











































































































































































































































































































































































































































































































































































































































































































































































Автор: Нейланд О.Я.
Теги: органическая химия химия органические соединения физическая химия издательство высшая школа учебник для студентов молекулы
ISBN: 5-06-001471-1
Год: 1990




Текст
О.Я. НЕЙЛАНД
ОРГАНИЧЕСКАЯ
ХИМИЯ
Допущено
Государстпеппым комитетом СССР
по народному образованию
в качестве учебника для студентов
химических специальностей
высших учебных заведений
Москва «Высшая школа» 1990
О.Я. НЕЙЛАНД
ОРГАНИЧЕСКАЯ
ХИМИЯ
Допущено
Государстпеппым комитетом СССР
по народному образованию
в качестве учебника для студентов
химических специальностей
высших учебных заведений
Москва «Высшая школа» 1990
ББК 24.2
Н45
УДК 547
Рецензенты: кафедра органической химии Ростовского государ-
государственного университета (зав. кафедрой проф. Л. Ф. Пожарский) и
проф.- |В. М. Потапов} (Московский государственный университет
им. М. В. Ломоносова)
Ней ланд О. Я.
Н45 Органическая химия: Учеб. для хим. спец. вузов. — M.i
Высш. шк., 1990.—751 с: ил.
ISBN 5-06-001471-1
В учебнике компактно и четко описаны основные классы органических соеди-
соединений, которые расположены по характеристическим группам. Рассматриваются
природа спялен, пространственное строение молекул, механн'-.мы реакции с исполь-
использованием представлении квантовой химии, современные методы физической органи-
органически химии. Основное пипмаипс уделено применению органических соединений
п народном хозяйстве, сырьевым ресурсам органической химии.
1705000000 D309000000)—009 г ББК 24.2
Н 001@1)—90 По—90 547
ISBN 5-06-001471-1 © Издательство «Высшая школа», 1990
ОГЛАВЛЕНИЕ
Предисловие 7
Введение 9
1. Определение органической химии и основные направления ее раз-
развития ..,.,..,,., , . . . 9
2. Сырьевые источники органических веществ .......... 13
3. Простейшие методы исследования органических веществ 15
1. Очистка органических веществ . . , , 15
2. Анализ органических веществ , 19
4. Литература по органической химии . 20
5. Развитие теоретических представлений органической химии , , . 22
1. Теория строения органических соединений , , , 22
2. Классические электронные теории химической связи , , , . . 25
3. Основные принципы квантовой органической химии 31
6. Физические методы в исследовании органических соединений . . 38
1. Рефрактометрия 39
2. Калориметрия 39
3. Измерение электрических дипольных моментов 42
4. Рентгенография и электронография 43
5. Электрохимические методы исследования 46
6. Спектроскопические методы исследования 48
7. Электронные спектры поглощения и эмиссии (люминесценции) 50
8. Инфракрасные спектры поглощения 53
9. Спектроскопия комбинационного рассеяния (Раман-спектро-
скопия) , 54
10. Электронный парамагнитный резонанс 55
11. Ядерный магнитный резонанс 57
12. Спектрополяриметрия , , 59
13. Фотоэлектронная спектроскопия 62
14. Л1асс-спектрометр1!я , 62
7. Основные понятия о реакционной способности органических со-
соединенней 63
1. Кинетика органических реакций 64
2. Типы реакций и реагентов 67
3. Взаимосвязь между строением и реакционной способностью ор-
органических соединений. Корреляционные уравнения 77
8. Классификация и номенклатура органических соединений ... 81
1. Классификация 81
2. Номенклатура 83
ЧАСТЬ 1
УГЛЕВОДОРОДЫ
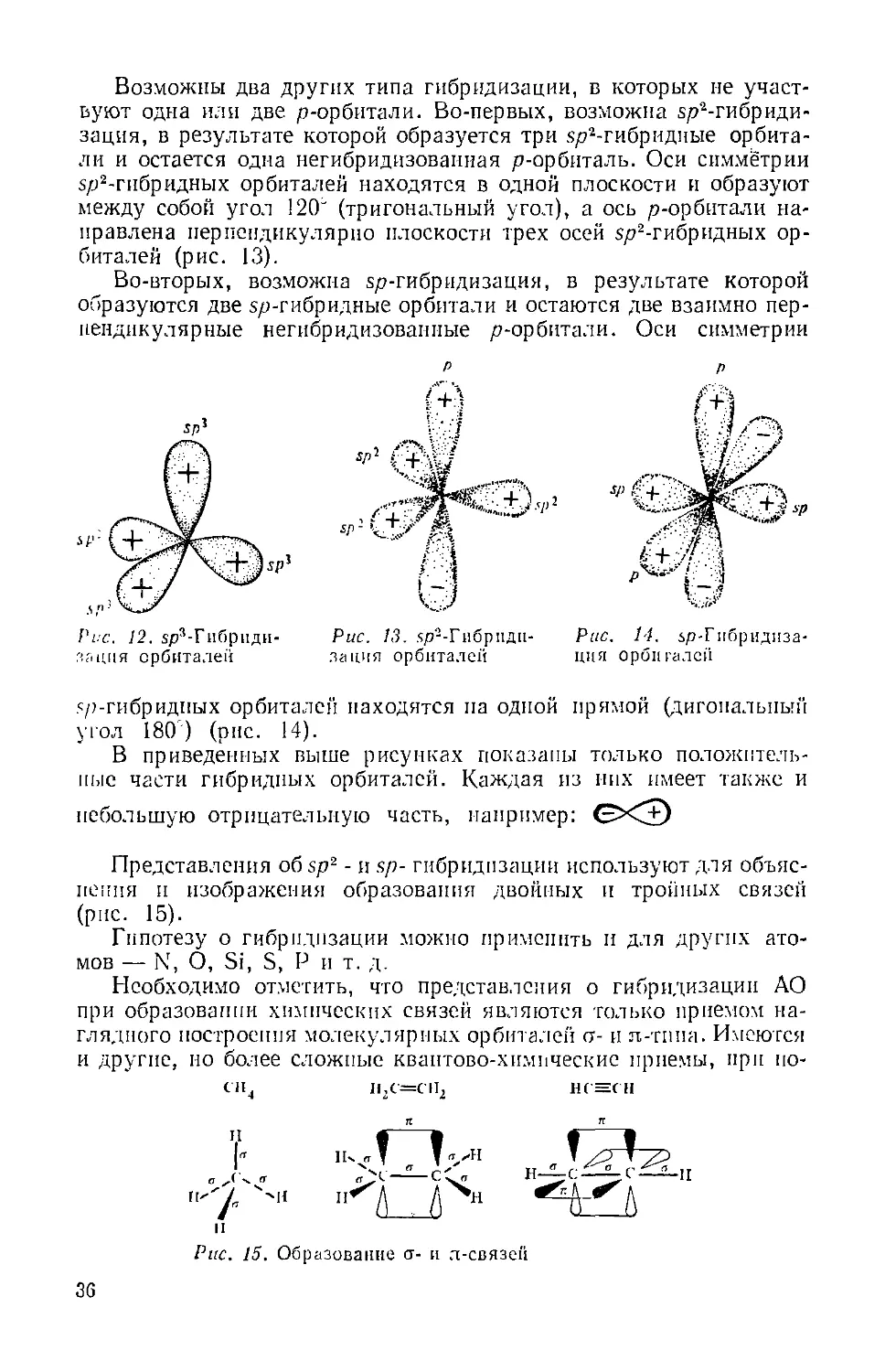
Глава 1. Алкпиы Я6
Глава II. Алкс:ы 102
Глава 111. Алкадиены 130
A. Алкаднены-1,2 (аллепы) 131
Б. Алкадиепы-1,3 132
B. Полнены 143
Глава IV. Ллкины 144
Глава V. Цнклоалканы, циклоалкепы и цнклоалкаднены 159
Л. Циклопропаны и циклопронены 162
Б. Цнклобутапы, цнклобутены и цнклобутадпеп 164
В. Циклопентаны, цнклопептены и цнклопситадиен 166
Г. Цнклогексаны, циклогексеиы н циклогексадиепы 168
Д. Циклогегттамы и цнклогептатриен 172
И. Циклооктаны и цнклооктатетрасн 173
Ж- Декалины 174
3. Циклопентапопергндрофенаптрепы 175
П. Адамаитан 175
Глава VI. Арены 176
A. Арены рада бензола 177
Б. Полициклические арены с изолированными циклами 196
B. Полициклические арены с конденсированными циклами .... 201
Г. Стабильность циклических сопряженных систем и понятие об аро-
ароматичности 212
ЧАСТЬ 2
ФУНКЦИОНАЛЬНЫЕ ПРОИЗВОДНЫЕ УГЛЕВОДОРОДОВ
Галогенпроизводные углеводородов 218
Глмоа VII. Галогеннроизводиые типа C(sp:l)--X 219
Глава VIII. Галогеппронзводные типа C(s/)-)--X 236
Глава IX. Галогенпронзнодные тина C(sp)—X. Галониевые соединения 245
Элементорганические соединения 248
Глава X. Металлорганические соединения 219
Л. Соединения металлов первой группы 249
IV Соединения металлов второй группы 253
В. Соединения металлов третьей группы 258
Г. Соединения металлов четвертой группы 261
Д. Органические соединения переходных металлов 262
Г.шва XI. Борорганические соединения 268
А. Производные борана и дибораиа 268
Б. Карбораиы 270
Глава XII. Кремннйорганические соединения 271
Глава XIII. Фосфорорганические соединения 275
Гндроксилпроизводные углеводородов 280
Глава XIV. Гидрокснлпроизводиые углсподородов со связью
C(.s-p:1)— ОН " 2S0
A. Алкянолы 281
Б. Алкеполы н алкпполы 297
B. Циклоалкаполы и циклоалкенолы 299
Г. Арилалкаполы 300
Д. Диолы 301
Е. Трнолы и полиолы 305
Глава XV. Гндрокснлиронзводиые углеводородом со связью C(sp-)—ОН 308
A. Енолы ' 309
Б. Фенолы 312
B. Арепдиолы и арентриолы 323
Глава XVI. Простые эфпры 327
Л. Диалкиловые эфиры 328
Б. Циклические эфиры 332
В. Виниловые эфиры 341
Г. Алкилариловые эфиры 342
Д. Диариловые эфиры 344
4
Глава XVII. Эфиры неорганических и элемепторганических кислот 345
A. Бораты 345
Б. Нитриты 346
B. Нитраты 347
Г. Гидрогтероксиды и пероксиды 348
Д. Эфнры кремниевой кислоты и кремнийоргани'шс.кнх кислот . . 350
Е. Эфиры фосфорных н фосфорорганически.х кислот 351
Ж- Эфиры серной кислоты 352
Сераорганические соединения 353
Глава XVIII. Сульфоиовые кислоты, их производные и сульфоны . . 354
Глава XIX. Сульфиповые и сульфенопые кислоты и их произподние.
Сульфоксиды 360
Глава XX. Тиолы, сульфиды и дисульфиды 363
Азоторгаиические соединения 3G7
Глава XXI. Ннтросоедипепия 368
A. Нитроалканы 369
Б. Нитроалкепы 377
B. Нитроарепы 378
Глава XXII. Нитрозосоедииення 385
А. Нитрозоалкапы 386
Б. Нитрозоарены и нитрозофеполы 387
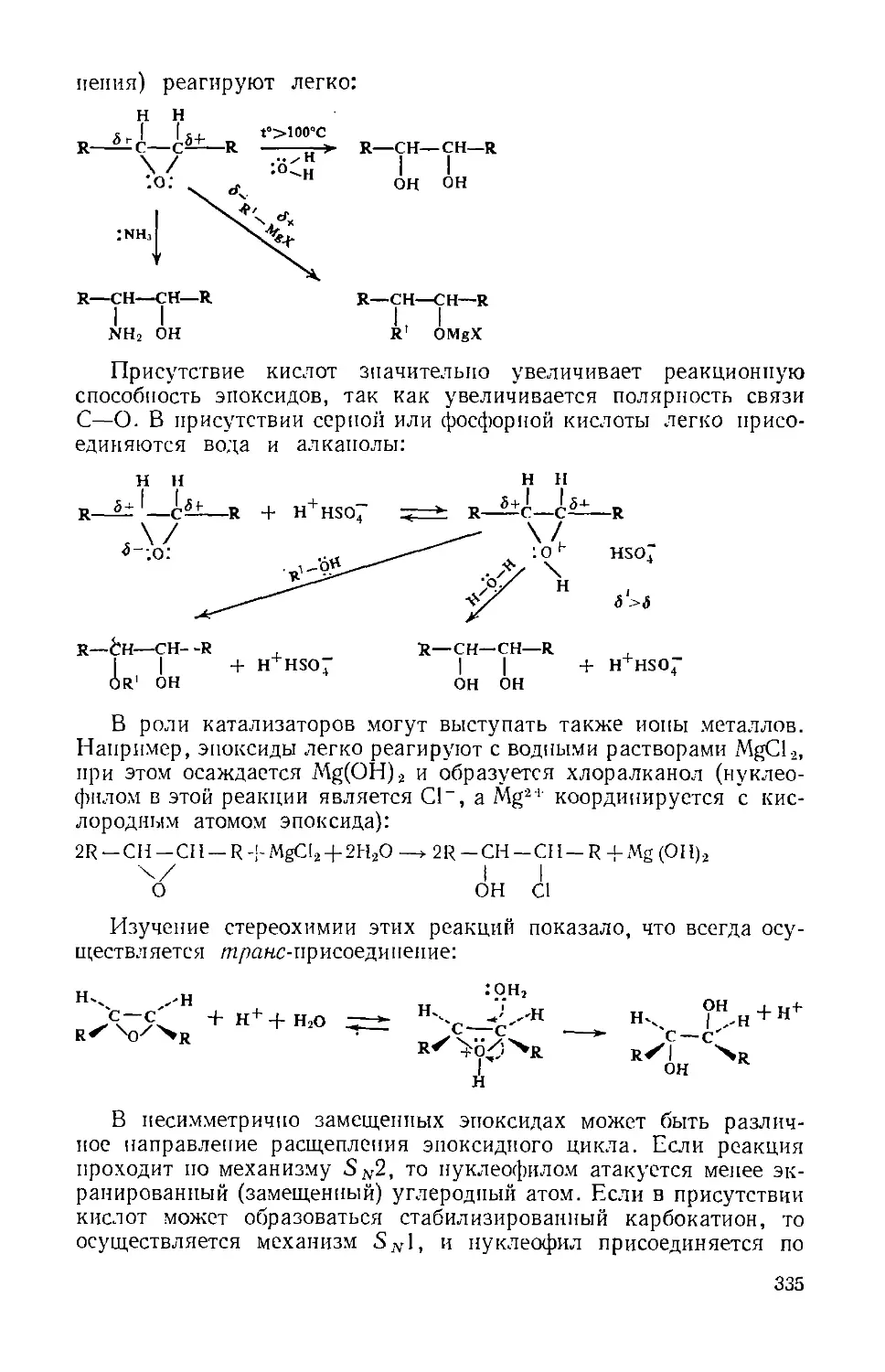
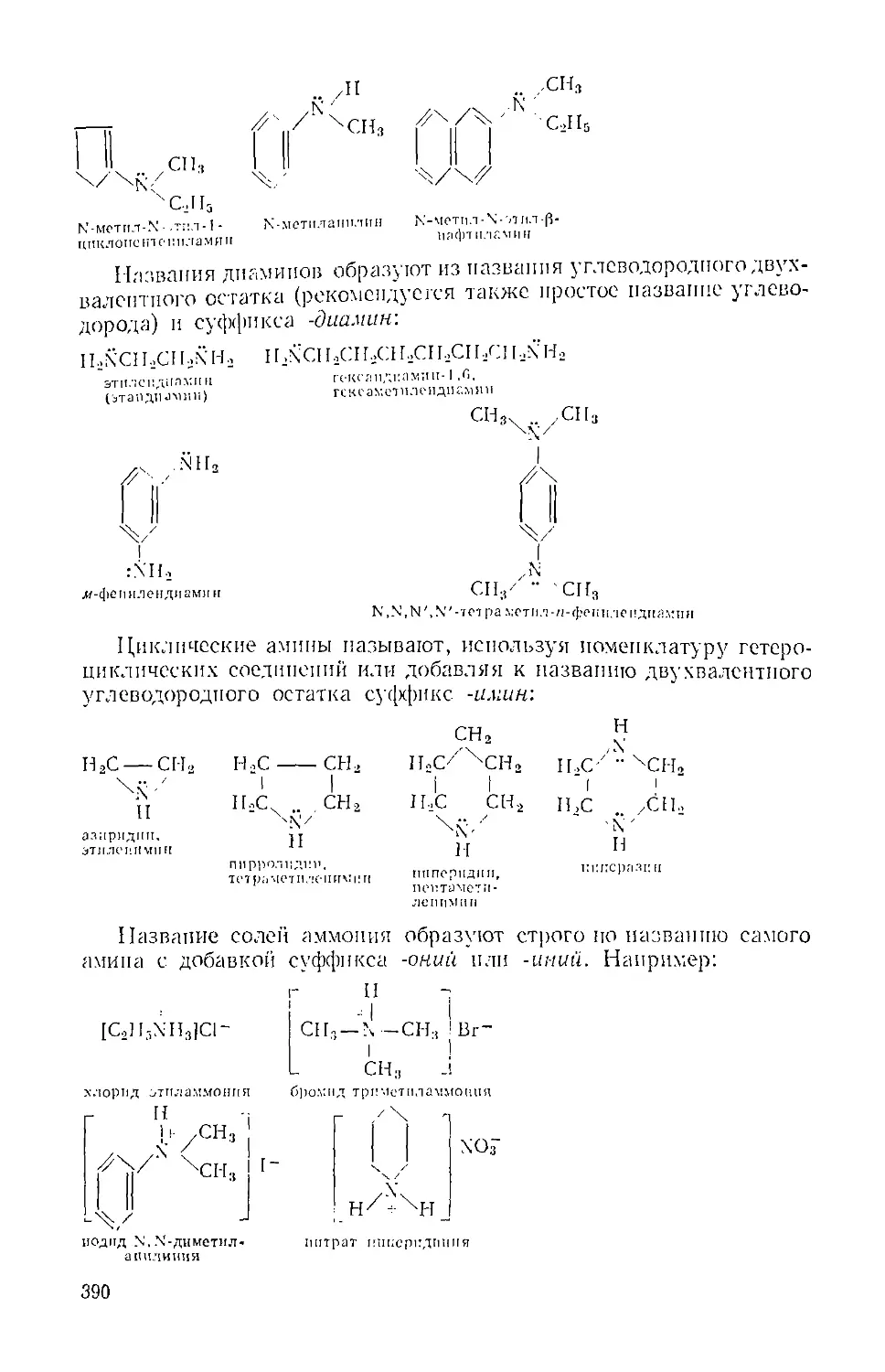
Глава XXIII. Амины и замещенные соли аммония 388
A. Алкиламииы, циклические амины и арилалкиламины 391
Б. Енамипы 404
B. Ариламииы 406
Глава XXIV. Производные гидразина 416
А. Алкилгидразины 417
Б. Арилгндразипы 418
Глава XA'V. Диазосоедипеиня 420
А. Соли арепдиа.юпин _ 421
Б. Диазоалкаиы i 428
Глава XXVI. А.юсоедмнения 430
А. Азоалканы 431
Б. Азоарепы 432
Карбонильные соединения и их производные 435
Глава XXVII. Мопокарбонильные соединения 436
A. Насыщенные карбонильные соединения 439
Б. Ненасыщенные карбонильные соединения 458
B. Кетепы 461
Г. Карбонильные соединения лренов 464
Д. Производные карбонильной группы 469
Глава XXVill. Дикарбонильпые соединения 474
A. 1,2-Дикарбоиильпые соединения 476
Б. 1,3-Дикарбопильные соединения 479
B. 1,4-Дикарбонильные соединения 490
Глава XXIX. Карбонильные соединения, содержащие другие функ-
функциональные группы 491
A. Галогепкарбонилъпые соединения 492
Б. Мопогидроксикарбонильпые и дигидроксикарбокильные соедине-
соединения 493
B. Углеводы 504
Г. Амипокарбонильные соединения 522
Д. а-Диазокарбонильпые соединения 521
Глава XXX. Хиноиы 526
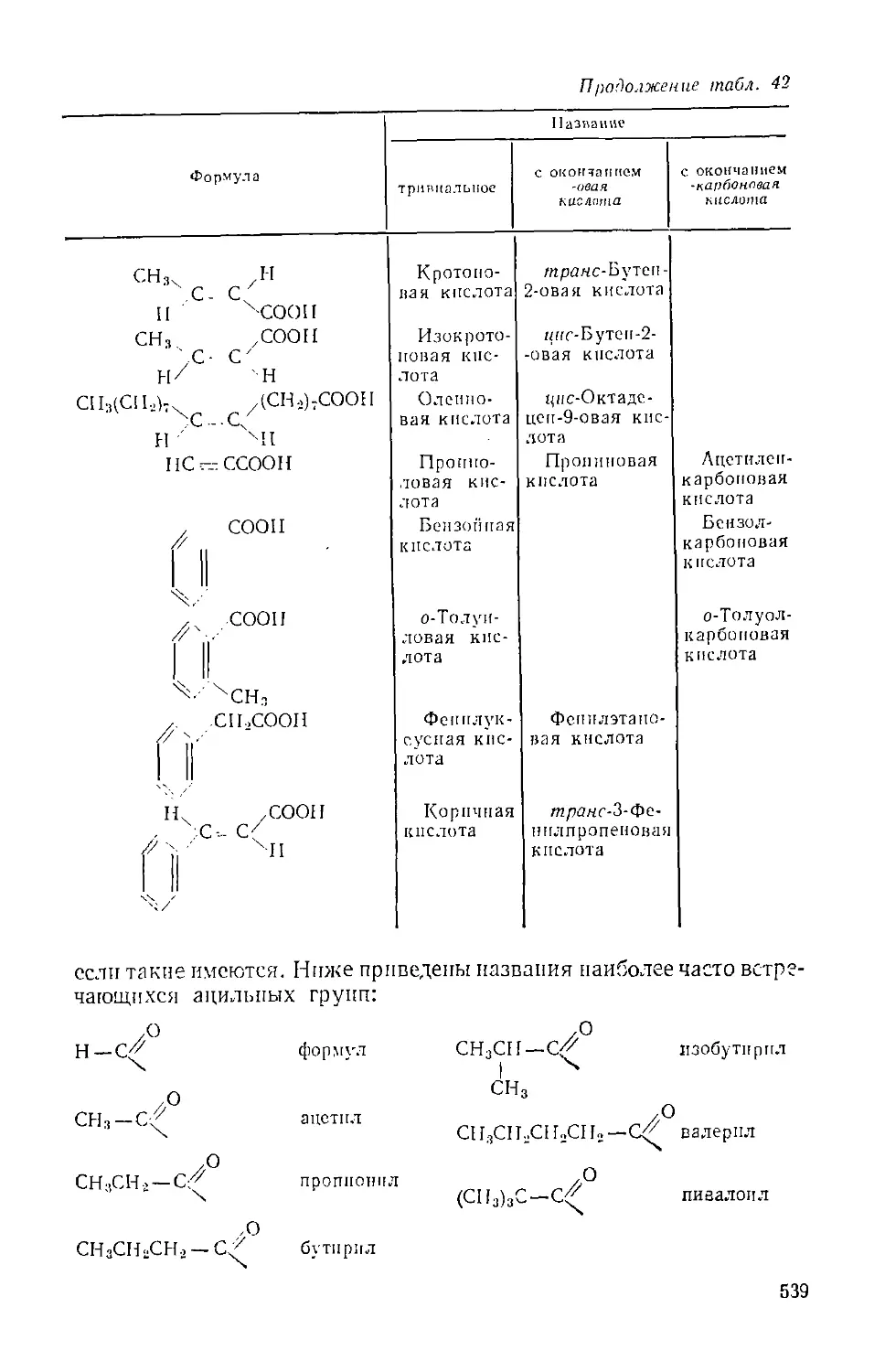
Карбоновые кислоты и их производные 536
Глава XXXI. Монокарбоновые кислоты 5.17
А. Насыщенные монокарбоновые кислоты 510
Б. Ненасыщенные монокарбоновые кислоты 550
В. Лренмонокарбоновые кислоты 553
Глава XXXI/. Дикярбоновые и поликарбоновые кислоты 556
Л. Насыщенные днкарбоновые кислоты 557
В. Ненасыщенные дикарбоповые кислоты 560
В. Лрендикарбонопые и аренполикарбоновыс кислоты 562
Глава XXX1I1. Функциональные производные карбонопых кислот 563
A. Ацилгалогепиды 567
Б. Ангидриды карбопопых кислот 570
B. Сложные эфиры карбоповых кислот и лактоны 573
Г. Ортоэфиры карбоповых кислот 582
Д. Амиды и имиды карбонопых кислот, ляктамы 582
Е. Гидразиды и азиды карбонопых кислот, гидрокспуопые кислоты 590
Ж- Нашдоэфиры карбоповых кислот и амидинн 592
3. Тиокислоты и днттокпелоты 594
И. Пероксикарбоновые кислоты и ацилпероксиды 595
К. Нитрилы (цианиды) 597
Л. Пзоциапнды (изонитрилы) 601
Глава XXXIV. Производные карболовых кислот, содержащие различ-
различные функциональные группы 602
A. Галогенкарбоновые кислоты 604
Б. Гпдрокспкислоты 007
B. Аминокислоты .* . . 615
Г. Велки и полипептпды 625
Д. Оксокнслоты 634
Глава XXXV. Производные угольной кислоты 640
A. Эфиры хлоругольной н угольной кислот 040
Б. Амиды угольной кислоты 643
B, Нитрилы угольной кислоты и кх изомеры 646
Г. Производные тиоугольпых кислот 649
ЧАСТЬ 3
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Гласа XXXVI. Питичленные гетероциклические соединения с одним
гетероатомом 658
А. Пиррол, фурап и тиофен 658
Б, Индол и карбазол 668
Глава XXXVII. Пятичлеппые гетероциклические соединения с дпумя
и более гетероатомачи 674
A. Пиразол и имидазол 674
Б. Соли 1,3-дитполия и производные 1,3 дитпола 679
B. Оксазол, тиазол и беизтиясол 681
Г. Триа.золы и тстразол 685
Глава XXXVIII. Шестпчлспные гетероциклические соединения с од-
одним гетероатомом 686
A. Пиридин 687
Б. Хниолин и изохинолнн 695
B. Пирилиепые соли и пироны 701
Глава XXXIX. Шестнчленные гетероциклические соединения с дпумя
гетероатомами 704
A. Пирпдпзпи, пиримидин и пиразин 704
Б. Пурин 709
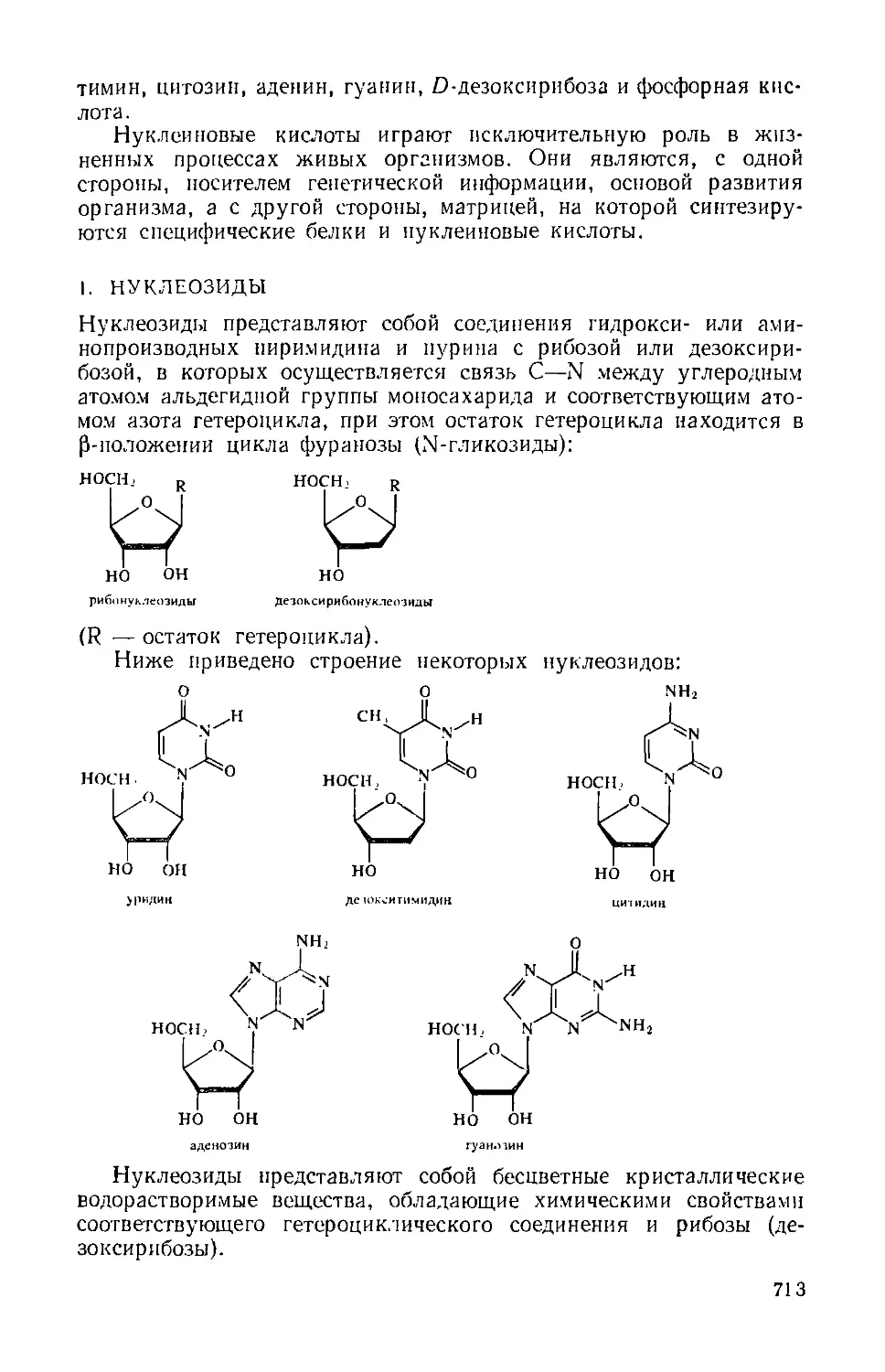
B. Нуклеозиды, нуклеотиды и нуклеиновые кислоты 712
Именной указатель 720
Предметный указатель 723
Книгу посвящаю памяти
своего первого учителя
органической химии,
профессора
Густава Яновича Ванага
ПРЕДИСЛОВИЕ
Органическая химия в наши дни бурно развивается. Число органи-
органических соединений ежегодно увеличивается па несколько сот ты-
тысяч. Химики открывают все новые реакции, совершенствуют уже
известные методы синтеза. Быстрому развитию органической химии
способствует широкое применение новейших физических методов
как для разделения сложных смесей веществ, так и для анализа
органических соединений, установления их строения, изучения
механизмов реакций органических соединении. На базе синтетичес-
синтетической органической химии выросли и стали самостоятельными разде-
разделами теоретическая органическая химия (физическая органическая
химия, квантовая органическая химия, стереохимия), органичес-
органический анализ, биоорганическая химия и др.
Органическая химия создает теоретический фундамент для раз-
развития таких исключительно важных технологических отраслей, как
нефтехимический синтез, основной органический синтез, топкий
органический синтез, промышленность полимеров, фармацевтиче-
фармацевтическая промышленность и др.
Органическая химия играет важнейшую роль в создании прин-
принципиально новых видов продукции и принципиально новых тех-
технологий, новых конструкционных материалов и физиологически
активных веществ для нужд медицины и сельскохозяйственного
производства.
В настоящее время все более сложной задачей становится изу-
изучение огромного материала органической химии: изменяются теоре-
теоретические представления о природе химической связи в органических
соединениях и о механизмах реакций, появляются новые соедине-
соединения, реакции, технологические процессы. Поэтому довольно трудно
в учебнике ограниченного объема изложить материал, соответст-
соответствующий требованиям программы для химических специальностей
высших учебных заведений.
Последовательность изложения материала в этой книге отлича-
отличается от обычно принятой в учебниках. Автор отказался от традици-
традиционного деления на алифатические и ароматические соединения.
В основу построения учебника положена классификация углеводо-
углеводородов и их производных по характеристическим группам. В начале
учебника изложен материал но углеводородам как основе построе-
построения любого органического соединения. Затем следуют функциональ-
ные производные углеводородов, заканчивается книга гетероцик-
гетероциклическими соединениями. Функциональные производные углеводо-
углеводородов расположены от соединений с простой связью С—X к кар-
карбонильным соединениям и карбоновым кислотам, т. е. в порядке
возрастания сложности (степени окисления углеродного атома)
характеристической группы.
Новым является подход к изложению свойств отдельных классов
соединений и характеристических групп в зависимости от типа
гибридизации электронных орбиталей углеродного атома, непосред-
непосредственно связанного с характеристической группой. Использованы
также представления квантовой органической химии и результаты
расчетов по методу Хюккеля.
Соединения с несколькими одинаковыми характеристическими
группами помещены непосредственно после монофункциональных.
Производные с различными (смешанными) характеристическими
группами рассматриваются в главах, посвященных основным клас-
классам органических соединений.
В книге нет специальной главы, посвященной элементам биоор-
биоорганической химии. Почти все необходимые данные но биологически
важным органическим соединениям приведены в главах, посвящен-
посвященных соответствующим классам соединений.
Во введение книги кроме исторического обзора включены крат-
краткие сведения о методах очистки и анализа органических соединений,
о классической электронной теории строения и стереохимии, о фи-
физических методах исследования, в том числе спектроскопических,
о реакционной способности, типах реакций и реагентов, о классифи-
классификации и номенклатуре.
Изучение курса органической химии не обязательно начинать
с подробного изучения материала введения. С ним можно знакомить-
знакомиться постепенно, в процессе изучения основного материала.
Преимущества книги, по мнению автора, кроются в применяемой
системе изложения материала, что позволяет весьма сжато, но на
достаточно высоком уровне дать исчерпывающую информацию о
всех важнейших классах органических соединений, их получении,
физических и химических свойствах, практическом применении.
Материал книги в основном соответствует программе курса ор-
органической химии для университетов и технологических вузов. Не-
Некоторые разделы рассмотрены более глубоко (элементорганические
и азоторганические соединения, карбоновые кислоты и их производ-
производные, производные угольной кислоты, гетероциклические соединения).
В основу книги легли курс лекций по органической химии, чита-
читаемый автором в Рижском политехническом институте в течение
последних 20 лет, и учебное пособие, написанное автором и издан-
изданное в Риге на латышском языке издательством «Звайгзне» в 1977 г.
Автор глубоко признателен рецензентам книги проф. В. М. По-
Потапову, проф. А. Д. Гарновскому и ст. науч. сотр. Ю. Е. Алексееву
за тщательный просмотр рукописи и ценные советы, немало спо-
способствовавшие улучшению книги.
Автор
8
Введение
I. ОПРЕДЕЛЕНИЕ ОРГАНИЧЕСКОЙ ХИМИИ
И ОСНОВНЫЕ НАПРАВЛЕНИЯ ЕЕ РАЗВИТИЯ
«При переходе от исследований неорганических веществ к иссле-
исследованиям органических веществ химик попадает в совершенно
новую область,— писал в 1808 г. известный шведский химик
Й. Берцелиус,— так как органическая химия является резко отли-
отличающейся отраслью науки». Берцелиус был первым, кто в курсе
химии выделил специальную главу «Органическая химия».
С органическими веществами человечество познакомилось в са-
самый ранний период развития. Органические вещества получали
из растений и животных организмов. Эти вещества обладали мень-
меньшей устойчивостью, чем вещества неживой природы, и имели более
сложный состав. Приготовление пищи и одежды были первыми хими-
химическими процессами, которые уже в древности привели к получению
первых индивидуальных органических веществ, таких, как сахар,
спирт, уксус, винный камень, красители и др.
До середины XVIII в. органические вещества систематически
не изучались. Первым химиком, который вплотную начал занимать-
заниматься ими, был К. Шееле (около 1770 г. До него были известны только
четыре органические кислоты: уксусная, муравьиная, бензойная и
янтарная. Шееле из природных продуктов получил винную, молоч-
молочную, лимонную, яблочную и другие кислоты, а также глицерин.
Берцелиус в своем курсе химии рассматривал органические
вещества в отдельной главе, при этом особенно подчеркивал боль-
большие различия между неорганическими и органическими вещества-
веществами. Он считал, что неорганические соединения можно получить в
лаборатории в результате различных химических превращений. Ор-
Органические же соединения образуются только в организмах в ре-
результате жизненных процессов, под влиянием таинственной «жиз-
«жизненной силы» (vis vitalis). Так утвердилась теория витализма, сог-
согласно которой органические вещества не могут быть получены нз
простых неорганических веществ. Эта теории в значительной мере
тормозила развитие исследований в органической химии. Но были
химики, которые пытались доказать, что органические вещества
могут быть получены в колбах из простых неорганических веществ,
т. е. могут быть синтезированы. Борьба между сторонниками вита-
витализма и химиками-синтетиками уже давно принадлежит истории
химии. Но эта борьба способствовала развитию таких важных сторон
органической химии, как органический синтез и органический ана-
анализ.
Первые синтезы органических веществ удалось провести немец-
немецкому химику Ф. Вёлеру. В 1824 г. он наблюдал образование щаве-
щавелевой кислоты из дициана, а в 1828 г.— образование мочевины
из цианата аммония. Были разработаны методы для элементного
анализа органических соединений: Ж- Дюма разработал метод
количественного определения азота, а Ю. Либих — метод определе-
определения углерода и водорода в органических соединениях. В середине
XIX в. быстро расцвел органический синтез. В 1845 г. Г. Кольбе
синтезировал уксусную кислоту, в 50-е годы М. Бертло из простых
неорганических веществ синтезировал муравьиную кислоту, этило-
этиловый спирт, ацетилен, бензол, метан, а из глицерина и жирных кис-
кислот получил жиры.
Русский химик А. М. Бутлеров в 1861 г. из метилениодида полу-
получил полимер формальдегида, а на основе последнего впервые полу-
получил сахаристое вещество «метиленитан», т. е. осуществил первый
полный синтез сахаристого вещества.
Одновременно развивались и укреплялись методы количествен-
количественного элементного анализа органических соединений.
Весьма скоро химики убедились, что органические вещества
подчиняются тем же закономерностям, что и неорганические. Но
деление химии на неорганическую и органическую сохранилось.
Критерием деления стал состав веществ. Л. Кекуле в 1851 г. опре-
определил органическую химию как химию соединений углерода. Одна-
Однако это определение не вполне последовательно. Есть группы сое-
соединений углерода, которые все-таки причисляют к неорганическим
(оксид и диоксид углерода, карбопилы металлов, карбонаты, кар-
карбиды). В то же время все металлорганические соединения могут
быть причислены к органическим. Определение, данное Кекуле,
упускает из виду принципы образования органичесьлх соедине-
соединений.
Более точное определение органической химии было дано К. Шо-
рлеммером в 1889 г.: «Органическая химия является химией угле-
углеводородов и их производных». Это определение тоже не проводит
резкой границы между органической и неорганической химией.
Так, например, оксиды углерода мы можем рассматривать как
неорганические и как органические (производные метана) соедине-
соединения. Большое семейство элементоргаиическнх соединений принад-
принадлежит одновременно как органической, так и неорганической хи-
химии («третья химия»)
Все органические соединения являются производными соедине-
соединений углерода и водорода — углеводородов. Углеводородов очень
много. При замещении одного или более водородных атомов други-
другими атомами или группировками получаем новые соединения. Анало-
Аналогично, замещая углеродный атом другими (гетероатомами), полу-
получаем новые соединения, в том числе гетероциклические. Принципом
построения органических соединений является замещение.
Органические соединения часто кроме атомов углерода и водо-
водорода содержат кислород, азот и другие элементы. Существует
также возможность различного соединения атомов друг с другом.
10
Каждое конкретное соединение имеет определенное расположение
атомов в молекуле, т. е. имеет определенное строение, или структуру.
Понятие о «троении (структуре) органических соединений и
соответствующая теория химического строения (структурная теория)
возникли в 1858—1861 гг. и большую роль в этом сыграли работы
трех ученых, имена которых вошли в историю химии,— немецкий
химик А. Кекуле, шотландский химик А. Купер и русский химик
А. М. Бутлеров. На основе теории строения стал возможным быст-
быстрый прогресс органической химии.
Развитие органической химии идет по двум основным направле-
направлениям: с одной стороны, это развитие теоретической и синтетической
органической химии, а с другой — развитие промышленного орга-
органического синтеза. Синтетическая органическая химия занимается
получением различных, в том числе новых органических соедине-
соединений и разработкой новых методов синтеза. Для успешного разви-
развития синтеза необходимы надежные методы анализа. Фактический
материал, накопленный синтетической органической химией, систе-
систематизирует и объясняет теоретическая органическая химия.
В свою очередь, новые теоретические выводы стимулируют поиск
новых типов реакций и новых классов соединений. В этом заключа-
заключается единство синтетической и теоретической органической химии,
и на этом твердом фундаменте строится многоэтажное здание ор-
органической химии. Сказанное иллюстрирует сам ход развития
химии.
Первый период — до рождения теории химического строения
A820—1860) — характеризуется интенсивным поиском в области
синтеза для получения природных органических веществ. Исследо-
Исследования еще разрознены и не объединены в общую систему, т. е. фак-
фактов много, а взаимосвязи уловить не удается. Этот период хорошо
описывает Ф. Вёлер в одном из своих писем Й. Берцелиусу A835):
«Органическая химия может сейчас кого угодно свести с ума.
Она представляется мне дремучим лесом, полным удивительных
вещей, безграничной чащей, из которой нельзя выбраться, куда
не осмеливаешься проникнуть». Этот период закончился созданием
теории строения, на основе которой и родилось неисчислимое коли-
количество новых идей.
Для периода, предшествующего возникновению электронных
теорий A860—-1910), характерен особенно интенсивный расцвет
синтеза, открытие новых классов органических соединений, синтез
сложных природных веществ. Удалось синтезировать такие веще-
вещества, как природные красители ализарин (К. Гребе, К. Либерман,
1869) и индиго (А. Байер, 1879), алкалоид никотин (А. Пикте, 1904).
Развивалась химия синтетических красителей, создавались первые
синтетические лекарственные вещества. Зародились основы стерео-
стереохимии A874).
Первые десятилетия XX в. отмечены внедрением в органическую
химию новых методов исследования — физических методов, про-
проникновением электронных теорий в теоретическую органическую
химию, проведением искусных синтезов природных веществ — ca-
call
харов, полипептидов, пуринов, дубильных веществ (Э. Фишер).
В 30-е годы создавалась квантовая органическая химия.
Современную синтетическую и теоретическую органическую хи-
химию отличает широкое применение физических методов, которые
облегчают выяснение структуры соединения и исследование меха-
механизма реакции. Современная органическая химия вооружена мно-
множеством специфических приемов для введения определенных групп
в органические соединения, эффективными методами для разделения
смесей и очистки веществ. Стабильной теоретической базой орга-
органической химии являются электронная теория и представления
квантовой химии. В настоящее время можно синтезировать почти
любое сложное органическое соединение, теоретически можно пред-
предсказать существование новых необычных соединений. Синтезиро-
Синтезированы природные соединения с очень сложной структурой: алкалоиды
стрихнин и морфин, зеленый пигмент растении хлорофилл, витамин
В12 (Р. Вудворд), полипептиды с более чем 30 остатками аминокис-
аминокислот: например, гормон инсулин человека, состоящий из 51 остатка
аминокислот (II. Зибер), рибонуклеиновые кислоты, состоящие из
50 и более нуклеозидов (Г. Корана).
На стыке двух наук — биохимии и органической химии — воз-
возникли новые научные направления —¦ молекулярная биология и
биоорганическая химия. Молекулярная биология — наука, ста-
ставящая своей задачей познание природы явлений жизнедеятельности
путем изучения биологических объектов и систем на уровне, при-
приближающемся к молекулярному, а в ряде случаев и достигающем
этого предела. В первую очередь это касается белков и нуклеиновых
кислот. Биоорганическая химия изучает органические вещества,
участвующие в процессах жизнедеятельности (белки, нуклеиновые
кислоты, ферменты, витамины, углеводы, липнды, гормоны, алка-
алкалоиды и др.), занимается моделированием основных биопроцессов.
С другой стороны, органическая химия в некоторых областях
стыкуется с физикой твердого тела. Синтезируются органические
соединения, которые в твердом состоянии обладают свойствами по-
полупроводников и металлов (органические полупроводники и «ор-
«органические металлы») *. Отмечены случаи, когда при низких тем-
температурах (ниже 11 К) кристаллы некоторых органических сое-
соединений приобрели свойство сверхпроводимости.
Органический синтез является не только основой органической
химии, самой необходимой частью ее прогресса, по и отраслью
увлекательной научной работы, которая полна приключений и
требует высокого мастерства.
Достижения органического синтеза непосредственно влияют на
развитие промышленного органического синтеза. В свою очередь
промышленный органический синтез и народное хозяйство в целом
выдвигают новые проблемы перед теоретической и синтетической
органической химией. Зарождение промышленного органического
* «Органическими металлами» называют органические соединения, об-
лядающие в твердом состоянии большой электрической проводимостью, воз-
возрастающей при охлаждении,
12
синтеза следует искать в начале XIX в., когда ученые начали ис-
исследовать продукты сухой перегонки каменного угля, открыли
бензол, другие ароматические углеводороды и их производные и
искали для них применение. Получение бензола в больших коли-
количествах из дешевого сырья способствовало изучению его химических
свойств. В результате был получен синтетический анилин (Н. Н. Зи-
нин, 1842) и на его базе синтетические красители. Производство
органических красителей (анилинокрасочная промышленность) бы-
было одним из первых промышленных органических синтезов. Деше-
Дешевые синтетические красители вытеснили более дорогие природные.
Это одна из характерных черт промышленного органического син-
синтеза — производство продукции, более дешевой по сравнению с
природными продуктами, и производство веществ, вообще не встре-
встречающихся в природе.
В настоящее время экономический и военный потенциал любого
государства оценивается, в частности, и по уровню развития хими-
химической промышленности, в том числе промышленного органического
и нефтехимического синтеза, так как он включает такие важные
отрасли промышленности, как переработка нефти, природного газа
и каменного угля, производство синтетического каучука, полимер-
полимерных материалов, органических красителей, взрывчатых веществ,
лекарственных веществ, средств для борьбы с сельскохозяйственны-
сельскохозяйственными вредителями (пестицидов) и т. д.
2. СЫРЬЕВЫЕ ИСТОЧНИКИ ОРГАНИЧЕСКИХ ВЕЩЕСТВ
Первыми источниками получения органических веществ были живот-
животные и растительные организмы V продукты их жизнедеятельности.
Каждый живой организм представляет собой своеобразную хими-
химическую лабораторию, в которой осуществляются как процессы
синтеза, так и распада. В растительных организмах из простых
исходных веществ (диоксид углерода, вода) под воздействием сол-
солнечной энергии синтезируются сложные органические вещества
(фотосинтез). В животных организмах, наоборот, сложные органи-
органические вещества (сахара, белки, жиры) распадаются на более про-
простые, часть из них как бы «сгорает», отдавая энергию и превращаясь
в СО2 и Н2О, но в то же время в организме также синтезируются
специфические белки, жиры и другие вещества. Растительный мир
является главным производителем органических веществ. Особое
место в этом отношении занимают деревья. Древесина и полученные
из нее целлюлоза и лигнин являются ценным сырьем для химичес-
химической переработки. Так, например, сухая перегонка древесины с
давних времен применялась для получения органических соедине-
соединений, таких, как уксусная кислота, метиловый спирт (древесный
спирт), ацетон, фенолы.
В настоящее время химическая переработка древесины использу-
используется в бумажной промышленности, в производстве целлюлозы,
искусственного волокна, кинофотопленки. Кислотный гидролиз дре-
13
весины дает смесь моносахаридов, используемую в производстве
глюкозы, этилового спирта, фурфурола и других органических
соединений.
В природе происходит непрерывный процесс превращения орга-
органических веществ. В организмах органические вещества распада-
распадаются (окисляются) быстро, а при соприкосновении с воздухом идет
медленное окисление и другие процессы (например, гниение), кото-
которые вызываются воздействием микроорганизмов. В результате этих
процессов образуются как простые вещества (СН4, СО2, НаО, NH3),
так и более сложные органические вещества. Если превращение
протекает без доступа воздуха, тогда очень медленно образуются
более богатые углеродом продукты (процесс обугливания). Таким
путем из остатков древних растений в течение миллионов лет обра-
образовался каменный уголь.
Продукты сухой перегонки каменного угля (коксования) — кок-
коксовый газ, каменноугольная смола, подтаольпая вода — являются
ценными источниками для получения различных органических соеди-
соединений: бензола, толуола, ксилола и других ароматических углеводо-
углеводородов, фенола и его гомологов, пиридина и других гетероцикличе-
гетероциклических соединений. Следует упомянуть и такие источники сырья,
как горючие сланцы и торф.
В XX в. с каменным углем успешно соперничают нефть и при-
природный газ. Согласно некоторым теориям нефть образовалась из
останков древних организмов. Следовательно, источники этого
сырья тоже следует искать в живой природе. В результате перера-
переработки нефти получают различные углеводороды, являющиеся не
только топливом для двигателей внутреннего сгорания и реактив-
реактивных, но и ценнейшим и незаменимым сырьем для промышленного
органического синтеза. Главной составной частью природного газа
является метан. В настоящее время используют метан и как ценное
топливо, и в органическом синтезе (например, для получения аце-
ацетилена).
В настоящее время мировог производство продуктов основного
органического синтеза базируется на использовании нефти и при-
природного газа (около 95%). Уголь, сланцы, торф, древесина обес-
обеспечивают не более 5% потребляемого сырья.
Химической переработке подвергается около 5% добываемой
нефти и ее продуктов. Остальная часть сжигается в различных дви-
двигателях и топках, в результате чего в воздух выбрасывается ог-
огромное количество СО2 (около 20 миллиардов тони в год).
Ресурсы нефти и газа ограничены. Ресурсы угля довольно вели-
велики. На его основе можно организовать выработку синтетических
нефти и газа.
Назрела проблема поисков нового сырья для органического и
нефтехимического синтеза. Это может быть продукт фотосинтеза —
самого крупного химического процесса на земле — растительная
биомасса, состоящая в основном из целлюлозы и более простых
углеводов (сахаров). Актуальными становятся промышленные син-
синтезы на основе СО2. Поэтому во многих лабораториях мира в на-
14
стоящее время ведутся исследования по использованию СО2, расти-
растительной биомассы, угля, сланцев, древесины для получения деше-
дешевого углеводородного сырья.
3. ПРОСТЕЙШИЕ МЕТОДЫ ИССЛЕДОВАНИЯ
ОРГАНИЧЕСКИХ ВЕЩЕСТВ
1. ОЧИСТКА ОРГАНИЧЕСКИХ ВЕЩЕСТВ
При изучении органических веществ химики уже давно столкнулись
с существенным вопросом: что считать чистым, индивидуальным
органическим веществом и как его получить?
Органические вещества, встречающиеся в природе, а также
получающиеся в лабораториях и на химических заводах, обычно
представляют собой смеси нескольких органических соединении.
Компонентами смеси могут быть и неорганические вещества (соли,
вода и др.)- Для оценки чистоты вещества выбирают такие физико-
химические характеристики, которые меняются в зависимости от
степени его чистоты и являются постоянными для чистого инди-
индивидуального вещества.
Для характеристики чистоты вещества используют следующие
константы и методы: температура плавления, температура кристал-
кристаллизации, температура кипения, коэффициент преломления света,
плотность, данные спектров поглощения (коэффициент интенсив-
интенсивности поглощения в электронных и инфракрасных спектрах), дан-
данные спектров ядерного магнитного резонанса (ЯМР), масс-спектро-
метрии, хроматографический анализ, люминесцентный анализ и др.
Получить чистое вещество — означает разделить данную смесь
веществ на индивидуальные вещества, очистить до желаемой степени
чистоты. Здесь необходимо различать две совокупности методов:
методы разделения смеси на компоненты, которые еще не являются
чистыми, и методы конечной очистки. Сегодня мы знаем методы
разделения, которые дают компоненты высокой чистоты: напри-
например газовая хроматография, жидкостная хроматография под вы-
высоким давлением.
Говоря о чистоте химических веществ, нужно отдавать себе от-
отчет в том, что абсолютно чистое вещество можно представить только
теоретически. Абсолютно чистых веществ нет и быть не может. В за-
зависимости от метода очистки вещество содержит определенное
количество примесей. Обычными методами очистки можно достичь
содержания основного вещества 99,9. . .99,95%. Специальными
методами глубокой очистки можно уменьшить содержание примесей
для органических веществ до 10~3. . . 10~4%.
Кристаллизация. Кристаллизация является классическим ме-
методом очистки кристаллических веществ. Метод основан на том, что
разные вещества имеют разную растворимость в определенном
растворителе, причем понижение температуры (за редкими исклю-
исключениями) приводит к уменьшению растворимости веществ. Фильтро-
15
ванием горячего раствора отделяют нерастворимые примеси, и после
охлаждения вещество выделяется из раствора в виде кристаллов.
Повторные перекристаллизации обычно уменьшают количество
примесей. Вариантом метода является кристаллизация из расплава.
Специальный вариант — зонная плавка—'Применяется для глубо-
глубокой очистки веществ.
Возгонка (сублимация). Многим кристаллическим веществам
свойственна способность к возгонке, т. е. к переходу в газовую
фазу, минуя жидкую, с последующей кристаллизацией из газовой
фазы. Этот метод позволяет отделить сублимирующиеся вещества
от несублимирующихся примесей и разделить смесь веществ с раз-
разными температурами сублимации или температурами кристаллиза-
кристаллизации из газовой фазы {градиентнаявозгонка). 1?сли вещества возгоня-
возгоняются трудно и при высоких температурах разлагаются, применяют
возгонку в вакууме или в высоком вакууме — до 0,0013 Па A0~5 мм
рт. ст.) *. Высоковакуумная возгонка в различных вариантах
применяется для глубокой очистки.
Перегонка (дистилляция). Для многих низкоплавких веществ
и большинства жидких хорошим методом очистки является фрак-
фракционная перегонка при условии, что разница в температурах кипе-
кипения компонентов смеси достаточно велика и не образуются
азеотропные смеси. Селективность (эффективнесть) фракционной
перегонки можно увеличить специальными приспособлениями:
дефлегматорами, дистилляционными колоннами и др. Для высо-
кокипящих веществ применяется вакуумная перегонка. Вариантом
метода является перегонка двухкомпонентных систем, которые при
охлаждении расслаиваются, например перегонка с водяным паром.
Хроматография. Методы хроматографического разделения ос-
основываются на различной способности веществ адсорбироваться на
поверхности сорбента или распределяться между двумя несмешиваю-
щимися фазами (жидкость — жидкость, жидкость — газ), из ко-
которых одна фаза (жидкая) находится на поверхности сорбента.
Поэтому различают разные виды хроматографии, а именно: жидко-
жидкостную адсорбционную и распределительную хроматографию, газо-
газовую хроматографию.
Жидкостная адсорбционная хроматография основана на различ-
различной способности веществ сорбироваться на поверхности сорбента и
десорбироваться при пропускании растворителя — элюента. В ка-
качестве сорбентов применяют оксид алюминия, кремниевую кислоту
и диоксид кремния (силикагели), гранулированные полисахариды
(например, декстраны) или другие полимеры, которые в раствори-
растворителе набухают, образуя гранулированный гель (гель-хроматогра-
(гель-хроматография).
Осуществляют разделение двумя способами: в хроматографпче-
ских колонках и в тонком слое сорбента (тонкослойная хроматогра-
хроматография). На рис. 1 дана принципиальная схема разделения смеси в
колонке.
* I мм. рт. ст.= 133,3 Па.
16
Элюент
/
На рис. 2 изображена схема установки для тонкослойной хрома-
хроматографии. На стеклянную пластинку нанесен тонкий слой сорбента.
Он может быть незакрепленным или закрепленным. На пластинке
помещают каплю раствора смеси веществ. Элюент на пластинку
поступает через капиллярную щель
или же конец пластинки просто
погружают в элюент.
На пластинке можно наблюдать
разделение смеси на компоненты
визуально непосредственно после
хроматографирования, если ве-
вещества окрашены, или после об-
обработки химическим проявителем
(рис. 3), или же, если разделяе-
разделяемые компоненты способны флуорес-
флуоресцировать, рассматривая пластинку
под ультрафиолетовой лампой.
Жидкостная распределительная
хроматография является разно-
разновидностью адсорбционной хрома-
хроматографии, в которой сорбент (но-
(носитель) покрыт тонкой пленкой какой-то жидкости. Элюентом обыч-
обычно является растворитель, который не смешивается с жидкостью
на сорбенте. При пропускании элюента происходит распределение
веществ между жидкой фазой и элюентом.
Рис. 1. Принципиальная схема
колоночном хроматографии
-••—-е-—
о
о о
о о
Рис. 2. Установка для тонкослойной хро-
хроматографии:
1 — сорбент; 2 — пластинка; 3 — элюент; 4 —
капиллярная щель; 5 — крышка
Рис. 3. Тонкослойная хро-
матограмма:
д-до элюацин; б —после
элюацни
Если сорбентом распределительной хроматографии является
бумага (вода сорбирована на целлюлозе), метод называется бумаж-
бумажной хроматографией. Применяются также крахмал и специальные
силикагели, обработанные водой. Метод осуществляется в колонке
или в тонком слое.
Ионообменная хроматография. В этом методе в качестве сорбен-
сорбента используют специальные материалы — иониты. Ионнты пред-
представляют собой полимеры с кислотными или основными группами.
Эти полимеры в воде не растворяются, а только набухают. Метод
применим только для разделения смеси органических кислот или
оснований. Вещества на сорбенте связываются химически (солеобра-
зование). В качестве элюентов применяются растворы кислот или
17
оснований с различными рН. Процесс осуществляется в колонках
или в тонком слое.
Тонкослойная хроматография является аналитическим методом.
Колоночная хроматография применяется в препаративных целях.
В последние годы развивается колоночная хроматография под дав-
давлением (элюент продавливается через колонку под высоким давле-
давлением и состав элюента анализируется в автоматическом режиме).
В этом случае можно использовать длинные колонки, что позволяет
достичь более эффективного разделения. Сконструированы специ-
специальные приборы, которые могут работать в аналитическом и пре-
препаративном режимах.
Газовая хроматография применяется для разделения смесей газо-
газообразных или легкоиснаряемых жидких и твердых веществ. Прин-
Принцип метода подобен жидкостной хроматографии. Разделяемую смесь
разбавляют газом-носителем (Н2, N2, He) и вводят в адсорбцион-
адсорбционные колонны. Газ-носитель является одновременно растворителем
и элюентом. В качестве сорбентов используют тонкие порошки сили-
силикатных материалов, которые могут быть чистыми (газо-адсорбцион-
ная хроматография) или покрытыми пленкой нелетучей жидкости
(газо-жидкостная хроматография). Используют также капилляры,
покрытые внутри пленкой нелетучей жидкости (капиллярная хро-
хроматография). Газ-носитель постепенно десорбирует компоненты
Рис. 4. Принципиальная схема газового хроматографа:
/-регулятор потока гам-носителя; 2-термостат; 3 -адсорбционная колонка;
4-детектор; 5 —самописец; б" —установка для улавлиндиия фракций
смеси и уносит с собой. Присутствие органических веществ в газе-
ноентеле и их количество обнаруживается при помощи специальных
детекторов и фиксируется самописцем. В препаративной хромато-
хроматографии газ-носитель затем пропускают через специальные приемни-
приемники, в которых органические вещества улавливают вымораживанием
(рис. 4).
Этим методом можно достичь полного разделения смеси. При ис-
использовании адсорбционных колонн повышенной мощности метод
применяется как препаративный для разделения небольших коли-
количеств веществ A. . .Юг).
Гель-фильтрация. При гель-фильтрации гель действует подобно
молекулярному ситу, разделяя молекулы в зависимости от молеку-
18
лярной массы и размера. Матрица представляет собой множество
пористых частиц, между которыми находится элюент. Если в верх-
верхнюю часть колонки внесена анализируемая смесь, то большие моле-
молекулы не могут войти в поры между частицами и элюируются пер-
первыми. Вещества с молекулами меньшего размера задерживаются на
некоторое время в порах геля. Поэтому при разделении смеси ве-
веществ компоненты смеси выходят из колонки в порядке уменьшения
молекулярной массы и размера молекул.
Метод осуществляется в виде колоночной хроматографии. В ка-
качестве гелей применяют гранулированные набухшие полисахариды
и другие полимеры. Гель-фильтрацию используют для разделения
смесей биологических объектов (белков и др.).
Электрофорез. Разделение компонентов смеси при электрофоре-
электрофорезе основано на различии в их подвижности в постоянном электри-
электрическом иоле на каком-то сорбенте, обычно в геле (например, в по-
лнакриламндном геле). Метод применим для разделения заряжен-
заряженных частиц (катионов или анионов).
Образец наносят в виде узкой зоны на поверхность геля. При
наложении электрического поля компоненты мигрируют в гель.
Зоны компонентов с различными подвижностями отделяются друг от
друга и передвигаются с различными скоростям;; через голь в
элюционную камеру (нижнюю часть колонки), отку, а они вымыва-
вымываются непрерывным потоком элюента — буферного раствора с
определенным рН.
2. АНАЛИЗ ОРГАНИЧЕСКИХ ВЕЩЕСТВ
После того как вещество получено в чистом виде, оно может быть
подвергнуто дальнейшим исследованиям.
Первой задачей является качественное и количественное опре-
определение элементного состава. Затем по данным элементного анализа
вычисляют простейшую суммарную формулу, определяют молеку-
молекулярную массу и вычисляют истинную молекулярную брутто-фор-
мулу. И наконец, заключительным этапом является определение
молекулярной структуры. Это является самой сложной задачей. Для
этой цели используют химические методы (постепенное расщеп-
расщепление, получение производных), а в последнее время все чаще
применяют физико-химические методы (масс-спектрометрия, рент-
геноструктурный анализ, спектроскопия во всех ее вариантах).
Количественный и качественный элементный анализ. Методы
анализа органических соединений были созданы в начале XIX в.,
но их усовершенствование продолжается до наших дней. В основе
методов анализа лежит полное расщепление органического веще-
вещества в результате окисления или другим путем и определение хи-
химических элементов известными методами. Углерод определяют в
виде СО.,, водород — в виде Н,О, азот — измерением объема N..
или определением ХН,, или NaCN (в зависимости от вида расщепле-
расщепления), галогены — в виде галогенид-иопов, серу — в виде сульфат-
ил и сульфид-иона, фосфор — в виде фосфат-иона и т. д.
19
Качественно углерод и водород определяют при нагревании с
СиО:
CnH2n + 3;tCuO—*.nCO2 + nH2O + 3nCu
и выделяющийся диоксид углерода обнаруживают пропусканием
газа в раствор Ва (ОНJ, а воду обнаруживают визуально на стенках
пробирки.
Азот, серу и галогены качественно определяют при сплавлении
с натрием. Образующиеся NaCN, Na2S и галогениды натрия обна-
обнаруживают в водном растворе обычными аналитическими реакция-
реакциями.
Для количественного анализа органических соединений суще-
существуют специальные приборы. Раньше обычно применялись уста-
установки для макроанализа (навеска образца 0,2. . .0,5 г). В наши дни
распространены различные приборы для микроанализа (навеска
0,001. . .0,01 г), для ультрамикроанализа (навеска 10~5. . .10~4г).
Для количественного определения углерода и водорода используют
приборы, в которых органическое вещество сжигают в токе кисло-
кислорода: СО2 улавливают раствором КОН, а Н2О — специальным
абсорбентом и определяют взвешиванием. Для количественного
определения азота используют сожжение вещества при нагревании
с СиО и объем выделившегося газа измеряют в азотометре над раст-
раствором КОН. Галогены и серу количественно определяют сожжением
образца в атмосфере кислорода, растворением газов в воде и титро-
титрованием галогенид-ионов или сульфат-иона.
Разработаны автоматические микроанализаторы с использова-
использованием принципа газовой хроматографии, в которых одновременно
определяют углерод, водород, азот и серу.
Молекулярную массу соединения обычно определяют масс-
спектрометрически.
4. ЛИТЕРАТУРА ПО ОРГАНИЧЕСКОЙ ХИМИИ
Число описанных в литературе органических соединений огромно, оно дости-
достигло 6 млн и продолжает увеличиваться. Интересно проследить динамику рос-
роста числа органических соединений. По приблизительным оценкам, к 1945 г.
был получен 1 млн соединений, за период 1945—1959 гг.— около 1 млн, а за
I960—1969 гг.— 1,2 млн новых соединений. К 1970 г. было описано уже 3,4
млн органических соединений, а к 1977 г. их стало уже около 5 млн. В послед-
последнее время ежегодно получают около 200—250 тыс. новых органических сое-
соединений.
Естрствсрпю, возникает вопрос, можно ли освоить этот огромный матери-
материал и ориентироваться в неисчислимом мире органических веществ? С чего
начинать?
Начинать следует с учебников и учебных пособий по органической хи-
химии, в которых систематизированы знания об органических соединениях.
Органическая химия находится в непрерывном процессе развития, меняется
и совершенствуется методика изложения материала органической химии,
поэтому здесь перечислены только фундаментальные издания по курсу орга-
органической химии, вышедшие в последние десятилетия.
1. Несмеянов А. Н., Несмеянов Н. А. Начала органической химии. В 2 т.
М., 1974.
2. Мэррисои Р., Бойд Р. Органическая химия. М., 1974.
20
3. Перекаляй В. В., Зонис С. А. Органическая химия. М., 1982.
4. Гауптманн 3., Грефе Ю., Ремаие X. Органическая химия. М., 1979.
5. Терией А. Современная органическая химия. В 2 т. М., Мир, 1981.
6. Кери Ф., Сандберг Р. Углубленный курс органической химии. В 2 т.
М., Химия, 1981.
7. Петров А. А., Бальян X. В., Трощенко А. Т. Органическая химия. М.,
Высшая школа, 1981.
8. Степаненко Б. Н. Курс органической хинин. М., Высшая школа,
1981.
9. Грандберг И. И. Органическая химия. М., Высшая школа, 1987.
При углубленном изучении органической химии и научной работе в об-
области органической химии каждый сталкивается со сложной проблемой ин-
информации. Как узнать, что полученное нами вещество является новым, ранее
неизвестным? Это очень существенно. Кроме того, химику в обыденной рабо-
работе часто необходимо узнать данные о свойствах тех или иных соединений. Где
их искать?
Для решения этой проблемы необходимо использовать специальную лите-
литературу. Она охватывает первичные (научные статьи, патенты и авторские
свидетельства, диссертации, научные отчеты), вторичные (реферативные жур-
журналы, сигнальная информация, справочники, обзоры, монографии), третичные
(указатели реферативных журналов, библиографические указатели, указа-
указатели справочников) и четвертичные (сводные указатели, указатели библио-
библиографий) источники.
Важнейшим первоисточником являются журналы, в которых химлки
публикуют статьи о новых веществах и их свойствах, о результатах теорети-
теоретических разработок.
Журналы и другие периодические издания, в которых можно найти ста-
статьи по органической химии, исчисляются тысячами. Каждый год появляется
более чем 200 000 публикаций в области органической химии. По данным Ин-
Института научной информации (США), 96% информации в области органиче-
органического синтеза можно почерпнуть из 100 главных журналов, две трети важней-
важнейшей информации — при чтении около 30 журналов. Около 60% важнейших
статей по органической химии публикуются на английском языке< 20% —¦
на русском языке, 10% — иа немецком.
Патентная литература является специфическим источником информации,
в которой главным образом описаны методы получения веществ и практиче-
практическое применение без данных теоретического характера. Описания патептоз
имеются в патентных библиотеках. Патенты реферируются в общих рефера-
тинных журналах, кроме того, издается специальная патентная информация.
В области синтетической органической химии патентная информация опере-
опережает публикации в научной периодике на 1—3 года.
Чтобы облегчить работу с этим огромным материалом, издаются рефера-
реферативные журналы (вторичные источники). В этих журналах оригинальные ;кур-
иальные статьи и патенты очень кратко пересказываются (реферируются), а
рефераты систематизируются. Наиболее важными реферативными журналами
являются: Chemisches Zentralblatt (до 1970 г.), Chemical Abstracts, рефератив-
реферативный журнал «Химия», Current Abstracts of Chemistry and Index Chemicus.
Фундаментальным справочником по органической химии является спра-
справочник Бейлынгейна (Beilstein's Handbuc.h der organischen Chemie). Инициа-
Инициатор его создания и первый составитель — русский академик Ф. Ф. Бсйль-
штейн. Задача справочника — собрать все существенные сведения о всех из-
известных органических веществах. В основном издании, состоящем из 31 тома,
собраны сведения о соединениях, опубликованные до 1 января 1910 г. Име-
Имеются четыре дополнения: первое дополнение охватывает литературу за 1910—
1919 гг., второе — за 1920—1929 гг., третье — за 1930—1949 гг. (еще не за-
завершено), четвертое — за 1950—1959 гг. Издается этот справочник Беиль-
штейновским институтом в ФРГ.
Подробно с проблемой химической информации можно ознакомиться в
книге: Потапов В. М., Кочетова Э. К. Химическая информация. Что, где и
как искать химику в литературе.— М.: Химия, 1979.
21
5. РАЗВИТИЕ ТЕОРЕТИЧЕСКИХ ПРЕДСТАВЛЕНИЙ
ОРГАНИЧЕСКОЙ ХИМИИ
Теоретическими основами органической химии являются теория
строения органических соединений и теория реакционной способ-
способности, т. е. учение о соединении атомов в молекуле, о взаимном
влиянии атомов в молекуле и о протекании реакций. Критериями
ценности теории являются:
а) способность систематизировать имеющийся фактический ма-
материал;
б) способность объяснить природу той силы, которая удерживает
атомы в молекуле (природу химической связи);
в) способность объяснить протекающие химические процессы,
физические и химические свойства веществ;
I') возможность предсказания новых химических реакций, новых
типов соединений и т. д.
1. ТЕОРИИ СТРОЕНИЯ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Теория радикалов. Исторически первой в органической химии была
теория радикалов. В создании этой теории важную роль сыграла
электрохимическая теория химической связи Й. Берцелиуса, гос-
господствовавшая в то время в неорганической химии. Согласно этой
теории («дуалистическая теория») все соединения образуются из
противоположно заряженных частиц (элементов) в результате сил
электростатического притяжения. В органических соединениях
роль таких заряженных частиц играют не только атомы, но и целые
группировки атомов, названные радикалами. Принималось, что ра-
радикалы в органической химии соответствуют элементам в неорга-
неорганической химии и способны переходить в химических реакциях от
одного соединения к другому в неизмененном виде. Первым таким
радикалом был бензоил, обнаруженный в бензойной кислоте, бен-
зоилхлориде и бензальдегиде (Ю. Лнбих, Ф. Вёлер).
Теория радикалов дала некоторую основу для систематизации
соединений, в некоторых случаях удалось объяснить свойства сое-
соединений. Теория имела некоторую силу предвидения — можно
было предсказать существование соединений, содержащих до тех
пор неизвестные комбинации радикалов.
Но вскоре догма о неизменяемости радикалов потерпела пораже-
поражение. Ж. Дюма показал, что водородные атомы в органических соеди-
соединениях легко могут быть замещены атомами хлора (явление мета-
лепсии). Так возникло необъяснимое в рамках теории радикалов
противоречие: как положительный водородный атом может быть
замещен отрицательным атомом хлора.
Теория типов. Теория типов была построена Ж- Дюма на руинах
радикальной теории с сохранением понятия о группировках атомов—
радикалах. Предлагалось классифицировать органические соеди-
соединения по типам. Например, уксусная и хлоруксусная кислоты
принадлежат к одному типу. Эта теория заложила основу для новых
22
химических поисков — синтеза новых соединений одного типа.
Так, А. Гофман и А. Вюрц, исходя из типа аммиака, впервые синте-
синтезировали аналоги аммиака — амины:
ми nJh 3 NicHg nJcii*
1н \н \н \сн3
Унитарная теория. Теорию типов дополнила унитарная теория.
Эту теорию создали Ш. Жерар и О. Лоран в 50-е годы XIX в. и наз-
назвали ее унитарной в противоположность дуалистической теории
Берцелиуса. В основе унитарной теории лежит принцип замещения.
Все органические соединения могут быть образованы из определен-
определенных типов при замещении водородных атомов органическими (угле-
(углеводородными) группировками — радикалами. Основными типами
являлись:
н\о
н/ и
тип воды
II1
Н .N
н|
тип аммиака
Н1
щ
тип водорода
и \
а)
тип хлорово
дорода
При помощи унитарной теории были предсказаны и синтезиро-
синтезированы новые классы соединений, например ангидриды кислот, много-
многоатомные спирты. Эта теория отрицала любую возможность познания
расположения атомов в молекуле. Она не была в состоянии объяс-
объяснить некоторые явления изомерии, например существование двух
углеводородов с формулой С4Н10. Такая изомерия называлась
«тонкой изомерией».
Теория строения. В ходе развития органической химии синтези-
синтезировались все новые и новые соединения, описание которых не было
иод силу унитарной теории. На основе работ Франкланда появилось
понятие о валентности элементов, т. е. способности химических
элементов присоединять только определенное число атомов других
н элементов. А. Кекуле первым выдвинул принцип четырех-
н! валентности углерода и доказал, что углеродные атомы спо-
нГ собны соединяться между собой и образовывать длинные
н цепи. А. Кекуле предложил новый тип соединений — тип
тип метана
В конце 50-х годов XIX в. в органической химии уже узакони-
лись некоторые принципы: в реакциях различные группировки мо-
могут переходить от одного соединения к другому, существует возмож-
возможность замещения одного атома другим, атомы могут соединяться
между собой только в строго определенных пропорциях. Но не было
ясности в том, существует ли какая-то взаимосвязь между составом
соединения и его химическими свойствами, существует ли определен-
о н ная последовательность соединения атомов в молекуле.
с| г В конце этого периода были сделаны попытки изобра-
: н зить в формулах последовательность соединения атомов в
с н1 молекулах. Первым такие графические формулы предло-
j жил А. Купер. Примером может служить приведенная
с н3 здесь формула пропилового спирта по А. Куперу.
23
19 сентября 1861 г. на съезде немецких естествоиспытателей и
врачей в Шпейере русский химик А. М. Бутлеров докладывал о
новых воззрениях в органической химии. Он выдвинул новое по-
понятие — структура, которое отражало последовательность строе-
строения атомов в молекуле. Так родилась структурная теория, или
теория химического строения.
Для изображения последовательности соединения атомов в
молекуле Бутлеров предложил использовать черточки между ато-
атомами. Примером может служить изображение формулы метана:
Н
н-с-н
Правильность теории строения А. М. Бутлеров неопровержимо
доказал, синтезировав три изомера углеводорода С5Н12, которые
были им теоретически предсказаны.
Теория строения состоит из следующих положений:
1. В молекулах веществ существует строгая последовательность
химического связывания атомов, которая называется химической
структурой (строением).
2. Химические свойства вещества определяются природой эле-
элементарных составных частей, их количеством и химическим строе-
строением.
3. Если у веществ с одинаковым составом и молекулярной мас-
массой различное строение, возникает явление изомерии.
4. Так как в конкретных реакциях изменяются только некоторые
части молекулы, то исследование строения продукта реакции помо-
помогает определить строение исходной молекулы.
5. Химическая природа (реакционная способность) отдельных
атомов молекулы меняется в зависимости от окружения, т. е. от
того, с какими атомами других элементов они соединены.
Теория Бутлерова дает принципиальную возможность познания
геометрии молекулы (микроскопических свойств) через познание
химических свойств (макроскопических свойств). Основные поло-
положения теории строения сохраняют свое значение и в наши дни.
Структурные формулы в руках химиков стали мощным оружием.
Они позволяют делать выводы о химических свойствах соединений,
дают возможность систематизировать огромный материал органиче-
органической химии, предсказать существование неисчислимого количества
новых соединений.
Но оказалось, что не все могла объяснить и теория строения.
Много сложностей возникло с выделенными из каменноугольной
смолы бензолом СвН6 и его гомологами, с так называемыми нена-
ненасыщенными соединениями — этиленом, ацетиленом и др. Как изоб-
изобразить их строение? Не было ясности и с пространственным строени-
строением молекул органических соединений.
24
Здесь уместно привести высказывание А. М. Бутлерова, которое
верно на любом этапе развития науки: «Факты, которые не могут
быть объяснены существующими теориями, наиболее ценны для
науки. При изучении именно этих фактов ожидается прогресс
науки в ближайшем будущем».
Было предложено соединения типа этилена писать с двойной
связью, соединения типа ацетилена — с тройной. Для соединения
типа бензола А. Кекуле написал структуру с шестичлешщм циклом
и тремя двойными связями:
Н
н ..н хс^ хс/
ЧС- С{ Н—С---С—Н I 11^
Н' ЧН ацетилен
н
бензол
Я- Вант-Гофф и независимо от него Ж- Ле Бель в 1874 г. выдви-
выдвинули гипотезу о пространственной конфигурации насыщенного уг-
углеродного атома — тетраэдрическом расположении заместителей.
Понятие об асимметричном углеродном атоме и оптической изоме-
изомерии (зеркальной изомерии) дало возможность объяснить существо-
существование изомерных оптически активных веществ с противоположным
углом вращения (Введ. 6.12).
Если у насыщенного углеродного атома находятся четыре раз-
А А личных заместителя (асимметрический
I | углеродный атом), то возможно двоякое
с^ с расположение этих заместителей в прост-
х -'у ^"в В''' ^-х ранстве, причем оба изомера различаются
y Y между собой как зеркальные изображе-
зиантиомеры цИЯ (зеркальные изомеры, энантиомеры).
Несмотря на резкие нападки со стороны ряда ведущих химиков того
времени, гипотеза была принята и легла в основу нового направле-
направления теоретической химии — стереохимии.
Однако на вопрос, почему атомы держатся вместе в молекуле
и почему молекулы имеют определенное пространственное строение,
никто не мог дать ответа. Успех в изучении природы химической
связи был достигнут только после открытия строения атома и созда-
создания электронной теории химической связи.
2. КЛАССИЧЕСКИЕ ЭЛЕКТРОННЫЕ ТЕОРИИ
ХИМИЧЕСКОЙ СВЯЗИ
Для органических соединений была приспособлена разработанная
Г. Льюисом и В. Косселем A916) теория образования неорганиче-
неорганических ионных соединений, т. е. принцип дублета—октета. И. Ленг-
мюр A919) ввел понятие ковалентная связь (в противопоставление
ионной связи)- В ковалентной связи электроны принадлежат одно-
25
временно обоим атомам, но в то же время входят в дублет пли октет
электронов соответствующего атома:
н-с—н с=с н—с^с—н
н
метан этилен ацетилен
Формулы с точечным изображением электронов называют форму-
формулами Льюиса.
На основе теории дублета — октета появилась возможность
классифицировать различные виды химической связи, в том числе
ковалентной. Ковалептная связь может образовываться двояко.
Первый случай — каждая частица дает по одному электрону:
Н. + -Ci: —* H:Cf:; Н3С- + -СН3 —> Н3С:СН3
Второй случай — одна частица дает электронную пару, а вто-
вторая предоставляет незаполненную орбиталь:
H3N: + H+ —* H3N:H (NH4); Н3С: +СН3 —* И3С:СН3
Второй случай образования ковалентной связи называется коорди-
координацией. Из этого определения возникло понятие координационная,
или донорно-ащепторная связь. Отличие координационной связи
от других типов связи очень хорошо видно на примере образования
N-оксидов аминов:
(CH3KN:+O:-^(CH3KN:O:
Здесь возникает координационная связь между атомами азота и
кислорода. Обе частицы нейтральны, и в результате образования
ковалентной связи происходит перераспределение электронной
плотности: атом кислорода приобретает отрицательный, а атом
азота — положительный заряд:
(CH3KN-6:
Иногда для обозначения координационной связи применяют
стрелку, показывающую направление смещения электронной плот-
плотности: (CH3KN^O.
В органической химии связь, образовавшуюся в результате
координации двух нейтральных частиц, называют семипо-
лярной. Фактически эта связь является ковалентной, соединяющей
атомы, на которых при возникновении связи образуются положи-
положительный и отрицательный заряды. При этом вторая ковалентная
связь (двойная) образоваться не может, так как нет соответствую-
26
щих незаполненных орбиталей. Наиболее известной группой с
семи полярной связью в органической химии является нитрогруппа
—NO 2.
Теория дублета — октета применима только в тех случаях, когда
не рассматриваются атомы высших периодов (S, Si, P и др.). Для
этих атомов известны стабильные соединения с участием как 10,
так и 12 электронов.
На основе классической электронной теории были созданы пред-
представления об электронных смещениях в химических связях. Элект-
Электронное смещение наблюдается почти во всех случаях, когда химиче-
химическая связь образуется из двух различных элементов. Каждый эле-
элемент имеет свой ядерный заряд и свой атомный радиус, которые
определяют способность притягивать электроны. Другими словами,
каждый элемент имеет свое характерное ядерное силовое поле, в
котором размещаются электроны. Чем больше способность атомного
ядра притягивать электроны, тем больше и смещение электронов
в химической связи в направлении этого атома. С целью характерис-
характеристики этого сродства к электрону была создана шкала значений
электроотрицательное™. Электроотрпцателыюсть — мера способ-
способности атома или группы атомов притягивать электроны из других
частей той же молекулы. Впервые такую шкалу разработал Л. По-
липг. Чем больше электроотрицательность атома, тем сильнее его
сродство к электрону.
Ниже приведены значения электроотрицательности для некото-
некоторых атомов:
Н 2,1
В 1,9 С 2,5—2,0 К 3,0 О 3,5 F 3,9
А1 1,5 Si 1,8—1,9 Р 2,1 S 2,5 С1 3,0
Р.сли химическая связь образуется из элементов с различной
электроотрицательностью, то отрицательный заряд концентрируется
на более электроотрицательном атоме — происходит поляризация,
химическая связь становится полярной. Химическую связь называ-
называют полярной, если в ней имеется неравномерное распределение
электронной плотности. Это явление изображается при помощи
эффективных (долевых) зарядов:
б- б* 64- Ь-
О —Н; С—С1
Эффективные заряды на атомах образуются в результате элект-
электронного смещения. Это можно изобразить с использованием элект-
электронных пар:
б- б+ б+ 6-
0:0 О: И С:С С :С1
При помощи эффективных зарядов указывают на асимметрию
распределения электронной плотности в связи. Эта асимметрия
распределяется по всей молекуле и характеризуется дипольным
моментом.
Электронное смещение в химической связи можно указывать не
только долевыми зарядами, но и стрелкой на связи:' с^-сГ
27
Электронные смещения осуществляются также в двойных и трой-
тройных связях, только в этих случаях поляризация больше, так как
электроны двойных и тройных связей более подвижны. Для обозна-
обозначения смещения электронов в двойных и тройных связях применя-
применяют изогнутую стрелку:
<5+ 5~ 6- <5-
<5+ /-ч <5— Ь '•¦ ^ч <5 —
С = О C=-N
at /-^ 5—
Сн= N
5<cS<cS"
Электронное смещение перераспределяется по всем связям и
атомам молекулы. Если в молекулу введен электроноакцепторный
(оттягивающий электроны) атом (группа атомов) X или электроно-
донорный (отдающий электроны) атом (группа атомов) Y, поляри-
поляризация связей распространяется на всю молекулу:
S+ S-
R-*-X R-«-Y
электроноакцепторное влияние электронодонорное влияние
заместителя X ( -эффект) заместителя Y Н эффект)
При рассмотрении электронных смещений необходимо четко раз-
различать эффекты в молекулах насыщенных и в молекулах с сопряжен-
сопряженными двойными или тройными связями.
Электронные смещения в насыщенных системах. Заместитель
вызывает поляризацию связей, которая распространяется на всю
молекулу, постепенно затухая при удалении от заместителя. Это
влияние заместителя обнаруживается экспериментально при изме-
измерении физических констант или при изучении реакционной способ-
способности.
Экспериментально наблюдаемый эффект передачи заряда по цепи
атомов за счет электростатической индукции называется индуктив-
индуктивным эффектом и обозначается /. Например:
U^T (< <<
— I -эффект атома хлора
Электронные смещения в сопряженных системах. В молекулах о
несколькими двойными или тройными связями (или с атомами с
неподелеппыми парами электронов) в сопряженном положении,
например:
X---Y —Z = Q X = Y—Z = Q N—X = Y
28
Очень сильное влияние на распределение электронной плотности
оказывают электроноакцепторные или электронодонорные атомы и
группировки. В сопряженных молекулах подвижность электронов
особенно велика. Во-первых, в любом случае действует индуктив-
индуктивный эффект /. Во-вторых, проявляются своеобразные эффекты
электронов двойных или тройных связей.
Первый пример — сопряженная система с электроноакцептор-
ным атомом кислорода: С::С:С::О или С=С—С=О.
С точки зрения классической электронной теории своеобразность
сопряженной системы заключается в том, что электроны двойных
связей легко могут переходитть на соседнюю связь или атом. При
этом происходит изменение распределения электронной плотности
(электронного заряда) в сторону электроноакцепторного атома:
+ _ ^
С • С ; С • О- С = С
электроны перешли полиостью частичное смещение
«з соседние связи и атом электронов
кислорода
Второй пример — сопряженная система, содержащая атом с не-
поделенной парой электронов N:C::C или N—С=С.
Согласно классической электронной теории электрон неподелен-
ной пары легко переходит на соседнюю связь. При этом происходит
изменение распределения электронной плотности, смещение от ато-
атома с неподеленной парой на двойную связь:
N ; С ; С" N С = С (
электроны перешли полностью частичное смещение
на соседние свмзи и атом электронов
у> лерода
Электронные смещения в сопряженных системах с участием
электронов кратных связей или неподеленных электронных пар
называют мезомерным эффектом (±М), эффектом сопряжения
(±С), резонансным эффектом (±/?).
Точнее, мезомерным эффектом называют экспериментально на-
наблюдаемый эффект заместителя в сопряженных системах, связанный
с взаимодействием электронов двойных или тройных связей или
неподеленной пары заместителя с двойными или тройными связями
остальной молекулы. Это взаимодействие вызывает перераспределе-
перераспределение электронной плотности (заряда), которая может перемещаться
к заместителю или от него.
Электроноакцепторная группа с двойной (Z=Q) или тройной
(Z=hQ) связью в сопряженной системе показывает электроноакцеп-
торное действие благодаря мезомерному эффекту —М и индуктивно-
индуктивному эффекту —/:
z. — ч:
29
Электроноакцепторными группами с —М-эффектом, например,
являются:
—с/ —С<? " — C==N —N = O
NR ' 4R
О
/О ||
—N02 — Sf — S—R
XR II
О
Атом с неподеленной парой электронов в сопряженной системе,
с одной стороны, действует как донор электронов благодаря своему
мезомерному эффекту +М, a g другой стороны, проявляет —/-эф-
—/-эффект:
—/-Эс})фект оказывает действие, направленное противоположно
действию -(-М-эффекта. Электронодонорнымн группами D— явля-
являются R2N—, R2P—, RO—, RS— и др.
Особое место занимают группы СНЯ— и RCH2—, которые соглас-
согласно экспериментальным данным оказывают -\-М- и в то же время
/-эффекты (см. гл. П.З)
Этот Э([х1>ект называется эффектом сверхсопряжения (гиперконъ-
(гиперконъюгации) .
Прямые и изогнутые стрелки в формулах условно обозначают не
только смещение электронной плотности, но и разные механизмы
электронного влияния — индуктивный и мезомерный эффект.
Представления об электронных смещениях (электронных эффек-
эффектах) в сопряженных системах и их классификацию разработал
К. Ингольд A926—1933). Для объяснения мезомерного эффекта
К. Ингольд разработал представление о мезомерии, согласно кото-
которому распределение электронной плотности в реальной сопряжен-
сопряженной молекуле является промежуточным между двумя структурами,
изображаемыми обычными структурными формулами. Полярные
сопряженные системы могут быть изображены при помощи несколь-
нескольких граничных структур со строго фиксированным положением
электронов («-> — знак мезомерии):
a-CH-C —О;
СН3О — CII -¦= СН2 <-» СН3О = СП — СНГ
30
Первые предположения о своеобразном распределении электро-
электронов в сопряженных системах (о мгзоформе) принадлежат русскому
химику В. Л. Измаильскому A915).
Представления Ипгольла о мезомерии вошли как составная
часть в теорию резонанса, разработанную в 1928—1938 гг. Л. По-
лингом. Согласно Полингу, молекулу можно описать как быстро
флуктуирующую между двумя электронными формулами (резони-
(резонирующими структурами) и приобретающую стабильность большую,
чем любая из этих формул, благодаря резонансной энергии этой
флуктуации. В настоящее время теория резонанса (концепция мезо-
мезомерии — резонанса) трактуется как способ качественного описания
распределения электронной плотности в молекулах органических
соединений с сопряженными связями. Это распределение электрон-
электронной плотности по связям н атомам изображают при помощи несколь-
нескольких классических структурных формул (канонических структур,
или резонансных граничных структур). Реальная молекула рас-
рассматривается как «резонансный гибрид», в котором распределение
электронной плотности является промежуточным между распределе-
распределением электронной плотности в резонансных граничных структурах.
Например, бензол может быть изображен пятью резонансными
структурами:
Основные резонансные структуры анилина следующие:
+Ntfj +NH2
Можно сказать, что концепция мезомерии — резонанса является
способом моделирования реального электронного строения молекул
с помощью граничных структур. Резонансные структуры — это в
большинстве случаев привычные валентные схемы. Резонансные
формулы обладают хорошей наглядностью и позволяют более четко
подчеркивать те или иные особенности электронной структуры.
Начиная с 1934 г. для характеристики сопряженных молекул
используются уже квантово-химические расчеты (см. гл. II. 4.9,
гл. III. Б.2, гл. VI. А.З).
Дальнейшее развитие представлений о взаимодействии связей
и перераспределении электронной плотности в сопряженных моле-
молекулах нашло свое воплощение в квантовой органической химии.
3. ОСНОВНЫЕ ПРИНЦИПЫ
КВАНТОВОЙ ОРГАНИЧЕСКОЙ ХИМИИ
Классическую теорию дополнила квантовая химия, которая ос-
основывалась на новых открытиях физики о двойственной,(корпус-
двойственной,(корпускулярной и волновой) природе электрона. Была создана механика
31
микромира — волновая механика. Э. Шредингер A926) вывел урав-
уравнение, связывающее волновую природу движущейся материальной
частицы с ее пространственными координатами и энергией:
" Т I " Ч* I " Ч'
(E — U)^ = 0.
0,14 нм
В уравнении Шредингера гр является волновой функцией электро-
электрона. Ее физический смысл: г|) (*, у, г) является функцией от простран-
пространственных координат, ее квадрат of2 характеризует вероятность на-
нахождения электрона в данной точке пространства; Е — полная энер-
энергия электрона; U—потенциальная энергия.
Решение уравнения дает значение энергии электрона ? (собст-
(собственные значения) и выражения для волновой функции г]) (собствен-
(собственные функции). При решении уравнения для электрона в атоме во-
водорода получается целый ряд возможных значений Е, характерных
для электрона. Каждому значению Е отвечают свои выражения
длягр игр2, а следовательно, и область пространства, где вероятность
нахождения электрона наибольшая. Так мы получаем представле-
представление о различных состояниях электрона (различных электронных
орбиталях) в атоме (атомарных орбиталях
АО). Ими являются известные нам из неорга-
неорганической химии состояния s, р, d (s-, p-, d-
орбитали и электроны). Эти состояния резко
отличаются друг от друга своими энергиями
и конфигурацией той части пространства, где
вероятность нахождения электрона наиболь-
наибольшая.
Для s-состояния (s-орбиталн) характер-
характерна симметрия шара (рис. 5). В отличие от
радиуса атома водорода 0,058 им, рассчитан-
рассчитанного Бором, здесь граница поверхности шара находится па расстоя-
расстоянии 0,И нм от атомного ядра (поверхность шара ограничивает ту
часть пространства, в которой вероятность нахождения электрона
составляет 90%).
Для ^-состояния (р-орбитали) характерна цилиндрическая сим-
симметрия. Имеются три /7-состояния —• рх, ри, рг (рис. 6).
Рис. 5. s-Орбиталь
УХ
Рис. 6. р-Обитали
/j-Орбиталь имеет две части, которые разделяет узловая плос-
плоскость (вероятность нахождения электронов в узловой плоскости
равна нулю). Волновая функция в узловой плоскости меняет знак,
32
поэтому различают + и — части. В схемах р-орбиталн часто изоб-
изображаются знаком Д .
Имеется 5с?-состояний, характеризующихся своеобразными очер-
очертаниями пространства орбиталей.
Зная состояния электронов в атоме и их энергии, можно изобра-
изобразить схемы распределения электронов (рис. 7).
Как в результате развития квантовой химии изменились пред-
представления об образовании химической связи? Классическая теория
ъ
¦
пш
It С
Рис. 7. Схема распределения электронов в атомах
н
t
t
N
¦1
и.т.д.
объяснила образование химической связи взаимодействием электро-
электронов атомов, в результате чего снижается энергия системы и частицы
удерживаются вместе. Используя уравнение Шредингера, можно
доказать это снижение энергии системы при взаимодействии элект-
электронов.
Чтобы охарактеризовать химическую связь при помощи уравне-
уравнения Шредингера, необходимы некоторые исходные постулаты., Во-
первых, нахождение электрона в химической связи (в простейшем
случае между двумя атомами) или, в общем случае, в молекуле ха-
характеризуется молекулярной волновой функцией электрона — мо-
молекулярной орбиталью (МО). Эти молекулярные орбитали подобны
атомным орбиталям электрона, только относятся к нескольким ато-
атомам одновременно. Если АО характеризует нахождение электрона
в силовом поле ядра атома, то МО характеризует нахождение элек-
электрона в силовом поле двух или более ядер.
Во-вторых, МО выражается функцией от АО (базисных функции).
Для расчета МО используют несколько приближений. Так, напри-
например, используют принцип линейной комбинации АО (ЛКЛО).
Для двухатомной молекулы
где т\\ и г1з.2 есть АО, а а н с-2 — коэффициенты (собственные векто-
векторы), характеризующие долю каждой АО в МО. Вначале с{ и сг
неизвестны. Выражение для if подставляют в уравнение Шрединге-
Шредингера и решают его, используя принцип минимизации энергии. Для
двухатомной молекулы получаются две МО с различными энер-
энергиями — меньшей и большей по сравнению с исходным состоянием.
На рис. 8 изображены низкорасположенный уровень энергии —
связывающая орбиталь, уровень энергии исходного состояния (АО)
и высокорасположенный уровень энергии — разрыхляющая орбн-
517
33
МО
АО
таль. Электроны в химической связи размещаются на связывающей
МО. Как АО, так и МО характеризуются энергией орбитали, кото-
которая является наименьшей для связывающей МО (значения энергии
имеют знак «минус», а абсолютная величина энергии связывающей
МО больше энергии АО).
Энергия орбиталей харак-
характеризуется энергией иони-
ионизации (потенциалом иони-
ионизации), соответствующей
той энергии, которую не-
необходимо приложить к мо-
молекуле, чтобы удалить
электрон от химической
связи в бесконечность. Энергия ионизации равна энергии орбитали,
взятой с обратным знаком. Отрывать электрон от связывающей
МО химической связи труднее, чем от АО соответствующего атома.
Из решения уравнения Шредингера следуют также сведения о
конфигурации той части пространства, которая соответствует МО
и в которой вероятность нахождения электрона равна 90%. В за-
зависимости от типа комбинирующихся АО получаются различные
Разрыхляющая орбиталь
Уровень энергии
исходного сосюяния
Связывающая орбиталь
Рис. 8. Сравнительные энергии орбиталей
(Г7).
Рис, 9, Образование сг-связи (<т* — разрыхляющая а-орбиталь)
Рис. 10. Образование л-связи (л* — разрыхляющая л-орбиталь)
34
МО. Одна из возможных МО обладает цилиндрической симметрией
и называется а-ербиталью или а-связью. Она образуется при ком-
комбинации по оси х двух s-орбиталей, s- и рх- или рх- и /7ж-орбиталей.
На рис. 9 изображена комбинация s-орбиталей (изображена только
связывающая орбиталь). Разрыхляющая а-орбиталь обозначает-
обозначается о*.
Второй возможной МО является орбиталь с плоскостной симмет-
симметрией. Такие МО могут образоваться при комбинации по оси х ор-
биталей ру и pv или рг и рг (рис. 10). МО этого типа называются
я-орбиталями или я-связями. Для них характерна узловая плос-
плоскость, волновая функция меняет знак при переходе из одной части
в другую.
В разрыхляющих орбиталях имеется узловая плоскость между
атомами, поэтому нахождение электронов в этих орбиталях не вы-
вызывает силы связывания.
В органических соединениях а-связи встречаются наиболее час-
часто. Между атомами первой образуется а-связь. В структурных фор-
формулах она обозначается одной черточкой, например С—С. Если
создаются условия для образования второй связи между атомами,
то ею может быть только д-связь. Обе связи в формулах обознача-
обозначаются черточками, например С=С. Третья связь тоже может быть
только л-связью. Все три связи в структурных формулах обозна-
чают черточками, например с = -С.
Рассмотренные выше теоретические рассуждения весьма трудно
применять к углеродному атому. Несмотря па то что он имеет н е-
эквивалентные орбита л и (две /7-орбитали и одну
s-орбнталь с двумя электронами), в большинстве насыщенных сое-
соединений углеродный атом образует четыре одинаковые -fv, я-
з и.
Это противоречие разрешают при помощи гипотезы о гибридиза-
гибридизации. Принимается, что из нескольких АО с различной энергией и
и
п
t
t
Вепбужлснма
t
и
t
t
t
с с*
Рис. 11. Распределение электронов в атоме уг«
.порода в основном и возбужденном состояниях
различной симметрией образуется такое же число гибридных орбн-
тален с одинаковой энергией и одинаковой симметрией. Гибриди-
Гибридизация допускается только при образовании химических связей, и
гибридизуются обычно частично заполненные (имеющие по одному
электрону) АО. Поэтому гибридизации АО углеродного атома пред-
предшествует возбуждение (рис. 11).
Так, при взаимодействии одной s- и трех /7-орбнталей образуется
четыре хр3-гибридпые орбитали — происходит 5р3-гибридизация
(рис. 12). Оси симметрии гибридных орбиталей образуют между со-
собой углы, равные 109с28' (тетраэдрический угол).
2* 35
Возможны два других типа гибридизации, в которых не участ-
участвуют одна или две р-орбитали. Во-первых, возможна 5/?2-гибриди-
зация, в результате которой образуется три 5/?4-гибридные орбита-
ли и остается одна негибридизоваииая р-орбиталь. Оси симметрии
5р2-гнбридных орбиталей находятся в одной плоскости и образуют
между собой угол 120' (тригональный угол), а ось р-орбитали на-
направлена перпендикулярно плоскости трех осей 5р2-гибридных ор-
орбиталей (рис. 13).
Во-вторых, возможна sp-гибрндизация, в результате которой
образуются две sp-гибридные орбитали и остаются две взаимно пер-
перпендикулярные негибридизованные р-орбитали. Оси симметрии
Рис. 12. spsTn6piifln-
зг.цкя србиталей
Рис. 13. яр-Тибриди-
зания орбиталей
Рис. 14. sp-Гнбридиза-
ция орбнгалей
?р-гибридных орбиталей находятся па одной прямой (дигональиып
угол 180) (рис. 14).
В приведенных выше рисунках показаны только положитель-
положительные части гибридных орбиталей. Каждая из них имеет также и
небольшую отрицательную часть, например: О*С+)
Представления об sp2 - и sp- гибридизации используют для объяс-
объяснения и изображения образования двойных и тройных связей
(рис. 15).
Гипотезу о гибридизации можно применить и для других ато-
атомов — N, О, Si, S, Р и т. д.
Необходимо отметить, что представления о гибридизации АО
при образовании химических связей являются только приемом на-
наглядного построения молекулярных орбиталей а- и л-тпиа. Имеются
и другие, но более сложные квантово-химпческие приемы, при по-
поен. н,с=сп, нс=сн
3G
гт Л\
Л] U
Рис. 15. Образование а- и л;-связей
мощи которых МО строят из негнбридизованных АО и получают ана-
аналогичные результаты.
Таким образом, в органических соединениях выделяют два типа
связей — а- и я-связи, и говорят об а-электронном остове, на кото-
котором образуются я-связи и л-электронные сопряженные системы.
Это является некоторым приближением, но очень полезным для ор-
органической химии.
Квантовая органическая химия показала, что в молекулах ор-
органических соединений электроны находятся на строго определен-
определенных уровнях энергии — молекулярных орбиталях.
Если заполнены все связывающие МО, то говорят, что молекула
находится в основном состоянии So. При поглощении энергии мо-
может происходить переход электрона на энергетически близлежа-
близлежащую свободную (разрыхляющую) орбиталь. Так молекула переходит
в возбужденное состояние St.
Например, молекула метана имеет четыре а-связи С—Н, четы-
четыре одинаковые связывающие орбитали (при более точном рассмот-
рассмотрении надо учитывать взаимодействие этих орбиталей и образование
двух энергетических уровней). Имеется также четыре разрыхляю-
разрыхляющие орбитали (а*). Может произойти электронный переход а^>~а*
(рис. 16).
В молекуле этилена имеется пять а-связей и одна л-связь, а зна-
значит, два типа орбиталей — а и я (рис. 17). От я-орбиталей легче от-
отрывать электрон (меньшая энергия ионизации). я-Электроппые
системы легче возбуждаются (переходы я->-я*).
От каждой органической молекулы может быть оторван электрон.
Электрон обычно уходит от высшей занятой МО (например, от
я-орбитали этилена). В результате образуется катион-радикал ^ас-
тица с неспаренным электроном и положительным зарядом). При-
Присоединение электрона приводит к анион-радикалу (рис. 18). Электрон
*о 5,
-ft- -ft- -fl- -И- -+- -tt- -ti- -И;
Рис. 16. Возбуждение молекулы метана (электронный
переход а-*-а*)
S,
4-
4-
Рис. 17, Возбуждение молекулы этилена (л->я*-переход)
37
помещается на низшей свободной молекулярной орбитали (напри-
(например, на я*-орбитали этилена):
л
А:
Отрыв электрона характеризуют энергией ионизации (ЭИ), ко-
которая выражается в единицах энергии (кДж/моль, эВ, ккал/моль).
Присоединение электрона характеризуется сродством к электрону.
Другими словами, энергия ионизации характеризует электроподо-
-tt- V
•#¦
[СН2—СН2]+ 2;
Рис. 18. Распределение электронов по орбиталям
катио.ч-радикала и анион-раднкала этилена
норные свойства частицы, а сродство к электрону — электроноак-
цепторные свойства.
Первых больших успехов квантовая органическая химия до-
достигла при объяснении свойств сопряженных систем, особенно бен-
бензола и других ароматических соединений (гл. VI.А.3).
Точное применение квантовой химии для сложных молекул тре-
требует огромных математических вычислений. Поэтому долгое время
применялись весьма грубые приближения, например рассматри-
рассматривалась только л-электропная система. В последнее время благодаря
интенсивному развитию вычислительной техники стали доступ-
доступными расчеты как п-, так и а-электронных систем. Это помогает луч-
лучше понять строение молекул, их qbnsiroecKne и химические свойства
и предсказать возможность осуществления химических реакций,
получения новых необычных соединений.
6. ФИЗИЧЕСКИЕ МЕТОДЫ
В ИССЛЕДОВАНИИ
ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Для изучения органических соединений и их реакций использу-
используют множество физических методов. Это помогает решать такие
фундаментальные задачи, как установление структуры и простран-
пространственного строения органических молекул, характеристика моле-
молекулярных орбиталей, изучение взаимодействия атомов и молекул,
исследование скоростей и механизмов реакций.
Ниже кратко рассмотрены важнейшие физические методы, т. е.
сущность метода и возможности применения.
38
1. РЕФРАКТОМЕТРИЯ
Рефрактометрический метод исследования известен давно. Свя-
Связывать значение коэффициента преломления света со структурой
органического вещества можно при номощи молекулярной рефрак-
рефракции (R). Согласно Лоренцу
п2-! М
где п — коэффициент преломления света для D-линии натрия
E89 нм); М — молекулярная масса вещества; р — плотность.
Оказалось, что молекулярная рефракция имеет аддитивные
свойства, т. е. молекулярная рефракция молекулы может быть по-
получена суммированием рефракций составных частей молекулы. Та-
Такими составными частями являются химические связи п совокуп-
совокупности связей и атомов. Эти рефракции вычислены на основе иссле-
исследований многих органических соединений и могут быть найдены в
справочниках. Например:
^CII4 = 4^C-1I' ^CIIjNOj = 3^С- II "I" ^С- N
Явление преломления света связано с поляризуемостью элект-
электронной системы молекул. Под влиянием электромагнитного поля
света происходит поляризация молекул, в основном их электрон-
электронных систем. Чем подвижнее электронная система молекулы, тем
больше коэффициент преломления света и молекулярная рефракция.
Исследования молекулярной рефракции могут быть использо-
использованы для установления структуры соединения. Так, для изучаемого
соединения экспериментально определяют молекулярную рефрак-
рефракцию и сравнивают с рефракцией, полученной суммированием реф-
рефракций связей по предполагаемой структурной формуле. Если ре-
результаты совпадают, то можно считать структуру доказанной, если
пет, то надо искать другую структуру. В некоторых случаях на-
наблюдают сильное увеличение молекулярной рефракции но сравнению
с ожидаемой (экзальтация рефракции). Это характерно для сопря-
сопряженных систем.
Значения молекулярной рефракции химических связей, атомов,
молекул и ионов могут быть использованы для качественной оцен-
оценки их поляризуемости. Поляризуемостью молекулы (иона, связи)
называют способность ее к поляризации, т. е. к изменению поло-
положения ядер и состояния электронного облака под влиянием внеш-
внешнего электрического поля. В основном происходит электронная
поляризация.
Значения R и сравнение поляризуемости различных связей при-
приведены при рассмотрении отдельных классов соединений.
2. КАЛОРИМЕТРИЯ
Калориметрия является методом исследования тепловых эффек-
эффектов химических реакции и процессов фазовых переходов (например,
плавления, кристаллизации, возгонки, конденсации)- Процесс
39
(реакцию) проводят в специальных приборах — калориметрах и
количественно оценивают выделенное или поглощенное тепло. Реак-
Реакции, протекающие с выделением теплоты, называют экзотермиче-
скими, реакции, протекающие с поглощением теплоты,— эндотер-
эндотермическими.
Часто тепловой эффект реакции обозначают просто как теплоту
реакции (теплоту сгорания, теплоту возгонки и т.д.). Правильнее
употреблять понятие энтальпия реакции. Для любого химического
вещества при стандартных условиях характерно определенное зна-
значение некоторой величины, называемой энтальпией (Я), а тепловой
эффект реакций выражают разницей энтальпий (ЛЯ) реагентов и
продуктов реакции и называют энтальпией реакции.
Теплота реакции и энтальпия имеют противоположные знаки.
Например, для экзотермической реакции теплота реакции положи-
положительна, а энтальпия реакции отрицательна:
СA-рафит) + О2 (г) —>С02(г) + 393,5кДж/моль
Так, для сгорания графита получается положительная теплота
реакции, а ДЯ'——393,5 кДж/моль (ДЯ° вычисляется для стандарт-
стандартных условий — 25 СС и 1 атм или 101,3 кПа).
Тепловой эффект выражают в килоджоулях па моль (или в ки-
килокалориях на моль) * .
Калориметрическим путем определяют молярные теплоты сго-
сгорания веществ. В свою очередь теплоты сгорания (W) используют
для вычисления теплоты образования вещества Е или стандартной
энтальпии образования ДЯ°. Теплота образования вещества может
быть вычислена, исходя из элементов в атомарном состоянии или
из элементов в «стандартном» состоянии (углерод в виде графита,
газообразный водород Н2 и т. д.), при этом полученные числовые
значения, естественно, отличаются. При рассмотрении табличных
данных на это надо особенно обращать внимание. Обычно теплоты
образования веществ для процесса вычисляются из атомов элемен-
элементов, а АН°—-из элементов в «стандартном» состоянии. Например,
теплота образования углеводородов из атомов:
¦ W,
где W — теплота сгорания; QCo2 — теплота образования СО2
C93,5 кДж/моль); Qu2o — теплота образования воды
B85,8 кДж/моль); S — теплота атомизации (возгонки) углерода
(графита) (—715 кДж/моль); Б\\г — теплота атомизации (диссо-
(диссоциации) молекулы водорода (—436 кДж/моль).
Чем меньше теплота сгорания, тем больше теплота образования
для соединений одинакового состава.
Теплота образования молекулы из атомов называется также
энергией образования молекулы. Эту величину используют для срав-
сравнения стабильности различных соединений и для вычисления
* 1 ккал=4,187 кДж,
40
Таблица /,
Спязь
с-с
X-N
О— О
С-Н
N" — 11
О —Н
Энергия некоторых химических связей (^3
Энергия,
кДж/моль
344
159
143
415
391
463
Связь
C-N
С-О
C-F
С — С1
С —Вг
С—I
Энергия,
кДж/моль
292
350
443
328
279
240
кДж/моль
Связь
С-С
N--N
C----N
С -О
с-с
C-^N
1
Энергия,
кДж, моль
615
418
615
725
812
890
термохимической энергии химических связей (энергии связей),
принимая, что энергия связей имеет аддитивные свойства.
Можно найти такой набор значений энергии связей, сумма кото-
которых по всем связям молекулы равна теплоте образования вещества
в газообразном состоянии из атомов соответствующих элементов.
Энергии связей являются в некоторой степени усредненными
величинами, и в литературе встречаются несколько отличающиеся
значения для одних и тех же связен *.
В качестве иллюстрации приводим значения энергии некоторых
связей (табл. 1).
Энергия связей С—С, С—Н и других может иметь несколько
различающиеся значения в зависимости от состояния гибридизации
и окружения атомов.
Энергии связей могут быть использовлны для оценки теплоты
образования молекулы путем суммирования:
Этот метод является весьма приближенным.
В отдельных случаях удается измерить энергию диссоциации
отдельных связей (методы спектроскопии, фотоиоиизацпи, электрон-
электронного удара). Эта энергия характеризует процесс разрыва связи
Таблица 2. Энергия диссоциации некоторых химических связей
Счкзь
F—F
С! — С1
Вг —Вг
I —I
Энергия
диссоциации,
к Д ж/моль
158
243
193
151
СПИ 3Ei
1ЦС-СН,
(СН3KС-СНЯ
C0HjCH2-CH3
Энергия
диссоциации,
кД ж/ моль
368
335
293
* В учебнике использованы данные из кн.: Полнит Л., Полинг П. Химия.
М., Мир, 1978.
41
с образованием свободных радикалов:
Л-В— «-A--I-B.—ЕА
АВ.
Для двухатомных молекул энергия диссоциации равна термохи-
термохимической энергии связи, но для многоатомных молекул это не так.
Например, энергия диссоциации связи С—С в большой степени
зависит от строения углеводорода (табл. 2).
Термодинамическую энергию связей и энергию диссоциации ис-
используют для сравнения и характеристики стабильности и реакци-
реакционной способности органических соединений.
3. ИЗМЕРЕНИЕ ЭЛЕКТРИЧЕСКИХ
ДИПОЛЬНЫХ МОМЕНТОВ
Электрические диполыгые моменты ц характеризуют асимметрию
распределения электронной плотности в исследуемой молекуле
или химической связи, т. е. характеризуют полярность молекул
или связей:
где / — радиус-вектор, соединяющий центры тяжести зарядов, м;
q — заряд в центрах тяжести; q—be. (здесь е — элементарный за-
заряд электрона, равный 1,6-10~1!) Кл; б может меняться от 0 до 1).
В единицах СИ дипольный момент измеряется в кулон-метрах. В ли-
литературе можно встретить дппольные моменты, выраженные в
единицах СГС — дебаях (D); здесь заряд электрона равен
4,8-10~10 эл.-ст. ед., а / измеряется в ангстремах (i,\=10~8 см).
В единицах СГС 10~13 единиц дииолыюго момента называется
дебоем: ID-3,34 • Ю"™ Кл-м.
В общем случае электрический дипольный момент (или просто
дипольный момент) является векторной величиной и направлен
от центра тяжести положительного заряда
к центру тяжести отрицательного заряда.
Чтобы определить динольиый момент,
измеряют диэлектрическую константу (на
специальном приборе—дпэлкометре) и
константу преломления света для раствора
вещества в неполярном растворителе (бен-
(бензол, диоксап).
Экспериментально определяют суммар-
суммарный днпольиый момент молекулы. Диполь-
Дипольный момент связи можно определить не-
непосредственно только для двухатомных мо-
молекул. Дипольный момент молекулы состо-
состоит из векторной суммы дипольных моментов
отдельных связей. Поэтому дипольные моменты связей можно рас-
считатьна основе большого числа значений дппольпых моментов мо-
молекул (подобно рефракции связей или термохимической энергии).
Рис. 19. Суммирование
дипольных моментов свя-
связей:
цлвс = 01лв л- И-дс"Ь
+ cos 41"'
42
На рис. 19 показано определение диполыюго момента для
трехатомной молекулы векторным суммированием дипольных мо-
моментов связей.
Значения днпольных моментов связей можно использовать для
оценки эффективных зарядов б на атомах в полярной связи. На-
6+ б—
пример, для связи А—В
лв, 7Г
Полярность связей определ.чет реакционную способность и па-
правление реакции органических соединений в реакциях с ионным
механизмом (см. Введ. 7.2).
Полученные экспериментально значения днпольпых моментов
молекул используются для проверки правильности принятых
структур. Для этого в справочнике находят дипольпые моменты
связей согласно выбранной структурной формуле и рассчитывают
их векторную сумму. Если расчетная величина совпадает с экспе-
экспериментальной, то правильность избранной структуры становится
весьма вероятной.
4. РЕНТГЕНОГРАФИЯ II ЭЛЕКТРОНОГРАФИЯ
Рентгенографический метод — рептгепоструктурпый анализ — ос-
основан на дифракции рентгеновских лучей в кристалле вещества.
Рентгеновские лучи (электромагнитное излучение с длиной волны
0,1 —10 нм) при прохождении через кристалл взаимодействуют с
электронными оболочками атомов. В результате этого взаимодейст-
взаимодействия происходит дифракция рентгеновских лучей и на фотопленке
получается дифракционная картина — пятна или окружности. Из
дифракционной картины при помощи сложных расчетов получают
сведения о размещении молекул в элементарной ячейке кристалла и
о расстояниях между атомами и углах между химическими связями.
Чем меньше число электронов в атоме, тем слабее рефлексы рентге-
рентгеновских лучей. Поэтому определить местонахождение атомов водо-
водорода весьма трудно.
Рентгеноструктурный анализ — очень трудоемкий процесс.
Для определения структуры средней сложности требовалось много
месяцев сложной расчетной работы. В наши дни благодаря созда-
созданию специальных реитгенодифрактометров, работающих автомати-
автоматически согласно программе ЭВМ, определение полной структуры тре-
требует меньше времени.
Электронографический метод подобен рентгенографическому
и основан на взаимодействии потока электронов с веществом. По-
Поток электронов при прохождении через вещество напоминает
электромагнитное излучение с очень небольшой длиной волны и дает
дифракционную картину. Эти дифракционные картины (электро-
нограммы) можно получить для веществ в газообразном состоянии
или для очень тонких пленок. Дифракция электронов обусловлена
взаимодействием электронов с атомными ядрами.
43
Существует также нейтронографический метод структурного
анализа, основанный на дифракции потока нейтронов в молеку-
молекулах вещества.
Эти методы структурного анализа дают возможность опреде-
определить полную структуру молекулы — межатомные расстояния, углы
между связями, т. е. точное пространственное расположение всех
атомов молекулы в кристаллической решетке или в газообразном
состоянии. Методом рентгеноструктурного анализа определена
структура таких сложных природных веществ, как сахароза, пе-
пенициллин, стрихнин, витамин Bt2, некоторые белки (например,
миоглобин) и нуклеиновые кислоты.
В результате структурных исследований в органической химии
были получены экспериментальные доказательства устойчивости
длин связей и углов между связями в соединениях близкого типа.
Расстояние между атомными ядрами в химической связи называет-
называется длиной связи /дв- Длина связи используется для получения ха-
характерной постоянной атома — его ковалептного радиуса гх- Если
связь образована двумя одинаковыми атомами, то ковалентный ра-
радиус равен половине длины связи. В общем случае длина любой
ковалентпой связи равна сумме ковалентпых радиусов соответст-
соответствующих атомов. Длина сильпополярных связей меньше, чем сумма
ковалентных радиусов:
'лв = гк + гв -0,09(хА -хв),
где "/.а и хв — электроотрицательностн атомов (Введ. 5).
Ковалентные радиусы атомов являются функцией состояния
гибридизации атома и типа орбитали. Обычно с усилением s-харак-
тера гибридной орбитали ковалентный радиус уменьшается. Зна-
Значительные особенности длин связей выявляются в сопряженных
-.¦•"'Л?''--:«чy^v~>, ,--,""^:-.-:.j.,-—-:r--,4 системах, в которых они могут
¦¦¦.•'¦•.¦¦"•¦¦¦¦¦¦• ¦•.'•'¦:..'\/х':-'."•.'•'¦ •¦'¦¦':'•'¦]¦¦¦ v.X сильно различаться в зависимо-
:-."ci' '" ci-'-V/'i; '.:¦(.•'. ci—:—ci .'• .-' i сти от типа сопряженной систе-
¦;'.•¦ -.'¦.¦>¦' мы («выравненные» н «нецелочис-
;Я^й^>;>/ ленные» связи, например, в бу-
бутадиене, бензоле и др.).
0]f Из рентгенографических, эле-
Рис. ^Радиус эффективного дейст- хронографических и ряда дру-
вия молекулы хлора гих физических исследовании мо-
можно получить некоторые другие
характерные величины. Так, например, можно установить наимень-
наименьшее расстояние, на которое при данной температуре могут сбли-
сблизиться непосредственно не связанные атомы или группировки, или
две молекулы (рис. 20).
Это расстояние обозначается// (dt); половина этого расстояния
называется радиусом эффективного действия атома (радиусом Ван-
дер-Ваальса). Эффективные радиусы всегда больше, чем ковалент-
ковалентные, и они уменьшаются при повышении температуры.
В табл. 3 приведены некоторые значения атомных радиусов и
углов. Следует отметить, что ковалентный радиус атомов при sp2-
44
Таблица 3. Ковалентные и эффективные радиусы атомов и углы
между связями
н
N,
N
N,
N,
0
О
s
s
F
Cl
Br
I
Atom
1
1 sp3, тетрагональный
t sp2, трнгональный
^t Cs"!~Csp'
Cs/>2 ~CspJ
f Л sp, дигональиый
» ^ г Г
.—С—" s/> SP
тетрагональный
(немоделепная пара электронов)
тригопальный
дигональпып
(R-.O)
(Х = О)
(RaS)
X = S)
г, им
0,077
0,067
(в бензоле
0,0695)
0,076
0 060
0|07
0,03—0,036
0,07
—
0,062
0,055
0,066
0,051
0,104
0,094
0,06
0,099
0, 114
0,136
г .
'эф
—
0,18
(л-связь)
0,18
0,1
0,15
0,16
0,15
0,14
0,16
0,17
0,185
0,135
0,180
0,195
0,205
Валет пый
угол
109°28'
A09-113°)
120°
A10—122°)
180°
106—109°
120°
105°
95—100°
и sp-гибридизации меняется в зависимости от типа связи, например
в двойной связи С=С (С4-^ — С6рг) ковалентный радиус атома уг-
углерода С.,^ меньше, чем в связи =С—С (CijD,_cJp.,). Ковалептный
радиус атома водорода зависит от типа соединения. Углы между
одинаковыми связями в различных соединениях меняются в пре-
пределах 3—6е.
Значения ковалентпых и эффективных радиусов и углов между
связями используют для конструирования моделей атомов и моле-
молекул, схем молекул. Наиболее известными являются атомные модели
Стюарта — Бриглеба. Принцип их конструкции очень прост.
Атом изображают в виде сферического или частично сферического
тела, радиус которого пропорционален гэф. Тело усекают плоско-
плоскостями соответственно числу связей и валентному углу. Плоскости
лежат на расстоянии от центра, пропорциональном ковалентно-
му радиусу (рис. 21, 22). При соединении этих атомных моделей по-
получаем модель молекулы (рис. 23, 24, 25). Модели молекул дают
хорошее представление о пространственном Строении молекулы.
45
Рис. 21. Модель
атол1а водорода
Рис. 22. Модель атома
кислорода
Рис. 23. Модель мо-
молекулы метана
Рис. 24. Модель молекулы
этилена
Рис. 25. Модель
ацетилена
молекулы
5. ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЯ
Электрохимические методы основаны на зависимости силы тока от
приложенного напряжения при прохождении тока через раствор
в электролизерах специальной конструкции. В результате получа-
получают кривые зависимости силы тока — напряжение (потенциал).
Эти вольтамперные кривые характеризуют процессы, проходящие
на электродах. На катоде происходит электрохимическое восста-
восстановление (присоединение электрона), а на аноде — электрохими-
электрохимическое окисление (отрыв электрона). В зависимости от типа изу-
изучаемого процесса (анодного или катодного) применяются приборы,
отличающиеся между собой соотношением площадей электродов,
материалом электродов и др.
Полярография. В основе полярографического метода лежат
катодные процессы (присоединение электрона к веществу па ртут-
ртутном капающем электроде). Полярографический метод создал чеш-
чешский химик Я. Гейровскнй A922), за что был удостоен Нобелев-
Нобелевской премии A959). Принципиальная схема полярографа очень
проста (рис. 26). Он состоит из капающего ртутного микроэлект-
микроэлектрода с непрерывно обновляющейся поверхностью и электрода срав-
сравнения (ртутный или другой нормальный электрод). Площадь ка-
катода значительно меньше площади анода, поэтому решающими
в этом случае являются процессы поляризации катода. Оргапиче-
46
ское вещество диффундирует к катоду и принимает электрон, про-
происходит деполяризация кагода (органическое вещество, которое
способно восстанавливаться, называется деполяризатором или
полярографичгски активным веществом). Деполяризация катода
начинается при определенном
потенциале ?гыд (потенциал
выделения или восстановле-
восстановления, характерный для данного
, MicA
Предельный ток
Рис. 26. Принципиальная схема поля- Рис, 27, Полярограмма
рографа
деполяризатора). В результате начинается электролиз и сила тока
круто возрастает. При постепенном увеличении напряжения устанав-
устанавливается некоторое стационарное значение силы тока (предельный
ток), которое уже не зависит от повышения напряжения. В резуль-
результате на графике НЕ появляется полярографическая волна (рис. 27).
Каждая полярографическая волна характеризуется потенциалом
полуволны ?\/2, который зависит от концентрации водородных ио-
ионов в растворе (рН) и температуры, но не зависит от концентра-
концентрации деполяризатора. Потенциал полуволны при определенном зна-
значении рН является константой вещества.
Потенциалы полуволны характеризуют сродство частицы к
электрону (СЭ), поэтому они могут быть использованы для срав-
сравнения электроноакцепторных свойств соединений. Сродство к элект-
электрону связано с потенциалом полуволны восстановления эмпириче-
эмпирическим выражением:
Следовательно, полярографию можно использовать для характе-
характеристики процесса:
А++е~ —> А.
+е~
Образование свободных радикалов можно установить методом
электронного парамагнитного резонанса (Введ. 6.10).
Метод полярографии широко используется для определения
концентрации веществ в растворах.
Анодная вольтамперометрия. В основе этого метода лежат анод-
анодные процессы (окисление органического соединения на платпно-
47
вом или графитовом аноде)'. С точки зрения экспериментального
осуществления этот метод подобен полярографии. Найденный экс-
экспериментально потенциал полуволны окисления Е?Ц характери-
характеризует электронодонориые свойства соединений и их энергию иони-
ионизации (ЭИ):
Постоянные а и Ь находят опытным путем.
Анодную вольтамперомстрию используют для изучения процес-
процессов окисления:
D: * D. + >¦ D2 +
Метод используют также для количественных определений ве-
веществ в растворах.
Разработан метод циклической вольтамперометрип, позволяю-
позволяющий исследовать процессы восстановления и окисления на одном
и том же электроде в одном растворе. Прибор рисует циклическую
вольтамперограмму, по которой оценивают потенциалы восстанов-
восстановления и окисления данного органического соединения.
6. СПЕКТРОСКОПИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЯ
В основе спектроскопических методов лежит взаимодействие ве-
вещества с электромагнитным излучением, что вызывает поглощение
излучения или его эмиссию. Взаимодействие возможно в очень ши-
широком интервале электромагнитных волн, начиная с v-лучей и
кончая радиоволнами. При воздействии электромагнитного излу-
излучения происходит изменение энергии молекул согласно уравнению
Бора:
АЕ = Е*—?0=Av,
где Ео— энергия основного состояния; Е*—энергия возбужден-
возбужденного состояния; h — константа Планка; v — частота излучения;
АЕ — изменение энергии системы. Данное уравнение характери-
характеризует поглощение. В случае эмиссии:
Электромагнитное излучение характеризуется как волновыми
параметрами, так и энергетическими. Волновыми параметрами яв-
являются длина волны X (м, см, им) и частота колебаний v (с, Гц),
которые связаны между собой уравнением v = cA, где с—скорость
света.
Очень часто пользуются величиной, называемой волновым чис-
числом (иногда ее неправильно называют частотой): v=^-cm-*.
48
Энергетические параметры характеризуют разницу энергии
между основным и возбужденным состояниями (энергия перехода);
их выражают в электрон-вольтах (эВ) или джоулях на моль
(Дж/моль) *.
Между волновыми и энергетическими параметрами существует
следующая взаимосвязь:
а[эВ]= — [нм]-=806Са[см-'].
В табл. 4 приведен полный спектр электромагнитного излуче-
излучения.
Таблица 4. Характеристика электромагнитного излучения
Излучение
Длина лопни, м
Энергия
кванта излу-
излучения, эВ
Характер изменении в веще-
веществе и название метода
•у-Лучи
Рентгеновские
лучи
Ультрафиолето-
Ультрафиолетовое и видимое из-
излучение
Инфракрасное
излучение
Микроволны
радиоволны
10-10—10-"
10-я—Ю-*
A0—1000 нм)
ю-6—ю-4
(v-И 0 000—
— 100 cm~j)
>ю-3
(v<3-1011 Гц)
107—104
104—102
102—1,2
1,2—0,012
ю-3
Энергетические состо-
состояния атомных ядер
Энергетические состо-
состояния электронов внут-
внутренних оболочек
Энергетические состо-
состояния электронов внеш-
внешних оболочек (электрон-
(электронные спектры поглощения
и эмиссии)
Энергетические состо-
состояния колебаний атомов
(колебательная спектро-
спектроскопия)
Энергетические состо-
состояния вращения молекул,
состояния спинов элект-
электронов и ядер (спектро-
(спектроскопия ЭПР и ЯМР)
В зависимости от области электромагнитного спектра приме-
применяют различные экспериментальные методы и приборы. Чтобы на-
наблюдать спектры поглощения, необходимы: источник излучения,
кювета с изучаемым веществом, установка для получения монохро-
монохроматического излучения (с определенной длиной волны) с призмами
или дифракционной решеткой, приемник для измерения интенсив-,
ности излучения (падающего и прошедшего через образец) и реги-
регистрирующая установка.
Поглощение излучения подчиняется определенным закономер-
закономерностям. Правило Бугера — Ламберта — Бера определяет зависи-
* 1 эВ=1,602-10-19 Дж (на
ХЮ23 Дж/моль=96,48 кДж/моль,
1 ыолекулу)= 1 ,G02 • 10~i9 -6,0225X
49
мость интенсивности прошедшего *е1зез вещество излучения от
ряда факторов:
где /„ - интенсивность падающего излучения; 7 - интенсивность
прошедшего через вещество излучен^ е - коэффициент интенсив-
интенсивности поглощения (коэффициент *кстинкции); Я - оптическая
плотность образца; с-молярная концентрация вещества; d-
толщина слоя вещества, см.
Это правило относится к так наз*1Ваемьш оптическим спектрам
поглощения (электронные спектры логлощепия и инфракрасные
спектры поглощения).
7. ЭЛЕКТРОННЫЕ СПЕКТРЫ
И ЭМИССИИ (ЛЮМИНЕСЦЕНЦИИ)
Электронные спектры относятся к ультрафиолетовой и видимой
части электромагнитного излучения (г*°™ощение и эмиссия ультра-
ультрафиолетового и видимого света). ОбЛ^сть Длин„ волн Д° 20? ™
F,2 эВ) относится к вакуум-ультРа4'иолетовои части' в этои об-
области поглощают атмосферные газы, поэтому исследования прово-
проводят в вакуумных установках. 0быч^ыои "и1?в™ *1ИН Волн для
электронных спектров 200-1000 нм @,2-1,24 эВ). Граница интер-
интервала 800-1000 им простирается уже в области ближней инфракрас-
инфракрасной и частью приборов не регистрируй- Схемы приборов Для по-
получения спектров поглощения i зобр*жепы на Рис' 28 и 29- Для
Рис. 28. Принципиальная с*ема однолучсво-
го прибора:
/-регулятор длины полны; ^^ы для стандартного
источник спета; -/-щель; 5-кюпе' т; 7-рсгистрирую-
пещестпа и образца; б — фотоэлел^
щая установка
3
4
6
lA/Ju!
„е.ма двухлучсво-
Рис. 29. Принципиальная с*6
го прибора:
2 — источник света;
/ — автоматический монохроматор: т-юпетг.! /мя стлп-
3-щсль; 4-прср:.татсль светп; г^1рег„стр*фующая
дартпого вещества и образца;
установка; 7 — самописец
50
с,
lg Г.
D
М.;кС.
Перегни
(инфлексия)
A(hmj или ДД',(эВ)
Рис. 30, Спектр поглощения
получения эмиссионных (люминесцентных) спектров можно ис-
использовать такие же или подобные приборы, только вместо источ-
источника света находится кювета с люминесцирующим веществом. Из-
Излучение непосредственно вводится в спектрограф.
Спектр'поглощения изображается графически в системе прямо-
прямоугольных координат, по оси ординат откладывают интенсивность
поглощения (в, lg в, D), а по оси абсцисс — длину волны % (им) или
энергию АЕ (эВ) (рис. 30). Современные двухлучезые приборы ри-
рисуют спектр поглощения автоматически на специальном бланке.
На полученном спектре обычно обращают внимание на макси-
максимумы поглощения (Дм;„(С и е), но важно отметить также перегибы
(инфлексин), которые обусловлены частичным перекрыванием мак-
максимумов поглощения. Подобным же образом изображаются эмис-
эмиссионные спектры.
Максимумы поглощения члето получаются уширенными. При
низких температурах (ниже —200 °С) или в газовой фазе максимумы
значительно уже.
Ультрафиолетовые и видимые спектры поглощения и эмиссии
связаны с возбуждением электронной системы вещества. При по-
поглощении кванта света (излучения со строго определенной энер-
энергией) частица переходит в возбужденное состояние вследствие
перескока электрона с одного энергетического уровня на другой
(со связывающих орбнталей на разрыхляющие орбитали). Возмо-
Возможен целый ряд возбужденных состояний, но самые важные из них
первые (So — основное состояние; 5,, S.,, S3 — возбужденные со-
состояния с суммарным спином, равным нулю,—¦ синглетные состоя-
состояния; 7\, Tj, Т3—возбужденные состояния с суммарным епшюм,
равным 1,— триплетные состояния).
На рис. 31 схематично изображены So, Si и Ti (приведены
только высшая занятая и низшая свободная орбитали). Так как
для каждого состояния (So, Siy 7\ и др.) характерна определенная
энергия, на энергетических диаграммах их изображают как само-
самостоятельные уровни (рис. 32). Если возбужденное состояние обо-
обозначить А*, то процесс поглощения света можно изобразить сле-
следующим уравнением:
Время жизни возбужденных состояний очень короткое: для
еннглетных возбужденных состояний оно равно 10~s—10"s с, а для
51
триплетных — 10~2—1 с. Дезактивация возбужденных состояний
может происходить тремя путями:
а) посредством излучения — эмиссии (люминесценция): А* ->
- S0-4iv;
б) возбужденное состояние отдает свою энергию в виде теплоты
(безызлучательпый переход): А* ~> So+Q;
в) может произойти разрыв одних химических связей и образо-
образование новых (фотохимические реакции):
А* —>¦ Продукты фотохимической реакции
Если эмиссия обусловлена возбужденным синглетным состоя-
состоянием, ее называют флуоресценцией (эмиссия моментально исчезает
Рис. 31. Основное состояние
и возбужденные состояния
+ 4-
4- 4-
Рис. 32. Схема дезактина-
ции возбуждения молекулы
Поглощение Флуоресценция Фосфоресценция о
Колебательные урошш 0,1, 2, 3
после снятия возбуждающего источника света). Эмиссию, обуслов-
обусловленную трпплетным состоянием, можно наблюдать и после снятия
источника света, и называется она фосфоресценцией (послесвече-
(послесвечением).
Синглетное состояние может перейти в триплетное путем оезыз-
лучательной конверсии <s,~—*-т,) .
При поглощении электромагнитного излучения в широком ин-
интервале может меняться не только энергия электронов, но и энер-
энергия атомных колебаний и вращения молекул. Для изменения энер-
энергии электронов требуются сравнительно большие кванты энергии
A 7'эВ), в то время как для изменений колебательной энергии —¦
значительно меньшие @,05—0,5 эВ). Каждому энергетическому
(
состоянию электронов (So
0
S2, 7j, Тг) соответствует целый ряд
2 б
состоянию р (o, l 2, j, г) у
колебательных уровней: 0, 1, 2, 3 (основное и возбужденные коле-
колебательные состояния) (см. рис. 32). При поглощении световой энер-
энергии молекула из основного состояния Sa переходит на различные
колебательные (и вращательные) уровни возбужденных состояний,
вследствие чего полоса поглощения получается размытой.
52
Как правило, максимум флуоресценЩй сдвинут в сторону бо-
более длинных волн по сравнению с максимумом поглощения, так как
возбуждение So -> Si более вероятно переходит на высокие колеба-
колебательные уровни Su а излучает молекула с основного колебатель-
колебательного состояния St (см. рис. 32).
Следовательно, поглощение в ультрафиолетовой и видимой
частях спектра определяется электронными переходами с одной
орбитали на другую — разрыхляющую. Так как орбитали бывают
различными, отличаются и электроЕшые переходы. Переходы мо-
могут быть между о-орбиталями (а—>¦ а*), я-орбиталями (я->я*),
о- и я-орбиталями (а-> я*), орбиталями неподеленной электронной
пары (я) и а- или я-орбиталями (п -> а*, п -> я*).
Электронные спектры поглощения (и эмиссии) используются
для характеристики энергетических уровней электронов в органи-
органических соединениях. При увеличении числа гс-электронов в сопря-
сопряженной системе появляется поглощение в видимой области спектра,
соединения становятся цветными (табл. 5).
Таблица 5. Длины воли поглощения и цвет соединения
Длина полны
поглощения, нм
430—480
490—500
580—595
730-760
Цвет поглощенной
спектральной области
Синий
Сине-зеленый
Желтый
Пурпурный
Наблюдаемый цест
Желтый, желто-оракжевый
Красный
Синий
Зеленый
Электронные спектры поглощения незаменимы для исследова-
исследования структуры органических соединений, так как для многих груп-
группировок химических связей характерны определенные ,x*Vk:j волн
максимумов поглощения и интенсивности поглощения. Эти спектры
незаменимы для изучения межмолекулярного взаимодействия —
образования комплексов с переносом заряда (л-комплексов). Они
могут быть использованы при количественных определениях орга-
органических соединений в растворах. Примеры электронных спектров
поглощения приводятся при рассмотрении определенных классов
соединений.
8. ИНФРАКРАСНЫЕ СПЕКТРЫ ПОГЛОЩЕНИЯ
Инфракрасные спектры поглощения (ИК-спектры) относятся к
инфракрасной части электромагнитного излучения. Обычно при-
применяемая область длин волн 2,5-10~°—2,5-10~s м или 2,5—25 мкм
(волновые числа 4000—400 см), энергия кванта 0,5—0,05 эВ.
Поглощение в этой области связано с возбуждением колебатель-
колебательных уровней атомов в химических связях. Так же как электроны
в молекулах располагаются на определенных уровнях'энергии,
атомные колебания в химических связях характеризуются дискрет-
53
пыми уровнями энергии. ^Ьреход от одного колебательного уровня
энергии на другой, более высокий, связан с поглощением энергии.
Поэтому ИК-спектры поглощения часто называются колебательными
спектрами.
ИК-спектры поглощения более богаты максимумами, чем элект-
электронные спектры (рис. 33). На оси абсцисс откладывают длину вол-
волны Я, (мкм) или волновое число v (см), а на оси ординат — интен-
интенсивность поглощения в единицах s или просто в процентах погло-
поглощения (рис. 33).
В общем случае картина колебаний молекул является сложной,
особенно для многоатомных молекул. Но некоторые факторы облег-
облегчают ее интерпретацию. Существуют характеристические длины
l\jV^\
Рис. 33. Изображение
инфракрасного спект-
спектра поглощения
4000 3000 2000 1000 1/Д ,см
-1
волн (характеристические частоты), которые соответствуют строго
определенным связям и атомным группировкам и мало меняются
при изменении структуры молекулы. Так, например, интервал
2000—4000 см характерен для валентных колебаний связей
X—Н (где X=rSi, P, S, С, N, О), а интервал 1500—1950 см (ин-
(интервал двойных связей) —для валентных колебаний связей С -О,
С^С, C=N, N —1NL Для интервала 600—1500 см характерно
большое число максимумов поглощения (интервал валентных и
деформационных колебаний простых связей X—Y). Часто этот
интервал называется «областью отпечатков пальцев», так как со-
содержит очень характерные максимумы, используемые для иденти-
идентификации веществ.
В органической химии ИК-спектры поглощения используют для
определения структуры соединений, так как каждая группа ато-
атомов имеет свое характерное поглощение. В этом отношении ИК-
спектры ценнее электронных спектров поглощения. ИК-Спектро-
скопию с успехом используют для количественных определений
органических соединений в различных смесях. Это необходимо при
изучении скорости и механизма реакций.
9. СПЕКТРОСКОПИЯ КОМБИНАЦИОННОГО РАССЕЯНИЯ
(РАМАН-СПЕКТРОСКОПИЯ)
Спектры комбинационного рассеяния (КР), так же как ИК-спектры
поглощения, являются колебательными спектрами. Спектры КР
являются эмиссионными спектрами, их получают, интенсивно ос-
освещая образец монохроматическим светом и распределяя отражен-
отраженный свет по длинам волн в спектрометре. В спектре отраженного
света наблюдают линию, соответствующую использованному (воз-
54
буждающему) монохроматическому свету v0, и кроме этого ряд сла-
слабых спектральных линий v,, v->, v?, .... vft. Разницы vft—v3 соогвет-
ствуют характеристическим колебательным частотам молекулы.
Расположение спектральных линий около v0 является симметрич-
симметричным (рис. 3 0-
Таким образом, использование спектров комбинационного рас-
рассеяния служит источником информации о характеристических час-
частотах колебаний связен. По-
Поэтому спектры КР использу-
используются для определения струк-
структуры соединения. В отличие
от ИК-сиектроскошги этот
метод требует больше време-
времени. В последнее время для
получения спектров КР применяют новые источники очень интен-
интенсивных монохроматических пучков света — лазеры. Развивается
лазерная спектроскопия комбинационного рассеяния, при помощи
которой можно получить КР-спектр как жидких, так и твердых об-
образцов, причем образцы могут быть бесцветными или окрашен-
окрашенными. Благодаря большой интенсивности возбуждающего света
(лазерный импульс) для регистрации спектра комбинационного
рассеяния требуется меньше времени, чем в обычном методе.
V7
»6
f5
ь
Рис. 34. Схема спектра
иого рассеяния
комбииацггон-
10, ЭЛЕКТРОННЫЙ ПАРАМАГНИТНЫЙ РЕЗОНАНС
Электронный парамагнитный резонанс (ЭПР) является радиоспек-
радиоспектроскопическим методом, при помощи которого можно исследовать
взаимодействие радиоволн (электромагнитного излучения с боль-
большой длиной волны, Х>10~3 м) с веществом. Фактически этот метод
имеет мало общего со спектроскопией, так как излучение не раз-
разделяется в спектр (по отдельным длинам волн) при помощи призм
или дифракционной решетки.
Так как для радиоволн характерны очень маленькие кванты
энергии (/tv<10~3 эВ), возникает вопрос, где в молекуле органиче-
органического соединения встречаются такие близко лежащие энергетиче-
энергетические уровни. Такие переходы возможны в электроне и атомах и они
связаны с энергетическими уровнями магнитного момента спина
электрона или атомного ядра (магнитный момеит возникает благо-
благодаря циркуляции заряда вокруг оси частицы).
Но отдельные энергетические уровни наблюдаются только в
случае, если вещество находится в сильном однородном магнитном
поле. В магнитном поле происходит ориентация магнитного момента
спина (в направлении, совпадающем с направлением магнитного
поля и противоположном ему). Эта ориентация вызывает расщепле-
расщепление энергетического уровня магнитного момента спина на два.
Заселены оба эти уровня, но они различаются числом частиц на
них. Обычно более заселен нижний энергетический уровень и воз-
55
Без поля
Рис. 35. Расщепление энергетических уров-
уровней спинового магнитного момента в магнит-
магнитном поле
можен переход на высший уровень, что связано с поглощением
электромагнитного излучения.
Расщепление спиновых магнитных моментов происходит только
в очень сильном магнитном поле, разницы энергий АЕ между уров-
уровнями маленькие, при этом АЕ зависит от напряженности магнит-
магнитного поля //0 (рис. 35): AE = kH0.
В основе метода ЭПР лежит расщепление энергетических уров-
уровней спинового магнитного момента нес паренного элект-
д?-ш0 о о и а, который обычно
"о >н„ имеется в свободных ради-
tr ,'' ' калах.
Разница в уровнях энер-
энергии спинового магнитного
момента неспаренногоэлек-
неспаренногоэлектрона в сильном магнит-
магнитном поле составляет 10—
10~4 эВ (длина волны
поглощаемого излучения
0,1—1 см, частота 3-10й—
3-101» Гц).
Поглощение электро-
электромагнитного излучения мо-
может наблюдаться, если об-
образец находится в сильном
магнитном поле и через
него пропускается излуче-
излучение. Так как разница энер-
энергетических уровней (энер-
(энергия поглощаемого кванта
излучения) зависит от на-
напряженности магнитного
поля, то, изменяя напря-
напряженность, можно достичь
такого момента, когда радиоволны определенной длины будут по-
поглощаться веществом и будет наблюдаться резонансный сигнал.
Основной частью аппаратуры радиоспектроскопии (рис. 36)
является сильный электромагнит с очень гомогенным магнитным
полем и изменяемой напряженностью магнитного поля (известны
также приборы с постоянным магнитом и изменяемой частотой
радиоволн). Ампула с веществом находится между полюсами маг-
магнита и одновременно подвергается воздействию магнитного поля и
радиоволн. Ампула с веществом быстро вращается. Резонансный
сигнал записывается самописцем при изменении напряженности
магнитного поля или частоты радиоволн как уменьшение интенсив-
интенсивности генерированных радиоволи.
Метод ЭПР применяют для обнаружения свободных радикалов,
которые, например, могут образоваться при гомолитическом рас-
расщеплении химической связи:
R:R'—>?
2
1
я
3 1
\ 1 Г
щ
щ
4
5
jJluJl
Рис. 36. Принципиальная схема методов
ЭПР и ЯМР:
/ — образец; 2 — генератор ради ополи; 3— приемник
радпогюлн; '/ — электромагнит; 5—блок управления;
б-сьмошгес-ц
56
I
°"
Рис.
37. Сигнал
IV
Рис. 38. Расщеп-
?г?рЫЙ сигиал
В спектре ЭПР можно наблюдать один мак-
максимум (обычно говорят — одну линию) или це-
целый набор линий, которые образуются в ре-
результате расщепления сигнала ЭПР. Обычно
спектр ЭПР записывают как первое производ-
производное от функции интенсивность сигнала — на-
напряженность магнитного поля (рис. 37, 38).
Расщепление сигналов ЭПР вызвано взаимо-
взаимодействием неспарепиого электрона с близлежа-
близлежащими атомными ядрами, которые имеют собст-
собственный спиновый магнитный момент (главным
образом с водородными атомами).
Спектроскопия ЭПР является очень чувст-
чувствительным методом для обнаружения свобод-
свободных радикалов (до концентрации 10~12 моль).
Кроме этого, анализируя сверхтонкую струк-
структуру спектра ЭПР, можно судить о делокализации неспаренного
электрона и о структуре изучаемой частицы.
11. ЯДГ.РНЫЙ .МАГНИТНЫЙ РЕЗОНАНС
Ядерный магнитный резонанс (ЯМР) является радиоспектроскопи-
радиоспектроскопическим методом. Он основан на измерении поглощения веществом
радиоизлучения определенной частоты вследствие энергетических
переходов атомных ядер в сильном магнитном поле с одного маг-
магнитного энергетического уровня на другой. Сигнал ЯМР могут
вызвать только ядра со спиновым квантовым числом, отличным от
нуля. Ядра, не имеющие магнитного момента спина, например
J-C, J':O, непригодны для экспериментов по ЯМР. Наиболее удобны
для ЯМР-спектроскопии ядра, имеющие полуцелый спин, напри-
мер 1П, 9 1 ,
15M
7 IN •
1, 9 о 7
Ядра с целым спином (,'N, [D) тоже активны в ЯМР-спектрах,
но дают малоинтенсивные по сравнению с ядрами \Н сигналы.
В настоящее время наиболее широко применяется протонный
магнитный резонанс (ПМР). При плотности магнитного потока
около 1,4 Тл A400 Гс)* область поглощения ядер водородных ато-
атомов (протонов) находится при 60-Ю6 Гц F0 МГц), что соответствует
энергии квантов -~2,5-10~? эВ (длина радиоволн ~5 м).
Самописец записывает спектр ПМР на специальных бланках,
где по оси абсцисс откладывается так называемый химический сдвиг
(б) сигнала ПМР. В принципе можно откладывать напряженность
магнитного поля или частоту радиоволн.
Сигналы ПМР водородных атомов органического соединения
очень чувствительны к структурным изменениям в молекуле, к
окружению атома (эффект экранизации). Можно сказать, что почти
каждый атом водорода в молекуле или группа водородных атомов
одного типа дает свой сигнал в спектре ПМР.
1 Тл (тесла) 101 Гс (гаусс).
57
1,48 3,57
CIljCH2CI
2H ^
j
1
Г
1
' 311
TMC
1
1 1 : I
6.0 5,0 4,0 3,0 2,0 J,0 0 a
Рис. 39. Спектр ПМР этилхлорида и растворе
Для определения хи-
химического сдвига водо-
водородных атомов в спект-
спектрах ПМР вводится стан-
стандарт — сигнал ПМР во-
водородных атомов тетра-
метилсилана (CH3LSi
(TMC). Для этого соеди-
соединения химический сдвиг
протонов принят равным
нулю (б- 0). Химичес-
Химический сдвиг определяется
выражением
140 Ш 100 <~ 80 60 40 20
Puc, 40, Спектр ЯМР J3C этнлхлорпда
о &,
где v — частота радио-
радиоволн.
Таким образом, хи-
химический сдвиг н ПМР
измеряется в безразмер-
безразмерных единицах — мил-
миллионных долях (м. д.).
Интенсивность сиг-
сигналов ПМР пропорцио-
пропорциональна числу однотип-
однотипных атомов водорода в
молекуле. Поэтому спектры ПМР служат источником информации
не только о типах водородных атомов, но и о их числе.
В современных спектрометрах ПМР с высокой разрешающей
способностью хорошо наблюдается так называемая топкая струк-
структура сигналов ПМР. Каждый сигнал ПМР может расщепляться на
несколько сигналов в зависимости от соседних атомов, активных
в ЯМР. Такое расщепление происходит, например, при взаимо-
взаимодействии с соседними водородными атомами (спин-спиновое взаимо-
взаимодействие). Если рядом находится одни водородный атом, то сигнал
расщепляется на два, если два — на три, если три — на четыре
сигнала (рис. 39).
На рис. 39 изображена также кривая автоматического интегри-
интегрирования для определения числа протонов. Приведенные значения
6 СН3- и СН,-груип относится к середине мультпплета.
В последнее время развивается также спектроскопия ЯМР па
ядрах 13С, которая использует естественное содержание изотопа
углерода в органических соединениях. Так как естественное содер-
содержание 13С в углероде около 1,1% и сигналы малоинтенсивны, не-
необходима специальная аппаратура. Разработана новая техника
получения спектров ЯМР с использованном фурье-преобразова-
теля. На образец подается импульс радиоволн п при помощи ЭВМ
58
отыскиваются поглощенные частоты, которые записываются в па-
памяти ЭВМ. Такие импульсы подаются один за другим и сигналы
ЯМР накапливаются по необходимой интенсивности при плотности
магнитного потока 2,35 Тл B3500 Гс). Область поглощения ато-
атомов 1ЯС находится при 25 МГц. Стандартом служит ТМС. Спектры
ЯМР 13С очень чувствительны к структурным изменениям (рис. 40).
Метод ЯМР является самым эффективным для определения
структуры органического соединения, особенно при применении
всей гаммы ядер ^Н, 13С и др.). Этим методом изучают не только
структуру, но и межмолекулярное взаимодействие, таутомерные
равновесия, кинетику реакций.
12. СПЕКТРОПОЛЯРИМЕТРИЯ
Поляриметрия основана на измерении оптического вращения плоско-
поляризованного света при прохождении света через образец. Оп-
Оптическим вращением называется изменение плоскости поляриза-
поляризации плоскополяризованного света (вращение плоскости поляри-
поляризации). Вещества, способные к изменению плоскости поляризации
нлоскополяризовапного света при прохождении света через обра-
образец, называются оптически активными.
Спектрополярнметрия занимается измерением величины опти-
оптического вращения вещества в зависимости от длины волны плоско-
поляризованного света.
Впервые явление оптической активности наблюдал французский
физик Мал юс в 1808 г. При прохождении обычного света через
кристалл исландского шпата (кристаллического СаСОя) возникает
два плоскополяризованных во взаимно перпендикулярных плоско-
плоскостях луча — происходит двойное лучепреломление. Для выделения
одного плоскополяризованного луча применяют специальную приз-
призму — призму Николя, состоящую из двух склеенных кристаллов
исландского шпата (рис. 41).
Обычный свет представляет собор! совокупность электромагнит-
электромагнитных волн, колебания которых расположены в различных паправ-
/68°
Рис. 41. Выделение плоскополяризоваипого луча
Рис. 42. Схема иращепия плоскости по.чя-
ризацип свеча
59
1 2 I
Рис. 43. CxeMaTH4FFoe изображение поляриметра:
/-призмы Николя; 2-трубка с веществом; 3-глаз наблюдателя
лениях в плоскости, перпендикулярной направлению распростра-
распространения луча. После прохождения через призму Николя свет стано-
становится плоскополяризованным.
Вращение плоскости поляризации света впервые наблюдал
французский физик Био A813) при прохождении плоскополярнзо-
ванного света через растворы скипидара и сахара (рис. 42).
Для обнаружения оптического вращения пользуются поляри-
поляриметром — прибором, состоящим из двух призм Николя и трубки
с образцом (рис. 43).
Если призмы Николя ориентированы относительно друг друга
под прямым углом, поляризованный свет из первой призмы не бу-
будет проходить через вторую призму (анализатор). Если трубка
пуста или заполнена оптически неактивным веществом, наблюда-
наблюдатель увидит темное поле. Если в трубке между призмами находит-
находится оптически активное вещество, происходит оптическое вращение
и свет наблюдается через вторую призму. При вращении второй
призмы (анализатора) на определенный угол вправо или влево
опять достигается положение призм, при котором поляризованный
свет не проходит. Так может быть измерен угол вращения а (в гра-
градусах) и определено направление вращения: вправо (-]-, d) или
влево (—, /).
Угол вращения а является функцией длины трубки, концент-
концентрации оптически активного вещества, природы растворителя, тем-
температуры раствора и длины волны поляризованного света. Обычно
применяют свет с длиной волны 589 им (D-линия натрия).
Для характеристики оптически активного вещества определяют
удельное вращение [а] или молярное вращение [М]:
где а — наблюдаемое вращение, град; / — длина трубки, дм;
с — концентрация оптически активного вещества в исследуемом
растворе, г/см3; М — молекулярная масса.
Удельное вращение зависит от длины волны к поляризованного
света. На спектрополяриметрах можно получить кривую зависи-
зависимости [а] или \М] от X, которую называют кривой дисперсии оптиче-
оптического вращения (ДОВ). Кривые ДОВ могут быть плавными пли
с пиками и впадинами. Появление экстремальных точек (пиков и
впадин) называют эффектом Коттона. Это наблюдается при дли-
длинах волн, где в видимой или ультрафиолетовой области оптически
активное вещество имеет максимум поглощении (оптически актив-
активная полоса поглощения). Эффект Коттона связан с различным по-
60
глощением левой и правой компонент плоскополяризовапного света
в оптически активной полосе (круговой дихроизм).
Спектрополяриметрия является важным методом установления
строения оптически активных органических соединений.
Но какие соединения могут быть оптически активными? Вант-
Гофф связал оптическую активность органических веществ с от-
отсутствием симметрии в их молекулах, например с наличием асим-
асимметрического атома углерода (см. Введ. 5). В этом случае возможны
два тетраэдрическнх расположения заместителей вокруг асимметри-
асимметрического атома. Обе пространственные формы нельзя совместить
никаким вращением. Одна из них является зеркальным отражением
другой. Получаются два стереоизомера (энантиомеры, зеркальные
изомеры, оптические изомеры). Оба стереоизомера составляют пару
оптических антиподов, которые отличаются друг от друга
знаком оптического вращения —[а] и -\-[а] при одинаковом
значении [а].
Асимметрическими могут быть и атомы других элементов —
Si, N, P, S. Возможна также асимметрия всей молекулы в целом
без асимметрического атома. Асимметрической молекулу называют
в том случае, если в ней пет ни одного элемента симметрии — ни
центра, ни осей, ни плоскостей симметрии.
Органические вещества с асимметрическими молекулами всегда
оптически активны (в случае, если имеется только один оптический
антипод или в смеси оптических антиподов один из них преоблада-
преобладает). Но нельзя утверждать, что .молекулы оптически активного ве-
вещества должны быть лишены всех элементов симметрии, например
оси симметрии. Поэтому вместо понятия асимметричности все чаще
применяют термин хиральность. Хиралыюстыо называют свойст-
свойство неидентичности объекта его зеркальному отражению. Моле-
Молекулу называют хиралыюй, если она не идентична своему зеркально-
зеркальному отражению. Пели молекула идентична своему зеркальному отра-
отражению, ее называют ахиральной. Все хиральиые молекулы являются
молекулами оптически активных соединений и, наоборот, молеку-
молекулы оптически активных соединений всегда хиральиы.
Асимметричные молекулы всегда хиральиы, тогда как хиральиые
молекулы могут иметь некоторые элементы симметрии (см. винные
кислоты, с. 614):
к*у\
тральная молекула не идентична ехиралыия молекула
ее зеркальному отражению
На вышеприведенных рисунках заместители ?*"-« н»~ нахо-
находятся над плоскостью бумаги, Y---за плоскостью, а А — в пло-
плоскости бумаги.
61
13. ФОТОЭЛЕКТРОННАЯ СПЕКТРОСКОПИЯ
Фотоэлектронная спектроскопия изучает электронную фотоэмис-
фотоэмиссию из веществ в газообразном или твердом состоянии. Вещество
в газообразном состоянии облучается монохроматическим УФ-из-
лучением большой энергии. Происходит возбуждение валентных
электронов или неподелепных электронных пар на высокие уровни
с последующим уходом электронов из молекулы (фотоэмиссия элект-
электронов). Так как электроны в молекуле имеют различную энергию
( находятся на различных молекулярных орбиталях), эмиттирован-
ные электроны имеют различную энергию.
Если разделить этот поток электронов по энергиям, получим
энергетический спектр фотоэлектронов.
Для получения фотоэлектронных спектров органических сое-
соединений используют гелиевую лампу с излучением 58,5 нм B1,21 эВ).
9,0
9,3
11,4
7 в 9 Ю П 12 Е,Л
Рис. 44. Фотоэлектронный
спектр толуола
7,0
А.
1
8,4
/^
f
9,55
1
10,25
/-
i
s 1 % 9 10 II F. ,эВ
Рис. 45. Фотоэлектронный
спектр тетрацена
Фотоэлектронный спектр характеризует энергии орбиталей валент-
валентных электронов и неподеленных электронных пар (рис. 44, 45).
Таким путем определяют эпергии иопизации (потенциалы иони-
ионизации) различных орбиталей, что используется для характеристики
электронодонорных свойств молекул.
Фотоэлектронная спектроскопия является одним из наиболее
точных методов определения энергии ионизации молекул.
14. МАСС-СПЕКТРОМЕТРИЯ
При бомбардировке молекулы органического соединения электро-
электронами средней энергии B5—70 эВ) происходит выброс электрона
и образование молекулярного иона М+. Молекулярный ион явля-
является нестабильной частицей, так как находится в высшем возбуж-
возбужденном состоянии, и распадается на различные части (фрагменты):
как на положительно заряженные ионы, так и на нейтральные час-
частицы. В масс-спектрометре заряженные частицы распределяются по
их массам, определяются массы частиц и их количество. Записы-
Записывается масс-спектр в единицах mlz.
В спектре наблюдается пик молекулярного иона М+ и пики фраг-
фрагментов с различной интенсивностью. С малой интенсивностью на-
62
блюдаются также пики М+1 и М+2, что связано с присутствием
изотопов углерода 13С и водорода ?Н (рис. 46).
Число и размер фрагментов в значительной степени связаны со
структурой соединения, поэтому по масс-спектрам можно судить
о его строении. Большое значение масс-спектры имеют в расшиф-
100-
ян
Is 60-
и в
I40:
5^20-
с
л/ =
92
ll,
СИ,
6
M
M+l
,M+2 ,
20 40 60
Puc, 46. Масс-спектр толуола
8Э
100 т/г
ровке структуры сложных природных соединений, так как требу-
требуются очень малые количества вещества (доли миллиграмма). Этот
метод позволяет точно определить молекулярную массу вещества
(по молекулярному иону) и его элементный состав.
7. ОСНОВНЫЕ ПОНЯТИЯ О РЕАКЦИОННОЙ СПОСОБНОСТИ
ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Исследования превращений органических соединений, их реакций
являются одной из важнейших составных частей органической хи-
химии. Химические реакции по своему существу являются процес-
процессами, в которых происходит перераспределение электронной плот-
плотности в реагирующей системе. В результате этого некоторые хими-
химические связи исчезают (разрываются) и образуются новые. Часто
это связано с переносом электрона от одного атома на другой, от
одной молекулы на другую. Процесс, в котором происходит исчез-
исчезновение одних и появление других химических связей, называется
элементарным актом реакции. Иногда элементарный акт реакции
связан с переносом электрона от одного компонента на другой.
Атом, у которого происходит разрыв или образование связей,
является реакционным центром. Иногда реакционных центров мо-
может быть несколько одновременно. Предполагают, что реакционными
центрами могут быть те атомы, у которых более выражен электро-
ноакцепторный или электронодопорный характер (в момент реак-
реакции на этих атомах образуются значительные эффективные заряды,
положительные или отрицательные). Это относится главным обра-
образом к реакциям с ионным механизмом. Схему, в которой изображены
элементарные акты реакции, показаны реакционные центры, ис-
исходные и конечные продукты, называют механизмом реакции. Точ-
Точнее, так называется детальное описание пути, ведущего от реа-
реагентов к продуктам реакции, включающее как можно более полную
характеристику состава, строения и других свойств промежуточных
63
соединений и переходных состояний, а также предположении, ка-
касающиеся смещения электронов в ходе последовательных превра-
превращений частицы.
1. КИНЕТИКА ОРГАНИЧЕСКИХ РЕАКЦИЙ
Химическая кинетика изучает скорость химических реакций и за-
зависимость скорости от концентрации реагирующих веществ, темпе-
температуры и давления, типа растворителя.
Скорость химической реакции v можно выразить двумя спо-
способами: либо как функцию от концентрации продукта реакции х
и времени г. v=-^, либо как функцию от концентрации исходного
вещества с и времени /: v=—-g^-- Ьсли концентрацию измеряют в
молях на литр, то скорость реакции измеряют в моль-л^-с.
Для многих реакций скорость пропорциональна определенной
степени концентрации реагентов.
Для реакций первого порядка (например, реак-
реакций типа А->-Х) скорость пропорциональна первой степени концент-
концентрации реагента: v=kc.
Для реакций второго порядка скорость пропор-
пропорциональна концентрации реагента во второй степени, если реаги-
реагирующее вещество одно (реакции типа 2А->Х),
v=kc2
или произведению концентраций двух реагирующих веществ (напри-
(например, реакции типа А+В->Х):
»=fccAcB.
Известны также реакции третьего порядка: (реак-
(реакции типа 2Л+В->Х). Для них
Такая реакция является реакцией второго порядка по отно-
отношению к реагенту А и первого порядка по отношению к реагенту
В, суммарный порядок реакции является третьим.
Понятие «порядок реакции» не следует отождествлять с молеку-
ляриостью реакции. Под молекулярностью реакции понимают чис-
число молекул, участвующих в элементарном акте реакции. По-
Порядок реакции не учитывает молекулы растворителя, так как они
находятся в большом избытке, но в элементарном акте они могут
участвовать. Все-таки очень часто молекулярность реакций (мо-
(мономолекулярные реакции, бимолекулярные реакции) совпадает
с порядком реакции (реакции первого и второго порядка).
Величина k в выражении v=kc%c% называется константой ско-
скорости реакции. Она является важной величиной, так как служит
для сравнения скорости различных реакций. Если концентрации
всех реагентов равны единице, константа равна скорости реакции.
64
Константа скорости реакции первого порядка измеряется в с,
а реакции второго порядка — в л-моль^.
Порядок реакции и константу скорости определяют эксперимен-
экспериментально. Для этого измеряют изменение концентрации реагирующих
веществ во времени при постоянной температуре и давлении. Кон-
Концентрацию веществ определяют при помощи химического анализа,
хроматографии и спектроскопии. Эти измерения проводят с различ-
различными концентрациями исходных веществ и результаты обрабаты-
обрабатывают математически. С целью определения порядка реакции необ-
необходимо проверить, действуют ли соответствующие уравнения. На-
Например, для реакций первого порядка
Ас . ёх , . .
~1Г~ или "ЗГ ^а~^'
где а — начальная концентрация исходного вещества; х — кон-
концентрация продукта реакции спустя время /.
После интегрирования получаем кинетическое уравнение
In = kt или а—х= ае~м.
а—х
Для реакций второго порядка
после интегрирования это выражение принимает вид
а{а — х) ~~" '
Многие реакции в органической химии являются обрати-
обратимыми (равновесные реакции). Как прямая, так и обратная реак-
реакции могут быть первого или второго порядка, и они характеризуются
разными константами: kx и k_t. Например:
*, *¦
А;^ X; A;zt X+Y.
*_, *_i
Такие реакции имеют сложные кинетические уравнения. Реак-
Реакции характеризуют константой равновесия К-
Константа скорости реакции зависит от температуры. Взаимо-
Взаимосвязь между этими величинами исследовали Я- Вапт-Гофф
н С. Аррениус A887). Это уравнение известно как правило Арре-
ннуса:
k= Ае
а
где Т — температура в шкале Кельвина; R — универсальная газо-
газовая постоянная; са — энергия активации (аррениусовская энер-
энергия активации); Л—константа (предэкспоненциальный множитель).
517
g5
При повышении температуры скорость реакции растет, констан-
константа скорости увеличивается.
Здесь введена новая величина — энергия активации. Чтобы
осуществилась реакция между двумя частицами (молекулами, ато-
атомами, ионами), они должны столкнуться. Число столкновений за-
зависит от кинетической энергии частиц, и оно увеличивается с рос-
ростом температуры. Однако не каждое столкновение вызывает реак-
реакцию между частицами. В реакцию вступают только частицы, обла-
обладающие достаточной энергией — активированные частицы, способ-
способные преодолеть энергетический барьер. В результате столкновений
частицы активируются, но не все. С повышением температуры уве-
увеличивается число тех столкновений, которые приводят к реакции.
При взаимодействии активированных молекул образуется ак-
активированный переходный комплекс (переходное состояние):
Реагирующие ,_ ЛктнЕшропанные _^_ Переходное ,. Продукты
молекулы "— молекулы *~ состояние * реакции
Энергия, которая затрачивается на образование активированного
комплекса, называется энергией активации. Время жизни переход-
переходного комплекса очень мало—около 10~12 с.
Процесс может быть изображен графически, если отложить
па оси абсцисс координату реакции, а на оси ординат — изменение
&О
Переходное состояние
AG
'Координата реакции
Переходное состояние
Координата реакции
а б
Рис. 47. Изменение свободной энергии в ходе реакции:
?д--энергия актипацин; АДС харакп-ризует тепловом эффект реакции; а — зк-
зотермнческии процесс; в — эндотермическим процесс
содержания свободной энергии системы AG. Понятие «координата
реакции» означает кратчайший путь, но которому исходная сис-
система превращается в продукт с наименьшей затратой энергии
(рис. 47). Изменение содержания свободной энергии связано с
энтальпией и энтропией системы фундаментальным уравнением:
где II — энтальпия; S — энтропия; G — свободная энергия.
Величина AG связана с константой равновесия реакции К (для
обратимых реакций): AG--—RT In К-
Величина Д# может быть оценена по тепловым эффектам реак-
реакции (Введ. 6.2).
6G
Теоретически для простого случая энергию системы можно пред-
представить как функцию межатомного состояния. Изменение энергии
в зависимости от межатомных расстояний реагирующих частиц
можпо изобразить поверхностью потенциальной энергии реакции.
Графики AG — координата реакции являются некоторым сечением
поверхности потенциальной энергии по наиболее выгодному пути.
2. ТИПЫ РЕАКЦИЙ И РЕАГЕНТОВ
Классификация реакций проводится по характеру химических
превращении и по способу разрыва связи в исходной молекуле.
По характеру химических превращений различают реакции:
а) одпоэлектроппого переноса (реакции одпоэлектронного окис-
окисления — восстановления): D:-rA->-D •J'+A •"
б) диссоциации и рекомбинации: А—В^Л
в) замещения R—X-f-A->-R—А+Х;
г) отщеплен и я: Y - R - Л -* Y - R -;- Л - в;
В
д) присоединения: Y — R + A—В —>• Y—-R;
В Л
е) циклоприсоедипения: y R у
Циклоприсоединением называются реакции, в которых две или не-
несколько ненасыщенных молекул соединяются с образованием од-
одного циклического аддукта, в котором имеет место общее уменьше-
уменьшение кратности связей. Реакции могут быть перициклическимн
(см. ниже) или многостадийными;
ж) реакции изомеризации н перегруппировки (пергмещение крат-
пых связен, миграция атомов и групп, замыкание цикла и др.):
Ачв/°
c4d
Во всех реакциях, кроме первой, происходит разрыв или обра-
образование химической связи. Реакции типа а и б представляют со-
собой элементарные акты. Реакции замещения, отщепления, присое-
присоединения, изомеризации и перегруппировки являются белее слож-
сложными и в общем случае состоит из нескольких элементарных актов.
По способу разрыва и образования химической связи реакции
подразделяются на:
а) гемолитические (свободпорадикальный механизм)
B;U A-+B.
б) гетеролитические (ионный механизм)
Своеобразным гибридом являются реакции одноэлектронного
переноса, в которых могут образовываться ион-радикалы, кото-
которые реагируют дальше как с ионами, так и со свободными радика-
радикалами. Обычно реакции ион-радикалов причисляют к гемолитиче-
гемолитическим реакциям.
в) перициклические реакции •— химические реакции, в которых
реорганизация связей происходит согласованно через цик-
циклическую последовательность непрерывно связанных атомов, т. е.
разрыв и образование связей происходит одновременно в полностью
сопряженном циклическом переходном состоянии.
К перициклическим реакциям относятся часть реакций цикло-
присоедипения (см. выше) и некоторые молекулярные перегруппи-
перегруппировки, в том числе электроциклические реакции и сигматропные
перегруппировки.
Электроциклическая реакция включает образование о-связи
между концами полностью сопряженной линейной л-электрон-
ной системы
Н Н
I I
Нч ,СЧ Нч /
I
а также обратный этому процесс.
Сигматропная перегруппировка включает образование новой
ст-связи между ранее не связанными атомами и разрыв существую-
существующей ст-связи, например:
Н Н
I I
С С
Нг/ NSPH Р Т-Г С/У ^ГТГО
2^- V_ilT. ^^ 1\ 11 -?•*-' V_il 1 ?\
2^4 // — ^ ^2^-\х /СН R
н
В гетеролитических реакциях участвуют различные реагенты:
ионы или поляризованные молекулы. Ионы могут быть различного
типа в зависимости от характера взаимодействия с нротивононом
и молекулами растворителя.
При ионизации коЕалентной связи сначала может образоваться
контактная (тесная) ионная пара А : В7—А:~В + .
68
Контактная ионная пара представляет собой пару противопо-
противоположно заряженных ионов, удерживающихся вместе за счет куло-
новского притяжения без образования коЕалентной связи. Такай
ионная пара ведет себя как одно целое, электропроводность раство-
раствора не увеличивает.
Контактная ионная пара при взаимодействии с молекулами раст-
растворителя (Sol) может образовывать свободную (рыхлую) ионную
пару:
. Sol
АГВ:+ ^ А:~ |Sol|B +
Свободная ионная пара символически изображается как А7ЦВ4";
ее составные части легко вступают во взаимный обмен с другими
свободными ионными парами в растворителе. Далее может следо-
следовать диссоциация свободной ионной пары с образованием сольва-
тировапных ионов:
Sol Sol Sol
а: в+ ^=s А:- + в+
Sol Sol Sol Sol
В результате в растворе появляются свободные ионы, электро-
электропроводность раствора увеличивается.
Концепция кислотности и основности. В 1923 г. Бренстед и
Лоури независимо друг от друга предложили протолитическую
теорию кислот и оснований. Кислотами называют соединения, спо-
способные отщеплять протон, а основаниями — соединения, способ-
способные присоединять протон:
R-H++ :Sol ^ R:- + HSol *
кислота протоно- сопря-
акцептор- женпое
ный раст- основа-
вор итель иие
R —O-H + :SoI ^ R-O:
Oil-кислота
R' R'
I I
R —С—Н + :Sol ^rt R — С: - + HSoI +
R" R"
СН-кислота
R' R'
I I
R —X —II + :Sol ^ R-N
R' R'
I I
R-N'-H + :Sol irf R-\': + HSol +
' 1
R" R"
NH-кислота о t. ipa-
.69
При ионизации нейтральных кислот образуется анион, который
представляет собой основание и называется сопряженным основа-
основанием соответствующей кислоты.
В то же время протоноакцепторный растворитель также является
основанием, а протононированный растворитель — кислотой.
Основание представляет собой нейтральную частицу или анион
с неподеленной парой электронов:
В: +IISoI+ ^ В4" — Н +=Sol
ocuoEiaimc сопряжен-
сопряженная кислота
Нейтральное основание после присоединения протона превраща-
превращается в катион—кислоту, сопряженную с соответствующим основа-
основанием.
Кислотность характеризуется константой кислотности Ка'-
к
Аа [RH]
Чаще пользуются показателем кислотности:
Основность оснований обычно характеризуют константой кис-
кислотности сопряженной кислоты /Свп + :
Г. Льюис расширил понятие «кислота». Кислотами, по Льюису,
являются все соединения и катионы, способные соединяться с ос-
основаниями (частицами с неподеленными парами электронов):
Л + :В Г-1Л--В +
кислота осно-
•Пьюпса ванне
Кислотами Льюиса, например, являются BF3, BC13, AIC13, FeCI3,
SnClj, SO3, NO+ (катион питрозония), KOt_ (катион нитрония) и др.
Типы реагентов в гетероциклических реакциях. Реагенты под-
подразделяются па нуклеофильные и элек/профильные в зависимости
от того, какие свойства выражены более ярко — электронодонор-
иые или электроноакцепторные.
Н у к л е о ф и л ь н ы м и реагентами называются час-
частицы с электронодонорными свойствами, которые образуют связь
со своим партнером в реакции (электрофилом), отдавая неподелен-
иую электронную пару или оба связывающих электрона полярной
связи. Такими являются основания Льюиса, например анионы с
неподеленными нарами электронов, ионные пары и нейтральные
соединения с сильно полярной, способной к ионизации связью.
К нуклеофильным реагентам принадлежат все нейтральные соеди-
соединения, содержащие атомы с неподеленными парами электронов или
обладающие достаточно низкой энергией ионизации.
Нуклеофильные реагенты можно классифицировать в зависимо-
зависимости от типа нуклеофилыюго атома, например:
70
Н-нуклеофилы — Н:~ (гидридпои), BHiM+,
б- б +
С-нуклеофилы — R:"M+ (карбанионы), R—М (металлоргани-
ческие соединения), алкеиы, алкадиепы, арены и др.
N-пуклеофилы — R'R"N:~M+ (амиды металлов и их производ-
производные), R3N:, R,NM, RNHS, NH3
О-нуклеофилы —HO:-M+, RO:-M+, H.,0, R— О — R*
Р-нуклеофилы — R3P:
S-нуклеофилы—HS:-M+, RS:-M+, НД R—S —Rf
галогенид-ионы — :F:~M+, :C1:~M+, :Br:~M+, :I:~M+
Исходя из концепции кислотности и основности все основания
должны быть нуклеофильными реагентами. Во многих случаях это
действительно так. Но необходимо подчеркнуть, что нет количест-
количественной взаимосвязи между основностью и нуклеофильностью (свой-
(свойством быть нуклеофильным). Основность характеризует сродство
к протону. Нуклеофильность является понятием более широким
и характеризует реакционную способность нуклеофильных реаген-
реагентов в различных реакциях, например в реакциях нуклеофильного
замещения у насыщенного углеродного атома (см. с. 231), т. е.
сродство к положительно поляризованному углеродному атому. По-
Поэтому слабое основание может выступать в роли сильного нуклео-
фила (например, соединения серы — сульфиды R—S—R'). Это в
значительной степени зависит от поляризуемости электронной си-
системы нуклеофильного реагента и реагирующего вещества.
Электрофильными реагентами называются
частицы с электроноакцепторными свойствами, которые образуют
связь со своим партнером в реакции (нуклеофилом), акцентируя оба
электрона партнера. Такими являются кислоты Льюиса. Например,
свободные катионы со свободной орбиталью, ионные пары и нейт-
нейтральные соединения с сильнонолярной, способной к ионизации
сеязью. К электрофильным реагентам принадлежат все нейтраль-
нейтральные соединения, содержащие атомы с незаполненной орбиталью или
обладающие высоким сродством к электрону:
Н-электрофилы — Н + Х~ (сильные кислоты)
В-электрофилы—BF3, BC13, BR3
С-электрофилы — R3C+X~ (карбокатионы), соединения с силь-
й1- е- а+ б- дн 6-
нополяриой связью R3C—X, R.,C — Y, RC^N, хиноны (высокое
сродство к электрону)
N-электрофилы — NOX" (соли питрозония), NO-.X" соли нит-
ft-r б-
рония), R — N2X~ (соли диазония), R2N — X
а) д-
О-электрофилы — R—О —X, R—О—О —R (пероксиды)
S-электрофилы —R —S—X, HSO3X", SOa
галогены—F,,, С12, Вг„, I.,
7Г
Электрофильпым можно сделать почти любой элемент периоди-
периодической системы, если соединить его с электроноакцепторным эле-
элементом (например, с фтором, хлором и др.).
Электрофилыюсть характеризует относительную реакцион-
реакционную способность электрофила по отношению к общему субстрату
(способность атаковать углеродный атом).
Нуклеофнльныс и электрофильные реагенты вступают в реакции
замещения. Реакция нуклеофильного замещения —
это гетсролитическая реакция, в которой реагент, поставляющий
входящую группу, действует как нуклеофил:
R_X + :N'u:- —> R — Nu: -\- :X"
R —X -!- :Ku —> R —Nu+ :Х"
нуклеофил нуклгофуг
Уходящая группа, которая уносит связывающую электронную
пару, называется ну/слеофугом.
Реакция электрофильного замещения — это
гетеролитическая реакция, в которой реагент, поставляющий вхо-
входящую группу, действует как электрофил:
R — Y+ Е+ — R —E+ Y+
электро- злектро-
фил фуг
Уходящая группа, которая не уносит связывающую электрон-
электронную пару, называется эдектрофугом.
В поляризованных молекулах и химических связях всегда можно
найти два реакционных центра — нуклеофильный и электрофиль-
ный:
„| Н
с = о: R-—с-*-Li
электро- нуклео- нуклео- элсктро-
ФИЛЬНЫЙ фИЛЬНЫЙ фИЛЬНЫН фи.'1Ы1ЫЙ
центр центр центр центр
Относительная реакционная способность электрофильных и
нуклеофильных центров обусловливает нуклеофилыюсть или элек-
трофильность всей молекулы в целом. Так, карбонильные соедине-
соединения являются электрофильными реагентами, а литийорганические
соединения — нуклеофильными реагентами.
Чтобы прошла гетеролитическая реакция, нужны оба реаген-
реагента — нуклеофильный и электрофильный. Концепция нуклеофиль-
ности — электрофильности в органической химии в значительной
степени напоминает концепцию окисления — восстановления в не-
неорганической химии. В каждой неорганической реакции окисления
есть компонент, который окисляется — отдает электроны, и компо-
компонент, который окисляет (восстанавливается) — присоединяет
электроны. В органических реакциях мы тоже можем формально
найти две реакции — нуклеофильную и электрофильную. Напри-
72
мер, реакция нуклеофилыюго реагента с карбонильной группой
R\6+- fi- Rl4 /б:-
^С г O: + :Nu:- -> )C( "
R/ " R' 4Nu:
представляет собой реакцию нуклеофильного присоединения к
углеродному атому карбонильной группы, по в то же время элсктро-
фпльную атаку реагента :Nu:~.
Электрофильная частица, или электрофильный центр, вступает
в реакции с нуклеофильными частицами (в]нуклеофильных реакци-
реакциях), а нуклеофильная частица, или нуклеофильный центр, вступает
в реакции с электрофильными частицами (в электр'офильных реак-
реакциях).
Нуклеофилыше и электрофильные реагенты (основания и кисло-
кислоты Льюиса) характеризуются различной поляризуемостью и ка-
качественно могут быть подразделены на жесткие и мягкие. Жесткими
реагентами называются основания и кислоты Льюиса, которые со-
содержат донорный центр или акцепторный центр с низкой поляризуе-
поляризуемостью. Например, нуклеофилы: Н2О, ОН", ROH, RO~, F~, NH3;
электрофилы: Н + , Li + , AICI3, BF3. Мягкими реагентами называ-
называются основания и кислоты Льюиса, которые содержат донорный
центр или акцепторный центр с высокой поляризуемостью. Напри-
Например, нуклеофилы R2S, RSH, RS~, I", R3P, R~; электрофилы Hg2+,
Cu+, Ag+.
Существуют также реагенты, занимающие промежуточное поло-
положение, например Cl~, C6H5NH2, R + .
Р. Пирсон A963) сформулировал принцип жестких и мягких кис-
кислот и оснований (принцип ЖМКО), заключающийся в том, что
более стабильная связь образуется при взаимодействии жесткой
кислоты с жестким основанием или мягкой кислоты с мягким осно-
основанием. На основе принципа ЖМКО можно весьма приближенно
оценить реакционную способность при взаимодействии различного
типа нуклеофилов и электрофилов.
Гемолитические реакции. В гомолитическнх реакциях участву-
участвуют частицы с неспаренным электроном — свободные радикалы.
В свободных радикалах имеется молекулярная орбиталь, в которой
находится только один электрон, например <?зС«^) . Существова-
Существование неспаренного электрона является причиной малой стабиль-
стабильности свободных радикалов и их высокой реакционной способ-
способности. Обычно свободные радикалы в реакциях возникают и не-
немедленно реагируют дальше. Только в отдельных случаях могут
образовываться стабилизированные свободные радикалы, если не-
спаренный электрон находится в сопряжении с другими связями
свободного радикала (делокализация неспаренного электрона) или
если свободный радикал сильно разветвлен, что мешает димериза-
ции или взаимодействию с другими частицами.
Так как свободные радикалы обычно являются электронейтраль-
нымп частицами (кроме ион-радикалов), то полярные факторы в
73
реакциях с другими частицами (полярность связен, величина эф-
эффективных зарядов) имеют меньшее значение, чем в гетеролити-
ческих реакциях.
Методы изучения механизмов реакций. Изучить механизм реак-
реакции — значит установить, какими путями из исходных веществ
образуется конечный продукт. Необходимо выяснить, что с чем реа-
реагирует, в каких пропорциях, в какой последовательности, какими
являются элементарные акты, промежуточные соединения, каки-
какими могут быть переходные состояния.
Одним из основных методов изучения механизмов является ки-
кинетический метод. В соответствии с ним измеряется скорость реак-
реакции в зависимости от концентрации реагирующих веществ, устанав-
устанавливается порядок и молекулярность (стехиометрия) реакции.
Для изучения механизмов реакции часто применяют изотопные
методы. Чтобы определить реакционные центры и реагирующие
связи, в данную связь вместо обычного атома вводят его изотоп
(например, вместо соединения со связью С—Н изучается соедине-
соединение со связью С—D). Если скорость реакции поданной связи изменя-
изменяется, говорят, что наблюдается первичный изотопный эффект, и
это служит доказательством местонахождения реакционного цент-
центра. Вторым способом применения изотопного метода является изу-
изучение изотопного состава продуктов реакции изотопно-обогащенных
(«меченых») соединений. Так можно определить место разрыва хи-
химических связей (см. с. 574).
Стереохимические методы изучения механизмов реакции заклю-
заключаются в определении пространственного строения продуктов реак-
реакции при использовании исходных с известным строением (см.
с. 230). Так можно судить о пространственном протекании реак-
реакции.
Катализ. Многие реакции ускоряются присутствием третьего
вещества — катализатора. Катализатором называется вещество,
которое участвует в определенной химической реакции и увеличи-
увеличивает ее скорость, но при этом общее количество этого вещества
не меняется.
Каталитические процессы могут быть гомогенными (катализатор
находится в растворе реакционной смеси) и гетерогенными (реак-
(реакция, протекающая в жидкой или газовой фазе, осуществляется на
поверхности катализатора). Роль катализатора заключается в ак-
активации реагирующих молекул. Это достигается либо присоедине-
присоединением катализатора к веществу, что вызывает дальнейшие реакции,
либо адсорбцией вещества на активных центрах катализатора, что
активирует определенные связи, вызывает их диссоциацию и т. п.
Особое место занимают биокатализаторы — ферменты, представ-
представляющие собой сложные белки. Ферменты ускоряют строго опреде-
определенные реакции.
Большое значение для технологического использования катали-
катализаторов, особенно биокатализаторов, имеет методика их нанесения
на поверхность мелкозернистых материалов (полимеров, силикаге-
ля). Катализатор связывается с поверхностью подложки, при этом
74
получаются иммобилизованные катализаторы, пригодные для мно-
многократного использования.
Особое место занимает межфазный катализ. Это явление заклю-
заключается в увеличении скорости реакции между веществами, находя-
находящимися в различных фазах (песмешивающиеся жидкости или твер-
твердое тело и жидкость), при добавлении небольшого количества аген-
агента — межфазного катализатора, который переносит один из реа-
реагентов через поверхность раздела в другую фазу, где осуществля-
осуществляется реакция. Такими катализаторами являются четырехзамещен-
пые аммониевые соли (см. гл. XXIII. А) или агенты, образующие
комплексные соединения с неорганическими катионами, например
краун-эфиры и кринтанды.
Катализ в органической химии играет исключительно важную
роль. Это относится как к реакциям в лабораторных условиях
(например, катализ в присутствии кислот и оснований, в присут-
присутствии соединений переходных металлов, межфазный катализ), так и
к крупнотоннажным промышленным процессам (каталитическое
гидрирование и окисление, дегидратирование, крекинг углеводо-
углеводородов, полимеризация и др.).
Способы изображения механизмов реакций. Для изображения
хода химической реакции можно довольствоваться простой схемой,
которая содержит только исходные вещества и продукты реакции,
можно указать статические электронные смещения, эффективные
заряды, неподеленные электронные пары и, наконец, можно дать
предполагаемый механизм реакции с указанием динамических эф-
(|>ектов, т. е. исчезновения и образования связей, перемещения
электронной плотности (связывающей электронной пары), образо-
образования комплексов в результате донорно-акцепторного взаимодейст-
взаимодействия (комплексы с переносом заряда, я-комплексы). Ниже приведен
ряд примеров от простейшей до более сложной схемы механиз-
механизма:
а) простые схемы
CEl3CI-j-Na+OH- —* C
O-Na +
/Р I
RCf -f Na+OH- —v RC —OH
Nl |
H
б) схемы с указанием статических электронных смещений
R±
75
в) схемы с указанием динамических эффектов
сна—а: + :он + Na+ «- У^Л^су.' —*¦ с +:а: + Na+
Na+ "О"'], " НО Н
Ло- .'~'6-~ :o:"Na+
R — С + :ОН + Na+ *- R— С -^ .. _ . *- R—С—ОН
\ ¦• \f -:OH Na+ |
Н " Н
Чтобы отличить статические электронные смещения от динами-
динамического перераспределения электронной плотности (перемещения
связывающей электронной пары), для изображения последнего ре-
рекомендуется применять пунктирные изогнутые стрелки.
При написании уравнения реакции реагирующие частицы сле-
следует помещать пространственно в такой геометрии, каким предпо-
предполагается строение переходного состояния.
Применение прерывистой стрелки можно дополнительно иллю-
иллюстрировать простыми примерами:
и+ + :х-
С А С С — С С * С С—С
II-' —*¦ I ПК *¦ II
с с с с
Весьма наглядно можно изобразить перицнклическне реакции,
где образование новых связей и исчезновение старых происходит
согласованно (одновременно):
н
с
НС У, [ CHj
II '' х"-
II
НС _ СН2
н
н
НС CHi
> 1 1
' 1 1
НС СН2
н
Донорно-акцепторпое взаимодействие и образование комплекса
с переносом заряда КПЗ (я-комплекса) изображается стрелкой:
о: Ч- A ^=rt вТ-»А~
КПЗ
Например, образование КПЗ между этиленом и бромом:
сна ^ *Lcn. а-
СНг "' "СИ.
76
Схематично взаимодействующие центры можно обозначить при
помощи волнистой стрелки, например:
..^- ,v..f- :о'.~ Na+
R—C*^ -p-':OH + Na+ *¦ R—С * Ч:ОН Na+ *- R — С—OH
ч " v~ i "
3. ВЗАИМОСВЯЗЬ МЕЖДУ СТРОЕНИЕМ
И РЕАКЦИОННОЙ СПОСОБНОСТЬЮ
ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ.
КОРРЕЛЯЦИОННЫЕ УРАВНЕНИЯ
Взаимосвязь между реакционной способностью и строением орга-
органических соединений в реакциях различных типов является одним
из центральных вопросов теоретической химии. Реакционная спо-
способность характеризуется константами скорости реакции и констан-
константами равновесия, в том числе константами кислотности.
Константы кислотности в значительной степени зависят от
строения органической кислоты и могут быть использованы для ха-
характеристики влияния различных заместителей на кислотность.
В частности, константы кислотности используются для оценки реак-
реакционной способности функциональных производных карбоновых
кислот.
Уравнение Гаммета. Л. Гаммет наблюдал, что константы ско-
скорости реакции алкилировапия сложными эфирами возрастают при
увеличении кислотности соответствующей кислоты. Изучалась
реакция RCOOCH3+ (CH3KN-^RCOO-(CH3LN + .
В 1935 г. Гаммет обнаружил, что существует взаимосвязь между
константами реакционной способности м- и /г-замещенных произ-
производных бензола и константами кислотности соответствующих м-
и /г-замещепных бензойных кислот. Эта зависимость была выражена
уравнением A937)
is
lAig?+plg
где К — константа кислотности производного бензойной кислоты с
заместителем в бензойном цикле; k — константа реакционной спо-
способности производного бензола; Ка, ?0— константы незамещенных
соединений; р—константа реакционной серии.
При обозначении '8^~=а получаем уравнение Гаммета
lg —=ра.
"О
/ /,
Это уравнение отражает изменение величины lg& —lg йо( или \gj-
при вариации заместителей в однотипных соединениях. Это измене-
изменение линейно связано с константой заместителя о и с изменением
свободной энергии, 'или свободной энергии активации реакций.
77
Константы Гаммета. Уравнение Гаммета содержит две новые
величины — р и о. Константа р характеризует саму реакцию, тип
реакции и поэтому называется константой реакционной серии.
Константа о отражает влияние строения молекулы —¦ эффект
группировки или заместителя; о является константой заместителя
п называется константой Гаммета.
Чтобы пользоваться уравнением Гаммета, необходимо выбрать
некоторые стандартные величины. В качестве стандартного замести-
заместителя, для которого а принята равной нулю, Гаммет выбрал атом
водорода. Для реакции ионизации замещенных бензойных кислот
принималось, что р=-1. Исходя из этого, была получена первая
шкала о-констант заместителей.
Экспериментально найденные зависимости между lg k и о обыч-
обычно изображаются графически в координатах lg-^—а. Тангенс угла
наклона прямой, проведенной через найденные экспериментально
точки, соответствует константе р.
Для заместителей в о-, м- и /г-положепиях константы Гаммета
имеют разные численные значения и даже разные знаки. Например,
для группы ОН Од—-1-0,12, о„~—0,37, для группы NH2 ам—
--—0,16, о„--—0,66, для группы КО.> ол=---г0,71, о„=+0,78.
Это явление можно попять только при рассмотрении электрон-
электронных эффектов заместителей. Сравнение значении констант Гаммета
показывает, что знак + у константы соответствует электроноакцеп-
торным свойствам заместителя, а знак — соответствует электроио-
допорным свойствам. Численные значения могут быть использо-
использованы для сравнения электронных эффектов заместителей.
Константы Гаммета отражают суммарный электронный эффект за-
заместителя (сумму эффектов индуктивного, мезомерного и простран-
пространственного). В о-, м- и n-положениях замещенных бензолов вклад
этих эффектов различен. Константы о0 учитывают все эффекты, в
том числе и пространственный, поэтому они мало пригодны для
практического использования, так как пространственный орто-эф-
фект меняется при переходе от одной реакционной серии к дру-
другой.
о.и в основном отражает индуктивный эффект, а ап— сумму ин-
индуктивного эффекта и мезомерного эффекта. Эффект индуктивный
(электронные смещения по о-связям) и мезомерны» (электронные
смещения по я-связям) могут быть противоположны по направлению
(см. В вед. 5). Это отчетливо проявляется в значениях о-констаит
групп ОН и NH2 для м- и п-положений.
Константы реакционной серии. Константа реакционной серии р
может иметь как положительные, так и отрицательные значения и в
основном меняется в интервале от —1 до ---4. Чем больше числен-
численное значение р, тем более данная реакционная серия чувствитель-
чувствительна к электронным эффектам заместителей и тем более пригодна для
изучения эф(()ектов заместителей. Знак константы указывает, ка-
какого типа электронные эффекты способствуют протеканию реакции.
Если р>0, реакцию ускоряют электропоакцепторные эффекты за-
78
местителей, а при р<0 реакции способствуют электронодонорные
эффекты.
Константы Гаммета имеют ограниченное применение, так как
они характеризуют суммарное влияние эффектов индуктивного и
мезомерного, а па реакционную способность эти эффекты влияют
по-разному, особенно при «-положении заместителя.
Константы Гаммета — Брауна. Известны реакции, у которых
особенно сильно проявляется действие мезомерного эффекта, и
поэтому непригодны обычные константы о„. Это наблюдается с
случаях, когда имеется полярное сопряжение между заместителем
и центром реакции.
Примером реакции, на протекание которой влияет полярное со-
сопряжение электропоакценторного заместителя с центром реакции,
служит ионизация n-замещенных фенолов:
+ : soi "т^-
(здесь U=NO2, COR1, С—N, N.!).
В этих случаях необходимо пользоваться новыми увеличен-
увеличенными константами заместителей R, так называемыми константами
прямого полярного сопряжения о .
Прямое полярное сопряжение электронодопорного заместителя
с центром реакции проявляется, например, в карбокатионах:
с х —'
•<ч1 Л 5 + н2о
(здесь D=OR, SR, NR2).
Константы прямого полярного сопряжения электронодонорпого
заместителя D обозначаются о+.
Константы о~ и о+ предложили и экспериментально определи-
определили Браун и Окамото A957). По абсолютному значению они значи-
значительно больше констант Гаммета он; например, для группы NH2
о„---— 0,66, а о --—1,3, для группы КО2 а„ = -|-0,78, а а"-4-1,27.
Константы Тафта. Для оценки индуктивного влияния замести-
заместителей в алифатическом ряду Р. Тафт разработал методику опре-
определения индуктивных констант о*. Для этой пели он изучал
скорость гидролиза сложных эфиров X—COOR в кислой и ще-
щелочной среде и получил ряд констант о* для группировок X.
Соединением сравнения Тафт выбрал уксусную кислоту, для ко-
которой принял Оспа = 0. Тафт вычислил коэффициент ослабления
индуктивного влияния через один углеродный атом: охсп2 =
х
о* характеризует величину и направление индуктивного эф-
эффекта заместителя. Например: Ор = 4-3,1; ос„н5 = 4-0.6; ос„н6 =
= —0,1; сг(сн.,,с = -0,3.
79
Уравнение для констант Тафта имеет вид уравнения Гаммета:
Проблема разделения эффектов заместителей. Для более коррект-
корректного применения принципа линейных изменений свободных энер-
энергий необходимо разделить эффекты заместителей на составные час-
части (индуктивный, мезомерный, пространственный) и каждый эф-
эффект характеризовать своей константой. Отчасти это можно сде-
сделать. Поэтому в настоящее время известно много различных кон-
констант заместителей, например:
о — константы Гаммета
о+, с~ — константы Гаммета — Брауна
с* — константы Тафта
Es — константы пространственного действия
а/ — индуктивные константы для производных бензола
(О/=0,16 а*)
ос — константы для характеристики мезомерного эффекта за-
заместителя (ос=о—а/) и др.
В табл. 6 приведены значения различных а-констант для не-
некоторых заместителей. Надо отметить, что в разных источниках
встречаются несколько различающиеся значения.
Таблица 6
Замести-
Заместитель
сн3
F
С1
ОН
NH2
N(CH3)-2
СООСНз
CN
NO,
. а-Константы заместителе»
о
п
—0,17
0,06
0,23
—0,37
—0,66
— 0,8.3
0,46
0,65
0,78
+
—0,31
—0,07
—0,4
—0,92
— 1,3
— 1,7
—
—
.—
0,64
0,88
1,27
а
м
—0,07
0,34
0,37
0,12
—0,16
-0,211
0,315
0,56
0,71
0
—0,14
0,25
0,2
-0,3
—0,35
0,8
0,52
0,47
0,10
0,32
0,58
0,63
о*
0
3,1
2,68
1,31
0,72
1,75
3,25
3,53
Применение корреляционных уравнений. Уравнения, связываю-
связывающие константы реакционной способности или другие физические
константы с константами заместителей, называют корреляционными
уравнениями. Простейшим из таких уравнений является уравнение
Гаммета.
Применяют корреляционные уравнения для характеристики но-
новых реакционных серий, изучения механизмов реакций, характе-
характеристики электронных эффектов новых атомных группировок, пред-
предсказания реакционной способности новых органических соединений.
а-Константы заместителей служат полуколичественной мерой для
оценки электронных и других эффектов.
80
8. КЛАССИФИКАЦИЯ
И НОМЕНКЛАТУРА ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Основной задачей классификации и номенклатуры является созда-
создание системы для подразделения органических соединений и наимено-
наименования их; при этом должно соблюдаться соответствие между систе-
системой номенклатуры и существующей классификацией.
1. КЛАССИФИКАЦИЯ
В основе классификации органических соединений обычно лежат су-
существующие теоретические представления об образовании соедине-
соединений и расположении атомов в молекуле. В раннем периоде развития
органической химии не было никаких основных принципов класси-
классификации. Названия органическим веществам давались по методу
получения, исходному сырью, внешним признакам и запаху.
В период теории радикалов органические соединения можно было
классифицировать по типам радикалов, которые входили в их мо-
молекулы. Унитарная теория дала возможность классифицировать ор-
органические соединения по их типам, например соединения типа ам-
аммиака, или амины, соединения типа воды, или спирты и эфиры, сое-
соединения типа метана, или насыщенные углеводороды.
Теория строения дала прочную основу для классификации ор-
органических соединений. Можно было классифицировать по опреде-
определенным структурным элементам и по расположению атомов в моле-
молекуле. Были выдвинуты два основных принципа: деление органиче-
органических соединений по расположению углеродных атомов в молекуле и
по характерным структурным злементам.
Структурными элементами являются различные заместители,
которые связаны с углеродными атомами в углеводороде, или типы
связей в цепи углеродных атомов. Предложено следующее под-
подразделение структурных элементов:
а) нефункциональные заместители (F, CI, Вг, I, КО2 и др.);
б) функциональные группы (NH2, ОН, SH, С=О, СООН и др.).
Часто их называют просто функциями. По номенклатуре
ИЮПАК структурные элементы называются характеристическими
группами.
В зависимости от расположения углеродных
атомов в молекуле органические соединения делятся на
несколько больших групп.
I. Соединения с открытой цепью атомов углерода — ацикличес-
ациклические, или алифатические, соединения:
сн,сн3 сн3сн2сн3 си3сн2снасн3 си3сн2снсн3
си3
СН2^СН2 СН3СН^СН, СН3СН-=СНСН3 СНа^СН —СН -СН2
ПС — CU СНзС^СН СН3С -ССЬ'з НС = ССН2СН2СН3 и т. д.
и их производные, содержащие различные функции.
81
II. Карбоциклические соединения с циклами из углеродных ато-
атомов:
а) алициклические соединения — различные циклические угле-
углеводороды и их производные с разной величиной цикла и числом
циклов, и разным числом двойных связен, кроме шестичленных
циклов с тремя двойными связями:
О
О О
О
и т.д.
б) ароматические соединения, или арены, и их производные:
циклические углеводороды и их производные, которые построены
из шестичленных циклов с тремя двойными связями:
II Т. Д.
III. Гетероциклические соединения и их производные: цикличе-
циклические соединения, циклы которых построены не только из углерод-
углеродных атомов, но содержат также гетероатомы (О, N, S и др.):
NX NX Ч/ч/ я
н
О 00 О О
о
н
о
82
Второй основной принцип классификации — деление по
функциям (характеристическим группам).
В зависимости от того, какая функция введена в молекулу углево-
углеводорода вместо атома водорода, получаем семейство органических
соединений определенного типа:
— галогеппроизводные углеводородов RC1, RBr, Arl
— спирты и фенолы ROH, АгОП
/°
— альдегиды R—С'/
— кетопы R —С —R'
II
О
— карбоновыс кислоты R— С^ и т.д.
(здесь R обозначает остаток углеводорода, т. е. часть молекулы без
водородного атома, Аг — остаток ароматического углеводорода,
арена).
Аналогичные соединения можно получить из гетероциклических
соединений HetH: HetCl, HetOH, HetCOOH и т.д.
В общем виде остаток органического соединения можно обозна-
обозначить Org (органил) или R и изобразить, например, все гидроксиль-
ные производные как OrgOH (или ROH) и т. д.
2. НОМЕНКЛАТУРА
В начале развития органической химии, когда не существовала клас-
классификация, органические соединения получали случайные назва-
названия по источнику получения (лимонная кислота, яблочная кисло-
кислота), цвету или запаху, реже — по химическим свойствам. Зги наз-
названия в данное время составляют тривиальную (историческую)
номенклатуру. Многие такие названия часто применяются и в на-
наши дни, и это в некоторой степени затрудняет освоение курса ор-
органической химии — их надо просто выучить наизусть. Например:
мочевина, толуол, ксилол, индиго, уксусная кислота, масляная
кислота, валериановая кислота, гликоль, алаппп и многие другие.
Унитарная теория создала возможность образовать название
соединения по соответствующему тину. Например: метиламин, ди-
метиламип, триметнламнп, метиловый спирт, этиловый спирт, нро-
пиловый спирт. Такие названия составляют рациональную но-
номенклатуру. Эти названия содержат название основного типа и
названия заместителей.
Теория строения А. М. Бутлерова дала строгую основу для клас-
классификации и номенклатуры органических соединений но структур-
структурным элементам и по расположению атомов углерода в молекуле.
Однако проблемы номенклатуры все же остаются достаточно слож-
сложными. Название каждого органического соединения должно содер-
содержать правильные названия функций (заместителей) и основного
83
скелета углеводорода, и должно быть таким, чтобы по названию
молено было написать единственную правильную структурную
формулу. К сожалению, задача создания идеальной номенклатуры
осталась нерешенной до наших дней.
Стремление создать единую химическую номенклатуру для ор-
органических соединений возникло в 80-е годы XIX в. Это осущест-
осуществилось в 1892 г. па международном съезде химиков в Женеве.
Съезд утвердил правила номенклатуры органических соединений,
которые были разработаны международной комиссией и представ-
представлены съезду. Эти правила вошли в органическую химию под наз-
названием женевская номенклатура или просто официальная номенкла-
номенклатура. Название «официальная» ей было дано потому, что сущест-
существовали и в наши дни еще существуют номенклатура тривиальная и
рациональная.
На основе женевской номенклатуры создан известный справоч-
справочник Бейльштейна.
С ростом числа и типов органических соединений номенклатура
все время осложняется и появляются новые предложения. Новый
номенклатурный съезд химиков состоялся в 1930 г. в Льеже. Были
приняты дополнительные правила, основанные па принципах удоб-
удобства н краткости.
В последние десятилетия усовершенствованием номенклатуры
органических соединений занимается Международный союз теорети-
теоретической и прикладной химии — ИЮПАК (International Union of
Pure and Applied Chemistry — IUPAK). Съезды ИЮПАК в 1957 и
в 1965 гг. рекомендовали разработанную специальной комиссией
номенклатуру, которая называется номенклатурой ИЮПАК- Эта
номенклатура широко используется как в научной литературе,
так и в учебниках. В этой книге использованы правила номенкла-
номенклатуры ИЮПАК *.
Материалы ИЮПАК публикуются па английском языке, поэто-
поэтому при переводе на другие языки правила должны быть адаптиро-
адаптированы к соответствующим языковым нормам. Но при этом должны
быть сохранены принципы международных правил.
Правила ИЮПАК рекомендуют для образования названий не-
несколько принципов. Первый из них — принцип замеще-
замещения. На основе этого разработана заместительная номенклатура.
Второй принцип ¦— использование одинаковых
ф у н к ц и й (характеристических групп) и углеводород-
углеводородных остатков (заместителей, радикалов). Поэтому она на-
называется радикально-функциональной номенклатурой.
Кроме этого, ИЮПАК разработана специальная номенклатура
карбоцнклическнх и гетероциклических соединений.
Приведем несколько основных правил заместительной номенкла-
номенклатуры.
* Номенклатурные правила ИЮПАК по химии. Т. 2. Органическая хи-
химия. М., ВИНИТИ, 1979; Т. 3. Органическая химия. Высокомолекулярные
соединения. М., ВИНИТИ, 1983,
84
1. В основе названия лежит родоначальная структура (главная
цепь ациклической молекулы, циклическая или гетероциклическая
система).
2. Характеристические группы и заместители (структурные эле-
элементы) обозначаются префиксами и суффиксами.
3. Атомы родоначальной структуры нумеруются от 1 до п, эти
номера называются локаитами.
4. Характеристические группы подразделяются по старшинству,
например группы
О
— СООН, —SO^II, —COOR, — CONH2, —C^=N, — C-f , ^C = O,
-OH, —SH, -ООН, —NH2
перечислены в порядке уменьшающегося старшинства.
В название в качестве суффиксов включают обозначения двой-
двойных и тронных связей и главную (старшую) характеристическую
группу, остальные характеристические группы называют в префик-
префиксах в алфавитном порядке. Нумерацию начинают с того конца уг-
углеродной цепи, к которому ближе расположена старшая характе-
характеристическая группа. Например:
6 5 4 3 2 1 ,0
СНз —СН-СН—СН-С-С//
II I чн
ОН СН3 Вг
2-бром-5-гидрокси-4-метил
ваместители и характеристические группы
в алфавитном порядке
г е кс
корень
(глап-
н а и
; цепь)
i
е н - 2 -
суффикс
двойной;
связи :
а л ь
суф-
суффикс
стар-
старшей
группы
ЧАСТЬ ПЕРВАЯ
Углеводороды
Глава I. АЛКАНЫ
Наиболее простыми органическими соединениями являются угле-
углеводороды, так как их молекулы содержат только углеродные и во-
водородные атомы. Углеводороды различаются числом атомов угле-
углерода, наличием простых, двойных и тройных связей и последова-
последовательностью соединения углеродных атомов (цепь или цикл).
Углеводороды с открытой цепью, содержащие только простые
ковалентпые связи, называют насыщенными углеводородами или
парафинами, по номенклатуре ИЮПАК — алканами.
Самым простым алканом является метан СН4. Другие ал капы
можно рассматривать как образованные из метана введением одной
или более метилеиовых групп СН2 между углеродным и водородны-
водородными атомами метана. Общая формула алканов С„НМ(.,.
1. ИЗОМЕРИЯ И НОМЕНКЛАТУРА
Алканы могут иметь перазветвленпую цепь углеродных атомов (нор-
(нормальные алкапы, н-алкапы) и разветвленную (изоалканы, разветв-
разветвленные алканы).
Если расположить н-алкапы в ряд, в котором молекула каждого
последующего алкаиа удлиняется па одну группу СН2, получится
ряд, называемый гомологическим рядом: первые члены ряда (С^—С4)
имеют тривиальные названия. Названия остальных гомологов об-
образованы от греческих и латинских числительных. Справа приве-
приведено сокращенное изображение формул:
CIIj метан
СНзСНз
СН3СП2СН3
СНзСН2СМ2СНз
СНэСН2СН2СН2СН..СНз
СНзСН2СН2СН2СН2СН2СН3
СНзСН2СН2СН.2СН,СН..;СН2СН3
СН3СНоСЫ.2С1Г..СН,СН2СН:.СЦ.2СНя
СИ3С1Г2С1[..СП2СН2СН2С1ЬСН2СН2СН3
этан
пропан
бутан
лептам
гексан
гептан
октан
нонам
декан
/Ч/\/\/
86
Начиная с бутана, возможна изомерия алканов. Изомерами назы-
называют соединения, имеющие одинаковую молекулярную формулу, но
различающиеся порядком связей атомов или расположением ато-
атомов в пространстве. Изомеры, различающиеся последовательностью
соединения атомов в молекуле, называют структурными изомерами.
Например:
CHjCHjCH;CH3 или s^S* CKj—С — CHj или
н
бутан изобутан B-метилпроиаи)
Число структурных изомеров алканов быстро растет с увеличе-
увеличением числа углеродных атомов. Так, пентан C5Hi2 имеет три изо-
изомера, гептан C7Hi9 — 9, октан CsHl4 — 18, декан СюН.,2 — 75,
додекаи С,2Н2в — 355, эйкозан С2оН42 — 366319, гектап СтоНм* —
около 5,921 -1010.
Число изомеров алканов еще увеличивается за счет возможных
стереонзомеров (изомеров, различающихся только расположением
атомов в пространстве). Начиная с С7Н,Г„ возможно существование
хиральпых молекул, которые образуют два эиаитиомера (см.
Введ. 6.12). Так, из девяти гептаиов два являются хиральными.
Номенклатура разветвленных алканов (ИЮПАК) основана на
следующих правилах:
а) выбирают наиболее длинную иеразветвлеиную цепь, назва-
название которой составляет основу (корень);
б) разветвления называют в качестве заместителей (групп, ра-
радикалов), при этом углеродные атомы перазветвленпой цепи нуме-
нумеруют по принципу наименьших локантов:
1 2 3 4 5 6
С11Я — СН — СН— СН2 — СИ — СГГз
I I I
СНз СН3 СН3
2, 3, 5-1 риметнлгокеап
(по не 2, 'I, 5-триметнлгексан)
Наличие нескольких одинаковых заместителей обозначается со-
соответственно греческими числительными: ди-, три-, тетра-, пента-
н т. д.
Если в молекуле алкапа имеются различные заместители (боко-
(боковые цепи), отличающиеся числом углеродных атомов и степенью
разветвления, то в названии алкапа эти заместители перечисляют
в порядке алфавита:
СНз
I 2 3 4 | 5 6 7 8 О
СНз —СН —СН. —СН —С—СН, —СН. —СН, —СН3
I "II
СНз СН, СН
СПз СН:; СНз
5- изопропи л-2,5-дпметил-4-зтнл1!оиаи
87
В зависимости от числа других углеродных атомов, с которыми
непосредственно связан рассматриваемый углеродный атом моле-
молекулы, различают первичные, вторичные, третичные и четвертичные
углеродные атомы:
СНз СН3
I I
СНз """" СН2 ¦ С ¦ ~~" С ' СНд
t \ J\J.\
первич- вторич- третич- че-п^р-
ный нмй иый точный
При отнятии от молекулы алкана одного водородного атома (фор-
(формально) получаем остаток алкама: R—Н "--->¦ R.
Обычно его называют алкильной группой (алкильпым замести-
заместителем), встречается также название алкилышй радикал. Последне?
название иногда путают с обозначением, относящимся к активным
частицам — свободным алкильиым радикалам.
Названия алкнльных остатков (алкильпых групп) образуют п :
названий соответствующих алканов замещением суффикса -ан на
-ил:
СН3 — метил
СН3СН2 — этил
СН3СН2СН2— пропил
СН3
^)СН— изопропил (в/пор-проппл)
сн3/
CIIaCH.CI-bCH,— бутил
Ctl3—СН—СН2— 1гзобутил
С
СН3—СН2 — СН — emop-бутил
СН3
СН,
СНз^С— mpem-бутил
СИ, '
Для названия разветвленных алкильпых групп используют так-
также нумерацию цепи:
4 3 2 1 5 4 Г5 2 1
СНз—СН2 —СН —СН2— СНз —СН2 —СП—СН-2 —СН2 —
I I
СН3 СН3
2-метилбутил 3-метилпеитил
Названия углеводородов лежат в основе не только названий
алкильпых остаткоз (алкнльных групп), но и названий различных
частиц, образующихся в реакциях,— ионов и свободных радикалов.
Имеются три возможности отщепления водорода от молекулы гл-
капа.
1. При гемолитическом расщеплении связи С—Н образуется
свободный алккльнын радикал, частица с песпареииым электроном,
83
обычно называемая алкильным радикалом или алкилом. Например,
СН3- —метальный радикал, или метил:
2. При гетерслитическом расщеплении связи С—Н может обра-
образоваться катион, а водород уходит в виде гидрид-иона. Образуется
карбокапшон, или алкил-катион, например СЩ — метил-катион:
R:H-+R++H:~
3. При гетеролнтическом расщеплении связи С—Н может об-
образоваться анион, а водород уходит в виде протона R : H->-R:~-H
гН1. Образуется карбанион, или алкил-анион, алканид-ион, на-
например :СН3" — метил-анион.
Алкил-катионы являются очень сильными электрофильнымп
реагентами, а алкпл-анионы — очень сильными нуклеофильными
реагентами.
При отнятии от молекулы алкана двух атомов водорода (формаль-
(формально) получают двухвалентные остатки. Их названия образуют, до-
добавляя к названиям соответствующих одновалентных радикалов
суффикс -ен или -иден (если обе свободные связи находятся у од-
одного атома углерода):
сн4 _"
метиле II-
СНзСНз-
-СН2СНц —
СН3СН2СП3 —
/
¦СНзССНз
I
¦СН3СПСН2—
этилен-
пропилндеп-
изопропнлиден-
1, 2-пропнлен-
>—СН2СН2СН2— I, 3-пропилен-
Двухвалентпым остаткам формально соответствуют встречаю-
встречающиеся в реакциях активные частицы — бирадикалы (частицы с дву-
двумя неспарепными электронами). Если оба электрона находятся у
одного атома углерода, бирадикалы называются карбенами:
:СН2 карбен, или метилен
:СНСН3 метнлкарбеи, или этилиден
/СН3
:С'' диметилкарбен, или изопропилиден
2. ПРИРОДНЫЕ ИСТОЧНИКИ И МЕТОДЫ ПОЛУЧЕНИЯ
1. Нефть и природным газ. Главными источниками алкапов явля-
являются нефть и природный газ, состоящий из метана с небольшой при-
примесью этана, пропана и бутана. Из продуктов переработки нефти
89
получают различные смеси алканов. Фракционной перегонкой
бензиновой фракции нефти можно получить индивидуальные алка-
алкаиы (см. гл. 1.5).
2. Гидрирование угля. Гидрирование (обогащение водородом)
каменного или бурого угля происходит под действием водорода толь-
только в присутствии катализатора (оксиды и сульфиды молибдена,
вольфрама, никеля) и при высоких температурах D50—470 °С).
Процесс проводят в специальных реакторах — автоклавах, выдер-
выдерживающих давление до 30 МПа C00 атм). Уголь и катализатор рас-
растирают в тонкий порошок и суспендируют в органическом раство-
растворителе (продуктах переработки нефти). Смесь нагревают и в авто-
автоклав вводят водород (Ф. Бергиус, 1925). Этот процесс часто называ-
называют методом сжижения угля. В результате получают смесь различных
алканов и циклоалканов, которые используются в качестве мотор-
моторного топлива.
3. Метод Фишера — Тропша. Каталитическое гидрирование
СО, и СО обычно дает метан. В качестве катализатора используют
никель (П. Сабатье, И. Сандеран, 1902):
400 °С
СО2 +4И2—,.-' СН4 + 2И2О
300 °С
СО + ЗНа —,.->- СН4 + П..О
Каталитическое гидрирование СО может протекать по-разному,
в зависимости от применяемого катализатора и температуры. В при-
присутствии катализатора, содержащего кобальт или железо, при тем-
температуре 180- ¦ -300 СС смесь СО и Н, реагирует с образованием ал-
алканов. В основном образуются алкаиы неразветвленные и с неболь-
небольшой молекулярной массой (Ф. Фишер, X. Троиш, 1913—1926):
290 °С
СО-1-2Н2 -Алканы
Со
В последующие годы метод был усовершенствован: изменены катали-
катализатор, температура реакции и давление (Ф. Фишер, X. Пихлер,
1936—1941).
В настоящее время методом Фишера — Тропша могут быть по-
получены как моторные топлива, так и отдельные углеводороды (ал-
каны, циклоалканы, арены). Механизм реакции сложен. В основе
реакции лежит сорбция СО на поверхности катализатора, в ре-
результате чего па поверхности образуются карбонилы кобальта. Во-
Водород присоединяется к сорбированному оксиду углерода, образуя
связанные с поверхностью катализатора карбен :С!12 и метнльный
радикал -СН3. Карбен может внедряться по связи Со—СН3, что
приводит к росту углеродной цепи, как это показано на схеме:
со со
1 1
катализатор
-HjO
СО
СН,—
II.
-но1
-аи
1
сн2
- II
СН,
II
со со
1
СП,
,„,
н.
-II О
—СН
СН, СН. СО
1 II
ск2
J
90
В качестве побочных продуктов могут образоваться кислород-
кислородсодержащие соединения (спирты, альдегиды).
4. Лабораторные методы получения, а) Р е а к ц и я метал-
лорганических соединений с водой. Соеди-
Соединения, содержащие связь металл — углерод, в большинстве слу-
случаев легко реагируют с водой, образуя углеводороды. Рассмотрим
наиболее простой случай — реакцию карбидов:
А14С3+ 12ГГ2О -> ЗСН, + 4А1(ОНK
СаС2, SrC2 и ВаС2 в реакции с водой образуют ацетилен (см.
гл. IV.2). Карбиды Fe:,C и Ni3C в реакции с кислотами образуют
смеси углеводородов. Механизм реакции рассмотрен в гл. Х.Л.
б) Реакция галоген алканов с натрием.
Галогекалканы легко реагируют с натрием (еще легче с калием),
образуя хлорид натрия и углеводород (А. Вюрц, 1855):
2R— Br + 2Na — *R — R
3R—Br-|-3R' —Br + 6Na-^R —R-f R—R4-R' —R'-i-6N'a'Br-
Механизм реакции Вюрца изложен на с. 229.
в) Электролиз солей к а р б о и о в ы х кислот.
При электролизе водных растворов солей карбоиовых кислот па
аноде выделяется СО2 и образуются алкаиы. Реакцию открыл
А. Кольбе в 1849 г.
В этой реакции (электросинтез Кольбе) анион карбоновой кис-
кислоты на аноде отдает электрон (окисляется), образуя свободный ра-
радикал, который разлагается па СО2 и свободный алкнльный ради-
радикал:
/Р
2RCOO--2c-—*2R-C^ —>2R.+2CO2
ХО.
Алкнльные радикалы связаны с поверхностью анода и там димерн-
зуются:
2R--+R-R
г) Декарбоксилирование карболовых кис-
л о т. При нагревании солей карбоновых кислот в присутствии ще-
щелочей при температуре 250—300 аС происходит разложение и вы-
выделяется алкай:
R —СОО-\а+ +ХаОН Д R -H-|- Na2CO3'
В этой'реакции образуется также СО2, который связывается ще-
щелочью. Отщепление от молекулы СО2 называется декарбоксилиро-
ванием. Реакция используется обычно для получения низших ал-
алканов — метана, этана.
д) Реакции гидрирования и восстановле-
восстановления органических соединений. Алканы полу-
получаются при каталитическом гидрировании алкепов (см. гл. П.4)
91
и алкннов (см. гл. IV.4), например:
H,/Ni
RCH-CH, у RCH2CF13
Восстановление галогенуглеводородов (гл. VII.4.1), альдегидов
и кетонов (гл. XXVIIА.3) также приводит к алканам.
3. ФИЗИЧЕСКИЕ СВОЙСТВА И СТРОЕНИЕ
Алканы представляют собой бесцветные вещества, в обычных ус-
условиях газообразные или жидкие. Ллканы с большим числом угле-
углеродных атомов являются твердыми веществами. .Многие жидкие
алканы имеют слабый характерный «бензиновый» запах. Алканы
намного легче воды.
Для разветвленных алканов характерна более низкая темпера-
температура кипения (табл. 7), чем для н-алканов с таким же числом угле-
Таблица 7. Температура плавления, кипения и плотности некоторых алканов
Ал кп и
сн4
аисн!снэ
СН,СН.,СН2С113
СН3СНСН3
СНз
CH,CHSCH..CH2CH3
сн,снснхн3
1
СНз
СНз-С-СНз
СНз
Н-С6Н|8
H-Ci0Hi2
Т. пл., "С
— 182,5
-183,3
—187,7
— 138,4
— 159,6
— 129,7
— 159,9
— 16,6
—94,0
—56,8
-29,7
Т. кип,°С
-161,6
—88,6
—42,1
—0,5
— 11,7
36,1
27,8
9,5
68,7
125,7
174,0
0,436
0,561
0,501
0,573
0,551
0,626
0,620
0,591
0.660
0,702
0,730
(при
(при
(под
(при
(при
(под
— I7C-Q
— 100'С)
Лавл.)
-2Г.'С)
—25 С)
давл'.)
родных атомов. Наоборот, температура плавления выше у сильно
разветвленных алканов, особенно у соединений с шарообразной
структурой молекулы (например, 2,2-диметилпропан, 2,2,3,3-тет-
раметилбутан).
О пространственном строении алкапов можно судить по данным
рентгеноструктурного анализа и электронографии. Молекула мета-
метана представляет собой тетраэдр, в вершинах которого находятся
водородные атомы. Соответствующие углы между связями являются
тетраэдрическими и равны 109 28'. Молекулы к-алканов с большим
числом углеродных атомов имеют зигзагообразное строение, при
92
этом углеродные атомы находятся, с небольшими отклонениями, в
одной плоскости:
н н н н
Н V S X у*
10,109 нм 'Н. /С\ C>W54hjJ,H
н
0,254 нм
н
Выше приведена конфигурация молекул алканов. Конфигура-
Конфигурацией называют расположение в пространстве атомов молекулы оп-
определенной структуры без учета различий, возникающих только
после вращения вокруг одной или нескольких ординарных связей.
Алканы могут быть хиральными (см. Введ. 6.12), например
3-метилгексап содержит асимметрический атом углерода с четырь-
четырьмя различными заместителями:
с ,с^
Н'~у "--СШСНз СНзСН2 \ "H
СН3СН2СН2 СН2СН2СНз
энантномеры (зеркальные изомеры)
Те же соединения при рассмотрении в направлении связи С—Н:
СНз СНз
с с
СН.,СН2СН2-^!>*СН,СНз СН3СН2^!>*СН2СНСНз
Н н
S-3-меги.тгехсая R-3-метилгексан
Каждый энантиомер имеет свою абсолютную конфигурацию
молекулы, т. е. пространственное расположение заместителей во-
вокруг хирального элемента. Имеется специальная система обозна-
обозначения абсолютных конфигураций энантиомеров, R, S-номенклатура,
которая рассмотрена в гл. VII.3.
В углеводородах вокруг связей С—С легко происходит вращение,
поэтому существуют различные формы молекулы, отличающиеся
расположением атомов в пространстве, т. е. различие конформа-
ции. Конформациями молекулы определенной конфигурации назы-
называются состояния молекулы с различным расположением ее атомоз в
пространстве, возникающие в результате вращения вокруг связей.
Конформации различаются между собой стабильностью. Более ста-
стабильные конформации, которые фиксируются физико-химическими
методами, называются конформерами, Конформер — это молекула
в ко;!формации, в которую ее атомы самопроизвольно возвращаются
после небольших сдвигов. Наиболее стабильными конформациями
являются те, в которых межатомное отталкивание наименьшее. Их
называют заторможенными конформациями. Наоборот, конформа-
цни, где атомы располагаются близко (в случае алканов атомы водо-
93
рода), являются нестабильными и называются заслоненными
(рис. 48). Фактически различные копформацин находятся в динами-
динамическом равновесии, и возможность перехода одной заторможенной
конформацин в другую через заслоненную копформацию определя-
определяется барьером вращения. Определение структуры и состава копфор-
меров и энергий вращения является задачами конформационного
анализа.
Для изображения процесса вращения по связи С—С очень удоб-
удобно пользоваться двумерными изображениями соединений, которые
предложил М. Ньюмен A955). Так называемые проекции Ньюмена
> \ >
" б в
Рис. 48. Вращение в молекуле этана:
вверху — боковые проекции; книзу — проекции Ньюмена; а, в — за-
заторможенные коиформации; 6 — заслоненная коиформация (барьер
вращения [2,57 кДж/моль)
получают, рассматривая соединение вдоль соответствующей связи
С—С. Фронтальный атом углерода изображают точкой пересечения
его связей, а тыльный атом углерода — окружностью. Вращение в
молекуле этана изображено на рис. 48 при помощи боковых проек-
проекций с клиновидными и пунктирными связями и формул Ныо-
мена. Барьер вращения в молекуле этана равен 12,57 кДж/моль
C ккал/моль).
Установление тетраэдрического строения молекулы метана с
четырьмя одинаковыми связями С—Н привело к гипотезе о spa-
гибридизации углеродного атома. Четыре связи С—Н являются
о-связями, которые образуются при взаимодействии 5р3-гибридпых
орбиталей углеродного атома и ls-орбиталей четырех водородных
атомов.
В молекулах алканов с несколькими углеродными атомами име-
имеются не только ст-связи С—Н, но и ст-связи С—С. Взаимодействие
между связями С—С и С—Н является минимальным, валентные
электроны локализованы в определенных связях. Но увеличение
числа о-связей в молекуле алкана все-таки изменяет свойства. Од-
Одним из наиболее чувствительных критериев свойств электронной
системы молекулы является энергия ионизации (ЭИ). Увеличение
числа ст-связей увеличивает электронодонорные свойства алканов
(понижает ЭИ). Энергия ионизации алканов:
Ллкак Метан Этан Бутан Гексан
ЭИ, эВ 12,5 11,6 10,5 10,3
Алканы обладают очень слабыми электронодонорными свойства-
свойствами.
94
В молекулах алканов имеются только or-связи, следовательно,
отрыв электрона затруднен, возбуждение молекулы (электронные
переходы а—>-ст*) требует большой энергии. Ллканы поглощают толь-
только в вакуум-ультрафиолетовой области A25—140 им), поэтому
жидкие алкапы являются очень подходящими растворителями для
снятия электронных спектров поглощения или испускания различ-
различных веществ.
В колебательиых спектрах (инфракрасные спектры поглощения
и спектры комбинационного рассеяния) для алканов характерно
поглощение в интервалах 2800—2960 см C,75—3,38 ц) (валент-
(валентные колебания связей С—Н) и 1360—1480 см G,35—6,75 fi) (де-
(деформационные колебания связей групп СН2 и СН3). ПМР-снектры
алканов сложны и трудно расшифровываются, так как химические
сдвиги различным образом расположенных протонов имеют близ-
близкие значения @,5—2 м. д.).
4. ХИМИЧЕСКИЕ СВОЙСТВА
Начальное название алканов «парафины» говорит об инертности
этих соединений в химических реакциях (от лат. parum — мало,
affinis — находящийся в сродстве). Действительно, алканы не взаи-
взаимодействуют с обычными кислотами и щелочами, окислителями. Но
фактически алканы в определенных условиях не так уж инертны.
Легко осуществляются реакции с активными свободными радикала-
радикалами, превращения в условиях высоких температур или в присутст-
присутствии катализаторов, в том числе сверхеильпых кислот. Ниже при-
приведена простая схема реакций алканов:
R-CH-CH —
термические
IF H
I I
R-C-C—R—
II у
аамотце- ' л
ипе | |
>R-C —C-R
I
R'+CH2=CH2+R]« 4- _l .1. КЗОИСРИ
H H
R
зация )
->R-C-CH3
1. Реакции замещения водородных атомов алканов при взаимо-
взаимодействии с активными свободными радикалами. Гемолитические
реакции алканов должны быть инициированы активными свобод-
свободными радикалами. Это достигается в основном фотохимическим,
термическим или каталитическим путем.
а) Галоген и рование ал капов. Ллканы очень
активно взаимодействуют с фтором, реакция с хлором происходит
при освещении. Взаимодействие с бромом осуществляется только
при освещении и нагревании. Иод с алканами не реагирует
(гл. VII.2.1).
95
С хлором алканы (метан, этан, пропан и др.) при обычной темпе-
температуре и в темноте не реагируют. Напротив, при освещении солнеч-
солнечным светом или еще лучше УФ-лучамн начинается бурная реак-
реакция, иногда заканчивающаяся взрывом. В такой реакции из ме-
метана образуются хлорированные метаны с различным числом ато-
атомов хлора:
CH3CI -'r О,-Л СН2С12 + HCI
Под действием света молекулы хлора активируются и распадают-
распадаются на свободные радикалы CI-. Атомы хлора обладают большим
сродством к электрону и способны отрывать атом водорода от алка-
па с образованием свободного алкильного радикала. Алкпльньш
радикал, в свою очередь, реагирует с молекулой хлора, отрывает
атом хлора и образует хлоралкан. В реакционной среде опять обра-
образуется атом хлора:
а2 -X.2CI-
a--f-ai4—8-снз- + на
сн3-+а2—.-снзО+а.
На каждой ступени реакции генерируется свободный радикал,
который служит продолжателем реакции. Такие реакции называ-
называются цепными реакциями. После инициирования реакции возникает
цепной процесс, который прекращается только после исчезновения
всех активных свободных радикалов. Цепи «обрываются» в резуль-
результате рекомбинации (димеризации) свободных радикалов (R- +
+CI--R—С1, R+R->R—R).
В реакции метана с хлором могут образоваться также ди-, три-
и тетрахлорметапы:
a • -ь сн3а ^Х. .сн2с1 + на
¦сн.,а+а2—*сн2а2+а-
С1-4-сн3а2—*-снс12-| на
— сна3+а-
Подобные реакции хлорирования происходят также с этаном,
пропаном, бутаном и другими алканами. В этих реакциях обычно
образуются смеси изомерных хлоралканов.
Необходимо отметить, что активность алкапов в реакциях га-
логепировання растет с увеличением числа углеродных атомов в
цепи н-алкана. Это еимбатно увеличению электронодонорных
свойств алкапов (уменьшению ЭЙ).
б) С у л ь ф о х л о р и р о в а и и е а л к а и о в. Алканы RH
реагируют с SO2 и С12 под действием УФ-свста, при этом образуют-
образуются хлорангидриды алкансул'..фоновых кислот RSQ..CI. Под действием
УФ-лучей образуются атомы хлора и свободные алкильпые радика-
96
лы. Последние взаимодействуют с SO2:
Cl2—.-2CI.
RH + -C1—*R--|-HCI
О
II
R- +SO., —* R —S«
II
О
О О
'I II
r_S.+CI2—*R—S-CI + CI.
!l !l
О О
Хлорангидриды алкансульфоновых кислот широко использу-
используются в производстве моющих средств (гл. XVIII.4).
в) Сульфоокисление алканов. Алканы RH реа-
реагируют с SO2 в присутствии О, при УФ-облучении, при этом обра-
образуются алкансульфоновые кислоты RSO3H. Для осуществления
реакции необходимо непрерывное освещение, так как только в ре-
результате этого воздействия из алканов генерируются свободные
радикалы. Далее свободные алкильные радикалы реагируют с
SO2 и О2:
О
II
R.+SOj—*-R —S«
II
О
О О
:1 II
R_S.-f-O,—*R — S — О — О.
'I II
О О
о о
II I!
R_S —О—O-+R — H—*R —S —О —OH + R*
il il
О О
алканпероксисуль-
алканпероксисульфоновая кислота
Алканпероксисульфоновая кислота легко распадается с образо-
образованием новых'свободных радикалов:
О О
,1 II
R— S — О — OH-L2R—Н—*2R-+R— S — ОН + Н2О
II il
О О
Конечным продуктом реакции сульфоокнслення является алкан-
сульфоновая кислота.
г) Нитрование алканов. При взаимодействии алка-
алканов е разбавленной азотной кислотой или оксидами азота при на-
нагревании происходит замещение водородных атомов нитрогруппой
4 № 517 97
и образуются нитроалканы (метод Коновалова):
HNO3; NO2
R-H > R-NO,
/
Более подробно механизм образования нитроалканов описан в
гл. XXI.A.1.
д) Реакции алканов с кислородом. В присут-
присутствии кислорода алканы легко сгорают с образованием СО2 и Н2О
и выделением большого количества теплоты D6 000—50 000 кДж/кг).
Смеси газообразных алканов с воздухом или кислородом взрыво-
взрывоопасны.
Окислением алканов в газообразной или жидкой фазе воздухом
или кислородом в присутствии катализаторов могут быть полу-
получены продукты частичного окисления, в основном карбоновые кис-
кислоты. Промежуточными продуктами в реакции окисления алканов
являются свободные алкильпые радикалы и пероксисоединения.
Ниже приведена схема окисления бутана до уксусной кислоты:
СН3СН2СН2СН3—*СН3СН2 —СН-СНз'Л.
II Н
-»с»лсн,-<1-сн, сн'сн'с"'с^сн,-сн.-(!:-сн, Л
О —О. о —О —Н
гндроперокснд
СН3СН2ОН + СНз — С^
I о2
/Р о* /р
f — CHS-Cf
/Р *
СНз-Cf — CHS-C
н чон
Промежуточными продуктами являются спирты и альдегиды.
При окислении других алканов (С,о—С20) можно получить
как спирты, так и карбоновые кислоты с большим числом углерод-
пых атомов в цепи (высшие спирты и высшие кислоты). Эта реакция
используется в промышленности.
2. Термические превращения алканов. При температуре выше
500 СС алканы становятся нестабильными и распадаются с выделе-
выделением водорода и образованием углеводородов с более низкой мо-
молекулярной массой. Присутствие катализаторов уменьшает темпе-
температуру распада. В этих реакциях происходит гемолитический раз-
разрыв связей С—Н и С—С. Термические превращения алканов назы-
называются крекингом. Известны термический крекинг и каталитиче-
каталитический крекинг, которые широко применяются в промышленности.
Труднее всего превращается метан:
800-1000° С
СН4 ¦* КС
Этан дегидрируется при более низких температурах:
575-G50 °С
98
Алканы с более длинной углеродной цепью образуют ненасыщен-
ненасыщенные углеводороды или распадаются на углеводороды с меньшей мо-
молекулярной массой:
я-Алкапы с шестью или более углеродными атомами в присут-
присутствии катализатора способны к циклизации и образованию бензола
и его производных (гл. VI.Л.2):
Н
СН3СН2СН2СН2СП2СН3—> | II -I 4Н2
H/SC/C\H
ы
Эта реакция называется ароматизацией или дегидроциклизацией
алканов.
Термические некаталитические и каталитические превращения
алканов осуществляются по свободнорадикалыюму механизму.
При этом происходит разрыв как С—Н-, так и С—С-связей, образую-
образующиеся свободные алкильные радикалы участвуют в различных даль-
дальнейших реакциях. Ниже приведены некоторые из возможных пре-
превращений алкана RCH2CH2CH3:
RCH3-! CH2=-CII2 RH-CH2=-CH2+.CH3
| | р-распад
RCH2- + -СН2СНз< RCH2CH2CH3 > RCH2CIF2- + -СН3
RCII2CH2CH3 ДНСПРОПОРЦИО- I I
•"Г ниропапне .-j. i.
RCH = CHCH3- RCH2CHCH3+-H RCII=CH2 + CH4
H2
3. Изомеризация алканов. Под действием сильных электрофиль-
ных реагеитсв (кислот Льюиса) я-алканы частично превращаются
в изоалканы, а изоалканы — в н-алкаиы. Образуется равновесная
, Кислота
смесь: я-Алкан^з^г^Изоалкан
В качестве электрофилыюго реагента могут быть использованы
А1С13 или А1Вг3 при повышенной температуре. Наиболее перспек-
перспективно применение очень сильных кислот, так называемых сверх-
сверхкислот (BF3+HF, SbFg+HF, SbF5+FSO3H), которые имеют не-
необычно высокую активность протона. Эти кислоты изомеризуют
алканы уже при обычных температурах.
Изомеризация алканов возможна начиная е бутана. Чем боль-
больше углеродных атомов в молекуле алкана, тем легче идет изомериза-
изомеризация. Это объясняется понижением энергии ионизации таких алка-
алканов, что облегчает взаимодействие q электрофильным реагентом.
4* 99
Алканы обладают очень низким сродством к протону, что объясняется
присутствием только насыщенных связей (ст-связей) и отсутствием атомов с
неподеленными электронными парами.
При взаимодействии алкана со сверхкислотой образуются карбокатио-
пы, способные к внутримолекулярным перегруппировкам. Первичным про-
продуктом является протоннрованный алкан (ион алканония). Где присоединя-
присоединяется протон, неизвестно. Предполагают, что атом углерода и два атома во-
водорода образуют трехцептровую двухэлектрониую связь. Ион алканония рас-
распадается с отщеплением молекулы водорода или метана и образованием кар-
бокатиона:
R—СНз + Н
н
1
R—С"
А
"'••н
->¦ RCH7 + Н2
->¦ R+ + СН4
Перегруппировку можно показать на примере втор-бутнл- и трет-бу-
тилкатионов:
+ * I—'
С
+ * I—
СН3—С—С—Н
н i
сн
СНз С С
Сн-'
н
с„,к
СНз С С—Н
"—н ¦*--
Большей стабильностью обладают третичные карбокатиопы, поэтому процесс
перегруппировки в основном направлен на образование разветвленных ал-
алканов.
Изомеризация алканов в присутствии сверхкислот может найти
практическое применение в промышленности для производства вы-
высококачественного моторного топлива.
4. О реакционной способности алканов в зависимости от их
строения и активности реагента и о стабильности свободных ал-
кильных радикалов. Реакционная способность алканов, начиная с
метана, при переходе на неразветвленные и особенно разветвлен-
разветвленные алканы увеличивается. Это относится в одинаковой мере к реак-
реакциям радикального замещения, реакциям дегидрирования, кре-
крекинга и превращениям в присутствии сверхкислот. Это может быть
объяснено, с одной стороны, некоторым усилением электронодонор-
'ТсОлица 8. Энергия диссоциации некоторых связей С — Н
СП
СП
сн
сн
Ре?
; - "> СНз
-сп,сн3
;,СНСН3
СНз
:<пг.н диссоциации
¦ -¦-U-
СНоСН,.-|-Н.
->СН,СНСН3-:-Н.
—>сн3ссеь-;-н.
сн3
АН (усредненные
кДж'моль
425
40G
395
375
р
змачепгя)
<кпл/.о,ь
102
97
94
90
100
пых свойств при увеличении числа а-связей (см. гл. 1.3), а с дру-
другой — уменьшением энергии диссоциации связей С—Н и С—С при
увеличении длины цепи и ее разветвленное™. Изменение энергии
связей С—С при изменении строения приведены во Введ. 6.2, а
изменение энергий диссоциации связей С—Н — в табл. 8.
В реакциях радикального замещения и термического расщепле-
расщепления промежуточными частицами являются свободные алкильные
* радикалы, стабильность которых может быть охарактеризована
энергией диссоциации С—Н-связи: R—H->R-+H-.
Из данных табл. 8 вытекает, что наименьшую стабильность име-
имеют свободные радикалы СН3- и CH3CH2v Среднее время жизни
их не превышает 10~л. . . 10 с. Разветвленные свободные радика-
радикалы более стабильны, что объясняется пространственными эффек-
эффектами и небольшой делокализацией неспаренного электрона. При этом
третичные радикалы стабильнее вторичных.
Таким образом, реакции со свободнорадикальным механизмом
должны протекать преимущественно у третичного углеродного ато-
атома алкапа или у вторичного, т. е. реакции могут быть региоселек-
тивными. Региоселективной называется реакция, в которой хими-
химические изменения происходят преимущественно в одной из несколь-
нескольких возможных положений в молекуле.
Например, при региоселективной реакции должна соблюдаться
последовательность замещения:
СН3 СНз СН3 Н
I х- I х. | |
СНз —С —СН2СНз -*СН3-С -СН2СН3 -^СНз-С-С-^СНз
I I II
Н X XX
В реакциях алканов строго это не соблюдается. Регноселектив-
пость (избирательность) зависит от активности реагента и скорости
реакции. Чем активнее реагент и больше скорость реакции, тем
меньше региоселективность. Так, при фторировании (гл. VII.2.1)
получаются полифторпроизводпые. Реакция бромирования более
региоселективна, чем хлорирование.
5. ПРИМЕНЕНИЕ АЛКЛНОВ
Алканы являются не только простым и относительно дешевым топ-
топливом, но и исходным сырьем для крупнотоннажного производства.
Полученные из нефти смеси алканов и других углеводородов
применяются в качестве моторного топлива для двигателей внутрен-
внутреннего сгорания и реактивных двигателей.
При разгонке нефти получают несколько фракций: бензин
(т. кип. 40. . .180 °С, углеводороды Св—С10), керосин (т. кип.
180. . .230 °С, углеводороды Сп и С12), дизельное топливо (т. кип.
230. . .305 °С, углеводороды С13—С,,). Остается мазут, из которо-
которого перегонкой под уменьшенным давлением или с водяным паром
получают соляровое масло (углеводороды d8—С23), смазочные мас-
масла (углеводороды C:s—Сзе), вазелин, твердый парафин.
101
Высшие фракции разгонки нефти подвергают крекингу для по-
получения высокосортных бензинов. Кроме того, получаются алке-
ны — этилен, пропен, бутены — важнейшее сырье для химической
промышленности.
Метан — бесцветный газ без запаха, слабо растворим в воде
(около 50 мл или 0,033 г в 100 г воды) при 20 °С. Встречается в при-
природе как болотный газ, рудничный газ. Наибольшее содержание
СН3С1 •<¦
CHjCl,
CHjNO
:с=о
CS,
с + н2о
СО + ЗН.
сажа для
pcnuiORoii
промышленности
для синтеза
Фишера-Тррпша
со2 + и.
волород для
гидрирования
Рис. 49. Схема превращений метана
метана в природном газе F0. . .99%). Значительные количества ме-
метана образуются при сухой перегонке каменного угля, в процессах
гидрирования угля.
Метан широко используется в качестве топлива с большой теп-
теплотворностью E0 000 кДж/кг). С воздухом образует опасные
взрывчатые смеси. Метан служит важным сырьем для химической
промышленности. Главные превращения метана показаны на схе-
схеме (рис. 49).
Глава II
АЛКЕНЫ
При отнятии от молекулы алкана двух водородных атомов образуют-
образуются углеводороды другого типа с общей формулой CnH2n. Самым
простым из этих углеводородов должен бы быть СН2. Такого угле-
углеводорода получить не удалось. Позже было показано, что это соеди-
соединение является нестабильным, ведет себя как активный бирадикал
(частица с двумя неспаренными электронами), который был назван
карбеном.
Простейшим алкеном является этилен С2Н4- В его структур-
структурной формуле между углеродными атомами пишется двойная связь:
Н,С: + :СН2—*Н2С^СН2
Углеводороды С„Н2Л рассматривают как производные этилена.
Исторически первое название этих алифатических углеводородов
102
с двойной связью было олефаны («маслообразующие», так как при
взаимодействии с хлором и бромом образовывали маслянистые
жидкости). Называют их и ненасыщенными углеводородами, так
как они способны присоединять различные реагенты.
В то же время формуле СПН2П. соответствуют и углеводороды
циклического строения — циклоалканы, обладающие совершенно
другими свойствами.
1. ИЗОМЕРИЯ И НОМЕНКЛАТУРА
По номенклатуре ИЮПАК углеводороды СпН2п. с одной двойной
связью называют алкенами. Названия алкеиов образуют от названий
соответствующих алканов, заменяя суффикс -ан на -ен. Суффикс
-ен обозначает присутствие двойной связи в цепи углеродных ато-
мсв. Для образования названия алкена выбирают самую длинную
цепь, в которой находится двойная связь, и углеродные атомы ну-
нумеруют таким образом, чтобы двойная связь получила наименьшую
цифру. Эту цифру принято ставить после суффикса -ен (допускает-
(допускается и перед названием главной цепи).
В табл. 9 приведены структурные формулы и названия первых
представителей гомологического ряда алкенов.
Число структурных изомеров у алкенов больше, чем у алканов,
так как одновременно с изомерией, обусловленной разветвлением
цени, возможна изомерия положения двойной связи. Кроме того,
для алкенов характерна пространственная (геометрическая) изо-
изомерия; например, существуют ^«с-бутен-2 и транс-бутеи-2. Так как
свободного вращения по двойной связи алкена в обычных условиях
не происходит, метальные группы в бутене-2 могут быть фиксиро-
фиксированы по одной стороне двойной связи (цш>изомер) или по противо-
противоположным сторонам (/пранс-нзомер). Для обозначения геометри-
геометрических изомеров предложены также буквенные обозначения: Z
для цис- (от нем. zusammen — вместе) и Е для транс- изомера (от
нем. entgegen — напротив).
Если у двойной связи имеется три или четыре различных угле-
углеводородных или других заместителя, то обозначения Z и Е выби-
выбирают по пространственному расположению двух самых старших
групп. Старшинство углеводородных заместителей увеличивается
в ряду
-СН3 -СН2СН3 -СН2СН,СН3 —CH2CH2CIbR
сн3
/СН, I
— си< —с-сна
ЧСН3 |
СНз
Правила старшинства различных заместителей и характеристиче-
характеристических групп изложены в гл. VII.3. Чтобы отнести изомер к Z- или
?-ряду, необходимо среди четырех заместителей у двойной связи
найти два самых старших. Если оба старших заместителя располо-
расположены но одну сторону плоскости, в которой лежит двойная связь,
103
Таблица 9. Простейшие представители алкенов
общая
Формула
структурная
Номенклатура
Сокращенно? изображе-
изображение формулы
с2н4
С4Н,
с.,н
5п10
СН2 = СН2
СН3СН = СН2
сн3сн2сн^сн2
сн3сн = снсн3
СНз — С = Cri2
СНз
СН3СН2СНаСН=СН5
СН3СНоСН = СНСНз
з
4 3 2 1
СН3СНСН-=СН2
СИ,
СН3С=-С11СН3
I
сн3
Этилен («этен» при-
применяют иногда для
образования назва-
названий производных и
остатков)
Пропеи (применяет-
(применяется также «пропилен»)
Бутен-1
Бутен-2, иис- (или
2) транс- (или Е)
2-Метилпропен-1
(применяется также
«изобутнлен»)
Пентен-1
Пентсн-2, цис- (или
Z) транс- (пли Е)
2-Метилбутец-1
З-Метплбутен-1
2-Л\стплбутен-2
изомер относят к Z-ряду, з противном случае к ?-ряду. Например:
СП.. СНоСНз
' С- С:7
СНзСН/ CH,CILCH3
Z-J-л'етил-4-этилгептеп-З
Названия остатков алкенов (алкенильных групп) образуют нри-
соединенчем к названию алкена суффикса -ил. Некоторые остатки
Ю4
алкенов сохраняют тривиальные названия:
СН2 —СИ— этеиил или винил
Л 2 1
СН2 = СНСНа—• пропенил-2 или аллил
2 1
СН2 —С— 1-метнлэтенил или изопропеш:л
СНЯ
3 2 1
СНцСН-СН— пропснил-1
2. МЕТОДЫ ПОЛУЧЕНИЯ
Реакции получения алкенов основываются на отщеплении атомов
или атомных группировок от алканов и их производных. Меньше
применяются реакции, в которых алкены образуются из соединений
с тройной связью или несколькими двойными связями, и реакции
конденсации.
Легко установить взаимосвязь между алканамн и их производ-
производными и алкенами, с одной стороны, и более ненасыщенными произ-
производными и алкенами — с другой, как это изображено на схеме:
RCH2CH2R
¦RCH = CHR
чн н '
I -НХ
RCHCH2R
1. Дегидрирование и крекинг ал капов (промышленный метод).
Алкены получают отщеплением двух атомов водорода от молекулы
алкана:
RCH2CH2R - "i RCH = CHR
Эти реакции идут при повышенной температуре н в присутствии
катализаторов (оксидов различных металлов), например:
СНяСН2СНаС11Я -^-^ CI13СН - CI ICHa +1 h
При температурах около 600 "С дегидрирование идет дальше
и образуется бутадиен-1,3 СН2 —СН—СН--СН2.
При более высоких температурах происходит также разрыв свя-
связей С—С и получаются смеси различных алканов и алкенов, у ко-
которых молекулярная масса меньше, чем у исходного алкана (кре-
(крекинг).
2. Отщепление воды от спиртов. С давних времен спирты (ал-
канолы) служат исходным сырьем для получения алкенов:
RCH,CH,OH —> RCH - CI I, • (- H2O
Отщепление воды достигается различными способами:
а) при нагревании .в присутствии сильных кислот, например
K.SO,:
IIj.SO,
СН3СН,ОН —^СН^-СН,. 4-11*0
105
Это обычный лабораторный способ получения этилена. Меха-
Механизм реакции рассмотрен в гл. XIV.А.4.
б) при повышенной температуре на катализаторе, обычно на
Л12О3:
Л1аО,
сн3сн2сн2он ^г^-0^ сн3сн = сн2 + н„о
Каталитический способ применяется в промышленности, например
для получения этилена (см. гл. II.5).
3. Отщепление галогенводорода или галогена от галогеналканов.
Алкены образуются при воздействии на галогеналканы концентри-
концентрированных растворов щелочей. Побочным продуктом являются спир-
спирты. В более разбавленных растворах щелочей спирты являются
основным продуктом реакции
RCH2C[I2Br + \'a ' ОН-—> RCH = CII2(+RCH2CH2OH)-;-Na + Br- + H2O
Механизм этой реакции рассмотрен в гл. VII.4.5.
Отщепление галогенводорода возможно также термически:
RCH2CH2CI -'-> RCH=.CH2-|-HC1
Алкены образуются также при обработке дигалогеналканов
цинком:
R— СН— СН — R-'-Zn—*RCH = CHR-i-2nBr2
I I
Br Br
4. Гидрирование диенов и алкинов. При использовании селек-
селективных катализаторов гидрирования удается гидрировать диены
и алкины до алкенов:
п.
СН2 = СН — CIU СНа- -СЫз — СН = СН— СН3
Ni
5. Реакции конденсации. Алкены и их производные получают
при взаимодействии двух соединений с активными группами (на-
(например, карбонильной, метиленовой и др.). Общая схема реакции:
R4 Y R* R .R»
}С-Х+ ЧС< -*. )С-С( -I-XYZ
Rl/ Z/ \R3 Rl/ \R3
Примеры реакции конденсации приведены на с. 278, 447, 458, 559.
3. ФИЗИЧЕСКИЕ СВОЙСТВА И СТРОЕНИЕ
Первые представители гомологического ряда алкенов (С2—С4)
при обычных температурах представляют собой газы, следующие
члены ряда — бесцветные жидкости или кристаллические вещества.
Этилен и пропен имеют слабый запах. При увеличении числа угле-
углеродных атомов и разветвлении цепи запах становится едким, разд-
раздражающим слизистую оболочку.
106
Плотность алкеноз выше по сравнению с алканами с тем же чис-
числом углеродных атомов, больше также коэффициент преломления
света и молекулярная рефракция (см. гл. II.5). Значительно разли-
различается рефракция связей, например: Rc-c=~-1,296, но Rc=c "-'¦
—4,17, что свидетельствует о большей поляризуемости двойной
связи.
Термохимическая энергия двойной связи ?с=с — 615 кДж/моль
A47 ккал/моль), что больше, чем энергия простой (ординарной)
связи ?с_с=344 кДж/моль (82 ккал/моль), но меньше, чем сум-
сумма энергий двух ординарных связей. Это означает, что одна из свя-
связей двойной связи может быть разорвана легче, чем связь С—С.
Динольный момент этилена равен нулю, соединение неполярно.
Но симметрично замещенные алкены обладают небольшим диноль-
ным моментом, их молекулы иолярны, например:
СН3ч
)С-СН2 A= 1,64-Ю-™ Кл-м @.49D).
CEI,/
Это означает, что метильные группы изменяют распределение элект-
электронной плотности в двойной связи. Метильная группа действует
как электронодонорная группа и проявляет свой донорный индук-
индуктивный эффект (+/). Обсуждается также своеобразное взаимо-
взаимодействие а-электронов связей С—Н с я-электронами двойной свя-
связи — так называемый эффект сверхсопряжения (а, я-сопряжепия):
и1
Поляризацию двойной связи под действием электронодонорного
(D) или электроноакцепторного (А) заместителя в общем виде можно
изобразить (см. В вед. ,5):
?,.Г\ ^-Ч 6~ б- у N «4-
-/ и -М
Введение алкильных групп у двойной связи термодинамически
стабилизирует молекулу алкена. Эго следует из значений стандарт-
стандартной энтальпии (теплот) сгорания АЯСГ (см. Введ, 6) алкенсв и
теплот гидрирования (см. гл. П.4). Чем меньше теплота сгорания
соединений одинакового состава, тем стабильнее соединение. На-
Например, для СН2=СНСНаСН3 ЛЯ„Г = — 2719 кДж/моль, а для
т^анс-СН3СН=СНСН-3 АЯСГ=—2707 кДж/моль. Аналогично
уменьшаются теплоты гидрирования.
Электронографическое изучение этилена показало, что геометрия
молекулы резко отличается ог проотранствеиного строения алканов.
107
н\о,ш н»/н Молекула этилена плоская, углы между свя-
\: = с'т П6* зями близки к 120° (тригональные углы); связь
н^/ ^н С—С намного короче связи С—С @,154 нм).
ч> Для объяснения строения молекулы этилена и
этилен других алкенов используют гипотезу о sp!-ni6pu-
дизации атома углерода. Углеродный атом в этилене образует три о-
связи с использованием трех 5/?2-гибридных орбиталей и одну я-
связь за счет перекрывания р-орбиталей (рис. 50).
Поворот по связи С—С в алкенах значительно затруднен, так
как он вызовет разрушение л-орбитали, а это требует затраты энер-
чн
или С^^С
н н
Рис. 50. Типы связей в молекуле этилена
гии. Этим объясняется существование стабильных цис- и транс-
изомеров, которые переходят друг в друга только при высокой тем-
температуре или при УФ-облучении. ^
Наличие особой связи — я-связи— объясняет такие свойства
алкенов, как повышенная рефракция, пониженная энергия одной
связи, легкая поляризуемость. Электроны л-орбитали находятся
дальше от атомных ядер, поэтому они более подвижны, энергия
л-орбиталей меньше, чем энергия а орбиталей. Отрыв электрона
от я-орбитали требует затраты меньшего количества энергии, чем
отрыв электрона от а-орбитали.
Энергия отрыва электрона характеризуется энергией иониза-
ионизации (ЭИ).
Энергия ионизации некоторых алкеиов
СН3 Н3С СН3
Я —С=--СН СНСС
Алкеи . . . СН2==СН2 СН3СН = СН2 СНЯ —С=--СН2 СН3-С=-С-СЫ3
ЭИ, эВ . . 10,5 9,7 9,2 8,3
Энергия ионизации характеризует процесс:
RCII^CHR—* [RCH_l.CHR]-i +e~
Алкены имеют на 1. . .1,5 эВ более низкие ЭИ, чем алканы о
таким же числом углеродных атомов, а следовательно, значитель-
значительно сильнее выраженные электронодонорные свойства. Введение
метальных групп особо благоприятствует понижению ЭИ.
Подвижность я-электронной системы отражается также в элект-
электронных спектрах поглощения. Максимум поглощения находится
при более длинных волнах A80. . .200 нм), чем у алканов. Погло-
Поглощение электромагнитного излучения в этом случае связано с воз-
возбуждением л-электроноз. Электрон со связывающей л-орбитали
переходит на разрыхляющую л-орбиталь. Такой электронный пе-
108
реход обозначается как я-*я*. В возбужденном состоянии резко
изменяется плотность л-электронов в связи и появляется возмож-
возможность свободного вращения по С—С-связи.
В колебательных спектрах кроме поглощения алкильных групп
наблюдаются валентные колебания двойной связи при 1640. . .
.1660 см'1 и связей =С—Н при 3000. . .3100 см, а также де-
деформационные колебания связей =С—Н при 890. . .980 см х.
В спектрах ПМР характерны сигналы при 4,5. . .бм.д. (=С—Н).
4. ХИМИЧЕСКИЕ СВОЙСТВА
Реакции алкенов обусловлены присутствием двойной связи, кото-
которая легко поляризуема и обладает электронодонорньши свойства-
свойствами. Кроме того, она значительно влияет на реакционную способ-
способность расположенной рядом связи С—Н (аллильное положение).
Для алкенов характерны различные реакции присоединения, олн-
гомеризации, полимеризации, окисления и в отдельных случаях
реакции замещения в аллильном положении. На рис. 51 приведе-
приведена общая схема превращений алкенов.
Nthr
I
X
^R H^ ^R
c=o
o=c-
L4I.R'
P. li R H R
! i I I I
— г—с— с— с — с
I I I I I
CH,RR CH.UR CH.R
I!
R
X Y
цис-г.рисое л мнение трпнс-ирисоелимемие
Puc. 5/. Схема превращений алкенов
>o<R
V CI!R
/ \
х \
109
t. Гидрирование алкеиов. Алкены присоединяют водород только
в присутствии катализатора:
Кат,
RCH = CHR -|- Н2 > RCH.,CH2R
Катализатором служит никель (П. Сабатье, Ж. Сапдеран, 1899)
или платина (PtO2 — катализатор Адамса).
Реакция является экзотермической (выделяется теплота). При
сравнении теплот реакции гидрирования алкснов одинакового со-
состава наблюдается такое явление, что чем больше алкильных групп
у двойной связи, тем меньше теплота гидрирования. Например, для
СН2=СНСН2СН3 это 126,8 кДж/моль, а для транй-СН3СН=СНСН3
115,5 кДж/моль. Это свидетельствует об увеличении термодинами-
термодинамической стабильности молекул (гл. П.З).
2. Взаимодействие алкенов с электрофильными реагентами. Ал-
Алкены принадлежат к электронодонорным, или нуклеофильпым,
реагентам. Взаимодействие их с электрофильными реагентами при-
приводит к образованию комплексов и продуктов присоединения к
двойной связи. В некоторых случаях протекает олигомеризация и
полимеризация. Алкены взаимодействуют с некоторыми сильными
кислотами (галогенводородами, серной кислотой), с карбокатио-
нами, галогенами и ионами металлов. Гидроборирование рассмот-
рассмотрено в гл. XI.А.
а) Реакции с Н-электрофилами (сильными
кислотами). Алкены реагируют с сильными кислотами. Во
многих случаях происходит присоединение кислоты по двойной
связи:
RCH-CHR-I-H — Х~—>RCII2 —CHR
X
где Х=С1, Вг, I, OSOaH и др.
Хорошо изучены реакции присоединения галогенводородов
(гидрогалогеиирование) и серной кислоты. Присоединение гало-
галогенводородов происходит как в газовой фазе, так и в растворах,
на скорость реакции значительно влияет присутствие солей тяже-
тяжелых металлов.
В случае несимметричных алкенов возможны два направления
присоединения кислоты. Эти реакции подробно изучил В. В. Мар-
ковников и пришел к выводу, что в большинстве случаев направле-
направление присоединения предопределяется строением алкена. Протон
присоединяется к тому углеродному атому, у которого меньше уг-
углеводородных заместителей (к «более гидрированному») — правило
Марковникова A870):
> RCH-CHa
Эта направленность легко объясняется классической электронной
теорией. Молекула несимметрично замещенного алкена является
поляризованной, а алкильные группы как электронодонорные за-
110
местители определяют наиболее вероятное место присоединения
протона:
. 8+ 8-
+ Н—X *¦ СН3 — СН—СН3
Так как алкильные группы являются слабыми донорами, на-
направленность реакции присоединения может меняться в зависимо-
зависимости от условий реакций, растворителя, соотношения концентраций
компонентов, температуры. Реакции со свободнорадикальным ме-
механизмом идут «против правила Марковникова» (см. с. 118), так жа
как реакции алкенов, содержащих электроиоакценторные замести-
заместители:
^ (А = CN, NO,, COR)
Механизм реакции присоединения обычно трактуется как ион-
ионный. Присоединение протона вызывает образование карбокатио-
на, который присоединяет анион:
й+ 6- +
RCH = CHR + H — X—>RCH,— CHR —^RCHo—CHR
X
Однако образование свободных карбокатионов ввиду их боль-
большой реакционной способности, за исключением тре/п-карбокато-
нов, является весьма маловероятным процессом.
Присоединение можно рассматривать как тримолекулярный
процесс, в котором одна молекула кислоты поляризует я-связь
алкена образованием я-комплекса (комплекса с переносом заря-
заряда — КПЗ), а вторая молекула кислоты дает аннон:
8+ 8- . ^ 8+ 8+ НХ
RCH = CHR + Н X :„ *. RCH=j=CHR *-
4
г-г. «+
ГХ Н н х н+
8V^—S+ I I
»- RCH=F=CHR • *- R С С R
I—-'' I I
г* «'Чб- н н х
н-1—х
Возможно также промежуточное образование ионных пар.
Присоединение сильных кислот в присутствии других анионов
или нуклеофильных частиц может привести к смеси продуктов (со-
(сопряженное присоединение):
RCH = CIIR+H —X + M-fY- —* RCIIoCHR-i RCH2CHR
I I
X Y
В случае реакций гидрогалогенирования возможен молекуляр-
молекулярный механизм присоединения, в котором полностью исключается
111
образование ионов и реакция проходит через циклическое переход-
переходное состояние:
RCH=CHR + 2НХ »> RCH=±=CHR *- RCH2
Механизм реакции определяется многими факторами, особенно
полярностью реакционной среды и структурой алкена. В поляр-
полярных растворителях доминируют ионные механизмы, а в неполярных
растворителях и в газовой фазе — молекулярные механизмы.
Большое значение имеет реакция алкенов с серной кислотой,
в которой образуются алкилсульфаты:
RCH = СН2 + Н—OSO3H —> R -СН-СН3
OSO3H
При взаимодействии алкилсульфатов с водой происходит гид-
гидролиз и образуются алканолы (спирты):
R—СН —СН3+Н2О —> R — СН — CH3 + H2SO4
OSO3H ОН
Таким образом, серная кислота способствует присоединению воды
к алкенам. Гидратация алкенов осуществляется также в присутст-
присутствии кислого катализатора. На этих реакциях основаны промышлен-
промышленные методы получения спиртов из алкенов (с.\283 и 295).
Действие сильных кислот на разветвленные алкены, способные
образовывать третичные карбокатионы, вызывает их олигомериза-
ипю и полимеризацию (см. с. 119).
б) Реакции с С-электрофилами. Алкены при-
присоединяют сильные С-электрофилы, например карбокатионы, ко-
которые генерируются в реакционной смеси обычно в виде ионных
пар:
Ri СН=СН2
В результате присоединения образуется новый карбокатион,
который претерпевает дальнейшие превращения. Такой процесс
лежит в основе полимеризации алкенов по карбокатионному меха-
механизму (см. с. 119).
в) Галогенирование. Алкены легко присоединяют
галогены Х2 (X = F, C1, Вг, I). Очень энергично, даже со взрывом,
реагирует F2, медленно реагирует 1а. Это согласуется со сродством
к электрону (СЭ) молекул галогенов:
Галоген F2 Cl2 Br2 h
СЭ, эВ 3,1 2,5 2,5 1,6
Кроме того, при образовании связи С—F высвобождается значитель-
значительно больше энергии (A#C_F --443 кДж/моль), чем при образова-
образовании связи С—С1 (Д#с-с1 = 328 кДж/моль) (см. Введ. 6.2).
112
В результате реакции образуются дигалогеналканы. Присоеди-
Присоединение в большинстве случаев происходит пространственно избира-
избирательно — апереоселективно. Стереоселективной называют такую
реакцию, в которой химические изменения приводят к преимущест-
преимущественному образованию одного из двух или нескольких возможных
продуктов, которые отличаются только своей стереохимией. Ато-
Атомы галогеьа присоединяются только в /плане-положении по отно-
отношению к плоскости молекулы алкена (т/заяс-присоединеиие):
н
+Xs
H
A
/^Ч
X7 ! XR
H
Более наглядно это видно на формулах Ньюмена:
X
н
Стереоселективность реакции объясняется пространственным
действием молекулы галогена. Доказано, что при взаимодействии
алкенов с галогенами образуется КПЗ (я-комплекс), в котором из-
изменено распределение электронной плотности по двойной связи.
Дальнейшие презращения комплекса менее ясны. Предполагают,
что я-комплекс может ионизироваться н после этого следует нук-
леофильная атака анионом галогена с противоположной стороны:
V
II + х2
с
Н
"КПЗ
V"
с'..-'
н/ \R
тесная ионная
пара
Ч /"
с •¦'¦•.
сольватиро-
ванные ионы
При ионизации образуется КПЗ алкена с положительно заря-
заряженным галогеном Х + , который иногда рассматривается как трех-
трехчленный галогенониевый цикл.
Если реакция присоединения проходит по свободнорадикаль-
ному механизму, то стереоселективпость не наблюдается, получа-
получаются продукты как транс-, так и циоприсоединения. Радикаль-
Радикальный механизм вызывается освещением, присутствием пероксидов
и др.
Галогенирование алкенов в присутствии других анионов или
нуклеофилов ведет к смеси продуктов реакции, содержащей как
113
дигалогенпроизводные, так и смешанные продукты присоединения
(сопряженное присоединение):
Н Н
R /H R4 | 7Х R | X
\с/ \с/ \с/
Y- -> | + | -1-М'Х-
W XR X/ | XR У/ | XR
Н Н
Например, при хлорировании алкенов в водном растворе обра-
образуются хлоргидрины (хлоралканолы):
6-
R— СН^СН24-С12 + Н2О —» R—СН—CHnCl + HCI
он
Эта реакция может быть рассмотрена и как присоединение хлор-
хлорноватистой кислоты, которая образуется в водном растворе хлора
(С1НОНОС1НС1
R—CH-CH2 + Il6 —О—,-R-CH.j—СН2С1
г) Взаимодействие с ионами металлов.
Алкеиы образуют я-комплексы с ионами металлов, содержащих
незаполненные орбитали, например:
CH2=CH2+PdCb.^±CH2=CH2.PdCl2
СН2- CH2 + K2PtCI4 —> KPtCl3(CH2 = CH,)-KKCI
соли Цензе
Образование таких комплексов объясняется доиорно-акцептор-
ным взаимодействием, где алкен выступает в качестве электроно-
донора, а ион металла — в качестве электроноак-
м+" цептора. В результате взаимодействия изменяется
характер связи С—С, на углеродных атомах появля-
я-комплекс ется некоторый эффективный положительный заряд.
Характер связи в ^-комплексах металлов сложен. Здесь осущест-
осуществляется также дополнительная дативная связь (гл. Х.Д.З).
В образовании я-комплексов с ионами, а также атомами пере-
переходных металлов алкены выступают в качестве двухэлектронного
лигапда (гл. Х.Д.2).
л-Комплексы алкенов могут быть промежуточными продуктами
во многих важных реакциях.
Особое место занимают реакции с солями ртути. В присутствии
воды образуется продукт сопряженного присоединения — ртуть-
органическое соединение (гл. Х.Б.З) с гидроксилыюй группой:
ОН
R—CII = CH2 + Hg(OOCCII3J+H2O —* R—CH—CII,ngOOCCH3+CH3COOH
3. Гидроформилирование алкенов (оксосинтез). Алкены реаги-
реагируют с оксидом углерода и водородом под давлением в присутст-
П4
вин кобальтовых катализаторов. В результате реакции образуются
альдегиды, в отдельных случаях кетоны:
Кят.; 90-150 "С /О
II, 1О.зомПа^СН-С112С<и
Реакция была открыта О. Роэленом при исследовании процесса
Фишера — Тронша в присутствии алкенов A938) и имеет важное
промышленное значение.
Катализатором оксосинтеза обычно является металлический
кобальт, осажденный на пористом носителе, или соли кобальта.
Показано, что действующим катализатором является тетракарбо-
нилгидрид кобальта НСо(СО).,, который обладает кислыми свойст-
свойствами (по кислотности находится между HNO3 и НО):
L^ Н*-Со(СО4)
Механизм реакции гидроформилирования сложен. Предполагают, что
тетракарбонилгидрид кобальта может присоединяться к двойной связи как
сильная кислота:
НСо(СОL + СН2--=СН2 ->СП3-СН2-Со(СОL
Второй возможный путь — образование л-комплекса и внедрение алке-
на по связи Н—Со:
НСо(СОL + СН2=СН2 Т—*: HCo(CO)j + СО • *• СНа—СН2—Co(CO)i + СО
сн2=сн2
Образуется кобальторгаиическое соединение, по связи С—Со которого может
внедряться молекула СО:
,О
СН3-СН2~Со(СОK+СО —> СН3-СНа —С
Это соединение (ацилпроизводное кобальттрикарбоиила) расщепляется тет*
ракарбонилгидридом кобальта:
,0 .О
СН8-СН»-С^ +НСо(СОL-*СНзСН2-С^ +Со8(СО),
Катализатор в условиях реакции регенерируется:
Со2(СО),+СО + Н2 -* 2НСо(СО)^
Реакция гидроформилирования широко используется в промыш-
промышленности для получения альдегидов, а также первичных спиртов.
Например, из пропена получают масляный и изомасляный альде-
альдегиды, гидрирование которых дает спирты:
со, iij /P /О
СН3-СН= СН2-— -> CH3CH.2CHaC<f +СН3СНС^
со , \и I \и
Nt СНЯ
i H'|Ni
сн3сн2сн2сн2он сн3снсп2он
бутиловый спирт |
СНз
ИЗобуТИЛОБЫЙ СПИрТ
115
При повышении реакционной температуры оксосинтеза можно
получить спирты без выделения альдегидов.
4. Реакции окисления алкенов. Алкены легко окисляются.
В зависимости от окислителя и условий реакции образуются раз-
различные продукты: двухатомные спирты (гликоли), эпоксиды, аль-
альдегиды, кетоны, карбоновые кислоты. Обычно окислители взаимо-
взаимодействуют с двойной связью. В определенных условиях окисление
затрагивает аллильное положение.
а) Окисление КМпО4 и СгО3. Алкены окисляются
КМпО4 в водных растворах в слабощелочной среде. Фиолетовый
раствор меняет окраску, выделяется коричневый осадок (МпО2)
(реакция «на ненасыщенность»). В реакции образуются гликоли:
КМпО,
RCH = CHR ——-RCH-CHR
ОН ОН
Реакция была открыта Е. Е. Вагнером A888). Эта реакция про-
происходит стереоселективно как цис-присоединение двух ОН-групп:
н
R4 ,Н R* У/Н R
|| + ^Мп " | Мп "
с о^ хо-к+ А—о' хо-к+
н/ 4r н/ Ч н
но\ ^°
2 Мп *¦ K;MnO, + МпО2 + 2Н2О
При взаимодействии с СгО3 в растворе уксусной кислоты алке-
алкены расщепляются по двойной связи с образованием альдегидов или
кетонов. В условиях реакции альдегиды окисляются дальше до
карбоновых кислот:
/R сю, R4 /R его, R
ЧСО + О С<
C--Cv ^ ЧС=О + О = С< ^ )C-O-!-RCOOH
R/ ЧН R XH R
б) Окисление перо к с и кис лотами и кисло-
кислородом. Алкены легко реагируют с нероксикислотами, например
с перокснбензопной кислотой (с. 597), и образуют эпоксиды: .
/°
RC[I-CHR-J-C0H-,C/ —* RCH—CHR+C0HjCOOII
-0он
0-о-н
\0
/
Реакцию открыл Н. А. Прилежаев A909). Пероксибензойиая кпе-
лота часто называется реагентом Прилежаева.
При взаимодействии алкенон с кислородом или воздухом в при-
присутствии катализатора тоже могут быть получены эпоксиды:
RCIb- CHR —-RCH —CHR
Кат. ч
о
116
Эта реакция нашла промышленное применение для получения
оксида этилена.
В качестве окислителей могут быть применены также гидропе-
рокскды в присутствии катализатора (с. 337).
в) Озонирование. Алкены очень легко реагируют с
озоном и образуют взрывчатые продукты присоединения — моль-
озониды и озониды:
I !
RCHr-CHR + Оз ->RCH — CUR
мсиьозонид
Мольозонид перегруппировывается с разрывом связи С—С в
озонид. В зависимости от типа заместителей озониды могут иметь
шести- или пятичленный цикл:
/°ч
R4 /0-0, yR R / \ /R
Н/ -О-О7 Н RL/ Ч) —О7 XRL
озонмды
Озониды при взаимодействии с водой гидролизуются с образо-
образованием карбонильных соединений (RCHO и RR'CO) и перокснда
водорода.
Реакцию озонирования используют практически для установле-
установления строения алкена, так как озон расщепляет молекулу алкена
селективно по месту двойной связи.
г) Окисление в присутствии солей палла-
палладия. Алкены в присутствии солей палладия реагируют с водой с
образованием карбонильных соединений (альдегиды или кетоны)
и металлического палладия. Фактически Pd(II) окисляет алкен,
превращаясь в Pd(O):
—> CHj —C<f +Pd -I-2HC1
Известно, что этилен с PdCla образует я-комплекс, который со-
содержит активированную к нуклеофильным реагентам двойную
связь. Возможно присоединение воды и образование палладнйор-
ганического соединения, который распадается на уксусный альде-
альдегид (через енольную форму — виниловый спирт) и палладий:
г+с с сн
сн,—с
Эта реакция усовершенствована до промышленного способа
получения уксусного альдегида из этилена. Добавлением к реак-
117
ционной смеси CuCl2 достигается возврат палладия в реакцию. Про-
Продуванием воздуха регенерируется CuCI2 из Cud:
Pd + 2CuCl2 -+ PdCl2 + 2CuCl
2CuCH-2HC14-1/2O2 —> 2CuCl2
Таким образом этилен окисляется кислородом в водном растворе
в присутствии солей Pd и Си.
5. Взаимодействие алкенов со свободными радикалами. Алксны
присоединяют свободные радикалы с образованием нового свобод-
свободного алкильного радикала, способного вступать в дальнейшие реак-
реакции:
CH2=CH+R. —> .СН2—CH2R
|СПг = СН2 JHR |R- ]-CIIjCM2R
•CH2CH2CH2CH2R CH3CH2R-|-R. RCH2CH2R RCH2CH2CH2CH2R
Может происходить рекомбинация радикалов, отрыв водород-
водородного атома от других молекул при присоединении алкилыюго ра-
радикала к молекуле алкена. Этим начинается процесс олигомери-
зации и полимеризации. Возможно также диспропорционироваиие
алкилыюго радикала: 2RCH2—CH2--HRCH=CH2+RCH2—CHa.
Направление присоединения свободного радикала к молекуле
несимметричного алкена зависит от стабильности образующегося
нового свободного алкильного радикала. Более стабильными яв-
являются те алкильные радикалы, которые имеют большую раз-
ветвленность. Так, присоединение НВг по свободнорадикальному
механизму к пропену происходит следующим образом:
Н —Вг—>-Н
СН3-СН =
(образование СН3СНСН2 менее вероятно)
Вг
СН3 — СН — СН2Вг-ЬПВг —* CFI.-1 — СН2 — СН2Вг + Вг<
Присоединение происходит против правила Марковникова.
В результате присоединения НВг к пропену по двум различным
механизмам — ионному и свободнорадикальному — образуется
два разных продукта присоединения: 2-бромпропан и 1-бромпро-
пан. Это яркий пример влияния механизма реакции на строение
образующихся продуктов.
6. Олигомеризация и полимеризация алкенов. Алкены способны
присоединяться к двойной связи, т. е. соединяться между собой с
образованием длинных цепей из углеродных атомов. Эти реакции
олигомеризации (димеризации, тримеризации, тетрамеризацни и
т. д.) и полимеризации осуществляются только в присутствии дру-
других веществ, инициирующих образование активных промежуточ-
промежуточных частиц (карбокатионов и карбанионов, свободных радикалов),
и в особых условиях.
Можно написать общую схему олигомеризации и полимериза-
полимеризации, но она является чисто формальной, так как не отражает меха-
118
иизма:
R-CH=C
R—СП=
Н
н
чн
-Н)с
r/
-н>
R7
R—СН = (
—СП-
R
= СН—СН—
i
R— СН=СН— СН— СП.—СН — СНа
/Н Н.
R — СП=С
ЧН
_ г—СН —
к
R—СН = СМ— Г— СН — СП2— 1 — СЫ —СНз
—1 — СН—СН.
[— СН — СН2— "I — CI
R \n-iR
R J» . R
а) Полимеризация в присутствии кислот.
В присутствии серной кислоты способны олигомеризоваться или
нолимеризоваться только алкены с несколькими донорными ал-
кильными группами. Впервые олигомеризацию изобутилена в при-
присутствии серной кислоты наблюдал А. М. Бутлеров. В зависимости
от количества серной кислоты и температуры можно получить ди-
мер, тример или полимер. Присутствие серной кислоты вызывает
образование т/?ет-5утилкатиоиа, который присоединяется по двой-
двойной связи изобутилена:
+
Н;С^ ^~ . СН +
Х=СН2 +^-—~~С— СНз
К,С^ СНз-^
CHj
СНз
СП,
;с—СН2—С—С1
-* Тример
CHj
I!
СНз
СН,
;С=СН—С—СНз + Н1"
llj- I
СНз
2,4,4-трим«тилпеитен-2
(диизобутилеи)
сн3
сн3
Дальнейшая полимеризация приводит к полиизобутилену:
СНз
— С—СН2 —
СН3
з
— С—
СН,
СНа
полиизобутилеи
Тргт-карбокатионы относительно более стабильны, чем вто-
вторичные и первичные вследствие электронодонорного действия трех
алкильных групп. Поэтому т^гт-карбокатионы могут образовать-
образоваться в реакционной среде в большей концентрации по сравнению с
другими карбокатионами.
119
б) Полимеризация в присутствии свобод-
свободных радикалов. Ал/еиы полимеризуются в присутствия
активных свободных радикалов, если созданы условия для продол-
продолжения цепи полимеризации:
R-CH-CH2X
+ RCH = CH,
R_CH-CH2-CH-CH2X >
R
(n-l) R-CH = CH,
RCH - CH2 — CH-CH2-CH-CH2X у
I I
R R
RCH —CH2 —f—CH—CH2— —CH—CH2X
R L R
Цепь полимеризации обрывается, если происходит димеризация
или диспропорционирование свободных радикалов:
CH2Ri
2R-CH-CH2-R1 -^ R-CH-CH-R
2R-CH-CH2R1 — R-CH
Труднее всего полимеризовать этилен. Полимеризацию осу-
осуществляют под высоким давлением —до 150 мПа A500 атм) в при-
присутствии небольших количеств кислорода как инициатора свобод-
свободных радикалов. Этилен в условиях реакции находится в жидком
состоянии. Так получают полиэтилен высокого давления с молеку-
молекулярной массой 20 000. . .40 000. Этот полиэтилен представляет
собой гибкую полупрозрачную бесцветную массу с температурой
размягчения 112. . .115 °С и плотностью 0,92. . .0,93 г/см3 (поли-
(полиэтилен низкой плотности):
(/i-i-2)CH2-CM2 — СН2==СН —[—СН2 —СН2—]„ —СНаСНз
Полиэтилен широко используется в технике в качестве электро-
электроизоляционного и упаковочного материала и для изготовления
различных изделий (пленка, трубопроводы и др.). Полиэтилен ус-
устойчив к воздействию сильных кислот и щелочей, но обладает низ-
низкой термической устойчивостью и под действием солнечных лучей
и кислорода воздуха постепенно становится хрупким (старение по-
полимера).
Аналогично полимеризуя пропей, получают бесцветную вязкую
жидкость с температурой застывания около —35 СС — полипропи-
полипропилен высокого давления:
(л + 2)СН3СН-СН2 --!-СНзС1Ь- СН—Г—СН —СНг——СИ—СНЯ
_г_сН-СНг-1-
L СНз J,
СНЯ
в) Т е л о м е р и з а ц и я. Теломеризацней называется реак-
реакция олпгомеризации алкенов в присутствии вещества — передат-
120
чика цепи (телогена). В результате реакции образуется смесь олн-
гомеров (теломеров), концевые группы молекул которых представ-
представляют собой части телогена. Примером может служить реакция эти-
этилена с СС14:
Ииицматор
СС14 > -СС13 зарождение цепи
CHj-CHa + .CCI, —» .СН2СН2СС13 1
сн2- сна+.сн2сн2сс13-*.сн2снгснаснасс1, / пРад°л>ке"»е *сп"
CCI4+-CH2CH2CII2CII2CCI3 — •СС1Н-С1СН2СН2СН2СН,СС1;, передача цепи
Продуктом приведенной реакции-является смесь тетрахлоралкаиов:
Инициатор
пСН2- СН2 + СС14 -С1(СН2СН2)ПСС13
Реакция осуществляется в промышленном масштабе, тетрахлор-
алканы С1(СН2СН2)ПСС13 при п^З—5 являются исходными для
гидрокси- и аминокислот (с. 605,617).
г) Полимеризация в присутствии метал-
лоргаиических соединений.
Алкены взаимодействуют с такими металлоргапическими соеди-
соединениями, в которых атом металла имеет свободные орбитали, на-
например алюминий, .титан:
R2AI —R-!-nCH2 = CH2 —> R2Al(CH2CH,,)nR
Алкены внедряются по связи С—А1 и цепь углеродных атомов рас-
растет. При более высоких температурах алюминшюрганические сое-
соединения распадаются с отщеплением молекулы алкена (гл. Х.В).
Алюмипийорганические соединения в смеси с TiCl4 образуют
более активную систему катализаторов, которая способна легко
полимеризовать алкены до полиалкенов с высокой молекулярной
.массой. Активными частицами здесь являются титанорганические
соединения (гл. X.D.1), например:
сн,=сн.
R —Т|'С1Я + СН2--.СН2 — RCH-.CH2TiCI3 : + RCH2CHaCII2CH2TiCl3 —>
(г1-2)СН.,-С11„
: -" R(CH2CH.>)nTiCI3 — RCCIIaCH-^n.tCHiCHi!- -|-TiCI3
I —>• Дпспропоршюнпроваппе,
рекомбинация
Эти катализаторы были открыты и Енедрены в промышленность
К. Циглером и Дж. Натта A933—1955), поэтому часто говорят о
катализаторах Циглера — Натта.
Полимеризация этилена на катализаторах Циглера — Натта
осуществляется легко при умеренном давлении и дает полиэтилен
с большой молекулярной массой A00 000—1 000 000) с температу-
температурой размягчения 125—130 СС и плотностью 0,95—0,97 г/см3. Этот
полимер называют полиэтиленом низкого давления (или высокой
плотности). По своим техническим свойствам он лучше полиэтиле-
полиэтилена высокого давления, но быстрее стареет.
Полимеризация пропена на катализаторе Циглера — Натта
дает полипропилен с температурой размягчения 160—170 СС. Та-
121
кое резкое различие в свойствах полипропилена низкого и высо-
высокого давления (температура застывания —35 °С) объясняется не
только увеличением молекулярной массы, но главным образом
пространственным строением макромолекулы полимера.
Дж. Натта предвидел, что полимеры с регулярным строением
макромолекулы должны обладать более высокими температурами
плавления. Полимеризация пропена под высоким давлением дает
макромолекулы полипропилена с пространственно нерегулярно
расположенными СН3-группами (атактический полимер). Полиме-
Полимеризация же на катализаторе Циглера — Натта осуществляется сте-
реорегулярно, СН3-группы расположены регулярно, по одной сто-
стороне углеродной цепи (изотактический полимер):
СН, Н СИ, Н Н СН3 Н СИ, Н СН3
V / \ / \ /' \ / V /'
Чн/ \н/Чн/Чн/Чн/Ч„/
атактический полипропилен
СНз Н СН, Н СНз Н СНз Н СН, Н
vc/ \с/ \/ \у v /
ЧЧЧЧЧ
иэотактическик полипропилен
7. Реакции циклоприсоединения. Алкепы способны вступать
в реакции, в которых образуется цикл — происходит циклоприсое-
динение (см. Введ. 7.2).
Реакции подразделяют в зависимости от числа атомов, вступаю-
вступающих в образование цикла (цифры в квадратных скобках):
а) Циклоприсоединение [2+1]. К этому типу мо-
могут быть отнесены реакции алкенов с карбепами, в которых обра-
образуются производные циклопропана (см. гл. V.A):
н /н
X R-C< /X
Отчасти к циклоприсоединению [2-J-l] принадлежат реакции
образования эпоксидов (гл. XVfI.B.2).
б) Циклоприсоединение [2-J-2]. Димеризация ал-
алкенов с образованием производных циклобутана происходит толь-
только под действием ультрафиолетового света (фотохимически):
II R
хс/ hv R-C-C-H
II -I- II -¦*• I I
,СЧ .С. R-C-C-H
W XH R/ ХН | |
И R
122
Возбужденная молекула алкена присоединяется к другой не-
невозбужденной молекуле (см. гл. V.B).
в) Циклоприсоединенне [2+4]. Алкеиы способ-
способны взаимодействовать с сопряженными диенами и давать произ-
производные циклогекссна (диеновый синтез, см. гл. III.Б.3 и V.T):
Н
Н
R
\H
8. Реакции алкенов в аллильном положении. Реакции алкенов,
протекающие по свободпорадпкалыгаму механизму, могут прохо-
проходить также у углеродного атома, который находится рядом с двой-
двойной связью (аллилыюе положение).
а) Автоокисление. Взаимодействие алкенов с кисло-
кислородом воздуха по свободнорадикальному ценному механизму, ко-
которое приводит к гидропероксидам и продуктам их дальнейшего
распада, называют автоокислением. Реакция инициируется воз-
возникновением свободных радикалов аллильпого типа и. продолжа-
продолжается до их исчезновения:
СН2--=СН—CII2RJ-A. —>CH2 = CH—CIIR + HA
СН2 = СН—CHR + O2 —> СН2^СН — CHR
О —О<
СН2=-.СН—CHR+CHa--=CII— CH2R —> CII2-CH-CHR -i-CHa-=CH—CHR
I | яллплып.ш ради-
O — Oi О — ОН кал продолжает
цеш, аптоокисле-
гидропероиепд 1[пя
{
Продукты распада
Процесс образования аллильных свободных радикалов может
быть конкурирующим присоединению по двойной связи:
А —СН2 —СИ —CH2R <— CII2 = CH— CH2R + A« —> CII2 = CH— CHR + НА
б) Изомеризация. При температуре выше 500 °С на-
наблюдается изомеризация алкенов:
СН2-СН —СНаСНз-1^—* СН3СН = СНС113-!-СН3С = СН2
СН3
В этих реакциях образуются свободные радикалы, например
СН2=СН—СНСН3, которые способны к перегруппировкам.
в) Аллильное г а л о г е н и р о в а п и е. В присутствии
специальных бромирующих реагентов и инициаторов свободпора-
дикальных реакций (УФ-света, температуры, пероксидов) достига-
123
ется бромированне алкена в аллнльном положении:
бромируюишИ реагент; fev; t°
CH2-CH-CH2R >CH2 = CH-CHR
Вг
Классическими реагентами аллилыюго бронирования являются соеди-
соединения, способные гомолитическп расщепить связь Вг—X, например К-бром-
еукцинимид. Реакция начинается образованием аллилыюго радикала, что
вызывается инициатором (светом, активным свободным радикалом, который
генерируется в реакционной смеси термически или светом):
ипипилтор
CII., = CH-CH2R > CH2==CH-CHR
о. ох
СН2=СН —CHR-f Br-X'x~
¦СН2 = СН —CHR-I- -N
ч
Вг
N бромсукиинимид
°ч °
CH2==CH-CH2R+ -N
., = CH-CHR-bH-N'
Источником атома брома может быть и молекула брома, возникающая
при термическом разложении N'-бромсукциннмнда:
CH2-CH-CHR |-Br2--*CH2-CH-CHR j-Br.
Вг
^ Аллилыюе хлорирование пропена происходит при высокой тем-
температуре хлором:
500 СС
СН2=-СН-СН3 + С12 'CH2-CH-CH.2CI
Аллильные радикалы являются более стабильными, чем обычные
алкильные свободные радикалы, они дольше живут, легче обра-
образуются и вступают в различные реакции.
Стабилизация аллильных радикалов обусловлена сопряжением
неспаренного электрона с двойной связью. Говоря языком ме-
зомерии — резонанса, структура аллильного радикала изобража-
изображается двумя резонансными (мезомерными) формулами:
СН2=-СЫ — СН2<-> СН, — СН = СН2
Весьма наглядно эту систему можно характеризовать методом
МО.
9. Представление аллильного радикала, катиона и аниона методом МО.
Аллнльный радикал (катион, аннон) представляет собой простейшую сопря-
сопряженную систему, состоящую из трех углеродных атомов и трех р-орбитален.
Для характеристики такой системы можно применять простейший расчетный
квантово-химический метод, разработанный Э. Хюккелем (метод МОХ —
метод молекулярных орбпталей Хюккеля). Для лучшего понимания метода
ниже рассмотрен самый простой случаи — образование двухцентровой
л-евязи молекулы этилена.
124
а) Результаты расчета методом МОХ для л - с в я-
з и этилен а. Молекулярные орбнталн (.МО) л-свячи У у образуются при
комбинации двух атомных р-орбнталей: i|;, и ф2. Используется принцип ли-
линейной комбинации, т. е. суммирование с соответствующими коэффициента-
коэффициентами Cjr при атомных орбиталях. Этот коэффициент Cjr называется собственным
вектором и характеризует долю участия
й б МО
р рру д у
атомной орбнтали в МО, индекс / ука- -?-
зывает номер МО, а индекс г — номер
атома:
¦ ' У^спЪ + СпЪ; »
СН2-СН2 T^
— *
Помещая выражения V,¦ в уранпе-
пне Шредингера и решая это'уравнепие,
получаем значения энергии МО Е, и р 52 урОвни энергии л-связи
значения сооственных векторов с]Г для значения энергии
каждой МО. Значения энергии выража- '
ют в условных единицах а и fi. Здесь
а — кулоиовский интеграл, fi — резонансный интеграл, оба имеют размер-
размерность энергии (кДж/моль, ккал.'моль, эВ). Обычно в выражениях они фигури-
фигурируют со знаком минус, например кулоповскнй интеграл углеродного атома
ас -—11,2 эВ, резонансный интеграл л-связи С—Срс—С ="-—5,35 эВ.
Для л-связи этилена получаем две энергии орбнталей ?t и ?2 " Два вы"
ражспня МО для XV1 и Ч'2:
Можно начертить диаграмму уровней энергии (рис. 52).
Исходя \и значении ?у можно рассчитать важную величину л-электрон-
ных систем — полную энергию л-электронов ?я:
где п — число электронов в орбнтали @, 1, 2).
Значения собственных векторов cjr используют для получения таких важ-
важных параметров, как л-электронная плотность qr и порядок л-связи p,.s:
Яг ="- 2 "-/'•: Prs =-" 2 cJ'rcls-
Эти величины получают при суммировании по всем заполненным МО
квадратов с1Г или умножением Cjr и с/3 рядом расположенных атомов г и s.
Для системы этилена получается, что <7i=<?2=' " /'12"— '•
Кроме того, можно рассчитать величину, которая нлзывягтся свобод-
нон валентностью />. Ке получают, исходя н.ч суммы порядков ."г-связеп, ко-
которые образует рассматриваемый атом:
Для молекулы этилена Fy-F2— Y~3 — I -0,782. Величина \^3 :п;ляетс![
максимально возможной суммой порядков л-евлзей, которые может образо-
образовать углеродный атом.
Таким образом можем изобразить квантово-хнмпческне параметры
л-связп этилена:
I 1 1
СН? = СИ:
f. 732 0,'31
Можем 1акже нарисовать очертания МО л-связи этилена (рис. 53),
125
б) Результаты расчета методом МО для а л л и -
л ь и о г о радикала, Аллилышй радикал является системой с тремя
я-элсктронами:
з 2 .1
При комбинации трех АО: tyi, ifj и % — образуются три МО:
и Чг3 — и три уровня энергии: ?lt Е2 и ?"а (рис, 54),
——_ ИЛИ I 1 »— —. ИЛИ I J
^ Л Л ^<;?> ft I
C±.J
4<j (связывающая орби!аль) ч*2 (разрыхляющая орбиталь)
Рис. 53. Очертания МО я->
связи этилена
=«
Выражения для МО имеют вид:
1 V 1
Рис. 54. Уровни значения
энергии я-электронной си-
системы аллильного радикала
Полная энергия л-электронов аллильного радикала:
Один электрон находится на несвязывающей орбитали (Ч^). Эта орби-
орбиталь относится только к неспаренному электрону радикала и характеризует
его делокализацию.
Для сопряженной системы можно рассчитать энергию делокализации
?ДдЛ! которая характеризует выигрыш энергии сопряженной системы по
сравнению с аналогичной несопряженной. ?д?л получают как разницу между
вычисленной Ел и энергией такой системы, в которой нет взаимодействия на
л-электронном уровне. В случае аллильного радикала такая система пред-
представляет собой двойную связь и один песиаренпый электрон (энергия равна а):
Д Е^л = /Гя — (?СН2=СНа_|_ а) =о,82р.
Эта энергия делокализации показывает, что аллилышй радикал является
стабилизированной частицей в результате делокализации электронов.
Можно рассчитывать кваптово-химические параметры аллильного ради-
радикала.
1 о,7О5 1 о,7О5 г Следует обратить внимание на порядок я-связей:
(-и '-'—ГН-—СИ. г/"-<~0,705. Это меньше, чем для нормальной двойной
2 о * связи (prs^l). Такое явление характерно для всех сопря-
1 ' ' женных систем и связи могут быть названы делокализо-
126
ванными или нецелочисленнымп. Большая свободная валентность на ато-
атомах 1 и 3 свидетельствует о том, что именно здесь будут протекать реакции.
Очертания МО аллильного радикала приведены на рис. 55. Здесь высота
соответствующих «восьмерок» пропорциональна квадрату собсгаонного век-
Рис. 55. Очертания МО аллильного радикала:
а — 4f, (спязывагащая МО); б — Чг2 (несняиывающая |_МО); а—Чг3 (разрыхляю-
(разрыхляющая МО)
тора с"г, что характеризует вероятность нахождения электрона в данной МО
в районе данного атома (одноэлектронная плотность).
Орбиталь V.2 показывает делокализацию неспарепного электрона. Веро-
Вероятность нахождения его одинакова как на первом, так и на третьем углерод-
пом атоме и равна 0,5. Плотность неспаренного электрона па втором атоме
углерода равна нулю.
Изображение сопряженных систем связано с определенными
трудностями. Пользуются изображением дслокализованных свя-
связей при помощи пунктирных линий и указанием плотности электро-
электронов:
0,50
СН2—СН—gCH2
Иногда пользуются изогнутыми стрелками, обозначающими
сдвиг электронной плотности:
сн2=сн—
Можно писать две резонансные структуры (с. 124).
Подобная модель применима для рассмотрения других простых
сопряженных частиц аллильного типа — аллил-катиона и аллил-
аниона.
Аллнл-катион содержит 2л-электрона, и положительный заряд
делокализован по первому и третьему углеродному атому:
0,54- 0,5 f
СН2—СН —СН2
СН2 = СН—СН2
2 = СН — СН2
сн2—сн=сн2
Аллнл-аиион содержит 4л-электрона, два из которых находят-
находятся на несвязывающей орбитали и делокализовапы по первому и
127
третьему углеродному атому:
0,5- 0,5- г">^^\ /Т'~
СН2 —СН—СШ СН->=СН—СН2
2=СН—СМ2
5. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ
Этилен. Этилен принадлежит к наиболее важным алкенам, его про-
производят и используют в огромных количествах.
Этилен представляет собой бесцветный газ со слабым запахом;
его т. пл. —169,5 СС, т. кип. —103,8 °С, плотность (при темпера-
температуре кипения) 0,570 г/см3. Незначительно растворяется в воде
@,25 л в 1 л), лучше — в этиловом спирте C,59 л в 1 л), хорошо раст-
растворяется в диэтиловом эфире.
В промышленности этилен выделяют из газов пиролиза и кре-
крекинга нефти, которые содержат до 17. . .20% этилена. Разработан
селективный метод гидрирования ацетилена до этилена:
НС S СН Pd (силккагель. ИШ...Э20 "С) СН'2 = CUi
Промышленное значение имеет также каталитическая дегидра-
дегидратация этилового спирта. В лабораторных условиях этилен полу-
получается из этилового спирта и серной кислоты (гл. XIV.А.4). Что
экономичнее: этилен получить из этилового спирта или этиловый
Тиокол СН2=СН—CI *~ По.швинилх.'юрид Эиноламииы
носпгсн2С1 -^U- си,—ciij —»¦ о
Полиакрилоиитрил
Рис. 56. Применение этилена
128
Глицерин
СИ,—СН—СИ,
CHj—СН—CHjOH
ОН
Полипропилен
СИ—-С—CH.
СП, —С—CH=Cff2
СИ,
Синтетический
Рис. 57. Применение пропена
спирт из этилена? Это зависит от доступности и цен нефти и этило-
этилового спирта.
Этилен служит исходным сырьем для множества синтезов, как
это изображено на рис. 56.
Пропен (пропилен). Пропен принадлежит к широко используе-
используемым алкснам. Пропен представляет собой бесцветный га:; со сла-
слабым запахом; его т. пл. —185,3 С, а т. кип. —47,7 С. Плотность
d\u- 0,5139 (при давлении насыщенного пара).
В промышленности пропен выделяют из газов крекинга и пиро-
пиролиза при переработке нефтяных продуктов, которые содержат 5—
8"о пропена. При фракционировании выделяют смесь пропана и
пропена. Чистый пропей получается при дегидрировании пропана.
Пропен используется для получения ряда важных продуктов,
как это показано на рис. 57.
Бутены. Бутепы образуются в процессе пиролиза и крекинга
продуктов переработки нефти. Их отделяют от других углеводоро-
углеводородов перегонкой при низкой температуре п выделяют в виде бутан-
Таблию 10. Физические константы бутенои
„„„.„¦„
Бутен-1
1<//с-Бутеп-2
;н/юнс-Вутем-2
.Метилпропеп
[пзобутнлеп)
Т. пл., С
— 185,4
— 1.38,9
— 105,6
— 140,4
Т. кпп
—6
+3
-:¦•
—6
., "С
,3
,7
,0
,9
d-no
d-7S,o
"Г
. -0,740
- 0,724
¦0,6044
= 0,694s
Ко.ффнцчонт
I; |1С10\1Л0111Ш
1 ,3792
1,3916
1,3-:62
1,3811
5 St 517
129
бутеновой фракции, которая содержит близкокипящие бутан и
бутены (см. т. кип. бутанов и бутенов —табл. 7 и 10).
Бутены представляют собой бесцветные газы со слабым, раздра-
раздражающим слизистую оболочку запахом.
Бутеп-1 и бутены-2 используются в основном для получения
бутадиена-1,3:
На основе их могут быть получены также кислородсодержащие
продукты: бутиловые спирты, бутандиолы и др.
При полимеризации изобутплена получают полинзобутплец.
Димер нзобутилепа является ценным исходным для получения изо-
октапа — высококачественного моторного топлива:
ч
Каг.
Глава III
ЛЛКАДИЕНЫ
Углеводороды с открытой цепью углеродных атомов, содержащие
две двойные связи, называют адкадиснами. Общая формула алка-
диепов СпНгп_2. Этой общей формуле соответствуют также угле-
углеводороды с одной тройной связью — алксны.
В молекулах алкадиепов может быть различное расположение
двойных связей. Двойные связи могут располагаться рядом у одного
углеродного атома. Такие соединения называются алкадиепами-1,2
пли алленамп: СН3^С^СН2 пропадиеи-1,2, аллеи, а расположен-
расположенные так двойные связи называют куму.шрованными.
Между двойными связями в молекуле алкаднепа может нахо-
находиться одна ординарная связь:
СИо = СП —CIU=CH2 бутаднси-1,3 (дивинил)
СН3- С —С11= СН2 2-мстплбутлдис11-1,3 (изопрен)
СНЯ
Такие алкадиены называются сопряженными диенами.
Известны различные несопряженные алкадпены, в которых двой-
двойные связи находятся в положениях 1,4-, 1,5-, 1,6- и др.:
СН2 = СИ-СН2-СИ=СИ.. пептадиеи-1,4
СН2 = СЦ —СН2 —СН2-СН = СН2 гексадиеп-1,5
130
А. АЛКАДИЕНЫ-1,2 (АЛЛЕНЫ)
Аллены могут быть получены из дигалогеналканов отщеплением
атомов галогена при помощи цинка:
СИ а- С —CHj— Br-LZn -* СН^С^СНо
Вг
2,Я-дпбромпро1ге11
Иногда аллепы образуются из производных ацетилена в реак-
реакциях перегруппировки в присутствии щелочи:
коп
СН3—СИ —С- :С—Н
•си-,—с_-с= аи
170- С
СПз • СН3
З-метилбутип-1 З-метнлбутадмен-1,2
Аллены представляют собой бесцветные газы (С3, С4) или бес-
бесцветные жидкости со слабым запахом.
Своеобразно пространственное строение молекулы аллена. Че-
Четыре атома водорода молекулы аллепа находятся не в одной плос-
плоскости, а в двух взаимно перпендикулярных плоскостях. Все
три углеродных атома аллена размещены на
одной прямой:
Н ч
sp sp1 /
Н
Рис. 58. Образование
Углеродные атомы в молекуле аллена на- я-епязей в молекуле
ходятся в различных состояниях гнбрнднза- аллеиа
ции, sp2, sp, sp2. Средний углеродный атом
образует две двочные связи (рис. 58). Благодаря строению двуза-
мещенных алленов для них возможна стереоизомерия, например:
Структура I является зеркальным изображением структуры II.
Эти молекулы хиральны (см. Введ. 12).
Аллепы активны в различных химических реакциях, главным
образом в реакциях присоединения и полимеризации. В зтом от-
отношении они подобны алкенам. Центром электрофилыюй атаки
являются «боковые» углеродные атомы (зр'--гиГ,ридизова1шые).
Ниже приведена схема превращений аллена:
Н, Н Иг
2 = СН-СП3-
СНз—С—СНз
II
О
Каг.
СН2 = С=СН2 >CHi = C —СН3
и2о
СН2 = С—СП3
он
Вг
цпклодгмерпзрцвя
151
При нагревании и особенно в присутствии щелочного катализа-
катализатора аллены могут перегруппировываться в ацетилены. Настукает
равновесие, положение которого определяется термодинамической
устойчивостью компонентов:
R К '": ОИ-
)С = С=С/ -"R-CH,—C-C-R
IK ХН
Б. АЛ КАДИ ЕНЫ-1,3
I. МЕТОДЫ ПОЛУЧЕНИЯ
Алкадиеиы-1,3 получают различными реакциями отщепления как
из алканов или алкенов, так и из спиртов, гликолей и галогепнро-
изводных углеводородов. Здесь подробно будут рассмотрены методы
получения наиболее важных алкадиенов — бутадиена-1,3 и изо-
изопрена.
1. Получение бутадиена-1,3. Первым промышленным методом
получения бутадиена было термическое превращение этилового
спирта на катализаторе (Al2O3-:ZnO):
¦130-'!.'О °С
2СН.-.ОН "-СН.- СН-СН- СН2.-; 21-ЬО + На
К я г.
Этот метод предложил С. В. Лебедев A927). Выход обычно не-
невысокий. Предполагают, что на катализаторе этиловый спирт де-
дегидрируется до уксусного альдегида, который подвергается аль-
долыгай конденсации (гл. XXVII.А.3). Потом следуют превраще-
превращения альдоля.
Для получения бутаднена-1,3 применяется дегидрирование бу-
таи-йутеиовой фракции переработки нефти:
CH;!—C1L —СН2 —СНЯ — „„
СП,- СИ—СИ, —СН3 —
СИ.-, —СН-СП—СНз —
(цис- и транс-)
Сг2О3
^СН — СН^СНа |-П2
Возможно использование двухатомных спиртов (гликолей), бу-
тяпдпола-1,3 и бутандиола-1,4 (метод В. Репне):
2Ь0 ''С
СП, — СН — СП2 — СП2 >С11,-~. СИ — СП г, СН2Ч 2П2О
• , | Кат.
он он
п+ н;с-сп2гх,.Ю1
носп,сн..сн2си,он-—> I I > сп., = сн— сп = сн2
" - - -п,оц2с СН2 -П2о
о
2. Получение изопрена. Для получения изопрена применяется
дегидрирование изопентан-изопеитеновой фракции переработки
132
нефти:
сн3—сн—сн2—сн3—
I2- C-CH= CH2-i-H2
СНз
CH;) — С--СИ— СН,—
СН3
СН,- С — СН2 —СН3—
СНз
Изопрен образуется также каталитическим расщеплением 2-ме-
тнлнентепа . (димера нропена):
СН,-С-СН,СН..СН3— ->¦ СН2-С-СН--СН2-;-СН4
Кат. |
СН3 СН3
Изопрен легко получают дегидратацией ненасыщенного спирта:
ОН
I
СН3 —С-СН-СН2 _L,
Кат.
2-1 11,0
|
сн3
сн3
Исходный 2-метнлбутеп-3-ол-2 получают гидрированием про-
продукта присоединения ацетилена к ацетону (см. гл. IV.4) — метод
А. Е. Фаворского.
В основе одного из промышленных методов получения изопрена
лежит расщепление гетероциклического соединения — 4,4-диме-
тил-1,3-диоксана, который получают из изобутилепа и формаль-
формальдегида:
сн3, н.. нь
;с сн.,-1 2 ,о.:0—»
СН,
;
з-- |
Оч
О
''СП г
Кат.
~ о
—*сн2-¦с-сн---сн2-;-н2о-|
I н/
сн3
Мировое производство бутадиена-1,3 и изопрена превышает
несколько миллионов тонн в год, так как они используются в про-
производстве синтетического каучука.
2. ФИЗИЧЕСКИЕ СВОЙСТВА
АЛКАДИЕНОВ-1,3 И СТРОЕНИЕ
Алкадиены-1,3 представляют собой бесцветные вещества. Первые
члены гомологического ряда являются газами или низкокнпящнмн
жидкостями (табл. 11).
133
Таблица 11. Физические константы некоторых алкаднсиов-1,3
СП
СП
сн
Структурная
СГГЯ
Н;,С СП
(' ор'-у.та
аи
3
Т. т., "С
— 103,1
— 1 j(i
—7G
Т. к"
— 1
lii
С8
г., 'С
,5
,1
,0
0,
0,
о,
CSi
7?С2
Ко-
1,4
1,4
1,4
фс]. ""
391
Г 11 Т
5-С)
Для алкадпенов-1,3 характерна повышений я молекулярная реф-
рефракция. Так, например, для изопрена СН2 --QCH;,)—СН—С112
R^cn — 25,22. При расчете получается меньшая величина:
-il-: 2/?c-c-i
т.е. обнаруживается экзальтация рефракции: /?эксп — /?г,ор =
- 4,33. Экзальтация рефракции связана с увеличением поляризуе-
поляризуемости электронной системы, что характерно для сопряженных сис-
систем двойных связей.
Интересный вывод получается при сравнении эксперименталь-
экспериментальных значений теплоты образования молекул алкадненов-1,3 с по-
полученной суммированием термохимических энергий связей. Экспе-
Экспериментальная величина получается больше на 13. . .16 кДж/моль.
Эта разница в энергиях образования свидетельствует о том, что
при образовании сопряженной системы выделяется больше энер-
энергии, чем при образовании несопряженной системы. Оказывается,
что сопряженные системы являются более стабильными. Эту энер-
энергию называют энергией сопряжения (делокализации, резонанса).
Структурные исследования молекулы бутадиена методом электро-
электронографии показывают, что все ее атомы ле-
лежат в одной плоскости (молекула планарна),
наиболее вероятным расположением двойных
i связей является т/?а«с-расноложепие по от-
отношению к ординарной связи. Длины С—С-
связей отличаются от таковых в этилене
@,133 им) и этапе @,154 им).
Важные выводы о строении сопряженной я-электронной систе-
системы г.юлекулы бутадиена можно сделать из квантово-химнческнх
расчетов даже в самом простом варианте — методом МОХ (см.
гл.II — расчет аллилыюй системы).
В сопряженную систему бутадиена каждым углеродный атом отдает один
л-электрои С --С.—С = С. МО образуют линеарной комбинацией АО:
134
* с
н
бутадиен
При комбинации четырех АО получаются четыре МО (/=1, 2, 3, 4). По-
После решения ура^кечмя Шредн.тсру получаем чешге значения ?,- и четыре
выражения для Vj. На рис. 59 и 60 приведены уровни энергии и очгрта-
ния МО.
Из значения ?л мемекулы бу1адигна ?л=4а+4,472р и двух молекул
этилена ?л=4а+4(з ыожно«оцени1Ь энергию делокалигацни:
Д?*ел = Ел — 2С?'1'« = 0.4Г213.
Это лок.итьпа'т ст^илтппига молекулы в результате сопряжения (взаимодей-
(взаимодействия л--лек1ро.чов) и ccoibL'iciiiyeT лаемым, полученным при определении
/Г4 = « - 1,618/3
Рис. 59. Уровни энергии, значения
энергии Л\О л--'лектроппой системы
молекулы бутадиена-1,3
Рис, 60. Очертания МО молекулы
бутадиена-1,3
а = a -i 0,6 IS/J
'*->& €%^ #%^^
с2 0,137 0,362 0J62 0.1J7 0,362 0,1.17 0,1.17 0,.!62 0,362 0,137 0,137 0.)i-2 0.137 0,362 C,.i(.: 0,137
теплоты образонанип бутадиена. Четыре л-элсктроиа молекулы рл.'.мс.цаются
на двух заполненных МО, дне МО остаются незаполненными. Первая МО
(V)) полностью делокализоцапа, относится ко всем четырем углеродным ато-
атомам. Вторая МО (Чг2) имеет узловую плоскость между С2 и С3 и участвует в
образовании связи только между атомами С и С2, С3 и С. Чг2 является ныешем
заполненной молекулярной орбнталью (ВЗМО). Третья МО С?2) является низ-
низшей споСодноп МО (ИСАЮ). Четвертая МО {Ч'4) имеет три узловые плоскости.
ВЗМО и НСМО называются фронтальными орбиталямп; они определяют
реакционную способность сопряженной системы. Энергия ВЗМО но абсолют-
абсолютному значению соотнетствует энергии ионизации орбнтали н определяет элект-
ронодопорные свойства, взаимодействие с злектрофильиыми реагентами. Рас-
Распределение плотности электронен в орбпталп обусловливает местп атаки элек-
трофильных реагентов. Энергия НСМО определяет сродстьо к электрону,
т. е. электронодкцепторные свойства, взаимодействие с нуклеофнльными
реагентами. Распределение плотности электронов на МО характеризуется c\f.
В молекуле бутадиена наибольшие одноэлектрониые плотности во фрон-
фронтальных орбиталлх находятся на концевых углеродных атомах (см. рис. 60).
Это означает, что атака как электрофнльпых, так и нуклеофнльпы.ч реаген-
реагентов начинается с концевых (С1 пли С) углеродных атомов.
Суммарная л-электронная плотность qr па каждом атоме раина единице,
порядок л-связи /;„ для двойных связей меньше единицы, а для ординарной
связи С2—С3 значительно больше нуля.
135
Qr i 1 11 Порядок л-связеп свидетельствует; что для
prs (ДО 0,47 еда системы бутадиена характерны" значительно вы-
сн-—сн си—СН2 равненные (делокализованные) связи. Это под-
Fr ",84 0,37 0,3? 0,8* тверждается длиной связей.
Свободные валентности Fr показывают, что большей реакционной спо-
способностью должны обладать концеиые углеродные атомы сопряженной си-
cTevbt. ¦¦¦¦/.¦'.-.I- ¦ -¦¦¦+-. -,-,-.- . .
Киантово-химпческие характеристики л-электронной системы бутадиена
могут быть использованы для оценки электроподопорпых свойств и энергии
возбуждения.
В сопряженных системах ВЗМО имеет более высокую энергию, чем в не-
несопряженных (значение энергии имеет знак «минус»). Так как энергия иони-
ионизации равна энергии орбитали с обратным знаком, она меньше энергии пони-,
зацни несопряженных систем:
Евзмо эи, эв
СП.2 = СН2 а + р 10,5
СН.2 = СН —СН=СН2 а + 0,618р 9,1
Энергия возбуждения определяется разностью энергий E~-?R3MQ—
.^С и характеризует переход молекулы в первое возбужденное состоя-
состояние S0->-Si. При поглощении кванта света происходит переход электрона
с ВЗМО на НСМО. Для сопряженных систем ЛЕ меньше, чем для песо-
пряжеппых, и поглощение наблюдается при больших длинах волн.
Поглощение в УФ-слектре
эВ им
СН,-СН2 2Р 6,9-6,2 180-200
СНг-=СН-СН-СН2 1,2360 5,7-5,6 217-220
Имеются определенные трудности в правильном изображении
строения сопряженных систем с помощью классических структурных
формул. Поэтому иногда используется пунктирное изображение де-
локалпзованных* связей или изогнутые стрелки:
СН2^СП— С1[^СН2 Формула не показывает, что связь С2—С3 при-
приобрела частичный я-характер
сн/—"сн—сн—:сн2 Формула с полностью^ делокалнаованными
связями не показывает, что длины связей не
одинаковы и я-порядки связей различны
СН2=сн—сн=сн2 Показано взаимодействие я-связей
—сн=сн2 Показан сдвиг электронной плотности по систе-
системе сопряженных связей, который происходит
под влиянием внешних поляризующих факто-
факторов.
Ни одно из графических изображений структуры не дает пол-
полной картины, отражающей характер и природу связей сопряженной
системы. Это было уже показано на примере аллильной системы
(см. гл. П. 4.9). Только использование квантово-химических па-
параметров позволяет дать исчерпывающую информацию.
136
3. ХИМИЧЕСКИЕ СВОЙСТВА Л Л КЛДИЕНОВ-1,3
Для алкадтгснов-1,3 характерны различные реакции присоеди-
присоединения, в том числе важная реакция полимеризации, которая при-
приводит к синтетическому каучуку:
СП,
НС
НС
Л
^LciL- CH-CH--CH-,
СП.2
циклоприсоедн-
пепис (диомоньи!
синтез)
1,2-
полимерплацпя
I, (-
—>ХСП,—СН- СИ—CH,Y
1. ;-прмсоед:;нЛ!ге
- >ХСН,—СН —СП- СН2
... — СИ — СИ, — СИ — СИ, — СП — СП2 — ...
I
СП
п
сн,
I
СИ
СН
сн2 сн2 !
...— СН2— СП--СП— СН,— СП, — СН^СН — СН2— ...
Своеобразность алкаднеиов-1,3 состоит в том, что присоедине-
присоединение происходит в двух направлениях: 1,2- и 1,4-прнсоедпнепне.
Соотношение изомерных продуктов присоединения определяется
температурой реакции, полярностью' растворителя, характером
реагента.
1. Гидрирование. Алкадисны-1,3 каталитически гидрируются
с образованием алкепов и алкапов:
„ |-^СН,^СП — Cil..CH3
СН, - СН —СИ - СН2 -vf
' 1-^СНа —СП- СП—СП.,
2. Присоединение электрофильных реагентов. К алкадпепам-1,3
присоединяются галогеноводороды, галогены и другие электро-
фнльные реагенты. Эти реакции часто сопровождаются побочным
процессом — полимеризацией. В результате присоединения об-
образуются как 1,2-, так и 1,4-продукты. Здесь будет рассмотрено
только Премирование.
Известно, то при бромировапии бутадиена получается смесь
3,4-дибромбутена-1 и 1,4-днбромбутена-2. Их соотношение опреде-
определяется температурой реакции:
\;
'- СП;,СП;СН..СНЯ
, = СП—СП- СП,-|-Вг2
и растпоре СС14
ВгСП2 —СН—СН =СН,- -BiCH, — СП=.-СП— С1!,Вг
При .10 СС . . . 20 "о
При —80 "С . . . 80 %
80 %
20 %
При низких температурах получается больше того продукта,
скорость образования которого выше (так называемый кинетиче-
кинетически контролируемый продукт реакции). При высоких температурах
137
образуется термодинамически более стабильный продукт (термоди-
(термодинамически контролируемый продукт реакции).
Понятие «кинетический контроль» означает, что соотношение
продуктов реакции определяется относительными скоростями па-
параллельных реакций, приводящих к их образованию. Понятие
«термодинамический контроль» означает, что соотношение продук-
продуктов реакции определяется константами разновесыя их взаимного
превращения или взаимного превращении промежуточных про-
продуктов.
Реакция присоединения идет через образование КПЗ, который
может превращаться в конечный продукт аналогично алкенам (см.
гл. II. 4). В таком случае может образоваться только продукт
1,2-присоединения. Возможно превращение КПЗ в стабилизиро-
стабилизированный сопряженный карбокатион, который дает продукты как
1,2-, так и 1,4-присоединения. Образование промежуточного кар-
бокатнона более вероятно при повышенной температуре и в поляр-
полярных растворителях:
Вг2 вр
/^1т I f л\л ^_^ f~*IJ ^~^ (~* IТ "~"—™~—^^~ ^Н * • • * *(~* |_] с* и —*^~ {~^\-J
КПЗ
Вг"
Вг
BrClb—СН — СН=СН2 *- СН.,— СН—СН = СН2
I ВГ Вг
BrClb — СИ = СН — СН2 *- ВгСН.— СН = СН— ClbBr
Строение промежуточного карбокатиопа аллплыюго типа
может быть изображено резонансными формулами, как в схеме,
или точечными делокализоваиными связями н дробными зарядами:
5+ 5t- ,
ВгСН2—СН—СИ—СН2 5 + 5=1
Аналогично могут быть рассмотрены реакции с другими элек-
трофильными реагентами.
3. Образование я-комплексов металлов. Алкадиепы-1,3
очень легко образуют комплексы с производными металлов (соля-
(солями, карбонилами). Например, легко получается л-комплекс бутадие-
бутадиена с солями платины: КЛ(СИ2=СН—CH=^CH2)Pt2Clcl.
Важное значение имеет комплекс с хлоридом меди (I) CuCl —¦
желтое вещество состава [СН2—СН—СН—СН21Си2С!2. Легкое обра-
образование этого я-комплекса используют в практике для выделения
бутадиена из газовых смесей. Прп разложении комплекса получают
чистый бутадиен. Аналогичный комплекс образует изопрен.
В упомянутых комплексах алкаднены-1,3 связаны с двумя ато-
атомами металла.
Необычно стабильный я-комплекс образуется при взаимодейст-
взаимодействии бутадиена с карбонилами железа: [СН2—СН—СН=СН2] Fe(COK.
138
со Ct° со Рентгенографические исследования показали, что в
\1/' этом комплексе бутадиен существует в цис-форме и все
J связи С — С стали равной длины @,145 им). Бутадиен
здесь выступает как че*ырехэлектронпый лиганд (гл.
«-комплекс X. Д.2). Группировка Fe(COK находится над плоскостью
молекулы бутадиена.
4. Полимеризация. Ллкадиены-1,3 легко полимернзуются в при-
присутствии спободных радикалов или некоторых металлоргаиических
соединений. Полимеризация обычно происходит по свободноради-
калыгому или по анионному механизму. В присутствии щелочных
металлов осуществляется анион-радикальный механизм. В прин-
принципе полимеризация может идти и по катнопному механизму.
Обычно полимеризация происходит как 1,4-присоед1шение, но
с примесью 1,2-иолимера. В приведенных ниже реакциях показано
образование только 1,4-полимера.
Активный свободный радикал присоединяется к молекуле ал-
кадиепа-1,3 и образует новый сопряженный свободный радикал ал-
лилыюго типа, который относительно стабилен, но способен при-
присоединиться к другой молекуле алкадпена-1,3. Так возникает цепь
полимеризации:
R.-!-CH2.- СН-С!1-СНг—> RCH.2->CM-СН-СН2 __Г_1_1_4.
—>RCII,-CH--CII-Cn2CH,-CII^-C[I-Cn2 С1Ь-с"-С"=снд
2-CH-CII —СНгСН2 —СН--СП-СН2СН2-СН---СК-СН2
В промежуточно образующемся свободном радикале происходит
делокалплация песпаренпого электрона:
RCH2 — СП = СН — СП, ¦*—»- RCII2—CH —СН=СН2
г(о «со
Или RCH..—CII1^^CHIJ^CH2 f где S-i-S'-l'
Свободные радикалы из реакционной среды исчезают в резуль-
результате димеризации или диспропорцпопирования. Полимерная цепь
может быть изображена следующим образом:
RCH.,—сгь сн—с;г.:—[—сн2—сп = сн—сн2—]„—сн=-си—сн=снг
В общем случае кенцевые группы могут быть разными.
Свободные радикалы в реакционной среде генерируют разло-
разложением перокендов.
В присутствии металлического натрия осуществляется полиме-
полимеризация по анпои-раднкальному механизму. Анион-радикал бута-
бутадиена образуется при взаимодействии натрия с молекулой бутадие-
бутадиена, при этом атом натрия выступает как сильнейший электронодо-
нор:
СН2= СН —СП=-СП2-;- .Na —> [СП2 = СП — СИ = СИ2]- Na +
Неспареппый электрсп использует НСДЮ бутадиена (Ч?9) и являет-
является делокалнзованпым. Наибольшая плотность его на перпем и чет-
четвертом атомах углерода Анион-радикал присоединяется к другой
139
молекуле бутадиена и вызывает полимеризацию по радикальному
механизму. Полимеризация обычно осуществляется на поверхнос-
поверхности металла, причем образуются значительные количества 1,2-по-
лпмера.
Полимеризацию инициируют металлорганнческие соединения.
Например, натрий- или литийоргаиические соединения инициируют
полимеризацию по карбанионному механизму. Активной частицей
продления цепи полимеризации здесь является карбанион:
CH^CH-CH-CI-b-l-Na^R--^ :CHo-CII-CH-CII2R
Na + СН2=СП-СН=СМ2
Ка-.СН,—CH=CH— CH2—C1L—CII=CH— CH2R —* ...
Карбанпоп является сопряженной частицей аллильного типа,
правильно его структура изображается делокализованнымп связями:
СН. —СН—СИ—CH2R
= О
Реакции полимеризации с карбаипопным механизмом принадле-
принадлежат к реакциям пуклеофнлыюго присоединения к алкадпенам-1,3.
На катализаторах Циглера — Натта (алюминий- и тптанорга-
иические соединения) алкадиены-1,3 полнмеризуютея очень легко,
при этом в основном образуются стереорегулярные полимеры.
Макромолекулы полиалкадиенов могут иметь различное прост-
пространственное строение:
:;>uiit:~ полни v,np<.'ic
цчс- гю.шичипрен
Здесь показаны етереорегуляриые цис- н /и/эанс-полналкаднепы.
В присутствии катализаторов Цнглера — Натта в основном обра-
образуются цыг-полиалкадиепы.
В процессе полимеризации по свободпораднкальному механизму
образуются полимеры с нерегулярной структурой.
При полимеризации алкадиепов-1,3 в смеси с производными
алкенов происходит совместная полимеризация (сополимернзация)
и образуются полимеры со смешанной цепью (сополимеры), напри-
например иолпбутадиенстирол (см. гл. VI. А.5).
5. Диеновый синтез. Алкадиены-1,3 могут присоединяться к
двойной (или тройной) связи с образованием циклического продук-
J40
та (циклоприсоединение [2-f4])'. Легко происходят реакции алка-
диенов-1,3 с соединениями, содержащими активированную двой-
двойную связь (акцепторные или донорнце заместители). Впервые та-
такую реакцию наблюдали немецкие химики О. Дильс и К. Альдер
A928) при взаимодействии бутадиена с л-бензохипопом или малеи-
новым ангидридом:
О
II О
С
1
с
+
НС
II
НС
ч
О
СН
li
СН
/
л-бснзохинок
Происходит Никлоприсоедипение. Такую реакцию называют
диеновым синтезом или реакцией Дильса — Альдера.
Предполагают, что первой ступенью реакции является образова-
образование КПЗ, затем следует согласованное циклоприсоединение (пе-
рицпклнческаи реакция, см. гл. V. Г):
Удается осуществить также диеновый синтез между двумя моле-
молекулами диенов-1,3. Например, изопрен может образовать цикли-
циклический днмер —дппентен:
•
1 -метил-4-изопропенил-
циклогексен (дипеитеи)
Иногда диеновый синтез может быть обратимым. При нагрева-
нагревании продукт присоединения распадается иа компоненты (ретродпе-
новын синтез).
4. КАУЧУК
Натуральный каучук представляет собой эластичную при низких
температурах, пластичную при более высоких температурах мас-
массу, которую получают из молочного сока латекса бразильской гевеи
(Hevea brasiliensis). В этом соке в виде эмульсии находится каучук.
Из эмульсии при нагревании получают эластичную массу — кау-
каучук-сырец. Это умели делать уже древние индейцы Южной Аме-
Америки. Первым европейцем, ознакомившимся с каучуком, был X. Ко-
141
лумб. Подробно свойства каучука описывал французский исследо-
исследователь Ш. Кондамин в 1735 г. Первое практическое использование
каучука нашел К. Макинтош A823)—при пропитывании ткани раст-
раствором каучука он получил водонепроницаемый материал. Наиболь-
Наибольшее значение имело открытие Ч. Гудьира A839). Он обнаружил,
что при обработке каучука серой или серусодержащпмп соедине-
соединениями получается материал с отличными механическими свойст-
свойствами — резина. Этот процесс был назван вулканизацией.
В настоящее время потребителями каучука и резины являются
шинная промышленность, производство изоляционных материалов,
обувная промышленность и т. д.
Исследования по установлению строения каучука и созданию
методов получения синтетического каучука длились более чем 100
лет. В 1826 г. М. Фарадей установил, что каучук состоит только из
углерода и водорода, а Г. Вильяме в 1860 г. при сухой перегонке
каучука получил изопрен. Строение каучука окончательно уста-
установил немецкий химик Г. Штаудингер A924). Оказалось, что кау-
каучук является полимером изопрена:
СН3—С = СН — СНо — [—СН.— С = СН —СИ.,—]„ —СИ =С—CIU- СИ..
I I I
сн3 сп3 сн3
где п==1500. . .2200 (молекулярная масса 100 000. . .150 000). На-
Натуральный каучук состоит из смеси молекул полппзопрепа раз-
различной длины. Позже на основе рештеноструктурного анализа было
установлено, что натуральный каучук имеет строение г^мс-полиизо-
преиа. В природе встречается также транс-нолинзонпен, называе-
называемый гуттаперчей, который имеет худшие механические свойства.
Уже начиная с 1878 г. изучалась полимеризация алкадиенов-
1,3 с целью получения синтетического каучука, так как было до-
достаточно данных о том, что каучук представляет собой высокомоле-
высокомолекулярный ненасыщенный углеводород. До 1916 г. были разрабо-
разработаны различные способы полимеризации в присутствии как Na
или К, так и свободных радикалов для неразбавленных алкадие-
иов-1,3 и для водных эмульсий. Но для обеспечения промышленного
производства синтетического каучука необходимы также промыш-
промышленные методы получения алкадиепов-1,3.
В 1916 г. в Германии был впервые разработан промышленный
метод получения синтетического каучука полимеризацией 2,3-
диметилбутадиена. Так называемый «метилкаучук» имел много
отрицательных свойств, и его в настоящее время не производят.
Первый промышленный метод производства бутадиенового кау-
каучука был разработан советским химиком С. В. Лебедевым в 1927 г.
Бутадиен он получал из этилового спирта, а полимеризация велась
в присутствии Na. В 1931 г. была выпущена экспериментальная
партия, а в 1932 г. началось промышленное производство. В период
с 1938 по 1942 г. крупное производство синтетического каучука
выросло в Германии и США. В Германии в основном получали со-
сополимер бутадиена со стиролом (впннлбепзолом).
142
Начиная с 1950 г. исследовалась возможность получения стерсо-
регулярпого каучука. Впервые стереорегулярную полимериза-
полимеризацию изопрена в присутствии лития наблюдал советский химик
A. Короткое. В последующие годы при использовании комплексных
катализаторов Циглера — Натта было разработано промышленное
производство стереорегулярпого полиизопрена. В СССР промыш-
промышленная партия этого каучука была выпущена в 1963 г. (каучук
СКИ-3). По свойствам этот синтетический каучук не уступал на-
натуральному.
В 1956 г. под руководством советского химика В. Долгонлоска
была разработана методика стереорегулярпой полимеризации бута-
бутадиена и получения quc-бутадиенового каучука (дивинилкаучук,
СКД). По эластичности СКД не уступает СКИ-3, но превышает его
в устойчивости при низких температурах и износостойкости.
В настоящее время производят не только изопреновый и бутади-
бутадиеновый каучуки. При полимеризации 2-галоген-1,3-бутадиепов по-
получают хлоропреповый или фторопреновый каучук. Известны и
другие сополимеры — бутадпеи-стирольпын, бутацпеп-нитриль-
ный каучук и др.
Мировое производство синтетического каучука превышает
5 млн. т. в год и имеет тенденцию к росту.
B. ПОЛНЕНЫ
Углеводороды с несколькими двойными связями в молекуле на-
называются полиснами. Одним из примеров полисное с песопряжеи-
иымн двойными связями является макромолекула каучука. Свое-
Своеобразными соединениями являются сопряженные полнены. Мето-
Методы их синтеза сложные. Многие полиены и их производные сущест-
существуют в природе.
В молекуле полнена с сопряженными двойными связями л-
электронная система становится подвижной. В результате понижа-
понижаются энергия ионизации и эиергня возбуждения молекулы. Многие
сопряженные полнены являются скрашенными соединениями. Это
связано с уменьшением энергии ВЗМО и сближением ВЗМО и
ИСАЮ (рис. 61).
В помидорах присутствует полнен ли:со::ин С.,„НГ,6. который при-
придает плоду красный цвет. Лнконпн найден также в других растениях
и в веществах животного происхождения. Ликопнп представляет
собой кряспо-фполетопое кристаллическое вещество с т. пл. 175 С.
Строение ликошша выяснил П. Каррер A931):
I II II /\
Можно отметить, что большие сопряженные системы весьма ус-
устойчивы к действию различных реагентов и полимеризация не па-
143
2/5-
-д-
-
ft-
Р-
4к
0,618
0,618
Ьаан
—
Г
-0,445
0,445
8*
• 0,347
0,150
0,347 ~
АЕ ... 1,236/? 0,890/? 0,6940 0,3000
эВ ... 5,7 4,65 4,1 2,75-2,6 ПЪгло'пептге в
им ... 217,8. 268 304 450-480 УФ спгкщах ¦,
Рис. 61. Диаграммы уровней энергии некоторЕлх полиенов и длипчоволновые
максимумы поглощения в электронш.гх спектрах
блюдается. В реакциях превращения происходят лишь на концах
цени. По-видимому, разрушение большой сопряженной системы
связано с определенным проигрышем в энергии.
Полнен ^-каротин С4оН5в распространен в природе, находится в
растениях, например моркови, и в веществах животного происхож-
происхождения. ^-Каротин представляет собой фиолетовое кристаллическое
вещество с т. пл. 183"С. Строение (J-каротина выяснил П. Каррер
A931):
\/\
\/ I
/Ч/Ч./ЧУЧ
Р-каротмн
ч
Максимумы поглощения каротина в УФ-спсктрах находятся
при 478 нм (е= 122 000) и 452 им (е--319 000).
Из ^-каротина в организме образуется витамин А (гл. X1V.B.3).
Глава IV
АЛКИНЫ
Углеводороды с тройной связью в открытой цепи углеродных ато-
атомов называют алкинами или ацетиленами. Их общая формула та-
такая же, как для алкадиенов,— СпИ.,п_.2.
144
I. ИЗОМЕРИЯ И НОМЕНКЛАТУРА
Названия алкинов образуют, заменяя в названиях алканов суф-
суффикс -ан на -ин. Для первого представителя ряда алкннов С2Н2
сохраняется тривиальное название ацетилен, поэтому иногда го-
гомологи ацетилена называют как замещенные ацетилены (рациональ-
(рациональная номенклатура) и тронную связь иногда называют ацетиленовой
связью. В табл. 12 приведены названия простейших алкинов.
Таблица 12. Простейшие представители алкинов
Структурная
СНз-С--С-
формула
н
СН,СН-> — С -.С— Н
СНз —С- С —СН3
сн3сн2сн2 —
СНзСНл-С^
СНз —СП —С
СНз
Си С — И
с-сиа
-=-=С-Н
Номенклатура
ИЮПЛК
Этнн, ацетилен
Пропни
БуТЕШ-1
Б\'ТШ1-2
rioEiTim-l
IIeiiTini-2
З-МетилОутпн-!
рационатьная
Ацетилен
МеПЕлацетилсн
ЭтплацетЕЕлен
Днметилацетплен
Пропилацстилен
Изопропилйцетнлен
Сокращенное
изображение
формул
у,:
Углеродная цепь алкпнов нумеруется таким образом, чтобы
тройная связь получила наименьшую цифру.
Названия остатков алкинов образуют, присоединяя суффикс
-ил, следовательно, алкшшл:
Н — Ст=С— эти пил
3 2 1
СП-) — Cs-nC— пропЕпн!л-1
3 2 I
Н —С ¦--¦iC —CHi— прош1пил-2 (пропаргнл)
2. ПОЛУЧЕНИЕ
Алкииы получают главным образсм реакцией отщепления. Ацети-
Ацетилен имеет несколько специфических методов получения. Ниже
приведена схема генетической связи между алканамн, алкепами и
алкинамн:
145
1. Прямой синтез. Для получения ацетилена может быть ис-
использован прямой синтез из элементов:
2С-ЬН2т=±Н-С=нС-Н
Равновесие этой реакции сдвинуто вправо только при очень
высоких температурах, выше 3000 СС. Реакция осуществляется в
электрической дуге между угольными электродами в атмосфере
водорода (М. Бертло, 1860). Реакция имеет только теоретическое
значение.
2. Реакции пиролиза (промышленный метод). Ацетилен полу-
получается при пиролизе метана или этапа (этилена):
СН3—СНз^-*СН2-СНа-!-Н2— >Н —С--С-П+2Н2
При нагревании метана до 1500°С образуется ацетилен. Время
нагревания должно быть очень коротким, так как ацетилен при
этой температуре быстро распадается на углерод и водород. По-
Поэтому необходимо быстрое охлаждение продуктов пиролиза.
3. Реакции карбидов (промышленный метод). В реакции кар-
карбидов металлов с водой образуются алкипы. Карбид кальция об-
образует ацетилен (Ф. Велер, 1862):
+11— С = С— Н— Са(ОПJ
Аналогично реагируют SrC2 и ВаС2.
Карбид кальция можно рассматривать как мсталлорганическое
соединение, содержащее сильно полярную связь углерод — металл:
•8 + S- S- Я f- S- S-
—Са— СЗС — Са — CSC —
s-,i ;«+¦
НО —11
Карбид магния Mg2C3 с водой образует проппн:
Mg2C3-!-4Ii20 —*CiI3 — С г.С— K-;-2Mg(OHJ
4. Реакции отщепления. Отщепление атомов галогена ^ от га-
логепалкаиов пли галогеналкенов является общей реакцией полу-
получения алкипов. Реакция происходит в присутствии концентриро-
концентрированной щелочи при повышенной температуре:
Н И
СНа —С—С —IH-2N'aJ0II-—+СЫ3—С^зС —H--2NaJBr--i-2H2O
I I
Вг Вг
1,2-дпСромггролагг
оп-
сн3—о си.л—>сн3-с —с—н
L
2-бромпропсн
Дигалогеналкаиы получаются из алкепов в реакциях присоеди-
присоединения. Так можно написать общую схему превращения алкепов в
146
алкины:
R-CH-CH-R^X,-
Н X
II <*н-
_C — C-R1 ^R-C^C-
I I
X II
Механизмы реакции отщепления описаны на с. 233, 242.
5. Алкилирование алкинов. При замещении водородного атома
в алкшгах могут быть получены замещенные алкины, удлинена цепь
углеродных атомов. Это возможно через металлорганичеекне про-
производные алкипов (ацетилениды) (см. гл. IV.4.3):
R -С = С—II —> R —С - . С- М —- R -С . --. C-R4-MX
(здесь М — металл, X — галоген).
В присутствии солей палладия происходит непосредственная ре-
реакция RC=eCH с R'X.
3. ФИЗИЧЕСКИЕ СВОЙСТВА И ПРИРОДА СВЯЗЕЙ
Алкины предстазляют собой бесцветные газы или жидкости. На-
Начиная с С17 алкины являются кристаллическими веществами
(табл. 13).
Таблица 13. Физические константы некоторых алкинэз
Структурная формула Т, пл., СС
I
Т. кип., СС
II—С-:С —II
СН3-С. С-Н
СИзСНо-С^С —II
СИ, —С-^С —СИз
—80,8A70 кПп)
— 101,5
— 130
—32,3
-83,8 (no2roiri;a)
-23,3
8,6
27,2
0,6200 (—8ГС
0,6785 (—27СС)
0,6080 (О'С)
0,6880 B5СС)
Для алкинон характерна большая молекулярная рефракция,
чем у алкенов:У?с=с—-4,17, Rc~c -—5,96. Зто свидетельствует о
большей поляризуемости тройной связи.
Термохимическая энергия тронной связи меньше, чем сумма
энергии трех ординарных связей (табл. 14).
Таблица 14. Термохимическая энергия
углсрод-углеродмых связей
Связь
С —С
с-с
с~ с
Энергии сиязи,
кДж/мо::ь
344
615
Ы2
Эпсрткя п расчете ка
одну сьязь, кДж/моль
344
307,3
270.7
147
Термохимические данные свидетельствуют о том, что тройная
связь менее стабильна, чем двойная. Сам ацетилен — термодинами-
термодинамически неустойчивое соединение. Жидкий ацетилен способен к
взрывообразному распаду на углерод и водород.
Энергия ионизации ацетилена больше, чем этилена. Это озна-
означает, что электрон на ВЗМО связан крепче, чем у этилена (энергия
орбнтали больше).
ЭИ ацетилена резк^ снижается при введении алкильных групп:
Углеводород . . СН3 — СН3 СН2^СН2 НС = СН Н3С—Сг-jCH
ЭИ, эВ 11,6 10,5 11,4 10,4
Молекула ацетилена пеполярна, но при введении одной алкиль-
ной группы появляется значительный дниольный момент. Это сви-
свидетельствует о большей поляризации молекулы, чем в случае за-
замещенных этиленов:
Соединение . . . СН3 —СН- СП2 СН3 — С = С — Н (СН3)зС— С = С — Н
ц, Кл-м-1030 . . 1,17 2,51 2,17
И, D 0,35 0,75 0,65
Алкильные электронодонорные группы поляризуют тройную
связь больше, чем двойную, углеродный атом в тройной связи имеет
большую электроотрицателыгасть, чем в двойной:
S +
V
Увеличение электроотрнцателыюсти углеродного атома np;i
переходе от этапа к ацетилену подтверждается увеличением днполь-
ного момента С1зязн С—II F<б'<;6"):
R
t-| 6+ RN Ь'- R г- f.-i-
Соединение R —С—Н ;С^СЧ л-+ R—C--C—II
: R/ N1
R
ц, Кл.м-10м 1,С0 2.10 3,51
|i,I) 0,3 0,03 1,05
Элсктроног|1афпческне исследования показали, что ацетилено-
ацетиленовая группировке1, имеет линейное строение (днгоиальный углерод-
углеродный атом):
0,105 нм P.I21 км 0,106 им
н с==с>-^ н
0,146 hv 0,121 нм 0,106 нч
СН; С^^^С II
Тронная связь значительно короче двойной связи.
Строение молекулы ацетилена объясняется исходя из представ-
представлений о s^-гибрпдизации углеродного атома (рис. 62).
В электронных спектрах поглощения алкины имеют полосу при
170—180 им (л->л*-переход), в обычной УФ-области они прозрач-
прозрачны.
J4S
ft юн ft'
Рис. 62. Обра ювание л-CBinciin молекуле ацетилена
В ИК-спектрах характерны валентные колебания связи С —С
при 2100—2260 см (обычно малоинтенснвиое поглощение) и
связи =С-П при 3270—3330 см.
В спектрах ПМР химический сдвиг атома водорода ^С—II ра-
равен 1,7—2 м. д.
4. ХИМИЧЕСКИЕ СВОЙСТВА
Алкины вступают в реакции присоединения как с электрофильпы-
Mii, так и иуклеофильньши реагентами, в том числе в реакции диме-
ризации, циклоолигомеризации, полимеризации и окисления (рис.
63). Важны реакции присоединения СО (карбонилнрование). В от-
ноос ^н ft хр ,ы
RCOOH + HCOOH
Рис. 63. Характерные реакции алкипов
лично от алкепов у алкииов с тронной связью на конце молекулы
(терминальные алкины) имеется реакцношюспособцая группировка
= С— Н н возможны реакции с участием этого «ацетиленового во-
водорода».
1. Реакции присоединения, а) Гидрирование. Алкины
присоединяют водород в присутствии катализатора (Xi, Pt, Pd).
Гидрирование осуществляется ступенчато:
И И
Клт. II. R1 .„ [ •¦
R-C^-C-R] + H2 >¦ .С- С —i-R-C-C-R1.
R- II Кат. | |
II И
149
Обычно реакция происходит нестереоселективпо и образуются как
цис-, так 11 т/мнс-продукты.
б) Взаимодействие с э л с к т р о ф и л ь и ы м и ре-
реагентами. Активность алкипов к электрофильным реагентам
(сильные кислоты, галогены, ионы тяжелых и переходных металлов
и др.) меньше, чем активность соответствующих алкенов. Наиме-
Наименее реакцпошюспособпым является ацетилен, его алкильные про-
производные обладают большей активностью. Это согласуется с вы-
высоким значением энергии ионизации ацетилена и более низкими
значениями ЭИ алкипов.
Здесь рассмотрены некоторые реакции с Н-электрофилами
(сильными кислотами) и присоединение галогенов. Гидроборирова-
ние см. гл_. XI.Л.
Реакции с Н-электрофилами. Взаимодействие с галогеноводо-
родами (гидрогалогепированпе) идет медленно, особенно для ацети-
ацетилена. Вероятно, это объясняется уменьшенным сродством тройной
связи к прогону:
Медленно II- , С1
н—с —с—н+п—а > ,с с
i I'' Н
Алкилпроизводные ацетилена реагируют быстрее и согласно
правилу Марковиикова (в соответствии с полярностью тронной
связи):
н3с-*-с=с—н + н — х
\
с
с=с
Как правило, происходит /иранс-присоедниепие.
Реакцию присоединения галогеноводородов к тройной связи
резко ускоряет присутствие солей одновалентной меди. Считают,
что в этом случае тройная связь активируется образованием я-комп-
лекса:
+
на
а,а
I
н
|
сГ н
н c.
v
|| н'
/\СГ
C1 H
и н
v
|| +
А
C1 H
аГсГ
Здесь фактически происходит пуклеофильпое присоединение к
тройной связи, активированной ионом металла. Поэтому многие
реакции присоединения к тройной связи, катализируемые ионами
металлов (Cu+, Hg2 + ), гогут рассматриваться как реакции нуклео-
филыгаго присоединения. Например, реакция с водными раство-
растворами серной кислоты происходит только в присутствии солей ртути
и в итоге получается продукт присоединения воды (см. ниже).
Присоединение галогенов. Реакция происходит медленнее, чем в
случае алкенов, при этом могут образоваться как транс-, так и
150
1{ыоднгалогеналкены. Например, бромировапие:
R. ,Вг R. yR1
" Г~ ~~* Вг/ Ч1^ В?/ хВг
Механизм реакции присоединения галогенов к алкниам до кон-
конца не выяснен. По аналогии с реакциями присоединения к алкенам
можно предположить, что сначала образуется КПЗ, в котором трой-
тройная связь активирована для атаки нуклеофилыгой частицей. При
этом происходит предпочтительно м/ганс-присоедипение:
с ^ «-ic f,- ^ с.---. с
с s+c Ег с '••¦•' с
I I У 6ь У \ г
i< r' R вг а
Не исключено, что в ходе реакции могут проявляться свободные
радикалы (например, в процессе Вг2->-2Вг-) н реакция присоеди-
присоединения пойдет по свободпорадикалыюму пути (см. ниже).
в) В з а и м о д с й с т в и е с н у к л е о ф и л ь п ы м и р е-
агентами. Ацетилен и его производные могут присоединять
такие нуклеофпльные реагенты, как вода, спирты, анионы карбо-
новых и других кислот, но в основном в присутствии катализато-
катализаторов— солей медн (I) или ртути (II). Только в отдельных случаях
при высоких температурах наблюдается пекаталитнческое присоеди-
присоединение нуклеофила (например, алкоксндов).
Ал кипы присоединяют воду в кислой среде в присутствии солей
ртути (М. Г. Кучеров, 1881). Ацетилен в зтон реакции дает уксус-
уксусный альдегид, а другие алгпшы дают кетопы:
Ilf;2- IK , R*
R—С—-С —R^'-IIaO—->¦ ЧС С — >RCH, — С — R1
ы^ я/ 'он , " п
о
Первичным продуктом присоединения является енол (гл. XV.А),
который быстро перегруппировывается в карбонильное соединение.
Вероятный механизм присоединения воды к ацетилену пред-
предполагает образование я-комплекса между ацетиленом и ионом рту-
ртути:
у
к—о: н
Н н Hgt х2- н н
<4 ,,+ „,- \/ н+ \/ СН]
.. 1!!^ g \ —*¦ II — II HgJ(-x2- —*- I'-Hg'+x'-
-о:> <с. н+ с с с
U " I " ¦¦/ \ •¦/ \ .// \ ,
н н н—о н Н~Я и 'Я- н н
н
151
Алкины присоединяют спирты как в присутствии солей ртути
или меди, так и в присутствии алкоксидов при нагревании. В этой
реакции образуются простые алкеннльные эфиры:
R-C- -iC-H-fR1—ОН —-г* /с--с(
11; io/ ХН
В случае ацетилена получаются виниловые эфиры R'OCH --CH:.
В присутствии алкоголятов или щелочи осуществляется чистое
нуклеофилыюе присоединение к тройной связи:
R4 .. Na' R'on
R — C^C — H + R1 — O-.-Na+ —X
* R4 ,H
—* ^C-C -|-R1O~Na +
XH
Ллкппы присоединяют карбоповые кислоты, при этом образуют-
образуются сложные алкенильные эфиры:
/О Пй2'- R4 H
R-C-C-H + R*-Cf —- A;-Cv
ЧОН П1" R"C— O/ ЧН
О
Уксусная кислота присоединяется к ацетилену с образованием
винилацетатаСН3СООСН=СН2 (см. гл. XXXIII.В.4).
В результате присоединения к ацетилену цианистого водорода
образуется нитрил акриловой кислоты (акрнлонитрнл):
в.- б- Нч Д1
Н —С--С—Н + П-С ..-\— > )С С
IV С- N
г) К а р б о н и л и р о в а и и с. В присутствии никелевых ката-
катализаторов алкнны, в основном ацетилен, реагируют с оксидом
углерода (В. Реппе, 1944—1949). В этой реакции в случае ацетиле-
ацетилена образуются акриловая кислота или ее производные (эфиры, ами-
амиды):
О
— С=^-С —H-1-CO-j-HX * ;С С.
NHCO), Н. ,С— X
IV
(здесь Х=ОН, ОС2Н5, NH2).
Реакцию катализирует тетракарбонил никеля, который активи-
активирует тронную связь в результате образования л-комплекса:
н н
III + Ni(COL ^Г=± -,ll|-*Ni(CO)j + CO
С г+С
152
Затем следует присоединение СО. Предполагают, что в качестве
промежуточного продукта образуется циклопроиенон, который реа-
реагирует с водой, спиртом или аммиаком, присутствующими в реак-
реакционной среде:
* - ^
'с ^c vx
те
д) Реакции а л к и н о в со свободными ра-
радикалами. Свободные радикалы легко присоединяются к трой-
тройной связи с образованием нового свободного радикала, который
способен вступать в дальнейшие свободнорадикальиые реакции:
R_CsC-R + X.-^ ХС--С"
ЧХ
^С:-С +Х —X— С- С7 4-Х-
¦¦¦х х/ хх
Промежуточным продуктом здесь является свободный радпкат
нового типа — алкенильный радикал, у которого неспарепнын
электрон локализуется в sp2-rA6-
ридпой орбитали (рис. 64).
В результате присоединения
свободного радикала образуется
смесь двух изомерных алкениль-
ных радикалов, которые приводят, Рис 64. Ичочеры свободного ал-
в свою очередь, к образованиюсме- кемильного радикала
си цис- и /п/;йнс-изо\теров конеч-
конечного продукта. В этом заключается принципиальное отличие гемо-
гемолитической реакции присоединения к тройной связи от элсктро-
фильного присоединения. В последнем случае более вероятно транс-
нрисоединение.
е) Реакции окисления тройной связи.
Сильные окислители (пермангапат калия, дихромат в кислой среде,
озон) реагируют с алкннамн. Конечным продуктом реакции явля-
являются карбоновые кислоты, происходит расщепление молекулы но
тройной связи. В отдельных случаях удается изолировать продукты
частичного окисления — а-дикетоны:
- >R— COOH4-R1 — СООН
О О
2. Димеризация, циклоолигомеризация и полимеризация алккнов.
В присутствии катализаторов алкипы могут образовывать димеры,
циклические тримеры и тетрамеры, линейные полимеры. Роль ката-
катализаторов заключается в активации тройной связи и пространст-
пространственной ориентации алкннов в реакциях циклизации.
153
Ацетилен легко димеризуется в кисло» среде в присутствии ио-
ионов Си+ (Ныоленд, 1931):
н—с=с—н + к—сннс—н
Си '
н1
Эта реакция рассматривается как присоединение молекулы аце-
ацетилена (пли ацетнленид-попа) к активированной тройной связи.
Образуется также дивинилацетилеп СН2—СН--О-С—СН СН2.
Циклотримеризация алкпнов приводит к бензолу и его производ-
производным. Реакция осуществляется термически, в присутствии концент-
концентрированной серной кислоты или лучше всего в присутствии метал-
лоргапичеекпх катализаторов (соединения Сг, Ni.Co); труднее всего
реагирует ацетилен:
СНз
I
I—С--^С—Н
H;iC ' ^ ХСН,
1, 3, О-триметплСспмл
R1 R1
3R— CiiiC — R1-
Кат.
R1
При циклотетрамеризации ацетилена па катализаторе Ni(CNJ
образуется циклооктатетраен и другие продукты (бензол и высшие
циклоолигомеры) (В. Репне, 1949);
4H-CSC-H
Кат; 80-120°С
1,0-1,5 МПа*
В присутствии инициаторов (свободных радикалов) или особен-
особенно легко в присутствии специальных металлорганнческих катали-
катализаторов алкипы полимеризуются. В результате образуются сопря-
сопряженные полиены (полисопряжепные системы):
(л+2) R—С = С—R1
Кат.
\
с = с
\
R Я2
\ /
,с=с
Такие полисопряжепные системы в виде порошка или пленки
обладают своеобразными электрофизическими свойствами: повы-
154
шенпой электропроводностью, способностью повышать электропро-
электропроводность при освещении (фотопроводимость). Такие материалы на-
называют органическими полупроводникам^. При обработке их пара-
парами галогенов или другими сильными электроиоакцепторами (AsF5)
электропроводность резко повышается и материалы приобретают в
определенном интерзале температур металлические свойства (ор-
(органические или синтетические металлы).
Своеобразная полимеризация возможна в случае ацетилена в
присутствии ионов Си+ и окислителей:
пП-С ^с-Н-^* Н-[-С^С-]„-Н
Молекула полимера состоит практически только из углерод-
углеродных атомоз в состоянии s/ьгибридизации. Доказано, что в поли-
полимерной цепи встречаются только кумулированные двойные свяли:
...С—О--С—С=С-=С--... Этот полимер, названный карбином, может
быть рассмотрен как новая аллотропная модификация углерода
(рядом с алмазом и графитом).
3. Реакции по связи —-С—Н. а) Образование ацети-
ацетиле и и д о в. Благодаря значительной полярности связи С—Н в
присутствии сильного основания возможна ионизация ацетилени-
дов с образованием ацетиленид-иона:
й- fiJ-
R— С —С — H-i-:B^lR— C = C7 Ц-НВ +
В роли сильного основания могут выступать амид натрия ХаХН,,
металлоргапичеекпе соединения, иногда концентрированные раст-
растворы щелочей:
R— C^-C— II —
О- Й-i-
fi-
_С = С-MgX
:R — C=C:~Na ! -1-H2O
В присутствии щелочен ацетилеииды образуются только в не-
незначительных количествах.
Реакцию ацетилена и алкинов с мегпнйоргапичеекпмп соедине-
соединениями открыл Ж- Иоцич A902).
С ионами некоторых тяжелых металлов образуются малораство-
малорастворимые ацетнлениды, которые осаждаются пз водных растворов.
Например, из водных растворов солей меди (I) при пропускании
ацетилена выпадает красно-бурый осадок, мопозамещепные ацгти-
лепы образуют желтые ацетилепиды меди; ацетнлеппды серебра
являются бесцветными веществами:
Н—С^С —Н + 2Си+—>Си—С = С—Си + 2Н +
R—С—-С —Н+Си+ —¦ R— C--C—Си + П +
R-C^--C-H + Ag+—^R— С ---С —ЛЯ +11 +
Аиетнленнды меди н особенно серебра япляются термически
нестабильными соединениями. Производные самого ацетилена
взрывчаты.
J55
В ацетнленндах, как в настоящих металлорганнческнх соедине-
соединениях, связь углерод — металл может иметь различную полярность
в зависимости от природы металла. В ацетиленидах натрия можно
говорить об ионной связи, в ацетиленидах магния (реагентах Иоци-
ча) имеется силыюполярпая ковалеитпая связь, а в ацетиленидах
меди и серебра связь малополярна. Поэтому их реакционная спо-
способность уменьшена и они образуются в водных растворах. Малая
растворимость ацетилепидов меди и серебра в воде объясняется
также сильным межмолекулярным взаимодействием донорпо-ак-
цепторного типа;
t
R—С = С —Си
t
R—С = С — Си
t
R —С = С —Си
t
Лцетилениды используются в различных синтезах.
б) А л к и л и р о в а и и с и а р и л н р о в а и и е. Ацетиле-
ниды реагируют с галогенпроизводпыми углеводородов как нуклео-
фпльные реагенты и в реакции образуются замещенные ацетилены:
R—С-^С —M + R1—X-^R—Сг-С —ИЧ-МХ
В реакции могут быть использованы также ацетилеииды меди,
особенно при взаимодействии с иодуглеводородами. Иодарены всту-
вступают в реакцию арилировання алкииов только с ацетиленндами
меди. Присутствие солей Pd позволяет осуществить реакцию непо-
непосредственно между RC_- CH и RlX.
1) Галоген ирован и е. Взаимодействие алкииов RC -СН
с галогеном в присутствии щелочи дает RCi-CX (гл. IX.1).
г) П р и с о е д и н е и и с к альдегидам и к е т о п а м.
Ацетилен и монозамещенные ацетилены в присутствии КОН или
ХаОН присоединяются к карбонильной группе кетоиов с образова-
образованием ацетиленовых спиртов (А. П.Фаворский). В этой реакции аце-
тпленид-иои действует как сильный нуклеофильный реагент:
R—С С —Н4-КОН —VR—С С:-К'+П,0
Пг)С СИ;, СПЯ
RC^-C:-K'+ г —>RC--C-C—СН3—--
О6- I
U О-К +
СН3
—>RC--C — С — CU3
I
ОН
156
Наиболее широко используется реакция ацетилена с формаль-
формальдегидом в водном растворе в присутствии ацетиленида меди под дав-
давлением (В. Репне):
н
Н Кят. Н'
HC^CH-i- ус---О MIC-C—СНоОН
И' пропаргпловыП спирт
—*НОСН2 —С^С —С1ШН
бутлп-2-диол-1,4
д) О к и с л е и и е. Ацетиленид-ион способен к окислению (от-
(отдаче электрона) с образованием свободного алкинильного радикала.
При дпмеризации получаются диины:
[О] R-C^C
R—С = С;-М*- -R— С = С- > R — С ^ С — С = С— R
— е~
Практически реакция осуществляется кислородом воздуха в
присутствии солей Си (I). Рассмотренная выше реакция получения
карбина из ацетилена принадлежит к этому же типу окислительной
димернзации.
5. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ
Ацетилен является, бесцветным, легко сжимаемым газом. Слабо
растворим в воде A,15 : 1), в спирте F : 1), умеренно — в ацетоне
B5 : 1) и диметилформамиде C3,5 : 1). Особенно хорошо ацетилен
растворим в ацетоне под давлением. Это используют для хранения
и транспортировки ацетилена в баллонах. Баллоны наполнены
пористой массой и ацетоном, в котором ацетилен растворяется под
давлением 1,5. . .2,5 МПа A5. . .25 атм). Сжиженный ацетилен без
разбавителя крайне опасен ¦— может взорваться с большой силой.
Для получения ацетилена использует два метода. Реакция кар-
карбида кальция с водой применяется не только в лабораториях, но и
для получения ацетилена в больших количествах для сварки и нужд
химической промышленности. Для крупнотоннажного производства
используют термоокислительный крекинг и электрокрекииг мета-
метана. В электрокрекинге метан пропускают через электрическую дугу
между металлическими электродами с большой скоростью (около
1000 м/с), при температуре 1500... 1600"С. Выходящие газы быстро
охлаждают до 150...200 С впрыскиванием воды. Газы содержат по
объему 13% ацетилена и 45% водорода, остальное составляет пе-
прореагировавший метай.
Термоокислптельнып крекинг основан на термическом разложе-
разложении метана в результате воздействия теплоты, .выделяющейся при
сгорании метана в присутствии недостаточного количества кислоро-
кислорода:
6СН4 + 4О2—* Н—С -С —Н-;-8Н2 + ЗСО-|-СОН-ЗН2О
Температура газов в печи около 1500 С. Выходящие газы также
подвергаются быстрому охлаждению. Газы содержат по объему
157
РАСТВОРИТЕЛИ
чон
С1СН=СНС1
С1,С=СНС1
СН3С'(
Н;С С1Т2
Н0С1Г,—С==С—CHjOH
H,/Ni
- HOCH2CH2CH2CH2O1I
i°/icaT.
СН2=СН—СН=СН,
I '
СЦ,=СН—С—СИ,
. CI
ПОЛИМЕРЫ
Пол:шим;1лхяор:зд
По;шакрилнитрил
По.рн:акрилаты,
/юлмакрилам!!д
эфмры
Поливиинлацмат
Clllipi
СИНТЕТИЧЕСКИЕ КАУЧУКИ
->" ХЛОРОПРС1;0!:ЫЙ Каучук
-•- Byi адисновый ка> ;i>'K
Рис, 65. Применение ацетилена
8% ацетилена, 54,5% водорода, 26% СО, небольшое количество
СО о и метана.
Ацетилен из газовой смеси выделяется растворением в диметпл-
фору.амнде под давлением 0,8...1 МПа (8...10 атм).
Ацетилен производится в больших количествах, его годовое про-
производство в мире превышает 5 млн. т. Для промышленного органи-
органического синтеза используют около 70%, около 30% ацетилена рас-
расходуется для сварт'п и резания металлов (температура ацетилен-кис-
ацетилен-кислородного пламени около 3150:'С). Использование ацетилена пока-
показано на рис. С5.
Глава V
ЦИКЛОАЛКАНЫ,
ЦИКЛОАЛКЕНЫ И ЦИКЛОАЛКАДИЕНЫ
Углеродные атомы в углеводородах могут располагаться не только
в прямой или разветвленной цепи, но и образовывать циклы. Самы-
Самыми простыми циклическими углеводородами являются циклоалканы
с одним циклом, которые образованы от алкапов отнятием двух во-
водородных атомов п замыканием цикла. Общая формула для моно-
моноциклических цнклоалканов такая же, как для алкенов — С„Н2п.
Но известны также циклоалканы, содержащие в молекуле два, три,
четыре и больше циклов, соединенных между собой различными
способами. Циклы могут иметь один, два, три или больше общих
углеродных атомов. В результате общее число водородных атомов
уменьшается, и общая формула мпогоциклпчсских цнклоалканов
меняется от Сп\-1.1п_„ до СПН„. Известны также циклические угле-
углеводороды с одной, двумя, тремя и более двойными связями в
цикле, которые называются цнклоалкенамп, циклоалкадиепамн,
циклоалкатрнепами и т. д.
КЛАССИФИКАЦИЯ, ИЗОМЕРИЯ И НОМЕНКЛАТУРА
Циклоалканы подразделяются в зависимости от величины цикла,
числа циклов и способа соединения циклов.
Названия мопоцнклических циклоалкапов образуют от назва-
названий алкапов с использованием префикса цикло-:
Н н ПН Н Н Н Н
\ / II / \ \ /
V н-с — с-н н-с—с-н нх ^с^ /Н
/ \ || К/ хс/н Тс\ СГН
н-с — с-н н-с — с-н / \ У \ Hvc с-"н
н н н нх ^н / \
н н
А ' ? О О
циклопропм яиклоЗумк никлопентак .цпк-тагексак
150
Циклопропан и цнклобутан иногда называют цпклоалкапамп с
малыми циклами. Число углеродных атомов в цикле не ограничивает-
ограничивается шестью. Циклоалкаиы с большим числом углеродных атомов в
цикле (более 14) называются макроцикличсскими. Наличие двойных
связей в цикле обозначают таким же способом, как в случае алкенов
и алкадиенов.
Из моноцпклических циклоалканов образуют замещенные цик-
циклоалкаиы:
lie тнл циклопропан
с2н5
lhC/-3 2
Этилциклопентан
1,.1-диметил-
диклобутац
СИ,
1-метил-2-этил-
циклопентан
Если в молекуле циклоалкаиа два или более заместителей, на-
называя соединение, углеродные атомы нумеруют.
Для дпзамещенпых нлапарпых цпклоалканов возможно различ-
различное пространственное расположение этих заместителей. Замести-
Заместители могут располагаться по одной стороне плоскости цикла (цис-
изомер) и на противоположных сторонах (транопзомер), например:
СНз
СНз
СН1
tywc-диметилцнклопропан
Диклоалканы с двумя общими циклами называются бицш:.шчес-
кими. Если два цикла имеют один общий углеродный атом, такие
соединения составляют класс спироалканов:
спиро[2.2]пентая
3 4 5
сги:ро[3.4]октац
Название сппроалкапа образуют из названия соответствующего
алкана и цифрами в квадратных скобках указывают, сколько угле-
углеродных атомов находится по каждую сторону от общего (узлового)
углеродного атома. Циклы в молекуле спироалкана размещены во
взаимно перпендикулярных плоскостях, как это предопределяет
s/;;l-гибридизация узлового углеродного атома. Нумерацию сппро-
алкапов начинают всегда с меньшего цикла, а узловой атом нуме-
нумеруют последним.
Если два цикла имеют два или больше общих углеродных ато-
атомов, такие соединения называются бициклоалканами. Иногда их
называют мостиковыми углеводородами, так как два углеродных ато-
160
ма одного цикла как бы образуют мостик:
бицикло [ 2.2.0 ]гексан
бицик-™[3.3.0|октан
бицикло[2.2.1!гегггая
В название бнциклоалкана в квадратных скобках включают три
цифры, означающие число углеродных а томов в цепях, которые как
бы тремя мостиками соединяют узловые углеродные атомы. Для наз-
названия замещенных углеродные атомы нумеруются, начиная с уз-
узлового углеродного атома. Сначала нумеруется главный цикл бнци-
бнциклоалкана (с наибольшим числом углеродных атомов), а потом мо-
стиковые углеродные атомы. Нумерация идет от одного узлового
углеродного атома ко второму узловому атому по наиболее длин-
длинному пути, например:
3-метилбн[{икло [3.2.1] октан
СИз
Возможны циклоалканы с тремя и более циклами (трипикло-, тетрац:!к-
ло-), которые имеют два и более мостикоп. Их номенклатура яяляе-тся слож-
сложной. Числительное три-, тстра- или понта- определяется числом спящей, кото-
которые нужно разорвать, чтобы циклоалкан прекращался в соединение с откры-
открытой цепью (в схемах эти связи отмечены знаком ~), например:
трицкклононан
трициклодекан
пеитацикжкжтан
Чтобы правильно состапить название, необходимо выбрать главны:!
цикл и главный мостик (с наибольшим числом углеродных атомов). В назва-
названии в скобках сначала указывают три цифры, относящиеся к бициклу, кото-
который образован главным циклом и главным мостиком. Следующие цифры от-
относятся к числу углеродных атомов в других мостиках. Здесь иногда необхо-
необходимо указать, какие именно углеродные атомы соединены другими мостиками.
Поэтому выбранный бицикл нумеруют по известному правилу, а номера сое-
соединенных мостиками углеродных атомов пишут у цифры, указывающей число
углеродных атомов в мостике:
трнцикло [4.2.1.О''5] но пан
трицикло [3.3.1. ] v] декаи,
адамантам
В углеводороде адамаптане главный цикл восьмичлснный, а не
членный.
6 .v, 517
161
Очень своеобразны циклические углеводороды CnHn, пространст-
пространственное строение молекул которых соответствует различным мно-
многогранникам (тетраэдру, кубу и др.)- Они называются полиэдра-
нами, многие из них еще не синтезированы. В качестве примера вы-
выше приведена структура пентациклооктана (кубана, С8Н8).
А. ЦИКЛОПРОПАНЫ И ЦИКЛОПРОПЕНЫ
Методы получения. Циклопропаны получают взаимодействием
1,3-дигалогеналканов" с металлами (Na, Mg, Zn). Эту реакцию мож-
можно рассматривать как внутримолекулярный синтез Вюрца:
СН2 СН2
R-CH СН —RJ + Zn-j-R-CH-CH —R4-ZnBr2
I I
Br Вг
¦ Вторым общим методом является взаимодействие карбенов с ал-
кенами или алкинами (циклоприсоединенне). Карбены генериру-
генерируются прямо в реакционной среде из дназоалканов (гл. XXV. Б.З)
или галогеналканов (гл. VIГ. 4.5):
R4/H
С /R2
R
С
Физические свойства и природа связей. Циклопропаны и цикло-
пропепы являются бесцветными газами или жидкостями, нераство-
нерастворимыми в воде. Молекула циклопропана представляет собой регу-
регулярный треугольник. Угол НСН необычайно большой и не со-
соответствует углу тетраэдрпческого углеродного атома A09с28')-
н н При образовании циклопропанового
\S цикла тетраэдрические углы должны умень-
Лшаться до 60°, происходит напряжение
валентных углов. В свое время А. Банер
из™ A885) именно напряжением валентных уг-
__н лов объяснял большую реакционную спо-
J ^пб-1180 собность циклопропана — теория напря-
н н жения Байера.
Природа связей в циклопропане не так проста, как в пропане
или пропене. При образовании связей С—С перекрыванием s/Я-
гнбрндных орбиталей в циклопропане возможно только частичное
перекрывание. Могут образоваться изогнутые орбнтали («банано-
(«банановые связи», рис. 66).
162
Если допустить, что углеродные атомы в циклопропане находят-
находятся в в/э^-гнбридном состоянии, то связи образуются частичным пере-
крьшанием р-орбиталей, а в центре цикл* взаимодействуют sp2-
гнбридные орбитали. По-видимому, в образовании связей цикло-
циклопропана участвуют атомарные орбитали, не имеющие «чистой»
II II
"\Л ,..,:??|\ /',/
н
Яне. 66. Образование связей в молекуле циклопропана
s/Я- или s/J-rH6pHAH3an,nn. О значительном вкладе р-орбптали п
образование связен циклопропана свидетельствует сравнительно
низкая энергия ионизации (для пропана 11,5 эВ, циклопропана—¦
10,5 эВ, иропена — 9,7 эВ).
Теплоты сгорания циклоалканов свидетельствуют, что цикло-
циклопропан имеет наибольший избыток энергии в расчете на одну
группу по сравнению с циклогексаном (табл. 15), т. е. связь
в циклопропане имеет меньшую энергию, чем в циклогексане.
Таблица 15. Теплота сгорания цкклоалканов
в расчете на одну метнленовую группу
С—С
Циклоалкаи
Циклопропан
Циклобутан
Цпклопентан
Цпклогексап
Ц1;клогептап
Теплотл
ния, кД
697
686
664
633
662
сгор,^-
><\: МОЛЬ
,1
,6
,3
Избыток энергии гю
сравнению с цикло-
гексатюм, кДж/но."ь
38,5
27,4
¦¦¦Л
О
3,7
Химические свойства. В реакциях циклопропаны проявляют
двойственную природу т. е. реагируют как алканы — с замеще-
замещением атома водорода и как алкены — с присоединением (раскры-
(раскрытием трехчленного цикла):
к\\ С!,
СМ,
/\
СНа—ЧС
6*
Н„; Pf
си,—сн—ci
си3сн2си3
CH3CH2C!f2Br
ВгСПоСН2СН2Вг
163
Направление реакции определяется как природой реагента, так п
температурой.
Циклопропены — весьма нестабильные соединения, легко полп-
мерпзуются. Их стабильность возрастает при введении заместителей
ко всем трем углеродным атомам.
Своеобразным свойством циклопропанов является образование
стабилизированного карбокатпопа — катиона цпклопроиенплпя:
С—R или |: + . С—R
Клтнон циклопропепилпя является самой простои циклической
сопряженной системой (см. гл. VI. Г) и он более стабилен, чем его
апаюг с открытой цепью — аллил-катион СН2^СН—СН?.
Важнейшие представители. Циклопропан является бесцветным
газом с т. кип.— 34,5° С. Получают его из 1,3-дибромпропапа. Он
применяется в медицине в качестве наркотического вещества для
общей анестезии.
Б. ЦИКЛОБУТЛНЫ, ЦИКЛОБУТЕНЫ И ЦИКЛОБУТЛДИЕН
Методы получения. Циклобутановый цикл может быть построен
из соединений с открытой цепью несколькими способами. Известна
циклизация 1,4-дигалогепалкапов и 1,4-дигалогеналкенов, цпкло-
днморнзацня замещенных алкепов:
+ ZnBr2
R-
R
R
С П.,
СП
-R1
С
Сч
^R1
-СН,
СП
R«
4 С
-т- '''
.С
R-'
-R1
R1
R1
-f-Zn
,1V R
~* R
Р
R
1
-С
|
-С
1
R1
СП, —СП
СН- СН
1 R
1
-C-R1
1
-C-Ri
1
R
Пнклоприсоедипение [2 [-2] двух молекул алкепа происходит
то/и-ко при освещении ультрафиолетовым светом, при нагревании
реакция не идет. В реакцию вступает молекула алкепа в возбужден-
возбужденном состоянии (см. гл. V. Г). Во многих случаях эту реакцию рас-
рассматривают как перициклпческую (см. Введ. 7.2). Например, из
Чыс-Сутоиа-2 образуются два изомерных 1,2,3,4-тетраметилцикло-
бутапа, так как две молекулы бутена в циклоприсоединенип могут
164
принимать два различных положения:
НзС
\
СНз
/ \
CHj СНз
V
СН3
НзС
СНз
\
СНз
СНз
сн,
Физические свойства. Строение. Простейшие цпклобутаны яв-
являются бесцветными газами или жидкостями, нерастворимыми в
воде. Молекула циклобутана подобна квадрату с весьма длинной
связью С—С @,157 им). Самой большой особенностью циклобутапов
является то, что четыре углеродных атома не находятся в одной
плоскости. Эта не план арность вызвана внутримолекуляр-
внутримолекулярным отталкиванием водородных атомов или заместителей и сильно
зависит от строения. Один углеродный атом может быть смещен из
плоскости остальных трех атомов даже до 25. . .30°. Это означает,
что монозамещенпые циклобутаны могут существовать в двух кон-
формациях:
Как доказано методом ПМР, между коиформашгямп осуществля-
осуществляется быстрое равновесие, которое удается «заморозить» только при
низких температурах.
Валентные углы в циклобутане напряжены меньше, чем в цик-
циклопропане, возможно большее перекрывание атомных орбпталей,
поэтому и стабильность увеличивается (см. табл. 15).
Химические свойства. Циклобутан и его замещенные во многом
подобны алкаиам. Наиболее характерны для них реакции замеще-
замещения. Тем не менее известны также реакции раскрытия циклобута-
нового цикла, например при каталитическом гидрировании:
СН2-СН„
I I —
сн2—сн.
сь-Av СН.» —СН —Cl -на СНо-СН
—-» I I —> I
сн—сн2 сн2—сн
Н2; кат
>СН3СН2СН2СН3
Своеобразны свойства циклобутаднепа. Это вещество является
очень реакционноспособпым н его нельзя получить в чистом виде.
В реакциях, где ожидается образование цпклобутадиепа, выделены
165
только продукты его дальнейших превращений, например димер:
-2НВг
тр!!цикло[4.2.0.0-':]октадиен-3,7
Важнейшие представители. Циклобутан представляет собой
бесцветный газ с т. кип. 12,5JC. Его получают из 1,4-днбромбутапа.
Применяется в органическом синтезе.
В. ЦИКЛОПЕНТЛНЫ,
ЦИКЛОПЕНТЕНЫ И ЦИКЛОПЕНТАДИЕН
Методы получения. Циклопентан и его гомологи входят в состав
некоторых нефтей (нафтены). Синтетически циклопентан получают
восстановлением его кислородсодержащих производных или гид-
гидрированием ненасыщенных производных:
//°
7.П/Н +
циклопе нтлеюн
циклонентадиен
Циклопентадиен получают из продуктов переработки каменного
угля или продуктов пиролиза нефти.
Циклопентановый цикл замыкается в некоторых реакциях кон-
конденсации дикарбоповых кислот (гл. XXXII. А.З игл. XXXIII. В.3).
Физические свойства и пространственное строение. Циклопен-
тапы и его ненасыщенные производные являются бесцветными жид-
жидкостями со своеобразным запахом, в воде пе растворяются.
Молекула циклопентана образует правильный пятиугольник,
внутренний угол которого A08°) близок к тетраэдрическому углу.
Однако молекула циклопентана и его производных непланарна.
Благодаря силам отталкивания между атомами водорода два ато-
атома углерода выходят из плоскости цикла. Отдельные углеродные
атомы циклопентана пе запнмают жестко закрепленного положения,
кольцо как бы находится в постоянном волнообразном движении —
псевдовращение:
Химические свойства. Для циклопентана характерны все реак-
реакции алканов, а для цнклочентена — алкепов. Циклопентадпеп яв-
166
ляется активным диеном-1,3, легко вступает в реакции диенового
синтеза, в том числе в реакцию димернзацпи*
эндо-
ди!у;клопептадиеи
Дицпклопентадиеп является обычной продажной формой, из
которой циклопептадпеп регенерируют нагреванием при 200—
250^. Дициклопептадиеп является ненасыщенным трициклическим
мостиковым углеводородом, существующим в формах двух прост-
пространственных изомеров (экзо- и зндо-), и его название трицикло-
[5.2.1.0-' декадиен-3,8.
Особым свойством циклопентаднена является его повышенная
СН-кислотность. Константа кислотности /<а«10~15 (рК„~ 15) не-
необычно большая для углеводородов. Поэтому с концентрированными
щелочами и алкокендами он образует соли —-.циклопентаднениды:
Нч ,Н н\ /Н
с-с/ ,?~\
// \ + Na+~OR :=== / \ + "OR
/С\ L\ Na*-
Н* Н Н
Циклопентадиенид-ион является сопряженным карбанно-
ном, его относительная стабильность объясняется циклической
делокализацией я-электропов (гл. VI. Г). Цпклопентадиеннды
являются сильными нуклеофильными реагентами, легко окисля-
окисляются. Очень своеобразны циклопентадиенпды переходных метал-
металлов — так называемые металлоцены, или сэндзичевые соединения
(гл. X. Д. 2).
Важнейшие представители. Циклопентан — бесцветная жид-
жидкость с т. кип. 49,5 °С. Может быть получен из продуктов переработки
нефти. В лаборатории его получают гидрированием цпклопептадие-
на. Используют в органическом синтезе.
Циклопентен —¦ бесцветная жидкость с т. кип. 44,2 ° С. Получают
дегидратацией циклопентанола. Его используют в органическом
синтезе для получения производных циклопептана.
Циклопентадиен — бесцветная жидкость со своеобразным запа-
запахом; т. кии. 41 °С. Получают из дициклопентаднена, который со-
содержится в продуктах переработки каменного угля и нефти. Цн-
клопентадиен используют в органическом синтезе для получения
инсектицидов, ферроцена и других металлоценов.
Система циклолептена входит в молекулы важных биологически
активных соединений — простагландинов (гл. XXXIV,Д.3).
N7
пплучС.-л- Циклогексан и его гомологи получают из про-
Методы по.i j ки нефти (нафтепы). Синтетически 0!!Н могут быть
дуктов пер v_ итцЧескпм гидрированием бензола н его гомологов
получены *¦ пением ццклогексанопов:
или восстанови
,R /ч /R
К
н ия^дат.- производные циклогексана па1учают обычными
гИ11теза алкенов и алкадиенов. Особым случаем является
методами с проп3водных бензола натрием в Жидком аммиаке в
таИп«.,1 фта (д. Берч, 1944) с ооразоваппем j 4-цпклогекса-
гл г,г^ место в получении производных циклогексана занимают
магические реакции: диеновый синтез и электроцпклическпе
перпцпклич од„ых Гексатриепа-1,3,5,
реапЦ™пПтШ синтез - цпклоприсоединение [2+4] _ рассмотрен
17П В отличие от цнклоприсоедииения [2+2] двух молекул
И3 о" я лиеновый синтез идет легко при комнатной температуре пли
алкена ди певаиии:
небольшом нагр
R
*[ Ra W '
Г-.2 I /К
Пппизвочные гексатриена-1,3,5 при нагреваии,,, а также при
освещении инклизуются в производные ЦиклогекСадиена.1|3:
цис - 5,6-диметилциклогексадиен-1,3
к»« правило, ПРИ термической циклизации Подучаются цис-
^пенные циклогексаднепы, а при фотохимической - - тринс-
Д"^мешениые. Это связано со свойствами молекуЛяр11ЫХ орбита.
ZI умствующих в циклизации.
1G8
В процессе образования а-соязп между концевыми углеродными атомя-
ии сопряженной цепи необходим поворот около соязс!! С—С, чтоое.1 p-opunia-
ли могли перекрываться своими одинаковыми по**знаку частями:
2 I
ВЗМО
диерога горное вращение
СНз
конротаторное вращение
При термической циклизации рассматривается ВЗМО сопряженной си-
системы гексатрпепа, а при фотохимической — НСМО (в возбужденном состоя-
состоянии молекулы один электрон помещается на НСМО). ВЗМО и НСМО отлича-
отличаются расположением положительных и отрицательных частей атомным р-ор-
биталей (отличаются орбитальной симметрией). Чтобы перекрывались доли
р-орбиталей с одинаковыми знаками, направление попорота но связям для
ВЗМО и для НСМО различное. Для ВЗМО это поворот в противоположные
стороны (дисротаторно), а для НСМО в одну сторону (конротаторно).
Объяснение хода электроциклическнх реакций дали Р. Вудпорд и Р. Хоф-
ман A965). Основная идея заключается в том, что легче осуществляется тот
механизм, в процессе которого достигается максимальное нерекрыианме ор-
бптален (максимальное связывание). Это получается только в том случае,
если совпадают знаки частей перекрывающихся орбиталей. Правило Вуд-
ворда — Хоф.мапа гласит, что в согласованных (перициклических) реакциях
сохраняется орбитальная симметрия. Если достигается максимальное пере-
перекрывание орбиталей, реакция имеет низкую энергию активации и она «раз-
«разрешена по симметрии». В противоположном случае реакция «запрещена по
симметрии».
Так, в случае термической циклизации октатриена-2,4,6 при дисротатор-
ном вращении процесс циклизации имеет низкую энергию активации и раз-
разрешен по симметрии.
Представления о сохранении орбитальной симметрии применяются так-
также для объяснения хода реакций циклоирисоедипепия, например образова-
образования системы циклобутапа из двух молекул этилена. Процесс рассматриваем
как взаимодействие ВЗМО одной молекулы этилена с НСМО второй молеку-
молекулы. Эти орбитали имеют различную симметрию и максимальное перекрывание
не получается, реакция запрещена по симметрии. В возбужденном состоянии
йтилеиа один электрон находится в НСМО, и она стапошпея ВЗМО, симмет-
симметрия взаимодействующих орбиталей одинакова, реакция pirpcuiciia по сим-
симметрии:
НСМО
ВЗМО
,1 5ч
НСМО
взмо
-IK
<акц-'я папрещеса
Диеновый синтез является разрешенным по симметрии термически стиму-
стимулированным процессом:
немо
взмо
2 ?
сн>
j 1
взмо
немо
Физические свойства и пространственное строение. Циклогек-
саны и их ненасыщенные производные являются бесцветными жид-
жидкостями со своеобразным запахом, в воде не растворяются.
Для плоского цикла регулярного шестиугольника внутренний
угол равен 120J, что больше тетраэдрического угла A09с28'). Бо-
Более выгодно образование непланарнои молекулы, что ведет к умень-
уменьшению внутренних углов и межатомного отталкивания рядом рас-
расположенных водородных атомов.
Известно несколько изогнутых форм циклогексана:
конформация „кресла"
Шиформация„ваш|ы"
твист-конформация
Более стабильной является конформация «кресла». Возможно
также образование «искаженной ванны», или твист-конформацип,
которая более стабильна, чем конформация «ванны».
Циклогексан в конформации «кресла» имеет два типа водородных
атомов, которые отличаются друг от друга пространственным рас-
расположением: На (аксиальное положение) и Не (экваториальное поло-
положение):
Для монозамещенных циклогексапов возможны два конформера
в форме «кресла» (и еще несколько в форме «ванны»):
Инверсия
цикла
зкв -метил циклот*ксан
I/O
Взаимный переход аксиальных и экваториальных конформеров
происходит легко инверсией цикла.
Непланарными являются также молекулы циклогексена и его
производных:
Ь( г
В производных циклогексана тоже возможно аксиальное и эк-
экваториальное расположение заместителей.
Химические свойства. Циклогексан по своим химическим свой-
свойствам аналогичен алкапам. Его можно галогеннровать, нитровать,
окислять. Каталитическое дегидрирование циклогексана приводит
к бензолу.
Для циклогексана характерны также реакции перегруппиров-
перегруппировки, приводящие к сужению цикла (Н. Д. Зелинский):
AICI,
(в этих же условиях метилциклопептан частично превращается
в циклогексан).
Циклогексен и циклогексадиены дают все характерные превраще-
превращения алкенов и алкадиенов.
Важнейшие представители. Циклогексан является бесцветной
жидкостью со слабым запахом, т. кип. 80,7 °С. Получают его ката-
каталитическим гидрированием бензола. Циклогексан широко приме-
применяют в качестве растворителя и как исходное для синтеза мономеров
(капролактама, адипиновой кислоты).
Циклогексен является бесцветной жидкостью с острым запахом,
т. кип. 83 °С. Получают дегидратацией цнклогсксанола. Для цикло-
гексена характерны все реакции алкенов. Своеобразна реакция тер-
термического каталитического днепропорционированпя (Н. Д. Зе-
Зелинский):
/\ м /*Ч Л
3
Кат.
Ч/
Циклогексен используют в органическом синтезе для получения
производных щжлогексана.
Терпены являются продуктами растительного мира, они со-
содержатся в эфирных маслах, живице хвойных деревьев, сшпидаре.
В основном терпены представляют собой моноциклические и бицик-
лические производные цпклогсксана с двойными связями в молеку-
молекуле. Обычные терпены принадлежат к продуктам димеризации двух
молекул изопрена. Природные углеводороды, скелет которых об-
• 171
ралован олнгомеризацней нескольких молекул изопрена, называют
изопрсноидами. Известны TepiieHbIj молекулы которых построены
из трех, четырех, шести молекул НЗопрена (сесквитерпепы, днтер-
пены, трптерпены):
си.,
(it ).lit:;eir.' я
(+)дипентеп (лимонен)
СН3
СНз
сн2
СНз
СНз
СН3
\
B'б'бтРи^етил6ищ1кло[3.1.1]гепте1!-2)
а-Пппеп является главной составной частью скипидара сосны.
Терпены (особенно а-пппеп) Ш1,рОКо применяются в лакокрасочной
промышленности и в качестве исходных продуктов для органичес-
органического синтеза. Некоторые тер[1С11Ы применяются в парфюмерной про-
промышленности.
Д. ЦИКЛОГЕПТАНЫ И ЦИ МоГЕПТАТРИЕН
Методы получения. Цпклогепта11 впервые был выделен из кавказ-
кавказской нефти (В. В. Марковников). Синтетически его получают вос-
восстановлением циклогептанона, например по методу Кнжнера (гл.
X X V11. Д.З).
О H,NNH2
Цнклогептатрисн получаЮт из бензола присоединением карбспа:
+ :сн2
Впервые нпклогснтатрпец получен при химической деструкции
алкалоидов.
Физические свойства^ и пространственное строение. Циклогеп-
тапы представляют собой беСцветные жидкости со слабым запахом,
в воде нерастворимы.
Молекула циклогептана, подобно молекуле пиклогексана, имеет
изогнутое строение, при этОм ВОЗможпо значительное число коп-
172
формаций. Ниже приведены четыре возможных конформера мопоза-
мещепиого циклогептана:
Химические сеойства. Цпклогептан и его гомологи по химиче-
химическим свойствам очень напоминают алкапы. Своеобразны реакции
перегруппировки в присутствии кислых катализаторов, в которых
происходит сужение цикла:
СНз
Для циклоалкапов характерны реакции сужения, а во многих
случаях и расширении цикла. В большинстве случаев они проходят
с промежуточным образованием карбокатпопов, которые способны
к различным перегруппировкам.
Циклогептатриен является типичным ненасыщенным соеди-
соединением, он легко присоединяет галогены, окисляется, полимери-
зуется. В присутствии электропоакцепторпых реагентов он пре-
превращается в катион цнклогентатрненилпя (при отщеплении гпд-
рид-иоиа):
r-h
Катноп цнклогептатрненилня (тропилпя) является необычно
стабильным карбокатпоном, стабилизированным в результате цик-
циклической делокализации я-электронов (гл. VI. Г).
Важнейшие представители. Циклогептан— бесцветная жид-
жидкость со своеобразным запахом, т. кип. 119 СС, в воде не растворя-
растворяется. Получают его переработкой некоторых нефтей. Используют
в органическом синтезе.
Циклогептатриен (тропнлидеи) является бесцветной жидкостью
(т. кип. 115,5 ГС). Его получают реакцией бензола с диазометапом
в присутствии медных солей. Используют для получения солей тро-
пнлия.
Е. ЦИКЛООКТАНЫ И ЦИКЛООКТЛТЕТРАЕН
Методы получения. Цнклооктаны образуются при каталитиче-
каталитическом гидрировании циклооктатетраенов.
Цнклооктатетраен получают тетрамеризацней ацетилена
(В. Реппе):
... .. Ni(CNb
173
Впервые циклооктатстраен был получен Р. Вильштеттером
A911) при деструкции алкалоида исевдопельтьсрниа.
Физические свойства и пространственное строение. Циклоокта-
ны являются бесцветными жидкостями, цнклооктатетраепы — жел-
желтыми. Молекула циклооктана является изогнутой, при этом воз-
возможно значительное количество копформаций. Ниже приведены
четыре из возможных конформеров монозамещешюго циклооктана:
Подобные конформеры имеет также циклооктатетрасп.
Химические скойства. Цшслооктан по химическим свойствам
подобен алканам. В присутствии кислых катализаторов происхо-
происходит изомеризация — сужение цикла:
AIC1,
СН,
Циклооктатетрасп является типичным полисном, высокореак-
цпониоспособен.
Важнейшие представители. Циклооктан —¦ бесцветная жид-
жидкость с запахом камфоры, т. кип. 145 °С. Его получают каталити-
каталитическим гидрированием цпклооктатетраепа. Используют в органи-
органическом синтезе.
Циклооктатетраен — желтая жидкость с т. кип. 142 ГС. Его
получают из ацетилена. Циклооктатетрасп легко полимерпзуется.
Ж. ДЕКАЛИНЫ
Декалины (декагидронафтглнпы, бицпкло[4.4.0! деканы) получают
каталитическим гидрированием нафталина.
Они представляют собой бесцветные жидкости со своеобразным
запахом, в воде не растворяются. Т. кип. цис-декалипа 194,6 °С,
т/занс-декалнна 185,5 СС.
Изомеры цис- и транс- определяются расположением атомов
водорода при мостике:
н
екалин ?«-;икгли.|
По химическим свойствам декалины подобны производным цпк-
логексана. При гидрированим няфгллчи .'-о.'учл'стгя елгес:-. иис-
и траис-нзомеров, которую используют в качестве растворителя.
174
3. ЦИКЛОПЕНТАНОПЕРГИДРОФЕНАИТРЕНЫ
Система циклопентапопергидрофспадетрсна
04'13.05110]гептадекаиа)
(тетрацикло[12.3.0.
сн,
J5
является основой важных природных веществ — стероидов. При-
Приведенная в формуле нумерация атомов углерода является принятой
в химии стероидов и не отвечает правилам систематической но-
номенклатуры циклоалканов.
В молекулах стероидов бициклы декалинов (АВ и ВС) имеют
траяс-строение, а у 10-го и 13-го углеродных атомов находятся
метильпые группы (ангулярпые метильные группы).
К стероидам принадлежатстерины (R'=OH)— холестерин, сте-
стероидные, или половые, гормоны (эстрон, тестостерон) и сердечные
гликозиды, которые играют большую роль в жизненных процес-
процессах.
И. АДАМАНТАН
Углеводород адамантан С10Н,„ представляет собой бесцветное крис-
кристаллическое вещество со слабым запахом камфоры; т. пл. 269 °С
(возгоняется), в воде нерастворим.
Молекула адамантапа содержит систему угле-
углеродных атомов, пространственное расположение ко-
которых соответствует кристаллической решетке ал-
алмаза. Кристаллы адамантана имеют форму октаэдра.
Адамантап впервые был получен при переработ-
переработке некоторых пефтей. Синтетически его получают
гидрированием дициклопентадиеца с последующей перегруппиров-
перегруппировкой продукта гидрирования:
Адамантан имеет весьма низкую энергию ионизации (9,25 эВ)
и для него характерны реакции с электрофнльпыми реагентами (на-
(например, Премирование). Промежуточной частицей может быть ада-
мантил-катиоп, образующийся уже в концентрированных растворах
175
кислот:
Br,,A!Br, || I
Химия адамантана быстро развивается, некоторые его производ-
производные обладают сильным противовирусным действием.
Глава VI
АРЕНЫ
Аренами (ароматическими углеводородами) называют богатые угле-
углеродом циклические углеводороды, которые содержат в молекуле
особую систему связей — циклогексатрнеиовый цикл и обладают
особыми физическими и химическими свойствами.
ИСТОРИЯ ОТКРЫТИЯ. КЛАССИФИКАЦИЯ
При изучении конденсированных остатков светильного газа, полу-
получаемого из каменного угля, М. Фарадей в 1825 г. выделил угле-
углеводород с температурой кипения 80°С и определил соотношение
углерода и водорода в этом соединении, равное 1 : 1. В 1834 г.
Э. Митчерли при нагревании солей бензойной кислоты (вещества,
выделяемого из природных ароматических смол) получил подобный
углеводород и дал ему название бензин. Позже Ю. Либнх пред-
предложил назвать это вещество бензолом.
Бензол имел своеобразный запах и обнаруживал странные хими-
химические свойства. Несмотря на свою глубокую «ненасыщенность»
(формула С6Нв), бензол трудно вступал в реакции присоединения,
но легко давал своеобразные реакции замещения водородных ато-
атомов. Поэтому бензол и его производные, которые были получены
вскоре после открытия бензола, выделили в специальный класс и
дали название ароматические углеводороды. Их производные назва-
назвали ароматическими соединениями.
Структурную формулу бензола как системы циклогексатрнена
впервые предложил в 1865 г. немецкий химик А. Кекуле:
И
I
НЧ/^С\ Л\
чс с-7
с с ил"
нАс/\ц
I
н
Однако Кекуле не мог не видеть глубоких противоречий между
формальной ненасыщенностью бензола (сопряженный трнен) и его
176
физическими и химическими свойствами (термическая стабильность,
реакции замещения). Он предположил, что в бензоле нет настоящих
двойных связен, и выдвинул идею осцилляции связей:
В связи с этим понятия «ароматические соединения», «аромати-
«ароматические свойства» приобрели особый смысл. Так стали называть
соединения с циклическим строением и характерными особенностя-
особенностями — тремя двойными связями в шестичленном цикле и специфи-
специфическими химическими свойствами.
Противоречие между формальной «ненасыщепностыо» и свое-
своеобразными физическими и химическими свойствами объяснить
смогла только квантовая органическая химия.
В последнее время понятие «ароматические углеводороды»
предлагают заменить на «арены», а «ароматические соединения» —
на «производные аренов».
Арены подразделяются в зависимости от числа циклов в моле-
молекуле и способа соединения циклов. Самыми простыми являются
моноциклические арены — бензол и его гомологи и производные,
которые можно называть аренами ряда бензола. Полициклические
арены подразделяются на арены с изолированными цик-
циклами и с конденсированными цикла м п.
А. АРЕНЫ РЯДА БЕНЗОЛА
1. ИЗОМЕРИЯ И НОМЕНКЛАТУРА
Арены бензольного ряда можно рассматривать как продукты заме-
замещения атомов водорода в бензольном ядре па алкпльпые (алкеннль-
ные, алкинильные) группы. Ниже приведены некоторые важнейшие
представители аренов.
СН3 СН3 СН3. СН3
SCH,
мгта-ксилол
A ..S-дпмстмл-
бспзо.т)
СП;,
бензол толуол орто-кси:юл
A,2-диметпл-
бензол)
СП-,СН3 СН3 СН3
1 ¦' сня I
сня
A,1-диметнл-
бонзол)
СИ,
бтилбензол
1,9,3-триме-
тклбен.^ол
3
1,2,1 -тримс-
тилбеизол
(псевдоку.мол)
1,3,5-триметмл-
бС II ЗОЛ
(мезитилен)
177
н3с
/СНз
сн
(
II
кумол
(изопропил-
бензол)
и3с
п,с
A.2
/
II
ДУрол
4,5-тетраметнл
беЕЕЗОЛ)
Арены бензольного ряда с ненасыщенными заместителями;
Н. СН2
о
сн = сн2
стирол
(вппил бензол,
ф)
этипплйензол
(фешы ацетиле и)
сн- сн2
1И -дпвннил-
бепзол
Следуя номенклатурным правилам ИЮПАК, названия аренов
должны иметь суффикс -ен. Предлагается писать не бензол, а
бензен, не толуол, а толуен. Такие названия употребляют в англий-
английском языке, по в других языках пока еще сохраняется суффикс -ол,
несмотря на то, что получается некоторая путаница с названиями
спиртов и фенолов, которые по правилам ИЮПАК имеют суффикс
-ол.
Двузамещенные бензолы образуют три изомера в зависимости
от положения заместителей: орто-, мета- и пара-производные (сок-
(сокращенно о-, м- и п-) или, соответственно, 1,2-, 1,3- н 1,4-замещепные
бензолы.
Названия остатков аренов .образуют с помощью суффикса -ил
(арил-). Для некоторых остатков сохранились тривиальные назва-
названия:
СП3
,сн2-
сп3
I
фенил- бензил» о-толил- Л!-тол:гл« н-толил«
С,НЙ
XH..CI-L —
2, 3-ксилил-
178
Названия двухвалентных остатков:
I I I
/ /
V
о-фенилен- jK-феиилен- л-фенилея-
2. МЕТОДЫ ПОЛУЧЕНИЯ
Арены бензольного ряда в промышленности получают при переработ-
переработке каменного угля и нефти н алкилпрованнем бензола. В лабора-
лаборатории используют алкнлпрованне и ацнлированнс с последующим
восстановлением ацилпродукга.
1. Получение из природных веществ. Исторически первым мето-
методом получения арепов является сухая перегонка каменного угля.
Каменный уголь нагревают при 900—1000°С без доступа воздуха.
Получают сухой остаток (кокс), газы и каменноугольную смолу.
В 1 м3 газа содержится около 30 г бензола и 10 г толуола. Смола
содержит небольшие количества бензола, толуола, а также ксило-
ксилолы и полициклическне арены, фенолы и гетероциклические соеди-
соединения. И в настоящее время каменный уголь является главным ис-
источником аренов.
Бензол, толуол и другие простые арены встречаются в некото-
некоторых сортах нефти. Их получают также при каталитическом кре-
крекинге нефтепродуктов.
2. Дегидрирование и дегидроциклизация. Важным синтетическим
методом получения аренов является дегидрирование алкаиов и цик-
лоалкапов. Циклогексан легко превращается в бензол:
/ч
ЗСО "С
Алканы над оксидными катализаторами цпклизуются и дегидри-
дегидрируются с образованием аренов:
СПз
4 50 - Г.00 'С ¦¦¦
НзС-СНо —СН2 —СН2-СН2-СН2-СН3 —
гептан
СггО3.'Л!2О3
СП3
Н3С-СН2-С1-12-СН.,-СН,-СН2-СН2-СНз
4 3 С - 5 С 0 °С
+4Н2
Этот процесс называется дегидроцнклизацией алканов или аро-
матпзацчен нефти. Большие заслуги в изучении этой реакции и внед-
внедрении ее в промышленность принадлежат советским химикам
179
(Р. Л. Молдавский, Б. А. Казанский, Н. Д. Зелинский и
А. Ф. Платэ).
3. Циклотримеризация. В присутствии металлорганических ка-
катализаторов алкины легко превращаются в арены (гл. IV. 4.2).
4. Алкилирование. В реакциях алкнлироваиия бензола получа-
получаются алкнлбензолы. Алкнлнрование проводится галогеналканами,
алкенамп и спиртами (см. с. 188).
5. Получение из кислородсодержащих производных, а) Восста-
Восстановлением арилкетоиов получают алкилпроизводпые бензола:
О
|
,СЧ
//¦
чсн3
ацетофспоп
б) декарбоксплпроваинем аренкарбоповых кислот при нагрева-
нагревании, лучше всего в присутствии катализаторов (оксидов металлов):
О
чон r "
Кат.
-СО-,
беп.'юпная
3. ФИЗИЧЕСКИЕ СВОЙСТВА И СТРОЕНИЕ
Бензол и его гомологи являются бесцветными жидкостями и кристал-
кристаллическими веществами со своеобразным запахом. Они легче воды
и с весьма большими коэ4>фицнентами преломления света (табл. 16).
1. Теплота образования, полярность молекул и энергии иониза-
ионизации. При образовании бензола из атомов выделяется больше теп-
теплоты, чем можно было бы ожидать па основе расчета суммы термохи-
Таблица К).
Пазоамис
Бензол
Толуол
о-Кснлол
м-Ксилол
л-Ксплол
Пссндокумол
Кумол
Мезптилеи
Дурол
Некоторые физические
Т. ил., еС
5,5
—95,0
—29,0
—53,6
13,2
—57,4
—96,9
-52,7
80,0
Т.
константы аренов ряда бенлола
кип., °С
80,1
110,6
144,4
139,1
138,4
169,4
152,4
164,7
195,0
0,8790
0,8069
0,8802
0,8641
0,8610
0,8758
0.8С18
0,8651
0,838
(S1 -"С)
Коэффициент пре-
преломления спета
"D
1,5011
1,4969
1,5054
1,4972
1,4958
1,50-1.8
1,4914
1,4993
1.С150
180
мических энергий ординарных и двойных связей. Разница составля-
составляет около 160 кДж/моль.
Необычайно большая разница энергий АЕ свидетельствует о
специфической стабилизации молекулы бензола в результате со-
сопряжения трех двойных связей в шестичлешюм цикле (энергия ре-
резонанса, сопряжения, делокализацпи). Аналогичные результаты
можно получить при сравнении теплот гидрирования бензола и
циклогексепа (см. гл. VI. А. 4).
Бензол —¦ неполярное соединение (ц — 0), но алкилбензолы име-
имеют электрические дипольные моменты. Это свидетельствует о зна-
значительной поляризации электронной системы:
СИ,
/МО'9, Кл-м ... 1,34
М , D 0,4
Днпольный момент направлен от алкилыюй группы на бензоль-
бензольный цикл.
О подвижности электронной системы бензола и электроиодонор-
пых свойствах свидетельствуют весьма низкие энергии ионизации.
Энергия ионизации арснев
ЭИ(аВ)
Бензол 9,24
Толуол 8,72
о-Ксплол 8,45
Псемокумол . .
Дурол
Гексаметплбензол
ЭИ <эВ)
8,5
8,1
7,9
2. Геометрия молекул и расчет МО. Электропографпческие ис-
исследования бензола показали, что молекула имеет высокую сим-
симметрию — она представляет собой правильный плоский шести-
шестиугольник.
Все связи С—С одинаковы и их длина не со-
соответствует ни ординарной, пи двойной связи.
Углы тригональные — 120'. Для гомологов бен-
бензола длины связей и углы изменяются незначи-
незначительно.
Углеродные атомы п системе бензола на-
находятся в состоянии .ч/^-гибридпзации, каждый
атом образует три а-связн и предоставляет од-
одну /з-орбиталь для образования сопряженной системы из шести
л-электропов.
Целесообразно привести некоторые результаты расчетов по методу МО
Хюккеля, которые подтверждают большую стабильность молекулы бензола
и особенности л-электронпон системы.
0,1399 нм
Н
К
181
1 \Д' ' ; ' ] ' " 5 6
c. 67. Диаграмма энергетических уровнен бензола и значения собственных
векторов заполненных МО
С. 6Я. Очертания заполненных МО бензола и значения
одноэлектропной плотности с)г
При комбинации шести атомных орбнталей г|:л образуются шесть молеку-
молекулярных орбиталеп Чj:
ib (/=1,2, 3, 4, 5, 6)
Расчет дает энергии орбнталей Ej и собственные векторы Cjr при АО
в выражениях V/ (рис. 67).
В бензоле имеются дне заполненные МО с одинаковой энергией (вырож-
(вырожденные орбиталн), но их волновые функции XV.2 и У^ совершенно различны.
Если у?1 относится полностью ко всем шести углеродным атомам и не имеет
узловой плоскости, то V, и XY3 имеют по одной узлоноц плоскости и различа-
различаются симметрией. На рис. 68 сделана попытка более наглядно изобразить за-
заполненные МО бензола.
Суммарная ^-электронная плотность ил каждом углеродном атоме qr = \t
порядок л-связи prs— 0,667, спободпая цалешность Fr -0,39.
1
.0,667
182.
Можно рассчитать полную энергию я-электронов системы бензола:
Исходя из ?л бензола и этилена можно рассчитать энергию дслока-
лизации, сравнивая бензол с тремя молекулами этилена:
Получается очень большая энергия ^локализации, что говорит о ста-
стабильности молекулы бензола.
Еще точнее характеризовать стабильность циклических сопряжэнных
систем можно, используя энергию циклической делокала--ации Д/-^'""л- дел,
т. е. сравнивая я-злектронную энергию бензола с энергией анало! ичион, но
нециклической системы СИ.,—СИ—СИ—СИ—СН=СН2 (QH8):
Рассчитанная энергия циклической делокализацни является очень боль-
большой. Это свидетельствует о том, что образование циклических МО дает зна-
значительный выигрыш в энергии. Зтим в большой степени об1>яспягтся высокая
стабильность и сопротивление реакциям присоединения, так как любое при-
присоединение вызывает разрушение стабильной сопряженной системы.
Молекула бензола имеет тенденцию сохранять свою сопряженную
систему шести я-электронов, поэтому во многих реакциях кратковре-
кратковременно нарушенная циклическая сопряженная система регенериру-
регенерируется (реакции замещения).
Предполагают, что благодаря образованию замкнутых молеку-
молекулярных орбиталей в молекуле бензола иод влиянием внешних фак-
факторов может возникать некоторый кольцевой ток вследствие дви-
движения электронов по кольцу.
Это наблюдается при действии магнитного поля и
выражается в уменьшении диамагнитной восприимчи-
восприимчивости бензола и в сдвиге сигналов протонов бензольно-
бензольного кольца в спектрах ПМР в более слабые ноля.
Так как все связи С—С в бензоле одинаковы, возникает вопрос
о правильном написании структурных формул. Существуют раз-
различные способы, например:
Для незамещенного бензола вполне оправдан способ написания
с полностью делокализованными и выравненными одинаковыми
связями (а). Но все-таки наиболее часто употребляют формулу Ке-
куле (а), подразумевая при этом делокализацию я-электронов и
выравнивание л-связей.
Системы замещенных бензолов имеют меньшую симметрию, чем
бензол, длины связей неодинаковы, изменились порядки л-связей.
Поэтому для них некорректным было бы применение формул
(в). Но и структурные формулы Кекуле (а) не способны наглядно
изобразить влияние заместителя на распределение л-электропной
плотности.
183
Как показывают расчеты МО, заместители вызывают изменения
я-электронной плотности на углеродных атомах бензольного коль-
кольца. Электронодонорные заместители D: (алкильные группы, атомы
с иеподеленпыми парами электрогюв) увеличивают плотность элек-
электронов на о- и /г-углеродггьгх атомах, напротив, электропоакцентор-
ные заместители А (группировки с гетероатомами О, N, содержа-
содержащие двойные и тройпые связи) уменьшают электронную плотность
на о- и /г-углеродных атомах. Это затрудняет изображение при по-
помощи изогнутых стрелок, так как влияние заместителя относится ко
всей сопряженной системе:
4,0 -1
3,0
2,0
1.0
190
210
230
2S0 X, нм
Спектроскопические характеристики. Бензол и его гомологи
имеют очень характерное поглощение в ультрафиолетовой части
спектра, интенсивное в области 170—210 им, и'малоинтенсивное
(е~200) в области 240—270 им, при
этом наблюдается тонкая струк-
структура. Малая интенсивность объяс-
объясняется высокой симметрией л-ор-
биталей и существованием вырож-
вырожденных орбиталей (первый элек-
электронный переход имеет малую ве-
вероятность). Тонкая структура свя-
связана с возникновением весьма от-
отличающихся друг от друга коле-
колебательных уровней возбужденного
состояния (рис. 69).
В ИК-спектрах бензол и его
гомологи поглощают в областях
3000—3050 см (валентные коле-
колебания С—Н), 1500—1600 см (валентные колебания С^С) и 700 —
900 см (деформационные колебания С—Н).
В спектрах ЯМР протоны бензольного кольца характеризуются
сдвигом в слабые магнитные поля (б —6,4. . .8,2).
4. ХИМИЧЕСКИЕ СВОЙСТВА
Самыми характерными для бензола и его гомологов являются ре-
реакции замещения при взаимодействии с электрофильными реаген-
реагентами. Для гомологов бензола характерны также реакции у угле-
углеродного атома заместителя (реакции в боковой цепи), которые всег-
всегда осуществляются по свободнорадикалыюму механизму. Менее
характерны реакции присоединения к системе циклогексатриена;
такие реакции проходят в особых условиях: при интенсивном осве-
Рис. 69. Электронный спектр по-
поглощения бензола в растворе ци-
клогсксаиа
184
щеппи или прг: взаимодействии с очень активными реагентами и
катализаторами.
1. Реакции электрофильного замещения, а) Механизм ы.
При взаимодействии с электрофильными реагентами бензол п его
гомологи сначала образуют я-ком.плекс (комплекс с переносом за-
'ряда):
+ Е+Х- ^— f\*.]xr
Олектрафпльпий -^
Г"генг л-комплекс
Это можно обнаружить при помощи электронных спектров пог-
поглощения, так как образование я-комплекса связано с появлением
новой полосы поглощения (гл. XXX.3).
л-Комплскс, поглощая некоторое количество энергии, может
преодолевать активационный барьер и превращаться в новую час-
частицу, где произошла перестановка связей и образовалась новая
а-свюь С—Е. Такую частицу называют а-комплексом:
:-х~ ^^
(Г-комплекс
а-Комплекс является сопряженным карбокатиопом, где поло-
положительный заряд делокализовап в системе пяти углеродных атомов,
а шестой углеродный атом находится в состоянии 5/?3-гибридизацин
и в сопряженной системе не участвует. Распределение плотности
электронов сопряженного карбокатиона можно также изобразить
тремя резонансными граничными структурами:
а-Комплекс — частица малостабильная, его образование связа-
связано с разрушением стабильной бл-электронпой системы бензола.
Поэтому а-комплекс легко превращается опять в стабильную сис-
систему бензола отщеплением Е+ или Н + . В большинстве случаев
легче происходит отщепление протона (а-комплекс выступает в
роли СН-кислоты) и образуется замещенный бензол. В качестве
промежуточного продукта может образоваться другой л-комнлекс,
в котором электрофильной частицей является протон. Таким обра-
образом, весь процесс замещения теперь можно изобразить одной схе-
схемой:
185
Электрофильные реакции замещения в бензольном цикле рас-
рассматриваются как обратимые реакции. Их направление определя-
определяется энергетическими факторами (энергией начального и конечного
состояний, энергией связи С—Е) и относительной активностью
fij- 6-
электрофила и электрофуга Е + Х~ и Н—X.
Заместители в бензольном цикле существенно влияют па обра-
образование я- и а-комплексов и направляют (ориентируют) вступающую
группу в о, п- или лг-положенне. Заместители облегчают или за-
затрудняют реакции электрофилыюго замещения и проявляют эффект
ориентации. Электронодонорные заместители (—D:) облегчают об-
образование л-комплекса, так как понижают ЭЙ, и стабилизируют
а-компл~екс. Электроноакцепторные заместители (—А) сильно за-
затрудняют образование л-комплекса, так как повышают ЭИ, а
о-комнлекс стабилизируют незначительно.
Стабилизация а-комплекса электроподонорпымп заместителями
возможна только при определенной ориентации групп в а-ком-
а-комплексе:
Е Н
Э.чектр^нодонорный ia честите ль
Лштельиого заряда с
ЛС КТрп ПОДО НО р (!Ы |1
участвовать в -e.iO
+ НХ
-ориентация
Стабилизация а-комнлскса достигается дополнительно"! дело-
калпзацпей положительного заряда с участием заместителя только
в о- или я-положениях. Это можно нагляднее показать при исполь-
использовании резонансных граничных формул. В этом случае реакция
идет быстрее, лг-изомер образуется только в незначительном коли-
количестве.
Электронодопорными заместителями D: являются алкпльные,
алкеннльпые, арильные группы (—СН3, —C1I2R,—СН = СН2,
—СоН5), группы—ОН, —OR, —NH2, — NHR, а также атомы галоге-
186
на (—F:, —CI:, —Br:, —I:). В случае галогенбензолов реакции идут
медленнее, чем для гомологов бензола, так как атомы галогена име-
имеют большой индуктивный эффект (—/). Их ЭИ сравнимы с ЭИ
бензола, о-, «-Ориентирующие заместители называют заместите-
заместителями (ориентантами) первого рода.
В случае электроноакценторных заместителей ввиду электро-
электростатических факторов электрофильный реагент ориентируется в
основном в лмюложс-!пге:
А А'
-г Е'Х~ =^=г
Реакции в этом случае идут медленнее, требуют более жестких
условий. Главным продуктом является лг-изомер.
Электроноакценторными заместителями А являются группы
—С" , — С^ , — КОо, —SOnli, —С sN. Их называют замесtnu-
ЧОН 4R
мелями (ориентантами) второго рода.
Существуют также заместители, которые не обладают выражен-
выраженным эффектом ориентации (например, —СНС14, —СС13) и в реак-
реакциях получается смесь о-, м- и п-изомеров.
б) Взаимодействие с И - э л е к т р о ф и л а м и. Силь-
Сильные кислоты с аренами образуют я-комплексы. В присутствии осо-
особо сильных кислот (сверхкислот HF-|-SbF5, HF+PF5, HF+BF3,
SbF5-|-FSO3H) происходит присоединение протона с образованием
сопряженного карбокатиопа — бензолонневого нона (а-комплекса):
R
н н
Так происходит обмен атомоз водорода, а в присутствии дей-
терировапных кислот — дейтерообмен.
в) Л л к и л и р о в а н и е. Галогеиалканы в присутствии га-
логепидов алюминия легко реагируют с бензолом и его гомологами с
образованием алкилзамещенных бензолов и галогеноводорода
(Щ. Фридель, Дж. Крафтс, 1877). Считают, что галогенид алюминия
при взаимодействии с галогеналкапом поляризует связь углерод —¦
галоген, увеличивая таким образом электрофпльность галогепал-
кана:
¦V R—Х~ -г А1Х] ^ri | -il*-R-X-*-AIXj -^г*":
-)(-¦> R+(AlX,)~ *- a-Комплекс •>. I |l -I-HX+AiXj
337
Бензол алкилируется труднее, чем толуол и ксилолы. В реак-
реакции алкилировання всегда образуются также ди- и триалкилпро-
дукты:
В реакциях алкилироваиия Фриделя — Крафтса в качестве ка-
катализаторов можно использовать и другие кислоты Льюиса, на-
например FeCl3, SnCI4, TiCl4, BFn.
Для алкилироваиия арепов можно использовать не только гало-
геналканы, но и алкепы или спирты в присутствии кислот. Электро-
фильным реагентом в этом случае могут быть карбокатионы или
близкие к ним ионные пары:
СНа-СН=С1Ь + Н+Х .^: СНз-
-1ЬО
СНз-СН —СНл + Н+Х-^ЗГ СНз-СН-СНз
он н-+0!{ х-
г) А ц и л и р о в а и и е. В присутствии катализаторов Фрнде-
ля — Крафтса ацилхлорнды реагируют с бензолом и его гомологами
с образованием ашглпроизводпых — арилкетопов:
51 о
R-С ь, + А1С13 =^ Г JLt
С [А1С1Л ¦ *- а -Комплекс »-|^ || + НС1 + AlCb
R
Трихлорнд алюминия поляризует молекулу ацплхлорида. О по-
получении арилкетонов п свойствах ацилхлоридов см. гл. XXXIII.
А.З.
д) Нитрование. Бензол и его гомологи при взаимодейст-
взаимодействии со смесью азотной и серной кислот (нитрующей смесью) об-
образуют нитросоединения. В реакции нитрования электрофильный
реагент образуется в результате протонирования азотной кислоты,
188
особенно в присутствии концентрированной серной кислоты:
IiNO3-t Ht-HSO4" г^1П2ХО3]>--. HSO7
н
ко," ; и2о
Электрофилыюй частицей является катион N0*' — катион пнт-
ропия или его ионные пары N02' Х~:
- |] + NOj"HSor*= \ H}-NO/HSO7—•-<т-Комплекс—»-f X + Hso<
Дальнейшее нитрование проходит трудно, так как mnporpyima яв-
является мощным электропоакцепторь'ым заместителем:
,N0, /-ч ,N0,
UNO,; I!.SO, Г|'
NO*
Толуол и ксилолы нитруются легче, чем бензол.
Подробнее о нитроарепах см. гл. XXI. В.
е) С у л ь ф и р о в а н и е. При взаимодействии с серной кис-
кислотой или олеумом бензол и его гомологи образуют ареисульфопо-
вые кислоты. Трудно сульфируется бензол, легче —¦ толуол, кси-
ксилолы и другие гомологи бензола. Электрофильпыми частицами з
реакции сульфирования могут быть триоксиД серы S03 или ирото-
нированный триоксид серы 1HSOJ ':
?н Hsor
-я-Комалекс-
При повышенной температуре в растворе олеума бензолсульфо-
новая кислота превращается в ж-бензолдисульфоиовую кислоту.
Об арепсульфоновых кислотах см. гл. XVIII.
ж) Г а л о г е п и р о в а и и е. Бензол и его гомологи хлори-
хлорируются, бронируются и иодируются. Реакции способствуют катали-
катализаторы, поляризующие молекулу галогена: галогениды железа,
алюминия и другие кислоты Лыоиса. Иногда к реакционной смеси
просто добавляют железные опилки. Так получают моногалогеп-
бензолы, дигалогенбензолы и полигалогецбензолы:
Вг.-Вг + ГеВг
¦—*¦ я-Комгшекс —*
Подробнее о галогепарепах см. гл. VIII. 2.2.
1S9
2. Влияние заместителей на реакционную способность бензоль-
бензольного кольца и на соотношение изомерного состава продукта реакции.
Кинетические исследования показывают сильнейшее влияние элек-
электронных эффектов (±/, ±М) заместителей на скорость реакций и на
соотношение в продуктах реакции о-, м- и n-изомеров. Это иллю-
иллюстрируют данные табл. 17 и 18. В табл. 17 приведены значения от-
относительной скорости бромирования С0Н5Х-|-Вг2->ВгСйН.,Х-;-НВг.
Таблица 17. Относительная скорость бромнроваиия производных
бензола, а-константы н электронные эффекты заместителей
Параметры реакции
и замес!ителей
Относительная
скорость
<
Электронный эф-
эффект
X
н
1
0
—
сн3
3-102
—0,3
! J
-\-м
ОС Из
10э
—0,6
—/
-\-м
NfCIhh
5-101;:
— 1,7
+м
С1
0,1
—0,1
—1
4-Л1
N0,
2-10-й
¦-0,78
— /
—Л!
В табл. 18 приведен изомерный состав продуктов нитрования
производных бензола CfiH5X-rHNO3-^O2NCeH4X-;H2O
Таблица 18. Содержание о-, м- и //-изомеров в продуктах
нитрования производных бензола, %
Изомер
орто-
мета-
пара-
X
СНз
58,5
4,5
37
(СПз)зС
16
4,5
72,5
ОСН.,
44
2
54
С1
29,5
1
69,5
\О2
6,0
93,5
0,5
Заместители с сильным электронодонорным эффектом (-гМ)
и весьма незначительным акцепторным индукционным эффектом
(—/) [ОСН3, N(CH;1) ] резко увеличивают скорость реакции. До-
норный эффект атомов галогенов вследствие сильного —/-эффекта
оказывает небольшое влияние на скорость реакции. Нитрогруппа
совсем пассивирует бензольное кольцо.
В случае электронодонорных заместителей в продуктах реакции
должно быть 66,7% о-изомера (имеются два о-положения) и 33,3%
л-изомера. Но такое соотношение никогда не достигается — о-изо-
о-изомера всегда меньше. Это объясняется как стерическими эффектами
(пространственное отталкивание, о/7/по-эффект), так и электронными
(в о-положении сильнее действует —/-эффект). О/7/ло-эффект осо-
особенно ярко виден на примерах Х=СН3 и (СН3KС (табл. 18).
190
В случае электрогюякцепторного заместителя в продуктах реак-
реакции преобладает .«-изомер.
Реакционную способность в о-, м- и «-положениях характеризу-
характеризуют факторами парциальной скорости (ФПС), Kqropbie рассчитывают
исходя из скоростей реакций о, м- и «-замещения по отношению к
незамещенному бгнзолу.
Чем больше ФПС для о- и «- и меньше для .«-положения, тем
более региоселективна реакция. Но селективность замещения за-
зависит также от активности реагента. Чем больше активность реа-
реагента, тем меньше селективность и наоборот. Это иллюстрируется
примерами бромирования толуола бромом (п-изомера 67%, .«-изоме-
.«-изомера 0,3%) и изопропилирования толуола (СН3JСНС1+А1С13 («-
изомера 46%, .«-изомера 26%). Реакция изопропилирования имеет
низкую селективность, так как реагент активный, а метильная груп-
группа показывает весьма слабый +М-эффект.
В реакциях электрофилыюго замещения переплетается очень
много различных действующих факторов, которые определяют об-
общую скорость реакции и соотношение о-, м- и «-изомеров.
3. Реакции гомологов бензола с участием боковой цепи. В такие
реакции вступает углеродный атом, который непосредственно свя-
связан с бензольным циклом. Обычно эти реакции носят свободнора-
дикальный характер (галогенирование, окисление). Промежуточной
частицей является свободный радикал бензилыюго типа (бензиль-
ный радикал), в котором неспаренный электрон значительно дело-
кализован вследствие сопряжения:
II +RH
Бензнльный радикал принадлежит к стабилизированным свобод-
свободным радикалам. Приближенные значения плотности нсснарепного
электрона приведены на схеме.
Время жизни бензилыюго радикала больше,
о.57(о чем обычных алкильных радикалов. По стабиль-
стабильности он сравним с аллильным радикалом. Реак-
Реакции в боковой.цепи инициируются нагреванием,
освещением, добавками веществ, генерирующих
свободные радикалы.
Так, хлорирование и бромировапие в боковой цепи толуола
осуществляется при нагревании и интенсивном освещении:
j-C12
,/СНз
C1.-J-
-f СЬ
Н-НС1 п т. д.
191
Свободные бензильные радикалы легко реагируют с кислородом
с образованием гидропероксидных радикалов, гпдропероксидов и
продуктов их дальнейшего превращения — альдегидов и карбоиовых
кислот:
+о
0 ,0
С"
Эти процессы окисления могут быть катализированы солями
металлов переменной валентности.
Окисление такими окислителями, как дихромат, пермангапат,
приводит к образованию карбоновых кислот:
,C001I
KMnO4
4. Реакции присоединения. Бензол и его гомологи гидрируются
трудно, при повышенных температуре и давлении и в присутствии
катализаторов гидрирования, например пористого никеля, так
называемого никеля Ренея:
180 е'
В реакции гидрирования удается выделить только конечный
продукт — циклогексан. Выделить промежуточные продукты (цпк-
логексадиепы, циклогексен) не удается, так как они гидрируются
быстрее, чем бензол. Это означает, что присоединение водорода к
стабильной сопряженной бл-электронпой системе бензола требует
больше энергии, чем присоединение к циклоалкадпенам и цпкло-
алкенам.
При гидрировании бензола выделяется около 208 кДж/моль
(теплота гидрирования), а при гидрировании циклогексена — 120
кДж/моль. Разница в теплотах гидрирования трех изолированных
двойных связей и бензола составляет 3-120—208=152 кДж/моль.
Из этого можно сделать вывод, что молекула бензола имеет энер-
энергию стабилизации около 150 кДж/моль (ср. гл. VI.А.3).
Хлор присоединяется к бензолу при интенсивном освеще-
освещении, лучше всего УФ-излучением. Впервые эту реакцию при
стоянии раствора хлора в бензоле на солнечном свету наблюдал
192
М. Фараден A826):
Cl
CL I
-3C13
/IV
Cl
SC1
О пространственном строении и использовании гексахлорцикло-
гсксана см. гл. VII.5.
Бензол и его гомологи присоединяют озон, при этом образуют-
образуются исключительно взрывчатые триозопиды, гидролизом которых
получают дикарбонильные соединения (днальдегнд глиоксаль, кето-
альдсгиды) и продукты их окисления — карбоновые кислоты:
° /С"Чсн
СН-0 >Hv
I о
о сн
з ^с-с^ + зн,о2-
НО
глиоксаль
но он
щавелевая кислота
.Ш,0
5. Фотохимическая изомеризация. При УФ-облучении бензола
обрат у юте я нестабильные валентные изомеры, легко превращающие-
превращающиеся снова в бензол:
беи .жалеи
hv .
бнцикло [2.2.0] -
гсксаднен-2,.5
(лдьюаролскип бензол")
призман
(„ бенюл
Ладенбургаи)
5. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ
Физические константы некоторых важнейших представителей были
приведены в табл. 15. Ниже рассмотрены главные методы их полу-
получения и практическое использование.
Бензол — наиболее широко применяющийся арен. Главным ис-
источником бензола являются продукты сухой перегонки каменного
угля. Из других методов можно упомянуть дегидроциклизацпю
гексана и деметилирование толуола и ксилолов. Так, в атмосфере
водорода при температуре 500...700 °С и давлении 4...6 МПа D0...
60 атм) толуол и ксилолы образуют бензол и метан.
7 N, 517
193
Хлорбензолы
(растворители)
Полимеры,
красители,
п;пывчатые
н.щества,
.лекарственные
препараты
NO,
Г
Полистирол,
синтешческий каучук
Рис. 70. Применение бензола
NH,
анилин
Красители,
лекарственные
препараты
:н=сн
^алеиноныц
ашилрнд
# an рол актам
Синтетическое
волокно
Полиэфиры
Бензол широко используется в качестве растворителя. Он рас-
растворяет жиры, каучук, различные масла, нефтепродукты, лаки,
полимеры. В 100 г бензола при 26 °С растворяется около 0,05 г воды.
Бензол с водой образует азеотропную смесь, кипящую при 69,2^0
и содержащую 9% воды. Поэтому бензол может быть исиользован
для отделения воды от различных смесей (отделение воды при по-
помощи азеотроппой перегонки).
Вдыхание паров бензола постепенно вызывает отравление орга-
организма.
Основное количество бензола используется в качестве исход-
исходного сырья для химической промышленности (рис. 70).
Толуол широко используется в химической промышленности.
Иго получают из продуктов сухой перегонки каменного угля п в
процессе дегидроциклизацпи гептана.
Толуол применяется в качестве растворителя в переработке
пластмасс, в производстве лаков, типографских красок, резины и
и качестве компонента для высокооктановых бензинов. Приме-
Применение толуола в химической промышленности изображено на рис. 71.
Ксилолы получают из продуктов сухой перегонки каменного
угля. При этом выделяют ксилольпую фракцию, состоящую из
о-, м- и «-ксилола и небольшой примеси зтилбепзола. Подобную
смесь ксилолов получают в процессе дегидроциклизаци" октанов.
Смесь изомеров разделяют на чистые ксилолы различными методами,
134
например кристаллизацией при низких температурах с последующей
фр<'н<Ц!:о:м:оп перегон коп.
Смесь ксилолов используют в качестве растзорптеля в лакокра-
comioii промышленности и в качестве компонент;! высокооктановых
бепзимоп.
В хпм пческол промышленности наиболее широко применяют о-
и «-ксилол, особенно для получения бензолдпкарбоповых кислот
(фталепых кислот — гл. XXXII.В).
Нитрованием ксилолов получают исходное сырье для производ-
производства красителем; трппптрокеилолы являются взрывчатыми вещест-
веществами.
Кумол (пзопропилбепзол) получают алкилироваиис-м бензола
пропеном. В настоящее время кумол является важным промежуточ-
промежуточным продуктом дли получения (фенола и ацетона из бензола и про-
ис-пл по к\мольному методу (гл. XV. Б. 2).
Стирол (впнплбензол) в небольших количествах содержится в
каменноугольной сюле. В настоящее время стирол получают в
промышленности в больших масштабах, главным образом дегидри-
дегидрированием этнлбепзола:
¦-/
СП- СИ,
-- п.,
Стирол представляет собой бесцветную жидкость со своеобраз-
своеобразным запахом, т. пл.— 30,G "С, т. кип. 145,2"С.
DjpbiD4.Mi.ii: [
Рас. 71 Применение io.iyo.ia
105
Стирол легко полимеризуется с образованием полистирола —
прекрасного электроизоляционного материала:
rCeH5-j C,Hs
Jn-2
Сополимер стирола и бутадиена — синтетический каучук СКС:
с6н3
nCH2=CH-CH = r« — /^
Б. ПОЛИЦИКЛИЧЕСКИЕ АРЕНЫ
С ИЗОЛИРОВАННЫМИ ЦИКЛАМИ
В полнциклических аренах этого типа бензольные циклы могут
быть соединены непосредственно (тип бифенила) или присоединены
вместо атомов водорода метана, этапа и др.
1. БИФЕНИЛ
Молекула бифенила содержит два бензольных цикла, соединенных
о-связью:
2 3 /СН*
3' 2
4 N
/ \==/ 3 \=/ \ /
fi> 6 5 3, 4'-Д11метилбифепил
бнфснил
Получение. Бифенил образуется во всех реакциях, где могут
возникать свободные фенильные радикалы, например при нагрева-
нагревании бензола до 700 °С:
2С6Ив-Сс„Н5-С6Н5-[-Н2
При нагревании галогенбензолов, особенно иодбензола, до
200. . .250 °С в присутствии медного порошка образуется бифенил
(реакция Ульмана):
2CuH5I-i-2Cu --+ СиН5-С0Н5-|-2Си1
В реакции Ульмана промежуточными продуктами могут быть
медьоргаиические соединения, например С6Н5Си, и свободные фе-
нильные радикалы СвН5-.
В последние годы открыта своеобразная реакция днмерпзацин
бензола и его гомологов в присутствии солен палладия:
-¦ PdX, -* {' V^ ^-bPd-; 2HX
196
В этой реакции выделяется металлический палладий. Если через
реакционную смесь продувают кислород, выделения палладия не
происходит, регенерируется Pd-1:
По-видимому, промежуточными продуктами в этой реакции явля-
являются иал,;;адийоргаинческне соединения.
Физические свойства и строение. Бифенил и его гомологи яв-
являются бесцветными кристаллическими веществами со слабым свое-
своеобразным запахом; т. пл. бифеиила 71 СС, т. кип. 254 СС. Бифенил
является термически очень устойчивым соединением.
Рентгенографические исследования свидетельствуют о том, что
молекула бифенпла является планариой: оба цикла находятся в
одной плоскости:
Если в положениях 2, 2', 6 и 6' имеются заместители, то ввиду
пространственных препятствий происходит поворот по связи 1—Г
и циклы ориентируются почти перпендикулярно. В случае различ-
различных заместителей в результате такого поворота молекула становит-
становится хиралыюй. Поэтому возможно существование двух эиантпоме-
ров, зеркальных изомеров:
в в В в
А А
А А
В ряду бифеиила такая изомерия называется атроппзомерией.
Если атропизомеры получены в чистом виде, они показывают опти-
оптическую активность (см. Введ. 6.12).
Переход одного атропизомера в другой происходит только при
еысоких температурах поворотом по .связи 1—Г.
Характерно, что молекула бпфеиила имеет более сопряженную
систему, чем бензол, поэтому электронодонориые свойства у пего
более ярко выражены, энергия ионизации меньше (8,2 эВ). Это
значительно облегчает реакции с электрофильиыми реагентами.
Химические свойства. Бифенил легко галогеиируется, нитрует-
нитруется и вступает в другие реакции электрофильного замещения. Глав-
Главным образом реакции идут в положениях 4 и 4':
// V_^ ^n и:<0\ // V_/^~
\=/ \=/ naso4 \^=/ х =
4-ш1тробифенил
4, 4'-дпнитробифепн.1
197
Применение. Бифенпл широко применяется в качестве теплопо-
смюля дтя обогревания химических реакторов и других установок.
Обычно используют смесь бифепила с дифениловым эфиром, так
нажигаемый даутерм. Производные бифепила (например, бензидии)
применяются в производстве красителей.
2. ДПФЕНПЛМЕТЛП
В молекуле дифенплмстана
с метиленовой группой:
н
два бензольных цикла соединены
Днфенилметан и его производные образуются в реакциях алки-
лирования по методу Фридсля — Крафтса из бепзнлхлорида и бен-
бензола пли их гомологов:
+ HCI
Дпфепилметаи и его гомологи являются бесцветными соедине-
соединениями со слабым приятным запахом. Дифенилметан плавится при
26—27°С, а кипит при 261— 262 °С.
В отличие от бифепила дифепилмстан содержит полностью изоли-
изолированные бензольные циклы, между ними пет сопряжения. По сво-
своим химическим свойствам днфс-пилмстан напоминает толуол. Он
легко реагирует с электрофильпыми реагентами, при этом в основ-
основном образуются 4,4'-дизамеш,еп11ые и 2,4,2'4'-четырехзамещенные
дифенплметапы:
,C\U,
IINO3
N0,
ChNT
/Л,
:/"\,
HN'Oj
H.SO,
В дифенилметанс легко окисляется метиленовая группа, при
этом образуется дпфенплкетоп (бепзофспоп):
О
,СН.,
198
[01
.Г\/
\
Дпфепплчетан используется в качестве добавки к растворителям
в лакокрасочной промышленности, а также для огдушки мыл.
3. ТРПФЕНПЛМЕТЛН
Трпфегплмстап является тризамещенпым метаном ji содержит три
изолированных бензольных цикла:
Получают трнфеиилметан реакцией Фриделя — Крафтса из
хлороформа и бензола:
CI
з-"~~4-! а-с-н—4<f V") си-1 зла
CI
Трпфеннлметан образует бесцветные кристаллы с т. пл. С2,5 "С
п т. кии. 36(ГС.
. Для трпфенплметлпа характерны реакции электрофилыюго за-
ммнения в бензольных циклах. Но самыми интересными являются
роакцпн у центрального атома углерода, связанные с образованием
!тпд;а стабильных карбаппона, свободного радикала и карбока-
тпона.
При взаимодействии с амидом натрия в растворе жидкого амми-
аммиака образуется трифенилметапид-ноп (красного цвета), происходит
ионизация связи С—II:
(/' ^
Несмотря на то что центральный углеродный атом в анноне на-
находится в состоянии ЬуО'--гибрндизан.ии, все три бензольных цикла
в одном плоскости разместиться не могут, так как мешает отталки-
отталкивание о-атомов водорода. Бензольные циклы вывернуты из плоско-
плоскости па 30. . .40" (структура «воздушного винта»):
Трифенилметан легко окисляется до трифенилметанола и гало-
генируется до трифенилгалогенметанов:
V) с-он
Эти реакции осуществляются по свободиорадикалыюму меха-
механизму. Свободный радикал трифенилметил принадлежит к стабили-
стабилизированным свободным радикалам. Наиболее просто его генериро-
генерировать из трифенилхлорметана и металлов (Zn, Na):
-) C-C\-'-Zn
3
\ \ /
Трифенилметил-радикал желтого цвета. В растворе он сущест-
существует в равновесии со своим димером. Впервые образование этого
радикала наблюдал в 1900 г. М. Гомберг, и это был первый случаи
получения относительно стабильного свободного радикала.
В равновесной смеси раствора существует 3—5% свободного
трифепилметил-радикала, он реагирует с кислородом и образует
пероксид, реагирует с иодом и образует трифен ил подметан.
Долгое время считали, что при димеризации трифенилметила
образуется гексафенилэтан:
Теперь установлено, что димер имеет совершенно другое строение:
О
По-видимому из-за пространственных затруднений образование
гексафенилэтана маловероятно, и димеризация осуществляется с
участием одного атома углерода бензольного цикла в /г-положеним.
Стабилизацию трифенилметила вызывает значительная делока-
лизация песпаренного электрона:
200
Трифенилметил-катиои (красно-оранжевого цвета) образуется
при ионизации трифенилхлорметана (раствор в жидком SO2, при-
присутствие А1С13) или трифенилметапола (раствор в концентрированной
H2SO4):
i-i Н,0
Трифенилметил-катион имеет большую сопряженную систему
и вследствие этого происходит значительная дслокализация поло-
положительного заряда, катион в определенных условиях является ста-
стабильной частицей.
С водой трифенилметил-катиои образует трифенилметаиол.
Практическое применение в качестве исходного для органическо-
органического синтеза нашли трифенилхлорметан, трифенилметанол, а также
трифенилметилтетрафторборат (C6H5KC+BF4~.
Большое значение имеют красители на основе трифенилметана
(гл. ХХХ.5).
В. ПОЛИЦИКЛИЧЕСКИЕ АРЕНЫ
С КОНДЕНСИРОВАННЫМИ ЦИКЛАМИ
Существует три основных типа конденсированных систем:
а) линейно конденсированные циклы:
нафтялпгг антрацен
тетрацен
б) апгулярно конденсированные циклы:
хрпзсн
201
в) пернкопдепагровапные циклы:
/^ /%. //Ч /%
Ч/Ч-/ Ч/Ч./
перплем коронсп
Полнцнклическпе арены с конденсированными циклами будут
рассмотрены но отдельным типам.
1. НАФТАЛИН
Впервые нафталин изолирован из продуктов сухой перегонки ка-
каменного угля в виде бесцветных кристаллов со своеобразным :!.-пд-
хом. Первым структурную формулу нафталина написал К. Грсбе
A866).
1. Изомерия и номенклатура. Углеродные атомы в молекуле на-
фталнпа неодинаковы; в ней имеются четыре одинаковых а-положе-
нпя и четыре р-ноложення:
а а 8 1
'\/Ч
Чт
Могут быть два мопозамещенпых нафталина:
СН3
I
а-четпл- р-ме тил нафталин
п.-.фтплпи
Дизамещепных нафталинов с одинаковыми заместителями может
быть десять.
2. Методы получения. Главным источником нафталина является
каменноугольная смола. Известны методы дегндроциклизацин ал-
кчиов Сю—С]2, в результате чего получаются нафталин и его гомо-
гомологи.
3. Физические свойства и природа связей. Нафталин и его го-
гомологи (табл. 19) представляют собой бесцветные вещества со свое-
своеобразным неприятным запахом.
Рентгенографические п электронографические данные свидетель-
свидетельствуют о том, что молекула нафталина планарна, оба цикла одина-
одинаковы, но длины связен различны. На рис. 72 приведены данные,
полученные методом электронографии A981).
2С2
Таблица 19. Температура плавления и кипения
нафталина и некоторых его гомологов
Соединение
Нафталин
1-Метплнафталин
2-Метилиафталги1
Й.б-Диметплнафтялин
Т. пл., СС
80
—22
32
ПО
Т. кип., 'С
218
(легко возгоняется)
241
241
261
Сопряженную л-электронную систему нафталина можно характе-
характеризовать расчетами по методу МО Хюккеля. Па рис. 73 и 74 приве-
приведены диаграмма энергетических уровней и некоторые квантово-
химлческие параметры. Энергия я-электронов нафталина Еп~-
— Юс. 1-13,684E; энергия делокализации Л??сл—3,684 р.
Энергия циклической делокализации нафталина получена срав-
сравнением с о-дивипнлбензолом, и она оказывается меньше, чем для
-2/J
Н
II
II
?s = « + 0,618 В
?j c« + 1.3ОЗД
Рис. 72. Длины Связей
(им) и иалентные углы
в молекуле нафталина
Ч- (ВЗМО)
/Г," =а +
Р«с. 7,?. Диа|-рамма энер-
энергетических уровнем нафта-
нафталина
(НСМО)
1,1804
0,0691
0,0691
порядок 71-соязей рг<
Рис. 74. Одиоэлектромная плотность но фронтальных орСигалях нафталина
и порядок л-связей
бензола: Д?^ш<л-дел= 0,844 р. Это означает, ч;о образование кон-
конденсированной системы из двух циклов дает меньший выигрыш энер-
энергии, и в реакциях легче будет идти присоединение и превращен;rj
системы нафталина в систему дизамещенного бензола.
Энергия ВЗАЮ нафталина значительно выше, чем для бензола,
что свидетельствует об увеличении электронодонориых свойств
(ЭП—8,13 эВ). Распределение плотности электронов в ВЗМО п
НСМО показывает, что наиболее уязвимым местом нафталина явля-
является а-положенпе.
203
о , , ^ваиы, чем в беп-
В молекуле нафталина л-снязп более локалнзб» '
золе, есть разница в длинах связей и prs. Поэтому А
применение обычных структурных формул Кекул
более оправдано
)
и , /А) и (в). Однако
Иногда встречаются структурные формулы \и кула нафталина
способ написания (б) малоонравдан, так как м°л зависимые с„сте.
никак не может быть идентифицирована как две не'
мы бензола. ,„ны р е а к ц и и
4. Химические свойства, а) Для нафталина типН , ,„, . _,„_»„„_
замещения при взаимодействии с электрофи.'* rovo-югов
ми, которые проходят легче, чем в случае бензола '
В тоже время легче протекают и реакции присое* д
Для нафталина характерны более сильные эл^r -fin-nv
свойства, чем для бензола и его гомологов, поэтому , ' ч , р'п
ет п-комнлексы. В отличие от бензола могут сгр
о-ксмплекса в положениях ее и |3:
н Е
Е X
Поэтому в реакциях могут получаться два изомера' * ' ' ь °
в реакциях замещения получаются а-изомеры. Эт^ - гс с
данными квантово-хнмических расчетов (илотносТ^ ~" с° 3
ВЗМО больше в а-ноложепии). Кроме того, 0 с"'дКе г]
гибридным углеродным атомом в ot-ноложенин боле '
этому реакции в а-иоложении идут быстрее. Но в vfi ° ,VX J • а~
ях, если реакция обратима, при повышенных темпер "¦ а' ° )ра У"
ется р-пзомер (сульфирование). Иногда роль играе^ ж Р"сг
рнтель. ,
б) Л л к и л и р о в а и и е и а ц и л и'р о в а ^ q5 насР™ли~
на происходят легче, чем в случае бензола и толуол^' , 1!О р
зуется смесь а- и E-замещенпых. Их соотношение M^riji|° p r^J 1|р
вать изменением растворителя и температуры
в) Нитрование легко осуществляется
NO2
fIN'Oj
КПС10тол.
а-ннтро-
нафталии
204
При более интенсивном нитровании получается смесь 1,5- и 1,8-ди-
пнтроиафталииов.
г) Сульфирование происходит при растворении нафта-
нафталина в концентрированной серной кислоте:
SO3H
I
/S ¦'%.
f.O 'С
-I H2SO4 -
1 ГО "С
SO,H
В зависимости от температуры реакции получаются преимущест-
преимущественно или а-, или р-нафталинеульфоиовые кислоты. а-Изомер при
нагревании в серной кислоте может превращаться в E-изомер. При
160 "С образуется равновесная смесь с преимущественным содержа-
содержанием |3-нзомера:
SO3H
/SO3H
H.SO,: 1G0-C '¦-'"'
У/
19%
81%
Р-Изомер термодинамически более стабилен, чем а-изомер, по-
поэтому в обратимой реакции сульфирования нафталина при высоких
температурах, где достигается термодинамическое равновесие, боль-
больше образуется E-изомера (термодинамически контролируемая реак-
реакция). При более низких температурах термодинамическое равнове-
равновесие не достигается, поэтому получается а-изомер, образование кото-
которого идет быстрее (кинетически контролируемая реакция) (см. гл.
III. Б.З).
д) Талогенирование проходит очень легко:
Вг
'V
-гВг2
^ '' V/
-;-нвг
с-бром-
В избытке брома получается смесь 1,4- и 1,5-дибромиафталинов.
Иногда при галогепировании образуются также продукты присое-
присоединения галогена к двойным связям одного бензольного цикла.
е) Для нафталина характерны также реакции присое-
присоединен и я. Легче, чем к бензолу, присоединяется хлор, извест-
известны реакции присоединения брома. При каталитическом гидриро-
205
n:iiiiiii иафтал1!на легко получается тетрагидронафталин (тстралпн).
Дальнейшее гидрирование до декалина идет в более жестких усло-
условиях:
/N
Тсо' с
Ч/Ч/
11. Ni
200-..00 С
докали;!
ж) Нафталин окисляется различными окислителями. Ко-
Конечным продуктом окисления является фталевая кислота:
о.
Кат.
ххюн
¦соон
о
li
о
Окисление осуществляют в промышленных масштабах в паровой
фазе над оксидными катализаторами, содержащими V2O3 при попи-
попиленной температуре. В этих условиях образующаяся фталевая кис-
кислота превращается во фталевый ангидрид.
5. Применение. Нафталин используется главным образом для
производства фталевого ангидрида. Продукты гидрирования нафта-
нафталина тетралпн и декалин применяются в качестве растворителей в
лакокрасочной промышленности. Нафталинсульфокислоты и нитро-
нафталин являются исходными веществами для производства кра-
красителе».
2. ЛНТРАЦПН
Впервые антрацен выделен из каменноугольной смолы в 18Л2 г.
Дюма и Лораном. Структурная формула с тремя конденсированны-
конденсированными бензольными циклами написана позже, в 1866 г.:
10
С1Г3
2, 9 димет!
¦спя
1. Синтетические методы получения. Антрацен и его гомологи
могут быть получены в реакциях алкилировапия по методу Фриде-
ля—Крафтса при использовании в качестве исходных веществ
бензола, его гомологов, галогенметнлпроизводных, полнгало-
геналканов. Например, при взаимодействии бензилхлорида с А1С13
образуется антрацен. Происходит алкилировапне бензилхлорч.ча
Сензилхлоридом, промежуточным продуктом является 9,10-дмгпдро-
206
антрацен, который легко отщепляет водород:
-II,
с;н,с/
fl, Ю-днгндроан-рот и
ampan.eu
Антрацены получают также восстановлением 9, Ю-аитрахиноиа
и его производных (гл. XXX.5).
2. Физические свойства и строение. Антрацен и его гомологи
являются бесцветными или бледно-желтыми кристаллическими ве-
веществами с высокими температурами плавления. Так, антрацен
плавится при 216,6 СС, а кипит при 351 СС. Для растворов и особо
чистых кристаллов характерна фиолетовая флуоресценция.
Антрацен имеет ярко выраженные электроиодоиорные свойства
(ЭИ = 7,4 эВ).
Длины связей в молекулах антрацена и нафталина близки. Углы
между связями 119—12Г.
Кваптово-химические расчеты показы-
показывают, что наибольшая свободная валент-
валентность наблюдается в положениях 9 и 10.
Там и ожидается наибольшая реакционная
способность.
3. Химические свойства. Химические свойства антрацена отли-
отличаются некоторыми особенностями. Взаимодействие с электрофиль-
ными реагентами проходит легко, по во многих случаях сначала
получаются продукты присоединения в положениях 9 и 10. При на-
нагревании продукты присоединения превращаются в замещенные
антрацены. Для антрацена более, чем для нафталина, характерны
реакции присоединения.
При галогеиированин могут быть выделены промежуточные про-
продукты присоединения (X--CI, Вг):
Я X
В реакции сульфирования при повышенной температуре удается
получить 1- и 2-антрацеисульфоновые кис.юты. Первичным продук-
продуктом является 9-антраценсульфопоБая кислота, которая перегруппи-
перегруппировывается:
SO3H
HSOt
Антрацен легко гидрируется водородом в присутствии катализа-
катализатора с образованием 9,10-дигпдроантрацепа.
Своеобразно присоединение активных диенофилов (соединений
с активированной двойной связью), когда антрацен выступает в ка-
качестве сопряженного диена:
ангидрид
Антрацен весьма легко окисляется. Первичным продуктом окис-
окисления кислородом при освещении является нестабильный фотоок-
фотооксид:
9,10-антрахннон
Конечным продуктом окисления является 9,10-антрахинон. Это же
соединение образуется при окислении антрацена другими окисли-
окислителями.
Все химические свойства антрацена свидетельствуют о том, что
это соединение во многом значительно отличается от нафталина и
тем более от бензола. Доминируют реакции присоединения, сильно
уменьшена тенденция регенерировать в реакциях сопряженную си-
систему антрацена.
Применение. Антрацен высокой чистоты (содержание примесей
ниже 10%) изучается как органический полупроводник и фоточув-
фоточувствительный материал. Большое значение имеет 9,10-антрахипол —
исходное для важных красителей (гл. XXX.5).
3. ФЕЫАНТРЕН
Фенантрен, так же как и антрацен, содержится в каменноугольной
смоле и является структурным изомером антрацена с аигулярно
конденсированными циклами:
С 3 4
фенантрен
Для мопозамещенных фепантренов существует пять изомеров
A-, 2-, 3-, 4- и 9-), для мопозамещенных антраценов — только три
A-, 2- и 9-).
208
1. Синтетические методы получения. Кроме извлечения из ка-
каменноугольной смолы известны также пути синтетического получе-
получения фенантрена и его гомологов. Эти методы основываются на де-
гпдроцнклизации о,о'-диалкплбифенилов или производных 1,2-дп-
фенилэтана:
о — СН-,
1, 2-дифен1Ы jT
\-
Ч-2Н2
2. Физические свойства и строение. Фенаптрен и его гомологи
представляют собой бесцветные кристаллические вещества с более
низкой температурой плавления и лучшей растворимостью, чем со-
соответствующие аналоги антрацена. Например, фенантрен плавится
при 100СС, а кипит при 340 С.
Молекула фенантрена является более стабильной, чем молекула
антрацена, разница составляет около 33 кДж/моль. Донорные свой-
свойства у фенантрена выражены слабее, чем у антрацена (ЭИ^7,9
зВ). Это свидетельствует о том, что в молекуле фенантрена осущест-
осуществляется более значительное взаимодействие л-электронов (делока-
лизация), чем в молекуле антрацена.
3. Химические свойства. Фенантрен легко вступает в реакции
с электрофильными реагентами. Реакционная способность у фенан-
фенантрена ниже, чем у антрацена. Наиболее активными являются поло-
положения 9 и 10, при этом в отдельных случаях можно выделить про-
промежуточные продукты присоединения. Например:
f Br2
+ НВг
При каталитическом гидрировании феиантреиа получается 9,10-
дигидрофеиантрен. В более жестких условиях может быть получен
тгтрадекагидрофеиантрен (пергидрофенаптрен).
Окисление фенантрена различными окислителями приводит к
фенантренхинону:
фенамтренхнпон
4. Применение. Фенантрен применяется в качестве исходного
сырья в органическом синтезе, например для получения фенантрен-
хинона, который, в свою очередь, используется для синтеза краси-
красителей. Производные фенантрена, особенно частично или полностью
гидрированные, содержатся в природных продуктах (алкалоиды,
стероиды).
209
4. ТЕТРАЦЕН П ПЕНТАЦЕН
Тетрацен впервые был выделен из технического антрацена методом
хроматографии. Это значит, что тетрацен в небольших количествах
содержится в каменноугольной смоле. В еще меньших количествах
там содержится пентацен:
i
10
1
7
М
/%
о'
12
5
тетрлцен
[
\У/
4
2 10
3 9
1 1
¦У N
8
12
/^
\у/
7
13
0
1 1
5
пентацен
1. Получение. Тетрацен и пентацен получают восстановлением
кислородсодержащих производных — хинонов и гидроксисоедине-
ний. Эти соединения соответственно получают различными реак-
реакциями ацилировапия и конденсации:
О ОН О
[И]
[Н]
|| I
о ом
дмгидрокситстрацеп-
XI! НО II
I
о
тетр.-]цсн\-1111он
2. Физические свойства и строение. Тетрацен и пентацен вы-
выделяются среди других аренов своеобразными физическими свойст-
свойствами. Тетрацен образует желто-оранжевые кристаллы с температу-
температурой плавления 357 "С, а пентацен — сине-фиолетовые кристаллы с
температурой разложения более 300 СС. При пониженном давлении
тетрацен и пентацен возгоняются.
При увеличении числа циклов в линейно конденсированных но-
лнциклических аренах значительно понижается как энергия иони-
ионизации, так и энергия возбуждения молекулы. В табл. 20 приве-
приведены значения энергии ионизации и максимумы поглощения, со-
Табмща 20. Энергии ионизации и длинноволновое
поглощение некоторых аренов
Соединение
Бензол
Нафталин
Антрацен
Тетрацен
Пентацсн
ЭИ, эВ
9,24
8,13
7,41
7,01
6,01
11огло;ценпе, соответствующее
П'. pes-оду So - , 6", @-0)
1. им
2Й1.5
315
374
473
580
е
-200
-300
-8 000
-10000
-12 000
210
птстствующие переходу в первое возбужденное состояние (S0-vS,).
В таблице приведен только один из нескольких максимумов ко.1й-
гятсльной структуры перехода Sa-+Slt соответствующий так назы-
гасмому 0—0-нереходу.
3. Химические свойства и применение. Благодаря низким энер-
энергиям ионизации тетрацеп и пептацен очень легко реагируют с элек-
трофмлыгыми реагентами, окисляются. Наиболее активными поло-
положениями являются 5, 6, 11, 12 для тетрацепа и 6, 13 — дтя пел i a-
цепа. При окислении получаются хитоны.
Тетрацеп и пентаце-н могут быть использованы как органические
полупроводники и фотопрогсодннки. При освещении тонкой пленки
этих веществ резко повышается их электропроводность.
На основе тетрацепа синтезируют его халькогенпронзводпые, ко-
которые используются для получения материалов с металлической
проьодпмостью (органических металлов):
Х-Х
I I
X—X
ч'п тькогомототрг.
(X:-S, Sc, Те)
5. ППРПКОНДГ.ПСПРОВ \ННЫЕ
ГЮЛПЦИК.ЛИЧ1-СКИЕ АРЕНЫ
Главным источником получения перикондепеированных аренов яв-
является каменноугольная смола.
ю 1 1. Пирсн является бесцветным кристаллическим
веществом с т. пл. 156 "С, его растворы имеют харак-
характерную голубую флуоресценцию. Энергия ионизации
пирсиа G,4 эВ) выше, чем тетрацена.
Пнрен легко вступает в реакции электрофилыюго за-
замещения (нитрование, сульфирование, галогеннрова-
нне). По химическим свойствам пнрен напоминаоу
нафталин, только имеет большую реакционную сис-
собность. При нитровании получается 1-ннтроннреп, при окисле-
окислении-— 1,6-пиренхинон:
П'фСН
NO.,
I
О
гот
!
О
211
Пирен является исходным сырьем для синтеза органических
красителей.
2. Бензпирен — вещество желтого цвета с
т.пл. 179 СС. В небольших количествах содержится
в каменноугольной смоле. Для бензпирена харак-
характерно сильное физиологическое действие — он
| || I принадлежит к канцерогенным с о е д н-
Nx/x.-^/ нениям, вызывая образование злокачествеи-
бемзпирен кых оиуХОЛСЙ, ГЛЭЕНЫМ обраЗОМ рЭКЭ КОЖИ. По-
добные свойства обнаружены и у некоторых других периконденси-
рованных аренов.
2 3. Перилен представляет собой золотисто-жел-
i[( з тые кристаллы с т. пл. 274 ГС. Впервые был вы-
12 || делен из каменноугольной смолы. Может быть по-
п/ >/Х V^V лучен из 1,8-дииоднафталнна нагреванием в при-
присутствии меди или термическим декарбоксилиро-
ванием 3,4,9,10-перилентетракарбоповой кислоты.
б7 Перилен является сильным электронодонором, его
ц7 энергия ионизации G,0 эВ) сравнима с ЭИ тетраце-
^/ на. Легко вступает в реакции с электрофиль-
черчлен ными реагентами, окисляется. По химическим
свойствам напоминает нафталин. При нитровании перилена полу-
получается 3,10-динитроперилен, при окислении — 3,10-периленхннон.
Производные перилена являются важными органическими кра-
красителями, в том числе люмннесцнрующими красителями (днимиды
перилентетракарбоновой кислоты).
Г. СТАБИЛЬНОСТЬ ЦИКЛИЧЕСКИХ СОПРЯЖЕННЫХ СИСТЕМ
И ПОНЯТИЕ ОБ АРОМАТИЧНОСТИ
Из рассмотренного выше экспериментального материала следует,
что для шестичленных циклических систем с тремя двойными свя-
связями (бензольная система) характерна выдающаяся стабильность.
Трудно протекают реакции присоединения, доминируют своеобраз-
своеобразные реакции замещения. Поэтому такие соединения выделили в
особый класс, дав им название ароматические углеводороды или
ароматические соединения (некоторые соединения такого типа обла-
обладают приятным запахом). Структурные особенности (система цик-
логексатриеиа) и особые химические свойства (легкое замещение,
трудное присоединение) стали называть ароматическими свойства-
свойствами. Впоследствии к ароматическим соединениям причислили также
все полициклические соединения с конденсированными циклами,
несмотря на то, что не все они отвечали основному требованию —
вступать только в реакции замещения.
Для соединений бензольного ряда почти во всех реакциях харак-
характерна тенденция сохранить тип (систему циклогексатриена), реге-
регенерировать систему циклогексатриена. Эта способность к регенера-
регенерации типа уменьшается при переходе к полициклическим системам
с конденсированными циклами. Особенно резко это наблюдается
в ряду линейно конденсированных соединений (нафталин->пента-
212
цен). Напротив, для периконденсированных аренов способность к
регенерации сопряженной системы изменяется меньше и напомина-
напоминает поведение системы нафталина.
Большую стабильность системы циклогексатриепа объяснил
Э. Хюккель в 1931 г. (гл. УЬ^А.З). Основываясь на результатах
кваптово-хнмических расчетов, он показал, что при образовании
циклических замкнутых сопряженных систем происходит цикличе-
циклическая делокализация я-электронов, что дает большой выигрыш энер-
энергии (энергия делокализации).
Для циклических сопряженных систем характерно образование
вырожденных о р б и т а л е й. Хюккель предполагал, что
стабильность системы зависит также от заполнения этих орбиталей
электронами. Расчеты показали, что наиболее стабильные системы
получаются в том случае, если обе связывающие вырожденные орби-
тали заполнены Dл-электрона). Кроме того, каждая циклическая
система имеет одну наиболее низко расположенную орбнталь с дву-
двумя я-электронамн. Исходя из этого, Хюккель впервые сформулиро-
сформулировал правило стабильности циклических сопряженных систем.
Правило Хюккеля гласит, что стабильными являются такие цик-
циклические пленарные замкнутые сопряженные системы, которые со-
содержат в цикле Dя+2) я-электрона, где я=0, 1,2,...
Таким образом, стабильными являются циклические пленарные
сопряженные системы с я—2, 6, 10 и 14 я-электронов.
Следует особо обратить внимание на то, что правило Хюккеля
относится только к моноциклическим системам, при этом они долж-
должны быть плоскими (очевидно, циклодекапентаен не будет стабилизи-
стабилизированным ввиду непланарности молекулы, несмотря на присутствие
десяти я-электронов). Для полициклических систем (нафталин,
антрацен, фенантрен и др.) нет вырожденных молекулярных орби-
орбиталей и нет общих правил для характеристики стабильности. Срав-
Сравнение стабильности этих аренов возможно только после проведения
расчетов для каждого соединения в отдельности.
На основе правила Хюккеля впервые удалось объяснить особен-
особенности реакционной способности и стабильность ароматических сое-
соединений бензольного ряда, а также, с некоторой погрешностью, по-
полициклических соединений.
Теоретически было показано, что эффект стабилизации присущ
не только сопряженным шестичленным системам, но и трех-, четы-
четырех-, пяти- и семичленным сопряженным системам, число я-элект-
я-электронов в которых соответствует правилу Хюккеля. Впоследствии та-
такие соединения были синтезированы и подробно изучены. Соедине-
Соединения такого типа были названы небензоидньши (небензольными) аро-
ароматическими системами.
Если число я-электронов в циклической сопряженной системе
не соответствует правилу Хюккеля, то обычно эти соединения мало-
малоустойчивы (например, системы с 4я-электронами, циклобутаднен).
Такие системы иногда называют антиароматическими.
Па рис. 75—79 приведены примеры трех-, четырех-, пят»-,
шести- и семичленных систем с различным числом я-электронов,
213
Цикпопропенил-катион Циклопропенил-
(„небензоидная радикал
ароматическая"
система)
Циклопропенид-нон-
Hapoviariiw
система)
-Р-
»
Р
2/4
= 2/5
0,18/?
О
-0,82/1
А
Рис. 75. Расположение МО и энергия делокалияаци» и цикличес-
циклической делокалнзацин трехчленных сопряженных систем
Циклобутгндиил-катион ЦиклоБуталигн Циклобутсилнид-иогг
Снебснзоилная („агп парома шческая" (..небенюидная
ароматическая" система) система) ароматическая" система)
{ f. _}
А Г
ЛОЛ
= 2
11ПКЛ.ДСЛ _я
п l
' ~>
i 2 +- 1
0
764Д -О,472|?
ч _-*
2/J
0.764/J
ч _ ^
Pi/;1. 7ff. Расположение МО н энергия делокализации и циклической
делокализации четырехчленных сопрял^ениых систем
214
Цик.чопснтлдпенил-
к;пио;т
(„ангиарима гическия
систсм;Г)
Цнклопентаднсннл- Циклоиентадкспид-но!!
радикал (<• небстоидпая
ароматическая'1
система)
-20-
-0-
1|
Ркг. 77. Расположение МО и энергия делокализацни и циклической
делокалпзацли нятичленпых сопряженных систем
Kai ион-радикал
бензола
„Ароматическая"
сиосма
-20-
Анион-радикал
бен юла
О
7л;
---* +¦ -
20 ЗР
],О120 0,457/?
Рис. 7>1. Расиоложе::не .МО и энергия дслокализацпи и цикличес-
циклической дслохализацни шестичленпых сопряженных систем
215
Катион троннлия
(..небенэондная Циклогетгатриенил- Цнклогептатриеннд-
арсмашческая" система) радикал нон
о<
-0.445
ЛГ?СЛ = 2,988/? 2,543/? ХШР
,.1;гкл. Д=л _ q^^
с. 79. Расположение МО и энергия делокализации и цикличе-
циклической делокализации семичленных сопряженных систем
даны энергии орбиталей и энергии делокализации и циклической
делокализации. На этих примерах отчетливо демонстрируется содер-
содержание правила Хюккеля.
Главной особенностью циклических замкнутых сопряженных
систем является их стабильность, что определяется циклической
делокализацией я-электронов. Поэтому наиболее точно такие си-
системы могут быть охарактеризованы энергией циклической делока-
делокализации (разницей между расчетными энергиями я-электронов цик-
циклической и аналогичной, но нециклической системы) (см. гл. VI.А.3).
Для рассмотренных систем энергия циклической делокализации
меняется в интервале от 1,18 р (катион циклопропенилия) и 1,012 р
(бензол) до —0,472 р (циклобутадиен) и даже до —0,82 р (цикло-
пропенид-ион). Совершенно ясно, что отрицательные значения энер-
энергии обозначают крайнюю нестабильность системы и, возможно, оп-
оправдывают определение «антиароматичность». Однако имеются зна-
значения энергии циклической делокализации меньше р, но больше
0,2 р. Какие системы считать достаточно стабилизированными?
Исходя из правила Хюккеля, такими системами могут быть сопря-
сопряженные циклические молекулы с Д??"кл' дсл>0,76 р.
Известен ряд небензоидных ароматических систем, обладающих
стабилизацией вследствие циклической делокализации, например
производные катиона циклопропенилия (см. гл. V.A), циклонепта-
дненнд-иопа (см. гл. V.B), катиона тропилия (гл. V-Д). Синтезиро-
Синтезированы аннулены с 14 и 18 л-электронами, которые выявляют харак-
характерные для ароматических соединений сигналы протонов в спектрах
ПМР, но стабильность их меньше ввиду внутримолекулярного от-
талкпванпя атомов водорода внутри цикла, что делает молекулу
2:0
непланарной:
н
н н
Стабильной является также своеобразная система из конденси-
конденсированных циклов циклонентадиена и цнклогептатриена —¦ азулен—
кристаллическое вещество синего цвета:
Но в то же время существуют циклические сопряженные системы
с небольшой энергией циклической делокализации (например, сво-
свободные радикалы циклопентадиенил, циклогептатриенил, ион-ра-
ион-радикалы бензола). Считать ли их ароматическими?
Поэтому необходимо уточнить, что именно в настоящее время
подразумевается под понятием «ароматичность». Одно из определе-
определений следующее: ароматический — это обладающий химическими
свойствами, аналогичными свойствам бензола. Считается, что цик-
циклическая молекула с сопряженной системой связей, устойчивость
которой значительно больше, чем устойчивость гипотетической клас-
классической структуры, обладает ароматическим характером.
По другому определению ароматическими являются циклические
сопряженные системы, которые более стабильны, чем аналогичные
сопряженные системы с открытой цепью, т.е. имеют достаточно
большую энергию циклической делокализацни л-электронов.
В этих определениях имеются противоречия. Не все стабилизи-
стабилизированные циклические системы, называемые ароматическими, обла-
обладают химическими свойствами бензола, т. е. не все вступают в ре-
реакции электрофильиого замещения.
Таким образом, понятие «ароматичность» уже давно потеряло
свое первоначальное значение и теперь применяется по традиции
для удобного и сугубо качественного обозначения некоторых специ-
специфических свойств циклических сопряженных систем.
К понятию «ароматический» вполне применима цитата из книги
Л. Кэррола «Алиса в стране чудес»: «Кот исчезал очень медленно,
начиная с кончика хвеста и кончая улыбкой, которая оставалась
еще некоторое время после того, как все остальное скрылось».
ФуНКЦ
иональные
Галогенпроизводиые
углеводородов
Наиболее простыми производными углеводородов являются гало-
геппроь::водпые, в которых один или более атомов водорода в угле-
углеводороде замещены атомами галогенов (F, C1, Вг, I).
Число галогеинроизводпых очень велико. Это определяется раз-
разнообразном самих углеводородов, расположением атомов галогена,
числом и типом атомов галогена.
В основе классификации галогеппролзводных углеводородов ле-
лежит не принадлежность углеводорода к тому или иному классу, а
тип атома углерода, с которым связан атом галогена. В каждой
группе мо;;;по выделить подгруппы в зависимости от строения угле-
углеводородного остатка, природы галогена и числа атомов галогена.
В основу этой классификации положена гибридизация углеродного
атома {sp3, sp* и sp) или, иначе говоря, пространственное располо-
расположение заместителей у этого углеродного атома (тетраэдричеекнй,
три тональный, дн тональный).
I. ГалогеппроизБодпые со связью C(sp3)—галоген:
R--C— X
(X-F, C1, Вг, I).
II. Галогеппроизводпые со связью С (sp2)—-галоген:
R\
Y= с'
III. Галогенпронзводные со связью С (sp)—галоген:
Зги группы галоген производных значительно различаются по
физическим и особенно химическим свойствам, поэтому такая клас-
классификация вполне оправдана.
?18
В классг галогегтроизводпых углеводородов рассмотрим
также специальные их производные — галониевые соединения
[R—X ' —КJ\ ~.
Глава VII
ГАЛОГЕНПРОИЗВОДНЫЕ ТИПА C(spa)—X
1. КЛАССИФИКАЦИЯ И НОМЕНКЛАТУРА
К галогеппронзводным углеводородов со связью Cfs/r)—X (тетра-
эдрпческий углерод — галоген) принадлежат:
а) галогеналканы С„Н.2п.мХ; CnU.,ni.2.iaXlll; X—-F, C1, Вг, I;
б) нергалогеиалканы CKX5nf«;
в) галогенциклоалканы;
г) галогепалкены с атомом галогена в заместителе у этиленовой
R. /R
связи /С--С
R/ ^С„Но„-Х;
д) галогеналкины с атомом галогена в заместителе у ацетилено-
ацетиленовой связи R- С =С- С„Н.,„— X;
е) галогенарены с атомом галогена в боковой цепи
Лг —С„Н2„—X; Ar —C,,H-i,i A-гаХ,л
В основе названия галогенуглеводорода лежит название самой
длинной неразветвленной цепи. Атомы углерода нумеруют таким
образом, чтобы меньший локант получил заместитель, которы» в
названии пишется первым, а сами заместители перечисляются в по-
порядке алфавита.
Цепь углеродных атомов в алкенах и алкинах нумеруют так,
чтобы меньшие локапты получили атомы углерода кратной свя-
связи:
5 4 3 2 1
СИ., —СН —СН2 —СН —СН3 2-метнл-4-хлорп?нтан
I I
С1 СИ;,
1 2 3 4 Г. С
СП, — СИ — СИ — СИ — СП — СП3 2-бром-4-м(-т1тл-5-фгор-3-хлоргексан
I I I I
Rr C1 СН3 F
S 4 3 2 1
СП;- СИ — СП^-С— CU3 2-метпл-4-хл!-р!юмтеп-2
I I
С! С Из
I 2 3 4 Г:
С!С:[,-С: С—СП,СП2С1 1,г-;1н.\лорпонтп1;-2
Cc!!,-Cn,Cn,F З-фс-ппл-ГНт.-рзтаи
Для ч.екоторых простейших галогенпрокзтзодных углеводоролов
сохраняются также названия, в основе которых лежит название
210
углеводородного остатка:
СНПС1 метилхлорид
СИЛ метилиодид
QHr,Br этилбромид
CH2C12 метилепхлорнд
CLei [5CH2Cl бензплхлорид
С6Н5СНС12 бензилиденхлорид
Для некоторых галогенпроизводных сохраняются тривиальные
названия:
СНС13 хлороформ
СНВга бромоформ
СН1Я йодоформ
Для названия полностью галогенированных углеводородов ис-
используется префикс пер-:
QF6 перфторэтаи
QCl,, иерхлорпропан
Для соединения СС14 применяется название тетрахлорпд угле-
углерода или четыреххлсристып углерод.
2. МЕТОДЫ ПОЛУЧЕНИЯ
Галогенпроизводные углеводородов получают прямым галогенн-
роваиием углеводородов, присоединением галогенов или галогсн-
водородов к алкенам и алкинам, замещением кислородсодержащих
или других групп на галоген (рис. 80).
1. Прямое галогенирование. Таким методом можно получить
фтор-, хлор- и бромуглеводороды. Прямое иодирование алканов
неизвестно. Методы являются промышленными.
RCH=CH
RCH,CHj
RCHCHj
ОН
RCIICH,
RCH=CH,
i
Рис. 80. Генетическая связь между углеводородами, их галогон-
производиыми и кислородсодержащими соединениями
220
а) Фторирование. Фтор с алканами реагирует очень
энергично, даже со взрывом, так как в реакции выделяется много
теплоты (большая энергия образования связи С—F). Поэтому для
фторирования алканов необходимы специальные условия: разбавле-
разбавление фтора азотом, специальная конструкция реакторов с медными
сетками для эффективного отвода теплрты реакции.
В качестве переносчиков фтора используют фториды металлов
(CoF2, AeF, MnF2). Под действием фтора образуются CoF3, AeF2,
MnF4, которые фторируют алканы. Так получают перфторалканы:
F,; CnF,
l
¦ 2
Реакция протекает по свободнорадикальному механизму.
б) Хлорирование. Алканы реагируют с хлором под дей-
действием УФ-облучепия или температуры, которые инициируют обра-
образование свободных радикалов. Эквимолярная смесь метана и хлора
при УФ-облучении может взрываться. Поэтому хлорирование осу-
осуществляют в избытке алкана в реакторах с УФ-лампами. Реакция
протекает по свободнорадикальному механизму. Хлорирование
алканов происходит постепенно:
С„Н2„1;.-ЬС1,-Л-1С„Н2„AС1 + НС1.^.С„Н2„С1Н^НС1^ и т. д.
ftV ttV
Наиболее легко замещается водородный атом у третичного ато-
атома углерода, труднее — у вторичного атома (см. гл. I. 4).
При хлорировании алкенов хлор обычно присоединяется к двой-
Hoff связи. Только при 400. . .600 °С осуществляется аллилыюе хло-
хлорирование (см. гл. 11.4.8).
Хлорирование алкиларенов при нагревании и освещении при-
приводит к хлоралкиларенам (см. гл. VI. А. 4.3).
в) Б р о м и р о в а н и е. Прямое бромирование для простей-
простейших алканов (метан, этан) малохарактерно. В принципе бромирова-
бромирование возможно при нагревании и интенсивном облучении УФ-светом.
Для гексана, гептана и других алканов возможно бромирование
при кипячении и освещении:
С„П2„ , 2 ~ С„Но„ , iBr + HBr
Замещенные алкены подвергаются аллилыюму бромированию
при взаимодействии со специальными бромирующими реагентами
в присутствии инициаторов — свободных радикалов (см. гл. II.
4.8).
NBS
R —CII=CH —CH2R' >R — CII=CH — CHR'
Инициатор I
Вг
Бромирование алкиларенов при нагревании и освещении при-
приводит к образованию бромалкиларенов с атомами брома в боковой
цепи.
2. Реакции присоединения к алкенам и алкинам. В реакциях
присоединения галогеповодородов и галогенов получают различные
221
галогеппротводпые углеводородов (гл. II. 4.2; гл. IV. 4.1):
R —СИ-- СП2-; II- X—, R — СП — СНЭ
I- i
R-CII- СП2
I I
X X
X
R-C--CII + H —X— >R-C-CH,--, R- С -GT3
I I
-х.
X X
х, I I
—>R —С -СИ - >R~C—С—Н
! I II
XX XX
Реакции идут хороню, если Х^ CI, Вг или I. Если Х2 Ь, ско-
скорость-реакции уменьшается. Присоединение фтора связано с иы,ч>
лепнем большого количества теплоты, поэтому процесс трудно уп-
ратляем. Присоединение HF в обычных условиях не дает хороших
результатов. По-видимому, это связано с малоп нуклеофнлыюстыо
попа HF.J, который обычно образуется в растворах HF. При исполь-
использовании безводной HF наблюдаются лучшие выходы.
3. Реакции замещения. Используя различные галогеисодержл-
щпе реагенты, можно замещать галогеном атом кислорода в альде-
альдегидах, кетонах, группу —ОН в карбоновых кислотах и спмртах.
Илсестпы также реакции замещения одного атома галогена другим.
а) П о л у ч е н и е галоген произвол пых из спир-
спиртов. Замещение гидроксильной группы па атом галогена в спир-
спиртах происходит под действием галогеповодородов или галогеипдоз
фосфора и серы (гл. XIV. А. 4):
RCHi—ОИ-ГН —X7liRCH2—Х + Н./)
К эффективным реагентам относятся PCU, РОС1Л, PC!-., РВг,,
РВг,-„ PI3, SOC12. Очень аффективным реагентом фторирования яв-
является SF.,:
2RCII..OH-; SF,—> 2RCH.F-bSOj-:-2HF
б) Получение галоген производных из аль-
альдегидов и к е т о и о в. Эта реакция осуществляется при дейст-
действии РС15, PBrs или SF4 при нагревании:
О X
I f I
R-C-R1-|-PX5-> R-C—R^'.-POXj
X
О F
R-C-Rl-'-SF4'°, R-C-R1-! SOF2
I,
Rl — это II, Alk, Ar.
222
в) Получение га п о г с н п р о п з в о д п ы х и ч к а р-
б о к о в ы х к и с л о т. Карбсновые кислоты пол действием PCi:,
г:|)оврап!,аются в хлорангндрниЫ (ацплхлорпды), которые только
i:jjи нагревании под давлением дают трихлорпроизводные:
R—Ccf — R-C<f _..,R-CC13
Пря нагревании карагановых кислот с SF4 под давлением образу-
образуются трифтернроизеедные:
г) В ¦? а к м и о е за м е гц е и и е атомов галогена п
г а л о г е ii п р о и з в о д и ы х. При взаимодействии гаюгеинро-
нзводных с галогенпдами металлов один атом галогена замещается
на другой:
RCH, — X + N'a -» X'" г— RCI12 — X' -| Na + X -
Реакция обратима и при изменении концентраций галогенид-
понов может быть направлена в сторону RCbhX или RCH2X'.
Практически реакция применяется для получения иод- и фтор-
нроизводных. В последнем случае используют фториды сурьмы.
Наиболее часто применяют смесь SbF:. и SbCl5:
SbF, i-SbCij
RCHXI -RCUa-F
SbFj-i SbCl5 SbKj-iSbCU SbF^SbCij
> RCFC12 * RCF2CI : RCF3
4. Галогенметилирование аренов. При обработке ареноз фор-
формальдегидом и галогеноводородом в присутствии катализаторов
(Л!С1 з, ZnCh) получаются галогенметиларены:
Нч Z-лСА,
Лг —II-;- ">С = О-| ИХ ——-">-Лг — СИ2Х-{ Н2О (X --С!, Вг)
Реакция галогепметилировация является еще одним примером
э^ектрофилыюго замещения у углеродного атома арена. Электро-
фнльным реагентом в этой реакции является гидроксиметнл-катиоп:
I! 6. б- е- б- U. - Н. ,ОН
С. о- II — X ;-^ С— Oil T^i С7
II ' П ' Х- IK ЧХ
3. ФИЗИЧЕСКИЕ СВОЙСТВА И СТРОЕНИЕ
1. Физические константы. Галогенпроизводчые углеводородов при
обычной температуре являются бесцветными газами или жидкостя-
жидкостями со своеобразным сладковатым запахом. В воде нерастворимы н в
ботынппстпе случаев тяжелее ее. Некоторые нолигалогенопронзвод-
v.'.ac образуют бесцветные кристаллы, полпнодсоедннеиия имеют
жс-лгую окраску (табл. 21).
221
Таблица 21. Физические константы некоторых галогенпроизводиых
углеводородов
Галогенуглеводород
CH,F
CI1-.C1
СНчВг
СЛ,1
CII^CHoF
СН,СН,С1
СН,СН,Вг
СН.,СП21
СИ ..Fa
CM Ft
CF4
СН2С1а
CIICU
СС14
СН213
cm,
СН.,СН2СН..С!
СН2 -CHCHjCl
СИ- ССП2С1
СвН5СНаС1
Т. гл., ГС
— 141,8
—141,8
—93,6
— 66,1
-143,2
— 138,7
— 119
— 108,5
— 103
— 184
-96.7
—63,5
—22,8
5,0
119
— 122,8
— 136,4
—48,0
Т. кип., "С
—78,6
—24,2
3,6
42,5
—37,7
12,2
38,2
72,2
-51,6
—82,2
— 128
40,1
61,3
76,8
180
210 (разл.)
47,2
44,6
65
179,3
0,877(—79 СС)
0,991 (-25 "С)
1,732@сС)
2,279
0,816 (—37 СС)
0,921 @'С)
1,430
1,993
1,96 (-184 С)
1,326
1,488
1 ,595
3,325
4,008
0,892
0,938
1,045
1,103
1,5293
,3057 (—10 'С)
, 4239
1,5222
1 ,4237
1,4455
1,4603
1,7425 A5 СС)
1,800
1,3886
'1,4154
,5405
Температура кипения возрастает при увеличении атомной массы
галогена и числа атомов галогена и углерода. Исключением являют-
являются полифторалкапы, температура кипения которых уменьшается
при увеличении числа атомов фтора в молекуле полпфторалкана.
Это означает, что при увеличении числа атомов фтора уменьшается
межмолекулярное взаимодействие. При увеличении атомного ра-
радиуса галогена увеличиваются коэффициенты преломления света.
Это свидетельствует об увеличении поляризуемости при переходе
от фторпроизводных к иодпроизводным.
2. Характеристика связей углерод — галоген. Энергия связи
углерод — галоген в большой мере зависит от типа галогена. Наи-
Наиболее прочной является связь С—F, самой слабой — связь С—I.
Таблица *
Связь
С—F
С —С1
С-Вг
С-1
22. Некоторые характеристики
Энергия
связи,
кДж/моль
443
328
279
240
Диполыгыи
для СН
Кл-м \0'°
6,05
6,11
5,98
5,34
СВЯ31
момент д
з-Х
1
1
1
1
D
,81
,83
,79
,60
С (sp:i) -
Дли и л
свянн.
нм
0,141
0,176
0.191
0,210
-X
Коралентпьщ
радиус ало-
гсна, нм
0,060
0,099
0,114
0,136
Ре<}
пня
3CI
1
6
9
14
Г.г:-
снм-
i R
,44
,51
,38
',й\
224
Моиогалогенпроизводные имеют значительные дипольные мо-
моменты (табл. 22), что свидетельствует о полярности связи углерод —¦
галоген. Атом галогена имеет большую электроотрнцателыюсть,
чем углеродный атом, поэтому плотность электронов сдвигается в
сторону галогена и на атомах появляются эффективные заряды:
б- б- «¦>• 6-
СНЯ- Cl RCH-. — Вг
Необходимо обратить внимание па отсутствие пропорциональ-
пропорциональности между дипольными моментами и электроотрицательностямн
атомов галогена. Для связи С—F следовало бы ожидать большего
днпольного момента. На дипольнын момент влияет не только вели-
величина электроотрицательности (индуктивный диполь), но и наличие
неподелепных пар электронов и сравнительные ковалентпые радиу-
радиусы атомов.
Рефракция характеризует поляризуемость связей, которая уве-
увеличивается в ряду от С—F до С—I.
Ввиду полярности этой связи углеродный атом приобретает не-
некоторый электрофпльный характер. Неподеленпые пары электронов
на атоме галогена придают молекулам слабые электронодонорные
свойства, особенно иодндам. Энергия ионизации для R—I: около
9,5 эВ, для R—Вг: 10,5 эВ.
3. Хиральные галогенпроизводные углеводородов и их номенкла-
номенклатура. Подобноалканам, галогелалканы могут содержать в молекуле
асимметрический атом углерода. Это возможно в случае различных
галогенов пли алкнльных групп у одного атома углерода. Напри-
Например:
Я-бро.мфгорхлор метав ^-бролфторхлорметан
ВГ
Д-Л-бромбутаи S-2-бромбутан
Для точного обозначения расположения заместителей у асим-
асимметрического атома углерода в пространстве (абсолютной конфигу-
конфигурации) применяется ^^-номенклатура (Кап, Ингольд, Прелог,
1956), которая вошла в правила ИЮПАК. Для этой цели вводится
ряд старшинства атомов. В основе определения старшинства на-
находится атомный номер элемента. Таким образом,
самым младшим атомом и заместителем является водород, затем
8 Xs 517
225
следуют Li, Be, В, С, N, О, F и т. д. В случае одинаковых атомов,
связанных с асимметрическим центром, старшинство заместителя
(группы) определяет «вторая оболочка» атомов. Например, старшин-
старшинство групп увеличивается в ряду:
—СН„ —СН2СН3, —СН(СН3).2, —CH2NH2, —СН,ОН, -CHoF
В случае групп с различным типом связей старшинство увеличивает-
увеличивается в рядах:
,0 ,0 ,0
-СН2ОН, -Cf , -C<f , -О."
ХН XR ' ОН
—CH,NH2, —Cf , —C = N
чн
Для применения ^^-номенклатуры молекула должна быть ори-
ориентирована в пространстве определенным образом. Глаз наблюдате-
наблюдателя смотрит по оси углерод — младший заместитель, обычно это
связь С—Н. После ориентации молекулы смотрят, как три замести-
заместителя располагаются в ряд в направлении от старшего к младшему.
В случае AJ(rectus)-KOHc})Hrypamin этот порядок соответствует на-
направлению движения по часовой стрелке, в случае S(sinister)-KOH-
фигурацин — против часовой стрелки.
В приведенных выше примерах молекулы ориентированы так,
что наблюдатель смотрит по оси С—Н. Для бромфторхлорметапа
порядок от старшего к младшему соответствует ряду Br, Cl, F, а для
2-бромбутана — Вг, СН3СЫ2, СН3. Поэтому эпантномеры, распо-
расположенные слева, соответствуют R-, а справа — S-конфигурацип.
Индивидуальные энантномеры оптически активны, направление
угла вращения может быть правое (+) или левое (—). Смесь равных
количеств энаптиомеров — оптически неактивное вещество — раце-
рацемат (рацемическая смесь).
Если при синтезе в молекуле возникает асимметрический атом
углерода, то, как правило, получается смесь энаптиомеров — раце-
рацемат (см. асимметрический синтез). Молекулу, которая при реакции
становится хиральнон, называют прохиральной, а два одинаковых
атома, при замещении одного из которых возникает хиралыгость,—
энантиотопными. Например, молекула фторхлорметана является
прохиралыюн, а атомы водорода в СН2-групие — энантиотопны-
энантиотопными. Так, при бромнрованни фторхлорметапа возникает хпральность,
но получается оптически неактивная смесь R- и S-бромфторхлорме-
таиов (при сравнении с формулами, приведенными выше, следует
учесть, что молекула повернута по связи С—С1 на 120 ):
.а а а
S- Я-
Для получения оптически активных R- и S-бромфторхлормета-
пов рацемат надо разделить на оптические антиподы (гл. XXXIV.
Г. 4).
226
В молекуле органического соединения могут быть и два асим-
асимметрических атома углерода, например в 2,3-дибромпентане
СН3СНВгСНВгСН2СНл (рис. 81). Число стереоизомеров в этом слу-
случае возрастает, так как у каждого асимметрического углеродного
атома могут быть конфигурации R и S. Для 2,3-дибромпентаиа воз-
возможны следующие комбинации конфигураций: R, R; R, S; S, Rl
S, S. Следовательно, имеется четыре стерёонзомера.
Вг
СН2СН3
R
Н
сн,
Вг
CHjCH,
сн3
сн,сн3
H-I-Br
Br-f-H
CH,
Нестабильная
заслоненная
Конформация
¦CH,CIIj
стабильный
конформер
СИ,
СТЛОИль/1! 1Й
конформер
В г,
CILCH,
н
СНгСН,
Вг—[— II
Вг-4— Н
СИ,
СН3
СН2С1Г3
н-4—вг
II—j—Вг
сн,
Рис, 81, Стереоизомеры 2,3-дибромпентана
Bf
227
На рис. 81 изображены все четыре стереоизомера в боковых
проекциях, соответствующих им плоских проекциях (проекционные
формулы Э. Фишера) и формулах Ньюмена. Приведены нестабиль-
ные заслоненные конформации, которые соответствуют изображен-
изображенным проекциям, и рядом один ил стабильных конформеров.
При определении конфигурации следует учесть, что порядок
заместителей у одного атома углерода соответствует последователь-
последовательности Вг, СНВгСНоСНз, СНЯ, а у другого атома — последователь-
последовательности Вг, СНВгСН,, СНгСН3.
Пары R,R и S,S, S,R и R,S представляют собой оптические ан-
антиподы (зеркальные изомеры). Но молекулы с конфигурацией R,R
и R,S (пли S,5 и S,R) не являются оптическими антиподами, они
представляют собой стереопзомеры, которые называются диастерео-
мерами, Диастереомеры отличаются друг от друга физическими
свойствами (температурами плавления, кипения, растворимостью)
и отчасти химическими свойствами. Диастереомеры получаются,
если в хиральной молекуле возникает новый асимметрический центр.
Атомы, при замещении которых в хиральной молекуле возникает
асимметрический атом, называются диастереотопными. Например,
в 2-бромпентане атомы водорода в метиленовой группе СП2 явля-
являются диастереотопнымн.
Диастереомеры иногда называются эритро- и трео-изомерами.
Их легче всего определить, пользуясь проекционными формулами
Э. Фишера. Эритро-изомерами называют те, у которых два одинако-
одинаковых боковых заместителя стоят в проекционной формуле по одну
сторону углеродной цепи. Следовательно, S,R- или /?,5-2,3-дибром-
пентан соответствует эритро-изомеру, a R,R- или 5,5-2,3-дибром-
пентан—трео-изомеру.
В общем случае молекула может содержать несколько асиммет-
асимметрических атомов углерода. Число стереоизомеров N определяется
выражением N—-2", где п — число асимметрических атомов угле-
углерода.
Если у двух асимметрических атомов имеется одинаковый набор
заместителей, из-за симметрии молекулы число стереоизомеров
уменьшается. /Молекулы с конфигурацией R,S и S,R идентичны и
имеют центр симметрии. Этот стереомзомер называется мезо-формой,
он оптически неактивен, так как молекула в целом ахиральна (имеет
центр симметрии). Например, 2,3-дибромбутан CH^CHBrCHBrCHa
образует три стереоизомера: оптически активные R,R- и S,S-2,3-
дпбромбутаны и оптически неактивную мезо-форму — /?,S-2,3-;ui-
бромбутан. Ниже изображено пространственное строение мезо-
формы при помощи формул Ныомепа; в одном из конформеров хо-
хорошо видно симметричное расположение заместителей:
Н СИ3
Вг
С Н3
228
4. ХИМИЧЕСКИЕ СВОЙСТВА
Для галогенпроизводных характерна высокая реакционная способ-
способность. Благодаря электрофилы гости они легко вступают в реакции
с металлами (М) и различными нуклеофчльными реагентами, что
ведет к реакциям замещения атома галогена, отщепления галогено-
водорода. Известны реакции замещения атома галогена водородом:
Н,/Кат. й (- 6-М й- 6 +
CRCirCHM
-их
(-Х-)
RCH--CIIa-
:NnH
¦RCH2-CH2-Nu:
1. Замещение галогена водородом. Восстановление галоген-
производных до углеводородов осуществляется водородом в присут-
присутствии обычных катализаторов гидрирования или нагреванием с
иодоводородной кислотой. В последнем случае происходит замеще-
замещение атома галогена на. иод и восстановление действием иона I":
RCH.-X —^
Кат.
+ 2HI-C.RCH3-| HX + I.,
2. Замещение галогена атомом металла. Галогенпроизнодные
углеводородов реагируют с металлами, в результате чего получа-
получаются металлорганические соединения или продукты их дальнейше-
дальнейшего превращения. Металлорганические соединения подробно рассмат-
рассматриваются в гл. X. Здесь рассмотрим два примера: реакцию с актив-
активным щелочным металлом — натрием и менее активным металлом —
магнием.
При взаимодействии с натрием происходит димеризация галоген-
углеводорода с отщеплением галогена — реакция Вюрца. Реакция
начинается с переноса электрона от сильнейшего электронодонора,
атома натрия, на галогенуглеводород. В качестве промежуточных
продуктов возникают свободные радикалы и натрийорганические
соединения:
RCH2-X~-| -Na—>-[RCH2-=-X]-Na+^RCH2-! Na + X"
RCH..+ -Na —> RCHa —Na
RCHii-Na + RCH2-X~—> RCH2-CH2R +Na+ X~
Взаимодействие галогенпроизводных с магнием в растворе дп-
алкиловых эфиров ведет к образованию мапшйорганических соеди-
соединений (реагентов Гриньяра, см. гл. X. Б. 1):
RCH2—X + Mg —* RCH2 —MgX
3. Замещение галогена при взаимодействии с нуклеофильнымн
реагентами. Воздействие нуклеофильных реагентов (анионов и
229
нейтральных молекул с неподеленпыми парами электронов) на гало-
генпроизводные углеводородов ведет к замещению атома галогена:
6+ б-
RCH4 —X: + ;Nu:-M+—> RCII2-Nu:-|-:X:-,Ч+
б+ е- +
RCII2~X:-f:NuH—*[RCH2 —NuH]:X:- 7i RCI-Ь — Nu: -'-HXr
Эту реакцию называют реакцией пуклоофггльпого замещения у
насыщенного углеродного атома н обозначают символом Sx (S —
от апгл. substitution — замещение). Необходимо заметить, что па-
параллельно могут протекать реакции отщепления (см. ниже).
Механизм этой реакции изучался в течение нескольких десятиле-
десятилетий. Изучение скорости реакции Sv в зависимости от концентра-
концентрации пуклеофнлыгаго реагента и строения галогелуглеводорода дало
интересные результаты. Оказалось, что существуют реакции Sy,
скорость которых не зависит от концентрации нуклеофилыюго реа-
реагента (реакции первого порядка). В то же время скорость многих
других реакций S.Y зависит от концентрации как нуклеофилыюго
реагента, так и галогенпроизводного (реакции второго порядка).
Так, было обнаружено, что возможны по меньшей мере два меха-
механизма S_Y,
' Реакция первого порядка протекает в две стадии. Первая —
ионизация галогенпроизводного— является медленной стадией. В ре-
результате ионизации могут образоваться ионные пары (тесные или
сольваторазделеппые):
й-i- б— Медленно
RCH2-X: J
Вторая стадия — взаимодействие с нуклеофильным реагентом
протекает быстро:
Бистро
RCII2 :Х:--;- =Nli:- > RCH2 — Nu:'+ :Х:~
Общую скорость реакции S_v лимитирует медленная стадия — иони-
ионизация. Поэтоглу скорость реакции не зависит от концентрации и
типа нуклеофилыюго реагента. Такие реакции называют мономоле-
мономолекулярными и обозначают символом S,vl.
Реакция второго порядка является более общим случаем. Оба
компонента — галогенпроизводное и нуклеофильный реагент —¦
вступают в реакцию одновременно:
R
1-^91 + :nu:
н
h
ч„
R
.с + :ХГ
Н
>Nu:
переходное состояние
Скорость реакции определяется как концентрацией и типом ну-
нуклеофилыюго реагента, так и концентрацией галогенпронзводпого.
Такие реакции называют бимолекулярными и обозначают символом
S.V2.
230
Важно понимать, что в реакциях SN2 происходит обращение
конфигурации у атома углерода. S-Копфигурация переходит в R-
конфигурацшо и наоборот. Например, из У?-2-бромбутана при дейст-
действии щелочи получается 5-бутапол-2, аиз5-2-бромбутапа — R-бута-
нол-2. Реакция SX2 принадлежит к стеревспецифическим реакциям.
Реакцию называют стереоспецифической, если исходные вещества,
отличающиеся только стереоизомерией, превращаются в стереоизо-
мерно разные продукты.
При реакции оптически активного 2-иодбутапа с Na + I~ оптиче-
оптическая активность исчезает — образуется рацемат. Это объясняется
тем, что Я-2-иодбутан при реакции с ионом I" дает S-2-иодбутан, а
S-2-иодбутап превращается в ^-2-подбутан, пока не наступит рав-
равновесие при 5О"о R- и 50% S-2-иодбутана (см. также вальденовское
обращение).
В каждой конкретной реакции нуклеофильное замещение осу-
осуществляется по обоим механизмам, только сравнительные скорости
реакций могут сильно отличаться. В отдельных случаях получается
не первый и не второй порядок реакции, а какой-то дробный, из
которого можно вычислить отдельно скорости по S\-l и SY2 (табл.
23).
Тш'лица 23. Константы скорости реакции гидролиза
бромалкаиов RBr + OH- —- ROH + Br-
CJfi этанол, 55 СС)
Соединение
CH,Br
CH-jCH,Br
(CH-,JCHBr
(CH3j3CBr
Константа скорости
реакции Syl
3,5-10-°
1,4-10 - ч
— ю-6
Константа скорости
реакции -Sy2
2.14-10-
1,71-Ю-з.
5- Ю-5
При рассмотрении реакций S_v обращают внимание на следую-
следующие факторы: пуклеофильность реагента, растворитель п сольвата-
сольватация реагента, ионизирующая способность растворителя, природа
нуклеофуга (уходящей группы) и пространственное строение сое-
соединения (стерические препятствия реакции, стабилизация карбока-
тмопа).
Нуклеофпльность увеличивается при увеличении поляризуемо-
поляризуемости реагента, размера аниона. Реакции способствуют полярные раст-
растворители, которые не образуют водородные связи (гл. XIV. А. 3)
с нуклеофилом и таким образом его не пассивируют, например аце-
ацетон, диметилсульфокенд, диметилформамнд.
Наилучшие уходящие группы — слабые основания. В ряду га-
логенид-ионов наиболее слабое основание I", за ним следуют Вг~,
С1- и F".
На реакцию S,V2 сильное влияние оказывают стерпческие пре-
препятствия. Алкильные группы у центра реакции мешают нриближе-
231
нию пуклеофила, поэтому третичные галогеналкапы не реагируют
по механизму SK2.
Механизм S.vl возможен в случае соединений, которые могут да-
давать весьма устойчивые карбокатиопы, таких, как третичные гало-
геиалкапы, аллил- и бензилгалогениды. Особенно ярко это видно
на примере производных фенилметанов. Если принять скорость ре-
реакции гидролиза по механизму SN\ беизилхлорида СвН0СН2С1
за 1, то для дифенилхлорметана (СвН5JСНС1 скорость равна 2000,
а для трифенилхлорметана (С0Нг,);)СС1 — 30 000 000.
Так как в реакциях S.vl промежуточно образуется карбокатион,
который атакуется нуклеофилом со всех сторон, стереоспецифичность
обычно не соблюдается. При реакции оптически активного соедине-
соединения получается рацемический продукт. Нсть редкие исключения,
когда соседняя группа стабилизирует конфигурацию (см. вальде-
новское обращение).
В органическом синтезе реакции нуклеофильного замещения ис-
используют очень широко. Перечислим некоторые из них:
1) гидролиз
RCI12Х -)- Н2О —> RCH2OH + ЫХ
RCII2X + \af-ОН- -
2) аммополиз (получение аммониевых солей и аминов)
rch2x-j-nh3 —> [rch.2\h3]+x- ^± rch2nh.2+hx
rcii2x-i-nrJ—* [rch2nrJ]+x-
3) получение простых эфиров
I2X + Na + -ORi—^RCHaOR^Na + X-
4) получение сложных эфиров
5) получение цианидов (нитрилов)
RCH.2X+Na + -CN—>-RCH2C>4-Na + X-
6) получение иодуглеводородов
RCH2X-i-NaI—cRCHal + Na + X-
Число примеров реакций пуклеофильного замещения можно уве-
увеличить при использовании других нуклеофильных реагентов (см.
с. 71).
Все S.v-реакции фактически представляют собой введение ал-
кильной (или алкенильной, алкинильной, арилалкильной) группы
у какого-то атома или группы. Поэтому галогенпроизводпые угле-
углеводородов называют алкилирующими реагентами.
Реакции нуклеофильного замещения могут быть интенсифициро-
интенсифицированы при помощи межфазного катализа (см. с. 75, 340, 400, 403).
Например, гидролиз водным раствором щелочи может быть прове-
проведен в двух несмешивающихся фазах (водный раствор щелочи и га-
логеналкан) при интенсивном перемешивании в присутствии меж-
232
фазного катализатора (четырехзамсщешюй соли аммония, краун-
эфира, кринтанда).
4. Активация галогенпроизводных и генерация карбокатионов.
Поляризацию связи углерод—галоген можно увеличить при помо-
помощи координации галогенпроизводного с электроноакценторамп.
Для этой цели используют кислоты Льюиса, которые вступают во
взаимодействие с пеподеленнымн парами электронов атома галоге-
галогена (Л1Х3, SnCl,, TiCl,, BF3, SbX3):
Л ' ft - 6;. б'_
RCH.—Х:-рА1Х3 ;_: RCII2—X: -* Л1Х3
6'>6
Таким образом удается алкилировать весьма слабые нуклсофплы —
арены (см. гл. VI. А. 4.1. в).;
Как доказано методом ПМР, этим способом при низких темпера-
температурах в растворах удается генерировать свободные карбокатионы
(Г. Ола). Лучше всего для этой цели подходят фторуглеводороды и
сильные кислоты Льюиса (BF,1( SbF5, PF5):
б >• л - <-
RCH,— F-j-SbF., :.± RCHaSbFo"
5. Отщепление атома галогена, образование ненасыщенных сое-
соединений и карбенов. а) Образование а л к е н о в (Р- э л и-
минирование). Реакция отщепления является конкурирую-
конкурирующей для реакции пуклеофилыюго замещения, например при взаи-
взаимодействии со щелочью:
I .
R-C-C-XJ OH- —
I I
H H
H H
f I
¦C-C-OH (S,v,
H H
Реакция отщепления обозначается символом EY (элиминирова-
(элиминирование пуклеофильное).
Как показали кинетические исследования реакции ?_v, возмож-
возможно мономолекулярное протекание реакции (первый порядок) —¦
ENl и бимолекулярное (второй порядок) — ?V2. В случае ?л-1
скорость реакции определяется медленной стадией — ионизацией.
Затем следует быстрый распад карбокатиона па алкен и протон,
который связывается пуклеофильиым реагентом:
Н II Н II
| ||^^. ft- МОДЛОННО [ [
R —С —С—X: """R—С—О:Х:-
I I " " I I "
НИ Н Н
н н
I I .. Быстро Н.„ /Н
R —С-С + :Х:- +:Nu:~ > ЧС---С< -i :X: " -j- HN'u:
I | " r- Ni ¦•
II H
233
Скорость реакции ?л-2 зависит от концентрации нуклеофильно-
го реагента. В этом случае возникает переходное состояние, в кото-
котором нуклеофильный реагент предположительно связан с водород-
водородным атомом при C-углероде:
н н
R-c-c —x: н
1 1
н н
- :nu: ^s
г н
I
R—С-
Н
Н
1
-С
1
н
-c-^c-" :x
н
"r/C=4h
is. XX
и
Nu
Образование алкепа возможно с одновременным уходом HNu:
и :Х:~ (синхронно) или с промежуточным образованием карбаниона.
Взаимодействию нуклеофилыюго реагента с водородным атомом
способствует индуктивный эс|хрект атома галогена, в результате че-
чего поляризуются связи С—Н.
В принципе атака пуклеофнлом может осуществиться как у а-,
так и у C-водородпого атома.
Обычно реакции ?д- осуществляют действием концентрирован-
концентрированных растворов сильных оснований, например щелочен.
Если в реакции ?л- могут образоваться несколько изомерных
алкенов, то преимущественно получаются те изомеры, в которых
наибольшее число заместителей у двойной связи (правило Зайцева):
СН3 Н
| | он- СП3. ,Н
CH.J-C — С-СНз >¦ уС^С/
I | CU/ ХСН3
II X
б) Образование карбенов (а-э л и м п и н р о в а-
п и е). Если при взаимодействии с сильным основанием отщепля-
отщепляется сс-водрроднын атом галогеппроизводного, возможно образова-
образование карбепа. Это доказано для трнгалогепметанов, например хло-
хлороформа:
)j-ccij + он" ^2г Тса3 + HjO -s^r J^c^ + сГ + но
Образуется трихлорметаиид-пон, который способен генерировать дн-
хлоркарбен выбрасыванием хлорид-нона. Образование днхлоркар-
бена доказывается дальнейшими реакциями (например, образова-
образованием циклопропанов в присутствии алкенов).
5. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ
Мстиленхлорид (хлористый метилен) CH..CL — бесцветная жидкость
со сладковатым запахом, мало растворим в воде A,3 г в 100 г воды
при 25°С). Соединение обладает наркотическими свойствами.
В промышленности его получают хлорированием метана.
Метилепхлорид применяют в качестве растворителя для поли-
полимерных материалов (полпвипилхлорнда, полистирола, хлоркаучу-
224
ков и др.). Смесь метиленхлорида с этиловым спиртом широко при-
применяется для растворения ацетилцеллюлозы (см. с. 521) в производ-
производстве кинофотопленок.
Хлороформ СНС1., — бесцветная жидкость со сладковатым за-
запахом, мало растворим в воде A г в 100 г воды при 15 С). Соедине-
Соединение обладает сильными наркотическими и анестезирующими свойст-
свойствами, применяется в медицине. В промышленности его получают
хлорированием этилового спирта хлорной известью (см. с. 494).
Хлороформ хорошо растворяет жиры, смолы, каучук. Его ис-
используют также в промышленном органическом синтезе для получе-
получения фреопов.
При соприкосновении с воздухом, особенно на свету, хлороформ
постепенно окисляется с образованием фосгена СОС12.
Тетрахлоркетан (четыреххлористын углерод) СС14 — бесцвет-
бесцветная тяжелая негорючая жидкость со сладковатым запахом. Мало
растворим в воде. С водой образует азеотропную смесь, кипящую
при 66 СС и содержащую 96% СС14 и 4% Н2О. В промышленности
тетрахлорметан получают хлорированием метана или сероуглерода:
CS.,-!-3Cl2—>CCl4-!-S2CI2
Используют тетрахлорметан в качестве прекрасного растворите-
растворителя, особенно для растворения жиров, масел и смол. Тетрахлорметан
является исходным сырьем для промышленного синтеза (фрсоны,
реакции теломернзацип), рекомендуется в качестве огнетушитель-
ного средства. Ядовит, вызывает повреждение печени и почек.
Дихлорэтан С1СН2СН.С1 — бесцветная жидкость со сладко-
сладковатым запахом, т. ил. —35,9 °С, т. кип. 83,5 °С, dlb----1,26, коэффици-
коэффициент преломления света Лд —1,4476. Мало растворим в воде @,8%
прн 20 С). С водой образует азеотроппую смесь, кипящую при
71,5 ;С и содержащую 83% дихлорэтана.
В промышленности дихлорэтан получают хлорированием эти-
этилена. Он широко используется в качестзе растворителя и исходного
сырья в органическом синтезе (внпплхлорид, этиленднашгн, тпо-
кольнып каучук и др.). Дихлорэтан обладает наркотическими свой-
свойствами, но ядовит, вызывает тяжелые поражения печени.
Фреоны (хладоны) — фторхлоралканы представляют собой бес-
бесцветные газы пли низкокипящие жидкости (табл. 24) и вследствие
Таблица 24.
Соединенно
CCI-.F
CC1..F..
CHC1F.,
C.Cl.,Fi
QC1F,
Фреоны
.'.'.арка
Ф-11
Ф-12
Ф-У2
Ф-Ш
Ф-115
Т. к иг;
23
—29
—40
-3fc
.. °с.
,8
,8
,8
Т.'плота иг
ПРИ Tt'MCi'p;1
[1ня (кДж
in?!
234,
137,
> г
1
1
I
3
пения
,¦ ки::е-
VI*.)
235
своих специфических термодинамических свойств применяются в
качестве хладагентов в холодильных машинах.
Фреоны получают из соответствующих хлоралканов (см. гл. VII.
2.3).
Дифторхлорметап используется также в органическом синтезе
для получения тетрафторэтилепа (см. гл. VIII. 5).
Гексахлорциклогексан — бесцветное кристаллическое вещество.
Известно восемь стереоизомеров гексахлорцнклогексапа с различ-
различным положением атомов хлора. Важным является изомер с тремя
рядом расположенными экваториальными и тремя аксиальными ато-
атомами хлора (у-изомер, «липдан»).
С1 Температура плавления этого изомера 112—
сA 113 "С. Смесь изомеров получают при фотохи-
^ мическом хлорировании бензола (см. гл. VI.
A. 4.4). Содержание 7-изомера 10—18%.
(j Смесь гексахлорциклогексаиов долгие годы
ли"дан широко применялась в качестве сильного инсек-
инсектицида («гексахлоран»). Самым эффективным является у-нзомер. в
последнее время применение значительно ограничивается ввиду его
ядовитых свойств и накапливания в почве и живых организмах.
Аллилхлорид C-хлорпропен) CH.^CHCH.Cl — бесцветная жид-
жидкость с резким запахом, вызывает слезотечение (лакриматор). По-
Получают при высокотемпературном хлорировании пропеиа (см. гл.
II. 4.8). Используют в качестве исходного вещества для синтеза
эпихлоргидрипа, глицерина, аллиловых сложных эфиров).
Бензилхлорид СйН5СНаС1 является бесцветной жидкостью с
раздражающим запахом, лакриматор. Получают хлорированием то-
толуола при УФ-облучении и нагревании (см. гл. VI. А. 4.3). Исполь-
Используют в органическом синтезе для получения бензилового спирта,
бепзилцианида и бепзиловых эфиров карбоповых кислот и целлю-
целлюлозы.
Глава VIII
ГАЛОГЕНПРОИЗВОДНЫЕ ТИПА C(sp2)— X
1. КЛАССИФИКАЦИЯ И НОМЕНКЛАТУРА
К галогеипроизводным со связью C(sp-)—X (тригональпый угле-
углерод — галоген) принадлежат:
1) галогеналкены с атомом галогена у углерода двойной связи
(випилгалогениды):
Rx /H Rx . 7R
)С=С< /с = с<;
R-7 ЧХ Х/ ХХ
2) галогепалкадиены СН2 = С—СН —СНа;
X
236
3) галогенарены
X
Принципы номенклатуры галогеппроизводных изложены были
ранее. Например:
СПоСН — СН-- С — Н СПзСН^С —СН=-С —СН3
"| I II
СНз С1 Вг С1
3-мстпл-1-хлорбутсн-1 4-бром-2-хлоргексадиен-2,4
сн3
A/CHtCHi Br\/'\/CI - - -Br
51 N3 II
1-брои-З-этилбензол I-бром-2-метил- 2,6-дибромиафталин
3,4-дихлорбензол
Для некоторых простейших соединений сохраняются тривиаль-
тривиальные или рациональные названия:
СН2 = СНС1 СН2 = С— СН--СНа СН2 = С —СН = СН2
винилхлорид хлоропрсн фторопрен
2. МЕТОДЫ ПОЛУЧЕНИЯ
Для получения галогеиалкеиов и галогенаренов применяют замет-
заметно отличающиеся друг от друга методы.
1. Галогеналкены получают двумя основными реакциями:
а) отщеплением галогеноводородов от
1,2- д и г а л о г е н а л к а н о в. Это достигается действием ще-
щелочи или термически над катализатором (см. винилхлорид):
Н П
I | он- Нх ,Ri
I I R7 ХХ
X X
б) присоединением галогеноводородов к
а л к и н а м:
Кат. II /RI
R_C-C_Ri-j-H-X > ХС = С(
R/ ЧХ
2. Галогенарены получают прямым галогепированием арепов
или замещепнем других групп галогеном.
237
а) Прямое галоген ирование является главным
методом получения галогенаренов (за исключением фтораренов).
Хлорирование происходит очень легко при взаимодействии аре-
арена с хлором в присутствии небольших количеств катализатора (Fe,
FeCla, другие кислоты Льюиса) (гл. VI. А. 4.1). При хлорировании
бензола получают хлорбензол, дихлорбепзол, трихлорбензол:
_E!l,
FcCIj
4
FeCb
FeCI.,
Легко хлорируются толуол, ксилолы, бнфеппл, нафталин.
Бромирование аренов происходит труднее, чем хлорирование, и
катализируется Fe, FeBr3, иодом:
СИ,
СИ,
CH3
СНз
Br,
Вг
Иодирование аренов требует специальных условий. Иод с аре-
аренами непосредственно не реагирует, только образует слабые комп-
комплексы с переносом заряда, так как имеет меньшее сродство к элект-
электрону, чем бром и хлор. Иодирование аренов осуществляют в присут-
присутствии окислителей (НЮ3, НХОГ!+Н25О4, Н2О2 и др.). Окислители
превращают иод в катион 1 + , который является более сильным элек-
трофильным реагентом:
HN0,/H,SO4
i, ——ч+nsor
IIN0a/H2SO4
HNOa,H2SO4
Для иодирования активированных аренов можно использовать
хлорид иода IC1.
б) Замещение других групп галогеном. Этот
метод применяется главным образом для получения фтораренов.
Известны реакции замещения хлора на фтор действием KF при вы-
высокой температуре под давлением:
Cl
Г]
F
I
к г
238
300 "С
Для получения галогенаренов используют арендиазониевые соли
(гл. XXV. А. 3):
+ СиХ
[Аг—N — N]X- > Аг— X-J-N2
[Лг—N = K]BF7 -^> Лг—F + N'i-f
3. ФИЗИЧЕСКИЕ СВОЙСТВА II ПРИРОДА СВЯЗЕЙ
Галогепалксны и галогсггареиы являются бесцветными газами или
жидкостями со своеобразным запахом, в воде практически пе раст-
растворяются (таГхт. 25).
Таблица 25. Физические константы некоторых галогепуглеводородов
Га л огсчг у глеподород
СП,- СП—С1
СН2^-СН —Вг
cix
Х=-СН—CI
ск
СН2--С-СН----СН2
С1
С6Н5С1
С0Н,Вг
СбНгД
o-Cr,HiCl2
п-Сг,Н4С12
Т. ил., °С
— 153,8
— 137,8
—86,4
—15,6
—30,6
—31,3
— 17,5
53
Т. кип., еС
— 13,8
15,8
87,2
59,4
131 ,7
150
188,4
182
173
0
1
I
0
1
1
1
1
I
°
,973 (—15 X)
,5286A1 °Q
4650
9585
1063
495
832
305
458
1
—
1,477
1,458
1,5248
1,5572
1,6214
1,552
,521 (80 "С)
Дипольные моменты у ряда галогепалкенов и галогенаренов
меньше, чем у подобных галогеналкапов. Это свидетельствует о
меньшей полярности свяли C(sp")—X:
CHa—CHa —CI
СН,--СН-С1
~Ч—Cl
6,68
4,81
5,28
0, Кл-м и.
2,0
1,44
1,58
Причиной уменьшения полярности связи является измеиеннд...
состояния гибридизации углеродного атома sp'1 на sp2, что вызывает
увеличение электроотриг[ателыюсти углеродного атома и взаимо-
взаимодействие неподеленпой пары электронов атома галогена с л;-связя-
л;-связями алкена или арена.
Неподеленная электронная пара вступает в сопряжение и это
вызывает смещение я-электронной плотности от атома галогена в
сторону углеродных атомов. Другими словами, атом галогена про-
239
являет +М-эффект (см. с. 29, 80).
(здесь б-заряды относятся к изменению плотности л-электроиов).
Вследствие сопряжения изменяется распределение плотности
л-электронов (что изображено на схемах) и изменяется характер
связи С—X.
Одновременно действует сильный электроноакцепторный ин-
индуктивный эффект (—/) по а-связям, поэтому суммарный эффект по
а- и я-связям качественно трудно предугадать и тем более изобра-
изобразить при помощи б-зарядов:
СН2 = С^
Расчеты подтверждают такое распределение зарядов.
Длина связей C(sp2)—X укорочена по сравнению со связями
C(sp3)—X. Ниже приведены электронографические данные для ви-
нилхлорида.
н н Изучение энергий ионизации показало, что
\ П|п„. ^ випилгалогениды обладают большим электроно-
донорпым характером, чем этилен. Если для
"•%' этилена энергия ионизации 10,5 эВ, то для ви-
нилфторида 10,4 эВ, а для винилхлорида 10 эВ.
Галогенбензолы по своим электронодонорным свойствам срав-
сравнимы с бензолом. Если для бензола ЭИ 9,24 эВ, то для фторбензола
9,2 эВ, а для хлорбензола 9,08 эВ.
Энергия ионизации характеризует ионизацию сопряженной си-
системы винилгалогенидов и галогенбепзолов и отчасти реакционную
способность с электрофильными реагентами.
4. ХИМИЧЕСКИЕ СВОЙСТВА
Для галогеппроизводных со связью C(sp'2) — галоген характерны
два типа реакций: по связи углерод—галоген (замещение) и в угле-
углеводородном остатке (присоединение, полимеризация, отщепление,
замещение).
1. Реакции замещения атома галогена. В отличие от уже рас-
рассмотренных галогеппроизводных со связью C(sp3)—X, в которых
атом галогена подвижен, галогену глеводороды со связью C(sp2)—X
в реакции замещения вступают труднее, особенно при взаимодейст-
взаимодействии с нуклеофильными реагентами. Атом галогена в этих соедине-
соединениях малоподвижен.
а) Замещение атома галогена водородом
происходит труднее. В условиях каталитического гидрирования
над Ni или Pd галогеналкены сначала насыщают двойную связь,
240
а после этого замещается галоген. Галогенарены реагируют трудно,
за исключением иодаренов.
б) Взаимодействие с металлами. Галогеналке-
ны с металлами реагируют по-разному. Активные щелочные метал-
металлы могут инициировать полимеризацию. Магний реагирует только
в специальных условиях — в растворе тетрагидрофурана ТГФ:
ТГФ
СНо^-СН —Cl + Mg >-CH,r CH —MgQ
Галогенарены с металлами реагируют как обычно:
QI Т5Вг -Ь Mg > CeH5MgBr
в) Взаимодействие с нуклеофильными ре-
реагентами. Реакции галогеналкенов и галогепаренов с нуклео-
нуклеофильными реагентами всегда проходят трудно, необходимы более
жесткие условия реакции, но при этом протекают побочные реакции.
Отчасти это объясняется изменением природы связи C(sp2) — гало-
галоген (см. выше).
Реакции SN галогепалкепов при взаимодействии с нуклеофиль-
нуклеофильными реагентами в большинстве случаев не удается осуществить.
При обычной температуре реакции со щелочью, алкоксидами, ам-
аммиаком в желаемом направлении практически не идут. При 100—
200 °С доминируют побочные реакции — отщепление, присоедине-
присоединение, полимеризация. Исключением могут быть реакции полигалоген-
алкепов с сильными нуклеофилами, например тиолят-нонами (RS~).
Галогенарены с нуклеофильными реагентами взаимодействуют
только при температурах выше 200 °С. Сама система бензола при
этих температурах стабильна:
,С1
+ Na+OH"
+ Na+Cl-
Так из хлорбензола может быть получен фенол. Эти реакции ката-
катализируют медь и соли одновалентной меди:
200-300 °С
аГ
Во многих случаях для реакций можно использовать соли Cu(I)
с нужным анионом:
II +CuY-^| I! +CuX
Легче других галогенаренов реагируют иодарены. Роль меди
или солей меди до конца не выяснена. Предполагают, что активной
241
частицей является ион Си+, который выступает в качестве акцепто-
акцептора и образует комплекс с переносом заряда либо с атомом галогена,
либо с л-электронной системой бензола:
Я48
-CuY
8+jK
+ CuY
В обоих случаях уменьшается плотность электронов на атоме угле-
углерода в связи С—X F'>6), что способствует реакции.
2. Реакции в углеводородном остатке, а) Р е а к ц и и присое-
присоединен!! я. Галогеналкены присоединяют галогены, галогеново-
дороды и другие элсктрофильные реагенты, Скорости реакций при-
присоединения ниже, чем для соответствующих реакций алкенов. Но
в реакциях присоединения галогеноводородов атом галогена дейст-
действует как электронодопорная группа:
X
СНз-СН
Y XX
6+
н
!
fi-
- Y
¦сн.
= сн-х-
^4-СН.,
X
1
-сн
i
б) Полимеризация галогеналкенов и галогеналкадиенов
происходит очень легко в присутствии инициаторов или катализа-
катализаторов (ср. полимеризация алкенов и алкадпенов, гл. II. 4.6, гл. III.
Б. 3):
(при X—Cl — полихлоропрен, при X = F — полифторопрен).
Эти реакции имеют исключительное значение в промышленности
для производства полимерных материалов и синтетического каучука.
в) Реакции отщепления. Галогеналкены, содержа-
содержащие водородный атом у углерода двойной связи, при взаимодейст-
взаимодействии с концентрированными растворами щелочей отщепляют гало-
геноводород и превращаются в алкпны. Предполагается, что реак-
реакция начинается отрывом атома водорода в виде протона:
•• -
+:.он
l
R—C=C-
242
Подобная реакция осуществляется также в случае галогенаре-
нов, только выделить продукт с тройной связью невозможно, он
слишком активен. Сильное основание (обычно литийорганическое
соединение) отрывает протон от галогепбензола и образует карба-
пиоп, который отщеплением галогенид-иоиа превращается в свое-
своеобразную нейтральную частицу — дегидробепзол (Г. Виттиг):
+HR
Структуру дегидробепзола трудно изобразить. Можно написать
мезомерные структуры с локализованными у соседних углеродных
атомов зарядами и бирадикал. Структура с обычной тройной свя-
связью менее вероятна, так как перекрывание двух /7-орбиталей в би-
ргдикале небольшое. Уловить дегидробензол в растворе можно
при помощи диенового синтеза. Дегидробепзол является активным
дпенофилом и присоединяется к 1,3-диепам и антрацену:
триптицен
г) Реакции замещения характерны для галогенаре-
нов. Известны обычные реакции электрофилыюго замещения: нит-
нитрование, сульфирование, галогенирование. Скорости реакции срав-
сравнимы со скоростями подобных реакций бензола. Атомы галогена в
этих реакциях выявляют свои злектронодонорные свойства на л-
злектронпом уровне и ориентируют вступающие группы в о- н п-
положения:
X
I
X
I
X
UNO.,
NO,
X
1 /NO,
X X
,SO3H
IINO3
N02 N02
so.
SO3H
5. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ
Винилхлорид СН.,-СИ—С1 представляет собой бесцветный газ со
слабым запахом хлороформа, растворяется в органических раство-
растворителях, нерастворим в воде.
В промышленности винилхлорид получают в крупных масшта-
масштабах. Основным процессом является присоединение хлороводорода
к ацетилену в газовой фазе над катализатором (активированный
уголь — HgCl2):
120-180 °С
-СН2- СН-С1
Вторым способом получения является дегидрохлорирование ди-
дихлорэтана в газовой фазе при температуре 400—500 СС. В послед-
последнее время этот метод вытесняет дегидратация этилепхлоргидрина
B-хлорэтанола):
НОСН2СН2С1 -^ СН2 - СП - С1 + Н2О
Ка г.
В обоих методах в качестве исходного сырья используется этилен.
Винилхлорид легко нолимернзуется. Поливинилхлорид широко
применяется для получения полимерных материалов и синтетиче-
синтетического волокна.
Трихлорэтилен СС12—СНС1 — бесцветная жидкость с запахом
хлороформа. В воде не растворяется, по образует азеотроппую
смесь, кипящую при 73,6 °С и содержащую 5,4% воды. Получают
его из ацетилена по схеме:
400-500 °С
НС -л СН + 2С12 —> 11СС12 — СНС12 >¦ СС12 = С11С1 + HCI
Трихлорэтилен — прекрасный растворитель жиров, масел, смол,
каучука. Его используют для обезжиривания тканей, кожи, по-
поверхности металла, для экстракции жиров и масел из природных
продуктов, для химической чистки одежды.
Тетрафторэтилен CF2—CF2 — бесцветный газ без запаха,
т. кип. —76,3'С, uf4~77~l,519. He растворяется в воде.
Тетрафторэтилен получают в крупных масштабах пиролизом
дифторхлорметапа (фреона-22):
П00-700 'С
2F2CHC1 >¦ CF2 =- CF2 + 2НС1
При полимеризации тетрафторэтилена в присутствии перокси-
дов получают политетрафторэтилен (фторопласт, тефлон) в виде
белого порошка с молекулярной массой 50 000—2 000 000. Поли-
Полимер используют для изготовления различных изделий, особенно для
нужд химической промышленности. Он устойчив к действию кис-
кислот, щелочей, окислителей, даже элементарного фтора (имеет «кожу
носорога и алмазное сердце»).
Хлоропрен СН2—-СС1—СН = -СН2 — бесцветная жидкость со
своеобразным запахом, в воде не растворяется. В промышленности
244
его получают в крупных масштабах из винилацетилена в НС1:
0-20 "С
не = с-сп =- ch2-j-HCi C|Cl, NH<c-j*сн2=-_ cci-сн =. сн.
Соединение легко полпмеризуется, в результате чего получается
хлоропреповый каучук — исходное сырье для кабельной и шинной
промышленности.
Хлорбензол — бесцветная жидкость со своеобразным запахом.
С водой образует азеотроиную смесь, кипящую при 90,2 СС и содер-
содержащую 71,6 % хлорбензола. В промышленности его получают хло-
хлорированием бензола в присутствии железных опилок при 75—85 СС.
Используют хлорбензол в качестве растворителя и как исходное
сырье для промышленного органического синтеза (получение фено-
фенола, анилина, нитрохлорбензолов).
Дихлордифенилтрихлорметилметан (ДДТ) — бесцветное крис-
кристаллическое вещество с т. пл. 108—109 °С, хорошо растворим в
органических растворителях:
Н
Его получают из хлорбензола и трихлоруксусного альдегида (хло-
раля) в присутствии серной кислоты. Дихлорднфенилтрихлорметил-
метап представляет собой сильнодействующий универсальный кон-
контактный инсектицид. Долгие годы его с большим успехом ис-
использовали в сельском хозяйстве при борьбе с вредными насекомы-
насекомыми. Однако последнее время отказываются от широкого применения
ДДТ, так как он накапливается в почве, растениях, организме жи-
животных, в молочных продуктах и вызывает опасность хронического
отравления.
Глава IX
ГАЛОГЕНПРОИЗВОДНЫЕ ТИПА C(sp) —X.
ГАЛОНИЕВЫЕ СОЕДИНЕНИЯ
К соединениям типа C(sp)—X принадлежат галогенацетилены, ди-
галогенацетилены и галогеналкпны:
н— с=с—а I—с —с—i сн3сн2—с==с—вг
хлорацетнле'н дииодацетилеп I -бромбутпн-1
1. МЕТОДЫ ПОЛУЧЕНИЯ
Галогепалкины получают реакцией непосредственного галогеппро-
вапия и реакциями отщепления.
Прямое галогеиирование. Галогенировать можно ацетилен и мо-
нозамещенные ацетилены галогеном в щелочной среде. Предполага-
245
ется, что галогенируется ацетилеиид-ион:
Пг2; N.iOH
R —С—С—Н *R_c = C-Br
Реакции отщепления. Исходными продуктами служат дпгало-
геиалкеиы и тригалогеналкеиы или тетрагалогеналканы, которые
при действии крепкой щелочи отщепляют галогеноводород:
ОН-; /"
I-ICX г= СНХ > МС = С— X
ОИ-; t° OH-; Г
СХ2 = СИХ >Х — С^С — Х< СИХ, —СНХ2
он -
R —СН=СХ2 >-R_c=C-X
2. ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
Галогепалкипы с атомом галогена у тройной связи представляют
собой бесцветные газы или жидкости. Например, днхлорацетилеи
кипит при 32 °С. Дипольные моменты значительно понижены по
сравнению с галогепалкенами. Хлорацетилеп НС = С—CI имеет
дипольный момент 1,47-10~30 Кл-м @,44 D). Это свидетельствует о
дальнейшем понижении полярности связи С—С1.
Первые представители ряда (хлорацетилен, дихлорацетилеи,
бромацетнлен) весьма нестабильны п являются ппрофорами, т. е.
при соприкосновении с воздухом самовоспламеняются. Тройная
связь сохраняет способность вступать в реакции присоединения:
Н-С=С-С1-)-Н_Х—s-CH^c/"
ЧС1
Нй2 +
R_Cr-C-Cl-!-H2O -R —ССН2С1
['
О
Известны реакции нуклеофилыюго замещения атома галогена
у тронной связи.
3, ГЛЛОНИЕВЫЕ СОЕДИНЕНИЯ
При координации неподеленион электронной пары атома галогена
с карбокатиопом могут образоваться соли галония (хлоронпя, бро-
мопия, иодопия):
R-X:+R+Y-—4R-X-R1+Y-
(здесь Y" — комплексный анион: BF4~, SbF7 н т. д.).
Соли хлороиия и бромоння очень нестабильны. Стабильность в
значительной степени определяется природой того углеродного ато-
атома, с которым связан атом галогена. Относительно более стабильны
соли галония, имеющие связь X—C(sp2) (соли диарилгалоиия).
Образование катионов диалкплхлорония и дналкилбромопня в
растворах обнаружено методом ПМР только при очень низких тем-
температурах (—80. . .—120 "С) в смесях R—F, R—Cl (R—Вг) и SbF
246
(Г. Ола):
—X + SbF,—*[R —X —R]+SbF7 (X = Cf, Br)
При —50 CC и выше эти катионы начинают разлагаться. Они яв-
являются сильнейшими алкнлнрующими реагентами.
Более стабильны соли днарилхлорония и диарнлбромония. Их
получают с низким выходом арилированием галогепаренов борфто-
рндамн ареидиазопия:
Ar-Х: + [Аг —N = N] + ВРГ —* [Лг— Х-Лг] + BF.7 + Na (X=C1, Вг)
Предполагается, что в этой реакции образуются арилкатноны Аг+,
которые арнлпруют галогепарен (А. И. Несмеянов).
Соли диарилхлорония и днарилбромония — бесцветные кристал-
кристаллические водорастворимые вещества, разлагающиеся при 80—100сС.
Они являются арилнрующими реагентами.
Соли иодопия — наиболее стабильные из всех соединений гало-
шгя.
Диалкилиодониевые соли получают при низких температурах:
R — I-f -RF-LSbF5 —^[R —I —R]+ SbF^
Некоторые диалкнлиодониевые соли могут быть получены в кри-
кристаллическом виде, например (CH3JI+SbF^. Однако они быстро
разлагаются при соприкосновении с влагой воздуха.
Наиболее изученными солями галония являются соли диарнлио-
дошгя. Они стабильны до температуры 200 °С.
Соли диарилнодопия получают специальными методами иодони-
рования арепов соединениями 1A11). Для этой цели пригодны
арилиодозилсоедннення ArIY2, триацнлаты пода I(OOCRK и нодо-
зплсоли 10+Y~.
Арилиодознлсоединення получают при окислении иодаренов
сильными окислителями (С12, Н2О2, нерокслкнслотами):
/CI
А •„ Лс
С И ,СООО11 (ди хлориод)бсн?ол
пероксиук- ijv
сусная кис- ОН" )' HCI
лота
О
.ОССНз он-
ЧОССН3 ciiaCOOH
Ю
фепилиодозилди- нодозилбен-
аи.ет;;Т (диацетокси-. зол
|;одHснзол
247
В присутствии сильных кислот арнлиодозилсосдииення арил-
иодонируют арены:
!-H2SO4-
+ 2СН3СООН
Эта реакция является электрофильным замещением, в которой элек-
трофильным реагентом служат Аг1+ООССН3 или Аг12 + .
При действии на арены КЮ3 и H2SO4 образуются также иодо-
ниевые соли:
кю,
2ArII --[Ar-I-ArrHSO-T
H2SO4
Соли днарилиодопня представляют собой бесцветные кристалли-
кристаллические вещества, растворимые в воде. При взаимодействии с нуклео-
фильиыми реагентами они выступают в роли арилирующих реаген-
реагентов:
Разлнчие в стабильности хлороиневых, бромонневых и иодоиие-
вых соединений объясняется различным сродством к электрону
(электроотрнцателыюстью) атомов галогена. Фторуглеводороды
вовсе не образуют ониевые соединения. Легче всего свою электрон-
электронную пару координирует атом иода.
Соли диарилиодоиия используют в органическом синтезе для
введения арильной группы (арилирование).
Элементорганические
соединения
В органической химии выделяют особую группу соединений —эле-
менторганические соединения. В этих соединениях углеродный атом
образует химическую связь с различными другими элементами, не
считая атомов водорода, галогенов, кислорода, азота и серы.
Обычно к элементорганическим соединениям относят:
а) металлорганнческие соединения (связь С—металл);
б) борорганические соединения (связь С—В);
в) кремиийоргаиические соединения (связь С—Si);
г) фосфорорганические и мышьякорганические соединения (свя-
(связи С—Р и С—As);
д) селенорганические и теллурорганнческие соединения (связи
С—Se и С—Те).
Необходимо отметить, что это деление органических соединений
на элементорганическне и другие нестрогое и является весьма
искусственным.
248
Глава X
МЕТАЛЛОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Металлорганпческие соединения образуют очень большое семействе
органических соединений. Здесь они рассмотрены в последователь-
последовательности размещения металлов в периодической системе элементов
Д. И. Менделеева: сначала соединения металлов первой группы (Li,
Na, Си), затем металлов второй группы (Mg, Zn, Hg), третьей (Al),
четвертой (Ge, Sn, Pb) и, наконец, органические соединения пере-
переходных металлов (Ti, Cr, Fe, Co, Ni, Pd, Pt).
А. СОЕДИНЕНИЯ МЕТАЛЛОВ ПЕРВОЙ ГРУППЫ
Названия металлоргапических соединений образуют от названий
углеводородного остатка и металла.
Наиболее изученными и важными соединениями этой группы
являются литии-, натрий- и медьорганические соединения:
СНз—Li C0Hs-Na CGH5CH2-Cu
меткллитик, фенил н атрий бен зил медь
1. ЛИТПЙОРГЛИИЧЕСКИЕ СОЕДИНЕНИЯ
1. Методы получения. Литийоргаиические соединения образу-
образуются при взаимодействии металлического лития с галогенуглево-
дородами в растворителях, которые взаимодействуют с ионом метал-
металла (сольватпруют металлоргаиическое соединение), например в тет-
рагпдрофуране, диэтиловом эфире и других эфирах. Реакцию про-
проводят в инертной атмосфере (N2, Аг), чтобы избежать реакции окис-
окисления кислородом воздуха:
R —X-|-2Li —»• R —Li + Li''Х- (Х=^С1, Вг, I)
Механизм реакции см. гл. VII. 4.2.
2. Физические свойства и строение. В чистом виде литийоргаии-
литийоргаиические соединения представляют собой бесцветные жидкости или
кристаллические соединения с высокой температурой кипения. Раст-
Растворимы в органических растворителях (эфиры, углеводороды).
В чистом виде и в концентрированных растворах литийоргани-
ческне соединения представляют собой олигомеры — димеры, тет-
рамеры, гексамеры (fi"R—Li6l)n. Образование таких ассоциатов
объясняется образованием многоцентровых молекулярных орбита-
лей, так как связь а~С—Lifil- является силыюполярной, а катион
Li+ имеет незаполненную орбиталь.
3. Химические свойства. Литийоргаиические соединения обычно
в чистом виде не получают и не используют, так как они очень энер-
энергично реагируют с кислородом, СО2, водой и может происходить
самовоспламенение.
Их получают в растворах и в растворах же используют для даль-
дальнейших реакций. В растворах олигомеры литпйорганпческих соеди-
249
нений находятся в равновесии с сольватированными мономерами:
й- 6+ 2nO(C,HsJ 6- 6t-
В сильно сольватирующих растворителях происходит ионизация
и образуется карбанион:
л- б+
R —Li-f-2:Sol^R:.-+Sol:*-Li + -i-:Sol
6~ 6 +
Связь С—Li очень полярна. При переходе от галогепуглсводородов
к лнтийоргапическнм соединениям происходит обращение
поляризации углеродного атома от карбокатионного типа
к карбанионному типу.
Все реакции лнтийорганических соединений определяются нали-
наличием сильной поляризации связи С—Li, граничащей с ионизацией
и образованием карбаниона. Эти соединения являются сильнейшими
С-оспованиями и С-пуклеофнлами. Для них характерны реакции с
различными кислотами и другими электрофильными реагентами.
Литийорганические соединения активно реагируют с водой,
спиртами, аммиаком, аминами и другими соединениями, имеющими
подвижный водородный атом, т. е. достаточно полярную связь
л <- б—
II—Y:
Л- б+ fi+ б-
R —Li |-Н —Y—.-R —H-l-LPY-
В этих реакциях образуются углеводород и соответствующая литие-
литиевая соль. Например, при реакции с водой образуется LiOH, с ами-
аминами образуются амиды лития LiNHR, с ацетиленом — ацетиленид
LiCCH
Реакции с галогенидамн различных элементов, в том числе ме-
металлов (SiCI4, РС1з, GeCI4, A1C13, ВС1:) и др.), приводят к различным
элементорганическим соединениям:
6-6 + К-1Л R-Li
R — Li + 3Xn-^R — 3Xn_i-j-LiX -R,3XH_2 >¦ и т. д.
Эти реакции осуществляются благодаря сильной полярности свя-
б+б—
зи ХЯ_,Э—X.
Важное место занимают реакции присоединения к полярным
двойным и тройным связям (альдегиды и кетопы, нитрилы):
R1
б- 6+ R\ 6+ 6- | б- 6+
R—Li+ )C=O —*Ю — С — О — Li
R2/ I
R
алко:;снды лития
6-^
Li
б- 6+ 6+ 6— R\ й- /
R-Li-; R1 — C^N—>- )C = Xх
r/
ими пат лития
250
В некоторых случаях присоединение происходит также и к ма-
малополярным или пенолярным двойным связям С=С. Это, как пра-
правило, характерно для сопряженных систем, имеющих повышенное
сродство к электрону; например, в случае сопряженных диенов (бу-
(бутадиен) такое присоединение 'вызывает полимеризацию по карба-
ниошгому механизму:
R —Lif-;.CH2.--CH — СН- = СП2 -^ R —СНа —СН = СИ —СНг —Li —>
си?=сн-сн-сн,
>• ... -¦> Полниор
При окислении литнйорганических соединений кислородом воз-
воздуха в растворе конечными продуктами являются спирты (фенолы)
в виде их литиевых солен. Промежуточными продуктами являются
гпдропероксиды и их соли:
л- л+ л- 6 +
R —Li-;-O2—,R—О —O-Li+ —>-.., —> R — O-Li
ПЛКОКСИДЫ
(феноляты)
:п1Т[1я
Литийорганические соединения широко применяются в органи-
органическом синтезе в качестве промежуточных продуктов. В промышлен-
промышленности применение нашел бутиллитий C4HaLi (в виде раствора в уг-
углеводородах) в качестве инициатора полимеризации бутадиена.
Литийорганические соединения применяют для промышленного
синтеза комплексных металлорганнческих катализаторов для стс-
реорегулярпой полимеризации алкеноп, алкадиепов и алкннов.
2. НЛТРИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Натрийоргапические соединения могут быть получены взаимодейст-
взаимодействием галогенуглеводородов с натрием (см. синтез Вюрца, гл. VII.
4.2). В чистом виде натрипорганические соединения получают реак-
реакцией натрия с ртутьоргапически.ми соединениями:
й- fi +
СН3 — Нд — СНз-t 2Ма —>¦ 2СН3 —\а -;- Ilg
диметЕглртуть
Натрийорганпческнс соединения образуются также в реакции
некоторых углеводородов с повышенной СН-кислотпостыо (ацетиле-
(ацетилены, трнфенилмстап, циклопептадпеп) с натрием:
R — II | Na —> R:-Na+-[-I/2FI2
Натрийорга1[ическне соединения — кристаллические вещества,
в ряду алкапов бесцветные, в ряду сопряженных систем окрашен-
окрашенные. Окраска соединения обусловлена природой сопряженного
карбаниона. Эти соединения очень полярны и обычно пх считают
ионными:
б- 6 +
R — i\a^.iR;-
251
Натрийорганические соединения очень активны — реагируют с
влагой воздуха и кислородом, может наблюдаться самовозгорание.
Обычно их получают и используют только в растворах.
Некоторые из натрийоргаиических соединений применяют
в качестве инициаторов полимеризации.
3. МЕДЬОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
t. Методы получения. Медь с галогепуглеводородами при обычной
температуре не взаимодействует. Медь является металлом малоак-
малоактивным, кроме того, ее поверхность всегда покрыта плотным ок-
оксидным слоем. Возможность взаимодействия меди с галогенпроиз-
водными при высоких температурах спорна, так как медьорганиче-
екпе соединения термически нестабильны. Для получения медьорга-
нических соединений используют другие активные металлоргани-
ческие соединения, например литийорганические:
6- 6+ СгН.ОСНцв- в +
R —Li+CuX _i^onc-R —Cu + LiX
Известны только органические соединения одновалентной меди.
Они представляют собой бесцветные или желтоватые малораствори-
малорастворимые порошки. Медьорганические соединения сильно ассоциированы.
2. Химические свойства. Медьорганические соединения терми-
термически нестабильны и распадаются но свободпорадикальному меха-
механизму.
о°с
С2Н5—Си—^-С2Н5. -->СНяСН3-| СН2-=СН2
Относительно более стабильны соединения ар ил меди, например
С6Н5Си распадается при 50. . .60 "С.
Органические соединения меди чувствительны к действию кис-
кислорода воздуха, воды, спиртов, кислот. Легко реагируют с галогеп-
галогепуглеводородами и другими галогепсодержащими соединениями
(например, ацилгалогенндами):
СН,Вг
:—>СН3—СНЯ
з —Си —
С„Н51
-с6н5-сн3
H5-Qf —*С„Н,-С^ +СиС1
ХС1 ХСН3
Особое сродство медьорганические соединения имеют к иодпро-
изводпым. Предполагается, что Cu(I) имеет большую тенденцию ко-
координироваться с атомом иода.
Атом меди в медьоргапических соединениях имеет тенденцию
координироваться с донорными частицами. Например, при взаимо-
взаимодействии с литийорганическими соединениями образуются медьор-
медьорганические соединения нового типа — с двумя связями С—Си:
е- fi+
R — Li-|-R— Cu^ri [R — Си — R]-Li +
252
Эти соединения называют лшпийдиалкилкупратами. Они хо-
хорошо растворимы и используются в синтезах вместо соединений
R—Си. При взаимодействии с галогенпронзводпымн происходит
замещение атома галогена на алкнльп^га или арпльпую груп-
группу. Легко происходит присоединение» к полярной связи С—С (см.
гл. XXVII. Б. 3):
[R_Cu-R]-Li+-; R^— Х~—> R-R1-; R-Cu '-LiX
Реакции с карбонильной группой альдегидов и кетопов обычно
идут медленно.
Особое место среди медьорганических соединений занимают аце-
тилениды меди (см. гл. IV. 4.3), реакционная способность которых
меньше, чем R—Си, где R — алкил или арил.
Б. СОЕДИНЕНИЯ МЕТАЛЛОВ ВТОРОЙ ГРУППЫ
Органические соединения металлов второй группы могут быть двух
типов: с одним углеводородным остатком (связь С—М) и двумя
(связи С—М—С):
C2H6-Mg-Br CeIIe-Mg-CeH, C4H9-Zn-C4H9 CHaIIgI
этилм.'!П1мГ|бромид дифенилмагнин дибутилцимк метплртутьнодпд
Наиболее изученными и важными являются магний-, цинк- и
ртутьорганические соединения.
1. МАГНИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
1. Методы получения. Магннйорганические соединения получают
прямым взаимодействием магния в виде стружки с галогенуглево-
дородами, обычно в растворе сухого диэтилового эфира или тетра-
гидрофурапа:
сан5осан,
r __ X -;- Mg * R - MgX
Эту реакцию разработал французский химик В. Грипьяр A901 —
1903) и она носит его имя — реакция Граньяра. Магпиноргапические
соединения часто называют реактивами Гриньяра.
Реакция происходит на поверхности металла, который служит
электронодопором, а связь углерод—галоген — электропоакцепто-
ром. Промежуточными частицами могут быть свободные радикалы:
R- X: -}- -Mg- —> |R^X:]-Mg- ^ -, R. -\- :X:.- -|- Mg + —> R"-Mg-'xl.
Впервые эта реакция была проведена в среде безводного (абсо-
(абсолютного) диэтилового эфира, в котором растворяются магинйоргани-
чеекпе соединения. Позже было показано, что применять можно
другие простые эфиры, в том числе циклические (тетрагидрофуран)
и третичные амины, например гм'.М-диметилапилип.
2. Физические и химические свойства. Применение. Магпинорга-
ипческне соединения используются исключительно в виде раство-
253
ров. Они могут быть получены также в сухом виде —> бесцветные
аморфные неплавкие порошки, чувствительные к действию возду-
воздуха. В твердом виде они являются полиассоциатами:
•-.. ..*.'.- ..*-.. ..-¦«•... ...*-.
'Mg ".Me Ms. .Ms.
•••" '¦•¦х-' '••*¦;* "•х-' " -к-*'
Полнассоциатная цепь здесь изображена весьма приближенно, так
как связь Mg- • -X отличается от связи Mg- • -R.
В растворах магнийорганичсские соединения сольватированы
молекулами эфиров или других электроподопорпых частиц:
8 , ..^ « )^
R—MgX + 2 :0. ^=S R—MgX
В действительности в растворах магннйорганические соединения
представляют собой более сложную смесь, состоящую из сольвати-
рованных RMgX, R2Mg, их олигомеров (RMgX)rt и (R2Mg)rt и про-
продуктов их взаимодействия с MgX2, например:
2R —MgX^iR— Mg —R + MgX2^iR2Mg.MgX2
Независимо от строения соединения в растворе пользуются обыч-
обычно простой структурой RMgX, где акцентированы енлыгаполярные
связи и карбонионная природа углеродного атома:
б- в+ 6-
R-Mg-X
Реакции магнийорганических соединений в основном подобны
реакциям литийорганических соединений, только активность RiHgX
меньше и они менее чувствительны к действию кислорода воздуха,
что позволяет проводить реакции в обычных условиях, без инертно-
инертного газа. Это значительно упрощает технику эксперимента и делает
магнийорганические соединения одним из известнейших реагентов
в органическом синтезе.
Легко идут реакции с соединениями, содержащими подвижный
атом водорода:
е- 6+ е+ е-
— Y—*R — H-j-Y— MgX
В этой реакции выделяется углеводород и образуются соли маг-
магния или другие производные магния.
Если в реакции используют CH3MgI, выделяется метан. По
объему выделившегося метана можно количественно определить
соединения с активным водородным атомом (воду, спирты, тиолы,
амины, ацетилены и др.) —метод Чугавва — Цвревитинова.
В случае ацетиленов и циклопентадиена эта реакция дает новые
магнийорганические соединения, которые могут быть использова-
254
ны для дальнейших синтезов:
R—MgX + R1— С=С—Н *- R'-CsC— MgX + R-H
реагенты Иоцича
R-MgX + $\ *• <^гЪ MgX +« R-H
иикяопмггаяиеияд
Легко осуществляются реакции магнийорганических соедине-
соединений с галогенидами различных элементов (ЭХП)> приводящие к но-
новым элемеиторганнческнм соединениям (ср. реакции литийоргани-
ческих соединений).
Реактивы Гриньяра присоединяются к полярным двойным и
тройным связям. В органическом синтезе широко используют их
реакции с диоксидом углерода, альдегидами, кетонамн, нитрилами:
6- fi-i-
R —MgX
6- 6 +
R-MgX
6- 6 +
R-MgX
6-
-i-0 =
r
6+ t
r
6 +
Э—^R— C"
мапшепые соли
карбоноиых кислот
Rl
E- |
Э —* R— С — OMgX
R2
алкокспды
магния
el R\ ,/Mg:
иминагьг магния
Реактивы Гриньяра принадлежат к наиболее важным реагентам
органического синтеза,
2. ЦИНКОРГЛНИЧЕСКНЕ СОЕДИНЕНИЯ
Впервые цинкоргаиические соединения были получены в 1849 г.
3. Франкландом при взаимодействии цинка с этилиодидом:
+ 2C2H5J — у С2Н5 — Zn — C2H5 -|-ZnI2
Диэтилцннк был первым металлоргаиическим соединением в исто-
истории органической химии.
Галогеналканы с цинком реагируют хорошо, но протекание ре-
реакции в сильной степени зависит от чистоты поверхности цинковой
пыли или стружки. Вииилгалогеннды и галогенарены с цинком не
реагируют.
Общим методом получения ципкорганическнх соединений явля-
является реакция литнйорганпческнх соединений с безводным ZnG2:
е- бь
—¦ R — Zn — R-;-2Li CI~
255
Простейшие щшкорганпческпе соединения представляют собой
бесцветные жидкости с весьма низкими температурами кипения;
например С2Н.^пС2Н5 кипит при 116,8 °С. Это свидетельствует о
том, что циикорганнческие соединения в отличие от мапшйоргагш-
ческих являются ^ассоциированными. Известны также соединения
RZnX (X = I, Вг).
Цинкорганические соединения очень легко реагируют с кисло-
кислородом и при соприкосновении с воздухом происходит самовозгора-
самовозгорание (пирофорные соединения). Эксперименты с чистыми цннкоргани-
ческими соединениями являются опасными. Несмотря на это, цннк-
оргапические соединения исторически занимают видное место в по-
получении других элементорганнческих соединений, например:
3R2Zn-i-2AsCl3—>¦ 2R3As-i-3ZnCI.2
3Ro2n-!-2BCK —>¦ 2R3B + 32nCl,
Казанская школа химиков (А. М. Бутлеров, А. М. Зайцев,
С. Н. Реформатский) широко использовала циикорганические соеди-
соединения для синтезов (см. гл. XXXIV. Б. 1):
е- , „
R—C<f +R'-2n — R'
XH
/О
R-Cf
4
OZnR'H+
R'
0
il
-C-R'
КС ТОНЫ
он
R—С-Н
R'
вторичные
спирты
ацнлхлорнды
Циикорганнческие соединения менее активны, чем магнийорга-
6— rt +
нические, связь С—Zn менее полярна. Поэтому цинкорганические
соединения трудно реагируют с СО2, кетонами, сложными эфирами,
3. РТУТЬОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
t. Методы получения. Впервые ртутьорганические соединения были
получены в 1853 г. Э. Франкландом при взаимодействии иодалканов
с ртутью на солнечном свету:
hv
R-I + Hg—R-Hgl
2R_HgI~>R2Hg-bHgI2
Сначала образуются алкилртутьиодиды, которые при воздействии
некоторых реагентов превращаются в диалкилртуть. Образование
алкилртутьиодида под действием света является типичной свободно-
радикалыюй реакцией:
ftv
R —I —> R- + I.
R-Hg.+R-I—>R-
R-Hg.+Ь—«.R-Hgl
256
Более эффективным методом получения является взаимодействие
галогснуглеводородов с амальгамой натрия:
Любое ртутьорганическое соедините можно получить, исполь-
используя другие, более активные металлоргаиические соединения (Li,
iMg, Zn, A1), например:
2RMgX :-IIgCl2—> R —Hg —R-j 2MgXCl
Известны также методы прямого введения атома ртутп вместо
атома водорода в электронодопорные арены или соединения с под-
подвижным атомом водорода (реакция мвркурирования), например:
D: D:
I I
-;-Hg(OoccH3J
%,
-| НООССП,
HgOOCCII3
Злектрофильным реагентом здесь является катион +Hg00CCH3.
Известны реакции присоединения солей ртути к двойным и
тройным связям (см. гл. П. 4.2.г):
И—О с — H-;-HgCI2— >ClCH^CII — HgCI
fi-хлорвинил ртуть-
хлорид
Производные арилртути получают реакцией А. Н. Несмеянова
ил арендиазонневых солей (гл. XXV. А. 3).
2. Физические свойства. Производные ртути с одним
углеводородным остатком — бесцветные кристаллические соедине-
соединения. Соединения диалкилртути являются бесцветными легколету-
легколетучими жидкостями. Все ртутьорганические соединения, особенно
легколетучие, чрезвычайно ядовиты.
Группировка С—Hg—С является линейной: угол С—Hg—С ра-
равен 180'\ Полярность связи С—Hg невелика.
3. Химические свойства и применение. Ртуть-органические
соединения ввиду малой полярности связи С—Hg инертны по
отношению к полярным реагентам, стабильны в водных средах.
Соединения RHgX в некоторой степени ионизированы и проис-
происходит обмен анионов: RHgX + Y-;—RHgY :-X~.
Меньшей растворимостью обладают иодиды.
Своеобразными являются реакции симметризации и десимметри-
::ащш под действием различных реагентов:
SR-IIgl ¦ 2KI —R-IIg-R-i-K.,HgI4
Я — Hg —R .-HgX., - >2R —IlgX
Реакции с электрофнльнымн реагентами, например взаимодейст-
взаимодействие с кислотами, идут медленно. Малоактивные соединения с одной
связью Hg--C реагируют с кислотами только при нагревании:
IICI
R — Hg —R-:.ji С\~ —, R—HgCI-j-R —H - ' 2R — H -,- UgCl,
9 № 517 257
В принципе можно осуществить реакции с такими реагентами,
как галогены, ацнлхлориды, галогениды элементов ЭХ„.
Металлы более активные, чем ртуть, вытесняют ее из соединении
ртути и образуют другие металлоргапические соединения (Ка, К,
AI и др.), например:
R ~- I lg- R~-i-2К —> 2R: "К :- -|- Hg
Ртутьорганические соединения могут быть широко использова-
использованы в органическом синтезе, по этому мешает их сильная ядовитость.
Некоторые ртутьорганические соединения используют в качест-
качестве фунгицидов, а также для дезинфекции и протравливания семян.
В. СОЕДИНЕНИЯ МЕТАЛЛОВ ТРЕТЬЕЙ ГРУППЫ
Известны три главных типа органических соединений металлов тре-
третьей группы: RMXo, R2MX и R3M, где М — Al, Ga, In, Tl, a X —
галогены, Н, HSO4, OOCCR', OR' и др. Известны также соединения
с четырьмя связями углерод—металл, например [R4AI]~Li + . Наибо-
Наиболее важными являются алюминийоргаиические соединения. Напри-
Например:
С113А1С12 метилалюминийдихлорид
(С2Н3JА1Н дпэтп л алюминий гидрид
(CjIIjiJaAl трибутилалгомнний
[(C2II5)IAl]~Li+ тетраэтнлалгампппилптий, тетраэтнлалюмипат лития
1. Методы получения алюминийорганических соединений. Впер-
Впервые алюминийоргаиические соединения были получены в 1859 г.
действием алюминия па галогеналканы:
3RX+2A1—>
Однако они не были выделены в чистом виде. Соединения типа три-
алкилалюмииня были впервые получены в 1865 г. действием алюми-
алюминия на ртутьорганические соединения:
3R2Hg-i-2Al —> 2R3Al-b3IIg
Эти соединения оказались чрезвычайно реакционноспособпыми,
бурно реагировали с кислородом воздуха и водой.
Для получения алюминийорганических соединений могут быть
использованы литий- и магпийоргапические соединения:
ЗШ+А1С13—> R.r<\I + 3Li-;Cl-
Революцию в химии алюминийорганических соединений вызвало
открытие К. Циглера A955), который реакцией алкенов и алюминия
в присутствии водорода при повышенных давлении и температуре
C—20 МПа, 60—100 "С) получил алюмшпшоргаипческие соедине-
соединения, например:
Реакции способствует добавление к реакционной смеси неболь-
небольшого количества готового триэтилалюминия. Предполагают, что в
258
этой реакции промежуточными продуктами являются гидрид алю-
алюминия и алкилалюминнйгидриды, которые присоединяют этилен'
СН,=;СП2
гСНо^СН^-ЛШ,.,^ (С,Н5JА1Н —-*—>¦ (С2Н5KЛ1
Подобные реакции происходят и с другими алкенами:
3R—СН--СН2--Л1 ¦:-• 1,5Н2 —> (RCnXH2K/\I
2. Физические свойства и строение. Алюмиинйорганические сое-
соединения — бесцветные жидкости и кристаллические вещества с от-
относительно высокой температурой кипения. Эти соединения обычно
перегоняют в вакууме (табл. 26). Алюминийоргапическис соедине-
Таблица 26. Физические константы некоторых алюминийорганических
соединений
Соединение
(СНгОяЛ!
(С2НзKЛ1
(С,Н,)>Л1Н
(адо-С4Н!));,Л1
Т. пл., «С
15
—52
—50
4
Т. кип., еС
126
207 (разл.)
48—49 A33 Па)
58—60 F7 Па)
32—34 F7 Па)
"Г
0,752
0,837
0,808
0, 7ьи
ння ассоциированы и образуют главным образом димеры Мономе-
Мономерами они являются только в разбавленных растворах.
Своеобразна структура димера трнметнлалюмипия. В одной плос-
плоскости находится четыре углеродных атома четырех метальных групп
и два атома алюминия, а две мешльпые группы лежат в перпенди-
перпендикулярной плоскости:
AI
СН,
7.5 '
сн,
/СНз
>Q ,2.
1 o,Jd им '
Образуется многоцонтровая связь между двумя углеродами н двумя
алюминиевыми атомами. Этому способствует незаполненная орби-
таль атома алюминия.
3. Химические свойства и применение. Алюминнйорганическиг
соединения обладают большой химической активностью, так как
они содержат полярную связь С—А1 и, кроме того, незаполненную
орбиталь атома алюминия.
Очень интенсивно протекает реакция алюминийорганических сое-
соединений с кислородом и влагой воздуха, что вызывает самовоспла-
самовоспламенение и взрыв. Алюмннийоргапнческие соединения пирофорны,
поэтому крайне опасны при неосторожном обращении. Несмотря
9* 259
на это, их производят в промышленности в крупных масштабах и
используют для синтезов.
/Можно выделить несколько групп характерных реакций алюми-
шшорганических соединений: реакции термического разложения,
реакции с электронодонорпыми соединениями (металлорганическпе
соединения, гидриды металлов, эфиры и амины, алкеиы и др.) и,
наконец, реакции с электрофильпыми реагентами н кислородом.
Ллюмипийоргапические соединения термически весьма неста-
нестабильны. Для них характерна реакция расщепления, протекающая
с выделением алкепа п образованием дпалкплалкг.шпийгидрида:
200 'С
(СоН.,);,.\1 >(С,П5),А1 — Н ; СЛ\,-~СЛ\,
Триизобутилалюминип распадается с выделенном нзобутплепа уже
при 100 С. При температуре выше 300 С все алюмнппйорганическне
соединения распадаются полностью с образованием чистого алюми-
алюминия (кроме триметилалюминпя).
Элоктронодопорпые соединения присоединяются к атому алю-
алюминия, используя его незаполненную орбиталь, например:
(С..Н5ЬЛ1 C2[[,,-Li —- [(C2[l5>1,\l]-I.i •
(СоП5ЬЛ1 :0(Coir,,U - * (C2H3bAI -- :О(СЛ[,-,),
Наиболее важной реакцией алюмииийоргаппчеекпх соединений
является взаимодействие с алкепами. Под давлением алкепы при-
присоединяются, как бы внедряясь между атомом углерода и атомом
алюминия. Предполагают, что сначала образуется комплекс с пере-
переносом заряда:
R.Al — R + CH. = CH,ii^-=r R.AI^ lOMIla^ R.,AiCH.CH,R.
f
сн.=сн..
КПЗ
Процесс может продолжаться:
RoAICI!,CH2R . Clh -C[[2 — R?A1CHXH2CH2C[[.,R '' ""^
—> R2AlCH2CII2C[[2C[[oC[[2CH;,R
При удлинении цепи соединение становится термически неста-
нестабильным и отщепляется а.ткеп:
RaAICHoCHXHXdoCIIoCPbR- - R.AIH -|-CH2^-CHC[I.,CH,,C[I,C[LR
Таким путем можно осуществить димеризацию и очнгомеризл-
цию алкепов. Например, димеризацпя пронена в присутствии три-
пропилалюмипия:
СН3
Ллкииы в определенных условиях образуют полимеры с сопря-
сопряженными двойными связями.
260
Реакции алюминийорганическнх соединений с электрофильны-
ми реагентами подобны реакциям литийорганических соединении.
Алюминийорганические соединения широко используются в про-
промышленности. Они являются дешевым исходным сырьем для полу-
получения других металлоргапических и элемепторганических соедине-
соединении, в том числе для получения комплексных катализаторов
стереорегулярпой полимеризации. Их используют для олигомериза-
ции алкенов, в результате чего получаются алкепыС,—С10 — исход-
ные вещества для синтеза высших спиртов и карбоиовых кислот.
Диалкилалюмипийгидрид1)[ являются сильными восстановителя-
восстановителями. При термическом разложении алюминийорганическнх соедине-
соединений получают алюминий высокой степени чистоты (99,999%).
Г. СОЕДИНЕНИЯ МЕТАЛЛОВ ЧЕТВЕРТОЙ ГРУППЫ
1. Номенклатура. Известны четыре главных типа соединений:
RMX,, R2MX.,, R3MX и RiM, где М — Ge, Sn, Pb; X — галоген,
Н, OR' и др.
Кроме обычной номенклатуры для названия этих соединений
предложена и другая, основывающаяся па названии гидридов:
GeH4 •-- гермап, SnH4 — стаинан, PbH.i — плюмбан. Например:
(C0H.0aSnCI2 (СЛ15LРЬ
мет мл герма ни i"i- дкфешглолоподп- тетр<г;тилс1шкец
трихлортгд (ме- хлорид (дифсшглди- (тстрачтилилюм-
тллтрихлоргер- члорстаннан) fian)
М<|11)
2. Методы получения. Обычно органические соединения метал-
металлов IV группы получают из других металлорганических соединений
и галогенидов германия, олова, свинца (GeXi, SnX.b PbX«):
GeCI4 ;-4CH3Li - * (CH;1LGe-; 4LiCl
2PbCb' + 4CilI5MgCl -> (C»H5LPb ' Pb-,-4MgCl2
В реакции с соединениями Pb(II) всегда образуются производные
Pb(IV) и выделяется металлический свинец.
Для получения свинецорганических соединений разработан спе-
специальный метод — реакция галогепалканов со сплавом натрия и
свинца:
4С2Н5С14-РЬ(\'а) -- > (С2Н5LРЬ
3. Физические и химические свойства. Применение. Органичес-
Органические соединения металлов IV группы — бесцветные жидкости или
кристаллические вещества (табл. 27). Они ядовиты, особенно про-
производные тетраалкилсвинца.
Молекулы этих соединений имеют тетраэдрпческую конфигура-
конфигурацию. Обычные реакции металлорганмческих соединений для них
мало характерны, так как полярность связей М — С невелика.
Моногалогенпроизводпые в присутствии воды и оснований обра-
образуют гермаполы (станнанолы, плюмбаполы), которые легко отщеп-
отщепляют воду и дают гермоксаны (сташюксаны, плюмбоксаны):
2R3M—X -- 2R3M — Oil —у R3M —О —MRa-l-HjO
261
Таблица 27. Физические константы некоторых германийорганическик,
оловоорганических и свинецорганическнх соединений
Соединение
(CH3),GeCl
(СПзЬОеН
(CHaLGc
(ClI,LSn
(CH3LPb
(C2H5LPb
Т. пл., °С
— 123
—27,5
— 136
Т. кип., °С
98
26
44
78
ПО
198—202 (разл.)
82 A,73 кПа)
«Г
(
,2435
,009
),9725
,2915
,995
,652
1,435
1,389
1,39
1,52
Дигалогеипроизводные при гидролизе образуют олигомерные
продукты:
н.о
R2MX2—>- R2M(OHJ
(R2MO)n
Эти превращения аналогичны реакциям галогенсиланов (гл. XII.
4.1).
Тетраалкилпроизводиые германия, олова и особенно свинца при
нагревании свыше 250—300 °С разлагаются с выделением металла
и образованием свободных радикалов:
(C2IIeIM-:-4CiHe.
2С2Н., ;-2С2Н0
Реакцию термического разложения используют для получения
топких пленок чистого металла. Широкое применение нашел тетра-
этилсвииец. Его добавляют к бензину для уменьшения детонации
в двигателях внутреннего сгорания (антидетонатор).
Оловоорганические соединения применяют п качестве стабили-
стабилизаторов н антиокендантов для полимерных материалов, в качестве
фунгицидов, антисептических веществ и антигельминтов в сельском
хозяйстве.
Д. ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ ПЕРЕХОДНЫХ МЕТАЛЛОВ
Переходные металлы (Ti, V, Cr, Mn, Fe, Co, Ni, Pd, Pt и др.) образу-
образуют весьма неустойчивые органические соединения с ст-связыо М—С.
Обычно они легко распадаются с образованием свободных углево-
углеводородных радикалов.
В принципе органические соединения переходных металлов е
а-связью М —С образуются в обычных реакциях между галогепида-
ми переходных .металлов и активными металлорганическими соеди-
соединениями при низких температурах. Однако затем следуют реакции
гемолитического распада по связи М—С:
3C0H5Li -[-СгОз —> [(C0H5KCrj —* Продукты дальнейших превращений
трифенилхром
262
1. ТИТЛПОРГЛНИЧПСКИЕ СОЕДИНЕНИЯ
С а-СВЯЗЫО Ti—С
Известны четыре типа соединений, в которых титан находится в сте-
степени окисления IV:
CH3TiCls метилтнтантркхлорпд
(C0H5JTiCl2 дифсиилтнта иди хлорид
(C2H6KTiCl триэтилтитаихлорид
(CH3LTi тетраметилтнтан
Могут существовать также органические производные ТI (III)
и Ti (II), например RTiCl2. Все эти соединения являются нестабиль-
нестабильными, особенно тетраалкил(арил)титаны. При увеличении числа
атомов хлора стабильность увеличивается. Например, (CH3LTi
разлагается при О °С, a CH3TiCl3 — при 100 °С. Более стабильными
являются соединения, в которых ато.м титана образует связи с од-
одним или двумя л-лигандами (см. ниже), например диметил-бис-цик-
лопентадиепнлтитан.
Титаноргапические соединения получают мегаллорганическим
синтезом, т. е. при взаимодействии TiCli [или Ti(OR).J с литий-,
магний- или алюмпиийорганическими соединениями при —80. . .
. . .0 °С:
KMfiCI
TiCI4-J R.VIgCl -+ RTiCl3-j- MgClo >• R2TiCb —> ii т. и.
Титаноргапические соединения при нагревании легко распада-
распадаются:
R.i'1'i -' R3Ti ; R- —> R-.Ti-;-2R.
Наиболее важными титапорганическими соединениями являются
комплексные мегаллоргапические катализаторы (катализаторы Циг-
лера—Натта), которые применяются для стерсорегулярной полиме-
полимеризации алкепов (гл. II. 4.6), алкадиенов и алкинов.
Для получения катализаторов используют триэтилалюмипий
или трипзобутнлалюминий и TiCl., [или другие производные титана,
например эфиры ортотитановой кислоты Ti(OR)J в инертном раст-
растворителе:
R3AI ;- TiClj -¦* RTiCI3 J-RoA
RTiCI3-> R.-i-TiClj
R3.M-;-TiCI3 —> RTiCl2-i
Катализатор в инертном растворителе (углеводороде) может
быть нерастворимым (гетерогенный катализатор). Фактически эта
гетерогенная система катализатора представляет собой смесь RTiCl3,
.RTiCb, R2A1C1 и RA1C12. Ллюминийорганические и титаиорганиче-
ские соединения ассоциированы и образуют днмеры. Наиболее ак-
активными катализаторами являются соединения Ti(III).
Известны также растворимые, гомогенные системы катализато-
катализаторов. Их получают из циклопентаднеиильпых производных титана
и триэтилалюминия или других производных титана.
263
2. я-КОМПЛЕКСЫ ПЕРЕХОДНЫХ МЕТАЛЛОВ
\. л-Комплексы ванадия, хрома, марганца, железа, кобальта и и;ь
келя. Переходные металлы могут образовывать стабильные соеди-
соединения с рядом органических молекул, свободных радикалов и ионов.
Их строение не может быть объяснено обычными представлениями
теории валентности. Поэтому такие соединения называют ^.-комп-
^.-комплексами. В них атом металла в различных степенях окисления явля-
является электропоакцептором, а органические молекулы, свободные
радикалы и ионы представляют собой электронодоноры, их называ-
называют лигандами. По в л-комплексах переходных металлов осуществля-
осуществляется не только донорпо-акцепторпое действие лигапд->-металл. Име-
Имеет место также обратное взаимодействие между электронами атома
металла и разрыхляющими орбиталя.ми лиганда (дативная связь).
В я-комплексах переходных металлов пет обычных локализо-
локализованных а-связей углерод — металл, а образуются многоцентровые
делокализоваипые связи с использованием заполненных и незапол-
незаполненных орбиталей лпганда (L) и металла или попа металла (М).
В общем виде л-комнлекс можно написать как ML,,, где п определя-
определяется числом электронов в металле и в лиганде, которые использу-
используются для связывания. В общем случае лигапды могут быть разны-
разными, например A\L.rL^L^.
Наиболее стабильные л-комплексы переходных металлов чет-
четвертого периода образуются в том случае, если общее число элект-
электронов атома металла и лигандов равно 36 (число электронов крип-
криптона). Если число электронов меньше 36 (число лигандов меньше),
л-комп.текс является координационно ненасыщенным и обладает
большой реакционной способностью.
а) Главные типы лига и д о в рассмотрены в табл. 28.
б) При м еры л - к о м п л е к с о в.
л-Комплексы V@) (в атоме металла 23 электрона)
V2(COI2
V 23
(СО) . 6
додекакарбонпл диванадня трикарбоппл цнкло-
(имеете я снизь V — V) п'пт;п рнопп/тсападня
л-Комплексы Сг(О) (в атоме металла 24 электрона)
Сг(СО)в
гексякарбопил хрома, бензолтрнкарбопилхром, дибЕ'Изолхро\т, Т('\тно-
беецрешые кристаллы желтый крмст.'плм коричневые кристаллы
(т. пл. IG1 'С) (т. шт. 282 СС)
264
Таблица 28. Основные типы лигандов
Тш! лиггнда
Формула
Двухэлектроннын
Трехэлектрониый
Четырехэлектронпын
Пяти электронны и
Шестиэлектронпыи
Семпэлектроннып
Оксид углерода
Ллкепы
Ллкпны
Аллнлышй радикал
Цпклопропспильным ра-
радикал
Циклобутадпеп
Циклоцентадиенильный
радикал
Цпклонентадпеп ид-поп
Г)СНЗОЛ
Тропплпп-кагнои
Цн к логе птатр не пильный
радикал
)(
R' R
Н—С—С—И
/\ „ли[/Ч]
л-Комплексы Мп@) (в атоме металла 25 электронов)
Мп,(СОI0
Мп
(СО).,
25
6
декакарбоннл димангяна
(имеется связь Мп—Мп)
циклогкчпадиенилтрикарбо-
л и л марганец (цпмащ pv'n)
205
л-Комплексы Fe@) (в атоме металла 26 электронов)
FeiCO)s
лентячарбопил желе.">;],
КОриЧНОИаЯ ЖИДКОСТЬ
(т. клп. 10 1 СС)
Fe
(СОЬ
циклобутадиоптри карбо-
п ил железо
5
26
дицнклопентадиенил-
желело (ферроцен), жел-
желто-оран жен ые кристаллы
(т. ил. 173 °С)
л-Комплексы Со@) (в атоме металла 27 электронов)
НСо(СО).,
тетракарбонилгидрид
кобальта
^с>^ з
Со 27
(СО) я б
аллилтрнкарбомилкобальт
и-Комплексы Ni@) (в атоме металла 28 электронов)
Ni(CO),
тетракл рбонм:!
беепыч и;!'/; ^:и
(Т. kllil. -l.i
28
б
ал ко птрикарбопил никель
1'слп вокруг атома металла находится недостаточное число ли-
гандов, л-комплекс является координационно ненасыщенным н име-
имеет тенденцию к присоединению новых лнгандов. Так, например, сое-
соединения переходных металлов с а-связью металл — углерод фор-
формально могут быть рассмотрены как координационно очень нена-
ненасыщенные л-комплексы. Соединение дналкилиикель R2Ni[Ni@) +
-r 2R -] может присоединить три двухэлектронтлх лиганда:
Этим присоединением может быть вызвана тримеризация или
другая олнгомеризация алкппа.
В атоме титана только 22 электрона, поэтому
л-комплексы титана наиболее часто являются ко-
' ii1s ординационно ненасыщенными. Например, в бис-
циклоиентадненилдиэтилтитане лиганды дают 12
электронов, а титан — 22.
Имеется возможность присоединить еще одни
Ti
N
c2hs
266
лиганд с двумя электронами (алкеп, алкпн). Это является нача-
началом олягомернзации или полимеризации.
Для многих стабильных я-комплексов с лигапдами — цикличе-
циклическими сопряженными системами — характерны реакции замеще-
замещения в лиганде. Например, ферроцен легко реагирует с электрофиль-
ньши реагентами (проявляет «ароматические»свойства, см. гл. VI.Г).
2. я-Комплексы платины и палладия. Получе н и е. Соли
Pt(II) и Pd(II) легко реагируют с алкенами, алкадиенами и ал-
кииами, образуя продукты присоединения (я-комплексы). Впервые
образование таких продуктов в водных растворах наблюдал
В. Цейзе в 1827 г.:
II..0
CII2~CH2J Ka[PtCI4] - '<¦ KlC2H4PtCl3l • 11ЙО - i- KCl
соль ЦеПзе, желтые
кристаллы
PdCIo с этиленом в безводных средах образует димерный л-ком-
плекс:
2PdC!2-| 2СН2---СН2 -* (C2II4PdCl2J
В водных растворах образуются гидратироваиные я-комплексы:
и,о
PdCI-.-i-CH2--Cn2-;-II2O -V C,H,iPdCl2-H,0
Строение. Как свидетельствуют данные рентгеноструктур-
пого анализа, л-комплексы платины и палладия имеют строение
плоского квадрата. В вершинах квадрата
размещены лигапды (галогеиид-поны, алкеп,
молекулы воды или другие электронодоноры),
а в центре находится ион металла. Связь
С- С размещена перпендикулярно плоскости
квадрата.
При образовании л-комплекса изменяется
природа двойной связи алкеиа, связь удли-
удлиняется от 0,131 до 0,14—0,145 им и умень-
уменьшается плотность л-электроиов. Эти изме-
изменения объясняются пе только донорпым эф-
эффектом алкеп->-М" + , но н дополнительным
взаимодействием между разрыхляющей орбн-
талью алкеиа и электронами металла (рис.
82). Такое дополнительное взаимодействие
называется дативным эффектом металла или дативной связью.
Химические свойства. В л-комплексах металлов
можег произойти обмен лигандов, одни алкеп может вытеснять дру-
другой по принципу: слабый эл^ктронодоиор вытесняется сильным.
Изменяется реакционная способность связанного алкеиа: он
более легко подвергается атаке нуклеофильпых реагентов. Облег-
Облегчаются реакции присоединения. Практическое значение имеет при-
присоединение воды к этилену в присутствии солей палладия, когда
одновременно происходит и окисление с образованием уксусного аль-
альдегида (гл. II.4.4).
Рис. 82. Образование
святи в л-комплекее
алкеп—металл (за-
(заполненные орбит я л и
заштрихованы, неза-
незаполненные- II р<•!.!()[.IX-
ляющне пелаштрихо-
вапы)
267
Глава XI
БОРОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
В настоящее время известно большое число борорганических сое-
соединений. Их формально можно рассматривать как производные
бороводородов ВН3 (борана), В2Н6 (дчборана), В5Н9 (нентаборана),
BhiH,., (декаборана).
Из борана образованы четыре типа соединений: RBX2, R2BX,
R3B и R4B-M+, где X — галоген, ОН, OR1, NR2 и др.:
СП3ВС12 метилбордпхлорпд
(С6Н5)оВВг дифенплборбромпд
(С2ПаKВ трпэтплбор
СЛ13В(ОН).. этплборная кислота
(С0Н6)аВОН дифеннлборная кислота
(C0II5LB~NIa '" тстрафеннлборат натрия
Из диборана образуются соединения RB2H3, R2B2H.,, R3B2H3,
R4B2H2, например (СН3JВоН.,—димстилдиборан.
Из боранов с большим числом атомов бора в молекуле образуют
карбораны BuC2Hni_,, например C2rl2BUiH10— декакарборан (кар-
боран-10).
А. ПРОИЗВОДНЫЕ БОРАНА И ДИБОРАНА
1. Методы получения. Для получении производных борана и дибо-
диборана в основном используют два метода: металлоргаинческий синтез
и реакцию гидроборирования.
а) М е т а л л о р г а и ч ч е с к и и синтез. При взаимо-
взаимодействии галогенидов бора ВХ:1 (X— Cl, Br, F) с металлорганнче-
скими соединениями (М--Li, Mg, Л1) образуются борорганические
соединения. Можно использовать также сложные эфиры борной
кислоты В (OR).,:
BXa-i-3RMgX ->
H'MrX R'MgX R'AlgX
B(ORK «¦ R'B(ORJ > RjBOR ——> R3B
При использовании BF3 и большого избытка металлорганпче-
ского соединения можно получить четырехзамещеиные производные
б
бора:
ВF3 : 4C(iH.-,.WgBr -* (CJI^jB"
Диборан при взаимодействии с металлорганнчески.ми соедине-
соединениями образует производные днборапа:
fit e-
Это объясняется своеобразной полярностью связи В—Н.
б) Г и д р о б о р и р о в а и и с. При взаимодействии дибо-
рапа или замещенных диборапов с алкенами образуются триалкил-
2G8
производные бора:
6RCH СИ, ;-В.И„ ~> 2(RCH,— СН,KВ
2RCH СИ, ; (CnU5).2B-1Ui->-2(RCU.,—CU.,JBC,\lb
В принципе реакция представляет собой электрофилыюе присое-
дппеппе ВН3 пли С,11Г)ВН:> к двойной связи; электрофильиым
центром реакции является атом бора со своей незаполненной орбп-
талыо: В,Н,.^'2ВН3.
В лабораторных условиях B2Hf, получают непосредственно в
реакционной среде из NaBH4 И Л1С13 или BF3.
Гпдроборировацие алкинов дает трчалкеннлнроизводные бора:
CRC--.CII ; В,Н„ —>2(RCH--CHKB
2. Физические свойства и строение. Борорганические соедине-
соединения со связями С—В пли В—Ы являются бесцветными веществами;
трнметилбор (СН3)зВ кипит при —21,8 С; триэтнлбор — при 95 ГС,
а трпфенплбор представляет собой бесцветные кристаллы с т. ил.
142 С . Они окисляются на воздухе, некоторые легколетучие борор-
борорганические соединения имеют способность к самовоспламенению.
Молекула производных триа.чкил(арил)бора плоская, соедине-
соединения электронодефицптные незаполненной орбпталыо па атоме бора,
что способствует ассоциации. Например, диборап и его производные
представляют собой димеры с двумя водородными мостиками:
н н н нч н н
X X X Хи
Н Н Н Н,С Н СНз
дпСирлк дичетиллибпран
Связь В••-Н • •-В принципиально отличается от обычной водо-
водородной связи —О —Н ••• :б —П (lvi. XIV. А.З). Они имеют проти-
в- 6- Р- в-
воположную полярность (В—Н и О—Н) и, кроме того, атом бора
имеет незаполненную орбиталь. Это позволяет образовать четырех-
центровую систему ВН2В с многоцентровыми молекулярными ор-
биталямн.
3. Химические свойства. Применение, а) О к и с л е и и е. Бо-
Борорганические соединения очень легко окисляются, некоторые из
них являются пирофорами. В мягких условиях окисление приводит
к эфирам пероксиборной кислоты, гидролиз которых дает спирты
и борную кислоту:
R3B-| О, —> R,B —OOR -°2. RBfOOR). -°Л B(OORK --?
—г B(OHKJ-3H,O.,-1-3ROH
Эта реакция является хорошим способом получения спиртов из ал-
кепов, при этом главным образом первичных спиртов, которые не
могут быть получены присоединением воды к замещенным алкеиам.
269
Например:
п„н» о,
RCH = CH, —* (RCHo—СН2KВ —^ 3RClbCH2OH
б) Взаимодействие с н у к л е о ф и л ь н ы м и
реагентами. Борорганические соединения являются элект-
рофильнымн реагентами, так как на атоме бора имеется незанол-
неиная орбиталь. Они легко образуют продукты присоединения со
спиртами, эфирами, аминами, металлорганическими соединениями:
^Н^з^: R3B^:6(C2HSJ
Г-t R3B-NR3
й- 6-
R3B-; R —Li ^[R4B]-Li +
Активно с нуклеофильными реагентами взаимодействуют борор-
борорганические соединения со связью бор — галоген:
RiB — Cl + HoO —> R,B—0U-I-HC1
в) П р и м e ii e ii и е. Борорганические соединения исполь-
используют в качестве промежуточных продуктов в органическом синтезе.
Производные диборана могут быть использованы как горючее
в реактивных двигателях. Тетрафепилборат натрия (CeH5LBNa
используют в качестве аналитического реагента для определения
калия.
Б. КЛРБОРАНЫ
Наиболее важными борорганическими соединениями являются
карбораны — производные дскаборапа (карбораны-10).
При взаимодействии В10Нм с ацетиленом при 90—100 °С обра-
образуется повое соединение 1,2-карборан-10, названный барсном:
В10Н14-[-П-С=С-Н -> С2Н,В10Н10-{-211а
Соединение необычайно устойчиво, плавится при 287 СС без разло-
разложения. Только при 500—600 °С происходит изомеризация в другие
карбораны—¦ 1,7-карборан-10 и 1,12-карборап-Ю.
Рентгеноструктурный анализ свидетельствует о своеобразном
строении карборанов: 10 атомов бора и 2 атома углерода размеща-
размещаются на вершинах правильного двенадцатиугольника (икосаэдра).
Связь между атомами бора осуществляется
10 водородными атомами при помощи водо-
водородных мостиков, как в диборане. Образо-
н вание молекулы карборана и его стабильность
не могут быть объяснены обычными валент-
валентными представлениями. Здесь неприменимы
классические структурные формулы, и не-
обходимо пользоваться представлением о мно-
гоцентровых молекулярных орбиталях и де-
локализации электронов. В приведенной фор-
270
муле молекулы карбсрапа водородные атомы пе изображены, кро-
кроме связей С—Н. Черточки между атомами (за исключением С—С
и С—Н) не обозначают а-связь, а просто ггозво/тяют нагляднее изо-
изобразить пространственное строение. В сокращенном виде молеку-
НС-^т-СН
лу барепа изображают так: \-~/
Вю Ню
Карбораны — необычайно устойчивые соединения, они не из-
изменяются при действии окислителей и кислот.
Глава XII
КРЕМНИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
1. КЛАССИФИКАЦИЯ И НОМЕНКЛАТУРА
Ниже приведены примеры основных типов кремнийорганических
соединений:
RSiX3, R2SiX2, R3SiX, R4Si
(где X = галоген, H.OR', OOCR', NR, и др.).
CH3SiCI4 метилтрихлорсилан
(Со11;,)г SiCl2 днэтнлдихлорсилан
(С>Н-,J Sillo дпэтилсилан
(СН3K SiBr трмметилбромсплан
(G,H:,K Sill трпэтилсилан
(Сс11-,K SiOH трифенилсплаиол
(CH3)j Si тетраметилсилап
2. ,\\ЕТОДЫ ПОЛУЧГЛШЯ
Для получения кремнийорганических соединений используют ме-
таллорганичеекпй синтез, непосредственное взаимодействие крем-
кремния с галогенпроизводными углеводородов и некоторые специальные
реакции (гидросилнрование).
1. Металлорганический синтез. Взаимодействие галогенпроиз-
водных кремния или сложных эфиров ортокремниевой кислоты с
металлорганическими соединениями ведет к образованию кремний-
органических соединений:
SiCI,-|-RMgCl —у RSiCl3-j-.MpCU
RSiCI3-!-R,\1»CI —> R.2SiCI,-l-.\\fiCl2
HSiCI3-|-3RLi - >¦ R3SiH + 3LiCl
Si (OC,H5)i + 4RMgBr -+ R4Si-|-4C2HsO.\\?'Br
2. Взаимодействие кремния с галогенпроизводными углеводо-
углеводородов. Для получения простейших алкилхлорсиланов, арилхлор-
силанов разработаны промышленные методы, основанные на взаимо-
взаимодействии кремния с галогенпроизводными в присутствии некоторых
271
катализаторов при повышенной температуре. Обычно используют
размельченный сплав кремния и меди:
/ CHSiCb + tCHSiCU-; (CH^ SiCl
СН3С1
Si Си
Si Си
—- CeH3SiCl3-| (C,H,JSiCIH (СЙН5K SiCI
В этих реакциях всегда образуется смесь продуктов, которая раз-
разделяется фракционной перегонкой.
3. Гидросилилирование. Галогенсилаиы и алкилсиланы присое-
присоединяются к алкенам или алкпнам, например:
IlSiCl3--RCH- CH2 -* RCHo —Clio —SiCb,
R3SiH+CH2-CH3 -r R3Si —CHo —CH3
Присоединение происходит в присутствии катализаторов (инициа-
(инициаторы свободных радикалов, соли платины).
Присоединение к многократным связям происходит согласно их
of И-
полярности и полярности связи R3Si—П.
3. ФИЗИЧЕСКИЕ СВОЙСТВА И СТРОЕНИЕ
Кремиийоргаиические соединения являются бесцветными жидкос-
жидкостями, реже — кристаллическими веществами (табл. 29).
Таблица 29. Физические константы некоторых кремнинорганических
соединений
Соединение
CH3SiC!.(
(CHabSi'Cl,
(CH,bSiCI
(CH3).,Si
(C.H-,)-,SiH
CfljSiCb
(C6H5)oSiCU
(C4H5)aSiCl
Т. 11Л., CC
-77,8
—76
—57,7
88
T. Kim., °C
65,7
70,0
57,3
26,0
107,0
201,5
305,0
378,0
1,06
0,854
0,646
0,751 @ "C)
1,325
Они обычно не имеют запаха, кроме соединений со связью Si—С1.
Последние на воздухе «дымят» и имеют острый запах, так как выде-
выделяют галогеиоводород. Обычные кремиийорганические соединения в
воде не растворяются, они водой даже не смачиваются, т. е. являют-
являются гидрофоб и ы м и. Пространственное строение кремний-
органических соединений является тетраэдр и чес к им (sp
зацпя), молекулы могут быть хиральными:
7 " 7
Yy ^Х X \ Y
R' R'
272
Полярность связей кремнийорганических соединений отличается от
полярности связей аналогичных производных углерода, так как
атом кремния имеет меньшую электроотрицателыюсть, чем атом уг-
углерода:
й- Л <• 6-й-
R.-iC—H R3Si — И
6- б-
R3C—CR-, R3Si—CR3
ft- ft- 6'- 6'-
R3C—Cl R3Si—CL б' > б
4. ХИМИЧЕСКИЕ СВОЙСТВА. ПРИМЕНЕНИЕ
Наиболее активными из кремнийорганических соединений в хи-
химических реакциях являются галогеппроизводные со связью
Si—галоген. Менее активны соединения со связями Si—О,
Si—N и Si—Н. Весьма инертны соединения со связями Si—С.
1. Реакции связей Si—С(. Связь Si— Cl (связь Si — галоген)
силыгополярна, поэтому легко протекают реакции даже со слабыми
нуклеофилами, например водой:
ft- • б-
R3Si— Cl-fOIlo --, R3Si—OH-I-HC1
Легко идут реакции со спиртами, алкоксидами, аммиаком и ами-
аминами, металдорганическими соединениями. Происходит реакция
нуклеофилыюго замещения у атома кремния (сравните с реакциями
•S.v У углеродного атома):
ft - ft- ft- 6 i-
R:!Si —CI-J-R1 — Li —> R3Si —R'-i-LiCI
Реакции соединении, имеющих два атома галогена у кремния,
с водой, аммиаком, первичными аминами могут проходить необыч-
необычно. Образуются олигомеры и полимеры, например:
-ОН олпгомеризацпя
R2SiCl.,i 21ЬО —> RoSi-;' +2HC! - >
ХОН полимеризация
С11ЛПНДИОЛ (Л
Силандиолы являются реакциошюснособными соединениями и в
зависимости от природы остатка R (алкил или арнл) быстрее или мед-
медленнее образуют циклоолигомеры и линейные полимеры:
О
3R2Si(OH)o -^
4R2Si (O1IJ -^
R.,Si
1
6
RoSi
1
6
1
R2Si
SiR.,
1
О
•si/
R2
— 0 —SiR
0
1
— 0 —SiR
273
R R R
I I I
nR2Si (OH)j —> — Si—O—Si—O—Si—O-
I I I
R R R
полисилоксапы (R2SiO)rt
При нагревании циклоолнгомеры превращаются в полисилокса-
ны. Полнсилоксаны представляют собой бесцветные вязкие жид-
жидкости или эластичные массы. Они термически и химически очень
стабильны, поэтому полиснлоксапы нашли широкое применение в
народном хозяйстве. В зависимости от природы R и молекулярной
массы полисилоксапы (силиконы) используются в качестве смазоч-
смазочных масел, каучуков (силиконовые эластомеры), тормозных жид-
жидкостей и др. Выдающейся является стабильность полисилоксанов.
Важное значение имеет гндрофобность полнсилоксанов.
При взаимодействии дихлорсиланов с аммиаком или аминами
образуются цнклоолнгомеры — силазаны, например:
Н
А.
(CH3)oSi Si(CH3)a
(CH3),SiCl2-|-NH3 — | |
/N N
H- \Si/4H
n3c чсн3
гексаметилцнклотрнсилазан
Соединения с тремя атомами галогена у кремния при взаимо-
взаимодействии с водой образуют силаптрнолы, которые при отщеплении
воды превращаются в полимеры:
RSiCI3 + 3H2O
/iRSi(OHK -->
Силантриолы и их олнгомеры растворяются в щелочи с образо-
образованием солей (аналогия с кремниевой кислотой):
R— Si (OH)H-Na'OH- -* R — SiOJXa *¦-\-2H-Xi
Такие растворы (R=CH:1, СвН5) используются для гндро4юбиза-
цпи различных изделий.
Ллкил(арил)галогенснланы под действием сильных восстанови-
восстановителей превращаются в алкнл(арил)силаны, атом галогена замеща-
замещается водородом:
Кп1Ш4
R:iSiCl >¦ R3Si — ГГ
LiAlH,
R,S;C!2 у RjSiMjj
2. Реакции связей Si—Н. Благодаря своеобразной полярности
6-1- fr—
связи Si—Н для снланов характерны особые реакции. Гидросили-
лирование (присоединение к кратным связям) было уже рассмот-
рассмотрено выше.
274
В особых условиях соединения R3SiH и R2SiH.2 могут служить
донорами гидрид-иона, т. е. действовать гндрнрующе. Так, в при-
присутствии сильных кислот силаны гидрируют разветвленные алке-
ны, карбонильные соединения и др., например:
СН3 СН3ч
,C--CH.) + H-X-4-R3SiH --> СИ—Ctb-l-RaSiX
сн.,-' " си3
11од действием кислоты образуется карбокатнон, который отры-
отрывает гидрнд-пон от силана. Поэтому метод называется ионным гид-
гидрированием.
Для соединений R3SiH известны и другие реакции, которые
не свойственны соединениям R3CH. Например, при взаимодействии
с алкоксидамн в присутствии катализатора (Pt) выделяется водород
и образуется алкокспсилан:
C,H3ONa
R3Si-H+C2H.,OH " pt »
Глава XIII
ФОСФОРОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
1. КЛАССИФИКАЦИЯ И НОМЕНКЛАТУРА
Семейство фосфорорганических соединений очень большое. Соеди-
Соединения могут быть произведены как от фосфина РН3 и иона фосфо-
ния PHJ", так и от фосфорной кислоты, фосфористой кислоты, тпо-
фосфориых кислот и других производных фосфора.
От PI 1а и PH.; образуют RPX2, R2PX, R3P и R4P+Y" (где X = H,
галоген). Например:
СН3РС].2 (С0И.-,KР ' [(СсП.-.)зРСИ31Вг-
метилдихлорфосфнп три(^елилфосфин бромид трпфеинлметилфосфэмня
От фосфористой кислоты образуют:
RP(OH)a R.POH
фосфонистая фосфинистая
кислота кислота
От фосфорной кислоты образуют:
/° О
RP^OH R2P^ R3P=O !
ХОП ХОН
фосфоповая фосфинопая фосфипоксид
кислота кислота
Аналогичные соединения могут быть произведены от тиофосфо
ристых кислот и тно({хзсфорных кислот.
275
2. МЕТОДЫ ПОЛУЧЕНИЯ
1. Получение фосфинов, фосфониевых солей и фосфиноксидов.
Исходными веществами для получения фосфпнов являются РС13
и металлорганические соединения:
Rfi R
PCIs-i-RMgCl -, RPCl,-{-MgCl, ' R,PCl > R3P
Фосфопиевые соли легко образуются при алкилнровапин фосфи-
фосфинов галогенуглеводородами:
R:IP: + R-Br —>¦ [R3P-R|Br-
Фосфиноксиды получают окислением ([юсфипов или из оксохло-
рпда фосфора РОС13:
Oi r?p . о
RMfiCI
POCI;, * R;,P : О
Разработан метод прямого алкнли]эования красного фосфора
подалканами RI, которые в реакционной смеси образуются из
спиртов ROH, иода и фосфора:
3ROH-1-L-I-2P -^ R.,PI,-f Р(ОНK —-* R.,p.-C)J-2HI-| P(OH):,
В результате реакции образуют фоа^ипоксиды.
2. Получение фосфонистых и фосфинистых кислот. Исходными
веществами служат алкил(арил)хлорфосфнны:
ОН
RPC1. + 2H..0 —. RP'' -1-2IICI
RoPCl-i-ri-jO - > R.,P—ОП-l-HCl
При взаимодействии алкил(арнл)хлорфос([эипов со спиртами в
присутствии основании образуются соответствующие сложные эфп-
ры.
3. Получение фосфоновых и фосфиновых кислот. Фосфоновые
и фосфнповые кислоты образуются при окислении фосфонпстых и
фосфипистых кислот. Распространенным методом является реакция
РОС13 с металлорганическими соединениями с последующим гид-
гидролизом:
Cl R R
Cl3P-O + RI.i -^ R-P---0 R-V R-P--0 -—-* R-P-O
I I I
Cl Cl ОН
| н,о
I ,0
R —P^OH
4 ОН
Если вместо С13Р0 использовать C1:!P---S, получаются соответ-
соответствующие тно({)осфоновые и тнофоа|)иновые кислоты.
276
Своеобразную реакцию получения сложных эфиров алкидфос-
фоновых кислот открыл А. Н. Арбузов. При алкилированин слож-
сложных эфиров фосфористой кислоты происходит расщепление продук-
продукта алкилирования н образуется сложный эфир алкилфосфоиовой
кислоты:
OCHj ОСКз
6+ 6- +1 I
:Р(ОСН.,K + R—Вг »- R—Р —ОСН, ¦—*¦ R—Р—ОСН, + СН,Вг
О —СН., \ О
Вг
Эта реакция называется перегруппировкой Арбузова.
3. ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА.
ПРИМЕНЕНИЕ
1. Фосфины представляют собой бесцветные жидкости или крис-
кристаллические вещества (триарплфосфипы) с неприятным запахом.
Фосфипы ядовиты, особенно токсичны легколетучие алкилфоефниы.
По своим свойствам триалкил(арил)фосфипы напоминают амины
(гл. XXIII.А). Молекула имеет пирамидальную конфигурацию: в
вершине пирамиды находится атом фосфора, угол С—Р-—С около 100°.
Фосфипы являются основаниями (сравните с аминами), с кисло-
кислотами обрafу ют соли:
R3P:-i-HsO+ ;_Г RaP —H-i-H.,0
при R-CH, p/Cun+ - 8,7; при R -QH, pKnn> - 2,8.
Фосфпны легко алкилируются с образованием солей фосфонпя,
реагируют с кислородом и серой:
RsP:-i-R'-X~-^ [RSPR4X-
R3P: + O -^ R3P--O
фосфинокенд
R3P:-f-S --> R:1P-S
фосфннтиоякснд
Фос(|)ииы применяют в органическом синтезе для получения
других фосфорорганических соединений.
2. Фосфониевые соли представляют собой бесцветные кристал-
кристаллические вещества, легко растворимые в воде.
Конфигурация иона фосфония является тетраэдрическон, подоб-
подобно конфигурации солен аммония, соединений углерода и кремния.
Своеобразной является реакция фосч|юшювых солей с сильны-
сильными основаниями (металлоргапическимп соединениями). Если рядом
с атомом фосфора находится связь С -И, основание отрывает про-
протон и образуется внутренняя соль (фосфопиевьш бетаин):
II Н
! 6-6 -
R3P— С —R-fCH3- Li —> R3P —C-—R-; CH,-i-Li-X-
x- н6+
277
Такие соединения Г. Виттиг назвал илидами. Их называют так-
также фосфоранами (производными РНГ)).
Фактически эти соединения содержат сильнополярную двойную
Й-!- Й —
связь (аналогия со связью Р—О):
метилнд трифенилфосфоння
(метиле нтрифенилфосфоран)
Фосфонийилиды являются сильными нуклеофильными реаген-
реагентами и их широко используют в синтетической органической химии.
Так, например, при взаимодействии с карбонильными соединениями
отщепляется фосфипоксид и образуются алкепы (реакция Витти-
га):
(CeE-I6Kpt=cTlH О^с/ ^ _* (С„Н5KР—СН2 (С6Н5KР-О
/R ~* + /R
о—с/ сн2^с/
\nl \pl
3. Фосфиноксиды являются бесцветными жидкостями (триалкил-
фосфиноксиды) или кристаллическими веществами (триарилфос-
.... - финоксиды), без запаха, мало растворимы в воде. Име-
°1 ют относительно высокую температуру кипения. Прост-
,s*^ ранственное строение близко к тетраэдрическому. Фос-
RV R финоксиды имеют большие дипольные моменты [ц--;
R -A3,4. . . 16,7)х 10-30 Кл-м или 4. . .5 D].
Большая полярность молекулы определяется связью V=^o~ •
Вследствие большой энергии связи Р--0 (около 544 кДж/моль)
фосфиноксиды являются стабильными соединениями, не гидроли-
зуются, трудно восстанавливаются до фосфинов.
Фосфиноксиды являются слабыми основаниями:
r3P=O+H36 ^r R3P-OH4-II2O
Легко образуются комплексы с нонами различных тяжелых ме-
металлов:
Поэтому жидкие фосфиноксиды широко применяются в качестве
экстрагентов для выделения тяжелых металлов (радиоактивных
и редкоземельных элементов) из смесей.
4. Фосфонистые и фосфинистые кислоты. Эти соединения явля-
являются бесцветными жидкостями или кристаллическими веществами,
которые умеренно растворимы в воде. Легко растворяются в щело-
щелочах с образованием солей.
Фосфонистые кислоты существуют в двух таутомерных формах,
которые переходят друг в друга через образование аниона (о зако-
278
номериостях таутомерпых равновесий см. гл. XV.А}:
../ОН -н+
R— PC" «~- R
Фосфонистые кислоты и их производные (сложные эфиры, гало-
генапгидриды) служат исходными веществами для синтеза других
фос.((юроргапических соединений.
Фосфипистые кислоты также существуют в двух таутомерных
формах, при этом таутомерпое равновесие сильно сдвинуто в сторону
двухзамещенного фосфинокснда:
н
R\.. .. -nf R\?rr>- +"f R^' ••
P —OH ^ -. P-^-O: ^ * ^P=O:
Oihi являются слабыми кислотами.
5. Фосфоновые и фосфиновые кислоты — бесцветные жидкости
или кристаллические вещества; растворяются в воде.
Фос(|хшовые кислоты являются сильными кислотами и имеют
две константы ионизации, образуя два ряда солей:
О О
! —P-OH + 21I2O , r_p_o--|-H3O--+ H20
| pKa ~ ^...-',j i pKa ~
OH OH
0
o-
Важны производные — сложные эфиры, тиоэфиры и амиды,
которг.1е получаются из хлорангидрндов этих кислот. Производные
фосфоновых кислот используются в качестве пестицидов, лекарст-
лекарственных веществ, экстрагентов и боевых отравляющих веществ.
Сложные эфиры (фосфонаты), содержащие водород при а-уг-
леродном атоме, под действием сильных оснований превращаются в
карбаиионы, которые легко реагируют с карбонильными соедине-
соединениями — реакция Хорнера:
о R4
,0 , )с=о
/У Clt,-Li - 'I Я1'
RCH2 —Р—ОСН3 >¦ RCH —Р —ОСН3 *
ОСН3 ОСН3
Rl /ОСН3
—>¦ RCH = c/ . -[-OP — ОСНз
6"Li +
279
Фосфиновые кислоты также являются весьма сильными кислота-
кислотами:
-° ,Р
R-2P;7 +н2о -:r R,Pr-7 +11,0- (Р/ча ~3)
он ' о-
Практическое применение нашли производные фосфшювых кис-
кислот — сложные эфиры, тиоэфиры, амиды.
Гидроксилпроизводные
углеводородов
Введением в молекулу углеводорода вместо атомов водорода одной
или нескольких гидроксильных групп ОН (гидрокси-, или оксп-
групп) получают гидроксилпроизводные.
Для рассмотрения гидроксилпроизводных углеводородов удобен
принцип деления но типу гибридизации углеродного атома, у ко-
которого находится гидроксильпая группа. Так, можно выделить две
большие группы:
1) гидроксилпроизводиые со связью C(.s/;3)— ОН;
2) гидроксилпроизводные со связью C(.s/;-)—ОН.
Соединения со связью C(s/;)—ОН не описаны, известны только их
производные — эфиры C(sp)—OR.
В этой главе совместно с гидроксилпроизводпыми углеводоро-
углеводородов будут рассмотрены некоторые их производные: эфиры R--0—R1
и сложные эфиры неорганических и элеменюрганических кислот
R—О-ЭХ„, где Э -В, N, О, Si, P, S, Cl, Ti и др.
Глава XIV
ГИДРОКСИЛПРОИЗВОДНЫЕ УГЛЕВОДОРОДОВ
СО СВЯЗЬЮ C(sp») — ОН
В группу гидроксилпропзводпых углеводородов со связью C(sp3) —
—ОН входят:
а) гидроксилпроизводные алканов — насыщенные одиоатом11ыо
спирты, или алканолы С„Н.:„ ,.1 ОН;
б) гидроксилпроизводные алкенов и алкинов — ненасыщенные
одпоатомпые спирты, или алкснолы и алкинолы:
R.2C С^-(СЯ,)„ОП RC C-(CR,)HOH
в) гидроксилпроизводные ннклоалканов и циклоалкенов —
циклоалканолы п циклоалкенолы:
ОН .011
280
г) гпдрокснлпроизводпые алкиларенов с гндроксилыюй группой
в боковой цепи—арилалканолы Лг (CR>),,OH;
д) дигмдроксилнроизводные — двухатомные спирты, или диолы:
СнН..,дОН), HO(CRo);;-Cr:C-(CR.2|,IOH HO(CR,)rt-C - C-(CR,),,OH
! I
R R
e) тригидроксилнронзводпые и нолпгндроксплпроизводпые —¦
трехатомные и многоатомные спирты, или триолы и полиолы:
С„П-Л« -1. (ОИ)Я С„Н2„.-:-„д0П)м
А. АЛКЛНОЛЫ
1. ПЗОЛШРНЯ 1! НОМКНКЛЛТУРЛ
Алкаиолы являются производными алкапов. Гидрокснльная груп-
группа может находиться в углеводородной цепи в различных местах, у
различных углеродных атомов:
М
i
R—С — ОН —первичные алкаполы, или первичные спирты
I
11
\\
I
R—С — Oil —пгорнчные алкаполы, или вторичные спирты
i
R—С — ОМ —третичные алкаполы, пли третичные спирты
R
Для алканолов применяют заместительную и раднкалыю-функ-
цпональпую тюмеиклатуру (табл. 30). Углеродные атомы цепи ну-
нумеруют таким образом, чтобы гидрокснльная группа обозначалась
меньшим локаптом. Гндроксильиан группа как старшая характе-
характеристическая группа обозначается суффиксом -см. Если в молекуле
имеются более старшие группы ( С- О, — СООН^ или молекула
имеет очень сложное строение, гидроксильную группу обозначаю)
префиксом гидрокси- (иногда ока;-). По радикально-функциональ-
радикально-функциональной номенклатуре алкаполы называют спиртами.
Изомерия алкаполов определяется местонахождением гндрок-
силыюй группы и разветвлением углеродной пгпн. Начиная с бута-
пола-2 имеет место также стереоизомерия, появляется асимметри-
асимметрический атом углерода и хиральпость молекул (помепклеггуру см.
гл. VII.3):
CH.CHj СН CHj
I I
н н
Л ~ бута нол -2 S -Сутзнол -2
281
Таблица 30. Примеры номенклатуры алканолов
Структурная формула
СНзОН
СНзСН2ОН
СН3СН,СН,ОН
СНзСНСНз
1
ОН
СНзСНоСНаСНоОН
СНзСНСН.,ОН
1
СНз
СН:1СНоСНСН3
1
он
СН3
1
СНзССНз
1
он
CH.CH.Cn.CIbCIbOH
СНзСПоСНСНаОН
г
1
СНз
сп3снсп.,сп2он
1
СП,
СН3СП.,С1ЬСПСП3
1
он
сн2сп,спсн..сн3
1
он
СНаСН— СМСПз
1 ]
1 1
СНз ОН
1
СНаСП,,ССИз
1
он
СИз
1
СНзССНзОН
СНз
номенклатура
Метанол
Этанол
Пропаиол-1
Пропанол-2
Бутанол-1
2-Метилнропаиол-1
Бутапол-2
2-Метилпропанол-2
Пентанол-1
2-Метплбутаиол-1
З-Метплбутапол-1
Пентанол-2
Пентанол-3
З-Метплбутаиол-2
2-Метилбутанол-2
2,2-Л11 мети лирой а-
но л-1
Ра дика л bruj-фу [1К цп опаль-
опальная номенклатура
Метплонын спирт
Этиловый спирт
Пропнловын спирт
Пзопропиловый спирт
Бутиловый спирт
Изобутиловый спирт
emop-Бутпловый спирт
/пре/п-Бутиловын спирт
Амиловый спирт
Пзоампловын спирт
/преяг-Лмнловый спщ-т
Пеонептпловый спирт
282
2. МЕТОДЫ ПОЛУЧЕНИЯ
Для получения алкаполов используют галогеналканы, алкепы,
карбонильные соединения, элемепторгапические соединения, оксид
углерода (рнс. 83). Главные промышленные методы заключаются в
присоединении воды к алкеиам, гидрировании оксида углерода и
карбонильных соединений и в ферментативной переработке угле-
углеводов.
Приведенная на рис. 83 схема иллюстрирует взаимосвязь между
алканолами, алканами, алкенами, карбонильными соединениями.
RCH=CH,
Н,о
СО;
н/.
и1-
ми,; ви.
RCHCHj
ОН
Н7О
ОН"
(RCH2CH2)j0
[01
RCH2CH,O[f
Н
Ni
RCH,CH;CH;OH
RCHCIL,
RCHCIf,
OMgX
t
Рис. S3. Генетическая связь между алканолами, алканами, алкенами
и карбонильными соединениями
1. Гидролиз галогеналканов. Галогеналканы при реакции с во-
водой или щелочью легко образуют алканолы (гл. VII.4.3):
R—Х + ОН- —» R—ОН + Х-
2. Присоединение воды к алкенам. Присоединение воды к алке-
нам (см. гл. II.4.2.а) происходит в присутствии кислых катализа-
катализаторов (серная кислота, фосфорная кислота, оксид алюминия или
другой носитель, обработанный кислотами).
Кислота дает активный протон, который присоединением к моле-
молекуле алкена способствует присоединению молекулы воды:
t> СН:^СН'> 4- Н Х^Л- Н О я.* R-*"CH—-CU'> ^
н н
Н V-
R-CH-СНз + Н+Х~
:ОН
В промышленности присоедннение воды осуи1ествляется в газо-
газовой фазе при пропускании смеси алкена и воды над катализатором.
283
3. Гидрирование карбонильных соединений. Каталитическое гид-
гидрирование альдегидов и кетонов может привести к алканолам:
R и RN
,С = О --4 ;С-ОН
Ri/ Кат. щ/ I
н
Катализаторами служат Mi, Pt, Pd.
Мягкими реагентами гидрирования являются комплексные гид-
гидриды металлов, например, боргидрид натрия Na^BH,r и алюмогпд-
рнд лития Li-A1H4" (см. гл. XXVII.А.3).
При гидрировании сложных эфиров карбоповых кислот получа-
получаются первичные спирты. Гидрирование осуществляется водородом в
присутствии катализатора (Н>; Ni) или водородом в момент выделе-
выделения, образующимся при растворении металлического натрия в
спиртах (Na--С2Н5ОН)—метод Буво — Блана A903) или комп-
комплексными гидридами металлов (LjAlH.i):
/° ПИ
R — С/у — *R—СН2ОН
4. Реакции элементорганических соединений. Легко происходит
присоединение металлорганических соединений к альдегидам и ке-
тонам (гл. Х.Б):
R л - 6- <>- 6^- RN и,о R4
С = О --R2 — МХ„ —> .С —О.МХ„ —->¦ /С —ОН
R1 ' ' Rl/ I u"* Rl/ ]
R2 R2
Вторым методом использования металлорганических соединений
для получения алкаполов является их окисление. В промышленности
применяют окисление алюминийоргапнческих соединений (гл. X.
В.1) (К. Циглер):
о "-«
R:1A1 - J- (ROKAI ^- 3ROH + AI(On);,
Используют также окисление борорганических соединений
(гл. Х1.Л.З).
5. Гидрирование оксида углерода и оксосинтез. Оксид углерода
и водород в зависимости от применяемого катализатора, температу-
температуры реакции и соотношения реагентов СО и Н„ могут давать различ-
различные продукты реакции, т. е. метанол, смесь различных спиртов
(сиптол), метай, смесь углеводородов:
ZnO; Zr.Cr.O,
:—>¦ СН3ОН
СО , 211..— '"
Со; Rh
; >¦ CH3CH,CH2OH-f CH3CHCH3-I-...
он
В оксоспптезе (гл. II.4.3) из алкснов получают альдегиды, ко-
которые гидрируют до алканолов.
6. Ферментативные реакции. Некоторые простейшие алкаполы
образуются в результате ферментативного расщепления углеводов
284
(полисахаридов, моносахаридов) — в процессе брожения. Таким
путем в больших количествах получают этанол. В продуктах бро-
жегшя в зависимости от исходного (вещества и использованных мик-
микроорганизмов обнаруживают также нроиапол, бутанол и 2-метил-
б\;танол-1.
з. фнзпчгхкнп: свойства п строение
Алканолы являются бесцветными жидкостями пли кристалличе-
кристаллическими веществами с характерным запахом. Первые члены гомологи-
гомологического ряда алкаполов имеют приятный запах, для бутаполов п
иентанолов запах становится неприятным, раздражающим. Выс-
Высшие алканолы имеют приятный ароматный запах.
Своеобразными являются весьма высокая температура кипения
спиртов (табл. 31). Она выше, чем для соответствующих иодалканов,
несмотря на то, что молекулярная масса у алкаполов значительно
меньше. Это явление вызвано значительным межмолекулярным
взаимодействием ¦— ассоциацией молекул.
ТаСлп.иа 31. Физические константы некоторых алканолов
Сорд'.г.'спиг:
СМзОИ
CHXIL01I
chxhxhxih
СНХПОН
сн3
СН:|С1ЬСН,СП,ОН
енхпашп
си.
С На
сн,—с—он
сн;)
СН.ХНХНХНХП.ОП
сп,оснхнсп..он
сн,
СН3СНСП..СП.,ОН
1
1
СИ,
сн,
С Н,XIIX — ОН
1. м :., С
-07,8
-117.3
— 127,0
—88,5
—89,Г1
— 108,0
25,5
-7?, 5
— 117.2
— 11,9
Т. к пи.. ' С
64.7
78,4
97.2
82,3
117,7
108,4
83,0
13°.,0
128.0
13.), 5
101,8
0,7924
0.789
0,804
0,785
0.8097
0,801
0,788
0.FI4
0,819
A,812
0,809
2S5
Алканолы являются полярными соединениями. Они содержат в
б_ й- б- б ;•
молекуле две полярные связи: С—О и О—П. Диполи связей С—О
и О—Н направлены в сторону атома кислорода. Суммарный ди-
польиын момент алкаполов составляет 5,3—6,0-Ю0 Кл -м A,6 —
1,8 D). На суммарный дшюльиый момент влияют также пеподелен-
ныс электронные пары па атоме кислорода:
,' " MH A'69D) ЛМ С^-о" 3 01 0 9
HjC"»7P= *? (газообрачноо nU_u" cM , с
р' \ / состояние) u " э'и1 J'°
Дипольпые моменты связей показывают, что полярность связи
О—Н значительно выше, чем связи С—О.
Неподеленные электронные пары придают алканолам слабые
электроподопорные свойства. Это можно охарактеризовать энер-
энергией ионизации ЭИ (эВ):
Соединение Н2О СН3ОН С2Н,ОН
Э11, эВ 12,0 10,8 10,0
ЭИ алкаполов по сравнению с водой меньше, здесь и проявля-
проявляется электронодопорпый эффект алкилгруппы (-|-/-эффект).
Энергия ионизации характеризует процесс:
R—ОП -* R — ОН+е-
Полярная связь О—Н и неподеленные электронные пары опре-
определяют возможность молекулярной ассоциации:
Ассоциация осуществляется в основном за счет электростатичес-
электростатического притяжения, в меньшей мере вследствие перекрывания орби-
талей пеподелешюй электронной пары кислорода и орбитали водо-
водорода. Такое взаимодействие называют водородной связью (В. Ла-
тимер, В. Родебуш, 1920).
Энергия водородной связи значительно меньше энергий обыч-
обычных химических связей:
Связь С—Н С —О О—II Nd-.-H —
Е, кДж/моль ,.,,.. 415 350 403 25—20
Рентгенографические и электроиографические данные свиде-
свидетельствуют, что у алкаполов сохраняются обычные длины связей
С—С и С—Н и углы между связями.
280
Н Углеродные атомы находятся п со-
состоянии х/г'-гибридизации, по трудно од-
однозначно решить вопрос о гибридиза-
гибридизации кислородного атома, т. е. о приро-
природе его неноделенных электронных пар.
Имеются две граничные возможности —•
чистая 5р3-гибридизацин (обе элект-
электронные пары находятся в одинаковых орбиталях; угол HOR ра-
равен 109°28') и состояние без гибридизации (одна электронная пара
находится в /г-орбитали, другая — в s-орбитали; угол HOR равен
90°):
О О О О
/ \ / \
Н R H R
чистая sp1-гибридизация состояние без гибридизация
Нет прямых методов доказательства одного или другого состоя-
состояния. Есть только косвенные данные, такие, как величина валентных
углов, расположение молекул в кристалле. Есть все основания пола-
полагать, что природа неноделенных электронных пар атома кислорода
меняется в зависимости от природы R, агрегатного состояния и меж-
межмолекулярного взаимодействия (образование водородных связей).
Спектры поглощения. Ллкаполы в электронных спектрах по-
поглощения являются «прозрачными». Слабое поглощение наблюда-
наблюдается только в дальней УФ-области A70—180 им), что связано с пе-
переходом /г—>-(т* пеноделенной электронной пары атома кислорода.
В ИК-снектрах характерные валентные колебания связи О—Н
в разбавленных растворах углеводородов или галогенуглеводородов
наблюдаются в области 3580—3650 см. В концентрированных
растворах образуются водородные связи О—Н • •-О, поэтому ва-
валентные колебания связи О—М смещаются в область 3200—
3500 см.
В спектрах ПМР сигнал группы ОН наблюдается в широком ин-
интервале: б--2. . .4,5 м. д., в зависимости от типа растворителя и
концентрации вещества. В присутствии D2O сигнал исчезает вследст-
вследствие дейтерироваиия OH->OD. Характерные сигналы протонов
Н—С—О при 6--3,5. . .3,8 м. д.
4. ХИМИЧЕСКИЕ СВОЙСТВА
Свойства алканолов определяются наличием полярных связей О—Н
и О—R и пеподелеппых электронных пар. Характерные реакции
алканолов приведены на рис. 84.
Т. Реакции с сохранением атома кислорода в молекуле алканола.
Эти реакции главным образом связаны с высокой полярностью свя-
связи О—Н, что ведет к ионизации. Атом кислорода сохраняется так-
287
RCTI.C
R
R R'
Реакции с сохранением аюма Реакции с опцеп.чеииеч
кислорода в молекула i илроксилыюй i руины
Рис. 84. Характерные реакции алкаполов
же в реакциях окисления и взаимодействия с некоторыми электро-
фильпыми реагентами.
а) Образование а л к а н о л я т о в и их р с а к-
ц и и. Алкаполы являются очень слабыми ОН-кислогами:
R —О —H-(-:SoI ^ R—O:-+HSoii-
Анион алкапола, называемый алканолят-ионом (алкоксид- или
алкоголят-ионом), представляет собой сильное основание (жесткое
основание, с. 73). Кислотность алкаполов невелика:
Ллкапол СН,ОН СН3СП.,ОН (СП3JС1ЮН (СН3KСОН
рКа (для полных
растворов) . . . 15,2 15,8 16,9 19,2
Относительно более сильной кислотой является метанол. Его
кислотность сравнима с кислотностью воды (рКа для воды в водном
растворе около 15,7). Наиболее, слабыми кислотами являются тре-
третичные спирты. Здесь отчетливо проявляется донорпый эффект
( '-/-эффект) алкильных групп, увеличивающий плотность электро-
электронов на кислородном атоме. Анионы третичных спиртов — наиболее
сильные основания.
В водных растворах кислые свойства алкаполов проявляются
слабо. Только в концентрированных водных растворах щелочей,
где рН>14, частично происходит ионизация алкаполов (например,
15%-ный водный раствор КОН имеет рН«15). Небольшие коли-
количества алкаполятов образуются при растворении ДЮН в алканолах.
Обычно для получения алкаполятов применяют реакцию актив-
активного металла с безводным алканолом:
2ROH ^N'a —> 2RO-Na:+H.2
6ROII+2A1 -> 2(ROKA1 + 3H2
Алканоляты являются не только сильными основаниями, но н
сильными нуклеофильными реагентами. Они легко алкилпруются
(реакция Вильямсона) и ацилируются с образованием простых и
288
сложных эфиров:
R_O-Na'+Rl—X —* R—О —RHNa + X-
X. 6+-
R_O-Ka;+ ^C — Ri -* R— О — С — R'+Na + X-
0
Алканоляты как сильные основания применяются для генери-
генерирования карбанионов и других анионов в реакциях конденсации
(см., например, гл. XXXIII.В.3) и для введения двойных и трой-
тройных связей отщеплением галогеноводородов, например:
! /
СНа —С—Br-i-C.HsO\a+ —> СП, = С( +C.IbOH+Na
I NCH3
Реакции S v здесь мешают пространственные факторы и алкано-
лят выступает не как нуклеофил, а как основание, отрывающее про-
протон от метпльной группы.
б) Окисление. Первичные и вторичные спирты окисляют-
окисляются. Продуктами окисления являются альдегиды или кетоны, карбо-
новые кислоты:
RCH2O'H 'Л R-C''° ЩК-/
чн хон
н
R-C-OH [^l R-C^
R1
Окисление осуществляется кислородом в присутствии катализа-
катализаторов (Си, СиО) или различными неорганическими окислителями
(KMnO4, Na2Cr2O7+H2SO4, CrO3 и др.). Возможно также катали-
каталитическое дегидрирование алкаполов в присутствии Си, Pt или Pd
без участия кислорода:
.о ,0
RCH2OH --^ R-C^ +H2
Кат. -.Ц
Окисление является гомолитическим процессом, в ходе которого
образуются свободные радикалы.
Предполагают, что при окислении псрмангапатом или хроматом
в кислой среде промежуточными продуктами являются сложные
эфиры соответствующих кислот, которые легко распадаются гомо-
литически и могут вызывать цепной процесс окисления, например:
2RCH2OH ——- (RCIbO),CrO2 -> RCH2O- + RCH2OCrO2.
Промежуточны?.! продуктом является алкоксильный радикал
RCH2O-, который может перегруппировываться в гидроксиалкпль-
Ю д-о 517 289
ный радикал:
Н Н
R-C-O- -> R -С-ОН
Н
Свободные радикалы при взаимодействии с другими свободными
радикалами превращаются в карбонильные соединения:
Н
I /О
R_C_O.+R2- -ь R-Cf +R2-H
I 4R1
Rl
R_C-OH + R2. — R-C<f + R2-H
| XRi
Rl
R?. -MnO3-, RRiCHOCrOa., RR'CHO-, RR'COH и др.
Третичные спирты окисляются только в кислой среде в более
жестких условиях. Из третичного спирта образуется алкен, кото-
который легко окисляется (гл. II.4.4).
в) Основность и взаимодействие с элект-
р о ф и л ь н ы м и реагентами. Неподеленная электрон-
пая пара на кислородном атоме взаимодействует с электрофиль-
ными реагентами с образованием донорно-акцепторной (семиполяр-
ной) связи, при этом кислородный атом приобретает положитель-
положительный заряд. Образуются оксониевые соединения. В общем виде это
изображается схемой
.II
6 ± Я — 6(
+ \Е-
Здесь Е — кислоты Льюиса, например BF3, A1CI3, PCI3, SOCI2|
TiCU, SnClu, H + , NOJ и др.
Алканолы являются слабыми основаниями:
+ /Н
R—OH+HSol<-^lR— O( +:Sol
В результате присоединения протона образуется катион алкил-
сксония. Основность алканолов определяется кислотностью ка-
катиона алкилоксония (сопряженной кислоты), например:
+2 ^± сн3-он-;-н3о+ (Рквн+ =-2,0)
Катионы алкилоксония являются очень сильными ОН-кислотами,
их образование возможно только в присутствии сильных кислот.
Например, 20% -пая серная кислота имеет рН=—1, в этом растворе
около 10% метанола превращается в катион метилоксония. В сла-
слабых растворах кислот (рН>0) концентрация катионов алкнлоксо-
290
ния составляет только доли процента. Большинство молекул алка,-
нола в этих условиях образует водородные связи:
,Н ,Н ч- /Н
R_O/ +П>-Х- ^± R — o/ izlR—O(
•и+х- чн
х-
Взаимодействие алканолов с электрофильньши реагентами очень
часто ведет к дальнейшим реакциям. Например, взаимодействие с
ацилхлоридами ведет к образованию сложных эфиров:
О
R-OH + ^.C-R1 —> R-O-C-R' + HCl
В реакциях алканолов с карбоиовыми кислотами в присутствии
сильной кислоты тоже образуются сложные эфиры (с. 546, 574).
При взаимодействии с карбокатионами образуются простые
эфиры:
R —ОЫ + R't-X- — > R—О —R'+H+X-
Во многих случаях продукты реакции алканолов с электрофиль-
ными реагентами служат источниками карбокатионов:
6 и ч- /Н /Н
ЧН Ml
8 и ч-/Н ,Н
R-O( ^± R /
Это приводит к реакциям с отщеплением гидроксилыгой группы,
к замещению ОН-группы другими атомами или группами.
2. Реакции с отщеплением гидроксильной группы. В этих реак-
реакциях образуются различные производные алканов или алкены. Осо-
Особое место занимают реакции с оксидом углерода — карбопилиро-
вание.
а) Взаимодействие с галогеноводорода-
м и. При взаимодействии алканолов с галогеноводородами полу-
получаются галогеналканы:
ROH-l H+-X~^i R —Х + ИаО
(здесь Х=С1, Вг, I).
Параллельно осуществляются реакции отщепления.
HF обычно не дает желаемых продуктов, она имеет меньшую кис-
кислотность (рКа для HI, НВг, НС1, HF в водном растворе соответ-
соответственно —10; —9,5; —7,4; 3,2).
Эту реакцию можно рассматривать как нуклеофильное замеще-
замещение протонированной гидроксильной группы:
вь 6.Г 6+ + /Н .. .. Л1
R-OH + H — X: ^± R — О{ —* R«
10* 291
Реакция может осуществляться как по механизму SNl, так и
по SN2 (см. гл. VI 1.4.3) в зависимости от строения R и природы X
(CI, Вг или I). Самым активным является нодид-ион.
В реакциях часто используют не сами галогеноводороды пли га-
логеноводородные кислоты, а реагенты, которые их генерируют:
IbSO4 + KBr —> HBr+KHSO4
2Р +312 + 6Н2О —>- 6Ш + 2Н3РО3
красны!1!
фосфор
б) Взаимодействие с другими неоргани-
неорганическими кислотами. При реакции с кислородсодержа-
кислородсодержащими неорганическими кислотами алканолы образуют сложные
эфнры этих кислот (см. гл. XVII). В случае слабых кислот реакцию
катализируют добавки серной кислоты:
а+ ?.- в+ + 7Н
R—ОН + Н—О —ЭХ„ ^± R —О( +-:О-ЭХ„ -* R—О-ЭХ„ + ОН2
чн
в) Образование простых эфиров. Алканолы в
присутствии сильных кислот образуют диалкнловые эфиры:
2R-OH ^ R—О —R + H2O
Эфнры образуются при условии, если кислота взята не в избыт-
избытке, а температура реакции достаточно низка. В противном случае
образуются алкены. Образование эфиров связано с алкилированием
алканола алкилнрующим реагентом, который образуется из того
же (или другого) алканола при взаимодействии с кислотой. Алкилн-
Алкилнрующим реагентом служит протонпрованный алканол или образую-
образующийся из пего карбокатион:
бг ¦»• Л1 .. И
R—ОП + Н + t=t R— O{ ^± R + J-O--
R - ОН
R- .. R R + R
А
Реакция идет с хорошим выходом с первичными алканолами.
В случае вторичных и особенно третичных алканолов в продуктах
такой реакции преобладают алкены.
г) А л к а н о л ы как а л к и л и р у ю щ и е реагенты
а р е и о в. Алканолы в присутствии сильных кислот, в том числе
кислот Льюиса, становятся элсктрофнльны.мн реагентами, способ-
способными генерировать карбокатиопы. Ниже приведен пример дейст-
действия BF3:
б- t Н
ROH-|-BF3 ;=±R—О. - ;z±R
BF3
Ar —H + R + HOBFj -* Лг —R + h\OBF3
292
д) Взаимодействие с галогенангидр ида-
идами неорганических кислот. При взаимодействии с
PCU, РВг3, РС15 и SOG2 алканолы образуют галогеналканы. В ка-
качестве побочных продуктов образуются сложные эфиры неоргани-
неорганических кислот. Например:
ROH+ SOC12 —> RC!-f SO.-f НС1
тионилхлорид
Промежуточным продуктом является сложный эфир хлорангид-
рида сернистой кислоты, который претерпевает внутримолекуляр-
внутримолекулярное нуклеофилыюе замещение у атома углерода с отщеплением SO2
(реакцию Si)
+. /Н б,- ..
ROH+SOC1.2 ^± R-O.'_ П —> R-O-. + HCl —>¦ R — CI + SO2^-HCl
4S/ |
В этой реакции ОН замещается на CI с сохранением конфигура-
конфигурации у углеводородного атома.
е) К а р б о н и л и р о в а н и е а л к а и о л о в. При взаи-
взаимодействие с оксидом углерода в присутствии кобальтовых катали-
катализаторов под давлением происходит внедрение молекулы СО по свя-
связи R—ОН с образованием карбоновых кислот:
НСо(СОL
R-OH + CO • <• RCOOH
Активным катализатором является тетракарбонилгидрнд
кобальта НСо(СОL, который имеет свойства сильной кислоты и
может из алканола генерировать карбокатион:
6 ьи- Гб+ .. /Н1+ с0
R ОН + НСо(СОL z=Z R - О.; [Со(СО),]" —t
о о
Цй+6- ;|
—ч- R —С—Со(СО),-|-Н..О —¦* R — С — ОН + НСо(СОL
Протонированный алканол алкилирует оксид углерода и после
гидролиза кобальторганического соединения образуется карбоно-
вая кислота. Реакция имеет промышленное значение.
ж) П о л у ч е и и е а л к е и о в. При отщеплении молекулы
воды от алканолов образуются алкены. Отщепление воды происхо-
происходит в присутствии сильных кислот или над катализатором при
повышенной температуре:
RCH2CH2OH H-t RCH = CH2 + H2O
В присутствии сильных кислот алканольт могут давать сложные
эфнры или карбокатионы, которые при повышенной температуре
293
распадаются с образованием алкена:
R-CH2-CH2-OH + H-Y =S=r R-CH--СН2-О. + :Y
\н
7 + .
R-C-CH2 + :ОН2*=Ьг R-C-CH2—Y + :ОН2
Н :Y- L Н
R-CH=CH2 + HY + Н2О
Наиболее легко реакция идет с третичными алканолами, так как
образуется более устойчивый карбокатион.
Для каталитической дегидратации обычно применяют А12О3.
з) Перегруппировки при реакциях а л к а-
н о лов с кислотами. В реакциях, протекающих по меха-
механизму ?1 через карбокатион, наблюдается перемещение двойных
связей (аллильные перегруппировки) и изменение углеродного
скелета (скелетные перегруппировки). Перегруппировки связаны
с образованием сопряженных карбокатионов и миграцией водо-
водородного атома или алкильных групп в карбокатионе:
аллильная перегруппировка
RCH-- СПСН2ОН + Н + Х- ^ RCH^--CHCH2
+ t +х- + н2о
RCII—CH=CH2 + RCH = CHCIf..X ч— RCH —СП = СН.,
I
X
ретропинаколиновая перегруппировка — миграция углево-
углеводородной группы:
СНа
| Кислота НзСч /СНз
СН8-С —СНСНз >¦ ЧС-С/ +Н2О
| | Н8С/ ХСН,
СНз ОН
перегруппировка Вагнера — Меервейна (изменение углерод-
углеродного скелета в бициклоалканах (гл. XIV.B.2);
пинаколиновая перегруппировка — превращение 1,2-гликолей
в карбонильные соединения (гл. Х1У.Д.2).
5. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ
Метанол (метиловый спирт) иногда называют также «древесным
спиртом», так как его получали в результате сухой перегонки дре-
древесины. В настоящее время метанол получается исключительно из
СО и Н2 в крупнопромышленных масштабах:
СО+2Н2 -^ СНзОН
J\3T.
Наиболее активными катализаторами являются соединения цин-
цинка и хрома, например смесь оксида и хромита цинка ZnO+/2ZnCr2O4
294
(п=0,3. . .1). Температура реакции 250—350 °C, давление 5—ЗОМПа
E0—300 атм).
Исходным веществом может служить также СО2:
со2+зн2 — ^ сп3он+н2о
В этой реакции требуется больше водорода, который расходуется
на образование Н2О.
Мировая продукция метанола превышает 9 млн. т в год, область
его применения непрерывно расширяется.
Метанол представляет собой бесцветную жидкость со слабым
своеобразным запахом, смешивается с водой. Метанол сильно ядо-
ядовит, прием внутрь 10 мл его может оказаться смертельным.
Используют метанол в качестве хорошего растворителя и исход-
исходного вещества для синтеза других растворителей (простых и слож-
сложных эфиров). Большие количества его используют для получения
Нч [01
ХС=О •* ; CHjOH *- Растворитель
у"
н
«rCH.CHjOH
R—N
С1Г,ОЯ
СН,ОСН,СН,ОН
Рис, S5. Схема использования метанола
формальдегида. На основе метанола возможен синтез этанола и
других алканолов при использовании СО и катализаторов (рис. 85).
Этанол (этиловый спирт). Древнейшим методом его получения
является сбраживание раствора Сахаров (углеводов). Сбраживае-
Сбраживаемые углеводы (моносахариды) содержатся в различных фруктовых
соках или образуются при ферментативном расщеплении более
сложных углеводов (дисахаридов, полисахаридов).
При фракционной перегонке (ректификации) жидкости сбражи-
сбраживания («бражки») получают смесь этилового спирта с водой, так на-
называемый спирт-ректификат с т. кип. 78,15 °С, который содержит
около 95,5% этанола и 4,5% воды. Получить безводный этанол мож-
можно химическим способом при связывании воды CaO, Na или азеот-
ропным методом. Так называемый «абсолютный спирт» имеет т. кип.
78,4 °С.
В промышленности этанол получают также каталитической гид-
гидратацией этилена. Одним из лучших катализаторов является си-
ликагель, обработанный фосфорной кислотой:
280-300° С
сн2=сы2+н2о Кат-| 7_& м- сн5он
295
В связи с уменьшением мировых запасов нефти актуальным ста-
становится вопрос о получении этанола из другого сырья, например из
метанола и СО:
снзоп-; со-;-2Н2 —¦-> сп3сн2он + н,о
Мировое производство эт.глюла в настоящее время достигает де-
десятков миллионов тонн в год и имеет тенденцию к увеличению.
Этанол является одним из самых дешевых растворителей, его
применяют для изготовления лаков, косметических продуктов, ме-
медикаментов, для проведения синтезов. Этанол является также ис-
исходным сырьем в промышленном органическом синтезе для полу-
получения бутадиена по методу Лебедева (гл. III.Б,1), уксусного альде-
альдегида, этилацетата и других соединений.
В будущем этанол может приобрести значение для промышленно-
промышленного получения этилена каталитической дегидратацией (см. гл. II.5).
Пропанол (пропнловый спирт) представляет собой бесцветную
жидкость с характерным «спиртовым» запахом. Смешивается с во-
водой; образует азеотропную смесь, кипящую при 87,7 СС и содержа-
содержащую 71,7% пропанола.
Образуется в качестве побочного продукта при производстве
этанола.
Пропанол используют в органическом синтезе.
Пропанол-2 (изопропиловый спирт) — бесцветная жидкость с
характерным запахом. Смешивается с водой; образует азеотроп-
азеотропную смесь, кипящую при 80,3 'С и содержащую 87,4"d нропанола-2.
В промышленности пропанол-2 получягот из пропепа присоеди-
присоединением воды в галогон фазе над катализатором:
Г: Кат.
СН2-СН — СНз + НоО >- СПз — СИ — СИ3
ОН
Пропапол-2 широко используется в качестве растворителя вмес-
вместо метанола и этанола и как исходное в органическом синтезе. Боль-
Большие количества нропанола-2 используются для производства аце-
ацетона каталитическим дегидрированием (окислением) над катализа-
катализатором.
Бутанол-1 (н-бутиловый спирт) — бесцветная жидкость с ха-
характерным «спиртовым» запахом». Умеренно растворяется в поде
(около 8% при 15 СС).
Образуется в качестве побочного продукта при производстве
этанола сбраживанием углево/юв, а также з процессе оксоспнтоза
карбонилпросаппем пропс-нп.
Бутанол-1 используется в качестве растворителя в лако-кра-
сочпой промышленности и как исходное сырье для получения слож-
сложных эфиров, в том числе пластификаторов.
Бутанол-2 (втор-бутловый спирт) представляет собой бесцвет-
бесцветную жидкость, умеренно растворимую в воде (около 11% при 15 °С).
Бутанол-2 имеет хнральную молекулу, известна абсолютная
296
конфигурация оптически активных энантиомеров: правовращаю-
правовращающий 5-(Н-)-бутанол-2 и левовращающнй — R-(—)-бутанол-2 (см.
гл. XIV.A.1).
Рацемический бутанол-2 получают присоединением воды к буте-
нам в присутствии кислот. Бута кол-2 применяют в качестее раство-
растворителя и в синтезе мстилэтилкетона.
2-Метилпропанол-1 (изобутиловый спирт) — бесцветная жид-
жидкость со «спиртовым» запахом, умеренно растворим в воде (около
9% при 15 °С).
Образуется в качестве побочного продукта при производстве
этанола сбраживанием углеводов. Его используют в качестве раст-
растворителя и как исходное сырье в органическом синтезе.
2-Метилпропанол-2 (/л/зет-бутиловый спирт) представляет со-
собой легкоплавкие бесцветные кристаллы с приятным запахом кам-
камфоры. С водой смешивается; образует азеотропную смесь, кипящую
при 79,9 °С и содержащую 88,2 % mpem-бутнлового спирта.
Получают из 2-метилиропена присоединением воды в присутст-
присутствии серной кислоты.
Тре/п-бутиловый спирт применяют в качестве хорошего раство-
растворителя в органическом синтезе и как исходное вещество для введе-
введения в органические соединения трет-бутилъпой группы.
Пентауол-1 (н-амиловый спирт) представляет собой бесцветную
жидкость с характерным «сивушным» запахом, вызывающим ка-
кашель, мало растворимую в воде. Образуется в качестве побочного
продукта при производстве этанола сбраживанием углеводов. Его
используют для получения сложных эфиров (например, амилаце-
амилацетата), которые являются хорошими растворителями, некоторые
имеют приятный запах.
З-Метилбутанол-1 (изоампловый спирт) — бесцветная жидкость
с «сивушным» запахом, вдыхание вызывает кашель. Образуется в
качестве побочного продукта при производстве этанола сбражива-
сбраживанием углеводов. Синтетически нзоамиловый, а также амиловый спир-
спирты могут быть получены в процессе оксосиитеза карбонилированием
бутенов. Изоамиловый спирт используют для получения сложных
эфиров, которые являются растворителями. Некоторые изоамило-
вые эфиры имеют приятный запах.
Б. АЛКЕНОЛЫ И АЛКИНОЛЫ
1. НОЛ1ПНКЛЛТУРЛ
В основе названии алкеколов и алкинолоз лежит наиболее длин-
длинная цепь углеродных атомов с двойными или тройными связями п
гидроксильной группой в цепи. Например:
3 2 1 4 :; 2 1 3 2 1
СН,= СН — СН,ОН СНзСН^С —СН2СН2ОП НС-С — СН2ОН
СИ,
прогтеи-S-o.i-I; 3-метил[.р;т.сп-3 ол-1 про1шп-2-ол-1, про-
аллплош.г,'! пшрт ппргилопый пшрг
Некоторые соединения имеют тривиальное название.
297
2. МЕТОДЫ ПОЛУЧЕНИЯ И ХИМИЧЕСКИЕ СВОЙСТВА
Для получения алкеполов и алкинолов применяются такие же ме-
методы, как для получения алкаполов. Некоторые алкинолы полу-
получают присоединением ацетилена к карбонильным соединениям
(гл. IV. 4.3):
R1
R\ он- I
н—с = с-н+ ;с-о—-не — с-с-011
R7 I
R
Некоторые соединения встречаются в природных эфирных мас-
маслах.
Химические свойства алкеполов и алкинолов определяются на-
наличием гидроксильной группы и двойных или тройных связей. Для
этих соединений характерны обычные свойства алкаполов, присут-
присутствие кратных связей не имеет принципиального значения. Однако
надо помнить, что под действием сильных электрофильных реаген-
реагентов в реакцию может вступать как гидроксильпая группа, так и
двойная или тройная связь. Двойные и тройные связи вступают в
реакции присоединения, полимеризации и окисления.
3. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ
Аллиловый спирт является бесцветной жидкостью с острым запа-
запахом; т. кип.' 96,9 °С, <^4°=0,852. С водой смешивается; образует
азеотропиую смесь с т. кип. 88,9 СС, которая содержит 72,3% алли-
лопого спирта.
В промышленности получают из аллилхлорида:
NaOH; H,O
СН2 = СНСНаС1 —jT^r+CH2
Аллиловый спирт используют в производстве синтетического
глицерина и для получения аллиловых зфиров карбоповых кислот,
которые способны полимеризоваться.
Пропаргиловыи спирт представляет собой бесцветную жидкость
с. запахом герани; т. кип. 115 СС, dio^=0,9715. С водой смешивается;
образует азеотроппую смесь, кипящую при 97 СС и содержащую
45% пропаргилового спирта. Его получают из ацетилена и формаль-
формальдегида. Используют в органическом синтезе.
Гераниол и линалоол
CH-j — С = СН — СП2 — СНа — С = СИ — CHjOII
СПз С Н3
сн,
СИ., —С = СН —СН.-СПг-С-СП = СНа
I I
СН3 ОН
298
представляют собой бесцветные жидкости с запахом фиалок. Их
получают из эфирных масел некоторых растений и используют в
парфюмерной промышленности.
В. ЦИКЛОАЛКАНОЛЫ И ЦИ КЛОАЛ КЕНОЛЫ
1. НОМЕНКЛАТУРА
Названия циклоалканолов и циклоалкенолов образуют по общим
правилам. Если гидрокпишшя группа находится в длинной боко-
боковой цепи, то в основе названия лежит углеводородная цепь. Напри-
Например:
/ОН Д'/ОН /х /СН.2СН2СН2СН2ОН
17
п- -п
\„
циклогокса- циклопсн- 4-циклогексилбутаиол-1
пол тсн-2-ол-1
2. МЕТОДЫ ПОЛУЧЕНИЯ И ХИМИЧЕСКИЕ СВОЙСТВА
Циклоалкаполы и диклоалкеполы могут быть получены обычными
методами: №дролизом галогенпроизводпых, присоединением воды к
двойной связи, восстановлением карбонильных соединений. Неко-
Некоторые соединения встречаются в природных продуктах. Для цик-
лоалканолов и циклоалкенолов характерны обычные реакции гид-
роксильной группы и двойных связей.
Циклоалканолы при действии кислот претерпевают различные
внутримолекулярные перегруппировки, что связано с перегруппи-
перегруппировками карбокатионов. Подробно изучена перегруппировка бор-
неола в камфен в присутствии кислот (перегруппировка Вагнера —
Меервейна):
сн2
-н' ^
борнеоя камфец
3. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ
Циклогексанол представляет собой бесцветное гигроскопичное крис-
кристаллическое вещество с запахом камфоры; т. ил. 25 °С, т. кип.
161 СС, растворимость в воде около 4%.
Получают циклогексапол гидрированием фенола и окислением
циклогексана.
Применяют для синтеза мономеров, в частности капролактама1
и в качестве растворителя.
299
он
сн
ют его из масла персчноГ:
тивен, левовращающий. И:
Применяют ментол в пи
промышленности.
Ретинол (витамин Л
гексенил)-нонатетраеп-2,4
64 СС:
^ /
/Ч УЧ. /Ч ^Ч. /Ч
ОН
Ментол B-изопропил-5-метилцик-
логексанол) — бесцветное кристалли-
кристаллическое вещество с характерным мят-
мятным запахом; т. пл. 43 СС. Получа-
мяты. Природный ментол оптически ак-
звестпы и другие стереон.чомеры ментола,
щевой. плрфюмериой п фармацевтической
, 3,7-д1:м-:т1;л-9-B,б,б-тримстил-1-цикло-
6,8-cvi-i) — желтые кристаллы с т. пл.
Получают из печени морских рыб и животных. Известны также син-
синтетические методы получения.
Витамин At необходим для нормального роста человека и мле-
млекопитающих и играет важную роль в фоточувствктельности сетчат-
сетчатки глаз. Применяют в мед.!::г.нпс и ж::ьотпогюдстае.
Холестерин — жемчужные пластинки с т. пл. 149 °С
СНз СН2
\ /\
СН СН;.
СНз
СН3
СН;
СН2 — СМ— СНз
Впервые выделен из желчпых камней. Содержится в тканях и кро-
крови животных и человека. Отложение холестерина и его производ-
производных на стенках кровеносных сосудов связано с заболеванием ате-
атеросклерозом.
Г. АРИЛАЛКАНОЛЫ
1. НОМЕНКЛАТУРА
Название образуют по известным принципам, обычно арнльные
группы называют в качестве заместителей. Например:
/ч /СН2ОН ОМ ,.. .СНХП.ОН
бе И 31! 10! 1.1 it
е;п:рт
.l V.CI ЛНОЛ
2-фенплатапол-1
300
2. МЕТОДЫ ПОЛУЧЕНИЯ И ХИМИЧЕСКИЕ СВОЙСТВА
Применяются обычные методы, например гидролиз галогеиугле-
водородов, металлорганический синтез, восстановление карбо-
карбонильных соединений.
Алкилирование арепов этиленоксидом ведет к 2-арил-1-этано-
лам:
AIC.I,
АгН + СН2 —СН2 ' АгСН2СН2ОН
По своим химическим свойствам арилалканолы напоминают
алканолы. Накопление арильных групп у одного углеродного ато-
атома при гидроксильной группе способствует образованию стабили-
стабилизированных карбокатионов:
Аг Аг
Аг —С —ОН + Н + Х- ;z± Ar —C + X-+H2O
R
3. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ
Бензиловый спирт — бесцветная жидкость с приятным запахом;
т. кип. 205,8 °С.
Содержится в эфирных маслах некоторых растении. Синтетичес-
Синтетически получают из бензилхлорида. Используют в парфюмерии и лако-
лакокрасочной промышленности.
2-Фемилэтанол-1 — бесцветная жидкость с запахом роз, т. кип.
221 °С. Является составной частью розового масла. Синтетически
получают из бензола и этиленоксида. Применяют в парфюмерии.
Трифенилметанол — бесцветное кристаллическое вещество с
т. пл. 162,5 СС, в воде не растворяется. Его получают из хлортрнфе-
нилметана или окислением трнфенилметана. Используют для вве-
введения в органические соединения трифенплметильной группы (три-
тильной группы) и для получения трифенилметановых красителей.
Д. ДИОЛЫ
1. ИЗОМЕРИЯ И НОМЕНКЛАТУРА
Диолы являются производными углеводородов с двумя гидроксиль-
ными группами. Названия диолов образуют по известным прави-
правилам. Применяют также тривиальные названия. Диолы часто назы-
называют глпколями (табл. 32).
Важнейшие дполы рассмотрим раздельно, так как имеются
определенные особенности у каждого днола.
301
Таблица 32. Примеры номенклатуры дно лов
Структурная формула
Иоченгслат, ра ИЮПЛК
Другие названия
НОСН2ОН
НОСН.,СН.,ОН
1ЮСН2СН,СН2СН..ОН
CH3CH3
I I
СНз —С —С —СНз
он он
НОСН,СП- СМСН2ОН
НОСН2С:=ССН2ОН
Метанднол
Этандиол-1,2
Буталдиол-1,4
2,3-Диметнлбутанди-
ол-2,3
Бутем-2-днол-1,4
Бутин-2-Д!ЮЛ-1,4
Гликоль, этилепглнколь
1,4-Бутиленгликоль
Пннакон
2. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ
Гликоль (этандиол-1,2). Исходным веществом для получения гли-
гликоля является этилен. Ниже схематически показаны возможные
пути получения гликоля:
. сн2—сн
'"Кг = СН2
КМпО,
н.,0
СН2 —СН2
I I
ОН ОН
Гликоль — бесцветная, умеренно вязкая жидкость слад-
сладкого вкуса, кипит при 197,6 СС, имеет плотность d? — 1,113.
С водой смешивается; безводный гликоль гигроскопичен.
Реакции гликоля в основном аналогичны реакциям алканолов
с тем отличием, что в молекуле гликоля имеются две гидроксиль-
ные группы и превращения осуществляются в одной или в обеих
группах.
Так можно получить алкаполяты (гликоляты). Образование мо-
ноалканолята частично происходит уже в водном растворе щелочи,
так как в результате электроноакцепторного эффекта (—/) второй
гидроксилыюй группы гликоль является относительно более силь-
сильной кислотой, чем этанол:
HOCH2CH2OH + :Sol ^± HOCH2CH20- + HSo|- (p/<a=15,l)
Обычно гликоляты получают взаимодействием металла (Na,
К, Mg, A1) с безводным гликолем.
Гликоль легко окисляется различными неорганическими окис-
окислителями. Конечными продуктами являются СО2 и Н.2О, по в от-
302
дельных случаях можно выделить промежуточные продукты, на-
например гликолевую кислоту, щавелевую кислоту. Известен метод
каталитического окисления гликоля до глиоксаля:
Ог; кат. СХ ,0
носн,сн2оп <- Чс—of
\v хн
Специфическими окислителями 1,2-диолов являются
РЬ(ООССН3L и периодаты М+Ю.г, которые разрывают связь С—С
и образуют карбонильные соединения (см. ниже — Пинаконы).
Гидроксильные группы могут быть замещены атомами галогена,
например бромом:
НВг НВг
НОСН2СН2ОН—> НОСН2СП2Вг —>¦ ВгСН2СН2Вг
Простые эфиры гликоля легко образуются при взаимодействии
с алканолами в присутствии сильной кислоты:
H0CH2CfI20H-!-CII30fI —-+ НОСН2СП2ОСН3+Н2О
2-метоксиэтанол
НОСНаСН2ОСН3Н-СИ3ОН ^Х СН3ОСН2СН2ОСН3Ч-Н2О
1,2-диметоксиэтан
При нагревании с кислотами гликоль образует циклический
эфир — диоксан:
сн2
1
сп2
Чон
[ ноч
сн2 11 +
+ 1 -*
сп2
[ но/
/¦
' 2|
Н2С
\
°ч
сн2
1
сн2
-о/
+ 2Н2О
¦При действии сильных кислот происходит перегруппировка
1,2-диолов с образованием карбонильных соединений (примеры
см. ниже).
С органическими кислотами или их производными (ацилхлорида-
ми, сложными эфирами) гликоль образует сложные эфиры:
/°
р _ г "
/О ,0 R lnx
HOCH2CH2OH+R—C(f —> HOCH2CH2O —C<f >
ЧХ 4R
_^ ^c — OCH2CH2O — Cf
\V XR
С дикарбоновыми кислотами или их производными он образует
высокомолекулярные соединения (полиэфиры).
Гликоль получают в промышленности в больших масштабах и
используют главным образом для получения полимеров (полиэфир
лавсан и др., см. гл. XXXIII.В.4). Большие количества гликоля
используют для получения растворителей (простые эфиры гли-
гликоля, диоксан). Смеси гликоля с водой имеют низкую температуру
замерзания (до —75 °С), поэтому их применяют для охлаждения
автомобильных и тракторных двигателей (антифризы).
303
Пинаконы. Пинаконами обычно называют диолы, у которых
рядом находятся две гидроксильные группы у третичных углеродных
атомов. Их получают восстановлением кетонов металлами (Mg, A1,
Zn, Na):
R1 Rl
R\ Mfi" h+ I i
2 >C = O > r_C-C-R
R7 II
OH OH
Пинаконам свойственны все реакции гликолеи. Кроме того, при
действии сильных кислот пинаконы претерпевают перегруппировку
в кетопы:
R R R О
II ц+ I :1
R-C-C-R _+R_C-C-R+H,0
II I
ОН ОН R
Эту реакцию называют пинаколиновой перегруппировкой, так
как впервые полученный этой реакцией кетон R—СНа был назван
пинаколином.
Пипаколиновая перегруппировка является типичной реакцией
перегруппировки карбокатионов, которые образуются при протони-
роваиии пинаконов:
СНз СН3
1 !
СНз —С —С-СНз
1 1
он он
лпнакон
СНз X-
СНз —С —С-СНз
1 1
ОН СНз
СНз СИ,
1 1
-f H + X- ^± СНз —С С —СН3 Z
он +он2х-
СНз
—>¦ СНз — С — С — СНз 4-— СНз'—С
1 1 :1
Х- ОН СНз О
i СНз
СНз
-С-(
СНз
СНз СНз
1
— С — С —СНз
он он. х-
¦>тт I ll l y-
-<Пз "Т" П ' Л.
пинаколин
Пинаколиповую перегруппировку могут претерпевать также
другие 1,2-диолы, которые содержат не только третичные, по и вто-
вторичные и первичные ОН-группы. Легче алкилыгай группы мигри-
мигрирует водородный атом.
При окислении тетраацетатом свинца и периодатами (реакция
Малапрада) разрывается связь С—С:
R1 R2
/R2 РЬ(ООССПз). I I Na+!O4-
RCCR3 >
I I
R-C-C-R3
Орглническии i i
растворитель ! I растр.оры
он ин
304
Бутандиол-1,3 получают гидрированием альдоля:
^ ,00 СН3СНСН2СН2ОН
он он
альдоль
Бутандиол-1,3 — бесцветная жидкость, т. кип. 207,5'С, d%& =
— 1,0059 (для рацемата). Бутандиол-1,3 — хиральное соединение,
известны (+) и (—)-изомеры и оптически неактивный рацемат.
Бутандиол-1,3 применяют в промышленности для получения по-
полиэфиров и бутадиена-1,3. Бутадиен получают дегидратацией бутан-
диола над фосфатным катализатором (см. гл. III.Б. 1).
Бутандиол-1,4 получают гидрированием ненасыщенных глико-
лей, например:
НОСН,СНаСН,СН,ОН
Это бесцветное вещество (т. нл. 20,9 °С, т. кип. 203 °С). Его ши-
широко используют в промышленности в качестве мономера для по-
получения полимеров и в качестве исходного сырья для органического
синтеза (см. с. 132, 506, 644).
Гександиол-1,6 в промышленности получают гидрированием ади-
пиновой кислоты:
Чссн2сн2сн2сн.,с^ ——-> носн,сн2сн*сн2сн2сн2он
но/ "он
Гексапдиол-1,6 представляет собой бесцветное кристаллическое
вещество (т. пл. 42 СС и т. кип. 250 СС), хорошо растворим в воде.
Широко применяют в полимерной промышленности для получения
полиэфиров (с. 580) и полиуретанов (с. 644).
Бутин-2-диол-1,4 получают из ацетилена и формальдегида
в присутствии ацетиленида меди под давлением:
Нч ,Н си +
>C-O-j-H-C— С-Н + О = С/ —>• НОСН,С-=ССН2ОН
Н-7 ЧН
Он представляет собой бесцветное кристаллическое вещество
с т. пл. 58 СС, хорошо растворим в воде.
Бутип-2-диол-1,4 применяют в промышленном органическом сип-
тезе для получения мономеров.
Е. ТРИОЛЫ И ПОЛ ИОЛЫ
1. НОМЕНКЛАТУРА
Трнолы и полиолы являются производными углеводородов с тре-
тремя или более гидроксильными группами. Их названия образуют по
общим правилам. Применяются также некоторые тривиальные на-
305
звания. Например:
Н0СН.2СНС1120Н IЮС1I,CE I — СНСЛ IoOH
I I I
он он он
пропантриол-1,2,3 бутаптетраол-1,2,3,4
(глицерин)
2. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ
Глицерин. Впервые глицерин был получен К. Шееле в 1779 г. при
обработке жиров щелочью в присутствии оксида свинца. Жиры яв-
являются сложными эфирами алкановых кислот С1в—Cia (высших
жирных кислот) и глицерина. В присутствии щелочи происходит
гидролиз сложного эфира: в осадок выделяются свинцовые соли
алкановых кислот, а в водном растворе остается глицерин.
В настоящее время в промышленности часть глицерина получа-
получают из жиров, а большую часть — синтетически нз аллилхлорида или
аллилового спирта:
HOCI
= СНСН2С1 » С1С112СНСН2С1 —
Н,0;ОН-
Н,О;ОН- ОН
И2О2; И2О
СН2--СНСН2ОН —-* НОСН2СНСН2ОН
60 — 70° С I
он
Глицерин — очень вязкая жидкость со сладким вкусом; т. пл.
17.9 СС, т. кип. 290 СС. Глицерин кристаллизуется очень трудно,
образуя переохлажденную жидкость. При температуре кипения
глицерин постепенно разлагается, поэтому его перегоняют в вакуу-
вакууме. Смешивается с водой. Безводный глицерин гигроскопичен.
Глицерину присущи все характерные реакции алканолов. Од-
Однако следует заметить, что одновременное присутствие в молекуле
глицерина трех гидроксильных групп влияет иа его реакционную
способность и, кроме этого, позволяет получить три ряда произ-
производных.
В щелочном водном растворе легко образуются моиоалканоляты
(мопоглицераты). Глицерин по кислотности превышает гликоль.
Предполагается, что ионизируется средняя гидроксильная группа
(влияние индуктивного эффекта двух соседних кислородных ато-
мое):
НОСН2СНСН2ОП-|Н2О 5=t НОСН2СНСН2О11 + НЯО +
он о-
Очень легко образуются глицераты тяжелых металлов, например
глицерат меди (II), в которых нон металла связан в виде внутренне-
внутреннего комплекса, поэтому гидрокенд металла щелочью не осаждается.
306
Na+
.0:
Na+
н.с"-ч
¦¦0~
НС
/
Си
,н
чсн2
I
-сн
\
сн,
глицерат меди (II)
При окислении глицерина образуется
целый ряд продуктов. При мягком окисле-
окислении образуется смесь дигидроксиацетона и
глицеринальдегида (см. с. 500).
Большое практическое значение имеют
сложные эфиры глицерина. При взаимодей-
взаимодействии со смесью азотной и серной кислот
образуется эфир азотной кислоты—-трпнит-
рат глицерина («нитроглицерин»), который
является взрывчатым веществом и в то же время применяется как
лекарственный препарат (гл. XVII.В).
С органическими кислотами и их производными получаются
сложные эфиры глицерина. На основе дикарбоновых кислот и их
ангидридов получают полимеры —¦ полиэфиры.
При нагревании глицерина до высокой температуры в присут-
присутствии слабокислых катализаторов происходит своеобразное отщеп-
отщепление двух молекул воды и образуется ненасыщенный альдегид
акролеин — вещество с острым раздражающим запахом:
Н Н Н
III П+ Г"ч /Ч 1
НО — С —С-С-ОН - ;С-С = С< -j-2H2O
III '" Lh7 4ohJ
H ОН Н
си—Ы -;-2н2о
акролеин
Глицерин применяется для производства взрывчатых веществ
(нитроглицерин, динамит) и синтетических высокомолекулярных
соединений (полиэфиры, алкидные смолы). В меньших количествах
он применяется в качестве мягчнтеля в текстильной и кожевенной
промышленности и как составная часть косметических препаратов.
Бутантетраол НОСН2СНСНСН2ОН называется эритритом. Мо-
Of ЮН
лекула эритрита содержит два асимметрических углеродных ато-
атома с одинаковым набором заместителей, имеется три стереопзоме-
ра (см. рис. 227):
но
сн:оч
носн2
но\
СП20Н
ЧУ
I
СИ2О'Л
СН2ОН
СН2ОН
СН2ОН
СНаОН
-(-)- эритрит
СН2ОН
S.S-M- эритрит
307
Для определения R- или S-конфигурации группы следует рас-
располагать в порядке падения старшинства: ОН, СН(ОН)СН2ОН,
СН,ОН.
Эритриты встречаются в природных растительных продуктах.
Пентаэритрит B,2-бис-гидрсжсиметилпро:1апдпол-1,3) получают
реакцией формальдегида с ацетальдегидо.м:
СН2ОН
НОСН2 — С—СН2ОН
2ОН
сн2
Он представляет собой бесцветное кристаллическое вещество с
т. пл. 263,5 СС; растворим в воде. Пентаэритриту свойственны все
реакции алканолов, кроме реакций отщепления.
Его применяют в промышленности для получения полиэфиров
и взрывчатых веществ (тетранитрат пептаэритрита).
Пентаолы и гексаолы. Эти соединении обычно называют пентн-
тами и гекситами:
НОСН2СН—СН —СНСНаОН НОСН-.СН—СИ—СН-СНСН2ОН
III III
он он он он он он
I I I
¦¦-" - он
Пентиты содержат два асимметрических атома углерода — С2
и С4, третий атом имеет одинаковые заместители СН(ОН)СН2ОН,
известны четыре стереоизомера.
Гекситы содержат четыре асимметрических атома углерода, но
число стереоизомеров меньше 16 и равно 10 благодаря особенно-
особенностям строения молекулы. Благодаря наличию одинаковых замести-
заместителей у С2 и О, и, соответственно, у С3 и С4 молекулы некоторых
стереоизомеров имеют плоскость симметрии, находящуюся между
атомами Ся и С4.
Пентиты и гекситы содержатся в ряде растений. Они имеют
сладкий вкус. Синтетически их получают восстановлением моно-
моносахаридов (с. 507, 511). Пентиты и гекснты применяют для получе-
получения полимеров, пластификаторов, поверхностно-активных веществ,
а также в пищевой промышленности.
Глава XV
ГИДРОКСИЛПРОИЗВОДНЫЕ УГЛЕВОДОРОДОВ
СО СВЯЗЬЮ C(sp2) — ОН
В группу гидроксилнроизводных углеводородов со связью
C(sp-)—ОН входят:
а) еполы
R\ Ra
С С
Rx Ч)П
308
б) фенолы, пафтолы и другие аренолы (гидроксиарены)'
/ОЫ /V У /°Н
в) арендиолы (дигпдрокеггарены, двухатомные фенолы)
.ОН
г) арентриолы (тригидрокспарены, трехатомные фенолы)
ОН
ОН
А. ЕНОЛЫ
1. НОМЕНКЛАТУРА
Названия енолов образуют из названия алкена и суффикса -ол.
Например:
СН2-СП-ОН СН2=:С —СНз
Ан
¦этеиол (рипило- пронеп-1-ол-2
выA спирт)
Атомы углерода в молекуле нумеруют таким образом, чтобы гид-
роксильмая группа имела наименьший номер.
2. МЕТОДЫ ПОЛУЧЕНИЯ
Енолы образуются в реакциях присоединения воды к алкннам:
Кат. R. /ОН
R_C = C-R-!-H-O-H—> /С = С(
Эта реакция подробно рассмотрена в гл. IV. Кроме того, енолы мо-
могут образоваться при кислотном гидролизе сложных эфиров
енолов:
R. /O-Y ц+ Rx ,,OH
")С = С- —> ,С = С(- +IIOY
Енолы образуются также в реакциях фотохимического пли тер-
термического расщепления веществ, например при термическом
309
отщеплении воды от гликоля:
900° С
СН2-СН2 ——:-* СН2 = СН
| | Z,OU 113
он он
Уже давно было замечено (правило Эльтекова — Эрмнмейера),
что енолы не удается выделить из реакционной смеси, так как они
перегруппировываются в карбонильные соединения:
Н
R ,О-Н R. | /О
Однако в отдельных случаях спектроскопическими методами уда-
удается уловить присутствие енола в реакционной среде и измерить
скорость превращения его в карбонильное соединение. Время жиз-
жизни (время полупревращения) енолов колеблется от 10 с до 30 мин
в зависимости от растворителя, температуры и структуры епола.
Получено также несколько стабильных енолов, например:
CF3—C-CFg,
Ан
пентафторпро-
rien-1 -ол-2
3. СТРОЕНИЕ
Методом микроволновой спектроскопии удалось оценить длины свя-
связей в молекуле этенола (винилового спирта):
н
Связи С—-С и С—Н и О—Н имеют обычную длину, характерную для
алкенов и алканолов. Связь С—О укорочена.
4. ХИМИЧЕСКИЕ СВОЙСТВА ЕНОЛОВ.
ТАУТОМЕРИЯ ЕНОЛОВ И КАРБОНИЛЬНЫХ СОЕДИНЕНИЙ
Енолы и соответствующие карбонильные соединения фактически
являются изомерами, которые различаются между собой располо-
расположением водородного атома и двойной связи. Переход епола в кар-
карбонильное соединение происходит миграцией водородного атома,
обычно в виде протона. Но в то же время принципиально возможен
переход карбонильного соединения в соответствующий енол (ено-
лизация). Так получается система изомеров, переходящих друг в
друга при миграции протона. Такое явление называется динамиче-
динамической изомерией или таутомерией.
Таутомерную систему карбонильное соединеннее енол (К^±Е)
характеризуют константой таутомерпого равновесия iCT или иног-
310
да процентным содержанием енола:
Н
R4 I /О R4 ,0-Н
Н/ 4R Н/ XR
К U
В зависимости от структуры и растворителя К^— Ю~*—10~8, т. е.
содержание енола от 10~2 до 10~в%. Необходимо подчеркнуть,
что эти данные относятся к системам, которые достигли термоди-
термодинамического равновесия.
Во многих реакциях образуются енолы в значительно больших
концентрациях и равновесие с карбонильным соединением устанав-
устанавливается медленно.
Положение равновесия определяется сравнительной термодина-
термодинамической стабильностью енола и карбонильного соединения, т. е.
теплотой образования соединений. Обычно карбонильные соедине-
соединения термодинамически более стабильны по сравнению с енолами.
Таутомерные превращения енол *..: карбонильное соединение
происходят с переносом протона (прототропия), при этом промежу-
промежуточно образуется анион.
Енолы, подобно алкаполам, являются слабыми ОН-кислота-
ми, при этом кислотность енолов больше, чем алканолов, вследствие
эффекта <опряжения неподелепных электронных пар кислородного
атома с л-электронами двойной связи:
? \^ >й +
) + :Sol =^= ;С = С^ + HSol
Образуется енолят-ион, который в отличие от обычного алкоксидно-
го иона обладает сопряженной системой — неподеленная электрон-
пая пара кислородного атома и л-связь С^С. Группа —ОН и тем
более —б:~ действуют как сильные электронодоноры.
Енолят-ион является сопряженным ионом, зарял в нем делока-
лизован между кислородным и углеродным атомами:
Енолят-ион имеет два реакционных центра (амбидептный, или
амбифункциональный, ион). Протон к еполят-иону может присоеди-
присоединяться в двух положениях — к атому кислорода или к атому
углерода:
с=с(
R7cc4 ^^ )сс( ^^ )сс(
Н R H R H R
311
Константы кислотности енолов — величина порядка 11—12
(для алканолов р/Са«15—19). Следовательно, енол сравнительно
легко диссоциирует, особенно п полярных растворителях, и обра-
образует сопряженный ашюн, который может присоединять протон к
углеродному атому с образованием карбонильного соединения —
кислоты другого типа (СН-кислоты). Константы ионизации карбо-
карбонильных соединений но связи С—И оцениваются в единицах
рКа^ 16-20.
Система енол —¦*- карбонильное соединение является равновес-
равновесной системой, состоящей из сравнительно сильной ОН-кислоты и
слабой СН-кислоты, константы ионизации их в единицах р/С от-
отличаются на 5—8 порядков. Обе кислоты при ионизации образуют
один и тот же анион — амбидентный еполят-ион.
В равновесной смеси всегда больше того таутомера, у которо-
которого меньше кислотность. Чем больше разница в константах иониза-
ионизации, тем больше равновесие сдвинуто в сторону большего содер-
содержания менее кислого таутомера, в данном случае карбонильного
соединения. Известна взаимосвязь между константами ионизации
карбонильного соединения (Кк) и енола (/Се) " константой тауто-
мерпого равновесия (/Ст):
Производные енолов — простые и сложные эфиры (с. 341, 578) —
не содержат подвижного водородного атома и в обычных условиях
они не перегруппировываются в производные карбонильных соеди-
соединений:
r ,O-Ri R4 .о—С—Ri
Н-7 4R Н/ XR О
ено.-тьные эфиры
Так как еполы содержат силыгаполяризованную двойную связь
с электронодоноропым заместителем, они реагируют с электрофнль-
ными реагентами, например с бромом:
^С = С + Бг — Вг *- С— С + НВг
Эти реакции более подробно рассмотрели в химических свойст-
свойствах карбонильных соединений (гл. XXVII.А.3).
При рассмотрении енолов надо помнить, что енолы в чистом
виде могут быть получены в крайне редких и специальных условиях.
Обычно они присутствуют в малом количестве в альдегидах и кето-
пах как таутомерная форма. Стабильные енолы характерны для
Р-днкарбонильных соединений (гл. XXVIII.Б).
Б. ФЕНОЛЫ
Фенолы (аренолы) являются гидроксильными производными аре-
нов, содержащих гидроксильпую группу у углеродного атома цикла.
312
1. ИЗОМЕРИЯ И НОМЕНКЛАТУРА
Названия фенолов образуют от названий соответствующих арепов,
а наличие гидрокснльной группы обозначают суффиксом -ол. Ну-
Нумерацию начинают от углеродного атома при гндрокснльной груп-
группе. Многие фенолы сохраняют тривиальные названия. Иногда в
сложных соединениях наличие гндроксплыюн группы обозначают
префиксом гидрокси- (окси-). Например:
ч ОН /, ,011 /v ,0Н у. ОН /ч /ч ОН
СНз у НзС/^/
СН3
о- м- п-
(|снол (гид- крезолы (метилфенолы) иафтол-2
роксибен- (^-нафтол)
зол)
2. МЕТОДЫ ПОЛУЧЕНИЯ
Фенол и его гомологи получают из продуктов коксования камен-
каменного угля (каменноугольной смолы и нодсмолыюп воды), а также
синтетически.
1. Реакции нуклеофильного замещения углеродного атома цикла.
а) При сплавлении солей а р е н с у л ь ф о н о в ы х
кислот со щелочью получаются спеноляты и сульфиты
(гл. XVHI.3):
О-Ха +
Ч N'a-zSO-
"^' ^ЗО-ЗОО'С
•КаОН
.6) Н у к л е о ф и л ь и о е замещение атома х л о-
р а в хлорбензоле на гидрокси ль и ую груп-
группу осуществляется в жестких условиях (гл. VIII.4.1).
в) При нагревании а р е н д и а з о и и е в ы х с о-
лей в водных растворах выделяется азот и получаются
аренолы (гл. XXV.A.3):
[Аг —N'=-.-N]X--! Н.,0 —> Аг —OH-;-i\2-;-HX
г) К у м о л ь и ы й м е т о д. В 1912 г. Р. 10. Удрис и
П. Г. Сергеев нашли, что при расщеплении гндроперокснда кумола
(изопропнлбензола) в присутствии серной кислоты образуются
фенол и ацетон:
СП,
С —О —ОН ,„ „ОН О
! h.so
Такие реакции с внутримолекулярной перегруппировкой харак-
терпи для органических пероксндов и рассмотрены в гл. XVII.Г.
313
Метод Удриса —Сергеева можно использовать также для полу-
получения гомологов фенола и других аренолов:
СНЯ СН3
сн,=си-сн3 | о2 | H.h
Аг-Н > Аг-С-Н —» Лг —С —ООН ->
Н+ К
Н+
Кат.
О
СН3
СН3
1
—¦* АгОН-|-СИз —С —СН3
Аналогичные реакции разработали также X. Хок н С. Ланг
в 1944 г.
д) Прямое гидроксилнрование. Известны ме-
методы прямого введения гидроксилыюй rpyiintii вместо атома водо-
водорода при действии пероксида водорода в присутствии катализатора:
Kst.
-'-НООП
Катализаторами служат солн металлов, например Fe(II), Cufll),
Ti(III).
3. ФИЗИЧЕСКИЕ СВОЙСТВА И ПРИРОДА СВЯЗЕЙ
Фенолы являются бесцветными жидкими или кристаллическими ве-
веществами (табл. 33) с очень своеобразным, сильным и устойчивым
Таблица 33. Физические константы некоторых фенолов
Соединение
(Г
сня
Н3С
Т. ил., °С
41,0
30,0
11,0
36,0
Т. кип., °С
182
191
203
202
1,072
1,0465
1,034
1,035
1,5425 D0 СС)
1,5453
1,5398
1,5395
314
запахом («карбольпый» запах). В воде растворимы мало. При хра-
хранении на воздухе постепенно темнеют (окисление).
Полярность и электронодонорные свойства. Фенол и его гомоло-
гомологи являются полярными соединениями, их дипольные моменты по-
порядка 5-Ю-30 —5,3-Ю0 Кл-м A,5—1,6 D). При этом дипольный
момент в отличие от алканолов и циклоалканолов направлен в сто-
сторону бензольного кольца. Это связано с взаимодействием неподе-
ленной электронной пары кислородного атома с я-электронной сис-
системой бензола, кислородный атом выступает в качестве электроно-
допора. -(-М-эффект кислородного атома в этом случае преобладает
над электропооттягивающим эффектом (—/-эффектом) по системе
о-связей:
Образование такой сопряженной системы увеличивает электро-
электронодонорные свойства соединений. Энергия ионизации фенола равна
8,5—8,6 эВ, что ниже ЭИ бензола, толуола и алканолов.
Результаты расчетов по методу МО молекулы фенола. В я-электронную
систему фенола входит восемь электронов — шесть из бензольного цикла и
два от атома^кислорода. Результаты расчета по методу МО Хюккеля (рис. 86)
свидетельствуют, что увеличивается плотность электронов на углеродных
атомах в о- и re-положениях бензольного цикла и уменьшается на кислород-
пом атоме. Лучше всего это видно при использовании эффективных зарядов
п—q, где п равно числу электронов, участвующих в сопряженной системе
(для атомов углерода ге= 1, для кислорода п— 2).
В фенолах связь С—О не является больше чистой а-связью, а приобрета-
приобретает некоторую я-электронную плотность. Энергия высшей запятой молекуляр-
1,970/Н 0,03jxH
• *О. -О.
-2/1-
к-
Р-
2р-
-^— Ei = « - 1,000 ?
1,010
= й.
= а
= а
0,880/?
1,0000
0,01"
0,01-
Эффсктнвные заряды
П-Яг
(ВЗМО)
> 0,137
2,50б>
0,297
Рис. 86. Значения энергии МО, одночлектронные плотности ВЗМО;
плотность я-;лектронов, порядки я-связей и эффективные заряды л-
електрониой системы фенола
315
ной орбитали (ВЗМО) по сравнению с бензолом повышается, что согласуется
с уменьшением потенциала ионизации. Локализация электронов в ВЗМО сви-
свидетельствует о повышенном реакционной способности о- и ге-положенин np;s
взаимодействии с электрофильными реагентами.
Спектры поглощения. я-Электронная система фенола легче воз-
возбуждается, чем система бензола, что выражается в сдвиге максиму-
максимумов поглощения в сторону более длинных волн, т. е. меньших энер-
энергий (батохромный сдвиг) (табл. 34).
В инфракрасных спектрах характерно поглощение группы ОМ
в интервале 3200—3600 см в зависимости от растворителя и кон-
концентрации вещества.
1 аблица
бензола
34. Характерное поглощение в электронных
и фенола в растворе гексана
Соединение
Бензол
Фенол
Максим}'-! поглощена, им
~200
~2С0 (центр тонкой структуры)
210
~275 (центр тонкой структуры)
спектрах
Интенсивность "
8СГ0
~220
6200
~2000
В спектрах ПМР сигнал протона группы ОН наблюдается при
6 — 9,0. . .9,3 (раствор в диметилсульфоксиде) и при 6--4,5. . .7,5
(раствор в СС14), значение 6 зависит от концентрации вещества.
4. ХИМИЧЕСКИЕ СВОЙСТВА
При рассмотрении химических свойств фенолов целесообразно най-
найти общее п различие с химическими свойствами алканолов п ено-
лов. Часть реакций определяется присутствием полярной группы
ОН, В основном это реакции с сохранением атома кислорода в мо-
молекуле. Но реакции с отщеплением атома кислорода от бензольного
цикла практически трудно осуществимы и
6+ _ поэтому малоизвестны, В то же время для
"¦Nu фенолов характерны реакции в бензольном
кольце при взаимодействии с различными
электрофнльными реагентами. Особенно легко
такие реакции проходят с фенолят-иопом.
1. Кислотность и реакции с участием атома кислорода. Фенолы
являются слабыми ОН-кпслотами, по значительно более сильными
по сравнению с алкаполами. Это определяется двумя факторами:
большей полярностью связи О—Н и образованием сонря'/:гнного
фенолят-попа, в котором отрицательный заряд значительно дело-
кализовап:
- п-
^ \.о^
316
Константы кислотности рКа для фенола и его гомологов равны
9,9—10,4 (для водных растворов).
В водных растворах щелочей фенолы образуют феноляты. В фе-
фенолят-ионе плотность электронов па углеродных атомах в о- и
я-положепиях значительно больше, чем в феноле.
Фенолы имеют очень характерную цветную реакцию: в водных
растворах с FeCl^ они дают красно-фиолетовое окрашивание, кото-
которое исчезает после прибавления сильной кислоты или этанола.
Предполагают, что окраска связана с образованием комплексных
фенолятов с участием иона Fe(III).
Феноляты легко алхилируются и ацилируются у кислородного
атома:
R - Вг
О.
-! Na-'-Br-
V
алкилфенилопме эфиры
о
-f Na
фениловые сложные ^фиры
Анплироваппю подвергаются также фенолы, обычно при взаимо-
взаимодействии с ангидридами карбоповых кислот или ацилхлоридами:
он
О
и
!— С.
Ч,/
ЧО'
О
С —R
О
R
II
О
+ RCOOII
Ацилкрование карбоновыми кислотами в присутствии серной
кислоты, что очень характерно для алканолов, в случае фенолов идет
медленно, что связано со значительным уменьшением плотности
электронов на кислородном атоме вследствие сопряжения.
2. Окисление. Фенолы способны окисляться сравнительно лег-
легко, при отрыве электрона образуется катион-радикал, который иони-
ионизируется на свободный фвноксильный радикал и протон. Очено легко
фенокснльпые радикалы образуются при окислении фенолят-ионов:
^н -.- V
\.
~\ +
н
О:
Фепоксилы представляют собой свободные радикалы с сопряжен-
сопряженной системой л-элсктронов, в которых песпарепнын электрон зна-
значительно делокалп.;о,;;и1. Мо плотность неспарепного электрона па
отдельных атомах достаточно большая, чтобы протекали дальней-
317
шие реакции (димеризация, олигомеризация):
2Q.H.6. _
4,4'-дигидроксибифеиил
Стабильность феноксилов увеличивается при введении замести-
заместителей, особенно объемистых в о- и п-положениях. Например, 4-ме-
тнл-2,6-бис-трет-бутилфепол (нонол) образует устойчивый фенок-
еильный радикал, который может быть получен в кристаллическом
состоянии:
CIT3
Объемистые заместители в значительной степени мешают реакциям
димеризации и другим превращениям.
3. Реакции с электрофильнымн реагентами. Фенолы взаимодейст-
взаимодействуют с электрофильными реагентами значительно легче, чем бен-
бензол, толуол, кислоты, что соответствует их энергиям иониза-
ионизации. Местом атаки электрофнлыюго реагента является бензольное
кольцо с легкополяризуемоп я-электронной системой (механизм
реакции см. с. 186).
Реакции рассмотрены в такой последовательности: взаимодейст-
взаимодействие с галогенами, с N-электрофилами, S-электрофилами, С-элек-
трофилами.
а) Легко происходит галоген и рование, которое не
требует катализатора:
сь Г II' . Г Г ci,
| о-хлорфенол
n-хлорфенол
2,4-дихлор-
фенол
Конечным продуктом хлорирования может быть пентахлорфенол.
Особенно легко происходит бромирование в разбавлен-
разбавленных водных растворах — сразу образуется 2,4,6-трибромфепол.
Предполагается, что активно бромируется фенолят-ион. В избытке
брома образуется конечный продукт бромирования — 2,4,4,6-тет-
рабромциклогексадиенон:
Вг Вг
318
б) При нитровании фенола получаются нитрофеиолы.
Конечным продуктом нитрования является тринитрофенол (пикри-
(пикриновая кислота):
.ОН
011
UNO,
в) Известны реакции н и т р о з и р о в а и и я (с. 387), а з о-
сочетания (с. 425).
г) Сульфирование фенола ведет к фенолсульфоновым
кислотам. Соотношение о- и n-изомеров определяется температурой
реакции. о-Изомер при температурах выше 100 °С перегруппиро-
перегруппировывается в я-изомер:
ОН , он л /ОН
ii,so4 f \/ n2so4
I00°C
I003C
д) Легко провести а ц и л и р о в а и и е
вии ацилхлоридов или ангидридов кислот в
Льюиса, например А1С13:
фенолов при дейст-
дейстприсутствии кислот
Я л.сь
ОН
ОН
ОН
О
Аналогичные ацилфенолы могут быть получены перегруппиров-
перегруппировкой сложных эфиров фенолов под действием А1С1а (перегруппиров-
(перегруппировка Фриса):
/О /R /ч /ОН /ч /ОН
ХС/ MCI,
II —-
о
О
Предполагается, что под действием A1CL, сложный эфир расщепля-
расщепляется до фенолята алюминия и комплекса ацилхлорида RCOC1 с
А1С13 (с. 568). Затем следует ацнлирование.
е) Широко применяются реакции а л к и л и р о в а н и я фе-
фенолов. Наиболее часто для алкилирования используют алканолы и
алкены в присутствии протонных кислот (H2SO4, H3PO4) или кислот
319
Льюиса (BF3):
,он
+ R-OH
ОН
HlC
Н3С
Кислоты при взаимодействии с алкацолами и алкенами генери-
генерируют карбокатионы, которые являются электрофнльными частица-
частицами в реакциях алкилирования:
R_OH-!-BF3
R-O<_ 7H
4BF3
СН3.
Г
L
[BF3OH]-
+ 1
С-СН3 X-
j
Фенолы являются столь активными компонентами в электро-
фильных реакциях замещения, что вступают во взаимодействие с
весьма слабыми электрофилами — альдегидам» и кетонамн — в при-
присутствии кислот или оснований.
Наиболее легко реагирует формальдегид. Если реакцию прово-
проводить в мягких условиях, удается выделить гидроксиметилфенолы:
ОН
)с=о
ОН
и + х-
он
о-гндрокснме- я-гидроксиметилфенол
тилфенол
В этой реакции электрофильным реагентом является гидрокспме-
тил-катион, который образуется при протонировапии формальдегида:
Н-7 ~" 'h~H/x- "
Гидроксиметилфенолы в ггрисутствии кислот могут выступать в
качестве алкилирующего реагента:
HOv\
+
4,4/-дип1Дроксид11фС1:нлметан
ноч ^ но
4H
x-
о
н н
320
«Тидроксифеинлметнл-катион является весьма стабилизированным
карбокатпоиом вследствие электроиодонорного действия гидрокси-
фсиплыюй группы. Наиболее просто это можно изобразить при по-
помощи резонансных структур:
В более жестких условиях фенол с формальдегидом образует вы-
высокомолекулярные продукты пол и конденсации — фенолформальде-
гидные смолы:
ОН ОН ОН ОН
он
регулярная цепь
ОН
СН2.
СН2
но
2
чон
нерегулярная цепь
Возможна также дальнейшая ио.тикондепсация с образованием
СН2-мостиков между отдельными цепями. Так образуется сетча-
сетчатая структура полимера.
В реакции с кетонами фенол образует производные дифенилме-
тана:
НО. „ .о НО. лч ,,, Д)Н
О
II
+ R-C-R1-
4. Гидрирование. При каталитическом гидрировании фенола
получается циклогексанол. В качестве промежуточного продукта
образуется циклогексаноп:
,о\\
2Н2; Ni
.ОН
о
[[s; Ni
ОН
Все рассмотренные выше превращения фенолов свидетельствуют
о том, что наибольшее число реакций проходит в бензольном кольце,
которое активирозапо электронодонорным действием гидрокенль-
ной группы. Гидроксильная группа только придает этим соедиие-
№ 517
321
ниям слабокислые свойства и способность образовывать простые и
сложные эфиры. В этом заключается принципиальное отличие аре-
полов от алканолов и некоторое подобие енолам.
5. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ
Фенол производится в больших количествах: мировая продукция
его достигает 1,5—2 млн. т в год. Из получаемого количества 90%
является синтетическим фенолом и только 10% — каменноуголь-
каменноугольным. Главным синтетическим методом является кумольньгй (метод
Удриса — Сергеева). Более 50% синтетического фенола получают
этим методом.
Наибольшие количества фенола используются для получения
фенолформальдегидных смол, которые применяются в производст-
производстве фенопластов. Большие количества фенола перерабатывают в
Капролактам,
синтетическое
волокно
Медицинские препараты ,
антисептические вещества,
красители
Стабилизаторы
полимеров,
моюшие срелстиа
Рис. 87, Применение фенола
Фенол-
формальдегидные
смолы
Пестициды
циклогексаиол, который необходим для промышленности синтети-
синтетического волокна. На рис. 87 дана схема использования фенола.
Крезолы получают из продуктов сухой перегонки (коксования)
каменного угля, бурого угля, торфа и горючего сланца. Получен-
Полученную смесь о-, м- и n-крезолов можно исггользовать для дальнейших
реакций неразделенную или можно разделить на составные части.
Известны также синтетические методы получения крезолов. Ис-
Исходным веществом для них является толуол.
Смесь крезолов используют для получения крезолформальдегид-
ных смол. Чистые крезолы применяют для синтеза красителей, ме-
медицинских препаратов, антисептических веществ, аитиоксидантов.
Ксиленолы (диметилфенолы) имеют шесть изомеров. Из продук-
продуктов сухой перегонки каменного или бурого угля и горючего слапца
получают смесь ксиленолов, используемую в производстве синтети-
синтетических смол. Выделить чистые ксиленолы из этой смеси трудно. Для
их получения необходимо применять синтетические методы.
322
Ионол D-метил-2,6-бис-/п?ет-бутилфенол) представляет собой
бесцветное кристаллическое вещество с т. пл. 70 СС. Получают его
алкилированмем «-крезола 2-метилпропеном или /?ф<?ш-бутиловым
спиртом в присутствии кислот. Ионол широко применяют в качестве
аитиоксиданта для стабилизации полимерных материалов, парфю-
парфюмерных изделий и др. Антиоксидантные свойства ионола объясняют-
объясняются тем, что он реагирует с активными свободными радикалами, прев-
превращаясь в стабилизированный феноксильный радикал. Это обрыва-
обрывает цепь свободпорадикального окисления.
4,4'-Дигидроксидифенилпропан (диап) представляет собой крис-
кристаллическое вещество с т. ил. 156—157 °С:
СИ3
-if Ч-он
НО -
- С
Его получают из фенола и ацетата в присутствии кислого катализа-
катализатора. Диан широко используется в качестве мономера для получения
различных синтетических смол (эпоксидных, фенолформальдегид-
ных, поликарбонатов) п в качестве исходного для синтеза аитнок-
сидантов и гербицидов.
Нафтолы. Нафтол имеет два изомера:
ОН
а-мафтол
(т. пл. Об "С, т. кип. 278 — 280 ГС)
fl-нафтол
(т. пл. 123 Г'С, т. кип. 285 —28G Т.)
Нафтолы — бесцветные кристаллические вещества со своеобраз-
своеобразным запахом. Их получают из нафталинсульфоновых кислот сплав-
сплавлением со щелочью при 280—320 °С. Возможно также получение из
изопропилнафталинов кумольным методом.
Для пафтолов характерны все реакции фенолов. Их используют
для синтеза красителей.
В. АРЕНДИОЛЫ И АРЕНТРИОЛЫ
1. ИЗОМЕРИЯ И НОМЕНКЛАТУРА
Названия арсидиолов и арентриолов образуют по общим правилам.
Для простейших соединений сохранились тривиальные названия:
ОН ОН ОН ОН ОН
•'
А/он
А
пирокатехин
(I .'J-fliii идро-
ксибспзол)
11*
резорцин
A.3-дш пдро-
ксибенгол)
н
гидрохинон
A,4-дигидрокСи-
бегзол)
пирогаллол
A,2,3-трг.[ ид-
рокенбензол)
флороглюц:'п
A,.),.)-тригнд-
рокенбензол)
323
2. ВАЖНЕЙШИЕ
ПРЕДСТАВИТЕЛИ
Пирокатехин. Впервые пирокатехин был получен при нагревании
некоторых природных смол. Синтетически пирокатехин можно по-
получить из о-хлорфенола или о-феполсульфоновой кислоты и щело-
щелочи при 300 СС:
ОН
ОН
,SO3Na
,О\а
X.iCHI; - 300 С
~О.\а
,он
Ч)Н
"V
В промышленности применяется реакция прямого гидроксили-
роваыия фенола Н2О2 или RCOOOH в присутствии кислот или со-
солен тяжелых металлов, в которой получается смесь пирокатехина н
гидрохинона.
Пирокатехин образует бесцветные кристаллы с т. пл. 104 'С,
хороню растворяется в воде.
Подобно фенолу, пирокатехин является слабой ОН-кислотой,
но имеет две константы ионизации:
:Sol
= 10.3
+
HSol
:Sol
Дианпогг пирокатехина образуется только в силыгощелочпой сре-
среде, ионизации метает внутримолекулярная водородная связь.
Характерна цветная реакция с FeCI3 — появляется зеленая
окраска, которая в присутствии ацетата натрия переходит в крас-
красную. Соль свинца в воде нерастворима н выпадает в виде белого
осадка.
Пирокатехин легко алкилпруется и образует простые эфнры,
в том числе циклические:
о-
-сп.,
I
,0
он
I
осн3
он
он
сил.
V
324
V
сил
ои-
СП.,1
ои-
ОСНз
X оси.
V
Пирокатехин окисляется легче, чем фенол, одним из продуктов
окисления является о-хннон. Еще легче окисляются щелочные раст-
растворы пирокатехина.
Легко проходят реакции электрофплыюго замещения в бензоль-
бензольном кольце (галогепирование, нитрование и др.).
Пирокатехин используется для различных синтезов, применя-
применяется в качестве сильного восстановителя в фотографии, как аналити-
аналитический реагент колориметрического определения некоторых метал-
металлов (Ti, Mo, W, Fe, Ge).
Резорцин — бесцветное кристаллическое вещество со слабым
запахом, т. пл. 110,8сС, хорошо растворимое в воде. Получают
резорцин из ж-беизолдисульфоновой кислоты:
SO.,Na ON а ОН
I
NaOH
Я О О СС
и + х-
SO,
4ONa
OH
Резорцин является слабой двухосновной кггслотой:
ЮН
рК, = 9
:Яо1
+ HSOI+
РК, а I I
-f-2HSol +
Характерна цветная реакция с Fed, — появление темио-фпо-
летового окрашивания, исчезающего в слабощелочной среде (при
добавлении раствора ацетата натрия).
Резорцин очень легко алкплируется, ацилируется, нитруется.
Эти продукты используют для получения красителей, антисепти-
антисептических веществ, антиоксидантов, взрывчатых веществ.
При каталитическом гидрировании в щелочной среде образуется
производное циклогексана — циклогексадион-1,3 (дигидрорезор-
цнн) (с. 484):
ОН O-Na+ ОН О
I I I II
/Чч
КаО[Г
ОН
<о
-Г
енольняя
форма
днксто-
форма
о
Гидрохинон представляет собой бесцветное кристаллическое
вещество с т. пл. 169—171 СС; возгоняется, растворим в воде.
Впервые гидрохинон получил Ф. Вс'лер в 1844 г. восстановле-
восстановлением «-хннона. я-Хинон получается при окислении различных орга-
органических веществ (с. 527).
Гидрохинон получают реакцией я-хлорфенола или я-сульфоно-
вой кислоты со щелочью при 280—320 'С и прямым гпдроксилнро-
325
ванием фенола:
ОН ОН он
/к ° /к /к/°П °
I H + RC-О —ОН —| || +| || + RC-OH
ОН
Гидрохинон является слабой двухосновной кислотой:
:ОН
I
:SoI ^
:So.
-|-HSol+ ¦
-| 2HSoI +
:ОН
О
В щелочной среде он легко алкилируется и образует простые эфиры.
Моноанион и особенно дианион необыкновенно легко окисляют-
окисляются. Промежуточным продуктом окисления является весьма ста-
стабильный анион-радикал (анион семихшгона), в котором неспарен-
ный электрон значительно делокализован:
•б-- -б- °
'Г V "
' /\
и
А
0
анион
семихинона
Конечггым продуктом окисления является п-хинон.
Гидрохинон легко окисляется даже раствором FeCl3; из его
водных растворов осаждаются черно-фиолетовые блестящие кри-
кристаллы, которые являются комплексом с переносом заряда между
электронодонором гидрохиноном и электроноакцеггтором хиноном
(с. 530):
Гидрохинон широко используют в качестве восстановителя в
фотографии (проявители), антиоксиданта и ингибитора полимериза-
полимеризации и как исходное сырье для синтеза красителей.
326
Пирогаллол представляет собой бесцветное кристаллическое
вещество с т. нл. 133—134 °С, возгоняется, хорошо растворяется
в воде.
Пирогаллол получают нагреванием галловой кислоты, которую
в свою очередь получают из растительных продуктов (танинов):
ОН ОН
АОн А
-ьсо.
Впервые этим методом пирогаллол получил К. Шееле в 1786 г.
Пирогаллол в щелочной среде чрезвычайно легко окисляется,
его растворы интенсивно поглощают кислород из воздуха. Этим
пользуются для анализа газовых смесей.
Применяется пирогаллол в качестве эффективного восстанови-
восстановителя и для связывания кислорода из газовых смесей. Применяют его
также в аналитической химии и для получения некоторых краси-
красителей.
Глава XVI
ПРОСТЫЕ ЭФИРЫ
Простые эфиры являются производными спиртов, енолов и фено-
фенолов, в которых водородный атом гидроксилыгой группы замещен
углеводородным остатком.
Рассматривать простые эфиры целесообразно в той же последо-
последовательности, что и гидроксилпроизводные углеводородов.
1. Простые эфиры, содержащие связь C(spa)—О:
а) диалкиловые эфиры С„Н2п+1—б—CmH2m+1, циклоалки-
ловые эфиры;
б) циклические эфиры
R2C — (СЙ2)„ :О—(CR2)n
V «Wfc Wh.§k
2. Простые эфиры, содержащие связь C(sp-)—О:
а) алкилалкениловые эфиры (виниловые эфиры)
R
и диалкениловые э<})иры (дивиниловые эфиры);
б) алкилариловые эфиры Аг—б—C,JJ2,1+1;
в) диариловые эфиры Аг—б—Аг1.
3. Простые эфиры, содержащие связь C{sp)— О, например алкил-
алкиниловые эфиры R—с = С—б— R1.
327
А. ДИАЛКИЛОВЫЕ ЭФИРЫ
I. ИЗОМЕРИЯ И НОМЕНКЛАТУРА
Образуя названия простых эфиров, называют оба заместителя у
кислородного атома и присоединяют слово эфир или же называют
эфир как производное углеводорода, рассматривая алкоксигруппу
OR как заместитель, причем в основе названия лежит наиболее
длинная углеводородная цепь. Например:
днметиловый эфир, пли метокснметап
днэтнловый эфир, пли этоксиэтап
пропнлпзопропнловый эфир, пли 1-нзопропоксппропан
CUsOCHj
СН5ОС2Н5
СН3СНОСНоСН2СН3
СНз
2. МЕТОДЫ ПОЛУЧЕНИЯ
Методы получения диалкнловых эфиров основаны на реакции
алкилирования алканолов или алкоксидов (см. гл. XIV.А.4).
Для алкилирования алкаиолов в промышленности применяют
также алкены в присутствии кислот:
R1
R-OH +
R1
k"«o"r_o-c-ch.
3. ФИЗИЧЕСКИЕ СВОЙСТВА И СТРОЕНИЕ
Простые эфиры являются бесцветными жидкостями (кроме димети-
лового эфира) со своеобразным запахом и весьма низкими темпера-
температурами кипения (табл. 35). Температура кипения простых эфиров
Таблица 35. Физические константы некоторых диалкнловых эфиров
Соединение
СН3ОСНЯ
СНЯСН2ОСН2СН3
СН3СНОСНСН3
1 1
СНяСНз
СНз
1
СНзОССНз
1
СН3
СН3ОСН2СН2ОН
СП3СН.,ОСН2СН2ОИ
сн3осн2сн2осн3
СН3ОСН2СН2ОСН2СН2ОСНз
Т. пл., °С
— 138,5
— 116,3
—60
—58
Т. кип., СС
—23,7
34,6
67,5
54
124,3
135,1
84,5
1C1
0,714
0,726
0,758
0,966
0,931
0,863
0,9514
328
Нч
Н''
ф?&^
с in0
л
н
с
1
н
значительно ниже, чем соответствующих алканолов, что свидетель-
свидетельствует о более слабом межмолекулярном взаимодействии, так как
отсутствуют группы ОН и межмолекулярные водородные связи.
Простые эфары имеют меньший диполыгый момент (п
- 410-;1° . . . 4,3- Ю-:|" Кл-м или 1,2—1,3 D), чем алканолы, но
обладают более сильными электронодонорпыми свойствами, о чем
свидетельствуют значения энергии ионизации (для этанола ЭИ-^ 10,6
эВ, а для диэтнлового эфира ЭИ-~9,6эВ).
Увеличение электронодопорных свойств объяс-
объясняется электронодонорным действием двух ал-
кпльных групп. Валентный угол СОС прнбш-
жается к тетраэдрическому и равен 109—112!.
4. ХИМИЧЕСКИЙ СВОЙСТВА
Реакции простых эфироз можно подразделить на три группы. Пер-
Первая группа охватывает реакции, происходящие у кислородного ато-
атома, вторая — реакции у а-углеродно го атома, третья —реакции
расщепления связи С—О.
1. Реакции у кислородного атома. Простые эфиры образуют про-
продукты присоединения с кислотами, в том числе кислотами Льюиса.
Известны продукты присоединения посредством водородной связи:
R .. 6+ й- Rv .. 6+ 6-
1
С сильными кислотами происходит протонировапие и образуются
диалкплоксониевые катионы:
R . .. Г R
",О: + Н4Х-—i О— II
х-
R1 1.R1
• Простые эфиры являются очень слабыми основаниями, т. е.
диалкилоксопиевые катионы представляют собой сильные ОН-
кислоты. Например, для диэтилового эфира рКви ¦ ~~—3,6. Это
означает, что диэтиловый эфир начинает протежироваться в ощу-
ощутимых количествах только в растворе 30—50%-ной серной кис-
кислоты.
С кислотами Лыоиса образуются весьма стабильные продукты
присоединения (эфираты), например:
(C.II5JO:->BF3; (C2H5J6:-^SnCl4; (C2II5)., 6:^MgCI2
В этих соединениях образуется донорно-акцепторная (координа-
(координационная) связь за счет иеиоделеиной пары электронов кислород-
кислородного атома.
Способность простых эфиров взаимодействовать с катионами
различных металлов (сольватировать катионы) имеет исключитель-
исключительное значение в получении металлорганических соединений в раст-
растворе диалкиловых эфиров или тетрагидрофурана.
В специальных условиях можно к кислородному атому присо-
присоединять карбонатном, в результате чего образуются соли триалкп-
329
локсоиия:
C2H5 .
)O:+C2H,-F-}-BFa-
>O-C2H5
тетрафторборат
трн^тнлокоония
Соли триалкнлоксоння являются бесцветными кристалличес-
кристаллическими веществами, при соприкосновении с влагой воздуха они рас-
расплываются и разлагаются (реакция с Н2О). Эти соли принадлежат
к наиболее сильным алкилирующим средствам. Они очень легко
реагируют даже со слабыми пуклеофнльными реагентами:
«+
R4-+ г+ R\ ¦ +
&( }О-*-К + :Nu »- )q: + R—Nu
R BF4~ K BFT
2. Реакции у а-углеродного атома. Простые эфпры могут и всту-
вступать в реакции свободнораднкалыюго хлорирования и автоокнсле-
нпя, причем, как правило, реакции проходят у а-углеродного ато-
атома. Свободные алкоксиалкильные радикалы более стабильны, чем
алкильпые, вследствие делокализации неснарепного электрона при
взаимодействии с неподелеппон электронной парой кислорода:
Н Н
..I ..I
R— О — С— Rl+-Y— >R — О — С — R'+HY
Н
hv
R—О — ClIaR^-Cli-^R — О — CHR' + HCl
С1
R—O — CHiR' + Oa—>R— О — CHR'
О —ОН
Продуктами окисления являются гпдропероксиды — нестабиль-
нестабильные и взрывчатые соединения. При хранении диалкиловых эфиров
с доступом воздуха, особенно на солнечном свету в прозрачных
бутылках, всегда образуется примесь гидропероксидов.
3. Расщепление связи С—О. Расщепить связь С—О в простых
эфнрах не так легко. Для этого требуются сильные основания или
кислоты и повышенная температура.
Практическое применение имеет расщепление при нагревании
с HI-кислотой, кислотами Льюиса (А1С13, А1Вг3, All3, ВС13, ВВг3,
SiCl3I) и (CH3KSiI. Наиболее активными реагентами являются
ВС13 и SiClJ (смесь SiCl4+NaI). Первая стадия реакции с ВС]3
включает образование эфирата и внутримолекулярное иуклеофиль-
иое замещение:
R C1
R1 <!,
330
В растворах сверхсильных кислот («сверхкислот» — см. гл.
1.4.3) при низких температурах генерируются карбокатиоиы, ко-
которые при более высоких температурах могут претерпевать различ-
различные перегруппировки и распад до алкенов:
R1
I
R_O —СН-СН3
R_O-CH-CH3
н
R1
I
;н ^сн.
Легче расщепляются простые эфиры, которые содержат развет-
разветвленные группы (изопронил-, трет-бутнл-).
5. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ
Диэтиловый эфир получают из этанола в присутствии концентри-
концентрированной серной кислоты при 140. . . 145'С (отсюда его старое на-
название «серный эфир») или каталитической дегидратацией спирта
над А1оО:! при 300 СС (при 370...400°С получается этилен).
Диэтиловый эфир представляет собой бесцветную жидкость со
своеобразным «эфирным» запахом, очень летуч, его пары тяжелее
воздуха. При неосторожном упаривании эфирных растворов пары
эфира могут накопиться над лабораторным столом или над полом и
воспламениться при соприкосновении с нагретой электроплиткой.
При хранении эфира в нем могут образоваться пероксиды.
Диэтиловый эфир плохо растворим в воде (до 6% при 20 °С),
а вода плохо растворима в диэтиловом эфире (до 1,2%). Если
необходим безводный эфир («абсолютный эфир»), то воду связывают
сначала безводным СаС12, а затем металлическим натрием.
Диэтиловый эфир хорошо растворяет различные органические
вещества, поэтому его широко применяют в качестве растворителя
для экстракции и для металлоргаиического синтеза. В медицине
диэтиловый эфир применяют для общего наркоза.
Диизопропиловый эфир получают алкилнровапием изопропнло-
вого спирта нропеном в присутствии BF3 (см. гл. XVI. А.2). Это
бесцветная жидкость с «эфирным» запахом. В технике его приме-
применяют в качестве добавки к бензину как антидетонатор.
Метил-трет-бутиловый эфир получают из метанола и 2-метил-
пропеиа (изобутилена) в присутствии кислого катализатора (см.
гл. XVI. А.2). Его применяют в качестве добавки к бензину (анти-
(антидетонатор).
Целлозольвы являются моноэфирамп гликоля B-алкоксиэта-
нолы). Получают их из этилоноксида и алкаполов:
R-OH + CHj —СИ,—¦ROCH2CH4OH
Y "
331
при R--CH3 — метплцеллозольв, при R—СоН:, — этилцеллозольв).
Целлозольвы являются хорошими растворителями, особенно
для растворения сложных эфиров целлюлозы.
1,2-Диметоксиэтан (диметиловый эфир гликоля) получают ме-
метилированием метилцеллозольва метанолом в присутствии кислоты:
СН3ОСН2С112ОН -'- СН3ОН — -* CI 13ОСН,СН,ОСН3 -;- Н.,0
Хороший растворитель, растворяет также некоторые неорганиче-
неорганические соли. Диметоксиэтап сильно сольватирует ионы металлов:
С Н ч ~~~ С Н 2.
/ \. CHj
м
Диглим (диметиловый эфир диэтиленгликоля, 2,2'-диметокси-
диэтиловый эфир) получают из метилцеллозольва и этиленоксида:
СН3ОСН2СН,ОН-|-СН2СН2 —* СН3ОСН2СН4ОСН4СН2ОН —>¦
\/
О
сн,он
--* СНяОС112СНоОСН2СН2ОСИ3
Диглим — хороший растворитель, сильно сольватирует ионы
металлов (донорный эффект трех кислородных атомов одновре-
одновременно):
сн., сн2
СН2 I" , СН2
\ i /
:о:-*-м*-:о:
Б. ЦИКЛИЧЕСКИЕ ЭФИРЫ
Циклические эфиры являются производными алкандиолов, в ко-
которых эфирная связь образовалась внутримолекулярпо.
Циклические эфиры подразделяются па несколько групп в за-
зависимости от величины цикла и числа кислородных атомов. Их
часто называют оксидами (окисями). Для некоторых циклических
эфиров применяют номенклатуру гетероциклических соединений:
R2C-CR2
\/ эпокснды (этиленоксиды, окспраим)
R.C-CR,
I I триметплепоксиды (оксетаиы)
R-iC CR. тетраметилснокснды (тетрагидрофу-
/ 2 рапы, оксолапы)
О
332
R,C-0
; i
R,C
ч/
о
CR2
R,C7 CR2
I I
RiC, /CR2
О
О О
R,C/4CR2 R
! I
R2C О R2C CR2
1,3-л,"оксоланм
пешаметиленокснды (тетрагпдропп-
рапы)
диоксаны
CR2
1,3-
О
1,4-
о
макроцпклпческие эфпры (краун-
эфчры)
[IS]-краун-6
Более подробно здесь рассмотрены эпоксиды и некоторые дру-
другие представители циклических эфиров.
1. ЭПОКСНДЫ (ЭТНЛЕИОКСИДЫ)
1. Номенклатура. Основу названия эпоксида составляет наименова-
наименование углероводорода, а па присутствие кислородного мостика ука-
указывает слово эпокси-. Для простейших соединений сохраняются
названия эпшленоксид и пропиленоксид. Применяется также но-
номенклатура гетероциклических соединении — «оксираиы». Напри-
Например:
СП3 —СН—СН —СН3 2,3-эпокспбутаи, 2,3-диметилоксираи
О
2. Получение. Эпоксиды получают методом внутримолекулярно-
внутримолекулярного алкилирования галогеналканолов и прямым окислением алкепов.
Галогеналкаиолы (галогепгндрины) в присутствии сильных
оснований превращаются в эпоксиды:
НИ Н Н ПН
II II II
1 RC1RCCR1
С1 ОН
II
Cl O-Na+
О
-J-Na + Cl-
333
Образование эпоксндов идет только при условии, что атомы
галогена н кислорода находятся в транс-положении. Эти стерео-
химические ограничения показаны на примере реакции 2-хлор-
циклогексанолов:
о + сг
:ОН
транс- 2-хлорциклогексанол
Гон]
Реакция отщаиения НО
+ НС!
ОН
цис - 2-июрци1слогексанол
Алкены окисляются иероксикислотами и другими органически-
органическими нероксидными соединениями (см. гл. II. 4.4). Известны методы
каталитического окисления кислородом воздуха (см. «Отдельные
представители»).
3. Физические свойства и строение. Эпоксиды являются бес-
бесцветными газами (этиленоксид) или жидкостями со слабым запахом.
Эпоксидный цикл представляет собой почти правильный тре-
треугольник со значительно деформированными валентными углами
(~60°). Поэтому происходит только частичное перекрывание атом-
атомных орбиталей и энергия связей уменьшается (сравните с циклопро-
циклопропаном):
Н2С CHj
;0
Эпоксидная группировка полярна, этиленоксид имеет дип'оль-
ный момент (д,=6,28-10~30 Кл-мA,88 D). Причинами этого являются
полярность связей С—О и небольшой угол СОС, тогда как в обыч-
обычных простых эфирах угол СОС равен 109—112° и |л=4-10~30. . .
. . .4,3-10-30 Кл-м A,2. . .1.3D).
4. Химические свойства. Химические превращения эпоксидов
определяются тем, что в молекуле имеются полярные связи С—О
и атом кислорода с неподеленнымн парами электронов. В принципе
реакции аналогичны многим реакциям простых эфиров с нулео-
фильными и электрофильными реагентами, только в случае эпокси-
эпоксидов эти реакции проходят очень легко. Связь С—О в эпоксидах раз-
разрывается легко, особенно в условиях кислотного катализа.
Со слабыми нуклеофнлами (например, Н2О) без катализатора
реакция идет только при повышенной температуре под давлением;
сильные нуклеофилы (аммиак, амины, металлорганические соеди-
334
t">100"C
пения) реагируют легко:
н н
R—*1С—с«±-R
:о:
:nh3
ОН
Н—СН—R
ОН
R—СН—СН—R
I I
NH2 ОН
R—СН-
1.
)MgX
Присутствие кислот значительно увеличивает реакционную
способность эпоксидов, так как увеличивается полярность связи
С—О. В присутствии серной или фосфорной кислоты легко присо-
присоединяются вода и алканолы:
н н
R—СН—СН- -R
| |
OR' ОН
h+hso7
R—СН— СН—R
I I
ОН ОН
+
В роли катализаторов могут выступать также ионы металлов.
Например, эпоксиды легко реагируют с водными растворами MgCl2,
при этом осаждается Mg(OHJ и образуется хлоралканол (нуклео-
фнлом в этой реакции является Cl~, a Mg'21' координируется с кис-
кислородным атомом эпоксида):
2R — СН — СН — R -|- MgCI2 + 2Н2О —¦ 2R — СН — СН — R + Mg (ОНJ
\/ I I
О ОН С1
Изучение стереохимии этих реакций показало, что всегда осу-
осуществляется т/адке-присоедипепие:
с—с
+ н++ н2о
:он2
J ,.
с'
Vd/i
А
он
I .
С —С
l
ОН
В несимметрично замещенных эпоксидах может быть различ-
различное направление расщепления эпоксидного цикла. Если реакция
проходит по механизму SN2, то нуклеофилом атакуется менее эк-
экранированный (замещенный) углеродный атом. Если в присутствии
кислот может образоваться стабилизированный карбокатион, то
осуществляется механизм SN\, и нуклеофил присоединяется по
335
карбокатионпому центру:
Clb
СНз
- Гн
V
СП.
с п.,о
П + ;
ОН
-Na +
СН,
\
¦С —СН..ОСН.,
сня/ |
он
ch3n + сн3ч
;с-С1ьон-> >с-сн.,он
сн3/ Clb/ |
НОСНз ОСНз
В безводных средах в присутствии кислот Льюиса легко проис-
происходит димеризаиия, олигомеризация и полимеризация:
Н Н Н Н .„/CHR r
л/
S--Q-
-R + BFj
?
BF3
R— СН—СН—Р — СН—СН—О— СН R
:o: - - 4
HR
"BFj
CHR
R—СН—СН—О—CHR
.о: +с1
BF3
R—]
R—НС
HR
H—R
+ BFj
1—R
R R
RCHCH+OCHCH-f-OCHCHOH
I I
OH L R R
5. Важнейшие представители. Этиленоксид — бесцветный газ
с эфирным запахом, т. кип. 10,7 "С, плотность dj--0,8909. Хорошо
растворяется в воде и органических растворителях.
Этиленоксид принадлежит к очень важным промышленным про-
продуктам — его производят более 3-10° т в год. В промышленности
используют хлоргидрипный метод и прямое окисление этилена:
С],; П.,О Na ЮН"
СП, =-. СН2 ——"-* СН2 — СН2 > СН2 —СН2
i | Н,О - /
ОН CI
300—4 00 СС; О,
Ag/A.,O3 -C"
О
о
Этиленоксид широко применяется в органическом синтезе,
особенно для получения цепных растворителей и поверхностно-
активных веществ (рис. 88).
336
н,с—сн2
сн^-сн,
ROCrf,CIT,OH
делло юльпы
RO(CH3CH:O)n—Н
повер xi юс тно-акн/вные
вещества
H0CH,CH:0CH,CHj0H
/ HOCHjCHjNH,
Растворители
НО—(СП,СН,О)П-Н
полиэптленоксид
СП-=СН—CN
акрИЛОМИТрИЛ
АгСН,СН,ОН
Синтетическое
волокно
(HOCH,CH:).N
этаио.шчины
Рис. 88. Применение этиленоксида
Пропиленоксид — бесцветная жидкость с т. кип. 34,5 °С и плот-
плотностью d\a--;0,859 (данные для рацемата). Молекула пропиленокси-
да хиральпа, содержит асимметрический атом углерода, известны
два энантиомера и рацемат.
Получают пропиленоксид из пропела по хлоргидринному
методу:
CII3-CH-CH2
CI.tlKO
Na + OH-
I I
Oil С1
СНЯ-СН —СН2
N/
О
Каталитическое окисление пропена кислородом воздуха над
серебряным катализатором не дает желаемого результата. Найден
метод окисления пропена гидропероксидамн в присутствии я-ком-
плсксов молибдена:
Кат.
СН3 -СП- CUH-R —О — Oil -
гидролероксид
СН —СН2 J-ROH
О
Производят пропиленоксид в крупных .масштабах (более 2• 106 т
в год).
При полимеризации в присутствии катализатора получают
иолнпропиленоксид с различной молекулярной массой. Благодаря
наличию в молекуле нолипропилеиоксида двух концевых ОН-групп
елнгомеры полипропиленоксида используются для получения дру-
других полимеров (полиуретаны, пенопласта). Нго используют также
м качестве поверхностно-активного вещества и как исходное для
различных синтезов (рис. 89).
Эпихлоргидрин C-хлор-1,2-эиоксипропаи) представляет со-
собой бесцветную жидкость с запахом хлороформа и кип. 110 "С,
плотность df—1,1807 (данные для рацемата).
337
сн.^сн—сн
но он
j—СН—СН2
V/
>- но-|-сн—сн—о-^-н
нропионовыи
альдегид
сн3
полипропиленоксид
сн2=сн—снрн
аллнловый спирт
ацетон
Неопреновый каучук
Рис. 89. Применение пропилепоксида
Глицерин
Получают эпихлоргидрин из аллилхлорида по хлоргидрин-
ному методу:
СН2=,СН-СН2С1
I
ОН
СИ2-СН-СП2С1
/
О
Молекула эпихлоргидрина содержит две активные группиров-
группировки — эпоксидную и связь С—С1.
Эпихлоргидрнн служит промежуточным продуктом в синтезе
глицерина. Его широко применяют для получения эпоксидных
смол. Сначала при взаимодействии с бис-фенолами получают ак-
активный мономер, который при взаимодействии с другими молеку-
молекулами бис-фенола дает полимер:
СП3
2СН2 —СН—СН2С1 + 1Ю—<f ^—С — <^_/ — ОН —
СНз
СНз
о
¦CH.-CH-CH,O-<f"
ч/ " \=
о
*СН3-СН—СИ—
ч/
о
СН3
СНз
сн3
СНз
-ОСНо-СН-СН,-...
сн.
•^_О-С112-СН-СИ2—
=/ I
он
-он
Этот полимер содержит активные эпоксидные группы, поэтому
в присутствии некоторых реагентов (диаминов) происходит сшива-
338
иие линейных макромолекул (процесс отверждения), что увеличи-
увеличивает термическую и механическую стойкость полимерного мате-
материала.
2. ТЕТРЛГИДРОФУРЛН
Тетрагидрофуран получают из бутиндиола-1,4 или каталитическим
гидрированием фурана (гл. XXXVI. А.З):
ПОСН2-С: .С-СН2ОН
»"f/N1 u,x- H2C-CHJn./NI НС-СН
НОСН2СН2СН2СН2ОИ > I I ^— !1 (¦'
> юо-с н2с СН2 НС СН
о о
Тетрагидрофуран представляет собой подвижную бесцветную
жидкость с запахом эфира, т. кип. 65,7 СС, плотность df- 0,8892.
Растворим в воде и органических растворителях. С водой образует
азеотроппую смесь, кипящую при 64 °С и содержащую 6% воды.
Тетрагидрофуран хорошо сольватирует ионы различных ме-
металлов и способствует образованию металлорганических соедине-
соединений. Он широко применяется в качестве растворителя п также как
исходное для синтеза некоторых важных мономеров:
i Окисление
"CH2=-CH —CH-CH.2-—
ноос/ хсоон
янтарная кислота
HCI; 180 "С
СО/Н2; /°; клт.
с,с„,сн!с„гсн,с, „оосс„,с1„,а,,с„1сооН
эдипиновая кислота
3. ДНО КС АН
Диоксан получают из этиленоксида или этиленгликоля в присут-
присутствии катализаторов:
О
С1ШНц + х-Н2С СП., ,СН.,
2 | " > | | " *— 2 О< |
сн2он-21^°н2с сн2 хсна
О
длоксан
Он представляет собой бесцветную жидкость со слабым запахом;
т. пл. 11,8 °С, т. кип. 101,3 СС, хорошо растворим в воде и органи-
органических растворителях. С водой образует азеотропную смесь, кипя-
кипящую при 87,8 СС и содержащую 18,4% воды.
339
Диоксан образует кристаллические донорно-акцеиторные ком-
комплексы с рядом неорганических веществ (Br2, SO3, IC1 и др.):
/ ч / •
:О. :О:— > Вг2 :О: :О: —> SO3
\ / -¦• /
дпсжеандибромпд диоксансульфотриоксмд
Эти комплексы являются хорошими реагентами для бромирования
и сульфирования. Комплекс диоксан->-1С1 служит для иодирова-
иодирования.
4. КРЛУН-ЭФИРЫ
Краун-эфнрами называют макроциклические полиэфиры с четырьмя
и более кислородными атомами в цикле. В большинстве случаев
они являются производными этиленгликоля. В названии соеди-
соединений цифра в квадратных скобках указывает число атомов в мак-
макроцикле, а вторая цифра — число кислородных атомов:
/-о-\
[15]краук-5 [18]краун-6 [21]краун-7
Краун-эфиры синтезируют алкилированием этиленгликоля,
диэтилен гликоля НОСН2СН2ОСН.2СН2ОН, трнэтиленгликоля
НОС112СН2ОСН2СИ.2ОСН2СН2ОН подходящими реагентами, на-
например 2,2'-дихлордиэтиловым эфиром О(СН2СН2С1J. Простей-
Простейшие краун-эфиры являются бесцветными густыми жидкостями или
низкоплавкими кристаллическими веществами, растворимыми как
в углеводородах, так и в воде.
Самое важное свойство краун-эфиров — образование комплек-
комплексов с ионами металлов:
Ион металла находится в полости макроцикла и крепко удер-
удерживается благодаря донорно-акценторным связям R26:-4-Mn<".
Диаметр полости в макроцикле зависит от структуры краун-эфира.
Например, для [15]крауп-5 он равен 0,17—0,22 им, для [18]крауп-6
0,26—0,32 нм, для [21]краун-7 0,36—0,43 нм. Поэтому краун-
эфиры с различными ионами металлов образуют комплексы с раз-
разными константами устойчивости. Чем ближе ионный диаметр ме-
металла к диаметру полости макроцикла, тем устойчивей комплекс.
340
Так, [151краун-5 больше подходит для ионов натрия, а [18]краун-6—
для ионов калия.
Краун-эфиры используют для улучшения растворимости неор-
неорганических солей в органических растворителях, в качестве меж-
межфазных катализаторов (см. с. 75), для генерации несольватирован-
ных анионов в органических растворителях. Например, при по-
помощи [181краун-6 можно растворить КОН в бензоле, ион ОН~
в этих условиях обладает большей активностью, чем в растворе
воды или метанола. Соединение типа краун-эфиров играют боль-
большую роль в биологических системах — они осуществляют тран-
транспорт ионов через биологические мембраны (см. криптанды, гл.
XXIII.А.4).
В. ВИНИЛОВЫЕ ЭФИРЫ
1. Номенклатура. Названия соединений образуют по общим прави-
правилам (см. гл. XVI. АЛ).
CII3CHaOCH--CHi этоксиэтилен (этоксиэтеп), випилэтиловый эфир
СН3ОС- СНСН.СНз 2-метоксипентен-2
1 I
СН3
2. Методы получения. Основным методом получения является
присоединение алканолов к алкинам (гл. IV.4). Можно использо-
йать также галогендиалкиловые эфиры:
t° NaOH (конц.) ROCH — СН3
ROH-I-CH--CH -ROCH-CH2- i
Кат. ^
3. Физические свойства и строение. Виниловые эфиры являются
бесцветными жидкостями со слабым раздражающим запахом. Ви-
Випилэтиловый эфир кинит при 36(С, <fi°^-0,7531, а бутилвиниловый
эфир — при 93,8 С, d\a---0,7792.
В отличие от диалкиловых эфиров виниловые эфиры в моле-
молекуле имеют простую сопряженную систему — л-электроны двой-
двойной связи и иеподелениая пара электронов кислородного атома:
,С С R
Н' Н
4. Химические свойства. Реакции виниловых эфиров определя-
определяются главным образом наличием поляризованной двойной связи.
Это в основном присоединение к двойной связи электрофильных
реагентов и полимеризация. Реакции присоединения облегчены
благодаря электроподонорному эффекту алкоксигруипы:
CHj —СН —OR -« СН2 = СН—OR >¦ СНо —СН —OR
I " " I ' I
х С1 С1
341
В реакциях присоединения кислот в качестве промежуточной
частицы может образоваться алкоксикарбокатион, который ста-
стабилизирован вследствие делокализации положительного заряда
при участии кислородного атома:
СНз-С/ R<->CH3-C;f "NR
При взаимодействии с кислотами, которые имеют малонуклео-
фильные анионы, и кислотами Льюиса происходит полимеризация
по карбокатионному механизму:
/т. 6+ :'or Г>-ь+
СН3—C^"^R +5ClT^=CH^pR ¦- СНз—С—СН2—C^"XR *¦
"
OR
OR
CH2=CHOR -HX I I
*¦ • • *- СНз—С- -CH2—CH- -CH=CHOR
Н L \п
Поливиниловые эфиры применяются в промышленности полимер-
полимерных материалов.
Реакции виниловых эфиров с кислотами в водных растворах
приводят к производным альдегидов — полуацеталям, которые
легко гидролизуются до альдегидов:
от
ch^c^9^r + н+х- + н2о =^=^ сНз-ic^-^R + :о^ -^
х- н . »
В растворах алканолов образуются ацетали (гл. XXVII. Д.4).
В заключение следует отметить, что реакции виниловых эфи-
эфиров аналогичны реакциям, характерным для алкенов. Обычные
реакции, присущие простым эфирам, для виниловых эфиров мало
характерны.
Г. АЛКИЛАРИЛОВЫЕ ЭФИРЫ
1. Номенклатура. Алкилариловые эфиры называют как производ-
производные аренов (алкоксиарены) или как алкилариловые эфиры, приме-
применяются также некоторые названия:
ыетоксибензол, метилфениловый эфир, анизол
осн3
//'\/осн'
| 1,2-диметоксибензол, вератрол (диметиловын эфир пирокате-
пирокатехина)
V
2. Методы получения. Основным методом является алкилиро-
вание фенолов галогепалканами или другими алкилирующими реа-
реагентами (диалкилсульфатами, см. гл. XVII. 7) в присутствии осно-
оснований. Известно также алкнлирование фенолов алканолами и ал-
кепами в присутствии кислых катализаторов, по в этом случае об-
образуются в качестве примеси алкилированные в цикле фенолы
(см. гл. XV. Б.4).
3. Физические свойства и строение. Алкилариловые эфиры яв-
являются бесцветными жидкостями или кристаллическими вещест-
веществами с приятным запахом.
Полярность связей, направление диполыю-
го момента, распределение электронной плот-
плотности в алкилариловых эфирах напоминает
фенол.
Алкоксиарепы, подобно фенолам, обладают
электроподопорными свойствами.
4. Химические свойства. Реакции алкила-
я=4,5-1о0 кл-и (i,35D) риловых эфиров определяются повышенной
полярностью связи О—R, что облегчает
расцепление эфирной связи, и повышенной электронной плотно-
плотностью в ареиовом цикле, что позволяет осуществить реакции электро-
фпльного замещения. В основе этого лежит сопряжение неподелеп-
ной электронной пары кислородного атома с я-электронной систе-
системой арена.
Основность алкилариловых эфиров сильно понижена по срав-
сравнению с диалкиловыми эфирами, рЛ^вн + ж—6. . .—7. Они начи-
начинают заметно протонироваться только в 60—70%-ной серной кис-
кислоте. При этом протонирование возможно как у атома кислорода,
так и у углеродного атома цикла:
+ н+х"
н н
В протонировашюй форме связь О—R становится еще более
полярной и облегчается ее гетеролитнческий разрыв:
R—X (Х = Вг,1)
343
Алкилариловые эфиры хорошо расщепляются при нагревании
с кислотами HI, НВг, с кислотами Льюиса (АПЯ, А1Вг3) ВВг3).
Алкилариловые эфиры очень легко галогенируются, сульфи-
сульфируются, нитруются, алкилируются, ацилируются в о- и «-положе-
«-положениях бензольного никла.
5. Важнейшие представители. Анизол (метилфениловый эфир) —
бесцветная жидкость с приятным запахом, т. кии. 155 С, df —
— 0,994, в воде не растворяется. Получают его метилированием фе-
фенола диметилсульфатом в присутствии щелочи. Используют в ор-
органическом синтезе и в качестве растворителя, особенно в парфю-
парфюмерной промышленности.
Гваякол (монометиловый эфир пирокатехина, о-метоксифенол) —¦
бесцветное кристаллическое вещество с т. нл. 98,4 С, т. кип.
205 С. Его получают метилированием пирокатехина. Используют
для синтеза душистых (ванилин) и лекарственных веществ.
Всратрол (диметиловый эфир пирокатехина) — бесцветное ве-
вещество с т. нл. 22,5 С и т. кип. 206,5 С. Его получают метилиро-
метилированием пирокатехина. Используют в органическом синтезе для
получения душистых веществ.
Д. ДИАРИЛОВЫЕ ЭФИРЫ
1. Номенклатура. Для диариловых эфиров применяют номенкла-
номенклатуру эфиров; иногда их называют диарилоксидами:
дифениловый эфир (днфенилокенд)
3,4,3',4'-тетраметилдифениловьш
эфир
2. Методы получения. Диариловые эфиры образуются при ари-
лировании фенолятов галогенаренами. Реакция требует весьма
жестких условий B00—300 С), ее ускоряет присутствие меди и
солей одновалентной меди (Ф. Ульман, 1903—1905):
200 — 300 °С
ЛЮ-Ха+-!-Аг' — X >ЛгОЛг' —Na + X- (X^I, Cl, Br)
Легче всего реагируют иодарены.
Диариловые эфиры образуются в качестве побочного продукта
в синтезе фенолов из галогепаренов и щелочи (см. гл. XV. Б.2).
3. Физические и химические свойства. Диариловые эфиры пред-
представляют собой бесцветные кристаллические вещества со слабым
приятным запахом и высокими температурами кипения. В воде
не растворяются. Являются термически устойчивыми соединениями.
В молекулах диариловых эфиров осуществляется значительное
взаимодействие неподеленной электронной нары кислородного ато-
атома с двумя я-электронными системами бензола.
344
Протежирование молекулы обычными кис-
лотами практически не происходит, расщепить
5- связь С—О не удается. Только при темпера-
туре выше 220 С при взаимодействии с А1С13
диариловые эфиры постепенно расщепляются.
Для диариловых эфиров известны реакции галогепирования,
нитрования, сульфирования, алкилировапия, ацилирования.
4. Дифениловый эфир — бесцветное кристаллическое вещество
с запахом герани, т. ил. 26 С, т. кип. 259 С, в воде не растворя-
растворяется. Термически устойчивое соединение, не изменяется при нагре-
нагревании до 400 С. Его применяют в качестве теплоносителя в раз-
различных отраслях промышленности, в основном в виде смеси с бифе-
нплом (даутерм).
Глава XVII
ЭФИРЫ НЕОРГАНИЧЕСКИХ
И ЭЛЕМЕНТОРГАНИЧЕСКИХ КИСЛОТ
Эфиры неорганических и элемепторгаиических кислот являются
производными спиртов и фенолов и образуются при замещении
атома водорода в гидроксилыюй группе остатком неорганической
или элемеиторгаиической кислоты: R—О—ЭХЯ. Здесь ЭХЯ — ос-
остаток кислоты, где X — галоген, кислород, сера, углеводородные
остатки, а Э — В, \', О, Si, P, S и др.
Ниже рассмотрены некоторые типы таких эфиров в последова-
последовательности, соответствующей увеличению атомной массы Э.
А. БОРАТЫ
Боратами называют эфиры борной кислоты:
OCI13
СН,О—В —ОСПз
три метилборат (трпметоксибор)
Бораты получают из борной кислоты и алкаполов в присутст-
присутствии сильной кислоты или из галогенидов бора и алкоксидов (фено-
(фенолятов):
[I _| Y —
B(OH):i-)-3ROH v
BCl2 + 3Ar0-Na+ —* В
Триалкилбораты — легколстучие жидкости, окрашивающие
пламя в зеленый цвет. Это явление используют в аналитической
химии.
Бораты способны присоединять нуклеофильиые реагенты, так
как атом бора имеет незаполненную орбиталь:
OR
I
OR
OR
RO —В—OR
I
OR
тетраалкилборат натрия
345
Четырехзамещенные соединения бора имеют тетраэдрическую кон-
конфигурацию.
Особенно легко тетраалкилбораты образуются из многоатом-
многоатомных спиртов — гликолей и глицерина:
сн2—сн
/ \
СН2ОН НОСН2
| + В(ОНK + I
сн.он носн2
сн2—сн2
В результате взаимодействия гликоля и борной кислоты обра-
образуется кислота, анионом которой является бис-этилепборат. Эта
кислота более сильная, чем борная, поэтому реакция ее образова-
образования используется в аналитической химии для количественного оп-
определения борной кислоты.
Б. НИТРИТЫ
Эфиры азотистой кислоты называются нитритами:
C2II3O — N-0 C4HUO — N--O
этилнптрит бутилнитрит
Нитриты легко образуются при взаимодействии алканолов или
других спиртов с неорганическими нитритами в присутствии кис-
кислоты при пониженной температуре:
ROH + Nai\O, H*S°,4 RO-N -О
о°с
Действующими реагентами здесь являются азотистая кислота
и катион нитрозоиия (сравните гл. XXIII. А.З, гл. XXIII. В.З):
HNOH-H+HSO- ^± [H2NO2] ь HSO- ^zi H2O + N'O <-HSO-
Катион нитрозоиия как электрофильная частица взаимодейст-
взаимодействует с алканолом:
1 +
R—OH + NO + HSOj—* R — О/ HSOr^lR— О — N = O + H2SO4
Простейшие нитриты являются легколетучими жидкостями
(например, этилнитрит кипит при 17°С) с приятным запахом.
Нитриты являются термически нестабильными соединениями,
при нагревании легко разрывается связь О—N (термохимическая
энергия связи только ~175 кДж/моль):
R — 0 — N = 0—>RO. + -NO
При взаимодействии нитритов с нуклеофилышми реагентами
могут образоваться два продукта реакции, так как молекула иит-
346
рита имеет два электрофильиых центра:
г+
Г R N
9. .p-
г+ *t>
М+КОГ + R-Y -* М+УГ R N *- Y-N=O + ROM
9
Нитриты могут быть как алкилирующими, так и нитрозирую-
щими реагентами (группа —N=0 называется нитрозогруппой).
Особенно легко нитрозирование идет в присутствии кислот,
которые вызывают гидролиз нитритов и образование производных
нитрозония:
R —б —N=O + H|-X- ^± fR— б — N-=01 + Х- ^ ROH-fNO+X-
fR— О — N-=01 +
[ A J
Нитриты (например, амилнитрит) применяют в органическом
синтезе в качестве нитрозирукщих реагентов. Они являются фи-
физиологически активными соединениями — их используют в меди-
медицине для расширения сосудов и резкого понижения кровяного
давления.
В. НИТРАТЫ
Нитратами называют эфиры азотной кислоты:
C2H5ONO2 — эти л п птр ат
Названия производных многоатомных спиртов образуют из назва-
названия спирта, например тринитрат глицерина.
Нитраты получают взаимодействием алканола (диола, триола
и др.) с азотной кислотой в присутствии серной кислоты. Нитру-
Нитрующей частицей здесь, как и в реакциях нитрования аренов,
является катион нитрония NO.,'HSO4~:
Г /н 1 +
R—бН-1-NO.^HSOr 7^ R — б( HSOr ^ R — б— NOo-fH,SO4
L 4N0j " "' "
Г /н
7^ R — б(
L 4N0
Нитраты — бесцветные или слегка желтоватого цвета вещества,
обычно жидкости с приятным запахом.
Нитраты термически нестабильны. Если в молекуле вещества
несколько нитратных группировок, оно разлагается со взрывом.
Первичным процессом разложения является гемолитический раз-
разрыв слабой связи N—О:
R —О—NO2 —>- RO- + -NO2
Тринитрат глицерина («нитроглицерин») легко образуется из
глицерина и смеси азотной и серной кислоты:
СНа—СН —СН2
ONO2 ONO2 ONOa
тричитрат глицерина
347
Это маслянистая бесцветная жидкость, ее плотность d;J —1,596.
Одно из мощнейших взрывчатых веществ, которое взрывается от
сильного нагревания н от удара. Непосредственное использование
нитроглицерина в качестве взрывчатого вещества очень опасно;
на его основе изготовляют динамит и другие взрывчатые материалы.
Нитроглицерином пропитывают пористые минералы, древесную
муку, древесный уголь, нитрат натрия. При растворении в нитро-
нитроглицерине нитроцеллюлозы получают желатинизированный нитро-
нитроглицерин, который тоже применяют для изготовления динамита
и пороха (А. Нобель, 1870 —1880).
Нитроглицерин применяется в медицине для расширения крове-
кровеносных сосудов и кратковременного понижения кровяного давле-
давления. В больших дозах он вызывает противоположный эффект и
сильные головные боли.
Тетранитрат пентаэритрита — бесцветное кристаллическое ве-
вещество с т. ил. 141... 142 СС. Одно из мощнейших взрывчатых
веществ, взрывается от сильного нагревания и от удара. Приме-
Применяют в военной технике.
СН2О\'О2
I
O2N'OCH2 — С — СН2ОМО2
CH,ONO2
Оказывает физиологическое действие, подобное действию нитро-
нитроглицерина.
Нитраты целлюлозы («нитроцеллюлоза») образуются из цел-
целлюлозы (гл. XXIX. В,5).
Г. ГИДРОПЕРОКСИДЫ И ПЕРОКСИДЫ
Гидронероксиды и нерокснды являются органическими производ-
производными неорганической кислоты — пероксида водорода НООН:
СНЯ —О—ОН метилпгдроперокспд, гпдроперокспметан
СНЯ
СН:1 — С— О — ОН /ирет-бутмлгпдроперокснд
СН3
СН3
<^ ^— С —О —ОН кумплгпдропероксид
СН3
СН3 СНЯ
I I
СН;; — С — О — О — С — СНЯ Д11-т/)<?т-бутплперокспд
I |
сн3 сн3
Гидроперокснды и нероксиды образуются при алкилировашш
пероксида водорода или его солей. В присутствии сильных кислот
алканолы алкнлируют 80%-ный или более концентрированный
348
пероксид водорода:
ROH-; ПООН — - ROOII j Н.,0
RO!I-r ROOII ' -ROOR-; Н.О
Наибольшей алкилирующей способностью обладают третичные
спирты, что согласуется с большей устойчивостью разветвленных
карбокатионов.
Удобно алкилировать пероксид водорода в щелочной среде
сильными алкилирующимн реагентами (например, диал кил суль-
сульфатам и):
д - й-
НОО-\а+-; R —OSO..OR --+ ROOII ;-\а" "OSO,OR
В некоторых случаях гидропероксиды удается выделить при
окислении углеводородов (см. с. 98).
Гидропероксиды и нероксиды представляют собой бесцветные
жидкие или кристаллические вещества, очень нестойкие. Многие
при сильном нагревании или ударе разлагаются со взрывом. Раз-
Разложение обычно начинается гемолитическим разрывом связи О—О.
Эта связь является наиболее слабым местом в молекуле. Энергия
связи только ~143 кДж'моль (слабее связи 1\г—О)
R — О —О — R1 - * RO-i-.OR1
Гидропероксиды являются слабыми ОН-кислотамн (сравнимы
по кислотности с фенолами), в щелочной среде их можно алкили-
алкилировать и ацилировать.
При термическом разложении гидропероксидов образуются
свободные радикалы, которые инициируют полимеризацию нена-
ненасыщенных соединений и другие реакции. Сами алкокснльные ра-
радикалы могут претерпевать различные дальнейшие реакции —
образование карбонильных соединений, перегруппировки:
II Н
! г I
R—С —О —ОН - у R—С —О- i-.OII ¦-* R—С -О-|-П2О
I I I
II Н Н
R R
I г I
R —С —О —ОН -^ R —С —О- ---ОН —>¦ R —С- О-: ROII
R R R
Интересные внутримолекулярные перегруппировки гидроперок-
ендов могут осуществляться в кислой среде:
СНз СНз СНл
Аг —С —О —ОН + H+HSOT ^ * Аг—'С. ^
U" " J.o->fн
н Hsor
CHj СНз
¦—*¦ Ar\p;/C^o- H+Hsor
н
34')
Эта реакция имеет важное значение для получения фенолов (ме-
(метод Удриса — Сергеева).
Трет-бутилгидропероксид —¦ бесцветная жидкость с острым за-
запахом, т. кип. 35—37 "'С (при 2,3—2,4 кПа). Получают его из трет-
бутилового спирта. Взрывается при температуре выше 60 СС. Ис-
Используют в качестве инициатора для реакций полимеризации и как
окислитель при получении эпоксидов.
Кумилгидропероксид — бесцветная жидкость с т. кип. 60 °С
B6,6 Па). При 170 СС взрывается. Получают окислением кумола
кислородом воздуха. Применяют главным образом для получения
фенола и ацетона по методу Удриса — Сергеева.
Ди-трет-бутилпероксид — бесцветная жидкость с т. кип. 111 °С.
Получают его из mpem-бутилгидропероксида. Широко применя-
применяется в качестве инициатора полимеризации.
Д. ЭФИРЫ КРЕМНИЕВОЙ КИСЛОТЫ
И КРЕМНИЙОРГАНИЧЕСКИХ КИСЛОТ
Кремниевая кислота и кремнийорганические кислоты образуют
ряд эфиров. Например:
ОСИЭ ОС2Н5 С6Н3
СН3О —Si-ОСНз СПз-Si —OG,H3 C6II., —Si-OCII3
ОСН3 ОС2Н3 ОСН3
тетраметоксисилан метнлтриэтоксиснлан дифеиилдиметоксисилаи
Получают эфиры этих кислот взаимодействием галоген производ-
производных кремния и гидроксилпроизводных углеводородов в присут-
присутствии оснований:
/iROH + XnSiRi_n + «:B —>¦ (RO)nSiR4-n + rtHB-* X~
Тетраэтоксисилан — бесцветная жидкость с т. кип. 165,5 СС,
плотность d40—0t933. В воде не растворяется.
В присутствии кислот или оснований тетраэтоксисилан посте-
постепенно гидролизуется, в результате чего образуются высокомоле-
высокомолекулярные соединения со связью Si—О—Si (силоксаны):
2(C2H3OLSi ^> 2(C2H5OKSiOH ->- (C2H5OKSi-O-Si(C2H5OK —
OC2H5i
(C2H3OKSi-
— О—Si —
ОС,Н, J
— O-Si(OC2H5K
При гидролизе в избытке воды образуются также кремниевая кис-
кислота и SiO2.
Тетраэтоксисилан является также исходным веществом для син-
синтеза различных кремпийорганических соединений.
Диметилдиэтоксисилан — бесцветная жидкость с т. кип. 111 °С,
плотность d|°=0,890. При гидролизе диметилдиэтоксисилана об-
350
разуются полисилоксаны:
СН3 СН3
| Н.О |
(я-|-2)С2Н5О—Si —ОС2Н5 —' С2Н5О —Si —
СН3
СН;
СН, -1
— О — Si—
I
сня
_O-Si-OC2H5
I
СН3 An СН,
Свойства полисилокеанов рассмотрены в гл. XII.
Е. ЭФИРЫ ФОСФОРНЫХ И ФОСФОРОРГАНИЧЕСКИХ КИСЛОТ
Возможны различные типы эфиров фосфорных и фосфорорганиче-
ских кислот. Наиболее важные из них:
а) фосфиты P(ORK, например триэтилфосфит Р(ОС2Н5K;
б) фосфаты O = P(OR)a, например трифенилфосфатО^=Р(ОС6Н5K;
в) тиофосфаты S=P(ORK, например триметилтиофосфат
S = P(OCH3K;
О
I!
г) алкилфосфонаты Alk —P —OR, например диметилметилфос-
X
фонат, или диметиловый эфир метилфосфоновой кислоты:
О
II
СНз-Р —ОСНз
осн.,
Эфиры этих кислот получают главным образом из галогенпро-
нзводных фосфора и гидроксилпроизводных углеводородов в при-
присутствии оснований, например:
3ROH'-! РС13-( 3:В -^ P(ORK-j ЗПВ'С!-
R'OII
ROH-rX, PCl3r=B -> Xr--PC12(OR) ;-HBlCl-— - X.--PC1(OR)(OR])
Эфиры алкнлфосфоновых кислот получают из фосфитов и гало-
гепалкапов перегруппировкой Арбузова (см. гл. XIII. 2.3). Ниже
рассмотрены некоторые наиболее важные представители.
Триэтилфосфит — бесцветная жидкость с т. кии. 57 С'С B,5
к11а). Это одно из основных исходных веществ для получения дру-
других соединений фосфора. При его окислении образуется триэтил-
фосфат, при взаимодействии с серой —¦ триэтилтиофосфат, в реак-
реакции с галогепалканами образуются эфиры алкилфосфоновых кис-
кислот.
Диэтил($-этилмерк.аптоэтил)тиофосфат (меркаптофос)
S=P(OC2I I5J(OCH2CH2SC2H3) является эффективным инсектицидом.
Изопропиловый эфир фторангидрида метилфосфоновой кислоты
(зарин)
О
СН3-Р--О-СН(СНз),
I
F
351
представляет собой бесцветную жидкость с т. кип. 151,5 °С, плот-
плотность di°-1,094. Растворим в воде. Является сильнейшим ядом
нервно-паралитического действия для всех теплокровных организ-
организмов. Смертельная концентрация для человека 0,2 мг в 1 л воздуха.
Пинаколиповый эфир фторангидрида метилфосфоновой кислоты
(зоман)
О СН,
I I
СНз-Р —О —СНС(СН3)з
F
бесцветная жидкость с т. кип. 42 °С B6 Па), плотность df= 1,013,
мало растворим в воде. Сильнейший яд нервно-паралитического
действия.
Ж. ЭФИРЫ СЕРНОЙ КИСЛОТЫ
Серная кислота образует два ряда эфиров — гидросульфаты и
сульфаты:
О
il
СН3О — S — ОН метилгидросульфат (метнлсульфат)
,1
О
О
1
СН3О — S — ОСН3 диметилсульфат
il
О
Образование кислых эфиров (гидросульфатов) происходит очень
легко при растворении спиртов в концентрированной серной кис-
кислоте (фенолы и другие аренолы так не реагируют, недостаточна
О-основность):
R-O/
L "чн
О
R — OH + H + HSO; ;r± R — O( HSO7 — >¦ R—O —S—OH -VUO
1 HJ
о
Гидросульфаты образуются также при растворении алкепов в кон-
концентрированной серной кислоте (гл. П. 4.2).
Алкилгидросульфаты являются сильными кислотами и обра-
образуют соли. При взаимодействии с нуклеофильными реагентами
они выступают в качестве алкилирующих реагентов, так как со-
содержат сильнополярную связь R—О:
о
R-*-O —S —O:"Naf -\ -.Nil *- R —Nu -\- SO,~Na+
352
Дпалкплсульфаты образуются при взаимодействии алкаиолов
с хлорсульфоновон кислотой или олеумом:
О О
;i
2ROH : О—S —ОН —, RO —S —CR-'-HCl ; 11,0
О О
Дпалкплсульфаты принадлежат к сильным электрофпльпым
реагентам, так как содержат полярную спязь С—О. Их исполь-
используют кук алкилирующпе реагенты:
б- .г><1? <-.. «+¦ ?
R-^O —S —O-^R + '-В *- ROSOi R — Bv
" ill "
¦Р/
Диметилсульфат — бесцветная жидкость с т. кип. 188,5 С,
плотность d\~' 1,3332. Мало растворим в воде B,8"и), хороню раст-
растворим в органических растворителях.
Получают дпметилсульфат из метанола и олеума. Он широко
применяется в качестве метилирующего реагента.
Диметилсульфат очень ядовит, проникает в организм как через
дыхательные пути, так и через кожу.
Диэтилсульфат представляет собой бесцветную жидкость с
т. кип. 210 С, плотность d\* -1,1812. В воде не растворяется. По-
Получают его из этанола и олеума или насыщая концентрированную
серную кислоту этиленом под давлением 1 .MI la A0 агм).
Дпчтнлсульфат широко применяют в качестве этмлпрующего
реагента. Он очень ядовит.
Сераорганические соединения
Соединения, содержащие в молекуле связь С—S, обычно называ-
называются сераорганпчеекпми соединениями.
Эти соединения рассмотрены здесь в последовательное] и от
высших степеней окисления атома серы к более низким.
1. Сульфоповые кислоты, их производные и сульфопы:
О О О
I
R —.S — ОН R—S — X R— S-R1
6 О О
2. Сульфнповые кислоты, их производные и сульфсжепды:
о о о
i И
R—S —ОН R-S —X R—S — R^
12 ль 5 17 353
3. Сульфеновые кислоты и нх производные:
R-S-OH R-S —X
4. Тиолы (меркаптаны): R—S—Ы
5. Сульфиды, дисульфиды и полисульфиды:
R_S-Ri r_s —S —Ri R — [— S— ]„ — Ri
Известны также сераорганические соединения со связью С—S,
например тиокарбонильные соединения, тио- и дитиокислоты и
пх производные — сложные эфпры, амиды (гл. XXXIII.3):
R. ,S /S
>C=.S R-Of R-C^
Глава XVIII
СУЛЬФОНОВЫЕ КИСЛОТЫ,
ИХ ПРОИЗВОДНЫЕ И СУЛЬФОНЫ
Сульфоповые кислоты и сульфоны можно рассматривать как про-
производные серной кислоты, в молекуле которой одна или обе гидрок-
сильные группы замещены углеводородными остатками. Произ-
Производными сульфоновых кислот являются их эфиры, амиды, хлоран-
гидриды.
Названия этих соединений образуют от названий углеводорода
и серусодержащей функциональной группы.
Номенклатура И1ОПАК рекомендует образовывать названия
на основе сульфоновые кислоты. Часто употребляется также назва-
название сулъфокислоты:
CH3SO3H метгшсульфоиооая кислота (метапсульфокпс-
лота)
СН3—У ^—SO3H я-толуолсульфоновая кислота (п-толуолсульфо-
кислота)
<^ у—SO3H 1, З-бснзолдисульфоновая кислота
C2Hr,SO,.\a+ этансульфонат натрия
(^ \ — SO..C1 бензолсульфонплхлорнд (Сснлолсульфохлорнд)
CaII7SO2OCM3 мстнлпропансульфонат (\!етнловыи эфир про-
пянсульфоповои кислоты)
СН3 —'/ ")"—¦ SO-j\H2 3, 4-дпмст1:лбспзолсул!>фоп;|мпд
CiH-.SO;. —(^ ^р; этилфспплсульфон (этплсульфоиплбсизол)
354
1. МЕТОДЫ ПОЛУЧЕНИЯ
1. Сульфоновые кислоты. Методы получения алкаисульфоновых
и арепсульфоновых кислот несколько различаются. Прямое суль-
сульфирование алкаиов и циклоа.чканов почт во всех случаях осу-
осуществляется по свободнораднкалыюму механизму (сульфоокнсле-
нпе, см. гл. 1.4.1):
SO,; О2
R—II—-- R—SO,H
/IV
Арены легко сульфируются концентрированной серной кисло-
кислотой или олеумом (см. гл. VI. АЛЛ):
Аг — Н — —t Ar —SO3H
Сульфонаты образуются при взаимодействии сульфитов с алкп-
лирующими и арилирующимп реагентами (галогенуглеводороды
с подвижным галогеном, эфиры неорганических кислот):
О
II й- Д+ :Оч .. 6+ й- Оч
Х~ —R 5 q- -<—х R -'¦- ^S 6:~-!-R X у ^S О R~X~
1! ' -ю/ " ' -q/
О
В качестве побочных продуктов могут образоваться соли кис-
кислых,, эфиров сернистой кислоты. Сульфит-поп является анионом
с двумя реакционными центрами (амбидентный анион).
Чистые сульфоновые кислоты могут быть получены простым
гидролизом сульфопилхлоридов, которые являются наиболее до-
доступными производными:
О О
[\ II
r^_s-ci-;-h2o—»¦ R-s-oii-fHd
'I :i
О О
2. Производные сульфоновых кислот. Сульфопилхлорнды по-
получают из углеводородов прямым методом. Алкапы и циклоалкапы
сульфохлорнруют (см. гл. 1.4.1):
50,; CI,
R-H — v ; R —SOoCI
Арены сульфохлорнруют избытком хлорсульфоновой кислоты:
Лг —H + 2C1SO3H -* Лг —SOaCI-f HC1-J-H2SO4
п* 355
Сульфопплхлорпды являются исходными для получения других
производных:
о
R-SO2Cl-j 2\[I;1 -» R_S-XH,^-\
6
о
R— SO-.C1 -i-r^OH-l :В - К— S — OR1 ; - Г IB 4 СЛ -
6
;.фмр>>| оут.фонопых
Ы'СЛчТ
3. Сульфоны. Сульфоны образуются в качестве побочных про-
продуктов в реакциях сульфировании. Пои повышении температуры
реакции н использовании более эффективных сульфирующих ре-
реагентов удается превращать арены в дпарплсульфоиы:
О
n,4vso., I;
2Ar—H ——• - Ar-S-Ar
(USOJI)
О
Сульфопы легко образуются в реакции сульфонплированпя (ана-
(аналог реакции ацилироваппя по методу Фрпделя — Крафтса):
Л IСI,
ArSOXl-j-Ar1 — И '- Аг — SO, — Лг1 \ ЫС1
Сульфопы являются конечными продуктами окисления сульфи-
сульфидов (гл. ХХ.З).
2. ФИЗПЧ1-СКИК СВОЙСТВА FF СТРОЕШПг
Су.тьфоновые кислоты и их щ^оплводныс — бесцветные вещества,
большинство из них кристаллические, растворимые в воде.
Сульфопат-пон имеет тетраэдрическое строение, углы .между
связями 108—110 :
(\' = ОП, О, CI, NN.. OR R )
Все три связи S—О одинаковые и отрицательный заряд дел окал и зо-
ван по всем трем кислородным атомам.
Какова природа связи S —О, является лп она чистой семпиолир-
iKiii сн5'.:ыо S—О или иолярг.он двойной с.улгмо^ Формально ее
структуру можно изобразить с помощью семпполярных связей,
например:
:б:~
R—S-Ri-I^O: —> R_S2+—R*
:О:-
В то же время атом серы имеет незаполненные Зя'-орбнталп, ко-
которые могут быть использованы для делокалпзащш электронных
пар кислородных атомов. Так образуется полярная двойная связь:
s = о , где 6--0.5. . .0,6. Это вытекает из значений днпольпых
моментов связей S —О (и«10-10~3" Кл-м или 3D). Если бы связь
была чисто семинолярпой, дппольный момент был бы равен 20х
Х10-30—23-10-яо Кл-м (~6—7D).
г,
— s —
IK
S-O
Группировка SO2 является сильным электроноакцептором, про-
проявляет —/- и —Д4-Э(|к|>екты.
3. ХИМИЧЕСКИЕ СВОЙСТВА
1. Сульфоновые кислоты и их соли. Сульфоповые кислоты явля-
являются очень сильными кислотами, в водных растворах они целиком
ионизированы:
R—s63H-!-I!o0 7^ R—SOT-; П:гО<
По кислотности сульфоповые кислоты сравнимы с серной кисло-
кислотой. Наиболее сильной из них является трпфторметансульфоновая
кислота CF;iSO;in.
Эти кислоты образуют устойчивые соли. В большинстве случаев
натриевые соли осаждаются из водных растворов сульфоновых кис-
кислот прибавлением хлорида натрия до насыщения («высаливание»
сульфоновых кислот):
! NaCl ;:-. RSO,NU ; НС1
Сульфогруппа связана с углеродным атомом не очень прочно
и может быть отщеплена двумя способами. Во-первых, при взаимо-
взаимодействии с электрофильнымн реагентами. Это главным образом
характерно для аренеульфоновых кислот:
Лг — SO.,II-: -К- Х- -> Лг — Е ; XSO3I!
Реакция связана с электрофилыюй атакой Лг—SO-,H, в результате
чего образуется а-комплекс (см. гл. VI. Л.4.1), который стабили-
стабилизируется отщеплением XSO3II. Например, в водных растворах кпе-
357
лот при 120—150'С аренсульфоновые кислоты образуют арены и
серную кислоту.
Во-вторых, сульфогруппа может быть замещена па другую при
действии пуклеофпльпых реагентов. Такие реакции давно известны
в ряду аренсульфоповых кислот, например сплавление со щелочью
для получения фенолов:
Лг —SO3Na-;-Na-:Y- —> Лг — Y :-Na,SO3
В результате электроноакцепторпы.х свойств сульфогруппы уг-
углеродный атом аренового остатка приобретает некоторый положи-
положительный заряд, что облегчает атаку нуклеофилыюго реагента и от-
отщепление сульфогруппы в виде сульфит-иона. Но для реакции
необходимы весьма жесткие условия, температура 250. ..300 С.
Так получаются фенолы (Y—ОН), могут быть получены нитрилы
(Y-CN), амины (Y^NH,).
2. Производные сульфоновых кислот. Сульфонилхлориды харак-
характеризуются выраженными элсктрофильпы.мн свойствами и легко
взаимодействуют с различными пуклеофилами. Они служат ис-
исходными веществами для получения эфиров, амидов, гидразидов
и других производных:
О
0 * R-S-Y:
1Ь- в-
R-S-C1-
О
О
6
HY:-:B ,
' R—S —Y:- IIB:CI-
'i
О
Сульфонаты (эфнры сульфоповых кислот) являются хорошими
алкилирующнмн реагентами, сравнимыми с дпметилсульфатом:
„о ?+
R—S—С) + :VH ¦ *-R SOj + R YH^-^R1 Y:
h "
o*
Их используют в органическом синтезе.
Сульфонамиды — весьма инертные соединения. Они представ-
представляют собой слабые ХН-кнслоты, растворяются в щелочах:
Ч о.х+ ^ Ч
-S N^ + '.Sol -« *¦ R S
'.Sol -« *¦ R S —Ml + IISol
Сульфонямид-апиоп является. делокалнзовпппым анионом и
имеет пуклеофнльиые свойства. Его можно алкнлнровать, га-
35S
логенировать:
—"-' RSO2\'/ -\-KaX
RSO.,"NH\a+ — П
c-i. /:i n, .ci
•¦ RSO,N< -j XaCl —n]v RSO,\( ^
3. Сульфоны, особенно диарилсульфоны,— химически весьма
инертные и термически стабильные соединения. Сульфоновая груп-
группа имеет сильные электропоакцепторпые свойства и способна дсло-
кализовать неподелепную электронную пару карбашгона. Алкил-
сульфоны являются слабыми СН-кислотамн (р/Сц«21. . .23):
О Н6+ О
II I II - ,Н
R — S — С— RM-:B- 7^ R—S —С<^ -ГНВ
о н^ о
4. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ
п-Толуолсульфоновая кислота — бесцветное кристаллическое ве-
вещество с т. нл. 104 С (моногидрат), очень хорошо растворимое в
воде. Ее получают сульфированием толуола или гидролизом п-
толуолсульфонилхлорида. Ее применяют в качестве кислого ката-
катализатора во многих органических реакциях, так как она растворима
во многих органических растворителях.
, ,SO.,OR Алкил-п-толуолсульфонаты (тозилаты) —
" бесцветные кристаллические вещества; т. пл.
для R-CH, 28 СС, для R--CH5 32 °С. В воде
не растворяются. Получают из тозилхлорида
и алканолов в присутствии щелочи. Используют в качестве
хороших алкилирующих реагентов.
п-Толуолсульфонамид — бесцветное кристаллическое вещество
с тл1л. 138 LC, мало растворимое в воде, растворяется в щелочи.
При хлорировании образует N-хлореульфонамиды, Широкое при-
применение нашла натриевая соль М-хлор-тг-толуолсульфопампда (хло-
(хлорамин-Т).
SOoNCIN.t -311,0
хлорамик-Т
Хлорамип-Т является дезинфицирующим и дегазационным средст-
средством, действует как сильный окислитель.
Синтетические моющие средства содержат смесь солей различ-
различных сульфоповых кислот. При сульфировании смеси алкапов с чис-
359
лом углеродных атомов С10—См получают смесь алкансульфоновых
кислот и их солей («мерсоляты»). Они являются хорошими поверх-
поверхностно-активными веществами и моющими средствами. При суль-
сульфировании алкилбензолов или алкилиафталииов с длинными ал-
кильнымн группами Се—С15 получают алкиларенсульфоповые кис-
кислоты, соли которых являются хорошими моющими средствами
(«сульфоиолы»).
Тетраметиленсульфон (сульфолан) — бесцветное кристалличе-
кристаллическое вещество. Ои плавится при 28 С'С и кипит при 283 СС, имеет
большой дппольный момент (|х—!6- 10~30 Кл-м или 4,8D) и диэлект-
диэлектрическую константу (f —44). С водой смешивается. Получают его
из бутадиена п SOr.
НС СИ Н2С —СН2
! | Н, кят. | |
СМ..-СН —CH--.CIV SO2 - , Н2С СП2 — -> Н2С СН2
\/ \/
S S
Тетраметиленсульфоп — отличный растворитель, растворяет так-
также некоторые неорганические соли. Он сольватирует катионы вслед-
вследствие электронодоиорного действия кислородных атомов. Служит
реакционной средой для проведения реакций нуклеофнлыюго за-
замещения.
Глава XIX
СУЛЬФИНОВЫЕ И СУЛЬФЕНОВЫЕ КИСЛОТЫ
И ИХ ПРОИЗВОДНЫЕ. СУЛЬФОКСИДЫ
Сульфнновые кислоты и сульфоксиды можно рассматривать как
производные сернистой кислоты, в которой одна или две гидрок-
спльные группы замещены углеводородными остатками.
Примеры номенклатуры:
CH.iSO.,H метансульфнновая кислота
C,)H,-1Sr0Xa4' бемзолсульфпнат натрия
CH3SCI \3 днмстплсульфокснд
О
Cn3SC,iM3 метилфеиилсульфокспд (мепмгульфши'лбетпл)
О
Сульфеповые кислоты и их производные сосньегстиуют форму-
формуле R—S—X. Например:
метансульфеновая кислота
C.,,I-I,-,S(.I беи зо л су л ьфенн л хлорид
300
1. МЕТОДЫ ПОЛУЧЕНИЯ
1. Сульфиновые кислоты получают мягким восстановлением суль-
фоннлхлорндов:
О
R—SOX1 [-"l R—S —ОН
Общим метод получения сульфиповмх кислот заключается в реак-
реакции металлорганпчеекпх соединении с SO.,:
о , о
S- h- >-^7>v~» I! H |!
R_MgX + O=S=O —- R—S-OMgX R— S-OH
2. Сульфоксиды получают окислением сульфидов, обычно раз-
разбавленной а.чочпои кислотой пли оксидами азота:
О
R_S_R'101, R-S- R1
3. Сульфеновые кислоты можно получить из сульфеннлхлоридов:
R— SC1-: Na-OH- - R—SOU , N':i;C|-
Сульфеповые кислоты являются слабыми ОН-кпслотамп, легко
окисляются.
Сульфенплхлориды получают хлорированием чполов или дп-
суль(|)пдов:
2R —S1I СЧ 2R —SCI "'- R—S —S —R
2. ФИЗИЧЕСКИЕ СЙОЙСТВЛ И СТРОЕНИЕ
Сульфиповые кпе.юты, их соли н сул|3фокснды нредстав.чяют собой
бесцветные жидкие или кристаллические вещества, растворяющиеся
в воде.
R Молекулы имеют пирамидальную конфигурацию.
' утлы CSO и OSX приближаются к тетраэдрпчеекпм
С7] 9 105
] (95 105).
х Н отличие от сульфопоных кислот и c\vi;«|)onon u \к-
лекуле сульфипопых кислот вместо одного кислородного атсмя на-
находится пеподеленная электронная пара. Поэтому атом серы явля-
является асимметрическим (если К=^=Х), а молекула — хпралыюп.
(]ульфпповые кпетоты и сульфокепды содержат полярную связь
S —О. Сулы|юкс1[ды обладают большими дппольпыми моментами
(и-12,7-10-30. . .13.4-10-а", плп 3,8. . .4.0D). Дппольные моменты
сулы|юксидов больше, чем дппольные моменты связи S- О(~1() '
¦ 1A ":1|) Кл-м. п.чп 3,(iD). что г;б"ьяспяетея .'loiio.innii'.isubij; э.чек;-
роподопоршлм де1"[С1внем уг.чеводородпых остатков.
3. ХИМИЧЕСКИЕ СВОЙСТВА. ПРИМЕНЕНИЕ
1. Сульфиновые кислоты являются более слабыми, чем сульфопо-
вые (рКа--2,5. • -3). В водных растворах они ионизируются, легко
образуют соли:
RSOoH-;-iLO z_-z RSOT-: ц3О +
Сульфиновые кислоты являются нестабильными соединениями.
Они легко окисляются и образуют сульфоповые кислоты.
При алкилнровании сульфипат-нона образуется два изомерных
продукта: эфир сульфиповой кислоты и сульфоп, так как сульфи-
пат-иоп является амбпдентным анионом с двумя реакционными
центрами:
| б- б !• ч/-.' 64- б- /О —R
X--UR—S.-O <-- X — R-,- :S -!-R —X -^R1—s/ т Х~
ll il О
О :О:
Обычно преимущественно образуются сульфопы.
2. Сульфоксиды. Сульфокснды обладают свойствами слабых ос-
оснований и очень слабых СН-кнслот:
,6
R
../?. Г + /6-Н1
-S<f -;-Н'Х- ^i R-S( - х-
\Ri ^ " -Rl J
/О б- б i /О
3 "CHILi '-
При действии металлоргапическпх соединений образуется ста-
стабилизированный карбапион.
Димепшлсульфоксид (CHaJS-0 — бесцветная жидкость. Он
кипит при 189 °С с постепенным разложением, поэтому его перего-
перегоняют в вакууме. Полярное соединение, дипольный момент \i--
= 13-10~3J Кл-м или 3,9D, диэлектрическая постоянная ?--48,9.
С водой смешивается неограниченно. Получают в промышленности
окислением диметилсульфида.
Диметнлсульфокспд широко используют в качестве раствори-
растворителя — он растворяет и неорганические соли. Ввиду большой по-
полярности его называют биполярным апротоппым растворителем.
Хорошо сольватирует различные катионы и тем самым активирует
анионы. В растворе дпметплсульфоксида хороню протекают реак-
реакции нуклеофильного замещения.
3. Сульфеновые кислоты являются соединениями нестабильными
и весьма мало изучегными. Некоторые из них встречаются в при-
природе. Пропен-1 -сульфеновая кислота СП;!СН —CHSOH содержится
в луке и вызывает слезоточивое действие.
Сульфепилхлориды содержат силыюполярную связь S—CI и
являются активными электрофильпыми реагентами:
6+ б-
R— S— C]-|-Na + Y- -^ R —S —Y-;-Na^C[-
362
Сульфепнлхлорпды присоединяются к алкенам и алкииам и да-
дают продукты т/;аяс-присоедниепня:
R CI
R, /Н л. л- i |
С=С:7 -: R1— S — C1 — > И— С— С — II
\V 4R ' II
R'S R
Глава XX
ТИОЛЫ, СУЛЬФИДЫ И ДИСУЛЬФИДЫ
Тиолы можно рассматривать как производные сероводорода, в ко-
котором один атом водорода замещен углеводородным остатком. Они
являются аналогами алканолов, алкеполов и фенолов, в молеку-
молекулах которых атом кислорода замещен серой.
Номенклатура тполов подобна номенклатуре гндроксилпроизвод-
пых, только вместо суффикса -ол применяется -пгиол или вместо
префикса гидрокси- (окси-) применяется меркаппю-. Иногда эти
соединения называют меркаптанами, тносннртамн, тиофенолами.
Номенклатура ИЮПЛК использование этих названии не реко-
рекомендует.
Примеры номенклатуры:
CH3SH метяптпол (метилмеркаптап)
C6n,-,SH мерклптобеизол (тнофснол)
HSCH..CH.,SH 1 .г-этйилитлол
//x/SH
1,2-д|[\;сркяптобепзол
CoH-S~.\'a"'J этаптиолят натрия (этплмеркяптнд натрия)
Сульфиды и дисульфиды являются производными сероводорода
HoS и ILSo, в которых оба атома водорода замещены углеводород-
углеводородными остатками. Они являются аналогами простых эфпров п гирок-
сидов, в которых вместо атома кислорода находится сера.
Примеры номенклатуры:
CHjjSQb дпметплсул:)|!л:д (метнлтиомстпи)
CoIlr,SSCoiIr) ДИЭТ!1ЛД11СуЛ1>ф|[Д
CH2 — СН2 зштюэшлен (зт|!лепсул:..ф::д)
XS /
Иногда сульфиды называют тиоэфирами, но это гтзпаине но-
номенклатура ИЮПАК не pcKovic-iiAjer.
363
1. .МЕТОДЫ ПОЛУЧЕНИЯ
1. Алкантиолы получают реакциями алкилнровапня сероводорода
и его солеи:
л - л -
R —X \а ' -SH - > R—SII ' .\'а ' X-
.,00' С
R — ОН-1 H..S — - R—SM- Н..О
ThO2
В лабораторной практике часто применяют реакцию алкплп-
роваппн тпомочевипы и расщепления полученных тнурснпевых
солен щелочью (гл. XXXV. Г.2).
2. Арентиолы обычно получают восстановлением аренсульфо-
ннлхлоридов:
ArSO.Cl -"i Аг-SH
3. Сульфиды получают алкплчроваппем алкан(ареч)тцолятов
или неорганических сульфидов:
Rf._X : R1— S:-Na H --> R —S—R1-,' Na-X"
6. 6-
2R —X ; Na + :S:--.\'a'¦ —* R —S —R-'¦ 2N!a • Х~
.100- I 00"^ С
R—OH ; I1,S —^- - R — S — R ,-IbO
Для получении днарплсульфпдов используют реакцию галоге-
цидов серы с аренами и присутствии кислот Льюиса:
\;ci,
2Лг—II ' SXI,— ¦ .\r--S -Лг ; S 2НП
4. Дисульфиды получают окислением тполяюн пли алкнтирова-
ппсм неорганических ;шсулы|)идов:
2R—S-.\j • - R -S — S — R
Л • й-
2R —X N.T -S —S~\.i ¦¦ - - R — S -S — R , 2\;i X~
2. ФП.'ШШ-СКИГ: СНОПГТВЛ II СТРОЕНИИ
1. Тполы ирс.н.чан.тяют собой бесцвешые соединения с чреаиычан-
но неприятным запахом, который обна|)ужпр.ается \же в ничтожных
концентрациях. Например, ла\м\\ метаптпола и воздухе чунетвует-
ся уже в разбавлении I : 4- 10*. Алкаптпо.ты содержатся в кишеч-
кишечных газах человека и животных, в зловонных выделениях некото-
некоторых животных.
("вязь S— И менее полярпа, чем связь О--Н, поэтому у тполов
слабее .межмолекулярные водородные связи и ниже температурь!
кипения, чем у соответствующих кислородных аналогов (табл. 36).
Дипольпые моменты алкаптпо.тов D,:!• !()""' -о- !(Г:<" Кл-м плп
1,3—1,5 D) незначительно меньше, чем днпольпые моменты алкаио-
364
Таблица Згк
СВОДИМО:! ПС
CH;;S[[
C,H.-,SII
h-C-,[FuS[[
Физические
Т. ил.,
— 123
— 121
¦-75,
—
константы некоторых тиолов
<С
7
Т. nut:
7,
34,
[2(i
169
6
7
°
0,868
0,810
0,857
1,078
"и
1,1305
1,4407
1,587
лов, но поляризуемость значительно больше. Например, 1иолярные
рефракции Ro 1,64, fts 7,69.
Электронная система атома серы значительно подвижнее. Это
отражается и на энергии ионизации. Тнолы являются более силь-
сильными электроподонорами. Например, ЭИ для CH3SH равен 9,44
эВ, для СН:1ОН — 10,8 эВ, для CJ]5SH — 8,25 эВ.
В молекулах тиолов угол CSII равен 100 . . . 104°, что меньше,
чем угол СОН п алканолдх.
2. Сульфиды и дисульфиды являются бесцветными веществами
с неприятным запахом, особенно у летучих дисульфидов. Их тем-
температуры кипения выше, чем у аналогичных простых эфиров и пе-
роксидов. Ди.метилсульфид кипит при38сС, а диэтплсульфид — при
92°С.
Сульфиды являются полярными соединениями, их полярность
подобна полярности простых эфиров (|д 4,7- 10~;io. . .5,3-10~30
Кд-м нлн 1,4. . .1,6 D), несмотря па то, что связь С—S менее по-
лярна, чем связь С—О. Предполагается, что причинами такого
большого днполыюго момента являются меньший валентный угол
связи CSC (-—¦ 100 ) и эффект неиоделенных электронных пар атома
серы.
Неподелеипые электронные пары являются весьма подвижными,
сульфиды характеризуются более сильными электроподонорпымп
свойствами. Энергия ионизации па 1 эВ ниже, чем у простых эфи-
эфиров. Например, С,Н,ОС,Н, имеет ЭИ 9,6 эВ, a CJisSCH;—
8,5 эВ.
3. ХИМИЧЕСКИЕ СВОЙСТВА. ПРИМЕНЕНИЕ
1. Тиолы. Реакции тполов обусловлены главным образом иониза-
ионизацией связи S—II н пуклеофильпымп свойствами атомы серы.
Тнолы являются SH-кислотами, при этом значительно более
сильными, чем аналогичные ОН-кпелоты:
R—S —П-; :В
R — S:--; ПВН
Например, для C.,H.-,SH pKa~ 10,6, а для Cr,H6SH рД'а=6,5.
Кислотность тиолов па 4. . .5 порядков выше, чем аналогичных
гпдроксплпропзводпых, несмотря па то, что связь S—Н является
355
менее полярной. По-видимому, повышенная кислотность объясня-
объясняется особенностями электронной структуры аниона—тиолят-
иона, в котором возможна большая, чем в алкоголят-попе, делока-
лпзация отрицательного заряда (атомный радиус больше, имеются
незаполненные d-орбиталп).
Тиолы образуют стабильные соли (тнолиты, меркаптпды). Ха-
Характерны тиоляты ртути, которые очень легко образуются и содер-
содержат ковалентную связь Hg—S.
Тполы и особенно тиоляты легко окисляются. Первичным про-
продуктом окисления является дисульфид, который может подвергать-
подвергаться дальнейшим превращениям. Характерным окислителем тнолов
до дисульфидов является иод:
'l + R—S — S — R
2RSH
2RSO3H
Тполят-иоп является сильным нуклеофилом, легко алкнлирует-
ся:
б I- 6-
R— S-\'ai--;-Rl — X —у R —S—R1-' NaX-
Тиолы служат важными исходными для органического синтеза.
Некоторые алкаптиолы в небольших количествах добавляют к
бытовому газу для обнаружения утечки.
2. Сульфиды и дисульфиды. Химические свойства сульфидов и
дисульфидов определяются главпым^образом большой подвижностью
электронной системы атома серы. Они легко образуют донорпо-ак-
цепторпые комплексы с различными ионами металлов, с галогена-
галогенами и другими кислотами Льюиса:
Но в то же время сульфиды являются очень слабыми основа-
основаниями: их основность примерно па два порядка меньше, чем соот-
соответствующих эфиров:
СП.,Ч .. ГСПз\-- V"
>S: '-Ы1 Х- г* /S— Н Х- (p/Cniii-^— 5,25)
СИ/ ' LCHs7 J
Алкплировапие осуществляется очень легко и образуются соли
сульфонал:
R ч .. 6+ е- IR ч .. 1 •-
;S:-f R — X —> >S—R4 Х~
R1^ U17 J
В этих реакциях резко выявляется несоответствие между основ-
основностью и нуклеофильностью атома серы в сульфидах. Он обладает
малым сродством к протону, но большим сродством к атомам, имею-
имеющим высокую поляризуемость.
Соли сульфония—бесцветные вещества, легко растворимые
в воде. Ион сульфонпя имеет пирамидальную конфигурацию, и в
366
случае трех различных заместителей атом серы становится асим-
асимметричным, т. е. возможно существование эпатномеров:
г ск. Г
Lch3—s—сн,.|
I 4
R'r ^ R3/ Vr'
Co.-ih су.чьфония в алкилыюй группе характеризуются повышетюн СП-
кислотностью. При в.чяимодействии их с металлоргяннческими соединениями
образуется амиоч. Так как одновременно в молекуле имеется положительны:!
заряд, образуется внутренняя соль (бетаин, или илнд) (сравните с фосфоппи-
ИЛ1ТД.1МИ — гл. XIII. 3.2).
СН3
Br--;-cTl3—Li-^СНз —S-—СН.,~СП4 !-LrBr-
бромид триметмл- мстил ид днметнл-
сульфоччя сульфония (мстн-
леидичетилсуль-
фуран)
Сулы|)опииилиды принадлежат к стабилизированным карбацпо-
нам и используются в органическом сшпезе.
Сульфиды легко окисляются с образованием сулы[юксидов и
сульфоиов:
О О
IO1 Г LOl »
R—S—R1—>R -S — R1-^ R— S— R1
О
Димстилсульфид представляет собой бесцветную летучую жид-
жидкость с неприятным запахом. Получают каталитически нз мета-
метанола и сероводорода. Используют для производства дпметилсуль-
фоксида.
БисB-хлорэтил)сульфид CICHoCHoSCHoCHoCl является бес-
бесцветной жидкостью с т. кип. 215 С. Ню получают реакцией этилена
с хлоридами серы. Очень токсичное вещество кожно-нарывного
действия. В годы первой мировой войны использовался как боевое
отравляющее вещество под названиями иприт, горчичный газ.
Азоторганические соединения
Соединения с простой связью С—\ принадлежат к азоторгапичес-
ким соединениям. Эти соединения рассмотрены лдесь в последова-
последовательности от высших степеней окисления азота до низших, включая
производные с двумя непосредственно связанными атомами азота:
нптросоедннення R — N0»
пптрозосопдипепня R — \0
гидрокспллмины R- \--OII
Н
307
R1
Г i 1
амины и соли аммония R —N'H2 R—N—R1 R — \—R1 R—\'+—R2 i Х~
Н R2 L R3 J
R, R2
гидразины \ —.\
R1 R3
дмазососдннсния R — N.X CN2
R1'
азосоединения R —N = \— Rl
Соединения со связями C--N и С^-.Х1 рассматриваются соответст-
соответственно как производные альдегидов, кетопов и карболовых кислот.
Глава XXI
НИТРОСОЕДИНЕНИЯ
Нитросоединения являются производными углеводородов, в моле-
куле которых один или несколько водородных атомов замещены
группой NO2 — нитрогруппой.
В основе классификации нитросоедииепий лежат тип углеводоро-
углеводорода, состояние гибридизации углеродного атома и число пнтрогрупп
в молекуле. Здесь рассмотрены три основные группы:
I) ннтроалканы и нитроииклоалканы (соединения со связью
C(s/;:1)-\TOJ: С„Но„м\02; СпН.,„..,_м(\'О2)и; (СН3)„ НС\Оа;
i 1
2) питроалкены [соединения со связью С isp1)—ХО.1:
R , /NOS
С С/
R1- -R2
3) пнтроарепы (соединения со связью C(sp-)— NOJ: АгХО-.;
NO
а также некоторые производные ннтросоедпненин — галогенпптро-
соедипеиия, нитроалкаиолы и питрофенолы.
Название нитросоединенпя образуют из названия углеводоро-
углеводорода и префикса нитро-. Нумерацию углеродных атомов начинают с
того конца цепи, к которому ближе расположена питрогруппа, если
пет более старших характеристических групп ОН, N'H2, SO3H,
двойных" it тройных связен пли других заместителей. Заместители
F, C1, Вг, I, алкильные группы, пнтрогруппу в названии соедине-
соединения располагают перед корнем в порядке алфавита, а нумерацию
начинают с того конца цепи, к которому ближе расположен
368
титель, упоминающийся в названии первым. Если есть возможность
выбора, руководствуются принципом наименьших локаптов.
Примеры номенклатуры:
СП3 CI
I I
СН3СПСП2СН,, СН,СНСНСН3 CH3CHCHNO2 СН3СПСН.,СИ,СН,ОН
I I I I
NO, N02 CII3 NO.2
2-ннтробутан 2 - мо г ил-.1-нитро- '2- метил-1 - нитро- -1-нитроне н га мол-1
бутан I -хлорпроП'Ш
\о2 си, он
) O2N4J4/NO2 0,N !N/NOl
сн3снсн-снко2
3
СИ.,
a
З-метн.т-1-пнтро- шпро- 4-x.iop-J, ri-днннтро- 2, 4. С -три нитрофенол
бутеи I Сенчол толуол (ппкрпнонг.я кислота)
Л. НИТРОЛЛКАНЫ
1. МЕТОДЫ ПОЛУЧЕНИЯ
11птроалкапы и нптроцнклоалкаиы получают прямым нитрованием
алканов и алкилировалием леоргаипческих нитритов и реже окис-
окислением других азотсодержащих соединений (пптрозосоедпнепий,
аминов).
1. Прямое нитрование алканов происходит при повышенной
температуре в жидкой или газовой фазе разбавленной азотной
кислотой пли оксидами азота.
Нитрование алканов в жидкой фазе осуществил М. И. Конова-
Коновалов A899) при нагревании ал капов с 10 -- 25'"о-пой азотной кисло-
кислотой в Зсшаяппых ампулах при МО -150 С:
UNO ; II,О
:ir no
-II
1 ю- i г.о' с.
R качестве побочных продуктов образуются кетопы, карболовые
кислоты.
Нитрование в газовой фазе разработано X. Гессом A930). Па-
Пары алкапа и азотной кислоты в специальных реакторах кратковре-
кратковременно @,2 -2 с) нагревают до -120—180 С и быстро охлаждают. Из
метана образуется питрометан. При нитровании этапа, пропала или
бутаиов происходит разрыв связей С—С и образуется смесь пнтро-
алканов, которая разделяется перегонкой:
SO,
UNO, i
СН3СН2СПз -CiI,\O,-{ CH,CII.,XO ' CH,CH2CH2\O,- CII3CHCH3
'20> С 9 % ' L'l. %" ' .4^'% * ' '.U % '
Реакция прямого нитрования происходит по свободпорадикаль-
ному механизму. Свободные радикалы возникают в результате
369
термического расщепления азотной кислоты:
3CoAO--:--NOo :- 1Ш
R _Н -)- -ONOj —> R - -{-IЮХ6.2
r. + .NO.,—>R-NO2
Активным радикалом в этом процессе является ОА'О-.
2. Алкилирование нитритов. При алкнлировапнн нитрита сереб-
серебра галогеналканом В. Мейеру A872) удалось получить питроалка-
ны в смеси с алкилнитритами:
2R-Br + 2.-\gNO2- >R-NO2-|-R-OXO ; 2AgBr
Алкплировать можно также другие нитриты, например NaXO...,
KNO2, нужно только подобрать соответствующие растворители.
Самыми лучшими растворителями для этой цели являются биполяр-
биполярные апротонные, в которых сильно сольватнруются катионы и, та-
таким образом, активируется ннтрнт-ноп. Таким растворителями
являются днметнлеульфокенд и диметилформамид (CH3Ji\CHO
(гл. ХХХШ.Д.4):
5 (- 6- j - :SoI
R — X + NaNO2- -'R —
X~
B таких условиях получаются главным образом ннтроалкапы с
незначительно!! примесью алкнлннтритов.
Нитрит-ион является анионом с двойственной реакционной спо-
способностью (амбидентпый анион с двумя реакционными центрами,
подобно сульфит-попу и еполят-нону):
й- 5+
Х-+ R-NO2 — X-R+:N=SP- + R-X
^6Г
5-
О—R
2. ФИЗИЧЕСКИЕ СВОЙСТВА И СТРОЕНИЕ
Ннтроалкапы являются бесцветными или желтоватыми жидкостями
со слабым запахом или кристаллическими веществами (табл. 37).
Таблица 37. Физические константы некоторых нитроалканов
Соединение
сн:1\о2
СН,СН2ХО2
СНПСН,СН,\О2
СП3СИСП3
ко2
О.,\СП.,С11.ДО..
ИС(\'О,K
С(ХО2L
Т. гл., °С
—28,0
—89,5
— 10-1,0
—91 ,3
•10,0
25,0
13,9
Т. КШ1., "С
101,2
114,1
131,2
120,3
135 @,8 к Па)
45 B,9 кПа)
125,7
"Г
1 ,1382
J.0506
1 ,0014
0,9884
1.45
1,615
1,6300
"Iй
1,3819
1,3919
1,4016
1,3944
1,4488
1,4381
370
Для моиопитроалкапов характерны большие дппо.тьпые моменты
A0,5- 10-;t0. . .12,6-Ю-™ Кл-м пли 3,15. . .3,7 D). Причина зна-
значительной полярности иитроалкапов кроется в электронном строе-
строении нптрогруппы. Ннтрогруипа содержит семпноляриую связь.
Образование семиполярной связи наглядно можно показать при
помощи реакции окисления ннтрозосоедииеипя:
r + r ::
С):-
(см. также выше реакцию алкилироваиия интрнт-иона). В резуль-
результате атом азота приобретает положительный заряд, а кислородный
атом — отрицательный заряд.
Между атомами в семипал яр пой связи - N— о=- не может об-
образоваться вторая связывающая орбпталь, так как атом азота не
имеет энергетически соответствующей незаполненной орбнтали.
В этом заключается принципиальное отличие связи =^'~9:" от
связи >S - ОБ сульфоновых кислотах, сульфонах и сульфоксидах,
где атом серы имеет незаполненные d-орбиталн.
Ннтрогруппа является простой сопряженной л-электронпой
системой, в которой атом азота \т + находится в состоянии s/J-rn6-
риднзащш, так же как один из кислородных атомов. Второй кисло-
кислородный атом дает неподелепную электронную пару. Так образует-
образуется 4 л-электронная сопряженная система и обе связи \—О вырав-
выравниваются, кислородные атомы становятся одинаковыми. Это изоб-
изображается различными способами:
Выравнивание связей X—О подтверждается рептгеноструктурпьш
анализом. Связь N—О в пптрогрунне короче связи N—О в гидро-
кснламнне и N-оксндах @,136 им), но длиннее связи и шггрозогруп-
пе N-0 @,115 им).
Ннтрогруппа содержит атомы с большим срод-
сродством к электрону (О, N+). Ннтрогрупна облада-
обладает значительным эчектропоакцепториым эффектом,
—/- и —/Vf-эффектами.
На рис. 90 приведены некоторые результаты расчета -кс-элзктроппой си-
системы нптрогруппы в приближении МО Хюккгля'.
Высшая занята:-: молекулярная орбигаль (V.,) расположена мпж?, чем,-
например, в этилене, поэтому отрыв электрона происходя г труднее?. Низшая
свободная МО (xYn) также расположена восьма ничко, ню свидегелымпусг об
электропоакцепторных свойствах.
Полная плотность л-5.тектропоп (аг) свипогл-1ьс|вуот об акцепторном дей-
действии атома кислорода (на каждом кислородном атоме избшок электронов на
R—Nfj 124-126
371
-2/1
-Р
а
/(¦
2Р-
0.40:
-ft"» r 1,2
-Й-
а+ 2,707/3
0,242
t
O
0
0.500
l
A
0,516
О
V
I
0,242
f
0.500
4
SC/HCMO)
1,516
1.516
Рис. 90. Значения энергии МО. одномлоктроинме плотности фрсяпа.и.пых ор-
биталеп, п.ютпость л-элсктронои и порядки л-спи.чей питрогруппы
0,016). Порядок л-сняли (/),-.,) свидетельствует °б обра.чонапи» выраппеммых
связей.
Для ннтроалкапов характерно слабое поглощение в области
270—280 нм с интенсивностью ^«10. . .15. Это связано с электрон-
электронными переходами типа и->-л* от иеноделеппой электронной пары
атома кислорода па НСМО. Переход л—>-л" находится в области
200 им (с»5000).
В инфракрасных спектрах наблюдаются максимумы поглоще-
поглощения, связанные с симметричными и антисимметричными колебания-
колебаниями двух связей
в областях 1370 см и 1550 см.
3. ХИМИЧЕСКИЕ СВОЙСТВА
Химические превращения питроалкапов определяются, во-первых,
реакциями питрогруппы (восстановление, гидролиз), во-вторых,
реакциями с участием «-углеродного атома:
И
R —С — NOa :
Н
н,so,
[П! IH]
RCILNO —* RCHAHOH —* RCHAH,
[ll;.N()Hi ['.SO,
R —C- —\(), ¦
R'-X
Л - 6-
"i - X
— R--C-XO,
I
Y
4 c^ о
I!
R - C- XO,
Ri_C- UH
i-
: О-
R -C-XO.+R-C - X.
Ri
И
-O-R1
1. Восстановление нитроалканов. При действии восстановите-
восстановителей (металл и кислота, соли металлов в ннгшшх степенях окисле-
окисления - Sn'2", Ti:'' в кислой среде, соединения серы AUS,., Na.jS.Oj,
водород в присутствии катализатора) пли па катоде электролити-
электролитической ячейки пнтроалканы восстанавливаются. Этот процесс сту-
ступенчатый. Первой ступенью является присоединение электрона н
образование малостабплыюго анпои-радпкала. Затем следует быст-
быстрое присоединение второго электрона с образованием дпапиоиа:
R- N0., : г-~ >[R—\О,|- ' ,|R- Х02]--
При протоппроваипи днапнопа образуется ннтрозосоединение:
. 0=- 2П г . ,0-11 -П2() /.б:
R_\ " _ „R. \/ " • -R — NT
б:" 0-Ц
В некоторых случаях ннтро:юсоедипенпе перегруппировывается в
изомерное окснминосоединение — окенм (гл. XXJJ. Л.З):
R -СМ. —N.. О — R — СИ-ХОН
Нитрозоалкапы в процессе дальнейшего восстановления пре-
превращаются в алкилгидроксиламины н далее в алкплампны:
R- Х- О—-*\г—Х — Ои—->П -XIL + ILO
¦2 ] I '• | 1> 11 '
II
2. Превращения нитроалканов в присутствии сильных кислот.
Первичные нитроалканы в растворе 80—95*'«-noii серной кислоты
образуют карбоиовую кислоту и соль гпдрокенламппа:
RCIIAO.-MbSOi-i-IV) , RCOOI! [H,XOI!] HSO,~
Это один п.ч промышленных методов получения гпдрокспллмппа
(из шпрометапа или 1,2-дпиптроэтаиа).
В реакции происходит протопиронанпе кислородного атома нпт-
рогрунны и ппутримолекуляриый окиси пел ыю-восстнпопптельный
процесс с переносом кислородного атома от азота па близлежащий
атом углерода и образованием гпдроксамовы.х кислот (гл. XXXIII.
I-.3).
В случае вторичных пптроалк;и;ов RR4JIX0. обргпуютим окси-
мы RR'C \0Н (гл. XXVII. Д.2).
3. Кислотность и таутомерные превращения нптроалкаяов.
Первичные п вторичные пптроалканы являются СЛ 1-кпслотамп:
IV R1
I I
R ¦ -С —XO.,-;-:Sol -^ R -С"- ХО, ' HSol <-
I
н
Кислотность моиопптроалкапов в водных растворах сраеппма
с кислотностью фенолов. Нслп у одного атома находятся две пли
три нптрогруины, кислотность резко возрастает:
373
Соединение CH3NO-> CH3CHoNTO, СН,(\'О,J CH(\'O,)a
p/vd(H2O) 10,2 8,5 4,0 -0
Ионизации сзя::! С —Н в нптроалканах способствует полярность
этой связи, но главным образом возникновение сопряженного анн-
аннона:
/ ^б" или'точнее> R/ ^о^-
B5 + Ь'=2)
Точнее строение бл-электронной системы аниона пнтроалкапа
можно охарактеризовать расчетом но методу МОХ (рис. 91). Как
показывают значения энергий МО, для аниона возрастают электро-
нодонорные свойства, а уменьшаются акцепторные свойства. Анион
иитроалкана имеет нуклеофильные свойства.
Во фронтальной МО (?3) наибольшая электронная плотность на
углеродном атоме. Это делает вероятной атаку электрофилыюго
-2/1-
2/3-
1.77
°f
,«-0,92620 °f 0,172
i I 0>27~
аГ R\0.32- 014^
- ¦ »• а'0Л72
¦а+0,52940 N^ NCA(? ллогносп. я-электронов q
—iL. я + ];2/? R Л Л^^г» и порядок я-сиз» '
л олкоэлскгронная. \Д).32-
Т| л | л on/Co Д т Р- V
|| «-Г/.5/О5Р ПЛОТНОСТЬ (Г;"г / \ 027-
п ВЗМОСЧ*",) R ^О '
Лстокалиипмя о грни.ч ге.и,иого
'•фя.ы ri JMM ню
Рис. 91. Значения энергии МО, одноэлектроппые плотности ВЗМО, плот-
плотность л-электроноп, порядки л-снязеп п делокалн.зация отрицательного за-
заряда в нптропат-иопе
реагента по углеродному атому. Но, тем не менее, реакционным
центром может стать также атом кислорода.
Анион иитроалкана является амбндентным анионом, подобно
енолят-иопу. Например, при протопированпн может образоваться,
кроме пнтроалкапа, также его таутомсрпая форма:
-NV ^ ^^о«- ^
ттктроновая кислота
Таутомерпую форму иитроалкана ппллвают аци-нитрпфррмои пли ни-
нитроновой кислотой, которая в чистом пиде не получена. Анион иитроалкана
может быть назван ннтронат-ионом. Нитроновая кпе.мт;! является ОН-кис-
лотой средней силы со значением кислотности между кнелотностямн аготи-
стой (р/Сл=-=3,2) и азотной кнглог. У лее приближенный расчет показывает,
что в тяутомерной системе пптроалкап v.1 пнтронопая кислота! после достиже-
достижения равновесия концентрация п.чтропопоп кислоты не превышает 10~"%.
374
Но это пе исключает потможмостн, что npir подкислепин растворов солеп ппт-
роалкамоп при низкой температуре выделяется смесь пнгроалкаиа п ннтропо-
Bcui кислоты с большим содержанием нитроновой кислогы с последующим пре-
вршцеппем нитроновом кислоты в менее кислый шпроалкам.
Соли нитроалканов могут быть получены в кристаллическом
состоянии. Они разлагаются при сильном нагревании, иногда со
взрывом.
Нитрононые кислоты в кислых водных растворах гпдролизуются
с образованием карбонильных соединений и Х.,0 (реакция Нефа,
1894):
j¦ ,-О~ н+ ^О j _н + ,0
Первичные питросоедипения образуют альдегиды, вторичные —
кетоны. Это означает, что при добавлении избытка сильной кислоты
к щелочным растворам нитроалканов образуются наряду с исход-
исходным нитроалканом продукты гидролитического расщепления нитро-
нитроновых кислот.
4. Реакции нитроалканов с электрофильными реагентами в при-
присутствии оснований. Нуклеофильной частицей в этих реакциях
является анион питроалкапа (ннтронат-ион).
Легко протекает галогенирование:
Н X
¦в I х !
R — CI I..NO., + X, — > R — С — \'0„ - i R — С - \02
I " :Б |
X X
(X -CI, Вг, I).
При нитрозпровании получаются ннтрозоннтроалканы:
Rl. hno' R\
¦¦С-КО., <¦ >С-ХО,
R ' I R / I
Н N0
Алкнлирование ведет к С-алкилпродуктам с примесью О-алкнл-
продукта (эфира нитроновой кислоты). Можно найти условия, при
которых в продуктах алкнлированпя доминируют эфиры нитро-
нитроновой кислоты:
Alk
Эфиры нитроновых кислот являются очень реакцно)пюспособнымп
соединениями.
Для нитроалканов характерны реакции с альдегидами и кето-
нами, в которых образуются нитроалкаполы или другие нитро-
гидроксильные производные. В некоторых случаях в реакции
участвуют две молекулы пнтроалкана и образуются динитроалка-
375
ны. Общая схема реакции:
4 :В ^= „^C—NO.-f HB+
R'J?
C<
Реакция подобна альдолыюй конденсации (гл. XXVII. Л .3).
При взаимодействии с формальдегидом часто образуются про-
продукты реакции с двумя или тремя мотекуламн альдегида:
СМ..ОН
11 -в !
CH3NO2 + 3O- С — .НОСИ., -С— \О..
¦и
сн,ои
2-м!:тро-2-г пдроксм -
метилпроп.шдиол-1.i
4. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ
Нит/юметан — бесцветная жидкость, мало растворим в воде (до
9%), с водой образует азеотроныую смесь, кипящую при 83,6 С
и содержащую 23,5% воды. Ядовит.
Ннтрометан получают в промышленности нарофазным нитрова-
нитрованием пропана.
Он используется в качестве растворителя, особенно в полимерной
промышленности, как исходное для дальнейших синтезов (нитро-
алкаполы, взрывчатые вещества), а также служит топливом для
реактивных двигателей.
Иитроциклогексан является бесцветной жпл-
O костью с т. кип. 206 С, в воде не растворяется.
- Получают прямым нитрованием циклогексап;:
нитроциклогексаи азотной кислотой при повышенных темпера-
туре и давлении.
Используют в качестве исходного для синтеза канролактамл —
мономера для получения синтетического волокна капрона.
Тстраншпрометан C(i\O2h представляет собой бесцветнук;
жидкость с острым запахом, в воде не растворяется. Получают вза-
взаимодействием уксусного ангидрида со 10096-пой азотной кислотой.
В это» реакции происходит деструктивное нитрование уксусного
ангидрида (СН3С0).,0 ацети.тиитратом СН:,СОО.\"О,.
Тетрапптро.метаи взрывчат, особенно опасны его смеси с орга-
органическими веществами, которые по взрывчатости не уступают ни-
нитроглицерину.
Используют в качестве окислителя во взрывчатых смесях и
ракетных топливах.
Трих.юрнитрометан (хлорпикрин) — бесцветная жидкость с
острым запахом, т. кип. 112,3'С, в воде не растворяется. Получают
его хлорированием нптрометана пли пикриновой кислоты.
Хлорпикрин — сильный лакрнматор, в больших количествах
действует удушливо. Его используют для дезинфекции н в борьбе
с вредителями сельского хозяйства.
Б. НИТРОАЛКЕНЫ
Здесь рассмотрены только питроалкены с нптрогруппон у двойной
связи —сопряженные пптроалкепы.
1. Методы получения. Питроалкены получают главным образом
из галогеппитроалкенов или питроалкаполов в реакциях отщепле-
отщепления. Наиболее часто используют пнтроалканолы:
R
R1 i nix- R1 '<
С-С -NO., -> С с J-1I..0
R-' I i ' Я2 КО,
ОН II
2. Физические свойства и строение. Питроалкепы -- желтова-
желтоватые жидкости с острым запахом, лакрпмагоры
Сопряженная система л-связен шпроалкепов является еиль-
пополярной.
н д. В молекуле пптроалкена одновременно действуют
^^Г индукционный ( — /) и мезомерпып (- -.11) эффекты.
н N=6: Эго действие сильно поляризует связь С - С. Нптро-
~:Ь_) алкепы обладают значительными электроноакцеп-
нчтроа.-кеи торными свойствами, электрофнльными центрами
являются углеродный адом и атом азота в пптрогрунпе.
Для ннтроалкенов как сопряженных соединений характерно
сильное поглощение в ультрафиолетовой части спектра B25. . .
. ..235 им, f-^4000. . .10 000).
3. Химические свойства и применение. Питроалкены активно
взаимодействуют с различными пуклеофильными реагентами. Ог-
чаетп это вызывает полимеризацию.
Для шпроалкепов характерны не превращения ннтрогруппы.
а реакции поляризованной С—С-свя.чн:
R i- ^-ч f- + ^ «'- «"- f
^C=CH-NO: _ :Nu:HB —~ Н-С-СН—NO., » H-C-CH-NO, 4- :В
Nu: HB' Nu:
Легко происходят реакции присоединения нуклеофильных анио-
анионов и других нуклеофильных частиц. Таким образом к пуклесфпль-
ному реагенту присоединяется нитроэтпльпая группа (незамещен-
(незамещенная нлп замещенная). Эти реакции могут быть названы пит/юэпш.ш-
/юванием.
Реакции ннгроэтнлпровапия определяют основное практичес-
практическое применение пнтроалкенов. Полученные продукты используют-
используются для синтеза лекарственных н взрывчатых веществ. Особенно
реакцпоипоспособпымп являются галоген питроалкены.
В. НИТРОАРЕНЫ
1. МЕТОДЫ ПОЛУЧЕНИЯ
Нитроарепы получают прямым нитрованием аренов и их производ-
производных. Меньшее значение имеют методы окисления других азотсо-
азотсодержащих соединений (питрозосоединений, аминов).
1. Прямое нитрование аренов и их производных. Нитрование
осуществляют азотной кислотой или нитрующей смесью (HNO,,+
+ H2SO.|). Впервые нитрование бензола провел Э. Митчерлих A834):
нко3
Аг-Н 4-,\r-NO2
Н2.ЧО,
Нитрование является реакцией электрофилыюго замещения у
углеродного атома арепового цикла (гл. VI. А.4.1). Электрофиль-
ным реагентом в реакции нитрования является катион нитрония
N0;:
h г- м •• 1 -
N—Y == O=N=OhY
Катион иитрония всегда связан с каким-то анионом Y". В за-
зависимости от природы аниона образуется более или менее полярная
связь N—Y и меняется активность нитрующего реагента. Самыми
активными солями нитропия являются соли с анионами BF4",
SbF6"", PFG", в которых существует свободный катион питрония.
Это подтверждается рентгеноструктурным анализом кристаллов
[N02]! BFf. Катион N0/ имеет линейную структуру.
В смеси азотной и серной кислот и в меньшей мере в концентри-
концентрированной азотной кислоте соли нитрония могут образоваться при
протонировапии азотной кислоты. Практически образуется слож-
сложная смесь протоиировапной азотной кислоты и гидросульфата нит-
нитропия. Гидросульфат питрония обычно изображают в ионной форме.
По всей вероятности это соединение имеет силыюполярпую кова-
6+ 6-
лептную связь 02N—OSO,,H
П\О3-!-И + Н5ОГ:^± [HaNOa]-1 HSO4" ^± [\Oa] '• HSO4' +1UO
Чем меньше в реакционной среде Н20, тем реагент активнее.
Поэтому 100%-пая азотная кислота и особенно ее смесь с концент-
концентрированной серной кислотой являются очень эффективными нит-
нитрующими реагентами.
Первым продуктом нитрования арепов является мононитроареп.
При более высоких температурах и с применением эффективных
нитрующих реагентов получают дшштроарены и тринитроарены
378
(о механизме реакции см. гл. VI. Л.4.1):
п\о3
н\о,
сн3
O..N
I
ХО.,
сн:1"
1 хХО,
h.vo,
Н.\'О.,
11г6О,
N0,
С применением обычных нитрующих реагентов тринитробензол
не образуется или образуется с очень маленьким выходом. Но тет-
рафторборат иитропия легко превращает бензол в тринитробензол:
Х02
Энергично нитруются ксилолы, нафталин и другие арены.
При нитровании галогенаренов получаются моно- и дннитро-
продукты:
CI
/CI
¦ хо„ o,v
l.'N'O,
на;-,О4
Очень легко нитруются фенолы, возможно получение трнпитро-
производпых.
2. Окисление ариламинов. Аминогруппа мо;кет быть превращена
в питрогрунпу де|ствием различных окислителей при условии, если
окислители не затрагивают ареновый цикл. В качестве окислителе;!
применяют перокспсоедпнеппя, например перокенсерную кислоту
U^SOf, и ИаО;.
При окислении пптропнплипов получаются такие нитроарены,
которые прямым шировапием получить нельзя. Например:
Ю|
Х02
/У
ХО,
и-нитро-
анилин
•/
ХО.
бензол
37Э
0:!N,
02N '
IIOIITMI
NH2
,1 /NO,
II lO
11
Y4NO,
xo.,
нтроанилин
2. ФИЗИЧЕСКИЕ
O,N
r
O.,N /
NO,
II
i
NO.,
ЛО,
NO2
гексанитробеизол
СВОЙСТВА
И СТРОЕНИЕ
Нитроарены — желтоватые вещества со своеобразным запахом.
Ди- и полинитроарены являются кристаллическими веществами.
Нитробензол и другие нптроарены пред-
представляют собой 10л -электронную систему,
которая сильно поляризована вследствие
электропоакцепторного влияния иптрогруп-
иы. Поэтому для них характерны большие
/I-M-IO- Кл.м ,4,240) днаолы[ые моменты.
Мопопптроарены являются слабыми электропоакнепторамн. Вве-
Введение нескольких интрогруип делает их более сильными акцепто-
акцепторами. Например, сродство к электрону 1,3-дииптробензола сос-
составляет 1,35 эВ, а 1,3,5-трпнптробензола— 1,75 эВ (сравните со
сродством к электрону хппонов — с. 530).
124, Рептгепоструктурпын анализ показывает,
о^~>о что у нитробензола длина связен незпачптель-
0 137нм Холмим" но изменилась по сравнению с бензолом.
ouihJI Низшая свободная орбигаль (НСЛ\О, Мг,;) в
. нм1 i нитробензоле расположена «ниже», чей в нитро-
0,136нм — ^^ ^ .
алканах. Это свидетельствует о солыпем сродст-
сродстве к электрону. Присоединяясь к пптроарену, электрон по-
попадает па ИСАЮ и почти целиком локализуется ::а пптрогруппе
(Угу, па пптрогрунпе в НС1ЛЮ составляет 0,793) (рпс. 92).
Эффективные л-электронпые заряды показывают, что в о- it n-
положепиях имеется недостаток электронов, что делает атаку элек-
трофнльпого реагента в этих положениях маловероятной.
Нитробензол и другие нитроарепы интенсивно поглощают в уль-
ультрафиолетовой области спектра. Некоторые иитроарепы, особен-
особенно с двумя и более питрогруппами в .\:о,чеку.тс, имеют слабое погло-
поглощение в видимой области, чем объясняется желтая окраска ннтро-
арепов.
Нитробензол поглощает при 252 им (.^10 0(H), 280 нм (f^IOOO)
и 330 им ((--«125). 11срв1,1С два максимума связаны с лг—>гт*-перехо ¦
дом. Ма.'юиитеисшзныи максимум связан с п->-л*-переходом в нп-
трогрупие.
3. ХПЛШЧЕСКНК СВОЙСТВА
Лля питроаренов характерны превращения пптрогруппы (восста-
новление). Кроме того, известны реакции элсктрофпльпого замеще-
замещения при денстнии сильных элек1ро(|)ильпых реагентов. Нн1|)оарепы
380
0.201
-I-
a-2.011/1
a-fi
a- O..-35/J
2= « x /»
a f 1.2/)
-4j— a - 1.481/?
41- a i 2,837/?
Рш". Р2.
T,-i.[!,[i[iiv
нитробен
0.03:
ЭффсКМШНЫС Z-4.ll-KIpoilllb[0 ЧЛря'(И
!' - (у,
Значения энергии МО, отио'алситршшыс плотности фроп-
орбптален, эффсктЕшмыс таряды л-электроиноп системы
зола
как электропоакцепторные соединения могут взаимодействовать с
(|уклеофи,"!Ы1ымп частицами. Эти реакции характерны для ди- к
iiojFHFFFfTpoapeFFOB. Особое место занимают реакции галогенпнтро-
ареиов и нитрофеио/юп.
1. Восстановление нитрогруппы. Первая ступень восстановле-
восстановления шпроарепон, как и ннтроалкапов, заканчивается присоедине-
присоединением двух электронов и двух протонов и образованием интрозоаре-
на:
Аг — КО, —- - Лг - NO -t-11,0
Потучпть чистые нптрозоареньг восстановлением [штроареиоз
весьма трудно. Нптрозоарепы легко восстанавливаются до арил-
гидроксиламппов:
ЛЛ
Аг— N0 -—>¦ Лг— N'
-'1- ОН
Лрнчгидрокспламииы легко восстанавлиВс-мотся дальше. Ско-
Скорость восстановления в значительной степени зависит от кислот-
кислотности среды. В кистой средо (рН<6) арплгпдроксп.та.мпны быстро
превращаются в гфнламишл:
Ar--M!OiI—- -Лг- XII, I-ILO
В кислых средах наблюдается также побочная реакция — пере-
перегруппировка арплгндроксиламипов в амипофенолы:
п +х-
Чистые арилгидроксиламнпы получаются при восстановлении
ннтроареиов в нейтральной среде (рНя^7).
В щелочных средах (рН>8) наблюдается образование азокси- и
азосоедииеиий вследствие конденсации арилгпдроксиламппов с
нитрозоарепами. В щелочной среде ннтрозоарены восстанавливают-
восстанавливаются медленно, поэтому в реакционной среде могут возникать доста-
достаточные для их конденсации концентрации арилгидрокспламинаи нит-
розоарепа:
а- I
ej- ,0 НОЧ on- ;\N —Аг
Аг— у// + yN—Ат *Аг— К'/ +НаО
азокеиареиы
В молекуле азоксиарена имеется типичная семиполярпая связь
К—О. При их восстановлении образуются азосоедпнепия и даль-
дальше — гпдразосоединения:
О- Н
;.N-Ar 2e_ /N-Ar Oe- /N-Ar
Аг — N^ >¦ Ar— N^ +ILOJ * Аг —\/
2Н' азоарепы 211+ I
н
гидразоарены
При восстановлении гндразоаренов (М,.\'-днарилгидразинов)
получаются арилампны:
ArX'HM IAr — -* 2Лг\'Н„
211 +
Таким образом, изменением рН реакционной среды и восстанав-
восстанавливающих реагентов можно получить различные продукты восста-
восстановления.
2. Реакции электрофильного замещения. Нитроарены малоак-
малоактивны по отношению к электрофнльным реагентам в связи с умень-
уменьшением электроподопорных свойств. Реакции электрофильного
замещения осуществляются только с сильными реагентами (нитро-
(нитрование, сульфирование олеумом при повышенной температуре). За-
Заместитель вступает преимущественно в ^-положение, которое яв-
является менее пассивированным:
382
3. Взаимодействие с нуклеофильными реагентами. Нитроаре-
ны с одной питрогрушюй являются весьма слабыми злектропоак-
цепторами и способны взаимодействовать только с сильными элек-
тронодолорами (иуклеофилами). Динитро- и особенно полппитро-
арены взаимодействуют уже с более слабыми электроподонорамн н
образуют комплекс с переносом заряда (л-комплекс):
NCh
NO? NO2
КПЗ
Образование комплекса с перекосом заряда обнаруживается
появлением нового максимума поглощения в ультрафиолетовой и
видимой областях, часто появляется окраска. Подробнее КПЗ
рассмотрены в гл. XXX.3.4.
С сильными нуклеофильными и основными реагентами (напри-
(например, со щелочью) монопитроарены взаимодействуют медленно.
В основном реакция затрагивает питрогруллы, образуются азокси-
и азосоедипения и другие продукты. В некоторых случаях при
действии щелочи и окислителей отмечается образование нитрофепо-
лов. Гндроксильпая группа вступает в о- и «-положения по отно-
отношению к нитрогруппе.
Более активны дннитроарены. Если питрогруппы находятся в
о- или /г-положениях, они активируют друг друга и возможно свое-
своеобразное нуклеофилыюе замещение иитрогруппы с уходом нитрит-
иона:
КО, КО.
-Ма-!-:ОН—+ +.\'а+КО,,
N0, ОН
Присутствие двух и особенно трех иитрогрупп в .w-положенни
способствует присоедипеппю нуклеофнлыюго реагента к углерод-
углеродному атому бензольного цикла. Образуется апиоп, в котором цикли-
циклическая сопряженная система бензола разрушена, по анион стабили-
стабилизирован вследствие сопряжения с иитрогрулпами:
NO2
КПЗ
Растворы этих продуктов присоединения имеют интенсивную
окраску.
4. Реакции галогеннитроаренов. Введение одной п особенно
двух питрогрупп в молекулу галогеплрепа резко изменяет их свой-
383
ства. Если простые галогепарепы с нуклеофплышмп реагентами вза-
взаимодействуют только в жестких условиях, то для нитрованных гало-
генаренов характерна высокая подвижность атома галогена. Здесь
выявляется сильное электроноакцепторное влияние пнтрогрупп и,
кроме того, способность питрогрупп стабилизировать промежуточ-
промежуточные продукты реакции. Действие питрогрупп проявляется только
в о- и n-положениях к атому галогена. Особо активными являются
2,4-динитро- и 2,4,6-трппитропронзводиые:
NO2
NO2
NO;
NO2
NO2
Реакции легко идут также с нейтральными реагентами — водой,
спиртами, аминами.
5. Особенности нитрофенолов. Введение питрогрупп в молеку-
молекулу фенола резко повышает его кислотность. Трииитрофенол уже
является сильной кислотой. Повышение кислотности объясняется
электроноакцепторным влиянием питрогрупп п их участием в дело-
кализации отрицательного заряда в фенолят-поие:
+
O2N
+ HS4ol
NO2
Соединение
Фенол /i-I [in рофепол
9,95 7,21
Тримптрофепол (пик-
ршюнаи кислот)
0,3а
4. ВД/КГ ГГ-ППГПГ-: ПРНДСТЛ1ЩТ1.-ЛИ
Нитробензол — бесцветная или бледно-желтая жидкость с запахом
горького миндаля. Т. пл. 5,8 С, т. кип. 210,8 С, плотность <?',5 ¦
~-1,2028. В воде растворяется плохо (~О,2(!о), вода растворяется в
нитробензоле тоже плохо (~0,25%). Легко перегоняется с водяным
паром (состав азеотропа при 98 С 12°-о нитробензола). Ядовит.
В промышленности нитробензол получают нитрованием бензола
при 40. . .50 С нитрующей смесью. Иго используют в органическом
синтезе. При восстановлении нитробензола получают главным об-
образом анилин -- основное сырье для производства органических
красителей.
Нитротолуолы. о-Нитротолуол — светло-желтая жидкость с
т. пл. 3,2 С и т. кип. 222 С, плотностью d} —1,1742. «-Нитротолу-
«-Нитротолуол — светло-желтое кристаллическое вещество, плавится при
52'С и кипит при 105'С A,2 кПа), плотность d'f 1,1226.
Получают нитротолуолы нитрованием толуола при 30—40 С.
В продукте реакции присутствует 60?о о-пзомера, 36% «-изомерг.
384
ii 4*N „u-изомера. Смесь изомеров разделяют вымораживанием п
фракционной перегонкой. Практическое применение находят о-
п «-изомеры.
Нитротолуолы используют в органическом синтезе. При восста-
восстановлении получают толуидпны — исходные для синтеза красите-
красителей. Окислением метплыюй группы получают интробензальдегпды п
иитробепзоппые кислоты.
Т ринитротолиол — желтоватое кристаллическое вещество с
т. пл. 80. . .81С,"плотность df- 1,6407.
Получают его нитрованием пптротлуолов или динптротолуола
смесью i00 -noif азотной и концентрированной серной кислот.
Тринитротолуол — одни из наиболее распространенных взрыв-
взрывчатых веществ (тротил, тол).
Нитрофенола.. 11рп нитровании фенола получаются только
о-и «-изомеры. Пмтрсфеполы образуют желтоватые кристаллы.
о-Ннтрофепол плавится при 45 С, а «-изомер — при 1Н С. Питро-
фепо.ты используют в органическом синтезе, главным образом для
получения красителей.
Пикриновая кислота (трипитрофепол) —желтое кристалличес-
кристаллическое вещество с т. нл. 122,5 С. Мало растворима в воде, образует
ярко-желтый раствор. Имеет очень горький вкус.
Получают пикриновую кислоту нитрованием фенола азотной
кислотой в растворе серной кислоты. Фактически нитруются фспол-
сульфоповые кислоты. Пикриновая кислота легко получается при
нитровании 2,4-дннитрофенола.
Пикриновая кислота использовалась в качестве желтого краси-
красителя. Не брпзмппые взрывчатые свойства открыты в конце XIX в.
Соли пикриновой кислоты называются ппкратамп. Это терми-
термически нестабильные вещества, при нагрсиашш взрываются.
Пикриновая кислота является сильным электропоякцептором и
образует кристаллические комплексы с переносом заряда с электро-
подопорнымп соединениями - -аренами, фенолами, аминами. Реак-
Реакция образования применяется для идентификации органических
соединений по точке плавления.
Глава XXII
НИТРОЗОСОЕДИНВНИЯ
Нптрозоеоедипения являются производными углеводородов, в
которых атом водорода замещен нитрозогруппой - ,V О. В зависи-
зависимости от типа гибридизации углеродного атома связи С— X возмож-
возможны различные тины питрозосоедипепий. Номенклатура ннтрозосое-
дннений подобна номенклатуре питросоедипений:
/A /N° /А /0Н
CH3NO II
^/ ^ NO
нитрозомстяп интрочобемэол с-нитрозофенол
^3 Ks 517 ЗЯ5
А. НИТРОЗОАЛКАНЫ
1. Методы получения. Прямое введение нитрозогрупиы в молекулу
алкапа не удается. Ннтрозоалкапы могут быть получены окислением
алкилгндроксиламинов, которые, в свою очередь, получают из
питроалкапов:
п„хо5
RXHOII ——'¦* RXO-I-1LO
2. Физические свойства и строение. Ннтрозоалкапы могут су-
существовать в виде мономера и днмера, что в значительной мере оп-
определяет их физические свойства. Мономеры являются зелеными
газами или жидкостями, днмеры — бесцветные вещества. При на-
нагревании димеры диссоциируют па мономеры. Ннтрозоалкапы с
сильными электропоакцепторпыми группами, например трнхлор-
нитрозометан, существуют только в мономерной форме.
Нитрозогрупна содержит полярную двойную связь Х~О и
неподеленную электронную пару па атоме азота.
Угол С—У.--0 близок к 120°.
Окраска мономерных питрозосоединений объясняется поглоще-
поглощением в видимой области спектра, что связано с /i—*-л*-переходом в
нптрозогруппе. Поглощение малоннтенсивно F60—690 им, к —
-20. . .30).
3. Химические свойства. Ннтрозоалканы являются химически
активными соединениями, реагируют как с электрофильнымп, так
и с пуклеофпльными реагентами.
При днмеризации образуются своеобразные соединения с двумя
семнполярпыми связями и связью X'-- X:
:ОХ ,R -:О , + К
/ ' 'Но— / ЧГ
Нптрозоалкапы, содержащие водородный атом рядом с пнтрозо-
групноп, способны перегруппировываться в изопптрозоеоедппення
(окснмы):
R1
L
R ГЬЛ .1.
R-C — N'
S'-fH
г R1
Г? R-
;c=n'
оксимы
Нптрозоалкапы и оксимы можно рассматривать как таутомер-
иые формы, которые образуют один сопряженный анион (сравните
еиолы и карбонильные соединения, нитроалканы и нитроновые кис-
кислоты)- Вол ее стабильной формой являются оксимы.
При окислении ннтрозоалканы превращаются в нитроалканы,
а при восстановлении — в алкнлгидроксиламины и алкилампны.
386
Б. НИТРОЗОАРЕНЫ И НИТРОЗОФЕНОЛЫ
1. Методы получения. Нитрозоарены образуются при мягком вос-
восстановлении литроарепов или мягком окислении арилгидроксиламл-
нов. Применяется также .метал/юрганическпй синтез. Нитрозофе-
нолы получают прямым питрозировапием фенолов:
ПИ [01
Лг— ХО, —¦> Лг— ХО <— ArNHOI I
6- 6 ' 6,6-
Лг — ,MgCI + О --• N — С! — > ЛгК'О -\- ЩС^
нптрозилмлпрнд
¦¦¦ ои /-/ои
\iiNO,
СНз' Ч/ А СИ;/ ч^/ " КО
о-пптрозо-п-крслол
2. Физические свойства и природа связей. Нитрозоарены явля-
являются бесцветными кристаллическими веществами, димерамн. В раст-
растворах образуются мономерные иитрозоарепы, окрашивающие |)аст-
воры в зеленый цвет.
- Своеобразно строение о-питролофенолов. В их
* ^ дсолекуле образуется прочная внутримолекулярная
водородная связь.
n-Нитрозофенолы в кристаллическом состоянии дн-
2-нитрозофе»ол мсриы
2. Химические свойства. Ннтрозоарены являются весьма актив-
активными соединениями. Они легко вступают в реакции с пуклеофиль-
ными реагентами. Например, при конденсации с ариламннами по-
получаются азосоедпнения (гл. XXVI. Б. 1), а с арплгидроксиламина-
мн — ааоксисоединения (гл. XXI. В.З).
о-Ннтрозофенолы с ионами тяжелых металлов
образуют окрашенные внутренние комплексы
Это явление используется в аналитической хи-
химии для определения некоторых металлов. На
основе нитрозонафтолсв и солей Fe(II)
получают зеленые красители. Для этой цели используют 1-пптро-
зо-2-пафтол и 1-нптрозо-2-наф1Ол-6-сульфоповую кислоту:
-V0 КО
рф
сульфоиоьая кислота
13* 3«7
Глава XXIII
АМИНЫ И ЗАМЕЩЕННЫЕ СОЛИ АММОНИЯ
Лмипы принадлежат к пропзиодпым аммиаки, в котором атомы во-
водорода замещены углеводородными остатками. Замещенные соли
аммония, I) свою очередь, являются производными попа аммония.
КЛАССИФИКАЦИЯ. ИЗОМЕРИЯ И НОМЕНКЛАТУРА
В зависимости от степени замешанности аммиака или иона аммония
амины и соли аммония подразделяются следующим образом: ;
R-MI,
R ' — X Н
R
R1- Х- R2
i
R
первичные
амины
вторичные
ямины
трети чные
амины
\'Н;,1Х-
RJ —XII,
I
R
И
M(l|iO.)i:\ICIHClni:,IO СОЛИ
x-
дпл;] мсщо
аммония
сели
-R-
I
R
I
R1 — N't — R'J
v- тризамсгцрнные с(-лн
аммония
аммония
_ четырехзамещснмие голи
аммония
В зависимости от типа гибридизации углеродного атома выде-
выделяют три группы аминов.
1. Соединения со связью С (s/Я) — N. К этой группе принадлежат
алкиламины и циклоалкиламииы, а также некоторые алкеннлами-
пы, алкппиламины, в которых двойная или тройная связь удалена
от атома азота, и арилалкиламины. К ним могут быть отнесены и
циклические амины, в которых атом азота находится в цикле. Цик-
Циклические амины принадлежат к гетероциклическим соединениям,
по, с другой стороны, они являются типичными вторичными н тре-
третичными алкиламинами:
с... и
Rl
R1
1
(СП,
1
(R
зт
in ¦ 1^'/
С С —
н
1
>., СИ —
1
II 11Л1
R
R
/
1
V
N
I
с„
R1
1
— С-
I
R1
R
R
Л1к;
н,„;
\
\
/ //
R1
\-'
- 1
\
н,
/R
NR
/R
R
AI
)
R
i
С
R
к
2
R!-C-
1 \
\
- -\
/
1 /п
. Лг).
с
R
R
R1
' - С
\ 1
R1
R1
\R'
- ) - \
J"
\
С - ' -
, п
\ ¦
\
R
R
/
;' —С
\
R'\
R1/»
2. Соединения со связью С (s/?-)—W К этом группе принадлежат
производные алкенов с атомом азота у углеродного атома, образую-
образующего двойную связь (енамииы или внниламины) и производные аре-
нов с атомом азота у углеродного атома цикла (арпламипы):
R* \
С С
R-> R-
R
R1
R:J
R1
R1
3. Соединения со связью С (sp)—W К этой группе принадлежат
производные алкннов с атомом азота у углеродного атома, образую-
образующего тройную связь (ипамппы):
/
R1
В этой главе рассмотрены также некоторые производные с уже
известными функциями — ампиоалканолы, амппофеполи, амино-
сульфоповые кислоты, амипопптросоедипеппя.
/ Номенклатура аминов основывается па названии углеводород-
углеводородных остатков или углеводорода и слова «амии». Нел и к атому азота
присоединены различные углеводородные остатки, за основу наз-
названия принимают название самой длинном углеродной цепи, не-
непосредственно связанной с атомом азота. Группа —NIL называет-
называется аминогруппой, группы—NHR и —\R2—а.ткплампно- и
дналкиламипогруппами, по в образовании названия ампня эти
обозначения применяются только п сложных структурах. Для мно-
многих арнламипов сохраняются тривиальные названия:
СН3 —NH
мот плямяп
И
СНЯ — N — СН3
дпмети.'inмим
с,н5
I
Три j
0 .") 1 1 2 1
СН.СН^СН.СНСНоСН,., Cll,- CH — \Hj
гекс;нимим-3 ьилнламки анилин (фоиллпмтн
1
CH;t
II 5 4 3 2] С П3
СНзСПаСН., — N-' СН,СН,СН..СП—N СИ
CH:J CI h
К-мотилпрош1л<'1\1ИП N,N-AHMeTiKniioinnii,'iMnii- 2 >
4 см.
-1М<''ТП.1 1 ¦ про-
11 с 111 ] л;: м; 111
389
,С1Ь,
хн3
-К '
с,п5
N-метил-\' ¦ -т;'.л-1 - N-метил анилин К-метпл-Ч-'лпл-р-
цнклопснтег.пламип п^фтплг.мп н
Названия диаминов образуют из названия углеводородного двух-
двухвалентного остатка (рекомендуется также простое название углево-
углеводорода) и суффикса -диамин:
ibxciioCHAHo h,nch.:CH./:h,ch,ch./;ii,xh;;
эти.чокдплмп ii гокггшдкямпн-1 ,П,
(дтапди JMini) гскеау.отплрндш'.мин
сн3ч .. /сп3
и
//\
:ХН2
//-фсимлендмами
си./ - сн3
Циклические амины называют, используя номенклатуру гетеро-
гетероциклических соединений или добавляя к названию двухвалентного
углеводородного остатка суффикс -имин:
: — сн, н.,с
"„ нгсч
¦ сн,
I
сн»
СН,
н
Л'
ГХ"' " ЧСН,
н
п п ррол т; Д1:т,
тет]);,метн.-..счш«|!н
П,С СН2 П,с
н
хп.
пиперидин,
пепт^метп-
лоппмпп
Название солен аммония образуют строго по названию самого
амина с добавкой суффикса -оний пли -иний. Например:
г П
[С2П.-,\:Нз1С1- СН,, —N—СН:1 Вг-
сн3 J
хлорид отплам.чонпя бромид три мот ил аммония
н
.сн.. ' I I i I
ХОГ
нитрат
иодггд N. N-днметил*
аиплиния
390
А. АЛКИЛАМИНЫ,
ЦИКЛИЧЕСКИЕ АМИНЫ И АРИЛАЛ КИЛАМИНЫ
I. МЕТОДЫ ПОЛУЧЕНИЯ
Для получения алкнламинов и их аналогов применяют реакции
алкилпроваппя аммиака и аминов, восстановление некоторых
азотсодержащих соединении и специальные методы.
1. Алкилирование аммиака и аминов. При взаимодействии ам-
аммиака с галогеналкапами или другими алкилпруклцими реагентами
(алкплсерпымн кислотами, диалкнлеульфатамн) иолучг.ют соли
аммония и амины. Реакцию открыл А. Го(|)мап в 1850 г. На первой
ступени реакции образуются соли алкпламмонпя, которые при взаи-
взаимодействии с аммиаком в равновесной реакции образуют алкнлампн.
Последний реагирует с галогепалкапом п образует соль диалкнлам-
М01ШЯ. Так постепенно образуются и соли триалкиламмопня и
тетраалкиламмония:
б •• л - •• mi,
П;1\: ЬК-Х -> [R-ХТУХ -;\.' R -\П,-:-ХП, Х~
л. л- mi,
R--NIL-;-R — X -> [R.XHolX- .-; R All -- XII i X~
fi ¦¦ Л- - MI,
RAII-; R-X -> [R,NII1X- ;. : R:iX:~XlU~X-
R3\:-LR-x"--> [R,\]X-
B избытке аммиака преимуществеппо образуется первичный амин.
Для получения солей тетраалкиламмопия лучше алкнлировать
третичные амины.
Для алкнлироваппя могут быть использованы алкаиолы и дн-
алкиловые или циклические эфпры. В этом случае применяют ка-
каталитический способ (над Л1оОя при 300 С):
ROH + NH,, -- >R— XHo !-НоО
2ROH-|-XII..! - у RoXII + HjO
ROII--RXJU - yR.jXn-i-ПоО
Этим способом получают также циклические амины, например пир-
пиррол иди и:
II
Очень активными алкплпрующпмп реагентами являются энок-
сиды:
чох /СИ..СН.,ОН
R.XFI.,-1- * ,, / - , RXHCHXHoOH "R-\< " "
U' XCHXH,OII
391
Известии также реакции внутримолекулярного алкплировання.
Например, из этаполамипа получают этплепнмин. Сначала при
действии концентрированной серной кислоты образуется соль амп-
поэтплеульфата:
При подтелачнваппм раствора 2-ампноэтплсульфонат цпклпзуется:
СН2-СН2 СН2
HSO«
и
При нагревании этаполамина с разбавленными кислотами про-
происходит взаимодействие двух молекул и образуется пиперазнп:
ОН Н
СИ, СП., М4.х._
I + I --
СИ. ,СН, '°
^NIU НО'
+ 2НаО
2. Реакции восстановления. При восстановлении азотсодержа-
азотсодержащего соединения со связью С—N и связями N—О, \- О, M--N,
N -N, K--.--N и также связями С- \, С--- N можно получить амины.
Например, восстановление нитросоедипепий, окснмов (с. 471),
нитрилов (с. 599), изоцнаппдов (с. 602):
R —NO
RCH
R-C -
R —XC
[HI
, - R
It
NOH
|H]
- К '
'"- R-
— XH_,
i|
- RCH..-
RCH..-
-X—CH
H
-NH
XHj
3. Специальные методы. Для получения алкиламппов разрабо-
разработано множество специальных методов. Например, чистые первичные
амины получают расщеплением амидов карбоновых кислот галоге-
галогенами в щелочной среде (Л. Гофман) (с. 586):
к-с'° -^!r_nh8
ХН-, Иг,
Первичные амины получаются при алкплировании it расщепле-
расщеплении фталимида (с. 588).
2. ФИЗИЧЕСКИЕ СВОЙСТВА И СТРОЕНИЕ
Алкпламины в обычных условиях являются бесцветными газами
или жидкостями со своеобразггым запахом, напоминающим аммиак.
При большом разбавлении запах напоминает запах рыбы. Лмпны с
39°
большим числом углеродных атомов пли двумя первичными и-и
вторичными аминогруппами могут быть также кристаллическими
веществами.
Амины легко растворяются в воде. При увеличении числа ато-
атомов углерода в молекуле растворимость в воде постепенно умень-
уменьшается (табл. 38).
Тив.тци j"<. Физические константы некоторых алкиламиноа
СН;,\П,
(СП:1)АП
(СП:,)А
ГЛАСИЛИ,
(С[1,СИ,!А1[
(СП,СИ,):1\
GJIAH,
(О,НТ)АП
(С;:НТ,,\
Т. т..
'С
-92,5
90,0
124,0
80,0
- 50,0
-114,8
—83,0
- 39.0
-93.5
Т. ни.,
°с
0,5
7,4
3,5
10,0
55.5
89,5
98,7
110.7
150,0
0,709
0.080
U,(i0'2
0,700
0,700
0,723
0,719
0,738
0,757
(
(О1
(....
@
B0
B5
B0
B0
B5
70 С)
С)
5 С)
С)
С)
С)
'Q
С)
С)
К" ¦м
4,9
3,9
2,9
4,3
4,0
3,0
4,3
3,7
2,5
I)
1 , Ю
1 .17
0.80
1 .3
1 .2
0.0
1 .3
1.1
0.75
1. Особенности температур плавления и кипения. Р.слн сравнить
температуры плавления п кипения первичных, вторичных п тре-
тпчпых аминов с примерно одинаковой молекулярной массой, го
можно наблюдать определенные закономерности. Как правило,
эти температуры у третичных аминов ниже, чем у вторичных п
первичных аминов со сравнимой молекулярной маессп. 1-hn объяс-
объясняется образованием межмолекулярпых водородных сик;;еп:
.. \
<
R-N
Ь :|4
S' *
-—н
R
ь-\
.... .^
t
,V. н
Ho yni межмолеку.тярпые водородные евя.чн слабее, чем в слу-
случае алкаполо», так как сня:;ь X ~11 менее полирпа, чем свягл, О И.
Г-)то очень бтчетлпво отражается при сравнении температур кипения
мегиламппа и метанола.
2. Полярность связей и электронодонорные свойства аминов.
А.ткнламипы менее полярпы по сравнению с ал капо/тми. Дппо.ть-
пыи момент связи С X (~1,5-10~:'° Кл-м или 0,45 D) меньше дп-
полыюго момеи-ia связи X—11 (~4,.3-1()":1" К.т-м или 1,.D).
Н аминах, так же как в аммиаке, у агома а.чога имеется пеподелен-
ная электронная на|)я, более подвижная, чем ч.тектроппая пара па
атоме кислорода, так как атом азота имеет меньший ядерный наряд
и меньшую чтектроогрппачелытсть.
3.93
Электродонорпые свойства можно охарактеризовать энергиями
ионизации. Для аминов СоН5ХН2, (С2Н5)Л'Н и (С2Н3)ЯХ ЭИ равны
8,9, 8,0 и 7,5 эВ. Как правило, ЭИ .меньше па 1,6—1,7 эВ по срав-
сравнению со спиртами и эфпрамн. Сравнительно низкие ЭИ свидетель-
свидетельствуют о том, что амины, особенно третичные, являются сильными
донорами электронов, легко окисляются:
К:Л': " - R3\' - -!-<-'-
3. Пространственное строение молекул аминов и гибридизация
атома азота. Амины, подобно аммиаку, образуют пирамидальную
молекулу с атомом азота в вершине пирамиды. Углы между связя-
связями RXR'1, R'XR- и RXR2 в среднем равны 106—108 , т. е. близки
к тетраэдрпческому:
О R1
R
Можно предполагать, что электронные орбпталп атома азота
гт'ридизпровапы подобно орбнталям атома углерода в состоянии
л/;:|-гпбриднзацпп и поэтому пеподелеппая пара электронов атома
азота занимает пространственно направленную орбиталь s/):1-THiia.
Длина связей С - X 0,145 им, X—Н 0,1 им.
Так как атом азота при различных заместителях становится
асимметрическим, возможно существование двух эпаптиомеров
(зеркальных изомеров), только роль четвертого заместителя у асим-
асимметрического атома углерода в случае атома азота выполняет пе-
пеподелеппая электронная пара. Но такая конфигурация является
подвижной и происходит быстрый переход одной конфигурации в
другую путем инверсии .молекулы.
В молекуле аммиака инверсия осуществляется очень быстро,
~103 раз в секунду. У алкпламипов скорость инверсии уменьшает-
уменьшается, т. е. энергетический барьер увеличивается, по все-таки переход
осуществляется легко и выделить оптически активные эпаптиомеры
невозможно. Только в случае аминов определенной структуры, в
частности некоторых производных этилепнмипа, удается «заморо-
«заморозить» инверсию п выделить оба эиаптпомера. Например:
Четырехзамещенпые соли аммония являются полными аналогами
в стереохпмпческом отношении углеродных соединении, т. е. могут
образовать устойчивые оптические активные эпаптиомеры:
4. Спектры поглощения. В электронных спектрах аминов по-
поглощение наблюдается только в далекой ультрафиолетовой области
при 195—215 им с е- -1500. . .4000, что связано с возбуждением пе-
поделенпой электронной пары (переход и-mj*).
В инфракрасных спектрах первичных н вторичных аминов ха-
характерно поглощение в районе 3200—ЗГЮО см, что связано с
валентными колебаниями связен N—Н. Поглощение пеассоцпнро-
ванпых аминов наблюдается в разбавленных растворах углеводоро-
углеводородов; первичные амины характеризуются двумя полосами при
~3500 и ~3400 см, вторичные амины — одной полосой при
3310—3350 см. Образование межмолекулярпых водородных свя-
связей уменьшает волновое число максимума поглощения. Аммониевые
соли поглощают при 3030—3300 см.
В спектрах ГШР химический едпиг б протона связи N—Н на-
находится в области 0,5—3 и значительно меняется от концентрации
раствора, растворителя, температуры.
3. ХПДШЧССКШ7 СВОЙСТВА
Реакции аминов определяются главным образом присутствием
подвижной неподелепной электронной пары у атома азота. В мень-
меньшей степени характерны реакции связей N—Н и \—С с пуклео-
фильпыми реагентами. Особое место занимают реакции аммоние-
аммониевых производных, в которых имеется полярная связь X—С.
1. Основность. Ллкиламины и их аналоги, амины со связью
С (л/?:1)—N являются сильными основаниями, легко присоединяют
протон и кислоты Льюиса (ЭХ„ и др.). Основность выявляется уже
в водных растворах:
RaX.' + n.O rz:: R:1Mr >-Oir-
R;.\":-!-3X,,r.7R:l\:--3Xn n.iir Rr.\ —ЭХК
Для характеристики основности используются константы кислот-
кислотности аммониевых попов /(Ы|Г в единицах р/<\ш + '~—lgA'Bllt-.
Соединение p/чццн- (ПЛ) Cof-дннйние.
Nn-i<; 9,25 С,Н,Х1Г, 10,5
СИзЧН» 10,6 (Colt-,)o\H 11,0
(CH-i)A'H 10,7 (CH-J-jX 10,8
(СН;1)Л' 9,8
11,3
it
По сравнению с аммиаком все алкпламипы и их аналоги явля-
являются более сильными основаниями (кроме эгпленимнна). При этом
дналкнламииы являются более сильными основаниями, чем алкнл-
395
амины. Здесь ясно выражается электроподонорный эффект алкпль-
ных групп. Можно было бы ожидать, что третичные амины будут
самыми сильными основаниями, однако в водных растворах этого
не наблюдается, очевидно, из-за специфической сольватации тре-
третичного амина молекулами воды. В неводных растворителях ряд
основности оказывается ожидаемым - самыми сильными основа-
основаниями являются третичные амины R3N.
Вторичные и третичные амины (диэтнламнп, ипрролидпн, пипе-
пиперидин, трнэтпламин) — самые сильные органические нейтральные
основания и их широко используют в органическом синтезе в ка-
качестве основных катализаторов.
2. Реакции с различными электрофильными реагентами. Амины
как сильные нуклеофилы легко взаимодействуют с различными
электрофилами.
Лмппы, подобно аммиаку, реагируют с ионами различных метал-
металлов п образуют допорпо-акцепторные комплексы, в которых амины
выступают в качестве донора (лпгапда с двумя электронами):
L, R,N. .NR;
Y 'Ч /
R—NH,
В общем случае не существует параллельности междч1 осповшхчыо и
пуклеофпльпымн свойствами. Но всегда ("олсе основные ямины кн.г.ио.ч-я :"о-
лео сильными пукле.офилами. Основность хпракториjyev термодинамическое
равновесие между амином и попом аммония, а иукле-о^нльность солиш; ха-
paKTepujyer кш;е шче.скне свойства, CKopocib реакции н .чавн-'П г or типа
субстрата (у какого атома происходит реакция, какие прострапет г.еппые
ьффикгы мешают реакции).
При рассмотрении реакции аминов необходимо обратить внима-
внимание па различие между первичными, вторичными п третичными
аминами. В случае первичных п вторичных аминов взаимодействие
с электрофилами в большинстве случаев заканчивается отщепле-
отщеплением протона, т. с. осуществляется замещение у атома азота. В слу-
случае третичных аминов это невозможно и образуется более или менее
прочная ковалептпая связь с атомом азота, на котором появляется
положительный заряд.
а) В з а и м оде » с т в п е аминов с С ¦ э л е к т р о ф н-
л а м п занимает видное место в органической химии. К таким
реакциям относятся алкплпрование аминов с га.тогепуглеводоро-
да.мп, зфпра.ми неорганических и эле.менторганпчески.х кислот:
Г н
Н
R — \ — CR'K-R-1 ¦!- Н — X
Н
Важны реакции с карбонильными соединениями и функциона-
функциональными производными карбоповых кислот (см. с.449, 568, 583;:
RL V R> Y
R- NM, !-СУ- -R—X—С - ...
оЛ- н он
Лмппы при реакции с карбепамп дают различные продукты. Ре-
Реакция начинается образованием семпполярной связи X—С, потом
следует перемещение протона:
С\ R' н 1 Н |
R-NH, +- J\ ., —- R-N-CC^ , —- R—N—CC^R,
& H H
Своеобразная дальнейшая реакция происходит при взаимодей-
взаимодействии с дихлоркарбено.м (R1 -R- С1), генерируемым из хлорофор-
хлороформа щелочью. Образуются пзоциаппды (изопитрилы) — ядовитые
соединения с отвратительным запахом (гл. XXXIII. /I):
R—XII, ;-HCCI:1 '-.iXaOII - R —\C---3\aCI [-ЗП2О
б) Р е а к и и и а м п н о в с X - э л е к т р о ф и л а м и слу-
служат для получения соединений со связью X—X. Реакция ннтрозн-
ровапия аминов происходит при взаимодействии с азотистой кисло-
кислотой в кислой среде. Нитрозпрующпм реагентом являются ннтрозил-
л л—
производные О X -X, где X Cl, Br, O3SOII, OXO. Предполагают
также возможность образования свободного катиона питрозоппя
1X01' X".
Первичные амины при иптрозировапии образуют катион дпазо-
нпя, который раз.чагается с выделением азота:
г- II
1
RXII,-; (Х()| Х- - - I R--N" —X О
— к-к
I. II J
, 11,0
Х- R —ОП-г\.,-;-П X-
В реакции образуются также соединения R —X (если X — CI.
Вг) п а.чкены.
Реакция называется дназотнроваппем, механизм образования
катиона д^иазония подробно рассмотрен и случае арпламиион (гл.
XXV. АЛ).
Вторичные амины образуют Х-пнтрозоамнпы:
. II
RRlXII-!-[X()J- Х- - - RR'X X-.-.-RR'X—X О-Н X"
. N О.
Третичные амины могут образовать продукты присоединения:
3''?
Амины могут вступать в реакцию нитрования при взаимодей-
взаимодействии с солями иптропия, например [i\O2];BF4":
П
R —XH.,-1-|NO.,]i-BI:7 —> I R —X —NO., ; BF7 C2 R — X —XO., i-II-BFJ
I ; I
II J II
Образуются Х-питроамниы.
Реакции аминов с солями диазопия Лг\.,Х~ рассмотрены в гл.
XXV. А.З.
в) Р е а к ц и п аминов с О - э л ектр о ф п л а м и обыч-
обычно называются реакциями окисления. В качестве окислителен при-
применяют пероксид водорода МООП и органические пероксиды ROOH,
ROOR1- Первичными продуктами окисления в мягких условиях
являются N-оксиде)! аминов. В случае первичных и вторичных
аминов они перегруппировываются в производные гидроксиламнна:
:б: "
I
RR'X — H-i-HOOH -, RR'X ¦¦ — H-|-H,0 --- RR'XOII \- 11,0
R3X:-i-HOOH " - R;1X—O:--|-H2O
При окислении в более жестких условиях возможно протекание
свободнорадикальпых процессов, которые начинаются образованием
катион-радикала. Потом следует отрыв атома водорода от углево-
углеводородного остатка и образование двойной связи С - \ имипа:
.. [О]
RClIoXIb - [RCHoXIb] -- RCII ХП
И/) С)
RCH XII - RC •
"И
В водной среде пмины гидролизуются и образуют альдегиды.
В общем случае можно сказать, что при окислении алкнлами-
нов н их аналогов можно получить карбонильные соединения и
карбоновыс кислоты, что подобно окислению алканолов, цпклоал-
каполов, арилалкаполов.
г) Р е а к ц и и с галогенами. Амины образуют с га-
галогенами допорпо-акцепторпый комплекс, который переходит в
соответствующий N.-галогенамин:
П II г И 1
R —X: + X.. —: R — Х:-+Х., Г- i R —X — X 1 X" ^ * R — N — X -|- И - X -
I I I I
II II L- Н J И
Фтор реагирует слишком энергично н для получения Г\-фтор-
аминов требуется специальная методика, \-x.iop-, N-бром- и N-
иодамипы являются соединениями весьма неустойчивыми.
д) П р п р е а к ц и я х с S - э л е к т р о ф и л а м и образу-
образуется связь N— S. Следует упомянуть о взаимодействии с SO3 и
сульфопплхлоридами.
398
Амины с S0:t образуют допоргго-акцегтторпьш комтекс, кото-
который is случае первичных н вторичных аминов переходит в Х-суль-
фоновые кислоты:
RR'X: ¦-SO3^±RR1.\:-*SO3 —> RR'N'—SO3H
>1
R7
л-
. X
6-1-
-H-
л -
¦ CH;i-
Л :
L> —
IR1
1
1 R
II II
Лмппы с сульфопнлхлорг.дамп образуют сульфопамнды (гл.
XVII 1.3).
3. Ионизация связи N—Н. Первичные и вторичные алкпламнны
и их аналоги являются очень слабыми \Н-кислотами, слабее амми-
аммиака, рК„~-33. . .35. Только взаимодействие с металлоргаппчеекпмн
соединениями ведет к ионизации связи и образованию производ-
производных металлов — алкллампдов и диалкнлампдов:
Li -I-C1I.,
Замещенные амиды металлов — сильные основания. Будучи сте-
рически затрудненными (например, диизопропиламид лития) для
нуклеофильной атаки, они являются сильными основаниями, по
слабыми нуклеофиламп. Их применяют в органическом синтезе для
отрыва протона и генерирования карбаииоиов.
4. Особенности реакционной способности четырехзамещенных
солей аммония. Эти соли являются бесцветными крнсталлически-
мы соединениями, хорошо растворяются в воде. Удается получить
также гидроксиды тетраалкиламмоиия, например, действием Ag..O
на галогеппды пли Ва(ОПJ па сульфаты или пропуская раствор
через колонку с ионообменной смолой (апнопитом);
2R,\- Br--|-.\g,()-i Г Г.,О -у 2R,N ¦ OH--'-2.\gBr
В водных растворах гидрокепды тетраалкнламмопня полностью
ионизованы и являются столь же сильными основаниями, как гид-
гидрокепды натрия и калия. Некоторые из гидроксидов тетраалкилам-
тетраалкиламмоиия удается получить в кристаллическом состоянии, обычно в
виде гидратов. При кипячении водных растворов гидроксидов они
постепенно распадаются па триалкпламин, алкапол пли алкеп:
/¦
Алкен.ы легко образуются из солей тетраалкиламмоиия с более
длинными и разветвленными заместителями:
R
i
(CH3KN-C[[-CH.,RiJ Br- 1 > (CH.,).iN-;-RCH - CHR1
X.-i-'ОП-
[-CH..R1J
Эту реакцию открыл н изучил А. Гофман A881). Реакция является
бимолекулярным отщеплением, /Гл-2, подобна некоторым реакциям
галогеналкапов. В этом случае роль электронооттягпвающего за-
заместителя исполняет триалкиламмопиевая группа.
399
Если в реакции отщепления могут образоваться два различных
алкена, то в противоположность правилу Зайцева (гл. VII.А. 4.5)
образуется наименее замещенный алкен (правило Гофмана). На-
Например, при расщеплении гидрокспда й/нор-бутплтрпметнламмония
преимущественно образуется бутен-1
,N— СНСН.2С1Ь.|ОН- ' (CI]:tKN' + CH,- CHCII,.CH,-i-IbO
Наиболее стабильным является гндрокспд тетраметпламмоппя.
В водных растворах он разлагается на трпметнлампп и метанол
только при нагревании выше 250 'С.
В связи с тем, что некоторые гпдроксиды тетраалкпламмония,
являясь сильными основаниями, растворяются в органических
растворителях, их применяют в качестве катализаторов органичес-
органических реакций, а также в качестве аналитических реагентов для не-
неводного титрования.
Важное значение имеют такие четырехзамещепные соли аммо-
аммония, которые содержат один или несколько длннноцепных
(С,—С,о) углеводородных заместителей. Они обладают поверхностно-
активными свойствами и, кроме того, способностью растворяться в
органических растворителях. Их используют в качестве межфазных
катализаторов, т. е. для переноса одного из реагентов из твердой
пли водной фазы в органическую фазу, например в реакциях нук-
леофильного замещения (гл. VII.А.4.3), в реакциях генерирования
карбанионов и карбенов (сравните с краун-эфпрамн и криптапдамп).
4. ВАЖНЕЙШИЙ ПРЕДСТАВИТЕЛИ
Метиламин — бесцветный газ с острым запахом, подобным запаху
аммиака, а при большом разбавлении напоминающим запах селе-
селедочного рассола. Хорошо растворяется в воде, насыщенный раст-
пор содержит 35. . .40% метиламина.
Хлорид мстил аммония образует бесцветные кристаллы с т. пл.
225 ;С.
В промышленности метиламин получают каталитическим гид-
гидрированием цианистого водорода или при взаимодействии метано-
метанола и аммиака над катализатором при повышенной температуре:
п2
И—С =N >СНя -ХН,
Кат., /° •'
Метиламин применяется в органическом синтезе для получения
лекарственных веществ, красителей н поверхностно-активных ве-
веществ.
Димстиламин — газ с неприятным запахом, хорошо раствори-
растворимый в воде. Хлорид диметнламмония — бесцветное кристалличес-
кристаллическое вещество с т. пл. 171 С.
Получают диметиламнн каталитическим алкилпрованнем амми-
аммиака метанолом:
з
Jv ят.
400
Его используют в синтезе лекарственных веществ и растворителем
(диметплформамид).
Триэтиламин — бесцветная жидкость с характерным запахом
аминов. В промышленности его получают каталитическим алкплп-
ровапнем аммиака этанолом:
ЗСЛ1,ОН + \П:, , - (СН.-ОяХ-рЗН/)
К.эт.
Трнэтпламин используют в органическом синтезе в качестве
исходного вещества, растворителя и основного катализатора.
Этилендиамин (этандиамнп) 11 А'СН ,СА IАН, представ тет собой
бесцветную жидкость с запахом аммиака; т. пл. 8,5 С, т. кип. 117 С,
хорошо растворяется в воде.
1:го получают из дихлорэтана и аммиака:
С1С1ЬС11,С1-; 1\П;, - HA'CHoCHoMlo-: 2\H,CI
Этплепдпампн является важным исходным веществом для синте-
синтеза аналитических реагентов (комплексопов) п фунгицидов. Соли
этплепдпамина с высшими карбоповымн кислотами применяются в
текстильной промышленности в качестве мягчителен.
Гсксамепшлендиамин (гексапдпампи-1,6) --бесцветное кристал-
кристаллическое вещество с т. пл. 42 С и т. кип. 204 С, растворяется в воде.
Связывает из воздуха СО... (на влажном воздухе «дымит»).
В промышленности его получают каталитическим гидрирова-
гидрированием дппитрила аднниповон кислоты:
rV:-=c-cw..ai..(:n..cn,-с \ ' н.,\си..сп.,си..сп>сн,сн,хи..
- - - \i" ------
Гексаметнлепднампн используют для получения полиамидов и
синтетического волокна (найлон).
Этаиоламины -- бесцветные вязкие жидкости, при более низ-
низких температурах — кристаллические вещества, хорошо растворя-
растворяются в воде:
н,\сн,сн/)н н\!(аьсн.,он)., \(СПоСн.,он)л
'.Til ИОЛ ЯМ [ИГ Д И'JT Л IIО'тМПМ Три )ТПМОЛ;1\1И И
т. ]]-i. 10,". С, т. llj. '11,М С. т. пл. 21.2 ''С,
т. кип. 171 С т. кнгг. 27| 'С т. Finn. i'jO "С
Этаноламин получают из этпленокепда и аммиака. Они являют-
являются основаниями, в то же время обладают всеми свойствами алкано-
лов.
Этаполампны используют в органическом синтезе для получения
эмульгаторов, моющих средств и косметических препаратов, как
абсорбенты кислых газов (H2S, CO.., HCX) п как ингибиторы кор-
коррозии.
Важным производным этаполамина является холин [гпдрокенд
трпметнл B-гидроксиэтнл) аммония I:
ЦСН3K\'СН2СН..ОН]ОН- --Л (CH,KNCH2CH.jO-.H2O
40J
Холпн — бесцветное гигроскопичное кристаллическое вещество,
является О-основанием, рН1И| ~9, его свойствам больше соот-
соответствует строение внутренней соли (бетаина), чем гндроксида.
Холии широко распространен в животных и растительных тка-
тканях, высоко его содержание в нервной ткани. Холи и играет
важную роль в обмене веществ, входит в состав фосфолипндов. Важ-
Важную роль в механизме передачи нервных импульсов (медиации)
играет ацетил холпн [хлорид триметилB-ацетокспэтпл)аммония]:
Ll~ — соединение, ооладающее очень
высокой физиологической активностью.
Этиленимин — бесцветная жидкость с т. кип. 55 С. Получают
его из этаиолампна (см. гл. XXIII.Л.1).
Этнлепимпн имеет более слабые основные свойства, чем вторич-
вторичные амины и даже аммиак (рКШ1 • ~7,У). Это свидетельствует о
том, что атом азота в этплеппмипе имеет другой характер гибриди-
гибридизации, чем в дпалкиламинах и пиперидине. Энергия связей С—\
в этиленимпне уменьшена вследствие неполного перекрывания ор-
бнталей (сравните с циклопропаном — гл. V.2 и этилепоксндом—
гл. XVI.Б.2).
При взаимодействии с нуклеофнльпыми реагентами у этнлени-
мипа и его производных в отличие от обычных вторичных аминов
цикл расщепляется, т. е. разрывается связь С—N:
it- S-t-
сн2—сн2
\..У + :NuH RNHCH,CH2Nu
N
R
Происходит амнпоэтилпрованпе пуклеофилыюго реагента. Реак-
Реакцию ускоряет присутствие кислоты.
В присутствии кислот Льюиса происходит полимеризация эти-
леппмнпа. В мягких условиях можно осуществить реакции у атома
азота этилеппмнпа — алкнлированне и ацилировапие.
Этилепнмпн и его производные применяют в текстильной про-
промышленности для обработки тканей и в качестве добавки к ракет-
ракетным топлпвам. Некоторые производные этилепимипа используют-
используются в медицине как активные противораковые препараты. Этплен-
нмнп очень ядовит.
Пиперидин — бесцветная жидкость со своеобразным амиппым
запахом; т. кип. 106 С. Растворяется в воде. Его получают катали-
каталитическим гидрированием пиридина:
/ч
У
Ni
н
Пиперидин обладает сильными основными свойствами (рКап --~
= 11,2). Его применяют в качестве основного катализатора для
402
проведения ряда органических реакции и как исходное для полу-
получения лекарственных веществ.
Пиперазин — бесцветное гигроскопичное кристаллическое ве-
вещество ст. ил. 10ГС и т. кип. 145 С. Легко образует гексагидрат
с т. пл. 44 С. Растворяется в воде (~15?о) и спиртах. Имеет две
константы основности: p/\JA,- --=10, pKf]i{ " 5,7.
F.ro получают нагреванием этаноламппа в присутствии кислот
или каталитической циклизацией производных этилендиамппа:
н
II
II.
.с-
,с
'¦дг
II
N.
СП,
1
СИ,
ЯЧГПЮДИ'Л U
!¦
Кат.
I-
II
н
:О
Ппперазпп и сто соли используют в медицине и ветеринарии в
качестве противопаразнгарных средств (аптпгсльмпиты).
Криптинды представляют собой бицпклпческпе макроцпкли-
ческпе аминополнофиры, синтезирующиеся аналогично крауп-
эфпрам (см. гл. XVI.Б.5). Здесь приведено строение наиболее изу-
изученного крпптапдн, содержащего 18-члеппые циклы.
Крнпта.чды образуют очень устойчивые комп-
компах лексы с попами металлов (крнптаты). Констаи-
.-Q ^ " ' ты стабильности в 100. .1000 раз больше, чем
:О: •°^ комплексов крауп-эфпров. Ион металла помеща-
. ется в полости внутри молекулы и крепкоудер-
4 ' "¦о- •'о^ Ж'113-4-^'-51 между шестью атомами кислорода и
'• , j ' .•' двумя атомами азота и не обнаруживается
^у обычными аналитическими реакциями. В кислой
среде комплексообразующая способность крнн-
тапда исчезает, атомы азота протоппруются и комплекс разрушается.
Применяют крпптанды как меж(()азпые катализаторы, как экст-
рагеиты металлов в аналитической химии н металлургии редких
металлов.
Производные Э-фенилэтилпмина. Ряд производных 2-фенплэтпл-
амина обладают высокой физнлогическом активностью. Некото-
Некоторые из них синтезируются в живых организмах и выполняют важ-
важные функции:
ПО
СМ,
си3
X /
4CUMHH A--.:оч:л-2- -jfHupii'.i С.-не-, in-1-мотмл-
403
он
,. снсн.днсня си3оч /• сн.сн.л п..
ПО '¦' СИ„О v '
он осн:)
j.-.i'c-'ii'.ni'ii f\-метил-2- москали и [2C,1, Г)-три-
рик.пфокпд)-г, iMavuui |
Фенамин снимает чувство усталости. Эфедрин проявляет апти-
спазматическпс свойства. Адреналин, вырабатываемый надпочеч-
надпочечными железами (гормон надпочечников), проявляет сильлешпее фи-
физиологическое действие: он сужает кровеносные сосуды, ускоряет
сердцебиение, оказывает мобилизлрующее действие на организм.
.Меекалпп — алкалоид некоторых сортов кактусов. При приеме
внутрь вызывает эйфорию, красочные галлюцинации.
Все эти амины возбуждают симпатическую нервную систему,
иногда их называют симпатомиметпчеекими аминами,
Те амины, которые вырабатываются живыми организмами п иг-
играют важную роль в жизненных процессах, называются биогенными
аминами. К ним, кроме производных фепплэтиламнна, принадлежат
также холнп, ацетплхолип н некоторые производные индола
(гл. XXXVI.П.4).
Б. ЕНАМИНЫ
Известны только такие енамппы, у которых пет водородных атомов
при атоме азота. Другие енамппы являются нестабильными п пе-
перегруппировываются в азометииы (пмнпы), подобно ено.там, кото-
которые перегруппировываются в карбонильные соединения:
V-R
R3
R-
С
[1
с
\
•R1
-R
-
R:i
-
R-
И
1
С-С
1. Методы получения. Пет прямых методов получения епамппов
из га.тогепалкепов или нитроалкеиов. Для получения епамннов ис-
используют реакцию карбонильных соединении со вторичными ами-
аминами (гл. XXVII.A.3) в присутствии кислого катализатора и средств,
связывающих воду. Примером могут служить реакции вторичного
амина с карбонильным соединением в присутствии н-толуолеульфо-
новой кислоты в растворе бензола при кипячении. Вода отделяет-
отделяется в виде азеотроппон смеси с бензолом:
401
Для катализа реакции и связывания воды успешно применяют
TiCI,:
R2 R;l
| О R' R- N
2R'-C -С/ -I-GJI-X. + TiCI, -> С С. R' +
I -R R1 Rl/ NR
II
-f- TiO.,-1-4
R
C|-
2. Физические свойства и строение. Пнампиы обычно являются
бесцветными жидкостями пли кристаллическими веществами.
Молекула епамппа представляет сооой простую сопряженную
систему — пеподелеппая электронная пара атома азота и л-элек-
л-электроны двойной связи.
Атом азота действует как донор электронов
Д-t-R.i в системе я-связен ( ' М-эффект). В то же время
'2ii>vNR< !|° системе ст-связей действует электроиоакцептор-
ный эффект атома азога ( /). .Лучше всего эту
енамин сопряженную систему чарактерпзонатъ А\0 рас-
расчетами (рис. iK).
Д\олек\-ла снамипов имеет злектронодпнорные свойства. Нукле-
офпльпым центром епампиов является углеродный атом. Распреде-
Распределение электронной плотности па B3.W0 свидетельствует о том, что
1,90
i2i,
-р-
0,50
- |,U77/?
0.2:
«+0,707 р
1,12 0
0.949 ^ч
tt.iomnc:b я-|.и.'кмроков
и порядок тг-свя1еи ;)
0,11
0.12- 0.02 >
ЗффСИ МННЧО '.'ГЯ.ИЛ
ii-4,
Рис. 93. Значения энергии МО, од|юэлсктропп[,чм1ло1 пост R3MO,
плотность д-электпоноп, порядки л-сия:1С1"| и Э(|:фсктмвпые заряды
л-электронно11 систс\;ы еплмннл
напиолее вероятным центром атаки электрофплыюго реагента яв-
является ("i-углеродпый атом.
3. Химические свойства. Применение. Енамппы легко реагиру-
реагируют с различными электрофильиымп реагентами.
а) II р о т о и и р о в а и и е. Пнамипы являются более слабыми
¦основаниями, чем алкпламипы, ввиду сопряжения пеподеленпой
электронной пары с двойной связью. При этом епамппы имеют два
центра протопироваппя: атом азота и (т-углеродпый атом. Как по-
показывает опыт, преобладает протоппровапне атома углерода, в
результате чего образуются соли пммонпя. В водных растворах
405
соли иммоиия гидролизуются на карбонильное соединение и вто-
вторичный амии:
.. /R г R - ,R-
и •! х-
R-
4R
R—С —С-'
R
i
н
Н
нд'.{ |х-
4R
X
п.о
б) Л л к и л и р о в а и и е. Епамнны алкнлпруются галогеиал-
канамп или другими алкилирующими реагентами и образуют
С-алкилпродукты:
*;: ж г R
>С С( 4r-;-r'-X
4
R— С — C:v
I 4R
L R1
и., о
х - -'-
-+R_C-C;f -
I R
Rl
В водных растворах происходит гидролиз солн нммопия и об-
образуются карбонильные соединения. Таким путем можно про-
алкилировать карбонильные соединения (получение епамина из
карбонильного соединения, алкилнроваине и гидролиз — реакция
Сторка).
Енампны также легко ацплпруются и в конечном счете получа-
получаются р-дикетопы.
Реакции енампиов широко используются в органическом син-
синтезе .
Все важные химические реакции енампиов фактически обуслов-
обусловлены превращениями не аминогруппы, а поляризованной связи
с-с.
В. АРИЛАМИНЫ
1. МКТОДЫ ПОЛУЧЕНИЯ
Основными методами получения ариламинов являются восстанов-
восстановление нитроаренов, алкилировапие арпламппов, арплировапне
аммиака или ариламинов. Некоторые специфические методы будут
рассмотрены при описании отдельных представителей.
1. Реакции восстановления. Исторически впервые восстановле-
восстановление нитробензола провел русский химик Н. II. Зиннн A842): он
восстановил нитробензол сульфидами аммония и получил анилин.
Позже A880) А. Гофман сказал: «Если бы Зиннн не научил нас
ничему более, кроме превращения нитробензола в анилин, и тогда
406
имя его осталось бы записанным золотыми буквами в истории хп-
мии».
Для восстановления нитроаренов до арнламппог? в лабораторной
практике используют металл (Fe, Zn, Sn) и кислоту, соли металлов
в низших степенях окисления (SnCl2, TiCL,), каталитическое гидри-
гидрирование водородом:
ml
Аг—\О., -- Лг — XII2
В промышленности главным образом используют каталитичес-
каталитическое гидрирование водородом в жидкой или газовой фазе.
2. Реакции алкилирования служат для получения N'-алкил-
п МД'-диаткплариламниов. Лрпламппы алкилируют галогеналка-
памп, алкаполамп в присутствии кислот или другими алкплпрую-
щимп реагентами:
Лг- -\U,-rR —X
Лг-Х
3. Реакции арилирования. Галогенарены с аммиаком и аминами
реагируют только в жестких условиях B00—300 С, давление), лег-
легче всего процесс идет в присутствии катализаторов — меди и ее
соединении:
,ч С1 ,ч ,ХН,
-г 2М13
г
Кат.
-\Н4'С!-
Пели в молекуле галогенарена в о- или «-положениях находятся
электроноакценторные группы (NO2, CN, COR), реакции нуклео-
с|)ильного замещения атома галогена проходят легче.
При реакциях галогенарспов с арилампнамн в присутствии
медного катализатора получают дпарпламппы н трнариламнпы:
200-250 -С
Лг-Х + Лг\11, -Лг—X—Лг-!-П ' X-
1 " Cii(CuX)
II
Лг—Х-;-Лг —N —Лг -^ Лг — N — Лг-1-11 ¦ Х~
Н Лг
2. ФИЗИЧЕСКИЕ СВОЙСТВА II СТРОЕНИИ
Арнламииы в чистом виде представляют собой бесцветные жидкос-
жидкости или кристаллические вещества со слабым, по своеобразным за-
запахом. При хранении они желтеют вследствие окисления кислоро-
кислородом воздуха.
407
1. Полярность связей и электронодонорные свойства. Ариламн-
ны характеризуются большем сопряженной системой, чем еплмнпы.
s* Дипольный момент направлен от ато-
Сма азота на ареновып цикл. Лмнпо-
¦ 2 группа выступает в качестве сильного
=5-ю"м Кл-м <i,5d; электроподопора, проявляя -;-/И-эф-
-;-/И-эффект. По сравнению с аренами и фе-
фенолами арпламнпы имеют более силь-
сильные электроподопорные свойства. При
этом электронодонорные свойства увеличиваются числом аминогрупп
н арильных групп в молекуле. Это хорошо видно при сравнении
энергий ионизации различных ариламппов:
Соединение ЭИ, эВ Соединение ЭИ, чВ
С0Н3ОН 8,4 (С0Нг,).,\ 0,0
СоП-ДНо 7,7 СН-, . СИ-
С0Н5Х(СН3Ь 7,1 ЧЛГ-:' ^-\ 0,75
С На ' Ч^= СН:.
В семье арнлеидиамипов находится очень сильные нейтральные
электронодоноры (например, Х,Х,\",.\"-тетраметил-л-фепплеидп-
амин).
2. Результаты расчетов молекулы анилина методом МО. Наиболее пол-
полное представление о распределении электронной плотности и молекуле ани-
анилина можисГполучить при рассмотрении результатов расчета по методу моле-
молекулярных орбиталей Хюккеля (рис. 94).
Видно, что уменьшилась плотность пеподеленной электронной пары n;i
атоме азота и увеличилась плотность на о- и я-углеродпых атомах. На ныешеп
запятой молекулярной орбитали (ВЗМО) л-электронная плотность спльнч
делокализонана, главным образом перераспределена между атомом ачота п
о- и «-углеродными атомами.
-20-
А'; = «- 2.017/i
Е(.. - а - 1.0610
/.'; - а - в
N 1,847
N 0.1 |
1,041
0,998
1,034
ГГ11ННПС11. JT-VICkipOHOB
м пчрядок 71-свя ten p
Et = a -r 0.6760
/•:, = а
[ . = а + 2.1840
0,205
ВЗМО
Рис. 94. Значения энергии МО, одноялоктроппые плотноеги
ВЗМО, плотность л-электроион. порядки летней и эффекгпнные
?пряды л-алектроппоп системы апм.шнл
408
3. Спектры поглощения. По сравнению со спектрами поглощения
фенола дляапнлина максимумы поглощения в УФ-спектрах сдвину-
сдвинуты в длинноволновую область. Это соответствует расчету но методу
МО. Ниже приведены максимумы поглощения и интенсивность по-
поглощения фенола п анилина для водных растворов. В этом случае
тонкая структура не наблюдается.
Соединенно
Фенол
Анилин
Максимум
поглощении, им
210,5
270
230
280
1 hiToncHfinocTti
L
0200
1450
8000
1430
3. ХИМИЧЕСКИ!7. СВОЙСТВА
Арпламипы являются сильными электроподопорпыми реагентами,
которые имеют в отличие от алкиламппов несколько реакционных
центров. Атака электрофпльного реагента может происходить как
у атома азота, так и у углеродных атомов бензольного цикла
в о- плп «-положениях. В качестве промежуточных продуктов могут
образоваться комплексы с переносом заряда. В ряде случаев про-
происходит полный отрыв электрона от молекулы арнламипа (окисле-
(окисление) п образование дальнейших продуктов превращения, обычно
глубокоокрашепных соединений.
1. Основность. Ариламнпы в отличие от алкпламшюв являют-
являются слабыми основаниями. Протон присоединяется к атому азота:
Г R "I ¦
Лг —XRR'4-Ц ¦ X - . :\ Лг-N-H | X-
L R» j
Ниже приведены константы кислотности аммониевых попов
метиламина н некоторых ариламипов.
(.оедпнгкпе рКвп1 (П2О) Соединенно
СН-,\Н.. К).С и-().Л'С011,\Н.. 1,0
С„Н-,\Н- -U' (C,;H-,),.\H 0,78
С,;Н;,\ (СН;[),, 5.1
Уменьшение основности анилина по сравнению с метиламином
почти в миллион раз связано со значительной делокализацней не-
поде.тенпой электронной пары атома азота. Присоединению про-
протона препятствует стабильность сопряженной системы, так как
протежирование атома азота ведет к нарушению сопряженной сис-
системы, пеподеленпая электронная пара связывается.
При введении электроиоакцепторной группы в бензольный цикл
или присоединении к атому азота арнльпон группы основность
ариламппов резко уменьшается.
Соли арп.таммоппн образуют бесцветные кристаллы, раствори-
растворимые в воде. В водных растворах они частично гидролнзуются. От-
409
носителыю мало растворимыми являются гидросульфаты арнл-
аммоння.
2. Образование комплексов с переносом заряда, окисление. Ари-
ламииы как электроподопориые соединения образуют с сильными
электроиоакцепторамн допорпо-акцепторпые комплексы (комп-
(комплексы с переносом заряда, КПЗ). Часто это обнаруживается по из-
изменению цвета раствора при смешении компонентов, так как при
образовании КПЗ появляется новая полоса поглощения в видимой
области спектра (гл. XXX. 3.4). В некоторых случаях удается изо-
изолировать КПЗ в кристаллическом состоянии. Например, образова-
образование КПЗ с днпнтро- или тринитроарспамн:
VRl
.. .
сыч
О
NO;
.. R1
O2N
NO,
NO2
При взаимодействии арпламппов с сильными окислителями
(NaOCl, CrO3 и др.) происходит отрыв электрона п образование ка-
тпон-радпкалов, которые реагируют дальше п образуют красители:
г-ХН,
ГО]; -""
X-
При увеличении элсктроподопориых свойств (уменьшении ЭМ)
арнламннов возможно получение стабильных катпоп-радикальпых
солен. Это характерно для производных я-фепплендпамипа, трн-
ариламнпов:
га I,
I
N
СИЛ'
Вг-
cmtiiii BiopCTOpd
3. Реакции с различными электрофильными реагентами. В зави-
зависимости от типа электрофильного реагента, условии реакции (тем-
(температуры, растворителя) и энергии ионизации ариламипа реакция
осуществляется у атома азота или углерода, или происходит окис-
410
леппе:
R,
Н ' X - +
Н
,Н
ЧХ
I,
-, г.- х- -
R — X — II
PR
L
II"
Х--1-Е-
J
+ 11 ' Х- ;.._:
х-
Реакции с С-э л е к т р о ф и л а м и обычно заканчива-
заканчиваются образованием связи С-Л'. Реакции N-алкплнровапия и \г-
ацилнровапия идут медленнее по сравнению с алкилашшами
ввиду уменьшения основности атома азота:
^—X
Г
1
-CRR4T- |X- ^1
J
L
II
6- 6
7R
4X
Лг— \-C<
II
O
11 —X
При реакции ариламниов с хлороформом и щелочью образу-
образуются изоцнаипды (гл. XXXIII. Л).
С альдегидами арнламнпы образуют имипы (гл. XXVII. Д. 1),
в отдельных случаях наблюдается реакция по бензольному циклу.
Из р е а к ц и й с N-э л е к т р о ф и л а м и важно шгтро-
знроваппе. Первичные арнламнпы образуют соли дпазоппя:
II
Лг — X — N= -О
) Х---¦ [Лг—!\'^=\]Х- + Н2'
II J
Эта'реакция называется диазотированисм и подробно рас-
рассмотрена в гл. XXV.A.1.
Вторичные арпламнпы при шпрозпровашш образуют
розоамнпы:
КО -1 ХО
ЛгХ
н
Аг — X — R
I
II
Х- ^±Лг —х —R + H-X
411
Третичные арнламипы дают продукты С-питрозпровапия:
N-OH
Нитрование обычными нитрующими реагентами не дает жела-
желаемых результатов. В кислой среде атом азота блокирован протоном
и активация бензольного никла не происходит. В более жестких
условиях осуществляются реакции нитрования, окисления и де-
деструкции. Для получения питроапилннов лучше пользоваться
нитрованием N-ацмларнламипов (N-защищенпых арпламинов).
Нитрование IXOJ'BF," при низких температурах дает
N-нптроарнламипы или продукты окисления.
Ариламимы очень легко реагируют с солями арепдпнзонпя
(гл. XXV. А.3).
О-Э л о к т р о ф и л ы (НООН, ROOR1) обычно вызывают
окисление арнламипов с образованием катион-радикалов. В мягких
условиях возможно образование N -оксидов. Ариламины с электро-
поакценторпыми группами (пнтроапп.пшы) более устойчивы к
окислению. При действии сильных окислителен в этом случае
происходит окисление аминогруппы в питрогруппу.
Г а л о г е н ы, взаимодействуя с арпламнпамп, при низких
температурах в присутствии щелочи могут образовать Х-галоге-
нарнламины. Эти соединения перегруппировываются в га'огепа-
рпламнпы. При бромпроваипи в водных растворах образуются
бромариламипы. Например, анилин дает трпбромапплшг.
\П., N4 [.,
A
II
I
Вг
Хлор в водных растворах обычно окисляет анилин с образова-
образованием синих красителей.
Р е а к ц н и S-э л е к т р о (|) и л о в с арплампплмп мсгут
давать соединения со связью N—S (например, реакции с R—S- О,
SOC1,, SO3l RSO.C1):
Лг —i\H,-f RSOX1 — > Ar.\HSOoR-|-H Cl-
Реакция с серной кислотой приводит к соли — гидрогенсуль-
фату ариламина. Но при нагревании до 200 С гидрогепсульфат
арпламниа превращается в сульфоповую кислоту. Предполагается,
что сульфируется неиротопировапный арнлампи, который нахо-
412
днтся в равновесии с аммониевым ионом:
NIL _, \H3HSO7
Nil,
L'OO" С.
ЛИ,
HO;,S Ч -CJ...S Ч"
4. Защита аминогруппы в реакциях электрофильного замещения.
Аминогруппа защищается ацнлировапием. Таким путем исклю-
исключаются реакции по атому азота и окисление. Но ацпламилогрупг.а
сохраняет электроподонорпые свойства и ориентирует вступающую
группу в о- п «-положения. После проведения реакции ашгльпую
группу отщепляют кислым или щелочным гидролизом. Эти реакции
используют для получения нгпроарплампнов. сульфонилхлорпдов
и др.:
И Н
(Г
I
\COCIb
П\О,
и,so,
.NCOCH..,
\О..
А'СОСНз
О.,\'
NCOCH,
Л'СОСИ;,
a.so.,n
C1O..S'
\/
5. Ионизация связи N—Н. Арпламины являются относительно
более сильными NH-кислотами, чем алкиламины и аммиак, что
объясняется большей полярностью связи N'—Н п делокалнзацней
отрицательного заряда в сопряженном анноне (аналогично стабили-
стабилизации феполят-иопа):
Li
4 СН,
^ Ь- й-
-fCH.l.i
Арплампды металлов обычно получают реакцией арнламипа
с металлорганическпмн соединениями.
NH-кислотность ариламппов значительно увеличивается вве-
введением в о- или «-положения или у атома азота электроноакцеп-
торпых групп (ХОо, COR), которые участвуют в делокалнзапни
заряда:
Спедшюнпе
РА',
н-О.,.\С„Н,.\!1..
18,¦(
2,4,r).(O.N|.1C,;It.,.\H..
12,2
4C
4. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ
Лпнлнн является бесцветной жидкостью со своеобразным запахом;
т. пл. —6 ^С, т. кии. 184,4 С"С, мало растворим в воде (~3,4/6),
вода незначительно растворяется в анилине (~5%). Хлорид анн-
лппия [СОН5\НЯ1С1- образует бесцветные кристаллы ст. пл. 198 СС.
Анилин впервые был получен и 1826 г. О. Унфердорбсном при нагрева-
нагревании природного синего красителя индиго с Са(ОНJ и назван «кристаллипом».
В 1834 г. Ф. Рунге выделил анилин из каменноугольной смолы и нлзиал это
пещестпо «кианолом». Ю. Фрппше в 1841 г., нагревая индиго с КОН, получил
соединение, названное им «анилином». В 1842 г. II. Н. Зпппп при восстанов-
восстановлении нитробензола сульфидами аммония по'лучил вещество, которое назвал
«бетпдамом». В 1843 г. Л. Гофман доказал, что все эгн вещества идентичны
и предложил остагпть название ашиин.
В настоящее время в мире производится более J млн т анилина
в год, главным образом каталитическим восстановлением нитро-
нитробензола водородом в газовой фазе над XiS/AIuO:! или Oi/SiOo при
200—300 "С. Его используют главным образом для синтеза
мономеров—диизоцнапатов - - при получении полиуретанов
(гл. XXXV. Б.З, XXXV.В. 1), для синтеза производных бензтиазола
(гл. XXXVII. В) — ускорителен вулканизации для резиновой про-
промышленности, для синтеза аптиокендантов, красителей, лекарст-
лекарственных веществ и т. д.
К,\-Димстиланилин — бесцветная жидкость со своеобразным
запахом; т. пл. 2,5 :С, т. кии. 193 '"С, мало растворим в воде (~1%).
Его получают, алкилпруя анилин метанолом в присутствии
серной кислоты под дгшлепием или днметнловым эфиром в газовой
фазе над катализатором.
М.М-Диметилапилип используют для синтеза красителей и
взрывчатых веществ. При нитровании получают 2,4,6-трннитро-
К-питро-М-метиланилип (тетрил) — бризантное взрывчатое веще-
вещество. При ацилнровашш фосгеном получают кстон Мнхлсра —
важное исходное для синтеза красителей:
СНз. /NO..
O,NN I Л'О.. О ..
X//x/ П.)СЧ Г ,__-. ,CH3
Ч\ —у/ ^-С — /х ^—Х/
ПзС/' \=/ Ч=/ '"Neils
кстоп Ми\лера
N0,
тетрил
$-Нафтиламин — бесцветное, темнеющее на воздухе кристал-
кристаллическое вещество; т. пл. 113СС.
Его получают, нагревая р-нафтол при 130—150 СС в автоклаве
с NH3 и SO» в водной среде:
MI.,; SO,
414
Реакцию открыл X. Бухерср A903—1904). Предполагают, что
гидросульфит аммония (N'H:!-SO,~H2O ^± NH'IISO") реагирует
с таутомерной кетопной формой E-нафтола:
-ОН
В свою очередь Р-аафтнламни, взаимодействуя с NaHSO3+IIoO,
превращается в f>-нафтол.
Р-Нафтнламин используют для синтеза красителей. В настоя-
настоящее время его широкое промышленное производство приостанов-
приостановлено, так как он обладает канцерогенными свойствами.
Большое значение имеет М-фенил-р'-пафтнламин, который по-
получают нагреванием [J-нафтола с анилином:
II
ЮН НА\
230...280 "С
Кат.
]¦'¦ Н.,0
Под названием «Неозон Д» его применяют в качестве антнокси-
дапта для стабилизации резиновых изделий и других полимерных
материалов.
о-Фсшшндиамин —бесцветное кристаллическое вещество ст.пл.
102 СС. Его получают восстановлением о-питроапнлипа:
,ч ,.\Н. /N ,\П»
^ Ч\О., ^ Ч\ца
о-Фепилепдиамип используют для синтеза гетероциклических
соединений.
п-Фвнилендаамцн ¦—• бесцветное кристаллическое вещество с
т. ил. 147 °С, па воздухе темнеет (окисляется).
Получают его восстановлением /г-иитроанилнпа. и-Фепнлен-
диамин и его МД'-диалкнлпроизводные применяются для синтеза
азиповых красителей.
Бснзадин D,4'-днамнпобифепил) — бесцветное кристаллическое
вещество, темнеющее па воздухе; т. ил. 127 СС.
Его получают из гидразобепзола в реакции бензнднповон пере-
перегруппировки (гл. XXIV.2.2):
п.„so.,
U <¦ НА -
?- NH,
415
Бензидин применяют для получения азокрасптелей н в каче-
качестве аналитического реагента.
Сульфаниловая кислота (я-амнпобензолеульфоповая кислота)
представляет собой бесцветное высокоплавкое вещество, малораст-
малорастворимое в воде (~1%), растворимое в щелочи. Ее получают взаи-
взаимодействием анилина и серной кислоты.
Сульфаниловая кислота существует в виде внутренней соли,
является слабой \Н-кислотой (рКа -4,2 в водном растворе):
-O3S— /~Ч— Nib-1 Н2О :i: -O3S—(^ Ч— NH,-i-н^О-
Она применяется в промышленности азокрасптелей.
Исключительно важное практическое'значение имеет амид суль-
фаниловой кислоты и его X-замещенные (у амндной группы) про-
производные. Получают их из л-ацетнламинобензолсульфопилхло-
рида:
II
I ч
СНЯСО.\ —^ ^— SO..CI + 2R — N Н2 —>
О
H2N— {
Эти соединения используются в медицине как эффективные
антибактериальные средства для лечения различных инфекцион-
инфекционных заболеваний. Химико-терапевтический эффект сульфанила-
сульфаниламидов был открыт в 1932 г. Г. Домагком. В медицинской практике
сейчас используют несколько препаратов, например: белый стреп-
стрептоцид (R — Н), норсульфазол (R ¦-- 2-тиазолил), сульфадимезин
(R—4,6-дпметил-2-пнримидпл) и др.
Глава XXIV
ПРОИЗВОДНЫЕ ГИДРАЗИНА
Производные гидразина образуются при замещении одного или
нескольких атомов водорода в молекуле гидразина HAXIL угле-
углеводородными остатками.
Производные гидразина классифицируются по степени заме-
замещения атомов водорода:
монозамещепкые RNHNH2
Ы^'-дизаметениые RNHNHR1
R\.... /R2
R/ ХН
416
тстразамещенные ,WS
соли гндразшшя [RNH2NHo]X-,
Номенклатура производных гидразина основывается на тех же
правилах, что и номенклатура аминов; к названию углеводород-
углеводородного остатка присоединяют суффикс -гидразин. Называя сложные
соединения, используют префикс гпдразино-. Для некоторых типов
производных сохранились исторические названия, например Х,Г\"-
диарпл гидра гиты называют г пдразоа репами:
СН,ХП\Пл С,П5\'НХНСЛ1.,
\iotv::: |,дралп1 Х.Х'-днэтплгндразпн фепплгпдралш
It It
ОН
*\ Л/с1
хнхп2
^'-длфепм.'П'идразип, 2-хлор--1 -гпдрлян-
LCH3
н
~х-хн2
Г нн
С1- ! Clb-N—N-СНз
J
II Н
Х.'ЮрПЛ .V.N-ДМ'.'ОТПЛ- Гул!)ф;1Т ХлЧ'-ДММОТИЛГНДрл.ШИПП
А. АЛ КИЛ ГИДРАЗИНЫ
1. Истоды получения. Алкилгидразппы получают реакциями ал-
кплпроваппя и восстановления. Эги методы подобны методам
получения алкпламнпов:
ft' в- ¦!¦
R—X-1-H,X—i\!I-, - -> [RN'H.,Xtb]X-
R\ ни R1,
;.\—N-- О- - ;Х'-ХН,
R7 \V
Для получения алкилгидразннов испольчуют также реакции
с \-хлора\пшамн:
R_\n2 I-CI— X— R1
Н
> R_X_\_Ri-:-\a-Cl-
Hit
2. Физические и химические свойства. Применение. Алкилгпд-
разины — бесцветные вещества, обычно жидкости. Они имеют
основные свойства и образуют соли. Основность их меньше, чем
14 .V. 517
417
алкнламппов, так как сам гидразин мопсе осповен (р/(вп--г"-7,9),
нежели аммиак (р/Сцц + =9,25). Это объясняется взаимодействием
двух рядом стоящих атомов азота.
Для N-алклл- и Г\т,Г\т-диалкилгндразшюв характерны все ре-
реакции первичных аминов. При иитрозировашш N-алкплгидразн-
пов получаются новые соединения — азиды:
.. .. 2
R N1 Н N Но — т- R - К - N -- N
н + \ ••
М,Г\"-Диалкилгндразины при взаимодействии с окислителями
превращаются в азосоеднненпя:
[01
R — i\r — N — Rl > R —N=- N — R*
I I
Н Н
Б. АРИЛ ГИДРАЗИНЫ
1. Методы получения. Моноарилгидразпиы получают при мягколт
восстановлении арендиазоннсвых солей (гл. XXV.A.3), например
GnCI2 в водных растворах:
Ннтроарилгидразины получают арплпрованием гидразина хлор-
питроаренами. К.К'-Днарилгндразины получают восстановлением
ннтроаренов в щелочной среде (гл. XXI.В.3). Триарилгидразипы
получают арилировапием К,К-диарнлгндразннов:
(CeII5JN'XH2+CI ~<(У~ N02 — (QHsbN -X \/~ N'Oa
02\/ Н02\т/
К,\-Д11фсш1л-К'-п«кр|[Лгидра:1ии
2. Физические и химические свойства, применение. А р и л г и д-
разимы. Простейшие армлгидразпны — бесцветные лаикостн
со своеобразпьум запахом, ннтроарилгидразнпы — кристалличе-
кристаллические вещества.
Арнлгидразины являются слабыми основаниями. Для фенил-
гпдразина р/Свм + =5,2 (для анилина р/<"ви+—4,6):
Арнлгидралппы используют для синтеза гетероциклических
соединений (пиразелы, ииразолоиы, ппрпдпзопы) и в качестве
аналитических реагентоп для идентификации альдегидов и кетонов
(гл. ХХУП.Д.З):
ArN'HNH2+ XC = O—* >C==N"N
418
2. N.N'-Д н а р и л г н д р а з и и ы (гидразоарены) — бесцвет-
бесцветные кристаллические вещества, при хранении окисляются кисло-
кислородом воздуха и становятся желто-красными.
Гндргзоарепы являются слабыми основаниями (р/\вц-«0,5. . .
. . . 1,5). В кислой среде быстро происходит своеобразная перегруп-
перегруппировка, в результате которой получаются производные бнфонила
(бопзпднп и его производные) — бензидиновая перегруппировка.
Впервые эту перегруппировку наблюдал Н. Н. Зиннн в 1845 г.
Реакция проходит впутрнмолекулярпо и ее механизм до конца не выяс-
выяснен. Предполагают, что перегруппировка происходит в результате протоии-
роваиия атомов азота и внутримолекулярного синхронного (одноплеменного)
разрыва связи \ — N и образования связи С—С во внутримолекулярном до-
норпо-акцепторпом комплексе. Скорость реакции пропорциональна квадрату
концентрации протона [HJ-
Н'
Если оба «-положения заняты, образуются о-ампподифгнпламины —
семиОипосая перегруппировка:
н н
R —
\ /
При восстановлении гпдразоаренов образуются арилампиы, а
при окнслепип — азоарепы:
Н И
[Щ ! I [О]
2Лг.\Н2 ^— Лг —\ —\ — Лг —' Лг — N ^Х — Л-
арилампи азогреп
3. Т р н а р и л г п д рази п ы замечательны тем, что при
окислении могут образовывать стабилизированные свободные ра-
днкал!.-! — трнарнлгпдразплы:
.. .. /Лг [О] Лг .. .. Лг
Аг/
¦II
/
Удивительно ст1бпльм1!м свободным радикалом является дпфепкд-
пшфплгпдразпл: пи в'растворах, ни в твердом состоянии он не
14*
419
димеризуется:
о.л, , л-0.2
Соединение используют для калибровки ЭПР спектрометров.
Глава XXV
/ЗИАЗОСОЕДИНЕНИЯ
Дпазосоединения содержат группировки — \\ к л и —\У X" (дна-
?orpymia, диазониевая группа), которые связаны с одним атомом
углерода углеводородного остатка.
К дназосоединениям относят:
с ¦' i л 11 диазопия [R — N ^\]
/X /N--\4
диазопроизводиые /У.---У./ Ц/ X
IV
ан/пи-(т ране-) cuh-(v.i'c-)
/(J-M1 N- N4
лпазотаты -N - j\r/ R Х)-Л1+
R7
Д|:азоалканы /С\2
Примеры номенклатуры:
хлорид беи- тстряфторборат Ссиэол-ан>пи-дппзоглд-
золдиазомпя п- и in ред^ен^олдмазопия рокепд
,о-\а;
/ Л' X.
Н,С
СП,
азотат натрия 3,1 -дк метил бон я о '-;"//'/ -дио зотат
кал;: я
СИ, ССН,Ч
СНА".. C.V., -CN.,
СП;/ СаПг,
^.чзомст;, и 2-дп n3onpuiK.il Лмфенплдп^нол'О'т
420
А. СОЛИ АРЕНДИАЗОНИЯ
1. МЕТОДЫ ПОЛУЧЕНИЯ
Главным способом получения солей дилзония является взаимодей-
взаимодействие первичных ариламинов с питрозирующими реагентами
(HNO2+H + X-, КОХ, [NO] + BF4-, NO-J-XO,, ROXO;-H'X-) —
реакция диазотирования. Реакцию проводят при пониженной тем-
температуре (О—5СС).
Реакцию открыл П. Грнс A858):
NaNO,;
Аг-ХН2
Мсхацизм реакции дназотировапия весьма сложен. Считают,
что промежуточным продуктом является \-пнтрозоампп:
Аг—NH2-j-H + X- 4--i Ar — КШх- N_()
NO + X- I !П-Х- к/ " -j-П-Х-
Н + ,N- O^Ar/ |
>/ II
XH
При взаимодействии NaNO2 с кислотой образуется азотистая
кислота №40.,, которая в кислой среде переходит в ннтрозильные
производные:
О-К — XiJrON-'X- (X-Br, CI, HSOj)
N-Нитрозоамии в кислой среде легко переходит в свою тауто-
мерпую форму — диазогидроксид:
и. Н х~
который в кислой среде превращается в катион диазония:
/N —ОН Г ,N-6-H] +
Ar-N-^ ••-I-H'-X- i"> Ar —N^ | X-;^-
L н J
Соли дн&юшш обычно получают в водных растворах и непо-
непосредственно используют для дальнейших реакций. Кристалличе-
Кристаллические соли арепдиазопия могут быть получены в растворе этанола
при диазотировании алкплнитритами RONO в присутствии соот-
соответствующей кислоты. Некоторые соли диазония малорастворимы
в воде и могут быть осаждены из водных растворов (ArNtX", где
X = BF4, CIO,,, HgClr, и другие комплексные анионы).
421
2. ФИЗИЧЕСКИЕ СВОЙСТВА И СТРОЕНИЕ
Соли арепдпазония — бесцветные кристаллические вещества, ргст-
воримые в воде. Соединения нестабильны, некоторые взрывчаты.
Особенно опасны перхлораты и нитраты, которые взрываются
как от трения, так и от нагревания. Более стабильными являются
тетрр.фторбораты и другие соли с комплексными анионами.
Ион арендиазопия является сопряженным ионом, в котором
осуществляется сильное взаимодействие между л-электронной си-
системой арена и тройной связью дпазопневой группы:
Sf
Оба атома азота находятся в состоянии яр-гибридизации, при
этом один из них имеет положительный заряд. Это вызывает-элект-
вызывает-электронное смещение в диазопиевой группировке:
Ar—N=N-
Второй атом азота приобретает некоторый положительный за-
заряд. Эю может быть изображено мезомерпой граничной структурой,
в которой положительный заряд полностью локализован иа этом
атоме азота. Второй атом азота является главным центром реакции
в превращениях диазопневых солен.
Поданным рентгеноструктурного анализа, группировка С—\—N
является линейной и связь X—\г по длине соответствует тронной:
:N_n.u,,,,._..
0,11мм' Jt-^" ' ^R . R-
Дпязонневая группа принадлежит к слитым сильным я-лс-ктро-
иоакценторны.м группировкам в органических соединениях, она
оказывает больший эффект, чем пптрогруппа. Сам нон арепдназо-
иия является сильным элсктропоакцептором.
3. ХП.чНЧЕСКПЕ СВОЙСТВА. ПРП.МЕПЕППП
Для солей дназопня характерны реакцмп двоякого т::п::: реакции
у атома азота с сохранением азота в молекуле и реакции с замо-
замощением диазопневой групп;.; п выделением ясота. Все эти реакции
проходят при взаимодействии с нуклеофильмымн реагентами (с.\г.
схему 1).
Катион дпазошгя как электрофнльная частица (кпг."отл Лыонсг)
с электронодонорными реагентами может образовывать комиле.тс
с переносом заряда и дальше — продукт присоединения по атому
азота. В случае сильных допоров зозможен полный перепое ;>1ект-
422
Схема 1. Превращение солей диазония
С.-Нг,—Z
и другие продукты
син-диа.юпроизЕОлнсе
или азосоедикенне
ропа с возникновением свободных радикалов п продуктов их раз-
разложения it взаимодействуя. Особое .место занимают термические
превращения, где возможно образование карбокатионов. Неко-
Некоторые замещенные соли диазония разлагаются фотохимически при
освещении ультрафиолетовым и интенсивным видимым светом.
Присоединение пуклеофнла и дназониевой группе всегда ведет
к образованию а/и-диазонроилводиого или азосоедннення. Это
сноеобразное явление до сих пор не нашло удовлетворительного
объяснения. Предполагают, что причина образования енн-нзомера
кроется в особенностях строения промежуточного комплекса с
переносом заряда и особенностях молекулярных орбнталей ка-
катиона диазония и сан- и ан/иы-дпазопроизводиых. а/я-Диазопро-
изводные и азосоединенпя легко изомеризуются в ян/л«-пзомсры.
1. Образование и строение диазотатов. При добавлении к раст-
раствору соли диазония щелочи выкристаллизовываются дназотаты:
ArX'-JX--; 2Na*OH- —i-ArX- \О~\'а -+ Na » Х~-[ ПаО
Дназотаты — относительно стабильные соединения, умеренно
растворимые в воде. Каждой соли дпазония соответствуют два
диазотата. Тот, что выделяется из раствора, сохраняет некоторые
свойства солей дпазония (например, в водных растворах дает ре-
реакции азосочетапия с фенолами, ариламинамн). После хранения
пли кристаллизации этого дназотата образуется другой дпазотат
(мзо-дназотат), который утратил способность к азосочетанию. При
подкислении растворов д![азотатов опять образуется соль диазо-
диазония. А. Пшч A894) объяснил существование двух дпазотатов син-
анпш-нзомерней. Строение и реакционная способность диазотатов
были объектом ожесточенных споров более 70 лет. Прав оказался
А. Гапч.
Ион диазония в щелочной среде (рН 10—!2) быстро (в сотые
доли секунды) присоединяет гпдроксилышп ион и превращается
в сын-диазотат, который медленно изомерпзуется в гшши-дпазотат.
423
Изомеризацию ускоряют нагреванием и присутствием электроио-
акцепторных групп в бензольном цикле. Например, при 20 "С
син-д-нитробензолдиазотат изомеризуется в анти-изомер в течение
нескольких секунд:
/N—-N4
[Лг-.\~. \]X--|-Na'OH-^ кх/ ХОН
Ма'Х-
—*- Лг | 4O
I медленно
Аг ¦'
Очевидно, син- и ак/ии-диазотаты значительно отличаются по
своему пространственному строению. сия-Изомер не должен иметь
илапарпой структуры ввиду отталкивания атома кислорода и
атома водорода в о-положении цикла. В этом случае система менее
стабильна.
Диазотат-ион, подобно енолят-иоиу (гл. XV.A.4), является
сопряженным амбидентным анионом, имеющим несколько нуклео-
фильпых реакционных центров. Атака электрофилыюго реагента
может происходить по атомам О или N:
Е + NaX
При нротонировагти диазотат-иона могут образоваться диа::о-
гидроксид и N-нитрозоамии, которые являются таутомериыми
веществами:
/ОН
Более стабильной таутомериой формой является, как прав[[ло,
та, которая имеет меньшую кислотность. В большинстве случаев
меньшую кислотрюсть имеет N-нитрозоамин. В избытке кислоты
ои превращается в катион дпазония.
2. Реакции с различными нуклеофильными реагентами. Соли
арепдназония взаимодействуют с пуклеофильными нонами пли
нейтральными нуклеа|)илами с образованием диазопроизводпых.
При реакции арендпазониевых солей с алифатическими и цик-
циклическими первичными и вторичными аминами образуются диазо-
амипосоедпнеппя — триазены, которые в кислой среде снова пре-
превращаются в диазопиевые соли. В случае ариламинов триазены
тоже могут быть получены, но стабильным конечным продуктом
является азосоединеиие:
424
+ ft
[Ar — N'^NJ
M + CN-
M + - О R
<,
Лг
Лг7'
Лг/'
Г
Л г'
Л';
N
GV
¦so
OR
Ч "
i-M
¦7 л а
-г^
- -R
X
+ ";
х
х-
J
1^;- Ar'
\' - \
.R1
+ H'X-
Особос место занимают реакции с фенолятами и ариламппами,
в которых образуются азосоедпиения, т. с. происходит электро-
фильпое замещение в бензольном цикле. Реакция называется азо-
сочетанием и имеет фундаментальное значение в получении азо-
красителеп:
г-
г-
Ar-N
ОН + М X
В реакции азосочетания сначала образуются сын-азосоеднне-
ния, которые затем быстро изомеризуются в ан/пи-Aпранс-)а:ю-
соедпнення.
При взаимодействии с мягкими восстанавливающими агентами
в кислой среде наблюдается образование арилгидразпнов:
Slid;
^ г' :~ " ' и.о; па ^ г' ' я'
3. Реакции с выделением азота. Для солей ареидиазоння ха-
характерны реакции, в которых выделяется азот и вместо группи-
группировки N2' вступает другой атом или группировка, в том числе
водородный атом. Эти реакции реализуются тремя способами:
а) при взаимодействии с сильными электродонора.мн, иногда
в присутствии катализатора;
б) при нагревании;
в) при ультрафиолетовом освещении, а в некоторых реакциях —
видимым светом.
а) Реакции с о л е и д и а з опия с и у к л е о ф и л ь-
н ими а н и о п а м и и а л к снами. Соли ареидиазоння с
иодпдамн образуют иодгфепы и кыдетяется а::от. Предполагается,
425
что реакция идет по свободнорадикалыюму механизму:
[Аг —N = К]Х- + SV I- — ,\г —N --= N• -| 1- -,-М ' Х~
Хлориды и бромиды с солями дназоння не реагируют, эти
копы являются более слабыми электронодонорами (труднее окис-
окисляются). Однако в присутствии солей меди (I) реакция проходит.
Катализ солями меди впервые наблюдал Т. Запдмейер A884):
[Ar —N = N]Y-+CuX+M' Х- —>Лг —X-[-M+Y-+CuX-:-N2
(X = Cl, Br, CM, i\O2 и др.).
Так могут быть получены хлорарены, бромарепы, цпаиоарены,
нитроарены и другие производные аренов.
Ионы Cu(I) здесь служат переносчиком электрона:
Аг— N =э N-L-СиХ + :Х- —* Аг—N =-= N• +СиХ2 —* Ar- + N2-j-CuX2 —*
-* Ar —X-f N2-|-CuX
Предполагают, что свободные радикалы образуются и реагируют
в реакционном комплексе соль дпазопия — соль меди и в раст-
растворе как кинетически независимые частицы не появляются.
Для катализа реакций солей диазоиия с иуклеофилышыи реа-
реагентами могут быть применены также соли других металлов с
переменной валентностью.
Аналогично реакции Зпндменера идет разложение солей дна-
дназония в присутствии солел ртути, олова, сурьмы, висмута и ме-
металла с образованием мсталлоргаиических соединений (дназометод
А. Н. Несмеянова, 1929). Примером может служить реакция полу-
получения ртутьоргапичеекпх соединений:
ArNtC|--!-HgCI2 —> Ar\2HgCi;r—> ArIIgC! + N2-;-HgCI2
Арепдиазониевые соли в присутствии солен меди арнлнруют
соединения с активированной двойной связью (X. Меервенн):
Кат. -ИХ
Ar-N/X--bRCH = CHRl >R_CH—CH —Ri R—C-CHR1.
~'Nj I I I
Ar X Ar
(R1 - COR2, CN, C,Hfi)
б) Р е а к ii и и термического разложения. При
нагревании солей диазоиия обычно получаются смеси продуктов.
При нагревании водных растворов солей дназония со слабопуклео-
фнльными или комплексными анионами главным продуктом ре-
реакции разложения является арснол:
[Ar—N =
-^Ar—OH + II + X--; N2
(здесь X = HSO4l BF4)
Предполагают, что реакция расщепления осуществляется по
ионному механизму. Но не исключен также сгаободнорадикалыплй
механизм.
Разложение ареидиазопиевых солей в слабощелочной среде в
присутствии ареков it их производных или гетероциклических
соединений ведет к продуктам арплировапия как воды (ареполы),
так it аренов (биарилы):
Н,0
ArN, X- + A'H
Нагревание солей диазопия в присутствии алканолов или про-
простых эфиров приводит к замещению диазониевой группы водоро-
водородом. Образуются арены с примесью алкилариловых эфиров. Ре-
Реакция является гемолитическим разложением солей диазония;
алканолы и их производные служат источником атома водорода:
[Аг — N = N]X- + HOCHaR ;=; Аг — N^N— ОСВД + Н^ Х~
Лг — N=-..N — OCH2R —> Ar-
Ar.-:..OCH2R,—
Ar—H-'-^C —R
Ar —OCH2R
Реакция имеет препаративное значение для удаления амино-
аминогруппы от молекулы ариламиноп.
Известен также другой метод замещения диазониесой группы
на водород —¦ восстановление гппофосфитами:
Nall.PO,
[Лг —X = N]X- :—~> Аг —Н-;-.\2
Своеобразно проходит термическое разложение борфторпдов
арепдпазоипя. При нагрепаннн выше 100 "С они разлагаются с
образованием фторареноз (реакция Вальца — Шнмапа):
[Лг —К i-\]CF,7 - - Лг —F-L-K2-;-EF3
Предполагают, что в ходе реакции образуется арилкатнок.
в) Фотохимическое р а з л о ж е и и е характерно
для арендиазонневых солей, имеющих в молекуле электронодоиор-
пые группы, например динлкиламнногруппы:
^ -^ CH>:f
сн/ \/ сн/
При оезещепни и поглощении молекулой определенной энергии
происходит внутримолекулярный перенос заряда от донорпой
группы па акцепторную диазог.невую группу, в результате чего
разрывается счязь С—\ и выделяется азот.
Такие соли дп:г.о;п:я пеноль.чуюг практически для записи ин-
информации (мете;; бессеребрян'оп (Ьот^г:;афин, дназотншгя).
427
При получении записей на диазопленку проецируют изобра-
изображение. В неосвещенных местах сохраняется соль дназония, которая
при проявления превращается в краситель и появляется изобра-
изображение.
Б. ДИЛЗОЛЛКАНЫ
Соли алкапдпазопия очень нестабильны. При дназотпрованпн
первичных алкяламинов выделяется азот и в продуктах реакции
обнаруживаются алкаполы и алкены.
Малую стабильность солей алкандиазония но сравнению с
солями арепдиазония можно понять при рассмотрении природы
связи С—N. Если для солей арепдиазония связь С—X в результате
сопряжения аренового цикла с диазонпевоп группой приобретает
некоторую л-электронную плотность (С—л), то в случае солей
алкандиазония такого стабилизирующего фактора нет, здесь име-
имеется только очень полярная ст-связь С—X, которая способна к иони-
ионизации и легко подвергается атаке даже слабых нуклеофпльных
реагентов.
Более стабильны диазоалканы, которые формально можно рас-
рассматривать как внутренние соли алкандиазония:
Г Rl "I
I I + -и 1-х- R'4 -
R—C-N=\ Х- ¦¦>¦ /C-X-- \
I !
Ы -1
После отрыва протона углеродный атом приобретает отрица-
отрицательный заряд я пеиоделенпую электронную пару, которая всту-
вступает в сопряжение с дназониевой группой. Углеродный атом при-
принимает 5/;--гябридизацию.
1. МЕТОДЫ ПОЛУЧЕНИЯ
Дпазоалканы не могут быть получены днязотпрованнем первичных
алкпламниоп. Только в специальных случаях, если в молекуле
имеются акцепторные группы, при днаютнрованин образуются
аамещепные диазоалканы (например, дназотнрование «-лминоке-
тонов пли эфиров «-амипокарбоновых кислот дает диазокетоны я
дназоэA)иры).
Для превращения алкиламипа п дпазоллкан его ацнлнруют,
например, метоксикарбонилхлоридом (гл. XXXV.А), после зтого
нптрозируют и отщепляют ацильную группу щелочью:
Н
R1^ /О Основание R
^C-NH-. + CI-Cf
R7 | -ОСП
И
N0
/О Nя + оп- R\
R\ I /О N-я + оп- R\
С
;С— N — 0."
R/ | ОСН., R-'
II
c-
R7 1
H
1
-X
Xa
-c:-
< -o
,0
/
ОС
0
1
-с
н\о
Н3 "' Х
-ОСИ,-!
>•
Л,0
428
Для получения диазометаиа используют N-нитрозо-К-метил-
мочевипу СНД'(\'О)СОХНо и М-ннтрозо-М-метил-/1-толуолсуль-
фонамид CH,N (NO)SO,CeH4CH3.
2. ФИЗИЧЕСКИЕ СВОЙСТВА II СТРОЕНИЕ
N
Дназоалканы — газообразные или жидкие вещества, окрашенные
в желтый цвет, они взрывчаты и ядовиты. Например, диазометап
СНЛ\ представляет собой газ с т. кип.—23°С,
в чистом виде легко взрывается. Поэтому ис-
пользуют растворы диязометана в эфире.
Основная группировка дпазоалканов С—
—X -N имеет линейное строение. Получает-
ся своеобразная сопряженная система, кото-
рую можно изобразить несколькими мезомер-
иымн структурами или одной структурой с
выравненными связями (рис. 95):
Рис 95 перекры-
нания л'-орбиталей
в диазоа.-шанах
f¦ - ft -
0,113
M
Электроиографическне исследования показали, что связь в
диазомегане родственна тройной связи, поэтому структура дпазо-
дпазоалканов в большой степени приближается к структуре внутренней
диазоииевой соли.
3. ХИМИЧЕСКИЕ СВОЙСТВА. ПРИМЕНЕНИЕ
Диазоалкапы по своей природе являются нуклеофнльпыми реаген-
реагентами (С-осиоваииями), что определяется структурой внутримоле-
кулярно стабилизированного карбаниона. ЭлектрО[юакцепторный
характер дназониевой группы ослаблен влиянием карбанионной
части молекулы. Известны также реакции биполярного циклопри-
соедннения к кратным связям. Диазоалкапы легко расщепляются
термически и фотохимически.
1. Реакции с кислотами и другими электрофильными реагентами.
Дназоалкапы как С-основания легко нротопируются, в результате
чего получается соль алкандиазония, которая немедленно всту-
вступает в реакцию с пуклеофилами с выделением азота:
;С-\ -N'
)С —N^
Х-
R
/C-X-l N,
II
L " j
Например, диазометан часто используется для получения ме-
метиловых Э(|)нров карбоиовых кислот.
В принципе можно осуществить реакции диазоалканов со мно-
многими элсктрофтамн. К присутствии оснований удается получить
429
новые дназссоеднпепня:
R—СН —N = N + E'-X--~:\'(C.H,K —> ,С —N=--N-i X-[HN(C,HaK]
R/ ¦•
Так можно осуществить ацилирование ацплхлорндамн RCOC1 и
получить диазокетопы (гл. XXIX.Д). Реакции с альдегидами и
кетопамн см. гл. XXVII.А.3.
2. Реакции биполярного циклоприсоединения. Диазоалканы
способны присоединяться к двоГшым или тронным связям с обра-
образованием гетероциклических соединений — производных пиразола:
^,6-h nl pi
3. Фотохимические и термические реакции. Диазоалканы при
освещении или нагревании разлагаются, при этом выделяется
азот и получаются реакциопноспособпые частицы — карбены:
Карбены являются очень активными частицами, их нельзя
выделить из реакционной смеси. Непосредственно в растворе после
генерации из диазоалканов карбены вступают в дальнейшие ре-
реакции: взаимодействие с другими компонентами, днсироиорцно-
нпрованпе, ольтомернлашпо. Например, известны реакции присо-
присоединения к алкепам и алкииа.м с образованием производных цик-
циклопропана (гл. V.A.1).
Глава XXVI
АЗОСОЕДИНЕНИЯ
Азосоединення содержат группировку N=N (азогруппу), которая
соединена с двумя углеводородными остатками.
Азосоедипенпя подразделяются в зависимости от природы уг-
углеводородного остатка, который соединен с атомом азота, на
¦• ,/R -, ¦¦
а) азоалкаиы R ' " R ' R
транс- цис-
б) азоарсны Лг' Лг' чЛг
п:ранс- цис-
Пространственное расположение заместителей по отношению
к дбойноп связи N = N может быть двояким: транс- (или анти-)
и цис- (или син-).
430
Названия азосоедгшсннй с одинаковыми заместителями образу-
образуют названия углеводорода и префикса азо-. Производные этих
соединений назыпагат как замещенные основные соединения; если
у азогруппы различные заместители, их называют раздельно:
СН.,' Coll/ чС0Н5
ягрякг-а."омстли ^г.т-тсбепзол
t
4'-ампноаюбензол-4-сульфонорая О-бонзолазонафталип
кислота B-фе1!илазо11афтални)
А. АЗОАЛКАНЫ
Азоалканы обычно получают мягким окислением К,К'-дналкил-
гидразннов, используя в качестве окислителен HgO, L, Вг^:
го|
r_N_\_ri- - R_N'.-n_Ri
I i
H H
Азоалканы представляют собой желто-оранжевые жидкие или
кристаллические вещества, весьма нестабильные. При хранении
они постепенно разлагаются, при нагревании разлагаются со взры-
взрывом.
Разложение связано с гомолитнческнм расщеплением связи С—\т:
R-X-. N-R-'- R--KVH--R
Цветность азоалкапов обусловлена особенностями строения
азогруппы. Это поглощение в видимой области спектра связано
с электронным переходом я-»-я* от пеподеленпоп электронной
пары атома азота па незаполненную орЗнталь азогруппы. Мак-
Максимум поглощения находится в области 350—450 им с малой ин-
интенсивностью (еж 10. . .100), как это характерно для всех п-^-а--
переходов.
Некоторые производные азоллканор,, например 1,1-днцнано-1,Г-
днметнлззоэтан (ззонзобутпроннтрнл)
С\
С(СН3),
.л ^ N-'
(СН3JС '
CN
нсполь.уют в к'^тестг-е шшцпатороп полимеризации (нсточнпкоз
свободных радпкалог;).
43!
Б. АЗОАРЕНЫ
1. МЕТОДЫ ПОЛУЧЕНИЯ
Главными методами получения азоаренов являются азосочетанпе
и восстановление. Известна реакция конденсации арпламнпов с
нитрозоаренами.
1. Реакции азосочеталия солей арендиазония рассмотрены. Азс-
сочетапие очень легко проходит с. фенолами, нафтоламн, арила-
мипамн и их производными (азокомпопентами). В случае аренолов
и их производных необходимо присутствие основания, которое
генерирует аренолят-иоп.
2. Восстановление нитроаренов в щелочной среде с цинковой
пылью или амальгамами металлов дает симметрические азоарепы:
ПП
Ar —NO- >-Ar —N--N —Аг
NaOH
3. Реакции конденсации ариламинов с питрозоаренами в присутст-
присутствии оснований приводит к азоаремам, в том числе несимметриче-
несимметрическим:
:В
Аг— NH2 + O--N — Ar' —»Лг —N^N —А
2. ФИЗИЧЕСКИЕ СВОЙСТВА И СТРОЕНИЕ
Азоарены и их производные являются окрашенными соединения-
соединениями: желтыми, красными, синими и др.
1. Характеристика сопряженной системы. Молекулы азоарепов
содержат большую сопряженную систему и, кроме того, неподе-
ленные электронные пары на атомах азота. Возможно существо-
существование двух пространственных изомеров.
г(ис-Азоарепы не могут быть плапариыми ввиду пространствен-
пространственных затруднении (рис. 96). ^«с-Форма является нестабильным
Рис. 96. Пространстпеиная изомерия азоарепов
изомером, который быстро превращается в транс-форму — ста-
стабильный изомер с максимальным сопряжением.
траяс-Азоарены имеют несколько поворотных изомеров (рис.
96). В изомере а неподелепные электронные пары атомов азота в
сопряжении не участвуют. Напротив, в изомере б одна электрон-
электронная пара находится в сопряжении с бензольным циклом, а двойная
связь N--N — в сопряжении с другим бензольным циклом.
432
При освещении ультрафиолетовым светом /н/?анс-нзомеры ча-
частично превращаются в цис-изомсры.
2. Спектры поглощения. Азоарены поглощают в ультрафиоле-
ультрафиолетовой и видимой части спектра. Азоарены, не содержащие элект-
ронодоиорпых групп, имеют в видимой области ыалоннтенспвное
поглощение, которое связано с л-*-я*-переходом. Интенсивные
я ->¦ я*-переходы находятся в ультрафиолетово!! области (табл. 39).
Таблица 39. Спектры поглощения некоторых азоаренов
\
ч
Соединение
/
ОН
NH2
хСНз
Переход
я - >¦ я *
Я., нм
318
350
400
410
е
21 G00
20 000
22 000
2G000
Переход
п — .-я *
X, нм
440
450
(перегиб)
Не набл
То
е
300
1000
юдастся
же
При введении электроиодопорных групп полоса поглощения
я-*¦ я*-перехода сдвигается в длинноволновую область и маски-
маскирует малоиптенсивное поглощение, связанное с п -*¦ я* -переходом.
Азосоеднпения, содержащие остатки нафталина и бифенпла,
поглощают при более длинных волнах (красные, фиолетовые и
синие вещества).
3. ХММИЧПСКИЕ СВОЙСТВА
Химическое поведение азоаренов определяется присутствием азо-
азогруппы— двойной связи X—N и пеподелепны.х электронных пар.
1. Основность. Азоарены являются очень слабыми N-оспона-
ниями. Здесь выявляется взаимодействие двух трнгопальпых
атомов азота:
Ar-N-X- Лг-;-1РХ- г
г-X^N1- Лг 1-Х"
I
Н
=—2,5. . .—3,0)
По введениеэлектронодопорпых аминогрупп резко увеличивает
основность атома азота азогруппы. Оказывается, что в л-амппо-
азоарейах протонируется азогруппа. Таков эффект сопряжения:
61- '¦ 6'!-
ссн5—
+ Н
н
433
Протонирозлнньге амнпоазоярены интенсивно поглощают в ви-
видимей части спектра, при этом по сравнению с поглощением
непротоппрозаппого соединения происходит сдвиг максимума по-
поглощения в длинноволновую область па 100. ..150 им — бато-
хромный сдвиг. Поэтому при подкислении растворов аминоазоарс-
нов наблюдается углубление цвета: желтый переходит в красный,
красный — в синий. Некоторые аминоазоарены применяют в
аналитической химии в качестве кислотно-основных ппдикатороз
(метиловый оранжевый, конго красный).
2. Особенности гидроксиазоаренов. Гидроксиазоарепы являют-
являются ОН-кислотами. При ионизации их углубляется окраска, осо-
особенно в случаях о- и /г-изомеров, где имеется прямое сопряжение
с азогруппой. Углубление окраски объясняется увеличением
электроподонориых свойств заместителя при переходе от —ОН
к —О-:
Аг—N^ \=/ +Na+OH~ ^Г
о-Гидроксиазоарены образуют устойчивые и окрашенные внут-
внутренние комплексы (холаты) с нонами тяжелых металлов, которые
нерастворимы в воде, но растворяются в органических раствори-
растворителях:
"н-о'
В молекулах о-гндроксиазоарснов образуется прочная внутри-
внутримолекулярная водородная связь.
4. Восстановление и окисление. При восстановлении азоареноз
получают гидразоарепы пли арпламнны:
[in
Лг — Nr^N—Лг1 >- Лг.\Н2 -i-ArVN'Ha
[Щ Hill
>Лг —MI —NH—Лг1 [
Окисление приводит к азокспарепам:
[О] +
Лг —N = N' —Аг—*Аг —.\ = Х —Аг
о-
4. ЛЗОКРЛСИТЕЛН
Многие азоарепы пптенспсио логлоиг'-ют в видимом области спектра
и могут быть использованы дли крашения тканей, бумаги, поли-
полимерных материалов п других веществ. Лгюкргсптелп содержат з
434
молекуле одну или несколько азогрупп п электронодонорные
группы ОН, NH2, N(CH3)o, N(CH5), и др. Кроме того, могут при-
присутствовать нитрогруппы, сульфогрупны и др.
Азокрасителеи известно огромное количество, начиная с желтых
и кончая синими, зелеными или почти черными. Азокрасители
могут быть водорастворимыми (сульфокислоты и их соли, соли
аминов), нерастворимыми в воде, но растворимыми в жирах, ма-
малорастворимым» (пигменты) и др.:
пиразолоновые красикля метиловый .оранжевый
.••н-оч
>« О
ХНслый оранжевый ы:с.я:,'[ Liiuiiii 2.K
Карбонильные соединения
и их производные
Карбонильные соединения являются производными углеводоро-
углеводородов, в молекулах которых два водородных /.тома, находящихся
у одного углеродного атома (гемпналъпые атомы), замещены ато-
атомом кислорода. Таким путем получается группа /С-О, которую
называют карбонильной группой или оксогруппой.
Если карбонильная группа связана с одним водородным ато-
атомом и одним углеродным атомом (в простейшем случае с двумя
водородными атомами), то соединения называются альдегидами,
а группа — С^ — альдегидной группой.
Если карбонильная группа связана с дг,ум.ч углсрот.чмп
атомами^ то соединения называются кмпона.ии, а группа
~ с\ — ке.'погрцппой:
R —C^ R—С^
альдегиды Kciouu
435
Карбонильные соединения подразделяют в зависимости от
числа карбонильных групп в молекуле и присутствия других
функциональных групп. Так образуются три подгруппы:
1) мопокарбопильпые соединения;
2) дикарбонильные соединения;
3) карбонильные соединения с другими функциональными
группами.
Каждую подгруппу подразделяют в зависимости от типа угле-
углеродного атома, соединенного с карбонильной группой, и от при-
природы других функциональных групп (галогенкарбопильпые сое-
соединения, гидрокспкарбоиильпые соединения, аминокарбонильпые
соединения и др.).
К карбонильным соединениям относят также хиноны — шести-
членные циклические соединения с двумя карбонильными группами
и двумя двойными связями в цикле.
Глава XXVII
МОНОКАРБОНИЛЬНЫЕ СОЕДИНЕНИЯ
КЛАССИФИКАЦИЯ, ИЗОМЕРИЯ И НОМПНКЛЛТУРЛ
Мопокарбопильпые соединения подразделяют в зависимости от
типа гибридизации углеродного атома, с которым соединена кар-
карбонильная группа.
1. Насыщенные карбонильные соединения
2. Ненасыщенные сопряженные (а,р-непредельные) карбо-
карбонильные соединения
О О
г ;:
R\ /С R\ ,CN
>С~С( U }С-- С( XR
RT/ \R R3/ \R1
,O ,O
R_C-^C-Cf Ri_C = C-Cf
К ненасыщенным карбонильным соединениям формально при-
причисляют кстсны, хотя здесь совершенно друга"' тип химической
связи:
R\
;С=---С--0
R'
Таблица 40. Примеры номенклатуры альдегидов
Структурная формула
Пя.чпл иле
по номенклатуре
ИЮПЛК
и-Ы
н
.о
/О
сн.,ен2сно—с^
/О
сп.,-сн—с/
I ' Н
СП-,
н
II
,о
СЫл -СИ — С
СИ:, —СП.- СП—С
Н —С= С
/О
.о
/О
Мстаналь
Этаняль
Про па и а ль
Бутаналь
2-Мстплпропа-
наль
Цпклогексаи-
карбальдегид
Пропепаль
Бутеи-2-аль
Прошшаль
Бепзальдегид
2-Нафтллпп-
карбальдегнд
Формальдегид, альдегид
муравыплт кислоты
Лцетальдсгнд, альдегид
уксусном кислоты
Про л ноноиы» альдегид
Бутиральдегид, масля-
масляный альдегид
Изомасляный альдегид
Акролеин
Кротоповый альдегид
Пропаргиловый альдегид
Бензойный альдешд
E-Пафтальдегнд
3. Карбонильные соединения аренов (ароматические альдегиды
и кетоиы)
/О
чн
/О
At-С*
437
Номенклатура карбонильных соединений весьма многообразна.
Используют как исторические пазвлнпя, так и систематическую
номенклатуру. Поэтому многие карбонильные соединения имеют
несколько названий.
Альдегидную группу по систематической номен-
номенклатуре обозначают суффиксами -аль, -карбальдсгид или префик-
префиксами оксо-, формил-. Если альдегидная группа является старшей
характеристической группой (см. Введ. 8.2), ее обозначают суф-
суффиксом. Если углеродный атом альдегидной группы входит в
родоиачальную структуру, применяется суффикс -аль, в других
случаях -карбальдегид. Префиксы оксо- или формил- применяются,
если d молекуле имеются более старшие группы, например —SO3H,
—СООН.
Многие альдегиды имеют тривиальные названия, которые
произведены из названий карбоновых кислот, получаемых при
окислении альдегидов и содержат слово альдегид (табл. 40).
Кетонную группу по заместительной номенклатуре
обозначают суффиксом -он или префиксом оксо- (ксто-). Префикс
применяется, если в молекуле имеются более старшие характери-
/ О \
стическне группы ( —с\ - —SO3H, —СООН j. По радикально-
функциональной номенклатуре названия кетснов состоят из
названий углеводородных остатков в порядке алфавита и суф-
суффикса -кетон. Некоторые кетопы сохраняют тривиальные названия
(фенопы, халькоиы, ацетон):
о
|' ацетон, пропаиоп, диметнлкетон
СНзССНа
О
|; бутанон, метилэтндкетоп
СН3СС112СН3
О
I: гоксапон-2, бутплметнлкетон
сн3ссн2сн2сн:си3
о
СН3ССН;СНСН2СН3 4-мстнлгскса;юп-2
СН3
цпклогексанон
Ч
О
|| псктен-З-он-2
СНзСНг-СНССНд
О
I!
СНаС112С = СССН3 гскс1:н-3-он-2
43S
метилфенилкстои, ацстофснои
1,3-Днфештлпропепои, халкон
А. НАСЫЩЕННЫЕ КАРБОНИЛЬНЫЕ СОЕДИНЕНИЯ
1. МЕТОДЫ ПОЛУЧЕНИЯ
Альдегиды и кетоны получают реакциями окисления различных
органических соединений, гидролиза дигалогеиалканов (RR'CXj),
гидратации алкинов, прямого карбонилироваиия алкенов и др.
Большинство реакций получения уже рассмотрены при разборе
химических свойств алкенов, алкинов, галогенуглеводородов, спир-
спиртов, гидропероксидов. Здесь эти реакции только упоминаются.
Важнейшие промышленные методы рассмотрены ниже.
\. Реакции окисления. Альдегиды и кетоны образуются:
а) в реакциях окисления алкенов (гл. 11.4.4):
гг\ 1 R ч / R"
/R2
н
:с-с<
П,О R/ | VO
и
б) в реакциях окисления и дегидрирования алканолов (гл.
XIV.A.4):
rm /°
RCII2OIl'-^iR—С<^
ОН
R—СН —R1—*R —С^
2. Гидролиз дигалогеналканов. При гидролизе геминальных ди-
дигалогеналканов получают альдегиды и кетоны:
Ri /X но Ri .ОН R1,
R/ ХХ 9сноиание R/ XOH R/
(X=F, С!, ВГ, I).
3. Гидратация алкинов (гл. IV. 4.1):
Кат. И 7Rl
7R
)C- С ->RCHoC
Rx Oil "
439
4. Прямое карбонилированис. Реакция алкенов с СО и Н2 в при-
присутствии тетракарбонилгндрпда кобальта при повышенной темпе-
температуре и под давлением даст альдегиды и кетопы (оксосиитез)
(гл. II.4.3).
5. Термическое разложение солей карбоновых кислот. Кетопы
получают нагреванием кальциевых солей карбоновых кислот при
200 СС и выше (см. гл. XXXI.Л.З):
О
t° II
(RCOO),Ca —>- R — С—R Ч-СаСО3
Метод используется также для получения цпклоалканопов:
/СОО- (С | 1
(СН,)„Са2'- —
чсоо-
При использовании солей тория удастся с удовлетворительным
выходом получить макроциклические кетопы (реакция Ружнчки,
1926), где я-7, 8, 9 и т. д.
6. Реакция Удриса — Сергеева. Кетопы получают реакцией
Удриса—Сергеева (гл. XVII.Г) при расщеплении гидропероксп-
дов кислотой:
/К1 н + х- R1.
Лг —Cf-OOH > Лг —OII + ;С^-О
4R R/
7. Пинаколиновая перегруппировка тоже дает карбонильные
соединении (гл. XIV.Д.2):
R R OR
I I in-х- Г I
R—С —С —R >R — С — С — R-I Н-,0
I I I
ОН ОН R
2. ФИЗИЧЕСКИЕ СВОЙСТВА И СТРОЕНИЕ
Насыщенные альдегиды и кетопы являются бесцветными жидко-
жидкостями со своеобразным запахом (формальдегид — газ с острых-;
запахом). Карбонильные соединения имеют более низкие темпе-
температуры кипения, чем соответствующие алканолы (табл. 41). Это
еще раз подтверждает сильное межмолекулярпое взаимодействие
молекул гидроксилпроизводных через водородные связи, что не
может иметь места в случае карбонильных соединений.
1. Полярность и поляризуемость карбонильной группы, геомет-
геометрия и электроноакцепторные свойства. Карбонильная группа яв-
является сплы;о полярной группой. Дипольиые моменты альдегидов
равны 8,3- 10-зп. . .8.7- Ю0 Кл-м B.5. . .2.6D), а кетопов — 9,Ох
хЮ0. . .9,4-Ю-30 Кл-м B,7. . .2,8D). В то же время карбониль-
карбонильная группа имеет значительную поляризуемость. Это можно ил-
иллюстрировать данными молекулярной рефракции связен. Если
440
Таблица 41. Физические константы некоторых альдегидов и кетонов
Соединение
/О
ЧН
/О
си,-с/
1 1
.0
СНзСН-. — С^
о
СН1С11.,СН..-С//
1 1
у О
СН:?Н— С/у
1 И
СПз
,0
СН;,-С^
СП,
/О
СНзСН., — Сч^
xCf-b
Т. пл,, ''С
-92
— 123,5
—81
—99
—95
-86,4
Т. кип
—21
21
48
75
62
56
79
, СС
8
7
5
6
«Г
0,815 (—20 С)
0,780
0,807
0,817
0,791
0,792
0,805
для связен С—О R —1,5, то для связи О—О R —3,3. . .3,5, Это
означает, что на атомах карбонильной группы не только имеются
значительные эффективные заряды, но они еще увеличиваются под
действием внешних факторов (атакующих реагентов).
Причины полярности и поляризуемости кроются в особенно-
особенностях строения карбонильной группы. Карбонильная группа об-
образована из двух атомов с сильно различающимися электроотри-
цательностями. В образовании связи участвуют две орбптали каж-
каждого атома и можно говорить о а- и п-связи (углеродный атом в
s/Я-гибридиаацнп). Электронные смещения в карбонильной группе
больше, чем в связи С—О. Связь С О короче по сравнению со
связью С—О:
Пет полной ясности о состоянии гнбрмдтацпн кислородного атома (см.
гл. XIV.Л.3), т. е. о природе пеподцлепных электронных пар. Вочможпы два
варианта*
а) ^р--гнбрнд1пац1Н1 и равноценность обеих электронных пар (]):
441
б) кислородный атом негибридизован,одна электронная пара находится
в р-орбитали, а другая — в s-орбитали (II).
В последнее время предпочтение отдается второму варианту.
Карбонильные соединения являются слабыми электроподоно-
рами (сравнимы с алканолами и простыми эфирами). В то же время
они обладают элекгроноакцепторпыми свойствами. Это можно
продемонстрировать присоединением электрона:
В результате присоединения электрона образуется аппон-
радикал, несиаренный электрон локализуется на низшей свобод-
свободной молекулярной орбптали, которая является разрыхляющей.
Поэтому уменьшается порядок я-связи в анион-радикале, и часто
такой аниоп-радикал изображается как частица с сильно локали-
локализованным неспаренпым электроном:
[R\ .Л
)С~О
L R' "J
>
R/
Анион-радикал карбонильных соединений образуется в реак-
реакциях с сильными воссстановителями (Гча, Zn) (см. с. 453 и 469).
2. Результаты расчетов методом МО и спектры поглощения. Рас-
Распределение электронной плотности и другие особенности кар-
R. R
\ 0,552 1;448 \ 0.443 +
R R '
' плотность я-электронов qf эффективные
и порядок я -связей P[i заряды п-д
-jf- л (а+0,3730) 0,724
R' ^ ~|
ВЗМО
2/5-)
Рис. 97. Значения э;:српьч МО, одноэлектронные плотности ВЗМО, плотность
л-электронои, порядок л-связн и эффективные заряды карбонильной группы
бопплыюп группы мо;кпо приближенно охарактеризовать расче-
расчетами по методу МО Хюккс-ля (рис. 97).
В отличие от МО этилена в карбонильной группе изменилась
энергия ВЗМО вследствие электроноакцепторпого действия атома
кислорода. Неноделеипая элеюропная пара занимает свою соб-
собственную орбиталь (п). Эффектнппые заряды исказ!>тают, что па
углеродном атоме большой дефицит электронов, здесь находится
элсктрофильпый центр карбонильной группы.
442
Возможны два электронных перехода: я->-л* и л^-гсч:, что
и наблюдается в эксперименте. В ультрафиолетовых спектрах кар-
карбонильных соединений два максимума поглощения: интенсивный
в районе 150—200 нм (л—>-я*) и малопптенсивный (е = 10. . .30)
в районе 270—300 нм {п->п*).
Очень характерны инфракрасные спектры карбонильных сосг
динений в области 5,7—5,9 нм A755—1695 см). Наблюдаются
четкие максимумы поглощения с интенсивностью в=200. . .400,
которые связаны с валентными колебаниями группы С—О. Как
правило, альдегиды поглощают при более коротких волнах, чем
кетоны.
Например, в зависимости от применяемого растворителя по-
поглощение карбонильных соединений наблюдается в следующих
интервалах:
Соединение ..... Формальдегид Другие альдегиды Кетоны
V, см-1 1740—1750 1720—1740 1705—1725
Инфракрасные спектры, а также спектры комбинационного
рассеяния являются падежными методами для обнаружения кар-
карбонильной группы.
В ПМР спектрах альдегидный протон имеет очень характерный
сигнал 6—8. . .10,5 м. д., который обычно не перекрывается с
сигналами других протонов.
3. ХИМИЧЕСКИЕ СВОЙСТВА
Химические свойства карбону "ьных соединений определяются
присутствием полярной электропоякцепторион карбонильной груп-
группы, которая способна присоединять различные пуклеофилы. Кроме
того, много реакций определяется подвижным а-водородным атомом
и еполизацией (ni. XV.A.4). Значение имеет возможность взаи-
взаимодействия сильных кислот с иеподелешюй электронной парой
кислородного атома.
В схеме 2 отражены основные направления превращений кг.р-
бонильных соединений.
Мри рассмотрении реакции карбонильных соединений необхо-
необходимо обращать внимание на некоторые отличия в реакционной
способности альдегидов и кетонов. Как правило, альдегиды яв-
являются более активными и, кроме того, они легчо кетонов окис-
окисляются до карболовых кислот.
Надо заметить, что имеются та:с;ке псеполн."пруго;дпеся карбо-
карбонильные соединения, не содержащие нодвпжисго а-водородного
О
атома. Нсслолпзпрующн.мися являются с'-ормальдегнд \\с(у и
ЧН
,О
соединения типа R3C—c(f (R^H пли треш-слиил).
1. Кислотность и ei-олмзация. Ллг дегиды п кетоны с тпдородиым
атомом в а-пелпкёпни к карбонпльно:'! группе являются слабыми
413
Схема 2. Химические свойства карбонильных соединений
О
{
J
Восстановление ^.
Нуклеоф.с-.ьное / "I j'X. ^lA™0*
присоединениеХ^н / \ F.X ^ «меилнне
ОН
он
RCH;CH,R' /O
RCH -C-Nu: RCHj-C—H RCH-C( +HX
СН-кислотамн, они способны образовывать сопряженный амбп-
деппгый анион н енол (гл. XV.A.4):
i
н
+
Енолизирующиеся альдегиды и кетоны являются таутомерными
системами, в которых одна таутомерная форма, енол, находится в
малом количестве, 10~2—10~в%. Но этого достаточно, чтобы не-
некоторые реакции прошли с участием енолыюй формы (галогени-
роваиие, нитрозированне п др.).
Альдегиды и кетоны являются очень слабыми СН-кислотами
(рК„«16. . .20), слабее алкаиолов. В водных растворах в присут-
присутствии основных катализаторов (рН 12—13) концентрация аниона
может достигать 10—10~5% от пеионизованного основного ве-
вещества. В растворах алкаиолов в присутствии алканолятов кон-
концентрация аниона достигает 1 —10%.
2. Основность. Благодаря неподеленпой электронной паре на
атоме кислорода карбонильные соединения обладают слабыми
основными свойствами и реагируют с кислотами. При взаимодей-
стшш с протонными кислотами (в зависимости от константы кис-
кислотности и концентрации кислоты) карбонильное соединение при-
присоединяет протон либо посредством водородной связи, либо
вследствие образования ст-связи О—Н. В результате возрастает
электрофилыюсть углеродного атома карбонильной группы:
+ н+х~
к
444
С кислотами Льюиса образуется продукт присоединения по-
посредством донорцо-акцепторной связи.
Альдегиды и кетоны являются очень слабыми основаниями,
рКви*- — —6. . .— 8 (для алкаполов и простых эфиров рКви + ~-
=—2. . .—4). Концентрация протонироваппых карбонильных сое-
соединений 0,1 — 1% достигается только в 60—80%-ной серной кис-
кислоте. Целесообразно помнить, что даже образование водородной
связи значительно увеличивает реакционную способность кар-
карбонильной группы со слабыми пуклеофилами.
3. Реакции у а-углеродного атома обычно протекают с участием
енола или аниона. Примером такой реакции может служить бро-
мнрование:
Н .. ._ П. ST .-7.-J'» Н С ¦, |.
R С{ C^R' ЯЛ ^R' RC' ^R' Br R
H Br^r Br-
Бромировапие является медленным процессом, скорость ко-
которого лимитируется скоростью енолнзации карбонильного сое-
соединения.
Карбонильные соединения с подвижным а-водородпым атомом
вступают и в другие реакции, которые в принципе можно рас-
рассматривать как реакции электрофильного замещения, проходящие
через енольную форму или анион карбонильного соединения.
К таким относятся, например, реакции нитрозирования HN'CL
или нитритами, нитрования нитратами, альдольная конденсация.
4. Реакции карбонильной группы с нуклеофильными реагента-
реагентами. Легко протекают реакции присоединения нуклеофильных ре-
реагентов к карбонильной группе. Присоединяются как ионы, так
и нейтральные частицы с электроподонорным характером:
RCH —С^°\ Л- -"N^r *=RCH,-C—Nu: ^~ RCH.-C—Nu:
as- /F ¦ An
-C- + :NuH —RCH.-C~N.iH — RCH.-C-Nui
Ниже рассмотрены конкретные примеры этих реакций, систе-
систематизированные по типу пуклеофилыюго атома, присоединяюще-
присоединяющеюся к карбонильной группе, т. е. Н-нуклеофилы, С-пуклеофплы,
N-, О-, S-нуклеофнлы, галогенид-ионы.
а) Присоединение Н-н у к л е о ф и л о в представ-
представляет собой гидрирование донорами гндрнд-иопов. Например, ггд-
рпроваппе комплексными гидридами металлов — боргидрпдамн,
М'ВН/, алюмогидрндамн М!А1М4":
б- ОПП;1\а +
л- О i ;.¦:-- /ОН
RC11.-C -|-Хл' ШГГ -» RCI1.--C-M КСП.-С-И
RL
415
В качестве донора гидрид-иона могут быть использованы трн-
алкилсиланы в присутствии сильной кислоты (CF3COOH):
/О ГР гппи /он
RCH2-Of -j-H-SiCCHJa 3 lRCH2-cAl -f CF3COOSi(C 1Ь)з
XR* XR*
В этой реакции прежде всего происходит протопированпе карбо-
карбонильного соединения и затем перенос гидрид-нопа на положи-
положительно заряженный углеродный атом (метод ионного гидрирова-
гидрирования). В отдельных случаях ионного гидрирования карбониль-
карбонильные соединения превращаются в углеводороды (превращение
RR1C--O-^RR1CH:). Другие методы восстановления рассмотре-
рассмотрены ниже.
б) Присоединение С-н у к л е о ф и л о в к двойной
связи углерод—кислород включает целую группу весьма различ-
различных реакций, так как различна природа С-иуклеофплов. Известен
ряд С-нуклеофилов карбапиониой природы. В основном здесь
идет речь о металлоргапическнх соединениях со связью С—Л1 раз-
й- 6'-
личной полярности (R2—М i- ~\<-:-fM+):
б- O-.W+
б. О б- б»- I
RCFb-C;/ -[ R2 —М -> RCH..-C- R2
'• Rl I
RL
Углеводородпые остатки R- могут быть различного типа (ал-
кильпые, алкенильпые, арильмые, алкиннльиыс).
В результате реакции присоединения образуются производные
алканолов или других гидроксилсоедппений — алканоляты (ал-
kokci/ды), это важный метод получения алканолов (спиртов). На
присоединении сопряженного карбапиоиа, генерированного из
альдегида или кетона, к карбонильной группе основывается важ-
важная реакция — альдольная конденсация (правильнее было бы на-
называть эту реакцию альдольпым присоединением):
" И^6 Na -С°
К1 R
+
^6 Na — лен,-с-сн-сС
- ( !
он
НО I JO i ,
===r RCH,-O-CH-C< , -1-NaOII
I I л
R1 R
В реакции альдолыгого прпсоедппепня образуются р-гмдрок-
сикарбонпльные соединения. Образование нрос!сГ;шсго соод'пг.-г.пя
этого типа — альдоля из ацетальдегпда впервые наблюдали в
446
1872 г. независимо друг от друга русским лп.ч;т
А. П. Бородин I! французский химик Ш. Л. В:срц:
ОН
/О кяои I /О
II
а-.ьдоть
р-гмлроксммасляный альдегид
В некоторых случаях альдольное присоединение происходит
в присутствии кислого катализатора. При этом нейтральный и
слабый С-нуклеофил — енол — присоединяется к активированной
карбонильной группе (см. ниже).
Продукты альдолыюго присоединения легко отщепляют моле-
молекулу воды и образуют ненасыщенные соединения (кротоновая кон-
конденсация, см. ниже).
Подобно альдольиой конденсации протекает реакция присое-
присоединения нитроалкапов к карбонильным соединениям (гл. XXI.A.3).
Известно присоединение цианид-иона, генерируемого из циа-
цианистого водорода: II—С-^ К ;=? Н + + -:С\. В этой реакции необхо-
необходимо присутствие как цнапид-иопа, так и протона:
6- O-Na+ ОН
б4- /Л^ I н+ х~ '
RCH2 —C\ -J-Na + 'iC^K^RCH-.— С — С-; N i-* RCHo — С—С -_- N
R "I • " I
R1 R1
В этой реакции образуются а-гидроксииитрплы (цнангидрипы).
Реакция обратима, в щелочной среде циангидрппы расщепляются
на цианид п карбонильное соединение.
К нейтральным С-иуклеофилам принадлежат некоторые фос-
фоннепые и сульфопневые внутренние соли (нлнды) и диазоалканы.
ФооЬонийгилнды при реакции с карбонильными соединениями
дают ;:.(кены (реакция Впттпга), а сульфопинплиды — эпоксиды:
б- О- R»
6 ¦¦ .О Rs б- б * I i i ,о
-С'' :- С г Р(С0Н5K-
Ri R-
Rl/ 4Rl
б- °-R3
&¦¦ /О 1^з а- л. | ; , ,0
RCH,— С'-' -|- >С- S(CII..J —>- RCII— С — C--SlCII3)., --
RCH2- /иЧ /R3
—>- ;с—с{ -j-s(ChsJ
Rl/ \R2
Диазометан и другие дпазоалкапы присоединяются к карбо-
карбонильным соединениям. Продукт присоединения распадается с
выделением азота и образованием лисо эпокснда, либо за счет изо-
447
мори.шиш кетопа:
+ +
+":СН,-\ —N —>• RCH.,-C-Cn,-\ =,- \
« ?
б1-/.О + ' + -\,
-C^ C
RCHo-C-Cn2 > RCH; —С
R1
R1
/
RCn2-C(f
Реакция катализируется кислотами Льюиса.
Нейтральными С-пуклеофплами являются алкены п арены с
электронодоиорпымн группами (енолы, епамппы, фенолы, арпла-
мнпы, в отдельных случаях замещенные алкпльцыми группами
алкены, например 2-метилнропен). Енолы и енамппы являются
нуклеофилами с двумя реакционными центрами С и О, С п N. Фе-
Фенолы н ариламппы имеют реакционные центры на о- и л-углеродных
атомах и О- или К-атомах.
Для осуществления реакций карбонильных соединений с нейт-
нейтральными С-нуклеофилами необходима активация карбонильного
соединения протежированием. В условиях кислотного катализа
карбонильные соединения реагируют с фенолами и арпламипамн:
, S- L _ s_ ОН
siCo н х W I
R W_ ^
(D -- Oil, NRi).
Если D является первичной или вторичной аминогруппой,
параллельно протекает реакция у атома азота.
Реакции с фенолами и ариламипами обычно не останавлива-
останавливаются на первой ступени, и происходит реакция со второй молеку-
молекулой нуклеофнла. Примеры таких реакций приведены в гл. XV.Б.4.
Реакции с нуклеофильнымн алкепамн можно изобразить схемой:
,, ^.6- , i _ , /-, НО Н Х~ . s ОН
^нх ^ ^$-4
R &
(D-OH, XR:!).
В случае D—-Alk пли CJI3 реакция протекает более сложно,
л продуктами реакции являются циклические эфиры— 1,3-дпок-
саны. Это особенно характерно для реакций с формальдегидом (ре-
(реакция Прннса, гл. ТП.Б.1). Но первая ступень реакции соответ-
соответствует вышеприведенной схеме (до образования карбокатиона).
в) П р и с о е д и '<[ е н и е К-и у к л е о ф плов к карбо-
карбонильной группе представляет собой различные реакции аммиака,
первичных и вторичных аминов, гидразина и его производных,
448
R1
ОН
1
RCH;-C-
R1
XII — R*
--> RCHo
Rl H
X-R*
—c.-'
гидроксиламина и других соединений с первичной пли вторичной
аминогруппой.
Реакции аммиака п соединений с первичной аминогруппой
можно изобразить схемой:
в_ О- II
6^.0 .. I -i I
II .0
Реакция почти всегда .закапчивается отщеплением молекулы
воды от продукта присоединения н образованием соединении с
двойной связью С-Л'. В ряде случаев происходят дальнейшие
превращения - - олпгомернзацня, образование гетероциклических
соединении. Подробнее это будет рассмотрено в гл. XXVII.Д.
Азотсодержащие производные карбонильной группы имеют
свои названия:
R- ] 1рои uio.iii'ir
II, \lk, А: Азометнп (И.мни)
ОН Оксн.м
\На Ридразоп
X'lICel I-, Фепнлгидра.эон
XlFCOXII-j Ссмикарбазои
Многие из них используются для идентш^пкаппп карбониль-
карбонильных соединений по температурам плавления.
При взаимодействии гидразина с двумя молекулами карбониль-
карбонильного соединения образуются азпны:
х — х
RCII— С:"' ч;С — CII..R
Rl R"
Реакцпи со вторпчнымгг аминами закапчиваются иначе. Здесь
связь <> ,\ образоваться не может. Происходит, особенно в при-
присутствии кислых катализаторов, отщепление молекулы воды и
образование епнмшюв (гл. XXIII.Б). В избытке амина можег за-
замещаться 011-группа, получаются диамины (ампналп):
ОН
.О .. .R:1 ! R:1 us.rw
RCM.-C- - I[X ,RC[[,-C-X >
¦Rl R- i -R-
/X
,Сг=Сч R- -| 11,0
R/ 'R1
R\ ,R2
'i R:J
-*RCI1., —С — X -S HoO
я-
R1
15 .v.. -,[7 410
Образование амнналей особенно характерно, если двойная связь
С—С образоваться не может (формальдегид и другие неенолизн-
рующпеся карбонильные соединения, в том числе аренкарбальде-
гиды).
г) II р и с о е д и н е п и е О-н у к л е о ф и л о в к карбониль-
карбонильной группе представляет собой реакции карбонильных соединений
с водой, алкаиоламп, пероксикнслотами, реакции олигомериза-
ции и полимеризации. Реакции с анионными О-иуклеофилами (гнд-
роксиды, алкаполяты) обычно вызывают другие превращения (аль-
долыюе присоединение, реакции диспропорционировання).
Вода и алканолы являются слабыми иуклеофилами, поэтому
реакция идет только с очень активными карбонильными соеди-
соединениями (формальдегид, галогензамещенные альдегиды и кетоны)
или с активированными вследствие кислотного катализа карбо-
карбонильными соединениями. Эти реакции можно изобразить схемой:
ОН
/о...п:х- .. I
RCII.. —Cf ~'-HOR2 —> RCH,—С —OR2-' IHX-
\R1 ¦¦ |
К1
(R2^-H, алкил).
В реакции с алканолами получаются полуацетали. В избытке
алканола нолуацетали превращаются в ацетали:
Off OR2
! 11-Х- I
C2
R* R1
Реакции с гидроксидами и алканолятами могут привести к
продуктам присоединения к карбонильной группе, но для еполи-
зирующихся карбонильных соединений параллельно протекают
реакции альдолЬного присоединения, а для нееполизирующихся
альдегидов — реакции днснропорционирования (гл. XXVII.Г.З):
О"М"
RCH
r-c;
,^() M
+
KOR2
~iOR"
» A
= R
л(>дольное
о~м'
-C-OR
прис
оединение
прсвраи
450
Альдегиды могут трпмеризоваться, а формальдегид'—даже
полимеризоваться:
r-c:
Со •н+х
ОН v-
R-C—O-C-R
А н
;c-'r
:он 0_
R-C-O-C-R
А A
л
но
R-C-
н
R
I
-о—с-
н
R
I
о-с—он
н
д) П р и с о е д и и е и и е S-н у к л е о ф и л о в к карбониль-
карбонильной группе представляет собой реакции карбонильных соединешп"!
с сероводородом, июлями (меркаптанами), гидросульфитами.
Реакции с сероводородом и тиолами осуществляют б присут-
присутствии кислот с целью активации карбонильной группы. При взаи-
взаимодействии с сероводородом получаются тнокарбоинльные соеди-
соединения плп продукты их дальнейшего превращения (олигомеры):
RCH,— С •••'''
• О-..Ц • X-
R1
SII
on
-; h'sh - - rcii..—c—sh-'-ipx- -*
Rl
s
— 11.s ^
-^ RCII, — C — SII -RCII, — C-''
i R
R1
Хорошим реагентом для нрепращення карбонильных соединении
в тиокарбопильные является P^S.-,.
Тполы (меркаптаны) реагируют с карбонильными соединениями
с образованием тпоацспален (меркапталей):
ОН
,О- • -II ' Х~ .. 1 I1SR2
RCIIL,-Cf -I IISR--^RCII, -C-SR--pII*-X- >
—>¦ RCII; — C — SR2-;-II,0
Rl
Своеобразным S-пуклеофнлом является анион гидросульфита
HSOj. Гидросулыриты (бисульфиты) присоединяются к альдсги-
15*
451
дам и котонам с образованием «бпсульфитпых соединении», кото-
которые представляют собой соли u-гпдрокснсульфоновых кислот:
й-
Q- ¦ Н-() ^^' Q
RCf(.-<(), -I NaHSO, RCH.-c^V-' Ч - RCH,-C-S=O
R ^R :S-0 , ,
O"Na; R: ° "*
1-Я" и соединения хороню кристаллизуются п нередко применяются
,'ыя очистки карбонил[IП.1.\ соединении. В слабощелочной среде они
расщепляются на карбонильное соединение и сульфит.
с) Ир II с о е д п и о и и с г л л о г е и - н у к л е о ф п л о п к
ыфСонпльпон группе представляет собой реакции карбонильных
и.едпнеппн с галогенндамп ф(хч[юр.'1, серы н др. Прямое в:;апмо-
де1!ствне карбонильных соединении с галотшд-поплмп не паблю-
длстея.
При пзаимодспггвин с галогеппдамп (|)ос(|«!ра и серы (сильными
•.¦пектрофпламп) сначала происходит активация карбонильного
соединения и затем реакция с галогеннд-поном:
6- л-
KCII..- С
о
R1
О-
I-PCI.-, ,
PCI,
: RC1I;
Й ¦
- • С
Cl
,()
R1
. I
(
ч::,
— RCH,—С —CI -л RCU,—С -CI-; О я
R1 R1
Аналогично протекает реакция с SF:,.
5. Восстановление карбонильных соединений. Карбонильные
'¦оедппення могу! быть восстановлены до соответствующих спиртов
или углеводородов. Восстановление комплексными гидридами и
станами как реакции пуклеофплыюго присоединения было рас-
рассмотрено выше.
В реакциях каталитического гидрирования получают спирты:
OIF
0 м !
RCH2—С. : RCII..-C —II
4R1 Ni - |
R1
Гидрирование в присутствии аммиаки или аминов ведет к по-
получению аминов (восстановительное амппярованп^ карбонильных
соединений или восстановительное алкчлпроваппе аммиака пли
аминов):
H\R-
.0 R2 N11,: Н., !
RCH-, — С:' ¦ >RCII.,—С —Н
\Rl к. " |
R1
Восстановление карбонильных соединении происходит при
в.ча.нмодействпи с активными металлами Gм, A\g) в водной среде.
Так, восстановление цинком в кислой среде в основном дает угле-
452
водороды (метод Клемменсена):
Н
R1
Своеобразно протекает восстановление амальгамированным пин-
пинком или магнием в нейтралкой водной среде. При этом получа-
получаются нинаконы. Первой стадией этой реакции является присое-
присоединение электрона к карбонильной группе, образовавшийся анион-
радикал затем днмеризуется и протонируется:
2RCH2-C<;ri -— [rC1.2-C4ri J2Zn- -
ОН ОН
н2о I I
> RCH2-C — C-CILR
R1 R1
Восстановление карбонильных соединений до спиртов проис-
происходит при взаимодействии с алканолятами (алкокспдамп) алюми-
алюминия, производными вторичных спиртов (пропанола-2, ппклогек-
санола). Эту реакцию открыли X. Меервейн н А. Верлей A929)
и независимо от них В. Пондорф A926):
Г Н ^
3RCH2 —C-" -|-Л1[ОСН(СНзJ1з
R1
СН
;- с- о Л1 ; зо=. сч
R1 -:|
; и ' х-
Н
I
RCH-j—С —ОН
R'
Реакция обратима. Поэтому необходимо отделение продукта
реакции — ацетона — из реакционной смеси, что достигается от-
отгонкой.
В реакции Меервейпа — Понндорфа — Верлея происходит пе-
перенос водородного атома от вторичного спирта на карбонильное
соединение. Это осуществляется в циклическом комплексе; предпо-
предполагается, что водородный атом переносится в виде гидрид-попа:
л- ^ ch,r
[(CH.O.CK-oI.aK0^"^ [<СН.);СН-О]/1-О-С-Я'
h ,•''—- н
снЛсн, о=с^сн.,
Реакция требует избытка алканолята плюмиппя. В присут-
присутствии малых количеств а.чкаполяга алюминия восстановление не
453
идет. Альдегиды с этих условиях диспропорционнруются с обра-
образованием сложного эфира. Одна молекула альдегида восстанавли-
восстанавливается за счет второй молекулы, которая окисляется:
XII XOC1I,R
Реакцию открыл В. Е. Тищенко (i906). В реакционном комплексе
происходит перенос водородного атома с одной альдегидной группы
па другую. Л1еханнзм реакции во многом подобен механизмам ре-
реакции днепропорцпоннровання аренкарбальдегидов (реакции Каи-
пиццаро и Клямзеня, гл. XXVII. Г.З).
6. Окисление карбонильных соединений. Альдегиды легко окис-
окисляются различными окислителями. Конечным продуктом окис-
окисления является карбоновая кислота
f --R--C."
Альдегиды окисляются рядом неорганических соединений
(КМпО„, СгО3), т. с. выступают в качестве восстановителен. На-
Например, альдегиды в щелочной среде способны восстанавливать
ион серебра до металлического серебра, при этом серебро осаж-
осаждается ка стенках сосуда в виде зеркала — реакция серебряного
зеркала. Реакция идет в присутствии аммиака, окислителем яв-
является комплексный iiofi 1Ая(\ПяJГ'" (реагент Толленеа):
.О
R— С7 ¦¦• 2Пй(\Н..),] -ОН- — R—COO-\Hi ' 2Ag + H,O-f 3\П3
Щелочная среда и присутствие аммиака вызывают также побоч-
побочные реакции (альдольное присоединение, образование иминов и
их олигомеров).
Некоторые альдегиды в щелочной среде при нагревании восста-
восстанавливают также реактив Фелинга, который содержит комплексно
связанный с винной кислотой (гл. XXXIV.Б.3) ион Cu2t\ При
этом выделяется оксид меди СиХ) красного цвета. Реакция имеет
ограниченное применение, например, ее не дают ацетальдегид
и аренкарбальдегнды.
Превращение альдегида в карболовую кислоту связано с от-
отрывом двух электронов и водородного атома. Процесс облегча-
облегчается присоединением гидрокснльной группы. Формально этот
процесс можно изобразить как отрыв гидрид-иона окислителем:
ом'
R-C^° + М'01-Г *= R-C-OH
ОМ*
'~~~Окисл.
454
Весьма вероятно, что процесс идет более сложно, многоступен-
многоступенчато.
Реакцию серебряного зеркала и реакцию Фелгшга дают также
и другие органические соединения, способные в щелочной среде
легко окисляться, например u-гидрокспкетоны (гл. XXIX.Б. I).
Кетоны к действию окислителей весьма устойчивы и окисля-
окисляются лишь сильными окислителями при нагревании. В процессе
окисления происходит разрыв связей С—С по обе стороны карбо-
карбонильной группы и в общем случае получается смесь четырех кар-
боновых кислот:
/>° 1°1
RCH, — C-/ — RCH.COOn-'-R'COOK + RCOOH + R'CHXOOH
4CHR1
Некоторыми окислителями, например SeO,, ос-.мстилеповая
группа ал[,деп1дов и кстопов превращается в карбонильную:
О
о ¦, о
RCII, — С:у -; SeO., - >RC—C-" П.,О- Se
4' 4'
Своеобразно кеточы окисляются перокситамн и пероксикпсло-
тамм (гл. XXXIII.11.3).
7. Особенности реакционной способности альдегидов. Альдеги-
Альдегиды в отличие от кетонов всегда более активны в реакциях нуклоо-
фильного присоединения. Самым активным альдегидом является
формальдегид, который занимает по реакционной способности
особое .место. При переходе от формальдегида к другим альдегидам
и далее к кетоиам реакционная способность надает с уменьшением
эффективного положительного заряда на углеродном атоме кар-
карбонильной группы. В этом сказывается электроподопорпый ин-
индуктивный эффект (-'-/) алькпльиых групп.
Высокая реакционная способность альдегидов выражается в
образовании циклических тримеров и даже полимера (формаль-
(формальдегид). Легко протекает альдолыгое присоединение, которое в
жестких условиях (концентрированная щелочь, повышенна;! тем-
температура) может привести к полимерным продуктам (альдегидные
смолы).
Особым отличием альдегидов является их способность к окис-
окислению. Эго связано со структурными особенностями альдегидов.
4. ВЛЖПГ.ЙШШ1 ПРГ-ДСТЛВПТЕЛН
Формальдегид представляет собой бесцветный газ с острым запа-
запахом, хорошо растворимый в воде и органических растворителях;
35-- 37%-нып рас тор формальдегида в воде, содержащий 5—lo"d
метанола для стабилизации, называют формалином.
455
Формальдегид в промышленности получают в крупных мас-
масштабах. Основной .метод — каталитическое окисление метанола:
Си (л,о /О е
СНзОН-Ь1/./}, >Н- Cf +HiO- СИ,.i-O.,
(или А(г) \j_[ Кат.
Применяется также каталитическое окисление метана.
Формальдегид — самый активный из альдегидов, он легко
присоединяет нуклеофильпые реагенты, олнгомеризуется и даже
полимеризуется, действует как восстановитель. Он принадлежит
к неепол тирующимся альдегидам.
В водных растворах формальдегид легко присоединяет ноду,
олигомеризуется и полимеризуется. Известны циклические и ли-
линейные олигомеры:
он I
НО(СН,О)„СН2ОН
Из водных растворов при упаривании в вакууме постепенно
выделяется бесцветный порошок, смесь олигомеров и полимера
с п -8—100, так называемый параформ. При нагревании иараформ
деполимеризуется. Его используют для синтезов вместо газообраз-
газообразного формальдегида.
Формальдегид широко используется для получения различных
полимеров (феполформальдегидные смолы, карбамидные смолы).
При полимеризации безводного формальдегида в инертных раст-
растворителях получают высокомолекулярный полиформальдегид
(СН2О)П1 гдеvn« КЮО. Полимер размягчается при 170—180'С,
его используют для получения волокон, пленок и конструкцион-
конструкционных деталей.
Формальдегид используют в органическом синтезе для введе-
введения гидроксиметильпых (НОСН2), хлорметильных (С1СН2) н ами-
нометильных (R2NCH2) групп, для получения 1,3-диоксанов, для
восстановления.
С аммиаком формальдегид образует полицнклнческое соедине-
соединение со структурой адамаитана—уротропин:
^ ^ NO2
уротропин гексогек
^'ротронин представляет собой бесцветное кристаллическое
вещество, хорошо растворимое в воде. В кислой среде разлагается
на СН.2О и Х!Н3. Его используют в органическом синтезе вместо
формальдегида и в медицине.
При нитровании уротропина получают мощное бризантное
взрывчатое вещеегно — гексаген.
Ацетальдегид представляет собой легколсчучую бесцветную
жидкость со своеобразным запахом, в больших концентрациях —
острым и удушливым.
Методы получения aneiальдегида уже рассмотрены выше. В про-
промышленности используют реакцию ирпсое/ишепня воды к ацети-
ацетилену (гл. IV.4.1), каталитическое окисление этанола кислородом
воздуха (гл. XIV.А.4) и окисление этилена в йодном растворе кис-
кислородом в присутствии PdCla н СиС12 (гл. 11.1.-4).
Ацетальдегид используют для получения уксусной кислоты,
пероксиуксуспой кислоты, бутадиена, пептаэрмтрпта и других
важных соединении.
Пентаэрнтрпт получают в процессе альдолыюго присоедине-
присоединения ацетальдегнда к формальдегиду с последующим восстанов-
восстановлением:
СП.,-)]]
ЗН--С/ ¦¦си.-с -носи,--с: —с _L
СН..ОН
СН2ОН
—>МОСИ, — С — СН,0!1
СНоОН
Ацетон — бесцветная жидкость с приятным запахом, смеши-
смешивающаяся с водой и органическими растворителями.
Ацетон в промышленности получают в крупных масштабах из
прочена двумя способами:
а) Пропен - Пропл1ю:1-2 -> Ацетон
б) Пропеп - - Кумой ¦- Гпдропсроксид кумола ¦ • Ацетон -[ Фенол
Ацетон широко используют н качестве раствори геля, особенно
для растворения нитроцеллюлозы и ацетилцел.тюлозы, ацетилен:-!.
В органическом синтезе из ацетона получают кетен, изопрен, окись
мезитила.
Циклогсксанон — бесцветная жидкость с приятным запахом,
он плавится при —40, а кипит при 199,6'С. Мало растворим б
Еоде (до 7%).
В промышленности цнклогексанон получают каталитическим
окислением циклогексапа или каталитическим дегидрированием
циклогексапола.
Циклогексанон является одним из основных исходных веществ
для синтеза важных мономеров. Из циклогексанона получают кап-
ролактам п аднпиповую кислоту.
Камфора A,7.7-трнметплбнциклоB.2.11гептапон-2) — бесцвет-
бесцветное кристаллическое вещество с характерным запахом; т. пл. 178 —
179 С, легко возгоняется, т. кип. 209,1 С.
457
Камфора широко распространена в природе,
входит в состав многих эфирных масел. Особен-
=о но много ее в камфорном масле, получаемом из
камфорного лавра. Из этого масла выделяют на-
натуральную камфору, которая оптически активна (молекула кам-
камфоры хиральнл, известны два энантиомера). В промышленности
камфору синтезируют из а-пинена. Синтетическая камфора опти-
оптически неактивна (рацемическая смесь).
Камфора применяется в промышленности в качестве пластифи-
пластификатора и стабилизатора нитроцеллюлозы и в медицине как сред-
средство, усиливающее сердечную деятельность.
Б. НЕНАСЫЩЕННЫЕ КАРБОНИЛЬН ЫЕ СОЕДИНЕНИЯ
I. МКТОДЫ ПОЛУЧЕНИЯ
Сопряженные, или и,р1-ненасыщенные (непредельные), карбониль-
карбонильные соединения получают двумя основными методами: отщепле-
отщеплением воды с;т продуктов альдолыюго присоединения и окислением
алкеполов или алкепов.
1. Дегидратация альдолей, кротоновая конденсация. Альдоли —
продукты альдольного присоединения ф-гпдроксикарбонильные
соединения легко отщепляют воду в присутствии кислого катали-
катализатора, иногда и в присутствии основного катализатора в реакции
альдолыюго нрпсоедпней11я):
Н
! ,0
СН-, — С-СП.— С7
I ЧП
он
СП;,'
о
i
к рот оно nun ;i. н.дс^пд
В результате дегидратации яльдоля пот\чается кротоновып аль-
альдегид, поэтому часто этот процесс называют «кротонпзацпей».
В ряде реакции карбонильных соединений, например ареи-
карбальдегндов с ко гонами:
О
Г I II
,0
,. , с/ о
N
,С--'
+ СН,-С-СП
Nii-i-OII -
п
чсня
продукт альдольного присоединения выделить не удается, сразу
получается ненасыщенное карбонильное соединение. Такие ре-
реакции называют кротоновой конденсацией.
2. Реакции окисления. Алкенолы и алкинолы легко окисляются.
Для окисления необходимо выбрать окислитель, который в данных
458
условиях не взаимодействует с двойной или тронной связью, на-
например СгО;) при пониженной температуре:
ОН О
r/ xh r/ Nf
он
I Crdj у О
I xRl
II
При использовании специальных катализаторов окисления
(СиО) при повышенной температуре можно окисли гь алкены, на-
например пропек:
о 'С-',о ,0
СП-г-СП —СП., > СН2- СП—С'/
..он- м>и н: Чц
2. ФИЗИЧЕСКИЙ СВОЙСТВА II СТРОЕНИЕ
Простые а.р'-пенасыщенпые карбонильные соединении представ-
представляют собой бесцветные жидкости с острым раздражающим запахом.
Соединения с арильпыми группами в молекуле являются желтыми
кристаллическими веществами.
r:o-J,- Соединения характеризуются полярной сонря-
41 г'г женкой системой. Ввиду значительного иидукцмоп-
_ " ного эффекта (—/) карбонильной группы и боль-
R^*" """^н шого мезомерного эфс|)екта (—М) двойная связь
сильно поляризована, динольнын момент таких соединений A0':
X 10~м —10,4-10~30 Кл-м или 3--3JD) больше, чем для наськцен-
ных карбонильных соединений.
Распределение л-электронной плотности приблизительно можно
охарактеризовать результатами расчета по методу МО Хюккеля:
Эффективный
заряд 0,21+ 0,05- 0,44+ A,6-
с=с—с=о
Порядок 0.882 0,462 0.72.1
п -связей Prs
а, р-Ненасыщенные карбонильные соединения в молекуле имеют
два электрофильных реакционных центра.
Максимумы поглощения в УФ-спектрах сдвинуты баюхро.\и:о
по сравнению со спектрами иесопряженпых карбонильных соеди-
соединений. Сопряженные алкепалн и алкеиопы поглощают при 2LJ0 —
260 пм (е«10 000) (л-^л*-переход) и при 310—ЗЗ;; '»м (г^К.'О)
(п->л* -переход).
R ИК-спектрах поглощение карбонильной группы появляется
при меньших потовых чи-:лах, чем в случае пссопряжепных кар-
карбонильных сседнпеппн. Например, соединение RCH = СИ — С^
459
имеет максимум карбонильной группы при 1680—1705 см, сое-
о
\г сп-с/-' ^ „р,, 1665-1685 см.
ДН11С1П1Я
Jtii изменения в спектрах сопряженных карбонильных соеди-
соединений связаны с особенностями сопряженной л-электропной си-
системы (изменение энергии молекулярных орбиталей, уменьшение
порядка л-свяли карбонильной группы).
3. ХН.ЧИЧКСКИП СВОЙСТВА
Для а.р-пепасыщеиных карбонильных соединений характерны
все реакции карбонильной группы. В то же время в молекуле
имеется второй электрофпльпый центр - ^-углеродным атом, по-
поэтому возможно образование двух продуктов реакции:
он
л -
(¦", >s' о
,С--СП — Су -\--.\v.W-
R1
Л-
()
R — С-.. С —С
"* /
-, И -
— R-
R
:.\и
С^
R
1.
1
С-
/С
СП
-СП
С —
CI
—с-
1
R1
(^
он
-N'li.
',°
С —N'u:
Rl
I-C
/О
XR'
Зги реакции широко используют в органическом синтезе. Напри-
Например, по р1-углеродному атому легко присоединяются медьоргапп-
чеекпе соединения:
RCII СИ—
R
CI
СП-СП,-С
,0
R1
Ненасыщенные карбонильные соединения вступают также в
реакции, характерные для С -С-связп, например реакции вос-
восстановления, полимеризации. Они вступают в диеновый синтез
как активные дпепофплы, а иногда и в роли сопряженного дпепа
Д\етильная группа в кротоповом а/ниегнде обладает повышен-
повышенной реакционной способностью и легко вступает в реакцию крото-
новон конденсации с альдегидами, т. е. она реагирует аналогично
метилыюй группе в уксусном альдегиде, несмотря па то, что от-
отделена от карбонильной группы двойном связью. Эк) объясняется
переносом электронного эффекта карбонильной группы через
двойную связь на метальную группу. Такое явление называется
460
винилогиеи:
.0 О ои_ О
СНяС^ -i-CII-.CII Cf ГС L , СП^СН СНСП СНС7 -4-Н..0
чп н •п "
4. ВЛЖШ-ПНШН ПРПДСТЛВНТПЛП
Акролеин СП-2 CM -CM О представляет собой бесцветную жид-
жидкость с острым запахом, вызывает слезотечение (лакриматор);
т. пл. —87,7 С, т. кип. 52,5 'С. Растворяется в воде и органиче-
органических растворителях.
В промышленности получают окислением нронена над СиО
npir 300—400 С. Акролеин легко полнмерпзуется, поэтому его
стабилизируют добав.кмшем гидрохинона.
Акролеин является важным промежуточным продуктом в ор-
органическом синтезе. Из пего получают аллиловып спирт, глицерин
и множество других продуктов. Акролеин вступает в диеновый
синтез пс только как активный дпепофнл, по и в качестве диена,
например:
Окись мезипшла D-метплпентен-3-он-2) — бесцветная жидкость
с острым запахом; т. кип. 128 °С. Ее получают из ацетона реак-
реакцией альдолыюго присоединения и дегидратации:
О ОН о
/О .' I I
СНз — Of -j-CII2 —С —СП3--СН3—С —СН2 —С —СН3 —»
ХСПз I
СН3
СПзх /О
— хс=сн—с^ +н2о
Окись мезптила применяют в качестве растворителя некоторых
полимеров и как исходное в органическом синтезе, например для
получения производных цнклогександпона-1,3.
В. КЕТЕНЫ
Кетепами называют соединении, содержащие карбонильную группу
в системе кумулированных двойных связей С--С--О. Карбониль-
Карбонильная группа в кетенах принципиально отличается от карбониль-
карбонильной группы в обычных карбонильных соединениях. В кетенах
углеродный атом карбонильной группы другой гибридизации —¦ sp.
1. Номенклатура. В основе номенклатуры кетенов лежит назва-
название основного соединения. Например:
1-1 CIU
С С О ч,с С О
Л СП-,'
4G1
2. Методы получения. Простейшие кетепы получают пироли-
пиролизом кеюнов нлп карбоповых кислот G00. . .800 С) в присутствии
({юсфатпых катализаторов. В реакции происходит гемолитический
разрыв связей:
сн,-с-'° 700-soo°cicir3-c- о-;-.сн,-> "ч с с о+сп,
сн., н
.О 700-Ь00=О . R-.
RCH,— СУ/ -RCH,—С- О+-ОИ -¦ С С- O-i-H.O
ОН лп'О, ' н
Общим методом получения кетенов в мягких условиях является
отщепление галогсноводорода от ацнлхлоридов в присутствии
сильного органического основания, например третичного амина:
R\ ,0 R1,
,С-С" +N(C.H5)s -* /С С О-; [11Х(С.ПГ1ЫС1-
R/ | XCl R/
И
Специальной реакцией является термическое нлп фотохимиче-
фотохимическое расщепление и перегруппировка а-дпазокетоиов:
R-c-c/- —-> ;c с о-;-х..
; xRl R17
3. Физические свойства и природа связей. Кетепы в обычных
условиях являются газами пли желтоватыми жидкостями с острым
запахом. Вещества малостабпльны.
В молекуле кетепов углеродные атомы кетеноной группировки
находятся в состоянии sp'-- и зр-гибрпдпзацнп (структура типа
алленов). Кетепы являются этоктрофпльнымп реагентами, элект-
рофильный центр находится на лум'ибрпдпзпронаппом углеродном
атоме:
4. Химические свойства и применение. Кетепы — высокореак-
циоиноспособные соединения. Для них характерны реакции с пук-
леофнльными реагентами, реакции димеризацпи и цпклопрпсос-
дипения.
При взаимодействии кетенов с нуклеофильиыми реагентами
получаются производные карбоновых кислот:
R^=c4 + :NulI -* R^=C^l -* R>-C<"
К к. NuH. "¦ Гчц»
н
462
Кетеп в жидком состоянии легко дпмеризуегся, образуя днкетсы:
н,с=с=6' J',-<L,-H?! г., ,~_^о
н.с=с—о
н с—с
" "с-о
их'
дикегегг ¦
Эта реакция г'рмнлдчожнт к реакциям циклопрнсоедпнения
[2-(-2|, которые свойственны активной двойной связи кетенов:
R>=C=O R-C-C^
R4*> ~~~ r!-(j-y
Дпкетеи представляет собой ненасыщенный Р-лактон, произ-
производное ацетоуксуспого эфира. Дикетен попользуют для синтеза
ацетоуксуспою эфира и его производных.
В промышленности широко используют кетен, бесцветный газ
с т. кии. - -41 "С. Из пего получают уксусную кислоту, сложные
эфпры уксусной кислоты, уксусный ангидрид, дпкетен:
и.,0
Н,С г С = О •
ROH
О
ОН
О
OR
О О
¦СП:.С —О —ССПз
Своеобрлзпым представителем кетенои является недоокснд уг-
ле]1ода CjOL1, который получают взаимодействием малоновой кис-
кислоты и Р265:
с и,спок
О. ,0 р г,
^C-CI-b-C' —-5-н
НО-' ' ХОП --'"/
о.- с с-. С- о
Недоокснд углерода представляет собой бесцветный газ с раз-
раздражающим запахом; т. кип. 7 С. Он активно, подоГпю котеиам,
реагирует с пуклсофиламн, образуя производные маюновой кис-
кислоты:
О- С- С С--- О-;-2:\и1Г -->
О
:\u
—СИ- —С
О
\V.::
4G3
Г. КАРБОНИЛЬНЫЕ СОЕДИНЕНИЯ АРЕНОВ
I. МЕТОДЫ ПОЛУЧЕНИЯ
Для получения арепкарбальдегидов, алкпларплкетопов и ди-
арплкетопов используют реакции прямого окисления алкпларепов
и диарилметапов, гидролиз дпгалогепалкнларенов, реакции фор-
мплпрования и ацплпроваппя арепов и др.
1. Окисление. При окислении алкпларепов или диарилметапов
получаются карбонильные соединения:
гог /° км ,0
ХН R
В качестве окислителя можно использовать кислород в присутст-
присутствии катализаторов (солей кобальта и марганца). Реакцию обычно
проводят в жидкой фазе (без растворителя или в растворе уксус-
уксусной кислоты). Для получения днарилкетонов применяют также
обычные неорганические окислители (КМиОь МпО,,, СгО:1). Окис-
Окислены могут быть также арплалкаполы н днарилалкаполы.
2. Гидролиз дигалогеналкиларенов. При хлорировании пли
бромнроваппи алкиларепов получают дигалогеналкпларены, ко-
которые гндролнзуются до карбонильных соединений:
CI
ArCHoR ~2> Лг— С— R -'-Аг — С^ (R? Н, Л!1<, Лг)
I "
CI
Практическое значение имеет получение арепкарбальдегидов (R-=
3. Формилирование и ацилирование аренов. В присутствии А1С1Я
или других кислот Льюиса арены ацнлируются при взаимодей-
взаимодействии с ацилхлоридами (ацилирование по методу Фрпделя —
Крафтса):
¦ О Aid, /,0
Arll-bR—С/; >Лг— С" -4-HCI (R - Л1к, Лг1)
4CI 4R
Механизм этой реакции рассмотрен в гл. VI.А.4.1.
Для введения в арены альдегидной группы (реакция формпли-
ровапия) необходим хлорапгидрид муравьиной кислоты (формпл-
хлорнд) HCOCI. Но это соединение очень нестабильно и разлага-
разлагается на СО и НС1. Форматировать можно смесью СО и НС1 в при-
присутствии А1С13 п CuCl (метод Гаттермапа — Коха, 1897):
()
CO + IICI ;* U—C-
С\
лил /О
ArH+CO-J HC1 iAr — С/ -j- IICI
Q.CI [.[
464
Сильным форматирующим агентом является формнлфторнд
HCOF в присутствии SbF5.
4. Получение аренкарбальдегидов из галогенметиларенов. Арен-
карбальдегпды получают реакцией галогенметиларенов с уротро-
уротропином (реакция Соммле, 1913):
г 1 "'О
ArCHjClJ-CH^N, -> [C,,H1:.\iCU2Ar]CI-—-
О
—>-Аг — Су -; fCH..,\H:f]a--j-.\4-I;1 + ILC- О
В это» реакции сначала образуется уротрошшпевая соль, при
гидролизе которой происходит окпелптелыю-воестаповптелыгое
превращение. Окислителем служит HN- СИ , а восстановителем
АгСН..\'Н...
5. Восстановление хлорангидридов аренкарбоновых кислот. Арен-
карбальдегиды получают из хлорангидридов аренкарбоновых кис-
кислот действием восстановителен:
,0 .... -О
CI II
Для восстановления хлораппирндов применяют водород в при-
присутствии патладпевого катализатора и другие восстановители.
2. ФПЗПЧНСКШ-: СПОПСТНХ II СТРОННПП
Карбонильные соединения арепов являются бесцветными жид-
жидкостями пли кристаллическими веществами с приятным ароматом,
вполне оправдывающим их название - ¦ ароматические альдегиды
и кетопы.
В молекулах карбонильных соединении арепов образовалась
сопряженная система, в результате чего изменилось распределе-
распределение rt-электропиой плотности в цикле. Соединения
имеют сравнительно большие дппотьиьге моменты
(9-10 :|" -1Ы()-:1" Кл-м, или 2,7 -3,.3D).
Карбонп.тьпая группа является электропоакцеп-
торпоп группоп, поэтому л-электроппля плотность
в о- и л-положеппях бензольного цикла умепыпает-
"+ ся. Электропоакцепторпый эффект карбонильной груп-
группы слабее эффекта пптрогруппы (см. a-констапты, Ввел. 7.3).
Максимумы в электронных спектрах поглощения карбонильных
соединений арепов сдвинуты батохромпо. Например, бензальдегид
в растворе гексана поглощает при 244 им (е- 16 000), 280 им (f.^
«1600, центр топкой структуры) и 330 пм (к«20, «^-л*-персход).
В инфракрасных спектрах поглощение карбонильных групп по-
подобно а,р-пеиасы1цеппым карбонильным соединениям.
•Ю5
3. ХИМИЧЕСКИЕ СВОЙСТВА
Реакции карбонильных соединений аренов подобны превращениям
насыщенных карбонильных соединений. Однако необходимо об-
обратить внимание на следующие отличия:
1) вследствие наличия сопряженной системы эффективный
положительный заряд па углеродном атоме карбонильной группы
уменьшен, особенно в случае диарнлкетоиов, поэтому реакцион-
реакционная способность с пуклеофильпымп реагентами уменьшена (не-
(некоторые примеры реакций приведены ниже);
2) арепкарбальдегиды и диарнлкетоны являются нееиолмзнру-
ющимися карбонильными соединениями (пет а-водородпого атома);
3) возможны реакции электрофнлыюго замещения в ареповом
цикле.
Здесь рассмотрены некоторые специфические реакции арсн-
карбальдегидов и реакции электрофилыюго замещения.
1. Бензоиновая конденсация. Эту реакцию при изучении свойств
бензальдегида открыли 10. Либих и Ф. Вёл ер A834). При взаи-
взаимодействии бепзальдегида с. цианидом калия было получено ве-
вещество, названное бензоином. Позже было установлено строение
бензоина как а-гндроксикетопа:
ОН О
/О ксх
2СЪН;-С:/ >c(iII5-C-C-C(iH5
ЧН |
н
бопЗОНН
Такую реакцию дают все аренкарбальдегнды.
В настоящее время известен и главных чертах механизм этоГ: споробраз-
ной реакции. Бензоиновая конденсация принадлежит к реакциям присоеди-
присоединения анионного С-пуклеофила к карбонильной группе. Соответствующий
нуклеофильнын реагент генерируется из аренкарбальдегида при взаимодейст-
взаимодействии с циаппд-поиом:
о-к+ он
.0 ! |
Лг — Су/ + К ' :CNT- +± Лг —С —С— N 7-- Лг —С —С - = N
ХН ¦ -К'
Н
О1Г К;0" ОН
/,0 ! ! i
Лг —С.'7 -1-Лг —С —С-_\-> Лг —С —С—Ar,--i
ХН -К1" II
н см
он о-к+ он о
I ! IP
^* Дг—С—С —Лг ^ Лг —С —С —Лг-f KVCN"
I I I
н сх н
2. Реакции диспропорционирования. Арепкарбальдегиды в при-
присутствии гидроксидов щелочных или щелочно-земельпых металлов
превращаются в соль аренкарбоновой кислоты и арнлметапол
4GG
1
—с—
1
н
(Канниццаро, 1853):
О О
2.\т-С// -• -М'ОН- ->Лг — С* -'-ЛгСН.,ОН
н о-м-
В реакции Капппццаро одна молекула аренкарбальдегида вос-
восстанавливается, а другая окисляется. В этом процессе участвует
гидрокснльный ион. В настоящее время считают, что в реакции
происходит перенос гидрпд-нона от продукта присоединения гид-
роксилыюго нона арилметапдиолята к аренкарбальдегиду:
о-м-'- о-.м -
Q M-OII-
Лг — С* j-.U-OH- : -Лг —С —ОМ
И I
II
о-м- о-.м"
О I ,0 ц,о
Аг —С//- -\-\х— С — О-М- -*Лг — С —11-( Лг— С^ Z-
Н ! : О-.М *
И II
О
^i Лг — СП.,ОН + Лг— С7/ -|-Л\ ОН-
Ч)-.М =
Аналогичную реакцию дают неенолпзируюлшеся альдегиды
алифатического ряда, например формальдегид и альдегиды R^C—
—СНО. Формальдегид как активный альдегид можег играть роль
восстановителя в перекрестной реакции Капшщцаро:
О .0 VOM-
Лг— С/' ¦ М—С' '¦ -->Лг —CILOir-1-IICOO-M---
II II
При оо:!;!.епс]впп па арсикарбальчегпд алкаполягов получаются
сложные Э(!)пры (Л. Кляйзен, 1887). Копцетрацпя алкаполята
составляет только 10 -2О'.'о эквн.мотярпого количества, в реякцпи
происходит не только днепропорцианирование, но и переэтерифп-
кацня:
о - >;,-) '¦
,о I
Лг —С7 -'- \а i--OR ;¦ i \r —С —OR
н
.о ; ¦ ,о
Лг— С ' -;-Лг — С — OR —, Лг— С — П-;-Лг— О? ^±
¦II ' i 40R
,-=t Лг—С;
и
. 0
¦ о—си-.
+ R1
Лг
II
О-Ха +
Для превращения альдегидов в сложные эфиры можно исполь-
использовать алкаполяты алюминия (В. Тпгцсико, 1906). Реакция Ти-
щепко протекает также с алифатическими альдегидами, так как
467
алканоляты алюминия в отличие от алкаиолятов натрия не вызы-
вызывают альдольпого присоединения.
3. Автоокисление. Аренкарбальдегиды очень легко окисляются
кислородом воздуха, па свету. Для алифатических альдегидов это
нехарактерно. Легкое окисление объясняется возникновением
весьма стабильного аронлыюго радикала:
Л) ,
Аг— Су -Лг —С О+',ЛЬ
н
О Лг-СНО .О
Аг —С О —О. --.Лг —С Лг-С -:-Л:--С О
О —О- О -ОН
, о о о
Лг —С- ¦, Лг —С'/ , 2Лг-С •'
ЧО—ОН ' 'И ОН
Поэтому арепкарбальдегиды при длительном храпении должны
быть стабилизированы антпокепдаптамп (гидрохинон, алкплпи-
рокатехипы и др.).
4. Реакции электрофильного замещения в ареновом цикле. Кар-
Карбонильные соединения арепов в реакции электрофнлыюго заме-
замещения вступают труднее, чем арены, так как оттягивающая элект-
электроны карбонильная группа пассивирует бензольный цикл. Реак-
Реакция электрофнлыюго замещения осущестнляется главным образом
в л-положеннн. Действие карбонильной группы подобно действию
нитрогруппы:
Е'Х; , „ «. +нх
Одновременно с реакцией электрофплыюго замещения могут
проходить реакции окисления альдегидной пли ацнльном группы.
4. ВАЖНЕЙШИЕ ШМгДСТАВНТЕЛИ
Бензальдсгид — бесцветная жидкость с приятным запахом горь-
горького миндаля. В воде растворяется мало; т. ил. -26 С, т. кип.
179,5 "С.
Бепзальдегид в свободном виде содержится в некоторых эфир-
эфирных маслах. В промышленности его получают из толуола прямым
окислением или хлорированием до бензальхлорида СйН5СНС12
с. последующим кислотным гидролизом. Бензальдегид использу-
используется как душистое и вкусовое вещество и как исходное для многих
синтезов, например для получения беизонлхлорнда и трмфеннл-
метановых красителей:
о о
I il x;f
С. ,, С г
' CI С!, *' ,, II X=>-R /'' | " //•
.. _ > II | , Краситель
-ИГЛ II Кпслоти Ц
ч;>/ -П.') ч - ¦• /•
N-/ " R
Ацстофенон СПН.-,СОСН:! - бесцветное вещество с т. пл. 20 °С
и приятным запахом черемухи. В промышленности его получают
апеллированием бензола пли окислением этилбензола. Исполь-
Используют в органическом синтезе и парфюмерной промышленности.
Бензофенон С«11.-.COCoIIr, - -бесцветное кристаллическое веще-
вещество с т. пл. 49 X. В промышленности его получают окислением
дпфеиилметапа. Используют в органическом синтезе и в качестве
фотосеиепбилизатора. Фотоеепспбплизатором называется вещество,
способное передавать спою энергию возбужденного состояния дру-
другому веществу п таким образом вызывать фотохимические реакции.
Бензофенон поглощает при 250 им (г>30 000), переход л->л*
я при 345 им (е^120), переход п—>-л". Бепзофеиоп имеет долг о-
ж и в у щ е е (~ 10 с) возбужденное т р п п л е т и о е
состоя и и е Т (см. В вед. 6.7), от которого возбуждение пере-
передается на молекулы другого вещества, вызывай фотохимические
реакции (например, г(»с-ш/мнг-пзомеризацию, дпмерпзацпю алке-
нов в производные циклобутапа). Кроме того, возбужденная моле-
молекула беизофепона вступает в химическую реакцию, например де-
дегидрирование пропаиола-2, и превращается в бспзппнакон:
ОН ОН
,,,, I :
(С,,п,)Х о-нспп)/:н--оп -(c,ji,),c: -C(C,iii;,i-i(Cu,),c о
Для бензофенона характерно образование стабильного анион-
радикала при взаимодепстнпп с натрием. Эти аппон-радпкальные
соли называются кепшлами, опм обычно голубого цвета. 15 присут-
присутствии протоподопорпы.х веществ происходит дпмерпзацмя радп-
каюв н образование бсизпшшкопа:
(СйПг,)..С- О + \а — (С,П.-,),С — С):" \.: '
Д. ПРОИЗВОДНЫЕ КАРБОНИЛЬНОЙ ГРУППЫ
Альдегиды и кетопы легко реагируют с производными аммиака
и образуют новые соединения (азометнны (импны), окспмы, гид-
разопы|, которые имеют специфические химические свойства и
используются в органическом синтезе. Применение находят также
продукты присоединения к карбонильной группе гпдрокспльпых
производных —• ацеталп.
4С9
1. АЗОМЕТИПЫ
При реакции карбонильных соединений с аммиаком или первич-
первичными аминами образуются азометнны RlR-C -MR:1. Названия
азометпнов образуют из названия двухвалентного остатка RM^-C
и суффикса -амин или -анилин, например:
CHrjCII - \—Clb — М-ЭТИЛПДСПМСТНЛЯМПН
С6П5СП -- X — СОНГ, — Х'-бси.чиь'шдсиапилин
Производные алифатических альдегидов п кетонов имеют вы-
высокую реакционную способность. Мешлснамин НоО-ХИ (мета-
пимнн) существует только в разбавленных растворах. Он легко
превращается в уротропин:
4Н..С NH+2ILC О - -C0Hi,N,-; -2Н..0
Этплпдепампн СНЯСН Х!1 тргмормзуется в производное гек-
сагидро- ] ,3,5-трпазпна.
Лренкарбальдегиды с аммиаком образуют бис (арнлидепамлшо)-
метиларенг,!:
,0 Лг. -N- СПЛг
ЗЛгС' Н-2\Н-,-* ")СЧ -I-3H-.0
ХН Н-7 \\:-. СНЛг
Стабильные азометпны образуются из первичных аминов, осо-
особенно арпламнпов п аренкарбальдегидов:
О А' —Лг1
Аг — С'' +Н.Л' — Лг1 —>Лг — С/ +Н..0
Ml ¦ S-H "
Эти соединения называют основаниями Шиффа.
Лзометпны являются слабыми основаниями. В их молекуле
имеется полярная двойная связь и электрофнльпын атом углерода:
н
8Г
При протоппрованип атома азота получаются соли пммоппя.
Азометнны и особенно соли нммоппя присоединяют пуклеофпльные
реагенты подобно карбонильным соединениям. В кислых водных
растворах они присоединяют воду и реакция заканчивается от-
отщеплением амина в виде соли и образованием карбонильного сое-
соединения, происходит гидролиз.
Азометнны и продукты их олпго.меризацни или конденсации
используют для получения гетероциклических соединений (про-
(производных пиридина и .хиполппа).
470
2. ОКСИЛШ
Оксимы легко образуются при реакции карбонильных соединений
с гидрэксиламином.
Названия оксимов образуют из названия карбонильного сое-
соединения и слова оксим, наг.ример:
СН3СН -NOH ацетальдексим
ССН5СН = -NOH Сснзэльдокси.м
я)г С - NOH океим ацетона
Альдегиды и несимметрические кетопы могут образовывать два
стереоизомерпых окспма:
ОП
I
.О ,N—OII , N
R-C7-' -i-Н.Д-ОН -, R—С*' -j-R-C''' + II..О
Н Н II
au!;iu-(nipafK-j-inoM-^p am-(цис-)-изомер
Более стабильна анти-форма. Изомеризация происходит при
нагрепаиии, оспещешш и обработке щелочами.
Оксимы являются слабыми ОН-кислотамп и N- и О-оспопапи-
ями:
,К — ОН Х-О:-.\а- -1-Н-.0
R-C;' - -f\a--OII ;_: R-C
Rl RL
и i-x- , *
H
¦- +/П
"X —ОМ .X—О.
R-C'7 " X--LR-C'' " ы X-
Ri Rl
Окснмы более слабые кислоты, чем фенолы (р/е„«10 —13).
Оспошюсть окснмов сравнима с основностью днарнламннов
(рЛ'нп-~ — 1- ¦ -—2), при этом могут быть два центра нротони-
роваппя — атомы азота и кислорода.
В водных растворах кислот происходит гидролиз окснмов до
карбонильных соединений н гндроксилампна.
В присутствии сильных кислот осуществляется своеобразная
перегруппировка окснмов. При действии концентрированной сер-
серной кислоты (полнфосфорноп кислоты, Р0С1:1) окснмы кетонов
перегруппировываются в амиды карбоновых кислот:
X —ОП Кпстотп .0 .О
R-C-' ¦ R-C;-' -uRi-c.'
Ri \\HRl XIIR
В случае несимметричных кетоксимов (R^R1) могут образо-
образоваться два амида. Оксимы альдегидов (R1 -II) в этой реакции
часто образуют продукты дегидратации — нитрилы r_с — N.
Реакцию перегруппировки впервые наблюдал Э. Бекман A886),
поэтому превращение оксимов в амиды называют перегруппиров-
перегруппировкой Бекмана.
471
Перегруппировка связана с мш рацией углеводородного ос-
остатка R пли R' or атома углерода к атому азота и перемещенном
атома кислорода от азота к углеродному атому. Обычно мигрирует
та группа R пли R1, которая может образовать более стабильный
карбаппон. Но перегруппировка является чисто внутримолеку-
внутримолекулярной, отщепление ионов пе наблюдается.
Предполагают, что кислота вызывает протежирование атома
кислорода и отщепление воды с образованием азении-катиона.
В этом катионе происходит внутримолекулярная миграция угле-
углеводородных группировок.
Перегруппировка Бскмапа может быть изображена следующей
схемой:
Большое значение имеет пространственное строение оксима.
Наиболее часто мигрирует та группа, которая находится в транс-
положеннн по отношению к группе ОН.
Перегруппировка Бекмаиа нашла промышленное применение.
При перегруппировке оксима цпклогексанона образуется канролак-
там — важное исходное для производства синтетического волокна:
N0H н^
3. ГПДРЛЗОНЫ
Гпдпазоны легко образуются при взаимодействии карбонильных
соединений с гидразином и его алкнл- и арилпроизводными. Гпд-
ра.юиы обычно являются бесцветными кристаллическими вещест-
веществами и обладают слабыми основными свойствами.
i\-\H, /N-NHs
R'-C'' '-f IPX- -~ R'-C^ X-
4R 4R
Гидразоиы могут быть использованы для идентификации аль-
альдегидов и кетопов по температурам плавления этих производных.
Больше всего для этой цели применяют замещенные гидразо-
гидразоиы — производные феннлгпдразпна (X — Y=H), 4-нптро-(Х ^N0
472
Y- H) и 2,4-дгпштрофеш[лпир.!3]ш;) (X — V— MUJ.:
Н
!
N--N -¦' ^--Х
R1-C';' ='
*R Y
Для гидразопов характерны также некоторые- важные химиче-
химические превращения.
Прп нагревании гпдразонов A50--200 О в прпсуктппн ще-
щелочи или алкаполяюв выделяется а:ют и образуется углеводород:
II
R1-C' — Ir1 -C--II -| N.
R
Эту реакцию открыли Н. Кпжпер A910) и независимо от пего
Л. Вольф A912). Реакцию расщепления гидразонон до углеводо-
углеводородов пли восстановление карбонильных соединений гидразином
и щелочью называют реакцией Кижнера - Вольфа. Предполагают,
что в этой реакции происходит изомеризация гпдразопа в произ-
производное дпазепа, которое прп нагревании распадается с выделе-
выделением азота:
II
X — NH., К"ЭП ,\ \ \.-.он
R, .с./ —^-R'-C " у
R
.Х- К:- X;i • ,,
Z-~ IV— Сч " +Н..0 -¦* R1— C\;i ' -j-N'_,-|-H,O --*
RNl R S"
H
— R1—C, -:-X,-|-XaOH
I ' -Ы
R
Прп окислении гидразопов оксидом ртути HgO или другими
мягкими окислителями образуются диазоалкапы:
X —XII.. \.
R1—С -|-11^0 - ^Rl — С ' -П.т-;-Ц.,0
-R 4R
4. ДЦСТЛЛП
Ацеталп образуются при взаимодействии карбонильных соеди-
соединении с алканолами или другими гидрокен.шропзводпымп в ири-
ирису 1СГВПН кислого катализатора. .Альдегиды реагируют легче ке-
топол.
Известен также ряд других методов получения ацеталем, па-
прн.\:ер присоединение спиртов к виниловым эфпрам:
OR Ron OR
СИ. С — > СП,—С—OR
Ь1 '
473
При использовании 1,2-дполов или пирокатехина образуются
циклические ацеталн — 1,3-диоксоланы:
О
ур ц + х- R1, / СИ,
Ri—О^ •i-HOCH-.СП..ОН > С ! -i-H.,0
R R' \ ,,СП-2
по, /ч оч .
о
4 //
И- х -
,с
1,3-Диоксолаиы образуются также, при взаимодействии многих
моносахаридов и дпсахаоидов с ацетоном:
сн-он сн он
6
V
Листали являются бесцветными жидкостями или кристалличе-
кристаллическими веществами, некоторые ацетали имеют приятный запах.
В кислой среде соединения гпдролнзутотся, но устойчивы к
воздействию щелочен пли других основных реагентов.
Ацеталн используются в качестве растворителей, например мс-
тилаль СН2(ОСЫ3)^, в качестве душистых нс'цеств в парфюмерной
промышленности, а также как промежуточные продукты в орга-
органическом синтезе.
Образование ацеталеп часто используют для защипы альдегид-
альдегидной или кетопнои группы в различных реакциях, например реак-
реакциях галогепиропанпя, окисления, с участием металлоргапмческнх
соединении. В то же время в виде ацетялеи могут быть защпмшпы
^ос-расположенные гпдроксильные группы, что важно в химии
моносахаридов.
Глава XXVIII
ДИКЛРБОНИЛЬНЫЕ СОЕДИНЕНИЯ
Классификация дпкарбоппльных соединений основана на положе-
положении карбонильных групп:
о О
а) 1,2-, или сс-дмкарбоннльньге соединения R —С —С —R1
о х о
!¦ I 'I
б) 1,3-, или р-днкарбопильные соединения R—С—С—С —R*
У
474
О Н R3 О
i1 ¦ I i;
в) 1,4-, пли 7-Лнкарбопилы1ые соединения R —С—С —С—С —R1
R- Н
г) 1,5-, 1,6- и другие дикарбонильные соединения.
В оспопе номенклатуры дикарбоппльных соединений лежат об-
общие правила номенклатуры карбонильных соединении. Для на-
названий днальдегидов используется суффикс -диаль, дикетонов —
-дион. Р.сли одновременно имеются альдегидная и кетониая группы,
название альдегида образуют с префиксом оксо-. Для некоторых
соединении сохраняются тривиальные и рациональные названия.
Например:
°\ /°
^С ОУ экшдпаль, глноксаль
бугаид[!()П-2,3, дпацстмл
Л [ [ c})f; [ 11Л -jth i 1Л11 о 11, бспгшл
И[ю1:я и т.иа л ь, м;1лоиош.:и альдегид
3-okl(;u_\ ;;;[;ал[], формилацгюн
з — С — СИ-. — С — СП3 пентапдцон-2,4, ацеч планетой
цпклогассандноп-1 ,3
.О
ипдапдиои-иЗ
/с
сп3-
QHr,-
0,
";с
н '
сн3-
-G
0
0
-с-
0
1;
- С —
0
о
С-СПз
о
-С — С..1Г-,
0
1,-С :
¦II
¦СЛЬ —С;
0
0
и
о
°х ^ '°
/С--СНиС1Ь — С'^ бутапдпаль, янтарный альдегид
О О
X д
СП.-1 —(. —СН.СН , —С—СНЭ гг'кспплиоп-2,г1, ацстопгит'цстоп
.0
О
II ¦' ^
475
А. 1,2-ДИКЛРБОНИЛЬНЫЕ СОЕДИНЕНИЯ
1. МЕТОДЫ ПОЛУЧЕНИЯ
1,2-Дикарбоиплы1ые соединении получают различными реакциями
окисления п гидролизом оксимов.
1. Реакциям окисления подвергаются 1,2-диолы, альдегиды п
кетоны со стоящей рядом СН.,-группой, а-гидрокспкарбонильные
соединения, в некоторых случаях также соединении с тройной
связью и дпарилэтапы. В качестве окислителя используют кисло-
кислород в присутствии катализатора, а также SeO2, KMnOj:
Off Of[ О
R-CH-CH-R1 —
ОН О
R —СН—С—Rl —
О
H'Ir-c-C-R^-
R-Cll.-C-R1
2. Гидролиз оксимов. При шгтрозпровании кетонов получаются
моноокспмы 1,2-днкетопов, которые гпдролпзуются до 1,2-длке-
тонов:
О \'()Н О О о
R-CH,-C-R! —1, R-C-C-R1 -—-* R-C-C-R1
П+Х- Н'Х-
2. ФИЗИЧЕСКИЕ СВОЙСТВА II СТРОЕНИЕ
1,2-Дикарбопплы1ые соединения представляют собой жидкости
или кристаллические вещества, окрашенные в желтый цвет, легко-
летучие соединения имеют приятный запах-.
Стоящие рядом (инициальные) карбонильные группы значи-
значительно влияют друг на друга, в результате чего эффективные
заряды на углеродных атомах карбонильных групп больше, чем
в монокарбоинльпых соединениях:
В молекуле 1,2-дпкарбошгльных соединении рядом находятся
две электропоакцепторпые группы, между которыми происходит
взаимодействие на уровне л-электропов (сопряжение). Поэтому
низшая свободная ДЮ распотагается значительно «ниже», чем i»
мопокарбонпльных соединениях. Это отражается также на элект-
электронных спектрах поглощении. Малоиптепсивнын электронный
переход /i-гя* сдвинут в видимую область, соединения окрашены
в желтый цвет. Например, диацетил в растворе гоптапа поглощает
при 4.30- -J50 им (к—20-.ЗО).
476
3. ХПМПЧНСКПП СВОЙСТВА
Для 1,2-дпкарбопклы!ых соединении характерны вес реакции
карбонильной группы, только они протекают энергичнее, чем в
случае мопокарбопнльпых соединений. Реакции с монофункцио-
монофункциональными нуклеофндьпымп реагентами осуществляются ступенчато:
сначала реагирует одна карбонильная группа, потом вторая. Дн-
функцнональпые нуклеофпльпые реагенты при взаимодействии с
1,2-д1!карбоппльпы\н1 соединениями могут образовывать цикли-
циклические соединения или полимеры. Известны также некоторые
специфические реакции перегруппировки.
1. Присоединение электрона к 1,2-дпкарбопнльпым соединениям
происходит под действием сильных электроподопоров (например,
щелочных металлов) или при электролизе (на катоде). Д in этих
соединении характерно образование весьма устойчивого апион-
радпкала, который обнаруживается методом ЭПР. При дальнейшем
восстановлении образуется дпаппоп ¦ - епдполят-ноп, производное
нестабильных епдполов:
о
о
Ас'"'
:i
О
с
о
Hfx~
но
он
I н
R/ \c/
он
2. Воздействие нуклеофильных реагентов. Активные 1,2-дикар-
бонильные соединении, особенно алифатические кетоальдегпды
и глиоксаль, легко реагируют даже со слабыми иуклеофильнымп
реагентами. Например, легко присоединяется вода с образованием
так называемых «гидратов»:
6
чон
Менее реакцпонпоспособны производные аренов.
,'1егко происходят реакции с аммиаком, аминами, гидрокенл-
ампном, гидразином. Например, 1,2-дпкарбопнлы1ые соединения
с аммиаком в присутствии альдегидов образуют производные
гетероциклического соединения — пмндазол;1., а с гпдроксиламп-
но.\: .-.егко образуют дпокспмы:
О KOU
|| 211. N СJ11
С R1— С. .R1
R/ "С- R' ЧС'-
О
НО\
47Г
Пространственное строение дноксимов может быть различным.
Диоксимы образуют стабильные внутренние комплексы (хелаты)
с ионами тяжелых металлов. Наиболее характерными являются
комплексы никеля — малорастворимые соединения, окрашенные
в красный цвет. Эти реакции используют в аналитической химии
(Л. Чугаев).
„ 1^ Как показывает рентгеноструктурный ана-
ЧС_(/ лиз, никелевый комплекс имеет очень своеоб-
•'* N^ \г "¦ разное строение. Атом никеля находится в
.-•'й~ •'•". .-•'• ••"*;, иоле четырех атомов азота. Длины всех че-
Ч; .. •• '•-.,. .-,,.• тырех связей Ni—N одинаковы, одинаковы
/*"••* также длины связей N—О. Атомы кислоро-
С
/ С\ да связаны водородным мостиком. В структур-
**¦ **¦ нон формуле связи Ni---N и Н---0 пока-
показаны пунктиром, чтобы подчеркнуть их делокализацию. В реак-
реакцию с ионом Ni-f фактически вступает моноаниоц диоксима.
При реакции с гидразином 1,2-дикарбонильные соединения
образуют продукты поликонденсацни —¦ полиазины:
- R
R
R
= С — Сг-_
R
При взаимодействии с одпамнпоарепамн 1,2-дякетоны обра-
образуют гетероциклические соединения — хинокса.шны:
.NH.. 0ч Л1- ,N ,N\
1
-: 2H..0
3. Перегрупировка бензила в бензиловую кислоту. В присутствии
щелочи бензил перегруппировывается в дифенилгпдрокснуксусмую
(бензиловую) кислоту A0. Лпбпх и Н. Зимин, 1838—1839)—бен-
зи.гова.ч перегруппировка:
О О ОН ОН
i " NnOII I II ' X ~ I
CUH3- С- C-CJI, "(С„П.-,),С-СОО-\я ! >(С„Н3I!С-СОО11
В этой реакции происходит внутримолекулярная миграция фениль-
hoi'i группы.
Бетиловая перегруппировка характерна для 1,2-дикетонов с
арильны.ми заместителями, в алифатическом ряду она осуществ-
осуществляется редко.
4. ВАЖНЕЙШИЕ ПРКДСТЛ1ШТ1-Л11
Глиоксаль представляет собой жидкость желтого цвета, со свое-
своеобразным запахом, т. кип. 50,4 :С, легко растворяется в воде с
образованием гидрата, в растворах постепенно полимеризуется.
478
Для храпения и использования в синтезах глискеаль превращают
в «бисульфитное соединение» —CoHL, (OH)L> (SOnNaJ.
Получают глпоксаль каталитическим дегидрированием гли-
гликоля или окислением тримера ацетальдегпда (паральдегнда):
г Оч О seo, .О
НОСН2СН2ОН --> Ч,С-С/ *—- CH3-C.f
Глпоксаль используют п органическом синтезе для получения
гетероциклических соединений и красителей.
Диацстил представляет собой жидкость желтого цвета со свое-
своеобразным запахом, т. кип. 88'С. Он содержится в некоторых
природных веществах и продуктах их переработки (эфирные масла,
сливочное масло, сыр) и придает им аромат и вкус.
Диацетил используют в пищевой промышленности. Диоксим
диацетила (димстилглноксим) является важным аналитическим
реагентом для определения никеля.
Бензил — кристаллическое вещество желтого цвета, т. пл.
95 СС. Его получают окислением бензоина:
ОН О 0 0
С„Н3 — СП —ССаПг, —i QH:,C — СС„П3
Бензил используют в органическом синтезе для получения
гетероциклических соединений. Диоксим бензила я:г:я чтся лнялн-
тическим реагентом.
Важны аналоги бензила с двумя а-дпкетонпыми группиров-
группировками, так называемые бие-а-дикетопы, например:
О О О О
/¦'' Ч г г —//; ^ г с '/ ^>
1, 1 -6пс-феЕ1илглпо1а:плои:.бен:юл
Пнс-а-дикетоны при реакции с тетрааминоаренами (например
3,4,3',4' —тетраамннодифепиловым эфиром) образуют высокомо-
высокомолекулярные продукты конденсации — полифенилхинокса.шны. Это
термоустойчивые полимеры, длительное время выдерживающие
нагревание при 300—400 "С.
Б. 1,3-ДИКЛРБОНИЛЬНЫЕ СОЕДИНЕНИЯ
1. МЕТОДЫ ПОЛУЧЕНИЯ
1,3-Дикарбоннльиые соединения обычно получают ацплнроваиием
кетопов. Для ацилирования используют ангидриды карбоповых
кислот в присутствии кислот Льюиса или сложные эфнры карбоно-
вых кислот в присутствии алканолятов или металлического натрия
479
(реакции сложноэфирной конденсации). Схема реакции проста:
О ОНО
.1 о ; i
R —С —C1LR1-- R-- С ••' —>¦ R- С —С —С—R2-; НХ
X |
R1
Реже используют реакции окисления 1,3-диолов или р'-гидрокспке-
тонов.
1. Сложноэфирная конденсация. При взаимодействии кетоиа
л сложного эфира карбоповой кислоты в присутствии алкаполята
натрия пли металлического натрия происходит ацплированне анио-
аниона кетоиа. Наблюдается некоторая аналогия с альдольным присое-
присоединением, однако при сложиоэфпрпон конденсации нуклеофпльный
анион присоединяется к карбонильной группе сложного эфира. По-
После присоединения следует отщепление молекулы спирта:
о
R-C-CIL-R" + R -с<"с н -Ь 'NaOC2H, »=»
5-
-~C5t" ; HOC..H,
?Н ff"N.^ $ <?*'"
— R/C^_OCjHi + H°t-^H, _* „АД, -I- 2НОСД,
R1 R' R'
В результате ацилироваппя кетопа образуется натриевая соль
р-дпкарбонпльпого соединения, представляющая собой сопряжен-
сопряженный апноп с выраипенпымп связями:
? г Г ^ ? н ?
С о- С С с Г
н
Г ^ ? ? f
- С
s v r/ ^-cr r c
A' A' R'
дикарбокильная енольная
форма форма
При подкнелеини реакционной смеси получают Р-дпкарбоппль-
нос соединенно обычно в виде смеси таутомерпых форм — дикарбо-
пилыюн и онольной (|)ормы.
Реакцию сложпоэфнрпой конденсации подробно исследовал
Л. Кляйзеп, поэтому ее часто называют конденсацией Кляйзена. Ре-
Реакцией конденсации является также реакция образования сложных
эфпров Р-кетокарбоиовых кислот при взаимодействии двух молекул
сложного эфира в присутствии алканолята натрия (гл. XXXIV.Д. 1).
Механизм этой реакции исследовали Ф. Арпдт и Б. Эйстерт A936).
480
г.е:м в реакции используют слоимые эфнры муравьиной кислоты
IICOOR, получаются р-оксоальдегпды.
Конденсация К.:япзепа прмголпя г:п;жсдля получения цикличе-
циклических Р-дикетопоп.
2. Ацилироваиис кетскюв п присутствии кислот Льюиса. Днкстс-
пы образуются i:p:i EU'v.Ciicvjnii hi :;ri'.-:n-.i ангидридов i!.:n тлогсн-
rjiii.'pjriOB к.-рбо.топмх кислое г. присутствии кис.гол „"I:;:oi!ca (BF.,,
АК i;):
О О О О О
CM..CGI-. : С! !¦;(:- О - ССП:, l-!V Г.1 i,(T.i! (>¦.![., ' СП;.СОПГ
11р;-диолг1гчстся, что ацетон iv.Tvuk'TU реакцию п еиольпоГ:
С}"ОП\ГО.
2. СЧТМЧКО;!!!; СР.пГК'.Т'' \ И CTPMNVMP
[)-,"Li!кпрСопи.'1Ы!!.!0 соедпи-.'ния — <:'i\:-: ;.>i оссшип iimc жидкости с
приятным ,::,!!П.\ом п.ш бест;^] ко".1 .чрисеалли^'^ск'ло нещесг:'!/!.
(j-AnK.-ipoouii.iMibi,1 av.imieiiiiii пр'.-дс;-- : .:::от coGoii тауто.мерпые си-
системы; как i". iK ;)ci.v'>;;!;.'iOinio-.i isii.;'1, 'i-.:-: :: :; рлстпорлх в piuniotsccj;;¦
ипходячея неси:, n.i.'o T.:yiXi\!CiMn,::-; ф. р\; ¦ дпг^оСом.'Льмая фор\;.:
и ^полымя фор/.л (|..;,1К! п.!!! нссК'л::,i:;). i !|-IIч;:!:;л ; ':; :тг.1српи ;:..:
смогрсчП)! iiii/i<t-. ^ оно.'ил! и пппо.чст :i:o/.\\ I С>,мл, ;.;:; ; :覦:kj^ нрестр.-'п-
С'П5> IliiCC СТрО(':1ПО.
1. Пространственное стрсепио е;;олоз. [|-71п;;г;ф':\;!!.;:;Пыо есс-
о о o--hv^o о R'
! 1 1 I
р
епольпыо ф'..л|).л.!'I --- ((.'/с- и транс-:
о o--hv^o
i ! =^^ 1 I + 1 I
С С" С С С С
R/ ^/L\R. R/ -^c^KRl ек-^с" -он
R1 R1 R1
В /(г/б'-(|юрмо образуется прочная внутримолекулярная водород-
водородная связь. транс-Улюл имеет два поворотных изомера.
Известны соединения, образующие только qwc-енольпую форму.
Они называются г(/г?-фиксированными р-дчкарбопнльпымн соеди-
соединениями, например:
о
2-ацетил1^клогексанон.
Циклические Р-дикетоиы с обеими карбонильными группами в
цикле образуют только одну транс-енольиую форму. Такие E-днке-
16 Д", 517 481
тоны называются транс -фиксированными fl-днкетонамн, например:
цмк-огсксан-
Дпоп-l, i)
Как ци-с-, так и транс-тол представляют собой сопряженную
поляризованную систему, состоящую пз 4л-электро:юв карбониль-
карбонильной группы и С—С-связи и пз неподеленной электронной пары ато-
атома кислорода:
В результате повышается плотность электронов на одном атоме
углерода связи С—С и на атоме кислорода карбонильной группы.
11овышается полярность спязи О—Н. В случае цис-опола образуется
прочная внутримолекулярная водородная связь. тракс-Еиохы мо-
могут образовать только межмолекулярные водородные связи.
2. Пространсткенное строение анионов. При ионизации р-днкар-
бопиль.чого соединения образуется сопряженный аннон, который
может существовать в нескольких пространственных структурах
(копформациях). цис-Аипои стабилизирован ионом металла, кото-
который координируется с атомами кислорода, особенно в концентриро-
концентрированных растворах. Степень координации зависит от типа мета via
(например, уменьшается в ряду Li, Xa, К, Rb, Cs) и природы раст-
растворителя:
6'о-^
! R R I о у+ о' Q\
R' R1 ' R1
стабилн-м»ропаниый мопом преимущественная
м^г^тла умс-аниои к.ч:формапчя
ПИОН HI ^ОСГ «V (Г -
Аппоп р-дикяпГюпИьТьиого соединения независимо от i:oiic|',op:.;a-
цпн является ссиряжеипой бл-электропнон системой с делокалпзо-
саппым отрицательны:,; зарядом. Распределение плотности электро-
электронов н шпионе прпГ.лпзптечы.'О можно охарактеризовать расчетами
по \чтсау МОХ (рис. 98).
11'ютностп я-электронов с;г свидетельствуют, что ил среднем атоме
углерода плотность электронов значительно увеличена, так что этот
482
.-С'-
.С.
-Р
2Р
0,8 2 О.Я2.
5-1 -X^U >Q-. 1.5
1,51
54
?.= <*_ 1 f
_LI_ /,'j = я + 0,5'Л-Д
0.041 О-''08 0 041
ол% rf~l' f5
2-512,4
Pr/C. 5.5. ЗпЯЧСНИЯ ЭПОрПШ МО, ОДИО'.'ЛОКТрОППЫС ПЛОТНОСТИ
ВЗМО, плотность я-элсктропов шранс-аиття fi-дпкетопа
атом может быть центром электрофпльноп атаки. Г.ще более ярко это
подтверждают одноэчектроппые плотности в высшей занятом моле-
молекулярной орбнтали (lF, на рис. 98).
3. ХИМИЧЕСКИЕ СВОЙСТВА
р-Днкарбопнльные соединения являются высокореакцнонноспособ-
ными веществами. Характерны реакции как с пуклосфпльнымн,
так п с электрофпльнымп реагентами. Особое место занимают кис-
кислотность, енолпзацпн н обрлзоипппе солей (внутренних комплексов)
этих соединений. Менее характерны реакции со свободнорадпкаль-
ным :> ехапнзмом.
1. Кислотность и енолггзацня. р-Дикарбопнльные соединения
представляют собой кислоты. Константы кислотности в зависимо-
зависимости от строения соединения и растворителя меняются в широком
интервале. Например, в водных растворах p/(,..«s4. . .10.
В растворах обычно существует равновесие таутомерных форм:
:Sol-
R'
СН-кислот;
/Y\
^\
Дикарбонильная форма является СП-кислотой, а епольпая форма —
ОН-кислотой, и каждая имел' свою константу поппз,'.:ции: /\'к и
Ке соответственно (гл. XV.А.4).
Константу кислотности таутомерией смеси /<"к.г можно опреде-
определить экспериментально. Чтобы оценить Кк и /(,., псосходп.мо знать
соотношение таутомерпых форм, т. е. константу чаутомернсто рав-
равновесия /Ст (/Ст —1ШДЮ, где IE] и [К,] — концентрации таутомер-
16*
483
n:.ix форм):
Л'- --
Л ¦
Так, например, для анстплацстоп;>. в водном растворе /\т 0.2
((.коло 16,5% епола), р/(к 8.9, а р/С,. 8.2.
В таутомерией смеси всегда преобладает (|юрма, имеющая мень-
меньше ю кислотность. В случае ацетплацетопа это днкетоформа.
При изменении растворителя меняются также константы кислот-
кислотности таутомерпых форм. Константы кислотности СЫ-кислот и ОН-
kik'iot меняются в разно» синен» (различные сольватационпие
эффекты), поэтому при переходе от одного растворителя к другому
!^.епястея соотношение таутомерныч форм. Например, в водном раст-
растворе для ицетплацетона Кг 0.2. и в рг.створе метанола 1\т -2.6
G2''и снола).
На кислотность н еполпзацию сильно влияет строение соедине-
соединения. Кислотность больше у циклических /л/ганс-фиксировешпых
р-дпкетопов. Шестнчлепные циклические соединения обычно больше
енолн:!прованы, чем пятичлешгые, например цнклогексапдпоиы-1,3
по cpriiiHOHino с ипдапдионами-1,3:
н.о .=••
ом
ОН
-г Н2О
2. Образование солей и комплексов. рЧДпкарбошгльные соедине-
соединения в цнс-енольпои форме образуют соли, в которых нон металла
может быть координирован с обоими атомами кислорода. Особенно
характерны соли тяжелых металлов, которые в растворах практиче-
практически не диссоциируют. Ион металла связан во внутреннем комплексе.
Внутрикомплекеные соединения часто называют хелаталш. Хелаты
в поде не растворяются, по растворимы в органических растворите-
растворителях (хлороформе, бензоле).
Как свидетельствует рептгспоструктурпыи анализ, в хелатах
ато.м металла расположен симметрично по отношению к атомам кис-
484
лорода. Возможны два способа изображения структуры хелата:
v. С s ¦ R (J ^ "¦
°
Ч? ' о" -о
| R "Г
н н
В первой структуре показано обработанпе двух внутримолеку-
внутримолекулярных допорпо-акцеториых свилей {',:->Си. Вторая структура
изображает молекулу с выравненными спящий.
3. Взаимодействие с электрофильными реггента>;и. Реагирует
енольпая (|)орма пли анион (поимся пара), иногда хелаты металлог.
Реакции Р-дпкарбопильпых coe.'iiniennii с п; ктро(|щлы!ымм реаген-
реагентами Тг':к же многочисленны, как реакции ф(полой. Важны реакнп::
алкплпровапня, аиплнрог.апп'л, копдепсамнп с а.пьлегпдамн, шпро-
зирования, нитрования, а^осочетят^л, сул:,'[1пров;п!ня, галегсчп:-
роваппя.
Как сиол, гак и анион являются ."мби/и-нтпымп частицами, т. е.
имеют несколько реакционных центром. Аистом тгакм электрофплг-
еюго реагента могут быть кислородные акг.лл или углеродным атсм
между карбоппльпымн группами (так на.'^.и-аемая ак!!:гл1ая мети.те-
мовая группа).
В з а и м оде и с т вне с С-э л е к т р о ф и л а м и вклю-
включает реакции алкилироваппя, ацплмрснакия, коидс-исаиин с ал!3дс-
гндамн и кстопамп. В эти реакции обычно гстумает аппон (i-лпкар-
бонпльного согднпсимя:
В общем случае получается смесь продуктов С- и О-алкплпрогмппя
или С- и О-ацнлировапня. В случае рЧчпкарбонпльпых соединении,
образующих 1{ш>енольпую форму, преимущественно образуются С-
485
продукты, так как атомы кислорода значительно блокированы попом
металла (ср. гл. XXXIV.Д.2).
В случае л'г/.жс-фнкепроваппых р-дпкетоноа направленно реак-
реакций алкплнроваиня и ацплпрованпя в значительной степени с-.улш-
сит от строения соединения, что определяет распределение электрон-
электронной плотности в анноне, пот типа растворителя и концентрации реа-
гептгз.
В реакции с альдегидами обычно образуются бис-продукты:
R R
+ Х-- R1 ¦>- H-C-R
1
R
4 ОН Н
1
н
Реакции такого типа используются для аналитического определения
альдегидов и для получения производных пиридина XXXVIII.Л.2.
Взаимодействие с различными эле.ктрофнльпымп реагентами (пит-
розпровапие, нитрование, азоеочетапне, сульфпропаппе, галогепи-
рованпе) может быть изображено одной общей с.челюп:
L * Ж X I
- с. л с —-> /С , с
о/у •¦с/ --оп -tn о- чс:- -он
II F
(Е- NO, NCX, Лг\„ SO..H, С1, Вг, I).
4. Взаимодействие с нуклеоф:>л:>пму,и реагентами. р-Днкарбо-
пильпые соединения при взанмодеНстнпп с сильными пуклеофиль-
пыми н основными реагентами образуют аппои. Во многих случаях
аннон находится в равновесии с педпесоцинрованными формами
(еполом, дикарбопилыюп формой). Поэтому часто становятся воз-
возможными дальнейшие реакции, в которых центром атаки пуклео-
филыюго реагента становится углеродный атом карбонильной груп-
группы. Слабонуклеофильпыс и слабоосновпые реагенты (вода, спирты,
тиолы) взаимодействуют с р-дпкарбопильпымп соединениями только
в присутствии кислого катализатора.
Важными являются реакции с Х-пуклеофпламп: аммиаком, ами-
аминами, гидроксиламнном, гидразином и его производными — и с
О-нуклеофиламп: получение ацеталей, гидролитическое расщепле-
расщепление.
а) В з а и м о д е й с т в и е с К - и у к л е о ф и л а м и вклю-
включает реакции с аммиаком и его производными R\H2. В этой реакции
486
образуются епамшюны (р>-амш:ор.пнил карбонильные соединения):
О' ХО 1 |i.t .. О-' \\К
II I Г.:., ,Д'.. , , cl --R'-NI'i —> .| |_ -; П.,О
R/ 4-C;'^4R II7' II ' v/ XG^ N7
Это пример для ({г.-с-еполобразующего р-дпклрбонплыюго соедине-
соединения. Здесь в епамниопе сбр;|?.уекя прочили Diiyip:..молекулярная
водородная связь. A\'ti.';-.j;o:i формой, ptan;pyioi;;efi с R'NH2, ио-в;;-
дпмому, являс-тся дпкарбопиль.пая фор.\!а. В приведенной схеме
не указаны все стадии это;"; реакции.
В случае реакции с пмге.-чепламнпом, гцдраапнйчн, аммдппгл:;!
образуются гетсроинк. iiiT:oi к::о. соедьпепья (п'Ю1''сг::о.ты, нпра ;ол1::,
пиримнднпы). Волее подробно эти реакции онисашл в гетероцикли-
гетероциклических соединениях.
б) В з а и м о д е и с т в и е с О - и у к л е о ф и л а м и г-клю-
чает реакции гидролитического расщепления и образован!:;; ацета-
лей, в отдельных случаях - -- образование епольиы.-; эфпров.
В водных растворах п присутствии щелочи или кислоты и при
нагревании р-дикарбонпльные соединения расщепляются по свяли
С—С н образуют кетой и карбоновую кислоту. Псч-пдпмо.му, ломи-
ломимо образования аниона происходит присоединение гпдроксн;.ыюп
группы к карбонильной, а затем следует разрыв связи С—С, чему
способствуют индуктивный эффект п з:[ч}:ект сопряжения располо-
расположенном рядом карбонильной группы:
И Н
IA и
H.O |
--.' \ /"
О ОН О ; v О О
II I Ц-' ¦ ! I \
и м и и p
I
—^K — c: CII, i- ;Ja' ,j;.v—R
Предполагается, что происходит расщепление связи С- С. а
циклическом комплексе п образуется кегои в епольпом форме. Сле-
Следует обратить внимание па то, что эта реакция является обратной
реакции получению р-дикарбопплыюго соединения (ретрояльдоль-
пый распад).
5. Гоглолитяческие реакции. Р-Дпкарбонильныо соединения спо-
способны образог.ывать в некоторой степени стабилизированные в ре-
4S7
зультате делокалнзацнн неспаренпого электрона свободные ради-
радикал hi.
Такие радикалы возникают при взаимодействии р-днкарбониль-
ных соединений с активными свободными радикалами или при окис-
леиин анионов.
Свободные радикалы Р-днкарбоннльных соединений немедленно
вступают в дальнейшие реакции, например присоединяются к алке-
п;;\:, алкадненам, алкнпам или димернзуются:
R R
I [
\
R1
О О
/ / \_R
-C^ / ^c R
О О
Особое место занимают 2-арплипдаидпоны-1,3, которые обра-
образуют наиболее стабильные в этом ряду соединений свободные ра-
радикалы.
4 [SA/КНЕЙШИС ПРЕДСТАВИТЕЛИ
Лцспшлацетон представляет собой бесцветную жидкость с приятным
запахом, умеренно растворяется в воде (до 15%). Ацетил ацетон в не-
неразбавленном виде является смесью таутомерных форм с содержани-
содержанием около 20% енолыюй формы.
В промышленности ацетилацетон получают ацилированпем аце-
ацетона ацетилхлоридом пли ацетапгидридом в присутствии BFr3. Его
используют в аналитической химии для определения некоторых
ионов металлов и как исходное в органическом синтезе, особенно для
получения гетероциклических соединений. Хелаты ацетилацетона
с Fe, Co, Ni, Сг и другими металлами используют в качестве катали-
катализаторов различных реакций.
Циклпгександионы-1,3 - ¦¦ бесцветные кристаллические вещества,
умеренно растворимые в воде.
Цпклогександиоп-1,3 (дигидрорезорцин)— вещество с т. пл.
ldo 106 С — получают каталитическим гидрированием резорцина:
[[О ^-'' -ОН СУ- ч^ "ЧОН Оу/ N- Ч-Ю
Его применяют в органическом синтезе.
5,5-Диметнл1Шклогександио11-1,3 (димедоп) — вещество с т. ил.
147 "С — получают из окиси мезитила и малонового эфира в при-
присутствии этоксида натрия. В этом реакции происходит присоеди-
присоединение аннона малонового эфира к двойной связи ненасыщенного
кетопа и затем следует внутримолекулярная сложпоэфирная кон-
4SS
денсация с ооразоваппем системы
Дпмедон используется в аналитической химии для мд'/нтпфпкл-
дни а льде г; гу; и, так к;::с альдегиды с днмедоном дают про;.у и::.!
копдеисяШш — кристаллические вещества с четкими точками ил<;В-
леиия:
- R - С
О
4I1
¦Р
¦он и по
Некоторые производимо. диклогексапдмопоп-1,3 (оксимы 2-гщнл-
пропзводны.ч, 2-арплнрои.->подпыс) обладают биологической ;\кт1Л1-
постью и рекомендуются для борьбы с сорняками и предмими n.av-
komiiImii в сельском хозяйстве.
ИндандионА,3 представляет собой бесцветное пли желпжлтие
кристаллическое вещество с т. ил, 129—130 сС, малорастворк.мск- в
воде. В кристаллическом состоянии существует в днкетоформл По-
Получают ипдамдмом-1,3 гидролизом и декарбокснлировакием 2 алко-
ксп карбон ил пи дан.!,; юна-1,3:
О
о
•с/
i;
о
COO
, 11-х- |
О
СН,
Натриевая с,':.:ь 2 -¦., i о i-;r t! к;:; i По 111 г л in i дл 11 д i ioi i я. -1.3 получаемся п
с.южпоэфнрпо!! i:o;!,4C4ic,:!:;;ii in диэтплопого jjinp-i <|>г..дег:он кпели-
ты и этпл;-1цоТ(!1а в п|)псу;сп5пп мет;],;;ч1чес[\(;:ч) п.при.::
о
i;
с
>^
. [ 1
. У
"\
ОС 11
2t H, ОН
Ипдапдноп является bi.icoK(!peai:;;!.oiM!(;ciioiоПмым coc.jinciiiR.!.
Дли его аппома wip.-iKii р:ч: выестчая С-in, кле:л|м| и.иссги.
Очень легко происходит самоконденсация индапдиона с образо-
образованием биндона:
Бипдон является специфическим реагентом па первичные ами-
амины (при нагревании реакционно."! смеси появляется cmjicj окраши-
окрашивание).
Индандпоп легко нитруется п образует сильную кислоту 2-нит-
ронпдзпдноп-! ,3 (рКA~—2):
он
™ NO;
С аминами он образует малораетворимые соли.
Ряд производных ипданднопа имеет практическое значение.
При окислении некоторых производных пидапдиопа получают
2,2-Д1;п1дрокс11Ш1дандноп-1,3 — ннигидрин.
Это соединение при реакции с и-амннокпелота-
мп дает синее окрглшпг'.ппе, поэтому нппгпдрил
широко применяется для определения сс-амппокис-
лот (гл. XXXIV.В.2).
2-Арплнндандпопы-1,3 и их производные при-
применяются в медицине как аптикоагуляпти крови,
например фе.нилин B-фепнлнпдап;шоп-1,3) и омс-
фин B-феппл-2-гпдрокеиметплп11дапдноп-1,3). Особенно сильные
аптикоагулирующпе свойства имеют некоторые 2-ацилкпдандио-
иы-1,3, например 2-дифсш1лацетилипдапдпоп-1,3. Эти соединения
применяют для борьбы с вредными грызунами (родептмциды).
На основе производных инданднопа синтезированы лекарствен-
лекарственные средства, действующие на центральную нервную систему, име-
имеющие аптиаллергпческое и противовоспалительное действие.
Большая заслуга в рпзенпш химии ипдапдпопа принадлежит
Г. Я. Ванагу.
Е. 1,4-ДИКАРГ>О:п:ЛЬ!!ЫЕ СОЕДИНЕНИЯ
'. Методы получения. Синтез 1,4-дпкзрГ:о::нлы1ых соод1.ч1С!;пн до-
довольно сложен. В принципе их можно получить окислением 1,4-
дмолов
RCilC.'LC.'.'XIIR f-.l RCCIIoCIIXR
I
ОН
490
ОН
|
6
'i
6
но и сами 1,4-дполы малодоступны. Псяому рг.лработапо много спе-
специальных методов, применяемых для отдельных соединении. Ii.'i-
пример, самое простое 1 ,-!-днкарбоп1:л:>ноо соединенно, янтарный
альдегид, может быть получено n:i дпхлорапгпдрнда янтарной кис-
кислоты:
,0
NC1
II., I'd
о о
4 ее п.. с п.. с;
и7 " " чн
а/
1,4-Дмкетопы получают димерилацчем р-дпкарбонпльпых соеди-
соединений пли алкг-глпрованнем солен р-дпклрболн.т.ч.чил соединении
а-галогепкетопам!!.
2. Физические и химические свойства. Важнейшие представи-
представители. 1L-Дцкарбопплы1ые соединения обычно предстазляют собой
бесцветные ароматные жидкости. Соединения в небольшой степени
енолнзировапы, возможно образование диопола:
RCCH2CH,CR 7- RC- CHCH.CRr* RC СП-СН- CR
II ': '¦ ; i
о о он о он он
Для 1,4-д1п:арбоиплы1их соединении характерны все реакции
карбонильных соодппенпй. При г.злнмодеиствпн с пуклеофпл1)пыл:и
реагентами образуются гетероциклические соединения:
11..V 1
и.
RCCH Cil.CR
li II
О О
С- С '
I
х сч
I IR
ОПОИ
R —
Y-- Nil, S
','- R
I! N'-M
Глава XXIX
КАРБОН ИоПЬН Ы Е СОЕДИНЕН И Я,
СОДЕРЖАЩИЕ ДРУГИЕ ФУНКЦИОНАЛЬНЫЕ ГРУППЫ
В зависимости от введенной функции карбонильные соединения под-
подразделяются ни:
а) галогенкарбопнльные соединения,
б) мопогидрокси- и дигндрокенкарбонильные соединения,
в) полигндрокенкарбоннльпые соединения (углеводы),
г) сульфокнелоты карбонильных соединении,
491
д) пптрозо- и нптрокарбонпльные соединения,
с) аминокарбоппльпые соединения,
ж) дназокарбоппльиые соединения.
В каждой пз этих групп существует еще более подробное деление:
в Зависимости от числа функциональных групп, их расположения
относительно карбонильной группы («-, f>-, у-, о-) п от типа углево-
углеводорода.
На ;!;;и;кя этих соединений образуют, используя номенклатуру
карбонильных соединений, присутствие других функциональных
групп обозначают исходя из общих правил. Для некоторых соедине-
соединении сохраняются исторические названия. Например:
О
О
en,o.;i: Fir С! .с—с
С
о
и
с
П
¦
II-
'"'¦¦'
О
\ci
¦•'Гг.'
1
I
I
.г-л
с;
I
о
и
три
¦' -I; ,'!
0 >
(,
.-Aр
СП
1
([\[
¦'¦¦¦(
," vi:
yi.i-w-ii
-СП
О!
О
1
с.
4 \ и
с.
0
— с -
I
II -'-¦¦
1 1
¦)!¦-
¦'
пц(- >
CI
":;:ч* ч- п-
0
i
с
¦ 1
н
МП
о
CCI1 V,
ПГ l'."TO:llC
l(i!T
л 1ле;!0ды имеют специальную номенклатуру.
А. ГАЛОГЕНКАРБОНИЛЬНЫЕ СОЕДИНЕНИЯ
В ряду алифатических соединении и алкпларепов наиболее нзучен-
Ип/мп являются A-галоге.пкарбо11плы1ые соединения. Другие гало-
пь'карбонПьТьпые соединения здесь не рассматриваются.
1. Методы получения. Для получения а-гало! епкарбопнль-
ных ctxvuineiiini г.ча.впым образом используют прямое галогепнрова-
пие карбонильных соединений, реже — окисление галогепалкано-
лов:
X X
О х I О ч. | ,0
КСП,-СУ' -4R-C —С; -! R-C-C-' (Х- CI, Вг)
R1 I Rl i ' R1
II X
Реакция га.тогенироваиия обычно проходит через енольпую форму
(гл. XXVII.А.З).
492
2. Физические свойства и строение. а-Галогенкарбонильные сое-
соединения являются бесцветными жидкостями или кристаллическими
веществами с острым запахом, нары их сильно раздражают слизис-
слизистую оболочку глаза, вызывая слезотечение (лакрнматоры). Если у
одного углеродного атома находятся два или три атома галогена,
лакрнматорпые свойства исчезают.
Молекула и-галогенкарбонилыюго соединения содержит две
полярные группировки —связи С~"О и С—X, где X --галоген.
Взаимное влияние этих групп увеличивает элекфофнлыюсть угле-
углеродного атома как карбонильной группы, так и связи С—X. Увели-
Увеличивается также полярность близлежащих связей С—Н.
3. Химические свойства. Для и-галогецкарбоннльных соединений
характерны реакции как галогеналканов, так и карбонильных сое-
соединений. Наблюдаются также некоторые специфические реакции,
особенно для полигалогепкарбоппльпых соединений.
Легко проходят реакции нуклеофнлыгаго замещения агама гало-
галогена в а-мопогалогенкарбоппльных соединениях. Так можно полу-
получить а-гндрокснкарбонильные соединения, и-аминокарпонпльпые
соединения и др.:
[[ н
i .О i
R- С С
О
R1
П
i_-R- С- С
.О
R1
U '¦ X -
1 :\иН -- R- C--C
I
X Х- -'ХпН :\n
(:\uH-:OH2, :\HR; и др.).
Однако при действии сильных оснований (NaOII, MaOR и др.)
происходит своеобразная реакция перегруппировки:
о ,0
Х-СИ.,- С -Р 2\\it-0II- -*R- СП-.- С/ -: Na + X--: ц.,О
R хО-\а*
При этом из галогепкетонов получаются соли карболовых кис-
кислот. Впервые такую реакцию наблюдал А. Н. Фаворский A895),
поэтому она называется перегруппировкой Фаворского.
Механнтм перегруппировки Фаворского окончательно не выяснен. О'-
суждпются мехяшимы черел ацмлкярГены и через цнклопропаноны. Эго мож-
можно покачать на примере 2-хлорцпклогонсапоня:
Na ОН
Н,0
N- С!
N- C1
О-
соок
оон
4ЭЗ
Карбонильная групп? даст обычные реакции, если им не мешают
реакции но связи С—галоген. В полпгалогепкйрбоппльпых соедине-
соединениях карбонильная группа обладает большой злектрофнлыюстыо и
способна присоединять воду с образованием устойчивых гидратов:
х
Г*-, <5- II х »Н
I R H ' I
В ;цолсчпоп среде легко происходит гидролитическое расщепле-
расщепление связи С—С п образуются трнгалогепметаиь; (галоформы):
он ^о
I f - о, I
I I/. ,
ОН О На
XjC: ¦
0 Ыа ,.
\
С R
+
•+
Na
OOCR
Механизм этого «галоформного»расщепления, по-видимому, подобен
расщеплению Р-дпкарбопильпых соединений.
4. Важнейшие представител::. Хлораль (С1;,СС110) — бее; ротная
жидкость с острым запахом, т. кип. 97,7 "С. Его получают хлориро-
хлорированием этилового спирта:
п ^ п '
ГЦ ГИ ПТ-Т 2 ПТ С-'' П Г Гх/
Хлораль очень легко присоединяет воду, образуя хлорильгидрат
Cl.iCCH(OH).. — бесцветные кристаллы со своеобразным запахом,
т. ил. 51,7 "С. При нагревании выше 100 СС диссоциирует на хло-
хлораль и воду.
Хлоральгидрат является промежуточным продуктом в произ-
производстве хлороформа:
С13ССН(ОНJ-;-М :ОН- -^ С1;,СН-
Хлоральгидрат используется также для получения биологически
активных соединений.
Бромацетофенон (CGHftCOClLBr) -—бесцветное кристаллическое
вещество со слабым запахом, т. ил. 51 "С, являющееся сильным
лакриматором. Его получают бромпрованием ацетофепона.
В органическом синтезе бромапетофенон попользуют для введе-
введения в органические соединения группы С011.-.COCI Ь (фенацнлыюй
группы).
494
Б. МОНОГМДРОКСИ КАРБОН ИЛЬНЫЕ
И ДИП1ДРОКСИКАРБОНИЛЬНЬ:Е СОЕДИНЕНИЯ
В этой группе рассмотрены следующие соединения:
он о
а) сс-гпдрокспкарбонильпыс r_c—С —R1
г
II
он r R1 1 о
II ¦
П) Р-, у-, б-гадрокснкарПо- R-C.-i -С— i -C-R" (n = l,2,3)
пильные | ! i j
ОМОН
в) дигидроксикарбонильпые R — с—С- С/^
I I Nr
я п
ОНО ОН
R-C- Г- C-Ri
II
H
г) фсполкарбоннльпые ¦'
OH OH
1. а-ГПДРОКСПКАРБОНИЛЬНЫЕ СОЕДИНЕНИЯ
1. Методы получения. Общим методом получения п'дроксякарбо-
нильпых соединешп"! является гидролиз ы-галогеикириоппльпых
соедппеннн:
Вг О ОНО
R_C-C—R1 ; ТГ.О —> R—С С -R1-; ПВг
IF II
а-Гпдроксикарбопильпые соединения образуются из нееполпзп-
ругощпхея альдегидов в реакции тина бензоиновой конденсации (гл.
XXVII.Г.З):
ОН О
/О Г\яС\' | Г
> R-C-C-R
2R— С.
II
Сложные эфнры карбоновых кислот в присутствии метг.тлнчес-
кого натрия своеобразно дпмернзуютен п образуют а-гидрокспхар-
боннльные соединения. Реакция называется ацилоиноюй конденса-
495
цией, так как а-гпдрскснкетоны иногда называют ацилопнамп:
011 О
2R--C- —¦- R-C-C-R
ОС,П5 : |
Н II
Таким путем можно получить также циклические aim ".сипы
(«--1. 2, 3. ! .):
CHCOOCHj
Na
/
(СН,)„ э (СН,)Л
CI 1..С00Г Н -, CHi ^* О
Механизм ацилопновоп конденсации р::ом<лреи в гл. XXXIII.В.3.
2. Физические свойства, пространственная изомерия. а-Гпдро-
кспкарбопильные соединения—бесцветные или слегка жслто:и1-
тые жидкости с приятным запахом или крпсталлпчс-.кие вещества
(бензоины).
Молекулы гпдрокспкарбюпнльпых соедниенн!1! i::o:ot ,'!С1!\".:ет-
рическнГг атом углерода (кроме R II), их молекург лчрлм.чы.
Самая простая хиральпая молекула в этом ряду соединении — моле-
молекула и-гидроксипропионового альдегида:
()П
I О
II
Существует два энаитномера (зеркальные или оптические пяоме-
ры), которые можно изобразить обмчш;1.1.г:[ аруктурпымп (ьир.му !,:мп
или проекционными формулами Э. Фишера:
Н О О.ч Н
Н,
i
он
R - л -гидроксипро -
пионовый альдегид
S - i -гидроксипро -
пионовый альдегид
Абсолютную конфигурацию энантномеров обозначают при помо-
помощи /?,5-номенклатуры (гл. VII.3). Заместители при асимметричес-
асимметрическом атоме углерода а-гидроксипропионового альдегида в порядке
496
убывающего старшинства располагаются в следующий ряд:
ОН, С^ ^, CU-j, U. При рассмотрении молекулы в направлении связи
С—Н расположение заместителей ОН, СНО, CI].. соответствует на-
направлению движения часовой стрелки для энантпомера, изображен-
изображенного слепа,— это ^-конфигурация. Втором эпашпомер соответствует
S-копфпгурации:
НО II ПГ)
п с
На проекционных форму, iny Фишера /?-ксч.ф;п \ р.мшя соответ-
соответствует распило/Кению ОН-группы спрана, a .S-Koi!(]):irypani!!i —
с:юва.
3. Химические свойства. а-Гн.^р-^спг.арП.л'и.ть:; ле со^'И'.пепня
обладают свойствами, харякторпымп д.т:! ciiiipion н клрбопчтьных
соедмпепш"!. ОП-КислимюсП) этих i4K'-.'.iii!C!i!:ii вмшо, чом обичг:г;1\"
спиртов, вследстпне мидуктивного влпямчя клрбомптьно;! группы.
В присутствии сп.тьмых осиовппш'г iiw.i;.io>iiiio образование ди-
аппрпа ла счет иеппзаимп снязп С II:
он о
I II ,
R С С R
Na О
О
Na ОН ^:=.^r R-
¦R' Н-
Н
Na'f п
I
Na+ ОН"
Na
R-
О О \'а
I; i!
- С i4-!-' С R
Образованием такого диапнона (епдполпт-поиа) объясняется лег-
легкое окисление о-гндрокепкарбонпльпых соеднпемш"! в щелочпоГг сре-
среде. Например, очень легко идет реакция Фелппга (гл. XXVII.
А.З.д), где окислителем выступает поп меди:
он а
. 1 II
R с —С-
!
к
-R.' ^11
ом
О "-
\
R — C — С R + 2Си
497
Конечным продуктом окисления является а-днкарбоннлыюе сое-
соединение. Другими окислителями можно уловить промежуточный
продукт окисления — аппон-раднкал.
При подкнслепни силыющелочпых растворов н-гпдрокспкар-
боннльиых соединении образуется ендиол, который превр:;;:и.слся
в смесь двух изомерных а-гндрокенкарбопнльных соединений (если
R)
о о
-,- • II II ,
он он он о о он
II. I II , II I 1
С=С J'^i! С С R 4 R С С R
I I
н к
Пндпол и а-гидроксикарбоннлыюе соединение являются двумя
таутомерпымн формами, так же как еиол и карбонильное соедине-
соединение. Поэтому возможна изомеризация одного а-гндрокснкзрбо-
ннлыюго соединения в другое, особенно в присутствии оснований.
Эти превращения важны в химии углеводов.
2. Р-, у-, й-ГИДРОКСИ КАРБОНИЛЬНЫЕ СОЕДИНЕНИЯ
1. Методы получения. Р-Гпдрокснкарбопнльпые соединения обыч-
обычно получают методом альдолыюго присоединения, у- и б-Гпдрокси-
карбонпльпые соединения могут быть получены селективным окис-
окислением диолов или другими методами.
ОН г Ri -, ОН О г R1 - ОН
| I I I I го, : i
R— С— j —С— | — С— R3'—iR— С— —С— —С —R3
II! i I !
II L R* JB II L R* .;„ II
2. Физические и химические свойства. f5-, y-t о-Гндрокснкярбо-
пнльпые соединения обычно представляют собой бесцветные
жидкости с приятным запахом. Обладают химическими свойствами
как алканолов, так и карбонильных соединении. Одновременное
присутствие в молекуле гидрокенлыюн п карбонильной групп при-
придает им некоторые специфические свойства.
Р-Гидроксикарбоипльные соединения легко отщепляют воду и
образуют а, р-непасыщенпые карбонильные соединения (гл. XXVII.
Б.1).
у- и б-Гпдрокспкарбопнльпыс соединения легко образуют продук-
продукты внутримолекулярного взаимодействия гидрокенлыюи группы с
карбонильной — циклические полуацеталп. Эти соединения могут
существовать в форме с открытой цепью и в циклической полуане-
тальной форме. Такое явление называется кольчато-цепной изомери-
изомерией. В отдельных случаях наблюдается равновесие .между цикличе-
циклическими и открытыми формами.
498
Циклические полуацетали в присутствии спиртов в кислой среде
легко образуют ацетглн:
сн2
с»,
:о
011
X
OR/
у-Гндрокспклрбопнлыпле соединения образуют производные тет-
рагидрофураиа. Мри образовании цикла наблюдается существенная
особенность: в молекуле появляется асимметрический атом углеро-
углерода, молекулы 2-:'.'1К!!.'1-2-п:дроксп(алкокси)тстрагпдрС(|)урапа явля-
являются хнралышмп и существуют в виде двух эпаптномеров (зеркаль-
(зеркальных, млн оптических, изомеров):
r'o ,,
Молекулы производных TCTp.".nu;)o:j;ypaiu иг:пнсопаиы с плос-
плоским циклом. Следует п;шо:.п;иг,>, что это не вполне точное- изобр.-жс-
пне, так как пятпчлеппые П?;сыи;е1:пые циклы печного изогнуты
(гл. V.B.2).
б-Гндро1;спк;",рбот!.".Ы1ые соедшюнпл oopaiyioi цикл тетрагпдро-
ппряпа, точнее пропзЕодные 2-Г1;дроксг1тетра!'пдрсшф;ша п 2-алко-
кситетрагндроппрана, в которых нояв.:яется а с i: л i :.ict p 11 ческ i r r'V атом
углерода. Молекулы 2-.г;лкпл-2-П!дрскср!(алкокс11)тетрап1дропнра-
иа являются хнраль.чычн:
сн._ сн.
/ \
СЬЬ СП.
V 4
V:
R ОН
ОН
Н S~O R
Молекулы тотрагпдропнрлпа существуют в (;.ормс кресл,", при
этом возможны два конИюрмера с группой OR' в аксиально:.! клп
экваториалыюм положениях:
or'
OR'
OR.1
OR1
\
•«<^I^
У производпсго тс-трагпдроппрапа с асимметрическим атомом уг-
углерода может быть два зеркальных изомера (энантпомера): а и б.
499
Каждый из них может существовать в двух копформациях: I и П.
Если энантиомер б повернуть на 180 вокруг оси, перпендикулярной
листу бумаги, получается изображение молекулы в. Это помогает
лучше сравнить al с бП и all с el (видно изменение положения OR1
и R в пространстве).
Кольчато-цеппые превращения и пространственная изомерия
циклических форм имеют исключительное значение в химии угле-
углеводов.
3. ДШ'ПДРОКСПКЛРБОППЛЬПЫГ. СОПДНННПИЯ
1. Методы получения. Дигцдрокспкарбсшнльпые соединения обра-
образуются при мягком окислении трполов. Например, при окислении
глицерина получаются глицериновый альдегид " 1,3-днгпдрокена-
цетон:
• О
--IIOCH..CII -С
н
ПОС11..СНСП-.СН1 —
о
он
_ лосиха [..он
2. Физические и химические свойства. Глицериновый альде-
альдегид — бесцветное сиропообразное вещество со сладким вкусом, лег-
легко растворяется в воде. Молекула глицеринового альдегида содер-
содержит асимметрический атом углерода и является хпралыюи. Известны
два энантиомера глицеринового альдегида, они оптически активны.
Один из них, (-: )-нзомер, вращает плоскость поляризации света
вправо, а другой, (—)-изомер, влево. Правовращающий изомер
обозначают буквой D, а левовращающпн - /-• Установлена аГч-о-
лютпая конфигурация D- и /.-изомеров глицеринового альдегида:
НО Он
\/ чс/н
I !
*'\
ОН НО i Н
С'Л:ОН СН.ОН
изомер L (—) -изомер
НС = О НС=О
! i
И j ОН НО j Н
с и он сн.он
проекционные формулы Фишера
Конфигурации D- и /.-изомеров глицеринового альдегида ис-
используются для классификации других хпральпых соединении
(углеводов, а-гпдрокенкарбоновых кислот, и-ампиокарбоносых кпе-
500
лот), т. е. их отнесения к D- или L-ряду. Принадлежность к D- или
L-ряду в общем случае определяется не направлением вращения
плоскости поляризации спета соединением, а совпадением прост-
пространственного расположения заместителей у асимметрического угле-
углеродного атома рассматриваемою соединения с расположением за-
заместителей в D- или /.-изомере глицеринового альдегида.
Применение /\\5-номспклатуры к эпаптпомерам глицеринового
альдында покачивает, что D-изомер соочнетсдвуст Л!-конфигурации.
Так, при рассмотрении молекулы Й-п::омгра в направлении связи
С-И видим расположение заместителей в ряду ОН. CI1O, СП .Oil
в направлении движения часовой стрелки.
Д 1Я глицеринового альдегида характерны cr.oikTDa альдегидов
и 1,2-глнколеГ:. Специфически протекает реакция с фепптгндрпзп-
по.:. В мягких условиях может Г>ып> получен обы'-чп;!:) гептгпд-
ра;;сп, который при пагрог.лпнп с и 'Гмптг.о.-.! фчпмгп/;:;.- .;::[:п'п[хвра-
Щг'Ч'-тея в бпе-феиплгпдрачоп Ke'io:v!:.;kri:,:.! ¦¦¦;•/:;•'.¦¦:
HO(.;i сп—с:' •-, c.ii.mixii., - , поа.'.сч- с
и и
Oil fill
м
>' —\ПС.;Н-, с.11-м!\::. С---—v
H0CIiC.II—С liOOl,—С v-rils
II \ ¦¦. ¦ i I
ОН
В зтоп репкцпп одна молекула (^спмлгпдралппл пмступает i: ка-
чегтпе окисли тел я.
Пни пзап>.:одеистп1Ч1 1.3-дпгпдр'.жспгн.етона с фешпгпдрамнном
of разуется тот же самый озазоп, чю и в случае гл:н!.е])ппог.ого аль-
дегн/а. Образование озазонов имеет пажное зпачепп-е в химии угле-
углеводов.
4. ФПНОЛКЛРБОПИЛЬНЬП- СОНДПШПП !Я
1. Методы получения. Феполкарбоппльпые соединения образуются
в реакциях прямого ацплированпя (Ьеполоа.
Для получения феполальдегидоа применяются специальные ре-
реакции форматирования. Первый метод ([юрмилиповання ();енола —
взаимодействие фенола с хлороформом в щелочной среде, был разра-
разработан К- Реймером и Ф. Тпманом A875):
9 оп
I
501
Предполагается, что форматирующим реагентом в этой реакции
является дихлоркзрбен, который возникает лз хлороформа и ще-
щелочи:
Н
ь-
В реакции образуется смесь о- и я-пзомероп.
Другим форматирующим реагентом является диметпламггд му-
муравьиной кислоты (диметилформамид) п присутствии кислот Льюи-
Льюиса, главным образом РОС13, SOCL (А. Вильсмайер, А. Хаак, 1927):
о
I
о
I
•R
СН-, ,0
\ — С Х/ + РОС1Я —>
СН3/ ЧН
\\/
-о
I
i- -;- на + H;ipot
п..о
. СН3
чсн3
PO-Ci:
н
•-о
В этой реакции преимущественно получаются л-нзомеры. Фор-
мнлпруются как фенолы, так гг их эфири. Формнлирующпм реаген-
реагентом в этой реакции является катион, который образуется из
(CH3JN = (
днметилформамида и Р0С13 (SOCU) (гл. ХХХШ.Д.З).
Аналогично проводят формилирование арепдиолов или их эфн-
ров с целью получения дигндрокенбензальдегидов и их производ-
производных
ных.
х.
2. Физические и химические свойства. Феполкарбонилыше со-
соединения представляют собой бесцветные жидкости или кристалли-
кристаллические вещества с ароматным запахом. Наибольшее практическое
значение имеют о- и /z-гндроксибензальдегиды.
502
о-Гндрокснбепзальдегнд (салициловый альдегид) обладает силь-
сильной внутримолекулярной подеродпои спгтлью. В эточ отнесении он
подобен р-дикарбонильпым соединениям. Он образует соли, снопа-
снопами тяжелых металлов — нераасоримыг в воде ппуфенпн- комп-
комплексы (хелатг.!), с попом Ге:1Ь дает характерную красно-фнолс-тозую
окраску:
Стабильные внутренние комплексы обрг.чу'г.тса также в случае,
нмппов салицилового альдегида, например производных этилен-
.о-н
3. Важнейшие представители. Салициловый альдегид — бесцвет-
бесцветная жидкость с приятным запахом горького миндаля; т. ил. —7 СС,
т. кип. 197'С. Содержится в эфирных маслах некоторых растений.
Синтетически его получают из фенола.
Особое значение салициловый альдегид и его и.мпньг имеют в
аналитической химии. Его применяют также в парфюмерной про-
промышленности и в органическом синтезе.
" q^/ Ванилин D-гидроксп-З-метокспбепзальдегид) яв-
I ляется бесцветным кристаллическим веществом с
//¦-. запахом ванили; т. пл. 81-—83 С. Содержится
il в виде глнкознда в плодах вапнлп. Синтетически
получают форматированием гваякола.
| Используют в пищевой и парфюмерной про-
OII мышлепности.
503
В. УГЛЕВОДЫ
Углеводы широко распространены в жшютиоми растительном мире,
они играют исключительную роль во многих жизненных процессах.
Углеводы составляют 80% от сухой массы растерши и 2% от cyxoi'i
массы животных организмов. Они являются одним из главных пи-
пищевых продуктов и служат исходным веществом для промышленно-
промышленного производства искусственного волокна, взрывчатых веществ, бу-
бумаги и этилового спирта.
Название «углеводы» этим природным веществам предложил в
18М г. К. Шмидт, так как элементный состав известных тогда угле-
углеводов мог быть выражен как С„ (Н2О)т. В настоящее время понятие
«углеводы» стало гораздо шире.
Углеводы обычно подразделяют на моносахариды и олнго- и
полисахариды (при гидролизе расщепляются до моносахари-
моносахаридов).
Моносахариды классифицируют но числу углеродных атомов
в молекуле п присутствию альдегидной пли кетогрунпы:
Лльдозм
О
TVrpmu СИ..—СИ—СП—С"' Кетозы СИ,—СИ —С —СП ОН
C,:uo4 | | | н | !
он он он он он о
о
Пемточи СП..— СИ — СН — СИ— С" СП-. — СП — СИ — С—СИ .ОН
С-.Н^Оя III! NH | • ,
он он он он он он он о
, о
Гексолм СП,—СП—СН—СН—СП—С' СП, — СН — СП — СН —С— СП,
С,;Н,;С>, | III1 "H| j i I
он он он он он он он он он о он
По строению моносахариды являются полпгпдроксиальдегпда-
мп или иолигндроксикетоиамп. Молекулы моносахаридов хпраль-
ны, имеют два пли более асимметрических углеродных атома. Это
определяет существование большого числа пространственных изо-
изомеров. Углеводы, полученные пз природных продуктов,— оптиче-
оптически активные вещества.
К углеводам формально могут быть причислены триозы QHoOj
(глицериновый альдегид и 1,3-дигпдрокспацетон). Ряд альдоз и
кетоз может быть дополнен гептозами CTHi407, октозами С8Н,„О9
и др.
Отдельные моносахариды сохраняют тривиальные названия, на-
направление оптического вращения обозначают знаками плюс или ми-
минус, а буквами D или L обозначают принадлежность данного моно-
моносахарида к D- или L-ряду, сравнивая расположение заместителей у
504
четвертого углеродного атома для
глицериновым альдегидом.
и У
Дли гексоз с
1. ПКПТОЗЫ
1. Пространственная и ксльчато-цепная изомерия. Наиболее рас-
распространенными в природе я'1 ымгея ал;,;опелтоз:л. Их чнгк-ку-
лы содержат трм асимметрических углеродных агама, полому но.1,-
можно существование^11 8стереопзо.мероп -четыре пары знаитпо-
меров (зеркальных изомеров):
Н О
н —:— он
н —;— он
Н —|- ОН
Г,6(,.ОП
о и
с
-И
¦И
н
п
о
но
ПО--
ПО -
поен,
с: ¦¦¦
но --i - п
НО -.-- И
И -.-;-¦ ОН
СП,ОН
• с
о
н
п
[¦!) !
'. i
и,
о
о
но
н
н
-(
- -.¦—
1
;п
п
он
он
¦ОН
• с
п -: -
но ; -
но -:—
носн
он
и
и
н-
но—
о
-. ом
- и
--он
/) КСП.Ч1
о п
С.
ПО-' ,:
I! ¦ .>U
ПС) . il
у I ¦ ¦ ¦ | • (-
Проекционные с|)ормулы Фишера (а) показывают плоскостное п;;о-
бражеппе пространственного расположения атомов Н п групп ОН
относительно углеродной цепи молекулы углевода (в данном случае
рибозы). Полее наг.чядио строение молекулы изображает формула
б н ее сокращенный вариант в, в которых показаны валентные
углы:
он
«он
СИ ОН
гн он "
WV V- —Г'
он он
б
сн он н
< Г'
он он
505
Альдопсптсмы являются одновременно v-гндроксп- п 6-гплрок-
сиальдегпдамп и легко образуют циклические полуацеталп".
СН.СН
ом о: г ,. он он
110
C.i
OH
При образовании цикла фурлпозы происходит поворот по свяли
3—4. При образовании цикла ппрапозы поворота ист.
В растворах альдопентоз сохраняется равноессме между откры-
открытой формой, фуранозами и пирапозамп. Эго является своеобразной
кольчато-цепной таутомсрпоГг системой.
В циклических структурах появляется новый асимметрический
углеродный атом (первый углеродный атом). Это означает, что из
одной определенной альдопептозы могут образоваться две фурапозы
и две ппрапозы. Две циклические изомерные формы называются
аномерами и обозначаются буквами а и |5. Принадлежность к а-
или р-форме определяет пространственное положение гидроксиль-
ной группы у первого углеродного атома п группы СН2ОП или ОН
у четвертого углеродного атома. Подробно это рассмотрено при опи-
описании гексоз. Каждый аномер имеет свое значение удельного опти-
оптического вращения.
Если в кристаллическом состоянии альдоиептоза существует в
одной определенной форме (фурапоза или пираноза, а- или р-фор-
ма), то при растворении в воде происходят тауто.мерпые превращения
до установления равновесия между и- и р1-формами. Это приводит к
изменению удельного оптического вращения с момента растворения
до установления равновесия. Это я:пеппе называется мути рота-
ротацией.
2. Химические свойства. Лльдопентозам свойственны почти все
реакции альдегидов и многоатомных спиртов. Они окисляются, да-
дают реакции серебряного зеркала и Фелипга; при мягком окислении
образуют карбоновую кислоту (в виде лактспа); при восстановлении
дают пятнатомные спирты пентиты; с фепплгидразппом образуют
озазопьг:
506
'НО'
но он
I I I i
НО ОН 1.0 CT-S
! -м,о
СП .ОН
ос;..
КО ОН
СНДН.И X
С Н: ОН
V ,-*¦
СН.-ОЧ
он
I V
.[21
КО
он
сн,осн3
он
н н
H
носн2-с-с-с:с ""Nnn-c5h5
но он
H.CO ОСНз
OCH3
HN—C5H3
Л.ткнлированпе альдопептоз [гппводит к образованию простых
эфиров. Сначала реагирует гпдрокспльпая группа у 1-го атома угле-
углерода -- глпкозндпая пли полуацетальная гидрокснльная группа.
Алкплпроваппе сильными реагентами (д'.шетплсульфятом) приво-
приводит к тетрамотп.ЮБому эфиру, у которого одна мстнльпая группа
(гликозпдпая) легко отщепляется.
Превращения «альдоза: ¦ кс-тоза» и «ал[)До:1ептоза->-альдоге1ссо-
за» рассмотрены ниже.
3. Важнейшие представители. Ксилоза (дрессспып са?:ар) входит
в состав полисахаридов кспллпов. Ксилапы содержатся в древеси-
древесине, лузге подсолнуха, кукурузных кочерыжках, ic\;c;:.;e. При пх
гидролизе получается кспло:>а.
Kcii.-юза образует бесцветные кристаллы с. т. пл. 1-1 Г) С; опти-
оптически активна, 1сс!л----! 93,6". Принадлежит к D-ряду, в кристалли-
кристаллическом состоянии существует в форме пнряпозы, просграпстцеппое
строение молекулы соответствует ос-апомеру:
но
но
он
fx-D( i- Ьксплопирл [io:;a
При растворении a-D ( ;-)-кснло1шрапо'зы в воде наблюдают
мутаротацпю, угол удельного вращения раипоЕеспом смеси а- н
р-форм Ice] „--- г18,8:, содерж;нп:е а-формы 33%.
При нагревании ксилозы в водных растворах серной кислоты
происходит циклизация и отщепление воды, образуется гетероцик-
гетероциклическое соединение — фурфурол (фуранкарбальдегнд) эта реак-
реакция имеет важное промышленное значение.
507
Рибоза входит в состав нуклеиновых кислот и может Сы.ь полу-
получена, например, гидролизом дрожжевых рибонуклеиновых к;:.лот.
Рибоза образует бесцветные кристаллы, легко растворяется в иоде,
т. нл. 87 СС; оптически активна, \а\ ,, —23,1 (для свежеприготов-
свежеприготовленного раствора), принадлежит к D-ряду. В кристаллическом со-
состоянии существует и (|юрме фурапозы, р'-аиомер. В растворе наблю-
наблюдается мутлротс'щкя, угол удельного вращения равновесной смеси
[с.|п —23,7, содержание а-формы - • :26г'о.
си он сн он
*~ А.и.лсги.шля
ОН ОН
ОН ОН
Рпбоза псполь.;\т;ея в синтезах пуклеолпдоп.
2-ДсзоксириО(оа производное D-рпбо.лы, у которси
poKCii.4i.noii группы при 2-м углеродном атоме. 1:.е ;:¦.• туч
лнзом дезокспрпбопуклепповых кислот. Она образует
кристял.ты, хорошо растворимые в иске; т. пл. 78 -82
-¦- !)Г (свежеприготовленный раствор'!. 1^ к;шстал-н1Ч'
2-дезоксирнбоза существует в виде фуранолы. В водных
устапавлш'.аетсч равновесие, между с- п |'>-формямн ф
а- и ^-формами ннранозы (для равповесноп смесп \а
си он сн он
о пег
;ц-.)т п'
Сосцгл
С; ic
сксм
гпд-
- - 58 ):
ОН
ОМ
Лезоксир;1оо.5с1 используется в синтезах нук.тс-лпдев.
2. П-КСОЗЫ
1. Пространственная и кольчато-ценная изомерия. Известны как
альдо-, так п кето!ексозы. В молекуле альдогексоз в альдегидной
форме присутствуют четыре асимметрических углеродных атома,
поэтому возможно существование 2" 16 стереонломеров, что дает
8 пар эпаптиомеров "(зеркальных изомеров):
Нч ,О О, ,11 Ич О (К Н
/у ч' -Ы N
Н--
-ОН 1Ю-
—он
-он
но-
но —
-он но__
Н —
СН,О) [
D -а л лоза
— н
— и
— н
— н
н-
НО —
н —
JF —
носн2
/--аллоза
— он
-и
-он
— он
СИоОН
но-
п -
ПО -
но-
-н
- ()И
-- П
- и
носи.
508
н
о
о
с
н —
но -:
но-
н —
- O
— II
¦ - ОП
СН,ОИ
н
И -
II
ПО -
- II
on
ОП
II
по
по
II
и
иосп
. о
с
.._;.. if
: - И
: - оп
! - ОП
сп,ои
О .
II -
II -
по
ПО -
с
ОП
ОП
II
н
В кристаллическом состоянии ал!-.догс:,счзы осуществляют и
циклических полуацеталышх ппранозных формах, в раечворах of,-
разуется равновесная с\;ос:> циклических форм и альдегидном фор-
формы.
Каждой альдогексозе соответствуют две стсреопзомерные форм!.;
фурапозы (<х- н |-)-амомсры) м две формы пирапозы (а- и р-аномеры).
Для изображения циклических форм могут применяться и про-
проекционные формулы Э. Фишера, но это не дает наглядной картины.
Лучше всего пользоваться перспективными формулами У. Хеуорсч
(W. Haworth, 1929), как плоскими, так и изогнутыми. Ниже приве-
приведены псе три способа изображения для ос-У-глюкоинряиозы:
" ^ J
(,;i
Рассмотрим кольчато-иенпую таутомерную систему D-глюкозы
(структуры ф\рамоз мало характерны и в схеме не приведены):
t M..OH
оьон
i И.ОН
он
СИ ОН ОН
-О'
Oil
При образовании циклической структуры происходит поворот по
связи 4 --5.
Оiнесение к а- пли |-)-форме зависит от того, каким обра:юм в
пространстве располагаются г.тпкозпдпая гпдроксильная группа
(у 1-го углеродного атома) и гндрокспльиая (пли гидроксиметиль-
509
пая) группа у атома углерода, определяющего принадлежность к D-
илп L-ряду (для пептоз— четвертого, для гсксо:;— пятого). Для
фурапозпых п плоских ппранозпых структур ице-расположешю этих
групп определяет E-форму, а «гракс-расположепие — ос-форму.
Для изогнутых ппрапозпых структур неодинаковая пространст-
пространственная ориентация групп (аксиальная — экваториальная, эквато-
экваториальная — аксиальная) определяет с.-форму, а одинаковая ориен-
ориентация (аксиальная — аксиальная, экваториальная — экваториаль-
экваториальная) определяет р-форму.
Для пирапозпых структур существует два копформера, которые
переходят друг в друга при инверсии цикла. Относительно более
стабильным является тот копформер, в котором больше экватори-
экваториально размещенных ОН-групп. Поэтому в ряду стереонзомеров D-
глюкозы наиболее стабильной является ^-.О-глюко; ираноза в кон-
формации с пятью экваториально расположенными ОН-групплмн.
Кетогексозы в кетоформе имеют три асимметрических углеродных
атома, поэтому возможной простр
тиомеров. Ниже приведены две и
CFI..OH
2! . О
НО —
н —
поен,
О -!
:— п
н —
— он по—
— он но —
— ОН И —
— Н ПО —
— II
II —
шстзеппых изомеров, 4 нары эпап-
ары:
СН..ОП
¦-= О
НОСИ.,
о =-.!
он но
II
н -
— ОН ПО -
— н
— он
-- II
сп,он носн, сп,оп поен,
7)-фру кто.чя /.-фру кто.'*л D -сорСсл;! /. -co;:Go.'iii
Для кетогексоз характерны как фурапозиые, так и пирапозпые
циклические структуры:
н
I
СН;ОН „,,
, о он
СН.ОН с\\.О\1
$• П ф[>:.к-гпиг.а,:гм 1-П-фг1кг.--1;.ра!|,-зл
При образовании цикла фураиозы происходит поворот по связи
4—5.
Для фурапозпых структур цис-расположепие глпкозпдпеп ОН-
групны и группы СН.ОН у пятого углеродного атома определяет
Р-форму, /п/;анс-расположеппе — а-форму. Для кресловпдпых пи-
510
рапозпых структур одинаковая пространственная ориентация глп-
козидпой ОН-гоуипы и группы СИ..ОН у няюго атома углерода
(аксиальная — аксиальная, экваториальная —¦ экваториальная) оп-
определяет р-форму. В случае с.-формы ориентация этих групп про-
противоположна (аксиальная —экваториальная, экваториальная —
аксиальная).
2. Физические и химические свойства. Гексозы являются бес-
бесцветными кристаллическими веществами, хорошо растворяются в
воде и имеют сладкий вкус.
Для гексоз характерны реакции карбонильных соединений п
многоатомных спиртов: они восстанавливаются до шестпатомных
спиртов текстов, окисляются до мопокарбоновых и днкарбоновых
кислот, с фенил гидразином образуют озазопы. Ниже приведены не-
некоторые реакции Z)-глюкозы:
110
он
ПН
—СН2ОН
CHjOll о
О _j_
ОН
СН2ОН
CHjOH
/
IIO-
Г) -г.гм;.(т>л
( зс ИЛИ /1 )
Л д сн;он
он
он
но'
он
сн.он о
СООН
-н.о тю
он хо
СООН
НО
он
/?-сах:риая .кислота
Поп мягком окислеш!!! пльдогоксоз образуются пепт;.:г;1дроксн-
кг.обо'ювые к-1слот!л — алыЪновыс кислоты, которые обычно су-
ничптллот в виде ^-ляктопа пли -улактопа (гл. ХХХШ.В.1).
Дяльпемшее окисление альдоповых кислот ведет к тетрагпдро-
KCii;ii;K-ip6onorsi.:\i кислотам (сахарные, или «аровые», кислоты).
При окислении глнкозидов альдогексоз (простых эфпров с груп-
ппропкоп R0 у нерного углеродного атома) могут быть получены
альдегпдокарбоповые кислоты, так называемые урановые кислоты:
(ООП о
••о 011
511
Для окисления альдогексоз до альдоповых кислот применяют
бром в водном растворе, соли серебра в аммиачном растворе (реактив
Толленса, реакция серебряного зеркала, см. гл. XXVII. А.3), реак-
реактив Фелиига (механизм реакции см. гл. XXIX. Б.1). Альдогексози
окисляются легче кетогексоз. По в щелочной среде эта разница
не наблюдается.
В присутствии щелочи происходит изомеризация альдегексо-
за. - кетогексоза через ендпольпую форму. Ниже приведены изо-
изомерные превращения D-глюкозы, D-фруктозы и D-маппсзы:
СНОН
л 'СН..ОН
но
Через емдкольпую форму D-глюкоза превращается г. D-.\K;i:hc, у
и D-маппоза в D-глюкозу, которые различаются только прсктраисг-
вепным расположением ОН-групны у второго углеродного атома.
Происходит эпимеризация, структурным изменением подвержены
только первый и второй углеродные атомы. Гексозы, которые пмс:../г
одинаковую конфигурацию у третьего, четвертого и пятого атомов
углерода, называют эпимерными.
При нагревании с фепплгидразппом гексозы образуют сзазоны—
желтые хорошо кристаллизующиеся вещества. Эпп.мерпые гексозы
образуют одинаковые озазоны:
D -Глюкоза -
D -Маниоза -
D -Фруктоза ¦
Эпимериые гексозы
с-л.
С алкилирующими и ацплирующими реагентами образуются про-
простые и сложные эфиры. Наиболее легко реагирует глнкозндная
ОН-группа, при этом образуется смесь а- и р"-гликозпдов:
512
О (CM,C0),0 ^IbOtlQ. iCK):SO< CHjOCHir
NjOH
OOCCK,
miir.i.inriiii.D. r „,„¦ .. .. .,„.,_«-
H X ||
CH.-OH
Гликозидная связь С—О легко образуется и легко расщепляется.
Например, в пентаацетилглюкозе легче всего гидролпзуется группа
у первого углеродного атома.
Гликозидная ОН-группа легко замещается атомом галогена:
Для некоторых гексоз характерно ферментативное расщепление.
Наибольшее значение имеет процесс спиртового брожения, проис-
происходящий под действием смеси ферментов зимазы. Эту смесь фермен-
ферментов вырабатывают дрожжевые грибы.
Процесс превращения гексоз в этиловый спирт длинен и сложен.
Суммарно он выражается уравнением
CHi.,0,1 — 2С,Н;,ОН , 2СО,
Ферменты, вырабатываемые дрожжевым грибом, расщепляют до
этилового спирта не все гексозы, а только D-глюкозу, D-манпозу,
D-фруктозу.
3. Важнейшие представители. Глюкоза (виноградный сахар)—
самый распространенный моносахарид. В свободном виде она содер-
содержится во фруктовых соках, в качестве составной части входит в мо-
молекулы дисахаридов (сахароза, лактоза, мальтоза, целлобноза) и
полисахаридов (крахмал, целлюлоза, гликоген, декстрины). Полу-
Полученная из природных продуктов глюкоза принадлежит к D-ряду.
В кристаллическом состоянии можно получить оба аномера D-глю-
копиранозы — а- и Р-формы. Оба являются бесцветными кристалли-
кристаллическими веществами с приятным сладким вкусом, оптически актив-
активны. В растворе наблюдается мутаротация. Удельное оптическое вра-
17 .V. 517 51.3
щение равновесной смеси [а\ п=4-52,7°, содержание а-формы 36%.
но
и
110 он
а-Е(-(-)-глюкопирлноза 0-D(-t-) -глюкапнраноза
a-D (>-)-глюкопираноза кристаллизуется из воды, т. пл. 83 °С
(моногидрат), 146'С (безводная); [а] „ --|-112,2" (НЮ). fi-D (¦[-)-
глюкопираноза кристаллизуется из пиридина, т. пл. 148. ..150 °С;
Чистую глюкозу получают гидролизом картофельного или куку-
кукурузного крахмала кислотами.
Глюкозу широко используют в пищевой промышленности, меди-
медицине, в качестве исходного для синтеза глюконовой и аскорбиновой
кислот, в качестве восстановителя в текстильной промышленности,
для получения этилового спирта.
Галактоза встречается в природе в свободном виде, в виде гли-
козидов, дисахаридов (лактоза), полисахаридов (галактаны, агар-
агар, растительные клеи).
но В кристаллическом виде получены как ос-,
так и Р-форма. а-Форма плавится при 167 JC,
[al 7,-- + 150,7J (Н.,О), C-форма плавится при
153—155'С, [а]п--—54,4е (Н2О).
он В растворах наблюдается мутаротация, удель-
«-п,+).галактопиРа,>п,а ]юе ВращСние равновесной смеси в водном раст-
растворе [а] ^-=81,Г7, содержание а-формы 27%.
Фруктоза (плодовый сахар, левулоза) распространена в расти-
растительном мире, содержится в помидорах, яблоках, больше всего фрук-
фруктозы в пчелином меде (до 50%). В качестве составной части входит в
молекулу днеахарида сахарозы (в форме р-фуранозы) и в молекулы
некоторых полисахаридов.
Получают фруктозу гидролизом сахарозы. D-Фруктозу получа-
получают также из D-глюкозы при обработке последней щелочью (эпиме-
ризация).
он Природная фруктоза принадлежит к D-ря-
' -енхш ДУ' в кристаллическом состоянии существует
^_ ' в форме р-пиранозы; т. пл. 102—104 С, \о.] D—
но он °" — —133,5 ' (НЮ). В отличие от других моноса-
моносахаридов вращает плоскость поляризации света
э-п(-)-ФРУкт,)Пир.„„п, влево. При растворении фруктозы наблю-
наблюдается мутаротация, удельное вращение равновесной смеси [а\1}—
=—92°.
Фруктоза является наиболее сладкой из всех Сахаров, она в
полтора раза слаще сахарозы и в три раза слаще глюкозы. Не ис-
используют в медицине в качестве цепного продукта питания.
Сорбоза и аскорбиновая кислота. Кетогексоза сорбоза содержит-
содержится в природных продуктах, принадлежит к /.-ряду. Получают ее
микробиологическим окислением D-сорбита. В кристаллическом
514
состоянии L-сорбоза существует в форме пиранозы. В водных раст-
растворах ее наблюдается мутаротация.
L-Сорбоза служит исходным веществом для синтеза аскорбино-
аскорбиновой кислоты (витамина С).
Окислением в специальных условиях L-сорбозы получают кар-
боновую кислоту, которая изомеризуется в L-аскорбиновую
кислоту:
и
СИ ОН СП ОН
110 / \он
[о]
Oil
сн.он —.-
он
он
L -сорбоэа
[о]
спои ,
он
см.он
СП ОН
I
неон
-о
I
он
L-Аскорбиновая кислота представляет собой бесцветное кристал-
кристаллическое вещество с т. пл. 190—192 С и интенсивно кислым вкусом
(рАГа--4,17), хорошо растворяется в воде, оптически активна [а]п=
-+23° (Н2О).
L-Лскорбиновая кислота (витамин С) содержится во многих
фруктах и овощах. Она необходима для жизнедеятельности организ-
организма человека и животных, L-аскорбиповая кислота катализирует
окислптелыю-восстановительные процессы в организме. Сама ас-
аскорбиновая кислота является сильным восстановителем. Эти свойст-
свойства ей придает епдиольиая структура. В отличие от других а-гидро-
ксикарбонильных соединений аскорбиновая кислота является фик-
фиксированным епдиолом.
3. НАРАЩИВАНИЕ
II ДЕСТРУКЦИЯ УГЛЕРОДНОЙ ЦЕПИ МОНОСАХАРИДОВ
В установлении строения моносахаридов большое значение имеет
постепенное наращивание цени углеродных атомов, т. е. переходы
триоза—>-тетроза—нгентоза->-гексоза—>- и т. д., и наоборот, постепен-
постепенное укорачивание цепи. Кроме того, значение имеет также окисли-
окислительный разрыв цепи у определенного атома углерода. В качестве
примера приведены синтетические переходы альдопепгоза—киьдо-
17*
515
гексоза—»-альдопентоза:
-сн.он
но
н
D -арабиноза
г Окисление и
дскарбиксилирование
11,0.
Fe-'
-СО-
л СН.ОН
/он ф
hoIL—^
он
(Гидролиз и
лактонизация
сн он
сн он
он он ,
но1
лактон D -глюкононой кислоты
1Восстапоаление
Na/"BiH.O
СН.ОН ,
Вг2;Н.О НО
С:а(ОНJ
ОН
ОН
Са-соль [) -глюконоаой
кислоты
IJ -глкжопираноза
1* к Q )
Углеродная цепь наращивается циангидринным методом. В ре-
реакции всегда образуются два изомерных циангидрина. Следующие
стадии процесса: гидролиз нитрила (гл. XXXIII. К-3) и восстанов-
восстановление лактона (Г. Килиани, Э. Фишер). В данном примере конечный
продукт превращений второго изомера — D-манноза.
Укорочение цепи достигается окислением альдегидной группы
до кислоты и окислительным декарбоксилированием специальным
реагентом H2O2-;-Fe:1+ (Руфф).
Окислительное расщепление углерод-углеродной цепи достига-
достигается метилированием и окислением. Например, окисление тетра-
метилглкжопиранозы
СН.0СН.1 Q
СН3О
'он
,ОСНз
ОСНз
5IG
ОСНз
ноосснснснсоон
I I
ОСН,OCHj
соон
ОСН,
ноосснснсоон
ОСН,
идет по неметилированной гидроксилыюй группе, которая была
связана в пиранозный цикл, с последующим расщеплением связен
С—С (см. гл. XXVII. А.3). В этом случае образуются триметоксиглу-
таровая и диметоксияптарная кислоты. Это свидетельствует о том,
что свободная ОН-групиа была у 5-го углеродного атома. Этот ме-
метод используется для определения величины оксидного цикла
(пнранозы или фуранозы).
4. ДНСЛХ.АРНДЫ
Молекула дисахарида содержит два остатка моносахаридов, которые
соединены между собой кислородным мостиком, гликозидной связью,
т. е. дисахариды являются гликозидами (простыми эфирами с учас-
участием гликозндпой гидроксильной группы).
1. Главные типы. Гликозидная связь в дисахарндах может об-
образоваться по-разному, в зависимости от типа гидроксильных групп,
участвующих в образовании связи.
Часть дисахарндов характеризуется гликозидной связью, кото-
которая образовалась из гликозидной гидрокснлыюй группы н обычной
гидроксильной группы второго моносахарида. В большинстве слу-
случаев гликозндную связь образует гидрокснльная группа у 1-го
углеродного атома. Таким образом, дисахарнд имеет свободную
гликозидную гидроксильную группу, обладает восстанавливающи-
восстанавливающими свойствами, в его растворах наблюдается явление мутаротацип.
Восстанавливающие дисахариды принадлежат к типу мальтозы.
Принципиально от других отличаются дисахарпды, в образова-
образовании которых участвуют две гликозидные гидроксильные группы.
Эти дисахариды не обладают восстанавливающими свойствами (не
дают реакцию серебряного зеркала), так как нет гликозидной гид-
гидроксилыюй группы и пет возможности образования открытой кар-
карбонильной формы. I^восстанавливающие дисахариды принадлежат
к типу трсгалозы:
он он он
гликозидная связь типа мальтозы гликозидная связь типа трегалочь!
При кипячении водных растворов дисахаридов в присутствии
кислоты происходит гидролиз и образуются моносахариды.
2. Важнейшие представители. Мальтоза в небольших количест-
количествах содержится в некоторых растениях. Ее получают при фермента-
ферментативной переработке крахмала.
В кристаллическом состоянии мальтоза получена как в а-, так
и fl-форме. В растворе наблюдается мутаротация. Молекула малыо-
зы содержит два остатка глюкозы, при этом один остаток находится
в фиксированной а-форме. Мальтоза может быть названа как 4(сх-
517
0-глюкопиранозидо)-(а или р)-0-глкжопираноза:
сн.он
а -мальтоза /? -мальтоза
т.пл.108°С;[а]п=-М73° т.пл.Ю2 ... ЮЗ°С ;Га];=+Ш,7"
Мальтоза легко растворяется в воде, имеет сладкий вкус. Ее
используют в производстве вина и пива. Ферментативно легко рас-
расщепляется до глюкозы. В организме человека также ферментативно
легко расщепляется до глюкозы.
Целлобиоза — бесцветное кристаллическое вещество, легко раст-
растворяется в воде, в растворах наблюдается мутаротация. Получают
ее расщеплением полисахарида целлюлозы. Молекула целлобиозы
содержит два остатка глюкозы, при этом один остаток находится в
фиксированной fi-форме. Целлобиоза может быть названа как 4-ф-
Ь-глюкопиранозпдо)-(сс или [5)-Д-глюкопираноза, известны а- и
fi-целлобиозы.
сн он он В молекуле целлобиозы один
ho^VV^O\ H°/r^--J^r4oH остаток глюкозы по сравнению с
oA^^*^^-^0v^^**^o мальтозой повернут на 180". Цел-
он сн2он лобиоза ферментами дрожжевых
грибов не расщепляется, снирто-
^пГиггс-Т =4 244' вое брожение не происходит (эти-
" " ' ловый спирт не образуется). Цел-
Целлобиоза не расщепляется в организме человека и в качестве пищево-
пищевого продукта не может быть использована. Целлобиоза расщепляет-
расщепляется до глюкозы только ферментом целлюлазой или кислотой.
При сравнении свойств целлоииозы и мальтозы ясно проявляется
огромное влияние пространственного строения молекулы па био-
биохимические реакции.
Лактоза (молочный сахар) содержится в молоке. Молекула лак-
лактозы построена из галактозы в фиксированной fi-форме и глюкозы.
Известны а- и р-лактозы, обычно кристаллизуется а-лактоза. Лак-
Лактоза может быть названа как 4-([-)-/,)-галактопиранозидо)-(с. или
fi) -D -глюкопираноза.
он ¦ П11 Пространственное строение лак-
лактозы подобно строению целлобио-
целлобиозы.
Лактоза меньше растворима в
сн2он воде, чем другие дисахариды, об-
а-лактоза ладает в 5 раз меньшей сладостью,
т.пл. 22з-с;о«]0=+ад' чем сахароза. Спиртовое броже-
брожение с лактозой не происходит. В присутствии специальных молочно-
молочнокислых дрожжей образуется молочная кислота.
Сахароза (тростниковый или свекловичный сахар) известна с
древних времен, впервые получена из сахарного тростника. Сахар
518
сн2он
носн
но
он
был известен уже в древней Индии за 300 лет до нашей эры, в Европе
сахар появился около 1500 г. В сахарной свекле сахарозу нашли в
1747 г. В настоящее время выращена сахарная свекла, содержащая
17—19% сахарозы.
Сахароза представляет собой бесцвем пое кристаллическое вещест-
вещество, хорошо растворяется в воде, имеет сладкий вкус; т. пл. 185 'С.
Не растворы оптически активны,
[ос] п ¦--. 66,5 , мутаротацпя не
наблюдается. Реакции серебря-
серебряного зеркала и с реактивом Фа-
лп'.гга сахароза не дает. Молеку-
Молекула сахарозы образована из а-
глпкопнрапозы и |">-фруктофура-
ta"l""a позы с глпкозпдной связью ти-
(ot ¦* /5 -глюкопирано шдо-^- О -фруктофураиошд ) jj^j ТПСГаЛОЗЫ.
Сахароза при кипячении водных растворов в присутствии кислот
гпдролпзуется и образуется смесь глюкозы и фруктозы (инверти-
(инвертированным сахар, искусственный мед).
Сяхароза легко расщепляется фермептатпвпо в организмах чело-
человека и животных. Сахароза является цепным пищевым продуктом
и исходным для получения этилового спирта.
5. ПОЛИСАХАРИДЫ
Полисахариды являются высокомолекулярными соединениями, мак-
макромолекулы которых образованы из многих молекул моносахаридов,
связанных между собой гликознднон связью.
Крахмал содержится в зернах растений и в картофеле. Обычно
его выделяют из картофеля или кукурузных зерен. Крахмал обра-
образует микроскопические зернышки B0—100 мкм). В воде они набу-
набухают, распадаются и образуют вязкие растворы, клейстер (гели).
Крахмал является неоднородным полисахаридом и состоит из
полисахаридов амилозы и амилопектина:
СН2ОН
НОСН;
сн2он
не
амилоза
И = 90...470: мол.м. 32 000 ... 160000
Большая неразветвленная макромолекула амилозы образована из
остатков а-глюкозы, основным повторяющимся структурным звеном
является с.-малыоза. Макромолекула амилозы свернута в спираль.
519
Макромолекула амилопектина разветвлена. В основе ее лежит
цепь амилозы, с которой сшиты другие цепи амилозы с участием
гидроксильной группы у б-го углеродного атома и гликозидной гнд-
рокснльной группы. Молекулярная масса амилопектииов 100 000—-
1 000 000.
Очень характерным свойством крахмала является цветная реак-
реакция с иодом — появление интенсивной синей окраски (максимум
поглощения при 620—650 нм). Считают, что появление окраски
связано со специфическим донорно-акцеиторным взаимодействием
между гидроксильными группами и молекулами иода, при этом иод
помещается внутри спирали макромолекулы амилозы. В присутствии
небольших количеств иодид-иона внутри спирали располагаются
цепочки [I- • ¦ 1- • • I- • • I- ¦ • 11~.
Крахмал широко применяется в различных отраслях промышлен-
промышленности. Из него получают сироп и глюкозу, он является главной со-
составной частью пищевых продуктов (хлеб, крупа, мука, картофель,
кукуруза). Из крахмала в ферментативных процессах получают эти-
этиловый спирт, н-бутиловый спирт, молочную и лимонную кислоты и
др. Используют крахмал в текстильной промышленности и для из-
изготовления красок и клеев.
Целлюлоза — широко распространенный в природе полисаха-
полисахарид. Она содержится в растениях. Примерами чистой целлюлозы
являются хлопок, вата, фильтровальная бумага.
Макромолекула целлюлозы состоит в основном из остатков (¦}-
глюкозы, основным повторяющимся структурным звеном является
(i-целлобиоза:
СН2ОН.. ~,t 1 СН2ОН
НОЧ А>»^_ • Го ч ~ -^к. \ .<-»! / ^к. — .е о ч ^к. \ он
целлюлоза
П= 60 . . . 1500;-мол.м. 21000. . .525 000
В дальнейшем для изображения макромолекулы целлюлозы бу-
будет применяться упрощенная формула [С0НтО2(ОНK1.^.
Макромолекулы целлюлозы расположены параллельно друг
другу и связаны между собой межмолекулярными водородными свя-
связями, образуя волокна. Поэтому в отличие от крахмала целлюлоза
в воде не набухает и не растворяется. Только при химическом взаи-
взаимодействии с гидроксильными группами целлюлозы достигается ее
набухание и в отдельных случаях растворение. Целлюлоза набуха-
набухает в растворах щелочей. Это связано с образованием алкоксидов:
Целлюлоза с меньшей молекулярной массой растворяется в ще-
щелочи.
520
Целлюлоза широко применяется в производстве искусственного
волокна, кинопленок, взрывчатых веществ, этанола.
Целлюлоза растворяется в смеси солей меди (II) и гидроксида
аммония (реагент Швейцера, 1857), где активным началом является
ICu (МН3),]-! 2ОН" . В этой реакции образуются комплексные мед-
медные алкоксиды целлюлозы, что характерно для всех многоатомных
спиртов. При иодкислении-этого раствора целлюлоза выделяется в
другой модификации и может быть использована для получения ис-
искусственного волокна (искусственный шелк.медно-аммиачный шелк).
Целлюлоза хорошо растворяется в щелочи в присутствии серо-
сероуглерода, образуя ксантогенаты (гл. XXXV.Г):
[C6I I7O,(OHK].V -f ЖаО11 -f xCS,
При иодкислеиии полученного раствора (вискозы) опять выделя-
выделяется целлюлоза. Если вискозу продавливать через тонкие отверстия
(фильеры) в водный раствор сильной кислоты, получаются топкие
нити, из которых прядением делают искусственное волокно — вис-
вискозный шелк. При продавливании вискозы через тонкие щели полу-
получают прозрачную пленку — целлофан.
При взаимодействии целлюлозы с уксусным ангидридом происхо-
происходит ацетилированме гмдроксильиых групп целлюлозы и образуется
ацетил целлюлоза:
[Св11702(ОНK1л.~Здг(СНзСО)оО-^[СиИ70,(ООССП3)з]А.-!-3*СН...СООН
Ацетилцеллюлоза растворяется в различных органических раст-
растворителях и ее используют для получения искусственного волокна—
ацетатного шелка и негорючих кино- и фотопленок.
Взаимодействие целлюлозы со смесью азотной и серной кислот
приводит к образованию сложных эфнров азотной кислоты — нитра-
нитратов целлюлозы (нитроцеллюлоза). В зависимости от условий реак-
реакции («нитрования») получают нитроцеллюлозу с различным содер-
содержанием азота A — 2,7 \О2-групп на один остаток глюкозы):
UNO, UNO-,
[С0П7О.ДОНЫЛ.—^ [С,;Н.О,(ОН),.ОХО,]Л. —-
UNO,
—г [С0Н7О,(ОП)(О\-О,J|л. —^ [С(!П7О,(О\О,Ъ]Л.
Нитроцеллюлоза растворима в органических растворителях.
Продукт с малым содержанием азота используют для изготовления
питроцеллюлозных лаков. Из нитроцеллюлозы с большим содержа-
содержанием азота (около двух NO«-rpynn на остаток глюкозы) получают
коллодий — вязкий раствор в смеси этанола п диэтплового эфира,
который после испарения растворителей образует пленку.
Нитроцеллюлозу с максимальным содержанием азота называют
пироксилином. Его используют для изготовления бездымного поро-
пороха п для других целей.
При гидролизе целлюлозы водным раствором серной кислоты
получают раствор глюкозы, который после связывания серной
521
кислоты используют для получения этилового спирта (гидролиз-
(гидролизный спирт).
В качестве пищевого продукта целлюлоза не может быть исполь-
использована, так как в организме человека отсутствуют ферменты, рас-
расщепляющие целлюлозу до глюкозы. В этом состоит принципиальное
отличие целлюлозы от крахмала.
Бактерии, живущие в пищеварительном тракте жвачных живот-
животных (коров, овец и др.), вырабатывают фермент, расщепляющий
целлюлозу до глюкозы. Поэтому жвачные животные могут питаться
продуктами, содержащими целлюлозу.
Хитин представляет собой амипополисахарид, являющийся
главным компонентом внешнего скелета насекомых и ракоподоб-
ных. Хитин составляет также клеточную стенку многих грибов.
По строению хитин подобен целлюлозе, только вместо гидрок-
енльнон группы у 2-го углеродного атома остатка р"-глюкозы нахо-
находится ацетиламипогрупна СН3СО\'П:
сн, о
Y
но
но
CHiOH
СНз
СН2ОН,
с
I
о
но
N
=С
СН2ОН
СН3
СН20Н„
NH
о=сч
Хитин не растворяется в воде, кислотах, щелочах, малораство-
малорастворим в муравьиной кислоте. При обработке хитина горячей щелочью
отщепляется ацетильная группа и получается амннополисахарид
хитозан.
Г. АМИНОКАРБОНИЛЬНЫЕ СОЕДИНЕНИЯ
Наибольшее практическое значение имеют а- и Р-аминокарбониль-
ные соединения.
1. а-ЛМПНОКЛРБОППЛЬНЫЕ СОЕДИНЕНИЯ
1. Методы получения. Для синтеза а-амипокарбонильных соеди-
соединений обычно используют а-галогенкарбонильные соединения, ино-
иногда в виде ацеталей, и аммиак или амины:
IF О
I И
R —С —С—R1-
С1
ОСЛ1-,
BrCII.C— FF-: 2-\Н3-
ОС,НГ,
522
Н
О
:|
'¦ — NH, —vR- С — С— RJ-[-[R2-
I
ос,п5
I
¦IIoNCH.jC-H~.\H,Br-
OC,H5
Второй метод заключается в восстановлении а-оксиминокарбо-
нильных соединений в присутствии кислоты.Образуются соли а-
аминокарбонильных соединений:
О Н О
li [[ц.-п'-х- I i;
R-C-C-R1 >R-C-C-R1
I I
X —OH J-NH3X-
2. Физические и химические свойства, применение. Аминокар-
бокильные соединения обычно представляют собой бесцветные ве-
вещества, жидкие или кристаллические. Оии растворимы в воде, лег-
легко образуют бесцветные кристаллические соли с кислотами. Сами
амииокарбонильные соединения (свободные основания) нестабиль-
нестабильны, дают различные реакции самоконденсации. Поэтому их получа-
получают и храпят в виде солей или ацеталей.
Для аминокарбонильпых соединений характерны реакции кар-
карбонильных соединений и аминов. Наиболее реакциоипоспособными
являются соединения с первичной аминогруппой, их используют
для синтеза гетероциклических соединений. Например, при само-
самоконденсации образуются дигидропиразипы:
Rs /О IfoN. R. ,N. R
ч:^ ' хсн2 Чс^ хсн, [0
\ + 1 — 1 I + 211*0.-'
дпгндропиралинм пирязннм
а-Лминокарбонильные соединения используются в органическом
синтезе, главным образом для получения физиологически активных
веществ.
2. р-ЛМШГОКЛРБОПИЛЬПЫЕ СОЕДИНЕНИЯ
1. Методы получения. Для получения C-аминокарбонильных сое-
соединений используют реакцию аминометилирования енолизирующе-
гося карбонильного соединения с формальдегидом и аммиаком или
аминами:
О . О II
R-C-СНя-!- )C--..O-;-H-N/ -i-IICI—>R_C-CH., — СН,— \/
\V XR2 " " С1-х R2
Обычно реакцию проводят в присутствии кислоты и получают
соответствующие соли. Эту реакцию открыл A912) и изучил К. Мап-
них, поэтому ее пазывают реакцией Манниха, а (^-амииокарбопиль-
пые соединения — основаниями Манниха. Механизм реакции точно
не установлен, по предполагают, что активной частицей амииомети-
лпровапия является гидрсксиметилпроизводное:
Нч .. /R1 .R1 пьх-,,. ., Ri
/C-O-I-H-n/ —>НОСН2 —\/ "Cll,=:\/ Х-
Н/ 4R' 4R2 Ч(^а
523
Карбонильное соединение реагирует в анионной или енольной
форме:
О (Ч>./Н «<• +/R' - О Н+ СГ
II М S_ CH2=nCR!C1 II ..^R'
R-C-CH1=== R-C = CH2 ^-~~ R-C-CH,-CH2-N^r2
В большинстве случаев в реакции Маиииха применяют вторич-
вторичные амины.
2. Физические и химические свойства, применение. р-Амипокар-
бонильные соединения и их соли — бесцветные вещества, раствори-
растворимые в воде. Для них характерны реакции карбонильных соединений
и аминов. При нагревании они расщепляются с образованием a, f>-
ненасыщенных карбонильных соединенней, например:
ОН О
R-C-CFI2CH,N+-R1 X--.R-C-CH-- СН., -|- [ H2\'RlR2J X"
a
При взаимодействии с нуклеофильпыми реагентами происходит
замещение аминогруппы. Особенно характерно это для соответст-
соответствующих четырехзамещенных аммониевых солей:
OR1 О R1
i Ч ¦ I
R —С —CH.,CH,N —R- + :Y-M<- , R — C-CH.CH., — Y-f :N — R--f MX
! X
Используют Р-аминокарбонильные соединения в органическом
синтезе, главным образом для получения физиологически активных
веществ.
Д. сс-ДИАЗО КАРБОН ИЛЬНЫЕ СОЕДИНЕНИЯ
1. Методы получения. ct-Диазокарбоннлыше соединения получают
различными методами. Можно диазсл ировать а-аминокарбопильные
соединения. Образующиеся диазоииевые соли стабилизируются
отщеплением протона:
ОН -I о
1 I - I
R-C-C-N - N 'ci---,R-C-CHN:;-;-IFCl
и
Г О I
| , UNO..
[r- С- CH,.\H;,JC1- >¦
Известно окисление гпдразопов а-днкарбонильпых соединений
(гл. ХХУП.Д.З).
Широко используется метод ацнлироваиия диазометана или дру-
других диазоалкапов ацилхлоридами в присутствии сильного органи-
органического основания:
О
R —C^ +CH2N
524
Реакцию ацилирования диазометана исследовали Ф. Арндт и
Б. Эйстерт A935).
2. Физические и химические свойства. Применение. а-Диазокар-
бонильные соединения представляют собой желтоватые жидкие или
кристаллические вещества, в воде растворяются плохо. Соединения
малостабильны, простейшие ct-диазокетоиы могут разлагаться со
взрывом.
а-Диазокарбонильные соединения можно рассматривать как
внутренние соли, молекула которых, содержит катион диазопия и
еиолят-ион. Между обеими сопряженными системами молекулы про-
происходит взаимодействие я-электронов и образуется общая сопряжен-
сопряженная система с особым распределением л-электропной плотности:
K/CVN - ^СЧ'
н н
Для а-диазокарбонильных соединений характерны реакции с
электрофильиыми реагентами и термическое, каталитическое или
фотохимическое расщепление.
При взаимодействии с сильными кислотами происходит протежи-
протежирование и отщепление азота, образуется гидроксикарбопильиое сое-
соединение и галогенкарбонилыюе соединение:
N»
Очень характерно каталитическое или фотохимическое расщепле-
расщепление, в результате которого выделяется азот и образуются карбопо-
вые кислоты или их производные:
RCOCHN', —> RCf Г.,СООН -|-\2
" Г, кат.
Происходит внутримолекулярная перегруппировка (перегруппи-
(перегруппировка Вольфа). Первичный продуктом перегруппировки является
замещенный кетен (Л. Вольф, 1912):
Н
Г, кат. |
RCOCHN;. R — С = С = О -}- Nj,
В качестве катализатора применяют соли серебра.
Предполагают, что при каталитическом или фотохимическом
расщеплении диазокарбонильные соединения превращаются в кето-
карбепы, которые претерпевают перегруппировку в кетены:
И
^^-Xj-yR-C -С О-| Х±
523
б-
q
н
-f- Н+Х ^=
О
II н +•*?
1 х
н
0'
—*- R-C—СН2Х
В кетсжарбене происходит внутримолекулярная миграция группы R
от атома углерода карбонильной группы к карбсповому углеродно-
углеродному атому.
Кетены легко реагируют с нуклеофильными реагентами и обра-
образуют карбоиовые кислоты или их производные (гл. XXVII.В.4):
и,о
' ->RCH2COOH
R-CIU С-О-
H.NR1
R'OH
>RCHoCOOR'.
Таким образом, при использовании диазометана можно удли-
удлинить цепь углеродных атомов карбоиовой кислоты на одни углерод-
углеродный атом:
/О CH.N, И,О/Л(г,О
R — Of > RCOCHX', — '-> RCH2COOH
Это превращение называется реакцией Арнд/па —¦ Эйстерта.
а-Диа;кжарбоиильные соединения широко используются в ор-
органическом синтезе.
Глава XXX
хиноны
Хинопамп называют циклические соединения с двумя двойными
связями и двумя карбонильными группами в шестнчлепиых циклах.
1. КЛАССИФИКАЦИЯ И НОМЕНКЛАТУРА
Хиноны подразделяют в зависимости от числа циклов в молекуле
и положения карбонильных групп. В основе названия лежит назва-
название соответствующего арена:
бензохиноиы
О О
:[ /л/Р счА/а
у
о ci
1,12-бСНЗОХ1|1Ю11, '
о-бензо\1ШО1[ О
I И-бе плох и но [Г, тетр;1\лор-1.! -
л-бснзохщюи бспзохмион, хлораннл
дифенохнноны
¦1,1 '-днфенох и нон
526
нафтохиноны
О
о
li
о
1,4-нафтохнпон,
а-пафтохинон
аптрахиионы
О
/О
о
1,2-нафтохи|1О||
о
О
О
и
О
А
2,И-иафтохино1| q
1,5-|1афт.)хн110н
1,2-антрахпнон
О О
] ,4-антрахшюп 0,1 О-антрахимо»
фенаитренхинопы
О
/*Ч/^/
А 2,7-фена111ре11хинон
Э.Ю-фенантренхинон
Группировку —/ у= часто называют хиноидной.
2. МЕТОДЫ ПОЛУЧЕНИЯ
Главным методом получения хипопов являются реакции окисления.
Окислению могут быть подвергнуты как арены, так и их гидрокси-
и амииопроизводиые.
1. Бензохиноны. Наиболее просто бензохиноны получают окис-
окислением гидрохинона, пирокатехина и их производных:
ОН О
! Dl A/R r\//x/oh Ч//ч//°
о
I
он о
В качестве окислителя могут быть использованы: КгСггС
FeCla, Ag2O и др.
В качестве исходных веществ могут быть использованы фенолы,
ариламипы, фенилендиамииы. При окислении производных и-фе-
527
нилендиамипа можно получить также азотсодержащие производные
бензохинона — хинонимины:
Н R r
N
[О!
л
хиноннмииы
При энергичном хлорировании анилина получают тетрахлор-
1,4-бензохинон (хлоранил):
NIF.2 О
I II
С1
сь, на х
н,о ' II ||
ci/y^ci
о
Хлорированием пирокатехина получают тетрахлор-1,2-бензохинон
(о-хлоранил).
2. Нафтохиноны. Окислением нафталина СгОя получают 1,4-наф-
тохинон:
О
"V ^/
I
о
1,4-Нафтохипоп образуется в качестве побочного продукта при
парофазном окислении нафталина в производстве фталевого анги-
ангидрида.
Для получения 1,4-нафтохинона и его производных используют
также диеновый синтез:
О О О
и и :
ч ^
м -4Н
о
I
о
1
о
3. Антрахиноны. Наиболее важным является 9,10-антрахинон,
который обычно называют просто аптрахинопом. Он образуется при
окислении антрацена, но промышленным методом является ацили-
рование бензола или его производных фталевым ангидридом с по-
528
следующей циклизацией о-бензофенонкарбоновой кислоты серной
кислотой:
О
'
AICI,
СООН
' XR
о
+ н2о
о
Для построения системы антрахинона может быть использован
диеновый синтез:
О О О
О
3. ФИЗИЧЕСКИЕ СВОЙСТВА И СТРОЕНИЕ
Хиноны являются окрашенными кристаллическими веществами:
n-бепзохиноны окрашены в желтый цвет, а «-бензохиноны — в крас-
красный; нафтохиноны, антрахиноны и фенантренхиноны — светло-жел-
светло-желтые вещества.
1. Строение молекулы бензохинона и полярность связей. Ше-
стичленпый цикл молекулы хинона не обладает такой симметрией,
как цикл бензола, ярко выражено отличие в длинах связей С—С:
| 0,12.1 нм
122"
0,1J2hm
116°
ь\
Карбонильные группы оказывают значительное электроноакцеп-
торное влияние, и на углеродных атомах появляется недостаток
электронов. Поэтому сопряженная система углеродных атомов
хинона обладает электроноакцепторными свойствами.
2. Сродство к электрону и редокс-потенциалы хинонов. Элек-
троноакцепторные свойства хинонов характеризуются сродством
529
к электрону (СЭ) и потенциалами восстановления (окислительно-
восстановительными или редокс-потенциаламп). СЭ определяют
различными методами, в том числе изучением электронных спект-
спектров поглощения комплексов с переносом заряда в растворах (см.
ниже), потенциалы восстановления определяют полярографией или
циклической вольтамнерометрией (см. Введ. 6.5). Для хинонов ха-
характерно образование стабильной обратимой редокс-системы: окис-
окисленная форма (хиной) -^. восстановленная форма (гидрохинон). Ни-
Ниже приведены структурные формулы некоторых хипопов и значение
СЭ (в эВ) и потенциала восстановления (в растворе ацетонитрила
относительно стандартного электрода — насыщенного каломельно-
каломельного электрода).
о
CI II CI CI
/\/\ . .
CI || CI CI || CN
О О О О
СЭ, эВ 1,55 1,75 1,85 2,45
?„0(.сг, В —0,94 —0,7 —0,51 -fO,01
Сродство к электрону хлоранила определено специальным точ-
точным методом, и СЭ других хинонов вычислены при сравнении с СЭ
хлоранила.
СЭ хинонов возрастает при введении электропоакцепторных
заместителей (галогены, К0.2, C==N) и уменьшается при увеличении
числа конденсированных циклов в арене. о-Хиноны более сильные
акцепторы, чем п-хиноны.
3. Электронные спектры поглощения. Хиноны являются цветны-
цветными соединениями, для них характерно поглощение в видимой обла-
области спектра. Но это поглощение малоинтенсивпо. Например, п-
бензохинон (раствор в гексане) поглощает при 246 им (ел 19 500),
276 ffm (е»340) и 435 и 458 им (ел20). Первые два максимума связа-
связаны с электронными переходами между я-орбиталями (я-кх*), а тре-
третий, малоинтенсивный, связан с переходом электрона неиоделенной
электронной ггары карбонильной группы па низшую свободную
МО (я->я*). Этот переход маловероятен вследствие незначительно-
незначительного перекрывания орбиталей.
4. Образование комплексов с переносом заряда и их спектры
поглощения. В растворах хинонов в присутствии электроподонор-
ных соединений в ряде случаев наблюдается появление окраски.
Это обусловлено появлением нового максимума поглощения в элек-
электронных спектрах, который обычно сдвинут в видимую область
спектра. Поглощение вызвано образованием комплекса с переносом
заряда (донорно-акцепторпого комплекса, я-комилекса) между хи-
ноном и электронодонорной молекулой (D:), например, полицнкли-
ческимн аренами, ариламинами, фенолами:
6+ б-
Р: -f Л(хннон) :ц± D: —> Л
Возбуждение комплекса происходит легче, чем нсходгшх ком-
компонентов, поэтому новый максимум поглощения наблюдается в ви-
530
днмой области спектра и называется максимумом переноса заряда.
При встрече донора D: и акцептора Л образуется комплекс, ко-
который существует в равновесии с исходными компонентами. Равно-
Равновесие характеризуется соответствующей константой.
В комплексе осуществляется перераспределение электронной
плотности, акцептор приобретает некоторую плотность электронов
за счет донора. Это перераспределение выражается степенью пере-
переноса заряда F). Обычно эта величина мала @,02—(),!).
При поглощении кванта света возникает возбужденное состояние
комплекса, которое имеет иное распределение электронной плотно-
плотности и во многих случаях соответствует полному переносу электрона
от донора к акцептору п образованию пары ноп-раднкалов:
6. 6-
D: , .Л -[- Лч- — •
6 Ь
D: -- - Л
пли
(V —-г 1)
Ьслн концентрации хппопа и донора в растворе высоки, выкрис-
выкристаллизовываются окрашенные комплексы.
Копстапга образования комплекса и свойства комплекса (сте-
(степень переноса заряда, энергия переноса заряда) зависят от свойств
компонентов, главным образом от СЭ акцептора и энергии иониза-
ионизации донора (ЭИ).
Чем меньше разность ЭИ—СЭ, тем легче происходит перепое
электрона и увеличивается степень переноса заряда. Возможно
одпоэлектроппое восстановление хипопа (или, в общем случае, ак-
акцептора) и одпоэлектронное окисление донора:
Г5+- 5 п
^ //
D- +-A
Образование комплекса с переносом заряда или ион-радикаль-
ион-радикальной пары (ион-радикальной соли) при встрече очень сильных доно-
доноров и акцепторов в значительной степени определяют природа раст-
растворителя, сольватационпые эффекты. Малополярные инертные раст-
растворители (углеводороды и др.) способствуют существованию комп-
комплекса с переносом заряда, а полярные — ион-радикалов.
4. ХИМИЧЕСКИЕ СВОЙСТВА
Хиноны — соединения с выраженными электроноакцепториымн
свойствами, т. е. это электрофпльные реагенты, окислители. Для
них очень характерно образование комплексов с переносом заряда.
При встрече с сильным электронодопором (восстановителем) обра-
образуются апион-радикалы (семихипоиы), а затем — гидрохипены.
В отдельных случаях наблюдается присоединение нуклеофильных
частиц к системе двойных связей хпнона пли карбонильной группе
xiriFOira. Очень характерно взаимодействие ряда хинонов с диенами
(диеновый синтез).
1. Восстановление и образование семихиноноЕ. Присоединением
эл-ктро.ча хиноны превращаются в анион радикал — анион соми-
хппопа:
531
о
г
R •"'
О
о
6
ИЛИ
Электрон может присоединяться на катоде при электролизе п
растворе или в результате воздействия сильных неорганических
либо органических восстановителей —электронодоноров.
Система аниона семихипона является сопряженной частицей, в
которой как отрицательный заряд, так и неспаренный электрон де-
локализованы. Анион семихинона способен присоединять еще один
электрон и превращаться в дианион гидрохинона:
к
к
R
Хинопы и гидрохиноны образуют обратимую редокс-снстему,
промежуточным продуктом которой является анион семихинона:
О ОН
R, Л Л
О ОН
2. Взаимодействие с нуклеофильными реагентами. Бензохино-
ны и нафтохиноны способны присоединять различные нуклеофиль-
ные реагенты. Присоединению способствует протонный катализ.
В результате образуются производные гидрохинона (в случае о-бен-
зохинона — производные пирокатехина):
:Nu;ri+
:О-Н
:О-Н
Nu+
:ОН
NuHJH'l
:О-Н
•-он
532
:KuH- :C1H, :BrH, ROH, ArNH,, RSH
В редких случаях наблюдается реакция с карбонильной группой,
например реакция гидроксиламина с бензохиноном:
О КОН N0
-! H..NOH >
\/ Ч/
Г II I
о о он
В этой реакции образуется моноксгш п-бензохпнона, который
является таутомером п-интрозофенола.
3. Диеновый синтез. Х1гноны являются активными дпенофнламп
н их часто используют для построения полициклических систем:
О
5. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ ХПНОНОВ И КРАСИТЕ III
СО СТРУКТУРНЫМИ ЭЛЕМЕНТАМИ ХПНОНОВ
пБензохинон представляет собой желтое кристаллическое вещество
со своеобразным запахом, напоминающим запах хлора; т. ал. 116СС.
Получают его окислением анилина оксидом хрома СЮ-,.
Используют n-бензохинон для получения гидрохинона в каче-
качестве промежуточного продукта для дальггенших синтезов и окисли-
окислителя органических соединений.
п-Хлоранил — желтое кристаллическое вещество со слабым свое-
своеобразным запахом; т. ил. 290 С, легко возгоняется. Получают хло-
хлорированием фенола или анилина в растворе серной кислоты.
Применяют гс-хлоранил в качестве сильного окислителя и дегид-
дегидрирующего реагента и как исходное для получения красителей.
1,4-Нафтохинон — желтое кристаллическое вещество с т пл
128'С. Получают окислением нафталина. Используют в синтезах
красителей.
Важное значение имеют некоторые производные 2-метил-1,4-наф-
тохинона. Эти соединения способствуют свертыванию крови (коа-
(коагулянты крови) и их называют витаминами К- Так, важное зпаче-
533
ние имеет витамин Кх, или фитохинон:
О
11 ,сп.
(здесь R —
= СНЯСН=С—СН2СН2СН2СНСН2СН2СН2СНСН2СН2СН2СНСН3)
9,10-Антрахинон представляет собой светло-желтое кристалли-
кристаллическое вещество с т. пл. 286 °С. Получают из бензола и фталевого
ангидрида. Используют для синтеза красителей.
Антрахиноновые красители представляют собой производные
антрахинона с электроподонорными группами (ОН, КН.,, NHR,
NR2). Например, 1,2-дигидроксиантрахипон (ализарин) является
протравным красителем, который с ионами металлов образует окра-
окрашенные внутренние комплексы. Соли алюминия образуют ярко-
красный комплекс, соли Fe (III)—фиолетовый:
о он о \н2 о кн..
/
I!
о
I !
О ХН2
дггсггероггын
ораиженьтГг
С)
алгпарнн
фполетошлгг
Амипоантрахиноиы используются в качестве дисперсных кра-
красителей и исходных для получения светостойких кубовых красите-
красителей, например индаптрона:
о
¦индантрон (кубовый синий О)
Красители со структурным элементом хинона. Известно мно-
множество практически важных красителей, в строении молекул кото-
которых формально можно разглядеть хиноиднуго структуру. К таким
красителям принадлежат хинопимиииые, дифеиилметановые, три-
фенилметаповые и ряд гетероциклических соединении (ксаптена,
акридина, оксазипа, тиазииа).
534
Хипониминные красители являются производными дифенилами-
дифениламина и содержат большую сопряженную систему, например:
СНз—N
/
СН3
N
Ч)
СГ^-СНз
^ CHi
зеленый Биндшедлера
Дифепилметаповые катиоппые красители являются производны-
производными дифенилметнлкатиона, стабилизированного электроподоиорными
аминогруппами. Для изображения их строения применяют хинонд-
ные структуры или пишут выравненные связи:
сн,|
х- +Г\
С1Г,
сн.
СНз
н
I
•Л\-
/1
сн3
сн,
\
CHj
синий Миклера
Возможен и третий способ написания — с катионным центром
на углеродном атоме метанового фрагмента, но это менее наглядно:
CHj
Трифепилметановые красители могут быть катиогпгыми и анион-
анионными и соответственно содержат в молекулах амипо- или гидрок-
сильные группы:
CHj'
5+„
СН!
г*
|^СНз
CHj
сн/
СНз
Т-сн„
сн,
мдлахиювый зеленый;
535
COO Na
Na+OH
HO
OH
бесцветный
фенолфталеин
Карбоновые кислоты
и их производные
Карбоновые кислоты R ~С\ являются производными углево-
углеводородов, содержащими в молекуле группировку СООН, называемую
карбоксильной группой.
Карбоксильная группа —¦ наиболее «окисленная» характеристи-
характеристическая (функциональная) группа. Это можно легко проследить в
ряду алкан — спирт — альдегид — кислота, получающемся при за-
замещении атомов водорода в алкане на гидроксильныг группы (уве-
(увеличивается число связей С—О):
R-CH, R-CH.OH
ОН
R-C —ОН
I
н
он
R_C—ОН
он
R -
- II,0
,Q
¦H
- Н2О
,0
R-C;'
Карбоновые кислоты подразделяются в зависимости от числа
карбоксильных групп в молекуле и природы углеводородного остат-
остатка. В классе карбоновых кислот обычно рассматривают также про-
производные карбоксильной группы (функциональные производные
карбоновых кислот и замешенные карбоновые кислоты — производ-
производные карбоновых кислот, содержащие и другие функциональные
группы).
Здесь рассмотрены следующие подгруппы:
1) монокарбоновые кислоты (насыщенные, ненасыщенные, арен-
карбоновые кислоты);
2) дикарбоновые и поликарбоновые кислоты (насыщенные, нена-
ненасыщенные, аренди- и поликарбоновые кислоты);
3) функциональные производные карбоповых кислот (ацнлгало-
гениды, ангидриды, сложные эфиры, амиды, иминоэфиры, амидн-
536
ны, тиокислоты и дитиокислоты, нероксикарбоповые кислоты, пит-
рилы, изоцианиды);
4) производные карбоновых кислот, содержащие различные функ-
циональные группы (галогеикарбоновые кислоты, гидроксикарбо-
новые кислоты, амннокарбоновые кислоты, оксокарбоновые кисло-
кислоты);
5) производные угольной кислоты, которые формально могут
быть причислены к производным карбоновых кислот.
Глава XXXI
МОНОКАРБОНОВЫЕ КИСЛОТЫ
КЛАССИФИКАЦИЯ, ИЗОМЕРИЯ И НОМЕНКЛАТУРА
Монокарбоновые кислоты обычно подразделяют в зависимости от
природы углеводородного остатка:
а) насыщенные монокарбоновые кислоты (производные алкаиов
и циклоалканов):
слн..я|,соон j (сн,)„
б) ненасыщенные монокарбоновые кислоты (производные алке-
нов, алкинов, алкадиенов и других ненасыщенных углеводородов):
С„На„-.СООН, С„Ноя_аСООН и др.;
в) аренмонокарбоновые кислоты ЛгСООН, АгСН2СООН,
АгСН-.-СНСООН.
Названия карбоновых кислот образуют от названий родона-
чальных углеводородов с тем же числом атомов углерода, считая и
атом углерода карбоксильной группы, и окончания -овая кислота.
Нумерацию начинают от атома углерода карбоксильной группы.
Если таким путем образовать название сложно, применяют второй
способ. Название карбоновой кислоты образуют от названия угле-
углеводорода, содержащего карбоксильную группу в качестве замести-
заместителя, и окончания карбоновая кислота. Первый или меньший номер
получает тот атом углерода, у которого находится карбоксильная
группа (табл. 42).
Многие карбоновые кислоты сохраняют тривиальные названия.
Отняв у карбоновой кислоты группу ОН, получают остаток кар-
О
боновой кислоты — ацильную группу R — С^ или RCO—. Назва-
Названия ацильных групп образуют из названий соответствующих карбо-
повых кислот, наиболее часто из тривиальных латинских названий,
537
Таблица 42. Примеры номенклатуры монокарбоковых кислот
Формула
I-I COO II
CII3COOH
сп3сп.,соон
сн:,сп,сп.>соон
ClbCHCOOII
i
СПзСМ2СП2СНаСООН
CfI:lCIICHXOOII
1
CH,
СИз
1
СП-.ССООИ
1
CH3
СПз(С11..LСООН
СНз1СН,MССОН
СИз yCIIJuCOOII
СП.-дСН2I0СООП
,x /COOH
/ -¦/
ч./
СН2--^СНСООН
C1L.-CCOOH
i
СНз
Название
Муравьи-
Муравьиная кислота
Уксусная
кислота
Проппоно-
ная кислота
.Масляная
кислота
11зомасля-
ная кислота
Валериа-
Валериановая кис-
кислота
Изовале-
рпаповая
к [[слота
Ппвало-
вая кислота
Капроно-
Капроновая кислота
Энамтовая
кислота
Пальмити-
Пальмитиновая кис-
кислота
Стеар[шо-
вая кислота
Акрило-
Акриловая кислота
Метакрп-
ловая кис-
кислота
с окончанием
кислота
Метановая
кислота
Этановая ки-
кислота
Пропаповая
кислота
Бутапопая
кислота
2-Метплпро-
пановая кисло-
кислота
Пентановая
кислота
3-Дк>тплбута-
повая кислота
2,2-Дпметпл-
npo/tai/овая
кислота
Гексановая
кислота
Гептановая
кислота
Гексадекано-
вая кислота
Октадекапо-
вая кислота
Пропеповая
кислота
2-MenuiiTpo-
пеповая кисло-
кислота
с окопчл[п'.ем
Мстгшкар-
бопоиая к[[С-
лота (назва-
(название по при-
применяют)
2-Метпл-
пропгшкар-
боповая-2
кислота
Цпклогек-
сапкарбоно-
вая кислота
Этплен-
карбоновая
кислота
Проиен-
карбопо-
вая-2 кисло-
кислота
538
Продолжение табл. 42
Формула
сн,ч н
С- С/
и Nzoon
СН, XOOFI
'.С- С'
и/ н
CII-,(CII.,K ДСЦ.,)-СООН
Х-Х/
н •' хя
НС--ССООН
, COOII
1 II
/ч СООН
ч II
,ч спхоон
II
Нч /СООН
г
\
Пазпанис
тривиальное
Кротопо-
ная кислота
Изокрото-
поная кис-
кислота
Олеино-
Олеиновая кислота
Прошю-
ловая кис-
кислота
Бензойная
кислота
о-Толуи-
ловая кис-
кислота
Фепнлук-
сусная кис-
кислота
Коричная
кислота
с окончанием
-овая
кислота
транс-Ьутси-
2-овая кислота
«ггг-Бутеи-2-
-овая кислота
ijiic-Октаде-
цеп-9-овая кис-
кислота
Проинновая
кислота
Фепнлэтано-
вая кислота
транс-3-фе-
нилпропеновая
кислота
с окончанием
-капбонавая
кислота
Ацетилеп-
карбононая
кислота
Беизол-
карбоновая
кислота
о-Толуол-
карбоновая
кислота
если такие имеются. Ниже приведены названия наиболее часто встре-
чагощихся ацильпых групп:
/О
/О
СН;,СН.д — С
/О
\
СН3СН2СН.г —
,0
формул
ацетил
проппонпл
бутприл
,0
изобутпрнл
I
сня
О
г—Q^ валерпл
,0
пиеалоил
539
О
СН3(СН2Iв-С^
0
II
/х/С\
стеаронл
/ X4
II
!|
it
циклогексапкар-
бонпл /ч СН2 —
f
0
\
бе и зоил
фепилацстпл
СН2 -СИ— С^ акрилоил
о
"
цинпамоил
Отнятием от карбоксильной группы водородного атома получают
ацилоксигруппу R—С<^
Остаток —СНХООН называют карбоксиметильной (оксикарбо-
нилметильной) группой.
А. НАСЫЩЕННЫЕ МОНОКАРБОНОВЫЕ КИСЛОТЫ
I. МЕТОДЫ ПОЛУЧЕНИЯ
Монокарбоповыс кислоты получают окислением органических сс.ели-
нений, гидролизом галогенпроизводных, путем превращения ме-
таллорганических соединений. Промышлснно важным методом яв-
является реакция карбонилиропання спиртов, эфиров, галогепугле-
водородов. Известны также многие специс^ичсскис методы получе-
получения карбоновых кислот.
Приведенная ниже упрощенная схема хорошо иллюстрирует ге-
генетическую связь между углеводородами, галогенпроизводнымп,
спиртами, альдегидами и карбоновыми кислотами:
RCH3
RCH2CH2R
RCH=CHR
RH
RCI
RCCb
R-ce
ROH
1. Реакции окисления. Конечным продуктом окисления многих
органических соединений являются карбоновые кислоты. Для окис-
окисления используют как кислород (воздух) в присутствии катализа-
540
торов (соли Со, Мп), так и другие неорганические (Н2О.,, CrO3,
КМпОд, МпО2, HNO;,, NOi) и органические (пероксикарбоновые
кислоты, гидропероксиды) окислители. Реакции рассмотрены в пре-
предыдущих главах.
2. Реакции гидролиза. Карбоновые кислоты обычно получают
гидролизом соединений, содержащих трихлорметнльную группу и
цнапогрупиу (нитрилы) (гл. XXXIII.К.3), иногда гидролизом слож-
сложных эфиров и амидов:
ОН
/О
R—СС1а.
НчО
ОН" или Н +
R_C-OH
он
И,О /
"ЧЖ
+Н,0
RC = N7T7rR
Н+ или ОН~
3. Металлорганический синтез. Активные металлорганические
соединения реагируют с СО2 и образуют карбоксилаты — соли кар-
карбоновых кислот:
н + х-
R_MgX+CO2 —..RCOOMgX >RCOOH + MgX2
4. Реакции карбонилирования. Оксид углерода (П) в присутст-
присутствии специальных катализаторов при повышенной температуре и
лучше под давлением реагирует со спиртами, галогенуглеводорода-
ми, простыми и сложными эфирами с образованием карбоновых
кнслот:
r _ X +СО -^Т- R —СООН
(Х=ОН, галоген, OR, OOCR1).
В качестве катализаторов применяют карбонилы кобальта
НСо(СОL и родия Rh(CO)L3, Rh(COJL,,, температура реакции
100—200 °С, давление от атмосферного @,1 МПа) до 20 МПа
B00 атм). Карбонилы родия являются более эффективными.
В присутствии катализатора происходит алкилнрование моле-
молекул оксида углерода СО (см. гл. XIV. А.4).
Алкепы в этих условиях также дают карбоповые кислоты. При
карбонилированин алкенов в присутствии водорода получаются
альдегиды (гл. II.4.3)
и,о /О
—-CIIaCHjC^
м, -О
—>¦ СНзСНоС/
Реакции карбонилирования являются промышленными способа-
способами получения ряда кислот (см. ниже).
Для получения карбоновых кислот применяется также ряд спе-
специфических методов, часть из которых уже рассмотрена (реакция
541
Арндта — Энстерта, присоединение воды к котенам, перегруппиров-
перегруппировка а-днкетоиоп, перегруппировка Фаворского). Карбоновые кисло-
кислоты образуются при гидролизе сложных эфиров и амидов, получен-
полученных специфическими реакциями (например, окисление кетонов по
Байеру — Виллигеру, с. 596, реакция Тищенко, с. 454, перегруппи-
перегруппировка Бекмана, с. 471).
2. ФИЗИЧЕСКИЕ СВОЙСТВА И СТРОЕНИЕ
Насыщенные монокарбоповые кислоты представляют собой бес-
бесцветные жидкие плн кристаллические вещества с острым своеобраз-
своеобразным запахом, высшие карбоновые кислоты (Ск,—С18) имеют слабый
запах стеарина.
Они имеют весьма высокие температуры кипения (см. табл. 43)',
что свидетельствует о значительной межмолекулярной ассоциации
Таилииа 43. Физические константы некоторых насыщенных
монокарбоновых кислот
Соединение
нсоон
СНлСООН
СН3СН,СООН
СН3СН2СН.СООН
СН3СН (СНз)СООН
СН3СН2СН2СНоСООН
СН3СН (СН3)СН2СООП
СН3(СНоI4СООН
СН3(СН,IвСООН
Т. пл., =С
8,4
16,6
—22,4
—7,9
—47
—34,5
—37,6
64
69,4
Т. кип., 'С
100,7
118,1
141,1
163,5
154,4
187
176,7
271 A3,3 кПа)
291 A3,3 кПа)
<v
1,22
1,049
0,992
0,959
0,949
0,942
0,937 A5 СС)
0,853 F2 СС)
0,847 F9 СС)
вследствие образования межмолекулярных водородных связей, при-
причем образуются как циклические димеры, так и линейные олигоме-
ры:
о... н-оч
R-Cf \C-R
No —н ••¦ о^
R R R
I I I
/с\ /с\ /СчЧ
...н-о/ ^о...н-о/ ^о-.-н-о^ чЧо-..
Электронографическое изучение карбоновых кислот показало,
что в их молекуле имеются карбонильная и гидроксильпая группы,
при этом связь С---0 длиннее, чем в кетоиах, а связь С—О короче,
чем в спиртах. Это свидетельствует о взаимодействии электронных
542
систем атома кислорода и С=О-группы:
„ 0,1085 им „
Q 0,015им I
Неподелениая пара электронов кислородного атома гпдроксиль-
нон группы взаимодействуете л-электроиами карбонильной группы,
поэтому проявляет донорный эффект (;-М). Это увеличивает по-
полярность связи О—Н, но в то же время в некоторой степени умень-
уменьшает положительный заряд но сравнению с карбонильными соеди-
соединениями на углеродном атоме. Одновременно действует электропо-
акцепторный индуктивный эффект (—/) кислородных атомов.
Следовательно, в карбоксильной группе имеются сильно поля-
поляризованные положительно водородный атом и углеродный атом,
которые являются электрофильными центрами, и неноделенные
электронные нары двух кислородных атомов, которые являются
нуклеофильными центрами. Предполагают, что кислородный атом
карбонильной группы имеет более нуклеофильный характер.
3. ХИМИЧЕСКИЕ СВОЙСТВА
Для монокарбоновых кислот характерна многосторонняя реакци-
реакционная способность. Главным образом это определяется реакциями
карбоксильной группы (отщепление н присоединение протона, ну-
клеофильиые реакции у карбонильной группы):
Н г*-б'
Свободные
радикалы
R—С — С;
Электрофилы
Нуклеофилы
Известны также реакции замещения у ос-углеродного атома.
Возможен термический разрыв связей (декарбоксилировапие, обра-
образование кетенов) и другого типа разрушения карбоксильной груп-
группы.
1. Кислотность и основность. Карбоповые кислоты обладают кис-
кислыми свойствами, что и отражено в названии. В растворах проис-
происходит ионизация с образованием сольватированпого протона и анно-
аннона — карбоксилат-иона:
R — C\^. 8+- + :SoI ^=r R — C(f + HSol
Карбоксилат-пон построен симметрично и имеет систему сопря-
сопряженных связей. Обычно его изображают проще:
-,._ илн R-COO
543
В строении карбоксилат-иона много общего со строением ннтро-
групиы.
Ниже приведены константы ионизации некоторых карбоновых
кислот в водных растворах в единицах рКа ( Н2О, 25 СС)
Кислота
нсоон
СНзСООН
СН3СН2СООН
рКа
3,75
4,76
4,87
Кислота
СНзЬССООН
\—соон
соон
рКа
5,03
4,83
4,90
СН3СН2СН2СООН 4,82 \/
Наиболее сильной из монокарбоновых кислот является муравь-
муравьиная. Введение алкильных групп уменьшает кислотность, что объ-
объясняется электронодонорным действием алкильных групп.
В водных растворах соли карбоновых кислот частично гидроли-
гидролизу ются:
RCOO~Na <- + Н2О ;zr RCOOH + Na ' ОН -
Для названия солей карбоновых кислот применяют тривиальные
латинские названия кислот. Если таких названий нет, используют
названия с суЛ^фиксом-карбоксилат (табл. 44).
Таблица 44. Примеры номенклатуры солей карбоновых кислот
Карбоксилат
НСОО-М+
СНзСОО-М4-
СНзСНгСОО-М*"
СНзСН.СНоСОО-М*-
СН3СН (СНЯ)СОО-М +
СН:,СН2СН2СИ2СОО-М +
(СНзKССОО-М +
,х СОО-М +
N/
Название co^ci'i
Формнаты
Ацетаты
Прогпюнаты
Бутмраты
Изобутираты
Валсраты
Пииалаты
Циклогексанкарбоксилаты
В кислой среде (рН<3) ионизация кислот практически не про-
происходит, а может осуществиться присоединение протона к карбо-
карбонильной группе:
R—
+ H+HSO,
O—Н
S+Uo-'"-H'HSO," 5+UO —H
R C(^ ^l R—Cv*> +
Nb—н о—н
544
Вначале образуются прочные водородные связи С—О:--'Н + ,
но при увеличении кислотности среды растет концентрация прото-
нированнон карбоновой кислоты.
Образование водородной связи и тем более иротонированной
формы значительно увеличивает положительный заряд на углерод-
углеродном атоме карбонильной группы и электрофильность этой группы.
Это можно изобразить при помощи резонансных структур:
б^-н ^ б-Н б-Н
R-C^ <->R_c/" <_>r_c/
ХО — Н NO— Н 9+~Н
В протонированной форме оба кислородных атома фактически
становятся одинаковыми:
R— C-" , B8 +28 + «"= 1)
\o«i_H8+
Основность карбоновых кислот (р/Свн + л?—6) сравнима с ос-
основностью кетонов (р/Свп + ~—6. . .—8), но значительно ниже ос-
основности простых эфиров (р/Свн+=—2...—4).
Так, в растворе L моль/л серной кислоты концентрация прото-
протонированной формы карбоновой кислоты не превышает 0,001%, а в
растворе 70%-ной серной кислоты протопировано около 50% карбо-
карбоновой кислоты.
Несмотря на малую концентрацию протонированной формы,
присутствие небольших количеств сильных кислот исключительным
образом влияет на реакционную способность карбоновых кислот
(образование сложных зфиров и др.). Очевидно, большую роль иг-
играет также образование водородной связи.
2. Реакции с нуклеофильными реагентами у атома углерода кар-
карбонильной группы. Нуклеофилы, являющиеся сильными основа-
основаниями, как правило, с карбоновыми кислотами образуют соли. Сла-
боосповные нуклеофильные реагенты могут присоединяться к карбо-
карбонильной группе и в конечном счете образуются производные кар-
карбоновых кислот, происходит ацилирование нуклеофильного реа-
реагента. Механизм присоединения подобен механизму реакций кето-
кетонов. В большинстве случаев эта реакция катализируется сильными
кислотами.
Н-Н уклеофилы (комплексные гидриды) при взаимодейст-
взаимодействии с карбоновыми кислотами выделяют водород и образуют кар-
боксилаты, которые в избытке реагента и при более жестких ус-
условиях восстанавливаются до первичных спиртов:
NaBII«
RCOOH + \aBH4 —>¦ RCOO-Na <- + H2 + BH3 >¦ RCH2OH
N-H уклеофилы (аммиак, амины, гидразин и др.) при вза-
взаимодействии с карбоновыми кислотами, как правило, образуют ам-
аммониевые карбоксилаты и только при повышенных температурах
происходит присоединение к карбонильной группе и получаются
18 № 517 545
амиды:
/О
R-Cf
О-Н уклеофилы нейтрального характера, например спир-
спирты, реагируют в присутствии кислого катализатора и образуют
сложные эфиры:
• *r HUR -г Н Л * *- R — C<2^ """-•n di
н
:'6-H
I I "" XOR' 2
:о-н н
Нуклеофилы-галогенид-ионы с карбоновыми
кислотами не реагируют. Только при использовании специальных
реагентов (РС13, РС15, PBr3, SOC12) удается осуществить реакцию и
получить ацилгалогениды. Примером может служить реакция с
SOC12, в которой вначале происходит присоединение к атому кисло-
кислорода:
о
+ и _
С1 , o-s-ci
- -х,. + O = S i=!r R-C-- —C|8~ —* R-C^ + ( S-Cl — SO2 + HC1
°.~H a noh
3. Взаимодействие с электрофильными реагентами. Карбоновые
кислоты как очень слабые нуклеофилы взаимодействуют только с
особенно сильными реагентами, присоединение происходит по кис-
кислородному атому.
Для осуществления реакции с более слабыми электрофильными
реагентами необходимо карбоксильную группу активировать, что
осуществляется превращением ее в карбоксилат-ион. Тогда можно
осуществить реакции алкилирования, ацилирования и др.
у
O-C-R'
4. Реакции у а-углеродного атома. Водородные атомы у а-угле-
рода более подвижны, чем другие водородные атомы углеродной
546
цели. Возможно его замещение, например, галогеном (гл. XXXIV,
А. 1).
5. Распад карбоксильной группы. При нагревании caiefl карбо-
новых кислот, особенно в присутствии щелочи, до 300 °С и выше об-
образуются углеводороды, происходит декарбоксилирование (гл. 1.2).
При нагревании солей Са, Ва, Th или Се насыщенных карбоно-
вых кислот в большинстве случаев образуются кетоны. В этом слу-
случае происходит внутримолекулярное ацилирование образующегося
карбаниона:
/О
R—СН2—С<^д_
^Са6'*-»- ^С-О + СаСОз
,0/ R-CH/
Декарбоксилирование происходит при окислении карбоксилат-
иона, которое осуществляется как электрохимически (гл. 1.2), так
и химически:
Свободный карбоксильный радикал (ацилоксил)' декарбокспли-
руется легче, чем карбоксилат-ион.
Декарбоксилирование происходит также при взаимодействии
серебряных или ртутных солей карбоиовых кислот с иодом или
бромом. Продуктом реакции является галогеналкан (реакция Хупс-
диккера):
RCOOAg + Br2 —
Предполагают, что промежуточным продуктом является гипога-
логенит, который распадается на радикалы:
/О ,0
R-C<f —>RC^ +-Br—>R.+CO.+ .Br-^.RBr+COi
\O — Br XO-
Реакцию можно осуществить без выделения чистых солей:
2RCOOH + HgO + 2Br2 —»¦ 2RBr + HgBr2+2CO24-H?.O
Пиролиз карбоновых кислот при 700—800°С дает кегены:
Н
Rv I /О р R-.
>c-cf -*
Ri/ ЧОН Ri
4. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ
Муравьиная кислота — бесцветная едкая жидкость с острым запа-
запахом, смешивающаяся с водой. Впервые выделена в XVII в. из крас-
красных муравьев перегонкой с водяным паром. В природе встречается
в свободном состоянии также в крапиве.
18* 517
Получают муравьиную кислоту из гидроксида натрия и оксида
углерода нагреванием под давлением:
100-10 3 СС H2SO4
Na » ОН- +СО ^ - HCOO-Na1" >¦ НСООН
Оксид углерода и спирты в присутствии алкоксидов образуют
сложные эфиры муравьиной кислоты:
RO-Na+, ЯО СС
CO+ROlt 3.0 мп, "HC00R
Муравьиная кислота в отличие от всех других карбоновых кис-
кислот содержит атом водорода у углеродного атома, формально груп-
/°
пу Н —С<^ в молекуле муравьиной кислоты можно рассматривать
как альдегидную группу. Вследствие этого муравьиная кислота
обладает восстанавливающими свойствами, при действии окислите-
окислителей превращается в СО2 и Н2О:
/Р [°I /Р
H-Cf —-HO-C(f ^tC
ХЖ ХОН
Своеобразной реакцией муравьиной кислоты является распад
на СО и Н2О при взаимодействии с концентрированной серной кис-
кислотой:
n.so4
НСООН >¦ СО + Н2О
Муравьиная кислота широко используется в органическом син-
синтезе, например для получения формамида, диметилформамида, ща-
щавелевой кислоты.
Уксусная кислота представляет собой бесцветную жидкость с
острым запахом и кислым вкусом, неограниченно смешивающуюся
с водой. Безводную уксусную кислоту называют «ледяной», так как
при 16сСоиа замерзает и образует кристаллы, подобные льду. Обыч-
Обычная уксусная кислота, содержащая 2—3% воды, замерзает при
температуре ниже 13 СС.
Уксусная кислота известна издавна. Ее разбавленные водные
растворы образуются при брожении вина. При перегонке водных
растворов получают приблизительно 80%-ную кислоту («уксусную
эссенцию»), которую применяют для пищевых целей.
Синтетическую уксусную кислоту для нужд химической промыш-
промышленности получают различными методами. Один из методов заклю-
заключается в окислении уксусного альдегида, который, в свою очередь,
получают из этилена окислением в присутствии PdCl3 или из ацети-
ацетилена:
PdCI2; CuCI2 /О Н,О; Кат.
СН2---.С1[2———- CH3-C<f «- >НС = СН
НаО; О2 . \j.j
|О2; Кат.
Кат.; Г О,; Кат,
СИ/-Ш-; СО > СН3СООН -" СН3СН2СН,СН3
548
Второй метод заключается в карбонилировании метанола. Тре-
Третий метод —¦ каталитическое окисление бутана (гл. 1.4).
Уксусную кислоту используют в качестве растворителя и как
исходное вещество для синтеза производных уксусной кислоты (аце-
тилхлорида, ацетангидрида, амидов, сложных эфиров). Соли ук-
уксусной кислоты (ацетаты) применяют в текстильной промышленности
в качестве протравителей и в синтезе как основные катализаторы.
Масляная кислота — бесцветная жидкость с острым запахом,
при большом разбавлении для нее характерен неприятный запах
старого сливочного масла и пота.
Масляная кислота встречается в природе в виде сложных эфи-
эфиров; эфиры глицерина м масляной кислоты входят в состав жиров
и сливочного масла. Масляную кислоту получают окислением бута-
нола-1 или карбонилированием пронанола-1. Используют в орга-
органическом синтезе для получения ароматных сложных эфиров.
Изовалериановая кислота — бесцветная жидкость с острым запа-
запахом, в разбавленном виде имеет своеобразный запах валери-
валерианы.
Встречается в природе в корнях валерианы. Синтетически ее
получают окислением изоамилового спирта или карбонилированием
изобутилового спирта. Используют в синтезе лекарственных ве-
веществ и ароматных эссенций.
Пальмитиновая кислота представляет собой бесцветное кристал-
кристаллическое вещество со слабым запахом стеарина, в воде не раство-
растворяется. Широко распространена в природе, в виде сложных эфиров
с глицерином входит в состав жиров.
Получают пальмитиновую кислоту обработкой жиров щелочью
(гидролиз, омыление). При этом образуются соли (пальмитаты),
после подкисления которых осаждается сама кислота.
Пальмитиновая кислота и ее производные используются в ка-
качестве поверхностно-активных веществ (моющих средств и др.).
Ее натриевая соль называется мылом.
Стеариновая кислота — бесцветное кристаллическое вещество
со слабым запахом стеарина. Ее эфиры с глицерином входят в сос-
состав жиров.
Получают стеариновую кислоту омылением жиров. Обычно об-
образуется смесь стеариновой и пальмитиновой кислот, которую мож-
можно разделить на составные части.
Стеариновую кислоту в смеси с пальмитиновой используют в
производстве свечей, их натриевые соли являются обыкновенным
мылом. В органическом синтезе стеариновую кислоту используют
для получения других поверхностно-активных веществ.
Производные пальмитиновой и стеариновой кислот принадлежат
к важным природным веществам — липидам.
Циклогексанкарбоновая кислота — бесцветное кристаллическое
вещество с запахом пота, т. пл. 31 'С, т. кип. 233СС:
Получают ее каталитическим гидрированием бензойной кислоты:
, 7соон / /хюн
Используют для синтеза капролактама (гл. XXXIII. Д. 4)'.
Б. НЕНАСЫЩЕННЫЕ МОНОКАРБОНОВЫЕ КИСЛОТЫ
1. МЕТОДЫ ПОЛУЧЕНИЯ
Для синтеза ненасыщенных монокарбоиовых кислот можно исполь-
использовать почти все методы получения насыщенных кислот. Известны
также специфические методы (карбонилирование алкинов, реакции
отщепления, реакция Виттига, Кневенагеля и др.).
1. Окисление. Ненасыщенные монокарбоновые кислоты получают
окислением ненасыщенных спиртов или альдегидов в мягких усло-
условиях:
[О]
RCH-CH(CH2)nCH2OH-
RCH =СН(СН2)„СООН
RCH = CH(CHa)nC<f" —
[О]
RC=C(CH2)nCH2OH ->¦ RC=C(CH,)nCOOH
В отдельных случаях удается окислить также алкеиы кислородом
воздуха в присутствии специальных катализаторов:
СН2=СН — СН3—^СН2^СНСООН
Кэт.
2. Карбонилирование. Алкины легко реагируют в СО в присутст-
присутствии карбонилов металлов в водной среде и образуют а, р-ненасыщен-
ные монокарбоновые кислоты (В. Реппе):
СО; Ni(COL
RC^CR1 ^RCH=C —COOH
н2о I
R;
3. Реакции отщепления. Галогенкарбоповые, гидроксикарбоио-
вые кислоты и их сложные эфиры могут быть превращены в ненасы-
ненасыщенные монокарбоновые кислоты реакциями отщепления. В основном
это относится к получению а, р-ненасыщенных карбоновых кислот:
Na + OH-
RCIICH2COOH > RCH = СНСООН (X - галоген)
X
Na+OH-
RCH- СНСООН > RC = ССООН
X i
1°
RCH2CHCOOH > RCH --СНСООН+ СН,СООП
| Кат.
ООССНа
550
4. Реакция Виттига. Алкоксикарбонилфосфораны легко реагиру-
реагируют с альдегидами и образуют сложные эфиры ненасыщенных моно-
карбоновых кислот (гл. XIII. 3):
Ri_C<f +(С6Н5)зР4"=ССООСНз->Я1С11 = ССООСН3+(СоН5)зРО
R R
Реакцию используют для синтеза природных ненасыщенных кис-
кислот.
5. Синтезы с малоновой кислотой. Альдегиды конденсируются с
малоновой кислотой в присутствии оснований (реакция Кневенаге-
ля, см. гл. XXXII. А.З).
2. ФИЗИЧЕСКИЕ СВОЙСТВА. СТРОЕНИЕ
Ненасыщенные монокарбоновые кислоты являются бесцветными
жидкостями или низкоплавкими кристаллическими веществами;
простейшие их представители растворимы в воде и имеют острый
раздражающий запах.
Особое место занимают a, |i-ненасыщенные карбоновые кисло-
кислоты, в молекулах которых имеется сопряженная система. В резуль-
результате этого происходит поляризация двойной связи:
Если двойная или тройная связь удалена от карбоксильной груп-
группы, сопряженная система не образуется.
3. ХИМИЧЕСКИЕ СВОЙСТВА
1. Кислотность. а,р-Ненасыщенные монокарбоновые кислоты, осо-
особенно с тройной связью, являются более сильными кислотами но
сравнению с насыщенными. Это объясняется образованием сопря-
сопряженной системы и изменением состояния гибридизации а-углерод-
ного атома.
Значение рКа (Н2О) некоторых ненасыщенных монокарбоновых
кислот (для сравнения: рКа (Н.2О) для СН3СН2СООН равен 4,87):
Кислота рКо (Н2О)
СН2^-СНСООН 4,26
НС^ССООН 1,84
СН3С-= ССООН 2,6
СН3СН-СНСН2СООН 4,51
2. Реакции карбоксильной группы. Ненасыщенные монокарбоно-
монокарбоновые кислоты образуют обычные функциональные производные.
3. Реакции в углеводородном остатке. Для ненасыщенных моно-
монокарбоновых кислот характерны все реакции алкенов (алкинов),
551
например реакции присоединения и полимеризации:
RCH =• СН —СООН + X — Y —* RCH — СН —СООН
X
R
-СН—СН —
R
— С^СНСООН
nRCH= CIICOOH —> RCH2 —СН —
I
COOHl- COOH-n-2
Наиболее легко нолимеризуются а, ^-ненасыщенные монокарбо-
новые кислоты, что связано с увеличенной стабильностью свобод-
свободного радикала RCH2CHCOOH ввиду делокализацни неспарепного
электрона в карбоксильной групгге.
4. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ
Акриловая кислота — бесцветная жидкость с острым запахом;
т. пл. 13'С, т. кии. 141 °С, хорошо растворима в воде. Ее получают
из ацетилена и Ni(CO), в водной среде или каталитическим окисле-
окислением нропена.
Акриловая кислота легко иолимеризуется, на чем основано ее
использование в промышленности полимерных материалов. Наибо-
Наиболее важными являются производные акриловой кислоты — слож-
сложные эфиры, амид, нитрил (гл. XXXIII. К-4).
Метакриловая кислота (а-метилакриловая кислота) — бесцвет-
бесцветная жидкость с т. кип. 160,5 С. Кислоту и ее сложные эфиры по-
получают из ацетона и цианистого водорода:
ОН
СН, —Cf +HCN'—f-СНт —С —C=-=N >
Ч:пз I -н'°
СНз
ROH
—>СН, = С —C-^N-—-->CH2--C — COOR (R = H, CH3)
| HSO г
Метакриловая кислота легко гюлимеризуется. В промышленности
полимеров применяется ее метиловый эфир.
Олеиновая кислота (г{«с-октадецен-9-овая кислота) представляет
собой маслянистую бесцветную жидкость; т. пл. 16"С, т. кип. 225 "С
A,33 кПа).
Распространена в природе, в виде сложных эфиров глицерина
входит в состав растительных масел. Например, оливковое масло
содержит 70—85% трнолеата глицерина.
Получают олеиновую кислоту гидролизом (омылением) расти-
растительных масел. Ее сложные эфиры используют в производстве лаков
и красок. Соли олеиновой кислоты являются мылами.
Линолевая кислота (ц«с-?{ис-октадекадиен-9,12-овая кислота) —
бесцветная маслянистая жидкость, т. пл.—5 С, т. кип. 230'С
552
A5 мм рт. ст.) В виде сложных эфиров глицерина входит в состав
растительных масел, например соевого и конопляного. Линолевая
кислота является также составной частью важных для жизнедея-
жизнедеятельности организма человека и животных веществ—фосфолипи-
дое, она является незаменимой жирной кислотой
Нч ХННЧ /Н
С = С' ХС -. С'
СзНц/ ХСН2/ Ч(СН2OСООН
Ненасыщенные кислоты с системой связей СН--СНСН2СН СН
организм человека сам синтезировать не может и должен получать
их готовыми с нищей, поэтому они называются незаменимыми. Эти
кислоты необходимы для построения клеточных стенок (мембран).
Липолевая кислота легко окисляется, при соприкосновении с
воздухом происходит постепенное автоокислепие (превращения в
группировке СН^ СН— СН,— СН- СН).
Сложные эфиры линолевой кислоты применяют в производстве
лаков, красок и эмалей. Эфиры на воздухе постепенно полимери-
зуются, что вызывается автоокислением, и образуют прозрачные
пленки — их называют высыхающими маслами.
Линоленоеая кисло/па (октадекатриен-9,12,15-овая кислота)
Н 7НН, /НН /Н
/ ЧСН2/ СН^/ Х(СН2OСООН
является бесцветной маслянистой жидкостью с т. пл. 11 °С. В виде
сложных эфиров глицерина содержится в льняном масле. Линоле-
новая кислота является незаменимой жирной кислотой. Сложные
эфиры липоленовой кислоты используют в производстве лаков и
красок (высыхающие масла).
В. АРЕНМОНОКАРБОНОВЫЕ КИСЛОТЫ
1. МЕТОДЫ ПОЛУЧЕНИЯ
1. Реакции окисления. Алкиларены, главным образом метиларены,
являются основным сырьем для получения арепкарбоповых кислот
с карбоксильной группой непосредственно у бензольного цикла.
Для окисления используют обычные окислители (КМпО,, СгО3)
или кислород в присутствии солей Со и Мп:
о,
АгСНз — - АгСООН
Кат.
Окисление кислородом в присутствии катализатора является сво-
боднорадикальньш процессом.
Окислить можно также метпларилкетоны, лучше всего гнпо-
хлоритами:
/О NaOCl
Аг - Cf > Ar - СООН
ЧСН3
553
2. Реакции гидролиза. Наиболее широко используют гидролиз
нитрилов, которые получают из галогенарснов:
CuCN H.2O
Аг — Вг -Лг —C^N——-"-Лг —СООН
KCN 11,0
АгСН2С1 > ЛгСП2С= N — ЛгСН.2СООН
Г / jSO4
Кроме упомянутых методов для получения арепкарбоновых кис-
кислот могут быть использованы также другие, в том числе металлорга-
нический синтез, карбонилирование и др.
2. ФИЗИЧЕСКИЕ СВОЙСТВА И СТРОЕНИЕ
Аренмонокарбоновые кислоты являются бесцветными кристалли-
кристаллическими веществами, некоторые из них имеют слабый приятный
запах. Для аренмопокарборювых кислот с карбоксильной группой
у арецового цикла характерна сопряженная система:
Карбоксильная группа действует как электроноакцепторная группа.
3. ХИМИЧЕСКИЕ СВОЙСТВА
1. Кислотность. Аренмонокарбоновые кислоты по своей кислотно-
кислотности превышает а, ^-ненасыщенные кислоты. Отдаление карбоксиль-
карбоксильной группы от бензольного цикла в некоторой степени уменьшает
кислотность.
Значение р/С„(Н.,О) некоторых аренмонокарбоновых кислот:
Кислота р/С Кислота рК.
С6Н6СООН 4,17
/ира«с-С6Н4СН = СНСООН 4,44 ^/ чу/
СООН
С6Н6СН2СООН 4,31 4,16
3,70
2. Реакции карбоксильной группы. Для аренмонокарбоновых кис-
кислот характерны все реакции насыщенных карбоновых кислот в
карбоксильной группе. Нагревание аренкарбоновых кислот в при-
присутствии медного порошка или их солей (Са, Си) выше 200 СС ведет
к декарбоксилированию:
АгСООН^-Аг —Н+СОа
554
3. Реакции в углеводородном остатке. Лренмонокарбоновые кис-
кислоты реагируют с электрофнльпыми реагентами и подобно аренам
дают продукты электрофилыюго замещения (нитрования, сульфиро-
сульфирования, галогеннровапня). Как правило, заместитель вступает в
л*-положение:
/у ,/С00Н
E-NO2, SO3H 11 др.
Е
Особое место занимают арилуксуспые кислоты АгСН2СООН,
которые имеют активированную метиленовую группу благодаря
влиянию групп Аг и СООН. Легко происходят галогеппровапие в
группе СН2 и различные реакции конденсации. Для арилуксусных
кислот характерно образование карбапионных частиц. Например,
с металлорганическнми соединениями R —М образуются производ-
производные АгСНСОО"М+ (Д. Иванов, 1931), которые активно реагируют
с различными С-электрофилами (например, альдегидами, кетонами,
ангидридами кислот).
4. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ
Бензойная кислота — бесцветное кристаллическое вещество с т. пл.
122 °С, хорошо кристаллизуется, легко возгоняется. Впервые была
пыделена из природных продуктов — бензойной смолы. В промыш-
промышленности ее получают окислением толуола.
БепзоЙЕ!ую кислоту используют как исходрюе вещество для по-
получения красителей, душистых и лекарственных веществ. Она обла-
обладает бактерицидными свойствами. Бензоат натрия используют при
консервировании пищевых продуктов.
Фенилуксусная кислота представляет собой бесцветное кристал-
кристаллическое вещество со сладковатым навязчивым запахом; т. пл.
77°С. Получают ее гидролизом бензилцианида C6Hf)CHjCN или кар-
бонилированием бепзилхлорида С6Н.-,СН2С1. Используют в органи-
органическом синтезе для получения душистых и лекарственных веществ.
Коричная кислота (траяс-З-фепилпропеновая кислота) — бес-
бесцветное кристаллическое вещество, с т. пл. 133°С, имеет слабый
запах.
В виде сложных эфиров содержится в эфирных маслах, смолах
и бальзамах. Синтетически ее получают из бензальдегида и уксус-
уксусного ангидрида нагреванием в присутствии ацетата натрия (У. Пер
кин, 1868). Механизм реакции изложен в гл. XXXIII. Б.З.
.О NaOOCCH.,
С0Н5—Cf +(CH3COJO |-С6Н6СН = СНСООН+СН3СООН
Используют коричную кислоту в органическом синтезе для по-
получения душистых и лекарственных веществ.
555
Глава XXXII
ДИКАРБОНОВЫЕ
И ПОЛИКАРБОНОВЫЕ КИСЛОТЫ
КЛАССИФИКАЦИЯ, ИЗОМЕРИЯ И НОМЕНКЛАТУРА
Ди- и поликарбоновые кислоты подразделяют в зависимости от
типа углеводорода. От алканов и циклоалканов производят насы-
щенЕ1ые ди- и поликарбоповые кислоты, от алкепов, алкинов и ал-
кадиенов —¦ ненасыщенные, от аренов — арендикарбоновые и арен-
поликарбоновые кислоты. Номенклатура аналогична номенклатуре
монокарбоповых кислот. Используют также тривиальные назва-
названия (табл. 45).
Таблица 45. Примеры номенклатуры ди- и поликарбоновых кислот
Структурная формула
ноос—соон
НООССН2СООН
НООССН2СН2СООН
НООССНоСН.СН.СООН
НООССН2СН.;СН;СН2СООН
н соон
с7
н 'чсоон
н соон
С '
¦
носе ' чн
сооп
СН2 -С —СII,СООН
ноосс . ссоон
/Л /СООН
\ -\
Название
тривиальное
Щавеле-
Щавелевая кислота
Малоно-
пая кислота
Янтарная
кислота
Глутаро-
вая кислота
Лдипимо-
вая кислота
Малепно-
вая кислота
Фумаро-
вая кислота
Итакоио-
вая кислота
Фталевая
кислота
с окончанием
¦овая
кислота
Этандио-
вая кислота
Пропан-
дповая кис-
кислота
Бутандио-
вая кислота
Пентан-
диовая кис-
кислота
Гексан-
диовая кис-
кислота
цис-Ъутеи-
диовая кис-
кислота
транс-Ъу-
тендиовая
кислота
Бутпндно-
вая кислота
с окончанием
-карпонобая
кислота
Метан дик а рбо-
новая кислота
Этандпкарбоно-
вая-1,2 кислота
Проиандпкарбо-
новая-1,3 кислота
Бутандикарбо-
новая-1,4 кислота
Чыг-Этилендн-
карбонопая-1,2
кислота
тра«с-Этиленди-
карбоновая-1,2
кислота
Пропен-2-дикар-
боновая-1,2 кисло-
кислота
Ацетнленднкар-
боновая кислота
1,2-Бензолди-
карбоиовэя кис-
кислота
556
Продолжение табл. 45
СтруктурЕ1ая формула
*
соон
"V
сс
ноос 7
ноос
ноос
ноос
он
/ч ,соон
/ ,СООН
" II
, .соон
7II
Название
трипнальное
Изофтале-
вая кислота
Терефта-
левая кис-
кислота
Трнмеллн-
товая кис-
кислота
Пнромел-
лнтовая
кислота
С OKO!r4aF[IIOM
-оеая
кислота
с окончанием
-карионовая
кислота
1,3-Бепзолдг;-
карбонозая кис-
кислота
1,4-Бензолди-
карбоновая кис-
кислота
1,2,4-Бекзолтри-
карбоновая кис-
кислота
1,2,4,5-Бепзол-
тетракарбоновая
кислота
А. НАСЫЩЕННЫЕ ДИКАРБОНОВЫЕ КИСЛОТЫ
1. Методы получения. Для синтеза дикарбоновых кислот могут
быть использованы многие из известных методов получения моно-
карбоновых кислот, например реакции окисления, гидролиз дини-
трилов, карбонилирование диолов:
[О]
НОСН2(С[-Г,)„СН.,ОН - ' НООС(СН2)„СООН
i СН2
I \ [О]
(СН2)„ у С = О —»¦ НООС(СН2)„СН2СООН
1 ол/
N ¦: С(СН.,)„С ^ К —г-- J ЮОС(СНЭ)„СООН
НСо(СО),
НО(СН2)„ОН ——- НООС(СН2)„СООН
Применяются также специфические методы, которые будут рассмот-
рассмотрены отдельно.
2. Химические свойства. Насыщенные дикарбоновые кислоты яв-
являются двухосновными кислотами, при ионизации образуют моно-
моноанион и дианион и, соответственно, два ряда солей. Кислотность
первых представителей ряда значительно выше, чем монокарбоно-
557
вых кислот. При этом первая константа ионизации резко отличает-
отличается от второй, особенно для щавелевой и малоповой кислот. Значении
р/Са(Н2О) некоторых насыщенных днкарбоповых кислот:
Кислота pKai pKa2 Г>Ка2-рКаЛ
Щапелепая 1,27 4,27 3,0
Малоновая 2,86 5,70 2,84
Янтарная 4,21 5,6-1 1,43
Глутарояая 4,34 5,27 0,93
Адиннновая 4,41 5,28 0,87
Наиболее сильной кислотой является щавелевая, кислотность
глутаровой и адипииовой незначительно отличается от кислотности
уксусной кислоты (р/Са--4,76). Это связано с уменьшением взаимно-
взаимного индуктивного влияния двух карбоксильных групп при их отда-
отдалении.
3. Важнейшие представители. Щавелевая кислота — бесцветное
кристаллическое вещество с т. ил. 189 °С, из воды кристаллизуется
в виде дигидрата с т. пл. 101 °С. Растворяется в воде и спиртах,
трудно растворима в углеводородах.
В виде солей встречается в природных продуктах. Специфичес-
Специфическим методом получения является нагревание формиата натрия, в
результате чего образуется оксалат натрия:
4оо-42о °с COO-Na +
HCOO-Na+ »| -|-Н2
COO-N a-i-
Специфическими свойствами щавелевой кислоты являются раз-
разложение концентрированной серной кислотой и окисление:
НООССООН —-' СО + СО2 + Н2О
[О]
НООССООН -^
Используют щавелевую кислоту в качестве протравы при
крашении тканей и как аналитический реагент. Возможно ее
использование для получения полимеров — полиэфиров (см.
гл. XXXIII.B.4).
Малоновая кислота — бесцветное кристаллическое вещество;
т. пл. 134 Т; (с разложением), растворяется в воде. Получают из
хлоруксусной кислоты:
KCN И„О
CICH2COO-Na I- *N = CCH2COO-Na+ — - HOOCCH2GOOH
H2SO4
Малоновая кислота легко декарбоксилируется при нагревании
выше 133—135°С или кипячении водных растворов в присутствии
кислот:
НООССН2СООН -'- СН3СООН-|-СО2
Малоновая кислота содержит особую группу СН2 — активную
метиленовую группу. В присутствии оснований малоновая кислота
способна образовать не только дианион, но и в небольших коли-
558
чествах трианион за счет ионизации активной метиленовой группы.
Поэтому возможЕ1ы реакции малоновой кислоты с альдегидами и
кетопами типа альдольной конденсации:
/СОО-НВ+ BPI + -0 СОО-НВ +
d 6r/> i ги I I н+х~
СОО-НВ+ н
!
н
/СООН t°
C< —>RCH = CHCOOH+CO2
ЧСООН
Реакцию используют для получения ненасыщенных карбоновых
кислот (Э. Кневенагель, 1896).
При нагревании малоновой кислоты с Р2О5 получается своеобраз-
своеобразное соединение: недооксид углерода С3О., (см. гл. XXVII. В).
Малоновую кислоту используют в органическом синтезе для
получения аминокарбоновых кислот, лекарственных веществ. Ши-
Широко применяется диэтиловый эфир малоновой кислоты (гл. XXXIII
В.4).
Янтарная кислота представляет собой бесцветное кристалличес-
кристаллическое вещество с т. пл. 183°С, растворяется в воде и спиртах.
Янтарная кислота найдена в природных продуктах (буром угле,
янтаре). В промышленности ее получают гидрированием малеино-
вой кислоты и карбонилировапием этиленгликоля:
НССООН н2 СН2СООН нсо(СОL СН2ОН
НССООН Ni СП2СООН со СН2ОН
Для янтарной кислоты характерны все реакции карбоксильной
группы. Легко образуются циклические производные:
КН., СН2СООН 230-240 '
I
СН2СООН
иыид янтарной ангидрид
кислоты, сук- интарноЛ
цииимид кислоты, сук-
цинангидрид
Молекула янтарной кислоты содержит две активированные ме-
тиленовые группы. Их активация уступает активации метиленовой
группы в малонозой кислоте, но достаточна для осуществления
реакций галогенирования, конденсации с альдегидами и ангидри-
ангидридами кислот в более жестких условиях.
Янтарная тслот'а и ее сложтрьге эфиры широко используются в
органическом синтезе и птептмерргой промышленности.
Глутаровая кислота — бесцветное кристаллическое вещество с
т. пл. 97,5°С, легко растворяется в воде и спиртах.
Встречается в природных продуктах, например в свекловичном
соке. Синтетически получают окислением циклопентанона разбав-
разбавленной азотной кислотой.
559
Глутаровую кислоту используют в органическом синтезе для
получения некоторых амипокарбоповых кислот.
Адипиновая кислота представляет собой бесцветное кристалли-
кристаллическое вещество с т. пл. 149°С, мало растворима в воде, лучше —
в спиртах.
В промышленности ее получают различными методами, напри-
например окислением циклогексапона разбавленной азотной кислотой,
карбонилированием тетрагидрофурана или бутандиола-1,4, гидро-
гидролизом динитрила адининовой кислоты.
Адипиновая кислота является исходным веществом для произ-
производства синтетического волокна — найлона. В меньших количествах
она используется в органическом синтезе. Например, при нагрева-
нагревании ее кальциевой соли образуется циклопентанон (ср. гл. XXXI.
А. 3.5):
СН2 С\ Н2С \
U cJ-Ca2+-^ И± ^О + СаСОз
Б. НЕНАСЫЩЕННЫЕ ДИКАРБОНОВЫЕ КИСЛОТЫ
1. Методы получения. Для получения ненасыщенных дикарбоно-
вых кислот чаще всего используют методы введения двойной или
тройной связи в молекулу насыщенной дикарбоновой кислоты или
введения карбоксильных групп в ненасыщенное соединение:
нооссн—сн2соон —->¦ нооссн = снсоон
I
Вг
НООССН —СНСООН —у НООСС se ССООН
Вг Вг
СНзСН = СНСН3 ^* НООССН = СНСООН
Некоторые ненасыщенные дикарбоновые кислоты встречаются в
природе.
2. Химические свойства. Ненасыщенные дикарбоновые кислоты
являются более сильными кислотами по сравнению с насыщенными,
так как взаимное влияние двух карбоксильных групп но системе
л-связей переносится сильнее.
Следует обратить внимание на значения второй константы ио-
ионизации малеиновой и итаконовой кислот. Низкая кислотность моно-
аниопа свидетельствует о сильном внутримолекулярном взаимодей-
взаимодействии неионизировашюй и ионизированной карбоксильных групп.
Значение рЛ^, (Н2О) некоторых ненасыщенных дикарбоновых кис-
кислот:
Кислота
Малеиновая
Фумаровая
Итакоиовая
Ацетнлендпкарбоиовая
Р«а, 1.
1,92
3,02
3,82
1,73
Р*Ч а
6,23
4,32
5,66
4,40
Р*а, 2-Р
4,31
1,30
1,82
2,67
560
3. Важнейшие представители. Малеиновая кислота — бесцвет-
бесцветное кристаллическое вещество с т. пл. 13ГС, легко растворяется в
воде и спиртах.
/Малеиновая кислота образуется при растворении в воде ангид-
ангидрида малеиновой кислоты —¦ продукта парофазного каталитичес-
каталитического окисления бензола, бутенов и фурфурола (гл. XXXIII. Б. 4),
и ее выделяют в чистом виде. При нагревании малеиновая кислота
отщепляет молекулу воды и превращается в чистый малешювый
ангидрид. Таким образом, малеиновая кислота является промежу-
промежуточным продуктом промышленного производства малеинового ан-
ангидрида:
.о н. .соон
^ н2о Ч/
II /О >
С-С/ <°:-н,о
^ / ХСООН
Ее используют для промышленного получения янтарной, яблоч-
яблочной и винной кислот.
При кипячении водных растворов малеиновой кислоты, содержа-
содержащих НС1, образуется фумаровая кислота.
Фумаровая кислота — бесцветное кристаллическое вещество,
плавящееся при 287 °С в запаянном капилляре, обычно возгоняется
без плавления, трудно растворима в воде.
В природе фумаровая кислота встречается во многих растениях,
особенно в грибах. Синтетически ее получают термической изомери-
изомеризацией малеиновой кислоты. При облучении растворов фумаровой
или малеиновой кислот УФ светом образуется равновесная смесь
обоих кислот — происходит фотохимическая изомеризация:
Нч .СООН Н. ,СООН
НОО(У ХН Ы/ ХСООН
25% 75%
Итаконовая кислота (пропен-2-дикарбоновая-1,2 кислота)
представляет собой бесцветное кристаллическое вещество, с т. пл.
167°С, растворима в воде. Образуется при нагревании лимонной
кислоты. В промышленности ее получают ферментативным синтезом
из глюкозы в присутствии плесневых грибов Aspergillus terreus.
Используют итаконовую кислоту в производстве синтетических
смол, волокон и поверхностно-активных веществ.
Ацетилендикарбоновая кислота — бесцветное кристаллическое
вещество с т. пл. 178°С, из воды кристаллизуется в виде дигидрата
с т. нл. 173°С. Получают ее из дибромянтарной кислоты и щелочи.
Ацетнлендикарбоновую кислоту применяют в синтезах благодаря
наличию активной тройной связи (реакции присоединения нуклео-
фильных реагентов, диеновый синтез). При кипячении водных раст-
растворов происходит декарбоксилирование и образуется пропиоловая
561
кислота:
100 °С
НООСС = ССООН -^- НС = ССООН + СО2
Широкое применение в синтезах как активный диенофил имеет
диметиловый эфир ацетилендикарбоновой кислоты:
СООСН3 Rv уч /СООСНз
+ 01
- СООСНз
В. АРЕНДИКАРБОНОВЫЕ
И АРЕНПОЛИКАРБОНОВЫЕ КИСЛОТЫ
1. Методы получения. Главными методами получения арендикар-
боновых и аренполикарбоновых кислот являются реакции окисле-
окисления. Методы основаны на каталитическом жидкофазном или паро-
фазном окислении диметил-, триметил- или тетраметиларенов:
соон
соон
СНз СООН
2. Химические свойства. Арепдикарбоповые и поликарбоновые
кислоты являются более сильными, чем монокарбоновые. Ниже при-
приведены значения р/Са(Н2О) некоторых аренкарбоновых кислот:
Кислота
Бензойная
Фталевая
Изофталевая
Терефталевая
Трнмеллитовая
Пиромеллнтовая
р/ч'а, 1
4,17
2,95
3,46
3,51
2,52
1,92
5
4
4
3
2
,41
,46
,82
,84
,87
р а,
5,2
4,49
5,63
3. Важнейшие представители. Фталевая кислота —• бесцветное
кристаллическое вещество с т. пл. 200°С (происходит ангидриди-
зация), мало растворяется в воде.
В промышленности ее получают окислением нафталина или о-
ксилота в паро-газовой фазе над катализатором, содержащим V2O5.
В условиях реакции образуется ангидрид фталевой кислоты, кото-
который легко присоединяет воду:
О
У
,СЧ /ч СООН
Кат. I U / 2&1РС; -Н,О _ _
чсоон
и
о
Широкое практическое применение имеют фталевый ангидрид
и сложные эфиры фталевой кислоты —фталаты.
Терефталевая кислота представляет собой бесцветное кристал-
кристаллическое вещество; при 300 °С она возгоняется, в запаянном капил-
капилляре плавится при 425СС. Мало растворима в воде и органических
растворителях.
Главным промышленным методом получения терефталевой кис-
кислоты является каталитическое окисление л-ксилола кислородом
воздуха в жидкой фазе:
СН3
Кат.
Терефталевая кислота — крупнотоннажный промышленный про-
продукт, используют ее в производстве синтетического волокна — лав-
лавсана (терилена) (гл. XXXIII. В.4).
Тримеллшповая кислота является бесцветным кристалличе-
кристаллическим веществом, плавится при 224—225 °С с образованием ангид-
ангидрида (ангидридизация). Растворяется в воде. Получают ее ката-
каталитическим окислением 1,2,4-триметилбензола (псевдокумола).
Ангидрид тримеллитовой кислоты применяют для получения
термостойких полимеров.
Пиромеллшповая кислота — бесцветное кристаллическое вещест-
вещество с т. пл. 275 СС (ангидридизация), хорошо растворима в воде.
Получают ее каталитическим окислением 1,2,4,5-тетрамети л бен-
бензола (дурола) в паро-газовой или жидкой фазе. В газовой фазе об-
образуется диангидрид:
О О
II II
НООС /ч 7С00Н ,СЧ ~ ,С
II II
О О
Диангпдрид пиромеллитовой кислоты используют для получе-
получения термостойких полимеров (гл. XXXIII. Д. 4).
Глава XXXIII
ФУНКЦИОНАЛЬНЫЕ ПРОИЗВОДНЫЕ
КАРБОНОВЫХ КИСЛОТ
КЛАССИФИКАЦИЯ, ИЗОМЕРИЯ И НОМЕНКЛАТУРА
Функциональные производные карбоиовых кислот подразделяют
в зависимости от того, какими другими атомами или группами за-
замещены кислородные атомы карбоксильной группы.
663
Номенклатура соединений основана на названиях соответству-
соответствующих карбоновых кислот или ацильных групп, к которым присоеди-
присоединено обозначение соответствующего функционального производного.
Ниже приведены главные функциональные производные карбо-
карбоновых кислот и даны примеры номенклатуры.
Ацилгалогениды (галогенангидриды карбоновых кислот)
,0
R —С--7 (X = F, C1, Вг, I)
ЧХ
О
СИ, —
/О /О
У СцПь — С?
4F ' ЧС1
ацетилфторпд бензоилхлорид
циклогегссанкарбонил-
бромпд
Ангидриды карбоновых кислот
OR' О
R-Cwc
О
о
о
сн3
сн3/
ангидрид уксусной
кислоты, ацетангидрид
о
ангидрид бензойной
кислоты
сйн5
,с
N5;/ ч С /
6
ангидрид фталепоA
кислоты, фталевьгн
ангидрид
Сложные эфиры карбоновых кислот, лактоиы
О
R —
,0
—R'
,COOR
Y< X
XCOOR
О
H-Cf
Ч0—СНз
метилопый :>фнр
M>'puBbimoii кислоты,
метилфорь:1:ат
,0
I |
Н2С —О
fj-пропиолактон,
3-пропанолид
с,н,-с^
ЧО-С2НГ,
этплоньгй эфир бензойной
кислоты, этилбеизоат
0
^бутиролактон,
та нолид
4-бу-
,СООС2Н5
н,с/
ХСООС2Н5
дизтиловый эфир малоионоЛ
кислоты, диэтилмалонат
сн.
н2с/
!
Н2С
О
I
О
сн2
в-валеролактон.
5-пентанолид
564
Ортоэфиры карбоновых кислот
R — С—OR* Н —С(ОС2Н5K этиловый эфир ортомуравьнной кислоты
OR'
Амиды и имиды карбоновых кислот, лактамы
R1
/О
¦ N-R
^О
о
О
,0
,сн3
сн.
II
о
—н
амид муравьи- диметиламид уксусной имид фталсрой кисто-
иои кислоты, кислоты, N, .N-днметн- ты, фталимид
формамид лацстамид
.0
7/
Н2С—С7/
I I
H2C-N.
Н.С.-''
i
CH..-N
си2 ,
I I
Н2С. ^Ч^
н сн2
и
ииолактам v-^УТНролактам, 6-валеролактам,
2-пирролидон 2-ииперидон
Гидразиды и азиды карбоновых кислот, гидроксамовые кис"
лоты
о
/О 1 .О .0
/ R1 /C—N —R R—С- R —С7/
4N — N{ Y/ | NN3 ЧК—ОН
| -R2 С —Х-Ri |
H !| H
О
О
сн,-с^
4NHNH2
гндразид уксусной
кислоты
1
О
фталгндразид
беизонлазид
,0
"NHOH
бснзогидроксамовая
кислота
565
Имидоэфиры карбоновых кислот и амидины
/N-R1
Г ,NH-Ril
R-Cf X-
[+ -1
/NH2
г и с// \t-\-
XOCH3 J
N — R1
/R2
XD3
,NH
этиловый эфир аце-
ТИМИДОВО11 КИСЛОТЫ,
этилацетимидат
.NH-R1'
-Ccf R2
[
хлорид мепглбензимидиния
x-
/NH2
cn3—c<f ci-
xnhJ
хлорид ацета.мидиння
Тиокислоты и дитиокислоты
"NH2
формамидин
/
О-Н
R
4S—Н
S-кислота
/О
4SH
S-тиоуксуснаи
кислота
R-C'
XS
О-кислота
CH3CH2CH2C
-H
//¦
чон
бутан-О-тновая кислота,
О-тиомасляпая кислота
СИ3СН2СН2СН2С^
S\SH
пентаидитионая кислота,
дитиокалерпановая кис-
кислота
Пероксикарбоновые кислоты и ацилпероксиды
О
ЧО-ОН
/О О
XC-R
/
/.О /О О
^ CeH3-Cf
NOOH ¦ ХО —0
пероксиуксусная
(перуксусиая)
кислота
566
бепзоилпероксид
фталоилпероксид
Нитрилы (цианиды)
СН3—C =
ацетонитрил,
этанонитрил
N —- CCH2CH2CH2CH2C -=N
адиподинитрил, гександииитрил
4C=N
CH3CH2CH2C =
бутироиитрнл, бута-
ионитрил, пропил-
цианид
фталодинитрил,
1,2-дшшанобеи-
зол
Изоцианиды (изонитрилы)
,NC
R-NC Y<
CN — CH2CH2CH2 — NC
1,3-диизоиианопронан
CH3 — NC
метилизо-
циаиид
CfiH5-NC
феииллпо-
циаиид
СН3
. изовалеропитрил, 3 -мс-
-мстил бутанонитрил
циклогексанкарбонитрил,
циклогексилцианид
А. АЦИЛГАЛОГЕНИДЫ
1. МЕТОДЫ ПОЛУЧЕНИЯ
При взаимодействии карбоновых кислот или их солей с галогеии-
дами серы (SOC12, см. гл. XXXI. А.З, SO2C12) и фосфора (РС13,
РОС13, РС15, РВг6) или фосгеном СОС12 образуются ацилгалоге-
ниды:
R-
хон
+SOC12
С1
Так получают хлориды и бромиды. Иодиды получают из ацилхлори-
дов реакцией замещения хлора на иод. Аналогично получают ацил-
ацилфториды. Фториды перфторкарбоновых кислот образуются при
электрохимическом фторировании кислот.
Простейший ацилхлорид — формилхлорид HOOCI — разлага-
разлагается на СО и НС1, формилфторид HCOF более стабилен.
2. ФИЗИЧЕСКИЕ СВОЙСТВА И СТРОЕНИЕ
Ацилгалогениды представляют собой бесцветные жидкие или крис-
кристаллические вещества с острым запахом, легколетучие — на возду-
воздухе «дымят». Простейшие ацилфториды газообразны.
В воде растворяются мало, но быстро реагируют.
567
ь'~ Для ацнлгалогенидов характерна большая поляр-
.V " ность связей, что вызвано электроноакцепторпым деп-
R~ \-;б- ствием атома галогена. На углеродном атоме карбо-
^¦' пильной группы сильно занижена электронная пло-
плотность. Лцилгалогениды обладают сильными электрофильнымп
свойствами.
3. ХИМИЧЕСКИЕ СВОЙСТВА
1. Реакции нуклеофильного замещения. Ацилгалогеииды легко реа-
реагируют с различными нуклеофильными реагентами:
в.- 0е'- | /О
R-C^ t,, +:NuH —у R-C — Cl —> R-C-^ +HC1
4CI6 - | '"-Nil:
о-м+
61 /О6 - I .О
R—С/ ... +:Y-M+ —* R-C-Cl —> R-GT +М"'С|-
Y
Реакции осуществляются почти с любым нуклеофилом (сравните
с реакциями карбонильной группы). Лцилгалогениды используются
для введения в молекулу органического соединения ацильнои груп-
группы, они являются ацилирующими реагентами.
В случае слабых нуклеофильных реагентов (например, аренов)
ацнлгалогенид должен быть активирован, что достигается присутст-
присутствием кислот Льюиса (А1ХЯ, ВХ3, SnX4, SbX5 и Др.). Реакция ацил-
галогенида с кислотой Льюиса приводит к донорно-акцепторному
взаимодействию (координации), в результате чего увеличивается
положительный заряд на углеродном атоме:
'¦ ^ С1:° ~ ^CII^-AICI,
Координация осуществляется как с кислородным атомом, так и с
атомом галогена. Предполагают, что в отдельных случаях может
образоваться ион ацилия RCO+:
6+ .О6'"
R—С^ --1 R-C-O[A1C141-
•С1-*А1С1а
Существование иона ацилия доказано получением кристалли-
кристаллического [CHnCO]"SbF6' и данными его рентгеноструктурного ана-
анализа:
568
Углеродный атом в ионе ацилия находится в состоянии sp-гиб-
ридизации, поэтому в структуре необходимо писать полярную трой-
тройную связь.
Ацилирование в присутствии кислот Льюиса широко применя-
применяется для получения ароматических кетонов (реакция Фриделя —
Крафтса).
2. Реакции а-водородного атома. Если в ацилгалогенидах име-
имеется а-водородпый атом, возможны реакции енолизации и отщепле-
отщепления галогеноводорода с образованием кетенов. Это вызвано повы-
повышенной полярностью связи С—Н
i
.i
1 Я'" «
)с = с=о + нв+с1-
нв+
Ионизация ацилгалогенида и отщепление галогеповодорода про-
происходит при воздействии сильных органических оснований, напри-
например третичных аминов.
4. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ
Ацетилхлорид — бесцветная жидкость с острым запахом, на возду-
воздухе дымит, кипит при 51,8°С.
Получают его взаимодействием ацетата натрия с сульфурилхло-
ридом (SO2C12) или из уксусной кислоты и фосгена в присутствии
MgCl,:
СН3СООН+СОС12 > СЫ3СОС1-1-СО2
t
Используют в органическом синтезе для введения ацетильной
группы.
Бензоилхлорид представляет собой бесцветную жидкость со свой-
свойствами лакриматора, кипящую при 197 °С.
Получают его из бензойной кислоты и фосгена, бензальдегида
и хлора (гл. XXVII. Г.4) или при неполном гидролизе трихлорме-
тилбензола (бензотрихлорида):
Fed.,
CHC > CeH5COCl-i-2HCl
Используют для введения бензоильной группы (бензоилирование).
569
Б. АНГИДРИДЫ КАРБОНОВЫХ КИСЛОТ
1. МЕТОДЫ ПОЛУЧЕНИЯ
Ангидриды получают из карбоновых кислот при их термической
ангидридизации или взаимодействием с сильными водоотнимающими
реагентами (Р2О5, ангидрид трифторуксусной кислоты, карбоди-
имиды). При нагревании легче всего получаются циклические ан-
ангидриды дикарбоновых кислот с пяти-, шести- и реже семичленным
циклом.
Ангидриды образуются также при взаимодействии ацилхлори-
дов с карбоновыми кислотами или их солями (гл. XXXIII. А.З) и
при реакции карбоновых кислот с кетенами (гл. XXVII. В.4).
Для некоторых ангидридов известны специфические методы полу-
получения:
О R
Р°5 _> R- С. ,СЧ -'-2НРО3
2R-C<f
Ч0Н
О R
II '
R —Сч ,СЧ +2CF3COOH
(CFaC0J'0 " !
7С00Н ,„ ,C<f
Y< —* \( )О
ХСООН N/
Первый представитель ряда — ангидрид муравьиной кислоты —¦
нестабилен.
2. ФИЗИЧЕСКИЕ СВОЙСТВА И СТРОЕНИЕ
Ангидриды карбоновых кислот являются бесцветными жидкостями
или кристаллическими веществами. Первые представители гомоло-
гомологического ряда имеют острый раздражающий запах. В воде раст-
растворяются мало и с ней постепенно реагируют.
Связи в молекулах ангидридов карбоиовых кислот сильно по-
поляризованы, но меньше, чем в случае ацилгалогенидов. По срав-
сравнению с карбоновыми кислотами ангидриды имеют более электро-
фильный характер, так как в этом случае на один атом кислорода
приходятся две ацильные группы:
3. ХИМИЧЕСКИЕ СВОЙСТВА
1. Реакции нуклеофильного замещения. Ангидриды карбоновых
кислот, подобно ацилгалогенидам, легко реагируют с различными
нуклеофильными реагентами. Скорость реакции меньше, чем в слу-
570
чае ацилгалогенидов:
О6" R O
HNu
,Ч /С< ,С—Nu:
у/ \o + :N"H —* Y' >O —> Y/
N / ЧС/ ЧСООП
С
||
При реакции ангидридов карбоновых кислот с нуклеофильными
реагентами выделяется молекула карбонопой кислоты, в случае
циклических ангидридов получаются монозамещенные производные
дикарбоновой кислоты.
Ангидриды карбоновых кислот, так же как ацилгалогениды,
активируются кислотами Льюиса и являются хорошими ацилирую-
щими реагентами.
2. Реакции а-водородного атома. Связь С—Н в ос-положении в
молекулах ангидридов карбоновых кислот, так же как в ацилгало-
генидах и карбонильных соединениях, поляризована.
В присутствии оснований (соли карбоновых кислот, третичные
амины) ангидриды карбоновых кислот конденсируются с аренкар-
бальдегидами, образуя ненасыщенные аренкарбоновые кислоты
(реакция Перкина). Механизм реакции подобен альдолыюй конден-
конденсации:
. О СНз <^О5~ СНз
Н3—С. XV + Na+~OOCCHa -у—>¦ СН?^—С XI- + HOOCCHj
о ^о Wa+ vo *^о.
\Н N;
ОН
Аг—С—СГ
Н
ЛО5+
Г4 I
1+ ^о
О
СНз
¦1,
Аг—СН=СН—СООН +
> Аг—
i
р
СН^СООН
O~Na+
1
Н
—сн—с
о
II
о
СНз
1 СН СООН
СНз
571
4. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ
Уксусный ангидрид ¦— бесцветная жидкость с острым запахом,
кипит при 139 СС. В воде растворяется мало, постепенно реагирует
с ней.
В промышленности уксусный ангидрид производится в больших
количествах. Основным методом получения является реакция кете-
на с уксусной кислотой (гл. XXVII. В.4). Известен способ каталити-
каталитического окисления ацетальдегида в присутствии ацетатов кобальта
и меди:
О
/Р °« II
2CH3Cf —-> СНзСОССНз
\н Кат. Ц
О
Уксусный ангидрид используют в качестве ацетилирующего
реагента (получение ацетилцеллюлозы, лекарственных веществ) и
как реакционную среду для проведения некоторых реакций конден-
конденсации.
Малеиновый ангидрид представляет собой бесцветное кристал-
кристаллическое вещество с т. пл. 52,8°С, растворим в воде (реагирует с
ней) и органических растворителях.
В промышленности малеиновый ангидрид получают в значитель-
значительных количествах каталитическим парофазным окислением бензола,
бутенов или фурфурола над ваиаднйсодержащими катализаторами:
сн3сн2сн--сн2
/О
о
II
/\
о,. /<
vao.
о
о
Малеиновый ангидрид широко используется для получения по-
полимерных материалов (сополимеры со стиролом, полиэфирные смо-
смолы), синтеза гербицидов и в качестве активного диенофила в дие-
диеновом синтезе.
Фталевый ангидрид — бесцветное кристаллическое вещество с
т. пл. 130,8°С, легко возгоняется.
о Получается в промышленности в очень больших
II количествах окислением нафталина или о-ксилола
/?\/\ ([л- XXXII. В.З).
Фталевый ангидрид широко используется для по-
получения полиэфиров (алкидных смол), пластификато-
пластификаторов (сложных эфиров фталевой кислоты) и красителей.
Он является также исходным веществом для синтеза
572
биологически активных соединений (производные инданднона-1,3,
фталазина и др.).
Тримеллитовый ангидрид -- бесцветное кристаллическое ве-
вещество, плавящееся при 162—!63'С, вводе гидролизуется до кис-
кислоты. Получают каталитическим окислением псевдокумола:
О
н3сч /уЧ сн3 ноос. //ч )
о„
о
Кат.
о
Применяют для получения термостойких полимеров (полиэфи-
(полиэфиров, полиимидов) и красителей.
Пиромеллитовый диангидрид представляет собой бесцветное
кристаллическое вещество с т. пл. 286 С, малорастворим в органи-
органических растворителях:
о/
Получают каталитическим окислением 1,2,4,5-тетраметилбензола
(дурола). В промышленности его используют в значительных коли-
количествах для получения термостойких полимеров (полиимидов).
В. СЛОЖНЫЕ ЭФИРЫ КАРБОНОВЫХ КИСЛОТ И ЛАКТОНЫ
1. МЕТОДЫ ПОЛУЧЕНИЯ
Сложные эфиры карбоновых кислот легко получают взаимодейст-
взаимодействием гндроксильных производных углеводородов с карбоновыми
кислотами, ацилгалогенидами и ангидридами или алкилированием
солей карбоновых кислот. Известны также специфические методы
получения (реакция Тищенко, гл. XXVII. Г.З; окисление кетонов
по Байеру — Внллнгеру, гл. XXXIII. И. 3). . Лактоны очень легко
образуются при внутримолекулярной этерификацин у- и 6-гндрокси-
карбоповых кислот (гл. XXXIV. Б.2).
1. Этерификация карбоновых кислот спиртами. В смеси карбоио-
вой кислоты и спирта медленно образуется сложный эфир. В реак-
реакции выделяется вода. Реакцию этерификацпи можно считать обра-
обратимой, так как сложные эфиры при взаимодействии с водой гидро-
лизуются на карбоповую кислоту и спирт:
/О
RCOOH + R'OH ^i R—C^ +H2O
ORi
Несравнимо быстрее происходит образование лактонов:
^^_— С ООН
<сн,>„ - (он,
4 он ^-—о
S73
Доказано, что реакция этерификации катализируется кислота-
кислотами. В присутствии кислот скорость реакции значительно возрастает.
Кислота активирует карбоксильную группу (гл. XXXI. А.З) п
облегчает атаку слабонуклеофильного реагента — спирта.
Обычно в реакциях этерификации отщепляется в виде молекулы
воды атом кислорода карбоновой кислоты. Это доказано использо-
использованием «меченых» атомов (изотопа 19О) (механизм реакции см.
гл. XXXI. А.З):
.О п + х- /О
R-C^ +R1—18OH "R—С^ +Н2О
ЧОН Ч1ЮЯ*
/О н+х- /О
R —Cf +ri_OH > R —Of +H2«O
Ч"ОН 4OR*
Взаимодействию карбоновых кислот со спиртами способствуют
также некоторые реагенты, например карбодинмиды (гл. XXXV.
В. 2).
2. Реакции гидроксильных производных с функциональными про-
производными карбоновых кислот. Спирты, фенолы и другие гидро-
ксильные производные легко реагируют с ацилхлоридами и ангид-
ангидридами кислот. Известны также реакции спиртов со сложными эфи-
рами карбоновых кислот, особенно в присутствии кислот или осно-
оснований (реакции переэтерификации):
+„х
3. Алкилирование. Соли карбоновых кислот алкилируются гало-
геналканами или эфирами неорганических кислот:
RCOO-Na + + R+1 — X —> R-C^ +Na- X-
2. ФИЗИЧЕСКИЕ СВОЙСТВА И СТРОЕНИЕ
Сложные эфиры карбоновых кислот являются бесцветными жид-
жидкостями, иногда и кристаллическими веществами с приятным запа-
запахом. Температура кипения сложных эфиров обычно ниже, чем тем-
температура кипения соответствующих кислот (иногда и спиртов). Это
свидетельствует об уменьшении межмолекулярного взаимодейст-
взаимодействия, что объясняется отсутствием межмолекулярных водородных
связей.
Полярность связей в молекуле сложного эфира подобна поляр-
полярности связей в карбоновых кислотах. Существенное отличие сос-
состоит только в том, что здесь нет подвижного протона, вместо него
67 4
находится углеводородный остаток:
ft—С
8+
активированные зфиры
Электрофильный центр находится на углеродном атоме карбониль-
карбонильной группы. Кроме того, положительно поляризован углеродный
атом в алкилыгой группе сложного эфира. Электрофильность слож-
сложного эфира зависит от строения углеводородного остатка у кисло-
кислородного атома. Реакционная способность увеличивается, если угле-
углеводородный остаток с атомом кислорода образует сопряженную
систему, как это имеет место в виниловых и ариловых эфирах (ак-
(активированные эфиры).
3. ХИМИЧЕСКИЕ СВОЙСТВА
Сложные эфиры карбоновых кислот реагируют с нуклеофилами,
при этом замещается алкоксигруппа — происходит ацилирование
нуклеофила. Иногда наблюдается алкилирование нуклеофила. Для
сложных эфиров с ос-водородным атомом характерны реакции с
участием этого атома (сложноэфирная конденсация). Известны
специфические реакции (ацилоиновая конденсация). Гидрирование
сложных эфиров до спиртов рассмотрено в гл. XIV. А.2.
Q о г. п
II II
rch2cchcor'
RCH.COOR1
NaOR
о он
RCH2C—CHCH2R
Na
RCH2CH,OH
IH]
\
RCH..C
)R'
Nu:
CHjC
NaOC2Hs
NR
о о
\r' II II ,
*- RCH,CCH,CRJ
>- RCH.COO'fR'NRj]
1. Реакции нуклеофильного замещения. Сложные эфиры реаги-
реагируют с нуклеофильными реагентами, образуя соответствующую кар-
боновую кислоту или различные функциональные производные этой
кислоты:
R_C<f . +:NuH ^ R —C—NuH —* R — (
,0
'\
ORI
OR1
+R'OH
Наиболее известны реакции с водой (гидролиз), спиртами (алкого-
лиз, или переэтернфикация), аммиаком и аминами (аминолиз), гид-
гидразинами (гидразинолиз).
575
Подробно рассмотрим реакции с О-нуклеофилами — щелочами,
алкоксидами, водой и спиртами. Реакцию с С-пуклеофилами см.
гл. XXVIII. Б.1, а с N'-нуклеофилами гл. XXXIII. Д.1.
При взаимодействии сложных эфиров со щелочами получается
соль карбоновой кислоты, т. е. происходит гидролиз (омыление)
сложного эфира:
=?rNa+
?
R—С—ОН * RС '
R— с'^ + :OH + Na+^zzt R—С—ОН —*• R—С ' Na+ + R'OH
X " i %
Взаимодействие с алкоксидами в растворе соответствующего
спирта ведет к обмену алкильных групп сложного эфира (иереэте-
(иереэтерификация). Реакция является обратимой:
O-Na->-
Я — Cf +-OR2Na+ 7^ R-C-OR2 ^ R — C^ + -OR1Na->-
OR1 ! х OR2
OR1
Реакция со слабыми нуклеофнлами — водой и спиртами — ката-
катализируется кислотами, которые активирует молекулу сложного
эфира:
Г-б- , Г*-1*- + - :о—н х~-
>°: н х" s+>0:-H х /" I +/«
^r-C + ю( ^i R-c— о= ^г±:
0H 40R' \rj I Ж2
=OR'
^i: r—c( +
NOR' ROH
(R2--=H, Alk). В присутствии воды идет гидролиз и образуется кар-
боновая кислота. В растворе спирта происходит нереэтерифпкация.
2. Сложные эфиры как алкилирующие реагенты. В отдельных
случаях сложные эфиры алкилируют пуклеофильный реагент. Это
характерно для реакций с третичными аминами:
R-C^ +:NRf. ->RCOO-[Ri-KR32]
xO-Ri
Алкилирующая способность увеличивается при усилении элек-
троноакцепторных свойств ацилыюго остатка (увеличении констан-
константы кислотности соответствующей карбоновой кислоты).
3. Реакции ос-водородного атома. Сложные эфиры карбоновых
кислот, имеющие а-водородпый атсм, при взаимодействии с силь-
сильными основаниями (алкоксидамн), подобно карбонильным соедине-
соединениям (гл. XXVII. А.З), образуют карбанноп.
Образование карбапиона вызывает дальнейшую реакцию кон-
конденсации со второй молекулой сложного эфира.
576
Ниже приведен пример реакции сложного эфира в присутствии
металлического натрия или алкоксида натрия, в результате которой
образуется соль эфира р-кетокарбоновой кислоты (сложноэфирнля
конденсация, конденсация Кляйзена, сравните гл. XXVIII. Б.1):
н р н /*8"*
I 8+/° ,_.. | л-
R—С-»-С + Na ЮС2Н5 ^—**: R—C-j-C + С2Н5ОН
5-+} NoeiH5 " -Ч(а^ос2н5
п. л, •-Q~Na+
2—С С—е
| I \
R—СН2—с' + R—C-jC^ v RCH2
ОС2Н5 К
—»- JRCH2—С==С—С + С2Н5ОН
^ ХОС2Н5
При подкнсленни реакционной смеси получают эфиры р-к-это-
карбоповых кислот (гл. XXXIV. Д.2).
4. Ацилоиновая конценсация. Сложные эфиры в присутствии мел-
мелкодисперсного натрия в инертной атмосфере реагируют своеобраз-
своеобразно, образуя а-гидроксикетоны (ацилоины). Предполагают, что об-
образование ацнлоинов связано с возникновением анион-радикалов и
их димеризацией:
6+ /О6" . /О-
П — С" -|-.Na —* R—C< Na +
OR1 OR1
+Na-0 O"Na+ О О
. 70-Na+ II II II
2R-C{ —у R-C-C-R ^t R-C-C-R + 2NaOR*
4ORi | |
| |
ORi OR1
О О +Na~O O-Na +
: [I
R— С — С — R-|-2.\a —* R—C^C —R
+ Na~O O-Na+ OH OH OHO
II II I J
R — CU-:C — R-! 2H+X- -* R—C-C —R-|-2Na + X- —+ R—С —С —R
A
В качестве промежуточных продуктов образуются а-дикетопы,
которые натрием восстанавливаются до ацилоипов.
Реакция имеет большое значение для получения соединений с
большими циклами:
,0
/COOR Na С"
(СН2)„/ i^. (СН2)„/ | /ОН п > 4
COOR ЧС/
ХН
19 & 517 577
4. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ
Этилацетат — бесцветная жидкость о приятным запахом; т. кип.
77 °С, умеренно растворим в воде (8%), вода малорастворима в
этилацетате C%).
В промышленности получают из уксусной кислоты и этанола в
присутствии серной кислоты.
Этилацетат широко применяют в качестве растворителя для
растворения нитроцеллюлозы, ацетилцеллголозы и других полимер-
полимерных материалов, для изготовления лаков. Используют также в пи-
пищевой промышленности и парфюмерии.
Бутилацетат — бесцветная жидкость с приятным запахом;
кипит при 126 °С, малорастворим в воде.
Бутилацетат получают из бутапола и уксусной кислоты в при-
присутствии серной кислоты. Используют в лакокрасочной промышлен-
промышленности как хороший растворитель нитроцеллюлозы и полиэфирных
смол.
Амилацетаты — бесцветные жидкости с приятным запахом,
н-Амилацетат СН3СОО(СНоLСН3 имеет т. кип. 149,2 °С, а изоамнл-
ацетат СН3СОО(СН2JСН(СН3J — 142 °С.
Амилацетаты — хорошие растворители для нитроцеллюлозы и
других полимерных материалов. Изоамилацетат используется так-
также в пищевой промышленности (грушевая эссенция).
Винилацетат СН3СООСН=СН2— бесцветная жидкость с
т. кип. 73 °С. Получают из ацетилена и уксусной кислоты (гл. IV.4)
в присутствии катализатора. Известен метод получения из этилена и
уксусной кислоты в присутствии кислорода и катализатора. Винил-
ацетат легко полимеризуется. Поливинилацетат широко применя-
применяется для приготовления клеев, лаков, лакокрасочных материалов:
СНз-СН Г— СН2 — СН 1 — СН -СН-ООССНз
I I
OOCCHaL OOCCFI3Jn
пол нои ни л ацетат
Гидролиз (омыление) поливипилацетата дает другой ценный по-
полимер — поливиниловый спирт, из которого изготовляют синтети-
синтетическое волокно, пленки, клеи, эмульгаторы.
Искусственные фруктовые эссенции. Многие сложные эфиры име-
имеют приятный запах и используются в пищевой промышленности и
парфюмерии.
НСООС2Н5 этнлформиат, ромовая эссенция
СНзСН2СН2СООС2Н,-, этплбутнрат, ананасовая эссенция
СН3СН2СН2СООСН2СН2СН(СН3J изоамилбутират, грушевая эссенция
(CH3JCHCH2COOR алкнлизовалераты, яблочные эссенции
СНСООСНСН бекзплацетат, жасминовая эссенция
Малоновый эфир [диэтиловый эфир малоновой кислоты,
Н2С(СООС2Н5J1 представляет собой бесцветную жидкость с прият-
приятным запахом, кипящую при 199 °С. Его молекула имеет активную
метиленовую группу, поэтому соединение проявляет повышенную
С—Н-кислотность и способность образовывать сопряженный
Б78
карбанион:
о
н соос2н5 _^с—ос,н5 t _/_ос2н,
С + 'Sol Т^*"' Г
С
Н' ХСООС2Н5
8-6
В растворе этанола в присутствии этилата натрия малоновый
эфир ионизируется и образует диэтилмалонат натрия. Это соедине-
соединение легко алкилируется, ацилируется и реагирует с другими элек-
трофильными реагентами:
R+-X~-|-H-C(COOC,H5).2 —* N>C(COOC.2H5J + NaX
Na+ R/
Малоновый эфир галогеиируется, нитрозируется. Его исполь-
используют в органическом синтезе для получения карбоновых кислот,
аминокарбоновых кислот (гл. XXXIV. В.1), гетероциклических со-
соединений.
Например, при гидролизе алкилмалоновых эфиров образуются
алкилмалоновые кислоты, которые легко декарбоксилируются и да-
дают карболовые кислоты:
н2о -со3
RCH(COOQH5J -y^ RCH(COOHJ >¦ RCH2COOH
Сложные эфиры фталевой кислоты представляют собой бесцвет-
бесцветные жидкости с высокими температурами кипения. В воде не раст-
растворяются. Их получают из фталевого ангидрида и соответствующего
спирта в присутствии серной кислоты.
COOR
-COOR
Д и а л л и л ф т а л а т (R = СН->СН=СНЛ используют в ка-
качестве добавки для различных полимерных композиций и как
пластификатор для ноливпнилхлорида, алкидных смол.
Дибутилфталат (R = CH2GH2CH2CH3) — одни из ос-
основных пластификаторов для ноливинилхлорида, нитроцеллюло-
нитроцеллюлозы, полиакрилатов и синтетических каучуков. Его используют
также как средство для отпугивания насекомых (репеллент).
Диоктилфталат(Я = С8НO) — хороший пластификатор
и репеллент.
Акрилаты и полиакрилаты. Метилакрилат СН2=
=СНСООСН3 представляет собой бесцветную жидкость с острым за-
запахом; т. кип. 80 °С. Получают из ацетилена, СО и метанола в при-
присутствии Ni(COL. Легко полимеризуется в присутствии инициато-
инициаторов свободных радикалов и образует бесцветную эластичную мас-
19* 579
1
— СН-- СНСООСН3
су — полнметнлакрилат:
СНз —СП _С1[,-СН
СООСНз л СООСНз
Используют для производства пленок, листов органического
стекла и клеев.
Метилметакрилат СН,==С(СН3)СООСН3 является бес-
бесцветной жидкостью с раздражающим запахом, кипит при 101СС.
Получают из ацетона, HCN и метанола (гл. XXXI. Б.4). Метил-
Метилметакрилат легко полимеризуется, образуя бесцветную эластичную
массу — полиметилметакрилат:
сня г сн3 ~] сн3
¦СП-- С —СООСНз
С113 —С
— СН2-С
СООСНз 1_ СООСНз jn
(п-- 200—300)
Полиметилметакрилат используют для изготовления прозрачных
листов (органического стекла).
Полиэфиры — полимеры, которые получают из дикарбоновых
кислот или их ангидридов и многоатомных спиртов (гликолей и
глицерина). Из диметилового эфира терефталевой кислоты и эти-
ленгликоля получают полиэфир полиэтилентерефталат:
, ч .О Г
N=/ \o-L-
— С?
О
— СН2СН2ОН
Применяют этот полимер для изготовления синтетического волок-
волокна — лавсана (терилена), которое широко применяется в текстиль-
текстильной промышленности.
Полиэфиры малеиновой кислоты (полималеинаты) получают из
малеинового ангидрида и гликолей:
но—с//==чсо — Г— сн2сн2ос/==чсо — — сн2сн2он
i; и и и
о о L оо
Их используют в качестве связующего при изготовлении стекло-
стеклопластов. В полималеииатах имеются двойные связи, которые при-
придают им способность взаимодействовать с другими способными по-
лимеризоваться соединениями, образуя «сшитые» структуры.
Фталевьгй ангидрид с гликолями, глицерином или пептаэритри-
том образует полнфталаты — алкидные смолы. В реакции с глице-
глицерином получаются глифталевые смолы:
<=/
ЧСО- — СНХНСН,ОС
НО —С-
II
О
о
L
О
он
— СН-СНСН..ОН
J'l
В реакции с пентаэритритом —пентафталевые смолы.
Алкидные смолы часто модифицируют добавлением других ком-
компонентов, например ненасыщенных жирных кислот и их эфиров.
Так получают высыхающие алкидные смолы.
Алкидные смолы широко используют для изготовления лаков и
красок и в качестве связующего в производстве линолеума.
Липиды — липофильиые (жирорастворимые) вещества, играю-
играющие большую роль в жизнедеятельности различных организмов.
Большинство липидов являются сложными эфирами, например
простые липиды — жиры и воски, сложные липиды — фосфорсо-
фосфорсодержащие соединения (фосфолипиды), производные моносахаридов
(гликолипиды). Сложные липиды являются составной частью кле-
клеточных мембран. В группу липидов входят также стероиды (на-
(например, холестерин, гл. XIV. В.З) и каротиноиды (например, лико-
пин, каротин).
Жиры и масла, встречающиеся в организмах растений и жи-
животных,— сложные эфиры высших жирных кислот, как насыщен-
насыщенных," так и ненасыщенных, и глицерина:
RCOCH2CHCH2OCR (где R--CnH2n + 1, С„Н2„_Ь С„Н2П_3, С„Н2„_6)
II I II
О OCR О
II
О
При обработке жиров и масел щелочью происходит их гидролиз
(омыление) с образованием глицерина и солей карбоновых кислот.
Отсюда и возникло название «жирные кислоты». Соли высших кар-
карбоновых кислот являются мылами.
Воск является смесью сложных эфиров высших карбоновых кис-
кислот и высших спиртов. В состав пчелиного воска входят сложные
эфиры RCOOR', где R=C23H47, 0,,Ни, a R'=C21H19, С30Нв1-, и др.
Фосфолипиды являются важными составными частями клетки.
Из яичного желтка выделен фосфолипид лецитин:
О
II +
С17 НГ,5СОСН2СНСН2О - Р - О-СН2СН2К(СН3K
II I I
о осс17н35 о-
II
о
Известно много других фосфолипидов.
Бутиролактон представляет собой бесцветную жидкость, с
т. кип. 204°С. Его получают каталитическим окислением 1,4-бутан-
диола и тетрагидрофурана или селективным восстановлением ан-
ангидрида янтарной кислоты.
Н2С СН2
1с
н
Используют в качестве растворителя и как исходное в органи-
органическом синтезе.
581
Г. ОРТОЭФИРЫ КАРБОНОВЫХ КИСЛОТ
Ортоэф1фы являются производными гндратной формы карбоновых
кислот и в этом отношении они сходны с ацеталями — производ-
производными альдегидов и кетонов.
1. Методы получения. Ортоэфнры образуются в реакциях трига-
логенпроизводных с алкоксидами и в реакциях солей имидоэфиров
с алканолами:
R —CCI3-L3\a+-OR1 —>• R—C(OR1K-!-3Xa4C(-
+2R'OH —>- R-C(ORlK-:-NH4CI-
Cl-
2. Физические и химические свойстза. Применение. Ортозфиры
обычно представляют собой жидкости с приятным эфирным запа-
запахом; НС(ОСН3)з кипит при 102—103 С, ИС{ОСгЩъ — при 145—
147СС.
Ортоэфиры устойчивы в нейтральных и щелочных растворах,
в кислых средах они гидролизуются:
RC(ORiK -??* R-C^ + 2R'OH -^* RCOOH-I-3R1OH
Ортоэфиры являются эффективными ацилирующими иалкилиру-
ющнми реагентами, особенно в присутствии кислот. Их применяют
для ацилировання соединений, содержащих активную метиленовую
группу, для превращения альдегидов и кетонов в ацетали (гл.
ХХУП.Д.4):
R-C(ORiK
о
р
p? _ г~ _
R2
я3 ¦•'"
I /OR1
4R
н2о R
-R'OH R
>c
3/
OH
R
ORl
R2-C-R3-i-RCOORt
OR*
/O
| R
H
Ортомуравьиный эфир НС(ОС2Н5)з применяется для введения
альдегидной группы (форматирования) и получения дпэтилацета-
лей.
Д. АМИДЫ И ИМИДЫ КАРБОНОВЫХ КИСЛОТ,
ЛАКТАМЫ
1. МЕТОДЫ ПОЛУЧЕНИЯ
Амиды и имиды карбоновых кислот получают взаимодействием кар-
карбоновых кислот или их функциональных производных с аммиаком
или аминами. Наиболее трудно реагируют сами карбоновые кисло-
582
ты. Вначале образуются аммониевые соли, которые при нагревании
выше 200 °С превращаются в амиды или имиды:
200 °С /О
RCOOH + NH3 :ц± RCOO-NHJ >¦ R —C<f +H2O
XNH2
О
/СООН /COO-NHj1" ,о ,СЧ
Y< +NH3 ^1Y( -!> Y<
XCOOH XCOOH Х
О
у- и б-Аминокарбоновые кислоты легко цнклизуются в лактамы:
Легко реагируют с аммиаком и аминами ацнлгалогениды, ан-
ангидриды, сложные эфиры:
Rt О-
а+/оа- I I +/R1 /О
R-C<f +--M-R2 -* R-C-N/ -> R-C^ /RM-HX
NX I II XR2 \\/
H X H XR?
(X=C1, Br, OOCR, OR3).
2. ФИЗИЧЕСКИЕ СВОЙСТВА И СТРОЕНИЕ
Амиды и имиды представляют собой бесцветные кристаллические
вещества или жидкости, растворяющиеся в воде и органических
растворителях. Амиды, в молекулах которых имеются связи N—Н,
ассоциированы вследствие образования межмолекулярных водо-
водородных связей и имеют более высокие температуры кипения.
В молекулах амидов осуществляется значительное взаимодей-
взаимодействие между неподеленной электронной парой атома азота и л-
электронной системой дзойной связи С---О. Образуется сопряжен-
сопряженная система связей, изменяются природа связей С—N и С -О и
распределение электронной плотности. В результате связь С—N
становится короче, а связь С^О несколько длиннее по сравнению
с несопряженными соединениями:
R— С' ,. R—С
583
Атомы амидпой группировки С (CO)N находятся водной плоскости,
вращение по амидной связи С—N в значительной степени заторможе-
заторможено, связь имеет некоторый характер двойной связи. Это можно
изобразить резонансными формулами:
б: /6:-
R2 R»
3. ХИМИЧЕСГ<ИЕ СВОЙСТВА
Амиды отличаются по своим свойствам от сложных эфиров. У них
более выражено взаимодействие с электрофильными реагентами,
fio уменьшена реакционная способность с нуклеофилами. Это объ-
объясняется уменьшением —/-эффекта групп в ряду Cl, OOCR, OR1,
NH2 (б^б^б'^б111) и увеличением электронодонорного
+М-эффекта F1V<6V<6V1<6V11):
Кроме того, для амидов характерен ряд реакций с участием
связей N—Н:
/О— Е E + Y- /О :NuH /О
R— f/j. Y- « >• R— С" >¦ R— С"
\\Н2 XNH2 "NH« XNu5
X,
/О ОН-
R-C = N R-C^f >¦ R — NH2
(здесь Х=С1, Br).
1. Основность. По сравнению с аминами амиды карбоновых кис-
кислот являются слабыми основаниями ввиду сильного взаимодейст-
взаимодействия неподеленной электронной пары атома азота с карбонильной
группой. Присоединение протона обычно происходит по атому кис-
кислорода:
Протонировапная форма амидов имеет структуру имидовых кис-
кислот и является сильной ОН-кислотой. Например, р/Свн+ аце-
тамида равен 0,1, бензамида —2, капролактама 0,4.
Имиды являются очень слабыми основаниями, в этом они срав-
сравнимы со спиртами и эфирами.
584
В протонирогсанной форме амидов положительный заряд па
углеродном атоме значительно больше, чем в ампдной форме. Это
способствует реакциям со слабыми нуклеофильными реагентами.
2. NH-Кислотность. Если в молекуле амида имеется связь
N—Н, возможна ионизация. Амиды являются слабыми NH-кис-
лотами. Повышенная кислотность амидов по сравнению с аминами
объясняется сильным влиянием карбонильной группы (эффект со-
сопряжении, делокализация отрицательного заряда в анноне):
С- Р- s-
Ч
Л н
Кислотность ацетгмнда (р/С„--15) сравнима с кислотностью ме-
метанола, кислотность бензамида р/С,г— 13,5.
Им иды являются более сильными NH-кислотамп, так как атом
азота находится под влиянием двух карбонильных групп:
№. + HSol
¦»—'
3. Реакции нуклеофильного замещения. В реакциях с нуклео-
фильными реагентами может произойти замещение группы NH.
№)••
W \ + /У /к
R—С^ +:NuH у—*- R—с—NuH ^Zt R—С + Н—N:
I / Ч
R2 R-/ NR'
Реакционная способность амидов меньше, чем сложных эфиров.
Наиболее известны реакции с водой (гидролиз) и спиртами (алкого-
лиз).
Гидролиз амидов легко происходит в присутствии щелочи или
кислоты и очень медленно — в нейтральной среде.
В присутствии щелочи амиды превращаются в соль карбоновой
кислоты и аммиак (амины):
/О |
R—C<^ -j-Na + OH- ;r_t R— С — ОН —> RCOQ-Ka+ +HNR1R2
585
Если Rl или R2 являются водородными атомами, промежуточно
образуется также анион амида.
Присутствие кислоты активирует молекулу амида (протониро-
вание), поэтому происходит реакция со слабыми О-нуклеофила-
ми — водой и спиртами:
N—R' ^N—R1 R3
&' R2
:?н н :?H >o
T*" R—С—б t * R—С—О—R3 *~ R—С + H:NR'Ra
(R3-H, Alk).
Имиды в реакциях с нуклеофилами более активны, так как влия-
влияние атома азота на одну карбонильную группу меньше.
4. Образование и превращения N-галогенамидов. N-Незамещен-
ные или N-моиозамещенные амиды легко галогепируются у атома
азота. Обычно действуют галогенами в присутствии оснований:
^ RCONHX 7Ш
N-Галогенамиды являются весьма нестабильными соединения-
соединениями и обладают свойствами окислителя. Их применяют в качестве
галогенирующих реагентов. Разрыв связи N—галоген может осу-
осуществиться гомолитически:
RCO\/ -U RCONR4 -Br
ЧВг
Особой реакционной способностью обладают N-галогенамиды
со связью N—Ы, которые в щелочной среде образуют соли:
Со:, *Г
R—C%-v + Na+OH" ^=±Г R—C^ Na+ + Н.О
N-»-X: N^X:
Н« +
Соли N-галогенамидов при нагревании превращаются в первич-
первичные амины:
/Р t°
R-Cf - + -I-H..0 —* R—NH2-|-CO2 + Na + X-
NNX Na
Впервые эту реакцию наблюдал А. Гофман в 1881 г. (расщепление
амидов по Гофману).
566
Промежуточными продуктами в реакции Гофмана являются изо-
циапаты, которые образуются в результате перегруппировки своеоб-
своеобразных частиц—ацилнитренов. Ацилнитрепы— аналоги кото-
карбспов — образуются из N-галогеиамндов:
A- r
R—Сч =?=*! Г ?с=О + =Х= Na+
N—X: Na
+
R_N=c=O + NaX
Изоцианаты очень легко реагируют с водой и образуют карбами-
новые кислоты, которые декарбоксилируются:
R —\=^С = О-[-НаО —> R —К —СООН —+ RNH, + CO2
к
5. Взаимодействие с электрофильными реагентами, дегидрата-
дегидратация. Электрофнльные реагенты атакуют кислородный атом. Силь-
Сильные алкилнрующие реагенты дают производные имидоэфиров (гл.
XXXIII. Ж-1). Сильные кислоты Льюиса вызывают дальнейшие
превращения. N-Незамещенные амиды превращаются в нитрилы
(гл. XXXIII. К.1):
/Р
/О /О — Р—С1
R—C<f +POCI3 —*¦ R— С^+ \С1 —> R—C= N4-HOPOC12 + HC1
N\H2 ' XNH2 Cl-
М,\т-Дизамещенные амиды с РОСЦ дают производные хлоран-
гидрида имидокислот — активные ацилирующие реагенты. На-
Например, N.N-диметилформамид с РОС13 дает соль N.N-диметил-
хлорформпмидиния — активный формилирующнй реагент (гл.
XXIX.Б.4):
/О Г /О —РОС12 Г /С1 1
-fPOClj—* НС/+ CI- —» НС/х РОСB"
bh ' L XN(CH3J J L ^N(CH3JJ
4. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ
Формамид HCONH2 — бесцветная гигроскопическая жидкость; ки-
кипит при 210,5 °С, смешивается с водой и спиртами.
Получают его из метилформиата и аммиака. Применяют в ка-
качестве растворителя и исходного в органическом синтезе.
N ^-Диметилформамид (ДМФА) — бесцветная жидкость со
слабым запахом; кипит при 153 СС, смешивается с водой и органи-
органическими растворителями.
В промышленности его получают в больших количествах из ди-
метиламина и оксида углерода под давлением.
ДМФА применяют в качестве растворителя для растворения аце-
ацетилена, полимеров, для проведения синтезов. ДМФА хорошо соль-
ватирует катионы за счет взаимодействия е кислородным атомом-
но не сольватирует анионы, нет подвижного водородного атома.
Такие растворители называются биполярными апротонными (ср.
587
гл. XIX.3). ДМФА применяется также для реакций формнлирова-
ния.
Сукцинимид (имид янтарной кислоты) — бесцветное кристалли-
кристаллическое вещество с т. пл. 126СС, растворимое в воде.
Получают его нагреванием аммониевой соли янтарной кислоты.
Сукцинимид используют в органическом синтезе. Важным его
производным является N-бромсукцииимид, который применяют
для бромирования и окисления органических соединений.
Фталимид — бесцветное кристаллическое вещество; т. пл. 238 СС,
легко возгоняется.
Получают его изфталевого ангидрида и аммиака при 170—240 °С.
Фталимид является слабой NH-кислотой (р^Га — 8,3). Он раст-
растворяется в водных щелочах, но постепенно происходит гидролиз с
раскрытием цикла и образованием соли фталаминовой кислоты. Соли
фталнмида получают в безводных средах. Их используют в орга-
органическом синтезе, например для получения первичных аминов алки-
лированием и расщеплением полученного N-алкнлфталимида
C. Габриэль, 1887)
8+ S- .v
IN: К' + R —X —
N—R - >¦ R—NH2
Для гидролиза N-замещеппых фталимидов используют щелочь,
кислоту. Лучше всего использовать реакцию с гидразином, в ре-
результате чего образуется гидразид фталевой кислоты и первичный
амин.
Фталимид используется для получения аитраппловой кислоты
и других аминокислот.
а-Пирролидон (-у-бутнролактам) представляет собой бесцветное
низкоплавкое (т. нл. 25,6 °С) вещество ст. кип. 245 °С, растворяет-
растворяется в воде. Получают его из бутиролактона и аммиака.
а-Пирролидон используют в качестве растворителя и
для получения N-вимилпирролидона, который является
важным мономером для получения различных полимеров.
О Большое значение имеет чистый поливинилпирролпдоп с
молекулярной массой около 20 000, который растворяет-
растворяетн
ся в воде. Его водный раствор используют в медицине как за-
заменитель кровяной плазмы и для дезинтоксикации организма.
Производное пирролидопа — N-метилпирролидон является хоро-
хорошим биполярным апротонным растворителем. Он отлично растворя-
растворяет также различные полимеры.
г-Колролактам—бесцветное кристаллическое вещество с т. пл.
68СС, растворяется в воде.
Производится в значительных количествах. Одним из основных
методов является перегруппировка Бекмана оксима циклогекса-
588
нона в присутствии 20—25%-ного олеума:
.о
л он
При 250—260 "С капролактам в присутствии слабокислых или
основных катализаторов полимеризуется с образованием полиами-
полиамида —¦ поли-е-капроамнд.
Полиамиды являются высокомолекулярными соединениями
(М« 10 000...30 000), макромолекулы которых содержат амидиые
группировки CONH.
Полиамиды получают двумя способами: из диаминов и дикарбо-
новых кислот или их производных и из со-аминокарбоновых кислот
или их лактамов.
В табл. 46 перечислены важнейшие полиамиды, которые исполь-
используются в промышленности синтетических волокон и других изделий.
Таблица 46. Важнейшие полиамиды
Полиамид
Полигск-
самегидец-
аднпямнд
Полр-е-
канролмлд
Полн-oj-
энакто-
амид
Поли-со-
ундекан-
амид
Формула
О г- О О -1
MOOC(CH2),CN- -(CH2),NC(CH2)!cN- -(CH2),NH2
1 1 1
н L н н An
Г ° п
НООС(СП2N —
ИООС(СНг),—
-NC(CH2)B- I-NH,
H Jn
О -,
-NC(CH2)a- -NH,
L H Jn
Г ° 1
II
HOOC(CH2)I0- -NC(CH2I0- -NH,
L H Jn
Т. пл.,
°C
250
225
223
180...
...185
Волокно
Анид
(США —
наПлон-
6,6)
Капрон
(США —
нанлон-6,
ГДР —пер-
—перлон)
Энант
(США-
найлон-?)
Ундекан
(США —
найлон-11,
Франция—
рильсан)
Структурой полиамидов формально обладают также полипепти-
полипептиды и белки (гл. XXXIV.P).
Полиамиды являются высокомолекулярными соединениями, мак-
макромолекулы которых содержат имидные группировки. Получают
их из днангидридоз тетракарбоновых кислот и диаминов.
Полиимидпые пленки нерастворимы и имеют большую термичес-
термическую стабильность C00—350°С):
О О Г ООП
II
©
li
о
о
— Y —N'
II
О
о
_Y —N
S39
Обычно для получения полиимидпых пленок применяют пиро-
/ vNн/^
меллитовый диаигидрид А= Iй 4,4'-диаминодифениловый
эфир
Используют полиимиды главным образом в качестве электро-
электроизоляционных материалов.
Е. ГИДРАЗИДЫ И АЗИДЫ КАРБОНОВЫХ КИСЛОТ,
ГИДРОКСАМОВЫЕ КИСЛОТЫ
1. Гидразиды карбоновых кислот (ацилгидразины). Их получают
взаимодействием функциональных производных карбоновых кислот
с гидразином или его производными (алкил-, арилгидразииами):
/О /О
R—Cf +H,N-NH2 -> R—Of + HX
ЧХ " 4NHNHa
(X=CI, OCOR, OR1, NH2).
Известны также N, N-диацилгидразины и циклические гидрази-
гидразиды:
О
II
0 ° Я
II ||
r_C-N—N-C-R
с
II
о
I I k/\/NH
н н
По своим свойствам гидразиды напоминают амиды. Отличия за-
заключаются в том, что обычные гидразиды имеют группу NH2, не
сопряженную с карбонильной группой. Поэтому они являются ос-
основаниями, образуют соли с кислотами, реагируют с карбонильными
соединениями и образуют N-ацилгидразоны, реагируют с азотис-
азотистой кислотой:
О О О
'! + 11+ X— II HNO, II
R-C-NHNH3X- R-CNHNH2 ——* R-C-Ns
IH + X ~
азиды
Ri-C(f
.О
R-C<f /RX
NNH —N = C<
\R?
Применяют гидразиды в органическом синтезе.
2. Азиды карбоновых кислот. Их получают гштрозированием
гндразидов. Механизм взаимодействия аналогичен диазотированию
590
аминов (гл. XXV. A.I)':
о ГО
R-C-N-NH;
NO + X-
Н L H
I! .. +
R— С — \: — NsN
О
Х-
Второй метод заключается во взаимодействии неорганических
азидов с ацилгалогенидами:
R-Gf +NaN3 -*¦ R-C^ -fNaCI
NC1 4N.,
Строение азидной группировки напоминает строение дназоалка-
нов (гл. XXV. Б.2), и ее можно изобразить с помощью нескольких
резонансных структур:
.6: ,6'. О:"
р С. + ( ) Р С 4- ^—^ R' d " А-
Азиды являются малостабильнымн сосдннениягуш, низшие гомо-
гомологи склонны к разложению со взрывом. Разложение азидов при
нагревании в водных растворах ведет к первичным аминам:
R— О/'' -|-Н2О -^ R —NH,-!-N2-fСО2
Реакцию открыл Т. Курциус A890). .Механизм перегруппировки
Курциуса подобен механизму реакции Гофмана. При распаде азида
может образоваться ацилнитрен, который перегруппировывается в
изоцианат:
У.О ,о ,0
Изоцианат при взаимодействии с водой дает карбаминовую кис-
кислоту, которая декарбоксилируется в амин.
Разложение азидов в растворе спиртов дает уретаны RNHCOOR1.
Разложение азидов в растворах можно провести без их предвари-
предварительного выделения в чистом виде. Одним из таких вариантов яв-
является реакция карболовых кислот с азотистоводородной кислотой
HN3 в присутствии сильной кислоты (серной, полифосфорной):
HjSO,
RCOOH + HN3 R-\'
Эту реакцию открыл К. Шмидт A923). Предполагают, что в реак-
реакции Шмидта промежуточными продуктами являются азиды карбо-
новых кислот.
Известны также реакции нуклеофплыюго замещения азидной
группы при взаимодействии со щелочами, алкоксидами, аминами
(гл. XXXIV. Г.4).
591
3. Гидроксамовые кислоты (N-ацилгидроксиламины). Эти соеди-
соединения получают взаимодействием гидроксиламина с функциональ-
функциональными производными карбоновых кислот:
,0 /О
R-СГ +H2NOH -* R —C<f -+ НХ
ЧХ ЧМ-ОН
&
(X-Cl, OCOR, OR1).
Гидроксамовые кислоты — бесцветные кристаллические ве-
вещества; они растворяются в воде, термически нестабильны.
В отличие от амидов они являются более сильными NH-кисло-
тами:
+ :SoI !^-»-
XN-^OH NR-OH
H ptfa = 5,5-7,5
Образуют комплексные соли с тяжелыми металлами: с Ре (III)—¦
красные, с Си (II) — зеленые.
Своеобразной реакцией гидроксамовых кислот является пере-
перегруппировка в первичные амины:
у.О Кислота
R-Cf * R-NH2+CQ2
4NHOH
Перегруппировка происходит в присутствии кислот Льюиса
(например, SOC12), полифосфорной кислоты, Р2О5, уксусного ан-
ангидрида. О-Ацилгидроксамовые кислоты перегруппировываются
в мягких условиях:
/О NaOll ,0 Г
4N —OCR* 4N — OCR^ н'° * 2Т
I !! I!
Н О Na+ О
Перегруппировку гидроксамовых кислот открыл В. Лоссен
A872). Механизм перегруппировки Лоссена подобен механизму
реакций Гофмана и Курциуса.
Ж. ИМИДОЭФИРЫ КАРБОНОВЫХ КИСЛОТ И АМИДИНЫ
1, МЕТОДЫ ПОЛУЧЕНИЯ
1. Имидоэфиры получают из нитрилов присоединением спиртов в
присутствии кислоты:
.NH2C1- NaOH ,NH
R-C= N + RiOH + HCl -> R—Gf R-Cf
592
В этой реакции вначале образуются соли имндоэфнров, из которых
можно получить сами имидоэфиры.
Исходными для получения солей имидоэфиров могут служить
также амиды, особенно N-замещенные. Их алкилируют сильными
алкилирующими реагентами, например диметилсульфатом:
Л.6-
R~c;O. + ch^osojoch5 —*- R~cv+ ch3so4-
N—R' %—R'
2. Амидины получают из имидоэфиров или нитрилов:
бд- ,NH ,NH
R-Of -|-:NH3 ->¦ R-C<f J-R'OH
4ORi 4NH
R-C=N + Na+-NH2 -> R —Cf —-R-Gf
\\'H2 4NH2
2. ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
1. Основность. Как иминоэфиры, так и амидины являются основа-
основаниями, при этом амидины принадлежат к самым сильным органи-
органическим основаниям (р/Свп + = 12,4. . .12,5). Катион N-незамещен-
ного амидиния является симметричным образованием:
(\ S+
r—с' + н+х =г2: r—с' 5+ х
NNH, 4NH2
BS+5' = I)
Соли имидоэфиров и амидинов представляют собой устойчивые
бесцветные кристаллические вещества, хорошо растворимые в во-
воде.
2. Взаимодействие с нуклеофильными реагентами. Имидоэфиры
и амидины, подобно сложным эфирам и амидам, реагируют с раз-
различными нуклеофильными реагентами. При этом реакция идет у
углеродного атома карбоксильной функции.
Имидоэфиры легко гидролизуются как в кислой, так и в щелоч-
щелочной средах, при этом образуются как сложные эфиры, так и амиды,
а в конечном счете — карбоновые кислоты.
Амидины в кислой среде весьма стабильны, а в щелочной среде
гидролизуются до амидов:
5+
R—сч + Н2О ¦; " R—с/ S+ ОН" •- R—С^ + NH3
3. Реакции с карбонильными соединениями. Амидины конденси-
конденсируются с альдегидами и кетонами подобно аминам. Важны реакции
с р-дикетонами, в которых получаются производные пиримидина
(гл. XXXIX. А. 1.2).
593
з. тиокислоты и дитиокислоты
1. МЕТОДЫ ПОЛУЧЕНИЯ
1. Тиокислоты и их функциональные производные получают из
производных карбоновых кислот. Ацилировапие гидросульфидов
или тиолятов ацилхлоридами дает тиокислоты или S-эфиры тиокис-
тиокислот:
,0 ris-m+ /О hs-m+ /О
R-C<f * R-Cf -R-Cf
4i xCl 4SH
Тиокислоты являются таутомерной смесью тиольной формы
(S-кислота) и тионпой формы (О-кислота):
,0 70Н
4S-H ^S
О-Эфиры тиокислот получают из имидоэфиров и H2S:
Тиоамиды образуются при взаимодействии амидов с P2S5:
/Р p2s5 ^S
XNH2 4NH2
2. Дитиокислоты в виде солей легко получают присоединением
металлорганических соединений к сероуглероду:
г- 5+ f\ s+/> /<s '
R—MgBr+ S=C=S . *¦ R— C< Mg+Br
s
2. ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА.
ПРИМЕНЕНИЕ
1. Тиокислоты являются соединениями с неприятным запахом; в
растворах они весьма нестабильны, в присутствии воды гидролизу-
ются с выделением сероводорода.
Важны производные тиокислот. Некоторые эфиры обладают фи-
физиологической активностью. Тиомиды используют для синтеза ге-
гетероциклических соединений — тиазолов.
2. Дитиокислоты в свободном виде являются нестойкими вещест-
веществами красного цвета. Соли дитиокислот практически используют в
качестве аналитических реагентов. С ионами тяжелых металлов
дитиокислоты образуют цветные водонерастворимые комплексы,
которые растворяются в органических растворителях и экстраги-
экстрагируются ими из водных сред:
S S
R—CS2~Na+ + М2++2Х~->- R—cf ''\М['' \—R + 2^Х~
594
И. ПЕРОКСИКАРБОНОВЫЕ КИСЛОТЫ
И АЦИЛПЕРОКСИДЫ
1. МЕТОДЫ ПОЛУЧЕНИЯ
1. Ацилирование пероксида водорода. При воздействии на пероксид
водорода ацилхлоридами или ангидридами в присутствии основа-
оснований или карбоповой кислотой в присутствии кислого катализатора
образуются как пероксикарбоиовые кислоты, так и ацилперсксиды:
6+ ,0 О
R_C<f' -! H-0-0-H -* R-Cf +HX
ЧХ ЧО-ОН
6+ ,0 О. ,0
R-C<f -f ^C-R -> R-Gf + НХ
\\ Н-О-О/ \O-O-C-R
il
О
Используют 30%-ный Н2О2 (ацилирование ангидридами), 90—
95%-ный НоО-. (ацилирование карбоновыми кислотами в присутст-
присутствии серной кислоты) или Na2O2 (ацилирование ацнлхлоридамн).
Ацилпероксиды в присутствии воды и оснований гидролизуют-
ся до нероксикарбоновой и карбоновой кислот.
2. Окисление альдегидов при пониженной температуре
(—10. . .—20°С) сухим кислородом дает перокснкарбоновые
кислоты:
R_C<f -I R-Gf
Nl ЧО-ОН
2. ФИЗИЧЕСКИЕ СВОЙСТВА И СТРОЕНИЕ
Пероксикарбоновые кислоты и ацилпероксиды являются бесцвет-
бесцветными жидкостями или кристаллическими веществами, нестабиль-
нестабильными и взрывчатыми.
р.5- Энергия связи О—О только 143 кДж/моль (энер-
г'Л^"-н5+ гия связи С—О равна 350 кДж/моль). В молеку-
R—с/^ } лах пероксикарбоновых кислот образуется внут-
^о-*' римолекулярная водородная сеязь.
3. ХИМИЧЕСКИЕ СВОЙСТВА
1. Кислотность. Пероксикарбоновые кислоты — значительно более
слабые кислоты, чем карбоновые, ввиду того, что полярность связи
О—II меньше и в анионе невозможна делокализация отрицательного
заряда:
.О н«+ .О
R-Gf | -)-Н,0 ^ R-Cf -f H3O+
ЧО—О ХО—О-
2. Термическое разложение. Перокснкарбоновые кислоты и ацил-
ацилпероксиды являются термически малостабильньши, при нагревании
595
происходит быстрое разложение, иногда со взрывом. Инициировать
рзрывное разложение могут также удар или трение. Взрывчатость
уменьшается при увеличении молекулярной массы соединения.
Разложение начинается с гемолитического распада связи О—О,
образуются свободные радикалы, которые вызывают дальнейшие ре-
реакции:
о-
^-H, R-C
\
3. Пероксикарбоновые кислоты как окислители. Соединения спе-
специфически реагируют с алкенами и образуют эпоксиды (Н. А. При-
Прилежаев, 1909). Предполагают, что пероксикарбоновые кислоты вы-
выступают в качестве электрофильного окислителя:
II—.1
о
;о + RfcooH:
R1
Пероксикарбоновые кислоты легко окисляют сульфиды до суль-
фонов, амины до N-оксидов и азосоединений, фосфины до фосфипо-
{<сидов, иодарены до иодозиларенов и др.
Своеобразно происходит реакция с кетонами, в которой образу-
образуется сложные эфнры или лактоны (А. Байер, В. Виллигер, 1899):
ур о
+ R'COOOH • > R—с: + R'COOH
Л—с
—R
R1 COOOH
+ R'COOH
В этой реакции (перегруппировка Байера — Виллигера) вна-
вначале происходит нуклеофилыюе присоединение аниона пероксикар-
боновой кислоты к карбонильной группе, после чего следует внут-
внутримолекулярная перегруппировка:
Л.6- U О<" о-С
+ н+"о
R—С
/
\
+ HOOCR'
OR
4. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ
Пероксиуксусная кислота (перуксусная кислота) — бесцветная жид-
жидкость; т. кип. 25 °С (при 1,6 кПа), плотность df= 1,226, хорошо раст-
растворяется в воде н органических растворителях. При 100—П0°С раз-
разлагается со взрывом.
596
Получают пероксиуксусную кислоту окислением уксусного аль-
альдегида кислородом. Растворы ее в уксусной кислоте получают вза-
взаимодействием уксусного ангидрида с 30%-иым раствором Н2О2.
Пероксиуксусную кислоту используют в качестве сильного и
специфического окислителя.
Пероксибвнзойная кислота (пербепзойная кислота, реагент При-
Прилежаева)— бесцветное кристаллическое вещество, с т. пл. 41 "С,
слабо растворяется в воде.
Ее получают из бензоилпероксида:
CeH5COOOCOCeH5-f-Na+-OCH3 —> СвН5СООО-\а+ + СвН3СООСН3
Пероксибензойную кислоту используют в органическом синтезе
для получения эпоксидов и для других реакций окисления. Такое
же применение нашли некоторые ее производные, например м-хлор-
пероксибензойная кислота.
Бензоилпероксид—бесцветное кристаллическое вещество, плавя-
плавящееся с разложением при 100 °С, нерастворимое в воде.
Получают из бензоилхлорида и Н2О2 в водном растворе в при-
присутствии щелочи. Используют в качестве инициатора полимериза-
полимеризации в различных реакциях полимеризации.
К. НИТРИЛЫ (ЦИАНИДЫ)
1. МЕТОДЫ ПОЛУЧЕНИЯ
1. Превращения карбоновых кислот и их производных. Для полу-
получения нитрилов часто используются амиды. При взаимодействии с
РОС13 или Р2О5 амиды отщепляют молекулу воды:
.О
R—Cf +РОС13 —> R—С = М^-НРО2С12+НС1
N NHa
В промышленности применяется также каталитическая дегидра-
дегидратация амидов над А1.2О3. Над катализатором пропускают смесь
карбоновой кислоты и аммиака. Происходит образование амида и
его дегидратация:
RCOOH + NH, ~~ R-CN + 2H.0
2. Дегидратация оксимов. Альдоксимы в присутствии кислот от-
отщепляют воду, образуя нитрилы:
. NOH кислота
R —Cf «• R-C = N-|-HnO
3. Получение нитрилов замещением других групп цианогруппой.
Нитрилы получаются алкилированием цианидов натрия или калия:
:C=N -> R —C =
(Х-галоген, OSO2RX)
597
Производные аренов получают арилированием цианида меди (Г)'
в растворе диметилформамида или гексаметилтрнамнда фосфорной
кислоты:
ДМФА
Аг— X+CuCN —-^-* Ar-C^N-fCuX
(X-I, Вг)
Исходными для получения нитрилов могут быть также арендиа-
зониевые соли (гл. XXV.A.3).
2. ФИЗИЧЕСКИЕ СВОЙСТВА И СТРОЕНИЕ
Нитрилы являются бесцветными веществами, первые представи-
представители ряда имеют слабый своеобразный запах и растворяются в воле.
Соединения содержат полярную электроноакцепторную циано-
группу. В цианогруппе оба атома находятся в состоянии sp-гпб-
ридизации:
5—
Если цианогруппа находится у двойной связи или бензольного
цикла, происходит сильная поляризация связей:
5+ /1 8— 5^.
Электронные эффекты (—/, —М) подобны эффектам нитрогруп-
пы, только выражены несколько слабее (см. Введ. 7.3).
3. ХИМИЧЕСКИЕ СВОЙСТВА
Нитрилы присоединяют нуклеофильные реагенты к тронной связи.
В случае слабых иуклеофилов необходима активация группы кис-
кислотами. Если в молекуле нитрила в а-положении имеется связь
С—Н, возможно образование карбаниона и дальнейшие реакции.
Известны реакции гидрирования нитрилов.
1. Присоединение нуклеофильных реагентов. Нитрилы легко реа-
реагируют с сильными нуклеофильными анионными реагентами (кар-
банионами, амидами металлов, щелочами, алкоксидами, тиоля-
тами):
6+ б- /\:-\'а+ „ 0 /NH
(Y=CHR1R2, NHR1, ОН, OR1 SR1)
Реакции нитрилов со слабыми нуклеофнлами (водой, спиртами)
катализируют кислоты, которые образуют водородные связи с ато-
атомом азота. Нитрилы являются очень слабыми N-основаниями
598
л- /NH /NH2X-
f и — R-Qf
(Ri=H, алкил).
Так, при гидролизе нитрилов в присутствии кислот получаются
амиды:
н-нх- JNH /NH»
r—C=N + H2O >R-Of _>R_of
ЧОН ЧО
2. Реакции ct-углеродных атомов. Нитрилы, подобно функцио-
функциональным производным карбоновых кислот, являются слабыми СН-
кислотами, в присутствии сильных оснований образуется сопря-
сопряженный карбанион:
н н
I /> - __ I л 6-
R—C->-C=N: + =В у-*". R—С—C^N:
1 —i/
Такой карбанион легко присоединяется к карбонильным соедине-
соединениям (реакции типа альдольной конденсации) и к нитрилам.
3. Гидрирование. Нитрилы обычно гидрируют каталитически и
получают первичные амины:
R_Cr-N -^ R—CH2NHa
Ni
4. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ
Ацетонитрил — бесцветная жидкость со слабым запахом, кипит
при 81,6 °С, хорошо смешивается с водой и органическими раство-
растворителями. С водой образует азеотропную смесь, кипящую при
76 °С и содержащую 16% воды.
В промышленности ацетонитрил получают каталитическим спо-
способом из уксусной кислоты и аммиака.
Ацетонитрил широко применяется в качестве растворителя. Он
хорошо растворяет органические соединения, а также некоторые
неорганические соли.
Акрилонитрил представляет собой бесцветную жидкость с
т. кип. 77 °С; растворяется в воде, ядовит.
В промышленности акрилонитрил получают каталитически из
пронена, аммиака и кислорода:
О„: NMa
СН2 -СИ — СНз -^СН2--СН — CeheN
Его получают также из ацетилена (гл. IV.-1.1) и дегидратацией
HOCH2CH.,CN.
599
Акрилонитрил легко полимеризуется и образует полиакрило-
нитрил (М—40 000—70 000):
СНа-СН — г —СН2-СН - -СН =CH-CN
CN
Н-1-
Полиакрилонитрил используют для получения синтетического во-
волокна (нитрон, орлон).
При сополнмеризации акрилонитрила и бутадиена получают бу-
тадиеннитрильный каучук.
Акрилонитрил является исходным веществом для различных ор-
органических синтезов, например реакции цианэтилирования:
N + Н—Y~ -?-» У—CHf-CHi—CS5N
Малонодинитрил — бесцветное кристаллическое вещество, пла-
плавящееся при 30—31 °С. Его получают из амида циапуксусной кис-
кислоты:
у {-) POP 1
N = C-CH,-Gf _^NS.--:C-CH2-C = N
NNH2
Малонодинитрил имеет активную метиленовую группу, является
СН-кислотой (рКа«11), легко образует анион, представляет собой
сильный С-нуклеофил. Малонодинитрил вступает в различные
реакции конденсации с использованием как активной метиленовой
группы, так и цианогруип. Его используют в органическом синтезе
для получения соединений с несколькими цианогруппами, а также
для получения гетероциклических соединений.
Адиподинитрил — бесцветная жидкость с т. кип. 295 °С.
В промышленности его получают электрохимической гидроди-
меризацией акрилонитрила:
+ 1с-; 2Н +
КС — СН = СНа -|- СН2 = СН — CN > NCCH2CHaCH2CH2CN
Известны также каталитические методы димеризации акрилонит-
акрилонитрила.
Адиподинитрил является одним из основных исходных веществ
для получения мономеров (адипиповой кислоты, гексаметилендиа-
мииа, капролактама).
Фталодинитрил — бесцветное кристаллическое вещество ст. пл.
141 °С.
В промышленности его получают из фталевого ангидрида и ам-
аммиака:
О
^/\. ,оо.
O + 2NH3.
-:-ЗН,0
Кат.
^/ XCN
Фталодинитрил используют для получения фталоцианнновых
красителей.
Полицианоуглеводороды. Ненасыщенные углеводороды и арены
с четырьмя н больше цнапогруппами в молекуле обладают особыми
свойствами, они являются сильными электропоакцепторами и лег-
легко присоединяют электрон с образованием устойчивых анион-ра-
анион-радикалов (сравните с хипонамн, гл. XXX.3.2):
NCVCK сч
КС. У ,CN
\/У ч/
NC ,CN Ч]/ Nc/^/4CM
NC/ 4-CN NC'-CN CM
тетрзипано стилен тетрэцпаиохииодимаан гексацианобензол
(СЭ=2,75 iB) (СЭ = 2,8 эБ) (СЭ=2,55эВ)
Полицианоуглеводороды с электр о подо норными соединениями
образуют комплексы с переносом заряда и ион-радикальные соли.
Многие комплексы тетрацианохинодиметана обладают металличе-
металлической электропроводностью (органические металлы, см. гл. XXXVII.
Б.2).
Л. ИЗОЦИАНИДЫ (ИЗОНИТРИЛЫ)
Изоцианиды (изонитрилы) только формально можно рассматривать
как производные карбоновых кислот. Фактически они являются
производными нитрила муравьиной кислоты HCN (цианистого во-
водорода) .
Т. Методы получения, а) Алкилирование циани-
цианидов. При алкилировании AgCN галогеналканами наряду с нит-
нитрилами образуются также изоцианиды (изонитрилы):
в+ б- ¦» -
R -X-|-Ag:C = K:
В этой реакции проявляется амбидентный характер цианид-иона,
б) Реакция первичных аминов с дихлор-
карбеном. Первичные амины при взаимодействии с хлорофор-
хлороформом и щелочью образуют изоцианиды. Предполагают, что атом
азота атакуется дихлоркарбеном:
Н с[
-N-ё/
Эту реакцию можно использовать для качественного обнаруже-
обнаружения (но запаху) первичных аминов (изонитрильная реакция,
А. Гофман, 1868).
601
2. Физические свойства и строение. Изоцианиды — бесцветные
ядовитые вещества, обычно жидкости с очень неприятным запахом.
Молекулы изоцианидов содержат полярную тройную связь, при
этом атом азота образует четыре связи, а на углеродном атоме име-
имеется неподеленная электронная пара. Изоцланидная группировка
является по электронной структуре некоторым аналогом оксида
углерода СО. Изонитрилы принадлежат к нуклеофильным реаген-
реагентам.
3. Химические свойства. Применение. Изоцианиды реагируют с
электрофильными частицами по атому углерода. Затем следуют
дальнейшие превращения.
Например, в водном растворе в присутствии кислот происходит
гидролиз изоцианидов до формамидов:
R-fcaE. +н,о+х-^: *-±^с'-±н —- я-й-с'" —.
X -,Оч " Л
-+- R—N—с' + Н+Х~
Н °
Окисление изоцианидов дает изоцианаты, гидрирование — вто-
вторичные амины:
[О] + - Н,
R —N = C = O *- R_N = C: —^R-N—СНЛ
R—N=C=S H
Изоцианиды благодаря высокой реакционной способности ис-
используют в органическом синтезе.
Глава XXXIV
ПРОИЗВОДНЫЕ КАРБОНОВЫХ КИСЛОТ,
СОДЕРЖАЩИЕ РАЗЛИЧНЫЕ
ФУНКЦИОНАЛЬНЫЕ ГРУППЫ
КЛАССИФИКАЦИЯ, ИЗОМЕРИЯ И НОМЕНКЛАТУРА
Наиболее известными из производных карболовых кислот явля-
являются следующие:
1) галогенкарбоновые кислоты,
2) гидроксикарбоновые кислоты,
3) аминокарбоновые кислоты,
4) оксокарбоновые кислоты.
Каждая подгруппа может быть еще подразделена в зависимости
от числа функциональных групп и их положения по отношению
к карбоксильной группе (а-, C-, у-, б-, ...).
602
Таблица 47. Номенклатура галогенкарбоновых, гидроксикарбоновых,
аминокарбоновых и оксокарбоновых кислот
Формула кислоты
С1СН2СООН
CF3COOH
ВгСН2СНСООН
1
Вг
СНзСНСООН
1
он
носнсоон
СН2СООН
носнсоон
1
носнсоон
соон
UL
СНзСНСООН
1
NHj
H2NCH.2CH2COOH
/ч /СООН
Jn,h
^С-СООН
О
|1
СНзССООП
о.
О
II
СН3ССН2СООН
/к /СООН
I о
Название
тркр.иа 1ыюе
Молочная
кислота
Яблочная
кислота
Винная кис-
кислота
Салициловая
кислота
а-Аланин
fl-Аланин
Аитранило-
вая кислота
Глиоксило-
вая кислота
Пировино-
градная кис-
кислота
Малональде-
гидная кисло-
кислота
Ацетоуксус-
ная кислота
Фталальде-
гидная кисло-
кислота
по рационально!!
!:пмемк.:атуре
Хлоруксусиая
кислота
Трифтор уксус-
уксусная кислота
сс,Р-Дибром-
пропноновая кис-
кислота
а-Гидроксппро-
пноиовая кислота
Гидроксиянтар-
иая кислота
Дигидроксиян-
тарная кислота
о-Гидроксибен-
зойная кислота
а-Аминопропио-
новая кислота
р-Аминопропио-
новая кислота
о-Аминобензой-
ная кислота
и-Кетопропионо-
пая кислота
Формилуксус-
ная кислота
Ацетилуксусная
кислота, р-кето-
масляная кислота
о-Формилбси-
зойная кислота
но пгстематимеской
|К)мсч!клагуре
Хлорэтановая
кислота
Трифторэтано-
вая кислота
2,3-Дибромгтро-
пановая кислота
2-Гидроксипро-
пановая кислота
Гидроксибутаи-
диовая кислота
2,3-Дигидрокси-
бутандиовая кис-
кислота
2-Аминопропа-
новая кислота
З-Аминопропа-
иовая кислота
Оксоэтановая
кислота
2-Оксопропано-
вая кислота
З-Оксопропапо-
вая кислота
3-Оксобутано«
вая кислота
еоз
Номенклатура этих соединений аналогична номенклатуре кар-
боновых кислот. Для многих гндрокси- н аминокарбоновых кислот
сохраняются тривиальные названия (табл. 47). Для обозначения
класса соединений часто применяют сокращенные названия —
гидроксикислоты (оксикислоты), аминокислоты, оксокислоты.
А. ГАЛОГЕНКАРБОНОВЫЕ КИСЛОТЫ
1. МЕТОДЫ ПОЛУЧЕНИЯ
1. а-Галогенкарбоновые кислоты получают галогенпрованием кар-
боновых кислот галогеном (кроме иода). Фторирование ве-
ведет к перфторкарбоновым кислотам. В промышленности это осу-
осуществляется электрохимически в растворе жидкого HF. Продук-
Продуктами реакции являются ацилфториды:
HF /О Н„О
СН3СООН > CF3 — С" —-* CF3COOH
электролиз \р
CnH2n + 1COOH ^^^ CnF2n + 1C^ -ScBF,Hf lCOOH
Хлорирование карбоновых кислот происходит при на-
нагревании и освещении реакционной смеси. При хлорировании ук-
уксусной кислоты образуются все три возможных продукта:
СН3СООН -1* С1СН2СООН —^ С12СНСООН —^ ССЛ3СООН
t° t t°
Хлорирование других карбоновых кислот (пропноновой, мас-
масляной и др.) дает не только а-, по также р- и -^-замещенные хлор-
карбоновые кислоты. Преимущественно в сс-положение карбоновые
кислоты хлорируются в присутствии РС13.
Бромирование карбоповых кислот идет в присутствии
красного фосфора (или РВг3). Продуктами реакции являются бро-
бромиды сс-бромкарбоновых кислот. В этой реакции вначале образу-
образуются ацилбромиды, которые затем легко бромируются. Предпола-
Предполагают, что бром реагирует с енольной формой ацнлбромида:
,0 р ,0 ,ОН ,,г .О
RCH2Gf --> RCH2C^ ^±RCH-C/ -Л RCHGf
4OHBf2 чВг ЧВг | Вг
Вг
Реакции бромирования и хлорирования карбоновых кислот
в присутствии красного фосфора открылн п исследовали К. Хелль
A881), Я. Фольгард A887), Н. Д. Зелинский A887).
а-Фтор- и сс-иодкарбоновые кислоты получают реакцией нук-
леофильного замещения атомов хлора или брома:
RCHCOORM Na-iX- -> RCHCOOR]
Cl X
(X = F, I; R^H, алкил).
604
2. р-Галогенкарбоновые кислоты получают из а,р-непасыщ,ен-
иых карбоновых кислот:
fi+ 6-
RCH=CHCOOH ;-H —X —*RCHCH2COOH (здесь X^Cl, Br, I)
X
3. V-, 6-, е- и (о- Галогенкарбоновые кислоты'имеют свои специ-
специфические методы получения. В лаборатории их можно получить из
соответствующих гндроксикислот.
Галогенкарбоповые кислоты С1(СН2СН;.)„СООН получают из
1,1,1 ,(о-тетрахлоралкапов:
н,о
С1(СН2СНо)исС1з
1,SO4
С1(СН2СП2)„СООН («=-2,3,4,5)
которые, в свою очередь, получают реакцией теломеризации
(гл. П.4.6).
2. ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
Галогенкарбоновые кислоты представляют собой бесцветные жид-
жидкие или кристаллические вещества, растворимые в воде. Растворы
имеют кислую реакцию.
1. Кислотность. Галогеикарбоповые кислоты являются более
сильными кислотами, чем незамещенные (табл. 48), и легко обра-
образуют соли:
RCHCOOII + H2O^r RCHCOO- + H3O +
I I
Cl CI
Таблица 48. Константы кислотности рКа некоторых карбоновых
и галогенкарбоновых кислот в водных растворах
Кислота
СНзСООН
FCH.COOH
CICH.COOH
ВгСН2СООН
1СН2СООН
СС13СООН
СНзСООН
4,76
2,58
2,86
2,90
3,17
0,65
-0,2
Кислота
СН3СН2СН2СООН
СНзСНаСНСООН
I
С1
СН3СНСН2СООН
1
CI
С1СН2СН2СН2СООН
Р'<а
4,82
2,85
4,05
4,60
В значениях констант кислотности галогепкарбоповых кислот
ясно прослеживается электроноакцепторное действие атомов гало-
галогена (индуктивный эффект —/) в зависимости от типа галогена,
числа атомов галогена и их положения (расстояния от карбоксиль-
карбоксильной группы). Наиболее сильной карбоновой кислотой является
трифторуксусная. Индукционный эффект атомов галогена увели-
увеличивается в ряду I, Br, Cl, F п его действие уменьшается с увеличе-
605
нием расстояния от реакционного центра (карбоксильной группы)
(см. индуктивные константы в Введ. 7.3).
2. Реакции в карбоксильной группе. Галогенкарбоновые кислоты
образуют все известные функциональные производные, например
ацилхлориды, сложные эфпры, амиды:
2sf 2 ,
ХС1 ОЦ ЧМН2
3. Реакции нуклеофильного замещения атомов галогена. а-Га-
логенкарбоновые кислоты и их производные легко реагируют с
различными нуклеофильными реагентами. Скорости реакций, как
правило, больше, чем в случае галогеналканов, что объясняется
индуктивным эффектом карбоксильной группы:
Н Н
в + 1 /О I /уО
R—С —О^ -L.-NuH -^R — С—С" -fHX
| XY | 4Y
X :Nu
(:NuH = H2O, NH3, HNR^2, HSR1 u др.).
В случае реакций карбоксилат-иоиа а-галогенкарбоновых кис-
кислот с нуклеофильными реагентами происходит внутримолекуляр-
внутримолекулярная стабилизация карбокатиона. Реакции обычно протекают со-
согласно механизму SNl (гл. VII.4.3), но с сохранением конфигура-
конфигурации:
R " R.
Конкретный пример приведен при рассмотрении вальденовского
обращения (гл. XXXIV.B.3).
сс-Галогенкарбоновые кислоты являются исходными вещест-
веществами для получения гидроксп- и аминокислот.
Аналогичные реакции протекают с Р-, у-, б- и со-галогеякарбо-
новыми кислотами. Для |3-галогенкарбоновых кислот характерны
также реакции отщепления, которые могут протекать одновре-
одновременно с реакциями нуклеофильного замещения:
RCH -CH2COOFI —-> RCHCHoCOOH +RCH = СНСООН
X l:Xu
4. Декарбоксилирование. а-Днгалогенкарбоновые кислоты и
особенно а-тригалогенкарбоновые кислоты при нагревании декар-
боксилируются:
СХ3СООН-*СМХ3 + СО2 (Х = С1, Вг)
606
Легко декарбоксилируются соли тригалогенуксусных кислот,
они служат источником дигалогенкарбенов:
CX3COO-Na -С X3C4
X3C:-Na+^:XnC: + Na+X- (X = Cl, Br)
3. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ
Хлоруксусная кислота — бесцветное кристаллическое вещество,
плавящееся при 61°С; хорошо растворяется в воде и органических
растворителях.
В промышленности хлоруксусную кислоту получают хлориро-
хлорированием уксусной кислоты в присутствии хлоридов фосфора или
серы. Применяют также метод гидратации и гидролиза трихлор-
этилена в растворе 90%-ной серной кислоты при 180 °С:
IUSO, Н2О
СС12 = СМС|-;-Н2О——>С12С-СНС1 -> НООССН2С1
I I
он н
Хлоруксусная кислота является важным исходным веществом
для получения красителей, нонитов, гербицидов, комплексонов
и др.
Трифторуксусная кислота — бесцветная жидкость с острым
запахом; т. кип. 72,4 °С, на воздухе дымит, при соприкосновении
с кожей вызывает раздражение и ожоги. Неограниченно смеши-
смешивается с водой и органическими растворителями.
В промышленности ее получают электрохимическим фторирова-
фторированием уксусной кислоты или уксусного ангидрида.
Трифторуксусную кислоту используют в органическом синтезе
в качестве растворителя и эффективного кислого катализатора. Из
производных трифторуксусной кислоты используют ангидрид
(CF3CO)aO — бесцветную жидкость с т. кип. 38,5 °С. Это эффек-
эффективный катализатор для реакции ацилирования и водоотнимающее
средство.
Б. ГИДРОКСИКИСЛОТЫ
К гидроксикислотам относятся гидроксикарбоновые, гпдрокепди-
карбоновые, гидрокситрикарбоновые, дигидроксикарбоповые, ди-
гидроксидикарбоновые кислоты и т.д., а также феполкарбоновые
кислоты.
1. МЕТОДЫ ПОЛУЧЕНИЯ
Т. а-Гидроксикислоты легко/получают из сс-галогенкарбоновых кис-
кислот:
RCHCOOH-f H2O-^ R—CHCOOH + HQ
Ct OH
607
Вторым методом является циангидршшый, который заключа-
заключается в присоединении цианистого водорода к карбонильному сое-
соединению с последующим гидролизом образовавшегося ос-гндроксн-
нитрила (циапгидрипа):
ОН ОН
COOH
R1 Rl
/О HCN | Н2О |
—->R_C-C = N——rR-C-
.^i KCN ¦ li+X I
R1 Rl
Некоторые а-гидроксикислоты найдены в природных продуктах.
Природные а-гидроксикислоты оптически активны. Синтетически
получаются оптически неактивные сс-гидроксикислоты (рацемиче-
(рацемические смеси).
2. р-Гидроксикислоты легко получаются по методу, разработан-
разработанному С. Реформатским A887). Реакция Реформатского заключается
во взаимодействии сложных эфиров сс-галогенкарбоновых кислот
с карбонильными соединениями в присутствии цинка:
/°
R — С" -fZn-f ВгСН2СООС2Н5—>
OZnBr ОН
-*R —С —CH2COOC2H8iil^lR—С—СН2СООН
| Н2О |
Rl R1
Предполагают, что промежуточными продуктами в этой реакции
являются цинкорганические соединения BrZnCH2COOC2H5, кото-
которые присоединяются к карбонильной группе.
К другим методам получения р-гидрокснкислот относятся при-
присоединение воды к а, р-ненасыщенным кислотам, гидролиз р-
галогенкарбоновых кислот, гидрирование р-оксокарбоновых кис-
кислот.
3. V- и б-Гидроксикислоты получают различными методами.
В качестве примера можно привести получение у-гидроксимасля-
ной кислоты:
НОСН2СН2СН2СН2ОН -
О2; Cu/SiO2
200- 250 "С
NaOil
= 0
HOCH2CHjCH2COO-Na +
n + x-
—11,0
НОСН,СН..СН-,СООН
Каталитическим окислением бутандиола-1,4 или тетрагидрофурана
получаются лактон у-гидроксимасляпой кислоты (у-бутиролактон,
бутанолид), щелочным гидролизом которого получают у-гидрокси-
кислоту в виде соли.
4. Дигидроксикарбоновые и дигидроксидикарбоновые кислоты
могут быть получены из ненасыщенных карбоповых кислот окисле-
608
нием двойной связи КМпО4 пли HiO2:
соон
н
(R = H, Alk.COOH)
Окисление КМпО4 происходит стереоселектнвно, как цис-
присоединение (Гл. II. 4.4).
5. Феполкарбоновые кислоты могут быть получены из производ-
производных ареиов известными методами введения карбоксильной группы
или гидроксилыюй группы в бензольный цикл. Широкое промыш-
промышленное применение нашло карбокенлировапие фенолов в виде со чей
(фенолятов) СО2. Реакцию открыл Г. Кольбе A860) и усовершен-
усовершенствовал Р. Шмптт A885) проведением процесса под давлением:
он он
ifft-CJ
,C00~v,-r !
9
COO Na+
В качестве побочного продукта образуется я-гндрокепбеп :он-
ная кислота. Реакцию обычно проводят при температурах 100. . .
. . .200 "С и под давлением I—10 МПа.
2. ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
Гндрокснкислоты являются бесцветными жидкими или кристалли-
кристаллическими веществами; они растворяются в воде, имеют кислый вкус.
1. Пространственная изомерия и оптическая активность. Моле-
Молекулы всех а-гидроксикислот, кроме гликолевой кислоты, содержат
асимметрический атом углерода, они хиральны. Чистые энантно-
меры оптически активны, их удельное вращение равно по величине,
но противоположно по знаку. Смесь равных количеств энантиоме-
соон носе ров оптически неактивна (рацемат,
I I рацемическая смесь).
н^^^он ho^jXh сс-Гидроксикислоты, подобно гидро-
r R кспальдегидам и моносахаридам, мо-
d-.r- l-,s- гут быть причислены kD- или L-ряду.
По /^.S-поменклатуре D-ряд обычных сс-гидроксикислот (R--=
—-алкил, арил) и а-гидроксидикарбоновых кислот (R=CH2COOH)
соответствует R-копфигурацни (см. гл. VII. 3.3, гл. XXIX.Б.1,
старшинство заместителей уменьшается в ряду ОН, СООН, R).
2. Кислотность. Все гндроксикислоты являются более сильными
кислотами по сравнению с обычными карболовыми кислотами
(табл. 49), здесь проявляется электроноакцепторпый индуктив-
индуктивный эффект (—/) гидроксилыюй группы.
20 К! 517 fO9
Таблппа 49. Константы ионизации рКа некоторых
гидроксикислот (Н2О, 25"С)
Соединение
СНаС1ЦОН)СООН
НОСН(СООН).
НООССН(ОП)СН(ОН)СООИ
// •¦ /
^ хсоон
¦•'VI
3,00
2,37
2,93
3,00
>
4
4
,74
,23
3. Реакции карбоксильной группы. Для гидрокенкпелот харак-
характерны обычные реакции карболовых кислот: ионизация карбоксиль-
карбоксильной группы, образование солен, сложных эфиров, амидов, ацкл-
хлоридов. Отчасти осуществлению этих реакций мешает присут-
присутствие гидрокенльной группы, которая тоже может вступать
в реакции. Поэтому ее обычно «защищают», превращая в простую
или сложноэфнрпую группировку:
sod. /.О
RCHCOOH —>. RCHCOOH >~ RCII-C^ _v Другие
| | | ХС1 производные
ОН OR1 OR1
4. Реакции гидроксилыгои группы. Гидрокснкнслотам свойст-
свойственны обычные реакции спиртов, например, они образуют простые
и сложные эфиры, окисляются н др. Проведению отдельных реак-
реакций мешает карбоксильная группа, поэтому ее защищают превра-
превращением в сложноэфирную группировку.
рТндрокснкислоты легко отщепляют воду н образуют ненасы-
ненасыщенные карбоповые кислоты.
5. Реакции, в которых карбоксильная и гидроксильная группы
участвуют совместно. Взаимодействие карбоксильной и гпдроксиль-
ной групп гидроксикислот может привести к образованию цикли-
циклических сложных эфиров — лаптопов или лактидоа.
а-Гндроксиккслоты образуют лактпды — циклические сложные
эфиры — из двух молекул кислот:
О
Н
— R
СН -!
/)
1!+ X
О
_
'¦-* II
О
он
Ч)Н
\с
' R
а-лактои лонтид
Стабильные а-лактопы до сих пор не получены.
610
Р-Гидрокспкислоты .могут быть превращены в р-лактоны только
специальными методами:
— СП —
1
он
Rl
i
1
Rl
,0 ,. о
ОН
Rl
|
r _СЦ —С—Rl
I I
о—сч
¦у.б-Гпдрокспкпслоты очень легко образуют стабильные у- и
б-лактопы:
R-cH-cn..-(Cii.o.,-c' —-2*
| " ' ' ОН СП С„
Oil R -. ' 4J
о
(здесь п 1, 2).
Своеобразно реагируют к-гпдгюкспкпслоты в растворе кон-
концентрированно!'! серной кислоты. Опп расщепляются с выделением
СО п Н;0, в результате с.-гндрокспкнслоты превращаются в альде-
альдегиды пли кетоиы:
R1
| м.яо, R1
R—С —СООП— > R —С;ч -fCO- H..0
| '° "-0
ОН
(R---1I, алкпл, арпл).
з. в.\;-кпг.йШ11Е пре;т^стлвптплн
Глпколсваа кислота НОСИ/100Н — бесцветное кристаллическое
ьещестБО с т. пл. 80 'С, хорошо растворимое в воде.
Она найдена в природных продуктах (свекловичном соке, не-
незрелом винограде). Синтетически ее получают из хлоруксусноп
кислоты и воды.
Гликолевую кпсюту применяют в органическом синтезе.
Мо.ючная кисло/па СН3СН(ОН)СООН существует в трех ((юр-
мах: две оптически активные (энацтпомеры) и отнчески неактив-
неактивная рацемическая смесь. Определена абсолютная конфигурация
правовращающей н левовращаю;ден молочных кислот: левовра-
щакхдая принадлежит к D-ряду (/^-конфигурации), правовращаю-
правовращающая— к L-ряду (S-Koii фигурация):
соои ноос
сн, си.,
К 1 )-мелочная кислига D (—) -молочная кислота
20* 611
Чкстля Ц-;-)-молочпая кислота — бесцветное, очемг> гигроско-
гигроскопичное кристаллическое вещество с т. пл. 26 С. Однако обычно
она \wncr вид вязкой жидкости. Удельный угол вращения [а]д: —
-----З.Й" A0пг)-иый водный раствор). Впервые она была выделена
из >.:ясного экстракта (мясомолочная кислота).
(_^)-\\олочпая кислота—вязкая жидкость (т. пл. 18 СС), об-
образуется в процессе молочнокислого брожения углеводов, содер-
содержится в кислом молоке, кзашепоп капусте, соленых огурцах.
Молочную кислоту используют в пищевой и кожевенной про-
промышленности, в крашении тканей.
Яблочная кислота НООССН(ОН)СН2СООН предстазляет собой
бесцветное кристаллическое вещество с приятным кислым вкусом,
хоромю растворимое в воде. Известны два энантномера яблочной
кислоты н оптически неактивная рацемическая яблочняя кислота:
соон
1
сн.соон
)-яГлочная кислота
ноос
1
1
с
сн.соон
?>( ; )-ябл1тш1!! кислота
L-Мзомер отвечает S-конфш-урации, так что можно писать:
5(—}-яблочная кислота и /?(-;-)-яблочная кислота.
Ц—)-Яблочная кислота пллвится при 99. . .100 'С, [a] D—
=—5,7 (в ацетоне). Ее получают из природных продуктов (содер-
(содержится в незрелых яблоках, ягодах рябины, махорке) или разделе-
разделением (-")-яблочной кислоты на энантио.меры. D(-\-)-Яблочпая кис-
кислота плавится при 99. . .100 С, \а\ п—-\-Б,Т (з ацетоне), получают
ее из (—)-яблочнон кислоты или восстановлением (-г)-винной
ь'нсюты иодоводородом. (-^)-Яблочная кислота плазится при
129. . .i30"C, получают ее гидролизом хлор- или бромянтарггых
кислот пли присоединением воды к малеиновой кислоте.
Яб"точную кислоту используют в органическом синтезе и ме-
медицине.
При изучении реакций яблочной кислоты П. Вальден A8f'6)
обнаружил явление, при котором из (-г)-изомеров можно получить
(—)-п:,омеры и наоборот, т. е. из (—)-яблочной кислоты можно
получить (- )->;лорянтарную ;снслоту, а из нее (-г)-яблочную кис-
кислоту. Соответствен по из (-г)-яблочноп кислоты можно получить
(—)-хлор янтарную кислоту, а затем (—)-ябло'-шую кислоту. Таким
образом Вальдену впервые удалось осуществить обращение конфи-
конфигурации при асимметрическом углеродном атоме. Это язлепне
названо вальдгнозским обращением.
Объяснение вальдеповского обращения стало возможным только
значительно позже, когда была создана теория о пространствен-
пространственном течении реакций у тетра^дрпческого атома углерода (механиз-
(механизмы S.vl н S.Y2, гл. VII.4.3, XXXIV. А.2):
612
JD(+) I
5,vl,
сохранение
конфигурации
соон
I
с.
CH.COOH
11.0
Ag.O
1О1Г)
COOH
CH.COOH
PCI,
СООН
5,\ 2, обращение
конфигурации
PC,
i ? .Cifpamfht.il
?(-0
CH,COOH
H.O I 5,vl.
Ар О 1 сохранение
(Gli~) I конфигурации
COOH
С
CH.COOH
Винные кислоты НООССН(ОН)СН(ОН)СООН — бесцветные,
растворимые в воде кристаллические вещества с приятным кислым
вкусом. Молекулы их содержат два асимметрических углеродных
атома с одинаковым набором заместителей; известны два оптически
активных изомера (-[-) п (—), оптически неактивная рацемическая
(^)-винная кислота и оптическая неактивная мезовинная кислота,
которую невозможно расщепить на оптические антиподы (см.
гл. VII. 3.3). Ниже изображено пространственное строение винных
кислот при помощи формул Фишера, клиновидных проекции и фор-
формул Ньюмена. Клиновидные проекции изображают два контрме-
контрмера, один из которых соответствует формуле Фишера. Для обозна-
обозначения стереонномеров применена ??,S-CHcreMa:
COOH
H —
но—
—OH
—H
COOH
coon
HO —
H—
—я
—OH
COOH
COOH
H—
H—
—OH
;--0H
LOOH
COOH
с
1
1
с
соон
r,r ( ;-)-аи
соон
с
1
1
с
Н ^' ' >^0Е1
соон
соон
с
1
1
f
н*^' ^он
соон
соон
с
с
Н-' ^ ~~0Н
соон
шая кислота
соон
с
с
Н0''^-Н
соон
ц|;ая кислота
соон
(
с
НС'''
'>
н>
НОч
(
но^
нч
соон
кн
—^ч
соон
соон
Л)
ЧН
соон
соон
к
JOOH
jRtS -винная кж.юха, ке^о?икная
613
/?,/?(-!-)-Вин»ая кислота плавится при 170 °С, [а]$ 4-12°
A0%-пый водный раствор). Ее получают из кислой калиевой соли
(виннокаменной соли), которая образуется и накапливается в про-
процессе производства вина. Впервые (-!-)-виппую кислоту получил
К. Шееле A769).
S,S(—)-Винная кислота плавится при 170 СС, [а]^ ——12\
lie получают из (±)-виниой кислоты разделением па оптические
антиподы (см. гл. XXXIV.Г.4).
(-?--)-Виш1ая кислота (виноградная кислота) плавится при
205 СС (безводная). Ее получают окислением фумаровой кислоты
пермапганатом калия.
Мезопинная кислота (^,5-вииная кислота) плавится при 140 °С
(безводная). Получают ее окислением маленновой кислоты перман-
ганатом калия.
Молекулы R,R- и S.S-Biunibix кислот хиральны, но не асиммет-
асимметричны, так как имеют ось симметрии (в центре молекулы), прохо-
проходящую перпендикулярно к плоскости чертежа. Молекула мезозин-
иой кислоты ахиральна, имеет центр симметрии. Следует обратить
внимание па то, что упомянутые элементы симметрии наблюдаются
не во всех коиформерах.
Соли винной кислоты называются тартратами.
Практическое применение нашли соли (-[-)- или (^)-вшшой
кислоты.
Калий-натрийтартрат NaKC4H4O6i4H4O, известный под на-
названием сегнетовой соли, кристаллизуется в виде ромбических призм,
обладает пьезоэлектрическими свойствами.
Винная кислота с ионами тяжелых металлов образует внутри-
комплексные соединения. Например, натрий-медьтартрат является
основной составной частью раствора Фелинга
Н
илтрии-'ледьтартрат
Лимонная кислота — беецзетпое кристаллическое вещество, пз
воды кристаллизуется в виде моногидрата, безводная кнелога пла-
плавится при 153 СС.
СН2СООН
I
ПО—С —СООН
СН2СООН
Лимонная кислота распространена в природе (лимонный сок,
табачные листья и др.). В промышленности ее получают фермента-
614
тивкой обработкой растворов глюкозы. При сбраживании раство-
растворов мелассы грибами Asper^illus ni-^r получают растворы лимояпоГ1
кислоты (лимоннокислое брожение).
Лимонную кислоту применяют в шнцеьоп промышленности и
фармакологии. Сложные эфир и лимонной кислоты используют
в качестве пластификаторов.
Сиищилоеая кислота — бесцнгтное кристаллическое есщссчпо,
плавящееся при 159'С, легко возгоняется, растворяегся в горячей
воде.
.ОН
Встречается в природных продуктах, в основном в виде метило-
метилового эфира. Синтетически ее получают из фенола.
Салициловая кислота дает характерное для фенолов фиолето-
фиолетовое окрашивание с FeCl3.
Салициловую кислоту п ее производные используют в меди-
медицине, например ацетилсалициловую кислоту (аспирин), метило-
метиловый эфир салициловой кислоты (противоревматическое средство).
Галловая кислота — бесцветное кристаллическое вещество с
т. ил. 220 2С (моногидрат), хороню растворяется в воде. Легко
окисляется, поэтому при хранении постепенно темнеет.
COOII
A
он
Галлогзая кислота содержится в природных продуктах (дубиль-
(дубильных веществах, танине), из которых ее и получают. Водные раст-
растворы дубильных веществ используют для денатурации белков (глав-
(главным образом при обработке кожевенного сырья). Дубильные ве-
вещества содержатся в чернильных (дубильных) орешках, листьях
чая и в дубовой коре. Главной составной частью дубильных: ве-
веществ являются танины. Некоторые танины представляют собой
гликозиды галловой кислоты.
Используют галловую кислоту для синтеза красителей, для по-
получения пирогаллола и в качестве аналитического реагента.
В. АМИНОКИСЛОТЫ
Производные карболовых и дикарбоновых кислот с одной или не-
несколькими аминогруппами в молекуле составляют важную группу
органических соединений — аминокарбоновых кислот, или просто
аминокислот.
615
1. МЕТОДЫ ПОЛУЧЕНИЯ
1. а-Аминокислоты получают из природных веществ и синтети-
синтетически.
Белки при гидролизе в водных растворах в присутствии кислоты
дают смесь а-аминокислот. Из этой смеси различными способами
можно выделить индивидуальные ос-аминокнслоты. Все получен-
полученные из белка а-амииокислоты (кроме ампноуксусной кислоты)
являются оптически активными.
Для синтеза аминокислот исходными веществами могут быть
а-галогенкарбоповые кислоты, альдегиды, галогенуглеводородьг.
а-Галоген кар боковые кислоты легко реагируют
с аммиаком и образуют соли аминокислот:
RCHCOOH +3NH3 ^-+ RCHCOO-NH; + NH*+Cl-
Cl NH2
Альдегиды реагируют с цианистым водородом и аммиа-
аммиаком, образуя нитрилы а-амипокислот. В качестве реагента приме-
применяют смесь KCN и NH4C1 (метод Штреккера — Зелинского, 190р):
/О /О
^ / 3-f KCl-*
OH NH,
-* R-C-C—N + NHj + KCI -»-R—C-C==-N + H..O + KC1
к i,
Гидролиз нитрила дает а-аминокислоту:
I н,о |
R —C —C^s N > R —С— СООН
H H
Галоген углеводороды алкплнруют ацетиламипо-
мллоиовын эфир. Синтез закапчивается гидролизом и декарбоксили-
ровапием продукта реакции:
О СООС2Н5 О СООС2Н5 NH2
64- а- И | Г I н.о I
R — Х-|-СН3С\Н— C-Na+ —»CH3CNH —С —R ---Х'аХ—:—-RCHCOOH
I | ы+х"
COOQH5 COOCjH5
В результате синтезов получаются оптически неактивные ра-
рацемические а-аминокислоты.
2. р-Аминокислоты получают присоединением аммиака к а,
Р-ненасыщеиным карбоновым кислотам. В. М. Родионов A926)
предложил удобным метод осуществления этой реакции, сочетая по-
получение ненасыщенной кислоты с присоединением аммиака в одну
стадию. Реакция осуществляется при взаимодействии альдегида,
G16
малоносои кислоты и ацетата аммония в растворе уксусной
.О СОО11 л+ COOII
r_C^ -'II..C' -^R—CH-C7
4 II " СООН COOII
MI, MI.,
:\H,
.R_CH-CH
COOII
"СООП
r— гиснхооп : со.
3. Получение у-, 6-, е- и о>аминок!:слот. Для получения nvur-
нокнслот с отдаленной от карбоксильном группы аминогруппой
применяют различные специфические методы. Часть этих методе:',
заключается в получении соответствующих циклических ампдоз
аминокислот — лактамов и их щелочном гидролиз':.
у-Бутпролактам (пнрролндоп) получают каталитическим гид-
гидрированием имида янтарной кислоты:
Н2С-СН2
HX-CEL
н„ м
\ .он
О=С С-О
\ .
N
Н
н,с с=о
\/
N
II
-ILO
- 11...\С1ЬС11,С1|.,СОО-\а +
I! ' X -
I
1..ХС[1ХН.,СНгСООН
Гидролиз у-бутнролактама щелочью дает соль •у-амнномасляпон
кислоты, из которой подкисленисм можно получить саму кислоту.
у-Аминомаслянан кислота легко циклизуется в у-бутнролактам.
6-Валеролактам и е-капролактам получают путем бекмановскоп
перегруппировки оксимов:
СН2
НоС-(СН.,)„
H2C C = NOH
Clb
(я-1. 2)
»,SOl
H2C (СН,)„
к?.он
¦ I
II2C C=O
H
t
¦ Н.>\СП,СП-.С[ 1„ (CI!..)„ СОО-\а +
-I! ,O
1-х-
Исходными веществами для синтеза 6- и ш-аминокпелог могут
быть также продукты теломернзацип этилена и CCI i:
. I! -X -
Cl (CII2CH,)n CCI3
(С1ЬС1Ь)„
Н,\
I.,)., COOII (л .--2-5)
4. Получение аминоаренкарбоновых кислот. В отличие от других
аминокислот в молекуле аминоарепкарГюповых кислот как амино-
аминогруппа, так и карбоксильная группа находятся у бензольного цнг:-
617
ла. Обычно применяют восстановление нитроаренкарбоновых кис-
кислот :
соон
Для получения о-амиполрепкарбонопых кислот часто используют
расщепление пмндов о-дпкарбоновых кпслог по методу Гофмана
(с. 586):
О
О
II
с
,С\Н2 /ч N11., ,.. . МЬ
\iiOH '' |
II i;.-.,
ч- \/ '^ ' ' COO-.Nj'- ^'COO-\a ^ ••' COOII
О
2. ФИЗИЧЕСКИ?: I! ХИМИЧЕСКИЕ СВОЙСТВА
Аминокислоты представляют собой бесцкегпые кристаллические
веществ?., растворяющиеся в воде, труднее — в органических раст-
растворителях, некоторые из них имеют сладкий вкус Для них харак-
характерны высокие температуры плавления (плавится с разложением).
1. Пространственная изомерия и оптическая активность. Все
«-аминокислоты, кроме амппоукеусной кислоты, содержат асим-
асимметрический атом углерода, их молекулы хнральпы. Это значит,
что каждая и-амнпокислота образует два оптически активных энан-
Т'юмера и оптически неактивную рацемическую смесь обоих эпан-
1 номеров:
соон ноос
Подобно гидроксикпслотам, аминокислоты причисляют к L-
нлн D-ряду. /--ряд соответствует 5-конфнгурацин (старшппсгБО
заместителей уменьшается в ряду NH_,, COOII, R).
2. Строение в кристаллическом состоянии, основность и кислот-
кислотность. Молекулы аминокислот обладают двумя функциональными
группами, противоположными по характеру: кислой карбоксиль-
карбоксильной группой и основной аминогруппой. Это создает возможность
образования внутренних солей — бетаинов:
+ !ЧНз
I +
R —С —СОО- НаМ(СН2)„СОО-
н
618
Как показывает рептгепоструаурный анализ кристатлов а-
аминокислот, карбоксильная группа находится в ионизированном
состоянии (длины связей С—О почти одинаковы):
н
В виде бетаинов существуют также и другие аминокислоты
(р*-, у-, б-, е-, со-). Для аминоаренкарбоновых кислот это менее ве-
вероятно ввиду меньшей основности аминогруппы.
В водных растворах, в зависимости от рН среды, существует
равновесие между различными количествами бетаина, аннона или
катиона:
NH2 NHa NH3
| -11 + j -'II- |
r_c_coo- -—;r-c-cooh- -—"r-c-cooh
I +h+' ! -и f '
H H H
анион бетаин вятион
t
R —С —COOH |<-
н +
H
аминокислота
Соотношение бетаина, аниона, катиона и аминокислоты г. равно-
равновесии определяется соответствующими константами ионизации.
Про тонированная а-аминокислота (катион) является сильной ОН-
кпслотои (р^„«2,34. . .2,35), поэтому происходит главным обра-
образом ионизация карбоксильной группы, образуется бетаин. Иони-
Ионизация группы NHu и образование аминокислоты происходит в
очень незначительном количестве (около 105 раз меньше), так как
p/<:NH., »7. . .8.
Бетаин является слабой NH-кислотой, при его ионизации об-
образуется апноп аминокислоты; р/(бст»9,7. . .9,9. Протонирсря-
пие аниона происходит у атома asoia как более основного иенiрэ
(константа кислотности рКа аминокислоты может быть около 3—4).
Нотами как иапменее кислая форма в водных растворах преоб-
преобладает в широком интервале рП. Концентрация аминокислоты со-
составляет только 10~3 —10~° от концентрации бетаннл.
Имеется определенное значение рН, при котором содержание
бетлппа наибольшее. Это значение определяется на основе измере-
измерения электропроводности раствора аминокислоты при различных
рН. Наименьшая электропроводность наблюдается при наиболь-
тем содержании бетаина. Это значение рН называется изоэлект-
рической точкой и обозначается рН,. Для а-амимокислот рН/~6,1-
При отдалении аминогруппы от карбоксильной группы изме-
изменяется и константа кислотности. Рассмотрим это явление на при-
примере Р-аланнна:
-11+ + 4Н+ +
IbNCH.,CH.,COO-' уПЛ'С11оСН,СОО- " Н3\'СНХН2СООН
J-H+ -Н-
рКбет-10,25 рЛ'ц = 3,55
И в случае р-аланина наименее кислой формой является бетаин.
Зто относится также ко всем другим аминокислотам, кроме произ-
производных ариламинов. Например, /г-аминобензойная кислота суще-
существует в основном в форме аминокислоты с константой ионизации
р/Сп—4,85. Ввиду малой основности аминогруппы бетаин легко
ионизируется по связи N—II (рК6С1~3. . .3,5).
Днаминокарбоновые кислоты и аминодикарбоновые кислоты
тоже образуют структуры внутренних солей (бетаинов), но в то же
время вследствие присутствия второй амино- или карбоксильной
группы сохраняют соответственно основную или кислую реакцию.
Диаминокпслоты по основности сравнимы с аммиаком, аминоди-
аминодикарбоновые кислоты по кислотности превышают уксусную.
3. Реакции карбоксильной группы. Аминокислоты существуют,
виде внутренних солей, с основаниями они образуют обычные соли:
\Н3 NH2
R—CH—COO--|-NaJOH- —>¦ R—CHCOO"Na++ Н.,0
С ионами тяжелых металлов аминокислоты образуют соли,
в которых осуществляется внутрикомплексная связь:
о
</ NNH, Na+~OOCCH, H'iC—СН->/СНаСОО Na+
N. S N N
Си Н-(/ 'Ч /' ^СН2
Н2 С—С
\
Ам;;!!од;и-:арбоновыс кислоты связывают ионы тяжелых метал-
металлов в комплексе, который растворяется в воде. Такие аминокис-
аминокислоты называют комплексонами. Наиболее известным комплексо-
ном является этилендиаминтетрауксуспая кислота (трнлон Б)
(HOOCCHohNCH2CH2N(CH2COOH),.
Аминокислоты легко образуют сложные эфиры, которые могут
быть превращены в амиды:
NH3
I
RCHCOO- -C
С20
+
NH3 Cl-
1
RCHCOOC2
NH,
NH.,
1
RCHC
/°
\"
4NH
В реакции этернфикацип образуются соли сложных эфнров, из
которых при действии щелочи получаются свободные эфиры.
Получить хлораягпдрпды и ангидриды непосредственно из ами-
аминокислот обычно не удается, так как в реакцию вступает и амино-
аминогруппа. Поэтому аминогруппу защищают, обычно ацнлированпем,
и после этого получают хлорангндрнды Х'-ацилампнокнслот.
4. Реакции аминогруппы. Аминокислоты дают все реакции пер-
первичных аминов. Лцилпровашг: п алкялнрованме проводится в ще-
щелочной среде, чтобы аминогруппа не была протоппрована:
G
NH2 H С
I /О X
R —CHCOO-Na*-- R1—
С1
R—СМСООН
-• \аС1
При реакции с азотистой кислотой происходит диазотнрование
и распад дназонневой соли с образованием гидроксмкпслоты:
NH3 \2 ОН
I I
RCHCOO- + HXO, — RCHCOO--i-2H2O -> RCHCOOH -;-Nr., --,- Н,0
В этой реакции выделяется азот, что используется для количест-
количественного определения а-амннокпслот в растворах (Д. Ван Сллкк,
1910).
Диазотированием сложных эфнров а-амипокислот получяют
диазоэфи'ры —¦ желтые взрывчатые жидкости, которые используют
в органическом синтезе:
Гч'Нп С1- \,
! н\о„
RCilCOOCoHj -R—С—СООСНд
Строение и свойства их аналогичны дназокетонам (гл. XXIX.Д).
5. Реакции с одновременным участием карбоксильной и амино-
аминогруппы. Одновременным присутствием в одной молекуле карбо-
карбоксильной и аминогруппы обусловлены некоторые специфические
реакции аминокислот: образование внутренних солен (бетаиноз);
образование амидной (пептидной) связи, как межмолекулярпом,
так и внутримолекулярной; и, наконец, распад а-ампнокнслоты иод
действием окислителен.
а) Образование пептидной связи. При нагрева-
нагревании а-ампнокислот можно получить амид, который образовывается
из двух молекул аминокислот (в химии аминокислот такое соеди-
соединение называют дипептидол):
NH.n R \Н. О R
RCHCOO-J-H3\ — СКСОО- -- RCI1.—С —\--CFICOOH !-1ЬО
н
621
В реакции образуется много побочных продуктов. Для получения
дипептидов, трппептидов, олнгонептидов разработаны специальные
методы (гл. XXX1V.T.4).
б) Образование д и к е т о п и п е р а з и и о в и л а к-
т а м о в. Нагревание сложных эфиров а-амипокнслот дает цикли-
циклические амиды — днкетоппперазипы:
MI-2 И\М/Ч^/К
О
2RCHCOOC.,H3 -'->
и
-!-2СгМ3ОН
iv чс/" хн
II
о
В зависимости от условии реакции могут образоваться также ли-
линейные олигопептнды и полипептнды.
Р-Амипокислоты термически нестабильны, при пагрепапии очи
отщепляют аммиак. Только в особых условиях при использовании
специфических реагентов, например карбодннмидов (гл. XXXV.
В.2) удается замкнуть цикл C-лактама:
\Ч!3
R—CHCIIXOO- ¦¦-> RCH^ CHCOOII-j-NM-Ia
И^м ^° О
R—С —СНа
Н
fi-лактамы
Очень легко циклические амиды — лактамы образуют у- и
6-амипокпслоты (гл. XXXIII.Д.1).
в) Действие окислителей. а-Амииокислоты окис-
окисляются сильными окислителями. При этом выделяются NH3, CO..
и образуется альдегид, который затем окисляется до карбоповоп
кислоты:
NH3
I О
RCHCOO- -I R—с/ +М1Э-! СО2
I I
г) Н и п г н д р и п о в а я реакция. Очень характерна
реакция а-амипокислот с ни и гидр ином (гидратом нидаптрпопа-1,2,3).
В водных растворах при нагревании появляется снне-фполетовая
окраска, интенсивность которой пропорциональна концентрации
а-аминокислоты.
622
Окраска нингидриновои реакции связана с образованием кра-
красителя — синего Руэмана.
Аминокислота реагирует с нингндрином и образует продукт
конденсации типа азом^тинов:
о о
ОН
/NoH
+ |
+HaN-CHCOO-
ч
R
I
= N— CHCOOII~1-II2O
\_ ..
О
ниш идр»и
и
о
Продукт конденсации путем перегруппировки и гидролиза образует
2-аминоиндандион, который, реагируя с кипгидрином, дает краси-
краситель:
о
3. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ
а-А.\шнокислоты являются основным элементом строения белков.
В состав белков входит около 20 различных аминокислот. Часть
аминокислот живой организм синтезирует сам, а часть должен
принимать в составе пищи. Эти аминокислоты называются неза-
незаменимыми. Для организма человека незаменимыми аминокисло-
аминокислотами являются: лизни, треонин, триптофан, метионип, феннлала-
шш, лейцин, ва.чнн и изолеицин.
В табл. 50 приведены важнейшие а-амннокислоты.
п-Аминобензойная кислота — бесцветное кристаллическое ве-
вещество с т. пл. 186 °С, мало растворима в воде. Получают восста-
восстановлением л-иитробепзоппой кислоты.
л-А.мииобензоГтая кислота способствует росту микроорганиз-
микроорганизмов, является витамином, обеспечивающим нормальный обмен ве-
С23
G'8E-
z'ee—
e'9+
9'6l.-i-
Vs—
O'G+
(DM IVS)
oi—
S'frC—
S'Zl-r
G'lH-
9'RI'I-
II —
9'G+
S'8S—
s'6—
(iDHWl)
6'I IS—
9' 91 —
s'z—
8' 1 +
¦o r-v;
делы
-ряда
ацня)
i— г- О
S >)•
d-Ч
U[Q HL4I
'" I i \ - ' ¦ 10
"ID
USy liLMI
dsy
^1
94d
8iy
UJQ
ЭП
ПЭ-]
\BA
J4.L
1
1
S — s-^D
"S
biv
aid
P
2x>
5 2
"i
i
о
?
H
.\-
/ 4
hood('hn)hd?i id——-M
II
N
II II
^IXOD'HD'HDl^H^IIDDOOH
hood'hd?-hd(shm)hddooh
rH.\;OD"-HD(rHN:)HDDOOH
HOOD'HDl'HXOl-IDDOOH
HOOD(r-H,V)HDi;IID*Hl'DOH-''
HOOD(^HN)HD?'HDsH9D
hood('H-\:)hdehd;:hd4idhn(hm = )dn'"-h
IIOOD('H\t)lIDr'HDr-HDrHDNrH
HOOD(rH\)HD7-HDi!HDr-HD?HDN!;H
HOOD(:n\0lID(DIID)HDrHDl;HD
HOOD(!!H\')HD!!HDHDi;(EHD)
HOODEHN)HDHDr(cHD)
HOOD(r-H\')HD(HO)HDDHD
HOOD(rH.V)HD?-HDi;HDSl;HD
hood(?hn:Hid'Hds—s?hd(?h\;)hddooh
HOOD(r-HNl)HDr-HDSII
HOODruHN)HDr-HDOH
HOODDIM)HDlHD
IIOODrHDX?H
nulfiiiairj
ивф
-oiuiidx
-uiAl-j
bxol\")::h
ВВЯОН
-iikbiAl-j
Ш1J
-BdBiiDV
ВХ0ГЛ1Н
^bujv
miEodiiX
НИНВ1Г
-BL-ИНОф
HUHiudv
iiiixiiiido
HIlElllf
Н11П
-ИЭЬ-ОЕЦ
НИПИЭ[,"
HIIL'BQ
HHHoadx
ниноихэуу
НИХЭПП
низину
undo;)
IIHHBL'y-73
H11HJ1L"J
I4XOL-3I1HUHH1VB-» 3i4H»odndu эитнэнжвд -дд vhnvnvj.
Продолжение табл. 50
Название
Пролмн
Оксипро-
лнн
Формула
\
II
НО
ч
соон
н
Сокращенное обоз-
обозначение
Pro
Нурго
Sg. з
^ *Г" га
-8G.2
-72,5
ществ. Сложные эфиры п-амш;обензойной кислоты проявляют боле-
болеутоляющие свойства, являются местными анестетиками (анесте-
(анестезин, новокаин):
з СООСН2СМ, NII(C,H3J
I ci-
NH2
анелрлчн
\н2
поьскоин
Лнтрани.говаякислота (о-амнпобензойная кислота) плавится при
144 СС, малорастворима в воде. Получают из фталимида реакцией
Гофмана.
Антраппловую кислоту применяют в аналитической химии, так
как с ионами многих металлов oi;a образует нерастворимые комплек-
комплексы. Сложные эфиры антраннловой кислоты применяют в парфюме-
парфюмерии.
Г. БЕЛКИ И ПОЛИПСПТИДЫ
Белки представляют собой биополимеры сложного строения, мак-
макромолекулы которых состоят из остатков аминокислот, соединен-
соединенных между собой амидной (пептидной) связью. Кроме длинных
полимерных цепей, построенных из остатков аминокислот (полиием-
тидных цепей), в макромолекулу белка могут входить также остат-
остатки или молекулы других органических соединений. Наряду с нук-
нуклеиновыми кислотами белки играют исключительно важную роль
625
в живой природе. Жизнь немыслима без различных но строению и
функциям белков.
Число белков очень велико, различны их функции в организме.
Например, в волокнах мышц имеется белок миозин, участвующий в
превращении химической энергии в механическую. Волосы, шерсть,
ногти, роговой слой эпителия состоят в основном из белка кератина
Ферменты и гормоны (катализаторы и координаторы протекающих
в живом организме реакций) также принадлежат к белкам или полн-
пептпдам.
1. КЛАССИФИКАЦИЯ БЕЛКОВ
Белки подразделяются на две большие группы: простые белки, или
протеины, и сложные белки, или протеиды.
При гидролизе протеинов в кислом водном растворе получают
только а-амииокислоты. Гидролиз протеидов дает кроме а-амшю-
к 1слот также другие неорганические или органические вещества.
Протеины. Ниже перечислены важнейшие протеины.
Альбумины хорошо растворяются в воде. Встречаются в моло-
молоке, яичном белке и крови.
Глобулины в воде не растворяются, но растворимы в разбавлен-
разбавленных растворах солей. К глобулинам принадлежат глобулины крови
и мышечный белок миозин.
Глутелины растворяются только в разбавленных растворах ще-
щелочей. Встречаются в растениях.
Склеропротсины — нерастворимые белки. К склероиротеинам
относятся кератины, белок кожи и соединительных тканей колла-
коллаген, белок натурального шелка фиброин.
Протеиды построены из протеинов, соединенных с молекулами
другого типа (простетическими группами).
Фосфопротеиды содержат молекулы фосфорной кислоты, свя-
связанные в виде сложного эфира у гидроксилыюй группы аминокисло-
аминокислоты серипа. К ним относится вителлин—белок, содержащийся в
яичном желтке, белок молока казеин.
Гликопротеиды содержат остатки углеводов. Они входят в сос-
состав хрящей, рогов, слюны.
Хромопротеиды содержат молекулу окрашенного вещества,
обычно типа порфииа (гл. XXXVI.А.4.1). Самым важным хромо-
протендом является гемоглобин — переносчик кислорода, окраши-
окрашивающий красные кровяные тельца.
Нуклсопротеиды — протеины, связанные с нуклеиновыми кис-
кислотами. О;ш представляют собой очень важные с биологической точ-
точки зрения белки—составные части клеточных ядер.
Нуклеопротеиды являются важнейшей составной частью виру-
вирусов — возбудителей многих болезней.
2. СОСТАВ И СВОЙСТВА БЕЛКОВ
Белки являются высокомолекулярными соединениями, их молеку-
молекулярная масса в отдельных случаях достигает сотни миллионов.
Меньшая молекулярная масса может быть у простейших ферментов
С26
и некоторых гормонов белковой природы. Например, молекулярная
масса гормона инсулина около 6500, а белка вируса гриппа —
о20 000 000. Вещества белковой природы (состоящие из остатков
аминокислот, соединенных между собой пептидной связью), имею-
имеющие относительно меньшую молекулярную массу и меньшую сте-
степень пространственной организации макромолекулы, называются
пола пептидами. Провести резкую границу между белками п полп-
иептидаыи трудно.
В большинстве случаев белки отличаются от других природных
полимеров (каучука, крахмала, целлюлозы), тем, что чистый инди-
индивидуальный белок содержит только молекулы одинакового строения
и массы. Исключением является, например, желатина, в составе
ко горой входят макромолекулы с молекулярной массой 12 000
70 000.
Для изучения аминокислотного состава белков используется
главным образом метод гидролиза, т. е. нагревание белка с 6 -10
моль-'л соляной кислотой при температуре 100—110 "С. Получают
смесь а-амнпокислот, пз которой можно выделить индивидуальные
аминокислоты. Для количественного анализа этой смеси в настоя-
настоящее время применяют ионообменную и бумажную хроматографию.
Сконструированы специальные автоматические анализаторы ами-
аминокислот.
Разработаны также фергоптативпые методы ступенчатого рас-
расщеп, тения белка. Некоторые ферлюмты расщепляют макромолекулу
белка специфически — только в местах нахождения определенной
аминокислоты. Так получают продукты ступенчатого расщепления —
пепгоны и пептиды, последующим анализом которых устанавлива-
устанавливают их аминокислотный состав.
В результате гидролиза различных белков выделено не более
30 а-аминокислот. Двадцать из них встречаются чаще других.
При образовании молекулы белка или иолипептпда а-ампнокпе-
лоты люгут соединяться в различной последовательности. Возмож-
Возможно огромное число различных комбинаций. Так же как, пользуясь
20. . .30 буквами алфавита, можно написать текст любой длины, так
и из 20 а-амнпокислот можно образовать больше 1018 комбинации.
Существование различного типа нолпиептидэв практически пеогра-
ипчепо.
Последовательность соединения аминокислот в том или ином
белки устанавливают путем ступенчатого расщепления пли рептге-
ноструктурным анализом.
Для идентификации белков и полппепшдов используют специ-
специфические реакции на белки. Например:
а) биуретовая реакция (гл. XXXV.Б,2);
б) ксантопротеиновая реакция (появление желтого окрашивания
при взаимодействии с концентрированной азотной кислотой, кото-
которое в присутствии аммиака становится оранжевым; реакция свя-
связана с нитрованием остатков феиилаланнна и тирозина);
в) реакция Миллона (образование желто-коричневого окраши-
окрашивания при взаимодействии с Hg(NO3)-2+HNO3-f-HNO2;
627
г) нингидриновая реакция (гл. XXXIV.В.2);
д) при нагреЕзании белков со щелочью в присутствии солсГг свин-
свинца выпадает черный осадок PbS, что свидетельствует о присутствии
сор у содержащих аминокислот.
Белки обычно образуют коллоидные растворы. Многие реаген-
реагенты вызывают осаждение белков — коагуляцию, которая может
быть обратимой и необратимой. Например, этанол п ацетон коагу-
коагулируют белки, но эта коагуляция является обратимой. В чистой
воле коагулированные этим способом белки снова образуют кол-
коллоидный раствор. Обратимую коагуляцию вызывают также раст-
растворы некоторых' солей (MgSO4, (NH4bSO4, Na2SO.i). Необратимую
коагуляцию (денатурацию) белка вызывает кипячение, а также деи-
CTBire минеральных кислот, пикриновой кислоты, солеи тяжелы,-:
металлов, танина.
3. СТРОЕНИЕ БЕЛКОВ
Макромолекула белка (протеина) состоит только из остаткоз «-ами-
«-аминокислот, соединенных пептидными (амидными) связями:
Ч / I Я ", /' Г
VW
! «< s« i 5
Все а-аминокислоты белка принадлежат к L-ряду (S-::oiKJinry-
рацин). Группировка С (О.О)—N имеет планарное строение, в;);;-
щепие по связи О- -С—N значительно заторможено (см гл XXX11I
Д.2).
Различают несколько степеней организации молекулы бел;:а.
Первичная структура соответствует линеарной последователь-
последовательности а-аминокислот в полипептидной цепи и часто называете:!
секвенцией. Можно привести несколько примеров строения гор.'.о-
iiod белковой природы.
Например, гормон ангиотензин II построен из 8 аминокислот-
аминокислотных остатков:
1' AsP ArS Va I Туг Val И is Pro Phe 01 i
СП, СИ,
H I H
HOOCH.C И О HC-CHJ О HC-C-H
N Г:
о
. - i^ ^ iiv- \
Y ? Y Y Y Y Y V
О СН H О bh H О C
Y Y
О СН2 H О bh H
NH
с
628
Строение гормона гипофиза окситоцина:
s s
I
Н — Cys — Туг — Не —Glu — Asp — Cys — Pro — Leu —Gly — \H2
NH,
H2 NIL.
(Gly—NH2 обозначает группировку NHCH2CONH2).
Строение гормона поджелудочной железы инсулина (бычий):
Val—Glu S S Cys — Ser—Leu NH2
III I I I
Val Tyr Glu—Asp AspNH2
Gly—He Glu—Cy»
I I I
NH2 Cys — Ala—Ser
S
NH2
l — Leu Tyr—Cys
S
His —Leu
Leu—Tyr—Leu S Glu—Arg
NH2 NH2 S
III II I I I I I
Asp—Glu Cys—Gly—Ser Val—Glu—Ala Val—Cys—Gly Gly
Phe—Val ' His—Leu
Ala—Lys —Pro —Phe —Tyr — Phe
Phe
H
Вторичной структурой называют копформацию, которую обра-
образует полипептидная цепь. Для высокомолекулярных белков харак-
характерна структура спирали. Впервые та-
такая структура па основе рентгенострук-
турного анализа была обнаружена при
изучении главного белка волос и шер-
шерсти— а-кератипа (Л. Полинг). Ее наз-
назвали а-структурой или а-спиралью.
Обычно в природных продуктах встре-
встречаются белки со строением правой спи-
спирали (рис. 99), хотя известна и струк-
структура левой спирали (коллаген).
На один виток спирали приходится
но 3,6—3,7 остатков аминокислот. Рас-
Расстояние между витками около 0,54 им.
Строение спирали стабилизируется внут-
внутримолекулярными водородными связями.
При растяжении а-кератина образу-
образуется вещество с другими свойствами —
Р-кератин. При растяжении спираль мак-
макромолекулы белка превращается в дру-
другую структуру, напоминающую линей-
линейную. Отдельные полипептпдные цепи
здесь связаны межмолекулярными водо-
водородными связями. Эта структура называ-
называется Р-структурой (структура складчато-
складчатого листа, складчатого слоя), Ниже приведен фрагмент Рструк-
туры белка с антипараллельным расположением полииептпдиых
цепей: одна цепь имеет последовательность группировок —
—NHCHRKXJNHCHR2—, а вторая —NHCOCHR^NHCOCHR1—).
R-1
Рис. 99. П|)азая а-сиирдль
полипептпда
629
H R rt OK Л5 FT
\/ I II \/ I
к
II I У
о к
R4 И О
^-Структура характерна также для белка натурального шел-
шелка — фиброина.
Третичная структура характеризует пространственное распочо-
женые цилиндрических сс-спиралей или других образований вторич-
вторичной структуры. Спирали могут свиваться в клубок, образовывавь
глобулы или располагаться рядом, образуя нитевидные структу-
структуры — фибриллы. В образовании третичных структур большое зна-
значение имеют дисульфидные мостики S—S между различными час-
частями макромолекул.
Именно третичная и четвертичная структуры определяют хими-
химические и биологические свойства белков.
Четвертичная структура осуществляется в некоторых белках,
для которых характерно образование олнгомерного белка из не-
нескольких полипептидных цепей. Ассоциация цепей обусловлена
межмолекулярными взаимодействиями между боковыми группа-
группами — образованием водородных связей, ионных пар и т. д.
4. МЕТОДЫ СИНТЕЗА ПОЛИПЕПТИДОВ
Искусственное получение белка было актуальным вопросом уже
в прошлом столетии, когда стало ясно, что белки построены из
ct-аминокислот с помощью амидиых (пептидных) связен. Первые
синтезы низкомолекулярных пептидов связаны с именем немецкого
химика Э. Фишера. В 1903—1907 гг. Э. Фишер синтезировал не-
несколько олигопептидов из глицина и лейцина.
Синтез пептидов связан с рядом существенных трудностей. Преж-
Прежде всего, необходимы чистые энантиомеры — оптические активные
изомеры L-ряда (S-конфигурации) а-аминокислот. Кроме того,
требуются специальные приемы для осуществления последователь-
последовательного образования пептидных связей а нужной нам последователь-
последовательности а-аминокислот: защита аминогрупп, активация карбоксиль-
карбоксильных групп, отщепление защитных групп, множество специальных
реагентов.
1. Синтез оптически активных соединений. Получить оптически
активные соединения из оптически неактивных возможно двумя пу-
путями: разделением рацематов на чистые энантиомеры (оптические
антиподы) или прямым асимметрическим синтезом.
а) Разделение рацематов. Наиболее общим мето-
методом разделения рацематов является разделение диастереомеров. Ра-
630
цемат в реакции с оптически актирным соединением образует произ-
производное, которое содержит больше асимметрических атомов, нежели
исходное соединение. Поэтому продукт реакции состоит v,i двух
диастерсомеров (гл. VI 1.3.3), которые различаются но физическим
свойствам и могут быть разделены кристаллизацией или хромато-
хроматографией:
Д1]::стсрсоморы
рацемат
Таким путем Л. Пастер в 1852 г. рялделил на оптические антиподы ( —)-
винную кислоту. Была прнготоп.1е;|;1 соль (:J-)-miiiiioi"i кис.юты с оптически
активным основанием — алкалоидом цимхоиньом. Соль (—)-r>uiiiio(i кнелеш
с цинхопином оказалась менее расгпорнмон :i иыкрнсталлиаовалась hj реак-
реакционной смеси.
Для разделения рацематов а-пмшюкнелот mo;ki:o использовать образо-
образование солен (-г)-д11бепзо11лв;шноп кислогы.
Используют также биологическое п ферментативное разделение
рацематов. Первое из них основано на том, чго многие микроор-
микроорганизмы обычно потребляют только одни эпантиомер, тогда как дру-
другой накапливается в растворе. Шире применяют ферментативные
методы. Специальные ферменты катализируют химические превра-
превращения только одного эиантиомера. Так, например, стереоселектпв-
но происходит гидролиз сложных эфиров аминокислот в присутст-
присутствии некоторых ферментов:
(i)-CH4CHCOOR l^lll (_).QI-!CHCOO--i-(-1 )-CIIaCUCOOR
I 1It0 I I
I
Гндролизуется сложный эфир только одного эиантиомера. Смесь
легко разделяется, так как растворимость сложного эфира значи-
значительно отличается от растворимости аминокислоты.
Аналогично осуществляют стереоселектнвпое деацнлнровапме
(гидролиз) (-^)-М-ацплампнокислот. Раствор веществ пропускается
через колонку, наполненную иммобилизованным (связанным по-
поверхностью инертного носителя) ферментом.
Для разделения рацематов применяют также хроматографию на
хиральных сорбентах.
б) А с и м м е т р и ч е с к и и синтез. Метод заключается
втом, что из оптически неактивного соединения получают оптически
активное, создавая в молекуле новый асимметрический (хиральный)
атом таким путем, что образуется только один энантиомер.
Теоретически это может быть достигнуто стереоснецифическим
замещением одного из двух энантиотопных атомов или групп про-
хиральной молекулы (гл. VII.3.3), стереоспецифическнм удалением
одной из групп или присоединением к двойной связи:
631
R
С
в:.-д ;- цлые атлмм
3<ij\ .итоины
R R
I. _?_^ L [ + f
к R1 -t со,
о ;
R'
Лсимметрическии синтез в живых организмах обеспечивается
высокоспецифическими хиральпымн катализаторами — фермента-
ферментами. В колбе такие синтезы осуществить трудно, и это удается толь-
только в присутствии оптически активных веществ — катализаторов.
Обычно выход оптически активного продукта мал, больше образу-
образуется рацемат (смесь обоих энантиомеров).
В особых случаях в присутствии оптически активных катализа-
катализаторов или реагентов гидрирования удается провести гидрирование
связен С—О, С С, С--•¦•¦ N с образованием оптически активных про-
продуктов гидрирования с почти количественным выходом. Например,
гидрирование 2-ацетнламинопропеновой кислоты в присутствии
оптически активного родиево-фосфипового катализатора дает оп-
оптически активный ацетиламипо-сс-аланнн:
СН.-С-СООН — > СНЯ-С[[-СООН
I оптически |
КИСОСНя кат1лиза"ор \HCOCH,
Специальным случаем является образование второго хиралыюго
атом:1, в уже хиральной молекуле. Тогда образуются дпастереомеры:
н н у
R, Т Л2 R. | ,R* R~, ! ,-R2
С С* *
Н
!
г
R1
R R'
водор^дмме атомы у
exupa.iiiHciro углерода
диастереэтопны
Диастереомсры не являются оптическими антиподами, отлича-
отличаются физическими свойствами и могут быть разделены обычными
методами.
Присутствие хнральпого катализатора в реакции образования
диастереомеров может повлиять на их соотношение и пызывать об-
образование только одного диастереомера.
2. Синтез пептидов. Образование пептидных связей происходит
в реакции между N-защищенном (с защищенной аминогруппой) ами-
аминокислотой пли ее функциональным производным и аминокислотой
в виде соли или сложного эфира (О-защпщенная аминокислота):
,0 /О
Y — MICH— С7 -1-HoNQlCOOR2 —> Y — NHCH— O7 •(-ИХ
-X ' ' | | NMHCHCOOR2
R R1 R I
R1
К-За'.г.'.нцающая группа обычно бывает алкоксикарбонильпон
[Y = Cr,H.-,CH2OCO, (СНя)яС0С(I, такие соединения получаются
при взаимодействии аминокислоты с хлоругольными эфирами
'ROCOCI.
Для реакции образования пептидной связи N-защшценную ами-
аминокислоту можно превращать в хлорангидрид, сложный эфир
(X =С1, ()С.Н5; Э. Фишер), азид (X --¦= N-,; Т. Курциус). Азндный ме-
метод удобен тем, что реакция может быть осуществлена в водном р л ст-
створе с солью аминокислоты:
-° п о
Y —MICH—С ' -f HoN'CHCOO-Xa-- I. YXIICH -COXHCHCOOII-i-XaN,
! "Na " I ' I
R Rl R R1
Для этой цели используют также так называемые активированные
зфнры, которые содержат полярные группы, например:
F. .-F
'ч
Х- -О —^~^—\0о, -O-N'-CH-^~^N, —О—vf ^—F
F ' ЧГ
Реакцию Х-защнщенных аминокислот с О-защнщепными осу-
осуществляют прямо в присутствии специальных реагентов карбо-
днпмндоз:
С-Н,, Х = С = NC=I Гц
VNnr[iCOOH-'-II.,\CHCOOR2 - YXHCMCOXHCHCOOR2
К Rl R R1
Снятие защитных групп часто связано со значительными труд-
трудностями. Необходимы специфические реагенты и методы. Снятие
м/уЛ'н-бутокспкарбоннльной группы IY-- (СИ3)»СОСО] происходиf
в мягких условиях в присутствии CF;;COOH.
Перспективным является твердофазной синтез пептидов
(В. Мерпфплд, 1962). ¦ В этом методе наращивание пептидной це-
цепочки происходит па поверхности твердого гранулированного по-
633
арного носителя. Полимер содержит активные группы (СН^О,
/JH), и N-защищепиая аминокислота «привязывается» сложно-
С' рной связью. Следует отщепление защитной группы, образов;:-
эф' пептидной связи с другой молекулой N-защнщенной амипокио
"".-{,1. отщепление защитной группы и т. д.
л0 обычно в качестве носителя используют хлормстнлировапкый
„.¦стирол, сополимер полистирола с дпвннилбепзолом
Ц0-"
я
Na+ OOCCII—N
cH.ci з*- ;а_сн ооссн—n' —>.
^ н
^(I.OOCCIINHi — >- а СН OOCCHNHCOCHN
I ^ ' I | \у
r r1 Y
ciiemianbiio химически обработанные полисахариды (целлюло-
lu'rI .^кстрапы), в которых активной группой является СН2ОЫ.
за'г1осле наращивания цепи достаточной длины нолипентпд от-
;1яют от носителя действия смеси НВг-[-СГаСООН.
^'^ прием синтеза может быть автоматизирован. Сконструп-
$\\ъ\ специальные авто.матнческпе синтезаторы полипептплов.
Р00,^! образом синтезирован гору.оп инсулин, фермент рибонуклса-
'i|f\i содержащий 124 аминокислотных остатка, и другие полипеп-
за.[' п простейшие белки.
^сокислоты
мСт°ДЫ получения
Оксокислоты (альдегпдо- п кетокислоты) получают тремя ос-
'• {!ыМ11 методами: гидролизом ее, сс-дшалогенкарбоповых кислот,
1Ю'рением а-гидроксиккслот, реакцией ацплгалогепидов с цпа-
0I<;1 ^и с последующим гидролизом а-оксопитрилов:
Cl ОН О
L/CGOH— °-> R-C-COOH -^1 R-C-COOH-; Н.,0
V I
;,
I СООН —> R —С —СООН
l
н
.0 СиСМ /О н,0. Н+ !|
// ^R-Gf — * R-C-COOH
CC1 ^CN
634
2. р-Оксокислоты могут быть получены гидролизом сложных
эфпров Р-оксокмслот или карбоксилпрозаннем енолятов:
О
Г \яо;г
R-C-CH-COOC,lb
0
1
_с—сп—сое:
н
) - \'а г —
О
-Х- \:
-- -RCCHCOOH
со, R1
р-Оксокислоты и их coin нестабильны, легко декарбоксилиру-
ются. Поэтому обычно используют сложные эфиры р-оксокнслот.
В сложноэфпрной конденсация (гл. XXXIII.В.3) получают соли
эфпров Р-оксокпслот, при подкислешш реакционной смеси выде-
выделяется сложней эфир Р-оксокнслоты в виде таутомерпой смеси
енолыюй и дпкарбопилыюп форм (сравните с дикетонами,
гл. XXVIII.Б.3):
О-\т! О
RCH2COOC2H5-[ RCII2COOC2[b-;-Nal-OC2H5^ I ^
RCH../ ^С-^ \0С2На
I
R
/Ы. ОНО
Н-.Х- о/ 'о г | ;.
>- | |1 Ч RCH2-C-C-C-OC2H8
RCH., — С С—ОСЛ15 |
\:/ R
R
Конденсация смеси сложных эфпров муравьиной кислоты н дру-
других кислот даст эфиры р-альдегпдокнелот:
HCOOCH,-!-RCH,COOC.Hs i!i^2NX ••• —^
0 Н
• С | ,СООС,1Ь,-(- О О
R Н-' ^С'' ХОС.,Н5
При конденсации сложных ьфпров лнк?.рбонспых кне-пот поту-
чают эфиры циклозлканопкарбоьовых кисют (конденсация Дик-
)
/ СИ ON'a 1!+X
сн,соос.н5
COOC.Hs ООН 5
(СН,1„
обычно п = 3 или 4.
G35
3. v- и б-Оксокислоты могут быть получены окислением соот-
г. дствующих гидроксикислот. Большинство из них получают спе-
специфическими реакциями.
2. ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
1. с-Оксокислоты—бесцветные вещества, растворимые в воде.
Д'я сс-оксокислот характерны все реакции карбоновых кислот п
карбонильных соединений. Только взаимное индуктивное влияние
карбоксильной и карбонильной групп увеличивает их реакционную
способность.
сс-Оксокислоты являются более сильными кислотами (р/<~2,5),
чем а-гидроксикнслоты. При нагревании они способны декарСо-
котироваться легче, чем незамещенные кислоты:
о
Г ,, ,0
RCCOOH -. R-C^ - СО2
ЧН
сс-Оксокислоты реагируют с нуклеофильными реагентами, лег-
легко происходит присоединение по карбонильной группе.
Специфическими реакциями являются окисление и декарбопн-
лнрование в присутствии концентрированной HoSO4:
RCOCOOH -°i RCOOH--CO2
RCOCOOtt к°"ц- 'S_V RCOOH ЬСО
2. р-Оксокислоты и их соли — бесцветные, термически неста-
нестабильные соединения. Кислоты декарбоксилируются быстрее их
солей. Предполагают, что декарбоксилированию способствует об-
образование внутримолекулярной водородной связи:
о .н
Ч о>
R—С—СН.СООН 7~>" If'
3. Сложные эфиры р-сксокислот — бесцветные ароматные жид-
жидкости. Обычно они представляют собой смесь двух таутомерных
форм: енолыюй и дикарбонпльной. Их химические свойства анало-
аналогичны свойствам C-дикетопов.
Эфиры р-оксокислот — более слабые кислоты, чем р-днкетопы.
Константы ионизации для водных растворов рКцт. =10,5. . .11.
Рассмотрим пример этилового эф/ира ,6-оксомаслянсй кислоты (аце-
тоуксуспого эфира):
|| Н РКк=!0,7 О5" О«- р g F-8,4 О^"'О
СН,ССН-,СОС;Н5 '* *" У- ¦ Л " >' I il
НО ^\.Sto.C\ НаО CV ?^
Y ОС.Н5 НзС С oC.
н +НоО<- н 0,8%
ело
В водном растворе ацетоуксуского эфира содержится 0,8% ено-
ла, в спиртовом растворе—8,7(;о, неразбавленное вещество содер-
содержит около 7% енола.
Анноны эфиров р-оксокислот содержат сопряженную систему
с выравненными связями и делокализовашгым отрицательным заря-
зарядом, они являются амбндентпыми анионами с несколькими реакци-
реакционными центрами. Их соли в неразбавленных растворах существуют
в виде ионных пар (сравните с р-дикетонами, гл. XXVIII.Ь 2).
С ионами тяжелых металлов они образуют окрашенные внуфс-п-
ние комплексы — хелаты. Раствор FeCl:! дает интенсивную красно-
фиолетовую окраску:
о^ ^ог" \:-о о~/
|: . -I н-с^ V ' ч
Н НаС-° \
Соли эфиров р-оксокислот, подобно солям Р-дикетонов, легко ал-
килнруются и ацилируются:
I I I
Н R Н
/О
(R-алкнл, R1—C-' ).
Ал!и!лированне и ацилировапье происходят главным образом у
углеродного атома (сравните с дикетонами), так как атомы кислоро-
кислорода в значительной степени блокированы ионом металла. Только
в случае применения солей с катионами, менее склонными (Rb+,
Cs') или неспособными к комплексообразованию [(CH3LN + ], и
эффективных алкилирующих агентов (диалкилсульфаты) получают-
получаются и О-алкилпродукты.
С-Алкилпродукты ацетоуксусного эфира, а также эфиров дру-
других р-кетокислот являются исходными веществами для получения
кетонов и карбоновых кислот.
Гидролиз замещенного ацетоуксусного эфира в водном растворе
кислотой или разбавленной щелочью заканчивается декарбоксили-
рованием и образованием кетона. Эта реакция ацетоуксусного эфи-
эфира называется кетонным расщеплением:
ООО
'. NaOII I,' П.,О
СН..ССНСООСН- CII3CCI[COO-N'aJ -> CII/XH.,R -[-NaHCO3
, - H,0 | i'
R R
При взаимодействии с концентрированной щелочью (~5 У\)
происходит гидролитическое расщепление связен С—О и С—С н
G37
образуется соль карбоновой кислоты. Эта реакция ацетоукеусного
Цщра называется кислотным расщеплением:
cCOO.. * CH,
R ft
О—И - v
[ __ .О Na +
^± СП.,—Сч/'* ^С >- CH3:Co6~Na+ + RCFbCOONa
Na+0- I
Механизм этой реакции (ретроальдольного распада) можно срав-
сравнить с расщеплением Р-днкетонов.
Конденсация эфпров [5-оксокислот с альдегидами происходит
аналогично реакции р-дпкетопов:
О
V i1
СН-;СОС1!.СООГп[Т5 СН3С-С11-СООС.,Н3
/О on- I
-]R-C<< H-C-R -]H/J
Н |
СИоСОСП.СООСЛЬ СП,С-СН-COOC.tr,
I
о
Аналогично реакции с р-дикетопамн идут реакции эфпров C-ок-
сокнелот с N-нуклеофиламп. Например, при реакции с аммиаком
образуются эф;фы (З-амипокротоноЕон кислоты:
н н
Ч/Чъ
СН5СОСН,СООС2Н5-|-\Я3 -* I I -[-I1..0
с с
СН3 С ОС2П5
I
н
В реакции с гидразинами получаются ппразолоны.
4. Y" и 8-Оксокнслоты обладают всеми сво;'!ствами карбоновых
кислот и карбонильных соединений. Одновременно возможны так-
также реакции внутримолекулярной циклизации за счет присоедине-
присоединения карбоксильной группы к карбонильной:
СП,-(СН,)„ R СИ»-(СИ2)„
R_c /L-O ,__
1| И —О НО
о
Такие кольчато-цепные изомерные превращения в некоторой сте-
степени аналогичны внутримолекулярным реакциям у- и б-гидроксн-
карбонпльных соединений (гл. XXIX,Б.2) и углеводов
(гл. ХХ1Х.В).
638
3. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ
Глчо;-счловая кислота известна в виде кристаллического гидра га
(НОЬСНСООН с т. ил. 98 "С, р/С«3,3.
Глиокснловую кислоту Г!олучают из дихлоруксуснон кислоты.
Благодаря высокой реакционной способности, объясняющейся при-
присутствием активной альдегидной группы, ее используют в органи-
органическом синтезе.
Пировиноградная кислота СН;,СОСООН — бесцветная жидкость
с запахом уксусной кислоты; т. кип. 165 "С (с частичным разложе-
разложением), обычно ее перегоняют в впкууме. Растворяется в воде.
ПнроЕнноградпая кислота содержится во всех тканях организма.
Она имеет важное биохимическое значение, так как участвует в
различных биохимических процессах. Пировиноградная кислота
обнаруживается как промежуточный продукт в процессах молочно-
молочнокислого и спиртового брожения, в биосинтезе аминокислот.
Синтетически ее получают нагреванием винной кислоты в присут-
присутствии KHSO,. В этой реакции происходит дегидратация винной кис-
кислоты до щавелсвоуксусной кислоты, которая декарбоксилпруется:
н, "соон /Оон
ИОСНСООП Knso NC7 П.,С I!
I ~-t li 7-- I + П2О-*СО2 (СИзССООИ
HOCHCOOM tc С ,С
гк/ чсоон о// Чсоои
Ацетоуксусный эфир СН/ЮСНиСООСгН.-, — бесцветная
жидкость с приятным запахом; т. кип, 181 °С. Он представляет
собой смесь двух таутомерных форм. Дпкарбонильную форму можно
получить в чистом виде, охлаждая растворы ацетоуксусного эфира
до —40. . .50 гС. Образуются бесцветные кристаллы, плавящиеся
при -39 °С. При комнатной температуре дикарбонильная форма
частично превращается в енольпую форму.
В промышленности ацетоуксуеш^й эфир получается сложноэфнр-
ной конденсацией нзэтилацетата или реакцией дпкетена (гл. XXVII.
В.4) с этанолом:
О О
п с.маои
// — >
СИ,'
дикстек
/С —СИ*
сн3с--сн2-с-осн0
О О
R N 11 г I
аг3с -си. -с —nur
При реакции аминов с дикетеном образуются амиды ацетоуксус-
noii кислоты.
Ацетоуксуспый эфир широко применяется в органическом син-
синтезе для получения лекарственных веществ (пиразолоны, акрихин),
красителей и производных пиридина.
Простагландины — природные вещества с чрезвычайно высо-
высокой биологической активностью — по своей химической структуре
G39
представляют собой ненасыщенные ппроксикетокислоты или гнд-
роксикислоты с 20 углеродными атомами, содержащие в молекуле
циклонептановый цикл:
но он
простагландпн R,
Простагландины содержатся п сперме человека и в очень малых
концентрациях по всех тканях млекопитающих. Строение установ-
установлено в 1962 г., первый синтез осуществлен в 1968 г.
Организм непрерывно синтезирует простагландины во всех мес-
местах, где они необходимы как биорегуляторы. По эффективности дей-
действия иростагландипы принадлежат к наиболее активным биоген-
биогенным веществам. Простагландины действуют на половую систему,
стимулируют роды, прекращают беременность и имеют раз чинное
другое биологическое действие. Большое значение они имеют в
ветеринарии и животноводстве.
Глава XXXV
ПРОИЗВОДНЫЕ УГОЛЬНОЙ КИСЛОТЫ
Угольную кислоту формально можно рассматривать как карбоно-
вую кислоту, которая вместо углеводородного остатка содержит
/О
гпдроксильную группу: НО—С^
Свойства производных угольной кислоты (табл. 51) в основном
подобны свойствам производных карбоновых кислот. Отличие кро-
кроется в том, что в угольной кислоте присутствуют две гидроксиль-
пые группы, которые одновременно являются составными частями
карбоксильной группы.
КЛАССИФИКАЦИЯ И НОМЕНКЛАТУРА
Из приведенных в табл. 51 производных угольной кислоты рассмот-
рассмотрены будут только некоторые.
А. ЭФИРЫ ХЛОРУГОЛЬНОЙ И УГОЛЬНОЙ КИСЛОТ
1. Методы получения. Хлоругольпая кислота С1СООН, которая
образуется при взаимодействии фосгена с водой, нестабильна и рас-
распадается на СО2 и НС1.
Эфиры хлоругольнон кислоты получают из
фосгена и соответствующих гидроксилпроизводных в присутствии
640
Таблица 51. Важнейшие
Формула
о
а
а
,0
RO-C^
Х-С1
X0R
RO-C^
OR
1
RO-C-OR
1
OR
0 0
RO —С —0—С—OR
\N —C00-M+
rr>-c<° .
H2N-C^ т
H2N-C^NH
HO-C--N
O-rC- N — R
H2N —C--N
R-N=,C=N-R
H2N —Cc^
R-S-C—N
производные угольной кислоты
Название
Монохлорангидрнд угольной кислоты, хлор-
угольная кислота, хлормуравьиная кислота
Днхлораттгидрид угольной кислоты, фосген,
карбоннлхлорнд
Эфиры хлоругольной (хлормуравьиной) кис-
кислоты, хлоркарбонаты, алкоксикарбонилхлориды
Алкил(арнл)карбонаты
ДналкIл(арпл)карбонаты
Тетраалкнл(арпл)ортокарбонаты
Дпалкил(арнл)пирокарбонаты
Карбамиповые кислоты
Карбаматы
Эфиры карбампновых кислот, уретаны
Дпамид угольной кислоты, карбамид, моче-
мочевина
Гуаниднн
Нитрил угольной кислоты, циановая кислота
Эфиры циановой кислоты, цианаты
Эфиры изоциановой кислоты, изоц.чанаты
Амид циановой кислоты, цианамид
Амид изоциановой кислоты, карбодиимнд
N, Ы-Диалкнл(арил)карбодиимиды
Диамид тиоуголыюй кислоты, тиокарбамнд,
тиомочевина
Тиоцианаты, роданиды
21 № 517
641
Формула
S-C--=N —R
RO-C^"
4SRl
R1/ XSR2
RS-C^S
XSR
Продолжение табл. 51
Название
Изотиоциапаты
O.S-Диэфиры дитиоугольной кислоты, ксаи-
тогепаты
Дитиокарбаматы
Эфиры тритиоугольной кислоты, тритиокарбо-
наты
оснований:
-J-HOR °i^!^ Cl-C^° +HC1
" XOR
Алкилкарбонаты получают из алкоксидов и СО2.
Они стабильны только в виде солей:
6+
RO-Na+-l-O = C=^O __>. ROCOO-Na +
Диалкилкарбонаты образуются при взаимодействии
фосгена и спиртов в присутствии оснований:
С1-С^ +2HOR 2!^!i™ RO-c/ -I-2HC1
ХС1 NOR
Исходными веществами могут быть также хлоругольные эфиры.
Из фосгена и фенолов образуются диарилкарбонаты.
Эфиры пироугольной кислоты образуются при
взаимодействии эфиров хлоругольной кислоты и солей алкилкарбо-
натов:
о О
/Р 'I II
RO— Of -f Na+-OOCOR —> RO —Сч ,C —OR-f NaCl
4ci xo/
2. Физические и химические свойства. Эфиры хлоруголь-
хлоругольной кислоты — бесцветные жидкости с острым запахом. Ис-
Используют в органическом синтезе. Важны бензилоксикарбопилхло-
рид С1СООСН2С6Н5 и m/jem-бутоксикарбонилхлорид, которые при-
применяют для защиты аминогруппы в аминокислотах.
Эфиры угольной кислоты являются бесцветны-
бесцветными жидкостями с приятным запахом. Ариловые эфиры — кристал-
кристаллические вещества. Для эфиров угольной кислоты характерны поч-
почти все реакции эфиров карбоновых кислот. В том числе они всту-
вступают в сложноэфирную конденсацию и могут быть использованы
642
для введения алкоксикарбонильной группы:
о О
/О || NaOCH3 HCI ii
RGf 4CH3OCOCH3 > ,., —- R-C-CH2-COOCH3
Эфиры пироугольной кислоты реагируют по-
подобно ангидридам карбоновых кислот (гл. XXXIII. Б.З) и могут
быть использованы для алкоксикарбонилирования нуклеофильных
реагентов вместо хлоругольных эфиров.
Поликарбонаты являются полиэфирами угольной кис-
кислоты и двухатомных фенолов. Они образуются из соответствующего
фенола и фосгена в присутствии оснований или при нагревании диал-
килкарбоната с двухатомным фенолом при 180—300 °С.
R
, ч | . v .О Основание
но—;7 j—c-v ~— oii + ci — c<f *¦
\:=/ | ^^^ \С1
[R О R °
-<fHVc-^HVo-c-o-<*~Vc-<f^>-o-c-o-
R1 R1 Jji
Поликарбонаты — бесцветная прозрачная масса с температурой
размягчения 180—300 °С (в зависимости от метода получения) и мо-
молекулярной массой 50 000—500 000. Используют для изготовления
различных изделий (труб, пленок и др.).
Б. АМИДЫ УГОЛЬНОЙ КИСЛОТЫ
1. КАРБАМАТЫ И УРЕТАНЫ
1. Методы получения. При взаимодействии аммиака или аминов с
СО2 образуются аммониевые карбаматы:
R v.. 6+ R ч +
2 >N —H + O-C = O-
Свободные карбамиповые кислоты нестабильны, распадаются на
СО2 и аммиак (амин).
Уретаны получают взаимодействием хлоругольпых эфиров с
аммиаком или аминами и присоединением спиртов к изоцианагам
(см. ниже):
CICOOR+R^NH —* R1R2\'COOR-|-HCI
R — N = C = O + H0Ri —> RNIICOORi
21* 643
2. Физические и химические свойства. Уретаны являются бес-
бесцветными жидкостями или кристаллическими соединениями. Они
проявляют свойства как эфиров карбоновых кислот, так и амидов.
Соединения нитрозируются, образуя N-нитрозоуретаны, которые
служат исходными для получения диазоалканов:
RCH2NCOOC2H5 ™2l RCH2NCOOC2H3 — °Л RCHN2
Н NO
3. Важнейшие представители. Полиуретаны — полимеры, содер-
содержащие в цепи макромолекулы уретановые группировки:
НО —А-
Н Н
I I
— О— С — N — В— N— С — О— Л
II I,
О О
н
— О —С—N—В —N = C^
II
"¦ О
Их получают из гликолей НО—А—ОН и динзоцианатов О=С==
= N—В—N=C=O IA=(CH,L или (СН2)в; В — см. с. 648].
Молекулярная масса полиуретанов в среднем 10 000—12 000,
температура размягчения, в зависимости от А и В, 100—270 аС.
По своим свойствам полиуретаны подобны полиамидам, только
более устойчивы к действию атмосферы и кислот. Их используют
для получения пенопластов, каучуков, клеев, волокон.
2. МОЧЕВИНА И ЕЕ ПРОИЗВОДНЫЕ
1. Методы получения. Первым методом синтетического получения
мочевины было нагревание цианата аммония (Ф. Вёлер):
NHJ-OCN -С H2NCONH2
В промышленности мочевину получают из аммиака и СО4:
6 + I 3 0 °С
2NH3-|-O = C=O —> H2NCOO-\'H4 —-la- H2NCONH2 f- H.O
Замещенные мочевины могут быть получены в реакции фосгена,
уретанов или нзоцианатов с аминами.
2. Физические и химические свойства. хОДочевина и ее производ-
производные представляют собой бесцветные кристаллические вещества,
растворимые в воде. М.М.М'.М'-тетраалкилмочевины— жидко-
жидкости, хорошие растворители.
По химическим свойствам мочевина похожа на амиды карбоновых
кислот с тем отличием, что у мочевины более выражены нуклео-
фильные (основные) свойства атома азота, так как в молекуле мо-
мочевины одна карбонильная группа приходится на два атома азота.
Мочевина является слабым основанием, протоннруется у атома
кислорода:
644
Мочевина алкилируется и ацилируется у атома азота. N-Алкил-
мочевины ннтрозируются, Ы-нитрозо-Ы-алкилмочевины служат для
получения диазоалканов (гл. XXV. Б.1).
Важное значение имеет реакция мочевины с альдегидами, в ко-
которой образуются полимерные соединения — карбамидные смолы.
Молекулы альдегида как бы «сшивают» молекулы мочевины. Ниже
приведен возможный механизм реакции мочевины с формальдегидом:
.О ,0 ,0
Н-С^ -l-HoN-G" —* IIOCH2NH—C-^
Н ^NH, NH2
.О н+х- о
HOCH2NH-C/ ~L1 СИ2: М-(У -fH2O
¦NH. " NH2
6+ л- /О б->- й- /О в> /О
CH2-N—C^' -1-CH2- N-C^' ~CH2=N-C^ + ...—>¦
NH2 XNH., V\H2
0 0 0
—>-...— CH2 — \H — C — NH— CH2 — NH— C— Nil— CH2—NH — C — NH—...
Нагреванием мочевины получают бнурет:
О О
/О /О ,„ ! 1|
H2N-Cf +H2N-C^ - > H2N-C-N-C —NH2 + NH3
XKH2 ^NH2 |
H
Биурет в щелочной среде с ионами меди Си(II) дает темно-си-
темно-синюю окраску (бнуретовая реакция).
Мочевина — бесцветное кристаллическое вещество с т. ил.
132,7 СС. Она является конечным продуктом распада белков в ор-
организме человека и многих животных и выводится из организма мо-
мочой. Впервые выделена из мочи в 1773 г.
Мочевину получают в промышленности в больших количествах
из NH3 и С02. Ее используют для получения карбамидных смол и
аминопластов, в синтезе гетероциклических соединений, в том чис-
числе лекарственных веществ (гл. XXXIX.А.4).
Мочевину широко применяют в сельском хозяйстве как высоко-
высококачественное удобрение и как кормовую добавку. Некоторые произ-
производные мочевины, например ArNHCON(CH3J, являются активны-
активными гербицидами, применяющимися для уничтожения сорняков.
Мочевину используют также для выделения алканов с нераз-
ветвленной цепью из смеси различных алканов. Оказывается, в
кристаллах молекулы мочевины располагаются таким образом, что
остаются пустые цилиндрические каналы, в которых могут помести-
поместиться длинные неразветвленные молекулы н-алканов (н-гексана,
н-пептана и др.). Объемистые молекулы разветвленных изоалканов
в эти каналы поместиться не могут. Таким образом, при кристал-
кристаллизации мочевины в присутствии алканов получают кристаллы, со-
содержащие мочевину и н-алканы. Такие образования называют соеди-
645
нениями включения или клатратами. При растворении в воде сое-
соединений включения мочевины получают чистые н-алканы и водный
раствор мочевины.
3. ГУАНИДИН
Гуанидин получают из цианамида и аммиака:
6+ .. /Ш
H2N— C = N+NH3 —>¦ H2N-Cf
XNH2
Гуанидин и его производные представляют собой кристал-
кристаллические соединения, растворимые в воде. Это сильные основания,
образующие стабильные соли:
=NH + Н2О
с—nh.
+ он"
В катионе гуанидиния положительный заряд делокализован, ос-
основность гуанидина значительно превышает основность аммиака
(^ =9,25), вторичных аминов (р/СВп+ «11) и амидинов
+ «12,4).
Гуанидин — бесцветная кристаллическая масса с т. пл. 50 СС,
на воздухе расплывается, так как связывает Н2О и СО2. Обычно
известен в виде солей, например нитрата.
В промышленности соли гуанидиния получают из цианамида или
его димера — дициандиамида (цианогуанидипа), аммиака и солей
аммония при нагревании A60 °С).
Гуанидин применяют в синтезе гетероциклических соединений
и взрывчатых веществ.
В. НИТРИЛЫ УГОЛЬНОЙ КИСЛОТЫ И ИХ ИЗОМЕРЫ
1. ЦИАНАТЫ И ИЗОЦИАНАТЫ
1. Методы получения. Алкилирование солей циановой кислоты —
цианатов может привести к образованию как органических циана-
цианатов, так и изоциапатов, так как цианат-ион является амбидентным
анионом с двумя реакционными центрами:
R-O—C^
В продуктах реакции обычно преобладают изоцианаты. Алкил-
цианаты являются нестабильными соединениями, легко тримери-
зующимися в производные 1,3,5-триазина (циануровой кислоты).
Органические циапаты получают специфическими методами, на-
например взаимодействием C1CN с фенолятами.
646
Изоцианаты синтезируют из первичных аминов и фосгена в при-
присутствии оснований:
° 1^ R_NH-C<f°+HCl 2!^1™
ЧС1
С1 ЧС1
—+ R — N = C=
Известны специфические методы синтеза изоцианатов, например
реакции Гофмана, Курциуса, Лоссена. Арилизоцианаты могут
быть получены карбонизированием нитроаренов.
2. Физические и химические свойства. Изоцианаты — обычно
бесцветные жидкости с острым раздражающим запахом. Молекула
изоциапата имеет структуру типа кетенов с кумулированными
двойными связями, углеродный атом находится в состоянии sp-тпб-
ридизации, группировка атомов N—С—О линейна или близка к ли-
линейной:
Изоцианатная группировка содержит полярные активные двой-
двойные связи и высокоэлектрофильпый углеродный атом.
Характерны реакции с нуклеофильными реагентами и реакции
циклоирисоединепия:
Н
6+ I 6- /NuH
R_N = C^O-[-:NuH-> R-N=C<
L ЧО-
(:NuH= НОН, HOR\ NH3, HpNR1, HNR^2 и др.).
Изоцианаты могут образовывать нестабильные димеры:
О- C=-N~R
^ -R
Образование карбодиимидов из изоцианатов в присутствии фос-
финоксидов протекает в две стадии — две реакции циклоприсоеди-
циклоприсоединения:
6+ ,л+ е- R'P-0 r ,
R-N = C-.O + RiP-0 -> | | -> RJp-NR-;-CO,
,N —Сч
,e r5pn
RjP--NR-|-R-N= = C=- О -> | | -* RSP---O + R—N = C=N—R
^XR
3. Важнейшие представители. Диизоцианаты, применяющиеся
для синтеза полиуретанов и пенополиуретанов:
647
OCN'(CH2NNCO 1,6-гексамети лендиизоцианат
—СН2—^ у—NCO 4,4'-диизоцианатодифенилметаЕ1
2,4-толуилендиизоцианат
2. ЦИАНАМИДЫ И КАРБОДИИМИДЫ
1. Методы получения. Цианамид получают из кальцийцианамида,
который в промышленности производится в больших количествах:
CaC2 + N2 -^ CaNCN + C
CaNCN + 2H + X- —> H2NCN + CaX2
Замещенные цианамиды получают из хлорциана и аминов:
Карбодиимиды являются изомерами цианамидов, известны толь-
только N.N'-дизамещенные карбодиимиды. В принципе они могут быть
получены алкилированием натриевых солей алкилцианамидов
RNHC=N.
Важным промышленным методом получения карбоднимидов яв-
является превращение изоцианата в карбодиимиды в присутствии нг-
которых фосфиноксидов:
2R-N = С- О — ~° R —N=.C-N-R-fCO,
2. Физические и химические свойства. Цианамиды и карбодиимн-
ды — бесцветные жидкие или кристаллические вещества.
Цианамид обладает амфотерными свойствами — является сла-
слабой NH-кислотой (р/(а«10,3) и слабым N-основанием (/?Д'ВН+ --
= 1,1). В присутствии кислот происходит присоединение слабых
нуклесфильных реагентов (воды, спиртов) к тройной связи.
Карбодиимиды по своему строению подобны изоцианатам:
Для карбодиимидов возможна такая же пространственная изо-
изомерия, как и для дизамещенных алленов (гл. III.А), соединения яв-
являются хиральными.
С нуклеофильными реагентами карбодиимиды реагируют по-
подобно изоцианатам, только их активность меньше. Реакции со
слабыми нуклеофилами катализируются кислотами.
При реакции карбодиимидов с карбоновыми кислотами образу-
образуются О-ацилмочевины, которые, в свою очередь, являются ацили-
648
рующими реагентами и служат для получения сложных эфиров,
амидов:
О
Н O-C-Ri
R-N-.C-N-R+R'-Ofe- б+-^ R-X-C
0 0 0
1 + HOR* II
Н O-C-R1 ¦-> RXHCNHR-I-R'COR2 .
R-N-C
+ H,NR2
О О
II II
4N —R
Таким путем карбодиимиды способствуют реакциям этерифика-
ции и образования амидов.
Некоторые ^-аминокислоты в присутствии карбодиимидов пре-
превращаются в C-лактамы.
Дициклогексилкарбодиимид CeHuN=C^-NCeHu — бесцветное
кристаллическое вещество с т. пл. 34. . .35 °С. В воде не растворя-
растворяется. При соприкосновении с кожей вызывает экзему.
Получают его из циклогексиламина через циклогексилизоцианат
или окислением N.N'-дициклогексилтиомочевины. Широко приме-
применяется для синтеза сложных эфиров, амидов, лактамов, ангидри-
ангидридов, особенно в химии природных соединений.
Г. ПРОИЗВОДНЫЕ ТИОУГОЛЬНЫХ КИСЛОТ
Производные тиоугольных кислот представляют собой важную
группу сераорганических соединений. Здесь кратко рассмотрены
только тиомочевина, тиоцианаты, ксантогепаты и дитиокарбаматы.
1. ТИОМОЧЕВИНА
1. Методы получения. Диамид тиоугольной кислоты (тиомочевину)
получают нагреванием роданида аммония:
NH4+-SCN Z a + [^
Первичные и вторичные амины при нагреванпи с роданидом ам-
аммония дают N-замещенные тиомочевины RR'NCSNH2.
2. Химические свойства. Нуклеофильным центром в молекуле
тиомочевины является атом серы. Тиомочевина — слабое основание
(РКби+ »—1), она легко алкилируется у атома серы с образова-
образованием соли S-алкилизотиурония:
NH2 '->4JSH2
649
Расщепление солей S-алкилизотиурония щелочью применяется
для получения тиолов RSH.
Ацилирование и реакции с альдегидами протекают с участием
атома азота.
Ы,Ы'-Дизамещенпые тиомочевины при взаимодействии со сла-
слабыми окислителями (например, HgO, PbO) превращаются в карбо-
диимиды:
RNHCNHR-j HgO —* RN=C=NR-bHgS'[ H2O
3. Важнейшие представители. Тиомочевина — бесцветное крис-
кристаллическое вещество с т. пл. 180—182 °С (при быстром нагрева-
нагревании), растворяется в воде.
В промышленности ее получают из цианамида и сероводо-
сероводорода.
Тиомочевину используют в органическом синтезе для получения
тиолов и гетероциклических соединений (производные пиримидина,
тиазола).
Тиомочевина, подобно мочевине, образует салканами клатраты
(соединения включения). Только в отличие от кристаллов мочеви-
мочевины в кристалле тиомочевины каналы больше и в них размещаются
и удерживаются разветвленные алканы, а н-алканы не удерживают-
удерживаются.
2. ТИОЦИАНЛТЫ
И ИЗОТИОЦИЛНАТЫ
1. Методы получения. Алкилированием неорганических роданидов
получают главным образом тиоцианаты (роданиды). Только при по-
повышенной температуре наблюдается образование изотиоцианатов.
При нагревании тиоцианатов происходит частичная перегруппиров-
перегруппировка их в изоцианаты:
+s~ .8-
8+5- У v Ь+ 6-
Д—S—C=N —¦ *- S=C=N~ R
Тиоциапаты получают также прямым роданированием при по-
помощи родана (SCNJ.
2. Физические и химические свойства. Тиоцианаты — бесцвет-
бесцветные вещества с нехарактерным слабым запахом. В противополож-
противоположность им простейшие изотиоцнанаты обладают раздражающим за-
запахом и свойствами лакриматоров (горчичные масла).
JXhh тиоцианатов характерны реакции присоединения по трой-
тройной связи. Строение изотиоцианатов подобно строению изоцпанатов.
Сходны и их химические свойства. Изотиоцианаты легко присоеди-
присоединяют нуклеофильные реагенты, образуя производные тиоугольной
С50
кислоты:
*¦ R—К—С^
H
3. Важнейшие представители. Тиоцианаты R—-S—С =з N приме-
применяются в качестве инсектицидов, фунгицидов, гербицидов и бакте-
бактерицидов.
Изотиоцианаты R—N—C=S встречаются в природных продук-
продуктах в свободном виде или в виде гликозидов. Например, в горчице
содержится аллилизотиоцианат СН2-СНСН,—N—C=S.
Изотиоцианаты используются в органическом синтезе и орга-
органическом анализе. Некоторые изотиоцианаты обладают биологи-
биологической активностью (инсектициды, фунгициды, бактерициды).
3. КСАНТОГЕНАТЫ И ДИТИОКАРБЛМАТЫ
1. Методы получения. Основным исходным веществом для получе-
получения производных дитиоугольной кислоты является сероуглерод
CS2. При взаимодействии с алкоксидами образуются соли О-алкил-
дитиоугольной (ксантогеиовой) кислоты — ксантогенаты:
6+ /S-Na+ r.-x /SRI
RO-Na+-J-S = C = S —> S=-C< >¦ S = C/
XO —R XOR
Алкилированием ксантогенатов получают эфиры ксантогеновой
кислоты. Сероуглерод при реакции с аминами дает аммониевые
соли дитиокарбаминовых кислот — дитиокарбаматы:
2RR'NH + S=cL
В присутствии NaOH образуются дитиокарбаматы натрия.
2. Физические и химические свойства. Соли О-алкнлдитиоуголь-
ной кислоты (ксантогенаты) — стабильные, окрашенные в желтый
цвет соединения, растворимые в воде. Ксантогенаты тяжелых ме-
металлов в воде не растворяются.
Эфиры ксантогеновой кислоты (алкилксантогепаты) термически
нестабильны, при нагревании они отщепляют алкен (реакция Чу-
гаева, 1899).
Предполагают, что реакция происходит в циклическом комплек-
комплексе с одновременным (согласованным) разрывом и образованием свя-
связей:
651
Дитиокарбаматы являются бесцветными кристаллическими ве-
веществами, растворимыми в воде. Соли тяжелых металлов окрашены
и в воде не растворяются.
3. Важнейшие представители. Ксантогенаты ROCSJ М+ явля-
являются важными промежуточными продуктами в производстве искус-
искусственного волокна из целлюлозы и в органическом синтезе.
N-Алкилдитиокарбаматы применяют в качестве аналитических
реагентов и как активные фунгициды. N.N-Диэтилдитиокарбамат
натрия легко растворяется в воде, с ионами тяжелых металлов об-
образует нерастворимые в воде комплексы, которые экстрагируются
органическими растворителями.
Дитиокарбаматы цинка — эффективные фунгициды, использу-
используются в борьбе с болезнями растений.
ЧАСТЬ ТРЕТЬЯ
Гетероциклические
соединения
КЛАССИФИКАЦИЯ
И НОМЕНКЛАТУРА ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ
Гетероциклические соединения могут быть образованы из циклоал-
канов (циклоалкенов, циклоалкадиенов) или аренов замещением
одного или более углеродных атомов цикла другим атомом — гетг-
роатомом. Обычно гетероатомами являются азот, кислород, сера:
H
н.
с
\
^ |_ч
H
Гетероциклические соединения подразделяются в зависимости
от величины цикла, а также от числа и вида гетероатомов в моле-
молекуле.
Присутствие гетероатома в цикле указывается специальными
префиксами: окса- (О), тиа- (S), аза- (N), фосфа- (Р), сила- (Si),
которые присоединяются к названию соответствующего карбоцик-
лического соединения. Согласно номенклатуре ИЮПАК величину
цикла обозначают особым суффиксом: трехчленный цикл суффик-
суффиксом -ирин (для циклов, не содержащих азот, -ирен), четырехчлен-
четырехчленный ет, пятичленный ол, шестичленный ин, семичлен-
ный епин, восьмичленный оцин. Тип гетероатома указыва-
указывается соответствующим корнем: оке-, ти-, аз- и др., а число одинако-
одинаковых гетероатомов — обычными префиксами: ди-, три-, тетра-
653
и т. д. Если в гетероцикле несколько различных гетероатомов, их
называют в определенной последовательности: окса-, тиа-, селена-,
теллура-, аза-, фосфа-, арса-, стиба-, висма-, сила-, герма-, станна-,
плюмба-, меркура-.
Гетероциклические соединения с двумя или более конденсиро-
конденсированными циклами, если все гетероатомы находятся в одном цикле,
а остальные циклы являются бензольными, называют бензологами.
К названию такого гетероциклического соединения присоединяют
префикс бенз- (бензо-) или дибенз- (дибензо-). Для указания поло-
положения бензольного цикла связи гетероциклического соединения
обозначают буквами а, Ь, с, d и т. д., начиная со связи гетероатом —
углерод, например бепзо [Ь] пиридин.
В названии гетероциклических соединений отражают и степень
гидрирования системы двойных связей гетероцикла. Основное наз-
название относится обычно к соединению с максимально возможным
числом двойных связей в цикле. Полностью гидрированное соеди-
соединение обозначают суффиксами -идин или -ан. Для частично гидри-
гидрированных соединений используют префиксы дигидро-, тетрагидро-,
при этом цифрами указывают атомы, у которых присоединены водо-
водородные атомы. Если водородный атом присоединен только к одному
атому, это обозначается буквой Н и цифрой, указывающей гидри-
гидрированный атом.
Для названия производных гетероциклических соединений ато-
атомы цикла нумеруются. Обычно нумерацию начинают от гетероатома.
Если имеется несколько гетероатомов, нумерацию начинают с того,
который в ряду О, S, Se, Те, N, P, As, Sb, Bi, Si, Ge, Sn, Pb, Hg
находится левее. Нумерацию атомов проводят в направлении, чтобы
гетероатомы получили наименьшие цифры. Для некоторых гете-
гетероциклических соединений сохраняется особый вид нумера-
нумерации.
Для многих гетероциклических соединений сохраняются триви-
тривиальные названия.
Ниже приведены некоторые группы гетероциклических соеди-
соединений их названия.
Трехчленные гетероциклические соединения с одним гетероато-
мом:
Х7 Х7 V
N X О
7 Х
N X
p вэнридия, ексирен,
Зэациклопропеп йтиленимин СКСациклопропеп
Х7 ^7 \7
оксиран, тиирен, тииран,
атиленоксид теациклопропен бтшкнеульфид
654
Четырехчленные гетероциклические соединения с одним гетеро-
атомом:
а зет,
а зацн клобутадиен
ц
а а, ?
Чг Ч,
1,2-дигидроазет 3,4-дигидроазет азетидин
а а
а
оксеган
-S:
тиегии
-S:
тпеган
Пятичленные гетероциклические соединения с одним гетероато-
мом:
Q Q <? Q
R
пиррол,
азациклов
N
К
ццрролццы
г.З-дитдра- 2,5-Д«гидро-.
виррод
диррод
Н
дарролидма
фуран,
фксацикдопентадяеа
тиофеи»
дибензотиофец
Пятичленные гетероциклические соединения с двумя гетероато-
мами:
N
Н
пиразол
1,2-диаюл
и мида зол
н
иидазод
14
>
N
Н
бензимидазод
соли 1,3-днтиолия
V'
1,2-оксазол, изоксазод
V
1,3-дитиод.
/Л
1,3-оксазол, оксазол
1,3-дитиолак
О
бензоксазод
0
1,3-тиаэол,тиазод
бензтиазод
Пятичленные гетероциклические соединения с тремя гетероато-
мамн (примеры с тремя атомами азота):
1,2,3-триазод
Н
1,2,4 триазол
н
бенкпрпазод
Пятичленные гетероциклические соединения с четырьмя гете-
роатомами (пример с атомами азота):
:N—N:
О
N
Н
теграэол
656
Шестичлеииые гетероциклические соединения с одним гетеро^
атомом
1.-0 О
Соли пирилия
4Н-пирак
тефагидропирай
О 00 0 0
пиридии
н
1,2-
)
н
1,4-
дигидроп
0
н
2,3-
иридины
3,4
0
Н
2,3,4,5- 1,2,3,4- 1,2,3,6-
тетрагидропиридикы
^sN:
хииолин,
беязо [Ь]пиридин
изохинолик,
бенчо [^пириди
р
дибензо|Ь,с1 пиридин
Шестичленные гетероциклические соединения с двумя гетероато-
мами (приведены примеры с двумя атомами азота и с атомами азота
и серы)
Г»
У
лирндазнн, 1,2-диазип
N:
N
пиримидин, 1,3-диазии
¦N;
О
пиразин, 1,4-диазии
цишюлин,
беию lei пнридэзкк
фгалазин,
беазо[6J иирида
хпназолин, хиноксалии,
беазопиримидик бензиинраэиц
феналш,
дибензолиразия
657
Семичленные гетероциклические соединения с одним гетероатомом
N
Н
н
?>ензо[Ь] азепин
дийензо [b,f] азспиц
Гетероциклические соединения с конденсированными циклами
могут содержать гетероатомы в нескольких циклах. В таких слу-
случаях эти соединения не являются бензологами. Названия таких ге-
гетероциклических конденсированных систем образуют по тем же
принципам, что и названия бензологов, только префикс бензо- за-
замещают названием соответствующего гетероцикла (фуро-, тиено-,
пирроло-, пиразоле-, имидазо-, тиазоло-, пиридо- и др.). За основу
названия принимают название цикла с наибольшим числом атомов,
а при равном их числе — с наибольшим числом гетероатомов. При
помощи цифр указывают, которые атомы второго цикла являются
общими с основным циклом, а при помощи букв — какая связь
основного цикла является общей для обоих циклов:
d^rvc 4 .3
О -О
Примеры номенклатуры:
N
фуро[2,3-Ь]пиридия фуро[3,4-с]пи|«|дии имидазо [4,5-Ь] пиридин. имидио [4,5-е] пиримидин
Глава XXXVI
ПЯТИЧЛЕННЫЕ
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
С ОДНИМ ГЕТЕРОАТОМОМ
Пятичленные гетероциклические соединения можно рассматривать
как продукт замещения в бензольном цикле одной группировки
СН —СН на гетероатом с неподелешюй парой электронов (N, О,
S, Se, Те, Р). Ниже рассмотрены соединения с гетероатомами N,
О и S (пиррол, фуран, тиофен, индол, карбазол).
А. ПИРРОЛ, ФУРАН И ТИОФЕН
Называя производные пиррола, фурана и тиофена, положение за-
заместителя указывают буквами а и |J или цифрами:
658
"П"
2 а
ч
СН3
Н
2,5-ди метил пир рол
2-фуранкарбаль«
дегид, фурфурол
]. МЕТОДЫ ПОЛУЧЕНИЯ
Пиррол, фуран, тиофен и их производные получают синтетически,
а также при переработке некоторых природных продуктов.
1. Общие методы получения пиррола, фуранаи тиофена. а) Ц и к-
лизация ^-дикарбонильных соединений. Ме-
Метод рассмотрен в гл. XXVIII.В.2.
б) Взаимные каталитические превраще-
превращения. Над катализатором (А12О3) при нагревании D00. . .500 °С)
фуран в присутствии аммиака превращается в пиррол, а в присут-
присутствии сероводорода — в тиофен. Возможны и противоположные
превращения:
Реакцию открыл Ю. К. Юрьев A936). Практическое значение имеют?
реакции получения пиррола и тиофена из фурана. Выход фурана и
тиофена из пиррола или пиррола и фурана из тиофена не превыша-
превышает 2%. При реакции с H2Se фуран дает селенофен.
2. Специфические методы получения, а) Получение пир-
пиррола и его производных. В небольших количествах
пиррол содержится в каменноугольной смоле. Он образуется также
в результате сухой перегонки некоторых белков.
Пиррол получают нагреванием аммониевых солей сахарных кис-
кислот, которые образуются при окислении альдогексоз(гл. XXIX.B.3):
н н
но | | он
ту \^ \^ TJT
ыиГоос—с с—cocTnh?—
--¦¦ он
НО
NH* ООС—-^ „ V-
>
-СО,; -N11,
659
б) Получение фурана него производных.
Альдопентозы в присутствии кислоты циклизуются, образуя фур-
фурфурол:
но он
„_/_„ Vй/н JL-L* * х>-сГ + ЗН2°
\ / С!
но он ^0
Альдогексозы в аналогичных условиях образуют 5-гидроксиме-
тилфурфурол.
Сахарные кислоты циклизуются в присутствии кислоты, обра-
образуя 2-фуранкарбоновую кислоту (пирослизевую кислоту):
NC С—СООМ —тг-*- НООС—< >— СООН
\ /
о<
<о,- - - -соон
Фурфорол является основным сырьем для получения фурана и его
производных.
в) Получение тиофена. Тиофен содержится в ка-
каменноугольной смоле и перегоняется совместно с бензолом. Его от-
отделяют от бензола химическим путем, например обработкой кон-
концентрированной серной кислотой (тиофеч сульфируется) или аце-
ацетатом ртути. В последнем случае образуется ртутьорганическое сое-
соединение, которое при действии кислот дает чистый тиофен:
IIij(OOCCH3),
н + х-
V V^HgOOCCH, V
2. ФИЗИЧЕСКИЕ СВОЙСТВА И СТРОЕНИЕ
Пиррол, фураи, тиофен и их простейшие производные являются бес-
бесцветными жидкостями с приятным запахом. Карбоновые кислоты
и производные с большей молекулярной массой — кристаллические
соединения.
Молекулы пиррола, фурана и тиофена содержат систему сопря-
сопряженных связей и атом с неподеленной парой электронов. Формаль-
но образуется циклическая сопряженная система с шестью я-элект-
s- s- ронами (четыре я-электрона от двух двойных свя-
?Й\ зей и два — от гетероатома). Поэтому можно считать,
что действует правило Хюккеля о стабильности цик-
лических сопряженных систем (гл. VI.Г) и эти соедн-
нения имеют «ароматический» характер.
Неподеленная электронная пара гетероатома действует как
электронодонор, поэтому на углеродных атомах цикла плотность
л-электронов увеличивается (я-избыточные гетероциклы).
660
1. Энергия делокализации, полярность и энергия ионизации.
Энергия делокализации (гл. VI. А.3) пятичленных гетероциклических
соединений меньше, чем для бензола (~150 кДж/моль). При этом
она уменьшается в ряду тиофен (~120 кДж/моль), пиррол
(~110 кДж/моль) и фуран (<~80 кДж/моль).
Дипольный момент направлен от гетероатома на цикл:
\
•осу
N
A.75D)
@.7D)
Пиррол, фуран и тиофен являются электронодонорными соеди-
соединениями, их энергия ионизации меньше, чем бензола (9,24 эВ).
Наименьшая ЭИ у пиррола (8,2 эВ). Для фурана ЭИ=8,9 эВ, а для
тиофена — 9,0 эВ.
2. Геометрия молекул и расчеты по методу МО. Длины связей
в пирроле, фуране и тиофене свидетельствуют об образовании со-
сопряженной системы, т. е. связи короче обычных (длина связи С—С
0,154 нм,С—N 0,145—0,147 нм, С—О 0,140—0,142 им, С—S0,18 им),
но длиннее двойных (длина связи С—С 0,134 нм):
н
Расчеты по методу МО в приближении Хюккеля свидетельствуют
о том, что все эти системы стабилизированы вследствие циклической
делокализации я-электронов. Энергия циклической делокализации
зависит от электроотрицательности гетероатома и резонансного
интеграла связи С—X. При увеличении электроотрицательное™
гетероатома энергия уменьшается. Энергия циклической делокали-
делокализации меняется приблизительно от 0,6 р (фуран) до 0,9 р (тнофен).
Для бензола это значение около 1 р.
В большинстве случаев в а-положении плотность электронов
больше, чем в р-положении. Наиболее наглядно это выражается в
распределении электронной плотности ВЗМО:
о,оз^_^,оз- 0,120,2
0,22 +
B3VIO,c';r
3. Спектры поглощения. Пиррол, фуран и тиофен являются прос-
простыми сопряженными системами и поглощают в ультрафиолетовой
661
области спектра. Картина поглощения во многом напоминает погло-
поглощение бензола (за исключением фураиа), т. е. наблюдается интен-
интенсивное поглощение в далеком ультрафиолете A80—210 нм) и ма-
малоинтенсивное поглощение — в интервале 230—270 нм со слабовы-
раженной тонкой структурой (табл. 52).
Таблица 52. Электронные спектры поглощения н ПМР бензола
н пятичленных гетероциклических соединений (в гексане)
Соединение
Бензол
Пиррол
Фурап
Тиофен
ЭСП- \nax- нм «=>
в интервале
180 — 210 нм
184 F0 000)
204 G 900)
209 F 730)
200 A0 000)
190
в интервале
230—270 нм
256B00) (центр
тонкой структуры)
240 C00)
235 D300)
ПМР связи С —Н, 6, м. д.
а
7,
6,68
7,40
7,30
Р
24
6,22
6,30
7,10
В ПМР спектрах наблюдаются сигналы протонов, сдвинутые в
слабые поля.
Молекулы пиррола, фураиа и тиофена стабилизированы в ре-
результате циклической делокализации (имеют «ароматический» ха-
характер). Стабилизация меньше, чем в случае бензола, меньше всего
стабилизирован фуран. Это связано с природой неподеленной пары
электронов гетероатома, которая в значительной степени связана с
атомом, участвует в делокализации только частично, особенно в слу-
случае оттягивающего электроны атома кислорода. Система тиофена бо-
более стабильна вследствие особенностей атома серы, который для
циклической делокализации л>электронов может предоставить спои
незаполненные d-орбитали. Тиофен по свойствам наиболее близок
бензолу.
3. ХИМИЧЕСКИЕ СВОЙСТВА
1. Общие свойства пиррола, фурана и тиофена. а) Каталити-
Каталитическое гидрирование. Водород присоединяется в при-
присутствии катализаторов (Ni, Pd, Pt) при нагревании и под давле-
давлением. Образуются тетрагидропроизводные:
(X--NR, О, S).
Кат.; t°
X
X
Труднее всех гидрируется тиофен и его производные. В специаль-
специальных условиях можно добиться расщепления цикла тиофена и обра-
662
эова*ия углеводорода и сероводорода:
б) Взаимодействие с сильными кислота-
кислотами, ацидофобность. Пиррол, фуран и тиофен взаимо-
взаимодействуют с сильными кислотами, в случае пиррола, фураиа и их
алкилпроизводных обычно происходит «осмоление» (олигомериза-
ция и полимеризация), тиофеп в присутствии серной кислоты суль-
сульфируется. Говорят, что пиррол, фуран и отчасти тиофен и их про-
производные «боятся» кислот, являются ацидофобными. Ацидофобность
связана с присоединением протона, в большинстве случаев к а-уг-
леродному атому, разрушением стабилизированной замкнутой со-
сопряженной системы с вытекающим отсюда следствием — дальней-
дальнейшими превращениями активной диеновой системы:
б'-р .S1- 8+ jr Дальнейшие
// %. + — , /-*J \ / превращения
5~>. y/S~~ + Н Y -ч Й+ЦЛ X. *" (олигомеризация-,
X X ^ полимеризация)
8+ Y-
(X = NR, О).
Наиболее легко протонируется пиррол, он является слабым ос-
основанием (р/Свп+ ~—0,3). Трудно протонируется тиофен, поэтому
он наименее ацидофобен.
Введение электропоакцепторных заместителей (карбонильные
группы, СООН, NO2) резко снижают способность производных пир-
пиррола, фурана и тиофена присоединять протон и тем самым снижает
ацидофобпость.
в) Взаимодействие с различными электро-
фильными реагентами. Пиррол, фуран, тиофен и их
производные реагируют с различными электрофилами, активность
повышается в ряду тиофеп<фуран<:пиррол, что согласуется с ря-
рядом понижения энергии ионизации. Нов зависимости от типа гете-
роцикла, типа электрофилыюго реагента и условий проведения
реакции образуются различные продукты; происходит или электро-
фильпое замещение, или присоединение, или проявляется ацидо-
ацидофобность.
Пиррол ввиду выраженного электронодонорного характера легко
реагирует со слабыми электрофилами, например диазониевыми соля-
солями, альдегидами. Бромирование и иодирование пиррола дает тетра-
галогенпирролы. Прямое нитрование пиррола невозможно. а-Нит-
ропиррол получают через соли пиррола. Сульфирование происхо-
происходит в мягких условиях при использовании комплексно связанного
SO3 (например, пиридинсульфотриоксида, А. П. Терентьев):
н н
663
Фуран ацилируется ангидридами кислот в присутствии мягких
кислот Льюиса, например SnCI4. Гладко идет сульфирование пн-
ридинсульфотриоксидом. Нитрование возможно в растворе уксус-
уксусного ангидрида действием безводной азотной кислоты. Нитрующим
реагентом здесь является ацетилнитрат CH3COONO2, и промежуточ-
промежуточно можно выделить продукт присоединения:
9,' . н __ н
jr- а II S— о-г \ / \ / Основание
f \ + СНзС-О— NO2 —*. Х.-Х '
N-/ СН3СОО< Np^ >NO2
Такие продукты 2,5-присоединения в реакциях фурана и его про-
производных наблюдаются часто, например при галогенировании. Это
свидетельствует о весьма низкой энергии циклической делокализа-
ции фурановой системы.
Тиофен легко галогенируется, ацилируется, сульфируется сер-
серной кислотой, нитруется ацетилнитратом.
Реакции электрофильного замещения почти исключительно осу-
осуществляются в положении 2. Это связано с энергетически более
выгодным строением промежуточного иона (а-комплекса) I, не-
нежели II, и большей плотностью электронов в ВЗМО в положении 2:
2. Специфические химические свойства, а) Пиррол и его
производные. Пиррол содержит полярную связь N—Н и
является слабой NH-кислотой (р/Са«16,5):
н2о
По кислотности пиррол сравним с этанолом. Соли пиррола по-
получают взаимодействием пиррола со щелочами, алкоксидами или
металлорганическими соединениями. Анион пиррола (пиррил-ион)
является сопряженным ионом, подобен циклопентадиенид-ноиу
и имеет несколько реакционных центров (атомы N, а- и (}-С):
S- 8+
+ R—X
Н
— алкил, ацил, X —галоген). ¦
Соли пиррола используются для введения алкила или ацила в
молекулу пиррола.
664
При пониженных температурах образуются преимущественно
N-замещенные продукты, при повышенных — а-С-замещеиные.
Взаимодействие с НСООС2Н5 дает пирролкарбальдегид, с
r_ONO2 — а-нитропиррол. Реакция пиррола с формальдегидом
дает диииррилметан, который может быть окислен в пиррометен:
о-
*-<? —
о
н н н •
дипиррклметэн пиррометек
Пиррометен образуется также при конденсации пиррола с муравьи-
муравьиной кислотой или пиррола с пирролкарбальдегидом. Пиррометены
'являются промежуточными соединениями в синтезах порфиринов.
б) Фу ран и его производные. Фуран во многих
реакциях реагирует как 1,3-диен. Он вступает, например, в реак-
реакции диенового синтеза с малеиновым ангидридом:
,0
ангидрид 3,6-эндоксо-
1,2,3,6-гетрагидро-
фталевой кислоты
i. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ И ПРОИЗВОДНЫЕ
1. Пиррол и его производные. Пиррол — бесцветная жидкость с
запахом хлороформа; кипит при 130 СС, мало растворим в воде
F%). При хранении темнеет.
Получают пиррол из фураиа и аммиака над катализатором. Ис-
Используют в органическом синтезе.
Пирролидинкарбоновые кислоты являются важными аминокис-
аминокислотами (гл. XXXIV.B.3).
Порфин — темно-красное кристалличе-
кристаллическое вещество. Его молекула представляет
собой макроциклическую сопряженную си-
систему, содержащую четыре пиррольных
кольца. В молекуле порфина имеется два
атома водорода у атомов азота, которые мо-
могут быть замещены металлом. Образуются
устойчивые комплексы, в которых ион ме-
металла находится в поле действия четырех
атомов азота.
Замещенные порфины называются порфиринами. Они лежат в
основе очень важных природных соединений.
Красное вещество крови гемоглобин, переносящий кислород из
легких в каждую клетку тела, является хромопротеидом, состоящим
из белка глобина и окрашенной в красный цвет небелковой части —
665
гема. Гем является порфирином, содержащим Fe(II). Продукт окис-
окисления гема — гемин содержит Fe(III). Строение гема выяснил и
осуществил синтез Г. Фишер A927):
н3с сп=с\и н:с
СНз
CHj
.СИ..СН.СООИ
CH.Ciij
CM:,
СНз
сн.оос
Хлорофилл
/О
(при R=CH3 — хлорофилл а, при R=-C^ —хлорофилл Ь).
Зеленый пигмент растений хлорофилл содержит в частично гидри-
гидрированном порфиновом цикле комплексно связанный магний. Один
остаток пропионовой кислоты этерифицирован спиртом фитолом
С20Н3;)ОН. Из растений выделены хлорофилл а (сине-черные крис-
кристаллы) и хлорофилл b (темно-зеленые кристаллы). Хлорофилл оп-
оптически активен. Структуру хлорофиллов установили Р. Виллштет-
Цен,
h,ncchT
CHjCONH2
CH,CH2CONH2
Рис. 100. Витамин
тер, М. Штолл и Г. Фишер A913—1938), полный синтез его был про-
проведен Р. Вудвордом с сотр. A956—1960).
Хлорофиллы играют исключительную роль в процессе фотосин-
фотосинтеза: они трансформируют световую энергию в химическую.
Витамин В12 (циаикобаламин) — темно-красное кристалличе-
кристаллическое вещество, растворимое в воде. Вырабатывается микроорга-
666
низмами. Строение цианкобаламина установлено рентгенографиче-
рентгенографически (Д. Кроуфут — Ходжкин, 1948—1956). В основе его структу-
структуры лежит макроцикл, состоящий из четырех частично гидрирован-
гидрированных пиррольных циклов (рис. 100).
Полный синтез цианкобаламина осуществлен под руководством
Р. Вудворда и А. Эшенмозера A960—1971), что явилось одним
из выдающихся достижений органического синтеза.
В промышленности витамин В,2 получают путем бактериаль-
бактериального биосинтеза. Витамин В!2 является активным средством про-
против анемии, его применяют для лечения злокачественного мало-
малокровия, заболеваний нервной системы и печени.
2. Фуран и его производные. Фуран — бесцветная жидкость
с запахом хлороформа; т. кип. 31,3 °С. При хранении постепенно
темнеет, осмоляется.
В промышленности фуран получают декарбонилированием фур-
фурфурола на цинкхромовом катализаторе:
400 °С й i
+ СО
В лабораторных условиях фурап может быть получен декарбок-
силированием 2-фуранкарбоновой кислоты в присутствии соеди-
соединений меди.
Фуран применяют в органическом синтезе, из него получают
тетрагидрофуран, пиррол, пирролидин, диальдегиды малеиновой
и янтарной кислот.
Фурфурол — бесцветная или слегка желтоватая жидкость с
приятным запахом свежеиспеченного ржаного хлеба; т. кип. 162 °С,
умеренно растворим в воде (9%).
Фурфурол получают из растительных продуктов, содержащих
полисахариды пентозаны, при обработке их кислотой.
Для фурфурола характерны все альдегидные реакции бензаль-
дегида, а также реакции фуранового цикла. Это делает его очень
важным исходным продуктом для органического синтеза. Окисле-
Окислением фурфурола в паро-газовой фазе над катализатором получают
малеиповый ангидрид. Декарбоиилирование фурфурола дает фу-
фуран. Нитрование ацетил нитратом приводит к 5-нитрофурфурол-
диацетату:
UNO, Оснояаппс
q(CH3CO),O
.ООССНз
XOOCCH3
5-Нитрофурфуролдиацетат (ацилаль нитрофурфурола)' явля-
является исходным веществом для синтеза производных 5-нитрофурфу-
рола, которые находят применение в медицине и ветеринарии как
667
сильные бактерициды:
О
при R= —NHCNH.. фурацнлнн
< *с^
?= —К
Н
О
фуразелндон
R= — N\ / NH фурадонпи
II
О
3. Тиофен. Тиофен представляет собой бесцветную жидкость,
по запаху напоминающую бензол; т. кип. 84,1 °С, в воде не раст-
растворяется.
Получают тиофен из каменноугольной смолы или синтезируют
из бутана и серы (или H2S), пропуская их над специальными ка-
катализаторами. Используют тиофен в органическом синтезе.
Б. ИНДОЛ И КАРБАЗОЛ
Индол и карбазол являются бензологами пиррола. Нумерацию
атомов в индоле начинают от гетероатома, в карбазоле — от угле-
углеродного атома бензольного цикла. Атомы углерода в гетероцикле
индола обозначают также буквами аир:
9 N
н
1. МЕТОДЫ ПОЛУЧЕНИЯ
Индол и карбазол содержатся в каменноугольной смоле, откуда
их и выделяют. Кроме того, индол н его производные в небольших
количествах встречаются в природных продуктах. Индол, карба-
карбазол и их производные могут быть получены также синтетически.
1. Синтез индола и его производных. Наиболее распространен-
распространенным методом является циклизация фенилгндразоиов в присутствии
кислот: используют H2SO4, HCI, ZnCl,, H3PO4) полифосфорную
кислоту, BF3 (Э. Фишер, 1883):
CH,R
N=G
668.
Предполагают, что в реакции Фпшера в присутствии кислот про-
происходит изомеризация гидразонов в гидразины с последующей
сигматропной перегруппировкой (согласованный разрыв связи
до—N и образование связи С—С) и замыканием пиррольного цикла:
R
2. Синтез карбазол а и его производных. Исходными веществами
для получения производных карбазола могут быть аминопроиз-
водные бифенила, например о,о'-диами[юбифенилы:
+ NH,+X~
H;.N
N
Н
2. ФИЗИЧЕСКИЕ СВОЙСТВА И СТРОЕНИЕ
Индол, карбазол и их производные — бесцветные кристалличе-
кристаллические вещества, не растворяющиеся в воде. Индол и его алкилпро-
изводные имеют своеобразный неприятный запах.
Индол и карбазол имеют циклическую сопряженную систему,
в которой участвует гетероагом со своей неподелепной электрон-
электронной парой. Если рассматривать пиррол как некоторый аналог
бензола, то индол может быть аналогом нафталина, а карбазол—
фенантрена. Соединения имеют «ароматический» характер.
В индоле и карбазоле проявляется электронодонориое действие
атома азота, на атомах углерода наблюдается повышенная плот-
плотность электронов, особенно в положениях 3, 5 и 7 для индола и
1, 3, 6, 8 — для карбазола:
8-—. ,«_
«+
*-С,Ч-К!'к.тм B,05 D)
Индол и карбазол являются электронодонорными соединениями,
их энергия ионизации соответственно 7,8 и 7,6 эВ.
669
3. ХИМИЧЕСКИЕ СВОЙСТВА
1. Индол. Как электронодонорное соединение и слабое основание
индол реагирует с различными электрофильными реагентами.
В отличие от пиррола в индоле реакционным центром обычно яв-
является углеродный атом в положении 3, что обусловлено влия-
влиянием бензольного цикла. В то же время индол является слабой
NH-кислоюй и дает соли.
а) Взаимодействие с сильными кислотами,
а ц и д о ф о б и о с т ь. Кислоты протонируют индол в положении
3, тем самым нарушается сопряженная система одного цикла,
появляется активная двойная связь С —N + , что ведет к дальнейшим
превращениям (димеризация, олигомеризация):
5-
-^ **.
Димеризапия,
олигомеризация
б) Взаимодействие с различными элект-
электрофильными реагентами. Ввиду выраженных элект-
ронодонорных свойств и ацидофобности индола реакции с силь-
сильными электрофилами обычно ведут к продуктам окисления и оли-
гомеризации. С более мягкими электрофилами осуществляется
замещение в положении 3. Если это место занято, атакующая
группа вступает в положение 2. Сульфирование возможно при
действии пиридинсульфотриоксида, бромирование — при дейст-
действии комплекса брома с диоксаном. При действии формальдегида
и вторичных аминов происходит реакция Манниха (гл. XXIX. Г. 2):
HN(CH3J —»- | N> +Н2О
Некоторые реакции электрофильного замещения (нитрование,
нитрозирование, алкилирование, ацилирование) осуществляются
в мягких условиях через соли (металлические производные) ин-
индола. Окисление индола мягкими окислителями дает синий кра-
краситель индиго (см. ниже).
в) Образование и реакции солей индола.
Индол является слабой NH-кислотой, по кислотности (р/Са— 16. . .
. . .17) сравним с пирролом. При действии щелочей, алкоксидов,
металлорганических соединений образуются соли (металлические
производные) индола:
+ н.о
670
. Соли индола проявляют высокую нуклеофильную реакцион-
реакционную способность, могут быть алкилированы, ацилированы, нит-
нитрованы алкилнитратами. Реакционными центрами могут быть
атомы азота и углерода в положении 3. Соотношение продуктов
N- и С-реакций определяется температурой реакции, природой
металла и растворителем:
no2 ,
s- s+ .. JT а+ 8-
Чч RO—NCb Г TfC^V R~X
"* " L jl ry
+ MX
2. Карбазол. Карбазол обладает значительными электронодонор-
ными свойствами, с электроноакцепторами (хинонами, полинит-
роаренами) образует окрашенные комплексы с переносом заряда.
По химическим свойствам карбазол весьма напоминает дифе-
дифениламин, является очень слабым основанием и слабой NH-кисло-
той. При взаимодействии с ацетиленом в присутствии щелочного
катализатора карбазол превращается в N-винилкарбазол:
ij (Г\Л ГТ-Т
4. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ И ПРОИЗВОДНЫЕ
Индол — бесцветное кристаллическое вещество с неприятным за-
запахом; т. пл. 52 °С, т. кип. 253 °С.
Индол используют в органическом синтезе для получения
физиологически активных веществ и в парфюмерии.
Индоксил C-гидроксииндол) — желтоватое нестойкое кристал-
кристаллическое вещество с неприятным запахом; т. пл. 85 °С. В растворах
проявляет способность к кето-енольной таутомерии:
В кристаллическом состоянии существует в кетоформе C-ок-
соиндолин). Кислотность соединения сравнима с кислотностью
фенола.
В виде гликозида индикана
СНоОН
НО
он
Индикаи
"ГУ
1—та
671
индоксил встречается в растениях рода индигофера. Фермента-
Ферментативным или кислотным гидролизом индикан превращается в пн-
доксил и глюкозу.
В промышленности индоксил получают циклизацией фенпла-
миноуксусной кислоты:
NaNH.
NaOH; КОН
О
11 >
н
Индоксил обычно в чистом виде не выделяется. Он очень легко
окисляется кислородом воздуха, особенно в щелочной среде. Про-
Продуктом окисления является синий краситель индиго:
о о
Индиго (индиготин) — темно-синее высокоплавкое и малораст-
малорастворимое кристаллическое вещество. Раньше его получали из ра-
растений рода индигофера, содержащих индикан. Синтетический
индиго получают из фениламиноуксусной кислоты A898).
Индиго является кубовым красителем, т. е. он нерастворим в
воде и его переводят в растворимую форму восстановлением. Этим
раствором обрабатывают ткань, после чего окислением фиксируют
краситель на ткани. Для восстановления обычно применяют ди-
тионит натрия Na2S2O4:
С сильными окислителями (КМпО4) индиго окисляется до иза-
изатина (индолиндиона-2,3).
Триптофан (индолилаланин) — важная а-аминокислота (гл.
XXXIV. В. 3).
672
ck,ch,nh2 Серотонин (З-аминоэтил-5-гидроксиин-
дол) — биогенный амин (см. гл. XXIII.
А. 4) ряда индола. Он обладает высокой
биологической активностью, участвует в
процессе передачи нервных импульсов в
центральной нервной системе. В организме образуется из трип-
триптофана.
Индольные алкалоиды. Алкалоидами называется группа ве-
веществ, выделяемых из растений и обладающих основными свой-
свойствами. Алкалоиды являются физиологически активными вещест-
веществами, многие из них сильные яды. Обычно алкалоиды имеют очень
сложную структуру, но почти всегда можно в их структуре найти
какой-то гетероцикл.
Индольная группировка входит во многие алкалоиды, напри-
например в рассмотренные ниже резерпин и лизергиновую кислоту.
Резерпин выделен из растений рода раувольфня A952). Его
строение установил и провел полный синтез Р. Вудворд A956):
СНзО'
н
СООСНз
ноос
Резерпин — бесцветное кристаллическое вещество с т. пл.
284. . .285 °С, в воде не растворяется. Сильное основание, с кис-
кислотами образует водорастворимые соли. Оптически активен, [a]D—
=—118° (СНС13).
Резерпин применяют в медицине как средство, понижающее
кровяное давление и успокаивающее (действует на центральную
нервную систему).
Лизергиновая кислота входит в состав ал-
алкалоидов спорыньи. Структура установлена в
1936 г., полный синтез осуществил Р. Вудворд
A954).
Лизергиновая кислота—бесцветное кристал-
кристаллическое вещество с т. пл. 238 °С, [a] D= +40°,
образует соли с кислотами и щелочами.
Амид лизергиновой кислоты —эргин — про-
проявляет высокую физиологическую активность: сужает кровеносные
сосуды, действует на гладкую мускулатуру матки, облегчает роды.
Диэтиламид лизергиновой кислоты — препарат ЛСД-25 — силь-
сильное галлюциногенное средство.
N-Винилкарбазол —бесцветное кристаллическое вещество ст. пл.
65 °С. Легко иолимеризуется, образуя поливинилкарбазол (ПВК),
который легко перерабатывается в прозрачные пленки. Эти пленки
нашли применение в электрофотографии. Для этой цели в пленку
ПВК вводят сильный электропоакцептор (хиноны, полинитросое-
диненпя), который с ПВК образует комплекс с переносом заряда.
22
517
673
Такая пленка является хорошим электроизолятором, но при ин-
интенсивном освещении ее удельное сопротивление резко умень
шается. Это связано с возбуждением комплексов с переносом зг
ряда и генерированием носителей заряда. Явление используется
для получения изображения на пленке.
Глаза XXXVII
ПЯТИЧЛЕННЫЕ
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
С ДВУМЯ И БОЛЕЕ ГЕТЕРОАТОМАМИ
Из большого класса пятичленных гетероциклических соединений
с двумя, тремя и четырьмя гетероатомами здесь рассмотрены пи-
пиразол, имидазол, соли дитиолия, оксазол, тиазол, бензтиазол,
триазолы, тетразол и их производные.
А. ПИРАЗОЛ И ИМИДАЗОЛ
Называя производные пиразола и имидазола, атомы цикла нуме-
нумеруют, начиная с атома азота N—Н (N—R) в направлении второго
атома азота по кратчайшему пути.
Могут существовать четыре монозамещенных пиразола A-,
4 3 4 з 3-, 4-, 5-) и имидазола A-, 2-, 4-, 5-), 3-
/.—\ /Г~\ и 5-замещенные пиразола и 4- и 5-за-
J\../N:2 5\..y2 мещенные имидазола образуют системы
n i Ni таутомерного равновесия,
н н
1. МЕТОДЫ ПОЛУЧЕНИЯ
1. Пиразол и его производные. Для построения цикла пиразола
или гидрированных производных (ниразолина, пиразолндина) не-
необходимы два компонента:
В качестве компонента, поставляющего группировку
используют а, Р-ненасыщенные карбонильные соединения и Р-
дикарбонильные соединения, а в качестве компонента N—N —¦
гидразин и его производные.
По второму варианту в синтезе участвуют алкены (алкины)
и диазосоединения.
а) Синтезы с гидразином, его производ-
производными и Р-д икарбонильными соединениями.
Гидразины очень легко реагируют с Р-дикетонами. Обычно в ре-
674
акцию вступает дикетоформа:
н о
+ H.N— Nit.
—н.о
н о
I //
иестаоильиая таутомериая
форма пира-io.ia
Реакция эфиров [5-оксокислот с гидразинами дает пиразолоны
(гидроксипиразолы):
н о
h-V/
+ H..N — NHR1
Н
>
N... -С,Н.,<Ж
* /
А-
;n—н
но
Эфиры малоновой кислоты при взаимодействии с гидразинами
образуют пиразолидиндионы:
н о н
Н \ / XN-R
— — / г и nv
б) Синтезы с диазоалканами. Ацетилен и его
производные реагируют с дказоалканамн. В результате 1,3-дипо-
лярного присоединения (циклоприсоединения 12+3J) образуются
пиразолы:
г>2
Ч Г
с—с—н
.к
N
н
22*
675
2. Имидазол и его производные. Построение цикла имидазола
или его гидрированных производных (имидазолина, имндазолк-
дина) возможно различными путями:
N
N
I С
а) Синтезы с а-д и к а р б о н и л ь н ы м и с о е д и н е-
ниям и, аммиаком и альдегидами:
/н -,ад V\7
нестабильная таутомерная
форма имидазола
б) Синтезы с а-г идроксикарбо пильным и
соединениями и ам и динам и:
нестабильная тауточерна я
форма имидазола
2. ФИЗИЧЕСКИЕ СВОЙСТВА И СТРОЕНИЕ
Пиразол, имидазол и их производные — бесцветные вещества, в
основном кристаллические (если имеется связь N—Н). Соедине-
Соединения, содержащие связь N—Н, образуют прочные межмолекуляр-
межмолекулярные водородные связи типа N—Н- • -:N —.
Пиразольный и имидазольный циклы представляют собой со-
сопряженные системы, в которых три я-электроиа дают три атома
углерода, один л-электрон — атом азота, образующий двойную
связь, а неподеленную пару электронов — второй атом азота. Об-
Образуется циклическая замкнутая система из 6 л-электронов, фор-
формально соблюдается правило Хюккеля, осуществляется стабили-
стабилизация системы в результате циклической делокализацни я-элект-
ропов. Неподеленная пара электронов атома азота выступает в
качестве электронодонора, молекулы пиразола и особенно имида-
676
зола имеют большие дипольные моменты:
^=13,3-10~мКл-м D,0 Д)
По сравнению с пирролом пиразол и имидазол имеют одно прин-
принципиальное отличие: вместо одного углеродного атома они содер-
содержат более электроотрицательный атом азота, который образует
двойную связь, а неподеленная пара электронов этого атома ори-
ориентирована в направлении вне цикла и в сопряжении практически
не участвует (сравните с атомом азота в пиридине).
Как свидетельствует рентгеноструктурный анализ, длины свя-
связей соответствуют сопряженной системе (сравните с пирролом,
гл. XXXVI.А.2):
н н
По сравнению с системой пиррола в пиразоле и имидазоле непо-
деленная пара электронов одного атома азота больше вовлечена
в образование циклических молекулярных орбиталей. В этом,
по-видимому, сказывается влияние второго атома азота, оттяги-
оттягивающего электроны. Поэтому пиразол и имидазол более стабильны,
чем пиррол (более «ароматичны»),
3. ХИМИЧЕСКИЕ СВОЙСТВА
1. Кислотность и основность. Пиразол, имидазол и их производ-
производные являются амфотерными соединениями — слабыми NH-кисло-
тами и средней силы основаниями:
Н pA-BI1* = 2,S H
катион пиразолпя
// Л ^_ Г\
n
к н
15 Р^вн+-7
«атион имидазолия
Относительно более сильным основанием является имидазол. В ре-
результате присоединения протона образуются стабильные катионы,
677
циклическая сопряженная система не нарушается в отличие от
протонирования пиррола. Положительный заряд в одинаковой
степени делокализуется по обоим атомам азота и частично дело-
кализован в положениях 3 и 5 пиразола или в положении 2 имидазола.
2. Взаимодействие с электрофильными реагентами. Центром ата-
атаки электрофильного реагента является атом азота; в пиразоле
второй, в имидазоле третий. Образующиеся катионы пиразолия
и имидазолия, в свою очередь, могут подвергаться действию силь-
сильного электрофила с образованием продукта электрофильного за-
замещения. Так может быть осуществлено нитрование, сульфиро-
сульфирование, галогенирование. В случае пиразола образуются продукты
замещения в положении 4, в случае имидазола — в положениях
4 или 5. Ацилирование и алкилирование обычно дают продукты
N-замещения.
Во взаимодействие со слабыми электрофильными реагентами
легко вступают металлические производные (соли) пиразола и ими-
имидазола, подобно солям пиррола и индола.
3. Особенности реакционной способности пиразолонов. Пиразо-
лоны (гидроксиниразолы) являются слабыми основаниями и в
то же время обладают свойствами кислот. Они представляют собой
таутомерные системы. Наиболее важными являются пиразолоны-5,
возможные таутомерные формы которых изображены ниже:
т.1 Rl
В кристаллическом состоянии наиболее устойчива форма III.
Таутомерные превращения и многие реакции пиразолонов осу-
осуществляются через сопряженный анион, который имеет три ре-
реакционных центра (О, С, N). Например, азосочетапие проходит
по атому углерода, алкилирование — по атому азота:
Пиразолоны по своей реакционной способности во многом
подобны циклическим Р-дикетонам.
678
СН,
4. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ И ПРОИЗВОДНЫЕ
J. Пиразол и его производные. Пиразол — бесцветное кристал-
кристаллическое вещество со слабым запахом пиридина; т. пл. 70 °С, т. кип.
187 °С, растворим в воде.
Получают его окислением 3,5-диметилпиразола до 3,5-пиразол-
дикарбоновой кислоты и декарбоксилированием последней.
Используют пиразол в органическом синтезе.
Антипирин A-фенил-2,3-диметилпи-
разолон-5) — бесцветное кристаллическое
вещество с т. пл. 114 °С. Это важное
исходное вещество для получения лекар-
лекарственных веществ. Его используют так-
также в аналитической химии, так как
оно образует нерастворимые комплексы
с некоторыми ионами металлов.
Амидопирин (пирамидон, 1-фенил-2,3-
диметил-4-диметиламипоииразоло11-5) —
бесцветное кристаллическое вещество
с т. пл. 107—109 СС. Его получают из
антипирина. Амидопирин применяют в
медицине в качестве болеутоляющего п
жаропонижающего средства.
Анальгин — бесцветное водораство-
водорастворимое кристаллическое вещество. Его
получают из антипирина. Применяют
как болеутоляющее, жаропонижающее
и противовоспалительное средство.
2. Имидазол и его производные. Имидазол — бесцветное крис-
кристаллическое вещество; т. нл. 90 СС, т. кип. 256 °С, растворимо в
воде. Получают имидазол из глиоксаля, аммиака и формальде-
формальдегида. Применяют в органическом синтезе.
Гистидин (имидазолилаланнн) — важная а-аминокислота (гл.
XXXIV.B.3).
Б. СОЛИ 1,3-ДИТИОЛИЯ И ПРОИЗВОДНЫЕ 1,3-ДИТИОЛА
Соли 1,3-дитиолия являются аналогами имидазола, содержащими
вместо атомов азота атомы серы:
N
1
G.H-,
антипирин
СНз
НзС-N
N
1
1
с„
^СН3
CFb
н5
амидопирин
N.+ -WCH
[,_>/ СН,
0А^/ЧСНз
1
Ъиальгми
4 -А
<х
679
1, МЕТОДЫ ПОЛУЧЕНИЯ
Построение цикла 1,3-дитиола или 1,3-дитиолана возможно раз-
различными путями:
С s '
При взаимодействии а-галогенкарбонильных соединений с про-
производными тритиоугольной кислоты образуются соединения, цик-
циклизация которых дает 1,3-дитиолтионы-2:
Na+ -s 4 к
) 4 C-S-C(CH3h ¦ _"^3
OS»
CH2=C(CH3)j
Окислением 1,3-дитиолтионов-2 перуксусной кислотой или пер-
оксидом водорода получают 1,3-дитиолиевые соли:
R4 R4 5
сн.сооон S
снасоон
HSO.
'4
R'"Y
2. ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА.
ВАЖНЕЙШИЕ ПРОИЗВОДНЫЕ
1,3-Дитиолтионы-2 — кристаллические вещества желтого цвета,
другие производные дитиола и соли дитиолия — бесцветные ве-
вещества.
л-Электропная система катиона 1,3-дитиолия формально ана-
аналогична системе катиона имидазолия, т. е. образуется замкнутая
бл-электронная система и возможна циклическая делокализация:
Это вызывает некоторую стабилизацию таких своеобразных систем
с двумя атомами серы в пятичленном цикле. Однако 1,3-дитиолие-
1,3-дитиолиевые соли весьма нестабильны и проявляют высокую реакционную
способность по отношению к нуклеофильным реагентам (место
680
атаки — атом углерода в положении 2). Стабилизирующе влияют
электронодонорные заместители в положении 2, например алкнл-
тио- и особенно диалкиламиногруппы.
1,3-Дитиолтионы-2 и 1,3-дитиолиевые соли главным образом
используются для синтеза своеобразной системы — тетратиа-
фульвалена B,2'-би-1,3-дитиолилидена). Это достигается димериза-
цией дитиолтионов в присутствии фосфитов или димеризацией
дитиолиевых солей в присутствии триэтиламина:
R R
V-S . ^ ^
л> ^ XXX
^ XXX
Тетратиафульвалены являются сильнейшими электронодоно-
рами (ЭИ—6,3. . .6,8 эВ) и при действии окислителей образу-
образуют устойчивые катион-радикальные соли. Тетратиафульвален с
сильнейшим электроноакцептором тетрацианохинодиметаном (гл.
XXXIII.К-4) образует ион-радикальную соль, которая в кристал-
кристаллическом состоянии обладает высокой электропроводностью, по-
подобной металлам. Эта ион-радикальная соль была первым пред-
представителем класса «органических металлов» A972), которые вы-
вызывают огромный интерес в физике твердого тела. К «органическим
металлам» принадлежат также катион-радикальные соли тетра-
тиафульваленов (ТТФ), содержащие в кристалле также молекулы
неокисленного ТТФ, обычно типа (ТТФ)?Х~, где X = BF4, C1O4,
PF6, ReO4 и др. Плоские молекулы ТТФ и его катион-радикала в
кристалле располагаются одна над другой и образуют стопки.
Наивысшая проводимость наблюдается в направ-
направок лении оси стопки.
Более выраженными свойствами «органиче-
-•R ских металлов» обладают катион-радикальные
соли тетраселенафульвалена.
Среди катион-радикальных солей тетраселенафульвалена, а
также тетратиафульвалена найдены такие, кристаллы которых
при температурах ниже 8,5 °К становятся сверхпроводниками
A979—1983), например иодид бис-этиленднтиотетратиафульвалена:
В. ОКСАЗОЛ, ТИАЗОЛ И БЕНЗТИАЗОЛ
Оксазол и тиазол являются аналогами фурана и тиофена, содер-
содержащими вместо атома углерода в положении 3 атом азота. В то же
время они напоминают имидазол, только вместо группы NH со-
631
держат атомы О или S:
1. МЕТОДЫ ПОЛУЧЕНИЯ
1. Получение оксазола и его производных. Построение циклов ок-
оксазола, оксазолина и оксазолидина возможно из гидрокснамино-
соединений, а также из а-аыинокарбонильных соединений:
С—N С—N С—N
С' + С *- С' .С -« С. С
хо хох о
Оксазолидипы образуются при взаимодействии альдегидов и
этаполамина:
.С R "v y'~-~R
Циклизация а-ациламииокарбонильных соединений в присут-
присутствии кислот дает оксазолы:
к ни:
HC-N + _ C-N X
2. Получение тиазола и его производных. Построение системы
тиазола, тиазолина, тпазолидина возможно из амнпотиолов, а
также из тиоамндов:
С—N С—N с N
гл' i е-\ ^ i~** j-i ,. / • Л*
^ / "Г" *^"
С с/
Тназолндины образуются из 2-амипотиолов и альдегидов:
2N^ о.
Н2с( + V—R
( + VR >- Vdc V
682
а-Галогенкарбонильные соединения реагируют с тиоамидами
и дают тиазолы (А. Ганч, 1888):
с—R2
+
О NH2
R-/ V-tf
H
\
но н
R—С—N (
Н У
-HO
3. Получение бензтиазола и его производных. Их получают из
о-аминотиофенола и карбоновых кислот или из анилина, сероуг-
сероуглерода и серы:
•NH3 Оч
+ \-К
SH НО^
+ 2Н2О
2. ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА.
ВАЖНЕЙШИЕ ПРОИЗВОДНЫЕ
Оксазолы и тиазолы представляют собой бесцветные вещества,
растворимые в воде. Подобно фурану, тиофену, имидазолу, в мо-
молекулах оксазола и тиазола образуется циклическая замкнутая
сопряженная система, содержащая бл-электронов. Возможна ста-
стабилизация сопряженной системы в результате циклической дело-
делокализации л-электронов (системы «ароматичны»):
В результате электронооттягивающего действия атома азота ос-
остальные атомы цикла несут некоторые положительные заряды.
Длина связей свидетельствует, что имеет место некоторое вырав=
нивание связей, характерное для сопряженных систем.
Энергия циклической делокализации для оксазола меньше, чем
для тиазола (сравните с фураном и тиофенсм).
€83
Оксазолы и тиазолы являются слабыми основаниями, с кисло-
кислотами они образуют соли оксазолия и тиазолия:
При нагревании с кислотами соли оксазолия расщепляются.
Алкилирование проходит по атому азота с образованием солей
N-алкилоксазолия и N-алкилтиазолия.
Тиазол — бесцветная жидкость с запахом пиридина; т. кип.
117°С. Его получают из хлорацетальдегида и тиоформампда.
Тиазол используют в органическом синтезе. Важны некоторые
производные тиазола, обладающие биологической активностью
(лекарственные вещества, витамины, антибиотики).
Норсульфазол — бесцветное кристаллическое вещество с т. пл.
198. . .200 °С. Получают из 2-амннотн
азола. Это эффективный сульфанила^
-nh2 мидный препарат, применяющийся как
антимикробное средство при инфекцион-
инфекционных заболеваниях.
Витамин Вх (тиамин) — бесцветное, растворимое в воде кри-
кристаллическое вещество. В качестве простетической группы он
входит в состав некоторых фер-
ферментов, способствующих про-
^N цессам декарбоксилироваппя.
~* ' Тиамин синтезируется в рас-
носн2сн2" \s/ h3n^ ^tr ^сн3 тениях и в некоторых микро-
с1 организмах.
В настоящее время тиамин получают синтетическим путем.
соон Пенициллины — антибиотики, эффек-
н с. V ° тивно действующие на различные мик-
а X/i^f^ Q роорганизмы. В основе структуры пени-
HjC/\ J—L^ II циллинов лежат конденсированные цик-
s NHC лы тиазолидина н р-лактама.
Пенициллины образуются в результате жизненных процессов
некоторых микроорганизмов (плесневых грибков). Известно не-
несколько природных пенициллинов, которые получаются биосин-
биосинтетическим путем, например бензилпенициллин (R = ССН5СН2),
феноксиметилпенициллин (R — ССН5ОСН2) и др., а также полу-
полусинтетические пенициллипы, которые получают из соли амино-
пенициллановой кислоты и ацилхлоридов:
4- NaCl
684
2-Меркап/побвнзтиазол (каптакс) — кристаллическое вещество
желтого цвета; т. пл. 179 °С. Таутомерное вещество, более стабиль-
стабильной таутомерной формой является бензтиазолинтнон.
В промышленности каптакс получают в крупных масштабах из
анилина, сероуглерода и серы. Применяют в качестве ускорителя
вулканизации каучука и для получения других ускорителей вул-
вулканизации.
Г. ТРИАЗОЛЫ И ТЕТРАЗОЛ
Пятичленные гетероциклические соединения могут содержать три
атома в двух различных положениях A,2,3- и 1,2,4-), а также
четыре атома азота:
>
ЗДЗ-трназол Д,2,4-гриазол уегриая
1. МЕТОДЫ ПОЛУЧЕНИЯ
1. 1,2,3-Триазолы получают реакцией 1,3-диполярного присоеди-
присоединения (циклоприсоедипепия 12+3]) между производными ацети-
ацетилена и азидами или HN3:
Н N:
/
С
Н N:
/ * '"///
н
2. 1,2,4-Триазолы синтезируют из гидр азидов имидокислот и
производных карбоновых кислот:
/R
Rl^C<1./f + /C~R —*" R'~C\ /N + ° + HX
н н
3. Тетразолы образуются в реакции 1,3-диполярного присоеди-
присоединения (циклоприсоединенмя 12+3!) между нитрилами (пли HCN)
685
и азидами (или HN3):
«- г +
N: v N:
/// ¦¦—/// N—N
-о i 4^ S.J.. N
2. ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА.
ПРИМЕНЕНИЕ
Триазолы, тетразол и их производные являются бесцветными
кристаллическими соединениями, растворимыми в воде.
Эти соединения стабилизированы в результате циклической
делокалнзации бл-электронов. В этом они подобны пиразолу и
имидазолу, но вследствие замены еще одного или двух углеродных
атомов на азот их электронодонорные свойства уменьшаются.
Триазолы и тетразол являются слабыми основаниями и сла-
слабыми NH-кислотами. Основность уменьшается в ряду 1,2,4-триа-
зол (р/(ви + --2,3), 1,2,3-триазол (рКви+ —1,2), тетразол. В таком
же ряду увеличивается NH-кислотность: 1,2,4-триазол, 1,2,3-
триазол (рКа--9,4); тетразол (рКп—4,9).
При взаимодействии с электрофильпыми реагентами можно
получить продукты N-замещения (алкилирование, ацилирование).
Известны реакции у атома азота через металлические производные
(соли).
1,2,3-Триазол— бесцветное кристаллическое вещество; т. ил.
23 °С, т. кип. 203 СС. Получают нагреванием 1,2,3-триазол-4-кар-
боновой кислоты. Используют в органическом синтезе.
1,2,4-Триазол — бесцветное кристаллическое вещество с т. пл.
120 СС и т. кип. 260 СС. Используют в органическом синтезе.
Тетразол — бесцветное кристаллическое вещество с т. пл.
156 °С, легко возгоняется. Получают из HCN и HN3.
Некоторые производные тетразола пспользу-
ют как взрывчатые вещества и лекарственные
средства. Например, препарат коразол стиму-
стимулирует деятельность центральной нервной систе-
коразол МЫ.
Глава XXXVIII
ШЕСТИЧЛЕННЫЕ
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
С ОДНИМ ГЕТЕРОАТОМОМ
Шестичлеиные гетероциклические соединения можно рассматри-
рассматривать как бензол, в котором один атом углерода замещен на гете-
роатом (N, О, S). В этой главе рассмотрены пиридин, пирилиевые
соли, некоторые их производные и бензологи (хинолип, изохинолин).
68$
А. ПИРИДИН
Образуя названия производных пиридина, используют нумерацию
атомов цикла и обозначения греческими буквами а, р1 и у. Неко-
Некоторые простые производные имеют тривиальные названия:
СН3 СН3
4V I I
р5м1зр mi Л mi
сн, СН /^
N N
1
а-пиколин 2,4-диметил- 2,4,С-триметил-
пиридин пиридин
B,4-лутидин) B,4,6-коллндин)
1. МЕТОДЫ ПОЛУЧЕНИЯ
1. Получение из природных продуктов. Пиридин и его алкилпро-
дукты содержатся в продуктах сухой перегонки каменного угля —
в каменноугольной смоле и подсмольных водах, откуда их полу-
получают в промышленности. Пиридин содержится также в продуктах
сухой перегонки костей.
Система пиридина входит в состав многих природных продук-
продуктов — витаминов и алкалоидов.
2. Синтетические методы. Построение системы пиридина, дигид-
ропиридинов возможно по нескольким схемам:
б-Дикарбонильные соединения легко циклизуются в 1,4-ди-
гидропиридины:
сн2 сн2
С. С — HjO с .. с
н
Синтез Ганча A882) заключается во взаимодействии
Р-дикарбонильного соединения, альдегида и аммиака, в резуль-
результате чего образуются производные 1,4-дигидропиридина. р-Дикар-
бонильные соединения в реакции с альдегидами образуют продукты
конденсации (гл. XXVIII.Б.3, гл. XXXIV.Д.2), которые содержат
структурный элемент б-дикарбонильного соединения. Взаимодей-
687
ствием с аммиаком замыкается цикл дигидропириднна:
О Ь* О О R2 О
* АЛА- -l ii! -1
ST ^N"^ ^R
H
\o\
Окисление производного дигидропиридина дает соответствую-
соответствующий замещенный пиридин. Если в реакции использован эфир Р-
кетокислоты (R1—OC2H5), гидролизом продукта можно получить
пиридиндикарбоновые кислоты, декарбоксилирование которых дает
2,4,6-замещенные пиридины.
Циклизация ацетилена и HCN происходит обычно
при высоких температурах:
„/"
W'
Т
с
1.1
с
H
О
Реакция подобна получению бензола и его производных из
ацетилена или замещенных ацетиленов (гл. IV.4.2). Получение
пиридина таким путем представляет только теоретический интерес.
Более перспективными могут быть циклизации такого типа в при-
присутствии катализаторов — комплексов переходных металлов.
Циклизация альдегидов и аммиака в при-
присутствии катализатора при нагревании обычно дает смесь различ-
различных алкнлпиридинов, например:
СН3
С П.
Л-
V
N
N
В этом процессе происходит альдольная конденсация, образо-
образование иминов, дегидратация и дегидрирование.
688
2. ФИЗИЧЕСКИЕ СВОЙСТВА И СТРОЕНИЕ
1. Сопряжение, полярность, энергии ионизации. Циклическая со-
сопряженная система, состоящая из бл-электронов, подобна бензоль-
бензольной системе, за исключением того, что один атом углерода замещен
атомом азота в состоянии 5/о2-гибридизации. Неподеленная элект-
электронная пара атома азота ввиду своего пространственного распо-
расположения, в сопряжении практически не участвует:
&+
Вследствие электроноакцепторного действия атома азота моле-
молекула пиридина является полярной и все атомы углерода приоб-
приобрели некоторые положительные заряды.
Электронодонорпые свойства пиридина по сравнению с бензо-
бензолом уменьшены. Отрыв электрона возможен от неподеленной пары
электронов атома азота (ЭИ=9,6 эВ) и от высшей занятой молеку-
молекулярной орбитали сопряженной системы (ЭИ-^=9,75 эВ). Из этого
обстоятельства вытекает, что электрофильные реагенты вначале
будут атаковать атом азота.
2. Геометрия молекул, МО расчеты. Данные рентгеноструктур-
ного анализа свидетельствуют о том, что длина связей С—С в
пиридине такая же, как в бензоле:
Я
0,096 +
0,018+
0,106 +
0,040 +
!
R
Расчеты МО в приближении Хюккеля показывают, что атомы
углерода системы пиридина действительно приобрели некоторые
положительные заряды (на схемах приведены эффективные заряды
и порядки я-связей). Если к атому азота присоединился электро-
фильный реагент, эффективные положительные заряды значи-
значительно возрастают. Наибольшие эффекты наблюдаются в а- и
^-положениях.
Система пиридина характеризуется значительной энергией цик-
циклической делокализации @,99р — 1,03E), сравнимой с энергией
циклической делокализации бензола. Это свидетельствует о боль-
большой стабильности («ароматичности») циклической системы пири-
пиридина.
3. Спектры поглощения. В ультрафиолетовой части спектра для
пиридина характерны два максимума поглощения (раствор в гек-
сане): 251 им (е-^-2000) и 270 им (е-----.450). Второй максимум связан
с возбуждением неподеленной электронной пары (л-кп;*-переход).
689
В водном растворе оба максимума сливаются в один при 257 нм
F=2750).
Катион пиридиния поглощает при 256 нм (е=5250)'.
В ПМР спектрах сигналы протонов пиридина, как и в случае
бензола, наблюдаются в слабых полях: а-протоны при 8,29 м. д.,
E-протоны при 6,77 м. д., Y-протоны при 7,15 м. д. (неразбавлен-
(неразбавленная жидкость).
3. ХИМИЧЕСКИЕ СВОЙСТВА
Для пиридина и его производных характерны реакции с электро-
электрофильными реагентами у атома азота и в положении 3, и с нуклео-
фильными реагентами в положениях 2 и 4, что следует из распре-
распределения электронной плотности. Специфическими реакциями об-
обладают N-оксиды пиридинов, катионы N-замещепного пиридиния
и а-метилпиридипы. Известны реакции восстановления (гидриро-
(гидрирования).
1. Основность. Пиридин и его производные являются основа-
основаниями, с кислотами они образуют соли пиридиния. Основность
пиридина сравнима с основностью анилина (р/<(вн + = 4,60):
R r
н
R Н а-СНз ¦у-СНз
рКвп+ (Н2О) 5,23 5,97 6,02
2. Взаимодействие с электрофильными реагентами. Электрофиль-
ные реагенты присоединяются к неподеленной электронной паре
атома азота. Такие продукты присоединения в некоторых случаях
могут быть изолированы в кристаллическом состоянии. Легко
осуществляется алкилировапие:
X I X-
SOS R
пиридин- соли N-алкил-
сульфо- пиридипия
триоксид
Электрофильное замещение в цикле пиридина происходит
только в жестких условиях B30. . .300 °С). Заместитель вступает
только в р-положение:
,SO3H
N
новая кислота
690
Можно также нитровать и бромировать в положение р. Осу-
Осуществить прямое электрофильное замещение в положения а и у
не удается. Это достигается при использовании N-оксида пири-
пиридина.
3. Взаимодействие с нуклеофильными реагентами. Пиридин и
его производные реагируют только с сильными нуклеофильными
реагентами.
Щелочи реагируют с пиридином только при температуре
около 400 °С. Образуются соли а-пиридонов (а-гидроксипириди-
нов):
+ -ОНК+ —»
N N и *
Амид натрия реагирует с пиридином при 130 °С, в ре-
реакции образуется а-аминопиридин (с небольшой примесью
нопиридипа) (А. Е. Чичибабин, 1914):
+ H.N: Na
t-
NHNa"'
+ 1h
На первой ступени реакции образуется натриевая соль а-амино-
пиридина и выделяется водород.
4. Реакции окисления. Цикл пиридина устойчив к действию окис-
окислителей. Алкилпиридпны легко окисляются до соотвеюгвующих
пиридинкарбогювых кислот:
СН3 ,.ч ..СООН
[О]
и
С пероксикислотами, а также пероксидом водорода окисление
происходит по атому азота и образуется N-оксид пиридина:
RCOOOII
N
N +
о-
5. Реакции N-оксида пиридина. Относительно легко соедине-
соединение нитруется с образованием N-оксида у-нитроииридина. Такая
ориентация замещения объясняется некоторым электронодонор-
691.
ным эффектом неподеленной электронной пары атома кислорода.
Эту реакцию используют для получения 7"замещенных пиридина:
NH,
HNO,
ГН1
6. Реакции катионов 1Ч-алкил(арил)пиридиния. Катион пири-
диния весьма легко взаимодействует с нуклеофильными реаген-
реагентами, при этом могут образоваться продукты присоединения или
размыкания цикла:
S't-
R'NH,
7. Реакции а-метилпиридинов. Метильная группа в а- и у
жениях обладает подвижными атомами водорода. Реакционная
способность метильной группы в сс-пиколине сравнима с реакци-
реакционной способностью метилкетонов.
В присутствии оснований или, в отдельных случаях, кислых
катализаторов (ZnCl^, полифосфорная кислота) происходит кон-
конденсация с альдегидами и кетонами:
Кат.
N
ЧСН,
N
ОН
CH2-C-R
Н
-н,о
N
8. Гидрирование. Пиридин гидрируется трудно. Каталитиче-
Каталитическое гидрирование водородом в присутствии N: под давлением
даст пиперидин:
N
692
Ni; Г' \ /
н
Производные пиридиния легко гидрируются комплексными
гидридами:
NaBII,
ч /
N
R
1
\/
N
R
R Л
4. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ II ПРОИЗВОДНЫЕ
Пиридин — бесцветная жидкость с неприятным («пиридиновым»)
запахом; кипит при 115,6СС, растворяется в воде, ядовит.
Получают пиридин при коксовании каменного угля. Исполь-
Используют в качестве растворителя и исходного для органического син-
синтеза (получения пиперидина, аминопиридина, лекарственных ве-
веществ и др.).
Пиколины — бесцветные жидкости с неприятным запахом;
а-пиколин кипит при 129,5 °С, Р-пиколнн —¦ при 144 СС и у-"»ко-
лин — при 145 СС. Легко растворяются в воде.
Пиколины получают из каменноугольной смолы. Используют
в органическом синтезе.
Гидроксипиридины — бесцветные водорастворимые кристаллы.
Их получают из соответствующих галогенпиридннов, аминопири-
динов или пиридинсульфоповых кислот, подобно получению фе-
фенолов. Используют также реакции а- или у-пиронов (гл. XXXVIII.
В.З) или прямое гидроксилирование пиридина (а-изомер).
а- и 7~Гидроксипиридипы являются таутомерными соедине-
соединениями и их более устойчивой (менее кислой) таутомерной формой
является пиридоиовая:
О
и
N
И
а-пири-
дом
N
Р-гидрокси-
пирилпм
N
н
V-пири-
дон
он
Р-Изомер по своим свойствам более похож иа (}>енол. Все гид-
гидроксипиридины являются бифункциональными соединениями, об-
обладают свойствами слабых кислот и оснований, в реакции вступают
как по атому азота, так и по атому кислорода, известны реакции
электрофилыюго замещения у атомов углерода.
Некоторые замещенные гндроксппиридины нашли применение
в медицине.
Лминопиридины — бесцветные водорастворимые кристалличе-
кристаллические вещества. Получают прямым ампнированием пиридина (а-
изомер) или из нитро- и галогеппиридинов подобно ариламинам
(р- и у-нзомеры).
693
а- и'у-Аминопиридины являются таутомерными соединениями,
вторая таутомерная форма называется пиридониминной, более ус-
устойчивой является менее кислая аминоформа:
NH,
J _ =
N *
а-аминопири-
ДИН
\/w
N м
II
i Ч/
j
1 N
р-амино-
ниридин
1
ч/
N
V-ами ко-
копи ри дин
Аминопиридины обладают основными свойствами, их основ-
основность выше основности пиридина, это особенно выражено у у-
изомера 1р/(вн+=6,9 (а); 6@); 9,2(yI.
Аминопиридины используют для синтеза лекарственных ве-
веществ.
Пиридинкарбоновые кислоты представляют собой бесцветные
кристаллические вещества, растворяющиеся в воде. Их получают
окислением производных пиридина:
СООН
,ч ,СООН
N
пиколинопая никотиновая изолико-
киелота кислота типовая
кислота
Пиридинкарбоновые кислоты, будучи аминокислотами, явля-
являются амфотерными соединениями и в кристаллическом состоянии
и отчасти в растворах существуют в форме внутренней соли (бе-
(бетаина).
Никотиновая кислота впервые была получена при окислении
алкалоида никотина. Сама кислота является провитамином, а ее
амид — витамином PP. Недостаток этого витамина вызывает за-
заболевание кожи, называемое пеллагрой.
Гидразид изоникотиновой кислоты и его производные исполь-
используют при лечении туберкулеза.
СН2ОН Пиридоксин (витамин В6) — бесцветное лег-
НОСН2 ' ОН корастворимое кристаллическое вещество. Он
^f"Su// содержится в различных природных продук-
продуктах (дрожжах, бобах, печени).
"Ч/Чгн Фактически пиридоксин является только ис-
N 3 ходным для биосинтеза витамина В6. В организ-
организме из пиридоксина образуется пиридоксал-5-фосфат, который с
некоторыми белками образует пиридоксалевые ферменты, катали-
катализирующие превращения аминокислот.
694
N
I .
3
у i n
н
Пиридиновые алкалоиды. Наиболее важными представителями
этой группы соединений являются никотин, анабазин и соедине-
соединения ряда тронапа (атропин, кокаин).
Никотин — бесцветное масло с запахом табака,
кипит при 246 СС, растворяется в воде, [а] д=
= - +102° (Н2О+НС1). Главный алкалоид табака. Его
получают из отходов табачной промышленности.
Впервые в чистом виде получен в 1828 г., структура
выяснена в 1893 г., синтез проведен в 1904 г.
Соединение очень ядовито, летальная доза для человека 40 мг.
В малых дозах возбуждает центральную нервную систему, повы-
повышает кровяное давление, в больших дозах вызывает паралич нерв-
/\ ной системы.
Никотин используют в качестве инсектицида.
Анабазин — бесцветное масло с т. кии. 276 СС.
Получен из некоторых растений Средней Азии. В не-
небольших количествах содержится также в табачных
листьях. Анабазин используют в качестве сильнодей-
сильнодействующего инсектицида.
Атропин — алкалоид ряда тропана, со-
содержит в молекуле конденсированные цик-
циклы пнрролидииа и пиперидина. Получа-
Получают из некоторых растений. Это бесцветное
кристаллическое вещество с т. пл. 115...
... 116 °С. Действует на парасимпатическую
нервную систему, вызывает расширение зрачка. Применяется в
медицине.
Кокаин — алкалоид, получаемый из листьев кустарника кока.
Это бесцветное кристаллическое вещество с т. пл. 98 °С. С кисло-
кислотами образует водорастворимые соли.
^,сн3 Кокаин стимулирует и возбуждает нерв-
нервную систему, вызывает эйфорию, а затем
торможение нервной системы. Вызывает
сильную местную анестезию. При частом
применении может развиться болезненное
кокаии пристрастие к нему (кокаинизм). Строение
кокаина послужило моделью для синтеза многочисленных более
простых местпоанестезирующих средств.
ocociicjis
атро"и"
соосн
3
осос и
Б. ХИНОЛИН И ИЗОХИНОЛИН
5 4
5 4
/
8 1
695
1. МЕТОДЫ ПОЛУЧЕНИЯ
1. Получение из природных продуктов. Хинолин и изохинолин по-
получают из каменноугольной смолы. В ней содержатся также не-
некоторые метилпроизводные хинолина. Структурные элементы хи-
нолнпа и изохиполина входят в состав молекул многих алкалоидов.
2. Получение хинолина и его производных. Основным исходным
веществом для синтеза хинолина служат ариламины.
а) С и н т е з С к р а у п а. Ариламины в реакции с глицерином
и серной кислотой в присутствии слабых окислителей (нитробензол,
As2O5 и др.) при нагревании образуют хинолин или его произ-
производные. Реакцию открыл 3. Скрауп A880).
Предполагают, что в этой реакции из глицерина образуется
акролеин, который взаимодействует с арнламином:
Ю1
1,2-ДЩ'ИД[К»Х1Ш0.'1НМи
Вначале ариламин присоединяется к С=--С-связи акролеина,
а потом следует циклизация.
б) С и и т е з Д е б н е р а—М и л л е р а заключается в ре-
реакции ариламинов с а, ^-ненасыщенными альдегидами в 'присут-
'присутствии НС1, ZnCl2 или других кислот. Обычно ненасыщенные кар-
карбонильные соединения получают непосредственно в реакционной
смеси из альдегидов и кетонов путем альдолыюй конденсации. Ме-
Механизм реакции подобен механизму реакции Скрауна. Окислите-
Окислителями служат альдгшипы C0H5N--- CHR:
о
NH2
СИ N -CHR
696
Реакцию открыли О. Дебнер и В. Миллер в 1881 г.
3. Получение изохинолина и его производных. В синтезе исполь-
используют производные бензола и другие соответствующие компоненты:
Одним из исходных может быть 2-фенилэтиламин, который аци-
лируют, циклизуют и продукт циклизации дегидрируют (А. Биш-
лер и Б. Напиральский, 1893):
Исходными веществами могут быть также аренкарбальдегиды,
которые конденсируются с ацеталями а-амииоальдегидов с после-
последующей циклизацией продукта конденсации (Ц. Померанц и
П. Фрич, 1893):
H.NCHCH(OC;Hsh
Rl r-
R^
20 OC2Hs
С
H^ ^CH-Rl
1
¦
1
К
H+X
-2C\.HSOH.
:N:
2. ФИЗИЧЕСКИЕ СВОЙСТВА И СТРОЕНИЕ
Хинолин, изохииолин и их простейшие алкилпроизводные явля-
являются бесцветными жидкостями или низкоплавкими кристалличе-
кристаллическими веществами со своеобразным навязчивым запахом, в воде
растворяются мало.
Хинолин и изохииолин — азааналоги нафталина, содержат со-
сопряженную л-электронную систему из 10 л-электронов, которые
находятся в циклических молекулярных орбиталях. Присутствие
атома азота значительно изменяет распределение л-электронной
плотности. Ниже приведены эффективные л-заряды на атомах,
697
полученные методом МО в приближении Хюккеля:
0,00 0,014 +
0,00i<
0,042 + 1;
0,032 +
0,18 +
"^>|0,00
Jo,22 +
N
0,535-
0,045 +
0,0(М +
0,042
0,232 +
Электронооттягивающее влияние азота главным образом рас-
распространяется на цикл пиридина. Углеродные атомы бензольного
цикла несут меньшие положительные заряды. Распределение
зарядов служит основанием для объяснения ориентации в реак-
реакциях электрофильного и нуклеофильного замещения.
3. ХИМИЧЕСКИЕ СВОЙСТВА
1. Основность. Хпнолин и изохинолин являются слабыми основа-
основаниями, образуют соли с сильными кислотами:
-1-Н + Х-
х-
N !-
Н
Константа кислотности для иона хинолипия рКчн +=4,94,
для иона изохинолиння—5,14.
2. Взаимодействие с различными электрофильными реагентами.
Центром атаки электрофильного реагента является атом азота.
В результате образуется соответствующий катион, и дальнейшие
реакции электрофильного замещения протекают в бензольном
цикле согласно распределению электронной плотности.
Катион хинолипия обычно реагирует в положениях 6 и 8 (иногда
5), а катион изохинолиния — в положениях 5 и 7, иногда 4:
Е+Х-
N
N
(Е = SO3H, NO2)
698
Алкилирующие реагенты легко образуют соли N-алкилхино-
линия и N-алкилизохинолиния:
N+X-
Х~
3. Взаимодействие с иуклеофильными реагентами. Центром ата-
атаки нуклеофильного реагента является положение 2 в молекуле
хинолина и положение 1 в молекуле изохинолина. Легко проис-
происходят реакция Чичибабина и взаимодействие со щелочью:
4. Гидрирование. Каталитическое гидрирование в первую оче-
очередь затрагивает пиридиновый цикл:
H1
Н
1,2,3,4-тетра-
гидрохииолин
1,2,3,4-тетра-
гпдроизо-
хинолин
5. Окисление. Сильные окислители (KMnO4, HNO3) при нагре-
нагревании окисляют хинолин и изохинолин, один цикл, обычно бен-
бензольный, разрушается и образуются пиридиндикарбоновые кис-
кислоты:
,
Н00
HNO,
ноос/\/
HNOj
хинолиповая
кислота
цин:;омероновая
кислота
При взаимодействии g пероксикиелотами и пероксидом водо-
водорода образуются N-оксиды:
i-
699
4. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ И ПРОИЗВОДНЫЕ
1. Хинолин и его производные. Хинолин — бесцветная жидкость
с весьма неприятным запахом; т. кип. 237 СС.
Получают хинолин при переработке каменноугольной смолы
и используют в качестве растворителя и исходного вещества для
получения аналитических реагентов и лекарственных веществ.
Хинальдин B-метилхинолин) — бесцветная жидкость с запахом
хинолнна; т. кип. 247 °С.
Получают из каменноугольной смолы. Метальная группа хи-
нальдина легко вступает в реакции конденсации. Используют для
синтеза красителей.
Оксин (8-гидроксихинолин) — желтоватое кристаллическое ве-
вещество с т. пл. 75 СС. Получают из о-аминофенола в синтезе Скраупа
или из 8-хинолинсульфоновой кислоты сплавлением со щелочью.
Оксин является слабой ОН-кислотой (рЛГа=9,7), об-
образует соли. С ионами многих тяжелых металлов
образует соли — внутренние комплексы (хелаты), раст-
растворимые в органических растворителях. Оксин широко
применяется в качестве аналитического реагента.
Тиоксин (8-хинолинтиол) — красное кристалличе-
кристаллическое вещество (дигидрат). Получают восстановлением
8-хиполинсульфонилхлорида. С ионами многих ме-
металлов образует окрашенные внутренние комплексы,
растворимые в органических растворителях.
Тиоксин — важный аналитический реагент для определения
следовых количеств некоторых металлов.
1=сн2 Хинин — алкалоид коры хин-
но ff ~у ного дерева. Бесцветный порошок
горького вкуса; т. пл. 177 °С,
сно " Т '" [a]D=-—158° (в этаноле). Легко об-
^ '^ разует соли с кислотами. Строе-
Строение хинина выяснено в 1908 г.
(К. Рабе), полный синтез проведен
в 1944 г. (Р. Вудворд, В. Дёринг).
Хинин применяется для лечения малярии. Строение хининл
послужило моделью для поиска и синтеза других противомаля-
противомалярийных препаратов.
2. Изохинолин и его производные. Изохинолин — бесцветное
вещество с горьковатым запахом; т. пл. 24,5 °С, т. кип. 242,5 СС.
Получают из каменноугольной смолы. Ис-
Используют в органическом синтезе.
Важнейшими из изохинолиновых алка-
алкалоидов являются алкалоиды опиума.
Папаверин — бесцветное кристалличес-
кристаллическое вещество с т. пл. 147 °С, образует мало-
растворпмый в воде хлорид с т. ил. 225 °С.
Используют в медицине в качестве спаз-
vocn3 молитического и сосудорасширяющего
средства.
Морфин (R -= R1 = H) —бесцветное кристаллическое веще-
вещество; т. пл. 254 СС, [a]D=—130" (СН3ОН); растворяется в щелочах
й кислотах.
В молекулу морфина входит структурный
элемент гидрированного изохинолина (цикл с
атомом N и цикл с двойной связью и группой
OR1). Строение морфина установил Р. Робин-
Робинсон A925), полный синтез провел Г. Тсуды
A951—1956).
Морфин — сильнейшее болеутоляющее сред-
\0Rl ctbo, наркотик, вызывает эйфорию. Регуляр-
Регулярное применение ведет к привыканию, развива-
развивается болезненное пристрастие к морфину (наркомания, морфи-
морфинизм). Ослабляет и парализует действие центральной нервной
системы.
Кодеин является метилированным морфином (R = СН3, R1 = Н).
В виде солей используют в медицине в качестве болеутоляющего
средства и для успокоения кашля. Имеет более слабое наркотиче-
наркотическое действие, чем морфин.
Героин — солянокислый диацетилморфин (R — R1 = СОСН3).
Сильный наркотик.
В. ПИРИЛИЕВЫЕ СОЛИ И ПИРОНЫ
4 5 4
СОЛИ ШфИЛИЯ
СОЛИ бсН1и[/)]пИрИ.'1ИЧ
1. МЕТОДЫ ПОЛУЧЕНИЯ
Для построения пирилиевого цикла могут быть использованы
ненасыщенные б-дикарбонильные соединения:
Hv Xv ^-Н
о
о он
сюг
Обычно пнрилисвые соли синтезируются из а,р-непасыщен-
ных карбонильных соединений и метилкетонов в присутствии ук-
уксусного ангидрида и сильной кислоты (обычно НС1О4):
я
СН3
I
o;liciQi
-Н:О;-2Н
сюр
701
Вначале происходит алкилирование кетона а, Р-ненасыщенным
карбонильным соединением, в результате чего образуется насыщен-
насыщенное б-дикарбонильное соединение. Затем следует замыкание цикла
и дегидрирование. Акцептором водорода может служить ненасы-
ненасыщенное карбонильное соединение.
Из фенолов и а,р-пенасыщеиных карбонильных соединений
получают соли бензопирилия:
2. ФИЗИЧЕСКИЕ СВОЙСТВА И СТРОЕНИЕ
Простейшие пирилиевые соли являются бесцветными кристалли-
кристаллическими веществами. Соединения с электронодонорными арильными
группами окрашены.
Строение катиона пирилия в большой степени подобно строе-
строению системы катиона пирндиния, здесь имеет место циклическая
делокализация бя-электронов и стабилизация системы согласно
правилу Э. Хюккеля. Единственное, но существенное различие
заключается в замене атома азота сильно электроотрицательным
атомом кислорода в оксониевой форме. Это вызывает сильнейшую
поляризацию связен и высокую реакционную способность по
отношению к нуклеофильпым реагентам.
катион пирилия
3. ХИМИЧЕСКИЕ СВОЙСТВА
Соли пирилия являются сильнейшими электроноакцепторами об-
образуют окрашенные комплексы с переносом заряда с различными
электроиодонорами.
Очень характерны реакции с нуклеофильными реагентами
Обычно местом атаки является углеродный атом в положении 2
но возможны реакции также по атому 4.
Слабые восстановители и доноры гидрид-иона превращают пи-
рилиевые соли в а- и у-пираны:
R R
^R
Нуклеофильные реагенты присоединяются в положение 2.
Возможно и расщепление цикла с последующим замыканием и
702
образованием другой циклической системы:
•• х"
+ н+х~
T>H
:NH.,
+ Н2О + Н X"
X"
4. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ
а-Пироны являются сопряженным основанием солей 2-гидрок-
сипирилия:
R
сс-Пироны — соединения реакционноспособпые, они являютс
одновременно ненасыщенными б-лактонами и сопряженными дие-
диенами. Известны их реакции диенового синтеза, реакции полиме-
полимеризации и гидролитического расщепления.
Более стабильны бензологи а-пиронов. Напри-
*°.: мер, кумарин является бесцветным кристаллическим
кумарин веществом с приятным запахом свежего сена.
у-Пироны являются сопряженными основаниями солей 4-гид-
роксипирилия:
_:о: :ОН
РКГВ„- ~ 0,2-0,4
у-Пироны имеют свойства слабых оснований. При действии
щелочи расщепляются, с аммиаком и аминами образуют произ-
производные пиридина —у-пиридоны. Бензологи у-пиронов широко рас-
распространены в природе (хромон, флавон и их производные):
6' 5"
703
3',4',5,7-тетрагидрокси- и З',4',3,5,7-пентагидроксипроизводные
флавона — желтые пигменты цветов.
Антоцианидины — гидроксипроизводные солей 2-фенилбензо! Ы
пирилия являются основой пигментов цветов, ягод и овощей.
В виде гликозидов антоцианов (R —
он остаток моносахарида) они входят в
состав цветов, ягод, овощей (R'-H,
ОН). Антоцианы меняют окраску в
зависимости от кислотности среды.
В кислой среде окраска сдвигается
в сторону красной, в нейтральной —
она фиолетовая, в щелочной сдвигается в стЬрону синей. Изменение
окраски связано с ионизацией гидроксильных групп при изме-
изменении рН среды.
Глава XXXIX
ШЕСТИЧЛЕННЫЕ
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
С ДВУМЯ ГЕТЕРОАТОМАМИ
Замещением в бензоле двух углеродных атомов на гетероатомы
(N, О, S) получают соответствующие гетероциклические соединения.
В этой главе рассмотрены только пиридазин, пиримидин, пиразин
и его производные, конденсированная система пурин, а также
важные природные продукты — нуклсозиды, иуклеотиды и нукле-
нуклеиновые кислоты.
А. ПИРИДАЗИН, ПИРИМИДИН И ПИРАЗИН
N
I. МЕТОДЫ ПОЛУЧЕНИЯ
1. Пиридазин и его производные. Для замыкания цикла пиридазнна
применяют гидразин и соответствующую четырехуглеродную ком-
компоненту, например малеиндиальдегид, малеинальдегидную кис-
кислоту:
704
н
оЛ '
Н /
H.N
„Л
N
h ./
пиридаюк З-гилроксипиридази I
2. Пиримидин и его производные. Для синтеза цикла пиримиди-
пиримидина применяют соединения, содержащие атомы азота в положениях
1 и 3 (амидины, мочевина, тиомочевнна и др.) и Р-дикарбонильные
соединения:
R
Н i
+ 2H2O
jJ~l N|-< p 1 Р"'" ^M"^ "^D 1
oA -C R R N R
К.
он
н
урацид 2,4-дигидроксипиримиди(г
При взаимодействии форыилуксусной кислоты и мочевины об-
образуется урацил. Необходимую формилуксусиую кислоту полу-
получают из яблочной кислоты и олеума непосредственно в реакцион-
реакционной среде. Малоновый эфир и мочевина дают барбитуровую кислоту:
Н\ ^с—ос2н5
барбитуровая кислота,
23 № 517 705
3. Пиразин и его производные. Цикл дигидропиразина образует-
образуется в реакции самоконденсации а-аминокарбонильных соединений:
О
-2Н,О
R\J\/R
,С
XNH2
2. ФИЗИЧЕСКИЕ СВОЙСТВА И СТРОЕНИЕ
Пиридазин, пиримидин, пиразин и их производные — бесцветные
вещества, растворимые в воде. Производные с группами С—О и
N—Н образуют прочные межмолекулярные водородные связи, что
повышает температуру плавления и понижает растворимость.
Эти соединения являются аналогами пиридина и бензола, моле-
молекулы стабилизированы в результате циклической делокализации
бл-электронов. Так как эти соединения содержат два атома азота,
имеются две неподелепные пары электронов.
Рентгеноструктурный анализ показывает, что длины связей
С—С близки к длинам связей в пиридине и бензоле:
3. ХИМИЧЕСКИЕ СВОЙСТВА
1. Основность. Пиридазин, пиримидин и пиразин — более слабые
основания, чем пиридин. Так выражается электроноакцепторное
действие второго атома азота:
Лйн
х-
/
N
N
Значения рЯвп + (Н2О): пиридазин — 2,33, пиримидин — 1,3,
пиразин — 0,6. Протопирование второго атома азота возможно
только в концентрированных растворах кислот.
2. Взаимодействие с электрофильными реагентами. Центром ата-
атаки электрофилыюго реагента является один из атомов азота. Силь-
Сильные алкилирующие реагенты образуют N-алкилониевые соли, на-
например соль N-алкилпиримидиния:
х~
706
Осуществить реакции нитрования, сульфирования, галогени-
рования, ацилирования практически не удается.
Более активно в реакции электрофилыюго замещения всту-
вступают гидрокси- и алкоксипроизводные.
3. Взаимодействие с нуклеофильными реагентами. Пиридазип,
пиримидин и пиразин реагируют с сильными пуклеофилами легче,
чем пиридин. Для них характерна реакция Чичибабина:
N N
||NaNII2
/ Ч/
N N
Активио с нуклеофилами реагируют галогенпроизводные, ко-
которые легко синтезируются из гидроксипроизводных:
о он a nh.,
I I I
NH A
4. Гидрирование. Пиридазин, пиримидин и пиразин гидрируются
легче, чем пиридин. Пиридазин расщепляет цикл, пиримидин об-
образует гексагидропиримидин, а пиразин — пиперазин:
-I H2NCH2CH2CHXH2NH,
5. Окисление. Пиридазин, пиримидин и пиразин стабильны к
действию сильных окислителей. Пероксикислоты и Н2О2 при взаи-
взаимодействии с этими гетероциклическими соединениями образуют
N-оксиды. Метилзамещенные производные при действии СЮ^
или КМпО., окисляются до карбоновых кислот.
4. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ
1. Пиридззин и его производные. Пиридазин ¦—бесцветная жид-
жидкость со слабым запахом; т. пл. ¦—8 СС, т. кип. 208 СС, растворяется
в воде.
Пиридазин используют в органическом синтезе, его произЕод-
ные являются важными гербицидами.
Феназон (пирамин) — бесцветное кристаллическое
'"Я*2 вещество с т. пл. 200 СС. Его получают из дихторма-
CI\J\ леинальдегидной кислоты и фенилгидразина. Это эф-
I || фективный гербицид для уничтожения сорняков на по-
I 11^ лях сахарной свеклы.
of \/ 2. Пиримидин и его производные. Пиримидин —
I бесцветное вещество со слабым запахом; т. пл. 21 °С,
С6н5 т. кип. 124 '"'С, растворяется в воде. Его получают
23* 707
каталитическим восстановлением 2,4-дихлорпиримидина, который,
в свою очередь, синтезируют из урацила.
Пиримидин используют в органическом синтезе.
Урацил B,4-дигидроксипиримидин) — бесцветное
малорастворимое кристаллическое вещество, т. пл.
335 °С. Его получают из гидролизатов нуклеиновых
кислот, синтетически — из яблочной кислоты и мо-
мочевины.
Урацил — таутомерное соединение, более стабиль-
стабильной является диоксоформа, представляющая собой
слабую NH-кислоту (р/Са=--9,4). Предполагают, что
первой ионизируется связь N—Н у первого атома
азота. Урацил вступает в реакции электрофильного
замещения у атома углерода в положении 5.
Урацил входит в состав важных природных ве-
ществ — нуклеозидов и нуклеиновых кислот. Произ-
водные5-фторурацила, например фторафур (С. А. Гил-
лер), являются эффективными противораковыми пре-
препаратами.
„ с || Тимин E-метилурацил) — бесцветное малораство-
3 \/\\н римое кристаллическое вещество ст. пл. 318 С. Ста-
II бильной таутомерной формой является диоксоформа.
\/ Тимин входит в состав важных природных ве-
N ° ществ — нуклеозидов, нуклеотидов и нуклеиновых
Н кислот.
Ци/позин B-гидрокси-4-аминопиримидин) — бесцветное мало-
малорастворимое вещество с т. пл. 320—325 °С.
Впервые был выделен из гидролизатов нуклеиновых кислот.
Синтетически его получают нз урацила превращением последнего
в 2,4-дихлорпиримидин и последовательным аммонолизом и гид-
гидролизом:
Cl NH3 NH, NH2
mi,
/
N
N
Н
О
Цитозин является слабым основанием, сравнимым с анилином
-f-^4,6) и очень слабой NH-кислотой (р/Са--12,2).
Цитозин входит в состав нуклеиновых кислот.
Барбитуровая кислота — бесцветное малорастоо-
римое кристаллическое вещество с т. пл. 245 °С:
X , Ее получают из малонового эфира и мочевины. Ста-
1 бильной таутомерной формой является триоксофор-
ма. Барбитуровая кислота является СН-кислотой
(р/Са---4,05), по свойствам она подобна циклическим
Н р-дикетонам.
оЛЛ°
708
В медицине применяются 5,5-дизамещенные барбитуровые кис-
кислоты — так называемые барбитураты. Они являются сильными
снотворными веществами:
фенобарбитал, люминал
3. Пиразин — бесцветное кристаллическое вещество со слабым
запахом; т. ил. 57 СС, т. кип. 118°С, растворяется в воде, образует
гидрат. Его получают каталитическим дегидрированием пипера-
знна. Используют в органическом синтезе.
Б. ПУРИН
В системе пурина сохранилась нумерация атомов, которая не со-
соответствует общим правилам, применяемым для гетероцикличе-
гетероциклических соединений. Пурин является таутомерной системой, в кристал-
кристаллическом состоянии нахождение водородного атома более вероятно
в положении 7:
"Ч4! •
3 9
1. МЕТОДЫ ПОЛУЧЕНИЯ
Для построения системы пурина обычно используют производные
пиримидина и «пристраивают» цикл имидазола:
N'
+ С
709
Пурин и его производные получают из 5,6-диаминопиримидина
или его гидроксипронзводных (В. Траубе):
CICOOCKj R'^^N NNHj
R »
N^^NHJ HOOCR. nA^\
.Л-Р-^
Исходными могут быть также аминогидроксипиримидины, на-
например аминобарбитуровая кислота (Э. Фишер):
мочевая кислига 2,6,8-тригидроксипуриН
2. ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
Пурин и его производные — бесцветные кристаллические веще-
вещества. Производные с гидрокси-(оксо-) и аминогруппами малораст-
малорастворимы и высокоплавки благодаря сильному межмолекулярному
взаимодействию посредством водородных связей.
Пурин содержит сопряженную Юя-электронную систему со
значительной энергией делокализацин, стабилен к действию окис-
окислителей.
Пурин является амфотерным соединением, представляет собой
слабую NH-кислоту (pAV 8,9) и слабое основание (р/Свнн —=2,4,
протонирование по имидазольному азоту). Алкилирование дает
9-алкилпроизводные.
Гидроксипроизводные пурина являются таутомерными соеди-
соединениями, существуют в таутомерией оксоформе, представляют
собой NH-кислоты.
Галогенпроизводные пурина служат исходными для получе-
получения гидрокси- и аминопроизводных путем реакции нуклеофиль-
ного замещения. Так, 2,6,8-трихлорпурин, получаемый из моче-
мочевой кислоты и РОС13, служит исходным для синтеза важных нро-
710
изводных пурина:
ci
J
гипоксантин
3. ВАЖНЕЙШИЕ ПРЕДСТАВИТЕЛИ И ПРОИЗВОДНЫЕ
Пурин — бесцветное кристаллическое вещество с т. пл. 216 СС,
растворяется в воде. Получают из 5,6-диамипопиримидина. Ис-
Используют в органическом синтезе.
Аденин F-аминопурин) — бесцветное кристаллическое веще-
вещество, плавящееся при 360—365 °С, мало растворяется в воде.
nh2 Получают аденин из гидролизатов дрожжевых
^i n нуклеиновых кислот. Известны различные методы
^ 1^ V синтеза, в том числе из 4,5,6-триаминопиримиди-
Ц^-к^/ на или 2,6,8-трихлорпурина. В промышленности
н разработан также метод получения из формамида
и РОС13 при нагревании под давлением.
Аденин является основанием, присоединяет два протона — у
первого (р/(вн+ = 9,8) и седьмого (р/Свн *- = 4,15) атомов азота.
Аденин входит в состав нуклеотидов, нуклеозидов и нуклеиновых
кислот (аденозин, аденозинтрифосфорная кислота). Используют
в качестве исходного соединения для органического и микробио-
микробиологического синтеза и в медицине, например в качестве консер-
консерванта донорской крови.
Гуанин B-амино-6-гидроксипурин) — бесцвет-
бесцветное кристаллическое вещество с т. пл. 365 °С.
Содержится в экскрементах птиц, чешуе рыб и
рептилий, в гидролизатах нуклеиновых кислот.
Синтетически может быть получен из 2,6,8-трихлор-
2,6,8-трихлорпурина или другими способами.
711
Стабильной таутомерной формой гуанина является оксоформа.
Гуанин—слабая NH-кислота (р/Са--9,2) и слабое основание
(рХвн + ^3,3).
Гуанин входит в состав нуклеотидов, нуклеозидов и нуклеино-
нуклеиновых кислот.
Кофеин — бесцветное кристаллическое вещест-
вещество; т. пл. 235 СС, возгоняется, растворяется в воде.
Кофеин является алкалоидом, встречается в
кофейных бобах и чайных листьях, из которых
он и получается.
Кофеин — основание, образует соли с кислота-
кислотами. Широко используется в медицине в качестве
стимулятора центральной нервной системы.
Мочевая кислота — бесцветное малорастворимое кристалли-
кристаллическое вещество. Мочевая кислота — продукт обмена веществ
живых организмов, в значительных количествах встречается в
птичьих экскрементах. Синтетически получается из барбитуровой
кислоты (с. 710).
Мочевая кислота является таутомерным соеди-
пением, более стабильна триоксоформа. Соедине-
ние является двухосновной NH-кислотой и об-
/ разует два ряда солей (уратов).
jj Г} При взаимодействии с РОС13 образует 2,6,8-
трихлорпурин, исходное для синтеза различных
производных пурина.
В. НУКЛЕОЗИДЫ, НУКЛЕОТИДЫ
И НУКЛЕИНОВЫЕ КИСЛОТЫ
Нуклеозиды и нуклеотиды являются продуктами гидролиза нук-
нуклеиновых кислот, но встречаются также в живом организме в
несвязанном состоянии.
Нуклеиновые кислоты являются составной частью всех живых
клеток, входят в состав вирусов. Впервые выделены из клеточных
ядер в 1869 г. Позже было установлено, что нуклеиновые кислоты
представляют собой высокомолекулярные соединения, в макромо-
макромолекулы которых входят производные пиримидина и пурина, фос-
фосфорная кислота и моносахарид—D-рибоза или ?)-дезоксирибоза.
Существует два различных типа нуклеиновых кислот — рибо-
рибонуклеиновые кислоты (РНК) и дезоксирибонуклеиновые кислоты
(ДНК), разница между которыми заключается в строении моно-
сахаридного остатка. В результате гидролиза РНК в зависимости
от условий получают соединения производных пиримидина или
пурина с рибозой и фосфорной кислотой — нуклеотиды или сое-
соединения производных пиримидина или пурина с рибозой — нук-
леозиды. Конечными продуктами гидролиза являются урацил,
тимин, цитозин, аденин, гуанин, D-рибоза и фосфорная кислота.
В результате гидролиза ДНК соответственно получаются де-
зоксирибонуклеотиды, дезоксирибонуклеозиды и в конечном счете
712
тимин, цитозин, аденин, гуанин, D-дезоксирибоза и фосфорная кис-
кислота.
Нуклеиновые кислоты играют исключительную роль в жиз-
жизненных процессах живых организмов. Они являются, с одной
стороны, носителем генетической информации, основой развития
организма, а с другой стороны, матрицей, на которой синтезиру-
синтезируются специфические белки и нуклеиновые кислоты.
I. НУКЛЕОЗИДЫ
Нуклеозиды представляют собой соединения гидрокси- или ами-
нопроизводных пиримидина и пурина с рибозой или дезоксири-
бозой, в которых осуществляется связь С—N между углеродным
атомом альдегидной группы моносахарида и соответствующим ато-
атомом азота гетероцикла, при этом остаток гетероцикла находится в
р-положении цикла фуранозы (N-гликозиды):
носн.
но он но
рибонуклеозиды дезоь-сирибо нуклеозиды
(R —остаток гетероцикла).
Ниже приведено строение некоторых нуклеозидов:
о о nh2
но он
аденозин
Нуклеозиды представляют собой бесцветные кристаллические
водорастворимые вещества, обладающие химическими свойствами
соответствующего гетероциклического соединения и рибозы (де-
зоксирнбозы).
713
2. НУКЛЕОТИДЫ
Нуклеотиды являются эфирами нуклеозидов и фосфорной кис-
кислоты — нуклеозидфосфатами, обычно этерифицирована гидроксиль-
ная группа рибозы или дезоксирибозы в положении 5':
0
НО—Р—О—СН2
1 1
он 4.L/°-
но
рнбонуклеотиды
R
\
ОН
о
II
но—р—о-
1
он
5'
-CHS R
3^S /ъ-
НО
Дезоксирибонуклеоткды
Известны также дифосфаты и трифосфаты:
0 о о о о
II II II II II
но—р—о—р—о—сн2 r но—р—о—р—о—р—о—сн
1 I A | III
он он L/ \J он он он
но он но он
рибоиуклеозид-5'-дифосфаты ри6оиу|!леозид-5'-трифосфати
Нуклеотиды являются составными частями нуклеиновых кислот
и некоторых ферментов. Многие нуклеотиды обладают физиоло-
физиологической активностью.
Уридин-5'-монофосфорная кислота — бесцветное кристалличе-
кристаллическое вещество с т. пл. 202 °С, хорошо
растворяется в воде; [a]D=9,5°.
Является кислотой (р/(а~6,4) вслед-
т,^ - „ ™, - ^^« ствие ионизации связи О—Н фосфор-
I I "о | пои кис-™, образует соли. Харак-
он j/' \J терны свойства урацила и рибозы.
Соединение содержится в живых ор-
организмах как в свободном состоянии,
так и в составе нуклеиновых кислот.
Аденозин-5'-трифосфорная кислота (АТФ)—бесцветное во-
водорастворимое вещество, образует стабильные соли. Связи Р—О—Р
легко гидролизуются, при этом выделяется значительное коли-
количество энергии C3—46 кДж/моль):
о и у/ м |
НО—Р—О— Р—О—Р—О—СН2 4N JT
III'
он он он
но он
714
АТФ играет важную роль в процессах обмена веществ в живых
организмах. Она является своеобразным аккумулятором энергии,
поставщиком химической энергии в различных процессах био-
биосинтеза и в таких физиологических процессах, как сокращение
мышц.
3. ПОЛИНУКЛЕОТИДЫ
и нуклеиновые кислоты
Нуклеотиды могут связываться друг с другом при использовании
фосфатной связи С—О—Р. Так могут образоваться динуклеотиды,
тринуклеотнды, олигопуклеотиды. Нуклеиновые кислоты явля-
являются полинуклеотидами, в которых нуклеотидные молекулы свя-
связаны между собой 3'-5'-фосфатной связью С—О—Р—О—С. В при-
Ееденной формуле тринуклеотида R, R1 и R2 — остатки урацила,
тимина, цитозина, аденина или гуанина.
Нуклеиновые кислоты — фос-
фосфорсодержащие биополимеры, ко-
которые построены из остатков пук-
леозидов (N-гликозидов рибозы или
дезоксирибозы и производных пи-
пиримидина и пурина), связанных
между собой фосфодиэфирными
связями (рис. 101).
Нуклеиновые кислоты, как и
белки, очень разнообразны, соот-
соответственно различны и их специ-
специфические функции в организме.
Молекулярные массы нуклеиновых
кислот также различны — от не-
нескольких тысяч до миллиардов. Это
соответствует числу остатков нук-
леозидов от 10 до 106.
Нуклеиновые кислоты, как и
белки, имеют первичную и втори-
вторичную структуру.
Первичная структура нуклеиновых кислот оп-
определяется последовательностью расположения нуклеозидов, свя-
связанных фосфодиэфирной связью, или просто последовательностью
производных пиримидина и пурина. Обычно эго относится к ура-
цилу (U), тимину (Т), цитозипу (С), аденину (А) и гуанину (G).
Другие соединения встрачаются очень редко. Первичная структура
схематично изображается совокупностью букв, например:
pL'pUpGpApGpCpUpG ...
(буквой «р» обозначают остаток фосфорной кислоты).
715
Вторичная структу-
р а нуклеиновых кислот пред-
представляет собой пространственное
расположение макромолекул. Это
определяется внутримолекулярным
II межмолекулярным взаимодейст-
взаимодействием, главным образом посредством
водородных связей.
Известны различные вторичные
структуры.
0=<- ^.N ^ Известны одноцепочечные (од-
' СНч нонитевые) нуклеиновые кислоты,
цепь которых может принимать
различные пространственные фор-
мы, в том числе спиралеобразные,
в которых плоские молекулы про-
изводпых пиримидина и пурина,
как правило, расположены одна
над другой в виде стопки, рассто-
яние между плоскостями около
г, ,п, т 0,34 им.
Рис. 101. Тринуклеотид из гуани- _.
на, цитозина и урацила, PGpCPU Основу макромолекулы нукле-
нуклеиновых кислот составляет поли-
полиэфир фосфорной кислоты и рибозы (дезоксирибозы). К этой мак-
макромолекуле присоединены производные пиримидина и пурина.
Но именно при помощи этих пиримидиновых и пуриновых остатков
осуществляются все многосторонние внутримолекулярные и меж-
межмолекулярные взаимодействия, которые обусловливают все функ-
функции нуклеиновых кислот в живом организме.
Для многих ДНК характерно существование в виде комплек-
комплексов, образующихся из двух полинуклеотидных цепей, которые
стабилизируются водородными связями. Такие нуклеиновые кис-
кислоты называются двухцепочечпыми (двухнитевыми). Как пока-
показали на основе рептгепоструктурного анализа и химических дан-
данных Д. Уотсон и Ф. Крик A953), двухцепочечные нуклеиновые
кислоты в пространстве образуют структуру двойной спирали
(рис. 102).
Две макромолекулы нуклеиновых кислот закручены в спи-
спирали, при этом спирали находятся одна в другой и связаны между
собой посредством водородных связей, которые образуются между
пиримидиновыми и пуриповыми остатками. Спираль образует
полиэфир фосфорной кислоты, а плоские молекулы производных
пиримидина и пурина находятся внутри спирали, образуя ее серд-
сердцевину.
Межмолекулярные водородные связи образуются только между
определенными парами производных пиримидина и пурина, ко-
которые называются комплементарными парами. Такими компле-
комплементарными парами являются урацил—аденин, тимин—аденин и
цитозин—гуанин (рис. 103).
716
Рис. 102. Схема
двойной спирали
ДНК
N
11 R
Рис. 103. Комплементаршле нарыти-
мии-адепии с двумя водородными
связями и цитозин-гуанин с тремя
Еюдородными связями
В схеме двойной спирали ДНК (рис. 102) лептами изображены
макромолекулы полиэфира фосфорной кислоты и дезоксирибозы,
а соединяющими прямыми — плоскости молекул производных
пиримидина и пурина, образующие комплементарные пары.
В особых условиях двойная спираль может раскручиваться
с образованием однонитевых нуклеиновых кислот.
Основные виды нуклеиновых кислот. ДНК входят в состав всех
живых клеток и многих вирусов и фагов. Большая часть вирусов
и фагов содержит ДНК с М«30-106. . .200-106. Одна вирусная
частица содержит одну молекулу ДНК или один двухцепной ком-
комплекс.
В состав клеточного ядра бактерий входят ДНК с М«2,8-109.
В ядрах клеток высших животных и растений находятся ДНК
большой длины (до 2 мм или больше), молекулярная масса кото-
которых превышает 5- К)9.
Известны также внеядерные ДНК, которые входят в состав
различных субклеточных частиц — митохондрий и хлоропластов.
Задачи ДНК — обеспечение возможности самовоспроизведе-
самовоспроизведения клетки, хранение и передача генетической информации, обес-
обеспечение синтеза РНК, участвующей далее в синтезе белков в клетке.
РНК, подобно ДНК, являются необходимыми компонентами
живых клеток. Разные типы РНК имеют различные биологиче-
биологические функции.
717
Известны вирусы, содержащие РНК, которая обеспечивает
генетическую информацию: Л1«Ь10и. . .3-10".
В клетке встречаются в основном три типа РНК: рибосомаль-
ные, транспортные и информационные. Рибосомальныо РНК имеют
Мт\&. . .10". Транспортные РНК способны связываться с амино-
аминокислотами, при этом каждый вид только с определенной амино-
аминокислотой. Их молекулярная масса 3-Ю4, что соответствует около
100 остатков нуклеотидов. Информационные РНК являются осо-
особого вида РНК, нуклеотидная последовательность которых опре-
определяет аминокислотную последовательность синтезируемого белка.
Генетическая информация и механизм ее передачи. Генетическая
информация — это набор данных, определяющих структурные и
-Л::::::::Т—
-Т::::::::А—
-Gi:::;:::c—
-C;;;:;::;G-
-Т::::::::А —
-g;:::::;;c—
—А::::::::Т —
-G::::;i::c-
Рис. 104. Схема репликации двуцепочечиого комплекса ДКН
(двойной спирали). Упрощенно обе цепи нарисованы рас-
растянутыми
функциональные свойства клеток. Генетическая информация (ге-
(генетический код) зашифрована в нуклеотидной последовательности
ДНК (в отдельных случаях РНК). При делении клетки эта ин-
информация должна передаваться от одного поколения клеток сле-
следующему. Для этого должно осуществляться точное воспроизве-
воспроизведение (репликация, дупликация, копирование) ДНК- Это является
очень сложным процессом. Матрицей для репликации служат од-
нонитевые ДНК, которые образуются при расхождении цепей
двойной спирали. На каждой из них синтезируется новая компле-
комплементарная цепь спирали. Схематично это показано на рис. 104.
Местонахождение каждого нуклеотида в цепи определяется
образованием комплементарных пар Т—А и С—G. Процесс осу-
718
ществляется с участием фермента—ДНК-полимеразы, которая
катализирует подстановку в синтезирующиеся цепи дезоксирибо-
нуклеотидных звеньев из присутствующих в системе дезоксирибо-
нуклеозид-б'-трифосфатов. Фактически процесс репликации имеет
еще более сложный характер. На одной из цепей ДНК под дей-
действием РНК-полимеразы происходит образование коротких фраг-
фрагментов РНК E0—100 нуклеотидов), которые служат основанием
построения фрагментов ДНК.
Так происходит транскрипция генетической информации от
ДНК на РНК и от РНК на ДНК. На основе ДНК-матрицы синте-
синтезируются РНК, служащие для передачи генетической информации
из ядра в цитоплазму.
В ДНК в форме специфической последовательности Т, А, С и G
закодирована аминокислотная последовательность всех клеточных
белков. Кодирование осуществляется триплетами из тимина, аде-
нина, цитозина и гуанина. Три основания (кодон) кодируют одну
аминокислоту. Тем самым ДНК действует как матрица для синтеза
белков в клетке. Определенные участки ДНК (гены) ответственны
за то или иное действие в клетке. Каждая клетка содержит полный
набор информации для строительства своих белков, ферментов.
Синтез белка включает перенос информации (транскрипцию)
от ДНК к молекуле РНК, которая синтезируется на ДНК-матрице
и комплементарна данной части цепи ДНК — гену. Эта информа-
информационная, или матричная, РНК точно отражает последовательность
нуклеотидов в определенной части ДНК- Так, информационная
РНК содержит остатки аденина там, где ДНК содержит тимин,
остатки цитозипа там, где в ДНК гуанин.
Триплеты из аденина, цитозина, урацила и гуанина на инфор-
информационной РНК (кодопы) действуют как специфические места
посадки для комплементарных триплетов (антикодонов), располо-
расположенных на молекулах транспортных РНК, у которых привязаны
соответствующие аминокислоты. Специфичность стыковки кодона
и антикодона обусловливается специфичностью образования водо-
водородных связей между адепином и урацилом и цитозином и гуани-
гуанином. Например, для алапипа кодоном является триплет GCA, сле-
следовательно, антикодопом—CGU. Фенилаланин кодируется три-
триплетами UUU или UUC, глицин—GGC или GGA, GGG, GGU.
Таким образом осуществляется последовательность соединения
аминокислот в специфические белки, которая первично записана
на определенных участках ДНК—генах.
ИМЕННОЙ УКАЗАТЕЛЬ
Адаме Р. ПО
Альдер К. 141
Арбузов А. Е. 277, 331
Аридт Ф. 4Ь0, 525
Арреннус С. 65
Fiaiiep A. II, 162, 596
Бальц ['. 427
Бальян X. В. 21
Бснльштсйи Ф. Ф. 21
Бекман Э. 471, 472
Берцелнус Й. 9, 11, 22
Берч А. 168
Бергиус Ф. 90
Бор М. 49
Бертло М. 10, 146
Бншлер А. 697
Блан Г. 284
Бонд Р. 20
Еор Н. 32
Бородин А. П. 447
Браун X. 79
Брёнстед Й. 69
Бриглеб Г. 45
Буво Л. 284
Бугер II. 49
Бутлеров А. М. 10, 11, 24, 25, ВЗ, 119, 256
Бухерер Г. 415
Вагнер Е. Е. 116, 299
Вальден П. 612
Ванаг Г. Я. 490
Ван-дер-Ваальс Й. 44
Ван Слайк Д. 621
Ваит-Гофф Я. 26, 61, 65
Вё'лер Ф. 10, II, 22, 146, 325, 466, 644
Верлей А. 453
Виллигер В. 596
Вильяме Г. 142
Вильямсон А. 288
Вильсмейер А. 502
Внльштеттер Р. 174, 666
Виттиг Г. 278
Вольф Л. 473, 252
Вудворд Р. 12, 169, 666, 667, 673, 700
Вюрц А. 23, 91, 447
720
Габриель 3. 588
Гамчет Л. 77
Ганч А. 423, 683, 687
Гаттерман Л. 464
Гауптмани 3. 21
ГеЛровскиЛ Я. 46
Геес X. 369
Гнллер С. А. 708
Гомберг М. 200
Грандбсрг И. И. 21
Гребе К. II, 202
Грефе Ю. 21
Гофман А. 23, 391, 392, 399, 406, 41 f, 586,
601
Грнньяр В. 253
Грис П. 421
Гудьнр Ч. I 12
Дебнер О. 697
Деринг В. 700
Дикман В. 635
Дильс О. 141
Долгоплоск Б. А. 143
Домагк Г. 416
Дюма Ж. 10, 22, 206
Жерар Ш. 23
Зайцев А. М. 256
Запд-vieiiep Т. 426
Зелинский II. Д. 171, 180, 601, 616
Знбер П. 12
Зинин Н. Н. 13, 406, 414, 419, 478
Зонис С. А. 21
Иванов Д. 555
Измаильским В. А. 31
Ингольд К. 30, 225
Иоцнч Ж. 155, 255
Казанский Б. А. 180
Каи Р. 225
Кашшццаро С. 467
Каррер П. 143, 144
Кскуле А. 10, II, 23, 25, 176, 177, 204
Kepi! Ф. 21
Килна ни Г. 516
Ки/kiicp П. М. 172, 17J
Клемменссн Э. J53
Кляйзсн Л. Hi/, 180
Кнспенагель Э. 551, 559
Колумб X. Ill
Кольбе Г. 10, 91. 609
Копдамнн III. 1 12
Коновалоп М. II. '.'8, 369
Корана Г. 12
Короткоп Д. Л. 113
Коптель В. 25
Коттои Л. 61
Кох Ю. -161
Кочет она Э. К. 21
Крлфтс Дж. 187, Н18, 199, 206
Крик Ф. 716
Кроуфут-Ходжкнн Д. 667
Кучеров М. Г. 151
Kynet) Л. II, 23
Курцнус Т. 591, 633
Ламберт П. 49
Ланг С. 31 1
Латимер В. 286
Лебедев С. И. 1J2, 1 12
Ле Бель Ж. 23
Ленгмюр П. 25
Либсрман К. II
Либнх Ю. 10, 22, 176, 166, 178
Лоран О. 23
Лоренц Л. 39
Логсен В. 592
Лоури Т. 69
Льюис: Г. 2.", 70
Макнптош К. 112
Маланрад Л. 301
Мании К. 523
Морковником В. В. 110. 172
Месрнсмн X. 299, 126, 153
Meiiep I). 370
Менделеев Д. И. 2 И]
Мернфнлд Р. 633
Мптчерлих Э. 176, 378
Миллер В. 697
Молдапскпй Р. Л. 180
Aloppiirmi P. 20
Наппральсм!!! В. 697
Натта Дж. 121, 122, I 10. I 13
Несмеянов А. П. 20, 2O, 257
Несмеянов Н. Л. 20
Неф Дж. 375
Нобель Л. 318
Мьюленд Дж. 151
Ньюмен М. 91, 228
Окамото К. 79
Ола Г. 233
Пслттер Л. 631
Перекалин В. В. 21
Псркнн У. 535
Петров А. А. 21
Пирсон Р. 73
Пнхлёр X. 90
Пикте А. II
Пл:,и Д. ф. 180
Полниг Л. 26, 31, 41, 029
Ноллмг II. И
Померанц К. 697
Попндорф В. 153
Потапоп В. ,\\. 21
Прелог В. 225
Нрнле',каев II. Д. IKi, 596
Рабе К. 7С0
PeiiMep К. 501
Ремане X. 21
Репей М. ПJ
Реппе В. 132, 152, 151, 137, 173, 5!
Реформатский С. II. 608
Робинсон Р. 701
Родсбуш В. 28A
Родионов I). М. 616
Ромен О. I 15
Руемапн С. 623
Ружнчка Л. 1H
Руиге Ф. -114
Руфф О. 316
Сабатье П. 90. ПО
Сапдберг Р. 21
Сандеран Ж. 90. ПО
Сергеев П. Г. ИЗ
Скрауп 3. 6S16
Соммле Л\. 165
Степаненко Б. II. 21
Сторк Г. -106
Стю: рт Л. 15
Тяфт Р. 79
Теренты'м Л. II. 663
Тнман Ф. о01
Тшнепко В. Н. 151, -tf,7
Тол.тенс Б. 15 1, 512
Триубе В. 710
Тропш X. 90, 115
Трощенко А. Г. 21
Тгудп Г. 701
Удрис Р. К). 313
Ульман Ф. 311
Унфердпрбеп О. (If
Уотсон Дж. 716
Фаворский Д. Е. 133, 156, -193
Фирадеп М. I 12, 176, 192
Фелппг Г. fill
Фишер Г. 666
Оишср Ф. 90, ИЗ
24 .V., 517
Фишер 3. [2, 227, 516, СЗО, СЗЗ, CG8, 710
Фольгард Я. OOt
Фрамкланд Э. 23, 2ЛГ>, 2oG
Фрпдель Ш. 137, 198, I'JO, 20G
Фрмс К. 319
Фрнцше Ю. 411
Фрич 1'. G97
Хаак Л. Ь02
Хсл.чь К. GOf
Хсуорс У. 509
Хок X. 314
Хорпер Л. 279
Хофман Р. 169
Хупсднккер X. 517
Хюккель Э. 121, 2t3, C60
ЦеПэс LI. 2G7
IlepcHHTiinofi Ф. В. 254
Цнглер К. 121, МО, 143, 258, 2G3, 284
Чичибабн» Л. Е. С91, G99, 70;
Чугасо Л. А. 254, 478, G51
Ше,:ле К. 9, 30G, 327, G14
Шнкан Г. 4 27
Шифф X. 470
Шмпдт К. 591
IH.MHTT P. G09
Шорл«!ммер К. 10
Л1рсд|мп-ер Э. 32
Штаудпнгор Г. 142
Штолл М С06
Штроккер Л. 616
ЭС;стерт Б. 480, 525
Эльтскоп А. П. 310
Эрлеимсйер 3. 310
Эшенмолер Л. 667
Юрьен Ю. К. 059
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Автоокислеппе Алнцикличссчне соединения 82
— алке.нов 123 Алкадпепы ПО
— арепкарбальдегпдов 408 — изомерия 1 ДО
— дналкилоных эфпроп 330 — номенклатура 130
Адлчаптап 1CI, 17.i, 17G Ал падпены-!,2 см. Алломы
Ддамаптпл-катноп 1 7G Алкадиепы-) ,3 KV2
A'lu.Kcu катализатор ПО — глло-епировлппе 137, 133
Ддепнн 711, 712 — гпдрлроваг.не 137
Адепозлн 713 — образование л-комплексов 1.18
Адепозии-З'-трифосфорпля кислота 714 — полимеризация 1 :'.<), 110
Дднниповая кислота .40,"), 339, 457, 55G, JS8, — получение 132, 133
.'Г'0 -- природа связей 131
Лдиподппитрмл 401, 567, 000 ¦— присоединение гллогешзодоролоч 137
Адреналин 101 — пространственное строение l.-.;i
Азлцнклобутадпен 6.">Г> -- реакции присоединении 137
Азацнклопентаднеп Cj3 _ спектры поглощения 130
Атлцнклопропен 651 — физические свопстпа 13'
Дзепип-катион -172 — фронтальные орбпт^лн 13>
Азепнп 058 .— -^кзальтацпл рефраьмиш 131
А-ютндин С Г. 5 Алкалоиды 12, 673, GS.'i, 712, 7С0, rfil
Азетппы 6.">5 Алкапд|г<1.юнневые соли 307, 428, 129
Алет «;">.") Алкаполяты см. Ллиокснды
Лзидиая группа 591 Ллкагалы 28A, 2S1
А-шды 418, 681, СРВ -- ас.-оцншыи 2ЙГ>, 2Si>
-- iwirConoijbix кислот 5Cj, Г*!H, j9t .— взаимодействие с азотпеточ кнслото|'г 34G
Алип1,1 9 _ — — .-потно;! кислотой 3:7
Л.Ч!ф1!Д|1" 390, 6.11 — — , ммнаком и амиш.м i ЗЯ1
A.'iipun G.i 1 — — борпоп кнелото1! 3:.)
A.so.L"j\.!![bl -i31 — — галогеаиодо'юд.-.мп 291
Азолрены ?.S2, 387, 419, 432 — -- галогепндамп цгорг.шпчес'лпх ьн-.-лот
— оспопность 433 233
— получение 432 -- — днлзонпевымн солями 427
— :|роетр.'1пстпеппое строение 432 — — и'.оцп. п.ггамп Gi7
— споктрм поглонс.ншя 433 -- -¦ кгфбопплыпимн соединепчячн 450
— хамнче.-кие CHoiicTiia 433 —- — :;арбоно';ымн кислотами ."По
Азобензол, ий- и транс- 431, 433 -- — кеоргакмческ.ч.чп кислотами 290
Л:!ои.;обутнроннтр:1Л 431 — — нитрилами 598
Лзо:руппа -',30 -- — оксидом углерода 293
Азокраснте.-л 'Г.'.'!, -fit _ _ серпол Kuc.ioTo.i 3.J
Азокснарепы 382, 387, 434 — — фосгеном G12
Азометан 431 -- — члектрофплъпьр.ш рспгглтлчн 200
Азо.метнпы -;01, 4!9, 470 — — ,по;;опдамн 3.">о
Ачосоедипеппя 418, 4'.)Г>, 430 ~— iv, opn'-ini.i- 2SI
Азосочетанце 4?."), 43:', 180, ('73 — гго'.ютрпн молекул 247
Азотистая кислота, 'яЬг.ры .'110, 121 ¦— генерирование кар'ок.лпопов 29'2
Азопшя кислота, эфнры 347 — дс-'-н.-.рпронеппо 2Й9
Алулсн 217 — дч|:ать:;ыо mumchti.i 28'J
Акридин Go? .— inoMCi'iiv: и помсп^латурт 221, 282
Акрнлаты 57') _ _ --- 1;лр5оннлп))опаппе 203
Акриловая кнелотл I "¦-?, Гкч", ,"i.>0, fi.il, Г>Г>2 — кислотности 25S
Акрнлопнтрнл 1.")', Х-.: я;I), 000 — о.^пелскно 2rt9, 4.J9
Акролеин 307, 437, :"•'.<, 4G1, 09о — u.-i;oi<nocTl. 290
Ai^taia.ibiibie з<1\:сст;:тел;| 170 — опцеклеппс- ги;ил 2G3
Активированные офщнл ГO5, ^i33 -— :jo.i>4C!iik' 2S3, 284, 117, 4.12
Активная Метилеповая группа 'ЗЗо, 555, — пр к'транствепное стро лл:-е .'iJl
Т>58, 5.">9, 578, 000 .— с:гса7;)!з[ па1ло(це;;пя :;87
Актлнпы;"! подородпыи атом, оиреде;:еп:1е — '.пергил по!1нз,.цнп 28G
2.>4 Алк^пюнпя пин 1С0
а-Длапип 003, G20, 621 Л.-;;ч;ли:\.-::,..'>оно;;ие \iu:.v.r, :-i /Гь", 1й0
fi-Лланип Gl'3 А.".;аксу:'!|('!1о:;;':!>:лор]!ДЫ 9G, 3j5
Алтари и 11, 534 д : ^-птполы ;:(¦!
А.т|ф,т;1чо.'м:е соедппешш 81 А.-кантполяты 3G4
24* 723
Ллканы 86
— ¦ ароматизации 99
— бронированно О Г)
— - взаимодействие с злектрофнльнымп реа-
реагентами !Л>
— дегидрирование 105
— дегпдроцнклнзанпя Оу
-- изомеризация ОУ, 100
— изомерия 87
— конфигурации 93
— - конформеры 93
— крекинг 98, 105
нитрование 07, 309
- номенклатура 87
образование клатратов 615
— окисление У8
— получение 90, 91
применение 101
- пространственное строение 92, 03
— спектры поглощении 95
сульфоокисленпе 07
-— сульфохлорнроваппе 96
— термические превращения 98, 99
— - физические свойства 92
— фторирование 93
хпральпые 93
хлорирование 93
энергии ионизации 91
Л "sein; льпые группы I 0Г>
Л "кичшльцые свободные радикалы 153
Л к.енплы 280, 297, 2!J8. -159
Л 'Кептрикарбони л никель 26G
Л.-жгиы 102
антоокпсленпе 123
аллпл ьное галогенироваппе 1 23, 1 '2 I
лрилироваппе 4 20
взаимодействие с алюминием 2П8, 239
— — алюминипоргпническпмп соединения-
соединениями 200
— галогенами 112, 113
- галогеннодородамп ПО
— нонами переходных металлов 11 1
-- карбенамп 122
-- — свободными радикалами 118
— серной кислотой 112
— С-электрофпламн 112
возбужденное состоя шю 108
геометрии молекул J 08
гндрпропанпр 01, 10'J, ttO, 273
гпдроборнронанпе I 10
— гндрогалогепнровалне 209
— • [¦пдросплплпронапио 272
гидроформилировапне 11 1
дегидрирование 146
— днмеризация 260
— дппольные моменты 107
-- изомеризация 1 2 3
— - изомерия ЮЗ
— молярная рефракция 107
номенклатура 101
— образование л-комплексов 111, til, 205
— окисление 116, 117, '130
— - олигомеризация 200
— полимеризация 118, 119, 120, 121, 122
— получение 10,1, 233, 293. 331, 309, 417,
051
— применение 128, 129
— природа снязей 108
— ¦ присоединение воды 283
-- присоединение сопряженное 111, 1II
— пространетценное строение 103
— спектры поглощения 108, 109
— термохимическая энергия 107
— физические спопстна 106
— цнклоприсоединение 122, 123
-- энергии ионизации 108
— эффект снер\сопряжеппн 107
Ллкпдные смолы 572, 530
Ллкмл?\-пды .\к*т<-1Л.'1'>б .399
Ллкпд,!\:п11и:'р\ ппа ; >Ч
Лткпламмпы 301
-- млuii/'.iipoH.'.niie viУО
— ассоциация 393
-- ацнлпрованне 397
— взаимодействие с азотистой кислотой 307
— — галогенами 398
— — нзоц1г;шатлм1[ Ь13, GJ7
— - - пзотпоциапатами 050
-- — карбепамп 397
— -- карбонильными соединениями 1-19,
470
— - — катионами металлом 39G
Х-хлорамппами 41 7
— — олектрофильпымп реагентами 390
— - дппольные моменты 393
— ппверсп-л 30-1
-¦ ионизация снязеп N —П
— нитрование 308
— образонапие пзоцнанпдон 307, 601
¦- окпеленпе. 308
— основность 3U5
— - полярность снязеп 303
-- получение 373, 391, 302
пространственное строение 39 1
спектры поглощения 395
— сульфирование 399
-- физические снопства 393
-- энергия ионизации 39-1
Ллкнл-апионы 89
Ал кил арены, каталитическое окисление 464
Длкнлармлоные ^фпры 312, 3 U, 127
Ллкил.-фнлкетопы -101, 405, -Ш?*
Ллкнлгидра шны -П 7, ПН
Длкплгидросульфаты 352
Длкилгпдроксилпммны 373, 39S
Ллкплдптиокпрбаммты 051
Ллкплпзпвалераты 378
S - Лл ы 1,ч п.ютп у ро пневые соли 6 19
N-Ллкплизохпно.чнпиевые соли 6U0
Лльилпровапие
ллканолов 292, 328
— ал 1ч а пол ятов 289
— амидов кпрбононых кислот 593
-- амппоу! ЗП1, ЗУо, 407, 4П
— ?1\п'П.;ка 391
— ареной 198, 292
— арнлампнои ]11
— ацетпламиномгпопового эфира GT6
— ан,етилеппдпв 117
— ацотоуксуоюго ьфирп (>J7
— бензола 187
— пиутримолекулирное 392
-- дпа;[:<нлоных уфнрон ,4;JO
— fi-д и карбонильных соединении 185
-- галoreи ид-попов 223
— гексоз 513
— - гидразина 417
— гидроперокендов ^49
— - енамннов 406
— пзохиполи на 609
— - и идола (u I
— карбокеилат-попа 516, 571
— карбпипльных соедшгепп!! -JOG
— - м<]лоиово['о 'эфира 579
— нитритов 370
— ннтроалканон 375
— поптоз 507
— пероксида водорода 319
— - ппргмолопои (O8
— пиридина 690
— пиримидина 700
— пирокатехина 324
— пиррола 661
— РОД<|||НДОП 0.U)
— сероводорода 361
— сульфидов 36G
— сульфитов 35Я
— тнолятов 304, 306
— тпомочевины 040
— уротропина 615
— фс-нолсв 320
— (реполятон -ЧI 7
724
г— фосфора 276
•— фтилнмида .138
— хиполпна 699
— цпанатов 646
— цианидов 397, 601
Алкилирующие редюиты 232
— алканолы 188, 292, 301
— алксны 188
— алки.чгидросульфаты 2о2
— альдегиды 448
— галогенуглеводороды 232
— диалкплсульфаты 253
— диазоалкапы 429
— дихлоркарбен 601
— а, Р-ненасыщенные- карбонильные соеди-
соединения 460
— нитриты 317
— триалкилокеонневые соли ЗЛО
— этилепимнн 402
— '^тиленокенд 335
— чфиры карбопоных кислот г, 76
— - - ортокарбомовых киг.лот 582
-- — сульфоповых кислот 338
Алкплкарбонаты 611, 642
Алкил-катионы 89, 100
Алкнлмалоиовые кислоты 579
Алкилпнтраты 671
Алкилпитрнты 370, 421
Алкилокеоииевые катионы 290
N-Алкилпирндппиевые соли 690, 602
Алкилсиланы 271
Алкилсульфаты 352
Алкнл-/1-толуолсульфонаты 3)9
Алкнлфенолы 322
Алкнлфосфонаты ЗГ»1
Алкплфталимиды .188
N-ЛлкилхпнолнппеЕЫе соли 699
Алкильнан группа 88
Алкилыше свободные радикалы 88, 98, 100,
t 01
Алкнполы 2S0, 297, 298, 119
Алкипы 14I
— бромнрованне 131
— гидрогалогепировапие 130
— гидрирование 106
дшюльные моменты 148
— взаимодействие с алкаполлми 152
- иодоК 1Г>1, 309, 439
— - гплогеповодородами 1.10
- диазоалканамп 67о
— - карбепами 162
— нуклеофильпыми репгонта\ч: 151
— - свободными радикалами 133
— солями тяжелых металлов 11,1
— уксусном кислотой 1,12
-- электрофильнычи реагентами
110
-- гндроборирование 269
— карбопнлированпе 152
— молярная рефракция 147
— номенклатура М5
образованно ацетпленидов 1.15
— образование лжомплексов 265
— окисление L13, 476
олигомери 5,'|ЦНЯ til, 266
— полимеризации Ml, 155
— - получение I 15, 212
— природа связей 148
присоединение к азидам (SS5
— — дназоалкапам 67,1
— - карбонильной группе 156
— пространственное строение 1 18
— термохимические энергии 117
— физические свойства t M
циклотетраморизацпя 154
— пиклотримеризацпя \Ъ\
— энергия ионизации t 1Q
Ал'логоляты ем. Ллкокспды
Алк(;голиз
— а.м;:дов 58fi
«— слои:ных эфиров 576
Алкокснды 250, 255, 2S8, 3 45, 453, 467, 520,
521
Алкокеигруппа 328
Алкоксикарбоиилирование 633, 613
Алкоксикарбонилхлориды 611
Алкоксикарбокатион 342
Алкоксильный радикал 289, 319
Алк'оксисиланы 271, 275
Аллепы 130
— перегруппировки 132
— получение 131
— пространственное строение и стересизо-
мернн 131
— реакции Kit
Аллилизотиоцнапат 651
Аллил-катнон 127
Аллилоные эфкры 298
Лллнловый спирт 297, 298, 306, 338, 461
Аллплтрнкарбопплкобзльт 266
Лллнлхлорид 124, 236, 2!'8, 306, 338
Аллильная перегруппировка 291
Аллпльпое бромпровапие 123
Аллпльпое положение 109
Аллильнып радикал 121, 126, 127, :'&5
— расчет методом МОХ 126
Аллоза 508
Алкшипийорганические соединения 258,
263
Альбумины 626
Альдегидная группа 13,1, 438
Р-Альдегидокислоты, сложные эфиры 635
Альдегиды ароматические 464
Альдегиды 433
— альдольпая конденсация 416
— «бисульфитные» соединения 432
— брочпрованне 443
— восстановление 452, 453
— галоге.нпровапие 492
— гидрирование 4 45, 446
— - дипольньге моменты 440
— диспропорционироиапие 151, 407
— енолизация 4 44
-- реакции с алкенами 448
— — flmo/7-амипами 401
— — 2-амппотиолами 682
— — арнламннами 418
-- галогепидами фосфора п серы 452
— — дпазометаном 448
— — Р-дикарбоннльпыми соединениями
485, 489, 638
-- — енамннами 4 (8
— — малоновой кислотой ,159, 617
- - а-метплпнриднном 692
— — с С-нуклеофнлами 446
- — П-пуклеофилами 415
— — N-нуклеофпламп 448
— - О-пуклеофилами 450
— — S-нунлеофиламп 411
— — сульфонийилндамн 4 17
— — фенолами 448
— — фосфоранамн 447, 451
— - - цнапоподородом 447
— — — и аммиаком 616
— — этаноламином 682
— окисление 451, .19.1
— основность 445
— особенности реакционной способности
15.1
— получение 308, 439, 4-10, 611, 622
— тримерпзация 451, 456
Альдегиды ненасыщенные 158
Альдогексозы 508, .109, 512
Альдоксимы 471
Альдоль 303, 446, 417, 458
Альдольнпя конденсация 146, 448, 450, 461,
Альдолыюе присоединение см. Альдольная
конденсация
Альдозы 501
Альдоновыо кислоты 511
Альдопентозы 505, 660
Амбидентпип ион 311, -182, 485
725
Амбнфуикцпоийльнып нон см. Амбндептныи — роск;1т;г! ямппогруштм 6°1
linn — цгк'Тп;",! реакция " 490, 023
Лмгдппы Г.00, Г»93 — ъфщш C21)
— н.чанмодс.'к-тпш1 г oi-гидроксикярбонплк- ft-Лмппокпслпты G17
Ц1ГЛЧ1 С\>'\ЦПМРШГЯЛШ в 70 — бСТ ИННЫ 1J0
— - |}-д::кйрбо11И/1Ы1ЫМП соединениями 7Q3 — получение 017
— гпдролп.} 593 — прототропиые рапнопоспл 020
~ номенклатура Г)О6 — химические спонЧтиа I»*J-J
— оср-тпоегь 593 V-. <5-. f- и @-,^тн1нжнслоты 617
— - пол}, -м-пае i'^Ki р-Амн но л.роти пиная к п.-лота, мфцры 038
Лмпд .-"птпя, дни ;пп!)олмл 2Г>0 Ампполм.1 сложных ьфироп 070
Ампяная групня Г»Я9 V-Амппомаслнная кислота 583, G17
Амнд::ая снизь 028 Амшго^гетилиропаппе 023
Амидопирин 679 Амнио\:отплымя груыы 4 jG
Амид еульфяпиловоп кислоты 416 Аминопелици^ляиои^я кле.юта GS4
Лмпды к.чрбоновых кислот -:7г. ПС5, Г»82 A\ni;iorr;i[:.'i:iF:n 707
— юаммодгПспше с нуклеофпльиымн реп- Амнион и рндппы G9I, G0-, G04
гентпми 58л 4-Дмг.иочирпмпдкп 707
— — РОС]л «г»87 Амшюпласты 6-1Г»
— — P2S й й 94 Л мп но пол и сахари ды Г>2?
— ггкчсгсипрои.'пшс П86 Лмнногп)оппо!н,''Aя кислота см. Лл.'.нпн
-- дегидратация 587 6-Лминппурпн 711
— кислотность v,8?> 2-AsumoTiip нкч 084
— - номенклатура 5G5 ^-Дмннотиг.лы в'6'2
— осмонипсть 381 _ о-Лмннотпофекал 613
— получении 54G, Г.83, 0-19 Лминпуксусиая кислота см. Глгт.нп
-- полярность сиязеп лЬЗ, Г>81_ Амииоуцсуспьи! альдегид см. Лминоацеталь-
— расщепление ип Гофману .136 Дс-гпд
Амилацетат 297, 578 Лмпнофепплы 382, 700
Амнл нитрит .14 7 Амин охи нолин G99
А мп л оные спирты 282, 28о, 397 Амппо^тнлпроьшше 402
Амилоз;! 519 ^-Ампнозтилс-ульфоилт 392
Амилопектнн 51!) Амины 23, 388, ГJ3
Амппалы 449 — бпог-спньге -Ю-t
Ам и пиров;) пне восстапоинтел ыюе 452 — нзанмодепстпие. с диоксидом углородоп
л-Аминоа:10Eепзол 433 G f 3
4'-А%1нно;13обензол-4ч-ульфопопая кислота — — рода ни дамп G19
¦131 — — сероуглеродом Сч
Амнпояптрахиионы .4 — — фосгеном 617
Лчииоаренкарбонопьк* кислоты G1 8 — — хлоругольпымн эфирамн 643
Амино:1Цетальд(>гид_ 492 -- дслепмс
— ди'ггнлацеталь 522, 697 — номенклатура
АмниобаРбитуровля кислота 710 — N-оксиды
rt-Дыпноиепзальдегпд 492 — первичные, получения 58G, "*88, Г»Я1, Г>9'2
о-Лмпиоб(Ч1301"шая кислота см. Литрам ил о- — цнетная реакция 4'JQ
пая кислота - - циклические ЗИ1
/;-Лмннобепзонняя кпелотп G20, 623 Аммопиеыые еолп, злмещеиные 388, 391, 394,
Н-Лмнн')бен:юлсульфо1!оиая кислота 41G 399
б-Дмнноиалсрпанопая кислота 617 - - прострачетисчшое строение 39 [
Р-Л\:пноппнилкарбоипл1)ные соединения 487 Aii,-,u,i3iin GO.)
а-Ампно1'л>таронля кислота Г>2 1 Лналы-пн 679
Аминогрупп;) 389 А и гидр г. д
а-ЛмнполикарбоиоРме кислоты 620 -- бензо1'1иоП кислоты Г>G1
о- Лмннодифеннлачнпы <М У — малсиноиоП кис-лоты Jol, П72
2 Д 13 6'3 П
007
о Лмннодифеннлачнпы <М У — малсиноиоП кислоты Jol, П72
2 - Дм и ной п да i д:и!П-1,3 6_'3 — тримоллитоиоП кислоты 503, 373
а-Лмннок;фбопиль:гыо соединения 493, Г>?2, — три фтору кс ус noli кислоты 570, 0
Г.1'3, 52 1, 706 — уксус поп кислоты об 1, 572
р-Л\тш1ОКярй.>1!Плы[Ы1' соедп!1сння Г>23, Г.'М — ф га л е:юГ. кислоты .'»О J, 504
Ампиокарооноиые кислоты см. Ami: и от-; и с- — яктарноп к;, слоты Ъо1.}
л^ты Ангидриды карбоноьых кислот ,rUi4, ,170
а-Лмппокпслоты (ИГ>, G27 ~ вэанмодеГ'стпие с нукле^фильнымн рея-
— ;:ктпип]")(Д1а щ.ые эфпры 033 рентами Ъ71
— амиды G20 ~ конденсация с аренкарбальдегпдлми 571
~ а цнл upon-in не G21 — номенклатура Г» 6 I
— бет.-.нпы (ннутрепние соли) 018, GI9 -- итучоппс 070
na/i^iieiiiiTiie мредст -иптсли G2-; — применение J72
— - дна 1огнроиян;!с ')¦_'! - - реакции а-иодпрпдног-о атома 571
-- N- и О-;*;1:цппленные С>.ЧЗ - - физические спо:':стиа и природа аппеп
кнелотпоегь, o-'jhobiiocti. и прототролные Г>70
рапнопеспя fil 9 Апгпогепзнп \Г б28
-- количественное определение 6:'1 Лиостснмп 62Г>
— незаменимые 623 Л песте, пруюн^.ю пещестна 02Г>, СОГЗ
— пнтр:г.чы П16 Лш'Д Oh9
— onpiKUJBaune is нутр и комплексных солоП Анизол 3 {2, 344
620 ' Анилин 13, 31, 101. 389
- - дмкетолрлтеразппов 022 — бромпронаппе 412
— — дппептпдои G21 - - дназотнро'!пнпе 4 П
— - окисление G'2-1 -- днпольиып момент 408
— оптическая актпппогть 018 - - пппизлцпм спя.'.еп Г\ II 413
— - получе-нне 01 и, 027 — катпоп-рпдт^пл -i 1 0
-- - чистых опг.нтпоыероп <K1 - окисление 410
— простраистиеипан изомерия 618 - ос-ноппость 4U9
72G
— птучешю ?Р1, 400, -lit — !\тс(:~цикличсскпс 177, 179
— применение 414 — — плкпл1ф-/ч>лпно 187, 292
-- (Ч).'ш 41 I — -- ap'i.'miiQwiiH'* Г'7
— I .к-чогы по методу МПХ -IC8 -- — лцилщюр,ii.no 1УЯ
-- ptviMj.iiя с cenuyi 1ероДсм и серой G83 - - - r-i логенпрспашл1 IS'.i, 100, 238
— ( сектры погап:це"П',1 -JC*J - - — rri/i^rt'hvcM ;'"nip-.a шик1 2"_\i
— сльфпроиашк' 4 1 .'.¦ ¦ ¦- - - гидрнпопа пне 1V '
— фл.шче.'кпе споттп,. -И!/ . — — дмнильпые М'.м.ч-гы \Sl
— ч'.чорнрошшие 3_8 — - ;kjтопi:v-o'l',i!ino :' ',>.
— тлергпн иигш^ацн51_ -1"S — — мерчурщкл .кП'.е 2.':7
Ашыч-р.-диклл 3JG, tj3J - - - нитронл ипо INS, U;'J. 373
— Оси ила 1?!.") — — ночомклптл pa 1 77. ! 73
-- иутлдпона K'9 — -- окисление" 1{).\ I U'i
~ l.-'-AiiK^rcMon 477, чГЯ — — получение 179
илиГюн ильных соедини',nil 4-1?, -1 53, 469 — — реакции и ookodo'i ц-ип I'Jl
— пптросоедмнепг.п 373 — — — :^и',;триф:1ЛЬ.;иг,) .,,i -.ктцсиня t S6,
сложных зфкроп Г>77 187
— тотрпинанолгнлоки С>01 —- — с S-jC.l? Л'31
T(.'Tp;iniKiiin\iiiio,T:iML-i ..iici 1;UI — — сг.ечт;.ы jг«jr .lO'j'.ciiiiii 181
— xh;i.)!Kji* V.yi — — (.T|,ui 1МЧ1 1S1
— 'П1'Л1Ч1=1 38 — — L'y."ifi4:p0Ufiii!H' 18!1, ',iZ~y
Апиулср.ы L46, '217 — - - сул!>ф(,\ло'.П!рои<1 чпе- 3.:5
А!тли..'А ги;лы<1\:!1ером[>] \улу, \7 — - - \inop:'ii и noiiii':. ;l;:ii I Ъ\
Лиомгры Г»06, ЬUЯ, Л 1 U - ¦ пом1-:м.л,:Т\рп 1 78
А.1тп..р..\и;тлч1ч--,чне сястгмы 213-213 — иолчц-ик-кпнч-лпе 177, tOU, 201, 202
Антибиотики ()81 — р'ч оис-[1мти;ч1с1'Л poai\!j,iio:!ii;r,i способ-
Лптнгсл L)M и и 1 iii !ПЗ ко'.тг. '_'!'?.
Лигидотоийторы 331 - ряд 01Ч\,олл 177, 179
Аиткко^гуляшы 190 Api uiuiii i\l\
Лнтгпжснд.щты 3JJ, ?,'2G, Л13, 468 Apri.i.'iaorpyiTiin 430
Аптипнри и 079 Л[)||;|;1лк,!по:1ы 281, 300, 201
Аитцфртм 303 Лп.1Л^',м'.Д|,1 ц. t;.;i;;ob -!. 13
Ачтощк'.мнднпы 704 Арплами и \ \ ;J8!, J8J, 10")
Аигоцигли.! 701 - хчкглпрпмлкпе 4 07, 4 11
Литряиилорпя кi слота 603, GI8, 023 — .'ipn.viij.^juiiino 407
0,10-Лкт|)ахипо:1 оЗО. Ъ[\\ — ацилирои^цке 411, -112
Антрнхмнипы 208, Ъ'17, ГI.4 8, Г) 31 — is ^апмод^/и' r:i:c с азопи ro;i кислотпГг -!1 I,
Аптрахпиоиопыо Kpiicirrowiii 534 4 1:.'
Аигрппси 201, 2 00 — - диазо-шепыми солял-н 4'23
— пол^цчше 206 — — и кщилмлтр.мп G-17
— С!Н'1чТрЫ ПОГЛотСННЯ 210 — - KfipOOllli.'Ibfibl.M]] 1"ОСДМ ПСМИЯМН С06
— erpcvuiic1 207 — — фотемом i\\'i
-- x;i vii'ii'L-iiio гпоГи-тна L'U7, 208, 243 — — АП^чти-фцльиыми рс.чгоитлми 410
— 'j.'-opnnr иошмацни 2A7 — гмло-еппион.щис ¦'¦> 12
Лпг[);|Ц^!Г1;ул1|(ЬоноГ)Ыс кислоты 207 - - дил:ютимолнию П 1
Арлбщюза .10./, j 1 6 — д Hi голыше моменты 4 08
A ff'yjotta перетру гн in poi'K;) 277, 3 31 - - in лип,: ц;: я сия. jo;; N IГ ¦' 1 3
А |) г и hi; и С) 2 4 — киндоншция с лрспкарСсп-.дегндамц -170
Арсидннзомии соли 23Я, 2-17, 313, -ЯМ — -- iiiirpojoapeii.'ivn 432
— а.н)спчетан1Н- 425 -- обр.клнглапо комплексов с переносом за-
— 1П(Ц!М'1де:"и'ТЦ]1е с иуклс'о^^н^ьнькмп рем- ряда 410
гсчп л ми 424, 42 Г» — — крпс-птолеч! 4 1 1
— восстапоилепие 418, 425, 427 — иптропямпе 412
— - oGpajoisainie дмазитагоп 423 — окле.юпие 379, 110, 4 12
— получение -I'M — oi'HuiiiiocTb -100
— ]Н'Лкц1ш с иыдслснпом а:юта 425 — полvic-iiu- 406
— строение 4 22 - расчеты m.i методу МОХ 4 08
— тс]шпческ<)е разлох^енно 42E, 427 — егк'ктры поглощенчя -ЮЧ
— физические cisoi'icTua 4 22 — с ульфипогикпк1 -112, II3
-- фотохимические реакции 427 — уд^л^аио аминогруппы 127
Арс-[Дг.;!ЗШ1НI тетрлфториораты 4'.'7 — физические cisoiicrna 4 08
АрAПд111\ариоцоги.к1 кислоты 5G2 - - энергия и пни зацни 408
Аре ид полы 309, 323 Армламмоипсиые соли 40'J
Аренклрбальдег-иды 4 58 Аралгпдра.щ пы 418, -И У
-- Л11тоокмсл1чше 408 Лрилгпдр.-иоиы 472
-- otMUOiinorui я wo идеи fa ци я 4 66 Арн.'Н'ндроксплпмнпь] 381, 38 \ 3S7
— нзнммодепгтино с нуклеофильнымм pea- Ар1!лнодопп]1<а!лinie 24 8
гечтлмп 1П6, 470, G97 2-Л|IПнидлндионы-1,3 483, 400
— Д111'11])О11орщ1Оп;1рпмг:пие 4G7 Арплирсмчпик1
— Конденсация с ацотапгидрпдом 571 — аминов 407
— пол у чечше 4D 4, 4 05 — алкеноп 4 20
— полярность спялен 4G~> — а л кило» I 5G
— спектры мш'.тощсняя -Ям — аммиак,! 407
— ^ле1\трофплы1ое замещение 408 — арепдпазоппепымп солями 42G, 427
Арснмонокпрбонпные кислоты 563, о34, 555 — аренон 427
Аренолы см. Фенолы — галогепарепамп 407
Арсапи.п'карпономые кгн-лпты 502, 563 — гпдразпча 4 18
Аренс/льфопплхлориды 301 — подопиепымп со/гамп 248
Арепсульфопомые кислоты 313, 355 — фенолятоп 344
Арсчтполы 3G \ — цц;:!1гдпп ?,\)Н
ЛР11Ятрполы 309, 32 3 Арил-катнпны .>-17
Арены 82, 177 Арил.л(Лы1Ы 180
727
Арилметэновые красители 535
Арилуксусные кислоты 55о
Арнлциа ниды 59S
2-АрилцМКЛогександионы-1,3 489
Арильны" остаток 1 7S
Арндта- Зйстерта реакции 526
Ароматизация нефти 179
Ароматические альдегиды и кстоны 465
Ароматические свойстпа 212
Ароматические соединения 177
— небекзоидные 21 3
— углеводороды, см. Арены 13, 212
Аромати чиость 21 7
Асимметрическая молекула 61
Асимчцгрическш! атом
_ _ азота 394
_ _ кремния 272
серы 361, 367
_ _ углерода 25, 01, 22Г.
Асимметрический синтез 631, 632
L-Аскорбнноваи кислота 515
Аспарагип 621
Аспарагиновая кислота 624
Аспирин 615
Ассоциация
— алка полов 285
— борорганических соединений 269
¦— кардоновых кислот 542
>— металлоргапическнх соединения 249, 254,
259
— посредством водородной снял! 285, 542
Атрочизомерия 197
Атропи н 095
Ахиральнаи молекула 61
АцеталИ 450, 474 478, 582
— циклические 474, 499
Ацетал ьдегид 437, 4 11
— аль,дольная конденсация 447
— конденсация с формальдегидом 457
— окисление 479, 518, 572
— пол учение 548
Ацетальдоксим 471
Ацетаилид 584
Ацеташидинии хлорид 566
Ацетацгидрнд см. Уксусный ангидрид
Ацетатный шелк 521
Ацетаты 514, 519, 555, 569
л- Ацетил а ми нобензолсульфоиил хлорид 416
Ацетнламиномалоновып эфир 6I6
АцетиЛацетон 475, 48I, 484, 488
Ацетилен I 0, 14, 25, 26, 46, 24 1, 245, 250, 548
— взаимодействие с карбонильными соеди-
соединениями 298, 305
— -- формальдегидом 157, "98
— днмеризации 154
— гидрирование 128. 119
-- карбонилировонне 152
— образование ацетпленидоп 155
— окисление 153, 155
— полимеризация 154
-- получение. 115, 146
_ применение 158
— срисоедипенне алкаполов 152
— - . поды 151
_ -. галогеноподородов 150
- галогенов IоО
.- — солей ртути 257
- уксусной кислоты 152
-- — цианистого водорода 152
— пространственное строение 1 18
-- физические свойства I 17
— циклизация в пиридин 6S8
— iul клотетрамернзации 154
— циклотрнмеризация 151
Лцетцлендикарбонопля кислота 556, 560, 561
_ — диметиловын эфир 562
Лцет«'леииды 155
Ацетилеикарбоноваи кислота 539
Ацетилнитрат 376, 6G1, 667
Аи.ет илсалицилопаи кислота (П5
Ащ'тчлфторнд 564
Ацегилхлорнд 561, 569
Ацстилхолин 402
Ацетилцеллюлоза 521, 572, 578
2-Ацетилциклогексанон 481
Ацетон 438, 457
— альдольная конденсация 458, 461
— апеллирование 481
-- образование диоксоланов 474
— окспм 471
— получение 13, 194, 29С, 313, 338
— реакция с цианистым водородом 552
— физические сионстна 441
Ацетонплацетон 475
Ацетоиитрил 567, 599
Ацетонцнапгидрин 55'2
Ацетоуксуспая кислота, амиды 639
Ацстоуксуснын эфир 463, 636
— алкилиропание и ацилироваиие 637
¦— взаимодействие с аммиаком и гидразином
638, 675
— кетоппое расщепление. 637
— кислотное расщепление 638
¦— кислотность 636
— конденсация е альдегидами 638
— получение 635, 639
— соли 637
— таутомерия 636, 639
— физические свойстна и (тримепение 639
Ацетофеион 439, 409, 494
Ацидофобные соединении 663, G70
N-Ацилампнокислоты 621, 631
Ы-Ацил-а-амипокарбонпльные. соединения
682
Ацилбромиды 601
Ацилгалогениды 564
¦— енолнзация 569
— получение 546, 567
— реакции с кислотами Льюиса 568
— — металлорганическими соединениями
252, 256
— — нуклеофильными реагентами 568, 569,
— — цианидами 634
Ацилгидрааины 590
К-Лцилгидразоны 590
N-Ацилгидроксиламииы 592
2-Ацилиндандионы-1,3 490
Ацилиронанпе
— алканолов 291, 546, 574
-- алкиламинов 397, 449, 568, 583
¦— алкоксидов 289
— аминокислот 621
— аминов 397, 411
— аминопеницнллановоп кислоты 631
-- анионов 1,3-дикарбоннльных соединений
485
-- аренов 464
— ариламинов 411
— - ацетона 481
-- ацетоуксусного эфира 637
— внутримолекулярное 517
-- диазометана 524
— гексоз 513
-- гидропероксидов 595
— карбоксилатов 546
— ¦ кетоноп 480
¦- пероксида водорода 595
— пиррола 664
— серонодорода 594
— - тиолои 594
-- фенолов 317, 319
— фенолятов ,117
— - фураиа 664
— целлюлозы 5-М
Ацилирующие. реагенты
— ангидриды карбоиопых кислот 571
— ацплгалогепидьг 5G.S
— О-ацнлмочевины 619
— дпкетеи 463, 639
— изоцнапаты 617
— карбонопые 1\пс:юты 510
— кетепы 4Г>3
— ортоэфпры карбо1?огн,?К кислот 582
728
— фосген 612 — илл(М1Тные изомеры 19;?
— эфиры карбомовых кислот 575 — взаимодействие с карбепом 1712
— эфиры хлоругольпоп кислоты 643 - постановление натрием J об
Ацилия ион ,".08 — (сьюгомиропяшге 189
Ацилкарбены 103 — геометрия молекулы 181
О-Ацилчо'и'нины 618, 019 -- гидрирование 168, 192
Ацилпитрены 387, 391 - димерпзацни 19G
Ацилоины 496, ГO7 -- дьюаровскнн \iXi
Ацилоинонан конденсация 1^-"'. 577 - кати он-ради кал 2\ 5
Ацилокснгруппа 310 -- Лиаенбур^а 19-1
Ацнлперокепды duG. ">93, 5У7 — jini анд 263
Ацплфторнды ;3;i7, 601 -- нитрование .479, 381
Ацильпая группа Л.47 — образование л-коммлексок 264
-- - h;i31!<iiiiim 339 -- о.юнировуние 193
Ацилфенолы 319 --- окис л 141 iu1 572
Ацилхлорпды 1й8. 461 -- особенности строения 183
Аци-нитрофораы 3*4 -- получение 180, 193
- применение 191, 195
Байера теория напряжении t 62 - - присоединение .v.-орл 193
Ьапс/нг - Ни.;.Hire pa uepei руиппровка C9f» — расчеты ио методу МОЧ 182, 183, 215
Ba.itn\Li •- Ш к ми на рс.чкция -]'J7 - спектры погломичшн 1«3, 181
Барбитураты 709 - - сульфирование 189
Барбитуроп;щ кислота 705, 708 -- фнзн'ичжне t'BOJit-тв.ч I /7, 180
Барьер RjKiu^'Hmi 9] - энергия ношмгщнп 181
Варен 270 2- Беизолазон;|фт;;лин '131
Бсяы.'злучлтсльныс переходы .VJ Ьензол-ач/лм-диазогпдроке-цд -):}.О
Бек мам а и с per pv и и и ройка 471, :~>S9 Ьсчп:юлдид;(ония соли -123
Белки I'J, 13, 616, G'Jj - хлорид -1^0
-- 1-мдро:ш.) б-'й Ьеп.юлдпкпрбоно11ьц> кислоты 5Л0
— денатурация 62S ^-Гнчоолдисульфомонаи мкло'П! 1125
— классификации 6'*6 (пмгзологк 6л t
— коагуляция 6^8 Бои.юлошнчшп и пи 187
— методы синтеза ti.40 Ьен юлсульфепнлхлорид ЗНО
— реакции 627 Ткчшэлсульфтыт и,ггрия .460
— структура нторичнг.я (>29 Бонзолсульфо^п/лхлорид .4"» 1
— -- иориичная U28 Ьен.чолс-ульфопои^н кислота
— - третичная 030 1 ,'Л I, о- Пе нзол тет р ;i к ;'.рбон пи пи кис.чот;! ГГ|7
— -- 4L4T')ej>Tii4iiaH 630 1,2,1- Г»ензолтрикирбош.^к'1Я кисюта .~>Ь7
Белый стрептоцид 41 l> У>снзо1Ч1[)я:щн 6Г>7
Бензальацотон 158 _ Ье1]зо1с||1прид;'.;(нн 657
Бепзальдсгнд 22, 1У1, 437, 4"iM, 165, 4G6, bL4i;ioUlJiiHp;i,T;i3iiii G37
4 68, v'fi.'i I>en:u)[h (пиридин 6.17
Бензальдоксмм 171 Уи.ч{3о1с\1ш\упдии 6Г>7
Бснзальхлорпд -U5S Р)ензо1Ь1|Шри;П!Я соли 701, 702
Бензамид 08 ; Бензоинримидни ii-'»7
Бепзвален I УЗ Ьензотна.юл G.iti, o\S 1, 6ШЗ, (iS5
Бензеи см. Bt4iu\i Ьен.кНЫтпофел 6."i,i
Бонзе пол см. Фенол Гнчиотриазол <Г,{;
Беизидиноаа» 1;ерегруи пи рок wu 419 Бензотрихлорнд .i'i9
Бензиди н_ 4 1 о, 119 Бсплифспоп 198, 169
Бецзил 4 / о, 4 7^, -I 79 а- Бензофенопкарбоззои;»!! кислота Г>29
Бецзилацет;)т п Г^ Бепзо^Лфуран бой
N- Беизилидеьл иилип А 70 п-Боп.чохпмон ."ЗУ
Ьензилопан км слота 178 -- пилим оденет и не с пуклоофнльнымл ре.-.-
Беидплован пере; рушшронка 178 геитями ЪЗй
Бензпловын спирт .300, J0I -- восстаиоиленпе fbi'J
Ке»зилоксиклрбош]лх:юрнд G ¦ - — дне новый синтез о 33
Бонзплпенпци.члпи 381 — образование комплексом с иер^по^;!1.! j;l-
Бснзилхлорид 198. 2U(i, 207, i'3G, 0\"j ряда Г»30
— - скорость ГAД|юлнза 232 — получение 327
Бензилцнанид 55о _ - строение молекулы Су>0
БензильнЫ]'! остаток I < $ -- счи'ктры иоглогцемпн ;1-<0
Беизильпып р;;дик;ь-1 [91 — сродстпо к электрону .Vid
1>еизимида;ю;1 ЬЗв г/-Ьепзохиноиа монокснм fi.i 5
Бензин 101 Бензохиноны Гг.!A, 5'J7, 03.1
Ьензо[Ь)азспи1[ 638 Ьон:*гшнйкоп itiy.)
Бепзоат натрия fi35 Ьеизпнреи 2{'2
Б(м1зогндрокс,-1\гогк!я ипелотя 36") Ьеичтпачол ft82
Ьеизоиллзпд 563 - .'-меркапто- (iM, 6»s"i
Ьеп^омлироиаппе 309 Ьс(к'пп. а метод ','0
Бензоилперокспд 566, 597 БессереГфяпая фогографпч t_7
Бензоилхлорид 22, 469, 364, Зй9 Ьетаппы 618, G19
Бензолы 4GG. 179, 496 Гятрилы -127
Бензоиновая конденсация 466, 19"» Бнндон 190
ЬеизоГшая кислота 9, 22, 25, 3I, 176, I SO, Бин^шсОлера зеленый 333
I!) 1, 539, .">."H, 354, 335 Ьио1 енные амины 40 I, 673
Ьенчопнып г'.льдегнд 437 IiuoKiiTa.'iiKUiTopbi 7!
Ь^ачокс^чол G3() Гшаорпш и веским хлмн ч I °
Ьоизол 10, 13, [ 1, 21, 172, 176. 1 УЗ, IIH1. )и'> ЬнипляриыГ! амротоннын расшорптелг. 301',
~- ^лкплпровииле 187 3^.-, .ihS
,i пион-радиKii:i 21 3 Ьн(>адпьа.'1Ы М). 2 IJ
— а'Л'.:Л1' iiOIULiilU' I ^s Г»1'.1 :)'.'.¦¦'¦•Ы «)oS
72J
Ьпс-'.т:|ЛС1.б');:.;т 3 Hi
Бисульфитныо соединения 152, 179
Биурет 6 15
Ьиуретовая реакции 627, 615
Бпфепнл 190
получение 196
— пространственное строение промзпо;
онантиоиернн И) 7
— химические снопегва 107
-jncpi ия понгкн-.ни 107
л о с1 л к а 1: ы 1GI, 17 1
[ 1
Ь
Б,;ци
b
I 7 1
193
гидрпро;»
диен(Н1ы;:
кло1 1. 1,О1Д|
[2, ',01-:
IS is цикл of ¦.'. .'.0 ]: ексан 16 1
ьпцнкло] 2.2.1 |i епган 101
Ьпцпк.ю[3.3.0|октап 101
Борнеол 299
Бороводороды 268
Ьороргапическпе соедннения 208
Брома л капы 221, 2 2 1
Ьромарены -126
Бромацетон 102
Бромацегофепон 49-1
Бромбеизол 2 10
2-Ьро\:б>тап 225, 231
Бромироваппе
— - альдегидов и кетопон 1 15
apt"iio;> 233
— карболовых кислот 001
про\зчод::ых бензола, относительные ек<
ро.тп IV'0
фон о. и: 318
0С-1)рс):,!к;фС).)!:овые кислоты 601-
а-Ьромп^фталпн 205
Ьромоипя если 2 i0
N- Бро.\;сукции1:мпд 588
Ьромуксуспая к не лог;, 605
Ьромфторчлормепщ, -^^пгпомери 2'25
Б ром я пт; ряая кислота 5i.'O
Iu/ea — Ij.ittHU метод г ;|Дрпрп1>а:п: л 28 t
Бутадиен-1,5 100, :'.il, I',U"), 300
— бромирон^ппе 137
;ип:е 137
сн нтез 1 11
— oCiptViGUtiUHC л-хомилексов 138
-- полнмор;: :..цня 1 ;^9
— получение 13 ', 157
-- пространствеиное строение f 34
— расчеты по методу ЛЮХ 131, 133
-- спектры по1'ло]цепия 136
-- физические своИстна 131
-- фрО|'т;1Л[>цые орбнт;:ли 135
— энергия дслокалп^ан!! п I 35
-- vnepi mi ионизации 136
Г.утаи 86, 87, 92, 01, 08, 132, 548
Бутаналь 437, 14!
Ьутапднампп 707
Ьутандиол-1,3 132, 305
Ьутанднол-!, I 132, 302, 305, 6Q8
Б\Т(Л1диоп-2,3 475
Ьутанопая кислота 538
— /чаитпо%к ры i\Sl, 297
Б>тапгетраол 300, 307
Ьутап-О-тмоповая кислот;)
Ьутепдиопая кислота 556
Ьутон-2-аль 437
13утеп-2-д ' ол-f, I 30'2
Бутоны 101, ];V.», 560, 572
Ьупгицетат ,578
Буг'1.лви н 1Л 3 i л:\ эфир 3 11
трет- В ути л гп дроперокенд 348, 350
1,3-Вутилепгликоль 302
1, 1-Г)Утилепглпколь 302
Ьутиллптий 251
Б утилметилкетой 438
Fутилит\}\\\ ^46
Бутчли^ые спирты 115, 282, 288, 2ij6, 207,
'520
506
трет- Бутил хлорка рбоп:. г в [2
(?:^о/?-1)утилтримет ил аммония п. др оксид 100
Ь ути иди он а я кислота 55')
Бутн»дпол-1, I 302, 305, ЗЗУ
Бутнп-2-овая кислота 5ui
Бутпны f 1Г)
Ьут)фальде['пд 137, 44!
Вутираты ;Н8
7-Б\тиролактам 5G5, П88, 017
7-Бутиролактоп 50 1, 381, G05
Бутироццтиил о(>7
m/?iv;/-fiyroiiciiK:)p6oi-:[b-] хлор и Ч ь ' 'I
тf)em-h\"fi.ib.i'{;\<.;\y,Ci{n\\w.\bi\;\\i j ^yi::;^ 033
Bijxo.pcpa реакция -i [5
Вагнера - - Мее ренин a nepei рукпиронка 239
liii.'U'iiTHhio пзо.чоры 1 (j3
Валера г ы 3-1 i
Валериановая кислота 5М8, ,А'2
п-Ь^леролактон 5(j i
6-1^ а л е р о л а кт а м 065, 01 7
Иалпн 624
Вальдеповекое обращение -.И, '2J'2t G()'\ GI2
Пянилнп 503
beparpo.-i 313, 31 1
B-jpbiii'i</rbi(? воинства 13, 191, 105, 318, 376,
385, 4 14, 456, Г.21
Jhi.ihHMcvna реакция 288
Ь||ипл,!мпп 38'.)
инпнлацетат 15 \ 578
Нипил;:н,гп1ле:1 151, -' 15
Ьнпилбепзпл су.. Стирол
Г)пнилб[)с)мид 239
Ьннпл! ;i:iti: епнды 23G
Ьипилироиглпе 15'1
К - В и н ил к a pGii:: ол 6 71, 673
ЬппилоныеЧфпры I..2. 31!. 312, !73
]ini:iK.iunbi:[ спирт 300, 5| 9
fii:пк.-ки 1:я -101
Х-Иннилп-.фоолидоп 58S
Ьипилчлоркд 150, 2:;iJ, "lO, 1.', 214
ВппплчгнловыП зфмр 3ll, 1CI
Ьпппокммеппа'.: соль 01 i
Винные кис.-юты 9, 361, ГО')
-- — до! идр;л.'ан.пя 0,)(.1
- - кислотность 01 0
— — с/пчн'оизомерг.м Ь]Л, G14-
Виноградная к:и\тоы_ 01 I
Вп11о:'|)йдиы;'[ c;i\ap 513
Ь[|С:-о:л1ыГ[ шел:-. 5 ' 1
Ьпт::лн:и: Q
Впт.-iviMi Л, 300
— Г., 08 1
— Вч 0У1
— В;- 1-'. U06
— С 515
— К\ УМ
— - \'\> 6<М
13]:теллгн 6_'6
Duiiuiiiua реакция 278, 117, 551
Внутренние комплекс!.! см. Халаты
Водородная евн -.ь
— ¦ впутрнмолскул-цч:,..-! liii. ~>0Л, 636
— — меж молекулярная _8(;, 393, Г) ','2
-¦ -- энергии 286
Водородный обмен I87
Возбужденное состояние- 5!
Воз] оика 16
В ((Л новая меха пи к а ?•']
Вплпоная функции 32
Волокно искусственное 1 .'*, 521
синтетическое .'НО, 58'.)
Вол ьтамперо-лч'рг -i гим'.л :.1,сч-:-.ая 18
fhi,u>ifhi nepei pvi;ii:ipcii;wi 5l'5
Воскп 581
Восстанпнленне
— ал1-.(н<с][дамп алюминия 153
— - альдо! сксоз ."¦ 1 I
— альдопонтоз 507
-- г.р^-пкарбмльде! пдов IG7
— - i :)ло1Ч'пуч'Л1.чюД|..;1ОДоп I'-'i)
— диазоппеиых солеи -118, 425, -127
730
— пзоппанпдов СО/ Галогепопгевы!1: цш;.1 ИЗ
— карбонильных соединении 4 ">2, 433, 473 Гя-ioi енкронзводпые углеводородов С (<;;>) -X
— карбо!!овы\' кислот 469, 515 1215
— ммтрм.ши 599 Гало| енпропзводпые углеводородов С (s/>2) —
— питроалханов 373 X 236
— пнтроарепов 007, 132 Галогепкрогс.водные углеводородов С (я/)-1)—
— N-niiTp(Uo;>Ml:iion 417 X 2)9
¦— окснмон 523 — -- дппольпые vomchtu 221
— пирпдпипрвых солен 693 — — i:ivi\ ч.чше :''O
— сукцппнм.чда 617 -- — применение 231
¦— сульфопилхлоридов 361 — -- физические свойства 221, 225
-- хипонов 632 — — химические свойства 229
Вращение удельное 60 ._ .. хпралыгые 2J.1
BytteopOa -- Хифмана правило 169 Галопиевые соединения 240
Вюрстсра сиiгиГ[ 410 П.-лофорчы 491
Вюрца реакция 229 Гапмета константы 78
- - уравнение 77
Габриэ./я метод 588 Ганча спите:* 687, 688
Газовая хроматография 20 Гаттермана — A'".ui метод 461
Галактоза 30U, IH4 Гваякол 31!
а-Л( ¦ )-Галакго[;ираноза 514 Гексаалкплцпклотрпсилазапы 271
\-{[\-D Галс:кто:1нра1103идо)-а-0-1 люкопира- Гексаалкплцик.гогрисплоксаны 273
ноза 518 Гексаг пдроипрпмиднп 707
Галловая ки-слота 327, 615 Гексакарбонилхром 261
Галогеналкаполы 333 Гексадекановая кпеюта 538
Галогепалкапы Гексачотнлбепзол ISI
— алкилировамис арепов 187 Гексамегп.^епдпампн 390, 401
— взаимодействие с металлами 91 Гб-Пчдсачетилепдппзоцпанат С18
— -- пуклео(^и.1Ы[ыми реагентами 230 Гексан 86, 92, 91
— получение 220, 292, 293 Гексанампп-3 389
— реакции отщепления 106, 233 Гсксандиамнн-1,6 390, 401
Галогоналкены Гександипнгрил 567
— номенклатура 237 Гексапдновая кпелога 556
— полимеризация 242 Гекс;'.ндно.|-1,6 305
— получение 237 Гоксандпоп-2,5 475
-— физические свойства 239 Гексаьптробопзол 380
— химические euoilCTBa 242 Гоксановая г;пслота 338
Галогеиалкпны 2-15, 216 Гексанон-2 438
Ы-Галогенамиды 586 Гсксаолы 308
К-Галогонамилы 398, 412 Гексафепилътан 200
]"алогенангпдридь[ карбопопых кггелот см. Г'ексахлоран 236
Ацилг алогениды )'ексахлорцнклогексап 193, 23G
Галог енареггы Гоксациапобензол 601
— BjaiiMoAolicTnue с металлами 211 Гексенал 709
¦— — нуклоофильпыми реагентамгг 241, 407 Гокспты 308, 511
— нитрование 213 Гексо:;ы 50 1, 508
— номенклатура 237 — алкилпрованне п анплнрпваши- 513
¦— получение 238 ¦— ьзапмодепстппе с фсчшлгидразппом 512
— сульфирование 243 — восст;-.пов.теппе Т>11
— физические свойства 239 — кзомерия кольчато-ц/чшап 509, 510
Галогепг'ндрипы 333 — ¦ - пространственная 308
N-1'алогенимиды 12 1, 538 — окгслегпге 511
Галогенировэшн! — ферментативное расщепление 513
— адамаптапа 176 Гексогеп 456
— алкадиепов 138 Гель-хроматографпя 16
— алканов 96, 22! Гем 66!)
— алкеиов 113, 118, 124 Гсмин 66G
— алкигюв 151, 216 Гении.гльпме дигатогепалканы 439
— амидов 586 Гемоглобин 626, С65
— арсчю» 238 1,Ч1ПШ 86, 195
— в аллнлыюм положении 121 Гептаповая кислота 538
— 0-дикарбопп..11,ных соединении 436 Гепго.ш 505
— карбсигнлыгых соединений 492 Гераппот 2'.)°,
— карбоггопых кислот 604 Гербнцпдг,) 615, 707
а-Галогенкарбопнльпие соединения 192,522, I'ep\ianni'opi\ i:i!'ieci-:i:o соедпмгчг'Я 26!, 26?
680, CS3 Гермаполы 261
— — образование гидратов 494 Ггропп 7С1
— ¦ - перегруппировка 493 Гетерошклп'ич'кне создинеппя 8С, 653
— — получение Н;2, 525 — л-пзСьлочны,' 6С0
¦— -- рсякцип пуклеофилыюго замепгчення — клас^ндшкаипя п но\:енкл-тура С53
493, 195 Г1!5рнл:!3<;Ш1я отечных орбнт;| icii
Галог-енкарбоновые киепты 601, 0 — — — a ,;vra :'.'M
¦— — докпрбокспли|)ов;;нне CG6, GU7 ¦— — — ki:;y;jp,.;ui 2.-7
¦— — кислотность 605 — — — > iVien'.-.":! 36, 3G
— -- получение 6CI, riO5 Гплрмзчд Ч к -у.-ца:1 кпе-иты 56.5
¦— — — фупкипоиа;и ных производные 60G Гпдраз^ды iimi'.'lo.wi.vot 0^,i
•— — реакции нуклеофил! роги йамещенпя Г;!Ч;>.!Ч|П.>! к.-.рбонл-ых к,..лот 5С5, 590
сое. 616 Гидра ,шгы 413
— -- — отщепления ,i50, CCO Гндр.ч imiiiisi с.;.т' '.!7
Галогенметпларепы 41»5 1'и а'т (!:¦¦ лип сл-,;-;"Ы\- зф:гров 575
Галог епметилнреванп" 223 1 !!-,¦":¦.-leii.J .,»', ¦!:,'. ¦',{^. \':i
Галогешштроарепы '681 1 ii.i;.:.j.,Gbi'Jo.i Ял, 417, 41J
731
Гидр:ионы -119, 172. 473
Гидра:юсоединеиия -119
Гидрид-ион, доноры 14 5
— - перепое 167
1 идрнрованне .(лкадиеиов-1, 3 1 37
— алкенон 109
— алкинон 119
альдегпдон 4 l.~i, Мб, 1У2
— - асимметрическое 032
бензола 1 92
— бон :t он noil кислоты 550
— диоксида углерода 90
галогенугленодородон 229
-- и.к)Хп ноли на 699
— п юциапидов 602
— ионное '27."i, 'l 16
-- карбонильных соединении 281
— - кетоцов 4 15, 116, 152
малеи повои кислоты 559
¦ ¦ нафталина 206
пнтрилон 599
— пнтросоедппеипн 381, lit
оксида углерода 90, 281
ппрази )\л 7U7
ппрнда.шиа 707
пиридина Ь9'2
пиримидина 707
пиррола 662
резорцина 323
--- сила па ми '217)
— ел о ж н ы х эф и р ов 284
¦ - сукцппнмида 617
— - тиофена 662
-- угли 00
¦ фенола 321
— фурана 662
хпнолина 699
Гндроборпронание 269
Гндроксамоиые кислоты 363, 592
1 ндрокемды тетраалкиламмония 399, 4 00
о- и //-Гидрокена «оарепы 43 I
ti'l и дроке и азобензол -133
Гндрокспалкпльпая группа 1Г>и
Гндроксиалкнльнып радикал '290
2-1 ндроисп- 1-амппопирн мидии 708
Гпдрокснацетон 192
Гидроксмбензальдегпды :92, 501, 502
е)-Гпдпокепбеи.ши пая кнелогл см. Салнцпло-
мая кислота
/?-Гндрокснб1чкюнпая кислота 609
("i-Гидроксибутнральдегид см. Лльдоль
Гидроксигрумпа 280
3-1 идрокспнпдол (i71
ct-1 идрокспкарбонильные соеди пения 495
кислот notTb 497
— - - обра.iou.i ипе ендполон 198
окисление 17Ь, 4 98
— — оптическая п.юмерии ИN
— получешн' 177, 403, 195, -196
— pea кии я с .пинднпами и 76
рМ"ндрокп[ карбон ильные соединении 4 16,
158, 4 98
V- и й- Гндрокснкирбоиильпые сосдинении
¦И18. 199
Гпдроксикарбоноиые кислоты см. Гпдрокси-
кпелогы
а-1'идрокснк(тины 4 35, 577
1 пдрокенкпглоты П07
а-1 идроксикислоты 607
— защита i н дроке ильной группы (ПО
защип* к„рбоксн.'1Ы1О11 i руппы 610
-- кислотность lil 0
нитрилы 4 1 7, '608
— окисление 631
-- получение 607, 608
— пространственная п оптическая пгюморня
— 609
—- расщепление серной кислотой 611
0-Гндпоксикислоты 008. 610
V- и о-Гидроксикнслоты 610
Гпдроксиламнны 381
1 ндрокенлнрование
— аренон 311
— фенолов 320, 324, 326
Гндроксильная группа 280
Гидроксильпые производные С fsc2) —C)H
308
Гидроксильные проиласщные С (sp3) --Oil
280
Гидрокспмалоновая кислота Л]0
7-Гндрокс и масляная кислота l!08
р-ГидроксимасляпыГ| альде]-цд -М7
Гндроксиметнлиропание 456
Гидроксиметпл -катион 223. 320
Гидрокснмегилфеполы 320
5-Гпдрокснмегилфурфурол 660
Гпдроксиметильпая группа 4.">i3
4-Гидрокси-3-метоксибеи.)альдегид 503
tt-Гидроксннигрнлы 14 7, 1H8
Гпдроксп инрь.к)лы 675
3-1 идрокеппирндалш 705
Гидроксипиридмны 691, fi93
I ндрокеи пнрили я соли 703
1-1 пд роке и пиримидин 707
ct-I ндроксипропноновая кислота см. Молоч-
Молочная кислота
1- Гидро копиром попоны п альдег'пд, стерго-
н.юмерия 196
а-1 ндрокеисульфоиовые кислоты 152
2-1 идрокситетра!'идромп[щны И)9
2-1 пдрокситетра1Ч1Д|)оф> раны 199
7-1пдрокенхнполнн 700
ее-Гид[)окснциклоалканоны 577
Гпдроксияптарнан кислотл см. Яблочнаи
кислота
Гидролиз галогеналканов :.32, 139
Гидролитическое рао теплен lie си sun С С
4 87, 191
Гндроиероксид кумола 313. .157
1 идроперокепды 251. :!30 348, 319
Гидрокснлиропанпе 2 72
Iпдросульфигы 352
1 идрофобные соеднпеппя 272, 274
Гпдроформнлиронниие 115
Гндрохн поп 323. 325
-- окисление 527. 532
Гппогйлогепнты кислот 5 17
Гппоксаптнн 711
1 исгпднп 621, 679
Глико.!ндп;ш гпдроксп льна и : pynn.i .7,
50У, .НО
Гл нко.шди;|Я сня.)ь 513, 517, >1 У
\-Глпко.)иды 713, 71 1, 715
Глико.н1ды 512, 517
1 „чикоколь. см. [л ицп и
1 лпколепан кислота 61 1
Елпколп 302. 380. 64 1
Iлнколяты 302
Гликоль 302, 303
— и.ынмодепстпие с борной к и ел ото ii 34 0
— - — - карбонильными соедпнения.мп 174
— дегндратап."»! я 310
— дегпдрирона пне 4 79
— карбопилпроваппе 357, 5,У)
1 ликопротенды 626
[лноксаль Ш, 303, 475. 179. о78
Глиоксилопан кислота 603, ПЗ1)
Глннераты 306, .J,07
Глицерин 9, 10, 30b
в синтезе ('краупи U9<i
— окисление 500
¦ ¦ сложные чфнры 5iO, 5"i.\ "SO, Г81
трннптрат iiO7, 3 1 7
Гл и пери новый альдегид 192, 5 СО, 5С1, 505
I лини и 62 1
I "лифт а левые смолы 580
Глобул и иы (>26
Глутамни 621
Глутаминован кислота 62 I
Г.чутароьан кнслота 550, 55 S, 9
1 лутелипы 626
Глюкоза 11. ."-,08, 511, 512. 51.', г2\
D- Глюк он она я кислота 311, .'>! 6
ос-Л-Глюкоппрлю.),! 5С9, 5 I 1, 516
732
р-Л-Глкжоппрапоза 509. 510. МЛ. 51!. 516
4-(Я-Л>-ГлюкопнраноЛ1ДО) и-П -1л ю кони ра-
рано ja 518
4-(а/>1'люкоП|:рано.!ндо) P-/J -глюконирано-
за 518
OS-/J-E люкоппрлпозидо (^ />-фруктофуранозид
51!)
?>-Глюкуроиоп,:я кпслотп 511
Гомологнческни ряд 81")
Гормоны Г.', 626, 627, 628, 629
Горчичные масла 651
Горчичный г;..( 367
Гофмана правило @0
Горючие сланцы 14
Гофмана реакция расщепления эмндон 586
Грин^яра реагенты 253
— реакция 253
Группы функциональные, характер и :тнче-
сык1 81
Гуапндпн 6 И. 6 16
Гуачндпппи ион tilu
Гу.'нпн 711. 712
Гуано.шн 713
Даутерм 198. .315
Дсбнера - Ми.иера chhtoj 6lJG
Дегидратация
— ал ка нолов 29.3
— диолоп 303, 310
Дегпдробеизол 2K
Догндроцпклпзатгл f 79
Деэакгпнацня побужденного состояния 32
2-Де_'.окспрпбо:1а 308. 712
До.чоксирибокукленпоныс1 кислоты 508, 716,
717
^¦Де.)Окси-/;-рнбофураноз;! 508
Демтерооб\геп 187
Декакарбопил днмапгана 263
Декалины 17). 20о
Декан 80. 92
Декарбоксплпрованпе 180. 5 17, 534 558
562, 57Ц, 587. 606. 607. 636
¦ окислительное 91, 316
Декарбонилнропание 636, 667
Декстраны U3-I
Д"Л1Жалидацпи энергия 126, 135, 2!)
Делокалпзацня ник.шческая 213, 217
Деметнлироваипе I iK
Дназена пронзиодпые 473
Дпалщы 657
Дна.юалканы 120, 128
— ^цитирование C0
-- иг.агшодеИстние с электрофнльнымп реа-
реагентами 429
430
тентами 429
— получение 128, 4 73, 64 4, 6 l.">
— фотохимические превращения
— цнклоприсоединеиие 130, 675
Дп.кюамииосоодииеиии 4213
Д|'а.шацетофенон 492
Дигмогпдроксид 4 21
Дня.югруппа 420
^-Дмазокарбопильные соединении ,124
«-Дназокетоны 430, 462
Дмтолы 636
Диязомстан 4'JO, 4 18, 5L4
Диа.^омстоД /1. /Л Нес меч нова А2ь
Дпя:и>н иония группа 4"_'2
Диазомин солн 71. 420
Диа.зоцця катион 397, 122
Дпазопроизводиые \20
Дна.тпропан 420
Диа;н)соодиг[еп!!я l?Q
Диазотаты 120, 123, 121
Липзотипня 4:?7
Дн'кютиротинин1 .''.97, 111. ; М, -Ijfc, 52 I, 021
Д,|,'\и>ц1];[Ц][Д|1[ -!2."i
Д
Д ''1
ДиалкилалюминнПгндриды 261
Дпалкнлиодония солн °17
К^'-Диалкнл1-идразнны 431
Диалкнлкарбонаты 611, 64 2
Диалкнлкикель 261
Диалкплопыс )фпры 328
Диалкклоксоггпеныи катион .329
Дпалкмлнирокарбоиаты Gil, 642
Дн ал к пл с ул 1,фаты 353
Диаллилфтялат Г>79
Днальдегпды 173, 067
Диамнд yi-олыюп кислоты см. Мочепипа
2, 2'-Днаминодм утмламмн ЮЛ
2,2'-Дпамипоб|1фепил 6G9
4,4'-Дпамн нобпфенил [ 15
1. Г-Дна.мшюдифениловыП )фпр 390
З.б-Дпаминопириммднн 710
Дна» 323
Дпарнлампны 107
Днарнлбромопня соли 217
N, \-ДнариЛ1]|д[)а,{ииы 118
f4N' Диарилгндра.шны 119
Дпарплнодонпя солн 247, 218
/1,нарилкарбопаты 612
Днарнлкетоиы 161
Дпарилметаны 161
Дпарплопые :>фиры 311
Днарилоксиды 311
Дпарилсульфиды 361
/1,парплсульфопы 356
Диарилхлороння соли 217
Дпасгереомсры 2^8. 6.П. 632
Днастереотопные атомы 228
Диацетпл 475. 576. 579
(Диацетоксниод)бе|пол 2 1 7
/i,n6eH:to[h,f la зен и н li.38
Днбензоилвнниая кислота 631
Дибен.юлхром 26 1
Дибенчопира.шп 637
Дпбензо1Ь,е]пиридин 657
Дибен.)о-4Н-1, 1-тназип 657
Дпбепчотиофен 633
Днбензофуран 655
Днборан 269
2,3-Дибромбутаи. стереои.юмерпл 228
Днбромкарбои 607
2,3-Днбромпент.'1||, стерсонзомерия 227
1,2-Дибромп[)Опан 116
а.р-Днбромпропиононая кислота 603
Дпбромянтарнап кислота 561
Дп-«фет-бутилпероксид 318, 330
/(нбутилфталат 579
Дивинил см. ГЬутадиен-1,3
Диьннилбен.юл 178
а.а-Дшалогенкарбоновые кислоты 634
9,1 0-Дпгидроаптрацеп L'07
3,4-Дигндронзохинолнп 697
1,2-Дпгидрокспантрахиион 531
!,3-Дпгидр<1КС1гацето1г 500, .о01, 50.)
Дигпдроксибензолы 323
4.1'-Ди!'идрокснбифснил 318
Днгндроксиднкирбоновые кислоты см. Вин-
Винные кислоты
¦1,1'-Ди1ИДрокспдифенилметан 320
1. Г-Дигидрокспднфеннлпронап 323
Дигидроксикарбопнльиые соединения ч95,
500
2, .)-Дпп1дроксилиринид1ш 70... .03
Дщ-пдрокснтетрацепхинон 21 0
Дигндроксиянтарная кнелотч см. Винные
кислоты
Дшидроазеты 635
Днгпдропира.шкы 52^3
Дхгпдропиридины 657
Дигидронирролы 655_
Днгидрорезорнпк 323, 48S
1.2-Днгидрохинолин 696
Дш-лнм 328. 332
ЦтчюпыГ: СИИТ1М 12.5. III. 170. 204. 21.1. 461,
328. 529. 533, j62. u6j
Диено-ы 491
733
р
Дпнптрометэн 37 1
L',-l-,"lii[iHTpoc()oiiii-TrMiflpJ3ii!i -173
2, 1-Дипгтрофенол 319
lVi-Диннтрохлорбен.юл 37!)
1>Д 33
Дизельное топлппо 101 Динамит 348
Дшиобутилен 119, 130 Диннтрплы 557, 560
Диизопропиламнд лития Г199 .и-Дннптробепзол 370, 380
Дпизопропплонын эфир 328, 331 л-Дпнптробен.юл J7U. 38:)
Динзоцпанаты -111, 6 11, 648 Дпнптрометэн 37 1
Днииы 137 'l"l(
Днкарбопильные соединения 17 1
— — номенклатура 173 lViДиннтрохлорбен.юл 37
--- -- классификация 471 1, :>-Дип:1тро уг..п 370, 373
1,2-Дикарбонплы1ые соединения 176 Днпуклеотиды 715
— --- получение 13;>. 476 Дноксап 303
— — реакционная способность 476, 177, Дпоксанднбромнд 340
¦178. 676 ДпоксЛ1суЛ!)фотр:к)кснд 310
— — спектры по[лощения 471) Дпоксапы 333, 330, 310
1,З-Дикарбоннльпые соединения 479 Дноксим дп;':;ет.".лл '70
--- — алкнлировапне 48,1, 1'Л Дноиспмы 177, 478
— — ацпл нрогищие 485 1,З-Дпоксолапы Л >,,*, 171
— — енолизацня 483, -1а I Дноктилфталат 37(.1
— --- квантоьо-хп.мнческпе расчеты аниона 1.2-Днолы 301', 1711
483 1..!-Лпол|,1 305
— — кислотность 483. 48; 1.1-Днолы 30.",, 190
— --- образование хелатоп 481 Дмолы 281, 30!
— — получение ,06, 17'J. 480 Днпектен 111. 172
— — пространственное строение епольпои Дммептнды 621
формы и аннона 481. 4SV2 Д-.ширрплметан 663
— — реакционная способность 486. 487, -;ч!юлы|ын момент 27, 42
675, 688. 705 -¦• — альдегидов 440
--- — таутомерии 480 — алканолоп 2,46
1,4- Дикарбоннльиые. соединения 400, 491, --¦ -- алчач"! иолоп ЙО 1
687, 688. 701 — ¦ ,1лммю-.1 107. I4S
Дпкарбоповые kiicjiotli 5 i[) алкп. [амшнж J'JJ
— -- ароматические см. Лрспдпкарбонопые -- -¦- алкпларилорых чфироп 343
кислоты _ — ¦¦ алкиноп 1 18
- насыщенные Гы7 - - -- анплппл 4 08
— — -¦¦ кислотность ,>53 — — аренон 181
-- — — получение .~57 - гллогеи'алкенон 230
— — --¦ СЛОЖНЫС чфпры 1K5 — — riiJKirelKIJlKHHOI) _'1б
пенаерзпценны!' jt)O гллогепаренон Vi't
— — ... кислотность 51H — ¦ диалтглопых чфнрещ 31'!)
----- получение 0G0 нмнд;..и)ла 677
Днкетен 163._ б.'3'j — — индола 669
Днкс-топы -i75 — --- карбонильных соединений 440, 459,
f.-Днкстоны 4,40 4E3
6пс-о:-Дп14сто!!!|1 4 79 — --¦ метанола 1'8<>
Днкетштиерлнщ-.и 6'-- — — пит]н>ал'.-;ано\1 '171
JLiiKMiiHu коьденслцпя (;35 — ... ц|!троарено:( 380
Jlu.ima — Лл',ис.ра реакция см. Дпгповьп) пиразола C77
сиитсл — -- пиридина iiS'.'t
Днмедон 1SS пиррола Ml
1.2 ДнмеркчптоОензол 363 — -enmei. С 11 118
Днметилампн 389. 3!K, 3'M. 100, ,jS7 ¦¦ -- --¦ С-О 1'Sti
н-Диметиллмпиоазпбензол 433 — -¦ — O--U '.86
Днметиламмог.пя хлорид 100 s -О 361
К.К-ДимеТ1:лан:1Лпн 253. 108. -100 -- — — N 11 М.)
N.N-AllMeTH.'Ii'.Hll.Mninsi ноднд 390 С --N ЗЯ.Ч
N. Х-Днметнлацетамнд Г)(>5 — — сульфидов 3(>5
-,3-Димстнлбутандиол-1',3 см. Пнпакоп -- - - сульфокендон 361
ДпчетнЛ1лпоксIМ 47У — -¦- фенолов .'! I j
Даметилднэгокспсиллп ЗГ>0 --¦ -- rl>ypa:ia 661
Диметплдпборап 2(>8, 26П — -¦- ^тпленокепда 331
¦'г.|-Дн.метил-1.3-дио1и'ан 133 Днпропнлами н 393
Днметнлднхлорснл.-.п 1'72 Д:1с;.х:;риды 317
Днметплкетем 461 Д.нчюрсня оптического вращения 60
Диметплметнлфогфонат З.Л ДнскерсныГ! орап.-кевыч 53 1
Д||.мстп.1овЫ|'| л\)ир З.'З -¦- фиолетовый ,K1
днчтплепглпколи см. Дпглпм: Дпсиропорциопнроваппе альдегидов 167
N.N-Дпметилпс'нгаг.амнп 2 38'J .Чпсрогаторпое пр.пчеппе \в'.)
З.Г.-Димотплннра юл 679 Дп.\ льфпды 361. 361, 365, 366
Дпмегпл:1прпдппь[ 6S7 Дмт^овалеркапонач кислпта .С.
2.2-Днметпл:.'ро:|апо.:-1 282, 285, 2Й8 Дпт ю^.рбаматы 642, 631, 652
Диметилсульфэт 352. 333, 507 д:. тпокислоты 566. 591
Дпметилсульфид 363. 36,\ 367 1,3-Дптиол 656. 6S0
Диметилсульфокспд 360. 362. 370 1,3-Дптнолан 636. 680
ДпметнлсульФоппя метил:>д 367 1,3 Дш нолия со.п 656. 680, 681
N.N'-Диметплформамнд 370. 518, 337. 598 1.3-Днтлолпюи-2 ,:S0. 5sl
— формнлнруюпи: ii реагент 502 д i гиоугольипя кислота, эфнры 612, 651
N. ^-Днмстплхлорфорчидипкн соли 587 Дифениламин 160
Г).Г)-Днмегилциклогекеандпон-1,3 4 88 2-Днфепилацетнлнндаидион-1,3 490
Днметилцнклопропап Н;0 Днфенилборбромнд 268
I .l'-Дпмегоксибепзол 31.3 Дифпцнлборпг.! К'ч-лот;. 8
1.2-Дпмстоксп-,т;:п 304, Ч.'8. 33'! N. Х'-Дифпмт и-:др;щеи 117. 419
Дпметочсиянтарпач кислота 336 Дцфенплдимстокспспл.п1 351)
734
Дифенилдпхлорг.план 27^
Д кфегг и лд и хл орет:1 jjh л п 261
Дифечилыетап 108. Hij
Дифенилмотяноркс ь р.жители
Дифеннлоиып гфир 1 У8. ; I ;.
ДифегШЛОЛПрОП;1 П СМ. Д;!,.!1
Дифенилп1'.К|-,и.'.п:дра им 11.1
Дифе»илтптамдпх.:о|Л!Д --63
Дпфеиил хлор метан 232
Дифенилзтан 20!)
Дифеп >хппоп 5 ?1>
ДифТ0рХЛ0рМ<'Т;1П 2-14
Дихлорацетилеп 216
о-Дихлорбеп::ол 239
л-Дих;1орбсн:»ол 230
Дихлордпфепнлт[;пхлор\;еТ1гл.м
245
2,2'-Дихлордн'Л'пловын эфир
(Дихлориод1бсн:|ол 2 17
Дихлоркарбеп '!;>',, 3i.'7. 302. 6
Днхлормукогопля кислота 707
2,4-Ди хлор пиримидин 708
533
З'.З
Изатин С72
И:кк'\:ц-;Г)\тпр,-'7 578
lbo.-.MH.-.oiii.iii сгн.-.т
И:Ю1..l.'icpn; по: ;ii; и:и
87
1, ?Я">. ?Э7. 3 13
тл ;i38. o,:j 312,
(lOif
310
(ДДТ)
330
607
Дихлорэтан 235. 2И, 401
Дициандпампд 616
Дициклог(Ч\силкарбодн1и"ид 6 49
Дициклопептадиеи 167, 17.)
Дициклопенг,.;1Н1мн!лжелело см. Ферроцен
Диэтаполамнп 401
Диэтплалюминш'пндрид 259, 260
Диугиламип 393, 393
Диэтнлдисульфнд 363
Днэт и лен гликоль 332
Диътилмалоьаг 561, 578
ДиэтнловыН чфи]> 2!Я, 253, 328. 329 331,
Ди'*тилсилап 271
Дитгплсульфнд 365
Дпэтилцилк 256
Д[готил(р>-угилмеркапто':)Тил)тпофосфат 331
Додекакарбоиил дпванадия 26!
Додекап 87
Донорно-акцепторные комплексы 320, 530
Дрепссина, сухая перегонка 13, 14
Древесным сяхлр 507
Дубильные ве;цеста 12, 615
Дурол 178, 180, 181
Цнамнны 101, 405, 406. 119
Енднолы 477, 1У8. ,'I2. 513
1:ндио;[ят-нон -177. 407
Кнолплация 310. 311. 114. 483, 481, 401, 300
Р.нолы 30S. 309
Енольная форма -"80, 481
Ннольныс эфиры 312
Жело.чо
— ."Т-КОМПЛСКСРзЕ 266
— понтакарбоннл 2(i6
Жирные кислоты 10
-- ¦- незаменимые 333
Жпрм 10. 13. 306, 3 59. Г.81
Желатина 621
Зайцева мраппло 23-1
Заместители, [трапило старишнетпа 83
ЗанО.:ич'ич>а реакция IL'C
Зарин 331
Защита
¦— ампnorpyinijii ИЗ, 521, 633
— гндроксплыюн группы (ПС
— к.-фбокгпльно'л группы 174, 610, 633
Зеленый lumui,,; O.u'pa 333
3li\i;i:-.;i 313
Зомап 332
Зомп.чя планка 16
у
Мтооутплеп 101. 119, 129, 1 ".3. ЗГИ
Изобутнловыь спирт 115, 282. 2S3. 2Э7, 510
И.чобутнр.лы ;И4
l-lj0KpoT0i;cr-r.я к;.с.-отл 530
11:шксп:;ол ь.ЗО
M.to."c;uuin 62 1
H.ujM,,c.;>iiiai- кислэта 338. 312
ИломасляныИ альдегид 113, <137, 4Н
11.зомср[[.чацпя
-- а.'.кипом 00
— карОокатн;>:юп 100. '_' 11J
— фо!охтгнчоская 561
— |;нклоа.;км;оп 171, 173, 174
— цис-транс- 108
Изомерия 21
— атропп.юмерпя 107
— г»ллг-нтн(;я 103
— геометрическая 423
— днначшчесьля см. Таутомерия
— зеркальцам 23, 93
— кольчато-цепная ;П8, 4ЯЯ, 633
— конформацнонпая 0t, 170
— оптическая 25
— структурная 87
-- з;и<1прочнр?1,- 228
Н;и)ник.Aтпно1)ая кислота 694
Нлонитрплы см. Ичоцилгшды
Н.чопитрнльпая реакция 601
Плонптролосордипения 3S6
Изооктап 130
Нлопснтам 1.43
И.чоиентены 133
Илопрен 133. 1 П, 171
НзопреноиыП каучук 110, 143
И.чопреноиды 172
Пзопропнлбеп.чол 105, 313
И.чопропплопьп! спирт 282. 285, 29G. 331
Шотиоцианэты 154 '. 630, 631
Н.чофталспаи кислота 357, 562
Илохнполмн (M7, 693
— алкилнронанке 690
— н.чаимоде11сгнне с. "электрофплыгымп рса-
гомтамгт 608
— гидрирование C!>9
— окислепно 609
— основность 608
— получение G(t7, 700
Пзохнполимия кагион 698
Илоцианаты 387, 301. fiO_'. (ill. 613. 614, (i(C.
647
Изоциншгды 397. 567. 601, 602
Пзочлектрическая точка аминокислот 620
11л иды
— сульфонин 367
— фосфомин 27S
Нмпда.юл 1M6. 1O4. 677. 678
Имида.чолплалатш li:!4
Пмпдазолия кагион 677
11мнда.1оD,5-Ь|пнридмц 658
Имнда.и)! 1.5 - с: | пи пи ми дин (J58
Нмидо':)ф!фы ка[)боно!>ых кислот 566, 532,
592, j 91
Лмнды карбоиопых кислот 363, 582, 383
Импнаты ИЗО. 255
Имипы 398, 104. 419. 469. 503
Иммониепые соли 406, 470
Инверсия молекул 170, 394, 500
Пндаэол 650
Индандиоп-1.3 175. 1S1. 489. 1Й0
Ппдаптрнон-1,2.3, гнд]>ат 490, 622
Индантрон 531
Ипдп.-о П. 11 1. П72
Индик;1!! 671
Индикаторы 13!
1Ь'Д>ь;';|Л 671. 672
Ипд.1.; <',,:,. г,68. 671
— ацпдофобноегь 670
733
— взаимодействие с злсктрофильиыми реа-
реагентами 670, 671
— кислотность 070
— получение 668, 669
— энергия ионизации 669
-- физические свойства 669
Индолилалалип 624, 672
Индуктивный эффект 28- 30, 107, 190, 2-10
Инсектициды 695
Инсулин 12, 627, 629.
Инфракрасные спектры поглощения 53
Подарены 4'2а, 4 26
Иодбензол 1Я6, 239
2-Иодбутйн 231
Иодирование аренов 238
Иодозилбепзол 2-17
Иодозилеоедипения 247
Иодоння соли 247
Иодопированпе 2-17
Иодуксуспая кислота 604, 605
Иониты 17
Ионные чары 68, 69
Иопол 318. 323
Иоп-раднкалы 67, 531
Иоцича реагенты 1п6, 255
Иприт 367
Итаконовая кислота 556, 560, 561
Казеин 626
Калнй-натрнитартрат 614
Калийоргаппчеекие соединения 258
Калориметрия 39
Каменноугольная смола 193, ПM. 202, 208,
209, 211, 212, 313, 314, 660, 6G8, 687
Каменным уголь 13, 14,
Камфеп 172. 299
Камфора 457, 458
Канниццара реакция 467
Канойviческие структуры 31
Канцерогенные соединения 212
Каи рол актам 194, 376, -157, 4 72, 350, 584,
588, 617
Капрон 589
Капроновая кислота 538
Каптакс 68Г>
Карбазол 655, 668, 669, 671
Карбаматы 641. 6-13
Карбамид см. Мочевина
Карбамндные смолы 456, 645
Карбамнновые кислоты 587, 6-11, 64 3
— эфиры 6-11
Карбаиионы 89
— образование из металлоргапическнх сое-
соединений 250
— трифеиилметапа 199
— цпклопентадиена 167
Карбоны 89. 90, 102, 234, 397, 430
Карбиды 91
Карбпи 155
Кйрбодннмиды 622, 633, 641, 6 17, 6 18, 649,
650
Карбокатионы 79, 89, 112, 173, 233
— адамаитиЛ-катион 176
— аллил-катиои 127
— треш-бутил-катноп 119
— перегруппировка 100, 294
— стабилизированные 173
— трифепилметил-катиои 200
Карбоксилат-иоп 5-13, 546, 54 7
Кнрбоксплироваине 60S), 635
Карбоксильная группа 536, 54 3
Карбоксильный радикал 547
Карбокспметильпая группа 540
Карбопилнроваиие
— алкаиолон 293, 541
— алкенов 541
— алкипов 541, 550
— галогенуглсводородоп 541, 555
— гл и колен 557, 559, 500
— диметиламина 587
— метанола 048
— тетрагидрофурана 560
736
— эфироп 5-i I
Карбонильная группа 435
— — ашюн-радикал 1-1'2
— — поляризуемость III
— — природа связи 1-Й, \\1
— ¦ - расчеты по методу МОХ 142
— — спектры поглощения 143
Карбонильные соединения
— — алкнлиронание 106
-- — ареной -137. -164
— — насыщенные 13C, 439
— пееполнзнрующпеся 113
— - ненасыщенные -136. '158
-¦ — а.^-непредел ьные -136, 158
— - - номенклатура -137, 138
-¦ — таутомерия 311_
Карбоповые кислоты .K0
— — бромнровапие 601
— — взаимодействие с аммиаком н амина-
аминами 516
— — -- галогенидамп серы н фосфора 223,
516
— — -- спиртами 516
— — высшие 54 9
— — декарбокенлиров.тние Г>-17. 55 1, _Г>58
-- — кислотность .>13, 5Г>1, Sol, 558, 562,
605
— — классификация 536
— — номенклатура 538
— ¦— пиролиз Са солеП 4 10
— — получение 293, 434, 155, 193, 52.", 526
— -- содержащие различные функциональ-
функциональные группы 602, tiO3
— — соли П44
— — функциональные производные 364
— — хлорирование 601
— — электролиз солей 517
Карбораи-10 270
Р-Каротин 141
Каротиноиды 581
Катализатор 71, 75
-- иммобнлпзнрованпып 7о
— межфазиьп1! 232, 74
— xnpajibiibjii 632
Катион-радикал анилина ПО
— арилампнои -111
— бензола 215
— N. N, N', .\"-тетраметил-/г-(Ье!1пленднамина
110
— тетратиафулызалена C81
— этилена 38
Каучук
— натуральны!'! I 11
-¦ синтетические 13, 1-12, 113, 196, 2-12,
245, 600
Квантовая органическая химия 12
Кератин 626, 629
Керосин 101
Кетеп 461, -163
Кетепы 136. 161, 547. 569
— взаимодействие с нуклеофлльнымп реа-
реагентами 162
— димеризацмн -163
— реакции циклонрпсоедписнин KJ3
— получение 462, 52о
Кетнлы -169
Кетоальдегнды -175
Кетогруппа -1:)."), 138
Кетогексозы 510. 512
Кетозы П04
Кетокарбены 525
Кетокислоты см. Оксокнслоты
Кетокспмы 171
Кетоп Мпхлера 11-1
Кетоны 135, 438
— ароматические -164
— ацплиропапие 480
— взаимодействие с алкппамп 150
'— — дшор-ачннамп 40-1
— галогепидамн серы и фосфора 152
г— металлоргапичесиимн соединениями
250, 255
— — иптроалк.шами 376
— — нуклеофнльыыми реагентами 445, 1-16,
449, 152
— — фенолом 3:21
— — фосфораиамн 278
— восстановление до пи и а к ом он 301
¦ - гндрпропаппе 284
— насыщенные, ольдольпая конденсация
4 16, 448
— лми пометилировапне 523
— — бромнрованпе -145
— — взаимодействие с нуклеофу1ЛЫЕЫчн
реагентами 4 15, <MG, 4-18, 452, 480
— — постановление 102, -103
— - гидрирование 445, 440
— — еполн.чацин 444
— - кислотность н основность 4 1 I, -143
-' — окисление 45.)
— - получение 250, 375, 439, -140, 5 17,
till, 637
— ненасыщенные, взаимодействие с нуклео-
фпльцыми реагентами 460
— - спектры поглощения '1,19
Кетопеитозы 50I
Кижнерп — Вольфа реакция 47.\
Кинетическми контроль реакции 138, 203
Кинетическое уравнение реакции 6^>
Кислота сопряженная 69
Кислот но-осноипые раиновое и я 69, 70
Кислотность, С 11 69, 251
— алкилфосфоииевых солен 277
— ацетиленом 1о5
— ацилоиноп ЛОТ
— барбитуровой кислоты 708
— - р-днкарбонпльцых соединении 483, 484
— карбонильных соединении 312, 4 11
— р-кетоэфнров 636
— малонового эфира 579
малонодннитрила 600
— нитрилов 599
— - пнтроалкапов 373, 374
— сульфоксидов 362
сульфопнепых солей 367
— сульфопов 359
-- трифеиил.метана 199
— цнклопептаднена [67
Кислот ность, N 11 69
— нденипа 711
— азоареиоп, иротонпрокаиных 433
— ал килями нон 399
— амидов 585
— амиднпиП-катиопа 593
— - .ЧМН1НЖИСЛОТ 61!)
— аммониевых катионов, замещенных 395,
116
— арпламнноп 4 13
— арплуидралнппевых катионов 418
— гидрокелмоных кислот 592
гуанндипиепого катиона 610
— гуанина Т112
— изохп ноли нин-катиона 698
— пмидазола 677
— импдов 585, 588
— - и идола 070
— ппрячола 1O7
пиридинни-катиона 090, (ИL
— пирнмидн ни ii-катиона 706
пиррола 6E1
— пурина 710
— сульфониммдоп 358
— тетIг-пола fifili
тна;юлип- к.тгиопа 68 I
— 1,2,3 трназола C86
— урацила 708
-- нпточиш) 708
Кислотность, О Н 69
— - алкаполон 288
— ллкилоксопин катионов JQO
амидов, протопи роп а и и ill х 58 |
— амппокпелот (И9
— аренкарбомоных кислот 5, 562
-— арсполоп 317, 324
— гмлогепкарбоновых кислот 6 '15
— гядроксикирбопоиых кислот 610
— гпдроиероксндоп 349
— гидрохинона 326
— глицерина 306
-- р-днкарбонильных соединении 183, 184
— дпьарбопоных кислот ооВ, 560, 562
— дполов 302
— енолов 3i1
карбонильных соединен п ii, протопиро-
пйпных 444
— кпрбопоных кислот 5A, 551
-- 'Х-кетокнелсл 6;Ui, 039
— ("i-кстоэфпров 63ь
— нитроновых кислот 374
— пптрофенолов 381
-¦ окснмов 4 71
— окспна 700
— пирокатехина 321
— - резорцина 325
— сульфнноных кислот 362
— - уридниТ/-монофоефорно]"[ кислоты 714
— фенолов 31 и, 317
— фосфпноньЕХ кислот 1'80
— ¦ фосфоповых кислот 279
Кислотность. S II
— тполов 3C5
Кислоты, апротонпые 70
— Льюиса 70, 71, 188
жесткие и мягкие 73
— иротонодонорпые снерхкислоты 99
— - С — Н см. Кислотность, С- Н
— N 11 см. Кислотность, N —11
- О — Н см. Кислотность, О Н
— S 11 см. Кислотность, S 11
Классификация 81
Клатраты 645, 616, 650
Клемменсена метод 153
Клпйзена конденсация 180, 577
Кнсвенагеля реякцн н 551, 559
Кобальта
— - л-комплексы 206
— тетракарбоннлгидрид 1 15, 293
Коиалептпая снизь 2.»
Коналентпый радиус атома 4 1, 15
Кодеин 701
Коками 695
Коллаген 626, 629
Коллндппы 687
Кол л од и ii 521
Кольбе — Шмшпта реакция 609
Кольбе электроенптеч !I
Кольчато-цеппия изомерия 4 98, 499, 506
— — таутомерии 50li, 509
Комплекс
— активированный переходный G6
— донор и о-акцепторный 530
— с переносом заряда (КПЗ) 53, 76. 113,
185, 200. 3'2b, 383, 385, -НО, 123, 530, 531,
001, 071. 073, 702
-- л- 76, 117, 185, 530, 531,
— а- 185
Комплексопы 620
л-Комплексы переходных мсталлоп 263 —
267
Комплементарные ntipbi 716. 717
Коп ротаторное пржцепне 1 <)9
Константа Ьрауна 79
¦ - Гаммета 79
-- индуктивная 80
— заместители SO, 19Q
кислотности 70, 77
— кочплекеообрачоваппя 5 41
— прямого полярного сопряжения 70
— равновесия 65
— реакционной серии 78
— скорости реакции (И
— тауточерного pannoiiecnn ЗМ
¦ Тафта 79
Конфигурации
г|(кч)лкл п.ы 93
— — номенклатура 220, 228, 2 31
737
— дпгопалиная 36, 43
— обращение 231
— тетра-)Д5)ическля 30, 13
-¦ трпгональная 36, 15
Когформаци;: 93
— алкэпов 01
¦— галогеналкалов 227
— заслоненные 91
— :>аторможе!шые 03
— кмраноз 50(i, 309
— ипклоалкг!нов Hi6, 168. 170
Конформеры 93, 227
Копформацпопиым анализ 9!
Коразол 086
Коричная кислота 539, Я5-1. 53.")
Короне» 202
Корреляционные уравнения 77, ВО
Кантона чффекг 61
Кофеин 712
Крас-мтели 212, 387. '135. 531, 53.1, С72
Краун-эфнры 7 1. 333, 310
Крахмал 517, 519. 520
Крезолы 313. 31 I. 322
"W lid'.' ч иг- uR
271
Д \ |U M \.Jd J i'l к/ L
Крекинг У8
Кремниевая кислот;!. -?фпры 350
Кремиикорга пическне соединения
Крипгаиды 7."), 403
Кристалл H3iiu.it и 1 IS
Кротоновая коидомслцмя ^"|8
Кротоионан кислота о'М)
Кротомовый пльдегид -137, 438, 4G1
Круговой дихроизм G1
Ксантпи 711
Кс а итоге паты 642, 651
-- целлюлозы Г»2 (
Ксаптопротенмопая реакции G27
Ксилаиы 507
К^мглонолы 322
Ксилоза 50Г1, 307
Ксилоза 505, 507
D{-t ЬКсплоиирапоза 507
«-Ксилол 177. 180. 181, 190, 195, 562
л-Ксилол 177, 18A, 193
я-Кснлол 177, 180, 195, 563
Куб in 162
30, 131
Лавсан 563, 580
Лакримлторы 493, 49*
Лактамы ГNГ>. 383
Р-Лактлмы 622, 084
Лактиды 610
Лактоза 518
Лактоны 463. 501, 573, 596, 010, 611
Левулоза 518
ЛеОцшг С24
Лецитин ;>81
Лнганды в л-комллекспх переходных метал-
металлов 264. 26d
Лнгпин 13
Лнзергииоипя кислота 673
— — дн'^пглампд (>73
Лизни 624
Лнкопми 1 13
Лпксоза 505
Лимонен 172
Лимонная кислота 9, 520, 5G1, ОН
Ли налом 298
Липдан 236
Лут1'Д1:ны G87
Люмипал 70
Люминесценция 52
Ма1'н)п"[ор1'ан1гческпе соединения 15,1 °'"i3
251. 23Г), 335, 361, 387. 541. 591
.Макроцпклмческпс ацплотны 577
Miuatipaila реактгя 304
Малахитовый зелены;! 535
ААалепнальдегчгдная кислота 705
Мглеппонам кислота :>5б. 559, ;>С;0. 561, Git
Малепновын лшндрп^ ПН, 208, Г>61 57'
580, С65, 667
Л\,.'лонал1.дег-|гдная кислота 6ПЗ_
Малонопая кислота 163, 551, ,'M6. 558
-- конденсация с альдегидами 617
Малоновып пльдегид 4 7П
Мало1!опый --Лир 488. 578
— — алкилировапие 579
— — кчгслотность 579
— - - синтез гетероциклических соедине-*
iinfl 675, 705
Ма.'юподпнитрил (i00
Мальтоза 517. 518, 31!)
Млнглпа л-коммлексы 263
А\анпн\а основания 523
-¦ реакция 523, 070
Мапно.ча 509. 512
Марковннкова правило 118
Масла
— - высыхающие 553
-¦ растительные 552, 553
Маслушая кислота 538, 5"B, 014, 519
Масляный альдегид 437
Мясс-спектрометрия 62
Медьоргапнческпе соединении 190, 252, 460
Мсарва'/на — ПопиОарфа — Всрлся реакция
153
Мезитнлен 177, 180
Мезовиннли кислота 613. 614
Мезомерип - - речонапса концепции 31
Мезоме-рпя 30
А1езомерные формулы 31
Ме.чомернын чффект '.'Э, 30, 190. 210
Мезоформа 31, 228
Мезочритрит 307
МржфаинЫ!1! катализ 232, 400, -103
Ментол 300
Меркаптали 151
Меркаптаны 363
Меркаптпды 360
Меркаптобеизол 363, 365
2-Меркаптобеизтназол 683, 68J
Мерклигофос 351
Мерку|)и[ювапне 257
Мерсоляты 360
Мескалин 401
Механизм реакции 63
Метакрилокая кислота 538. 552
Металепсия 22
Л\еталлоргапнческие соединения 10, 91, 229,
249, 284, 110. 5,55
-- — переходных металлов 262, 261, 26Г>
Металлоцепы 107
Металлы органические. 211
Метан 10. 11, 21, 20. 37, 86, 89. 91. 92 91,
98, 102
Метапдпол 302
Метанол 13, 282. 281, 286, 288, 290, 2!И,
2!M, 331. 101). 11 !. 156. 318
Метансульфопован кислота 351
Мегантнол 363. ,'Hi-i, 365
Метплакриллт 57!)
Мстил а люмининтри хлорид 258
Метнлаль 4 74
Метиламин 389. 393. 395. 400, 109
Метпламмонпя хлорид 400
К-Метиланплчн 390
Метплбордихлорид 268
МетмлОромпд 22 1. 231
2-Метплбутадиен 1,3 см. 1 Lumpen
2-Метнлбутапол-1 282, 283
3-Мсти л бута пол-1 _8 .', _8 i — — л ре поп 16 I
2-Мстплбутапо;;-2 282, 285 — — насыщенные -130
Метмл-ш/^т бутиловый '^фнр 328, 331 — — ненасыщенные 4 58
З-Метилгекс^п, энаптиомч1;)!,! 93 - - - - номенклатура -137, 438
Метил герм a пиптри хлорид 2bl Мйиокарбоповые кислоты 537
Метплгпдразп-! -Я 7 — ароматические см. Лренкарбонопые
Метилщдрсперокспд 3-18 кислоты
Метилгидросульфат 352 — ¦ кллссифпкаци',1 1337
Метил-^-глкжышр.ию.чпды Г> 1 3 - - — насыщенные .1 1П
Мпнлдихлорфосфип 275 — - - ненасыщенные 550
Метиле и 8!) — — номенклатура .'>37
Метиле на мин 4 70 Моносахариды 285, ¦17-1, 504
Метиле и поди д 1 0 — деструкция 51 б
Мсти лепит tin 1 0 - - кольч^то-цепная^пчомерпн 7H0, 509, 510
Метплепован группа актгшнан -18.";, 555, ."о8, — копформлцпи .109
559, о7й, (J00 — мут;| ротация Г>Oti. Г) 1 0
Метилентрпфснплфосфоран -78 - - ii;ip;i кипы пне углеродной' цепи 7Л (>
Мотилопфтор1:д 2.1! -¦ прос ср-тетненпаи изомерия 505, 5CS
Метилен хлорид 231 Морфин 701
Метилиодпд 2'.1 i А'оетпконые у1\'!е1юдороды 1 GO
Метиллитин il5l. 77. 302, 3G7, 309, -113 Моченая кислот,i 710, 712
Мети л мета к рп лат .180 Мочении а 10. (HI. ОК. (МП
Метил нафталины 02. ЧП. 1 - обрлзопанпе кллтрлтои G13
Мет)глопы,'г орап/кеиып !3.1 — основность 61 I
Метиловый спирт, см. Метанол — применение. С5 iГ»
2-Мот)г.чие!|теп 133 - - синтез гетероциклических соединении 70П
4-Мотплпентен-З-он-:! -1G1 Л\(ло|цпе средств л, спнтегические ЗГИ), 3iiO
Метплпирндппы 092 Мур.тышал кислота 9, 10, 533, 7»Л2, ."i-M,
N-Метил пир рол пдоп .8 5-17. 5 18
2-Метилпро1:;имль '137 — — амиды ГK7
2-Метил проча иол-1 282, L'85 ... ^фнры 5и !, 635
2-Метил;1рО1:;тол-2 2S2 .Мураиьинып ал<здег"ид см. Формлльдспгд
Метилпропеп см. II ;обутилен Л\утлротацин_50(), 507, 508, ,А I, 518
К-Метплпроппламин 3S'J Мыло 5-!9, о5-, 381
Метилтитпнтрпхлорид 2G3
Метнлтрпхлоргсрман 201 Наилоп-О 589
Метплтрнхлорсилап 271_, 272 11н{|ЛОН-7 580
Мегнлтризтоксисилпп 350 I Ьтмлон-11 ")89
5 Мегнлурацн-'! 708 11;: .*: л он- 0 ti 589
Мегилформнат ЛЫ. 587 Плгрипмедигпртпят G1 -I
Мотилфенплоиьп"| ъфн р 312, 3-11 I laTpni'iopr.inn'iecKne соединения 229, 251
Мепыфешису льфпд 3G3 11 афта л и и 17-1, 201, 202
1 -Мет и л-2-фен н лчтп л а мн н 103 — бромиронмние 205
Мегилфто1)пд 22 1 — п'дрп ронян ire 20li
МетплфосфинопоГ! кислоты фторангпдрндп — n:iTpoi>ainie 201
изопропиловы:"! -)фнр 3,) 1 _ - - окисление 2(Mi, 5:'8, 562
----- нннлколлловыП ^фир 352 — сульфнроиаппе 20"
2-Мети л хи пол им 700 — мпергпя иони.чялшн 203
Мстил хлорид 224 2-ПчфгалпнкарС'.ль.'ичид ^37
М^гплцеллю.чольв 328. 331 1-Ь'|фт,1Лнпх;,1)бо1;о[п»1е к г. слеты .".5 f
Мегплътилкетон 438, •] И Плфгплиисульфоношие кислоты 203
Метионин G24 [',- 1-[;-,фт;)льдегпд 137
Л\етоксп карбон ил хлорид 128 Нлфтены 106, 168
Механизм реакции 03, 7 ! 11 афт и л.) мм мы 411, 115
— - способ изоброженп >i 75 г/.-11;н[ш)Л ilOf», З'^З
Мнллона реакция 627 ft-Нпфтол 309, .'ИЗ, 32.4. 414, 415
Миозин С26 ПасЬгохиноиы j'7, о28. 530. 533
Михлеря кетон 41 I Небеп.юпдпы" я i)OMjr:i4ec:;i:o системы 213
Михлера cnniiii 535 Псдооксид углгрода 4(j.i, 559
Модели атомов и молекул 43 Незаменимые аминокислоты U23
Молекулярная биология 1:! - - ,м<рпые кпе.логы 553
Молекулярные орбитллп 37. 38 \ 1ср"по-11яри.'штического депстппя яды J51,
-- • аллпльнoii системы 12G ,152
— — анилина 408 Н.'о.юн Л И5
-- - ;i ни она [^-дпкетопа 483 Но кичнилош,!!! спирт :?8'!
— — бензол^ 182 _ // •_.ч:мн--ьа днпзо^ч-тод 426
— -¦ Сутлднена 13.) fi'фи речкнии 375
— -- епамииои 405 11сфг:> 13, 89, 101
— — карбонильной группы 442 Пп:<ечя
— ¦ - нафталина 203 — л-ком!гтс:<сы L'0'З
— — нитробензола 381 — '.ч^рМхГ'бопил 2D6
— нптро! руины 372 Никотин 11, 691, 6'J5
— — нитрона;-нина 3>7-\ Пт':о1 иись.'.я кислот;) CL4
-- -¦ фенола 315 Ичпгпдрип '190. (J2H
— ->- от плена 1 25 i Ii:nri:i|)Hiioniin реякцн и 02 'Л, 628
Молочная кислота 9, 520, 010, GM Нитраты 347, 3 18
Мо:.о'1пып cax;ip518 -- т^"[пюло:'.ы ,г-21
Мслнрноэ iipauieniie GO Нпгрклы 2'Z'J, 567. 507
Мол1,о'зоиид 117 — 1 пдрнрои.'мше .:>90
М-люгидрокс и карбонильные соодннеппя, — гид|)^л]м 55 ', 5."; 5, 557
классификация 495 — кпелот.ю.ть 509
Мчнокгфбонпльпые соединения 436 ^- оси^иность 59U
739
— получение 471, 587, ."97, 598
— присоединение нуклеофнльных реаген-
реагентов 250, 253, 592, 593. ."98. 599
— реакции ot-углеродного атома 599
— циклопрнс.оединенпе 080
Нитриты 316, 3G
Питроалканолы 377
Пптроалкапы 368, 3G9
— - анион-радикал 373
восстановление 373
-- галогешфоваиие 374
-- кислотность 371
— получение .4G9
--- расгцеплснпе кислотами 373, 37."
— реакции с карбонильными соединениями
37 1, 373
— спектры поглощении 372
Интроалкены 308, 377
N-Пптронммпы 39Й
о- Нитроанплн н 115
и-Питроаннлин 379. 109, 413, 413
11,'Проарсны 308. 378
— взапмодет-твпе с иуклеофнльнымн реа-
реагентами 3S3
— восстановление 381. 107, 132
— получение 378. 420
-¦ полярность спидеи 380
— спектры г-.о1 лощения 380
-- сродство к электрону 380
Мнтроли:таШ1лиды А 1 IS _
Питробензальдегпды 38.~>
Нитробензол ЗОЯ, 379, 380. 381. 100
н-Нитробен:юлдпазопия тетрафторборат 420
и- Митробе1иолдиазотаты 12 t
¦t-Нитробнфенпл 1 97
Нитрование алкапов 309
алканолов, дполоп и триолов S'l7
— алкиламппав 398
--- ацетанплнда 413
-- арепов 378, 381
— бензойной кислоты 385
— ¦ бнфенила 197
— галогенбенаолов 213
--¦ р'-дикетонои -180
— ннданднона-1,3 490
— индола 071
-- N-оксида пиридина 692
— пиррола 065
уротропина !30
— фенола 319
— фурана 66-1
— фурфурола 067
— хнполнпа 698
¦ - хлорбензола 213, 379
целлюлозы 521
— диклогексана 370
Нитроглицерин 307. 347
Нптрогруппа 27. 268. 371
— природа связен 371
расчеты по методу МОХ 372
Мптрозирование алкаполоь 316
— злккламинов 397
— алкнлгпдразипов 418
— ct-аминокислот 021
— ариламиноь 411
— N-ациламнно» 428
-¦ гидразидов 390
— пнтроалкапов 375
— ¦ уретапов G44
— фенолов 587
Нптрозирующпе реагенты 317
Иптрозоалканы 373, 386
N-Пнтрозоалкплмочевины 429
N-Интрсноалкилсульфонамнды 429
N-Иитрозоамипы 397. 411. 421. i.'C1
Нитрозоарены 381. 382. 387, 1J2
Нпт))озогруппа 317. 385
п-Пнтро-М.Гч-днметпл-аннлпп 112
Нптрозокраснтсли 387
1 -11птро-2-пафтол 387
Интрозон1!троалканы :$7.">
Нптрозоиня катион 71, о 16
ещества
Нитрозосоеднненпя 371, 373
N-Нитрозоуретаны 014
Нптрозофенолы 385, 387, 533
2-Питроипдандноп-1,3 190
3-Нитронндол 071
Нитрометан 369, 370, 373, 371, 376
Нитрон 600
Ннтронат-нон 37 1, 375, 370
а-Нптропафталнп 201
Нитроннн катион 71. 317. 378
- - тетрафторборат 379
Ннтроноиьге кислоты 37 1, 373
-¦ ¦ эфпры 373
1-Ннтропирсн 211
ее- Пмтроппррол 603. 665
Нптропропаны 369. 370
Иптросоедннения 308
Пптроалканолы 370, 377
Ннтротолуолы 381
п- Нптрофеинлгидразнн 172
Питрофеполы 319, 383, ['.81. 385
Пнтрофуранопые лекарственные вен
(.08
З-Питрофурфурола днацетат GG7
Пнтрохлорбепзолы 379
Нитроцеллюлоза 318. 521. 378
Нптроцмклиалканы 30S, 369
Ннтроцнклогексан 370
Нитроугап 369. 370, 371
Ннтро.)тплрн 377
Пптроугплпрованне 377
Поноклни 023
Номенклатура 83, 8t
гтс-реохнмпческая 225, 497. З'Л
Поили 80
Норсульфазол 11G, 081
Нуклеиновые кислоты 12. 712, 715
— - днухцепочечпые 71G
— — ицформацнонпыс 717
— - одпоцепочечные 710
— — структура 710. 717
— -- транспортные 717
Нуклео;шд-3'-дифосфлты 71 4
11уклео:шд-3'-трнфо"фаты 7 1 4
Нуклеознды 712, 713
Иуклеопротеиды 026
Нуклеотнды 712, 71 1
Пуклеофилыюе замещение 230. 293. ЗЗЭ
— - бимолекулярное 230
-- — внутримолекулярное 333. 331
— -¦ гндроксильноп группы 291
— — мономолекулярное 230
- — с сохранением конфигурации GOG,
6П
— - у .чр'-гибрндного углеродного атома
24 2
— — - - углеродного атома карбонильной
группы 51)8. 571. 57.1, 585
П> клеофплыюсть 71, 231
F 1>леофнльные реагенты 70, 71, 250
Пуклеофнлыюе присоединение к гюмид-м
3:C
Пуклсофуг 72, 231
Чью mi на формулы 113, 227. 2.S
Одпоэлектронпая плотность 127
Озлзоны 501, 507, 512
Озоннды 117
Очопнроваппе 117, 193
Окисление
— азоарепов 431
— алканов 98
— алкаполов 289, 139
— алкенов 439, 590
— алкеполоп 550, 439
— алкиламипов 398
-- алкпларенои 404, 55'J. 502
— алкнлппридипов 6J1
— алкипов 476
— алкннолов 459
— альдегидов 451, 593
— альдогексоз 51I
740
- альдопентоз о07
- .1 л ючн и им органических соединении 28 i
- а-а м иноки с л от 622
антрацена 208
- аренкарбальдегидов 168
- ¦ «фплзмниоп 379. 110, A2, 527
--- ацетальдегида 572
- ацнлоиноп 197
бензоина 179
- - бензола ГO2
- ; борорганических соединений 269, 281
- иутеноп 500, 572
¦ гпдразоарепои -119
гидразонов -4 73
гидрокси карбонильных соединении -176
- г/-гид роке н кислот 63-1
гидрохинона 326, 53 2
гли колеи -170. -С90. 557, 608
-- глицерина 500
N. V-диалкилгидразинов 1 И), 131
-- днарнлчетапон 1П8, 16 t
диарнлуганов 476
- диолоп 303. 30), 176, 490, 557, 008
- - дурола 363
изохинолина 699
- индиго п71
индоксила 672
- карбокенлат-иопа 91. 5G
каталитическое 553. 562, 563
кетопоп 135, 176, 596
- - к с и л ол о и Г) 6 2. Г) 6 3
кумоли 31 1, 350
- лит и и органических соединении 251
чалеиповоИ кислоты 60'J, 614
метила рил кетон он 553
- - муравьиной кислоты 5*18
- - нафталина 206. 528. 502
- - ннтрозосоедипепип 386
а-оксокнелот 636
- пентоз 507
пи ремj 211
-- пиридин.ч 091
пирогаллола 327
- - пирокатехина 325
пропена 139
псевдокумола 503, 373
солями палладия 11 7
- сорбозы П13
сульфидов 307
-- тетрагмдрофурана 608
ТМО.1ОН 366
- фенаитрена _'О9
фенолов 31 7, 327
фоефпнов 277
фумароноп кислоты 61 \
- фУРФУРола 572
хиполипа 009
цнклоалкапонон 5.'7, 53'J, 360, 596
- щавелевой кислоты 558
этилена 336
Ок1К'Ь мезитила 161. 488
Онгазолпдп пы 68'.'
Окггиол 636, 081
Оксазолни соли 681-
Оксалаты 358
Оксепп и *'M8
Оксетаи 'Mil, 6
Оксетии 655
Окенгруппа с:ч. Гндроксигруппа
Оксид уч'лерода 2G5
N-Оксид нлкнлнчипов 398
- - пиридина 091
хниолииа 699
Оксикислоты см. Гидроксикислоты
Оксич ацетона 471
Оксиметилированне см. Гидроксиметилиро-
п а пне
сс-Оксиминокарбонильиые соединения 523
О^сич циклогексаиома 472. .189
Окснмы 373, 386, 119, -171-
- кислотность и основность 171
- перегруппировка 171, 17:?, 017
— пространственная изомерия 471
Оксмн 700
Оксппролн н 025
Окси ран 332, 65 t
Окси реп 05 (
Окснтоции 629
р-Оксоальдегнды 181
З-Оксобутаналь 17.i
Оксогруппа 4 35, -138
Оксокис.чоты 631
получение 631 ИЗО. (ЯЧ
--- хичическне cboi'ictb.) 6.10 li.!8
р-Оксокнслот сложные эфиры GTj
— — — алкнлироваине и i-n\i шреванис
6Л7
— - - - - - гидролиз 635
— - - - котонное расщепление 0 !7
кислотное расщепление C38
— - - — кислотность и таутомерия 636,
637
— копденсаци я с альдегидами 038
----- реакции с нуклеофпламн ti,'i8, 675
Оксоланы 332
Оксониевые соединения 290, 3l'9, 330
а-Оксонитрилы 034
Оксосинтез 11 1. *>rt I
Октадеканопая кислота 5118
/(//г-()ктндецен-9-оная кислота 539, 352
/(нс-((нг-Октядекндиен-0,1 2-опач кислота 51J
()ктадекатриеп-9,12.1 3-овая кислота 553
Октан 92. 179, \cJ7i
Октатриен-2, 1.6 168
Олефниы см. Ллксны
Олеиновая кислота 539, 552
Олигопуклеотпды 715
Олоноорганическне соединения 201, 262
Очефип -190
Омыление 576, .>81
Опия алкалоиды 700
Оптическая активность 59, (>1
Оптические антиподы 228
Оптическое вращение 59
Орбиталь
атомарная 33. 1 25
--- вырожденная 213
гибридная Зо
— молекулярная 33. 37, 125
— чолекул яр и а я выснгмн *а и лт<зн М, 1 35
-- --- низшая свободная 135
— ¦ неподеленпоп пары чле1.тропов 1 12
— песвязывающая 12()
разрыхляющая 33, 35, .'17
-- связывающая 33. 37
фронтальная 1 ,^>
— циклическая 182
— л- 35. 37
— а- 35, 37
Орбитальная спмметрн я. иранило сохране-
сохранения 169
Органические вещества 9
— кислоты 9
— красители 13
— металлы 1 _', 155, 601. 681
— полупроводники 12. 15."», 20S, Li 1
— сверхпроводники 1.'. li.Sl
Органическое стекло 580
Орнепташюинып эффект 1ЪЯ
Орлон 600
Орлптнп 02 I
Ортомуравьн пая кислота, vt:i ()'.,] 5S2
Ортоэфиры 505, 582
Орто-'аффект 190
Основания жесткие и мягкие 73
--- Манниха 523
— сопряженные 70
Основания
— О- 70. 290, 329, 115
— S- 70, 360
— - С- 70, '?30, 129
- N- 70
Основное состояние 37, ¦>[
Основность 70, 71
741
— лдсппна 711
— ачоарсноп 433
— ллкаполоп 290
— алкплампг.оп 395
-- алк:1л;1р:1ЛО1«ых эфироп 313
— ампдигоп 5уЗ
— амидов 58 I
— - ямппоккслот E1Q
— амшюпкрпдпноп 69-1
— - црилачпмюп -109
— арплгидрн.'шиов 418
— гул |П1Д1.гп;' 616
— гуанина 712
— дпалкилоиых чфнров 329
— пэохпполппа 698
— - лмлд-'кюлл 077
— имидоэфпров 5931
-- карбонильных соединении И 5
— карбонопых кислот 51j
— лактамоп 584
— метанола 290
— моченииы 014
-- нитрилов 599
— окснмоп 471
— пнпера чипа -103
— пиперидина Ш2
— пиразола 677
— ппрчдинов 690
— пиримидина 706
— ¦у-ппроноп 70.1
— пурина 710
— сульфидом 366
— тиазола G&(
— трназолоп 086
— - фосфннон 277
— хпнолпна 608
— цитопииа 708
— этилонимина -102
ПалладнП, л-комплексы 197
Пальмитсггы МУ
Пальмитлнопая кислота 538, 5-12, 549
Папаверин 700
Паральдегпд 179
Парафины 66, 101
Паруформ '1JG
Пеиициллпны 081
Пектаацепы-Ю-глюкоза 313
Пе.нтаднеи-2. 3, хпральиость 131
Пеитаметилспнмнн 390
Пснтаметпл-/)-глюкоза 513
Пинтам 80, 92
Пе.итан, '2,3-дибром-, стергоизомерпн 227
Пентлпднонаи кислотп 3jG
Пе.нтанитроанилпи 380
Понтановая кислота 538
Пентаноли 28,>
Пснтаитиол 305
Помтаолы 308
Пентафтлленыс смолы 580
Пептафтор пропс н-1-ил-2 310
Пеитацеи 201, 210
11« пт ацикл оокт аи 161
Пеитаэрмтрит 308. 157, .-.S0
— тстранитрат 3 18
Г1ентоиы 10Г.
Пеитнты 308, Л0(»
Пентоды Ь0\, Г>05
Пептидная спизь 021, 628
-- — об р яз о и яп ие 033
Пептиды 027
— синтез 033
— тпердофпзнмп синтез G34
Пептоны 627
Первичные амины 388
Перегруппировка
— пллпльпая 29-1
— БайРра — Виллигсра 396
— арилгпдрокг-.ыдмипон 382
— бепзпдпноиая 4 1 9
-- Ескмана 171. 477, 017
— бепзилоная -178
Перегруппировка Вагнера — Мссрвсйиа 299
— Памира 323
-- гпдроперокендоп 319
— К урипуса 391
—¦ „'/аса: на 592
— иинаколипопг.я 301. 410
—¦ ретропипаколиновая 294
— - сем иди нон;! я 11 <!
— сигматрогшая 08, 069
—¦ Фаворского -193
— Фг-иса 319
Перенос заряда Г>30
Переходное состояние 06
11ере')терифи1{.ап.нн ">7 I, 576
Пернлеп L'02, 212
Пернцинлнчсскпе реакции G8
Перки на реакция 371
Перлон ПЗУ
Пероксибепзокиня кислота \1G, 397
Перокснды 3-18. 319. 398
Порокепкарбононые кислоты ЗОО, 595, 500
Иероксчуксуспли кислота 217, 'I."i7. 500, л96
Перфтормлкапы 221
11ерфто;1карбо!ЮНЫ1; кислоты G0-I-
ПепНпорметаи 22 1
I \\-.-\ ПШ1ДЫ 1 3
11|:пал;п ы 5 И
11ми-ии.ная кислот,! 538. 544
II п кол и нова я кислота 69*1
П;;колппы 6_87, 693
Ппкряты 3S5
Пикриновая кислота 319, 369, 376, 38!. Зй5
Ппиакилнн 301
Пинаколииовая перегруппировка 301
Пниакои 302
Пннакопы 301, 433
а-Г1инеи 1 72
Пинеразнп 390. 399, -103. 707
Пиперидин 390, -102, 657, 692 ф
2-1 Ьшерндом 5ti5
Пиразин Ь'2Л, 657, 701, 700, 707, 709
11ирааол 036, 07 1-
•— взанчодопстние с электрофплыгымп реа-
реагентами 678
— кислотность и основность 077
— получение 4 30. 67-1. 073, 079
—¦ природа связен 077
3.5-11празолд!1карбопопая кислота G79
Пиразолидиидиони 675
Пиразолин катион 077
1 [иразолопоиые красители 435
Ппразолопы 075, 07Й
Пирамидон 679
Пнрампн 707
Ппраноэы о0(). 507, 509. 510, 517
4Н-Пирап 057
Пнрапи 702
Ппргп 202. 21 I
] ,6-Пнрепхинон 'Л 1
Инридлзпп 191. 057. 701, 707
Пиридин 1 1. 657, G87
— - ампно- (iO-1
—¦ гпдрлроинин
—¦ пзаимод^я'ц'т
гентамп 091
-t0?., 092
е с иуклеофильпымп
реа-
•— — 'лчектрофплышми реагентами 690
—¦ гидрокси- в'^3^
— - методы получения 087, 088. 093, 703
— окисление 091
—¦ ос нон ноеть 690
— расчеты по методу МОX 089
— спектры поглощении 689
Ппрпдннкарбононые кислоты G91. 09-1, 099
Пирндинсуиьфотржжсид 663, 0E1, 090
Пнридиния катион 090
Пирндоксаль-5-фоефат G91
Пнрпдокспн 69I
Пнридоиы 691, 693. 703
Пирилнн соли 057, 701. 703
Пиримидин 037. 70 1. 705. 707
Пирпиииоградпая кпсло'1 а 603>, G39
Пирогаллол 323, 327
742
Пирокатехин 323, 321, 171, 327
— дпм^тплоешП ъфпр 314
— мопометилоиым эфир 3-М
Пироксилин 321
Пнромоллнтопая кис юта ГM7, 362, 563
Пмромсл.читонып дпсшгпдрмд 503, ol;0
Ct-Ппроиы 703
V-Пиропы 703
Ппроелизеги-Я к'не л ста 660
Пирсугольнг.и кислот;!, э<1шры G12, 613
Пирофорные соединения 236, 2j9, 26(J
I 1иррил-поп 60-1
Пиррол 033, ОЗЯ
— ацпдоф..бп;к ть 0ti3
— гндрирот. ппо 6t>2
— методы получения 491, 609, 60")
— строение iitH
-- химические сноГк-тна ОН Л, 665
Пир рол иди и МЮ, ;*(Л, З'м. (i.).). G67
Лпрролпдпнкарбопоиаи mi ел от а 625, 663
2-11цррол1:до11 305, 588
Ни рролппы б о Г)
Пиррометен 6иГ)
Пицеп 201
Г1']астнф|[[;нторы 379, til ""•
Платина, .-[-комплексы 207
Плодоньк! сахар 31 1
Плюмбаполы 261
Плюмбоксакы 261
Попсрх постно-а ктпшше вещества 377, 360,
400. 3 Р.)
Полназпны 478
Полнакрнл^чы 079
Пол на крилонптрнл G00
Полиамиды ,;8У
Полпбутадиен МО
Пол и ни пил ацетат 378
Полним нилкирбазол 073
1К)лн1Я1нпло:1ЫС эфпры .1-12
Поли ни пилонып с 11 j 11 п "O8
11ол1]Ш1нпл1111 р рол и дои ."S8
Пол и ни !'.ил хлорид .'0:!
Пплигекслметил1.ч';1Д;11;;1\111Д 389
11олиеиы. conpn:heиные 1 4 \
Пплккзоб^тилем 1 1 [)
1 lo.'iin! ior:pt'ii ! :0
Полппч'плы 38A
11олн-1--К?1Проамид 389
Полi[K;ip6on<'T[ii 643
I Io.1MlK(lp6oi!(iHUA КI If ЛОТЫ 336, Jtj'2
П;?л[1Мс!леп]!;п ы 380
Нолпмсрпг'нипя
-- алк;)дпенои-1,^ \?Л), 1-10, 212
— ллкоион 118
—¦ нппплпг.ых оф:1рон ') 1.'
-- в npiicVTCTHiui кисло! 1 1 Г)
—¦ - Mt'T.'iJi.io'jM -I ::n4t'cb;ix соедппеппП
121, 231
— сь(Т)од|и.[.х радикалов 120
— га,чогчт:;|Л Keiiort 2 12
Г If ¦ Е! ;:СЫЛ tril ".i 111 V К! ГС Л ОТ 3.".?
—¦ сч e;ieopri \ л vp:ii!M 122, 110
фопмлльтогмп, I --3G
Полимеры
— СШфЯ.:^!!!:!:!? I 34
— стероорггуляр1':.:о \1>.2. 1 10
термопопкле 38U, 30j
11(Ъ11:мо-1Пл;!Кр]гл;'Т 3hO
ПAЛИЖ;т;:лмп;-.:фп,-;;-1 ,"8t)
Пол ниш p(j;'pe:!!ii 'Mil). 3S^. '-!ЬЗ
По.1]и пуклеотпды 7i .i
П ол и о к с I: м i 'Т ] i .4 е и 1 3 и
Полполы :\S1. лО»
ГГол ипепт1;д}>1 1:'. 027. 6'.10
11ол 1:пр(л;нло!1 1 '0, 1 2 ?
— lUiicbKoro дан.-ii'Hii я 120
—¦ и л от ,i кт; i ч ее к; i: i I 2 2
-- нп:!К(Л'о да иле;ni я 1 22
—¦ стероорегулярпыГ! 1 2 2
ГГол ]: пропн л (.чюкемд 3;',7, -i!i8
Полмс.1Х;.рпды 2S3, о07, 311, п 19
-— амино- 522
Полиснлокслпы 7-1, 330, 331
Полнсонряжеппыо молекулы 134
11ол и стирол 1 Г)б, б;П
Политетрафторэтилен 242, J i \
Полм-м-умдекамзмпд 389
Полиуретаны 337. 6D
Полтгфенилхпиок^а.'шчы 17У
Пол ифор м ал h,i,cгид 4 j f>
Пол пфта латы Г) 80
11олифторонреп 2 У.2
Полихпноксалипы 4 79
Поличлоропреп 212
Полнпиаггочт'ленол^род!)! 601
11олн-и)-эп<-нгтоампд 3S9
Пол1:эдрап1Л 162
Полн^тнлсчт
- высокого даьлепня 1 '20
- - шгжого даплеппн 120, 121
Поли'зтилепоич'лд -437
11олн vTiuienTi'iietjjTJ^TfiT 3S0
Шип-афнры 30V. ^Ofi. Уоъ, 372, 380
11ол > ацетали ;М2. -130
- - циклические- 109. оОП
Полчпронодппк!:, органические 12, 13"), 208,
4 1
ру
— - алкаптполон 303
— дном по;: еиязп 1 07
— - к;:рбопнл tiiioii группы -it!
Пол яриметрнн 1 60
Полярность снятии -'Л
1 Голярографпчеекп акти иное негцостно 47
Пол i-'porpdijupiecNii ii потенциал пол у полны
11ол',-рографи я 46
Порфи.п О6.">
Порфгпы (iti,)
11отопдиа.']Ы иосстаконлепи.я 47
— — хп попоп 330
Поте иди a;i ионизгииш см. '-Энергия нопиза-
П|):'3\|,||[ 193
Прилсжаеьа реагент 39(i, 597
I tpa родпып г'аз I .'3, 8!)
Пролпн tj 3
Пропан 86, 9°. 309, 370
11ропапс1ль АЛ7, 4 41
11ропандиол-1,2 338
П рола пол-1 '2.4, 28 ', 2S3, •"'90
Ппо:1апол-2 2б:>. '285, 288, 2(J0. 133, 437, 469
'Л-l 1ропа][олид bb'i
1 Гр(Л:аргилоиыП ?,л[»дегпд \";7
Пропаргилоиы:! спирт 137. \.\O, 208
Прогкгм 101, 129. ЗЛ1, 337, 157
— гпдроформплм [клешне 113
— - дпмерп^ацпя 260
— OMic:UMi:ie 330
-- полммерпзсЩп я 1 — 1
— получеши1 \Ч\)
--- iipi'np-iueiiiio о я^рилшгптрил 30'J
— присоединение там 2У)
— хлорпронаппе 124
npo:ieii-2-o.-i-l ЗОН
Пропои- [-еульфепо.чан кислота 3G2
11ропил-'.ма п 3<:3
I lpoi:ik-ie:i см. I Iponen
Прог'плс'юьспд i<V.<, ,'¦37
I1 jionr, и по и по им л (J n ui' v,l>::p 328
I Громил (ты"! fMitpT см. Up-.:[,-] !:и.[-1
Промплхлорид 22 1
11роми .i lib
1 l|)fn:niionaw к г сил а о-Л)
Г Ipomi n-2-o.'i-l 2'j7
(J,-t 1ро.1пол,-.:п ам 60 ?
[VI 1ро:1полакто:; ГИ'. 1
1 Тропи олоия я к и с л i.'i а ЗЛУ, П31, 362
Пропиопаты 5 I I
11ро:шопона я кисюта ЗЛО. 342, ','•> 1 !
Проппопоньг! ;:.¦]ьдсг":д !:3S. 1.47. I i 1
— - а-гпдрокгп-, стг'»ео1: юмо:п:я 490
П роста гл,1 пдм |;ы 1 (i7. nil)
Простетпчоскне гр> :п:ы G26
743
Протеиды 626 Рибонуклео.чнды 713
Протеины 626 Рибонуклеотиды 714
Протонныи магнитныП резонанс 57 D-Рибопираноды 506
Прототропич 3t I D{ -)-Рибофураноза 50G, 508
Прохнральная молекула 2'J6 Рмльсаи 589
Псевдокумол 177. 180, 181, 5G3, 573 Роданиды 641
Пурин 12, 700, 710, 711 Роданировиние 6.10
Пьезоэлекгрпкн 614 Родентицпды 100
Ртутьорганичеекиесоедппення 251, 25G. 126,
Радикалы fibO
— свободные -12, 56, 73, УС, П8 Pyi'Mtmn синий 623
— стабильные -00, 420 Ружички реакция 4 10
Радиус
— ковалейгнып 4 5 Салицп/юнан кислота 603, 60У, 61 И. Ыо
— эффективного действия атома -15 — — метиловый эфир 61 Г»
Рацемат '2'2E Салициловый альдегид 192, 1301, 503
— разделение па оптические антиподы 631 Сахар виноградным 513
Реагенты, типы 71,73 — древесный 307
Реакции бимолекулярные: 64, 230 — молочный 518
— второго Порядка 64 — плодолын 51 \
— ] етеролитпчеекпе 68 -- свекловичный 518
— гидролипа 232, 439, 576, 583 — тростниковый 518
— гомолитичеекпе С8Г 73 D-C;ix;ipua» кислого 51 1
— диссоциации 67 Сахарные кислоты 63VJ, 660
— замещения G7 Сахароза 511, MU
— изомеризации 67 Сперхкислоты 9У, 187, 331
— конденсации 106 Сверхпроводники органические 12, 681
— мономолекул яр и ые 6 L 230 Сгшнецоргапические соединения '261, J^-
— иуклеоф|гльные 72, 7,1 Свободная валентность 125
— обратимые 65 Снизь
— одноэлектронного переноса 67 — «бгпижопак» 162
— отщепления 67, 223 -- водородная 269, 286, 512
— первого порядка 64 — дативная 261, 267
— перегруппировки G7 — делокализироваппая 126
— пер и циклические 68, 76, 141 — донор но-акцепторная 1*6, 329
— присоединения 67 — длина <1<1
— равполесные Gj — ковалентная 26
— репгоселективпые 101, 191 — координационная 26
— рекомбинации fi7 — мпогоцентровая 259, 12G0, 270
— скорость 6 \ — нецелом мечен и A я 126, 127
— стереоселектпвные 113, 116 — порядок 11'5
— стереоспецпфмчес1\пе 231 — полярная 27
— фотохимические 52 — семиполирная 26, 371, 382
— цпклопрпсоедппепия 67, 122, 123, Ml — S =0 357
— экзотермические 10 — сопряженная 130, 136
— электрофнльные 72, 73 — я- и о- 34, 36, 37
— электроцмкличеекпе 68 Сегнстоиа со;н> 61 I
— эндотермические -10 Секвенция аминокислот 6L>8
Реакционный центр Семидинонан перегруппировка -119
— — иуклеофмльныП 72 Сечикарбгшопы 149
— -- электрофпльиыП 72 Семи х и нона анион 326, 532
Регенеративная реакционная спосоСеюсть Сенсибилизаторы <i69
212 Серии G2 1, 626
Региоселсктивные реакции 101, 191 Серебряного *еркала реакция -151, 155
Редокспотенциалы 530 Серная кислот;), "зфпры 352
Резерпин 673 Серотонин 073
Резина IV2 Сш-матронная перегрушшроньа 6s, ou9
Резонансны Л интеграл 12j Си л азаны 17 \
Резонанса - мезомерии концеггцпк 31 Спландиолы 273
Резорцин 323, 'Л2Тз Силантриолы 27-1
Рсймера - Тилшна реакция 501 Силаньг 271
Ройся никель 192 Силиконы _>74
Рентгеноструктурпый анализ 4 3 Си л океаны 273, 271, 350, .451
Репелленты 370 Синий Михлера ЪЗо
Ретроальдолыши распад 487, ь.Ц Синий Руэмана 623
Регродиеповып стгтез \67 Сииглетное состояние 51
Ретронинаколпновая перетрут иг pii'Ms.:; 294 Сннтол 281
Ретинол 300 Синтетические моющие, средств;! 3,Vj\ lii"i0
Рсфармашсно.'о ренкцня 60S Склеропротеины 6:26
Рефракция Скорость реакции 64
— ;1Л кадке нон-1,3 131 - — или я кие электронных ^ф(Ьеитои 77
— злкомо» 107 Скраупа синтез 696, 700
— алмшов 117 Счожиоэфпрная конденсация '-80, 577, 633,
— атома кислорода 3G5 6-13
— — серы 3(jj С'южпые эфиры карбоновых кислот 56], 573
— молекулярная 39 — активированные ГO5, E33
— .экзальтация 39 — — - и цп леи нова я конденсация 4 95,
Рефрактометрия 39 577
Рпбозл 505, 506, 508, 712 .... взаимодействие с м\*кл?-)фпль-
Рпбопукле,'!за Л 63 [ иыми реагентами 575. 5Го, 577
Рпбинуклепновые кислоты 12, Г06, 712, 71G. -- • - получение 151, 4G7, 5hi, 573,
718 571, 586, 5!)G
744
— — — — полярность связей 575
Смазочные масла 101
Соединения включения 6А5, G4G, 650
Собственные векторы 125
Сольватация 329, 33l\ 362, 370
Соммле реакция 165
Сополимеризацпя МО
Сопряженные двойные связи 134, 136
Сорбенты 16
— хиральные 631
?)-Сорбпт 511, 514
Сорбоза 510, иИ
Спектропол,яриметрия 59
Спектроскопия инфракрасная 33
— колебательная 51
— комбинационного рассеяния 54
— ПМР 57
— фотоэлектронная 62
— электронная ,0
— Э11Р Г>:>
— ЯМР 57
Спектры переноса зарндл 530
а-Спираль полипептпда 629
Спироалканы 1 A0
Спирты см. Л л канолы
Сродство к электрону 38, 47
— галогенов 1 1 2
— гексацнанобешола 601
— нитроарепол 380
— тетрациаиочиноди четаиa 601
— тетрацианоэтплена 601
— хинойон j30
Стаиианы 261. '262
Станноксаны 261
Стеариновая кислота о38, 5-12, 519
Степень переноса заряда ,331
Стерео][зомеры 01, 87
Стереоселектпвные реакции I I 3, 116
Стереоспецпфическпс реп к [(.ни 231
Стереохимия 11, 25
Стерины 17 Г)
Стероиды 1 75. Г* S1
Стирол 178, 193
Спюрка реакция 100
Стрептоцид 416
Стрихнн н 1 2
Структура родоиачальная 83
Структурная
— изомерия - I, 87
— теория 11, 21
Структурные формулы 24
Сукцпна игидрид 559
Сукцппичпд Г>30. 585, 588, 617
Сульфадимезин 4IП
Сульфа пиламиды 4 10
Сульфаннловая кислота 116
— - - амиды 4 I E
Сул[.фенилхлориды 361, 362
Сульфеновые кислоты 3(П, 3E2
Сульфиды .461, 361, 36j, 360
Сульфппомыс кислоты 361, 362
Сульфирование ал к иламиион '199
--- аптрацепн 207
— ареной 4 3
— арнлимппоп 4 12, 413
-- ацетлпнлпда ИЗ
— h-дцкетопон ^80
-- индола 070
— нафталина 20Г>
пиридн пи 0110
— пиррола Об'»
-- тпофепа( 6tjO
фенола 31 9
— фурапа 681
-- хиполппа G98
Счльфогруппа 354
Сульфоксиды -461, 362, 367
С\ ..btjio.'uin 300
Су. п. фонами ды ЗГ»Г), 358
С\ тьфонаты 358
f.v т'.-Ьнмч-плнД!,- 307, 1 J7
Сл ;|,ф >п;!ЛН1)ои;.пне 3,>0
Сульфонм л хлориды 355. 356, 358, 3(з1
Су л ьфо и 11 я с о л и 366, 367
Сульфоновые кислоты 351, 355, 357, 358
N-Сульфоновые кислоты 3;(9
Сульфонолы 360
Сульфотгы 356, 359, 307
СулыЬоокислепие 97
Сульфохлорпропанне 355
Танины 327, 615
Тартрагы 014
Таутомерия 31 0
— ампкопиридинов 69I
-- ацилбромидов 604
— fi-дикарбонильных соединении 48') 483,
48 (, 636
— епдполов и ct-i идрокепкарбонпльных сое-
соединении 498
— енолои и карбонильных соединений 444
— нчпдазолов 676
— индоксила 671
— кольчато-цепная 506, 509, 510
— 2-меркаптобензтиа;юла 685
пптроалкапов и нитрономых кислот 373
— |[птро:юалканов и оксимов 386
— К-пнтрозоампнов и дпизо[пдроксидов 421»
4 24
— пиразолов 675, 678
-- ппра:)олопов 675
— пирпдонов 693
— пурина 709
тиокислот 5У4
— урацпла 708
-- цитонина 708
Телочернзацпя 120
Теория
— дуалистическая 22
— дублет-октетпая 25
— радикалов 22
— резонанса 31
— структурная II, 22
— типов 22
— унитарная 23
— химического строения 1 1, 23, 24
— электронная 25
Теплота
— гидрирования 192
— образования молекулы 4 0
— сгорания цпклоалкапов 163
Терефталевая кислота о57, 562, 563
— -- диметнлоиып чфир 580
Терефталевын альдегид 4 75
Терилен 580
Термодинамический контроль реакции -05
Терпены 171
а-Терпипеп 172
Терпи нолей 172
Тестостерон 175
Тетраалкиламмония
-- гидроксиды 399
— соли 391
Тетраалкилбораты 315
Тетраа пеги л-Л-гл юкипираноз,! Г. П
2,41416-Тетраброми,иклогек1;адП(Ч1ии 318
Тетрагпдропирапы 333, 4 99
Тетра гн д роли риди ны 657
Тетрагндрохинолин 699
Тетрагидрофуран 211, 24 0, 253, Xi2, 339,
199, 608
Тетразол 650, 685, 686
Тетра карбон и лги дрид кобалыл 266, 2!K
Тетракарбоп(ил никеля 266
1етралип 206
Тетраметпламмония гидрокепд 400
1,2, 1,5-Тетраметилбепзол 1 78
Тетрачетиленимин 390
Тетрачетплепоксиды 332
Тетра мет плен с ульфон 360
Тетраметплгермаи 262
TfTp.'iметилгглюмбаи 20 '
'I'ci р.;моги.-си 'ia и о?, ¦_¦;'!, 272
Г45
5 30
Тетр^мстплсташи ii 2G2
Тет р;.мет|[лтнт.1|[ 203
N, N, \'',1\'-ТеТ|)('1мстпл-/<'-фс1::1Ле1:Л1: JM
390, toe. -но
Тетранптрометап 37п
Те1раселоа;.фуль!-.;:;Н'И 'о81
Тетратлафул1,п;)л<.ч1 08 I
— 6nc-jTi:.'itM[,i;jTHO- 081
Тетратпотет;а,чеи 210
Тстрафе!'ил борат натрия 2(.>8, 270
Тстрафтс рм.т;' и '21)
Тетрафтор/г :лсн :2-t2t 2 1 1
], 1,1 ,tu-1 ет|'а хлорал капы 005, 01 7
Тетрахлор-1, 2-бепзохипоп d'J8
Тетрахлор-1,-1-бепзохппоп 5'JO. ."US,
1,1,1,7-Тетрахлоргспт;}1] 605, 01 7
Тот p;i хлор мета и 231, О A3
1,\, 1.5-Тсл рахлориептап 005, 01 7
Тетрацеп 201, 210
Tei рацепхппом '2\ 0
Тетргыигшохшюдимотун 001, 081
Тетрацмапо^тнлен GO I
Тетра'л плал юмп пат литии 258
Тетра отплетшей, 2til, 202
Тетр;|.-,токс1:оилаи 350
Тетрил 4\ -5
Тетрты :Ш7, 308
Тот розы ГH t
Тефлон 2 12, 2 1 1
Тиазол 67*6. 081, 682, 084
Тналолидпиы G">G, t»8"J
Тназолп кы Got)
Тиазол ця соли G8 I
Тиамин i;8 I
Тиепин ti.'d
Тиотаи (> 1л
Тиотии G,"),")
Тмируц 61) I
Тииреп 0,11
Тммнм 708, 712
Тноамнды -;(.M, G83
Tiioaner.uin 4 51
Тиокпслоты .300, 501
Тпокс-пи 700
Тиолы 3Gt, 303, 305, 431, о J0
Тиолят-ноп 300
Тпомочеиппа G 11, 0 !9, о."О
Тиоуюльмая кислота и со кро!'ЗТ!оД'.г.1е 0'У
Тпосппрты ом._ Тиолы
Тнофсн Gl»5, 65Й
— в.ишмоденетипо с элект рофпльнымп реа-
реагентами (JtiO, 06,3, 60 [
— гпдрпропанпо 0G2. 063
— получение -МИ, 059, (>П0. 008
-- физические ст.опстпа 001, G02
Тиофенолы 'Аьл
Тноформнмпд G8\
Ти-офоефнты 35\
Тиоци ;jn:n ы Oil, G50, G.'i 1
Тпоофиры 303
ТироЛ!Н Ol11
Тит аи органические (-оед:и:спп и 203
Тшцснко реакция 1G7
Тозилаты 3.'I>
To;»i л хлорид ЗГ)9
То.испса рлп опт -15 I, 5 1 2
Толуидпиы 385
2,4-Толупленднпзоп.и,гл.1Т 0 18
ТолуН.ТОНЬК1 К!!СЛОГЫ <;.>л.)
Толуол 177, 180, 1я!, ПР. 37:), 3SI, -!'J8
«- 'Гол у оле >.' i ьф;', п;: м i • д ¦ > ; Ч
л-То;|уолсу.'!),(})опопа/1 кселчта 3 >!, 3.;lJ
Торф \\
mpco-l 1зоме[)'.г 228
Треонин 02I
рцие л
Тр( [ ало:(а Г» I
Tj-'n.-.-'oKt.! _"¦ .Г»
Трн;;.;ол 0"ч>,
010
.'88
Трп,1ЛК[1Локсг':;нл соли jJO
TpiI.ipil.Ti.MH !!Ы * 07
Трн;.р^гл.'11Дра:лп:ы i 10
Трпброманп.шн 11 2
Тркбро\"фс:юл 31 8
2,6,Ь-Триг идрока:л[;ур;:п 710
Tpin::io6yTiu"UL'iKi\uiu:i"i "'5Ч -J
р| ;
ТрикарОоннл цпьлсбу7
Трпкарбоппл цп к л t
20
:е.!и."/Гйь':р:1и 206
TJT1)n.i'.LL.ib1i,.;iAi.fl
ТрнкарСопил мич.'.оиеяг.-дг.енилм^р-
2ti I
Три к урбопилы алкепкпке;!'-! 200
Трилоп К 020
Тримеллиторая к и глота 5 о 7, 5G2, 503,
- - — ацгадрид 503
Три мет ил ал ю мп n;i:i 2~;), ^:G0
Три метил;! мп и 3'J3, 'Л[).->
Триме! и л аммония соли 3(J0
Т|) и мети л бензолы 1 77
Тпимети-.бор 2G0
Три мети.': бор ат 3-М
Т))нметнл;ермпп 202
Тркметкленокепды 332
Три метил г.ц[)ндпны 087
1 риметклсульфснШ'Л голм 3G7
'Гра метил хл open л а и 27'.!
Тринитрат глпцорч и i 5 17
--- иегта'/рптритн о !и
2, 1Д)-Т[)ПН1!Т])О,1 МИЛИ И 313
\,3,r,-Tpiuii!Tpo6e-.r.H>;i 379, 330, ЗЬЗ, ¦!
Три иm ро.метлп 570, 37 1
2, 1,0-Tpriii: гро-\-П1П]К)-\-ме1Пл;.пилпп
2, 1,0-1 рпмлтрсполуол ,57У, -\&о
Три нитрофенол 50(), 381, J83
Тричуклеотнды 7 1 .">
Tpiiou-i 50->
1.3, 5-Трноксацы
Т:;ао.;е,:т глиперш:;! ",52
1 р::олы 2Ы, 3A5
''"ряилет^лн1 состоя:и;о ¦•!
Т;)п кронn.'!.iмн :i JCQ, J'j.^
Трпгтпгеч 213
1 puiiTOtlKai 02 1, 07:1
J73
10
-11 I
puTiib.r p ;o
Tpn i ion а [н'кш лты t> \ _', 080
ТрафспплСс'р -bl.i
T p i тфе и i: .'i мета 11 1 09, ! 0 0
T
535
Об"
pp р
7 мцфепнлмот.лп'Д-поп Н)9
Т|Щфеппл\1етн1:о.; 200, 201. 300, 301
Трпфеиилметпл-и.'тпоп 'J01
'I рпфепплметпл -[Радикал 200
Трпфепплфосфнп :'75. 1'77
TprApcuu.".4ioet')oiiH'/i митилид 278
Тр1:1|)ем;1лхлирмет..1[ '-00, 201, 232
1 рлфокнл хлореи::;: " 272
7 рифтирметап^ульфопо^а!: кислота 3~>7
'1 рпфторукс'^елач и полога 003, CU.\ C07
Трс\:\орМ1:Т..п'11Д-ио11 °3 ', 23 t
1 рпхлорпптромета:! 370, 371
Т])!!\'лориу;)п:| 711, 712
¦ [ч: x.'iopyKi'V спаи кислота 605
7':>п x.'iopyucyciit.i.i алt,let ид 2 15, 192
1 \у,\ хлор sjv. ie:i '2-'Л). л \ 1, 007
Тр'пм^лодекаД1:!1!! 107
'1 :4U[tiIK-*ItVXt'i4;. ii I 4.) 1
7"рпцпкл(;[и)[и:п t1-) I
Три jt,::kvi:imh:i -101
'i ри ;Т1!л.).пом1Ггп"'1 2.:8, 2.i9, 203
Трц-миламми ЗИП. 3'j3, ,)V,.j. '-01, \62
1 г::'лч:.-:бо;) 20'J. :;8u
Tpr ,тплг1 :лц\оли .510
ТрП'/( i loKcoiM ci co.wi 5,?0
Т-.ул :i:.4'i:;i..n 271. J7:'. ; IG
7 |\гт:'Л(>^Ф-1т 5.il, 081
чг'М.ч.ч;!1.: k..ti.o;i 173, 2 Id, 265
7 \\y.ii; ¦ ;.1л'1 1 /3
Тро." п ¦ Mid'i caxiip ."I8
746
Углеводороды 10, 81, 8G Феиплтрич'лорсплщ 272
— алпцнклпчсскнс 82 Феннлуксуспая кислота 339, 551, 555
— ароматические S'J 2-Феннл\-ро:.:он 703
— ациклические 81 Феннльпая группа 178
— карбоцнклпческне 82 2-Феппл лапол-1 300, 301
— насьнцещгые 80 2-Феппл,тпллм1!п, производные 103, 697
— ненасыщенные 103 Фенобарбитал 709
¦— С ОТКРЫТОЙ ЦСиЬЮ 80 ФсПОКСНЛЫ 317
Углеводы 12, I:-, 29о, 501 Фепокепметплпепицнллпп 084
Углеродный атом Фег;ол 13, 14, 3:>2
— — асимметрически Г: 25, G1 — ал кпл ированпе 320
— •- вторичный SS _ — арп.тиронакне 311
— - дпгопальпы:": 1.» ~- анплпровапне 310
— -- перппчкы.; 88 — тиапмедепсттге с дпааоппепымп солями
¦— -- тетраг of.алкиып (тотра'.«дрпческин) 45 425
— •- третнч i 88 -- кетонами 320, 321
— -- трпгопальпыи -.5 Фенол
— ¦- узловой 1(;1 -¦ в.1апмодеЛ(лвне с формальдегидом J°0
— — четкертпчпып 88 — г алог енпрова пне 318
Угольная кислота — гидрирование 321
— — амиды 0:3. 0 18 - - карбокенлпроваппо 009
— ¦ - нитрилы (ill, (JIG. G'8 — кислотность 310, 381
— — прон.,ко.т.пыг, к.:;..-;'пфилацпя и но- — тппронанпе 319
мепклатура б'1, 012 — нитро'жрование 387
— — тпопропзнодпые 0 19 — окисление 317
— -¦ хлора п:;-дрпдь1 011 — получение 211, 313, oil, 350, 358, 420,
— — эфпры 0 10, 612 457
Удрта -(Сергеева реакция .ИЗ, 322, 349, -¦ применение 322
350, 4',0 — расчеты но методу МОХ 31".
Узловая плоскость 32 — спектры поглощения 316, 409
Уксусная кислота ;), 10, 22, 98, 538 — сульфирование 319
— -- кислотность 5I1 — физические споПства 314, 315
— — получение i.,7. :03 — форматирование 501. 502
— — применение 518 --- "энергия ионизации 315, 408
— — сложные '^фпры 403 Фенолкарбоппльные соединения 501
— -¦ физические снолетпа 512, 5 18 Фснолкарбоновые кислоты 009
Уксусный алг.дегнд см. \н.ст альдег ид Фсполсульфоповые кислоты 319. 32 1
Уксусный аи; пдрпд 3/0. 463, 181, 521, 555, Фенолформальдсгндггые смолы 321. 15G
561, 571, 572 Феполфтален н 53G
Ульмана реакция 196 Фенолы 79, 309, 312
Ундекан 389 гн/с-Феполы 321, 323
Ураппенпе Фенокснльпып радикал 317
— liopa 18 Фснолят-нон 310
— Гаммета 77 О.-политы 317. 311, 315, 125
— - Шредпшч'ра 33 Фенопласты 3'2
Ураты 7[2 Фемтпязпп 057
Урацпл / 05, 7С8, 712 Фептназиния соли 057
Уретапы 5!П, (ill, 613, 014 Ферментатппные реакции 285, 513
Урпдпн 713 Ферменты 12, 620, 031
Урпднп-5'-мопофосфоппая кислота 714 Ферроцен 106, 260, 207
Уронены? кислоты 311 Фппронн 626, 630
Уротропин чаб, 10.1, 470 Фишера проекционные- формулы 227, 223
<i'iitttcpa реачцн-,! 06;)
<!>1Ш1сра - ¦ Тропши синтез 90
Фис..ра<о,-о перегруннироикп 493 Фптол ььО
Факторы нлрцпнлг.пок скорости 191 Фптохппш1 531
I'e.iim.'a реактпп 451, 01 I Флаг.г.и 703
— реакция 15 1, ¦i.j.i, -)'j~, 512 Флороглюцнн 323
Феллапдреп 172 флуорсспепипя 52, 53
Фепазпгг с,.7 Формалин 155
ФормаЛ',яе1 пд 10, 4',1, 455
— !13,"имояепстпп'> с лмунаком 455
— - - alU'T ::Л1.де]]:лом 457
— — ацетиленом 157, 305
-- ¦-- нк'рпчнымн аминами 523
— — гаобутплепом 133
— — и:долом 070
-- -- мочепшюп 6.15
— — натромет;:п:-м 370
— — гмррол^м '.d.'i
— - ¦ ф чюлом 320
— днм-.'тнлапетглг, 171
— г.очпмерпзацпя ^31
— г.;Лучение 295, .155
— реакция К:.г: ';ц1.аг~ *.(-7
Ф ¦руамид 518, 555, Г.''Г, t:02
2-Фспплппда|:дпоп-!,5 190 С-. ,,i-i:':;-.t'i'j 5i;,"rr.R
Фе!п;.п:одозилдн:.цет,:Т 2!7 Фо via.'ianei or; -575
Фелглаодозплднх-лоргд :: ; 7 Ф ):.м::;:нроп-м!пс ;р;-поп AM
К-11\-:ш.-|-Г'-пафтп аамнп 115 - - ч ¦ акси;¦ pi'fi;.p 503
747
Фена
Фена
Фена
Феаа
Фоки
Фепп
Ф.-:-м
1, ;-бг
ф(,,,1:
/!-Ое
О-<'Ч'1
Фет:!.
Ф -пи
зон
мин
птре
НПЛГ
лаце
707
!03
чг 201.
.па-а г
ЛИ.!! И
лгпдр;..(;:н
чг пдразош,
гс-Ф
i: "п
ипл;
:п те
¦г'.ч'ч
.ГМК'
.тип
енилгл
¦л'пн о;
Ч1Д.: \:i
; дн;:-.:|г
.|,и.,мп
:оцнапг
4Г,0
178
11 7, 4
j -r.ii.
нок;-:.'Л
"о
м .190
н 415.
II ¦' 1 ¦;
гд 507
:-09
i'o 1
7'. Г.01. 512,
501, 01 S
:>плиспоол -179
4 73
79
707
— фенолов 501, 502
Формилуксуспая кислота 603, 705
Формнлфторид -165, 567
Формилхлорид 464, 567
Фор ми льна я группа 4 38
Фосген G-П, 642, 613, G-J7
Фоефяты 351
Фосфииистыс кислоты 273, 27G, 78
Фосфн новые кислоты 275, '276, 279
Фосфи«оксиды '273- 278, 617
Фосфпнтиооксиды 277
Фосфи ны '2 7Г) - 77
Фосфиты За \
Фосфодиэфнрпая связь 715
Фосфолпппды 533, 381
Фосфонаты 279
Фосфонкевые соли 273- '277
Фосфои и Лил иды -278, 1 47
Фоефонистые кислоты '275, '276, 278
Фосфоновые кислоты 275, 276, 270
Фосфопротеиды 626
Фосфоршш 277, 278, 'I 17
Фосфоресценция 52
Фосфорорганические соединения 275
Фотосеисибили;1атиры -1С9
Фотосинтез 13, 66G
Фотоэлектронная спектроскопия 62
Фрсоны 235
ФрцОеля — Крафтса реакция 35G, 161, 5G9
Фриса перегруппировка 3\9
Фруктовые эссенции, искусственные 578
Фруктовый сахар 514
Фруктояа 510, 512, 514
/J-Фруктопиргтноаы 510, 51 1
D-Фруктофуранозы 31 I
Фтала;шн 657
Фталальдегпдиаи кислота G03
ФталгидразиД 565
Фталевая кислота 206, 556, 562
-- сложные эфнры -189, 579
Фта левый ангидрид 564, 372
— - - ацилировлппе аминов 571
— - - — арепов 529
— -- — спиртов 571
-- образование полиэфиров 580
— получение 502
— - применение 572, 580, п88, 600
Фталимпд 565, 588, 618
Фталодм нитрил 567, G00
Фталоилперокеид 566
Фталоцианипопые красители 601
Фторалкамы 223
Фторарены '238, 127
Фторафур 708
Фторбеизол 2Ю
Фторирование
— алкапоп 221
— ¦ карбоновых кислот 604
Фторопласт 2J2, 244
Фторопрен 242
Фтор углеводороды 233
Фтороформ 22 1
Фторхлоралканы 235
фтор уксусная кислота 601, 003
5-Фторурнцил 708
Фумаровая кислота 556, 560, 5G1, (ill
Фунгициды 652
Функциональные группы 84
Фурадоним C(i8
Фураюлидои 668
Фурам ЛМ1 G55, G58
— ацпдифобность 663
— бромировачие 6Г>3
— гидрирование G62
— диенопын синтез 665
— нитрование 663
— получение (91
— превращение а пиррол и тиофеп 659
— сульфирование 663
-- физические свойства 661
2-Фу рапклрбальдегид см. Фу])фу])Ол
2-Фурапкарбоиоп;г/1 кислота Г.60, 667
Фура позы 306, 508, 510, 517
Фурацнлии 668
Фуропиридины 658
Фурфурол 14, 660, G67
— декабоннлированпе И, C67
— нитрование 6(i7
— ¦ окисление 572
— получение 307, ЬСО
Халкоиьг 43U
Xарактернстнческая группа 81, 85
Характеристические частоты 54
Хелаты
— ализарина 531
— fj-дикетопов 485
— 1,2-диокснмов 178
-¦ дитиокислот 591
— р-кетоэфиров 637
-- о-нптрозофенолов 387
— ¦ окспна 700
— порфина Gt36
— - салицилового альдегида ";03
тпокси1{а 700
X н ми чес к и i\ сдн и г 5 7
Хина.10лнн 657
X инальдпн 700
Хингндрон 326
Хинин 700
Хиной дна я группировка 5'27
Хи ноксалин 4 78, 657
Xи нолин 657, 693
— - взаимодействие с пукле».Сильными реа-
реагентами 6У9
взаимодействие с электрофнльнымн реа-
реагентами 698
— гидрирование G9'J
— окисление 609
— основность fi9S
— получение 69G, 700
физические свойства ti98
ХинОиШпня катион G98
Хинолинован ннс/юта 69*J
а-Хпполон 690
о- Хи нон 325
п- Хиной 325, 320
X и пони мины 528, 335
Хи попы
— классификация и номенклатура Г»26
— ¦ комплексы с переносом io.v-дц 530
— получение 527
— редокспотешшялы 530
— сродстно к электрону 71, '-, -.0
— химические cnoiicTBa 5-JJ
X и р ал ьм а 'л мол ек у л а 6 \, 2 2 3
Хиральность G1
Хитин 522
Хладоиы 235
Хлоралканы 2J1
Хлораль 215. \№, Ш
Хлоральшдрат 191
Хлорамнн-Т 359
N-Хлорамппы 417
Хлорапгилриды карбоновых кислот см.
Липл хлориды
/)-,Хлоранмл 7J8
//-Хлорапил 5°*">, о30, 7K$
Х.'юрареиы 126
Хлорй нет альдегид 681-
Хлоргтетилек 2@
Хлорбензол 239, 2H, 211, '[о
ft- Хлорин пилртутьхлорпд 2о7
Хлоргпдрииы 111, 333
Хлорирование а л капов 2:М
— ¦ алкенов 1 \ 3
— анилина 528
— ярсноп L'38
— бе и.гола '238
— ¦ карбоновых кислот с.01
— пирокатехина о 2 К
— толуол ;i 1 У 1
— уксусной кi[слоты f-04
— фенола 3\8
748
— этанола 4 94
ос-Хлоркарбоновые кислоты 601
Хлормасляпые кислоты 605
X лор м ет и л а ре н ы 223
Хлорметнлирование -'23
Хлорметильная rpvnna E6
Хлоропия соли 2 lG
Хлоропрсн -39, 242, 214
Хлоропремовын каучук 243
Хлорофиллы 666
Хлороформ 1У0. 231, 235, 397, 49-1, .1, G01
Хлорпикрин 376
•у-Хлорин ри дни 692
4-Хлоринримидин 707
N-Хлореульфонампды 35У
Хлоругольпая кислота 011
— эфмры 633, 610, 642
Хлоруксусштя кислота ГM8, 603, 605, G07
Хлорфенолы 318, 321
2-Хлорцпклогексапол 331
2-Хлорциклогексанон 193
3-Хлор-1,2-э[юкс[Ш])опан 337
2-Хлората пол 2 11
Холестерин 300
Холип 401, 102
Хорчсра реакция 27')
Хризен 201
Хром
— гек^акарбопнл '201
~ .i-KOMN.-lfKL-U 261
Хроматография
— адсороцкопппя жидкостная 16
— -- б\мп;кпая 17
— --- газона я 1Н
~- ¦ - га.чо-жпдкоетная 18
— — капиллярная 1 8
-¦ ¦ - распределительная 17
— — тонкослойная 1G, 17
Хромопы 70J
Хрочопротепды G?G
ХусОикксра реакция Г> \7
Хюккеля метод МО 125, 126, 13t
— правило '21.1, 6G0
Цензе сол!' '267
Неллобиолн Г) 18, 5l'O
Целлозольпы .'J28, 331, 337
Пеллофл и Ъ'2\
Целлюлоза I :i, I \, Г/20
— - ацетаты Г>->1
— ксанто! епяты oi'l
нитраты 521
Цепная реакция 0G
Цианамид ОН, ti'.H
Цмапаты 611, GlG
Цнйншдриннып метод 01G
Ннангндри п ы М7, G08
Цианиды см. Нитрилы
Цпанкобнламин 666
Цианоарсны 4'2G
Циановая кислота Gil
Цпгтуксусиаи кислота 5,"8
Цпапуровая кислота G(G
Цпличтнлирование 600
Hiuwena - ¦ Hamtna катализаторы 21, I 10,
U3. 2G3
Циклическая делокалпзацня 183
Циклические '^фнры 332
Цнклоалкаполы 280, 290
Цпклоалка1{ои-2-карбоповых кислит сдрпры
635
Цпклоалкапопы 440
Циклоалканы, бицнклпчоекпе 1G0
Пиклоалкаиы 1 ЗУ
— изомеризация 17], 173, 174
— изомерия 1 о У, 1G0
— класенфпкпци я 1 Г)9
— MiiHpoiurii.'inwec'Ktie I GO
— номенклатура 15У
— пространственное строение 16,'j, I6u, 1 ГО,
171, 17L1, 171
—теплоты сгорания 163
Циклобутадпен 1G3, 16G, 213, 214, 26Г)
— трикарбоннлжелело '266
Циклобута и 1 59, 1 G3 1 G5
Циклобутен 161
Цнклобутенднпд-пои 21 1
Цнклобутендинл-катпоп 21 \
Цнклогексадиеи-1,4 1G8
Цпклогексаи 159, 1G3, 1G8
— дегидрирование 179
— коиформацпп 170
— получение 168
— реакции 171, 376, 157
Цпклогекс-аидпон-1,3 325, 175, 482, 481, <1^
Циклогексапкарбальдешд 137
Цн к л огекса и к а рбоксн латы 5-1 {, 558
Цпклогексанкарбонилбромид 561
Циклогексаикарбоннтрил 5G7
Цнклогексанкарбоповая кислота 1У 4, 538.
5-И, 5-19
Циклогексанол 249, 321, 453, 457
Циклогексанон 168, 32 \, 334, 438, 157
— - окспм 47L'
Циклогекс-атрпен см. бензол
1Диклогексеп 1G8, 171
Пнклогептян 16.3, I 72, 1 73
Цпклогептатрпен \ 72, 1 73
I-Цтлогептатриеппд-пон 16
Цнклогептатрпенил-катноп 173, 216
Цнклогеитатрнепил-радикал 216, 263
Циклооктан 1 73, 1 7 i
I(нклооктатетраен 173
Циклопептадпен 166, 167
Цпклопеитадпепид-иоп 215, 255, 205
6ис-\ 11[клопептадненилдиалкилтптлн 263,
266
Цнклоггектадискггл-кйтиогг 2 \5
Циклопептадиеннл-радикал 215, 265
Цнклопентадненплтрнкарбоипл марганца
265
Цпклопечтан 159, 163, 166, 167
Цпклопептапдиоп-1,2 176
Цпклопентаноп 166, 559
Цпклонептанонокспм 617
ЦиклопентAпопе])[1ндрофенацтрен 175
Цнклопоптеп 167
Циклонрнсоедппенпе 67, 122, 123, 14 1, 162.
164, 169, 2U3. 67,5, 685, 686
Циклопропан 1 59, 162 - 164
— - получение 162
— природа связен 162,163
— - реакции 163
— энергия ионизации 163
Циклопропанони 4 93
Пиьлопропен 153, 162, 164
Цпклопропенид-нон 214
Ц] i клоп ропеинл-катион 161, 211
Цнклопропепнл-радпкал 214, 265
Циклотрпсилазаны 271
Цикл отри си л океаны 273
Цпмаптреи 265
Цпнкоргапичеекие соединения 2^5, 60S
Цпннолин 657
Ци и хоме. ромовая кислота 699
Цпстенп 624
Цпстин G2 !
1.[птиднн 71 3
Цптозпп 708, 712
Четырех,! а метенные соли аммония ЗПГ'
Четырсххлорнстып углерод см. 'Гетрачир-
мета и
Чичибабина реакция 6У1, 699, 707
Чугаева pea к ци я 651
Чугаева — Церевшпипова метод 251
fff'i,\''Hepu реагент 521
lib,- чк аиетятпып 52 I
'чк-чо:л1Ы11 521
— .\год п о а мм i г а ч, i ы -i 321
749
Шиффа основания -170 — резонансная 31, 131, 181
Шмидпт реакция 591 — свободная GG
Штрсим'ра — Зе.ишскаго метод C1G — связи, термохимическая -11
— сопряжения \'Л\
Щавелевая кислота 10, 193, Г>:8, 536, 538 — циклической делокалпзацим 183, 214,
Щавелсвоуксусная кислота 639 215, CGI, G89
— электрона Л2
Эйкопаи 87 Эппмеризацпя 512, 314
Экваториальные заместители 170 Эпнмерня 512
Электромагнитное излучение, изапмодеЯст- Эппхлоргидрнп 3:17, 338
вне с веществом '19 Эпоксиды 333
Электрон, неспарепнын 5G — взаимодействие с нуклеофпль мн реа-
я-Электроппая 1]лотпость 123 гептамн Лз.">, 330, 391'
Электронные переходы AT, '18, 53 — получение 334, 447, 4-18. 596
я-Электропные_спстемы, характеристика 31, - стереохимия реакции 331, 335
124, 12G, 13j, 181 Эпокспцпклогексан 334
Электронные смещения 27, '28, 30, 73 Эрптрат 307
Электронные спектры fiocлощриия 50, 53 Эршмро-ияомер 2'28
Электронные 1*ффеК'П|1 30 Эр1-цп G73
Электронный парамагнитный, резонанс 55, Эстроп 173
5G Этерпфпкацпя Г1 J.G, 573, G49
Электроноакцепторпые свойства 28, 30 Этап 86, 92, 91, 98
Электронография 43 Этапаль см. Лцет;!.-:ьдегпд
Электронодонорпые свойства 28, 30 Этапдиэмнн см. Этплондиамнп
Электроотрицательное^ атомов 27, 44 Г-)тапдпол-1,2 см. f"jiпкс:/1ь
Электрофилыюе замещение 72 Этанол 10, 11, 132, 282, 283, 331, 101
Элскфофилыюсть 72 — геометрия молекулы 287
Элск'1 рофнлыше реагенты 71 -- кислотность 288
Электрофоре:! 19 — получение 293, :?%, 513, 520, 321
Электрофото! рафп я 673 — хлорирование -194
Электрофуг 72, 18G -¦ .анергия nonH:ioi;i:n '.'(")
Электроцнклнчсскме реакции 68 Эт;\поламипы 337, 392, -101. 682.
Элементарпып акт реакции G3 Этаптпол 303
Элементный аналил 19 Этепол 309
Элемепторганнческие j\lic;iOTbl, эфиры 345 Этилацетимидат 5GG
Элемеиторганпческне соединения 10, 2 18 Этиламнп ЗУЗ, ЗУЗ
«-Элиминирование 231 Этпламмоппи хлорид 390
Р-Элнминнроваппе 233 Этнлацстат 469, 378
-¦ бимолекулярное 234 Этнлбеп.чоат 5G4
¦- мопомоле!;улярпое 233 ~>[нлбепзол 195, 'G!)
Элюент 16, 17 ЭтплброммД 22 1, 231
Э.иппекава — Эр.и'нмейера правило 310 Этплборпая кислота 2G8
Эпант 589 Г-)тплбутир;.т 378
Энантиомеры 25, 01, 93, 226 Этилвнппловы;: чфпр 311
Эпаптнотоипые атомы 22G Этп.-ien 23, 37, <6. 102, 101, 128
Энантопая кислота 538 — л-комплдксы 117, 207
3,0-;-)идоксо-1,2,3,6-тетрагнд])офтэлевьп"| ан- — окис.:еннс 11G, 318
гидрид GG5 — получение 128
Энергетические уровни МО см. Модскуляр- -¦ нолпмерпзацп:: 118
иые орбнталн — применение 1:!,ч
Энергия -- природ;. свячеП 108
— aiiTiieaun:' 05, GG — присоединение воды 293
— водородной овм:ш 28G -- тело.мери.з.чцп я 121
— делокализацг.п 12G, 131, 183, 214, 215 — фпчнчеекпе сиопства ГЧ8
-- диссоциации il -- XHNn:llocrLiie eBoiicTna 109
— иопи.чацип 3 1, 38, -18, 62 Этплепдпами и 3!H, -;01, 503
— — алкаднепов 13G Этплепдна.мп гпет раукс\сная кислота 620
— — ал:;апов 94 ЭтплС:|Глнко:!ь 302, S3", 3 10. ЗНО
-- — алка.полов 280 Этнлепнмнп ЗУО, ЗУ2, 39». 402, G51
— — алкенон 108 Эти.^епоксид 331, 332, 337, 6;>4
— -- алкпламппоп 394 — ваапмоде/н'шпе с нуклеофнлънымп pea-
— — алкнпои 1:8 гейтами 333
-- -- ареиоп 210 ^ полимеризация 336
— — ариламнпов 408 — получение 333, 33G
— — впп^лгалогеиидов ?-10 -- ]1схг1.'.;рпость Ciui3eii 334
— -- галогенаЛКапол .!'5 — прчмепепке 301, 337
— — галогепаремс:.1! 2iO Этп.'|е!:гу.п,(})|;д 3u!i, 03 1
— — днуги.ювого эфира 329 Эшлен.х.юршдрпи 2 1-1
-- — индола G69 N<->i н;1]1Д1Ч!ЛН'Т ила.мнп 170
-- -- карбапола 6иП Этплподпд 224
— — пяридпн;: G89 Этилмегплпотоп 411
— — пиррола 601 йтилиатрат 317
— — сульфидов 365 Э-[плнпт;>мт з;С
— — тетргтпафульиалопоп 684 Этпловьп1! с-пмрт см. Этанол
— — тполов Збо Этило'льч11! '-)ф;г.> ojiTOMypaBbiinoii кпелоть] 365
— — тиофепа 661 Этплформпат 578
— — фенолов 315 Йтнлхлорпд 221
— — фурапа 061 Этилн.еллюлозол1> 32В, 331
¦- молекулы '!0 Эфедрин !03
— орбиталп 34, 48 Эфараты }-<¦.!. 330
— переноса заряда 531 Эфпры
— полная, :1-1*лектронов 125 — — азотистой кислоты 316
750
— ,-потпоп кислоты .317
¦— борной кислоты 3-1 Г»
— опнилопые 311, 31.', АТЛ
— гликоля 328, 332
¦— дпалкиловые. 3^8
— КрСМНИСПОЙ КИСЛОТЫ "¦ С
— МаКрОЦПКЛПЧеаШе 3-1.'., .'НО
¦— нитронопых кислот 375
— ортокпслот 5G5, 582
¦— пероксидл подорода 5S2
— пирокатехина 3J4
— простые 327
— серпом кислоты 252, 2Г>3
— сложные 454, 4G7, 481
— сульфопопых кислот 330, 338
— фенолоп У1 7
— фосфорных кислот 351, 352
— циклические 3.VJ
.— элемеиторгапичеекпх кислот 350,
352
Эффект
351,
— динамический 70
— пидуктнпмы]! 28, 7S
— мезоморпый ?9, 78
.— ориентации 186
— орто- 78, 170
— резона пены)! 29
— сверхсопряжеппя 30, 107
¦— сопряжения 29
— злектропоэкцегпорнын 28
— электронный 30
Эффект клектроподонорпьп"; Z3
Эффективные заряды -27
Яблочная кислота 9, 501, 01''. 703
Ядерный магнитный реюнамс 37
Янтарная кислота 9, 55G, ~>о&, 339
— ангидрид 559
— днальдегнд -173, -1!)[
— дихлорапгпдрид -191
— получение 339, 501
— 11 мид 5^9
Учебное издание
Нейланд Ояр Янович
ОРГАНИЧЕСКАЯ ХИМИЯ
Зав. редакцией С. Ф. Кондрашкова
Редактор А. В. Бородина
Мл. редакторы С. М. Ерохина, Л. С. Макаркина
Художник В. В. Гарбузов
Художественный редактор Е. Д. Косырева
Технический редактор Г. А. Фетисова
Корректор С. К- Завьялова
ИБ Хя 6812
Изд. № ХИМ—771. Сдано п набор 19.1J.88. Подп. п печать 2fi.07.89. Формат ?0X90'/,,.
Бум. тип .\ч 1. Гарнитура литературная. Печать пысокаи. Объем 47.0 усл. печ. -ч. -5 7,0
уел, кр.-отт. 50,44 уч.-изд. л. Тираж оо 000 экз. Заказ № 517. Цена '2 руб.
Издательство «Высшая школа». 101430, Москпа, ГСП-i, Псглинная ул., д. 29.1-1
Ордена Октябрьской Реполюцми и ордена Трудопого Крлспсо Знамени МПО *Перьля
Образцопая типография» Государственного комитета СССР по делам издательств, нолшра-
нолшрафии и кнпжноЛ торговли. 11305-1, Москпа, Валовая, 2А