























































































































































































Автор: Коровин С.С.
Теги: металлургия основы химической технологии химия геология гидрогеология химические технологии издательство мисис редкие и рассеянные элементы
ISBN: 5—87623—012—X
Год: 1996




РЕДКИЕ
И РАССЕЯННЫЕ
ЭЛЕМЕНТЫ
химия и технология
Под общей редакцией
проф. докт. хим. наук
С. С. КОРОВИНА
Рекомендовано Министерством общего и профессионального
образования Российской Федерации в качестве учебника для
студентов высших учебных заведений, обучающихся по
направлению «Материаловедение и технология новых
материалов», специальности «Химическая технология
редких элементов и материалов на их основе»
МОСКВА -МИСИС* 1996
Авторы: С.С. КОРОВИН, Г.В. ЗИМИНА, А.М. РЕЗНИК,
В.И. БУКИН, В.Ф. КОРНЮШКО
Рецензенты: кафедра редких металлов и порошковой металлур-
гии МИСиС; член-корр., проф. А.М. Чекмарев
УДК 669.7/.8:66.01
Редкие и рассеянные элементы. Химия и технология. В 3-х книгах.
Книга I: Учебник для вузов /Коровин С.С., Зимина Г.В., Резник А.М.,
и др. /Под ред. С.С. Коровина — М.: •МИСИС», 1996. — 376 с.
Приведен обширный материал по количественным и качественным характеристикам
элементов и их соединений. Дается физико-химическое обоснование технологических
процессов, рассматриваются эффективные технологические схемы переработки мине-
рального и вторичного редкоэлементного сырья, важнейшие области применения, а также
перспективные методы и способы производства Большое внимание уделено методам
очистки и разделения близких по свойствам элементов.
Рекомендован в качестве учебника для студентов вузов, обучающихся по направлению
«Материаловедение и технология новых материалов», по специальности «Химическая
технология редких элементов и материалов на их основе». Может быть полезен студентам
родственных специальностей, а также специалистам, работающим в области химии,
металлургии и технологии редких и рассеянных элементов. Ил. 85. Табл. 51. Библиогр.
список: 51 назв.
При издании учебника финансовую поддержку оказали:
Московская государственная академия тонкой химической
технологии им. М.В. Ломоносова
А О «Соликамский магниевый завод»
Институт химических проблем микроэлектроники
А О «Красноярский завод цветных металлов»
2605000000
ISBN5—87623—Oil—I
укр."' сжато
ДЕР/’ !.». ИХ
УНШСГКИТЕТУ
Коровин С.С., Зимина ГВ.,
ISBN 5—87623—012—X
Резник А М идр., «МИСИС* 1996
ОГЛАВЛЕНИЕ
Обозначения и сокращения 7
Предисловие 9
Введение...... 12
Раздел I. РЕДКИЕ ЭЛЕМЕНТЫ! А ПОДГРУППЫ. ЛИТИЙ, РУБИДИЙ, ЦЕЗИЙ 16
Общая характеристика элементов 16
Глава 1 Литий....... 21
Химия лития....... 21
1.1. Оксиды и гидроксиды............................................ 21
1.2. Соли кислородсодержащих кислот 26
1.3. Галогениды лития.... 29
1.4. Взаимодействие лития с неметаллами. 33
1.5. Сплавы лития с металлами 37
Технология лития........ .... .40
1.6. Важнейшие области применения лития и его соединений... .40
1.7. Сырьевые источники лития... 43
1.8. Общие вопросы технологии лития.. 48
1.9. Переработка сподумена ... 49
1.10. Переработка лепидолита . 56
1.11. Извлечение лития из солевых растворов. ..... 58
1.12. Переработка карбоната лития на гидроксид и хлорид. 59
1.13. Получение металлического лития.... ......... 62
Гл а в а 2. Рубидий и цезий 68
Химия рубидия и цезия............................................. 68
2.1. Физико химические свойства .... 68
2.2. Оксиды и гидроксиды............................................ 70
2.3. Соли кислородсодержащих кислот 74
2.4. Галогениды..... 79
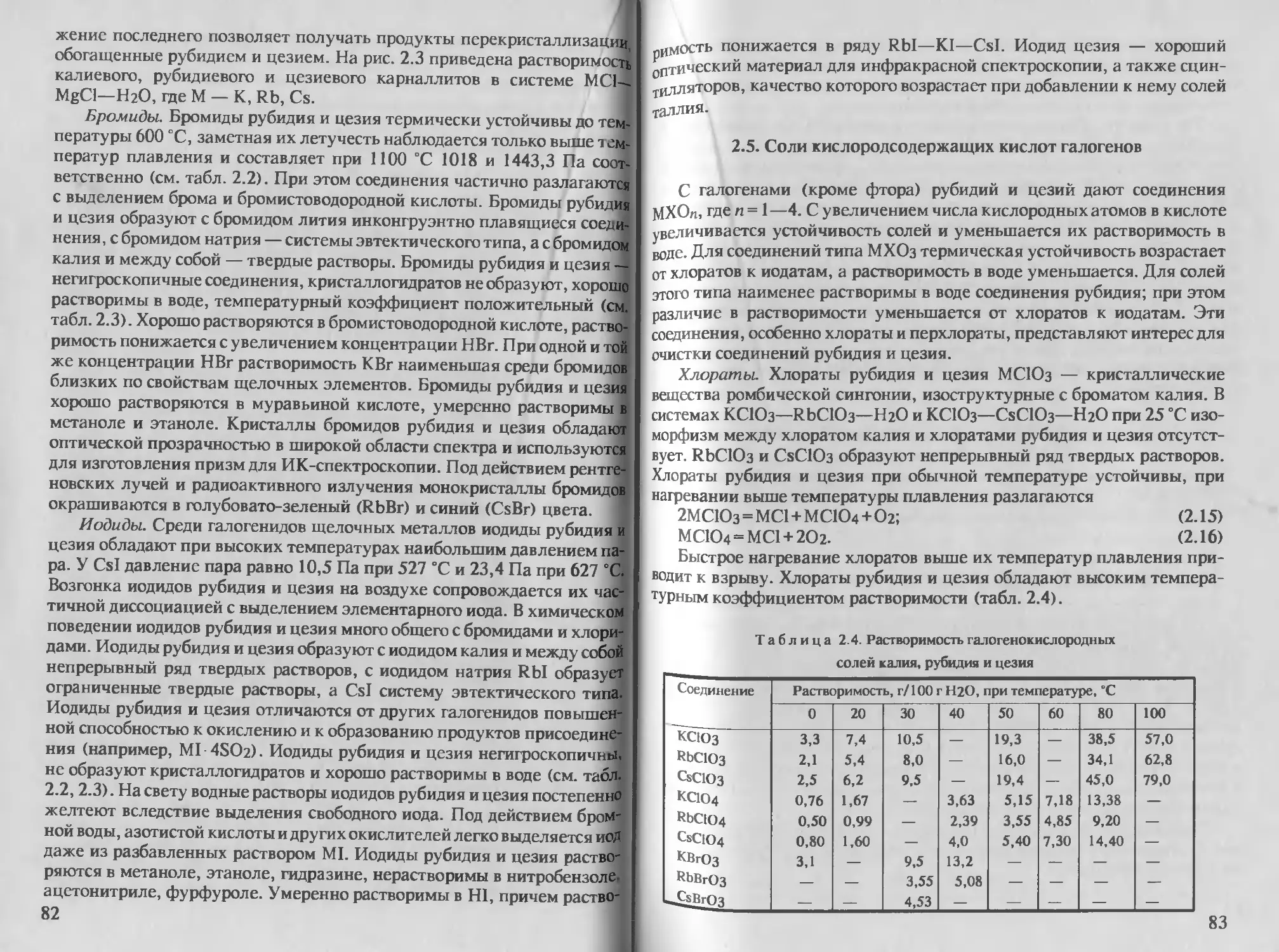
2.5. Соли кислородсодержащих кислот галогенов 83
2.6. Соединения рубидия и цезия с неметаллами . . 85
2.7- Рубидий и цезий в комплексных соединениях 88
Технология рубидия и цезия 93
2 8- Важнейшие области применения рубидия и цезия и их соединений . . 93
2.9. Сырьевые источники рубидия и цезия. 95
2.10. Переработка природного минерального сырья 99
2.11. Извлечение рубидия и цезия из отработанного ядерного топлива 107
2.12. Извлечение рубидия и цезия из рапы соляных озер и
рассолов морского типа .....108
2.13. Получение соединений рубидия и цезия различной степени чистоты. 110
2 14. Получение металлических рубидия и цезия.....................118
3
Раздел II, РЕДКИЕ ЭЛЕМЕНТЫ IIА ПОДГРУППЫ. БЕРИЛЛИЙ, СТРОНЦИИ 121
Общая характеристика элементов . 121
Г л а в а 3 Бериллий 123
Химия бериллия . 123
3.1 Физические и химические свойства бериллия 123
3.2. Оксиды и гидроксид бериллия . ... 127
3 3 Соли кислородсодержащих кислот . 132
3 4 Соли органических кислот 133
3.5 Галогениды бериллия .... .135
3.6. Сплавы и интерметаллические соединения бериллия . 138
Технология бериллия . .140
3.7 Важнейшие области применения бериллия и его соединений . .140
3 8- Сырьевые источники бериллия...... 143
3 9. Технология извлечения бериллия из минерального сырья . . 145
3.10 Технология получения важнейших соединений бериллия . 153
3 11. Получение металлического бериллия 155
3.12. Техника безопасности в бериллиевом производстве .... 161
Глава 4 Стронций 162
Химия стронция 162
4.1 Общая характеристика . 162
4.2. Оксиды и гидроксид стронция .... 163
4 3. Соли кислородсодержащих кислот 164
4 4 Галогениды стронция . 165
4.5. Соединения стронция с неметаллами . 166
Технология стронция. . ........ .............. 167
4.6 Важнейшие области применения стронция и его соединений 167
4 7 Сырьевые источники стронция . 169
4.8. Производство соединений стронция . . ... 172
4 9. Переработка апатита 17 6
4.10 Получение металлического стронция 180
Раздел III РЕДКИЕ ЭЛЕМЕНТЫ III Б ПОДГРУППЫ.
РЕДКОЗЕМЕЛЬНЫЕ ЭЛЕМЕНТЫ И СКАНДИЙ 184
Общая характеристика элементов . 184
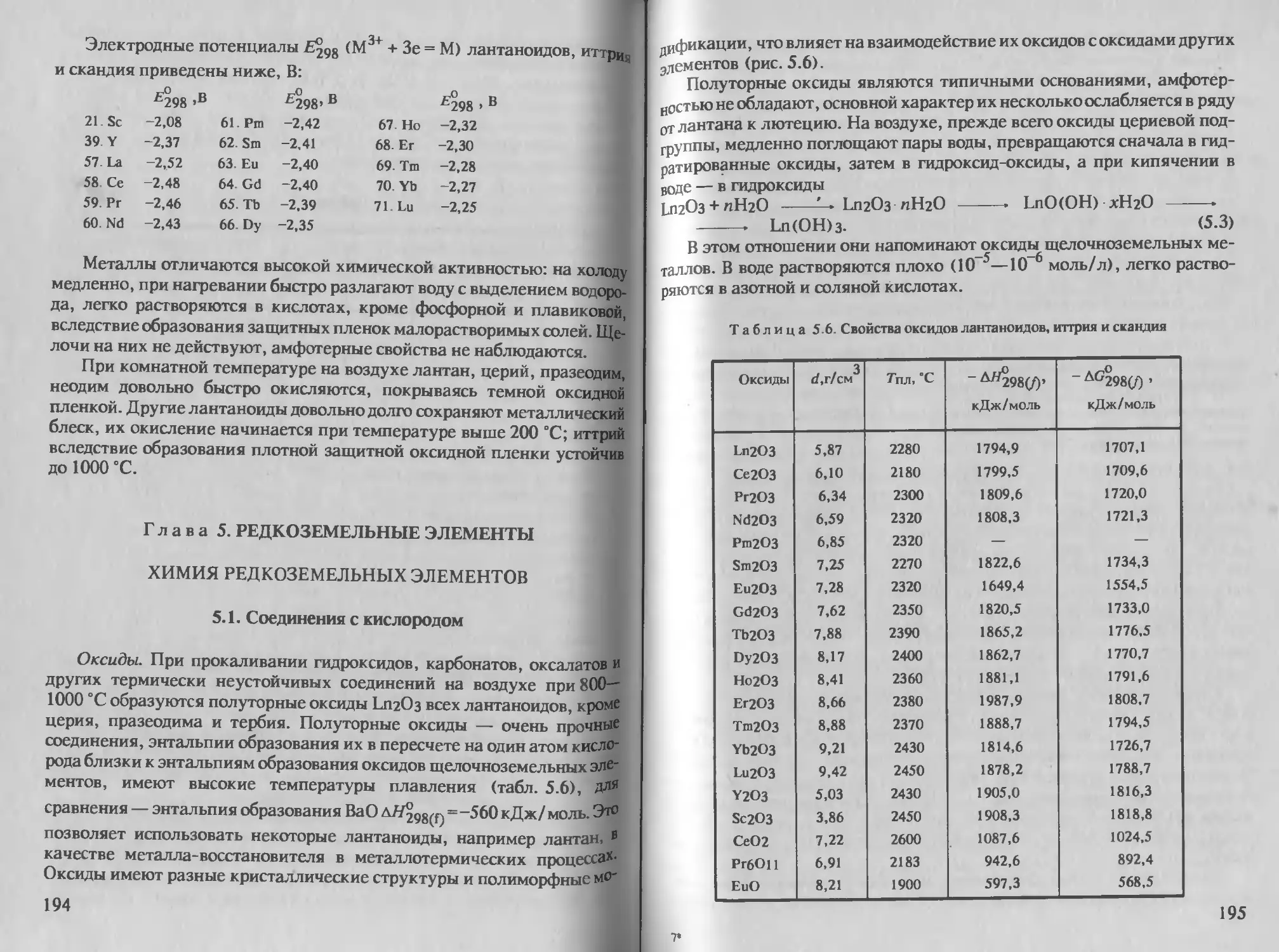
Глава 5 Редкоземельные элементы. . .... 194
Химия редкоземельных элементов. .... 194
5 1 Соединения с кислородом 194
5 2 Соли кислородсодержащих кислот ... 201
5.3 Галогениды .... . 208
5 4 Соединения с халькогенами 213
5 5 Соединения с неметаллами 216
5 6 Комплексообразование РЗЭ 219
Технология редкоземельных элементов..................................227
4
5 7 Важнейшие области применения РЗЭ и их соединений . . 227
5 8 Минералы РЗЭ, руды и месторождения 232
5 9 Общие вопросы технологии РЗЭ 236
5 10. Переработка монацита . 237
5 11. Переработка бастнезита 243
5 12. Выделение РЗЭ и тория из растворов сложного солевого состава 244
5.13. Разделение РЗЭ методами селективного окисления и восстановления . 245
5 14 Разделение РЗЭ методом ионообменной хроматографии 252
5 15. Экстракция. Общие закономерности 259
5 16. Разделение РЗЭ экстракцией 266
5 17. Принципиальные схемы разделения РЗЭ ... 275
5.18. Производство оксидов РЗЭ высокой степени чистоты 279
5.19. Получение полирующих материалов . 281
5.20. Металлотермия. Общие закономерности 282
5.21. Металлотермическое получение РЗМ 290
5.22. Электролитическое получение РЗМ . 293
5 23. Рафинирование РЗМ . 296
Глава 6. Скандий 297
Химия скандия..................................................... 297
6 1. Физические и химические свойства скандия . 297
6.2. Соединения скандия с кислородом. 298
6 3. Соли кислородсодержащих кислот . 301
6.4. Соединения с галогенами 304
6.5. Органические производные скандия ... 306
6 6. Соединения с неметаллами 308
6.7 Сплавы, легированные скандием 309
Технология скандия .... 310
6 8. Важнейшие области применения скандия и его соединений 310
6 9 Сырьевые источники скандия 313
6.10. Общие вопросы технологии скандия... ......314
6 11. Методы первичного концентрирования скандия .... 315
6 12 Экстракционный метод концентрирования и очистки скандия 320
6.13. Извлечение скандия из различных видов сырья . . 323
6 14 Получение металлического скандия . 329
Ра 3д е л IV. МЕТОДЫ МАТЕМАТИЧЕСКОГО МОДЕЛИРОВАНИЯ В ХИМИИ
И ТЕХНОЛОГИИ РЕДКИХ МЕТАЛЛОВ 332
Глава 7 Статистические методы обработки экспериментальных данных 334
Глава 8. Моделирование сложных физико-химических
равновесий в многофазных системах 338
8 1 Термодинамический подход 339
8.2. Методы оценки термодинамических характеристик
кристаллических соединений . 343
5
8 3 Химико-термодинамическое приближение
8 4 Прямая задача химического равновесия
350
351
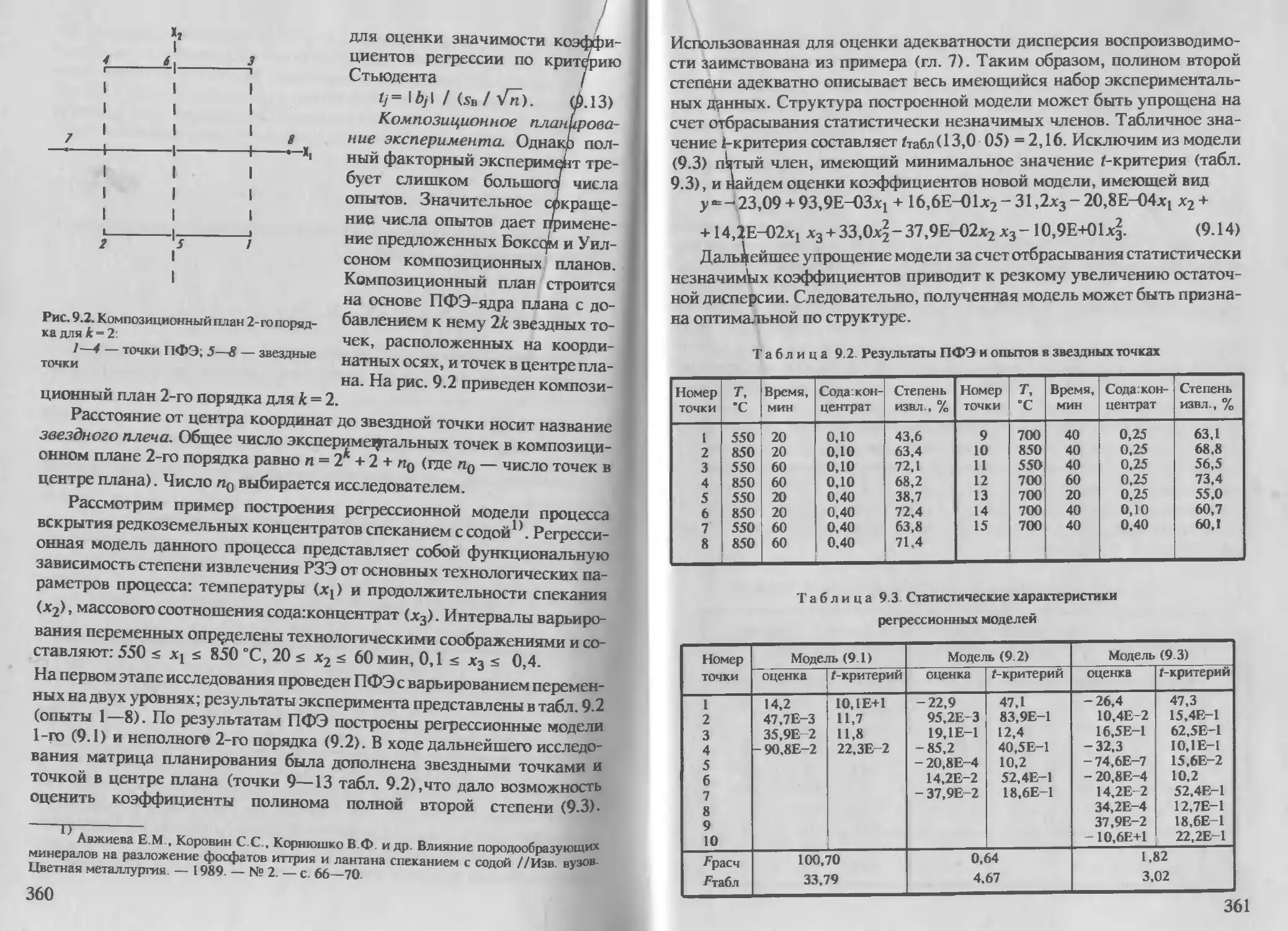
Г л а в а 9 Линейный регрессионный анализ и планирование эксперимента 354
9 1 Элементы множественного регрессионного анализа . 354
9 2. Планирование экспериментов ... . 357
9. 3. Планирование эксперимента на симплексе . 362
9. 4 Методы экспериментальной оптимизации 364
Контрольные вопросы и задачи . . 369
Рекомендательный библиографический список .............................375
6
ОБОЗНАЧЕНИЯ И СОКРАЩЕНИЯ
Порядковый номер элемента .
Температура
Температура плавления, кипения
Давление
Z
Т, °C, к
rri rrt
1 ПЛ» 1 кип»
Р, Па, кПа, МПа
Энтальпия реакции в стандартных условиях .. Л //298 , кДж/моль
Энтальпия образования соединения из элементов
в стандартных условиях и при 298 К . ... - Л //298(f) > кДж/МОЛЬ
Энергия Гиббса образования соединения из элементов
в стандартных условиях и при 298 К
AG^9g(f) , кДж/моль
Энтропия при 298 К . . . .
Энергия активации реакции
Энергия ионизации атома . .
Сродство к электрону
Работа выхода электрона
Ширина запрещенной зоны
>$298 , Дж/ (К МОЛЬ)
. А/Такт , кДж/моль
/1, /2, эВ
J , эВ
. А, эВ
. д Е, эВ
Электродный потенциал /?298 i В
Радиусы:
орбитальный • - Го, НМ
атомный (металлический, ковалентный) . . .. Гат (Гм , гк) , НМ
ионный (по Шеннону и Прюитту), к.ч 6 ги , НМ
Температура точки Кюри . . 0, К
Температура перехода в сверхпроводящее состояние .... 7с, К
Период полураспада радиоактивных изотопов Т1/2
Сечение захвата тепловых нейтронов... .1 барн =10 СМ^
Растворимость ..........................S, моль/л; г/л; %
7
Степень превращения
Коэффициент распределения. D
Коэффициент разделения уЗ
Металл М
Галоген.... . X
Лиганд .. . L
Лантаноиды ... . Ln
Масса атома или молекулы ... М
Алкильная или арильная группа ... ... R
Время. .... Г
Вода гидратная. ...... .... aq
Координационное число ... . . К.Ч.
Фаза (индексы) — твердая, жидкая, пар, газ . (тв), (ж), (п), (г)
Кристаллическая решетка
гранецентрированная куб. ГЦК
объемноцентрированная куб.. . . ОЦК
Структура плотноупакованная гексагональная . ГПУ
Твердый раствор . . ТР
Сегнетоэлектрик . . СЭ
Антисегнетоэлектрик АСЭ
Оптический квантовый генератор.... ОКГ
Тепловыделяющий элемент . ТВЭЛ
Химический источник тока ... .............ХИТ
Атомная орбиталь.... АО
Молекулярная орбиталь МО
Валентная связь................................ .ВС
Модифицированная теория кристаллического поля . МТКП
Алкильная или арильная группа........... . . R
8
Светлой памяти К. А. Большакова посвящается
ПРЕДИСЛОВИЕ
Без преувеличения можно сказать, что только благодаря широкому
применению редких элементов и их соединений стали возможны успе-
хи, достигнутые во многих областях современной техники: авиации и
космонавтике, атомной энергетике, полупроводниковой и квантовой
электронике, производстве конструкционных материалов, катализато-
ров и др. В настоящее время в промышленности в тех или иных масшта-
бах производятся все редкие элементы и потребность в них непрерывно
возрастает. Однако потенциальные возможности их использования да-
леко не исчерпаны. Ярким примером этого является открытие в конце
1986 г. высокотемпературной сверхпроводимости у сложных оксидов, в
состав которых входят редкие элементы — лантаноиды, иттрий, строн-
ций, таллий. Прогнозируется, что редкие элементы будут играть важ-
нейшую роль в решении глобальных проблем, по своим масштабам
сопоставимых с атомной и космической программами, — овладения
термоядерной энергетикой и использования возобновляемых источни-
ков энергии — солнечной, ветровой, океанической.
Возрастание потребности в редких элементах выдвигает новые на-
учные и технические проблемы: создание технически и экономически
эффективных процессов комплексной переработки редкоэлементного
минерального сырья и вторичных материалов с учетом требований ох-
раны окружающей среды, совершенствование методов получения ред-
ких элементов и их соединений высокой степени чистоты, создание на
их основе новых материалов, новых методов контроля и т.д. Решение
всех этих проблем невозможно без глубокого понимания и знания
химии и технологий редких элементов, а следовательно, и без специа-
листов, владеющих этими знаниями.
Учебное пособие «Химия и технология редких и рассеянных элемен-
тов» под редакцией члена-корр. АН СССР К.А. Большакова, предназ-
наченное для студентов, специализирующихся в области химии и тех-
нологии редких элементов, изданное в 1976 г., в значительной степени
устарело. За годы, прошедшие с момента его издания, объем научных
исследований в области химии и технологии редких элементов резко
возрос, появились новые данные, новые технологические процессы, из-
менился во многих случаях и подход к созданию новых технологий и
Управлению технологическими процессами, поэтому назрела настоя-
тельная необходимость в издании нового учебника на современном
Уровне.
9
В основе учебника лежит курс лекций, читающийся в течение мно-
гих лет на кафедре химии и технологии редких и рассеянных элементов
МИТХТ им. М.В. Ломоносова, он сохраняет известную преемственность
с пособием под редакцией К.А. Большакова. В частности, в нем сохранен
принцип изложения материала в соответствии с положением элементов
в периодической системе Д.И. Менделеева. Рассеянные элементы,
вследствие специфичности их технологии, рассматриваются отдельно.
Это позволяет дать глубокую групповую характеристику элементов на
основе электронного строения их атомов, выявить особенности химиче-
ского поведения, причины сходства и различия в свойствах.
В учебнике приводится довольно обширный материал по количест-
венным характеристикам элементов и их соединений — это необходимо
для выработки у будущих специалистов потребности оперировать не
качественными представлениями, а количественными данными, кото-
рые в определенном объеме должны быть доступны студентам. В то же
время учебник не является справочником. В нем дается физико-хими-
ческое обоснование технологических процессов, рассматриваются толь-
ко наиболее типичные и эффективные схемы переработки редкоэле-
ментного сырья и перспективные способы производства. Этому благо-
приятствует краткое рассмотрение теоретических основ процессов, име-
ющих общее значение в технологии редких элементов (твердофазные
реакции, ионный обмен, экстракция, металлотермия и др.).
Структура разделов, в каждом из которых рассматриваются элемен-
ты одной подгруппы периодической системы, примерно одинакова. По-
сле общей характеристики элементов данной группы рассматриваются
химия и технология отдельных элементов. В тех случаях, когда элемен-
ты обладают особо близкими свойствами, они рассматриваются совме-
стно.
Учебник включает раздел, в котором рассматриваются вопросы ма-
тематического моделирования в химии и технологии редких элементов,
а также раздел по проблемам переработки вторичного редкоэлементного
сырья.
Учебник состоит из трех книг. В кн. I — изложены химия и техноло-
гия лития, рубидия, цезия, бериллия, стронция, лантаноидов, иттрия,
скандия, математическое моделирование; во II кн. — химия и техноло-
гия титана, циркония и гафния, ванадия, ниобия, тантала, молибдена и
вольфрама; в III кн. — химия и технология рассеянных элементов —
галлия, индия, таллия, германия, селена, теллура, рения и проблемы
переработки вторичного редкоэлементного сырья.
Справочные данные, приведенные в учебнике, взяты из справочни-
ка под редакцией В.П. Глушко и других изданий, помещающих данные,
согласованные с государственной службой стандартных справочных
10
данных. Пересчет численных значений величин в единицы системы СИ
выполнен авторами.
Авторы глав книги I: введение, гл. 5 — С.С. Коровин, гл. 1 — С.С.
Коровин совместно с Г.В. Зиминой, гл. 2,4 — Г.В. Зимина, гл. 3 — В.И.
Букин, гл. 6 — А.М. Резник, гл. 7, 8, 9 — В.Ф. Корнюшко.
Авторы приносят благодарность за благожелательную и полезную
критику рецензентам: профессору А.М. Чекмареву и коллективу кафед-
ры металлургии редких металлов МИСиС во главе с профессором В.С.
Стрижко, а также старшему научному сотруднику кафедры химии и
технологии редких и рассеянных элементов МИТХТ Е.М. Авжиевой и
доценту кафедры информационных технологий О. А. Морозовой, прини-
мавшим активное участие в подготовке рукописи учебника.
Учебник выходит под общей редакцией заслуженного деятеля науки
и техники, проф., докт. хим. наук С.С. Коровина.
11
ВВЕДЕНИЕ
С исторической точки зрения небесполезно от
личать от общеизвестных... химических эле
ментов так называемые «редкие элементы»,
которые открыты гораздо позднее обычных
элементов уже по той причине, что или мате
риалы, служащие для извлечения, встречают
ся лишь в немногих местностях, или отделени
и очищение — по малости содержания — пред
ставляют большие трудности.
Д.И. Менделеев
Деление элементов на редкие и нередкие в какой-то мере условно i
не является однозначным. Высказывания многих ученых о целесообраз
ности и критериях такого деления разнообразны и противоречивы
Предлагалось в качестве основного критерия оценивать «степень изу
ченности, освоения в промышленности и использование в технике», а пс
мере роста промышленного производства переводить данный элемент и
«редких» в «обычные». Принятая сейчас точка зрения во многом совпа-
дает с соображениями по этому поводу Д.И. Менделеева. К числу редки}
относят элементы на основании совокупности критериев, поэтому гра-
ница между редкими и нередкими элементами остается размытой .
Важнейший критерий выделения редких элементов — геохимиче
ский; в соответствии с ним к редким следует относить малораспростра
ненные в природе элементы. Эта характеристика неизменна, но не абсо-
лютна. Условились считать «нередкими» все широко распространенные
элементы, содержание которых в земной коре (литосфере, гидросфере i-
атмосфере) >0,1 %. Таких элементов 15, на их долю приходится при
близительно 98 % массы земной коры. Кроме того, к числу нередки}
причисляют ряд элементов, содержание которых в земной коре находит
ся на уровне n 10-2 % (азот, барий, хром, никель, кобальт, бор).
К числу нередких относят элементы, известные с глубокой древно-
сти; таких элементов 12, из них только 3 (железо, сера, углерод) входяп
в группу широко распространенных элементов. Содержание же некото
рых из остальных 9 элементов (Золото, серебро, ртуть, свинец, олово
сурьма, цинк, медь, мышьяк) в земной коре меньше на несколько поряд
ков, чем многих элементов, признанных как редкие. Таким образом
В западной научной литературе используется термин «less common metals» — мене*
обычные металлы.
12
появляется историко-технический критерий .То, что эти элементы были
известны древним, не является случайностью — все они встречаются в
природе либо в самородном состоянии, либо образуют минералы и руды,
из которых их можно получить относительно легко. Редкие же элементы
входят, как правило, в состав сложных многокомпонентных минералов,
отличающихся большой устойчивостью к действию химических реаген-
тов. Это и предопределяет трудности их выделения и отделения от со-
путствующих элементов.
Все остальные природные элементы (их 62) следовало бы относить к
редким. Однако на основании общности и специфичности некоторых
свойств, выделили особые группы элементов. Так, вполне закономерно
выделена группа радиоактивных элементов (11); работа с ними требует
особых условий, сложилась самостоятельная отрасль промышленности,
занимающаяся их получением. Прослеживается вполне определенная
тенденция относить к редким только металлы, поэтому обычно в их
число не включают благородные газы (5) и галогены (3).
Таким образом, собственно редких остается 43 элемента, но титан,
занимающий по распространенности 10-е место и по геохимическому
критерию никак не может быть отнесен к редким элементам; по своим
свойствам, нахождению в природе, сложности технологии он весьма
близок к таким бесспорно редким элементам, как цирконий и гафний, и
обычно рассматривается вместе с редкими элементами. Имеет место
неопределенность относительно 5 элементов (стронций, кадмий, вис-
мут, селен, теллур), принадлежность их к редким элементам определя-
ется различными авторами произвольно.
Редкие элементы не обладают какими-либо общими физическими и
химическими свойствами, так как располагаются во всех группах Пери-
одической системы элементов Д.И.Менделеева:
I группа — литий, рубидий, цезий;
II группа — бериллий (стронций, кадмий);
III группа — скандий, иттрий, лантан, лантаноиды, галлий, индий,
таллий;
IV группа — (титан), цирконий, гафний, германий;
V группа — ванадий, ниобий, тантал (висмут);
VI группа — молибден, вольфрам, (селен, теллур);
VII группа — рений;
VIII группа — рутений, родий, палладий, осьмий, иридий, платина.
В группе редких элементов выделяют редкие платиновые металлы
(РПМ) очень близкие по свойствам, технология которых отличается
большим своеобразием. Иттрий, лантан и лантаноиды образуют группу
редкоземельных элементов (РЗЭ). Некоторые редкие элементы нахо-
дятся в природе в чрезвычайно рассеянном состоянии, не имеют про-
мышленных месторождений собственных минералов и руд. Это — руби-
13
дий, скандий, галлий, индий, таллий, германий, селен, теллур и рений.
Содержание их в земной коре от 1 10 7до1 10~4 %, только содержание
рубидия значительно выше — около 9 10~3 %. Рассеянность этих эле-
ментов нельзя объяснить неспособностью их образовывать собственные
минералы.
Для всех перечисленных элементов, кроме рубидия, известны (хотя
и редко встречающиеся) собственные минералы. Например, для скандия
— тортвейтит Sc2 [Si20? ], галлия — галлит CuGaS2, индия — индит
CuInSi и т.д. Рассеянность их объясняется тем, что у этих элементов есть
электронные аналоги и близкие по размерам атомов и ионов широко
распространенные элементы; наиболее яркие примеры: рубидий — ка-
лий, галлий — алюминий, селен, теллур — сера. При образовании
минералов широко распространенных элементов родственные им мало
распространенные элементы включаются в их состав в виде изоморфных
примесей. Это и является причиной их рассеяния. Содержание рассеян-
ных элементов в минералах-концентраторах может превышать их сред-
нее содержание в земной коре на 1—2 порядка, но редко бывает >0,1%.
Поэтому извлечение их возможно только как попутное при переработке
минерального сырья с целью получения основных компонентов.
К числу рассеянных элементов следовало бы отнести и гафний,
который не имеет собственных минералов. Но от других рассеянных
элементов он отличается тем, что встречается только в минералах, со-
держащих цирконий. В технологических операциях переработки цир-
кониевого сырья гафний всегда следует за цирконием, кроме специ-
альных операций по их разделению.
Производство редких элементов, в особенности с учетом современ-
ных тенденций получения на их основе разнообразных материалов,
металлов и соединений высокой степени чистоты, относится к числу
наукоемких. Особенности технологии редких элементов обусловлены
рядом факторов. Содержание редких элементов в рудах обычно не пре-
вышает нескольких процентов, поэтому для получения концентратов
используют сложные схемы обогащения рудного сырья. Месторождения
часто имеют комплексный характер, и на стадии обогащения получают
коллективные концентраты, состав которых изменчив как по содержа-
нию минералов редких элементов, так и сопутствующих минералов
горных пород. Это приводит к необходимости вводить коррективы в
технологию, а следовательно, усложняет задачи управления технологи-
ческими процессами, что во многих случаях осуществимо только на
основе применения компьютерной техники. Разделение близких по
свойствам редких элементов — редкоземельных, циркония и гафния,
ниобия и тантала и др., получение их и их соединений высокой степени
чистоты осуществимо в разделительных каскадах, имеющих десятки и
14
даже сотни ступеней разделения. Управление такими процессами, их
оптимизация требуют соответствующей научно-технической базы.
Большинство минералов редких элементов отличается высокой тер-
модинамической устойчивостью, и разложение их представляет собой
сложную задачу и требует, как правило, высоких температур. В то же
время многие редкие элементы отличаются высокой химической актив-
ностью, особенно при повышенных температурах. Это предопределяет
необходимость проведения многих технологических процессов в защит-
ной атмосфере, вакууме, безводной среде, применения специальных
контейнерных материалов и т.д. Технологические схемы переработки
редкоэлементного минерального и вторичного сырья отличаются много-
стадийностью и разнообразием.
В настоящее время в промышленных масштабах производятся прак-
тически все редкие элементы, причем промышленность редких элемен-
тов практически полностью была создана после второй мировой войны.
Это удалось сделать только благодаря необычайно широкому развитию
научных исследований в области химии и технологии редких элементов,
разработке новых принципов концентрирования, разделения, восста-
новления соединений редких элементов и создания на этой основе эф-
фективных технологических процессов.
Дальнейшее развитие промышленности редких элементов связано с
решением ряда сложных научно-технических проблем: создания новых
эффективных технологических схем переработки минерального сырья с
учетом его ухудшения, снижения в нем содержания ценных компонен-
тов; вовлечения в переработку новых нетрадиционных видов сырья, в
том числе и вторичного; совершенствования существующих произ-
водств с учетом требований экологии. Ряд этих проблем может быть
решен путем создания гибких технологических схем и модульного по-
строения технологии.
Особое значение наряду с расширением потребления и применения
редких элементов и их соединений в традиционных областях приобре-
тает проблема создания различных функциональных материалов на
основе их соединений (сегнето- и пьезоэлектриков, высокотемператур-
ных сверхпроводников, пироэлектриков, преобразователей излучений
и т.д.), а также различных конструкционных материалов. Значение их
для техники будущего трудно переоценить, но решение этой проблемы
связано с успехами комплекса наук, начиная с физики и химии твердого
тела, решения практических задач, прежде всего получения веществ
высокой степени чистоты, разработки методов синтеза материалов оп-
ределенного состава с высокой химической и фазовой однородностью и
воспроизводимыми свойствами.
15
Раздел I. Редкие элементы IA подгруппы.
Литий, рубидий и цезий
ОБЩАЯ ХАРАКТЕРИСТИКА ЭЛЕМЕНТОВ
Из шести элементов главной А подгруппы I группы Периодической
системы Д.И. Менделеева три (Li, Rb и Cs) — редкие элементы, Fr —
редкий радиоактивный элемент и только натрий и калий относятся к
обычным широко распространенным элементам. Все они — щелочные
элементы, их атомы имеют по одному валентному s-электрону сверх
оболочки соответствующего благородного газа. Это предопределяет
большое сходство всех щелочных элементов, хотя и не исключает опре-
деленных различий в их свойствах. Химия этих элементов является
наиболее простой по сравнению с химией элементов любой другой груп-
пы.
Вследствие низких энергий ионизации Ji атомы этих элементов лег-
ко образуют ионы М , другие степени окисления для них не известны
из-за высоких значений Ji (табл. 1.1). Они — самые электроположи-
тельные металлы, при нагревании непосредственно реагируют с боль-
шинством других элементов и многими соединениями. При движении
по группе от Li к Cs обнаруживаются общие закономерности, связанные
с усилением металлических свойств. Но при этом литий — первый
элемент подгруппы, проявляя свойства щелочного металла, имеет зна-
чительные отличия от своих аналогов.
Аномальные свойства лития определяются главным образом неболь-
шими размерами его атома и иона. Атом лития поэтому характеризуется
большей силой притяжения электрона, что видно из сравнения энергий
ионизации для него и остальных щелочных элементов. Ион лития, как
и ионы других щелочных элементов — сферический, но в отличие от них
практически не поляризуется, а поляризующая способность является
наивысшей среди ионов щелочных элементов. Литий обладает меньшей
реакционной способностью, чем его аналоги — Na, К, Rb, Cs. Для него
характерна тенденция к образованию соединений со значительной ко-
валентностью связей. В растворах ион лития сильно сольватирован,
энтальпия гидратации и гидратное число для него значительно больше,
чем для других щелочных элементов (см. табл. 1.1). Это обусловливает
сходство лития с магнием, причем это явление, носящее название «диа-
гональной периодичности», наблюдается у всех элементов 2-го малого
периода и заключается в большом сходстве их с элементами 3-го периода
следующей группы.
16
Таблица 1.1. Свойства Li, Na, К, Rb, Cs
Свойства Li Na К Rb Cs
Атомный номер Z 3 11 19 37 55
Атомная масса 6,94 22,99 39,10 85,47 132,91
Плотность d, г/см 0,53 0,97 0,86 1,52 1,89
Температура плавления 7пл, “С 180,5 99,7 63,7 39 28,6
Температура кипения 7кип< 'С 1317 880 762,2 698 670
Энергия ионизации, эВ: Л 5,39 5,18 4,35 4,15 3,96
J2 75,66 47,26 31,60 27,26 23,11
Работа выхода электрона А, эВ 2,36 2,33 2,26 2,13 1,93
Атомный радиус га. нм 0,155 0,189 0,236 0,248 0.268
Ионный радиус (к.ч.6) г_ , нм М “ 0,090 0,116 0,152 0,166 0,181
Радиус гидратированного иона aq, нм * 0,340 0,276 0,232 0,228 0,229
») + Гидратное число иона М 25.3 16.6 10,5 10,0 9,9
Энтальпия гидратации иона М+ — AH^-j , кДж/моль 520 406 322 301 277
*Сечение захвата тепловых нейтронов, барн 71 4 3,7 12 50
Электродный потенциал (расплав) ^298 ’ ® —2,10 —2,43 —2,61 -2,74 —2,91
Электродный потенциал (водн. раствор) ^298 ’ в —3,05 —2,71 —2,92 —2,93 —2,92
Удельная теплоемкость (20 ”С), Дж / (г град) 3,39 1,20 0,74 0,33 0,21
Приближенные значения.
Природный литий имеет два изотопа Li (7,52 %) и Li (92,48 %), для которых
сечение захвата тепловых нейтронов соответственно равны 912 и 0,033 барна. Ба^>н —
единица эффективного поперечного сечения ядерных взаимодействий, равная 10 см
Она характеризует вероятность ядерных реакций, для многих атомов она приб-
лизительно равна геометрическому сечению атомного ядра, которое находится в пределах
(0,5—2,0) 10“24 см2 _
Б I Б Л 1 О Т Е К А
Я»‘ТНГ».ХГ»-о 17
ДЕГ*. XIM-TEX
VkHMCb^lJTCTV
Различия лития и его аналогов, сходство с магнием можно оценить,
используя соотношение: заряд иона/ионный радиус (ионный потенци-
ал) , количественно характеризующее поляризующую способность ка-
тиона
Ф = г+/ги, (1.1)
где Z — заряд иона; ги — радиус иона.
Некоторые значения ионных потенциалов приведены ниже, нм-1:
Li+ . . . . 17 Na+ .... 10 К+ . . . . 8
2+ 2+ 2+
Ве ...64 Mg ...31 Са . ..20
Чем больше ионный потенциал, тем выше поляризующая способ-
ность иона; в первом приближении можно считать, что с увеличением
поляризующей способности катиона увеличивается ковалентная сос-
тавляющая ионной связи. Из приведенных данных видно, что Li+ и Mg2+
обладают более сильной поляризующей способностью, чем натрий и
другие щелочные ионы. В то же время разница в значениях Ф для пар
Li —Na и Li —Mgz , выраженная в относительных единицах, почти
одинакова, поэтому этот фактор не является единственным достаточ-
ным для объяснения диагональной периодичности.
Остановимся на некоторых свойствах лития и его соединений, отли-
чающих его от других щелочных элементов и сближающих с магнием.
Разница в величинах ионных радиусов Li+ и Na+ > 26 % (для других
щелочных элементов еще больше), для пары же Li+ и Mg + она < 10 %.
Поэтому взаимное изоморфное замещение ионов 1л и Na+ маловероят-
но, взаимный изоморфизм ионов Li+ и Mg2+ наблюдается достаточно
часто.
В отличие от всех щелочных элементов (но подобно магнию) литий
непосредственно реагирует с азотом, образуя нитрид при комнатной
температуре.
Растворимость фторида, фосфата и карбоната лития в отличие
от аналогичных солей его аналогов очень мала, и в этом он напо-
минает соли магния. Реакции термического разложения некоторых со-
лей лития протекают при относительно невысоких температурах, когда
аналогичные соединения других щелочных элементов термически ус-
тойчивы (например, термическое разложение IJ2CO3 начинается при
550—600 °C).
Карбонат магния также оказывается менее стойким по сравнению с
карбонатами других элементов его подгруппы. Он начинает разлагаться
при 500, а при 650 °C давление диссоциации становится равным атмос-
ферному. Менее термически стойки по сравнению с аналогичными сое-
динениями других щелочных элементов гидроксид и гидросульфид ли-
тия, которые довольно легко распадаются соответственно на оксид и
сульфид.
18
В раду Li—Cs увеличивается способность стабилизировать некото-
рые большие анионы, низкая же стабильность солей лития с такими
анионами, по-видимому, является следствием его малых размеров и
очень высокой поляризующей способности. Это и является одной из
причин термической неустойчивости карбоната лития. Вследствие того,
что ионы щелочных элементов мало склонны к образованию ковалент-
ных связей, они (кроме лития) практически не бывают центральными
атомами комплексов. Но они принимают участие в образовании комп-
лексных соединений в качестве внешнесферных ионов, образуя своеоб-
разные ионные пары, причем литий в качестве внешнесферного иона
выступает крайне редко. Устойчивость таких комплексов возрастает от
лития к цезию.
С увеличением размеров ионов щелочных элементов изменяются
координационные числа и структуры кристаллических солей. Так, в
хлоридах крупный ион Cs окружен восемью ионами хлора. Такая ОЦК
структура стабильна при условии 1,0 < г /г+ < 1,37; если это соотноше-
ние > 1,37, то стабильны кристаллические структуры с к.ч.б. При этом
реализуется структура ГЦК, которую имеют КС1, NaCl и LiCl. Верхним
пределом стабильности этого типа структуры является соотношение
2,44; к нему очень близко соотношение для хлорида лития, и структура
типа ГЦК для него находится на грани метастабильности.
Различия в химическом поведении, связанные с размерами атомов
и ионов щелочных металлов, проявляются в реакциях с кислородом. На
воздухе все компактные щелочные металлы горят. При этом литий
образует только оксид LiiO со следами пероксида IJ2O2. Натрий образу-
ет обычно пероксид ИагОг, но при нагревании под давлением кислорода
он способен поглощать его с образованием надпероксида NaC>2- Рубидий
и цезий образуют оксиды R ЬгО и СвгО только при недостатке кислорода,
на воздухе — пероксиды РЬгОг и CS2O2, в кислороде — надпероксиды
КЬОг и СвОг, а при действии смеси кислорода и озона — озониды RbO3
и СвОз. Литий, натрий и калий можно обрабатывать на воздухе, хотя
они быстро тускнеют вследствие образования оксидной пленки, рубидий
и цезий обрабатывают только в среде аргона.
Изменение реакционной способности в раду щелочных элементов
можно проследить на многих реакциях. Например, литий реагирует с
водородом в интервале температур от 400 до 600 °C, в то время как для
остальных щелочных элементов эта реакция полностью завершается
при 350—400 °C. Сравнение свойств солеобразных гидридов щелочных
элементов МН показывает, что вследствие большей ковалентности свя-
зи Li—Н, чем связи М—Н в гидридах других щелочных элементов,
гидрид LiH обладает большей термической устойчивостью, он не разла-
гается, когда, например, NaH полностью диссоциирует на Na и Нг.
19
Аналогичную закономерность можно проследить и на реакциях ще-
лочных элементов с водой. Литий при комнатной температуре лишь
медленно взаимодействует с водой с выделением водорода, натрий —
бурно реагирует, калий — воспламеняется, а рубидий и цезий взаимо-
действуют со взрывом.
Усиление реакционной способности в ряду Li—Cs четко прослежи-
вается по значениям электродных потенциалов для расплавов — их
отрицательные значения закономерно увеличиваются (см. табл. 1.1). В
кажущемся противоречии с этими данными находится аномально высо-
кое значение электродного потенциала лития для водного раствора. Это
связано со значительно большей гидратацией иона лития, вследствие^
чего равновесие реакции очень сильно смещается в сторону образования
акваиона лития
Li(TB) X Li+ aq + e. (1.2)
В первой гидратной оболочке иона лития находятся 4 молекулы
воды. Тетраэдрическое окружение ионов лития наблюдается и в кри-
сталлических солях лития. Ионы Na+ и К+ в первой гидратной оболочке
также содержат по 4 молекулы воды, а рубидий и цезий, по всей вероят-1
носги, координируют по 6 молекул воды. Электростатическое взаимо-1
действие между поляризованными молекулами воды первой гидратной!
оболочки и последующими слоями молекул изменяется обратно пропор- >
ционально размерам негидратированного катиона. Это приврдит к
уменьшению гидратного числа и размеров акваиона и, как следствие,I
увеличению его подвижности в растворе, способности проходить через!
мембраны, увеличивается способность сорбироваться на ионообменных!
смолах вследствие увеличения прочности связи со смолой.
Все щелочные металлы имеют объемноцентрированную решетку!
(к.ч.8). Энергия связи в металлах невелика, поскольку их атомы отдаютI
на образование металлической связи только по одному электрону, поэ-
тому все щелочные металлы имеют низкие температуры плавления и
кипения и отличаются большой пластичностью. Вследствие малых раз
меров атома лития его кристаллическая решетка обладает наибольшей!
прочностью. По этой же причине литий имеет более высокие темпера-I
туры плавления и кипения и большую твердость, чем остальные щелоч-1
ные металлы (см. табл. 1.1).
20
Глава 1.ЛИТИЙ
ХИМИЯ лития
1.1. Оксиды и гидроксиды
Литий в соединениях с кислородом имеет степень окисления +1, но
образует оксиды, различающиеся по составу и заряду анионов О ,
О2- , отвечающие формальным степеням окисления кислорода -2 и -1.
Все остальные щелочные элементы образуют такие же оксиды и, кроме
того, для них известны оксиды с анионами О2 и О3 (степени окисления
кислорода -0,5 и -1/3). Объяснение наблюдаемой закономерности не
так просто, поскольку здесь действуют различные факторы, причем
противоположно направленные: повышение стабилизирующей способ-
ности для крупных анионов в ряду Li—Cs, невысокая стабильность
аниона О2 в свободном состоянии, повышение относительной устойчи-
вости многоатомных кислородных анионов при увеличении числа ато-
мов кислорода и уменьшении эффективного отрицательного заряда,
приходящегося на один атом кислорода.
Термодинамические характеристики (хотя и многие из них требуют
уточнения) позволяют сделать вывод о том, что усиление тенденции к
образованию оксидов типа М2О2, МОг у рубидия и цезия отражает не
повышение их устойчивости, а увеличение их конкурентной способно-
сти по отношению к оксиду типа МгО соответствующего металла (рис.
1.1, табл. 1.2, 1.3). Это происходит прежде всего за счет уменьшения
устойчивости последних в ряду Li—Cs, что отражается в значительном
уменьшении энтальпии их образования. Зависимость энтальпии обра-
зования оксидов от номера щелочного металла не является монотонной,
она распадается на два участка
Li—Na и К—Rb—Cs. Аналогично
и энергия ионизации Ji изменяет-
ся более значительно при переходе
от Li к Na, чем для других пар эле-
ментов. Немонотонность затруд-
няет интерполяцию свойств соеди-
нений щелочных элементов при
их прогнозировании, а также яв-
ляется основанием деления ще-
лочных элементов на две подгруп-
пы.
Li No К КЬ
Рис. 1.1. Энтальпия образования оксидов
щелочных элементов (на 2 г-атома метал
ла):
1 — М2О; 2 — М2О2; 3 — МО2; 4 —
МОЗ
21
Таблица 1.2. Оксиды щелочных металлов
Оксиды Li2O Na2O К2О Rb2O Cs2O
Пероксиды L12U2 I Na2O2 К2О2 Rb2O2 Cs2O2
Надпероксиды L1O2 № 1 КО2 RbO2 CsO2
Озониды — NaO3 'KITS ‘ R5U3 Ls(J3
Примечание. Ниже жирной черты — неустойчивые соединения.
Оксиды типа М2О всех щелочных элементов характеризуются очень
высокими значениями энтальпии сублимации; для наиболее прочного
U2O эта величина равна 440 кДж/моль. Поэтому для перехода их в
газовую фазу нужны высокие температуры, а это приводит к термиче-
ской диссоциации. Область существования твердого U2O — от комнат-
ной температуры до температуры плавления, которая определена раз-
ными авторами от 1400 до 1600 °C, в расплавленном состоянии — до
температуры кипения (~ 2600—2700 °C). Но уже при температуре выше
1000 °C оксид начинает сублимировать с разложением, при 1400 °C в
парах содержатся главным образом Li, О2, около 10 % U2O и только ~
0,2 % Li2O2.
Оксид лития Ы2О можно получить при непосредственном окислении
металлического лития при температуре выше 200 °C разложением
IJCO3, L1NO3, LiOH в токе сухого водорода — выше 800 °C. Пероксид
Ы2О2 получают косвенным путем, при температуре > 195 °C он практи-
чески полностью разлагается на U2O и Ог-
Таблица! 3. Энтальпия и энергия Гиббса образования
кристаллических оксидов щелочных металлов,
кДж на 2 г-атома металла
Ok- - д #298(f) - д c298(f)
СИД Li Na К Rb Cs Li Na К Rb Cs
M2O M2O2 2MO2 2MO3 **д 597 416 362 331 318 634. 511 494 425 402 611** 520. 566 570 580. 527** 456*’ 519 439** 439*’ анные, требующие уточнения. 561 393 322 301 318*) 565. 435 418 314 289 469.* 377. 427 427. 435. 301 * 230** 293 289** 289**
Оксид 1дгО взаимодействует как с жидкой водой, так и с ее парами
с образованием гидроксида LiOH, но менее энергично, чем оксиды дру-
22
гих щелочных элементов; из воздуха поглощает СО?, образуя Ы2СО3.
Пероксид лития, так же как и пероксиды других щелочных элементов,
является сильнейшим окислителем. При взаимодействии с водой они
образуют соответствующие гидроксиды, пероксид водорода и газообраз-
ный кислород.
Оксид лития с оксидами переходных элементов IV и V групп перио-
дической системы образует многочисленные соединения. Из них наи-
больший интерес представляют соединения LiNbOa и ЫТаОз. Оба соеди-
нения принадлежат к группе кислородно-октаэдрических сегнетоэлек-
триков АВОз, хотя их структура не является перовскитоподобной. Нио-
бат и танталат лития обладают уникальным комплексом разнообразных
свойств; их можно вырастить в виде больших монокристаллов оптиче-
ского качества. Известны также и более сложные соединения, например
LiBaiNbsOis, КзЫгМЬ5О15, КзЫгТа5О15 и др.
Не меньший научный и практический интерес представляют соеди-
нения оксидов лития и алюминия. Из водных щелочных растворов вы-
деляется плохо растворимый в воде алюминат лития, которому обычно
приписывают стехиометрический состав ЫАЮг. Изучение же системы
ЫгО—AI2O3—НгО при 50 °C показало, что равновесная твердая фаза
является фазой переменного состава и близка к гидроалюминату лития
состава (1-х)ЫгО 2AI2O3 иНгО, где х = 0,12—0,21, число молекул
воды 8, растворимость ~ 0,14 %. Кристаллический моноалюминат ли-
тия ЫАЮг, видимо, можно получить нагреванием эквимолекулярной
смеси Ы2СО3 и AI2O3 при 900—1200 °C.
В безводных системах, образованных оксидами щелочных металлов
и алюминия, существуют полиалюминаты с общими формулами
(1+х)МгО IIAL2O3, гдех=0,10—1,07и (1-х) МгО 5А1гОз, х=0—0,45.
Оба соединения являются типичными нестехиометрическими фазами и
обозначаются соответственно как fl- и /?"—А1гОз-глиноземы. Природа
обеих фаз до конца не выяснена, нет надежных данных о температурных
интервалах существования фаз, значения х — разные у различных
авторов. Здесь приведены значения для натриевых/?- и/?"-глиноземов.
Оба типа полиалюминатов при температуре > 1500 °C вследствие испа-
рения теряют оксиды щелочных металлов и претерпевают необратимые
превращения.
Для алюминатов стехиометрического состава определены энталь-
пии образования их оксидов, кДж/моль: A//^98(f) = -105 для ЫАЮг;
ДЯ$98(П = для LiAbOe; A//298(f) = -16,7 кДж/моль для, видимо,
термодинамически нестабильного Ы5АЮ4-
Структура /3-глиноземов слоистая, в ее основе лежат двумерные
шпинельные блоки, перпендикулярные оси с. Связь между блоками
осуществляется через атомы кислорода, в щелях между блоками нахо-
23
дятся ионы щелочных элементов или водорода, обладающие большой
подвижностью. Наибольшей подвижностью обладает ион Li*. Это пред-
определяет использование/3-глиноземов в качестве твердых электроли-
тов для химических источников тока (ХИТ) и датчиков для определения
соответствующих металлов в водных растворах и металлических рас-
плавах. Твердый электролит на основе 1л+/3-глинозема имеет одну из
самых высоких электропроводностей 2 10-3—3,7 Ю30м' см1 при
25 °C. Нагревание до 675 °C не оказывает существенного влияния на
электропроводность. Высокой электропроводностью обладает^"-глино-
зем, содержащий Li : Na в атомном отношении 1:1. Известен также и
Cs ^"-глинозем.
Оксид лития активно взаимодействует с большинством металлов и
со многими соединениями. Ниже 1000 °C по отношению к нему устой-
чивы только Ni, Au и Pt, выше 1000 °C устойчив только сплав Pt с 40 %
Rh; Al, Mg, Si восстанавливают Li2O до металла; он разрушает также
большинство коррозионностойких неметаллических материалов.
Гидроксиды щелочных элементов относятся к истинным гидрокси-
дам; их кристаллическая структура имеет две подрешетки: катионную
и анионную, в узлах последней находятся ионы ОН-. Они плавятся и
кипят при относительно высоких температурах без разложения, кроме
гидроксида лития, который недостаточно термически устойчив (табл.
1.4). Это иллюстрируется данными по энтальпиям образования гидро-
ксидов (рис. 1.2). Расплавленный LiOH при температуре > 500 °C начи-
нает диссоциировать на твердый Li2O и пары Н2О. Давление паров воды
достигает атмосферного при 927 °C, т.е. при температуре более низкой,
чем температура кипения, определенная экстраполяцией (~ 1500 °C под
давлением). Это говорит об обратимости реакции образования LiOH при
относительно невысоких температурах
1л2О(тв) + Н2О(г) 2 21дОН(тв);- дН^98 = 138 Дж. (1.3)
Гидроксид лития остается не-
устойчивым до 1900 °C, а затем в
интервале температур 1900—
3200 °C снова становится устойчи-
вым. Это происходит вследствие
того, что образование газообраз-
ного LiOH, идущее с большим эн-
доэффектом, становится более ве-
роятным при повышении темпера-
туры. Меньшая термическая ус-
тойчивость гидроксида лития при
относительно невысоких темпера-
турах является свидетельством
24
Рис. 1.2. Энтальпия образования гидрокси-
дов щелочных металлов МОН из твердого
М2О и жидкой Н2О (на 1 моль МОН)
сходства Li и Mg, гидроксид которого также менее стоек по сравнению с
гидроксидами его аналогов.
Таблица 1.4. Свойства гидроксидов щелочных металлов
Гидроксид Гпл. °C 7кип, °C , / 3 а, г/см S(15°C), моль/л “А «298(0 > кДж/моль
LiOH 471 1550#) 2,54 5,3 69,0
NaOH 328 1388 2,13 26,4 100,4
КОН 380 1324 2,12 19,1 121,3
RbOH 328 1200 3,20 17,9 117,2
CsOH 346 1100 3,68 25,8 113,0
*) Под давлением
Для LiOH характерна более низкая по сравнению с гидроксидами
других щелочных элементов растворимость в воде, которая увеличива-
ется при повышении температуры (см. табл. 1.4, рис. 1.3). Из вод-
ных растворов гидроксида лития кристаллизуется моногидрат LiOH
НгО, который теряет кристаллизационную воду только при температу-
ре > 600 °C. Гидроксид лития менее гигроскопичен, чем NaOH и КОН,
но на воздухе легко карбонизуется, превращаясь в IJ2CO3. При обычной
температуре гидроксид лития и его концентрированные растворы раз-
рушают стекло и фарфор, его можно хранить в парафинированных или
во фторопластовых сосудах. В расплавленном состоянии он разрушает
все металлы, кроме Au, Ag и Ni.
Гидроксид лития можно получить электролизом на ртутном като-
де, в результате чего получается литие-
вая амальгама, которую разлагают во-
дой. Наибольшее же практическое зна-
чение имеют обменные реакции типа
1д2СОз + Са(ОН)2
2 2UOH + СаСОз |. (L4)
В качестве реагентов для этой цели
могут быть использованы гидроксиды К,
Ва, Са, в качестве исходных соединений
лития — сульфат или карбонат. На прак-
тике же наиболее часто используют гид-
роксид кальция, а поскольку реакция
проводится в присутствии воды, то и
СаО. Такой выбор реагентов диктуется
Рис. 1.3. Политерма растворимо-
сти LiOH в воде
25
тем, что реакция типа (1.4) может пройти до конца только при
выведения одного из продуктов реакции в осадок.
1.2. Соли кислородсодержащих кислот
Сульфат лития. Сульфат U2SO4 — наиболее устойчивая соль ли-
тия ( -Д//298(f) = 1434 кДж/моль), хорошо растворяется в воде, при
температуре выше О °C имеет отрицательный коэффициент раствори-
мости (рис. 1.4). Из водных растворов в широком интервале температур,
выделяется в виде моногидрата U2SO4 Н2О , который можно обезво-
дить при ~ 500 °C. Температура плавления U2SO4 859 °C. В органиче-
ских растворителях (спирте,
ацетоне и др.) не растворяется. С;
сульфатами других щелочных ме-
таллов образует двойные соли!
(MUSO4, Na3Li(SO4)2 • 6Н2О и
др.), но в отличие от сульфатов
других щелочных металлов в обы-
чных температурных условиях не
образует соединений типа квас-
цов. Алюмолитиевые квасцы (до-
декагидрат сульфата алюминия
лития) LiAl(SO4)2 12НгО суще-
ствует только в узкой области кон-
центраций сульфата лития и алю-
Т,‘С
Рис. 1.4. Политерма растворимости L12SO4
в воде
миния при температуре < —2 °C.
Нитрат лития. Нитрат LiNOa — термически неустойчивое со-
единение ( - Д//298(f) = 428 кДж/моль), имеет температуру плавления!
254 °C, при 600 °C начинает разлагаться с выделением кислорода и
оксидов азота. Очень гигроскопичен, хорошо растворяется в воде, рас-
творимость увеличивается с повышением температуры, образует пере-|
сыщенные растворы:
Температура, °C.... 10,5 22,1 29,9 50,5 80 100
Растворимость
LiNO3, %......... 37,9 42,6 52,6 61,3 67,1 70,1
В водном растворе LiNOs сильно диссоциирован, степень диссоциа-
ции в 0,1 молярном растворе 84 %, а в 0,001 молярном 97,5 %. Из водных
растворов при температуре ниже 30 °C кристаллизуется LiNO3
ЗН2О, при более высокой — безводный LiNOs. Для выделения твердого
нитрата лития растворы упаривают предпочтительно в вакууме. Раство-
ряется в спирте, ацетоне и других органических растворителях.
26
Фосфат лития. Фосфат Ы3РО4 — термически устойчивое соедине-
ние, плавится без разложения при 837 °C. Это одна из наименее раство-
римых солей лития; при О °C растворимость равна 0,022 г/ 100г НгО, а
при 18 °C 0,034 г/100г НгО. В присутствии аммиака растворимость
Ы3РО4 уменьшается, а в присутствии аммонийных солей, например
NH4CI, — увеличивается. Из водных растворов при обычной темпера-
туре осаждается LJ3PO4 2НгО, который после длительного высушива-
ния при 60 °C превращается в L13PO4 0,5НгО, а выше 120 °C обез-
воживается полностью.
При действии на Ы3РО4 избытка фосфорной кислоты, сильных кис-
лот (соляной или азотной) происходит образование гидрофосфата
L12HPO4 и дигидрофосфата L1H2PO4. Гидрофосфат лития в чистом виде
выделить не удается, так как равновесие реакции сдвинуть влево трудно
21лгНРО4 2 U3PO4+UH2PO4. (1.5)
Дигидрофосфат можно выделить в твердом состоянии. Оба гидро-
фосфата лучше растворяются в воде, чем средний фосфат, поэтому для
полноты выделения последнего из растворов необходимо добавлять
NaOH для нейтрализации и предотвращения образования гидрофосфа-
тов.
Малая растворимость Ы3РО4 используется в технологии для доизв-
лечения лития после, например, осаждения U2CO3. Так как сам фосфат
лития не имеет самостоятельного применения, то его перерабатывают
на гидроксид обработкой оксидом кальция (известью) в водном растворе
или на LiCl в расплаве при 850 °C обработкой хлоридом кальция:
2ЫзРО4 + ЗСа(ОН)2 г Саз(РО4)2 + 6Ь1О; (1.6)
2Li3PO4 + ЗСаС1г Z Саз(РО4)2 + 61дС1.
Карбонат лития. Карбонат U2CO3 —
термически неустойчивое соединение, пла-
вится при 732 °C, но уже при этой темпера-
туре становится заметной его термическая
диссоциация
П2СОз 2 Li2O + СО2.
Давление СОг становится равным ат-
мосферному при 1270 °C, карбонат лития
менее стоек, чем карбонаты натрия и калия
(рис. 1.5); его термическая диссоциация ус-
коряется в вакууме, а также в присутствии
Углерода вследствие восстановления СОг до
СО и смещения равновесия реакции. Выше
температуры плавления реакционная мас-
са, содержащая U2CO3 и U2O, необычайно
(1.7)
Рис. 1.5. Зависимость давления
СО2 при термической диссоци-
ации карбонатов от температу-
ры:
1 — L12CO3; 2 — Na2CO3;
3 — К2СОЗ
27
агрессивна — разрушает корунд, диоксид циркония, платину.
Карбонат L12CO3 — одна из плохо растворимых солей лития: <
растворимость значительно ниже растворимости карбонатов натрия
калия, которая для них соответственно равна 17,7 и 52,5 %
Температура, °C......... О 10 20 50 75 100
Растворимость L12CO3, %. 1,54 1,41 1,30 1,18 0,87 0,73
Карбонат лития не образует двойных или комплексных соединений
с карбонатами щелочных металлов, поэтому при добавлении в раствор
их избытка вследствие высаливающего действия одноименного иона
растворимость карбоната лития уменьшается.
При пропускании СОг через водную суспензию U2CO3 происходит
его растворение вследствие образования более растворимого гидрокар-
боната
U2CO3+CO2 + H2O ? 2ЦНСОз. (1.8)
Гидрокарбонат может существовать в водном растворе в равновесии
с СОг при температуре < 90 °C, при нагревании в обычных условиях он
разрушается. Растворимость гидрокарбоната уменьшается при повыше-
нии температуры:
Температура, °C.. 2 25 90
L1HCO3, г/л...... 126,2 75,9 24,6
На практике U2CO3 осаждают из нитратных, хлоридных и сульфат-
ных растворов, хотя в присутствии сульфатов щелочных металлов его
растворимость несколько увеличивается. Для этой цели обычно исполь-
зуют твердый карбонат натрия или его насыщенный раствор, реже
К2СО3. Карбонат лития выделяется в виде безводной соли.
Силикаты лития. В системе Ы2О—SiO2 установлено образование
следующих силикатов лития: Li4SiO4, 1дг£Юз и Li2Si2O5, соответствен-
но имеющих Температуры плавления 1250, 1201 и 1032 °C. Силикаты
Li4SiC>4 и Li2Si2O5 плавятся инконгруэнтно. В отличие от силикатов
натрия Li4SiC>4 и калия K4SiC>4 они в воде практически не растворяются,
но разлагаются кислотами с выделением SiO2 хНгО. Вследствие избыт-
ка щелочного компонента ортосиликат Li4SiO4 разлагается и водой
(1.9), метасиликат LuSiCh разлагается водой также, но гораздо медлен-
нее
3Li4SiC>4 + 4НгО ЫгБЮз + Li2Si2O5 + 8LiOH. (1.9)
В связи с этим из водных растворов получить Li4SiO4 не удается.
Имеются данные о существовании других силикатов, например, под
давлением из смеси Li4SiC>4 и U2CO3 может быть получен силикат
L16S1O5.
Безводные орто- и метасиликаты можно получить сплавлением при
— 900 °C карбоната лития и чистого кварцевого песка. Метасиликат
28
L12S1O3 можно получить также при взаимодействии гидроксида лития и
гидрата S1O2 хНгО нагреванием при 80 °C.
Введение в систему ЫгО—SiO2 оксида AI2O3 приводит к образова-
нию силикатов лития, алюминия и алюмосиликатов лития. В качестве
примеров можно привести силикат LiAl(Si2O6) — в природе минерал
;подумен, алюмосиликат Li(AlSiO4) — в природе минерал эвкриптит.
1.3. Галогениды лития
Галогениды лития, как и других щелочных элементов, являются
соединениями элементов с самыми ярко выраженными металлическими
свойствами и самыми типичными неметаллами. В группе галогенов в
соответствии с общей закономерностью первый элемент группы — фтор
отличается от своих аналогов. Он самый электроотрицательный элемент
из всех элементов периодической системы элементов, но в то же время
сродство атома фтора к электрону меньше, чем у хлора (табл. 1.5). Фтор
исключительно химически активен, что в значительной мере обуслов-
лено относительной слабостью связи F—F в молекуле фтора (159
кДж/моль). Для молекулы хлора энергия связи С1—С1 равна 243
кДж/моль. Кроме того, энтальпия и энергия Гиббса для реакций ато-
марного фтора с щелочными элементами имеют более высокие значе-
ния, чем для других галогенов.
Таблица 1.5. Свойства галогенов
Элемент X Гат, нм г*-, нм /, эВ Л эВ Энергия диссоци- ации молекул Х2, кДж/моль ЭО (по Полингу)
F 0,060 0,115 20,86 3.50 159 4,0
С1 0,099 0,167 15,03 3,82 243 3,2
Вг 0,119 0,182 13,10 3,54 190 3,0
I 0,136 0,206 12,67 3.23 149 2,6
Фториды — наиболее прочные галогениды, например энергия связи
Ь—F 573, a Li—Cl 506 кДж/моль (табл. 1.6).
Все галогениды щелочных металлов — ионные соединения с относи-
тельно небольшой долей ковалентной составляющей. Степень ионности
в них по одним данным изменяется от 0,7 до 0,9, по другим — для
некоторых соединений может достигать 1,0. Для галогенидов какого-ли-
бо щелочного металла уменьшение ковалентности наблюдается в ряду
—Cl—Вг—I, для щелочных металлов аналогичный ряд предположи-
29
тельно может быть следующим: Cs—Li—Rb—К—Na. Последователь-
ность здесь зависит от поляризующей способности и поляризуемости как
катионов, так и анионов.
Таблица 16 Свойства галогенидов лития
Соединение Т'пл. "С Т'кип. “С о - AH298(f), кДж/моль d, г/см^
LiF 848 1681 617 2.2
LiCl 607 1385 405 2,07
LiBr 550 1310 349 2,9
Lil 469 1171 271 3,5
Фтор в соединениях проявляет степень окисления только -1; хлор,
бром, йод — от -1 до +7. Фториды имеют, как правило, более высокие
температуры плавления и кипения; из растворов кристаллизуются в
виде безводных солей или с меньшим содержанием молекул кристалли-!
зационной воды, чем другие галогениды щелочных элементов; некото!
рые фториды плохо растворяются в воде.
Фторид лития. Фторид лития — самое тугоплавкое соединение
из всех галогенидов щелочных элементов (см. табл. 1.6). Однакц
при температуре 1100—1200 °C, т.е. значительно ниже точки кипения
(1681 °C), начинается быстрое его испарение. Фторид LiF не гигро-
скопичен, в воде растворяется плохо, при 25 °C его растворимость 0,13
г/ 100г НзО. Из водного раствора кристаллизуется в виде безводной соли
с кристаллической решеткой ГЦК типа NaCl (к.ч. лития 6). Раствори-1
мость LiF понижается в присутствии аммиака и особенно NFUF, в азот-
ной и серной кислотах растворяется при комнатной температуре. В
плавиковой кислоте образуется гидродифторид LiHF2, который раство-
ряется в воде несколько лучше LiF. При нагревании он разлагается на
LiF и HF. Фторид лития не растворяется в спиртах, ацетоне и других
органических растворителях.
Хлорид лития. Хлорид лития — бесцветное кристаллическое веще-]
ство с решеткой ГЦК типа NaCl, имеет довольно высокие температурь!
плавления и кипения (см. табл. 1.6). Заметное испарение LiCl начина!
ется при температуре > 880 °C, при этой температуре давление паров
LiCl равно 260 Па.
Хлорид лития в отличие от NaCl и КС1 гигроскопичен, на воздухе
расплывается сильнее CaCh, хорошо растворяется в воде, при повыше^
нии температуры растворимость увеличивается:
30
Температура, °C... О 25 50 75
Растворимость, % . . . 40,89 45,85 49,06 52,18
Из водных растворов в отличие от LiF, NaCl и КО выделяется в виде
кристаллогидратов, из которых в интервале температур от комнатной
до 94 °C наиболее устойчив кристаллогидрат LiCl НгО. Выше 94 °C
может быть выделен безводный LiCl.
В водных растворах LiCl сильно диссоциирован, в 0,1 молярном
растворе степень диссоциации равна 84,1 %, а в 0,002 молярном 97 %.
Причина этого — необычайно высокая энтальпия гидратации иона Li+,
равная 520 кДж/моль; вследствие этого равновесие приведенной ниже
реакции (1,10) весьма трудно сдвинуть влево
LiCl 2 Li+ а</+СГ. (1.10)
Поэтому LiCl нельзя высолить из раствора ни добавлением концен-
трированной соляной кислоты, ни пропусканием че^ез раствор газооб-
разного НС1. Вследствие этой же способности иона Li к гидратации LiCl
является сильным высаливателем и дегидратирующим агентом.
Растворы LiCl поглощают аммиак вследствие образования комплек-
сных ионов [Li(NH3)n J+; твердый безводный LiCl поглощает из воздуха
пары аммиака с образованием продуктов присоединения LiCl /1NH3,
где п изменяется от 1 до 4. Аналогично взаимодействует LiCl с метила-
мином, этиламином и другими соединениями.
Хлорид лития растворяется в спиртах, ацетоне, пиридине, сложных
эфирах и других органических растворителях. Растворение в этиловом
спирте сопровождается выделением тепла и образованием сольватов
LiCl ЗС2Н5ОН и LiCl 4С2Н5ОН, которые можно выделить в виде
кристаллов. Причиной повышенной растворимости хлорида лития в
спиртах является высокая гидратация и сольватация иона Li+. Образу-
ющиеся сольваты, очевидно, аналогичны гидратам и представляют со-
бой своеобразные ионные пары.
Взаимодействие LiCl с хлоридами щелочных металлов в безводных
системах определяется главным образом соотношением ионных радиу-
сов. В системе с хлоридом натрия — ближайшим аналогом лития —
по данным различных авторов существуют непрерывные твердые рас-
творы или твердые растворы на основе LiCl и NaCl с довольно широки-
ми областями гомогенности, содержащие соответственно 25 % NaCl
и ~ 24 % LiCl. По-видимому, ближе к истине точка зрения об образова-
нии ограниченных твердых растворов.
Система LiCl—КС1 имеет эвтектическую фазовую диаграмму (эв-
тектика 58 % LiCl, температура плавления 348—358 °C) с небольшими
областями твердых растворов на основе LiCl и КС1. В то же время в
31
tr
400
350
300
250
200
380
4
№
100
ВО
Li+Ж
93 4
Li + Na
Ди жидки фоки
171
963
i 10 20 30 40 50 60 70 80 90 No
Na, % (ат.)
системе NaCl—КО образуют-
ся непрерывные твердые рас-
творы. Причем все три хлорида
изоструктурны.
Бромид лития. Бромид ли-
тия — кристаллическое веще-
ство со структурой ГЦК типа
NaCl, имеет температура
плавления и кипения ниже,
чем LiF и LiCl (табл. 1.6). На-
гревание LiBr выше температу-
ры плавления приводит к за-
метному разложению LiBr;
расплавленный LiBr разруша-1
ет стекло, фарфор и платину. I
Бромид лития более гигро}
скопичен, чем LiCl, хорошо
.6. Фазовая диаграмма системы Li—Na
Рис. 1
растворим в воде, при повышении температуры растворимость увеличи-
вается (при 20 °C 177 г/100г воды). Из водных растворов выделяется в
виде кристаллогидратов с 2; 3 и 5 молекулами воды, обезвоживается с
трудом. Растворяется в спиртах, ацетоне, пиридине и других органиче-
ских растворителях, поглощает NH3 с образованием продуктов присоег
динения LiBr /1NH3 (n = 1—4).
Иодид лития. Иодид лития — кристаллическое вещество, как и
остальные галогениды лития имеет кристаллическую решетку ГЦК ти-
па NaCl. Самый неустойчивый галогенид лития имеет самую низкую
энтальпию образования, температуры плавления и кипения (см. табл
1.6). Расплавленный Lil разрушает стекло, фарфор, платину; очень
гигроскопичен, хорошо растворяется в воде (при 19 °C 164 г/ 100г НгО)
Из водных растворов выделяется в виде кристаллогидратов с 0,5; 1; 2
3 молекулами воды. Обезвоживание достигается нагреванием в токе Н1
пропусканием через расплавленную соль сухого водорода либо возгон»
кой в вакууме. Растворяется в спиртах, пиридине; при действии жидко®
аммиака образует продукты присоединения типа Lil пЬ1Нз (и= 1—7)
При взаимодействии расплавленного безводного Lil с I2 образуются по-
лигалогениды Liln (и = 3—9). При добавлении к растворам Lil соляной-
азотной или серной кислоты выделяется элементарный иод; выделение
иода ускоряется при облучении раствора солнечным светом.
Соли кислородсодержащих кислот галогенов. Литий образует солй
со всеми кислородсодержащими кислотами хлора: LiCl’o гипохлорит
лития, 1дС1шО2 хлорит лития, LiClvC>3 хлорат лития, LiClVIIO4 перхло-
рат лития.
32
Все соли являются сильнейшими окислителями. Из них наиболее
устойчив перхлорат, его температура плавления 236 °C; выше 400 °C он
начинает разлагаться на LiCl и Ог. Перхлорат IJCIO4 гигроскопичен,
очень хорошо растворяется в воде:
Температура, °C........... 0 20 40 65 89
Растворимость, г/100г Н2О ... . 42,7 56,3 72,4 100 150
Очень хорошо растворяется в спиртах и эфирах, например при
25 °C его растворимость в этиловом спирте равна 60,3 %.
Для брома и иода известны только некоторые представители соеди-
нений этого типа. Из них наиболее интересен с практической точки
зрения иодат лития 1дЮз- Имеет температуру плавления 420 (по другим
данным 435 °C); известны три его кристаллических модификации, хотя
о структуре и температурах полиморфных превращений данные проти-
воречивы; видимо, при комнатной температуре существует ромбиче-
ская модификация. Обладает уникальным комплексом свойств — оп-
тических, акустических, пьезоэлектрических. Имеет хорошую раство-
римость в воде — при 15 °C 125г/ 100г НгО; из водных растворов выра-
щивают крупные монокристаллы оптического качества, обладающие
пропусканием, близким к 1 при длине волны 0,28—1,5 мкм. Использу-
ется в акустооптике и других направлениях электроники.
1.4. Взаимодействие лития с неметаллами
Литий активно взаимодействует во всеми неметаллами: с некоторы-
ми из них при обычной температуре, с другими — при нагревании.
Образование соединений с ними предопределяет использование лития в
качестве дегазатора сплавов черных и цветных металлов.
Литий — водород. При непосредственном взаимодействии рас-
плавленного лития с газообразным хорошо очищенным от примесей
водородом образуется твердый гидрид лития LiH. Реакция начинается
при 400 °C и заканчивается при 700 °C
21д(ж)+Нг(г) 2 21дН(тв) ; аЯ298 = ~181,4 кДж, (1.11)
Молекула LiH по характеру химической связи приближается к типу
ионных соединений Li+H~, в ней имеет место частичная ионизация во-
дорода, эффективный заряд которого -0,35 (Н-0,35). Он в известном
смысле может рассматриваться как аналог галогенидов, а ион Н — как
аналог галогена. Не случайно LiH имеет кристаллическую решетку ГЦК
типа NaCl. В атмосфере благородного газа гидрид плавится при 680—
700 °C практически без разложения; при более высокой температуре
интенсивно разлагается, давление диссоциации достигает атмосферного
при 850 °C. В вакууме сублимируется при 220 °C, а заметная диссоциа
ция начинается при 450 °C. Гидрид лития термически довольно устой-
чивое соединение с относительно малой реакционной способностью
Различия в термической устойчивости LiH и гидридов Na и К, которые
подвергаются термической диссоциации при 420 и 247 °C, используюта
при получении чистого лития.
С кислородом на холоду LiH не реагирует, взаимодействие начина-
ется при нагревании до 500—600 °C. При нагревании с азотом образуете!
нитрид LiaN. С хлором, серой, углеродом, кремнием и фосфором реаги-1
рует также при повышенной температуре. Чистый LiH белого цвета, нс
при хранении на дневном свете, а особенно при действии ультрафиоле-
тового облучения, изменяет цвет, становится голубым за счет частично-
го разложения с выделением лития в тонко распыленном состоянии. С
водой LiH реагирует очень бурно
1дН(тв) + HzO(r) г LiOH(тв)+Н2(г);-дЯ298 = 155кДж. (1.12)
При гидролизе 1 кг LiH выделяется 2,8 м3 газообразного водорода.
Это предопределило использование его в качестве легкодоступного и
простого аккумулятора водорода для автономных потребителей неболь
шой мощности.
С жидким аммиаком при комнатной температуре, а с газообразнык
при 440—460 °C образуется амид
L1H + NH3 t LiNH2 + H2.
При высокой температуре LiH активно взаимодействует с металла
ми, SiO2 и силикатами, разрушает аппаратуру из стекла и фарфора.
Гидрид лития вступает в реакции обмена почти со всеми галогени-
дами металлов и неметаллов. Реакции обмена используются для полу-
чения многих гидридов; таким образом можно получить, например,
моносилан
SiCl4 + 4LiH г SiH4 + 4LiCl. (1.13)
Гидрид LiH обнаруживает склонность к образованию двойных гид-
ридов, из которых наибольший интерес представляют LiAlH4 и LiBH4.
Реакция получения LiAlH4 протекает при температуре от 0 до —4 °C в
эфирном растворе
41дН+А1С1з 2 LiAlH4+3LiCl; - ДЯ298= 101 кДж. (1.14)
Тетрагидридоалюминат лития LiAlH4 — твердое нелетучее соедине-
ние с плотностью 0,92 г/см3. В сухом воздухе начинает медленно разла-
гаться при нагревании до 120 °C, водой бурно гидролизуется
2LiAlH4 г 2LiH + ЗН2 + 2А1. (1.15)
Хорошо растворяется во многих органических растворителях, что
имеет большое значение для его использования в различных реакциях.
34
С его помощью по реакциям обмена были получены летучие гидриды В,
д1, Si, Ge, Sn, As и Sb
4MX + LiAlH4 г 4MH + LiX+AlX3. (1.16)
В органическом синтезе он используется как гидрирующий агент и
как восстановитель. Реакции восстановления с его использованием про-
текают количественно, а продукты реакции отличаются высокой чисто-
той, так как не сопровождаются побочными реакциями. При действии
L1AIH4 альдегиды, кетоны, ангидриды, сложные эфиры легко превраща-
ются в спирты.
Тетрагидридоборат IJBH4 получается при взаимодействии диборана
В2Н6 с LiH в среде этилового эфира при температуре его кипения 34,6°С.
Это вещество с плотностью 0,66 г/см , довольно термически устойчивое,
плавится при 275 °C с незначительным разложением, при длительном
нагревании при 275—280 °C разлагается с выделением водорода
B2H2+2UH 2IJBH4; - ЛЯ298 = 185кДж. (1.17)
Тетрагидридоборат L1BH4 имеет структуру ионных солей типа
Li+BH4-, образованных борогидрид-ионом, в котором связи между В и Н
ковалентны. Борогидриды металлов можно также рассматривать как
двойные гидриды, образованные бором и металлом, каждый из которых
способен образовывать гидриды.
Водой и спиртами LiBH4 легко разлагается; при его полном гидроли-
зе из 1 кгобразуется 4,1 м3 водорода. В гидриде лития водород составляет
12,5 %, в борогидриде лития 18,2 %, т.е. как аккумулятор водорода он
более эффективен, чем гидрид.
Оба гидрида LiH и LiBH4 обладают высокой теплотой сгорания, их
добавляют к ракетному топливу с целью повышения его эффективности
и стабильности горения. Вообще теплота сгорания борогидридов значи-
тельно выше углеводородов. Например, теплота сгорания керосина рав-
на 43 МДж/кг, а пентаборана В5Н9 71 МДж/кг.
Особое значение гидриды имеют для термоядерного синтеза, одной
из основных реакций которого является реакция (1.18), хотя в смеси
2 з
1D и 1Т могут идти идругие ядерные реакции. Для поддержания реакции
(1.18) необходимы реакции производства трития
2 3 4 1
1D+1T- 2Не+оП+ 17,6 МэВ; (1.18)
зЬ+in- гНе + 1Т + 4,8 МэВ. (1.19)
Для осуществления термоядерного синтеза в качестве топлива ис-
пользуют дейтерид лития-6. При температуре 10е—10 К протекают
одновременно реакции синтеза трития и образования ядер гелия с выде-
лением энергии 22,4 МэВ на одну молекулу LiD (2,16 109 кДж/моль
LD).
г»
35
Гидрид лития-6 используют для нейтронной защиты ядерных реак-
торов. Перспективно использование для охлаждения термоядерных ре-
акторов и воспроизводства трития таких соединений, как ЫгВеРд, обо-
тащенных изотопом 6Li. Они оказывают меньшее коррозионное дейст-
вие, чем металлический литий. Отсюда проблема разделения изотопок
лития. По всей вероятности, наиболее эффективным методом является
центрифугирование водных растворов LiCl, позволяющее получить на
одной ступени высокие степени обогащения.
Литий — углерод. С углеродом литий образует одно соединени!
Ы2С2, являющееся производным ацетилена и содержащее ион
[С = С]2 . Карбид образуется при непосредственном взаимодействий
лития и углерода при 650—700 °C, а также при пропускании над рас-
плавленным литием ацетилена или этилена. По своим свойствам карбид
лития напоминает карбиды щелочноземельных элементов, при дейст-
вии воды он разлагается со взрывом, с парами воды медленно выделяется
ацетилен.
Литий — кремний. Предполагается, что литий при 185—200 °C
образует с кремнием ряд соединений с общими формулами Li4nSin и
LilnSin- Однако достоверно установлена индивидуальность соединения
Li4Si и LizSi, имеющих температуры плавления соответственно около
720 и 850 °C. На воздухе при обычной температуре оба силицида доволь-1
но устойчивы, но во влажном воздухе гидролизуются; при повышенной
температуре являются сильнейшими раскислителями. Получение ин-
дивидуальных силицидов — весьма сложный процесс. Лучшие резуль-£
таты дает прямой синтез из элементов, взятых в стехиометрическим
количествах при 600—700 °C. Но обычно в этом случае получают сплав!
состоящий из смеси силицидов и непрореагировавших лития и кремния!
Литий — фосфор. В системе Li—Р установлено образование ряда
фосфидов: L13P, U2P, LiP, L12P5- Все это твердые вещества, некоторые'
из них термически устойчивы, например U2P5 не разлагается до 650 °С|
a LiP в вакууме при температуре ниже 600 °C распадается с выделением
фосфора. Фосфиды образуются при непосредственном взаимодействии!
лития с фосфором в вакууме или атмосфере аргона при 600—700 °C.
Водой и ее парами мгновенно разлагаются с выделением токсичной!
фосфина РНз.
Литий — азот. При непосредственном взаимодействии лития С
азотом образуется нитрид U3N. Во влажном воздухе реакция протекает
при комнатной температуре, причем в токе сухого азота она ускоряется
в 10—15 раз. Кислород, а также водород являются ингибиторами реак-1
ции; при высоких их концентрациях в газовой смеси реакция может
полностью прекратиться. При нагревании до 450—460 °C интенсивность!
реакции сильно возрастает. Данные о температуре плавления нитрида]
36
противоречивы: от 845 до 2275 °C. Нитрид — соединение неустойчивое,
водой энергично разлагается
Li3N + Н2О - 3LiOH + NH3. (1.20)
При повышенной температуре расплавленный нитрид очень агрес-
сивен по отношению ко многим металлам (Fe, Ni, Со, Си, Pt и др.) и
неметаллическим материалам. С некоторыми он образует соединения
типа LJ7MN4 (М—V, Nb, Та) и U9MN5 (М—Сг, Mo, W).
Литий — сера. Сульфид L12S образуется при нагревании расплав-
ленного лития и элементарной серы или при восстановлении сульфата
лития углеродом или водородом. Сульфид лития довольно устойчивое
соединение; кислород и другие окислители окисляют его до сульфата
при нагревании до 300 °C. Он гигроскопичен, хорошо растворяется в
воде, но при этом гидролизуется
U2S + H2O-» LiHS + LiOH. (1.21)
Кислоты разлагают его с выделением токсичного газообразного H2S.
Аналогичные соединения литий образует с селеном и теллуром.
1.5. Сплавы лития с металлами
Литий — самый легкий металл, имеет малые атомные размеры и
относительно прочную кристаллическую структуру. Со многими метал-
лами (Al, Mg, Си, РЬ, /пи др.) образует ограниченные твердые растворы
и интерметаллиды, отличающиеся большой прочностью и тугоплавко-
стью. Литий не взаимодействует с железом и никелем, что имеет боль-
шое практическое значение, так как позволяет для работы с ним ис-
пользовать тигли из высокочистого никеля и некоторых нержавеющих
сталей. До 900 °C в расплавленном литии устойчивы титан, ниобий и
тантал.
Сплавы Li—Na (К). В системах, образованных литием с натрием и
калием, имеет место расслаивание в жидком состоянии. На рис. 1.6
приведена фазовая диаграмма системы Li—Na. Предполагается, что в
твердом состоянии на основе лития и натрия существуют ограниченные
твердые растворы с очень небольшими областями гомогенности. Для
системы Li—К область несмешиваемости в жидком состоянии еще более
обширна. Такой характер взаимодействия лития с натрием и калием
предполагает наличие факторов, препятствующих смешиваемости в
жидком состоянии и образованию твердых растворов, несмотря на то что
все три металла являются щелочными. Такими факторами могут быть
для лития и натрия: разница атомных (> 21 %) и ионных радиусов
( > 26 %), разный тип кристаллической структуры (литий — ГЦК,
натрий — ОЦК), различия в параметрах кристаллической решетки
(Ап ~ 23 %). Для жидкого состояния, видимо, наибольшее значение
37
имеет склонность лития к образованию химических связей с большой I
долей ковалентности. Это подтверждается обнаружением молекул Li2 в
его парах (~ 1 %), чего нет у натрия. Можно предполагать, что такие
кластеры могут существовать в большем количестве и в расплаве.
Из систем, образуемых литием с другими металлами, наибольшее
внимание привлекают сплавы лития с алюминием, магнием и медью.
Сплавы Li—Al. В системе Li—Al установлены интерметаллиды AlLi
и А11д2, предполагается также образование метастабильного AI3LL Рас-
творимость лития в алюминии имеет максимальное значение при тем- *
пературе эвтектики (600 °C) 9,9 % или [~ 30 % (ат.) ], при понижении
температуры она довольно быстро уменьшается и при 15 °C составляет!
6,8 % (ат.). Растворимость алюминия в литии ничтожна. Интерметал-
лид AlLi имеет довольно широкую область гомогенности [45—56 %
(ат.)Li ], а интерметаллид AIU2 образуется по перитектической реакции
(рис. 1.7). Довольно высокая растворимость лития в алюминии объясня-
ется главным образом близостью атомных радиусов (Дгат > 10 %), оди-
наковостью структуры (ГЦК).
Литий принадлежит к числу металлов с наибольшей растворимо-
стью в алюминии (Ag, Zn, Mg). Сплавы Ct-(Al, Li) обладают интересным
сочетанием пониженной плотности с повышенным модулем упругости,
отличаются большей пластичностью. Такой эффект не может быть по-
лучен при легировании алюминия любым другим металлом (рис. 1.8).
U, % (ат.)
Рис. 1.7. Фазовая диаграмма системы
Li—Al
Рис. 1.8. Плотность сплавов алюминия,
легированных разными металлами:
1 — Li; 2 — Mg; 3 — Be; 4 — Си, С —
концентрация легирующей добавки
38
Литий, кроме того, обладает удивительной способностью сообщать алю-
миниевым сплавам повышенную устойчивость против коррозии. Наибо-
лее устойчив против нее сплав, содержащий 2,94 % (ат.) (0,77 %)
лития, отличающийся также повышенной пластичностью. Уменьшение
растворимости лития в алюминии позволяет для упрочнения сплавов
использовать закалку, а образование интерметаллидов, имеющих, как
правило, более высокую твердость, позволяет получать двухфазные
сплавы с нужными механическими свойствами.
Добавки меди к двойным сплавам А1—Li приводят к значительному
уменьшению растворимости лития (при 515 °C до 1,5 %), в тройной
системе образуются интерметаллиды CU4L1AI7, CuLiAb и СиИзА1б- Раз-
работаны и сложнолегированные сплавы, обязательным компонентом
которых является литий, с суммарным содержанием легирующих ме-
таллов (Li, Си, Zr, Mg) 5—6 %; такие сплавы имеют исключительно
высокие эксплуатационные характеристики.
Сплавы Li—Mg. В системе Li—Mg образуется один интерметаллид
LiMg2 с температурой плавления около 600 °C и очень широкой областью
гомогенности [29—95 Li % (ат.) ]. Металлический литий и магний име-
ют разные кристаллические структуры (литий — кубическую, магний
— ГПУ), но размеры их атомов и ионов очень близки (разница соответ-
ственно равна 3 и 5 %). Поэтому растворимость лития в магнии довольно
велика, поданным различных авторов она составляет от 16до21 % (ат.).
Магний, как и алюминий, является одним из важнейших металлов, на
основе которых создаются легкие сплавы для авиакосмической техники.
Но магний, как и все металлы с гексагональной кристаллической струк-
турой, обладает недостаточной ковкостью, хрупок при низких темпера-
турах. Так, чистый магний и его сплавы при 78К дают хрупкий излом.
Сплавы же, содержащие 4,6 % лития, при той же температуре дают
Удлинение до 12,2 %, в 4 раза больше, чем чистый магний. Кристалли-
ческая структура а-ТР (Mg, Li) также ГПУ, но растворенный литий
производит изменение в параметрах кристаллической решетки: умень-
шается отношение с/а. Уменьшение величины с/а, по-видимому, изме-
няет характер сдвига в плоскости, перпендикулярной оси с, в результате
чего сплав приобретает ковкость при низкой температуре. Другие леги-
рующие металлы, дающие уменьшение с/а, имеют малую раствори-
мость в магнии, а следовательно, мало влияют на ее понижение и, как
следствие, не улучшают пластичность металла. Поэтому литий занима-
ет особое место среди легирующих металлов для магния.
39
Технология лития
1.6. Важнейшие области применения лития и его соединений
Традиционные области применения. Одной из наиболее важных
областей применения лития является электролитическое получение
алюминия; так, США для этого используют до 45 % потребляемого
лития. Добавки Ы2СО3 в электролитическую ванну позволяют снизить
температуру плавления электролита, уменьшить выделение фтора!
увеличить электропроводность электролита, уменьшить расход анода и
криолита, а следовательно, уменьшить расход электроэнергии и соот-
ветственно снизить себестоимость металла или увеличить производи-
тельность. На 1 т алюминия расходуется 2,5—3,5 кг L12CO3. В связи с
ростом производства алюминия потребление лития для этих целей будет
возрастать.
Высококачественные концентраты литиевых минералов и химиче-
ские соединения U2CO3, ИгБЮз и другие широко используются в
производстве стекла и керамики; на эти цели в США расходуется около
40 % потребляемого лития. Добавки лития улучшают качество фарфора!
и керамики, повышают их химическую стойкость и термостойкость.]
Глазури и эмали с литием более плотны и блестящи, более устойчивы к
атмосферным воздействиям. Добавки лития используются в производ-1
стве стекла для катодно-лучевых трубок, телевизионных кинескопов,!
стекла с электроизоляционными свойствами, светочувствительных!
легкоплавких и других специальных стекол. При варке стекла добавки!
лития снижают температуру плавления и вязкость расплава, позволяют!
повысить производительность труда и уменьшить расход топлива.
В цветной и черной металлургии металлический литий и его сплавы]
с кремнием и кальцием применяются в качестве необычайно активного]
раскислителя, десульфуризатора и дегидрирующего агента, что позво-
ляет значительно улучшить качество многих металлов и сплавов за счет
удаления нежелательных примесей в шлак. Например, обработка элек-
тротехнической меди при ее выплавке сплавом лития с кремнием (1:1)
в количестве s 0,025 % от массы меди повышает ее плотность и элект-
ропроводность. Добавка таких же количеств сплава лития с кальцием
при плавке белого и серого чугуна, хромоникелевых сталей повышает
их жидкотекучесть, твердость и временное сопротивление разрыву. Вве-
дение в металлы и сплавы небольших количеств лития в качестве леги-|
рующей добавки позволяет улучшить их эксплуатационные характе-
ристики.
В авиации и военной технике широко применяются литиевые кон-
систентные смазки, содержащие до 10 % стеарата лития, они сохраняют1
40
уникальные физические свойства в температурном интервале от -50 до
+150 °C.
Хлорид и бромид лития благодаря их способности поглощать дымы,
пары Н2О, СО2, NH3, органические вещества используют для кондици-
онирования воздуха в различных помещениях. В щелочных аккумуля-
торах используют добавки LiOH к электролиту, которые повышают их
емкость и срок службы в 2—3 раза.
Значительные количества металлического лития и металлооргани-
ческих соединений используют в качестве катализаторов при синтезе
синтетического каучука и других органических соединений.
Легкий изотоп 6Li — единственный промышленный источник для
производства трития, используемого в водородной бомбе; он необходим
для термоядерных реакций.
Перспективные области применения. До 2000 г. прогнозируется
ежегодный прирост потребления лития приблизительно на 5 % ежегод-
но, главным образом за счет традиционных областей применения. Но в
XXI веке он особенно быстро будет расти за счет новых перспективных
областей: в производстве алюминийлитиевых сплавов (АЛС), химиче-
ских источниках тока (ХИТ), топливных элементах урановых ядерных
реакторов и термоядерном синтезе. Из них наиболее емкой, по-видимо-
му, будет термоядерная энергетика.
Особенно эффективны стандартные алюминиевые сплавы, дополни-
тельно легированные 1—3 % лития, для использования в авиаракетно-
космической технике. Известны, например, составы таких сплавов, %:
94,98 AI; 2,2 Li; 2,7 Си; 0,12 Zr и 95,2 Al; 2,5 Li; 1,3 Си; 0,9 Mg; 0,1 Zr.
Плотность этих сплавов меньше плотности стандартных сплавов на
8—12%, они более устойчивы к коррозии, прочностные характеристики
сохраняются или улучшаются. Применение их в самолетостроении по-
зволяет снизить массу самолета на 20 %, Это дает возможность увели-
чить число посадочных мест либо обеспечить экономию топлива, кото-
рая может достигать 1 тыс.т на один авиалайнер в год. Применение этих
сплавов сдерживается высокой стоимостью, она в 6—10 раз выше сто-
имости конкурирующих композитных материалов. Предполагается, что
их стоимость при массовом производстве будет только в 2,5—3 раза
превышать стоимость серийных сплавов.
Металлический литий наряду с другими металлами (Bi, Hg, К, Ва,
Pb, Ga) перспективен для использования в качестве жидкометалличе-
ского теплоносителя атомных реакторов. Жидкометаллические носите-
ли имеют ряд достоинств, важнейшим из которых является возможность
создания высокотемпературного контура (450—650 °C) при атмосфер-
ном давлении и то, что они обеспечивают интенсивный отвод тепла от
I ВЭЛов. При использовании в качестве теплоносителя воды требуется
41
высокое давление, кроме того, вода замедляет быстрые нейтроны. Рас-
плавленный литий имеет широкую область жидкого состояния (1 SO-
1336 °C), его удельная теплоемкость больше удельной теплоемкости
натрия в 4 раза, калия — в 5,6 раза, а ртути, свинца и висмута — в 28—30
раз, теплопроводность выше только у натрия. Для этой цели пригоден
7
изотоп з!д, который практически прозрачен для тепловых нейтронов.
Литий можно использовать также в реакторах на быстрых нейтронах.
Однако расплавленный литий более агрессивен, чем Hg, Bi, Pb и эвтек-
тика К—Na. Поэтому создание сплавов, имеющих низкие температуры
плавления и менее агрессивных в расплавленном состоянии, имеет при-
нципиальное значение.
Изотоп 6Li — основной компонент термоядерных реакторов (ТЯР),
а сплавы Си—Li, Al—Mg—Li, по всей вероятности, будут использовать*
ся в качестве конструкционных материалов ТЯР. В области ХИТов
наиболее перспективными для создания безопасных батарей с высокой
удельной энергией являются системы, основанные на использовании!
металлического лития, его сплавов и различных соединений. Можно
привести, например, систему из электродов Li / FeS и расплавленного
электролита КВг—LiBr—LiCl. В качестве новых эффективных элект-
родных материалов ХИТ предполагается использование LixCoOz, I
1дхМоО4 и других литийсодержащих соединений, перспективны также
сплавы алюминия и магния с большим содержанием лития.
В ряде стран (США, ФРГ, Швейцария, Япония) существует и раз-
вивается относительно небольшая по объему область применения нио-
батов и танталатов лития, имеющая очень большое значение в новей-
ших отраслях техники: нелинейной и интегральной оптике, опто- и
акустоэлектронике и т.д. На их основе создаются устройства для моду-
ляции и отклонения лазерного луча, устройства для обработки и отобра-
жения информации, запоминающие устройства и т.д. Пироэлектри
ческие свойства кристаллов используются в приемниках и преобразовав
телях ИК-излучений в СВЧ-диапазоне. Эпитаксиальные пленки ниоба-
та и танталата лития все шире используются в микроэлектронике, II
волоконно-оптических линиях связи, линиях задержки на поверхност-
ных акустических волнах и т.д.
Разрабатываются новые материалы на основе использования соеди-
нений лития. Так, в США получена высокопрочная керамика на основе
алюмината и силиката лития, затвердевающая при комнатной темпера-
туре и предназначенная для использования в термоядерной энергетике,
металлургии и военной технике. В рамках программы СОИ введется
разработка литий-алюминий-силикатного стекла, упрочненного волок-
нами карбида кремния, для космических конструкций.
42
По объему потребления лития на первом месте стоят США; на их
долю приходится около 40 % мирового потребления лития, далее идет
Западная Европа 27 и Япония 18 %.
1.7. Сырьевые источники лития
Содержание лития в земной коре 6,5 10 3 %; он более распростра-
нен, чем Pb, Sb, Со, Hg, Cd, Cs и другие элементы. Литий входит в состав
многих горных пород, содержится в глинах и почвах, каменных углях,
морской воде, рапе соляных озер, подземных водах, растениях и живых
организмах. Он обнаружен в 150 минералах, из которых 28 являются
собственными минералами лития.
Литий является типичным литофильным элементом, концентриру-
ется совместно с натрием в продуктах остаточной кристаллизации маг-
мы — гранитных пегматитах натролитиевого типа. Тесная ассоциация
лития и натрия в природе обусловлена большой близостью энергетиче-
ских характеристик их ионов, хотя оба элемента не отличаются большой
склонностью к взаимному изоморфному замещению. В то же время
вследствие большой близости ионных радиусов Li и Mg, Fe(II), Al он
легко входит в состав многих минералов, замещая эти элементы. При
кристаллизации расплавов, обогащенных летучими компонентами
(пневматолитические процессы), образуются литиевые минералы, со-
держащие фтор. Наиболее важные минералы лития (сподумен, лепидо-
лит, петалит, циннвальдит, амблигонит) большей частью встречаются
в редкометальных пегматитах, содержащих минералы других редких
элементов — поллуцит, берилл, циркон, колумбит—танталит, а также
касситерит и др. По существу их месторождения являются полиметал-
лическими.
Литиевые минералы отличаются довольно высокой устойчивостью к
действию химических реагентов, однако при гидротермальных процес-
сах и процессах выветривания они, за исключением слюд, довольно
легко трансформируются. Эти процессы сопровождаются выносом из
них лития и концентрированием его вследствие адсорбции в глине,
почве и рапе соляных озер.
Минералы лития
Сподумен — силикат лития и алюминия LiAl [Si2O61; теоретическое
содержание 1дгО 8,1 %, однако фактически оно всегда меньше (s 7,5
%) вследствие замещения его магнием, железом (П), марганцем и,
возможно, натрием. Плотность 3,10—3,20 г/см3, сингония моноклин-
ная, температура плавления 1430 °C.
43
Сподумен относится к цепочечным силикатам, основу структуры
которых составляют непрерывно связанные между собой кремнекисло-
родные тетраэдры [S1O4 ], образующие цепочки, вытянутые вдоль оси с.
Ионы Li , Аг и другие располагаются между цепочками. Свойства
сподумена находятся в полном соответствии с его структурой. Неполяр-
ная связь Si—О—Si более прочна, чем связь кремнекислородных тетра-
эдров с ионами Li+, А13+, поэтому эти ионы обладают определенной
подвижностью; связь же между тетраэдрами [SiC>4 ] разрушить довольно
трудно. Кристаллы сподумена обладают совершенной спайностью вдоль
оси с, на него не действуют кислоты.
При нагревании сподумен монотропно переходит в высокотемпера-1
турную модификацию. Переход сопровождается увеличением удельно-
го объема минерала на 24 % и уменьшением плотности до 2,4 г/см ,
вследствие возникновения термических напряжений минерал рассыпа-1
ется в порошок. В зависимости от состава переход природного ОГ-споду-*
мена в высокотемпературную ^-модификацию происходит при*
950—1150 °C. Высокотемпературная модификация — алюмосиликат с
тетрагональной кристаллической решеткой; в нем в каждом третьем
кремнийкислородном тетраэдре кремний замещен атомами алюминия!
Возникают связи Si—О—AI, которые, видимо, менее прочны, чем связи|
Si—О—Si; это находит отражение в том, что/2-сподумен довольно легка
разрушается кислотами.
Под действием гидротермальных растворов, обогащенных натрием,}
происходит превращение сподумена в альбит (силикат натрия и алюми-
ния) и литиевый минерал эвкриптит (алюмосиликат лития), часть ли-
тия переходит в водорастворимую форму
4LiAl[Si2O6]+Na2O £ 2NaAl[Si3O8]+2Li[AlSiO4]+Li2O. (1.22)|
Теоретическое содержание U2O в эвкриптите 11,9%, сингония гек-|
сагональная, встречается вместе со сподуменом, относительно легко
разлагается кислотами.
Петалит — алюмосиликат (Li, Na) [AlSi40iol; теоретическое col
держание U2O 4,9 %, плотность 2,3—2,5 г/см3, сингония моноклинная.
Относится к каркасным алюмосиликатам, в которых тетраэдры [SiO41
и [AIO4 ] образуют трехмерный каркас, где каждая вершина тетраэдра
соединена с четырьмя другими тетраэдрами. Эта особенность структу-)
ры, а также то, что только в одном из пяти кислородных тетраэдром
кремний замещен алюминием, делает структуру петалита довольно
прочной, кислоты на него не действуют. Но для него характерна реакция
Li[AlSi40ioJ 2 /2-Li[AISi2O6 ] + 2SiO2. (1.23)
Процесс распада петалита на кварц и/2-сподумен обратим, при 680—
700 °C он сдвинут в сторону образования сподумена.
44
Лепидолит — водный алюмосиликат из группы литиевых слюд
KLii,5Ali,5[AlSi30io]<F,OH)2; содержание 1дгО 1,20—5,90 %, плот-
ность минерала 2,8—3,3 г/см3, сингония моноклинная. Как и во всех
слюдоподобных минералах тетраэдры [S1O4 ]и [AIO4 ] образуют плоские
слои с гексагональными кольцами, расположенные перпендикулярно
оси с. Алюминий в структуре лепидолита играет двоякую роль: часть его
атомов замещает атомы кремния в кремнекислородных тетраэдрах, а
часть располагается вместе с другими катионами (Li+, Mg2+, Fe2+ и др.)
между слоями. Между слоями располагаются также анионы ОН и F и
другие, иногда лепидолит содержит до 3,73 % CszO и RbzO. Минерал
обладает совершенной спайностью, легко расщепляется на тонкие лис-
точки. Связь между слоями, осуществляемая через катионы и анионы,
несомненно слабее связи между тетраэдрами [SiO4 ] и [AIO4 ], но тем не
менее лепидолит довольно устойчив в зоне выветривания, с трудом
разлагается кислотами.
Циннвальдит — водный алюмосиликат из группы литиевых слюд
KLiFenAl[AISi30io](F,OH)2; содержание 1дгО 1,0—5,0 %, плотность
2,9—3,2 г/см3, сингония моноклинная. Кроме обычных примесей содер-
жит в небольших количествах Cs20 и ИЬгО, менее распространен и
менее устойчив, чем лепидолит, разлагается кислотами.
Амблигонит — фторсодержаций фосфат лития и алюминия
LiAl[PO4](F,OH); теоретическое содержание U2O 10,10 %, фактиче-
ское 7—9,5 %, плотность 2,98—3,15 г/см3, сингония триклинная. Раз-
лагается серной кислотой.
Месторождения, запасы литиевого сырья
За рубежом в качестве основных промышленных источников лития
используют редкометальные пегматиты и литиеносную рапу. На долю
концентратов пегматитовых минералов приходится > 60 % производст-
ва лития; из них на первом месте находится сподумен, дающий 90 %
общей добычи этого вида сырья. Минимальное содержание 1дгО в руде
эксплуатируемых месторождений 1 %.
Вторым по объему источником производства является литиеносная
Рапа, из нее извлекается более 1 /3 добываемого лития. О форме нахож-
дения лития в промышленных месторождениях рапы сведения ограни-
чены. По-видимому, это хлорид (США, Боливия) и сульфат (Чили).
Содержание 1дгО в эксплуатируемых месторождениях рапы > 0,04 %.
Помимо лития рапа содержит К, Na, Mg, Са, В. Благодаря простоте
производства лития, возможности использования сопутствующих эле-
ментов, высокой рентабельности роль этого вида сырья будет возрастать.
Повышается интерес и к другим видам нетрадиционного литиевого
СыРья — некоторым глинам, буровым водам, геотермальным рассолам.
45
В 1989 г. условно рентабельные ресурсы лития (в пересчете на U2O)
оценивались приблизительно в 48 млн.т. Из них, по данным Горног®
бюро США, извлекаемые запасы составляли 2,2 млн.т (на начало 1991
г. эти цифры соответственно равнялись 100 и 4,7 млн.т). По масштаба^
подтвержденных запасов ведущим типом является литиеносная рапа
(78 %), немного более 20 % приходится на редкометальные пегматиты
Данные по запасам в разных странах представлены в табл. 1.7.
Месторождения литийсодержащих рассолов встречаются в пустын{
ных областях или соляных озерах континентальных районов. Наиболее
крупные из них сосредоточены в Южной Америке в районах цент-
ральных Анд. В Боливии имеются соляные озера «салары» площадью до
10 тыс.км2 и содержанием LizO в рапе от 0,02 до 0,3 %, а в отдельных
зонах до 0,9 % (среднее содержание 0,05 %). Салары в Чили харак-
теризуются исключительно высоким средним содержанием Ы2О — до
0,3 % и максимальным — до 2,1 %. Соляные озера США, расположен-
ные в западных штатах, имеют среднее содержание L12O в рапе от0,0075
до0,015 %.
В 1989 г. основными производителями лития из рапы были Чили и
США, дававшие соответственно 19,5 и 10 % мирового производств
лития.
Таблица 1.7. Подтвержденные запасы лития на начало 1991 г. (L12O)
Страна Запасы Li2O в руде, % Страна Запасы Li2O в РУде. 1
тыс.т % тыс.т %
Боливия 11650 57,4 0,05 Зимбабве 58 0,3 1,3
Чили 2912 14,4 0,3 Мозамбик 32 0,2 —
США 874 4,3 1.5 Намибия 15 <0,1 3,0
Канада 777 3,8 1,0—6,0 Финляндия 30 0,2 1.3
Бразилия 1942 9,6 1,5 Португалия 2 <0,1 1.5
Австралия 1300 6,6 0,7—2,9 Аргентина 11 <0,1 —
Заир 681 3,4 1,3 Всего 9268 100
Производство концентратов литиевых минералов из пегматитовы
месторождений в 1988—1990 гг. несколько увеличилось, но особен»
быстро оно растет в Канаде — в 19 раз и в Австралии — в 4 раза, и она
занимает первое место по производству этого вида литиевого сырья. Все
производители, кроме различных литиевых концентратов, получают
концентраты минералов других металлов. В Австралии получают в ос-
новном высококачественные сподуменовые концентраты (95 %) с со-
46
держанием LizO 7,3—1,5 % и ЕегОз 0,04—0,1 %. Попутно извлекается
небольшое количество минералов Na—Та и касситерит. Канада наряду
с высококачественными концентратами сподумена производит концен-
траты лепидолита, петалита, поллуцита, колумбита-танталита и берил-
ла. В США из литиевых минералов производят свыше 90 % сподумена.
В Зимбабве, а также в Намибии основным литиевым концентратом
является петалитовый (95 %), попутно производятся поллуцит и кон-
центраты других редких элементов. Из редкометальных пегматитов
литиевые минералы в Австралии, США, Канаде, Зимбабве, Намибии,
Бразилии добывают преимущественно открытым способом. Для обога-
щения литиевых руд используют флотацию, магнитную сепарацию, а
также гравитационные методы и ручную рудоразборку при добыче особо
крупных кристаллов сподумена. Промышленные концентраты содер-
жат LizO, %: сподуменовые 4—7, петалитовые 3,5—4,5, амблигонито-
вые 6—8, лепидолитовые 3,0—3,5, эвкриптитовые 5,5—6,5. Следует
отметить, что из экологических соображений уменьшается переработка
лепидолита для получения лития, так как при разложении этого мине-
рала выделяется токсичный фтор (табл. 1.8).
Ресурсы лития в Европе (Финляндии, Португалии, Франции) и Ав-
стралии очень невелики, так же невелико и производство.
Таблица 18 Производство L12O в концентратах и рапе
Страны 1988 г 1990 г
т доля в мировом производстве, % т доля в мировом производстве, %
США 8600 58,7 8600 49,2
Канада 930 6,4 942 5,4
Чили 3100 21,1 3850 22,0
Бразилия 87 0,6 128 0,7
Аргентина 4 0,1 4 0,1
Зимбабве 1090 7,4 685 3,9
Намибия 39 0,3 64 0,4
Австралия 780 5,3 3200 18,3
Португалия 30 0,2 нет св
Всего 14660 100 17473 100
Известны месторождения: Завитинское в Забайкалье (сподумен и
лепидолит), Липовское на Урале (лепидолит), Калбинское в Восточном
Казахстане (сподумен, лепидолит, амблигонит, поллуцит), Туркестан-
ское в Средней Азии (сподумен, петалит, лепидолит, поллуцит).
47
Цены на химическую литиевую продукцию возрастают ежегодно на
2—5 %. В 1990 г. цена на африканские петалитовые концентраты (3,5-1
4,5 % LizO) составляла 135—140 фунтов стерлингов за тонну, 5 %-
ный сподуменовый концентрат стоил 168 фунтов стерлингов за тонну, а
7,25 %-ный 385 долларов/т. Цены за 1 кг ЫгСОз 4, LiOH 5, и
99,9 %-ного металлического лития 56,2 долл. 1
1.8. Общие вопросы технологии лития
Все минералы лития характеризуются низким содержанием ценного
компонента; еще меньше содержание лития в рудах (обычно 1—3 %).
Это вызывает необходимость обогащения литиевого сырья. В результате
обогащения получают концентраты минералов лития, в которых ценно-]
го компонента содержится в несколько раз больше, чем в руде, хотя и
меньше, чем в чистом минерале.
Основной метод обогащения литиевых руд — флотация. Флотация
осуществляется как в щелочной среде с использованием анионных кол-
лекторов — жирных кислот и их производных, так и в кислой среде с
применением катионных коллекторов — сульфированных масел. В пер-
вом случае в пенный продукт выделяются минералы лития; во втором —
в него попадают минералы пустой породы, а минералы лития депресси-i
руются и выводятся в хвосты. В результате флотации обычно получают
концентраты, содержащие 4—6 % U2O. Большое значение для техно-]
логии соединений лития имеет метод термического обогащения (декри-
питация) сподуменовых руд. Метод основан на монотропном а -» пе-|
реходе минерала при нагревании. В процессе обжига руд вследствие
значительного увеличения объема кристаллов сподумена происходит
разрушение пустой породы и выделение/8-сподумена, который довольна
хорошо отделяется воздушной сепарацией или грохочением. Для обога-
щения литиевых руд возможно использование и других методов: обога-
щение в тяжелых суспензиях и магнитная сепарация (для циннваль-1
дита). Часто используют сочетание нескольких методов.
В связи с низким содержанием лития в минералах, а тем более в их
концентратах, современные методы переработки литиевого сырья ти-
пично гидрометаллургические. В гидрометаллургической переработке!
можно выделить два основных технологических этапа: 1) разложение
сырья, в результате которого литий переводится в водорастворимое или
летучее соединение, и 2) концентрирование лития химическими мето-
дами и отделение его от сопутствующих примесей. Определяющей ста-1
дней технологической схемы является разложение концентрата. По
типу используемых для этой цели реагентов все способы переработки]
литиевых концентратов можно разделить на кислотные, щелочные, ще-
48
лочно-солевые и способы, основанные на взаимодействии со средними
солями.
Кислотные методы. Наибольшее значение для переработки лити-
звых концентратов имеет серная кислота. Она позволяет проводить
разложение минералов при относительно высокой температуре, когда ее
действие наиболее эффективно. Использование фтористоводородной
кислоты связано с большими техническими (аппаратурными) трудно-
стями и, кроме того, экономически нецелесообразно. Применение силь-
нолетучих кислот не дает положительных результатов, так как ми-
нералы лития (в основном силикаты и алюмосиликаты) требуют для
разложения достаточно высокой температуры.
Щелочные методы. В щелочных методах переработки литийсодер-
жащих концентратов используют оксиды и гидроксиды металлов, а так-
же карбонаты щелочных и щелочноземельных металлов. В результате
разложения минералов выделяется оксид лития, который в дальнейшем
обычно извлекается в виде гидроксида.
Щелочно-солевые методы. Эта группа методов предусматривает
использование смеси оксидов (карбонатов) или гидроксидов и средних
солей; анион последних определяет природу образующегося при разло-
жении соединения лития. Из возможных компонентов таких смесей
практическое значение имеют соли и оксид кальция. При этом обычно
используют известково-сульфатные и известково-хлоридные смеси.
Методы переработки, основанные на взаимодействии со средними
солями. В процессах разложения минералов лития средними солями
возможно использование лишь соединений, термически устойчивых в
температурных условиях технологического процесса. Наиболее подхо-
дящими для этой цели являются сульфаты щелочных или щелочнозе-
мельных металлов, из них наиболее эффективен сульфат калия. Это
один из старейших и в технологическом отношении наиболее изученных
методов переработки литиевых минералов, но в настоящее время он
практически не используется.
1.9. Переработка сподумена
Для разложения концентрата сподумена возможно использование
всех перечисленных выше методов разложения литиевых минералов.
Наиболее распространенными среди них являются сернокислотный и
щелочной методы переработки.
Сернокислотный метод переработки сподумена. Сернокислотный
метод переработки предполагает использование /^-модификации мине-
рала, которая химически значительно более активна. Это определяется
49
тем, что силикатная структура а -сподумена переходит в алюмосиликат-
ную
(Li, Na)Al[Si2O6] -» Li (Na) |AlSi2O6].
Образующийся /3-сподумен обрабатывают небольшим избытке»
серной кислоты при 250 °C. Подробное исследование реакции сульфати-
зации /3-сподумена позволило установить схему взаимодействия по-
следнего с серной кислотой
Li|AlSi2O6] + H2SO4-» LJ2SO4 + Н [ AlSi2O6 L (1.24)
При сульфатизации происходит ионный обмен между алюмосили-
катным ядром /3-сподумена и серной кислотой, в результате которого
литий в сподумене замещается водородом, причем структура минерала
практически не нарушается. Новое минеральное образование
НгО AI2O3 4SiO2, или Н[А181гОб] известно только как синтетическое
и в природе не найдено. Вода в этом соединении связана с решеткой
минерала так же, как оксид лития в сподумене. Это соединение в воде
нерастворимо. Реакция сульфатизации обратима — выше 700 °C литий
может вытеснить водород, образуя /3-сподумен. Принципиальная тех-
нологическая схема сернокислотного способа представлена на рис. 1.9.
Исходным сырьем служит концентрат сподумена с содержанием
3—5 % U2O. Перед сульфатизацией концентрат подвергают обжигу в
трубчатой печи. При 1100 °C за 10—20 мин степень а -» /3 перехода
сподумена 99—100 %. Из печи материал поступает в холодильник, где
охлаждается до 95—120 °C, а затем на измельчение до 0,074 мм. Измель-
ченный спек смешивается с 93 %-ной серной кислотой, взятой с 35—
40 %-ным избытком по отношению к теоретическому. Температура
сульфатизации 250 °C. Затем реакционную массу выщелачивают водой
в реакторе при непрерывном перемешивании; там же производится ней-
трализация избытка серной кислоты карбонатом кальция до pH 6,0—
6,5. Нейтрализованная масса поступает на фильтр, на котором нераст-
воримый остаток промывают водой, а промывные воды используются
для выщелачивания новой порции спека. Затем остаток выводят из
процесса как отвальный продукт, потери лития с ним не превышают 1 %
Раствор после выщелачивания содержит около 100 г/л сульфата
лития и примесей (Mg, Са, Al, Fe), которые до выделения лития должны
быть удалены. Вначале раствор очищают от магния, который обычно
сопутствует литию в руде. Для выделения магния в виде гидроксида
раствор нейтрализуют известью до pH 12—14. Затем осаждают кальций
в виде СаСОз при обработке раствора кальцинированной содой. После
удаления осадков Mg(OH)2 и СаСОз фильтрованием раствор остается
загрязненным алюминием (из рудного сырья) и железом (в результате
коррозии стальных трубопроводов). Поэтому при последующем упари-
вании в выпарной аппарат подается серная кислота до pH 7 для осажде-
50
Рис. 1.9. Технологическая схема переработки сподумена сернокислотным способом
51
ния гидроксидов железа и алюминия. Упаривание раствора проводят д
содержания сульфата лития — 200 г/л. После упаривания в раство,
добавляется некоторое количество газовой сажи (с целью обесцвечива
ния раствора), которая удаляется вместе с осадком гидроксидов. Кон
центрированный раствор перекачивают в реактор, в котором лити!
осаждается в виде карбоната насыщенным раствором кальцинирован
ной соды при 90 °C. U2CO3 осаждается в виде мелких белых легк<
фильтрующихся кристаллов. После отделения маточного раствора кри
сталлы U2CO3 промывают деионизированной водой; две промывки даю'
96—97 %-ный карбонат лития. Для получения сухого технической
продукта влажный карбонат подвергается вакуумной сушке (70—85
кПа). Сухой карбонат лития содержит следующие примеси, %:
2-
Na2O + K2O........0,18 SO4 ......0,35
СаО...................... 0,04 С1 ..........< 0,005
Fe2O3.............. 0,003 Н2О.......< 0,01
Тяжелые металлы . . . . < 0,001
При переработке концентратов, содержащих 4—5 % U2O, извлече-
ние лития 85—90 %; выход лития в карбонат из руды 50—55 %.
Несмотря на то что сернокислотная схема применима только к fl-
сподумену, она обладает рядом несомненных достоинств. Это, прежде
всего, резкое сокращение энергоемких операций (стадии декрипитации
и сульфатизации требуют для своего завершения 10—20 мин) и исполь-
зование в процессе нелетучей серной кислоты.
Достаточно близким по физико-химическим основам к сернокислот-
ному является способ переработки сподумена, заключающийся во вза-
имодействии его с сульфатом калия. В интервале 1050—1150 °C споду-
мен взаимодействует с K2SO4; при этом основными продуктами реакци»
являются сульфат лития и искусственный лейцит (алюмосиликат ка-
лия)^. Очевидно, что в условиях длительного нагрева при спекание
осуществляется Ct -» fl переход сподумена и фактически с сульфато*
калия взаимодействует /8-сподумен. Таким образом, процесс можнс
описать следующими реакциями
/8-(Li, Na) [AlSi2O6 ] + K2SO4 -» (Li, Na)2SO4 + Ct-K [AlSi2O6 J;
a-K[AISi2O6] ? fl-K [AlSi2O6 ]. (1.25)
Эти реакции описывают взаимодействие /8-сподумсна с K2SO4 с об-
разованием растворимых сульфатов лития, натрия и кубического СС-
лейцита, который при охлаждении переходит в тетрагональный/8-лей-
цит. Внешняя аналогия формул сподумена и лейцита давала основание
рассматривать реакцию (1.25) как ионообменную. Однако из-за боль-
П Плющев В Е ДАН — 1959 —Т 24 —С 642
52
того различия в величине ионных радиусов лития (0,09 нм) и калия
(0,152 нм) простой обмен ионами невозможен. Фактически в данном
случае происходят молекулярные преобразования, в результате кото-
рых образуется алюмосиликат новой структуры К [AlSi2O6 ]. В опреде-
ленных условиях можно, используя этот метод, извлекать из сподумена
на первой стадии 95—98 % лития. Однако при этом расход сульфата
калия составляет 250 ’% от теоретического. Это объясняется тем, что
реакция (1.25) обратима.
Щелочной способ переработки сподумена. В основу способа поло-
жено спекание литиевых минералов с известью или известняком (прин-
ципиального различия в химизме взаимодействия СаО и СаСОз со спо-
думеном нет, так как при высоких температурах СаСОз диссоциирует с
образованием СаО и СОг) с последующим выщелачиванием спека во-
дой. По аналогии с хорошо изученным взаимодействием алюмосиликата
натрия с оксидом кальция процесс можно представить следующим урав-
нением:
LiAl[Si2O6]+4CaO- LiA102+2CaSi04. (1.26)
Таким образом, при спекании образуется не свободный оксид лития,
а малорастворимый алюминат. Для перевода алюмината лития в рас-
твор необходим избыток оксида кальция в шихте, который при обработ-
ке водой образует Са(ОН)г, последний же взаимодействует с алюми-
натом лития
21дА1О2(тв) + Са(ОН)2(р—р) -* LiOH(P—Р) + СаА12О4(Тв). (1.27)
В сподумене в качестве примесей содержатся алюмосиликаты на-
трия и калия, которые при спекании дают соответствующие алюминаты,
а при обработке известью — гидроксиды натрия и калия и алюминат
кальция. Выделение гидроксида лития из такого раствора происходит в
процессе упаривания, при этом вначале выделяется моногидрат гидро-
ксида лития, имеющий наименьшую растворимость. Известковая схема
переработки сподумена представлена на рис. 1.10.
Исходным сырьем служат богатые сподуменовые концентраты, со-
держащие 5—6 % оксида лития. Для спекания применяется известняк
с минимальным количеством кремния. Перед шихтованием концентрат
и известняк (соотношение 1 : 3) измельчаются до минус 0,25 мм в
шаровой мельнице, а затем они совместно доизмельчаются при шихто-
вании до 0,074 мм. Спекание проводится в трубчатых вращающихся
печах при 1150—1200 °C, причем необходим жесткий контроль за тем-
пературой. Выходящий из печи спек охлаждается водой или оборотными
Растворами; при этом происходит его частичное выщелачивание. Затем
спек измельчают до 0,147 мм и выщелачивают в три стадии (противо-
ток) при Т : Ж = 1 : 3 и температуре 90 °C. Остаток от выщелачивания
<Шлам) отфильтровывается и поступает на многократную промывку по
принципу противотока. Содержание оксида лития в отвальном шламе
53
Рис. 1.10. Технологическая схема переработки сподумена спеканием с известью
(известняком)
54
не превышает 0,10—0,15 %. Раствор выщелачивания (10 г/л 1дгО)
направляется на вакуумное упаривание, которое проводится в две ста-
дии. На первой стадии раствор упаривается до уменьшения объема в
8—10 раз. При этом выпадает осадок кальциевых соединений и карбо-
ната лития, который отфильтровывают и направляют на выщелачива-
ние. Затем раствор упаривается до 1,17—1,20 г/см3 и охлаждается до
40—50 °C; при этом выделяется моногидрат гидроксида лития. Кристал-
лы LiOH Н2О отделяют центрифугированием и промывают. Для полу-
чения более чистого продукта LiOH Н2О очищают перекристаллиза-
цией. Для этого кристаллы растворяют в дистиллированной воде при
нагревании до содержания оксида лития -160 г/л. Горячий раствор
фильтруют и кристаллизуют моногидрат гидроксида лития при 40 °C.
Маточный раствор (120 г/л U2O) используют для растворения новой
порции кристаллов. Извлечение лития — около 70 %; с учетом потерь
при обогащении — не более 50 %.
Известковая схема обладает рядом несомненных достоинств. Это,
прежде всего, возможность прямого получения гидроксида лития, так
как никакая другая схема такой возможности не дает. Другие достоин-
ства — универсальность, доступность и дешевизна применяемых реа-
гентов. Переработка сподумена по этой схеме может быть осуществлена
на базе цементных заводов, так как для спекания шихты пригодны
цементные печи. Схема имеет и ряд серьезных недостатков. Для ее
осуществления нужны богатые (не менее 5—6 % 1дгО) концентраты,
так как в шихте они разубоживаются большим количеством известняка.
Многие технологические операции энергоемки — тонкий помол компо-
нентов шихты и спека, упаривание больших объемов растворов.
Заслуживают внимания попытки заменить трудную стадию разло-
жения сподумена известью при высокой температуре разложением из-
вестковым молоком в автоклаве. По этому методу измельченный до -0,1
мм концентрат /3-сподумена смешивают с оксидом кальция (в весовом
отношении 1 : 2) и водой и нагревают пульпу 2 ч при 190—200 °C в
автоклаве. При этом идет реакция
Li(AlSi2O6) + 2 СаО + 2Н2О - ПОН+А1(ОН)з + 2Са8Юз. (1.28)
Образующийся разбавленный раствор гидроксида лития очищают от
примесей карбонизацией. При этом гидроксид натрия переходит в
ИагСОз, a NaAlO2 гидролизуется, образуя NaOH и А1(ОН)з, кальций
же осаждается в виде СаСОз. После фильтрования раствор повторно
карбонизуют для осаждения карбоната лития, который после промывки
и сушки является готовым продуктом. Выход лития в продукт > 80 %.
Описан очень простой вариант щелочного метода переработки /3-
сподумена, предусматривающий получение карбоната лития в резуль-
тате автоклавного выщелачивания минерала раствором соды при 200 °C.
Продуктами реакции являются карбонат лития и алюмосиликат натрия
55
2Li[AlSi2O6] + Na2CO3- Li2CO3 + 2Na [A!Si2O6 ]. (1.29)1
Полученную смесь обрабатывают под давлением углекислым газом;
при этом карбонат лития переходит в растворимый гидрокарбонат, к»,
торый отделяют от осадка и декарбонизуют при 90 °C. Этот вариант
щелочного метода используется в промышленных масштабах. i
Щелочно-солевые методы. Наибольший интерес вызывает испол J
зование щелочно-хлоридных смесей; как правило, применяется смесь
оксида (карбоната) кальция с хлоридом кальция или аммония. На осно-
ве взаимодействия сподумена с СаО и СаС12 создано несколько вариан-
тов технологической схемы; наиболее интересен из них вариант, преду-
сматривающий возгонку образующегося в результате реакции хлорида
лития и улавливание его в электрофильтрах. Взаимодействие сподумена
с СаО + СаС12 описывается суммарной реакцией
4 [Li2O А12О3 4SiO2 ] + ЗОСаО + 4СаС12 - 8LiCl +
+13 [у- 2СаО SiO2 ] + 5 СаО ЗА12О3 + ЗСаО А12О3 3SiO2. (1.30)
Показано, что при 1100—1200 °C можно перевести в хлорид до 98 %
лития, содержащегося в сподумене. Несмотря на чрезвычайную агрес-
сивность парообразного хлорида лития, метод следует считать перепек-'
тивным при условии успешного решения аппаратурных вопросов, так
как он позволяет получить технический LiCl в одну стадию с выходом
до90 %.
1.10. Переработка лепидолита
Для переработки лепидолита применимы те же методы^ что и для
сподумена, но взаимодействие лепидолита с реагентами протекает в
более мягких условиях, что определяется его большей химической ак-
тивностью. На ранней стадии развития литиевой промышленности ле-1
пидолит был основным сырьем для получения различных соединений!
лития. Описаны многочисленные методы его переработки. В результате!
обогащения получрют концентраты лепидолита с содержанием 3—4 %]
оксида лития. Лепидолит в качестве изоморфных примесей содержит!
рубидий и цезий, извлечение которых должно быть предусмотрено схе-
мой. Из различных способов переработки концентратов лепидолита от-
дается предпочтение сернокислотному, реализуемому в различных ва-
риантах.
По одному из вариантов тонкоизмельченный (до 0,09 мм) лепидолит!
смешивают в стальном реакторе с концентрированной серной кислотой,
количество которой составляет ПО % от массы минерала. Смесь вы-
держивают 30 мин, затем медленно нагревают в течение 8 ч от 110 до
340 °C. При этом степень разложения минерала составляет >94 %.|
Сульфатизированную массу в теплом состоянии выщелачивают водой.
Выделившийся осадок диоксида кремния отфильтровывают. В раствор
56
переходят соли всех щелочных металлов (Li, Na, К, Rb, Cs), алюминия,
железа, марганца. Для удаления алюминия в раствор вносят сульфат
калия в расчете на образование алюмокалиевых квасцов, с которыми
выделяются также рубидий и цезий в виде соответствующих квасцов.
Маточный раствор после отделения квасцов нейтрализуют карбонатом
кальция для отделения остатка алюминия в виде гидроксида. После
этого осаждают кальций, магний, железо и марганец щавелевой кисло-
той и раствором аммиака. Таким образом получают чистый раствор
сульфата лития, из которого с помощью карбоната калия осаждают
технический карбонат лития, который промывают и сушат при 60 °C.
Извлечение лития в карбонат составляет не более 70 %. Из раствора
после отделения карбоната лития выделяют рубидий и цезий. Лепидо-
лит перед сульфатизацией рекомендуют сплавлять в стеклообразную
массу при 1090 °C и измельчать.
Хорошие результаты были получены при использовании для разло-
жения лепидолита сульфата калия. Установлено, что при 1090 °C раз-
лагается не весь комплекс минерала, и в растворе после выщелачивания
спека горячей водой содержатся только сульфаты лития, калия и неболь-
шое количество сульфата марганца, который легко удаляется гидрокси-
дом калия. Из этого раствора выделяют карбонат лития. Практически
весь рубидий и цезий остаются в остатке, который специально перера-
батывают для извлечения этих элементов.
При переработке руд, содержащих минералы лития, основные поте-
ри приходятся на стадию их обогащения. Так, при содержании в концен-
трате 4—5 % Li2O извлечение лития в карбонат по сернокислотной
схеме составляет 85—90 %, а выход лития в готовый продукт из руды
50—55 %, так как при ее флотации в концентрат извлекается только
60—70 % Li2O. Это обстоятельство явилось стимулом для изучения
вопроса о применимости ряда технологических схем к переработке не-
посредственно сподуменовой руды. Лучшие результаты были получены
Для сернокислотной схемы и отчасти при использовании известково-
хлоридных смесей. Была создана технологическая схема переработки
сподуменовой руды на основе ее взаимодействия с серной кислотой.
Руду, содержащую 25 % сподумена (~ 1,5 % Li2O), обжигают при
1095 °C, продукт охлаждают до 65 °C, измельчают до минус 0,07 и
сульфатизируют при 230 °C концентрированной серной кислотой. Реак-
ционную массу выщелачивают водой; раствор нейтрализуют известня-
ком, фильтруют и обрабатывают последовательно гидроксидом кальция
и карбонатом натрия для осаждения гидроксида магния и карбоната
Кальция. Отфильтрованный раствор упаривают до содержания 150 г/л
Li2SO4 и обрабатывают кальцинированной содой. Выход лития в карбо-
нат — до 80 %; схема применима к рудам, содержащим не менее 1 %
L12O.
57
Известны работы по непосредственному хлорированию минералов
лития (сподумена, лепидолита, петалита) с получением хлорида лития.
Это направление изучено крайне мало, однако оно представляется пер-
спективным, так как применимо для извлечения из комплексных руд,
содержащих бериллий, ниобий, тантал, олово, хлориды которых, так же
как и LiCl, летучи.
1.11. Извлечение лития из солевых растворов
В последние годы интенсивно разрабатываются способы получения
литиевых солей из нетрадиционных видов сырья. Литий производится
из природных рассолов в США и Чили. В Чили на базе подземным
рассолов месторождения в пустыне Атакама построен комбинат. Состав
подземных рассолов различных месторождений приведен в табл. 1.9. I
Таблица 1.9. Содержание полезных компонентов в
подземных рассолах, г/л
Компо- ненты Месторождение, страна
Атакама, Чили Сильвер Пик, США Грейт Солт- лейк, США Дид Сиа, Иордания
к 22,4 Нет св. 6,3 7,0
2- SO4 21,7 — 20,0 0,6
Li 1,7 0,3 0,05 0,02
Н3ВО3 4,4 — — —
Известные способы переработки природных рассолов (содержание
лития 0,32—1,7 г/л) имеют, как правило, общую начальную стадию —
их концентрирование. Концентрирование достигается за счет солнечно-
го выпаривания. Например, подземные рассолы пустыни Атакама кон-
центрируются в испарительных бассейнах по содержанию лития от 0,17
до 4,3 %. Выделение лития из концентрированного рассола может быть
осуществлено различными путями. Представляют интерес два способа
извлечения лития: процесс высаливания и литий—калий сульфатный
процесс. Способ высаливания заключается в выделении сульфата лития
сульфатом магния из рассолов с содержанием лития > 1 % при 35 °C-
Полученный гидратированный сульфат лития переводят в карбонат об-
работкой кальцинированной содой при нагревании. Литий—калий
сульфатный процесс основан на кристаллизации двойной соли LiKSO4
при 18—35 °C. Эта соль растворяется в рассоле, содержащем КС1, ®
58
результате чего выделяется в осадок сульфат калия, а литий остается в
рассоле. Обычно извлечение лития не превышает 70 %.
Оценивая дальнейшие пути развития технологии лития, необходи-
мо учитывать, что структура перерабатываемого литиевого сырья неми-
нуемо будет изменяться. На смену традиционным рудным видам сырья
придут нетрадиционные — рассолы, попутные нефтяные воды, литий-
содержащие глины, так как более 70 % разведанных запасов лития
сосредоточено именно в этих источниках. В результате изменения сырь-
евой базы, вероятно, произойдут изменения и в технологии. Перспек-
тивным направлением, возможно, станет разложение концентратов в
гидротермальных условиях. Несомненно, повысится роль ионообмен-
ных процессов. Это потребует проведения значительного объема науч-
но-исследовательских работ, направленных на поиск принципиально
новых решений в технологии лития.
1.12. Переработка карбоната лития на гидроксид и хлорид
Рассмотренные выше методы переработки литийсодержащего сырья
в качестве товарного продукта предусматривают получение карбоната
лития. Исключение составляет известковый метод. Карбонат лития ис-
пользуется непосредственно и, кроме того, он служит источником полу-
чения различных соединений лития, основными из которых являются
гидроксид и хлорид.
Получение гидроксида лития. Единственным промышленным спо-
собом получения гидроксида лития является каустификация известью в
растворе
1д2СОз + Са(ОН)2- 2ЫОН + СаСОз. (1.31)
Приведенные ниже данные по растворимости при 20 °C компонентов
реакции (1.31) показывают, что равновесие реакции должно быть сдви-
нуто вправо:
Соединение............Li2CO3 Са(ОН)2 LiOH СаСОЗ
Растворимость, г/100 г Н2О . . 0,13 0,165 12,8 1.3- 10 $
В то же время из данных по растворимости в системе U2CO3—
Са(ОН)г—Н2О при 75 °C следует, что максимальная концентрация
LiOH в растворе не может быть выше ~ 36 г/л, т.е. можно получать
только разбавленные растворы LiOH.
Технологическая схема получения гидроксида лития представлена
на рис. 1.11. Исходным продуктом при каустификации является влаж-
ный карбонат лития. Карбонат лития и гидроксид кальция замешивают
в реакторе; известь берется в количестве 105 % от теоретического.
Реакционная масса нагревается до кипения. По окончании процесса
°Ульпу отстаивают и осветленный раствор декантируют. Он содержит
59
Влажный L12CO3 ----1
[ Каустификация ~|
Ca(OH)j -»— Приготовление
известкового
__________ ыопока
| Раствор LiOH (30 г/л) |
Декантация
1Л ,
I Шлаы |
Упаривание
Охлаждение
и кристаллизация
| ! проыывка |
Декантация
Центрифугирование
I lli/таы |
~г
| Раствор
Маточный
раствор
I И проыывка
Декантация
*| Раствор |
Коистаппы
LiOH-HjO
| Шлаы |
f—Вода
| III проыывка |
•| Растворение j
-| Декантация [
-| Фильтрование ~|—
Шлаы
(в отвал)
| Метеор |
Нерастворииый
остаток
Рис. 1.11. Технологическая схема получения гидроксида литня каустификацией
60
28,7—35,9 г/л LiOH. Шлам (в основном карбонат кальция) подвергают
трехстадийной противоточной промывке для доизвлечения лития. Ос-
новной раствор упаривается до содержания LiOH 166,6 г/л. Затем тем-
пература понижается до 40 °C. Гидроксид лития выделяется в виде мо-
ногидрата LiOH НгО. Кристаллы LiOH Н2О отделяют от маточного
раствора центрифугированием. Для получения чистого соединения пер-
вичный продукт перекристаллизовывают. Выход лития в готовый про-
дукт — около 85—90 %. Основной недостаток метода — высокие требо-
вания к чистоте исходных продуктов. Карбонат лития должен содержать
минимальное количество примесей, особенно хлоридов. Известь также
должна быть чистой, не должна содержать алюминия во избежание
образования малорастворимого алюмината лития.
Получение хлорида лития. Промышленный способ получения хло-
рида лития основан на растворении карбоната или гидроксида лития в
соляной кислоте, причем обычно используют карбонат
Li2CO3+2HCl > 2LiCl + H2O+CO2 | ; (1.32)
LiOH + HCl* LiCl + НгО. (1.33)
Так как технические карбонат и гидроксид лития содержат значи-
тельное количество примесей, их необходимо предварительно удалять.
Карбонат лития обычно очищают переводом его в хорошо растворимый
гидрокарбонат с последующей декарбонизацией и выделением IJ2CO3.
После очистки карбоната лития, содержащего 0,87 % SOj" и 0,5 %
щелочных металлов, получают продукт, содержащий следы серы и
0,03—0,07 % щелочных металлов. Для очистки гидроксида используют
или перекристаллизацию, или осаждение U2CO3 карбонизацией рас-
твора. Принципиальная схема получения хлорида лития из карбоната
представлена на рис. 1.12.
Процесс получения хлорида лития связан с двумя трудностями —
упариванием растворов и обезвоживанием соли. Известно, что хлорид
лития и его растворы обладают высокой коррозионной способностью, а
безводная соль—большой гигроскопичностью. Хлорид лития при нагре-
вании разрушает почти все металлы, кроме платины и тантала, поэтому
Для упаривания растворов LiCl применяется аппаратура из спецспла-
вов, а для обезвоживания — керамическая.
Для получения хлорида лития используется влажный карбонат,
который обрабатывают 30 %-ной НС1. Полученный раствор содер-
жит - 360 г/л LiCl (плотность 1,18—1,19 г/см3). Для растворения дают
небольшой избыток кислоты и после перемешивания хлоридом бария
осаждают сульфат-ионы. Далее раствор нейтрализуют карбонатом ли-
тия и добавляют еще LiOH для получения 0,01 н раствора по LiOH.
Раствор кипятят для выделения Са, Ba, Mg, Fe и других примесей в виде
ГиДрокисдов, карбонатов или основных карбонатов.
61
Li2CO3 HCI
После фильтрования полу-
чают 40 %-ный раствор LiCl,
часть которого находит непос-
редственное применение, а
большая часть перерабатыва-
ется на безводную соль. Без-
водный хлорид лития получа-
ют в последовательно соеди-
ненных выпарной башне и суч
шильном барабане. Содержа-
ние примесей в хлориде лити:
приведено ниже, %:
NaCl+KCl.........0,5
СаС12............ 0,15
ВаС12............0,01
so3-.............0,01
Fe2O3............ 0,006
Н2О.............. 1,0
Нерастворимый
остаток..........0,015
Рис. 1.12. Технологическая схема получения
хлорида лития
1.13. Получение металлического лития
Металлический литий получают двумя способами: электролизом и
вакуумтермией. Вследствие высокой химической активности лития и
термодинамической устойчивости его соединений процессы его получе-
ния связаны с рядом трудностей. Они заключаются в ограниченности
выбора возможных способов получения, в необходимости защиты по-
лучающегося металла от действия кислорода, азота, углекислого газа и
водяных паров, в подборе конструкционных материалов, устойчивых к
действию лития и его соединений при повышенной температуре.
Электрохимическое получение лития. Электрохимические хараК"|
теристики показывают, что литий можно получить только электроли-
зом расплавов, электролиз водных растворов невозможен. В водных
растворах электродный потенциал лития имеет очень большое отрица!
62
Рис. 1.13. Зависимость давления пара щелочных ме-
таллов и магния от температуры:
1 — Cs; 2 — Rb; 3 — К; 4 — Na; 5 — Mg; б — Li
тельное значение, вслед-
ствие чего на катоде при
электролизе будет выде-
ляться водород (см. табл.
1.1). Для электролиза
обычно используют рас-
плавы галогенидов, од-
нако индивидуальные
галогениды лития LiCl и
LiF для этой цели непри-
годны вследствие высо-
ких температур плавле-
ния, при которых давле-
ние паров лития стано-
вится довольно большим
(см. табл. 1.6, рис. 1.13).
Для снижения темпера-
туры плавления элект-
ролита используют смесь LiCl и КС1 в соотношении 1:1; такой состав
близок к составу эвтектики в системе LiCl—КС1 [58,5 % (мол.) LiCl,
Тпд = 361 °C ]. Электролиз проводят при 400—430 °C. Напряжение раз-
ложения хлоридов магния, кальция и натрия в расплавах меньше напря-
жения разложения хлорида лития при 700 °C, В:
MgC12 CaC12 NaCl LiCl КС1
2.60 3,38 3,39 3.41 3,53
Эти элементы будут восстанавливаться на катоде раньше лития и
поэтому будут полностью переходить в металл. Напряжение разложе-
ния хлорида калия лишь очень немного больше напряжения разложения
хлорида лития (ЛЕ = 0,11В). Такая небольшая разница сохраняется в
широком интервале температур: так, при 405 °C значения напряжения
разложения для LiCl и КС1 соответственно равны 3,78 и 3,89В. Выделе-
ние калия на катоде увеличивается при обеднении расплава электроли-
та хлоридом лития, поэтому состав электролита постоянно нужно кор-
ректировать, добавляя LiCl и не допуская снижения его концентрации
ниже 55—57 %. Для уменьшения загрязнения металлического лития
загнием, кальцием и натрием необходимо использовать исходный хло-
рид лития высокой степени чистоты.
Для электролиза используют электролизеры с разделенными анод-
ным и катодным пространствами (рис. 1.14). Корпус электролизера
стальной, футерован графитом, материалом, наиболее устойчивым по
отношению к расплавленному хлориду лития. Футеровка из графита
служит около трех месяцев, так как постепенно разрушается вследствие
образования карбида лития. Более чистый металл получают в электро-
63
Рис. 1.14- Электролизер для получения метал-
лического лития:
1 — стальной кожух; 2 — камера для ра-
зогрева ванны; 3 — газовая или нефтяная фор-
сунка; 4 — футеровка; 5 — графитовый анод; 6
— стальные катоды, 7 — диафрагма из желез-
ной сетки; 8 — диафрагма из химически- и
термостойкого материала; 9 — жидкий литий;
10— бункер для загрузки солей; 11 — отверстие
для удаления жидкого лития
лизерах со стальными водоох-4
лаждаемыми стенками, на ко-
торых образуется гарниссаж из
затвердевших солей электро-1
лита. Аноды графитовые, каточ
ды — из малоуглеродистой ста-
ли, но могут использоваться и
более стойкие металлы, такие
как ниобий и тантал. Катодное
и анодное пространство разде-
ляют диафрагмой из железной
сетки. При электролизе литий
вследствие меньшей плотное!
ти, чем расплав солей (см. табл.
1.1, 1.6), собирается на поверх-
ности расплава электролита в
катодном пространстве. Рас!
плавленный литий защищен от
соприкосновения с воздухом
тонкой пленкой расплавлен*
ных солей. По мере накопления
расплавленного лития он уда-
ляется вручную. Хлор из анод-»
ного пространства отсасывает-
ся вентиляторами и направля-.
ется на нейтрализацию извест-
ковым молоком.
Электролиз характеризуется следующими показателями: выход по
току 90—93 %, расход электроэнергии на 1 кг лития до 144 кВт ч,
извлечение лития до 95 %, напряжение на ванне 6—6,6В. Содержание?
примесей в электролитическом литии, %: 0,3—2,5 Na; 0,02—1,50 К;
0,002—0,12 А1; 0,003 Mg; 0,001—0,04 Са, Fe, Си; 0,004—0,8 Si; 0,01 Cl.
Электрохимический метод может быть использован для получения
сплавов лития с другими металлами — магнием, алюминием, кальцием!
свинцом и др. При получении сплавов с легкоплавкими металлами с
невысокой плотностью возможны два варианта проведения процесс!
электролиза. По первому — хлорид соответствующего металла вводится
в состав электролита; по второму — из этого металла изготовляется
катод, который по мере выделения лития растворяется в нем. В обоих
случаях образуется жидкий сплав, собирающийся на поверхности рас-
плава электролита.
Электрохимический метод является достаточно эффективным Я
применяется в промышленных масштабах. В то же время он не лишен
64
недостатков, важнейшие из которых следующие: необходимый для него
безводный хлорид лития высокой чистоты является дорогим продуктом;
получающийся металлический литий загрязнен примесями, прежде
всего натрием, что требует дополнительной его очистки; выделяющийся
при электролизе хлор необходимо обезвреживать.
Вакуумтермическое получение лития. Термодинамический анализ
реакций типа приведенной ниже показывает, что большинство из них
характеризуется положительными значениями д 0^98 J особенно велики
они для восстановления фторидов и хлоридов
LiX + M7 Li + MX, (1.34)
гдеХ —F,C1,O.
Это является следствием того, что соединения лития с кислородом и
особенное фтором и хлором термодинамически более прочны, чем соот-
ветствующие соединения наиболее активных металлов-восстановите-
лей (Na, Са, Mg, Al и др.). И только реакции восстановления оксида
лития кальцием и магнием характеризуются значениями Д G298 порядка
10 кДж/моль, что свидетельствует об их обратимости (рис. 1.15). Сам
литий вследствие большой термодинамической прочности его соедине-
ний используется как металл-восстановитель. При таких термодинами-
ческих характеристиках осуществление металлотермического
процесса становится возможным при условии удаления получаемого
металла из зоны реакции или
связывания получаемых про-
дуктов каким-либо компонен-
том, дополнительно вводимым
в исходную шихту. В результа-
те равновесие реакции (1.34)
смещается вправо, этому бла-
гоприятствует относительно
низкая температура кипения
лития. Низкая температура ки-
пения металлического кальция
и магния исключает их исполь-
зование для этих целей.
Наиболее подходящими
Для восстановления оксида ли-
тия оказались элементарный
кремний и металлический алю-
миний, имеющие высокие тем-
пературы кипения. Реакция
восстановления оксида лития
Рис. 1.15. Зависимость энергии Гиббса образо-
вания некоторых оксидов от температуры
(на 1 моль кислорода)
3 —- 691
65
кремнием характеризуется положительными значениями энтальпии и
изменения энергии Гиббса
2Li2O+Siz 4Li + SiO2, (1.35)
А Й298 = + 285 кДж, Л 6§98 ~ + 246 кДж.
Реакция (1.35) эндотермическая, поэтому смещению равновесия
вправо кроме удаления лития способствует повышение температуры.
Однако процесс восстановления осложняется протеканием вторичной
реакции (1.36), приводящей к образованию термодинамически очень
устойчивого ортосиликата лития Li4SiO4 (A6298(f)= ~ 2183 кДж/моль).
В ортосиликате связывается 50 % лития
4Li2O + Siz 4Li + Li4SiO4; а(?298 = + 61 кДж. (1.36)
В шихту вводят оксид кальция, который образует также термодина-
мически устойчивый ортосиликат Ca2SiC>4 (л 6298(f)=_2192 кДж/моль).
В присутствии в шихте оксида кальция значительная часть первично
образовавшегося оксида кремния взаимодействует с СаО, а образовав-
шийся силикат лития сам может восстанавливаться по реакции
Li2SiO4 + Si + 4СаО » 4Li + 2Ca2SiO4; A 639s = +95 кДж. (1.37)
Суммарная реакция процесса может быть выражена уравнением
2Li2O+Si+2CaOz 4Li+Ca2SiO4, (1.38)
а6з98 = + 108 кДж, a6?oook = -351 кДж-
Оксид лития, необходимый для процесса, получают термичес-
ким разложением карбоната лития. Для этого нагревают в вакууме при
850 °C брикетированную смесь IJ2CO3 и СаО, взятых в соотношении
1:1,5. Оксид кальция добавляют для предотвращения плавления IJ2CO3
и облегчения удаления СОг. Полученную смесь оксидов U2O и СаО
измельчают, шихтуют с кремнием, который берется с избытком 10 %,
нагревают в вакуумной печи при давлении ~ 0,1 Па и температуре
1000—1300 °C. Извлечение лития при 1000 °C составляет 75 %, а при
1300 °C увеличивается до 93 %. Основные примеси в металлическом
литии: 0,01 % Si и 0,04 % Са.
При восстановлении оксида лития алюминием часть лития связыва-
ется в алюминат ЫАЮг, для которого A 6398(f) = - 1126 кДж/моль
41лгО + 2А1^ 6Li + 2LiA102? Абз98 = + 13 кДж. (1.39)
Для предотвращения потерь лития в шихту добавляют СаО, который
образует алюминат Са(А10г)2 (a 6398(f) = -2208 кДж/моль)
ЗЫ2О + 2А1 + Са ~z 6Li + Са(А10г)2; А 6398 = +81 кДж. (1.40)
66
Лучшие результаты по получению лития повышенной чистоты дает
восстановление алюмината лития, которое не требует введения в шихту
СаО
6I1A1O2 + 2A1X 6Li +AI2O3; ДG298 = +428 кДж. (1.41)
Исходный алюминат получают путем взаимодействия стехиометри-
ческих количеств L12CO3 и AI2O3 в вакууме при 900—1000 °C. Процесс
восстановления проводят при давлении ~ 10 Па и температуре 1150—
1200 °C. Извлечение лития —до 95—98 %.
Металлотермическое восстановление оксида лития имеет некоторые
преимущества перед электролизом расплавов: в качестве исходного
сырья используется карбонат лития, являющийся основным продуктом
большинства современных технологических схем переработки литиево-
го сырья. Способ позволяет получать из технических продуктов более
чистый литий, чем электрохимический метод; восстановителями слу-
жат кремний и алюминий, относительно дешевые и доступные.
Вакуумтермическое получение лития из сподумена. Способ являет-
ся одним из перспективных направлений в совершенствовании техноло-
гии лития. Сущность предложенного метода заключается в нагревании
в вакууме смеси сподумена, ферросилиция или алюминия в присутствии
СаСОз. Химизм процессов в обоих случаях близок к химизму восстанов-
ления оксида лития кремнием или алюминием.
Компоненты шихты измельчаются до крупности частиц < 0,1мм,
смешиваются в соотношении: сподумен : ферросилиций (75 % Si) :
известняк = 1 : 0,28 : 2,3. При использовании алюминия соотношение
сподумен: алюминий: известняк =1:0,15:1,5. Процесс восстановления
проводят при давлении 1—4 Па и температуре 1050—1200 °C в реторте,
снабженной конденсатором. При использовании ферросилиция в кон-
денсат извлекается 85—90 % лития, содержание лития в нем 90 %.
Основные примеси — магний, переходящий из известняка, натрий (до
2 %), калий (до 1 %), переходящие в металл из сподумена. При исполь-
зовании в качестве восстановителя алюминия извлечение лития может
достигать 93—94 %. В производственных условиях, по-видимому, из-
влечение лития в металл не будет более 70 %.
Прямое получение лития из сподумена позволяет уменьшить число
технологических операций, количество различных отходов и сбросов.
Наиболее серьезный недостаток способа — большая загрязненность ме-
талла и необходимость проведения довольно сложных операций по его
рафинированию.
Рафинирование лития. Металлический литий, полученный тем или
иным способом, содержит примеси щелочных, щелочноземельных и
тяжелых металлов, неметаллические примеси, прежде всего кислород и
азот, твердые частицы. Примеси обладают различными свойствами,
з*
67
поэтому универсального метода их удаления нет. При получении лит»
высокой степени чистоты прежде всего удаляют твердые частицы мет|
дами отстаивания расплавленного металла под слоем парафинового или
трансформаторного масла или фильтрования через сетчатые фильтрь
из железной, титановой или молибденовой проволоки. Кислород и азся
удаляются с помощью геттеров — порошкообразных или губчатых ти-
тана и циркония. При повышенной температуре они активно взаимо-
действуют с кислородом и азотом, но в литии растворяются мало. После
такой обработки при 800 °C в течение 24 ч литий содержит лишь следы
этих примесей.
Натрий и калий эффективно удаляются методом, основанным на
различной устойчивости гидридов. При нагревании лития в водороде
при 700—800 °C гидриды NaH и КН не образуются, так как при темпе-
ратуре выше 427 °C NaH и КН полностью диссоциируют. Гидрид лития
разлагают нагреванием в вакууме. Наибольшее же значение среди ме-
тодов рафинирования лития имеет вакуумная дистилляция, позволяю]
щая производить очистку от щелочных, щелочноземельных и тяжелы»
металлов. Процесс проводят при остаточном давлении 10 1—10 3 Па.
Сначала при 450 °C отгоняют натрий и калий, затем при 600—800 °C
испаряют литий; температура в конденсаторе поддерживается на уровне
340—420 °C. При извлечении лития в конденсат 85—90 % содержаний
в нем примесей снижается до п-10 3 %. Наибольшие трудности вслед-
ствие близости значений давления пара представляет удаление магнш
(см. рис. 1.13). Для очистки сильно загрязненного лития может быт1
использован метод электрохимического рафинирования.
Качество очистки лития во многом зависит от конструкционны»
материалов, использованных для изготовления аппаратуры. Для пере-
плавки лития рекомендуются тигли из ZrOi, футерованные LiF, устой-
чивые к действию расплавленного лития и его паров до 800 °C, высоко-
хромистые стали (до 17 % Сг), чистые железо, титан, молибден, ниобий.
Глава 2. РУБИДИЙ И ЦЕЗИЙ
ХИМИЯ РУБИДИЯ И ЦЕЗИЯ
2.1. Физико-химические свойства
Рубидий и цезий, как и все щелочные элементы, имеют по одному
валентному электрону. Электронную структуру их атомов можно пред-
ставить соответственно— [Кг 15s1 и [Хе]б№.Электроны5sи6sэкрани-
рованы большим числом электронов внутренних оболочек, поэтому
68
связь их с ядром атома слабее, чем у других щелочных элементов.
Вследствие этого рубидий и цезий имеют самые низкие энергии иониза-
ции, легко образуют ионы типа М+, которые имеют электронную струк-
туру соответствующих благородных газов (см. табл. 1.1). Они являются
самыми электроположительными элементами, их соединения характе-
ризуются наибольшей долей ионной составляющей химической связи по
сравнению с аналогичными соединениями всех других элементов. Руби-
дий и цезий имеют наименьшие коэффициенты поляризации и наиболь-
шие — поляризуемости, практически никогда не бывают центральными
атомами комплексных соединений, но во многих случаях выступают в
качестве внешнесферных катионов. Устойчивость таких комплексов
возрастает в ряду Na -» Cs. Их нормальные электродные потенциалы
имеют наибольшие отрицательные значения как в водных растворах,
так и в безводных средах, поэтому получение их электролизом водных
растворов невозможно, а электролиз безводных сред связан с большими
трудностями и неэффективен.
Рубидий и цезий обладают интересными оптическими свойствами.
Эти металлы прозрачны в ультрафиолетовой части спектра. Показатель
преломления в прозрачной области меньше единицы, наблюдается пол-
ное внутреннее отражение. Границы прозрачности рубидия и цезия
расположены в области длин волн 360 и 440 нм соответственно. Среди
всех физических свойств рубидия и цезия наиболее характерными яв-
ляются фотоэлектрические. Фотоэффект основан на способности метал-
лического катода испускать поток электронов под действием света. Для
щелочных металлов порог фотоэффекта, т.е. наибольшая длина волны,
вызывающая электронную эмиссию, расположен в длинноволновой об-
ласти спектра. Рубидий может испускать электроны под действием на-
иболее длинных красных лучей видимого спектра, а цезий — даже под
действием невидимых инфракрасных тепловых лучей.
Рубидий и цезий имеют серебристо-белый цвет и на свежем срезе
металлический блеск. Это наиболее тяжелые и легкоплавкие щелочные
металлы (см. табл. 1.1). Рубидий и цезий — наиболее реакционноспо-
собные металлы. На воздухе они мгновенно окисляются с воспламене-
нием; с водой бурно реагируют с образованием гидроксидов и выде-
лением водорода, который немедленно воспламеняется. Эта реакция
протекает даже при температуре -100 °C. С галогенами, диоксидом
Углерода и четыреххлористым углеродом рубидий и цезий взаимодейст-
вуют со взрывом.
Образование гидридов и амидов рубидия и цезия происходит при
нагревании их расплавов в атмосфере водорода или аммиака. Рубидий
и Цезий бурно реагируют с ртутью с образованием амальгам или интер-
металлических соединений. Они непосредственно не реагируют с азо-
том; с углеродом, фосфором, кремнием взаимодействуют только при
69
температуре выше 300 °C. Рубидий и цезий растворяются в жидц!
аммиаке, некоторых алкиламинах и полиэфирах с образованием рас!
творов синего цвета, обладающих электронной проводимостью; бурц ।
реагируют со всеми кислотами с выделением водорода и образований]
соответствующих солей. ]
Рубидий и цезий образуют сплавы с другими металлами, в том числ I
и щелочными, а также многочисленные интерметаллические соедиие!
ния. В системах К—Rb; К—Cs; Rb—Cs образуются непрерывные твер
дые растворы; в системах же Rb—Li и Cs—Li имеет место расслаиваем
в жидком состоянии, что объясняется прежде всего существенной разни
цей в их атомных и ионных радиусах. Система Rb—Na эвтектической
типа, в то время как в аналогичной системе с цезием установлено суще-
ствование интерметаллического соединения Na2Cs. Образование интер-
металлических соединений чаще всего наблюдается у тех металлов
свойства которых (в частности, удельные объемы и электрохимические
характеристики) сильно различаются. Состав интерметаллических со-
единений не определяется обычной валентностью металлов (например
RbCdi3). В интерметаллических соединениях элементы не утрачивают
своих металлических свойств, а только несколько изменяют их, чтс
обусловливает появление сингулярных точек на изотермах свойств. I
Из сплавов рубидия и цезия с другими элементами особый интерес
представляют соединения с висмутом, сурьмой и ртутью. У рубидий-
дивисмута и цезий-дивисмута кубической структуры обнаружена
сверхпроводимость при 3,2 К. Интерметаллические соединения — чер-
но-фиолетовый RbsSb и черные RbsBi, CssBi и CssSb — обладают высо-
кой фотоэлектрической чувствительностью. Этим соединениям отве-
чают сингулярные точки на изотермах свойств и индивидуальные кри-
сталлические структуры с упорядоченным расположением атомов и об
ласти гомогенности. При избытке атома щелочного металла соединение
имеют p-тип проводимости, при дефиците — n-тип проводимости. В
системе Rb—Hg установлено существование восьми меркуридов руби-
дия RbnHgm, а в системе Cs—Hg — четырех, прочность которых возра-
стает от рубидия к цезию. Меркуриды рубидия и цезия легко окисляют^
на воздухе, образуя черно-коричневую массу — смесь надпероксидЗ
щелочного металла и оксида ртути (I).
2.2. Оксиды и гидроксиды
Рубидий и цезий образуют ряд соединений с кислородом — оксид#
М2О, пероксиды М2О2, надпероксиды МО2, озониды МОз (см. табл. 13»
рис. 1.1).
Оксиды. Оксиды рубидия и цезия представляют собой прозрачные
иглы или листочки бледно-желтого (Rb2O) или коричнево-красное
70
(Cs?O) цвета, расплывающиеся на воздухе. Оксид рубидия имеет куби-
ческую решетку типа СаРг, оксид цезия — гексагональную, содержа-
щую сильно поляризованные ионы цезия. Оксиды рубидия и цезия раз-
лагаются под действием света с выделением металла. В вакууме оксид
рубидия сублимирует без разложения при нагревании до 500 °C. Повы-
шение температуры приводит к диспропорционированию Rb?O на ме-
талл и RЬгОг, который не испаряется до 630 °C. Оксид цезия возгоняется
при 350—450 °C; при 500 °C образуется CS2O2, который полностью
сублимирует при повышении температуры. Оксиды рубидия и цезия
энергично взаимодействуют с водой
Rb(Cs)2O + H2O = 2Rb(Cs)OH, (2.1)
с влажным диоксидом углерода с воспламенением, при 150—200 °C — с
водородом, фтором, хлором. Бурно реагируют с расплавленной серой по
реакции
4M2O+7S=M2SO4 + 6MS. (2.2)
При обработке оксидов рубидия или цезия жидким аммиаком обра-
зуется эквимолярная смесь гидроксида и амида.
Оксиды не являются характерными соединениями для рубидия и
цезия. Получаются они с большим трудом только в вакууме или атмос-
фере благородного газа или азота при недостаточном доступе кислорода.
Для получения чистых оксидов над металлическими Rb или Cs в лодоч-
ке из особо чистого родия медленно пропускают кислород в количестве
~ 65 % от необходимого для полного окисления металла. Реакционный
сосуд охлаждают. Металл постепенно превращается в жидкое вещество
медно-красного (Rb2O) или коричнево-черного (CS2O) цвета, которое
затем затвердевает. После завершения реакции при 180—200 °C отгоня-
ют остаток металла.
Пероксиды. Пероксиды рубидия и цезия (РЬгОг и CS2O2) — бледно-
желтые кристаллические очень гигроскопичные вещества, устойчивые
в сухом воздухе. Это — ионные кристаллы ромбической сингонии, в
которых каждый пероксидный ион окружен восемью катионами. Они
являются производными пероксида водорода. РЬгОг и CS2O2 плавятся
инконгруэнтно при 570 и 594 °C соответственно. Ледяная вода (0 °C)
Растворяет М2О2 без выделения кислорода, но выше 25 °C протекает
Реакция
2М2О2 + 2Н2О = 4МОН+О2. (2.3)
Пероксиды рубидия и цезия взаимодействуют с кислотами с выделе-
нием пероксида водорода
M2O2 + H2SO4 = M2SO4 + H2O2. (2.4)
Они очень агрессивны в расплавленном состоянии — взаимодейст-
вУют со стеклом, никелем, серебром, платиной и даже с золотом вслед-
ствие выделения атомарного кислорода. Устойчивым к действию рас-
Плава пероксидов является алюминий. При нагревании пероксиды ру-
71
бидия и цезия диссоциируют. Давление диссоциации ИЬгОг при
~ 1000 “Ссоставляет 10,2 кПа, CS2O2—17,3 кПа. Существует несколько
способов получения пероксидов рубидия и цезия. По одному из нщ
надпероксид МОг обрабатывают при 0 °C тетрахлоридом углерода, со-
держащим диоксид хлора СЮг, до белой окраски реакционной смеси I
2МО2+2СЮ2=М2О2+С12+ЗО2. (2.5)
По окончании реакции М2О2 отфильтровывают и промывают ох-
лажденным тетрахлоридом углерода для удаления избытка диоксида
хлора. I
Надпероксиды. Надпероксиды рубидия и цезия, RbO2 и CsO2 —
желтые парамагнитные вещества тетрагональной сингонии, плот-
ность их соответственно 3,06 и 3,80 г/см3, температуры плавления 412
и 432 °C. При нагревании выше 180—200 °C надпероксиды обратимо
меняют цвет на оранжевый, начинают разлагаться ниже температуры
кипения. В кристаллах надпероксидов установлено существование иона
Ог , имеющего один неспаренный электрон, который участвует в обра-
зовании химической связи М+— Ог. Сильные окислители, способные
реагировать со взрывом. Легко окисляют многие органические вещества
Надпероксиды являются для рубидия и цезия наиболее характерными
кислородными соединениями. Получить их можно несколькими спосо-
бами, наиболее простой — сжигание чистых металлов в избытке очи-
щенного кислорода.
Озониды. Озониды — мелкокристаллические вещества оранжевого
(ИЬОз) или оранжево-красного (СяОз) цвета. Устойчивость озонидД
возрастает от натрия к цезию. Озонид рубидия неустойчив при комнат1
ной температуре, озонид цезия устойчив при 17—19 °C несколько дней.
СбОз разлагается при 70—100 °C, образуя пероксид и кислород. С водой
реагируют бурно
4CsO3+2H2O=4CsOH+5O2. (2.6)
Озониды рубидия и цезия — очень сильные окислители. Д ля получи
ния озонидов порошок безводных гидроксидов обрабатывают сначала
озонированным кислородом (8—9 % Оз) при -30 °C, а затем жидким
аммиаком
2МОН + 5Оз“2МОз + Н2О + 5О2. (2.7)
Выделяющаяся вода связывается с непрореагировавшим гидрокси*
дом. Продукт после испарения аммиака содержит 56—67 % МОз и
29—41 % МОН.
Гидроксиды. Гидроксиды RbOH и CsOH — бесцветные кристалл»
ческие вещества, имеющие кубическую гранецентрированную решетку
типа NaCl (см. табл. 1.4). С водой образуют серии кристаллогидратов
различной устойчивости. Для рубидия известно пять гидратов, содержа'
щих одну, две, три, четыре и пять молекул воды. Моногидрат
72
RbOH-НгО плавится конгруэнтно при 145 °C, дигидрат RbOH 2112О
плавится при 47 °C с образованием моногидрата. У цезия наиболее
устойчивым среди известных трех кристаллогидратов, содержащих три,
две и одну молекулы воды, является также моногидрат CsOH Н20,
температура плавления которого 226 °C. Дигидрат гидроксида цезия,
CsOH 2НгО, претерпевает при 2,5 °C полиморфное превращение в р-
CsOH 2H2O, который при 10 °C теряет молекулу воды и переходит в
моногидрат. Гидроксиды обоих элементов растворяются в воде, их рас-
творимость выше растворимости гидроксида калия, но ниже раствори-
мости гидроксида натрия. Температурный коэффициент растворимости
отрицательный. При 15 °C растворимость RbOH и CsOH 64,17 и 79,41
%, а при 30 °C — 63,39 и 75,18 соответственно. Хорошо растворяются в
этаноле и заметно в жидком аммиака: при -40 °C растворяется 0,9 г
RbOH и 1,0 г CsOH в 100 мл NH3.
Гидроксиды рубидия и цезия — сильнейшие основания, очень актив-
ные в химическом отношении вещества. На воздухе расплываются и
карбонизуются, образуя М2СО3. При 400—500 °C взаимодействуют с
кислородом, образуя М2О2, и с СО с образованием формиатов и оксала-
тов. Гидроксиды рубидия и цезия и их концентрированные растворы при
обычной температуре разрушают стекло; расплавы разрушают железо,
кобальт, никель, платину, корунд и диоксид циркония, постепенно рас-
творяют даже серебро и золото. Устойчив против действия МОН родий
и его сплавы. Гидроксид рубидия образует непрерывный ряд твердых
растворов с гидроксидом калия и ограниченные твердые растворы с
карбонатом рубидия.
BcncTeMeRbOH—NaOH наблюдается расслаивание расплава [8,8—
37,6 % (ат.) RbOH ] и образование соединения RbOH 2NaOH, что яв-
ляется следствием существенного различия параметров кристалличес-
ких решеток. В вакууме при -21 °C водные растворы гидроксидов руби-
дия и цезия взаимодействуют с пероксидом водорода с образованием
молекулярных соединений — пероксигидратов Rb2O2 4,5НгО2 0,7НгО
иСягОг 4,15НгО2 1,90НгО. Пероксигидраты рубидия и цезия распада-
ется при 50—55 °C с образованием гидроксидов и выделением кислоро-
да, в вакууме же распадаются с образованием надпероксидов
М2О2 4Н20г = 2МОг + 4Н2О + Ог. (2.8)
Для получения гидроксидов рубидия и цезия обычно используют
обменные реакции в водном растворе сульфатов, карбонатов, квасцов с
ГиДроксидом бария
M2SO4 + Ba(OH)2=2MOH + BaSO4. (2.9)
Для получения гидроксидов высокой чистоты используют электро-
лчтический метод с применением жидкого ртутного катода. Электроли-
т°м являются концентрированные растворы М2СО3, к которым
73
постепенно добавляют твердые М2СО3. Полученные амальгамы разла-
гают водой. Этот способ позволяет получить 6,5 н. растворы МОЦ
содержащие не более 0,01 % М2СО3. Из раствора кристаллизуются
гидроксиды упариванием под вакуумом.
2.3. Соли кислородсодержащих кислот
Сульфаты рубидия и цезия. Сульфаты рубидия и цезия — бесцвет-
ные кристаллические вещества, изоструктурные друг другу и своему
ближайшему аналогу — сульфату калия. Сульфат рубидия имеет две
модификации: при обычной температуре устойчива ромбическая, кото-
рая при 650 “С обратимо переходит в гексагональную. Сульфат цезия,
вероятно, триморфный с температурами перехода 660 и 712 °C. Плот-
ность Rb2SO4 и CS2SO4 при 20 °C 3,615 и 4,246 г/см3 соответственно.
Сульфаты рубидия и цезия — термически устойчивые соединения, их
температуры плавления 1074 и 1019 °C. Заметная летучесть без из-
менения состава наблюдается выше 1400 °C. При прокаливании (700—
800 °C) в токе водорода или аммиака восстанавливаются до сульфи-
дов, M2S. Сульфаты рубидия и цезия хорошо растворяются в воде,
температурный коэффициент растворимости положительный, кристал-
логидратов не образуют. Наблюдается значительное увеличение рас-
творимости в ряду сульфатов калия, рубидия и цезия (табл. 2.1).
Таблица 2.1. Растворимость в воде солей кислородсодержащих кислот
калия, рубидия, цезия, г/100 г Н2О
Соеди- некие Температура, "C
0 20 25 40 50 60 80 100 I
K2SO4 7,3 11,2 — 14,8 — 18,2 21,3 24,1
R62SO4 36,4 48,2 — 58,5 — 67,4 75,0 81,8
CS2SO4 167,1 178,7 — 189,9 — 199,9 210,3 220,3
KA1(SO4)2 3,0 6,0 — 13,6 — 25,5 70,6 154,0
RbAl(SO4>2 0,7 1,5 — 3,3 — 7,5 21,6 140,9
CsA1(SO4>2 0,2 0,5 — 0,9 — 2,0 5,5 22,9
KNO3 13,9 — 38,0 61,3 — 106,2 166,6 245,0
RbNO3 19,5 — 69,0 116,7 — 200,0 309,0 452,0
CsNO3 9,3 — 28,8 47,2 — 83,8 134,0 197,0
KH2PO4 14,8 — 25,1 33,6 40,8 50,2 70,4 —
RbH2PO4 43,3 — 78,9 103,7 123,8 137,2 162,9 —
CSH2PO4 105,9 — 147,5 169,5 185,3 199,8 259,0 —
K2CO3 106,3 118,8 — — 121,4 — — —
Rb2CO3 234,7 249,3 — — 301,1 — — —
CS2CO3 273,0 308,3 — — 347,0 — — —
74
Сульфаты рубидия и цезия образуют с сульфатом лития несколько
соединений; в системе Rb2SC>4—№286)4 существует непрерывный ряд
твердых растворов, а система CS2SO4—Na?SO4 характеризуется образо-
ванием соединения CS2SO4 №286)4 и наличием ограниченных твердых
растворов на основе CS2SO4. С сульфатами металлов в степени окисле-
ния +2 и +3 образуют многочисленные двойные соединения, из которых
лаибольшее значение имеют квасцы. Для получения сульфатов исполь-
зуют реакцию нейтрализации 50 %-ной серной кислоты 30—40 %-ным
водным раствором карбоната или гидроксида рубидия или цезия (до pH
8—9) с последующим упариванием.
Для рубидия и цезия известны гидросульфаты MHSO4, дисульфаты
M2S2O7, пероксосульфаты M2S2O8, а также тиосульфаты.
Квасцы. Квасцы — это двойные сульфаты с общей формулой
MMnI(SO4)2 12Н2О, где М — Rb или Cs; МШ — Al, Ga, In, V, Мп, Fe,
Со. Они представляют большой интерес для технологических процессов
концентрирования, первичного разделения этих элементов и отделения
от калия. Алюморубидиевые и алюмоцезиевые квасцы — прозрачные
кристаллы, имеющие кубическую гранецентрированную решетку типа
NaCl. При нагревании квасцы сначала плавятся при 109 и 122 °C в
кристаллизационной воде, а потом постепенно обезвоживаются. Полное
обезвоживание RbAl(SO4>2 12НгО происходит при 225—230 °C, а раз-
Рис. 2.1. Политермы растворимости алю-
моквасцов рубидия, цезия, калия и аммо-
ния в воде
ложение с выделением серного ан-
гидрида — при 710 °C: соединение
цезия обезвоживается при 235, а
разлагается при 780 °C. Раствори-
мость квасцов рубидия и цезия в
воде незначительна и уменьшает-
ся от калия к цезию, причем алю-
моцезиевые квасцы имеют
наименьшую растворимость среди
известных квасцов (табл. 2.1, рис.
2.1). Твердые растворы квасцы об-
разуют только в тех случаях,когда
соответствующие им простые
сульфаты также образуют твер-
дые растворы. Алюморубидиевые
и алюмоцезиевые квасцы образу-
йся при взаимодействии водных
слабокислых растворов сульфатов рубидия и цезия с сульфатом
алюминия. Для перевода квасцов в простые соединения рубидия и цезия
^пользуется взаимодействие их с гидроксидом или карбонатом бария
Реакции
mA1(SO4)2 12H2O+2Ba(OH)2=MOH+Al(OH)3+2BaSO4+12H2O. (2.10)
75
Нитраты. Нитраты рубидия и цезия — бесцветные кристалличе-
ские вещества гексагональной сингонии при обычной температуре. При
164 °C RbNOa и 154 °C CsNOa переходят в кубическую модификацию,
Для RbNOa известны еще две модификации с температурами переходу
219 и 291 °C. Термическое разложение нитратов с выделением кислоро-
да происходит выше их температур плавления и начинается у нитрата
рубидия при 430, а у нитрата цезия — при 490 "С
2MNO3=2MNO2+O2. (2.11)
Выше 700 °C происходит одновременное выделение азота и кислоро-
да
2MNO3 = M2O + N2 + 5/202- (2.12)
В вакууме (— 5,1 Па) нитраты возгоняются без разложения; в связи
с этим вакуумная дистилляция может быть использована для выращи-
вания монокристаллов из газовой фазы. Расплавленные нитраты -I
сильные окислители, они разрушают кварц, платину и многие металлы
Очень хорошо растворяются в воде, температурный коэффициент рас-
творимости положительный (см. табл. 2.1).
Нитраты рубидия и цезия хорошо растворяются в азотной кислоте с
образованием сольватов MNO3 HNO3 и MNO3 2HNO3- Растворимость
в азотной кислоте не подчиняется периодичности свойств щелочных
металлов — она возрастает в ряду NaNOs < RbNCh < CsNO3 < KNO3, что
объясняется комплексообразованием в растворе. Нитраты рубидия
цезия получают нейтрализацией М2СО3 иди МОН разбавленной азот-
ной кислотой с последующим упариванием раствора и нагреванием су-
хого остатка до плавления.
Фосфаты. Рубидий и цезий образуют ряд фосфатов, в том числе
цепных, кольцевых и разветвленных полимеров на основе тетраэдров
[РО4 ]- Трехзамещенные фосфаты рубидия и цезия М3РО4 выделяются
из водных растворов в виде гидратированных кристаллов М3РО4 4HzO,
которые хорошо растворяются в воде; при этом реакция раствора щелоч-
ная. Соединения чрезвычайно гигроскопичны и быстро расплываются во
влажном воздухе. Получают трехзамещенные фосфаты при нейтрали-
зации водных растворов карбонатов рубидия и цезия с концентра-
цией 3 моль/л одномолярной фосфорной кислотой при 70 °C С
последующим упариванием до сиропообразного состояния и выдержкой
длительное время в эксикаторе над серной кислотой.
Дигидрофосфаты рубидия и цезия МН2РО4 — термически неустой-
чивые соединения, при нагревании теряют часть конституционной вод#
(250—350 °C) и превращаются в метафосфаты МРО4 и дигидродифос-
фаты М2Н2Р2О7, а при дальнейшем нагревании — в дифосфаты М4Р2О7-
Дигидрофосфаты очень хорошо растворяются в воде; растворы имеют
кислую реакцию (pH 4—5) (см. табл. 2.1).
76
Дигидрофосфаты рубидия и цезия получают нейтрализацией фос-
форной кислоты карбонатами этих металлов и упариванием раствора до
начала кристаллизации. Дигидрофосфаты рубидия и цезия (тетраго-
нальная сингония) обладают пьезо- и сегнетоэлектрическими свойства-
ми. В точке Кюри их диэлектрическая проницаемость возрастает до
аномально высокого значения. По механическим и пьезоэлектрическим
свойствам они находятся между кварцем и тартратом калия—натрия.
По сравнению с кварцем их пьезоэффект в семь раз больше; в отличие
от тартратов они более устойчивы по отношению к влаге.
Карбонаты. Карбонаты КЬгСОз и СзгСОз — белые кристалличе-
ские вещества, в атмосфере диоксида углерода плавятся без разложения
соответственно при 873 и 792 °C. При 900 °C карбонат рубидия начинает
заметно диссоциировать, для карбоната цезия диссоциация наступает
при 800 °C. Давление диссоциации при 1000 °C равно 183 Па (КЬгСОз)
и 448,8 Па (СзгСОз). В расплавленном состоянии разрушают кварц,
стекло и многие керамические материалы. Карбонаты рубидия и цезия
очень гигроскопичные вещества и отличаются высокой растворимостью
в воде, которая увеличивается с повышением температуры (см. табл.
2.1). Водные растворы карбонатов имеют щелочную реакцию вследст-
вие гидролиза, максимальная степень (15 %) которого достигается при
концентрации 0,009—0,07 моль/л.
У карбоната рубидия существуют четыре кристаллогидрата с 9; 8;
1,5 и 0,5 молекулами воды; при комнатной температуре устойчив кри-
сталлогидрат ДЬгСОз 1,5Н2О, который при 162 °C переходит в полугид-
рат, а выше 190 °C полностью обезвоживается. Для карбоната цезия
описаны октагидрат CS2CO3 8Н2О, который при -15,6 °C переходит в
тригидрат, а при 84 °C — в CS2CO3 1,5НгО. Полная дегидратация про-
исходит при 150 °C. В отличие от карбонатов рубидия и калия карбонат
цезия растворяется в спирте. Карбонаты рубидия и цезия обычно полу-
чают прокаливанием гидрооксалатов или по реакции между M2SO4 и
Ва(ОН)2 с последующей карбонизацией образующихся МОН. Карбона-
ты являются исходными соединениями для получения многочисленных
солей кислородсодержащих кислот. При пропускании диоксида углеро-
да в раствор карбонатов образуются гидрокарбонаты рубидия и цезия
МНСОз. Эти соединения менее устойчивы и менее растворимы, чем
М2СО3. При нагревании выше 170—180 °C они разлагаются, выделяя
СО2.
Соли органических кислот. Особенностью органических производ-
ных рубидия и цезия является значительная растворимость их средних
солей многих органических кислот при малой растворимости соответст-
вующих им кислых солей. В связи с этим кислые соли органических
кислот (гидрооксалаты, гидротартраты) используются для выделения
Рубидия и цезия из растворов. Достоинство этих соединений заключа-
77
ется в том, что от них достаточно легко перейти л карбонатам рубидия
и цезия в результате термического разложения.
Гидрооксалаты МНз(СгО4)2 — бесцветные кристаллические веще-
ства, из раствора выделяются в виде дигидратов. На воздухе гидроокса-
латы устойчивы, при нагревании до 100 °C начинают терять воду, а при
180—195 °C полностью обезвоживаются. Гидрооксалаты переходят в
нормальные оксалаты в интервале 240—245 °C, выше 450 °C образуются
карбонаты М2СО3
2 [МНз (С2О4) 2 2Н2О ]=М2СО3 + 4СО+ЗСОг + 7Н2О. (2.12)
На практике гидрооксалаты прокаливают при 600—650 °C. Раство-
римость гидрооксалатов калия, рубидия и цезия при 21 °C соответствен-
но равна 2,46; 3,03 и 4,34 г/100 г воды. Гидрооксалаты часто используют
как промежуточный продукт в процессах очистки различных соедине-
ний рубидия и цезия. Для их осаждения в нагретый раствор соли рубидия
или цезия добавляют твердую щавелевую кислоту из расчета, чтобы
после выделения осадка раствор был насыщен ею.
Гидротартраты рубидия и цезия МНС4Н4О6 — наименее раствори-
мые соли винной кислоты; растворимость гидротартратов калия, руби-
дия и цезия при 25 °C 0,645; 1,18 и 9,66 г/100 г воды соответственно. В
то же время растворимость среднего тартрата рубидия в этих же услови-
ях ~ 66 %. Соединения устойчивы только до 100 °C, выше 500 °C
разлагаются с образованием соответствующих карбонатов
2МНС4Н4О6=М2СО3+5С+2СОг+5Н2О. (2.13)
Монохроматы. Монохроматы рубидия и цезия М2С1Ю4 — термиче-
ски устойчивые вещества, их температуры плавления 994 и 982 °C
соответственно. При 705 °C Rb2CrO4 и 770 °C СзгСгО4 претерпевают
полиморфные превращения. Растворимость их мало меняется с темпе-
ратурой и возрастает от калия к цезию. Получают монохроматы на
основе реакции между нагретым 25 %-ным водным раствором хромово-
го ангидрида и 30 %-ними растворами МОН.
Дихроматы. Дихроматы рубидия и цезия М2СГ2О7 — оранжево-
красные кристаллические вещества; дихромат рубидия диморфен с тем-
пературой перехода 63 °C. Переход полиморфных форм R ЬгСггО? опре-
деляется не только температурой, но и pH растворов: из кислых раство-
ров выделяется преимущественно триклинная модификация, а из сла-
бощелочных — моноклинная. Температура плавления 390 °C. У ди-
хромата цезия полиморфный переход отмечен при 352 °C. Дихроматы
получают по реакции между солями рубидия и цезия и дихроматом
натрия в растворе
M2SO4 + ЫагСггО? = М2СГ2О7 + Na2SO4. (2.14)
Хроматы и дихроматы рубидия и цезия используются для получения
металлов.
78
2.4. Галогениды
Галогениды рубидия и цезия, прежде всего хлориды, имеют большое
значение в технологии этих элементов и являются наиболее изученны-
ми соединениями, для них характерна ОЦК решетка (тип CsCl). Проч-
ность галогенидов рубидия и цезия может быть оценена энтальпией их
образования в сопоставлении с данными для галогенидов калия (табл.
2.2). Как видно из таблицы, для одного и того же элемента энтальпии
образования галогенидов уменьшаются с повышением порядкового но-
мера галогена; энтальпия образования хлоридов, бромидов и иодидов
повышается с увеличением порядкового номера щелочного элемента (у
фторидов зависимость обратная). Галогениды рубидия и цезия характе-
ризуются высокими температурами плавления и кипения и незначи-
тельной летучестью, их термические свойства близки к свойствам соот-
ветствующих соединений калия.
Общий способ получения MX (X — Cl, Вг, I) — нейтрализация
М2СО3 соответствующей галогеноводородной кислотой.
Таблица 2.2. Физико-химические свойства галогенидов рубидия и цезия
Соединение 3 d. г/см (при 20 °C) Тил. "С /кип, “С A//298(f)> кДж/моль
KF 2,48 865 1502 563
RbF 2,88 790 1408 549
CsF 3,59 694 1251 531
КС1 1,99 790 1407 436
RbCl 2,80 719 1381 431
CsCl 3,98 645 1302 433
KBr 2,75 734 1382 392
RbBr 3,35 690 1352 389
CsBr 4,43 636 1300 410
KI 3,12 681 1324 328
Rbl 3,55 655 1304 328
CsI 4,51 634 1280 337
Фториды. Фториды рубидия и цезия — соединения, образованные
самыми электроположительными и электроотрицательными элемента-
ми, поэтому они сильно полярны и в них преобладают ионные связи,
ермически это самые устойчивые соединения среди галогенидов руби-
дия и цезия (см. табл. 2.2). Заметное испарение их наблюдается только
выше 800—900 °C, в парообразном состоянии молекулы частично диме-
ризованы. Фториды рубидия и цезия очень гигроскопичны и очень хо-
79
рошо растворимы в воде; из водных растворов выделяются в виде к]
сталлогидратов различного состава. При 25 °C в 100 г воды растворяе
289,8 г RbF и 530 г CsF. При 50 °C растворимость повышается до 573 и
608 г соответственно. Фториды рубидия и цезия растворяются в метано-
ле, гидразине, трифториде брома, не растворяются в эфире, ацетоне,
нитробензоле. Водные растворы фторидов имеют щелочную реакцию
вследствие образования гидрофторида
2MF + H2O^ MHF2 + MOH.
Гидрофториды с различным числом молекул HF образуются при ее
избытке. Они представляют собой бесцветные кристаллические вещест-
ва с невысокими температурами плавления. Соединения, содержащие
более двух молекул фторида водорода, легко расплываются и разлага-
ются на воздухе. Получают MF по реакции между карбонатами и избыт-
ком плавиковой кислоты с последующим упариванием раствора досуха
и прокаливанием образующихся гидрофторидов при 300—600 °C до
постоянной массы.
Хлориды. Хлориды рубидия и цезия — термически устойчивые сое-|
динения, плавятся без разложения с незначительным улетучиванием,
которое наступает немного ниже их температур плавления (см. табл.
2.2). Выше 454 °C хлорид цезия обратимо переходит из ОЦК в модифи-j
кацию ГЦК, которая образует твердые растворы с КС1 и RbCl. Хлориды
рубидия и цезия выделяются из водных растворов в виде негигроскопич-
ных безводных кристаллов, хорошо растворяющихся в воде; темпера-
турный коэффициент растворимости положительный (табл. 2.3. рис.
2.2). Значительное увеличение растворимости RbCl и CsCl с температу-
рой в отличие от NaCl благоприятствует отделению натрия от хлоридов
рубидия и цезия. В соляной кислоте растворимость хлоридов рубидия и
цезия понижается с увеличением концентрации хлорида водорода. MCI
хорошо растворяются в муравьиной кислоте и практически нераствори-
мы в пиридине, ацетоне, хлорбензоле.
Хлориды рубидия и цезия образуют с хлоридом натрия системы
эвтектического типа, с хлоридом калия и между собой — твердые рас-
творы, а с хлоридом лития — соединения типа иМСГ mLiCl pH2O.
Хлориды рубидия и цезия образуют многочисленные двойные сое-
динения с хлоридами многих элементов. Их них наибольший интерес
представляет карналлит. Карналлит Rb(Cs)Cl MgClz 6Н2О — соедине-
ние, в форме которого рубидий и цезий как изоморфные примеси нахо-
дятся в природе в калиевом минерале карналлите. Этот минерал имеет
большое значение для получения магния и солей калия. Рубидиевый и
цезиевый карналлиты — малорастворимые соединения, причем цезие-
вый карналлит более растворим, чем рубидиевый. В отличие от природ-
ного калиевого карналлита, рубидиевый и цезиевый карналлиты раст-1
80
Таблица 2.3. Растворимость в воде галогенидов
калия, рубидия и цезия
Соеди- нение Растворимость, г/100 г Н2О, при температуре, °C
0 25 40 60 80
KCl 28,15 35,98 40,3 45,6 —
RbCl 76,16 94,41 103,5 115,6 —
CsCl 162,29 191,87 207,7 229,4 —
RbBr 89,6 114,1 132,0 155,0 —
CsBr 45,00 55,20 60,81 66,10 —
KI 127,6 148 160 176 192
Rb! 124,8 163 183 219 271,8
CsI 43,1 85,6 122,9 160 171 *’
2 При 50 °C. > При 75 °C. ***) В процентах
воряются в воде конгруэнтно. Эту особенность используют при промыш-
ленной переработке природного карналлита, который легко разлагается
водой с выделением КС1 и образованием обогащенного рубидием и це-
зием так называемого искусственного карналлита. Многократное разло-
Рис. 2.2. Политермы растворимости хлори-
дов натрия, калия, рубидия и цезия в воде
Рис. 2.3. Изотермы растворимости в систе-
мах MCI—MgC12—Н2О, где М — Cs, Rb, К
при 25 °C (инвариантные точки El, Е2, Р
Нанесены условно):
м 1 — CsCl—MgC12—Н2О; 2 — RbCl—
М8С12—Н2О; 3 — КС1—MgC12—Н2О
81
женис последнего позволяет получать продукты перекристаллизации
обогащенные рубидием и цезием. На рис. 2.3 приведена растворимость
калиевого, рубидиевого и цезиевого карналлитов в системе MCI—
MgCl—Н2О, где М — К, Rb, Cs.
Бромиды. Бромиды рубидия и цезия термически устойчивы до тем-
пературы 600 °C, заметная их летучесть наблюдается только выше т ем-
ператур плавления и составляет при 1100 °C 1018 и 1443,3 Па соот-
ветственно (см. табл. 2.2). При этом соединения частично разлагаются
с выделением брома и бромистоводородной кислоты. Бромиды рубидия
и цезия образуют с бромидом лития инконгруэнтно плавящиеся соеди-
нения , с бромидом натрия — системы эвтектического типа, а с бромидом
калия и между собой — твердые растворы. Бромиды рубидия и цезия -L
негигроскопичные соединения, кристаллогидратов не образуют, хорошо
растворимы в воде, температурный коэффициент положительный (см.
табл. 2.3). Хорошо растворяются в бромистоводородной кислоте, раство-
римость понижается с увеличением концентрации НВг. При одной и той
же концентрации НВг растворимость КВг наименьшая среди бромидов
близких по свойствам щелочных элементов. Бромиды рубидия и цезия
хорошо растворяются в муравьиной кислоте, умеренно растворимы в
метаноле и этаноле. Кристаллы бромидов рубидия и цезия обладаю!
оптической прозрачностью в широкой области спектра и используются
для изготовления призм для ИК-спектроскопии. Под действием рентге!
новских лучей и радиоактивного излучения монокристаллы бромидов
окрашиваются в голубовато-зеленый (RbBr) и синий (CsBr) цвета.
Йодиды. Среди галогенидов щелочных металлов иодиды рубидия и
цезия обладают при высоких температурах наибольшим давлением па-
ра. У CsI давление пара равно 10,5 Па при 527 °C и 23,4 Па при 627 °C.
Возгонка иодидов рубидия и цезия на воздухе сопровождается их час-
тичной диссоциацией с выделением элементарного иода. В химическом
поведении иодидов рубидия и цезия много общего с бромидами и хлори-
дами. Иодиды рубидия и цезия образуют с иодидом калия и между собой
непрерывный ряд твердых растворов, с иодидом натрия Rbl образует
ограниченные твердые растворы, a CsI систему эвтектического типа!
Иодиды рубидия и цезия отличаются от других галогенидов повышен-
ной способностью к окислению и к образованию продуктов присоединен
ния (например, MI 450г). Иодиды рубидия и цезия негигроскопичны,
не образуют кристаллогидратов и хорошо растворимы в воде (см. табл.
2.2, 2.3). На свету водные растворы иодидов рубидия и цезия постепенно
желтеют вследствие выделения свободного иода. Под действием бром-
ной воды, азотистой кислоты и других окислителей легко выделяется иоЛ
даже из разбавленных раствором ML Иодиды рубидия и цезия раство-j
ряются в метаноле, этаноле, гидразине, нерастворимы в нитробензоле
ацетонитриле, фурфуроле. Умеренно растворимы в Н1, причем раство-
82
пимость понижается в ряду Rbl—KI—CsI. Иодид цезия — хороший
оптический материал для инфракрасной спектроскопии, а также сцин-
тилляторов, качество которого возрастает при добавлении к нему солей
таллия.
2.5. Соли кислородсодержащих кислот галогенов
С галогенами (кроме фтора) рубидий и цезий дают соединения
МХОп, где п = 1 —4. С увеличением числа кислородных атомов в кислоте
увеличивается устойчивость солей и уменьшается их растворимость в
воде. Для соединений типа МХОз термическая устойчивость возрастает
от хлоратов к йодатам, а растворимость в воде уменьшается. Для солей
этого типа наименее растворимы в воде соединения рубидия; при этом
различие в растворимости уменьшается от хлоратов к йодатам. Эти
соединения, особенно хлораты и перхлораты, представляют интерес для
очистки соединений рубидия и цезия.
Хлораты. Хлораты рубидия и цезия МСЮз — кристаллические
вещества ромбической сингонии, изоструктурные с броматом калия. В
системах КСЮз—ДЬСЮз—НгО и КСЮз—СвСЮз—НгО при 25 °C изо-
морфизм между хлоратом калия и хлоратами рубидия и цезия отсутст-
вует. КЬСЮз и СвСЮз образуют непрерывный ряд твердых растворов.
Хлораты рубидия и цезия при обычной температуре устойчивы, при
нагревании выше температуры плавления разлагаются
2МС10з = МС1 + МС104+02; (2.15)
МСЮ4 = МС1 + 2О2. (2.16)
Быстрое нагревание хлоратов выше их температур плавления при-
водит к взрыву. Хлораты рубидия и цезия обладают высоким темпера-
турным коэффициентом растворимости (табл. 2.4).
Таблица 2.4. Растворимость галогенокислородных
солей калия, рубидия и цезия
Соединение Растворимость, г/100 г Н2О, при температуре, °C
0 20 30 40 50 60 80 100
КСЮз 3,3 7,4 10,5 — 19,3 — 38,5 57,0
ИЬСЮз 2,1 5,4 8,0 — 16,0 — 34,1 62,8
СьСЮз 2,5 6,2 9,5 — 19,4 — 45,0 79,0
KCIO4 0,76 1,67 —- 3,63 5,15 7,18 13,38 —
КЬС1О4 0,50 0,99 — 2,39 3,55 4,85 9,20 —
CSCIO4 0,80 1,60 — 4,0 5,40 7,30 14,40 —
КВгОз 3,1 — 9,5 13,2 — — — —
КЬВгОз — — 3,55 5,08 — — — —
L£sBrQ3 — — 4,53 — — — — —
83
Хлораты рубидия и цезия получают обменной реакцией межД
M2SO4 и хлоратом бария или нейтрализацией МгСОз(МОН) хлорнов»
той кислотой. Кристаллизацию хлората рубидия используют для очисъ
ки технической соли от примесей калия и цезия.
Перхлораты. Перхлораты рубидия и цезия МСЮ4 изоморфный
перхлоратами и перманганатами калия и аммония и с сульфатом бария,
основные их свойства приведены в табл. 2.5. Соединения устойчивы на
воздухе и в водных растворах. Разложение RbCICU начинается при
408 °C, при 473 °C разложение сопровождается взрывом и заканчиваетс!|
около 530 °C. CsC104 полностью теряет кислород при 677 °C.
Перхлораты рубидия и цезия незначительно растворяются в мета-
ноле, этаноле, пропаноле (при 25 °C в среднем 0,01—0,15 г/100 г рас-
творителя). Низкая растворимость CSCIO4 в воде и органических раст-
ворителях определила возможность использования этого соединения
для определения макроколичеств цезия. Для получения перхлорате»
можно использовать обменную реакцию между M2SO4 и перхлоратов
бария или нейтрализацию раствора МгСОзСМОН) хлорной кислотой|
другие способы.
Таблица 2.5. Физико-химические свойства перхлоратов
калия, рубидия и цезия
Свойство КС1О4 RbClO4 CsClO4
з Плотность при 16 “С, г/см 2,52 3,01 3,32
Температура плавления, "С 610 606 575
Энтальпия образования -A //298(f). кДж/моль 434 435 —
Броматы. Броматы рубидия и цезия МВгОз, — бесцветные кристал-
лические вещества гексагональной сингонии, они не изоструктур™ I
бромату калия, который имеет ромбическую решетку. Температуры I
плавления КЬВгОз и СзВгОз — 430 и 420 °C соответственно, плотность I
при 20 °C — 3,68 и 4,11 г/см3. Плавление соединений происходит! I
незначительным разложением, в результате которого выделяется кис- г
лород и образуются соответствующие бромиды. Под действием ионизи- I
рующего облучения броматы рубидия и цезия разлагаются на бромитЫ
и бромиды с выделением кислорода. Данные по растворимости броматов
калия, рубидия и цезия приведены в табл. 2.4.
Обычный способ получения броматов — нейтрализация бромнова-
той кислоты водными растворами М2СО3 или МОН. Перброматы рубй'
дия и цезия неизвестны.
84
Иодаты. Иодаты рубидия и цезия МЮз — бесцветные кристаллы
псевдокубической структуры типа перовскита. Иодаты рубидия и цезия
изоморфны только с йодатом калия, с броматами и хлоратами твердых
растворов не образуют. Иодаты рубидия и цезия — соединения более
устойчивые, чем броматы и хлораты. Они, так же как хлораты и брома-
ты, являются хорошими окислителями. Плотность ИЫОз и СяЮз при
14—16 °C равна 4,56 и 4,83 г/см3 соответственно. Иодаты рубидия и
цезия мало растворимы в воде. Растворимость при 25 °C составляет 2,41
и 2,6 г на 100 г воды. В отличие от хлоратов и броматов иодат рубидия
склонен к присоединению йодноватой кислоты с образованием гидроди-
иодатов RbICh НЮз или ИЫОз 2Н10з; иодат цезия образует соедине-
ния с йодноватым ангидридом, например 2СзЮз I2O5. Для получения
йодатов обычно используют обменную реакцию между M2SO4 и йодатом
бария или нейтрализацию НЮз растворами М2СО3 или МОН.
Перйодаты. Перйодаты рубидия и цезия MIO4 — бесцветные кри-
сталлы, имеющие различную структуру, что встречается редко у про-
стых солей рубидия и цезия. Перйодаты термически мало устойчивы,
при нагревании разлагаются, часто со взрывом. Растворимость в воде
соединений рубидия (г/100 г воды) 0,65 (13 °C), цезия 2,15 (15 °C).
Минимальной растворимостью (5,58 10-2 г/100 г воды) обладает пер-
йодат цезия в 20 %-ном этаноле при 0 °C и pH 3,5.
Перйодаты получают при пропускании хлора в нагретый до 70—
80 °C сильнощелочной раствор йодатов или нейтрализацией концентри-
рованного водного раствора H5IO6 водными растворами М2СО3 или
МОН.
2.6. Соединения рубидия и цезия с неметаллами
Гидриды. Гидриды рубидия и цезия МН представляют собой белые
блестящие вещества с кубической гранецентрированной решеткой типа
NaCl. Гидриды рубидия и цезия — солеобразные соединения, которые
содержат анион Н~. Существование структуры М+—Н объясняется
большей энергией ионизации атома водорода по сравнению с энергиями
ионизации рубидия и цезия и небольшим сродством к электрону у атома
водорода (табл. 2.6).
Таблица 2.6. Физико-химические свойства гидридов
калия, рубидия и цезия
Показатели КН RbH CsH
з Плотность при 20 "С, г/см 1,45 2,60 3,42
Энтальпия образования -A //298(f) .кДж/моль 61 54 56
85
При нагревании гидриды диссоциируют, образуя водород и щелоч-
ной металл. Давление диссоциации достигает атмосферного при 389 и
364 °C для RbH и CsH соответственно. По увеличению давления диссо-
циации гидриды образуют ряд КН < NaH < CsH < RbH, который не
совпадает с периодичностью свойств этих элементов. Гидриды рубидия
и цезия — чрезвычайно химически активные вещества; бурно разлага-
ют воду и этанол, образуя гидроксиды и алкоголяты, под действием
паров воды воздуха гидриды окисляются с воспламенением, воспламе-
няются они в атмосфере фтора и хлора, при этом образуются MF и MCI.
При нагревании с азотом или аммиаком образуют амиды, с фосфором —
фосфиды. Гидриды рубидия и цезия нерастворимы в органических рас-
творителях. Они обладают сильными восстановительными и каталити-
ческими свойствами. RbH и CsH образуют двойные гидриды, из которых
наиболее известны боргидриды МВЩ. Получают гидриды рубидия и
цезия гидрированием при 300—350 °C расплавленных металлов водоро-
дом под давлением 5—10 МПа или в присутствии катализаторов.
Соединения с азотом. Нитриды рубидия и цезия M3N — малоустой-
чивые серовато-зеленые очень гигроскопичные вещества, на воздухе
воспламеняются. При нагревании взрываются, выделяя азот, легко вза-
имодействуют с хлором, фосфором и серой. Водой разлагаются по реак-
ции
M3N + 4H2O=3MOH + NH3 Н2О. (2.17)
Нитриды рубидия и цезия можно получить в жидком азоте при
электрическом разряде между электродами, изготовленными из руби-
дия или цезия. Нитриды не характерны для рубидия и цезия.
Азиды рубидия и цезия MN3 — желтовато-белые кристаллические
вещества. Температуры плавления азидов в глубоком вакууме состав-
ляют 321 и 326 °C для соединений рубидия и цезия соответственно,
энтальпия образования 0,293 (RbN3) и 9,92 (CsN3) кДж/моль. При
ударе азиды рубидия и цезия не взрываются. Растворимость азидов в
воде значительна и составляет: для CsN3 224 и 307,4 г при 0 и 16 °C, для
RbN3 107,1 г при 16 °C в 100 г воды. Водные растворы азидов имеют
щелочную реакцию. Азиды нерастворимы в абсолютном эфире, в этано-
ле растворимость составляет при 16 °C 0,182 (RbN3) и 1,037 (CsNs) г в
100 г этанола. Нагревание азидов в высоком вакууме выше температур
плавления (396 °C для RbN 3 и 390 °C для CsN3) приводит к разложению
соединений на металл, который оседает в виде зеркала на стенках реак-
тора, и азот. Разложение азидов используется для получения чистых
металлических рубидия и цезия. Установлено, что азиды рубидия и
цезия обладают фотоэлектрическими свойствами. Для получения ази-
дов используется реакция между азидом бария и сульфатом соответст-
вующего щелочного металла. Для синтеза чистых азидов рубидия и
86
цезия используется взаимодействие амида щелочного металла с моно-
оксидами азота
MNH2+N2OMN3 + H2O. (2.18)
Амиды рубидия и цезия MNH2 — белые расплывающиеся на воздухе
кристаллические вещества с температурами плавления 309 °C (RbNH2)
и 262 °C (CsNH2); расплавленные амиды разрушают стекло и фарфор,
испаряются при 400 °C. С влагой воздуха амиды взаимодействуют с
воспламенением, выделяя аммиак и образуя гидроксиды
MNH2+H2OMOH + NH3. (2.19)
В присутствии кислорода амиды, растворенные в жидком аммиаке,
превращаются в смесь гидроксидов и нитритов
4MNH2 + ЗО2 = 2МОН + 2MNO2 + 2NH3. (2.20)
Общепринятый способ получения амидов — взаимодействие рас-
плавленного щелочного металла с сухим газообразным аммиаком
М + NH3 = MNH2 + 1/2Нг. (2.21)
Соединения с углеродом. С углеродом рубидий и цезий образуют
соединения двух типов: М2С2 — ацетилиды, которые можно рассматри-
вать как продукты замещения атомов водорода в ацетилене, и соедине-
ния графита с общей формулой СПМ.
Ацетилиды рубидия и цезия М2С2 — белые порошкообразные веще-
ства с неявно выраженной кристаллической структурой. В вакууме при
500—600 °C разлагаются на металл и аморфный углерод. Устойчивость
ацетилидов уменьшается в ряду К > Rb > Cs > Na > Li. Ацетилиды
рубидия и цезия разлагаются водой со взрывом; образующийся ацетилен
распадается, выделяя углерод. Их получают при взаимодействии ацети-
лена с рубидием и цезием и последующим нагреванием в вакууме при
300 °C образовавшегося однозамещенного ацетилида МНСг. Присутст-
вие примеси углерода часто придает продукту серый или черный цвет.
Соединения графита с рубидием и цезием СПМ образуются в зависи-
мости от температуры или в виде темных порошков CgRb и CsCs или в
виде соединений сине-стального цвета с металлическим блеском C24Rb
и C24CS. Соединения относятся к категории кластеров. Кластер состоит
из структурных элементов, представляющих собой кольца из шести
атомов углерода. Все они обладают разрыхленной решеткой графита, в
которой слои атомов щелочных металлов чередуются со слоями атомов
Углерода и характеризуются отсутствием термических эффектов плав-
ления и кристаллизации и обладают химическими свойствами как ще-
лочного металла, так и графита. На воздухе они самовоспламеняются,
Разлагаются водой с выделением водорода. Кластеры СбоМ, С70М,
CsoM, получившие общее название «фуллерены», обладают свойствами
втсп.
Соединения с кремнием. Моносилициды MSi — кристаллические
вещества темного (RbSi) или желтого (CsSi) цвета с кубической решет-
87
кой, содержащей 32 молекулы в элементарной ячейке. В глубоком ва-
кууме моносилициды разлагаются с образованием MSis (350—360 °С)|
Моносилициды самовоспламеняются на воздухе с образованием гидро-
ксида и SiO2 хНгО; при взаимодействии с водой воспламеняются со
взрывом. Для получения моносилицидов смесь порошкообразного крем-
ния и металлического Rb или Cs длительное время нагревают в атмос-
фере аргона при 600 °C с отгонкой избыточного металла в глубоком
вакууме при 150—180 °C.
Соединения с серой. Рубидий и цезий образуют с серой моносульфид
M2S и полисульфиды (ди-, три-, пентасульфиды). Все сульфиды гигро-
скопичны, хорошо растворимы в воде, из водных растворов выделяются
в виде кристаллогидратов. Для получения сульфидов используют взаи-
модействие металлических рубидия или цезия с сульфидом ртути с
последующей отгонкой избытка щелочного металла и ртути или реак-
цию с серой в жидком аммиаке.
Соединения с фосфором. Рубидий и цезий образуют с фосфором ряд
фосфидов, например М2Р5. Пентафосфиды рубидия и цезия плавятся'
около 650 °C, выделяя при этом фосфор; в среде водорода плавление и
разложение наступает около 300 °C с образованием черного фосфида!
неизвестного состава. С водой Rb2?5 и CS2P5 реагируют при -15 °C с
выделением РНз и Нг, растворяются в жидком аммиаке. Для полученияI
пентафосфидов рубидий и цезий нагревают с красным фосфором или его»
парами при 400—430 °C в реакторе, из которого предварительно удален*
воздух. Продукт реакции подвергают вакуумной дистилляции для уда-
ления непрореагировавших металла и фосфора. Можно также получить!
пентафосфиды при взаимодействии расплавленного фосфора с гидрида-
ми рубидия и цезия.
2.7. Рубидий и цезий в комплексных соединениях
Рубидий и цезий обладают слабой комплексообразующей способно-
стью. Это связано со стабильностью их электронных оболочек и большой!
поляризуемостью их атомов и ионов. Однако они широко представлены
в различных классах комплексных соединений, где всегда выполняют!
функцию внешнесферных катионов, причем влияние катионов рубидия
и цезия на процесс комплексообразования очень велико. Особенно это!
проявляется в таких классах комплексных соединений, как анионгало-
генааты.
Галогенметаллатные соединения. Галогениды щелочных метал-
лов входят в состав большого числа комплексных соединений, которые!
относятся к типу ацидосоединений, в комплексном анионе которых ли-1
гандами являются кислотные остатки исходных солей. Наиболее устой-1
чивыми среди щелочных металлов являются ацидогалоидные соедине- !
88
ция рубидия и цезия. Термичеркая устойчивость ацидогаллоидных сое-
динений обычно возрастает от лития к цезию, а растворимость умень-
шается; исключение составляют фториды, у которых растворимость
меняется в обратном направлении.
Гексахлорплатинаты Мг[РхС1б] — золотисто-желтые мелкокри-
сталлические вещества кубической сингонии, термически устойчивы,
начинают разлагаться выше 420 °C. Соединения характеризуются ма-
лой растворимостью в воде, которая понижается в ряду К -» Rb -» Cs. В
виде этих соединений рубидий и цезий впервые были выделены в чистом
виде. В настоящее время из-за высокой стоимости платины практиче-
ского значения не имеют. Образуются при действии платинохлористо-
водородной кислоты Hz [Р1С1б ] на растворы солей рубидия и цезия.
Гексахлоростаннаты Mz[SnC16] — бесцветные мелкокристалличе-
ские вещества кубической сингонии. При нагревании на воздухе частич-
но окисляются, а в интервале 410—650 °C разлагаются с выделением
тетрахлорида олова и хлора. Растворимость Mz[SnC16] заметно выше,
чем гексахлороплатинатов; при 20 °C она составляет, г/100 мл раствора:
CszISnCU] 4,4; Rbz[SnC16 ] 10,0; Kz[SnC16 ] 64,0. В то же время в смеси
спирта с соляной кислотой растворимость Csz [SnC16 ] и Rbz [SnCle ] рез-
ко падает и становится ничтожной. Для получения Mz[SnC16] исполь-
зуют реакции между MCI и SnCU в солянокислом растворе.
Гексахлороплюмбаты Мг[РЬС1б ] — желтые мелкокристаллические
вещества, устойчивые только в солянокислом растворе, содержащем не
менее 20 % НС1, мало растворимые в соляной кислоте и гидролизующи-
еся в водном растворе. При нагревании MzIPbCU] переходят в
Мг [РЬСЦ ] с выделением хлора. Гексахлороплюмбаты рубидия и цезия,
в отличие от калия, не разлагаются разбавленной соляной кислотой
и 96 %-ным этанолом; это позволяет осуществить предварительное
отделение рубидия и цезия от остальных щелочных элементов, в том
числе и калия.
Галогенокомплексы сурьмы и висмута Mn[AmXn+3m], где М — Cs,
Rb или К; А — Sb (III) или Bi (III); X — галоген. Отдельные представи-
тели этого класса соединений известны давно и были рекомендованы для
выделения цезия из растворов, содержащих другие щелочные элементы.
в системах MX—Sb(Bi)X3—НХ—HzO(25 °C), где М — К, Rb и Cs; X —
в> С1, Вт или I, установлено существование и синтезированы галогено-
комплексы калия, рубидия и цезия различных составов и определены их
Физико-химические характеристики. Растворимость соединений пони-
жается в ряду калий—рубидий—цезий (кроме фторидов), а термиче-
ская устойчивость и прочность комплексов повышается. Все соединения
Устойчивы только в растворах соответствующих галогеноводородных
кислот, в водных растворах гидролизуются, причем устойчивость к гид-
ролизу повышается в ряду калий—рубидий—цезий. При гидролизе об-
89
разуются фазы переменного состава — /пМХ и8Ь(ВОХз pSbOX, при-
чем фазы, содержащие цезий, растворяются меньше других.
Эннеахлородистибиат цезия Сзз [SbzCl 9 ] — бледно-желтое мелко-
кристаллическое вещество ромбической сингонии; плотность при 25 °C
3,45 г/см3, температура плавления 540 °C. На воздухе соединение ус-
тойчиво, разлагается с выделением SbCh при нагревании в вакууме до
450 °C. В воде соединение гидролизуется
Сзз [Sb2Cl9 ] + 2Н2О = 2SbOCl + 3CsCl + 4НС1. (2.22)
В растворе аммиака гидролиз протекает по уравнению
Cs3 [Sb2Cl9 ] + 6NH3 Н2О = Sb2O3 + 3CsCl + 6NH4CI + ЗН2О. (2.23)
В результате гидролитического разложения и отделения осадка*
SbOCl или 8Ь20з получают чистые растворы хлорида цезия. Раствори-
мость Сзз [Sb2C19 ] в соляной кислоте мала и незначительно меняется с
изменением концентрации кислоты. Соединение образуется при взаи-
модействии растворов CsCl и твердого SbCh или его солянокислого раст-
вора.
Эннеаиододивисмутат цезия Свз [Bi2l91 — ярко-красное кристалли-
ческое вещество гексагональной сингонии, негигроскопичное, устойчи-
вое на воздухе, разлагающееся водой. В кипящей и нагретой разбав-
ленной азотной кислоте разлагается, выделяя иод, а в концентрирован-
ной HNO3 переходит в йодаты цезия и висмута. В 0,5М растворе HI
гидролиз Cs3 [Bi2l9 ] не происходит. Растворимость Свз [Bi2l9 ] при 25 °C,
г на 100 г растворителя: 0,17 (0.1096М HI), 0,099 (0,472М HI), 0,0007
(абс. C2HsOH). Осаждение Cs3 [Bi2l9] используется в аналитической
химии для количественного определения цезия. Соединение образуется
при взаимодействии раствора иодида цезия с раствором иодида висмута’
в иодистоводородной кислоте.
Цианометаллатные соединения. Цианиды рубидия и цезия с соля-
ми некоторых металлов, чаще всего в степени окисления +2, образуют
металлатные комплексные соединения типа М4[Мп(СМ)б] и
M2[Mn(CN)4], где М — Rb, Cs; М1* — Zn, Ni, Со, Си, Fe(II). Практиче-
ское значение среди этой группы соединений имеют гексацианоферра-
ты. Гексацианоферраты рубидия и цезия M4[Fe(CN)6] ЗН2О выделя-
ются при нейтрализации концентрированного раствора H4[Fe(CN)6]
соответствующим карбонатом щелочного металла; соединения хорошо
растворимы в воде; при нагревании сначала удаляется гидратная вода
(100—200 °C), затем происходит разложение ферроцианидного аниона
по реакции
M4|Fe(CN)61 • 4MCN + Fe(CN)2 (2.24)
и далее происходит распад цианида железа (II) с выделением азота.
Разложение Rb4(Fe(CN)6] начинается около 635 °C; Cs4[Fe(CN)6] —
590 °C. Выше этих температур продукты распада цианида железа (II)
катализируют разложение цианидов рубидия и цезия, которое на воз-
90
духе заканчивается образованием карбонатов. Гексацианоферраты ще-
лочных металлов склонны к образованию смешанных соединений с ме-
таллами в степени окисления +2 и +3 типа M2Mu[Fe(CN)6,
M2Mn3[Fe(CN)6]2, M4Mn4[Fe(CN)6]3, MMin[Fe(CN)6]. Практиче-
ский интерес из-за их незначительной растворимости представ-
ля- ют смешанные гексацианоферраты рубидия и цезия с магнием и
кальцием. Растворимость в воде при 25 °C Mi2Mge[Fe(CN)6]7 12НгО
составляет 0,22, а аналогичной соли цезия 0,10г/л. Растворимость
M2Ca[Fe(CN)6] иНгО в тех же условиях: соединения рубидия 0,18;
цезия 0,038 г/л. Такая низкая растворимость может быть использована
как в технологических, так и в аналитических целях. Наименее раство-
римы среди этой группы соединений смешанные гексацианоферраты
рубидия и цезия с никелем. Для получения смешанных гексацианофер-
ратов используют взаимодействие гексацианоферратов магния или
щелочноземельных элементов с солями рубидия или цезия в растворе.
Нитрометаллатные соединения. Эта группа соединений чрезвы-
чайно многочисленна, что объясняется тем, что в нитритных комплек-
сах химическая связь центрального атома с лигандами — NO2”
осуществляется через атом азота. Это усиливает ковалентность связи и
ее прочность. Практическое значение имеют гексанитрокобальтаты ру-
бидия и цезия типа Мз [А(Ь40г>6 1, где М — Cs, Rb, Li, Na или Ag и A —
Co.
Гексанитрокобальтаты рубидия и цезия — желтые мелкокристалли-
ческие вещества, которые имеют переменный состав у соединения ру-
бидия — RbJCNa3-jr[Co(NO2)6] ОД, причем х зависит от концентрации
рубидия в исходном растворе и от pH осаждения и находится в интервале
1,92—2,94, и постоянный у соединения цезия Cs3[Co(NO2>6]. При на-
гревании соединение рубидия теряет воду и выделяет NO2. Остаток от
разложения (смесь RbNOs, NaNO3 и оксид кобальта) устойчив до
550 °C. Соединение цезия теряет воду при 110 °C, выше — разлагается
с выделением NO2, остаток представляет собой смесь CsNO3 и
оксида кобальта Растворимость при 17 °C (г/100 г НгО):
КЬхКаз-х[Со(МО2)б] од —5,0 Ю3, Cs3|Co(NO2)61 Н2О —4,97 10 .
Гексанитрокобальтаты рубидия и цезия получают при взаимодействии
Растворов MCI или M2SO4 с водным раствором Na3[Co(NO2)6]. Они
используются для осаждения рубидия и цезия вместе с калием с целью
отделения отлития, алюминия, железа, марганца и щелочноземельных
металлов.
Гетерополисоединения (ГПС) рубидия и цезия — соединения с
комплексными анионами сложного строения. Общая формула ГПС —
МХ(АгО7)б] од.гдеМ — NH4,K+,Rb ,Cs+;A — V,Mo,W;X — Si,P,
As, Bi и другие элементы. Строение многих ГПС еще не выяснено, состав
91
их зависит от условий осаждения. Более изучены ГПС рубидия и цезия
в которых комплексообразователями являются кремний и фосфор,И
лигандами — оксиды молибдена и вольфрама. Характерная особенное^
этих соединений — незначительная растворимость в воде и в разбавлен*
ных кислотах; они могут быть использованы для выделения рубидияL
цезия из разбавленных технологических растворов и для разделения
калия, рубидия и цезия.
Кремнемолибдаты МзН5[8ЦМо2О7)б]-<«7 — зеленовато-желтые
кристаллические вещества, производные кремнемолибденовой кислоты
Не [Si(Mo2O7>6 ], которая является реактивом на рубидий и цезий. Рас-
творимость М3Н5 [Si(Mo2O7>6 ] в воде мало изменяется с температурой
но уменьшается в растворах НС1. Растворимость при 25 °C, г / 100 г водJ
у соединения рубидия 0,48, цезия 0,123. Выше 40 °C соединения в
водных растворах разлагаются. Кремнемолибдаты рубидия и цезия по-
лучают обменной реакцией солей Rb и Cs с кремнемолибдатом натрия.
Разложение кремнемолибдатов рубидия и цезия протекает при 400-1
450 °C в токе нагретого хлористого водорода
M3H5[Si(Mo2O7)6]+27НС1=ЗМС1+12(МоОз 2HCl)+SiO2+4H2O. (2.25)1
Кремневольфраматы M8[Si(W2O7)6]a^ или 4M2O S1O2- 12WO3-
иНгО — белые кристаллические вещества, плохо растворимые в воде,
нерастворимые в спирте и разбавленной соляной кислоте. При 20 °C
растворимость рубидиевой и цезиевой солей соответственно 0,65 и 0,005
гв 100 г воды. Получают их при взаимодействии кремневольфрамовой
кислоты с хлоридами рубидия и цезия. Разлагают соединения или в ток!
НС1 (см. кремнемолибдаты), или при 18—25 °C гидроксидом бария или
кальция, взятыми в вид е суспензии с 20 % -ным избытком от стехио-мет-
рического количества
М8 [Si(W2O7)6 ]+13Ba(OH)2=8MOH+BaSiO3+12BaWO4+5H2O. (2.2(4
Осадок солей бария отфильтровывают, а в раствор пропускают СЯ
для образования карбоната.
Фосфоромолибдаты МзН4[Р(Мо2О?)б] а^ или ЗМгО-РгО5-4МоОз
— желтые кристаллические вещества, соли фосфоромолибденовой ким
лоты Н7 [Р (МогО?) 6 ]. Растворимость в воде незначительная и составляв
ет при 25 °C, г/100 гводы: соединения рубидия 6,2-10 4ицезия5,6-10 I
Получают при взаимодействии Н7[Р(МогО7)б]с разбавленными (1-г
2 %) растворами RbCl и CsCl.
Анионгалогенааты — комплексные соединения типа М [ ХпХ” Х^п I
где М — катион щелочного металла, X1, X11, X111 — О, Вг, I, которД
образуют комплексный однозарядный анион, п + т + к = 3; 5; 7 или
Анионгалогенааты, содержащие фтор, неизвестны. Молекула анионга
логенаата содержит легко поляризующийся внешнесферный катион
ион щелочного металла и легко деформируемый ион—комплексообрЗ' 92
92
з0Ватель. Характерная особенность таких соединений: в комплексном
аНионе и центральный атом, и лиганды — галогены. Образование ани-
Одгалогенаатов обусловлено взаимной поляризацией иона галогена и
присоединяемых им молекул галогенов. Наиболее изучены анионгало-
^нааты с тремя атомами галогена. Они образуют последовательный
ряд, в котором устойчивость соединений возрастает с увеличением атом-
ной массы составляющих галогенов. При этом соединения, содержащие
в качестве наиболее тяжелого галогена иод, образуют одну серию по
устойчивости и кристаллической структуре, а соединения, содержащие
бром, — другую. Соединения серии иода вообще более устойчивы, чем
серии брома. Анионгалогенааты рубидия и калия менее устойчивы, чем
соответствующие соединения цезия.
Дииодиодааты М[1(1г)] — кристаллические вещества красно-ко-
ричневого Rb [KI2) ] или сине-черного Cs [KI2) ] цвета, устойчивые на
воздухе при комнатной температуре. Температуры плавления — 194 и
214 °C для соединений рубидия и цезия соответственно; давление диссо-
циации (200 °C) — 1754 и 1285 Па. Важным в практическом отношении
является то, что растворимость соединений рубидия в 300 раз больше
растворимости соединений цезия. Получают М [I (1г) ] при действии на-
гретым раствором MI на иод с последующей кристаллизацией при ох-
лаждении.
Бромиодиодаат цезия Cs [I(IBr) 1 — единственное соединение ще-
лочных металлов такого типа, выделенное в свободном состоянии в виде
красно-коричневых кристаллов из водно-спиртовых растворов; в воде и
эфире подвергается гидролизу и сольволизу. Получают действием на-
гретого водно-спиртового раствора CsBr на элементарный иод с после-
дующей кристаллизацией соединения при охлаждении. Перевод сое-
динения в CsBr осуществляется прокаливанием Cs [I (1Вг) ] при 400 °C.
Хлорбромиодааты М [I (ВгС1) ] — кристаллические вещества грана-
тово-красного цвета, устойчивы при хранении в закрытых сосудах, на
воздухе разлагаются, теряя 1Вг и переходя в MCI. Такая же схема раз-
ложения наблюдается при нагревании соединений. Для получения
М[1(ВгС1) ] горячий концентрированный водный раствор MCI растворя-
ют в жидком бромиде иода (IBr) и выделяют кристаллы соединения при
последующем охлаждении.
ТЕХНОЛОГИЯ РУБИДИЯ И ЦЕЗИЯ
2.8. Важнейшие области применения рубидия
и цезия и их соединений
В течение длительного периода, прошедшего со времени открытия
Рубидия и цезия (1860 г.) до тридцатых годов XX столетия, рубидий,
93
цезий и их соединения почти не имели определенных областей прик,
нения и представляли преимущественно научный интерес. В настояцц
время рубидий, цезий и их соединения применяются в электрониц,
радио-, электро- и рентгенотехнике, химической промышленности,!)
тике и медицине. Близость свойств рубидия и цезия определяет их вза)
мозаменяемость во многих областях применения.
Наибольшее практическое значение имеют фотоэлектричесж
свойства рубидия и цезия, благодаря которым они применяются 1
изготовления фотоэлектрических приборов — фотоэлементов, фотоул
ножителей — в качестве светочувствительного слоя фотокатодов. Сред
всех металлов цезий обладает самой низкой работой выхода электрону
(см. табл. 1.1), следовательно, требуется минимальная затрата светово
энергии при получении электрического тока с помощью цезиевых фок
элементов. Последние обладают высокой чувствительностью и мало,
инерционностью и в этом значительно превосходят селеновые фотозйе
менты, ранее пользовавшиеся наибольшей известностью.
Особо чувствительные фотоэлементы обычно имеют смешанные®:
токатоды (серебряноцезиевые и сурьмяноцезиевые). Они восприняв»
ют излучение не только видимой части спектра, но также инфракрасно;
и ультрафиолетовой. Такие фотоэлементы применяются в современна
установках для автоматизации производственных процессов, регулирс
вания осветительных систем, ночной сигнализации. Фотоэлементы»
ляются также неотъемлемой принадлежностью звукового кино и теле
видения, используются в радиолокации, в аппаратах связи и устройст
вах, применяемых в технике безопасности. Рубидий также использует
ся в фотоэлементах, однако они уступают цезиевым по чувствительно
сти и диапазону действия. Наибольшее значение для этих целей имею
металлический цезий и его двойные сплавы с кальцием, стронцием ил
барием или тройные сплавы с барием и алюминием. Эти сплавы устой
чивее чистого металла, и их удобнее использовать.
Кристаллы иодида цезия, активированные таллием, обладают спс
собностью сцинтиллировать под действием падающего излучения, бла
годаря чему применяются в сцинтилляционных счетчиках, испольЗ)
емых для регистрации частиц в атомной технике, геологии, медицин*1
космических исследованиях. Чувствительность цезия к инфракрасна^
и ультрафиолетовым лучам используется в специальной оптике®
иодид и бромид цезия применяются в качестве оптических материя®
инфракрасной аппаратуры, например в приборах ночного видениа
прицелах и т.д.
В электротехнике рубидий и цезий применяются в произволе®
светящихся трубок, содержащих инертные газы при пониженном давЛе
нии. При их изготовлении рубидий и цезий вводят в состав люминесцС®
94
тцых веществ в виде метацирконатов MzZrCh или ортостаннатов
M4SnO4-
Цезий, рубидий и их соединения используются в качестве катализа-
торов и промоторов в органическом и неорганическом синтезе. Их ката-
литическую активность можно использовать в процессах получения
аммиака, серной кислоты, синтезе бутилового спирта, муравьиной кис-
лоты и в различных реакциях дегидрогенизации. Большое будущее в
катализе, вероятно, будет принадлежать рубидию и его соединениям.
Рубидиевые квасцы в отдельности или в смешанных катализаторах при-
меняют в процессах синтеза бутадиена из бутиловой фракции нефти;
промотируя катализатор (оксид железа), соли рубидия по сравнению с
солями калия вдвое увеличивают скорость реакции, повышают селек-
тивность и устойчивость катализатора. Такое же действие оказывает
ацетат рубидия в катализаторах для синтеза метанола и высших спиртов
из водяного газа.
Радиоактивные изотопы Cs-137 и Rb-86 широко используются в
гамма-дефектоскопии, измерительной технике, при стерилизации ле-
карств и пищевых продуктов.
В медицине препараты рубидия применяются в качестве снотвор-
ных, болеутоляющих средств и для лечения эпилепсии и сифилиса;
соединения цезия — для лечения язвенных заболеваний, шоков и диф-
терии. Радиоактивный изотоп Cs-137 применяется в радиотерапии для
лечения злокачественных опухолей.
Перспективной сферой применения цезия и рубидия являются на-
учные исследования в новой энергетике (магнитогидродинамические
генераторы, термоэмиссионные преобразователи энергии, ионные дви-
гатели для космических кораблей), где используется их самая низкая
Для всех металлов энергия ионизации (см. табл. 1.1). Благодаря высоким
теплофизическим свойствам цезий и особенно рубидий испытывают в
качестве теплоносителей и рабочей среды в высокотемпературных тур-
боэнергетических установках.
2.9. Сырьевые источники рубидия и цезия
В рассеянном состоянии рубидий достаточно широко распространен
в природе. Его содержание в земной коре (литосфере) составляет
,1 Ю 2 %, т.е. больше чем содержание Ag, Au, Hg, Sn, Pb, As, Sb, Bi.
Содержание цезия в литосфере 7 IO-4 %. Эта величина сопоставима
с Удержанием Мо и Th, но несколько выше содержания ртути.
Рубидий и цезий — типично литофильные элементы. В незначи-
тельных количествах рубидий и цезий обнаружены в многочисленных
Разцах горных пород — базальтах, гранитах, диабазах, габбро, нефе-
95
линах, полевых шпатах и глинистых сланцах. При этом рубидий и цезж
преимущественно концентрируются в кислых изверженных и осадЛ
ных породах.
В процессах выветривания горных пород и минералов рубидий ц
цезий вымываются и попадают в минеральные воды. Они обнаружении
минеральных источниках Германии, Бразилии, Японии (0,20—0,10
мг/л). В меньших количествах рубидий и цезий присутствуют в морской
воде. Из минеральных источников и морской воды рубидий и цезий
переходят в соляные рассолы и отложения, поэтому они содержатся в
поташе, селитре и, особенно, в сильвине и карналлите. Тесная геохими-
ческая связь рубидия и калия, основанная на близости кристалл ох им*
ческих характеристик их соединений, предопределяет постоянное на-
хождение рубидия в широко распространенных минералах калия, глав-
ным образом, в алюмосиликатах. Цезий не способен к такому четком}
и широкому изоморфизму с калием, как рубидий, ввиду заметного раз-
личия в их ионных радиусах, хотя является иногда заместителем калия
в изоморфном ряду его соединений, однако вхождение цезия в кристал-
лические решетки минералов калия ограничено.
К минералам, в которых содержание рубидия и цезия достигает
относительно высоких концентраций и которые можно считать минера-
KLii.sAli ,5 [S13AIO1 о ] [F,OH ]2
K(Mg, Fe2+)3[Si3A10io][F,OH ]2
(К, Rb, Na)2[A12Si60i61
(Li, Na) [Si2A10io]
ВезА1г[81бО18 ]
KLi Fe2+Al[Si3A10io][F,OH ]2
K[AlSi2O61
лами-концентраторами, относятся:
Лепидолит
Биотит
Амазонит
Петалит
Берилл
Циннвальдит
Лейцит
и др. Все они являются алюмосиликатами.
Рубидий собственных минералов не образует. Практическое значе-
ние для извлечения рубидия попутно с литием имеют лепидолит и цин-
нвальдит (см. гл. 1). В лепидолите содержание рубидия может достигать
3,7 % в расчете на оксид, обычно 0,6—0,8 %; в циннвальдите макси-
мальное содержание Rb2O не превышает 0,8—0,9 %. Важнейшее про
мышленное месторождение лепидолита находится в Юго-Западной Аф-
рике, циннвальдита — в Чехословакии. Практическое значение ДД*
попутного извлечения рубидия имеет также карналлит, несмотря М
невысокое содержание в нем рубидия. Карналлит — двойной хлорид
калия и магния КС1 MgCh 6Н2О, в котором рубидий и цезий присутШ
вуют как изоморфные заместители калия. Среднее содержания рубиДЧ*
в карналлите 0,015—0,040 %, считая на RbCl, цезия — на поряДО*
меньше. Карналлит перерабатывается в огромных количествах с цель*®
96
получения металлического магния и соединений калия. Крупнейшие
месторождения карналлита находятся в России (Соликамск) и Герма-
нии (Страссфурт) (табл. 2.7).
Таблица 2.7 Состав Соликамского карналлитового концентрата
Минералогический состав Химическая формула Содержание соединений, %
Карналлит КС1 MgC12 6Н2О 75,2
Галит NaCl 22,2
Ангидрит CaSO4 0,6
Полевой шпат — 1.1
Кальцит + магнезит СаСОЗ+ MgCO3 0,2
Сильвин КС1 0,3
В отличие от Соликамского, карналлит Страссфуртского месторож-
дения содержит 13 % кизерита MgSO4 НгО и значительно большее
количество галита (32,3 %). Содержание рубидия в карналлитовом
концентрате колеблется от 0,003 до 0,012 %; содержание брома — от
0,16 до0,20 %.
Практическое значение для извлечения цезия попутно с литием
имеют лепидолит и циннвальдит. В лепидолите среднее содержание
CszO 0,15—0,20 %; максимальное — до 1,5 %; в циннвальдите содер-
жание CszO не превышает 0,5 %.
Собственных минералов цезия известно два — авогадрит и поллу-
цит.
Авогадрит К, Cs[BF4] — борофторид калия, в котором калий час-
тично изоморфно замещен цезием. Химический состав непостоянен;
“держание CsBF4 может достигать 20 %, среднее же — не более 9,5 %.
Авогадрит — редкий минерал и промышленного значения не имеет.
Поллуцит (Cs, Na) [AlSizOe ] лНгО— водный алюмосиликат цезия.
Содержание оксида цезия находится в пределах 29,8—36,7 %, так как
в результате процессов выветривания происходит замещение Cs на
Na +Н2О (явление объемного изоморфизма). В поллуците в качестве
примеси присутствуют рубидий [0,1—0,7 % ИЬгО ] и таллий (I). Плот-
ность поллуцита 2,86—2,90 г/см3, твердость 6—7. В настоящее время
поллуцит — единственный промышленный минерал цезия. Месторож-
дения поллуцита известны в Юго-Западной Африке, США, Швеции,
Канаде. Поллуцит ассоциируется с петалитом, лепидолитом, амблиго-
нитом, бериллом, кварцем.
«
691
97
Сырьевая база рубидия исключительно благоприятна. Рубидий вме-
сте с литием, стронцием, редкоземельными элементами, цирконием
ванадием и ниобием входит в группу редких металлов, запасы которьц
способны обеспечить промышленные нужды на много лет даже с учетом
роста потребления. Выявленные мировые ресурсы рубидия (в расчете на
RbiO) составляют 770тыс.т (данные 1975 г. без учета СССР).
Цезий входит в группу элементов с ограниченными запасами вместе
с кадмием, бериллием, танталом и гафнием. Выявленные ресурсы (без
учета СССР, 1975 г.) — 180тыс.т.
Производство рубидия и его соединений по данным 1978 г. (без учета
СССР) составило < 10 т в год. По данным 1985 г. мировая потребность в
металлическом цезии и его соединениях составляла ~ 30000 т в год (без
учета потребности СССР).
Получение соединений рубидия и цезия
Несмотря на то что основное минеральное сырье рубидия и цезия —
достаточно реакционноспособные алюмосиликаты, получение соедине-
ний рубидия и цезия из него — трудная задача. Сложность технологии
определяется в основном природой и составом перерабатываемого
сырья. Сырье обычно комплексное, рубидий и цезий извлекаются по-
путно (это, разумеется, не относится к извлечению цезия из поллуци-
та). Так как оба элемента практически всегда сопутствуют друг другу,
необходимо решать задачу их разделения и отделения от близкого к ним
по свойствам и преобладающего в сырье калия.
У рубидия, который не имеет собственных минералов, рудная тех-
нология не существует. Технология его получения сводится к разделе;
нию близких по свойствам щелочных элементов (калия, рубидия и це-
зия) в процессе переработки промежуточных концентратов. Аналогия*
ную задачу приходится решать и при получении соединений цезия,
причем она осложняется тем, что содержание цезия в любом комплекс-4
ном сырье в десятки раз меньше рубидия.
Рудная технология соединений цезия базируется на переработке
поллуцита. В технологии поллуцита чаще всего имеют дело со штуфныМ»
минералом, который обогащают ручной рудоразборкой, в результате
чего получают концентрат поллуцита с содержанием CszO 10—30 %.
Помимо ручной рудоразборки для обогащения поллуцитовых руд при-
меняют флотацию, которую проводят в кислой среде; в качестве флото-
реагента используют аминоацетат кокосового масла. В пенный продукт
попадает пустая порода (кварц, полевой шпат, слюда), а поллуцит ос-
тается в хвостах (обратная флотация). Рубидий, содержащийся в слюде,
переходит в пенный продукт и на 80 % концентрируется в его первой
фракции.
Соединения цезия в настоящее время получают из лепидолита и
поллуцита; возможно извлечение цезия из карналлита, а также из це-
98
зийсодержащих бериллов, перерабатываемых ради получения соедине-
ний бериллия, и из некоторых слюд, содержащих наряду с цезием и
рубидий. Основной источник получения рубидия — лепидолит. Рубидий
извлекается также из карналлита и поллуцита.
2.10. Переработка природного минерального сырья
Переработка поллуцита. Все способы переработки поллуцита по
характеру стадии разложения минерала можно разделить на две группы
— кислотные методы и методы спекания и сплавления. Выбор того или
иного метода определяется рядом факторов: составом конечного продук-
та, его чистотой и экономическими факторами.
Кислотные методы. Эти методы наиболее многочисленны и
основаны на использовании фтористоводородной, соляной, бромистово-
дородной и серной кислот.
Наиболее распространенным в технологии переработки поллуцита
является солянокислотный способ. Сущность способа заключается в
обработке поллуцита соляной кислотой при нагревании; при этом про-
текает реакция
(Cs, Na) [AlSi2O6 ] nH2O + 4НС1 =
= (Cs, Na)Cl + А1С1з + 2SiOz «Н2 + 2H2O. (2.27)
По одному из вариантов метода концентрат, измельченный до
0,14—0,25 мм, обрабатывают при непрерывном перемешивании 6—12
моль/л НС1 в реакторе с конденсационной колонкой при 90—110 °C в
течение 8—30 ч. В процесс вводят двукратный избыток соляной кисло-
ты. При кипячении смеси отгоняются летучие хлориды германия,
мышьяка, селена, теллура и бора, которые в небольшом количестве
присутствуют в сырье. Для материала реактора рекомендуется исполь-
зовать фарфор, стекло, фторопласт-4, графитопласты. По окончании
реакции смесь охлаждают до 75 °C и фильтруют на вакуумном фтороп-
ластовом или керамическом фильтре. Из солянокислого раствора осаж-
дают цезий в виде эннеадистибиата Cs3[Sb2C19], для чего в раствор
вводят трихлорид сурьмы SbC13 в виде раствора или твердого продукта.
Оптимальные условия осаждения соединения: концентрация НО
б моль/л и остаточное после осаждения Cs3 [Sb2Cl<> ] содержание SbCh
20—25 г/л. Осадок отфильтровывают, промывают НО (6 моль/л) и
подвергают гидролитическому разложению (2.22, 2.23).
Недостатком гидролитической переработки Cs3 [Sb2C19 I на CsCl яв-
ляется необходимость работы с большими объемами растворов (5—10 л
на 1 моль CsCl), что вызывает необходимость упарки в последующем.
Был предложен метод вакуумного разложения Cs3 [Sb2C19 ] при 450 °C и
102—204 Па. Так как SbCh кипит при 220 °C, a CsCl только плавится
4*
99
при 646 °C, в условиях процесса SbCh отгоняют и конденсируют. Неко-
торое количество SbC13 (до 1,0—1,5 %) при этом остается в хлор Л
цезия и должно быть удалено с помощью сероводорода после растворе,
ния CsCl в воде. Метод до настоящего времени не получил промышлен-
ного воплощения из-за чрезвычайной агрессивности паров SbC13-
Разложение поллуцита соляной кислотой считается наиболее деше-
вым способом получения хлорида цезия. Он обеспечивает переход в
раствор 95—98 % цезия и позволяет получить CsCl чистотой до 99,9 %
после дополнительной очистки.
Для разложения поллуцита рекомендована также бромистоводород-
ная кислота, что связано со стремлением получить в качестве конечного
продукта бромид цезия, который применяется в специальной оптике.
Это достигается осаждением эннеабромдистибиата цезия Cs3 [Sb2Brg ],
растворимость которого немного ниже аналогичного хлорида, с последу-
ющим переводом его в CsBr.
Основные этапы технологической схемы переработки поллуцита с
помощью фтористоводородной кислоты заключаются в следующем.
Поллуцит предварительно высушивают при 130 °C, измельчают до
0,20—0,25 мм и замешивают с небольшим количеством воды для полу-
чения густой пульпы. Затем к пульпе добавляют порциями 50—60 %-
ную фтористоводородную кислоту из расчета 2,18—2,20 кг кислоты на
1 кг поллуцита. Смесь нагревают до кипения и выдерживают 1 ч для
удаления S1F4. Поллуцит взаимодействует с HF по реакции
(Cs, Na) [AlSizOb ] пНгО + 36HF =
= (Cs, Na)2[SiF6] + 2H3[AlF6] + 3H2[SiF6]+ (n+12)H2O. (2.28)
По окончании реакции к реакционной массе добавляют при нагре-
вании 96 %-ную H2SO4 (1,2 кг на 1 кг поллуцита) для разрушения
фторокомплексов, в результате чего удаляются HF и S1F4 и образуются
гидросульфаты цезия и алюминия. Избыток H2SO4 удаляют выпарива-
нием и обрабатывают сухой остаток кипящей водой (12,5 л на 1 W
поллуцита), при этом протекает реакция
CsHSO4 + A1H(SO4)2+ 12H2O = CsA1(SO4)2 I2H2O + H2SO4. (2.29)
Нерастворившийся остаток отфильтровывают, а фильтрат нейтра-
лизуют алюминиевой стружкой до слабокислой реакции, очищают от
примесей тяжелых металлов с помощью H2S и упаривают до начал2
кристаллизации алюмоцезиевых квасцов. Квасцы отфильтровывают И
перерабатывают на карбонат цезия с помощью карбоната бария. Извле-
чение цезия в карбонат — около 93 %. В оптимальном варианте, испоЛЬ'
зуя значительный избыток плавиковой кислоты, можно разложить пол-
луцит на 99 %. Однако метод имеет существенные недостатки — сил*>'
ное загрязнение растворов примесями и сложность аппаратурного офор'
мления ввиду агрессивности реагентов и продуктов реакции.
100
Для разложения поллуцита возможно использование серной кисло-
ты. Сернокислотный метод переработки включает следующие основные
операции. Концентрат поллуцита, измельченный до 0,04—0,07 мм,
смешивают с водой до пастообразного состояния, затем обрабатыва-
ют 96 %-ной серной кислотой (на 1 кг поллуцита 0,73 кг H2SO4 и 0,25
кг воды). Разлагают поллуцит в реакторе при 120—150 °C в течение 4 ч;
смесь охлаждают и выдерживают 1—2 ч для более полного разложения
поллуцита. Затем продукты реакции выщелачивают водой (на 1 кг пол-
луцита 11,7 л воды) и при непрерывном перемешивании нагревают до
кипения. Реакции разложения и выщелачивания поллуцита протекают
по следующей схеме:
(Cs, Na) [AIS12O6 ] пНгО + H2SO4
—’ CsHSO4 + А1Н (SO4) 2 + Н2О + SiO2 + NaHSO4. (2.30)
Затем продукты реакции выщелачивают водой при температуре ки-
пения (2.29).
Присутствие в растворе сульфата алюминия способствует коагуля-
ции кремневой кислоты, которую отфильтровывают при 100 °C через
полихлорвиниловую ткань. Фильтрат охлаждают до 0—10 °C для кри-
сталлизации алюмоцезиевых квасцов, которые отфильтровывают и
промывают горячей водой. Сернокислотный способ переработки поллу-
цита имеет ряд преимуществ перед солянокислотным: меньше коррозия
аппаратуры и загрязнение воздуха, снижение затрат на реагенты для
осаждения цезия, так как алюминий содержится в самом минерале.
Выход цезия в готовый продукт такой же, как и в солянокислотном
методе переработки.
Для разложения поллуцита можно использовать также более раз-
бавленную (50 %) серную кислоту, однако при этом замедляется скоро-
сть реакции и увеличивается степень коррозии аппаратуры.
Методы спекания. В качестве реагентов в этой группе методов
были опробованы многие вещества, однако промышленное воплощение
получил практически единственный способ, основанный на спекании
концентрата поллуцита со смесью СаО + СаСЬ (рис. 2.4). Концентрат
поллуцита с размерами частиц 0,04—0,07 мм тщательно смешивают с
карбонатом (оксидом) и хлоридом кальция (20 % поллуцита, 66 %
оксида кальция и 14 % хлорида кальция) и спекают во вращающейся
печи при 800—900 °C. Продолжительность спекания зависит от массы
Шихты и типа оборудования. Взаимодействие поллуцита с СаО и СаСЬ
протекает без образования растворимых алюминатов и силикатов по
Реакции
(Cs, Na) [AIS12O6 ] пН2О + СаО + СаС12 =
а (Cs, Na)CI + СаО AI2O3 2SiO2 + шСаО £SiO2 + иНгО. (2.31)
Таким образом, в результате взаимодействия образуются раствори-
Мыс хлориды цезия и натрия и нерастворимые алюмосиликаты и сили-
101
Поллуцит
} CaCI; |
Шихтование
Спекание
Измельчение опека
Рис. 2.4. Известково-хлоридная схема переработки поллуцита
102
каты кальция, о составе которых нет единого мнения. По окончании
реакции спек охлаждают, измельчают и обрабатывают в автоклаве ки-
пяшсй водой. При этом происходит дополнительное разложение непро-
реагировавшего поллуцита в результате его взаимодействия с Са(ОН)2
под давлением. Пульпу фильтруют, остаток проверяют на полноту из-
влечения цезия и в случае необходимости повторяют водную обработку,
фильтрат упаривают досуха с серной кислотой для образования сульфа-
та кальция. Остаток-выщелачивают в автоклаве горячей водой. Такой
режим выщелачивания уменьшает переход CaSO4 в раствор, так как в
этом случае он остается в виде ангидрита, который имеет отрицатель-
ный температурный коэффициент растворимости. Фильтрат после вы-
щелачивания, содержащий сульфаты цезия, рубидия, натрия и примесь
сульфата кальция, обрабатывают соляной кислотой и осаждают цезий в
виде Cs3Sb2Cl9. Далее эннеахлордистибиат цезия перерабатывают од-
ним из изложенных выше способов.
Схема достаточно проста, экономична; реагенты, используемые на
стадии спекания, дешевы. Серная кислота расходуется только на осаж-
дение части кальция, SbCh возвращается в процесс. В оптимальных
условиях поллуцит разлагается практически полностью (~ 98 %), из-
влечение цезия в хлорид 85—90 %, а чистота получаемого CsCl 99,9 %.
Все другие методы переработки поллуцита, основанные на спекании
(сплавлении) с различными реагентами, не дают столь высоких показа-
телей. Заслуживает лишь внимания щелочной метод, предложенный
для извлечения цезия из комплексных поллуцито-сподуменовых руд.
По этому методу рекомендуется водно-известковое выщелачивание из-
мельченной руды в автоклаве при 220 °C и 20 МПа. При этом поллуцит
разлагается, а СГ-сподумен остается в нерастворимом остатке, который
отделяют от раствора. Цезий из раствора выделяют в виде алюмоквасцов
и их же используют для очистки цезия от примесей щелочных элементов
в процессе фракционированной кристаллизации. Квасцы разлагают
гидроксидом или карбонатом бария и в результате получают гидроксид
или карбонат цезия. Прямое извлечение цезия 74 %; чистота соответ-
ствует квалификации «чистый».
Переработка лепидолита. Несмотря на существование многочис-
ленных методов разложения лепидолита, разработанных в связи с тех-
нологией лития, лишь немногие содержат сведения о попутном извлече-
нии рубидия и цезия. К ним относятся сернокислотный метод и метод,
°снованный на сплавлении лепидолита с сульфатом калия (см. гл. 1).
В результате разложения лепидолита серной кислотой получают
Раствор сульфатов щелочных металлов (Li, Na, К, Rb, Cs), из которого
Рубидий и цезий выделяют в виде алюмоквасцов. От квасцов переходят
к Карбонатам, а затем можно выделить рубидий и цезий в виде любого
Малорастворимого соединения в зависимости от конкретной задачи.
103
Примером комплексной переработки лепидолита с извлечением ли«
тия, рубидия и цезия является метод, основанный на сплавлении его с
сульфатом калия. Лепидолит сплавляют с K2SO4 при 1090 °C и плав
обрабатывают водой. При этом в раствор переходит весь литий и лицц
частично рубидий и цезий. Основная часть рубидия и цезия содержит»
в остатке, который разлагают серной кислотой при 100 °C и обрабатыва-
ют водой. Из раствора осаждают алюмоквасцы калия, рубидия и цезия
и проводят несколько перекристаллизаций квасцов для обогащения их
рубидием и цезием.
Переработка карналлита. Карналлит КС1 MgCh пНгО — ценное
сырье для получения калийных удобрений, металлического магния,
брома и пищевой соли. Переработка карналлита представляет интерес,
главным образом, для России и Германии, которые располагают боль-
шими запасами этого минерала.
При переработке карналлита с целью извлечения магния и хлорида
калия рубидий или остается в отработанном электролите, или попадает
в хлорид калия и соответственно в удобрения. Извлечение рубидия при
переработке карналлита ограничено. Это связано с небольшим спросом
на соединения рубидия и их высокой стоимостью. Однако в масштабам
мировой переработки карналлита ежегодно теряются значительные ко-
личества этого элемента.
Разработаны многочисленные варианты технологических схем пе-
реработки карналлита, предусматривающих попутное выделение руби-
дия. Однако в конечном итоге все методы переработки сводятся к раз-
делению рубидия и цезия и отделению их от калия, что и составляет
основную трудность в осуществлении процесса. В настоящее время мож-
но говорить о двух принципиально различных подходах к решению
задачи комплексной переработки карналлита: 1) получение обогащен-
ного рубидием искусственного карналлита и 2) выделение рубидия из
«отработанного электролита» магниевого производства.
Получение искусственного карналлита. Метод основан на инконг-
руэнтном характере растворимости калиевого карналлита в отличие от
рубидиевого и цезиевого карналлитов, которые растворяются конгруэн-
тно (см. п. 2.4). При переработке природного карналлита горячей водой
в количестве, недостаточном для полного растворения, он разлагается»
выделением в осадок менее растворимого хлорида калия; рубидиевый и
цезиевый карналлиты остаются в растворе. После отделения КС1 и ох-
лаждения раствора кристаллизуется 1 -й искусственный карналлит, обо-
гащенный рубидием и цезием. Содержание RbCl в 1-м искусственном
карналлите 0,025—0,11 %. Кристаллизация искусственного карналли-
та начинается при содержании в растворе более 300 г/л MgCh, причем
при концентрации MgCh > 305 г/л растворимость хлорида рубидия и>
вероятно, хлорида цезия меньше растворимости КС1 (табл. 2.8).
104
Таблица 2.8. Растворимость KCI и RbCl в растворах MgC12 при 20 *С
Концентрация MgC12, г/л 50 100 150 200 250 300 350 400
Растворимость'^ КС1, г/л 245 190 148 107 73 52 35 30
2) Растворимость RbCl, г/л 625 550 450 310 168 68 19 6
° Система KCl—MgC12—H2O2 2) Система RbCl—MgC12—H2O
Маточный раствор после отделения искусственного карналлита на-
правляют на извлечение хлорида магния и брома. Для дальнейшего
обогащения рубидием 1-й искусственный карналлит растворяют в воде
при 60—70 °C до плотности горячего раствора 1,31 г/см3. Выпавшие на
этой стадии КО и NaCl отделяют, а маточный раствор охлаждают для
кристаллизации 2-го искусственного карналлита, состав которого сле-
дующий, %:
RbCl.....1,5 NaCl......12,0
KCI..... 38,5 MgSO4......0,2
MgC12-• • . 17,6 H2O........30,2
Второй искусственный карналлит снова растворяют в воде при 60—
70 °C, охлаждают и отфильтровывают хлорид калия, а в нагретом до
кипения маточном растворе растворяют новые порции 2-го искусствен-
ного карналлита до насыщения. Операцию повторяют дважды. После
Удаления осадка и охлаждения раствора кристаллизуется 3-й искусст-
венный карналлит, содержащий суммарно до 9,5 % RbCl и CsCl.
Для переработки 3-го искусственного карналлита на соли рубидия и
Цезия предложены различные методы: гексахлоростаннатный, квасцо-
вый и др. Наиболее применимым представляется квасцовый метод. Ис-
кусственный карналлит растворяют при нагревании в воде, отфильт-
ровывают осадок, фильтрат смешивают с раствором сульфата алюминия
и охлаждают до 20 °C. Выпавшие алюмокалиевые квасцы, обогащенные
Рубидием и цезием, отфильтровывают, растворяют в горячей воде, ох-
лаждают до 40 °C и снова кристаллизуют квасцы. Эту операцию повто-
ряют три—четыре раза; затем квасцы растворяют в воде и раствор ох-
лаждают до 5 °C (см. табл. 2.1). Маточные растворы используют для
Растворения квасцов предыдущей стадии. Для выделения соединения
^езия рубидиево-цезиевые квасцы переводят в CsaSbiClg после раство-
Рсния в соляной кислоте и обработки солянокислым раствором БЬС1з.
та операциядает возможность отделить рубидий от цезия. Дальнейшая
105
переработка комплексной соли с получением хлорида цезия изложена
ранее. Из раствора после выделения Cs3Sb2Cl9 рубидий может быц
выделен в виде гидротартрата ИЬНСдЩОб или гидрооксалата
RbH3(C2O4)2 2Н2О с последующим прокаливанием при 400—450 °C
(см. п. 2.12).
Для извлечения рубидия из искусственного карналлита могут быть
использованы и другие способы — ферроцианидный, кремнемолибдат-
ный и др.
Описанные выше методы были разработаны для извлечения рубидия
из страссфуртских карналлитов (Германия). Переработка Соликамских
карналлитов (Россия) несколько отличается, так как последние харак-
теризуются отсутствием растворимых сульфатов, малым содержанием
хлорида натрия и значительно большим содержанием калия.
Извлечение рубидия из отработанного электролита. При перера-
ботке Соликамских карналлитов целесообразнее извлекать рубидий не
из искусственного карналлита, а из отработанного электролита — остат-
ка после электролитического выделения магния из безводного искусст-
венного карналлита. В отработанный электролит переходит ~ 90 %
рубидия, содержащегося в карналлите, он содержит следующие компо-
ненты, %:
КС1....... 76 80 MgO.........0,3—0,5
NaCl...... 8—10 Rb2O .... 0,03—0,06
MgC12..... 4—10
Наиболее рациональный метод извлечения рубидия из отработанно
го электролита предложен И.В. Тананаевым. Он заключается в способ-
ности осадков смешанных ферроцианидов железа, цинка, никеля и
калия извлекать из растворов незначительные количества рубидия и
цезия. Метод включает следующие операции. Измельченный отрабо
тайный электролит выщелачивают водой при комнатной температуре и
Т : Ж = 1 : 2. В раствор переходит до 85 % рубидия, концентрат®
которого составляет 0,14—0,17 г/л. Раствор подкисляют НС1 до pH 5—6-
добавляют 0,5М раствор ферроцианида калия K4[Fe(CN)6] ЗН2О 11
FeCh 6Н2О, соответственно 11,3 кг и 3,1 кг на 1 т электролита. Коэф
фициент обогащения осадка K^Rba-x) Fe[Fe(CN)6 ] рубидием и цезием
100—500 в зависимости от условий осаждения.
Осадок отстаивают в течение суток, раствор сливают, пульпу филь'
труют на центрифуге и промывают водой, содержащей солянокисл!»
раствор хлорида железа. В фильтрате остается 0,013—0,017 г/л руо|'
дия. Осадок высушивают при 100—200 °C и прокаливают при 7(ХИ
800 °C; при этом выделяется азот и образуются карбонаты щелочив
металлов. Остаток выщелачивают водой, содержащей соляную кислот?
упаривают и обрабатывают раствором ферроцианида магния или цин^3.
Осадок отфильтровывают, промывают водой и прокаливают при бОО""
106
700 °C. Остаток выщелачивают водой, содержащей НС1, и упаривают.
Конечный продукт содержит, %: RbCl 80—90; КС1 10—15; NaCl 1—2;
CsCl 2; примеси железа, магния, кальция, алюминия.
Различные варианты метода И.В. Тананаева касаются прежде всего
стадии фильтрации осадка и его прокаливания. Наиболее интересным
является использование гранулированных осадков ферроцианидов же-
леза или никеля. Гранулирование производят путем их замораживания
и оттаивания. В результате этого плотность осадков возрастает в 10—60
раз. В процессе замораживания пространственная структура коагулян-
тов разрушается и после оттаивания приобретает зернистый характер.
Такие осадки разрушаются медленнее, а их фильтрующая способность
сильно возрастает.
2.11. Извлечение рубидия и цезия
из отработанного ядерного топлива
Работа энергетических атомных реакторов связана с образованием
высокорадиоактивных продуктов деления урана, используемого в каче-
стве ядерного топлива. Радиоактивные отходы создают серьезную эко-
логическую проблему. Основное количество радиоактивных отходов
(~ 99 %) отправляют на установки регенерации для извлечения урана,
тория и других ценных элементов, в том числе и некоторых продуктов
деления. Важнейшая проблема ядерного топливного цикла — проблема
обезвреживания радиоактивных отходов, их безопасного хранения и
надежного захоронения с гарантией предотвращения контакта с биосфе-
рой.
При делении урана-235 в каждой тонне ядерного горючего обра-
зуется 352 г изотопов цезия и 54 г изотопов рубидия. Среди них особое
место занимает долгоживущий радиоактивный изотоп Cs—137 (Ti/2 =
33 года), который выде ^ется в относительно большом количестве при
Реакции
137 т И7 Р 137 137 у 137 <2-33)
531- * 54 Хе ' 55 Cs ” 56 Ва 5бВаСтаб.
71/2-23сек 7'1/2“3,9мин Г1/2~ЗЗгода Т^/г-г.бмин
Он определяет до некоторой степени активность продуктов деления.
Выделение цезия из радиоактивных отходов, помимо решения вопросов
безопасности хранения и экологии, связано также с потребностью в
Радиоактивном цезии различных отраслей науки, техники и народного
х°зяйства (медицина, дезинфекция, пастеризация, стерилизация, по-
ЛиМеризация, вулканизация, возбуждение химических реакций).
107
Для удаления продуктов деления из урановых стержней их раство-
ряют в азотной кислоте, в полученный раствор уранилнитрата добавляй
ют нитрат натрия и экстрагируют трибутилфосфатом в непрерывно!
противоточном процессе. Радиоактивные продукты деления, в том чис-
ле цезий и рубидий, концентрируются в водной фазе, а уран и плутоний
— в органической. При осуществлении промышленной технологии из-
влечения цезия и рубидия из водной фазы после отделения U и Рц
необходимо учитывать высокую радиоактивность водной фазы. В связи
с этим схемы переработки должны быть максимально простыми, исполь!
зуемая аппаратура — надежной. Из известных методов промышленного
выделения изотопов цезия и рубидия указанным требованиям наиболее
отвечает ферроцианидный.
Ферроцианидный метод. Метод основан на использовании ионооб-
менных свойств сложных ферроцианидов железа, никеля, цинка и дру-
гих металлов. Он применяется в США и России. На рис. 2.5 представлена
схема переработки радиоактивных отходов с применением ферроцианиЛ
да цинка и калия. Сбросные растворы обрабатывают аммиаком до pH
2—3, отфильтровывают осадок гидроксида железа с примесью плутЛ
ния, циркония и ниобия. Фильтрат нейтрализуют NaOH до pH 12—13
для выделения диураната натрия и гидроксидов стронция и редкозе-
мельных элементов, которые удаляют, а раствор подкисляют азотной
кислотой до pH 1—2 и при непрерывном перемешивании добавляют
Zn(NO3>2- 6Н2О и K4[Fe(CN)e]. Смесь отстаивают в течение несколь-
ких суток, так как осадок образуется медленно. Осадок ферроцианида,
содержащий рубидий и цезий, центрифугируют, промывают водой, вы-
сушивают при 100 °C и измельчают. Потери цезия с маточным раство-
ром — не более 5 %. Осадок ферроцианида прокаливают при 550 °C, при
этом образуются ZnO, РегОз, ЙЬгСОз и CS2CO3. Продукты выщелачи-
вают водой и отделяют нерастворимые оксиды цинка и железа, а филЛ
трат упаривают с соляной кислотой и кристаллизуют хлориды рубидиИ
и цезия.
2.12. Извлечение рубидия и цезия из рапы соляных озер
и рассолов морского типа
Природные воды, рапа соляных озер, рассолы и грязи морского типа
являются потенциальными источниками рубидия и цезия. В морской
воде содержание рубидия и цезия составляет в среднем 1,210-4 и 5 • 10 I
г/л соответственно.
Главная трудность при извлечении рубидия и цезия из морской водД
заключается в первичном концентрировании, которое требует больших
затрат энергии и сопровождается заметными потерями рубидия и цезия
108
Рис. 2.5. Технологическая схема ферроцианидного метода переработки
радиоактивных отходов
с солями натрия, калия и магния, кристаллизующимися при упарива-
нии. Об извлечении рубидия и цезия из этих источников можно говорить
лишь как о попутном процессе при комплексной переработке соляных
Рассолов на соединения калия, магния, натрия и лития. Только в этом
случае выделение рубидия и цезия из различного типа рассолов и мор-
ской воды будет экономически целесообразно.
Была сделана попытка извлечь рубидий из воды Мертвого моря
(0.007 % Rb). При выпаривании сначала отделяли хлориды натрия и
калия, а затем осаждали 1-й искусственный карналлит; в результате
Фракционированной кристаллизации которого получали 2-й, 3-й, 4-й и
109
5-й искусственные карналлиты с сг держанием 0,045; 0,14; 0,47 и 0,52 %
RbCl. Однако соединения рубидия выделены не были, что объясняется
большими потерями его с промежуточными продуктами и маточными
растворами.
Наибольший интерес представляют методы селективного выделения
рубидия и цезия непосредственно из морской воды или рассолов, полу-
ченных естественным упариванием морской воды до начала кристалли-1
зации хлоридов натрия и калия. Для этих целей рекомендуется ис-
пользовать соосаждение рубидия и цезия с носителем, например дипик 1
риламинатом, кобальтинитритом или ферроцианидом. Ферроцианид!
ный метод извлечения рубидия и цезия из морской воды в принципе
мало отличается от описанных выше ферроцианидных методов извлеч. -
ния Rb и Cs из карналлитов и радиоактивных отходов, поэтому пред-
ставляется целесообразным описание другого рекомендуемого метода —
кобальтинитритного.
Кобалътинитритный метод. В этом методе осадителем является
раствор кобальтинитрита натрия в уксусной кислоте. Осадитель готовят
смешиванием растворов нитрита натрия и ацетата кобальта в ледяной
уксусной кислоте непосредственно перед употреблением и вводят в мор-
скую воду при температуре ниже 10 °C из расчета на 1 л воды 20—30 мл
реагента. Осадок отделяют и термически разлагают при < 500 °C, при
этом образуются следующие продукты:
3Cs2Na[Co(N02)6]- 6CsN02+3NaN02+Co304+4N0+5N02. (2.33а)
После прокаливания нитриты щелочных металлов выщелачивают
водой, а остаток С03О4 используют для приготовления ацетата кобаль-
та. Из водного раствора рубидий и цезий извлекают в виде дипикрила-
минатов, для чего раствор нейтрализуют до pH 8—9, нагревают до
60—70 °C и обрабатывают раствором дипикриламината магния. Осадок
растворяют в ацетоне и добавляют 6 н НС1. При этом дипикриламинат
разрушается, выделяя в водную фазу хлориды щелочных металлов..
Смесь хлоридов перерабатывают одним из описанных ранее способов (п.
2.10).
2.13. Получение соединений рубидия
и цезия различной степени чистоты
Конечными продуктами в процессах переработки цезий- и рубидий-
содержащего сырья являются цезиевые или рубидиевые концентраты,
содержащие значительное количество примесей (калий, натрий, маг-
ний, кальций, кремний, алюминий, железо и др.). В осуществление
дальнейшей очистки концентратов и получении соединений рубидия и
цезия реактивной квалификации основное внимание уделяется калию
ПО
__примеси, наиболее трудно удаляемой из соединений цезия и особенно
рубидия. В связи с этим процессы получения чистых соединений руби-
дия и цезия сводятся к проблеме разделения калия, рубидия, цезия.
Для разделения калия, рубидия и цезия предложено несколько тех-
нологических процессов, использующих небольшие различия в услови-
ях образования и физико-химических свойствах некоторых соединений
этих элементов, — фракционированная кристаллизация, осаждение,
ионообменная хроматография и экстракция. Перечисленные процессы
не являются равноценными для получения чистых солей и веществ
высокой степени чистоты.
Фракционированная кристаллизация. Близость свойств большин-
ства простых солей калия, рубидия и цезия и их высокая растворимость,
как правило, исключают возможность их применения для разделения.
Использовать небольшие различия в свойствах соединений разделяе-
мых элементов более всего позволяют некоторые двойные и комплекс-
ные соединения, применяемые поэтому для фракционированной крис-
таллизации и осаждения. Наибольшей сложностью отличается фракци-
онированная кристаллизация солей рубидия, при которой в зависимости
от порядка изменения растворимости в ряду К—Rb—Cs в твердой фазе
происходит накопление либо примеси калия, либо цезия, причем для
очистки соединений рубидия наибольший интерес представляют те его
соли, растворимость которых в ряду К—Rb—Cs наименьшая.
Для разделения рубидия и цезия широко используется фракциони-
рованная кристаллизация алюмоцезиевых и алюморубидиевых квасцов
(квасцовый метод) и хлората рубидия. Это хорошо изученные и наибо-
лее доступные методы как в препаративной, так и в технологической
практике. В результате трех—четырех перекристаллизаций из водного
раствора технического хлората рубидия RbClCh (0,5 % калия, 0,005 %
натрия и 0,16 % цезия) можно получить препарат с содержанием, %:
натрия 0,002; калия 0,03; цезия 0,02; железа и тяжелых металлов 0,001
С выходом хлората рубидия после каждой ступени кристаллизации
90 %.
Квасцовый метод разделения и очистки соединений рубидия и цезия
привлекателен тем, что при разложении алюмосиликатных минералов
серной кислотой или сульфатом калия квасцы образуются за счет содер-
жащихся в минерале элементов. Состав алюморубидиевого и алюмоце-
зиевого концентратов, выделенных из растворов после разложения алю-
мосиликатов типа литиевых слюд, зависит от содержания калия, руби-.
№я и цезия в руде и обычно составляет, %: KA1(SO4)2- 12НгО 76—90;
RbAl(SO4)2 12Н2О 6—15; CsA1(SO4)2- 12НгО 2—8. При переработке
п°ллуцита алюмоцезиевый концентрат содержит 85—93 % цезиевых,
4~-5 % — рубидиевых и 1—2 % калиевых квасцов. Присутствие в
Растворе калиевых квасцо в значительно уменьшает растворимость
111
квасцов рубидия и цезия, что способствует более полному их извлече-
ний.
Фракционированная кристаллизация проводится разделением на
две фракции: концентрат (твердая фаза) и маточный раствор. При этот
концентрат постепенно обогащается цезием, а маточный раствор —
рубидием. Обычно после разделения маточного раствора на две фракци»
твердая фаза в цикл не возвращается. В каждой стадии кристаллизаций
концентрат растворяют в 4-кратном количестве воды при 80—90 °C
полученный раствор охлаждают только до 50—70 °C, что дает значи-
тельно более высокую степень обогащения концентрата цезием. Сте-
пень кристаллизации квасцов 0,6—0,8. Используя изложенный режим
кристаллизации, из рубидиево-цезиевого концентрата, содержащего
70 % RbAl(SO4)2 12НгО, 8 % CsA1(SO4)2 12Н2О и 19 X
KA1(SO4)2- 12Н2О, в результате семи перекристаллизаций получаемся
продукт с содержанием 82,5 % алюмоцезиевых квасцов. Оценивая при-
менимость квасцового метода для разделения калия, рубидия и цезия,
необходимо учитывать способность квасцов образовывать твердые рас-
творы вследствие изоморфизма. Так как алюмокалиевые и алюмоцези-
евые квасцы твердых растворов не образуют, основная трудность при
использовании метода — разделение пары рубидия и цезия и пары
рубидия и калия. Основные недостатки метода — малая эффективность
и трудоемкость. Квасцовый способ следует рассматривать как удобную
форму первичного выделения рубидия и цезия из технологических рас-
творов и получения обогащенных цезиевых и рубидиевых концентра-
тов, дальнейшая переработка которых осуществляется другими спосо-
бами.
Осаждение малорастворимых соединений. Для разделения калия,
рубидия и цезия используется осаждение гексахлоростаннатов, гекса-
хлороплюмбатов и комплексных галогенидов сурьмы с рубидием и це-
зием.
Осаждение гексахлоростаннатов используется в технологических
процессах разделения рубидия и цезия на стадии, следующей за выде-
лением квасцов, в основном для очистки солей рубидия от примесей
калия и натрия. По сравнению с квасцовым гексахлоростаннатный ме-
тод дает более высокую кратность очистки соединений рубидия от ка-
лия, обусловленную большим различием в растворимости соединений.
Осаждение гексахлороплюмбатов используется в основном для уда-
ления примеси калия из соединений рубидия и цезия. Этот метод имеет
ряд преимуществ по сравнению с гексахлоростаннатным. Хлорид свинца
— более дешевый реагент, чем SnCl4, гексахлороплюмбаты рубидия и
цезия менее растворимы в 35 %-ной соляной кислоте, гидролиз гекса-
хлороплюмбатов осуществляется полнее и легче, чем аналогичных сое-
динений олова. Для очистки соединений рубидия и цезия
112
(gb—Cs-концентрат) от калия к раствору хлоридов этих элементов
добавляют равный объем 35 %-ной НС1. Осадок хлоридов натрия и
калия отфильтровывают, к охлажденному фильтрату добавляют порци-
ями суспензию РЬС12 в 35 %-ной НС1 при О °C (5 г РЬСЬ в 100 мл
кислоты) и пропускают хлор. Образующийся желтоватый осадок
(Rb, Cs)2PbC16 практически не содержит калия. Осадок отфильтровы-
вают, промывают охлажденной 35 % -ной соляной кислотой и разлагают
зрячей водой, содержащей небольшое количество аммиака
(Rb, Cs) 2PbCl6+ЗНгО - 2 (Rb, Cs) Cl + РЬОг + 4 НС1. (2.34)
Диоксид свинца РЬОг отфильтровывают, фильтрат упаривают досу-
ха для удаления НС1, сухой остаток растворяют в нагретой до 70—80 °C
воде, подкисляют соляной кислотой и обрабатывают сероводородом для
удаления остатков свинца. Осадок сульфида свинца отфильтровывают,
а из раствора осаждают Свз [SЬгС1д ].
Осаждение комплексных галогенидов сурьмы и висмута. Эннеах-
лордистибиат цезия Csa [Sb2C19 ] как форма выделения цезия из техно-
логических растворов и отделения от остальных щелочных элементов
занимает, пожалуй, главное место среди других комплексных соедине-
ний, содержащих рубидий и цезий. Однако в присутствии в растворах
значительного количества рубидия и калия реакция осаждения
Cs3[Sb2Cl9 ] не является специфичной, так как RbCl и КС1 образу-
ют аналогичные соединения, растворимость которых уменьшается в
ряду К — Rb — Cs. То же относится и к аналогичным соединениям
висмута. Различие в растворимости указывает на возможность исполь-
зования комплексных галогенидов сурьмы и висмута для их разделения.
Например, трехкратное осаждение РЬзВ1гС19 позволяет из Rb-концен-
трата, содержащего ~ 80 % RbCl и ~ 20 % КС1, получить технический
хлорид рубидия с содержанием основного вещества ~ 98 %. Применение
тех или иных комплексных галогенидов сурьмы и висмута для разделе-
ния калия, рубидия и цезия зависит от поставленной задачи.
Ионообменная хроматография. Этот метод до настоящего времени
не нашел широко применения ни в препаративной, ни, тем более, в
технологической практике. Описан способ непрерывной противоточной
ионообменной хроматографии, на основе которого разработана техно-
логическая схема, которая на нашла промышленного применения.
Более перспективным представляется направление исследований,
евязанное с использованием обменной сорбции К , Rb и Cs на ионитах
Минерального происхождения — цеолитах, анальциме, фосфате и мо-
либдате циркония. Минеральные иониты обладают более высокой се-
лективностью и устойчивостью к действию высоких температур по срав-
нению со смолами.
Методы хроматографического разделения и очистки соединений ру-
бидия и цезия имеют один общий недостаток: в результате процесса
113
получают более разбавленные растворы, чем исходные, а это влечет за
собой значительные энергетические затраты на упаривание больших
объемов растворов.
Экстракция. Рубидий и цезий, как и другие щелочные элементы, не
склонны к комплексообразованию. Они образуют хорошо диссоцииру.
ющие в водных растворах ионные соединения и существуют в раствор,*
в форме гидратированных катионов (Rb , Cs ) ад. Для перевода гидра-
тированного иона калия рубидия или цезия из водного раствора в орга-
нический растворитель необходимо затратить энергию, равную, к*
минимум, сумме энергии гидратации иона, ориентации и поляризации
растворителя. Компенсация этой энергии энергией комплексообразоЛ
ния и сольватации иона приводит к тому, что образовавшийся гидрофоб-
ный комплекс нарушает структуру воды и переходит в органическую
фазу. Так как энергия сольватации значительно меньше энергии гидра-
тации, а способность близких по свойствам щелочных элементов к ком-
плексообразованию с органическими лигандами ограничена, экстрак-
ция в технологических процессах разделения калия, рубидия и цезия не
имеет того значения, как, например, в технологии разделения редкозе-
мельных элементов, циркония и гафния, тантала и ниобия и др.
Наиболее эффективные экстрагенты для разделения рубидия и це-
зия — различные замещенные фенолы (алкил- или арилфенолы). Их
можно использовать для извлечения и разделения рубидия и цезия из
щелочных технологических растворов, для извлечения Cs137 из продуй
тов деления и в аналитических целях. Замещенные фенолы, как слабые
кислоты (константа диссоциации 10—9—10 10), экстрагируют К, Rb, Cs
только из щелочных растворов, в которых отсутствуют элементы, гид-
роксиды которых в этих условиях выпадают в осадок. Экстракционная
способность фенолов определяется природой заместителя и его положе»
нием в бензольном кольце, а также типом разбавителя. Установлена
что при экстракции рубидия и цезия наиболее высокие коэффициенты
распределения О достигаются при использовании замещенных фенолов
с заместителем в параположении, причем D растете увеличением числа
углеродных атомов в цепи заместителя. Так, при возрастании числа
атомов углерода в цепи заместителя от Ci до Cs Des увеличивается от
20,3 до 124 (pH 12). При экстракции щелочных металлов фенолами
экстрагируемый элемент переходит в органическую фазу в виде фено-
лята, обычно сольватированного свободной молекулой фенола
CsB+ ад + [m(RH)n ]о + ОН" X [CsR(RH)mn-i ]о + Н2О. (2.35)
Обычно экстрагируемый фенолят цезия сольватирован тремя моле-
кулами фенола. Однако при высокой щелочности раствора может обра-
зоваться фенолят, не сольватированный молекулами фенола. В отличие
от фенола феноляты рубидия и цезия являются поверхностно-активнЫ-
114
мл веществами и при достаточной их концентрации (в области с высо-
ким содержанием гидроксидов щелочных элементов) образуют мицел-
лы обратного типа, резко увеличивающие растворимость воды в органи-
ческой фазе и способные растворять неорганические соли, например
CS2SO4, на чем основан мицеллярный механизм экстракции щелочных
элементов фенолами. Наличие мицеллярной экстракции снижает се-
лективность процесса и сильно увеличивает объем органической фазы,
что в ряде случаев нежелательно. В связи с этим для экстракции из
сильнощелочных растворов предложено использовать 2-<7-метилбен-
зил-4-вторбутил фенол (БАМБФ)
ОН СНз
С4Н9
который менее склонен к мицеллообразованию и обладает относительно
низкой растворимостью в водном растворе (при pH 12,8 она составляет
0,3 г/л). Обычно используют 1 М раствор БАМБФ в керосине. Коэффи-
циенты распределения Cs, Rb и К увеличиваются с ростом pH. При pH
~ 13 Des ~ Ю, D«b < 1. При pH 12,5 коэффициенты разделения/? для
пары Cs+/M+, где М — Rb и К, равны 20 и 300 для рубидия и калия
соответственно. Для экстракции рубидия и цезия применяются также
олигомерные производные алкилфенолов, обладающие рядом преиму-
ществ по сравнению с исходными алкилфенолами: большой эффектив-
ный объем молекулы делает экстрагент гидрофобным и тем самым сни-
жает его растворимость в водной фазе; более кислые свойства олигоме-
ров позволяют экстрагировать Rb и Cs из менее щелочных растворов;
Разбавители оказывают меньшее влияние на экстракционные свойства
олигомерных экстрагентов по сравнению с алкилфенолами; феноляты
менее склонны к мицеллообразованию, чем алкилфенолы, что сущест-
венно снижает взаимное растворение фаз и потери экстрагента. Химизм
экстракции при использовании этих реагентов, как и алкилфенолов,
катионообменный.
Получение соединений рубидия и цезия особой чистоты. Техноло-
гия соединений рубидия и цезия располагает достаточным количеством
Методов, позволяющих получать реактивные и химически чистые сое-
динения рубидия и цезия. Гораздо сложнее обстоит дело с достижением
115
более высокой квалификации по чистоте. При получении особо чистых
соединений рубидия и цезия наибольшую трудность представляет раз-
деление калия и рубидия (микрокомпонент — калий) и в меньшей
степени рубидия и цезия (микрокомпонент — рубидий или цезий).
Основное препятствие при удалении микропримесей из соединений ру,
бидия и цезия — изоморфная кристаллизация. Известно, что на изомор.
фную или изодиморфную кристаллизацию большое влияние оказывае!
поляризация ионов. Если степень поляризации значительно различает-
ся, то даже при близости ионных радиусов микро- и макрокомпонентоИ
можно избежать образования изоморфных смесей. Поляризуемость эле-
ментов оказывает весьма существенное влияние на их химическое по-
ведение и прежде всего на устойчивость соединений.
У калия, рубидия и цезия среди ограниченного числа различающих-
ся между собой физико-химических характеристик величина поляризу-
емости их атомов и ионов занимает одно из ведущих мест. Поэтому для
глубокой очистки соединений рубидия и цезия особый интерес представ!
ляют комплексные соединения, анион которых содержит легко дефор-
мируемые ионы или атомы. Комплексные соединения этого рода ха-
рактеризуются, как правило, значительной молекулярной массой, на-
личием полярных групп в анионе, небольшой устойчивостью в раство-*
рах и особенно при нагревании. Всем этим требованиям в полной мере
отвечают анионгалогенааты рубидия и цезия, которые используются в
процессах получения соединений рубидия и цезия высокой степени чи-
стоты в лабораторных условиях. Достоинствами способа являются высо-
кая кратность очистки, простота перевода анионгалогенаатов в простые
соединения рубидия и цезия, образование хорошо фильтрующихся осад-
ков, возможность полной регенерации галогенов и межгалоидных сое-
динений. Ряд устойчивости анионгалогенаатов щелочных элементов
следующий: Cs > Rb > К » (Na, Li). В связи с этим для очистки следует
выбирать те соединения рубидия и цезия, которые или не существуют
для калия, или сильно различаются по растворимости в воде и констан-1
там нестойкости.
Получение особо чистого хлорида рубидия. Для этой цели рекомен-
дуется использовать хлорбромиодаат рубидия Rb[I(ClBr) ], наиболее
устойчивый из группы соединений, для которых неизвестны калиевые
аналоги. Очистка проводится следующим образом. К растертому иоду
при непрерывном перемешивании добавляют бром в количестве, на 5 %
выше стехиометрического. Через полученную смесь пропускают сухой
азот для удаления избытка брома и приливают при непрерывном пере-
мешивании концентрированный водный раствор (70—80 °C) техниче-
ского хлорида рубидия. По окончании реакции охлаждают смесь до
0 °C. Выпавшие оранжево-красные кристаллы Rb [I (ВгС1) ] отфильтро-
вывают, промывают ледяной водой и высушивают на воздухе или в
116
ракууме. Рекомендовано дважды переводить RbCl в Rb [I (BrCl) ] снача-
да в водном растворе, а затем в 0,5 М растворе СНзСООН с промежуточ-
ным прокаливанием при 400 °C. Способ позволяет получить из тех-
нического RbCl (2—5 % КС1) особо чистый RbCl с содержанием 2 10 4
% КО. Для удаления примеси цезия, который накапливается в осадке
Rb[I(BrCl) ], первые фракции кристаллов (5 % от массы осадка) можно
выводить из цикла. Осадок растворяют при 80—90 °C в минимальном
количестве воды и охлаждают до 0 °C. При этом выпадают кристаллы
Cs [I (BrCl) ], а маточный раствор, содержащий рубидий, направляют на
стадию растворения технического RbCl.
Удаление микропримеси цезия из соединений рубидия. Для этой
цели используют дииодиодаат рубидия Rb [KI2) ]; растворимость анало-
гичного соединения цезия в триста раз меньше. Изоморфизм этих сое-
динений приводит к обогащению кристаллов Rb [I (1г) ]цезием. Вданном
случае соединение рубидия выполняет роль носителя для удаления мик-
роколичеств цезия. Суть метода состоит в следующем. В водном раство-
ре Rbl при 60—80 °C растворяют иод из расчета выделения ~ 10 % Rbl
в виде Rb [I (I2) ], т.е. первой фракции, обогащенной цезием. Смесь ох-
лаждают, отделяют осадок и таким же образом осаждают вторую фрак-
цию дииодиодаата рубидия. Обычно проводят 3—4 осаждения. Остав-
шийся раствор упаривают при 120—130 °C досуха. Остаток прокалива-
ют сначала при 150 °C для удаления основной массы иода, затем при
300—350 °C. Полученный иодид рубидия содержит ~ 0,01 % CsI при его
исходном содержании 0,25—2,5 %. Выход Rbl ~ 55 % в прямом цикле;
остальное его количество содержится в обогащенном цезием остатке и
возвращается в голову процесса после прокаливания.
Получение особо чистого бромида цезия. Лучшие результаты полу-
чены при использовании бромиодиодаата цезия Cs [I (IBr) ] — единст-
венного соединения с анионом [I(1Вг) ], известного для щелочных
элементов. Для очистки к нагретому до 70—80 °C водно-спиртовому
раствору бромида цезия (CsBr : Н2О : спирт =1:2:2) добавляют 1,2
части иода, охлаждают до 5 °C. Кристаллы Cs [I (IBr) ] отфильтровывают
и прокаливают сначала при 150 °C, затем при 400 °C. Полученный таким
образом CsBr повторно переводят в CsfKIBr) ]. После второго прокали-
вания бромид цезия содержит не более 0,02 % RbBr и не более 0,005 %
КВг при содержании в исходном CsBr 5 % RbBr и до 1,5 % других
Щелочных элементов.
Достоинства анионгалогенаатов при получении особо чистых соеди-
нений рубидия и цезия неоспоримы с точки зрения физико-химических
основ процесса. Однако успех в осуществлении процесса глубокой очи-
стки веществ зависит и от решения чисто технологических и инженер-
ных задач, прежде всего создания особо устойчивой аппаратуры.
117
Поэтому говорить о широком использовании метода в технологическом
плане пока еще рано.
2.14. Получение металлических рубидия и цезия
Для получения металлических рубидия и цезия используют метод!
вакуумтермического восстановления и термического разложения солей.
В принципе металлы могут быть получены электролизом расплавов
Разрабатываются также способы прямого получения цезия из поллу-
цита.
Вакуумтермическое восстановление. Первые исследования процес-
сов металлотермического восстановления рубидия и цезия были осуще-
ствлены Н.Н. Бекетовым, который получил металлические рубидий и
цезий действием алюминия на их гидроксиды. В дальнейшем в качестве
исходных веществ для получения металлов была опробована большая
группа соединений (галогениды, гидроксиды, карбонаты, хроматы!
алюминаты, силикаты, бихроматы) и различные восстановители (маг-
ний, кальций, барий, натрий, алюминий, железо, цирконий, кремний),
выбор которых определялся сопоставлением энергии Гиббса для реак-
ций восстановления. Удовлетворительные результаты получены при
восстановлении алюминатов рубидия и цезия магнием, хроматов и бих-
роматов — цирконием. Фториды рубидия и цезия могут быть восстанов-
лены магнием и кальцием, хлориды — кальцием. Реакции восстанов-
ления фторидов имеют лучшие термодинамические характеристики,
однако из-за высокой гигроскопичности фторидов они применяются ре-
дко. Наиболее простой и доступный способ получения рубидия и цезия
— восстановление их хлоридов кальцием
2Rb(Cs)Cl + Са = 2Rb(Cs) + СаС12- (2.36)
Для реакций с RbCl и CsCl — ДЯ298 соответственно равны 15 и 20
кДж, что свидетельствует об их обратимости. Сместить равновесие
вправо можно, удаляя получающиеся металлы из зоны реакции. В дан-
ном случае это возможно, так как температуры кипения рубидия и цезия
значительно ниже температур кипения остальных компонентов процес-
са восстановления (см. табл. 1.1, 2.2, 5.18, рис. 1.13).
На рис. 2.6 представлена схема лабораторной установки для получе-
ния рубидия и цезия. Процесс восстановления хлоридов рубидия и цезия
протекает в вакууме (0,1—100 Па) при медленном подъеме температу-
ры до 700—800 °C. Извлечение металла 90—95 %. Метод можно исполь-
зовать и для восстановления других галогенидов рубидия и цезия.
Термическое разложение соединений. Метод имеет ограниченное
применение, в основном для получения небольших количеств спект-
рально чистых не содержащих газов металлов, для определения термо-
118
динамических и физических кон-
стант. Лишь немногие соли руби-
дия и цезия (гидриды, азиды, фер-
роцианиды) при нагревании в ва-
кууме разлагаются с выделением
металла. Лучшие результаты по-
лучены при вакуумтермическом
разложении азидов. Азиды руби-
дия и цезия диссоциируют в ваку-
уме (~ 102 Па) при 390—395 °C
2Rb(Cs)N3=2Rb(Cs)+3N2. (2.37)
Разложение протекает очень
медленно: 6—7 г RbNa или C5N3
диссоциируют в течение 3—6 сут.
Нагревание азидов до более высо-
ких температур может привести к
взрыву реактора и увеличивает
степень возгонки неразложив-
шихся азидов. Выход рубидия и
цезия 60 и 90 % соответственно.
Электрохимический метод.
Электрохимический метод в ме-
таллургии рубидия и цезия почти
не используется из-за высоких
значений потенциалов разложе-
ния галогенидов (см. табл. 1.1),
низкой температуры кипения ме-
Рис. 2.6. Схема лабораторной установки для
вакуумтермического получения рубидия и
цезия:
1 — реактор; 2 — шихта; 3 — рабочий
патрон; 4 — крышка реактора; 5 — крышка
патрона; 6,13 — сухие камеры; 7 — ловуш-
ка, 8 — стакаи с охлаждающей смесью, 9 —
термопары; 10— гальванометр; II — элек-
трическая печь; 12 — вакуумный кран; 14
— приемник жидкого металла; 15 — холо-
дильник приемника
таллов и их высокой растворимо-
сти в солевых расплавах (раство-
римость цезия в расплаве CsCl при
646 °C составляет 7,2%). Эти фак-
торы снижают выход металла и со-
ответственно экономическую эф-
фективность метода. Электрохи-
мический метод представляет ин-
терес для получения сплавов ру-
бидия и цезия с рядом металлов
(Pb, Sn, Bi, In, Tl, Cd), которые используются в ваннах в виде жидкого
катода. Например, электролизом расплава CsCl с жидким свинцовым
катодом при катодной плотности тока 0,31 А/см2 был получен свинцо-
воцезиевый сплав. Из таких сплавов рубидий и цезий легко удалять
Дистилляцией в вакууме.
119
Методы прямого получения цезия из поллуцита. Эти методы нахо-
дятся на стадии лабораторных исследований, однако они представляя»!
несомненный интерес, так как позволяют получать металл непосредс*
венно из руды. Цезий может быть получен прямой возгонкой из поллу!
цита при нагревании гранулированной смеси поллуцита с избытком
извести и восстановителя (алюминия, кремния или их сплавов) при
1050—1150 °C и давлении 102—1,02 Па. По другому методу поллуцщ
нагревают при 900 °C для удаления влаги, а затем нагревают при той же
температуре в вакууме в смеси с металлическим кальцием. При этом
достигается довольно высокое извлечение цезия — 85 % при расходе
кальция 3 весовых части на 1 часть поллуцита, содержавшего 35 %
Cs2O. При использовании в качестве восстановителя металлической
натрия при 700—800 °C в инертной атмосфере выход цезия достигая
75—95 %, однако металл содержит ~ 10 % примеси натрия.
Рафинирование рубидия и цезия. Металлические рубидий и цезий,
полученные одним из описанных выше методов, содержат механические
включения и целый ряд растворенных в металле примесей. Для очистки
металлов разработано несколько способов: фильтрование, переплавка
под слоем масла, нагревание с геттерами, зонная плавка, гидрирование
и вакуумная дистилляция. Для удаления механических примесей жид-
кие металлы фильтруют через перфорированную железную, молибде-
новую или титановую жесть. С этой же целью переплавляют металлы
под слоем парафинового или вазелинового масла при температуре, не-
много превышающей температуру плавления металлов. Очистку жид-
ких металлов геттерами применяют для удаления кислорода и азота. В
качестве геттеров используют порошкообразные титан и цирконий, ко-
торые не растворяются в жидких металлах. Наибольшее практическое
значение среди методов рафинирования рубидия и цезия имеет вакуум-
ная дистилляция.
Рубидий и цезий сохраняют в ампулах из термостойкого стекла в
атмосфере аргона или в стальных баках под слоем обезвоженного вазе-
линового или парафинового масла. Однако перед употреблением обычно
возникает необходимость в их дополнительной очистке. В связи с этим
стремятся получать металлы на месте их использования, а не на основ-
ном производстве. Этим устраняется дополнительная очистка и ликви-'
дируются связанные с ней потери дорогостоящих металлов.
120
Раздел II. Редкие элементы ПА подгруппы.
Бериллий, стронций
ОБЩАЯ ХАРАКТЕРИСТИКА ЭЛЕМЕНТОВ
Ко II группе Периодической системы относятся Be, Mg, Са, Sr, Ва и
Ra. Из них редкими элементами являются бериллий и стронций.
Элементы II группы йа внешнем электронном уровне содержат два
электрона ns2. Общим свойством для них является способность образо-
вывать двухзарядные катионы, имеющие электронную структуру бла-
городного газа. Первая степень окисления, отвечающая образованию
ионов М , для элементов IIА подгруппы на практике не осуществляется,
что сильно упрощает их химию. Свободные ионы М образуются и су-
ществуют только при высоких температурах; химические соединения,
содержащие ионы М , обычно диспропорционируют на М и М . Для
отрыва третьего электрона требуется очень большая энергия (табл. 3.1),
поэтому катионы М3+ в химических соединениях элементов II А под-
группы также не существуют.
Хотя свойства бериллия и магния значительно отличаются от
свойств других элементов подгруппы, известны некоторые закономер-
ности, свойственные всему ряду Be—Ra. Из их числа можно назвать
следующие: литофильность всех элементов рассматриваемого ряда; уси-
ление основных свойств гидроксидов; увеличение энтальпии их образо-
вания и растворимости в воде; уменьшение тенденции к гидратации
кристаллических солей; понижение растворимости в воде сульфатов и
галогенидов в этаноле; повышение в том же порядке термической устой-
чивости карбонатов; способность всех элементов подгруппы в отличие
от Zn, Cd, Hg давать нитриды M3N2 при непосредственном взаимодей-
ствии с азотом в условиях повышенных температур.
Первый член II группы элементов — бериллий — имеет совершенно
особые химические свойства. Бериллий образует преимущественно ко-
валентные соединения. У величение заряда ядра атома бериллия по срав-
нению с зарядом атома лития наряду с тем, что 25-электроны только
частично экранируют друг друга, приводит к уменьшению атомного
Радиуса бериллия (0,113 нм) по сравнению с атомным радиусом лития
(0,155 нм) и увеличению энергии ионизации бериллия (см. табл. 3.1),
п°этому полной потери обоих электронов с образованием иона Bez не
пРоисходит; связи ВеХ (даже Be—F) в значительной степени носят ко-
8алентный характер. В водных растворах ионы бериллия гидратируются
11 сидролизуются, образуя [Ве(НгО)4 Г и более сложные гидролизован
йЫе и полимеризованные частицы.
121
Таблица 3.1. Свойства Be, Mg, Са, Sr, Ва и Ra
Свойства Ве Mg Са ! sr Ва Ra 1
Атомный номер Z 4 12 20 38 56 88
Атомная масса 9,01 24,31 40,08 87,62 137,34 226,0
Плотность d, г/см 1,84 1,74 1,55 2,63 3,63 5,5
Температура плавления Тил, °C 1287 650 842 768 727 969
Температура кипения ТКип, °C Энергия ионизации, эВ: 2507 1095 1495 1390 1860 1500
Л 9,32 7,64 6,11 5,69 5,21 5,28
18,21 15,03 11,87 10,98 9,95 10,10
J3 153,85 78,20 51,21 43,60 37,10 (34)
Работа выхода электрона А, эВ 3,92 3,64 2,80 2,35 2,49 3,2*4
Атомный радиус Гат, нм 0,113 0,160 0,197 0,215 0,221 0,2351|
Ионный радиус г 2+ , нм 0,034 0,074 0,104 0,120 0,138 0,144,
Ионный потенциал Z/r, нм 64 31 20 18 15 14
Гидратное число иона М2+ Энтальпия гидратации иона — 13,2 12,0 10,7 7,7 —
М2 , —А//0) , кДж/моль Сечение захвата тепловых 2516 1955 1616 1447 1329 1297
нейтронов, барн Электродный потенциал 0,009 0,059 0,43 — — — 1
(расплавхлоридов), В Электродный потенциал -0,90 -1,59 — — — — 11
М2+-а0 + 2г-М, В Температура перехода -1,85 -2,36 -2,87 -2,89 -2,90 -2,92
в СП состояние Тс, К Теоретическая оценка 0,026 — — — — — 1
При движении по группе от Be к Ra закономерно увеличиваются
радиусы ионов и атомов и уменьшается величина ионного потенциала
(отношение z/ги), характеризующего поляризующую способность ка-
тиона. Поэтому для Mg , хотя в меньшей степени, чем для Ве2+, прояв-
ляется тенденция к образованию ковалентных связей. Остальные ионы
М являются менее поляризующими, поэтому соединения Са, Ba, Sr и
Ra по существу все ионные. Магний проявляет химические свойства,
промежуточные между свойствами бериллия и близких ему по свойст-
вам Са—Ra. Например, как и в случае бериллия, гидроксид магния
122
можно осадить из водных растворов, тогда как гидроксиды остальных
элементов заметно растворимы в воде.
Электронное строение атомов других элементов II группы подобно
строению бериллия, однако больший размер этих атомов уменьшает
влияние заряда на валентные электроны, поэтому их энергия ионизации
ниже, чем у бериллия (см. табл. 3.1). Они более электроположительны,
что подтверждается их высокой реакционной способностью в свободном
состоянии. Компактный бериллий окисляется кислородом воздуха толь-
ко при температуре выше 800 °C; при нормальных условиях он покрыт
оксидной пленкой. Магний также устойчив из-за тонкой оксидной плен-
ки, но при нагревании до 650 °C сгорает.
Щелочноземельные металлы (Са, Sr, Ва) на воздухе неустойчивы,
легко окисляются, покрываясь рыхлой пленкой, содержащей МО, МОг
и M3N2, которая не предохраняет их от дальнейшего окисления, поэто-
му их хранят под слоем керосина. Аналогичная зависимость в свойствах
элементов наблюдается при их взаимодействии с водой. На бериллий
вода не действует. Магний почти не взаимодействует с холодной водой,
но медленно выделяет из нее водород при кипячении. Щелочноземель-
ные металлы легко вытесняют водород из воды, причем при переходе от
Са к Ra энергичность взаимодействия увеличивается.
Еще одним отличием в свойствах элементов II группы является
различная склонность элементов к комплексообразованию. Бериллий
образует достаточно прочные комплексы, например, фторидные, аце-
тилацетонатные, термически устойчивый тетрамин; магний и особенно
кальций образуют весьма непрочные комплексы. Так, для магния изве-
стен комплекс с ацетилацетоном; для Са аддукты со спиртом и весьма
непрочные комплексы с аммиаком; Sr, Ва и Ra комплексов не образуют.
Они могут входить в состав комплексного соединения только в виде
внешнесферного катиона.
Глава 3. БЕРИЛЛИЙ
ХИМИЯ БЕРИЛЛИЯ
3.1. Физические и химические свойства бериллия
Бериллий — четвертый элемент Периодической системы Д.И. Мен-
делеева. Атомная масса 9,01, электронная конфигурация lsz2sz.
Большинство простых соединений бериллия имеют ковалентные
Связи, для образования которых необходимо разъединение его спарен-
нЫх 2s электронов, один из которых переходит на подуровень 2р. В
123
случае свободных мономерных молекул ВеА2 (например, газообразны^
галогенидов при высокой температуре) образуются две эквивалентны^
sp-гибридные орбитали, т.е. молекулы линейны, к.ч. равно 2. Атокщ
бериллия для образования связи имеют четыре энергетически устойчи
вые орбитали (2s и три 2р), поэтому они не могут образовывать более
четырех двухэлектронных связей и в валентном уровне никогда не могу7
иметь более восьми электронов (правило октета), поэтому максимами,
ное к.ч. у бериллия равно 4. Однако у бериллия имеется всего лишь два
электрона, поэтому он имеет сильную тенденцию использовать незаня-
тые орбитали.
Бериллий обычно достигает к.ч., равного 4, путем образования двух
донорных связей в дополнение к двум простым ковалентным связям.
Поэтому те же галогениды при более низкой температуре, а тем более в
твердом состоянии, полимерны, их координационная сфера — искажен-
ный тетраэдр, вытянутый вдоль оси цепи атомов Be. К.ч. 4 реализуется
в большинстве соединений бериллия, например эфиратах C12Be(OR2)2.
В ВеО существуют тетраэдры ВеО4- Необычное положение среди орто-
силикатов щелочноземельных металлов занимает Bc2SiC>4, в котором
атомы Be имеют тетраэдрическое окружение из атомов кислорода. Все
остальные ортосиликаты этой группы имеют структуру с октаэдриче-
ской конфигурацией окружения иона металла. Be с F часто дает соеди-
нения, изоморфные кислородным соединениям кремния. Так, BeF? изо-
морфен кристобалиту S1O2; BaBeF4 изоморфен BaSO4 и NaBeFa изомор-
фен CaSiCh. Известны также пять различных кристаллических форм
Na2BeF4 и Ca2SiO4- В комплексных соединениях бериллия образуются
четыре $р3-гибридные связи в значительной мере полярные, т.е. комп-
лексы в этом случае носят ионный характер, например ион BeF^ . I
По ряду свойств бериллий близок к цинку — элементу побочной
подгруппы, а также к алюминию — элементу III группы. Это вытекает
из близости вторых энергий ионизации Be и Zn (18,21 и 17,89 эВ) и таких
характеристик, как отношение заряда к радиусу иона (z/r) Ве2+ и АЖ
(64 и 60 нм 1). Так, Be и Zn образуют летучие соединения с карбоновыми
кислотами, что затрудняет их разделение, хотя комплексы цинка ®
отличие от соединений бериллия быстро гидролизуются водой.
Сходство в свойствах бериллия и алюминия является наилучше
примером «диагонального соотношения». Оксиды и гидроксиды как Бе
так и А1 являются амфотерными. Бериллий подобно А1 растворяется»
растворах сильных оснований, образуя тетрагидроксобериллат-ион I
Ве+2ОН-+2Н2О= [Ве(ОН)4]2+Н2. (3.1’
ВеО напоминает AI2O3 необычно высокой температурой плавлени»
и низкой летучестью, а также чрезвычайной твердостью. ПрироднЬ?
124
бериллий состоит из одного стабильного изотопа бериллия 9Ве, что от-
личает его от других четных элементов Периодической системы.
Бериллий — один из наиболее легких металлов и самый легкий из
устойчивых на воздухе. Это хрупкий твердый металл светло-серого
цвета с металлическим блеском. Бериллий, имея малую плотность, до-
статочно тугоплавок, что имеет практическое значение. Имеет две кри-
сталлические модификации: СИ-Ве — решетка гексагональная типа Mg и
В-Ве — решетка кубическая типа Ct-Fe. Температура перехода а —► /8
1277 °C.
Бериллий имеет высокие электро- и тепловые характеристики.
Электропроводность бериллия в зависимости от способа получения со-
ставляет 35—42 % от электропроводности меди; ниже 11 К он обладает
сверхпроводимостью. Из всех металлов он имеет самую высокую
энтальпию плавления. Его теплоемкость резко возрастает с повышени-
ем степени чистоты. Термический коэффициент расширения при 25—
100 °C примерно в два раза меньше, чем у алюминия и магния.
Бериллий отличается особыми ядерно-физическими свойствами,
обусловившими его применение в атомной технике. Для него характер-
на сравнительно небольшая энергия связи нейтронов (1,666 МэВ), что
позволяет использовать его в качестве источника нейтронов. Он облада-
ет необычайно малым сечением захвата тепловых нейтронов и высокой
способностью их рассеивания (поперечное сечение рассеивания 7,54
барн). Исключительно велика и способность бериллия пропускать рен-
тгеновские лучи (в 16—17 раз больше, чем у алюминия).
По удельной прочности (частное от деления величины любой проч-
ностной характеристики на удельную массу) бериллий превосходит все
конструкционные металлы и сплавы. Однако механические свойства
бериллия в значительной степени связаны со способом его изготовления,
в основном они определяются его гексагональной кристаллической ре-
шеткой, характеризующейся высокой степенью ориентации. Это приво-
дит к анизотропии механических свойств бериллия, что проявляется в
изделиях, полученных обработкой давлением. Так, предел прочности
при растяжении для выдавленного пруткового бериллия в продольном
Направлении в 1,5—2 раза выше, чем в поперечном.
Современные методы получения бериллия, такие, как литье и по-
рошковая металлургия, не позволяют полностью избежать анизотропии
ею свойств, но металл, полученный металлокерамическим методом от-
личается высокими механическими свойствами: высоким пределом
прочности при растяжении и большим модулем упругости (30000
Кг/мм2). Модуль упругости бериллия в четыре раза больше, чем у алю-
миния, и на 40 % больше, чем у стали, что в сочетании с легкостью
°еРйллия — качество весьма ценное. Высокая прочность сохраняется
fla*e при 800 °C. Бериллий — самый твердый элемент II группы, но он
125
недостаточно пластичен. Основной причиной хрупкости бериллия явля-
ются особенности его электронной структуры. Межатомная связь в ба-
зисной плоскости является в основном металлической, но вдоль оси с она
имеет большую ковалентную составляющую, определяющую склон-
ность бериллия к сколу.
Компактный бериллий в обычных условиях устойчив к окислению
хотя образования оксида велика. Это объясняется тем, что объи
оксида бериллия превышает объем металла, из которого он образуется.
Поэтому поверхностная оксидная пленка защищает металл от дальней
шего окисления. При комнатной температуре полированный берилл®
только слегка тускнеет. С повышением температуры активность метал-
ла повышается, но даже при 500 °C окисление ограничивается поверх-
ностным слоем; только выше 800 °C идет окисление в массе металла, а
при температуре 1200 °C он сгорает, образуя оксид бериллия ВеО. Ус-
тойчивость к окислению зависит от метода получения. Порошкообраз-
ный бериллий значительно активнее: при нагревании загорается не
только в атмосфере кислорода, но и на воздухе.
С водой бериллий практически не взаимодействует, довольно устой-
чив к действию кислот (если только он не тонко измельчен) вследствие
образования на его поверхности инертной и непроницаемой пленки
оксида, хотя величина электродного потенциала бериллия (см. табл.
3.1) должна свидетельствовать о быстрой реакции с разб вленными
растворами кислот (и даже с водой). Скорость реакции сильно зависит
от способа получения металла. В случае очень чистого металла относи-
тельные скорости растворения изменяются в следующем порядке: HF >
H2SO4 > НС1 > HNO3. Разбавленная HNO3 в обычных условиях, а также
концентрированная при нагревании, взаимодействуя с бериллием, вы-
деляют оксиды азота и аммиак. Концентрированная серная кислота
восстанавливается бериллием при нагревании до SO2 и даже до H2S. 1
Бериллий растворяется не только в кислотах, но и в щелочах, обра-
зуя комплексы анионной) типа (3.1). Взаимодействует с расплавами
щелочей и карбонатов, образуя бериллаты
2NaOH + Be = Na2BeO2 + Н2;
Na2CO3 + Be=Na2BeO2 + CO. О2’
Растворяется в водном растворе гидродифторида аммония
2NH4HF2 + Be = (NH4)2BeF4 + Н2.
Эта реакция применяется для извлечения бериллия из шлака. I
Бериллий восстанавливает до металлов оксиды магния, бария, адН’"
миния и титана; оксид кальция в аналогичных условиях не восстань'
ливается.
126
Галогены взаимодействуют с бериллием лишь при нагревании, об-
разуя галогениды ВеХг. Исключением является фтор, реагирующий с
бериллием на холоду.
При нагревании порошкообразного бериллия в атмосфере азота при
500 °C образуется нитрид ВезЫг. В такой же атмосфере компактный
металл образует нитрид только выше 900 °C. Частично взаимодействует
с азотом и при нагревании на воздухе. Нитрид образуется также при
взаимодействии порошкообразного бериллия с аммиаком (1000 °C) и с
цианом (800 °C).
При взаимодействии расплавленного Be с углеродом образуется кар-
бид ВегС. Имеются сведения о получении дикарбида бериллия ВеСг —
продукта взаимодействия Be с С2Н2 при 400 °C.
С серой, фосфором и мышьяком образует сульфиды, фосфиды и
арсениды. Силицидов не образует, что отличает бериллий от других
элементов подгруппы. С водородом непосредственно не реагирует даже
при нагревании до 1300 °C. Гидрид ВеНг образуется при разложении
выше 200 °C некоторых бериллийорганических соединений.
В расплавленном состоянии бериллий растворяет почти все метал-
лы. Однако до температуры 600 °C он устойчив в расплавленных литии,
натрии и калии, при более высокой температуре он растворяется в ще-
лочных металлах за счет окисления кислородом, присутствующим в
них.
3.2. Оксиды и гидроксид бериллия
Оксид бериллия ВеО получают при прокаливании Ве(ОН) 2 или тер-
мическом разложении нитрата, оксиацетата, гидроксикарбоната и др.
при температуре от 500 до 1000 °C в зависимости от исходного соедине-
ния. Оксид бериллия, полученный прокаливанием солей или гидрокси-
да, представляет собой аморфный порошок. В виде кристаллов может
быть получен различными методами, связанными с нагреванием до вы-
сокой температуры, в частности при кристаллизации из расплавленных
карбонатов щелочных металлов. Оксид бериллия имеет гексагональную
кристаллическую решетку типа цинкита, при 2050 °C переходит в тет-
рагональную модификацию. Ниже приведены физические константы
оксида бериллия:
Тпл, ’С........................................~ 2550
Гкип, ’С................................4000 ± 200
Плотность d, г/см^.......................3,01—3,025
—5 —4
Растворимость в воде при 25 °C, г/л......5-10 —2• 10
кДж/моль........................... 599,6
Давление пара при 2000 °C, Па...............8,4- 10
127
Коэффициент теплопроводности (образец с d- 2,87 г/см3), Вт/(м • °C):
50 "С...................250
1300 "С..................18
Коэффициент линейного расширения, 1/ ‘С:
50 “С....................5,0 ю"6
2000 *С.................10,9- 10^
Как видно из приведенных данных, оксид бериллия обладает высо-
кой теплопроводностью и незначительным термическим расширением.
Сочетание этих свойств делает его превосходным термостойким матери-
алом.
Реакционная способность ВеО зависит от способа получения и осо-
бенно от температуры прокаливания. Повышение температуры прока-
ливания ведет к увеличению размера зерен, т.е. к уменьшению удель-
ной поверхности, а следовательно, и к уменьшению химической актив-
ности. По своим химическим свойствам оксид бериллия занимает про-
межуточное положение между оксидами алюминия и магния Хими-
ческая устойчивость ВеО превосходит химическую устойчивость боль-
шинства оксидов металлов.
В воде оксид бериллия почти не растворим. Слабо прокаленный (не
выше 500 °C) ВеО растворяется в кислотах, даже разбавленных, и в
водных растворах щелочей, образуя соответствующие соли и тетрагвд-
роксобериллаты. ВеО, прокаленный при 1200—1300 °C, еще может рас-
творяться в минеральных кислотах, прокаленный при 1800 °C, или после
плавления растворим только в плавиковой кислоте. Газообразные гало-
гениды водорода не реагируют с ним даже при сильном нагревании.
Растворяется в расплавленных щелочах, карбонатах и пиросульфатах
щелочных металлов. Оксид бериллия взаимодействует с гексафторсили-
катом NaiSiFe и гексафторферратом натрия NaiFeFe, что используется
при регенерации урана и бериллия из отходов производства или облу-
ченного ядерного топлива.
Оксид бериллия при нагревании взаимодействует с целым рядом
хлористых соединений (хлором в присутствии углерода, фосгеном, тет-
рахлоридом углерода и др.). Например, реакция с фосгеном начинается
при 500 °C
ВеО+COCh=BeCh+СОг. (3.3)
Взаимодействие с бромом проходит труднее, сведений о взаимодей-
ствии с иодом нет. В связи с большим сродством бериллия к кислороДУ
ВеО восстанавливается далеко не всеми обычно применяемыми восста-
новителями. Кальций может быть использован в качестве восстанови
теля при температуре ниже 1700 °C и атмосферном давлении. По дан-
ным некоторых авторов, для этой цели может быть применен и тита®
(при 1,33 10 2 Па и 1400 °C). Но металл в обоих случаях получается
128
загрязненным, так как разделить продукты реакции технологически
чрезвычайно трудно.
Давление пара ВеО незначительно. В отсутствие паров воды это
наименее летучий из тугоплавких оксидов. Примесь таких оксидов, как
MgO, СаО, AI2O3, SiO?, еще больше понижает летучесть ВеО из-за
химического взаимодействия между ними. В присутствии паров воды
при 1000—1800 °C летучесть сильно возрастает в связи с образованием
газообразного гидроксида
ВеО(тв) + НгОц) = Ве(ОН)г(г).
Оксид бериллия устойчив до 800 °C по отношению к расплавленным
щелочным металлам — литию, натрию и калию — и почти совсем не
реагирует с Се, Pt, Mo, Ga, Ni и Fe, только при 1800 °C взаимодействует
с Nb, Si, Ti, Zr и Th.
Оксид бериллия (I) ВегО. При исследовании процесса испарения
бериллия в высоком вакууме было установлено, что при температуре
1400 °C в вакууме идет реакция
Be + ВеО = ВегО, = + 41 кДж/моль,
продукт которой — оксид бериллия (I) — устойчив при нормальных
условиях. Оксид бериллия (I) обладает большей летучестью, чем оксид
бериллия (II) и является одним из источников загрязнения бериллия
кислородом при дистилляции в тиглях из ВеО. Оксид бериллия (I) был
обнаружен с помощью масс-спектрометра. Существование соединения
ВегО было подтверждено также рентгенографически. При нагреве бе-
риллия в вакууме (т.е. в условиях недостатка кислорода) на поверхности
металла образуется оксидная пленка. Количество ВегО в пленке возра-
стает с повышением температуры.
Гидроксид бериллия Be (ОН) г — типично амфотерное соединение,
мало растворимое в воде (2 10~3 r/л при 25 °C). Известен в виде трех
модификаций: аморфной, метастабильной а-формы и кристаллической
/3-формы. Аморфный гидроксид Be (ОН) г хНгО получается действием
растворов щелочей и аммиака на растворы солей бериллия. Это белый
студенистый осадок, содержащий до 95 % неконституционной воды.
Осаждение гидроксида бериллия из растворов солей, за исключением
Фторидов, происходит при pH 6, что близко к условиям выделения
А1(ОН)з (pH 5) и Fe(OH)3 (pH 3). Поэтому разделение алюминия,
железа и бериллия дробным осаждением их гидроксидов неэффективно.
Из растворов фторида бериллия и особенно фторобериллатов гидроксид
бериллия осаждается при pH 11, что позволяет из таких растворов отде-
лять Be от Fe и А1 дробным осаждением их гидроксидов.
Свежеосажденный аморфный гидроксид хорошо растворим в кисло-
тах и щелочах. В отличие от гидроксидов алюминия и железа растворя-
ется в растворах карбоната аммония и гидрокарбонатов щелочных эле-
5-69! 129
ментов, а также в растворах ряда солей бериллия (фторида, сульфата и
др.), образуя комплексные соединения. При поглощении из воздуха COj
образуется гидроксокарбонат Ве4(ОН)бСОз. Благодаря большой удель-
ной поверхности аморфный гидроксид может сорбировать из растворов
различные примеси, в том числе соли аммония и щелочных металлов.1
а-Ве(ОН)2 (плотность 1,920 г/см3) образуется в результате старе-
ния аморфного гидроксида при хранении на воздухе и в воде. Менее
активен, на холоду медленно растворяется в щелочах и кислотах, при
нагревании растворимость улучшается. Не сорбирует примеси и не по-
глотает диоксид углерода и аммиак; /?-Ве(ОН)2 (плотность 1,9244
г/см"5) выделяется из растворов бериллатов при кипячении и введении
затравки гидроксида.
Кристаллическая структура Д-Ве(ОН)2 подобна структуре
Zn(OH)2; группы ОН в ячейке тетраэдрически расположены вокруг
катиона Bez , что отличает гидроксиды бериллия и цинка от большин-
ства кристаллических гидроксидов, имеющих слоистую структуру. Это
связано с высоким ионным потенциалом Ве2+, вследствие чего образу-
ется структура с более плотной упаковкой, чем слоистая. Подобно а-
форме/J-гидроксид бериллия трудно растворяется в кислотах и щелочах,
не поглощает СОг и NH3 и почти не сорбирует примеси из растворов, что
имеет существенное значение в технологии.
Аморфный гидроксид бериллия с трудом обезвоживается. При
100 °C даже после 2 ч высушивания удаляется не более 50 % влаги. Лишь
повышение температуры до 180 °C позволяет получить безводный
Be (ОН) 2. Кристаллическая jS-форма начинает разлагаться до оксида
выше 200 °C, но получить безводный оксид удается лишь прокаливанием
при 800—900 °C.
Состояние бериллия в водных растворах. В концентрированных
кислых растворах бериллий, по-видимому, существует в виде
[Ве(Н2О)4Г -ионов, и эти ионы встречаются в некоторых солях. Вода»
таких солях связана довольно прочно: [Ве(Н2О)4]С12, например, не
теряет воду над Р2О5.
Растворы всех бериллиевых солей имеют кислую реакцию вследст-
вие гидролиза. Они могут растворять значительные количества оксида
или гидроксида бериллия из-за образования бериллием оксо- и гидро-
ксокомплексов с Be—О—Be и Be—ОН—Be мостиками. При осаждении
Be (ОН) 2 растворы остаются прозрачными, если n-число ионов ОН I
добавленных в расчете на один ион Ве2+, меньше 1. Осаждение начина-
ется при п > 1. В случае солей галогеноводородных кислот полное осаж-
дение достигается при п = 2, а с остальными ионами полное осаждение
осуществляется при п = 1,8—1,9, что свидетельствует о том, что осадки
«гидроксида» бериллия содержат SO4 , NO3 или С1О4-ионы.
130
Тот факт, что осаждение происходит только тогда,
позволяет утверждать, что существуют раствориц. огда п > 1 >
гг ОЛ Че частицы
[Ве(ОН) ]п . Наличие таких частиц с п = 3 было подтве^
циометрическим титрованием разбавленных растворов ( по^ен~
Была оценена константа равновесия их образования моль/л).
ЗВе2+ + ЗНгО = [Ве(ОН)]з+ + ЗН+, рК = - 8,66.
Бериллий преимущественно образует соединения
/„тт\,з+ . 4 к.ч. 4; так,
[Ве(ОН ) имеет структурную формулу
н
О
Ве(0Нг)2
(Н2О)гВе
О— Н
Ве(ОНг)2
н
Для очень низких концентраций бериллия известны
ные о существовании частиц ВегОН +иВе(ОН)2. ыед
В разбавленных щелочных Р^ворах образу-
ется полиатомныи ион типа [Веп(ОН) 2n+m J • повыщ^ концен-
трации щелочи полиатомный ион постепенно превращу
гидроксобериллат-ион [Ве(ОН)4 ] , который затем, отщ^^ водуТоб-
разует ион BeOj. Практически полное превращение 1ц
соотношении Na2O: ВеО=8:1. Тетрагидроксобериллат-ij
но устойчивее полиатомного, поэтому при высокой конц„
лочи даже кипячение не приводит к разрушению тетрад пил-
лат-ионов и выделению гидроксида бериллия в осадок.
Бериллаты. В общем случае это соединения, в кото^^ бериллий
входит в состав комплексного аниона, являясь компле^^ _
лем. Если лигандами являются атомы кислорода, соедин^^ называют
просто бериллатами. Бериллаты образуются в различи^
результате высокотемпературного синтеза при взаимоде^^^^^8^1^’ &
бериллия с щелочными металлами или их оксидами полу^ ви^ ок
ты: М2ВеО2, М4ВеО3, МгВегОз, Na6Be2O5 и Rb2Be3O4. ‘
ВеДена реакция между расплавом щелочи и бериллием ппико-
Цящая к образованию бериллата Na2BeC>2. При раствоц» • ’
'Мроксида бериллия в щелочном растворе получается те^рагидроксо^е_
Риллат натрия
2NaOH + Ве(ОН)2 = Na2 [Ве(ОН)41-
5»
131
Бериллаты менее устойчивы к гидролизу, чем алюминаты. При гид-
ролизе образуется гидроксид бериллия
Na2BeO2 + 2Н2О = Ве(ОН)2 + 2NaOH;
Na2Be2O3 + ЗН2О = 2Ве(ОН)2 + 2NaOH.
На разной устойчивости бериллатов и алюминатов основан один из
методов разделения бериллия и алюминия.
3.3. Соли кислородсодержащих кислот
Сульфат бериллия BeSCU "4Н2О — четырехводный гидрат, устойчи-
вый на воздухе. Выделяется из растворов, полученных обработкой окси-
да или гидроксида бериллия разбавленной серной кислотой, после их
упаривания и охлаждения. Кристаллы сульфата псевдокубические,
плотность их 1,713 г/см3.
Безводный сульфат бериллия получают термическим обезвожива-
нием четырехводного гидрата при 400 °C. Процесс протекает путем,
образования ряда промежуточных гидратов при 115, 200 и 250 °C. Суль-
фат бериллия устойчив до температуры 550 °C, при которой он начинает
разлагаться, выделяя БОз- Полное разложение наступает при 1031 °C.
Метод термической диссоциации сульфата используют в промышленно-
сти для получения оксида бериллия. Следует отметить, что скорость
разложения сульфата бериллия значительно меньше, чем сульфата
алюминия (при 750 °C давление пара SO3 над BeSCU 4,87 кПа, над
А12 (SO4) з — 12 кПа). Это можно использовать для частичного их разде-
ления.
Растворимость сульфата бериллия в воде при 25 °C 42,4 г/ 100г Н20,
при 100 °C — 100 г/ 100г Н2О. На уменьшении растворимости с пониже-
нием температуры основана его очистка перекристаллизацией. Серная
кислота «высаливает» сульфат бериллия из растворов, при концентра-
ции серной кислоты 66 % растворимость сульфата уменьшается прибли-
зительно до 0,9 %. С повышением концентрации H2SO4 уменьшается и
содержание воды в осадке. BeSC>4 в отличие от Al2 (SO4) 3, MgSCU раство-
ряется в присутствии (NH4)2SO4 (рис. 3.1).
Сопоставление кривых растворимости позволяет сделать вывод о
возможности использования сульфатов для отделения бериллия от маг-
-р
ния и алюминия. Даже при незначительной концентрации ионов NH4
образуются труднорастворимые двойные соли с магнием и алюминием,
в то время как двойные соли бериллия с аммонием хорошо растворимы-
Это следует из ветвей кристаллизации Е\ Е2 соответствующих двойных
солей. Поэтому растворимость BeS04 (29 % при 25 °C) практически не
меняется вплоть до концентрации (NH4)2SO4 25 % (ветвь кристаллиза-
ции АЕ\). В присутствии сульфата бериллия растворимость алюмоаМ-
132
РЛС. 3.1. Кривые растворимости в суль-
фатных системах BeSO4—(NH4)2SO4—
S2O (1); MgSO4—(NH4)2SO4—Н2О
(2), A12(SO4)3—(NH4)2SO4—H2O (3):
1: AE1 — ветвь кристаллизации
BeSO4'4H2O, £1£2 — ветвь кристалли-
зации BeSO4'(NH4)2SO4‘6H2O; E2B
_ ветвь кристаллизации (NH412SO4; 2:
jig] — ветвь кристаллизации
MgSO4 ‘7Н2О; U1-E2 — ветвь кристалли-
зации MgSO4‘(NH4)2SO4'6H2O; Е2В
_ ветвь кристаллизации (NH4J2SO4; 3:
— ветвь кристаллизации
AJ2<SO4)3 "18Н2О; Е1Е2 — ветвь кри-
сталлизации A12<SO4)3 '(NH4)2SO4‘
24Н2О; Е2В — ветвь кристаллизации
(NH4)2SO4
монийных квасцов еще более уменьшается, что увеличивает эффект
разделения элементов. Способ отделения алюминия от бериллия в виде
алюмоаммонийных квасцов один из самых надежных.
Карбонат бериллия. Известны карбонат ВеСОз‘4НгО и гидроксо-
карбонаты различного состава. Плохо растворимый Ве(ОН)гСОз обра-
зуется при взаимодействии водных растворов карбонатов щелочных
металлов или карбоната аммония с солями бериллия. Из кипящих рас-
творов выделяются в виде плотного осадка гидратированные гидроксо-
карбонаты переменного состава: Ве<п+1> (ОН)2пСОз 'шНгО, где п = 2, 3,
4,5, 7; т -1,2, 3.
В избытке карбонатов щелочных металлов и карбоната аммония
гидроксокарбонаты и гидроксид бериллия образуют растворимые комп-
Ве(ОН) 2 + 2 (NH4) 2СО3 - (NH4) 2 [Be (СОз) 2 ] + 2NH4OH.
еакция представляет большой интерес в связи с возможностью ее
ользования для отделения бериллия от алюминия и железа, так как
роксиды этих элементов малорастворимы в карбонате аммония.
Карбонат бериллия образуется из тех же растворов, что и гидроксо-
бонат, но в присутствии большого избытка СОг- Чтобы выделить
бонат, раствор упаривают в атмосфере СОг. Несколько выше 100 °C
бонат начинает разлагаться, выделяя СОг.
3.4. Соли органических кислот
Оксалат бериллия. В виде тригидрата ВеСгО4 ’ЗНгО получается при
ривании раствора гидроксокарбоната с небольшим избытком щаве-
ой кислоты. Эта соль интересна тем, что она единственная из окса-
133
латов металлов в степени окисления +2 обладает значительной раство-
римостью (при 25 °C 24,85 % в расчете на безводный оксалат). Отмечена
относительно низкая электропроводность ее растворов, равная одной
четвертой электропроводности эквивалентного раствора сульфата бе-
риллия. Это объясняется комплексообразованием бериллия в растворе,
где он частично находится в виде комплекса [Ве(СгО4)2]2 . При нагре
вании тригидрат неустойчив и около 50 °C переходит в моногидрат,
который выше 225 °C разлагается до оксида. Термическое разложение
оксалата предложено использовать как одну из стадий получения оксида
бериллия особой чистоты.
Оксиацетат бериллия Ве4О(СНзСОО)б может быть выделен из
раствора гидроксида бериллия в концентрированной уксусной кислоте
после его охлаждения. В обычных условиях кристаллизуется в кубиче-
ской модификации, которая при температуре около 148 °C переходите
высокотемпературную моноклинную. Горячей водой гидролизуется, об-
разуя ацетаты переменного состава. В минеральных кислотах, за иск-
лючением соляной, растворяется, как и другие аналогичные соедине-
ния, разлагаясь. Растворяется в ацетоне, хлороформе, нитробензоле;
почти не растворяется в эфире и мало — в этиловом спирте.
Подобно всем органическим производным бериллия оксиацетат —
ковалентное соединение. Плавится и кипит при низкой температуре
(Тпл = 283 °C, Ткип = 325 °C), возгоняется почти без разложения. В
вакууме может быть перегнан при температуре плавления. При прока-
ливании образуется оксид бериллия. Подобное летучее соединение есть
лишь у цинка, поэтому возгонкой оксиацетата бериллия и последую-
щим его разложением можно получить оксид бериллия высокой степени
чистоты.
Комплексонат бериллия. Нормальные комплексонаты элементов П
группы образуются в водных растворах в широком диапазоне pH: от 4 до
12. По мере увеличения ионного радиуса устойчивость комплексов из-
меняется не монотонно: Са2+ > Ве2+ > Mg + ~ Sr2+ > Ва2+ > Ra2+. При
переходе от кислых растворов к нейтральным последовательно образу-
ются комплексонаты [ВеНгМ, [ВеНЬ] и [BeL]2-. Последний при pH
6—8 претерпевает оксоляцию, заканчивающуюся полным разрушени-
ем при pH 8 с образованием Be (ОН) 2. При попытке выделения нормаль-
ного комплексоната упариванием комплекс [BeL ]2 в твердую фазу не
переходит, вместо него из раствора выпадает гидроксид бериллия. В
твердой фазе удается выделить только протонированные комплексона-
ты следующего состава: KBeHL’2H2O и BeH2L'2H2O. Таким образом,
ЭДТА (H4L) образует с бериллием комплекс, значительно менее устой-
чивый (особенно в нейтральной и щелочной средах), чем соответствуй3'
щие комплексы с рядом элементов, сопутствующих бериллию (Al, Fe,
Са и др.). Это свойство комплекса используется в технологии и аналИ'
134
т11ческой практике для осаждения бериллия в присутствии ЭДТА в виде
рлдроксида из растворов, содержащих алюминий и железо.
3.5. Галогениды бериллия
Свойства фторида бериллия отличаются от свойств остальных его
галогенидов. Это различие определяется разным характером связи бе-
риллий—галоген. Связь Be—F на 80 % носит ионный характер, связи
Be с другими галогенами по преимуществу ковалентные. Ионные со-
ставляющие связей Be—Cl, Be—Вг и Be—I соответственно достигают
только 42; 35 и 25 %. Этим объясняется устойчивость чисто фторидных
комплексов бериллия и способность хлоридов, бромидов и иодидов (но
не фторидов) образовывать стабильные комплексы с нейтральными ли-
гандами.
Фторид бериллия ВеРг — бесцветное гигроскопичное вещество.
Расплавленный, он при застывании образует стекловидную массу. Это
объясняется тем, что по структуре ВеРг аналогичен SiOi. Может быть
получен упариванием раствора Be (ОН) 2 в плавиковой кислоте и даль-
нейшим высушиванием в токе HF. Другой способ получения безводного
фторида бериллия — фторирование газообразным фтором, а также гид-
рофторирование оксифторида или карбида бериллия
ВегОгРю + 4HF = 7ВеРг + 2НгО,
ВегС + 2Рг = 2ВеРг + С.
В промышленности пользуются термическим разложением тетраф-
торобериллата аммония. Разложение (МН4)гВеР4 с достаточной скоро-
стью протекает при 400 °C, но получающийся продукт — тонкий поро-
шок, легко гидролизуется. Поэтому практически применяют высоко-
температурное разложение (900 °C)
(NH4)2BeF4 = BeF2 + 2NH4F.
Фторид бериллия — одно из самых устойчивых его соединений (эн-
тальпия образования из элементов -1009 кДж/моль), вследствие чего
не все применяемые на практике восстановители восстанавливают его
До металла, в частности ВеРг нельзя восстановить водородом.
Фторид бериллия хорошо растворим в воде (табл. 3.2), частично при
этом гидролизуясь. В растворе фторид бериллия находится главным
образом в виде комплекса. Это подтверждается низкой электропровод-
ностью и малой криоскопической константой раствора ВеРг.
Если упарить раствор и сухой остаток прокалить, то образуется
°ксифторид бериллия ВеуОгРю — пушистый белый порошок. В рас-
плавленном состоянии фторид и оксифторид бериллия ничтожно элек-
тропроводны. Подобно фториду бериллия оксифторид хорошо растворя-
ется в воде.
ВеРг не растворяется в плавиковой кислоте, но со фторидами щелсж
ных металлов и аммония образует растворимые в воде комплексные сол>
— фторобериллаты.
Фторобериллаты. В зависимости от соотношения F: Be и знамени,
pH в водных растворах могут присутствовать различные фтороберцц
латные комплексные ионы: BeF+, ВеРз и BeF^. Эти ионы гидратирова-
ны, например ион BeF+ имеет вид [ВеР(НгО)з]+ в соответствии с к.ч
бериллия 4. Комплексообразование усиливается в избытке F~ и, в част-
ности, в растворах фторидов щелочных и щелочноземельных металле^
Образующиеся в растворах комплексные анионы дают с катионами со-
ответствующие соли. Известны, например, фторобериллаты N^BeFj
M2BeF4 , MjBeFs , MHBeF4. На использовании фторобериллатов осно
ваны вскрытие бериллиевых руд фторидными методами и металлотер-
мическое получение бериллия. В связи с этим несомненный интерес
представляют данные о плавкости в системе ВеРг—MgFz (рис. 3.2). В
расплавленном состоянии ВеРг и MgF2 неограниченно взаимно раство-
римы, но они не образуют ни соединений, ни твердых растворов, что
позволяет получить чистый металлический бериллий.
Наибольшую роль в технологии играют соли типа M2BeF4 , очень
близкие по структуре к силикатам, так как соединения типа М!ВеРз
устойчивы лишь в определенных условиях и растворяются инконгруэн-
тно, образуя МгВеР4 - Для всех тетрафторобериллатов, за исключением
ЫгВер4 и Na2BeF4, характерен изоморфизм с сульфатами, так как ионы
2- 2—
Ве?4 и SO4 кристаллохимически близки. Это подтверждается образо-
ванием непрерывного ряда твердых растворов в системе (NH4>2SO4—
(NH4)2BeF4—Н2О. В отли-
чие от ВеРг все тетрафторо
бериллаты в расплавления1
состоянии электропровод-
ки. Электропроводны и рас
творы тетрафтороберилла-
тов, чем они также отлича-
ются от растворов BeF2
Na2BeF4 можно получить,
упаривая раствор с соотнО"
шением NaF: ВеРг = 2:1.0я
растворяется в воде конгрГ
энтно. Растворимость устой'
чивой ромбической у-форМ^
при 20 °C приведена в табл-
3.2.
Рис. 3.2. Диаграмма плавкости системы BeF2—
MgF2
136
Таблица 3.2. Свойства фторидов бериллия
Свойство BeF2 Na2BeF4 NH4BeF3 (NH4)2BeF4
Тпл> ’С 800 578 — 230*)
Укип, ’С 1327 — — —
-Л#298(у) > кДж/моль 1009 — — —
Растворимость в воде, г/100г Н2О Температура разложения В граммах на 1 л ~84б’ 0,26 32 54
Тетрафторобериллаты натрия используют в технологии для отделе-
ния от алюминия, комплексные фториды которого растворимы еще
меньше. В присутствии фторида натрия растворимость y-Na2BeF4 зна-
чительно уменьшается, эвтонический раствор в системе NaF—
Na2BeF4—Н2О содержит лишь 0,19 % тетрафторобериллата натрия.
Фторид бериллия, напротив, увеличивает растворимость Na2BeF4.
Фторобериллаты аммония также получают упариванием растворов,
содержащих фториды аммония и бериллия в соответствующих соотно-
шениях. Известны два фторобериллата аммония: (NH4)2BeF4 и
NH4BeF3. Оба хорошо растворяются в воде (см. табл. 3.3). При нагрева-
нии тетрафторобериллат аммония разлагается по реакции
230 270 °C
(NH4)2BeF4 • NH4BeF3---------------• BeF2(+NH4F).
Хлорид бериллия — белые или слегка зеленоватые игольчатые кри-
сталлы, расплывающиеся на воздухе из-за сильной гигроскопичности;
плотность 1,9 г/см3. ВеС12 можно получить хлорированием ВеО различ-
ными хлорирующими агентами. Наиболее часто используется реакция
между оксидом бериллия и хлором в присутствии угля, идущая при 800
С. При 500 и 450 °C идет хлорирование соответственно фосгеном (3.3)
и тетрахлоридом углерода. Температура плавления BeCh 415 °C, тем-
пература кипения 550 °C. Но уже при 400 °C начинается сублимация с
образованием волокнистых кристаллов полимерной структуры, относя-
щейся к ромбической сингонии. Кроме нее существуют еще три моди-
фикации ВеС12, из которых наиболее стабильна)?-форма, относящаяся
Такжек ромбической сингонии. Полиморфизм хлорида бериллия связан
с в°зможностью различной упаковки тетраэдров [ВеСН ]. В парах при
Температуре от 500 до 600 °C хлорид существует в виде димера ВегС14,
^я которого предложена следующая структурная формула
С1
Cl —Be Be—Cl
^^~С1—""
137
Рис. 3.3. Зависимость давления пара S1C14
(/), А12С16 (2), FeC13 (3), ВеС12 (*) от
температуры (х — точки кипения)
Повышение температуры ве
дет к диссоциации, т.е. к образова
нию линейных мономеров BeCh
Летучесть хлорида берилл^
ниже, чем хлоридов алюминия
железа и кремния (рис. 3.3). Эт
используется для отделения бе
риллия от указанных элементов
при хлорировании берилла.
Хлорид бериллия — менее
прочное соединение, чем его фто.
рид (д//^98^ ~ 471,4 кДж/моль),
но и он не восстанавливается водо-
родом. Как и фторид, восстано-
вить его можно такими металлами, как натрий, магний, кальций.
В воде BeCh легко растворяется, выделяя значительное количество
тепла. Склонен к гидролизу, но в меньшей степени, чем FeCh и А1С1з
Степень гидролиза зависит от pH раствора. Присутствие соляной кисло-
ты подавляет гидролиз. В концентрированных солянокислых растворах
образует с НС1 катионные комплексы, например [BeCl ]+ (IgX - -0,66
где К — константа устойчивости), но тем не менее бериллий мало скло-
нен к образованию хлоридных комплексов. Подтверждением этому слу-
жит хорошая электропроводность растворов BeCh (в отличие от раство-
ров фторида бериллия).
Из водных растворов хлорид бериллия выделяется в виде устойчи
вого гидрата ВеС1г 4НгО. При нагревании тетрагидрата удаляется часть
соляной кислоты и образуются труднорастворимые в воде гидроксосоля
переменного состава. Гидроксосоли выделяются также при упаривании
водных растворов хлорида бериллия. BeCh хорошо растворим в спиртах
— метиловом, этиловом, амиловом.
В отличие от ВеРг хлорид бериллия склонен образовывать комплек'
сы с нейтральными аддендами. Так, известны четыре аммиаката хлори
да бериллия: BeCh 12NH3, BeCh 6NH3, BeCh 4NFh, BeCh 2NH3
причем низшие аммиакаты достаточно устойчивы. Аналогичные комП'
лексы получены со многими органическими аддендами (пиридином,
ацетоном, эфиром, нитрилами и др.).
3.6. Сплавы и интерметаллические соединения бериллия
Бериллий не образует обширных областей твердых растворов в двоЙ'
ных сплавах ни с одним из изученных металлов. В твердом состояни*’
все металлы имеют ограниченную растворимость в бериллии. ТоДь1СС
138
,ледь, никель, кобальт и серебро растворяются в бериллии более 1 %
(ат.)- Низкая способность бериллия образовывать растворы замещения
обусловлена обширным электронным перекрытием вдоль базисной пло-
скости. Внедренные атомы также плохо растворяются в бериллии. Меж-
доузельные пустоты ГПУ структуры аналогичны пустотам ГЦК струк-
туры. Диаметр тетраэдрических пустот (0,025 нм) недостаточен для
размещения предполагаемых атомов внедрения. Бериллий и сам плохо
растворим в других металлах. Наиболее растворим бериллий (%) в Си
2,75; Ni 2,7; Сг 1,7; растворимость сильно уменьшается с понижением
температуры, в результате чего сплавы, содержащие бериллий, способ-
ны к дисперсионному твердению.
Для большинства бериллиевых систем энергия образования сплава
отрицательна. Другими словами, существует определенная вероятность
образования соединения. Действительно, большинство металлов обра-
зуют с бериллием двойные интерметаллические фазы. Известны соеди-
нения с металлами — представителями всех групп Периодической сис-
темы, кроме щелочных (за исключением калия). Щелочноземельные
металлы образуют фазы типа МВщз- Переходные металлы образуют
один или несколько бериллидов.
Возможность образования той или иной фазы определяется в основ-
ном соотношением атомных радиусов г. При г больше 1,33 образуется
только фаза MBei3, а при меньшем отношении радиусов — фаза MBei2-
Фазы МВе?2 будут появляться при отношении радиусов до 1,225, а
MzBei 7 — при значениях г, лежащих в области 1,301—1,455. С переход-
ными металлами бериллий всегда (исключением является калий) обра-
зует фазы Лавеса, причем их тип определяется объемом атома элемента
И. При малом объеме атома М возникает структура типа MgZn2. При
промежуточных значениях объема атома формируется структура типа
MgCu. Большой объем атома М способствует образованию соединения
А1В2.
Интерметаллические соединения с точкой плавления около 1500 “С
были обнаружены в двойных системах бериллия с кобальтом, железом
и никелем, а интерметаллические соединения с температурой плавле-
ния 1600—2000 °C — в двойных системах с ураном, цирконием, торием,
Молибденом, вольфрамом и бором. Интерметаллические соединения со-
става MBei 3, где М — U, Th, Се и Zr, имеют решетку гранецентрирован-
Пого куба; они весьма тугоплавки. Так, температура плавления UBei3
составляет 2000 °C, ZrBei3 1600 °C и ThBei3 1700—1800 °C.
Для получения интерметаллических соединений бериллия с цирко-
вом и молибденом применяется холодное и горячее прессование смесей
п°Рошков, содержащих до 10 % указанных металлов. Лучшие резуль-
Таты при получении соединений были достигнуты плавлением или вза-
имодействием порошков с последующим повторным прессованием
139
смеси интерметаллического соединения с основным металлом, приме
няемым для изготовления керметов.
ТЕХНОЛОГИЯ БЕРИЛЛИЯ
3.7. Важнейшие области применения бериллия
и его соединений
Для бериллия характерен значительный разрыв между временем ел
открытия Л. Вокеленом в 1798 г. и началом широкого промышленное
применения в 30-х годах текущего столетия. Причина тому — трудно-
сти, связанные не только с переработкой бериллиевого сырья, но и сс
сложностью получения чистого металла, с его химической активностью
особенно большим сродством к газам, в первую очередь — к кислород)
и азоту. Отсутствие чистого бериллия как объекта исследования не по-
зволяло долгое время оценить его замечательные свойства, а следова-
тельно, и с наибольшей полнотой определить области его применения
Долгое время применение бериллия было связано лишь с использовани-
ем свойств его оксида, употреблявшегося для изготовления огнеупорных
изделий, высококачественного фарфора для электроизоляторов, азока-
лильных колпачков и специальных стекол.
В настоящее время бериллий стал серьезным конкурентом для обыч
но применяемых авиационных материалов. Облегченные конструкции
самолетов с применением бериллия увеличивают дальность и потолок
полетов. Он отличается малой удельной массой (1,84 г/см3), к ысокой
прочностью, высоким модулем упругости (в 1,5 раза большим, чем
стали, и в 2,5 раза, чем у титана), высокими жесткостью, теплопровод-
ностью, удельной теплоемкостью (в 2,5 раза выше, чем у алюминия,в
в 8 раз, чем у стали), хорошей коррозионной стойкостью, способность^
сохранять точность и стабильность размеров деталей, выдерживать
большие температуры. В американском «Шатле» из бериллия выпалке
ны каркас, обшивка, диски тормозов, приборы.
Несмотря на высокую стоимость, бериллий стараются использовав
во всех критических случаях, особенно на космических аппаратах, П1
скольку происходит непрерывное ужесточение требований по габар”
там, надежности и условиям их эксплуатации.
В условиях механических и тепловых нагрузок бериллий способе-
обеспечить самый высокий уровень точности оптических систем (откЛ0'
нение между элементами не более 0,005 мм).
Сплавы с бериллием, резко повышающим механические свойст
многих металлов, применяются для производства деталей и констрУ*
140
ций, подвергающихся длительному интенсивному напряжению или
трению при переменной или высокой температуре. Наиболее широко в
промышленности используются Be—Си сплавы (бронзы), сочетающие
высокую прочность и выносливость с коррозионной устойчивостью и
высокой тепло- и электропроводностью. Пружины из них выдерживают
миллиарды циклов при значительной нагрузке. Современный автомо-
биль содержит десятки, а самолет — тысячи деталей из бериллиевых
бронз.
Около 65 % потребления бериллия в США приходится на Be—Си
сплавы, в которых содержится в среднем 2 % Be. Ленты из этих сплавов
используются для пружин, соединительных деталей и переключателей
в автомобилях, самолетах, спутниках, радарах, телекоммуникациях,
промышленной автоматике, компьютерах, бытовых приборах, инстру-
ментах и контрольно-измерительных приборах. Трубы большого диа-
метра находят применение в нефтегазовых буровых инструментах,
втулках и вкладышах в тяжелом машиностроении и системах, обеспе-
чивающих посадку самолетов. Главное применение стержней из Be—Си
сплавов — переключатели в волоконно-оптических системах, а брусков
и плит — в производстве литейных форм для металла, стекла и пласти-
ков.
Сплавы А1—Mg—Be и Be—Al, отличающиеся большой легкостью,
применяются в самолетостроении и ракетной технике. Известны корро-
зионностойкие сплавы на бериллиевой основе, содержащие до 2 % Са,
V, Ni, Zr. В последнее время большое внимание уделяется интерметал-
лическим соединениям бериллия с тугоплавкими металлами, в первую
очередь — с танталом и цирконием (ZrBei3 и ТагВер). Тугоплавкость
бериллидов, легкость и устойчивость к окислению до 1650 °C делает их
идеальными конструкционными материалами для ракет, управляемых
снарядов и спутников.
В атомной технике бериллий и его оксид используются как источни-
ки, замедлители и отражатели нейтронов, а также как конструкцион-
ный материал для бериллизации изделий с целью придания им высокой
поверхностной твердости. Из бериллия изготовляют окошки рентгено-
вских трубок, так как он проницаем для рентгеновских лучей.
В начале XXI века очень крупным потребителем бериллия может
стать термоядерная энергетика, в которой он исследуется как компонент
горючего и материал для теплопередачи, размножения и замедления
нейтронов. По расчетам американских экспертов, использование его в
составе расплава Li—Be—F для воспроизводства трития и размножения
нейтронов обеспечит снижение себестоимости производства энергии на
12 %. Оксид бериллия (~ 15 % потребления США) используется как
субстрат для интегральных схем, систем зажигания в автомобилях, в
лазерах, радарах, микроволновых устройствах. В традиционных обла-
141
стях применения значение оксида бериллия не только сохранилось, но
и увеличилось; как огнеупорный материал ВеО в ряде случаев незаме-
ним. Это касается, в частности, изготовления тиглей для плавки метал-
лов (Be, U, Th, Ti), где используется такое уникальное свойство ВеО,
как необычайно высокая теплопроводность наряду с огнеупорностью.
Оксид бериллия широко используется при конструировании индук-
ционных печей и вакуумных нагревательных приборов. Весьма перс-
пективным огнеупорным материалом является пористая керамика из
оксида бериллия, получаемая пенометодом и выдерживающая темпе-
ратуру 1750 °C. В связи с высокой устойчивостью к тепловому удару ВеО
находит применение в авиации для изготовления лопастей газовых тур-
бин и деталей реактивных двигателей. Важная область применения
оксида бериллия — получение меднобериллиевой лигатуры, использу-
емой в производстве бериллиевых бронз. Применяется ВеО и как ката-
лизатор в некоторых органических синтезах.
Прогнозы потребления бериллия противоречивы. К середине 80-х
годов предсказывалось увеличение спроса на 8—12 % в год. Прогноз был
основан в первую очередь на увеличении потребления Be—Си сплавов
в электронике. За последние пять лет количество областей применения
бериллийсодержащих материалов в автомобилях удвоилось, особенно
большим спросом пользуется наиболее дешевый сплав аллой 174 (0,15—
0,5 % Be; 0,35—0,6 % Со). Продолжается рост его потребления в таких
новых областях, как сенсоры систем безопасности в самолетах и конт-
рольные устройства для электронных автоматических коробок передач.
В то же время некоторые специалисты высказывают сомнения в
увеличении потребления бериллия в ближайшем будущем, поскольку в
гражданских сферах применения широко рекламируемые прогнозы ро-
ста спроса пока не оправдываются. Помимо токсичности металла пре-
пятствием к его более широкому использованию является возможность
замены на более дешевые, а иногда и более эффективные материалы. .
Различным областям применения соответствует разнообразие то-
варных форм бериллия. Американские фирмы выпускают бериллий в
виде порошков различного гранулометрического состава, кусков, брУ'
сков, проволоки, труб, профилей различной формы и размеров. Кроме
того, производят оксид, фторид, сульфат и нитрат бериллия достаточной
степени чистоты. Мировое потребление бериллия в 1988 г. ориентире"
вочно составляло 300 т (830 т ВеО).
Цены на бериллиевые продукты на начало 1990 г. составляли,
долл/кг (для сплавов за 1кг содержания Be):
Слитки вакуумного производства (98,5 %-ной чистоты) . . . 495,6
Порошок (98,5 %-ной чистоты).....................431,7
Be—АГ сплавы.................................... 572,7
142
Изделия из Be—Си сплавов:
стержни, прутки, проволока..............21,7
ленты.................................. 19,6
лигатуры............................... 352,4
литейные сплавы........................12,1—13,9
3.8. Сырьевые источники бериллия
По распространенности бериллий занимает 32-е место среди других
элементов; его содержание в земной коре составляет 6*10 4 %. Это
типично литофильный элемент. Он встречается в виде собственных
минералов или входит в состав других минералов в качестве изоморфной
примеси. Известно 54 собственно бериллиевых минерала, большая часть
которых мало изучена. Преобладающее значение имеют силикаты; из-
вестны фосфаты, оксиды, антимонаты, бораты, арсенаты и карбонаты.
Основные минералы бериллия в связи с малым содержанием этого
элемента образовались на поздних стадиях кристаллизации магмы. Все
известные месторождения бериллия — постмагматические образова-
ния, связанные с поздними стадиями пегматитового или различными
этапами гидротермально-пневматолитического процесса.
В настоящее время за рубежом используются два промышленных
минерала: берилл и бертрандит.
Берилл ВезА12[51бО18] — силикат кольцевой структуры, содержит
10,5—14,3 % ВеО. Своеобразие такой структуры заключается в образо-
вании из колец силикатных групп [SibOis 1 сквозных каналов, внутри
которых располагаются примесные катионы и молекулы воды. Как и все
кольцевые силикаты, берилл с трудом поддается разложению. Обычные
примеси — щелочные элементы Li, Na, К, Rb, Cs; меньшее значение
имеют Mg, Мп, Fe, Сг, НгО. Чистый берилл бесцветен; окраска его
обусловлена примесями, главным образом железом и хромом. Окрашен-
ные и хорошо закристаллизованные разновидности берилла используют
как драгоценные камни: изумруд — зеленый, аквамарин — зеленовато-
голубой, воробьевит (содержит до 3 % СвгО) — розовый, гелиодор —
*елтый.
Берилл встречается почти во всех минеральных образованиях, за
исключением собственно магматических. Но для промышленного ис-
пользования до последнего времени разрабатывали лишь месторожде-
ния крупнокристаллического берилла, связанные с гранитными пег-
матитами, позволяющие применять ручную рудоразборку. Большая по-
требность в бериллии привела к совершенствованию методов обогаще-
ния и к использованию других типов месторождений, содержащих
143
мелкокристаллический берилл, в частности комплексные сподумен-бе-
рилловые руды.
Интенсивные исследования, проводившиеся в последние годы во
многих странах, привели к открытию новых промышленных типов мес-
торождений гидротермально-пневматолитического характера. Основ-
ные минералы этих месторождений — бертрандит, фенакит, хризобе-
рилл, а в некоторых случаях вместе с ними присутствуют бехоит, гадо-
линит, гельвин и эвклаз.
Бертрандит Вед [SizO? ](ОН)г — минерал бесцветный, иногда блед-
но-желтый, содержит 39.6—42,6 % ВеО; в качестве примесей иногда
присутствуют А13+ и Fe3 . Кристаллизация его происходит в широком
диапазоне условий минералообразования, но при явно выраженном де-
фиците алюминия.
Фенакит Bei [SiO4 ] — островной силикат, содержит 45,5 % ВеО.
Обычно бесцветен, но иногда окрашен примесями в винно-желтый или
розовый цвет. Генетически родствен бертрандиту. В ближайшей перс-
пективе планируется освоение его месторождений.
Хризоберилл АЬВеОг содержит 18,1—20,7 % ВеО. Окрашен в цвета
от зеленого и зеленовато-желтого до буровато-желтого. Изумрудно-зе-
леная драгоценная разновидность хризоберилла известна под названи-
ем александрита. Окраска александрита вызвана примесью Сг3*. Об-
разование хризоберилла связано с пегматитовым и гидротермально-
пневматолитическим процессами в условиях большого дефицита S1O2.
Вновь открытые месторождения бериллиевых руд обладают преиму-
ществами по сравнению с пегматитами, являвшимися до последних лет
основным источником бериллиевого сырья. Это возможность их разра-
ботки открытым способом и большее содержание ВеО в минералах ука-
занных месторождений.
Содержание ВеО в разрабатываемых рудах — от 0,02 до 0,6 %. В
большинстве пегматитовых месторождений берилл входит в состав ком-
плексных руд наряду с другими редкометальными минералами, берт-
рандитовые месторождения относятся к монометальным. Подтверж-
денные запасы бериллия за рубежом оцениваются в 900 тыс.т ВеО.
Около 80 % запасов сосредоточено в четырех странах: Бразилии (380
тыс. т), Индии (180 тыс. т), Аргентине (70 тыс. т) и США (55 тыс. т). По
западным оценкам в бывшем СССР запасы бериллия оцениваются в 186
тыс.т.
Бериллиевые руды относятся к труднообогатимым вследствие бли-
зости свойств минералов бериллия и пустой породы.
Основные методы обогащения бериллиевых руд следующие:
1. Ручная рудоразборка. Применяют для крупнокристаллических
берилловых руд (минимальный размер кристаллов должен быть — 1ч
мм) пегматитовых месторождений, что дает возможность извлекать
144
зять 30 % содержащегося в руде бериллия. Разработаны методы авто-
матизированной радиометрической рудоразборки по наведенной радио-
активности при облучении у-лучами.
2. Избирательное измельчение применяют для руд, содержащих
мягкие породы (слюдистые сланцы, тальк). Твердые минералы берил-
лия отделяют на грохотах или классификаторах от минералов пустой
породы.
3. Флотация. Применяют для руд с мелкой вкрапленностью берил-
ла. При обогащении сподумен-берилловых руд вначале производят фло-
тацию сподумена. Ее хвосты, представляющие собой черновой берил-
ловый концентрат, флотируют затем по кислотной или щелочной схеме,
флотационные методы дают возможность извлечь ~ 85 % берилла. При
флотационном обогащении пегматитов, содержащих менее 0,1 % ВеО,
удалось получить концентраты с 8—11,5% ВеО с 70—80 %-ным извле-
чением.
При обогащении фенакитовых и бертрандитовых руд разработаны
схемы, в которых используют гравитацию в тяжелых жидкостях (для
средне- и крупновкрапленных руд) и флотацию для мелковкрапленных.
Кондиционный берилловый концентрат 1-го сорта должен содер-
жать не менее 10—12 % ВеО (при ручной рудоразборке) и не менее 9,72
% (флотационный). Концентрат 2-го сорта — не менее 6,16 % ВеО,
содержание влаги не более 3 %. Примерный химический состав берил-
лового концентрата после ручной переработки приведен ниже, %:
ВеО SiO2 А12ОЗ Fe2O3 СаО MgO К2О Na2O
11.35 62 22,50 0,41 3,40 0,30 0,22 0,13
3.9. Технология извлечения бериллия из минерального сырья
Берилл весьма устойчив по отношению к химическим реагентам.
При обычной температуре с ним взаимодействует только плавиковая
кислота и то в едва заметной степени, поэтому все применяемые в
настоящее время процессы извлечения бериллия связаны с высокотем-
пературной обработкой руды. Кроме того, наличие в его составе алюми-
ния, имеющего сходные с бериллием химические свойства, обуслов-
ливает сложность процесса их разделения.
В настоящее время применяют только два вскрывающих агента —
серную кислоту и гексафторсиликат натрия, которые позволяют не
т°лько извлечь бериллий, но и отделить его от алюминия. Оба способа
включают в себя плавку или спекание руды, благодаря чему в обоих
Случаях бериллий в виде соли переводится в водные растворы. В первом
Из них растворимой солью является сульфат, во втором — фторид берил-
лия. Отделение бериллия от алюминия в сульфатном процессе основано
145
на различной растворимости сульфатов алюминия и бериллия: в
время как сульфат алюминия имеет ограниченную растворимость, бе-
риллий образует хорошо растворимый сульфат. Во фторидном процессе
разделение достигается за счет ничтожной растворимости гексафтор,
алюмината щелочного металла и хорошей растворимости соответству-
ющей соли бериллия.
Эти методы были разработаны в свое время применительно к берил-
лу, но они могут быть использованы и для переработки других минера-
лов бериллия, имеющих в настоящее время промышленное значение,
так как, за исключением хризоберилла, все эти минералы являются
силикатами и в достаточной степени однотипны по основным примесям.
Переработка берилловых концентратов с помощью фторирую-
щих агентов. Наиболее старый из существующих методов, основанный
на взаимодействии берилла с гексафторсиликатом натрия, в различных
вариантах используется и до настоящего времени. Реакция гексафтор-
силиката натрия с бериллом при спекании компонентов (Т = 750 °C)
протекает предположительно по схеме
BeaAhlSibOiel + ONazSiFb —-
—♦ 3Na2BeF4 + 2Na3AlF6 + 3SiF4 + 9SiO2. (3.4)
Единственный растворимый продукт, образующийся в результате
этой реакции, — тетрафторобериллат натрия, извлекают из реакцион-
ной массы выщелачиванием водой.Однако эта схема не отражает истин-
ного направления реакции. Значительная часть оксида алюминия не
фторируется вследствие улетучивания S1F4. Таким образом, продуктом
взаимодействия является не столько криолит, сколько AI2O3. Уле®учи-
вание SiF4 — факт неблагоприятный, так как теряется фтор и корроди-
рует аппаратура.
Можно значительно сократить количество гексафторсиликата на-
трия, если ввести в реакцию карбонат натрия
Be3A12[Si6Oi8] + 2Na2SiF6 + Na2CO3 -—•
—• 3Na2BeF4+8SiO2+AI2O3+ СО2. (3.5)
Частичная замена гексафторсиликата натрия железным криолитоМ|
действующим избирательно на оксид бериллия и регенерируемым в
технологическом процессе, приводит к конечному уравнению реакции
Be3A12[Si6Oi81 + 2Na3FeF6 —•
—- 3Na2BeF4 + РегОз + AI2O3 + 6SiO2. (3.6)
В некоторых из действующих технологических схем железным кри-
олитом заменяют при шихтовании 60 % гексафторсиликата натрия.
Стехиометрическое соотношение компонентов дает выход бериллия
около 92 %. Учитывая существующую в настоящее время высокую ценУ
на берилловый концентрат, выгодно применять избыток фторидов с тем.
чтобы поднять выход до 100 %.
146
Температура шихты колеблется в различных вариантах метода от
750 до 800 °C. Высокая температура нагревания не рекомендуется из-за
возможности сплавления шихты и образования нерастворимых соедине-
ний бериллия. При более низкой температуре фторирование берилла
протекает с недостаточной полнотой, что уменьшает извлечение берил-
лия и ведет к загрязнению растворов кремнием за счет непрореагировав-
щего NazSiFb- Кроме того, в этих условиях отмечено образование
растворимых соединений железа и алюминия, что также отрицательно
сказывается на качестве готового продукта.
Чаще всего берилловый концентрат шихтуют с фторирующими
агентами (NaiSiFb: Na3FeF6=2:3) и содой, которую берут в количестве
25 % от теоретически необходимого для связывания всего бериллия в
тетрафторобериллат натрия. Спрессованные из шихты брикеты спекают
2 ч при 750 °C.
На следующем этапе технологического процесса выщелачивают по-
лученный измельченный спек, наиболее растворимый компонент кото-
рого — тетрафторобериллат натрия. Основное количество кремния,
алюминия и железа остается в кеках от выщелачивания. Условия выще-
лачивания должны максимально переводить бериллий в раствор и пре-
пятствовать растворению примесей. В связи с этим процесс ведут обычно
без нагревания и с минимальной продолжительностью, так как длитель-
ное взаимодействие SiOz с фторидными растворами способствует пере-
ходу его в растворимую форму, а повышение температуры, почти не
сказываясь на извлечении бериллия, также увеличивает растворимость
SiOz. Выщелачивание осуществляется водой, имеющей комнатную тем-
пературу, в несколько стадий. После отстаивания пульпы раствор де-
кантируют и в агитатор повторно добавляют воду. Эту операцию
повторяют до тех пор, пока из спека не будет извлечено 90—92 %
бериллия; обычно достаточно трехкратной водной обработки. Содержа-
ние бериллия в объединенных растворах колеблется в пределах 4—5 г/л
ВеО.
Полученные при выщелачивании фторобериллатные растворы со-
держат значительно меньше примесей, чем растворы от выщелачивания
продуктов сульфатизации берилла. Поэтому их обычно не подвергают
специальной очистке, а сразу же направляют на дальнейшую перера-
ботку. В схемах, предусматривающих получение бериллия в виде окси-
Да или гидроксида, следующий этап — гидролитическое осаждение бе-
Рмллия, в процессе которого может быть произведено попутное отделе-
ние от таких примесей, как железо и алюминий. При осаждении гидро-
ксида необходимо учитывать его способность выделяться в зависимости
°т условий осаждения в аморфной или кристаллической, хорошо филь-
тРуемой /3-форме.
147
Кристаллический гидроксид часто получают, разлагая тетрагидрЛ
собериллат натрия. Для этой цели в раствор тетрафторобериллата за
гружают избыточное количество гидроксида натрия. Образующий
тетрагидроксобериллат натрия при разбавлении водой и кипячении р!
лагается, выделяя/3-форму гидроксида
Na2BeF4 + 4NaOH = Na2[Be(OH)4 ] + 4NaF. (3.7)
Расход гидроксида натрия, значительно больший, чем при непосред.
ственном осаждении, окупается хорошей фильтруемостью осадка; к
тому же некоторое количество щелочи поддается извлечению и регене
рации
Na2BeF4 + 2NaOH = 4NaF + Be(OH)2- (3.8
При этом способе осаждения Ве(ОН)г примесь железа можно отце
лить в виде гидроксида после введения во фторобериллатный раствор
избытка NaOH; в противоположность гидроксидам бериллия и алюми
ния гидроксид железа Fe(OH) з не растворяется в избытке щелочи. Алю-
миний отделяется при последующем кипячении раствора, оставаясь в
виде алюмината в растворимом состоянии; менее устойчивый бериллат
натрия разлагается с выделением Be (ОН) 2. В осадок переходит 97 ’/
бериллия.
Для получения гидроксида бериллия в /3-форме используют и метод
зародышей: перед осаждением вводят затравку ранее полученных кри-
сталлов /3-Ве(ОН)2- Чтобы удержать при этом в растворе алюминий и
железо, используют соответствующие комплексообразователи, напри
мер этилендиаминтетрауксусную кислоту (ЭДТА).
После выделения гидроксида бериллия в растворе остается фтори,
натрия, который извлекают в виде железного криолита действием суль-
фата железа при pH 4
Fe2(SC>4)3 + 12NaF = 2Na3FeF6 + 3Na2SO4-
Для увеличения кислотности к маточным растворам добавляют сер-
ную кислоту. Хорошо фильтруемый осадок железного криолита аправ-
ляют в смеситель для шихтования. Отжатый и отмытый от NaF гидро-
ксид прокаливают при 1000 °C до ВеО. Общее извлечение бериллия из
концентрата составляет 85—90 %. На рис. 3.5 приведена технологиче-
ская схема производства оксида бериллия фторидным способом.
Содержание примесей в гидроксиде бериллия, полученного по при-
веденной схеме указано в табл. 3.3.
Таблица 3.3. Состав гидроксида бериллия, %
Процесс AI Fe Mg Si Са Мп Na Li Со
Фторидный 0,01 0.03 0,05 0,008 0,4 0,04 0,05 0,02 <0,002
Сульфатный 0,22 0,04 0,3 0,04 0,5 0,15 0,25 0,02 0,002
Экстракционный 0,04 0,005 0,005 0,25 0,005 0,001 0,06 <0,002 <0,0001
148
Преимущество фторидного
процесса по сравнению с сульфат-
ным состоит в том, что образую-
щиеся растворы совершенно не
вызывают коррозии аппаратуры.
Применяемое во фторидном про-
цессе оборудование, в том числе и
довольно крупные емкости, изго-
товляют главным образом из ма-
лоуглеродистой стали. Поэтому
все оборудование дешево и сохра-
няется в течение длительного вре-
мени. Другим преимуществом
фторидного процесса перед суль-
фатным является простота анали-
тического контроля процесса. Не-
достаточное внимание со стороны
обслуживающего персонала боль-
ше отражается на извлечении, не-
Рис. 3.4. Диаграмма плавкости системы
ВеС12—NaCl
жели на загрязнении продукта.
Сульфатный метод переработки берилловых концентратов. Этот
способ основан на переводе бериллия вместе с алюминием и железом в
сернокислый раствор с оставлением основной массы диоксида кремния
в нерастворимом остатке и на последующем разделении бериллия и
алюминия, основанном на различии в поведении их сульфатов в раство-
ре сульфата аммония. В связи с тем, что берилл взаимодействует с
серной кислотой очень медленно, проводят подготовительные операции
сплавления концентрата с щелочами или термическое активирование
берилла.
Механизм щелочной обработки изучен недостаточно. По-видимому,
он зависит от природы щелочи и от температуры процесса. Так, сплав-
ляя берилл со щелочами при температуре не выше 750—800 °C, получа-
ют соединения типа Na2BeSiO4, не растворимые в воде, но взаимо-
действующие с серной кислотой
Na2BeSiC>4 + 2H2SO4 = Na2SO4 + BeSO4 + SiCh + 2НгО.
После обработки серной кислотой бериллий можно перевести в рас-
твор водным выщелачиванием. При более высокой температуре сплав-
ления (1000—1200 °C) образующиеся Na2BeSiO4 или CaBeSiO4 (в зави-
симости от применявшегося щелочного реагента) разлагаются на оксид
бериллия и растворимый силикат
Na2BeSiO4 = ВеО + Na2SiO3-
В этом случае при водном выщелачивании в раствор переходят при-
неси. В настоящее время чаще применяют термическую активацию
149
берилла, изменяющую его кристаллическую структуру. Концентрат
берилла плавят при 1700 °C, затем резко охлаждают, выливая плавы в
закалочную ванну с водой; при этом берилл распадается, выделяя оксид
бериллия. Классификация на грохоте стекловидных агломератов, по-
лученных при закалке, позволяет отделить куски размером более 12 мм,
в которых возможна рекристаллизация берилла, затрудняющая после-
дующее взаимодействие с серной кислотой. Эти куски направляют в
начало процесса. Однако в этом случае только 50—60 % оксида способно
раствориться в серной кислоте. Остальная часть — твердый раствор
оксида бериллия в кремнеземе, для разрушения которого требуется
вторичный нагрев до 900 °C. Отсеянный спек подвергают вторичной
термической обработке во вращающейся печи. После двукратной тер-
мической обработки в раствор удается перевести 90—95 % бериллия.
Большая экономичность метода по сравнению с щелочной обработкой
заключается в отсутствии затрат на какие-либо реагенты при почти
равноценных энергетических затратах.
Для дальнейшей переработки берилла независимо от способа акти-
вирования обычно используют одинаковые схемы, так как в обоих слу-
чаях при обработке серной кислотой бериллий, а также А1 и Fe переходят
в раствор в виде сульфатов. Различие заключается лишь в том, что
термически активированный берилл поддается воздействию только кон-
центрированной H2SO4, а продукт щелочной обработки берилла раство-
рим и в разбавленной кислоте. На первый взгляд, выгоднее использовать
продукт щелочной обработки, но в действительности в этом случае при-
ходится тратить значительно больше кислоты, так как часть ее идет на
нейтрализацию щелочи, что видно из уравнений реакции для термооб-
работанного берилла
ВезА12[81бО18 ]+6H2SO4=3BeSO4+A12(SO4)3+6SiO2+6H2O (3.9)
и для продукта после щелочной обработки
ВезА12[51бО18 ] + иМ(ОН)2 + (6+п)Нг8О4 =
= 3BeSO4 + A12(SO4>3 + nMSO4 + 6S1O2 + (6+2п)НгО.
При щелочной обработке берилла на 1 кг руды тратится 13 кг карбо-
ната натрия и 40 кг серной кислоты, в то время как на 1 кг термообрабо-
танного берилла только 26 кг серной кислоты.
Спек измельчают в шаровой мельнице, которая работает в замкну-
том цикле с воздушным классификатором. Мокрое измельчение не при-
меняют, чтобы при сульфатизации не разбавлять серную кислоту. Из-
мельченный спек через дозатор поступает в стальной аппарат предвари-
тельного смешения. Туда же поступает серная кислота (93 %-ная) в
количестве, несколько превышающем то, которое необходимо для обра-
зования сульфатов бериллия и алюминия. Избыток серной кислоты
нужен в дальнейшем для получения сульфата аммония при взаимодей-
ствии с аммиаком. Кислая пульпа впрыскивается тонкой непрерывной
150
струей в стальной барабан, нагретый до 250—300 °C. Пульпа попадает
на его раскаленные стенки. При этом почти мгновенно сульфатизиру-
jotch ВеО и А1гОз- Полнота сульфатизации 93—95 %. Такой метод
значительно продуктивнее одновременной сульфатизации больших ко-
личеств оксидов. Отходящие газы пропускают через циклон, где оседа-
jot тонкие частицы унесенной пыли. Затем они проходят скруббер с
насадкой, где SOz и SO3 поглощаются маточником от осаждения гидро-
ксида, содержащим NaOH и некоторое количество дисперсного
Be (ОН) 2.
Продукт сульфатизации выщелачивают водой по принципу проти-
вотока с целью извлечения растворимых сульфатов и отделения от SiO2.
Кек (в основном SiO2) отделяют на непрерывно действующей центри-
фуге, применяя двукратную репульпацию. Это обеспечивает извлече-
ние бериллия в раствор на 99 %. Получают сульфатный раствор состава,
г/л: Be 13; Al 15; Fe 2; Si 0,1; H2SO4 0,5 моль/л, который очищают от
алюминия и железа. Чтобы удалить большую часть алюминия в виде
дисульфата алюминия, аммония (квасцов) (наиболее распространен-
ный метод), горячий раствор сульфатов, содержащий серную кислоту,
смешивают с аммиаком, который берут в количестве, необходимом для
образования квасцов (NH4)A1(SC>4)2’12H2O.
Пульпа квасцов поступает в кристаллизатор, охлаждаемый до 20 °C.
Там ее выдерживают до тех пор, пока не удалится из раствора 75 % А1
в виде квасцов (см. рис. 3.1). Квасцы отделяют на центрифуге непрерыв-
ного действия. Оставшийся в растворе алюминий, а также железо (после
его окисления до Fe3+) выделяют в виде гидроксидов при pH 3,8—4,2. Из
очищенного раствора выделяют Ве(ОН)г теми же способами, что были
описаны во фторидном методе.
На рис. 3.6 приведена технологическая схема производства гидро-
ксида бериллия сульфатным способом. Общий выход бериллия в процес-
се 90 %. Состав полученного продукта представлен в табл. 3.3.
Переработка бертрандита. Кроме кристаллизационно-осадитель-
ного метода для извлечения бериллия из сульфатных растворов можно
применять экстракцию. Процесс получения ВеО основан на экстракции
бериллия из сульфатных растворов с помощью ди-2-этилгексилфосфор-
ной кислоты (Д2ЭГФК). Он предназначен для извлечения бериллия из
вулканического туфа, где он находится в виде бертрандита.
Бертрандитовую руду, содержащую, %: ВеО 0,5—1,5; S1O2 60—80;
AI2O3 10—15; РегОз 1—2, подвергают мокрому помолу до минус 1,3 мм,
а затем выщелачиванию 35 % -ной серной кислотой при 65 °C в течение
24 ч. При этом растворяется 95 % бериллия. Твердый остаток отделяют
07 раствора, содержащего сульфат бериллия, декантацией. В получен-
ном растворе обычно содержится, г/л: Be 0,4—0,7; Al 4—7; Mg 3—5 и Fe
151
Концентрат берилла
Рис. 3.5. Технологическая схема производства оксида бериллия фторид-
ным способом
1,5. Бериллий извлекают из такого раствора экстракцией с помо1
0,2—0,5 моль/л Д2ЭГФК в керосине по реакции
Ве?Л + (ROH)2(oprr=
где ИОН = Д2ЭГФК.
Экстракцию проводят при pH < 1 в восьмиступенчатом противо
ном экстракторе. Так как в исходном водном растворе содержатся зна-
(RCOzBetopr) + 2Н0,
(3.10)
чительные количества алюминия, он экстрагируется первым, а за'
постепенно вытесняется из органической фазы бериллием.
Поэтому рас-
твор после экстракции (рафинат) содержит большую часть алюминия и
весь магний, имевшийся в исходном растворе. А органическая фаза —
бериллий, часть алюминия и железо. Время достижения равновесия на
каждой ступени — не менее 20 мин. Медленное достижение равновесия
в данном случае объясняется гидролизом бериллия с образованием ус-
тойчивых анионных гидроксокомплексов [Be (ОН) 412- и низкой скоро-
стью гетерогенной химической реакции (ЗЛО).
152
Скорость экстракции можно увеличить нагреванием растворов. Для
р кстракции используют растворы карбоната аммония, при этом бе-
риллий переходит в водный раствор в виде комплекса
(NH4)2 [Ве(СОз>2 ]• Железо также выделяется вместе с бериллием. Од-
нако после нагревания до 70 °C оно вместе с оставшимся алюминием
выпадает в виде гидроксида или гидроксокарбоната и затем отфильтро-
вывается. Органический экстрагент регенерируют, обрабатывая серной
кислотой; при этом аммонийная соль Д2ЭГФК превращается снова в
органическую кислоту и ее возвращают в производственный цикл.
Раствор после отделения железа и алюминия вновь нагревают, но
уже до 95 °C. При этом происходит разложение бериллиевого комплекса
с выделением гидроксокарбоната бериллия Ве(ОН)2СОз. Газообразные
СОг и NH3 улавливают для повторного использования. Дальнейший
нагрев гидроксокарбоната бериллия до 165 °C приводит к его разложе-
нию на СОг и гидроксид бериллия. Продукт, полученный противоточ-
ной экстракцией, содержит 99 % ВеО по сравнению с 92—95 % ВеО при
сульфатном способе. Извлечение из руд, содержащих 0,5 % ВеО, состав-
ляет 85 %. Товарный гидроксид (см. табл. 3.3) используют для перера-
ботки в металл, меднобериллиевую лигатуру или в оксид бериллия для
производства керамики.
3 10. Технология получения важнейших соединений бериллия
Получение оксида бериллия. Технический оксид бериллия получают
из гидроксида. Гидроксид высушивают на противнях в сушильных печах
при 100—150 °C, затем прокаливают при 850—1000 °C во вращающихся
печах с наружным газовым обогревом. Но для некоторых отраслей тех-
ники, в первую очередь для ядерной энергетики, требуется оксид берил-
лия высокой чистоты. Для его получения технический гидроксид очи-
щают, используя следующие способы.
Карбонатно-аммонийный способ очистки основан на растворении
гидроксида бериллия в насыщенном растворе карбоната аммония, тогда
как гидроксиды алюминия и железа нерастворимы в нем. Дополнитель-
ная очистка от следов тяжелых металлов достигается осаждением
сульфидов. При кипячении раствора комплексный карбонат
^Н4>2[Ве(СОз)2] разлагается с выделением основного карбоната бе-
риллия. Прокаливанием последнего получают оксид бериллия.
В некоторых схемах чистый оксид получают через кристаллогидрат
сульфата бериллия BeSO4 '4Н2О. Кристаллы BeS04 '4НгО очищают пе-
рекристаллизацией. При этом используется уменьшение растворимости
сульфата бериллия с понижением температуры (при 100 °C она
Равна 45 %, а при 25 °C 29,9 %). Термическим разложением сульфата
пРи 1100 °C получают оксид бериллия.
Эффективен ацетатный способ очистки, который заключается в воз-
гонке оксиацетата бериллия ВедСЦСНзСООб при 360—400 °C. Этот
способ дает продукт, удовлетворяющий по чистоте (более 99 % ВеО)
требованиям, предъявляемым к материалам для ядерной техники. Ок-
сид бериллия получается в тонкодисперсной форме, что имеет исключи-
тельно важное значение в производстве изделий из него, так как спо-
собствует увеличению их плотности.
Получение фторида бериллия. Фторид бериллия — исходный мате-
риал для производства бериллия. Его получают термическим разложе-
нием тетрафторобериллата аммония (NH4>2BeF4- Гидроксид бериллия
растворяют в растворе гидродифторида аммония так, чтобы в растворе
содержалось 20 г/л Be при pH 5,5. Затем в этот раствор добавляют
твердый карбонат кальция и смесь нагревают до 80 °C; при pH 8,3
происходит полное выделение из раствора примесей железа и алюминия
без осаждения Ве(ОН)г. При добавлении в образовавшуюся пульпу ди-
оксида свинца окисляются и затем осаждаются марганец и хром в виде
МпОг и РЬСгО4. Медь, никель и свинец можно осадить из отфильтро-
ванного раствора с помощью полисульфида аммония (NH4)2Se. Из рас-
твора со стехиометрическим соотношением NH4F : BeF2 = 2 : 1 в про-
цессе вакуумной выпарки кристаллизуется тетрафторобериллат. Кри-
сталлы (N Н4) 2BeF4 разлагают при 900— 1100 °C (выше точки плавления
ВеРг) в графитовых тиглях, помещенных в индукционную печь. В ре-
зультате получают стекловидный фторид бериллия. Газообразный фто-
рид аммония улавливают в скруббере и электрофильтре. Растворы, со-
держащие фторид аммония, используют для растворения гидроксида
бериллия.
Получение хлорида бериллия. Его получают хлорированием оксида
бериллия хлором в присутствии углерода или взаимодействием ВеО с
тетрахлоридом углерода.
При 1200 °C оксид бериллия реагирует с хлором
2ВеО + 2Ch = 2BeCh + О2.
Однако реакция обратима при высоких температурах, поэтому для
получения хлорида бериллия добавляют уголь, который связывает кис-
лород (С +1/202 = СО) и направляет реакцию в сторону образования
BeCh:
900—950 ’С
ВеО + Ch + С ---------♦ BeCh + СО, лО°оооК = - 53 кДж.
В интервале 450—700 °C хлорирование тетрахлоридом углерода
идет в соответствии с суммарным уравнением
2ВеО + СС14 = 2BeCh + СОг, aGiqqqk = - 64,7 кДж.
В действительности хлорирование осуществляется продуктами про'
межуточных реакций — фосгеном или смесью хлора и оксида углерод3,
154
Одорированию подвергают брикетированную шихту ВеО + С (при хло-
рировании хлором) или брикеты ВеО (при хлорировании ССЦ) в хлора-
тОрах шахтного типа. Образующийся при хлорировании газообразный
хлорид бериллия улавливают в конденсационной системе. Поддерживая
различную температуру в последовательно установленных конденсато-
рах, можно осуществить фракционную конденсацию BeCh и летучих
хлоридов примесей в соответствии с их температурами кипения.Однако
отделение примесей при этом неполное. Поэтому хлорид бериллия очи-
щают повторной дистилляцией при 500—550 °C в атмосфере водорода.
Последний восстанавливает FeCh до нелетучего FeCh. Прямой выход
при хлорировании ССЦ составляет 85,7 %, с учетом переработки отхо-
дов 96 %.
Хорошие результаты дает солевая очистка хлорида бериллия барбо-
тированием через расплав солей, % (мол.): LiCl 55, КС1 36, NaCl 9.
Температура плавления указанной смеси 346 °C. Это позволяет вести
возгонку хлорида и барботаж при температуре (возгонка при 435 °C и
остаточном давлении 133 Па), исключающей коррозию аппарата и за-
грязнение очищаемого продукта.
Ниже приведены температуры кипения хлоридов:
Хлорид..... ВеС12 FeC13 А12С16 S1CI4
Гкип.'С.... 492 319 183 58
3.11. Получение металлического бериллия
Металлотермические методы. Эти методы были испробованы по
отношению к оксиду и галогенидам бериллия. Для восстановления ок-
сида бериллия из обычно применяемых металлов пригоден лишь Са. Это
следует из данных табл. 3.4, в которой приведены изменения энергии
Гиббса для некоторых реакций. Однако продукт восстановления загряз-
няется кальцием вследствие образования соединения СаВщз- Неудачна
и попытка использовать для восстановления Ti и Zr. В данном случае
Реакция проходит в твердой фазе (температуры плавления компонентов
очень высоки), поэтому выход во многом зависит от степени контакти-
рования ВеО с восстановителем, в связи с чем брикетирование произво-
дили под давлением 101 МПа. Этот процесс, проводившийся в глубоком
вакУУме (133 Па) и при 1785 °C, оказался слишком дорогим, чтобы
Получить широкое применение.
Для восстановления фторида и хлорида бериллия применимы все
°бычные металлы-восстановители, что подтверждается термодинами-
ческими характеристиками соответствующих реакций (см. табл. 3.4).
Упомянутых галогенидов менее пригоден BeCh- Его труднее обезво-
Днть, процесс восстановления можно проводить лишь ниже 500 °C (тем-
155
пература кипения BeCh). Это намного ниже температуры плавление
металла, поэтому бериллий получается в виде тонкого порошка, кото-
рый легко окисляется при выгрузке и с трудом отделяется от шлака. 1
Таблица 34 Изменение энергии Гиббса некоторых реакций
восстановления оксида, хлорида и фторида бериллия
Реакция Изменение энергии Гиббса, кДж/моль, при температуре, К
298 1000 2000
ВеО + Н2 “ Be + Н2О +360,0 +334,1 +292,1
ВеО + С-Be + СО2 — +294,0 —
ВеО + 2Na - Be + Na2O +214,3 +208,0 —
ВеО + Mg - Be + MgO +17,2 +23,3 +96,5
ВеО + Ca - Be + CaO +15,9 -13,0 +16,3
BeC12 + H2 “ Be + 2HC1 +245,9 +145,4 +100,3
BeC12 + 2Na - Be + 2NaCl -338,5 -285,2 -85,6
BeC12 + Mg - Be + MgC12 -157,5 -138,6 -34,8
BeC12 + Ca - Be + CaC12 -318,1 -306,3 -234,9
BeF2 + H2 - Be + 2HF +360,1 +251,1 +628,3
BeF2 + 2Na - Be + 2NaF -179,8 -572,5 —
BeF2 + Mg - Be + MgF2 -137,4 -118,5 —
BeF2 + Ca - Be + CaF2 -260,1 — —
Фторид бериллия (Ткип = 1327 °C) позволяет вести процесс с получЫ
нием расплавленного бериллия, образующего корольки металла. Из
восстановителей наиболее подходит магний, так как щелочные метал-
лы, например Na, обладают низкой температурой кипения, кроме того,
в процессе восстановления образуется соединение NaF’BeFi, которое
далее не восстанавливается натрием, что существенно снижает выход
бериллия.
Магнийтермическое восстановление фторида бериллия. Это са-
мый распространенный метод получения металлического бериллия. Ре-
акция восстановления фторида бериллия магнием протекает быстро уже
цри температуре около 900 °C, однако для разделения продуктов реак-
ции они должны быть нагреты до 1300 °C, т.е. выше температур плавле-
ния бериллия и фторида магния. В процессе восстановления выделяется
большое количество тепла, которое идет на испарение магния, что со-
здает возможность выброса материала из зоны реакции. Указанные
осложнения устраняют добавлением флюсов, на плавление которых это
тепло и расходуется. Наиболее целесообразным оказалось использовать'
156
качестве флюса избыток BeFz, что исключает загрязнение металла
посторонними добавками. Система MgFz—BeFz (см. рис. 3.2) позволяет
убрать наиболее рациональный состав шлака. Хорошие результаты
^гут быть достигнуты при введении магния в количестве не более 75 %
стехиометрически необходимого. При таком соотношении бериллий
дегко отделяется от шлака за счет растворения с поверхности частичек
восстановленного металла оксидной пленки, мешающей их слиянию.
Плотность шлака больше плотности бериллия, поэтому он всплывает на
поверхность ванны.
Восстановление ведут в графитовых тиглях, нагреваемых в индук-
ционных печах. Расплавленный бериллий накапливается на поверхно-
сти шлака. При охлаждении тигля металл кристаллизуется раньше шла-
ка, что позволяет извлекать слиток бериллия из расплава солей. Шлак
в расплавленном состоянии сливают из тигля в графитовую изложницу.
Соли с поверхности слитка бериллия растворяют в воде. Фторид берил-
лия извлекают из шлака в процессе выщелачивания раствором фторида
аммония. Получаемый раствор поступает на стадию производства фто-
рида бериллия. Нерастворимый осадок, состоящий из фторида магния,
посте переработки используют в качестве флюса в процессе восстанов-
ления. Частицы и куски восстановленного металла содержат 97 % Be, а
также некоторое количество шлака и непрореагировавшего магния.
Суммарное извлечение бериллия в процессе восстановления 96 %, про-
должительность цикла 3,5 ч.
Электролитическое производство бериллия. Бериллий не может
быть получен электролизом водных растворов его солей, так как имеет
высокий отрицательный электродный потенциал (см. табл. 3.1), поэто-
му при электролизе на катоде выделяется водород. Электролитическое
производство можно осуществить, используя расплавы солей, в частно-
сти расплавы галогенидов бериллия. Однако их расплавы не проводят
тока, поэтому электролиз возможен лишь в присутствии второго компо-
нента , обладающего достаточной электропроводностью и более высоким
по сравнению с галогенидами бериллия напряжением разложения. Эти-
ми свойствами обладают галогениды щелочных металлов.
Соответствующий состав электролита в принципе допускает ис-
пользование и хлоридной, и фторидной ванн. Но высокая температура
Ставления ВеРг (800 °C) обусловливает проведение высокотемператур-
ного процесса. Это влечет за собой конструктивные затруднения и спо-
с°бствует окислению выделяющегося металла. Предпочитают низко-
ТеМпСраТурНЬщ электролиз из ванны, содержащей хлориды бериллия и
НатРия. Оптимальный состав электролита был найден в результате изу-
Чения плавкости в системе BeCh—NaCl (рис. 3.4). В системе обнаруже-
На низкотемпературная эвтектика (215 °C), содержащая 50 % (мол.)
157
[ Концентрат берилла |
;
Плавление а ругоаоа печи (1700°С)
4
Грануляция в воре
I
fepKX^patfowa(900-950°C)
4
Изиельчение
[ 93% HgSOj [---------, Смешение
4
Сульфатизация
I
I Н;0 |--------> Выщелачивание
I
Центрифугирование --------» | Кек (SiOg) |
4____________ I
| Раствор сульфатов | опал)
I NHA° |-----------* Сыешение
{Сброс)
|~ N^AIlSO^tt^O |
4
(Товарный продукт)
Рис. 3.6. Технологическая схема производства оксида бериллия сульфат-
ным способом
BeCh и инконгруэнто плавящееся соединение NaiBeCU- В этом соеДИ
нении бериллий координационно связан с хлором, но связь неустойчив
Электролиз проводят в сварных никелевых ваннах, снабжений
электрическим нагревателем. Анод изготовляют из плотного графи13'
что снижает выкрашивание анода с поверхности и тем самым у меныВае1
158
возможность загрязнения электролита. Катодом служит сама никелевая
ванна. В этом случае по окончании электролиза электролит перекачи-
вают в другую ванну, а металл вычерпывают перфорированным ков-
ром. На некоторых предприятиях применяют съемные катоды в виде
дерфорированных никелевых ящиков, вставляемых в ванны. Ванну и
катод перед электролизом бериллируют для уменьшения загрязнения
никелем.
При загрузке ВеСЬ и NaCl берут в массовом соотношении 1:1, что
позволяет получить в электролите в начале электролиза 54 % (мол.)
BeCh- Загруженную смесь расплавляют в атмосфере хлора при 350 °C.
Большая электроположительность многих примесей (Си, Fe, Pb, Ni) по
сравнению с бериллием дает возможность освобождаться от них элект-
ролитическим путем. Предварительно, чтобы очистить электролит,
электролиз проводят при пониженной плотности тока (2,8 с
цилиндрическим никелевым катодом, на котором выделяются примеси.
После окончания очистки катод сменяют перфорированным и далее
электролиз ведут при катодной плотности 6—7 А/дм2. Электролиз про-
должают, пока содержание ВеСЬ в ванне не снизится до 45 %. На это
требуется около суток. Затем катод вынимают и заменяют новым. Перед
сменой катода температуру ванны повышают до 380 °C для увеличения
текучести электролита. Состав ванны корректируют, добавляя ВеСЬ до
исходной концентрации его 54 % (мол.). Выход по току 50 %. Осажден-
ный в виде дендритов бериллий промывают водой, затем раствором
NaOH, разбавленной HNO3 и спиртом. В крупных кристаллах содержа-
ние бериллия 99,966, в мелких 99,937 %. Полученный электролизом
бериллий можно подвергнуть переплавке в вакууме или направить не-
посредственно на металлокерамический передел.
Электролитический бериллий чище металлотермического (табл.
3.5). Это объясняется тем, что и электролиз, и предшествующее ему
хлорирование ВеО — рафинирующие операции. Указанное преимуще-
ство делает электролитический метод конкурентоспособным, несмотря
на значительно меньший выход металла.
Плавка и литье. Для удаления шлака и магния из магниетермиче-
схого бериллия, а также включений электролита из катодного продукта
Металл подвергают вакуумной плавке в индукционных печах в тиглях
Из ВеО при 133—267 Па и 1500—1550 °C. При этом свободный магний,
Фторид бериллия и электролит испаряются, а нелетучие примеси ВеО,
М^?2, Ве2С в виде шлака всплывают на поверхность либо оседают на дно
Тигля. Очищенный металл выливают в изложницу, в которой он кри-
сталлизуется в слиток массой 75—100 кг. Иногда вакуумную плавку
с°четают с центробежным литьем, что способствует удалению шлака и
?3°в из отливок. Металл, полученный вакуумной плавкой, содержит
’5 % Be. Выход бериллия из корольков в отливку 90%.
159
Таблица 3.5. Содержание основных примесей в бериллии, %.
Металл Металлотермический после вакуумной плавки Электро- литный После вакуум- ной дистилляции После электро, рафинированна
Fe 1,1 -io-1 -з 7’10 -3 6’10 -3 I 3’10 °
Си — 1,7 ’10~3 -4 5’10
В 1.2 ’10“4 1 ю-5
Мп -3 740 ° 2’10 3 -3 2’10 6’1(Г4
Ni -2 1,3‘Ю -з 3’10 ° -3 1 -10 2’10~4 1
Al -2 6"10 -3 3’10 -3 2’10 8’10~4
Si 1,2 -2 2,2’10 -2 1 ’10 2,5'10'3
Cr -з 9’10 ° -4 8’10 -3 4’10 —
Наиболее чистый бериллий получают вакуумной дистилляцией тех
нического металла в интервале 1300—1400 °C при остаточном давлении
порядка 1,3’10 4 Па. Бериллий конденсируется на молибденовомэкра
не, расположенном внутри трубки из оксида бериллия, обогреваемой
электрическим током, который пропускают через молибденовуюобмот
ку. Примеси конденсируются в различных температурных зонах,
наиболее чистый бериллий — в зоне с температурным интервалом
1120—1150 °C. В верхней части экрана, в температурном интервал
900—1000 °C конденсируется основная масса примесей алюминия а
магния. Дистилляция в вакууме позволяет получать бериллий высокое
чистоты.
В настоящее время основным методом получения качественных бе-
риллиевых изделий является метод порошковой металлургии. Удобен
метода применительно к бериллию определяется его хрупкостью, сп(
собству ющей получению порошка, и достаточным давлением пара вбли
зи температуры плавления. Это облегчает спекание.
Металлокерамическое производство бериллия. Для получения бе-
риллиевых изделий можно применять различные варианты поров1
ковой металлургии: 1) холодное прессование, спекание и тепловую об
работку полученных заготовок под давлением; 2) горячую обработку
давлением непосредственно порошка, обычно помещаемого для этого
стальные сварные оболочки (контейнеры); 3) горячее прессование в
вакууме с применением небольших удельных давлений; 4) горячее пр#
сование на воздухе с применением больших удельных давлений; Р
прессование в вакууме при умеренных температурах и относитель11’
небольшом удельном давлении (так называемое теплое прессование)'
160
Технологический процесс начинается со стадии измельчения берил-
дИя. Электролитические чешуйки измельчают в шаровой мельнице
мокрого помола. Затем порошок обрабатывают щавелевой кислотой для
извлечения примесей хлора и хлоридов. Попытки приготовить порошки
бериллия непосредственно из магниетермического металла оказались
деудачными из-за выраженной интеркристаллитной коррозии в издели-
ях, поскольку в таких порошках имеется небольшое количество шлака.
С целью получения доброкачественных слитков для изготовления бе-
риллиевого порошка современная техника производства заготовок обя-
зательно включает вакуумную переплавку чернового металла для уда-
ления из него остатков шлака и магния. Для изготовления порошка
переплавленный в вакууме слиток обтачивают на токарном станке, а
полученную стружку измельчают в атмосфере сухого азота в дисковом
истирателе, водоохлаждаемые диски которого футерованы бериллием.
Большую часть металлокерамических заготовок получают горячим
прессованием в вакууме при 1000—1150 °C и давлении 0,5—1,0 кПа,
после чего они могут быть обработаны горячим выдавливанием, прокат-
кой или ковкой в стальных контейнерах.
Прочность и пластичность бериллиевых изделий, получаемых мето-
дом металлокерамики, регулируется главным образом величиной час-
тиц исходного порошка. Одним из основных критериев качества готовых
бериллиевых изделий является их плотность, которая составляет 1,83—
1,845 г/см3 (теоретическая плотность бериллия, содержащего обычные
примеси, 1,845—1,855 г/см3).
3.12. Техника безопасности в бериллиевом производстве
Бериллий и его соединения в высшей степени токсичны и обладают
поливалентным действием: общетоксическим, аллергическим, канце-
рогенным и эмбриогенным. Они вызывают тяжелые заболевания, иног-
да со смертельным исходом. В связи с этим на предприятиях берил-
лиевой промышленности остро стоит вопрос о безопасности работаю-
щих.
Строго соблюдаются нормы допустимых концентраций бериллия в
воздухе. ПДК бериллия в воздухе рабочих помещений составляет 1 • 10 J
мг/ м3, в атмосферном воздухе 1 10 5 мг/м3. Концентрацию бериллия в
воздухе контролируют на предприятиях непрерывно. В помещениях
Действует интенсивная вентиляция. В зданиях поддерживается некото-
рое разрежение, чтобы предотвратить возможное просачивание берил-
лия наружу. На всех операциях, связанных с пылеобразованием, пре-
дусмотрена возможно полная герметизация и местная вентиляция.
6'691 161
Работы с сухими порошками бериллия и его соединениями ведут 8
герметических рукавных боксах, работающих при разрежении 267 Па
Плавят бериллий в герметичных печах, установленных в кабинах с
вентиляцией. Воздух из вентиляционных систем перед выбросом в ат-
мосферу пропускают через стекловолокно и фильтры Петрянова. Филь-
тровальные ткани уничтожают.
Рабочие снабжены респираторами и полным комплектом спецодеж-
ды, в том числе и белья, которое должно меняться два раза в неделю, в
подвергаются регулярному медицинскому освидетельствованию. От са-
мих работающих требуется исключительная аккуратность и вниматель-
ность при проведении операций и безусловное выполнение всех без
исключения правил техники безопасности.
Глава 4. СТРОНЦИЙ
ХИМИЯ СТРОНЦИЯ
4.1. Общая характеристика
Стронций — элемент II группы Периодической системы Д.И. Мен-
делеева — входит в подгруппу щелочноземельных металлов вместе с
кальцием, барием и радием. Основные физико-химические характери-
стики стронция приведены в табл. 3.1.
Электронная конфигурация атома стронция — [Кг ]5s2. Природный
стронций состоит из смеси четырех стабильных изотопов с массовыми
числами 84,86, 87 и 88, из которых наиболее распространен 88Sr (82,56
%). Искусственно были получены радиоактивные изотопы стронция с
массовыми числами от 80 до 97, в том числе 9°Sr, образующийся при
делении урана. Изотоп 9°Sr (71 /2=27,7 года) относится к числу наиболее
опасных радиоактивных изотопов. Помимо большого периода полурас-
пада, имеет свойство прочно удерживаться в организме, в основном в
костях, и медленно выводится из него, являясь источником постоянного
облучения костного мозга. Стронций — мягкий, серебристо-белый ме-
талл, ковкий и пластичный, триморфен. При комнатной температуре
имеет ГЦК решетку (СГ-Sr), при 215 °C переходит в гексагональную
модификацию (fl-Sr), а при 605 °C — в ОЦКмодификацию (y-Sr)-
Стронций парамагнитен.
По химическим свойствам стронций сходен с кальцием и барием,
занимая промежуточное положение между ними. В соединениях имеет
степень окисления +2. В ряду напряжений стронций находится среди
наиболее электроотрицательных металлов, что обусловливает его высо"
162
кую химическую активность. Стронций является сильным восстанови-
телем: в соответствии с величиной электродного потенциала стронций
легко вытесняет водород из разбавленных кислот и тяжелые металлы из
растворов их солей.
Металлический стронций активно окисляется на воздухе, образуя
на поверхности желтоватую пленку, в которой наряду со SrO содержится
пероксид SrO2 и небольшое количество нитрида SnN?; при нагревании
на воздухе воспламеняется, а порошкообразный металл самовоспламе-
няется при комнатной температуре. Стронций энергично разлагает воду
с выделением водорода и образованием гидроксида; при повышенных
температурах взаимодействует с водородом (выше 200 °C), азотом (вы-
ше 400 °C), фосфором, серой и галогенами, образуя соответствующие
бинарные соединения. С кальцием и барием стронций образует непре-
рывные ряды твердых растворов. В расплавленном состоянии стронций
дает со многими металлами (Mg, Zn, Pb, Cd и др.) однородные расплавы,
в твердом же состоянии — интерметаллические соединения, например
SrMg2, SrMg4, SrMg9. В системе Sr—Al существуют интерметаллиды
SrAl и SrAU и ограниченные твердые растворы на основе А1 и Sr с малой
протяженностью. Стронций практически не подвергается действию
концентрированных растворов щелочей; концентрированные кислоты
(HNO3 и H2SO4) на стронций действуют слабо; разбавленные — раство-
ряют стронций с образованием соответствующих солей. Соли минераль-
ных кислот с анионами СГ, Вг~, Г и NO3 хорошо растворимы в воде, с
анионами F~, SO4 , СОз- и РО4- — малорастворимы. Соединения строн-
ция, как правило, изоморфны соответствующим соединениям кальция
и бария.
4.2. Оксиды и гидроксид стронция
Оксид стронция SrO — белое кристаллическое тугоплавкое ве-
щество кубической сингонии, плотность 4,7 г/см3, температура плавле-
ния 2430 °C, энтальпия образования = 590,4 кДж/моль. Легко
взаимодействует с водой с выделением большого количества тепла и
образованием гидроксида Sr (ОН) 2. Получают оксид стронция при про-
каливании карбоната или нитрата стронция при 1100 °C.
Пероксид стронция SrO2 имеет энтальпию образования -ЛЙ298(/) =
"° 640,9 кДж/моль. Пероксид стронция образуется при взаимодействии
оксида с кислородом при высоком давлении. В виде октагидрата SrO2
можно получить при действии пероксида водорода на гидроксид строн-
ция по реакции
Sr(OH)2+4H2O2=SrO2 8Н2О. (4.1)
Октагидрат полностью обезвоживается при 100—130 °C, при нагре.
вании до 700—800 °C выделяет кислород и переходит в оксид SrO.
Гидроксид стронция Sr (ОН) 2 представляет собой бесцветные кри-
сталлы, расплывающиеся во влажном воздухе; плотность 3,625 г/см3-
температура плавления 375 °C. Энтальпия образования -ЛЯ2ОВ., а
= 959,4 кДж/моль. Гидроксид стронция — более сильное основание, чем
Са(ОН)2- Образуется при взаимодействии оксида стронция с водой
реакция экзотермична. С избытком воды образует гидраты, из которых
наиболее богатым водой является октагидрат Sr (ОН) 2 8Н2О. Октагид-
рат гидроксида стронция образует бесцветные тетрагональные кристал-
лы; плотность 1,90 г/см3. В сухом воздухе Sr(OH)2 8Н2О отщепляет
воду и переходит в моногидрат Sr(OH)2 Н2О, который полностью
обезвоживается при 100 °C; выше 400 °C образуется SrO. Октагидрат
Sr (ОН) 2 8Н2О растворяется в воде с отрицательным тепловым эффек-
том (—61,1 кДж/моль), в то время как при растворении в воде
безводного гидроксида стронция выделяется 48,5 кДж/моль тепла. Рас-
творимость в воде Sr(OH)2- 8Н2О сильно растет с температурой и сос-
тавляет 0,90 (0 °C) и 47,71 % (100 °C). Водные растворы гидроксида
стронция поглощают СОг, при этом выпадает осадок 8гСОз.
4.3. Соли кислородсодержащих кислот
Сульфат стронция SrS04 образует бесцветные ромбические кри-
сталлы; плотность 3,96 г/см3, энтальпия образования -лН^^ = 1444,7
кДж/моль; диморфен, при 1152 °C переходит в другую, вероятно, моно-
клинную модификацию; при 1580 °C разлагается, не плавясь, с выделе-
нием SO2. Сульфат — одна из наименее растворимых солей стронция.
Растворимость составляет 0,0132 % (20 °C), что выше растворимости
BaSO4> но меньше растворимости CaSO4. Сульфат стронция образуется
при взаимодействии растворов солей стронция с серной кислотой. Он
образует двойные соли с сульфатами щелочных металлов и аммония.
Карбонат стронция S1CO3 образует бесцветные ромбические кри-
сталлы, изоморфные арагониту СаСОз и витериту ВаСОз; плотность 3,7
г/см3; энтальпия образования -дН^98, = 1218,4 кДж/моль. При 929 “С
переходит в гексагональную модификацию, которая плавится при 1497
°C под давлением СО2 6 МПа. Давление СОг при диссоциации карбоната
стронция на SrO и СО2 равняется атмосферному при 1211 °C. В воде
S1CO3 растворяется плохо, растворимость при 20 °C равна 1,1 мг в 100 г
НгО. Раствор вследствие частичного гидролиза имеет щелочную реак*
цию. Присутствие в растворе избытка диоксида углерода значительно
повышает растворимость S1CO3 вследствие образования гидрокарбона-
164
а $г(НСОз)2. Карбонат стронция растворим в растворах аммонийных
сОдей, легко разлагается кислотами. Получается карбонат стронция при
ращении карбонатом аммония из растворов стронциевых солей.
Цитрат стронция Sr (NОз) 2 выделяется в виде бесцветных кубиче-
ских <ристаллов; плотность 2,986 г/см3; температура плавления 645 °C.
Энтальпия образования _а//298(/) = 975,7 кДж/моль. При нагревании
Jbnne температуры плавления выделяется кислород и нитрат превраща-
ется в нитрит SrNO2, 'который при дальнейшем прокаливании разлага-
ется на оксид стронция и оксиды азота. Растворимость в воде, %: 39,3
(19°С),58,1 (98 °C). Ниже 31,3 °C из водных растворов кристаллизуется
тетрагидрат Бг(МОз)2• 4Н2О — моноклинные кристаллы с d=2,2 г/см3,
которые легко теряют воду при нагревании. В отличие отСа(МОз)2
нитрат стронция плохо растворим в спирте, еще менее растворим в смеси
равных объемов этанола и этилового эфира. Это свойство нитрата строн-
ция используют для отделения его от кальция. Получают нитрат строн
ция взаимодействием гидрокисда с азотной кислотой и последующей
кристаллизацией из раствора.
4.4. Галогениды стронция
Галогениды стронция SrX2 образуются при взаимодействии строн-
ция с галогенами, а также при растворении SrO или S1CO3 в галогено-
водородных кислотах. Среди галогенидов особое место занимает фто-
рид, так как он почти нерастворим в воде и в разбавленных кислотах.
Свойства галогенидов стронция приведены в табл. 4.1.
Таблица 4.1. Свойства галогенидов стронция
Показатели SrF2 SrC12 SrBr2 SrI2
- Д//398(/), кДж/моль 1209,2 828,4 715,5 560,7
Тпл» 'С 1190 873 643 402
Ткип, °C 2460 2030 — —
d, г/см3 4,94 3,05 4,22 4,55
Фторид стронция SrF2 — белый объемный осадок, кристаллы при-
надлежат к кубической сингонии; очень плохо растворяется в воде:
"1173 г/л при 17,4 °C; растворяется в горячей соляной кислоте. Кри-
сталлогидратов не образует.
Хлорид стронция SrC12 — кристаллы кубической сингонии; хорошо
Растворим в воде. Растворимость, %: 34,6 (20 °C), 50,2 (100 °C). Раство-
Ры имеют горький вкус. При температурах ниже 61,34 °C из растворов
Вь*Деляется SrCh 6Н2О, изоморфный СаС12 6Н2О в виде гексагональ-
165
ных игл, расплывающихся на воздухе. SrCh'bHzO в отличие
СаС1г -бНгО с трудом растворяется в этиловом спирте. Выше 61,34 и,
145 'С кристаллизуется моноклинный SrCh ’2Н2О и, наконец, выше Ц;
°C и при давлении выше атмосферного образуется БгСЬ'НгО. Послед
ний при нагревании выше 250 °C полностью обезвоживается, при этс
наблюдается частичный гидролиз. Хлорид стронция подобно СаСЬ при.
соединяет аммиак.
Бромид стронция БгВгг образует бесцветные ромбические кристал
лы, хорошо растворимые в воде: 50,6 % при 20 °C. Из водных раствор»
ниже 88,6 °C кристаллизуется гексагидрат БгВгг'бНгО, а выше этой
температуры — дигидрат 8гВг2‘2НгО. Бромид стронция растворим в
этиловом спирте.
Иодид стронция Srl2 — бесцветное кристаллическое вещество, хо-
рошо растворимое в воде: 0 64 % при 20 °C. При температурах ниже 83,6
°C из водных растворов выделяется гексагидрат БгЬ’бНгО, при более
высоких — дигидрат. Так же как и бромид, иодид стронция растворим в
этаноле.
4.5. Соединения стронция с неметаллами
Из соединений стронция с неметаллами представляют интерес суль-
фид, гидрид и нитрид.
Сульфид стронция SrS выделяется в виде бесцветных кубических
кристаллов, плохо растворим в воде, но постепенно гидролизуется по
реакции
2SrS+2H2O=Sr(SH)2+Sr(OH)2. (4.2)
Получают SrS при восстановлении SrS04 углем. Особо чистый
SrS для люминофорных составов получают действием H2S на SrCCh при
900 °C.
Гидрид стронция SrH2 — бесцветные кристаллы с плотностью 3,27
г/см3; разлагается до плавления при 899 °C на стронций и водород; при
нагревании в атмосфере азота превращается в имид SrNH желтого две'
та. Получают SrH2 взаимодействием металла с водородом при 200 °C-
Нитрид стронция Sr3N2 выделяется в виде черных кристаллов,
которые легко разлагаются водой
Sr3N2+6H2O = 3Sr(OH)2 + 2NH3. <4.3)
Растворы стронция в жидком аммиаке имеют синий цвет, после
отгонки из них аммиака получается аммиакат [Sr(NH3)6]2+ медН°'
красного цвета с золотистым блеском. Последний в присутствии катаЛИ'
затора (например, платины) разлагается с образованием белого амиЛ
стронция Sr(NH2)2, который при нагревании в вакууме превращается®
имид. При нагревании имида стронция в высоком вакууме получает^
166
красновато-коричневый кристаллический пернитрвд Sr2N4, разлагае-
мЫи кислотами с выделением азота.
С углеродом стронций образует карбид SrC2, который, как и СаСг,
разлагает воду с выделением ацетилена.
Помимо указанных соединений стронций образует бинарные соеди-
нения, такие как бориды, силициды, арсениды и др. Ион стронция обра-
зует комплексы с большим числом органических соединений. Подробно
изучено взаимодействие стронция с аминополикарбоновыми кислотами
__ этилендиаминтетрауксусной, иминодиуксусной и ее производными,
константы устойчивости которых представлены ниже:
т, °C igK
Этилендиаминтетрауксусная кислота .... 25,3 8,53
Нитрилотриуксусная кислота.............26,3 4,91
Иминодиуксусная кислота............... 20,0 2,23
ТЕХНОЛОГИЯ СТРОНЦИЯ
4.6. Важнейшие области применения стронция
и его соединений
Стронций был открыт в 1790 г., но длительное время его применение
было ограничено. В настоящее время все новые и новые отрасли народ-
ного хозяйства испытывают потребность в этом металле. Стронций ис-
пользуется в основном в виде солей, главным образом — карбоната. Ос-
новными областями применения стронция и его соединений являются
пиротехника, электроника, радиотехника, стекольная промышлен-
ность, металлургия, химическая и нефтеперерабатывающая промыш-
ленность, пищевая промышленность и медицина.
До недавнего времени значительная доля выпускаемых зарубежной
нромышленностью соединений стронция использовалась для изготовле-
ния пиротехнических составов. Применение соединений стронция в
»К)Й области обусловлено характерным карминово-красным цветом,
которым окрашено пламя солей стронция и который виден на большом
Расстоянии. В пиротехнике используются азотнокислый, углекислый,
Клоритный стронций, а также оксалат и гидроксид стронция. Содержа-
нИе этих соединений в пиротехнических смесях достигает 75 % от соста-
ва смеси. Пиротехнические составы используются для производства
Сигнальных, осветительных и зажигательных ракет различных типов,
^единения стронция применяются также в производстве сигнальных
красок для железных дорог и шоссе, а также в световой рекламе на
Улицах и в помещениях.
167
Соединения стронция и целестиновый концентрат применяются
керамической, стекольной, фарфорово-фаянсовой промышленной
Целестиновый концентрат используется при производстве строитель,
ной и бытовой керамики. Карбонат стронция входит в состав низкоте^.
пературного фаянса и обычной бытовой посуды, тонкого фарфора
фаянсовых электроизоляторов, обеспечивая изделиям повышенную
прочность и малую растворимость в воде. Карбонат стронция широк
используется в стекольном производстве, способствуя повышению хи
мической и термической устойчивости стекла, уменьшает склонность к
кристаллизации и значительно понижает вязкость стекломассы.
Соединения стронция применяются в составе эмалей и глазурей
вместо ядовитого оксида свинца. Эмали, содержащие до 30 % оксид»
стронция, используются для покрытия магния и его сплавов. Эти эмали
применяются также для покрытия сталей и жаропрочных сплавов. Та
кие покрытия выдерживают температуру выше 1000 °C.
Добавки гидроксида стронция к нефтемаслам способствуют умень-
шению их дезинтеграции в воде, снижают окисляемость и значительно
повышают стабильность смазок. Содержание стронция в них колеблется
от 20 до 30 %.
Разнообразно применение стронция в химической и пищевой про-
мышленности. Стронций используется для очистки растворов гидрокси-
да натрия от алюминия и железа. Целестиновый концентрат применя-
ется при производстве каустической соды высокой чистоты, которая
необходима для изготовления белых сортов искусственного волокна,
бумаги, целлофана. Целестин или карбонат стронция при этом адсорби-
руют примеси железа и марганца.
Оксид стронция используют как катализатор в процессах обезвожи-
вания бутенов крекинга нефти; гранулированный SrO необходим при
производстве водорода для связывания углекислого газа. В производстве
сахара соединения стронция служат для выделения сахара из мелассы в
результате образования нерастворимых сахаратов стронция. Это обес-
печивает больший выход сахара по сравнению с применяемой для этой
цели известью.
Соединения стронция и целестин применяют в качестве наполните-
ля при вулканизации каучука. Целестиновый концентрат является эф-
фективным заменителем барита при изготовлении красок, сургуча и
аккумуляторных батарей. Белые краски, изготовленные на его основе,
не уступают по качеству баритовым белилам. Сернистый стронций вхо-
дит в состав светящихся красок.
В металлургии стронций как раскислитель и флюсующая добав*2
при выплавке стали улучшает ее механические свойства и повышав
хладостойкость. Уменьшению хрупкости титана и его сплавов при вЬГ
соких температурах способствует введение в них 0,1 % стронция. Кар"
168
0онат стронция применяют при рафинировании цинка, меди, свинца,
тйТана, индия. Силикостронций используется в качестве модификатора
чугуна-
В последние годы отмечается резкий подъем стронциевой промыш-
деНности, который вызван применением стронция в цветном телевиде-
нии и при производстве ферромагнитных материалов. Способность ок-
сида стронция более эффективно, чем оксид бария, поглощать рентге-
новское излучение позволила использовать его при производстве стекол
кинескопов цветных телевизоров.
В новых ферромагнитных керамических материалах карбонат бария
заменяют карбонатом стронция, благодаря чему значительно улучша-
ются магнитные свойства. При производстве электродвигателей строн-
циевые ферромагнетики обеспечивают значительное уменьшение мас-
сы и размеров последних, а также способствуют их удешевлению и
увеличению надежности.
Соединения стронция отличаются очень хорошими светотехниче-
скими характеристиками, в связи с чем находят применение в электро-
технике, радио- и светотехнике. Оксидные покрытия со стронцием в
электронно-лучевых трубках, лампах СВЧ, газоразрядных трубках, ра-
диолампах значительно повышают эмиссионные свойства ламп и про-
длевают срок их службы. Сульфат стронция входит в состав наиболее
эффективных и энергоэкономичных люминофоров. На основе оксида,
цирконата и гафната стронция производится высокотемпературная ке-
рамика, на основе других соединений стронция — конденсаторная кера-
мика с высокой диэлектрической проницаемостью. Фторид стронция —
перспективный материал для получения оптической керамики, отлича-
ющейся большой механической прочностью и устойчивостью к резким
перепадам температур.
Некоторые соли стронция (бромид, иодид, арсенид, фторид и сали-
цилат) входят в состав фармацевтических препаратов, в частности про-
тив малярии, некоторых нервных заболеваний, а также используются в
качестве болеутоляющих средств.
4.7. Сырьевые источники стронция
Содержание стронция в земной коре 4,0'10 2 %. Стронций находит-
ся в природе как в виде собственных минералов, так и в виде изоморфной
пРимеси в других минералах. В магматических породах стронций не
^Разует самостоятельных минералов, а находится в рассеянном состо-
”Нии как изоморфная примесь в различных кальциевых минералах. В
Пегматитах, связанных со щелочными породами, стронций содержится
8 апатите, нордите, юксюрите и др. Всего известно около 20 стронциевых
Минералов, не считая тех, в которых он находится в виде изоморфной
169
примеси, например баритоцелестин, кальцитостронцианит, лопарит
др. Морская вода содержит 0,013 % стронция.
Промышленное значение имеют два минерала стронция — целестщ
и стронцианит. Кроме того, представляется перспективным попутное
выделение стронция при переработке апатита.
Целестин SrS04 распространен довольно широко. Он встречаетсе
преимущественно в осадочных породах как продукт химического оса*,
дения в замкнутых бассейнах. Ассоциируется с гипсом, кальцитом, ан
гидритом, баритом, флюоритом, иногда серой. Окраска целестина голу-
бовато-белая или голубовато-серая с желтоватым оттенком; обладает
стеклянным блеском. Кристаллы целестина относятся к ромбической
сингонии; плотность 3,9—4,0 г/см3; твердость 3,5—4,0; целестин кру-
пок. Кристаллы целестина таблетчатые, столбчатые или призматиче-
ские. Химический состав: 56,4 % SrO и 43,6 % SO3; содержит примеси
сульфатов кальция и бария. Температура плавления 1660 °C.
Наиболее крупные месторождения целестина за рубежом находятся
в Англии (Бристоль), Германии (Вестфалия), Италии (Сицилия), Ис
пании, США (Калифорния, Аризона, Техас), Мексике и Индии.
Стронцианит БгСОз встречается преимущественно в гидротер-
мальных образованиях в виде жильного минерала полиметаллических
месторождений в ассоциации с сульфидами, кальцитом, баритом и флю-
оритом. Бесцветен или имеет зеленоватый, желтоватый или сероватый
оттенки; обладает стеклянным блеском, в изломе жирный. Относится к
ромбической сингонии; плотность 3,6—3,8 г/см3; твердость 3,5—4,9
хрупок. Кристаллы тонкоигольчатые, призматические. Химический со-
став, %: 70,2 SrO, 29,8 СО2. Почти всегда в стронцианите присутствует
СаО, реже ВаО и РЬО. Стронцианит является второстепенным источни-
ком стронция. За рубежом в значительных количествах стронцианит
находится в Германии (Вестфалия), Шотландии, Англии (Бристоль),
США (Калифорния) и Перу.
Апатит Ca5(PO4)3(F,Cl) теоретически должен содержать 55,4 %
СаО, 42,1 % Р2О5 и 2,4 % F. Однако состав его подвержен сильны^
колебаниям вследствие изоморфных замещений: F- СГ, ОН , СО?
Р #Si; Са Sr, Na, Ln. В связи с этим существует много разновидностей
апатита, в том числе содержащих 0,4—1,0% ЬпгОз и до 3 % SrO. Апатит
— единственный природный фосфатный минерал, который содер*иТ
стронций. Апатит — широко распространенный минерал пегматитовог0
происхождения, его спутники — магнетит, кальцит, полевой шпат
нефелин. Уникальные месторождения апатита имеются на Кольской
полуострове.
Обогащение стронциевых руд. Содержание целестина в рудах
леблется в широких пределах. При преобладании пластовых и линзоо0
170
а3цых залежей оно составляет от JU до Уи /о. в месторождениях с
преобладанием прожилковых и вкрапленных руд содержание целестина
не более 15—30 %. Содержание барита в рудах обычно не превы-
шает 1 %, но иногда увеличивается до 5—10 %.
Целестиновые месторождения обычно разрабатываются карьерным
способом. Крупные куски руды дробят в щековой дробилке, затем под-
вергают измельчению до ~ 0,04 мм и воздушной сепарации. В результа-
те получают концентрат, пригодный для дальнейшей переработки. Не-
которые целестиновые руды обогащают рудоразборкой и гравитацион-
нЫм способом. Обогащение низкосортных целестиновых руд (~ 60 %
SrS04> включает обесшламливание, концентрацию на столах и флота-
цию. На дальнейшую переработку поступает целестиновый концент-
рат, содержащий более 90 % SrS04.
Высококачественный целестиновый концентрат содержит не менее
95 % SrS04, например, %: SrS04 98,6; BaSO4 0,25; CaSO4 0,11; РегОз +
Д120з 0,26; SiO2 0,68; СаСОз 0,01.
Стронцианитовые месторождения разрабатываются не только карь-
ерным, но и более дорогими подземными горными способами. Руда
обычно содержит 10—15 % минерала, поэтому ее подвергают обяза-
тельному обогащению, включающему ручную рудоразборку, мокрую
отсадку и концентрацию на столах. В результате обогащения получают
концентрат с содержанием 85 % SrCCh, который по качеству уступает
целестиновому. Типичный состав стронцианитового концентрата (Гер-
мания), %: БгСОз 90,9; СаСОз 7,5; ВаСОз 0,1; РегОз 0,05; AI2O3 0,1;
SiO2 0,04; S 0,1; Р 0,005.
Запасы стронция в пересчете на оксид в зарубежных странах на
начало 1990 г. оценивались в 14,3 млн.т, в том числе извлекаемые 8
млн.т. Первое место в мире по выпуску целестиновых концентратов
занимает Мексика; в 1990 г. он составлял 30 % мирового, что почти
вдвое выше уровня 1988 г. В табл. 4.2 приведена динамика производства
Целестиновых концентратов.
Таблица 4.2 Производство целестиновых концентратов, тыс.т. SrO
Страна Год Доля, % (1990 г)
1985 1987 1990
Мексика 12,9 25,1 48,3 38,3
Турция 14,1 14,1 29,0 23,0
Испания 11,0 13,9 20,3 16,1
Великобритания 6,8 6,0 12,7 10,1
Иран 1,9 8,9 11,2 8,9
Алжир 2,2 2,2 2,7 2,1
Прочие 1,8 3,8 1,9 1.5
—Всего 50,7 74,0 126,1 100,0
171
Цены на 92—94 % -ный целестиновый концентрат с 1985 по 1989
возросли с 27,6 до 84,9 долл, за 1 т. Цена на карбонат стронция в С1Щ
за тот же период оставалась стабильной на уровне 771 долл, за 1 т.
Ведущими потребителями стронциевой продукции являются Яц
ния и США. В 1988 г. в Японии использовано 55 тыс.т БгСОз. Основной
объем потребления приходится на цветное телевидение. Структура цд,
требления стронция в США в 1988 г. приведена ниже, %:
Цветное телевидение.......68
Ферриты...................11
Пиротехника...............10
Пигменты и красители.......4
Прочие.....................7
4.8. Производство соединений стронция
Переработка целестина. Сущность карботермического методам
ключается в переводе сульфата стронция в растворимый в воде сульфщ
с последующим выщелачиванием его и выделением из раствора соле
стронция (рис. 4.1). Целестиновый концентрат смешивают с углем в
соотношении 67 % концентрата и 33 % угля; шихту обжигают во вра
щающейся трубчатой или шахтной печи при температуре 950—1100 'С.
При этом сульфат стронция восстанавливается до сульфида
SrSO4+2C=SrS + 2CO2; (4.4)
SrSO4 + 4С = SrS + 4СО. (4.5)
Продукт восстановления (спек) представляет собой серую пористую
массу, содержащую, %: 70—80 SrS, 3—5 SrSO4,2,5 РезОз+AI2O3, 5—Ю
SrCCh, 2—5 угля и 8—13 SrSiOa. Для получения гидроксида стронция
спек выщелачивают водой (90—95 °C) при перемешивании; при это
сульфид стронция в растворе подвергается гидролизу
2SrS+2H2O=Sr(OH)2+Sr(HS)2. (4.6’
Пульпу отжимают на фильтре и промывают остаток водой. Он со-
стоит в основном из сульфата и силиката стронция. После повторного
выщелачивания остаток направляют в отвал. Раствор охлаждают №
кристаллизации гидроксида стронция, который центрифугируют и о*
мывают водой или разбавленным раствором щелочи от гидросульфит
стронция. Из маточного раствора выделяют карбонат стронция, для че
через раствор пропускают углекислый газ
Sr(HS)2+CO2+H2O=SrCO3+2H2S. (4-
Образующийся при этом сероводород улавливают и окисляют >
серы или используют для получения серной кислоты. Карбонат стр0*1
ция отжимают на центрифуге или вакуумном фильтре, промываю*
сушат при 120 °C. Он является исходным продуктом для получе®*’’
других солей стронция. Полученный таким способом карбонат cTpoHfl*’
172
Целестиновый Уголь
Sr(NO3)2
Рис. 4.1. Технологическая схема переработки целестиновых кон-
центратов карботермическим способом
обычно загрязнен сульфидными соединениями. Иногда в технологиче-
скую схему включают стадию очистки от серы, для этого сульфидный
Раствор стронция после выщелачивания обрабатывают азотной кисло-
той при 60—70 °C. Выпавшую в осадок серу отделяют, а из раствора
осаждают карбонат стронция, как было описано выше. Извлечение
стронция составляет 80—85 %.
Содовый способ заключается во взаимодействии сульфата стронция
и Углекислого натрия
173
SrS04 + NaCOa = S1CO3 + NazSCU. (4.8)
В результате образуются нерастворимый в воде карбонат стронция
и растворимый сульфат натрия. Реакция сдвигается вправо при избытке
соды по сравнению с теоретическим количеством (рис. 4.2). Существуют
два примерно равнозначных варианта содового метода переработки це-
лестина, которые отличаются первой стадией процесса — условиями
взаимодействия сульфата стронция с содой. По первому варианту целе-
стиновый концентрат спекают с содой во вращающихся трубчатых пе-
чах (соотношение компонентов 1 : 1 по массе) при — 800 °C. Спек
измельчают и выщелачивают водой; при этом в раствор переходит суль-
фат натрия, а технический карбонат стронция направляют на очистку
от примесей.
Второй вариант содовой схемы предусматривает обработку целести-
нового концентрата раствором соды. Для этого концентрат, измельчен-
ный до — 4 мм, обрабатывают горячим раствором соды (200 г/л ЫагСОз,
90—95 °C) при перемешивании в течение 30—40 мин. После отстаива-
ния пульпы раствор сливают, а остаток обрабатывают свежим раство-
ром соды при нагревании (90—95 °C) в течение 1,5 ч; соды при этом
берут 50 % сверх теоретически необходимого, что соответствует отно-
шению целестин : сода = 1 : 0,8. Образующийся карбонат стронция
отфильтровывают, отмывают горячей водой от сульфат-ионов и раство-
ряют в азотной кислоте. Содовый раствор поступает в оборот. Для очи-
стки азотнокислого стронцийсодержащего раствора от железа и алюми-
ния pH его доводят аммиаком до 5—6 и отделяют гидроксиды этих
элементов фильтрованием. Раствор осветляют и упаривают примерно
на 80 % от первоначального объема; выпавшие кристаллы азотнокисло-
го стронция отжимают от маточного раствора, промывают и высушива-
ют. Для получения чистой соли азотнокислый стронций перекристал-
лизовывают из водного раствора. Маточный раствор после кристаллиза-
ции нитрата стронция направляют на осаждение карбоната стронция.
Карбонат стронция отжимают на центрифуге, промывают и сушат при
120 °C. Конечные продукты схемы — карбонат и нитрат стронция дол-
жны удовлетворять следующим требованиям, %:
Азотнокислый стронций
Sr(NO3>2............. > 99,0
Ca_(NO3)2............ <0,2
Cl".................. < 0,01
Тяжелые металлы...... < 0,01
Нерастворимый остаток . . < 0,2
Fe.................... <0,1
Н2О....................<0,5
Углекислый стронций
SrCO3................>95
СаСОз................<3
С1 ..................<0,05
Тяжелые металлы......< 0,05
Нерастворимый остаток . . < 0,25
Н2О..................<0,5
174
Вариант 1
Вариант 2
Технический
&С03
Рис. 4.2. Технологическая схема переработки целестиновых концентратов
содовым способом
175
Извлечение стронция при переработке целестиновых концентратов
содовым методом ~ 80 %.
Переработка стронцианитовых концентратов. Для переработки
стронцианитовых концентратов можно использовать несколько мето-
дов, так как стронцианит гораздо более реакционноспособен, чем целе-
стин. Один из способов переработки стронцианита — термическое
разложение. Стронцианит обжигают при 1200 °C, а затем оксид строн-
ция выщелачивают водой
SrCO3=SrO + CO2; SrO + H2O = Sr(OH)2< (4.10
Температура диссоциации карбоната стронция значительно снижа-
ется в присутствии угля (до 900 °C).
Рекомендуется разлагать стронцианит лид давлением 100 кПа пере-
гретым до 500—600 °C водяным паром; образующийся при этом гидро-
ксид стронция путем обработки соответствующими кислотами пере-
водят в нужные соли стронция.
Стронцианит можно разлагать и соляной кислотой; при этом обра-
зуется хлорид стронция
Si-СОз+2HCl=SrC12+НгО. (4.11)
Затем в раствор добавляют щелочь и охлаждают до начала кристал-
лизации Sr (ОН) г ’8 НгО, который отделяют от раствора
SrC12 + 2NaOH = Sr(OH)2 + 2NaCl. (4.12)
В результате обработки стронцианита азотной кислотой образую-
щийся 8г(ЫОз)2 выделяется из раствора или кристаллизацией при упа-
ривании, или добавлением избытка HNO3 (высаливание).
4.9. Переработка апатита
Кольский апатитовый концентрат — перспективное и уникальное
сырье для комплексной переработки, позволяющее получить, помимо
основного продукта — фосфорных удобрений, соединения стронция, 3
также редкоземельных элементов и фтора. Комплексная переработка
апатита позволяет извлекать стронций без дополнительных расходов на
разработку месторождений и добычу сырья. Тем более, что при азотно-
кислотной переработке апатита стронций может быть выделен в bjW
нитрата, переработка которого в карбонат существенно проще перера-
ботки целестина.
Апатитовый концентрат перерабатывают по двум принципиально
отличающимся технологическим схемам — сернокислотной (— 80 %
перерабатываемого сырья) и азотнокислотной.
В основе различных вариантов сернокислотной схемы переработки
апатита лежат следующие реакции разложения минерала:
176
Са5(РО4>зР + 5H2SO4 + 5лН2О----
—r— 5CaSO4-nH2O+3H3PO4 + HF; (4.13)
Ca5(PO4)3F + 7НзРО4 = 5Са(Н2РО4)2 + HF; (4.14)
4HF + SiO2 ----- SiF4 + 2H2O; (4.15)
SiF4+(л+2)Н2О -----» 2H2SiF6 + nH2O. (4.16)
При разложении апатитового концентрата смесью серной и оборот-
ной фосфорной кислот стронций полностью переходит в осадок сульфата
кальция (фосфогипс); РЗЭ распределяются между осадком фосфогипса
(60—80 %) и фосфорнокислым раствором (20—40 %), из которого затем
переходят в удобрения. Выделяющийся при разложении апатита фтор
взаимодействует с содержащимся в сырье кремнеземом, образуя тет-
рафторид кремния SiF4 и гексафторсиликат водорода H2SiF6, который
остается в удобрениях, переходя в малорастворимые соли — гексафтор-
силикаты. Из общего количества фтора, содержащегося в сырье, в удоб-
рения попадает 55—60 %, остальное удаляется с вентиляционными
газами в виде S1F4 и частично HF.
Разновидностью сернокислотного способа переработки апатита яв-
ляется разложение его фосфорной кислотой1). При этом стронций и РЗЭ
оказываются вместе с кальцием в фосфорнокислом растворе и впослед-
ствии попадают в удобрения.
Предложены способы извлечения стронция из фосфогипса, однако
все они предполагают полное разложение фосфогипса, что экономиче-
ски нецелесообразно. Выделение РЗЭ из фосфорнокислых растворов
после осаждения фосфогипса тоже не представляется экономически оп-
равданным, так как в результате распределения их между раствором и
фосфогипсом, извлечение РЗЭ будет очень мало. Представляется более
рациональным извлечение стронция и РЗЭ из фосфорнокислых раство-
ров, полученных после разложения апатита фосфорной кислотой. Одна-
ко такие сведения в литературе отсутствуют.
Азотнокислотный способ. Несмотря на явную экономическую и
экологическую целесообразность комплексной переработки апатита, в
настоящее время существует единственная промышленная схема, пре-
дусматривающая попутное извлечение РЗЭ, стронция и утилизацию
фтора, основанная на разложении апатита азотной кислотой. При пере-
работке апатита по азотнокислотной схеме РЗЭ вместе с кальцием пе-
реходят в раствор; 80—85 % содержащегося в апатите фтора также
переходит в азотнокислотную вытяжку; около 1 % фтора остается в виде
не разложившегося фторапатита, остальное количество выделяется в
газовую фазу в основном в виде тетрафторида кремния
1 Этот способ применяется при производстве двойного суперфосфата.
177
Ca5(PO4)3F + 10HNO3 = 5Ca(NO3)2+3H3PO4 + HF. (4.17,
Комплексная технологическая схема переработки апатита включа-
ет следующие основные стадии (рис. 4.3):
— разложение апатитового концентрата азотной кислотой;
— кристаллизация и отделение нитрата стронция;
— кристаллизация и отделение нитрата кальция;
— извлечение фтора и РЗЭ из азотнофосфорнокислого раствора;
— переработка азотнофосфорнокислого раствора на удобрения.
Апатит Азотная кислота
Рис. 4.3. Технологическая схема комплексной переработки апатита
178
Разложение апатита азотной кислотой протекает легко в широком
диапазоне технологических параметров. Известно несколько режимов
разложения апатита азотной кислотой. Обычно используют 58—60 %-
ную HNO3, которую берут в количестве 110—115 % от стехиометрии,
при температуре 50—60 °C в течение 90 мин. Продуктами взаимодейст-
вия являются фосфорная кислота, фторид водорода, нитраты кальция,
стронция и РЗЭ. Степень разложения апатита 99 %. При увеличении
концентрации HNO3 переход нитратов в раствор уменьшается.
В описанных выше условиях разложения апатита большая часть
нитрата стронция высаливается из раствора и оказывается в твердой
фазе вместе с нерастворимым остатком от апатита. Отделение его осу-
ществляется фильтрованием, которому обычно предшествует сгущение.
Извлечение стронция в концентрат составляет — 60 % по отношению к
содержанию в апатите. Нитрат стронция хорошо растворим в воде, и его
ограниченная растворимость в азотнофосфорнокислом растворе объяс-
няется высаливающим действием соединений с одноименными ионами
— HNO3 и Са(ЫОз)2. Увеличение концентрации HNO3 от 5 до 46 %
приводит к уменьшению содержания в растворе нитрата стронция от
39,55до 1,15 %.
Раствор после отделения стронциевого концентрата направляют на
кристаллизацию нитрата кальция, которую осуществляют «выморажи-
ванием» его при -15 °C; при этом ~ 90 % Са(ЫОз)2 '4Н2О выделяется в
твердую фазу. Кристаллы тетрагидрата нитрата кальция отделяют от
маточного раствора фильтрованием или центрифугированием и промы-
вают 58—60 %-ной азотной кислотой. Промывной раствор направляют
на стадию разложения апатита.
Маточный раствор после отделения Са(МОз)2 '4Н2О содержит ~ 97
% Р2О5; 85 %F и 93 % РЗЭ (от введенных с апатитом). Он поступает на
стадию выделения фтора. С этой целью раствор обрабатывают содой или
Другими солями натрия для осаждения фтора в виде Na2SiF6. Для отде-
ления осадка кремнефторида требуется предварительное сгущение сус-
пензии. Из полученного раствора выделяют фосфаты РЗЭ путем час-
тичной нейтрализации их аммиаком до pH 1,4 при 80—90 °C. Степень
ведения РЗЭ около 85 % по отношению к содержанию в растворе.
Раствор после отделения РЗЭ направляют на получение удобрений.
Концентрат РЗЭ содержит 25 % суммы РЗЭ (в пересчете на оксиды),
12,5 % СаО и 33,0 % Р2О5, а также соединения железа и алюминия. При
Переработке 1 т апатита можно получить около 30 кг концентрата.
Извлечение РЗЭ в концентрат ~ 75 % по отношению к содержанию в
апатите.
Стронциевый концентрат, выделенный из апатита, может быть пе-
реработан на различные соединения стронция после очистки от приме-
Сей. В концентрат попадают сопутствующие апатиту минералы, сос-
179
тавляющие нерастворимый остаток апатитового концентрата ильме
нит, сфен, титаномагнетит; кроме того, он содержит железо, алюминщ
фториды, фосфаты, а также соли кальция. Для очистки от примесей
концентрат Sr выщелачивают водой; при этом нерастворимые в вод(
минералы остаются в твердой фазе, и их отделяют от раствора. Зате-
водный раствор нитрата стронция обрабатывают аммиаком: при pH
6—7 выпадает осадок, содержащий фосфаты и фториды железа, алюмц
ния и стронция, гидроксиды железа и алюминия.
После отделения осадка получают раствор нитрата стронция с при
месью кальция, который обрабатывают азотной кислотой с целью выса
ливания кристаллического нитрата стронция. Кристаллы Sr(NO3)2
растворяют в воде и полученный раствор нитрата стронция направляют
на выделение карбоната, для чего через раствор пропускают диоксид
углерода. Маточный раствор со стадии кристаллизации Sr(NOs)2 на-
правляют на разложение апатита. Полученный таким образом карбонат
стронция содержит 98,3 % основного вещества и ~ 1 % СаСОз.
При переработке РЗЭ-концентрата необходимо обеспечить возврат
в основную технологическую схему фосфора, потери которого составля-
ют - 3 % по отношению к апатиту. Переработка РЗЭ-концентрата
может быть осуществлена несколькими путями. Наиболее распростра
ненный основан на растворении его в азотной (реже соляной) кислотах
с последующим осаждением оксалатов РЗЭ. Возможна также обработка
РЗЭ-концентрата раствором щелочи при нагревании с получением осад
ка гидроксидов РЗЭ и раствора фосфата натрия. Осадок затем растворя-
ют в кислотах (азотной или соляной) и получают раствор РЗЭ, сво-
бодный от фосфат-ионов. Из этого раствора РЗЭ могут быть извлечены
экстракционными или осадительными методами.
При создании комплексной технологии переработки апатита необ-
ходимо учитывать, что апатит является главным образом источником
фосфора, и технология его переработки должна обеспечивать макси-
мальное извлечение этого элемента в удобрения. Следовательно, техно-
логия выделения стронция и РЗЭ не должна существенно изменять и
усложнять процесс переработки апатитового концентрата на удобрения,
ухудшать их качество и, таким образом, всегда должна рассматриваться
как побочный процесс.
4.10. Получение металлического стронция
Для получения металлического стронция можно использовать раз*
личные методы — термическое разложение некоторых соединений*
электролиз и металлотермическое восстановление соединений строН'
ция.
180
Термическое разложение соединений. Для этих целей наиболее под-
ходящими являются гидрид SrH2 и нитрид SraN2. Разложение SrH2
проводят в стальной трубке в вакууме. Процесс начинается при 675 °C
и заканчивается при 1100 °C. Чистота полученного продукта составляет
- 98,8 %. При разложении нитрида стронция в трубке из йенского
стекла в вакууме при 140—150 °C был получен черный мелкокристал-
лический порошок металлического стронция. Эти способы получения
металла не нашли промышленного применения вследствие ряда причин:
при разложении получается мелкодисперсный легко окисляющийся по-
рошок стронция, исходные материалы — SrH2 и 8гз1Чг — получают или
из чистого металла, или из его соединений высокой чистоты.
Электролиз. Металлический стронций можно получать электроли-
зом расплава смеси 80—85 % SrCh и 20—25 % КС1 с жидким катодом.
Процесс ведут при 650—700 °C; анодом служит пакет графитовых бло-
ков. Получающийся металлический стронций загрязнен солями — нит-
ридом и оксидом, кроме того, выход по току очень мал. В связи с этим
метод не получил широкого распространения. В промышленности элек-
тролизом на жидком катоде получают сплавы стронция, например с
оловом.
Металлотермия. Для получения металлического стронция приме-
няют преимущественно металлотермические методы. В качестве метал-
лов-восстановителей были в разное время предложены почти все ме-
таллы первых четырех групп Периодической системы элементов Д.И.
Менделеева. Практическое значение получил лишь метод алюмотерми-
чсского восстановления оксида стронция. Сущность метода заключает-
ся в восстановлении оксида стронция алюминием с последующей дис-
тилляцией полученного металла и конденсацией его на охлаждаемой
поверхности. Оксид стронция получают путем термического разложе-
ния его нитрата или карбоната при 1050—1100 °C. Содержание SrO в
полученном оксиде 96—98 %. Оксид стронция смешивают с порошком
алюминия и брикетируют. Брикеты загружают в стальную реторту и
помещают ее в вакуумную электрическую печь. Восстановление оксида
стронция протекает по реакции
4SrO+2Al=3Sr+SrO*Al203. (4.18)
Реакция начинается при 850—900 °C с образованием сплава строн-
ция с алюминием. Дальнейшее нагревание в вакууме при давлении
10,2—20,4 Па и температуре 1100—1150 °C приводит к возгонке строн-
ция. Пары стронция конденсируются на охлаждаемой поверхности
вставленного в реторту конденсатора. По окончании восстановления
реторту заполняют аргоном и расплавляют конденсат, который стекает
в изложницу. Продуктами реакции являются парообразный стронций и
твердый моноалюминат стронция.
181
рис. 4.4. Электровакуумная печь для получения металлического стронция:
1 — термопара; 2 — колпак; 3 — центральный нагреватель, 4 — патрубок вакуумный;
у — лампа вакуумметра, 6 — прокладка резиновая; 7 — предохранительный стакан, 8 —
реторта; 9 — печь; 10 — изложница; 11 — нагреватель; 12 — корзина для шихты; 13 —
окно смотровое; 14 — токоввод; 15 — патрубок контрвакуумный
Металлический стронций получают в промышленных электропечах
с верхней или нижней конденсацией металла (рис. 4.4). Она состоит из
собственно печи, реторты, корзины для шихты, изложницы и колпака с
цилиндрическим конденсатором. Внутри конденсатора помещается на-
греватель. Оксид стронция, измельченный до — 0,07 мм, и порошко-
образный алюминий, взятые в мольном отношении SrO : Al = 2 : 1, пе-
ремешивают в барабанном смесителе. Смесь прессуют под давлением
30 кПа в брикеты плотностью 3,1—3,3 г/см3 и загружают в корзину. В
реторту, установленную в печь, помещают корзину с шихтой и излож-
ницу, реторту закрывают колпаком. Необходимое давление в реторте
10,2—20,4 Па. Печь имеет три температурные зоны — зона восста-
новления оксида стронция и дистилляции образовавшегося металла
(1200 °C), зона конденсации стронция (400—500 °C), его плавления
(850—900 °C) и зона изложницы, охлаждаемая водой. Печь нагревают
до заданной температуры (1200 °C) 4—5 ч. По мере нагрева алюминий
взаимодействует со SrO, стронций возгоняется и осаждается на конден-
саторе. По окончании процесса восстановления (5—6 ч) реторта запол-
няется аргоном. Для расплавления стронция конденсатор перемещается
в зону нагрева. При 850—900 °C металл плавится и стекает в
изложницу. Вследствие образования алюмината теоретический выход
металла до 75 %. Остатки после восстановления (алюминат стронция)
содержат ~ 50 % SrO, их переводят в карбонат или нитрат для извлече-
ния стронция. Чистота металла ~ 99 0 %. Содержание примесей в ме-
таллическом стронции, %: Fe 2-10 ; Zn 2-10 ; Pb 4-10 4; Cr 2-10 4;
Си 6 10 4; Cd 2 10 4. Для получения стронция высокой чистоты его
повторно возгоняют в вакууме при 1000 °C.
Раздел III. Редкие элементы III Б подгруппы.
Редкоземельные элементы и скандий
ОБЩАЯ ХАРАКТЕРИСТИКА ЭЛЕМЕНТОВ I
Электронная конфигурация. Степени окисления. Лантаноидами
т.е. подобными лантану, называются элементы III Б подгруппы Перио-
дической системы Д.И. Менделеева, у которых происходит заполнение
4/-подуровня. Свое название они получили от лантана, как элемента-
прототипа, хотя он и является d-элементом. Для обозначения этой груп-
пы элементов вместе с иттрием, так же d-элсментом, но имеющим очень
близкие свойства с лантаноидами, используется термин «редкоземель-
ные элементы» (РЗЭ). Иногда в группу РЗЭ включают и скандий, одна-
ко между ним и лантаноидами больше различий, чем сходства.
При увеличении порядкового номера элемента электронные оболоч-
ки К, L, М, а также подуровни s, р, d оболочки N заполняются в нор-
мальной последовательности. Но затем она нарушается: вместо под-
уровня 4/ заполняются 5s и 5р, затем и бя-подуровни. Только с 58-го
элемента — церия — начинается заполнение 4/-орбиталей. Однако
вследствие энергетической близости 4/ и 5с/-орбиталей заполнение 4/-
подуровня протекает нерегулярно — у церия, гадолиния и лютеция
появляется по одному d-электрону (табл. 5.1).
При возбуждении атома лантаноида, что всегда имеет место при
химическом взаимодействии, один из /-электронов, реже — два, пере-
ходит в d-состояние. В результате /—d-перехода возникает электронная
конфигурация 5d'6s2, которая в основном и определяет химические
свойства лантаноидов и их исключительное сходство. Этим объясняется
и то, что наиболее устойчивая степень окисления всех лантаноидов
равна +3. Остальные 4/-электроны, экранированные от внешнего воз-
действия двумя электронными оболочками, на химические свойства
лантаноидов оказывают значительно меньшее влияние. Существенной
является повышенная устойчивость электронных конфигураций 4/°.
4/7, 4/14; с ней связана вторичная периодичность в группе лантаноидов
и относительная стабильность степеней окисления +2 и +4 в тех случаях,
когда реализуются такие или близкие к ним электронные конфигура-
ции. Необычные степени окисления связаны с возможностью /—d-пере-
хода, которая зависит от заселенности 4/-подуровня и качественно мо-
жет быть оценена из рассмотрения зависимости разности энергий
состояний/* 'd's2 и fxs2 (рис. 5.1). Положительные значения лЕ харак-
теризуют уменьшение вероятности /—d-перехода, а следовательно, по-
явление степени окисления +2, отрицательные — свидетельствуют °
большей вероятности таких переходов и степени окисления +4.
184
Таблица 5.1. Электронные конфигурации атомов РЗЭ и скандия
Элемент Уровни и подуровни
К L 3s Зр 3d 4s 4р 4d 4/ 5s 5р 5d 6s
21. Sc 2 8 2 6 1 2
39. Y 2 8 18 2 6 1 2
54 Хе 2 8 18 18 2 6
57 La 2 8 18 18 2 6 1 2
58. Се 2 8 18 18 1 8 1 2
59 Рг 2 8 18 18 3 8 2
60. Nd 2 8 18 18 4 8 2
61. Pm 2 8 18 18 5 8 2
62. Sm 2 8 18 18 6 8 2
63. Eu 2 8 18 18 7 8 2
64 Gd 2 8 18 18 7 8 1 2
65 Tb 2 8 18 18 9 8 2
66 Dy 2 8 18 18 10 8 2
67 . Ho 2 8 18 18 11 8 2
68 . Er 2 8 18 18 12 8 2
69 Tm 2 8 18 18 13 8 2
70 Yb 2 8 18 18 14 8 2
71. Lu 2 8 18 18 14 8 1 2
Энергия ионизации — количественная характеристика, непосредст-
венно связанная с электронной структурой атома, — позволяет судить
о возможных степенях окисления
атомов данного элемента и их от-
носительной устойчивости (табл.
5.2). Так, энергия отрыва 3-го
электрона Оз) имеет наибольшие
значения для европия, иттербия и
самария, так как в этом случае
нужно удалить один электрон из
полностью или наполовину запол-
ненного 4/-подуровня. Для этих
элементов легче будет реализо-
ваться степень окисления +2.
нергия отрыва 4-го электрона
Минимальна для церия, тербия и
Празеодима, у них и реализуется
Степень окисления +4; менее веро-
< 20
53 10
О
-10
-20
-30
Рис. 5.1. Разность энергий электронных
конфигураций У*- d s —- f*s в зависимо-
сти от числа 4/электронов х:
х-1(Се), ...,Х14 (Yb)
185
ятна она у неодима, диспрозия и гольмия и совершенно исключается у
лантана, гадолиния и лютеция. Лантаноиды в таких степенях окисления
могут быть преимущественно в твердых соединениях, наблюдается их
стабилизация в сложных комплексных соединениях.
Таблица 5.2. Энергия ионизации атомов и катиона РЗЭ
и скандия 21,.., эВ. Работа выхода электрона А, эВ
Элемент /1 22 /3 24 A ГА I 3+ Окраска иона Ln
21. Sc 6,54 12,80 24,75 73,76 3,23
39. Y 6,38 12,23 20,51 61,77 3,07
57. La 5,58 11,06 19,18 49,95 3,33 Бесцветная
58. Се 5,47 10,85 20,20 36,76 2,84 Бесцветная
59. Рг 5,42 10,55 21,62 38,98 2,70 Желто-зеленая
60. Nd 5,49 10,73 22,10 40,41 3,30 Красно-фиолетовая
. 61. Pm 5,55 10,90 22,30 41,10 Розовая
62. Sm 5,63 11,07 23,10 41,40 3,20 Желтая
63. Eu 5,67 11,24 24,92 42,60 2,54 Бледно-розовая
64. Gd 6,14 12,09 20,63 44,00 3,07 Бесцветная
65. Tb 5,85 11,52 21,91 39,79 3,09 Бледно-розовая
66 Dy 5,93 11,67 22,80 41,47 3,09 Желтая
67. Ho 6,02 11,80 22,84 42,50 3,09 Коричнево-желтая
68. Er 6,10 11,93 22,74 42,65 3,12 Розовая
69. Tm 6,18 12,05 23,86 42,69 3,12 Бледно-зеленая
70. Yb 6,25 12,19 25,03 43,74 2,59 Бесцветная
71. Lu 5,43 13,90 20,96 45,19 - J 3,14 Бесцветная
Для получения соединений лантаноидов в степени окисления +4 в
качестве окислителей обычно используют кислород под давлением и
повышенной температуре либо дифторид ксенона XeF2. Такими мето-
дами были получены высшие оксиды, соединения типа БгТЬОз и
Rb(Cs) 3NdF?. Соединения лантаноидов в степени окисления +2 получа-
ют восстановлением щелочными металлами либо соответствующими
металлическими лантаноидами. Процессы восстановления галогенидов
лантаноидов протекают очень медленно (5—7 сут) при температуре Д°
850 °C. Так были получены соединения типа NdCh и K(Li)Ln2C15- ПрЯ'
мым синтезом из элементов были получены халькогениды типа NdS и
ТтпТе, однако они могут считаться соединениями лантаноидов в степени
окисления +2 только формально, так как содержат кластеры Ln—Ln-
Соединения европия, самария и иттербия в степени окисления +2 в
186
водных растворах можно получить восстановлением порошком цинка,
амальгамой натрия и электрохимически.
Теоретически не исключена возможность существования лантанои-
дов в степени окисления +1. при этом должны реализоваться электрон-
ные конфигурации 4/*+16sI. По спектрам люминесценции п^и повы-
шенной температуре было обнаружено присутствие ионов Ей и Sm в
системах Eu(Sm)—КС1—К, а масс-спектрометрически — соединения
типа LnCl для многих лантаноидов над смесями Ln—BaCl? при 1100 К.
Твердые соединения LnCl имеют слоистую структуру, изоструктурны
ZrCI, проявляют свойства металлов, содержат кластеры Ьпб- Их можно
только формально считать соединениями Ln 1 (табл. 5.3).
Таблица 5.3. Степени окисления лантаноидов и лантана
La Се Рг Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Yb Lu
3 4 3 4 3 (4) 3 3 3 3 3 4 3 (4) 3 (4) 3 3 3 3 3
1 (2) 1 (2) 1 (2) 1 2 2 1 (2) 1 (2) 1 (2) 1 (2) 1 (2) 2 1
Атомные и ионные радиусы. Важнейшими характеристиками эле-
мента являются размеры его атома и иона. У лантаноидов, имеющих
практически одинаковые конфигурации валентных электронов, разли-
чия в свойствах отдельных элементов в значительной степени будут
определяться размерным фактором, т.е. соотношениями орбитальных,
атомных и ионных радиусов.
Орбитальный атомный радиус г0, характеризующий свободные ато-
мы и определяемый расчетным путем, равен расстоянию от центра ядра
атома до максимума электронной плотности внешней электронной ор-
битали. При возрастании числа электронов на любом подуровне из-за
кулоновского притяжения оболочка сжимается и орбитальный радиус
Уменьшается. При возникновении новой оболочки орбитальный радиус
Увеличивается скачком, а затем при возрастании Z оболочка снова стя-
гивается к ядру. У лантаноидов заполняется глубоко лежащий подуро-
вень 4/, и в ряду от церия до лютеция го уменьшается монотонно. Из
последовательности выпадают только гадолиний и лютеций, имеющие
несколько меньшие радиусы, чем следовало бы ожидать (табл. 5.4).
Атомные радиусы гат получают экспериментально из межатомных
Расстояний в структурах простых веществ — эта величина равна поло-
вине кратчайшего межатомного расстояния. В зависимости от типа свя-
зи в простом веществе получают либо металлические гм, либо кова-
лентные радиусы гк- Значения гат и г0 не совпадают вследствие того, что
187
в реальном веществе имеет место перекрывание орбиталей. Лантаноид^
— металлы, их атомные радиусы могут называться также металличе-
скими. Они монотонно уменьшаются от церия к лютецию, и только у
европия и иттербия монотонность нарушается; у них радиусы значи-
тельно больше, чем следует из последовательности (рис. 5.2). Можно
предполагать, что вследствие устойчивости конфигураций 4/7 и 4/14.
больших значений энергии перехода /— d-валентное состояние атомо|
этих элементов в кристаллической структуре металлов будет опреде-
ляться конфигурацией 6s2 или 5d*6s , для остальных же — 5а 6.<,2
Полагая, что атомы, находящиеся в узлах кристаллической решетки,
отдают все валентные электроны в зону проводимости, можно сделатж
вывод о том, что европий и иттербий будут находиться в виде двухзаряд-
ных ионов. Радиус же двухзарядного иона должен быть больше, чем
трехзарядного. Для различных кристаллических модификаций атом-
ные радиусы также будут различными, для металлов обычно приводят
радиусы для к.ч.12, для других к.ч. их значения будут меньше: к.ч.8 —
на 2 %, к.ч.6 — на 4 %, к.ч.4 — на 12 %.
Таблица 5.4. Орбитальные, атомные и ионные радиусы РЗЭ и скандия
(ионные радиусы — по Шеннону и Прюитту)
Эле мент Fq.HM гат.нм I 3+ Гц Ln ,HM Эле- мент ГО.НМ гат.нм 1 3+ Гц Ln ,HM
К.Ч.6 к.ч.8 К.Ч.6 к.ч.8
21.Sc 0,157 0.164 0,089 0,101 64.Gd 0,171 0,179 0,108 0,119
39.Y 0,169 0,181 0,104 0,116 65.Tb 0,178 0,177 0,106 0,118
57.La 0,192 0,187 0,117 0,130 66.Dy 0,175 0,177 0.105 0,117
58.Ce 0,198 0,183 0,115 0,128 67.Ho 0,173 0,176 0,104 0,116
59.Pt 0,194 0,182 0,113 0,127 68.Er 0,170 0,175 0,103 0,114
60.Nd 0,191 0,182 0,112 0,125 69.Tm 0,168 0,174 0,102 0,113
61.Pm 0,188 0,181 0,111 0,123 70 Yb 0,166 0,193 0,101 0,113
62.Sm 0,185 0,180 0,110 0,122 71.Lu 0,155 0,174 0,100 0,112
63. Eu 0,183 0,202 0,109 0,121
Из межатомных расстояний катион-анион для серии изоморфных
ионных соединений строится система ионных радиусов ги. Систем ион-
ных радиусов предложено большое число; в табл. 5.4 приведены ионное
радиусы по Шеннону и Прюитту, которые для построения своей системы
исходили из межатомных расстояний Ln—О, радиус иона кислорода О
был принят равным 0,126 нм. По их данным ионные радиусы Ln в ряДУ
188
1аНтаноидов монотонно уменьшаются и их можно рассчитать по фор-
мулам (5.1,5.2)
к.ч.6ги=0,1178 — 0,0014л; (5.1)
к.ч.8ги=0,1298 —0,0014и, (5.2)
где и ~ 1 Для церия, п = 14 для лютеция.
Ионные радиусы изменяются в зависимости от лиганда, и в особен-
ности при изменении координационного числа. При переходе от соеди-
нений с к.ч. 6 к соединениям с к.ч. 8 ионный радиус центрального атома
увеличивается приблизительно на 8 %, а при увеличении к.ч. до 12 —
на 12 %-
На свойствах лантаноидов, изменяющихся немонотонно при увели-
чении Z (атомный радиус, удельный объем, плотность, температура
плавления и кипения и др.), обнаруживается вторичная периодичность.
Особенно четко она выявляется на зависимостях разности между интер-
полированными и экспериментальными значениями тех или иных
свойств (рис. 5.2—5.5). В соответствии с ней лантаноиды, а вместе с
ними и всю группу РЗЭ, делят на две подгруппы — легкую (цериевую)
и тяжелую (иттриевую). На ряде таких зависимостей свойств, измерен-
ных прецизионно, можно видеть не только «гадолиниевый угол», но и
разбить лантаноиды на 4 «сегмента»: лантан—неодим, самарий—гадо-
линий, тербий—гольмий, эрбий—лютеций. Закономерно изменяется и
окраска ионов Ln3+ в растворах: цвета первых семи ионов повторяются
у последующих семи, но
расположены они в обрат-
ном порядке. Одинаковую
или близкую окраску обна-
руживают ионы, имеющие
одинаковое число неспарен-
ных 4/-электронов. Бес-
цветны ионы с электронны-
ми конфигурациями /°, /7,
f , а также /, /13. Так как
Окраска обусловлена /—f пе-
реходами, а 4/-подуровень
ГлУбокий, то она мало зави-
сит от внешнего окружения
(см. табл. 5.2).
Ион иттрия имеет раз-
меры, практически совпада-
вшие с ионными радиусами
Г°ЛЬМИЯ И диспрозия, ПОЭТО-
МУ свойства иттрия очень
Л11зки к свойствам тяже-
Рис. 5.2. Атомные гат (Ли ионные радиусы ги (2)
3+ 3+
лантаноидов (Ln ), иттрия и скандия (М , к.ч.6)
189
Рис. 5.3. Температура плавления (/) и плотность
(2) лантаноидов, иттрия и скандия
Рис. 5.5. Экспериментальные «
вычисленные значения эффектив-
. 3+
ных магнитных моментов Ln при
300 К:
ооо — экспериментальные
La Я Яя [и П На Тт Lu
Се Ш Sm Cd Dr £г П
Рис. 5.4. Разность между экспериментальны-
ми и интерполированными значениями
свойств в функции порядкового номера ланта-
ноида (
1 — ионные радиусы, линейная интерпо-
ляция La—Nd, Pm—Gd, Gd—Ho, Er—Lu, 2 —
удельный объем, линейная интерполяция La—
Gd, Gd—Lu
значения;---значения, рассчи-
танные для основных /-состоянии
с учетом аномалии для Sm и Ей; -
— значения, рассчитанные без уче-
та аномалий Sm и Ей
лых лантаноидов. В природе
иттрий ассоциируется преиму-
щественно с тяжелыми ланта-
ноидами, поэтому эту под-
группу часто и называют итт-
риевой.
Несмотря на некоторые на-
рушения монотонности, основ-
ная закономерность в измене'
нии орбитальных, атомных и
ионных радиусов заключается
в их уменьшении при увеличь
нии Z; эта закономерность на
зывается «лантаноидным с*а"
тием». Вследствие лантаноиД"
ного сжатия пары элементов IV, V, VI групп Периодической систем1*’
стоящие до группы лантаноидов и после нее, имеют практически оди113
ковые атомные и ионные радиусы, а следовательно, и очень близки
свойства, разделение их — сложнейшая задача химии и техноло1*^
Атомные радиусы элементов IV, V и VI групп Периодической систем1*’
стоящих до и после группы лантаноидов, приведены ниже, нм:
190
40. Zr. . 0,160 72. Hf. .0,159
41. Nb. 0,145 73. Ta .0,146
42. Mo . .. 0,139 74. W. .. 0,140
Скандий и иттрий являются элементами, их невозбужденные ато-
1ДЫ имеют электронную конфигурацию as, наличие в ней только од-
лого J-электрона обусловливает ее малую устойчивость, поэтому оба
элемента проявляют только одну степень окисления +3. Низшие степени
окисления не реализуются вследствие неустойчивости электронной
конфигурации а , а степень окисления +4 практически невозможна, так
как ионы Sc3* и Y3+ имеют электронные конфигурации благородных
газов — аргона и криптона.
Ион скандия Sc3+ относительно небольшой, его размеры не попадают
вобласть значений ионных радиусов лантаноидов, что является важным
фактором, обусловливающим отличие скандия от лантаноидов (см.
табл. 5.4). В то же время скандий обнаруживает большое сходство с
алюминием, несмотря на то что их электронные структуры различны
(соответственно—3 J14s2 и 3s23p*), но электронные структуры их ионов
соответствуют структуре благородных газов неона и аргона. Химия
скандия относительно проста, она близка к химии соединений алюми-
ния. Но вследствие того, что ион скандия больше иона алюминия, его
соединения меньше подвержены гидролизу. По этой же причине иттрий
и лантаноиды обладают более основными свойствами, чем скандий.
Магнитные свойства. Лантаноиды и их трехзарядные ионы имеют
уникальные магнитные свойства, которые определяются 4/-электрона-
ми и вследствие этого мало зависят от внешнего окружения. Все они,
кроме лютеция, а также лантана и иттрия, имеют неспаренные элект-
роны и обладают парамагнетизмом. По магнитным свойствам лантано-
иды можно разделить на две группы: легкие от церия до европия —
слабомагнитные и тяжелые — от гадолиния до иттербия — сильномаг-
нитные (см. рис. 5.5). Некоторые тяжелые лантаноиды в температурном
интервале ©1—©2 образуют сложные антиферромагнитные структу-
ры; при температуре Т < ©1 (точка Кюри) антиферромагнитное состо-
яние переходит в ферромагнитное, а при Т > ©2 (точка Нееля) возни-
кает парамагнетизм. Гадолиний имеет точку Кюри 290 К, выше этой
ТеМпературы он парамагнитен.
Температура магнитных превращений некоторых лантаноидов при-
ВеДена ниже:
Точка Кюри Точка Нееля Точка Кюри Точка Нееля
61 , К 62 , К 61 , К 62 , К
64. Gd 290 — 67. Но 20 133
65. ТЬ 219 230 68. Ег 20 53
66. Dy 85 174 69. Тт 25 65
191
Между лантаноидами образуются твердые растворы замещения ь
широких интервалах концентраций. Это позволяет создавать разнооб.
разные магнитные сплавы, в том числе и с немагнитными металлами
скандием, иттрием, лютецием. Известно также большое число интерме
таллических соединений лантаноидов с ферромагнитными металла^
— железом, никелем, кобальтом, которые используются в качестве ма
териалов для изготовления постоянных магнитов с высокой энергией
например SmCos, NdzFeuB, Pri7Fe76,5B5Cui,5 и др. Некоторые интер-
металлиды, например TbFe?, обладают гигантской магнитострикций
— анизотропным удлинением при намагничивании. Удлинение может
достигать величины 10 3, т.е. 1 мм на 1 м длины. Это явление использу-
ется при изготовлении различных приборов: магнитострикционных дат
чиков, преобразователей, резонаторов, ультразвуковых устройств. <
Ядерные свойства. Природные лантаноиды состоят из большого
числа изотопов, и только празеодим, тербий, гольмий и тулий имеют по
одному стабильному изотопу. Некоторые изотопы лантана, церия, нео-
дима, самария и лютеция радиоактивны, все они имеют большие пери-
оды полураспада — от 1 1011 до 5 1015 лет. Прометий не имеет ста-
бильных изотопов, наибольшие периоды полураспада имеют изотопь
Рг (18 лет) и 14'Рг (2,6 года) —/3-излучатель, поэтому в природе он
встречается в ничтожно малых количествах. Прометий был открыта
1947 г. в продуктах деления 235U в атомном реакторе.
Большинство лантаноидов имеет высокие значения сечения захвата
тепловых нейтронов; особенно велики они у самария, европия, гадоли-
ния (табл. 5.5). При делении урана в атомных реакторах легкие ланта
ноиды и иттрий составляют около 22 % всех продуктов деления. Многие
образующиеся изотопы обладают необычайно высокими сечениями зах
вата тепловых нейтронов: у149Sm 66000 и у 157Gd — более 150000 барн
Этих изотопов образуется соответственно 1,3 и 0,5 % от суммы всех
осколочных элементов, они являются наиболее опасными ядерными
«ядами». Накопление их и других осколочных элементов приводит к
затуханию ядерных реакций, поэтому ядерное горючее приходится на
правлять на переработку, когда 235U полностью еще не израсходован (не
«выгорел»).
Металлы. Лантаноиды — металлы серебристо-белого цвета, толь*0
празеодим и неодим имеют желтоватый оттенок. Лантаноиды — един
ственные металлы, для которых характерно явление политипии, все они
имеют полиморфные модификации. Большинство их, а также скандии
и иттрий имеют плотноупакованную двухслойную гексагональную кР®
сталлическую структуру (ГПУ), лантан, неодим и празеодим — четН'
рехслойную, самарий — девятислойную, что позволяет представить
кристаллическую решетку либо как гексагональную, либо как ромбоэД
рическую. Церий и иттербий имеют кубическую плотноупакованнУ10
192
структуру типа ГЦК, европий — ОЦК решетку. Высокотемпературные
модификации в основном имеют ОЦК решетки. Многие металлы имеют
гексагональные, ГЦК или ОЦК решетки, поэтому лантаноиды могут
широко использоваться как легирующие добавки. Металлы высокой
степени чистоты пластичны, легко поддаются ковке, твердость их не-
сколько увеличивается от лантана к лютецию. Сплавы церия с другими
лантаноидами и железом пирофорны.
Таблица 5.5 Свойства РЗЭ и скандия
Элемент Кристал- лическая решетка , / 3 G,r/CM Т’пл.’С 7"кип,“С Сечение захвата тепловых нейт- -24 2 ронов, 10 см
21. Sc Гексагональная 2,90 1539 2630 13
39. Y — 4,47 1525 3025 1,4
57. La — 6,15 920 3454 8,9
58. Се ГЦК 6,77 798 3257 0,7
59. Рг Гексагональная 6,77 931 3212 11,2
60. Nd — 7,01 1018 3127 44
61 Pm — 7,26 1168 2460 —
62. Sm Ромбоэдрическая 7,52 1072 1778 6500
63. Eu ОЦК 5,24 822 1597 4500
64. Gd Гексагональная 7,90 1311 3233 44000
65. Tb — 8,23 1360 3041 44
66. Dy — 8,55 1409 2335 1100
67. Ho — 8,80 1470 2720 64
68. Er — 9.07 1522 2510 166
69. Tm — 9,32 1545 1727 118
70. Yb ГЦК 6,97 824 1193 36
71. Lu Гексагональная 9,84 1656 3315 108
На ряде свойств металлических лантаноидов обнаруживается четко
выраженная вторичная периодичность, наибольшие отклонения от мо-
нотонности имеют свойства европия и иттербия. К сказанному ранее
следует добавить, что по химическим свойствам эти металлы очень
близки к щелочноземельным. В отличие от других лантаноидов они
Растворяются, как и барий, в жидком аммиаке.
Электродные потенциалы РЗЭ и скандия имеют отрицательные зна-
чения и лишь немного отличаются от электродных потенциалов щелоч-
ноземельных металлов, их нельзя получить электролизом водных раст-
воров.
193
7-691
Электродные потенциалы £298 (М3+ + Зе = М) лантаноидов, иттрИя
и скандия приведены ниже, В:
£298 >В £298> в £298 > в
21. Sc -2,08 61. Рш -2,42 67 Но -2,32
39 Y -2,37 62. Sm -2,41 68. Ег -2,30
57 . La -2,52 63. Ей -2,40 69. Тш -2,28
58. Се -2,48 64. Gd -2,40 70. Yb -2,27
59 Рг -2,46 65 . Tb -2,39 71 Lu -2,25
60. Nd -2,43 66. Dy -2,35
Металлы отличаются высокой химической активностью: на холоду
медленно, при нагревании быстро разлагают воду с выделением водоро-
да, легко растворяются в кислотах, кроме фосфорной и плавиковой,
вследствие образования защитных пленок малорастворимых солей. Ще-
лочи на них не действуют, амфотерные свойства не наблюдаются.
При комнатной температуре на воздухе лантан, церий, празеодим,
неодим довольно быстро окисляются, покрываясь темной оксидной
пленкой. Другие лантаноиды довольно долго сохраняют металлический
блеск, их окисление начинается при температуре выше 200 °C; иттрий
вследствие образования плотной защитной оксидной пленки устойчив
до 1000 °C.
Глава 5. РЕДКОЗЕМЕЛЬНЫЕ ЭЛЕМЕНТЫ
ХИМИЯ РЕДКОЗЕМЕЛЬНЫХ ЭЛЕМЕНТОВ I
5.1. Соединения с кислородом
Оксиды. При прокаливании гидроксидов, карбонатов, оксалатов и
других термически неустойчивых соединений на воздухе при 800—
1000 °C образуются полуторные оксиды ЬпгОз всех лантаноидов, кроме
церия, празеодима и тербия. Полуторные оксиды — очень прочные
соединения, энтальпии образования их в пересчете на один атом кисло-
рода близки к энтальпиям образования оксидов щелочноземельных эле-
ментов, имеют высокие температуры плавления (табл. 5.6), ДлЯ
сравнения — энтальпия образования ВаО ~ “560 кДж/ моль. Это
позволяет использовать некоторые лантаноиды, например лантан, ®
качестве металла-восстановителя в металлотермических процесса*-
Оксиды имеют разные кристаллические структуры и полиморфные мо-
194
длфикации, что влияет на взаимодействие их оксидов с оксидами других
элементов (рис. 5.6).
Полуторные оксиды являются типичными основаниями, амфотер-
лостью не обладают, основной характер их несколько ослабляется в ряду
от лантана к лютецию. На воздухе, прежде всего оксиды цериевой под-
группы, медленно поглощают пары воды, превращаются сначала в гид-
ратированные оксиды, затем в гидроксид-оксиды, а при кипячении в
воде — в гидроксиды
ЬпгОз + пНгО ---ЬпгОз иНгО ---------• LnO(OH) хНгО -------♦
—-—. Ьп(ОН)з. (5.3)
В этом отношении они напоминают оксиды щелочноземельных ме-
таллов. В воде растворяются плохо (10 5—10 6 моль/л), легко раство-
ряются в азотной и соляной кислотах.
Таблица 5.6. Свойства оксидов лантаноидов, иттрия и скандия
Оксиды , / 3 G,r/CM Тля, "C “ АЯ298(/)’ кДж/моль “ AG298(/) ’ кДж/моль
Ln2O3 5,87 2280 1794,9 1707,1
Се2Оз 6,10 2180 1799,5 1709,6
Рг2ОЗ 6,34 2300 1809,6 1720,0
Nd2O3 6,59 2320 1808.3 1721,3
Рш2Оз 6,85 2320 — —
Sm2O3 7,25 2270 1822,6 1734,3
Еи2Оз 7,28 2320 1649,4 1554,5
Gd2O3 7,62 2350 1820,5 1733,0
Tb2O3 7,88 2390 1865,2 1776.5
Dy2O3 8,17 2400 1862,7 1770,7
Ho2O3 8,41 2360 1881,1 1791,6
Ег2ОЗ 8,66 2380 1987,9 1808,7
Тш20з 8,88 2370 1888,7 1794,5
Yb2O3 9,21 2430 1814,6 1726,7
Lu2O3 9.42 2450 1878,2 1788,7
Y2O3 5,03 2430 1905,0 1816,3
Sc2O3 3,86 2450 1908,3 1818,8
CeO2 7,22 2600 1087,6 1024,5
Pr6Ol 1 6,91 2183 942,6 892,4
EuO 8,21 1900 597,3 568,5
195
7»
При прокаливании на воз-
духе соединений церия, празе-
одима и тербия образуются ди-
оксид церия СеОг и оксиды
приблизительно отвечающие
эмпирическим формулам
РгбОц (РгО1,83) и ТЬ4О7
(ТЬО1,75) - Восстановлением
этих оксидов водородом при
600 °C получают оксиды типа
ЬпгОз-
Диоксид церия по своим
свойствам сходен с диоксидом
тория, имеет такую же кри-
сталлическую структуру (типа
СаРг), так же плохо растворя-
ется в кислотах. Растворяется в
кипящей серной кислоте, а в
других кислотах — в присутст-
вии ионов фтора. С оксидами
тяжелых лантаноидов, имею-
Рис. 5.6. Схема фазовых превращений оксидов
лантаноидов Кристаллические модификации:
X — структура неизвестна, Н — высоко-
температурная гексогональная, А — гексаго-
нальная, В — моноклинная, С — кубическая,
производная от структуры CaF2
щих кубическую решетку, дает непрерывные твердые растворы.
Оксиды РгОх с х < 2,0 при обработке кислородом под давлением > 10
МПа при температуре 500 °C в течение 8—10 ч превращаются в стехи-
ометрический оксид РгОг с такой же кристаллической структурой, как
и СеОг. На фазовой диаграмме диоксид РгОг имеет область гомогенно-
сти 1,7 -—- 2,0; оксиды, попадающие в эту область, имеют структуру
СеОг, дефектную по кислороду. Аналогично диоксид ТЬОг может быть
получен обработкой оксидов ТЬОх с х < 2,0 атомарным кислородом при
450 °C. Область гомогенности этого оксида 1,75 -—-2,0. Оксиды празео-
дима и тербия с х > 1,5 сильные окислители, разлагают воду; при раство-
рении в соляной кислоте выделяется хлор.
Имеются данные, что при длительном хранении оксидов РггОз и
ИбгОз на воздухе при определенных условиях (температура, влажность
и др.) могут быть получены оксиды РГ2О5 и NdzOs- Предполагается, что
празеодим и неодим в них имеют степень окисления +5. Пентаоксиды
термически неустойчивы и при 360—380 °C превращаются в оксиды с
эмпирическими формулами РгбОц и NdbOn. При термическом разло-
жении карбоната празеодима (III) получена смесь оксидов РггОз и
РггОб, а при разложении гидрокарбоната неодима (III) — NdbOii и
Nd2O5.
Оксиды типа LnO для лантаноидов, наиболее устойчивых в степени
окисления +2 — европия, самария и иттербия, получены восстановлени-
196
£|Л оксидов ЬпгОз. Для этого могут быть использованы различные вос-
становители — металлический лантан, углерод, гидрид лития и др., но
методом, обеспечивающим получение наиболее чистого продукта, явля-
йся восстановление тем же металлом. Процесс проводят при > 1000 °C
в вакууме или атмосфере аргона. Монооксиды имеют структуру NaCl,
низкие температуры плавления, по свойствам очень близки к оксидам
щелочноземельных металлов, а также MgO. Из оксидов наиболее устой-
чИв ЕиО, но и он на воздухе медленно окисляется с выделением тепла.
Сложные оксиды. Оксиды лантаноидов взаимодействуют с оксидами
элементов всех групп Периодической системы с образованием большого
числа соединений. Важное практическое значение имеют сложные ок-
сиды со структурой типа перовскита (СаТЮз) и граната (MgsAlzSiaOi 2).
Соединения типа ЬпРеОз образуют все лантаноиды, они имеют иска-
женную перовскитоподобную ромбическую структуру. Основой струк-
туры перовскита в полиэдрическом представлении являются кислород-
ные октаэдры [ТЮб], в центрах которых находятся ионы титана, в
пустотах между октаэдрами — ионы кальция. Гетеровалентное замеще-
ние этих ионов и приводит к образованию ферритов РЗЭ: ион кальция
(Л+ =0,114 нм) замещается ионами лантаноидов и иттрия, ионы же
са
титана — близкими по размерам ионами железа (г^4 = 0,069 нм, = =
0,075 нм). Классическим методом получения ферритов является твер-
дофазный синтез из тонко измельченных оксидов РЗЭ и железа, взятых
в стехиометрическом соотношении. Взаимодействие оксидов начинает-
ся при 700 °C и завершается при 1300 °C. Температуры плавления
ферритов лежат в пределах 1700—1900 °C. Все они являются ферромаг-
нитными полупроводниками. Электрическое сопротивление их прибли-
зительно в 109 раз больше сопротивления ферромагнитных металлов;
°ни находят широкое применение в магнитных цепях устройств СВЧ-
техники.
Сложные оксиды со структурой, аналогичной структуре природного
Минерала граната, имеют кубическую решетку, элементарная ячейка
к°торой содержит 8 формульных единиц (160 атомов). Оксиды РЗЭ и
Железа образуют феррогранаты LnaFesOiz, структура которых может
возникнуть только при соотношении ионных радиусов л 4 : Л+ < 1,7.
Ln re
^тому соотношению удовлетворяют все ионы РЗЭ, кроме ионов церия,
пРазеодима, лантана и неодима. Феррогранаты относятся к классу фер-
Рн , находят широкое применение в гидроакустике. Ионы алюминия
11 галлия имеют близкие размеры с ионами железа, поэтому железо
^ожет быть полностью замещено ими и получены алюмоиттриевый
197
(ГГГ)Т ¥зА15°12 (АИГ) и галлий-гадолиниевый гранат GdsGasOj
Алюмогранаты получены для всех тяжелых лантаноидов и иттр!
таллиевые гранаты — для всех лантаноидов и иттрия, кроме лантана
церия. В структурах гранатов возможно частичное замещение основны*
ионов, как изовалентное, так и гетеровалентное, вследствие чего станс*
вится возможным получение гаммы материалов с различными свойст
вами. Так, частичное замещение галлия в ГГГ на скандий и
Gd3Ga5-xScxOi2 (х до 2,0) (ГСГГ) позволяет повысить к.п.д. ОКГ в
несколько раз. Все гранаты имеют высокие температуры плавления
(1700—2000 °C), синтез их достаточно сложен из-за образования раз-
личных промежуточных фаз. Лучшие результаты по продолжительно-
сти синтеза, химической и фазовой однородности получаемого мате-
риала дает использование метода совместного осаждения гидроксиД
или синтез через смешанные гидроксиды. По сравнению со спеканием
смеси оксидов температура синтеза снижается с 1300—1600 до 1000-
1100 С. Для выращивания монокристаллов используют в основном
метод Чохральского, реже — кристаллизацию из раствора в расплаве.
Высокотемпературные сверхпроводящие материалы. СовертпеЛ
уникальное значение для прогресса современной науки и техники име-
ют сложные оксиды на основе лантаноидов, щелочноземельных элемен-
тов и меди, обладающие высокотемпературной сверхпроводимости
(ВТСП). Первое сообщение об этом швейцарских ученых Мюллера и
Беднорца появилось в научной печати в конце 1986 г. К этому времени
максимальная температура перехода в сверхпроводящее состояние была
получена для интерметаллида NbsGe (Тс = 23 К); прогнозировал®
достижение Тс = 25 К к 2000-му году. Действительность же превзошла
все прогнозы высокотемпературная сверхпроводимость была открыта
«преждевременно». Первыми материалами были сложные оксиды с об-
щей формулой La2-xMxCuO4-x, где М — Sr, Ва и х ~ 0,2, Тс = 30—40 К,
кристаллизующиеся в структуре типа K2NiF4, производной одновре-
менно от структуры перовскита и NaCl. Затем весьма быстро было уста-
новлено, что в к ваз и тройной системе Y2O3—ВаО—СиО образуется фаза
УВагСизОт-х («123») со структурой дефектного по кислороду перовски-
та и Тс = 90—93 К. Замена иттрия в соединении на самарий, гадолиний,
диспрозий, гольмий и лютеций не вызывает изменения в сверхпроводя-
щих свойствах. В то же время с лантаном и неодимом, имеющими ион-
ные радиусы значительно больше, чем у иттрия, такая структура не
реализуется, возникают фазы с Тс = 33—40 К.
В настоящее время установлено, что в указанной квазитройной си-
стеме существует пять сверхпроводящих фаз: УВагСизф-х-
У2Ва4Си7О15-х, УВа2Си4О8 («124»), УВагСи^и УВагСибОю, образу-
ющих гомологический ряд с общей формулой Y2Ba4Cu6+nOi4+n. Фзза
198
Рис. 5.7. Один из вариантов триангуляции
диаграммы квазитройной системы Y2O3—
ВаО—СиО при атмосферном давлении и
950 °C
124» имеет Тс = 77—80 К и мо-
* еТ быть получена при давлении
(,иСЛорода Ю2 МПа. Остальные
три фазы обнаружены в виде
^кродефектов в эпитаксиаль-
gjjx слоях фазы «123» и при раз-
ложении фазы «124». Кроме
лтих фаз в системе образуются
й другие двойные и тройные фа-
зы, не обладающие сверхпрово-
димостью. Один из вариантов
триангуляции изотермического
разреза этой системы при атмос-
ферном давлении и температу-
ре 950 °C приведен на рис. 5.7. В
фазе «123» содержание кислоро-
да определяется тем, что медь в
ней может находиться в степе-
нях окисления +1, +2 и +3, что
зависит от давления кислорода в равновесной газовой атмосфере и тем-
пературы. Коэффициент х у кислорода может принимать значения от 0
до 1; сверхпроводимостью обладает фаза с х=0,1—0,7. При нагрева-
нии материала на воздухе наблюдается поглощение кислорода до 420
460 °C, затем кислород начинает выделяться, значения х становится
> 0,7, и сверхпроводящая ромбическая фаза претерпевает полиморф-
ный переход в несверхпроводящую тетрагональную модификацию.
Сверхпроводящая фаза плавится инконгруэнтно при 970 С, плавление
же всей системы завершается при 1000 °C. Фаза «123», кроме нестехио-
метрии по кислороду, может иметь и некоторую нестехиометрию по
металлам вследствие образования твердых растворов с двойными окси-
дами: ВаСиОг и Y2CU2O5.
Для синтеза фазы «123» используются те же методы, что и для
получения сложных оксидов, однако получение однофазного продукта
связано со значительными трудностями — необходимо очень точно вы-
держивать режимы термообработки исходной реакционной смеси, дли-
тельность процесса, давление кислорода в газовой атмосфере. Большое
значение имеет выбор исходных компонентов.
Гидроксиды. Гидроксиды Ьп(ОН)з «НгО образуются в виде гелеоб-
Разных осадков при действии аммиака или щелочей на растворы солей,
PH осаждения их зависит от концентрации солей, характера аниона,
температуры. При этом наблюдается закономерность: pH уменьшается
в Ряду лантан—лютеций приблизительно на 1,5—2,0 единицы (табл.
5-7). В осадок сначала выделяются гидроксосоли ЬпАз-х(ОН)х, где х -
= 0,4—0,6. При стоянии под маточным раствором гидроксосоли превра
щаются в гидроксиды. У легких лантаноидов и иттрия этот процс
длится несколько часов, а у тяжелых (иттербия, лютеция) может про
должаться несколько месяцев, нагревание ускоряет его. Гидроксид
рия Се(ОН)4 осаждается при pH 0,7—1,0, т.е. при условиях, близких (
условиям осаждения гидроксидов титана и других элементов IV группЬ|
Периодической системы. Разница в pH осаждения гидроксидов церия ц
других лантаноидов используется для их разделения. Все гидроксиды
воздухе поглощают СО2, превращаясь в гиДроксид-карбонаты и даже >
карбонаты.
Таблиц а 5.7 Растворимость в воде при 20 'С и pH начала
осаждения гидроксидов скандия, иттрия и лантаноидов
из раствора нитратов NaOH при 25" С
Элемент S- 10^,моль/л pH Элемент S 10 ,моль/л pH
Sc — б.Ю*’ Gd 0,9 6,83
Y 0,9 6,95 Tb
La 8,8 7,82 Dy — 6,37*)
Се 4,1 7,60 Но — —
Рг 2,2 7,35 Ет 0,6 6,76
Nd 1,9 7,31 Тт — 6,40
Pm — — Yb 0,5 6,30
Sra 1,3 6,92 Lu 0,5 6,30
Eu 0,8 6,82
Осаждение из раствора ацетата при 20 “С.
Гидроксиды лантаноидов, как и других элементов, являются удоб-
ными исходными веществами для синтеза различных соединений: он»
обладают более высокой реакционной способностью по сравнению '
оксидами, не содержат кислотных остатков. Это позволяет снизить тем*
пературу синтеза различных оксидных соединений, уменьшить его пр0'
должительность и т.д. Однако большим неудобством их использования
является гелеобразное состояние, обусловленное большим содержанием
воды — до 40 молекул на 1 атом металла. Поэтому разработаны способа
получения гидроксидов с малым содержанием воды. Методом г идротер"
мального синтеза получены кристаллические гидроксиды лантана, нт5'
рия и лантаноидов (обработкой солей 7—10 моль/л раствором NaOH®
автоклаве). При 160—260 °C состав гидроксида соответствует эмпир”'
ческой формуле Ьп(ОН)з, а при 400 °C — LnO(OH).
200
Более простым является гетерофазный метод, сущность которого
включается в обработке при комнатной температуре кристаллогидра-
трр солей растворами аммиака или щелочей определенной концентра-
ции. Взаимодействие протекает в твердой фазе; так как соль не
растворяется, гидроксид образуется в виде плотного, большей частью
аМОрфного, хорошо фильтрующегося порошка. Содержание металла
колеблется от 48 до 65 %, а воды — всего 2—3 молекулы на 1 атом
металла. Маловодные гидроксиды медленнее стареют, лучше растворя-
ются в кислотах, обладают хорошими ионообменными свойствами.
5.2. Соли кислородсодержащих кислот
Редкоземельные элементы в степени окисления +3 образуют соли со
всеми кислородсодержащими кислотами при растворении оксидов или
гидроксидов. Они довольно хорошо растворимы в воде, кроме карбона-
тов, фосфатов и оксалатов. Из водных растворов выделяются в виде
кристаллогидратов с различным числом молекул воды (от 2 до 16).
Термически устойчивы только сульфаты и фосфаты, которые обезвожи-
ваются без разложения при 500—600 °C.
Сульфаты. Средние сульфаты РЗЭ из водных растворов выделяются
преимущественно в виде кристаллогидратов с 8 молекулами воды —
Ln2(SO4)3 8Н2О, которые довольно хорошо растворяются в воде, однако
ясно выраженной зависимости растворимости от порядкового номера
лантаноида (Z) не наблюдается. Так, растворимость сульфатов в воде
при 20 °C в пересчете на безводную соль равна для Се 9,4; Рг 10,2; Ег
13,0; Yb 28,2; Lu 37,8; Y 7,03, для остальных лантаноидов — от 2 до 6
г/100 г Н2О. Сульфаты РЗЭ склонны к образованию пересыщенных
растворов, имеют отрицательный температурный коэффициент раство-
римости, при повышении температуры до 75 ”С растворимость умень-
шается в 2—2,5 раза. В водных растворах сульфатов наряду с катиона-
ми Ln3+ aq существуют и катионные комплексы LnSO4 . При
пРокаливании сульфатов образуются сначала оксид-сульфаты
^20(80^2 (1000 °C), которые не растворяются в воде и разбавленных
кислотах; полностью разложение завершается при 1500 °C.
При добавлении к сульфатным, нитратным, хлоридным растворам
ЗЭ растворов сульфатов щелочных металлов, аммония и магния обра-
3У*отся двойные соли с общей формулой xLn2(SO4)3 yM2SO4 иН2О, где
*:У=1 :1, 1 :5,2:3.
В отличие от простых сульфатов у двойных прослеживается четкая
За®исимость их растворимости от Z. Двойные сульфаты легких РЗЭ
п^охо растворяются в воде и в особенности в растворах сульфатов ще-
чных металлов. Растворимость двойных сульфатов РЗЭ и калия типа
201
1 : 1 в насыщенных растворах K2SO4, г/л: Sm 0,5; Gd 7,7; Y 46,7. ЭТс
используется для разделения РЗЭ на подгруппы — цериевую и иттрцс
вую.
Церий, кроме сульфата Ce2(SO4>3, образует сульфат Ce(SO4>2npl,
обработке СеОг или Се (ОН) 4 концентрированной серной кислотой прр
нагревании. Из раствора он выделяется в виде тетрагидрата,изоморфц(
го Th (864)2 4НгО и Zr(SO4)2 4НгО; в воде сульфат церия (IV) хорощ,
растворяется, но при этом гидролизуется.
Самарий, европий и иттербий образуют сульфаты типа LnSO4 при
восстановлении ацетатных растворов амальгамой натрия или электро-
химическим методом и осаждении серной кислотой. Европий может
быть также восстановлен цинковой пылью. Сульфаты LnSO4, как и
сульфаты щелочноземельных элементов, плохо растворяются в воде, на
воздухе, кроме сульфата европия, который более устойчив, легко окис-
ляются.
Нитраты. При упаривании нитратных растворов РЗЭ кристалли-
зуются соединения типа 1л1(МОз)з хНгО, где х = 1—6. Нитраты очень
хорошо растворяются в воде. При 25 °C растворимость их в пересчете на
безводные соли равна 51—62 %. Температурный коэффициент раство-
римости — положительный. Кристаллогидраты гигроскопичны, на воз-
духе расплываются, при нагревании дегидратируются с разложением,
превращаясь в малорастворимые гидроксид-оксид-нитраты, а затем в
оксиды
400 С” Ьп(МОз)з 500-600 “С 800—900 "С
Ln(NO3) хН2О * LnONO3 Ln2O3. (5.4)
~~ - Ьп(ОН)2МОз) уНгО —
Нитраты тяжелых РЗЭ термически менее устойчивы, так при 400 °C
образуются оксид (гидроксид) —нитраты Er, Tm, Yb, Lu и Y; у остальных
лантаноидов при этих условиях существуют или безводные, или гидра-
тированные средние нитраты.
В водных растворах'ионы Ln3+ образуют с ионом NO3 малоустой
чивые катионные и анионные комплексы с общей формулой
[Ьп(МОз)п]3 ”, где п= 1—6. Анионные комплексы могут образовывать-
ся только при концентрации азотной кислоты > 6 моль/л, устойчивое1*
катионных комплексов возрастает от La до Ей, а затем уменьшаете®
Анионным комплексам Ln(NOa)4, Ln(NO3)5~ и Ln(NO3)|- соответству-
ют кристаллические соли, двойные нитраты с щелочными металлам*1'
аммонием, магнием, цинком и др. Разница в термической стойкости*1
растворимости двойных нитратов, которая возрастает от La к Gd, мо*
быть использована для разделения РЗЭ на подгруппы.
Карбонаты. При действии на растворы солей РЗЭ растворов кар**0
натов аммония, натрия калия в зависимости от соотношения pear6”
202
тОв, температуры, концентрации и pH образуются аморфные или кри-
сталлические осадки средних карбонатов [1д12(С0з)з хНгО, pH 4,7—
5,6], гидроксидкарбонатов [1л(ОН)СОз хНгО, pH > 12] и двойных
карбонатов. При стехиометрическом отношении реагентов на холоду
преимущественно образуются средние карбонаты, повышение темпера-
туры благоприятствует образованию гидроксид-карбонатов. Карбонаты
плохо растворяются в воде; в избытке осадителя растворимость не-
сколько увеличивается вследствие образования анионных комплексов
[Еп(СОз)и]3 2”, где п = 2—4 (табл. 5.8). При увеличении Z усиливается
тенденция к образованию гидроксид-карбонатов и комплексных
ионов.
Свойства карбонатов РЗЭ сильно отличаются от свойств карбонатов
тория и урана, часто сопутствующих РЗЭ в минералах и рудах. Уран и
торий образуют прочные, хорошо растворяющиеся в избытке осадителя
комплексы типа ]иО2(СОз)и]2 2" и [ТЬ(СОз)л]4 2”, гдеи= 1—5. Так,
растворимость (ПН4)4[иОг(СОз)з] почти в 1000 раз больше раствори-
мости карбоната уранила UO2CO3 хНгО соответственно в пересчете на
уран — 23,0 (15 °C) и 0,028 (30 °C).
Таблица 5.8 Растворимость карбонатов некоторых РЗЭ в растворах
карбонатов щелочных металлов, г/л Ьп2Оз
Элемент К2СОЗ,г-экв/л Na2CO3 ,г—эк в/ л (NH4) 2СОЗ,г-экв/л
0 4,0 0 3,5 0 6,0
La 0,092 0,29 0,010 0,040 0,039 0,079
Nd 0,059 1.12 0,051 0,143 0,620 0,170
Y 0,221 15,90 0.071 2,920 — —
При нагревании карбонаты переходят в гидроксид-карбонаты, ок-
сид-карбонаты и при 900 °C превращаются в оксиды ЬпгОз, карбонат
Церия — в СеОг
Ьп2(СОз)з хНгО-----1л(ОН)СОз уНгО—"-1л2О(СОз)2-------*
----* Ln2O3; (5.5)
Сег(СОз)з хНгО-----* СеОг. (5.6)
Соединения с карбоновыми кислотами. Соединения РЗЭ с карбоно-
ВЬ|Ми кислотами получают растворением карбонатов, оксидов и гидро-
Ксидов в соответствующих кислотах и последующей кристаллизацей.
Соединения РЗЭ с простейшей карбоновой кислотой — муравьиной
^Формиаты) — кристаллизуются из раствора с разным содержанием
кристаллизационной воды, в воде растворяются плохо. Так, раствори-
203
мость Се(НСОО)з при 13 °C равна 1,45 10 2 мол/л. С увеличением
растворимость несколько увеличивается, растет и число молекул водц Е
кристаллогидратах; у формиата иттрия оно равно 2. Нагреванием в
обезводить не удается, так как они разлагаются.
В отличие от формиатов соединения с уксусной кислотой — ацета
— растворяются в воде довольно хорошо, из водных растворов выдел я.
ются в виде кристаллогидратов с одной молекулой воды. В водных рас
творах обнаружены малоустойчивые комплексы [Еп(СНзСОО)]2
[Ьп(СНзСОО)21+ и [Ьп(СНзСОО)4] . С увеличением Z растворимость
несколько понижается; так, при 25 °C растворимость в пересчете на
безводную соль равна для Ьа(СгНзО2)з- НгО 14,47, а дл?
¥(СгНзО2)з НгО 8,28 г/100 г раствора. Температурный коэффициент
растворимости, как правило, отрицательный.
Оксалаты. Из соединений РЗЭ с двухосновными карбоновыми кис
лотами наибольшее значение имеют оксалаты. При добавлении к нейт-
ральным или слабокислым (pH 2—3) растворам солей РЗЭ щавелевой
кислоты или ее растворимых солей выпадают творожистые осадки окса-
латов Ьп2 (С2О4) з хНгО, где х = 9—10, которые при стоянии под маточ
ником или нагревании становятся кристаллическими. Оксалаты—одн]
из наименее растворимых в воде соединений РЗЭ, растворимость их прв
25 °C в пересчете на безводную соль равна 0,40—3,3 г/л. Растворимость
их уменьшается при добавлении избытка щавелевой кислоты, но не-
сколько увеличивается в растворах оксалата аммония и щелочных ме-
таллов. Повышение растворимости связано с образованием комплек
сных анионов [ЬпССгСМпГ 2n, п = 2, 3 (а возможно, и 4, и 5), причем,
растворимость тяжелых лантаноидов увеличивается несколько больше,
чем легких, что согласуется с общей тенденцией усиления комплексо-
образования у тяжелых лантаноидов. По сравнению с анионами одноос-
новных карбоновых кислот и карбонат-ионом оксалат-ион образует бо-
лее прочные комплексы вследствие образования замкнутых колец ке-
латного типа. Однако вследствие малой способности РЗЭ к комплексо-
образованию оксалатные комплексы не очень существенно отличаются
от карбонатных. В частности, это обнаруживается при сопоставлени
растворимости оксалатных комплексов РЗЭ, урана и тория, образую"
щих аналогичные комплексы, но отличающиеся высокой растворим0"
стью. Так, растворимость оксалата тория в растворе оксалата аммони4
при концентрации последнего 300 г/л равна 391 г/л, а соотношенй1
растворимостей этого оксалата и оксалатов РЗЭ при таких условия
равно 2660: (1—10).
Растворимость в воде оксалатов Ln2(C2O4>3 ЮНгО при 25 °C в
ресчете на ЬпгОз приведена ниже:
La Се Pr Nd Sm Gd Tb Y
Ln2O3, г/л 0,37 0,25 0,45 0,45 0,43 0,34 2,10 0,51
204
Средние оксалаты хорошо раство-
ряются в неорганических кислотах,
причем хуже растворяются оксалаты
тяжелых лантаноидов и тория. Пред-
положительно это можно объяснить
образованием комплексов с анионом
НС2О4 ; комплекс [EUHC2O4 ]2+ об-
наружен в растворе с концентрацией
HNO3 0,5 мол/л. Концентрирован-
ной азотной кислотой оксалаты раз-
рушаются. Осаждение оксалатов РЗЭ
является весьма эффективным мето-
Рис. 5.8. Растворимость оксалатов РЗЭ
и тория в серной кислоте
дом отделения их от всех хорошо растворимых оксалатов — урана,
тория, хрома, циркония, железа, магния, кальция и других элементов
(рис. 5.8).
При нагревании оксалатов на воздухе происходит их обезвожи-
вание с одновременным разложением и выделением СО и СОг. Разло-
жение проходит через стадию образования оксокарбоната IJ12O2CO3
(550—725 °C), полностью завершается выше 800 °C.
Обработкой твердых оксалатов концентрированными растворами
щелочей (25—40 %) может быть осуществлена конверсия их в гидро-
ксиды, а оксалат щелочного металла вновь использован.
Фосфаты. С анионами фосфорных кислот РЗЭ в зависимости от
соотношения реагентов, концентрации, pH образуют в водных раство-
рах огромное число соединений малорастворимых в воде.
Ортофосфаты. С анионом РО4 образуются средние, гидроксо-,
гидро- и двойные соли. Средние фосфаты LnPC>4 хНгО (х = 1—3) осаж-
даются из растворов солей РЗЭ в виде гелеобразных осадков при дейст-
вии фосфорной кислоты, одно- и двузамещенных фосфатов щелочных
металлов при pH 1,5—2,5. Растворимость их в воде равна (3—4) 10 4
мол/л, но они хорошо растворяются в минеральных кислотах, в
том числе и фосфорной. Растворимость в концентрированной фос-
форной кислоте увеличивается в ряду от лантана (~ 2,0 %) до лютеция
10,0 %). В этом же ряду возрастает склонность к образованию гидро-
голей типа ЬпНз(РО4)2 Н2О. Поэтому осадки фосфатов при осаждении
Фосфорной кислотой не образуются при соотношении Ln3+: РО4 =1:1;
Фосфорной кислотой можно осадить только фосфаты легких лантанои-
дов, иттрия и скандия. Образование осадков фосфатов происходит легче
при использовании в качестве осадителей фосфатов щелочных метал-
лов. Однако при большом их избытке (более чем четырехкратном) об-
разуются двойные соли, например, типа 2YPO4 КН2РО4 4НгО.
205
Осадки рентгеноаморфных средних фосфатов при pH ~ 2,3 и ком-
натной температуре постепенно переходят в кристаллическое состоя-
ние. Образующиеся кристаллогидраты по структуре делятся на дВа
типа: с тетрагональной решеткой — для Но, Er, Tm, Yb, Lu, Y и гекса-
гональной решеткой — для остальных лантаноидов. Приблизительно
при 450 °C кристаллогидраты обезвоживаются, а выше 600 °C фосфаты
I группы переходят в структуру минерала ксенотима (YPO4), имеющею
также тетрагональную решетку, а фосфаты II группы — в структуру
минерала монацита — (La...Gd)PO4 с моноклинной кристаллической
решеткой. Ксенотим изоструктурен с цирконом (ZrSiCU) .
Безводные фосфаты отличаются высокой химической и термической
стойкостью, не растворяются в воде, незначительно растворяются в
кислотах, имеют высокие температуры плавления (2000—2200 °C),
только у ScPO4 она равна 1780 °C.
Конденсированные фосфаты. Конденсированные фосфаты, т.е. со-
ли фосфорных кислот, содержащих связи Р—О—Р, образовавшиеся в
результате объединения вершинами тетраэдров [РО4 ], включают боль-
шую группу соединений с цепочечным и циклическим строением анио-
нов, которые могут содержать до 8 атомов фосфора. Конденсированные
фосфаты РЗЭ в отличие от ортофосфатов в природе не встречаются.
Связи Р—О—Р термодинамически нестабильны в водной среде. Под
действием воды, повышенной температуры и кислотности происходит
гидролитический распад сложных конденсированных анионов, при ко-
тором образуются простейшие анионы. В нейтральной среде довольно
устойчивы конденсированные фосфаты щелочных металлов, и их мож-
но использовать для синтеза соответствующих фосфатов РЗЭ.
Наиболее гидролитически стоек анион дифосфата Р2О7 — простей-
ший из фосфатов цепочечного строения. Он состоит из двух связанных
вершинами тетраэдров [РО4 ]. Средние дифосфаты Еп4(РгО7)з хНгО
осаждаются из водных растворов дифосфатами щелочных металлов при
pH 2,3—4,0 и в строго стехиометрическом соотношении Ln3+: РгО^
= 4 : 3. В воде они мало растворимы (~ 10 2 г/л), свсжеосажденные
гелеобразные осадки хорошо растворяются в минеральных кислотах;
при выдерживании под маточным раствором переходят в кристалличе-
ское состояние и растворимость в кислотах уменьшается. При увеличе-
нии в растворе концентрации дифосфата щелочного металла выпадают
осадки двойных дифосфатов — ML11P2O7 ХН2О, M5Ln(₽2O7)2 xHzO’
растворимость которых в избытке осадителя существенно увеличивает-
ся. Все реакции по получению как ортофосфатов, так и конденсирован'
ных отличаются большой продолжительностью, образованием проме-
жуточных метастабильных форм, одновременно протекающими пр0'
цессами превращения аморфных осадков в кристаллические; получение
206
соединений определенного состава требует соблюдения жестких усло-
вий. Следует отметить, что малорастворимый дифосфат тория осажда-
ется при pH 1,0, что позволяет отделять его от дифосфатов РЗЭ.
Безводные конденсированные фосфаты различного состава, в том
числе и двойные, например, типов LnPsOu, MLnP40i2, могут быть
получены спеканием или сплавлением соответствующих компонентов.
При введении в них активаторов (ионов неодима, самария, европия и
др.) они могут быть использованы для получения фосфатных стекол для
твердотельных ОКГ.
Молибдаты. При взаимодействии ЬпгОз и МоОз образуется боль-
шое число соединений с различным соотношением оксидов (1 :1, 1 : 2,
1:3, 1 : 4, 1 :6 и др.), причем в ряду лантаноидов с увеличением Z число
соединений уменьшается. Для них характерно многообразие структур-
ных типов и полиморфизм.
Соединения типа ЬпгМоОб (1:1) —оксимолибдаты — известны для
все лантаноидов. Они могут быть получены спеканием смесей оксидов,
причем процессы, протекающие при этом, сложны. Первоначально по-
лучаются тетрамолибдаты, которые при повышении температуры взаи-
модействуют с оксидом лантаноида, образуя молибдаты с большим его
содержанием. Для эквимолекулярной смеси последовательность пре-
вращений может быть представлена схемой
500—530 "С 540—670 “С
ЬпгОз + МоОз--------» ЬпгОз 4МоОз---------»
540—670 °C 790—830 ”С
-----------* ЬпгОз ЗМоОз +ЬпгОз---------► ЬпгОз МоОз. (5.7)
Оксимолибдаты имеют высокие температуры плавления (1700—
1900’С).
Соединения типа Ьпг(МоО4)з (1:3) — нормальные молибдаты —
известны также для всех лантаноидов. Они могут быть получены твер-
дофазным синтезом из смесей оксидов, взятых в соотношении 1 : 3 при
440—700 °C. Температуры плавления их находятся в интервале 990—
1350 ’С. Основой их структуры являются тетраэдры [МоО4 ].
Сплавлением молибдатов лантаноидов и щелочных металлов могут
быть получены двойные молибдаты двух типов: MLn(MoO4>2 (1 : 1) и
M5Ln(MoO4)4 (1 : 5). Двойные молибдаты лития, имеющие шеелитопо-
Добную нецентросимметричную структуру, обладают сегнетоэлектри-
ческими свойствами. Двойные молибдаты натрия (1 : 1), активирован-
ные ионами неодима, перспективны в качестве рабочих тел ОКГ; молиб-
даты (5 : 1) — высокоэффективные люминофоры.
Вольфраматы. В системах ЬпгОз—WO3 число соединений меньше,
Чем в аналогичных системах с оксидом молибдена. Основными типами
Являются соединения 1 : 1, 1 :3, 3: 1.
207
Соединения типа Ln2WO6 (1:1) — оксивольфраматы — известно
для всех лантаноидов. Оксивольфраматы получают твердофазным син-
тезом в две стадии при 850 и 1200 °C с промежуточным перетиранием
смеси. Продолжительность синтеза около 6 сут. Однако для полной
гомогенизации продукта синтеза и доведения его до равновесного состо-
яния необходим длительный отжиг (до 10 сут) при 1400 °C. Некоторые
оксивольфраматы обладают фотолюминесценцией, например Y2WO6
дает яркое голубое свечение.
Нормальные вольфраматы Ln2(WO4)3 (1:3) также известны для
всех лантаноидов. При твердофазном синтезе взаимодействие между
ЬпгОз и WO3 начинается при 500—600 °C и до температуры полиморф-
ного превращения WO3 (750 °C) протекает медленно. По достижении
750 °C скорость образования вольфрамата возрастает, при 900 °C реак-
ция завершается за 6—8 ч. Температуры плавления нормальных воль-
фраматов возрастают в ряду от лантана к лютецию (1090—1580 °C).
С вольфраматами щелочных металлов вольфраматы лантаноидов
образуют двойные соли типа MLn(WC>4)2 (1 : 1) и MsLn(WO4)4 (5 : 1).
Первые плавятся конгруэнтно при 1150—1265 °C (La * Lu), вторые —
инконгруэнтно при 735—760 °C. Спектрально-люминесцентные харак-
теристики Na5Nd(WO4)4HTBepat>ix растворов на его основе показывают,
что он является высокоэффективным лазерным материалом. Высокая
яркость люминесценции характеризует также вольфраматы европия и
тербия.
5.3. Галогениды
Фториды. Фториды лантаноидов довольно существенно отлича-
ются от хлоридов, бромидов, иодидов. По сравнению с ними они
термодинамически устойчивее, не подвергаются термическому разло-
жению до ~ 800 °C, имеют более высокие температуры плавления и
кипения, менее гигроскопичны (табл. 5.9). Безводные фториды исполь-
зуются для получения металлов.
Безводные фториды РЗЭ можно получить фторированием оксидов
газообразным фтороводородом, гидрофторидом аммония или обезвожи-
ванием кристаллогидратов
1л120з + 6HF - 2LnF3 + ЗН2О; (5.8>
Ln2O3 + 6NH4HF2 X 2LnF3 + 6NH4F + ЗН2О. (5.9)
Фторированием оксидов газообразным HF при 600—700 °C можно
получить фториды высокого качества с содержанием кислорода ^0,1%’
но при реализации его имеются значительные трудности: реакция (5-8)
обратима, поэтому нужен большой избыток HF (до 250 %). Во-вторы*’
нужны специальные конструкционные материалы, чаще используй
208
сплавы на основе никеля. Гидрофторид аммония более реакционноспо-
собен по отношению к оксидам вследствие образования на первом этапе
взаимодействия фторметаллатных комплексов типа МНдЬпГд (La—Tb)
и NH4Y2F7 (Dy—Lu и Y) при 60—110 °C. При 240—380 °C комплексы
распадаются на I.nFa и NH4F. Избыток NH4F и NH4HF2 отгоняется при
450 °C. Избыток фторирующего агента не превышает 30 %, но трудности
с конструкционными материалами те же, что и при использовании HF.
Таблица 5.9. Свойства фторидов РЗЭ, иттрия и скандия
Фто- рид 7пл, •c -zV/298(f)> кДж/моль ^98(0 • кДж/моль
LaF3 1493 1731,8 1652,7
CeF3 1432 1732,6 1654,8
PrF3 1400 1712,1 1634,3
NdF3 1377 1712,9 1637,2
PmF3 — 1648.5 1569,8
SmF3 1305 1707,1 1630,4
E11F3 1276 1169.4 1157,3
GdF3 1232 1713.3 1637,2
TbF3 1177 1707,9 1631,3
DyF3 1157 1719,6 1642,6
H0F3 1143 1714,2 1637,2
ErF3 1117 1722,6 1646,4
TtoF3 1158 1694,5 1617,9
YbF3 1162 1656,9 1581,6
L11F3 1184 1700,8 1623,4
YF3 1155 1718,4 1644,3
ScF3 1552 1648,9 1570,3
В расплавах фторидов щелочных металлов образуются фторметал-
латные комплексы разных типов; MLnF4, M2LnF5, МзСпРб, устойчи-
вость которых возрастает в ряду Li —• Cs.
Фториды РЗЭ относятся к наименее растворимым соединениям, на-
пример растворимость LaFa в воде при 20 °C равна 0,25 мг/л. В водных
Растворах образуются устойчивые комплексные ионы типа LnF , толь-
ко для иттрия установлено образование ионов YFn ” , где п может при-
нимать значения от 1 до 4. Поэтому фториды лантаноидов не раст-
еряются в избытке плавиковой кислоты; они могут быть получены
каждением плавиковой кислотой или фторидами щелочных металлов
209
практически из любых растворов в виде кристаллогидратов LnF3 хНгО
где х < 0,5.
Обезвоживание кристаллогидратов привлекает доступностью рея.
гентов, относительной простотой аппаратуры. Удаление воды протекает
в интервале 60—450 °C, но при этом происходит частичный пирогидро,
лиз с образованием нерастворимых в воде оксофторидов LnOF. Про
ведение процесса в вакууме позволяет снизить температуру до 200—
250 °C и получить материал с содержанием кислорода < 0,02 %. Фтори-
ды, обезвоженные в атмосфере HF, также содержат < 0,02 % кислорода
Недостаток этого метода: при осаждении фторидов из растворов
получаются гелеобразные осадки, плохо фильтрующиеся и плохо отмы-
вающиеся от анионов. Этот недостаток можно устранить, используя для
конверсии твердый карбонат РЗЭ, имеющий псевдокристаллическую
структуру. Фториды получают путем обработки карбоната плавиковой
кислотой; реакция протекает в твердой фазе, и осадок фторида получа-
ется плотным хорошо фильтрующимся
1л12 (СОз) 3+6HF+xH2O=2LnFз хНгО + СОг+ уНгО. (5.10)
При недостатке плавиковой кислоты образуются малорастворимые
фторокарбонаты. Так, в природе довольно широко распространены ми-
нералы, относящиеся к этому типу соединений — бастнезит Ln (СОз) F,
иттросинхизит ЕпСа(СОз)гР и другие.
Хлориды. Безводные хлориды РЗЭ — соединения менее термодина-
мически устойчивые, имеют более низкие температуры плавления и
кипения, чем соответствующие фториды (табл. 5.10). Хлориды хорошо
растворяются в воде, очень гигроскопичны, из водных растворов выде-
ляются в виде кристаллогидратов 1л1С1з • хНгО, где х = 6—7.
Безводные хлориды могут быть получены хлорированием оксидов
газообразным хлором, однако для реакции (5.11) значения энергии Гиб-
бса в интервале 200—600 °C имеют положительный знак, а константа
реакции от п 10 'доп 10 4, поэтому хлорирование можно осуществить
только в присутствии уТлерода при 800—900 °C
ЬпгОз + ЗС12 г 2LnC13 +1,5Ог; (5.11 >
ЬпгОз + ЗС12 + 2С х 2LnCl3 + СО (СОг). (5.12)
Кроме того, для хлорирования оксидов РЗЭ может быть использован
ряд соединений, которые являются более активными хлорирующими
агентами, чем газообразный хлор, например тетрахлориды углерод2’
титана и кремния. Эти соединения содержат второй компонент, активно
взаимодействующий с кислородом и выводящий его из зоны реакции-
смещая ее таким образом вправо
400—600 "С
2Ln2O3 + 3CC14 Z 4ЬпС1з + ЗСО2; (5.13)
2Ln2O3 + 3TiCl4 ? 4ЬпС1з + ЗТЮг.
210
Таблица 510. Свойства хлоридов РЗЭ, иттрия и скандия
Хлорид Тпл/С Т’кип, "С “ A//298(f)> кДж/моль ~AG298(f)’ кДж/моль
LaCl3 862 1710 1071,1 995,8
СеС13 882 1651 1058,1 973,2
Рг€13 786 1627 1051,7 981,6
NdC13 760 1624 1040,6 964,8
PmC13 720 1628 1025,1 949,8
SmC13 678 1471 1028,4 941,8
E11CI3 624 1453 939,3 854,0
GdC13 605 1595 1005,4 925,5
TbC13 582 1522 988,7 919,6
DyC13 653 1539 995,8 898,7
H0CI3 720 1517 1006,3 931,4
TmC13 828 1487 988,3 911,3
YbC13 875 1471 961,1 881,2
L11CI3 925 1522 954,0 881,6
YC13 721 1482 1000,0 920,5
NdCl2 835 — 686,2 661,1
SmC12 858 1950 816,3 766,5
EuC12 854 2062 811,7 772,4
YbCl2 702 — 757,3 732,3
ScCl3 967 975 943,8 850,0
Тетрахлорид углерода используется в лабораторной практике, при-
менение его сильно упрощает эксперимент. Участие тетрахлорида тита-
на в хлорировании оксидов РЗЭ необходимо учитывать при хлорирова-
нии комплексного сырья, например лопарита. В сложной системе воз-
можны вторичные реакции, так при 700 °C тетрахлорид титана хлори-
рует оксиды РЗЭ на 96 %.
При хлорировании оксидов РЗЭ неизбежно образуется некоторое
количество оксохлоридов LnOCl, энтальпия образования которых не-
сколько больше, чем энтальпия образования трихлоридов. Очистка три-
клоридов от оксохлоридов, необходимая при получении из них метал-
лов, может быть осуществлена дистилляцией в вакууме при давлении
0>13 Па и температуре 900—1000 °C. Процесс длится десятки часов, для
его осуществления необходима сложная аппаратура из коррозионно-
стойких материалов.
Водные растворы трихлоридов можно получить растворением гидро-
ксидов и карбонатов в соляной кислоте. Вследствие малой склонности
211
иона хлора к комплексообразованию, хлоридные комплексы РЗЭ в вод
ных растворах малоустойчивы, и данные о них не очень надежны. Прц
концентрации НС1 6 моль/л хлоридные комплексы обнаружены не бы-
ли, образуются они только в растворах с более высокой концентрацией
соляной кислоты. В растворах хлорида неодима существуют комплексы
при концентрации соляной кислоты 1 моль/л — Nd(H2O)g+, 8 моль/л
— N dCl( НгО)2+, 13моль/л — N dCb( Н 20) у . Т олько при очень высоких
концентрациях соляной кислоты координированные молекулы воды мо-
гут частично вытесняться ионами хлора. Константы устойчивости хл0.
ридных комплексов очень невелики — от 10 2 до 1. Имеются данные об
образовании анионных комплексов типа LnCl? в растворах, содержа-
щих LiCl (8—11 моль/л) и НС1 (до 1 моль/л), из которых иттрий,
лантан, иттербий и лютеций поглощаются анионообменной смолой. С
их образованием связывают экстракцию хлоридов РЗЭ солями органи-
ческих аминов из растворов такого же типа. Из водных растворов были
выделены только цезиевые соединения типа СхзЬпОб 5НгО с La, Pr, Nd
и Sm. Устойчивость их несколько выше благодаря большому внешне-
сферному катиону цезия.
В расплавах хлоридов щелочных металлов образование анионных
комплексов более вероятно вследствие отсутствия конкуренции за ко-
ординацию иона хлора с молекулами воды. В системах с натрием соеди-
нения либо не образуются, либо образуются комплексные соединения
типа NaLnCU, плавящиеся инконгруэнтно при s 500 °C. В системах с
хлоридом калия образуются соединения типа КгЬпСЬ и Кз1лС1б, пла-
вящиеся конгруэнтно. В этом проявляется общая закономерность повы-
шения устойчивости комплексных соединений при увеличении разме-
ров внешнесферного иона щелочного металла. Комплексы с хлоридом
калия тем не менее не отличаются прочностью, энтальпия их образова-
ния находится в пределах от-25 до —45 кДж/моль, комплексы довольно
легко разлагаются на компоненты при нагревании. Они менее гигроско-
пичны, чем простые хлориды, что закономерно, так как ионы хлора
экранируют атомы РЗЭ. С комплексными хлоридами удобнее работать
при получении металлов.
Безводные хлориды можно получить обезвоживанием кристалло-
гидратов в токе НС1 или в присутствии NH4CI при температуре от 130
до 300 °C при атмосферном или пониженном давлении. Однако это1
простой метод дает удовлетворительные результаты только для легки*
РЗЭ (от лантана до неодима). При термическом обезвоживании кри'
сталлогидратов хлоридов тяжелых РЗЭ вследствие большей их склонно-
сти к гидролизу получается продукт с большим содержанием кислород3
Эффективным способом получения безводных хлоридов РЗЭ явЛ®
ется метод хлорирования их оксидов или оксохлоридов хлористым а*1
212
монием в присутствии хлорида калия. Реакция хлорирования различ-
ных оксидов РЗЭ протекает в интервале 220—260 °C с образованием
промежуточных соединений типа (NH4>2LnC15 для легких лантаноидов
и типа (NH4)3LnC16 для тяжелых лантаноидов
ЬпгОз + 1ONH4CI = 2 (NH4) 2LnC15 + 6NH3 + ЗН2О. (5.14)
Промежуточные соединения разлагаются при 400—420 °C. До этой
температуры хлорид калия участия в реакции не принимает, интенсив-
ное формирование кристаллической структуры КзЬпОб происходит при
500—550 °C. Его образование способствует большей полноте хлориро-
вания. Продукт, получающийся в результате хлорирования, представ-
ляет собой гигроскопичный мелкокристаллический порошок, плавле-
нием его переводят в компактный вид и удаляют избыток хлористого
аммония. Конечная температура процесса определяется температурой
плавления комплексного соединения, которая повышается от соедине-
ния самария (745 °C) до 830 °C у соединения иттрия и 815 °C для
лютеция.
Нагревание элементарных лантаноидов с ВаС12 или ЕиС1г в ячейке
Кнудсена до 1100—1500 °C приводит к образованию в газовой фазе LnCl
и ЬпОг. Масс-спектрометрическими измерениями установлено образо-
вание соединений типа LnCh для всех лантаноидов, кроме гадолиния.
2Ln + ВаС12(EuCh) X LnCh + Ba(Eu). (5.15)
Рассчитаны константы этой реакции для различных Ln; величины
их находятся в интервале от п 10 3 до 2.
Методом ампульного синтеза в контейнере из тантала нагреванием
до 800—900 °C смесей трихлоридов и металлических лантаноидов полу-
чены дихлориды европия, самария, иттербия, неодима, диспрозия, пра-
зеодима, эрбия и тулия. Определены температуры плавления (700—
800 °C), энтальпии и энергии образования Гиббса (см. табл. 3.14). Эти
данные показывают, что в расчете на один атом хлора для
дихлоридов большем, чем для трихлоридов, что свидетельствует о до-
вольно высокой их устойчивости. В особенности это относится к евро-
пию, самарию и иттербию, что препятствует получению этих металлов
методом металлотермии. При восстановлении их трихлоридов получа-
ют не металлы, а устойчивые дихлориды.
5.4. Соединения с халькогенами
Халькогениды РЗЭ — обширная группа, насчитывающая более 3000
соединений. Среди них—диэлектрики, полупроводники, люминофоры,
ферро- и антиферромагнетики, сверхпроводники.
Особенности этого класса соединений РЗЭ — тип химической связи,
Физико-химические свойства — определяются тем, что для атомов халь-
213
2 4
когенов, имеющих валентные электроны s р , характерна акцепторная
способность, стремление к достройке до более энергетически выгодной
конфигурации — s р . Это обусловливает значительную долю ионной
связи Ln—S и образование ковалентно связанных групп халькогенов
-Х-Х- в полихалькогенидах. В ряду S—Se—Те происходит ослабление
неметаллических свойств, уменьшаются электроотрицательность (со-
ответственно 2,6; 2,4; 2,1) и ионность связи, возрастает ковалентная
составляющая, увеличивается число соединений каждого лантаноида
более широкими становятся области гомогенности. К халькогенидам в
подавляющем числе случаев неприменимы классические представле-
ния о валентности, так как многие из них являются бертоллидными
фазами.
Из халькогенидов лучше изучены сульфиды. Все РЗЭ и скандий
образуют сульфиды, которые можно представить следующими стехио-
метрическими формулами: LnS, LJ12S3, LniChS, кроме того, для всех
легких лантаноидов, диспрозия и эрбия — LJ13S4 и LnS?.
Моносульфиды LnS кристаллизуются в структуре типа NaCl (ГЦК)
с преобладанием ионной связи, но и значительной металлической со-
ставляющей, что обусловливает их высокую электропроводность и тем-
пературу плавления (2000—2450 °C). У некоторых из них установлены
области гомогенности: LaSo,75—-1 о, TbSo,75~i ,0, LuSo,75^-1 о и др. (табл.
5.11).
Сульфиды типа LniSs кристаллизуются в кубической структуре ти-
па TI13P4. Основа ее — тетраэдры из атомов серы, в центрах которых
находятся атомы РЗЭ. Тетраэдры имеют общие атомы серы. В соедине-
ниях преобладает ионная связь, но одновременно возникает и ковален-
тная связь между атомами серы, серы и атомами лантаноида. Соедине-
ния LnsS4 имеют ту же структуру, и их, очевидно, можно рассматривать
как нижнюю границу области гомогенности LnSi,33 «-Л.5- Фазы Lnz$3
являются полупроводниками, имеют высокие значения ТДС, относи-
тельно широкие запрещенные зоны (> 2 эВ), более низкие теплопровод-
ности и температуры плавления, чем моносульфиды. В них два атома
серы могут быть замещены кислородом с образованием оксосульфи-
дов ЬпгОгВ, имеющих тетрагональную симметрию и являющихся полу-
проводниками с хорошими фоточувствительными свойствами.
Дисульфиды кристаллизуются преимущественно в тетрагональ-
ной сингонии, относятся к полисульфидам, в слоистой структуре ко-
торых чередуются слои из атомов серы и лантаноида. У некоторых из
них установлены двусторонние области гомогенности, например
NdSi.g. .2,33- У селенидов и теллуридов более отчетливо выражена спо-
собность к образованию полисоединений с большим содержанием ато-
мов халькогена, чем в сульфидах, например — La4Ten, LaTe3, NdTe3
и др.
214
Таблица 5.11. Температура плавления сульфидов некоторых РЗЭ
и ширина запрещенной зоны ( ЛЕ) сульфидов типа Ln2S3
Элемент Тпл, °C ДЕ, эВ
LnS Ln2S3 LnS2 Ln2O2S Ln2S3
La 2,200 2130 165о\\ 1940 2,66
Се 2450 1890 >720*** 1950 2,06
Рг 2230 1795 — 2,20
Nd 2200 2210 >720 ’ 1990 2,12
Sm 1940 1900 1980 3,57
Y ») * Данные тр Температ 2060 ебуют уто», •ура разлом 1600 [нения <ения >600 ’ 2120
Халькогениды относительно устойчивы в сухом воздухе, а некото-
рые (сульфид церия) устойчивы к действию расплавов солей и металлов.
Большинство же халькогенидов при температуре ~ 1 000 °C подвергается
термической диссоциации, причем равновесное давление паров серы
может достигать нескольких атмосфер. Полисульфиды (LnSx с х > 1,5)
разлагаются при более низкой температуре. При высокой температуре
пары серы и редкоземельные металлы взаимодействуют практически со
всеми контейнерными материалами, удовлетворительно устойчивы по
отношению к ним только тантал и молибден.
Все халькогениды разлагаются разбавленными кислотами
Ln2S3 + 6НС1 = 2LnCl3 + 3H2S. (5.16)
Для синтеза халькогенидов предложено большое число способов,
которые можно разделить на две основные группы: прямые и косвенные.
К прямым относятся методы, в которых в качестве исходных компонен-
тов используются металл или его гидрид и сера. Прямые методы обычно
реализуются в ампульном варианте. Ампула откачивается предвари-
тельно до давления ~ 1,3 10 д Па либо заполняется чистым аргоном.
Компоненты берутся либо в стехиометрическом отношении, либо вво-
дится избыток серы для того, чтобы создать в ампуле давление паров
серы, соответствующее температуре синтеза. Прямой синтез обычно
проводят в две стадии: первая при температуре 400—600 °C, а затем
длительный (40—80 ч) гомогенизирующий отжиг при 1000—1200 °C.
В косвенных методах в качестве исходных соединений используются
оксиды, хлориды и другие соединения РЗЭ, а в качестве сульфидизиру-
ющих агентов — сероводород, сульфид натрия, сероуглерод. Реакцию
215
проводят при 600—1200 °C. Косвенные методы более производительны
и технологичны, но чистота продуктов, как правило, невысокая.
Оксосульфиды получают разными методами, из которых наиболее
просто осуществляется метод, основанный на реакции между оксидом и
полуторным сульфидом
2Ln2S3 + ЬпгОз X 3Ln2O2S. (5.17)
5.5. Соединения с неметаллами
Соединения металлов с неметаллами (Н, В, С, Si, N), атомы которых
имеют небольшие размеры, по характеру химической связи чрезвычай-
но разнообразны; различны и их свойства, которые закономерно изме-
няются по периодам и группам Периодической системы. Их можно раз-
бить на три основные группы:
солеобразные стехиометрические, как правило, неустойчивые сое-
динения с преимущественно ионным характером связи, которые обра-
зуют электроположительные элементы — щелочные и щелочноземель-
ные металлы;
ковалентные соединения, как правило, стехиометрического состава
образуют непереходные элементы III—V групп Периодической систе-
мы, имеющие низкие температуры плавления и кипения, термически
неустойчивые, легко гидролизующиеся;
металлоподобные твердые растворы и нестехиометрические соеди-
нения, относящиеся к фазам внедрения, с широкими областями гомо-
генности d- и /-элементов. Химическая связь — преимущественно ме-
таллическая с некоторой долей ковалентности или ионности. Как пра-
вило, устойчивы, имеют высокие температуры плавления и высокую
твердость.
Гидриды. Водород — первый элемент Периодической системы эле-
ментов ; в соединениях он может иметь степень окисления +1, что роднит
его со щелочными металлами, и степень окисления -1, как и галогены.
Соединения, образуемые им, различны по характеру химической связи
и множеству промежуточных ее типов. Все соединения металлов с водо-
родом называются гидридами, несмотря на то, что по характеру связи
они могут сильно различаться.
Солеобразные гидриды образуют щелочные металлы (МН) и щелоч-
поземельные металлы (МНг). Они состоят из катионов металлов и гид-
рид-ионов Н , легко разлагаются водой и на воздухе, имеют структуру
NaCl. Ковалентные гидриды — А1Нз, СеЩ, SnH4 и другие — при обыч-
ных условиях жидкости или газы, легко гидролизуются.
РЗЭ образуют гидриды двух типов — ЬпНг (ГЦК) и, кроме Ей и Yb<
ЬпНз (ГПУ), для них характерно сочетание металлической и ионнои
216
Г, С
1200
1000
800
600
400
200
0 0,5 1,0 1,5 2,0 2,5 3,0
Рис. 5.9. Схематическая диаграмма
системы Gd-H (заштрихованы об-
ласти гомогенности)
сВязей. Гидриды хрупки, по внешнему
виду напоминают порошки металлов.
Оба гидрида имеют широкие области го-
могенности (рис. 5.9). Они более устой-
чивы, чем гидриды щелочных и щелоч-
ноземельных металлов, но тем не менее
на воздухе превращаются в гидроксиды
и карбонаты, растворяются в кислотах и
разлагаются щелочами
21лНз + 6Н2О = 2Еп(ОН)з + 6Н2. (5.18)
Гидриды образуются при гидриро-
вании металлических РЗЭ при 300—
400 °C.
Бориды. РЗЭ образуют стабильные
бориды ЬпВд и ГлВб, которые близки со-
ответствующим боридам щелочнозе-
мельных металлов по структуре, на-
пример СаВб со структурой, родственной структуре типа CsCl. Особен-
ность строения гексаборидов заключается в наличии октаэдрического
каркаса из атомов бора, атомы металлов находятся в его пустотах и
почти не влияют на прочность связи. Следствием этого является высокая
температура плавления гексаборидов РЗЭ (2210 °C для ЬаВб, 2190 °C —
СеВб, 2300 °C — УВб), довольно высокая твердость, стойкость к воздей-
ствию разбавленных кислот при комнатной температуре и щелочей при
нагревании. Но бориды хрупки. Вместе с тем для них характерны и
металлические свойства: они обладают высокими электропроводностью
и теплопроводностью. Так, удельное сопротивление ЬаВб равно 27 10-6
Ом см — меньше, чем у металлического лантана (59 10 6 Ом см).
Тетрабориды имеют преимущественно ионный характер связи, поэтому
металлические свойства у них выражены гораздо слабее, ниже темпера-
туры плавления и т.д.
Бориды РЗЭ, как и бориды других элементов, получают непосредст-
венным синтезом из порошка элементарного бора и стружки РЗМ
при ~ 1500 °C в атмосфере аргона или нагреванием порошка бора,
Углерода и оксидов РЗЭ до 1500—1800 °C
Гп20з + 12B+3C = 2LnB6 + 3CO. (5.19)
Карбиды. В ряду В—С—N увеличивается число валентных р-элек-
тРонов; в связи с этим растут тенденция к акцептированию электронов
и электроотрицательность. Поэтому в соединениях лантаноидов с угле-
родом по сравнению с боридами возрастает ионность связи. Все ланта-
ноиды и иттрий образуют карбиды ЬпСг со структурой изоморфного им
Тетрагонального карбида СаСг, для которого характерно преобладание
ионной связи и отсутствие электропроводности. В структуре СаСг суще-
217
ствует ион [С=С ]2 ; предполагается, что у карбидов РЗЭ также возмож-
но существование такой пары, а третий валентный электрон атома лан-
таноида, видимо, может переходить на нее, но одновременно несом-
ненно участие его в образовании связи металл—металл.
Кроме карбидов ЬпСг лантаноиды образуют карбиды и других соста-
вов. Скандий и церий образуют карбиды LnC со структурой типа NaCl
(ГЦК), а лантаноиды, начиная от самария — карбиды ЕпзС с той же, но
дефектной структурой NaCl, иттрий и лантаноиды от лантана до эрбия
— карбиды ЬпгСз.
Двойственный характер химической связи в карбидах РЗЭ опреде-
ляет и такой же характер их свойств. Карбиды лантаноидов имеют
высокие температуры плавления (большинство—выше 2000 °C) и элек-
тропроводность; для некоторых карбидов установлены области гомоген-
ности, они непрозрачны и т.д. В то же время ряд свойств указывает на
их определенную близость к солеобразным карбидам — как и карбид
кальция они разлагаются водой с образованием углеводородов (преиму-
щественно ацетилена), разрушаются разбавленными кислотами и ще-
лочами.
Для синтеза карбидов РЗЭ используются различные способы, осно-
ванные на нагревании при высокой температуре стружки металлов или
их соединений с углеродом. Наиболее доступным материалом являются
оксиды РЗЭ, но синтез с их использованием требует наиболее высокой
температуры (1800—1900 °C) и проведения процесса в вакууме. Кроме
того, в этом случае конечный продукт почти всегда бывает загрязнен
кислородом. При использовании в качестве исходного соединения гид-
ридов РЗЭ температура синтеза снижается до 1000 °C, карбиды получа-
ются более чистые
ЬпгОз + 7С = 2ЬпСг + ЗСО;
2Ьп Нз + ЗС = 2ЬпС2 + ЗНг (5.20)
Силициды. Кремний является аналогом углерода, поэтому, естест-
венно, силициды должны иметь много общего с карбидами. Однако
кремний имеет атомный радиус значительно больший, чем углерод
(0,134 и 0,077 нм), и меньшую электроотрицательность (1,9 и 2,6),
слабее акцептирует электроны. РЗЭ образуют силициды типа LnSi и
LnSi2- В структуре последнего предполагается наличие пары Si—Si, так
же как и у карбидов, но у силицидов больше доля ковалентности в связи
металл—кремний. Поэтому силициды обнаруживают большое сходство
с боридами, но они имеют относительно невысокие температуры плав-
ления; для дисилицидов иттрия, лантана и других она равна 1520-"
1540 °C. Силициды хрупки, имеют металлический блеск; некоторые из
них являются полупроводниками. Довольно устойчивы на воздухе, с
водой не реагируют, медленно разлагаются минеральными кислотами,
при нагревании активно взаимодействуют с азотом и галогенами. И3
218
физических свойств следует отметить ферромагнетизм при низкой
температуре. Сложные силициды LnMnSi, где Ln — Y, La, Gd, имеют
точку Кюри при 275—330 К. Большое значение имеют лигатуры марки
«Симиш» , содержащие до 25 % РЗМ, 10 % А1 и 50—60 % Si.
Силициды получают непосредственным взаимодействием металлов
с элементарным кремнием: либо спеканием при температуре ниже тем-
пературы плавления (800—1000 °C), либо плавкой в дуговых печах.
Дисилициды можно получить также восстановлением оксидов РЗЭ эле-
ментарным кремнием при 1350 °C в вакууме с удалением летучего
моносилицида SiO. Для получения лигатур типа «Симиш» лучшие ре-
зультаты дает плавка в электропечи (~ 1600 °C) смеси оксидов РЗЭ и
восстановителей — порошков алюминия и кремния.
Нитриды. Нитриды во многих отношениях тесно примыкают к кар-
бидам, они близки к ним по структуре и ряду свойств. Однако сущест-
вуют и различия, которые прежде всего связаны с разницей в числе
валентных электронов. У атома азота на оболочке L 5 электронов
(2s22p3), она более достроена до октета и поэтому у него преобладает
акцепторная способность; по электроотрицательности азот уступает
только фтору и кислороду. Поэтому в соединениях с РЗЭ он больше
склонен к степени окисления -3, и для него характерно наложение двух
типов связи — ионной и металлической. РЗЭ образуют нитриды типа
LnN со структурой NaCl, имеют высокие температуры плавления
(2670 °C YN, 2650 °C ScN), довольно высокую твердость, и в то же время
разлагаются на влажном воздухе, растворами щелочей, растворяются
минеральными кислотами
LnN + 3H2O = Ln(OH)3 + NH3. (5.21)
Нитриды получают при взаимодействии аммиака или азота с метал-
лами или гидридами при 800—1200 °C либо нагреванием оксидов с
углеродом в атмосфере азота при более высокой температуре
Ln2O3 + ЗС + N2 = 2LnN + ЗСО. (5.22)
5.6. Комплексообразование РЗЭ
В настоящем разделе обсуждаются некоторые закономерности, об-
разование и свойства комплексов РЗЭ с органическими лигандами. Ко-
ординационные числа в комплексных соединениях РЗЭ могут прини-
мать значения от 6 до 12, высокие значения их обусловлены тем, что
помимо 5d, 6 s и бр-орбиталей в комплексообразовании принимают, хотя
и малодоступные, орбитали 4/. При увеличении Z уменьшаются разме-
ры атомов лантаноидов и, по-видимому, затрудняется участие 4/-элек-
тРонов в образовании химической связи, поэтому к.ч. в ряду
________
«Симиш» — сокращенное от кремний, мишметалл; производились в СССР.
219
Рис. 5.10. Энтальпия гидратации ионов лантанои-
дов (Ln )
220
Lt Pr Ра Л П Не Та It
Се Hd Sa Ы Of b П
уменьшается. Для легких лантаноидов по спектральным данным вклад
4/-орбиталей в химическую связь составляет 10—15 % от общей энергии
связи. Этим же объясняется то, что в комплексах РЗЭ связь преимуще.
ственно ионная; ковалентная составляющая, связанная прежде всего с
наличием 4/-электронов, невелика. В некоторых комплексах эффектив-
ный положительный заряд центрального атома равен 2,7—2,8.
С другой стороны, уменьшение размеров атома при увеличении Z
приводит к усилению взаимодействия между центральным атомом и
лигандом и повышению прочности комплексов. Устойчивость комплек-
сов зависит от многих факторов, в том числе и от свойств лигандов
поэтому одинаковой зависимости характеристик разных комплексов от
2, не существует. Для наиболее достоверных количественных характе-
ристик комплексов — констант устойчивости, которые нужно сравни-
вать для однотипных комплексов с одинаковым к.ч., можно указать на
общую тенденцию их изменения в ряду La—Lu в сторону увеличения,
иногда на 3—4 порядка. Но эти зависимости не являются линейными,
на них имеются максимумы и минимумы, но наиболее характерен для
большинства из них «гадолиниевый угол» — специфический излом.
На поведение ионов РЗЭ, взаимодействие с различными лигандами
в водных растворах весьма существенное влияние оказывает гидрата-
ция, т.е. по существу комплексообразование с молекулами воды. К на-
стоящему времени можно считать достоверно установленным, что лег-
кие РЗЭ в аквакомплексах могут иметь к.ч. 9 как в некоторых солевых
растворах, так и кристаллах. Для тяжелых РЗЭ, начиная с эрбия, к.ч. в
аквакомплексах всегда равно 6. Энтальпия гидратации ионов РЗЭ при
увеличении порядкового номера почти монотонно возрастает с некото-
рыми отклонениями в сторону увеличения. Это свидетельствует об уси-
лении связи иона с каждой молекулой воды (рис. 5.10).
Комплексообразование
в водных растворах связано
с конкуренцией лиганда и
молекул воды за координа-
цию центрального атома.
Уменьшение активности
воды в растворе при высо-
ких концентрациях соот-
ветствующих солей щелоч-
ных металлов способствует
образованию комплексов. В
безводной среде, в том числе
и расплавах, комплексооб-
разование протекает более
активно.
Комплексы РЗЭ с неорганическими лигандами в водных растворах
^устойчивы, они диссоциируют на исходные компоненты, хотя их об-
иаружено довольно большое число. Наиболее устойчивы комплексные
соединения с органическими лигандами, имеющими несколько функци-
ональных групп, т.е. относящихся к типу внутрикомплексных, или хе-
латных соединений.
В безводной среде РЗЭ образуют с органическими соединениями
различных классов металлоорганические соединения (МОС). Истинные
МОС со связью Ln—С долгое время не были получены вследствие их
неустойчивости, которая обусловлена тем, что разность электроотрица-
тельности Ln и С недостаточна для образования чисто ионной связи, а
слабое участие 4/-орбиталей препятствует образованию прочной кова-
лентной связи. Первыми соединениями со связью Ln—С были цикло-
пентадиенильные производные Ln (СзН5)з с преобладанием ионной свя-
зи. Циклопентадиен является слабой кислотой, с металлами он образует
соли, содержащие циклопентадиенил-анион С5Н5; эти соединения лег-
ко гидролизуются
^^СН^
СН
%.
Н2С-----СН
Более многочисленны соединения РЗЭ, в которых связь между ато-
мом металла и органическим лигандом осуществляется через кислород.
При непосредственном взаимодействии хлоридов РЗЭ в безводной среде
СО спиртами образуются сольваты 1лС1з nROH, где п = 2—4, кристал-
лосольваты могут быть выделены из насыщенных спиртовых растворов
изотермическим упариванием при комнатной температуре. Обменной
реакцией между алкоголятами щелочных металлов и безводными хло-
ридами РЗЭ в спиртовой среде можно получить соответствующие алко-
соляты Ln(OR)3- Алкоголяты устойчивы только в неводных растворах,
водой они разлагаются с выделением гидроксидов.
Соединения с оксикарбоновыми кислотами. Оксикарбоновые кис-
л°ты были первыми комплексообразователями, использованными для
ионообменного разделения РЗЭ. Сейчас их значение уменьшилось, од-
нако в некоторых случаях они эффективнее широко применяемых ком-
Г1лексонов. Из многих оксикарбоновых кислот наибольшее значение
иМеют лимонная и винная. Лимонная кислота является трехоснов-
ной с одной гидроксильной группой НООС—СН2—С(ОН)(СООН)—
^2—СООН (2-оксипропан-1, 2, 3- трикарбоновая кислота). Винная
кислота имеет две карбоксильные группы и две гидроксильные НООС—
LH(OH) —СН(ОН)—СООН (1, 2-ОКСИЭТИЛ-1, 2-дикарбоновая кисло-
221
та). Комплексные соединения с оксикарбоновыми кислотами значи
тельно устойчивее комплексов с карбоновыми кислотами вследстви
координации иона РЗЭ с недиссоциированной гидроксильной группой и
замыкания циклов.
При взаимодействии растворов солей РЗЭ и цитратов щелочных
металлов осаждаются нормальные цитраты Ln (С6Н5О7) иН2О, раство-
ряющиеся в кислотах, щелочах и избытке реактива. В кислых растворах
образуются комплексы типа LnH2A2+ и LnHA+, а при избытке цитрата
— комплексный анион LnA$. Растворение LnA в щелочной среде, по-
видимому, связано с нейтрализацией иона водорода гидроксильной
группы и образованием комплекса LnA .
Взаимодействие ионов РЗЭ и аниона винной кислоты, как и в других
случаях, определяется соотношением концентраций компонентов и pH
раствора. С виннокислым аммонием ионы РЗЭ образуют осадки средних
солей, малорастворимые в воде — Дп2(С4Н40б)з иН2О. Растворимость
их в кислотах, щелочах и избытке осадителя связана'с образованна!
комплексных ионов: [Ln(C4HsO6) ]2+, [Бп(С4Н40б) ]+, [Ln (С4Н3О6) ]°,
[Ы(С4Н?Об) ] и др. Относительное содержание комплексов увеличи-
вается в этом ряду с повышением pH. Комплексы с винной кислотой
довольно устойчивы; в щелочной среде, когда в комплексообразовании
принимают участие обе гидроксильные группы, они не разрушаются в
1—2 М растворах щелочи, и осаждение гидроксидов не происходит.
Комплексы с фосфорорганическими соединениями. Известно боль-
шое число комплексов РЗЭ с органическими производными фосфорной
кислоты, из которых наибольшее практическое значение для разделе-
ния РЗЭ методом экстракции имеют комплексы с ТБФ и Д2ЭГФК. И те,
и другие лучше растворяются в органических растворителях, на чем и
основано их использование в экстракции. Комплексообразование ионов
РЗЭ с ТБФ осуществляется за счет донорно-акцепторного взаимодейст-
вия с фосфорильной группой [(C4H9O)3P=O....(Ln3+)i/3 ].
Комплексы с алкилфосфорными кислотами значительно прочнее
комплексов с НФОС вследствие того, что происходит замещение атома
водорода гидроксогруппы и замыкание цикла в результате образования
донорно-акцепторной связи иона РЗЭ с фосфорильным кислородом
Вследствие этого при экстракции фосфорорганическими кислотами воз-
никают определенные трудности в проведении реэкстракции. Для экст-
ракции РЗЭ широко применяется ди-2-этилгексил-фосфорная кислота
R О \
R О- -Р = О,гдей СН2----- СНз
Н — О—" |
СНз СН (СН2)4-——
222
фрагмент комплекса с ней: R-----О
R---
Р = О
Комплексы с fl-дикетонами. Типичным представителем уЗ-дикето-
йов является ацетилацетон и его производные. В водной среде уЗ-дике-
тоны в енольной форме реагируют с ионами металлов, в том числе и РЗЭ,
отшспляя протон и образуя хелатное соединение I .пАз. Кето-енольное
равновесие ацетилацетона:
НзСС СН2-С-СН3 Н3С С-СН= С СНз
II II 7--------> II I
0 0 о он
Фрагмент комплекса суЗ-дикетоном: ... R-С — О.
И \ з+
НС Ln3+------------
R----С = О /
Ацетилацетонаты РЗЭ образуются при добавлении к раствору хло-
ридов РЗЭ раствора ацетилацетоната аммония с 50 %-ным избытком от
стехиометрии и нейтрализации раствора аммиаком до pH несколько
меньше, чем pH осаждения соответствующего гидроксида. В растворе
находятся в равновесии ионы: Ln3+, LnA2+, Ln A J. При большом избытке
ацетилацетоната щелочного металла возможно образование комплекса
IJ1A4.
Ацетилацетонаты растворяются в органических растворителях зна-
чительно лучше, чем в воде, причем растворимость увеличивается в
Ряду La—Lu. На этом основано использование их для экстракционного
Разделения РЗЭ, экстракция/3-дикстонатов усиливается в присутствии
алкилфосфатов, сульфоксидов и других экстрагентов. Это явление, по-
лучившее название синергизма, объясняется образованием комплексов,
содержащих два типа органических лигандов, довольно широко исполь-
зуется на практике.
Из водных растворов ацетилацетонаты могут быть выделены только
с гидратной водой. Обезводить их нагреванием нельзя, так как они при
зтом разлагаются. Некоторые уЗ-дикетонаты РЗЭ обладают летучестью
пРи температуре ниже 100 °C, что дает возможность их использования
в Различных напылительных процессах.
Комплексонаты РЗЭ. Термин «комплексоны» первоначально рас-
пространялся на полиаминоуксусные кислоты — производные различ-
Нь*х аминов, в которых два или три атома водорода при атомах азота
223
замещены карбоксильными группами. В дальнейшем в их число стали
включать органические кислоты, содержащие гидроксоэтильные, ал
килфосфоновые, алкилсульфоновые и другие группировки. Характер
ным признаком этих соединений является сочетание в них основных и
кислотных центров: с катионами металлов они образуют комплексы
хелатного типа, включающие большое число циклов. Для РЗЭ известны
соединения примерно со 100 комплексонами, изучение их сыграло боль
шую роль в развитии химии РЗЭ и решении практических задач по их
разделению.
В качестве примеров будут рассмотрены комплексонаты, нашедшие
наибольшее применение для разделения РЗЭ методом ионного обмена
Это производные моно- и диаминов, имеющие следующие названия,
обозначения и символы для написания реакций с их участием:
.. СНгСООН
иминодиуксусная кислота (ИДА) H?L
СНгСООН
нитрилотриуксусная кислота (НТА) H3L
. СНгСООН
N-— СНгСООН
\ СНгСООН
этилендиаминтетрауксусная кислота (ЭДТА) H4L
НООССНг СНгСООН
-N—СНг СН2—N
НООССН2/ \ СНгСООН
В комплексах с этими лигандами связь ионов РЗЭ осуществляется
через атомы азота и атомы кислорода карбоксильных групп; комплексы
имеют трехмерную конфигурацию, причем длины связей иона РЗЭ с
кислородом карбоксильных групп и азотом довольно близки между со-
бой. Так, в комплексе лантана с ЭДТА длины связей La—Осоо одинако-
вы и равны 0,254 нм, а расстояния La—N равны 0,286 нм. Связь лап
таноид—азот частично ковалентная, а связь металла с кислородом кар
боксильных групп носит преимущественно ионный характер. В комП'
лексе ИДА занимает 3 координационных места, т.е. является трех'
дентантным лигандом, НТА — трехзарядный лиганд и может проявлять
дентантность 4, а для ЭДТА дентантность может быть 6.
Состав комплексов, образующихся в водных растворах, зависит ст
pH и соотношения концентраций компонентов, участвующих в комО
лексообразовании. Области существования комплексов различного со
става довольно широки и находятся в пределах значений pH от 1,5 Д°
Для низких значений pH характерно подавление диссоциации комплот
224
сона и образование протонированных комплексов, при pH <1,5 комп-
дексы разрушаются.
В щелочной среде образуются гидроксокомплексы; в разных систе-
мах образование их начинается при разных pH: в системах с ИДА — при
pH > 8, а в системах с ЭДТА — при pH > 10. В присутствии комплексонов,
как и других сильных комплексообразователей, осаждение гидроксидов
при обычных значениях pH не происходит. Представление о характере
комплексообразования и возможных равновесиях в зависимости от pH
раствора, концентрации ионов лантаноида и комплексонов в системе
Ln—ЭДТА дает схема:
Увеличение концентрации лиганда
LnH2L+-----; Ln(HL)l
Уве-
LnHL° '=^=- Ln Н 1.2
личе-
Ln2L2+ : . LnL~------------LnlJ
II X
Ln2L(OH)+ LnL(OH)2
pH ‘ ---------------------------------------—
Увеличение концентрации Ln
На рис. 5.11 дано распределение европия между различными комп-
лексами в системе Ей3*—ЭДТА—NaOH. Устойчивость комплексов по-
вышается с увеличением в комплексоне числа карбоксильных групп,
способных к координации. Количественно оценку ее можно дать путем
сравнения констант устойчивости, значения которых для рассматрива-
емых комплексов даны на рис. 5.12.
Растворяются комплексоны в воде плохо. Так, растворимость ЭДТА
ШдЬ) при 22 °C равна 2 г/л, несколько лучше растворима НТА — до 30
г/л Растворимость их уменьшается в кислой среде, при pH 2,2 начина-
ется область высаливания ЭДТА в форме H4L. Гораздо лучше раствори-
мы их соли с аммонием и щелочными металлами. Двузамещенная
Натриевая соль ЭДТА (трилон Б) — Na2H2L имеет растворимость при
^2 °C, равную 108 г/л. Еще лучше растворяется натриевая соль НТА
(трилон А). Из растворов путем медленного упаривания и последующей
длительной кристаллизации могут быть выделены кристаллогидраты
225
8 " 691
Рис. 5.11. Относительное содержа
ние комплексов европия N в систе-
ме Еи3+—ЭДТА—NaOH:
1 — Ей ; 2 — EuHL; 3 —
EuL, 4 — Eu(OH)L2“
Ct Hd Sa Gd Of Er П
Рис. 5.12. Константы устойчивости различных
комплексов лантаноидов и иттрия (1 : 1):
1 — ЕпЕ+(ИДА); 2 — LnL° (НТА); 3 -
LnL- (ЭДТА)
Рис. 5.13. Растворимость комплексных со-
единений лантаноидов и иттрия с ЭДТА в
воде при 25 °C:
1 — К [LnL] хН2О; 2 — Na [LnL]
хН2О
различного состава: из раствора
ИДА — NdLCl ЗН2О, из раство-
ров ЭДТА — LaHL 7Н2О,
NdHL 6Н2О и др., которые отли-
чаются относительно высокой рас-
творимостью. При взаимодейст-
вии водной суспензии ЬпгОз и
H4L получено соединение
Ьп4Ьз пНгО, растворимость кото-
рого не превышает 2 г/л. Из рас-
творов, содержащих щелочные
металлы, упариванием или выса-
ливанием органическими раство-
рителями выделяются кристалло-
гидраты комплексных солей раз-
ных типов: MsNdLa ЗН2О (ИДА)’
K3LaL2 6Н2О (НТА), MNdL Y2O
(ЭДТА), большинство из них хорошо растворимы в воде. Различия в
растворимости комплексных солей в ряду РЗЭ весьма существенны и
могут быть использованы для их разделения (рис. 5.13).
226
ТЕХНОЛОГИЯ РЕДКОЗЕМЕЛЬНЫХ ЭЛЕМЕНТОВ
5.7. Важнейшие области применения РЗЭ и их соединений
Практическое использование РЗЭ началось во второй половине XIX
века. Соединения РЗЭ применяли в производстве газокалильных кол-
пачков для осветительных газовых и керосиновых фонарей. Длительное
время колпачки и кремни для зажигалок оставались практически един-
ственной сферой применения РЗЭ. Быстрый прогресс в этой области
произошел во второй половине XX века в связи с развитием авиакосми-
ческой, электронной, нефтехимической, атомной и других отраслей
промышленности. Особенно быстро растет потребление индивидуаль-
ных РЗЭ (до 20 % в год), многие из которых обладают уникальными
свойствами. Этому способствовала разработка технически и экономиче-
ски эффективных способов разделения РЗЭ, в первую очередь экстра-
кции.
Общее потребление РЗЭ (в пересчете на ЬпгОз) в мире (без СССР)
в 1984—1987 гг. составило 25—30 тыс.т в год. В 1990 г. только в трех
крупнейших потребителях оно достигло 27,7 тыс.т: США 17,5; КНР 6,77
(КНР вышла на второе место с 1987 г.), Япония 3,35. Прогнозируется
рост спроса и потребления в текущем десятилетии на 4—6 %, а индиви-
дуальных РЗЭ высокой степени чистоты до 10 % в год (суммарно до 58
тыс.т к 2000 г). Структура потребления РЗЭ в разных странах различна:
в США до 53 % используется в производстве катализаторов, в Японии
— до 70 % для изготовления керамики и стекол, причем Япония зани-
мает первое место в мире по потреблению высокочистой редкоземельной
продукции в электронике и электротехнике (до 20 % общего потребле-
ния ее в мире). КНР использует до 67 % РЗЭ в металлургии и значи-
тельную их часть в качестве микродобавок к удобрениям, что позволяет
(по их данным) повысить урожайность на 15—20 %. Общие данные по
структуре потребления РЗЭ приведены ниже, %:
Катализаторы (в основном крекинг нефти)..39
Металлургия...............................20
Стекло и керамика.........................35
Люминофоры, лазеры, магниты, гранаты и т.д. ... 6
Однако по стоимости на первом месте стоят люминофоры (30 %),
так как для них нужны индивидуальные РЗЭ ОСЧ. В металлургии
используются главным образом дешевые смеси легких РЗЭ с природным
Удержанием элементов; их суммарная стоимость составляет около 6 %
°т общей стоимости (данные 1987—89 гг.).
227
8*
Металлургия. Использование РЗМ в металлургии основано на вц.
соком их сродстве с неметаллами, вследствие чего они применяются в
качестве раскислителей и десульфуризаторов сталей. В расплавленную
сталь они вводятся в виде мишметалла или сплавов типа «Симищ»
(приблизительно 2 кг на 1 т стали). Соединения РЗЭ с неметаллами
переходят в шлак, за счет этого происходит очистка расплава и улучше-
ние механических свойств стали. РЗЭ образуют также тугоплавкие со-
единения с As и Р, интерметаллиды с Bi, Sn, Pb (Trul > 1000 °C), что
позволяет производить очистку меди и ее сплавов от этих элементов.
РЗМ широко используются как легирующие добавки, например дЛя
получения высокопрочного чугуна. В обычном чугуне графит присутст-
вует в виде пластинок, легированный же РЗМ чугун содержит графит в
шарообразной форме; за счет этого его износостойкость и другие меха-
нические свойства возрастают в несколько раз. Добавки РЗМ эффектив-
ны в производстве высокопрочных низколегированных сталей для авто-
мобилестроения и трубопроводов большого диаметра. Для этих целей
разработана марка стали, содержащая 0,1 % Nb и 0,05 % РЗМ. В
последнее время усилился интерес к легированию металлов индивиду-
альными РЗМ. Особенно эффективным оказалось использование итт-
рия; чугун с его добавками («иттриевый чугун») обладает высокими
механическими и литейными свойствами, что позволяет использовать
его для изготовления наиболее ответственных деталей двигателей внут-
реннего сгорания. Имплантация поверхности деталей иттрием повыша-
ет их износостойкость по сравнению с хромированием в 100 раз.
В цветной металлургии РЗМ наиболее широко применяются для
легирования магниевых сплавов, их добавки повышают прочность, пла-
стичность, устойчивость против окисления при нагревании. Для этого
используют мишметалл, но наилучшие показатели получают при при-
менении неодима. Легирование технического алюминия повышает его
прочность при сохранении высокой электропроводности, имеются све-
дения, что электропроводность может и увеличиться, в особенности при
легировании иттрием.
Катализаторы. Соединения РЗЭ, прежде всего оксиды, входят в
состав многокомпонентных катализаторов различного назначения. На-
ибольшее распространение получили катализаторы для крекинга неф-
ти, при котором использование катализаторов на цеолитовой основе с
оксидами РЗЭ позволяет увеличить на 60 % выход высокооктановых
бензинов, уменьшить выход газообразных продуктов, увеличить время
работы катализатора. Катализаторы, содержащие смеси оксидов РЗЭ, а
также индивидуальные соединения (чаще всего церия и лантана), шИ'
роко применяются для проведения реакций алкилирования, изомериза-
ции, дегидрирования и др. Разработаны и применяются катализаторы в
виде шариков из оксида алюминия с покрытием, основным компонентом
228
которого является оксид церия, для дожигания токсичных компонентов
выхлопных газов двигателей внутреннего сгорания. По эффективности
очистки выхлопных газов автомобилей они не уступают платино-пал-
ладиевому катализатору. Имеется возможность создания многокомпо-
нентного антидетонатора для бензинов вместо токсичного тетраэтил-
свинца.
Стекло и керамика. В оптической промышленности используются
индивидуальные высокочистые оксиды РЗЭ для обесцвечивания и окра-
ски стекол, придания им способности пропускать или поглощать различ-
ные излучения (ИК, УФ и др.), повышать их термостойкость. Так,
оптическое лантановое стекло, содержащее от 5 до 40 % ЬагОз, обладает
высоким коэффициентом преломления и малым светопоглощением, ис-
пользуется для изготовления линз и призм различных оптических сис-
тем, в кино- и фотоаппаратуре. Неодимовое стекло (3—5 % Nd?O3)
используется в мощных твердотельных ОКГ. Желто-зеленая окраска
стекол, вызываемая присутствием РегОз, снимается добавлением к
стеклу — 0,02 % оксида неодима или эрбия, которые имеют сильную
полосу поглощения в желтой части спектра.
Для окраски стекла добавляют оксиды РЗЭ — от десятых долей до
нескольких процентов. Окраска стекла соответствует окраске тех или
иных ионов лантаноидов. Так, неодимовое стекло имеет фиолетовую
окраску, эрбиевое — бледно-розовую, празеодимовое — бледно-зеле-
ную, а стекло, окрашенное смесью СсО? и Т1О2, — от светло-желтой до
оранжевой. Стекла, окрашенные оксидами РЗЭ, имеют высокую про-
зрачность, повышенную термостойкость и стабильность, устойчивость
к рекристаллизации, очень чистые цвета. Окрашенные стекла исполь-
зуются в оптическом приборостроении и производстве предметов широ-
кого потребления. Добавки до 5 % фторидов лантана или неодима поз-
воляют получать высококачественное стекловолокно для волоконной
оптики.
Различные мощные излучения (рентгеновское, ультрафиолетовое,
электронные пучки и др.) вызывают изменение структуры стекла, кото-
рое сопровождается его окраской от желтого до темнокоричневого. Ста-
билизация стекла достигается добавкой 1—2 % оксида церия (IV). Це-
рий (IV) действует как электронная ловушка (Се4 + е —• Сел ). Та-
кое стекло применяется для телевизионных трубок, иллюминаторов
ЯЦерных реакторов и т.д.
Оксид иттрия широко используется для получения стабилизирован-
ной керамики на основе диоксида циркония, выдерживающей темпера-
туру до 2200 °C (10—16 % Y2O3), а также конструкционной керамики
на основе частично стабилизированного диоксида циркония (5—6 %
W.
229
Хромиты лантана (ЬаСгОз) и иттрия (YCrO3), легированные кал
цием, стронцием, магнием, обладают высокой огнеупорностью и термп
стойкостью, электронным типом проводимости. Керамика из них нащл
применение для изготовления нагревательных элементов электрич/
ских печей, работающих в окислительной атмосфере и темпеоатупр Г
1600—1900 °C. Р 2,0
Электроника. В электронике используется большое число материа-
лов, в которых РЗЭ являются или основными компонентами, или леги-
рующими добавками, за счет которых материал получает необходимые
электрофизические свойства. Все материалы перечислить не представ-
ляется возможным. Так, титанат бария, легированный лантаном, и ма-
териал на основе титаната кальция и алюмината лантана используются
для керамических ВЧ конденсаторов, производство которых исчисляет-
ся миллиардами штук. На основе твердого раствора РЬТЮз и РЬ7гОз
легированного лантаном (6—12 %), производится электрооптическая
керамика (система ЦТСЛ), отличающаяся высокой плотностью (до
99 ^), высокой оптической однородностью для модуляторов, спектраль-
ных фильтров, устройств запоминания, обработки и отображения ин-
формации. Позисторная керамика на основе ВаТЮз, легированного La,
Y и другими РЗЭ [0,1 0,5 % (ат.) ], нашла применение для изготовле-
ния датчиков температуры, в схемах термостатирования элементов ра-
диоэлектронной аппаратуры и т.д.
Широкие области применения имеют соединения РЗЭ со структурой
граната — от мощных твердотельных ОКГ до ювелирных изделий. Мо-
иокристаллы их с высокой степенью совершенства (плотность дислока-
ций 2—5/см ) выращивают диаметром более 100 мм и массой более 8 кг.
Эпитаксиальные пленки редкоземельных феррито-гранатов (например,
YsFesOi?) на немагнитной монокристаллической подложке из галлий-
гадолиниевого граната (GchGasOi?) перспективны для запоминающий
устройств (ЗУ) на цилиндрических магнитных доменах (ЦМД) для ЭВМ
нового поколения. Такие ЗУ обладают сочетанием целого ряда уникаль-
ных свойств механической прочностью, вибростойкостью, надежно-
стью в эксплуатации, Стойкостью к воздействию агрессивной среды,
малым энергопотреблением и высокой плотностью записи информации
(до 10 бит/см ). Железо-иттриевый гранат предложено использовать
для высококачественных магнитных головок видеокамер и других уст-
ройств.
Люминофоры. Известны сотни люминофоров, содержащих РЗЭ. Их
можно разделить на две основные группы: люминофоры для люминес-
центных ламп и люминофоры для телевидения, экранов осциллографи-
2 Позисторный эффект заключается в резком росте сопротивления керамики (в
10 10 раз) при достижении температуры точки Кюри
230
иеских и радиолокационных приборов, приборов ночного видения и т.д.
пдя люминесцентных ламп преимущественно используются люмино-
форы на основе ортованадата и фосфатованадата иттрия, активирован-
ные европием. С их помощью корректируется спектр излучения ламп
(люминофоры дают красное свечение). Светоотдача таких ламп в 3 раза
вЫше обычных ламп накаливания.
В цветном телевидении для красной компоненты наилучшие резуль-
таты получены для люминофора на основе оксосульфида иттрия, акти-
вированного европием (Y2O2S : Ей), для черно-белого телевидения —
люминофор Y2O2S : ТЬ. Все большее значение приобретают антисток-
совские люминофоры, для которых длина волны излучаемого света
меньше длины волны возбуждающего излучения. Это позволяет преоб-
разовывать ИК-излучение в видимое. Разработаны люминофоры с крас-
ным, зеленым, голубым свечением на основе оксосульфидов, оксо-
хлоридрв, фторидов РЗЭ; голубое свечение дает, например, люминофор
Yo 64Ybo,35Tmo,oiF3. Во многих типах люминофоров Eu, Sm, Tb, Nd
используются как активаторы. Для производства высококачественных
люминофоров необходимы соединения индивидуальных РЗЭ высокой
степени чистоты (99,99—99,999 %).
Магнитные материалы. Для постоянных магнитов с высокой энер-
гией наиболее эффективными оказались интерметаллические соедине-
ния легких РЗЭ. Магнитные материалы класса SmCos (разработаны в
1968 г.), в которых Sm может быть частично заменен на Се, Рг, Nd,
позволили расширить области применения постоянных магнитов, со-
здать устройства нового типа, миниатюризировать изделия (микродви-
гатели, динамики, СВЧ-генераторы, магнетроны и др.). В 80-х годах
разработаны новые семейства материалов на основе интерметаллидов
РЗЭ (Nd2Fei4B, Pri7Fei6,5B5Cui,5 и др.), в которых также могут иметь
место частичные замены компонентов. Магнитная энергия этих матери-
алов в несколько раз превышает магнитную энергию лучших сплавов на
основе железа, не содержащих РЗЭ, которая равна, кДж/м : 199 для
SmCo5, 277 для Nd2Fei4B и 95 для сплава Fe—Ni—Со—Al. Материалы
этих групп находят разнообразное применение, в том числе для сепара-
ции слабомагнитных руд (ильменит — рутил, апатит лимонит и др.),
классификации гранатов. Их использование позволяет повысить произ
водительность в десятки раз и уменьшить потребление электроэнергии.
Второй группой высокоэффективных материалов являются магни-
тострикционные материалы на основе интерметаллидов тяжелых РЗЭ с
Железом — ЬпЕег; наиболее эффективны материалы на основе Tb, Dy,
Er.
Гидриды РЗЭ, и особенно их интерметаллоиды, перспективны как
аккумуляторы водорода. Они поглощают на единицу массы больше во-
дорода, чем индивидуальные РЗЭ, они дешевле и позволяют создавать
материалы, работающие в различных интервалах температур
(GdFe2H4, YFeiFU, LaNisH*, где 2 < х < 6,8 и др.).
Ядерная техника. РЗЭ используются в ядерной технике для изць
товления материалов регулирующих стержней, защитных устройств
Из них наиболее перспективен европий. Реакция захвата тепловых
нейтронов протекает по типу п, у (у бора — по типу п, а), вследствие
этого облегчается проблема охлаждения стержней, так как рассеяние
энергии у-излучением протекает в большем объеме, чем СХ-излучением
кроме того, у европия образуются дочерние изотопы с большим сечени-
ем захвата тепловых нейтронов. Материал для стержней представляет
собой двухфазный кермет, в котором дисперсная фаза ЕигОз (до 38 %)
распределена в металле — нержавеющей стали и других сплавах. Пер-
спективно использование керметов с ЕигОз и в реакторах-размножите-
лях на быстрых нейтронах. Оксиды самария, европия, гадолиния ис-
пользуются в красках для покрытий, защищающих от радиации.
Некоторые изотопы, образующиеся в ядерном реакторе, находят
применение: 147Pm — в миниатюрных электрических батарейках для
стимуляторов сердца, слуховых аппаратах; изотоп 170Tm (Ti/г 129
дней), дающий мягкое у-излучение (84 кэВ) — для портативных меди-
цинских и технических «рентгеновских» аппаратов.
Полирующие материалы. Полирующие материалы на основе окси-
дов РЗЭ (полириты) обеспечивают высокое качество обрабатываемых
поверхностей при меньшем расходе материала и большей скорости по-
лирования по сравнению с другими материалами (ZnO, ZrO2 и AI2O3 и
др.). Полириты применяются для полировки листового стекла, телеви-
зионных экранов, оптических стекол и др. Производятся различные
виды полиритов на основе смесей оксидов легких РЗЭ, содержащих от
50 до 80 % оксида церия и размерами частиц от 1 до 5 мкм. Увеличение
содержания в полирите СеОг повышает эффективность полировки, по
всей вероятности, за счет химического воздействия СеОг, поскольку
процесс полировки по сути является процессом механохимическим. По-
явились марки полиритов с содержанием СеОг до 99 и даже 99,9 %.
5.8. Минералы РЗЭ, руды и месторождения
Суммарное содержание РЗЭ в земной коре довольно велико, их
общий кларк равен 0,15—0,16 %, их больше, чем В, Си, Со, ZnH
Ni. Кларки же индивидуальных РЗЭ находятся в пределах п Ю $"
п 10 4 %, и только для Тт и Lu они соответственно равны 3 10 й
7 10~5 %. Но даже тулий распространен больше, чем Sb, Bi, I, Cd, SeH
др. Содержание индивидуальных РЗЭ, % от суммы: Се 28, Y 18, Nd 16,
La 12, Gd 6, Sm 4, Eu 0,7, Tm 0,5.
232
Известно больше 200 минералов, содержащих РЗЭ > 0,01 %, но
собственных минералов, в которых ЬпгОз больше 5—8 %, около 60. Для
рЗЭ характерно образование селективных минералов, когда в одних
минералах преобладают цериевые, а в других — иттриевые РЗЭ. Мине-
ралы , в которых содержание РЗЭ приблизительно соответствовало бы
их кларкам, очень редки. По соотношению цериевых и иттриевых РЗЭ
минералы делятся на три группы:
цериевые с соотношением СегОз : Y2O3 >15;
иттрий-цериевые с соотношением СегОз : Y2O3 = 7—15;
иттриевые с соотношением СегОз : Y2O3 < 7.
Это явление связано с уменьшением ионных радиусов и основности
в ряду La—Lu. Элементы цериевой подгруппы изоморфно замещают
преимущественно ионы Са, Sr, Ва и др., иттриевые РЗЭ присутствуют в
минералах Zr и Мп, в урановых рудах накапливаются элементы середи-
ны ряда. Различия в основности РЗЭ также приводят к дифференциации
их содержания в основных и кислых горных породах: легкие РЗЭ накап-
ливаются в основных породах, тяжелые — в кислых1 . Для РЗЭ харак-
терно также присутствие в минералах других редких элементов — Th,
Ti, Nb, Та, Be.
Основными промышленными минералами являются бастнезит, мо-
нацит, ксенотим. Бастнезит образует собственные месторождения, мо-
нацит и ксенотим являются компонентами ильменит—ру-
тил—цирконовых и других россыпей. В последние годы важными источ-
никами РЗЭ, и в особенности иттрия, стали так называемые «ионные
руды», которые представляют собой глинистые материалы, образующи-
еся при выветривании гранитов. Иттрий и другие РЗЭ находятся в них
в виде адсорбированных ионов. Значительное количество иттрия посту-
пает из отходов уранового производства, содержащих до 1,5 % БпгОз, в
ТОМ числе до 1 % Y2O3. Перспективным сырьем является апатит, извле-
чение РЗЭ из которого при промышленной его переработке на удобрения
осваивается во многих странах. В нашей стране промышленное значение
имеет комплексный минерал — лопарит.
Бастнезит (Се, La ...)СОзР относится к фторокарбонатам, содер-
жит до 40 % СегОз, 36 % (La, Nd...)2O3, 20 % СО и 8 % F. Кроме
бастнезита, известно более 10 минералов, относящихся к этой группе и
образовавшихся при выветривании горных пород и разрушении силика-
тов РЗЭ. Для большинства из них характерна ассоциация с кальци-
ем: Са(Се,Са...)2(СОз)зРг — паризит, Ca(Ce,La...)(СОз)гР —
оинхизит, Ca(Ce,La...Y) (СОз)гР — иттросинхизит и др.
Горные породы делят на указанные типы в зависимости от содержания кремнезема:
основные — от 40 до 55 % SiO2 (базальты, диабазы), кислые — до 70 % SiO2 (граниты)
233
Монацит (Ce,La...Th)P04 имеет суммарное содержание ЬпгОз д0
68 %,втомчислеСегОз 20—30%, (La,Nd)2O3 30—40 %, до5 %УгОз
до 12 % ThO?, 30 % РгО5,до7 % 7гОгиО,1—0,3 % U. Минерал устойчив
и при выветривании горных пород накапливается в россыпях. В корен-
ных месторождениях основные спутники — циркон и апатит, в россыпях
— циркон, ксенотим, ильменит и др.
Ксенотим YPO4 с теоретическим содержанием Y2O3 62,13 % фак-
тически содержит от 30 до 60 %. Встречается в пегматитах гранитного
типа, может кроме РЗЭ содержать Th, Si. Во вторичных месторождениях
накапливается, как и монацит, в россыпях разного типа.
Апатит Са5(РО4)з(Ь,С1) теоретически должен содержать 55,4 %
СаО, 42,1 % РгО5и2,4 % F. В действительности же состав его подвержен
сильным колебаниям вследствие изоморфных замещений: F- X СГ,
ОН ; Р X Si; Са J Sr, Na, Ln. Вследствие этого существует много
разновидностей апатита, в том числе фторапатит, практически не содер-
жащий хлора; хлорапатит, не содержащий фтора и т.д. В среднем апатит
содержит 0,4—1,0 % ЬпгОз, основную массу которых представля-
ют легкие РЗЭ: (Се,Ьа)гОз 45—70 %; наиболее ценны РЗЭ: Nd20j
- 10—15 %, ЗгпгОз ~ 3 %, Е112О3 ~ 0,4 % и Y2O3 1,0 %. Апатит —
широко распространенный минерал пегматитового происхождения, его
спутники — магнетит, кальцит, полевой шпат и др.
Лопарит (Ce,Nd,Ca) (Ti,Nb)O3 имеет химический состав, %: ЬпгОз
29—33; ТЮг 37—40; NbzOs 9—11; ТагО5 0,8; ТИОг до 0,6; кроме того
Fe, Si, Sr. Относительное содержание редкоземельных элементов, %:
(Се, Ьа...)20з> 70; Nd2O3 ~ 10; 5тгОзО,6; ЕигОзО,!; УгОзО,2. Лопарит
— минерал нефелиновых сиенитов и пегматитовых жил. Спутники —
эвдиалит, нефелин, сфен.
Месторождения руд по относительному содержанию легких и тяже-
лых РЗЭ можно также разделить на три основных типа. Основной тип
месторождений легких РЗЭ — бастнезитовые и монацит—бастнезито-
вые руды с содержанием ЬпгОз 5—9 %. Запасы их исчисляются десят-
ками миллионов тонн (КНР, США). Второй тип — руды, в которых
соотношение между отдельными РЗЭ приблизительно соответствует их
кларку. В основном это комплексные прибрежно-морские, погребенные
россыпи и небольшие коренные месторождения, содержащие также
минералы титана, циркония, ниобия, тантала. Содержание ЬпгОз в
рудах достигает нескольких процентов, запасы — миллионы тонн (Авс-
тралия, Индия, США и др.). Месторождения тяжелых РЗЭ в основном
содержат ксенотим, самарскит, гадолинит и другие довольно редкие
минералы, для которых характерна ассоциация с минералами титана,
циркония, ниобия, тантала, олова и других элементов. Содержание в
рудах < 0,5 %, содержание Y2O3 30—60 % (от суммы ЬпгОз); запасы
234
„ сотни тысяч тонн. К этому типу следует отнести также и ионные руды,
открытые в КНР в начале 80 годов.
Запасы ЬпгОз по данным Горного бюро США на начало 1991 г. (без
СССР) приведены ниже (в числителе — в тысячах тонн, в знаменателе
_ в процентах):
КНР......... 48000/50,7
США......... 13990/14,8
Австралия .. . 6950/7,3
Индия........2560/2,7
Канада....... 1047/1,1
Бразилия .... 314/0,3
Малайзия . 35/0,1
Таиланд.......1/—
Другие 21784/23
Всего . 94681 /100
Производство концентратов РЗЭ довольно быстро растет, так, за 5
лет (с 1985 по 1990 г.) оно выросло почти на 40 % (табл. 5.12). Концен-
траты РЗЭ имеют разное содержание суммы ЬпгОз. Так, бастнезитовые
концентраты содержат до 60 % ЬпгОз, монацитовые 55—60 %, ксено-
тимовые 25—30 %, а после химического обогащения — до 60 %, содер-
жание в них Y2O3 — до 65 % от суммы ЬпгОз. Цена на концентраты
определяется содержанием в них РЗЭ, но главным образом наличием
наиболее ценных тяжелых РЗЭ. Так, на начало 1990 г. в США цена
бастнезитового концентрата составляла 2,65 долл, за 1 кг содержания в
нем суммы ЬпгОз, а 1 кг Y2O3 в 60 %-ном иттриевом концентрате стоил
32 долл., 1 т монацитового концентрата — 656 долл. Бастнезитовые
концентраты получают преимущественно методом флотации, монаци-
товые — при добыче из россыпей — комбинацией гравитационных,
магнитных и электростатических методов, основанных на различиях в
плотности, магнитных свойствах и электропроводности минералов (см.
табл. 5.12).
Таблица 5.12. Производство РЗЭ в рудном сырье, тонны ЬпгОз
Страна 1985г., т 1990 г. Страна 1985г., т 1990 г.
т % т %
США 13878 16500 28,9 Таиланд 135 1500 2,6
Австралия 10304 7500 13,1 Шри-Ланка ПО 100 0,2
Малайзия 3191 1700 3,0 Канада — 100 0,2
Индия 2200 2200 3,8 КНР 11150 25000 43,8
Бразилия 667 2500 4,4 Всего 41635 57100 100,0
235
5.9. Общие вопросы технологии РЗЭ
Особенности технологии РЗЭ в значительной степени определяются
комплексностью руд, разнообразием состава собственных минералов
РЗЭ, многообразием минералов вмещающих горных пород, присутству-
ющих в концентратах. Последние могут сильно влиять на процессы
разложения концентратов вследствие протекания вторичных реакций
Во многих случаях концентраты РЗЭ содержат уран, торий и продукты
их радиоактивного распада, поэтому необходимы операции по их удале-
нию на первых этапах переработки минерального сырья.
Минералы РЗЭ (в основном фосфаты и фторокарбонаты) не отлича-
ются высокой устойчивостью к воздействию различных реагентов. Для
их разложения предложено большое число способов, но в промышлен-
ности используются лишь немногие. Например, для разложения мона-
цита и других фосфатных минералов используют два основных метода:
сернокислотный и щелочной. Выбор способа разложения концентрата
определяется главным образом задачами производства конечных про-
дуктов и способами переработки реакционной массы, получаемой при
операции разложения.
В развитии и совершенствовании технологии РЗЭ прослеживаются
некоторые тенденции. Широкое развитие экстракционных методов раз-
деления РЗЭ и очистки их от примесей привело к существенному сокра-
щению традиционных операций, таких, как дробная кристаллизация,
дробное осаждение различных солей и гидроксидов и т.д. Это позволило
не только сократить число технологических операций, повысить эффек-
тивность технологии РЗЭ вообще, но и решить многие задачи, которые
не могли быть решены традиционными способами. В некоторых случаях
это повлекло и изменение способа разложения концентратов.
Вторая очень важная тенденция связана с изменением приоритетов
в использовании РЗЭ и их соединений. В областях, в которых до послед-
него времени использовали смеси соединений РЗЭ (полирит) или спла-
вы (мишметалл), растет потребление индивидуальных РЗЭ. Непре-
рывно расширяются области применения высокочистых индивидуаль-
ных РЗЭ (люминофоры, материалы ОКГ, магниты и др.), поэтому все
большее развитие получают процессы разделения и получения веществ
ОСЧ.
Значительные количества РЗЭ производят и еще большие количест-
ва могут быть получены при переработке комплексного сырья. В каче-
стве примера можно привести лопарит, который в нашей стране пере-
рабатывался прежде всего с целью получения ниобия и тантала серно-
кислотным способом и методом хлорирования в расплаве солей. В каче-
стве побочных продуктов при хлорировании получали тетрахлорид ти-
236
танаи плав хлоридов, содержащий 37—45 % хлоридов преимуществен-
но легких РЗЭ
Перспективным видом сырья для получения РЗЭ в нашей стране
ивляется апатит Са5(РО4)з(Р,С1), уникальные месторождения которого
имеются на Кольском полуострове. Апатит добывается как сырье для
производства фосфорных удобрений, но он содержит до 0,4— 1 % ЬпгОз,
до 3 % SrO, и его нужно рассматривать как комплексное сырье для
получения фосфора, РЗЭ, стронция и фтора. При добыче апатита в
несколько миллионов тонн потенциальные возможности извлечения
РЗЭ составляют десятки тысяч тонн ежегодно. Однако до настоящего
времени апатит в основном перерабатывается сернокислотным спосо-
бом, который исключает попутное извлечение РЗЭ и стронция. Хотя
уже давно (1936—1939 гт.) в нашей стране разработан способ разложе-
ния апатита азотной кислотой, позволяющий извлекать попутно и РЗЭ,
и стронций. РЗЭ при этом получают в виде 30—35 %-ного концентрата,
который можно перерабатывать известными методами.
Основные переделы в технологии РЗЭ: разложение концентрата;
отделение радиоактивных элементов; отделение основной массы обыч-
ных примесей; разделение РЗЭ на группы; разделение и получение
индивидуальных соединений РЗЭ; получение целевых продуктов, в том
числе ОСЧ; получение металлов, сплавов и их рафинирование.
5.10. Переработка монацита
Сернокислотный способ (рис. 5.14). Способ основан на реакциях
2LnPO4 + 3H2SO4 X Ьп2(8О4)з + 2НзРО4;
ТЬз (РО4) 4 + 6H2SO4 Z 3Th (SO4) 2 + 4НзРО4. (5.23)
Присутствующий в концентрате минерал торит также разлагается
ThSiO4 + 2H2SO4 г Th(SO4)2 + SiO2 nH2O. (5.24)
Монацит, измельченный до 0,1 мм, можно разложить серной кис-
лотой с концентрацией более 50 %, однако на практике используют
93 %-нуюкислоту. Сульфатизациюпроводят при 180—210°С, повыше-
ние температуры выше 250 °C приводит к образованию безводного ди-
фосфата тория ТЬРгО?, не растворяющегося в сульфатных растворах.
Расход кислоты составляет 1,5—2,5 т на 1 т концентрата, что превышает
теоретически необходимое количество в 2,5—3 раза. Процесс проводят
периодически в чугунных реакторах с мешалками или трубчатых вра-
Щаю- щихся аппаратах в течение 2—4 ч с непрерывной загрузкой и
выгрузкой реакционной массы. При этом достигается практически пол-
ное разложение монацита. Сульфатизация сопровождается выделением
большого количества паров SO3 и радиоактивного газа торона, поэтому
237
I
I
I
I
1
I
1
I
I
I
I
нн3нго
Осаждение Th
(1ОО'С,рН1)
Концентрат
2-6 вариант
1-0 вариант
NagSO^
Раствор
, ^Н3"2°
Осаждай в в РЗЗ
(pH 2,3)
NaOH
Раствор
Концентрат U
HNO3 Н;0
Осаждение U
(pH 4)
[ Раствор
—1 (сброс)
Раствор (сброс)
КоОН
Кониерсов
в твдровсиды
Осаждение двоО-
нив сглвфитов
Конверсии
i тидровсиды
pb. h°?C°3
т^(40%-нон)
Осаждение
РЗЗ ______
| Раствор
U, П
Растворение
тидровсидея
Раствор (сброс)
\КМг(40%-нои)
Растворение
твдровсидов
Растворение
варбонетен
Раствор
РЗЗ
на ратделении
Раствор
РЗЗ
на рас деление
Раствор
РЗЗ
на ратделенае
Рис. 5.14. Принципиальная схема переработки монацита сернокислотным способом
для обеспечения нормальных условий труда и устранения вредного воз-
действия на окружающую среду необходима не только хорошая венти-
ляция, но и надежная система улавливания и обезвреживания газов.
Реакционную массу выщелачивают водой при Т : Ж = 1 : (9^12) с
целью полного перевода в раствор сульфатов РЗЭ и тория в освинцован-
ных реакторах. Так как растворимость сульфатов РЗЭ понижается при
повышении температуры, то выщелачивание проводят холодной водой,
в конце процесса температура не должна превышать 20—25 °C. В рас-
твор кроме РЗЭ и Th переходит Н3РО4, избыточная H2SO4, примеси,
находившиеся в концентрате: Mg, Al, Ti, Fe, U и др.; в нерастворившем-
ся остатке — ZrSiO4, ТЮ2, S1O2 иНгО, неразложившийся монацит-
Вместе с этим осадком удаляют мезоторий (изотоп 228Ra, Ti/2=6,7 года)
путем добавления к пульпе ВаС1г и осаждения его на носителе BaSO4-
238
Следующей задачей является выделение тория, так как присутствие
еГ0 на последующих переделах крайне нежелательно вследствие его
радиоактивности и высокой токсичности.
Для переработки серно- и фосфорнокислых растворов предложено
большое число методов, из которых наиболее распространен был метод
ступенчатой нейтрализации. Он основан на различии pH осаждения
фосфатов тория и РЗЭ. Раствор, полученный при выщелачивании, раз-
бавляют до концентрации по РЗЭ 2 г/л, нагревают до кипения и при
интенсивном перемешивании нейтрализуют аммиаком до pH 1,0. В
осадок выпадает 98—99 % тория в виде гидратированного дифосфата
TI1P2O7 2Н2О; вместе с ним осаждается до 5—8 % РЗЭ, содержавшихся
в растворе. После отделения осадка тория раствор нейтрализуют амми-
аком до pH 2,3, при этом в осадок выпадает около 90 % РЗЭ в виде
пирофосфатов Ьп2(НРО4)з хНгО и двойных сульфатов с аммонием
NH4Ln(SO4>2 хНгО. Фильтрат, содержащий небольшое количество
РЗЭиуран, нейтрализуют до pH 6,0 для получения осадка (NH4)2U2O7,
вместе с которым осаждаются и все оставшиеся в растворе РЗЭ.
Операции ступенчатой нейтрализации требуют жесткого регулиро-
вания pH растворов и создания таких условий, которые исключали бы
местные пересыщения. Обычный способ приливания раствора осадителя
даже при интенсивном перемешивании и подаче раствора распылением
не создает необходимых условий. Лучшие результаты дает метод слива-
ния двух растворов. Осаждение газообразным аммиаком, который бар-
ботируется воздухом через раствор, позволяет избежать разбавления
раствора, которое происходит при обычном способе осаждения, и почти
полностью исключает возникновение местных пересыщений. Идеаль-
ным же является метод «гомогенного» осаждения. Сущность его заклю-
чается в том, что осадитель образуется в результате медленно про-
текающей реакции во всем объеме раствора. Для этой цели используют
карбамид CO(NH2)2 (мочевину), который является очень слабым осно-
ванием. При комнатной температуре он почти не гидролизуется, но при
90—100 °C гидролиз идет с достаточной для технологии скоростью
CO(NH2)2+H20- CO2+2NH3. (5.25)
Этот способ и был предложен для селективного осаждения гидрокси-
дов РЗЭ, тория, урана.
Осадок фосфатов и двойных сульфатов РЗЭ подвергают конверсии в
'и.щоксиды обработкой концентрированным раствором NaOH; гидро-
ксиды растворяют в азотной кислоте и направляют на экстракционное
Разделение. В этом способе торий довольно полно удаляется за одну
операцию, но РЗЭ «размазываются» по фракциям, что вызывает необ-
ходимость проведения дополнительных операций по извлечению из
осадков.
239
Метод осаждения двойных сульфатов РЗЭ и натрия позволяв про
вести грубое разделение РЗЭ на цериевую и иттриевую подгруппу
К раствору после выщелачивания добавляют Na2SO4 в количестве 50-~
55 % от массы монацита; в осадок при этом выпадают двойные сульфа^
цериевой подгруппы NaLn(SO4>2 хНгО и 50 % тория в виде сульфата
Th(SO4>2 хНгО. Осадок обрабатывают концентрированным раствором
NaOH для перевода их в гидроксиды, которые для окисления цсрия
сушат на воздухе при 120 °C, затем растворяют в азотной кислотен
экстракцией ТБФ селективно удаляют торий и церий.
Раствор, содержащий иттриевые РЗЭ и торий, можно перераба-
тывать разными способами: осадить РЗЭ в виде карбонатов
Ьп2(СОз)з хНгО и Ln (ОН) СОз хНгО добавлением \агСОз; торий и
уран, образующие растворимые комплексы [ТЬ(СОз)5Г и
[иО2(СОз)4 ] , остаются в растворе; осадить оксалат тория, РЗЭ
при этом остаются в растворе, так как их оксалаты лучше растворяются
в серной кислоте. Осадок оксалатов в пересчете на оксиды содержит -
90 % ThO? и ~ 10 % ЬпгОз (см. рис. 5.14).
- При высоком содержании в концентратах урана используют ме-
тод осаждения оксалатов. Раствор после выщелачивания разбавляют до
15 г/л РЗЭ, нейтрализуют аммиаком до pH 1,5 и осаждают торий и РЗЭ
10 %-ным раствором Н2С2О4. Горячим раствором NaOH оксалаты пе-
реводят в гидроксиды, которые растворяют в HNO3 и направляют на
экстракцию ТБФ. Большая часть оксалата натрия направляется в обо-
рот, им частично заменяют щавелевую кислоту. Этим способом торий
извлекается полностью, РЗЭ — на ~ 98 %, уран — на 90 %; последний
извлекают ионным обменом.
Щелочной метод. Метод (рис. 5.15) основан на реакциях
LnPO4 + 3NaOHx Ln(OH)3 + Na3PO4;
Th3(PO4)4 + 12NaOH - Th(OH)4 + 4№зРО4. (5.26)
Вместо NaOH можно использовать соду №гСОз, принципиального
различия в химизме взаимодействия этих реагентов с монацитом нет,
разница заключается лишь в температуре процессов и аппаратурном
оформлении. При работе с растворами NaOH температура в предо*
лах < 200 °C, процесс периодический в стальных коррозионностойких
реакторах. При использовании №гСОз, имеющей меньшую реакцион-
ную способность, температура должна быть более высокой, но ниже
плавления №гСОз (852 °C) процесс может быть осуществлен как непре-
рывный.
Тонкоизмельченный монацитовый концентрат (0,043 мм) обра-
батывают 45—50 %-ным раствором NaOH при 140—150 °C в течение
3—4 ч, количество NaOH — 1,5 кг на 1 кг концентрата, что в 3 ра-
за превышает теоретически необходимое. Разложение монацита
240
Концентрат NaOH (15%-ный)
Hi, U
и разделение
Рис. 5.15. Принципиальная схема переработки монацита щелочным способом
96,5 %. В раствор количественно переводится РО4 , а торий и РЗЭ — в
осадке в виде легкорастворимых гидроксидов. Для их растворения чаще
используют соляную кислоту. Полного разложения монацита можно
достичь повышением температуры до 200 °C, однако при этом гидрокси-
ды тория и РЗЭ частично дегидратируются и трудней растворяются в
кислотах. Поэтому неразложившийся концентрат после растворения
гидроксидов предпочитают направлять в голову процесса.
Для уменьшения расхода NaOH предложен двухстадийный процесс
разложения в шаровых обогреваемых мельницах (130 °C), в которых
процесс вскрытия совмещается с механическим активированием мате-
риала, устранением с поверхности зерен монацита пленки гидроксидов
и улучшением условий диффузии NaOH. В этом случае концентрат
Может быть измельчен до частиц > 1 мм. На первой стадии расходуют
150 % от теоретически необходимого количества NaOH. Гидроксиды
Растворяют в соляной кислоте, а неразложившиеся остатки от 10 партий
241
вторично обрабатывают NaOH. При обработке в течение 2 ч мойац^т
разлагается на > 99 %, расход NaOH уменьшается почти в 2 раза.
Реакционную массу после завершения реакций разложения мона-
цита разбавляют водой и нагревают до 100—110 °C для полного раство-
рения тринатрийфосфата, растворимость которого сильно увеличива-
ется при повышении температуры (при 20 °C 9,9 и при 80 °C 44,8 %)
При этой температуре пульпу выдерживают 1 ч для образования хорошо
фильтрующихся осадков гидроксидов и фильтруют при 80 °C. Осадок
гидроксидов тщательно промывают водой для удаления №зРОд (содер-
жание Р2О5 должно быть < 0,4 %) и направляют на растворение соляной
кислотой, а промывные воды — на разбавление реакционной массы.
Фильтрат упаривают, а затем кристаллизуют NaaPO4 12Н2О, который
является товарным продуктом. Тринатрийфосфат выделяется очень по-
лно, так как при упаривании в растворе повышается концентрация
NaOH (до — 45 %), что приводит к уменьшению его растворимости (в
36 %-ном растворе NaOH она равна всего 1,3%). Около 50 % фильтра-
та, содержащего в основном NaOH, направляют на операцию разложе-
ния, полностью его утилизировать не удается вследствие накопления в
нем примесей, прежде всего силиката натрия.
Для переработки гидроксидов наибольшее применение нашли два
варианта растворения их в соляной кислоте. По одному из них смесь
гидроксидов обрабатывают соляной кислотой до pH 3,5—4,0 для селек-
тивного перевода в раствор РЗЭ. По другому варианту смесь гидроксидов
растворяют полностью в соляной кислоте, которая берется с избытком
25 %. Затем раствор разбавляют и нейтрализуют до pH 5,8; при этом в
осадок извлекается 99,7 % тория, ~ 99 % урана и ~ 3 % РЗЭ, содержав-
шихся в растворе. При дальнейшей нейтрализации раствора осаждают
гидроксиды РЗЭ.
Сравнение сернокислотного и щелочного методов переработки мо-
нацитовых концентратов показывает, что сернокислотный метод явля-
ется более универсальным, им можно перерабатывать различные кон-
центраты, в том числе и бедные, не требуется тонкого их измельчения,
метод более экономичен. К числу недостатков его следует отнести «раз-
мазывание» тория и РЗЭ по различным фракциям, большее число опе-
раций и значительные экологические вредности. К преимуществам ще-
лочного метода относится выделение NagPCM I2H2O в виде товарного
продукта, получение довольно чистых концентратов тория и РЗЭ на
ранней стадии производственного процесса. Метод более дорог, но ему
отдают предпочтение при получении чистых соединений РЗЭ.
242
5.11. Переработка бастнезита
Бастнезит в рудах ассоциирован с кальцитом (СаСОз), кварцем
(SiOz) > баритом (BaSC>4), флюоритом (CaFz). Содержание ЬпгОз в рудах
< Ю %. После обогащения (преимущественно флотацией) получают
35- и 60 %-ные концентраты. Обработкой 10 %-ной соляной кислотой
из 60 %-ного концентрата можно получить 72 %-ный. Выход богатых
концентратов не превышает 75 %. Поэтому предложены способы пере-
работки как концентратов, так и необогащенной руды. Бастнезит, как и
другие фторокарбонаты, термически неустойчив, и в большинстве
методов первой операцией его переработки является обжиг при 400—
800 °C. При обжиге разрушается кристаллическая структура бастнези-
та, образуются оксиды и фториды РЗЭ,масса становится пористой, что
облегчает последующее кислотное выщелачивание. При обжиге проис-
ходит также окисление церия, что позволяет уже на начальной стадии
провести селективное его выделение.
Основная реакция, протекающая при обжиге бастнезита,
ЗЬпБСОз Z ЬпгОз + ЬпРз + ЗСОг. (5.27)
Известны многочисленные варианты переработки реакционной мас-
сы. По одному из них массу после обжига богатого концентрата выще-
лачивают 5 %-ной соляной кислотой для перевода в раствор РЗЭ; в этих
условиях СеО? не растворяется. Получают осадок — цериевый концен-
трат с содержанием СеОг 80—90 % и раствор всех остальных РЗЭ. Из
фильтрата осаждают гидроксиды РЗЭ, в которых содержание Се(ОН)4
не превышает2—4 %.
Для извлечения РЗЭ из 10 %-ной бастнезитовой руды проводят
обжиг при 800—900 °C и выщелачивают огарок 57 %-ной азотной кис-
лотой. После отделения осадка из фильтрата экстрагируют РЗЭ трибу-
тилфосфатом. Извлечение РЗЭ составляет > 98 %. Около 80 % HNO3
Регенерируют обработкой рафината концентрированной серной кисло-
той (96 %) и ее экстракцией ТБФ.
Разработан сернокислотный метод переработки бастнезитового кон-
центрата. Концентрат нагревают с H2SO4 (Т > 100 °C) до полного уда-
ления фтора, затем реакционную массу обжигают при 650—750 °C. При
этом большинство компонентов концентрата, кроме РЗЭ, переходит в
Нерастворимые в воде соединения. Затем сульфаты РЗЭ выщелачивают
из огарка холодной водой, фильтруют и из фильтрата осаждают гидро-
ксиды РЗЭ. Разработан также метод хлорирования бестнезитового кон-
центрата в смеси с углеродистым восстановителем при 1000—1200 °C.
243
5.12. Выделение РЗЭ и тория из растворов
сложного солевого состава
В технологии переработки минерального сырья удовлетворительно
отработаны методы его разложения и перевода РЗЭ в водный раствор.
Значительные трудности представляет извлечение РЗЭ из растворов
отделение их от обычных примесей и радиоактивных урана и тория.
Широко используемые методы ступенчатой нейтрализации, осаждения
двойных сульфатов и другие по своей сущности не могут обеспечить
четкого и эффективного отделения РЗЭ и характеризуются «размазы-
ванием» по фракциям или РЗЭ, или тория и урана. Последнее в связи с
ужесточением требований к содержанию радиоактивных элементов в
продукции совершенно недопустимо. Наиболее перспективным спосо-
бом решения этой задачи является экстракция, однако практическая ее
реализация также сопряжена с рядом трудностей. Например, для экст-
ракционного извлечения и разделения РЗЭ ТБФ необходимы нитрат-
ные растворы. В реальных растворах, получаемых при выщелачивании
реакционной массы, содержатся ионы SO4, РО4, а во многих случаях
и F , затрудняющие экстракцию вследствие образования в водной фазе
прочных комплексов с РЗЭ, ураном и торием.
Наиболее прочные комплексы РЗЭ, торий и уран образуют с ионом
F ; удалить полностью его не удается и при конверсии их соединений,
например в гидроксиды. Так, константы устойчивости комплексов типа
LnF2* для лантана и церия соответственно равны 1,6 103и0,6 103, для
комплексов тория типа ThF3*, ThF3+ и ThF3+ соответственно 4 107;
3 1013и1 1016, для комплексов урана UO2F+hUO2F3— 4 104иЗ 1О10
Фосфатные комплексы менее прочны; для [ТЬНгРО413+ и
[ТЬ(Н2РО4>2]2+они равны 2 104 и 1,4 108. Мешающее действие ионов
фтора можно устранить добавлением к раствору нитрата алюминия,
образующего с ионом F- более прочные комплексы. Для комплексов
алюминия с общей формулой AlF„~n при изменении п от 1 до 6 констан-
ты увеличиваются от 1 • 106 до 7 1019.
Такой прием представляется целесообразным, так как помимо свя-
зывания ионов фтора ионы алюминия наряду с ионами щелочных ме-
таллов, магния, железа, кальция экстрагируются плохо, но являются
высаливателями по отношению к РЗЭ, торию и урану. При определен-
ных условиях в противоточном экстракторе (12—15 ступеней) практи-
чески полностью извлекаются РЗЭ, торий и уран с одновременным
уменьшением содержания обычных примесей в 20—100 раз.
Сравнение коэффициентов распределения урана, тория и РЗЭ пока-
зывает, что при концентрации HNO3 в водной фазе 2—4 моль/л коэф'
244
фициенты распределения соответственно равны п 10, п, п 10 2 при
экстракции ТБФ, где п = 1—9. Оптимальные условия для разделения
рЗЭ и тория — концентрация HNO3 4,0—4,5 моль/л. При такой кон-
центрации азотной кислоты коэффициент разделения /Зть^зэ / /^рзэ в
зависимости от общей концентрации РЗЭ и тория в растворе изменяется
от 60 до 200. Это позволяет в 12-ступенчатом каскаде полностью извлечь
торий. Относительное содержание РЗЭ (Nd, Sm, Gd) после промывки
органической фазы 4 моль/л HNO3 не превышает 2 10 4 % каждого
элемента. Уран экстрагируется вместе с торием, и разделение их может
быть проведено промывкой экстракта.
При экстракции Д2ЭГФК при одинаковых условиях лучше всего
экстрагируется торий, соотношение коэффициентов распределения то-
рий : уран : РЗЭ равно приблизительно 400 : 4 : 1, что позволяет также
эффективно разделять эти элементы. Имеются также данные об исполь-
зовании экстракции этих элементов ТБФ непосредственно из пульп,
получающихся при выщелачивании реакционной массы. Это позволяет
устранить такую трудоемкую операцию, как фильтрация, но связано со
значительными потерями экстрагента.
5.13. Разделение РЗЭ методами селективного
окисления и восстановления
Методы разделения, основанные на способности Се окисляться, а
Sm, Ей и Yb — восстанавливаться, отличаются большой эффективно-
стью вследствие возрастания различий в свойствах соединений лантано-
идов в аномальных степенях окисления и соединений лантаноидов с
обычной степенью окисления — три. По химическим свойствам церий
(IV) приближается к Th и Ti, a Sm, Ей и Yb в степени окисления (II) —
к щелочноземельным элементам, причем наибольшее сходство наблю-
дается у Yb и Са, a Sm, и особенно Ей, являются аналогами Sr.
Отделение церия. Отделение церия, наиболее распространенного
из РЗЭ, предусматривается на начальных стадиях переработки мине-
рального сырья. Окислительно-восстановительный потенциал системы
Се3* / Се4+ зависит от природы минеральной кислоты и ее концентра-
ции. В кислых растворах он равен 1,3—1,8 В, в слабокислых (pH г 5) и
Щелочных он уменьшается до 0,07 В. Вследствие этого для окисления
Церия можно использовать слабые окислители, например кислород воз-
духа. В промышленности используют кислород, озон, хлор, КМпО4,
КВгОз, Н2О2 и др. В последнее время все шире применяется безреаген-
тный метод электрохимического окисления. После окисления церий
отделяют осаждением малорастворимых соединений, ионным обменом,
экстракцией.
245
Широко используется метод, основанный на меньшей растворимо,
сти Се(ОН)4 в разбавленных кислотах (ПР = 2 10 48), чем Ln(OH)3
(ПР = 1,0 10 19—1,0 10 24), и метод, использующий разницу в рц
осаждения гидроксида и основных солей Ce(IV) 0,7—1,0 и гидроксидов
Ln(III) 6,3—7,8. Этот метод позволяет удалить основную массу церия
вместе с торием.
Для удаления церия ионным обменом используют катионообменные
смолы и сульфатные растворы. Церий (IV), образующий сульфатоком-
плексы, в отличие от остальных РЗЭ смолой не сорбируется. Для экст-
ракционного разделения можно использовать спирты, кетоны, эфиры и
др.; в промышленности же в основном применяют ТБФ и Д2ЭГФК.
Промышленный метод окисления церия кислородом воздуха может
быть осуществлен в разных вариантах. Так, он может быть окислен в
процессе сушки гидроксидов при 120—130 °C в сушильных аппаратах в
течение 2—6 ч. Высушенную смесь гидроксидов выщелачивают 5 % -ной
азотной или соляной кислотой. Нерастворившийся остаток содержит ~
95 % гидроксида церия. Способ прост, но недостатком его является
неполнота окисления церия и значительные потери РЗЭ вследствие
пылеуноса, велики энергетические затраты.
Другой способ окисления церия кислородом воздуха заключается в
пропускании его через пульпу смеси гидроксидов. Скорость окисления
увеличивается при повышении температуры и давления. В оптималь-
ных условиях (при pH 10, температуре 130 “С, давлении 5 105 Па) церий
окисляется на 90—95 % за 30 мин. Гидроксиды остальных РЗЭ удаляют
растворением в разбавленных кислотах. Небольшая добавка озона в
воздушную смесь сильно повышает эффективность процесса окисления:
уменьшается время окисления, удается получить 98—99 %-ный кон-
центрат церия. Оптимальные условия: pH 10—12, температура 50 °C.
Окисление церия перманганатом калия является довольно сложным
процессом, связанным с введением в систему нового реагента и необхо-
димостью последующего удаления марганца. Окисление проводят в сла-
бокислых или почти нейтральных растворах при 70—80 °C. Применение
этого способа целесообразно для очистки РЗЭ от небольших количеств
церия. Подготовленный раствор нитратов РЗЭ, в том числе и индивиду-
альных, нейтрализуют аммиаком до pH 6 и обрабатывают раствором
окислительной смеси, состоящей из КМпО4 и Иа2СОз или NH3,
3Ce(NO3)3 + КМпО4 + 8NH3 + 2Н2О Z
t ЗСе(ОН)4 + МпО2 + KNO3 + 8NH4NO3. (5.28)
Из раствора одновременно с Се(ОН)4 выделяется МпО2. Затем дл®
восстановления избыточного КМпСЦ до МпО2 в пульпу добавляют не-
большое количество этилового спирта. Этим способом удается снизить
содержание церия в оксиде лантана до 1 10 4 %.
246
Пероксид водорода для окисления церия можно использовать в ней-
тральной или слабокислой среде (pH 5—6), в кислых растворах (pH < 1)
пероксид водорода почти мгновенно восстанавливает Ce(IV) до Се(Ш).
Окисление проводят при комнатной температуре: к слабокислому рас-
твору нитратов РЗЭ добавляют пероксид водорода и аммиак
2Ce(NO3)3 + H2O2 + 6NH3 Н2ОХ 2Ce(OH)4 + 6NH4NO3. (5.29)
Часть пероксида водорода (берется избыток 50 %) расходуется на
образование пероксида церия СеО2(ОН)4, который при нагревании
пульпы превращается в гидроксид Ce(IV). Осадок содержит некоторое
количество гидроксидов других РЗЭ; для их удаления осадок обрабаты-
вают ТБФ, насыщенным HNO3; при этом нитрат Ce(IV) переходит в
экстракт, а нитраты РЗЭ (III) остаются в рафинате. Обработкой экстрак-
та разбавленной азотной кислотой с добавлением пероксида водорода
извлекают церий. Таким способом можно получить довольно чистый
оксид церия, однако процесс длителен, требует большого количества
реагентов, извлечение церия в готовую продукцию невелико.
Электрохимическое окисление церия в сочетании с жидкостной эк-
стракцией является наиболее перспективным методом отделения и по-
лучения церия высокой степени чистоты. Для электролиза используют
нитратные растворы. Электролиз хлоридных растворов осложнен выде-
лением хлора и неустойчивостью в них Ce(IV). Растворимость сульфа-
тов мала, в них возможно образование осадков основных солей. И хло-
ридные, и сульфатные растворы менее пригодны для экстракционного
разделения. При электролизе нитратных растворов на аноде протекают
следующие процессы:
Се3+-е — Се4+ (5.30)
2Н2О-4е------• О2 + 4Н+.
Процесс у анода контролируется диффузией к нему ионов Се3+, при
затруднении диффузии на аноде усиливается выделение кислорода.
Уменьшить выделение кислорода можно повышением температуры и
интенсивной циркуляцией электролита в анодном пространстве.
Основная реакция протекает на катоде (5.31); кроме того, возможно
образование азотистой кислоты и оксидов азота
2Н+ + 2е — 2Н----* Н2. (5.31)
NO3 + ЗН+ + 2е---• HNO2 + Н2О. (5.32)
Преимущественное развитие той или другой реакции зависит от
концентрации азотной кислоты: при концентрации HNO3 <1,5 моль/л
Протекает основной процесс — выделение водорода, при более высокой
— усиливается образование азотистой кислоты и оксидов азота. В этом
*е направлении действует повышение температуры выше 50 °C. Опти-
мальные условия электрохимического окисления церия: СняОз = 1—1,5
Моль/л, температура 45—50 °C.
247
Рис. 5.16. Схема электролизера с диафрагмой
для окисления церия:
I — корпус, 2 — кольцевой трубопровод с
отверстиями для подачи раствора к аноду; 3 —
катод; 4 — кольцевой приемник для слива рас-
твора в коллектор 5,6 — патрубок для отвода
газов, выделяющихся на катоде; 7 — штуцер
для залива раствора в катодное пространст-
во; 8 — крышка электролизера, 9 — анод; 10 —
диафрагма
Вследствие растворещ^
продуктов, образующихся Ва
катоде и аноде, в электролит»
развиваются процессы восста-
новления церия, снижается
выход по току. Поэтому наибо-
лее эффективны электролизе-
ры с разделением анодного и
катодного пространства, в ко-
торых достигается степень
окисления церия 98—99 %, в
бездиафрагменных электроли-
зерах степень окисления церия
не превышает 90—95 %, выход
по току на 30—40 % ниже. Ис-
пользование их обусловлено
простотой конструкции; для
них необходима циркуляция
только одного раствора — ис-
ходного электролита и харак-
терна довольно высокая произ-
водительность и относительно
низкие энергозатраты. Приме-
няются они в тех случаях, ког-
да не требуется высокая сте-
пень окисления церия.
На рис. 5.16 представлен
схематический чертеж элект-
ролизера, в котором катодное и анодное пространство разделены диаф-
рагмой. Корпус электролизера обычно выполняется из титана, анод —
из платины или платинированного титана, катод — из графита или
титана; электроды снабжены системой водяного охлаждения. В состав
установки для электролиза кроме электролизера входят баки для исход-
ного раствора, циркуляционные насосы, которые обеспечивают раз-
дельную циркуляцию растворов через анодное и катодное пространство,
устройства для отвода газов из электролизера. По достижении в анолите
степени окисления церия 98—99 % его перекачивают в бак исходного
раствора для экстракции.
Отделение самария, европия и иттербия. Для восстановления Ей,
Sm и Yb можно использовать различные методы: восстановление амаль-
гамой щелочных металлов и цинка, цинковой пылью, электролиз на
ртутном или твердом катоде. Мягкие восстановители — порошок цияка
и цинковая амальгама — восстанавливают в водном растворе селектив-
248
flo только европий. Для отделения восстановленных РЗЭ от остальных
лспользуют осаждение малорастворимых сульфатов, гидроксидов и эк-
стракцию.
Для выделения Eu, Sm и Yb довольно широкое распространение имел
иетод восстановления их амальгамой натрия. Восстановление ведут в
ацетатных, солянокислых и азотнокислых растворах амальгамой, со-
держащей 0,2—0,5 % Na, при pH 3—5. При взаимодействии амальгамы
с раствором происходит ступенчатое восстановление Eu , Sm и Yb
до металлов и одновременное растворение их в ртути, причем способ-
ность к образованию амальгамы у всех трех элементов примерно одина-
кова
Yb (СНзСОО) з+Na (Hg) „ X Yb (СНзСОО) 2+CH3COONa+Na (Hg)
Yb(CH3COO)2 + Na(Hg)n- Yb(Hg)n + 2CH3COONa. (5.33)
Амальгама натрия восстанавливает также легкие РЗЭ — La, Се, Рг
и Nd, которые переходят в амальгаму. Способность к ее образованию
убывает в указанном ряду и при этом она существенно меньше, чем у
Eu, Sm и Yb. Легкие РЗЭ прочно удерживаются в амальгаме; так, при
обработке амальгамы, содержащей Sm и Nd, разбавленной кислотой в
раствор Sm извлекается практически полностью и только 0,1 % Nd.
Извлечь его можно только более концентрированной кислотой. Осталь-
ные РЗЭ, в том числе и иттрий, амальгамы не образуют.
После отделения от раствора амальгаму обрабатывают смесью раз-
бавленных уксусной (~ 1 %) и серной кислот. При этом в осадок выпа-
дает E11SO4, содержащий до 20 % SmSO4, большая же часть самария
окисляется и остается в растворе. Осадок сульфатов обрабатывают раз-
бавленной азотной кислотой (— 0,5 моль/л), при этом происходит изби-
рательное окисление самария и переход его в раствор. В осадке остается
90—95 % -ный концентрат европия (при содержании в исходном мате-
риале 0,1 % европия получают 80 %-ный концентрат). Из объединен-
ных растворов, содержащих самарий, тем или иным способом осаждают
концентрат, содержащий до 99 % самария. Если в исходном материале
содержался иттербий, то он следует вместе с самарием. Способ эффек-
тивен, но имеет очень крупный недостаток — использование очень
токсичной ртути (рис. 5.17).
Метод восстановления европия цинковой пылью нашел широкое
Применение в промышленности. Восстановление проводят из хлорид-
ах растворов при pH 2—4 в присутствии серной кислоты при 0—5 °C
Аля увеличения растворимости сульфатов невосстановленных РЗЭ и
лучшего их отделения. Из концентратов с высоким содержанием евро-
Пия (3—5 % от суммы РЗЭ) E11SO4 можно осаждать без носителя. В этом
Случае в осадок выделяется от 75 до 95 % европия. Фильтрат и промводы
6°звращают на вторичное восстановление. При низком содержании ев-
249
Рис. 5.17. Принципиальная схема выделения
Sm и Ей восстановлением амальгамой натрия
ропия в исходном материя
(< 0,25 %) осаждение EuSn
ведут на носителе, луЧщ 4
из которых является SrSO^-*
осадок при этом выделяется’'
95 % европия 3
2ЕиС1з + Zn Z 2ЕиС1г + ZnCl2
u - (5’34)
Чистый оксид европия из
богатых концентратов получа-
ют также осаждением гидро-
ксидов. К раствору после вос-
становления цинком добавля-
ют NH3 до pH 11—12. Все РЗЭ
осаждаются в виде Еп(ОН)з, в
растворе же остается раствори-
мый гидроксид Ей (ОН)2 и ком-
плексный аммиакат цинка
[Zn(NH3>4 |2+. Отфильтрован-
ный раствор обрабатывают не-
большим количеством HN03
для окисления европия, при этом он выпадает в осадок в виде Еи(ОН)з.
Осадок промывают раствором аммиака, растворяют в HNO3 и снова
осаждают гидроксид европия. Процессы растворения и осаждения гид-
роксида повторяют до полного удаления цинка. На последней стадии
осаждают оксалат европия, прокаливают и получают оксид европия с
чистотой 99,9 %. Одновременно с получением чистых соединений евро-
пия метод позволяет решить и проблему очистки от европия соединений
других РЗЭ. В этом отношении весьма эффективна экстракция ТБФ.
которая характеризуется высокими коэффициентами разделения
= 105—10”) и позволяет получать материалы с содержанием европи
от 0,1 до 1 10 4 %. Операции с восстановленными растворами проводят
в защитной атмосфере азота, углекислого газа, аргона.
С помощью электролиза на ртутном катоде цитратных растворов
эффективно отделяются европий, самарий и иттербий. Метод основан
на способности РЗЭ образовывать комплексы [LnHCit 1+ и [Ln(Cit)2 ] •
которые не разрушаются при высоких значениях pH, поэтому при элек'
тролизе при pH 7—10 выпадение гидроксидов не происходит. Недоста-
ток способа тот же, что и при использовании амальгамы натрия Д'151
восстановления.
Электрохимическое восстановление европия на твердом катоде свя-
зано с определенными трудностями, так как вследствие невысокого от-
250
ицатедьного потенциала европия одновременно с его восстановлением
происходит выделение водорода.
Для того чтобы уменьшить обратный процесс окисления Eu(II),
необходимо изолировать катодное пространство от кислорода воздуха,
продуктов анодного окисления, исключить смешивание анолита и като-
лита- Восстановление проводят в герметизированной ячейке, катодное
и анодное пространства которой разделены ионообменной мембраной.
ддя удаления из электролита растворенного кислорода через него бар-
ботируют инертный газ. Анод выполняют из гранита, катод — из гра-
фита или титана; при их использовании достигается наибольшая ско-
рость и степень восстановления европия.
Лучшие результаты получены при электролизе хлоридных раство-
ров. На катоде протекают процессы
Еи3+ + е---* Еи2+;
2Н+ + 2е----*Н2. (5.35)
В начале электролиза при относительно высокой концентрации Еи3+
преимущественно идет его восстановление, в конце преобладающей ста-
новится реакция восстановления водородя, вследствие чего увеличива-
ется pH электролита. На аноде протекают процессы выделения хлора и
кислорода. Соотношение между скоростями этих реакций зависит от
концентрации соляной кислоты
2СГ~2е ------Ch;
4ОН -4е------- О2 + 2Н2О. (5.36)
Повышение pH электролита может привести к выпадению гидрокси-
дов; для того чтобы избежать этого, необходимо поддерживать опти-
мальную кислотность (pH 1—2), непрерывно добавлять соляную кис-
лоту. При указанных условиях, начальной концентрации ЕигОз5—
10 г/л и температуре 30—50 °C степень восстановления европия прибли-
жается к 100 %. Выделение европия после восстановления обычными
химическими и экстракционными методами малоэффективно вследст-
вие неустойчивости Eu (II) в таких растворах — он легко окисляется
кислородом воздуха и ионами-окислителями, находящимися в растворе.
Наиболее эффективен метод электрохимического гидролиза: после
окончания процесса восстановления европия в католит прекращают
Подачу соляной кислоты, pH увеличивается до 9—11- При этом выпада-
Ют крупно- или псевдокристаллические осадки гидроксидов Ьп(ОН)з;
Ойдроксид Ей (ОН) 2 не выпадает и при pH > 10. В результате получают
Чистый европий и другие РЗЭ с содержанием европия 0,1—0,01 %.
Метод характеризуется малым расходом реагентов, небольшим объемом
Точных вод и довольно простым аппаратурным оформлением.
251
5.14. Разделение РЗЭ методом ионообменной хроматографии
Разделение основано на различии в сорбции ионообменной смолой
ионов Ln3+, которая протекает в соответствии с общими закономерно-
стями ионного обмена. Они отличаются большой сложностью, мы оста-
новимся в самой общей форме только на некоторых из них
Сорбируемость катионов, прочность их связи с отрицательно заряжен-
ными функциональными группами смолы, по-видимому, определяется
в основном электростатическим взаимодействием, поэтому при погло-
щении катионов с одинаковым зарядом сорбция будет уменьшаться с
увеличением размеров гидратированного катиона. При сорбции разно-
зарядных катионов преобладающее значение приобретает заряд, поэто-
му в большинстве случаев многозарядные катионы вытесняют из смолы
катионы с меньшим зарядом. Ионный обмен используется для разделе-
ния близких по свойствам элементов, различия в сорбируемости кото-
рых очень невелики, вследствие чего при сорбции разделение
практически не происходит. Основной же эффект разделения достига-
ется в процессе селективной десорбции, при которой кроме различий в
сорбируемости используется разная устойчивость комплексных соеди-
нений разделяемых элементов. Различные варианты динамичных ионо-
обменных процессов получили название элюентной, вытеснительной и
фронтальной хроматографии.
При элюентной хроматографии сначала происходит сорбция разде-
ляемых катионов, а затем селективная десорбция (элюирование, вымы-
вание) раствором, содержащим комплексообразователь и катион, сор-
бирующийся слабее любого из разделяемых ионов. Чаще всего это кати-
он, которым первоначально была насыщена смола (Н+, NH4 , Na+)- При
непрерывной подаче элюирующего раствора сначала в верхних слоях
смолы, а затем и нижележащих происходит вытеснение ранее сорбиро-
ванных катионов. В первую очередь будут вытесняться катионы, обла-
дающие меньшим сродством к смоле и имеющие большую устойчивость
комплексов. Вследствие'этого катионы перемещаются вдоль колонны0
разной скоростью, и в смоле образуются зоны (полосы), преимущест-
венно занятые тем или другим элементом и накладывающиеся друг на
друга. Концентрация того или иного элемента по высоте слоя смолы
сначала увеличивается, а затем уменьшается. Эта зависимость имеет
куполообразную форму, что в полной мере отражается на выходны*
кривых1 * (рис. 5.18). При очень малой скорости и очень большой высоте
Выходные кривые — кривые, выражающие концентрацию разделяемых элементов
(в абсолютных или относительных единицах) в вытекающем из колонки растворе
функцию объема пропущенного раствора или как функцию времени
252
Рис. 5.18. Выходные кривые вымывания
при разделении двойной смеси методом
элюентной хроматографии (пара Nd—
Рг). Десорбция 0,5%-ным раствором
ЭДТА (pH 3,3):
V — Объем элюирующего раствора;
С — концентрация оксидов лантаноидов
в растворе, выходящем из разделитель-
ной колонки
Рис. 5.19. Кривые вымывания при разделе-
нии ионов методом вытеснительной хрома-
тографии:
С — относительная концентрация
ионов в растворе, выходящем из раздели-
тельной колонки; V — объем элюирующего
раствора; А — ион, которым первоначально
была заряжена смола; Г— ион-вытеснитель;
Б, В — ионы разделяемых компонентов; Vc
— объем чистой зоны; Vn — объем зоны
перекрывания
колонны полосы могут полностью разделиться; таким образом все раз-
деляемые вещества могут быть получены в чистом виде. Это возможно
при разделении микроколичеств веществ, например радиоактивных
изотопов, со 100 %-ным выходом. Однако этот метод недостаточно эф-
фективен для разделения больших количеств веществ из-за низких кон-
центраций растворов, длительности процесса, больших размеров ко-
лонн и др.
Для осуществления разделения методом вытеснительной хроматог-
рафии после сорбции разделяемых катионов в колонку должен пода-
ваться элюирующий раствор, содержащий катион, сорбируемость ко-
торого и концентрация должны быть настолько велики, что позади
Фронта иона-вытеснителя все другие катионы отсутствуют. При этом на
смоле образуются полосы отдельных катионов, имеющие довольно рез-
кие передние и задние границы, перекрывающие частично друг друга
Фис. 5.19). В стационарном состоянии полосы передвигаются по колон-
ке, сохраняя постоянную ширину. Ширину полос можно регулировать
Подбором диаметра колонн, высотой слоя смолы, размерами частиц смо-
лы, скоростью подачи элюирующего раствора, концентрацией и pH
Раствора. Метод можно рассматривать как вариант метода элюентной
Хроматографии, по сравнению с которым он более производителен, рас-
поры характеризуются более высокими концентрациями, процесс ме-
нее продолжителен. При этом, однако, несколько меньше возможности
253
Рис. 5.20. Выходные кривые при разделе-
нии ионов методом фронтальной хроматог-
рафии. Смола в Н -форме, раствор с иона-
ми Са^+ и NH4+:
1 — кривая для Н ; 2 — для NH4 ; 3
— для Са ; х — количество катионов,
пропущенных через колонку; С — относи-
тельные концентрации катионов в элюате
Рис. 5.21. Простейшая ионообменная ус-
тановка
J — сорбционная колонка; 2 — разде-
лительная колонка; 3 — вентили; 4 —
распределительные решетки; 5 — смола; 6
— подача рабочего раствора; 7 — подача
элюирующего раствора
в однократном процессе достигнуть полного разделения многокомпо-
нентной смеси.
Фронтальный метод может быть успешно реализован на стадии сор-
бции для разделения катионов, очень сильно различающихся по своей
сорбируемости, например однозарядного и многозарядного. При пропу-
скании через колонку раствора, содержащего их смесь, будет происхо-
дить сорбция их обоих. В вытекающем из нее растворе будут содер-
жаться только ионы Н , если смола первоначально была в водородной
форме. Через некоторое время многозарядные катионы начнут вытес-
нять из смолы однозарядные, и они появятся в вытекающем растворе,
концентрация их будет расти. Затем наступает момент, когда в вытека-
ющем растворе появляются многозарядные катионы, их концентрация
будет расти, а однозарядного — падать до тех пор, пока состав вытека-
ющего раствора станет одинаков с поступающим (рис. 5.20). Таким
способом можно получить определенное количество чистого компонен-
та, обладающего наименьшей сорбируемостью. Этот метод может быть
использован и для разделения близких по свойствам катионов; для этого
в раствор вводится комплексообразователь, образующий устойчивые
комплексы с одним или несколькими катионами при разделении много-
компонентной смеси, в результате чего резко снижается их сорбируе"
254
цость. Метод наиболее пригоден для разделения многокомпонентных
с[лесей на фракции и отличается высокой производительностью.
Простейшая ионообменная установка состоит из двух колонок, со-
единенных последовательно, — сорбционной и разделительной (рис.
5.21). В промышленности же для разделения близких по свойствам
элементов используются установки, состоящие из десятков колонн, со-
единенных последовательно и последовательно-параллельно, что по-
зволяет реализовать различные варианты разделительного процесса.
Соотношение между числом сорбционных и разделительных колонн
может быть различным — от 1 : 3 до 1 : 10.
В ряду La—Lu вследствие усиления гидратации и увеличения разме-
ров акваионов Ln3+- aq, сорбция уменьшается, причем различия в сор-
бируемости очень невелики. Например, при сорбции на катионите
КУ-2, содержащем функциональную группу SO3H, коэффициенты раз-
деления равны/Зьа/се = 1.012, /3sny£u = 1,009, /?YtyLu = 1 >03 и разделение
провести невозможно. Разделение происходит при селективной десорб-
ции, которая основана на различии в константах устойчивости комплек-
сов РЗЭ; для крайних членов ряда La—Lu они могут различаться на 2—4
порядка. В качестве комплексообразователей были опробованы органи-
ческие вещества, принадлежащие к различным классам: карбоновые
кислоты, оксикислоты, аминополикислоты и др. По эффективности раз-
деления РЗЭ их можно расположить примерно в следующий ряд: уксус-
ная кислота < винная кислота < лимонная кислота < аминоуксусная
кислота < нитрилотриуксусная кислота (НТА) < этилендиаминтетраук-
сусная кислота (ЭДТА). Наиболее эффективным оказались реагенты,
дающие комплексы хелатного типа, из них наиболее популярна ЭДТА;
при ее использовании для соседних РЗЭ /ЗуьГц = 1,8—3,7. Значение
констант устойчивости иттрия с различными комплексонами может за-
нимать в ряду констант для РЗЭ разное положение: с ЭДТА константа
Ку близка к Хть, а в случае с НТА — к Asm- Поэтому при разделении
РЗЭ на группы иттрий может оказаться либо с тяжелыми, либо с легки-
ми РЗЭ.
Рассмотрим разделение РЗЭ с использованием ЭДТА. В сорбцион-
ные колонки подают раствор хлоридов или нитратов РЗЭ с концентра-
цией ЬпгОз 3—5 % и pH 2,5—3. Сорбцию проводят так, чтобы смола
была полностью насыщена ионами Ln3+ в соответствии с ее обменной
еМкостью. Если смола находится в водородной форме, то ионы LnJ
замещают ионы Н+, которые переходят в раствор, кислотность его уве-
личивается, условия сорбции ухудшаются. Поэтому часто смолу для
сорбции используют в аммонийной форме, для чего ее предварительно
обрабатывают растворами солей аммония, pH среды при сорбции не
изменяется
255
3(RnH) + Ln3+ - (Rn)3Ln + 3H+, (5.37)
3Rn(NH4) + Ln3+; (Rn)3Ln + 3NH4+. (5.38)
Легкие РЗЭ сорбируются лучше, поэтому в верхней части сорбцион-
ной колонки происходит некоторое обогащение смолы ими. После завер-
шения сорбции колонку сначала промывают водой, а затем промывныц
раствором. При прохождении промывного раствора через сорбционную
колонку в ней устанавливается новое ионообменное равновесие. В про-
мывной раствор будут извлекаться прежде всего тяжелые РЗЭ, хуже
сорбирующиеся и образующие с ЭДТА более прочные комплексы (см
рис. 5.21). Раствор, обогащенный тяжелыми РЗЭ, поступает далее в
разделительные колонки. Процесс десорбции можно представить реак-
цией
(Rn)3Ln + H4L X 3RnH + HLnL, (5.39)
где H4L — условное обозначение ЭДТА.
Комплексная кислота HLnL — более сильная, чем Н4Ь, может дис-
социировать, поэтому pH раствора понижается
HLnL- H++LnL-.
Комплексный анион LnL в свою очередь диссоциирует, и в растворе,
поступающем в разделительные колонки, будут находиться незакомп-
лексованные ионы Ln3*
LnL- - Ln3++L4-
Такой процесс будет протекать интенсивнее для менее прочных
комплексов, т.е. для легких РЗЭ. В разделительной колонке в первую
очередь и будут сорбироваться незакомплексованные ионы, в результа-
те чего равновесие в растворе нарушится, для его восстановления будут
диссоциировать новые комплексы. Таким образом, по мере продвиже-
ния раствора в колонке он будет обогащаться тяжелыми РЗЭ.
При использовании в разделительных колонках смолы в водородной
форме происходит уменьшение pH и, как следствие этого, понижение
растворимости таких комплексообразователей, как НТА и ЭДТА, кото-
рые могут выпасть в осадок и нарушить процесс. Во-вторых, повышение
кислотности способствует разрушению комплексов, а следовательно,
уменьшает эффективность разделения
LnL- + H*X Ln3+ + HL3-.
Для предотвращения этого можно смолу в разделительных колонка*
также переводить в аммонийную форму, а для элюирования использо-
вать растворы указанных кислот, нейтрализованные аммиаком или
NaOH до pH 7,5—8. В таких растворах содержатся хорошо растворимые
гидросоли приблизительного состава NaiHiL (трилон Б). Далее оказа-
лось, что предварительное насыщение смолы ионами некоторых тя*е'
лых металлов повышает эффективность разделения за счет замедление
256
продвижения ионов РЗЭ через колонку. В качестве таких ионов-«замед-
дителей» используются Си , Ni2+, Zn+и др. Они должны образовывать
прочные комплексы с комплексоном, который используется в конкрет-
ном процессе. Для перечисленных выше ионов константы устойчивости
комплексов с ЭДТА соответственно равны 6,3-1018, 1,6-1018, 3,2 1016
(константы устойчивости комплексов Ln3+ с ЭДТА — от 5-1014 до
3,2 Ю19). Попадая на катионит с ионом-замедлителем, например с
Си2*, ионы Ln3+сорбируются на нем, вытесняя Си2+, которые в растворе
конкурируют с закомплексованными ионами Ln. В первую очередь,
естественно, будет происходить вытеснение из комплексов ионов легких
РЗЭ
3(Rn)2Cu + 2Ln3+x 2(Rn)3Ln + 3Cu2+, (5.40)
HLnL + 2Cu2+7. Cu(CuL) +Ln3+ + H+, (5.41)
HLnL + Cu2+ + H+ H2(CuL) + Ln3*. (5.42)
Вытесненные ионы легких РЗЭ будут задерживаться смолой, и
именно их продвижение в колонке будет сильнее замедляться, чем тя-
желых.
Ионы Си2+ могут образовывать плохо растворяющееся соединение
Cu(CuL), которое может выпасть в осадок. Для предотвращения это-
го в разделительной колонке используют смолу в смешанной медно-во-
дородной форме (на одну часть смолы в Н - форме две части смолы в
Си2+- форме). В таких условиях образуется хорошо растворимое соеди-
нение H2(CuL).
В качестве примера можно привести результаты, полученные при
разделении смеси, содержащей 53 % ЬигОз, 33 % Y2O3, 11,9 % ТтгОз
и 2,1 % ЕггОз, на катионите в Си -форме и элюировании 2 %-ным
раствором (NH4)2HL (НТА) при pH 8,5. При отборе 25 фракций раство-
ра только в фракциях № 1 и № 2 содержание ЬигОз составляло 99,8 и
99,5 %. Во всех остальных фракциях содержались смеси элементов.
Использование ионов-замедлителей можно рассматривать как реа-
лизацию одного из вариантов механизма вытеснительной хроматогра-
фии. Этот метод применяется в промышленных масштабах как для
Получения РЗЭ высокой степени чистоты, так и для разделения их на
группы. Он более производителен по сравнению с элюентным методом,
растворы характеризуются более высокими концентрациями РЗЭ, про-
цесс менее продолжителен. Смола может предварительно насыщаться
ионами Fe3+, Cu2+, Mg2+, Са2+, которые при использовании ЭДТА проч-
нее ионов РЗЭ удерживаются на смоле.
При использовании фронтальной хроматографии предварительная
сорбция РЗЭ не производится, в колонку непрерывно подается раствор,
содержащий смесь РЗЭ, и комплексон, который добавляется в таком
количестве, чтобы связать только часть РЗЭ (например, от Lu до Dy),
257
1 ,
присутствующих в смеси. При пропускании через колонку такого pat
твора сорбируются только незакомплексованные ионы, т.е. — легкие
тяжелые проходят через колонку. Со смолы РЗЭ десорбируются кисл^
той (HNO3, НС1), в полученный раствор снова добавляют расчетное
количество комплексона и снова проводят разделение. Таким образу
получают фракции, каждая из которых далее разделяется методом элю-
ентной хроматографии.
Фронтальный метод ускоряет разделение РЗЭ на группы. Он обес-
печивает более устойчивую работу разделительных колонок, так как
первоначальное комплексообразование происходит вне их и его можно
регулировать, исходя из состава разделяемой смеси. Возможно также
предварительное обогащение смеси в простых реакторах с мешалками.
Методом фронтальной хроматографии возможно разделение РЗЭ в не-
прерывном противоточном процессе. Недостаток этого метода — невоз-
можность разделения многокомпонентных смесей с получением чистых
компонентов, поэтому его используют для разделения их на фракции и
для разделения двухкомпонентных смесей.
Ионообменный метод в начале 60-х годов являлся основным техно-
логическим методом получения значительных количеств соединений
почти всех РЗЭ. Однако в связи с развитием более производительного
метода разделения РЗЭ — жидкостной экстракции — ионообменную
хроматографию в настоящее время используют в промышленности пре-
имущественно для разделения РЗЭ на легкие и тяжелые и получения
индивидуальных РЗЭ высокой степени чистоты на финишной стадии
очистки. Низкая производительность ионообменной технологии была
обусловлена малой скоростью элюирующего раствора (< 1 см/мин),
низкой концентрацией рабочих растворов, в особенности после элюиро-
вания (< 1 г/л), необходимостью многократного повторения циклов
сорбция-десорбция. Промышленные установки для разделения состоят
из десятков и даже сотен колонн, что затрудняло контроль и управление
процессом. Однако в настоящее время разработаны и применяются в
промышленности более совершенные колонные аппараты (вибрацион-
ные, тарельчатые, с аэролифтами), пригодные для использования в
непрерывных противоточных разделительных процессах и извлечения
РЗЭ из пульп. Установлено положительное влияние повышенной тем-
пературы (до 60 °C) и давления на эффективность разделения компо-
нентов.
258
5.15. Экстракция. Общие закономерности
Экстракцией называется извлечение вещества из одной жидкой фа-
зы в другую, с ней не смешивающуюся Экстракция — сложный
физико-химический процесс, связанный с различными взаимодействи-
ями и реакциями в растворах, диффузией и переносом вещества через
поверхность раздела фаз.
В технологии редких и цветных металлов практическое значение
имеет экстракция неорганических веществ — кислот и соединений ме-
таллов из водных растворов. С водными растворами не смешиваются
малополярные органические жидкости, но в них большинство неоргани-
ческих веществ растворяется плохо. В водном растворе они диссоцииро-
вании на ионы, гидратированные молекулами воды, и переход их в
органическую фазу становится возможным, если все или большая часть
молекул воды, координированных ионом, будут удалены и получен
нейтральный комплекс.
Уменьшение степени гидратации и образование нейтральных комп-
лексов или соединений возможно при взаимодействии извлекаемых
ионов с органическими веществами, дающими более прочные связи, чем
их связи с молекулами воды, и сопровождающемся уменьшением энер-
гии Гиббса. Эти вещества, содержащие определенные функциональные
группы, можно разделить в первом приближении на три основных клас-
са. Они и являются собственно экстрагентами.
Первый класс — нейтральные экстрагенты. Их молекулы содержат
активные атомы кислорода, азота, серы и способны к донорно-акцептор-
ному взаимодействию с извлекаемым ионом. К ним относятся спирты
R—О—Н, простые эфиры R—О—R, кетоны
R1\ R1\
С = 0, сложные эфиры (RO)3^ Р = О, сульфоксиды S = 0 и др.
r2x r2z
Они образуют сольваты, замещая молекулы воды в гидратной обо-
лочке катиона, либо присоединяясь к ним.
Второй класс — органические кислоты. К ним относятся карбоновые
Кислоты с числом углеродных атомов более 7, фосфорорганические кис-
(RO)2x
лоты Р = О, хелатообразующие реагенты типа/3-дикетонов,
НО — С = СН— С— .
Имеющих две функциональные группы
I II
ОН О
1) Это определение относится к жидкостной экстракции. Экстрагирование полезных
компонентов из твердых веществ в данном случае не рассматривается.
‘/2 9*-691
259
При взаимодействии ионов с ними образуются соответствующие
соли или внутрикомплексные соединения.
Третий класс — соли органических аминов (RNH2, R2NH, R3|\j
[R3N]A) или четвертичных аммониевых, фосфониевых, арсониевых
оснований. Экстрагируемое соединение образуется, если извлекаемый
металл входит в состав аниона.
Экстракционная способность нейтральных экстрагентов в основном
зависит от полярности функциональной группы и доступности атома
кислорода. Так, кислород кетонов и сложны^ эфиров доступнее кисло-
рода простых эфиров. Полярность группы Э = О определяется не только
характеристиками центрального атома (Э)л но и тем, с какими замести-
телями он связан. Последнее можно прдследить на эфирах фосфорной
кислоты:
С4Н9 — О \
С4Н9----- О Р = 0 —трибутилфосфат (ТБФ),
С4Н9----- О '
С4Н9---- О
С4Н9 —— О ---- Р = О — дибутилбутилфосфонат,
С4Н9/
С4Н9-- О \
С4Н9--
С4Н9^
Р = О — бутилдибутилфосфинат,
С4Н9 \
С4Н9-----Р = О — трибутилфосфиноксид.
С4Н9 /
В молекуле ТБФ электронная плотность смещается от центрального
атома фосфора к функциональном атому — атому кислорода, который
и образует донорно-акцейторную связь с катионом, и в сторону атомов
кислорода эфирных групп. При замене эфирных групп (С4Н9—О—) на
алкильные (С4Р9—) смещение электронной плотности в сторону агома
кислорода фосфорильной группы (Р = О) увеличивается, растут ее по-
лярность и эффективный заряд на атоме кислорода. Это приводит I
повышению прочности образующихся сольватов и в значительной сте-
пени определяемой этим параметром экстракционной способности.
Экстрагенты могут быть как жидкими, так и твердыми веществами-
Жидкие органические вещества, которые сами не экстрагируют и не
смешиваются с водой, но применяются для растворения экстрагентов
или для уменьшения их плотности и вязкости, называются разбавите-
лями. Для этих целей используют предельные углеводороды алифати-
260
ческого ряда, некоторые ароматические углеводороды (бензол, толуол),
хлорированные углеводороды (хлороформ, тетрахлорид углерода). В
промышленности же преимущественно используют осветительный или
гидрированный керосин, не содержащий непредельных углеводородов и
кислородсодержащих соединений, как наиболее доступный и дешевый.
Экстракционные равновесия могут рассматриваться как с точки зре-
ния термодинамики, так и закона действующих масс. Растворенное
вещество распределяется между фазами в определенном закономерном
соотношении. Закон распределения, открытый М. Бартло и Юнгфлей-
шем и обобщенный В. Нернстом, можно сформулировать так: растворен-
ное вещество распределяется между двумя несмешивающимися фазами
так, что отношение равновесных концентраций вещества в обеих фазах
не зависит от общей концентрации, если в каждой фазе вещество имеет
одну и ту же молекулярную массу. Закон Нернста не является строго
термодинамическим и выполняется в частных случаях для разбавлен-
ных растворов (1 10 3—1 10 5 моль/л)
Kd—Co! Св, (5.43)
где Kd — константа распределения; Со и Св — концентрации вещества
в органической и водной фазах.
Условием равновесия при распределении вещества, как и в любой
другой гетерогенной системе, является равенство химических потенци-
алов (парциальных моляльных энергий Гиббса) всех компонентов сис-
темы в обеих фазах
(5.44)
Химические потенциалы распределяемого вещества в водной и ор-
ганической фазах (ив и/40) равны
/zB=^B + ATln ав,
Ц0=[Л0 + RT\n ао,
где /лв и fl0 — химические потенциалы в стандартном состоянии. За
стандартное состояние принимают либо чистое вещество, либо, как в
данном случае, гипотетический раствор, обладающий некоторыми свой-
ствами бесконечно разбавленного раствора (у = 1 при С—• О); ав и Оо —
активности в водной и органической фазах. Из (5.44) получим
fiB + RT In ав =[io + RT In До,
До I aB = exp(/zB-/Z0) / RT. (5.45)
При данной температуре правая часть уравнения (5.45) величина
постоянная; обозначив ее через К, получим
До / Дв = X; До= Хдв. (5.46)
Отсюда термодинамическая константа распределения есть отноше-
ние активностей распределяемого вещества, а активность вещества в
органической фазе пропорциональна активности в водной.
261
9 — 691
В подавляющем большинстве случаев активности в органической
фазе не известны; заменив ао на Со, получают эффективную константу
(к), а при замене и ав на Св — кажущуюся (К).
К таким же выводам можно прийти, рассматривая экстракцию как
химическую реакцию и используя закон действующих масс
Мв++vA + nS X МАр nS, (5.47)
{МА„ и5} / ({Мг+}{АТ{5}") =
= ([МА„ nS |ус) / ([Mv+ ] [A ]Y±+ 1 [5 ]>S) , (5.48)
где в фигурных скобках — активности ионов или соединений, в квадрат-
ных — концентрации.
Уравнение (5.47) — для экстракции нейтральными экстрагентами,
при которой образуются сольваты.
Используя уравнение (5.48), можно выразить термодинамическую
константу распределения через эффективную (А) и кажущуюся (.К)
К=к(ус1уп0) = К(ус1 (У±+1Уо>> (5 49)
Зная термодинамические константы, можно рассчитать коэффици-
енты распределения и концентрации компонентов в органической фазе.
При расчете по эффективным и кажущимся константам возникают по-
грешности, допустимые только при решении некоторых практических
задач.
Экспериментально определяемая характеристика экстракционного
процесса — коэффициент распределения
£> = С0/Св;В= [М\ nS]/ [М1'+]. (5.50)
Коэффициент распределения — отношение аналитических концен-
траций, т.е. включающих все виды соединений данного компонента в
органической (Со) и водной (Св) фазах при равновесии. Это сложная
функция концентраций и коэффициентов активностей, а поэтому в
общем случае — переменная величина. Подставив выражение для D в
уравнение (5.48), получим
П=А [А" Г/±+ 1 [S ]>5 / ус. (5.51)
Если в системе несколько экстрагируемых компонентов, то каждый
распределяется в соответствии с термодинамической константой. Но
из-за взаимного влияния компонентов и конкуренции за свободный
экстрагент экстракция протекает независимо, только если компоненты
присутствуют в микроконцентрациях.
Уравнениедля коэффициента распределения (5.51) позволяетопре-
делять принципиальные пути управления им и создавать оптимальные
условия для экстракции. Наиболее простой и часто применяемый способ
— введение высаливателей в водную фазу электролитов, которые не
262
экстрагируются или экстрагируются очень плохо. Обычно это соли ще-
лочных или щелочноземельных металлов. Высаливание в водной фазе
складывается из двух основных эффектов, увеличивающих коэффици-
ент распределения.
1. Высаливатель, имеющий общий ион с экстрагируемым соедине-
нием, увеличивает коэффициент распределения вследствие возраста-
ния концентрации одноименного иона. В уравнении (5.50) член [А ] —
суммарная концентрация анионов, образующихся при диссоциации
экстрагируемого соединения и высаливателя. Этот эффект не зависит от
природы высаливателя. Действие высаливателя с одноименным ионом
увеличивает концентрацию экстрагируемого нейтрального комплекса и
наиболее сильно проявляется при малой концентрации экстрагируемого
вещества.
2. Второй эффект более сложен и связан с природой высаливателя.
Гидратация ионов высаливателя уменьшает концентрацию несвязан-
ной воды, а следовательно, увеличивает эффективную концентрацию
(активность) экстрагируемого вещества. Коэффициент активности уве-
личивается тем больше и высаливатель действует тем эффективнее, чем
сильнее он гидратирован. Связывание воды высаливателем способству-
ет дегидратации катиона экстрагируемого соединения и его сольватации
молекулами экстрагента. Этот эффект не зависит от того, имеет или не
имеет высаливатель общий ион с экстрагируемым соединением. Крите-
рием высаливающей способности электролита могут быть гидратные
числа, однако он недостаточно строг, так как гидратные числа, опреде-
ленные различными способами, сильно различаются между собой. По
высаливающей способности их солей катионы можно расположить при-
мерно в следующий ряд:
К+ < NH4+ < Na+ < Li+ < Н+ < Ва2+ < Sr2+ < Са2+ < Mg2+ < Al3+
При использовании в качестве высаливателей кислот необходимо
Учитывать, что многие из них
хорошо экстрагируются.
Экспериментальная (или
Расчетная) зависимость между
концентрациями вещества в
органической и водной фазах
При изменении его общей кон-
центрации, полученная при
Постоянной температуре, на-
зывается изотермой экстра-
кции (рис. 5.22). Обозначив
Концентрацию в водной фазе х,
а в органической у, из уравне-
ние. 5.22. Изотермы экстракции в системе элек -
тролит—неэлектролит
1 — без высаливателя; 2 — с высаливате-
лем
263
ний (5.49, 5.50) получим уравнение изотермы экстракции
у= Кх[А Г/±+ 1 [5 ]>g / ус. (5.52)
Вид изотерм экстракции может быть различным. В данном случае
дана изотерма экстракции, соответствующая наиболее типичному рав-
новесию электролит—неэлектролит, описываемому уравнением (5.47).
На изотерме (кривая /) можно выделить три участка: начальный А —
параболический, для которого вследствие очень малой активности экс-
трагируемого вещества коэффициент распределения стремится к нулю;
участок насыщения В, который характеризуется практически постоян-
ной концентрацией вещества в органической фазе вследствие того, что
весь экстрагент связан в комплекс. Между этими участками находится
участок Б, ход изотермы экстракции на котором определяется соотно-
шением коэффициентов активности в водной и органической фазах.
Коэффициент распределения в этой области сначала возрастает, а затем
падает. Кривая 2 — изотерма экстракции с высаливателем, в присутст-
вии которого особенно значительно изменяется начальный участок изо-
термы, так как [А ] в основном определяется его концентрацией.
Насыщение экстрагента достигается при значительно меньшей концен-
трации вещества в водной фазе.
Экстракция — не только способ извлечения и концентрирования
веществ, но и один из наиболее эффективных способов разделения близ-
ких по свойствам элементов. Для разделения компонентов необходимо,
чтобы коэффициенты их распределения различались в достаточной сте-
пени. Эффективность разделения характеризуется коэффициентом
разделения
ft = D / D- = (Со / С*) : (Со / Св) = (С6 / С6') : (Св / Св'). (5.53)
Коэффициентом разделения называется отношение коэффициен-
тов распределения компонентов — всегда большего к меньшему. Коэф-
фициент разделения показывает, во сколько раз меняется отношение
равновесных концентраций двух компонентов в органической фазе
по сравнению с таким же отношением в водной фазе.
Полное извлечение экстрагированного вещества из органической
фазы в водную называется реэкстракцией. Ее проводят водой или воД'
ными растворами, содержащими реагенты, разрушающие экстрагирО"
ванный комплекс.
Экстрагенты, применяющиеся для извлечения и разделения метаЛ'
лов, должны удовлетворять ряду физико-химических, технологически*
и экономических требований. Экстрагент должен обладать хорошей экС'
264
трагирующей способностью и избирательностью. Вещество наиболее
полно экстрагируется при больших значениях К и D, т.е. когда энергия
взаимодействия вещества с экстрагентом велика. Однако при высоких
значениях D трудно получить эффект разделения компонентов. Кроме
того, затрудняется реэкстракция, что наблюдается при экстракции кис-
лыми алкилфосфатами. При очень слабом взаимодействии вещества с
экстрагентом значение D мало и экстракция недостаточно эффективна,
полностью извлечь вещество можно при D > 1. Наиболее приемлемы
экстрагенты, энергия взаимодействия которых с экстрагируемым веще-
ством имеет средние значения = 10—40 кДж/моль); к ним отно-
сятся нейтральные экстрагенты.
Экстрагент должен легко регенерироваться, мало растворяться в
воде, иметь плотность, отличную от плотности воды, малую вязкость,
быть относительно нелетучим и невоспламеняющимся, дешевым. ТБФ
(трибутиловый эфир фосфорной кислоты), получивший большое рас-
пространение, отвечает большинству этих требований, за исключением
плотности, мало отличающейся от плотности воды, и высокой вязкости,
вследствие чего его используют в виде раствора. Разбавители не должны
взаимодействовать ни с экстрагируемым комплексом, ни с экстраген-
том, хорошо растворять экстрагируемый комплекс (в противном случае
образуется третья фаза), иметь низкую вязкость и плотность, быть не-
токсичными и негорючеопасными и т.д.
В реальных технологических процессах за одно контактирование
водной и органической фазы (одну ступень) в подавляющем большин-
стве случаев не удается получить необходимое извлечение вещества и в
особенности разделение компонентов. Поэтому применяются полупро-
Рабочий
раствор
Прошивной Ршстрагяруюциа
раствор
раствор
Рис. 5.23. Схема каскадов экстракционной установки
265
тивоточные и противоточные процессы, для чего экстракторы объединя-
ются в каскады таким образом, что каждый единичный экстрактор
работает как прямоточный аппарат (соответствует одной степени)
Группа ступеней, в которой компоненты извлекаются из водного раство-
ра, называется экстракционным каскадом; группа ступеней для извле-
чения их из органической фазы в водную — реэкстракционным
каскадом. При разделении компонентов для дополнительной очистки
извлекаемого вещества вводится дополнительно промывной каскад, в
котором из органической фазы удаляют другие вещества. Иногда вво-
дится дополнительный четвертый каскад для регенерации экстрагента
— удаления из него продуктов деградации и гидролиза (рис. 5.23).
5.16. Разделение РЗЭ экстракцией
Экстракция применяется для извлечения РЗЭ, отделения примесей
других элементов, но особое значение она имеет как наиболее эффек-
тивный промышленный способ разделения и получения индивидуаль-
ных РЗЭ, в том числе и высокой степени чистоты. Для экстракции РЗЭ
используются экстрагенты, обычно применяющиеся в системах жидко-
стной экстракции других элементов.
Наиболее широко для этих целей используются нейтральные фос-
форорганические соединения (НФОС), среди которых первостепенное
значение имеет ТБФ, применяемый для экстракции из нитратных рас-
творов с различной концентрацией азотной кислоты. Экстракция ТБФ
из хлоридных растворов малоэффективна, но все же возможна при вы-
сокой концентрации соляной кислоты ( > 8 моль/л).
Вторым типом экстрагентов являются фосфорорганические кисло-
ты, среди которых наибольшей популярностью пользуется ди-2-этил-
гексилфосфорная кислота (Д2ЭГФК), которая обладает хорошими ха-
рактеристиками в качестве экстрагента для РЗЭ, хорошо изучена, до-
вольно доступна. Алкилфосфорные кислоты могут использоваться в
нитратной, хлоридной, сульфатной и других средах.
Реже и в меньших объемах используются такие экстрагенты, как
органические основания (преимущественно соли четвертичных аммо-
ниевых оснований), монокарбоновые кислоты, сульфоксиды, смеси
НФОС с алкилфосфорными кислотами, смеси различных экстрагентов
с комплексонами, хотя использование последних позволяет повысить
селективность экстракции.
Экстракция ТБФ. При экстракции трибутилфосфатом в органиче-
скую фазу извлекаются трисольваты, и уравнение экстракционного рав-
новесия можно записать следующим образом:
Ln3+(B) + 3NO3 (b) + ЗТБФ(о) г Ln(NO3)3 ЗТБФ. (5-54)
266
Рис. 5.24. Зависимость коэффици-
ента распределения от Z при экст-
ракции микроконцентраций РЗЭ
100 %-ным ТБФ при различных
концентрациях азотной кислоты:
1 — 0,98 М; 2 — 3,89 М; 3 —
5,81 М; 4 — 10,1 М; 5 — 15,5 М; 6
— 18,5 М
Из уравнения следует, что экстра-
кция РЗЭ сильно зависит от концентра-
ции ионов NO3 в растворе, постав-
щиками которых могут быть: азотная
кислота, высаливатели и сами нитраты
РЗЭ. На рис. 5.24 представлены зависи-
мости коэффициентов распределения
индивидуальных РЗЭ при различных
концентрациях азотной кислоты для ря-
да La—Lu. Из него следует, что более
высокой концентрации азотной кислоты
соответствуют большие значения коэф-
фициента распределения и что послед-
ний изменяется в зависимости от поряд-
кового номера элемента. При высоких
концентрациях азотной кислоты (> 6
моль/л) он увеличивается, однако, на-
чиная с области примерно 64-го элемента
(Gd), наблюдается замедление роста.
При низких концентрациях азотной кис-
лоты, начиная с того же 64-го элемента,
коэффициент распределения уменьша-
ется. Это явление можно объяснить толь-
ко качественно, поскольку здесь
действует ряд факторов, трудно поддающихся количественной оценке:
высаливающее действие азотной кислоты и одновременно ее конкурен-
ция за ТБФ, комплексообразование Ln3+ с молекулами воды (гидрата-
ция), препятствующее экстракции, и комплексообразование с ТБФ
(сольватация), способствующее переходу вещества в органическую
фазу.
В ряду La—Lu константы устойчивости комплексов увеличиваются,
что в одинаковой мере относится и к гидратации, и к сольватации,
причем первый процесс характерен для водной фазы, второй — для
органической. В органической фазе вода практически отсутствует, и
состояние компонентов ее мало зависит от состава водной фазы, а про-
цессы в ней не будут сильно влиять на коэффициент распределения. При
низкой концентрации азотной кислоты в водной фазе будет доминиро-
вать процесс гидратации, понижение коэффициентов распределения
будет наиболее сильно выражено у тяжелых РЗЭ, так как для подавле-
ния гидратации легких РЗЭ необходима меньшая концентрация NO3
(-дЯ/’^енф < ~а#й(тяж)) • При высокой концентрации HNO3 уменьшает-
267
Рис. 5-25- Зависимость коэффициентов
распределения микроконцентраций лег-
ких РЗЭ и иттрия от концентрации HNO3
при экстракции ТБФ
Рис. 5.26. Зависимость распределения
микроконцентраций некоторых тяже-
лых РЗЭ от концентрации HNO3 при
экстракции ТБФ
ся активность воды в водной фазе, а следовательно, уменьшается гидра-
тация и воздействие ее на коэффициенты распределения.
Зависимость коэффициентов распределения индивидуальных РЗЭ
от концентрации азотной кислоты представлена на рис. 5.25, 5.26. На
кривых для легких РЗЭ в области концентраций HNO3 3—9 моль/л
происходит уменьшение коэффициента распределения, которое более
существенно у элементов с меньшим номером. Уменьшение коэффици-
ента распределения, по-видимому, можно объяснить конкуренцией
HNO3 за ТБФ, а рост — при высоких концентрациях HNO3 — возмож-
ным образованием анионных комплексов НхЬп(ЫОз)з+х ЗТБФ, хотя
этой точки зрения придерживаются не все. Для тяжелых РЗЭ ход этой
зависимости несколько иной; их экстракция с повышением концентра-
ции азотной кислоты увеличивается непрерывно, но при концентрации
HNO3 около 5 моль/л происходит инверсия коэффициентов распреде-
ления. До концентрации HNO3 5 моль/л коэффициент распределения
увеличивается в ряду Yb—Er—Dy—Но, а при более высокой — порядок
обратный. Разница между коэффициентами распределения соседних
элементов увеличивается при увеличении концентрации азотной кис-
лоты, поэтому при низких концентрациях HNO3 разделение не проис-
ходит, эффективность же его увеличивается с повышением концент-
рации кислоты.
При экстракции 100 %-ным ТБФ, исходя из молярной концентра-
ции чистого ТБФ 3,66 моль/л и перехода в органическую фазу трисоль-
ватов, максимальная концентрация РЗЭ в ней около 1,2 моль/71
(170—210 г/л в зависимости от того, какой элемент экстрагируется)-
268
f акая концентрация соответству-
ет полному насыщению экстраген-
та и может быть достигнута при
очень больших концентрациях
рЗЭ в водной фазе Gio 500 г/л)
(рис. 5.27). Максимальное значе-
ние D = 0,7 достигается при кон-
центрации РЗЭ в водной фазе
□коло 150 г/л; при дальнейшем ее
увеличении вследствие уменьше-
ния концентрации свободного
ТБФ коэффициент распределения
уменьшается, но возрастает коэф-
фициент разделения. При разбав-
лении ТБФ инертным
растворителем содержание РЗЭ в
органической фазе и соответствен-
но коэффициент распределения
Рис. 5.27. Изотермы распределения суммы
РЗЭ при экстракции 100 (/) и 70 %-ным
(2) ТБФ (исходный концентрат, %: 20 La,
40 Се, 8,75 Рг, 30 Nd, 0,85 Sm, 0,3 Gd)
уменьшаются.
В присутствии высаливателей насыщение органической фазы насту-
пает при меньших концентрациях РЗЭ в водной фазе. Например, при
экстракции в присутствии нитрата алюминия этому отвечает равновес-
ная водная фаза, содержащая около 2 моль/л А1(МОз)з, 150 г/л РЗЭ и
0,2 моль/л НЫОз, которая добавляется для предотвращения гидролиза.
При экстракции смесей легких РЗЭ широко распространенные и хуже
всего экстрагируемые La и Се могут выступать сами как высаливатели
(самовысаливание). Эго позволяет, не вводя в систему новых реагентов,
выделять из смеси легких РЗЭ самарий и гадолиний, а также концентрат
PrnNd (рис. 5.28).
При экстракции смесей тяжелых РЗЭ из нейтральных растворов в
; присутствии высаливателей и без них наблюдаются другие закономер-
ности, связанные с инверсией коэффициентов распределения (рис.
5.29). На кривых, характеризующих коэффициенты распределения
элементов при экстракции из смесей с преобладающим содержанием
тяжелых РЗЭ, отчетливо виден излом на Gd. Уменьшение коэффициен-
тов распределения иттриевых РЗЭ таково, что они приближаются к
коэффициентам распределения легких лантаноидов — Рг и Nd. Такие
Данные получены для полного насыщения органической фазы. Наблю-
даемые при этом коэффициенты разделения малы, что препятствует
использованию нейтральных растворов для разделения иттриевых РЗЭ.
Коэффициенты распределения РЗЭ при экстракции ТБФ зависят от
многих факторов, поэтому однозначную и общую картину по коэффи-
циентам разделения соседних пар элементов дать невозможно. Однако
269
Рис. 5.28. Зависимость коэффициентов
распределения легких РЗЭ от равновесной
концентрации в водной фазе (в пересчете
на оксиды) при экстракции 100 %-ным
ТБФ (исходный концентрат см. рис. 2.27)
Рис. 5.29. Зависимость коэффициентов
распределения РЗЭ от атомного номера
при экстракции 100%-ным (7) и 70%-
ным ТБФ (2). Водный раствор: 2 моль/л
A1(NO3)3,0,1 моль/л HNO3, 150г/лсум-
мы РЗЭ Исходный концентрат, %: 11,0
La; 12,5Се; 1,6Рг;5,0Nd; l,4Sm; 7,5Gd;
1,4 Tb; 8,7 Dy, 1,3 Ho; 0,15 Tm; 1,1 Yb; 0,15
Lu; 45,8 Y
можно указать на некоторые закономерности. Коэффициенты разделе-
ния увеличиваются при повышении концентрации азотной кислоты или
высаливателей. Так, при концентрации 6 моль/л ЫЫОз или ~ 2,5
моль/л Al (NO3) 3 (что эквивалентно по высаливающей способности 15—
16 моль/л HN Оз) и концентрации РЗЭ в водной фазе 90— 160 г/л /? для
пар Рг—La и Sm—Nd равны соответственно 4,6 и 3,5. При повышении
концентрации в водной фазе коэффициенты разделения легких РЗЭ
увеличиваются; fl для пары Sm—Nd при 60 и 460 г/л в водной фазе
соответственно равны 1,4 и 2,26. Для тяжелых РЗЭ в большинстве слу-
чаев зависимость обратна^ — для концентраций 60 и 460 г/л/? пары (50
% ТБФ) Dy—Y равны соответственно 3,96 и 1,30; для пары Но—Y 3,0
и 1,20; для пары Но—Ег 1,43 и 1.04.
При повышении температуры/J увеличивается, например при 60 °C
и концентрации РЗЭ в водной фазе 335 и 600 г/л пары Nd—La соот-
ветственно равны 4,1 и 7,2. Разбавление ТБФ предельным углеводоР0"
дом (керосином) снижает вязкость, уменьшает плотность экстрагента,
увеличивает скорость расслаивания органической и водной фаз, н°
уменьшает концентрацию РЗЭ в экстрагенте; fl снижается незначитель-
но.
Экстракция фосфорорганическими кислотами. Для экстраки101
РЗЭ используют двузамещенные фосфорорганические кислоты, прежДе
270
всего Д2ЭГФК в виде растворов в неполярных растворителях (бензол,
толуол, гептан, керосин и др.), для снижения вязкости, а в некоторых
случаях и коэффициента распределения. В таких растворах молекулы
фосфорорганических кислот димеризуются за счет образования водо-
родных связей, причем с повышением концентрации фосфорорганиче-
ской кислоты степень димеризации увеличивается
R--- О О ................ НО/ ^OR
/’х /р\
R--- О ОН .................... О OR
На эффективность разделения РЗЭ, величину коэффициентов рас-
пределения и разделения мало влияют длина и природа углеводородного
радикала. Большее значение имеет замена алкоксильной группы на
алкильную. Экстракция фосфоновыми и фосфиновыми кислотами ха-
рактеризуется большими значениями коэффициентов распределения, а
в некоторых случаях и коэффициентов разделения. Уравнение экстра-
кции РЗЭ фосфорорганическими кислотами при низкой концентрации
Ln3* а^(в) + ЗНА X Ьп(А)з(о)+ЗН+(в>; (5.55)
при высокой
Ln3+ а<?(в) + ЗН2А2£ Ьп(НАг)з(о)+ ЗН+(В), (5.56)
где НА — мономерная кислота; Н2А2 — димер.
Из уравнений следует, что анион минеральной кислоты, присутст-
вующий в водном растворе, в органическую фазу не переходит, а в
процессе экстракции в нем увеличивается концентрация ионов водоро-
да, что не благоприятствует экстракции.
При экстракции Д2ЭГФК коэффициенты распределения в ряду La—
Lu увеличиваются. При этом график зависимости IgD = f(Z) в первом
приближении можно аппроксимировать прямой линией, и тогда средние
значения коэффициентов разделения пар соседних РЗЭ будут равны
~ 2,5, т.е. несколько больше, чем при экстракции ТБФ. В зависимости
от условий экстракции (концентрации минеральной кислоты, природы
кислоты, концентрации Д2ЭГФК, типа разбавителя, который также
может принимать участие в экстракции за счет образования водородных
/0Н
связей с группой 4 Р ) коэффициент разделения пары Lu/La
О
будет равен 2,5 104—1 105. Коэффициент распределения иттрия имеет
среднее значение между значениями его для Но и Ег. Однако при деталь-
ном изучении зависимости IgD = /(Z) было установлено существование
«тетрадного эффекта», что четко видно на рис. 5.30. Вследствие этого
271
Рис. 5.30. Зависимость IgDот Z при экстра-
кции хлоридов РЗЭ Д2ЭГФК
Рис. 5.31. Зависимость коэффициента распределения эрбия при экстракции Д2ЭГФК (1
моль/л) от концентрации в водной фазе HNO3 (/) и HCJ (2)
коэффициенты разделения пар элементов, находящихся в начале «тет-
рады», будут больше, чем для последних. Например, для пар Се—La,
Tb—Gd, Тт—Ег они соответственно равны 2,98; 4,92; 2,49, а для пар
Nd—Pr, Gd—Eu и Lu—Yb они значительно меньше: 1,38; 1,43; 1,86.
При повышении концентрации соляной или азотной кислоты коэф-
фициенты распределения РЗЭ сначала уменьшаются, а затем увеличи-
ваются, и на кривых lgf>=/(СКисл) имеются минимумы (рис. 5.31). Для
нитратных растворов минимумы размыты (концентрация кислоты 1—5
моль/л), для хлоридных растворов минимумы выражены более четко
при концентрации соляной кислоты 6—8 моль/л. В обоих случаях уве-
личение экстракции связывают с переходом в органическую фазу ком-
плексов другого состава, например Ьп(КОз)з ЗНА, LnCLA? и др. При
одной и той же концентрации кислоты наиболее сильное уменьшение
коэффициентов распределения наблюдается у легких РЗЭ. При экстра^
кцки кз сульфатных раствори» увеличение концентрации ионов sv4
приводит к противоположному эффекту — увеличению коэффициента
распределения. При низкой концентрации серной кислоты коэффици-
енты распределения РЗЭ меньше соответствующих коэффициентов ДЛЯ
нитратных растворов, что однозначно объясняется большей устойчиво-
стью сульфатных комплексов. При повышении же концентрации суДЬ'
фатных ионов подавляется вторая ступень диссоциации H2SO4’
увеличивается концентрация ионов HSO4-, уменьшается концентрации
ионов Н . Эффект понижения кислотности водного раствора оказывает
272
более сильное воздействие на распределение, чем усиление комплексо-
образования лантаноидов с ионом SO42 .
Комплексы РЗЭ с фосфорорганическими кислотами хелатного типа
отличаются большой прочностью и высокой энергией связи, что вызы-
вает серьезные затруднения при реэкстракции металлов из органиче-
ской фазы, особенно тяжелых РЗЭ. Легкие лантаноиды можно ре-
экстрагировать из органической фазы при экстракции из нитратных
растворов азотной кислотой с концентрацией > 0,5 моль/л. Элементы
иттриевой подгруппы, в том числе и иттрий, при экстракции из хлорид-
ных растворов реэкстрагируют 6—8 М НС1. Однако при этом Yb и Lu не
выделяются полностью из органической фазы, их можно извлечь 20 %
HF или 10 % NaOH.
Коэффициенты разделения различных пар РЗЭ при изменении ус-
ловий экстракции изменяются неодинаково. Например, коэффициент
разделения пар Sm—Nd и Nd—Се не изменяется заметно в интервале
концентраций соляной кислоты от 0,3 до 1,0 моль/л и при изменении
концентраций металлов в водной фазе от 0,05 до 2,0 моль/л. Для пар
Ег—Но, Y—Но и Ег—Y в интервале концентраций азотной кислоты
0,1—16 моль/л fl уменьшается; но при экстракции из хлоридных рас-
творов для первых двух пар fl сначала остается постоянным, а затем
уменьшается, для пары Ег—Y начиная с концентрации 4 моль/л НО —
увеличивается (рис. 5.32). Для тяжелых РЗЭ увеличение содержания в
бинарной смеси более тяжелого элемента приводит к уменьшению fl.
Например, при экстракции смеси Yb и Тт из 5,0 моль/л НО раствором
1 моль/л Д2ЭГФК и концентра-
ции металлов 0,2 моль/л при со-
держании в смеси Yb 5 % /^Ytrrm"
- 5, а при содержании более 95 %
Ybfl уменьшается до ~ 3. Для пары
Nd—Рг при изменении соотноше-
ния между элементами fl остается
практически постоянным.
Д2ЭГФК применяют для экст-
ракции как микроконцентраций
РЗЭ и разделения радионуклидов,
так и макроконцентраций. Ре-
зультаты, полученные при экстра-
кции микроконцентраций, не
всегда совпадают с результатами
экстракции макроконцентраций,
при которой могут быть получены
Не только иные зависимости, но и
Рис. 5.32. Зависимость коэффициента раз-
деления /? от концентрации НС1 в водной
фазе при экстракции Д2ЭГФК для пар:
/ — Ег—Но; 2 — Y—Но; 3 — Er—Y
273
наблюдаются другие явления, например образование третьей гелеобра3_
ной фазы. Не всегда целесообразно применять и высокие концентрации
Д2ЭГФК в экстрагенте, так как получение очень высоких коэффициен-
тов распределения ведет к уменьшению эффективности разделения, с
другой стороны, уменьшение концентрации Д2ЭГФК снижает емкость
экстрагента.
Экстракция карбоновыми кислотами. Для экстракции РЗЭ исполь-
зуют промышленные смеси монокарбоновых кислот с общей формулой
СпН2п+1СООН и числом углеродных атомов 6—9. Кислоты с меньшим
числом атомов углерода довольно заметно растворяются в водной фазе
а соли кислот с числом атомов углерода больше 9 плохо растворяются в
углеводородных растворителях. Радикал кислоты может быть неразвет-
вленным, например у капроновой кислоты СНз—(СНг)4СООН, и раз-
ветвленным. Смеси последних у нас в стране называют высшими
изомерными карбоновыми кислотами:
За рубежом аналогичные смеси называют версатовыми кислотами.
Как экстрагенты для РЗЭ несколько более эффективны кислоты с
разветвленным радикалом. Нафтеновые кислоты, получаемые попутно
при переработке нефти, содержат одну группу —СООН и пятичленный
цикл, число углеродных атомов 12—13. Монокарбоновые и нафтеновые
кислоты дешевы, используются в виде растворов в углеводородных рас-
творителях (керосине и др.), экстракция ими возможна из нитратных,
хлоридных и сульфатных растворов.
Уравнение экстракции карбоновыми кислотами
Ln3+ а#(в) +3HR(0) X LnRato) +ЗН+(В). (5.57)
При высокой концентрации кислоты в экстрагенте возможно обра-
зование комплексов типа LnRa3HR. Комплексы карбиновых кислот
менее прочны, чем комплексы РЗЭ с Д2ЭГФК, поэтому их экстракция
очень сильно зависит от pH водной среды. При изменении значения pH
на единицу D может уменьшиться или увеличиться в 100 раз. ДлЯ
количественной характеристики и сопоставления экстрагируемостй
различных ионов пользуются значением рНо,5, при котором D = 1; при
Vo Vb= 1:1в органическую фазу переходит 50 % вещества от исходного
его количества.
Экстрагируемость ионов металлов уменьшается с возрастанием
рНо,5 и, как правило, снижается в ряду М3+ > М2+ > М+. Значения рНо.5
274
ддЯ РЗЭ 3,7—4,2. Меньшие значения (0,99—3,19) имеют Fe, Ti, Al, Си,
pb, Сг идр., а следовательно, лучше экстрагируются, чем РЗЭ; значения
л} > 4,0 характерны для ионов Zn, Са, Ni, Со, Мп, Mg и всех щелочных
Металлов. Коэффициент распределения в ряду La—Lu увеличивается,
причем на графике lg£>=f(Z) четко прослеживаются «тетрады», как при
экстракции Д2ЭГФК (см. рис. 5.30). Различие в значениях рНо,5 для
соседних РЗЭ составляет 0,15—0,02 единиц pH, соответственно различ-
ны и коэффициенты "разделения. Для пары Lu—La/З в зависимости
пт использованного экстрагента равны 300—600, т.е. на 2—3 поряд-
на меньше, чем при экстракции ТБФ и Д2ЭГФК (среднее значение
« 1,4). Емкость экстрагентов по сумме оксидов РЗЭ при содержании
кислот в нем 50—75 % составляет 40—100 г/л.
Карбоновые кислоты поэтому целесообразно использовать для очи-
стки суммы РЗЭ от других элементов, конверсии одних солей в другие и
концентрирования растворов. Конверсия солей осуществляется путем
экстракции, например их хлоридных или сульфатных растворов, а ре-
экстракция из органической фазы — азотной кислотой. Концентриро-
вание растворов достигается при экстракции из разбавленных растворов
при Ко < Ев, а реэкстракция при Ео > Ев. Это позволяет уменьшить
энергозатраты по сравнению с упариванием раствора. Так, при экстра-
кции из раствора, содержащего 10—25 г/л ЬпгОз, смесью синтетических
жирных кислот (СЖК), содержащих 7—9 углеродных атомов, можно
получить растворы с концентрацией ЬпгОз 150—200 г/л. Экстракцию
проводят 40—50 %-ным раствором СЖК в керосине при pH 4,8—5,2 и
отношении Ео: Ев=1:1, реэкстракцию 8—10моль/л HNO3 и отношении
Уо: Ев=(8—12) : 1.
5.17. Принципиальные схемы разделения РЗЭ
Для получения индивидуальных РЗЭ используется большое число
технологических схем, что обусловлено многообразием по составу кон-
центратов, поступающих на разделение, и задачами получения конеч-
ных продуктов. Ими могут быть, например смеси легких РЗЭ для про-
изводства полирита или мишметалла. Для производства люминофоров,
постоянных магнитов, активаторов ОКГ и др. требуются соединения с
Содержанием основного вещества 99,99—99,9999 % (Nd, Sm и др.).
Среди различных способов разделения первостепенное значение
Имеет экстракция, причем первоначально довольно широко применяв-
шиеся полупротивоточные и другие нестационарные и периодические
процессы в значительной степени вытеснены противоточными.Для по-
лучения индивидуальных РЗЭ вследствие малых значений коэффици-
ентов разделения пар соседних элементов необходимы каскады в 50—90
275
ступеней, состоящие из последовательно работающих аппаратов. Прц
таком большом их числе важнейшим требованием к ним является на-
дежность в эксплуатации. Поэтому в производстве РЗЭ получили рас.
пространение горизонтальные смесители-отстойники ящичного типа
Их достоинством является простота конструкции, высокий к.п.д., при-
годность к работе при изменении концентраций растворов и соотноше-
ния объемов фаз, сохранение распределения компонентов по ступеням
при остановке и быстрый вход в режим после пуска.
На рис. 5.33 представлен один из возможных вариантов принципи-
альной схемы переработки плава хлоридов РЗЭ, полученного при хло-
рировании лопарита в солевом расплаве. Плав имеет примерно
следующий состав, %: ЬпгОз 37—45; СаО 7,7—9,2; SrO 1—2; NazO
7,5—9,2; К2О0,14—0,24;MgO0,9—1,0; FeO 0,3—0,5; U 0,02; Th 0,15-
0,25. Плав растворяют в воде с небольшой добавкой соляной кислоты при
60—70 °C до получения раствора с концентрацией оксидов РЗЭ 160—
Рис. 5.33. Принципиальная схема разделения РЗЭ из плава хлоридов, полученных
при хлорировании лопарита
276
200г/л. Для отделения тория в виде Th(OH)4 раствор нейтрализуют
5 %-ным NH3 до pH ~ 4,0. Вместе с гидроксидом тория осаждается
ре(ОН)з, причем находящийся в плаве FeCh предварительно окисляют
пероксидом водорода. Для выделения мезотория к раствору добавляют
(NH4)2SC>4 и ВаС12, (последний добавляют в том случае, когда в плаве
содержится недостаточно хлорида стронция). Радиоактивный осадок,
захватывающий 2—3 % РЗЭ от содержащихся в растворе, направляют
на промывку разбавленной соляной кислотой, а затем — на захороне-
ние.
Отфильтрованный хлоридный раствор направляют на получение
индивидуальных РЗЭ. На первом экстракционном каскаде с помощью
Д2ЭГФК РЗЭ делят на две группы. Разделение проводят по паре Nd—
Sm, так как вследствие отсутствия между ними прометия коэффициент
разделения этой пары элементов больше, чем для любой другой пары
рядом стоящих в таблице Д.И. Менделеева лантаноидов.
Из экстракта самарий, европий, гадолиний и очень небольшие ко-
личества тяжелых РЗЭ можно реэкстрагировать любой кислотой. Если
предусмотрено электрохимическое восстановление европия, то реэкст-
ракцию нужно проводить соляной кислотой. После восстановления ев-
ропия аммиаком осаждают гидроксиды Sm(OH)3 и Gd(OH)3, которые
затем растворяют в соляной или азотной кислоте в зависимости от того,
какой далее используется экстрагент — ТБФ или Д2ЭГФК. Удаление
европия, стоящего между самарием и гадолинием, облегчает их разде-
ление.
Из рафината, полученного на первом экстракционном каскаде, в
первую очередь выделяют церий тем или иным способом, основанным
на окислении его до Ce(IV). По одному варианту церий окисляется
перманганатом калия и экстрагируется Д2ЭГФК (раствор хлоридный),
а затем проводят конверсию хлоридов в нитраты. По другому варианту
конверсию хлоридов в нитраты проводят до окисления церия, который
в этом случае окисляют электрохимическим способом и экстрагируют
ТБФ.
Из оставшихся в рафинате La, Pr, Nd прежде всего экстракцией ТБФ
выделяют лантан — он экстрагируется хуже празеодима и неодима. В
Растворе его концентрация наиболее высокая, что позволяет использо-
вать эффект самовысаливания. Коэффициент разделения для пары
Та—Рг вследствие отсутствия между ними церия выше, чем для пары
Pr—Nd. После отделения лантана экстракцией ТБФ делят празеодим и
Неодим.
При переработке концентратов, содержащих все РЗЭ, их предвари-
тельно делят на подгруппы — легкие и тяжелые. В некоторых случаях
Удается получить за один процесс не два, а большее число промежуточ-
ных концентратов. В качестве примера приводится (рис. 5.34) принци-
277
Рис. 5.34. Один из возможных вариантов разделения РЗЭ из сульфатных раство-
ров, образующихся при переработке фосфатных концентратов с получением груп-
повых концентратов РЗЭ
пиальная схема экстракционного разделения РЗЭ из сернокислых
растворов, полученных при переработке фосфатных минералов. Экст-
ракцию проводят Д2ЭГФК: в водной фазе остаются лантан, церий,
празеодим и неодим; в органическую извлекаются: иттрий, все осталь-
ные РЗЭ и небольшое количество неодима. Дальнейшее получение про-
межуточных концентратов происходит на промывной части каскада-
При промывке экстракта азотной кислотой с концентрацией 1,5 мсль/л
в водную фазу извлекаются неодим, самарий, европий, гадолиний и
тербий. Затем при второй промывке азотной кислотой концентрацией 6
моль/л реэкстрагируются диспрозий, гольмий, иттрий, эрбий и тулий;
содержание иттрия в этом концентрате > 75 %. Наконец, иттербий "
лютеций извлекаются 10 %-ным раствором NaOH. Полученные кон-
центраты перерабатываются по своим схемам.
278
5.18. Производство оксидов РЗЭ высокой степени чистоты
В оксидах РЗЭ высокой степени чистоты содержание красящих при-
месей (Fe, Си, Мп, Cr, Ni, Со, Ti, V и др.) не должно превышать
(0,2—5) • 10 5 %. Жесткие требования предъявляются также к сод^зжа-
лию щелочных и щелочноземельных металлов, анионов (Cl , SO4 , F~
и др-). Для глубокой очистки соединений РЗЭ применяют химические
методы, сорбцию примесей на материалах с сильно развитой поверхно-
стью (например, активированном угле), экстракционные методы.
Химические методы используют в основном для удаления щелочных
и щелочноземельных примесей. Они заключаются в осаждении гидро-
ксидов РЗЭ, растворении их в HNO3, осаждении оксалатов, их отмывке,
сушке и прокалке. Оптимальные условия осаждения гидроксидов: кон-
центрация раствора 20—30 г/л ЬпгОз, концентрация аммиака 25 %,
температура 80 °C, трехкратная промывка гидроксидов горячей водой
при Т : Ж = 1 : 1. Осадок гидроксидов растворяют в HNO3 так, чтобы
получить раствор с концентрацией 50—60 г/л ЬпгОз и pH 1 —2. Из этого
раствора осаждают оксалаты насыщенным раствором или твердой
Н2С2О4 при 80—90 °C. Щавелевую кислоту добавляют небольшими
порциями для получения крупнокристаллических и хорошо фильтрую-
щихся осадков. При pH 1—2 повышается растворимость оксалатов Са,
Fe и других примесей, а следовательно, улучшаются условия очистки от
них. Осадок оксалатов промывают на фильтре горячей водой, содержа-
щей 0,2 % Н2С2О4 при Т : Ж = 1 : 10. Фильтрат и промводы нейтрали-
зуют до pH 3—4; выпадающие при этом оксалаты РЗЭ содержат при-
меси, поэтому их направляют в голову процесса.
Осадок оксалатов подвергают термическому разложению, которое
протекает через стадии потери внешнесферной воды (40—150 °C), по-
тери кристаллизационной воды и образования безводных оксалатов
(150—400 °C), образования карбонатов (280—500 °C), образования ок-
сидов (600—800 °C). В производственных условиях процесс заканчива-
ют при 800—1000 °C, сушку и прокалку оксалатов проводят R
электрических муфельных печах в кварцевых кюветах. Длительность
Процесса прокалки 4—6 ч. Получаемый при этом продукт содержит до
0,5 % углерода. Лучшие результаты можно получить при прокали-
вании оксалатов в плазменной струе с температурой 2500—4000 °C,
оксиды при этом содержат < 0,01 % углерода. Частицы их приобретают
сферическую форму, выравнивается гранулометрический состав. Этот
Метод позволяет получать оксиды РЗЭ с содержанием примесей щелоч-
ных и щелочноземельных металлов (0,5—ВЮ-3 %, анионов (0,2—
О 10~2 %, красящих примесей (0,5—3) 10 4 %.
279
Предложен довольно эффективный метод очистки от красящих при
месей осаждением их органическими реагентами, например диэтилди-
тиокарбаминатом натрия (С2Н5) — N----(ДДТК). Он об
\S Na ЗН2О
разует в кислой среде устойчивые комплексы с переходными металла-
ми, к которым принадлежат все красящие примеси; РЗЭ таких комплек-
сов не образуют. Так, для очистки иттрия осаждение проводят 2 %-ньщ
раствором ДДТК при pH 2—3,5.
Для более полного осаждения примесей и лучшей фильтра-
ции к раствору добавляют 0,1 %-ный водный раствор полиакрилами-
да. Этим способом получают ЬпгОз с содержанием красящих
примесей (0,2—3) 10 4 %. По сравнению с ранее описанным методом
этот способ позволяет получить те же результаты за меньшее число
операций, его недостаток — высокая стоимость реагентов.
Широко используется для очистки РЗЭ от нередкоземельных при-
месей экстракция ТБФ и карбоновыми кислотами (СЖК). Большинство
нередкоземельных примесей экстрагируется из кислых растворов ТБФ
хуже, чем РЗЭ. Это позволяет очистить растворы РЗЭ от основной массы
щелочных и щелочноземельных элементов, алюминия и красящих при-
месей и получить оксиды с содержанием примесей (0,5—3) 10-4 %.
Особенно эффективна экстракция ТБФ для удаления кальция, который
плохо удаляется при осаждении оксалатов. При экстракции СЖК (С7—
С9) часть примесей экстрагируется хуже, чем РЗЭ, другая часть —
лучше. Поэтому отделение их происходит в процессе экстракции, про-
мывки и реэкстракции. Результаты аналогичны получаемым при экст-
ракции ТБФ.
Для получения более чистых оксидов РЗЭ (например, для волокон-
ной оптики) конечней операцией является адсорбционно-комплексооб-
разовательная очистка. В качестве комплексообразователей ис-
пользуют диметилглиоксим, дибромоксин, ДДТК и др., в качестве сор-
бентов — активированный уголь, более эффективные аниониты с пер-
вичными. вторичными аммониевыми группами или четвертичными
аммониевыми основаниями (АН-31, АВ-17 и др.). При прохождении
раствора РЗЭ через слой сорбента наряду с основными реакциями ион-
ного обмена протекают процессы молекулярной сорбции, комплексооб-
разования. Аниониты не сорбируют катионы РЗЭ, но сорбируй1
нейтральные комплексы переходных металлов и избыток комплексооо-
разователя; на поверхности частиц анионита создается естественный
барьер, препятствующий «проскоку» примеси. Раствор после осаждения
диэтилтиокарбаминатов, содержащий избыток комплексообразоват6"
ля, пропускают через колонку с анионитом АН-31. После осаждения
оксалатов и прокалки получают оксиды с содержанием примесей, /°'
280
5 10 5 Fe, < 3 10 6Cr, 1 10 6Cu, < 1 10 5 Ni, Co, V, Мп (каждого),
для отделения Co и Ni наиболее эффективно применение диметилгли-
оксима, позволяющего снизить их концентрацию до (2—5) 10 6, %. Из
полученных растворов осаждают оксалаты, которые и прокаливают.
5.19. Получение полирующих материалов
Производство высококачественных полирующих материалов на ос-
нове оксидов РЗЭ, главным компонентом которых является СеОг (от 50
до 99 %), должно обеспечить получение материалов с определенными
химическим и фазовым составами, содержанием примесей и дисперсно-
стью.
Состав смеси оксидов РЗЭ оказывает существенное влияние на ка-
чество полирующего материала. Чистые оксиды La и Nd не обладают
полирующими свойствами, что определяется их гексагональной кри-
сталлической структурой и малой твердостью. Полирующие свойства
определяются диоксидом церия и его твердыми растворами с оксидами
РЗЭ с кубической структурой типа флюорита. Кроме химического со-
става на полирующие свойства порошка влияют гранулометрический
состав, твердость и механическая прочность частиц и др. Нередкозе-
мельные примеси отрицательно влияют на свойства полирита, напри-
мер 5 % СаО снижают его полирующую способность на 25 %, неже-
лательно присутствие хлора более 0,2 %. В то же время присутствие
ионов фтора, вступающего во взаимодействие со стеклом, оказывает
положительное влияние на процесс полирования.
Физические и физико-химические свойства полирита зависят также
от условий получения промежуточных соединений. Лучшие свойства
имеют порошки, получаемые при прокаливании оксалатов, однако в
промышленности преимущественно используют более дешевые карбо-
наты.
Оптимальные условия осаждения карбонатов, позволяющие полу-
чать порошки с однородным гранулометрическим составом: концентра-
ция растворов 1.пгОз 50—100 г/л, ИагСОз 50—60 г/л, температура
50—60 °C, pH 4,7—4,9, время кристаллизации 2—4 ч. PeaitHTiM берут-
ся в строго стехиометрическом соотношении для получения
Ьп2(СОз)з 8Н2О. Оптимальная температура прокаливания карбонатов
при доступе воздуха 950—1000 °C. При температуре 450—500 °C проис-
ходит окисление церия, при 600 °C — начинает формироваться тверды»
раствор оксидов РЗЭ и СеОг. По гранулометрическому составу порошо»
Достаточно однороден — более 90 % его имеет частицы с размерами 1 —1
мкм. Повышение температуры приводит к образованию конгломерате)
О — 691
28
частиц, повышению их твердости и хрупкости, появлению царапающих
примесей.
5.20. Металлотермия. Общие закономерности
Металлотермическими называются процессы восстановления ме-
таллов из их соединений другими, более активными металлами, проте-
кающие с выделением тепла. Металлотермический процесс можно запи-
сать в виде обычной окислительно-восстановительной реакции
МХ + М'-М + М'Х. (5.58)
Металл может быть восстановлен из соединения, если оно менее
устойчиво, чем соответствующее соединение металла-восстановителя.
Устойчивость соединений характеризуется величиной изменения энер-
гии Гиббса при образовании его из элементов. Металл может быть вос-
становлен в том случае, если убыль энергии Гиббса при образовании
восстанавливаемого соединения меньше, чем аналогичного соединения
металла-восстановителя при данных температуре и давлении, в пере-
счете на 1 моль или 1 г-атом неметалла. В этом случае Дб^ реакции (5.57)
имеет отрицательную величину. Целесообразность применения метал-
лотермического процесса определяется, во-первых, принципиальной
возможностью или невозможностью применения карботермического
процесса, т.е. восстановления металла углеродом, во-вторых, требова-
ниями, которые предъявляются к содержанию углерода в металле. Уг-
лерод во многих металлах является одной из наиболее трудно
удаляемых примесей.
Зависимости дб^ =/(Т) для реакций образования оксидов металлов
из элементов показывают границы применимости карботермического и
металлотермического методов и их взаимосвязь (рис. 5.35). В принципе
углеродом можно восстановить оксиды всех металлов из-за особого ха-
рактера изменения энергии Гиббса образования СО при повышении
температуры, однако для многих металлов температуры, при которых
возможны такие процессы, практически недостижимы. писСТаНОВЛёНН"
ем углеродом могут быть получены металлы, для оксидов которых вели-
чина (по модулю) меньше дб? реакции
2С + О-2СО. (5.59)
Выше линии Дб^г) = /(Г) для СО лежит область карботермии, ниже
— область металлотермии. Карботермией могут быть получены: сереб-
ро, ртуть, медь, свинец, никель, кобальт, железо при относительно
невысоких температурах. Если линия зависимости дб^ = f(T) оксида
282
Рис. 5.35. Зависимость энергии Гиббса образования оксидов от температуры (в
пересчете на 1 моль 02)
пересекается с линией =f<T) для СО, то металл выше температуры
точки пересечения может быть получен карботермией, ниже — только
металлотермией. Например, для FeO эта точка лежит приблизительно
283
10»
при 973 К, a Аб° реакции восстановления его углеродом при 673 К равна
+45 кДж; при 1473 К она становится равной минус 72 кДж. Карботермия
непригодна для получения металлов, образующих карбиды, — марган-
ца, хрома, ванадия, вольфрама и др.
Очевидно, что чем ниже на диаграмме находятся значения дбф =
=/(Г) соединения металла-восстановителя, тем активнее он восстанав-
ливает все соединения, Абф = f(T) которых лежат выше. Разница в
значениях aG^q должна быть достаточно большой; при малой разнице
реакция восстановления вследствие ее обратимости полностью не завер-
шается. В то же время металлотермический процесс может быть осуще-
ствлен, если Аб° реакции восстановления при данной температуре
имеет не очень большую положительную величину. Для смещения рав-
новесия в сторону образования металла необходимо удалять его из зоны
реакции, например испарением. Еще больший эффект достигается про-
ведением процесса восстановления в вакууме.
Термодинамический анализ, определяющий принципиальную воз-
можность осуществления металлотермической реакции, недостаточен.
Необходим учет ряда факторов, касающихся выбора исходных соедине-
ний для восстановления, металла-восстановителя на основе их физико-
химических характеристик, энтальпии реакции восстановления и т.д.
Наиболее доступными соединениями редких и нередких элементов
являются оксиды, однако их использование связано с рядом ограниче-
ний. Многие металлы, к которым относятся практически все редкие,
образуют низшие оксиды и твердые растворы с кислородом. Низшие
оксиды характеризуются большей прочностью связи металл—кислород,
поэтому восстановление может закончиться получением не металла, а
низшего оксида. В твердых растворах при уменьшении концентрации
кислорода также возрастает энергия связи металл—кислород. При не-
которой концентрации парциальная энергия Гиббса Дбт, характеризу-
ющая взаимодействие растворенного кислорода с металлом, становится
равной лб-^Q оксида металла-восстановителя. и дальнейшее уменьше-
ние концентрации кислорода становится невозможным. Равновесная
концентрация кислорода в металле может достигать нескольких долей
процента, что, как правило, ухудшает его свойства. Поэтому использо-
вание оксидов для получения высококачественного металла не рекомен-
дуется.
Более предпочтительно использование хлоридов и фторидов, кото-
рые, хотя и менее доступны, имеют ряд преимуществ перед оксидами-
Прежде всего соотношение энергий Гиббса образования хлоридов и фто-
284
Рис. 5.36. Зависимость энергии Гиббса образования хлоридов от температуры (в
пересчете на 1 моль С12)
Ридов таково, что позволяет расширить круг металлов-восстановителей.
Они являются бескислородными соединениями, имеют более низкие
температуры плавления и кипения. Важнейший недостаток хлоридов и
фторидов — высокая агрессивность при повышенной температуре. Кро-
ме того, хлориды гигроскопичны, легко гидролизуются, фториды менее
гигроскопичны, но некоторые из них не растворяются в воде и др. (рис.
5.36).
При получении чистых металлов необходимо, чтобы восстановитель
не образовывал твердых растворов или интерметаллидов, чтобы металл,
Шлак и избыток восстановителя легко разделялись. Восстановитель не
Должен вносить загрязнения в получаемый металл. Выбор металлов-
восстановителей расширяется при получении сплавов (например, титан
— железо, никель, медь, алюминий), так как при образовании твердых
285
растворов, так же как и при образовании интерметаллидов, свободна
энергия системы уменьшается (табл. 5.13). 4
В качестве восстановителей наиболее широко применяются алю^
ний, магний, натрий, кальций, кремний. Использование каждого из них
имеет свои особенности,поэтому различают алюминотермию, магние-
термию, натриетермию и т.д. В зависимости от теплового баланса вос-
становления металлотермические процессы делят на два типа
внепечные и печные, которые могут быть реализованы в различных
вариантах:
Четкой границы между внепечной и печной техникой не существу-
ет, так как тепловой баланс металлотермического процесса зависит от
масштаба, состава шихты и т.д. Один и тот же процесс может быть
осуществлен как тем, так и другим способом.
Важнейшей характеристикой металлотермического процесса, по-
зволяющей предварительно оценить его тип, является удельный тепло-
вой эффект реакции восстановления, или термичность шихты, которая
равна количеству тепла, выделяющегося при реакции, отнесенному
к 1 г шихты или к 1 г исходного соединения
Q>,= ("A^98(f)M-x“AH2g8(f)MX) / (2 (Л/мх+Л/м>), (5.60)
где Л/мх — молекулярная масса восстанавливаемого соединения; Мм "
атомная масса металла-восстановителя.
286
Таблица 5.13 Свойства некоторых металлов-восстановителей
и их соединений
Показатели Li Na Ca Mg Al Si
Тпл,"С 181 98 843 650 659 1410
Ткип» 1324 882 1483 1105 2450 3280
d, г/см 0,53 0,97 1,50 1.74 2,55 2,35
1.120 Na2O CaO MgO AI2O3 SiO2
7пл,‘С 1727 920 2527 2825 2050 1720
Ткип, °C 2600 1350 3500 3260 2980 2950
d,г/см 2,01 2,31 3,37 3,65 3,97 2,65
кДж/моль 598 415 635 602 1676 911
, кДж/моль 561 376 374 569 1581 856
LiF NaF CaF2 MgF2 A1F3 SiF4
Тил» °C 848 996 1418 1263 Субл. -90* >
7'кип, °C 1681 1787 2510 2332 1291 -98*^
d, г/см 2,64 2,77 2.18 — 3,10 —
кДж/моль 618 577 1228 1124 1210 1615
-AGjggpy кДж/моль 590 546 1176 1062 1131 1572
LiCl NaCl CaC12 MgC12 A1C13 S1C14
Тил,°C 610 801 772 714 193#) -70
7’кип,°C 1382 1465 2000 1418 181 58
d, r/at3 2.07 2.17 2.15 2,32 2,41 1,52
-ДЯ^эд^кДж/моль 409 411 796 644 584 662
-AGjQg^Q, кДж/моль 384 384 746 595 570 583
*)„ Под давлением. Сублимация.
287
При получении металлов и сплавов внепечным способом возможна
три случая.
1. Удельный тепловой эффект достаточен для самопроизвольно^
протекания реакции. После разогрева одного участка шихты внешним
нагревом или с помощью запальной смеси реакция распространяется по
всей ее массе (по другой терминологии СВС — самораспространяющий-
ся высокотемпературный синтез).
2. Если удельный тепловой эффект недостаточен, то используют
подогревающие добавки, которые реагируют с металлом-восстановите-
лем с большим выделением тепла (КСЮз, NaNO3, ВаОг, CaSCU и др.).
Например, для реакции КСЮз + AI тепловой эффект равен 9730 Дж на
1 г шихты.
3. Удельный тепловой эффект чрезмерно велик, реакция протекает
бурно, может сопровождаться выбросами шихты. Используют нейт-
ральные добавки (флюсы), не реагирующие ни с получаемым металлом,
ни с металлом-восстановителем (СаО, CaFi и др.).
Печные металлотермические процессы характеризуются тем, что
удельный тепловой эффект реакции совершенно недостаточен для под-
держания реакции, возникает необходимость внешнего нагревания
шихты. Большей частью для этой цели используют электрические печи.
Химически активные металлы получают в защитной атмосфере благо-
родных газов, преимущественно аргона, или в вакууме.
Аппаратурное оформление металлотермических процессов самое
разнообразное. Для выбора конструкционных материалов большое зна-
чение имеет температура, при которой проводится процесс — ниже
температуры плавления получаемого металла (Т<Тпл) или выше
(Т>Гпл).
Важнейшей характеристикой металлотермического процесса явля-
ется температура, при которой он может быть осуществлен и от которой
зависит, в каком агрегатном состоянии будут находиться продукты ре-
акции. Для этого предложен ряд эмпирических и полуэмпирических
формул
Т— (Qy- X (2-пл + 2-кип) / S Ср, (5.61)
где Z-пл и -£кип — скрытая теплота плавления и кипения продуктов
реакции; 2 ср — сумма теплоемкостей продуктов реакции.
Расчеты по этой и другим формулам являются приближенными, что
обусловлено неполнотой термохимических характеристик соединений
I при высоких температурах, а также сложностью расчета потерь тепла в
окружающую среду, которые непостоянны во времени, зависят от массы
восстанавливаемой шихты, продолжительности процесса, т.е. в конеч-
ном счете — от скорости реакции. Например, термичность шихты оксид
288
н11келя — алюминий около 3350 Дж/г, расчетная температура восста-
новления около 3000 °C, фактическая 2700 °C.
Наблюдается корреляция, хотя и не очень строгая, между энергией
Гиббса образования восстанавливаемого оксида и удельным тепловым
эффектом, при котором восстановление возможно. Чем более отрица-
тельно значение aG^q> тем больше должна быть величина Qy. Если при
восстановлении какого-либо оксида реально выделяющегося тепла ока-
зывается меньше, то для его осуществления необходим дополнительный
подвод тепла. Напротив, если термичность шихты чрезмерно велика,
для снижения температуры процесса необходим отвод тепла или введе-
ние в шихту инертных добавок. Эти обстоятельства имеют принципи-
альное значение для практического осуществления метал-
лотермических процессов; именно на основании необходимости отвода
или подвода тепла они делятся на печные и внепечные.
Ниже приведены удельные тепловые эффекты, требующиеся для
восстановления некоторых оксидов алюминием, Дж/г оксида:
Оксид Qy
Li2O ... 5530
ВеО... • 5230
SrO. .. 5430
La2O3 6310
Оксид Qy
TiO2 • 2480
ZrO2 ... 3520
HfO2 • • -3960
V2O5 .. 2740
Оксид Qy
Nb2O5 • 2920
Ta2O5 •. 3210
MoO3 - - • • 1930
WO3 • 2100
Оксид Qy
CuO .... 1440
PbO2 .. • 1360
FeO .... 2040
S1O2 3330
Сопоставление этих данных с термодиначескими характеристи-
иками оксидов (см. рис. 5.35) действительно показывает, что чем более
отрицательно значение
AG?(f)
оксида, тем больше должен быть Qy. Ока-
зывается также, что для оксидов [лгО, SrO, ВеО, ЬагОз и НЮг выделя-
ющегося реально тепла меньше, чем требуется, поэтому при восста-
новлении их алюминием возникают дополнительные сложности.
Скорость металлотермического процесса при одном и том же удель-
ном тепловом эффекте зависит от температуры. Повышение ее вызыва-
ет увеличение скорости реакции, что, в свою очередь, увеличивает вы-
деление тепла в единицу времени. Происходит самоускорение реакции,
причем оно тем более выражено, чем больше тепловой эффект реакции
и выше температура.
Металлотермические процессы относятся к твердофазным реакциям
типа Т—Т и Т—Ж, для которых определение скоростей реакций вообще
весьма сложно. Более того, от других типов твердофазных процессов
(например, спекания, проходящего при постоянной температуре) ме-
таллотермические отличаются тем, что температура изменяется во вре-
мени, меняется агрегатное состояние компонентов шихты. В начале
289
процесса ее компоненты — большей частью твердые вещества, в ходе еГТ|
происходит плавление одного или обоих продуктов реакции. Эти фазе
вые превращения, протекающие с поглощением тепла, замедляют рост
температуры, а следовательно, и скорости реакции. Может измениться
и лимитирующая стадия реакции, что еще более усложняет ее расчет
С точки зрения полноты разделения продуктов реакции более вы-
годны процессы, приводящие к плавлению получаемого металла и шла-
ка. Однако это возможно не всегда — при восстановлении тугоплавких
металлов или в тех случаях, когда температура процесса ограничена
например, развитием нежелательных вторичных реакций при высокой
температуре. Для снижения температуры плавления тугоплавких ок-
сидных шлаков в шихту добавляют известь, флюорит и другие флюсы.
Флюсы понижают вязкость шлаков, повышают их текучесть, что благо-
приятствует расслаиванию реакционной массы. Кроме того, например
при алюминотермическом восстановлении, оксид кальция связывает
образующийся оксид алюминия и сдвигает равновесие в сторону образо-
вания металла.
5.21. Металлотермическое получение РЗМ
Все соединения РЗЭ отличаются высокой химической и термической
устойчивостью, металлы обладают высокой реакционной способностью.
Это затрудняет выбор металла-восстановителя и материала аппаратуры
для проведения процесса восстановления. По свойствам, определяющим
выбор наиболее эффективного метода получения металлов, все РЗЭ
можно разделить на три группы.
Легкие лантаноиды (La, Се, Рг и Nd) имеют температуры плавления
< 1000 °C, а галогениды — высокие температуры плавления и кипения.
Наиболее распространено получение металлов электролизом распла-
вов.
Элементы, склонные к. образованию соединений со степенью окис-
ления +2 (Sm, Eu и Yb), имеют сравнительно низкие температуры плав-
ления и кипения. Металлы получают методом лантанотермии из ок-
сидов.
Тяжелые лантаноиды, а также гадолиний и иттрий, имеют высокие
температуры плавления ( > 1400 °C), но их галогениды плавятся и кипят
при пониженных температурах. Применимы различные методы метал-
лотермии.
Получение металлов из оксидов. Теоретически из термодинамиче
ской оценки следует, что оксиды РЗЭ можно восстановить до металла
только кальцием (см. рис. 5.35). Другие металлы-восстановители при
менимы, когда имеется возможность сдвига равновесия путем удален
290
металла из зоны реакции. Кальциетермический метод практически не
используется вследствие высокой температуры плавления оксида каль-
ция.
Металлические самарий, европий и иттербий можно получить толь-
ко восстановлением оксидов, так как при восстановлении фторидов или
хлоридов образуются галогениды типа LnXz, которые далее до металла
не восстанавливаются. Оксиды этих элементов можно восстановить
кальцием, церием, цирконием; обычно восстановление проводят ланта-
ном, что позволяет получить наиболее чистый металл
5шгОз + 2Еа^ Бш + ЬагОз; aG2qb =+7 кДж/моль. (5.62)
Z>O
Процесс проводят в вакууме 1 К) 2 Па при температуре 1400 °C. В
этих условиях образующийся самарий, имеющий температуру кипения
1778 °C, испаряется и выводится из зоны реакции, а металлический
лантан (Ткип = 3454 °C) и оксиды самария и лантана (ТКип > 3000 °C)
остаются в твердой фазе, что приводит к смещению равновесия приве-
денной реакции в сторону образования металлического самария. Таким
способом можно получить металлический европий при 900 °C и иттер-
бий при 1350 °C. Применять для восстановления оксидов этих металлов
кальций нельзя из-за его низкой температуры кипения (1483 °C). При
использовании в качестве металлов-восстановителей циркония, алюми-
ния и церия получают более загрязненные металлы.
На принципе смещения равновесия путем связывания продуктов
реакции основаны процессы получения из оксидов РЗЭ лигатур, пред-
назначенных для раскисления и легирования чугуна, стали и других
сплавов. Лигатуры марки «Симиш» содержат 10—35 % РЗМ, 5—15 %
алюминия, 50—60 % кремния и до 10 % железа. Алюминий и кремний,
взятые по отдельности, не восстанавливают оксиды РЗЭ до металла, что
видно из термодинамической оценки реакций восстановления в широ-
ком интервале температур
ЬагОз + 2А1X 2La + AI2O3;
Л^?200-2400К = + 60 -5- + 40 кДж/моль Ог. (5.63)
Примерно так же изменяется и AG? для реакции восстановления
L12O3 элементарным кремнием. При одновременном использовании
Для восстановления алюминия и кремния в результате связывания лан-
тана в силицид термодинамика реакции существенно меняется
ЬагОз + 4Si + 2А1 X 2LaSi + AI2O3;
А(7?ЭПА_, .mil. = ~260 -г- -320 кДж/моль Ог. (5.64)
Таким способом получают лигатуры, не содержащие кислород.
Практически в шихту вводятся алюминий и ферросилиций с содержа-
нием кремния около 75 %. Для понижения температуры плавления
291
шлака в шихту добавляют известь, флюоритовый концентрат, поварен-
ную соль. Процесс проводят в электродуговой печи при 1750 °C. Такая
температура необходима для плавления и прогрева шлака для лучшею
расслаивания, температура же плавления лигатур в зависимости от
состава 1180—1470 °C. Расход электроэнергии при получении лигатур
«Симиш» металлотермическим методом в 7—8 раз меньше, чем при
электрохимическом получении мишметалла: соответственно 2200 и
16000 кВт ч/т.
Получение металлов из фторидов. При использовании в качестве
исходных соединений для восстановления фторидов выбор металлов-
восстановителей более широк. Но некоторые из них — алюминий и
магний — дают с РЗМ твердые растворы, что, как правило, нежелатель-
но. При использовании в качестве восстановителя кальция возникают
сложности с отделением металла от шлака (CaFi). Гидрометаллургиче-
ский метод нельзя использовать, так как CaFi не растворяется в воде,
невозможна и вакуумная дистилляция из-за его высокой температуры
кипения (2510 °C). При получении продуктов реакции в расплавленном
состоянии температура должна быть выше температуры плавления
CaFi (1418 °C), а при получении РЗМ с высокими температурами плав-
ления (иттрий, лютеций, тулий и др.) температура должна быть выше
1750 °C. Это неизбежно осложняет проблему контейнерного материала
— фториды при такой температуре весьма агрессивны. При получении
чистых металлов возникает необходимость в дополнительной очистке
кальция вакуумной дистилляцией, так как при хранении он активно
поглощает из воздуха пары воды и азот, образуя нитрид.
Несколько лучше для восстановления фторидов литий
LnFs + 3Li X Ln + 3LiF. (5.65)
Для реакций этого типа изменяется в пределах 115—200
кДж. Принципиально возможно понижение температуры кальциетер-
мического или литиетермического восстановления РЗЭ. Например, при
получении иттрия в шихту вводят металлический магний и СаСЬ- В
результате восстановления получают сплав Y—Mg (Mg 24 %), плавя-
щийся приблизительно при 1000 °C. Температура плавления шлака,
состоящего из СаРг и СаСЬ, понижается примерно до 950 °C. После
разделения сплав подвергают вакуумной дистилляции для удаления
магния и остатков кальция при давлении — 10~3 Па и 900—950 °C, а
затем переплавляют в электродуговой печи. Содержание кислорода в
металле 0,1—0,2 %. При использовании в качестве восстановителя ли-
тия в шихту добавляют металлический магний, восстановление прово-
дят при — 1000 °C, отделяют шлак, состоящий из LiF, с температурой
плавления 848 °C и обрабатывают сплав иттрия с магнием так же, как и
292
при восстановлении кальцием. Содержание кислорода в металлическом
иттрии 0,05—0,15 %.
Для уменьшения содержания кислорода в металле предложено до
проведения вакуумной сепарации экстрагировать его из расплавленного
иттриймагниевого сплава при 950 °C смесью фторида иттрия и хлори-
стого кальция в атмосфере благородного газа. Содержание кислорода в
металле может быть снижено до 0,05 %. Некоторые трудности при
осуществлении этого способа получения РЗМ представляет получение
безводных фторидов, а также фторидов, не загрязненных посторонними
анионами. По всей вероятности, наиболее чистые фториды могут быть
получены обработкой твердых карбонатов РЗЭ плавиковой кислотой с
последующим обезвоживанием в токе HF при 600—700 °C.
Получение металлов из хлоридов. Хлориды в качестве исходных
соединений для металлотермии имеют ряд преимуществ перед фторида-
ми. Они имеют более низкие температуры плавления и кипения, водо-
растворимы, что позволяет применять гидрометаллургические методы
переработки реакционной массы, несколько менее агрессивны при вы-
соких температурах; взаимодействие их с металлами-восстановителя-
ми, как правило, начинается при более низкой температуре. Наи-
большую сложность их использования вызывает получение безводных
хлоридов, что являлось главным препятствием их практического ис-
пользования. В известной степени эта трудность преодолена благодаря
использованию двойных хлоридов с калием типа Кз1лС1б, которые ме-
нее гигроскопичны и более устойчивы к гидролизу. В качестве восстано-
вителя обычно используют литий, взаимодействие хлоридов с которым
начинается при 200 °C. Процесс восстановления ведут при температурах
ниже температуры плавления получаемого металла (максимальная
температура 1000 °C). Шлак — LiCl, имеющий температуру плавления
614 °C и кипения 1360 °C, удаляется сливом, а остатки — последующей
вакуумной дистилляцией. Тигель для восстановления может быть вы-
полнен из ниобия, в то время как при восстановлении фторидов единст-
венным материалом является остродефицитный тантал.
5.22. Электролитическое получение РЗМ
РЗМ имеют высокие отрицательные нормальные электрохимиче-
ские потенциалы, вследствие чего получить их электролизом водных
растворов невозможно. Промышленное значение имеет электролиз рас-
плавов, разработаны и используются различные варианты процесса с
использованием хлоридных, фторидных и фторидно-оксидных электро-
литов. В состав электролита, как правило, входят соответствующие соли
Щелочных и щелочноземельных металлов (Li, Na, К, Са, Ва), что позво-
ляет снизить температуру его плавления до 700—750 °C. При использо-
293
вании хлоридных ванн введение в электролит этих солей позволяет
уменьшить образование оксохлоридов РЗЭ при взаимодействии распла-
ва с влагой воздуха, не растворяющихся в расплаве хлоридов, препятст-
вующих образованию слитков металла и снижающих извлечение
металла на 15—25 %. Щелочные и щелочноземельные металлы на
катоде не выделяются, так как напряжение разложения их солей значи-
тельно больше напряжения разложения солей РЗЭ (табл. 5.14).
В промышленных масштабах получают индивидуальные РЗМ цери-
евой подгруппы, имеющие температуры плавления > 1000 °C, и мишме-
талл — сплав, содержащий 45—52 % Се, 24—24 % La, 18—19 % Nd,
5—6 % Pr. Тяжелые РЗМ иттриевой подгруппы, кроме иттербия, имеют
высокие температуры плавления, при которых становится заметным
испарение галогенидов, возникают проблемы с подбором материалов
для электролизеров, устойчивых к действию расплавленных РЗМ. Име-
ется принципиальная возможность снижения температуры электролиза
путем проведения его на жидком катоде, в качестве которого могут
использоваться расплавы магния, цинка, алюминия, кадмия, с которы-
ми РЗМ дают легкоплавкие сплавы. Для различных РЗМ получают
сплавы с содержанием металла от 5—6 до 48 %. Например, сплавы Y—А1
(28 % Y) и Y—Mg (49 % Y) можно получать при температуре электро-
лиза 827—927 °C. Для удаления легкоплавкого металла используется
вакуумная дистилляция, однако и с ее помощью не всегда удается пол-
ностью произвести очистку. Это сильно усложняет процесс получения
металла и препятствует широкому использованию электролиза.
Таблица 5.14. Напряжение разложения Ео хлоридов
некоторых РЗЭ и других элементов
Соединение Т, °C £b,B Соединение T,'C Eo, В
СеС13 738 2.1 LiCl 650 3,58
LaC13 783 1,66 NaCl 877 3.35
NdC13 800 1,55 KCI 768 3,41
PrC13 737 1.45 BaC12 700 3,62
Электролиты для получения металлов цериевой подгруппы и миш-
металла из расплавов безводных хлоридов содержат 50—60 % LnC13>
остальное — смеси хлоридов эвтектического состава или близкие к ней,
например 50 % КС1 + 50 % CaCLz (температура плавления 660 °C),
смесь50 % NaCl + 50 % КО (температура плавления658 °C). Электро-
лизеры могут быть различных конструкций с внутренней футеровкой из
огнеупорной керамики, анод графитовый, катод может быть выполнен
из различных материалов, в том числе и из железа. Температура элек-
294
тролиза 850—950 °C, катодная плотность тока 2—10 А/см2, выход по
току < 75 %, прямое извлечение металла до 70 %, расход электроэнер-
гии ~ 16 кВт ч на 1 кг металла. Большая часть потерь связана с образо-
ванием шламов, в основном состоящих из оксохлоридов РЗЭ. При
электролизе на аноде выделяется хлор, поэтому необходима хорошая
вытяжная вентиляция и устройства для улавливания хлора. Электроли-
зом расплава хлоридов получают преимущественно мишметалл или
церий, используемые'в качестве раскислителей и для которых допуска-
ется значительное содержание примесей. В полученных этим способом
металлах оно может составлять 1—6 % (углерод, кальций, алюминий,
кремний, железо и др.).
Индивидуальные РЗЭ цериевой подгруппы повышенной чистоты
могут быть получены электролизом из хлоридных расплавов в электро-
лизерах, футерованных молибденом, танталом, с катодами из этих же
металлов в атмосфере благородных газов.
Для получения индивидуальных РЗМ хорошие результаты получа-
ются при использовании фторидно-оксидных расплавов. Метод основан
на том, что в ванне, содержащей ~ 75 % LnF3 до 15 % BaFz и 10 % LiF,
растворяется до 5 % ЬпгОз при температуре порядка 800 °C. Электро-
лиз, который фактически протекает за счет разложения оксида, прово-
дят при 900—1000 °C. При электролизе оксидов значительно уве-
личивается выход по току и прямое извлечение металла: на 10—15 %
по сравнению с электролизом хлоридов. Облегчается питание ванны
оксидом.
Электролитический метод получения мишметалла и многих инди-
видуальных РЗМ, получивший широкое распространение, характери-
зуется довольно высоким расходом электроэнергии и большим со-
держанием примесей в получаемых металлах. Поэтому в последнее
время с ним успешно конкурирует металлотермический способ получе-
ния РЗМ.
Электролитическое рафинирование РЗМ. Анод изготавливается из
чернового металла, процесс ведут при температуре ниже температуры
плавления рафинируемого металла, который осаждается на катоде в
виде крупных кристаллов. Электролитическим методом удаляются пре-
имущественно металлические примеси (железо, тантал и др.), очистка
от примесей внедрения практически не происходит. Для рафинирования
могут использоваться как хлоридные, так и чисто фторидные ванны.
Например, для иттрия применяют электролит,состоящий из 94,6 % LiCl
и 5,4 % YC13; температура электролиза 750 °C. Для гадолиния рекомен-
дован электролит: 75 % LiF, 10 % BaFi и 15 % GdFs, температура
электролиза 850 °C. Рафинированный гадолиний содержит металличе-
ских примесей — 0,005 % и каждой из неметаллических примесей (Н,
С, N, OhF) по 0,01 %.
295
5.23. Рафинирование РЗМ
Технические РЗМ, получаемые электрохимическим и металлотер-
мическим способами, содержат значительное количество примесей
Суммарное содержание неметаллов (кислород, азот, углерод, фтор или
хлор) может превышать 1 %. Эти примеси вследствие малой раствори-
мости в металлах присутствуют главным образом в виде включений
оксидов, карбидов, оксогалогенидов. Загрязнение РЗМ материалами
контейнеров и металлами-восстановителями также может быть значи-
тельным. Примеси сильно изменяют физико-химические свойства РЗМ,
уменьшают их пластичность, коррозионную стойкость и др. Для ряда
случаев, например получения магнитных материалов, необходимы ме-
таллы с малым содержанием примесей. Для очистки РЗМ преимущест-
венно используют вакуумную дистилляцию, а также зонную плавку и
электроперенос в твердом состоянии.
Вакуумная дистилляция. Метод вакуумной дистилляции является
эффективным методом рафинирования РЗМ, позволяющим проводить
глубокую очистку их от примесей внедрения и металлов, у которых
давление пара выше или ниже, чем у рафинируемого металла. К числу
первых относятся Са, Mg, Na, Li, к числу вторых — Nb, Та, W и др. (см.
табл. 5.5, 5.13). Такие металлы, как Fe, Ti, Сг имеют давление пара,
близкое к давлению пара большинства РЗМ, поэтому очистка от них
практически не происходит.
Дистилляцию проводят в высоковакуумных печах (давление s 10 л
Па) преимущественно с индукционным нагревом. Тигель и конденсатор
выполняют из тантала. Температуру в тигле и конденсаторе поддержи-
вают в зависимости от температуры кипения рафинируемого металла.
Например, для Dy, Но, Ег они соответственно равны 1600—1700 и
900—1000 “С, для Y, Tb, Lu 2000—2200 и 1300—1400 °C. Методом
дистилляции получают металлы с содержанием основного вещества до
99,95 %, содержание некоторых примесей, например N, Н, Са, Мп, Zn
и др., может быть на уровне п 10 4 %.
Электроперенос в твердом состоянии (ЭТС). Сущность метода
заключается в пропускании постоянного тока через пруток металла,
нагретого до температуры, соответствующей 80 % от температуры плав-
ления. Под действием тока ионы примесных элементов мигрируют к
аноду, вследствие чего происходит очистка катодного конца прутка. Для
исключения взаимодействия нагретого металла с атмосферой процесс
проводят в глубоком вакууме (10 5—10 7 Па). Метод оказался наиболее
эффективным для очистки металлов от примесей внедрения (О, N, С),
содержание которых снижается более чем в 10 раз по сравнению с на-
чальным (до 0,01 % и менее). Наиболее полно метод изучен для туго-
296
лдавких РЗМ (Y, Gd, Lu). Анализ РЗМ такой чистоты, какой обеспечи-
вает этот метод, затруднен йз-за опасности загрязнения проб металла
при их отборе, поэтому качество очистки контролируют по электросоп-
ротивлению при 4,2 и 300 К.
Вместе с тем метод малопроизводителен (использовали прутки диа-
метром 6 и длиной 100 мм), процесс продолжается 100 ч и более. Поэто-
му метод может быть рекомендован для получения металлов специ-
ального назначения, например создания магнитооптических уст-
ройств.
Зонная плавка. Метод зонной плавки широко применяется для глу-
бокой очистки полупроводников, ряда металлов, однако применение его
для РЗМ сдерживалось отсутствием контейнерных материалов. Исполь-
зование его для рафинирования РЗМ стало возможным после создания
водоохлаждаемых тиглей-лодочек и высоковакуумного оборудования,
обеспечивающего остаточное давление 10 8 Па. Зонную плавку РЗМ с
высоким давлением пара (Sm, Eu, Tm, Yb и др.) проводят в атмосфере
высокочистых благородных газов. Имеющиеся данные по зонной очист-
ке La, Gd и Tb говорят о том, что очистка их от Н, О и N протекает не
очень эффективно, очистка же от металлических примесей характери-
зуется высокими показателями. Например, содержание в начале слитка
тербия Ni и А1 равно п 10 4 %, Си, Fe, Si, Pb и Ti — п 10 5 %, что в
50—200 раз меньше их содержания в конце слитка. Эти данные свиде-
тельствуют об эффективном перераспределении примесей.
Глава 6. СКАНДИЙ
ХИМИЯ СКАНДИЯ
6.1. Физические и химические свойства скандия
Чистый скандий имеет серебристый блеск, на воздухе тускнеет,
сравнительно мягок (твердость по Бринеллю 0,157 МПа), хорошо обра-
батывается. Наличие примесей (1 —2 %) делает металл твердым и хруп-
ким; a-Sc имеет структуру, построенную на основе плотнейшей двух-
слойной гексагональной упаковки, которая при нагревании переходит в
объемноцентрированную кубическую (уЗ-Sc). Температура полиморф-
ного перехода 1337 °C.
При температуре 1400—1450 °C в вакууме (~ 1 Па) металлический
скандий возгоняется, что используется для получения металла высокой
степени чистоты. Он имеет слабые парамагнитные свойства, на что
указывает величина атомной магнитной восприимчивости (К=236 10
297
при 20 °C). Физические и химические свойства сильно зависят от чисто
ты скандия. Этим объясняется заметное расхождение физических Кон
стант в опубликованных работах, так как до последнего времени не
могли получать достаточно чистый металл (> 99,99 %).
Металлический скандий химически активен. Он, как и РЗМ, взаи-
модействует с кислородом, серой, углеродом, азотом, галогенами. При
хранении на воздухе поверхность металла покрывается оксидной плен-
кой, препятствующей его дальнейшему окислению. При нагревании
взаимодействует с водой с выделением водорода, растворяется в мине-
ральных кислотах, за исключением хромовой и плавиковой; медленно
реагирует с концентрированным раствором гидроксида натрия.
Соединения скандия в большей мере, чем соединения РЗЭ, склонны
к гидролизу, что связано с разменами их трехзарядных ионов. Различия
в размерах ионных радиусов Sc3 и Ln3+ определяют и другие отличия в
их химическом поведении. Ион скандия имеет ионную конфигурацию
(f и, как все ионы этой конфигурации, имеет относительно низкую
склонность к комплексообразованию, хотя она и выше, чем у РЗЭ.
Сводка основных свойств скандия и некоторых его соединений в
сопоставлении со свойствами РЗЭ дана в гл. 5.
6.2. Соединения скандия с кислородом
Оксид скандия. Известен единственный оксид скандия SC2O3. Его
можно получить, прокаливая гидроксид, карбонат, оксалат, сульфат
или нитрат скандия. При окислении металлического скандия возможно
первоначальное образование оксида SC2O3-X (где х=0,0—0,11), который
при дальнейшем окислении переходит в SC2O3. Система Sc—О показана
на рис. 6.1.
Система характеризуется образованием эвтектики между /З-Sc и
SC2O3 при 1560± 10 °C и содержании ~ 35 % (ат.) кислорода, а также
образованием эвтектоида между CT-Sc и SC2O3 при 1130+ 10 °C и содер-
жании 16 % (ат.) кислорода. Максимальная растворимость кислорода в
/З-Sc достигает ~ 33 % (ат.) при 1560 °C и - 11 % (ат.) при 1130 °C в
ct-Sc. При понижении температуры до комнатной растворимость кисло-
рода в скандии снижается до 2,5 % (ат.). Монооксид скандия при изуче-
нии системы не обнаружен.
Оксид скандия практически не растворяется в воде, плохо раствори'
ется в холодных разбавленных кислотах, хорошо — в концентрирован-
ных.
Оксид скандия взаимодействует с оксидами большинства элементов
Периодической системы. С оксидами шелочных металлов образуются
термически неустойчивые, гигроскопичные соединения типа LiScOz, в
298
системах с оксидами элементов
ц группы образуются соединения
различных составов, но преиму-
щественно с соотношением 1 : 1
(CaSczCU) и ограниченные твер-
дые растворы. С легкими РЗЭ ок-
сид скандия образует соединения 1
: 1 (LnScCh) с температурой плав-
ления > 2000 °C и ограниченные
твердые растворы, с тяжелыми
РЗЭ — образует кубические твер-
дые растворы; в системе с Y2O3 ку-
бический твердый раствор при
температуре ниже 1700 °C распа-
дается с образованием соединения
У&Оз. Соединение 8сА10з устой-
чиво только выше 1730 °C. Оксид
скандия не образует соединений
типа алюминиевого (R3AI5O12) и
галлиевого гранатов (КзСа5О|2),
Рис. 6.1. Система скандий—кислород
но в известных пределах может замещать в галлий-гадолиниевом гра-
нате галлий (см. гл. 5). С оксидами элементов IV группы (Ti, Zr, Hf)
оксид скандия образует соединения различных составов. Наибольший
интерес представляет система с ZrO2, в которой образуется несколько
соединений и твердые растворы (ТР), в том числе ТР с кубической
структурой типа флюорита, что позволяет использовать SC2O3 для ста-
билизации ZrC>2. С оксидами элементов V группы (V, Nb, Та)гО5 обра-
зуются соединения 1 : 1 (ScMO4), из которых ортотанталат БсТаОд
плавится конгруэнтно при 2340 °C. Соединение Са5сгО4 получено из
эквимолярной смеси оксидов при 1200—1400 °C. Твердофазный синтез
цирконата скандия протекает при 1400—1800 °C. Для получения моно-
алюминатов скандия необходима температура 1600—1700 °C, однако
процесс взаимодействия оксидов алюминия и скандия начинается при
900—950 °C (Тпл 8сА10з = 1870 °C). Оксид скандия не реагирует с
ниобием, молибденом и вольфрамом до 2300 °C, с танталом до 2250 °C,
с углеродом — до 1900 °C.
Гидроксид скандия — амфотерное соединение. Из раствора выделя-
ется в виде белого студенистого осадка (аморфного или кристаллическо-
го — в зависимости от условий) при действии аммиака или разбавлен-
ного раствора гидроксида натрия на растворы солей скандия; pH начала
выделения осадка 4,8—4,9. В начале осаждения образуются малораст-
воримые основные соединения, которые при увеличении концентрации
ОН- переходят в Sc(OH)3- Значения pH осаждения этих соединений
299
100 200 300 400 500 000 700 000
NaOH, r/n
РИС. 6.2. Растворимость в системе
Sc(OH)3—NaOH—Н2О при 25 °C
по данным различных авторов:
1 — Б.Н. Иванова-Эмина и др.,
2 — Л.Н. Комиссаровой и др.; 3 —
Л В. Фаворской и др.
зависят от природы аниона, определяю-
щей прочность связи скандий—анион
кислоты.
Кислотная диссоциация гидратиро-
ванного иона [Sc(H2O)e]3+ -»
X [Sc(OH)(H2O)5]2+ + Н+ сопровож-
дается полимериза-
цией \2[Sc(OH)(H2O)5]2+ *
£ [Sc2(OH)2/H20)io]4+ образованием
полиионов [Sc(OH)2Sc]n<n+4)+, нахо-
дящихся в равновесии с мономерными
ионами [Sc (ОН) ]2+и [Sc(OH)2]+. Амор-
фный гидроксид скандия, обработанный
концентрированным раствором NaOH,
переходит в кристаллическую форму.
Этому же способствует длительная вы-
держка гидроксида при нагревании.
Кристаллическая форма Sc(OH>3 хоро-
шо фильтруется.
Гидроксид скандия малорастворим
(рис. 6.2) ~ 7 10 5г/л SC2O3; произведе-
ние растворимости Sc(OH)3 2,0 10 3 .
При нагревании на воздухе гидроксид скандия ступенчато обезвожива-
ется: при 200—260 °C образуется ScO(OH), при 450—500 °C — SC2O3.
Первый легко растворяется в концентрированном растворе NaOH. Из
раствора кристаллизуется Na [Sc (ОН) 4] 2НгО. Растворимость гидро-
ксида скандия в водных растворах некоторых щелочей и карбонатов
представлена в табл. 6.1.
Гидроксид скандия в щелочном растворе частично присутствует в
коллоидной форме, причем пептизатором может быть скандиат-ион.
Ряд примесей — SiO2, NH4OH, NaCl — при концентрации NaOH до 400
г/л увеличивает переход гидроксида скандия в раствор. В присутствии
карбоната, фторида и оксалата аммония полнота осаждения гидроксида
скандия уменьшается из-за образования растворимых комплексных со-
единений скандия. Малорастворимые гидроксиды Fe(OH)3, Са(ОН)2
при любой щелочности резко уменьшают растворимость гидроксида
скандия.
300
Таблица 6.1. Растворимость гидроксида скандия
в водных растворах МОН, М2СОЗ, МНСОз при 25 'С
Реагент Концентрация, моль/л Содержание Sc(OH)3 в растворе, г/л Состав твердой фазы
NaOH NH4OH (NH4)2CO3 NH4HCO3 Na2CO3 NaHCO3 K2CO3 Na2CO3 + NaOH KOH -2 8,1 10 0,22—0,29 -2 1,4 10 — -2 —4,2 10 13,7 10‘2— —0,36 4,16 io'4— -2 —15,5-10 -3 2,02 10 — -2 —15,7 10 -3 6,6 10 — —1,81 2 8,8 10 — —0,92 5.7210'2— —2,52 Na+: СОз2'- -(1,5—2,5) 1 Na+: CO32'- - (3—3,5) . 1 13,5 -2 1,6 10 0,5—3,2 5 10'5 (0,1-0,2) 10'3 0,15—11,23 _2 0,9 10 —1.15 -3 3 10 —1,50 0,14—16,7 0,13—2,9 5,32—2.76 -3 (2,2—5,8) 10 2,12 (Sc203) Sc(OH)3 пН2О Na3Sc(OH)6 2Н2О Sc(OH)3 nH2O Sc(OH)3 пН2О NH4Sc(CO3)2 nH2O Тоже Основные карбонаты скандия переменного состава Не опред
6.3. Соли кислородсодержащих кислот
Сульфат скандия Sc2(SO4)3 иНгО выделяют упариванием серно-
кислых рас 1 воров, полученных растворением оксида., гидроксида или
карбоната скандия в серной кислоте. Из раствора выделяются кристал-
логидраты, содержащие 2; 4 и 6 молекул воды. При нагревании он
сначала обезвоживается, а затем разлагается:
100 °C 250 °C 550 °C
Sc2(SO4)3 6Н2О-------* Sc2(SO4)3 2Н2О-------► Sc[Sc(SO4)31---•
550"С 600 "С
-----’ Sc2O(SO4)2-----* Sc2O3. (6-1)
В растворе в зависимости от концентрации сульфат-иона образуют-
ся катионные [ScSO4]+ и анионные [Sc(SO4) 2] h[Sc(SO4)3]3 комплек-
301
сы. Анионные комплексы появляются при небольшом избытке ионов
[SO42 ], с повышением их концентрации анионные комплексы стано-
вятся доминирующими. Об этом свидетельствует сорбция скандия из
растворов с концентрацией серной кислоты более 0,25 моль/л, хотя при
дальнейшем увеличении ее концентрации из-за конкуренции с суль-
фат-ионами сорбция скандия прекращается. Катионные формы комп-
лексов наблюдаются при концентрации серной кислоты не более
1 моль/л.
При растворении сульфата скандия в воде происходит процесс авто-
комплексообразования
Sc2(SO4)3X Sc3+ <7<?+[Sc(SO4>313 - (6.2)
Растворимость Sc2(SO4)3 в серной кислоте приведена ниже:
H2SO4, г/л. . .. 0,0 24,5 49,0 121,5 243,3
Sc2(SO4)3, % 28,53 29,50 19,87 8,36 1,32
Сульфат скандия образует комплексные соединения М [Sc(SO4) 2 ] с
сульфатами лития и натрия и Мз[8с(5О4)з] — с сульфатами калия,
рубидия, цезия и аммония. Комплексные сульфаты скандия и аммония
или натрия хорошо растворяются в воде и концентрированных раство-
рах (NH4)2SO4 и Na2SO4- Комплексные сульфаты скандия и калия,
рубидия, цезия малорастворимы в воде и растворах соответствующих
M2SO4- Это используют для отделения скандия от элементов иттриевой
подгруппы: Кз[У(5О4)з] растворим в 30 раз лучше соответствующего
соединения скандия.
Основной тиосульфат скандия Sc(OH)S2O3 получают действием
тиосульфата натрия на растворы солей скандия
ScC13 + Na2S2O3 + Н2О---• Sc(OH)S2O3 + NaCl + HC1. (6.3)
Осадок представляет собой хорошо фильтруемые желтые кристал-
лы. Это эффективный метод отделения скандия от лантаноидов, при pH
2 лантаноиды остаются в растворе.
Нитрат скандия Sc(NO3)3 4НгО выделяют путем упаривания
азотнокислого раствора. Это кристаллическое гигроскопичное вещест-
во, хорошо растворимое в спирте, эфире и других органических реаген-
тах. В системе SC2O3—N2O5—Н2О при О °C и 25 °C в зависимости от
концентрации HNO3 из водных растворов выделены
ScOH(NO3)2 3H2O;Sc(NO3)3 4НгО; Sc(NO3>3 ЗН2О; Sc(NO3)3 2НгО;
Sc(NO3>3 2HNO3. Все соединения хорошо растворяются в воде. Основ-
ной и средний нитраты скандия при растворении в воде гидролизуются.
Растворимость 8с(ЫОз)з 4НгО в воде:
7, °C 0 15 25 30 40 50
Sc(NO3)3, %......56,34 61,27 62,36 64,25 66,96 67,60
302
Остальные кристаллогидраты растворяются инконгруэнтно.
Нитрат скандия имеет невысокую термическую устойчивость; при
нагревании происходит его термическое разложение с выделением ок-
сидов азота и образованием оксонитратов, а затем и оксида. Стадии
этого процесса:
120 °C 160 "С 190 °C
Sc(NOa)3 * [Sc2(NO3)4]O ' Sc(NO2)O *
190 °C 435 °C
* Sc4<NO3)2O5 ’ SC2O3. (6.4)
В нитратных и азотнокислых растворах скандия образуются катион-
ные и нейтральные комплексы типа Sc(NO3)/3 1 , где i= 1, 2, 3. Анион-
ная сорбция из азотнокислых растворов не наблюдается.
Карбонаты скандия. Скандий образует основные карбонаты пере-
менного состава [8с(ОН)т]2(СОз)з-/п ЗН2О, представляющие собой
белый объемный осадок, выделяющийся при действии (ЬШ^гСОз или
№гСОз на растворы солей скандия. Так, Sc(OH) СОз ЗН2О получен при
добавлении раствора №гСОз к раствору ScC13 или путем взаимодейст-
вия SC2O3 и растворов М2СО3. Основные карбонаты скандия растворя-
ются в растворах (NH<i)2CO3 или №гСОз лучше, чем аналогичные
соединения РЗЭ, образуя комплексные соединения, которые мало рас-
творимы в воде и слабокислых растворах
Sc3+ + 4Na2CO3 Z Na5[Sc(CO3)4]- «Н2О + 3Na+; (6.5)
Sc3++2(NH4)2CO3Z NH4[Sc(CO3)2] nH2O+3NH4+. (6.6)
На этом основан метод карбонатной очистки скандия от РЗЭ, тория,
железа, марганца и кальция. Устойчивость карбонатных комплексов,
состав и кинетика образования зависят от природы внешнесферного
катиона, pH раствора и температуры, соотношения СОз : Sc .
Фосфат скандия. Ортофосфат скандия ScPO4 2НгО получают дей-
ствием на водные растворы солей скандия фосфорной кислоты. В отли-
чие от фосфатов РЗЭ фосфат скандия выделяется из раствора только в
виде дигидрата. Его дегидратация происходит при температуре выше
300 °C. Известен дигидрофосфат Sc(H2PO4>3, разлагающийся при на-
грсвзнии
200 °C 1200 °C
Sc(H2PO4)3-------° [Sc(PO3)3]n-------' ScPO4. (6.7)
Скандий образует также труднорастворимое соединение с фитино-
вой кислотой СбНбО[ОР(ОН)з ]б. Фитат скандия осаждается из раство-
ров, содержащих 30 мг/л Sc. Нерастворим в растворах соляной и азотной
кислот и в царской водке, устойчив по отношению к растворам щелочей
и карбоната аммония, в то время как фитаты тяжелых металлов в них
растворяются. Для перевода скандия в раствор из этого соединения его
сплавляют со щелочью.
303
Молибдаты и вольфраматы. При взаимодействии растворов солей
скандия с растворами нормальных молибдатов и вольфраматов аммония
(NH4)2MoO4 и (NH4) WO4 (оба этих соединения в твердом виде сущест-
вуют только в виде парамолибдатов и паравольфраматов аммония —
(NH4)6Mo(W)7C>24 иНгО) образуются молибдат Sc2<MoO4) и вольфра.
мат скандия Sc2(WO4) при pH 2,5—5,0. Их можно получить также
спеканием соответствующих оксидов. С нормальными молибдатами и
вольфраматами щелочных металлов образуются двойные молибдаты и
вольфраматы M[ScMo(W)O4)2], где М — Li, Na, К, Rb (Тпл 990, 660 и
1170 °C соответственно). В форме вольфрамата скандий входит в состав
некоторых видов сырья.
Свойства сульфатов, хроматов, молибдатов, вольфраматов скандия
изменяются в зависимости от природы анионов. Так, растворимость
в воде и склонность к гидролизу соединений скандия уменьшается в ряду
СгО4 > SO4 > МоО4 > WO4 . Термическая устойчивость этих
соединений увеличивается в обратной последовательности. В этой
же последовательности (обратной) увеличивается и прочность связи
Sc3+— (L—СгО42- WO42").
Свойства двойных вольфраматов (молибдатов, сульфатов, хрома-
тов) изменяются не только от природы аниона, но и от природы катиона.
В случае однозарядных катионов устойчивость этих соединений увели-
чивается в ряду от Li к Cs.
6.4. Соединения с галогенами
Скандий взаимодействует непосредственно со всеми галогенами.
Однако его галогениды (кроме фторида) получают в основном косвен-
ными методами. Галогениды выделяются из раствора в форме кристал-
логидратов.
Фторид скандия ScFa получают действием плавиковой или кремне-
фтористоводородной кислоты, фторидов щелочных металлов на рас-
твор, содержащий скандий, действием фтора на оксид или металли-
ческий скандий.
При нагревании на воздухе ЗсРз выше 650 °C переходит в ок-
сид. Гидролиз в токе влажного азота приводит к образованию ScOF при
800 °C.
ScFa — слабо растворим в воде и минеральных кислотах (FIPscF3 г
3 1 0 20). При обработке ScFa концентрированной серной кислотой при
200—300 °C он превращается в сульфат; при кипячении ScF3 в 20—30
%-ном растворе NaOH или сплавлении с NaOH — в гидроксид.
В плавиковой кислоте, растворах фторидов щелочных металлов и
аммония ScF3 растворяется, образуя комплексную кислоту Нз|ЗсРб)
304
йли комплексные фториды М [ScF4 ], М2 [ScFs ], М3 [ScFe ]. Фторсканди-
аТы натрия и калия слабо растворимы в воде, но растворимы в разбав-
ленных кислотах. Растворимость ScFa в воде и минеральных кислотах
меньше, чем фторидов РЗЭ. Для отделения скандия от РЗЭ и иттрия
используют реакцию
ScF3+3NH4F=(NH4)3ScF6. (6-8)
Безводный фторид скандия — основное исходное соединение для
получения металла.
Хлорид скандия ScCh может быть получен нагреванием оксида скан-
дия в присутствии углеродсодержащего восстановителя в токе хлора,
взаимодействием SC2O3 с СС14 или NH4C1 или обезвоживанием
ScCh (6—9)НгО в токе сухого HCI; ScCh — белое кристаллическое,
гигроскопичное вещество, на воздухе расплывается. ScCh 6Н2О при
нагревании на воздухе превращается в оксохлорид ScOCI, плохо раство-
римый в воде, кислотах и щелочах.
При 800—850 °C ScCh возгоняется, что может быть использовано
для его отделения от других хлоридов.
ScCh растворяется в воде, спирте и растворах НС1, что используют
для очистки скандия от алюминия, иттрия, лантаноидов.
Растворимость хлоридов в 100 мл насыщенного хлористым водоро-
дом раствора при 0 °C и 0,1 МПа приведена ниже, г/100 мл (в пересчете
на оксид, указанный в скобках):
Элемент Водный раствор НС1
А1 0,0015 (А12ОЗ)
Sc..... >1,5(Sc2O3)
Y......... 0.024 (Y2O3)
Эфирно-водный раствор НС1
0,0003 (А12ОЗ)
> 4 (Sc2O3)
0,0015 (Y2O3)
Из солянокислого раствора выделяются кристаллогидраты
ScCh 6Н2О и БсС1з 9НгО при 6,8 и 2,13 моль/л НС1 соответственно
(рис. 6.3). Солянокислые растворы скандия используют для его экстра-
кционного извлечения и очистки различными органическими раствори-
телями Безводный ScCh образует комплексные соединения с хлорида-
ми щелочных металлов — МзБсОб (М = К, Na, Rb, Cs). Комплексные
хлориды скандия менее гигроскопичны, чем его хлориды, и могут быть
ж пользованы для получения металла.
Бромид и иодид скандия ScBf3, Sch — кристаллические весьма гиг-
роскопические вещества. Сублимация происходит соответственно
при 933 и 912 °C, плавление — при 970 и 953 °C.
Безводные соли получают действием элементарных брома и иода на
металлический скандий или галогенводородов на ScCh при нагревании
ScCh + ЗНХ------- ScX3 + ЗНС1, (6.9)
где X = Br, 1.
305
рис. 6.3. Изотерма 298 К раствори-
мости в системе ScC13—НС1—Н2О
Йодат скандия 5с(Юз)з 1,5НгО об-
разуется при добавлении раствора нит-
рата скандия к разбавленному раствору
йодноватой кислоты. Используют в тех-
нологии для очистки скандия от тория и
циркония, йодаты которых
Th(Zr) (Юз) 4 в отличие от йодата скан-
дия, плохо растворимы.
Тиоцианаты скандия Sc(NCS)3 по-
лучают взаимодействием сульфата
скандия и тиоцианата бария в раство-
ре. Известно соединение HSc(NCS>4,
используемое для экстракционной очи-
стки скандия. С тиоцианатами щелоч-
ных металлов и аммония тиоцианат
скандия образует комплексные соедине-
ния типа M3[Sc(NCS)61, которые полу-
чают по реакции
Sc2(SO4)3+3M2SO4+6Ba(NCS)2------ 2M3[Sc(NCS)6]+BaSO4. (6.10)
Мз [Sc(NCS>6 ] — гигроскопические соединения, связь Sc—NCS осу-
ществляется через азот. В ряду Li+—Na+—К+—Rb+—Cs+ с увеличением
радиуса внешнесферного катиона растворимость комплексов в воде
Уменьшается (с 78 до 70 %), а термическая устойчивость возрастает.
। 6.5. Органические производные скандия
Скандий, как и РЗЭ, образует соединения с органическими кисло-
тами (щавелевой, уксусной, винной, лимонной и др.). Карбоновые и
Ьксикарбоновые кислоты были первыми комплексообразователями,
примененными для разделения скандия, иттрия и РЗЭ. В настоящее
время используют полиуксусные кислоты: нитрилуксусную, этиленди-
аминтетрауксусную и др. (см. гл. 5).
Окса пот гглиЛ/ я .§С2(С2О4)з- лН2О (.•г=3,4,5,6,18) образуется ПрИ
Действии щавелевой кислоты на нейтральные или слабокислые растворы
далей скандия. Осаждение при 60 °C дает устойчивый при обычных
условиях Sc2(C2O4)3 6Н2О; при длительном кипячении образуется во-
;смнадцативодная соль, осаждение при 8 °C приводит к образованию
григидрата. Как правило, снижение температуры кристаллизации при-
водит к повышению гидратного числа, например для РЗЭ. Для гидратов
Оксалата скандия обнаружена обратная зависимость.
Безводный оксалат — бесцветное, кристаллическое, гигроскопиче-
ское соединение, получаемое дегидратацией кристаллогидратов
06
100—190 "С 190—280 °C
SC2(C2O4>3 6Н2О ------------* SC2(C2O4>3 2НгО *
190—280 "С 280—600 "С
----------- 5с2(СгО4)з----------- SC2O3. (6.11)
Оксалат скандия, как безводный, так и кристаллогидрат, мало рас-
творим в воде: 60 мг/ л (в пересчете на SC2O3) при 25 °C. Оксалат скандия
образует комплексные оксалаты с оксалатами аммония, натрия и калия
Мз [Sc (С2О4) з 1 5НгО. Эти соединения растворяются в воде, легко гид-
ролизуются.
Растворимость оксалата заметно повышается в присутствии мине-
ральных кислот, оксалата аммония и избытка щавелевой кислоты (табл.
6.2). При этом в растворе образуются комплексные ионы [8с(СгО4) ]+,
[Sc(C2O4)2]~, [5с(СгО4)з]3-, [SCC2O4SO4]~. Так, из растворов с кон-
центрацией скандия 1 г/л увеличение количества Н2С2О4 от теоретиче-
ского до 20-кратного приводит к уменьшению степени осаждения
скандия от 88,9 до 59,1 %. При недостатке С2О42 по отношению к
стехиометрии могут быть получены основные оксалаты, например
SCOHC2O4 1,5НгО; его растворимость 44,2 мг/л (20 °C).
Таблица 62. Растворимость оксалата скандия
Концентрация кислоты, % 7, °C Растворимость (в расчете на 5с2Оз),г/л
В воде 25 0,061
1 ВНС1 25 0,413
10 25 7,540
20 25 13,870
15 60 21,600
10 В HNOQ 25 8,170
20 25 17,360
10 50 32,800
5 В H2SO4 25 0,861
15 25 9,730
5 50 7,050
307
Ацетилацетонаты скандия (ААС) 5с(СНзСОСНСОСНз)з осажда-
ют из водного раствора соли скандия добавлением к нему спиртово-
го раствора ААС с дальнейшей нейтрализацией аммиаком. ААС —
устойчивое кристаллическое соединение, плавится без разложения при
— 187 ”С. Хорошо растворим в органических растворителях (спирты
эфиры, ацетон, хлороформ, бензол).
ААС возгоняются без разложения при ~ 200 °C. В этих условиях
соответствующие соединения лантаноидов разлагаются. По термиче-
ским свойствам ААС отличается от ацетилацетонатов Be, Zr, Hf, Th.
Указанные различия используют для очистки скандия. ААС можно ис-
пользовать для получения пленок SC2O3 или соответству ющих компози-
ций.
6.6. Соединения с неметаллами
Халькогениды скандия. Сульфид скандия SC2S3 получают сплавле-
нием элементов, прокаливанием SC2O3 в токе H2S, обработкой безвод-
ных сульфатов или ScCh сероводородом при повышенной температуре.
SC2S3 — желтое кристаллическое вещество, плотность 2,80 г/см3, тем-
пература плавления 1700 °C. До 100 °C устойчив на воздухе. При высо-
кой температуре на воздухе сульфид разлагается
2SC2S3 + 2IO2------* 2Sc2O3 + 2Sc2(SO4)3 + 3SO2- (6.12)
Горячая вода разлагает SC2S3 с выделением H2S
SC2S3 + 6H2O ------- 2Sc(OH)3 + 3H2S| . (6.13)
Селенид скандия Sc2$c3, теллуриды 8сгТез, ScTe получают сплав-
лением скандия с селеном или теллуром. Селенид и теллурид — кри-
сталлические тугоплавкие вещества: ВсгЗез — красно-фиолетовый,
5сгТез — черный. Нерастворимы в воде, при действии минеральных
кислот разлагаются, выделяя НгБе или НгТе.
Соединения скандия с углеродом, азотом, водородом, бором и крем-
нием следует отнести к фазам внедрения, закономерности образования
которых рассмотрены в гл.5.
Карбиды скандия. Известны SC3C, Sc2C, SC4C, ScC и др., из которых
более подробно изучен ScC, получающийся восстановлением БсгОз У1”
леродом при 2000 °C или синтезом из элементов
Sc2O3 + 5С------ 2ScC + ЗСО. (6.14)
Карбид скандия — темно-серое с металлическим блеском вещест-
во, кристаллизуется в кубической решетке, температура плавления
1800 °C, микротвердость 26,7 ГПа. ScC склонен присоединять кислород-
образуя оксокарбиды состава SC2C2O; растворяет углерод, образует
твердые растворы с карбидами других элементов. Твердые растворы на
основе системы TiC—ScC отличаются высокой микротвердостью, РаВ'
308
ной 53,4 ГПа, существенно превышающей микротвердость карбида ти-
тана — 29,5 ГПа.
Нитрид скандия ScN получают взаимодействием скандия с азотом
при температурах до 900 °C. Нитрид скандия — твердое темно-синее
вещество, имеет ГЦК решетку, температура плавления 2600± 50 °C,
имеет полупроводниковые свойства. Микротвердость ScN, содержащего
1,2 % (мол.) ScC, составляет 11,5 ГПа. Нитрид устойчив на воздухе до
600 °C; при дальнейшем нагревании быстро окисляется; устойчив в
холодной и горячей воде, разбавленных растворах минеральных кислот
и щелочей. Концентрированные кислоты быстро разлагают ScN.
Силицид скандия ScSi получают взаимодействием SC2O3 с Si в ваку-
уме в интервале температур 1000—1900 °C
2Sc2O3 + 7Si------ 4ScSi + 3SiO2. (6.17)
Гидрид скандия 8сНз получают при нагревании металлического
скандия в атмосфере водорода. Это твердое кристаллическое вещество,
обладающее при повышенной температуре заметным давлением пара.
Например, при 600 и 900 °C давление пара равно соответственно
7,34 10и 34,10-10“3 МПа.
Диборид скандия ScB2 — серое кристаллическое вещество
Гпл = 2250 °C; нерастворим в воде и органических растворителях. Его
получают взаимодействием Sc2O3 с В или с В4С. Используют как ком-
понент легких жаропрочных сплавов.
6.7. Сплавы, легированные скандием
Скандий используют как легирующую добавку для легких сплавов,
главным образом на основе алюминия и магния. Дороговизна скандия
нс позволяет использовать его как основу сплавов. Фазовые диаграммы
систем, образованных скандием с другими металлами, построены для
ограниченного числа случаев. Тем не менее известны многие интерме-
таллические соединения скандия с металлами IB, ПВ, VIIB, VIIIB, ПА,
IIIA, IVA, VA подгрупп. Из числа известных интерметаллических сое-
динений скандия около 70 % имеют температуру плавления ниже, чем
скандий. Выявлены и тугоплавкие соединения — ScjRc4 (2575 °C),
ScRe2 (2035 °C), ScRu2 (1840 °C), ScFe2 (1600 °C).
Особый интерес представляют сплавы А1—Sc, Mg—Sc, Mg—Sc—Li,
Mg—Y—Sc. Добавление десятых долей процента скандия к алюминию
и его сплавам обусловливает повышение прочностных и пластических
свойств, рост сопротивления коррозионному растрескиванию, улучше-
ние свариваемости. При кристаллизации расплавов большая часть скан-
дия входит в пересыщенный твердый раствор, а оставшаяся выделяется
в форме AI3SC (рис. 6.4), который обусловливает измельчение зерна.
309
Рис. 6.4. Участок диаграммы плав-
кости А1—Sc
Легирование сплавов Al—Mg (2—8,5
Mg) 0,4 % скандия увеличивает времен
ное сопротивление на 20—35 %, а пре
дел текучести на 60—80 %. Относи,
тельное удлинение при этом остается до.
статочно высоким (15—20 %).
Скандий оказывает заметное влия
ние и на свойства других сплавов. Добав-
ка скандия к хрому (0,6 %) увеличи-
вает его устойчивость к окислению д0
1200 °C, а к меди и цирконию — их твер-
дость. Введение в железо 0,3—0,6 %
(ат.) Sc увеличивает его термический ко-
эффициент расширения. Присадки
скандия к ванадию повышают темпера-
туру перехода ванадия в сверхпроводя-
щее состояние. Для циркония
установлена обратная зависимость. По-
ка не выявлено сколько-нибудь общих
закономерностей влияния скандия на свойства легированных им спла
вов, однако имеющиеся данные показывают возможность регулирова-
ния некоторых свойств сплавов легированием скандием.
ТЕХНОЛОГИЯ СКАНДИЯ
6.8. Важнейшие области применения скандия
и его соединений
Возможности применения скандия ограничены объемами его произ-
водства и высокой ценой. Тем не менее ряд уникальных свойств скандия
и его соединений постоянно расширяет сферу его применения.
Лазерная и вычислительная техника, электроника. Скандий вхо-
дит в состав ряда материалов структуры граната, используемых в лазер-
ной технике: гадолиний-скандий-галлиевый гранат (ГСГГ).
алюминий-скандий-галлиевый гранат (АСГГ), иттрий-скандий-галли-
евый гранат (ИСГГ). Указанные материалы содержат значительные
количества скандия (до 30 % в пересчете на SC2O3). Они использу-
ются в качестве лазерных и подложечных материалов для запоминаю-
щих устройств. Лазерная техника на основе гранатов, включаюШиХ
скандий, применяется в средствах связи, лазерных устройствах ДлЯ
310
1аВКи металлов, системах противоракетной обороны, низкоэнергети-
цеских лазерных системах (медицина, электроника).
Важной областью применения скандия было и остается его исполь-
зование в производстве ферритов с малой индукцией для ЭВМ. Она
примерно в 3 раза меньше, чем у ферритов на основе оксидов железа,
магния, марганца. Скандиевые ферриты меньше нагреваются при пере-
магничивании, что увеличивает быстродействие ЭВМ. Скандий и его
оксид — перспективные материалы для применения в эмиссионной
электронике, позволяющие повысить эмиссионную способность като-
дов.
Скандий входит в состав ряда люминофоров, он может быть актива-
тором и компонентом основы. Для использования в электронике и ла-
зерной технике необходим оксид скандия высокой чистоты.
Стекло и керамика. Оксид скандия используется для производства
специальных видов керамики и стекла, где он является модификатором
и стеклообразователем. Добавка оксида скандия (десятки процентов)
повышает показатель преломления силикатного и боросиликатного
стекла, увеличивает дисперсию, что существенно влияет на конструк-
' цию и качество приборов. Используют оксид скандия и для производства
германатных стекол.
Оксид скандия обладает рядом преимуществ по сравнению с тради-
ционными керамическими материалами: оксидами алюминия, бария,
магния и циркония. Он обладает максимальным значением прочности
при 1000 °C. Коэффициент теплового расширения и модуль упругости у
SC2O3 минимальные; в то же время оксид скандия имеет самую низкую
теплопроводность в ряду указанных оксидов. Оксид скандия обладает
высокой термостойкостью.
Керамика на основе оксидов циркония и гафния, модифицированная
оксидом скандия, может быть использована при высоких температурах.
Она обладает лучшими характеристиками, чемкерамика, стабилизиро-
ванная Y2O3. Превосходными свойствами для высокотемпературных
систем обладает керамика из скандата иттрия (YSc-Оз). Предложено
использовать оксид скандия как компонент ВТСП-керамик, особенно
системы Y(La)—Ва—Си—О.
Светотехника. Разработаны металлогалогенидные ртутные лампы
с добавками скандия. Они успешно зарекомендовали себя в осветитель-
ных установках промышленных и общественных зданий и стали наибо-
лее употребительны среди ламп этого типа. Предложено использовать
оксид скандия в ртутных лампах как один из компонентов герметизиру-
ющего материала.
311
Сплавы. Скандий перспективен как конструкционный материал ддя
самолетостроения, астронавтики, производства снарядов, так как он
обладая в 2,5 раза более высокой температурой плавления, чем алюми-
ний, имеет близкую плотность. Особый интерес в этом отношении пред,
ставляют легкие сплавы, главным образом на основе алюминия и
магния. Весьма перспективным при снижении цены на скандий может
оказаться использование низколегированных скандиевых сплавов в ав-
томобилестроении. Указывается, что Ga—Sc-связка может найти при-
менение для металлических клеев и покрытий. Для использования
в конструкционных сплавах не нужен скандий высокой степени чисто-
ты. Однако использование скандиевых сплавов реально пока лишь для
особо ответственных деталей из-за высокой стоимости скандия.
Другие области применения. Для контроля ряда химических, ме-
таллургических и других процессов и исследований применяют радио-
активный изотоп 46Sc в качестве метки, а также для лечения раковых
опухолей. Предложено использовать изотоп 47Sc как излучатель пози-
тронов. Есть данные об использовании соединений скандия как катали-
заторов при получении некоторых органических продуктов. Легиро-
вание карбида титана карбидом скандия позволяет получить материал,
по твердости приближающийся к алмазу. Диборид скандия ScB2 предло-
жено использовать как компонент жаропрочных сплавов, а также в
материалах катодов электронных приборов. Гидрид скандия использу-
ют в ядерной технике как высокотемпературный замедлитель нейтро-
нов, в нейтронных генераторах.
В Японии структура потребления оксида скандия оценивается сле-
дующим образом, %: металлогалогенные осветительные лампы 50;
цветные кинескопы 20; лазерные материалы 20; исследования (автомо-
бильные фильтры, ВТСП и др.) 10.
Данные об объемах производства скандиевой продукции не публи-
куются. Основные ее формы — оксид скандия и металл. Оксид скандия
выпускают чистотой 99,9—99,999 %, металлический скандий 99,9—
99,999 %.
Ня основе отдельных данных о потреблении можно предположить,
что производство оценивается сотнями килограммов в пересчете на
БсгОз. Производство оксида скандия в мире по данным 1988 г., кг/год:
КНР 50—60; Франция 100; Норвегия 120; США 500; Япония — более
30.
Цены в 1991 г. по данным Горного бюро США, тыс. долл/кг: Sc?03
99,9 % — 3,5; 99,999 % — 10,0; металлический порошок крупностью
250 мкм: дистиллят 99,9 % — 296; куски дендритов 99,9 % — 248.
312
6.9. Сырьевые источники скандия
Скандий — типичный рассеянный литофильный элемент. Содержа-
ние его в земной коре 6 10 4 % (поданным А.П. Виноградова). Несмотря
на то что скандий более широко распространен в природе, чем Sb, Bi, Ag
и Au, значительных скоплений он не образует. Основная его масса
рассеяна в изверженных горных породах; содержание ЗсгОз в них
0,0002-0,0003 %.
Собственно скандиевых минералов известно лишь два: тортвейтит и
стереттит. Тортвейтит SczfSizO? ] — дисиликат скандия, содержит 41—
42 % SC2O3. Скандий в нем обычно замещается иттрием и РЗЭ иттриевой
подгруппы, цирконием, гафнием и торием. Оба минерала крайне редки
и найдены в виде отдельных скоплений в Норвегии, на о. Мадага-
скар, на Урале и в Восточной Сибири. Стереттит ScPO4 2НгО содер-
жит до 39 % SC2O3.
Известно более 100 минералов, в которых скандий присутствует в
виде изоморфной примеси с содержанием 0,005—0,3 % SC2O3. Для боль-
шинства скандийсодержащих минералов характерно присутствие ионов
Ln3+, Fe2+, Mn2+, Mg , Са , Zr4+, U4+. Большинство этих ионов и ион
Sc3 имеют близкие ионные радиусы при одинаковом для всех коорди-
национном числе (6) и близкие значения других констант. Отсюда —
замещение скандием в природных условиях указанных элементов.
В гранитных пегматитах скандий накапливается вместе с редкозе-
мельными элементами иттриевой подгруппы, входя в состав титано-
тантало-ниобатов (эвксенит, самарскит, хлопинит и др.) и силикатов
(иттриалит, гадолинит) РЗЭ. В пневматолито-гидротермальных про-
цессах Sc концентрируется вместе с Be, W, Мо и Sn, входя в минералы
вольфрамит, касситерит, ферримусковит, цинвальдит, берилл.
Месторождения указанных минералов крайне невелики и не обеспе-
чивают его сырьевую базу. Проблема промышленного производства
скандия может быть решена при комплексной переработке руд редких и
цветных металлов, содержащих рассеянный скандий (табл. 6.3). Эти
руды являются основными для производства скандия как в России, так
и за рубежом. Потенциальным источником скандия следует считать
каменные угли, в золе которых его содержится около 0,001 %.
Заметными запасами скандия располагает Норвегия, существенно
большими Китайская Народная республика и США. Указанные страны,
а также Франция и Япония имеют мощности по производству скандия.
При оценке сырьевых источников для извлечения скандия следует
оценивать не столько их запасы (по запасам первое место занимают
природные угли), сколько освоенность технологии переработки сырья,
распределение скандия по операциям и точки его концентрирования в
технологических схемах производства основных металлов.
313
И — 691
Таблица 6.3. Характеристика важнейших
сырьевых источников скандия
Минеральное сырье Объем переработки, тыс т/год Содержание SC2O3, % Максимальное содержание SC2O3, % — Общее количество Sc2O3 в сырье, т/год
Бокситы 71000 0,001—0,002 0,01 710—1420
Урановые руды 50000 0,0001—0,001 0,01—0,08 50—500
Ильмениты 2000 0,001—0,002 0,1 20-40
Вольфрамиты — 0,01 0,1—1.0 —
Касситериты 200 0,01 0,22—0,02 20
1 [ирконы 100 0,005—0,01 0,08 5—10
6.10. Общие вопросы технологии скандия
Получение соединений скандия — крайне сложная технологическая
задача, так как скандий извлекается попутно из комплексного сырья,
где концентрации сопутствующих элементов многократно превышают
концентрацию скандия. Рациональность попутного извлечения скандия
определяется его содержанием в исходном сырье, масштабами производ-
ства основного металла, степенью концентрирования в полупродуктах
и отходах производства. Делать выводы о поведении скандия в тех или
иных процессах на основании свойств его соединений не всегда возмож-
но, поскольку на его поведение оказывают влияние другие элементы,
присутствующие в сырье в значительно больших количествах.
При организации производства скандия как сопутствующего эле-
мента большое значение имеет распределение его по продуктам основ-
ного производства, выявление «точек» концентрирования. При кон-
центрировании скандия в отходах и промпродуктах производства созда-
ются благоприятные условия для его извлечения без нарушения основ-
ной технологии.
Содержание скандия в продуктах переработки минерального сырья
составляет от сотых до десятых долей процента. Поэтому вначале в
большинстве случаев из исходного сырья получают концентраты скан-
дия, которые затем перерабатывают на чистые его соединения. Природа
отходов различна — растворы, шлаки, шламы, фторидные продуктЫ’
314
ддавы хлоридов, гидроксиды. В случае твердых продуктов перевод скан-
дия в раствор осуществляют различными методами.
Для извлечения скандия из шлака в раствор необходимо его тонкое
измельчение. Для выщелачивания преимущественно используют соля-
ную кислоту, так как солянокислые растворы предпочтительны при
последующем осаждении оксалата, фторида или фосфата скандия, а
также при его экстракции нейтральными экстрагентами, особенно три-
бутилфосфатом (ТБФ). Сернокислотное разложение применяют реже.
Фторидные скандийсодержащие продукты сульфатизируют серной
кислотой при 150—180 °C или разлагают 15—30 %-ным раствором
едкого натра при 80—90 °C; фторид скандия переходит соответственно
в сульфат или гидроксид. При этом существенных потерь скандия со
щелочными растворами не происходит, несмотря на некоторую раство-
римость в них гидроксида скандия. Гидроксид скандия задерживается в
осадках гидроксидами железа, кальция и других элементов. При суль-
фатизации возможны потери скандия с гипсом.
Ряд скандийсодержащих продуктов разлагают методом хлорирова-
ния в присутствии восстановителя. При этом в зависимости от темпера-
туры хлорирования скандий в форме БсС1з может оставаться в огарке от
хлорирования или возгоняться. Способ применяли для разложения цир-
коновых концентратов, минерала тортвейтита, шлаков, содержащих
0,12 % Бс20зиО,82 % ВеО.
Для выделения скандия из растворов и отделения его от примесей
используют следующие основные способы: осаждение в составе малора-
створимых соединений; экстракция органическими реагентами, ионо-
обменные способы. При переработке твердых продуктов можно исполь-
зовать сублимацию и конденсацию. Однако ни один из указанных спо-
собов не является строго селективным для скандия. Получить богатые
скандиевые концентраты, и особенно соединения скандия высокой сте-
пени чистоты, можно, лишь сочетая указанные способы.
6.11. Методы первичного концентрирования скандия
Методы осаждения просты и позволяют достаточно полно, особенно
в присутствии соосадителя, выделить скандий из растворов. При низких
концентрациях скандия (50 мг/л) в отсутствие соосадителя методы не-
достаточно эффективны. Полнота выделения малых количеств скандия
из растворов, содержащих большие количества примесей, часто не до-
стигается. Высокая концентрация примесей заметно меняет поведение
скандия. Это явление называется «потеря химической индивидуально-
сти» элемента. Причиной этого явления являются процессы комп-
лексообразования в растворах, образование смешанных комплексов.
315
и*
При осаждении скандия в присутствии элементов, образующих рас-
творимые комплексные соединения с осадителем, а также при избытке
осадителя, уменьшается полнота его выделения.
Осаждение фторида. Для выделения скандия из бедных растворов в
качестве осадителя рекомендованы фториды и плавиковая кислота. До-
статочно полное осаждение скандия достигается при большом избытке
осадителя. При осаждении фторида скандий можно отделить от элемен-
тов, образующих хорошо растворимые фторидные комплексные соеди-
нения, например железа, титана, циркония, марганца.
Способность фторида скандия растворяться в растворе фторида ам-
мония с образованием фторскандиата в отличие от плохо растворимых
фторидов РЗЭ и тория можно использовать для разделения. Эффектив-
но проходит разделение при обработке суммы фторидов РЗЭ, Th и Sc
плавиковой кислотой: скандий переходит в раствор в форме Нз[ЗсРб],
РЗЭ и Th (фториды) остаются в осадке
SCF3 + NH4F---. (NH4)3[ScF]6- (6.16)
Осаждать скандий из растворов с высокой концентрацией алюминия
нецелесообразно, так как из-за осаждения алюминиевого криолита кон-
центрация скандия в осадке будет низкой.
Для осаждения скандия из циркониевых растворов рационально
использовать кремнефторид или фторид калия, так как образующийся
в этих условиях фторцирконат калия имеет высокую растворимость в
воде (~ 25 % при 100 °C). Недостатком методов осаждения скандия в
форме фторидов является трудность перевода скандийсодержащего
осадка в растворимое состояние.
Осаждение оксалата. При осаждении оксалата скандия
5с(СгО4)з 6Н2ОI возможно его отделение от железа и алюминия
Sc(NO3)3 + 2Н2С2О4 + 6Н2О----* SC2(C2t)4)3 6H2O| + 6HNO3. (6.17)
Однако при избытке Н2С2О4, солей аммония и щелочных металлов
осаждение скандия неполное из-за образования комплексного аниона
[Sc(C2O4)313 - Более полному осаждению оксалата скандия, особенно
из бедных растворов, способствуют соли кальция, являющегося коллек-
тором. Оптимальные условия: pH 2—3, Т=90 °C, продолжительность —
не менее 4 ч. Количество щавелевой кислоты и соосадителя определяют
экспериментально в зависимости от состава раствора.
Для отделения скандия от РЗЭ используют различие в устойчивости
комплексных соединений. Оксалаты скандия и РЗЭ растворяют в вод-
ном растворе ЭДТА (H4L) при pH 6
Sc2(C2O4)3 + 2H4L------ 2HScL + ЗН2С2О4. (6-18)
При кипячении разрушают менее прочные комплексы РЗЭ. После
охлаждения раствора добавляют 10 % Н2С2О4 и осаждают оксалаты
РЗЭ, комплекс скандия остается в растворе.
316
Осаждение карбонатов. Карбонат скандия растворяется в избытке
растворов МагСОз или (NH4)2CO3 с образованием комплексных соеди-
нений, что используют для очистки от РЗЭ, железа, марганца, кальция.
Карбонатный комплекс скандия разрушается кипячением — в осадок
выпадает малорастворимый карбонат скандия переменного состава. Со-
став карбонатных комплексов, выделенных в твердом состоянии, при-
веден в табл. 6.4.
Таблица 6.4. Условия образования комплексных
гидроксокарбонатов скандия
Исходные соотношения СОЗ2-: Sc3* pH Состав осадка
1,0 4,69 Sc4(OH)10CO3 пН2О
1,5 5,20 Sc4(OH)6(CO3)3 «Н2О
2,0 7,77 Sc4(OH)4(CO3)4 иН2О
Основные карбонаты полностью растворяются с образованием
карбонатных комплексов типа [8с(СОз)п](2п 3) (п > 2) при СОз2 :
Sc3+ > 8,35. В зависимости от условий могут существовать комплексы
[8с(СОз)гГ> [8с(СОз)з]3~, [8с(СОз>4]5 и [8с(СОз)б]9-. Увеличение
концентрации Sc3 в растворе приводит к стабилизации более сложных
полимерных форм гидроксокомплексов.
В ряду карбонатных комплексов соединения скандия проявляют
максимальную растворимость по сравнению с соединениями иттрия и
других РЗЭ.
Для наиболее полного извлечения скандия рекомендуется исходный
раствор с концентрацией 15—25 г/л (в пересчете на SC2O3) нейтрализо-
вать содой или аммиаком до pH 2, а затем постепенно при перемешива-
нии вливать его в равный объем 20 % -ного раствора соды при комнатной
температуре. После отделения осадка примесей раствор подкисляют
соляной кислотой, кипятят для удаления СО2 и осаждают с помощью
NH3 НгО гидроксид скандия. Недостаток карбонатной очистки: необ-
ходимость использования больших объемов растворов №гСОз или
(ЫН4)гСОз из-за низкой растворимости соединений скандия в них и
образование труднофильтруемых осадков.
Осаждение гидроксида. Осаждение Sc (ОН) з дает возможность отде-
лить скандий от щелочных и щелочноземельных элементов. При нейт-
рализации кислых растворов скандий начинает осаждаться из 0,1
моль/л растворов при pH 4,8—4,9 вначале в форме основной соли, а
затем гидроксида. Значительные потери скандия с фильтратом наблю-
317
даются при осаждении его аммиаком из растворов, содержащих карбо-
нат или фторид аммония. Поэтому возможно отделение скандия от
магния и кальция путем его выщелачивания из осадков гидроксидов
этих элементов 20 %-ным раствором (МН4)гСОз, а не только за счет
различий в pH осаждения гидроксидов этих элементов:
Осаждаемое соединение pH
Осаждаемое соединение pH
Sc(OH)3.............. 4,9—5,5
Ln(OH)3............. 6,3 и выше
Се(ОН)4................1,2
Се(ОН)3................7,4
ZrO(OH)2 хН2()........1,9—2.6
TiO(OH)2 хН2О..........0,7
Th(OH)4................> 3,0
SiO2' хН2О.......Сильнокислая среда
АКОН13...............3,6—5,1
Fe(OH)3...............1,6—3,5
Fe(OH)2..............6,6—9,3
Mg(OH)2.............. 8,3—11,3
Основываясь на различиях pH, частично можно отделить скандий от
циркония, титана, тория, церия (IV) (гидроксиды последних выделяют-
ся при более низких значениях pH, чем скандия), а также от РЗЭ
цериевой подгруппы, железа (II), гидроксиды которых осаждаются при
более высоких значениях pH, чем гидроксид скандия. Реализовать ме-
тод можно лишь для разбавленных растворов при тщательном регули-
ровании pH. Осаждая гидроксиды, невозможно разделить Sc3+ и Fe3+,
особенно при высокой его концентрации, так как скандий соосаждается
с железом, причем в этом случае Sc(OH)3 осаждается при более низком
значении pH. Близость значений pH осаждения гидроксидов скандия и
алюминия не позволяет их разделить. Однако задача может быть решена
при использовании избытка едкого натра, в котором А1(ОН)з растворя-
ется. Преимуществом этого метода является простота, существенный
недостаток — трудная фильтруемость осадков и недостаточный эффект
отделения многих сопутствующих элементов.
Метод ионного обмена используют для первичного концентриро-
вания скандия при очень низкой его концентрации (< 20 мг/л) и для
глубокой очистки соединений скандия на конечных операциях. Ионный
обмен можно применять для любых скандийсодержащих растворов. В
| зависимости от ионного состояния скандия в таких растворах могут быть
применены различные классы ионообменников. Используют главным
образом органические смолы — катиониты, аниониты и амфолиты.
Сульфокатиониты — сильнокислотные смолы — используют весьма
широко, хотя они недостаточно селективны, в том числе и для скандия.
(Поэтому эффект разделения скандия и примесей достигается при ис-
1318
пользовании комплексообразователей на стадии элюирования, особен-
но для трудноотделяемых примесей РЗЭ, Y, Th, Zr, Ti. В качестве
элюэнтов используют лимонную кислоту, ЭДТА, гидразинуксусные
кислоты и другие комплексообразователи. Одним из наиболее эффек-
тивных элюэнтов скандия является аммонийная соль СГ-гидроксиизо-
масляной кислоты (tZ-ГИМК). Она превосходит другие гидроксикарбо-
новые кислоты (молочную, лимонную) по коэффициентам разделения
и ЭДТА — по скорости элюирования. Устойчивость комплексных сое-
динений с элюэнтами повышается в ряду La < Y < Yb < Sc, так что при
элюировании в первую очередь вымывается скандий. Для элюации мо-
гут быть использованы минеральные кислоты, особенно HF aq, а также
фториды щелочных металлов и аммония.
Использование фосфорсодержащих смол, обладающих повышен-
ным сродством к скандию и селективностью, связано с трудностями при
элюации скандия. Для сорбции используют катиониты марок КУ, КПФ,
Амберлит, Дауэкс, Цеокарбидр. Большинство сопутствующих скандию
двух- и трехзарядных ионов металлов сорбируются на катионитах хуже,
чем скандий.
Сорбция скандия на анионитах определяется его способностью обра-
зовывать анионные комплексы. Поэтому сорбцию на анионитах различ-
ных типов проводят из фторидных, сернокислых, оксалатных и тарт-
ратных, а также из щелочных растворов. Для этого используют различ-
ные марки ионообменных смол — ЭДЭ, АВ, АН-2ФГ, Дауэкс, Амберлит
и др.
Таким образом, вне зависимости от типа используемой смолы про-
цесс ионного обмена включает:
1) стадию сорбции — пропускание раствора, содержащего скандий,
через колонну, ионообменный фильтр, например, через анионит
(Rn)2SO4 в сулъфоформе:
(Rn)2SO4 + [Sc(SO4)2]~----• Rn[Sc(SO4)2] + SO42-; (6.19)
2) стадию элюирования — раздельное вымывание примесей и скан-
дия комплексообразователем (лимонной, винной кислотами, ЭДТА и
т.п.) — в данном случае серной кислотой разных концентраций
2Rn [Sc(SO4)2 J + H2SO4---• (Rn)2SO4 + 2H [Sc(SO4)2 j. (6.20)
Недостатки метода — малая производительность, образование про-
межуточных фракций.
Фракционная сублимация. Метод имеет важное значение для очист-
ки хлорида скандия в связи с использованием в промышленности хлор-
ных методов переработки минерального сырья. Метод основан на
различиях в температурах сублимации (кипения) или конденсации
хлоридов скандия и сопутствующих элементов:
319
Хлорид . . Ткип, °C. . . . ScC13 . 1077 LnC13 1456—1747 YC13 1452 FeC13 319 NaCl 1765 KCI 1407 BeC12 547
Хлорид .. . Т1С14 ZrC14 ThC14 SnC14 NbC15 NbOC13
Ткип, °C.. . 136 331 922 113 248 400
Этим методом из оксида скандия, содержащего (%) 9 ZrO2, 12
ЬпгОз, при хлорировании S2CI2 в присутствии восстановителя получен
спектрально чистый хлорид скандия. Наиболее трудно отделим в этом
процессе хлорид тория.
Весьма эффективно можно очистить скандий сублимацией его аце-
гилацетоната 8с(СНзСОСНСОСНз)з, который сублимируется без раз-
ложения в вакууме. В этих условиях ацетилацетонаты титана,
ци^кония^гафция и тсцэия разлагаются, а ацетилацетонаты Y , РЗЭ3*,
Mg , Caz , Мп , UO2 существенно менее летучи. Процесс проводят
з вакууме (— 10 3 Па) при 170 "С. Недостатки процесса — невозмож-
ность очистки от Be , Al , Fe3*, высокая цена ацетилацетона.
6.12. Экстракционный метод концентрирования
и очистки скандия
Экстракционный метод занимает важное место в химической техно-
югии скандия. Высокая селективность, большая производительность,
тсутствие промежуточных фракций делают его весьма перспективным,
(.ля жидкостной экстракции скандия опробовано более 50 экстраген-
ов различных классов. Наибольшее применение нашли нейтральные
ислородсодержащие и катионообменные экстрагенты и существенно
(еньшее анионообменные. Выбор того или иного экстрагента определя-
тся ионным состоянием скандия в исходном технологическом растворе,
кстракция может быть использована на любом этапе технологической
кемы, кроме первичного концентрирования. При концентрации скан-
я менее 20 мг/л применение экстракции экономически неоправдано
з-за потерь экстрагента за счет его растворимости и эмулымрова-
ИЯ.
Избежать таких потерь позволяет использование твердофазных экс-
эагентов (ТВЭКС). Это — твердые пористые вещества, полученные в
эоцессе сополимеризации компонентов матрицы, чаще всего стирола
дивинилбензола, в присутствии экстрагента. Молекулы экстрагента
аходятся в микропорах, углеводородным радикалом они связаны смат-
ацей, а полярной группой (Р = О, С = О, S = О и др.) ориентированы к
и капилляра. При малом радиусе капилляра (50—80 нм) это приводит
заметному увеличению концентраций группы Р = О и других в средней
:0
части капилляра, что эквивалентно увеличению активности экстраген-
та в ТВЭКС. В результате коэффициенты распределения металлов уве-
личиваются в 1,3—3,0 раза. ТВЭКСы, содержащие НФОС—ТБФ,
фосфиноксид разнорадикальный (ФОР), перспективны для извлече-
ния, и особенно для первичного концентрирования редких металлов —
U, Zr, Hf, Th, Sc, Ln, Mo, W и других при содержании их меньше 20мг/л.
Технология их применения подобна сорбционным процессам и может
быть реализована в статических, динамических и противоточных усло-
виях.
Для экстракции скандия чаще других применяют ТБФ и ДААФ
(диалкилалкилфосфонат). Экстракцию ими можно проводить из силь-
нокислых растворов (НС1, HNO3, H2SO4, НСЮ4) или в присутствии
большого количества высаливателей — соответствующих солей кальция
и магния. Высокая экстрагируемость скандия НФОС в присутствии вы-
саливателей является характерной его особенностью, позволяющей от-
делять его от других элементов.
Экстракция ТБФ из солянокислых и азотнокислых растворов пред-
ложена для отделения скандия от многих примесей. Экстракция описы-
вается уравнением
Sc3 + ЗА" + иТБФ--------- ScA3 иТБФ, (6.21)
где А = Cl , NO3 , п = 2, 3.
Коэффициент распределения скандия при экстракции из 6 моль/л
НС1 составляет 3,2, иттрия 0,001, т.е. j5sc/y = 3200. Весьма селективны
по отношению к скандию системы ТБФ—H2SO4 и ТБФ—НСЮ4, где £>sc
на два порядка выше, чем Z>Ln,Ai,Zr (рис. 6.5, 6.6).
Разработан селективный метод тиоцианатной экстракции скандия
нейтральными кислородсодержащими реагентами в форме
H[Sc(CNS)4]. Экстракция протекает по гидратно-сольватному меха-
низму.
Коэффициенты распределения при экстракции скандия и сопутст-
вующих элементов диэтиловым эфиром из тиоцианатных растворов (pH
— 3,5) приведены ниже:
Элемент D Элемент D Элемент D
Sc^ 2,9 Ве2+ 1 Zr4+ 0,00001
-0,0007 Mg2+ 0,0002 Ti4+ 0,025
^La3* -0,00001 Fe34" 0,75 Al3* 0,59 > Иттриевая подгруппа. > Цериевая подгруппа. Са2+ Мп2+ 0,0004 0,0014 Th4+ 0,0006
321
Рис. 6.5. Зависимость коэффициентов распределения скандия и сопутствующих
элементов от концентрации H2SO4 в равновесной водной фазе при экстракции 100
%-ным ТБФ (в скобках — концентрация элементов в равновесной водной фазе,
моль/л): -3
1 — Sc (7,25 10 ). 2 — Zr (0,0244), 3 — Ti (0,0238); 4 — Yb (5,08 10-3), 5
-3 -3 -3 -3
— Er (5,23 10 ),6 — Dy (5,36 10 );7 — Y (8,86 10 );8 — Eu (5,68 10 );
9 — Nd (5,95 10 3); 10 — Fe (0,0125); 11 — Al (0,0392)
Рис. 6.6. Зависимость коэффициентов распределения скандия и сопутствующих
элементов от концентрации хлорной кислоты в равновесной водной фазе при
экстракции 100 %-ным ТБФ:
1 — Sc (0,0111); 2, 3 и 4 — Ln (0,030); 2 — La; 3 — Y, 4 — Yb, 5 — Zr (0,080)
Тиоционатную систему используют, как правило, для глубокой ко-
нечной очистки соединений скандия. Диэтиловый эфир из-за его высо-
кой пожароопасности используют лишь в препаративных целях. В тех-
нологической практике используют менее горючие экстрагенты, напри-
мер метилизобутилкетон (МИБК), ацетофенон и др.
Среди катионообменных экстрагентов наибольшее применение на-
шла Д2ЭГФК. Она имеет меньшую селективность, чем НФОС (ТБФ) и
вместе со скандием экстрагирует титан, торий, цирконий, железо, уран
и др. Поэтому ее используют для извлечения скандия из бедных нейт-
ральных или слабокислых растворов при малых соотношениях органи-
ческой (Vo) и водной (Св) фаз: Со / Св < < 1. Реакция экстракции идет
по катионообменному механизму
Sc3* + 3nHR-------. ScR3 nHR + 3H+, (6.22)
где HR — Д2ЭГФК.
322
Но реэкстракцию скандия можно осуществить лишь гидроксидами
натрия или калия, фторидами или плавиковой кислотой.
Карбоновые кислоты для экстракции скандия используют сущест-
венно меньше, чем АФК. Анионообменные и хелатные экстрагенты пока
не нашли применения в технологии скандия. Однако их использование
в будущем может быть перспективным.
6.13. Извлечение скандия из различных видов сырья
Переработка промпродуктов уранового производства. Урановые
руды перерабатывают кислотными или щелочными методами. Данные
о распределении скандия при щелочных методах переработки отсутст-
вуют, но можно предположить, что при такой переработке скандий
останется в кеке после выщелачивания.
Для извлечения скандия используют главным образом кислотные
схемы переработки руд, которые разлагают серной или азотной кисло-
той. При этом скандий переходит в раствор. В США нашел промышлен-
ное использование способ извлечения скандия из сбросных серно-
кислых растворов, содержащих ~ 10’3 г/л SC2O3. При экстракции урана
0,1 моль/л раствором додецилфосфорной кислоты в керосине и последу-
ющей реэкстракции 10 моль/л НС1 скандий остается в органической
фазе. Органическая фаза является оборотной, и в ней накапливаются
скандий (до 0,1 г/л), а также торий, титан, РЗЭ. После реэкстракции
HF и обработки концентрата щелочью полученные гидроксиды раство-
ряют в НО и гидролизом отделяют основную массу примесей Ti, Zr
и Fe. Из фильтрата, содержащего скандий с примесями урана и железа,
осаждают оксалат скандия. Окончательную очистку проводят экстра-
кцией ТБФ из раствора НО или диэтиловым эфиром (или МИБК) из
хлоридно-тиоционатного раствора. В результате получают SC2O3 чисто-
той 99,5 % (рис. 6.7).
В технологической схеме Горного Бюро США в качестве исходного
продукта используют непосредственно ураново-железистые кеки, со-
держащие 0,14% SC2O3. Разложение исходного сырья и перевод скандия
в раствор проводят раствором H2SO4 концентрацией 5 моль/л. Концен-
трирование скандия осуществляют экстракцией 10 %-ным раствором
первичного амина (Праймен JMT или АНП) в керосине. Степень обога-
щения получаемого продукта по скандию ~ 30, степень его извлече-
ния ~ 98 %. При этом скандий отделяется от Fe(II), Ti, Мп, Al, Zr.
Реэкстракцию скандия ведут НО концентрацией 2моль/л. После
осаждения гидроксидов раствором аммиака и растворения их в НО
следует ионообменное отделение урана на анионите. Заключительная
очистка состоит в экстракции скандия из хлоридно-тиоционатных рас-
творов МИБК, осаждении оксалата скандия из реэкстракта и прокали-
323
вторлдны) глг($с. Th, Ti, Zr, Ft, Si, U)
HCI ——| Рагпорпч |
I З/гстрапкокиал счлгтка |
ННдОН-----| Опждит 5с(ОН)з ~|
| Прочлчанл» (ВОО'С) |
Рис. 6.7. Технологическая схема из-
Sc2O3 (99,S%)
влечения скандия из скандиево-то-
риевого кека
вания. Схема обеспечивает получение оксида скандия чистотой 99,6 %
при извлечении 90 %.
Переработка промпродуктов титанового производства. Титано-
вое сырье перерабатывают в основном двумя методами: сернокислотным
с получением диоксида титана и хлорированием с получением тетрах-
лорида титана. В процессе производства пигментного диоксида титана
сернокислотным способом с гидролитическим выделением титана скан-
дий концентрируется в растворе гидролизной кислоты (на 76 %), откуда
его можно извлечь методами экстракции или ионного обмена.
При хлорировании титановых шлаков в расплаве солей скандий из
шлака переходит в расплав на ~ 77 %, и его концентрация в расплаве
составляет 0,01—0,03 %. Около 23 % скандия переходит в парогазовую
смесь и конденсируется в разных аппаратах системы конденсации.
Основным источником скандия является отработанный хлоридный
плав титановых хлораторов. Плав обрабатывают раствором НС120—40
г/л, при этом скандий переходит в хлоридный раствор. Раствор фильт-
руют и в нем корректируют концентрацию Fe , поскольку его присут-
ствие способствует экстракции скандия за счет соэкстракции с железом.
324
В то же время высокая концентрация FeCh вызывает загрязнение скан-
дия и повышение вязкости экстракта, что приводит к интенсификации
эмульгирования. Содержание железа (III) в исходном растворе необхо-
димо поддерживать в пределах 7—12 г/л, что достигают восстановлени-
ем Fe3+—. Fe2+
2FeC13 + Mg----. 2FeCh + MgC12. (6.23)
Экстракцию скандия из такого раствора проводят 70 %-ным ТБФ;
экстракт, содержащий скандий и сопутствующие элементы, отмывают
от них раствором НС1 (220—240 г/л), после чего реэкстрагируют скан-
дий 7 %-ной НС1. Из реэкстракта осаждают оксалаты скандия и сопут-
ствующих элементов; осадок фильтруют, сушат и прокаливают при
- 700 °C, получают концентрат, содержащий 40—60 % SC2O3.
Концентрат растворяют в НС1 и повторяют экстракционный цикл
(экстракция 70 %-ным ТБФ, промывка, реэкстракция). Из реэкстракта
после многократных переосаждений йодатов (очистка от Th* и Zr ),
осаждения гидроксидов и оксалатов получают товарный оксид скандия,
содержащий более 99,9 % БсгОз-
Замена 1-й экстракции 70 %-ным ТБФ на экстракцию ТВЭКС—
ТБФ снизило потери экстрагента и повысило извлечение скандия.
Содержание SC2O3 в первом скандиевом концентрате возросло до
50—80 %.
Переработка отходов вольфрамового и оловянного производств.
При переработке вольфрамитовых концентратов гидрометаллургиче-
ским методом скандий практически полностью остается в кеках от вы-
щелачивания содового спека и его содержание в них составляет
п 102—п 10-1 %. В процессе восстановительной плавки оловянных
концентратов скандий переходит в шлак. Кеки переработки вольфрами-
товых концентратов, шлаки производства ферровольфрама или олова
разлагают соляной или серной кислотами. Первичное концентрирова-
ние скандия достигают осаждением его фторида, карбоната или экстра-
кцией АФК.
При выщелачивании шлаков оловянной плавки соляной кислотой
получают раствор, содержащий 0,15—0,3 г/л Sc и 1—2 моль/л НС1.
Экстракцию проводили 0,1—0,3 моль/л Д2ЭГФК в керосине. После
реэкстракции концентрированной HF и последующей сульфатизации
фторидного продукта получен скандиевый концентрат с содержани-
м ~ 72 % БсгОз при извлечении — 82 %. Основной недостаток этой
схемы — образование на границе раздела фаз при экстракции твердых
алкилфосфатов титана, циркония, олова. Кроме того, процесс экстра-
кции Д2ЭГФК характеризуется значительными потерями экстрагента
с рафинатом из-за его разрушения кислыми растворами.
Для дальнейшей очистки используют комбинацию различных опе-
раций: осаждение оксалатов, их прокаливание и перевод скандия в со-
325
лянокислый раствор, осаждение тиосульфата скандия, его растворение
в НС1, осаждение йодатов титана, циркония и тория, осаждение
гидроксида скандия из йодатного раствора, растворение его в НС1
вторичное осаждение оксалата и его прокаливание до SC2O3 чистотой
99,7 % (марка ОС-99). Извлечение скандия в ОС-99 из вольфрамитово-
го кека 70,4 %, из шлаков оловянной плавки 55,4 %.
Для переработки вольфрамитовых кеков предложена следующая
технологическая схема. Кек разлагают 98 %-ной H2SO4. Карбонатной
очисткой отделяют от примесей железа, марганца и титана. Оконча-
тельную очистку проводят тиоционатной экстракцией диэтиловым
эфиром и осаждением оксалата скандия. При его прокаливании получа-
ют SC2O3 чистотой 99,99 % при извлечении скандия ~ 88 %. В качестве
экстрагентов можно использовать изоамиловый спирт, бутилацетат и
ацетофенон.
Возможны еще два варианта конечной очистки: БсгОз и примеси
растворяют в НС1, железо удаляют экстракцией эфиром, затем из этого
раствора скандий экстрагируют ТБФ, ScCh реэкстрагируют водой,
осаждают тартрат скандия и, прокаливая его, получают SC2O3. Второй
вариант связан с переводом Fe3^— Fe2+ с помощью SO2 и экстракцией
скандия первичным амином. Далее проводят реэкстракцию скандия
раствором НС1 (2 моль/л) и получают SC2O3. По этому варианту пол-
учают наименее чистый оксид скандия. По первому и второму вариан-
там оксид скандия содержит кремний и алюминий. От кремния оксид
скандия может быть очищен обработкой HF. Типичные технологиче-
ские схемы извлечения скандия из отходов вольфрамового и оловянного
производств показаны на рис. 6.8, 6.9.
Другие виды сырья. Из других видов следует выделить переработку
циркониевого сырья, в первую очередь по известково-хлоридной схеме.
Скандий концентрируется главным образом (~ 50—70 %) в маточных
растворах после выделения основного сульфата циркония (ОСЦ). Часть
скандия (14—23 %) соосаждается с ОСЦ. Важнейшее преимущество
этой схемы — отсутствие необходимости разлагать промпродукт, так
как скандий находится в сбросном растворе, направляемом на нейтра-
лизацию, и этот раствор пригоден для непосредственного извлечения
скандия. Извлечение может быть осуществлено комбинацией описан-
ных выше методов.
При переработке цирконовых концентратов хлорированием в рас-
плаве скандий концентрируется в отработанном расплаве хлоридов, иди
распределяется приблизительно поровну между огарком и ПГС (при
хлорировании брикетированной шихты). В этом случае для извлечения
скандия могут быть применены методы, используемые в титановом про-
изводстве.
326
При перера-
ботке бокситов на
глинозем по мето-
ду Байера или спе-
канием скандий
(~ 95 %) наряду с
титаном, цирко-
нием, ванадием,
РЗЭ и частично
галлием остается в
отвальном про-
дукте — «красном
шламе». Красные
шламы являются
перспективным
сырьем для извле-
чения редких ме-
таллов, однако
вследствие слож-
ности этого про-
дукта и специфич-
ности физико-хи-
мических свойств
(мелкодисперс-
ный сильно пы-
лящий продукт)
работы по их ис-
пользованию оста-
ются на уровне по-
лупромышленных
испытаний. В ре-
зультате комплек-
сной переработки
г~
Ков
I
В опал
Ж1
Плав
Z3_______1_
Вывюлачавовав
h2so4
н2о
НоОН
HCI
Н2СгО4
на
НН4ОН
Раствор |—
Осаждот ScFj
Ков
-----! !________
Срльфатлзацлл
Распорот
Осаждояяв тидровсвдов
Осадоа
Распорот
Осожденве ополотое
Проваллваллв (бОО'С/
Распоровлв
Осождат пдровпдоо
Проволввапве
------f------
Sc203 (30%)
Ln2O3 (70%)
Ho2SiF6
Раствор
В отвал
Раствор
В отвал
Рис. 6.8. Технологическая схема переработки вольфрамитовых
кеков сернокислотным способом
1
1
Т
красных шламов
могут быть получены: алюможелезный коагулянт, оксиды титана, цир-
кония, ванадия, кремния, РЗЭ и скандия. Комплексная переработка их
весьма трудоемка и энергоемка.
Необходимо иметь в виду крупнейший по запасам скандия источник
— каменные угли. Однако извлечение из них скандия разработано не-
достаточно, чтобы говорить о возможности его промышленного исполь-
зования.
Возможна переработка скандиевого минерала тортвейтита, хотя его
запасы невелики. Для его разложения используют различные методы:
327
на
Клак
Алилфосфорпея
каспаза
[ Вы^алочааоааа -----------------
Литлр „ , ♦ ,
| Как (а откол)
| Зкстрокцкк |_________Ллкклфосфорааа ислота
Оргояаческая фаза
Вадкап фаза (а опал)
Hf
Оргоааческок фаза
РОзкстракдаа [.
Водям фаза
Водяоя фаза
Ортаапчасиафд,,^ H?so<
Востаор КторадкыЯ продукт
NaOH
Дотлиаголыюе
азалачеааа St
на
[ОВроВоткл едкая погрел]
—~ г —'
——I facnopenaa]
Н2С2О4
| Пракалаааапа |
St2O3
Рис. 6.9. Технологическая схема извлечения скандия из шлаков оловянного про-
изводства
кислотные (обработка НС1, H2SO4, NH4HF2, HF), щелочные (сплавле-
ние с NaOH, спекание с ИагСОз), карбидный и хлорный. Кислотные
методы в промышленности не применяют, так как для полного извлече-
ния скандия необходима многократная обработка концентрата. Доста-
точно эффективно разложение спеканием с дифторидом аммония при
375___лол»£
SC2O3 2SiO2 + I4NH4HF2 =
= 2ScF3 + 2SiF4 + 14HF + HNH3 + 7Н2О. (6.24)
Спекание с содой также требу ет двух-трехкратного повторения про-
цесса. Полно извлекается скандий из тортвейтита после его сплавления
со щелочью
SC2O3 2S1O2 + 2НагСОз = SC2O3 + 2Na2SiO3 + 2СОг; (6.25)
SC2O3 2SiO2 + 4NaOH = Sc2O3 + 2Na2SiO3 + 2H2O. (6.26)
328
Плав обрабатывают водой, и скандий остается в твердом остатке,
который растворяют в НС1. Из солянокислого раствора скандий можно
экстрагировать в одной из описанных систем (НС1—ТБФ, НО—
NH4CNS—МИБК и т.п.) и затем осадить в форме оксалата или тартрата.
Осадок прокаливают до БсгОз-
Карбидный способ состоит в спекании тортвейтита с углем (1 : 1,2)
при 1800—2100 °C. Образовавшийся продукт обрабатывают НО; при
этом в раствор переходят Sc, РЗЭ, Ti, Zr, Al, Fe. Карбид кремния прак-
тически не растворяется. Солянокислый раствор перерабатывают для
извлечения Sc и РЗЭ по описанным выше схемам.
Возможно хлорирование тортвейтита в смеси с углем газообразным
хлором при 900—1000 °C. Различие в температурах кипения и конден-
сации хлоридов элементов, образующих рудный концентрат, позволяет
их разделить при конденсации: SiCU, TiC14, AICI3, FeCfe и ZrCh конден-
сируются ниже 400 °C, ScC13 — в интервале 600—900 °C. Степень извле-
чения скандия — 87 %. В зоне хлорирования остаются РЗЭ и частично
скандий.
Рассмотрение ряда методов извлечения и очистки скандия при пере-
работке различных видов минерального сырья показывает, что это
сложные и многооперационные процессы. Выбор сырья дЯя извлечения
скандия должен в первую очередь определяться распределением Sc в
технологической схеме переработки основного элемента, отработанно-
стью и простотой извлечения скандия из этого промпродукта без нару-
шения основной технологической схемы. В этом аспекте рассмотренные
сырьевые источники следует расположить в следующий ряд: урановые
руды > титановое сырье > цирконы > вольфрамиты > касситериты >
бокситы > природные угли.
6.14. Получение металлического скандия
Металлический скандий получают методами электролиза и метал-
лотермии. Впервые металлический скандий был получен в 1937 г. элек-
тролизом из расплава хлоридов скандия, калия и лития при 700—800 °C
на жидком катоде из цинка. Получали сплав Zn—Sc с 2 % Sc, который
подвергали дистилляции при 1250 °C в вакууме, при которой удаляли
цинк, калий и литий. Полученный металл содержал — 20 % SC2O3,
который отделяли фильтрацией расплава через вольфрамовый фильтр
в атмосфере водорода. Полученный металл представлял собой спекшу-
юся светло-серую массу с содержанием, %: 94—98 Sc, 0,2—0,5 Fe,
0,3—0,52 (Si, Al).
Другой способ электролитического получения скандия состоит в его
выделении из расплава NasScFb—SC2O3 (2 % SC2O3) при 800 °C. По мере
329
Рис. 6.10. Температурная зависимость
AGt образования оксида, фторида и
хлорида скандия:
J — ScCe3; 2 — ScF3; 3 — Sc2O3;
М — температура плавления; В — тем-
пература кипения
снижения в электролите содержания
скандия в него добавляют SC2O3. Этот
способ аналогичен способу получе-
ния металлического алюминия элек-
тролизом глинозема (AI2O3) в
расплаве алюминиевого криолита
(K3AIF6) • Электролиз проводят в гра-
фитовом тигле, являющемся анодом.
Графитовый (катод) помещен в цен-
тре тигля. Процесс проводят в атмос-
фере йргона или гелия. Металл
получают в форме гранул. Выход по
току ~ 50 %. Преимущества процесса
перед металлотермией: нет необхо-
димости в вакуумной дистилляции
для отделения металла-восстанови-
теля; скандий не загрязняется мате-
риалом тигля. Возможно получение
алюминия, легированного скандием,
при использовании жидкого алюми-
ния в качестве катода.
Тем не менее основной метод по-
лучения металлического скандия —
это металлотермия. Восстанавливают безводные ScF3 или ScCh кальци-
ем или магнием в инертной атмосфере. Температурные зависимости
дб^ образования оксида, фторида и хлорида скандия представлены на
рис. 6.10
2БсС1з + ЗСа = ЗСаСЬ + 2Sc; (6.27)
2ScF3 + 3Ca = 3CaF2 + 2Sc. (6.28)
Предпочтение отдается фториду — он негигроскопичен и его легче
Получить в чистом видё. Восстанавливают ScF3 при 1550—1600 °C в
индукционной печи, кальций берут с избытком 10 % по отношению к
необходимому по реакции (6.28) количеству. Шлак (CaF/) легко отде-
ляется от металла после охлаждения танталового (молибденового) тиг-
ля. Возможен вариант, когда восстановление ведут при 850 °C, а затем
гемпературу поднимают до 1600 °C и отделяют расплавленный металл
эт шлака. Это позволяет повысить чистоту металла, который переплав-
ляют в вакууме (10 3 Па) для удаления летучих примесей, затем при
1700 °C в вакууме возгоняют. Выход чистого металла составляет 95 %•
Получение более чистого металла (особенно по танталу) возможно
лри проведении восстановления при 1100 °C добавлением в шихту LiF,
юразующего с СаРг эвтектику, и цинка для образования легкоплавкого
(30
сплава Sc—Zh—Са. Восстановление протекает в атмосфере аргона по
схеме
1100 "С
2ScF3 + ЗСа + 8Zn + 12LiF = 2(Sc 4Zn) + 3(CaF2 4LiF). (6.29)
Кальций и цинк отгоняют в вакууме, скандиевую губку переплав-
ляют.
При получении скандия из фторида возможно применение алюми-
ния; реакция начинается при 840 °C.
Восстановление ScCl3 кальцием (6.27) протекает весьма бурно при
850 °C (в аргоне). Металл загрязнен примесями Са, CaO, СаС12, Si,
ScCl3. Все примеси, за исключением кремния, отмывают водой, а крем-
ний — 10 %-ным раствором NaOH. Высушенный на воздухе, а затем в
вакууме (10 2 Па, 500—600 °C) металл переплавляют (1500—1600 °C)
ввакууме (103—10 4 Па). Такая обработка позволяет получить металл
чистотой ~ 97 %.
Для получения металла высокой степени чистоты его либо сублими-
руют в вакууме, либо используют исходные вещества (в первую очередь
Sc2O3) максимально доступной степени чистоты. Глубокую очистку
скандия можно осуществить электролитическим рафинированием из
расплава или вытягиванием монокристалла.
В настоящее время разработаны методы получения металлической
пленки и фольги из скандия и скандийсодержащих лигатур. Пленки
(толщина 0,0003—0,3 мм) получают осаждением паров металлического
скандия на алюминиевую или графитовую подложку.
Имеющиеся данные говорят о том, что скандий и его соединения не
представляют сколько-нибудь серьезной опасности для здоровья чело-
века. Показано, что скандий менее токсичен, чем гадолиний, самарий,
ниобий или гафний. Опасности и вредности при производстве скандия
связаны главным образом с веществами, используемыми в технологии
его получения (плав хлоридов, кислоты, щелочи, аммиак, ТБФ и т.д.).
331
Раздел IV. Методы математического
моделирования в химии и технологии
редких металлов
Развитие вычислительной техники и методов математического мо-
делирования, появление высокопроизводительных персональных ЭВМ
поставило перед исследователями вопрос о разумном использовании
этих средств на всех стадиях создания и отработки технологических
процессов. При проведении исследований квалифицированный техно-
лог должен сочетать теоретические представления и натурный экспери-
мент с методами математического моделирования и реализацией их с
помощью компьютера. Появление персонального компьютера позволя-
ет не только совершенствовать традиционные задачи, связанные со сбо-
ром, обработкой и хранением экспериментальных данных, построением
математических моделей и их применением для исследования, управле-
ния и проектирования технологических процессов, но и решать принци-
пиально новые задачи, которые были недоступны экспериментатору. К
ним относятся, например, изучение предельных режимов технологиче-
ских процессов, экологическая оценка последствий аварий и нарушений
ПДК, оценка вероятности взрыво- и пожароопасности процесса, оценка
влияния технологии на здоровье человека и т.д.
Для этих исследований используются методы имитационного моде-
лирования, вычислительного эксперимента, искусственного интеллек-
та. Однако в химии и технологии редких и рассеянных элементов при-
менение компьютеров и методов математического моделирования до сих
пор не получило широкого применения. Это связано со следующими
особенностями отрасли: 1) многообразием технологий, применяемых
для получения редких металлов, вызванным многообразием используе-
мого сырья; 2) слабым парком контрольно-измерительной аппаратуры
в научных лабораториях и на предприятиях, что вызвано сложностью
организации контроля в присутствии характерных для перерабатывае-
мого сырья соединений железа, кремния, радиоактивных элементов, а
также агрессивностью реагентов, высокотемпературными режимами
проведения многих процессов; 3) отсутствием надежных математиче-
ских моделей для большинства технологических процессов, что объяс-
няется сложным характером физико-химических явлений в процессах,
их нестационарностью и высокой инерционностью.
Математические модели, применяемые для исследования, управле-
ния и оптимизации технологических процессов, достаточно условно
можно можно разделить на три класса: физико-химические, экспери-
ментально-статистические и смешанные.
332
Физико-химические модели строятся на основе уравнений, отража-
ющих общие физико-химические закономерности процесса (например,
на основе законов термодинамики, сохранения массы и энергии, урав-
нений, описывающих химические превращения, массообмен, кинетику
реакций и т.д.). Параметры физико-химических моделей находят из
справочной литературы или с помощью специального эксперимента.
Такие модели применяются для проведения широкого круга физико-хи-
мических исследований (например, термодинамической оценки воз-
можности протекания реакций в данной системе, расчета равновесных
составов многокомпонентных систем, расчета кинетических кривых и
Т.Д.).
Экспериментально-статистические модели отражают корреляцион-
ные или регрессионные соотношения между входными и выходными
параметрами процесса и не используют информацию о физико-химиче-
ских превращениях в процессе. Статистические модели используются
для оценки влияния входных переменных на показатель качества про-
цесса, для оценки значимости переменных, для решения задач управле-
ния и оптимизации процессов.
Смешанные модели сочетают оба описанных подхода. Структура
этих моделей выбирается обычно на основе уравнений физической хи-
мии, а для оценки параметров используется статистическая обработка
данных эксперимента.
Задачи, решаемые при построении математических моделей, а так-
же задачи, решаемые с помощью моделей, крайне разнообразны. К
числу наиболее популярных проблем можно отнести: 1) получение,
обработку и хранение информации (планирование и статистическая
обработка экспериментальных данных, обработка данных, получаемых
с помощью приборов физико-химического анализа, расчеты на основе
правочно-литературной информации, запись и хранение информации
в электронных базах данных); 2) построение моделей процесса (выбор
типа и структуры модели, оценка ее параметров, статистическая оценка
качества модели, дискриминация моделей); 3) математическое модели-
рование процесса (решение задач исследования, оптимизации и управ-
ления на модели, постановка вычислительного эксперимента для
прогнозирования технически нереализуемых ситуаций).
333
Глава 7. СТАТИСТИЧЕСКИЕ МЕТОДЫ ОБРАБОТКИ
ЭКСПЕРИМЕНТАЛЬНЫХ ДАННЫХ
Одной из проблем, с которой приходится сталкиваться каждому
исследователю, является статистическая обработка экспериментальных
данных. Можно выделить две наиболее часто встречающиеся задачи:
получение статистических характеристик по выборке эксперименталь-
ных данных и математическая обработка экспериментальных зависимо-
стей. Поскольку все измерения производятся с некоторой случайной
ошибкой, то результаты их можно рассматривать как различные значе-
ния случайной величины.
Оценки статистических характеристик случайной величины. На
практике исследователь не может найти все допустимые значения слу-
чайной величины (так называемую генеральную совокупность) и имеет
дело с некоторой выборкой длиной п элементов из этой совокупности.
По выборке рассчитываются выборочные статистические характеристи-
ки, которые являются оценками соответствующих генеральных стати-
стических характеристик (математического ожидания тх, дисперсии
(Jx и т.д.). Для каждой числовой характеристики случайной величины
можно получить несколько оценок. Из них наибольший интерес пред-
ставляют состоятельные и несмещенные оценки.
Состоятельные несмещенные оценки математического ожидания
и дисперсии имеют следующий вид:
х=(1/п)^х1, (7.1)
1=1
Э "о
s‘=[l/(n-l)]£ (xt-x)2, (7.2)
i=l
где х — среднее значение; s2 — выборочная дисперсия.
Оценка коэффициента корреляции двух случайных величин х и у
носит название выборочного коэффициента корреляции и определяется
кяк
п
глу-12 ( х,-х) (у,-у) 1/[(n-l)sxSy), (7.3)
i=i
где sxk sy — квадратные корни из выборочных дисперсий.
Расчет доверительных интервалов. После вычисления состоятель-
ных и несмещенных оценок числовых характеристик случайной вели-
чины необходимо оценить их точность. А так как любая оценка также
представляет собой случайную величину, то необходимо найти откло-
нение найденной оценки от истинного значения, задавшись вероятно-
334
стью этого отклонения. Для этой цели определяются доверительные
интервалы. Пусть по результатам ряда измерений величины X = ( jq,
х2 ,..., хп) найдено среднее значение этой величины, представляющее
собой состоятельную и несмещенную оценку х . Тогда, задавшись веро-
ятностью/3, можно записать
Р [ I х-х* I < £ ]=/3, t (7.4)
где £ — заданное отклонение I х - х I , т. е. с вероятностью Р можно
сказать,что ошибка при замене х на х равна ±Е. При этом ошибки,
большие чем х ± Е, будут появляться с вероятностью р = 1 -/?, называ-
емой уровнем значимости, а вероятность/? носит название доверитель-
ной вероятности. В соответствии с этим интервал х ± £
х-£ < х < х + Е (7.5)
носит название доверительного интервала. Его границы х + Е и х - Е
носят название доверительных границ. Величина доверительного ин-
тервала с доверительной вероятностью определяет точность полученной
оценки случайной величины. Чаще всего задаются значениями довери-
тельной вероятности 0,9; 0,95 или 0,99.
Найдем доверительный интервал для оценки значения случайной
величины. При малом числе наблюдений, а именно с этим случаем чаще
всего приходится сталкиваться на практике, для построения довери-
тельного интервала оценки математического ожидания используется
критерий Стьюдента ((-критерий)
t = ['Гп (х-тх)]/ sx, (7.6)
где п — число экспериментов; sy — оценка среднеквадратического от-
клонения. Для критерия Стьюдента доверительная вероятность опреде-
ляется как вероятность того, что случайная величина t попадает в ин-
тервал (tp/2, tl-p/2)
Р [tp/2 * t< tl-p/2 ] = 1-р=/?, (7.7)
где р — заданный уровень значимости. Отсюда с учетом симметрии
распределения Стьюдента, т.е. tp/2 = - tl-p/2, находим
— ti-p/2 (х-тх) / (sxyTn )< П-р/2-
Проведя несложные преобразования, запишем окончательную фор-
мулу определения доверительного интервала для оценки математиче-
ского ожидания
x-tj^SxVn s тх< x + tx_(nsxyTn. (7.8)
Величины t табулируются и имеются в любом справочнике и учеб-
нике по математической статистике.
Оценка дисперсии воспроизводимости. Рассмотрим определение
доверительного интервала для оценки дисперсии. Вначале кратко оста-
новимся на вычислении дисперсии воспроизводимости. Каждый
экспериментатор для повышения точности вычислений обычно ставит
335
несколько параллельных опытов в каждой экспериментальной точке.
Причем в каждой точке число этих параллельных опытов в общем слу-
чае различно.
Под дисперсией воспроизводимости понимается выборочная диспер-
сия, вычисленная в каждой z-й точке. Если число параллельных опытов
в точке i равно т, то
mi
sb = s?= 1 7 < тГ 1) 2 (Xj-Xi)2 . (7.9)
/=1
Ошибка воспроизводимости опыта sB = Если в общем случае
анализируется п проб и в каждой сделано различное количество парал-
лельных опытов тп , то общая дисперсия воспроизводимо-
сти эксперимента находится как
^=(^/i+s2/2 + ...+s2/n)/ (/1+/2+ (7.10)
2
где$. —z-я частная дисперсия (z= 1,2,..., и);Д— число степеней свободы
в z-м опыте. Так как при вычислении дисперсии число степеней свободы
fi вычисляется как ft = rm - 1, то
sb~ + s2^2 + — +snfJ I + m2 + — + mn~n^
n mi
C2 = V У ! r (7-11)
Sb Д 2Л xij xi ) ✓( mi n) ’
i=lj=l
где Xjj — значение /-го параллельного опыта в i-й экспериментальной
точке; xt — среднее значение zfly-x параллельных опытов в i-й экспери-
ментальной точке.
В частном случае, если число параллельных измерений в каждом
опыте одинаково,
и п mi
sb 2 Ц)/” = Е S ( xy-JC)2/l n( 1 )L (7-12)
i=i i=i;=i
где n — число опытов.
Доверительный интервал для оценки дисперсии строится на основе
распределения Пирсона, или %2 (хи-квадрат), т.е. показано, что для
выборки из п независимых наблюдений х2, —> хп величина
%2=£ [(л,-х)/ах]2=л2/а2 (7.13)
1=1
имеет распределение %2 с f= п- 1 степенями свободы.
2
Для критерия / доверительная вероятность определяется из усло-
вия
336
P[fi< fs2xlo2x<x22]~fi-\-P, (7.14)
2 2 2 2
raexf=X^;zhXi-^-
Подставив выражение для/2 из (7.13) в условие (7.14) и проведя
некоторые преобразования, находим доверительные интервалы для дис-
„2
Персии Ох
<7-15)
2 2
Значения критерия Пирсона уи/. находятся для задаваемого
исследователем уровня значимости р и заданного числа степеней свобо-
ды /из справочников или учебников по математической статистике.
Рассмотренная методика обработки экспериментальных данных при
малых объемах выборки п может быть реализована вручную, а при
больших п для расчетов целесообразно использовать компьютер.
Пример. Контроль за процессом вскрытия бастнезитовых концен-
тратов осуществляется путем лабораторного анализа на содержание
суммы РЗЭ гравиметрическим методом.
Необходимо определить по текущим измерениям, включающим па-
раллельные опыты, ошибку методики для данной выборки результатов
и доверительный интервал для ошибки. Кроме того, необходимо найти
среднее значение и доверительные интервалы для каждой пробы. В табл.
7.1 приведены значения оценок математического ожидания и довери-
тельные интервалы для них, а также дисперсии воспроизводимости для
каждой из проб.
Таблица 7.1. Исходные данные для расчета и оценки
математического ожидания и дисперсии воспроизводимости
Номер опыта Номер пробы
1 2 3 4 5 6 7 8
1 78,5 85,5 90,2 79,8 83,2 87,2 81,2 82,7
2 77,9 83,2 91,2 78,1 84,1 85,8 80,0 81,5
3 79,2 84,1 — 80,5 81,8 — 83,1 82,0
mi 3 3 2 3 3 2 3 3
xi ±f^i 78,5 84,3 90,7 79,5 83,0 86,5 81,4 82,1
1,09 1.95 3,45 2,07 2,04 4,41 2,63 1,01
2 si 0,42 1,34 0,5 1,52 1,34 0,98 2,44 0,36
337
Для вычисления доверительных интервалов задана доверительна®
вероятность /3 = 0,9 (уровень значимости р = 0,1). Для нахождения
статистической ошибки методики найдем дисперсию воспроизводимо!
сти по 8 опытам по формуле (7.11). Общее число степеней свободы
8 8
/общ = £Л =£ m/-n = 22-8 = 14;s2=l,16;sB = l,07.
i=i i=i
Доверительные границы для ошибки воспроизводимости найдем,
задаваясь доверительной вероятностью/? = 0,9. Используя статистиче-
ские таблицы критерия/2 для числа степеней свободы /общ - 14, имеем
Яо,О5 = 23’7;Хо,95 = 6’6; тогда °’68 ~ ах - 2’44; °’82 ~ °х ~ 1'56-
Таким образом, ошибка методики определения РЗЭ с использовани-
ем гравиметрического метода на основе проведенных опытов составляет
1,07 с доверительным интервалом (0,82; 1,56). Из приведенного резуль-
тата видно, что границы доверительного интервала несимметричны от-
носительно центра. Это связано с несимметричностью распределения
2
критерия/ .
Глава 8. МОДЕЛИРОВАНИЕ СЛОЖНЫХ
ФИЗИКО-ХИМИЧЕСКИХ РАВНОВЕСИЙ
В МНОГОФАЗНЫХ СИСТЕМАХ
Моделирование физико-химических равновесий включает разнооб-
разные задачи, сводящиеся к нахождению количественных взаимосвя-
зей между тремя группами термодинамических данных: составом, ус-
ловиями равновесия и термодинамическими свойствами (которые, в
свою очередь, являются функциями состава и условий).
В зависимости от того, какие данные являются исходными, а какие
получаются в результате решения, множество задач по расчетам равно-
весий принято делить на прямые и обратные задачи химического равно-
весия. Под прямой задачей химического равновесия понимается расчет
равновесного состава системы на основе исходной термодинамической
информации и (или) данных по исходным концентрациям, а также
стехиометрической матрицы системы. Обратная задача заключается в
оценке термодинамических свойств (констант) по равновесным экспе-
риментальным данным.
Можно выделить два подхода к решению задач моделирования рав-
новесий: это «термодинамический» подход, основанный на минимиза-
ции свободной энергии системы, и «химико-термодинамическое
приближение», основанное на законе действующих масс (ЗДМ). Отме-
338
тим, что «термодинамический» подход используется на практике наибо-
лее продуктивно для расчета равновесий в системах твердое—газ, в то
время как «химико-термодинамическое приближение» удовлетвори-
тельно для систем типа жидкость—жидкость и жидкость—газ.
8.1. Термодинамический подход
Термодинамический подход основан на решении задачи минимиза-
ции свободной энергии системы (или на максимизации энтропии систе-
мы) при ограничениях на состав в виде уравнений материального ба-
ланса. Функция, описывающая зависимость свободной энергии (энтро-
пии) системы от состава и термодинамических свойств ее частей, назы-
вается характеристической функцией системы. Основное преиму-
щество термодинамического подхода при моделировании равновесий
заключается в том, что характеристическая функция практически лю-
бой системы может быть сконструирована на основе свойств подсистем.
Для теоретического расчета сложной характеристической функции ее
представляют в виде суммы характеристических функций отдельных
частей; при этом в модель должны быть включены все возможные формы
существования веществ в системе. Какие из этих форм могут присутст-
вовать в системе,показывает расчет равновесия. С математической точ-
ки зрения задача формулируется как задача поиска условного экст-
ремума характеристической функции при ограничениях на перемен-
ные.
Так, для многокомпонентной изолированной гетерофазной систе-
мы, содержащей газовую фазу с произвольным числом молекулярных
форм и набор конденсированных фаз (индивидуальные вещества, жид-
кие и твердые растворы) с произвольным числом химических реакций,
характеристическая функция записывается
I I LS
G~'£ni[ci+ ln(nz/2 «/)+ + ns [ с* +
1=1 1=1 /=ls=l
s к
+ ln(ns / £ ns) + ln Vs'+ 2 «* ck - (8Л)
s=l k=l
Уравнения материального баланса и неравенства, используемые в
виде ограничений:
1 L s , , к
Е ац + aiS ns+l ajk nk=bi </=1 > 2> ; (8-2)
1=1 /=ls=l k=\
nt> 0 G = l,2,...,/); nls> 0 (s=l,2,..., 5; 1= 1,2, ...,£);
и* > 0 (k = 1,2,..., Ю;
339
где <pt и уг — соответственно коэффициенты фугитивности и активности
для газовой фазы и растворов; индексы г, s, к относятся соответственно
к газовой фазе, конденсированным растворам и индивидуальным веще-
ствам; щ — число молей; а — термодинамические функции, которые
рассчитываются по следующим выражениям:
ci = ^98(7) <*> / RT ~ ФГ / R + InPi, Ф** — ( (?г - Яг98(/)) / Г,
c(s, k)=f^298(f) *> / RT-ФГ IR, (8.3)
где л#298(/) — стандартная энтальпия образования соединения из про-
стых веществ, Ф — приведенная энергия Гиббса.
Преимущества этого подхода особенно ярко проявляются при расче-
тах равновесий в сложных системах, которые состоят из частей с различ-
ными свойствами (гетерогенные многокомпонентные системы). Рав-
новесие в подобных системах рассчитывают сегодня исключительно
численными методами. При этом термодинамические расчеты являют-
ся, как правило, частью более общего анализа проблемы, а практическое
значение имеют не термодинамические свойства непосредственно, а
зависящие от них характеристики процессов. Особенно ценную инфор-
мацию дают такие расчеты для объектов, недоступных для непосредст-
венного экспериментального исследования. Термодинамический под-
ход позволяет выявить предельные направления превращений, т.е.
принципиальную возможность получения тех или иных веществ Варь-
ируя состав и внешние условия, принципиально возможно найти усло-
вия оптимизации технологических процессов по различным критериям,
таким, как выход целевого продукта, минимальное содержание нежела-
тельных примесей, состав отходов и т.д.
Пример. Технология извлечения РЗЭ и иттрия из ксенотимовых
и монацитовых концентратов включает их высокотемпературное спека-
ние с содой с целью перевода РЗ-фосфатов в форму оксидов и последу-
ющее выщелачивание спека азотной кислотой. Состав концентратов
достаточно сложен, так как помимо собственных РЗ-минералов они
содержат такие породообразующие минералы, как кварц, гематит,
флюорит. Особенностью данного процесса является значительная чув-
ствительность полноты извлечения РЗЭ к минеральному составу сырья
и условиям высокотемпературной переработки (температуре и концен-
трации соды в шихте), обусловленная протеканием разнообразных по-
бочных реакций с участием породообразующих минералов и соединений
РЗЭ.
Использование аппарата классической термодинамики для описа-
ния реального процесса возможно только при учете ряда модельных
допущений. При решении поставленной задачи важнейшими идеализи-
рующими допущениями являются: 1) использование равновесного опи-
340
сания для неравновесного процесса (такой подход полезен для выявле-
ния предельных закономерностей и направлений самопроизвольного
развития взаимодействий); 2) использование понятия изолированной
системы для реально открытой системы (избыток газовой фазы имити-
рует осуществление процесса в открытой системе); 3) выделение среди
множества присутствующих в системе веществ термодинамически зна-
чимых.
Задача моделирования процесса спекания сформулирована в виде
прямой задачи химического равновесия. Расчет равновесного фазового
состава выполнялся для многокомпонентной гетерогенной системы, со-
стоящей из идеальной газовой фазы (СО2), индивидуальных конденси-
рованных фаз и идеальных твердых растворов с использованием ав-
томатизированной системы термодинамических данных и расчетов рав-
новесных состояний (АСТРА). В качестве исходных данных использо-
ваны термодинамические характеристики виртуальных фаз системы.
Равновесный фазовый состав рассчитывался при вариациях температу-
ры, давления и исходного фазового состава смеси. Анализ полученных
закономерностей разложения фосфатов иттрия и лантана в присутствии
основных породообразующих минералов приводится ниже.
В присутствии гематита разложение LaPO4 происходит в интервале
температур 573—673 К, YPO4 — в интервале 873—973 К и приводит к
образованию ферритов типа LnFeOa-
В присутствии кварца вероятно полное связывание соды в силикаты
натрия различного состава (NazSiCh, NazSizO?, Na4SiO4). Очевидно,
вследствие этого заметное разложение LnPO4 в исследованном диапазо-
не температур не достигается. YPO4 устойчив в интервале температур
473—1173 К и исходном содержании соды, более чем в 30 раз превыша-
ющем стехиометрическое количество, фосфат лантана в избытке соды
разлагается до NaLaSiO4. Отметим также тенденцию к образованию
LazSizO? при температуре выше 1173 К.
В присутствии флюорита резко снижается термическая устойчи-
вость фосфатов. Так, фазы YPO4 и LaPO4 полностью отсутствуют в
равновесной системе уже при 473 К. В интервале температур 473—
773 К РЗЭ существуют в форме фторидов. Причем, если для иттрия
характерно образование фазы NaYF4, то лантан находится в виде
ЬаБз NaLaF4, устойчивом в достаточно узком температурном интерва-
ле 673—873 К. При температурах 773—873 К наблюдается взаимодей-
ствие фторидов иттрия и лантана с содой, приводящее к последова-
тельному образованию LnOF и ЬпгОз (рис. 8.1).
При совместном присутствии гематита,кварца и флюорита равно-
весный состав смеси YPO4 — LaPO4 — РегОз — SiOz — CaFz — NazCOa
существенно зависит от исходной концентрации соды. При стехиомет-
рическом количестве соды наблюдается образование широкой области
341
Рис. 8.1. Зависимость равновесных концентраций: NaYF4 (/), LaF3 (2), NaF (3),
Са4₽2О9 (#), Na2CO3 (5), CaF2 (6), LaOF (7), Y2O3 (S), La2«3 (9). CaO (10) от
температуры в спеке смеси YPO4—LaPO4—CaF2—Na2CO3
Рис. 8.2. Зависимость равновесных концентраций: YPO4 (/), LaF3 (2), LaFeO3
(3), YFeO3 (4), NaFeSi2O6 (5), NaF (6), Ca4P2O9 (7),.CaFe2O4 (8), Na2CO3 (9),
Na2SiO3 (10), Ca3Fe2[Si€>4]3 (ID, Fe2€>3 (12) от температуры в спеке смеси
YPO4- LaPO4—SiO2—CaF2—Fe2O3—Na2CO3
устойчивости кальциевого апатита, гетеровалентные изоморфные заме-
щения в котором (типа ЗСа2+ = 2Ln3+) могут приводить к накоплению
РЗЭ в силикатапатитных фазах. Связывание гематита в устойчивый до
973 К СагРез IS1O4 ]з предотвращает образование ферритов РЗЭ; иттрий
и лантан концентрируются при этом в фазах NaLnF4. Использование
сверхстехиомстрического количества соды исключает образование апа-
тита и соединений типа-НаЬпр4 во всем рассмотренном диапазоне тем-
ператур. Однако получение РЗЭ в форме оксидов маловероятно даже в
этом случае, так как усиливается тенденция к кристаллизации ферри-
тов. Использование большего избытка соды нерационально, так как
сопровождается взаимодействием соды с породообразующими минера-
лами (перераспределенье РЗЭ между равновесными фазами при этом не
происходит) (рис. 8.2)
На основании проведенных расчетов можно выделить следующие
причины неполного извлечения РЗЭ в азотнокислый раствор при пере-
работке минерального сырья переменного состава методом спекания с
содой: неполное разложение фосфатов РЗЭ; концентрирование РЗЭ в
фазах ЬлРеОз, NaLaF4, ЬпРз, La2Si2O?, трудно выщелачиваемых из
спека азотной кислотой; гетеровалентный изоморфизм между иона-
342
ми Ьп3+и Са2+, приводящий к образованию твердых растворов на основе
силикатов кальция и кальциевого силикатапатита.
Анализ равновесной модели дает возможность качественно обосно-
вать приемлемые режимы спекания с содой сырья различного минераль-
ного состава. Так, для ассоциации минералов с высокой концентрацией
флюорита можноснизить температуру процесса и использовать количе-
ство соды, не превышающее стехиометрическое. При переработке сме-
сей, обогащенных минералами железа, не следует значительно уве-
нчивать температуру и продолжительность спекания, а также исполь-
зовать большой избыток соды, поскольку при этом усиливается тенден-
ция к кристаллизации ферритов РЗЭ, трудно выщелачиваемых из спека
азотной кислотой. В присутствии значительных концентраций кварца
принципиально невозможно полностью разложить редкоземельные
фосфаты в исследованных условиях.
8.2. Методы оценки термодинамических характеристик
кристаллических соединений
Для большой группы соединений редких металлов необходимые для
расчетов равновесий термодинамические характеристики не определе-
ны экспериментально. Это связано с трудностью достижения равнове-
сий в системах тугоплавких соединений, а также с малоизученностью
этих систем. Поэтому для получения исходной термодинамической ин-
формации широко используются полуэмпирические методы оценки
термодинамических свойств. Выбор подходящего метода для определе-
ния термодинамических характеристик конкретных соединений явля-
ется в какой-то степени искусством, так как заранее нельзя дать ре-
комендаций для его реализации. Кроме того, исследователь лимитиро-
ван ограниченностью и недостоверностью данных, необходимых для
расчета.
Методы оценки кРисталлических соединений. Большин-
ство методов полуэмпирического расчета стандартных энтальпий обра-
зования кристаллических соединений основано на оценке параметров
линейных регрессионных уравнений, устанавливающих связь между
аЯ298(/) и каким-либо свойством в ряду родственных соединений (мето-
ды 1—4 в табл. 8.1). По существу, это примеры использования 2-го
метода сравнительных расчетов Карапетьянца, возможности которого
не исчерпываются рассмотренными зависимостями. Однако, несмотря
на простоту и надежность (следующую из физико-химического обосно-
вания) , методы сравнительного расчета неработоспособны при отсутст-
вии данных о А#298(у) однотипных соединений.
343
Таблица 8.1. Методы оценки кристаллических соединений
№ пп Название Расчетная формула 11римечания |
1 2 3 4 1
1 11равило термохимической логарифмики Л^298(/) / w а In Z + b, где w - валентность элемента в соединении, Z — порядковый номер элемента Результаты надежны для 1 соединений металлов с 1 низкой валентностью
2 Зависимости стандартных энтальпий от энергии ионизации а + где IwiaHs — к-я энергия ионизации и энергия сублимации атома металла
3 -о//экв + *>, где 7экв=(/а+ /к )/М /а, 7к — /-е энергии ионизации аниона и катиона, N — номер группы металла Данные по 7а (особенно для сложных по составу и строению анионов) отсутствуют либо малонадежны
4 Зависимость стандартных энтальпий от стандартных электродных потенциалов ЛЯ298С0" а~ЬЕ° Е° — стандартный электродный потенциал катиона Погрешность минималь- на для соединений с труднополяризуемыми анионами
5 Первый метод сравнительных расчетов Карапетьянца ДЯ298(У)(П) ~а Л//298(»(1)+ + Ь, где ДЯ^98(у)(1), Л/У298(/)(11) — энтальпии образования соединений в двух рядах однотипных веществ Точность зависит от монотонности измене- ния Л/^98(/) в рядах однотипных соединений
344
Продолжение таблицы 8 1
1 2 3 4
6 Инкрементные методы vAB(xB "xA)2+vA>A + vB>B’ где Уде — число единичных связей А—В, Уд Vg —число катионов и анионов, ХА' УА’ ХВ’ УВ ~ оцениваемые параметры 11редложен для оценки стандартных энтальпий образования галогенидов
7 Л//298(У) " VAB(XA ~ ХВ)2+ + Wa + Wb + + W^A /и,в) ’ таехА^А-и'АихВ->В’и,В- оцениваемые параметры катиона и аниона Предложен для оценки стандартных энтальпий образования кислород- содержащих соединений
8 ^К = -С^КГА.)П1, где К( — параметр <-го кати- она. Aj , rij— параметры /-го аниона; c.j — стехиометри- ческий коэффициент Для оценки энтальпий реакций образования сложных оксидных сое- динений С- из оксидов 2?; и С
Другая группа методов основана на оценке характеристических па-
раметров атомов или групп атомов, образующих соединение (методы
6—8 в табл. 8.1). Характеристические параметры (так называемые ин-
кременты), постоянные для каждого из атомов, катиона или аниона,
входят в полуэмпирическое уравнение, которое описывает зависимость
лЯ2ОВ/а сложного соединения от его состава. Используемые в данном
zye(Z)
случае зависимости, как правило, нелинейны, что исключает их про-
стую геометрическую интерпретацию и приводит к использованию бо-
лее сложного математического аппарата для оценки инкрементов.
Методы оценки 5°98 кристаллических соединений. Стандартные
энтропии образования кристаллических соединений могут быть найде-
ны на основании универсальных принципов сравнительных расчетов
Карапетьянца, а также с использованием ряда методов, базирующихся
на аддитивной схеме расчета (методы 1—5 в табл. 8.2). Кроме того, для
345
12
расчета ионных кристаллов часто используются различные систе-
мы ионных инкрементов энтропии, в которых каждому виду ионов при-
писывается постоянный инкремент энтропии. Энтропия соединения вы-
числяется в данном случае как сумма инкрементов энтропии ионов с
учетом числа ионов каждого вида в молекуле (методы 6—8 в табл. 8.2).
Таблица 82. Методы оценки S^gg кристаллических соединений
№ пп. Метод Расчетная формула- Примечания
I 2 3 4
1 1-й Киреева п Л298 = S vi Si +AS’ 1=1 где v. —число однотипных атомов в соединении; S.— энтропии атомов; AS —изменение энтропии реакции образования соединения из элементов; п — число раз- личных атомов в соединении с° ^298 опРеДеляетСЯ по данным для однотипных соеди- нений
2 2-й Киреева AS- S + v(S. -s.) , ат i k шд i7 ’ где 5ат — атомная энтропия обра зования соединения; 5^ид —энтропия атома в состо- янии идеального газа Преимущество метода в том,что значения затабу ли рованы для всех элементов
Q о ШарОВа п оО V Гт °298 Z/ vi ani %,ат + 1=1 п + Я1пу£ v, (1) 1=1 где S —энтропия поступательного движения частиц определяется по формуле Сакура—Тетроде: S„ = 6,861g А + 25,99, (2) п где А — атомная масса элемента. Представляет собой усо- вершенствованный 2-й метод Киреева. Учиты- вает изменение атомной энтропии образова- ния соединения в ряду однотипных
346
Продолжение таблицы 8.2
1 2 3 4
входящего в состав соединения; V — мольный объем соединения ASy ат —вычисляется для одного однотипного соединения по формулам (1) и (2) соединений
4 Филиппова *^298 ™ X - 21g М-Igg; М —молекулярная масса вещества; g — его плотность; а и Ь — рег- рессионные коэффициенты Основан на использова- нии регрессионной зави- мости в ряду однотипных веществ
5 Цагарей- швили Если сложный оксид представить в форме А п-^В , где Л и/? — оксиды-компоненты; Л[ и —сте- хиометрические коэффициенты, тогда 5298 = "1 Л298 t7™77™ > + + п2 "$298 7гн 7 7т > ’ где S^9g , S298,7^ , 7^ — молекулярные стандартные энтропии и температуры плавления оксидов Л и В; 7^ — температура плавления сложного оксида Для расчета стандартной энтропии сложных оксидных соединений по их температурам плавления и $298 окси- дов - компонентов. Ошибка метода 8Дж/(моль град). Метод пригоден для расчета интерме- таллидов, карбидов, бо- ридов, силицидов и дру- гих тугоплавких соеди- нений
6 Яцимир- ского Для расчета энтропии одно- и двухвалентных ионов в кристал- лическом соединении Sz = 1,5 Я1пА-1,5 Z2 /г, где: — энтропия иона; Z — заряд иона; А — атомная масса элемента; г — радиус иона 11реимущество метода — учет влияния заряда и размера иона на энтро- пию соединения. Не да- ет надежных результа- со тов при оценке *^298 кристаллических со- единений с сущест-
347
Продолжение таблицы 8 2
1 2 3 4
венным вкладом ковалентной связи.
7 Келли п S298 = S vi si • i=l где s.— инкременты энтропии Наиболее полная на сегодня сис тема инкрементов. Недостаток — не учитывает ионность связи (поляризующее действие и по- ляризуемость соседних ионов)
8 Латимера п S298 = 2 vi si i=l где s. — инкременты энтропии (инкременты катионов не зависят от их валентности, а инкременты анионов зависят от формальной величины заряда положительного иона) Метод пригоден для кристалли- ческих соединений со значи- ной долей ковалентной связи. Погрешность не превышает 5—10 Дж/(моль град) и растет с увеличением радиуса иона и его заряда
Методы расчета теплоемкости кристаллических соединений.
Метод Ландия пригоден для расчета высокотемпературной теплоем-
кости всех типов кристаллических неорганических веществ по стандар-
тной энтропии, точность метода 5%. Идея метода заключается в том,
что вначале оценивается изохорная теплоемкость Cv, а затем вносится
поправка на разность Ср - Cv. Метод использует различные формулы для
оценки Су в зависимости от природы соединений и температуры. Алго-
ритм расчета различен для двух типов соединений — бескислородных и
сложных кислородных (если число атомов кислорода больше числа ато-
мов других элементов). Кроме того, при выборе расчетных формул учи-
тывается температура, для которой рассчитывается теплоемкость.
Возможны два случая: температура ниже характеристической темпера-
туры 0 (0 оценивается как 0 = 2535 / 5ат, где 5ат — атомная энтропия)
и температура выше характеристической.
Переход от изохорной к изобарной теплоемкости осуществляется по
формуле
I <^[ = (ЛТ+^7ЗП,
348
где (5 = 1,24 / Тпл ( C*T29g )3/2 Ю 3; Гпл — температура плавления
соединения.
Пересчет атомной теплоемкости в теплоемкость соединения:
сР=пс;т.
Метод Ивановой для расчета высокотемпературной изобарной теп-
лоемкости использует зависимость между величиной теплоемкости и
температурой первого фазового перехода (температурой плавления,
возгонки, фазового перехода), которая выражается формулой
Ср = и (5,283 + 1,987 Т/ 7фп),
где п — число атомов в молекуле; Тф.п — температура фазового перехо-
да.
Метод Ивановой применим для простых веществ и неорганических
соединений различной сложности.
Таблица 8.3. Формулы для оценки теплоемкости по методу Ландии
Темпе- рату- ра Тип соединения
бескислородные кислородсодержащие
Низкая т<е СуТ -6,6- [2200/ (5’атП] т< в С*т-б,б-[4400/(5атТ)1- [(«1 + n?)/n
Высо- кая где п — чис вычисляете и анионе сс Т>в с®т = 6,6 - 2200/{5ат[ в + + к(Т-6)]} к - в/Тпл, при As 0,34 принимают к - 0,5 амия: — доля энтропии на о; ло атомов в соединении; Т — темп я теплоемкость; nj и л2 — число а (единения. е/т< 1,6 С®т = 6,6 - { a/\bbk(T-th\} а - 507+1070/5ат; b - 0,8 а; к-(а- 298) / (5070/5ат - 298) 1ин атом; 5ат ” S-jgg / п> ература, для которой томов в катионе
Рассмотрим пример оценки стандартной энтальпии образования
иодида тантала с использованием 7-го метода сравнительных расчетов
Карапетьянца.
Сопоставим изменение стандартных энтальпий образования в рядах
галогенидов двух родственных элементов Nb и Та. Исходные стандарт-
349
ные энтальпии образования галогенидов, использованные в расчете,
представлены ниже, кДж/моль:
MJ5
-269,4
9
Оценим коэффициенты
Металл
Nb . ...
Та.
MF5
-1813,76
-1903,59
МС15
-797,47
-857,93
МВг5
-556,05
-598,31
NbXs, кДж/чопь
Рис. 8.3. Графическая интерпретация оценки стан-
дартной энтальпии образования Та15 методом срав-
нительных расчетов (X - F, С1, Вт)
в уравнении регрессии:
- ДЯ^98(^) (Та) =
— а0 - at (Nb)
методом наименьших квад-
ратов. Получим <зо = 50,75,
=0,9995. Рассчитаем с ис-
пользованием полученной
зависимости значение -
АЯ298(/)ДЛЯ Taj5
— A ^298(/) (TaJs) =
= 50,75-0,9995 269,4 =
= 218,5 кДж/моль.
Графическая интерпре-
тация метода показана на
рис. 8.3.
8.3. Химико-термодинамическое приближение
Этот подход тесно связан с термодинамическим, но его применение
основано на других исходных данных. В соответствии с физико-химиче-
скими соображениями условие химического равновесия в матричной
форме можно представить
Ад=0, (8.4)
где А — стехиометрическая матрица системы с элементами о*/, Д —
матрица химических потенциалов, присутствующих в системе веществ.
[ Согласно определению Льюиса для г-го вещества
Hi = рЯ+RT\nxi + RT\nyi, (8-5)
где/z ? — стандартный химический потенциал; yt — коэффициент актив-
ности г-го вещества; xt — его концентрация. Тогда, подставляя (8.5) в
(8.4), находим после преобразований
-
1пА=А1пХ, (8.6)
где In К = - A In у — логарифм вектора концентрационных констант
равновесия; К° = ехр[(-1/ЯТ)А/л0\ — термодинамическая константа
равновесия. Выражение (8.6) — это выражение ЗДМ в матричной фор-
ме. Для решения прямой задачи химического равновесия оно должно
быть дополнено уравнениями материального баланса (МБ). Сформули-
руем окончательно в матричном виде прямую задачу химического рав-
новесия в системе, где протекает г линейно независимых реакций меж-
ду х веществами
Ln К = A In X; (8.7)
q = В In X,
где А — стехиометрическая матрица размерностью (г > х) (задана);
(In А")т = (In , In к2,..., In Лг) — вектор логарифмов констант (известен);
(1пХ)т = (In Х[, In х2, In xs) — вектор равновесных концентраций х
веществ (неизвестен); q = (qlt q2, ..., qs) = (x10, x2o> —> -W — вектор
исходных концентраций (известен); В — балансовая матрица размер-
ностью (т к х); х = т + г; т — символ транспонирования.
Прямая задача химического равновесия формулируется как реше-
ние системы нелинейных уравнений (8.7) относительно вектора равно-
весных концентраций 1пА при известных значениях коэффициентов
матриц А и В и векторов 1пА и q. Найдя вектор равновесных концентра-
ций, можно далее вычислить любое измеряемое свойство системы Y,
зависящее от исходных и равновесных концентраций компонентов сис-
темы (это широко используется в уравнениях состав—свойство, приме-
няемых в спектрофотометрии, калориметрии, ЯМР-спектроскопии и
Т.Д.).
Расчет равновесий в многокомпонентных системах жидкость—жид-
кость осложняется во многих практически важных случаях необходимо-
стью учета неидеальности фаз. Отклонение от идеальности принято
описывать с помощью коэффициентов активности. Проблема расчета
коэффициентов активности компонентов в многокомпонентных раство-
рах и их зависимостей от состава до настоящего времени не решена
окончательно. Существующие методы расчета коэффициентов актив-
ности являются полуэмпирическими и существенно различаются для
растворов электролитов (водные растворы) и неводных растворов (орга-
нические растворители).
8.4. Прямая задача химического равновесия
Отметим, что для решения прямой задачи химического равновесия
нет необходимости проводить экспериментальные исследования, если
351
где^1 = b0 + />i ; fl2 = b0 + Ь2 ; /З3 = Ьо + Ь3 ;
б) квадратичная
У = fi\X2 + fi2X2 + Р3 Х3 + /?12 Xj Х2 + /З13 X! х3 + fl23 х2 х3 =
~ 2 flj xj + S fl ij xi xj ' (9.6)
1 s i < j < к
где flj = b0 + bj + Z>y , = by — b^ — bjj.
Уравнения (9.5)—(9.6) носят название регрессионных уравнений в
канонической форме Шеффе для трехкомпонентных систем.
Оценка адекватности регрессионных уравнений. После построения
уравнения регрессии (выбора структуры и определения коэффициен-
тов) необходимо оценить адекватность найденного уравнения и прове-
рить значимость всех коэффициентов в уравнении регрессии.
Адекватность уравнения, когда переменные Xj , х2 , ..., х^ являют-
ся независимыми, проверяется по критерию Фишера. Если известна
2
оценка дисперсии воспроизводимости sB , то значение критерия Фише-
ра рассчитывается как
F = ss£.T / sB , (9 7)
2
где sSqct — остаточная дисперсия;
п
SSOCT =^(>?—З'?) / /ост •
1=1
где yf — экспериментально измеренные значения функции отклика в z-й
точке; уР — значения отклика в z-й точке, рассчитанные по модели;
/^ — число степеней свободы остаточной дисперсии, определяемое как:
/ост =/= « “ <7, где q — число коэффициентов в уравнении регрессии; п —
число опытов.
Отметим, что число степеней свободы остаточной дисперсии имеет
формально тот же смысл, что и число степеней свободы в физической
химии (число компонентов минус число связей). Здесь роль числа ком-
понентов играет число опытов, а число связей равно количеству коэф-
фициентов регрессионного уравнения. Число q может быть вычислено
заранее: для линейной модели q = А+1; для линейной модели с дополни-
тельным включением в нее эффектов двойных взаимодействий типа
Xj Xj{j^ z) q = k(k + 1) I 2 + 1, для полного квадратного уравнения q ~
= (k + 1) (k + 2) / 2, где k — число независимых переменных.
Оценка дисперсии воспроизводимости зависит от постановки парал-
лельных опытов [см. формулы (1.9—1.12) ]. Расчетное значение крите-
рия Фишера сравнивается с табличным значением Етабл при заданном
уровне значимости fl и числе степеней свободы Д = /ост ; f2 = /в. Если
356
Fp < Ртабл, то уравнение адекватно. Отметим, что речь идет не об адек-
ватности модели процессу, а об адекватности модели описываемым экс-
периментальным данным.
Если дисперсию воспроизводимости не оценивали, то расчетное зна-
чение критерия Фишера определяется с использованием дисперсии от-
носительно среднего
F = Sy / ss^-j, (9.8)
где$^= 1 /(л - 1) У (jf->?) —дисперсия относительно среднего; уэ—
1=1
среднее значение функции отклика в п экспериментах. Далее находится
табличное значение критерия Фишера при заданном уровне значимости
b и числе степеней свободы f\=n-\vif2-n-q. В этом случае для
адекватности модели необходимо, чтобы Fp > Ртабл-
Проверка значимости регрессионных коэффициентов. Проверка
значимости коэффициентов bj регрессионного уравнения производится
по критерию Стьюдента. При этом расчетные значения /-критерия вы-
числяют по формуле
tj= IZ>yl /^slcjj , (9.9)
где Cjj — диагональные коэффициенты ковариационной матрицы.
Найденное расчетное значение /-критерия сравнивается с таблич-
ным /табл, взятым при заданном уровне значимости и числе степеней
свободы /=/в Если расчетное значение / для некоторого /-го коэффици-
ента регрессии будет меньше /табл, то этот коэффициент со статистиче-
ской точки зрения незначим, и его можно вывести из уравнения. Так как
все коэффициенты уравнения регрессии коррелированы между собой,
то после исключения незначимого коэффициента все остальные должны
быть пересчитаны, т.е. вся процедура оценки коэффициентов модели (с
учетом исключения незначимых) и регрессионного анализа результатов
должна быть повторена.
9.2. Планирование экспериментов
Плохая обусловленность информационной матрицы, возникающая
при произвольном выборе плана эксперимента приводит к большим
ошибкам определения коэффициентов регрессионного уравнения и тем
самым к ошибкам моделирования изучаемого процесса. Эффективность
регрессионного моделирования может быть значительно увеличена с
помощью применения методов математического планирования экспе-
римента. Методы планирования экспериментов широко описаны в лите-
ратуре, поэтому мы остановимся только на некоторых из них.
357
Рис. 9.1. Полный факторный эксперимент для:
а) п - 2, к - 2; б) п - 2, к - 3
Полный факторный эксперимент (ПФЭ). Одним из наиболее часто
применяемых планов, используемых как самостоятельно, так и в соста-
ве других планов является ПФЭ. Полным факторным экспериментом
называется эксперимент, реализующий все возможные неповторяющи-
еся комбинации уровней независимых переменных. Общее число экспе-
риментов N при ПФЭ определяется как N=mk, где т — число уровней,
на которое разбивается значение независимой переменной; к — число
независимых переменных.
Для построения плана эксперимента удобно от физических пере-
менных перейти к безразмерным. На практике очень часто имеют дело
с планами 1-го порядка, когда варьирование переменных идет на двух
уровнях, т.е. значение переменной х, определяется как Хипах или хдшп.
Тогда переход к безразмерным переменным xt выполняется по форму-
лам
х, = ( х( - xOi) / Л х(, (9.10)
ГД® -*0| = ( ’•imax + \min ) /2, А Xj = ( Х(шах - Хг1Пjn ) /2.
Точка с координатами ( х01 , х02 , ..., хдо ) носит название центра
плана, а Ах(- — интервал варьирования по оси х{.
Легко заметить, что при переходе к новым координатам х( (г = 1. 2,
..., к) центр плана окажется в начале координат, а безразмерные пере-
менные xt будут принимать только два значения +1 или -1. На рис.
9.1 в качестве примера приведены планы ПФЭ для случаев п = 2, £ = 2
(рис. 9.1, а) и и = 2, к = 3 (рис. 9.1, б).
В табл. 9.1 приведены матрицы планирования для двух указанных
случаев. Точки плана в таблице соответствуют точкам плана на рисун-
ках.
358
Из рис. 9.1 и табл. 9.1. видно, что ПФЭ несложно достроить для
любого числа переменных. Использование ПФЭ позволяет не только
снизить число экспериментов по сравнению с интуитивным планом про-
ведения эксперимента, но и значительно упростить и повысить эффек-
тивность регрессионной обработки данных. Применение ПФЭ приводит
к ортогональности информационной матрицы, т.е. ее члены вычисляют-
ся следующим образом:
и I п
У Лу = п для/ = /, У xi:Xik = 0 для/# к. (9.11)
<=1 \ 1=1
Запишем информационную и ковариационную матрицы для линей-
ной регрессионной модели при к = 3
8 0 0 0 1/8 0 0 0
0 8 0 0 0 1/8 0 0
(Хт X) = 0 0 8 0 (Хт X) 1 = 0 0 1/8 0
0 0 0 8 0 0 0 1/8
Любой из коэффициентов уравнения регрессии определяется не из
решения системы нормальных уравнений, а ищется раздельно по фор-
муле
fy=(l/n) У xijfi , (9.12)
где Ху — безразмерные значения переменной ху, взятые из матрицы
планирования, а у? — экспериментально измеренные значения целевой
функции у.
Таблица 9.1. План ПФЭ для двух и трех факторов
Но- мер то- чки Значения переменных Но- мер то- чки Значения переменных
2 ПФЭ 2х з ПФЭ 2 2 ПФЭ 2 з ПФЭ 2
*1 х2 *1 х2 х3 -Ч х2 *1 х2 х3
1 -1 -1 -1 -1 -1 5 — — -1 -1 + 1
2 + 1 -1 + 1 -1 -1 б — — + 1 -1 + 1
3 -1 + 1 -1 + 1 -1 7 — — -1 + 1 + 1
4 + 1 + 1 + 1 + 1 -1 8 — — + 1 + 1 + 1
Так как ковариационная матрица превратилась в диагональную, то
коэффициенты уравнения регрессии некоррелированы между собой и их
можно вычислять отдельно. Тогда при оценке значимости коэффициен-
тов по критерию Стьюдента незначимые можно отбрасывать без повтор-
ного пересчета всех коэффициентов. Так как диагональные элементы
ковариационной матрицы равны между собой, упрощаются формулы
359
для оценки значимости к<
4
для оценки значимости коэффи-
циентов регрессии по критерию
Стьюдента /
Рис. 9.2. Композиционный план 2-гопоряд-
ка для к- 2:
1—4 — точки ПФЭ; 5—8 — звездные
точки
tj=\bj\ / (sB/У/п). (£.13)
Композиционное планирова-
ние эксперимента. Одн;
ный факторный эксперимент тре-
бует слишком большого числа
опытов. Значительное сокраще-
ние числа опытов дает примене-
ние предложенных Боксдм и Уил-
соном композиционных планов.
Композиционный план строится
на основе ПФЭ-ядра плана с до-
бавлением к нему 2к звездных то-
чек, расположенных на коорди-
натных осях, и точек в центре пла-
на. На рис. 9.2 приведен компози-
ционный план 2-го порядка для к = 2.
Расстояние от центра координат до звездной точки носит название
звездного плеча. Общее число экспериментальных точек в композици-
онном плане 2-го порядка равно и = 2* + 2 + пц (где п0 — число точек в
пол-
7
8
1
центре плана). Число м0 выбирается исследователем.
Рассмотрим пример построения регрессионной модели процесса
вскрытия редкоземельных концентратов спеканием с содой1*. Регресси-
онная модель данного процесса представляет собой функциональную
зависимость степени извлечения РЗЭ от основных технологических па-
раметров процесса: температуры (хр и продолжительности спекания
(х2), массового соотношения сода:концентрат (х3). Интервалы варьиро-
вания переменных определены технологическими соображениями и со-
ставляют: 550 < xj s 850 °C, 20 s х2 < 60 мин, 0,1 < х3 s 0,4.
На первом этапе исследования проведен ПФЭ с варьированием перемен-
ных на двух уровнях; результаты эксперимента представлены в табл. 9.2
(опыты 1—8). По результатам ПФЭ построены регрессионные модели
1-го (9.1) и неполного 2-го порядка (9.2). В ходе дальнейшего исследо-
вания матрица планирования была дополнена звездными точками и
точкой в центре плана (точки 9—13 табл. 9.2),что дало возможность
оценить коэффициенты полинома полной второй степени (9.3)-
Авжиева Е.М., Коровин С.С., Корнюшко В Ф и др. Влияние породообразующих
минералов на разложение фосфатов иттрия и лантана спеканием с содой //Йзв вузов.
Цветная металлургия. — 1989. — № 2. — с. 66—70.
360
Использованная для оценки адекватности дисперсия воспроизводимо-
сти заимствована из примера (гл. 7). Таким образом, полином второй
степени адекватно описывает весь имеющийся набор эксперименталь-
ных данных. Структура построенной модели может быть упрощена на
счет отбрасывания статистически незначимых членов. Табличное зна-
чение (-критерия составляет (табл (13,0 05) = 2,16. Исключим из модели
(9.3) пятый член, имеющий минимальное значение (-критерия (табл.
9.3), и найдем оценки коэффициентов новой модели, имеющей вид
у = -23,09 + 93,9E-03xj + 16,6Е-01х2 - 31,2х3 - 20,8E-04jq х2 +
+ 14.2Е-02Х! х3 + 33,0х2-37,9Е-02х2 х3- 10,9Е+01х|. (9.14)
Дальнейшее упрощение модели за счет отбрасывания статистически
незначимых коэффициентов приводит к резкому увеличению остаточ-
ной дисперсии. Следовательно, полученная модель может быть призна-
на оптимальной по структуре.
Таблица 9.2. Результаты ПФЭ и опытов в звездных точках
Номер точки Т, "С Время, мин Сода кон- центрат Степень извл., % Номер точки 5 Время, мин Сода кон- центрат Степень извл., %
I 550 20 0,10 43,6 9 700 40 0,25 63,1
2 850 20 0,10 63,4 10 850 40 0,25 68,8
3 550 60 0,10 72,1 11 550 40 0,25 56,5
4 850 60 0,10 68,2 12 700 60 0,25 73,4
5 550 20 0,40 38,7 13 700 20 0,25 55,0
6 850 20 0,40 72,4 14 700 40 0,10 60,7
7 8 550 850 60 60 0,40 0,40 63,8 71,4 15 700 40 0,40 60,1
Таблица 9.3 Статистические характеристики
регрессионных моделей
Номер точки Модель (9.1) Модель (9.2) Модель (9.3)
оценка /-критерий сценка /-критерий оценка /-критерий
1 14,2 10,1Е+1 -22,9 47,1 -26,4 47,3
2 47.7Е-3 11,7 95.2Е-3 83.9Е-1 10.4Е-2 15.4Е-1
3 35.9Е2 11,8 19.1Е-1 12,4 16.5Е-1 62.5Е-1
4 - 90.8Е-2 22.3Е2 -85,2 40.5Е-1 -32,3 10.1Е-1
5 - 20.8Е-4 10,2 — 74.6Е-7 15.6Е-2
6 14.2Е-2 52.4Е-1 - 20.8Е-4 10.2
7 - 37.9Е-2 18.6Е-1 14.2Е-2 52.4Е-1
8 34.2Е-4 12.7Е-1
9 37.9Е-2 18.6Е-1
10 - 10.6Е+1 22.2Е-1
Трасч Ттабл 100,70 33,79 0,64 4,67 1,82 3,02
361
9.3. Планирование эксперимента на симплексе
Все рассмотренные планы применяются в случае, если измеряемые
переменные независимы. Выше отмечалось, что при исследовании про-
цессов в технологии РЗЭ часто приходится иметь дело с зависимыми
переменными (иссле-
дование составов, по-
строение диаграмм
состав—свойство и
т.д.). В этом случае
используются регрес-
сионные модели спе-
циального симплекс-
ного вида. Существу-
ет несколько видов
симплексных планов.
Наиболее эффектив-
ными являются симп-
Рис. 9.3. Симплекс-решетчатые планы для построения мо-
делей Шеффе 2-го (с) и 3-го (б) порядка
лексно-решетчатые планы, или планы Шеффе. Эти планы являются
насыщенными, т.е. число точек плана равно числу определяемых коэф-
фициентов модели. В планах Шеффе экспериментальные точки симмет-
рично располагаются по симплексу в интервале уровней от 0 до 1 и
принимают значения 0, \/q, 2/q, ..., q/q (где q — заданная степень
полинома). На рис. 9.3 и в табл. 9.4 приведены примеры построения
симплексно-решетчатых планов для трехкомпонентных систем для мо-
делей 2-го и 3-го порядка.
Таблица 9.4. Симплексно-решетчатые планы 2-го и 3-го порядка
Номер . точки Значения переменных для модели 2-го порядка Значения переменных для модели 3-го порядка
*1 х2 х3 х\ х2 х3
1 1 0 0 1 0 0
2 0 1 0 0 1 0
3 0 0 1 0 0 1
4 1/2 1/2 0 1/3 2/3 0
5 1/2 0 1/2 1/3 0 2/3
б 0 1/2 1/2 0 1/3 2/3
7 2/3 1/3 0
8 1/3 0 1/3
9 0 2/3 1/3
10 1/3 1/3 1/3
362
Математические модели при этом имеют вид:
модель 2-го порядка
3
y=2Pixi+ 2 Pijxixi> (9Л5)
i=l lsz</s3
модель 3-го порядка
3
yS0ixi + 2 fiijxixj + 2 Yijxiixj(.xt~xj) +
1=1 l<i<j<3 1SK/S3
4 2 Pijlxixjxl’ <916>
lsi</<Zs3
где выполняется условие У xt = 1 , а коэффициенты /3 и у находятся
либо из решения системы нормальных уравнений, либо по специальным
формулам.
Пример. Построение математической модели поверхности ликви-
дуса тройной системы As—Sb—InAs.
Поскольку из априорных сведений известны диаграммы плавкости
всех бинарных систем, образующих данную тройную систему, то для
повышения информативности планируемых опытов эксперименталь-
ные точки целесообразно сдвинуть внутрь симплекса. Был выбран сим-
плексно-решетчатый план эксперимента, позволяющий построить мо-
дель третьего порядка. Расположение точек в концентрационном треу-
гольнике As(X])—Sb(x2)—InAs(x3) представлено на рис. 9.4, результа-
ты опытов — в табл. 9.5. Температуры ликвидуса определяли тер-
мическим анализом. В качестве оценки дисперсии воспроизводимости
опыта приняли инструментальную ошибку использовавшейся методики
4" 100.
Таблица 9.5. План эксперимента на симплексе
и результаты опытов
Номер опыта Точки на симплексе Тликв Номер опыта Точки на симплексе Тликв
jcj (As) x2(Sb) jc3 (InA) xj (As) х2 <Sb) x3(InAs)
1 0,90 0,05 0,05 800 6 0,05 0,69 0,26 680
2 0,05 0,90 0,05 604 7 0,26 0.69 0,05 610
3 0,05 0,05 0,90 936 8 0,26 0,05 0,69 900
4 0,69 0,26 0,05 735 9 0,05 0,26 0,69 870
5 0,69 0,05 0,26 755 10 0.33 0,33 0,33 770
363
В ходе математиче-
ской обработки полу-
чена регрессионная
модель поверхности
ликвидуса тройной
системы. Адекват-
ность построенной
модели проверена по
контрольным опытам
по t- и F-критериям.
Модель адекватна
при 5%-ном уровне
значимости. Линии
уровня для расчетной
поверхности ликви-
дуса представлены на
рис. 9.4. Вблизи сто-
роны As—Sb доста-
точно четко выделя-
ется линия кристал-
лизации двойной эв-
тектики в тройной си-
стеме.
Рис. 9.4.1 Товерхность ликвидуса системы As—Sb—InAs(a)
и план эксперимента (б)
9.4. Методы экспериментальной оптимизации
Главной задачей физико-химических исследований, управления и
проектирования технологических процессов является достижение и
поддержание наилучших в некотором заданном смысле значений пока-
зателя качества процесса, т.е. оптимизация процесса. Для решения
задачи оптимизации существует несколько подходов. Из них два явля-
ются наиболее известными:
1) на основе физико-химических соображений построена математи-
ческая модель процесса. В этом случае для решения задачи используют-
ся известные аналитические или численные методы оптимизации;
2) математическая модель процесса отсутствует, а задача оптимиза-
ции решается с помощью экспериментально-статистических методов.
364
Пусть зависимость показателя качества (целевой функции) процес-
са у от переменных, характеризующих процесс X] , х2, •- > хп имеет
вид
у= /( X] , х2...хл) (9.17)
Тогда при первом подходе необходимо найти частные производные
от цеЛевой функции по переменным X] , х2 , ..., хп и приравнять их к
нулю. Решение полученной системы уравнений аналитически или чис-
ленными методами даст значения х* , х2, ..., х*п такие, которые обес-
печат оптимум целевой функции (9.17). Однако во многих прак-
тических случаях получить модель процесса в виде (9.17) не удается. В
этом случае целенаправленно изменяя значения переменных
X] , х2 , ..., хп и измеряя при этом значения целевой функции у, опти-
мизацию выполняют на основе второго подхода. По существу этот под-
ход близок к задаче построения регрессионной модели на основе методов
планирования экспериментов. Возможны два пути решения оптимиза-
ционной задачи на основе такого подхода. При первом в предполагаемой
(или предварительно найденной) зоне оптимума строят регрессионную
модель и решают задачу оптимизации стандартными аналитическими
или численными методами. Второй путь не предполагает построения
регрессионной модели, а основан на постановке специальных дополни-
тельных экспериментов. Первый путь достаточно широко освещен в
литературе, поэтому мы остановимся ниже только на втором пути реше-
ния оптимизационной задачи.
Для определенности сформулируем задачу оптимизации как задачу
поиска максимума целевой функции при отсутствии ограничений на
варьируемые переменные. Практически нельзя встретить задачу опти-
мизации, где физические переменные не имели бы ограничений. В тех-
нологии всегда есть ограничения, ниже или выше которых техноло-
гический параметр (температура, давление, расходы и т.д.) не может
изменяться, не приводя к нарушению технологий. Однако во многих
случаях исследователю удается выбрать начальную точку поиска опти-
мума достаточно далеко от границ области, заданной ограничениями
4 mm 5 -4 < 4 max , *2min 5 х2 < *2тах- т(,гда ПРИ правильной организа-
ции поиска и достаточно гладкой поверхности функции отклика влия-
нием ограничений можно пренебречь.
Метод крутого восхождения (метод Бокса—Уилсона). В методе
крутого восхождения движение организуется оптимальным образом по
пути наибольшего возрастания целевой функции, т.е. по градиенту.
Метод сочетает в себе определение направления движения на основе
методов планирования эксперимента и организацию самого движения
365
по выбранному направлению (направлению градиента) до достижения
частного максимума, как в методе Гаусса—Зайделя.
Определение направления движения основано на том факте, что
коэффициенты линейного уравнения регрессии представляют собой ча-
стные производные от целевой функции (составляющие градиента) по
соответствующим переменным. Так, для функции двух переменных в
регрессионной модели вида
У = + ^1 *1 + ^2 х2 > (9.18)
Z>0 = /(х° , %2) , Ьг = д у/д xj , b2 = <5 у/д х2 , (9.19)
О о
где xj , Х2 — начальная точка поиска.
Вычисление коэффициентов Ьц , by , b2 производится методом на-
именьших квадратов, а экспериментальные данные для вычислений
находят с использованием методов полного или дробного планирования
экспериментов. Рассмотрим общую процедуру организации поиска мак-
симума экстремума методом крутого восхождения.
1. Выбирается нулевая точка Mq . Задание этой точки достаточно
произвольно. Если область оптимума приблизительно известна, то це-
лесообразно задавать ее в центре области, если нет, то где-нибудь в
середине плоскости, ограниченной технологическими ограничениями
переменных х, и х2 , X]mjn s Xj < Xjmax , Х2Ш,П — x2 < X2max .
2. Выбирается интервал варьирования для каждой переменной А Х|
и А х2 . Интервал варьирования Д х, должен экспериментально подби-
раться так, чтобы изменение целевой функции у на этом интервале было
значительно больше, чем величина ошибки измерения, т.е. необходимо
выделить значение целевой функции на фоне помех.
3. Определяются точки плана эксперимента на основе нулевой точки
Мо методами ПФЭ и ДФЭ и в одной из точек, например, в центре плана
ставится несколько параллельных опытов для оценки дисперсии восп-
роизводимости Sg .
4. Измеряются значения целевой функции у в точках плана.
5. Рассчитываются на основе МНК значения коэффициентов
bo , bi , b2 .
6. Рассчитывается дисперсия воспроизводимости и на основе кри-
терия Стьюдента производится оценка значимости коэффициентов bj.
Если хотя бы один из коэффициентов bi или Ь2 окажется незначимым,
то необходимо повторить процедуру сначала, меняя начальную точку
или интервал варьирования, либо проверив гипотезу об однородности
дисперсии воспроизводимости в нескольких точках плана.
366
7. Вычисляются рабочие шаги движения. Рабочие шаги движения /у
по выбранному направлению в реальном масштабе определяются как
Лу= bjbXj (J = 1 , 2) . (9.20)
8. Вычисляются координаты рабочих точек движения к частному
экстремуму по найденному направлению в реальном масштабе.
Xj = Ху + kXj , (к = 1, 2, ...). (9.21)
9. Путем постановки проверочных опытов через 3—4 расчетных
шага движения измеряются значения целевой функции по выбранному
направлению.
10. Проверяется по
заданному критерию
достижение частного
экстремума. Наиболее
простым из таких кри-
териев является усло-
вие уменьшения зна-
чений целевой функ-
ции. На рис. 9'5 пока-
зано движение из точ-
ки Мо (х^°\х^ ) в точ-
ку Л/4 (х^). Точка
М4 принята за точку
экстремума, так как
/(х(3\х<3))</(х<4\44))и
/(х(5\х(5))</(х<4\х<4)).
11. Точка М4 при-
Рис. 9.5. Организация поиска максимума функции двух
переменных х] и х2 методом крутого восхождения (Л/о.
Л/4 — точки центров плана; Ау (/-1,2,...) — рабочий шаг
движения)
нимается за новый центр плана (новую нулевую точку), и весь цикл,
начиная с п. 1, повторяется уже из этой точки. Отметим, что, так как во
время первого цикла движения мы приблизились к экстремуму, для
второго цикла величину дху следует выбирать меньше, чем в первом.
12. После окончания второго цикла движения, если есть необходи-
мость, организуется третий, четвертый и т.д. Критерием достижения
оптимума является статистическая незначимость всех коэффициен-
тов bj.
Пример. Оптимизация процесса электрохимического окисления
церия методом крутого восхождения.
Для выделения церия из азотнокислых растворов, содержащих лег-
кие редкоземельные элементы, широко используется метод электрохи-
мического окисления. Процесс проводится в электролизерах; управ-
367
ляющими параметрами процесса являются исходная концентрация
трехвалентного церия и сила тока. Контролируемая величина — кон-
центрация четырехвалентного церия на выходе из аппарата. Интервалы
изменения параметров процесса определяются технологическими сооб-
ражениями и составляют: для силы тока (хр 70—110 А, для исходной
концентрации Се (III) (х2) 90—160 г/л.
В качестве начальной точки для реализации метода крутого восхож-
дения выберем точку с координатами: X] = 90, х2 = 130. Зададим интер-
валы варьирования переменных axj = 10 и дх2 = 20. Проведем полный
факторный эксперимент вокруг центральной точки. Результаты ПФЭ
приводятся в табл. 9.6.
По результатам опытов построена линейная регрессионная модель
следующего вида: у = 85,5 + l,8xi + 2,4х2. Значения целевой функции
при движении по выбранному направлению и результаты проверочных
опытов представлены в табл. 9.7.
Таблица 9.6 Результаты ПФЭ
№ *1 х2 Уэксп
1 95,0 140,0 89.8
2 85,0 140,0 86,2
3 95,0 120,0 85,0
4 85,0 120,0 в. 81,4 —
Таблица 9.7. Трассировка поиска
Шаг *1 Х2 ^эксп Урасч
2-й 93 136 89,1 88,1
3-й 96 142 89,9 90,6
4-й 99 148 89,4 93,1
5-й 102 154 87,8 95,6
Оптимальное значение параметра оптимизации в первом цикле кру-
того восхождения у = 89,9% достигается при следующих значениях
факторов: Х| = 96, х2 = 142. Координаты новой начальной точки крутого
восхождения xj = 96, х2 = 142. Интервал варьирования 1-го фактора 10.
Интервал варьирования 2-го фактора 5. По результатам опытов второго
цикла оптимизации построен линейный полином вида: у =89,3 +
+ 0,72х] - 0,12х2. Оптимальное значение параметра оптимизации во
втором цикле крутого восхождения у = 90,1 % достигается при следую-
щих значениях факторов: Xj = 99, х2 = 142.
368
КОНТРОЛЬНЫЕ ВОПРОСЫ И ЗАДАЧИ
К главе 1
1. Чем обусловлено отличие свойств лития от свойств остальных щелочных элементов’’
2 Как изменяется характер химической связи в соединениях для ряда литий — цезий’’
Какие свойства лития и его соединений сближают его с магнием?
3 Какие количественные характеристики отличают поведение иона лития в водных
растворах от ионов других щелочных элементов?
4. Почему литий очень редко бывает центральным атомом комплексных соединений
и практически никогда внешнесферным ионом?
5. Какой характер имеет химическая связь и какие свойства характерны для соеди-
нений лития с водородом, углеродом, кремнием и другими неметаллами?
6 Какие фазы образует оксид лития с оксидами элементов IV и V групп Периодической
системы и каковы их свойства?
7. Какие свойства обусловливают применение лития и его соединений в традиционных
и перспективных областях?
8. Какая разница и почему имеется во взаимодействии а- и b-сподумена с различными
реагентами?
9. Сопоставьте сернокислотный и известковый способы переработки сподумена (число
операций, расход и стоимость реагентов, энергозатраты, конечные продукты, использова-
ние отходов, проблемы экологии).
10. В чем заключаются достоинства и недостатки электрохимического получения
металлического лития?
11. Обоснуйте с точки зрения термодинамики возможность получения металлического
лития вакуумтермическим способом.
12. Каковы перспективы развития литиевой промышленности (сырье, применение,
технология)?
К главе 2
1. Свойства каких соединений рубидия и цезия наиболее сильно отличаются от свойств
аналогичных соединений остальных щелочных элементов?
2. В состав каких комплексных соединений входят рубидий и цезий, каков характер
химической связи в них?
3. Почему рубидий не образует собственных минералов, имеющих промышленное
значение, в отличие от цезия, хотя его кларк почти в 10 раз меньше?
4 Какие свойства рубидия и цезия определяют их главные области применения?
5. Какие способы могут быть использованы для разложения поллуцита? Сопоставьте
кислотные методы и методы спекания.
6. От каких примесей и почему наиболее трудно очистить рубидий и цезий? Какие
способы наиболее эффективны?
7. Какие способы могутбыть использованы для получения соединений рубидия и цезия
квалификации ОСЧ?
369
8 Какие особенности имеет вакуумтермический метод получения металлического
цезия9 Какие другие методы могут быть предложены?
К главе 3
1 Как изменяются свойства элементов IIА подгруппы по группе? В чем причина этих
изменений?
2. В чем причина резкого отличия в химических свойствах бериллия от свойств других
элементов II А подгруппы?
3. Что такое «диагональный эффект» и как он проявляется у бериллия?
4 Что такое бериллаты и как их можно получить?
5. Назовите важнейшие минералы бериллия и дайте им характеристику (состав,
физико-химические свойства).
6 Назовите основные области применения бериллия. Какие физико-химические
свойства определяют его использование в этой или иной области? Каковы масштабы
производства бериллия9
7 Охарактеризуйте основные принципы технологии переработки берилла сульфат-
ным и фторидным способами. Каковы достоинства и недостатки обоих методов?
8. Какие методы можно использовать для получения металлического бериллия?
9. Используя данные по Д6° реакций восстановления оксида, фторида и хлорида
бериллия выберите систему для металлотермического получения бериллия. Что кроме
AG° реакции влияет на выбор системы?
10. В чем причина сложности получения металлического бериллия с высокими
конструкционными свойствами?
К главе 4
1. Какие химические свойства стронция отличают его от бериллия9 В чем причина
отличия?
2 Назовите способы, позволяющие отделить стронций от кальция
3. Какие можно предложить способы обезвоживания Sr(OH)2 8Н2О9
4. Какие концентраты экономически целесообразнее перерабатывать — целестино-
вые или стронцианитовые? Изложите преимущества и недостатки переработки каждого
концентрата.
5. Какие металлы можно использовать для восстановления SrO и почему?
К главе 5
1. На основании какого фундаментального свойства выделяется группа лантаноидов?
Каковы особенности электронного строения атомов элементов этой группы9 Адекватны ли
термины «лантаноиды» и РЗЭ?
2. Объясните причины, приводящие к уменьшению атомных радиусов лантаноидов
(«лантаноидное сжатие») при увеличении Z. К каким следстиям оно приводит?
79
3. Как изменяются свойства лантаноидов и их соединений при увеличении *-•
Объясните различия в ходе кривых е -f(Z). Обоснуйте разделение РЗЭ на две подгруппы-
370
4. Исходя из электронного строения атомов лантаноидов объясните, какие из них могут
иметь степени окисления, кроме основной +3? С возможностью каких электронных
переходов они связаны?
5. При окислении на воздухе Рг и ТЬ образуют оксиды с эмпирическими формулами
РгбО11 иТЬ4О7. Можно ли эти соединения рассматривать как стехиометрические? Какова
их структура, какие дефекты преимущественно она имеет?
б. Охарактеризуйте ядерные свойства РЗЭ- Исходя из данных по эффективным
сечениям захвата тепловых нейтронов дайте рекомендации по применению Sm, Ей и Gd в
ядерной энергетике.
7 Назовите важнейшие минералы РЗЭ и дайте им характеристику (состав, структура,
физико-химические свойства, происхождение, «селективность»). Охарактеризуйте важ-
нейшие месторождения, концентраты и их получение.
8. Назовите основные области применения РЗЭ. Какие физико-химические свойства
определяют использование их в той или иной области? Каковы масштабы производства
РЗЭ и каково распределение их по областям применения?
9. Какие атомы и почему замещает скандий в галлий-гадолиниевом гранате
Gd3Ga5O|2 (ГГГ)? Число формульных единиц в элементарной ячейке граната Z- 8, а
максимальное число атомов скандия в ГСГГ равно 16. Напишите формулу ГСГГ в общем
виде и при максимальном содержании скандия.
10. Охарактеризуйте основные принципы технологии переработки монацитового
концентрата щелочным и сернокислотным способами. Каковы достоинства и недостатки
обоих методов.
11- В чем заключается сущность метода выделения малых количеств веществ из
растворов на носителе? Как можно использовать этот метод для выделения малых коли-
честв Ей из смеси РЗЭ?
12. Какие способы используются в промышленности для разделения РЗЭ на группы
и для получения индивидуальных РЗЭ?
13 . В чем заключаются основные принципы разделения близких по свойствам элемен-
тов, в том числе и РЗЭ, методом ионного обмена''
14. Напишите уравнение экстракции нитратов РЗЭ ТБФ. Выведите формулу для
термодинамической константы экстракции К и для коэффициента распределения D. Как
изменяется D при увеличении концентрации HNO3 и введении высаливателей?
15 Какие преимущества имеет экстракционный метод разделения РЗЭ перед ионо-
обменным9
1 б. В интервале температур от 400 до 2400 К АС реакции восстановления Еа2ОЗ
алюминием имеет значение от +60 до +40 кДж/моль. Каким образом можно модифици-
ровать этот процесс, чтобы получить лантан в виде продукта, не содержащего кислорода9
17. Металлические РЗЭ получают восстановлением фторидов или хлоридов, самарий
(также Eu, Yb) — только восстановлением оксидов. Почему? Выберите и количественно
обоснуйте наиболее подходящий восстановитель и определите условия проведения про-
цесса получения самария.
371
18. При каких условиях теоретически возможно получение практически всех метал-
лов, в том числе и РЗЭ, методом карботермии?
19. Какие способы используются для рафинирования металлических РЗЭ? Какова их
эффективность?
К главе 6
1. За счет чего возникают потери скандия при растворении его фторпроизводных в
растворе NaOH?
2. Почему не достигают полного осаждения скандия из растворов в форме
Sc2(C2O4)3 6Н2О?
3. Напишите химические реакции, описывающие очистку скандия при карбонатной
обработке его гидроксида, содержащего РЗЭ, Fe, Мп.
4. Можно ли разделить алюминий и скандий, осаждая их гидроксиды? Почему?
5. Каким образом можно очистить скандий, находящийся в солянокислом растворе,
от Fe, U, Си, Zn?
6. Какими экстрагентами можно заменить диэтиловый эфир при тиоцианатной
очистке скандия?
7. Как экстракционным методом отделить Sc от Zr? От Th?
8. За счет чего могут возникнуть потери Sc при его извлечении из отработанных
хлоридных плавов титанового производства?
9 Как влияет железо, присутствующее в плаве хлоридов титанового производства, на
последующее экстракционное извлечение скандия?
10. Предложите технологические схемы извлечения и очистки скандия: при перера-
ботке циркона по известково хлоридной схеме, при переработке цирконового концентрата
хлорным методом.
11. Как влияет температура восстановления ScF3Ca на чистоту конечного продукта?
12. Предложите методы получения металлического скандия повышенной чистоты.
13. Почему для извлечения скандия при переработке уранового сырья используют
экстракцию, хотя концентрация скандия в исходном растворе составляет всего 10 г/л?
К главе 9
I. Что называется доверительным интервалом и доверительной вероятностью?
2. При извлечении Y2O3 раскислением содовых спеков РЗЭ в открытой системе (/>
и в автоклаве (2) были получены следующие результаты: степень извлечения Y2O3
способом /, %: 89.2; 93,8; 91,7; 87,3; 95,3, 91,8, степень извлечения Y2O3 способом?, %:
90,3; 95,0; 94,3, 87,1; 92,2, 90,7. Необходимо найти оценки математического ожидания и
дисперсии для обеих выборок и, задавшись доверительной вероятностью 5-0.9, опреде-
лить, различимы ли статистически полученные результаты.
3. Как определяется дисперсия воспроизводимости при параллельных опытах?
4. Вывести расчетные формулы для оценки коэффициентов уравнения Y - bQ + b\ X
методом наименьших квадратов.
372
5 Как формулируются прямая и обратная задачи химического равновесия? На чем
основаны различные подходы к описанию равновесных систем?
6. Какие основные методы применяются для полуэмпирической оценки стандартных
энтальпий образования, стандартных энтропий и теплоемкости кристаллических соеди
нений?
7. Как записываются в матричной форме основные уравнения ЗДМ и МБ ? Как
проверить единственность решения прямой задачи химического равновесия?
8. Задана следующая схема образования некоторого комплекса А2В из молекул А и В:
2А - А2:
А + В-АВ
АВ+А-А2В
А2 + В - А2В
с константами равновесия реакций соответственно ki, к$, к^ и исходным составом
лА0 и пВ0 (в молях). Проверить единственность решения прямой задачи химического
равновесия для этой системы и с учетом результатов проверки записать в матричной форме
математическую модель данной системы.
9. Какие основные задачи решаются методом линейного регрессионного анализа?
Каковы основные предпосылки применения метода?
10. При гравитационном обогащении редкометаллических щелочных метасоматитов
исследовалось влияние класса крупности руды на степень извлечения Та2О5 и Nb2O5
Построить регрессионные зависимости содержания Та2О5 и Nb2O5 в обогащенной руде
от класса крупности и проверить адекватность полученных моделей на основе следующих
исходных данных: класс крупности : -6; -3,5; -1,0; -0,5; -0,25; -0,1; содержание Та2О5.
%: 0,03; 0,023,0,014; 0,01; 0,05,0,05; содержаниеNb2O5, %: 0,49; 0,39; 0,22; 0,13; 0,091;
0,075.
11. В чем особенности построения регрессионных моделей при использовании взаи-
мозависимых переменных?
12. Составить план полного факторного эксперимента для линейной четырехфактор-
ной модели.
13. Сопоставьте число опытов при построении полной квадратичной модели для двух
независимых переменных при использовании полного факторного эксперимента Зк и
композиционного плана 2-го порядка.
14. Поверхность ликвидуса системы Ni—N13A1—Ni3Nb может быть описана полино-
мом третьей степени. Построить план эксперимента на симплексе для оценки коэффици-
ентов полинома.
15. Как выбрать направление при движении к оптимуму в методе крутого восхожде-
ния?
373
РЕКОМЕНДАТЕЛЬНЫЙ БИБЛИОГРАФИЧЕСКИЙ СПИСОК
Общая и справочная литература
Вольдман Г.М., Зеликман А Н. Теория гидрометаллургических процессов. — М.:
Металлургия, 1993. — 400 с.
ГольдшмидтХ.Дж. Сплавы внедрения. Кн.1 и 2: Пер,сангл. — М.: Мир, 1971. — 424;
404 с.
Зеликман А Н., Коршунов Б.Г. Металлургия редких металлов. — М.. Металлургия,
1991—431 с.
Коршунов Б.Г., Стефанюк С.Л. Введение, в хлорную металлургию редких металлов.
— М.: Металлургия, 1970. — 343 с.
КребсГ. Основы кристаллохимии неорганических соединений: Пер. снем. — М.: Мир,
1971. — 304 с.
Кубашевский О., Олкок С.Б. Металлургическая термохимия: Пер. с англ. — М.:
Металлургия, 1982. — 390 с.
Меретуков М.А. Процессы жидкостной экстракции в цветной металлургии. — М.:
Металлургия, 1985. — 222 с.
Новое в развитии минерально-сырьевой базы редких металлов. — М.: ИМГРЭ РАН,
1991. —227 с.
Основы металлургии. Т.4 Редкие металлы /Под ред. Н.С. Грейвер, Н.П. Сажина,
И.А. Стригина. — М.: Металлургия, 1967. — 644 с.
Проблемы применения хлорных методов в металлургии редких металлов /Под ред.
Д .В. Дробота. — М.: Металлургия, 1991. — 190 с.
Свойства элементов /Под ред. М.Е. Дрица. — М.: Металлургия, 1986. — 762 с.
Смольянинов Н.А. Практическое руководство по минералогии. — М.: Госгеолтехиз-
дат, 1955. — 430 с.
СоколовИ.П., Пономарев Н.Л. Введение в металлотермию. — М.: Металлургия, 1990.
— 134 с.
Термодинамические свойства индивидуальных веществ. Т. 1—4/Под ред. В.П.
Глушко. — М.: Наука, 1978—1982.
Термодинамические константы веществ. Вып. 1—10 /Под ред. В.П. Глушко. — М.:
АН СССР, ВИНИТИ, 1965—1979.
Третьяков Ю.Д. Твердофазные реакции.—М.: Химия, 1978. — 359 с.
Химия и технология редких и рассеянных элементов. Т. 1 —3 /Под ред. К.А. Больша-
кова. — М.: Высшая школа, 1976. — 368, 359; 320с.
Ягодин Г. А, Синегрибова О. А., Чекмарев А.М. Технология редких металловв атомной
технике. — М.: Высшая школа, 1974 — 344 с.
К разделу I
Никифоров А.С., Куличенко В.В., Жихарев М.М. Обезвреживание жидких радиоак-
тивных отходов. — М.: Энергоатомиздат, 1985. — 186 с.
ОстроушкоЮ М., Бучихин П.И., Алексеева В.В. и др. Литий, его химия и технология.
— М.: Атомиздат, 1960 — 197 с.
Плющев В.Е., Степин Б.Д. Химия и технология соединений лития, рубидия, цезия.
— М.: Химия, 1970. — 405 с.
Урсу И. Физика и технология ядерных материалов: Пер. с рум. — М.; Энергоатомиз-
дат, 1988. — 476 с.
Шамрай Ф.И. Литий и его сплавы. — М.: АН СССР, 1952. — 234 с.
374
К разделу II
Беляев Р.А. Окись бериллия. — М.: Атомиздат, 1980. — 224 с
Бериллий. Наука и технология /Под ред. Д. Вебстера, Г.Дж. Лондона, Д.Р. Флойда
и Дж.Н. Лоува: Пер. с англ. — М.: Металлургия, 1984. — 624 с.
Бериллий /Под ред. Д. Уайта Дж. Берка: Пер. с англ. — М.: ИЛ, I960. — 616с.
Бериллий. Токсикология, гигиена, профилактика, диагностика и лечение бериллие-
вых поражений: Справочник /Под ред. А.И. Бурназяна. — М.: Атомиздат, 1980. — 115с.
Гольдинов А.Л., Копелев Б.А., Абрамов О Б., Дмитриевский Б.А. Комплексная
азотнокислотная переработка фосфатного сырья. — Л.: Химия, 1982. — 206 с.
КоганБ.И.КапустинскаяК.А., ТопуноваГ.А. Бериллий. — М.: Наука, 1975. — 371с.
Основы металлургии. Т.З /Под ред. Р.И. Беляева, Н С. Грейвера. — М.: Металлургия.
— 1963 — 591 с.
Папиров Н.И. Бериллий в сплавах: Справочник.—М.: Энергоатомиздат, 1986.—
184 с.
К разделу III
Бандуркин Г.А., Джуринский Б.ф., Тананаев И.В. Особенности кристаллохимии
соединений редкоземельных элементов. — М.: Наука. 1984. — 230 с.
Браун Д. Галогениды лантаноидов и актиноидов: Пер. с англ. — М.: Атомиздат, 1972.
— 272 с.
Коршунов Б.Г., Резник А.М., Семенов С.А. Скандий. — М.: Металлургия, 1987. —
184 с.
Костромина Н.А. Комплексонаты редкоземельных элементов. — М.: Наука, 1970. —
220 с.
Михайличенко А.И., МихлинЕ. Б., Патрикеев Ю.Е. Редкоземельные металлы. — М.:
Металлургия, 1987. — 282 с.
Портной К.И., Тимофеев Н.И. Кислородные соединения редкоземельных элементов:
Справочник. — М.: Металлургия, 1986. — 480 с.
Редкоземельные элементы. Технология и применение: Пер. с англ. /Под ред. Ф
Виллани. — М.: Металлургия, 1985. — 375 с.
Соединения редкоземельных элементов. Карбонаты, оксалаты, нитраты, титанаты
/Комиссарова Л.Н., Шацкий В.М., Пушкина Г.Я. и др. — М.: Наука, 1984. — 235 с.
Ярембаш Е.И., Елесеев А.А. Халькогениды редкоземельных элементов. — М.: Наука,
1975.— 258 с.
К разделу IV
Ахназарова С.Л., Кафа ров В.В. Методы оптимизации эксперимента в химической
технологии. — М.: Высшая школа, 1985. — 327 с.
Воронин Г.В. Основы термодинамики. — М.: — Изд-во МГУ, 1987. — 192 с.
Глазов В.М., Павлова Л.М. Химическая термодинамика и фазовые равновесия. — М.:
Металлургия, 1988. —580 с.
Горский Г.В. Планирование кинетических экспериментов. — М : Наука, 1984.—
241 с.
Драйпер Н., Смит Г. Прикладной регрессионный анализ: Пер. с англ. — М.: Финансы
и статистика, 1987. — 351 с.
Евсеев А.М., Николаева Л.С. Математическое моделирование химических равнове-
сий. — М.: Изд-во МГУ, 1988. — 192 с.
Кабанова О.В., Максимов Ю.А., Рузинов Л.П. Статистические методы построения
физико-химических моделей металлургических процессов. — М.: Металлургия, 1989.
215с.
375
Математика в химической термодинамике /Под ред. Г.А. Коковина. — Новосибирск:
Наука, 1980 — 193 с
Морачевский А.Г., Сладков И.Б. Термодинамические расчеты в металлургии: Спра-
вочник — М.: Металлургия, 1985. — 136с.
Новик ф С Планирование эксперимента на симплексе при изучении металлических
систем. — М.: Металлургия, 1985. — 256 с.
Рузинов Л II, Слободчикова Р. И. Планирование эксперимента в химии и химической
технологии. — М Химия, 1980 — 280 с.
Учебник для вузов
КОРОВИН Сергей Сергеевич
ЗИМИНА Галина Владимировна
РЕЗНИК Александр Маркович
БУКИН Вячеслав Иванович
КОРНЮШКО Валерий Федорович
РЕДКИЕ И РАССЕЯНЫЕ ЭЛЕМЕНТЫ
Химия и технология. Книга I
Редактор Л.М. Цесарская
Художественный редактор Л.В- Коновалова
Корректор Т.М. Подгорная
Переплет художника ВЮ. Яковлева
ИБ№10
ЛР № 020777 от 13 мая 1993 г.
Подписано в печать 28.10.96 Формат бумаги 60*88 1/16 Бумага книжн.-журн.
Печать офсетная Печ л.23,5 Уч.-изд. л.24,8
Тираж 1000 экз. Зак. 691 Изд. № 188/О9А
•МИСИС*, 117936 Москва, Ленинский проспект, 4
Отпечатано в московской типографии № 2 РАН
121099, г. Москва, Г-99, Шубинский пер , 2