku, 18,
596 (1958).
21. Р. А. Кульман, Коллопдн. ж., 31, 305 (1969).
22. А. А. А б р а м з о н, Е. В. Г р о м о в, Коллопдн. ж., 31, 163 (1969).
23. J.M. G. Lankveld, Adsorption of Polyvinylalcohol, Dissertation, Wage-
ningen, 1970. '
24. Е.В.Громов, Автореф. канд. днсс, ЛТИ им. С. М. Кирова. 1970.
25. Е. A b d е г h а 1 d e n, W. W б 1 t z, Hoppe-Seylers Z. Physiol., 59, 13 (1909).
26. V.S torch, Analyst, 22, 197 (1897).
27. А. А. А б р а м з о н, Н. В. А б р а м о в а, Коллопдн. ж., 34, .№ '.', (1972).
28. Л. Я. К р е м н е в, ДАН СССР, 79, 655 (1951).
29. В.Н.Измайлов а, Автореф. докт. дпсс, МГУ им. М. В. Ломоносова,
1971.
30. А. А. Т р а п е з н и к о в, К.В.Зотова, Коллопдн. ж., 27, 614 (196о).
31. E.H.Lucassen-Reynders, M.van denTempel, Proceedings
of the IV International Congress on Surface Activity, vol. 2, Brussels, 1964.
32. A. R e h b i n d e r, Biochem. Z., 187 (1927); Z. phys. Chem., 129, 161 (1927).
33. А. Б. Т а у б м а н, С. И. Б у р ш т е и н, Коллопдн. ж., 20, 535 (1958).
34. А. А. Ж у х о в и ц к и й, ЖФХ, 18, 214 (1944).
35. А. А. А б р ам з он, Коллопдн. ж., 29, 467 (1967).
36. А. А. А б р а м з о н, А. А. С л а в и н, ЖФХ, 44, 564 (1970).
37. J.A. Batler, D.W.Thomson, J. Chem. Soc, 1933, 674.
38. А. А. А б р а м з о н, Е.Е.Малахов а, Коллопдн. ж., 30, 475 (1968).
39. Л. А. Ш н ц п др., ДАН СССР, 180, № 2, 412 (1968).
40. А. А. П е т р о в, Г. Н. П о з д н ы ш е в, Коллопдн. ж., 28, 7о8 (1966).
41. Г. Н. П о з дн ы ше в, А.А.Петров, Коллопдн. ж., 29, 589 (1967).
42. J. J. J a s р е г, R. D. van D е 1 1, J. Phys. Chem., 69, 481 (1965).
43. A. F.H.Ward, Surface Chemistry, London, 1949.
44. А. А. А б р а м з о н, Н. В. А б р а м о в а, Е. Е. М а л а х о в а, Коллопдн.
ж., 33, 475 (1971).
45. P.Eehbinder, Z. phys. Chem., 121, 103 (1926); 129, 161 (1927).
46. А. А. А б р а м з о н, Ю. Б. М о и с е е в а, Коллоидн. ж., 30, 79о (1968).
47. W. G i 1 1 а р, N. W e i n е г, М. G i b a 1 d i, J. Am. Oil. Chem. Soc, 44,
71 (1967).
48. А. А. А б р а м з о н, Коллоидн. ж., 32, 475 (1970).
49. J. W. М с В a i n, Nature, 137, 659 (1936).
50. L. Z. Р о 1 1 а г a, J. Chem. Phys., 46, 1163 (1942).
51. С. А. В о й т к е в и ч, ЖФХ, 37, 1029 (1963).
52. Э-. М.Спивакова, ЖФХ, 14, 406 (1940).
53. I.R. Partington, Advanced Treatise on Physical Chemistry, vol.2,
London, 1951.
54. С. О н о, С. К о н д о, Молекулярная теория поверхностного натяжения
в жидкостях, ИЛ, 1963.
55. А.И.Русанов, Фазовые равновесия и поверхностные явления,
Изд. «Химия», 1967.
56. А. А. Абрам з он, II. И. В о л ь ф е н з о н, В. М. К и с е л е в а, ЖПХ,
57. L.'Osipow', Surface Chemistry, New York, 1962.
58. П. М. К р у г л я ко в, А. Ф. К о р е ц к и й, ДАН СССР, 197, № о,
1106 (1971). '
59. P. Becker, Emulsion Theory and Practice, New York, 1966.
60. Н.Шенфельд, Неионогенные моющпе средства, Изд. «Химия», 196о.
61. Nonionic Surfactants, Ed. by M.J.Schick, London, 1967.
Агрегация 106, 108. 231, 245 ел., 407 ел.
зависимость от скорости сдвига 232 ел.,
246 ел.
Адгезия 44, 80, 173, 178 ел.
Адсорбционные слои
вязкость 90, 91, 271, 291 ел.
высокомолекулярных ПАВ НО ел.,
423 ел.
перемещение молекул 292
разрушение 68, 143, 302
от низких температур 78, 126 127
при ультрацентрифугировагаш 'l30
структура 425, 426
Адсорбция
высокомолекулярных ПАВ 86, 423, 424
изотерма
Гиббса 84, 85, 174, 187
Лэнгмюра 418, 421
скорость 97
Аэрозоли
методы получения
акустический 45
конденсационный 11, 12, 59
электрический 58
УКАЗАТЕЛЬ стабильность 108
Броуновское движение 76, 106 ел., 227 ел.
Вискозиметр(ы)
капиллярные 201, 202 ел., 260 ел.
конус-пластина 201, 202
межфазной вязкости 91
пьезоэлектрический 208, 214
с коаксиальными цилиндрами 201 ел,
255 ел.
с падающим шариком 201, 202, 207, 213
с соплом 213
Водородная связь 175, 178
Вращение
пары капель 238
сфер
в коаксиальных цилиндрах 255
в круглых трубках 260
Время релаксации в структурированных
системах 390, 391
Вязкость
динамическая 222
мгновенная 201
остаточная 227
относительная 204
равновесная 201
суспензий 266, 267, 280 ел.
тонких жидких пленок 286
удельная 263
характеристическая 263
эмульсий 198, 200 ел., 254 ел.
взаимодействие капель 248, 251, 255
влияние флокуляции 223, 226 ел.
деформация капель 272, 293
изменение при старении 300 ел.
образование пар капель 239, 240
обращение фаз 289, 290
пластичность 224, 225
псевдопластичность 226 ел.
Гидратация эмульгатора 83, 268
Гидрофильно-липофильный баланс (ГЛЕ1
132 ел., 420, 439 k '
влияние на тип и стабильность эмтльсий
132 ел., 420
групповые числа 139 ел.
оксиэтилированных ПАВ 133
определение
калориметрическое 135
по температуре помутнения 134
титрованием 134
хроматографическое 135, 136
смеси ПАВ 133, 143
Гидрофильное отталкивание 83
Гидрофобная ассоциация 83
443
Гистерезис в тиксотропных эмульсиях 200
Гомогенизатор(ы) 16, 22, 26
Гуи — Чепмепа теория 96 ел.
Движение сфер
в круглых трубах 260, 261
влияние на вязкость эмульсий 261
Двойной электрический слой 92, 93, 96 ел.,
247 ел.
внутри капли 98
плотность заряда 97, 102, 297
симметрия 294
энергия взаимодействия 97, 186
Девая параметр 97
Дебая — Хюккеля теория 85
Дерягина, Ландац, Вереея, Овербега (ДЛВО)
теория 92 ел., 247 ел.
Деформация
капли
влияние скорости сдвига 257 ел.
в круглых трубках 260, 2В1
в электрическом поле 69
при эмульгировании 37, 38, 76
ползучести 216 ел.
запаздывающая 2!6 ел.
мгновенная 216, 217, 220
механическая модель 219 ел.
Ньютоновская 217
полная 218, 220
Деэмульгирование 65 ел.
отстаиванием 68
температурным воздействием 70
химическое 68, 69
центрифугированием 69
электрическое 69, 70
Дзета-потенциал 83, 101, 102, 250
Динамические измерения реологических
свойств 221 ел.
Дисперсная фаза
вязкость
влияние на деформацию и циркуляцию
капли 256, 257, 259
кажущаяся объемная концентрация 303 ел.
объемная доля 130, 262 ел.
роль в емкостных свойствах эмульспй
326 ел.
Диффузия 63, 77
Диффузный слой
внутренний 98, 250
толщина 97, 247, 295
Диэлектрическая дисперсия 316, 335 ел.
Диэлектрическая проницаемость 94, 314,
316, 324 ел.
влияние динамического состояния и
температуры 373 ел., 401, 407 ел.
гелей желатины 399
гидрозолей 400
комплексная 320, 334, 338 ел.
определение 321 ел.
солюбилизированных систем 400 ел.
суспензий 394 ел.
биологических клеток 382 ел.
теория 325 ел., 409 ел.
частотная зависимость 351, 355 ел.
Емкость 326, 334, 335
комплексная 319, 335 ел.
конденсатора 316, 321 ел.
поверхностных пленок 260, 261 ел.
суспензий 366, 367
биологических клеток 384, 385
частотная зависимость 335, 347 ел.
эмульсий 366, 367
Закон
Гука 220, 269
Ламберта — Вера 147
Стокса 202, 207
Замораживание и оттаивание эмульсий
125 ел.
Индукция электростатическая 314, 315, 326
Кавитация 52, 53
Капиллярные волны 50
Капли
внутренняя циркуляция 256, 257
влияние адсорбционных слоев 259 271
вязкость фаз 257, 258
вращение временных пар 258, 270
зависимость от скорости сдвига 238 ел
деформация
в круглых трубках 260, 261
влияние скорости сдвига 257, 258, 275
в электрическом поле 69
в эмульсии 76
межфазное натяжение 271
при сближении двух капель 78, 79. "
81. 98
при эмульгировании 37, 38, 76
образование 23 ел.
распад 22, 25, 41 ел., 259
растворимость 77
рекомбинация 41, 43 ел., 05
течение
асимметричное гиперболическое 40
зависимость от вязкости фаз 39 ел.
Куэтта 40, 41, 255, 256
ламинарное 40
турбулентное 41 ел.
Коагуляция, см. Флокуляцпя
Коалесценция 9, 23, 25, 66 ел., 288, 300
влияние адсорбированного эмульгатора и
примесей 78 ел., 90, 110 ел., 249,
420
кинетика 67, 79, 114 ел., 301, 303
константа скорости 114, 417, 4IS
ограниченная 114
Когезня 114, 173, 186, 262
Коллоидные мельницы 15, 22
Константа
Гамакера — Лондона 84, 93, НО
Лондона — Ван-дер-Ваальса 251 ел.
скорости коалесценции 114, 417, 418
скорости течения 24Г
К оэффициент
активности 85, 428
неоднородности 158
отражения 146
поправочный при измерении натяжения
166 ел.
преломления 147
растекания 137, 174 ел.
трения 265
эластичности 87 ел.
Краевой угол смачивания 174 ел.
порошка 181, 18 2
Кривые
напряжение сдвига — градиент скорости
198 ел., 224, 252
потенциальной энергии 98, 248 ел.
псевдоплаетпческого течения 225, 226
сила — площадь 184, 185
Критерий
Вебера 26, 41, 42
Рейнольдса 29, 42, 205
стабильности капли 43, 44
Критическая концентрация мицеллообразо-
вания (ККМ) 85, 128 ел., 288
Кцитта течение 40, 41, 255, 256
Массообмен
при самоэмульгировании ь2, 75
при эмульгировании 189, 190, 271 ел.,
296
Межфазное натяжение 165, 173 ел., 437
динамическое 96, 166 ел.
методы определения
Вильгельми 167
висячей каплей 171
лежачей каплей 172
444
Межфлзное натяжение
методы определения
осциллирующей струей 86
по высоте поднятия в капилляре 170,
171
по объему (весу) капли 85, 168, 169
по отрыву кольца (дю Нуи) 85, 165,
166
связь с поверхностным натяжением 104,
175
статическое 86
Методы определения
ГЛБ 134 ел.
натяжения 167 ел.
размера частиц 145 ел.
скорости флокуляции 103 ел.
типа эмульсии 190
Микроскопия 76 ел., 144, 163, 189, 325
Микроэмульсии 188
Мицеллы 287, 288
светорассеяние 149, 150
Множественные эмульсии 188, 189
Монослои нерастворимые
виды пленок 185 ел.
кривые сила — площадь 183 ел.
поверхностное давление 185
электрические свойства 360 ел.
Напряжение сдвига
зависимость от скорости сдвига 198 ел.
определение коническим пенетрометром 209
' предельное 199, 200
Напряженность поля 314, 315
Нестабильность
гидродинамическая 29 ел., 49 ел.
капиллярных волн 50
Образование льда 125 ел.
Обращение фаз 66 ел., 189
влияние ГЛБ 142, 143
зависимость от состава фаз и температуры
129
Оптическая плотность 147
Оптические свойства эмульсий
мутность 147 ел.
отражательная способность 146
светорассеяние 147 ел.
Отталкивание
Борна 82
гидрофильное 83
Пенетрометры 209, 214
Пленки
вязкость 282
динамика утончения 82, 88, 131, 132
между каплями 79, 116, 143, 238, 417
мыльные 80
толщина 131
минимальная 424
равновесная 116, 117
черные 80, 82, 117
Поверхностная вязкость 90, 91, 271, 292
Поверхностное давление 184 ел.
градиент 271 ■
Поверхностное натяжение
влияние фаз 433 ел.
градиент 88, 91
методы определения
Вильгельми 167, 168, 182, 183
висячей каплей 171
лежачей каплей 172
осциллирующей струей 85
по высоте поднятия в капилляре 170, 171
по объему (весу) капли 85, 168 ел.
по отрыву кольца (дю Нуи) 85, 165, 166
Поверхностный потенциал 98 ел., 138, 139
Поверхность эмульсий
предельная 421
удельная 25, 26
Поглощение света 146 ел.
Получение
' аэрозолей 11, 12, 45, 58 ел.
эмульсий 11, 12 ел.
Поляризация поверхностная 315 ел., 347
385
Поправочный коэффициент при измерении
натяжения 166 ел.
Порошки
краевой угол смачивания 113, 181 182
стабилизация эмульсий 76, 113, 114
Потенциал
отталкивания 98, 138, 139, 247 ел.
притяжения 93, 95, 248
Правило
Антонова 437
Банкрофта 142, 425
Псевдопластичность 227 ел.
Пьезоэлектрический
генератор 46, 47
эффект 46
Работа
адгезии 44, 173 ел.
адсорбции 63, 418, 419
влияние строения ПАВ 426 ел.
Размер частиц
определение
микроскопией 144, 145
нефелометрическое 146 ел.
по отражательной способности 146
по светорассеянию 150 ел.
счетчиком Коултера 155, 156
распределение 107, 158 ел.
гистограмма 158
средний 108, 156 ел., 252, 274 ел.
Расклинивающее давление 80 ел., 97, 131
Светорассеяние 103, 150 ел.
Свободная энергия
испарения 428
образования водородной связи 437
растворения 428 ел.
Сдвиг
напряжение 199 ел., 241
зависимость от скорости сдвига 198 ел
224 ел. ■ '■
определение коническим пенетрометрощ
предельное 199, 200
скорость
в капиллярном вискозиметре 206
влияние на вязкость эмульсий 261 293
300 ел.
в пьезоэлектрическом вискозиметре 208
межфазных пленок 293
определение 198, 199, 224, 241
при деформации капли 257 ел.
Седиментация 66, 75, 115
Силы
отталкивания 96 ел., 117, 247
притяжения 80
влияние адсорбционных слоев 250, 252
замедление 95, 248
Лондона — Ван-дер-Ваальса 81 ел
92 ел., 131, 248 ел.
Скольжение
в капиллярном вискозиметре 204
на межфазной границе 263, 269, 293
Смесители 14, 22
Солюбилизация 137
Стабилизаторы 75, 76, НО ел.
Стабильность
коллоидов 92 ел., 116, 247 ел.
пей 79, 131
черные пленки 80, 82, 117
эффект Марангони — Гиббса 89, 9п
эмульсий 13, 19, 75 ел.
" в зависимости от температуры 125
126 ел.
влияние ГЛБ и объемной доли дисперс*
ной фазы 130, 136 ел.
445
Стабильность
эмульсий
дзета-потенциал 101, 114, 116
при ультрацентрифугировании 77, 119,
130
роль природы эмульгатора 75, 76, 136
Старение эмульсий 300 ел.
Структура воды в поверхностном слое 83,
138, 186
Счетчик Коултера 105, 106, 155, 156
Твердое тело
деформация 197
растекание жидкости 178 ел.
краевой угол смачивания 179, 180
Теория
абсолютной скорости 241 ел.
Гуи — Чепмена 96, 97, 102
Дебая — Хюккеля 85
Дерягина, Ландау, Вервея, Овербека
(ДЛВО) 92 ел., 116, 247 ел.
импульсная 227 ел.
кинетики агрегации 237, 238
кинетическая 245 ел.
ориентированных клиньев 419
Течение
ламинарное 205
неньютоновское 198, 199, 205, 224 ел.
ньютоновское 198, 199, 202
пластическое 199, 226 ел.
псевдопластическое 198, 199, 205, 224
Тиксотропия 199, 200
влияние сдвига 228
время жизни связи 228
гистерезис 200
Типы эмульсий
влияние геометрии молекулы ПАВ и
ГЛБ 142, 420
методы определения 190
Турбулентность 29, 42 ел.
поверхностная 61, 63, 64
Ультрамикроскопия 105
Флокуляция
быстрая 106 ел.
влияние на вязкость и структуру
эмульсий 223, 225 ел., 285
медленная 108 ел.
ортокинетическая 76, 107, 108, 115, 116
скорость 67, 99, 104, 114, 116, 251, 285
вторичный минимум 100, 108. 114, 115,
249, 253
определение 103 ел.
Флуктуация
давления 41
плотности 30
Эластичность поверхностная 86, 111, 112
Электрическое смещение 314
Электровязкостный эффект 294 ел.
Электропроводность 318 ел., 334
гидрозолей 400
дисперсий твердых сфер 364 ел., 378, 379
желатиновых гелей 399
методы измерения 321, 323, 324
солюбилизированных систем 400 ел.
суспензий биологических клеток 382 ел.
теория 333 ел.
частотная зависимость 335, 355, 363, 367,
373, 377 ел.
Электропроводность
эмульсий 363, 365 ел.
Электрофорез 160 ел.
Электрофоретическая скорость 101, 160,
163, 250
Элемент
Кельвина — Войгта 220, 221
Максвелла 220 '
Эмульгаторы 19, 20, 21, 75, 76
влияние на стабильность эмульсий 19,
72, 136 ел.
высокомолекулярные 76, 112, 422 ел.
гидратация 83, 268
ГЛБ 20, 132 ел.
двойные 189
необходимое количество 21, 441
ПАВ — мыла, детергенты 76
поверхностная вязкость 90, 91, 271, 275
понижение поверхностного натяжения 19
порошки 76, 113, 114
растворимость в фазах 134, 181, 271, 280,
420
способ введения 21, 271
Эмульгирование
влияние вязкости и плотности фаз 25, 26
деформация капли 37, 41, 76
зависимость от температуры 26, 27, 420
максимум Донана 418 ел.
методы
акустические 45 ел.
дисперсионные 12, 14 ел.
электрические 55 ел.
необходимая энергия 17, 18
самопроизвольное 18, 60 ел., 75, 188
Эмульсии
вязкость 198, 200 ел., 254 ел.
деформация ползучести 216 ел.
диэлектрические свойства 362, 363, 368 ел.
концентрированные 115, 201, 272, 286
влияние эмульгатора 287
предельная площадь поверхности 418,
421, 422
течение 224, 227, 228, 255, 264
толщина пленок 286
оптические свойства 144 ел.
стабильность 13, 19, 75 ел., 130 ел.
тип 142, 420
определение 190
удельная поверхность 421
электропроводность 363, 365 ел.
Эмульсии обратные 129, 287, 288, 297
вязкость 272, 273, 275, 278 '
диэлектрическая проницаемость 164,
373 ел.
получение 20 ел., 53, 91, 112
потенциальная энергия 249, 250
электропроводность 378, 379
электрофорез 164, 165
Энергия
адсорбции 63, 292, 418, 419
свободная
испарения 428
образования водородной связи 43
растворения 428 ел.
Эффект
Вейзенберга 214
Вернера 107 ,
магнитострикшюнный 47
Марангони—Гиббса 79, 84, 87, 89'сл.
Мюллера 115
пьезоэлектрический 46
Предисловие редактора перевода 5
Предисловие редактора книги ... 7
Глава I. Принципы получения
эмульсий (Е. С. Р. Гопал) .... 9
Основные методы приготовления
эмульсий 11
Физико-химические факторы,
влияющие на образование
эмульсий 18
Изучение различных моделей
эмульгирования 27
Эмульгирование звуком и
ультразвуком 45
Получение эмульсий
электрическими методами 55
Самопроизвольное
эмульгирование 60
ОГЛАВЛЕНИЕ Проблема деэмульгпрования . . 65
Литература 70
Дополнительная литература .... 73
Глава II. Теория стабильности
эмульсий (Дж. А. Китченер,
П. Р. Массельвайт) 75
Поверхность раздела фаз и
стабильность эмульсий 75
Применение теории устойчивости
лиофобных коллоидов к
эмульсиям 91
. Устойчивость к коалесценцин,
обусловленная механическими
свойствами межфазных слоев 110
Некоторые выводы из
исследования стабильности эмульсий 114
Литература 117
Дополнительная литература .... 122
Глава III. Основные свойства
эмульсий и их строение (Ф. Шер-
ман) 125
Стабильность эмульсии при
замерзании и оттаивании .... 125
Стабильность эмульсий при
повышенных температурах . . . 128
Устойчивость к
ультрацентрифугированию 129
Влияние ГЛБ на образование
эмульсий 132
Распределение шариков по
размерам 144
Электрофорез 160
Поверхностное и межфазное
натяжение 165
Смачиваемость 173
Свойства пленок эмульгатора
на поверхностях раздела
масло — вода 182
Микроэмульсии 188
Множественные эмульсии . . . 188
447
Определение типа эмульсии 190
Литература 191
Дополнительная литература 194
Глава IV. Реология эмульсий (Ф. Шерман) • . 197
Основные типы течения 197
Методика изучения реологических свойств 200
Анализ неньютоновской вязкости 223
Реология модельных дисперсий 255
Факторы, влияющие на реологические свойства эмульсий 261
Изменение реологических свойств эмульсий при старении 300
Литература 308
Дополнительная литература 312
Глава V. Электрические свойства эмульсий (Т. Хапай) 313
Основные свойства диэлектриков 314
Экспериментальные методы изучения диэлектрических свойств . . 321
Теория диэлектрической проницаемости и электропроводности
гетерогенных систем 325
Теория комплексной диэлектрической проницаемости гетерогенных
дисперсных систем. Поляризация поверхности раздела,
обусловленная гетерогенной структурой 334
Экспериментальные исследования диэлектрической проницаемости
и электропроводности эмульсий. Подтверждение поляризации
поверхности раздела 360
Объяснение диэлектрической дисперсии, обусловленной поляризацией
поверхности раздела 385
Некоторые виды частотной зависимости диэлектрической
проницаемости 389
Диэлектрические свойства некоторых дисперсных систем 394
Диэлектрическая теория гомогенных систем. Молекулярные растворы 402
Сравнение диэлектрических проницаемостей различных дисперсных
систем 404
Диэлектрические свойства систем в динамическом состоянии . . . 405
Литература 413
Дополнительная литература 415
Дополнение. Об эмульгирующих свойствах ПАВ (А. А. Абрамзон) 416
Стабилизация эмульсий низкомолекулярными ПАВ 417
Стабилизация эмульсий высокомолекулярных ПАВ 422.
Зависимость адсорбции в системе жидкость — жидкость от строения
молекулы ПАВ 426
Зависимость поверхностного натяжения от строения молекул фаз 433
Выбор ПАВ для стабилизации эмульсии 439
Литература 441
Указатель ' . . 443
Издательство «Химия»
Ленинградское отделение
Невский пр., 28 "
ЭМУЛЬСИИ Редактор Л. Ф. Травина
Техн. редактор 3. Е. Маркова
Переплет художника Б. А. Быкова
Корректор Л. А. Любой и ч
Сдано в набор 31/ХП1971г. Подписано к
печати 24/1111972г. Бумага типогр. №2, 60X90'/i«.
Уч.-изд. л. 30,29. Печ. л. 28. Тираж 4600 экз.
Цена 3 р. 27 к. Заказ К» 1070.
Ленинградская типография JM4 6 Главполпграф-
прома Комитета по печати при Совете
Министров СССР. Московский пр., 91.
ЗАМЕЧЕННЫЕ ОПЕЧАТКИ
Страница
49
04
146
162
163
174
170
176
182
210
213
Строка
21 снизу
3 снизу
18 сверху
2 сверху
10 сверху
16 сверху
11 спилу
10 снизу
1 снизу
1 снизу
12 сипзу
Напечатано
звуков
факторов
1 мкм > D ]> 30 мкм
RVl
осматпческого
~~аМ (В)
а™ „ af.T
aMV
водяному
комбинированной
дптчпка
Должно быть
звуком
фактов
1 мкм < D < 30 мкм
R/V2
осмотического
+ стм/в
Fvr F\-
°в и V
°м
водоструйному
калиброванной
датчика
Зап. 1(17»