



















































































































































































































Текст
химическая
метрология
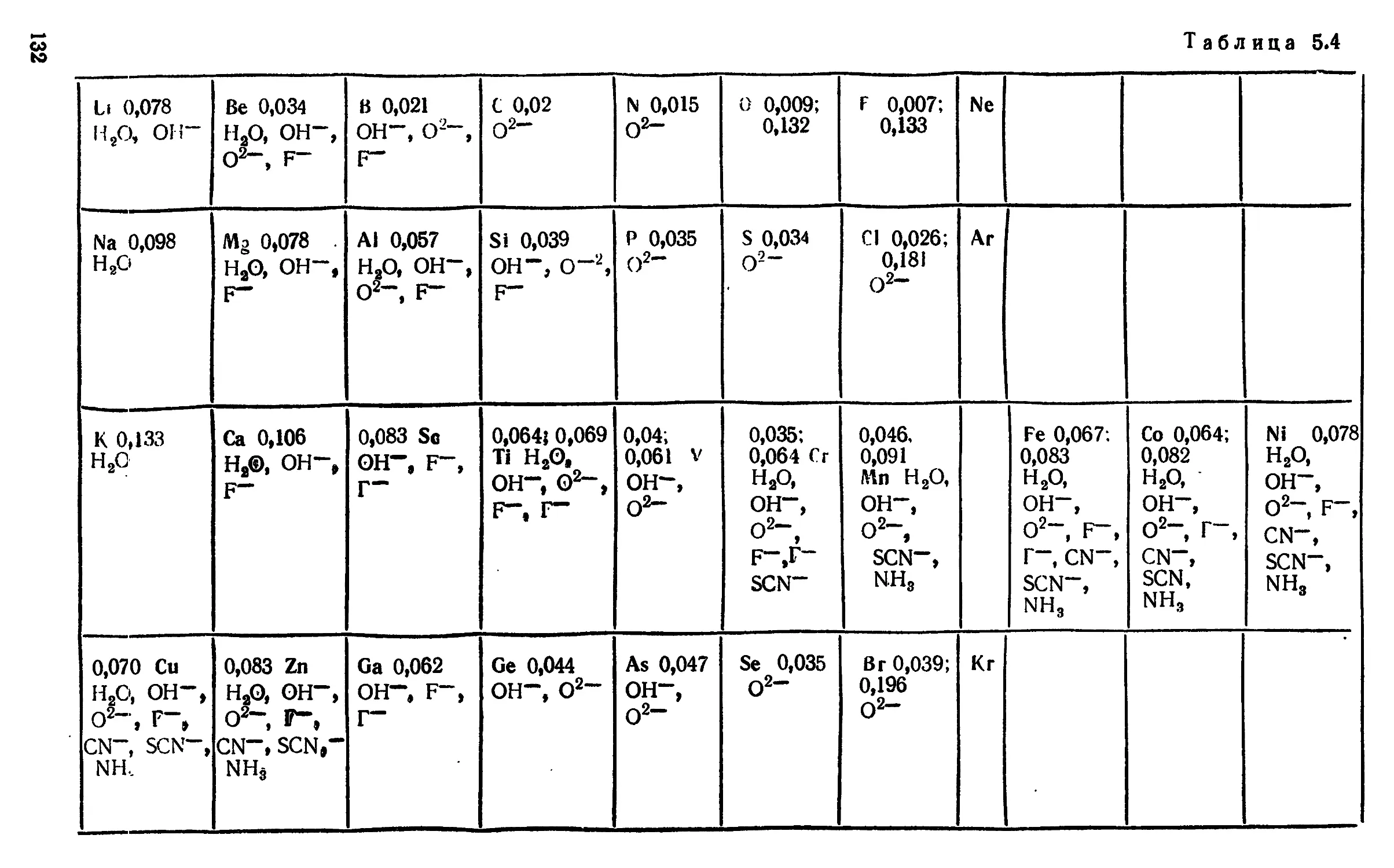
Н.П. Комарь
ГОМОГЕННЫЕ
ИОННЫЕ
РАВНОВЕСИЯ
УДК 543/545+541.122
Химическая метрология. Гомогенные ионные
равновесия. Комарь Н. П. — Харьков: Вища школа. Изд-во при
Харьк. ун-те, 1983.—208 с.
В монографии излагаются идеи и основные
принципы химической метрологии — нового оригинального
направления в современной аналитической химии.
Подчеркивается необходимость последовательного
метрологического подхода к химико-аналитическим проблемам.
Рассмотрены основные положения теории растворов и
теории ионных равновесий, изложен единый подход
к расчету равновесий любой степени сложности.
Теоретические положения иллюстрируются большим
числом конкретных, примеров расчета систем,
используемых в качественном и количественном химическом
анализе.
Нормативные материалы приведены по состоянию
на 1 января 1983 г.
Для химиков-аналитиков, научных работников и
специалистов. Табл. 46. Ил. 7. Библиогр.: 69 назв.
Научный редактор: д-р хим. наук, проф. Л. Я. Адамович
Рецензенты: чл.-кор. АН УССР д-р хим. наук, проф.
В, А. Назаренко, д-р хим. наук, проф. А, И. Бусев
Редакция естественнонаучной литературы
И. о. зав. редакцией Е. Я. Иващенко
© Издательское
1804000000-070 объединение
К М226(04)—83 ^ «Вища школа», 1983
ПРЕДИСЛОВИЕ
Монография «Гомогенные ионные равновесия» была
задумана Н. П. Комарем как первая из цикла книг, объединенных
общим названием «Химическая метрология». Он мечтал о
превращении рецептурной аналитической химии в науку об
измерении химической формы движения материи, основанную на
строгом учете физико-химических и метрологических
закономерностей, и вместе с возглавляемым им коллективом первой в
стране кафедры химической метрологии много сделал для того,
чтобы эта мечта стала реальностью.
Основной задачей химической метрологии, по мнению
автора, является обеспечение единства и правильности измерений
химического состава, для чего необходимо иметь: одну
основную химическую единицу, производные химические единицы и
систему мер, их воспроизводящих; набор эталонных методик
измерения, обеспечивающих связь между химическими мерами;
сокращенное до минимума число стандартных методик
измерения химического состава, сведенное к небольшому количеству
(за счет сохранения наиболее достоверных данных) число
констант, используемых при химических расчетах.
Основными разделами химической метрологии автор считал
теорию гомогенных и гетерогенных ионных равновесий,
измерение параметров равновесий, гравиметрию, титриметрию,
инструментальные методы измерения химического состава, теорию
и технику подготовки систем к исследованию химического
состава, применение математической статистики для оценки и
представления результатов измерений химического состава.
Указанные вопросы Н. П. Комарь предполагал осветить в
отдельных книгах задуманного цикла.
К сожалению, смерть помешала автору полностью
осуществить этот грандиозный замысел. Он успел дописать первые три
книги, приблизился к завершению четвертой, остались также
отдельные главы и наброски последующих книг. Но даже
законченные Н. П. Комарем книги не вышли в свет при жизни
автора. Почетная, хотя и нелегкая задача подготовки
рукописей к печати выпала на долю его сотрудников и учеников.
Данная монография по существу является первой частью
руководства по теории ионных равновесий и объединяет главы,
относящиеся к гомогенным равновесиям. Вторая часть,
посвященная гетерогенным равновесиям, готовится к печати. В
редактировании и подготовке к печати этой монографии приняли
участие сотрудники кафедры химической метрологии Харьков-
ского университета и аналитической лаборатории ВНИИмонокристаллов (г. Харьков) А. Б. Бланк, Т. В. Васильева, С. И. Вовк,
Л. Л. Лукацкая, Л. Е. Никишина, И. Г. Перьков, М. И. Рубцов.
Благодаря энергии и неистощимой вере в успех учениц Н. П. Комаря Т. В. Васильевой и С. И. Вовк замысел автора стал
реальностью. Изданию книги и устранению имевшихся
погрешностей в значительной мере способствовали ее рецензенты — член-
корреспондент АН УССР В. А. Назаренко, доктора химических
наук А. И. Бусев и К. П. Столяров.
Многое сделал для того, чтобы книга увидела свет,
профессор, доктор химических наук Л. П. Адамович, осуществивший
научное редактирование.
Молярные концентрации растворов, приводимые в книге,
выражены в моль/л*. В тех же единицах выражена и ионная
сила растворов (в молярной шкале). В расчетных схемах и при
перечислении нескольких веществ единицы концентраций
опущены. В соответствии со сложившимися традициями не
приведены единицы используемых констант равновесий (их легко
можно установить из стехиометрии соответствующих реакций),
а также коэффициентов молярной экстинкции (моль-1 • л•см-1).
Использование единицы СИ для давления — паскаля (Па)
вместо нестандартизованной единицы — физической атмосферы
(атм)—при расчетах равновесий с участием газообразных
веществ связано с пересмотром принятой водородной шкалы
электродных потенциалов и пересчетом соответствующих
констант равновесий. Пока это не сделано, и поэтому в данной
книге использована прежняя единица давления с
одновременным - указанием ее взаимосвязи с единицей СИ.
Принципиальных затруднений такое положение не вызывает, поскольку
рассчитываемые концентрации получаются выраженными в
моЛЬ•Л-1.
В отдельных расчетных примерах иллюстративного
характера учтены не все указанные в литературе равновесия (главным
образом, из-за неоднозначности имеющихся данных). Двойные
стрелки в уравнениях реакций указывают на наличие
химического равновесия (в термодинамическом смысле), а не на
обратимость процесса, который может и не быть. Б большинстве
расчетных примеров, приведенных в книге, автор использовал
константы равновесий из учебного пособия А. А. Бугаевского [3].
Все вопросы, замечания и пожелания, возникшие у читателя
при знакомстве с монографией, просим направлять по адресу:
310077, Харьков-77, пл. Дзержинского, 4, Харьковский
государственный университет им. А. М. Горького, химический
факультет, кафедра химической метрологии.
* Эта общепринятая единица концентрации растворов является
производной единицей, составленной из основной единицы СИ (моль) и
внесистемной единицы (л), допускаемой к применению наряду с единицами СИ
и связанной простым соотношением с одной из них (1 л= 1 дм3).
1. СИСТЕМЫ, ИЗУЧАЕМЫЕ В ТЕОРИИ
ИОННЫХ РАВНОВЕСИЙ
1.1. ОСНОВНЫЕ понятия
Теория ионных равновесий — учение о применении законов
стехиометрии, т. е. «той части химии, которая изучает весовое
и объемное количественное отношение между реагирующими
веществами» [24, с. 44], для расчета равновесий в системах,
содержаш^их электролиты (растворах, расплавах).
Системой условимся называть совокупность веществ,
находящихся в данном ограниченном элементе пространства.
Например, в качестве системы можно рассматривать раствор какого-
нибудь вещества, находящийся в закрытой склянке в контакте
с воздухом, который над ним находится, кристаллами
растворенного вещества и стенками сосуда. Очевидно, что система
может состоять из нескольких фаз.
В тех случаях, когда раствор практически не
взаимодействует со стенками сосуда и воздухом, который с ним
соприкасается, часто считают, что изучаемая система состоит только из
двух фаз: раствора и кристаллов, которые в нем находятся, или
только раствора, если твердой фазы в нем нет. В дальнейшем
газообразную фазу будем обозначать буквой г в круглых
скобках, стоящих за формулой соединения, образующего газовую
фазу или входящего в ее состав, например, NO (г); твердую
фазу — (т), например, BaS04 (т). При формулах частиц,
находящихся в растворе, никаких дополнительных обозначений
вводить не будем. Наличие какого-либо вещества, находящегося в
контакте с водным раствором в виде отдельной жидкой фазы,
будем отмечать символом (ж). Если начальный состав системы
известен, то, пользуясь законами стехиометрии, его всегда можно
записать в форме, наилучшим образом представляющей
особенности системы.
Записи эти должны отображать состав каждой системы с
наибольшим приближением к истине. Для этого надо ввести
соответствующую символику, ряд определений, полные и
сокращенные формы записи равновесий для любых изучаемых систем,
в нашем случае растворов.
1.2. РАСТВОРИТЕЛИ
Любой раствор считают гомогенной системой, состоящей из
жидкого растворителя и растворенных в нем веществ, которые
сами по себе могут быть газообразными, жидкими и твердыми.
Растворители делятся на протолитические и апротонные.
Протолитическими считаются растворители — электролиты,
молекулы которых находятся в равновесии с соответствующими
лионий- и лиат-ионами. Как правило, эти равновесия
представляют в виде схемы
2HL«H2L+ + L- (1.1)
Апротонные растворители (СеНе, CCI4,...) лионий- и лиат-ионов
не дают.
Если учесть, что, согласно Бренстеду, любое вещество,
дающее протоны (р), называется кислотой, акцептор этих
протонов— основанием, а система кислота :<=fcp+основание —
сопряженной кислотно-основной парой, то для любого протолитиче-
ского растворителя можно записать два равновесия:
H2L+<=±p + HL (1.2), HL^p+L~ (1.3),
из которых следует, что лионий-ион — всегда кислота, лиат-
ион — всегда основание, а растворитель может быть и
акцептором и донором протонов, т. е. является амфолитом, для^
которого характерно равновесие диспропорционирования (1.1).
Наиболее интересный растворитель — вода — порождает
три сопряженных кислотно-основных пары*.
НзО+5=^р + Н20 (1.4), H20;=tp + 0H- (1.5),
0Н-вр. + 02-. (1.6)
Записывают же равновесие растворителя воды в виде
2Н20вНзО+ + ОН- (1.7)
Еще в 1894 г. Кольрауш и Гейдвайлер показали, что в чистой воде
оксоний-ионы *НзО+ и гидроксил-ионы ОН- находятся в очень
незначительных и практически равных количествах. ,
1.3. СИЛЬНЫЕ И СЛАБЫЕ КИСЛОТЫ
При растворении в протолитическом растворителе кислот
или оснований количественное соотношение между лионий- и
лиат-ионами изменяется. Сильными в данном растворителе
считаются кислоты (мы припишем им общую формулу НУ),
способные протонизировать растворитель HL, практически
полностью диссоциируя при этом:
Некоторые авторы называют HgO"^ гидроний-ионом.
HY ^p + Y-
HL + p ^H2L+
HY ^p + Y-
H2O + P »НзО+
HY + HL5z=t Y- + H 2L+ HY + H2O « Y- + H3O+
Для этих равновесий характерны равенства
[HY] - О и [H2L+] - [Y-] - chy; / .
[HY]-0 и [H30+]^[Y-]^chy, ^ ' ^
согласно которым сразу же после растворения сильной кислоты
в протолитическом растворителе в нем возникают практически
равные концентрации лионий-иона и аниона сильной кислоты.
Важнейшие сильные кислоты в водных растворах — НзРе(СМ)б,
Н4ре(СЫ)б, HCIO4, НСЮз, НС1, НВгОз, НВг, HI, НМПО4, H2SO4,
H2Se04, HNO3.
При этом многопротонные кислоты, как правило, оказываются
сильными только на первой ступени диссоциации. Это значит,
например, что, приписывая символ HY серной кислоте, мы считаем
У"" = HSO4.
Для слабых кислот, общую формулу которых запишем в виде
НаА, где а может принимать различные значения, начиная с
единицы, можно записать такие же равновесия, как для HY:
НаА 5=±p + Ha-iA- НАс вр + Ас-
P + HL ?=±H2L+
НаА + HL <=± Ha-iA- + H2L+
Р + Н2О ^НзО+ . (1.9)
НАс + Н20»Ас-+ Н3О+
Но в этом случае для однопротонной кислоты, например уксусной
(СН3СО2Н = НАс), в водном растворе [НАс] ФО,а [Ас-] = [Н3О+],
где формулы в [ ] — равновесные концентрации. Для НаА также
[НаА]#0, а [Ha-iA-] [H2L+], если учесть дальнейшую
диссоциацию.
Это значит, что в водном растворе слабой кислоты ее молекулы
всегда сосуществуют с Н2Ь+-ионами.
1.4. СИЛЬНЫЕ И СЛАБЫЕ ОСНОВАНИЯ
Если основание, введенное в растворитель HL, способно
практически полностью превратиться в сопряженную кислоту,
депротонизуя эквивалентное количество HL, то его считают
сильным основанием. Схема этого процесса выглядит так:
HL ^p+L-
Осн.+ р 5=гКисл. (1.10)
НЬ + Осн.;=±Ь- + Кисл.
При этом характерны равенства [Осн.] ^ О и [Кисл.] ^ [L-J ^ Оосн.
Сильными основаниями в водных растворах являются амид-ион
(NHr) и оксо-ион (О^-). Растворение натрий-амида и натрий-оксида
в воде изображается, например, следующими схемами:
N320 2Na+ + 02-
Н2О вр + ОН-
р + 02- 5=±0Н-
NaNHs Na+ + NHf
Н2О ^р + ОН-
p + NH2~ :^NH3
H20 + NHr^OH- + NH3 H2O + О 2-^ OH- + OH- = 20H-
При этом сразу же после растворения [NHF] ^ О и [О^-] ^ 0.
Во втором случае можно считать, что оксо-ион порождает один
ОН--ион как сопряженную ему кислоту, а другой ОН--ион —
в результате депротонизации растворителя.
Растворение слабого основания, например NH3, в воде
описывают схемой
Н2О 5=±р + 0Н-
NH3 + P ^NHt
Н 2О + NH3 z=± ОН- +NHt
При этом О, а [ОН-] ^ [NHt].
Из всего сказанного следует, что единственной сильной
кислотой, которая может существовать в данном растворителе, является
лионий-ион H2L+, так как все более сильные кислоты при
растворении в HL депротонизуются. Подобным же образом самым сильным
основанием следует считать лиат-ион L-, так как все более сильные
основания протонизуются за счет растворителя.
Сильными основаниями считают также гидроокиси с общей
формулой ХОН, где Х+= К"*", Rb+, Cs+, Fr+, так как при их
растворении в воде выполняются условия [ХОН] ^ О, [Х+] ^
^ [ОН-] = схон.
Гидроокиси лития и натрия можно считать сильными основаниями
только в их разбавленных растворах, так как акволитий- и акво-
натрий-ионы вляются очень слабыми кислотами. К сильным
основаниям относятся также Са(0Н)2, Sr(0H)2, Ва(0Н)2, для которых
Х+=МеОН+.
Все основные кислоты и основания, с которыми мы будем
встречаться, считаются слабыми, т. е. способными сосуществовать
с Н3О+- и ОН--ионами в водных растворах.
1.5. СОЛЬВАТАЦИЯ (ГИДРАТАЦИЯ) ИОНОВ
Соли катионов щелочных металлов (Х+) и NH| обычно считают
сильными электролитами. Это значит, что их растворение можно
представить схемой
XY(T) eX+ + Y-
X+ + XH2O . 5=tX(H20);^
Y-+yH20 5=±Y(H20)7
XY (T) + (x + y) H20« X (H20)++Y {H2O)/
учитывающей гидратацию ионов, образующихся при разрушении
кристаллической решетки XY (т). Электролит считается сильным,
если в его растворе [XY] ^ 0. Последнее выражение указывает
на отсутствие комплексообразования.
При растворении в воде сильных кислот (HY) и сильных
оснований (ХОН) подобным же образом получаем
HY + (4 + у) Н2О ^ Н (H20)t + Y (Н20)7, (1.12)
ХОН + (X + 3) H20<=t X (Н2О)? + ОН (Н20)з"", (1.13)
где числа гидратации протона и ОН~-иона соответствуют
литературным данным и считаются достаточно точными [38, 40, 68]. При
учете тех же чисел гидратации диссоциация воды должна
выражаться схемой
8Н20^Н (H20)t + ОН (Н20)Г. (1.14)
При растворении солей остальных катионов, которые мы
обозначим М»^+, и анионов сильных и слабых кислот, т.е. Y~ и А*-,
всегда следует учитывать гидратацию, возможность
комплексообразования в растворе между катионом и анионом растворяемой соли,
ионизацию гидратированного металло-иона, т. е. аквометалло-комп-
лекса M(H20)S"*", и иротонизацию аниона, если он не является
анионом сильной кислоты.
Запомнить все возможные в таких сложных системах
превращения нельзя. Соответствующие сведения обычно находят
в таблицах констант комплексообразования [66], где
приведены не только значения констант или их логарифмов, но и
указаны равновесия, которым та или иная константа отвечает.
Наличие константы в таблице указывает, что данное
равновесие обнаружено и его следует учитывать. Отсутствие
константы чаще всего говорит о тохм, что данное равновесие не
изучалось. В тех случаях, когда в [66] есть указание, что комплексы
не обнаружены, можно смелее пренебречь их возможным
существованием.
Если равновесия записываются с целью их последующего
использования при расчетах, то очень важно представить их
в том виде, которому отвечает табличная константа, и в форме,
облегчающей последующие вычисления.
Первое требование связано с тем обстоятельством, что песо-
ответствие между схемой равновесия и используемой
константой ведет к ошибкам в расчетах.
Второе требование связано с естественным желанием
представить схемы равновесий так, чтобы последующая запись
закона действующих масс прямо давала значение активности
наиболее сложной частицы, участвующей в равновесии, в виде
произведения константы равновесия на активности более
простых частиц, из которых образуется сложная.
1.6. НЕКОТОРЫЕ ПРИМЕРЫ ЗАПИСИ РАВНОВЕСИЙ
Пример 1. 1. Равновесию диссоциации уксусной кислоты отвечает константа
диссоциации К = 10"^'^^, Можно записать поэтому
НАс«Н+ +Ас- /С. (1.15)
Сложной частицей здесь является НАс, но схема (1.15) позволяет выразить
(НАс) через /С» (Н"^) и (Ас") лишь после преобразования соответствующего
выражения закона действующих масс.
Та же схема, переписанная в виде Ас" +Н+5=±НАс, а=/С~^ (1.16),
т. е. как равновесие протонизации, позволяет сразу написать: (НАс) =
= а (Ас~) (Н^") (1.17). При этом используется правило: активность' продукта
равновесия, формула которого стоит справа, равна константе равновесия
образования этого продукта, умноженной на произведение активностей исходных
веществ, возведенных в степени, равные соответствующим стехиометрическим
коэффициентам.
Пример 1. 2. Равновесию диссоциации врды отвечает схема НоОт-^Н"^ 4-
4-ОН", W (1.18); где мы будем считать ш равным ионному произведению воды-
Если считать ОН"-ион более сложной частицей, то полезно переписать (1.18)
в виде HgOН"^ 0Н~ (1.19). Тогда по изложенному выше правилу
(ОН-)==ш(Н+)-^ (1.20).
Пример 1. 3. Подобным же образом для равновесия ионизации аквбме-
талло-иона
+ iHoO^M (ОН)^-^ + Ш+, 7)., (1.21)
где фигурной скобкой объединены металло-ион и диссоциирующие молекулы
воды из его гидратной оболочки, более сложной является частица М (ОН)f"'^
Если переписать (1.21) в виде
Mf^+-iH+5z-M(0H)f-\ 7,j, (1.22)
где М'^'^=М (НгО)^"^, а в силу этого молекулы воды, входящей в гидратную
оболочку, при расчете константы if)j не учитывались, то
(М (OH)f= т). (М^+) (H+)-i. (1.23)
Записи подобного рода дают ясное представление о
равновесиях, константы которых нужны при вычислениях. Составители
таблиц (см. работу [66]) этому обстоятельству внцмания не
уделяли. В таблицах работы [4], специально составленных
для учебных целей, все условия, связанные с ' расчетами,
учтены.
10
1.7. АЛГЕБРАИЧЕСКАЯ ФОРМА ЗАПИСИ РАВНОВЕСИЙ
Выражениями многоступенчатых равновесий в алгебраической
форме, подобными (1.22) и (1.23), мы будем пользоваться часто,
так как они обеспечивают компактность записей. И в этом случае
следует предпочесть константы образования. Например, для Н3РО4
обычно учитывают три ступени диссоциации с cooтвeтcтвyющимиJ
им константами:
НзР04«Н+ + Н2РОГ, Ки
Н2Р07^Н+ + НРО^Г,
НРО^Г«Н+ + РОГ, /Сз.
Обобщить эти уравнения можно только путем сложения, но это
поведет к недоучету Н2РОГ и НР04'"-ионов, безусловно,
существующих в системе.
Записи можно сократить, воспользовавшись обратимостью
процесса диссоциации слабых электролитов. Так, считая исходными
частицами Н+-и Р04"~-ионы, можно полностью учесть все частицьу,
образующиеся при диссоциации фосфорной кислоты, с помощью
записи
РО^- + jH+^HjPOr\ где j =Т:з, (1.24)
причем черта над цифрами 1, 3 обозначает, что j «пробегает»
все значения от 1 до 3, т. е. 1, 2 и 3.
Эта запись вполне законна, так как гомогенные ионные
равновесия устанавливаются за время, измеряемое
микросекундами (с-^).
Записи усложняются при образовании многоядерных
комплексов. Так, если учесть все равновесия диссоциации акво-
плюмбо-иона, приведенные в работе [66], то для записи и;с
понадобится три строчки, причем записи эти могут включать
и формулу для расчета активности наиболее сложной частицы,
участвующей в данном равновесии:
Pb2+-iH+5=tPb(OH)?"'^ 7|И,
(РЬ (OH)f-O = (РЬ2+) (H+)-^ (1.25)
gPb2+-4H+^Pbg(OH)4^^^ i3g4,
(Pbg mfr') = ^g4 (Pb2+)s (H-b)-4, (1.26)
6РЬ2+-8Н+»РЬб(ОН)|-*-, -^68,
(РЬб (ОН)!"^) = i(]68 (РЬ2+)б (H+)-». (1.27)
Фактически формулы (1.25)—(1.27) изображают систему из шести
уравнений, так как i = 1. 3 и g = 3^ 4, т. е. i пробегает значения
1, 2, 3, а g —3 и 4.
и
к сожалению, не всегда сведения о равновесиях, в которых
участвуют одни и те же ионы, полученные разными авторами,
совпадают. Так, согласно работе [47] ионизация аквовисмути-
иона приводит к равновесиям
Bi„OS±^i + Bi3+ + Н20« Bin-biOS"^' + 2Н+, n = ГГЗ, (1.28)
а в работе [59] показано, что, по-видимому, реализуются только
равновесия
Bi3+_H+»BiOH2+ (1.29), 6Bi3+—12Н+«В1б(ОН)12^ (1-30).
В монографии Я. Вьеррума [5] принято, что при взаимодействии
меркуро-иона с аммиаком наблюдается ступенчатое образование
HgNH^з+-, HglNHs)!"^-, Hg(NH3)|+-h Hg(NH3)'4'^-hohob, а в [69]
показано, что опытные данные Вьеррума могут с таким же успехом
. служить доказательством существования только комплексов
Hg(NH3)r и Hg(NH3)'4+
О множестве трудностей, с которыми химики сталкиваются
при попытках точно представить очень многие системы, можно
получить представление, рассматривая многочисленные схемы
реакций в работе [19]. Для их изучения используются
различные химические и физико-химические методы (см., например,
[9, 30, 31, 35]) и делаются попытки математической оценки
результатов истолкования опытных данных [48, 69].
Таким образом, выбор правильных схем равновесий и
значений констант, им отвечающих, не всегда однозначен. Угадать,
какая из схем и констант лучше, не всегда легко, и, как
правило, только путем сопоставления расчетных и опытных данных
можно получить правильную оценку достоверности схем и
значений констант.
1.8. ПРИМЕНЯЕМЫЕ СИМВОЛЫ
Для унификации всех записей введем следующие обобщенные
обозначения различных частиц:
Х+— гидратированный катион любого сильного основания XL
или ХОН, например, К+, Rb+, Cs+, ВаОН+, Na+ (условно);
М«^+ — любой гидратированный катион; У- — анион любой сильной
кислоты, например, С1-, NOi", НЗОГ; А*- —анион а-протонной
слабой кислоты, например, 0С1-, S0|~ CN~, SO4""; А
—молекула (незаряженная) основания, например NH3, N2H4.
Начальные концентрации различных веществ будем обозначать
строчной буквой с с формулой соответствующего вещества в виде
подстрочного индекса или с помощью заглавной буквы (без всяких
индексов), отвечающей природе данного соединения. Например^
Сдс, = У, так как НС1—сильная кислота; Cj^qj^ = X — КОН—
12
сильное основание; 0^^^^= Л-Н3РО4- слабая кислота; ^г^н^а = ^>
так как 1 моль NH4CI эквивалентен 1 молю NHs; сщ^^^^^ =Л1,
так как M(C104)ix — соль катиона МУ-^; cj^^^ = М = Л = с: в
зависимости от того, какая из вводимых с MaAj^ частиц нас интересует,
но при этом, естественно, с^=^ аМ = аА== ас, а = (хМ = [хЛ =
= Р-с в зависимости от выбранного нами первичного обозначения
^MaAj^' Активности частиц мы выразим соответствующими
формулами в круглых скобках, а чаще — строчными латинскими
буквами, отвечающими приведенным выше обобщенным символам: (Х+)=
= x, (М^+) = т, (Y-) = у, (А«-) - а, (А) = а, (Н+) = А, (ОН-) =
= О). Рав новесные концентрации покажем формулами, взятыми
в квадратные скобки: [Х+1, т^Ч [У-], [А--], [А], [Н+], [ОН-],
[Н2РО7], [HS071 и т.п.
Для констант простейших равновесий, определяющих
состояние любых равновесных систем, используем следующие
обозначения, принятые и в таблицах [4]: W" —константа автопротолиза
любого растворителя HL, в том числе и воды; w — ионное
произведение растворителя HL, Н2О и т. п.; ш = (Н+)(Ь-),
= (Н+)(ОН-), ш=И^(НЬ)2, а; = Г (Н20)2; _ константа
ионизации аквометалло-иона М^*+, ведущей к образованию М(ОН)Г'"\
где i = 1, (х' и 1а' > (X, что отвечает образованию гидроксоанионов
•;до= 1: о\ — константа протонизации А*-, ведущей к образованию
HjA^-*, где j = 1, а', а' > а, если может образоваться катионная
кислота На'А^*'-*>+, оо= 1; Рп —константа комплексообразования,
ведущего к образованию МА^~"'", MAS"^, MYJ;""", если М^"+-ион
координирует А*-, ¥--ионы или молекулы основания А. Здесь
п = 1, N, N — координационное число; Ро ^ 1. Символом Рп
обозначим константы комплексообразования, ведущего к появлению
частиц Anlfi, например Si"", или У^+ь когда анион координирует
п атомов соответствующего элемента; К\ — константа на 1-й
ступени любого процесса диссоциации, например, К2 для Н3РО4
отвечает равновесию Н2РОТ5=^Н+ + НРОГ; /( — константа сопря-
женной окислительно-восстановительной реакции Ок + ve Вое,
где Ок — окисленная и Вое — восстановленная формы данной
сопряженной системы и V —число электронов; Р— произведение
растворимости труднорастворимого соединения, для МаА^д, всегда
\ — константа распределения незаряженной частицы между
раствором и газовой фазой, раствором и какой-либо жидкой фазой,
раствором и твердой фазой, если распределяющиеся частицы
образуют с ней твердый раствор; % — константа любого
простейшего равновесия, не относ.яшегося к упомянутым выше^ например;
HF + F-^HFi".
13
1.9. МАТРИЧНАЯ ЗАПИСЬ СИСТЕМ РАВНОВЕСИЙ
Для записи систем равновесий лучше всего использовать
матричную форму fl.25] с учетом всех сделанных выше замечаний
и принятой символики.
В первой строке матрицы условимся записывать простейшие
ионы, входящие в систему, начиная с тех катионов и анионов,
которые в данной системе ни в. какие взаимодействия не
вступают. Вслед за ними записывают, не придерживаясь особого
порядка, анионы и основания, подвергающиеся протонизации, аквоме-
талло-ионы, ионизирующиеся как кислоты, и т.д. В самом конце
первой строки помещают Н+-ио№. Молекулы воды и ОН--ионы
не вписывают никогда, так как в водных растворах вода есть
всегда, а ОН--ион — продукт равновесия Н2О — H+5=tOH-.
Матрицу можно записать для простейшей системы, например,
раствора КС1, а можно —и для системы очень сложной, полученной,
например, сливанием растворов двух и более реактивов. В
следующих строках записывают под символами соответствующих ионов
стехиометрические коэффициенты равновесий, в которых эти
ионы принимают участие. Если коэффициенты записываются в
буквенной форме, то справа от матрицы дается их расшифровка.
Формулы продуктов равновесий записывают в конце
соответствующих строк в последнем столбце матрицы. В сложных системах
могут возникать (например, за счет реакций
комплексообразования, окислительно-восстановительных процессов и т. п.) новые
частицы, способные к последующим взаимодействиям. Формулы
таких частиц записывают не только в столбце продуктов, на
пополняют ими первую строку, записывая их вслед за Н+-ионом,
и учитывают их взаимодействие с другими ионами, вписывая в
матрицу по уже указанным правилам стехиометрические
коэффициенты и продукты.
Очевидно, что такая матрица представляет собой компактную
запись всех частиц, сосуществующих в системе, и всех
равновесий, в которых эти частицы принимают участие. При
составлении матриц сложных систем нельзя полагаться на память, так
как для правильной постановки последующих расчетов
необходимо со всей доступной полнотой учесть равновесия,
возникающие в системе, и обнаруженные для них стехиометрические
соотношения. Здесь необходимо внимательно использовать таблицы
констант, монографическую и текущую литературу.
В последующем, когда дело дойдет до расчетов, можно будет
правее столбца продуктов помещать столбец констант равновесий.
Рассмотрим несколько примеров.
Пример 1. 4. Записать матрицу раствора КС1:
К+С1- I Н+
I -1
ОН-
14
Здесь, учитывается только одно равновесие растворителя HgO, ведущее за счет
потери (—1) Н+-иона, к образованию продукта ОН"".
Пример 1. 5. Записать матрицу системы, образовавшейся при сливании
растворов К2НРО4 и K2SO4:
к+
sol- POl" н+
0
1
0
0
0
1
—1
1
j
он-
HS07
HyP0{-3
а
0/ / = Пз.
Здесь при сливании растворов не появляется новых соединений, происходит
лишь их разбавление.
Пример 1. 6. Записать матрицу системы, образующейся при сливании
растворов KCN и AgNOg:
K+NO^
Ag+ CN- Н+
Ag(CN)P-p
0 0—1
1 0 —i
0 1 1
1 1 0
1 n 0
0 0 j
•
1
OH-
Ag(OH)}-^
HCN
AgCN (t)
Ag (CN)i-»
H^.Ag(CN)i:j:P
Ш
с
P
Pn n 2,4
J=Tp
P = l,3.
В трех строках матрицы, начиная со второй, учтены равновесия, Бозникаю-
щие в каждом из двух растворов до их сливания. В трех последующих записаны
процессы, ведущие к образованию новых продуктов. Здесь при
соответствующих концентрациях может появиться твердая фаза — новое качество. Один из
продуктов приходится вписать в верхнюю строку, так как он способен прото
низоваться.
Новые частицы, способные к реакциям диссоциации и ассоциации, чаще
всего возникают в системах, осложненных окислительно-восстановительными
процессами. Процессы этого рода сводятся к тому, что акцептор электронов
(е) — окисленная форма какого-либо элемента — превращается в
соответствующую восстановленную форму. Необходимые электроны получаются при этом
от донора электронов — восстановленной формы другого элемента, которая
сама при этом переходит в отвечающую ей окисленную форму.
Число электронов, участвующих в каждой из полуреакций, может быть
одинаковым и разным. Это может повлечь необходимость в специальном
столбце матрицы для учета числа взаимодействующих электронов и включения
электрона как своеобразной частицы в первую строку.
Однако появления этого столбца можно избежать, если условиться
заранее приводить все окислительно-восстановительные процессы к одному электрону.
Ради упрощения записей надо стараться записывать
окислительно-восстановительные равновесия так, чтобы образующиеся восстановленная к
окисленная формы были простейшими частицами, из которых затем образуются более
15
сложные. Для этих частиц надо, как всегда, предусмотреть отдельные столбцы.
Число таких столбцов уменьшается, если, например, образуется окисленная
форма, вроде N0^, или восстановленная, вроде С1", так как такие частицы
не протонизуются. Однако и для таких частиц приходится выделять особый
столбец, если в системе окажутся металло-ионы, способные их координировать.
Пример 1. 7. Записать матрицу системы, получающейся при сливании
растворов ферросульфата, соляной кислоты и натрийдигидроарсената. ,
Система включает окисленную форму мышьяка — арсенат-ион и
восстановленную форму железа — ферро-ион; следовательно, можно ожидать появления
Fe^"*"- и AsO^-ионов. Необходимо проверить возможность координации SO^-,
С1""-, As05~, AsO^- ионов Fe^+ и Fe^"*"-ионами, а также возможность
образования малорастворимых арсенатов и арсенитов. После такой проверки мож-
но начать запись первой строки матрицы, а затем последовательное
заполнение остальных строк и столбцов.
Na+ С1-ре2+ ASO4- Н+
реЗЧ- As02 FeCl4 HSO7
О
О
О
О
О
О
О
о
о
о
о
п
о
о
о
о
1
о
1
о
1
о
о
о
о
о
о
о
о
о
о
о
1
1
о
о
о
о
о
1
о
о
о
о
о
о
о
о
о
1
о
о
о
о
о
о
о
о
1
-1
—1
1
о
j
о
2
—1
—2
о
о
о
1
1
о
о
о
о
о
о
о
1
о
о
о
о
о
о
1
о
о
о
о
о
1
о
о
о
о
он-
FeOH+
HSO-
FeSO^
H|As04^-3
Fe3+
V2ASO7
FeOH2+
Fe2(0H)|+
FeS04+
FeHS042+
FeCl
|3—n
HFeCl4
HAsOg
FeAs04(m)
w
j=1.3
К
К
^22
1—
n=l,4
с
P
Первая строка, как всегда, отводится равновесию растворителя. В
последующих трех строках записаны все равновесия, отвечающие равновесиям в
растворе ферросульфата, а в пятойтри равновесия протонизации арсенат-иона.
В шестой и седьмой строках ' учитываются сопряженные
окислительно-восстановительные системы. Остальные строки матрицы заняты дополнительными
равновесиями, в которых участвую! образовавшиеся частицы. Последняя
строка отведена возможному в этом случае гетерогенному равновесию.
Пример 1. 8. Записать матрицу системы, получающейся при приливании
хлорной воды (водного растворе хл ора) к твердому ртуть меркурох лор иду
При записи считаем, что H^oCU частично растворяется в воде, посылая.
в нее Hg2"*"-H0Hbi, а эти последние дают Н£^"^-ионы и Hg,
16
CI- Hgl"^ Hg Hg2+ Cl2 H+
HgCl^j.p
0
2
0
0
n
0
0
0
0
1
1
0
0
0
0
0
0
0
0
0
0
0
0
V2
0
0
0
1
1
0
0
0
0
0
0
0
0
0 j
V2 0
0 0
—1
0
—1
—i
0
OH-
Hg2Cl2(T)
Hg20H+
Hg(0H)2-i i=T73
HgCl2-" n=l,4
HjHg-Cli+P p = l,2 ] = Ц
cr
Ч2 Hg2+
Просмотр матрицы показывает, что введение в систему, состоящую из-
Hg2Cl2(T), хлорной воды не ышяет на ее качественный состав, так как в ней
не появляется при этом новых ртутьсо держащих частиц.
Пример 1.9. Записать матрицу системы, получающейся при сливании
растворов натрий-иодида, серной кислоты, калий-перманганата и бензола..
Вписываем в таблицу сначала равновесия, отвечающие изолированным
начальным растворам, а затем равновесия, возникающие после их смещения.
Подставную фазу в первую строку не вписываем, считая бензол апротонным
растворителем.
Na+ К+ I SO4 МПО4 Н+
Мп2+ I,
0 0 0 —1
0 10 1
0 0 Vg %
10 0 0
0 0 0 -i
0 10 0
10 0 0
0 0 0 0
1 0
1 0
0 1
0 1
он-
HS07
Mn(OH)M i = l,3
MnSO^
ir
Приведенные примеры охватывают практически все случаи, с которыми
приходится сталкиваться в 1еории ионных равновесий.
2- СИСТЕМЫ УРАВНЕНИЙ, НЕОБХОДИМЫХ
ДЛЯ РАСЧЕТА РАВНОВЕСИЙ
2.1. ОЦЕНКА ВОЗМОЖНОСТИ РАСЧЕТА РАВНОВЕСИЯ
С ПОМОЩЬЮ МАТРИЦЫ системы
Рассмотрим матрицу какой-нибудь очень простой системы,
например, раствора средней соли ХаА, кристаллическая решетка
которой включает катионы Х+ сильного основания и анионы А"-
а-протонной слабой кислоты Н«А. В этом случае
Х+ А«- Н+
j = 1, а'.
0 —1 ОН- \w
1 j HjAi-« I a,
Наша матрица включает (а' + 4) частицы: Х+, А«-, Н+, ОН-
и а' j-гидроанионов. Расчет равновесия сводится к вычислению
равновесных концентраций всех этих частиц, а для этого надо
иметь (а' + 4) уравнений.
Применяя закон действующих масс ко всем (а' -f 1)
равновесиям, представленным в двух последних строках матрицы, можно
получить (а' + 1) уравнение (ОН-) = wh-^ (2.1), (HjA^-") = aj-aAj,
j = 1, a' (2.2). Ho уравнения эти дают лишь значения
активностей ОН- и а' различных продуктов протонизации А"--иона,
выраженные через неизвестные активности аниона и оксоний-иона.
Необходимых нам значений [ОН-] и [HjAJ-*] с помощью одного
закона действующих масс получить нельзя. Здесь необходимо
еще (1 + а') уравнений, обеспечивающих переход от активностей
к равновесным концентрациям. Тогда три последних уравнения
можно записать, используя законы сохранения.
2.2. СВЯЗЬ МЕЖДУ АКТИВНОСТЯМИ И РАВНОВЕСНЫМИ
КОНЦЕНТРАЦИЯМИ
По определению можно записать:
(i) = A[i]; [i] = /r40 = cpi(i), (2.3)
где (i) — активность; [i] — равновесная концентрация;
/i — коэффициент активности; cpi — величина, обратная
коэффициенту активности i-й индивидуальной частицы.
Применять в этом случае значения относительных активностей
[10] нельзя, так как в уравнения законов сохранения входят
абсолютные значения концентраций,
18
Теперь уравнения (2.1), (2.2) можно лредставить в
подходящем для расчетных целей виде
[ОН-] = ^oHwh-' (2.4), [HjAi-«] = срн^да^а^ (2.5).
К сожалению, точные значения ф1 известны в настоящее
время лишь для немногих частиц. Особенно дефицитны эти
данные для ионов, так как долгое время считали, что измерения
ф1 для отдельных ионов без дополнительных незаконных
предположений вообще невозможно, а порой даже полагали, что
само понятие коэффициента активности отдельного иона бес-
cмыQлeннo [56, с. 271].
В большинстве случаев приходится поэтому для
приближенного вычисления ф1 использовать различные теоретические
и эмпирические уравнения.
2.3. ТЕОРЕТИЧЕСКИЕ УРАВНЕНИЯ ДЛЯ ВЫЧИСЛЕНИЯ
КОЭФФИЦИЕНТОВ АКТИВНОСТИ
Теоретических уравнений для расчета коэффициентов
активности предложено очень много. Первое из них было получено
в предположении, что ионы являются точечными зарядами:
Ig/j = -Az]VT или Igcpi = Az'iVT, (2.6)
где А — постоянная, значение которой зависит от температуры
раствора и относительной диэлектрической проницаемости
растворителя е; Zi — число зарядов, которое несет i-й ион.
После внесения поправки, учитывающей конечные размеры
ионной атмосферы, «предельное» уравнение Дебая — Хюккеля;
(2.6) принимает вид
Az?VT Az^VT
где В — постоянная, включающая значения температуры и
относительной диэлектрической проницаемости; ai — радиус ионной
атмосферы, определяющий расстояние наибольшего сближения
ионов (ионный параметр).
Величина /, входящая в теоретически выведенные формулы
(2.6), (2.7), оказалась точно совпадающей с «ионной силой»,
предложенной Льюисом и Рендаллом (1936 г.) в качестве
функции, вызывающей расхождения между величинами
равновесных концентраций ионов и их активностями (активными
массами по Гульдбергу и Вааге), входящими в закон
действующих масс;
/ = 0,5Scu2L (2.8)
где Ск — концентрация к-то иона, находящегося в системе, z^ —
число зарядов, которое несет этот ион.
19
Ниже приведены значения констант А и В в уравнениях
(2.6), (2.7) при условии, что ионные параметры щ выражены в
нанометрах (1 нм = 10-"^ м), а концентрации в моль • л""^ [55].
в 10»
в 102
15
20
0,5002
0,5046
3.267
3,276
25
30
0,5092
0,5141
3.286,
3,297
Уравнение (2.7) нельзя считать точным, так как при его
выводе (1923 г.) Дебай и Хюккель считали, что ионная
атмосфера вокруг данного иона образуется независимо от
образования ионных атмосфер вокруг других ионов; что
диэлектрическая проницаемость раствора совпадает с диэлектрической
проницаемостью чистого растворителя; тепловое движение
ионов, усиливающееся с ростом средней температуры системы,
должно способствовать разрушению ионной атмосферы.
Непосредственное использование уравнения (2.7) оказалось
невозможным, так как Дебай и Хюккель не указали метода
расчета или измерения параметра
Только после того как в [42] было показано, что величины
ui можно рассматривать как действительные диаметры гидра-
тированных ионов, Кайленд [51] использовал различные
методы оценки этой величины и опубликовал сводную таблицу
полученных им значений ai для ряда ионов (табл. 2.1).
В табл. 2.1 не включены многие часто встречающиеся ионы
и не указан интервал ионных сил, на котором можно
пользоваться уравнением (2.7), подставляя в него значения ui из
табл. 2.1. Все эти обстоятельства ограничивают применимость
уравнения (2.7) и параметров ai, приведенных в табл. 2.1.
2.4. ЭМПИРИЧЕСКИЕ УРАВНЕНИЯ ДЛЯ РАСЧЕТА
КОЭФФИЦИЕНТОВ АКТИВНОСТИ
При / < 10-3 моль • л"-Ч когда BaJ^^^ < 1, можно
пользоваться предельной формулой Дебая-Хюккеля: Igcpi = 0,53f/^/2* (2.9),
где для комнатной температуры принято А ^ 0,5. Из этого
уравнения следует, что при / < 10-^ моль • л"-^ значения Igcpi не
зависят от индивидуальных свойств i-ro иона, характеризуемых
параметром щ. Здесь сохраняет значение лишь число зарядов 3i,
которое несет ион.
Гуггенгейм, отрицавший возможность измерения коэффициентов
активности отдельных ионов [10], все же считал их полезными
в случае, когда их оценка не требует высокой точности. Совместно
* В дальнейшем везде даются преимущественно значения Ig f^, = =-lg
20
Таблица 2.1
Ион
Ы+
Rb+, Cs+, NH+ Т1+ Ag+
к+ С1-, Br- I- CN- N0^-, mj-
GH- F-, CNS-. NC0-, HS-. ClO^, CIO7. BrOj", Ш7, MnO^
Na+. CdCl+ aOj, IO3, HCO3, H2P07, HS©^-, HjAs©^,
Co(NH3)4 (N02)^
Hg2+. S0|- S2OI- SjOi- S2GI- Se02- СГ02- HP(^-
Pb2+. C0|-, SG|-. MoO^-, Co(NH3)5C12+ Fe(CN)5N©2-
Sr2+. Ba2+ Ra2+ Cd2+ Hg2+ S2- S202- W02-
Ca2+. Cu2+. Zn2+. Sn2+, Mn2+. Fe2+ Ni2+, €0^+
Mg2+ Be2+
РОЗ- Fe(CN)i- Сг(ЫНз)|+ Со(ННз)|+ Co (ЫНз)5Н2®'+
СоепЗ+
Al3+ реЗ+ с,з+ sc3+ La^+, ln^+. Ce'+ Рт^+, m^.
Fe(CN)r
Co (S203) mt
Th<+. Zr<+, Ce*+. Sn*+
Co(S03)2(C.N)5-
HCO^, HgCit-, CHgNH^, (CH3)2NH^
NH3CH2CG2H+ (СНз)2ЫН+ C2H5NH+
Ac- CH2CICO7, (СНз)4Ы+ (C2H5)2NHt, NH2CH2CGi-
CHCljCO^. CCljCO^, (С2Н5)зЫН+ (C3H7) NH^
СбН4 (OH)CO^, C6H4CICO7, C6H5CH2CO7, CH2=CH.CH2C0i-
(СНз)2С-СНСО^, (C2H5)4N+ (СзН7)2НН^, C^HfO^
ОС^Щ (ЫОз)Г, (СзН7)зЫН+ СНзОСбН^СОГ
(СбН5)2СНСОГ, (СзН7)4Ы+
0,6
0,25
0,3
0.35
0.4-0.45
21
продолжение табл. 2.1
Ион
C202-.HCit2-
СН2 (COg)!-, (CHaCOg)!-, (СНОНСОз)!"
CgH^CCOg)!-, СН2 (CHgCOg)!' (СНзСНзСОг)^-
(СН2)з (CH2C02)^- (CH2)4(CH2C02)^- (Congored)^-
Cit^-
0,45
0.5
0,6
0,7
0,5
Примечание: Ac — ацетат, Cit — цитрат, Congored — конго красный,
en — этилендиамин.
с Шиндлером он предложил в связи с этим приближенное
уравнение, получаемое из уравнения (2.7) при условии, что Вгц = 1:
0,5 2? УТ
Igcpi =
(2.10)
Это предположение использовали в дальнейшем авторы ряда
других приближенных формул. Считают, что уравнение (2.10)
пригодно при / < 0,1 моль • л-^.
Кайленд [51] для всех неорганических ионов, кроме Fe (CN)!"",
Fe(CN)6~, Со(КНз)б''" и им подобных, предложил фармулу,
получающуюся из (2.7) при Ва\ zi:
М,,."!^. (2.11)
Для указанных выше комплексных ионов и всех органических
ионов, не вошедших в табл. 2.1, он рекомендует формулу
считая, что в этом случае Вщ = 2.
Сведений об этом интервале, ионных сил, для которого
пригодны уравнения (2.11) и (2.12), нет.
Из формул (2.6), (2.7) следует, что \gfi должен принимать
тем большие по модулю отрицательные значения, чем выше /.
Это должно вести к монотонному снижению fu а значит, и
активности, так как (1)=Д[1]. Ряд наблюдений и измерений*
показывает, однако, что с ростом / значения Д в действительности
сначала убывают, а затем, пройдя через минимум, начинают
возрастать. Согласно Хюккелю (1925 г.) эти изменения f\ связаны
* Вчастности^ измерений средних коэффициентов активности [53, с. 152, 169].
22
с тем, что при повышении /, а значит, и cxy все большее
количество молекул воды координируется ионами, находящимися в
растворе. Это ведет к .снижению концентрации свободных
молекул растворителя, росту диэлектрической проницаемости системы
в непосредственной близости к ионам, снижению кулоновских
сил, действующих между ионами, и соответствующему
возрастанию активности. Для компенсации влияния этих факторов на
значение Ig/i Хюккель ввел в формулу (2.7) дополнительное
слагаемое, пропорциональное /, и она приняла вид
Формулы (2.13) не используются на практике из-за отсутствия
методов измерения параметров ai и bi. Упрощенный вариант
формулы (2.13) дал Дэвис [46]:
1еТ1=^г?(тт^==-0.2/). (2.14)
Сопоставление уравнений (2.13) и (2.14) показывает, что Дэвис
принял ради упрощения два условия: Ва{= 1, bi = 0,2 Az]^
~0,l2f. Яцимирский и Тетюшкина считают [37], что уравнение
(2.14) дает удовлетворительные значения при /< 0,8 моль . л~^.
При 2ii = 0, т. е. для незаряженных частиц, уравнение (2.13)
имеет вид \gfi = bj (2.15). При 6i>0 Ig/i и /i- растут с /, т.е.
с возрастанием концентрации соли XY. Поскольку при этом
концентрация i-й незаряженной частицы снижается, множитель
6i > О называют коэффициентом «высаливания». По аналогичным
соображениям множители 6i < О называют коэффициентами «всали-
вания».
Все приведенные выше упрощенные формулы (2.9) —(2.14)
нельзя считать приспособленными для вычисления
индивидуальных коэффициентов активности, так как вместо параметров а\ и
&i, учитывающих в какой-то мере химическую природу ионов,
они* содержат либо постоянные величины, например ai = JB~"^
либо величины, пропорциональные z\.
В результате каждая из этих формул должна давать
одинаковые значения для всех ионов, несущих одинаковое число
отрицательных или положительных зарядов. При / > 0,8 моль • л-^ ни
одна из предложенных формул не применяется.
2.5. УРАВНЕНИЯ И ПАРАМЕТРЫ, ПРИСПОСОБЛЕННЫЕ
ДЛЯ ВЫЧИСЛЕНИЯ ИНДИВИДУАЛЬНЫХ КОЭФФИЦИЕНТОВ
АКТИВНОСТИ НА ШИРОКОМ ИНТЕРВАЛЕ ИОННЫХ СИЛ
Показано, что путем одновременного применения к системам,
достигшим равновесия, закона сохранения и закона действующих
масс, можно измерить не только константу равновесия, но и эм-
23
лирические коэффициенты ai, 6i, си «2, «з» «к, входящие в
расчетные уравнения:
Ig ср, = -IgA = -т^Щ^ + bil + cj^'\ (2.16)
Ig = —Ig/i = a, ]// + аг/ + «зЯ,
(2.17)
(2.18)
Эти уравнения оказались наиболее удобными для выражения
зависимости Igcpi и lg/нАс от ионной силы [14].
В табл. 2.2 приведены параметры ai, bi и в формуле (2.16)
для вычисления Ig^i при ^ = 25°С и 0<j<3 моль • л~^ для Н+,
0Н~, Bz- — бензоат и Ас — ацетат-ионов.
Таблица 2.2
Фон
9Н
9рН
9Ас
в*
«о
в*
<г
в*
&
о
?г
м
О
Ь
^Г*
NaCl
КС1
KNO3
NaC104
NaNOg
4,20
6,10
8,02
9,30
22
11.3
9.2
16.0
0
0
0
0
1,08
1.50
1.34
1.53
3.7
12,4
11.3
3,3
0
0
0
0
6,68
6.61
6,90
10,9
—6,55
—8,09
2,93
-8,1
6,3
3,65
8,1
14
3,39
3,89
2,41
1,72
-1,56
-11.4
-17,7
-8.5
—0,86
-1,0
3,1
В табл. 2.3 приведены параметры аь аг, аз в формуле (2.17)
для вычисления cpi при ^ = 25°Си 0<j<3 моль • л~^ для ади-
пиновой кислоты (НгАс!), ионов HAd— и Ad^-.
Таблица 2.3
п
Фон
NaCl 1 KCl KNO,
NaClO^
«2
«3
—0,003e
0,140;
-0,0022
—7,2-10-^
-0,083б
7,9.10-^
0,01l2
0,0135
—0,003i
0,005i
0,014i
-0,0115
"^HAd—
Ol
(h
0,2958
-0,2264
0,023i
0,247o
—0,1534
—0,0008
0,3142
-0,141»
0,0165
0,2669
—0,07l2
0,0001
at
0,9759
—0.336t
o;o2i;
0,8537
—0,324з
0,874e
—0,293,
0,0144
0,943з
-0,240»
0,011i
24
Значение параметров ак в формуле (2.18) для расчета lg/нАс
при ^ = 25''С и 0</<3 моль • л~^ приведены в табл. 2.4.
Таблица 2.4
NaCl
KCl
NaNOa
KNO3
^1
^2
аз
0,0126
0,0936
—0,0098
0,0162
0,0462
0,0467
0,0018
0,0392
—0,0204
0,0117
2.6. СРАВНЕНИЕ ЗНАЧЕНИЙ КОЭФФИЦИЕНТОВ АКТИВНОСТИ,
ВЫЧИСЛЕННЫХ ПО РАЗНЫМ ФОРМУЛАМ
В разделах 2.3 и 2.4 приведен ряд уравнений для
вычисления коэффициентов активности определяются природой не толь-
. Поскольку верхняя граница применимости этих формул обычно
указывается лишь ориентировочно, представляет интерес
сравнение значений fu вычисленных с помощью этих уравнений,
с теми же значениями, полученными, скажем, по формуле
(2.16), с учетом значений параметров, приведенных в табл. 2.2.
Сопоставление значений fi (для однозарядного иона), /н и /он,
полученных таким обр^азом, приводится в табл. 2.5.
Т а бл ица 2.5
/ моль-л
—1
0.04
0,64 1.00 2,89
Использованные уравнения
ft
fi
fi
fu
Jh
fn
/он
foH
/он
0,794
0,826
0,834
0,863
0,849
0,877
0,826
0,805
0,811
0,398
0,600
0,695
0,757
0,985
0,964
0,605
0,509
0,537
0,316
0,562
0,708
0,743
1,014
1,082
0,579
0,458
0,494
0,141
0,484
0,942
0,715
2,382
2,099
0,509
0,369
0,425
(2.9) Ig/i = -J),5K/
(2.10)lg/i = -0,5// (l+/7)-^
(2.14) Ig/i = - 0,5 IV7 (1 + V~I)-'-
-0,20/]
lg/н = - Л /7 (1 + Ван % =
= 0,9 HM из табл. 2,1
(2.16), табл. 2.2 для NaCl
(2.16), табл. 2.2 для NaC104
Ig/OH == - ^ У7 (1 + Баон VT)-K
ctQi^ = 0,3b HM из табл. 2.1
(2.16), табл. 2.2 для NaCl
(2.16), табл. 2.2 для NaCI04
Данные табл. 2.5 позволяют сделать следующие выводы:
1. Формула (2.9) дает результаты, далекие от полученных
опытным путем даже при / = 0,04.
25
2. Значения /н и /он, полученные на фоне NaCl и NaC104,
не очень сильно отличаются от /ь вычисленных по уравнениям
(2.10) и (2.14) только при 7=0,04. В остальных случаях
расхождения очень велики.
3. Значительные расхождения между значениями /н и /ош
полученными на фоне NaCl и NaC104, показывают, что
значения коэффициентов активности определяются природой не
только того иона, к которому они относятся, но и тех ионов,
которые создают ионную силу. Влияние природы фоновой соли
заметно увеличивается с ростом ионной силы.
4. Применение зфавнения Дебая — Хюккеля (2.7) с
одновременным использованием параметров ai, рассчитанных Кайлендом,.
в случае расчетов /н и /он дает результаты, далекие от
правильных, уже при /==0,64 моль • л~^
5. Формула Дэвиса в случае /н и /он оказывается
несостоятельной уже при /<0,64 моль • л~Ч значительно меньших тех,,
которые указаны в [37].
6. Очевидна необходимость измерения параметров щ, bi, ...
для возможно большего числа ионов, так как' только при
наличии этих данных станут возможными вычисления при высоких
ионных силах.
2.7. ЗНАЧЕНИЯ КОЭФФИЦИЕНТОВ АКТИВНОСТИ ПРИ
ИСПОЛЬЗОВАНИИ ЗАКОНА ДЕЙСТВУЮЩИХ МАСС
В табл. 2.2 и 2.4 приведены параметры, с помощью которых
можно рассчитать коэффициенты активности молекул НАс- и
Ас-ионов при различных ионных силах в растворах, содержащих
разные фоновые соли. С помощью этих величин рассчитаны
значения активностей и равновесных концентраций в растворе НАс
при снАс = 1,000 • 10-3 i^Qjij^ . л-1 и разных / на интервале 0,1 <
< / < 3,0 моль • л~» (табл. 2.6).
В столбце 5 табл. 2.6 приведены значения Igo, вычисленные
по формуле (НАс)/(Н+) (Ас-) = о, а в графе 10 — значения Ig о^.
Таблица 2.6
I
x
x
4-
x
<
±
0,1
0,5
1,0
1,5
2,0
2,5
3,0
1,32
1,51
1,91
2,12
2,54
2,95
3,72
11,8
11,1
9,85
9,73
9,04
8,58
7,57
0,870
0,933
1,04
1,15
1,28
1,41
1,57
4,747
4,746
4,743
4,746
4,746
4,746
4,746
0,988
1,044
1,138
1,247
1,370
1,496
1,646
1,55
1,72
1,62
1,64
1,52
1,44
1,23
1,55
1,72
1,63
1,64
1.52
1,42
1,23
8,45
8,28
8,38
8,37
8,48
8,57
8,76
4,545
4,446
4,501
4,494
4,566
4,622
4,762
1,000
1,000
1,001
1,001
1,000
0,999
0,999
26
отвечающие выражению [HAcJ/fH+] [Ас-]. Сопоставление этих
данных служит хорошей иллюстрацией факта, что в закон
действующих масс входят активности, а не равновесные концентрации
ингредиентов равновесия и что, действительно, как того требует
формулировка закона действующих масс, произведение
активностей ингредиентов равновесия при постоянной температуре и
давлении— величина постоянная, не зависящая от ионной силы.
В 6-м и 11-м'столбцах табл. 2.6 приведены значения 2(i) =
= (Ас-) + (НАс) и 2[i] = [Ас-] + [НАс]. Из сопоставления этих
данных очевидно, что только сумма равновесных концентраций
Ас--ионов и недиссоциированных молекул НАс остается равной
^нАс при любых /, тогда как сумма активностей тех же частиц
может отличаться от этой величины значительно.
Данные табл. 2.6 еще раз подтверждают необходимость
измерения параметров уравнений, приспособленных для расчета
истинных значений коэффициентов активности.
Z8. РАЦИОНАЛЬНОЕ ИСПОЛЬЗОВАНИЕ СУЩЕСТВУЮЩИХ
ПРИЕМОВ РАСЧЕТА КОЭФФИЦИЕНТОВ АКТИВНОСТИ
Из данных табл. 2.5 следует, что все расчетные формулы
дают значения коэффициентов, тем больше отклоняющиеся от
истинных, чем выше ионная сила. В связи с этим всюду, где
это возможно, следует использовать значения Д и cpi,
вычисленные по уравнениям (2.16) — (2.18) с помощью
параметров, отвечающих соответствующей i-й частице.
К сожалению, параметры эти измерены лишь для небольшого
числа частиц. Приходится поэтому всюду, где нет другого
выхода, пользоваться формулами, учитывающими только
различия в числе зарядов и приспособленными в то же время к
учету небольших ионных сил. Такой формулой является, в
частности, формула Дэвиса (2.14). Поскольку для однозарядного
иона она записывается в виде
lg<p, = л(J^-0.2/j, (2.19)
а для z-зарядного —в виде
lg<f, = A(^j^-0,2iy, (2.20)
очевидно, что Igcp^ = Ig^i (2.21), т.е. имеет место следующая
зависимость:
Z 1 2 3 4 "
41g<pi 91g<pi 161g<pi
Указанное соответствие очень упрощает расчеты. В то же
время оно допускает возможность приближенной оценки расхож-
27
дений между значениями активностей и равновесных
концентраций частиц, несущих определенное число зарядов z, при данной
/. Так, при / = 0,64 моль • л-^ вычисленный по уравнению (2.19)
Igcpi = 0,158. Тогда Igcpg = 9 • Igcpi = 1,422, а срз = 26,4. Это
значит, что [ионЗ+] = 26,4 (ионЗ+) и (ионЗ+) = 3,75 • 10-2 [ионЗ+].
Иначе говоря, при / = 0,64 моль • л-^ «активны» лишь 3,75%
любых трехзарядных ионов, присутствующих в растворе.
2.9. НЕСКОЛЬКО ФОРМУЛИРОВОК ЗАКОНА СОХРАНЕНИЯ
МАТЕРИИ
В разделе 2.1 сказано, что для расчета равновесных
концентраций всех частиц системы, кроме уравнений, основанных
на законе действующих масс, придется использовать уравнения,
связанные с законами сохранения. С этими законами и их
применением необходимо познакомиться.
В 1756 г. в рапорте конференции Академии наук М. В.
Ломоносов сообщил о результатах, полученных им при
повторении опытов Р. Бойля: «Делал опыты, в заплавленных
накрепко сосудах, дабы исследовать, прибывает ли вес металлов от
чистого жару. Оными опытами нашлось, что славного Роберта
Бойла мнение ложно, ибо без пропущения внешнего воздуха
вес сожженного металла остается в полной мере».
Так впервые опытным путем был установлен закон
сохранения материи, о вечности и неуничтожимости которой уже
догадывались ученые и философы древности, например Эмпе-
докл. Эти опыты и ряд соображений по другим вопросам
привели Ломоносова к обобщенной формулировке закона
сохранения, опубликованной им в 1760 г. «Но как все перемены,
в натуре случающиеся, такого суть состояния, что, сколько
чего у одного тела отнимется, столько присовокупится к
другому, так, ежели где убудет несколько материи, то умножится
в другом месте; сколько часов положит кто на бдение,
столько ж сну отнимет. Сей всеобщий естественный закон
простирается и в самые правила движения» [22, с. 341].
Поскольку прямо из вводной ч^сти формулировки
очевидно, что она опирается на количественные результаты измерений,
выполненных при проверке опытов Бойля, можно утверждать,
что Ломоносову принадлежит честь неоспоримого опытного
открытия одного из важнейших законов природы.
Опытные данные и формулировки Ломоносова долгое время
замалчивались или были зйбыты, и только после появления
изысканий Б. Н. Меншуткина (1904) и издания работ
Ломоносова приоритет его в части научного обоснования закона
сохранения материи стал общепризнанным.
Менделеев, работая над «Основами химии» и опираясь на
экспериментальные исследования Лавуазье, дал следующую
формулировку закона сохранения массы: «вещество не тво-
2а
рится и не пропадает, или материя вечна, или общая масса
(вес) веществ при химических изменениях (реакциях)
сохраняется, т. е. остается постоянной» [24, с. 24], Таким образом,
Менделеев считал, что в зависимости от решаемой задачи
словесная формулировка закона сохранения материи может
принимать различную форму. Этим прецедентом нам придется
воспользоваться позже.
2.10. ПОСЛЕДУЮЩИЕ ПОПЫТКИ ОПЫТНОЙ ПРОВЕРКИ
ЗАКОНА СОХРАНЕНИЯ МАССЫ
В 1893 г. Ландольт опубликовал свою первую работу,
посвященную экспериментальной проверке закона сохранения
массы. Опыты проводились с водными растворами веществ,
которые помещались в отдельных коленах П-образных сосудов.
В каждом опыте использовались два сосуда. Их массы и
объемы были примерно одинаковы. После установки сосудов. на
разные чашки специально сконструированных для этого
исследования весов проводились следующие измерения: 1)
измеряли разницу масс сосудов Л и Б до реакции; 2) перемешивали
растворы, находившиеся в разных коленах сосуда Л, и после
завершения реакции снова измеряли разницу масс сосудов
Л и Б; 3) после перемешивания растворов в сосуде В и
завершения в нем реакции опять измеряли разницу в массах
сосудов А и В.
Исследовали системы, в которых исходное вещество,
записанное на втором месте, всегда брали в избытке:
Ag2 SO4 + 2Fe SO4 = 2Ag + Fes (804)3; (2.22)
HIO3 + SHI = 61 + ЗН2О; (2.23)
21 + 2Na2S203 = 2NaI + Na2S406; (2.24)
CCI3COH . H2O + KOH = CCI3H + HCO2K + H2O; (2.25)
CCI3COH . H2O + H2O (процесс растворения). (2.26)
Таким образом, почти через 140 лет после Ломоносова
Ландольт повторил опыты «в заплавленных накрепко сосудах»,
используя новую технику и большее число разных процессов.
Было установлено, что реакции (2.22), (2.23)
сопровождаются уменьшением массы на величины, значительно
превышающие погрешности измерения. Для реакций (2.24) в одном
случае наблюдалось возрастание массы, а в двух — небольшие
отклонения, лежащие в пределах погрешностей измерения.
Процессы (2.25), (2.26) проходили без заметных колебаний массы.
Сенсационные результаты Ландольта способствовали
появлению ряда статей, авторы которых повторяли измерения
Ландольта или искали причины погрешностей, которые могли
вызвать кажущееся нарушение закона сохранения. Сам Ландольт
29
продолжал исследования и в 1906 г. опубликовал второе
сообщение. Он использовал еще более усовершенствованные весы
и на основании специальных измерений предположил, что
изменения массы, превышающие по абсолютной величине 0,03 мг,
не могут быть отнесены за счет погрешностей измерения.
Схема измерений осталась прежней.
Вторично были использованы процессы (2.22) — (2.26),
реакции
3AgN03 + 3FeS04 = 3Ag + Рез (504)з + Fe (КОз)з;
Ее -f CUSO4 = Си -f FeS04;
АиС1з + 3FeCl2 = Au + ЗРеСЦ;
I2 + NaHS03 + Н2О = 2HI + NaHS04;
2UO2 (N03)2 + 6K0H = K2U2O7 + 4KNO3 + ЗН2О,
a также электролиз водного раствора Cdh переменным током и
некоторые процессы растворения веществ.
Суммируя результаты своих опытов и измерений Хэйдвайлера,
Ландольт отмечает, что для исследованных реакций, кроме вос-
<:тановления Ag+-ионов Fe2+-ионами и HlOi" — 1--ионами, закон
сохранения массы выполнялся с точностью до 0,03 мг.
Из 75 опытов, отклонения в которых превышали 0,03 мг,
Ландольт и Хэйдвайлер зарегистрировали 61 случай снижения
начальной массы. Для реакции Ag++ Ре2+= Ag(T) + РеЗ+было
зарегистрировано 9 измерений, при которых потеря массы
составляла от 0,068 до 0,199 мг, а для равновесия
НЮз + 5Н-ь + 51-<=± 61 + ЗН2О
также в 9 случаях — от 0,047 до 0,177 мг.
Поскольку в четырех опытах, поставленных Ландольтом
для системы Ag+, Ре^+ в парафинированных изнутри сосудах,
потеря массы обнаружена не была, он даже не пытается
поставить под сомнение закон сохранения массы, а лишь ищет
возможные причины систематических ошибок при измерениях.
Менделеев очень спокойно отнесся к результатам,
полученным Ландольтом и другими учеными, включившимися в
проверку закона сохранения массы. Он писал [24, с. 357]:
«Ландольт... нашел также или полное сохранение веса, или столь
ничтожное его изменение (например, 0,2 мг), что его, можно
приписать неизбежным погрешностям взвешивания. В
настоящее время можно, однако, взвешивать гири, подобные платино-
иридиевым, весящие от 1000 до 400 г, с полной точностью
до стомиллионных долей килограмма, как это достигнуто
в Главной палате мер и весов, но такая высокая (высшая,
чем в каких-либо иных непосредственных измерениях)
точность достижима только при соблюдении множества
предосторожностей и только для тел такой твердости и неизменяе-
30
мости, как платиноиридиевые гири. В обычных же химических
исследованиях, особенно когда взвешивают стеклянные
сосуды и жидкости или газы, едва достижима уверенность в
десятых долях миллиграмма, потому что разного рода поправки
на взвешивание (приведение к пустоте), стирание
поверхности, гигроскопичность и другие более или менее влияют на
результат». Менделеев, незадолго до этого опубликовавший
специальное исследование о приемах точных взвешиваний,,
больше, чем кто-нибудь другой, мог оценить создавшееся
положение.
В дальнейшем сообщений о попытках прямой проверки
закона сохранения массы не появлялось.
2.11. СОВРЕМЕННАЯ ОЦЕНКА ТОЧНОСТИ ЗАКОНА
СОХРАНЕНИЯ МАССЫ
Ландольт проводил свои опыты со всей тщательностью,
-которой мог добиться при современной ему технике измерений.
Он не располагал при этом теоретическими сведениями, с
помощью которых можно было бы заранее предвидеть возможность
отклонений от закона сохранения массы, оценить их вероятную^
величину и установить на этом основании чувствительность
методов, необходимых при таком исследовании. Ясность в этот
вопрос вносит специальная теория относительности А.
Эйнштейна. Согласно этой теории энергия свободной частицы (е)
не обращается в нуль при скорости v=0, а остается
конечной величиной, равной
е„=о = тс2, (2.27)
которую называют энергией покоя. В уравнении (2.27)
т—-масса частицы (масса покоя) и с —скорость света в вакууме
(^ 3 • 10» м . с-»).
Формула (2.27) применима и к любой системе, масса
которой равна т. Она выражает закон взаимной связи массы й
энергии.
Поскольку хим1^ческие процессы, происходящие в системе,,
сопровождаются изменением энергии, например выделением
тепла, закон сохранения массы, как это следует из (2.27),
должен выполняться с точностью до
Am = Аб/с2. (2.28)
Полезно с помощью формулы (2.28) найти оценку Am для
а8=1 кДж-моль-Ч Она составляет приблизительно 10-^* кгХ
Хмоль-Ч Следовательно, даже для очень экзотермической
реакции, сопровождающейся выделением 1 мДж-моль-^ тепла. Am
составит всего около 10"^^ кг•мoль-^ т. е. 10-^мг • моль-^ Эту
величину можно считать исчезающе малой по сравнению с
чувствительностью лучших современных весов, рассчитанных на
31
сравнительно большую нагрузку, и со случайными
погрешностями, неизбежно возникающими при взвешивании сравнительно
больших сосудов.
Очевидно, что при химических процессах закон сохранения
массы выполняется с точностью до --10-^ мг на моль реагиру-
юшего вещества.
2.12. РАЗЛИЧНЫЕ ВЫРАЖЕНИЯ ЗАКОНА
СОХРАНЕНИЯ МАССЫ
В «Основах химии» [24, с. 24] приводится следующая
математическая формулировка закона сохранения массы: «Если
через Л, В, С и т. д. означим вес взятых, а через М, Л^, О и
т. д, вес происходящих, то Л + В + С+... =M+iV+0 + ... (2.29)-
И далее указывается, что, «прилагая закон вечности вещества
и производя взвешивания, химик может не упустить из виду ни
одного из действующих и происходящих тел».
Уравнение (2.29) выражает неизменность массы всей
замкнутой системы, в которой происходит процесс. Поскольку при
этом вместо веществ Л, В, С, ... возникают новые вещества М,
N, О,выражение (2.29) называют уравнением материального
баланса.
Для водного раствора соли ХаА (см. раздел 2.1) уравнение
(2.29) пришлось бы записать в виде
о а'
тх^А + тн,о = гпн,о + той + + тн|А, (2.30).
j=0
о
где mi—масса i-ro компонента; тн^о — начальная масса воды.
Использовать (2.30) совместно с уравнениями (2.4), (2.5) очень
трудно. Еще большие затруднения возникают в связи с тем, что
при этом подходе очень трудно осуществить получение за счет
законов сохранения еще двух уравнений, необходимых для
решения системы, указанной в разделе 2.1.
При решении задачи надо учесть, что большинство
необходимых уравнений записывается с помощью закона действующих
масс. Они могут быть представлены в виде уравнений (2.4),
(2.5), дающих значения равновесных концентраций всех более
сложных частиц, находящихся в системе. Это дает повод к
замене масс исходных веществ их начальными концентрациями.
Например, если в рассматриваемом примере в 1 кг воды
растворено G (г) соли ХаА с молярной массой Мхад, то начальная
моляльность (моль-кг-^)
тх,л = :^^. (2.31)
Если то же количество граммов соли ХаА содержится в одном
32
литре раствора, то можно записать, что начальная концентрация
(моль • л~0 соли ХаА в этом растворе
СХаА = 55^^ = Л. (2.32)
В последнем случае (в дальнейшем всюду будут
использоваться d) уравнение (2.32) дает право записать два равенства: сх = аЛ,
са = А (2.33) и получить два уравнения:
= аЛ = [Х+]; (2.34)
СТА = Л = [А^-] + 2 [HiAi-«] = V [HjAM. (2.35)
j-i j=o
Уравнение (2.34) показывает, что после достижения
равновесия [Х+] остается равной начальной концентрации этого катиона,
если Х+-ИОН не координирует А*--ион.
Из уравнения (2.35) следует, что начальная концентрация
аниона А*-, введенного в систему в виде соли ХаА, оказывается
равной сумме равновесных концентраций самого А*"--иона и всех
частиц, получающихся из него за счет протонизации. При этом
сам анион А**- можно считать за Н}А^-°' при j = 0, что дает
право упростить уравнение (2.35).
Записи типа (2.35) вполне законны, так как в них
учитывается не сохранение массы, а сохранение значения начальной
концентрации. В силу этого закон сохранения,
отвечающий такому методу счета и размерности моль • л-^, следует
называть законом сохранения начальной концентрации,
В случаях, когда начальная концентрация отвечает
одноядерной частице, дающей после достижения равновесия ассоциаты,
равновесные концентрации этих последних входят в закон
сохранения начальной концентрации с соответствующими
коэффициентами. Например, известно, что в сильно кислых растворах
Сг04"-ион дает НСгОГ-, СггС?"- и СгзО?Г-ионы. Если с 2^ = Л,
ТО закон сохранения начальной концентрации хромат-иона
придется записать так:
А = [СгОГ] + [НСгОГ] + 2[СГ207~1 + 3 [СгзО?Г], (2.36)
поскольку, например, на образование одного СгзО?о'-иона должно
расходоваться три СгО^^-иона.
В некоторых случаях, например, при расчетах равновесий,
возникающих при исследовании экстракционных процессов,
удобнее использовать закон сохранения масс в виде закона
сохранения начального количества вещества (числа молей).
Рассмотрим систему, состоящую из у л водного раствора, в
котором СрессЮд), = М И Снс! = У, И У Л насыщенного водой изо-
пропилового эфира. В этом случае матрица системы имеет
следующий вид:
2 2-436 3^
СЮл
CI— Fe
|3+ H+
FeCl7 FeGla HFeGI ^
0
0
0
n
0
0
0
0
1
2
1
0
0
0
—1
-1
0
1
0
0
1
0
0
OH-
КеОН2+
Fe2(OH)^2"^
HFeCl4
Ре*С1з
HFe*Cl4
В ней учтены обе способные распределяться нейтральные
частицы: РеСЦ и HFeCl4. В этом случае с^е = М, с^ю, == ЗМ,
Cqi =z у (2.37), но еистема двухфазная и частицы, происходящие,
от Fe^+- и СН-ионов, находятся в разных фазах, имеющих разные
объемы. Здесь целесообразно ивпользовать закон сохранения
массы в виде закона сохранения начального количества вещества
Fe^"<-- и С1""-ионов, т. е. величин Mv и Уи. Уравнения запишем так:
Mv = {[РеЗ+] + (РеОН2+1 + 2 [Fes (0Н)^2-^] + S [РеС1Г"] +
п—I
+ (HFeCUll V + {[Feth] + fHFeCU]} 'v, (2.38)
= (ЮН + S n IFeCl^'']+ 4(HFeCl4]}v+{3[FeCbl +
+ 4[HFeCl4l} 0,
(2.39)
где звездочками отмечены частицы, перешедшие в изопропило-
вый эфир, а коэффициенты перед равновесными
концентрациями учитывают многоядерность.
2.13. ЗАКОН СОХРАНЕНИЯ ЭЛЕКТРОНЕЙТРАЛЬНОСТИ
Применение к равновесию в растворе соли Х.А закона
сохранения начальной концентрации дало два из трех недостающих
уравнений: (2.38) и (2.39). Последнее недостающее уравнение
можно записать, используя закон, выражающий
электронейтральность системы.
Согласно этому закону сумма всех положительных и
отрицательных зарядов, которые несут все находящиеся в системе
ионы, должна быть равна нулю.
Очевидно, что для подсчета зарядов равновесную
концентрацию каждого иона надо умножить на число Авогадро, чтобы
получить число отдельных ионов данного вида, и на
коэффициент, выражающий число зарядов, которое несет ион, со знаком
«+», если речь идет о катионе, и со знаком «—» для анионов.
34
Сумма всех таких произведений приравнивается нулю.
Поскольку число Авогадро является общим множителем и при том не
равным нулю, производят сокращение и уравнение закона
сохранения электронейтральности получают в виде суммы
равновесных концентраций всех ионов с соответствующими
положительными и отрицательными коэффициентами.
Так, используя матрицу раздела 2.1, записываем
[Н+1 - [ОН-1 + [Х+] + (j - а) [HjAi-l == 0. (2.40)
При составлении уравнения закона сохранения
электронейтральности первые два места отводят Н+- и ОН--ионам, затем
учитывают заряды ионов, не принимающих участия в
равновесиях системы, и лишь после них заряды остальных частиц.
Используя это правило для матрицы системы, где 6>e(ci04)s =
= М и Сна = У, получаем ,
[Н+] - [ОН-] - [CI071 — [СЬ] + 2 [FeOH2+] + 4 [Peg (OH)^^l +
+ S (3-п)[РеС1^"] = 0. (2.41)
п= 1
Вопрос о совместном решении получаемых систем уравнений
рассмотрим в главах, посвященных соответствующим
равновесиям.
3. КОНСТАНТЫ РАВНОВЕСИЙ И СВЯЗЬ МЕЖДУ НИМИ
3.1. ВАЖНЕЙШИЕ ВИДЫ РАВНОВЕСИЙ И ИХ КОНСТАНТЫ
В главе 2 показано, что. компактность записей равновесий
в алгебраической форме можно обеспечить лишь в том случае,
если мы всегда будем изображать их как процесс образования
сложных частиц из простейших. Соответствующие константы
чаще всего называют константами устойчивости [36], хотя, ве-
poiiTHO, больше подходит для них.название констант образования.
Символы констант образования различных по природе
частиц приведены в разделе 1.8.
Наряду с константами образования нам придется
пользоваться ступенчатыми константами диссоциации сложных
частиц—кислот, комплексов и т. п. Эти константы в дальнейшем
обозначим символом Ки где i — номер ступени диссоциации.
В таблицах констант равновесий, составленных для учебных
целей [4] *, приводятся:
* Как уже указывалось во введении, эти таб.чицы будут пркссдспг
в виде приложения во второй части настоящей книги.
2*
для кислотно-основных равновесий — логарифмы констант
протонизации (Ig а), показатели ступенчатых констант
диссоциации кислот (р/С), показатели общих констант ионизации акво-
металло-ионов (рт]) и показатели ступенчатых констант
диссоциации гидроксометалло-комплексов (р/Сосн);
для равновесий комплексообразования—логарифмы
(общих) констант образования (IgP) и ступенчатых констант
образования (Igft);
для гетерогенных равновесий — показатели произведений
растворимости (рР) и логарифмы постоянных закона Генри и
распределения между водой и органическими растворителями
для окислительно-восстановительных полуреакций —
логарифмы констант (lg/() и значения стандартных потенциалов
3.2. СООТНОШЕНИЯ МЕЖДУ РАЗЛИЧНЫМИ ВИДАМИ
КОНСТАНТ, ОТНОСЯЩИХСЯ К ОДНИМ и ТЕМ ЖЕ
РАВНОВЕСИЯМ
При выяснении соотношений между константами
исследуемых равновесий используются два важных равенства.
Первое выводится путем прямого приложения закона
действующих масс к любому одиночному равновесию, например,
равновесию протонизации А- + Н+5=^НА (ЗЛ). Если константу
протонизации, т. е. процесеа, идущего слева направо, обозначить а,
то константа обратного процесса должна быть равна ее обратной
величине, т. е. о-^ как это следует из записей закона
действующих масс для равновесия (3.1) по стрелкам -> и
(НА) (A-)(H-i-) , 3 2V
Это означает, что для однопротонной кислоты НА должно
выполняться равенство Ig о = р /(, так как по определению рК =
= —lg^» а —lg/iC = lgo, поскольку /( = 0-4
Смысл второго очень важного равенства легко устанавливается
с помощью примера. Рассмотрим две ступени процесса комплексо-
0 бразования:
МЧ-А«МА (3.3); МА-ЬА;=±МА2 (3.4).
Если равновесию (3.3) отвечает константа ku а равновесию (3.4)—
k2j то по закону действующих масс
перемножая эти два уравнения, находим, что
(М)(А)2
= klk2. (3.6)
36
Очевидно, уравнение (3.6) представляет собой выражение
закона действующих масс для равновесия
M+2A.=sMA2,
получающегося путем сложения уравнений (3.3), (3.4). Это
дает право утверждать, что константа равновесия, которое может
быть представлено в виде суммы двух или более равновесий,
равна произведению констант уравнений — слагаемых, а
логарифм ее равен сумме их логарифмов.
Равенства вида (3.6) являются равенствами точными, так
как перемножаемые уравнения (3.5) относятся к одной и той
же равновесной системе и величина (МА), на которую
сокращается произведение, не равна нулю.
Разобранный пример дает зависимость между IgP и Ig^ в
таблице констант равновесий комплексообразования. Например, в
этой таблице для бромовисмути-ионов находим запись:
Вг-
BiA7
BiAs
BiA^
BiA2+
при / = 3,0
за счет
2NaC104 -f 1Н+
IgP
7,80
6,18
4,26
2,26
1,62
1,92
2,00
2,26,
которая расшифровывается следующим образом:
Bi^+ + 4Вг- 5=£ BiBrT, Ig Р4 = 7,80;
BiBr3 + Br-s:sBiBr7, Ig^4= 1,62;
Bi^+ -f 3Br-s2 BiBrs, Ig p3 = 6,18:
BiBrt + Br~ 7=t BiBra, Ig^a = 1,92
Bi^+H-2Br~«BiBrt, lgp2 = 4,26;
BiBr^+ -f Bp- BiBr?, Igk2 = 2,00;
Bi^+ + Br- BiBr^+ Ig Pi = 2,26;
Bi^+ + Br- BiBr''+, \gki = 2,26.
Кроме того, в таблице указано, что константы равновесий для
данной системы измерены при ионной силе / = 3,0 моль-л"' на
фоне перхлората натрия (2 моль-л-') и хлорной кислоты
(1 моль-л-').
Из этой расщиренной записи следует, что индекс при (общей)
константе комплексообразования р„, т.е. п, обозначает число
присоединяющихся лигандов (Вг--ионов), а индекс при ki, т. е. i,
номер ступени комплексообразования. Легко видеть, что
lgpi = lg*i, lgp2 = lgA:i+lgA:2
37
и т. д. в общем случае
п п
IgPn = S Ig^i или р„ = П ki. (3.7)
1=1 i=i
Сложнее связь между Iga и р/С. Найдем сначала
соответствие между о к К для четырехпротонной кислоты Н4А, записывая
равновесия в том порядке, который принят в [4], например, для
этилендиаминтетрауксусной кислоты:
(1) А^- + 4Н+^Н4А, 04-, Н4А^Н++НзА~, Кх\ (5)
(2) А4-+ЗН+^НзА-, аз; НзА~;=гН++Н2А2-, К2\ (6) (3.8)
(3) А^~+2Н+вН2А2~, а2; H2A^-»Н-ь + НА^-, Къ\ (7)
(4) А^- + Н+«НА^-, а,; НАз-«Н-ь+А^-; /(4. (8)
Обратим, прежде всего, внимание на тот факт, что индекс
в любом К\ равен числу отрицательных зарядов, которое несет
образующийся на i-й ступени анион, и в то же время числу
образовавшихся от начала диссоциации Н+-ионов. В то же время
число протонов, входящих в диссоциирующий гидроанион,
равно а —(i —1), если а —основность исходной кислоты НаА. Тогда
в общем виде равновесие диссоциации гидроаниона можно
представить следующим образом:
Ha«o~.)A(^-^^-^H++Ha«iA^- Ки
Теперь, сопоставляя Значения а и /С в (3.8), устанавливаем,
что 01=/(7\ откуда, учитывая равенство 4= а в
рассматриваемом примере, получаем для общего случая
01 = /СГ^ и Iga, == -Ig/Ca = р/Са. (3.9)
Учитывая, что в (3.8) сложение равновесий (8) и (7) дает
равновесие, обратное (3), сложение равновесий (8), (7), (6) —
равновесие, обратное (2), и сложение равновесий (8), (7), (6) и (5)—
равновесие, обратное (1), находим, используя правила (3.2), (3.6):
для Н4А для Н4А = НаА
04= Al «Л? 'Аз 'Ад » «4 == Аа--3*Аа--2*Аа--] •Да—О,
аа=П КТ\
1=1
<J3 = КТ^ 'КТ^ •Ка ^; ^3 = Ka-^2'K7l\ • ^а—о; (3.10)
<J2 = КТ^ 'КТ^; 02 = K7l\ 'KZ^o'y
01 = К7^: а\ = К71о\ СП =
Чтобы получить общее выражение для oj, учтем, что
значение любой константы о выражается произведением величин
обратных ступенчатых констант диссоциации, причем индекс у
38
первого сомножителя равен а — (j — 1), у второго — а — (j — 2)
и т. д., а у последнего — а — (j — j). Тогда
I
0|= П или lgaj=. S py(a^(,_i). (ЗЛ1)
Сопоставляя выражения с\ через произведения /СГ' для Н4А,
приведенные в левом столбце (ЗЛО), можно написать
; А2=-=т—; ^3 = -: = —; ^4 = -
Очевидно, что в общем случае
Ki - ; p/Ci = lgaa-.(i-,) - Ig са^и (ЗЛ2)
^«—(1—1)
В таблице показателей общих констант ионизации аквокомп-
лексов (pig)* и показателей ступенчатых констант диссоциации
соответствующих гидроксокомплексов (р/Сосн = рК) сразу
учитывается два ряда равновесий:
Mi^+ + H205=tMO№-»4-H+ или М»^-ь —H+вMO№-^ -qi
М^'+Ч- 1Н20«М(0Н)Г*+ iH"^. или М*'-*-— 1Н-*-вМ(0Н)Г', Tji
(ЗЛЗ)
где 1 = 177;
M(OH)j!i'-"^^-^M(OH)^'JJi^-*^>- + OH- Ki
М(ОН)^?Л^"*'^-:^ M(0H)i'JJ5'"^^"" + ОН^, К2
mOHt'Zr^i'^Щ0Н)1^/^1^-''^ + ОН-, /(| (3.14)
M(OH)f^-J^M»^H- + OH- К^^,
где индекс i при К указывает общее число ОН""-ионов, которое
должно уйти из исходного комплекса, чтобы получился комплекс,
образующийся на этой ступени. Например, формуле комплекса,
образующегося на второй ступени, —М{ОНУ^'^^''^^'^. Он образуется
из исходного за счет потери двух ОН--ионов, значит, i = 2.
Между равновесиями (3.13) и (3Л 4) нет прямой связи. Можно,
однако, системе равновесий ступенчатой диссоциации
гидроксокомплексов (ЗЛ4) противопоставить систему равновесий их образо-
вания:
Mt--H + OH-«MO№-J, Pi
+ 20Н- в М(ОН)Г^,
* Константу ионизации аквометалло-иона i\ в литературе часто называют
константой гидролиза, а равновесие (3.13) — равновесием гидролиза иона М**"*".
39
(3.15)
М'^++пОН-.=.М(ОН)Г", р„
м^"^+|х' ОН- <=± м(он)|;;г^',
Равновесия комплексообразования (3.15) и ступенчатой
диссоциации гидроксокомплексов (3.14) находятся в такой же
зависимости, как ранее рассмотренные равновесия протонизации и
ступенчатой диссоциации кислот (3.8). Поэтому, используя формулу
(3.11), можно написать
Рп = П /Cjl^i.(n—к)» (3.16)
Для дальнейшей увязки констант ig, р, Косп учтем, что любое,
например i-e, равновесие ионизации аквометалло-иона (3.13) можно
рассматривать как суммарный процесс:
М'^"^+ЮН-вМ(ОН)Г'
Н205:±Н+ +0Н-|
Pi
(3.15')
i Н20вМ(0Н)Г^ + iH+ Tji = ^iwK
или
- i H-^ 5=± М(ОН)Г-^i = (3.130
константа которого y;n = ^iW^ (3.17), так как равновесие
диссоциации воды входит в суммарный процесс i раз.
Сопоставляя (3.17) и (3.16), находим, что
i
711 = 22;* П /C-,L(i«k). (3.18)
Теперь, используя сходство (3.18) и (3.11) и заменив в
равенстве (3.12) а на р и а — на fi.', получаем
Ai(ocH) = = -iTTTi -z = zr^ ^. (3.19)
Соотношения между общими pn и ступенчатыми константами
комплексообразования ki уже рассмотрены. Зависимость между
Рп и значениями ki выражается формулой (3.7). В то же время
А, = ^ и Igfei =^ IgPi — lgPi«b
Pi—1
40
3.3. ПЕРЕХОД ОТ РАЗЛИЧНЫХ ТАБЛИЧНЫХ ЗНАЧЕНИЙ
КОНСТАНТ К ЗНАЧЕНИЯМ, ПРИМЕНЯЕМЫМ ПРИ РАСЧЕТАХ
Сборники констант ионных равновесий*. Наиболее полным
собранием констант диссоциации органических кислот в водных
растворах является справочник Кортюма, Фогеля и Андрусова
[54] и его продолжение [61]. В нем приводятся либо константы
ступенчатой диссоциации Ki, либо их показатели pKi. Переход
к Oj здесь осуществляется по формуле (3.11). Например, ДЛЯН4А
2
lg02 = S рК4—(2-i) = рКг + рКл-
В справочнике Перрина [60] в большинстве случаев
приводятся значения показателей констант ступенчатой диссоциации
сопряженных с соответствующими основаниями катионных
кислот. Эти константы отвечают равновесиям:
На'А*'+ -f Н205=± На _lA«'-l -f Н3О+ Kl
Ha'-i+lA-'-^+l + Н205=± Ha'-iA-'-* + H3O+, Kt
HA+ + H2O e A + H3O+, Ka',
где индекс i при К равен полному числу оксоний-ионов, образе»
вавшихся в результате диссоциации катионной кислоты На'А*'+
на всех предыдущих и на i-й ступени.
Показатели констант этого типа обозначаются в справочнике
[60] иногда символом рКа, а чаще просто рКи где i имеет смысл,
указанный выше.
Зависимость между константами Ki и aj выражается и в этом
случае формулами (3.11) и (3.12) после замены в них а на а'.
Для некоторых органических оснований в справочнике [60]
приводятся показатели констант диссоциации оснований pKbif
отвечающр.е равновесиям вида
А + j Н2О 5=± HjА^+ + JOH-, Кы. (3.20)
Исходя из (3.20) получаем
А + j Н2О « Hj Ai+ -f j 0H-| 1
H+ + OH-5=tH20
]
w-i ^ (3.21)
A + JH+ «HiAi+ KbiW-i
Суммарное равновесие (3.21) представляет собой равновесие
протонизации j оксоний-ионами основания А. Этому равновесию
должна отвечать константа Oj, вычисляемая, как следует из (3.21),
по формуле Oj = Kbpr-K
* Обозначения констант равновесий, принятые в справочниках [54, 60,
66], но не используемые в этой книге, в разделах 3.3 и 3.4 набраны
полужирным курсивом.
41
Одним из первых справочников, посвященных реакциям
комплексообразования в растворах, является книга К. Б. Яцимир-
ского и В. П. Васильева [36]. В справочнике [26] представлена
большая сводка констант? ионизации аквокомплексов с
образованием моноядерных продуктов гидролиза.
Наиболее полной сводкой констант равновесий
комплексообразования, кислотно-основных и
окислительно-восстановительных является продолжающееся издание [64—66]. Во введении
к каждому тому этого справочника составители приводят
схемы равновесий, наиболее часто встречающихся в справочнике,
и символы отвечающих им констант. Для остальных равновесий
объяснения даются в тексте таблиц. Унифицировать и
пересчитать константы составители этого справочника не пытались.
Четырехтомное издание [44] содержит критически отобранные
значения констант равновесий протонизации и
комплексообразования с участием ряда неорганических и органических
лигандов.
Рассмотрим методы расчета интересующих нас констант из
данных, приведенных в [65, 66].
Ступенчатые константы протонизации и комлексообразова-
ния, В справочнике [65, 66] константы протонизации и
комплексообразования обозначаются одной и той же буквой К.
Для реакций протонизации частицы L приводятся два
равновесия и рядом с ними символы констант:
Н+ + L « HL, Kl Н+ + Hn-iL 7=t HnL, /Г(3.22)
Здесь индекс 1 показывает «одноядерность» продукта
протонизации, а индекс п — число Н+-ионов, присоединившихся к L
на п-й ступени. Очевидно, что при сложении, например, j
равновесий (3.22) получится суммарное равновесие
L-bjH+^HjL, /fi./f2- ... -А-, =a|.
Для расчета интересующих нас а\ по данным, пр;1веденным в
справочниках [65, 66], надо пользоваться уравнением
. lgoi=SlgA-„, (3.23)
так как в [65, 66] во всех случаях приводятся логарифмы
констант.
Надо помнить, что значения /Гь, приведенные в [65, 66], не
совпадают с константами ступенчатой диссоциации. Для расчета
этих величин следует пользоваться значениями Igoj,
вычисленными по формуле (3.23) и уравнением (3.12).
Равновесия комплексообразования в [65, 66] рассматриваются
в виде последовательных ступеней:
ML„«j-f L«MLn, /f„. (3.24)
42
в этом случае Кп^К, значения Рп и Igpn с учетом (3.7) можно
вычислить по уравнению
IgPn = .S Ig/fi, где Ig/fi = IgAi. (3.25)
Обозначение констант ступенчатого комплексообразования
сохраняется и в тех случаях, когда металло-ион координирует
гидроанионы. Так, для равновесия
М (HpL)„_, + HpL « М (HpL)n, /fn
константа обозначается тем же символом, что и для равновесия
(3.24). И в этом случае для расчета интересующего нас IgPn
приходится пользоваться уравнением (3.25). При этом, конечно, не
учитывается кислотная диссоциация комплекса M(HpL)n, оценить
которую можно, лишь зная IgPn для равновесий образования
комплексов MLn.
В таблицах [65, 66] наряду с константами ступенчатого
комплексообразования приводятся и общие константы
комплексообразования Рп. В случае образования многоядерных по отношению
к центральному атому комплексов, например, для равновесия
mM + nL^MmLn
константа Pmn включает два индекса: первый, отвечающий числу
центральных атомов, и второй —числу лигандов.: Как всегда,
индекс ш = 1 опускается.
Присоединение протонарованного лиганда (HL) с выделением
протона. В справочнике [65, 66] приводится большое число
значений логарифмов *Кпу относящихся к равновесиЗям
комплексообразования
MLn_i + HL;=iML„ + Н-ь, */f„. (3.26)
Суммарное уравнение, получающееся при сложении уравнения
(3.26) с уравнением протонизации лиганда L, т. е. с
равновесием H++L?=tHL, о (3.22), имеет вид
MLn_i + L^MLn, Кп = *А-па = К.
Дальнейший расчет по (3.25) дает значения Ig Pn.
Звездочкой слева отмечают составители справочника [65, 66]
и общие константы комплексообразования в случае
взаимодействия с лигандом HL, которое сопровождается выделением протона.
В этом случае рассматривается равновесие
шМ + nHL^MmLn -f пН+,*р^п.
Прибавляя к нему п раз равновесие пр(3(гонизации (3.22),
находим выражение для константы суммарного процесса:
Pmn = *Pmna" ИЛИ Ig Р^п = Ig *Pmn + nig а.
По этой формуле из значений *Pmn, приведенных в [65, 66],
получают необходимые для расчетов значения Pmn.
43
Для комплексов более сложного состава, например тройных^
в [65, 66] используют символ р (формула).
Для равновесий
Fe'+ + 2S0'r + SCN-« Fe(S04)2SCN'-
и
Th'+ + H3PO4 в ThH3P0j+
константы образования записываются так:
p(Fe^+L|-"SCN-) и ИТЬ'+НзЬ),
поскольку логарифм первой из них находится в таблице суль-
фато-, а второй —в таблице фосфато-комплексов.
Кислотные и основные константы. Ступенчатые константы
равновесий ионизации аквометалло-ионов в справочнике Силлена—
Мартелла обозначаются символом */Гп, например, для равновесия
РеЗ+ з=± ЕеОН2+ + н+ Ig* Ki = —2,17.
Тогда для получения значений pig и рК следует пользоваться
формулами p/ifi = —Ig */fi и pTf]i = —S lg*/fn.
В ряде случаев для некоторых комплексных кислот в [65, 66]
приводятся логарифмы констант ступенчатой диссоциации,
обозначаемых Ка (формула), где в скобках стоит формула
диссоциирующей на данной ступени частицы. Например, для
комплексной кислоты Н2ре(СО)4, в которой СО = L, в [65,66]
находим
H2FeL4 5=iН-*- + HFeLT; Ig(Н2реЬ4) = -4,4;
HFeLT ^ Н+ + FeU~; Ig/ГЛНРеЬГ| = -13,4.
Очевидно, используя символы, принятые в этой книге, можно
записать
Ig Ка (H2FeL4) = Ig Ki; Ig Ка f HFeLr) = Ig K2.
a тогда no (3.9) и (3.11):
p /С, = _lg Kl = 4,4; p /С2 = -Ig K2 = 13,4;
lgai = 13,4 и lga2=: 17,8.
Для «основных» констант, отвечающих равновесиям вида
NH3«NHt+ 0Н", в [65, 66] приводятся lg/Гб, например:
lg/fb(NH3) = —4,79. Простое преобразование
NHs + Н2О « NHt + ОН- Ig Kb = -4,79
Н+ + ОН-^ Н2О -Ig ^ = 14
NH3+H+ «NHt lga=lg/f6 —Iga; = 9,21
позволяет перейти к равновесию протонизации, для которого
lgo^]gKb — lgw==pK.
44
Константы растворимости. Все константы равновесий
растворимости твердой фазы обозначаются в таблицах [65, 66]
символом /fs. Обычное произведение растворимости, отвечающее
равновесию МаЬв (т)» аМ + bL, обозначается /fso. В нем индекс о
показывает, что речь идет о растворении в чистой воде. В этом
случае Р = Kso и р Р = —Ig/fsc
К той же группе констант растворимости Ks Силлен и Мар-
телл относят константы равновесий растворения твердых
соединений MaLb за счет комплексообразования, т. е. взаимодействия
МаЬь(т) с лигандом L и за счет взаимодействия с оксоний-ионами.
В первом случае общим символом константы является /fsmm где
индексы пит отвечают формуле образующегося растворимого
комплекса MmLn. Во втором случае Силлен и Мартелл приняли
обозначение */fsmn. В обоих случаях индекс m = 1 опускается.
Если во втором случае в результате взаимодействия с Н-Ь-ионами
образуется металло-ион, то вместо */ifsmr. пишется */fso.
Далее будет показано, что все равновесия, которым отвечают
константы Ksmn и */fsmm ОТНОСЯТСЯ К числу суммарных и
константы их могут быть всегда получены с помощью правил (3.2)
и (3.6) из констант простейших равновесий.
3.4. ВЫЧИСЛЕНИЕ КОНСТАНТ, ОТСУТСТВУЮЩИХ
В ТАБЛИЦАХ, С ПОМОЩЬЮ ИЗВЕСТНЫХ КОНСТАНТ
СМЕЖНЫХ РАВНОВЕСИЙ
Используя правила (3.2) и (3.6), можно с помощью констант,
приведенных в таблицах и статьях, вычислять константы
суммарных равновесий, а иногда и равновесий простейших.
В разделе 3.3 уже говорилось о том, что константы */fsmn
и Ksmn можно в таблицы не включать, так как они являются
константами суммарных процессов. Рассмотрим два равновесия,
приведенных в справочнике [66, табл. 2, с. 41]:
Ве(0Н)2 (т) + 2Н+ 5=t Ве2+ + 2Н2О, Ig* К^ = 6,86; (3.27)
ЗВе(0Н)2(т) + ЗН+?=^Вез(0Н)з+ ЗН2О, lg*/fs33 = 11,67. (3.28)
Равновесие (3.27) получается при сложении равновесий
Ве(0Н)2 (т) « Ее2+ + 20Н-
Н+ + 0Н- вНгО
—2\gw
(3.29)
Ве(0Н)2 (т) + 2Н+ « Ве2-Ь + 2Н2О, Ig *Kso =
= lgP —21gz2;=6,86
Из равенства (3.29) следует, что р Рве(онь == 21,14. Эту
константу и следовало внести в таблицы, так как с ее помощью
можно рассчитать любые сложные равновесия, включающие
твердую Ве(ОН)о,
45
Гораздо сложнее равновесие (3.28). Его можно представить
в виде суммы равновесий:
Ве(ОН) 2 (т) 5=t Ве2+ + 20Н-
Ве2+ + Н 20 ^ ВеОН+ + Н+
ЗВеОН-^ «Вез(0Н)1+
Н + ОН- ^НзО
31gP = —63,42
31g7j, =-10,89
Igx
—61gK/ = 84
ЗВе(ОН)2 (т) + ЗН+ ^ Вез(ОН)^"*- + ЗН2О lg*irs33 = 11,67
и, используя значение Ig^ji = lg*/fi = —3,63, взятое в том же
справочнике [66], рассчитать логарифм константы образования
тригидрооксотрибериллий-иона, имеющий гораздо большее право
на включение в справочник: Igx = 1,98.
Лишними можно считать и константы равновесий растворения
за счет комплексообразования, например А'збг, для равновесия
2AgI (т) + 4Г ^ Ag2l6", 1ё A-S62 = -3,0,
так как значение это легко вычисляется с учетом первичных
р;авновесий
Agl (т) ^ Ag+ + Ig Р = -16,35;
2Ag-^ + 61« Ag2r6'"", Ig Рб2 = 29,7,
константы которых приведены в одной строке с Ig/rs62 в [66,
табл. 75, с. 339].
Используя те же приемы, можно найти много полезных
констант, значения которых отсутствуют в таблицах. Например,
в таблицах можно найти значение pig =9,10 для равновесия
2Cd2+ + H20<=±Cd20H3+ -f Н+, \gn = —9,10,
хотя трудно себе представить, чтобы два аквокадмий-иона могли
сообща диссоциировать, освобождая один оксоний-ион. Гораздо
вероятнее равновесие комплексообразования:
Cd2+ + CdOH+« Cd20H3+, Igjc,
HO значения Igx в таблицах нет. Найти его можно, используя
равновесия
Cd2-b + Н2О «CdOH+ + Н+ Igig = -10,2
Cd'^ +CdOH+;=±Cd20H3+ Igx
2Cd2+ + H^ 5=iCd20H3++H+ lgig2i = —9,lo'
В результате находим lgx=l,l.
Подобным же образом рассчитываются Igx для А12(ОН)2"^р
Cu2(0H)l'*', НЕГ и им подобных.
В справочнике [65, 66] не выделены специально константы
протонизации комплексных анионов, но иногда встречаются кон-
46
станты комплексообразования для равновесий, в которых один
и тот же металло-ион координирует и анионы, и гидроанионы
одной и той же кислоты. В подобных случаях возникает
возможность расчета соответствующих констант протонизации.
Так, в [66, табл. 836, с. 635] находим следующие значения
логарифмов констант для равновесий:
Са2+ + L^<=±CaL2-, Ig pi = 10,59 (3.30)
Са2+ + HL3-^ HCaL- Igx = 3,51, (3.31)
где — этилендиаминтетраацетат-ион.
Используем эти данные для расчета логарифма константы
протонизации СаЬ2---иона, т. е. равновесия
CaL2- + H+^HCaL- а,.
Общий подход к решению таких задач сводится, как уже было
показано, к составлению системы равновесий, в которой
равновесие с определяемой константой должно быть или одним из
слагаемых, или суммой. Если. константы всех равновесий, кроме
одного, известны, то задача решена.
В данном случае следует начать запись с равновесия (3.30),
но записать его надо в обратном порядке:
CaL2- -:,са2+ + L^-, —Ig р, = —10,59. (3.32)
В левой части равновесия (3.32) у нас не хватает Н+-иона.
Его можно ввести, прибавляя к равновесию (3.32) равновесие
протонизации L"*"". Это вдвойне выгодно, так как ведет к
устранению из правой части слагаемого L*-:
L^- + Н+ i=t HL3-, Ig а, = 10,26, (3. 33)
причем значение Igai найдено в той же таблице справочника
[66]. Теперь остается заменить в правой части сумму Са2+-(тНЬ^-
на HCaL"~. Очевидно, что для этого к сумме равновесий (3.32)
и (3.33) достаточно прибавить равновесие (3.31). В результате
CaL2-;=tCa2H- + — Igpi = —10,59, (3.32)
+ Н+ «HL3- Ig Gi = 10,26, (3.33)
Ca2H- + HL3- ^HCaL- Igx =3,51, (3.31)
CaL2~ + H+ ^ HCaL- Igo =3,18.
Далеко не всегда все необходимые для расчета искомой
константы равновесия можно найти в пределах одной таблицы. В
качестве более сложного примера найдем константу образования
комплексного тиосульфат-иона из сульфит-иона и серы.
Решению такой задачи должен предшествовать поиск
подходящих равновесий в справочниках. Поскольку справочники [65,
66] построены по схеме «анион-металло-ионы», необходимо
47
просмотреть таблицы, относящиеся к 520з~-и501""-ионам.В этих
таблицах интересующего нас равновесия
S0|-+S(T)«S20i- (3.34)
и соответствующего значения Igp нет.
Тогда остается просмотреть таблицу констант сопряженных
окислительно-восстановительных равновесий. Здесь [66, табл. 1,
с. 28] находим два обнадеживающих равновесия:
2H2SO3 + 2Н+ + 4е- в SiOt + ЗН2О Ig Kl = 27,0; (3.35)
H2S03 + 4H+ + 4e-«S(T) + 3H20 lg/f2 = 30,4. (3.36)
Если равновесие (3.36) переписать в обратном порядке и
сложить с равновесием (3.35), то получится суммарное равновесие:
Н250з + 5(т)«520з'~+2Н+, lgK= lgKi-lgK2 =-ЗА. (3.37)
Теперь для получения искомого равновесия (3.34) к равновесию
(3.37) достаточно прибавить равновесие протонизации 50з"~:
SOf- + 2Н'^ 5=> H2SO3 Ig 02 = 8,97.
В результате находим, что искомый lgp = 5,57.
Конечно, подобные расчеты можно осуществить лишь в тех
случаях, когда удается подобрать все необходимые равновесия.
3.5. О ПОЛНОТЕ И ДОСТОВЕРНОСТИ СПРАВОЧНЫХ
ДАННЫХЛРИВЕДЕННЫХ В ТАБЛИЦАХ КОНСТАНТ
ХИМИЧЕСКИХ РАВНОВЕСИЙ
Справочники [54, 60] — специализированные таблицы констант
диссоциации нейтральных и катионных органических кислот.
В них нет сведений о дисперсиях, характеризующих точность
приведенных в таблицах значений, но все константы делятся на
«очень надежные», «надежные», «относительно надежные» и
«неточные». В первую группу авторы отнесли те р/С, для которых,
по их мнению, Ар/С ^ ± 0,0005, во вторую —те, для которых
Ар/С ^ ±0,005, в третью — с Ар/С ^+0,04, в четвертую — все
остальные. В первом случае Ар/С<0,1%, во втором — <1%,
в третьем — несколько процентов, а в четвертом — еше хуже.
В [54] приведены константы для 1056 кислот, а в [60]—для 3790
органических оснований.
Справочник [65, 66] включает, помимо констант
кислотно-основных равновесий, константы комплексообразования,
окислительно-восстановительных процессов, произведения растворимости и
константы многих суммарных равновесий. В справочнике совсем
нет констант распределения и коэффициентов активности, нет
дисперсий и лишь изредка можно увидеть вопросительный знак, которым
составители таблиц отмечают особенно подозрительные значения.
48
Для оценки достоверности справочных данных проще всего по--
знакомиться с отзывом о них Силлена [62]. В подобном докладе
о подготовленных к первому изданию таблицах констант Силлен
отмечает ряд основных положений.
Составители решили табулировать все значения, найденные
в литературе, в том числе и старые, так как на особенно
надежные результаты при измерениях констант рассчитывать не
приходится и новейшие результаты далеко не всегда лучше старых,
поскольку доля неряшливо выполненных работ в последние годы
сильно возросла.
Основными причинами ненадежности данных Силлен считает
отсутствие точных сведений о составе комплексов (так, в одном
и том же 1951 г. при изучении хлорокомплексов кадмия авторы
работы [18] обнаружили только CdCir и CdCl^"" " измерили
lgp3 = 0,8 и lgp4 = 0,2, а авторы статьи [6] обнаружили CdCl+,CdCl2
и CdCle"", которым отвечают Ig Pi = 2,19, lgp2 = 2,47 и Ig рб=7 2,59;
измерение констант без учета коэффициентов активности в
среде с изменяющейся температурой и переменным солевым
составом;
трудности, связанные с обнаружением и исследованием частиц^
существующих в сколько-нибудь достаточных концентрациях лишь
на узком интервале значений показателя координируемой
частицы или рН.
В результате Силлен приходит к выводу, что даже очень
опытные исследователи вряд ли могут обеспечить ДрК'<±0,1,
или даже ±0,2 логарифмической единицы. В справочнике [65^
66] в связи с этим интервальные значения не приводятся даже
в тех случаях, когда они указываются в цитируемых работах.
В качестве примера огромных расхождений в значениях
констант, полученных для одного и того же равновесия, можна
привести значения Pbi^s, = /Tso (В125з), указанные в [66, табл. 53,
с. 222]. Здесь находим Ig/fso, равные —90,5,—71,8 (при этом
значении стоит ?), — 88J, —60,15, —96, —97. В таблице нет
указаний, какое из значений, разбросанных на интервале в 37
порядков (!), следует предпочесть.
Скоре всего именно отсутствие рекомендаций составителей
относительно достоверности табличных данных вызвало
замечание Ирвинга [49], сводящееся к мысли, что каждому
пользующемуся справочником, придется самому решать, какие
значения заслуживают доверия.
Сам Силлен [62] считал, что «выжимать» из данных
таблиц слишком много теорий не следует.
Такое положение нельзя признать нормальным. Константы
равновесий применяются не только в учебной работе. Ими
пользуются при научных и технических расчетах и здесь, особенно
при оптимизации процессов, очень важно располагать
настоящими стандартными справочными данными.
49
Данные сомнительные могут привести к ошибкам и крупным
экономическим потерям. Так, согласно [58], в США два
химических завода, предназначенных для получения соединений
бора, оказались непригодными, так как проекты были рассчитаны
по ошибочным и неполным справочным данным. Потери
составили 76 млн. долларов. Этот случай и ряд других экономических
потерь по аналогичным причинам привели к тому, что в 1963 г.
в США была срочно организована Национальная система
стандартных справочных данных (НСССД). У нас такое
учреждение под названием Государственная служба стандартных
справочных данных СССР (ГСССД) была организована двумя
годами позже.
Главной обязанностью этих учреждений является проверка
существующих и отбор надежных данных, получение новых и
представление их в виде, удобном для использования в науке
и технике. Это —сложная, ответственная и очень трудоемкая
работа. В частности, проверка всегда должна осуществляться
путем сопоставления расчетных и опытных данных.
В качестве примера можно рассмотреть вопрос, какое из
двух крайних значений pPsi^s^ — 60 или 97 — ближе к истине.
Здесь в качестве критерия можно использовать тот факт, что
Bi2S3 практически не растворяется в НС1 <• молярной
концентрацией 3 моль-л-"Ч Простейший расчет сводится к учету
равновесий:
В125з(т) «2Bi3+ + 3S2-
S2-+2H+ «H2S
IgP, = —60 lgP2 = —97
31ga«60 31ga;^60
В128з(т) + 6Н+ »2Bi3+-f 3H2S Ig/fi^O Ig/f^^—37
с — 3 — —
[ ] _ 3 —6a: 2x 3x
Очевидно, начальная концентрация Н+-ионов равна сиа =
= 3 моль-л-^ равновесные концентрации [В1^+] = 2л:, [Н25]=3д:
и [Н+] = (3 —6л:) моль-л-Ч По приближенной формуле закона
действующих масс
(3.38)
2^ • 3^
Сокращая уравнение (3.38) на и подс^тавляя вместо lg/ifo6a
испытуемых значения, получаем два уравнения:
1 —102,7. ^
(0,5~^,)б ' (0.5^;с,)«
= 10-34,3.
Первое приходится решать методом подстяновки. Он дает
a:i~0,35 и 2ху = [ВР+] =; 0,7 моль-л-^о Во втором можно пред-
50
положить, что Х2«0,5. Тогда Х2= Ю"^'*^ .(0,5f;^I0"-^^*\ л:2~
~ 10-7.2_ 6.10-8, а [ВР+]^ 1,2.10-7 моль-л-!.
В результате значение pPi = 60 признается несостоятельным,,
но это еще не значит, что значение рР2=97 достоверно.
Внутреннее согласование всех наличных констант,
характеризующих различные равновесия, в которых может участвовать
В125з, могут обеспечить наилучшее приближение. Если расчеты,,
которые должны вести специалисты с помощью ЭВМ, не
позволят выяснить достоверное значение, необходимо
дополнительное опытное исследование и новые расчеты.
При. оценке достоверности литературных значений констант
устойчивости перспективна предложенная В. Н. Кумоком
методика прогнозирования устойчивости комплексных соединений,
основанная на статистическом анализе больших совокупностей
термодинамических характеристик реакций
комплексообразования [20].
Отобранные достоверные данные должны быть представлены
в единообразном виде, приспособленном к простейшему вводу
в современные ЭВМ.
Возникает вопрос, стоит ли разрабатывать более
совершенные методы расчета, если у нас еще нет настоящих
достоверных стандартных справочных данных. Ответ может быть
только положительным, так как требования научно-техническога
прогресса неизбежно ускорят наведение порядка в этой
области, да и само наведение порядка возможно лишь при
правильной организации расчетной работы.
4. КИСЛОТНО-ОСНОВНЫЕ РАВНОВЕСИЯ
4.1. РАСЧЕТ РАВНОВЕСИЙ В ВОДНЫХ РАСТВОРАХ ИНЕРТНЫХ
СОЛЕЙ
Для водного раствора соли XY, в котором Cxy = с, матрица
равновесий системы имеет вид
Н+
-1 I ОН-.
В этой системе четыре неизвестных величины: [Х+], [Y-],.
[Н+] и [ОН-]. Значения [Х+] и [У-] сразу же определяются
с помощью уравнений закона сохранения начальной концен*
трации:
сх = с = [Х+] (4.1); cy = с =.[¥-] (4.2).
51
Уравнение закона сохранения электронейтральности записывается
в этом случае в виде
[Н+] - [ОН-] + [Х+] -IY-] = [Н+] - [ОН-] = О (4.3)
с учетом равенств (4.1) и (4.2).
Дальнейши й расчет активности (Н+) = (Н3О+) связан с
оценкой зависимости [ОН-] от (Н?0+), выражаемой с помощью
закона действующих масс и соответствующего коэффициента
активности.
В каждом конкретном случае выражение закона действующих
масс и соответствующая ему константа равновесия должны быть
увязаны с формой записи последнего.
Если равновесие и выражение закона действующих масс записаны
fio Бренстеду-Лаури, то
2Н20^НзО-Ь + ОН- W и [OH-] = cpoj^lJ7(H20)2/i-i (4.4)
и (4.3) записывается в виде
^нЛ-?онВ^(Н2О)2А-> = 0, (4.5)
где \gW ^ —17,49 при 25°С—логарифм константы автопротолиза
воды; (Н2О)—активность воды, выраженная в моль . л-^ Тогда
h, = ^20)УТ^^. [Н+], = ср^й, = {^20)У^^^\ (4.6)
(ОН-) 1 = 0)1 = (Н 2О) 1/срн/он«^' ('^•'^)
[ОН-], = ср^н">1 = (Н20)|/ср^ср^^Ц7.
Из сопоставления (4.6) и (4.7) следует, что в любом растворе
инертной соли [Н+] = [ОН-]. Значения А и (о в растворах солей
будут всегда разными и будут сближаться по мере снижения
так как при этом (Н2О) будет стремиться к (Н2О) в чистой
воде, а /н и /он —к единице.
Если принять, что активности оксоний-иона и гидроксил-
иона связаны ионным произведением ш = 10-^^ при 25 °С, то
Таблица 4.1
pU7= 17,49
XY
pH
pOH
[H+] = [OH~3
pH
pOH
[H+]=[OH—
NaCl
KCl
0,0
1,4
3,0
0,0
1.4
3,0
7,000
6,787
6,636
7,000
6,965
6,948
7,000
7,260
7,475
7,000
7,080
7,101
1,00.10-7
1,22.10~^
8,39.10""^
1,00 10-^
1,16.10-"^
1,93.10-®
7,000
6,764
6,580
7,000
6,943
6,902
7,000
7,236
7,420
7,000
7,057
7,098
1,00.10-7
2,30-10-^
7,25.10-^
1,00.10-^
1,2110"-^
8,81 • 10-^
pai = 14
S2
{ОН-] = <Ро„ш-> и (4.3) переходит в ^f^h — ^fQ^^wh-^ =0 (4.8).
В этом случае
^2 = K/h<Poh^' [Н+Ь = <Рн^2 =/ТнТон^; (4-9)
(ОН-)2 = «.2 = -К<Рн/он^. [ОН-]2 = <Рон«'2 = У?нЧ'он^- (^-Ю)
в табл. 4.1 приведены результаты расчета Л, ш = (ОН-),
IH+] и [ОН-] по формулам (4.6), (4.7), (4.9) и (4.10) в
растворах, содержащих разные с^у
Значения lg<pjj и IgcpoH вычисляли по формуле
. 0.5115, . ,
(4.11)
где / = с^у, а параметры щ и 6t принимают следующие значе -
ния:
XY
«он
NaCl
KCl
6,87
4,89
—0,207
-0,129
3,25
—0,0303
—0,089
Для вычисления (Н2О) использовали уравнение
где при 25''С qi равны:
1 = 0
(4.12)
XY
^0
Я\
Я2
Яз
NaCl
55,273
—1,791
—0,0521
—0,0186
KCI
55,303
—1,773
0
0
Из табл. 4.1 видно, что вычисления по уравнениям (4.6) и
(4.7) с учетом константы автопротолиза растворителя дают
результаты, существенно отличающиеся от полученных с учетом
ионного произведения w. Таких же отклонений от более
правильных значений можно ожидать и при использовании
современных табличных значений констант других равновесий. Поскольку,
однако, другой информации у нас нет, в дальнейшем будут
использоваться сущесавующие табличные значения.
Отметим еще, что, как следует из данных, приведенных в
табл. 4.1, в растворах «нейтральных» солей рН отличается от 7.
4.2. РАСЧЕТ РАВНОВЕСИЙ В ВОДНЫХ РАСТВОРАХ СИЛЬНЫХ
КИСЛОТ И ОСНОВАНИЙ
Для водного раствора сильной кислоты HY, в котором Chy~
матрица равновесий системы имеет вид
53
Н+
1
—1
0Н~
w 1
I [OH-]=cpoH^/i-i.
(4.13)
Сама матрица представляется тремя первыми столбцами слева.
В последнем столбце дано выражение [ОН-], записанное с
помощью закона действующих масс. В системе три неизвестных
величины: [Y-], [Н-Ь] и [ОН-]. Для их расчета необходимо,
кроме (4.13), еще два уравнения: с^у = Y = [У-] (4.14);
[Н+] — [ОН-] - [Y-] = ср^Л — 9онШЛ-1 —7 = 0 (4.15).
Сравнение (4.15) и (4.8) показывает, что в (4.15) появляется
дополнительное слагаемое —7 = —chy> которого в уравнении (4.8)
для раствора соли XY не было.
Для водного раствора сильного основания ХОН, в котором
f ХОН
х+
н+
—1
он-
W
[ОН-] = ^ронКуЛ
-1.
^хон
= Х = [Х+];
(4.16)
(4.17)
[Н+] — [ОН-] + [Х+] = c^^h — + Х = 0. (4.18)
Из сопоставления (4.8) и (4.18) следует, что в уравнение
.закона сохранения электронейтральности для водного раствора силь^
ного основания, кроме разности cp^/i — ?он^^""*» входит
слагаемое Х = Схон-
Чем больше У, тем строже неравенство (p^h^ (p^^^wh-^ (4.19),.
Если (4.19) выполняется, то
срн/1= У, ft = /HY. (4.20)
Остается вычислить [ОН-] по (4.13). Подобным же образом в
растворах ХОН
^onwh-^ > срнй, ^oHwh-^ = X, Л ь= сронауХ-^ (4.21)
Если речь идет о водном растворе, содержащем р сильных
кислот: НУ(.1), НУ(|), ..., НУ(р), причем chy^i^ = Yx, ..
CHY(i) = yi, Сн¥(р) = Ур, то в первой колонке матрицы (4.13)
появится р элементов: У^), ..., У(7), ..Y^). Вместо (4.14)
придется записать р уравнений
CHY(i) = Yi = [Y(i)J, Р»
(4.22)
определяющих р искомых [У(1)]. Уравнение (4.15) сохранит свой
вид, если принять, что SYi = У (4.23).
54
Сказанное справедливо для водного раствора, содержащего q
различных сильных оснований X(j)OH. Здесь сохраняет силу
<4.18), если принять, что j:X,=X (4.24).
Расчеты проводятся в порядке, указанном для раствора одной
сильной кислоты или одного сильного основания.
4.3. СИСТЕМЫ УРАВНЕНИЙ, ХАРАКТЕРИЗУЮЩИЕ РАВНОВЕСИЯ
В РАСТВОРАХ СЛАБЫХ КИСЛОТ И ИХ СОЛЕЙ
К числу слабых кислот относятся: обычные а-протонные
кислоты НаА, отвечающие аниону А""", несущему а отрицательных
зарядов; а'-протонные катионные кислоты На'А*'+, отвечающие
незаряженному основанию А; аквометалло-ионы M(H20)n*" =
^Mf^+; средние соли ХаА, МаА^., HaAYa, MYf,. и кислые соли
Ха—рНрА.
Растворы этих кислот и их солей являются гораздо более
сложными системами, чем рассмотреные в 4.1 и 4.2. Поэтому
в каждом случае прежде всего придется установить число
искомых равновесных концентраций, проверить возможность
составления достаточного числа уравнений, записать их и
установить порядок последовательного исключения нс-известных.
Рассмотрим примеры таких систем.
4.3.1. Для водного раствора НаА, в котором Сн^а=са = Л,
если в системе нет ассоциаюв.
0 -1
1 ]
он-
[ОН-] = ^o^wh"^ (4.25)
Н,.А^-Ч = срн.А<^1«^^» 1 = (4.26)
Общее число искомых равновесных концентраций совпадает с
числом отдельных частиц, содержащихся в матрице. Здесь у
нас две частицы А" ~ и Н+ в верхней строке, ОН- и а'
j-гидроанионов в третьей колонке, т. е. всего (а'-ЬЗ) неизвестных. Мы
уже имеем а' уравнений (4.26) и одно уравнение (4.25), т. е.
(а'-Ь1) уравнение, отвечающее закону действующих масс.
Одно недостающее уравнение можно получить, применяя закон
сохранения начальной концентрации к заданной Са=Л. Для
записи последнего уравнения используется закон сохранения
электронейтральяости. Таким образом, получение необходимого
для расчета равновесия числа уравнений возможно. Наличие
этой .возможности легко прослеживается при рассмотрении всех
остальных задач этого раздела.
В данном случае уравнение закона сохранения начальной
концентрации в подробной записи имеет вид
са = Л = 1А«-] + V lHjAJ-«] = V [HjAi-«]. (4.27)
55
Последнее уравнение характеризует электронейтральнЬсть системБв
[Н+] - [ОН-] + 20- а) [HjAi-«] = 0. (4.28)
j=0
Сразу видно, что, используя уравнения (4.25), (4.26), можно
легко исключить из уравнений (4.27) и (4.28) [ОН-] и все [HjA^-^].
Если одновременно учесть, что [Н+] = срнй, то сразу получим
Л = а I; cpH^-AOj/ii; (4.29)
j=0
а'
9нй — сронО^Л-^ + а 5] (j — а) срн^да^й^ = 0. (4.30)
j=o
Так система, включавшая (а'+З) уравнения, без всяких
ухищрений превращается в систему из двух уравнений с двумя
неизвестными а и Л.
В силу принятого в самом начале условия об отсутствии ас-
социатов уравнение (4.29) линейно относительно а. В таком
случае
\j=0
-1
(4.31)
Подстановка этого значения в уравнение (4.30) приводит к
уравнению с одним неизвестным Л:
* срнЛ — (foHwh-^ + А -i-^^p = О,
j=0 ^
(4.32)
если считать, что все cpi могут быть вычислены с помощью
соответствующих уравнений.
4.3.2. Катионная кислота На'А"'+ может быть введена в
раствор только в виде соли Ha'AYa'. Если сн^.АУа^ = Л и в системе
нет ассоциатов, то в первой строке матрицы придется записать
Y-, А и Н+, Здесь А — незаряженное основание. Его формулу
можно выразить символом А*—, считая, что а = О, тогда имеем
н+
0
1
—1
j
он-
[ОН-] = ^oH^h-^
[H,Ai-«
(4.33)
Здесь одна лишняя искомая равновесная концентрация, [Y-],
но она непосредственно определяется из уравнения cy = л'А =:
= [Y-] (4.34). Уравнение закона сохранения начальной концен-
56
трации незаряженного основания можно записать следующим
образом:
= Л == 2 [HjAi-b] = 2 [HjAi-«]a=.o = а 2 щаО',Ы, (4.35)
j=0 j=0 j=0
гда а= (А).
Используя тот же прием, получаем уравнение закона
сохранения электронейтральности:
[Н+] - [ОН-] - [Y-] + f j [HjAJ+] = [Н+] - [ОН-] - [Y-] +
j=0
+ 2 (j_a)[HjAi-«] = 0, (4.36)
которое после подстановки всех значений переходит в
уравнение, приспособленное для вычисления h в растворах Ha'AYa'
при а = 0:
?нЛ - сронО^А-» - а'Л + Л = 0. (4.37)
Расчет а проводят по уравнению (4.35), а остальных
неизвестных—по '(4.25) и (4.33).
4.3.3. Для водного раствора соли ХаА, в котором сх^а = Л,
сх = лА и Са = Л, если нет ассоциатов.
0
1
—1
j
он-
Н.А'*-*
W
[он-] = <fonwh-^
HjA'-"] = fii^A^iOhh S =ТГТ'.
Здесь Сх = аЛ = [X+J (4.38),
[Н+] — [ОН-] + [X+J + 2 а - а) [Н)А1-«] = срнй - <ронаЛ-' +
1=0
+ аЛ + а (j — а) <pH,AejW = 0.
1=0
(4.39)
Закон сохранения Са = Л выражается уравнениями (4.27), (4.29),
а уравнение для расчета h имеет вид
а'
<рнЛ - <Роншй-' + аЛ + л Ь2_^ 0. (4.40)
1=0 '
■ft'
57
4.3.4. Для водного раствора соли Ха-рНрА, в котором
Сх^_рНрА = А, 6А = Л и бх = (а Л, матрица равновесий
системы имеет тот же вид, что для раствора соли ХаА (см. 4.3.3.),
но Сх = (а —Р) Л = [Х+] (4.41). Это приводит к уравнению для
расчета А:
а'
9нЛ — tfoHwh-^ + (а — р) Л + Л '-
1=0
==0. (4.42)
4.3.5. Для водного раствора соли MY^,, в котором Cf^y^ =
а значит, См = М и = {лУИ при отсутствии ассоциатов и
комплексов MYS^",
Y-
н+
0
1
—1
— к
он-
W
Здесь
[ОН-] = <роншЛ->
(4.43)
(4.44)
(4.45)
См = S [М (OH)g ] = m 2 ?м(он)к1|}кЛ-'';
к=0 к=0
[H+]-[0H-]-[Y-]4- S ({.-к)[М(ОН)Г''
к=0
= (рнА —
сронг(уА-» — [аМ + m Д (р. — к) срм(0Н)к'»]кА-*' = 0. (4.46)
Уравнение для расчета А получается после подстановки в (4.46)
значения т из (4.45):
S (1^ - к) 9м(ОН)к'^к^'"^
к=0
2 9м(ОН)1^^к^"
= 0. (4.47)
к=0
4.3.6. Для водного раствора соли МаЛ^,, в котором См^а^ =
c^^ = ас, Са = [х«, если в нем нет ассоциатов и М'^^ не
координирует А*--ионы,
да-
0
1
0
0
0
1
—1
— к
j
он-
Л)
^к
[он-] = .рони-Л-'
М(ОН)Г''] = <Pм(OH)fc^k'"'^-^ к=
[Н)А'-»] =<Рн,АО|аА*, j = rT».
58
Используя дважды закон сохранения начальной концентрации,
получаем
См = ас = S [М (0Н)Г1= т 2 ^щощ^пФ-^; (4.48)
к==0 к=0
а' а'
СА = К = 2 [HjAi--] = а 2 ?H:AajA4 (4.49)
j=0 j=0 ^
Закон сохранения электронейтральности приводит к выражению
ФиА — (^oHwh-^ + т 2^ (р- — к) срм(он)кТ(1кА-^ -f
+ ^ (j — а) срн| AajA^' = 0. (4.50)
Уравнение для расчета h получается после исключения т и
а из (4.50). Оно имеет следующий вид:
2({^-к)^д,(ОН)к^к'^"^
ТнА—Сронг(УЛ-^+ ас!^-^-;;;; Ь
S ^^М(ОН)ь%^""^
к=0
S(i-»)<?H,A»i'''
+ \У'С'^. • = 0. (4.51)
4.4. ХАРАКТЕРИСТИКА ОТДЕЛЬНЫХ БЛОКОВ, ВХОДЯЩИХ В
РАСЧЕТНЫЕ УРАВНЕНИЯ КИСЛОТНО-ОСНОВНЫХ РАВНОВЕСИЙ,
И ИХ ИСПОЛЬЗОВАНИЕ
Основные блоки. Сопоставление уравнений (4.8), (4.15), (4.18),
(4.32), (4.37), (4.40), (4.47), (4.51) позволяет сделать ряд выводов:
а) все перечисленные уравнения обязательно включают
величину срнА —cpoHtiyA-i = [Н+1 —[ОН-], (4.52) которую можно
назвать «блокбм воды»; *;
б) в уравнении (4.15) для расчета h в растворе сильной
кислоты HY появляется блок сильной кислоты —Y, численно
равный Chy;
в) подобным же образом в уравнении (4.18) появляется блок
сильного основания + X, численно равный схон;
* Очевидно, что аналогичные уравнения для любого прото л итичес кого
растворителя HL будут включать блок этого растворителя
9h(H2L+)-<Pl'«hl(H2L+)-'-
59
г) в уравнения для систем, включающих нейтральную а-про-
тонную кислоту НаА, катионную а'-протонную кислоту На'А»'+
и их соли, обязательно входит блок слабой кислоты:
а'
+ А^^. —, (4.53)
д) подобный же блок для аквометалло-иона M**'"*" = M(H20)n**,
являющегося катионной кислотой, имеет вид
р.*
+ М ^ ; (4.54)
к==0
е) блоки катионов и анионов, вносимых в растворы в
составе солей слабых нейтральных, катионных и анионных кислот,,
всегда выражаются через начальные концентрации этих
последних:
блок X"*" в ХаА равен + аЛ, (4.55)
блок Х+ в Ха-рНрА равен +(а^Р)Л, (4.56)
блок Y"" в На'АУа' равен — а'Л, (4.57)
блок в MYt, равен — [хМ. (4.58)
Блок воды, выраженный разностью (4.52), равен нулю для
воды и растворов нейтральных солей XY. Он больше нуля, если
[Н+] > [ОН-], т. е. в кислых растворах. В подобных случаях
блок воды дает вклад в равновесную концентрацию оксоний-
иона за счет всех источников этих ионов в системе, кроме воды,
так как из равновесия H20<=tH+ + 0H- (4.59) следует, что
при диссоциации воды Н+ и ОН--ноны образуются в равных
количествах, а тогда [ОН-] становится мерой той части [Н-Ь],
которая образовалась за счет воды. По таким же соображениям
при [H+J < [ОН-] блок воды определяет вклад в [ОН-] за счет
всех источников ОН—-ионов, кроме воды. Здесь роль поправки,
учитывающей ОН--ионы, которые дает вода, играет величина срнй.
Система блоков и строгое соблюдение принятой символики
позволяют записать общее уравнение для расчета в любой
системе без записи промежуточных уравнений, а затем
конкретизировать окончательное выражение.
Запишем в качестве примера такое расчетное уравнение для
системы, в которой £?NaH,P04 = 0,3 И c^^eso, = 0,1 МОЛЬ • л-^ В общем
виде имеем сх^_^н^а = А = 0,3 моль • л-' при а = 3 и ci^^a^ =
= 0,1 моль • л-^ при а = [JL = 2. Сведений относительно фосфато-
феррокомплексов в таблицах констант нет.
60
Поскольку формула ферро-сульфата FeS04 = MA, задача
сводится к записи уравнения для системы, в которой сх^^рирл = А
и Смл = с. Здесь [Х+] = (а — Р) Л и можно сразу написать
а'
<Рнй-<Рона'Л-Ч-(а-р)Л +
1=0
|l' a'
S (i^ -1^) 'fmoH)^^^'^ So- ei) <ph .A-'i'''
Л-с^. + ^ = 0. (4.60)
k=0 j=0 ^
Для перехода к конкретному в этом случае уравнению нада
учесть, что для Н3РО4 а' = а = 3, для Ре^"^-иона [х = 2, к =U
для 504""-иона ai = 2 и j = 1. Тогда при р = 2, а — р = 1
уравнение (4.60) переписывается так:
?нА- сроншЛ-! + Л + Л i:^^^ +
S ?H,A^j^^
j=0 '
(2 - к) 9м(0Н)к^к^-^ S (i - 2) ^H,. A->^
S ^M(OH)i,^k^'"'' S ^H.A^'i^^
k=0 1=0 '
+ c'S^T + ^=0. (4.61)
Некоторые указания о численном решении таких уравнений бу-
дут даны позже. Здесь же отметим, что значительное
расхождение между значением А, вычисленным по уравнению (4.61) к
полученным для той же системы путем прямого измерения рН,
может возникнуть за счет систематических погрешностей в
значениях, использованных при расчетах параметров, а также за?
счет недоучета каких-либо равновесий, например образования
феррофосфато-комплексов.
4.5. ФУНКЦИИ БЬЕРРУМА И ИХ ПРИМЕНЕНИЕ ПРИ
СОКРАЩЕН НОЙ ЗАПИСИ РАСЧЕТНЫХ УРАВНЕНИЙ
КИСЛОТНО-ОСНОВНЫХ РАВНОВЕСИЙ
Существенное значение имеет физический смысл множителей
при Л и ЛГ в блоках (4.53) и (4.54).Он становится ясным, если
Числитель и знаменатель в первом блоке умножить на а, а во»
втором — на т.
61
в первом случае получаем выражение
i—а"
= пТ. А, (4.62)
2 (j ~ «) Тн,А^/«^^ S (i -«) [Н|А^-«
i=0 ' 1=0
где Пг,к — средний заряд на одну частицу, происходящую от
брёнстедовского основания A°^- (в частном случае а = 0).
Действительно, в знаменателе выражения (4.62) находится
сумма равновесных концентраций всех j-гидроанионоБ,
происходящих от А''--иона, а в числителе— сумма произведений
равновесных концентраций [HjAJ-*] на числа их зарядов (j —а).
Подобным же образом во втором случае
2^ ((х - к) TMCOHjk^k^-''" - к) [М(ОН)Г'^
к=0 - к=0
2 ^M(OH)kV^-'^ S [М(ОН)Г^
к=0 к=0
= п., м. (4.63)
Введение функций вида п^, т. е. средних зарядов и им
подобных величин, которые носят общее название функций
Вьеррума [2—5], обеспечивает существенное упрощение записей. Т. к,
уравнение (4.60) переходит в выражение
срнЛ — срон^Л-^ + (а — Р) Л + ЛПг, А + (Мг, м + СПг, А, = 0. (4.64 )
Развернутые значения Пг легко записываются по их смыслу.
Для относительно простых систем, используя уравнения типа
(4.63), можно получить дополнительные оценки функций Бьер-
рума^Так, уравнение (4.32)^ переписанное в видесрнй —срон^Л-^ +
+ Лпг. А = О (4.65), дает n,:^ = — (срнА — сронК^Л-') А'^ (4.66).
Подобным же образом находим,, что для раствора Ha'AYa' из
уравнения (4.37)
/г,, А = — (срнЛ — сронгсЛ-^ — а'А) А-\ (4.67)
Для раствора соли Хс^А из уравнения (4.40) находим
Пг, А = — (срнЛ — cpoHtc^/i-^ + 0.А) А-'. (4.68)
Для раствора кислой соли Ха-рНрА уравнение (4.42) дает
П2.А = — [срий —сронг2^/1-^ + (а-Р)ЛМ-Ч . (4.69)
Накрнец, из уравнения (4.47) следует, что для соли MY,x
Пг. м = — (?нй — cpoHt(y/i-i — (хМ) 1\/Г\ (4.70)
Если представить теперь уравнения (4.15) и (4.18) в форме,
сходной с уравнением (4.32) и другими ему подобными, то они
примут вид
52
<РнЛ — tpoHwh-^ + Y (-p^) = 0; (4.71)
<РнЛ -сронгг^й-! + X = 0. (4.72)
Это значит, что для растворов HY любой концентрации
^z. Y = — 1 = — (срнЛ — сронм^Л-О Y-\ (4.73)
а для растворов ХОН
Пг, X = 1 = — (срнй — сронПуЛ-^ (4.74)
4.6. ВЫЯСНЕНИЕ УСЛОВИЙ, ДОПУСКАЮЩИХ УПРОЩ.ЕНИЯ ПРИ
РЕШЕНИИ РАСЧЕТНЫХ УРАВНЕНИЙ КИСЛОТНО-ОСНОВНЫХ
РАВНОВЕСИЙ, КРИТЕРИИ, ОПРЕДЕЛЯЮЩИЕ ПРАВОМЕРНОСТЬ
УПРОЩЕНИЙ
Приближенные вычисления в растворе НаА. Разворачивая
суммы, находящиеся в числителе и знаменателе Ъг, а уравнения
(4.32) для случая а' = а, получаем
<рнЛ — (pouwh-^ —
^ ?На,,А<^.^1^^-'Ч2<Рн,^,да^2Н-Ч.»+«<Рд ^
Согласно Полингу [29] для ступенчатых констант диссоциации
неорганических кислот /Ci: /С2: /Сз = 1 * Ю""^: 10-^° ...;
табличные данные [44, 54, 61, 64—66] показывают, что всегда К\>
>К2> Кь> ..., хотя и не всегда выполняется соотношение,
приведенное в [29].^
Следовательно, наибольшее количество Н-Ь-ионов должно
появляться за счет первой ступени диссоциации, меньше — за счет
второй, еще меньше —за счет третьей и т.д. Соответствующие
равновесия ступенчатой диссоциации имеют вид
НаА^Н+ +Ha-iA-, /Сг, (4.76)
На-, Л- 5=г НИ- + Iia-2A2-, /Сз', (4.77>
На-2А2- ^ Н+ + На-зАз- Кз. (4.78>
Допустим, что каждое из таких равновесий устанавливается
независимо от остальных, тогда по уравнению (4.26)
[Н+], = [Ha-lA-] = <рН,^,А0а-1аЙ«-1,
[Нн-]2 = [На-гАМ = ^^^.AOa^.ah--^ (4.79)
и т. д., где iH+]i — приближенная оценка вклада в общую [Н+}
системы за счет i-й ступени диссоциации.
Умножение числителя и знаменателя последнего слагаемого в
<4.75) на а превращает его в
[нП'+2[НН-],-Ь ...+а,да ^^^^^
[Н,А] + [H+]i + IH+L + ... +срда'
Если предположить, что [11+]^ превосходит по величине сумму
всех остальных [H+]i, оставить в связи с этим в числителе и
знаменателе последнего слагаемого (4.75) только величины с
индексами а и а—1, а заодно отбросить слагаемое срон^^""^ <?нЛ,
поскольку речь идет о растьоре кислоты, то (4.75) превратится в
9иП — А —Т7=Г= О» (4.81)
которое после сокращения на h^-^ приводится к уравнению 2-й
степени относительно Л.
Поскольку снижение степени достигнуто за счет
предположения, что [H+ji > S [H+ji или, менее строго, за счет условия
jH+]i > 2 [H+J2, сразу же после вычисления необходимо
проверить, выполняется ли условие
?н«_1Аа«-1Л«-^ » 2срн„_2А^«-2
Критерии такого рода обычно преобразуют с таким расчетом,
чтобы в одной части неравенства стояла вычисляемая величина
Л, а в другой — сравниваемое с ней выражение. Деление
данного неравенства на 9Ha__iAOa-i^'*-2 дает Л > 2 "^""""^^ .
Для дальнейшего упрощения правой части учтем, что lgcpHa_2A=
= lgcp2 = 41g<pi, algcpH„_,A= Igcpi,4Tooa-2 = /Cr^/C7^ ... К7^ и
<sa^\ = KJ^KT^KT^ .../C7^ После подстановки этих значений,
полученных с помощью формул (2.21), (3.14), находим
окончательное выражение критерия: А>2- Ю^^б'Рх/Сг (4.82).
Преобразование уравнения (4.81) начинают с сокращения на
Са^КТКТ^ ... К7^ и h'"'\ Это приводит к уравнению
Ун/^--^. TrJ^^' /г =Q> (4.83)
^H„A^ + ^H„_iA^I
где Kl—константа диссоциации НаА на 1-й ступени. Смысл
множителя при А устанавливается следующим образом. Согласно
предположению, подавляющее количество Н+-ионов образуется
на первой ступени диссоциации. Тогда из равновесия
протонизации На-lA--иона следует
Ha-iA-+ Н-^ ^ НаА, КТ\ [НаА] ^и.аКТ' (Ha-iA")ft. (4.84^
64
Закон сохранения си^а = Л сводится к уравнению
А = [НаА] + [На-,А~1 = (Ha-iA^) {<?и,аКТ'Н + срн^ИК (4-85)
из которого
(На«1А-) = Л и lHa-lA-] =
где множитель при А—степень диссоциации НаА на первой
ступени. Теперь ясно, что уравнение (4.83) может быть записано
в виде [Н+1 — [Ha-iA-]. = О, т. е. представляет собой выражение
закона сохранения электронейтральности.
В приведенной форме оно имеет вид
+ 9Ha«iA/HaAKl/l —/н9Н^1А/НаА/С1Л = 0. (4;87)
Упрощенное решение
h = V ^Ho-iA/HaA^i (/нЛ —/г) = }/>нсрн^1/наА/С,Л (4.88)
можно использовать, если выполняется критерий fnA^h (4.89).
Если учесть, что /н9н^1А/наА =/i?i/o« 1, то простейшее
решение отвечает формуле Н^УКхА (4.90).
Если значение А, вычисленное по формуле (4.90), условию
(4.89) не удовлетворяет, то вычисляют h из (4.87), используя
расчетную формулу, исключающую «потерю точности за счет
вычитания:^:
h= (4.91)
где Р == ?He_,A/H„A/Cl и Ч = /н?На«1А/НаА/С1Л, ПрИЧСМ
/н?На_1А/НаА ~ L
Только В тех случаях, когда вычисленное значение А удовлет-
воряет критерию (4.82) и критерию А> v7h?h^;^ lO-^ (4.92),
вычисление А можно считать законченным. После этого
вычисляют а по формуле (4.29), а остальные искомые равновесные
концентрации — по формулам (4.25) и (4.26).
Приближенные вычисления А в растворах солей катионных
кислот. Если учесть, что в этом случае а=0, и сложить два
последних слагаемых уравнения (4.37), то получается уравнение
а
2 о — «') <Рн ,А«.Л'
ТнЛ ~ <ронаЛ-' + А = 0. (4.93)
3 ?.4зь 65
в этом уравнении (j — а) — число зарядов, которое приходится
на долю иона HjAJ"*" и а' Y~-hohob, которые были связаны с
На'А^'-ионом в исходной твердой соли Ha'AYa'. Тогда множитель
при А — среднее число заря дов на один ион, появляющийся при
диссоциации соли Ha'AYa'."
- = аГ..а. (4.94)
S ?h^.a^jfl'
Реакция в растворе Ha'AYa' должна быть кислой. Снижение
степени h в (4.93) может-- осуществиться лишь в том случае, если
большую часть Н+-ионов дает первая ступень диссоциации
На'А*'+. Решающую роль здесь играют равновесия
На'А«ч-«Н+ + Ha'-iA<«'-')+, Кх\ (4.95)
На'- lA<«'"-l)+ ^ Н+ + На'-2А(»'-2)+, ^2. (4.96)
Дальнейшее исследование ведется по схеме, использованной для
уравнения (4.32). Слагаемыми сроно^Л-' можно пренебречь и здесь
в случае выполнения критерия (4.92).
Учет только первой ступени диссоциации На'А возможен,
если выполняется условие
А » 2 Z!!£z±L . If^ = 2 К2 = 2 . 10(3-2a'),gcp./^ (4.97)
так как На'-гА^"''"^^"^ несет (а' —2) зарядов, а Ha'-iA<*'-i>+
соответственно (а' — 1).
Учитывая только первую ступень диссоциации, можно
переписать (4.93) в виде
, г = 0. (4.98)
Коротко ЭТО уравнение можно записать так:
1Н+] —[Ha'-iA(«= 0.
Его физический смысл сводится к тому, что за счет первой
ступени диссоциации На'А*'+, если она играет основную роль,
должна возникнуть 1Н+], численно равная [Ha'-iA^*'~^^+].
Формула упрощенного решения, аналогичная уравнению (4.88),
в этом случае имеет вид
А = l/"/h?Ha'_,AfHa,AiClJ (4.99)
Ее нельзя свести к уравнению (4.90), так как
66
Расчетная формула (4.91) переходит при этом в выражение
(4.100)
Отметим, что рассуждения и формулы, приведенные в этом раз-
деле, полностью относятся и к солям таких катионных кислот,
которые образует, например, этилендиаминтетрауксусная кислота:
НбАУг, но при этом надо учитывать, что а' = 2, а а = 4.
Приближенные вычисления h в растворе соли ХаА. В такой
системе анион А*- будет протонизоваться за счет Н+-ионов
растворителя— воды. Это поведет к накоплению ОН-'-ионов и
щелочной реакции, причем слагаемым срнЛ можно будет пренебречь, если
(0Н~) S «) > Ксрн/онш~ 10-7. (4 101)
Отбрасывая в (4.40) объединяя в нем два последних
слагаемых и заменяя h = ш<1)~*, получаем уравнение
— ?он<о + А -Ь^ = — ?oH<i) + Лпн, ХаА = О, (4.102)
где пн, ХдА — среднее число протонов на одну частицу,
появляющуюся в растворе соли ХаА за счет протонизации ее аниона.
Таким образом, преобразование уравнения (4.40), которое
выражало закон сохранения электронейтральности, превратило его
в уравнение (4.102), физический смысл которого сводится к
утверждению, что в растворе соли ХаА возникает (ОН"-] === сронш,
а'
численно равная У]/[Н,Л^-"«1.
При Э10М можно ожидать, что после умножения числителя
и знаменателя множителя при Л на а соотошение между
отдельными слагаемыми в числителе определится неравенствами
[НА^-«]= cpHAoiGyao)-' > 2 fH2A2-»] = 2срН2А02ш2аа) -2> ..
а в знаменателе соответственно:
[НА»-«] = cpHAO,ta;aa)-J > (НгА^-»] = ^н,мЧ<л-'^ ...
Такое предположение оправдывается тем, что наибольшего выхода
ОН^-ионов можно ожидать за счет суммарного равновесия
А«~ + Н+ «НА1-- , аь
Н2О Н+ + ОН- , ш, (4.103)
А»- + Н2О ^ ОН- + НА1-% ода,
3* 67
причем оценкой [ОН-]i, вклада в общую [ОН-] = сроно), может
служить здесь [НА^-*] = cpHAOi^A = срнА01шао)-^
Разворачивая в (4.102) сумму и оставляя в ней слагаемые
G / = О, 2, получаем
сроно) = А ; -1 . rzif- • (4.104)
Снижение степени будет оправдано, если срнА<31гг;о)-^ > 2срн,А02йу2а)-2.
После деления неравенства на срнА01Ши)-^ находим, что это возможно,
если О) > 2^^ . — ш. Наконец, учитывая, что oj = 02 =
^^Kr^^K"^^ и что (а—I) и (а — 2) — число зарядов при НА^-"
и Н2А2""*-ионах, получаем искомый критерий в виде
ш > 2 . 10(3-2a)lg^, ^ ^ 2 . 10<з-2«>»?911^ ш. (4.105)
После отбрасывания всех слагаемых с индексами, превышающими
единицу, приводим (4.104) к виду
Тоно) - А , ^'^j^T. г. = О- (4-106)
Используя (4.103), можно показать, что второе слагаемое в
уравнении (4.106) представляет собой (НАмножитель при А —
степень протонизации А*--иона, а смысл всего уравнения (4.106)
в том, что [ОН-] совпадает в [НА^-*], если в основном накопление
ОН--ионов происходит за счет 1-й ступени протонизации. Тот же
вывод можно сделать из правой части уравнения (4.103).
Дальнейшие расчетные формулы и критерии получаются, как
в предыдущих случаях, и в пояснениях не нуждаются.
Из (4.106) в приведенной форме
+ /а?на<'1^ —Wa9ha<'i^^ = О (4-107)
получаем простейшее решение:
^ - V^WaTha<'i^^ ^ (4.108)
которое справедливо, если выполняется критерий /онЛ > «> (4.109).
В противном случае значение ш вычисляется по формуле
•^^ОН^УНА°1^^
«> = ООО (4.110)
Приближенные вычисления h в растворе кислой еоли. Объединяя
два последних слагаемых в уравнении (4.42), находим, что в
растворе соли Ха-рНрА
68
срнА — <foHwh-'^ + A Ь2 = 0. (4.111)
j=0
Физический смысл множителя при А можно выяснить, если учесть,
что соль Ха-рНрА диссоциирует в растворе на Х+- и НрАР-""-ионы.
Поскольку в растворе из последних должны образоваться все
частицы от А*"~ до На'А, очевидно, что множитель (j — р) указывает
число Н+-ИОНОВ, обеспечивающее за счет равновесия
HpAP-« + (j-p)H+^H|AJ-
при j = о, а образование любой из частиц. Тогда
— = Пн.нрА (4Л12)
i]?HiA^i^^
1=0 '
среднее число протонов на одну частицу, происходящую от НрАР~" -
иона. _ _
Возможны три случая: Пн = О, /гн > О, пи < О (4.113). В первом
случае все Н+, которые получаются при диссоциации НрАР~*-ио-
нов, ведущей к образованию частиц Hp^iAP-*-",... НА^-'', А"-,
полностью расходуются на образование частиц, более протонизо-
ванных. В результате срн^ — <foиwh-^ = [Н-^] — [ОН-] = и и раст-,
вор будет нейтральным.
Во втором случае, когда Ян > О, общее число Н+.ионов,
расходуемое на иротонизацию НрАР"""-ионов, больше того, которое может
дать их диссоциация. Недостающая часть Н+-ионов получается
за счет диссоциации воды при одновременном накоплении ОН--ионов.
Реакция системы оказывается щелочной, как это и следует из
(4.И1), которое можно записать в виде [ОН-]-[Н+] + Апн (4.114).
Здесь [Н+] эквивалентна той доле ОН--ионов, которые дает не-
прсредственно растворитель.
В случае лн < О (4.111) имеет вид [H+J = [ОН-] + Апи (4.115).
Здесь реакция кислая, так как, кроме Н+-ионов, которые дает Н26,
в систему поступают избыточные Н+-ионы за счет диссоциации
р-гидроаниона.
Поскольку исходным в системе является НрА<*-Р>--ион,
важнейшим источником Н+-ИОНОВ является равновесие
HpA<«-P)-5=tH+ + Hp«iA(«-P+»)-. (4.116)
В то же время наибольшее количество Н+-ионов будет
потребляться за счет равновесия протонизации
НрА^«-Р>- + Н+ » Нрн. ,Ai«-P-J)-. (4.117)
Степень при h в уравнении (4.111) может в связи с этим
снизиться в том случае, если количества Н-Ь-ионов, получающихся
на второй ступени диссоциации НрА^-^-иона и расходуемых на
второй ступени его протонизации, будут намного меньше
получающихся и расходуемых на первых ступенях тех же процессов.
Если, как это следует из равновесия (4.116), оценкой количества
Н+-ИОНОВ, образующихся на каждой ступени, можно считать
равновесную концентрацию одновременно возникающего аниона, то доля
[Н+] за счет равновесия (4.116) [H+Ji ^ [Hp«iA^*-^-b^>-] и по тем
же соображениям [Н+]2 ^ [Нр^гА^^-Р+з)-].
Подобным же образом из равновесия (4.117) следует, что доля
[Н+], потребляемой на каждой ступени протонизации, примерно
совпадает с равновесной концентрацией образующегося при этом
/идроаниона и будет выражаться в нашем случае величинами
[Нр-ыА(«-Р-^>-], [Нр+2А(«-Р-2)-1, ...
Развернем суммы в уравнении (4.111), оставляя слагаемые с
индексами, равными (? —2), (Р —1), Р, (Р+1) и (Р + 2), которые
соответствуют двум первым ступеням диссоциации и протонизации
НрА(*-Р)--иона. Множитель при А переходит при этом в выражение
' '■"^УНр,2А^Р-2^^'"^
'^^^^^^^^^ ^^-"^^
Если и числитель и знаменатель его помножить на а, то окажется,
что ОН0 эквивалентно выражению
... + 2 [Hg+2A<"-g-^>-] + [Hp+iA'°-g-»-1 - [Hp-iA"-P+'>-] -
.[Нр+2А<«-Р-2>-1 + [Hp+iA<»-P-»-l+ [НрА("-Р>-1 +
-2[Нр-2А('-Р-Ь^)-]^...
+ [Нр_.А<«-Р+1>-1 + [Нр_2А<«-Р+2>-1 + ... *
Если учесть ранее сделанные предположения, то станет ясно, что
степень при h в выражении (4.118) может быть понижена только в том
случае, если одновременно будут выполняться два условия:
После сокращения первого на гсрНр^гА'з+з^^"*"' " второго на
'PHp_iAap-ift^~^ получается объединенный критерий
1!!^.!Ш»Л»2!^-^. (4.120)
Если учесть заряды при (pi, то (4.120) переходит в
i-. lO[2<«-P)-3]ig9. "-^ > Л > 2 . 10l2(«-P)+31ig9. !!г£. (4.121)
70
Наконец, переходя от констант протонизации 0| к константам
ступенчатой диссоциации с помощью формулы (3.14)
Oj = П /CZl.(j-i),
можно получить тот же критерий в виде
^ . 10t2(«-P)-3]to,/(^_p_j > Л > 2 • 10f2(«-P)+3iig9i/C„_p+2. (4.122)
Поскольку аир всегда известны из формулы соли Ха-рНрА,
индексы при а и К вычисляются, без труда. Если такой подсчет
дает индекс, не существующий для данной НаА, то это значит,
что «отбрасываемых» частиц просто не существует. В таких случаях
критерий становится односторонним. Например, для NaH2P04
Р = 2 и а = 3. Здесь op^.i = 03, = 04, /(a-p-i == Ко\ ясно, что
левая часть неравенства (4.122) отпадает, так как H4P0t в водных
растворах не существует. В то же время ар_2 = == I, op_i = 01,
/Са-р+г = ■'Сз и правая часть критерия должна выполняться.
В общем случае после отбрасывания в множителе при Л в
уравнении (4.111) всех слагаемых, которые по предположению
малы по сравнению с остающимися, получаем уравнение
и X . А ^Hp^.,A^P+l^^"*"'-?Hp.,A<^P-l^^"^
срнА-сронОУЛ-^ + А ?±——- PL = О,
^Hp+lA^P+l + 9НрА^р^^ + ^Hp^iA<^p-l^^
(4.123)
а после сокращения на c^+ih^"^ уравнение
^ "PHp^lA^^ + ^НрА^о-р^ + ?Hp_iA^a-P^a-(P-l)
(4.124)
Анализ этого уравнения приводит к следующим результатам.
1. Если [Н+] = [ОН-] или если [Н+] и [ОН- настолько
близки, что разностью срнЛ — (^obwh-^ можно пренебречь, то
после законного сокращения уравнения на произведение А х
X (cpHp^iA/i^ + ТИрА^Са-рЛ + <pHp_lAi(a-pЛ:a-(p-i7^ КРТОрОС ВССГДа
ОТЛИЧНО от нуля, получаем решение в виде
/1 = 1/ /Са-.рКа-(р-1) ^ Kl04(«-P)>8<P./(a-pXa-(P-l). (4:125)
г ^Hp^lA
2. Если приближенное решение даст такое А„ что ,срн Л > срои X
X wh-\ то считают, что при диссоциации НрАР-'^-иона
образуется значительно больше Н+-ионов, чем расходуется на его прото-
71
-V
низацию. Тогда, пренебрегая величиной сронгг;А-*, получают после
обычных преобразований кубическое уравнение
+ (?НрА/нр_,А/(«-Р + /Н^4) Л2 + срНр_1А/нр^1АКа-.р/Са-.(Р-1)Л —
— /н?Нр_, A/Hp+lA^:a-p/Ca-(P-l)^ = 0. (4.126)
Если в этом уравнении h<^fwA^ то первым и третьим
слагаемым можно пренебречь. Тогда
Если срнрА/нр^1А^а-р</н^, получается уже найденное выше
простейшее решение.
3. Если приближенное решение дает такое значение Л, что
срнЛ < сронгг;/г-^ то расход Н+-ионов на протонизацию НрЛР-«-иона
превышает их поступление за счет его диссоциации. Тогда,
пренебрегая величиной срнА, получают после обычных преобразований
и перехода к новой неизвестной о = (ОН-) = wh"^ кубическое
уравнение
+ (/Hp^iACpHpA 1^^^^^^^^ + /онЛ) 0)2 + /нр_,АСрНр+,А д^^^ X
Х.^-^(о-/он/нр.,Асрнз+,А-^^ (4.128)
Если (оС/онЛ, то можно пренебречь первым и третьим
слагаемыми. Тогда
W W
Ю =
^Нр_,А?НрА /С._(р_,, +^ОН
(4.129)
Если /нр_,А<рнрА ^—j^-j-</онЛ, то
Как это и следует, произведение Лш = ш, что подтверждает
пранилbHoci'b выкладок.
Уравнение (4.124) дает решение, выражаемое формулой (4.125),
в тех случаях, когда Н+-ионы, получающиеся при
диссоциации НрА*-"Р--ионов, полностью расходуются на образование
Hp«iA^*-P-'>~.hohob. Запись соответствующих равновесий и их
суммирование приводит к системе, которую можно рассматривать
как диспропорционирование р-гидроаниона:
НзА<—H-^ + Нр^, А^»-''^»'-, (4.131)
72
НрА(«-Р)- + Н + » А<«-Р-^)-, К7\ (4.132)
2НрА(«-Р)-^Нр_,А(«-Р+»)- + Нр+,А<«-Р-1), /Ca-p+i/Crip. (4.133)
Индексы при константах записываются автоматически, так как
простого просмотра ряда последовательных констант диссоциации
достаточно, чтобы установить, что индекс при константе
совпадает с числом зарядов у продукта диссоциации. Приближенное
решение получается очень просто. Из условия известно, что а
= Л. Из равновесия (4.133) очевидно, что [Hp-iA] = [Hp4.iA];
если принять, что каждая из них равна л:, то [HpAj = Л — 2д:.
Не совсем законное использование закона действующих масс
приводит к уравнению хУ(А —2х)^ =Ка-^^\К7^^, откуда х=: [Hp_iAj=:
= [Hp^iAl = АК/(1 + 2/), где / = VKaJ^K7l^ и [НрА] = А-
— 2x = A/(l+2V). Теперь, применяя закон действующих масс
к уравнению (4.131), находим, что А =/Ca-p+i [HpA]/[Hp_iA] =!
= |/'/Ca-p/Co-p4-N Это значение h меньше более точного,
получаемого по формуле (4.125), в Ю^^^-Р)»^ *pi раз.
Приближенные вычисления h в растворе соли MY^. После
объединения двух последних слагаемых уравнение (4.47) переходит
в уравнение
S К^М(ОН)к^кЛ''''.
S ^М(ОН)к^кЛ
к>=0
где
(I*
2 «?М(ОН)«''к'' "
= (4.135)
функция Вьеррума, равная среднему числу Н+-ионов,
образующихся при иоцизации М (Н20)^"*"-иона.
Реакция в растворе MY^ должна быть кислой, и при
выполнении условия (4.92) слагаемым (fouwh-^ в (4.134) можно
пренебречь.
Полагая, что основной вклад в [H+j дает первая ступень
ионизации аквометалло-комплекса, разворачиваем суммы в (4.134)^,
учитывая i = 0,2. Тогда
и М ^МОН'П!^""^ + 2УМ(ОН)»^2^""^ + - - ^
^ ?M + ^MOH^l^ +?М(ОНЬ^2'^ +•••
И все слагаемые g индексами к = 2, |х' можно отбросить, если
73
(p^^oj^irii/i-i > 2<pM(oh)^2^'"^. критерий после сокращения на
фмонтй!^"'^ и учета числа зарядов при частицах принимает вид
Л » 2 . ^ == 2 . 10(3-2.)igf. ^, (4.137)
Упрощенное уравнение, как легко показать, имеет вид
.«К-М - |Н-Ч-|МОН>-]-0. ,4.138)
Смысл уравнения (4.138) сводится к тому, что для случая, когда
основную роль играет первая ступень диссоциации, [Н+] =
= [М0№~4. Тогда множитель при М — степень ионизации акво-
металло-иона на той же первой ступени.
Уравнение (4.138) в приведенной форме +/мcpмoн1(|lA —
—/н/м^мон-гцМ = О (4Л39) дает при условии /г</нЛ1 (4Л40)
h = K/h/mTmoh^iM ^. 10-^'8^« (4.141)
Если условие (4.140) не выполняется, то по (4.139)
^m'Pmoh^i +V /m'Pmoh^i + 4/нУм9мон^1^
приближенные вычисления h в растворах солей МаА^.
Уравнение (4.51) может быть сведено к уравнению более низкой
степени относительно ft, если можно предположить, что большая
часть Н+-ионов получается на первой ступени ионизации
М(Н20)й^-иона.и в то же время большинство этих ионов
расходуется на первой ступени протонизации А*--иона. Первое
предположение справедливо, если выполняется условие h > 10(3-2ii)igcp,^i,
второе—при выполнении неравенства Ш'^^-^)^^'^^-^^^h. Оба
неравенства получаются обычным путем из сопоставления
слагаемых, стоящих в знаменателях уравнения (4.51). Совместно эти
выражения дают для уравнения (4.51) двухсторонний критерий:
10(2«--3)igcpill ^ Л ^ 10(3--2ix)igcp^ (4.143)
Разворачивая в уравнении (4.51) суммы и оставляя только
слагаемые с индексами к = 0,1 и j = 0,l, получаем
. . 1 , ^^Ум + ({^--^),Умон^1^""^
<рнЛ — <fov\wh-^ + ас ; —1 |хс X
?м + умон^!^
, , ; а9А + (а->1).НА-1/^
. ?А + ^НА^1^ ^ '
Причем (4.144) является таким же, как и (4.51), уравнением
закона сохранения электронейтральнрсти.
\
74
Выделяя в двух последних слагаемых уравнения (4.144) целую
часть, получаем после обычных преобразований уравнение
- ,oHwh-^ - ас ^ + ^"f= 0. (4.145)
Нетрудно показать, что dc^ouni/imh + ^рмон-^дО = (М0Н«^^-^1
и что множитель при ас — степень ионизации М (Н20)Й^-иона на
первой ступени. Подобным же образом оказывается, что
последнее слагаемое в (4.145) равно [НА^-"], а множитель при —
степень протонизации А*-. Тогда (4.145) можно представить
в виде
[Н+] — [ОН-] — [М0Н«^-1] + [НА1-«] = О,
или (4.146)
[М0Н«^-1] — [НА1-«] = [Н+1 — [ОН-].
Ясно, что преобразования, проведенные в уравнении (4.144),
изменили его смысл. Уравнение (4.145) уже не является
уравнением закона сохранения электронейтральности. Смысл его, как
видно из (4.146), сводится к следующему: если [МОН]>[НА],
то при ионизации аквометалло-иона получается больше Н+-ионов,
чем идет на иротонизацию А"--иона, и [Н+] — [ОН-] определяет
этот избыток с поправкой на долю [Н+], которую обеспечивает
диссоциация НгО; если [НА^-*] > [МОН«"-^], то разность [ОН-]—
—.[Н+] определяет избыток ОН--ионов, накапливающихся в
системе за счет участия в протонизации Н+-ионов воды.
Наконец, из (4.146) следует, что разность [Н+] —[ОН-]
является мерой разности между [МОН»^-^] и [НА^-**].
Если разность |срнА — ^oHwh-^\ много меньше любого из
остальных слагаемых, то ею можно пренебречь. Тогда после
сокращения на с и обычных преобразований с учетом ах = К7
получаем
!мШонп\ {l—jjh —jh^fAOH^^AhAyixKa = о, (4.147)
где /м9мон - /.9.-1 = 10 • 10 = l0-(2.-u.g.. (4,148) и
Wmoh?a/a =^ 102(»-i^)ie?. (4.149).
Если
Л « f ЫиАКа ^ -J- 10(2«-»'g<p. iC„ (4.150)
TO
A= У^ЫшонЫнАщКа^. '/•^102(«-^)'в91Ч1/(.. (4.15.1)
75
в противном случае
^-^/m^moh^a'ha'^i^.
(4.152)
При а = [л, как следует из (4.151), Л = V-^iKa (4.153). Если
вычисленное значение /г, каким бы упрощенным методом оно ни
было получено, удовлетворяет условию Л^-Ю-^ и критерию
(4.143), то с его помощью вычисляют
"^'^'°-.„'.rw,'l"°"'-'-"-'"'-^l-
Если окажется, что каждая из этих равновесных концентраций
много больше разности (9нА — ?онйуА-^), то вычисленное значение
h считают правильным и заканчивают расчет равновесия. Если
9он^
[Н+] > lOH-J, то считают ^nh > —^ и получают уравнение,
которое в приведенной форме имеет вид
+ (/m9MOHTQ1 + ?а/на/Са + /нЮ + [/м?мон<РА/нАТ(]1Л'а +
+ /н/м?монСт(11 ([Л — а)] Л — /н/м?мон?а/нАасто i/Ca = 0. (4.154)
Если [ОН-] > [Н+], то отбрасывают (рнЛ и после подстановки
А = а;а)-1, где ш = (ОН-), приводят уравнение (4.145) после
обычных преобразований к виду
+ (тм/мон ^ + /а9на ^ + /он ас j (о^.+ 9м/мон/а?на ~- X
X + /он/а9наС (а — [х) ^ о) — /онсрм/мон/А9наК :Г" • F" = ^•
По найденному значению о) вычисляют А, а затем, убедившись,
что критерий удовлетворяется, рассчитывают значения т, а,
[МОНГ*"] и [HjA^^"].
При a = [jL уравнения. (4.154) и (4.155) несколько упрощаются.
Ту же роль, что аквометалло-ионы, могут играть NH^-ионы
и их органические производные, КН+-ионы, относящиеся к клас-
. су катионных кислот. Например, в растворе соли (HA(i))aA, если
^(ha(i)^a= А, учитывается равновесие A(i) + Н+5=± НА^), а, [НА^)] =
= ?ha(i) <3aiA, уравнение закона сохранения начальной концент-
i радии cha(i) = аЛ = щ (<^HA(,)oh + 9а(о), а вместо слагаемого, опре-
76
деляющего сумму зарядов, которую дают и М (ОН)|^'"^'-ионы
в уравнение закона сохранения электронейтральности войдет вы
ражение
где /С = — константа диссоциации НА^.
4.7. ПРИМЕРЫ НЕКОТОРЫХ РАСЧЕТОВ, КОТОРЫЕ МОЖНО
ПРОВОДИТЬ БЕЗ ПРИМЕНЕНИЯ ЭЦВМ
Пример 4.1. Рассмотрим расчет равновесия в водном растворе соляной
кислоты, если в нем с^^^ = У = 0,04 и c^q^ = с^у =0,45 моль.л""^ В
разделе 4.2 было показано, что в этом случае для расчета h надо использовать
уравнение (4.15). Поскольку К = 0,04 > 10"~^ моль-л""', (4.15) сводится к
выражениям ^^h — Y = Q и h^fy^Y (4.157). Ионная сила, необходимая для
расчета /д, равна в этом случае /=0,49 моль.д""^» и, если использовать
формулу Дэвиса (2.13), то ^
Ig/j = _ 0,5 -0,2.0,49j = -0,157. = 0,697. h = 0,697.4-10-2=.
c= 2,788.10~2, pH = 1,55, [H+]=4.10-2 моль-л""*, pOH =12,45. (0H-) =
= 3,55.10"-*^, [ОН-] = 5,Ы0~*^ моль-л-^
Подсчет по формуле (4.11) дает при / = 0,49 на фоне КС1 значения lg<PH =
= 0,1046 и lg<PoH = ^'16^2. В этом случае Л = 3,144.10~^ рН = 1,50, [Н+]=
= 4.10-2, pOH = 12,5,(OH~) = 3,l7.10""^ 10Н~1 = 4,66.10~'^ моль-л-'.
Сравнение результатов, полученных при подсчете lg<Pj, по Дэвису и по
уравнению (4.11), учитывающему природу ионов, показывает, что значительные
расхождения наблюдаются лишь в значениях активностей и показателей
активности оксоний- и гидроксил-ионов.
Пример 4.2. Рассчитаем равновесия в системе, где c^qi =7 = 1,8 • Iq""^
и Ci^ci=^»49 моль-л-*. Здесь /=0,49 моль-л""', а в уравнении (4.15). все
три слагаемых — величины одного порядка и пренебречь ни одной из них
нельзя. Уравнение (4.15) в приведенной форме имеет вид — f\^^ ~ /н^он^—
= 0 (4.158).
После подстановки в него значений <pj, вычисленных по Дэвису и по
уравнению (4.11), получаем соответственно —0,697*1,8-10""^ h —10"" =
v^O и 0,786-1,8-10-"^ Л —1,158-10-'^ =0.
В первом случае h = 1,43-10-^ (ОН"") = 6,95-10-*, [Н+1 = 2,05.10-^
(ОН-] = 1,00.10-^ моль-л""' и рН = 6,84.
Во втором случае h = l,65•l0-^ (ОН"") = 6,08.l0-^ [Н+] = 2,09.10-^,
[ОН-] = 8,95-10""^ моль-л-' и рН=6,78.
Результаты, конечно, различаются, но в дальнейшем нам придется
пользоваться формулой Дэвиса, так как пока нет еще достаточного числа
параметров для расчета различных <f
77
пример 4.3. Для расчета равновесия в системе, в которой c^pQ^ == Л =
= 0,02 и Cj^Qi = У = 0,4 моль-л""^, используются указания, содержащиеся в
разделе 4.6, и следующие значения констант протонизации (диссоциации):
Н3РО4 21,68 2,15
Н2РО4 19,53 7,21
НР02- 12,32 12,32
Критерии (4.82), (4.89) и (4.92), допускающие упрощение расчета h:
^>2.10^'^^« /(2- Ю^'^^^Ю-^'^^ (4.159); /„^4 =/„-0,02 =/д.lO-^'^^^ »/г
(4.160); fi> 10"^^ (4.161),
Расчеты удобно вести в виде таблиц. В табл. 4. 2 в первых четырех
столбцах представлен расчет по уравнению (4.90). Под ними проведена проверка
условия (4.160). Поскольку она дала отрицательный результат, в последующих
столбцах проводится вычисление Н^^^ по формуле (4.91), отвечающей критерию
4.82), но при /| = I, так как в данном случае / лишь немного больше 0,04.
Здесь же дана проверка условий (4.159) и (4.161), давшая в обоих случаях
положительный результёт, расчет /(^ с учетом диссоциации Н3РО4 и Igcp,,
отвечающего /(ij.
В-табл. 4.3 приводится повторный расчет h в системе Н3РО4 —КС1—HgO
с учетом Ig^j =0,085 (пример 4.3) по формуле (4.91), где теперь р = 10®*°^^ X
_ iQ-2,065 и q = KjA = JO-3'^^
На этом вычисление h заканчивается: так как 1^2) совпала а /^j^,
дальнейшие изменения значения ig(pj реального емысла не имеют.
Последующие вычисления а == (РО^""!, [Н/РО^~^] и [0Н~] (пример 4.3)
приведены в табл. 4. 4.
Несколько замечаний, относящихся к табл. 4.2—4.4
1. Расчеты проводятся с четырехзначными таблицами логарифмов.
2. Поскольку условие h < t^A не выполняется и /гц^ составляет ^^60% Л,
первый расчет проводится по уравнению +/C^/i —/Cj Л =0 без учета
коэффициентов активности, так как очевидно, что при подсчете / надо принять
во внимание
3. Повторный расчет проводится с учетом /=0,049 моль^л""^- О" Д^^'^
^1(3) = 8,33«10"'^. Это значение принято за окончательное, так как с' его
учетом /^2) = 0»048 моль«л'~Ч что почти не сказывается на значении Ig<fi,
подпитанном по формуле Дэвиса.
2^10—2
4. в табл. 4.4 0=-^ , значения [HjPOJ~^] вычисляются по
обшей формуле [Н^РО^"^] = 9н^А^\аН^ и [ОН*-] — по формуле [OR-] = ^он^^""*-
Стрелки в таблицах указывают последовательность получения отдельных
значений. Если расчеты ведутся в помощью настольной вычислительной машины,
таблицы упрощаются соответственно возможностям данного вычислительного
устройства.
Массовые расчеты равновесий рассмотренного типа имеет смысл проводить
78
Таблица 4.2
^1
а
К
^1
4/С»Л
/
Кг +/
1
7,08 10-^
10-''^
2.10-2
10-3,85
1
1,413.10-^
10-1.925
10—3,55
10-4,30
\
5,012.10-5
10-3,25
\
5,624-10-^
10-3,2118
t
6,125-10-^
10-1,6064
1
2,475-10-2
,0-1.497
3,183'10-2
10-2,053
8,85-10-^
Примечание: Af = Ю-'-^^З/ю-з-^о = io'-"5_ 60% Л; Л,> 10^ 'sf-. IQ-^-^'. так как Ю-^'-э'/Ю-^-''^ = 10-2'«^=
= 1.4 10-3% ftjj.
= -i- f(K+] + [СП + [Н+] + IHaPOri) = 4" <^ • + • + • ^^"^ + ^'^^ '°~^^=
= 0.049 моль л~'. '
Jg <pi = 0,5 (-у||- — 0,2 . 0,049J = 0,0901 - 0,0049 = 0,085.
Т а бл ица 4.3
р
2q
4q
P=' + 4q
A(.)
JQ—2,066
8.61 • 10-3
10-3.55
10-4.13
7,413 10-*
10-3,25
5,624. 10-^
10-3,1962
t
6,365 10-^
10-1.5981
i
2,523 • 10-2
10-1.4706
3,384 10-2
10-2.0794
8,33 • 10-3
/(2) = 0,048 моль . л
—i
lg(pi = 0,085
A(3) = 10-2'0794 моль-л-'
Пример 4.4. Для приближенных расчетов равновесий часто используют
описанный ниже простой прием, который можно применить и к только что
рассмотренной задаче.
В первой строке записывают изучаемое равновесие, например равновесие
диесоциации НдРО^ на первой ступени, а рядом с ним — логарифм константы.
Используя значение константы, устанавливают, в какую сторону сдвинуто
равновесие. Так, в случае HgPO^ рК = 2,15. Это значит, что ^'
(Н3РО4)
= 10-2.15^ откуда следует, что произведение (Н+) (^г^^Г) ^ ^^^'^^ 140 раз
меньше (Н3РО4) и что продуктов диссоииапии много меньше, чем исходной
кислоты.
'^атем среди меньших равновесных концентраций выбирают наименьшую и,
считая ее основным неизвестным, обозначают через v;c, где v —- стехиометри-
ческин коэффициент при соответствующей формуле в данном равновесии.
В нашем случае [Н+1 и [HgPOT"] равновелики, поэтому в качестве первой
неизвестной можно ^ыбрать любую из них, например, приравнять [Н"^] =^1-Л =
= h, так как здесь v = 1.
Затем через выбранную неизвестную выражают равновесные концентрации
остальных частиц. В нашем случае при этом рассуждают так: как следует из
равновесия, на 1 моль Н"'' иона образуется 1 моль Н^РО^-иона, тогда на h
молей Н"*" иона должно получится ^-молей Н2РО7 -иона и [HgPO^ =Л. Далее
из равновесия следует, что на образование 1 моль Н''"-иона должен
расходоваться 1 моль Н3РО4; на h моль их расходуется h моль; следовательно, [Н3РО4] =
= 2-10^^—'i, если с^зРо, моль-л'"^
Записи делают в следующем юрядке: в первой строке записывают
равновесие и IgA', во второй — начапьные концентрации, отвечающие условию
задачи. 8 третьей — равновесные концентрации, выраженные через наименьшую.
В нашем случае
Н3РО4 «н-*- -t p/Ci = 2,I5
с 2.10-2 — —
[ J 2.10-^-Л Л
(4.162)
Тогда по закону действующих масс —j- = 10""2.15^ Если h <
<2 10-%^ТО 4^ = 10-2.' ЮО.З _ 10-3.85, 10-1.92 ^ ^^^^^
< 2 10 ^ моль л ' не выполняется.
Решение квадратного уравнения 4-10~^'*^ /1 — 10-^*^^=0 дает Л =
= 8,85 10-\ i. е. [Н+1 = (Н,РО-^] = 8,85 . IQ-^ и [Н,РО4[=0,011 моль л"^-
Исходя из формулы fH^PO^l = ^2^^^ можно получить приближенное значение
а и рассчитать все остальные равновесные концентрации. Однако из сопостав.
ления результатов такого расчета i = 8,85 • Ю""^' [Н2РО7] = 8,85 - 10"^
и iH-jPO^J = 0,011 моль • л""^ со значениями, приведенными в табл. 4.4
(h = 1,013 . 10-2, [Н2РО7] = 1,014 . 10-' и [Н3РО4] = 9,87 • 10-^ моль л-*),
становится ясно что, даже в таком относительно простом случ|ае упрощенный
метод дает лишь приближенные оценки искомых равновесных концентраций.
Несмотря на это, он может принести пользу, так как приближенные оценки
могут служить основой для последующих уточнений.
Отметим, что при составлении схемы (4.162) использовались -^акон
сохранения начальной концентрации (в записи H3PO4I 2 . 10"~^ —/tj, закон
80
9t
h
9»
s
a
100,035
10—2,0794
10lS.44U
2,766 . 10
1015.4662
4
2,859 lO'*
1010,5806
3,807-lO'"
,00,765
1
5.82
,0'5.75
5,625-10
,0-17.46
IH+J
IH3PO4)
[ЩРОГ]
IHPO2-]
[POM
IOH-]
10-1,9944
1
1,013 . 10-2
10-2,0882
1
9,87 • 10-3
10-1.993?
1,014 10-2
jO-6,87
1,3 • 10-^
10-16,68f
2.1 10-"
2.001.10-2
10-11.8356
1,46 J0-'2
9i
h
«р,сг,Л
93
в
100.167
1017.48
3,020 • lo'^
1016,887
4
7,74 . 10'^
1011.548
3.532. lo''
1
25.88
1017,5791
t
3,794 . lO'^
J0i6,336i
Зтз
S
A
0
0
1016.887
1
7,74 • lO'^
1011.849
7,07 10*'
.10^.S9
t
77,6
10l6,887
7,74^. 10'® -
10—0,5509
1
0.282
сохранения электро нейтральности (когда было введено обозначение h = [Н"^]=
= HgPOT" I и закон действующих масс.
В дальнейшем простейшая схема будет вводиться со ссылкой на раздел 4.7
и схему (4.162),
Пример 4.5. Рассчитаем начальную концентрацию c^^pQ^ = Л, которую
необходимо создать в растворе, содержащем с^^^ = 0,45 моль • л
обеспечить рН = 1,4.
Н2РО7 ^ 0,04, /
В этом случае
lg?i = 0,157.
Из уравнения (4.32), где при рН = 1,4 (f^h > <PoH^^~^
расчетное уравнение в виде
3
А ^
чтобы
0,49 моль • л"~'и по Дэвису
прямо получается
?1<J2^ +2cp2Ci/l + 3(p3
(4.163)
где индексы при <р в развернутых суммах равны числу зарядов при
соответствующих частицах.
Расчеты по формуле (4.163) приведены в табл. 4.5.
Для проверки полученного результата можно сделать следующие неслож.
\-1
/ 3
ные выкладки. Из
(4.29) а = (РОМ = А (^2 ^H^A^i^^
10
1—0.5509
X
X
10-17.5791 _ 10-18.13. [НоРО;] ^^^c^h'a = 101б'«»7 . ю»
-18.13.
^ 10-1.243 ^5 5.10-2. Той же величине равна [Н+] = (pj/i = 100''57 х
X 10—= 10—'«243 j^Qjjb , jj-1. Хотя теперь / = 0,505 моль • л"', вторичного
пересчета делать не следует, так как расчеты <pj по Дэвису, безусловно,
включают серьезные погрешности.
Для упрощенного подсчета, зная / и lg<Pi =0,157, находим, что при
рН = 1,4 [Н+1 = 10-''243 моль . л-'- Тогда
Н3РО4 5=> Н+ + HgPO- рК = 2,15
с Л - -
[]Л —10-''243 10-1.243 10-1.243
10-2,486
и по закону действующих масс -ю—К243 = Ю"2.1^ и Л = 0,507 моль • л—',
что в 1,8 раза больше найденного в расчетной табл. 4.5 более точного
значения (пример 4.5).^
Пример 4.6. В растворе гидразиний дихлорида, в котором Ch^ayj—
= 0,02 п С1^^^=0АЬыолъ * л"^^* /=0,49 и по Дэвису lg<pi = 0,157, lgC2 =
= 8,21, lgoi = 7,94. Критерии (4.82), (4.89) и (4.92), определяющие возмож-
ность упрощения расчетов по (4.93), имеют вид
Л > 2 . 103»^^«/(2= Ю^'З . 10^'^^ • 10-7'94= 10-^''^ (4 164)
/нЛ = 10-0-'57 . 2 . 10-2 ^ 10-1.857 ^ ^ (4.165); h > Ю"^- (4.166)
Все решение сведено в одну табл. 4.6 (пример 4.6).
82
Таблица 4.6
i Ki
KtA
"(.)
P
q
P'+ iq
V
u(2)
,0-0,27
! 0,537
10-1.7
2 . 10-2
10-1.97
1,072 . 10-2
10-0.985
10-0.7.
i
0.182
10—2.598
2,523.10-^
10-1.48
Ф
0,03311
10-1.3646
t
0,0432
. ^Q-.0,6822
\
0,2078
J 0—0,4092
t
0.3898
10-1.8878
1
1,295 . 10- 2
l^^2) > и 10"^
"Pi
h
a
100,157
00
10-1.8878
j05,0624
1,154 . 10^
106,2092
1
1,619 10^
10^
1
1
106.2391
1,7344 • 10^
10-7,9391
[H+J
In^hh-]
IN2H4]
S
[OH-J
JO—1.7308
1,86. 10-2
10-2,8767
1
1,328 . 10-^
10-1.7299
i
1,863 . 10-2
10-7.9391
1,15 10-^
1,996 . 10-2
10-11.9552
1,1 10-12
в ней р и q отвечают формуле (4.91), а для остальных расчетов
используются уравнения (4.25), (4.33).
Контрольная сумма 1,996 • Ю"^, приведенная в конце табл. 4.6, харак'
теризует точность расчетов, она всего на 4 • 10~^ моль • л~~^ отличается от
Л = 2 . 10~2 моль . л~*.
Говорить о правильности результатов здесь трудно, так как она зависит
от правильности значений Oj и особенно «Рр которые рассчитывались по Дэвису,
т. е. без учета химической индивидуальности частиц.
Упрощенный расчет по схеме (4.162):
Н2А2+5=±Н++НА+, р/с = 0,27,
с 2. 10-2 ~ -
П 2. 10-2-^ Л.
Дальше по закону действующих масс
10-0-27, ^2_^ 1о-о.27./г _ 10-^-97^ О, А = 1,922 . 10-2=[НА+1.
10-1'7^Л
[НА+1 = о,а/1, а = [NHgNHg] = а]"* = Ю-^'»^ = 1,15 • 10-«.
[Н2А2Н-] = a^ah^ = I0-«'2l . 10-^.94 . ^io-1.7l63J2 _ е,88 • lO^S
[Н+] = [НА+] = 1,922 . 10-2 моль . л-^-
Эти значения, конечно, существенно отличаются от полученных в табл. 4.6.
Пример 4.7. Рассмотрим полный расчет равновесия в системе, где
^NaiPO. = ^ХзА = ^= 0,02 = 10-^'' и СКС1 = 0.37 моль • л-^-
Б такой системе / ~ 0,49 моль • л—' и по Дэвису (2.13) lg9i' = 0,157»
Ig аз = 21,68, Ig 02 = 19,53^ Ig = 12,32.
Вычисление со = (ОН—) может быть проведено по формуле
О) = 10-**^^*/^^ (4.108)
пцй а = 3, если по (5.109) toH^ > или по формуле
где р = 10^^—2*>l^^ioia/ и q = Ю""2« ^кф^ пр„ а = 3, если одновременно
выполняются условия: (4.101), со » Ю"^ и (4.105), со > 2 . 10<3-2a)lg <p,f2^ ^
= i0-«-9^
Расчет равновесий в системе ЫазР04 — КС1 — HgO (пример 4.7) приведен
в табл. 4.7.
Контрольная сумма 1,996 • 10—2 моль • л—^ мало отличается от Л =
= 2 . 10—2 моль'л—^ Следует отметить, что активность трехзарядного
РО4—-иона а = 4,711 • 10—при / = 0,49 моль • л—* оказалась в 25,88 раз
меньше равновесной концентрации.
Упрощенное решение по схеме (4.162):
дЗ- + Н+?=2: НА2- Ig af = 12,32
HgO ^ Н"^ + ОН- lga; = —14
АЗ-4-Н20^НА2~ + 0Н- lgK = -l,68
с А — — —
[ ]Л — О) со со
• и по закону действующих масс со2/(Л — со) = /( и — К А = 0.
84
Таблица 4.7
9iwA
/
<•>(!)
P
q
P>-b 4q
Р+/
^(2)
JOO.157
i-
JQ—3,38
10-1.69
,0-2.161
,0-2.465
1
3,428 10"-^
10-4.322
\
4,764.10-5
10-4.930
1,172 10-5
10—3.6940
2,0228.10-^
10-1.847
1,427-20-^
10-1,7521
t
1,7698 10-2
,0—2.2689
fonA = 10-1.857 ^ ^ _ jo-2,I6. ^ ,0-2.2689 » ,o-6.96 „
h
Ъ
2
a
,0—11,7311
,0—13,5133
1
3,07-10-"" •
,0-3.7752
1
1,68 10-^
,01.2169
1
16.48
101.4130
25,88
101.6269
t
42,36
10-3,3269
4,711.10-^
IH+]
[HsPOJ
(H2PO7]
[HPOM
[POM
2
[OH-J
,0-11.5741
2,67 10-'2
,Q-16.8402
1,44 • 10-^^
,0-7.1021
\
7,91 . 10-^-
10—2,1100
7,761.10"^
10-1.9139
1
1,2196.10-2
1,996 . 10-2
-
,0-2.1119
1
7.73 • 10-3
•
Решив это
: А —О) = а..
Последующие расчеты дают
уравнение, получаем со
н+
он-] = [НР02-] и [РО^
— дао)
.—1.
Н2РО7] = z^ah^ и
Н3РО,
Схема упрощенного расчета равновесий в системе ЫазР04 — КС1 — HgO
(пример 4.7) приведена в табл. 4.8. Расхождения между результатами табл. 4.7
и полученными упрощенным способом достаточно велики. Они близки к 40 %
по концентрации HgPO^""- и РО^" -ионов и приводят к такой же погрешности
в оценке [ОН—]. Ясно, что в ответственных случаях следует вести расчеты
по точным формулам. Очевидной становится и необходимость в более точных
значениях Ig 9 для всех ингредиентов равновесия.
Пример 4.8. Рассчитаем равновесия в системе, где Cj^g^^PO^ = Л =
= 2 . 10""^ = Ю"*'^» ^КС1 = 0.47 моль • л.
Здесь /=0,49 мoль•л"■^ Ig<pi = 0,157, а=3, р = 2, lg03 = 21,68,
lga2 = 19,53, lgci==: 12,32 и критерий (4.121), допускающий вычисление h по
формуле (4.125), принимает вид
^ , l0-ig9i ^^^^2. 10^^^^^ . (4.167)
Поскольку в водных растворах H4POJ" не образуется, критерий (4.167)
превращается в одностороннее условие: /г > lO^'^-lO^'^^^ .io-12,32^1q-11,24
(4.168).
Решение по (4.125) дает
h = |/"io^(-P)»g-Pi я,^р/(,^^р_1) = ]/io^^g-p^/(i/c, =
= Y10°'^^^ • 10-2.23 . 10-7.21 _ 10-4.406. (4 Igg^
Найденное значение Л, безусловно, удовлетворяет условию (4.168), но
в то же время оказывается, что h > (ОН""). Это значит, что для вычисления
h надо использовать уравнение (4.126), начиная с простейшей расчетной
формулы (4.127), которая здесь превращается в
У ^oKi + hA =]/
10-0,157. 100.628 . 10-2,23 . io-7,21 . iq-1,7
,00,157 . 10-2,23 ^10-0,157.10-1.7
10-4,5092. (4.170)
Этот результат можно считать окончательным, так как f^^A = Ю""^'^^^ ^ ^^g.
денное значение h составляет всего 0,22% от этой величины.
Расчет равновесий в системе NaH2P04 — НС1 — Н2О (пример 4.8)
приведен в табл. 4.9.
Решение задачи по упрощенной схеме сводится к системе
Н2РО7 <=t Н+ + НР02- 10-^-21
Н2Р04 + Н^"5=±НзР04 102И5
2Н2РО-
с
п
10
^-1,7
(10-Ь7_2х)2
10-1'^«2х
10-'
-5,06.
10
X
86
Р07
— 2х
= 5,89
1,989 .Ю-^ моль-л-*-
НР02-+ НзРО^ 10-5*06
НР02-
X
=[НзРО,]=
10-*.
Таблица 4.8
К
К*
V
0) « [ОН-]=
= [НР0М
А = [Н+]
а = [роЗ-]
(Н3РО4]
10-1.68
\
2,089.10-2
10-3,36
\
4,36 . 10-2
10-2.6786
2,096 . 10-^
10-1.3393
4,578! 10-2
10-1.176
6,667 . 10-2
,0-1.904
1,25 • 10-2
,0-12.096
8,02 . 10-'*
,0-2.1249
7,5 • 10-*
,0-4.662
2,18 . 10-^
,0-14,603
1
2,5 Ю-'-*
Таблица 4.9
ь
9ов,А«
9,»,А
ъ
S
а
J0—4.5092
,08.1524
1,420; 10*
,010.6686
4.662^. 10'"
108.4388
1
2,747 • 10^
101.413
25,88
1010,6725
4,704^. 10*°
,0-12.3725
fH+1
IH3P04]
[Н2РОГ]
[РОМ
S
[ОН-]
,0—4,3522
4.44 • 10-^
,0-4.2201
6,024 10-*
,0-1.7039
Ф
1,977 • 10-2
10—3,9337
\
1,165 • 10-^
1Q-. 10,9595
\
1,1 .10-^^
1,995 • 10-2
,0—9.3338
4,64 Ao-'"*
SS
Расчет [Н+] по первому из равновесий дает
Н"^] = • 10-^*2* = 1,987 . 10-2 . 10-7.21 (6,45 . 10-5)^1 _ 1 9 . ^-s^
Наконец, [НР02-] = с^ак, откуда )i = [РО^""] = 1,6 • 10""*2 j^q^^ . л-*.
Разница в результатах достаточно велика, чтобы показать преимущества
более точного способа расчета.
Пример 4.9. Рассмотрим полный расчет равновесия для системы, в кото.
^Сг1СЮ4)§ = ^ = ^NaClO^ = -«^ = 0,58 моль • л~К Здесь / =
= Y Jo,Ol . 32 + 0,03 + 0,58 + 0,58J = 0.64, lgcpi= 0,5 {j-^^ ~ 0,2 . 0,64^ =
= 0,158 no Дэвису, если рт)1 = 3,8, pif)2 = 10,0 и рщ = 26.
Здесь следует учесть, что при выполнении условия (4.92) Л > 10—^ можно
пренебречь [ОН—] и дальнейшие значительные упрощения получаются при
выполнении критериев (4.137) и (4.140).
Используя (4.141), находим, что Л 10-^'s?i |/-^^д| ^ 10-0'474 ^
X ]/l0-3'8 . 10-2 ^ 10-3'^^^ составляет от f^M = Ю-^'^^^ • 10-2= 10-2'15&
примерно 6,1%. Это значит, что условие (4.140), h<^f^M не выполняется,
тогда как критерий (4.92) можно считать выполненным.
Более точное значение h получается по формуле (4.142), если
выполняется условие (4.137), которое в данном случае имеет вид
Л> 2 . 10<3-2l^)lS^. J2 _ ЮО.З , 10-0.474 . io-10'O . 103'8 = 10-6.374 (4 j^j^
Формулу (4.142) можно записать в этом случае в виде/i=2q(p-|-]/*pa+4q)—**
где Р = ЫЫонт = Ю-'- ■ 10°-«". 10-*'» = 10-*.s«.
q = /h/m?moh1iM = Ю-»''*» . Ю-"-*» . 10-2 ^ ,0-6.74.
Вычисления Л, проведенные по формуле (4.142) с помощью указанных
выше значений р и q, и последующий расчет равновесий в системе Сг(С104)з—.
— NaC104 —HgO (пример 4.9) даны в табл. 4.10.
Упрощенное решение проводится по схеме
Сг^++Н20?=^СгОН2+ + Н+, р7]1 = 3,8 .
о 10-2 - -
1\ 10-2—Л h П
• = Ю-З'^Л = [Н+] = [СгОН2+] = l0-2.^з = 1.18 • Ю"^
10-2 _Л
Из равенства [СгОН2+] ^ ^^^^^^-i, ^ ^ [Сг^^] = 10-2'0б=8,71 о Ю^^ моль X
ХД—1. Дальнейшие расчеты дают
Сг (ОН)^] = 7|2тЛ-2 = 10-^'2 ^ 3 3 . 10-7 и
Сг (0Н)7 = 7)4тЛ-^ = 10-*^'3^ = 4,6 • 10-^^ моль . л-'
Как следует из сопоставления результатов, при упрощенном методе счета
получаются значения, достаточно сильно отличающиеся от данных,
приведенных в табл. 4.10. Результаты табл. 4.10 также нельзя считать вполне
правильными, так как при расчетах использовались коэффициенты активности,
вычисленные по приближенной формуле Дэвиса.
88
Таблица 4.10
q
2q
P
p'
p'+4q
V
Р + /
fl
,0-S.748
1.786 I0-'
,0-6.447
,0-4.59
2.570 . 10—
,0-9,18
6,608 . lO-'''
IQ-6.1455
t
7.153 • 10-'
10-3.0728
8,455 • 10-^
10-3.0599
t
8,712 . 10-^
10-3.3871
^ 0-3.3871 ^>j0-6,374 (5 , 7,)
'l
ъ
m
|Q_3,3«7)
IQ—12,2936
1
6.09 10-"
,0-3.0678
1
8.56 10-^
,00.2191
1
1,656
,01.422
1
26.426
10^4484
t
28,083
10—3,4484
[H+J
(Cr(OH)7j
[Cr(OH)t]
(CrOH+)
lCr3+l
[OH-j
jQ_3.0861
8,20 10-^
,0-15.7420
i
1.8 10-'^
JQ—6.5162
3.046 10-'
,0-3,2293
1
5,896 10-"
10—2,0264
1
9,410 • 10-^
9,9996 10-3
Ta(
jQ-10.4549
3,51 • 10-**
5 Л И Ц a 4.11
q
2q
p
P'
Р + /
ft
10-8.65I1
2,233 \0-^
,0-8,3501
,0-4.7561
1,754 10-5
,0-9,5122
3,075 10-'"
lQ-8.0343
t
9.2395 10-^
10-4.0172
1
9,612 . 10-5
10-3.9444
t
1,1366 .10-^
10-4,4057
пример 4.10. Полный расчет равновесия в растворе хроми-формиата надо
проводить по формулам раздела 4.6. Пусть Ссг(нс02)з =0,02 и Cj^^^.q-
= 0,69 моль • д—^ В таблицах нет констант образования формиатохроми-комп-
лексов. Это дает право считать, что / = 0,81 и lgcpi = 0,156 (по Дэвису).
Находим в таблицах для диссоциации аквохроми-иона P'^i = 3,8, р7)2 = 10,0,
PYJ4 = 26 и для протонизации формиат-иона — Igoj = 3,75. Учтем, кроме того,
что (JL = 3 и а = 1.
Критерий (4.143), допускающий упрощенный подход к решению
уравнения (4.51)/ для однопротонной муравьиной кислоты (og не существует)
превращается в односторонний: Л > 2 • Ю-^-^'^^б . jq-IO . |q3.8 ^ |q~6.367 (4 172).
Дальнейший расчет по простейшей формуле (4.151):
h = ]/1. 10-4 • О' • 10-^'^ • 10 -3.75 = 10-4'325б ^
(4.173)
но принять это значение нельзя, так как оно не удовлетворяет условию (4.150).
В табл. 4.11 приводится более точный расчет h по уравнению (4.152)
(пример 4.10): Л = 2q (p + /p2+4q)-^ где q = -J/мЧ^МОнТл/нА-^з^а =
= 10-S'6511 и р = fM4>MOH^i{l = 10-4'75б1.
Найденное значение h > 10—^ моль • л—*- Это значит, что для
окончательного решения придется использовать уравнение (4.154). Дальше приво-
h
9iti4a-4
9i^2^-2
98
а
10-4.4078
10-8.2128
i
6.13 .10-^
10-1.0284
9,367-10-2
101.2318
17,058
101.404
Ф
25,35
10Ь6284
t
42,502
[Сг (OH)rJ
[Сг (OH)t]
[СгОН2+]
[СгЗ+1
2
10-4.2518
5.60. 10-5
10-И.5412
2,88 .\о-*2
1Q~4.3568
4,397 • 10-5
10-2.0966
8,005-10"^
10-1.9244
Ф
1,1903.10-2
1,9952 . 10-2
дится подробный расчет коэффициентов этого уравнения. Индексы при / и ср
указывают число зарядов, которое несет соответствующая частица. Так находим
tsmi + НоКа + AiHtC = 10-4'5^ + 10-3-594 + g . 10-2.156 _
= 4,218 • 10-2; fmnfo^i^a + fitm<^i • 2 = 7,413 • Ю*"^ и /i/a^PaTi/o • 1 • 2 X
X 10-2 . 10-3.8 . 10-3.75 _ g 354 . 10-11.
виде Ri=/i3 +
одна перемене
Ч-4
90
В результате получаем расчетное уравнение (4.154) в вид
.218 . 10-2л2-I-7,413 . 10-7/1—9,354 • 10-lUo.(4.174).B Ri-c
знака, значит, только один положительный корень. Решая по схеме Хор-
нера, преобразуем его путем умножения коэффициента при на 10°, при /гЗ—
на 10^, при /I — на 10^ и при — на 10^ столько раз, сколько потребуется для
получения уравнения, корень которого окажется больше 1. Так, в случае
данного Rt после пяти указанных преобразований получится уравнение Re =
= + 4218/i2 + 7413.^. — 93 540 = О, при подстановке в которое h = 1 левая
часть окажется < 0. Поскольку каждое преобразование снижает значение
подставляемой единицы в 10 раз, единица, подставляемая в Re, является
фактически единицей минус пятого порядка, а из этого следует, что значение /г,
удовлетворяющее Ri, должно начинаться с пяти нулей.
Дальнейшее решение по схеме Хорнера
I 1
4 218 7 413 —93 540
4 222
4 221
4 224
4 2271
24 301 3 664
20 076 -33 312
32 748 i
36976 и ^||1|= 0,900
36 976
(4.175)
показывает, что 3 • Ю ^ < /г < 4 • Ю^^, так как при подстановке в Re Л =
= 4 остаток 3664 > О, а при подстановке /г = 3 остаток < 0. Отношение
Таблица 4.12
т
<Po»ft
"Pi
a
10-3.3284
j0-b,6578
0.2199
100.156
1
1.4323
,00,2181
t
1.6522
,0-1.4399
[HCOjH]
[HCO7I
[0H-]
10-2.0977
7,986.10-3
10-1.2839
5.201-10-2
5,9996-10-''
,0-9.4362
3,662.10-'°
33312 ,
^g^^=: 0,900 показывает, что Л 3,900 • 10—^(4.176). Окаймленные уступом
цифры в схеме (4.175) являются коэффициентами нового уравнения,
получающегося из Re путем подстановки в него значения /г = 3 + л;, где —
неизвестная дробная часть единицы минус пятого порядка. Переходя путем уже при-
менявшегося преобразования к единицам минус шестого порядка, получаем
R7 = /i3 42 270Л2-1- 3 274 800/i — 33 312 ООО = О, где Л > 1, и, судя по
приближению (4.176), должно быть равно -9.
Используя схему Хорнера, находим
91
9
10
1 42 270 3 274 800 —33 312 000
1 42 279 3 655 311 -414 201 и .-illM. = 0.102,
1 42 280 3 697 600 3 664 000 4 078 201
что h = 3,91 . 10-5 = 10-^'^°^*-
Проверка счета путем подстановки значения h = 3,91 в Re по схеме
Хорнера подтверждает правильность расчета, так как левая часть Rg
оказывается равной — 10,2
1 4218 7413 -93540
3,91
1 4221,91 23 920,6681 —10,2
что составляет лишь '^1,1-10 % от 93 540.
Последующий расчет равновесий в системе Сг (НС02)з — KNO3 —
(пример 4.10) дан в табл. 4.12.
Упрощенный метод расчета той же системы проводим по схеме
СгЗ+ + Н20^СгОН2++Н+, 10-3'«о (4.177)
HCO^-l-H+^rtHCOgH, 10^(4.178)
СгЗ+ + Н2О + НСО7 5=t СгОН2+ + НСО2Н, 10-°'05 (4.179)
с 0,02 0,06 — —
[ ] 0,02 — X 0,06 — х X X
По закону действующих масс откуда 2 ^
+ 10-0'*^з х-' 10-2,0072 ^ Q Решение дает х = [CrOH^+J = [НСО2Н] ==
= l,467.l0-^ [CгЗ+1 = 0,02-;c = 5,ЗЗ.lO-^[HCO^] = 0,06-;с = 4,533 X
X 10-2 моль-л-*.
Теперь, например, из закона действующих масс для (4.177) [Н+] =
= IO-3'S [СгЗ+1 [СгОН2+Г* = 1о-4'2398 _ 5j6.10-^
Тогда [ОН-] = lO-^•^^°^ = 1.7-10-*°, а разность [Н+] — [ОН-] =5,76-10-^
Поскольку эта разность составляет всего -0,4% от [СгОН2+] = [HCOgH] =
= 1,467-10-2 моль-л—*, использование приближенного равновесия уравнения
(4.179) считается на основании критерия (4.147) обоснованным.
Наконец, [Cr(OH)t] =^2lCr^+] [н+1-2=1о-з.7937_1 ^61.1^-4 и [Сг(ОН)-]=
= ^4lCr3+j[H+]-4 _ 1о-**.зИ1 _ 4,85.10-12 моль-л-*.
Сопоставление результатов расчетов и в этом случае обнаруживает
значительные расхождения, и поскольку расход времени на точный расчет
сравнительно невелик, нет смысла пользоваться упрощенным путем при рН,
заметно отличающихся от 7.
Пример 4.11. В качестве примера сложной системы используем раствор,
в котором ^нсОвН = '4 = 0,08, CQr(Qio.)5 = = 0,12, ^^^^104 = = 0,002 и
^H^NOg = ^1 = 0,278 модь-л—*. Считая, что НСО2Н — слабый электролит,
находим / = ^ (O.I2.32 4- 3-0,12 -|- 2-0,002 -|- 2-0,278) = 1, а тогда по Дэвису
lg?l =- 0,15.
Начальные концентрации простейших частиц, образующих систему,
выражаются следующим образом:
92
^HCOi - = = ^ = 0.08, сегЗ+ = c„ = 0,12,
tior = + К = 0.362, Cnhj = л, = о ,278,
Cf^Qj = Л J = 0,278 моль-л—
Матрица системы имеет вид:
Аг ЗМ + У
0.278 0.362
AM л,
0.08 0.12 0.278
N07 С107
НС07 СгН NHg Н+
0 0 0 —1
1 0 0 1
0 1 0 —к
0 0 11
он-
НСО2Н
Сг (0Н)3-«
NH+
lga; = -14
Ig а = 3,75
P^i = 3,8
РЪ=10
рт)4 = 26
Ig ci = 9,24
Пользуясь матрицей и указаниями в разделах 4.4 и 4.5 относительно
записи расчетных уравнений с использованием блоков, получаем в общем виде:
"fH^ — "fOH^^"^ - ^ + ^^2.Н60Г — 3iW+iW^z . СгЗ+ - ^ 1 + ^4 NHi = О-
(4.180)
Полагая далее, что ^он^^"^^ * ^ используя выражение (4.53) при
а* = а = 1 для HCOgH и при а = О и а'= 1 — для NH3, приводим (4.180)
к виду:
к=0
+ л, 1^21 ^ = о, или — Г—.4
?а
Тна^Л+та
j=0
S ?М(ОН)к'^к^"
к=0
и, наконец,
9н^^У^А
тна^^ + ^ а
Ё КТм(ОН)«Т)к^""
— М —1 — у4
Та,
V
(4.181)
93
Таким образом, для получения точного значения h приходится решать
уравнение седьмой степени. Предварительно следует, конечно, оценить
значения отдельных слагаемых, входящих в уравнение (4.181), и, если это
окажется возможным, исключить некоторые из них.
Приближенная оценка роли HCOgH = НА с учетом CHCIO4 = К = 2 X
X 10"*^ моль-л""*.
НА « Н+ +А- lg/C = ~3J5.
с 0,08 2-10"^ —
[ 1 0,08-а 2.10-3+0 "
По закону действующих масс (2-10-3 + а) д.(0,08 — а)—* = Ю—^'^^ Из этого
квадратного уравнения 0=^2,9-10—3. Это значит, что третьим слагаемым
пренебречь нельзя.
Приближенная оценка роли аквохроми-иона с учетом
^НС104 = ^ = 2-10-3 моль-л"*.
СгЗ-Ь + Нр 5=t Сг 0Н2+ + Н+, IgT,, =-3,8
с 0,12 2-10-3
[ ] 0,12-д: X 2-10-3+;с
По закону действующих масс ;г (2-10-3 + а:)-(0,12—;г)-* =10-3'*. Из этого
квадратного уравнения с-3,33.10—з. Это значит, что пренебречь четвертым
слагаемым также нельзя. Найденное значение [Н**"] = 5,33-10—3 моль - л~*.
Условие (4.137), допускающее учет только первой ступени ионизации
аквохроми-иона, имеет здесь следующий вид:
h > 2.l0^3-2{i.)lg9. 2L ^ 100.3.,0-0.45^jq-10.0.1q3.8 ^ jQ-6,35 ^ jggj
так как {1 = 3 и lg<Pi =0,15. Поскольку Ю—^'35 составляет лишь .^0,01% от
/I = 5,33-10—3, условие (4.182) выполняется и для значений к = О и У
четвертое слагаемое левой части уравнения (4.181) принимает более простой вид:
^MOH^i^""^ Ыoн^^ (4.183)
Приближенная оценка роли NH^-иона с учетом Cj^qq^ = К =2-10—3 мольх
X Л-*.
NHt« Н+ +NH3, lg/Ci = «9,24
с 0,278 2И0-3 —
[ 1 0,278 —а, 2-10-3+^1 а,.
По закону действующих масс Qi(2- 10—3 _|_ (0,278—а,)—*= 10-^•^^
Если < 2-10-3 < 0.278, то находим = 8-10-^ < 2-10-3, а это
значит, что при расчете h вкладом в эту величину за счет NH^f-иона можно
пренебречь. Тогда с учетом (4.183) уравнение (4.181) существенно упрощается:
S4
в приведенной форме уравнение (4.184) выглядит так:
+ (Та/на^ + /мТмон^1 -/н^)^2+Ына/мТмон^1^-/н9а/на^ (У +
+ А) - /н/м?мон^ \{У + Щ^- /h^a/ha/mTmoh^j^ (Y + A+M) = 0,
(4.185)
Подстановка в (4.185) значений = «р^^. 9а = /на = /о = Ь /м = ь\
Ыон = Ъ^ /С,т)1,У,Ли дает ^ 1^133.Ю^Ч^ ^ иоиЮ^Ч^
—1,013.10-^ = 0.
Решая это уравнение по схеме^^Хорнера, приведенной в разделе 4.7,
находим.
что /1 = 4,754.10-3=10-
2.3229
моль.л
1
Дальнейшие расчеты т= (СгЗ+),а=(НС02 ) , = (NH3) и равновесньх
концентраций этих исходных и всех происходящих от них частиц проводятся
по схемам, указанным в табл. 4.4 и 4.10 с учетом уравнений (4.29) и (4.45)
при заданных значениях j, а, а', к, р. и fx'. Результаты расчета равновесш г)
состава в системе Сг (0104)3 — HCIO4 — HCOgH — NH4NO3— HgO (пример 4.11>
приведены в табл. 4.13.
Они полностью подтверждают все предположения, использованные при
переходе от уравнения (4.181) к упрощенному уравнению (4.184).
Примеры расчетов равновесий, осложненных присутствием многоядерных
комплексов.
Пример 4.12. Для начала можно взять систему, в которой ^вксю*)» ~
= М = 5-10-2» %С104 = Н = 0,375 моль-л'^^ и ионная сила поддерживалась
постоянной (7 = 3,0 моль.л—О за счет добавок NaC104. Согласно исследова.
нию [59], эту систему можно представить матрицей равновесий:
CIO7 |Bi3+ н+ 1
0 —1
1 —1
6 —12
он-
ВЮН2+
til
pijj = 1,58
=0,33
[BiOH^+J^iiimr
[BieO|+]=,em^^-^'
В этом случае значения <pj входят в значения констант, так как послед-
ние измерялись при / = const.
Используя закон сохранения начальной концентрации, можно записать:
М = т + T)im/i-i + erjg/i-i^m^ (4.186); h = H + fi^mh-^ + 12-ц^к''^^т^ (4.187).
При записи уравнения (4.187) учтем тот факт, что [Н+] должна,
превышать введенную Chqjq= Н, так как при образовании каждого моля BiOH^"*'
выделяется 1 моль Н'*"-ионов, а при появлении одного моля Bie
Oq^—двенадцать.
Вычислить значения hum, удовлетворяющие этой системе, можно
итерационным методом и лучше —; с помощью ЭВМ.
В Данном частном случае можно легко, выразить т через h. Для этога
достаточно умножить уравнение (4.186) на 2 и вычесть из него (4.187). В
результате, оказывается, что
2M + H — h
^= 2 + 7),/i-i • . (4.188>
9S
Таблица 4.13
А
ъ
m
IQ-2,3229
10—16,5584
\
2,8 ■ 10-"
,0-5,2042
6,25 10-'
,0-0,8771
\
0,1327
,01.35
22,39
,01.3527
t
22,523
10-2.2735
IH+J
(Сг(ОН)7
Сг (ОН)^
[СгОН2+)
[СгЗ+1
2
[0H-]
jO-2.1729
4
6.72 10-^
,0-18.8310
1,5 10п'9
,0-7.4777
3,33 10-»
,0—3,1506
7,07 10-<
J0-0.9235
0.11925
0,11996
10~П.6771
1
2,1 10-^2
«Pl
а
«Ро
S
,01,4271
26,736
,00.I5
+
1,413
,01.«94
t
28.149
,0—2,5463
,07.0671
1,167 10^
10°
1
107.0671
t
1,167 10^
10-7.6231
[HCOgHJ
[нсоп
S
[NHt]
[NH,J
2
,0-1.1192
7.600. 10-2
,Q-2.3963
4,02 10-3
8,002 • 10-2
,0—0,556
0.278
,0-7.6231
2.4 10-*
0,278
Для дальнейших расчетов приходится использовать уравнение закона со.
хранения электронейтральности, в котором [Н"^] > [ОН""]'
[Н-^] ^ [С107]-1- 3 (Bi^+l + 2 [BiOH^-^j -Ь6 [В1бОв+] =0 или .
/7 - (3 Л1+ Н) + Зт Ч- 27,1 tnh-\ ^ 67,6^^/1-*2 = 0. (4.189)
Подстановка в это уравнение значения т из (4.188), конечно, приведет
к уравнению с одним неизвестным Л, но затрата сил и времени может
оказаться при ручном счете даже большей, чем при решении системы (4.186)—
(4.187) с помощью какого-либо итерационного процесса.
Расчеты в этом случае чрезвычайно упрощаются за счет дополнительных
измерений. Так, согласно опытным данным [47], в рассматриваемой системе
/i =г 0,393 моль-л"*» а чначит, рН = 0,4056. Подстановка этого яначения в
уравнении (4.188) сразу дает m = 3,966-10""^ моль-л""*, т. е. значение, очень
близкое к найденному в [47] с помощью амальгамного В1-электрода: т =
= 3,96.10""2 моль-л""'. В дальнейшем, используя константы из (59|, находим,
что [ВЮН2+) = 7,,тЛ-* = 2,655- ХО"^ и [BigO^+l = •»1б^^И-"*2 = 1,342 х
X Ю""* моль-л""**.
Пример 4.13. В качестве второго примера системы с многоядерными
частицами возьмем систему, в, которой Cpj^^^iQ^^^ = М = ЫО"*^ и с'хон ~ ^«^^
X 10~"3 моль-л~* при / =0,3 за счет фоновой соли NaClO^, для которой с =
= 0,3 моль-л"*. Эту систему можно представить матрицей равновесий:
Na+ Х+С1071рь2+ н+ 1
0 -I
1 —к
4 —4
3 —4
6 -8
он-
РЬ (0Н)2-'<
(0Н)*+ •
РЬз (ОН)2+
РЬб(ОН)|+
ртц 7,8, pir]2 =
Р^34 =23,4
>1б8 = 42,7
17,2 pii3 = 28,0
Отсюда следует, что
4=0
(4.190).
Пользуясь той же матрицей, получаем уравнение закона сохранения
электронейтральности в виде
3
^н^ - ?он«^Л""^ Н- X - 2М + m 5] (2 к) •n^h-'' -h 4Y,44m*/i-"* + 2yi^,m^
-h47,g8m6/,-e=0. (4-191)
В уравнениях (4.190) и (4.191) коэффициенты активности входят в
константы, так как последние получены при /-const на фоне NaC104.
• Обеспечить каким-либо путем переход от уравнений (4.190) и (4.191)
к уравнению линейному относительно т или h нельзя. В связи с этим
.следует вести расчет с помоихью ЭВМ.
* К сожалению, подстановка этих значений в уравнение (4.186) дает М
= 4,312-10"^, т. е. величину очень сильно отличающуюся от 1(0104)5,
= 5.10"^ моль-л"*. В связи с этим вначения yjj, ijg, приведенные в [59;,
нельзя считать достоверными и окончательными.
4 ?-43б
97
Есть, однако, еще одна возможность упростить решение: прямое
измерение активности т или h.
Такое измерение при указанных начальных условиях дает, например,
рН = 8, h= Ю—^моль.л""^ и позволяет обеспечить решение путем расчета m
из уравнения (4.190), так как (4.191) становится излишним. Расчет
коэффициентов уравнения (4.190), рН-8 (пример 4.13) приведен в табл. 4.14.
В первой строке табл. 4.14, начиная с 7-го столбца, даны коэффициенты
в (4.190) при т, т^у и т^. Их значения в логарифмическом виде после
деления на Gijeg/i—^ даны в предпоследней строке, а в виде десятичных
дробей с выделенным порядком — в последней. В результате имеем
+ 4,207.10-^т* Н- 9,974 . 10"^V + 2,211 . 10-^2 ^ — 8,351 . Ю^^б = о.
(4.192)
Решая это уравнение по схеме Хорнера, приведенной в разделе 4.7,
находим, что т = 5,944.10-5 = io-4.2250 1^оль.л-^
Расчет равновесного состава системы РЬ (0104)3 — ХОН — NaC104 — HgO
(пример 4.13) приведен в табл. 4.15. В первых трех строках даны
результаты проверки правильности найденного значения т. Поскольку полученная
сумма, равная 9,994.10""^, практически совпадает с М = 1,00-10"~^ моль.л""*,
расчеты проведены правильно.
В следующих двух строках даны результаты расчета равновесных
концентраций рь2+-иона и всех гидроксоплюмбо-ионов, т. е. решение задачи.
Наконец, в последних двух строках приводится расчет расхода ХОН на
образование гидроксокомплексов. Здесь
^ХОН « ^ = [ОН- ] - [Н+] + S к [РЬ (0Н)2--] + 4 [Pb4(0H)J+] +
+ 4 [РЬз(ОН)2+] + 8 [РЬб (0Н)|+]. (4.193)
Примеры упрощенных расчетов некоторых равновесий и контроля
получаемых оценок.
Пример 4.14. Расчет рН в растворе, где с^НзОН = 0»1 ^ в таком же
растворе при Ci^ei=0j49 моль'Л""^
В первом случав / Ю'"^ моль-л"как в воде;
NH20H + H+5=iNH30H+ <j 5,98
HgO ^Н+ + ОН- оу—14
NHgOH + HgO 5=i NH3OH+ + OH- K==aw lO-^*^^
с 0,1 — —
[ 1 0,1 —«) (J) (0
rio закону действующих масс
f ^ io-8>02, ^«;o,l, a)2=iO-^Л a)=10-**5l моль-л-^ условнее)«
0,1—О)
< 0,1 выполнено и рН = 14 — ро) = 9,49.
При / ^ 10~7 моль'Л—^Ti = 1 и решение можно считать правильным.
Здесь О) = VawA, т. е. выражается формулой (4.108), в которой Ю""^^'р» = 1.
Поскольку для NHgOH а = О и при c^q^ = 0,49, т. е. при / = 0,49 моль-л—! значение
U) и рН остается тем же самым.
98
Таблица 4.14
м
10"?
IQ-25,0782
8.351 10-2^
10^
10-^
1
0,0001
10-ь2
0,0631
1
1,5847
ф
1,0000
j00,4228
2,6^79
10—21,6554
2,211.10-22
1012.7021
10-9,3761
4,207-10"*°
109>0771
10-13,0011
9,974 10-*''
1q22,0782
lO'^
1
Таблица 4.15
Tl2mA~"2
6^68''-^
10-4.225P
\
5,94 10-^
jQ*.4,025S
9,42 -\o^^
10—5,426®
1
3,75- 10-^
,0-s.225^
5,9. 10-^
,0^-4,201.
1
6 287- 10-'
10~3,6006
2,509. 10-'
10-3.2772
1
5,283-10-*
9,994 10-^
[PbOH-f ]
[РЬЮН)2)
[Pb (0h)7i
1 Pb^ (0h)J-+
[РЬз (0h)2+1
fPbe(OH)M
5,94 . lO-'^
9,42. 10-^
3,75 • 10-6
5,9. 10-
1,572. 10-'
8,363. 10—'
8,805. 10-^'
[OH-j
[Pb 0h+]
2[Pb (OH2)]
3[Pb (OH)-]
4[Pb4 (0h)4^-bj
4 [РЬз (OH)2+]
«[РЬб(ОН)^+]
10-^
9,42. 10-^
7,50. 10-^
1,8. 10-^
6,288 • 10-^ •
3,345. 10-^
7,044. 10-^
1,20466.10-^
Пример 4.15. Рассчитаем равновесия в водном растворе, если ^нАс ~ ^»^^
и Сдс! =0,05 моль-д—^ В подобных случаях можно получить
приближенное решение, рассматривая равновесие слабой кислоты, в присутствии = с^у.
Тогда
кк
НАС «Н+ + Ас- —4,76
с 0,01 0,05-
[ ] 0,01 —X 0,05+ х X
По закону действующих масс
^^Q%'tl\' = 10-*-'«. X « 0.05. 0.05Х = 10-в-7в. X « 10-'-*^ = 3.5.x
X 10""^ моль-л—^
Значит, с достаточной точностью [Ас"] = 3,5-i0'"^ [НАс] = 0,01, [H+]=s
= 0,05 и [CI-] =0,05 моль.л"^
Пример 4.16. Расчет рН смеси из 30 мл раствора НАс (0,1 моль-л^^) и
50 мл раствора КАс (0,3 моль-л""*). Решение таких задач начинается с
перехода к начальным концентрациям, выраженным в моль.л""^ Здесь с/^д^ =
= 0,03 л, ^кАс = 0.05 л, t;HAc + ^KAc = O.OS л, Сндо = ^^^^ = 0,0375
0,3.0,05 ,
" ^КАс = Q Q3 = 0,1875 моль-л"-*-
Тогда, рассматривая диссоциацию НАс в присутствии Ас"-ионов, полу,
чаем
НАС 4=iH-*- + Ac- ' lg/C= —4,76
с 0,0375 — 0,1875
[ ] 0,0375 —л: X 0,1875
По закону действующих масс
i^^-±~L 10-4'7^ X « 0.0375 < 0,1875, х = 10"^'^^ т. е. составляет
1Q-5.66
— =10~^'^^* ^ 5,8-10"^ % от величины 0,0375. Поскольку усло-
10^1.426.10-2
вие X < 0,0375 выполняется, можно считать, что х = [Н+] вычислен
правильно. Но pH = -lg(H+) =-lg/H[H+]- При СкАс = 0.1875 / = 0,1875 моль х
Хл""*.
Тогда по Дэвису
Igcp, =0,5- fpJ^^-0,2/j = 0,1324, lgfi=-. 0,1324 и рН = - (Ig/j +
+ lg[H+]) =5,79.
Пример 4.17. Рассчитаем количество твердого NaAc, которое должно быть
растворено в 50 мл раствора НАс (0,04 моль-л""*), чтобы обеспечить рН =
= 5,43; рН — интенсивное свойство, а это значит, что его численное значение
не зависит от объема раствора, поэтому надо вычислить ^дс ^NaAc ^» '^Р"
которой будет обеспечена [Н*^] = Ю""^'^^ при Сдд^ =0,04 моль-л""*. Здесь
приходится вычислять А при заданных с^д^ и [jfi+]:
НАс Н+ + Ас- lgK = -.4,76
с . 0,04 — А
I ] 0,04—10-^''*^ Ю""^'^ А + Ю"^*'*^
100
По'закону действующих масс
lO-'-'HA + iO-^-^') _ ,„_4де „ ^ _ 10-''-^П0.04-10-^-''^) ,.,3 ^
0,04-10-5-« ""^^ 10=-5-''з ="
= 0,186 моль-л~'.
Такому значению у4 отвечает / = 0,186 моль-л~', Ig9, = 0,1371 и при
рН = 5,43 [Н+] = 10-^•2^. В этом случае [А[с-'] = Л + Ю-^-^^ [НАс] =
= 0.04- 1о-«'28, А = i°l^:!^i2^1;^l^l!f!L_io-5.29 ^. 0,135моль-л-'.
Такие пересчеты приходится продолжать, пока У перестанет заметно изменяться.
Пример 4.18. Рассчитаем с^^^, которую надо создать в водном растворе,
чтобы при пропускании через него HgS обеспечить (S^") = Ю""*^ моль-л "~*.
Заданная {S^'~) должна поддерживаться за счет протонизации S^""-ионов
в водном растворе, насыщенном HgS. Здесь необходимо учитывать
одновременно равновесие протонизации S^—- иона и распределения HgS между водной
фазой и атмосферой. Тогда при 25 °С
+ 2Н+ ^ H2S Igo = 19,95
H2S ^H2StlgX = -0,99
s2- + 2H"*"5=±H2St aX = lO*«•^б
(52—) (н+)2
По закону действующих мисс i L1 i-^l0~l8.86^ где jt?^^—парциаль"
ное давление HgS над раствором. При пропускании его = 1 атм =
= 1,01325-10^Па. Тогда для расчета рН, поддерживающего заданную (S^—)»
получаем формулу
рН =V2[ 18,96+ Ig(s2-)J.
При (s2-) = lo'-ie моль-л-* рН = 1,48, (Н+) = 10-*'^»иснс1 - 0,033 мольх
хл-*.
Дальнейших пересчетов здесь можно не делать, так как при / =
= 0,033 моль-л-* <Pi - 1.
Пример 4.19. Приближенная оценка [NH^], [nh3 с^—]^ [HCN] и рН
в следующих случаях: cj^h.ci 0»l»^NaCN = О'Ь %H4CN = ^»1 моль-д-*.
1) Используя равновесие диссоциации NH^-иона, получаем
NH+ 5=t Н-Ь + NH3 Ig/C = ~ 9,24
с 0,1 - —
[ ] 0,1-/1 h h
Л «0,1, /i2^lO-*0'2S
/1= 10-5'*2, рН =-5,12, [NHs] = 10-^'*2, [NH+1 - 0,1 моль-д-*.
2)cqjsj=0,1 моль-д—*. Используя равновесие протонизации CN—-иона
101
за счет Н"*"-ионов воды, получаем
CN- Ч- H+;=tHCN \ца = 9,35
CN- + Н.р 4=t HCN -f OH-lgaш = — 4,65
с 0,1 — —
f j 0,1 —О) О) О)
2
^ Ю-'^-б^ ft) < 0.1, О) = 10-2'^2\т. е. 1,5% от 0.1. Тогда [HCN] =
= [а)Н-1 = 10-2»^^'^ - 1.5.10-^ ICN-] = 9.85.10-2 моль.л"-*, рОН = 2,825
ирН=ПД
2)^NH4eN = ?nh^ = ccN— ~ 0,1 моль-л""*. Учитывзя одновременно
диссоциацию NHJ^ и протонизацию СК^-иона, получаем
NH+ « Н**- -I- ЫНз IgK = — 9,24
CN- Н HCN Igo = 9,35
NH^-bCN-«NH3-i-HCN /(о = 10°''"
с 0,1 0,1 - -
f ] 0,1 — А' 0,1 X X
По закону действующих масс
(ОТ^ = 10^'*^ оЛ^^ ;с = [ЫНзЫНСЫ1 = 5,3.10-2, 0,1 ^х =
= iNH^}ICN—] = 4,7* 10—2 моль ♦л—*. Для проверки законности решения
б помощью критерия (4.147) необходимо знать [Н"*") и (ОН""]. Значение [Н"*"]
получается из равновесия диссоциации NH^-иона или протонизации CW-hона
С помощью уже вычисленных равновесных концентраций, входящих в них ча-
етиц.
Так, из первого равновесия по закону действующих масс нолучаем
1Н+1 = 10-^*2^^^^ ^ 10-^*2^ ъ!з!\0-^ ^ ^^"''^ моль.л-*, рН ^ 9Д
10Н-1 — 1Н+] = 3.10-^ -.5И0-*° = 3.10-'^ моль.л-^
Поскольку найденная разность (ОН""] — [H'^l = 3tl0"'^ моль.л—* ео*
ставляет всего 0,064% от значений [NHJ*"] и [CN~], вадачу можно считать
решенной.
Отметим, что взаимодействие NH^-и CW-hohob приводит к рН = 9,3,
тогда как в растворе NHj'-иона pH=5,12a а в растворе, содержащем только
NaCN, рН=П,2.
Формула (4.151) прямо дает значение ='(/'ю-э'г^ • Ю"^»^^ « 10""^'295
и рН = 9,295.
4.8. РАВНОВЕСИЯ В РАСТВОРАХ ЦВЕТНЫХ
КИСЛОТИО-ОСНОВНЫХ ИНДИКАТОРОВ
Общие приемы измерения интенсивных свойств.
Кислотность— свойство раствора, численное значение которого не
зависит от массы (объема) системы, в которой проводится соот-
102
ветствующее измерение. Подобные свойства называются
интенсивными. Их измерение осуществляется путем совмещения
исследуемой системы с другой — индикаторной — системой.
Индикаторная система должна сама обладать измеряемым
интенсивным свойством и, кроме того, каким-либо
дополнительным свойством, изменение которого может служить мерой
интенсивного свойства в объединенной системе. Так, при измерении
температуры, которая также является интенсивным свойством,
исследуемую систему совмещают с термометром, т. е.
приспособлением, в котором изменение температуры сопровождается
изменением объема индикаторной жидкости или газа,
возникновением электродвижущей силы и т. п. Такая индикаторная
температурная система калибруется по опорным температурным
точкам и снабжается шкалой, деления которой отвечают
определенным значениям интенсивного свойства.
При совмещении исследуемой системы с температурной
индикаторной системой может быть три случая: 1) заметного
сдвига на шкале нет; 2) обнаруживается сдвиг, указывающий на
повышение температуры в индикаторной системе; 3) возникает
сдвиг в сторону снижения температуры индикаторной системы.
При совмещении систем происходит усреднение
температуры. Тогда в первом случае температура исследуемой системы
совпадает с температурой индикаторной системы, во втором —
измеряемая температура выше температуры индикаторной
системы. Усредненная температура окажется ниже истинной и
погрешность будет тем больше, чем больше масса
индикаторной системы и ее теплоемкость. В третьем случае
наблюдается обратная картина и измеренная температура оказывается
выше истинной.
В связи со сказанным размеры и свойства индикаторной
термометрической системы выбирают в каждом случае таким
образом, чтобы снизить до минимума разницу между
измеряемой температурой и усредненной температурой совмещенной
системы.
Это требование выполняется автоматически, если
индикаторная система много меньше исследуемой. В некоторых
случаях это условие оказывается необязательным. Так, например,
при измерении температуры кипения жидкости термометр,
собственная температура которого вначале совпадает с
комнатной, постепенно нагревается за счет скрытого тепла испарения
до температуры соответствующей системы жидкость — пар
и результат измерения температуры оказывается правильным.
Такой же «буферностью» обладают двухфазные системы,
возникающие в расплавах, содержащих твердую фазу, и в любых
системах, термостатируемых специальными способами.
Измерение кислотности с помошрю цветных индикаторов.
Кислотность системы является ее интенсивным свойством,
измеренным в единицах рН: рН =—lg{H30+) =—lg(H+) (4.194).
103
Из уравнения (4Л94) следует, что рН = О возникает в растворах,
где (Н+) = 1 моль • Л-*.
Для измерения (Н+) системы ее приходится объединять
с другой (индикаторной) системой, какое-либо свойство
которой, например, окраска, заметно зависит от рН.
При этом свойство измеряют после того, как в
объединенной системе установится кислотно-основное равновесие.
Погрешность же при измерении будет тем меньше, чем меньше
дополнительная система по сравнению с исследуемой и чем рН
в ней самой ближе к рН исследуемой системы.
В качестве цветных индикаторов используют нейтральные
кислоты и соли катионных и анионных кислот, окраска
растворов которых изменяется в зависимости от рН.
Поскольку индикаторная система вводится в измеряемую,
необходимо, чтобы окраска частиц индикаторной системы была
как можно более интенсивной, так как только при этом
условии концентрация индикатора может быть исчезающе малой
по сравнению с концентрациями частиц в исследуемом
равновесии. Этому требованию удовлетворяют индикаторы-красители,
так как уже очень малой концентрации такого вещества
достаточно, чтобы сообщить заметную окраску большому объему
раствора.
Индикаторы-красители имеют сложное строение.
Изменение окраски их растворов с изменением рН объясняют не
только смещением кислотно-основного равновесия индикатора, но
и дополнительными перегруппировками внутри частиц
индикатора, ведущими к изменению окраски. Строго поставленных
исследований соответствующих равновесий не проводилось.
Нет и общего мнения о процессах диссоциации и изменениях
окраски [11, 13].
Рассмотрим некоторые из предложенных моделей.
Метиловый оранжевый, или натрий — м-диметиламиноазобензолсуль-
фонат относится к числу двухцветных индикаторов. Он
окрашивает растворы, в которых рН>4,4, в желтый цвет, при
рН<3,1—в красный, на интервале 4,4>рН>3,1 изменяет
окраску от желтой к красной через оранжевую. Все эти
изменения с точки зрения координационной теории [13, с. 57]
объясняются следующим.
Если записать формулу метилового оранжевого в виде NaA,
где
А- ^ (CH3)2N - <( У - N = N - <( У-SOT, (4.195)
о желтую окраску следует приписать этому аниону. При
снижении рН начинается протонизация А- +Н+«НА, а (4.196),
сопровождающаяся одновременной перестройкой молекул НА:
НА5=»НА, X (4^197), в красный амфотерный ион НА:
104
(CH3)2N-/"
>_\=N-<:
(СНз)гМ-
-<( ^=N-NH-<( У-SOT
(4.198)
С этой точки зрения в растворе, содержащем метиловый оран -
жевый, при 4,4>рН>3,1 должно находиться три вида частиц:
А-, НА и +НА"~. Первой из них приписывают желтую окраску ^
последней — красную. Относительно окраски НА никаких
сведений нет. Не приводятся и спектры поглощения этих частиц
Представитель одноцветных индикаторов — фенолфталеин.
В кислых растворах он бесцветен, на интервале 8,2<рН<10
его растворы меняют свою окраску от бледно-розовой до
красно-фиолетовой, а при рН>10 они обесцвечиваются. Дальше
приводится ряд равновесий, объясняющих изменения окраски
растворов, содержащих фенолфталеин, с точки зрейия
координационно-ионной теории [32]:
ОН ОН ОН ОН
XX
с —О+НгО:
/\-со
^Х
\
(
х\
с—он
-СООН, или H2R+H20»H4R^i=?
\Х \/
H2R (лактон) бесцветный H4R бесцветная
ОН ОН ОН ОН
х\ /\. х\ х\
(4.199)
V \х
\/ \х
/\ -СООН /\ -С00-, или H4R« н+ + Нзк , /с, =^?
\х
H4R бесцветная НзК"" бесцветный
(4.200)
105
он он
о- о-
ч/у
с—0Н^Н+ + Н20+ с
—COQ-
COQ-
или H3R-;=2H++H20+f?2-
4/^2 = ?
(4.201)
H3R- бесцветный R^'- красно-фиолетовый
О О
о о
C + 0H-3=i СОН
/\—СОО- /\-C00 % илиR2-^-OH-»ROHЗ-,^fe2=?
(4.202)
R2- красно-фиолетовый R (ОН)з- бесцветный
В этом случае красно-фиолетовую окраску приписывают кар-
о
бониевому аниону R2~, остальные частицы считаются бесцветными,
хотя они могут поглощать в УФ.
Нейтральный красный или 2-метил-3-амино-6-диметил-амийО-
феназиний хлорид диссоциирует в растворе, образуя С1 -ион и
катионную кислоту RH+:
сн,
С1 С1- -ь
(НзС)2
f
Иг
(4.203)
106
обеспечивающую индикаторные свойства этого вещества. Этот
индикатор при рН<6,8 окрашивает растворы в красный цвет,
при рН>8,0—в оранжевый, а на интервале 6,8<рН<8,0—г
постепенный переход от красного к оранжевому.
И в этом случае в руководствах по теории индикаторов
указывают, что интенсивность поглощения света связана с
внутримолекулярными перегруппировками в самой катионной
кислоте HR+ и в сопряженном с ней основании R.
Существуют и многоцветные индикаторы. К их числу
принадлежит, например, представитель сульфофталеинов — тимол-
сульфофталеин или тимоловый синий. Его формула
ОН ОН
/\-СзН7 СзН7-/\
НзС
\
у
СНз
с -о
(4.204)
1H2R
похожа на формулу фенолфталеина. Общепринятой теории
изменений окраски нет и в этом случае. Опыт показывает, что
этот индикатор окрашивает растворы при рН<1,2 в красный
цвет, при 2,8<рН<8,0 —в желтый, при рН>9,6 —в синий,
на интервале 1,2<рН<2,8 цвет растворов изменяется от
красного к желтому и на интервале 8,0<рН<9,6 —от желтого
к синему.
И в этом случае предполагается наличие
внутримолекулярных перегруппировок, ведущих к повышению
интенсивности поглощения света. Константы отдельных равновесий,
описывающих механизм индикации, не изучались, не измерялись
и коэффициенты молярной экстинкции (81) соответствующих
частиц. В результате — большое число теорий цветности
индикаторов и практически полное отсутствие данных,
необходимых для их рационального использования.
Рассмотрим на примере метилового оранжевого
первоочередные расчеты, выполнение которых желательно при
использовании любой индикаторной системы, а заодно установим пере-
^ень всех необходимых для такой работы параметров.
107
Выше было указано, что в системе метиловый оранжевый —
вода должно находиться три вида частиц: А- НА, +НА-.
Если ^NaA=A, то, используя равновесия (4.196) и (4.197) и
отвечающие им константы, можно рассчитать для любого рН
значения [А-] =<9kCiy [НА] = ^ндааА и [+НА-] = ^на^<з«Л с помощью
уравнения
Л = а [ТА + а (s^HA + ?нАх)/1]. (4.20 5)
Дальнейшие расчеты по формуле
Д, = (ex.А [А-] + SX.HA [НА] + ех+нд- [+НА-]}/, (4.206)
где Dxj— оптическая плотность при длине волны Xi,- ех-к —
коэффициент молярной экстинкции при той же длине волны для ^-й
частицы и / — длина слоя, см, могли бы дать все данные для
расчета кривой поглощения (D, X) при заданных Л и рН.
Подобным же образом в случае фенолфталеина можно пред- ^
о
ставить три первых равновесия в виде схем протонизации ROH^—-
аниона, а последнее —в виде внутримолекулярного превращения
(в приведенных ниже равновесиях молекулы воды пропущены).
Тогда без учета внутримолекулярных перегруппировок,
сопровождающих иротонизацию:
R0H3- + Н+« R2-; ai (4.207)
ROH3-+2H+«H3R-; 02 (4.208)
R0H3- + ЗН+ и H4R; аз (4.209)
H4R^H2R; X (4.210)
В этом случае, полагая (ROH^-) = г, можно записать:
[ij2-] = ^o^airA, [HsR-] = 9h,r^2^^^. [H4R] = ?h.rV^' « fH2R] = ,
«= 9н,К^^ЗГЛЗ- ^^^^ ^HgR = ^. TO
(4.211)
В этом случае оптическая плотность системы равна Dx. =
3=eXjO^[R2-] (4.212). К сожалению, в работах и монографиях,
посвященных цветным индикаторам, нет надежных значений
0, X, 8, ф даже для таких широко используемых индикаторов,
как метиловый оранжевый и фенолфталеин. При этом очень
часто предположения относительно равновесий и частиц,
играющих роль в индикаторном процессе, расходятся с опытными
108
данными. Так, прямыми спектрофотометрическими
измерениями установлено [40], что, за малыми исключениями, изменение
рН в растворе, содержащем индикатор (фталеиновый или азо-
краситель), не приводит к появлению новых полос поглощения,
а лишь изменяет интенсивность поглощения на участках
спектра, отвечающих кислой и щелочной форме данного индикатора.
Но тогда, например, в случае метилового оранжевого
приходится либо признать, что существуют лишь две частицы А-
и НА, либо допустить, что равновесие (4.198) сдвинуто
целиком вправо и устанавливается с огромной скоростью. Тогда
метиловому оранжевому будет отвечать суммарное равновесие
A-+H+qF±H*A, gk (4.213),константу которого ах в теории инди-
•каторов называют «кажущейся». Отметим, что в связи с
появлением работы [40] были признаны мгновенно
устанавливающимися все внутримолекулярные перегруппировки,
предусматриваемые для индикаторов различными теориями. Константы
равновесий для всех таких индикаторов считаются кажущимися.
Обычно записывают поэтому равновесие диссоциации любого
индикатора в виде HId;f±:H++Id-, К (4.214), гле/С = (ax)~i, счи
тая, например, что красные амфотерные ионы метилового
оранжевого (Hid) дают Н+-ИОНЫ и желтые анионы Id".
Многоцветные индикаторы, .вроде тимолового синего,
рассматриваются при этом как многопротонные кислоты, константы
диссоциации гидроанионов которых имеют сильно
различающиеся значения. Так, считают, что в случае тимолового синего
красную окраску растворам при рН<1,2 сообщают молекулы
двухпротонной кислоты НгЫ, желтую на интервале 2,8<рН<
<8,0—гидроанионы Hid-, синюю при рН>9,6—анионы Id^-
Для соответствующих равновесий диссоциации приводятся
кажущиеся константы:
H2ld^H++ Hid, pKi = 1,65 (4.215)
к ж
HId-=tH+ + Id2-, pKi = 8,9. (4.216)
Ж С
Буквы к, ж, с под символами указывают видимую окраску
соответствующих частиц.
. К сожалению, такая же упрощенная форма равновесий
применяется и для таких индикаторов, как фенолфталеин, хотя из
уравнения (4.201) следует, что в этом случае бесцветный гидро-
0
анион НзК"" превращается в красный анион R^-. Такое
пренебрежение зарядами может привести к ошибкам в оценке
коэффициентов активности и влияния ионной силы.
Во всех случаях связь между окраской раствора,
содержащего индикатор, и рН можно вполне удовлетворительно выразить,
рассматривая равновесие Hid 5=±Н+ + Id-, /С, где Hid — кислая
и Id"" —щелочная формы, имеющие различные окраски, и К —
кажущаяся константа диссоциации индикатора.
109
при оценке окраски считают, что она возникает за счет смешения
окрасок, присущих кислой и. щелочной формам, причем раствор
кажется нам имеющим чистую окраску кислой формы, если
IHId]> 10[Id-l (4.217). В тех случаях, когда [Id-]> 10|HIdl
(4.218), наблюдается чистая окраска щелочной формы.
Переходная окраска соответствует случаям, когда 0,1 [Id-] < [Hid]<
< 10 [Id-] и 0,1 IHId] < [Id-] < 10 [Hid] (4.219), и наконец,
промежуточная наблюдается при [Hid] = [Id-] (4.220).
По закону действующих масс
= д. ,Н.. = РН = р^- ,g и . ,4.22,,
Поскольку далее при [Hid] > 10 [Id-] Ig > 1, а при [Hid] <
lid J
< 0,1 [Id-] lg~^ > —1, можно утверждать, что чистую окраску
(Id J
кислой формы любой индикатор имеет при рН < р/( — 1,
щелочной— при рН > р/с + 1, промежуточную — при [Н = рК и
переходную — на интервале р/Г — 1 < рН < рК + 1. Эта упрощенная
теория индикаторов основана на теоретических соображениях»
впервые сформулированных В. Оствальдом.
В табл. 4.16 приведен набор индикаторов, располагая
которым можно оценить значения рН на интервале 1,2<рН<10,5,
даны также молярные массы (ЛГ), значения показателей
кажущихся констант и установленные опытным путем интервалы
перехода. Как видно из табл. 4.16, длина интервала перехода
лишь в одном случае совпадает с теоретическим значением
равным (p/C+l) — (р/С—1) =2 единицам рН.
Таблица 4.16
^aзвaниe
и~1
Интервал
перехода (рН)
Окраска
кислая
форма
сцелочная
I форма
Тимоловый синий
Метиловый оранжевый
Бромфеноловый синий
Метиловый красный
Бромкрезоловый
пурпурный
Азолитмин (лакмус)
Нейтральный красный
Бромтимоловый синий
о-Крезоловый красный
л-Нафтолфталеин
Фенолфталеин
Тимоловый синий
Тимолфталеин
466,27
327,2
669,8
269,14
540,02
215,21
288,65
642,09
386,5
422,26
318,11
466,27
741,23
1,65
3,52
4,10
5,0
6,40
6,5
6,85
7,3
8,46
8,4
9,77
9,20
9,7
1,2—2,8
3,1—4,4
3,0-4,6
4,2—6,3
5,2—6.8
5,0-8,0
6,8-8,0
6,0—7,6
7,2—8,8
7,0-9,0
8,2—10,0
8,0-9,6
9,3-10,5
к
к
ж
к
ж
к
к
ж
ж
ж
б
ж
б
ж
ж—о
п
ж
а
о
о
G
П
3
К
в
с
* к —красная, о —оранжевая, п —пурпурная, ж —желтая, з — зеленая,
с —синяя, б — бесцветная.
ПО
Для объяснения этих расхождений^ явления дихроматизма
и изменений окраски смешанных и универсальных индикаторов
необходимы элементарные сведения по теории зрительного
восприятия.
Теория зрительного восприятия. При оценке зрительного
восприятия окраски какого-либо раствора надо учесть, что для
каждой длины волны (X) ее восприятие (Lx) выражается через
произведение относительного значения спектральной излучатель-
ной способности источника
(t^x/t^555) и коэффициента
твора:
пропускания
относительной видности
(тх) поглощающего рас-
^555. Тс ^555
(4.222)
где Тс — цветовая температура источника, а абсолютные значения
спектральной излучательной способности {е\т^ и видности (t^x)
отнесены к значениям тех же величин при X = 555 нм,
отвечающей максимуму на кривой относительной видности глаза
среднего наблюдателя.
Логарифмируя (4.222), получаем
lgLx = IgaxT,-Dx, (4.223)
где
ахг, = --^.^, (4.224)
^ ^555.7^ ^^555
а Dx = —Ig tx — оптическая плотность наблюдаемой системы при
длине волны X нм.
Значения vjvsbb, необходимые для расчета ахт^» приведень
в табл. 4.17, а график этой функции —на рис. 4.1,
чувствительность к свету с X = 555 нм принята за^ единицу. Значения
спектральной интенсивности излучения «серого тела» для длины волны
Т аб л и ца 4.17
X
X
X
«Х/^ббб
X
400
410
420
430
440
450
460
470
480
0,0004
0,0012
0,0040
0,0116
0,023
0,038
0,060
0,091
0,139
490
500
510
520
530
540
550
555
560
0,208
0,323
0,503
0,710
0,862
0,954
0.995
1,000
0,995
570
580
590
600
610
620
630
640
650
0,952
0,870
0,757
0,631
0,503
0,381
0,265
0,175
0,107
660
670
680
690
700
710
720
730
740
0,061
0,032
0,017
0,0082
D,004l
0,0021
0,00105
0,00052
0,00025
111
X и цветовой температуры
формуле Планка:
серого тела Тс вычисляются по
6xT, = CiX-5
где Ci = 3,70-10-12 Вт-смз и С2
К'ш (К—обозначение кельвина-
цы термодинамической температуры).
= 1,43
-едини-
(4.225)
т /?нм
Рис. 4.1 Рис. 4.2
Значения exxjessb, г^, вычисленные по (4.225) для дневного
света {Тс = 6000 /Г), вольфрамовой лампочки накаливания {Тс =
= 2800 Л*) и излучателей с раскаленным углеродом (Гс = 2000/(Г),
приведены в виде графиков на рис. 4.2. Они были использованы
для расчета значений Ig а\тс Для разных X и Тс, приведенных
в табл. 4Л8.
Таблица 4.18
6000
2870
2000
6000
2870
2000
400 4,59 5,74 6,94 555 0',000 0,000 б.ООО
420 3,60 4,90 4,38 580 1,928 0,001 0,055
440 2.38 3,80 3,37 600 1,776 1,915 0,021
460 2,799 2,341 3,995 620 1,543 Т,744 Г,898
480 1,167 2,815 2',550 640 1,190 Т,450 Т,648
500 1,530 1,278 1,087 660 2,716 1,031 1,271
520 Г,868 Т,70б 1,589 680 2,146 1,505 2,790
540 1,989 1,911 Г,852 700 3,50 3,923 2,241
Используя значения
твора, можно получить
от X для данной системы
При использовании кривых (L, X) следует помнить, что из
всей совокупности длин волн глаз ощущает лишь часть, для
которой IgLi имеет максимальное значение; оценивает как примесь
(оттенок) длины волн, для которых I^L, удовлетворяет условию
IgLi — lgL2 < 1; не ощущает совершенно остального излучения.
Iga и D для любого окрашенного рас-
зависимость зрительного восприятия L
112
в качестве примера можно привести кривую (IgL, X) для
бромфенолового синего при дневном свете (рис. 4.3),
рассчитанную с помощью табл. 4.18, и значений Z)x = exd, вычисленных
при 1=1 см, 6= 1,3-10^4 моль-л-' по значениям Ige,
приведенным в [17]. Значения lO-^g для кислой (Hid) и щелочной
(Id-) форм индикатора нанесены, соответственно, пунктирной
и прерывистой линиями на рис. 4.3. Расчеты выполнены для
водного раствора индикатора и рК = 3,85. Как видно из
графика, при дневном свете
окраска индикатора будет фиоле-
то вой, так как над
пунктирной прямой, ограничивающей
область (IgLi—1), находятся
отрезки кривых (Ig L, X),
расположенные в синей и
красной частях спектра. Тот же
раствор при вечернем
освещении будет казаться
красным, так как в случаях (б)
и (в) lgL2<(lgLi-l).
Так, с помощью кривых
(IgL, К) можно оценить
видимую окраску любого
цветного индикатора при любом
освещении. В частности,
рис. 4.3 объясняет
дихроматизм бромфенолового синего.
Оценка рабочей концент-
рации индикатора. В
разделе (4.8.2) было показано,
что измерения рН можно
провести тем точнее, чем
меньше концентрация индикатора. Значение же Снм
определяется способностью глаза обнаруживать окраску
соответствующей его формы. Точных сведений по этому вопросу в
литературе нет. Нет и указаний относительно методов оценки
чувствительности глаза к окраске индикаторов.
В. В. Кисилевский впервые применил для оценки
минимальных, надежно обнаруживаемых глазом среднего наблюдателя
концентраций индикаторов статистический метод [15, 16],
Результаты выражались в виде значений концентрации
соответствующей формы индикатора (с), которую обнаруживают 50%
наблюдателей; вероятного отклонения от нее (г)—такого, что
концентрацию (с — г) обнаруживают 25% {о+г)—75
наблюдателей, и надежно открываемой концентрации,
обнаруживаемой практически всеми наблюдателями.
Результаты измерения чувствительности некоторых
кислотно-основных индикаторов приведены в табл. 4.19. Определения
Рис. 4.3
113
чувствительности проводились в колбочках на белом фоне,
освещенном рассеянным дневным светом, при объеме раствора
100 мл.
Таблица 4.19
индикатор
м
п-1
рн
окраска
„-1
.-1
с + 4г
опк.п— *
Метиловый
оранжевый
Бромфеноловый
синий
Фенолфталеин
327,05
669,8
318,11
2,35
3,65
6,10
2,6
2,6
2,6
3,8
5,95
8,4
9,3
10,3
к
к —р|
ж
ж
ж
ж
3
с
р
р
р
1,6.
2,2.
3,6.
1,9.
2,3.
1,6.
3,2;
5,3.
7,7.
1,2.
1,0.
>-7
г'
Ю-'
10-^
т-8
1-7
7
^-7
1—7
0,7.10'
0,9-10
2,7-10
9,9-10-^
7,5.10-*
7
,-8
7
1-6
7
7,8.10'
1,9-10
5,3.10-*
1,0.10-^
1,5.10-^
2,5.10-*
4,4.10
5,8-10
1,4.10"
5.9.10
5,3.10
4,7.10
1,1.10
2,6.10
1,2.10-
1,8.10"
2,0.10-
-7
>—7
1-7
г'
.—7
1—7
* Cii. подстрочное примечание к табл. 4.16; р — розовая окраска.
Даны три значения (с+4г) для кислой формы бромфеноло-
вого синего. Первое получено в пасмурный день, второе — в
туманную погоду, третье — в солнечный день. Они различаются
не очень сильно. Для
одному? нм
4
3
2
цветного индикатора
фенолфталеина чувствительность
растет с рН, т. е. по мере
увеличения концентрации
окрашенной анионной
формы индикатора. Это
позволяет использовать фенолфта-
/1^1 " I7T "—ГГГ\—' 1 «^ин для
дифференцированной оценки значений рН.
Так, если раствор
окрашивается в розовый цвет при ci^=
Рис. 4.4 =2.10-6 моль-л-* и
остается бесцветным при меньших
начальных концентрациях индикатора, то рН'-9,3.
Различная чувствительность глаза к разным по окраске
формам индикаторов объясняется тем, что наблюдения проводятся
на разных участках кривых видности и различительной
способности (рис. 4.4). Последняя характеризует способность
нормального глаза распознавать разницу в оттенках окраски. В
частности, глаз лучше всего распознает примесь света посторонних
• -
\
\
л
1 !
0 к
114
длин волн при и ---585 нм, т. е. на границах перехода от
синей к сине-зеленой окраске и от желто-зеленой — к желтой;
хуже —в зеленой области и еще хуже — на краях видности.
Различительная способность имеет существенное значение при
наблюдениях изменения рН системы по изменению оттенка
окраски индикатора.
Смешанные индикаторы. Точность, с которой можно
обнаружить появление перехрдной окраски для обычного
двухцветного индикатора, в значительной мере определяется
различительной способностью глаза, так как переход этот (например^
от красного к оранжевому для метилового оранжевого)
происходит постепенно через ряд трудно различимых оттенков.
Резким этот переход может быть только в том случае, когда кислая
и щелочная формы окрашены в дополнительные цвета.
В табл. 4.20 приведены интервалы длин волн, отвечающие
основным цветам спектра, и дополнительные цвета, дающие
вместе с основным нейтрально серую окраску.
Таблица 4.20
к нм
Основной
цвет
Дополнительный цвет*
Л нм
Основной
цвет
Дополнительный цвет
400—435
435—480
480—490
490—500
500—560
Ф
6
С—3
3—С
3
Ж —8
Ж '
0
К
п 1
560—580
580-595
595-605
605—750
Ж—3
ж
0
к
Ф
С
С—3
3—G
* См. подстрочное примечание к табл. 4.16.
Среди индикаторов (табл. 4.16) только бромтимоловый
синий и тимоловый синий можно отнести к числу красителей,
кислая и щелочная формы которых окрашены во взаимно
дополнительные цвета. В ряде других случаев того же эффекта
добиваются, используя смеси индикаторов или добавки к
индикаторам подходящих красителей. Так, в качестве индикатора
можно использовать водный раствор, содержащий в 100 мл 0,05 г
метилового оранжевого и 0,125 г индигокармина. Этот
индикатор окрашивает растворы с рН<4,1 в фиолетовый цвет, срН>
>4,1—в зеленый, а на интервале 4,07<рН<:4,13—в
нейтральный серый. Наличием узкого переходного нейтрально-серого
интервала характеризуются все смешанные индикаторы [11,
с. 191]. Надо только помнить, что значения рН, отвечающие
середине нейтрально-серого интервала каждого смешанного
индикатора, будут иметь при разных ионных силах несколько
различающиеся значения.
Универсальные индикаторы, В очень многих случаях более
точной оценке рН с помощью двухцветного индикатора,
дающего возможность сравнительно точно оценить интервал в две
единицы рН, или смешанного индикатора, определяющего
интерна
вал в 0,06 единицы рН, должна предшествовать
ориентировочная оценка рН системы. Для такой оценки используют
универсальные индикаторы [И, с. 227]. Несколько капель такого
индикатора, добавленных к используемой системе, достаточно для
ориентировочного измерения ее рН, так как универсальному
индикатору отвечает не один, а ряд последовательных изменений
окраски, каждая из которых отвечает своему интервалу рН.
Эти же универсальные индикаторы могут использоваться
для наблюдения за постепенным изменением рН в исследуемой
системе.
В качестве универсальных используют смеси обычных
индикаторов, подобранные таким образом, что в системе с
постепенно возрастающим рН окраска меняется в порядке изменения
цветов в видимой части спектра от красной к фиолетовой.
В табл. 4.21 дан состав трех универсальных индикаторов (в
Г'раствора в 60 %-ном этиловом спирте).
Таблица 4.21
Индикатор
№1
Кя2
№3
Метиловый оранжевый
Метиловый желтый
Метиловый красный
Бромтимоловый синий
Нафтолфталеин
Крезолфталеин
Фенолфталеин
Ализариновый желтый G
0,03
0,15
0,30
0,35
0,02
0,03 '
0,40
0,40
0,20
0,20
0,01
0,013
0,16
0,24
0,16
0,40
0,30
Индикатор № 1 подходит в большинстве случаев. Растворы^
содержащие этот индикатор, имеют следующую окраску при
разных рН.
рН
10
Окраска
к—о
ж
ж—3
3—с
Ф
Окраски, отвечающие интервалу рН 3—8, устойчивы. Синяя
окраска при рН 9 достаточно устойчива, а фиолетовая при рН
10 быстро выцветает. В случае, если необходим стандарт*-для
оценки окраски при рН 10, индикатор прибавляют к очень
разбавленному раствору NHs. Недостатки индикатора № 1: пло-
jxo заметный переход окраски между рН 7,5 и 8,5 и
сравнительно тусклая окраска в области рН 9 — 9,5.
Для устранения этих недостатков в индикаторы №2 и №3
введен а-нафтолфталеин, окраска которого изменяется от
желтой До синей на интервале рН 7,3 —8,7. Но в присутствии а-наф-
116
толфталеина красная окраска при рН<:3 становится оранжевой.
Введение ализаринового желтого G в индикатор № 3
расширяет его применение до рН 12. В то же время замена метилового
оранжевого на метиловый желтый, желтая окраска которого
в щелочной области значительно бледнее, меньше искажает
синюю окраску индикатора. К сожалению, благодаря низкой
растворимости кислой формы метилового желтого в воде он
сравнительно быстро выпадает из кислых растворов.
В качестве дополнения к описанным универсальным
индикаторам для щелочной области рекомендуется смесь из обычного
раствора тимолфталеина (4 части) и тропеолина О (тимолового
8
Ю
12 14
1 \ПГК
2
3
4
5.
6
7
'Ж
5 7 S
Рис. 4.5
// 15
фиоУ1етового) (1 часть). На интервале рН 9—12 она дает
переходы: ж-)-з->-с->-ф.
В работе [45] предложен еще один универсальный
индикатор, пригодный для оценки рН на интервале от рН О до рН 14.
Состав универсального индикатора Чута и Камена (раствор
в метаноле) приводится в табл. 4.22, а на рис. 4.5 нанесены
интервалы перехода отдельных индикаторов, входящих в
универсальный, и окраски, которые каждый из этих индикаторов
' сообщает системе при различных значениях рН. Интервалы
Таблица 4.22
Индикатор
Концентрация,
(20 ч:)
Интервал
перехода
(рН)
1. Пентаметоксил
красный
2. Метиловый оранжевый
3. Метиловый красный
4. Бромтимоловый синий
5. о-Крезолфталеин
6. Фенолфталеин
7. Тринитробензол
50
8,5
22,0
100,0
30
35,5
125
1,86
3,50
5,04
7,24
8,8
9,4
8,94
9,55
13,0
1,2-3,2
3,0-4,5
4,4—6,2
6,0-7,6
8,0-10,0
8,1—10,2
12-14
117
перехода заштрихованы параллельными линиями. Стрелки
указывают область сохранения окраски.
Рассматривая на рис. 4.5 окраски отдельных индикаторов,
отвечающие различным целочисленным значениям рН, можно
получить некоторое представление о суммарных окрасках всей
индикаторной системы в целом.
Подобные схемы можно составить и для любого другого
универсального индикатора.
Расчеты, основанные на простейшей теории цветных
индикаторов. С помощью значений р/с и сведений относительно
интервалов перехода и окраски кислой и щелочной формы
различных индикаторов, приведенных в табл. 4.16, можно решать ряд
практически важных вопросов. Сюда относятся грубая оценка
рН системы по показаниям нескольких индикаторов, расчет
окраски индикатора в заданной системе, подбор наиболее
подходящего к данным условиям индикатора, вычисление
концентрации одноцветного индикатора, обеспечивающей
появление окраски при заданном рН.
Пример 4.20. Установить, какой рН имеет раствор, если бромтимоловый синий
дает в нем чисто-синюю окраску, а фенолфталеин остается бесцветным.
Решение: бромтимоловый синий окрашивается чисто-синий цвет, если
рН > 7, 6, фенолфталеин остается бесцветным при рМ < 8, 2, значит, искомый
7,6 < рН < 8,2.
Пример 4.21. Установить окраску водного раствора бромкрезолового пурпур-
ного, если сн,^=:2 . 10*"^ моль • л~~^
Решение: система включает
Hid ^ Н++ Id-, р/с = 6,4
в 2-10-^ - ~
;] 2* 10-^ —л h h
По закону действующих масс ——^ = 4.10 Попустим, что ^ <
<2 • 10""^ моль. л-*; тогда: ^ 8 • 10""** = 80 • 10*"'^, откуда Л = ^ 10"^^
чю составляет всего 9.10""б . (2 . Ю""*. Ю"^^^ = 4,5 % от 2 10'"'* моль .л^^
Поскольку Л == 9 - 10"-^ > 10—7 = (н+] в чистой воде, диссоциацию воды
можно не учитывать. Тогда [ Id~] = А = 9 . IQ-^, [Н Id) = 2 . 10~^ — 9 . Ю^^ „
= 2,00 • 10-^ — 0,09 . 10-^ = 1,91 • 10-^ моль • л"^
[HIdl/[IdI = 1,91 . 10--V9 . 10-^ = 21
и, следовательно, окраска раетвора будет чисто-красной.
Пример 4.22. Установить окраску раетвора, в котором «нАс — ^NaAc =
= 0,9 и Chjj = 4 . 10""'' мoль.л""^ причем Hid s бромфеноловый синий.
Решение. Поскольку с^д^ = i4 = 0,5 > в^^^^^ ^ ■ моль.л""^в можн®
считать, что основным источником Н'^'-ионов является равновесие
HAg»H+ + Ag- /С=10-'^'^^
о 0,5 — 0,9
11 0,5-/1 h 0,9+ &
118
По закону действующих масс ^ ^o,i^h^ Ю""^'^^. Если h С 0,5, то fi =
« 0,5Л0-^'^^/0,9, Igh =1,70 - 4,76 -1,95 = - 5,01 ='б,99, h = 9,8 10-^ мольХ
X л""^ - Найденное значение h является окончательным, так как
предположение, что h < 0,5 моль . л"~^ оправдалось. Оправдано и пренебрежение
Н+-ионами, которые дает HgO, так как 10""^ моль • л""' составляет примерно 10~^/(9,8х
ХЮ"^ . 10""^) 1 % от [Н+], которую дает НАс.
Поскольку далее Igh = — 5,01, рН = 5, то раствор будет казаться
окрашенным в чисто-пурпурный Цвет, так как рН =5 > 4,6—щелочной границы интервала
перехода бромфенолового синего.
Пример 4.23. Установить окраску водного раствора, в котором с^^^^^ = 0,3,
Cj^j^^ = 0,6, ^Hjid ~ • ^^""^ моль.л""^ если Hg Id = тимоловый синий-
Решение. Если за счет процесса протонизации аммиака [Н"^] заметно
снизится, а [ОН""] станет много больше, чем в чистой воде, то ОН"~-ионами, которые
дает HgO, можно будет при оценке рН пренебречь. Тогда
NH3 + Н+ « NH+, а = 10^'^^
или
NH+ 5=^ Н+ + NHs, К = 10-^'2*
с 0,3 — 0,6
[] 0,3 —Л h Ofi + h
По закону действующих масс ^ ^q^^j^^ = 10""^'^^.' Если /i < 0,3* to/i =
= ^10"^*^^= 10""^'^'*; значит, задача решена, так как действительно Л= 10~■^'^^<
<0,3 моль.л""^ Тогда рН = 9,54 попадает на второй интервал перехода
тимолового синего (8,0 — 9,6). Из равновесия Н Id""5=^ Н+ + Id^~, К = 10""^*^°
находим, что ттйЛ= - рН = 9,20 - 9,54 = - 0,34. Тогда
Iglld J
[Hid-] / [Id2-] = 0,46 или [Hid-] = 0,46 [Id2-J.
откуда = 0,46/ [ (1 + 0,46) . 10^^] = 31,3o/o и = 68,7 %. Что
касается Hgid, то, используя равновесие Hgid ^rt Н"^ + HId"~, для которого
р/с = 1,65, находим, что Ig [^^=у = р/С - рН = 1,65 — 9,54 = — 7,89 =8; 11,
откуда [Hgldl/[Hid-] = 1,3 . 10""^ |H2ld]=l,3 • 10-^[НId^J, что составляет-
- 1,3 ОО""^ .31,3 = 4,1.10""^ %-
Таким образом, результирующая окраска будет отвечать смеси 68,7 %
синих анионов Id^-, 31,3 % желтых гидроанионов Hid"- и 4 • 10-*^ %
красных нейтральных молекул НгИ; очевидно, что окраска ее будет зеленой.
Пример 4.24. Какой р/С должен иметь индикатор, чтобы обеспечить про'
межуточную окраску в растворе, где Cj^^^Cl = 0»^ и %Яз = 2 . ^ моль . л—^*
119
Решение. Полагая,'что доминирующую роль играет равновесие NHI" NHzf
находим
NH;J- —H+ + NH3, /C=10-'^'^^
с 0,8 — 2 . 10-^
[] 0,8—Л h + h
По закону действующих масс (2-^0 ^ -\'h) h ^ iq-9M, При условии /i<2 • 10""^
0,8 — h
2 . 10-2
Поскольку найденное значение h = 10—^'^^ одного порядка с [Н"^], кото*
рую дает вода, сразу пренебречь равновесием диссоциации HgO нельзя.
Записывая все равновесия, получаем по закону действующих масс:
NH3+H+;=tNH+ o = 10^'2^ [НН|] = oc/i,
НаО 5=i Н+ + ОН- ' W = lO-^^t [ОН-] = te/i-*-
По закону сохранения начальной концентрации имеем
^NH+ + ^NH, = 0,8 + 2 . 10-3 ^Q=A=aah+a=a{ah+ I)
По закону сохранения электронейтральности
[Н+] - [он-] + [NH+] _ [С1-] = 0. h-\ + -^-0.8 = О
ИЛИ o/i3_|-[(^-«0,8)o+l|/i2 —(0,8+сш)/1 —су==:0. После деления на с
и подстановки значений имеем /1» + 2 • lO^^fiZ— 4,57- lO-^^/i — 5,75 . 10"^^ = 0.
Из упрощенного решения известно, что h^^lO"^, Тогда л2<2. 10 ""^^2
и 4,57 . 10—^6^ > 5,75 • 10~^^ Отбрасывая первое и последнее слагаемое,
находим, что h = 2,285. 10"^ и рН = 6,64. Значит, искомый индикатор, дающий
промежуточную окраску, должен иметь р/<'=6,64. Этому требованию приближен-
н о удовлетворяет нейтральный красный, для которого р/С = 6,85.
Пример 4.25. В табл. 4.19 указано, что при рН 8,4 розовая окраска появляется
при начальной концентрации фенолфталеина (Hid) 1,2 • 10—^ моль • л—^ а при
рН 9,3 тот же результат достигается при c^^i^=z 1,8. 10"^ моль • л"^ Оценим
[Id"]» обеспечивающую получение надежно обнаруживаемой розовой окраски.
Используем равновесие Hid5=± Н+-|-Id"", р/С = 9,77. В первом случае
^Hid = 1э2 • 10-5 = 10"^'^^ h = 10-**^ моль . л"^ Полагая [Id-] = jc, нахо-
10—^*^х
ДИМ, что [Hid] == 10—^'^^—.jc. По закону действующих масс^^_4^92 _^^.= 10—^'^^^
откуда jc= [Id""] = 4,9 . 10—^ моль . л"^ Во втором случае с^^^ = 1,8 .10—^»
h = l0-^'3 моль • л^К Полагая [Id-] = t/, находим, что [Hid] = 10""^'^*—У-
Тогда 10-5,74^^ =10-^'77 и [Id-] =4,0. 10"^ моль . л"*-
120'
Полученные значения достаточно близки и хорошо согласуются с ранее
высказанным объяснением снижения надежно открываемой концентрации
фенолфталеина при возрастании рН.
Кривые диссоциации индикаторов. Полезную информацию дают
кривые диссоциации индикаторов в зависимости от рН (Н. Бьер-
рум, 1914).
Для любого индикатора
HId;=±H++Id-, К
при сиы = с можно принять, что при данном рН, а значит, и
/i[Id-] = ac, где а —доля Hid, превратившегося в Id". Тогда
[Hid] = (1 — а) с и по закону действующих масс
-^^^^K и \ga = \gK-\gih+K).
Если, наконец, Л = q /С, то Ig oi = —Ig (q — 1).
q
20
10
5
4
2
1,5
1
0,7
0,5
0,4
0,2
0,1
0,05
a
0,05
0,09
0,17
0,20
0,33
0,40
0,50
0,59
0.67
0,71
0,83
0,90
0,95
Абсциссы, отвечающие этим ординатам, получаются по
формуле рН = р/( — Igq. На рис. 4.6 приведены построенные таким
образом кривые диссоциации метилового оранжевого (/), бром-
крезолового пурпурного (2) и фенолфталеина (3).
121
5.
РАВНОВЕСИЯ КОМПЛЕКСООБРАЗОВАНИЯ
5.1. РОЛЬ КОМПЛЕКСООБРАЗОВАНИЯ В ХИМИИ
Комплексообразование широко применяется при химических
исследованиях и измерениях. Приведем основные случаи
использования.
За счет комплексообразования простой ион, не дающий
характерных реакций, можно перевести в комплексный, обладающий
характерными свойствами. Так аквоферри-исн при взаимодействии
с роданид-ионами дает ярко окрашенные родано-ферри-ионы:
РеЗ-ь+SCN-?=iFeSCN2+. Образование изо- и гетерополикислот
подобным же образом обеспечивает открытие и измерение
небольших концентраций РО4"', ASO4"', 8Юз~-ионов. Так, при
взаимодействии фосфат-иона с молибдатами в кислой среде
образуются тетрамолибдатофосфоранат- ионы
РО'Г+ 12Н2Мо04«Р(Моз01о)'Г+ I2H2O,
образующие с NHt-ионами малорастворимый желтый осадок.
Часто комплексообразование используют для «маскировки»
ионов, мешающих проведению какой-либо реакции. В качестве
примера можно привести осаждение желтого сульфата кадмия
в присутствии купро-ионов, замаскированных введением в систему
избытка цианид-ионов:
Си'++ 4CN~:piCu(CN)^.
Концентрация Си^^-ионов снижается при этом так сильно, что
образование черного осадка CuS становится невозможным даже
в насыщенном сероводородом растворе. Подобным же образом
введение Р04"~-ионов в раствор, содержащий одновременно Fe^"^-
и Со^"^-ионы, обеспечивает маскировку Ре^"^-ионов:
Ре'+ + НР04~ :г=± FeHPOt,
предотвращает появление красной окраски Ре5С№+ в присутст -
ВИИ SCN"--HOHOB и дает, таким образом, возможность
обнаруживать и измерять концентрацию Со^^-ионов, образующих с SCN"--
ионами комплекс голубого цвета.
Осаждение Ni^^-nona с помощью диметилглиоксима, Со^-Ь-
иона с помощью 1-нитрозо-2-нафтола — хорошо известные примеры
селективных реакций с органическими реагентами.
Используются методы разделения, основанные на отгонке
летучих комплексных соединений, например SiF4; экстракции,
например FeCls, HFeCU, различных дитизонатов, оксихинолина-
тов и т. п.
122
Комплексообразование используют для группового и
селективного растворения малорастворимых соединений. Примерами могут
служить следующие реакции:
HgS(T)+ S^-^HgSl",
А1(0Н)з (т) + OH~;=tAl(OH)T,
Hgb(T)+2I~^Hgir.
Приведенных примеров достаточно, чтобы показать важную
роль комплексообразования не только в химическом анализе,
но и в химии вообще.
5.2. РАВНОВЕСИЯ КОМПЛЕКСООБРАЗОВАНИЯ И ИХ
ПРЕДСТАВЛЕНИЕ С ПОМОЩЬЮ ХИМИЧЕСКИХ УРАВНЕНИЙ
Комплексообразование обычно считают избирательным
процессом замещения молекул растворителя (HL) в сольватной
оболочке металло-иона на какие-либо другие незаряженные или
заряженные частицы:
M(HL)?,-^ + пА ^ M(HL)n-nAr + nHL; (5.1)
M(HL)^n"^ + nY- ^ M(HL)n-nYr" + nHL; (5.2)
A\(HL)U^ + nHa-iA*" ^ M(HL)n^n (Ha^iA)r" + nHL. (5.3)
Записи и здесь упрощаются, евли принять, что
ЩНЬГ^^М!'^ (5.4)
Они переходят в уравнения
M'^-^+nA^MAS^; (5.5)
М'^-^ + пУ-^МУГ"; (5.6)
+ nHa^lA- <=± М(На-1 А)Г". (5.7)
В этих уравнениях А —любое незаряженное основание: nh3,
пиридин и т. п.; У~" — анион любой однопротонной сильной
кислоты: С!"", Bf"", N07 и т. п.; Ha-.iA" — (а — 1)-гидроанион а-
протонной кислоты НаА: HS07-, Н2РО7-, Н8А'"-(тригидроэтилен-
диаминтетраацетат)-ион и т. п.
Уравнения (5.3) и (5.7) нельзя считать уравнениями
комплексообразования, так как они, наряду и комплексообразованием,
включают процесс протонизации. В этом случае, например, (5.7)
составляется из уравнений
М'^^+пА^-^^-МАГ""; (5.8)
МАГ"" + п (а — 1) Н+ ^ М (На.,А)Г". (5.9)
где (5.9) является уравнением равновесия протонизации комп-
123
лексного MAn""" -аниона. Поскольку протонизация —процесс
ступенчатый, правильнее заменить (5.9) системой уравнений:
д^д(па^,)- ^ jH+ « HjMAjr^""^"\ (5.10)'
где]*=1, n(a—1). Так мы будем поступать в дальнейшем.
В качестве примера можно npiffiecm равновесия:
Cu2+ +A3-5=±CuA-, CuA- + H+«HCuA,
где А^- — цитрат-ион, а HCuA — цитратокупроатная кислота.
В рассмотренных случаях предполагалось, что при
ступенчатом комплексообразовании происходит последовательное
замещение молекул растворителя на отдельные лиганды (координируемые
частицы) в соотношении 1:1. Этот порядок может, однако,
нарушаться в случаях, когда замена частиц растворителя лигандом
сопровождается изменением структуры комплекса. Так, при
исследовании процессов образования HgCln^"-комплексов
установлено [57, с. 140—142], что HgCl+(aq)-HOH и HgCl2(aq) получаются
путем обычного замещения воды на С1~-ион без изменения
конфигурации. При последующем же переходе от трихлоро- к тет-
рахлорокомплексу изменяется структура от планарной для HgCir
к тетраэдрической для HgCl^"", что сопровождается освобождением
большего, чем обычно, числа молекул воды:
Hg(H20)3Cir + СГ ^ HgCl'r + ЗН2О
и возрастанием изменения энтропии.
Оксалаты, ацетаты, различные амины координируются
подобно галогенид-ионам и аммиаку. Иначе ведут себя
органические реагенты, способные давать внутрикомплексные
соединения. К их числу относятся: 1-нитрозо-2-нафтол, диметиглиоксим
и другие глиоксимины, 8-оксихинолин, дитизон,
этилендиаминтетрауксусная кислота и многие другие.
Все эти реагенты обязательно содержат по соседству друг
с другом: 1) солеобразующую группу (-СООН, -ОН, -SOsH^
= NOH), а в некоторых случаях — NH2 или -NRH, которая после
отщепления Н+-иона может сочетаться с металло-ионом за счет
главной валентности;
2) группу, включающую атом элемента (N, О, S) с неподелен-
ной парой электронов, т. е. парой электронов, не участвующих
во внутримолекулярной связи этого атома с молекулой
реагента. Эта электронная пара становится общей для атома-донора,
который делится ею с металло-ионом — акцептором. Этот вид
связи, который иногда обозначают стрелкой, направленной от
донора к акцептору, называется донорно-акцепторным или
координационным, в отличие от обычного, осуществляемого с
помощью электронов, поставляемых обоими соединяющими ато-»
мами на равных началах.
124
Наличие этих двух групп обеспечивает возможность
образования с металло-ионом так называемой клешневидной связи,
характерной для всех внутрикомплексных (хелатных —от
хела — клешня) соединений.
Высказано предположение [7], что органические реагенты,,
образующие с металло-ионами внутрикомплексные соединения,
сами по себе уже являются соединениями циклическими,
которым можно приписать общую формулу:
-Н
/
где R — радикал или молекулярный остаток; А — лиганд (ад-
денд), координируемый в данном случае входящим в молекулу
атомом водорода. Дополнительная связь, указанная стрелкой^
но после ее образования, не отличимая от ковалентной,
приводит к заметному возрастанию константы протонизации (а) для
реагента и констант комплексообразования для комплексов (Р)»
Образцом реагента, дающего внутримолекулярные
соединения, может служить 8-оксихинолин (оксин);
5 4
'ЧА/
8 N
I I
он
Он содержит оксигруппу и
особенно легко реагирует с металло-ионами,
гидроокиси которых плохо
растворимы. Так, с А13+ он дает
малорастворимое соединение темно-зеленого
цвета, расположение отдельных
элементов которого в пространстве
показано на рис. 5.1.
В этом случае равновесию
комплексообразования отвечает схема
А1(ОН2)б^ + ЗА~« А1Аз + 6Н2О
или А1з+ + ЗА-;=^А1Аз,
где вода не учитывается.
Рис. 5.1
\2Ъ
к комплексным соединениям относят также ионные ассоциаты,
образующиеся в растворителях с низкой относительной
диэлектрической проницаемостью (е < 40), например
(С2Н5)20Н+, FeCbf(C2H,),0]r.
Частицы, несущие отрицательные заряды, также могут стать
центрами, координирующими незаряженные частицы и катионы:
r+l2^ir р.
S4nS«Sn+, р„.
Но тогда и протонизацию А°^-+ nH+<=t НпА"-« можно
рассматривать, согласно Усановичу [34], как координацию оксоний-
иона различными основаниями А°^, среди которых ьозможны
и такие, у которых а = 0.
5.3. КОНСТАНТЫ РАВНОВЕСИЯ КОМПЛЕКСООБРАЗОВАНИЯ
Применение закона действующих масс к равновесиям
комплексообразования позволяет выражать активности и
равновесные концентрации сложных комплексных частиц через
активности более простых, из которых они образуются. Константы
многих равновесий комплексообразования измерены и табулированы.
В основных справочниках [36, 44, 64—66] значения констант
сгруппированы в отдельные таблицы, каждая из которых
включает константы равновесий координации какого-либо лиганда,
например аммиака или ацетат-иона, оксоний-ионом и
различными металло-ионами.
В [36] приводятся константы нестойкости (Ki), т. е. константы
ступенчатой диссоциации комплексных соединений, аналогичные
константам диссоциации кислот.
В [4, 44, 64—66] даны значения полных фп) и ступенчатых
(^п) констант образования.
Связь между этими величинами можно установить,
сопоставляя их значения для равновесий комплексообразования при
п=1,4, т. е. для координационного числа N=4:
7(4 М +A^tAA kl М+ А^МА pi
Къ МА +A<=iMA2 k2 М+2А^МА2 Рг
К2 МА2+А^МАз ks М+ЗА^МАз Рз ^ ^
Kl МАз+А^МА4 k. М + 4А^МА4 Р4.
В системе (5Л1) символы констант, стоящие справа, отвечают
верхним стрелкам равновесий, стоящие слева—нижним.
Поскольку любое равновесие M + nA^^tMAn может быть получено
путем суммирования п равновесий последовательного
комплексообразования
Pn=nA>j, (5.12)
т
константы комплексообразования p« и константы нестойкости К
должны быть связаны такой же зависимостью, как константы
протонизации оснований и последовательной диссоциации кислот:
Рп= П /(nVo, (5.13)
где N — координационное число. *
С помощью формул (5.12) и (5.13) всегда можно перейти от
значений и Ki к более удобным при расчетах полным
константам комплексообразования Рп.
Анионы, образующиеся при комплексообразовании, могут,
конечно, протонизоваться и координироваться. К сожалению,
процессы эти почти не изучались, а соответствующие константы
приводятся в справочниках лишь для немногих частиц,
например для Fe(CN)6'"- и Ре(СМ)б^-ионов.
По большей части процессы координации и протонизации
не разделяют, а изучают совместно и дают в статьях и
справочниках константы координации уже протонизированных частиц.
Так, в [66] в одной и той же таблице приводятся равновесия
и логарифмы констант:
Fe2++ Cit3-5=tFeCit-, lgPi = 3,08; (5.14)
Fe2+ + HCit2-^ HFeCit, Igx = 2,12. (5.15)
Комбинируя эти равновесия с равновесием протонизации
цитрат-иона, можно получить значение константы протонизации
цитрато-ферриат-иона:
Fe2+ + HCit2-;;=± HFeCit 2,12 Igx
Cit3- + H-b «HCit2- 5,39 Igai (5.16)
FeCit- «Fe2+ + Cit3- —3,08
—^gPi .
FeCit-+H-b :^HFeCit 4,43 lga2,
которое и следовало привести в таблицах. Из сопоставления
Igai и lga2 следует, что цитратоферриатная кислота сильнее
моногидроцитрат- иона.
Распределение информации о константах
комплексообразования по лигандам полезно в тех случаях, когда необходимо
быстро выяснить отношение того или иного комплексообразова-
теля к возможно большему числу металло-ионов. Для этого
можно использовать бланки таблицы Д. И. Менделеева с
нанесенными на них характеристиками образующихся комплексов.
Так, в табл. 5.1 даны значения IgPi для этилендиамин-тетра-
ацетато-комплексов различных металло-ионов, взятые из [66].
Этилендиаминтетраацетат-ион координируется почти всеми
металло-ионами, образуя достаточно прочные комплексы даже
с катионами щелочно-земельных металлов. Становится далее
127
Таблица 5.1
I
и
111
IV
V
VI
VII
vm
1
Ik
II
111
IV
V
VI
VII
VIII
J
2
з'
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
I
H
10,23
16,39
19,06
21,05
H
He
2
Li
2,79
Be
В
С
N
0
F
Ne
3
4
Na
1,66
Mg
9,1
Al
16,13
Si
P
s
CI
Ar
К
Ca
11,0
So
23,1
Ti
21,3
V
12,7
25,9
Cr
Mn
13,6
Fe
14,3
25,1
Co
16,21
36,0
Ni
18,6
Cu
18,8
Zn
16,3
Ga
24
Ge
As
Se
Br
Kr
5
Rb
Sr
8,8
Y
17,98
Zr
19,4
Nb
Mo
6,36
Ru
Rh
Pd
18,5
7^32
Cd
16,46
In
24,95
Sn
Sb
Те
1
X
6
Cs
Ba
7,8
H\
Та
W
Re
Os
Ir
Pt
Au
Hg
21,8
Tl
22,5
Pb
18,3
Bi
Po
Rn
7
Ra
Ac
Th
17,3
23,2
Pa
и
Np
Pu
Am
Cm
ясным, что добиться здесь некоторой селективности можно
лишь в случаях, отвечающих большим разницам в значениях
Igp. В то же время значения Igaj, приведенные в клетке Н,
указывают на дополнительную возможность регулировки
селективности за счет изменения рН. Если в клетке две цифры,
то нижняя отвечает металло-иону, несущему большее число
зарядов.
В табл. 5.2 взяты в рамку элементы, дающие дитизонато-
комплексы, извлекаемые из водного раствора в ССЦ.
Значения IgPjiX, приведенные в соответствующих клетках
таблицы, отвечают суммарным процессам:
+ [xHDz-<=iM(HDzV Pp.,
ЩНРг)^ ^M(HDz)t, X
Mf^++[xHDz-^M(HDzV р^Х,
где HDz —ридродитизонат-ион и значок 0 отвечает раствору
в ecu.
Сводки по лигандам очень удобны также при сопоставлении
прочности комплексов, образуемых разными лигандами с одной
и той же группой металло-ионов. В табл. 5.3 проводится такое
сопоставление для ацетато- и аминоацетато-комплексов ряда
металло-ионов [66, с. 364, 377]. Большие и на первый взгляд
неоправданные различия в значениях IgPn, наблюдаемые здесь,
относят как за счет «хелато-эффекта» (ацетат-ион дает
обычные, а анион амино-уксусной кислоты
Н2
НгС
\
\С
внутрикомплексные соединения), так и ряда других, в частности
квантово-химических причин, рассматриваемых в теории
координационных соединений [19, с. 177—186].
Лиганды, включающие только один донорный атом, например
галогенид-ионы, RCOO""-, ЗО^'^'-ионы, молекулы Н2О, R3N
называются монодентатными (однозубыми). Аминоацетат-кон (биден-
татный) относится к числу полидентатных ионов, т. е. ионов,
обладающих несколькими донорными группировками. Хелатными
такие ионы называют лишь в том случае, если их строение
допускает возможность образования цикла за счет одновременного
5 2-436 1 29
Таблица 5.2
1
11
m
IV
v
VI
VII
VIII
II
III
IV
V
VI
VI1
vm
)
7
8
10
11
12
13
14
15
16
17
18
1
H
He
2
Li
Be
В
С
N
0
F
Ne
3
Na
Mg
Al
Si
P
s
CI
Ar
4
Ca
Sr
Ti
Cr
Mn
Fe
Co
17,30
Ni
16,77
Cu
26,96
Zn
20,10
Ga
Ge
As
Se
Br
Kr
5
Rb
Sr
Y
Zr
Nb
Mo
Ru
Rh
Pd
1^64
Cd
19,54
In
30,94
Sn
15,35
Sb
Те
1
X
6
Cs
Ba
Hf
Та
W
Re
Os
' Ir
Pt
Au
Hg
44,15
Tl
5,36
Pb
18,66
Bi
36,96
Po
Rn
7
Ra
Ae
Th
Pa
и
Np
Pu
Am
Cm
Таблица 5.3
Ацетато-комплексы
Аминоацетато-комплексы
Ig а
Ig Pt
Ig P«
A Ig r<
Д Ig P,
Ag+
Ba2+
Co2+
Zn2+
4,7560
.0,36
0,41
1,70
1,36
2,24
1,80
157
9,7796
3,51
0,77
4,80
5,23
8,62
6,18
5,03
5,02
6,89
8,83
9,25
15,59
11,14
9,30
3,15
0,36
3,10
3,87
6,39
4,38
3,46
соединения двух или большего числа донорных группировок
данного лиганда с одним и тем же мёталло-ионом. Возможны,
однако, случаи, когда хелато-лиганд взаимодействует с металло-
ионом только одной какой-либо из своих донорных группировок,
т. е. ведет себя как монодентатный лиганд. Может быть, этим
и объясняется небольшое значение AlgPi для аминоацетато- и
ацетатобарий-иона.
В справочнике [4] значения IgPn и Ig^n представлены для
отдельных металло-ионов. Это дает возможность сразу
установить, какие лиганды координирует данный М^+-ион и какова
их стойкость. В результате можно всегда выбрать наиболее
подходящий вариант маскировки, выделить комплексы, наиболее
подходящие для выделения данного М^+-иона путем экстракции
и т. п. Наконец, с помощью таких таблиц гораздо легче
составлять иллюстративные схемы, характеризующие возможности
комплексообразования в отдельных группах и рядах таблицы
Д. И. Менделеева. В качестве примера в табл. 5.4 показаны
комплексные соединения с некоторыми неорганическими
лигандами по группам и рядам периодической системы элементов.
В первой строке каждой клетки вписан символ элемента, а
рядом с ним длина радиуса иона в нм. Если рядом стоят две
цифры, то первая относится к высшей, а вторая — к низшей
валентности. Так для С1 0,026 нм — кристаллографический радиус
CF+, а 0,181 нм —CI". Для Fe и Со даны, соответственно,гмЗ-Ь'
и Гм^+. Наличие формул НгО, ОН-, ... во второй строке клетки
указывает, что ионы соответствующего элемента координируют
эти частицы. Такой же смысл придается формулам третьей
строки. В ней символ Г означает —«галогенид-ион». Формул
в строке нет, если в справочниках соответствующие Igp
отсутствуют, т. с. комплексы не обнаружены или процесс
комплексообразования еще не изучался.
131
Таблица 5.4
Li 0,078
HA Oil'-
Be 0,034
HgO, OH-,
B 0,021
t 0,02
N 0,015
0^
0 0,009;
0,132
F 0,007;
0,133
Ne
Na 0,098
Mg 0.078 .
HaO, 0H~,
Al 0,057
H2O, 0H-,
Si 0,039
0H-, 0~s
F-
P 0,035
S 0,034
CI 0,026;
0,181
Ar
K 0,133
H2O
Ca 0,106
Ha®, 0H-|,
0,083 Sc
OH- F-,
r-
0,064; 0,069
Ti ha
0H-, 02-,
F-. F-
0,04;
0,061 V
OH--,
0^
0,035;
0,064 Cr
H2O,
0H-,
F-,r~
SCN-
0,046,
0,091
Mn H2O,
0H-,
0^-
SCN~,
N.H3
Fe 0,067:
0,083
H2O,
0H~,
0^~, F~,
r- CN-,
SCN~,
NH3
Co 0,064;
0,082
H2O,
0H~,
o2- r-,
CN-,
SCN,
NH3
Ni 0,078
H2O,
0H~,
02-, F~,
CN~,
SCN-,
NH3
0,070 Cu
H2O,
CN-, SCN-,
NH.,
0,083 Zn
CN-, SCN,-
NH3
Ga 0,062
OH- F-,
r-
Ge 0,044
0H-, 02-
As 0,047
OH-,
02-
Se 0,035
02-
Br 0,039;
0,196
q2-
Kr
Продолжение табл. 5.4
Rb 0,149
Н2О
Sr 0,127
HgO OH-
0,106 Y
0H-, F-
Г-
0,087 Zr
H2O, 0H~,
0,069 Nb
0,065 Mo
©2-
Tc
Ru 0,065
©2-
Rh 0,065
©2-
Pd 0,064
F-^
0,113 Ag
Н2О,0Н-,
CN-,
SCN-,
NHg
0,103 Cd
«2О, 0H~,
r~, CN-,
SCN-
NHg
In 0,092
0H~,
Г-, SCN-
Sn 0,074;
0,102 H2O,
0H-, ©2-,
SCN-
Sb 0,062
0H-,
©2-, F-
Te 0,056
©2-
1 0,050;
0,220
©2-
Xe
Cs 0,165
Ba 0,143
H2O, 0H-,
F~
0,122 La
0,082 Hf
0H-, o2-,
0,066 Та
0,065 W
©2-
Re
Os 0,067
©2-
Ir 0,066
©H-,
©2-
Pt 0,064
CN-,
NHg.
0,085; 0,137
Au
CN-,
SCN-,. NHj
0,112 Hg
H2O, 0H-,
Г- CN-,
SCN-,
NH^
Tl 0,105
HaO, 0H-,
CN-,
SCN-, NHg
Pb 0,084;
0,132 H2O
©H-", 02-,
SCN-
Bl 0,074;
0,120
Ha©.
©H-o
©2- F-
Po
At
Rn
На первый взгляд, табл. 5.4 дает даже меньше сведений, чем
таблицы значений IgPn- Но это не так. Сопоставление сведений
о комплексообразовании в рамках периодического закона с
привлечением данных о размерах радиусов ионов приводит кряду
важных заключений.
Из табл. 5.4 следует, что ионы практически всех элементов
координируют воду и продукты ее диссоциации, а некоторые —
ОН-- и 02-- или только 02--И0НЫ. Только Н2О координируют
однозарядные катионы щелочных металлов, у которых г>0,1 нм;
только 02--И0Н — многозарядные ионы с небольшими по
размерам радиусами, а значит, большими ионными потенциалами.
Увеличение радиуса при снижении числа зарядов всегда
приводит к исчезновению оксо-комплексов. Но тогда гидроксо- и
оксо-комплексы следует рассматривать как акво-комплексы
ионов отдельных элементов, подвергшиеся частичной или
полной ионизации:
Э(0Н2Гы-^^ Э(0Н2)ы^п(0Н)Г° ^ эсонГы-"" ^
- Э(ОН)ы^шОГ'^^'" - ЭОГ'^
В двух первых периодах в табл. 5.4, кроме акво-комплексов
в широком смысле этого слова, встречаются лишь фторо-ком-
плексы и при том только для элементов, ионы которых дают
гйдроксо-соединения. Это позволяет утверждать, что другие моно-
дентатные лиганды не могут конкурировать с молекулами воды,
активность которой даже в растворах средней концентрации
огромна. Особое положение р--иона связано, по-видимому, с
известной его способностью заменять ОН--ионы в минералах.
Однако тогда образование фторокомплексов нужно приписать
конкуренции р--ионов с ОН--ионами, а не с водой.
В ионах высокоэлектроотрицательных элементов О^- и р-
электроны связаны очень прочно. Они дают поэтому, в
основном за счет электростатических сил, очень прочные комплексы
с ионами шаровой симметрии коротких периодов таблицы
Менделеева. Ионйые оболочки непереходных элементов длинных
периодов гораздо сложнее, склонность к образованию гетеро-
полярных связей здесь убывает, возникают условия,
благоприятствующие возникновению гомеополярных связей,
координации, указанной неравенствами
1->Вг->С1->Р-, S2->02- NH3>H20 (5.17)
и возрастанию числа различных координируемых лигандов.
Наконец, как видно из табл. 5.4, для ионов переходных
элементов с ^-вакантными оболочками склонность к образованию
комплексов с легко поляризующимися лигандами становится
законом уже в первом большом периоде, подчиняясь неравенствам
(5.17). Разницы в значениях IgPn, обнаруживаемые при этом
134
для комплексов одного и того же лиганда с разными металло-
ионами, объясняют обычно доступностью d-орбиталей
координирующего иона, стехиометрическим расположением лигандов
вокруг него и другими квантово-химическими факторами
В фундаментальных справочных изданиях [36, 44, 64
очень часто вместо значений IgPn встречаются замечания:
«комплексы не обнаружены», «комплексов, видимо, нет»... и далее
следуют формулы. В разделах, относящихся к
кислотно-основным равновесиям, таких неопределенных указаний почти не
встречается.
Все недомолвки в таблицах Igpn следует отнести за счет
сложности процессов комплексообразования и трудностей,
возникающих при их изучении. Здесь, прежде всего, надо учесть,
что основной процесс комплексообразования сводится к замене
молекул Н2О в акво-ионе частицами лиганда:
м(0Н2)&+ 5=tM(OH2)St, + H20 рг/
М(0Н2)Й -Ь А « M(0H2)n-iA^+ Р21
в условиях, когда (Н20)>(А). Очевидно, что при p2i<Pii
обнаружить комплексообразование очень трудно. При Р21,
сравнимых с Рп, результаты могут оказаться сомнительными, и лишь
при P2i>Pib т. е. при высокой конкурентоспособности лиганда,
по сравнению с молекулами Н2О, координация становится
несомненным фактом. Очень часто, однако, этот первичный
процесс сопровождается ассоциацией, протонизацией образующихся
комплексных анионов, диссоциацией аминов [5]. Недоучет этих
процессов исключает возможность получения достоверных
значений констант равновесий, приводит к ошибкам и сомнениям.
5.4. МАТРИЦЫ СИСТЕМ, ВКЛЮЧАЮЩИХ РАВНОВЕСИЯ
КОМПЛЕКСООБРАЗОВАНИЯ. РАСЧЕТНЫЕ УРАВНЕНИЯ
Беглый просмотр справочников [4, 36, 44, 64, 65, 66] сразу
же приводит к заключению, что важнейшими равновесиями
комплексообразования в водных растворах являются
взаимодействия аквометалло-ионов с анионами слабых и сильных кислот
и незаряженными частицами.
Системы, в которых Mj^-^-uoh координирует Ha-iA--wo«6/.
Допустим, что CfAY = И что М^^^-ион не координирует ¥^)-ионы.
Предположим далее, что Сх^а = А. Тогда, кроме равновесий
комплексообразования, придется учесть равновесие растворителя,
ионизацию аквометалло-иона и протонизацию А*--иона и
комплексных анионов. Вся равновесная система точно описывается
с помощью матрицы:
135
м а
M^^+ А»- Н+
Ha_lA-
0 0—1
1 0 —к
0 1 j
1 0 0
0 0 j
п 0
0 1
он-
М(0Н)1^-''
М|Н._,А)Г"
HjMfH._,A|J-P
w
К
'и
В верхней строке матрицы записаны начальные концентрации
частиц, которые дают исходные вещества. Во второй строке —
отдельно Х"^ и YО)-ионы, не принимающие участия в
равновесиях; М^^+, А*- и Н+-ИОНЫ, из которых комбинируются все более
сложные частицы, и комплексные Ha-iA"^ и M(Ha-iA)J:j:p-HOHbi,
образующиеся из простейших, а затем выступающие как единое
целое в дальнейших процессах. В трех следующих строках
матрицы записаны равновесия ионизации растворителя и аквометал-
ло-ионов, а также протонизации А*--иона. В двух последних —
равновесия координации и протонизации комплексных анионов.
В последней колонке приведены обозначения констант равновесий.
Как и всегда, к = 1, ji'; j = 1, и п = 1, N. Для оценки общего
числа гидроанионов HjM(Ha-iA)J:j:p, а значит, и числа
соответствующих равновесий надо учесть, что комплексный M(Ha-iA)j;;:j.i-
шн может дать (при отсутствии катионных кислот) только
частицу кислоты HM(Ha-.iA)jxH-r, M(Ha«iA)^Iil2 — моногидроанион
HM(Ha~|A)jr+2 и ДВуХПрОТОННуЮ кислоту H2M(Ha-lA)j,^.2, т. е.
ш:его две частицы. У комплекса M(Ha-iA)S""" при п, равном N,
X е. координационному числу, p==N —[а. Такой комплексный
анион в результате протонизации даст N — |а — 1 гидроанионов
и (N — 11.)-протонную кислоту, т. е. всего N — продуктов
протонизации.
Эти соотношения можно представить в виде следующей схемы,
где в столбце j даны значения, которые пробегает этот индекс
при соответствующем значении р, а в столбце Шр — число
образующихся при этом протонизованны X частиц:
Формула комплекса р j
^т.^хк)^:^, 1 1
M(Ha^iA)^+2 2 1,2
Шр
1
2
336
M(Ha-lA)P+p
1. P
Из схемы следует, что для металло-иона, несущего jj. зарядов
при координационном числе N, р пробегает все значения от 1 до
N — [i, а соответствующие значения j изменяются для каждого
такого аниона от 1 до соответствующего р. Общее число
образующихся гидроанионов и р-протонных кислот равно
Smp= 1 + 2+ ... + Р+ ...-f(N —т) =
= t(N-t*)(N-f<^+l).
(5.18)
Теперь очевидно, что полный расчет равновесия в нашей
системе сводится к вычислению
{6+ix' + a'+N + l(N-|x)(N-ix+l)
равновесных концентраций ионов
-к
он-
[х' равновесных концентраций [М(ОН)к
N - [М (На-1 А)Г1 и 1 (N - (N - IX + 1) - [HjM (На-1А)Й51,
Для решения задачи можно записать 1 -f [х' + а' + N +
+ Y (N — jx) (N — |х + 1) уравнений, применяя к равновесиям,
записанным в шести строках матрицы, закон действующих масс;
4. уравнения получаются за счет закона сохранения начальных
концентраций, и, наконец, последнее дает закон сохранения
элёктронейтральности.
Используя приемы, применявшиеся при расчетах кислотно
основных равновесий, получаем четыре выражения закона
сохранения начальных концентраций:
с^^ = аА [Х+] (5.19); = (хМ = [у^,] (5.20);
См = М = 2 [М (0Н)Г1 + S [М (На-,А)Г1 +
к=0
п=1
Л4 = m
+ Д Д [H,M(H._,A)i-:j?p
P N-pi
(5.21)
137
а' N
0л = ^ = [Н|А1-«] 4- Е| п [М (На_,А)Г"] +
+ t 2' ((* + Р) [Н,М (На-1А)Й?р];
1=1 р=1
а' Г N
+ S (!* + Р) ?H,M(H,_iA)^^.pOi,p^+p (oa-iah'-y+Phi^ (5.22)
и выражение закона сохранения электронейтральности:
[Н+] - [ОН-] + [Х+] - [ Yo)] + (I. - к) [М (ОН)Г'] +
+ S (l*-n)[M(H«_,A)S-"] +
п=1
+ i X - ["'^ (H-iA)l.-?p] + 2 (j - а) [HjAJ-«] = О
или
<РнЛ - <роншй-14. аЛ - (iM 4- m (р, - к) mm)i,Vkh-^ +
N
p N-it
+ S (j — P) TH,M(H._,A)^+pO„p^+p (а._,аЛ«-1)1Ч-ра1
a'
+
+ ag(j —а)срн,Ав,Л' = 0.
(5.23)
Наконец, подставляя значение m из (5.21) в (5.22) и (5.23),
получаем систему с двумя неизвестными — а и Л:
n
Л = а |] cpHjAajAJ + М
1=0
ng,"V,-.A)„P°(°»--"^'-')° +
^ ?М(ОН)1,1Г)кЛЧ
к=0
р n—{X
+ S, <^ + 'fH,M(H,_,A),+p-,j^+p X
+ S ?M(H,_,A),;„("a-l«ft'-')"
Р N-p.
X (а,_,аЛ'"-')''+РА'
-; (5.24)
138
S (!^-к)9„(он)к%Л~'' +
(fah — (poHttiA-i ^a.A — \i.M + M
k=0
+ S ((^ - ") ?m(h._,a)„Pn C=.-l'"»"-'|"+
n=l
N
S
n=l
+ «S(J-a)cpH:AajAi-0. (5.25)
Для решения системы уравнений (5.24), (5.25) надо
использовать ЭВМ.
Если при ионизации аквометалло-иона (М»*+) образуются
многоядерные относительно М гидроксо- и оксо-комплексы, то
уравнение (5.21) станет нелинейным относительно т и исключение т
из (5.22) и (5.23) становится невозможным. В таких случаях
приходится решать с помощью ЭВМ систему уравнений (5.21),
(5.22) и (5.23). Найденные таким путем значениям, т и А
должны удовлетворять начальным условиям. Подставляя найденные
значения а, m и Л в соответствующие выражения закона
действующих масс, рассчитывают равновесные концентрации всех
остальных частиц, участвующих в равновесии.
Системы, в которых М^-^-ион координирует У^-ионы.
Допустим, что CmY(1)j^ = ^1 и Mi*+-hoh не координирует У-^-ионы.
Предположим далее, что лиганд вводитх^я в виде соли XY и
CxY = Тогда матрица, описывающая систему, имеет вид
y
у м
х+ УТ
0 0—1
0 1 —к
п 1 0
0 0 1
1
он-
м(он)г^
w
■%
р«
«и
138
в этом случае можно прямо использовать систему уравнений
^5J9)_ (5.25). При этом надо учесть, что
сх = У = [Х+], (5.26)
а'
а Y"- — анион сильной кислоты и заменить в (5.22) а ^w^pj^}^
на фу^/ = й что координируется У--ион, а в связи с этим
все произведения Oa-ifl^A"""^ должны быть заменены на у = (Y~).
Тогда (5.25) переходит в
S ({^-к)?м(ОН)|,^к^""^ +
?Hft — сронга^Л-1 + Г — [хМ + Л!
n
+ 2 (t^~n)<PMY„M" +
й >
р N—{1.
^"'7n-. ^у^^'О' (5.27)
а (5.24) в
n
S n?MY„P„J'" +
r = <p,t/+M^^^^ =^1—
к=0 п= I
(5.28)
И в этом случае уравнения (5.27) и (5.28) нелинейны
относительно неизвестных h и у; для решения такой системы
необходима ЭВМ.
Системы, в которых М^^-ион координирует незаряженные
частицы А. В системе, где ^my^ij^ = М и М^+-ион не коорди^хи-
рует У^)-ионы, а Л, матрица имеет вид
МО
а м
а м1*+н +
0 0—1
1 0 j
0 1 —к
п 1 0
©н-
h,aJ+
м(он)г''
w
"!
Чк
Здесь отпадают двойные суммы, вместо у появляется а,
исчезает блок катиона, число зарядов на всех комплексах остается
одним и тем же, равным {х.
В результате (5.25) упрощается и переходит в уравнение
а'
9нЛ — сронйуЛ-^ —\i.M + a J>HjA ^i^^ +
k=0
n=l
= 0, (5.29)
k=0 n=l
a вместо (5.24) появляется уравнение
N
a'
n=l
j=0
|^М(ОН,Л^-'+|^ТмА„Р„«"
. (5.30)
Таким образом, и в этом случае получается система
уравнений, решать которую надо на ЭВМ.
Упрощение записи конечных уравнений за счет использования
функций Я. Бьеррума. В качестве примера рассмотрим систему,
в которой М^^^-ион координирует ¥~-ион, представленную в виде
матрицы в разделе 5.4. Используя эту матрицу, можно сразу
записать уравнение закона сохранения электронейтральности
в виде алгебраической суммы блоков, начиная, как всегда,
с блока воды:
[Н+] - [ОН-] + [Х+] - [YJT)] - [Y-] + Мп., м = 0. (5.31)
Произведение начальной концентрации металло-иона (М) на
функцию Бьеррума металло-иона /Zz, м заменяет здесь алгебраическую
сумму всех зарядов, которые несут частицы, содержащие М^^^-ион.
По определению
141
где Zi —число зарядов, которые несет i-я частица, а [i] —ее
равновесная концентрация. В нашем случае все комплексы M'^+'Hona
одноядерные, выражения их равновесных концентраций содержат
(М^^+) = m в первой степени и после сокращения на т этой
величины Пг. м не содержит. Теперь, пользуясь обобщенными
формулами, приведенными в матрице, можно сразу записать:
- k=0 n=l
p N—(i
p N-ix -
(5.33)
Подс1ановка значения aZz, м из (5.33) в (5.31) сразу, минуя
стадию, аналогичную (5.23), дает уравнение (5.27).
5.5. НЕКОТОРЫЕ ЗАМЕЧАНИЯ
Даже при расчетах относительно простых равновесий
комплексообразования приходится использовать системы уравнений,
пугающие своей сложностью. В разобранных случаях
осложнения вносили сопутствующие равновесия: диссоциации аквометал-
ло-ионов и протонизации лигандов. Еще больших осложнений
можно ожидать для систем, в которых одновременно с кислотно-
основными равновесиями и комплексообразованием будут идти
окислительно-восстановительные процессы и возникать
гетерогенные равновесия.
При вычислениях на ЭВМ необходимость в результирующих
уравнениях типа (5.22) —(5.25) отпадает.
Для программирования достаточно исходных данных: сте-
хиометрической информации, значений параметров,
фиксированных значений начальных или равновесных концентраций, т. е.
сведений, которые могут быть представлены с помощью
обычной несколько расширенной матрицы равновесной системы.
В этом случае технические трудности, связанные с выводом и
преобразованием громоздких уравнений, устраняются.
Правильность результатов расчетов на ЭВМ в основном
определяется при этом достоверностью используемых
параметров- К сожалению, качество этих данных вызывает сомнения, а
142
многих параметров вообще нет. В связи с этим в качестве
первичного этапа при планировании экспериментальных
исследований сохраняют свое значение различные методы приближенных
вычислений. Среди них особенный интерес представляет метод
итеративного уточнения состава, успешно разрабатываемый
А. А. Бугаевским и его сотрудниками [3].
Новые перспективы в части расчетов ионных равновесий
открывает бурное равитие ионометрии, т. е. измерение активности
различных ионов с помощью рИон-метрических установок с ион-
селективными электродами. С каждым днем их число растет,
и, может быть, в недалеком будущем любую т= (М^+) и а=
= (А"-) можно будет измерять подобно (Н3О+) .Тогда даже
для систем с многоядерными гидроксометалло-комплексами
достаточно будет создать систему, начальные условия которой
совпадают с заданными для расчета, и измерить в ней значения
рН, рМ, и рА, а затем рассчитать равновесные концентрации
всех более сложных частиц с помощью выражений закона
действующих масс. Вывод сложных уравнений здесь исключается,
так как прямое измерение одной из активностей (Л, т, а)
приводит к снижению на единицу числа уравнений в системе.
К сожалению, рИон-метрические установки могут давать
правильные значения измеряемых активностей только после
соответствующей калибровки. Но химики пока не имеют
надежных методов, мер и стандартных справочных данных. В
результате ионометрия в данный момент может лишь упростить
расчеты, отнюдь .не обеспечивая возрастания правильности.
Упрощения при расчетах равновесий комплексообразования за
счет измерения рН. Пусть речь идет о самой сложной из
рассмотренных систем, когда М«^+-ион координирует Ha-iA--HOH.
Предположим, что до начала расчетов был приготовлен раствор,
в котором ^MY(i) ~ ^ХдА = ^> и в нсм измсрен рН. Это
значит, что стало известно значение h. В результате, вместо двух
уравнений (5.24) и (5.25), необходимых для расчета а и А, можно
оставить только (5.24). Если значения cpi известны или могут быть
вычислены, то Можно написать
S срн,АО,й^ = F и 2 Тм(он)к'^ЗкА""^ = G (5.34)
i=0 к=0
и считать эти величины известными. Если заменить далее
формульные индексы при ср индексами, указывающими число зарядов,
которые несет частица, то вместо (5.24) можно написать
A=^Fa+ М-
п=1
N ^
п=1
143
р N—|i
^^^715=;: (5-35)
+ S S 9,-р»иР.+р('«-.Л'-Г-'''А'а'^+''
j=l P=l
Это уравнение (Л^+1)-й степени относительно а всегда может
быть решено по Хорнеру. Дальнейший расчет равновесия при
известных h и а трудностей не представляет.
^ 5.6. НЕКОТОРЫЕ ТИПИЧНЫЕ ЗАДАЧИ, СВЯЗАННЫЕ С
РАВНОВЕСИЯМИ КОМПЛЕКСООБРАЗОВАНИЯ
Рассмотренные задачи относились к расчетам равновесий
в системах, где комплексообразование происходит 'при каких-
то случайных начальных условиях.
Гораздо чаще приходится проводить расчеты, связанные с
выяснением условий, которые необходимо создать, чтобы
равновесная концентрация той или иной частицы приняла некоторое
наперед заданное значение.
В качестве примера можно указать на расчет Су- = Y или
Сда- = Ау достаточной для снижения Сд^ц.+ = М до заданной
1МУ-+] = срм/п. Такая задача возникает при расчетах
маскирующего действия координируемого лиганда. Подобным же образом
приходится рассчитывать c^^j^ = М, которую надо создать при
фиксированном рН для снижения Су- == Y или Сда- = Л до
заданного значения (руу или (рд^.
Важной задачей является, расчет начальных условий,
обеспечивающих получение заданной равновесной концентрации какой-
либо частицы, например, нейтральной частицы МаАц. или МУц..
Такие расчеты необходимы при экстракционных методах
разделения, которые широко используются в химическом анализе и
современной технологии.
Расчет концентрации лиганда, достаточной для снижения
концентрации металло-иона в заданное число раз, при
фиксированном рН. Допустим, что надо найти Сда- = А или Су- = Y,
или сд = А такую, что при заданном рН (в условиях,
приведенных в 5.4) начальная концентрация металло-иона М снизится до
значения [M^^+J = (р^^т.
Здесь число неизвестных снижается на единицу, так как h
о
и т даны, а значения Л, Y или Л = сд надо найти. В связи
с этим уравнение закона сохранения электронейтральности не
используется и решение для всех трех случаев проводится по одной
и той же схеме.
144
Подставляют в соответствующее уравнение закона сохранения
М значения hum. При этом (5.21) переходит в
g + S ?1х-пРп(аа-1Л*"-0"«" +
п=1
+ t <Pj-p»ijP.+p (aa-ift-O'^+PAla^+pj; (5.36)
j=l p=l J
для комплексов М (Ha-iA)S"'", превращается в уравнение
M = m\G+ ^ ?,-пРпУ" + S S ?j-P^ijP.+pr+Pft^' (5.37)
I n==l j==l p==l J
для лиганда Y-, и в уравнение
М = m {g + cpix-npna"} - (5.38)
для лиганда А.
Во всех этих уравнениях G вычисляется по (5.34) и индексы
при ср указывают число зарядов на соответствующей частице.
Каждое из них легко решается относительно активности а =
= (Л"^-'), у или а = (А), а последующая подстановка найденного
значения и заданных величин hum в-выражения закона
сохранения Сда-, Су- и Сд, например в (5.22), дает искомые значения
этих величин.
Расчет рН, при котором равновесная концентрация металло-
иона или какого-либо его комплекса принимает заданное значение
при фиксированных М, А и Y. Если в системе, где cmYj, = М,
схаА = Л и лигандом является Ha-iA--HOH, задана [МУ-^] = ср^ш
или
;М (Ha-iA)rl = cp,_rPraLimarA<«-^>^
и надо найти рН, обеспечивающий заданную срмт, то число
уравнений снижается на единицу, а затем сводится к двум уравнениям:
V k=0 n=l
+ S Y ?j-paijP,+paa^±lV+P/l(«-U(.-l-P)+J j; (5.39)
j=l p=l J
■a" f N
Л = a S cpj«,ajW + m S ncpj,.npnaa-ia"W«-J>" +
j=0 I n=l
+ S ? (t^ + P) cpi-paijp,+paS±?a^+PA(«~0(i^-l-P)+J 1 (5.40)
j=l P=i J
145
Если речь идет о достижении определенной [M(Ha-iA)? "^l, то в
уравнения (5.39) и (5.40) вместо т войдет его значение
lM(Ha^iA)rn
т =
(5.41)
Полученная система нелинейна относительно а и А. Найти искомый
рН можно по Ньютону — Рафсону или на ЭВМ.
Та же задача в случае, когда лигандом является анион сильной
кислоты Y"-, сводится к системе уравнений:
off"' ^
lk=0 n=l
1=1 p=I J /
(5.42)
Y. = <pi«/ + m
(5.43)
+ S д (\^ + P) ?j-paiip,+pr+pft^}.
Здесь получить искомое решение, не пользуясь ЭВМ, можно лишь
в частных случаях. Если М^^+-ион не дает с Y~ анионных
комплексов, то двойные суммы, в (5.42) и (5.43) не понадобятся. Тогда
в (5.43) остается лишь одна неизвестная величина у. После ее
вычисления и подстановки в (5.42) вычисление h и рН трудностей
представить не может.
Второй случай реализуется для систем с р = I. Здесь вместо
двойных сумм в уравнение (5.42) войдет слагаемое сроарыу^А, а в
(5.43) —NcpoapNy^A. Подставляя выражение Н из (5.43) в (5.42),
можно рассчитать у, а затем —Л и рН.
Наконец, в случае, когда лигандом является основание Л,
получается система уравнений:
, S ф^-к^кЛ-" + S Тн^РпО" ; (5.44)
I к=0 п=1 J
а' ' N • i
а 2 (fioihi + m 2 ncpi^Pnan, (5.45)
1=0 п=1
М = т
А
решать которую в общем случае надо с помощью ЭВМ. Только
для систем, где основание Л дает одну катионную кислоту НА+^
уравнение (5.45) становится линейным относительной. Подстановка
выражения Н из (5.45) в (5.44) позволяет вычислить а, подставить
его в (5.45) и закончить вычисление Н и рН. Наибольшие
алгебраические трудности возникают здесь при подстановке значения h
в уравнение закона сохранения м.
146
при решении задач этого рода дополнительные измерения
активностей с помощью рИон-метрических установок помочь не могут,
так как создать равновесную систему, в которой равновесная
концентрация комплексной частицы или аквометалло-иона имеет
заданное значение, без предварительного расчета нельзя.
Расчет рН, nj^u котором «выход», т. е. равновесная концентрация
частицы заданного состава, достигнет максимального значения.
Рассмотрим схему решения для относительно простой системы,
в которой лигандом является ¥~-ион. Допустим, что все cpi= 1,
и что образующиеся анионные комплексы не протонизуются. Будем
искать рН, обеспечивающий максимальный выход незаряженной
частицы MY^. Такие задачи имеют существенное значение при
планировании экстракционных процессов.
Обозначим [MY^] = г = ^^ту^ (5.46) и запишем уравнения
закона сохранения начальных концентраций См = М и = Y:
М = m^S 7|кЛ-^ + m S Рпу" + m S РпУ" + г; (5.47)
л=0 п=1 ^+1
|х-1 N
Y = у + m S пРпГ + /п S прпГ + ]у^г. (5.48)
п=1
Значение т связано с h уравнением (5.47), а у и z—^ерез т.
Дифференцирование всех трех уравнений по h дает систему^
включающую соответствующие производные:
/п> + \imy^-^y = (5.49)
О = т' S ^кЛ-^ - m S кщк-^-^ + m'2 p„r/" + ту S пр„г/п-> + ,
к=0 к=0 п=1 п=1
N N
+ т' S р„(/" + ту S nPnf/"-^ + z; (5.50)
р.—1 |х—1 N
О =t/' + S пРпУ" + ту S п2рп(/п"-» + т' S пРпГ +
п=1 п=\
N
+ ту S п2рпГ/"~» + [хг. (5.51)
Наконец, уравнение 2=0 (5.52) определяет возможность
максимального выхода MY^.
При Z = 0 (5.49) дает зависимость между производными т' и у г.
/п' = -5^. (5.53)
Подстановка значений г' = О и т' из уравнения (5.53) в (5.51)
после сокращения на у фО и обычных преобразований дает
зависимость между тиув экстремальной точке:
147
m =
1 I
ix-l N N
. (5.54)
n=l ix-i-l n=l
так как слагаемое для л = [л равно нулю. Физический смысл
формулы (5.54) становится ясным после следующих
преобразований, которые легко записать с учетом условия <pi = 1,
эквивалентного равенству (i) = [i]:
У = [Y-] = S (l^ - п) nPn/nyn = S (l^ - n) n [MYr"].
n=l nel
Поскольку уравнения (5.46) — (5.48) справедливы и для
экстремальной точки, их можно использовать для дальнейшего решения
задачи. В частности, в уравнении (5.48) можно заменить на
^^^ту^ и записать его в виде
Y^y + m S nPn^". (5-55)
n==l
После подстановки значения т из уравнения (5.54) в (5.55)
получается уравнение, позволяющее вычислить значение у,
отвечающее максимальному значению z. Подстановка этого значения
у в (5.54) позволяет рассчитать т. Наконец, искомое значение
h дает уравнение (5.47).
В более сложных случаях, например в присутствии протонизу-
ющихся анионных комплексов, решение также возможно, но
расчеты приходится вести на ЭВМ.
5.7. КОНКРЕТНЫЕ ПРИМЕРЫ РАСЧЕТОВ РАВНОВЕСИЙ
В СИСТЕМАХ С КОМПЛЕКСООБРАЗОВАНИЕМ
В этом разделе рассматриваются расчеты равновесий,
указываются приемы предварительного исследования, выясняющие
возможность упрощения вычислений, приводятся примеры
ориентировочных оценок.
пример 5.1. Рассчитать равновесие в водном растворе, если в нем
^hno3 = ^h=^no. = ^=«'25hc„^,^=.„^ = M=0.1 моль.л-'. ■
Матрица равновесий системы может быть записана так:
0.25
0.1
0.2
\4 kl
NO-
Hg2+
GI—
н+
а
It
\
X
X
1
2
3
4
0
1
1
0
0
0
n
0
—I
-к
0
3
0
0
0
1
он-
Hg(0H)2-^
HgCl2-"
HjHgciiqiP
w
Pn
—14
-3,65
5,27
—7,72
12,78
-22,6
13,92
14,92
148
Из условия задачи (0^=0,25 моль . л""^) следует, что [ОН ] < [Н+]-
Из матрицы видно, что значения не измерялись; это исключает возможность
учета [HHgClj], [HHgCl7] и [H2HgCl4] и ведет к систематическим
погрешностям в значениях равновесных концентраций остальных частиц. Эти погрешности
могут быть относительно большими, так как при = 0,25 моль • л""' можно
ожидать существенного сдвига равновесий в сторону возрастания [HHgClg]
и [HgCI^], [H2HgCl4], [HHgCl7] и [HgCI^]. В кислой среде должна подав-
ляться ионизация аквомеркуро-иона. Для ориентировочной оценки учитывают
1-ю ступень ионизации при с^^ =0,1 и = 0.25 моль . л~^
Hg2+<=±HgOH-b + Н+ = -3,65
е 0.1 — 0,25
[] ОЛ — хх 0,25+x
По ЗДМ "^Qf^"'^''^ = l0-2•^^ при условии ;с < 0.1 < 0.25 х = Ю-^'^^- Ю'^Х
Х100'^= 10~^'^^ = 10^''^5 = 9 . Ю"^ или 0.09 % от М = 0.1 моль . л^^ Это
значит, что на начальной стадии расчета ионизацией аквомеркуро-иона можно
пренебречь. В нашей матрице остается только третья строка. Основные
погрешности возникают из-за вынужденного отбрасывания четвертой строки.
При всех сложных расчетах очень важно иметь предварительные
ориентировочные сведения о значениях вычисляемых величин.
В данном случае обращает на себя внимание высокая стабильность (lgP2 =
s=: 12,78) HgCl2. Интересно хотя бы приближенно оценить способность этого
соединения диспропорционировать, образуя HgCl"** и HgCl^-noHbi. Используя
набор значений Igp^, приведенный в матрице, и не раз описанные методы
пересчета констант равновесий, находим
HgCl2 ^HgCl+ + Cl-" -7,51
HgCr2 + Cl-5=tHgCl7 1,14
2HgCl2 5=i HgCl+ + HgCI^ - 6,37
e 0,1 — —
EI 0,1—2a; x x '
По ЗДМ ,7т4гГ2= 10~'*''. ;ТтЧг=10^'*^'^; 10-2's«5)_10-4.185.
(u,l — » U. 1 -~2^
x= [HgCl+] = [HgCIi"] = 10-^'>85 = 6.5 . 10-5, T. e. ~ 6.5 • IQ-^ % от M
Наконец, из выражения ЗДМ для первого из равновесий
Иногда таких результатов достаточно. Для получения более точных результатов
надо записать уравнения закона сохранения = М = 0,1 и c^j =2M =
= 0,2 моль . л""Ч
149
М = S [HgCl^-"] = m 2 (5.56)
n=0 n=0
2M = [cl-]+ S n[hgc\^-"] = y + m 5] (5.57)
n=0 n=0
Подстановка значения m из C5.56) в (5.57) дает после обычных преобразовании
уравнение
S РпУ""^' + S М (п -2) ?У = О, (5.58)
п==0 п=0
ИЛИ
^5 + 0,3/+ 1,725 . 10-V +2*238. 10-^V-2»238. IQ-^V-
-2.404.10-^^ = 0. (5.59)
Найденная выше оценка [С1"~] = 4,7 . 10""^ моль • л""^ позволяет утверждать,
что в уравнении (5.59) у < 0,3 и у < 1,725 . 10'^ 0,3-* = 5,75 . Ю^^. С учетом
этих неравенств (5.59) переходит в
1,297 . 10-V- Ь297 . lO-^y — 1,394 . Ю"-^^ = 0. (5.бо)
Решение уравнения (5.60) по Хорнеру дает t/== [С1"~] = 4,05 • 10""^ =
= 10-4.3925^ Дальнейший расчет т по (6.56) и [HgCI^-"] = p„m^" представим
в виде табл. 5.5. Как видим, ]^ [HgCl^-"] = 0,10000.
п=0
Это совпадение говорит лишь о том, что в вычислениях нет грубых
арифметических ошибок, значения же [HgCl^"""] нельзя считать достоверным
так как из-за недостатка информации не учитывалась протонизация хлоромер-
куроат-ионов.
Пример 5.2. Рассчитать равновесие в водном растворе, если в нем Cpe(ci04)s
— ^FeYe = = 1 • 10-2 и Cj^aF = ^NaA = ^ = 2,7 • 10-2 моль . л-1. Запишем
матрицу равновесий системы:
1
I
со
L
1
о
oT
х+
Y—
Fe3+
F—
H+
К
1
2
3
0
1
2
0
1
o'
0
0
1
n
—1
-1
—2
1
0
OH-
FeOH2+
Fe2(0H}|+
HF
FeF3-n
w
^1
0
Pn
—14
-2,17
-2,85
3,17
5,28
9,30
12,06
Начальные условия дают
= [X+] = л =2,1 . 10-2, ^ [CIO7I == 3M = 3 . 10-2 моль . л-^; (5.61)
^Fe=^ = (TFeOH^l^ Ч Tpe)+ 2?Fe,(OH).^2^^^ ^ +
3
(5.62)
150
Таблица 5.5
м
у
^2У'
m
0,1 .
lQ-4,3925
10—2,6500
1
2,24.10-^
100.7425
5,528
103,9950
\
9884
100.8775
7,541
10°
1
1,0
103.9956
t
9898
10-4,9956
[HgciM
|HgC17]
[HgCl2]
[HgCl+]
[Hg2+]
IQ—7,6456
2,3.10-^
10—4,2531
\
5,58.10-^
10-1,0006
9,986.10-2
10-4.1181
1
7,62.10-^
10—4.9956
1,01 10-^^
0,10000
6.10-2
NaH-
СЛ
2.10-2
Ы0-2
Cu2H-
6.10-2
ci-
0
1
2
1
0
0
0
0
n
0
—1
—к
—2
0
CuClP-
0
0
0
0
Cu(0H)2-^
Cu40H)i+
CuCl2-"
w
—14
-8,0
—10,95
2,80
?
-13,7
4,40
?
-27,3
4,89
-40,4
5,62
?
/
1
?
Ср = л = (9нр о л + <Рр) а + m S " fPeFn Pn«"- (5-63)
Наконец, по закону сохранения заряда
^fH^^ — <Р0Н«'Л~' +А-ЗМ + (2<PpeOHllft~' + ^Fe) « +
3
+ 4<pFe2(OH)2l2'«^A~^ - <Pf« + m S <3 - n) <РреР„Рп«" = (S'^^)
n=l
Из трех последних уравнений только (5.63) линейно относительно кит. Это
допускает исключение одной из этих неизвестных из двух остальных. Остается
нелинейная система.
Решение упрощается, если возможно непосредственное измерение хотя бы
одной из неизвестных величин.
Для заданной системы с помощью рР"-метрической установки найден pF =
= 2,5 и а = 10-2'^ моль . л""^
Теперь для решения достаточно уравнений (5.62) и (5.60). Допустим, что все
= 1. Тогда уравнение (5.63) можно использовать для оценки т в виде
т ■
А^а — aah
(5.65)
если выполняется условие ааН<^А^а (5.66). Уравнение (5.2) используется
для расчета h. Его удобно представить в виде
{[М - /п [ 1 -f ^ (l)]}/i2 - 7)1тЛ - 27)2т2 = О, (5.67)
где S(1)=SM" (5.68).
"TgP3= 12,06 I Ig Pa = 9.30 I lgPt = 5,28
a —a
10-2,5
104,56
36 310
104.30
19 950
102,78
602,6
ha
108 930
[Р^Рз]
39 900
[FeF2+]
602,6
56 863
149 433
105,1745
[FeF2+]
10-
,-5,9127
10-
2,2372
5,791.10"
1-3
10-
2,4972
3,183.10"
10-
4.0172
9,61.10
,—5
2,384 X
X 10
10-
,—2
,-1,6227
3,16.10-31
152
1,737.10-2 I 6.366.10-3 9,61.10-5
Схема расчета равновесного состава Fe (СЮ4)з — NaFHgO (пример 5.2)
представлена в виде табл. 5.6. Приведенное в ней значение h вычислено с
помощью уравнения (5.67), которое после подстановки значений М, т, 2 (1),
и 7]^ и обычных преобразований приняло вид — 10~■^'^^^^/l-—10~^^'^^^^ =а
= 0 (5.69.) Поскольку оказалось, что
aah = 5,72 . 10-® < (Л — а) = 2,384 . 10
,-2
(5.70)
найденное значение m можно считать правильным. Правильность расчетов
подтверждают также значения сумм (S), приведенные в 3-й и 1-й снизу строках
таблицы, так как первая практически совпадает с М в уравнении (5.62), а
вторая —с А в уравнении (5.63).
Пример 5.3. Рассчитать равновесие в водном растворе, если в нем ^^„(N03)2 =
= М = iQ-^ и Cj^aEi = У = 6 • 10—^ моль . л—^ Записываем матрицу
равновесий системы: (см. стр. 71).
Используя начальные условия и равновесия матрицы, сразу записываем
уравнения закона сохранения четшрех начальных концентраций и
электронейтральности:
^Na = [Na+] = F, с^оз = [NOi"] = 2М;
4 4
= М = m 2 ^к^^^Ч- ^22 f^^h-^ +/^11 ?n^";
k=0
(5.71)
(5.72)
n=l
(5.73)
Таблица 5.6
m
jQ—6,7972
[Fe3+]
[Fe0H2+]
[Fe2(OH)4+]
s
10-6,7972
JQ—3,0545
10-4.61^0
1,6.10-^
8,82.10-^
2,40.10-^
9,975.10-3
[HF] S
5,72.10-^
2,6998.10-2
153
к-о
+ mY, (2-n)
(5.74)
При составлении уравнений принято, что все <Pi = Протонизация хло-
рокупроат-ионов не учитывалась из-за отсутствия констант. Используя
уравнение (5.73), можно исключить иЗ системы т. Последующее решение
осложняется из-за нелинейности. Решение упрощается за счет измерения рС1,
осуществляемого с помощью рС1-метрической установки. При этом уравнение
(5.74) заменяется результатом измерения:
рС1 = 1,35, (С1-) = г/= 1о-1»35 ^ 4,467.10-2 (5.75), а уравнения (5.72) и
(5.73) записываются в следующем, приспособленном для дальнейших расчетов
виде:
т
Y^y _ 6.10-2-4,467.10-2
io-
1,8145
Snpy S(n) 2(n)
{M - m [1 + Е(1)1}т->Л^ - 10-«-V- (10-^3.7 _^ io-io.95^);,2
-10-
40.4
^0,
(5.76)
.10-27,3;j_
(5.77)
где
S (n) == Б n pjjt/" и S (1) = Spn^" (5.78). Схема расчета равновесного состава
системы Си(ЫОз)2 — NaCl — НоЬ (пример 5.3) приведена в табл. 5.7.
Таблица 5.7
у
Ну*
2
т
w(l+2)
1Q-1.35
1.8145
100.22 •
1,66
Х4
6,64
100.«4
6,92
хз
20,76
101.70
50,12
Х2
100,24
101.45
28,19
XI
28,19
86,89
155,83
10-4,0071
10-2,0632
Уравнение (5.74) принимает после подстановки в него значения т
следующий вид:
^4 _ 1о-«»^390дЗ 10-14.4158^2 _ 1о-28.439^_10-^^'539 _ о.
(5.79)
в нашей системе аквокупро-ион — слабая кислота—донор протонов, а
неучтенные, но существующие хлорокупроат-ионы — их акцепторы. В этих
условиях надо ожидать рН - 7. Но тогда fl» 10-13.100 ^ 10-13.6232 ^5 go) и
уравнение (5.79) переписывается в видеЮ-^'^ЗЭО;^ _ 1^-14.8158^0
моль-л""^ (5.82). Дальнейший расчет рав-
откуда h = 3,946.10-* = 10-^-^^38
новесия приведен в табл. 5.8.
Расчет закончен. Суммы, приведенные в последней колонке таблицы tj4-
но совпадают со значениями М и У, но рН = 7,40, отвечающий измеренному
рС1 = 1,35, мог возникнуть лишь за счет протонизации хлорокупроат-ионов.
Учесть этот процесс нельзя, так кяк он до сих пор не изучен — значит,
арифметическую правильность нельзя считать подтверждением правильности най*
денного равновесного состава.
154
Таблица 5.8
lgP4 = 5.62 1дРз = 4,89
Ig р2 = 4.40
Ig i3i = 2.80
у
т
h
1Q—1,35
4,467.10-2
10-4,0071
100,22
100.84
101.70
101.45
10-7.4038
CuCl|-]
[CuCl^]
[CuCl^]
[CuCl+]
1Q—4,0071
9,84.10-^
10-3.7871
1,633.10-^
Х4
6,532.10-4
10-3,1671
6,806-10-4
ХЗ
2,042.10-3
10-2.3071
4,931.10-3
X 2
9,862.10-3
10—2,5571
2,773.10-3
Xl
2,773.10-3
Продолжение табл. 5.8
Ig^li = —8,0
lg?l2--13.7 lgr)3 = -27.3 1 lg7), = -40,4 j Ig г)^^ =-10,95 |
Tl»/i-l
^2/»-2 1 ti3h-3 1 y^^h-^
^1«Л-2 1 2
10—0,5962
101.1076
10—5.0886
10-10.7848
1q3.8576
[CuOH+]
[Cu(0H)2]
[cu(0H)i:j
140")4-]
[Cn,{OH)l+]
iq—4,6033
2.493.10-^
10-2.8995
1,260.10-33
10-9.0957
8.10-'"
10-14,7919
1,6.10-^^
10-4,1566
6,97.10-^
0,01001
0,06000
Si
пример 5.4. Рассчитать равновесие в водном растворе, если в нем
^Zn(n05)i = 2.10-2 М, cnh3= 6-10-2= А и Cnh.n03= 0«5 = у моль.л-^ .Считая
все <Pi,= l записываем матрицу равновесий системы;
0.52 J2.IO-2
0,5в
NOJ-
Zn2-h
нн-
1
3
а
0
1
1
0
0
0
п
—1
0
1
он-
Zn0H+
w
h
—14
-8,96
2,18
9,24
4,43
6,74
8,70
Начальные условия и равновесия матрицы позволяют сразу записать
уравнения закона сохранения трех начальных концентраций {ci^q., c^f^t суммы с^щ
и и электронейтральности:
%Os = [NOr] - 2УИ + К = 0,52, (5.83)
4
= = + 1) m + m ]2 М" = 2.10-2 моль^л"^; (5.84)
h^wh-^^ — (2М + n + ("П^""* + 2) m + 2m Б Рп^" +
(5.85)
(5.86)
При последних уравнения линейны относительно т, но после исключения
т остается система из двух уравнений, нелинейных относительно неизвестных
а и /г.
W Р. = 8.70
Igr = 6,74
P, - 4.43
Ig = 2,18
h
а
Pefl*
Pifl
lQ-6,8
IQ—2,7006
10-2.1024
7,900.10-3
IQ—1.361
4,347.10-2
,0-e.97i2
0,10685
0,5206
1
0,30157
[NH3J
[Zn(NH3|2-i-]
[Z4NH3I2-H]
[Zn(NH3|i+]
[ZnfNHgf+l
1,922. Ю-''
10-3.9678
1,077-10-4
Х4
4,308.10-4
10-3.2271.
5 927.10-'
X3
1,778.10-3
10-2.8366
1,4567.10-3
X2
2,913.10-3
10-2.3860
4,112-10-3
Xl
4,112.10-3
156
Решение упрощается после измерения рН. В этом случае уравнение (5.86)
исключается, из (5.84) находим
подставляем в (5.85) и получаем расчетное уравнение
A+Y=Ga+M 2(п) [F + (5.88)
где F ^ tih-^+h G = а/г + 1, 2(1) = Ерпв", 2(п) = 2пр„а". (5.89)
В данном случае измерение дало рН = 6^8, h = 10""^'^ (5.90). После
подстановки этого значения, значений констант и обычных преобразований
уравнение (5.88) принимает вид:
^5 + 9,232. iQ-^a^ + 3,386.1(Г"^с^ + 2,009* V + 1,419.10-^Q — 4,070х
X 10-*2 = 0 (5.91). Решение по Хорнеру дает а = 1,9925^0-3 = 1о-2'700о
(5.92). Схема расчета равновесного состава системы Zn (N03)2 — NH3—
— NH4NO3 — HgO (пример 5.4) дана в табл. 5.9. Контрольные суммы в
последней колонке таблицы практически совпадают с начальными Ми A + Y
Пример 5.5. Рассчитать начальную концентрацию аммиака s= Л,
необходимую для снижения с^^ до [Ag+] = m == lO^^. если Са« mo. = М
р1 = 1,зап1
10""2 моль^л—1. Считая все 9I
:т = ш , если CAgNor
> записываем матрицу равновесий системы:
10-2
10-2
А
N07
Ag+
0
1
0
1
0
0
1
п
н+
—1
—к
1
0
он-
NH+
AgCNH3}+
i
1
2
w
0
к
—14
-П,7
9,24
3,32
—23,8
7,23
Таблица 5.9
i.Tj = —8.96 Ig a== 9.24
— 1
о Ы
0,45979
100.1664
t
1,46679
10-
1,8654
[Zn2+1
10-'
.2,16
102.44
|ZnOH+]
1,363.10-
L—2
4.0255
10-
9,43 10-
[NHf]
JO—0,2606
0,5487
0i01999
0,5599
157
Начальные условия и равновесия матрицы приводят к следующим
уравнениям законов сохранения:
^ыОз = ^= [NO^] = lO-2; (5.93)
2 2
Сд^ = Л1 = m 2 -^Ji-^ + m S Pn«" = ^0~'»
k=0 n=l
2
^ынз = Л = (ah -f 1) a + m 2 np„an. (5.95)
n=l
2 2
— суЛ-^ — Л1 + m 2 (I — k) + m E Pn«" + = 0-
k=0 n=l
-1
В уравнении (5.96) [Н+] = h опущена, так как в аммиачной среде wh'
= [0Н~" ] > /i. Хотят= Ю"^ известно, простого пути к решению нет.
Необходим предварительный анализ системы с химических и математических
позиций. Анализ можно начать с приближенной оценки А, необходимой для
снижения М = 10—2 ДА т= IQT^ моль.л—^ и расчета Л, если образуется только
Ag(NH3)+:
Ag+ + 2NH3 :?=±Ag(NH3)+ lgP2 = 7,23
о Ж =10-2 ^ —
I ] m=lO-^ Л —2.10-2 10-2
-.(10-2-10-5) ,^10-2
10—5 (л— 2.10—2)2
По закону действующих масс ^^^2 = Ю-^'23^ [NHsl а= (Л—
-2.10-2) = 10-2-^^5 ^7,7.10-3, Л-2,8.10-2 мoль.л-^
Для оценки h используем в качестве с^щ найденную [ЫНз]= 7,7.10—^
и равновесия:
NH3 + н+ « NH+ Iga = 9,24
ЩО :^Н-^ +0Н- Iga; = — 14
NH3 + H20eNH+ + OH- ig/C = —4,76
с 7,7.10-^ * - -
I ] 7,7.10-^-0) О) О)
0)2
По закону действующих масс |q-3 _ =10-^'^®, ш = [ОН"] = 10-3.4475
h =[Н+] = 10-^°'552,
Приближенные расчеты привели к оценке значения а и показали, что
чем больше сдвинуто равновесие в сторону образования Ag (NHg)^, тем
ближе друг к другу значения [NHJ^] = oah и [ОН—] =wh~^ и тем меньше
разность между этими величинами.
Разность aah — wh"^ входит в уравнение (5.96). Если в то же уравнение
подставить значение М из (5.94), то оказывается, что
2
гп У k7)J= oa/i —< Ю-^'^^ (5.97)
к=о
158
так как разность двух величин одного порядка всегда много меньше каждой
из них, взятых порознь. Но тогда
т
к=о
к=0
(5.98)
и первым слагаемым в правой части можно пренебречь. Само (5.94)
превращается в
, Pi М
= 0, 10-3.91 а-10-^*23 _0
(5.99)
и приводит к значению а = lo^^'^l^^ = 7,612.Ю^З.
Для уточненного расчета h используем (5.94). После обычных
преобразований оно переходит в аа^з — + tm\— 2if)2m = О, или 1^ — lO""^I'l^^^A _
_ lQ-35.6205 ^ Q (5^00). Согласно предварительной оценке Л - Ю-"^^'^^ >
> 10-^*'^^^^ Тогда из (5.100) h = io-iO'5604^ ^^j^ _ Iq-3.4389 ^ 3.540. io~4^
whr^ = 10-3.4396 ^ 3 534. jo-io, cah — whT^ = 6-10-7 и условия (5.97) и (5.98)
можно считать оправданными.
Дальнейший расчет А по формуле (5.95) приводится в табл. 5.10.
Уточненное значение Л = 3.23-10—2 намного превышает предварительную оценку
Л =2,8.10-2 моль-л—!^ но и его нельзя считать достоверньин, так как было
принято, что все <Pi = 1.
В этом примере задано значение т, входящее во все уравнения в первой
степени. Это не привело к немедленному упрощению решения. Заметные
упрощения получаются, как правило, в тех случаях, когда известны или заданы
вначения величин, входящих в уравнения в высоких степенях.
Пример 5.6. Рассчитать начальную концентрацию KI,Cj^l = К,
необходимую для снижения c^j^j До [Cd2+] = m = Ю-®, если ^сА{С\О^Уг =М = 2- lO^^
моль.л—Считая все <pj = 1. записываем матрицу равновесий системы:
1"
О
t
3
о
1
1
о
—1
-к
о
j
о
о
о
1
он-
|cd(0H|r
Cdl„2-"
•^к
-14
1-9,30
2,28
?
Л9,17|
3,92
?
|-32,48|
5,0
?
е \
а еатем уравнения законов сохранения:
= К = [К+] (5.101); ср,о^ =2М =[С107] = 4-10-2; (5Л02)
Ccd = ^^fn у t)i,/i-^ + w У Рр£/ = 2 . 10-2 моль . л-1; /5.103)
к=0 .^1
159
Таблица 5.10
Ig а = 9,24
Ig p2 = 7.32
Ig P, = 3.32
т
k
а
aft
ah-i-l
(ah+l) a
2p2ma2
Pima
A
10-5
lQ—10,5604
10-2.1185
jO-1.3204
1,782.10-2
JOO.0202
t
1,04782
10—2.0983
7,973.10-3
IQ-1,6160
1
2,421.10-2
10-3.7985
1
1
1,59.10-4
0,0323
Та бл ИЦ a 5.11
lg4i = —32,4
|g,,j =-19.17
Ig 1), = —9.30
lift-'
s
m S
10-^
10-11,4
4.10-'2
10-5.17
6,76.10-^
10-2.30
5,01.10-2
10'
1,000
1,005
1,005-10-»
Cj = K = ^ + m 5] np„i^";
(5.104)
3 4
Я — cy/i-^ +r — + m 5] (2 — k) ti^^h-^ + m ^ (2-n) p = 0. (5.105)
k=0 n«=l
Система уравнений (5.102) — (5.105) линейна относительно заданной
[C(j2+] = m, но это не облегчает решения. Как и в предыдущем примере,
можно обойтись без ЭВМ, если окажется, что
к=0
-к
< М = 2 с 10-2 моль, л-^
(5.106)
Для доказательства правильности (5.106) необходима оценка Л. Поскольку
в данном случае ожидать отклонения от рН = 7 можно лишь из-за ионизации
аквокадмий-иона, а его
Cd2+
„—1
: m == 10"~* моль • л""'^ очевидно, что для
рассматриваемой системы рН 7.' Несколько нагляднее можно это доказать путем
вычисления степени ионизации аквокадмий-иона иа первой ступени:
Cd2+ 5=tCdOH++H+ lg^i = -9,30
сМ=10-«
11(1—a)iW аМ аМ
По закону действующих масс
ЧТО при М = Ю-* моль • л—^ приводит к а = 10""°»^^ ь,23.
В этом случае вклад в общую (Н**"] за счет ионизации Cd2"*'(aq) составит
всего - 2,5 . 10""^ моль • л~^-
Теперь, считая h= 10""^, можно проверить (5.106). Схема расчета
равновесной концентрации С^2+-иона системы Cd(C104)a—KI —НаО (пример 5.6)
3
приведена в табл. 5.11. Найденное значение m ]^ ^к'*"^ = ^»^^^ • ^^""^ ^ ^ ~
=2 • IQT^ моль • л-—^ Это позволит использовать (5.103) для расчета у. После
подстановки значений М, /к, lgP„ и обычных преобразований получаем
у'^ + 7,943 . 10-2 уЪ ^ g . iQ-3^ ^ 1 . 1q-4^ _ 1^5gg ^ q. (5.107)
Расчет no Хорнеру дает ^ = 1,1016 = 10°»°^2°моль • д-^ Схема расчета
начальной концентрации KI в системе Cd (СЮ4)2—KI — HgO приведена
в табл. 5.12.
Таблица 5.12
Ig Р4 =6,10
Ig Ра = 5.0
Ig Р2 = 3.92
Igp, = 2.28
у
ЗР,А,»
^гУ
2
m 2
1q0,0420
Ы016
106.8701
1
7,415.10^
105.6031
1
4,010.10^
104.3050
2,018.10*
102,3220
1
2,10.102
7,8364.10^
0,0784
1.18
б 2-436
161
Найденное значение У нельзя- считать правильным, поскольку из-за
отсутствия данных не была учтена протонизация Cdl^IjIp-noHOB и было принято, что
все 9i == 1. В этой части лучшей оценки значения Y ЭВМ обеспечить не сможет.
Пример 5.7. Рассчитать. рН, который надо создать в системе, где
^Fe(Ci04)8 = ^ = 0,1, CNajHPO* ~ МОЛЬ • л^1, чтобы обеспечить FeHPO'^] =
= aAf, для случая q = 3, а = 0,9. Считая все = 1, записываем матрицу
равновесий системы:
0.6
0,3
0.1
0,3
Na+
(SIO*:
н+
0
1
2
О
1
О
О
О
1
о
—1
—1
—2
j
о
о
о
о
о
1
он-
РеОН2+
Fe2(0H)|+
н^ро^-з
FeHPO+
"^22
Р
—14
1-2,17
—2,85
12,32
9,35
19,53
21,68
Поскольку [peHPCj*] задана, для решения задачи достаточно двух
уравнений закона сохранения и уравнения образования FeHPO^l
3
Cpo^ = qAf = а 5] a^hJ + аМ;
j=0
[РеНРО+] = =: рс,таЛ.
Подставляя а из (5.110) в (5.109), находим, что
(5.108)
(5.109)
(5.110)
т •■
i=0
(5.111)
Тогда из (5.108) и (5.111) после обычных преобразований получаем уравнение
3
(1 _ a)(q - afrnf^y^ а (q - а) ^o.h^-^i + Л) Ш +
1=0
(5.112)
переходящее в + 10-^*589 10-Ьб434;^4^ 10-11.5294 ;,3^1о-21.6249д2 ^
-Ь 10-^3,6464 10-46,2675 _ о (5.113).
в (5.113)—две перемены знака и, следовательно, два положительных
корня. Можно ожидать при этом, что/г < 10""^'^^89 ^ joO.2155^
таких; низких рН [НРО^""] ничтожна. Тогда^ отбрасывая два первых слага-
162
емых, приводим (5.113) к виду Л* — 1,3003 • Ю^^^ —Л,0437 • Ю'^^ h^ —
— 9,932. 10"-32/i_ 2,376. 10-45^0 (5.114).
Расчёт по Хорнеру дает ответ: h = 1,$61 • 10""^° моль • л~'; рН = 9,73.
Пример 5.8. Иногда приходится вести расчеты, связанные с разрушением
образовавшихся комплексов или предотвращением комплексообразования.
В качестве примера можно рассчитать Сдсю^г^К, которую надо создать
в системе, где €^^^00^)2 = = 2 • Ю^^^ ^kcn = 4 • М = 8 • 10-2 моль-л""Л
чтобы обеспечить [Cd2+] = m = 1,2 • 10""2 моль-л""'- Считая все ср. = 1,
записываем матрицу равновесий системы:
if
о
о
S
x
о
1
о
1
О
-*1
—к
1
о
О
6
О
о
1
Cd(0H)2-^
HCN
Cd |CN)
HiCd{CN)i-P
.2—n
n
w
a
К
—14
1-9,301-19.17|
9,35
5,54,
?
10,62
|-32,48|
15,30
?
18,85
?
Поскольку m = [Cd2+] = io--l'9208 j^Qjjj^. JJ 1 задана, неизвестными
являются m, Л и У. Для их расчета достаточно трех уравнений закона сохранения:
•■3 4 . . .. ' _
= М = m 2 fi^h-^ + m 5] Рпа"; (5.115)
к=0 п=1
4
CcN=4M = (a/i+l)a + m2nPn«"; (5.П6)
n=l
3
c„ = r=r(o/ia4-/i)—m У Ь^Л-*^.
к=о (5.117)
В (5.117) учтен расход Н"*'-ионов на протонизацию CN"" ионов и Н+-ионы,
поступающие в систему за счет ионизации аквокадмий-ионов.
На первый взгляд, решение системы надо вести на ЭВМ. Можно
предположить, однако, что в кислой среде ионизация аквокадмий-ионов будет
подавляться и в результате окажется, что
3
! m у; Yii,/i-^<M. (5.118)
к=0
Тогда'(5.115) можно записать в виде ^40* + РзО^ + Pg^^ + Pl^—^'"""^ = О
или а* + 2,818.. 10^V + 5,624 • 10-^02 + 4,898 . 10"— 2,355 • lO"= О
(5.119)
6*
163
2
Таблица 5.13
igp4 = ie.8
= 15,30
10,60
!!b7
а
ЗРзО»
1(я)
4Уи - 1 •
oa
h
3,407 10-®
10-2.4183
3,817.10-^'
10-0,62.^7
0,2367
10-0.0342
0,9242
100,0724
1.1813
100.3703
2,з\б
10—1.5505
i
2,815 10-"^
2,815 10-2
10-1.2853
5,185.10-2
,03,вв24
10-5. J677
Таблица 5.14
•g 4i = —32,48
Ig t|, =-19,17
lg4i = -9,30
—ft
aah
3tijft-3
24.ft-2
2
m 2
Y
10-6,167?
e,8.io-^
10-l.2»53
i
0,05185
10-16,4998
1
3,164.10-'^
10-8.6338
2,927.10-»
10-4,1323
\
7,373-10-^
10-4,1323
7,373 10-*
10-6,0531
8,85-10-'
0,052
Решая (5.119) по Хорнеру, находим, что
а = 3,4066 . 10-^ = 10-5.4676 ^^^^ ^
Последующий расчет h с помощью (5.116), переписанного в виде
4M —
fl + m5](n)
(5.120)
4
где 2(п) = 2"Рпа" и Ig 3,8824, приводится в табл. 5.13.
п=1
Расчет Y по (5. 117) дан в табл. 5.14 (например 5.8). Найденное в ней
значение 8,85 - 10~^ < М = 2 • 10""^ моль • л~*, и, поскольку условие
(5.118) выполняется, вычисление можно считать законным.
5.8. Краткие выводы. Принятая система матриц равновесий
и записи уравнений в виде сумм специфических блоков
оправдала себя при рассмотрении систем, в которых наряду с
кислотно-основными устанавливаются равновесия
комплексообразования.
Показано, что применение ЭВМ обеспечивает возможность
любых расчетов и в этих более сложных случаях. Оказалось,
что дополнительные ионометрические измерения значительно
упрощают решение некоторых (но не всех) задач.
Отсутствие сведений о константах протонизации для
большинства комплексных анионов и о коэффициентах активности
исключает возможность проведения точных расчетов
рассмотренного типа для технически важных систем.
6.
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ
РАВНОВЕСИЯ
6.1. ОБЩИЕ СВЕДЕНИЯ ОБ ОКИСЛИТЕЛЬНО-
ВОССТАНОВИТЕЛЬНЫХ РАВНОВЕСИЯХ
Окислительно-восстановительные равновесия очень часто
используются при химических измерениях. Так, очень близкие по свойствам
А13+ и СгЗ+-ионы можно легко разделить, используя возможность
окисления СгЗ+ в СгО^^-ион. Практически бесцветный Мп^^-ион
можно окислить, а затем открыть и даже измерить его
концентрацию в виде пурпурного МпОТ-иона. Прибавляя окрашенный
раствор КМПО4 к раствору, содержащему легко окисляющееся
вещество, можно оценить концентрацию последнего, зная
концентрацию раствора КМПО4 и объем его, пошедший на окисление.
Во всех таких случаях надо подобрать реагенты-окислители
и восстановители, действующие избирательно, и, по
возможности, количественно. Конечно, решить такую задачу можно
165
путем ряда проб, но при наличии огромного числа веществ,
которые можно использовать в качестве окислителей и
восстановителей, этот, путь нельзя считать рациональным. И здесь полезны
предварительные расчеты, связанные с общей теорией
окислительно-восстановительных процессов.
История развития этой теории дана в работе [27]. Мы же
используем лишь важнейшие соображения и работы, ставшие
основой современных представлений в этом очень важном
разделе теории ионных равновесий.
Прежде, всего отметим, что химики долгоевремя
рассматривали реакции замещения или вытеснения, не связывая их с уже
известным их применением в электрохимии. Так, А. Вольта,
опровергая виталистическую точку зрения Гальвани о «животном
электричестве», подчеркивал, что изобретенный им столб
(1800 г.) является «искусственным электрическим органом».
Он не подметил, однако, связи между возникновением зарядов
и тока и химическими процессами, а ряд напряжений Риттера
(1805 г.) использовал только при выборе металлов для столба.
На протяжении первых тринадцати лет 19 в. Г, Дэви, поль^
зуясь вольтовым столбом, получил большинство щелочных и
щелочно-земельных металлов, а М. Фарадей к 1834 г. открыл
законы электролиза. Но химическая наука не достигла еще
к этому времени такого развития, которое позволило бы
установить что-то общее между выделением металлического калия
на катоде и вытеснением одного металла из водного раствора
его соли другим.
На существование связи между вытеснением и электролизом
впервые указал в 1863 г. профессор Харьковского
университета Н. Н. Бекетов. Он пришел к выводу, что «всякое
восстановление одного металла другим совершается в виде электролиза;
токи же, происходящие при этом, суть только следствие
химического действия, которому они дают только известное
(полярное) направление».
Бекетов не привел опытных доказательств правильности
своих предположений, поскольку основной своей • задачей считал
изучение выхода вытесняемого, в зависимости от массы
вытеснителя. В частности, он показал, что под давлением в
несколько атмосфер водород способен вытеснять Ag и Hg из растроров
некоторых их солей. Эта работа не вызвала должнЬго отклика,
так как была опубликована за четыре года до появления
монографии Гульдберга и Вааге, посвященный закону действующих
масс (1867 г.).
Последующие электрохимические работы, связанные с
теорией С. Аррениуса (1883 г.), были предприняты в основном ради
объяснения того факта, что одни металлы, будучи
погруженными в раствор их собственной соли, заряжаются положительно,
а другие — отрицательно.
166
в. Нернст искал противоборствующие силы, вызывающие,
с одной стороны, стремление металла выйти из раствора, а
с другой —уйти в раствор (1889 г.). В качестве первой силы
он избрал осмотическое давление, второй — электролитическую
упругость растворения, которую он изобрел. Последующие
исследования показали, что представления Нернста об
электродном процессе далеки от истины, подход же его к самому
выводу зависимости между потенциалом на электроде и жидкой
фазой был правильным и различные варианты его формулы
используются до сих пор.
Уравнение окислительного потенциала в привычной для нас
концентрационной форме впервые получил Р. Петере (1898 г.).
Формула Петерса была важным достижением, так как впервые
связывала потенциал отдельного электрода с уже принятым
всеми химиками законом действующих масс. Однако
электрохимики не смогли еще объяснить, почему при растворении
металла в раствор переходят его ионы, а не атомы, и как могут
металло-ионы выделяться на поверхности металла, сообщая ей
положительный заряд. Отсутствие ясности в этом вопросе
давало повод к сомнениям в формулах и даже в самой теории
электролитической диссоциации.
Так, в последнем прижизненном издании «Основ химии»
(1906 г.) Д. И. Менделеев относит к реакциям замещения или
двойного разложения взаимодействие между железом и
раствором медного купороса, ведущее к выделению металлической
меди й переходу синего (медного) купороса в зеленый (железный).
Получение тока в гальванических элементах, выделение
металлов на электродах при пропускании тока через растворы
давно стали обыденными и технически важными процессами,
а механизм давно известных реакций вытеснения металлов
металлами и галогенов галогенами оставался тайной. В
частности, оставалось не ясным, действительно ли всякое
восстановление одного металла другим совершается в виде электролиза
и действительно ли при этом возникают токи, как утверждал
Бекетов.
Поскольку факт вытеснения одного металла другим
обнаруживался прямым наблюдением, для подтверждения
предположений Бекетова достаточно было обнаружить возникающие при
этом токи. Первую и единственную попытку обнаружить токи,
возникающие в гомогенных системах, содержащих окислители
и восстановители, сделал профессор Харьковского
технологического института А. И. Щукарев (1915 г.). Он думал, что
процесс любого химического взаимодействия эквивалентен элек-
TpH4ecK0jviy разряду, совершающемуся между реагирующими
частицами. Поскольку разряды тождественны с токрм, в среде, где
идет химическая реакция, такие токи возникают во всем объеме
и по самым разнообразным направлениям; обнаружить их в
таких хаотических условиях невозможно, так как они взаимно
167
компенсируются. Щукарев считал, что, поместив раствор, в
котором идет реакция, между полюсами сильного электромагнита,
можно направить часть этих элементарных токов в направлении,
перпендикулярном к магнитному полю и обнаружить в связи
с этим некоторую разность потенциалов между двумя
электродами, соответствующим образом размещенными в исследуемом
растворе.* Щукарев получил указанным способом много
опытных данных, но ему не удалось их объяснить и использовать
для теоретических выводов.
Начало современной теории
окислительно-восстановительных процессов положил профессор Екатеринославского
горного института Л. В. Писаржевский, которому удалось объяснить
с единой точки зрения как явления, возникающие при
погружении металла в раствор его соли, так и результаты опытов Щу-
карева. Ему же принадлежит исследование и объяснение
явлений, происходящих на электродах из благородного металла
(платины), погруженных в раствор, в котором происходит
окислительно-восстановительная реакция [28].
Согласно теории Писаржевского, в любом куске металла
часть его атомов разложена на ионы и электроны за счет
обратимого процесса ионизации, который приводит к состоянию
равновесия между атомами металла, его ионами и электронами:
Ионизация атома сводится к потере одного или нескольких
валентных электронов. При этом радиус иона сокращается по
сравнению с радиусом исходного атома не только за счет
потери периферических валентных электронов, но и за счет уплотнен
ния оставшегося электронного облака по направлению к
положительному ядру иона, действие которого распространяется
теперь на меньшее число электронов.
При погружении куска металла в воду в раствор должны
переходить преимущественно ионы металла, так как они
подвергаются не только кинетическому воздействию ударяющих
о поверхность металла молекул воды, но и гидратации, которая
облегчается как наличием положительного заряда, так и тем,
что ориентированные по отношению к этому заряду диполи
воды могут подойти к нему значительно ближе, чем к
незаряженному атому. Таким образом, согласно теории Писаржевского
взаимодействие между куском металла и раствором его соли
выражается равновесиями
М^М^^+Ч-р-е,
М«^+ + пНгО М (H20)|;+,
в которых участвуют атомы металла, электроны, ионы, их
гидраты и молекулы воды. При этом, согласно позднейшим данным,
168
можно считать, что в разбавленных растворах число молекул
воды, координируемых металло-ионом, равно его обычному
координационному числу [30, с. 13—79; 8].
После погружения куска металла в раствор его соли на
нем обычно возникает отрицательный или положительный
заряд. Отрицательный заряд указывает на наличие в куске
металла избытка электронов, а этот избыток может появиться
только в том случае, если часть ионов решетки перейдет в
раствор в виде гидратированных ионов. Этот процесс, как
показывают наблюдения за скоростью возникновения потенциала
между металлом и раствором, заканчивается очень быстро и
сопровождается растворением ничтожного количества металла.
Объясняется это тем, что по мере перехода ионов в раствор кусок
металла заряжается отрицательно. В результате затрудняется
дальнейший выход положительно заряженных металло-ионов
из металла, а также диффузия гидратированых металло-ионов.
от поверхности металла в толщу раствора. Электроны,
оставшиеся в металле и удерживаемые ими у поверхности
последнего гидратированные металло-ионы образуют так называемый
двойной электрический слой. Этот слой в разбираемом случае
состоит из избыточных электронов, находящихся в металле и
гидратированных катионов, находящихся в окружающем металл
растворе. Только часть катионов вплотную подходит к
поверхности металла, образуя так называемый «плотный слой».
Остальные катионы, участвуя в общем для раствора
молекулярном движении, движутся в окружающем металл пространстве.
При этом концентрация их у поверхности металла значительна
превышает концентрацию ионов другого знака, в данном
случае анионов, и лишь на достаточном расстоянии от поверхности
концентрации анионов и катионов становятся такими, что
число положительных и отрицательных зарядов в единице объема
раствора оказывается практически одинаковц[м.
Часть двойного электрического слоя, в которой катионы
сохраняют подвижность, носит название диффузной. В тех случаях,,
когда металл, погруженный в раствор соли, заряжается
положительно (например, в случае медного электрода,
погруженного в раствор, содержащий Си^^-ионы), также образуется
двойной электрический слой. Внутреннюю обкладку такого слоя
составляют дегидратированные и внедрившиеся в металл Cu2+-
ионы, а анионы притягиваются к поверхности меди, образуя
плотный и диффузный слои.
Полная толщина двойного слоя в. очень разбавленных
растворах близка к одному микрометру (1 мкм=10-^ м), при
повышении концентрации она быстро сокращается, составляя
в концентрированных растворах всего несколько десятых
нанометра (lнм=10-^ м). Даже наибольшая из этих величин
весьма мала по сравнению с толщиной металлического электрода
169
ш окружающего его раствора, т. е. тех двух фаз, на границе
между которыми возникает двойной слой.
Совокупность сил, приводящих к образованию двойного
<:лоя, называется'электродвижущей силой (э. д. с.) границы
фаз [12].
Типичными окислителями, как давно известно, являются
галогены. В процессе окисления они захватывают электроны и
превращаются в галогенид-ионы: Г2+2е:?=^2Г-, где Г — любой
галоген.
Было известно также, что платиновая пластинка,
погруженная до половины в раствор какого-нибудь хлорида, заряжается
положительно, если верхняя половина ее окружена
газообразным хлором. Но если платина заряжена положительно, то
прилегающий к ней раствор должен содержать избыток
отрицательно заряженных хлорид-ионов, которые могут получиться в этих
условиях только из газообразного хлора, растворенного в воде.
Как происходит это превращение, прежде не знали. Писаржев-
ский устранил все вопросы, показав, что и в этом равновесии
принимают участие атомы, ионы и электроны индифферентного
электрода. Например, часть электронов, входящих в состав
решетки платинового электрода, может присоединиться к
растворенному в воде хлору, образуя хлорид-ионы. Поверхность
платины при этом заряжается положительно, хлорид-ионы
удерживаются у поверхности металла, возникают двойной
электрический слой и отвечающее ему динамическое равновесие.
Результат можно представить в виде следующей схемы:
Pt^pt2-h + 2е, CI2 + 2е&2СЬ.
Но если это так, то к такому «хлорному электроду» и другим
ему подобным применимы все рассуждения, подробно изложенные
при рассмотрении системы, состоящей из куска металла,
погруженного в раствор его соли. Очевидно также, что при замене платины
другим индифферентным материалом новое равновесие будет
отличаться от равновесия на платине, так как вместо равновеси5|
Pt«Pt2+-|-2e в него войдет равновесие, отвечающее новому
металлу: M5=tMt^++ |j.e.
Наконец, Писаржевский тщательно исследовал ряд гомогенных
окислительно-восстановительных систем в присутствии платиновых
электродов и показал, что в зависимости от условий реакции
платиновая пластинка может заряжаться положительно, теряя
часть своих электронов, или отрицательно, перехватывая часть
электронов, которыми обмениваются участники
окислительно-восстановительного процесса. Так, нарример, было найдено опытным
путем, что платиновая пластинка, погруженная в раствор, в котором
концентрация Т1^+-ионов превышает концентрацию Ti^^-noHOB
больше, чем в 4, 7 раза, заряжается положительно. Та же пластинка
заряжается отрицательно, если С^.^4^/С^13-{- < 4, 7, и остается
нейтральной, если это отношение будет равно 4,7.
170
• Рассмотрим теперь реакцию между куском
металла-восстановителя, например цинка, и раствором, содержащим ионы другого
металла, стоящего в ряду напряжений ниже первого, например
меди. В этом случае электроны, остающиеся на поверхности цинка
после гидратации и перехода в раствор части Zn^^-noHOB, сразу
же захвааываются находящимися у поверхности металла гидрати-
рованными Си2+-ионами, обладающими большим сродством с
электронами, чем 2п2+-ионы, а 2п2+-ионы получают воз1Ложность
свободно диффундировать вглубь раствора, так как поверхность
металла разрядилась. На месю ушедших ионов цинка за счет
ионизации атомов появляются новые Zn^+ и новые электроны,
разряжающие дальнейшие количества ионов меди. Появление атомов
меди на поверхности цинка при наличии свободных электронов
приводит к юму, что начинается дегидратация ионов меди,
находящихся в непосредственной близости к атомам меди, и
образование решетки металлической меди. Перечисленные процессы можно
представить в виде следующих схем:
Zn — 2е 4=i Zn Си (Н20)п « Си" + /гНгО
Zn" + тНгО7=±Zn(Н2О);;; Си" + 2е <=±Си
Zn —2e4=tZn2+ Cu^+ + 2e<=±C\i
В этих схемах Zn*' и Си* *—не гидратированные ионы цинка и меди,
а Zn (НгО)^ = Zn2+ и Си (НгО)^ = Cu2+ — гидратированные ионы
тех же элементов. Поскольку константы ионизации и гидратации
неизвестны и измеряемые разности потенциалов между металлом
и раствором его ионов характеризуют суммарные процессы (Cu^'^2e:*=t
«Си, Zn2+ + 2e«Zn и им подобные), в дальнейшем будем
пользоваться только уравнениями, этих суммарных процессов.
При таком способе изображения перехода в раствор
металлического цинка и одновременного выделения металлической меди ясно,
что речь идет об окислительно-восстановительной реакции,
состоящей из двух/ полуреакций:
Zn —2е «Zn2+
Cu2^+2e«Cu (glj
Zn + Cu2+eZn2+ + Cu,
при суммировании которых получается обычное уравнение
восстановления Си2+.ионов металлическим цинком. .
При погружении металла-восстановителя в раствор,
содержащий ионы другого металла, обмен электронами происходит
непосредственно на границе раздела между металлом и
раствором. В таких условиях, как указали А. Н. Щукарев и Л. В.
Писаржевский, следить за процессом передачи электронов нельзя,
можно лишь констатировать, как это было сделано уже очень
давно, факт «вытеснения» одного металла другим,
171
Почти одновременно с работой Л. В. Писаржевского [28]
была опубликована теория кислотно-основных равновесий Брен-
стеда [41] (см. гл. 4). В ней каждому акцептору протонов р-
основанию (Осн.) противопоставлялась сопряженная кислота
(Кисл.)—донор протонов. Поскольку сама вода является
акцептором протонов, переходящим в Н3О+-ИОН, любой кислотно-
основной процесс в водном растворе комбинируется из двух
полуреакций:
НзО+
± р + Н2О
Осн. + р 5=t Кисл.
Н3О+ + Осн. в Н2О + Кисл. а = kr^k2
(6.2)
или в краткой записи: Н+ + Осн.вКисл.
Сопоставление (6.1) и (6.2) привело к мысли, что каждой
окислительно-восстановительной полуреакции также отвечает
сопряженная пара, состоящая из окисленной формы (Ок) —
акцептора электронов и восстановленной формы (Вс)—донора
этих частиц. В простейшем случае Ok + ve5=±Bc (6,3).
Записывая (6.2), мы учли, что вода может быть акцептором
протонов. Она же может стать и их донором (H20:?:fcp + 0H-),
причем оба равновесия относятся к числу простейших процессов.
Наш растворитель — вода может также играть роль
акцептора и донора электронов. Однако соответствующие процессы
гораздо сложнее, как видно из следуюпдих записей, которые
всегда надо начинать с ионизации воды:
Н2О
?=tH++ОКОН- вН+ + 02-
02- —2евО-
20- ?=t02
(6.4)
2Н2О—4е5=±4Н+ + 02
Н2О вН+ + ОН-
Н++е5=±Н.
2Н. ^Нг
2Н2О+ 2е«Н2 + 20Н-
(6.5)
В системе (6.4) Н2О — донор электронов. Здесь она играет
роль восстановленной формы (Вс). Окисленной формой (Ок)
является растворенный в воде кислород, так как именно он
образуется, когда, оксо-ионы теряют электроны. В системе (6.5)
Н2О — окисленная форма (Ок), а восстановленной (Вс)
является растворенный в воде Н2.
Существование равновесий (6.4) и (6.5) должно вести к
окислительно-восстановительному диспропорционированию воды:
172
2Н20 —4е<=±4Н+ + 02
2H20+2e<=tH2+20H-
Н+ + 0Н-«Н20
4 (6.6)
2Н2О 5=t2H2 + 02
Из (6.6) следует, что в любом водном растворе всегда
должны находиться растворенные Н2 и О2.
Сложность системы (6.4) и (6.5) приводит к тому, что лишь
в редких случаях окисляющее действие воды сопровождается
заметным выделением газообразного Н2, а
восстанавливающее— газообразного О2. В качестве примеров можно привести
окисление водой хромо-ионов и восстановление фтора. В
случаях, когда газообразные Нг и О2 не выделяются, считают, что
вода своих окислительно-восстановительных способностей
вообще не проявляет.
В таких системах окислительно-восстановительное
равновесие для какой-либо сопряженной
окислительно-восстановительной пары может сдвинуться только в том случае, если в том
же растворе окажется какая-то другая
окислительно-восстановительная пара. Окислительно-восстановительное равновесие
описывается тогда суммарным процессом, например:
а Oki + rR + ve ?=i aBci + qQ Ki
b Bc2 — ve«pOk2 KT^ (6.7)
a Oki + rR + b Bc2 5=taBci + qQ+pOk2 A' = A'i/(i"^'
константа которого равна произведению констант двух
полуреакций. В системе (6.7) R и Q — формулы частиц, которые сами
по себе акцепторами или донорами электронов не являются, а
лишь сопровождают процесс обмена. Поскольку активности
частиц подобных R и Q должны входить в выражение закона
действующих масс и экспериментатор может изменять их по своему
желанию, их можно считать регуляторами процессов.
6.2. ПРИМЕНЕНИЕ ЗАКОНА ДЕЙСТВУЮЩИХ МАСС
К ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫМ РАВНОВЕСИЯМ
Кислотно-основные равновесия сопровождаются изменением
рН системы. За равновесиями комплексообразования можно
следить, измеряя тем или иным способом выход комплекса.
Наблюдение за окислительно-восстановительным процессом
становится возожным, если контакт между двумя сопряженными
окислительно-восстановительными системами осуществляется
через полупроницаемую перегородку, предотвращающую
смешение жидких фаз, окружающих погруженные в них
металлические электроды, а передача электронов происходит по
металлическому проводнику, соединяющему электроды.
173
Установки такого типа, рассчитанные на получение
электрической энергии за счет химических процессов, называют
гальваническими элементами; в лабораториях, где ими пользуются
для исследовательских целей, их чаще называют цепями.
Используя систему (6.7), можно собрать цепь:
Pt|Ok,(^kO, К(ак), Вс,(авс,), Q(aQ)||Bc2(aBcJ, Ok2(aokJ| Pt,
(6.8)
о
где Pt — платиновые индифферентные электроды; щ —
неравновесная активность i-й частицы, двойная вертикальная черта —
полупроницаемая перегородка, а вертикальная черта
обозначает границу между поверхностью электрода и жидкой фазой.
Знак «—» над правым электродом поставлен потому, что в
правой части Вс2 генерирует электроны.
Э. д. с. цепи (6.8) выражается формулой Вант-Гоффа-Нерн-
ста:
£ = £°-¥ln°^/^. (6.9)
Смысл формулы (6.9) сводится к тому, что по мере стремления
системы (6.7) к равновесию начальные (неравновесные) активности
Gi превращаются в равновесные ai, а £ ~> 0. Но тогда
где R — универсальная газовая постоянная, равная 8,31441 ±
±0,00026 Дж/моль-* • К""^; Г —термодинамическая температура,
К*; F = (96484,56 ± 0,27) К • моль-^ — постоянная Фарадея**, v —
число электронов и /(-константа суммарного процесса (6.7).
Из формулы (6.10) следует, что для измерения К какого-либо
суммарного окислительно-восстановительного равновесия надо
о
составить цепь типа (6.8) при произвольно выбранных Дать ей
разрядиться до £ = 0, измерить а\ и вычислить /С, используя
закон действующих масс.
Рассмотрим в качестве примера реакцию между Fe^+ и окто-
циано-молибдеат-ионами, выражаемую уравнениями
Fe3+ +e;=-Fe2+
Mo(CN)|~-e<=±Mo(CN)8~ (6.11)
Fe^-*- + Mo (CN)r ^ Fe^+ + Mo (CN)|
3—
* К — сокращенное обозначение Кельвина — единицы термодинамической
температуры.
** Постоянная Фарадея равна отношению количества электричества
в кулонах (Кл) к количеству вещества эквивалента (моль), выделенного
этим количеством электричества, при электролизе.
174
Составим цепь:
Pt; Fe^+(c,,3+). Fe^+(ср^2+) | Mo (GN)|-(с^„с^,.-).
Mo(CN)i-(c^^CN,t-)i Pt (6.12>
и замкнем ее через милливольтметр.
При начальных условиях
^РеЗ+ = ^ре2+ = ^Mo(CN)4- = ^Mo(CN)3- = О Л МОЛЬ . Л"!
В первый момент после замыкания напряжение на клеммах
окажется равным ^40 мВ, электрод справа будет заряже1|[
отрицательно, слева — положительно. Это значит, что справа идет
процесс Mo(CN)8"'-> Mo(CN)8""+е, сопровождающийся падением
Сд4- и ростом Сдз— Тогда слева должны восстанавливаться Fe^+-
ионы: РеЗ+ + е -> Fe2+.
В результате для цепи (6.12)
^ ^РеЗ+. ре2+ + -^Mo(CN)J-. Mo(CN)3-'
о 8
где Е с индексами — потенциал на отдельных электродах. Оба
процесса будут продолжаться до тех пор, пока установится
равновесие (6.11). В этот момент Е снизится до нуля, т. е.
станет равной нулю алгебраическая сумма потенциалов на
отдельных электродах.
Как показывает анализ, при Е = 0 [Fe^+] = 6,37 • IO-2,
[Fe2+] = 0,136, [Мо (CN)i-] = 0,136, ^
Mo(CN)8"] = 6,37 . 10-2 моль . л-1.
и для суммарного процесса
fFe2+][Mo(CN)|-] ^ (0Л36)- ^
[Fe3+]|Mo(CN)^-] (6,37 • 10-2)2 ~ » •
Описанный метод применим лишь для равновесий с небольшими
значениями К, так как при больших К равновесие сдвигается
очень сильно, а измерение очень маленьких равновесных
концентраций приводит к недопустимым погрешностям.
Еще важнее другое обстоятельство. Как следует из (6.7),
константа любого суммарного равновесия равна произведению
констант двух полуреакций. Имея п таких констант, можно
рассчитать константы для общего числа суммарных
равновесий, равного] числу сочетаний из п элементов по два:
С^
2! (п — 2)-
Например, при п=100 С2„ =4950. Значит, нет смысла
измерять константы суммарных процессов, а все внимание надо
направить на измерение констант полуреакций.
175
Как следует из формулы Нернста-Петерса,
£ = £° _ In f2!i = ^ In /( - ^ In . (6.13)
«Вс «Вс
значение К для любой сопряженной
окислительно-восстановительной системы можно найти из измерений потенциала Е на
электроде, погруженном в эту систему, и активностей ее
компонентов. К сожалению, методов измерения потенциалов на
отдельных электродах пока нет, поэтому возник вопрос об оценке
констант полуреакций по результатам измерений э. д. с.
гальванических цепей.
Для цепи (6.8), если пренебречь потенциалом на
полупроницаемой перегородке,
£ = £ок.вс, + £вс,.ок,- (6.14)
В (6.14) индексы записаны в последовательности, ясной из
схемы (6.7), где по соглашению переходу Ок-bve всегда
отвечает прямое, а превращению Вс — ve — обратное значение
констант полуреакций. Теперь, заменяя Ei их значениями, находим,
что
f In ^Bc,«Q , r.° rt V «?)к.
t = iiOK.Bct — ;^ln-;—^ + ^Bc,. Ok, — ;;^ in-^— =
«Ok/r «Bc, .
^^Uk^ ^T1^ «Вс, ' IrT.j. «ВсД
«OK,-«r \ «?)кУ
Ясно, что для оценки констант, относящихся к разным
полуреакциям, например константы Ки необходимо использовать
цепи типа (6.8), составленные из двух полуэлементов,
соединенных через полупроницаемую перегородку. Первый полуэлемент
должен состоять из электрода, погруженного в сопряженную
систему, для которой ищут константу; в случае, например,
измерения К\ такой полуэлемент будет таким же, как левая
сторона цепи (6.8). Второй полуэлемент должен быть стандартным,
т. е. общепринятым и всюду одинаково воспроизводимым.
Потенциалу между его электродом и находящейся с ним в
контакте сопряженной окислительно-восстановительной системой, а
значит, и константе соответствующего равновесия должны быть
приписаны общепринятые значения при навсегда принятых фик-
а
сированных начальных активностях а{.
Если бы стандартной была принята система, отмеченная
индексом 2, входящая в цепь (6.8), и величинам /Сг, аок, и авс^
были приписаны определенные значения, то выражение в
скобках в уравнении (6.15) стало бы постоянной величиной, опреде-
176
ляющей возможность измерения К\ и констант любых других
полуреакций в этой условной системе отсчета.
В настоящее время в качестве стандартного принят
водородный полуэлемент, состоящий из платинированного платинового
электрода, погруженного в раствор с единичной активностью
ионов водорода (в соответствующей шкале концентраций),
находящийся в равновесии с газообразным водородом при его
парциальном давлении 1,01325*10^ Па (1 атм).
Для измерения Ki можно использовать цепь
Pt|Ok,(aoO, R(aR), Вс^авс,), Q Ы Ц Н+(ан+= 1),
Н2(г)(/7= 1 атм)|Р1. (6.16
Соответствующие равновесия выглядят так:
а Oki-f rR + ve<=taBci+^Q|
Н2(г) —2е^2Н+
2 K^ к!
V КГ (6.17)
2а Oki + 2rR + vH2;=t 2aBci + 2qQ + 2vH"*- К = KIKT"
a Э. Д. c. будет выражаться формулой
Для стандартного водородного полуэлемента "принято, что
при любой температуре его стандартный погенциал = О и,
следовательно IgjK2 = 0. Тогда формула (6.18) переходит в
(6.18)
Я, = -1п/С._-1п25£^, (6.19)
«ок,4
где Ен — э. д.с. цепи (6.16), одним полуэлементом которой
является сопряженная система, отмеченная индексом 1, а другим —
стандартный водородный полуэлемент.
Для такой цепи при оок, = o^r = аво, = ад = 1 моль • л-^
уравнение (6.19) переходит в
£?, = ^ln/C, = ,^lgK, (6.20)
где е — основание натуральных логарифмов. Величину Ей
называют стандартным потенциалом данной сопряженной
окислительно-восстановительной системы.
Если в цепи (6.16) в условиях, приводящих ее э. д. с к
величине Ен, платиновый электрод заряжен отрицательно, то
величине £н приписывают знак плюс, в противном случае — знак
минуа
7 2-436 177
Из (6.20) следует, что
V • 96485 . £н п 1
IgK = 0,43429 . S 3^eggy " = 5038,2v£?,T-^, (6.21)
причем знак перед IgK определяется знаком при £н.
Следует отметить, что описанный метод измерения в
настоящее время практически не используется из-за присущих ему
неопределенностей. Последние обусловлены невозможностью
строгого учета вклада в жидкостного потенциала, возникающего
на границе раздела двух жидких фаз и невозможностью строгого
расчета термодинамических активностей отдельных ионов (в том
числе и водородных). Обычно эти величины измеряются и
вычисляются другими способами [21].
0.3. 1АБЛИЦЫ КОНСТАНТ СОПРЯЖЕННЫХ
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫХ РАВНОВЕСИЙ
В таблицах работы [43], изданных «под эгидой Комиссии по
электрохимическим данным секции аналитической химии ШРАС»,
приведены значения Ей для большого числа полу реакций.
В наиболее полной современной сводке [65, 66] приведены
только значения IgK сопряженных равновесий, которые
рассматриваются как процессы координации электронов
соответствующими окисленными формами. Основанием для такого подхода
могут служить многочисленные сообщения о существовании
сольватированных электронов [67], появление которых
непосредственно наблюдал еще Писаржевский [28, с. 100].
В наших учебных таблицах [4] в первой колонке приводятся
символы элементов. Они расположены в алфавитном порядке.
Во втором широком столбце представлены уравнения
полуреакций. В третьем —даны соответствующие Ig/C, взятые из [66] или
[44, 65] или вычисленные по (6.21) с помощью значений £н,
приведенных в четвертом столбце. Наконец, в последней колонке
даны сведения о среде, которой отвечают значения Ig/C и £н,
находящиеся в той же строке.
Пользуясь учебными таблицами IgK окислительно-востано-
вительных полуреакций, необходимо помнить о некоторых их
особенностях.
1. Все полуреакции записаны в таком порядке, что в левой
их части находится акцептор электронов, т. е. окисленная
форма, а в правой — восстановленная форма. Поэтому при
отыскании констант полуреакций, в которых роль исходных веществ
играют восстановленные формы, следует искать нужные
вещества в записанных справа частях равновесий.
2. В таблицах приведено минимальное число равновесий
полуреакций для каждой окисленной формы. Значения констант,
178
соответствующих более сложным, а иногда и более простым
полуреакциям для тех же Ок- и Вс- форм, можно всегда
получить по правилам суммирования равновесий.
6.3.1. Найти IgK равновесия
С10Г+ 2Н-^ + 2евС10- + Н2О. (6.22)
В таблицах констант полуреакций точно такого равновесия нет,
но в одной из строк находим равновесие между частицами,
родственными нашим, и соответствующий Ig/C:
HCIO2 + 2Н+ + 2е 5=- нею + Н2О, Ig /С = 55,6. (6.23)
Очевидно, что от (6.23) можно перейти к (6.22), прибавляя
равновесия
СЮГ + Н-*"?=± НСЮ2 Iga = 1,97,
нею 5=± Н+ + ею- Ig /С == —7,53.
Значит, для (6.22) Ig/C = 55,(j + 1,97 — 7,53 = 5о,04.
6.3.2. Найти Ig/C полуреакции окисления Ре''^"*"-иона и
превращения его в твердую ферригидроокись, если предполагается,
что окисление будет происходить в аммиачном растворе.
В таблицах дана полу реакция Fe3+ + e«Fe2+ и ig/C= 12,37.
Очевидно, что заданную полуреакцию и Ig/C для нее можно
получить суммированием следующие! равновесий и отвечающих
им IgK:
Fe2+ _e5=±Fe3+
Fe3+ + ЗОН- « Fe (ОН)з (т)
Н2О 5=± Н+ + ОН-
NH3 + H+ «NHt
_lg/( = _12,37
-lgP = 37
3 3Iguy = —42
3 3 Iga =27,72
Fe2+ + ЗН2О + 3NH3 — e « Fe (ОН)з (т) + 3NH4"*" Ig К = 10,35
6.3.3. Найти Ig/С полуреакции, отвечающей переходу As05"" —
— ASO2"". Прежде всего надо записать полуреакцию так, чтобы
она соответствовала условиям задачи. Первый шаг приводит к
равновесию ASO4'" + 4Н"^ + 2еч=± AsOF + 2Н2О, которое этим
условиям не удовлетворяет, так как введение Н+-ионов должно
вести к образованию продуктов протонизации ASO4'". Выход в
обеспечении равновесия Н+-ионами за счет диссоциации воды:
AsO^ + 4Н+ + 2е « As07 + 2Н2О
Н2О «Н-н +0Н-
AsO?"" + 2HiO + 2е^ AsOT + ЮН"
Но в таблицах нет IgA для такой полуреакции, а приведены
лишь IgK для переходов H3ASO4As(т) и HASO2As(т).
7* 179
Используя эти полуреакции и другие нужные нам
равновесия, получаем
H3ASO4 + бН-^- + 5е ;=iAs(т) + 4H2OI
As (т) + 2Н2О — Зе вН As02 + ЗН+ '
АвО^-^^+ЗН-*- вНзА804
HAsO? eAsOr + Н-»"
НЮ «Н+ + ОН-
lg/( = 31,09
— Ig/С = — 12,39
lga3 = 20,73
-lga = -3,7
4 Ig ш == — 56,0
AsOr + 2H2O + 2e ^ AsOr + 40H- = — 20,27
6.3.4. Для равновесия газ .=t газ (г), где слева — насыщенный
раствор газа в воде, еправа — находящаяся с ним в равновесии
газообразная фаза, по закону Генри выполняется соотношение
(газ)
= X, (6.24)
где ргаз — парциальное давление данного газа над раствором
X—константа закона Генри, зависящая от температуры. Отсюда
(газ)-Х-»/7р,. (6.25).
После подстановки выражения типа (6.25) в уравнение
закона действующих масс для полуреакции с участием
газообразного вещества величину X включают в константу полуреакции.
При расчетах ионных равновесий с использованием таких
констант полуреакций для систем, насыщенных газами, в
уравнения закона действующих масс вместо активности газообразного
вещества надо подставлять его парциальное давление,
равное атмосферному.
Если система не насыщена газом и он из нее не выделяется,
такой подход неприменим. В этом случае табулируемые
константы полуреакций преобразуют обратной подстановкой в
выражение закона действующих масс величин /7газ=Л • (газ) (6.26Х
•6.3.5. Найти IgK для полуреакции 2СО2 + 2Н++2ег^Н2С204
(6.27), если в (4] для полуреакции при 20 ^C
2СО? (г) + 2Н+ + 2е « Н2С2О4, Ig /С = — 15,9. (6.28)
В знаменатель выражения закона действующих масс для
равновесия (6.27) вмевто pgo, придется подставить
рс®,вХ .(002)= 101-42(СО2).
Отсюда искомый Ig/C, отвечающий выражению закона дейвт-
вующих масс в его обычной форме, равен lg/C = —15,9+2 х
X 1,42= —13,06.
6.3.6. Найти !g /(для полуреакции 2N0 + 2Н+-f 2eeN20 +
+ Н2О (6.29), если в таблицах констант полуреакций мы
находим 2NO(г) + 2Н+ + 2е« N2O (г)+ Н2О, Ig /( = 53,8.
В этом случае в числителе выражения закона действующих
масс вмеато р j^^q появится множитель Xn,o(N20) = 10*'^^(N?O),
180
а в знаменателе вместо рмо — множитель Xno(NO) = [102'6^(NO)p/
и после переноса постоянных в правую часть окажется, что
искомый Ig /С = 53,8 — 1,57 + 2 • 2,68 = 57,59.
6.4. СУММАРНЫЕ ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ
РАВНОВЕСИЯ И ИХ ОСОБЕННОСТИ
6.4.1. Общий мегод упрощенных расчетов
окислительно-восстановительных равновесий сводится к объединению заданных
сопряженных полуреакций в одно равновесие, в записи
которого уже должны отсутствовать электроны; расчету константы,
выбору наименьшей равновесной концентрации, выражению
через нее всех остальных и расчету наименьшей концентрации
с помощью уравнения закона действующих масс.
Возьмем в качестве примера окисление Ре^'*"-ионов МпОГ-
ионами в кислой среде. Комбинируя две полуреакции и
логарифмы их констант, получаем
Мп07 + 8Н"^ + 5е » Мп^"^ + 4Н2О
Fe2+ -евРез-ь
Ig/Ci = 127,4
_51g/C2 = —64,8
MnOT + 8Н-*- + 5Ре^-^ в Mn'"^ + 4Н2О + SPe'"^ = 62,6
Здесь при lg/(=62,6 следует ожидать, как мы это видели при
расчетах кислотно-основных равновесий и процессов
координации, что наше суммарное окислительно-восстановительное
равновесие сильно сдвинуто вправо. Заметим еще, что в [4] для
трех разных солевых фонов приведены три разных значения
IgK: 12,97; 11,39; 12,37, отвечающих разным «реальным» потен-
диалам. Использовано первое. Результаты округлены, так как
при наличии трех разных значений IgK рассчитывать на
особую точность не приходится.
Допустим теперь, что Смпо^ = сре^-ь ^ 5У14 „ ^ jj^
Если У > 5Л1 и Н так велика, что диссоциацией аквоферро-
аквоферри- и аквомангано-ионов можно пренебречь, то
наименьшей можно считать [Ре2+]. Тогда при обычной форме записи
МпОГ +8Н-ь -Ь5Ре^-»" ^
о У Н ЪМ
[ ]Y — M + m Н — Ш+8т Ът
5=> Мп2+ -Ь4Н20 +5Рез+ IgK = 62,6
М — т — Ъ{М — т)
т
и по закону действующих масс для обратной реакции
(Y^M + m)(H^SM + Smf(bmf ^ jq_62,5 /g 39)
(M^m)\5(M^m)f
Здесь, судя по значению /С, т^М. Поскольку в
последующем для расчета потенциала при равновесии интересно иметь
значение отношения (Fe3+]/[Fe2+] = (М — m)/m, можно
пренебречь т только в первом множителе знаменателя и в двух
первых множителях числителя (6.30). Тогда
= 10-12.62 (У _ (Н — 8Af)-81i/5
и, например, при М = IO-2, Г = 2.10-2 и Я = 2 моль • л-*
т(М —m)~i = 10-i2-»7 и т= 10-12.97^ М = Ю-^^.эу, Поскольку
условие т<^М выполняется, приближенный расчет можно
считать удовлетворительным, так как обычные расчеты показывают,
что при Я = 2 моль . л-1 ионизацией всех аквометалло-ионов
можно пренебречь.
Для равчета потенциала используем приближенное выражение
где Ян == 0,767В при 25 «С, 6 = 0,05916 Дж • Кл-' и iFe^+j х
X [Fe2-»-]-i = (М — m) m-i = Ю^^'»?. В результате Е = 0,767 +
+ 0,05916 . 12,97 = 1,534В.
Равновесие устанавливается очень быстро. Пока в растворе
есть ощутимые количества Ре^-ь-ионов, каждая новая капля
раствора МпОТ-иона практически мгновенно обесцвечивается. Первая
7^е избыточная капля окрашивает весь раствор в розовый цвет.
6.4.2. Приближенные раечеты в более сложных тстемах.
Рассмотрим равновесие ежду хромо- и перхлорат-ионами:
Сг2+ — e.=tCr^+ из 14] Ig/C = 6Д
Для С107 -иона там же приводится ряд полуреакций, но какую
из них следует учитывать при взаимодействии ^ Сг^^-нонами,
указаний нигде нет. В евязи е этим интересно выписать патрицу
всех возможных реакций с учавтием С107-иона, считая, что он
может, присоединяя электроны, превращаться в С10Г, СЮг,
СЮГэ СЮ"", СЬ и СР. Логарифмы констант соответствующих
полуреакций частично приводятся в таблицах таких констант,
а частично могут быть подсчитаны на основании приведенных
там данных. Для записи соответствующей матрицы равновесий
надо вводить столбец, в котором указано число электронов,
участвующих в равновесии. Проще давать
окислительно-восстановительные равновесия в приведенном к одному электрону виде:
ш
ег2+
ао7
н+
0
0
0
0
0
0
1
1/2
1/3
1/4
1/6
1/7
1/8
0
1
4/3
5/4
7/6
8/7
1
0
1/2 CIO5-
1/3 ClOjCr)
1/4 HCIO2
1/6 нею
1/14 012 (г)
1/8 С1-
СгЗ+
20.1
19,9
20,3
22,8
20,6
20,9
6.9
Суммируя первую и последнюю строки матрицы, умножая
сумму на 2 и используя полученное равновесие и константу для
приближенного расчета, находим
СЮГ +2Н+ +Хг2+ .=s
с Ш н т
I 13М + т Н —2Л1 +2т 2т
s=t СЮГ + Н2О + 2Сг'+ lg/C = 54,0
М — т 2{М—т)
_t)^(2r^^
(iM —т)12(Л1—m)j2
^ = 10-27[3-1 (Я — 2Al)-2j'/2 = 10-27.24 Щ _ 2Л1)-1.
m
М — т
дает
Сложение и умножение на 8 двух последних строк матрица
СЮГ + 8Н+ +8Сг2+ S3
о \т Н Ш
[ ] \Ш + т H — SM + 8m 8т
« С1- +4Н2О +8СгЗ+, lg/C = 222,4
М—т 8(М — т)
л8 /о^ч8
(Л1-т)(8(М —т)1«
m
^-.10-2г.«ЧЯ-8М)-'.
Как видно, оба решения приводят к очень маленьким и
практически одинаковым значениям т. Это значит, что СЮТ-ион
183
должен практически полностью окислять Сг2-Ь-ион. Факты
расходятся с этим результатом. Так, в работе [39] при исследова-
Бии восстановления хромо-ионами HN3 применяли как наиболее
стойкие растворы Сг (0104)2. Подобным же образом в [63]
установлено, что растворы Na2C204 в HCIO4 (0,1 моль-л-^) гораздо
устойчивее водных, хотя для равновесия окисления Н2С2О4
перхлорат-ионом Ig/С'= 56,1 и расчет обещает практически полное
завершение процесса.
Общепринятой теории, позволяющей предсказать, в каких
случаях рассчитывать на достижение равновесия не приходится,
пока не существует. Современные сведения изложены в [23].
Считают, что равновесие устанавливается медленно в тех
случаях, когда в соответствий с суммарным уравнением прямой
процесс может происходить лишь за счет одновременного
взаимодействия большого числа частиц. Однако использовать это
правило следует .с большой осторожностью. Так, при окислении
щавелевой кислоты перманганатом суммарный процесс
описывается уравнением
2Мп07+ бН;^ + 5Н2С204<=± 2Мп'"*' + 8Н2О + IOCO2 (г),
lg/C = 334,3
и, на первый взгляд, может показаться, что равновесие
устанавливается за счет единовременного взаимодействия тринадцати
частиц, тогда как реакции даже третьего порядка считаются
очень редкими. В действительности реакция между МпОТ и
С2О4" идет достаточно быстро и широко используется в титри-
метрий.
Согласно общепринятому мнению, суммарные процессы да-
•ют лишь представление остехиометрических соотношениях
реагирующих веществ, ничего не говоря о механизме процесса.
Исследования кинетики сложных процессов позволяют
установить или предположить существование отдельных стадий.
Например, сведения о механизме взаимодействия Мп07- и СгО^""-
ионов [331: 1) непосредственно Мп04'" и СгО^"" взаимодействуют
очень медленно; 2) быстро идет процесс 2МпОГ + ЗМп^"*" +
+ 16Н"*"->5Мп^'^'+8H2Q; 3) быстро й обратимо устанавливается
равновесие
Л1п^++А1п2+ ?=±2МпЗ+;
4) дальше предполагаются следующие стадии:
Мп'^- + C20f- Мп'+ + СО2 + СОГ;
Мп'"*- + COF Мп^+ + СО2;
Мп^"^ + 2С2О!" « Мп (C204)F;
Мп'"^-f СзО^Мп'-*" + СО2 + СОГ;
'184
Mn'+ + COF -> Mn'-*" + CO2,
причем первые три стадии считаются быстрыми.
Очевидна роль ионов Мп2+, которые медленно получаются
при прямом взаимодействии МпОГ и Н2С2О4 или вводятся как
катализатор.
Ясно, что при записи любой матрицы равновесий системы,
включающей равновесия окисления-восстановления, в
серьезных случаях нельзя полагаться только на сводки констант
равновесий и ставшие привычными со школьной скамьи
представления, а необходимо пользоваться справочниками типа [33] и
другими литературными источниками. Например, по
установившемуся мнению, HCIO4 в растворах, где ее массовая доля ниже
70 Vo, вообще не проявляет способности окисления. Однако, как
теперь известно, Ti^+, V^^, способны восстанавливать С104~-
ион в разбавленных растворах [33, с. 77], хотя Сг^^-ионы
с С104~-ионами сосуществуют.
6.4.3. Рассчитать равновесие в системе, где Сре(С104)з = ^=
= 2 . 10-2 и = F = 6 . 10-2 моль.л-1.
В литературе нет значений IgPn Для Feln^" и Fein""". Это
дает нам право не учитывать комплексообразование. В работе
[33, с. 64] описан механизм взаимодействия Fe3+ и 1--ионов;
все стадии процесса обратимы и среди них нет медленно
идущих. Из условия задачи видно, что равновесие должно
устанавливаться при избытке 1--И0Н0В, т. е. вести к прямому превра-^
щению 1--И0Н0В в иодиодид-ионы.
Используя начальные условия и ^известные Ig/C, получаем
матрицу равновесий:
6- 10—2
6.10—2
2.10—2
6.10-2
У
ЗМ
М
у
к+
сю-
Fe3+
I—
н+
Fe2+
к
\gK
0
. 1
2
1
0
0
0
0
0
0
3/2
0
—1
—1
—2
0
0
—1
1
он-
РеОН2-Ь
Fe2+
1/217
FeOH+
w
-Hi
Кг
К,
-14
-2,17
-2,85
12,97
18,11
-5,92
Закон сохранения начальных концентраций дает четыре
уравнения: ск = [К+] = Y (6.31); Сею. = [СЮТ] = ЗМ (6.32);
Ср^з+ = М = т{1 + -Qih-i) + 27)22/п2Л-2 + Ь{1 + -qh-^y, (6.33)
ci = Y = y+3[ll
(6.34)
185-
и по закону сохранения нейтральности
Л—оЛ-» 4- Y-3M + т(3 + 2■nlh-^) + 4-Q22m^h-2 +
+ b{2 + 'Qh-^) — y — [IT] = 0. (6.35)
При составлении этих и последующих уравнений принято,
что все f I s= 1 и (Fe2+) = b. Три последних уравнения включают
5 неизвестных: т, К 6, у и [li"]. Еще два уравнения дает
суммарное окислительно-восстановительное равновесие
Fe3++ e;=iFe2+
2 2 lgiCi = 12.97.2
2 ■ -\gK2 18,11 , (6.36)
. 2Fe'+ + 31- « 2Fe'+ \gK = 7,83
используя которое можно написать:
/г] =/CmVb-2; (6.37)
[/Г] = 4 (fFe^+l + (FeOH+l) ^\b{\ + (6.38)
Система уравнений (6.33)—(6.38) нелинейна относительно /га,
й и у. Быстрое и хорошо согласованное решение здесь возможно
лишь с помощью ЭВМ. Точных результатов оно все же дать не
может из-за отсутствия данных о yi.
Рассмотрим возможность приближенного решения и оценки
его правильности.
Используем (6.36) для приближенного расчета:
2Fe3+ + 31- =2Fe'+ +1Т lg/(=7,83
с 2. 10-2 6-10-2
[ ] 2х 3 • 10-2 + Зх 2 (10-:^ _ X) (10-2—л:)
_2(10-2 —X)
(2х) 2 13( 10—2_| и]^
По закону действующих масс —^^з"^- Полагая а;<
< 10-2 и пренебрегая им только в одном из множителей 10~2— л;,
находим, что
—Л =р 10-2.6306 10-4.6315 _ 2,336 . 10-5 моль-л-!.
10^^—д:
Условие X = 2,336 . 10-^ < 10-2 выполнено и, следовательно,
т = lFe3+l = 2х ^ 4,672 • 10-^ = 10-4,ззо5.
b = (Fe^+l, = 2(10-2 —X) ^ 0,01995 = Ю-^.бэээ.
[it] = 0,009977 = 10-2,0010 ^^j^^ .
Результаты расчета показывают, что Рез+-ионов остается очень.
шло. Это значит, что рН определяется диссоциацией аквоферро-
иона. Тогда
186
Fe2+ 5
с 0,01995
[ J0,01995—А
FeOH+ +H+lg4s=—5,92
и no закону действующих масеуоТ995"13'==='10-5;92^ А < 0,02 t
h ^ 10-3.81 = 1,549 • 10-4 j^o^b • Л-1.
Теперь, пользуясь матрицей равновесий, находим, что
[FeOH2+l 10-2.6905 ^ 2,04 • 10-3;
[Fe2(OH)2'^)^10-з.s^lo= 1,285-10-4 моль • л-^.
Подстановка найденных значений тиНв (6.33) дает b ^ [Ре2+] ^
^ 10-1,7564 ^ 0,01853 и [FeOH+l 10-^.8664 ^ 1,36-10-4 моль • л-».
Последующий расчет по (6.38) дает (1Г1??^ 9,333 . 10-з = 10-2.озоо
моль . л-1, а из (6.38) у = ^ 0,032 = 10-i'4949 ^оль • л-^
Наконец, из (6.37) [1Г] ^ 10-ь»029 = 1^574 . ю-г ^оль • л-^ и
[ОН-] ^. 10-lO'^^ = 6,46 • lO-^i моль • л^К
Подстановка полученных значений в (6.33) вместо' 0,02 дает
0,02101 моль • л-*, а такая же подстановка в (6.35) вместо нуля
дает 2,9 • 10-** моль • л-^.Для [1ГЗ получено три разных значения:
0,00998, 0,00933 и 0,01574 моль • л-^; для &=[Fe2+] — два: 0,01995 и
0,01853 моль - л-!, последнее значение для [1Г] включает все
погрешности, накопленные в т, у и 6, и вызывает наибольшие
сомнения.
6.4.4. Рассчитать равновесие в системе, где ch.aso. =^ А = 10-з,
^Fe(cio.)g = М = 0,2 и снсю. = у = 2 моль - л-!.
в работе [33] нет сведений о механизме взаимодействия Fe2+
и H3ASO4 и замедленных стадиях этого процесса. Матрица
равновесий системы H3ASO4 — Fe (€104)2 — HCIO4 — Н2О записана
в табл. 6.1.
Таблица 6*1
2.4
2Л1+К
'о-
0.2
м
10-3
4г
+
X
+
Cl.
О
1
о
1
о
о
о
о
о
о
1
о
о
о
о
о
~1
—1
j
о
о
—1
—2
1
1/2
О
о
о
он-
FeOH+
H,AsO'-^
Fe3+
1/2 HAsOg
FeOH2+
Fe2(0H)^H-
HASO2
w
^22
—14
1-5,92
11,50
12,97
18,9
L-2,17
-2,85
9,29
18,44
20,63
18?
Записываем также уравнения закона еохранения начальны х
концентраций:
сею. = 2,4 = 2Л1 + У = [СЮГ]; (6.39)
Сре2+ = 0,2 УИ = т(1 + ^Л-О + Ь{1+ nih-^) +
+ 2у^22Ь^к-^\ (6.40)
OH,Aso. = 10-^ = Л = а ][; ф + г (1 + а/г); (6.41)
1 = 0
£:н = 2 = У = Л—(3 — ])o^hi—arh — 'qmh-^ —
— '^i&A-^ — 2^22Ь2Л-2 + 2r (1 + оЛ). (6.42)
В уравнениях (6.40) — (6.42) принято, что все = 1. В них
ш = (Fe2+), b = (Fe'^-+-), а = (As04^, г = (AsOF) и значения
активностей совпадают со значениями соответствующих равновесных
концентраций. При доставлении уравнения (6.42) из Л = (Н+)=
== [Н+] вычитаются слагаемые, выражающие вклад в значение h
за счет дисеоциации кислот и аквометалло-ионов. Слагаемое
2г(1 + оЛ) выражает расход Н+-ионов в равновесии:
H3ASO4 + 2Н+ + 2е и HAsO? + 2Н2О
Ре^ч- —ев Fe^-^-
1 lg/(2= 18,9
2 —21g/Ci= —25,94
H3ASO4 + 2Н-ь + 2Fe2+eHAs02 + 2Н2О + 2Fe3+ lg/(= —7,04
(6.43)
Действительно, из (6.43) следует, что при образовании 1 моль
HASO2 расходуется вдвое большее количество оксоний-ионов.
Если учесть теперь, что образующаяся НАзОг частично
диссоциирует, образуя АзОГ-ион, то станет . ясно, что расход Н+-ионов
определится выражением 2(lAsOr] + [НАзОг]) = 2г (1 + о/г).
Уравнения (6.40) — (6.42) включают пять неизвестных: т, 6,
/г, а, г. Недостающие два уравнения можно записать, используя
Х6.43):
&=^2г(1 + о/г); (6.44)
-W-2==--4l-= (6.45)
Система нелинейна, но высокое значение си позволяет думать,
что ионизация кислот и аквометалло-комплексов будет практически
подавлена за счет небольшого расхода Н+-ионов. Это позволит
использовать приближенный расчет.
Используя (6.43), получаем
НзАзО' + 2НН' -Ь 2Fe2+ «нАзОг + 2Н2О -f 2Fe3+, 10-^.04
с 10-3 2 0,2 — —
ПЮ-з—2 —2а:0,2—2;с х 2х
10-^ — x
2х
2(0.1 -x)
1
= 10-^'°^ и при условии
Пользуясь методом подстановки, находим
x = [HAsOj] = arh 10-3.8356 ^ j^g . jQ_4.
10-^ — л; = IH3ASO4] = ОЗОЛЗ ^ 10-3.0685 ^ 3^54 . jq_4.
2л: = [Fe3+] = b 10-з.534б 2,92 • 10-*;
0,2 — 2л; = [Fe2+] = 0,1997 lo-o-ssss.
2— 2л: = [Н+] = А 2,000 = ioo-a"'" моль • л^».
Из (6.50), (6.51) и (6.54) находим
[AsOri = г <==«.10-'з.42бб ^ 3J44 . iQ_i4.
[AsO^l = а « 10-24.6016 _ 2,503 • 10-2^ моль • л-К
(6.46)
(6.47)
(6.48)
(6.49)
(6.50)
(6.51)
(6.52)
С помощью этих первичных значений и матрицы равновесий
системы можно рассчитать все остальные равновесные концентрации.
Для проверки правильности можно сделать подстановку в
(6.40) — (6.42) и (6.45). Результаты М = 0,199996, А = 1,0025 ■ Ю-з
У = 2 + 1,43 .10-^ и Л = Юо-зобз = 2,024 моль • л-' вполне
удовлетворительны, но на достоверность в химическом смысле они
претендовать не могут.
6.4.5. Раесчитать равновевие в сиетеме, где сре(а©,), = М =
= 5 . 10-^ Сре(С1о,), = 5 = 5. 10-S снсю. = Н= 10-^ моль • л-',
причем раствор насыщен воздухом при комнатной температуре,
т. е. содержит растворенный киелород.
В работе [33] нет сведений о механизме взаимодействия Fe^+-
иона с растворенным О2. Записываем матрицу равновееий, ечитая,
что в 1 л раствора, находящегося в контакте с воздухом, со,=
== 1,4 . 10-^ моль • л-':
0,2Ь
5.10-2
1.4.10-3
l.lO-iJ
М
В
0
н±
1
0
0
I
0
0
1
2
1
0
0
0
0
0
1/4
—1
^2
0
1
FeOH-*^
FeOH^H"
1/2 ЩО
•П22
к,
-5.92
-2Д7
-2.85
12,97
83,10
189
Для оценки равновесных концентраций простейших частиц»
входящих в систему [СЮГ], [Н+], [Fe^+l, [Fe^+j и [О2],
необходимо иметь 5 уравнений. Если учесть, что [Н+]>[ОН-],
принять, что <Pt = 1 и значения равновесных концентраций
совпадают с активностями [СЮГ] = (СЮГ), (Н+] = (Н+) =Л, [Fe2+]=
= (Fe2+) = m, [Fe3+] = (ЕеЗ+) = fe, [O2] = (O2) = g, то, используя
законы сохранения и закон действующих масс для окислительно-
восстановительного равновесия
Fe2-b — е 5=±Fe3+
О2 + 4Н++ 4e?=t2H20
—12,97.4 = —51,88
83,10
4Fe2+ + O2 + 4H+?=t 4Fe3+ ^ 2H2O Ig /( = 31,22,
получаем систему уравнений
CC104 = Ш + 35 + Н = 0,26 = [СЮГ]; (6.53)
сре = М + В = 0,1 = m (1 + -^Л-») + b (1 + -^i/i-i) +
+ 21322Ь2/г~2; (6.54)
сн = Н = 0,01 = h —qmh-^ — 7]i&ft-i — 2ig226%-2 +
+ 4(G-gr); (6.55)
h — {2M + 3B + H) + m{2 + -qA-O + b(3 + 2-^^-^) +
+ 4n22b"h-^ = 0; (6.56)
= Km'gh^ {IgK = 31,22). (6.57)
В (6.55) CO знаком «—» стоят слагаемые, эквивалентные
вкладу в Н+ за счет ионизации аквоферро- и аквоферри-ионов,
а со знаком «+» слагаемые, характеризующие расход
введенных Н+-И0Н0В. Так, из суммарного окиСлительно-восстано-
вительного равновесия следует, что при снижении со2 = 0 до
[02]=^ снижается на 4 (G — g). Этот расход Н+-ионов и
учитывается в (6.55). В свою очередь, коэффициенты 1, 2, 3
и 4 в (6.56) отвечают числу зарядов, которые несут
соответствующие ионы.
Нелинейную систему (6.54) — (6.57) можно решить на ЭВМ.
Предварительно, однако, следует получить приближенную
оценку равновесных концентраций системы и выяснить
возможность упрощений. Начать можно с изолированного расчета
суммарного окислительно-восстановительного равновесия:
4Fe2+ + О2 + 4Н+ в2Н20+ 4¥е^-^ lg/C=31,22
е 5.10-2 1,4.10-3 10-2 5-10-2
[] 4,44. 10-24.4g 4,4.10-44g 5,56.10-2-4g
190
По закону действующих масс
4(1.11 • 10-^+ g)
4(1.39. 10-2-g)
g[4(l,l • 10-^+g)]* = 10-31.22 (6.58)
и, если < 1,1 . 10-3, g ^ [O2] ^ 10-21.4032 = 3^95 . 10-22, (Ре2+] =
=m^4,44 10-2, [Fe3+]=6==?5,56.10-2,[Н+]=/г^10-2.з5б5моль.л-1=
=4,4-10-3.
Полученные результаты нельзя считать окончательными,
поскольку ионизация акво-ионов во внимание не принималась.
Можно, однако, получить несколько очень важных качественных
оценок:
1) безусловно, g<^G и G — g — G;
2) -^m/t-i = [FeOH+]~ 1,2 • lO-s,'Qi6/t-i=[FeOH2+l~8,5.10-2;
7)22б%-2 = (Fe, (OH)t+] ~ 0,23, -Qh-^ ~ 2,7 • 10-* « 1 моль • л-i.
С учетом этих данных система (6.58) — (6.60) принимает вид:
М + В = т+Ь(1 + тцЛ-') + 2'qnb^h-\ (6.59)
H = h — -Qjbh-^ — 2i322&%-"^ + 4G; (6.60)
A — (2уИ + ЗБ + Я) 4- 2/п 4- Ь (3 + 2-5,/i-i) + ЦгФ^Н-^ = 0. (6.61)
Суммируя (6.59) и (6.60), находим, что M + B+H — 4G =
= m+b + h (6.62).
Подстановка значения т из (6.59) в (6.61) приводит к
равенству B + H^b + h (6.63).
Из (6.62) и (6.63) следует, что m = М — 4G = fFe2-i-] = 4,44х
X 10-2 jjojjb • л-' в полном согласии с приближенным расчетом.
Подставляя в (6.59) m = Л! — 4G и 6 = В Ч- Я — Л из (6.63),
приходим после обычных преобразований к уравнению
— (2-(i22 — ni+H — 4G)h^—{B + H){nx — 4-522)h — 2^22 X
Х{В+ ну == 0. (6.64)
После замены всех символов их значениями получаем
уравнение /гз — 4,640 • — 6,660 . 10-5/г — 1,0175 • 10-» = 0.
Решение по Хорнеру дает Л = 2,285 • IO-2 ±= lO-'-^^n моль • л-'.
Последующий расчет по (6.63) дает b = 3,715 • IO-2 = 10-1.4300 ^
по (6.57) —g=\,\- 10-''5 моль • л-1.
В табл. 6.2 приведены результаты расчета равновесного сое.
тава системы Fe (€104)2 — Fe (С104)з — HCIO4 — О2 — Н2О и
проверки их согласованности с уравнениями (6.54) — (6.56).
Согласованность достигнута. Тем самым подтверждается
пригодность принятого хода решения. Результаты, конечно, имеют
приближенный характер, так как получены при <Pf = 1.
Равновесный потенциал системы при 2 > "С и условии <pi = 1
Е = е=р,з+,ре2+ - 0,05916 = 0,767 + 0,0047 = 0,772В.
191
Таблица 6.2
Уравнение
[н+]
[Fe2+]
[реОН+]
[Ре3+]
[реОн2+]
[ре2(ОН)2"^]
g
2
2,285-10—2
0.26
4,44-10—2
2.34.10—6
3,715.10—2
1,099-10—2
3.734-10-3
1,4.10-3
7.58
[Fe2+]
[FeOH+]
[Fe^+]
[FeOH2+]
2[Fe2
(0Н)|+]
4,44-10-2
2,34.10~^
3,715-10~2
1,099.10-2
7,468-10-^
0,10001
0,1
7.59
[Н+]
~[РеОН+]
-[FeOH2+]
(он)2^-Ь]
4G
Н
2,285.10-2
-2,34.10-"^
—1,099.10-2
-7,468-10-^
5,6.10-3
9,99.10-3
1.10-2
7.60
[н+]
-[СЮ7]
2[Fe2+]
[РеОН+1
3 [Fe^+l
2 [FeOH2+]
4[Fe2
2,285-10-2
-0,26
8,88 10~2
2,34.10~^
0,11145
2,198 10-2
1,4936-10-2
1,83 10""^
0
6.5. ЦВЕТНЫЕ ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ
ИНДИКАТОРЫ
6.5.1. Общие сведения. Все кислотно-основные равновесия
включают оксоний-ион. Он же входит в равновесие любого
цветного кислотно-основного индикатора: Hid + Н2О 5=^ Id- + Н3О+.
Именно поэтому (Н3О+) = (Hid) • (Id-)"-^ • К и интервал
перехода. рН оказывается равным р/С ± 1.
Окислительно-восстановительные равновесия устанавливаются
в подавляющем большинстве случаев лишь при наличии двух
сопряженных окислительно-восстановительных полуреакций,
ведущих к суммарному равновесию:
К?
Ок1 + пе 5=:±Bci I
Вс2 —те;=±0к2|
тОк1 + пВс2^ mBci + пОк2 К = ЛГ/СГ" ^^'^^^
При расчетах систем, включающих такие равновесия, чаще
всего ищут не отдельные активности, а отношения активностей
участников каждой из сопряженных полуреакций. На эти
отношения должны, очевидно, отзываться и соответствующие
цветные окислительно-восстановительные индикаторы.
Допустим, что сопряженной системе такого индикатора
отвечает равновесие
ОкИ+ре«ВсИ,/Си.
Эта система должна одновременно взаимодействовать
основными. Тогда
\К1
(6.66)
обеими
ОкИ+ре5=±ВсИ
Bci — пе«0к1
пОкИ+ pBci ^пВсИ + рОк1 Ki = KIKT^
(6.67)
ОкИ+ ре гйВсИ
Вс2 — те .=г Окг
2
тОкИ+ фс2^тВсИ+ рОкг Км = КЧ^КТ^ ^^'^^^
Из (6.67) и (6.68) следует, что по закону действующих масс для
любой сопряженной i-й системы OKj + vessBci,
взаимодействующей с цветным индикатором,
Г(ОкИ)
(0«i)
(ВсИ)
К.
(6.69)
Если индикатор двуцветный, то, как и для
кислотно-основных индикаторов, при отношении = Ю, если око близко
19а
К ^g^t ВСЯ система имеет окраску ОкИ; если то же отношение
равно единице, система будет окрашена в промежуточный цвет,
а при отношении, равном j^y примет окраску чистой ВсИ. ,
Для одноцветного индикатора, окисленная форма которого
окрашена, а восстановленная — бесцветна, окраска будет
обнаруживаться, естественно, тем раньше, чем выше начальная
концентрация индикатора.
Путем логарифмирования уравнения (6.69) и обычных
преобразований находим для разных значений (ОкИ)/(ВсИ)
следующие выражения:
ОкИ
(ВсИ)
(Око
(ВС.)
Окраска
10
1
0.1
рл.-| р/с. +7
рл.-урл,.-у
ОкИ
промежуточная
ВсИ
Допустим, например, что система, содержащая Fe^^- и Ре^-Ь-
ионы, окрасилась в синий цвет после введения в нее
дифениламина. Что можно сказать относительно величины (РеЗ+)/(Ре2+)?
Дифениламин — одноцветный индикатор. Синяя окраска
возникает, если (ОкИ)/(ВсИ) > 0,1. Для дифениламина р = 2 Ig Ки =
= 25,7 (см. табл. 6.3). Ig/<'реЗ+,ре2ч-= 12,97. ^
В этом случае
>0,24.
lgg^> 12,97- 1(-25,7)-0,5=-0,62,
(Fe^-b)
4Fe2-»-) ^ 2^ » (Fe2+) "
Для расчета (OKi)/(Bci) можно воспользоваться
непосредственно значениями стандартных потенциалов полуреакций.
Соответствующая формула получается путем логарифмирования (6.69) и
подстановки значений Ig/Ci и lg/Си по (6.20):
. (0«i) v ,рО c-Ov, v,(OkH)
(6.70)
•.71)
Например, для только что разобранного примера при f = 25 °С
,(Fe^-<^) 1 .(0,76 -0.767) 1 ^ g^g
Ту же формулу (6.69) можно преобразовать в выражение для
расчета lg/Си, т. е. оценки константы окислительно-восстанови-
194
тельного индикатора, который заметно изменит окраску, когда
отношение (OKi)/(BCi) примет заданное значение. Искомая
формула имеет вид
(6.72)
Подберем, к примеру, одноцветный индикатор с р = 2, заметно
изменяющий окраску в системе, где (Fe^'*+)/(Fe2+) = Ю^. Для
Fe3+ + e5=iFe2+ Ig/Ci = 12,97 и v = 1. Положим, что заметное
изменение окраски происходит при (ОкИ)/(ВсИ) = 0,2. Тогда
искомый lg/(„ = 2 (12,97 + 0,7 + 5) = 37,3. -
Все окислительно-восстановительные процессы связаны с
переносом электронов. Это значит, что и окисленная, и
восстановленная формы индикатора, или, по крайней мере, одна из них, могут
принимать участие в кислотно-сснпвных равновесиях, ведущих к
изменению значений (Oki), (Bci), а значит, и окраски системы.
Но тогда практически во всех расчетах, связанных с
применением окислительно-восстановительных индикаторов, надо учитывать
влияние рН. Пусть Си = и. По закону сохранения гг = Соки + ^вси,
где d — аналитические концентрации. Если 9i= 1, то
«ОкИ «ВсИ
г/= сокисвси = (ОкИ) 2 с^1.А^+(ВсИ) £ a,iAi (6.73)
и окраску системы определяет отношение (ОкИ)/(ВсИ). В общем
случае
«ВсИ
(ОкИ) --Оки jio
(ЬСИ) Свеи «Оки . '
НО
(6.74)
где индекс 1 относится к ОкИ, а 2 —к ВсИ. При заданном рН
уравнение (6.73) включает две неизвестных величины—(ОкИ),
(ВсИ). Вычислить их можно лишь при полном расчете равновесия
в данной конкретной системе, обязательно включающей второе
сопряженное окислительно-восстановительное равновесие: Ок^ +
+ ve5=tBci.
Однако некоторое представление о влиянии рН можно
получить с помощью уравнения (6.74). Допустим, что формула
окисленной формы Ок-, а восстановленной — Вс^-*. В этом случае
авси = 2, аоки = 1 и (6.74) можно переписать в виде
(Ок-) ^ОкИ ^22^4^21^ + 1 .^7^.
(i?=) ^ ^ • ^uh + l • ^^-^^^
Подстановка значений с22 = KTi^KT2 9 <'2i = К22 и ац = КТх
приводит к выражению
195
(Ьс2-) С^,^ ' /(21/C22 * '^+^11 •
Если учесть, что К\—ступенчатые конвтанты диссоциации
соответствующих кислот и что всегда /C2i>K22, то можно
утверждать, что при всех А, удовлетворяющих условиям
Л</(2. и h<^Kiu (6.77)
окраска системы не будет зависеть от рН. В случаях, когда эти
условия не выполняются, окраска от рН зависит и это надо
учитывать.
6.5.2. Табличные данные. Из раздела 6.5.1 следует, что для
каждой окислительно-восстановительной* индикаторной системы
надо знать стандартный потенциал (£«,) или константу (/(и),
число электронов (р), а также константы протонизации (o2j) и (aij)
или ступенчатые константы диссоциации (/(21 и К и). В работе
14J таблиц окислительно-восстановительных индикаторов нет.
В сводной табл. 6.3 приведены их основные характеристики
{М — молярная масса (г/моль), окраска: б — бесцветная,
к — красная, ж — желтая, з — зеленая, г — голубая, с — синяя,
ф — фиолетовая). Из уравнений (6.67), (6.68) следует, что
изменение отношения (ОкИ) / (ВсИ) до значения, отвечающего
изменению окраски в ту или иную сторону, связано с расходом
окисленной или восстановленной форм основной системы.
Расход этот тем меньше, чем интенсивнее поглощают свет в
видимой области окрашенные формы индикатора. В связи с этим
в качестве окислительно-восстановительных индикаторов, как
видно из табл. 6.3, используются в основном красители и
комплексные ионы.
6.5.3. Применение цветных окислительно-восстановительных
индикаторов. Если система с введенным в нее кислотно-основным
двуцветным индикатором имеет ту же окраску, что и молекулы
НЫ, ее рН < р/с — 1; для интервала перехода рК — 1 < рН <
< р/с + 1 и при окраске, совпадающей с окраской Id-, система
может иметь любой рН > р/( + 1.
Путем обычных преобразований можно привести формулу
(6.72) к виду
р (ОкО = р (BCi) Н Ig ^щщ. (6.78)
При (Bci) = 1 для значений (ОкИ) (ВсИ)-^, равных 0,1 и 10,
формула (6.78) превращается в выражение
p(OK.) = ^ii^^if^''±l. (6.79)
Оно позволяет рассчитать p(OKi)j при котором происходит
изменение окраски индикатора при данных Ig/Cip IgKnt р и v, еели
в системе, где (Bqi) = 1, создаются различные по величине (Oki).
196
Таблица 6.3
Название
Т ри- 2 >2' -д ипи рид ил о рутених ло-
рид
Три-нитро-о-фенантролинофер-
ро-хлорид (нитроферроин)
Ы«Фенилантраниловая кислота
Три-1,10-фенантролиноферро-
хлорид (ферроин)
дг-Этоксихризоидиний ;слорид
Три-2,2'-дипиридилоферро-хло-
рид
Три-5,б-диметил-1,10-фенан-
тролиноферро-хлорид
0-Дианизидин
М. г-моль"""'
Приготовление раствора
676,0
802,4
213,2
667,4
292,8
595,3
335.0
244.)
В разбавленном растворе
HCI
0,025 моль.л~^; в воде
0,2% раствор в Воде
0,025 мольл-»; 1,624 г
фенантролиний хлорида
и 0,695 г FeS04 в 100
мл HgO
Водный раствор
В разбавл. НС1
0,025 моль.л~* в воде
В разбавл НС1
Окраска
Вс И Ок И
Ж
ф
Ж—3
1,33 22,5
1,25
1,08
1,06
1,00
0,97
0,97
0,85
21,1
36,5
17,9
33,8
16,4
16,4
28,7
10
со
1
Дифениламинсульфонат натрия
Дифенилбензидин
Дифениламин
2,6-Дибромбензолиндофенолят
натрия
2,6-Дихлорфенолиндофенолят
натрия
'0-Крезолиндофенолят натрия
Диаминофенотиазиний-хлорид
(тионин, фиолетовый Лаута)
Метиленовая синяя
Индиготетрасульфоновая кис«
лота
Индиготрисульфоновая кислота
Индигодисульфоновая кислота
(индигокармин)
Индигомоносульфоновая
кислота
Феносафранин
Сафранин Т
Нейтральный красный
1-Нафтол-2-сульфоновая
кислота
272,3
336,4
169,2
379,0
290,2
234,3
257,7
283,7
554,5
474,4
394,3
314,3
317,8
345,8
288,8
224,2
Продолжение табл. 6,3
0,05% в воде
1% в конц.. H2SO4
То же
0,02% в воде
То же
0,05% в 60%-ном спирте
0,05% в воде
0,05% в воде
0,05% раствор в воде
То же
»
0,01% раствор в 60%-
ном спирте
0,05% раствор в воде
6
б
б
б
б
б
б
б
б
б
б
б
б
к—ф
Ф
Ф
с
с
с
к
ф—к
к
2
2
2
0,84
0,76
0,76
0,64
0,64
0,62
0,56
0,53
0,37
0,33
0,29
0,26
0,28
0,24
0,24
0,65
28,4
25,7
25,7
21,6
21,6
21,0
18,9
17,9
12,3
11,2
9,8
8,9
9,5
S,l
8,1
21,7
8,7
10
6,95
7,3
7.8
9,05
Так, при введении РеЗ+-ионов в систему, где (Fe2+)=1, с
учетом lg/CFe3+,Fe2+= 12,97 и v= 1 будут возникать окраски,
отвечающие ОкИ разных индикаторов, приведенных в табл. 6.3,
при р (Ре2+), указанных в табл. 6.4 (р = 2).
Таблица 6.4
Индикатор
Переход
окраски
Р(реЗ+)
Сафранин Т
Индиготетрасульфоновая кислота
Метиленовая синяя
Натрий-о-крезолиндофенолят
Дифениламин
о-Дианизидин
п-Этоксихризоидиний хлорид
N-Фенилантраниловая кислота
8,1
12,3
17,9
21.0
25,7
28,7
33,8
36.5
б-^ ф —к
б-»- С
б-^ С
б-^С
б-^С
б-^К
ж-^ к
б->ф
8.9
7.8
5.0
3.5
1,1
-0,4
-2,9
-4.3
Как следует из табл. 6.4, возможна дифференцированная
оценка р(РеЗ+) с помощью ряда цветных
окислительно-восстановительных индикаторов.
Для системы МпОГ» Мп2'*" осуществить такую оценку нельзя,
так как в этом случае Ig/Ci = 127,4, v = 5 и даже для N-фени-
лантраниловой кислоты переход окраски б->'ф осуществляется
при р(МпОГ) =37,2.
В некоторых случаях в исследуемой системе можно создать
значения р (Bci), существенно отличающиеся от нуля, и таким
путем расширить интервал р (Oki), которые можно оценить с
помощью данного набора индикаторов. Соответствующие значения
p(Bci) должны быть фиксированными. Этого можно добиться,
применяя труднорастворимые соединения, в решетку которых
входит Bci, достаточно прочные комплексы или вторую жидкую
фазу, насыщенную Bci.
Для системы Ре (CN)6~ + е ;=tpe (CN)!"", v = 1, Ig /d = 6,02.
Тогда окраску ОкИ примут все системы, содержащие
индикаторы, для которых р == 2 и
lg/CH=2[lg/Ci+ l-pPe(CN)^].
(6.80)
Поскольку практический интерес представляет оценка рРе(СН)б"">
> О, нас могут устроить в данном случае все индикаторы, для
которых lg/Си < 2(6,02+ 1)~14. Таких индикаторов в табл. 6.3
всего 7. С их помощью можно охватить значения pPe(CN)e~ от
0,87 для индиготетрасульфоновой кислоты до 2,97 для сафранина Т.
В той же системе можно создать путем введения в нее твердой
соли Zn2pe(CN)6, для которой произведение растворимости Р =
= lO-^s'-*, фиксированное значение р Ре (CN)6~ = 5,3. Тогда, как
следует из (6.78), при pPe(CN)i- = О и (ОкИ) (ВсИ)-1 = 0,1 ока-
199
жутся пригодными индикаторы, для которых при p=21g/CH<
< 24,6.
Подобным же образом, используя в качестве второй жидкой фазы
раствор 12 в ecu или СНСЬ, можно несколько увеличить число
индикаторов, пригодных для систем, в которых h оказывается
восстановленной формой.
Попробуем применить теперь (6.78) к равновесию
НзО+ + 1/2 Н2 (г) + Н 20. (6.81)
Для него по условию lg/C = 0, рн, = 1 атм (1,01325- Ю^Па), р =
= 1 и уравнение (6.78) превращается в известное уравнение:
р (НзО+) = рКи ± 1 (6.82) для интервала перехода двуцветного
кислотно-основного индикатора. Оказывается, что уравнение
(6.82) можно рассматривать как частный случай уравнения (6.78).
Цветные окислительно-восстановительные индикаторы
используют в качестве систем, отмечающих наличие
эквивалентности в случае взаимодействия двух сопряженных окислительно-
восстановительных систем. В подобных случаях необходимо
вычислить отношение (Oki) (Bci)""^ для ддной из систем в момент
эквивалентности, рассчитать подходящее значение Ig/(и и выбрать
индикатор.
Рассмотрим в качестве примера окисление Ре^^-иона.
Используя методы приближенного расчета при рН = О, получаем
к К
Сг207^ + 14Н+ + бее 2СгЗ+ + 7Н2О
Fe2+ — е;=гРеЗ+
136
(—12,97)6
Сг20|"+ 14H++6Fe2+ «2СгЗ+ + 7Н2О + бРе^н- 58
с А 6Л - -
[] а рН=0 6а ^ 2(А—а) 6(Л —а)
Пусть А = 0,02 моль . л-* и, судя по значению Ig/C, а = [Сг207~]<
< Л. Тогда по закону действующих масс 2—10-57.4 j^c-
пользуя неравенство а<Л только в одном из восьми
сомножителей знаменателя, находим оценку для а(Л — а)-^ = (Ре2+) х
X (РеЗ+)-1= ]/10-57.4^ ^ 10-8'44 соответствснно, (РеЗ+)х
X (Ре2+)-1 = 108'44.
Тогда согласно (6.72) из числа индикаторов, для которых
р = 2, пригодны все с lg/Си = 2(13 + 1 + 8,44) = 44,9, а для
случая р = 1 — все с lg/Си = 22,4.
Среди индикаторов, приведенных в табл. 6.3, только три-2,2'.
дипиридилорутенихлорид имеет подходящее значение Ig/C. К
сожалению, изменение окраски от желтой к бесцветной в системе,
200
где оранжевая окраска СггО?"" сменяется зеленой окраской
хромикомплекса, отчетливого сигнала о достижении эквивалент,
ности дать не может.
Выход из положения следует искать в этом случае в
снижении значения (РеЗ+) за счет комплексообразования. Этот прием
можно назвать подгонкой системы к индикатору с заданными
значениями р и lg/Си. Так, в работе [52] было показано, что при
добавлении раствора К2СГ2О7 к сернокислому раствору Ре^^-ионов,
содержащему соответствующее количество Н3РО4 или фторида,
индикатор дифениламин окрашивает систему в синий цвет как
раз в момент достижения фактической эквивалентности. Для
дифениламина р = 2, lg/<' = 25,7. Тогда, как следует из (6.72),
Ig [(РеЗ+) (Ре2+)-1] = 1/2 . 25,7 — 13 — 1^ = —1,15. Это соотноше.
ние достигается, по-видимому, в случае приливания раствора
К2СГ2О7 к раствору Ре2+-ионов в Н3РО4, за счет превращения
РеЗ+-ионов в РеНРО^-ионы, а при наличии Р--ионов—в Н2рер5.
Расчеты здесь провести трудно из-за отсутствия всех
необходимых данных.
список ЛИТЕРАТУРЫ
1. Арис Р. Анализ процессов в химических реакторах: Пер. с англ.—Л.; Химия,
1967.—328 с.
2. Бугаевский А. А. Исследование равновесий, описывающих системы со
ступенчатыми равновесиями: Автореф. дис.... канд. хим. наук. —X., 1965. — 16 с.
3. Бугаевский Л. Л. Расчет химических равновесий.—X.: Изд-во Харьи. ун-та,
1980.— 136 с.
4. Бугаевский А. Л. Таблицы констант важнейших равновесий, имеющих
значение в аналитической химии. — X.: Изд-во Харьк. ун-та, 1967. — 67 с.
5. Бьеррум Я» Образование аминов металлов в водном растворе: Пер. с англ.—
М.: Изд-во иностр. лит., 1961.—308 с.
6. Васильев Л. М., Троухина В, Я. Полярографическое изучение устойчивости
хлористых и бромистых комплексов кадмия и свинца.—Журн. аналит.
химии, 1951, 6, вып. 4, с. 218—222.
7. Вознесенский С. Л. Внутрикомплексные соединения и их значения для
аналитической химии.— М.; Л.: ГОНТИ, 1938.— 131 с.
8. Грей Г. Электроны и химическая связь: Пер. с англ.—М.:Мир, 1967.-—227 е.
9. Гринберг А. Л. Введение в химию комплексных соединений.—М.; Л.:
Химия, 1966.-631 с.
10. Гуггенгейм Е. Л. Современная термодинамика, изложенная по методу
У. Гиббса: Пер. с англ. — М.; Л.: Госхимиздат, 1941.—188 с.
11. Индикаторы/Пол ред. Э. Бишопа: Пер. с англ.—М.: Мир, 1976.—Т. 1. 476 с-
12. Кабанов Ё. Я., Темкин М, И. К вопросу о знаке электродного потенциала
и электродвижущей силы.—Журн. физ. химии, 1954, 28, вып. 12,
с. 2258—2261.
13. Карякин Ю. В. Кислотно-основные индикаторы. — М.; Л.: Госхимиздат,
1951. —198 с.
14. Комарь Н, П. Измерение параметров равновесий в растворах. — Журн.
•аналит. химии, 1975, 30, вып. 3, с. 421—442.
15. Комарь Я. Я. Математическая оценка чувствительности химических реакций.
Сообщение 1. — Уч. зап. Харьк. ун-та, 1951, 37. Тр. хим. ф-та и НИИ
химии, 1951, 8, е. 143—148.
16. Комарь Я. Я., Кисилевский В. В., Бехер Р. М. Математическая оценка
чувствительности химических реакций .Сообщение 3. Реакция на Ре^+-ион
с роданидами.—Уч. зап. Харьк. ун-та, 1951, № 12, с. 120. 37. Тр. хим.
ф-та и НИИ химии, 8, с. 159—165.
17. Комарь Н. Я., Толмачев В, Я., Рыбина О. В. Спектрофотометрическое
исследование бромфенолового синего (тетрабромфенолсульфофталеина) (!).—
Уч. зап. Харьк. ун-та, 1951, 54. Тр. хим. ф-та и НИИ химии, 1954, 12,
«. 215—224.
^8. Коршунов Я. Л., Малюгина Я. Я., Балабанова О. М, Полярографическое ис-
следование комплексов кадмия с некоторым» одновалентными анионами
Журн. общ. химии, 1951, 21, вып. 4, с. 620-625.
202
19. Коттоя Ф., Уалкиясон Дж. Современная неорганическая химия: Пер.
с англ.—М.: Мир, 1969 —Т. 1—3.
20. Кумок е. Н. Закономерности в устойчивости координационных соединений
в растворах, Томск: Изд-во Томск, ун-та, 1977.—230 с.
21. Латимер В. Окислительные состояния элементов и их потенциалы в водных
растворах: Пер. с англ.—М.: Изд-во иностр. лит., 1954.—400 с.
22. ЛолсояособМ. В. Избранные философские произведения.—М.: Госполитиздат,
1950.—758 е.
23. Марк Г., Рехниц Г. Кинетика в аналитической химии: Пер. с англ.— М.»
' Мир, 1972.—368 е.
24. Менделеев Д. Я. Основы химии. — М.; Л.: Госхимиздат, 1947, —Т. 1. 624 с.
25. Мухина Т. Я., Рудная Л. Е., Бугаевский А, А. Расчет равновесий в сложных
системах. Сообщение 4. Оценка буферных свойств системы.— Жури, аналит.
химии, 1970, 25, вып. 4, с. 640—645.
26. Назаренко В, Л., Антонович В. Я., Невская Е. М. Гидролиз ионов металлов
в разбавленных растворах.—М.: Автомиздат, 1979.—192 с.
27. Оксредметрия / Пол ред. Б. П. Никольского, В. В. Пальчевского. — Л:.
Химия, 1975.-304 е.
28. Писаржевский Л., Розенберг М, Электрон в химии растворов и в электро
химии.— X.; Гос. изд-во Украины, 1923.—137 с.
29. Полинг Л. Общая химия: Пер. с англ.— М.: Мир, 1964—583 с
30. Современная химия координационных соединений/Под ред. Дж. Льюиса,
Р. Уилкинса: Пер. с англ.— М.: Изд-во иностр. лит., 1963.— 445 с
31. Спектроскопические методы в химии комплексных соединений / Под ред.
8. М. Вдовенко, М.; Л.: Химия, 1964—268 с.
32. Теренин А. //. Фотохимия красителей и родственных органических
соединений.—М.* Л.: Изд. АН СССР, 1947.—353 с.
33. Терпи Т. Механизмы реакций окисления-восстановления: Пер. с англ—
М.: Мир, 1968.—238 с.
34. Усанович М. И. О кислотах и основаниях.— Журн. общ. химии, 1939,
9, выи. 2, е. 182—192.
35. Химия координационных соединений/ Под ред. Дж. Бейлар, Д. Буш: Пер.
о англ.— М.: Изд-во иностр. лит., 1960.—695 е.
36. Яцимирский К. Б.^ Васильев j5. Я. Константы нестойкости комплексных
соединений.—М.1 Изд. АН СССР, 1959.—206 е.
37. Яцимирский /С. 5., Тетюшкина В. Д. О влиянии ионной силы на
константы нестойкости галогенидных и пеевдогалогенидных комплексных
соединений.—Журн. неорг. химии, 1957, 2, вып, 2, с 320—329.
38. Ackermann Th, Hydration or Н"^" and ОН~ ions in water from heat
capacity measurements.—Disc. Faraday Soc, 1957, № 24, p. 180—191.
39. Ardon M., Mayer B. E. The oxidation of cromous perchlorate with sodium
azide.—Joum. Cham. Soc. 1962, JVb T—?, p. 2816—2818.
4a Br ode W. R. The. determination of the hydrogen-ion concentration by a
spectrophotometric method and the absorption spectra of certain indica^
tors. — Joum. Amer. Chem. Soc, 1924, 46, h^? 3, p. 581—596.
41. Hrdnsted J, N. Einige Bemerkungen uber c!en Begriff der Sauren und Ba-
sen.—Rec. trav. chim. fays—Bas, 1923, 42, S, 718—728.
203
42. Brüll L. Activity in solutions of zinc chloride.—Gazz. chim. ital., 1934,64,
p. 261-270.
43. Chariot G., Bezier Z)., Courtot /. Constantes selectionnees. Potentiels d*oxydo-
reduction.—Paris: Pergamon Press, 1958.—41 p.
44. Critical stability constants.— New York; London: Plenum Press, Aminoacids/
CompL by A. E. Martell, R. /I. Smith. 1974.— V. 1. 469 p.
45. Cuta f.. Kamen /C. A universal indicator for determining pH in the range
1,2—12,7 and for use in volumetric analysis.—Coll. trav. ehim. tehee, 1936,
8, p, 395—407.
46. Davies C. W,^ Hoyle /. The dissociation constants of calcium hydroxide.—
Journ. Chem. Soc, 1951, K9 1, p. 233—234.
47. Graner F., Stilen L. G. — Acta chem. Scand., 1947, 1, p. 631.
48. Gryder J. W. The estimation of the validity of stepwise association
constants.—Joura Amer, Chem. Soc, 1955, 77, Kg 23, p. 6196—6197.
49. Irving H M. International Conference on co-ordination chemistry, London-
April 6th-11th 1959, Special publication .N'9 13. London, The Chemical
Society, Burlington House, 1959, 1, p. 13.
50. Jo^il C, Anbar M. The effect of ionic hydration on rate and equilibrium
in concentrated alkaline solutions. I. The H function in aqueous alkaline
solutions and the hydration on the OH ion.—J oil rn. Amer. Chem. Soc.,
1963, 85, Ko 16, p. 2376—2380.
51. Kielland J. Individual activity coefficients of ions in aqueous solutions.—
Journ. Amer. Chem. Soc., 1937, 59, p. 1675—1678.
52. Knop y. Diphenylamine as indicator in the titration oí iron with dich-
romate solution.— Journ. Amer. Chem. Soc., 1924, 46, 2, p. 263—269.
53. Kortum G. Lehrbuch der Elektrochemie. 3 Aufl., Weinheim /Bergstr.: Chemie,
1962.—564 S.
54. Kortum G., Vo^el W.^ Andrussow K. Dissociation constants of organic acids
in aqueous solution. London, Butterworths, 1961.—345 p.
55. Manov G. G., Bates R. G., Hammer W. Acree S. F, Values ef the
constants in the Debye-Hückel equation for activity coefficients.—Journ. Amer.
Chem. Soc., 1943, 65, Ke 9, p. 1765—1767.
56. Mattock G. pH- Measurement and titration. London, Heywooda. a. Co LTD,
1961.-406 p.
57. Nancollas G. H. Interactions in electrolyte solutions. Elsevier Publ. Co,
Amsterdam, London, New-York, 1966.—214 p.
58. National standard reference program.— NBS Technical Note, 1963, 19\
59. Olin i4. Studies on the hydrolysis of metal ions. 19. The hydrolysis of wis-
muth (III) in Perchlorate medium.—Acta Chem. Scand., 1957, 11, p. 1445—1456,
60. Perrin D. D. Dissociation constants of organic bases in aqueous solution. —
London, Butter-worths 1965.—524 p.
61. Serjent E. P., Dempsey B. Ionisation constants of organic acids in
aqueous solutions. Oxford, Pergamon Press LTD, 1979.—998 p.
62. Sillen L G. International Symposium on the chemistry of the coordination
compounds.—R icerca sclent., 1958, anno 28, p. 3—22.
204
63. Smith О. f., Duke F. R. Cerate oxidimetry. Determination of glycerol.—
Ind.Eng. Chem., Anal. Ed., 1941. 13, p. 558—560.
64. Stability constants of m.etal-ion complexes. Oxford, Pergamon Press.— Part
A. Inorganic ligands / Compl. by Hogfeldt F., 1980—1200 p.
65. Stability constants of metal-ion comllexes. Suppl. 1, Special publication
№ 25/b. G. Compl. by Sillen, A. E. MartelL—London, Chem. Soc, 1971.—
865 p.
66. Stability constants of metal-ion complexes. Specia' publication ^^ l7/Compl.
by Sillen L. G., Martell A. E., London, Chem. Soc, 1964. 754 p.
67. Solvaied electron/Ed. Hart E. J. Ad van. Chem. Ser. v. 50. Washington.
AmeF. Chem., Soc. 1965.—304 p.
68. Tuck D. G., Diamond R. M. The primary solvation of the proton in the
solvent exstraction or strong acids.—Journ. Phys. Chem., 1961. 85, № 2,
p. 193—198.
69. Wormser J. Remarques sur theorie des reactions reversibles par degres de J.
Bjerrum.-='BuU. soc ehim., France, 1954, № 3. p. 387—392.
ОГЛАВЛЕНИЕ
Предисловие 3
206
1. Системы, изучаемые в теории ионных равновесий
1.1. Основные понятия .••.•»»•••• 5
1.2. Растворители ....«••»••»*• 6
1.3. Сильные и слабые кислоты 6
1.4. Сильные и слабые основания • « 7
1.5. Сольватация (гидратация) ионов 8
1.6. Некоторые примеры записи равновесий . . • 10
1.7. Алгебраическая форма записи равновесий . • 11
1.8. Применяемые символы . . • 12
1.9. Матричная запись систем равновесий . . » 14
2. Системы уравнений, необходимых
для расчета равновесий
2.1. Оценка возможности расчета равновесия с
помощью матрицы системы 18
2.2. Связь между активностями и равновесными
концентрациями 18
2.3. Теоретические уравнения для вычисления
коэффициентов активности 19
2.4. Эмпирические уравнения для расчета
коэффициентов активности . 20
2.5. Уравнения и параметры, приспособленные для
вычисления индивидуальных коэффициентов
активности на широком интервале ионных сил ... 23
2.6. Сравнение значений коэффициентов активности,
вычисленных по разным формулам 25
2.7. Значения коэффициентов активности при
использовании закона действующих масс 26
2.8 Рациональное использование существующих
приемов расчета коэффициентов активности . . . 27
2.9. Несколько формулировок закона сохранения
материи . . , 28
2.10. Последующие попытки опытной проверки закона
сохранения массы 29
2.11. Современная опенка точности закона сохранения
массы . . . . . , 31
2.12. Различные выражения закона сохранения массы. 32
2.13. Закон сохранения электронейтральности . . 34
3. Константы равновесий и связь между ними
3.1. Важнейшие виды равновесий и их константы . ^ 35
3.2. Соотношения между различными видами констант,
относящихся к одним и тем же равновесиям . . 36
3.3. Переход от различных табличных значений
констант к значениям, применяемым при расчетах ^ 41
3.4. Вычисление констант, отсутствующих в таблицах,
с помощью известных констант смежных
равновесий , . 45
3.5. О полноте и достоверности справочных данных,
приведенных в таблицах констант химических
равновесий ' , . 48
4. Кислотно-основные равновесия
4.1. Расчет равновесий в водных растворах инертных
солей , 51
4.2. Расчет равновесий в водных растворах сильных
кислот и оснований 53
4.3. Системы уравнений, характеризующие равновесия
в растворах слабых кислот и их солей .... 55
4.4. Характеристика отдельных блоков, входящих в
расчетные уравнения кислотно-основных
равновесий, и их использование 59
4.5. Функции Бьеррума и их применение при
сокращенной записи расчетных уравнений
кислотно-основных равновесий 61
4.6. Выяснение условий, допускающих упрощения при
решении расчетных уравнений кислотно-основных
равновесий. Критерии, определяющие
правомерность упрощений 63
4.7. Примеры некоторых расчетов, которые можно
проводить без применения ЭЦВМ 77
4.8. Равновесия в растворах цветных
кислотно-основных индикаторов .102
5. Равновесия комплексообразования
5.1. Роль комплексообразования в химии 122
5.2. Равновесия комплексообразования и их представление с помощью химических уравнений ... 123
5.3. Константы равновесий комплексообразования . . 126
5.4. Матрицы систем, включающих равновесия комплексообразования. Расчетные уравнения ... 135
5.5. Некоторые замечания 142
5.6. Некоторые типичные задачи, связанные с равновесиями комплексообразования 144
5.7. Конкретные примеры расчетов равновесий в системах с комплексообразованием 148
5.8. Краткие выводы • • 165
6. Окислительно-восстановительные
равновесия
6.1. Общие сведения об окислительно-восстановительных индикаторов 165
6.2. Применение закона действующих масс к окислительно-восстановительным равновесиям . ... 173
6.3. Таблицы констант сопряженных окислительно-восстановительных равновесий 178
6.4. Суммарные окислительно-восстановительные
равновесия и их особенности ... 181
6.5. Цветные окислительно-восстановительные
индикаторы , , . 193
Список литературы 202
НИКОЛАЙ ПЕТРОВИЧ
КОМАРЬ
химическая
метрология
ГОМОГЕННЫЕ
ИОННЫЕ
РАВНОВЕСИЯ
Редактор Л. Ф. Кизилова
Переплет художника В. Б, Мартыняка
Художественный редактор В. Е, Петренко
Технический редактор Г. П, Александрова
Корректор Л. М. Забродина
Информ. бланк № 7097
Сдано в набор 15.12.82. Подп. в печать 09.08.83. БЦ 17028.
Формат 60X90/ie. Бумага, типогр. № 3. Лит. гарн. Выс. печать,
13 печ. л. 13,25 кр. отт. 16 уч.-нзд. л. Тираж 2000 экз. Изд.
№ 989. Зак. 2-436. Цена 2 р. 60 к.
Издательство при Харьковском государственном университете
издательского объединения «Вища школа». 310003, Харьков-3,
ул. Университетская, 16
Харьковская книжная фабрика «Коммунист», 310012, Харьков-12,
ул. Энгельса, 11
химическая
метрология
Н.П. Комарь
ГОМОГЕННЫЕ
ИОННЫЕ
РАВНОВЕСИЯ
харьков
издательство при харьковском
государственном университете
издательского объединения
«вища школа»
1983