














































































































































































































































































































































































































Автор: Хмельницкий Р.А.
Теги: химия физическая химия химическая физика коллоидная химия
ISBN: 5-06-001257-3
Год: 1988




Текст
P. А. ХМЕЛЬНИЦКИЙ
ФИЗИЧЕСКАЯ
И КОЛЛОИДНАЯ
ХИМИЯ
Допущено Министерством высшего
и среднего специального
образования СССР
в качестве учебника
для студентов сельскохозяйственных
специальностей высших
учебных заведений
Москва „Высшая школа“,1988
ББК 24.5
Х65
УДК 541.1
Рецензенты:
кафедра органической, физической и коллоидной химии Кубанского
сельскохозяйственного института (зав. кафедрой В. Д. Зиновьев)
и проф. В. М, Глазов (Московский институт электронной техники)
Хмельницкий Р. А.
Х65 Физическая и коллоидная химия: Учеб, для с.-х.
спец, вузов. — М.: Высш, шк., 1988. — 400 с.: ил.
ISBN 5—06—001257—3
В учебнике изложены основные разделы физической и коллоидной
химии: состояние вещества, химическая термодинамика, термохимия, 3d-
кон Гесса, устойчивое химическое равновесие, кинетика, v'.тализ, фото-
химия. Рассмотрены основные характеристики коИ*.. ' стем, их
классификация и методы их получения. .АД. . t .,
1805000000(4309000000) —230 | КБК 24.5
X------:------------------77—88 ।
001(01)—88 541
ISBN 5—06—001257—3 ©Издательство «Высшая школа», 1988
ПРЕДИСЛОВИЕ
Для реализации Продовольственной программы на всех ее эта-
пах необходимо обеспечить повышение квалификации специа-
листов, осваивающих быстро обновляющиеся в настоящее время
интенсивные технологии. Это возможно лишь при значительном
увеличении роли фундаментальных дисциплин, в первую очередь
химических, в профессиональной подготовке современного работ-
ника сельскохозяйственного производства.
В условиях перестройки образования, намеченной решения-
ми XXVII съезда КПСС, возрастает необходимость значительно-
го повышения научно-теоретического уровня химических дис-
циплин и одновременно с этим усиливается роль фундаменталь-
ных химических исследований в научных сельскохозяйственных
разработках. Это способствует экономически эффективному ис-
пользованию всех средств химического воздействия на сельско-
хозяйственное производство и повышению его качества.
Создание и рациональное применение новых и высокоэффек-
тивных удобрений, разработка и внедрение пестицидов, улучше-
ние физических и физико-химических свойств почвы невозможны
без знания основ физической химии. Изучение почвенно-погло-
щающего комплекса и гумуса почв, так необходимое для раскры-
тия способов повышения плодородия, прежде всего осуществля-
ется с выявления физико-химического механизма возникновения,
изменения и деградации этих систем. Глубокое исследование про-
цессов фотосинтеза на основе знания механизма фотохимических
реакций позволит в будущем повысить коэффициент использо-
вания солнечной энергии культурными растениями.
Физическая химия позволяет создать прочный фундамент для
изучения таких специальных дисциплин, как биотехнология, аг-
рономическая и биологическая химия, почвоведение, физиология
растений и животных, земледелие и многие другие.
В настоящем учебнике рассмотрены все основные разделы
физической и коллоидной химии с традиционным расположением
материала. Исключен раздел физической химии, посвященный
строению атомов и молекул, так как он подробно излагается в
курсе общей химии и частично в курсе физики. Не рассматри-
ваются также физико-химические методы исследования и анали-
за, ибо даже короткое обсуждение основных инструментальных
методов значительно перегрузило бы учебник. Желающие могут
ознакомиться с этими методами по учебному пособию
Р. А. Хмельницкого «Современные методы исследования агроно-
мических объектов» (М., Высшая школа, 1981).
Автор надеется, что учебник будет полезен студентам не толь-
ко сельскохозяйственных, но и других вузов, программа которых
близка к программам сельскохозяйственных вузов страны.
Главы книги «Электрическая проводимость растворов элект-
ролитов» и «Электрохимические процессы» написаны совместно
с канд. хим. наук, доц. В. А. Кончицем. \
Автор благодарен доц. В. А. Кончицу и доц. Е. В. Храповой
за прочтение и обсуждение всех глав.
Автор приносит глубокую благодарность проф. В. М. Глазову
и доц. В. Д. Зиновьеву за весьма ценные замечения, сделанные
в процессе рецензирования, а также канд. биол. наук А. Ю. То-
мащук и С. Н. Булычеву за большую помощь при подготовке
учебника к изданию.
Замечания и пожелания по книге можно направлять по адре-
су: 101430, Москва, ГСП-4, Неглинная ул., д. 29/14, издательство
«Высшая школа».
Автор
ВВЕДЕНИЕ
Более двухсот лет тому назад один из основоположников физи-
ческой химии — гениальный русский ученый Михаил Василье-
вич Ломоносов — дал следующее определение этой науки: «Фи-
зическая химия — наука, которая должна на основании положе-
ний и опытов физических объяснить причину того, что происхо-
дит через химические операции в сложных телах». С тех пор фи-
зическая химия как наука прошла долгий путь, непрерывно раз-
виваясь, усложняясь и охватывая все большее число химических
объектов и методов исследования, однако определение М. В. Ло-
моносова не утратило своего значения и даже сейчас поражает
точностью формулировки.
Физическая химия характеризует связи между физическими и
химическими явлениями, между физическими и химическими
свойствами веществ. Установленные закономерности этих связей
позволяют сформулировать общие принципы протекания хими-
ческих реакций, необходимые для достижения конечной цели —
предсказания направления реакции и получения нужного про-
дукта.
Возникновение и развитие физической химии диктовалось
прежде всего необходимостью создания теоретических и практи-
ческих основ важнейших технологических процессов, определяю-
щих прогресс цивилизации. Управление современными процес-
сами с целью получения продуктов с заданными характеристи-
ками потребовало создания современных физико-химических ме-
тодов исследования, анализа и производственного контроля ве-
ществ и их смесей. Глубокое изучение характеристик веществ
и обобщение химических явлений привели к сильному проникно-
вению в химию физики с ее уже развитым математическим ап-
паратом и наиболее прогрессивным объяснением сложных хими-
ческих превращений на атомно-молекулярном уровне.
Физическая химия включает следующие основные разделы.
Строение вещества. В этот раздел входит учение о строении
атомов и молекул и учение об агрегатных состояниях вещества,
где рассматривается зависимость строения и важнейших свойств
веществ, находящихся в газообразном, твердом и жидком состо-
яниях.
5
Создание модели идеального газа обеспечивает получение
фундаментальных уравнений, которые затем с определенными
поправками используются для описания широкого круга реаль-
ных (и не только газообразных) систем.
Химическая термодинамика. Этот раздел применяет законы
термодинамики к изучению химических реакций и процессов, в
результате которых происходят глубокие изменения физико-хи-
мических свойств взаимодействующих веществ при одновремен-
ном выделении либо поглощении теплоты. Химическая термоди-
намика устанавливает фундаментальное значение химического
состава как одного из важнейших факторов, определяющих пове-
дение и состояние вещества в конкретных условиях.
Изучение тепловых эффектов, сопровождающих химические
реакции, а также некоторых термических характеристик реаги-
рующих веществ позволяет установить критерии самопроиз-
вольного течения реакции, а также критерии равновесия. В ре-
зультате выводится один из важнейших законов химии — закон
действующих масс, определяющий концентрации компонентов
системы в условиях равновесия.
Химическая кинетика. В задачи кинетики входят определение
скорости реакции в гомогенной и гетерогенной среде, исследова-
ние зависимости скорости от концентрации реагирующих ве-
ществ, температуры, давления, а также влияния излучения и ка-
тализаторов. Особенно важную роль в жизнедеятельности орга-
низмов играют биологические катализаторы белковой природы
(ферменты), присутствующие во всех без исключения живых
клетках и обеспечивающие протекание почти всех биохимиче-
ских реакций в любом организме. Конечной целью кинетических
исследований является установление механизма изучаемой ре-
акции.
Фотохимия. Задача фотохимии — установление связи между
поглощением световой энергии и химическими процессами. Ис-
следование фотохимических реакций необходимо для понимания
сложного механизма процессов фотосинтеза, который является
непременным условием жизни растений и животных и самым
крупномасштабным синтетическим процессом на Земле.
Растворы. В этом разделе рассматривают природу растворов
неэлектролитов и электролитов, их важнейшие свойства, зависи-
мость свойств растворов от концентрации и химической природы
компонентов и вопросы растворимости. Это весьма важная об-
ласть физической химии, поскольку большая часть химических,
биохимических и биологических процессов протекает в жидкой
фазе.
Электрохимия. В этот раздел входит изучение некоторых
особенностей растворов электролитов, электрической проводи-
мости растворов, электрохимических элементов и электродвижу-
щей силы.
Поверхностные явления. Здесь рассматриваются явления,
происходящие на поверхности раздела фаз в гетерогенных сис-
6
темах. Особенно важную роль играют процессы, связанные с
адсорбционно-десорбционным равновесием.
Коллоидная химия. В учениях о коллоидах рассмотрены
структура, свойства и поведение систем, включающих, частицы
относительно больших размеров, часто не взаимодействующих
с окружающей средой (лиофобные коллоиды) или образующих
растворы, близкие к молекулярным (растворы высокомолеку-
лярных соединений). Коллоидная химия выделилась в самостоя-
тельный крупный раздел физической химии благодаря бурному
развитию в последние десятилетия этой области науки, ее боль-
шой роли практически во всех процессах, связанных с жизнедея-
тельностью организмов, и во многих природных процессах.
Изучение физической и коллоидной химии дает возможность
получить более глубокие знания об окружающем мире и, в част-
ности, позволяет на более высоком уровне решать проблемы, свя-
занные с развитием научных основ ведения сельского хозяйства.
Физико-химический подход позволяет понимать процессы, иду-
щие в такой сложной системе, как почва, улучшать производство
новых удобрений, внедрять более эффективные методы разработ-
ки и вводить химические средства борьбы с вредителями и бо-
лезнями растений. Исследования фотохимических реакций, столь
блестяще начатые К. А. Тимирязевым, позволяют глубже понять
сущность сложных процессов фотосинтеза. Исследование почвен-
ных коллоидов — необходимое условие повышения плодородия.
Необходимо иметь в виду, что такие специальные дисциплины,
как агрохимия, почвоведение, физиология растений и животных,
химия защиты растений, биохимия и микробиология, на совре-
менном уровне не могут развиваться без знания фундаменталь-
ных положений физической химии.
Глава I
АГРЕГАТНЫЕ СОСТОЯНИЯ ВЕЩЕСТВА
1. КЛАССИФИКАЦИЯ СОСТОЯНИЙ ;
В зависимости от расстояния между частицами и от степени их
взаимодействия вещество может находиться в твердом, жидком
или газообразном состоянии. Эти состояния называют агре-
гатными состояниями вещества (рис. 1).
Определяющим фактором в получении того или иного агре-
гатного состояния при постоянном давлении является температу-
ра, которая изменяет соотношение средней потенциальной энер-
гии частиц вещества и их средней кинетической энергии.
При достаточно низкой температуре все вещества находятся
в твердом состоянии. Расстояния между частицами вещества при
этом по порядку равны размерам самих частиц, а их средняя по-
тенциальная энергия выше средней кинетической энергии. Дви-
жение частиц ограничено, и силы, удерживающие частицы в
определенном положении, обусловливают наличие собственной
формы и объема.
Повышение температуры приводит к изменению агрегатного
состояния, и в результате плавления твердые вещества переходят
в жидкие. Средняя кинетическая энергия частиц становится
примерно равной их средней потенциальной энергии, а расстоя-
ния между ними различны для разных частиц. Некоторые части-
цы находятся примерно на тех же расстояниях, что и в твердом
веществе, однако часть частиц удалена на большие расстояния.
Вещество в жидком состоянии не имеет собственной формы и
принимает форму сосуда, в который оно помещено. Снижением
температуры обычно можно вернуть вещество в прежнее твердое
состояние (рис. 1).
Дальнейшее повышение температуры жидких веществ приво-
дит к испарению (кипению} и к переходу в газообразное состоя-
ние. Средняя кинетическая энергия частиц газа становится значи-
тельно больше их средней потенциальной энергии. Расстояния
между частицами намного больше их размеров, а силы взаимо-
действия между частицами чрезвычайно малы. В результате это-
го частицы газа стремятся занять как можно больший объем.
Переход вещества из твердого состояния в жидкое называ-
ется плавлением, а из жидкого в газообразное — паро-
образованием. Обратные процессы называют затвер-
деванием и сжижением. Некоторые вещества (напри-
8
мер, иод) могут переходить, не-
посредственно минуя жидкое,
из твердого состояния в газо-
образное. Такой процесс назы-
вают сублимацией, а
обратный — десублима-
цией.
В последние годы глубоко-
му изучению подвергается чет-
вертое состояние вещества —
плазма. Переход в это сос-
тояние из газообразного осу-
ществляется разрушением ато-
мов и молекул вещества и пре-
вращением их в смесь положи-
тельно и отрицательно заря-
женных частиц. Плазму можно
получить воздействием, напри-
мер, на газ электрическим раз-
рядом или высокими темпера-
турами (105... 106 К)-
ПЛАЗМА
ЖИДКОЕ СОСТОЯНИЕ
Рис. 1. Различные агрегатные состоя-
ния
2. ГАЗООБРАЗНОЕ СОСТОЯНИЕ
В газообразном состоянии частицы удалены друг от друга на
гораздо большие расстояния, чем в твердом или жидком состоя-
нии. Экспериментальные данные, позволяющие описать газооб-
разное состояние, могут быть получены довольно легко, однако
корректное описание даже такой простой системы требует введе-
ния определенных упрощений или, иначе говоря, создания моде-
ли. Такой моделью является идеальный газ. Как и всякая мо-
дель, идеальный газ не может быть отождествлен с реально су-
ществующими газами; такого газа, как идеальный, не существует
в природе. В то же самое время следует подчеркнуть, что созда-
ние модели обеспечивает получение фундаментальных уравнений,
которые затем с определенными поправками используются для
описания широкого круга реальных (и не только газообразных)
систем.
Рассматриваемая модель была получена при следующих
условиях:
1) идеальный газ образован большим числом молекул или
атомов, расстояние между которыми намного больше, чем раз-
меры самих частиц;
2) молекулы этого газа характеризуются определенной мас-
сой, однако их собственным объемом можно пренебречь;
3) молекулы находятся в непрерывном беспорядочном дви-
жении;
4) столкновения молекул между собой, а также молекул со Й
стенками сосуда — упругие. Это значит, что кинетическая энер-j
9
Гия может передаваться от одной молекулы к другой, но она не
переходит в другие формы энергии, например в теплоту;
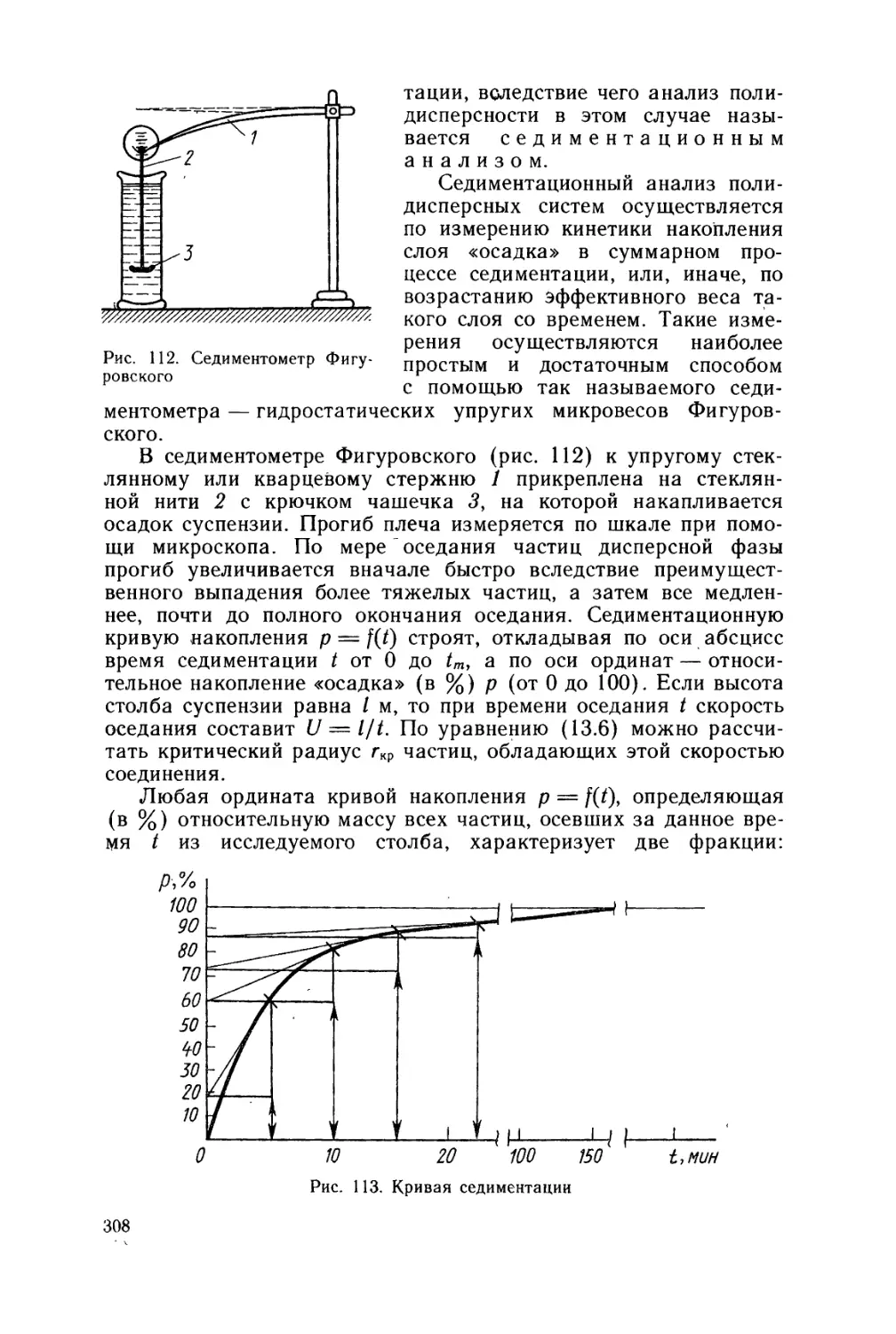
5) между молекулами не существует взаимодействия, будь то
притяжение или отталкивание.
2.1. Законы идеальных газов
2.1.1. ЗАКОН БОЙЛЯ —МАРИОТТА
Рассмотрим массу т идеального газа, который при температу-
ре Г и давлении р\ занимает объем Vi. Переменные р, V, Г,
позволяющие определять состояние газа, называются пара-
метрами состояния. Таким образом, состояние газа с
массой т может быть описано следующим образом:
p\V\T (состояние 1).
При этой же температуре Т состояние 2 будет
P2V2T (состояние 2).
Если переход из состояния 1 в состояние 2 происходит при по-
стоянной температуре, то такой процесс называется изотер-
мическим.
По закону Бойля — Мариотта
р\Vi = P2V2 = РзКз-.-
При постоянной температуре для массы т идеального газа произведение
объема газа на его давление есть величина постоянная:
pV = const.
Закон Бойля — Мариотта можно представить, графически в ко-
ординатах pV и р прямой, параллельной оси давлений, а в ко-
ординатах р и V — ветвью гиперболы (рис. 2, а, б).
Рис. 2. Графическое изображение закона Бойля — Мариотта
2.1.2. ЗАКОН ШАРЛЯ
Рассмотрим два состояния идеального газа с массой т\
T\Vip (состояние 1),
T2V2P (состояние 2).
10
Переход из состояния 1 в состо-
яние 2 осуществляется при постоян-
ном давлении; такой процесс назы-
вается изобарным. Из экспе-
риментального изучения зависимо-
сти изменения объема от температу-
ры Шарль установил, что при посто-
янном давлении объем газа с массой
т линейно изменяется с температу-
рой (рис. 3). Коэффициент пропор-
циональности равен 1/273,16. Это
значит, что при каждом повышении
температуры на один градус объем
газа увеличивается на 1/273,16 его
первоначального значения. Из экс-
периментальных данных следует
также, что
Закон Шарля формулируется
следующим образом:
Отношение объема к абсолютной темпе-
ратуре при постоянном давлении постоянно:
V /Т = const.
Рис. 3. Графическое изображе-
ние закона Шарля
Рис. 4. Графическое изображе-
ние закона Гей-Люссака
2.1.3. ЗАКОН ГЕЙ-ЛЮССАКА
Рассмотрим массу т идеального газа в двух следующих состоя-
ниях:
T\Vp\ (состояние 1),
T2VP2 (состояние 2).
Переход из состояния 1 в состояние 2 происходит при постоянном
объеме; такой процесс называется изохорным. Гей-Люссак
экспериментально установил (рис. 4), что при постоянном объеме
давление газа с массой т, так же как и в случае закона Шарля,
линейно изменяется с температурой с тем же коэффициентом
пропорциональности (1/273,16). Это значит, что при каждом
повышении температуры на один градус давление газа увеличи-
вается на 1/273,16 его первоначального значения. Было также
показано, что
Р2 Р1 ,
-^ = -7F- = const.
12 1 1
Закон Гей-Люссака формулируется следующим образом:
отношение давления к абсолютной температуре при постоянном объеме
постоянно:
р/Т = const.
И
2.1.4. УРАВНЕНИЕ СОСТОЯНИЯ ИДЕАЛЬНОГО ГАЗА
Уравнения, которыми выражаются законы Бойля — Мариотта,
Шарля и Гей-Люссака, описывают соотношения между давле-
нием, температурой и объемом некоторой массы т идеального
газа, причем один из параметров в каждом случае остается
постоянным. Следовательно, эти законы можно объединить и
описать состояние некоторой массы идеального газа, если задать
две величины из трех изменяющихся величин.
Запишем три состояния идеального газа с массой т\
T\V\p\ (состояние 1);
TV\p2 (состояние 2);
TVp (состояние 3).
Используем законы идеальных газов для описания перехода из
одного состояния в другое. Переход из состояния 1 в состояние 2
происходит при постоянном объеме. По закону Гей-Люссака
Р2 _ Р'
Т Т\ '
Переход из состояния 2 в состояние 3 осуществляется при по-
стоянной температуре. По закону Бойля—Мариотта
р2Г1 = рУ.
Величина р2, выведенная на основании закона Гей-Люссака,
равна
Т
Р2 = Р'1Г
Используя это значение р2, получим
Pl -i- V1 = pV ИЛИ •
11 111
Величина этого отношения зависит от массы исследуемого газа.
Предположим, что количество вещества, участвующее в про-
цессах, равно одному молю. Поскольку, согласно закону Авогад-
ро, равные объемы различных газов при одинаковой температуре
и одинаковом давлении содержат одинаковое число молекул,
объем, занимаемый молем газа в условиях нормальной темпе-
ратуры и давления (Г = 273,15 К, р = 101,3кПа), будет одинако-
вым для любого газа. В этих услрвиях объем, равный 22,414 л
(22,414-10“3 м3), называется стандартным молярным
объемом.
Для моля идеального газа величина pV/Т обозначается че-
рез R — универсальную газовую постоянную. Следовательно,
можно записать
рУ Р1Г1
12
Таким образом, для 1 моль идеального газа уравнение состоя-
ния имеет вид
pV = RT,
а для п моль
pV = nRT.
Это уравнение называется уравнением состояния
идеального газа.
Значение R можно получить следующим образом. Из эксперимента известно,
что 1 моль идеального газа при давлении 101,3 кПа (1 атм) и температу-
ре 273,15 К (стандартная температура и давление) занимает объем 22,414 л.
Отсюда
D 1 атм-22,414 л ААООПС ,,
R = —:------П7О-, т? = 0,08206 л • атм / (моль• К).
1 моль-273,15 К
По определению 1 атм — это давление, создаваемое столбиком ртути высо-
той 76 см в точке Земли, где ускорение силы тяжести составляет 980,67 см/с2.
Поскольку плотность ртути равна 13,595 г/см3, имеем 1 атм = 76 смХ
X 13,595 г/см3-(980,67 см/с2) = 1,0 1 33-106 дин/см2, 1 дин = 1 г-см/с2.
В СИ атмосферу нужно перевести в Н/м2. Так как в СИ единица давления
паскаль (Па) и размерность Н/м2:
QHg = 13,595-103 кг/м3,
£ = 9,8067 м/с2.
Таким образом,
1 атм = (0,76 м)(13,595-103 кг/м3)(9,8067 м/с2) =
= 101325 кг/(м-с2) = 101 325 Н/м2 « 101,3 кПа, 1 Н = 1 кг-м/с2.
По уравнению состояния идеального газа получим
„ pV 101325 Н/м2-22,414.10-3 м3 ОЪ1/1 „ ,,
R=—f~ =-------7-t----г TZX---= 8,314 Н-м/ моль-К) =
Т (1 моль) (273,15 К)
= 8,314 Н-м/(моль-К) = 8,314 Дж/(моль-К).
Физический смысл газовой постоянной можно выяснить, рас-
сматривая процесс нагревания на 1° при постоянном давлении
1 моль идеального газа, первоначально имевшего температуру Г,
т. е. до температуры (Т + 1). Уравнения для конечного’и началь-
ного состояний газа будут иметь вид
pV2 = R(T+ 1),
pVi = RT.
Объем Vi соответствует температуре Т, а объем V2 — темпе-
ратуре на 1° выше, т. е. Т -f- 1.
Вычтем из первого уравнения второе:
pV2 - pV, =7?(Т+ 1)- RT,
p(V2- Vl) = pbV=R.
Произведение pAV, а следовательно, и R — работа расшире-
ния 1 моль идеального газа при повышении температуры на 1°
при условии, что давление остается постоянным.
13
2.2. Парциальное давление. Закон Дальтона
Если в ограниченном пространстве объема V смешать несколько идеальных
газов, то каждый газ будет оказывать свое собственное давление, называемое
парциальным давлением, такое, как если бы он один занимал весь объем. Об-
щее наблюдаемое давление будет равно сумме парциальных давлений каждого
газа.
Так формулируется закон Дальтона, который можно дока-
зать, исходя из уравнения состояния идеального газа..
Предположим, что в ограниченном объеме V при температу-
ре Т содержится смесь идеальных газов А, В, С, количество ве-
щества (молей) которых равно соответственно пд, лв, пс. Общее
измеренное давление составляет робщ-
Тогда состояние смеси газов определяется общим количест-
вом вещества (молей) па + пв + лс при температуре Г, объ-
еме V и давлении робщ. Используя уравнение состояния, получим
РобщТ = (Яд + Яв 4“ Яс) RT. (1.1)
Если па молекул А занимают весь объем V при температуре Г,
газ А оказывает давление рд, которое является парциальным
давлением газа А в смеси.
Запишем уравнение состояния газа А:
PaV = nAR Т.
Таким же образом для газов В и С имеем
p3V = n3RT\ pcV = ncRT.
Суммирование этих трех уравнений дает соотношение
(Ра + Рв + Pc)V = (пА + яв + яс) RT. (1.2}
При сравнении (1.1) и (1.2) получаем
Робщ — Ра + Рв + Рс = 2 парциальных давлений — /гобщ.
Исходя из приведенных уравнений, можно легко получить
значение молярной доли, показывающей, какую часть от общего
количества (молей) смеси составляет данный газ. Так, например,
RT
для газа А парциальное давление равно рА = ПА-у-, а для
RT
суммы газов рОбщ — лОбШ -у- - Поделив одно равенство на дру-
гое, получим
Ра Яд v
----=-----= Ад,
Робщ Яобщ
где Ха — молярная доля газа А; Хв и Х& — соответственно мо-
лярные доли газов В и С.
Естественно, сумма молярных долей всех газов, входящих в
данную смесь, равна единице, т. е. для рассматриваемого случая
Ха + Хв + Хс = 1.
14
2.3. Кинетическая интерпретация абсолютной температуры
Из курса физики известно, что давление газа
р = п'ти2, (1.3)
О
где rt' — N/V—число молекул в единице объема; N— общее
число молекул; V_— объем, занимаемый газом; т — масса одной
молекулы газа; v2 — среднеквадратичная скорость молекул, ко-
торая равна
Подставляя значение п' в уравнение (1.3), имеем
1 N -2
откуда
17 1 А/ ~2 2 Л7 mv2
pV = — Nmv2 = -у N —•
Для моля идеального газа N равно постоянной Авогадро Na:
.. 2 .. mv2 пТ
= —pV = RT,
откуда
Отношение R/Na = 1,38- 10 23 Дж/К=к (к — постоянная Боль-
цмана). Тогда
3 . 1 ~2 j-,
~2кТ==~2 = £кин’
где l/zmv2 — средняя кинетическая энергия перемещения моле-
кулы идеального газа.
Полученное выражение показывает^ что
абсолютная температура пропорциональна средней кинетической энергии
перемещения молекулы идеального газа.
2.4. Закон Авогадро
Рассмотрим два идеальных газа при одинаковой температуре Г,
давлении р и объеме V, Пусть т\ и — массы молекул, а п'\
и /12 число молекул в единице объема.
Согласно уравнению (1.3) для двух газов при одинаковом
давлении имеем
1 , “2
р = — rtf/niuf,
О
15
p = -x- ri'2m2v2.
о
Температура обоих газов одна и та же, поэтому
ьт 1 “2 1 ~2
у кГ = у mw\ = у tn2V^-
Отсюда п'\ — п'2 и, поскольку газы занимают одинаковый
объем,
п\ V — n2V.
Следовательно,
Ni = N2.
Любой газ при одинаковых температуре, давлении и объеме содержит
одинаковое число молекул.
В частности, при р = 101,3 кПа, Т — 273,15 К V = 22,414 л
Wj = = = 6,023’1023.
Этот результат, полученный из кинетической теории газов,
ранее был введен А. Авогадро как постулат.
2.5. Скорость молекул. Диффузия Эффузия
Рассмотрим два идеальных газа при одинаковой абсолютной
температуре. Пусть т\ и — массы их молекул. Имеем
3 !Т 1 - 1 -2
К/ = — tniVt = — m2v2.
Пусть vc = — средняя квадратичная скорость, которая свя-
зана со средней скоростью уравнением vm = 0,92ус, где vm — сред-
няя скорость. Тогда
/ vj = /^2 VC\ ~ У mi j 4)
/Й //Л? VC2 Vm2
Таким образом, средняя квадратичная скорость молекулы
обратно пропорциональна корню квадратному из массы молеку-
лы. Это находится в согласии с эмпирическим законом диффу-
зии Грэма. Отсюда следует, что
средняя скорость молекул газа увеличивается с его абсолютной температу-
рой и уменьшается при увеличении массы молекул.
Закон диффузии Грэма. Эффузия. Явление диффузии газов
экспериментально подтверждает существование молекулярного
движения. Если убрать непроницаемую для молекул газа стенку
между двумя сосудами, заполненными газами, или заменить ее
на проницаемую пористую стенку, то газы легко, быстро и само-
произвольно смешиваются. В 1829 г. Т. Грэм показал, что ско-
рости диффузии газов обратно пропорциональны квадратным
корням из их плотностей, при постоянных давлении и температу-
ре. Таким образом, для двух различных газов
16
— = -^, (1.5)
U2 /V
где vi и у2 —- скорости диффузии газов 1 и 2.
Уравнение (1.5), известное под названием закона диф-
фузии Грэма, можно переписать в виде уравнения (1.4),
поскольку отношение плотностей двух идеальных газов при оди-
наковой температуре и давлении равно отношению их относи-
тельных молекулярных масс. Как было показано, закон Грэма
является логическим следствием кинетической теории газов, так
как скорость диффузии должна быть непосредственно связана
со скоростью движения молекул.
Поскольку равномерное распределение вещества в данном
объеме наиболее вероятно, процесс диффузии является самопро-
извольным. Согласно закону Фика количество вещества dm, про-
диффундировавшего за время d/ через площадь S, пропорцио-
нально изменению концентрации на единицу расстояния или гра-
dC
диенту концентрации :
— dm = DS ( Д"4) dt.
Знак минус перед dm указывает, что процесс идет в направ-
лении уменьшения концентрации. Коэффициент пропорциональ-
ности D называется коэффициентом диффузии ве-
щества; он численно равен количеству вещества (в киломолях),
проходящему через единицу площади (1 м2) за единицу време-
ни (1 с) при градиенте концентрации, равному единице (т. е. из-
менению концентрации в 1 кмоль/м3 на расстояние в 1 м). Легко
видеть, что коэффициент диффузии D имеет размерность
__ кмоль-м-м3 м2
L с-м2-кмоль с
Диффузия играет большую роль на многих стадиях процесса фотосинтети-
ческого включения углерода СОг в углеводы. При этом углекислый газ диффунди-
рует из атмосферы, достигая поверхности листа, а затем проходит через усть-
ичные отверстия. Войдя в лист, СО2 диффундирует по межклеточным воздухо-
носным пространствам, а затем через клеточные оболочки и плазму клеток мезо-
филла листа. Далее углекислый газ, по-видимому, в форме НСО.Г диффунди-
рует через цитоплазму и достигает хлоропластов. Затем СО2 оказывается в хло-
ропласте и попадает в зону действия ферментов, участвующих в образовании
углеводов. Как видно, одну только эту сторону фотосинтеза можно расчленить
на много стадий, в каждой из которых важную роль играет диффузия. Если бы
с помощью ферментов фиксировался весь углекислый газ, находящийся в сфере
их действия, и не происходила бы диффузия новых количеств углекислого газа из
атмосферы, окружающей растение, процесс фотосинтеза прекратился бы. Диф-
фузия важна также для многих других аспектов физиологии растений, осо-
бенно для проникновения веществ через мембраны.
Эффузия (истечение) газов — процесс, тесно связанный с
диффузией. Однако между ними есть некоторое отличие. Диффу-
зией называется поток газа из области с более высоким давле-
нием в область с более низким давление^ через: дористую. утенку
17
Рис. 5. Схема центрального и
бокового соударений молекул
газа
столкновения до другого
или капилляр, тогда как эффузия .
означает прохождение отдельных
молекул через отверстие малого
, диаметра. При низких давлениях
в условиях, когда средняя длина
свободного пробега молекул вели-
ка по сравнению с диаметром от-
верстия, скорость эффузии также
определяется уравнением (1.5).
Свободный пробег молекул газа. 1
Молекулы газа движутся от одного
прямолинейно. После столкновения в*
общем случае изменяется направление прямолинейного дви-
жения.
Путь молекулы от одного столкновения до другого называется свободным
пробегом.
Если считать, что молекула движется с некоторой средней
арифметической скоростью и при этом сталкивается с другими
молекулами, то длина свободного пробега будет равна скорости,
деленной на число столкновений в единицу времени.
Пусть в 1 м3 находится п молекул, представляющих собой
абсолютно упругие шары с радиусом г и диаметром d.
При столкновении молекул удар может быть центральным,
когда столкновение происходит по линии, проходящей через
центры тяжести молекул, и боковым. Это показано схематичйо
на рис. 5, а. При положении б произойдет боковой удар, при по-
ложении в молекулы, пролетая одна около другой, коснутся друг
друга одной точкой своей поверхности.
Предположим, что все молекулы, за исключением одной,
остановились, а движущаяся молекула обладает удвоенной сред-
ней арифметической скоростью 2vm. Если вместо молекулы дви-
жется диск, радиус которого равен диаметру молекулы, то в 1 с
диск вырежет в пространстве объем, равный 2ш/2ут. В этом объ-
еме может содержаться 2nd2vmn молекул.
Если бы двигалась одна молекула, а остальные были непо-
движны, то в одну секунду она встретилась бы с другими моле-
кулами и столкнулась с ними 2nd2vmn раз. Тогда длина свобод-
ного пробега I будет равна:
/ — Vnt — 1
2nd2vmn 2nd2n *
В результате более строгого подсчета числа столкновений
можно получить
/ = —1—.
/Т nd2n
Введем в уравнение плотность газа. Так как q = тп, то, под-
ставив вместо п плотность, получим
18
ngd2
Как видно, средний свободный пробег обратно пропорциона-
лен плотности газа.
Закон распределения скоростей. Можно доказать, что ни одна
из молекул газа не может сохранять все время постоянную
скорость. При каждом столкновении между двумя молекулами
кинетическая энергия распределяется между ними самым раз-
личным образом в зависимости от условий соударения, причем
только сумма этих энергий остается постоянной. Поэтому для
более подробного рассмотрения движения молекул необходимо
выяснить распределение их по скоростям. Максвелл и Больцман
определили, сколько молекул при данной температуре движется
с какой-либо определенной скоростью. Они получили уравнение,
которое характеризует распределение молекул по скоростям;
О,A — mv2
т-4Ч-25г)
где N — общее число молекул; dN/N— доля молекул, движу-
щихся со скоростями в интервале от v до у + с1у.
Уравнение (1.6) известно под названием распределе-
ния Максвелла или распределения Максвел-
ла — Больцмана по скоростям.
Это распределение можно представить графически, отклады-
вая по ординате число молекул п,, имеющих скорость vt (рис. 6).
График соответствует температуре Т и общему числу молекул N.
Площадь, заключенная между кривой и осью абсцисс, отвечает
общему числу молекул N. Площадь, ограниченная кривой, осью
абсцисс и двумя линиями, параллельными оси ординат, с абс-
циссами Vi и ^2 равна числу молекул, скорость которых заключе-
на между и V2. Скорость vp, соответствующая максимуму на
кривой, — наиболее вероятная скорость: этой скоростью обла-
дает большинство молекул.
На рис. 7 показано распределение молекул Н2 по скоростям
Рис. 6. Распределение Максвелла —
Больцмана по скоростям
Рис. 7. Распределение молекул Н2 по
скоростям при 300 и 1000 К
19
при разных температурах. При низкой температуре кривая рас-
пределения характеризуется узким максимумом, который с рос-
том температуры становится шире и сдвигается в сторону боль-
ших скоростей. Это значит, что все> большее число молекул
движется с более высокими скоростями (такие молекулы иногда
называют «горячими»). Температурная зависимость кривой рас-
пределения имеет важное значение для скоростей химических
реакций. Чтобы молекулы могли вступить в химическое взаимо-
действие, они должны обладать некоторой избыточной кинетиче-
ской энергией, называемой энергией активации, кото-
рая превышает их среднюю кинетическую энергию. При низкой
температуре число быстро движущихся молекул мало, поэтому
многие реакции при низких температурах идут чрезвычайно
медленно. Повышение температуры увеличивает число «горячих»
молекул, что приводит к ускорению реакции.
Используя уравнения (1.3 и 1.6), можно получить формулы
для расчета средней квадратичной vc, среднеарифметической vm
и наиболее вероятной скорости vp:
^/Srt ^HiRf ^/~2RT
* M ; Vm~ ' лМ ; Vp~ V M ’
где M — молярная масса.
В качестве примера вычислим vpy vm и vc для Ог при 300 К, зная, что R =
= 8,314 Дж/(К-моль), М = 0,032 кг/моль, Г = 300 К-
, / 2-8,314 Дж/(К-моль)300 К л 7
vp= у--------------------------= У ^56-10& Дж/кг =
т 0,032 кг/моль
= /1,56-105 кг - м2/(с^-кг) =394 м/с.
Точно так же можно показать, что vm = 445,5 м/с, ис=483,6м/с. Таким образом,
Vc V m '^> Up-
В табл. 1 приведены наиболее характерные значения моле-
кулярных постоянных для различных газов. Частота столкнове-
Таблица 1. Молекулярные постоянные некоторых газов
Вещество Диаметр молекул, нм Средняя скорость моле- кул- 10\ м/с Частота столкнове- ний- 109 Средняя длина сво- бодного про- бега • 10-4, м
273 К 293 К
Аргон Диоксид угле- 0,29 3,81 3,95 4* 9,88
рода 0,33 3,62 3,76 6,1 6,15
Водород 0,24 16,96 17,55 10,0 17,44
Неон 0,26 5,38 5,57 — —
Азот 0,28 4,54 4,71 5,07 9,29
Кислород 0,29 4,25 4,44 4,43 9,93
Вода — 5,66 5,87 — —
Ксенон 0,35 2,10 2,18 — —
Криптон 0,31 2,63 2,72 — —
20
ний представляет собой среднее число столкновений одной моле-
кулы за единицу времени (секунду) при 293 К и 101,3 кПа.
2.6. Реальные газы
2.6.1. УРАВНЕНИЕ ВАН-ДЕР-ВААЛЬСА
Экспериментальные исследования показали, что реальные
газы не подчиняются законам идеальных газов. Максимальные
отклонения от идеального поведения наблюдаются при высоких
давлениях и при низких температурах. При этих условиях объем
системы становится относительно малым и собственный объем
молекул составляет заметную часть общего объема. Кроме того,
когда молекулы находятся на близких расстояниях друг от друга,
экспериментально измеренное давление оказывается значительно
меньше расчетного идеального значения; это происходит в ре-
зультате увеличения сил межмолекулярного притяжения. Харак-
тер и степень отклонений в поведении различных газов от идеаль-
ного различны (рис. 8). Для идеальных газов произведение дав-
ления на объем pV при постоянной температуре остается по-
стоянным. Поэтому на графике зависимость pV от р при постоян-
ной температуре изображается прямой линией, идущей парал-
лельно оси абсцисс (р). Поведение водорода, кислорода и ди-
оксида углерода отклоняется от поведения идеального газа,
причем характер отклонения для этих трех газов различен. Как и
следовало ожидать, особенно сильные отклонения происходят
при высоких давлениях. В точности такой же по виду график по-
лучается, если в качестве ординаты взять не просто рУ, а отно-
шение pV/(nRT}— так называемый коэффициент сжи-
маемости. Различие состоит лишь в следующем: если на
рис. 8 все кривые пересекаются при значении 22,4 л-атм, то на
графике коэффициента сжимаемости (рис. 9) кривые пересе-
каются при значении ординаты, равном единице, так как для
идеального газа pV/(nRT) = 1,0.
Ван-дер-Ваальс (1879), пытаясь устранить причины, из-за
которых уравнение идеального газа практически нельзя исполь-
Давление объем
Рис. 8. Отклонения в поведений реальных га- Рис. 9. Зависимость коэффици-
зов от идеального ента сжимаемости от давления
21
зовать для описания поведения реальных газов, предложил вве-
сти в это уравнение два дополнительных члена: константу а,
добавляемую к р для того, чтобы скомпенсировать уменьшение
давления из-за межмолекулярного притяжения, и константу Ь,
представляющую собой эффективный объем молекул газа и вы-
читаемую из общего объема V. Обе константы подбирались
эмпирически. Для 1 моль газа уравнение приобретает следую-
щий вид:
(р+-£т) (V~b) = RT.
Поправка к давлению принята равной а/V2 по следующей при-
чине: молекулы на поверхности газа притягиваются к молеку-
лам, находящимся внутри. Сила этого притяжения пропорцио-
нальна как плотности газа на поверхности, так и плотности газа
во внутренних слоях. Чем выше эти значения плотности, тем
больше силы притяжения. Поскольку плотность — это отношение
массы к объему, влияние плотности можно учесть умножением
константы а на 1/V2. Поправка b в реальном случае примерно
в четыре раза превышает собственный объем молекул одного
моля газа и иногда называется исключенным объемом, так как
соответствует пространству, реально исключенному одним молем
плотноупакованных молекул. Поправки а и b для любого газа
приводятся в расчете на 1 моль. Если количество вещества газа
больше или меньше 1 моль, то нужно пользоваться следующим
уравнением:
(р + п2а/V2) (V - nb) = nRT.
Численные значения констант уравнения Ван-дер-Ваальса для
нескольких газов приведены в табл. 2, а в табл. 3 сопоставлены
значения наблюдаемых и вычисленных по уравнению Ван-дер-
Ваальса значений pV для этилена при 293 К и разных давле-
ниях.
Исследования многих газов показали, что и уравнение
Ван-дер-Ваальса не применимо для точного описания их состоя-
ния. Для решения задач повышенной точности используют либо
поправки к уравнению Ван-дер-Ваальса, либо применяют другие
уравнения, составленные на основе степенных рядов.
Таблица 2. Численные значения констант уравнения Ван-дер-Ваальса
Газ а ь- ю2 Газ а Ь-102
н2 0,245 2,67 НС1 3,8 4,1
Не 0,034 2,36 NH3 4,0 3,6
n2 1,38 3,94 С2Н2 4,4 5,1
о2 1,32 3,12 С2Н4 4,5 5,6
со 1,49 4,00 С12 5,5 4,9
СО2 3,60 4,28 SO2 . 6,7 5,6
22
Таблица 3. Экспериментальные и вычисленные значения pV
для этилена при 293 К
р, кПа Эксперимен- тальные Вычисленные р, кПа Эксперимен- тальные Вычисленные
3200 0,914 0,895 11 106 0,454 0,456
4600 0,781 0,782 17605 0,643 0,642
8420 0,399 0,392 28230 0,941 0,940
9460 0,413 0,413 33000 1,067 1,067
40000 1,248 1,254
В настоящее время известно более 150 видов уравнений со-
стояния. Так называемое вириальное (от лат. vires — силы)
уравнение состояния в общем виде записывается с помощью
степенного ряда:
рУ _ < . В(Г) , с(Т) d(t)
RT V V2”"1 V3”’
Вириальное уравнение состояния является единственно теоре-
тически обоснованным уравнением. Коэффициенты В(Т), С(Т),
D(T) и т. д., зависящие от температуры, называют вторым,
третьим, четвертым и т. д. вириальными коэффициентами. Пер-
вый вириальный коэффициент равен единице. Для идеального
газа все вириальные коэффициенты, начиная со второго, равны
нулю.
2.6.2. КОНДЕНСАЦИЯ ГАЗОВ И КРИТИЧЕСКОЕ СОСТОЯНИЕ
Явление конденсации и существование критических констант
характерны для реальных газов. Лучше всего рассмотреть это
на конкретном примере исследования отношений между давле-
нием и объемом в процессе конденсации диоксида углерода. На
рис. 10 показаны изотермы диоксида углерода при нескольких
температурах. При высоких температурах изотермы имеют гипер-
болическую форму, что соответствует
выполнению закона Бойля — Мариот-
та. Однако при понижании темпера-
туры становятся все более заметны-
ми отклонения от идеальности, а при
температуре Т4 поведение системы
резко отличается от идеальных га-
зов. При движении вдоль этой изо-
термы по мере повышения давления
объем уменьшается, пока изотерма
не достигнет точки пересечения с
пунктирной линией. Если этот про-
цесс наблюдать визуально, то в точ-
ке пересечения можно заметить об-
разование жидкого диоксида угле-
Рис. 10. Изотермы диоксида уг-
лерода при различных темпера-
турах
23
рода. Далее, при поддержании постоянного давления объем
системы уменьшается, пока весь пар не сконденсируется в жид-
кость. После этого даже очень сильное повышение давления
почти не приводит к изменению объема ввиду малой сжимае-
мости жидкости. Длина горизонтального отрезка внутри области,
ограниченной пунктирной линией, т. е. области, в которой жид-
кость и газ сосуществуют, уменьшается с ростом температуры.
Имеется температура, при которой изотерма только соприкаса-
ется с этой областью в одной точке. В этой так называемой
критической точке существует лишь одна фаза, которая
не является ни жидкой, ни газообразной. Соответствующие этой
точке температура, давление и объем называют критической тем-
пературой Тс, критическим давлением рс и критическим объ-
емом Vc. Выше критической температуры конденсации не проис-
ходит, каким бы высоким ни было давление в системе. Значения
критических констант для ряда газов представлены в табл. 4.
Таблица 4. Критические константы некоторых веществ
о ч • о
о S о S
Газ СО Газ СО
к С "s
о. * g. о- о. о. о.
Ci. Ь- с».
Гелий 5,2 2,7 58 Метан 190,7 46,3 95
Водород 33,2 12,9 65 Д иоксид, уг лерода 304,5 73,9 96
Азот 126,0 33,9 90 Аммиак 405,5 112,9 72,4
Оксид углерода 133,0 34,9 93 Хлор 417,1 77,0 124
Аргон 150,7 48,6 75 5 Диоксид серы 430,3 78,7 123
Кислород 154,3 50,3 74 5
Описываемое явление можно легко объяснить, рассматривая
природу межмолекулярных сил, действующих в реальных газах.
Когда молекулы находятся на достаточно большом расстоя-
нии друг от друга, между ними действуют силы притяжения. На
близком расстоянии, например, в жидкости под давлением моле-
кулы, наоборот, отталкиваются друг от друга, что обусловлено
электростатическим отталкиванием электронов и ядер. Обычно
силы притяжения проходят через максимум при некотором
конечном расстоянии между молекулами. При температурах ни-
же Тс газ можно сжать таким образом, чтобы молекулы оказа-
лись на расстояниях, соответствующих притяжению между ними.
Тогда происходит конденсация. Выше Тс кинетическая энергия
молекул газа такова, что притяжение между ними оказывается
недостаточно эффективным, и конденсация невозможна.
24
2.6.3. СООТВЕТСТВЕННЫЕ
СОСТОЯНИЯ
Ранее было указано, что по-
ведение газов все более прибли- *
жается к поведению идеальных
газов по мере повышения темпе-
ратуры и понижению давления.
Один из наиболее эффективных
способов еще более упростить
математическое описание пове-
дения газов основан на том,
что все параметры газового сос-
тояния относятся к критическим
условиям: давление выражают
относительно критического дав-
ления, температуру — относи-
тельно критической температуры
и объем — относительно крити-
ческого объема. Эти относитель-
ные величины называют приве-
денными значениями (рпр, Тир
и Vnp):
1,0
о Пропан • Вода
ь Зтилен А Диоксид
углерода
8
?пр
Рпр_77; г"р~У7;
v у
Рис. 11. Зависимость коэффициента сжимае-
мости z от приведенных параметров состоя-
ния
На рис. 11 изображены графики зависимости коэффициента сжимаемости
(2 = pV/nRT) от приведенного давления при различных приведенных темпе-
ратурах. Эти графики показывают, что при переходе к приведенным параметрам
состояния для всех газов с весьма удовлетворительной точностью выполняется
общая закономерность поведения коэффициента сжимаемости. О веществах с
одинаковыми значениями приведенных параметров состояния говорят как о на-
ходящихся в соответственных состояниях; эти вещества, как правило, обладают
сходными свойствами.
2.6.4. МЕЖМОЛЕКУЛЯРНЫЕ ВЗАИМОДЕЙСТВИЯ
Рассмотрение поведения молекул реальных газов приводит к
выводу о наличии некоторых нехимических взаимодействий —
так называемых межмолекулярных и межатомных взаимодейст-
вий. Роль их велика хотя бы потому, что первая стадия любой
химической реакции — это межатомное или межмолекулярное
взаимодействие, возникающее при сближении молекул.
Сближение двух молекул, происходящее благодаря тепловому
движению, приводит к их взаимному притяжению, которое
вызывается силами различного происхождения. Эти силы объеди-
няются под общим названием сил Ван-дер-Ваальса (или ван-дер-
ваальсово взаимодействие). Истинные физические причйны, по-
рождающие это притяжение, ранее не были известны и только в
последнее время расшифрованы составляющие сил, вызывающих
ван-дер-ваальсово взаимодействие.
Первый тип — это дипольное или ориентацион-
25
Рис. 12. Ориентацион-
ное взаимодействие мо-
лекул
Рис. 13. Индукционное
взаимодействие моле-
кул
ное взаимодействие. Оно вызывается ориентированием (при-
тяжением «плюса» одной к «минусу» другой) молекул с ди-
польной структурой при их сближении (рис. 12).
Второй тип взаимодействия называется индукционным.
Это взаимодействие возникает, когда молекула, обладающая
диполем (дипольная молекула), сближается с нейтральной моле-
кулой, у которой имеются заряды, равномерно распределен-
ные по молекуле (рис. 13, а). Под влиянием дипольной мо-
лекулы у нейтральной молекулы происходит перераспределе-
ние заряда и возникает (индуцируется) дипольный момент
(рис. 13, б).
Третий тип — самое главное взаимодействие — называют
дисперсионными силами или силами Лондона
по имени немецкого физика Ф. Лондона, который в начале
30-х годов XX в. обнаружил их существование и предложил
теорйю взаимодействий. Это взаимодействие вызывает притяже-
ние неполярных молекул и атомов, лишенных электрических
зарядов (например, метан и аргон).
Дисперсионные силы универсальны; они действуют между
любыми молекулами и атомами, независимо от их полярности и
реакционной способности. Величина дисперсионных сил возраста-
ет с увеличением массы взаимодействующих молекул и атомов,
причем, что особенно важно, их дальность действия на два по-
рядка больше, чем дипольных и индукционных сил (до 2000 нм).
Хотя почти всегда энергия дисперсионных сил больше, чем ди-
польная или индукционная, однако для полярных молекул эта
энергия сопоставима с дипольным взаимодействием, а у воды
дипольное взаимодействие выше дисперсионных сил.
Физическая сущность дисперсионных сил трудно объяснима.
Наиболее понятное объяснение принадлежит Полингу: притяже-
ние происходит благодаря притяжению атомных ядер .одной мо-
26
лекулы и электронов другой, которое в значительной мере, но не
полностью компенсируется отталкиванием электронов одной мо-
лекулы от электронов другой и взаимным отталкиванием ядер.
3. ТВЕРДОЕ СОСТОЯНИЕ
3.1. Аморфное и кристаллическое состояния. Полиморфизм
В отличие от газообразного состояния твердые вещества
характеризуются высокой степенью упорядоченности. Силы взаи-
модействия между частицами, слагающими твердое вещество
(атомы, ионы, молекулы), велики, и благодаря этому твердое
вещество обладает определенной формой, не изменяющейся при
перенесении их из одного объема в другой. Существуют две ос-
новные формы твердых веществ: кристаллическая и аморфная.
Первая из них обладает гораздо большей степенью упорядочен-
ности. Большей частью аморфные и кристаллические формы яв-
ляются лишь различными состояниями одного и того же вещест-
ва. Таковы, например, кристаллический кварц и различные
аморфные формы кремнезема. Перевод аморфной системы в
кристаллическую можно часто осуществить, например, длитель-
ным выдерживанием при высокой температуре или другими
путями.
Аморфные вещества отличаются от кристаллических прежде
всего изотропностью. Изотропность характеризуется
одинаковыми значениями данного свойства при измерении в
любом направлении внутри вещества. Вторым характерным свой-
ством аморфного вещества является то, что переход аморфного
вещества из твердого состояния в жидкое не сопровождается
скачкообразным изменением свойств. Так, в отличие от кристал-
лического вещества, имеющего определенную температуру
плавления Тпл, при которой происходит скачкообразное из-
менение свойств (рис. 14, а), аморфное вещество характери-
зуется интервалом размягчения (Та — Ть) и непре-
рывным изменением свойств (рис. 14,6). Этот интервал в зави-
симости от природы вещества может иметь значение порядка де-
сятков и даже сотен градусов.
Рис. 14. Изменение объема веществ при нагревании:
а — кристаллические вещества; б — аморфные вещества
27
Аморфные тела менее устойчивы, чем кристаллические. Лю-
бое аморфное тело, в принципе, должно кристаллизоваться, и
этот процесс должен быть экзотермическим. Поэтому теплота
образования аморфного вещества всегда меньше теплоты обра-
зования кристаллического (из одних и тех же исходных ве-
ществ).
Одно и то же вещество иногда оказывается способным су-
ществовать в нескольких различных кристаллических формах,
называемых модификациями. Само это явление назы-
вается полиморфизмом. Примером его могут служить ал-
маз и графит, являющиеся различными кристаллическими моди-
фикациями углерода, или кварц, тридимит и кристобаллит —
различные кристаллические модификации кремнезема.
Явление полиморфизма часто встречается в природе, особен-
но при образовании различных минералов. Так, минералы кальцит
и арагонит обладают одним и тем же химическим составом
(СаСОз), но различным кристаллическим строением. Анатаз и
рутил — минералы, образованные диоксидом титана, также об-
ладают разным кристаллическим строением.
В природе наблюдается явление, в какой-то мере противо-
положное полиморфизму. Оно заключается в том, что ряд ве-
ществ различного химического состава может образовывать
кристаллы совершенно одинаковой формы и довольно близкие
по структуре. Так, например, очень близким кристаллическим
строением обладают соли КН2РО4 и KH2ASO4, карбонаты СаСОз,
MgCOs и МпСОз, сульфаты MgSO4-7H2O, ZnSO4-7H2O,
NiSO4-7H2O. Это явление называется изоморфизмом.
Благодаря изоморфизму в природе возникает большое разнооб-
разие минералов, обладающих сложным составом (силикаты,
алюмосиликаты).
От полиморфизма и изоморфизма следует отличать алло-
тропию — явление, когда данный элемент способен существо-
вать в виде различных простых веществ.
Твердое кристаллическое состояние характеризуется наиболее
упорядоченной структурой. Эта упорядоченность выражается
правильным геометрическим расположением частиц, из которых
состоит твердое тело. Каждый кристалл образует плоские грани;
относительные длины ребер, так же как углы между гранями, яв-
ляются характеристиками типа кристалла. Для
его описания пользуются системой трех коор-
динат осей, направленных вдоль ребер крис-
талла и имеющих длины а, Ь, с и углы а, (3, у,
заключенные между этими осями (рис. 15).
Семь систем координатных осей характери-
зуют семь кристаллических систем (рис. 16,
табл. 5).
Одной кристаллической системе могут соот-
ветствовать несколько элементарных
ячеек. Куб с ребром а и частицей в каждой
28
Рис. 15. Система
координатных осей,
принятая в кри-
сталлографии
Рис. 56. Ориентация осей семи кристаллических систем
Q
yC? a
a / Ромбоэдрическая
Рис. 17. Элементарные ячейки простой кубической (а), гранецен-
трированной (б) и объемно центрированной (в) систем
вершине образует элементарную ячейку простой кубической си-
стемы (рис. 17, а). В кубической гранецентрированной системе
частица расположена в каждой вершине и в центре каждой
грани куба (рис. 17,6). Наконец, в объемно центрированной
системе частица расположена в каждой вершине и в центре куба
(рис. 17, в).
Кристаллическая решетка образуется при много-
кратном повторении элементарных ячеек (рис. 18). Внутри решет-
ки путем перемещения возможен переход от одной элементар-
ной ячейки к другой.
Узел решетки — это место, где находится частица.
Параллельные и равноотстоящие плоскости, проходящие через
29
узлы решетки, называют-
ся плоскостями
решетки. Естествен-
ные грани кристалла об-
разованы некоторыми из
этих плоскостей.
Кристаллические ре-
шетки обычно классифи-
цируют в зависимости от
типа частиц, образующих
кристалл, и от природы
сил притяжения между
ними. Если в узлах ре-
шетки расположены ионы,
образующие ионные свя-
зи, то такая решетка на-
зывается ионной. Если в
Рис. 18. Строение простой кубической ре- узлах решетки — аТОМЫ,
шетки соединенные ковалентны-
ми связями, то решетка
называется ковалентной. Молекулярную решетку образу-
ют молекулы, связанные ван-дер-ваальсовыми силами. И, нако-
нец, если решетка образована одинаковыми катионами, окру-
женными электронным облаком, то. говорят о металлической
решетке.
Таблица 5. Геометрические характеристики семи
кристаллических систем
Система Длина между осями Углы между осями
Кубическая Квадратичная или тетра- а = b = с а = р = 7 = 90°
гональная а = b =# с а = р = у = 90°
Орторомбическая а =/= b =/= с а = р = 7 = 90°
Моноклинная а #= b =£ с а = у = 90°; р #= 90°
Триклинная а =/= b =/= с а #= Р #= У 90°
Гексагональная а = b с а= р = 90°; 7== 120°
Ромбоэдрическая а = b = с а = р = 90°; 7 #=90°
3.2. Ионные решетки
Ионные решетки построены из ионов, между которыми дей-
ствуют электростатические силы притяжения (между ионами
противоположного заряда) и отталкивания (между ионами оди-
накового заряда). Ионные решетки образуют только те соеди-
нения, у которых химические элементы, входящие в их состав,
сильно различаются по величине сродства к электрону. Так, на-
пример, решетка, образованная хлоридом натрия, является ион-
30
ной: атом Na находится поблизости
от атома С1; при этом осуществля-
ется перенос электрона и образуется
электровалентная связь между дву-
мя ионами: Na+ и С1“. Число
ионов Na+,_ которое может размес-
титься вокруг иона С1“, определяет-
ся отталкиванием между положи-
тельными ионами натрия. Чем боль-
ше кулоновское притяжение между
анионами и катионами и чем меньше
силы отталкивания между ионами
одинакового знака, тем устойчивее
Кристалл. Рис. 19. Кристаллическая решет-
Способ образования ИОННЫХ ре- ка хлорида натрия
шеток приводит к тому, что они об-
ладают компактной структурой. Кристаллическая решетка хлори-
да натрия построена как бы взаимопроникновением гранецентри-
рованных кубических систем, одна из которых содержит только
катионы Na+, а другая — анионы С1“ (рис. 19).
Электронейтральность кристалла хлорида натрия обусловли-
вает наличие Na+ на каждый ион С1~; в кристалле хлорида
магния на один ион Mg2+ приходится два иона С1“.
Ионные соединения при нормальной температуре и давлении
представляют собой твердые тела и характеризуются сильной
электровалентной связью. Разорвать эту связь и разрушить
кристаллическую решетку очень трудно. Поэтому соединения с
ионной кристаллической решеткой имеют высокую температуру
плавления. Поскольку ионные решетки обладают компактной
структурой, плавление обычно происходит с увеличением объема.
Кристаллическая решетка разрушается также при растворе-
нии. Соединения с ионной решеткой растворяются в жидкос-
тях, состоящих из полярных молекул (например, в воде). Ион,
расположенный на поверхности кристалла, «окружается» моле-
кулами растворителя и «отделяется» от кристалла.
3.3. Ковалентные решетки
Ковалентные решетки состоят из атомов. Ковалентная связь
возникает между атомами, обладающими неспаренными электро-
нами. Например, атом углерода может образовать четыре оди-
нарные ковалентные связи, направленные к четырем вершинам
правильного тетраэдра, в центре которого располагается атом
углерода. Следовательно, с этим центральным атомом могут
быть связаны четыре других атома углерода. Каждый из них
обладает еще тремя неспаренными электронами, которые могут
образовывать связь с тремя атомами углерода.
Таким образом строится трехмерная решетка, составленная
исключительно из атомов углерода. Все связи эквивалентны,
31
Рис. 20. Решетка алмаза
как и углы, образующиеся между
атомами (рис. 20). Полученная
структура является структурой
алмаза; понятие молекулы здесь
несостоятельно, поскольку нельзя
выделить молекулярную ячейку.
Ячейка алмаза характеризуется
совокупностью 6,023 • 1023 атомов
углерода. Атомы углерода могут
быть замещены атомами крем-
ния, как в случае карборунда —
разновидности карбида крем-
ния SiC.
При возникновении, кристал-
лической решетки атомы разме-
щаются вокруг центрального атома так, чтобы их число было
равно валентности центрального атома. Направление связей
определяется валентными углами этого атома.
Плавление таких кристаллов связано с разрывом множест-
ва ковалентных связей и поэтому температуры плавления их
велики. Такой кристалл практически невозможно разрушить ра-
створением. Природа ковалентной связи препятствует возмож-
ности взаимодействий между атомами ковалентной решетки и
молекулами растворителя независимо от того, полярный он или
неполярный.
Ковалентные решетки — плохие проводники электричества.
Ионная проводимость в них невозможна, так как они построены
из атомов. Электронная проводимость также исключена, потому
что все электроны внешней оболочки использованы для обра-
зования связей и свободных электронов нет.
3.4. Молекулярные решетки
В узлах молекулярной решетки находятся молекулы, кото-
рые связаны между собой ван-дер-ваальсовыми силами. Эти
силы гораздо слабее, чем силы, определяющие связь в ионных
или ковалентных решетках, и температуры плавления молеку-
лярных решеток намного ниже.
Ван-дер-ваальсовы силы быстро уменьшаются с увеличе-
нием расстояния между молекулами; следовательно, притяже-
ние между молекулами сильно зависит от их формы. Оно тем
сильнее, чем более «компактна» молекула. Для неона, молекулы
которого имеют сферическую форму, характерна компактная
гранецентрированная кубическая структура кристалла.
Роль формы молекул можно проследить при сравнении физи-
ческих свойств н-гептана и триметил-2,2,3-бутана. Эти два соеди-
нения, обладающие одинаковой относительной молекулярной
массой, имеют температуры плавления, отличающиеся на 65 К.
Компактная форма триметил-2,2,3-бутана позволяет его молеку-
лам находиться ближе друг к другу, чем молекулы н-гептана.
32
Следовательно, ван-дер-ваальсовы силы для триметил-2,2,3-бу-
тана в твердом состоянии больше, а температура плавления
выше.
Если молекулы содержат полярные группы, то межмолеку-
лярные силы имеют большие значения по сравнению с неполяр-
ными молекулами. Температуры плавления и кипения таких ве-
ществ выше. Свойства полярных молекул промежуточны между
свойствами неполярных молекул и ионных.
Растворимость соединений, образующих молекулярные решет-
ки, зависит от природы молекул, из которых они построены, и,
в частности, от полярности этих молекул. Если решетка построе-
на из полярных молекул, то она растворяется в полярных раст-
ворителях. Размер полярной группы по отношению к остальной
молекуле определяет большую или меньшую растворимость в
полярных растворителях. Например, уксусная кислота СН3СООН
растворяется в любых соотношениях в воде, тогда как раство-
римость стеариновой кислоты С17Н35СООН составляет 0,03 кг на
100 кг воды при 298 К. Если молекулы не имеют полярной груп-
пы, то они практически не взаимодействуют с полярными раст-
ворителями. Ароматические соединения, построенные из двух
или нискольких бензольных ядер (нафталин, фенантрен), раст-
воряются в бензоле.
Таким образом, можно сделать вывод, что для органиче-
ских веществ, образующих молекулярную решетку, раствори-
мость в жидкости тем больше, чем ближе структура решетки к
жидкости.
/
В последнее время значительный интерес вызвали исследования, посвящен-
ные белковым кристаллам. Ботаники, исследуя растительные клетки,
уже давно обнаружили в них кристаллы самой разнообразной формы: куби-
ки, шары, ромбы, нити и даже звезды. Исследования, проведенные в Бота-
ническом институте АН СССР, позволили выделить в белковом кристалле от-
дельные субъединицы различной формы. Анализы подтвердили, что кристаллы
действительно состоят из белков. Их обнаруживали в любой части клетки —
в ядре, в цитоплазме, в пластидах и в митохондриях. Было показано, что
для некоторых растений число кристаллов в ядре — характерный устойчивый
признак. Для других — насыщенность ядра кристалликами белка меняется.
Причины, вызывающие кристаллизацию белка в естественных условиях, пока
еще не выяснены. Не ясна и роль их в жизнедеятельности растений.
3.5. Металлические решетки
Структура четвертого типа решетки обусловлена особым
видом связи — металлическим. Эта связь возможна и в жидком,
и в твердом состояниях.
Экспериментальное изучение металлических решеток показы-
вает, что металлы имеют простую плотную структуру и что
каждая частица окружена шестью или восемью частицами. Та-
кая структура не может быть образована ионной связью, так
как присутствует только один химический элемент. Ковалентная
связь также невозможна, поскольку металл (например, натрий,
который имеет только один внешний электрон) может образо-
2—696
33
вать только одну ковалентную связь, а он связан с восемью
одинаковыми частицами. Физические свойства (высокая темпе-
ратура кипения, большая электрическая проводимость) исклю-
чают возможность связи в решетке за счет ван-дер-ваальсовых
сил.
Итак, единственный валентный электрон натрия должен
принимать участие в восьми связях, и значит, этот электрон не
локализован. Каждый электрон может перемещаться в кристалле.
И каждая частица окружена электронами, которые не принадле-
жат исключительно и постоянно ей. Эту металлическую решетку
можно представить в виде решетки из ионов Na-1", которая по-
гружена в облако свободных электронов. Связь обеспечивается
электростатическим притяжением между положительными иона-
ми и электронным облаком. Такая модель позволяет качествен-
но объяснить некоторые свойства металла.
Металлическая связь слабее ковалентной связи и металличе-
скую решетку можно деформировать. Как известно, металлы
обладают тягучестью и ковкостью. Однако вырвать атом из та-
кой решетки трудно; об этом свидетельствуют, в частности, вы-
сокие температуры кипения металлов: 630 К (Hg), 1153 К (Na),
3273 К (Fe) и т. д.
Электронное облако внутри решетки легко привести в движе-
ние с помощью электрического поля; следовательно, металл —
хороший проводник электричества. Однако трудно вырвать элек-
трон из металла, поскольку между ансамблем положительных
ионов и электронами действуют силы притяжения.
Большая теплопроводность металлов обеспечивается наличи-
ем облака подвижных электронов. Если часть металла нагрета,
то кинетическая энергия электронов в этой области возрастает.
Электроны распространяются по всему металлу, вызывая рост
температуры во всей решетке.
4. ЖИДКОЕ СОСТОЯНИЕ
Жидкое состояние изучено менее полно, чем газообразное
или твердое. Поэтому представляется целесообразным провести
сравнение жидкого состояния с твердым и газообразным состоя-
ниями. У жидкости, как и у газа, нет собственной формы, но есть
собственный объем, в то время как молекулы газа занимают весь
представленный им объем. В жидком состоянии вещества теку-
чи и принимают форму сосуда, в который они помещены. Вещест-
ва, находящиеся в жидком состоянии при 298 К и более низкой
температуре, называются жидкостями. Если же вещество
переходит в жидкое состояние при температурах, превышающих
298 К, то такое состояние называют расплавленным или
расплавом.
Жидкое состояние вещества обусловлено особым характе-
ром расположения частиц и особым характером связи между
ними. В отличие от газов в жидкостях и расплавах частицы
84
находятся на расстояниях того же порядка, что и размеры са-
мих частиц. В газе силы, обусловливающие тепловое движе-
ние, гораздо больше сил притяжения. В результате этого га-
зообразное состояние — неупорядоченное, частицы распределены
случайно. Путь пробега частицы между двумя столкновениями
очень большой по сравнению с диаметром этой частицы. Сжи-
маемость газа велика, плотность его мала и меняется с темпе-
ратурой.
В твердых телах силы притяжения превосходят силы, опре-
деляющие тепловые движения; поэтому частицы не могут пере-
мещаться или вращаться, они могут только колебаться с огра-
ниченной амплитудой вокруг некоторого среднего положения.
Сжимаемость твердых тел незначительна, плотность их велика и
слабо меняется с температурой.
В жидкостях силы притяжения соизмеримы с силами, обус-
ловливающими тепловые колебания молекулы. Частицы могут
колебаться, перемещаться и вращаться. Если сила, вызывающая
тепловое колебание частйцы, превышает силу ее связи с другой
частицей, то она может переместиться на расстояние порядка
ее диаметра и образовать новую связь с другой частицей.
Сжимаемость жидкостей мала, их плотность близка к плот-
ности твердых тел, но более чувствительна к изменениям тем-
пературы.
При понижении температуры при постоянном давлении веще-
ства переходят из газообразного состояния, которое характе-
ризуется отсутствием связи между частицами и формой, в жид-
кое состояние, где частицы взаимосвязаны, но форма не опре-
делена, а затем в твердое состояние с правильной фиксирован-
ной структурой. Однако при понижении температуры жидкость
может замораживаться без упорядочения структуры. Тогда обра-
зуется аморфное вещество, структура которого приближается к
структуре жидкости.
Наличие в жидкости пространственного упорядочения моле-
кул подтверждается экспериментальными данными по рассеянию
света, дифракции рентгеновского излучения, нейтронов и элект-
ронов. Рентгеноструктурные исследования показали, что в жид-
костях, состоящих из многоатомных молекул, наблюдается не
только упорядоченное расположение молекул, но и известная
закономерность во взаимной ориентации частиц. Эта ориентация
усиливается для полярных молекул и при формировании водо-
родной связи. Однако, как видно на рис. 21, только в окрест-
ности данной частицы наблюдается закономерное расположение
соседних частиц. При удалении от рассматриваемой частицы А
на расстояние порядка 10 атомных расстояний закономерное
расположение частиц нарушается.
Таким образом, жидкое состояние характеризуется законо-
мерным расположением частиц в небольших объемах и неупоря-
доченным — во всем объеме. Такое структурное свойство жид-
кого состояния выражают терминами «ближний порядок» и
2*
35
Рис. 21. Расположение частиц:
а — в твердом теле; б — в жидкости
«дальний порядок» и говорят, что в жидкостях и расплавах
имеется ближний порядок и отсутствует дальний порядок.
Следует отметить, что для жидких веществ отсутствует удо-
влетворительное общее уравнение состояния. Если для газов вы-
ведено несколько рабочих уравнений состояния (уравнение
идеального газа, уравнение Ван-дер-Ваальса для реальных га-
зов и т. д.), то для жидкостей аналогичных обобщений не су-
ществует.
4.1. Плотность и молярный объем
Плотностью вещества называют величину q, которая харак-
теризует массу т, содержащуюся в единице объема V вещества:
q = т/У.
Полагая tn = 1 кг и V = 1 м3, получают единицу плотности с
размерностью
[q] — кг/м3 = кг-м~3.
В химии часто плотность жидкости принято выражать отношением мас-
сы тела при 293 К (20°С) к массе равного объема воды при 277 К (4°С). Не-
обходимость выбора определенной температуры вызывается тем, что плотность,
как правило, уменьшается с повышением температуры. Д. И. Менделеев по-
казал, что зависимость плотности от температуры выражается уравнением
Q? = ео (1 — аТ),
где qt — плотность при температуре Т\ — плотность при 273 К; а — коэф-
фициент; Т — температура, К.
36
Объем, занимаемый 1 моль жидкости, называется моляр-
ным объемом. Он равен отношению молярной массы жид-
кости к плотности:
Vm = M/Q.
Молярный объем жидкостей обладает свойством аддитивно-
сти сложения:
молярный объем приблизительно равен сумме атомных объемов групп, вхо-
дящих в молекулу жидкости.
Например, молекула гексана СеН14 состоит из 6 атомов угле-
рода и 14 атомов водорода. Молярный объем гексана, соглас-
но свойству аддитивности, равен сумме 6 объемов углерода Vc и
14 объемов водорода Ун:
С6Н14 = 6Vc + 14VH = 6-11,0 4- 14-5,5 = 143 мл.
Опытное значение молярного объема гексана равно 139,9 мл.
Другой аддитивной величиной, характеризующей свойства
жидкости, является молярная рефракция MRd
п2 — 1 М п2 — 1
MRd = 2 , -о---=- $ гтг
п + 2 q /г 4- 2
где п — показатель лучепреломления; индекс D указывает, что
относится к D-линии натрия (желтая линия).
Молярная рефракция — структурно чувствительное свойство
жидкости, и ее чаще применяют для характеристики жидкостей,
чем молярный объем.
4.2. Электрический дипольный момент
Известно, что электронная плотность в молекуле АВ может
неравномерно распределяться по отношению к атомам А и В.
Когда электронное облако концентрируется у одного из атомов,
то центры тяжести положительного и. отрицательного зарядов
в молекуле в целом не совпадают и раздвинуты на некоторое
расстояние /. Таким образом, хотя молекула остается электро-
нейтральной, в ней имеются как бы два полюса электричества:
положительный и отрицательный. Молекулы с несовпадающими
центрами тяжести электрических зарядов называются поляр-
ными.
Полярность молекул характеризуется дипольным мо-
ментом ц
н = qi,
где I — расстояние между центрами положительных и отрица-
тельных зарядов; q — абсолютное значение этих зарядов.
Чем более полярны молекулы, чем значительнее смещены
валентные электронные пары к одному из атомов, тем больше ц.
Наоборот, если электрическая асимметрия молекул незначитель-
на, то ц невелик.
37
Рис. 22. Сложение дипольных моментов свя-
зей в молекулах HCN (a), SO2 (б) и
СНзС! (в)
Так как расстояние
между атомами в молеку-
лах есть величина поряд-
ка 1О-10 м, а заряд элект-
рона равен 1,6-10-19 Кл,
то дипольные моменты
молекул или отдельных
химических связей имеют
величину порядка 1О-30
Кл-м. Дипольные момен-
ты некоторых молекул
приведены в табл. 6. Часто
в справочной литературе дипольные моменты приводят в едини-
цах дебая (D): 1D = 3,336-Ю-30 Кл-м. Эта единица названа
в честь известного физика Дебая, внесшего большой вклад в
изучение электрических свойств молекул.
" Если в молекуле несколько полярных связей, то дипольные
моменты суммируют как векторы. Поэтому возможны случаи,
когда отдельные связи в молекуле полярны, а суммарный ди-
польный момент равен нулю (т. е. молекула в целом неполярна).
Так, в молекуле СО2 дипольные моменты двух полярных свя-
зей С = О взаимно уничтожаются, в ССЦ сумма четырех ди-
польных моментов связей С—С1 также равна нулю. Молекула
же' Н2О полярна, так как дипольные моменты двух полярных
ОН связей суммируются и дают дипольный момент, направлен-
ный по биссектрисе валентного угла НОН. Векторное сложение
дипольных моментов связей показано на рис. 22; для всех при-
меров принято, что вектор направлен от плюса к минусу.
Дипольный момент обычно определяют экспериментально,
измеряя связанную с ним диэлектрическую постоян-
ную е вещества при различных температурах. Если вещество
поместить в электрическое поле, создаваемое конденсатором,
то емкость последнего возрастает в 8 раз, т. е. 8 = с/со (где
Со и с — емкость конденсатора в вакууме и в среде вещества).
Конденсатор в среде вещества имеет больший запас энергии,
чем в вакууме. Это обусловлено тем, что под действием поля
происходит ориентация диполей.
Наличие дипольного момента у молекулы эквивалентно
электрическому полю, поэтому, когда в непосредственной близо-
сти от полярной молекулы, как это имеет место в жидкости, на-
ходится другая молекула, то она испытывает действие электри-
ческого поля. Электрическое поле, воздействуя на частицу, вы-
зывает смещение в ней электрических зарядов, называемое
поляризацией. Поляризация проявляется в возникнове-
нии у частиц индуцированного дипольного момента рИНд вслед-
ствие смещения электронов и ядер. В первом приближении
индуцированный дипольный момент можно считать пропорцио-
нальным напряженности электрического поля Е:
Нинд — <xF.
38
Таблица 6. Дипольные моменты некоторых молекул
Молекула р. - 1030, Кл-м Молекула |LL - 1 О30, Кл-М Молекула lx- 1030, Кл-м
Н2О 5,52 n2o 0,51 CsF 24
Н2О2 6,77 NO 6,48 CsCl 31,2
HF 5,73 no2 0,87 CsI 36,8
НС1 3,24 HNO3 6,47 CH3OH 5,13
НВг 2,97 F2O 0,90 C2H5OH 5,01
HI 1,14 BrF 3,87 CHCI3 3,03
NH3 4,44 Оз 1,59 HCOOH 4,05
РНз 174 H2S 3,30 CH3COOH 5,19
AsH3 0,39 NaCl 30 C6H5CH3 1,H
Коэффициент пропорциональности а называют поляри-
зуемостью частицы. Поляризуемость измеряется в Кл • м2/В.
Для ионов поляризуемость приблизительно пропорциональна ку-
бу их радиуса.
4.3. Вязкость
Когда какое-нибудь вещество движется по поверхности, то
этому движению препятствует трение. Если этим веществом яв-
ляется жидкость, то трение вызывает эффект, называемый
вязкостью.
Рассмотрим жидкость, находящуюся между двумя большими
параллельными пластинами (рис. 23, а), одна из которых не-
подвижна, а другая движется в направлении х со скоростью v.
Движению бесконечно тонкого слоя жидкости, прилегающему к
каждой пластине, препятствует трение. Движущаяся пластина
заставляет жидкость двигаться в направлении х со скоростью,
приблизительно равной v, а слой, прилегающий к неподвиж-
ной пластине, движется очень медленно. Если представить, что
жидкость состоит из большого числа слоев, то каждый слой
будет скользить вдоль соседнего и сопротивление за счет трения
между прилегающими слоями приведет к появлению градиента
скорости. Деформация жидкости, вызванная градиентом ско-
рости, называется сдвигом. И. Ньютон показал, что сила со-
противления между слоями f пропорциональна площади слоев £
и градиенту скорости между ними dv/dy:
"7)
Обычно т] называют коэффициентом вязкости
или вязкостью; f/S — F— напряжением сдвига;
dv/dy=G — градиентом сдвига или скоростью
сдвига. Если величина т] постоянна, жидкость называется
ньютоновой, если т] — функция F или G, то жидкость — не-
ньютоновая.
39
Сила сдвига
Рис. 23. Движение жидкости между под-
вижной и неподвижной пластиной (а) и
по трубе (б):
1 — пластина, движущаяся со скоростью v\
2 — неподвижная пластина; 3 — стенка трубы
Если жидкость находится
не между пластинами, а те-
чет по цилиндрической труб-
ке, трение возникает у сте-
нок трубки; в этом случае
скорость максймальна вдоль
оси трубки и минимальна у
ее стенок (рис. 23,6), так
что градиент скорости стано-
вится параболическим, а не
линейным.
Условия течения, пока-
занные на рис. 23, соответ-
ствуют ламинарному
течению; оно существует до
тех пор, пока значение гра-
диента сдвига не слишком
велико. Высокие значения
градиента сдвига связаны с
возникновением турбу-
лентности.
Если принять S = 1 м2,
dv/dy = 1 м/с на 1 м и F —
— 1 Н, то из уравнения (1.7)
можно получить единицы
вязкости:
, , Н-М-С Н
1т]| =—к---=—5- с = Па-с.
м -м м
Вязкость, равная 1 Па-с, соответствует вязкости такой
жидкости, у которой при градиенте скорости 1 м/с на 1 м сила
трения между слоями площадью в 1 м2 равна 1 Н. Значения
вязкости некоторых жидкостей приведены в табл. 7.
А. И. Бачинский установил, что вязкость многих жидкостей
выражается формулой
с
Ууд Усобств
1
Л =-j7---------- ИЛИ — = <р---------,
Гуд Усобств Л С
где С — постоянная величина; Ууд — удельный объем; Кобств —
собственный объем, занимаемый частицами жидкости; ср — теку-
честь.
Таблица 7. Вязкость некоторых жидкостей при 293 К
Жидкость Вязкость, мПа-с Жидкость Вязкость, мПа-с
Вода 1,005 Глицерин 1490
Бензол Этанол 0,649 1,200 Тетрахлорид угле- рода 0,969
Хлороформ 0,570 Сероуглерод Ртуть 0,365 1,554
40
Вязкость жидкости уменьшается с увеличением температу-
ры, так как с повышением температуры увеличиваются средние
расстояния между молекулами жидкости, ослабляется взаимное
притяжение между ними, и, следовательно, уменьшается сила
трения между движущимися слоями (табл. 8).
Таблица 8. Вязкость воды при разных температурах
Температура, К Вязкость, мПа-с Температура, К Вязкость, мПа «с
273 1,792 333 0,469
283 1,308 343 0,406
293 1,005 353 0,356
303 0,801 363 0,316
313 0,656 373 0,284
323 0,549
Для многих жидкостей зависимость вязкости от температуры подчиняется
уравнению де Гузмана
L = Ae-E/RT,
ч
где 1 /т| — текучесть; Е — энергия активации текучести.
Исходя из современных представлений о строении жидкости и особого
механизма передачи количества движения между движущимися слоями жид-
кости за счет прилипания частиц, находящихся в разных слоях, Г. М. Пан-
ченков получил уравнение, выражающее зависимость вязкости от температуры:
л = Зт/Л }/ ^°6ств M~lbdl3Tl\eEIRT + e~EIRT — 2),
/уд
где R — универсальная газовая постоянная; Гсобств — собственный объем моле-
кул; Ад — постоянная Авогадро; М — молекулярная масса; d — плотность; Т —
температура; Е — энергия связи между молекулами жидкости.
Приведенное уравнение хотя и громоздко,
однако не содержит произвольных постоянных
и дает лучшее согласие с опытом.
Для измерения вязкости пользуются прибо-
рами, называемыми вискозиметрами.
Широко распространены капиллярные вискози-
метры, в которых вязкость определяется по
времени вытекания определенного объема
жидкости через капилляр. Один из капилляр-
ных вискозиметров показан на рис. 24. При ра-
боте с вискозиметром этого типа определяют
время вытекания жидкости, заключенной в
объеме между метками 1 и 2.
Для расчета вязкости применяют формулу
Пуазейля:
Рис. 24. Капилляр-
ный вискозиметр
яг4
41
где г — радиус капилляра; I — длина капилляра; V — объем
вытекающей жидкости; р — давление, под которым жидкость
протекает через капилляр; t — время вытекания.
На практике обычно определяют относительную
вязкость, т. е. отношение вязкости испытуемой жидкости т)
к вязкости какой-либо другой жидкости т)о, которая известна.
Тогда для одного и того же вискозиметра из уравнений Пуа-
зейля можно получить отношение
\ Л_ pt
Л о pot о
Если обе жидкости протекают через капилляр под давле-
нием собственного веса, то отношение р/ро можно заменить от-
ношением их плотности q/qo:
/ &
откуда
Л = Т,О^Г'
Вязкость можно определить и по скорости свободного паде-
ния в жидкости шарика известного объема и массы. В этом
случае коэффициент вязкости вычисляют по уравнению Стокса:
где г — радиус шарика; р и р0 — плотность материала шарика
и жидкости соответственно; v — скорость падения шарика; g —
ускорение свободного падения.
4.4. Давление насыщенного пара жидкости
Рассмотрим жидкость, находящуюся в открытом сосуде, на-
греваемом извне. Процесс испарения может быть упрощенно объ-
яснен следующим образом. В объеме жидкости молекулы взаи-
модействуют между собой больше, чем в газовой фазе, и, чтобы
перейти из жидкой фазы в газовую, надо затратить определен-
ную энергию. Однако, согласно распределению молекул по энер-
гиям, среди них имеются молекулы, обладающие кинетической
энергией, в 5—15 раз большей средней кинетической энергии
молекул жидкости. Эти молекулы, составляющие малую долю от
всех молекул, могут покинуть жидкую фазу и «вылететь» из
объема в газовую фазу. Средняя энергия молекул, оставшихся
в объеме, при этом понизится, и для повышения ее необходимо
будет подвести теплоту извне. Таким образом, процесс испаре-
ния эндотермичен, но так как происходит компенсация энергии
путем притока теплоты от окружающей среды, жидкость в откры-
том сосуде может быть испарена полностью.
42
Если жидкость находится в закрытом сосуде, то испарив-
шиеся молекулы постепенно накапливаются в газовом слое. Пе-
редвигаясь в объеме парообразного слоя и ударяясь о стенки
сосуда или о поверхность жидкости, молекулы газовой фазы
могут поглотиться жидкостью. Таким образом, осуществляется
экзотермический процесс, обратный испарению, — процесс кон-
денсации пара в жидкость.
Эндотермический процесс испарения обратим. Поэтому одно-
временно с ним протекает экзотермический процесс конденсации.
Следовательно, при определенных условиях устанавливается рав-
новесие. Равновесное состояние системы жидкость — пар при
данной температуре характеризуется давлением насы-
щенного пара. Эта величина не зависит от количества
взятой жидкости, пара и от наличия и концентрации воздуха
или другого газа, инертного по отношению к данному пару.
Таким образом, давление насыщенного пара данного вещества
при неизменной температуре является величиной постоянной и
характерной для него.
При повышении температуры кинетическая энергия молекул
возрастает и, следовательно, большая доля молекул окажется
обладающей энергией, достаточной для перехода в пар. Число
молекул, вылетающих из жидкости в 1 с, при повышении тем-
пературы сильно увеличивается и равновесие пара с жидкостью
достигается только при более высоких концентрациях пара. Та-
ким образом, с повышением температуры давление насыщенного
пара увеличивается (рис. 25).
Переход вещества из
жидкого состояния в па-
рообразное называют в
общем случае парооб-
разованием. Различа-
ют две формы этого про-
цесса: испарение —
когда парообразование про-
исходит со свободной по-
верхности жидкости, и ки-
пе н и е — когда парооб-
разование происходит не
только с поверхности, но и
внутри жидкости путем об-
разования пузырьков пара
во всем объеме жидкости
и выделения их. Испаре-
ние жидкости с той или
иной скоростью происхо-
дит при любой темпера-
туре. Кипение происходит
только при температуре,
при которой давление на-
Зависимость давления насыщенного
температуры для некоторых жидко-
Рис. 25.
пара от
стей
43
сыщенного пара достигает (или превосходит) величины внешнего
давления.
Кривые давления пара выражают не только зависимость
давления насыщенного пара от температуры, но и зависимость
температуры кипения от внешнего давления. Повышение давле-
ния всегда увеличивает температуру кипения и, наоборот, пони-
жение давления жидкости уменьшает температуру кипения.
Нормальной температурой кипения называется температура кипения при
нормальном атмосферном давлении,
т. е. температура, при которой давление насыщенного пара
становится равным нормальному атмосферному давлению:
101,3 кПа.
4.5. Вода и лед
Вода является основным компонентом любого живого орга-
низма и большинства растительных клеток. Содержание воды в
клетках меняется в зависимости от типа клетки и физиологи-
ческих условий. Например, в корне моркови содержится около
85% воды, тогда как молодые листья салата-латука на 95% со-
стоят из воды. В некоторых сухих семенах и спорах содержа-
ние воды достигает всего лишь 10%; однако для того чтобы они
стали метаболически активными, содержание воды в них должно
существенно увеличиться.
Наличие водной среды является необходимым условием для
многих биохимических реакций. Только в водной среде могут
протекать химические процессы с той интенсивностью, какая ха-
рактерна для растения и живого организма.
Так, изучение интенсивности дыхания зерновых в зависимо-
сти от процента влажности в семенах показало, что если в
начале увеличение влажности повышает интенсивность дыхания
на незначительную величину, то затем, начиная приблизительно
с 15%, повышение влажности на 1% усиливает дыхательный
процесс на 150%; последующее увеличение влажности уже повы-
шает дыхание на несколько сот процентов. Следовательно, чем
выше содержание воды в зерне, тем интенсивнее процесс ды-
хания.
Вода также непосредственно участвует в метаболизме.
Она служит источником кислорода, выделяемого в ходе фото-
синтеза, и водорода, используемого для восстановления угле-
кислого газа. При образовании АТФ — важного микроэнерге-
тического соединения — из АДФ и фосфата происходит отщепле-
ние воды; иными словами, фосфорилирование есть не что иное,
как процесс дегидратации, происходящий в вйдном растворе в
биологических условиях. Таким образом, знание многих уни-
кальных свойств воды имеет громадное значение для общего
понимания физиологии растений и животных.
Вода является средой, в которой происходит диффузия
растворенных веществ по клеткам растения.
44
Минеральные питательные вещества, необходимые для роста,
и органические соединения, образующиеся в ходе фотосинтеза,
транспортируются по растению в виде водных растворов. У на-
земных растений существует непрерывный водный путь из почвы
через тело растения к листьям, где вода испаряется через
устьица.
Вода довольно мало сжимаема при давлениях, существующих
в организме, и поэтому важна ее роль в поддержании струк-
туры растения. Вода обладает высокой теплоемкостью и высокой
для жидкости теплопроводностью; оба эти свойства делают ее
идеальной жидкостью для поддержания теплового равновесия.
Энергия, необходимая для перевода молекулы из жидкости
в газовую фазу без изменения ее температуры (теплота испа-
рения), в расчете на 1 г при 373 К для воды является самой
высокой теплотой испарения для всех известных жидкостей.
Высокое значение теплоты испарения обусловлено громадным
числом водородных связей, имеющихся в водных растворах.
При 298 К испарение 1 моль воды требует 43 кДж, а это озна-
чает, что испарение воды в ходе транспирации сопровождается
значительной потерей теплоты растением. Большая часть этой
энергии испарения идет на то, чтобы разорвать водородные
связи и дать молекулам воды возможность существовать в га-
зовой фазе порознь. Потеря теплоты при испарении воды яв-
ляется одним из главных средств регуляции температуры у на-
земных растений. Тем самым рассеивается много теплоты, при-
шедшей в виде излучения, а также полученной в результате
метаболической деятельности.
Изучение структуры воды и особенно рассмотрение возник-
новения и распада водородных связей позволяют понять свой-
ства этой удивительной жидкости.
Чтобы разобраться в свой-
ствах воды, сначала следует
выяснить структуру льда. Из-
вестно девять кристалличе-
ских форм льда. Большая
часть их устойчива только
при высоких давлениях. На-
иболее распространенная
структура льда I (рис. 26)
изучена особенно подробно.
Эта кристаллическая форма
льда характеризуется плот-
ностью 0,924 кг/м3 при 273 К
и 101,3 кПа. В этой форме
льда расстояние между ато-
мами кислорода равно 0,276
нм. Длина связи О—Н лежит
в пределах 0,096—0,102, а
длина водородной связи
Рис. 26. Структура льда I
45
0...Н — между 0,174 и 0,180 нм. Таким образом, лед явля-
ется координированной кристаллической структурой, в которой
практически все молекулы воды соединены водородными
связями.
Благодаря такой структуре лед имеет меньшую плотность,
чем жидкая вода, и этот факт существенно важен для экологии.
Если бы не особая структура, обусловленная водородными
связями, то лед был бы плотнее, чем жидкая вода, как это
обычно бывает у большинства твердых веществ. В этом случае
при замерзании лед опускался бы на дно водоемов, которые
промерзали бы до дна. В таких условиях большинство живых
организмов не смогло бы перенести зиму. Однако вода дости-
гает максимальной плотности при 277,15 К, т. е. на четыре граду-
са выше температуры замерзания. Охлаждение ниже этой тем-
пературы приводит к уменьшению плотности воды, в результате
чего более легкая холодная вода поднимается на поверхность
и здесь замерзает. Слой льда, образующийся на поверхности,
не тонет и, что особенно важно, действует как теплоизолятор
по отношению к находящейся под ним жидкой воде.
Структура жидкой воды выяснена не столь детально, как
структура льда. Для структуры воды было предложено много
моделей.
Так, В. К. Рентген высказал гипотезу, что вода представляет
собой раствор льда в воде. Отсюда следовал вывод о наличии
двух состояний молекул воды: в виде льда и в «истинно жид-
ком». Гипотеза об изменении соотношения этих двух состояний с
температурой хорошо объясняла многие необычные свойства
воды, в частности, уменьшения объема при плавлении и макси-
мума плотности при 277 К.
Было показано, что сильные взаимодействия между молекула-
ми воды обусловлены структурой молекул этого соединения
(рис. 27). Расстояние между ядром кислорода и ядрами каж-
дого из двух атомов водорода равно примерно 0,099 нм, а угол
между связями Н—О—Н равен примерно 105°. Атом кислорода
обладает сильной электроотрицательностью и стремится оттянуть
электроны от атомов водорода. Благодаря этому на атоме кисло-
рода возникает частичный
отрицательный заряд, в то
время как два атома водо-
рода приобретают частич-
ный положительный заряд.
Несущие положительный за-
ряд атомы водорода воды
испытывают электростатиче-
ское притяжение со стороны
отрицательно заряженных
атомов кислорода соседних
молекул воды. Это приводит
к возникновению водород-
Рис. 27. Схематическое изображение
структуры молекул воды.
46
ных связей между молекулами воды. В результате такого
связывания молекул воды друг с другом возникает большая
упорядоченность в водных растворах. Действительно, на от-
дельных участках жидкая вода приобретает почти кристалли-
ческую структуру, что чрезвычайно важно, поскольку может
играть определяющую роль во взаимодействиях и ориентации
молекул в водных растворах.
Дж. Д. Бернал (1933) предположил, что водородные связи,
существующие во льду, сохраняются и в жидкой воде, хотя
упорядоченная трехмерная структура, характерная для кристал-
лического состояния, при плавлении исчезает.
Таким образом, при нагревании некоторые из межмолеку-
лярных водородных связей разрываются. Теплота плавления
льда при 273 К равна 6,05 кДж/моль. Полное разрушение
межмолекулярных водородных связей, в которых участвуют все
атомы водорода, потребовало бы 40,5 кДж на 1 моль воды.
Определение теплоты плавления показывает, что при плавлении
льда разрушается только около 15% всех водородных связей.
Соответственно в жидкой воде при 273 К остается 85% ненару-
рушенных водородных связей. Нагревание воды от 273 до 298 К
потребует примерно 2,1 кДж/моль. Если бы даже вся эта энергия
была затрачена на разрушение водородных связей, то свыше
80% из них все еще оставалось бы ненарушенным при 298 К-
Возникающие свободные мономерные молекулы воды могут
занимать пустоты в остальной «льдоподобной» решетке. Этот
факт позволяет объяснить повышенную плотность воды по срав-
нению со льдом. С ростом температуры разрывается все боль-
ше водородных связей, но одновременно увеличивается кинети-
ческая энергия молекул. Первый фактор способствует увеличе-
нию плотности воды, тогда как рост кинетической энергии при-
водит к уменьшению плотности, поскольку в последнем случае
каждая молекула занимает все больший эффективный объем.
Суммарный результат этих двух тенденций проявляется в том,
что плотность достигает максимума при температуре 277 К,
выше которой плотность воды монотонно понижается с ростом
температуры.
Согласно другой теории при плавлении льда в действитель-
ности разрывается лишь очень небольшая доля водородных
связей.
Дж. Франк и П. Вин предложили весьма близкую модель
так называемых мерцающих кластеров. Они предпо-
ложили, что эти кластеры постоянно разваливаются и вновь
образуются, причем среднее время их жизни составляет около
1О“10 с. На рис. 28 дана схема структуры воды по Франку
и Вину, модифицированная Г. Н. Немети и Г. Шерага. Каждая
молекула воды здесь может образовать 4, 3, 2, 1 водородную
связь или совсем не участвовать в образовании водородных
связей. Образование водородных связей протекает специфически:
наличие у данной молекулы воды одной водородной связи об-
47
Рис. 28. Строение воды согласно модели
мерцающих кластеров
легчает образование других
водородных связей. Если
одна из водородных связей
данной молекулы воды /ра-
зорвана, то это облегчает
разрыв и остальных водо-
родных связей. /
Одна из последних [ тео-
рий, разработанная Г. Эй-
рингом, предполагает цали-
чие в жидкой воде двух| кри-
сталлоподобных структур,
причем одна из них плотнее
другой на 20%. Между дву-
мя структурами существует
динамическое равновесие.
Кроме того, в жидкой
воде имедотся мономерные
молекулы и подвижные по-
лости.
4.6. Жидкие кристаллы
Обычно между высокоупорядоченным кристаллическим состоянием и менее
упорядоченным жидким состоянием существует резкий переход. Однако молекулы
некоторых веществ имеют настолько сильную тенденцию к упорядоченности,
что при плавлении кристалла сначала образуется мутная жидкость, которую
называют мезоморфным или паракристаллическим состоя-
нием. Вещества в этом состоянии сохраняют некоторые признаки кристал-
лических свойств. При более высоких температурах мутная жидкость резко
переходит в прозрачную, обладающую уже свойствами обычных жидкостей.
Вещества в мезоморфном состоянии называют жидкими кристаллами.
Молекулы,"образующие жидкие кристаллы, как правило, имеют вытянутую,
прямолинейную? палочкообразную форму. Примером является 4,4'-диметоксиазо-
ксибензол
который имеет следующие точки перехода (или плавления) между тремя со-
стояниями:
391 к . 409 к
кристаллическое------► мезоморфное-------► жидкое
В мезоморфном состоянии молекулы располагаются параллельно друг другу,
причем допускается вращение только вокруг длинных молекулярных осей. Веще-
ства такого типа называют анизотропными, поскольку они характеризуются
различными свойствами по различным направлениям. В обычных жидкостях
преимущественной ориентации нет, вещества в этом состоянии изотропны.
Известны два типа жидких кристаллов: термотропные и лиотропные. Тер-
мотропные жидкие кристаллы образуются при нагревании вещества, и их
можно, в первую очередь, подразделить на смектические, нематические и холе-
стерические жидкие кристаллы (рис. 29).
В смектических жидких кристаллах длинные оси молекул перпен-
48
дикулярны плоскости слоев. Слои могут
свободно скользить друг относительно
друга, так что вещество имеет механи-
ческие свойства двумерного, кристалла.
Оптические свойства смектич'еских жидких
кристаллов похожи на свойства таких
трехмерных кристаллов, как кварц. Н е-
матические жидкие кристаллы наи-
менее упорядочены. В них молекулы ори-
ентированы таким образом, что их длин-
ные оси параллельны, однако они не
образуют слоев. Холестерические
жидкие кристаллы похожи на смектиче-
ские тем, что в них молекулы также
образуют слои, в пределах каждого из ко-
торых длинные оси молекул параллельны,
но в соседних слоях оси молекул ориенти-
рованы под некоторым углом друг к другу.
Лиотропные жидкие кристаллы
образуются при смешении двух или не-
скольких соединений, одно из которых —
вода или какой-нибудь другой полярный
растворитель. Этот тип жидких кристал-
лов еще изучен слабо, хотя и предпола-
гается, что они встречаются в живых
IIIIIIIIIIIII
IIIIIIIIIIIIH
IIIIIIIIIIIIIII
а)
в)
Рис. 29. Схема расположения моле-
кул в термотропных жидких кри-
сталлах:
а смектические; б— нематические;
в — холестерические жидкие кристал-
лы
системах.
Термотропные жидкие кристаллы нашли широкое применение в науке, тех-
нологии и медицине. Разработаны методы использования холестерических
жидких кристаллов в качестве чувствительных температурных датчиков, осно-
ванные на том, что цвет холестерических кристаллов изменяется при очень сла-
бых изменениях температуры.
Под действием электрического поля нематические жидкие кристаллы вызы-
вают интересные оптические эффекты, называемые динамическим рассеянием.
При этом прозрачная пленка становится мутной. Это свойство используется во
многих карманных вычислительных машинах, часах с цифровой индикацией и
наручных часах, индикаторные устройства которых выполнены на основе таких
жидких кристаллов.
Свойствами жидких кристаллов обладают в определенном температурном
интервале некоторые индивидуальные органические соединёния — сложные
эфиры холестерина, фосфатиды, цереброзиды, а также и растворы многих био-
логически важных соединений.
Можно сделать вывод, что с жидкокристаллическим состоянием связаны
важнейшие функции живого организма: восприятие, размножение, движение,
синтез и энергетический обмен.
Контрольные вопросы
1. Идеального газа в природе не существует. Как вы думаете, для чего
создана эта модель? Каким условиям она отвечает?
2. При переходе из одного агрегатного состояния в другое происходит
изменение расстояния между частицами. Чем оно обусловлено?
3. Почему в уравнении Фика в левой части стоит знак минус?
4. Почему длина свободного пробега молекул газа уменьшается с увеличе-
нием давления?
5. Чем вызвано наличие критического давления и критического объема в
реальных газах?
6. Почему ионные решетки характеризуются высокой температурой плав-
ления?
7. Какие структурные особенности воды делают ее столь важной для биологи-
ческих процессов?
Глава II
ХИМИЧЕСКАЯ ТЕРМОДИНАМИКА
1. КЛАССИЧЕСКАЯ И СТАТИСТИЧЕСКАЯ ТЕРМОДИНАМИКА
Термодинамика изучает законы, которые описывают обмен
энергией между изучаемой системой и внешней средой и, в част-
ности, превращение тепловой энергии в другие формы энергий.
Химическая термодинамика — это раздел физической химии,
изучающий превращения энергии в химических процессах и
энергетические характеристики различных веществ. Она основы-
вается на положениях, законах и теоретических методах термо-
динамики. Применение химической термодинамики позволяет
рассматривать процесс, не вдаваясь в механизм взаимодействия
отдельных составных частей вещества. Зная законы химической
термодинамики, можно предвидеть, возможна ли данная реак-
ция при данных условиях или невозможна, какие условия необ-
ходимо создать, чтобы реакция быда возможной и чтобы выход
продуктов был максимальным. Для решения этих вопросов
термодинамическим методом необходимо знать только начальное
состояние системы и те внешние условия, в которых она нахо-
дится.
Классическая (феноменологическая) термодинамика занима-
ется изучением макроскопических динамических систем, облада-
ющих большим числом степеней свободы. Однако любое макро-
скопическое состояние можно описать с точки зрения микро-
скопических свойств вещества. Этот подход лежит в основе ста-
тистической термодинамики, которая позволяет установить связь
между физическими законами макро- и микромира. В учебнике
все вопросы (за редким исключением) будут рассмотрены с по-
зиции классической термодинамики и ее приложений.
2. СИСТЕМА И ВНЕШНЯЯ СРЕДА
Системой называют ограниченную каким-либо образом часть
физического мира, всякий материальный объект, обособленный
физическими или воображаемыми границами раздела от окру-
жающей среды и подвергнутый теоретическому и (или) экспери-
ментальному изучению.
Системы, в которых исключен какой бы то ни было обмен
веществом или энергией с окружающей средой, называют изо-
лированными. Систему называют закрытой, если она
50
может обмениваться с окружающей средой энергией и не может
обмениваться веществом. В качестве примера можно привести
запаянную ампулу с веществом, которая попеременно помеща-
ется в горячую и холодную среду. Масса содержимого ампулы
остается постоянной, хотя онал будет получать и отдавать энер-
гию. Системы, которые могут'Обмениваться веществом и энер-
гией с окружающей средой, называют открытыми. К ним
относятся, например, все живые организмы.
Следует заметить серьезные осложнения, которые возникают
при применении термодинамики к биохимическим процессам.
Это связано с тем, что, как правило, термодинамика имеет дело
с закрытыми системами, находящимися в равновесии, а живые
организмы относятся к открытым системам, в которых равнове-
сие обычно отсутствует. Они находятся в так называемом стацио-
нарном состоянии, когда концентрация частиц поддерживается
постоянной за счет непрерывного притока и оттока веществ из
системы; любая клетка в равновесном состоянии — это уже мерт-
вая клетка. Для рассмотрения открытых систем требуются мето-
ды термодинамики необратимых процессов, обсуждение которых
выходит за рамки данного учебника. Однако следует особо под-
черкнуть, что в пределах термодинамики обратимых процессов
возможно решение многих важных частных теоретических и при-
кладных задач биохимии.
Обмен энергией между системой и внешней средой может
проявляться в различных формах. Механическая, тепловая,
электрическая энергии и энергия излучения прямо или косвенно
превращаются друг в друга. В 1843 г. Дж. Джоуль осуществил
первую количественную проверку эквивалентности тепловой
энергии, или теплоты, и механической энергии. При этом рас-
сматривались превращения, в которых участвуют только тепло-
вая энергия Q и механическая работа (энергия) Л; эти превра-
щения называются термомеханическими.
Единица работы (энергии) А определяется на основании
уравнения
где F — сила, действующая на пути I и совпадающая с ним по
направлению. Полагая силу, равную 1Н, и путь, равный 1 м
(F = 1 Н, 1= 1 м), получают единицу работы в СИ, которая назы-
вается джоулем (Дж). Таким образом, джоуль — это рабо-
та, совершенная силой в 1 Н на пути в 1 м:
[4] = [F] [/] — кг • м • с“2 ♦ м = кг • м2 • с-2.
Так как количество теплоты Q эквивалентно некоторой работе
Л, теплота выражется тоже в единицах работы, т. е. в джоулях.
Для проведения расчетов необходимо ввести знаки, учиты-
ваемые при рассмотрении обмена между системой и внешней
средой. Приток энергии или ее отдачу можно рассматривать как
«с позиции» системы, так и с «позиции» внешней среды.
51
В термодинамике приняты следующие условия знаков. Если
система обменивается энергией с внешней средой, то в какой бы
форме это ни происходило, количество энергии, полученное си-
стемой, обозначается знаком «+», количество энергии, отданное
системой, — знаком «—». Следовательно, Q и А — алгебраиче-
ские величины. Если величина Q положительна, система полу-
чила тепловую энергию, если величина Q отрицательна, система
отдала тепловую энергию. Таким образом, знаки рассматрива-
ются с «позиции» системы. В термохимии, где знаки рассматри-
ваются с «позиции» внешней среды, они меняются на обратные.
3. ТЕРМОДИНАМИЧЕСКИЕ ПАРАМЕТРЫ СОСТОЯНИЯ СИСТЕМЫ
Состояние системы может быть определено совокупностью её
свойств. Все величины, характеризующие какое-либо макроско-
пическое свойство рассматриваемой системы, называются тер-
модинамическими параметрами. Опытом установ-
лено, что для однозначной характеристики данной системы необ-
ходимо использовать некоторое число определенных параметров.
Такие параметры называются независимыми парамет-
рами. Остальные параметры рассматриваются как функции
независимых параметров.
Независимыми термодинамическими параметрами чаще всего
выбирают параметры, поддающиеся непосредственному измере-
нию, например температуру, давление, молярный или удельный
объем, концентрацию и т. д.
Температура — один из важнейших термодинамических
параметров, который характеризует тепловое состояние данного
тела или системы. Экспериментально установлено, что при сопри-
косновении двух тел с разной температурой теплота переходит
от тела с более высокой температурой к телу с более низкой
температурой. z
Теплота, количество теплоты — энергетическая характе-
ристика процесса ^теплообмена, измеряемая количеством энергии,
которое получает или отдает в этом процессе тело или система.
Единицей температуры является кельвин (К), измеряемый
по термодинамической температурной шкале. В 1954 г. X Гене-
ральная конференция установила термодинамическую шкалу с
одной реперной точкой — тройной точкой воды, температура ко-
торой принята 273,16 К (точно), что соответствует 0,01°С, так
как в шкале Цельсия отсчет ведется от точки таяния льда. По-
этому соотношение между температурами по шкале Цельсия и
абсолютной термодинамической температурной шкалой следую-
щее: Т К=/°С4-273,15 К.
Давление — также важнейший параметр состояния и за-
висит лишь от внутренних свойств системы, характеризует взаи-
модействие системы с внешней средой и определяется отноше-
нием силы (равномерно распределенной по нормальной к ней
поверхности) к площади этой поверхности. Единица давления —
52
паскаль (Па). Давление газа с молекулярно-кинетических по-
зиций определяется ударами молекул на стенки заключающего
этот газ сосуда и, следовательно, зависит от кинетической энер-
гии их теплового движения.
Объем характеризует макроскопические свойства системы
и, следовательно, также является параметром состояния. Свой-
ства тел могут характеризоваться удельным (объем единицы
массы) либо молярным (объем одного моля) объемом. Единица
объема — м3.
Для описания системы не обязательно требуются знания всех
параметров системы, если они зависят друг от друга.
Например, для закрытой системы — смеси идеальных газов —
достаточно определить количество вещества (молей) щ каждого
из компонентов, суммарное давление р и температуру Т. Тогда
известен объем V, поскольку параметры состояния связаны урав-
нением состояния идеальных газов:
рУ=(2п^Л
а парциальные давления pi каждого из компонентов даются
выражением
PiV — niRT.
Различают интенсивные параметры (или факторы интенсив-
ности) и экстенсивные (или факторы емкости). Интенсив-
ными называют такие параметры и определяемые ими свой-
ства, величина которых не зависит от массы, например темпера-
тура, давление. Они могут иметь одно и то же значение во всей
системе или изменяться от точки к точке; величины этих свойств
не аддитивны. Интенсивные свойства — это специфические свой-
ства системы в данном состоянии. Поэтому их часто используют
в качестве независимых термодинамических параметров.
Экстенсивными называют такие свойства, величина
которых пропорциональна массе и обладает свойствами аддитив-
ности. К числу экстенсивных свойств можно отнести такие пара-
метры, как объем системы, ее массу или количество вещества
(молей) и т. д.
4. СОСТОЯНИЕ РАВНОВЕСИЯ. ОБРАТИМОЕ И НЕОБРАТИМОЕ
ПРЕВРАЩЕНИЯ
Система находится в состоянии термодинамического равновесия, если зна-
чения параметров одинаковы в любой точке системы и остаются одинаковыми
во времени.
Когда температура, давление и состав системы соответственно
одинаковы во всех точках системы и постоянны во времени, то
одновременно осуществляются тепловое, механическое и химиче-
ское равновесия.
Химическое равновесие является равновесием динамическим.
Так, например, когда реакция этерификации достигает состоя-
53
ния равновесия, химические реакции продолжают идти, а не
останавливаются. В каждый момент времени образуется столько
молекул эфира и воды, сколько молекул эфира распадается с
образованием молекул кислоты и спирта. Таким образом, в со-
стоянии равновесия концентрация каждого компонента (кислоты,
спирта, эфира и воды) постоянна во времени, хотя молекулы
компонентов продолжают реагировать друг с другом.
Если изменить значение, по крайней мере, одного из пара-
метров системы в состоянии равновесия, то система претерпевает
превращение и достигает другого состояния равновесия, харак-
теризуемого другими значениями параметров состояния.
Превращение называется обратимым, когда система
бесконечно медленно переходит из одного состояния равновесия
в другое через непрерывный ряд промежуточных равновесных
состояний. Между двумя соседними состояниями равновесия
параметры состояния изменяются на бесконечно малое значение,
и поэтому можно вернуть систему в предыдущее состояние рав-
новесия бесконечно малым изменением параметров состояния.
При этом в каждый момент времени параметры состояния систе-
мы отличаются от параметров внешней среды (например, давле-
ние) лишь на бесконечно малое значение.
Обратимое превращение — идеальный случай, неосуществи-
мый на практике, поскольку продолжительность такого превра-
щения должна быть бесконечно большой. Поэтому в качестве
обратимых рассматривают процессы, происходящие в ко-
нечное время через непрерывный ряд равновесных состояний,
очень близких друг к другу.
Когда в результате протекания процессов в прямом и обрат-
ном направлениях в системе или в окружающей среде останутся
неисчезающие изменения, то процесс называется необра-
тимым. Такой процесс возможно реализовать в обратном
направлении только с применением внешних воздействий, как
правило, оставляющих изменения в системе или среде. Напри-
мер, если масса газа, содержащегося в закрытом цилиндре,
подвергается обратному превращению при движении поршня,
то при этом внешнее давление, которое равно для каждого со-
стояния равновесия внутреннему давлению, изменяется очень
медленно. Каждое положение поршня соответствует состоянию
равновесия. Если же, наоборот, быстро переместить поршень,
приложив большую силу, то давление газа изменится быстро;
оно будет неодинаково во всех точках системы в течение этого
процесса, который называется необратимым превращением.
Необратимые процессы обычно идут самопроизвольно и толь-
ко в одном направлении — в сторону приближения к равновес-
ному состоянию — и прекращаются, когда такое состояние будет
достигнуто. Например, переход теплоты от более нагретого тела
к менее нагретому, кристаллизация переохлажденной жидкости
или испарение перегретой жидкости, взаимная диффузия газов
или жидкостей.
54
5. ЭНЕРГИЯ. РАБОТА. ТЕПЛОТА
Энергия (греч. cnergeia — действие, деятельность) — общая качественная
мера движения и взаимодействия всех видов материи.
По определению М. Планка, энергия есть измеренная в едини-
цах механической работы величина всех действий (механический
эквивалент), которые материальная система производит в своем
внешнем окружении, когда она любым образом переходит из
некоторого определенного состояния в произвольно фиксирован-
ное нулевое или стандартное состояние, имеет однозначное
значение и является, следовательно, независимой от способа
перехода.
Энергия является неотъемлемым свойством системы. Разли-
чают кинетическую энергию, или энергию движения, и
потенциальную, или энергию положения и взаимодейст-
вия частиц системы. Разность потенциальных энергий двух со-
стояний системы равна работе гравитационных, упругих, электро-
статических или других сил, взятой со знаком минус. Следова-
тельно, физический смысл понятия работы имеет только разность
потенциальных энергий двух состояний или двух уровней сис-
темы.
Работа в системе может совершаться за счет изменения
энергии, содержащейся внутри данной системы, т. е. так назы-
ваемой внутренней энергии.
Внутренняя энергия вещества представляет собой его полную
энергию, которая суммируется из кинетической и потенциальной
энергий, составляющих вещество атомов и молекул, а также
элементарных частиц, образующих атомы и молекулы. Она
включает: 1) энергию поступательного, вращательного и колеба-
тельного движения всех частиц; 2) потенциальную энергию взаи-
модействия (притяжения и отталкивания) между ними; 3) внут-
римолекулярную химическую энергию; 4) внутриатомную энергию;
5) внутриядерную энергию; 6) гравитационную энергию; 7) лу-
чистую энергию, заполняющую пространство, занятое телом, и
обеспечивающую внутри тела тепловое равновесие между отдель-
ными его участками. Внутренняя энергия не включает потен-
циальную энергию, обусловленную положением системы в про-
странство, и кинетическую энергию движения системы как це-
лого.
Процессы, происходящие без обмена энергией с внешней сре-
дой, сопровождаются изменением внутренней энергии. Когда
система под действием силы производит работу, то ее энергия,
т. е. способность совершать работу, постепенно уменьшается.
Энергия измеряется в тех же единицах, что и работа.
Работа, как уже указывалось, определяется как произведе-
ние пути, пройденного системой под действием какой-то силы,
на эту силу. Представим себе цилиндр с подвижным поршнем
площадью S (рис. 30). Поршень нагружен, он создает давле-
ние р. Тогда, очевидно, сила, действующая на поршень, будет
55
------------const-------------------
равна pS. Если поршень соверша-
ет элементарное перемещение dx,
то при этом совершается элемен-
тарная работа d4, которая, оче-
видно, равна
= — pSdx,
но Sdx==dV, следовательно,
— -..................... М = —pd V. (2.1)
Рис. 30. Расширение газа в цилинд- Для описания конечного процес-
ре в изотермических условиях са работа д проделанная систе-
мой, определяется интегрированием уравнения (2.1):
А = - f рЛх,
VA
где Va и Vb — объемы, соответствующие начальному и конеч-.
ному состояниям.
Итак, работа расширения газа выражается произведением pdV. Однако
система может производить и другие виды работы. Так, если в системе увели-
чивается поверхность двух фаз, например жидкости и газа, то работа против
сил поверхностного натяжения выразится произведением odS, где о — поверх-
ностное натяжение, a dS — изменение величины поверхности. Если работа свя-
зана с изменением электрического состояния тел системы, то ее можно выра-
зить в виде произведения вектора напряженности электростатического поля на
вектор электростатической индукции DdH. Работа, обусловленная переносом
заряда е против разности потенциалов d£, выразится произведением edE и т. д.
Следовательно, работу можно представить в виде произведения некоторой
величин^ X, которая не зависит от количества вещества (т. е. является факто-
ром интенсивности), на dx— дифференциал величины X, являющийся факто-
ром экстенсивности:
Xdx=M.
Величину X называют обобщенной силой, ах — обобщенной координатой.
Для всех форм энергии, за исключением теплоты, возможна
обратимость передачи энергии, связанной с работой. Так, элект-
рическая, механическая и другие формы энергии могут быть пол-
ностью превращены в работу.
Качественно и количественно иной формой передачи энергии
является теплота. Если система теряет энергию путем тепло-
проводности или излучения, то она отдает теплоту. Теплообмен
не связан с изменением положения тел, составляющих термо-
динамическую систему, а состоит в непосредственной пере-
даче энергии молекулами одного тела молекулам другого тела
при их контакте:
теплота и работа являются единственно возможными неравноценными фор-
мами передачи энергии, зависящими от способа перехода системы из одного
состояния в другое.
Как будет показано далее, теплота не может быть ни при
каких обстоятельствах полностью превращена в работу.
56
Таблица 9. Факторы интенсивности Д и экстенсивности fe
для различного вида работ
Виды работы f, fe
Механическая (Дж) Расширение (Дж) Электрическая (Дж) Гравитационная (Дж) Изменение поверхности (Дж) Сила (Н) Давление (Па) Разность потенциалов (В) Гравитационный потенци- ал (м2-с-2) Поверхностное натяжение (Н-м_|) Путь (м) Изменение объема (м3) Количество электриче- ства (Кл) Масса (кг) Изменение поверхности (м2).
6. ФУНКЦИЯ СОСТОЯНИЯ
Функция F параметров состояния (р, V, Т...) является функцией состояния,
если значение этой функции зависит только от параметров состояния и не
определяется процессами, приводящими к этому состоянию.
Математическая формулировка функции состояния выражает-
ся следующим образом: при бесконечно малом превращении из-
менение dF функции состояния F(x, у) выражается полным
дифференциалом:
Таким образом, если о какой-либо функции F(x, у) говорится,
что dF — полный дифференциал, то это значит, что функция F
есть функция состояния.
Так, например, потенциальная энергия представляет собой
функцию состояния. Рассмотрим массу т, находящуюся на вы-
соте h\ над землей и подверженную действию силы тяжести.
Состояние 1 определяется величиной h\ параметра состояния h.
Потенциальная энергия Е[ равна
Е\ ~mgh\ +k,
где k — постоянная; g — ускорение силы тяжести.
На высоте /12 потенциальная энергия £2 будет
Е2 — mgh2 + k.
Разность потенциальных энергий £2 —£1 составляет
Е2 — E\~mg(h2 — /11).
Она не зависит от способа, которым эта масса m перемещается
с высоты /ii на высоту h%.
Иначе говоря, каким бы способом, например, ни был достав-
лен камень на вершину горы — вертолетом, по извилистой тро-
пинке или канатной дороге, — потенциальная энергия этого кам-
ня будет определяться начальным и конечным положением кам-
ня, т. е. высотой горы.
Другой пример функции состояния — это произведение pV.
57
Данная масса идеального газа в начальном состоянии харак-
теризуется величинами р\ и V\ параметров состояния р и V.
После ряда превращений этот газ достигает конечного состояния
равновесия, при котором давление и объем соответственно равны
р2 и V2. Функция pV последовательно принимает значения p\V\
и P2V2, зависящие только от значений параметров состояния р
и V в состояниях 1 и 2. Следовательно, pV — функция состояния,
а разность P2V2 — p\V\ не зависит от способа перехода из состоя-
ния 1 в состояние 2.
Внутренняя энергия вещества зависит только от его физического состояния
и не зависит от способа или пути, которыми данное вещество приведено в данное
состояние, т. е. внутренняя энергия есть функция состояния.
Это следует непосредственно из закона сохранения энергии. В самом деле,
обозначим цифрами 1 и 2 два произвольных состояния системы. Пусть си-
стема переходит в состояние 2 по определенному пути, причем на этот переход
затрачивается энергия U'. Теперь пусть система переходит из состояния 1 в
состояние 2 по некоторому другому пути с затратой другого количества энергии
U". Если затем система совершает первый переход в прямом направлении, вто-
рой— в обратном, то она окажется снова в прежнем состоянии 1. При первом
переходе будет затрачена энергия (/', при втором отдана £/", следовательно,
внешние тела, окружающие систему, получают энергию Д(7 =(/' — U", причем
никаких изменений в самой системе не происходит. Будет ли положительна
или отрицательна, безразлично; во всяком случае наше рассуждение привело
нас к противоречию с законом сохранения энергии. Отсюда следует, что U' = U",
г. е. внутренняя энергия во всяком состоянии системы не зависит от пути, по ко-,
горому система пришла в данное состояние, а определяется однозначно только
самим состоянием системы.
Как функция состояния внутренняя энергия может быть выражена через
независимые параметры, характеризующие состояние системы. Для простой си-
стемы (например, чистого вещества) внутренняя энергия определяется одно-
значно, если заданы любые два параметра системы в данном состоянии. На-
пример, если состояние определяется независимыми параметрами Т и V, то для
внутренней энергии можно записать
U = f(T, V).
Тогда полный дифференциал внутренней энергии имеет вид
д1/=(4т-) аг+(4г-)dv-
\ дТ Jv \ OV Jt
В противоположность этому механическая работа Л, совер-
шаемая против сил давления, обычно не является функцией
состояния.
Рассмотрим, например, систему, состоящую из массы m
идеального газа, находящегося в закрытом цилиндре с поршнем;
вся система помещена в термостат так, чтобы все процессы про-
исходили при постоянной температуре. Основание и стенки ци-
линдра образуют границу с внешней средой, поршень сечения S
образует подвижную границу. Положение поршня в состоянии 1
определяется его абсциссой х по отношению к основанию ци-
линдра (см. рис. 30).
В состоянии 2 поршень смещается на бесконечно малое рас-
стояние dx и его абсцисса равна % + dx. Работа А при переходе
из состояния 1 в состояние 2 равна произведению силы, проти-
водействующей перемещению поршня на величину этого переме-
58
щения. Сила, противодействующая перемещению dx, обусловлена
внешним давлением на внешнюю стенку поршня с поверх-
ностью S. Поскольку это перемещение очень мало, внешнее
давление предполагается постоянным. Тогда
6Д — Рвнеш^ dx
ИЛИ
Sdx = dK
Итак,
6Д = Рвнешн d V •
При соблюдении условий знаков, принятых ранее, 6Л поло-
жительно, если работа производится над системой, и отрица-
тельно, если работа производится системой над внешней средой.
Для предыдущей схемы
х<х-|- dx,
откуда dV положительно: объем системы увеличивается. В этом
случае система совершает работу против сил внешней среды,
следовательно, 6Л должно быть отрицательным и можно за-
писать
6Д = РвнениЛ V.
Если dV отрицательно, т. е. имеет место уменьшение объема и
в соотношении
6Д == РвненлЛ V,
6Л положительно.
Для конечного превращения работа выражается соотноше-
нием
Пути, позволяющие перевести систему из начального состоя-
ния в конечное, могут быть различны. Так, если процесс осу-
ществляется необратимо, внешнее давление постоянно и равно
атмосферному давлению ратм. Тогда
2
Днеобр^^ Ратм d V= Ратм(^2 V1).
1
Если же процесс обратим, то внешнее давление практически
равно в каждый момент внутреннему давлению газа:
Рвнешн ==Ргаз-
Поскольку рассматривается идеальный газ, уравнение состоя-
ния позволяет выразить давление как функцию объема и тем-
пературы, причем последняя предполагается постоянной:
I/ п'г п%т
Ргаз^—nt\T, Рвнешн— tr
59
Тогда работа ЛОбР будет равна
2
•^обр 5 Рвнешн^ 1^>
с dV
Лобр = -\п^Т—,
Vq
Лобр = — nRT\n~y~.
Таким образом работа, сопровождающая процесс перехода
системы из одного состояния в другое (при постоянной темпе-
ратуре), различна в зависимости от того, является ли этот про-
цесс обратимым или необратимым. Механическая работа зависит
от рассматриваемого процесса, следовательно, она не является
функцией состояния.
7. ПЕРВОЕ НАЧАЛО ТЕРМОДИНАМИКИ
Первой машиной, которая превращала теплоту в механиче-
скую работу*в больших масштабах, был паровой двигатель,
изобретенный в начале XVIII в. Т. Ньюкоменом. В конце века
двигатель, усовершенствованный Дж. Уоттом, нашел практиче-
ское применение. Так как паровая машина производила работу
путем передачи энергии в форме теплоты из горячего резервуара
с паром в холодный резервуар с водой, наука о взаимопревра-
щениях энергии и работы была названа термодинами-
кой — от греческих слов, означающих «движение теплоты».
Первая ясная формулировка первого начала термодинамики
приписывается обычно Ю. Майеру, который вычислил механи-
ческий эквивалент теплоты; результаты этой работы были опуб-
ликованы в 1842 г. Примерно в это же время независимо от
Ю. Майера к тем же выводам пришел Дж. Джоуль. Он опубли-
ковал (1843) точные измерения механического эквивалента теп-
лоты. Любопытно, что первое начало термодинамики было уста-
новлено намного позднее второго.
Итак, многочисленными опытами было показано, что различ-
ные виды энергии переходят друг в друга в эквивалентных коли-
чествах. В результате обобщения этих исследований был открыт
и сформулирован закон сохранения энергии, являющийся одним
из важнейших всеобпшу законов природы:
в замкнутой системе сумма всех видов энергии постоянна; при их взаимо-
превращениях энергия не теряется и не создается вновь.
Этот закон был назван Р. Клаузиусом первым нача-
лом термодинамики.
Закон сохранения энергии как объективный закон природы
подтверждает положение о вечности и неуничтожаемости движе-
ния, поскольку энергия есть мера движения материи при ее пре-
вращениях из одной формы в другую. Этим подтверждается
60
также положение о вечности и неуничтожаемости материи, по-
скольку движение неотделимо от материи, являясь формой ее
существования.
Из закона сохранения энергии вытекает невозможность созда-
ния такой машины, которая совершала бы работу, не затрачивая
на это соответствующего количества энергии. Иначе говоря,
вечный двигатель (перпетум мобиле) первого рода невозможен.
Это еще одна формулировка первого начала термодинамики.
Первое начало устанавливает связь между количеством теп-
лоты, полученной или выделенной в процессе, количеством произ-
веденной или полученной работы и изменением внутренней энер-
гии системы.
Ряд следствий, вытекающих из него, имеет большое значение
для физической химии и для решения различных производст-
венных задач. Применяя этот закон, можно осуществить расчеты
энергетического и, в частности, теплового баланса, расчеты теп-
ловых эффектов различных процессов. Достоверность первого
начала термодинамики подтверждается многолетней практикой
исследований. Все экспериментальные данные, полученные до
настоящего времени, находятся в согласии с первым началом.
Согласно первому началу термодинамики количество энергии,
которое выделяется или поглощается в форме теплоты Q и рабо-
ты Л, постоянно для любого процесса; энергия не может ни воз-
никнуть, ни уничтожиться, и, следовательно, Л-f-Q равно изме-
нению полной энергии системы, называемой внутренней
энергией U. Первое начало выражается соотношением
= Л + Q,
где AU — изменение внутренней энергии системы при переходе
из одного состояния в другое*.
Первое начало термодинамики может быть сформулировано
иначе следующим образом:
внутренняя энергия U является функцией состояния. Изменение внутренней
энергии Л(7 системы, переходящей из состояния 1 в состояние 2, — алгебраиче-
ская сумма всех энергий, обменивающихся с внешней средой.
Эта формулировка включает утверждение, что внутренняя
энергия U системы зависит только от состояния системы. Отсюда
следует, что изменение энергии AU системы при переходе из со-
стояния 1 в состояние 2 всегда одинаково вне зависимости от
того, какой путь выбран для этого перехода; изменение равно
разности величин Uz и U\ внутренней энергии системы в со-
стояниях 2 и 1:
MJ=U2-U^
Следует особо отметить, что оценить абсолютное значение
внутренней энергии невозможно; во всех практических приложе-
* Такого рода условие для записи первого начала термодинамики рекомен-
довано Международным Союзом теоретической и прикладной химии взамен
ранней записи: \U = Q — А.
.61
ниях термодинамики фигурирует лишь разность значений внут-
ренней энергии.
Если для перехода системы из состояния 1 в состояние 2 вос-
пользоваться двумя процессами А и В и если Ад, Qa и Ав, Qb —
энергии, обменивающиеся в виде работы и теплоты в процессах
А и В, то получим
AU~ U2 — U1 = Дд -f- Qa = Дв + Qb-
Отметим, что из равенства
А а + Qa = Д в + Qb
не обязательно следует, что
Дд = Дв И О А = Qb-
Действительно, А и Q не являются функциями состояния,
поскольку величины А и Q зависят от способа перехода системы
из состояния 1 в состояние 2.
Бесконечно малое изменение системы, при котором изменения
механической и тепловой энергии равны соответственно 6Л й
6Q, можно выразить соотношением
dU = SA +6Q,
где только dU — полный дифференциал.
Тогда изменение внутренней энергии At/, сопровождающее
конечный переход t/ из состояния 1 в состояние 2, будет равно
2 2 2
At/=jjdt/=$64 + jj6Q.
1 1 1
Процессы при постоянном объеме. Если объем системы оста-
ется постоянным в ходе процесса, то работа не совершается;
dV = 0,
следовательно,
6Д = —pdV=0,
откуда
dU = 6Q + SA = 6Q
и
\U = \dU = $6Q = Qv,
где Qu — тепловая энергия процесса при постоянном объеме;
она равна изменению внутренней энергии и, следовательно, за-
висит только от начального и конечного состояний рассматри-
ваемой системы..
Процессы при постоянном давлении. Энтальпия. Рассмотрим
процесс превращения при постоянном давлении; к ним, напри-
мер, относятся реакции, идущие при атмосферном давлении.
Система в состоянии 1 характеризуется массой т, давлением
температурой Т\ и объемом Vi. В состоянии 2, в закрытой систе-
62
ме при постоянном давлении, меняются только температура и
объем, которые будут равны соответственно Т2 и У2. Изменение
внутренней энергии будет равно
\U=U2-Ui=A + Qp,
где Qp — тепловая энергия процесса при постоянном давлении.
Для такого превращения.
Л = —p(V2— Vi),
откуда
f/2-^l = Qp-p(V2-Vl),
0p=U2-Ul+p(V2-V^
тогда
Qp=(i724-pV2)-(t/i+pVi)-
Функция Н= U-\-pV называется энтальпией. Энталь-
пия есть функция состояния, так как U и pV — функции состоя-
ния, а значит, и их сумма — функция состояния. Энтальпия
имеет размерность энергии.
Итак, U2 + PV2 и Ui+pVi — величины энтальпий для состоя-
ний 1 и 2 системы соответственно:
{72 + рУ2 = Я2; Ut+pV^Hi,
тогда
Qp = H2-Hi = AH.
Таким образом, Qp — тепловая энергия превращения системы
при постоянном давлении — равна изменению энтальпии в про-
цессе этого превращения и, следовательно, зависит только от
начального и конечного состояний, поскольку энтальпия явля-
ется функцией состояния.
8. ПРИЛОЖЕНИЕ ПЕРВОГО НАЧАЛА ТЕРМОДИНАМИКИ
К ХИМИЧЕСКИМ ПРОЦЕССАМ
8.1. Теплота реакции (тепловой эффект)
Рассмотрим закрытую’термодинамическую систему, в которой
происходит химическая реакция по следующему стехиометриче-
скому уравнению:
ак 4- ЬВ-- сС 4" dD
В начальном состоянии, внутренняя энергия которого U\,
имеется а моль реагента А и b моль реагента В. В конечном со-
стоянии с внутренней энергией U2 образуется с моль продукта
С и d моль продукта D. Теплота этой реакции при температуре
Т — это тепловая энергия, которая выделяется или поглощается
при взаимодействии реагентов А и В в стехиометрических про-
63
порциях с образованием продуктов С и D; реагенты и продукты
реакции находятся при одной и той же температуре Т,
Теплота реакции при постоянном объеме (Qv). Это тепловая
энергия, которая выделяется или поглощается в ходе реакции.
В этом случае Qv соответствует изменению внутренней энергии
между начальным и конечным состояниями:
MJ=U2-lh = Qv.
Если величина Qv положительна, то реакция эндотерми-
ческая; система поглощает тепловую энергию в ходе реакции,
внутренняя энергия реагентов (А и В) меньше внутренней энер-
гии продуктов (С и D).
Если величина Qv отрицательна, то реакция экзотерми-
ческая: система выделяет тепловую энергию в ходе реакции,
внутренняя энергия реагентов выше внутренней энергии про-
дуктов.
Реакции при постоянном объеме могут идти, например, в сле-
дующих условиях: 1) в закрытом сосуде; 2) между твердыми те-
лами и жидкостями без выделения газа; 3) между газами, если
число молекул остается постоянным; например,
н2 (г) +С12 (г)-> 2НС1 (г)
Многие химические реакции осуществляются при постоянном
давлении, как правило, при атмосферном давлении.
Теплотой реакции при постоянном давлении Qp при температуре Т назы-
вается тепловая энергия, которая выделяется или поглощается в ходе реакции
при постоянном давлении; реагенты и продукты реакции приводятся в стехиомет-
рических количествах при температуре Т начального состояния.
Теплота реакции равна изменению энтальпии АЯ:
QP = \H.
Если величина Qp положительна, то реакция эндотермиче-
ская: энтальпия продуктов реакции выше энтальпии реагентов.
Если величина Qp отрицательна, то реакция экзотермическая:
энтальпия продуктов реакции ниже энтальпии реагентов.
Стандартное состояние. Чтобы сравнить между собой тепло-
ты разных реакций, необходимо точно указать условия, при кото-
рых эти реакции протекают. Прежде всего предполагают, что
реагенты и продукты реакции взяты в стехиометрических коли-
чествах. Физическое состояние каждого компонента реакции
должно быть точно задано.
Чтобы установить некоторую шкалу отсчета, вводят понятие
так называемого стандартного состояния. Для любо-
го чистого вещества это физическое состояние, в котором веще-
ство наиболее устойчиво при давлении 101,3 кПа и определенной
температуре (чаще всего 298 К). Для твердого тела стандарт-
ное состояние, например при 298 К, является кристаллическим,
оно наиболее устойчиво при этой температуре и давлении
101,3 кПа. Углерод в стандартном состоянии при 298 К пред-
64
аА + ЬВ ’
(Г,р. И,)
Д1/ — С
V — const
р= const
ди=о
(по закону
Джоуля)
сС + dD
(Ttp\Vx)
31. Превращение идеальных
Рис.
газов в изохорном и изобарном про-
цессах
сС + JD
ставлен графитом, а не алмазом,
так как графит более распро-
странен в природе.
Символ АН (298 К) обозна-
чает изменение энтальпии реак-
ции при 298 К; реагенты и про-
дукты реакции находятся в стан-
дартном состоянии при 298 К.
Таким образом, можно сделать
вывод, что стандартное состояние
имеет условный характер. Напри-
мер, в повседневной жизни все время применяются подобные
же условные правила, хотя, привыкнув к ним, редко задумыва-
емся над их сущностью. Например, когда говорится о высоте
горы, равной некоторому числу метров, то тем самым дается
не очень точное определение, и если его уточнить, то надо
указать, что речь идет о высоте над средним уровнем
моря.
Соотношение между Qp и Qv для реакции между идеальными
газами. Если реакция происходит между идеальными газами,
то можно установить соотношение между Qp и Qv. Рассмотрим
реакцию
аА + дВ-и сС + dD
где реагенты и продукты находятся при одинаковой темпера-
туре и в стехиометрических количествах (рис. 31).
Если реакция идет при постоянном давлении р и если V2 и
Vi — соответственно объемы конечного и начального состояний,
то изменение внутренней энергии АС/ равно
bU = Q + A = Qp-p(V2-Vi).
Если реакция протекает при постоянном объеме и образующиеся
продукты занимают объем V при температуре Т и давлении р',
то изменение внутренней энергии реакции будет
АС/' =
Согласно закону Джоуля внутренняя энергия идеального газа
зависит только от температуры. Иначе говоря, внутренняя
энергия продуктов, образующихся в ходе реакции при постоян-
ном объеме, равна внутренней энергии продуктов, образующихся
в ходе реакции при постоянных давлении и температуре. Отсюда
следует, что АС/" = О.
Согласно первому началу термодинамики
следовательно,
Так как реакция осуществляется между идеальными газами, то
pV2 = (c + d)RT
65
3—696
pVi=(a + b)RT,
откуда
QP= Qy + [(c + d)-(a + d)]/?T,
что можно записать в виде
Qp == Q v Ч~ Лл/? Т,
где Ап — разница между количеством вещества (молей) образо-
вавшихся газообразных продуктов и количеством вещества (мо-
лей) газообразных реагентов; R — универсальная газовая по-
стоянная.
Это соотношение устанавливает связь между теплотой реак-
ции при постоянном давлении (Qp или А//) и теплотой реакции
при постоянном объеме (Qr или АС/).
8.2. Теплоемкость. Зависимость теплоты реакции
от температуры
Для количественной оценки теплоты, которую получает тело
при нагревании, используется понятие теплоемкости.
Теплоемкостью называют количество теплоты, соответ-
ствующее изменению температуры единицы количества вещества
на 1 К. Количество теплоты, необходимое для нагревания 1 г
вещества на 1 К, называют удельной теплоемкостью
Суд. Количество теплоты, необходимое для нагревания 1 моль
вещества на 1 К, называют молярной теплоемкостью
сМол- Связь между ними выражается уравнением
^мол == ^удЛ/,
где М — молярная масса.
Поскольку величина Q зависит от вида процесса, от него так-
же будет зависеть и теплоемкость. Чаще всего пользуются тепло-
емкостью при постоянном давлении — изобарическая теплоем-
кость ср и теплоемкостью при постоянном объеме — изохориче-
ская теплоемкость су.
Различают среднюю теплоемкость для интервала температур
Q
с=-т _ут и истинную при данной температуре, соответствую-
щую бесконечно малому приращению количества теплоты при
бесконечно малом приращении температуры. Таким образом,
истинная теплоемкость равна
r Q dQ
итдт-^о—= с.
В настоящее время за единицу количества теплоты принят
джоуль, который равен работе, производимой силой в 1 Н при
перемещении точки ее приложения на 1 м по направлению этой
силы.
Следует отметить еще одно определение джоуля, связанное
66
непосредственно с представлением о
количестве теплоты. Джоуль — это та-
кое количество теплоты, которое необ-
ходимо для нагревания 1/4,184 г воды
на 1 К в интервале температур от
287,81 до 288,81 К. Это определение
иллюстрирует взаимосвязь джоуля с
калорией, которая в настоящее время
для определения количества теплоты
не рекомендуется.
Следовательно, единицей теплоем-
кости является Дж/К.
Теплоемкость в значительной сте-
Рис. 32. Зависимость тепло-
емкости от температуры
пени зависит от температуры, при которой ее определяют. Эта
зависимость характеризуется довольно сложной функцией. Так,
вблизи абсолютного нуля теплоемкость изменяется весьма
резко с температурой, в то время как в промежутке 273...773 К
эти изменения не столь значительны (рис. 32). В последнем слу-
чае эта зависимость для большинства веществ может быть выра-
жена уравнением
с = с$-\-аТ -\-ЬТ2сТ\
где а,Ь,с — постоянные коэффициенты (табл. 10).
В условиях постоянства объема в температурном интервале
Т\ — Т<2 тепловая энергия Qv равна
т2
Qv=\U = $cvdT.
Таблица 10. Коэффициенты температурной зависимости
в уравнении с = с0 + аТ + ЬТ2 + сТ6
Вещества а, Дж/(моль-К) Ь, 10 3 Дж/(моль Ж) с, 10 5 Дж/(моль -К)
Газы (298—2000 К) Не, Ne, Аг, Кг, Хе 20,78 0 0
н2 27,28 3,26 0,50
о2 29,96 4,18 -1,67
n2 28,58 3,77 -0,50
С12 37,03 0,67 -2,82
со2 44,23 8,79 -8,62
Н2О 30,54 10,29 0
NH3 29,75 25,10 -1,55
СН4 23,64 47,86 -1,92
Жидкости (ТПЛ-^ТКИП) Н2О 75,48 0 0
Твердые вещества С (графит) 16,86 4,77 -8,54
Си 22,64 6,28 0
А1 20,67 12,38 0
РЬ 22,13 11,72 0,96
67
3*
Чтобы повысить температуру 1 моль чистого вещества при
постоянном давлении от температуры Т\ до температуры Г2,
нужно затратить тепловую энергию Qp, равную
г2
Qp=bH=\cp&T.
Т|
Если в температурном интервале Т\ — Тг молярную теплоем-
кость ср считать постоянной, то
Qp= ср(Т2-Т^
Соотношение теплоемкостей при постоянном объеме и по-
стоянном давлении можно получить при использовании первого
начала термодинамики для идеальных газов и уравнения со-
стояния.
Это соотношение выражается уравнением
cp—cv = R.
Теплоемкость при постоянном давлении ср всегда больше, чем
теплоемкость при постоянном объеме су, так как нагревание
вещества при постоянном давлении сопровождается работой
расширения.
Тепловой эффект химических реакций и фазовых превращений
зависит от температуры. Эта зависимость может быть получена
в общем виде при рассмотрении перехода системы из состояния
А в состояние В различными путями (рис. 33).
Рассмотрим химическую реакцию
ак + ЬВ -+ сС + dD
Пусть теплоемкости реагентов будут соответственно равны ct, cf,
а теплоемкости продуктов — с2, с2; суммарная теплоемкость
реагентов Sci, а продуктов Sc2.
Пусть тепловой эффект реакции, протекающей при темпера-
туре Г, будет равен Q; тогда тепловой эффект реакции, проте-
кающей при температуре T-\-dT, будет равен Q + dQ.
Данную реакцию можно провести двумя путями, каждый из
которых можно осуществить двумя последовательными дейст-
виями.
I путь:
1) осуществляется процесс при температуре Г; выделяется Q;
2) осуществляется нагрев продуктов на величину Г; затра-
чивается теплота Sc2dT\
Суммарный эффект Q — Zc2dT
II путь:
1) осуществляется нагрев реагентов на d?; затрачивается
теплота Sc id Г;
2) осуществляется процесс при T-\~dT; выделяется Q + dQ
Суммарный эффект Q-J-dQ — ScidT
68
Так как оба пути приво-
дят к одному и тому же со-
стояни, очевидно, получен-
ные эффекты равны:
Q— Hc2dT = Q4-dQ— XcidT,
dQ = ScidT — Sc2dr,
1£=sC1-sC2.
al
Это уравнение называется
законом Кирхгофа
и формулируется следую-
щим образом:
-Zc2iT
dr
dr
-Zc^r
Г+dr
Рис. 33. Схема про-
цессов с различными
тепловыми эффектами
/I
Т
В
+0+d«
температурный коэффициент теплового эффекта равен разности сумм тепло-
емкостей реагентов и сумм теплоемкостей продуктов.
Рис. 34. Схема кало-
риметра:
1 — термостат; 2 — со-
суд Дьюара; 3 — медная
крышка; 4 — термометр;
5 — пробирка с вещест-
вом; 6 — мешалка
Для определения тепловых эффектов, сопровождающих хими-
ческие реакции, применяются специальные приборы, называемые
калориметрами (рис. 34). Калориметрическое определение
ведется так, чтобы вся химическая энергия выделялась в виде
теплоты или частично затрачивалась на совершение работы
расширения газа, которая также учитывается. В простейшем
случае химическая реакция проводится в сосуде Дьюара, который
представляет собой стеклянный сосуд с посеребренными изнутри
двойными стенками, из пространства между которыми выкачан
воздух, вследствие чего стенки сосуда почти не проводят теплоты.
Для более равномерного теплообмена с
окружающей средой сосуд помещают обыч-
но в*большой термостат, наполненный во-
дой. Во время опыта температура термо-
стата поддерживается постоянной. Сосуд
покрыт медной крышкой с тремя отверсти-
ями: для термометра, мешалки и пробирки.
В сосуд и пробирку помещают навески
реагирующих веществ, где рни находятся до
тех пор, пока не уравняется температура
всех частей прибора. Определив темпера-
туру реагирующих веществ До начала реак-
ции, проводят реакцию и определяют тем-
пературу в калориметре после реакции.
Зная теплоемкость системы (которая опре-
деляется предварительно), можно вычис-
лить количество теплоты, приобретенной со-
держимым калориметра во время реакции,
и отсюда — тепловой эффект реакции.
69
8.3. Скрытая теплота изменения состояния
Когда при постоянном давлении чистое вещество подвергается
изменению состояния — плавлению, затвердеванию, испарению,
конденсации или возгонке, происходит обмен тепловой энергией
с внешней средой. Температура при изменении состояния
остается постоянной.
Переход из более упорядоченного состояния в менее упоря-
доченное всегда происходит с поглощением энергии из внешней
среды. Так, например, при плавлении твердого вещества происхо-
дит поглощение теплоты, так как энергия связи между частицами
твердого вещества больше, чем между частицами жидкого веще-
ства, и для уменьшения взаимодействия частиц следует затратить
энергию. То же самое происходит при переходе вещества из жид-
кого состояния в газообразное.
При переходе 1 моль чистого вещества из жидкого в газообраз-
ное состояние при давлении р и температуре Т тепловая энергия,
поглощаемая из внешней среды при изменении состояния, назы-
вается скрытой теплотой парообразования (или энтальпией паро-
образования) и обозначается через Lp. Необходимо знать значение
Lp, если рассматривается процесс, включающий изменение состоя-
ния, например процесс нагревания воды с переходом ее в газо-
образное состояние. Так, например, чтобы вычислить тепловую
энергию, необходимую для теплового превращения 1 моль жид-
кой воды при температуре 298 К и атмосферном давлении в 1 моль
газообразной воды при температуре 400 К и том же давлении,
необходимо учесть:
1) количество энергии, поглощенной 1 моль жидкой воды для
повышения температуры с 298 до 373 К,
373 К
ЛЯ1 = § Ср(Н2О,ж) dr ;
298К
2) энтальпию парообразования 1 моль воды при 373 К и
атмосферном давлении
ДЯ2 = Ьр ;
3) количество энергии, поглощенной 1 моль газообразной воды
при повышении температуры с 373 до 400 К,
400 К
ЛЯз=$ 6>(Н2О, г)dr.
373 К
Суммарная тепловая энергия, необходимая для проведения
указанного превращения, равна
373К 400К
Qp = АН 1 4~ ДЯ2 -|- ДЯз Ср (Н2О,ж) dr -J- Lp 4- j c(P(H2O,r)dr .
298K 373K
70
8.4. Энтальпия образования
Энтальпия образования соединения равна изменению энтальпии, сопровож-
дающему реакцию образования 1 моль этого соединения из элементов или простых
веществ при постоянном давлении.
Так, например, энтальпией образования этилового . спирта
С2Н5ОН называют изменение энтальпии при реакции углерода,
кислорода и водорода при постоянном давлении:
2С + ЗН2 -и 1 /2О2-> СНз - СН2ОН
Значение энтальпии образования не всегда можно определить
непосредственно. Так, например, энтальпия образования КСЮз
будет отвечать тепловому эффекту реакции
К (крист) + 7зС12 (г) + 3/2О2 (г)-КСЮз (крист)
Эту реакцию получения КСЮз нельзя провести. Однако закон
Гесса (см. раздел 8.5) дает возможность вычислить ДЯ данной
реакции из энтальпий других процессов.
Стандартная энтальпия образования соответствует стандарт-
ным условиям как для образования продуктов, так и для элемен-
тов: давление 101,3 кПа, элементы находятся в наиболее устой-
чивой форме, температура 298 К.
Так, стандартная энтальпия образования диоксида углебода
в газообразном состоянии равна изменению энтальпии реакции
С (графит) 4-О2(г)-> СО2(г)
протекающей при давлении 101,3 кПа (температура специально
оговаривается). Во всех рассматриваемых примерах температура
равна 298 К.
Стандартная энтальпия образования диоксида углерода обо-
значается символом Д/Я°(СО2); при 298 К Д/ЯЬ(СО2) =
= — 393,50 кДж/моль.
Из определения энтальпии образования следует, что энтальпия
образования простых веществ (элементов) в стандартном состоя-
нии будет равна нулю, т. е. Д^Я°(О2) = 0, Д^Я° (С, графит) = 0.
Стандартные энтальпии образования многих молекул можно
измерить или вычислить. Некоторые их значения приведены
в табл. 11.
Поскольку стандартные энтальпии образования элементов
равны нулю, значения энтальпий образования позволяют сравни-
вать устойчивость различных молекул по отношению к элементам
и между собой. Если стандартная энтальпия образования отрица-
тельна, то соединение более устойчиво, чем элементы, из которых
оно состоит; и, наоборот, если она положительна, то соединение
менее устойчиво. Если рассматриваются соединения, принадлежа-
щие к одному классу, то
из различных молекул более устойчивы те, энтальпия образования которых
меньше.
71
Таблица 11. Стандартные энтальпии образования некоторых соединений
при 298 К
Элементы в стандартном состоянии Образовавшиеся соедине- ния Д/Я°(298), кДж/моль
н2 + '/2Ог Н2О (г) —241,84
н2 + 1 / 2О2 Н2О (ж) -285,95
7гН2+ 72С12 НС1 (г) -92,30
S(t) +О2 SO2 (г) -296,90
S(t) + 3/2O2 SO3 (г) -395,18
n2 + 3/2Н2 NH3 (г) —46,19
С (графит) 4г 1/2О2 СО (г) — 110,54
С (графит) + О2 СО2 (г) -393,50
С + 2Н2 СН4 (г) -74,85
2С (графит) + ЗН2 С2Н6 (г) -103,85
2С (графит) + 2Н2 С2Н4 (г) 4-52,30
2С (графит) + Н2 С2Н2 (г) 4-266,73
6С (графит) + ЗН2 , С6Н6 (г) 4-49,04
S (т) + Н2 H2S (г) -20,17
РЬ (т) + О2 РЬО2 (т) -276,65
Na (т) + 72С12 NaCl (т) —411,00
Например, этан А^Я° = —103,85 кДж/моль более устойчив,
чем этилен А/Я° = + 52,30 кДж/моль, который, в свою очередь,
более устойчив, чем ацетилен А///6 = + 266,73 кДж/моль.
Так как переход из одного состояния вещества в другое связан
с затратой или выигрышем 'Энергии, то очень важно указать
физическое состояние, в котором находится образовавшийся
продукт. Например, разница в 44 кДж/моль между стандартной
энтальпией образования газообразной и жидкой воды обусловлена
изменением энтальпии при изменении агрегатного состояния:
Н2О (ж)--> Н2О (г); = —44,01 кДж/моль.
8.5. Закон Гесса
Если известны энтальпии образования реагентов и продуктов,
то можно определить изменения энтальпий, сопровождающие
реакции, не прибегая к прямым измерениям. Так, любые продукты
можно получить двумя путями:
1) непосредственно из элементов или простых веществ; соот-
ветствующее изменение энтальпии равно А/Я° (продуктов);
2) сначала получить реагенты, соответствующие изменению
энтальпии Af//° (реагентов), затем превратить реагенты в про-
дукты; для этой реакции изменение энтальпии составляет АЯ
(298 К).
Во всех реакциях элементы (простые вещества), реагенты и
продукты находятся при одинаковой температуре, например при
298 К.
72
AfH® (продуктов)
Простые вещества - — >• Продукты
ДгЯ ° (реагентов) /
X? /дЯ(298К)
Реагенты
Термодинамический баланс этих реакций
Д//7° (продуктов) = (реагентов) Д/Д298 К),
откуда
ДЯ(298 К) = Д/Я° (продуктов) — Д/Я° (реагентов).
Полученное уравнение связывает тепловой эффект реакции,
протекающей при постоянном давлении, лишь со стандартными
энтальпиями продуктов и реагентов.
Это положение было впервые экспериментально открыто рус-
ским ученым Г. И. Гессом в 1840 г. и известно как закон
Гесса, являющийся основным законом термохимии — раздела
физической химии, посвященного изучению тепловых эффектов
химических реакций. Закон Гесса устанавливает, что если из
данных исходных веществ можно различными путями получить
заданные конечные продукты, то независимо от путей получения,
т. е. от вида промежуточных реакций, суммарный тепловой эффект
для всех путей будет одним и тем же. Иначе говоря,
тепловой эффект химической реакции не зависит от пути перехода (промежу-
точных реакций), а зависит только от вида и состояния исходных веществ и
конечных продуктов.
Например, при 298 К изменение энтальпии, соответствующее
реакции гидратации этилена в этиловый спирт
СН2 = СН2 + Н2о(ж) > СНз—СН2ОН
равно
ДЯ(298 К) = Д/#°(СНзСН2ОН)— Д^0(СН2=СН2)— Д^°(Н2О, ж)
Энтальпии образования молекул, участвующих в этой реакции,
составляют:
Д^°(СН3СН2ОН) = - 277,61 кДж/моль,
Д^(СН2 = СН2) = + 52,30 кДж/моль,
Д^°(Н2О, ж) = — 285,95 кДж/моль.
Следовательно, изменение энтальпии реакции равно
Д/7 = — 277,61 — 52,30 + 285,95 = — 43,96 кДж..
Закон Гесса обязателен только для процессов, протекающих
при постоянном давлении либо при постоянном объеме. Строгость
соблюдения закона Гесса в процессах, происходящих при постоян-
стве внешнего давления, состоит в том, что в этих условиях тепло-
вой эффект реакции определяется величиной изменения энтальпии
системы Qp = АН. Энтальпия является функцией состояния систе-
73
Рис. 35. Тепловые эффекты получения ве-
ществ -различными путями
мы, ее изменение не зависит
от пути совершения процес-
са, а определяется лишь на-
чальным и конечным состоя-
нием системы. Аналогично и
для процессов, идущих в ус-
ловиях постоянства объема,
согласно равенству Qv=AUy
тепловой эффект реакции за-
висит от характера измене-
ния опять-таки функции со-
стояния — внутренней энер-
гии системы.
Представим себе процесс превращения исходных веществ Аь
Аг, Аз, ... в продукты Bi, Вг, Вз, ..., причем превращение это
может быть осуществлено различными путями (рис. 35):
1) непосредственно одной реакцией, тепловой эффект которой
равен A/Zi;
2) реакциями, тепловые эффекты которых равны соответствен-
но АЯг, АЯ3 и А//4;
3) другими реакциями, тепловые эффекты которых равны
АЯ5, АЯ6, АЯ7 и ЛЯ8.
Согласно закону Гесса эти тепловые эффекты связаны между
собой соотношением
д/7, = дя2 + дя3 -р д/у4 = дя 5 + дяб + д н7 4- дя8.
Закон Гесса широко применяется при различных термохими-
ческих расчетах. Он дает возможность вычислить тепловые эффек-
ты процессов, для которых экспериментальные данные отсутст-
вуют, а во многих случаях — и для таких, для которых они не могут
быть измерены в нужных условиях, или когда процессы еще не
осуществлялись. Это относится как к химическим реакциям, так и
к процессам растворения, испарения, кристаллизации, адсорбции
и др.
Из закона Гесса можно вывести несколько следствий, имеющих
важное практическое значение для термохимических вычислений.
Первое следствие. Тепловой эффект разложения какого-либо
химического соединения точно равен и противоположен по знаку
тепловому эффекту его образования (закон Лавуазье — Лапла-
са). Это утверждение непосредственно следует из того, что тепло-
вой эффект кругового процесса должен равняться нулю.
Второе следствие. Если совершаются две реакции, приводящие
из различных начальных состояний к одинаковым конечным
состояниям, то разница между их тепловыми эффектами пред-
ставляет собой тепловой эффект перехода из одного начального
состояния в другое.
Второе следствие закона Гесса позволяет точно вычислять
тепловые эффекты процессов, которые зачастую даже не имеется
возможности практически осуществить. Рассмотрим классический
74
пример определения теплового эффекта превращения графита в
алмаз путем расчета реакций их горения по закону Гесса:
С (графит) — С (алмаз)
При горении графита и алмаза получается оксид углерода (IV)
и выделяются соответствующие количества теплоты:
С (графит) + О2 >- СОг + 393,51 кДж
С (алмаз) -j-O2 * СО2 4- 395,34 кДж
С (графит) ► (алмаз) = — 1,83 кДж
Таким образом, переход графита в алмаз сопровождается погло-
щением сравнительно небольшого количества теплоты.
Третье следствие. Если совершаются две реакции, приводящие
из одинаковых начальных состояний к различным конечным сос-
тояниям, то разница между их тепловыми эффектами представля-
ет собой процесс перехода из одного конечного состояния в другие.
Так, например, образование 1 моль воды при горении водорода в
зависимости от ее конечного физического состояния сопровождает-
ся следующими эффектами:
Н2+72О2= Н2О (г)+241,83 кДж
Н2 + ’/2О2 = Н2О(ж) +285,84 кДж
Н2 + 1 /2О2 = Н2О (т) + 291,67 кДж
Следовательно,
Н2О(т) = Н2О(ж) —5,83 кДж
Н2О(ж) = Н2О(г) — 44,01 кДж
Н2О(т) = Н2О(г) -49,84 кДж
Четвертое следствие. Тепловой эффект реакции равен разности
между суммой теплот сгорания исходных веществ и суммой теплот
сгорания конечных продуктов с учетом коэффициентов' веществ в
уравнении реакции.
Для многих соединений не удается осуществить реакцию их
образования из простых веществ и тем более измерить теплоту
образования. Однако в большинстве случаев можно осуществить
реакцию полного сгорания. Поэтому термохимические расчеты
весьма часто производят с использованием теплот сгорания
исходных веществ и продуктов реакции. Теплотой сгора-
ния называется тепловой эффект реакции полного сгорания
1 моль данного соединения до образования высших оксидов.
8.6. Энергия связи
Энергия, выделяющаяся в процессе образования ковалентной связи между
двумя атомами, взятыми в газообразном состоянии, называется энергией связи.
Пусть атомы А и В находятся в газообразном состоянии. Тогда
энергия связи А — В равна изменению энтальпии АЯ(А—В)
75
--------нчс
Энергия связи H-CL
-----— /Л?Н2+//Ш2
,, Энтальпия образования
---------- HCL
Рис. 36. Сравнение энергии связи
Н—С1 и энтальпии образования HCI
следующей реакции, идущей
при давлении 101,3 кПа и опре-
деленной температуре, как пра-
вило, 298 К:
А(г)+В(г)---. (А—В) (г)
ЛЯ (А— В) соответствует об-
разованию одного моля связей
А — В и выражается в
кДж/моль.
Энергия связи АЯ (А—В)
всегда отрицательна; на рис. 36
показаны относительные энер-
гетические уровни начального и конечного состояний для мо-
лекул НС1. В табл. 12 приведены некоторые средние значения
энергии связей.
При температуре 298 К энергия связи А—В обозначается
символом
4/7(298 К) (А—В)
Энергия диссоциации соответствует обратной реакции, т. е.
гомолитическому или радикальному разрыву по схеме
АДВ----► А* Н-В*
Энергия диссоциации равна по значению и противоположна по
знаку энергии связи А—В. Это энергия, необходимая для разрыва
ковалентной связи; она положительна. Иногда эту энергию диссо-
циации можно измерить экспериментально, например
Н—Н-^Н • + Н • , АН = 435,97 кДж
С1—С1^СГ +С1\ 4/7 = 242,67 кДж
Когда молекула содержит больше двух атомов, сумма энергий
связей молекулы равна изменению энтальпии в процессе реакции
nA (г) +тВ (г)-к A„Bm (г)
Например, изменение энтальпии для деакции
2Н(г) +О(г)-->Н2О(г)
атомы атом молекула
равно удвоенной энергии связи О—Н, поскольку в ходе реакции
образуются две связи О—Н
АН(298 К) = 24/7(298 К) (О—Н).
Подобным образом при реакции
С(г)+4Н(г)--->СН4(г)
образуются четыре связи С—Н и, следовательно,
4/7(298 К) = 44/7(298 К) (С - Н)
76
Чтобы лучше понять разницу между значениями энтальпии
образования и энергии связи (см. рис. 36), рассмотрим следующие
реакции:
Н*н-С1* . Н—С1, ДЯ1 = -431,79 кДж
72Н2+72С12 ,н—С1, Д#2^-92,30 кДж
Первая реакция характеризует образование молекулы Н—С1 из
атомов водорода и хлора. Изменение энтальпии АН\ соответст-
вует энергии, выделяющей при образовании ковалентной связи
Н—С1.
Вторая реакция характеризует образование молекулы НС1 из
простых веществ, т. е. из молекул Н2 и CI2- В этом случае изме-
нение энтальпии ДЯ2 соответствует разнице в устойчивости между
ковалентной связью Н—С1 и ковалентными связями в Н2 и CI2-
Действительно, образование 2 моль НС1, 1 моль Н2 и 1 моль
CI2 включает в себя:
1) разрыв связей Н—Н и С1—С1 в реагентах Н2 и С12
(что требует некоторых затрат энергии):
Н—Н-------кН* +Н*, Д#= + 435,97 кДж
Cl—С1-----> СГ + СГ , ДЯ = + 242,67 кДж
2) образование 2 моль НС1 из атомов (при этом выделяется
энергия, равная удвоенной энергии связи Н—С1):
Н* 4-Н* +сГ +сГ ----к 2НС1, ДЯ= — 863,58 кДж
Баланс этих процессов экзотермичен и равен — 184,94 кДж.
Можно вычислить энергию связи Н—С1, если известны энталь-
пия образования НС1 и энергии связей Н—Н и С1—С1. Рассматри-
ваемые процессы суммированы в схеме
дя,
2Н + 2С1 -2НС1
ДЯ^\. /ДЯ3
н2+ С12
А7/1 равно удвоенной энергии связи Н—С1; ДЯ2 представляет
собой сумму энергий связей Н—Н и С1—С1; ДЯ3 равно удвоен-
ной энтальпии образования соляной кислоты.
Согласно первому началу термодинамики
ДЯ1 = ДЯ2 + ДЯ3
= 2ДЯ(298 К) (Н—С1).
Из экспериментальных данных известно, что
ДЯ2 = ДЯ(298 К) (Н—Н) + ДЯ(298 К) (Cl—С1) =
= (-435,97) + (-242,67) = -678,64 кДж,
ДЯз = 2Д/Я0(НС1) = (—92,30)2= -184,60 кДж,
откуда величина энергии связи Н—С1 равна.
2ДЯ(298 К) (Н—С1) = -678,64 - 184,60= -863,24 кДж^
77
~ —863,3 кДж;
ДЯ(298 К) (Н—С1) = -431,7 кДж.
Часто вычисляют энергии связей из экспериментальных изме-
рений энтальпий сгорания и гидрогенизации. Так, энергия связи
С—Н в метане может быть рассчитана, если известны, например,
энтальпии сгорания метана, углерода (графита) и водорода.
Если известны энергии связи реагирующих молекул, то мож-
но приблизительно вычислить значение теплоты данной реакции.
Она будет равна разности энергий связей молекул продуктов
и молекул реагентов.
Рассмотрим этот случай на примере оценки энтальпии реак-
ции гидрирования ацетилена в этан:
Н Н
I I
Н—С=С—Н + 2Н2 -> Н—С—С—Н
I I
н н
Для осуществления этой реакции необходимо разорвать одну
тройную связь С=С и две связи Н—Н. Затем надо образовать
одну простую связь С—С и четыре связи С—Н.
В соответствии с этим (см. табл. 12)
\н = ЬН (С—С) + 4ДЯ (С—Н)—ДЯ (С=С) - 2Д/7 (Н—Н),
ДЯ = — 347,69 — 4-413,38 + 811,70 + 2-435,97= — 317,57.
Таблица 12. Энергия связи
Молекула - ДЯ(А-В), кДж/моль Молекула - ДЯ(А-В), кДж/моль
Н—Н 435,97 С—С1 328,44
н—с 413,38 с—в 275,73
Н—Si 294,55 С—I 240,16
Н—N 390,79 Si—Si 176,56
Н—Р 319,66 Si—О 369,03
Н—О 462,75 N—N 160,67
H-^S 339,32 N=N 418,40
Н—F 563,17 N=N 946,42
Н—С1 431,79 N—F 269,87
Н—Вг 366,10 N—C 199,58 -
Н—1 298,74 P—P 214,64
С—С 347,69 O—O 138,91
С=С 615,05 0—F 184,93
с=с 811,70 O—Cl 202,92
С—Si 289,95 s—s 212,97
С—N 291,62 F—F 153,13
C=N 615,05 F—Cl 253,55
C=N 891,19 Cl—Cl 242,67
С—О 351,46 Cl—В г 218,22
с=о 719,65 Cl—I 210,46
с—S . 259,41 Br—В г 192,88
(2—-S 476,98 Вг—I 177,82
С—F 484,51 I—I 151,04
78
9. ВТОРОЕ НАЧАЛО ТЕРМОДИНАМИКИ
В рамках представлений первого начала термодинамики воз-
можны и равновероятны любые процессы, в которых происходит
эквивалентный обмен различных форм энергии, в частности,
внутренней энергии, теплоты и работы. Так, например, первому
закону термодинамики не противоречит передача тепловой энер-
гии от более холодного тела к более теплому, ибо этот процесс
означает лишь перераспределение энергии внутри системы. Пер-
вое начало термодинамики не исключает, например, поднятие
камня над землей за счет охлаждения окружающего воздуха
или процесса самопроизвольного сжатия газа. Иначе говоря,
первое начало термодинамики ничего не говорит о направленности того
или иного процесса, о его самопроизвольности.
В то же самое время наблюдение за процессами, проис-
ходящими в природе, говорит о том, что физические и хими-
ческие превращения совершаются в определенном направлении:
газ самопроизвольно заполняет весь имеющийся объем, тепло-
вая энергия передается от более холодного до тех пор, пока тем-
пература этих двух тел не сравняется. Все эти превращения
являются самопроизвольными, иначе говоря спон-
танными.
И наоборот, обратные превращения самопроизвольно не
происходят. Газ не сжимается самопроизвольно; холодное тело
не отдает тепловую энергию горячему телу.
Следовательно, все процессы, происходящие в природе и осу-
ществляемые человеком, можно разбить на две группы:, на
самопроизвольные процессы, протекающие сами собой,
и несамопроизвольные. Для осуществления последних
необходимо затратить работу в количестве, пропорциональном
происходящему изменению.
Рассматривая химические превращения, можно в качестве
самопроизвольного процесса привести пример взаимодействия
цинка с раствором медного купороса CuSO4‘5H2O. При опус-
кании цинкового стержня в раствор происходит экзотермиче-
ская реакция вытеснения меди:
Zn 4- Cu2+ Zn2+ 4“ Си
Обратный эндотермический процесс не идет самопроизвольно.
Используя гальванический элемент Даниэля, можно, приложив
внешний источник электрической энергии, осуществить обратный
несамопроизвольный процесс выделения цинка из раствора меди:
Zn2+ 4- Си -> Zn + Си2+
Рассмотрение многочисленных экспериментальных данных по-
казывает, что изменение внутренней энергии или энтальпии,
которое сопровождает химическую реакцию, не может служить
критерием, позволяющим предвидеть направление реакции. Са-
мопроизвольные (спонтанные) химические реакции могут быть
79
Теплоотдатчик
| Теплоприемник
Рис. 37. Принцип работы
тепловой машины
цесс — превращение
как экзотермическими, так и эндотермиче-
скими. Очень важцо иметь критерии, поз-
воляющие предвидеть, может ли химиче-
ская реакция происходить самопроизволь-
но, и если может, то уметь определить
количества образовавшихся продуктов.
Это требует введения новой термодинами-
ческой функции. Введение этой функции
начнем с рассмотрения термомеханиче-
ских процессов.
В то время как механическую энергию
можно полностью превратить в теплоту
(например, путем трения), обратный про-
теплоты в работу — возможен лишь при
Л
определенных ограничениях.
Периодически действующее устройство для превращения теп-
лоты в работу — тепловая машина — может работать только при
наличии разности температур.
Работа тепловых машин включает, в принципе, следующий
процесс (рис. 37): выполняя один рабочий цикл, «рабочее ве-
щество» машины отбирает некоторое количество теплоты Qi от
резервуара при температуре Л и за этот же цикл отдает мень-
шее количество Q2 резервуару, находящемуся при температуре
Т2 (Т\>Т2). После этого рабочее тело возвращается в исход-
ное состояние. Согласно первому закону термодинамики механи-
ческая работа Л, производимая рабочим телом за один такой
цикл, равна
Я — Qi — Q2.
При этом, как показывает опыт, всегда выполняется нера-
венство: Qi > Q2 или, другими словами, невозможно с помощью
циклически действующей машины превратить теплоту Qi, полу-
ченную при температуре Л, в работу, не передав часть теплоты
Q2 системе с более низкой температурой Т2. Именно в таком
виде второе начало термодинамики впервые сформулировал
Карно.
Коэффициент полезного действия (КПД) указанного цикла
определяется как отношение величины работы Л, совершаемой
машиной, к количеству теплоты Qb получаемой от более нагре-
того резервуара:
кпд ^Т--2 .
41
В таком случае, согласно второму началу термодинамики, КПД
всегда меньше 1:
КПД= 1 1.
41
Следовательно, при выполнении цикла только часть теплоты
может быть использована для получения работы.
80
Если ввести абсолютную шкалу температур, то максималь-
ное значение КПД (или идеальная отдача) будет равно
кпдмакс= Г| ~ Гг .
Если горячий резервуар имеет температуру 400 К, а холод-
ный 300 К, то идеальная отдача будет равна 0,25. Другими сло-
вами, в работу может быть превращена в лучшем случае лишь
1 /4 теплоты, содержащейся в паре, тогда как 3/4 ее просто не
используются.
Имея только горячий резервуар (400 К), получим, согласно
уравнению Карно, КПД, равный нулю. В паре много энергии,
но ни одна часть ее не может быть превращена в работу, если в
устройстве не предусмотрена разность температур.
Так же обстоит дело и с другими формами энергии: например, большой
камень, лежащий на краю обрыва, может произвести работу, если его пере-
местить из положения с большей потенциальной энергией в поле силы тяжести
в положение с меньшей потенциальной энергией, например на дно обрыва. Чем
меньше разность потенциальных энергий (чем менее глубок обрыв), тем мень-
ше работы может, падая, совершать камень. А если вообще обрыва нет, а есть
просто плоскогорье, то камню некуда падать, совершая при этом работу, даже
если плоскогорье находится на высоте 1000 м над уровнем моря. Следова-
тельно,
ни одно устройство не может извлечь работу из системы, которая целиком
находится на одном энергетическом уровне.
Это одна из формулировок второго начала термодинамики.
Существует много других эквивалентных формулировок второго
начала, например:
теплота не может самопроизвольно переходить от менее нагретого тела
к более нагретому телу (формулировка Клаузиуса);
невозможно построить периодически действующую машину, единственным
результатом действия которой было бы совершение механической работы за
счет охлаждения теплового резервуара (формулировка Кельвина
и Планка).
Рассмотрим снова два резервуара в изолированной системе
По мере перекачки энергии горячий резервуар охлаждается, а
холодный нагревается. Следовательно, разность температур го
рячего и холодного резервуаров уменьшается в течение всего
периода времени, когда совершается работа. Но это означает,
что количество энергии, которое может быть потрачено на рабо-
ту (а оно зависит от разности температур), должно все время
уменьшаться.
Наоборот, количество энергии, которую уже невозможно пре-
вратить в работу, т. е. количество как бы связанной энергии,
должно все время расти. Это увеличение недоступной для нас
энергии есть следствие потока теплоты, отображенное вторым
началом термодинамики:
в любом самопроизвольном процессе количество недоступной энергии уве-
личивается со временем*
81
Немецкий физик Р. Клаузиус для учета этой недоступной
энергии в 1865 г. ввел величину, выражающую отношение из-
менения теплоты к температуре, и назвал ее энтропией.
(Энтропия по-гречески означает превращение.)
9.1. Энтропия. Математическая формулировка второго
начала термодинамики
1. Существует функция состояния, которая называется энтро-
пией и обозначается буквой S. Следовательно, dS — полный
дифференциал.
2. Для обратимого бесконечно малого превращения, во время
которого система обменивается тепловой энергией 6Q06P с внеш-
ней средой при температуре Г, энтропия изменяется согласно
уравнению
dS = ^Qo6p/T.
Если в результате обратимого превращения система перехо-
дит из состояния 1 в состояние 2, то соответствующее изменение
энтропии будет равно
2
as = s2-s,=$2^L. (2l8)
Если обратимое превращение осуществляется при постоянной
температуре, то изменение энтропии равно
ДЗ = QoCp/T,
где Qo6P — полное количество тепловой энергии, выделенное или
поглощенное системой.
3. Для необратимого превращения, т. е. самопроизвольного
процесса, идущего при постоянной температуре, изменение энтро-
пии выразится как
д5>0необр^ (29)
Процессы, для которых AS < при постоянной темпе-
ратуре, никогда не реализуются спонтанно.
Нужно отметить, что поскольку энтропия — функция состояния, измене-
ние энтропии ДЗ, которое сопровождает превращение системы, обусловлено
только начальным и конечным состояниями системы и не зависит от пути про-
цесса и, в частности, от того, является ли процесс обратимым или необрати-
мым. Для превращения, осуществляющегося при постоянной температуре (изо-
термический процесс), изменение энтропии ДЗ независимо от характера про-
цесса дается выражением
в котором обязательно присутствует тепловая энергия Q06P, выделяемая или
поглощаемая системой во время обратимого процесса.
82
Из соотношений (2.8) и (2.9) следует, что тепловая энергия, выделяемая
или поглощаемая системой, не одинакова в зависимости от того, осуществляется
ли процесс при одной и той же температуре, обратимо или необратимо.
Таким оразом, для любого превращения, переводящего систему из состоя-
ния 1 в состояние 2, имеем:
(*$2 — 51)обр = ($2 - 5|)необр —
(S2 - S,)o6p = (S2 - ХОнеобр
Следовательно, согласно второму началу термодинамики,
Т Т
или
Рнеобр < Собр.
При вычислении изменения энтропии AS, сопровождающего спонтанный
(необратимый) процесс, учитывают тот факт, что энтропия — функция состояния.
Для этого нужно рассмотреть обратимый процесс, начальное и конечное со-
стояния которого одни и те же, и вычислить изменение энтропии AS из выра-
жения
КОН. COCT. tn
AS= $
нач. сост.
Можно сделать вывод, что при переходе системы из состоя-
ния 1 в состояние 2 изменение энтропии AS для обратимых про-
цессов зависит только от исходного и конечного состояний сис-
темы и по своему значению больше AS для любого реального
процесса, связанного с необратимым переходом системы из со-
стояния 1 в состояние 2:
2 2
Если рассматривается изолированная система, то в этом слу-
чае нет обмена тепловой энергией с внешней средой. Когда эта
система подвергается обратимому превращению, то изменение
энтропии равно нулю. Тогда Q06P = 0, AS = 0. Если процесс
протекает самопроизвольно, то изменение энтропии положитель-
но. ^необр “— 0, AS 0.
Для изолированных систем процессы, для которых изменение энтропии
отрицательно, запрещены.
Таким образом, для изолированной системы второе начало
термодинамики можно сформулйровать следующим образом:
существует функция S, называемая энтропией, которая является функ-
цией состояния, такая, что
dS = dQogp//1.
Для обратимого процесса энтропия системы постоянна, а для необрати-
мого процесса возрастает. Энтропия системы не может уменьшаться.
Или, иначе говоря,
83
в изолированных системах самопроизвольно могут совершаться только такие
процессы, при которых энтропия системы возрастает, и процесс может идти
самопроизвольно только до такого состояния, при котором энтропия обладает
максимальным для данных условиях значением.
Таким образом, можно дать следующую объединяющую фор-
мулировку первого и второго начала термодинамики:
в любой изолированной системе полная энергия остается постоянной,
а полная энтропия со временем только повышается.
9.2. Статистическая интерпретация энтропии
Физический смысл энтропии не столь очевиден, как, напри-
мер, физический смысл внутренней энергии. В характерном
свойстве энтропии — монотонно возрастать во всех естественных
процессах, — как и в самом втором начале, есть нечто недоступ-
ное нашему пониманию, основанному на механических представ-
лениях.
Решение этой важной проблемы дал Л. Больцман в 1877 г.
Он ввел в теорию теплоты статистические представления,
приписав каждому состоянию системы термодинамиче-
скую вероятность, которая тем больше, чем более бес-
порядочным или неопределенным является это состояние с точки
зрения распределения параметров механического движения моле-
кул. При таком подходе возрастание энтропии означает, что
система, предоставленная самой себе, переходит из одного со-
стояния в другое, термодинамическая вероятность которого
больше.
Возьмем, например, два различных газа, разделенных мемб-
раной. Если мембрану убрать, то газы немедленно начнут пере-
мешиваться посредством диффузии, которая является типичным
необратимым процессом. Разумеется, весьма вероятно, чтобы
после того, как произошло перемешивание, все молекулы каждо-
го сорта вернулись в первоначально занятые ими половины
объема, которые заполнены теперь смесью газа. Следовательно,
если система вначале находится в таком маловероятном состоя-
нии, то молекулы сразу же начнут перемешиваться. Таким
образом, необратимые процессы будут продолжаться до тех пор,
пока не будет достигнуто наиболее вероятное состояние — со-
стояние, характеризующееся максимумом энтропии.
Статистические представления базируются на использовании
других, отличных от классических параметров системы. Если
в классической терминологии, которая рассматривалась ранее,
использовались так называемые макроскопические па-
раметры, то в статистической термодинамике используют
микроскопические параметры.
Макроскопическое состояние системы определено, если из-
вестны такие макроскопические параметры, как давление, тем-
пература, химический состав системы. Эти параметры в опреде-
ленном смысле дают некоторое усредненное выражение, не
84
рассматривая раздельно каждую частицу и систему, а харак-
теризуя их общее поведение.
Микроскопическое состояние системы определено, если из-
вестны относительные характеристики каждой из частиц.
Как правило, данному макроскопическому состоянию соот
ветствует очень большое число микроскопических состояний,
которые неразличимы в макроскопическом масштабе. Кроме того,
в разные отрезки времени макроскопические параметры могут ос-
таваться неизменными, несмотря на изменчивость микроскопичен
ских параметров. /
Таким образом, определяя, например, значение давления,
температуры и химического состава системы, считают установ-
ленным макроскопическое состояние системы, хотя в микроско-
пическом масштабе скорость и положение каждого атома или
молекулы системы неизвестны.
Например, в момент времени t\ п молекул обладают ско-
ростью v, а в момент /2 эти п молекул могут иметь скорость
и у2, в то время как другие молекулы будут обладать скоростью
v. Общая энергия системы сохраняется постоянной, ее мак-
роскопические параметры могут не измениться, хотя энергия
каждой молекулы меняется в любой момент времени.
Л. Больцман (1877) создал статистическую термодинамику
и ввел в теорию теплоты статистические представления. Со-
гласно этой термодинамике число микроскопических состояний
или, иначе говоря, термодинамическая вероятность, соответ-
ствующая макроскопическому состоянию, определяется точными
величинами параметров состояния и обозначается через Й.
Например, две одинаковые монеты можно расположить обе
«орлами» вверх с помощью единственной комбинации. Следо-
вательно, число состояний, позволяющих реализовать такое рас-
положение, равно 1. Реализовать же состояние «орел + реш-
ка», т. е. такое расположение монет, когда одна лежит «орлом»,
а другая — «решкой», можно двумя способами. Однако в резуль-
тате получим то же состояние «орел + решка», поскольку мо-
неты между собой не различаются. Следовательно, для состоя-
ния «орел + орел» й равно единице, а для состояния «орел +
+ решка» й — двум. Если бросают монеты, то более вероятно
реализовать «орел + решка» (две возможности из четырех),
чем «орел + орел» или «решка + решка» (одна возможность из
четырех). Число таких примеров можно увеличивать до бесконеч-
ности. Следовательно, можно сказать, что
система самопроизвольно стремится к состоянию, которому в микроскопи-
ческом масштабе соответствует наибольшее число возможностей реализации.
Точно так же молекулы растворенного твердого вещества рас-
пределяются случайно среди молекул растворителя. В растворе
возможна реализация гораздо большего числа микроскопиче-
ских состояний, чем для растворителя и нерастворенного ве-
щества. Макроскопическое состояние, отвечающее скоплению
85
всех молекул растворенного вещества в какой-то части объема
раствора, маловероятно, поскольку оно соответствует гораздо
меньшему числу микроскопических состояний.
При расширении идеального газа в пустоту конечное состоя-
ние (с большим объемом по сравнению с начальным состоя-
нием) включает гораздо большее число микроскопических со-
стояний просто потому, что молекулы могут принимать боль-
шее число положений в пространстве.
Когда в изолированной системе происходит самопроизвольный процесс,
число микроскопических состояний Q возрастает; то же самое можно сказать
об энтропии системы.
Процессы, протекающие в изолированной системе, сопровож-
даются ростом энтропии; в свою очередь все естественные про-
цессы заключаются в переходе системы из менее вероятного в
более вероятное состояние, т. е. в постепенном переходе к со-
стоянию равновесия, отвечающему наибольшему значению тер-
модинамической вероятности. Исследуя это, Л. Больцман показал,
что энтропия системы есть функция вероятности ее состояния:
S = f(Q).
Чтобы найти вид этой функции, представим себе систему
из "Двух тел, например двух газов, находящихся в двух раз-
личных состояниях. Энтропии их пусть будут Si и $2, а термо-
динамические вероятности Qi и й2. Рассматривая эту систему
в целом, надо считать ее энтропию S равной сумме обеих энтро-
пий, так как из определения энтропии следует, что она является
аддитивной величиной, т. е. что энтропия системы равна сумме
энтропий, ее составляющих:
S = S1
Согласно известной теореме теории вероятностей вероятность
одновременного наступления двух независимых событий равна
произведению вероятностей каждого из них:
Q — QiQ.
Единственная функция, которая одновременно удовлетворяет
рассматриваемым выше уравнениям, есть
S = k In Q,
где к — постоянная Больцмана.
Таким образом, можно сделать основной вывод статистиче-
ской термодинамики:
энтропия системы пропорциональна логарифму вероятности ее состояния.
Формула Больцмана раскрывает статистический смысл энтро-
пии, связанной с вероятностью состояния системы. Второе на-
чало термодинамики можно сформулировать следующим об-
разом:
86
изолированная система стремится достигнуть наиболее вероятного со-
стояния, т. е. макроскопического состояния, соответствующего наибольшему
числу макроскопических состояний.
Второе начало термодинамики, выраженное в понятиях вероятностей, не
исключает, например, процессы перехода теплоты от холодного тела к горяче-
му, но расчет вероятностей показывает, что протекание такого макроскопиче-
ского процесса столь маловероятно, что практически он неосуществим. Воз-
можны самопроизвольные процессы, сопровождающиеся уменьшением энтропии.
Например, для малых объемов газа с содержанием в них небольшого числа
молекул наблюдается нарушение равномерного распределения плотности. Дру-
гим примером возникновения в системе процессов, протекающих с наруше-
нием второго начала термодинамики, можно назвать броуновское движение.
В микрообъемах коллоидных растворов могут наблюдаться изменение числа
частиц во времени, связанные с неравномерностью молекулярного давления
на коллоидную частицу. Однако в макрообъемах эти нарушения утрачивают
значение.
Нетрудно привести и другие примеры понижения энтропии, если речь
идет не о всей системе, а о части ее. Например, известно, что металлы извле-
кают из руд и создают машины исключительной сложности из металлических
болванок. Можно видеть, как скользят вверх лифты, катят в гору автомобили,
строятся в колонны солдаты и т. п. Все эти и очень многие другие действия,
связанные с понижением энтропии, вызываются жизнедеятельностью людей.
Возникает впечатление, что разумная жизнь способна «повернуть вспять энтро-
пию».
Однако тут не все учтено. Люди поглощают пищу и поддер-
живают свою жизнеспособность благодаря энергии, извлекаемой
в результате химических реакций в организме. Чтобы дать энер-
гию машинам, сжигаются уголь и нефть, используется энергия
гидроэлектростанций, чтобы получить из руды металл. Вся дея-
тельность человека, направленная на понижение энтропии, осу-
ществляется за счет процессов, повышающих энтропию. Дейст-
вия, повышающие энтропию, всегда больше действий, понижаю-
щих энтропию. Общий конечный результат — повышение энт-
ропии.
Таким образом можно сказать, что
чем больше беспорядок, тем больше энтропия системы;
энтропия является мерой беспорядка в системе.
Статистическое толкование второго начала термодинамики
дает энтропии конкретное физическое содержание как меры
вероятности термодинамического состояния тел и системы.
10. ТРЕТЬЕ НАЧАЛО ТЕРМОДИНАМИКИ
Третье начало термодинамики также является постулатом,
утверждающим, что
энтропия чистых веществ, существующих в виде идеальных кристаллов,
при температуре абсолютного нуля равна нулю.
Это значит, что при абсолютном нуле достигается полная
упорядоченность. В рамках статистической интерпретации энтро-
пии нулевое значение энтропии при абсолютном нуле соответ-
ствует макроскопическому состоянию, которое реализуется един-
87
ственным микроскопическим состоянием, что выражается соот-
ношением Больцмана;
S = klnQ = 0 при Q = l.
Абсолютная энтропия. Третье начало термодинамики позво-
ляет определить абсолютную энтропию всех чистых веществ при
любой температуре.
Предположим, что температура 1 моль вещества при постоян-
ном давлении увеличивается от абсолютного нуля (где вещества
находятся в состоянии «идеального кристалла») до температу-
ры Т (где вещество находится в газообразном состоянии). Тогда
должны существовать следующие процессы:
плавление
ОК р °> Гпл -
тв. тела
нагрев
жидк.
кипение
нагрев^ гр
Чип
газы
Изменение энтропии без учета
равно
полиморфных превращений
z4 Т I
а Т Ч—-—
* пл
Гил
AS = Sr-So=$
ок 1
Поскольку энтропия чистого кристаллического вещества при
абсолютном нуле равна нулю, имеем
+ $ A^df,
где 5/ — абсолютная энтропия чистых веществ при температуре
Т и атмосферном давлении.
Таким образом, для любой температуры Т можно вычислить
абсолютную энтропию чистых веществ, если известны теплоем-
кости в твердом, жидком и газообразном состояниях, а также
изменение энтальпии при изменениях состояния (плавлении,
испарении, возгонке).
Абсолютная энтропия в основном приводится при температуре
298 К и давлении 101,3 кПа (стандартные условия) и выража-
ется в Дж/ (моль-К). Необходимо отметить, что для твердых тел
абсолютная энтропия в среднем равна 40...60 Дж/ (моль-К).
Значение 2,4 Дж/(моль-К) для алмаза свидетельствует о весьма
жесткой и упорядоченной структуре. Для жидкостей и газов
энтропия выше; она заключена между 60 и 380 Дж/(моль-К).
Так как можно рассчитать абсолютную энтропию чистых
веществ, то, естественно, используя эти значения, можно оценить
изменение энтропии AS, которое соответствует химической реак-
ции. Энтропия есть функция состояния, и изменение энтропии
88
будет равно разности абсолютных энтропий продуктов и реаген-
тов. Так, для химической реакции, протекающей при темпера-
туре Г,
дА -|- ► сС -|- rfD
изменение энтропии равно
ASr = cSr(C) 4-dSr(D)-aST(A) -6Sr(B)
или в общем виде
AST=XST (продуктов) — SSt (реагентов).
11. СВОБОДНАЯ ЭНЕРГИЯ
Для изолированной системы изменение энтропии, сопровож-
дающее процесс, позволяет предсказать, будет ли реакция необ-
ратимой или обратимой. Как было показано, изменение энтропии
положительно, если процесс самопроизвольный, и равно нулю,
если процесс обратимый. Эти критерии не могут быть использо-
ваны для закрытых систем, для которых вводятся две новые
термодинамические функции: свободная энергия Гиббса (G) и
свободная энергия Гельмгольца (У7).
Рассмотрим закрытую систему, в которой осуществляется
процесс при постоянной температуре Г.
Согласно второму началу термодинамики разрешены следую-
щие превращения:
обратимый процесс необратимый процесс
^s=^, дз>-^ч
Qo6p-TAS = 0, QHeo6p-TAS<0.
Если процессы протекают при постоянном давлении, то
обратимый процесс необратимый процесс
Qp —TAS = о Qp - tas < о,
или
A//—TAS = 0, (2.10) АН-TAS <0. (2.11)
Функция H—TS обозначается буквой G и называется свобод-
ной энергией Гиббса:
G = H—TS.
Функция G=H — TS также называется изобарно-изо-
термическим потенциалом или изобарным по-
тенциалом.
Свободная энергия G — функция состояния, поэтому изме-
нение ДО этой функции при конечном процессе равно
AG — AH—TAS — SAT.
При постоянной температуре ДТ=0, откуда
AG = AH—TAS.
89
Сопоставляя данное уравнение с (2.10) и (2.11), получаем
обратимый процесс необратимый процесс
Аб=о, Дб<0.
В этих условиях AG=0— критерий равновесия, a AG<0—
критерий необратимости процесса.
Если процесс идет при постоянной температуре и объеме, т. е.
A[/=Qy, то можно записать
обратимый процесс необратимый процесс
A/7-7AS = 0, MJ-TbS<0.
Функция U — TS обозначается буквой F — это функция состоя-
ния; она называется свободной энергией Гельм-
гольца:
F—U—TS.
Функция U—TS играет большую роль при изучении равно-
весия в изотермических процессах; ее называют также изо-
хорно-изотермическим потенциалом, изохор-
ным потенциалом или энергией Гельмгольца.
При постоянных температуре и объеме имеем
обратимый процесс необратимый процесс
AF=0, А/?<0.
В этих условиях AF=0 — критерий равновесия, a AF<0—
критерий необратимости процесса.
12. Свободная энергия и направление химических реакций
Необратимому процессу, т. е. самопроизвольной реакции, соответствует от-
рицательное изменение свободной энергии. Если изменение свободной энергии
равно нулю, то начальное и конечное состояния могут существовать в равно-
весии. Если изменение свободной энергии положительно, то самопроизвольно
реализуется обратная реакция.
Таким образом, в общем виде для химической реакции
лА -|- &В q=t гС -|- (Hl)
2
.можно записать:
1) AG=0; система находится в динамическом равновесии,
т. скорость реакции, идущей в направлении 1, равна скорости
реакции, идущей в направлении 2;
2) AG<0; реализуется самопроизвольный процесс в направ-
лении 1;
3) AG>>0; реализуется самопроизвольный процесс в направ-
лении 2.
Исходя из сказанного, можно сделать вывод, что определе-
ние знака AG позволяет без эксперимента установить направ-
90
ление данной химической реакции. Однако при этом нужно знать
значение ДЯ, AS и Г, так как \G = \H — TS.
Рассмотрим на ряде примеров, как меняется знак Д6 (или AH—T&S)
при изменении знака &Н и As.
1. Экзотермические реакции, протекающие с возрастанием энтропии. Эти реак-
ции протекают необратимо (самопроизвольно). К ним относится, например,
разложение молекулы на несколько молекул. Число микроскопических состоя-
ний системы, связанных с данным микроскопическим состоянием, растет.
В качестве примера приведем распад пероксида водорода при 298 К и атмо-
сферном давлении:
2Н2О2 (г)-> 2Н2О (г) + О2 (г)
(ДЯ=-211,43 кДж, Д5=129Дж, Д6 =-250,37 кДж).
При сгорании любого органического вещества образуются диоксид углерода
и вода — это экзотермическая реакция. Реакция сопровождается значительным
изменением энтальпии, при этом изменение свободной энергии отрицательно.
2. Эндотермические реакции, протекающие с понижением энтропии. Эти
реакции не протекают необратимо. В качестве примера приведем реакцию азота
с кислородом при 298 К и атмосферном давлении:
N2(r) 4-2О2--,2МО2(Г)
(ДЯ=-67,83 кДж, Д5=-122 Дж, ДО= 103,83 кДж).
3. Экзотермические реакции, протекающие с понижением энтропии. Так как
изменение энтальпии мало зависит от температуры, реакции такого типа более
вероятны при низких температурах, т. е. когда член TAS не значителен. Дейст-
вительно, при |ДЯ|> |ТД5| изменение свободной энергии ДО становится отри-
цательным.
Так, реакция синтеза аммиака возможна при 298 К и атмосферном дав-
лении:
3H2(r)+N2(r)---->2NH3(r)
(ДЯ=— 92,53 кДж, Д£= —195 Дж, ДО =—33,49 кДж).
Повышение температуры не благоприятствует реакции образования аммиака.
4. Эндотермические реакции, протекающие с возрастанием энтропии. Они
могут осуществляться при высокой температуре; в этих условиях член TAS может
быть больше, чем ДЯ.
Так, при 298 К и атмосферном давлении реакция
N2O4(t)---,2NO2(r)
будет сопровождаться положительным изменением свободной энергии, так как
ДЯ=58,20 кДж, Д£=177Дж, Д6 = 5,44кДж. При 298 К величина Д6 отрица-
тельна и реакция протекает спонтанно. В табл. 13 обобщены рассмотренные
примеры.
Стандартная свободная энергия образования. Для расчета
свободных энергий необходимо, как и при расчете энтальпий, вве-
сти шкалу отсчета, т. е. дать определение свободной энергии
образования соединений в стандартных термодинамических усло-
виях.
Стандартная свободная энергия образования соединений
Д/G0 (298 К) равна изменению свободной энергии реакции обра-
зования этого соединения из элементов или простых веществ,
причем соединения и элементы (простые вещества) рассматри-
ваются в их стандартном состоянии при температуре 298 К. Из
91
T а б лица 13. Направление химических реакций различных типов >
Тип реакции Знак Вывод
ДЯ AS AG
1 — + — Самопроизвольный процесс
2 + — + Процесс не самопроизвольный
3 — — Процесс самопроизвольный при низкой
температуре
4 + + Процесс самопроизвольный при высокой
температуре
этого определения следует, что для любого элемента A/G°
(298 К) =0.
Из значений стандартных свободных энергий образования
соединений, которые обычно даются в виде табличных данных,
можно определить изменение стандартной свободной энергии.
Эти величины характеризуют химическую реакцию, протекаю-
щую в стандартных условиях. Она равна разности стандартных
энергий продуктов и реагентов.
Изменение свободной энергии при изменении температуры и
давления. При изучении химических реакций иногда необходимо
установить, как влияет на величину свободной энергии измене-
ние температуры и давления. Поскольку G = H —TS = U'+ pV—
— TS, при термомеханическом превращении дифференциал сво-
бодной энергии запишется в виде
dG = dU + pdV + Vdp—TdS — SdT; dU = dA + 6Q.
Так как 6Д = — pdV и, если процесс обратим, 6Q = TdS,
d G = —pd V + TUS + pd V + Vdp - TdS - Sd T,
dG = Vdp — SdT.
Если процесс происходит при постоянном давлении, то dp=O;
тогда изменение свободной энергии при изменении температуры
равно
(£),=-*
Если процесс изотермический, то dT=O; изменение свободной
энергии при изменении давления равно
==v-
X др /т
Рассмотрим термомеханическое обратимое превращение 1 моль
идеального газа при постоянной температуре и при изменении
давления от pi до р2. Изменение свободной энергии составляет
2
Л6=0г — G\ — § Vdp.
92
Газ предполагается идеальным, следовательно,
V=RT/p,
откуда
J
= #Лп.
Pl Р Р'
Изменение свободной энергии при изотермическом термоме-
ханическом превращении 1 моль идеального газа можно запи-
сать в виде
\G = RT\n-^.
Рнач
Выражение, полученное для обратимого изотермического про-
цесса, справедливо для необратимого изотермического процесса,
так как свободная энергия — функция состояния. Это значит, что
начальные и конечные состояния для этих двух процессов оди-
наковы.
Если начальное давление равно давлению, соответствующему
стандартному состоянию, то при температуре Т будем иметь
\G = GPT-GQT = RT\n-^-t
где Gt — свободная энергия 1 моль газа в стандартных условиях
при температуре Г.
Следовательно, свободная энергия 1 моль идеального газа
при температуре Т и давлении р равна
G?=GQT + RTinp. (2.12)
Для п моль получим следующее соотношение:
G^ = nGr + nRT In р.
13. ПОНЯТИЕ О ХИМИЧЕСКИХ ПОТЕНЦИАЛАХ
В общем случае изменение внутренней энергии системы происходит не
только вследствие отдачи или получения теплоты или совершения работы, но
и изменения масс входящих в нее компонентов.
Изменение (удаление или получение) массы вещества системы вызывает
изменение внутренней энергии системы, что выразится уравнением
U = f(V, S, m2, ... , mi),
где U — внутренняя энергия; V — объем; S — энтропия; mi — масса компонентов
системы.
Если единица массы вызывает изменение внутренней энергии системы на р,
то изменение внутренней энергии, вызванное изменением массы dm, будет равно
pdm. Тогда суммарное изменение внутренней энергии будет равно
d^T’dS-pdV'-pdm. (2.13)
При условии, что V и Т постоянны, получаем
—(TdS—d U) = pdm,
откуда
93
-(TdS — <W)=dF,
Отсюда следует, что ц при постоянном объеме и температуре есть изменение
энергии Гельмгольца, соответствующее изменению массы системы на единицу.
Аналогично при постоянном давлении и температуре
\ dm Jp> т
где р. — изменение энергии Гиббса, соответствующее изменению массы системы
на единицу.
Из уравнения (2.13) следует:
\am/S,p
Величина р, которая определяет изменение внутренней энергии системы,
связанной с изменением массы входящих в нее компонентов, называется
химическим потенциалом.
• Если масса компонентов выражается в граммах, то химический потенциал
называется удельным цуд, а если в молях, то молярным рм.
Химический потенциал есть функция, определяющая направление и предел
самопроизвольного перехода данного компонента нз одной фазы в другую при
соответствующих превращениях (путем испарения, растворения, кристаллизации
и взаимодействия).
Химический потенциал является функцией состояния и значение его зависит
от температуры, давления (или объема) и концентрации. JaK, для 1 моль
идеального газа
р=цо + ЯГ1пХ„
где Xi — молярная доля компонента i или парциальное давление р компонента i
в смеси газов; ро— константа, численно равная р при Х/==1.
Общим условием возможного самопроизвольного течения процесса служит
неравенство
2 ptdzn<;0,
а состояние равновесия отвечает равенству
2 pidn/ = 0,
где 2 — алгебраическая сумма произведения химического потенциала (р) ком-
понентов на изменение количества вещества (молей) их (dn).
Контрольные вопросы
1. Закон Гесса был сформулирован на основании экспериментальных ре-
зультатов. Как осуществить доказательство термодинамически?
2. Известно, что Q не есть функция состояния. Докажите, что Qp и Qy суть
функции состояния.
3. Выведите все возможные соотношения между термодинамическими функ-
циями //, U, G, F, S.
4. Как изменение свободной энергии определяет направление химической
реакции?
5. Вся разумная деятельность человечества связана с процессами, происходя-
щими с уменьшением энтропии. Как доказать, что при этом не нарушается
второе начало термодинамики?
6. Увеличение энтропии в изолированной системе показывает, что процесс
осуществляется самопроизвольно. Зачем вводятся такие термодинамические
функции, как свободная энергия Гиббса и Гельмгольца?
7. К каким термодинамическим системам относятся живые организмы?
Глава III
ХИМИЧЕСКИЕ И ФАЗОВЫЕ РАВНОВЕСИЯ
1. ХИМИЧЕСКИЕ РАВНОВЕСИЯ
Равновесным состоянием называется такое термодинамическое состояние
системы, которое не изменяется во времени, причем эта неизменяемость не
обусловлена протеканием какого-лйбо внешнего процесса.
Это значит, что существующее для данной системы состоя-
ние равновесия не поддерживается извне каким-либо процессом.
Любые состояния, смежные с равновесным, являются менее
устойчивыми, и переход к ним из состояния равновесия всегда
связан с необходимостью затраты работы извне. Иначе говоря,
к равновесному состоянию принципиально можно подойти с двух
противоположных направлений.
При равновесии всякое бесконечно малое воздействие вызы-
вает только бесконечно малые изменения в состоянии системы.
Оно не может вызвать течения необратимых процессов. По пре-
кращении такого воздействия система стремится вновь восстано-
вить прежнее состояние.
Равновесие является динамическим по характеру и сохраня-
ется во времени не вследствие отсутствия или прекращения
процесса, а вследствие протекания его одновременно в двух про-
тивоположных направлениях с одинаковой скоростью. Именно
равенство скоростей прямого и обратного процессов является
причиной сохранения равновесия системы при неизменности
внешних условий.
Например, процесс этерификации с образованием сложного
эфира связан с противоположной реакцией омыления эфира
водой:
СНзСООН + С5Н5ОН Z СНзСООС2Н5 + Н2О
В каждый данный момент большое число молекул уксусной кис-
лоты и этилового спирта взаимодействуют между собой и одно-
временно с этим также большое число молекул уксусноэтилового
эфира разлагаются водой. Скорости этих процессов вначале раз-
личны, но потом наступает момент, когда Vi становится равной v?
и система приходит в состояние равновесия, хотя и в этот момент
продолжают идти реакции как образования сложного эфира, так
и его омыления.
Таким образом, можно сказать, что в равновесной системе
95
нет ни продуктов, ни реагентов, все вещества одновременно
участвуют в создании равновесия. В равновесной системе веще-
ства называются продуктами или реагентами только формально
по написанию уравнения реакции. Так как скорости реакции
в обоих направлениях равны, в каждый момент времени наряду
с образованием продуктов из реагентов происходит образование
реагентов из продуктов.
Вычисление константы равновесия при строго контролируе-
мой температуре заключается в экспериментальном определении
равновесных концентраций каждой из частиц, входящих в дина-
мическое равновесие, за исключением растворителя и чистых
конденсированных фаз. Доказательство постоянства константы
равновесия было получено в исследовании р!авновесных смесей
при реакции водорода и иода, тщательно выполненном Тейло-
ром и Кристом (1942).
Исследовались равновесные системы, образованные при со-
вместном нагревании водорода и иода, либо при нагревании
иодоводорода:
2Н1^Н2(г)+12(г),
к_ [Н2] [12]
Л [HI]2
Некоторые из этих данных приведены в табл. 14. Следует
отметить, что вычисленные по полученным данным значения
констант действительно постоянны: экспериментальная погреш-
ность составляет ±1%. Одинаковые константы получаются неза-
висимо от того, нагревался иодоводород или смесь водорода
и иода.
Все имеющиеся экспериментальные доказательства, получен-
ные в большом числе опытов, так же, как и все современные
химические теории, подтверждают идею динамического харак-
тера равновесия:
1) каждая реакция при равновесии строго сбалансирована
противоположной реакцией, протекающей с той же скоростью;
2) при равновесии не происходит никаких суммарных изме-
нений концентраций.
Таблица 14. Равновесие между водородом, иодом
и иодоводородом при 698,6 К
[Н2] • Ю 3, моль/л [I2] • Ю 3, моль/л [HI] -10 3, моль/л к [На] [Ы .0-2 Л— [Н1р 10
1,8313 3,1292 17,671 1,835
2,2523 2,3360 16,850 1,853
2,9070 1,7069 16,482 1,827
3,5600 1,2500 15,588 1,831
4,5647 0,7378 13,544 1,835
0,4789 0,4789 3,531 1,840
0,4953 0,4953 3,655 1,832
1,1409 1,1409 8,410 1,840
Средняя ... 1,837-10-2
96
1.1. Влияние изменения внешних условий
на равновесие
Ле Шателье в 1884 г. сформулировал общий принцип, каче-
ственно отражающий влияние изменения различных факторов на
положение равновесия, — при н ц ип смещения равно-
весий, называемый иначе принципом Ле Шателье,
суть которого сводится к тому, что система, находящаяся в рав-
новесии, реагирует на внешнее воздействие таким образом, что-
бы уменьшить это воздействие. Более полная формулировка
принципа Ле Шателье следующая:
если на систему, находящуюся в устойчивом равновесии, воздействовать
извне, изменяя какое-нибудь из условий, определяющих ^положение равновесия,
то в системе усилится то из направлений процесса, течение которого ослабляет
влияние произведенного воздействия и положение равновесия сместится в том
же направлении.
Равновесие всегда отвечает равенству скоростей прямого и
обратного процессов, а смещение равновесия происходит тогда,
когда произведенное воздействие неодинаково влияет на скоро-
сти прямого и обратного процессов. Нарушение равенства ско-
ростей приводит к смещению равновесия и переходу системы
в новое состояние равновесия, при котором скорости прямой и
обратной реакции опять станут одинаковыми между собой, но
будут отличаться от первоначальных.
Таким образом, если в реакции
(jA -j- /?В сС -|“ t/D
V<2
наступил момент, когда скорости прямой и обратной реакции
равны между собой (хм = ^2), это значит, что наступило равнове-
сие. Воздействуя на систему извне, можно изменить соотношение
скоростей реакции, так что Тогда в системе произойдет
смещение равновесия до нового равновесного состояния
(jA -j- £>В сС -J- dY)
V2
при котором скорости обеих реакций опять станут равны между
собой: vf = U2, хотя v\=£v'\ и иг 7^2.
В качестве примера рассмотрим сначала качественное смеще-
ние равновесия в системе, возникающей при повышении темпе-
ратуры реакции
N2 + 3H2^2NH3
Реакция образования аммиака сопровождается выделением
теплоты (экзотермическая реакция). Тогда очевидно, что обрат-
ная реакция протекает с поглощением теплоты (эндотермическая
реакция). Если повысить температуру системы, находящейся -
в равновесии, подводя к ней теплоту извне, то такое воздействие
вызовет усилие того из направлений реакции, которое сопровож-
дается поглощением теплоты, т. е. диссоциацию аммиака. Это
4-696 97
смещение равновесия ослабит влияние оказанного внешнего воз-
действия. Экспериментальная проверка подтверждает это поло-
жение. Зная теплоемкости азота, водорода и аммиака, рассчи-
таем для какой-нибудь смеси известного состава, находящейся
в равновесии, количество теплоты, которое нужно подвести к
этрй системе для повышения ее температуры, например, на
100°. Однако данные расчета не совпадут с экспериментальными.
Это объясняется тем, что часть полученной теплоты будет израс-
ходована на диссоциацию аммиака, и в результате произойдет
повышение температуры не на 100°, а соответственно в меньшей
степени.
Этот факт является следствием неодинакового увеличения
скорости прямого и обратного процессов с повышением темпе-
ратуры. Скорость эндотермической реакции возрастает сильнее,
чем скорость экзотермической реакции, что и вызовет смещение
равновесия в сторону образования азота и водорода. С пониже-
нием температуры равновесие, наоборот, будет смещаться в сто-
рону образования аммиака.
В общей форме влияние изменения температуры можно выра-
зить таким образом:
повышение температуры всегда благоприятствует накоплению тех веществ,
которые образуются в данной реакции с поглощением теплоты, т. е. усиливает
эндотермическую реакцию процесса. Понижение температуры способствует усит
лению экзотермической реакции процесса.
Рассмотрим влияние изменения давления. В реакции из 1 моль
азота и 3 моль водорода получается 2 моль аммиака, т. е. течение
реакции в прямом направлении сопровождается уменьшением
объема в два раза (из четырех объемов получается два). Следо-
вательно, обратная реакция сопровождается таким же увеличе-
нием объема. Повышение давления при постоянной температуре
и постоянных количествах реагирующих веществ можно осуще-
ствить, только уменьшая ее объем. Такое воздействие вызывает
ускорение в системе того процесса, который сопровождается
уменьшением объема, т. е. в данном случае образование аммиа-
ка. Понижение давления должно, наоборот, способствовать дис-
социации аммиака.
Здесь, как и ранее, можно провести эксперимент, подтверж-
дающий факт смещения равновесия. Уменьшим объем реакцион-
ной системы в 2 раза. Если бы не было смещения равновесия,
давление в ней должно было бы увеличиться тоже в два раза.
Однако эксперимент показывает, что давление увеличивается не
в два раза, а в меньшей степени. Это происходит потому, что
равновесие сместится в сторону образования аммиака, который
займет меньший объем, чем сумма газов водорода и азота. Сле-
довательно, произойдет относительное уменьшение объема суммы
газообразных продуктов и реагентов по сравнению с тем, кото-
рое было бы без смещения равновесия. Соответственно с этим,
давление тоже относительно уменьшится.
98
В общем виде влияние изменения давления на положение
равновесия можно выразить следующим образом:
повышение давления благоприятствует образованию веществ, занимающих
в данных условиях меньший объем, т. е. усиливает ту из реакций процесса,
которая сопровождается уменьшением объема. Понижение давления усиливает
реакцию, сопровождаемую увеличением объема.
В табл. 15 приведены экспериментальные данные, подтверж-
дающие приведенные выводы. Изменение состава равновесной
смеси с температурой и давлением показывает, что оптималь-
ными из приведенных параметров являются минимальная тем-
пература (573 К) и максимальное давление (100 МПа). В этих
условиях выход аммиака максимален.
Таблица 15. Состав равновесной смеси при синтезе аммиака
в зависимости от температуры и давления
г, к Содержание аммиака в смеси (в %) при давлении (в МПа)
0,1 10 30 100
573 52,0 71,0 92,0
673 0,44 25,1 47,0 79,8
723 0,23 16,4 35,8 69,8
773 0,13 10,6 26,4 57,5
823 0,08 6,8 19,1 41,2
873 0,05 4,5 13,8 31,4
Заметные изменения объема связаны, естественно, с реакция-
ми, включающими газовые компоненты. В процессах, происхо-
дящих в конденсированных фазах, где ни один из компонентов
реакции не находится в газообразном состоянии, значительных
изменений объема происходить не может. Поэтому в таких про-
цессах изменением давления, не удается достичь значительных
смещений равновесия.
На смещение равновесия могут воздействовать кроме пере-
численных и другие факторы. Если равновесие системы зависит
от внешних электрических, магнитных полей или поля тяготения,
то при изменении их усиливается то из направлений процесса,
которое уменьшает влияние произведенного воздействия. Точно
так же влияет и введение дополнительных количеств одного из
компонентов реакции. Химическая термодинамика позволяет
количественно выразить все эти влияния.
Изменение свободной энергии химической реакции между
газами в зависимости от их парциальных давлений. Рассмотрим
смесь двух идеальных газов, которые могут реагировать между
собой.
Пусть реакция
аА + ЬВ —> сС + сЮ
протекает при постоянной температуре.
4*
99
Начальное 1 и конечное состояние 2 этой системы определя-
ется следующим образом. Состояние 1 соответствует а моль А и
b моль В; их парциальные давления будут рд и рв при темпера-
туре Т. Состояние 2 соответствует с моль С и d моль D; их пар-
циальные давления — рс и ръ при той же температуре Т.
Изменение свободной энергии этой реакции равно
> = GT (состояние 2) — GT (состояние 1).
Поскольку газы предполагаются идеальными, для каждого
состояния свободная энергия равна сумме свободных энергий
различных компонентов:
GT (состояние 1) = aG^A (A) (В),
GT (состояние 2) = cGp (С) (D).
Определим значения свободных энергий начального и конеч-
ного состояний согласно соотношению
G$=G*T + RT\np.
Таким образом
GT (состояние 1) =aG°(A) + aRT\npA +bG?(B) bRT\npB,
GT (состояние 2) =cGr(C) + + + dRTlnp^.
Изменение свободной энергии, соответствующее реакции,
будет
GT (состояние 2) — GT (состояние 1) => = [cGr(C) 4-dG°(D)] —
— [aG°(A) 4-6Gr(B)] +/?Г(с1прс4-^lnpD~ alnpA — b In pB) •
Объединив члены, связанные co стандартными значениями сво-
бодной энергии
[cG?(C) 4-rfG°r(D)]-[aG°T(A) + Z>G°r(B)] = AG?,
получим
AGr = AG? + /?Tln-44-- (31)
РаРв
Соотношение (3.1) позволяет определить изменение свобод-
ной энергии AGr, сопровождающее реакцию, если при одной и
той же температуре известны парциальные давления реагентов,
парциальные давления продуктов и изменение стандартной сво-
бодной энергии AGr, которое можно вычислить при использова-
нии таблиц термодинамических данных.
1.2. Закон действующих масс
При достижении равновесного состояния при температуре Т
скорости прямой и обратной реакции равны; свободная энергия
становится минимальной и ее изменение равно нулю:
tzA -J- ЬВ:«=► сС -j-z/D, AGr = 0.
Из уравнения (3.1) получим
юо
1„ PcpD _ AG?
П na П'Г »
PaPb Кi
где pa, рвг pc, рэ -^парциальные давления компонентов А, В, С,
D при равновесии?'7
Так как AG? — изменение свободной энергии в стандартных
условиях постоянно, то при равновесии получим
РсРр
РаРв
е RT =К,
где КР — константа равновесия при постоянном давлении.
При данной реакции КР зависит только от температуры и
связана с изменением стандартной свободной энергии AG? вы-
ражением
ДО?=-ЯГ1пКр. (3.2)
В тех случаях, когда реакция осуществляется при постоянном
объеме системы, состав реакционной смеси удобнее выражать
через концентрацию. По определению концентрация компонен-
та А равна количеству вещества (молей) компонента А (па)
в единице объема системы, или, иначе, количеству вещества (мо-
лей А), деленному на общий объем системы V\
[А]=па/К
Поскольку речь идет об идеальных газах
рАК=ПлЛТ, рА=-^-₽Т,
следовательно,
рА= [А]РГ.
Отсюда остальные парциальные давления рв, pc, ро можно вы-
разить в виде
Рв=[В]РЛ pc=[C]RT, pD==[D]RT.
Тогда для КР можно записать
_ [C]W]c[D]W]d
Ар [А]а[Т?Г]а[В]
или
При температуре Т произведение постоянно;
обозначим его через Кс.
Таким образом, при равновесии концентрации различных
компонентов связаны соотношением
_ [C]W
с [Aja[B]* ’
где Кс — константа равновесия при постоянном объеме.
101
Если количество вещества (молей) реагентов и продуктов
одинаково, то КР—Кс.
Полученные соотношения выражают закон действую-
щих масс или закон Гульдберга и Вааге.
Итак, для идеальных газов получено соотношение между кон-
центрациями различных соединений при равновесии и термодина-
мическими функциями. Таким образом, классическая термодина-
мика позволила определить направление реакции и количест-
венно оценить концентрации компонентов системы в состоянии,
когда свободная энергия минимальна.
Представленная схема (рис. 38) характеризует свободную
энергию системы как функцию отношений реагентов или про-
дуктов. Вне зависимости от исходного состояния — берутся ли
только реагенты или продукты — система самопроизвольно при-
ходит к состоянию равновесия, из которого она без внешнего
воздействия не может выйти, поскольку это соответствует уве-
личению свободной энергии.
Ст(реагенты)
(^(раМесие)
^(продукты)
Реагенты,10010-
0%
0%
% при равновесии 100%,продукты
Рис. 38. Изменение свободной энергии системы в
процессе химической реакции
При использовании закона действующих масс для реальных
газов и жидкостей нужно вместо концентраций ввести активно-
сти — произведения концентраций на множитель, учитывающий
взаимодействия между молекулами!в жидком состоянии.
Химическая термодинамика позволяет описать смещение рав-
новесия, т. е.; количественно оценить изменение константы равно-
весия с изменением температуры, давления и концентрации.
1.3. Количественная оценка смещения равновесия
с изменением температуры и давления
Из уравнения (3.2) следует, что
1пКР=—(3.3)
1x1
При дифференцировании получим
dlnXp _ d /AG?\_ 1 d /А6?\
dT “ dT I RT )~ R dT I T J
102
Принимая во внимание, что Дб? — функция температуры,
d /Дб?\_ Дб? 1 адб?
df к Г )~ "Т2 Н Т dr ’
Ранее было показано, что (—туг) =—S, откуда
\ U / /р
d /Дб?\_ Дб? ДЗ? _ &Gt-{-T&St
dr \ Г ) ~Т!~ Т ~ Г2
Поскольку Дб?=Д//?—ГДЗ? при температуре Г, выражение
d / Дб? \
dr \ Г )
примет вид
d / Дб? ЛЯ?
dr \ Г )~ ~Т7~'
Это уравнение для стандартного состояния — частный случай общего урав-
нения
d Mi
АТ\т)~
(3.4)
Используя уравнения (3.3) и (3.4), можно записать закон изменения 1пКр
с изменением температуры:
din Л,. ДЯ?
(3’5)
Это уравнение Вант-Гофф а. Оно показывает, что при положитель-
. Z7o , ч din/С
ных значениях ДЯУ (эндотермическая реакция) —- положительно и др уве-
личивается с повышением температуры. Если реакция экзотермическая, то зна-
чение ДЯ° отрицательно и Кр уменьшается с повышением температуры. Следо-
вательно, в обоих случаях концентрации соединений при равновесии меняются,
как это и следует из принципа Ле Шателье.
Интегрируя уравнение (3.5), можно получить отношение между констан-
тами равновесия при двух различных температурах:
ьн*
АГ, т
dT.
Если ДЯ? практически постоянно в рассматриваемом температурном интервале,
то можно записать
In
Кг2
Кт,
ДЯ? г 1 У2
R L Т Jr/
откуда
Кт2 _ 1 ДЯ? / 1 1 \
gKr,~ 2,303 К \Т1 Tj’
Это уравнение позволяет рассчитать константу равновесия при заданной темпе-
ратурё Т2, если известны константа равновесия при температуре Т\ и значение
ДЯ реакций. При больших интервалах температур величину ДЯ надо выразить
как функцию температуры. Для этого надо знать зависимость от температуры
всех веществ, участвующих в реакции.
Для выяснения влияния температуры на концентрацию продуктов реакции
изобразим при двух различных температурах (7'2>7’i) кривые концентраций
продуктов в зависимости от времени для эндотермической (рис. 39) и экзотер-
103
[Продукты]
Рис.' 39. Предельные концентрации продуктов
при протекании эндотермической реакции
[Продукты!
Предельная _
концентрация лриТ,
Предельная
концентрация приТг
[Продукты!? -
[Продукты!^
Время
мической (рис. 40) реакций.
В случае экзотермической ре-
акции предельная концентра-
ция, достигнутая при Твыше
предельной концентрации
при ТУ И наоборот, концен-
трация продуктов в момент
времени t (рис. 40) больше
при Т2, чем при Г1. Таким
образом, повышение темпера-
туры всегда увеличивает ско-
рость реакции, даже если это
в конечном счете приводит
к уменьшению образовавше-
гося продукта.
На смещение равновесия
влияют также давление и
концентрации реагирующих
веществ.
Пусть система аА4-6В^=*
*=zcC-\-dD находится в рав-
новесии при постоянной тем-
пературе с изменением толь-
ко у:
О , t
Рис. 40. Предельные концентрации продуктов
при протекании экзотермической реакции
у—Кр>=
РсРр
РаРь
а величина КР, характеризующая равновесие, остается постоянной.
Выразим у как функцию общего давления р:
Hi
Pi=-P,
откуда
л*
У=Кр)= /П-4-/П $ р(^-(°+Ч
(v) (v)
Если количество вещества (молей) продуктов равно количеству вещества
(молей) реагентов (c-f-J — a-\-b), то у не зависит от р, т. е. изменение общего
давления в этом случае не приводит к смещению положения равновесия.
Если количество вещества (молей) продуктов больше количества вещества
(молей) реагентов (c-\-d)>(a~\-b), то величина у при возрастании р становится
больше значения Система приходит к новому состоянию равновесия, при
котором у снова будет равно Кр.
При повышении давления система переходит в состояние, соответствующее
уменьшению общего количества вещества (молей): происходит дополнительное
образование веществ А и В. При понижении давления система переходит в со-
стояние, соответствующее увеличению общего количества вещества (молей):
образуются С и D. Если (a-\-b)7>(c-\-d'), то увеличение давления благоприят-
ствует образованию соединений С и D, уменьшения давления — соединений
А и В.
При изменении концентрации одного из компонентов равновесия система
реагирует так, чтобы уменьшить произведенное воздействие. Увеличение кон-
центрации одного из компонентов способствует изменению состояния равновесия
системы; последняя смещается в направлении, соответствующем уменьшению
концентрации этого компонента.
Если, например, добавить спирт, когда реакция этерификации уже достигла
104
состояния равновесия, то возникает новое состояние равновесия, при котором
часть введенного спирта под действием кислоты снова превращается в эфир и
воду. Если же удалять воду по мере ее образования, можно провести реакцию
этерификации целиком, так как в этом случае состояние равновесия не будет
устанавливаться в течение всего процесса.
2. РАВНОВЕСИЕ МЕЖДУ ФАЗАМИ И ДИАГРАММА СОСТОЯНИЯ
2.1. Правило фаз
Одно и то же вещество может при изменении температуры и
давления переходить в различные агрегатные состояния. Эти
переходы, осуществляемые без изменения химического состава,
называют фазовыми переходами. Если рассматрива-
ется гетерогенная система, в которой нет химического воздейст-
вия, а имеются лишь фазовые переходы, то при постоянстве тем-
пературы и давления существует так называемое фазовое
равновесие. Это равновесие характеризуется некоторым
числом фаз, компонентов и числом степеней термодинамической
свободы системы или числом степеней свободы.
Фазой называется такая однородная часть системы, обладающая одинако-
вым составом, физическими и химическими свойствами, которая может быть
удалена из системы чисто механическим путем. Эта часть системы отделена от
других частей видимыми поверхностями раздела.
Так, система лед + вода имеет две фазы, система соль + на-
сыщенный раствор + пар—три фазы и т. д. Очевидно, газооб-
разная фаза в системе может быть лишь одна, жидких может
быть несколько (две-три) и твердых — сколько угодно.
Компонентом называется такая химически однородная составная часть
системы, которая может быть выделена из системы и может существовать
вне ее.
Так, например, в водном растворе хлорида натрия компонен-
тами являются вода и хлорид натрия, но ионы натрия и хлора
не могут считаться компонентами, так как они не существуют
как отдельные вещества.
Число степеней свободы определяется как число параметров системы (тем-
пература, давление), которые могут быть произвольно изменены в некоторых
пределах без изменения числа и природы фаз в системе.
У системы, состоящей лишь из газа, можно менять два пара-
метра, ибо если известны два параметра, то третий определяется
из уравнения состояния; иными словами, можно выбрать два
параметра, и тогда система произвольно устанавливает третий.
Число степеней свободы определяется правилом фаз.
Правило фаз установлено Дж. Гиббсом (1876):
число степеней свободы равновесной термодинамической системы, на кото-
рую из внешних факторов влияют только температура и давление, равно числу
независимых компонентов системы минус число фаз плюс два:
С = К- Ф + 2,
105
вы ми диаграммами,
Р С|
Рис. 41. Диаграмма состояния воды:
ОВ—кривая сублимации; ОС—кри-
вая плавления; ОА — кривая давления
пара
где С — число степеней свободы; К — число компонентов; Ф —
число фаз; 2 — число независимых параметров, например темпе-
ратура и давление. Если один из параметров постоянен, то
цифра 2 заменяется цифрой 1.
По классификации систем их принято разделять по числу фаз
(однофазные, двухфазные и т. д.), по числу компонентов системы
(однокомпонентные, двухкомпонентные и т. д.) и числу степеней
свободы (инвариантные, моновариантные, дивариантные и т. д.).
В однокомпонентной гетерогенной системе фазы состоят из
одного вещества. Исходя из правила фаз, при К=1 и при усло-
вии одновременного изменения двух параметров (р и Т) число
степеней свободы системы равно
С=з —Ф.
Для трехфазной однокомпонентной системы число степеней
свободы С=1—3-|-2=О. Отрицательного значения С иметь не
может, значит, максимальное число равновесных фаз в одноком-
понентной системе не может быть больше трех. Для однофазной
однокомпонентной системы число степеней свободы будет равно
двум. Это значит, что здесь можно изменять без нарушения рав-
новесия два параметра — температуру и давление. Если же чис-
ло фаз станет равным двум, то число степеней свободы окажется
равным единице и произвольно можно будет изменять лишь один
параметр (либо температуру, либо давление) без нарушения
равновесия. Соответственно при наличии трех фаз число степеней
свободы станет равным нулю и тогда равновесие будет характе-
ризоваться определенными, жестко фиксированными значениями
давления и температуры.
Обычно рассматривают графическую зависимость состояния
системы от внешних условий. Такие графики называют ф а з о-
диаграммами состояния
или р — Г-диаграммами.
Рассмотрим диаграмму состо-
яния воды и проанализируем ее
с позиции правила фаз (рис. 41).
Поскольку вода является единст-
венным веществом, присутствую-
щим в системе, К=1. Вода может
существовать в трех фазах: твер-
дой (область ВОС), жидкой
(ДОС) и газообразной (ДОВ).
При этом число фаз, которые
могут существовать в равновесии,
зависит от температуры и давле-
ния. Кривая ОД представляет
собой кривую зависимости насы-
щенного пара воды от температу-
ры. Вдоль этой линии при всех
температурах, от точки О, вода
А
Т
106
и пар находятся в равновесии друг с другом. Кривая ОВ является
кривой сублимации льда. Во всех точках, лежащих на кривой ОВ,
имеет место равновесие лед—пар; поэтому ее можно назвать кри-
вой зависимости давления насыщенного пара над льдом от тем-
пературы. Выше этой кривой вода существует в виде льда, а ни-
же — в виде пара.
Кривая ОС описывает зависимость температуры плавления
льда от давления; лед и жидкая вода вдоль этой линии находят-
ся в равновесии. Наклон кривой ОС относительно оси ординат
указывает на то, что при увеличении давления температура плав-
ления льда понижается. Три кривые имеют обычную точку пере-
сечения О, известную под названием «тройной точки», в которой
в равновесии находятся три фазы.
Итак, фазовая диаграмма содержит области, кривые и трой:
ную точку. Чтобы показать соответствующие степени свободы,
рассмотрим табл. 16.
Таблица 16. Значения К, Ф, С в различных
частях плоскости диаграммы воды
Части плоскости
Области
Кривые
Тройная точка
1
1
1
Внутри области (С = 2) можно одновременно варьировать
без изменения числа фаз и температуру, и давление. На кривой
(С=1), описывая систему, можно изменить либо температуру,
либо давление.
Вдоль кривой ОВ твердое вещество — пар, так же, как и
вдоль кривой ОА жидкость — пар, при любом значении темпе-
ратуры давление пара может иметь только одно значение, а при
любом давлении пара для льда может существовать только
одна температура плавления (кривая ОС). В тройной точке не
существует степеней свободы; три фазы могут существовать в
равновесии только при одном определенном значении темпера-
туры и давления. Экспериментально было найдено, что в тройной
точке давление пара равно 6,1 кПа, а температура 273,16 К.
Кривая сублимации ОВ теоретически продолжается до абсо-
лютного нуля, а кривая давления пара ОА заканчивается при
критической температуре 607,46 К и давлении 19,5 мПа. В верх-
ней части кривой плавления ОС характерно появление различ-
ных новых кристаллических форм льда (см. гл. I), плотность
которых в отличие от обычного льда больше, чем у воды. Эти
новые фазы на рисунке не показаны.
107
2.2. Уравнение Клапейрона
Рассмотрим чистое вещество, находящееся в равновесии меж-
ду двумя различными фазами (например, равновесие твердое
тело — жидкость или жидкость — газ), что можно изобразить
следующим образом:
* Фаза А Фаза В
Изменение свободной энергии, соответствующее этому равнове-
сию, равно нулю, т. е. свободная энергия Ga 1 моль чистого ве-
щества в фазе А равна свободной энергии Gb 1 моль чистого
вещества в фазе В, когда температура и давление равны соот-
ветственно Т и р:
&Gpt=Cq—Ga = Q.
Если рассмотреть новое состояние равновесия, соответствую-
щее температуре Т' = Т -\-dT и давлению p' = p + dp, то
A Gfy'> =Gq — Ga = 0,
Предполагая Gb = Gb + cIGb и Ga = Ga + c1Ga, получим
GB 4~ d Gq — G\ — d Од = 0,
откуда
dGB = dGA.
В общем случае для обратимого термодинамического процесса
можно записать
dG = Vdp —SdT.
Введем выражение для 6Ga и 6 Gb
d6A= VAdp — SAd7\
dGB === V^Bdp — SBdT.
При равновесии между фазами имеем
dGA = dGB,
откуда
VAdp-SAdT==VBdp-SBdT
или
(VA-VB)dp = (SB-SA)dT.
Перепишем это в виде
dp _ SB — SA
dT ~~ Va-W
Величину нужно знать при данных температуре и давлении,
а именно при равновесии. Известно, что
108
отсюда
\ dp _ ЬН
dT ~ TAV ’
где ЛЯ— скрытая теплота изменения состояния; Т — темпера-
тура, при которой происходят изменения состояния и молярного
объема AV (ЛЯ и AV — функции температуры и давления). Это
уравнение известно под названием уравнения Клапей-
рона.
Следовательно, можно установить связь между изменением
энтальпии, сопровождающим изменение состояния, и тангенсом
угла наклона кривой равновесия p=f(T):
ЬН=ТЬУ&
а/
В частном случае, когда одна из фаз, например В, является
газовой фазой, т. е. для преобразования или возгонки, имеем
AV=VB-VA=Vra3-VA.
Объем 1 моль газа значительно больше объема 1 моль вещества
в жидком или твердом состоянии, тогда
vra3» vA.
Уравнение Клапейрона примет вид
dp _ A#
dT ~ 7Vra3 ’
Если газ предполагается идеальным, то для 1 моль чистого веще-
ства
Vra3 = RT/p,
откуда
dp _ Atf _ pAtf
dr ~ TRT~~RTtr'
Р
Разделив переменные, получим
_dp__ АЯ dT
или Р К*
,i AtfdT
dlnp=-^--
Для многих чистых веществ скрытая теплота парообразо-
вания или возгонки постоянна в большом температурном интер-
вале. Тогда можно проинтегрировать уравнение
109
Итак, получим уравнение, которое устанавливает связь между
давлением пара в равновесии с жидкостью, и изменением энталь-/
пий при температуре равновесия:
. АЯ 1 ,
lgp=~ ^303₽T+const
Контрольные вопросы
1. С ростом температуры равновесие реакции SO2 +{/2Q2 -+ SO3 смещается
влево. Сделайте вывод о знаке теплового эффекта реакции.
2. В каких рамках выполняется закон действующих масс?
3. Как экспериментально показать, что равновесие в условиях химической
реакции является динамическим?
4. Как связана константа равновесия с изменением стандартной свободной
энергии?
5. Как использовать закон действующих масс, если рассматриваются не газо-
образные, а жидкие реагенты и продукты?
6. Используя диаграмму состояния воды (см. рис. 41), опишите все кривые,
области и тройную точку. Какую роль играет тройная точка воды в определении
стандартного состояния?
7. Напишите уравнение, устанавливающее связь между давлением пара в
равновесии с жидкостью и изменением энтальпии при температуре равновесия.
Глава IV
ХИМИЧЕСКАЯ КИНЕТИКА
1. КИНЕТИКА И МЕХАНИЗМ РЕАКЦИЙ.
РЕАГЕНТЫ И ПРОДУКТЫ
Известно, что некоторые химические реакции проходят мгно-
венно, тогда как другие — крайне медленно, и что некоторые
реакции начинаются бурно, а затем замедляются, в то время как
другие, наоборот, начинаются медленно, а затем ускоряются.
Законы протекания химических реакций во времени -- предмет
кинетического исследования химических реакций.
Химические реакции редко протекают в одну стадию так, как
их обычно представляют в учебниках химии. В записи химиче-
ской реакции указывается только начальное и конечное состоя-
ния, эти уравнения являются символическим выражением мате-
риального баланса. В действительности реакция протекает через
ряд промежуточных стадий или, как говорят иначе, через ряд
элементарных процессов. В большинстве случаев идентифициро-
вать эти элементарные процессы сложно из-за больших трудно-
стей выявления промежуточных продуктов, через образование
которых протекает реакция.
Так, метан реагирует с хлором с образованием хлороформа и
соляной кислоты по схеме
СН4 + ЗС12-- СНС1з 4- ЗНС1
Химическая кинетика позволяет понять механизм этой реакции,
т. е. установить, как разрываются связи С1—С1 и С—Н, а затем
соединяются водород с хлором в Н—С1 и углерод с хлором
в СНС13.
Таким образом, можно сделать вывод, что химическая кине-
тика — один из возможных путей выявления элементарных про-
цессов и установления механизма реакции.
На первой стадии изучения реакции надо прежде всего опре-
делить скорость протекания изучаемой реакции. Эта область
кинетических исследований называется формальной кинетикой.
Она включает также изучение зависимости скорости реакции от
концентрации реагирующих веществ, температуры и давления.
Вещества, вступающие в процесс химического превращения,
называются исходными веществами или реаген-
тами.
Вещества, образующиеся в процессе химического превраще-
111
ния и не претерпевающие в ходе этого процесса дальнейших хи-
мических изменений, называются продуктами реакции.
Вещества, образующиеся в одних стадиях процесса химиче-/
ского превращения и расходующиеся в других стадиях этогб
же процесса, называются промежуточными вещест-
вами, а реакции с участием промежуточных веществ называ-
ются промежуточными реакциями.
Химическая реакция, протекающая в пределах одной фазы,
называется гомогенной химической реакцией.
Химическая реакция, протекающая на границе раздела фаз,
называется гетерогенной химической реакцией.
Примером гомогенной реакции может служить любая реакция
в растворе, а гетерогенной реакции — любая из реакций, идущих
на поверхности твердого катализатора (гетерогенная каталити-
ческая реакция).
Существуют различные способы наблюдения за развитием
химической реакции во времени. Выбор оптимального метода
анализа зависит прежде всего от природы реагирующих веществ
и физических свойств системы в целом. Если представляется
возможным приостановить реакцию в нужный момент времени,
то производят химический анализ системы в разные моменты
остановки реакции. Реакцию останавливают быстрым охлажде-
нием или внесением в такую среду, в которой тормозится проте-
кание исследуемых процессов. Для наблюдения за реакциями,
которые сопровождаются изменением концентрации кислот, ще-
лочей, галогенидов, применяют титрование.
Задача определения состава значительно упрощается при
использовании инструментальных методов, позволяющих прово-
дить измерение без изменения состояния системы. Основной
метод, применяемый для этого, — поглощение или рассеяние
электромагнитного излучения от радиочастотной до далекой
ультрафиолетовой области. Например, за бромированием бензола
можно легко следить, наблюдая спектрофотометрически (в види-
мой УФ- или ИК-областях) исчезновение брома и (или) бензола.
, Реакции в растворах, сопровождающиеся образованием мало-
растворимых газов — азота, кислорода, диоксида углерода,
исследуются по давлению выделяющегося газа.
При изучении кинетики реакций в растворах учитываются
следующие физические свойства растворов: окраска, вращение
плоскости поляризации, электрическая проводимость, коэффи-
циент преломления, изменение температуры замерзания. Доволь-
но часто продукты образуются в наномолярных (10-9 моль) ко-
личествах. В подобных исследованиях применяют чувствитель-
ные физические методы, такие, как газовая хроматография,
масс-спектрометрия и изотопные метки.
2. СКОРОСТЬ ХИМИЧЕСКОЙ РЕАКЦИИ
Скорость химической реакции определяется как изменение концентрации
веществ в единицу времени и в единице объема.
Такое определение, однако, не может быть однозначным, так
как в реакции могут принимать участие в качестве реагентов,
продуктов и промежуточных веществ несколько соединений.
Поэтому следует говорить не о скорости химического процесса
вообще, а о скорости превращения какого-либо компонента или
о скорости по некоторому компоненту.
Скорость химической реакции по некоторому компоненту
может быть легко определена, если процесс идет при постоянном
объеме и известной зависимости концентрации от времени для
этого компонента.
В химической кинетике широко используется графический
метод изображения функциональных зависимостей. Кривая, от-
ражающая изменение концентраций какого-либо вещества во
времени в ходе химического превращения, называется кине-
тической кривой.
Рис. 42. Кинетические кривые
для реагентов и продуктов
реакции
Рис. 43. Кинетическая кривая
для продуктов реакции
Получив кинетическую кривую для какого-либо компонента,
можно легко определить скорость его накопления или расходо-
вания графическим дифференцированием кинетической кривой.
На рис. 42 приведен простой вид кинетической кривой для исход-
ного вещества (реагента) А, концентрация которого убывает со
временем, и кинетической кривой продукта В реакции, концент-
рация которого растет до некоторого предельного значения.
Рассмотрим подробнее изменение концентрации продукта В
(рис. 43). Если в моменты времени t\ и /2 концентрации про-
дукта равны Ci и С2, то среднюю скорость vm в интервале вре-
мени t\ и можно выразить как
_ С2 — С| _ АС
Vm~ t2-h —ьГ
Если, например, в интервале времени, равном 5 мин, кон-
центрация продукта, образовавшегося в ходе реакции, изменяет-
113
ся от 0,15 до 0,25 кмоль/м3, то соответствующая средняя ско-
рость будет
от = 0,10/5 = 2-10 2 кмоль/(м3-мин).
Чтобы выражение для скорости было всегда положительным,
при убывающей концентрации реагента необходимо правую часть
уравнения записывать с отрицательным знаком:
ДС
Vm~~ М '
Если же скорость реакции определяется при увеличивающейся
концентрации продукта, то уравнение будет записано с положи-
тельным знаком:
Vm
ДС
м *
Таким образом, в общем случае выражение скорости должно
быть представлено в виде
дс
Vm~± м '
Средняя скорость реакции неодинакова в интервале времени t.
Величина скорости в каждый момент времени (мгновенная
скорость) определяется пределом, к которому стремится выра-
жение AC/Af при А/->0, т. е/ производной концентрации по вре-
мени AC/At. Следовательно, мгновенная скорость равна
/ДСх de
i>/ = hm(-I = ±
В каждый момент времени она равна тангенсу угла наклона
кривой в данной точке:
dC ,
-dT=tga-
3. ПОРЯДОК И КОНСТАНТА СКОРОСТИ РЕАКЦИИ
Скорость химической реакции зависит от целого ряда факто-
ров. При заданных внешних условиях (температура, давление,
реакционная среда) скорость является функцией концентраций
реагирующих веществ. Чтобы установить соотношение между
скоростью реакции- и концентрациями реагентов или продуктов,
производят точные экспериментальные измерения при постоянной
температуре. Например, чтобы найти выражение скорости реак-
ции образования КВг в зависимости от концентраций реагентов
СНзВг и КОН в реакции
СНзВг + кон СНзОН 4- КВг
экспериментально измеряют скорость этой реакции как функцию
концентрации каждого из реагентов.
114
Сначала измеряют концентрацию СНзВг, оставляя концент-
рацию КОН постоянной. Для этого создается очень большой
избыток КОН по отношению к другому реагенту, в результате
чего изменение концентрации КОН в ходе реакции становится
пренебрежимо малым. В этой серии измерений определяют
скорость образования КВг как функцию концентрации СНзВг.
Затем изменяют концентрацию КОН, оставляя на этот раз неиз-
менной концентрацию СНзВг, и измеряют скорость образования
КВг как функцию концентрации КОН.
Для реакции гидроксида калия с метилбромидом экспери-
ментально устанавливают, что при удвоенной концентрации КОН
скорость увеличивается вдвое, а при утроенной концентрации
КОН — втрое и т. д. Следовательно, скорость пропорциональна
концентрации КОН:
Если в выражении для скорости концентрация КОН дана в пер-
вой степени, считают, что кинетический порядок реакции равен 1
по отношению к КОН или что эта реакция первого порядка
относительно КОН.
Аналогично экспериментально показывают, что скорость этой
реакции пропорциональна также концентрации СНзВг; значит,
это реакция первого порядка по СНзВг:
^==-^^-=МснзВг].
Скорость реакции в целом пропорциональна [КОН] и
[СНзВг]; следовательно, можно записать
о = 6 [КОН] [СНзВг].
Из приведенного эксперимента следует, что реакция КОН
с СНзВг — это реакция первого порядка относительно КОН и
СНзВг; в целом же она является реакцией второго порядка,
или, иначе, порядок ее равен 2.
Общий порядок представляет собой сумму парциальных порядков реакций
по отношению к различным реагентам.
Рассмотрим теперь реакцию, в которой участвуют три моле-
кулы:
C124-2NO->2NOC1
Экспериментальное измерение скорости этой реакции показы-
вает, что она пропорциональна [С1г] и [NO]2:
и|==^юсц_=й1[СЬЬ
г,2 = .а!КО9.1 = *2[NQ]2;
откуда
v = k [С12] [NO]2.
115
Реакция имеет первый порядок по С1г и второй по NO, общий
порядок равен 3. Реакции третьего порядка редки.
Множитель k, показывающий, с какой скоростью идет хими-
ческий процесс при концентрации реагирующих веществ, равных
единице, называется константой скорости химиче-
ского процесса. Наряду со скоростью константа скорости
химического процесса является основной величиной в химической
кинетике. Чем больше скорость процесса, тем больше константа
скорости реакции. Для разных реакций эта величина может
изменяться в весьма широких пределах. Например, для обычных
реакций органического синтеза k ж 10 м3 • кмоль-1 • с-1, а для
реакции нейтрализации (одного из самых быстрых химических
процессов) fe^lO11 м3-кмоль-1 «с-1. Соответственно константы
скорости очень медленных химических реакций ничтожно малы.
На первый взгляд может показаться, что кинетический поря-
док реакции можно определить из стехиометрических коэффициен-
тов реакции. На самом деле это не так. В качестве примера
рассмотрим реакцию
2HI4-H2O2—2Н2О + 12
Экспериментальное изучение этой реакции показывает, что
она имеет первый порядок по HI, хотя в ней участвуют две
молекулы HI, и первый порядок по Н2О2; следовательно, общий
порядок реакции равен 2:
v=-^-=fe[HI] [Н2О2].
Рассмотрим еще один пример, иллюстрирующий несовпаде-
ние стехиометрических коэффициентов с порядком реакции. Это
реакция взаимодействия бромида третичного бутила с водой:
(СНз)зС - Вг + нон (СНз)зС - ОН + НВг
Скорость этой реакции как функция времени может быть легко
измерена титрованием образующейся бромоводородной кислоты.
Скорость образования НВг оказывается пропорциональной лишь
концентрации бромпроизводного:
d [НВг] , .
v =— dt ' = k I (СНз) зС—В г].
Экспериментально показано, что эта реакция имеет первый
порядок по (СНз)зСВг и нулевой порядок по Н2О, поскольку
опытным путем установлено, что скорость реакции не зависит от
концентрации этого реагента. Общий порядок реакции равен 1,
хотя из уравнения реакции видно, что в ней участвуют две мо-
лекулы.
Таким образом, если зависимость скорости реакции от кон-
центрации реагирующих веществ записывается в виде
и = ^[А1]Л1 [А2]Л2...[А1] \
то величины гц принято называть порядком реакции по
веществам А/.
116
Величины щ, «2, .. могут быть равны нулю, иметь дробные
значения, но часто они оказываются целыми числами.
Сумму порядков реакции по всем реагирующим веществам называют по-
рядком реакции.
Важно подчеркнуть, что порядок реакции определяется толь-
ко из экспериментальных данных и что кинетический порядок
может не зависеть от коэффициентов уравнения, описывающего
эту реакцию.
Иначе говоря, стехиометрическое уравнение реакции, устанав-
ливающее пропорции реагентов и продуктов, не определяет
характера протекания этой реакции во времени. Это является
причиной, по которой экспериментально найденный порядок
реакции не всегда согласуется с уравнением, описывающим
реакцию.
Все реакции подразделяются на две группы: элементар-
ные и сложные.
4. ЭЛЕМЕНТАРНЫЕ (ПРОСТЫЕ) РЕАКЦИИ. МОЛЕКУЛЯРНОСТЬ
РЕАКЦИИ
Элементарные реакции классифицируют по числу молекул,
которые, согласно экспериментально установленному механизму
(а не по написанному уравнению), участвуют в одном элемен-
тарном химическом акте.
Мономолекулярными (одномолекулярными) назы-
ваются реакции, в которых происходит химическое превращение
одной молекулы (изомеризация молекулы, ее диссоциация на
несколько молекул, содержащих меньшее число атомов, и т. д.).
Бимолекулярные реакции — это такие реакции,
элементарный акт которых осуществляется при столкновении
двух молекул (различных или одинаковых).
В тримолекулярных реакциях элементарный акт
осуществляется при столкновении трех молекул.
В качестве примера рассмотрим три элементарные реакции:
(4.1)
СНзВг 4- КОН -> СНзОН + КВг (4.2)
О2 + NO + NO -> 2NO2 (4.3)
Реакция (4.1) происходит при распаде одной активирован-
ной молекулы 12; это мономолекулярная реакция. Реакция
(4.2) осуществляется при столкновении молекулы СН3Вг с моле-
кулой КОН. При этом элементарный процесс включает столкно-
вение двух молекул; это биомолекулярная реакция. Реакция
(4.3) происходит при тройном столкновении молекулы кислоро-
да с двумя молекулами NO. Эта элементарная реакция, в ко-
торой участвуют три частицы, тримолекулярна. Таким образом,
для элементарной реакции (и только элементарной реакции)
молекулярность определяется из стехиометрического уравнения.
117
Для элементарной реакции мажно теоретически рассчитать ско-
рость этой реакции и показать идентичность величин порядка и
молекулярности, т. е.
кинетический порядок элементарной реакции равен ее молекулярности.
Поэтому реакция (4.1) будет реакцией первого порядка, реак-
ция (4.2) — второго порядка, а реакция (4.3) — третьего по-
рядка.
5. СЛОЖНЫЕ РЕАКЦИИ
Сложными реакциями называют реакции, протекающие более
чем в одну элементарную стадию. Рассмотрим простейшую
сложную реакцию, уравнение которой записывается следующим
образом:
2HI + Н2О2 -> 12 + 2Н2О
Согласно уравнению в этой реакции участвуют три молекулы,
но, однако, проведенный эксперимент показывает, что эта реак-
ция второго порядка.
Тщательное изучение механизма позволило установить, что
она протекает в две последовательные стадии:
- HI + Н2О2 НЮ + Н2О (медленная стадия) (4.4)
НЮ 4-HI м- 124-Н2О (быстрая стадия) (4.5)
2Н14-Н2О2-^ 124-2Н2О.
Скорость стадии (4.5) настолько велика, что измерить ее не
удается. Поэтому эта элементарная реакция не вносит свой
вклад в величину общей скорости сложной реакции. В то же
самое время экспериментально измеренный порядок медленной
элементарной реакции (4.4) оказывается равным 2. Именно эта
медленная бимолекулярная стадия характеризует скорость слож-
ной реакции.
В общем, когда порядок реакции, найденный эксперименталь-
но, не соответствует стехиометрическому уравнению реакции,
то эта реакция не является элементарным процессом, а проте-
кает по сложному механизму. В частности, процесс может
включать медленную стадию, молекулярность которой равна ус-
тановленному на опыте общему порядку.
Если экспериментально определенный порядок реакции соот-
ветствует стехиометрическому уравнению реакции, можно счи-
тать ее элементарным процессом. Таким образом, элементар-
ная бимолекулярная реакция всегда имеет второй порядок, но
в противоположность этому реакция второго порядка, проис-
ходящая между двумя молекулами, не обязательно должна
быть элементарной бимолекулярной; она может быть сложной
реакцией, порядок которой будет определяться самой медлен-
ной элементарной стадией, называемой поэтому лимитирую-
щей стадией.
118
Итак, если реакция протекает в одну стадию, то порядок
равен молекулярности; если реакция протекает в несколько
стадий, то порядок определяется скоростью медленного эле-
ментарного процесса и отражает его молекулярность. Другими
словами, кинетический порядок каждой из стадий реакции ра-
вен молекулярности этой стадии.
Последовательные (консекутивные) химические реакции. Ранее был рас-
смотрен частный случай сложной реакции, когда при установлении порядка
реакции одна из двух слагающих ее элементарных реакций может не учиты-
ваться из-за ее большой скорости. Такие реакции довольно распространены,
однако в общем случае пренебрегать скоростью одной из элементарных реак-
ций нельзя.
Рассмотрим реакцию превращения молекул А в молекулы В с образова-
нием некоторой промежуточной частицы Р:
Ap*V В
(4.6)
Р — называется промежуточной частицей, пртому что она может быть как
устойчивой молекулой, так и ионом или стабильным радикалом. Цепочка пре-
вращений, описанных уравнением (4.6), называется последовательной, а вся ре-
акция — последовательной химической реакцией.
В рассматриваемой последовательности реакции промежуточная частица Р
образуется из реагента А по реакции первого порядка с некоторой констан-
той скорости k[. Одновременно с этим Р является реагентом второй реакции
образования по первому порядку продукта В с некоторой константой скорости k2.
Исходя из сказанного, кинетические уравнения можно записать в виде
[А!.
d Р
^-=МА] -МР].
[Р].
(4.7)
(4.8)
(4-9).
Для получения уравнений кинетических кривых нужно решить систему
дифференциальных уравнений, что довольно сложно, поэтому ограничимся ка-
чественным анализом вида кинетических кривых последовательной химической
реакции (рис. 44).
Так как соотношение (4.7) представляет собой уравнение для реакции
первого порядка, то кинетическая кривая, соответствующая расходу вещест-
ва А, характеризуется монотонно убывающей кривой А (рис. 44). В начальный
момент времени концентрация промежуточных частиц равна нулю, но в процес-
се превращения А концентрация вещества Р бу-
дет расти до тех пор, пока два члена в правой
части уравнения (4.8) будут равны между собой,
а производная d[P]/d/ окажется равной нулю.
Поскольку концентрация вещества А будет про-
должать уменьшаться и второй член по абсо-
лютному значению станет больше первого, про-
изводная d [Р] /d/ в этом случае будет отрица-
тельной и концентрация вещества Р начнет
убывать. Таким образом, кинетическая кривая Р
проходит через максимум. Концентрация В бу-
дет непрерывно возрастать, причем по мере
накопления Р и скорость образования В воз-
растает.
Параллельные реакции. Реакции называют-
ся параллельными, если в каждой из них при-
Рис. 44. Кинетические кривые
последовательной химической
нимает участие одно и то же исходное вещество реакции
НО
(реагент). Данный реагент или реагенты одновременно по различным направле-
ниям превращаются в разные продукты. В общем виде простейшая параллельная
реакция имеет вид
В качестве примера можно привести реакцию разложения, которая при
умеренных температурах может совершаться парараллельно в двух направле-
ниях:
BKCIOq—Г2КС1 4" ЗО2
|_>ЗКСЮ4 + КС1
Наиболее часто параллельные реакции встречаются в органической химии.
Так, например, при нитровании фенола азотной кислоты нитрогруппа может
занимать, или орто-, или пара-положение.
Если в данных условиях термодинамически возможны два или три направ-
ления химической реакции, то преобладание того или другого из них и относи-
тельные количества получаемых продуктов реакции определяются всецело соот-
ношением скоростей этих реакций, а не соотношением термодинамической
устойчивости конечных продуктов.
Если реакции значительно различаются по скорости, то обычно реакцию,
обладающую большей скоростью, называют главной (основной), а остальные
побочными. При не слишком большом различии в скоростях реакций главной
называют реакцию, приводящую к получению нужного продукта, хотя бы она
и обладала меньшей скоростью.
Сопряженные реакции. Так называются две реакции, одна из которых, буду-
чи самопроизвольной, вызывает (индуцирует) протекание в этой же системе
второй химической реакции, неосуществимой в отсутствие первой.
Так, например, иодоводородная кислота не окисляется непосредственно
хромовой кислотой, но если в систему ввести FeO, который может непосредст-
венно взаимодействовать с хромовой смесью, то одновременно с окислением
FeO начинается окисление HI.
Явление, лежащее в основе протекания сопряженных реакций, называется
химической индукцией. Следует подчеркнуть, что обе сопряженные
реакции должны быть сложными, так как, по определению, элементарная
реакция не может быть индуцирована другой реакцией.
Сопряженные реакции протекают через общие активные промежуточные ве-
щества, которые ответственны за явление химической индукции.
Например, бензол в водном растворе не окисляется пероксидом водорода,
но при добавлении соли Fe (II) происходит превращение его в фенол и дифе-
нил. Механизм реакции следующий. Сначала образуются свободные радикалы
по реакции
Fe2+ + Н2О2 Fe3+ + ОН + ОН~
которые реагируют с Fe2+
Fe2+ + ОН Fe3+ + ОН~
а также с бензолом е
С6Н6 + ОН С6Нб* + Н2О
Образующиеся радикалы рекомбинируют:
С6Н5 + ОНС6Н5ОН
2СбНб +С6Н5—С6Н5
Таким образом, обе реакции окисления СеНб и Fe2+ протекают с участием
общего промежуточного свободного радикала (5н.
120
Обратимые (двусторонние) реакции. Очень часто химические
реакции являются двусторонними или, как часто говорят,
обратимыми. Понятие обратимая реакция в из-
ложенном выше смысле следует отличать от термодинамического
понятия обратимый процесс (см. гл. II). Последний характери-
зуется бесконечно малым различием скоростей прямого и обрат-
ного процессов и, следовательно, бесконечно малой скоростью
результирующего процесса и бесконечно малым отклонением си-
стемы от положения равновесия.
Двусторонняя химическая реакция, естественно, обратима в
термодинамическом смысле только в непосредственной близости
к состоянию химического равновесия. В состоянии же, далеком
от равновесия, когда скорости прямого и обратного процессов
существенно различны и суммарная (результирующая) скорость
реакции значительно отличается от нуля, она термодинамически
необратима. Область применения понятия двусторонняя реакция
шире, чем термодинамическое понятие обратимая реакция. По-
этому для реально протекающих реакций следовало бы при-
держиваться первого термина. Однако термин обратимая реак-
ция в широком, не термодинамическом смысле укрепился и при-
ходится использовать его.
Рассмотрим обратимую (двустороннюю) реакцию
А + В^С + D
&2
Скорость уменьшения концентрации вещества А при протека-
нии прямой реакции определяется уравнением
-^L = MA] [В] (4.10)
а скорость возрастания концентрации вещества А при протека-
нии обратной реакции — уравнением
[С] [D] (4.11)
Если в начальном состоянии системы в ней содержатся
только вещества А и В, то скорость прямой реакции уменьша-
ется по мере течения ее в соответствии с уменьшением этих
концентраций. Таким образом, зависимость этой скорости от вре-
мени представляется кривой 1 (рис. 45); если бы не было обрат-
ной реакции, эта кривая достигла бы оси абсцисс при израсходо-
вании одного из исходных веществ.
Кривая 2, относящаяся к скорости обратной реакции, начи-
нается в начале координат (так как по условию в начальный
момент [С] = [D] = 0) и повышается по мере течения прямой
реакции с увеличением этих концентраций.
Общая скорость реакции в любой данный момент равна разности скоростей
прямой и обратной реакций:
= 1Д1 IB] — *2 [С] [DJ.
121
Рис. 45. Кинетические кривые
прямой и обратной реакций
При дальнейшем течении реакции кри-
вые 1 и 2 должны где-то пересечься,
т. е. должно быть достигнуто такое
состояние, при котором скорости пря-
мой vi и обратной ^2 реакций станут
равными. После достижения этого кон-
центрации каждого из компонентов
реакции будут постоянными. Таким
образом, в системе будет достигнуто
состояние равновесия, когда скорости
прямой и обратной реакций будут рав-
ны между собой по абсолютному зна-
чению. Следовательно, должны быть
равны и правые части уравнений (4.10) и (4.11):
[А] [В] = [С] [D],
откуда
_ [С] [D]
[А] [В] '
Правая часть этого равенства представляет собой константу
равновесия К данной реакции и, следовательно,
Ас =
т. е. константа равновесия равна отношению констант скоростей
прямой и обратной реакций.
Этот вывод четко показывает динамический характер равно-
весия в химических реакциях.
Гетерогенные реакции. Когда реакция совершается между
веществами, находящимися в различных фазах, то законы кине-
тики гомогенных реакций уже не могут быть применены.
В гетерогенных реакциях роль промежуточных продуктов
обычно играют молекулы, связанные с поверхностью химическими
силами, т. е. химически адсорбированные на поверхности. В хи-
мическом механизме гетерогенной реакции существенны обычно
те или иные адсорбционные стадии (см. гл. XI). Поэтому ге-
терогенный химический процесс можно подразделить на следую-
щие стадии:
1) диффузия частиц реагентов к реакционной зоне, находя-
щейся на поверхности раздела фаз;
2) активированная адсорбция частиц реагентов в реакцион-
ной поверхностной зоне;
3) химическая реакция в этой зоне;
4) десорбция продуктов реакции, образовавшихся в реак-
ционной зоне;
5) диффузия продуктов реакции из реакционной зоны.
Стадии 1 и 5 называются диффузионными, а стадии
2, 3, 4 — кинетическими.
122
При низкой температуре кинетические стадии, как правило, определяют
общую скорость реакции, так как они в этих условиях протекают медлен-
нее процесса диффузии (кинетическая область гетерогенного процесса). При
этом возможен любой порядок реакции. Скорость гетерогенного процесса
сильно зависит от температуры и от величины поверхности раздела фаз.
Скорость процесса не зависит от разности концентраций, плотностей и давле-
ний в различных частях системы. Иначе говоря,, скорость гетерогенного
процесса не зависит от факторов, влияющих на диффузию.
При высокой температуре скорость химической реакции возрастает быст-
рее, чем скорость диффузии, и поэтому суммарная скорость процесса будет
определяться диффузионной стадией (диффузионная область гетерогенного
процесса). При этом гетерогенная реакция характеризуется первым порядком
реакции, слабой зависимостью скорости процесса от температуры и незначи-
тельным влиянием на скорость процесса величины поверхности раздела фаз.
Скорость во многом начинает определяться факторами, влияющими на диффу-
зию.
Универсального определения скорости для гетерогенного про-
цесса не существует. Так как суммарная скорость определяет-
ся как чисто диффузионной, так и кинетической стадией, то при
оценке скорости гетерогенного процесса необходимо точно знать,
в какой области — диффузионной или кинетической — соверша-
ется данная реакция.
Цепные реакции. Они отличаются от обычных взаимосвязан-
ностью элементарных актов, когда каждый осуществляемый в
системе акт генерирует один или несколько других элементар-
ных актов. Цепная реакция протекает благодаря наличию частиц
с повышенной химической активностью.
В создании современного учения о цепных реакциях основ-
ная роль принадлежит работам лауреатов Нобелевской премии
по химии 1956 г. — Н. Н. Семенова и С. Хиншельвуда.
Цепные реакции являются очень распространенными. По цеп-
ному механизму, например, могут совершаться многие реакции
окисления углеводородов. Процессы сгорания горючего в цилинд-
рах двигателей внутреннего сгорания во многом обусловлены
цепным механизмом. Процессы полимеризации также большей
частью протекают по типу цепных реакций. Значительную роль
эти реакции играют в биологических процессах.
Для всех цепных реакций характерны три стадии: 1) за-
рождение (возникновение) цепи; 2) развитие цепи; 3) обрыв
цепи.
Зарождение цепи является первичной реакцией, которая свя-
зана с получением в системе или введением в нее химически
активных частиц или, как их еще называют, активных центров.
Зарождение цепи может происходить под воздействием света,
радиоактивного излучения или высокой температуры, а также
осуществляться благодаря введению в систему атомов. Напри-
мер, если в смесь Нг и С1г ввести пары натрия, то они будут при-
чиной возникновения активных центров. Возникновение цепей мо-
жет происходить вследствие самого акта химической реакции,
при которой образуются радикалы.
В последних своих работах Н. Н. Семенов и его школа сде-
лали открытие (1976), что генерирование активных частиц воз-
123
можно и в молекулярных реакциях при условии, что реагирую-
щие молекулы обладают избыточной энергией.
Цепи, возникающие при помощи свободных атомов и ради-
калов, называются химическими или радикальными. Примером
может i служить фотохимическая реакция образования хлоро-
водорода. Первичными активными центрами данной цепной реак-
ции являются атомы хлора, возникающие в результате расщеп-
ления молекул хлора под воздействием квантов света, обладаю-
щих высокой энергией. Атомы хлора вступают в реакцию с
молекулами водорода; возникающие атомы водорода, в свою оче-
редь, реагируют с молекулами хлора, образуя атомы хлор^.
и т. д.
Cl2 -|- hv -> 2С1 * (зарождение цепи)
Н2 + Cl * НС1 + Н* i (развитие цепи)
Cl2 + H* +HC1 + CI* J
СГ + СГ ^С12 (обрыв цепи)
Таким образом, реакция хлора с водородом осуществляется
при помощи двух активных центров: атомов хлора и водорода.
Число молекул продукта реакции, приходящееся на одну ак-
тивную молекулу, называют длиной цепи.
Обрыв цепи может происходить не только при воздействии
свободного радикала с другим свободным радикалом в так на-
зываемом процессе рекомбинации радикалов. Очень часты взаи-
модействия свободного радикала с материалом стенки сосуда,
где протекает реакция, что также приводит к обрыву цепи.
Если в каждом звене цепи на каждый исчезнувший активный
центр приходится не более одного вновь возникающего, то такие
реакции называются простыми цепными реакция-
м и. Если в одном звене цепи на один исчезнувший актив-
ный центр в среднем возникает больше одного нового центра,
то такие цепные реакции называются разветвленными.
Таким образом, в разветвленных цепных реакциях один ради-
кал вызывает появление более чем одного нового радикала, так
что число радикалов по мере протекания реакции возрастает
в геометрической прогрессии.
В качестве примера рассмотрим реакцию взаимодействия Н2
и О2 при 923 К. Стадией инициирования реакции является
процесс
Н2 + О2->Н2О + Ое (4.12)
Генерированные атомы кислорода приводят к следующим трем
реакциям:
О* + Н2-> ОН* + Н* (4.13)
Н* + О2 + ОН* + О* (4.14)
ОН* +Н2-^Н2О + Н* (4.15)
Получается разветвленная цепь, поскольку на каждый атом
О, возникающий в реакции (4.12), образуется избыточный
124
атом Н* в результате протекания
реакций (4.13) — (4.15). Эти реак-
ции дают снова атом кислорода.
Если простую цепную реакцию
условно можно представить в виде
зигзагообразной линии (рис. 46),
каждый прямоугольный участок
которой обозначает одно звено це-
пи, а число таких участков — чис-
ло звеньев, т. е. длину цепи, то
Рис. 46. Схемы простой (а), раз-
ветвленной ,(б) и сплошь разветв-
ленной цепной реакции (в)
разветвленную цепную реакцию можно представить разветвлен-
ной зигзагообразной линией. В предельном случае разветвление
осуществляется на каждом звене цепи. Такие цепи называют
сплошь разветвленными.
Скорость простой цепной реакции с течением времени стремится к некото-
рому постоянному значению
аУо
У ==-7----- ,
1 — еа
где а — вероятность продолжения цепи; е — среднее число активных центров,
возникающих в одном звене цепи; и0 — скорость создания активных центррв,
т. е. число активных центров, появляющихся в 1 с в 1 м3.
Для простой реакции 8=1. Следовательно, для простой цепной реакции
еа < 1.
Для разветвленной цепной реакции еа> 1, и скорость таких реакций вы-
ражается уравнением
где <р связано с числом актов взаимодействия одной частицы, происходящих
в 1 с, т. е. частоты соответствующих процессов.
Через сравнительно короткое время скорость разветвленной реакции
может достичь такого значения, что реакция приобретает характер взрыва.
Однако если значение <р не будет положительным на протяжении всей реак-
ции и изменит знак, то взрыв не произойдет.
6. КОЛИЧЕСТВЕННЫЕ СООТНОШЕНИЯ МЕЖДУ СКОРОСТЬЮ
НЕКОТОРЫХ РЕАКЦИЙ И КОНЦЕНТРАЦИЯМИ РЕАГЕНТОВ
Реакции первого порядка. В реакции первого порядка ско-
рость пропорциональна концентрации одного реагента (концент-
рации выражены в кмоль/м3). Для такой реакции, как
А -> Продукты
выражение скорости как функции концентрации имеет вид
d [Д] А> FA1
v =----h-=*(A]-
Концентрация реагента А в момент времени t = 0 равна а
кмоль/м , в момент времени t х кмоль/м3 реагента А превраща-
ется В| продукты. Следовательно, остается (а — х) кмоль/м3
реагента А в предположении, что объем реакционной среды не
меняется.
125
Общее выражение скорости реакции первого порядка
d [А] А ГЛ1
v=---------------------£-*-= fe lAJ
at
переходит в выражение
d (а — х)
v=------di—
или
>=*(a-x).
Разделив переменные, получим
Запишем интеграл, взяв пределы концентрации а и х в интервале
времени 0 и t:
Проинтегрировав, получим
In (a — x)lo = Ifcflo
или
— In (а — х) + In а = kt,
что иначе можно записать так:
In—-— = kt. (4.16)
а — х
Воспользовавшись десятичными логарифмами, перепишем это
выражение в виде
а ___ kt
ё а - х ~ ЛЗОЗ-’
ИЛИ
ki
lg(e_x)=_—_+lga,
где k — константа скорости реакции первого порядка, с-1.
Если построить график зависимости 1g(а — х) от времени, то
получим прямую, имеющую отрицательный наклон с тангенсом
угла, равным ~k/2,303; эта прямая отсекает на оси ординат от-
резок, равный Iga (рис. 47).
Если при экспериментальном исследовании реакции неизвест-
ного порядка график 1g (а — = получается в виде пря-
мой, то отсюда следует, что реакция имеет первый порядок.
Для реакции первого порядка характерной величиной являет-
•126
ся время полупревращения /»/2
реакции, т. е. время, которое необходимо,
чтобы концентрация реагирующих веществ
уменьшилась вдвое по сравнению с исход-
ным значением. Приняв а—х = а/2, из
уравнения (4.16) получим:
In 2 = kt^ ti'^ = In 2/ky k — 0,6932//y2.
Время полупревращения реакции перво-
Рис. 47. График зави-
симости 1g (а—х) от
времени для реакции
го порядка не зависит от начальной кон- первого порядка
центрации вещества. Для уменьшения кон-
центрации от 1 до 0,5 кмоль/м3 потребуется
ровно столько же времени, сколько для 0,5 моль до 0,25, от 0,25
до 0,125 моль. К реакциям первого порядка относятся многие
реакции радиоактивного распада (рис. 48). .
Реакции второго порядка. Для такой реакции, как
А + В Продукты
выражение скорости имеет вид
v^k [А] [В].
Если концентрации реагентов в момент времени t = 0 соот-
ветственно равны а и & и если в момент времени' / х кмоль/м3
реагента А взаимодействует с х кмоль/м3 реагента В, то оста-
ется (а — х) кмоль/м3 реагента А и (Ь — х) кмоль/м3 реаген-
та В.
Скорость реакции пропорциональна концентрациям двух ре-
агентов в каждый момент времени, и выражение
и = = А 1А| [В1
превратится в выражение
Рис. 48. Зависимость изменения
концентрации реагентов от времени
для реакции первого порядка
-jp = k (а — х) (6 — х).
Если начальные концентрации а
и b равны, то
-^-=fe(a —х)2.
Разделив переменные, получим
Td* 5 =fedt.
V (а —х?
.127
Интеграл в тех же пределах, что и для реакции первого порядка,
запишется
Проинтегрировав, получим
I——|* = |Ш
I а — х ' о
1 1
что можно записать в виде
, 1 X t 1 X
kt = —7---г- ИЛИ k = — 7----С- .
а(а — х) t а{а — х)
Как видно из уравнения, константа скорости реакции второго
порядка выражается в м3/(кмоль-с).
Если построить график зависимости от времени величины,
обратной концентрациям, т. е.
то получим прямую, отсекающую на оси ординат отрезок, рав-
ный 1/а, и имеющую положительный наклон с тангенсом угла,
равным константе k (рис. 49).
Если в качестве реагентов участвуют два разных вещества с различными,
начальными концентрациями, то уравнение, определяющее скорость, будет
следующим:
v = = k (а — х) (Ь — х).
После разделения переменных получим
(а — х)(Ь — х)
а интегрированием
Рис. 49. Зависимость 1/(а—х)
от времени для реакции второго
порядка
—L- U--------=
а — 6 “ \ а — х b — х / j
приведем к выражению
а b 11п (а — х) — In (b — х)1о = Ifcdo
или
а]_ b [In (а - х) - In (b — х) -
— Ina + In 6] = kt,
что можно переписать в виде
128
или, используя десятичные логарифмы,
1 b (а — х) kt
а — b g а (Ь — х) 2,303
Отсюда
2,303 Ь(а-х)
t(a-b) g a(b-x)
. b(a — x)
Рис. 50. Зависимость 1g—--------*-
a(b—x)
от времени для общего случая
реакции второго порядка
от
г , , ь (а — х)
График зависимости 1g—--------г
а (Ь — х)
через начало координат (рис. 50), с тангенсом угла наклона, равным —— •
Реакции нулевого порядка. Скорость этой реакции не зависит от концентра-
ции исходных веществ. Рассмотрим схему
А Продукт реакции
времени дает прямую, проходящую
k (а — Ь)
Имеем
y = (4.17)
где k — константа скорости реакции нулевого порядка.
Преобразуя уравнение (4.17), получим
d [А] = — k&t.
Интегрирование от t = 0 до t = t дает
[aj t
$ d [А] = [A] - [Ao] = - J Ш=- kt,
[Ao] 0
или
[A] = [Ao] - kt,
где [Ao] и [A] — концентрации при t = 0 и t = t соответственно.
График зависимости [Ao] — [А] от t представляет собой прямую линию,
тангенс угла наклона которой равен k.
Реакций нулевого порядка очень мало, большая их часть является гетеро-
генными реакциями, протекающими на поверхности металла.
7. ВЛИЯНИЕ ТЕМПЕРАТУРЫ НА СКОРОСТЬ РЕАКЦИИ.
ЭНЕРГИЯ АКТИВАЦИИ
Скорости всех одностадийных реакций или элементарных стадий увеличи-
ваются обычно вдвое при повышении температуры на каждые 10°.
Таким образом, реакция, которая при температуре Т, напри-
мер, длится 1 ч, при (Т — 100) пройдет приблизительно
1-210 ч, т. е. 1000 ч, или 40 дней. И наоборот, при (Г + 100) вре-
мя прохождения реакции составляет примерно 1-2“10 ч, т. е.
3,6 с (рис. 51, а).
Скорости сложных реакций зависят от температуры по-раз-
129
5—696
ному. Большинство реакций ускоряется при повышении темпера-
туры, но некоторые замедляются. Кроме того, во многих слу-
чаях зависимость скорости от температуры не монотонна
(рис. 51, б, в).
Снижение скоростей ферментативных реакций при высоких
температурах (рис. 51, в) объясняется просто: выше температу-
ры, соответствующей максимуму на кривой, происходит деструк-
ция фермента и кажущаяся скорость реакции при этом умень-
шается. Снижение скорости, показанное на рис. 51,6, происхо-
дит из-за существования обратной реакции, скорость которой
повышается с увеличением температуры быстрее, чем скорость
прямой реакции. Таким образом, кажущееся уменьшение скоро-
сти с ростом температуры на рис. 51,6 и 51, в происходит в
обоих случаях вследствие существования конкурирующих реак-
ций.
а) S) S)
Рис. 51. Зависимость скорости различных типов реакции от температуры:
а — обычная одностадийная реакция; б — обратимая реакция; в — ферментативная
реакция
В отличие от химических процессов зависимость скорости
биологических процессов от температуры более сложна, так как
течение биологических процессов осуществляется в довольно
узких температурных интервалах. За пределами этих интервалов
жизненные процессы останавливаются и наступает смерть.
Этот температурный интервал обычно заключен в пределах
273...323 К, хотя для некоторых представителей животного и
растительного мира имеется достаточно исключений из этого пра-
вила.
При рассмотрении температурной зависимости жизненной кривой можно
установить три температурные точки: температурный минимум, оптимум и мак-
симум. В границах температуры от минимума до оптимума интенсивность
жизненного процесса растет, и здесь в основном наблюдается приблизитель-
но такая же зависимость, как и в обычном химическом процессе. Темпера-
турный оптимум у животных колеблется в пределах 308...315 К (у растений
он даже выше). Дальнейшее повышение температуры приводит к быстрому
снижению процесса, и по достижении максимальной температуры наступает
гибель, что связано с денатурацией белка и инактивацией ферментов.
Энергия активации. Теория Аррениуса. Для осуществления
химической реакции необходимо, чтобы молекулы реагентов во-
130
шли в контакт. Химическое взаимодействие часто совершается
при столкновении двух или более частиц, но не любое соударе-
ние частиц ведет к химическому превращению. Согласно тео-
рии Аррениуса соударения будут эффективны (т. е. будут при-
водить к реакции) только тогда, когда встречающиеся моле-
кулы обладают некоторым избытком энергии по сравнению со
средней энергией молекул в рассматриваемой системе при дан-
ной температуре. Молекулы, несущие в себе эту избыточную
энергию, называются активными, а сам избыток энергии —
энергией активации.
Энергия активации равна разности между наименьшим избыточным запасом
энергии, необходимым для взаимодействия, и средней энергией молекул.
Чем выше в системе активных молекул, тем выше скорость
реакции. Чем выше энергия’ активации, тем меньшее число моле-
кул будет обладать этим избыточным запасом энергии и тем ни-
же будет скорость реакции.
Согласно распределению молекул по их скоростям при любой
температуре имеется некоторое число молекул, обладающих за-
пасом энергии, необходимым для реакции. С повышением тем-
пературы на 10° число таких молекул возрастает в среднем в
2—4 раза.
В химических реакциях происходит разрыв одних связей и
образование других, однако измерение энергии активации пока-
зывает, что она всегда меньше энергии связей. Для протека-
ния реакции нет необходимости полностью разрывать связи
атомов в молекуле, нужно только их несколько ослабить. Такое
ослабление связей происходит при образовании так называемого
переходного состояния или активированного комп-
лекса, дальнейшее превращение которого приводит к продук-
там реакции.
Энергия активации есть разность энергии активированного комплекса и сред-
ней энергии исходных молекул.
Константа скорости реакции связана с энергией активации
сложной зависимостью, описанной уравнением Аррениуса:
k = Кое RT ,
где k — константа скорости реакции; Ко — постоянный множи-
тель (предэкспоненциальный множитель); Еа — энергия актива-
ции; R — универсальная газовая постоянная; Т — абсолютная
температура.
Чтобы понять, что такое энергия активации, рассмотрим хи-
мическую реакцию в виде диаграммы энергии. Если по оси орди-
нат отложить значение энергии, а по оси абсцисс — координа-
ту реакции, то полученная кривая отразит энергетическую схему
реакции (рис. 52). Диаграмма характеризует процесс взаимодей-
ствия реагентов А и В, обладающих некоторой средней энер-
131
5*
Рис. 52. Энергетическая диаграмма реак-
ции
гией с образованием более
устойчивых продуктов С и D,
средняя энергия которых
меньше средней энергии ре-
агентов А и В. Средняя
энергия процессов ниже
энергии реагентов, т. е. энер-
гия выделяется в процессе
реакции и, значит, реакция
экзотермична (АЯ < 0).
Энергия активации изобра-
жена в виде так называемо-
го энергетического барьера. Столновение молекул реагентов А и
В будет эффективным (т. е. при этом столкновении образуются
продукты C + D), если молекулы реагентов достигнут вершины
энергетического барьера, т. е. уровня активированного комплек-
са, которые благодаря большому запасу энергии является не-
устойчивым. Он не может быть отождествлен с химическим сое-
динением и к нему неприменимы такие понятия, как межатомные
расстояния, валентные углы и т. д. Находясь на высоком энерге-
тическом уровне очень малый промежуток времени, комплекс
распадается, образуя продукты С и D.
Если рассмотреть обратную реакцию (см. рис. 52), т. е. реак-
цию молекул С + D, в результате которой образуются молеку-
лы А + В, то тогда энергия активации Еа будет большей, по-
скольку средний уровень энергии молекул С + D ниже, чем
молекул А 4- В, но активированный комплекс будет одним и тем
же как для прямой, так и для обратной реакции.
Поскольку энергия активации — это тот энергетический
барьер, который отделяет активированный комплекс от вступаю-
щих в реакцию веществ, очевидно, что разность энергий акти-
вации прямой (Еа) и обратной (Е0 реакций равна тепловому
эффекту:
\Н^Еа~ Е'а.
Если АЯ >> 0 (эндотермический процесс), то Еа — Еа > 0, т. е.
Еа > Еа’, если АЯ < 0 (экзотермический процесс), Еа — Еа < 0,
т. е. Еа < Еа. Естественно, что превращение активированного
комплекса в продукты реакции всегда является процессом экзо-
термическим/
Проиллюстрируем конкретным примером понятие активиро-
ванного комплекса. Рассмотрим классическую реакцию заме-
щения
НО- + СНз - Вг НО - СНз + Вг-
Известно, что реакция происходит в результате атаки иона
ОН“ на атом углерода со стороны, противоположной брому.
Встреча молекулы СН3Вг с ионом ОН” приводит к образова-
132
нию активированного комплекса, который представляет собой не
промежуточный продукт реакции, а определенную конфигурацию
атомов, создание которой требует затраты энергии.
В активированном комплексе А...В связи НО—С—Вг начи-
нают образовываться при приближении иона ОН- к углероду.
Одновременно происходит удаление атома брома и удлинение
связи С—Вг, которая, однако, еще не разорвана:
. ?/Вг
но + с -->
/\
н н
А + В -► А—В —> С + D
Если энергия активации достигнута, то столкновение эффек-
тивно, и бром отделяется с электронами связи, образуя Вг-,
тогда как гидроксил соединяется с углеродом, образуя метиловый
спирт.
Определение энергии активации. Значение энергии актива-
ции можно определить несколькими методами. Первый метод —
> графический с использованием уравнения Аррениуса:
k = Кое
£ai
RT
ИЛИ
Еа
In k = In Ко----.
А 1
Переходя к десятичным логарифмам, имеем
lgft— 2,3/? Т + ,£Л'°-
Если построить график зависимости экспериментальных величин
Igfe от \/Т (рис. 53), то получим прямую, отсекающую на оси
ординат отрезок, равный 1gКо (откуда можно определить Ко),
Р
и имеющую тангенс угла, равный-----(откуда можно оп-
ределить £а). Прямая, изображенная на рисунке, соответствует
реакции
2HI —>- Н2 4- 12
Она позволяет определить энергию
активации этой реакции, составляю-
щую 177,5 кДж/моль.
Чаще энергия активации опреде-
ляется вторым методом: измеряется
скорость этой реакции при двух раз-
личных температурах. При темпера-
турах Т\ и имеем соответственно
Га Га
= RT' и ^=Кае кт*
Рис. 53. График зависимости
1g К от обратной абсолютной
температуры
133
Разделив первую из этих величин на вторую, исключаем множи-
тель Ко:
Еа Еа Еа
kr е~~™'
—— =-------= е
k2 _ _£1_
е
или
. k\ Еа / Т\ — Т2Х
,п^ = -/Н~т7Н’
RT\T2 г 1 \
Еа =-=--— (In ki — In k2y
I \ — 1 2
Энергия активации для подавляющего большинства процес-
сов находится в пределах от 50 до 250 кДж/моль. Лишь для
реакций с участием атомов и радикалов (в соответствии с боль-
шей их реакционной способностью) она меньше 50 кДж/моль
и составляет иногда несколько кДж/моль, а для взаимодействия
ионов близка к нулю.
Энергии активации некоторых реакций приведены в табл. 17.
Таблица 17. Энергия активации некоторых реакций
Реакция Еа, кДж/моль Реакция Ea, кДж/моль
СНЧ-Н2^СН4 + Н* 42 2HI + Н2 + 12 184
СН4 -> с + 2Н2 335 2NH3 N2 + ЗН2 326
СНзСНО с2н4 + со 190 2NO2-^2NO + O2 111
С2Н6Вг -> СН4 + НВг 226 2N2O -> 2N2 + О2 245
2С12О 2С12 + О2 88 2SO2 + 02 + 2SO3 251
8. ТЕОРИИ СТОЛКНОВЕНИЙ И ПЕРЕХОДНОГО СОСТОЯНИЯ
На скорость реакции влияет не только энергия активации, но и размеры,
форма и, самое главное, взаимное расположение атомов и реакционных цент-
ров молекул. При невыгодном их расположении столкновение молекул с энер-
гией, равной или большей £а, может не привести к химическому взаимодействию.
Рассмотрим столкновение двух атомов водорода. Электронная плотность в
атомах Н распределена равномерно по шаровой поверхности и поэтому без-
различно, какими местами столкнутся реагирующие атомы водорода. Перекры-
вание орбиталей произойдет в любом месте (рис. 54).
Если же сталкиваются атомы водорода и галогена, то не безразлично,
каким образом они ориентированы по отношению друг к другу. Электронная
плотность валентных орбиталей галогена распределена в пространстве сложным
образом. На рис. 55 зачернены две р-орбитали атома галогена, имеющие по
два спаренных электрона, и не зачернена одна р-орбиталь, имеющая 1 элект-
рон. Только последняя может перекрываться с шаровой сферой электронной
134
плотности атома Н, образуя химиче-
скую связь. Если же атом Н подходит
к двум остальным орбиталям, перекры-
вания орбиталей не происходит и
реакция не имеет места.
Если для простоты сделать очень
грубое допущение о тождественности
математической и термодинамической
вероятности, то вероятность ориента-
ции молекул, необходимая для взаимо-
действия при их столкновении, может 61
Рис. 54. Перекрывание орбиталей при-
взаимодействии атомов водорода
вычислена по формуле
Число способов ориентации, приводящих к взаимодействию
Общее число возможных способов ориентации
Натуральный логарифм величины 1Г, помноженный на постоянную Больцмана,
называется энтропией активации:
Sa*= fcln W.
Помножив S* на число Авогадро, можно получить значение энтропии актива-
ции для одного моля:
Sa = S*Wa = Мь k In W = R In W.
Отсюда вероятность взаимной ориентации, необходимой для осуществления
химического акта, равна
W __ esa//?
Рис. 55. Перекрывание орбиталей при взаимодействии атомов галогена
и водорода
Чем больше вероятность необходимой для реакции ориентации, тем выше
скорость реакции и соответственно константа скорости. Последняя, таким обра-
зом, пропорциональна es*IR\
k ~ eSa,R.
Согласно уравнению Аррениуса константа скорости также пропорциональ-
на e~EIRT.
Если ввести в уравнение Аррениуса фактор взаимной пространственной
ориентации с некоторым коэффициентом пропорциональности Z, можно получить
уравнение для константы скорости реакции
k = Zes&,Re~ E^lRT.
Отсюда следует, что предэкспоненциальный множитель Ао в уравнении
Аррениуса равен
= Ze5**.
135
Иначе говоря, согласно теории соударений, Ко характеризует выгодную для
реакции пространственную ориентацию взаимодействия активных молекул. Он
называется стерическим фактором. В него не входит температура, так как она
не влияет на ориентацию молекул.
Теория переходного состояния, В этой теории делается допущение, что
активированный комплекс представляет собой не простую группировку атомов,
а истинный промежуточный продукт, существующий в течение очень короткого
времени в реакции. Этот промежуточный продукт называют переходным
состоянием. Предполагается также, что между реагентами и переходным
состоянием устанавливается, истинное равновесие:
А + В^ (А- В)
Тогда константа этого равновесия Д* равна
_ переходное состояние [А ••• В]
реагент [А] [В]
Затем переходное состояние разрушается с образованием продуктов реак-
ции. Скорость этого разрушения и, следовательно, образование ' продуктов
исключительно велика, поскольку связи в переходном состоянии очень слабы.
Скорость образования продуктов, т. е. скорость v реакции, пропорциональна
числу Р переходных состояний, разрушающихся за единицу времени в единице
объема, т. е. их концентрации, что выражается уравнением
v = Р [Активированный комплекс].
Из статистической механики известно, что число Р зависит только от темпера-
туры. Эта зависимость имеет вид
Р = kT/h,
где к — постоянная Больцмана; h — постоянная Планка; Т—абсолютная тем-
пература.
Следовательно, для данной температуры число Р одинаково для всех
переходных состояний, а скорость какой бы то ни было химической реакции
зависит только от концентрации активированных комплексов:
v — -fa- Т [Переходное состояние].
Однако концентрация переходных состояний связана с концентрацией
реагентов соотношением
[Переходное состояние] = К* [Реагент] = Д5^ [А] [В].
Тогда уравнение для скорости примет вид
кТ кТ
v=—^-K^ [Реагент] =—^~ К* [А], [В].
Из обычного выражения для скорости реакции
и = к [Реагент] = к[А] [В].
видно, что константа скорости k в теории переходного состояния равна
fe = (4.19)
h
Итак, при данной температуре она пропорциональна константе равновесия Д^
между реагентами и переходным состоянием.
Термодинамические данные позволяют связать константу Д* со стандартной
свободной энергией образования переходного состояния AGf, называемой также
свободной энергией активации:
AGf = -/?Т1пД*. (4.20)
136
Как и в теории Аррениуса, мож-
но схематически изобразить реак-
цию как функцию стандартной
свободной энергии Go (рис. 56).
Из уравнения (4.20)
- Д6^
К* = е RT
> Поскольку
Переходное состояние
Реагенты
_______Продукты
Координата реакции
AG* = -TbSf.
константу К* можно представить
в виде
Рис. 56. Энергетическая схема сво-
бодной энергии активации
* RT
Подставляя значение /С* в уравнение (4.19), получим константу скорости
реакции по теории переходного состояния:
AS* А/Г
, к Т R RT
k= h е
Это выражение можно записать в виде
1 т &S* _ Д//^
k = —-— е е RT
п
При сравнении последнего выражения с классическим уравнением Арре-
ниуса
k = Ков
видно, что в теории переходного состояния
К.
Здесь вводится энтропийный фактор ASf — это стандартная энтропия актива-
ции; она заменяет стерический фактор теории столкновений, а энергия активации
Ег аналогична теории переходного состояния стандартной энтальпией актива-
ции AH;f.
Эта теория в большей мере, чем теория столкновений, позволяет подойти
к решению проблемы сложных реакций. Она применима к реакциям; протека-
ющим в растворе, тогда как теория столкновений хорошо описывает, как правило,
реакции в газовой среде.
9. ПОНЯТИЕ О КИНЕТИКЕ РЕАКЦИЙ В ОТКРЫТЫХ СИСТЕМАХ
Все рассмотренные в этой главе кинетические закономер-
ности относились к.закрытым системам, т. е. к системам, в кото-
рых отсутствует материальный обмен с окружающей средой.
В закрытую систему в начале процесса вводится некоторая
масса исходных веществ, которые далее претерпевают ряд хими-
ческих превращений, переходя в промежуточные вещества и
продукты реакции, но все эти вещества до окончания процесса
остаются в пределах системы, т. е. не выводятся из реакцион-
ного сосуда.
137
Для открытой системы характерно изменение состава реаги-
рующей смеси не только за счет химической реакции, но и допол-
нительного введения исходных веществ и продуктов реакции из
соседных объемов. Важнейшим примером открытых систем явля-
ются живые организмы, у которых непрерывный материальный
обмен с внешней средой (обмен веществ) является необходимым
условием их существования.
К открытым системам неприменимо понятие скорости реак-
ции, выведенное ранее. Это связано с тем, что к изменению
массы вещества в открытой системе приводят не только химиче-
ские процессы, но и процессы материального обмена со средой.
Под скоростью реакций в открытой системе следует понимать
изменение количества вещества в единицу времени в единице
объема за счет идущих в системе химических процессов (а не
полное изменение количества вещества в единицу времени в еди-
нице объема). При таком определении скорость химической
реакции в открытой системе является функцией концентрации
реагентов, присутствующих в системе, причем вид функциональ-
ной зависимости аналогичен соответствующей зависимости в
закрытой системе.
Однако в отличие от закрытых систем для определения
скорости реакций в открытой системе недостаточно знать харак-
теристики изменения объема открытой системы во времени и кон-
центрации вещества в системе. В частности, при постоянном
объеме открытой системы скорость реакции не может быть
определена непосредственно графическим дифференцированием
кинетической кривой накопления данного реагента. В отличие
от закрытых систем, в которых число линейно независимых
дифференциальных уравнений равно числу линейно независимых
химических реакций, для открытых систем все дифференциаль-
ные уравнения линейно независимы.
Таким образом, число линейно независимых уравнений,
описывающих кинетику реакций в открытой системе, равно числу
компонентов в системе. Это сильно усложняет количественное
рассмотрение скоростей реакции и подробное обсуждение дан-
ного вопроса не может быть осуществлено в настоящем учеб-
нике.
Контрольные вопросы
1. Можно ли по написанному уравнению химической реакции установить
кинетический, порядок реакции?
2. Кинетический порядок реакции может быть выражен не целым числом.
Когда это бывает?
3. Какая разница между молекулярностью и кинетическим порядком реак-
ции? Когда кинетический порядок реакции равен молекулярности?
4. Как изменяются скорости прямой и обратной реакции при смещении
равновесия?
5. Как связаны константы скоростей прямой и обратной реакции с констан-
той равновесия?
6. Какая разница между прямой и разветвленной цепной реакциями? ;
Глава V
КАТАЛИЗ
1. КАТАЛИТИЧЕСКИЕ ПРОЦЕССЫ И КАТАЛИЗАТОРЫ
Подавляющее большинство химических реакций осуществля-
ется в результате активации молекул и образования активиро-
ванного комплекса, распад которого в основном определяет
скорость протекания реакции. Широкий круг так называемых
каталитических процессов связан с образованием
активированного комплекса, в который кроме реагентов входят
вещества, стехиометрически не являющиеся реагентами. При
этом после распада активированного комплекса эти вещества,
так называемые катализаторы (Кт), регенерируются в неизмен-
ном виде
A + B-JSU [(А + В) Кт] —> СВКт
Катализ — ускорение химической реакции, благодаря присутствию в системе
некоторого вещества, состояние и масса которого в конце реакции остаются
неизменными. Вещество, ускоряющее реакцию, называется катализатором.
Каталитические реакции отличаются от сопряженных или
индуцированных реакций (см. гл. IV) тем, что в последних про-
исходит необратимое превращение вещества — индуктора, в то
время как возможные изменения катализатора при каталитиче-
ских реакциях являются результатом побочных процессов, не
обусловливающих каталитическое
действие.
Влияние катализаторов на ско-
рость химических реакций в ос-
новном заключается в осущест-
влении процесса, энергия актива-
ции которого ниже, чем энергия
активации, соответствующая не-
катализируемой реакции. Кроме
того, катализаторы увеличивают
энтропию активации (см. гл. IV),
т. е. способствуют ориентации мо-
лекул в пространстве, удобной
для химического взаимодействия.
На рис. 57 приведена диаграм-
ма энергий некаталитической и
каталитической реакций, из кото-
Рис. 57. Диаграмма энергий акти-
вации некаталитической 1 и катали-
тической 2 реакций
139
е RT
р RT
рой видно, что энергия активации некаталитической реакции
(Еа) много больше энергий активации каталитических реакций
(Еа и Еа9. Это связано с тем, что активированный комплекс
некаталитической реакции отвечает гораздо большему уров-
ню энергии по сравнению с комплексами каталитической ре-
акции.
Если предположить, что для рассматриваемых пар некатали-
тической и каталитической реакций значение предэкспонен-
циальных множителей (Ко) в уравнении Аррениуса близки, то
можно, зная значения энергий активации для некаталитической
(Ei) и каталитической (Е2) реакций, подсчитать величину
ускорения реакции (у):
_ Скорость каталитической реакции
Скорость некаталитической реакции
В табл. 18 приведены результаты такого сопоставления для
некоторых реакций в газовой фазе с использованием в качестве
катализатора иода. Из таблицы видно, что уменьшение энергии
активации на 17...50% вызывает соответствующее увеличение
скорости протекания реакции в 102—105 раз.
Катализатор изменяет скорость термодинамически возмож-
ной реакции, но он не влияет на истинное равновесие, т. е. не
меняет константу равновесия и термодинамически равновесные
концентрации. Он в равной степени ускоряет и прямую, и обрат-
ную реакции. Это происходит от того, что катализатор не может
служить источником свободной энергии, так как в результате
реакции он остается в неизменном состоянии и, в частности/
сохраняется его свободцая энергия. Если повышение темпера-
туры не только убыстряет процесс, но и смещает равновесие, то
катализатор лишь изменяет время его достижения; это время тем
меньше, чем активнее катализатор.
Часто присутствие катализатора вызывает не только измене-
ние скорости реакции, но и направление процесса, что связано
с изменением механизма реакции под влиянием катализатора.
Таблица 18. Ускорение некоторых реакций в газовой фазе
(катализатор — молекулярный иод)
Распадающаяся молекула Энергия активации, кДж/моль Д£ = Е1 -£2, кДж/моль Ускорение при 700 К
для некаталити- ческой реак- ции Е\ для каталити- ческой реак- ции £2
СН3ОС2Н5 193 159 34 102
С2Н5ОС2Н5 212 142 70 ю4
СзН7ОС3Н7 252 119 133 105
СНзСНО 190 137 53 104
140
Так, например, мономолекулярный некаталитический пиролиз ди-
этилового эфира приводит к образованию оксида углерода,
метана и этилена:
С2Н5ОС2Н5 2СН4 4- 1/2С2Н4 4- СО
В присутствии катализатора (иода) скорость распада эфира
увеличивается примерно в 10 000 раз, но в результате образу-
ются оксид углерода, метан и этан:
С2Н5ОС2Н5 С2Нб + СН4 + со
Каждый катализатор способен катализировать только вполне
определенные химические реакции. Иначе говоря, для катализа-
тора характерна специфичность (избирательность) дейст-
вия. Например, два различных катализатора, действуя на одно
и то же вещество при одной и той же температуре^ могут привес-
ти к образованию различных продуктов:
СНз - СН2ОН - А|2°3-. сн2 = СН2 4- Н2О
673 к
г
СНз—СН2ОН СНз—сг
673 к \Н + н2
Специфичность действия особенно сильно проявляется у фер-
ментов, которые являются биологическими катализаторами.
2. КИСЛОТНО-ОСНОВНОЙ И ОКИСЛИТЕЛЬНО-
ВОССТАНОВИТЕЛЬНЫЙ КАТАЛИЗ
По характеру химического взаимодействия при катализе
различают две основные группы каталитических процессов:
кислотно-основной и окислительно-восстановительный.
При кислотно-основном катализе возникновение активного
комплекса, связано либо с переходом от катализатора протона
к реагенту, либо с отдачей реагентом протона катализатору. На
стадии восстановления состава катализатора протон перемеща-
ется в обратном направлении.
Так, при гидролизе сложного эфира катализатор (кислота),
передавая сложному эфиру протон, переводит его в протони-
зованную форму, обладающую более высокой реакционной спо-
собностью. Протонизованная молекула сложного эфира
значительно легче гидролизируется водой по реакции
R' 4- н2О
Н
X)
R—СГ 4- R'OH 4- Н+
ЧЭН
141
чем исходная молекула эфира. В результате этого в присутствии
кислот гидролиз сложных эфиров ускоряется, т. е. имеет место
кислотный катализ.
Активная промежуточная частица образуется в результате реакции
А + Н3О+ АН+ + Н2О
(предполагается, что процесс идет в водном растворе).
Иногда активная промежуточная чабтица образуется в результате при-
соединения к молекуле исходного вещества двух протонов, т. е. в результате
двух последовательных реакций:
А + Н3О+ АН+ + Н2О
АН+ + Н3О+ AHi+ + Н2О
Известны примеры, когда активная промежуточная форма образуется
в результате отщепления молекулы воды от протонизованной формы исход-
ного вещества. Для этого исходная молекула должна содержать гидроксил, и ее
можно обозначить как ROH. Активная промежуточная частица образуется
здесь в результате двух последовательных реакций:
ROH + Н3О+ ROH^+ Н2О
ROH^- R+ + Н2О
При реакциях окислительно-восстановительного катализа образование актив-
ного комплекса связано с электронными переходами между катализатором и
реагентом. Так, например, распад пероксида водорода ускоряется ионами
железа. Процесс этот сложный и протекает в несколько стадий.
Первая стадия приводит к образованию очень реакционноспособного
гидроксильного радикала, несущего один неспаренный электрон:
Fe2+ + Н2О2 — Fe3+ + ОН" + ОН*
Этот радикал взаимодействует с другой молекулой пероксида, получается новый
радикал:
ОН* + Н2О2 Н2О + НО2
Этот радикал распадается на протон и анион-радикал кислорода:
НО2 -> Н+ + от*
Последний взаимодействует с ионом железа (III), восстанавливая его, а сам
превращается в молекулу кислорода:
Fe3+ + ОГ — Fe2+ + О2
Таким образом, железо после окончания процесса оказывается в первона-
чальном состоянии в результате протекания окислительно-восстановительной
реакции.
3. ГОМОГЕННЫЙ И ГЕТЕРОГЕННЫЙ КАТАЛИЗ
В зависимости от фазового состояния реагентов и катализа-
тора различают гомогенный и гетерогенный катализ. При гете-
рогенном катализе химическая реакция идет на границе раз-
дела фаз, образуемых катализатором и реагирующими веще-
ствами. В гомогенном катализе катализаторы образуют
единую фазу с реагирующими веществами. Наиболее распро-
страненными гомогенными катализаторами являются кислоты и
основания, ионы переходных металлов и их комплексы и биологи-
ческие катализаторы, так называемые ферменты или энзимы.
Гомогенный катализ. Для гомогенных реакций установлено,
что катализатор образует в той же фазе промежуточные реак-
142
ционноспособные продукты. Рассмотрим гомогенную реакцию,
медленно протекающую при отсутствии катализатора:
А + В — АВ
В присутствии катализатора (Кт) осуществляются две быстро
протекающие стадии
А Кт — АКт
и
АКт + В — АВ + Кт
в результате которых последовательно образуются частицы
промежуточного соединения АКт (затем активный комплекс
АВКт) и конечные продукты с регенерацией катализатора.
Примером такого катализа может служить реакция разложения
уксусного альдегида
,0
СНз—с< (г) СН4 (г) + СО (г)
для которой Еа = 190 кДж/моль. В присутствии паров иода этот
процесс протекает в две стадии:
СНзСНО + ь -* СНз1 + hi + со
СНз1 + hi — СН4 4- 12
Уменьшение энергии активации этой реакции при введении
катализатора составляет 54 кДж/моль (ускорение в 105 раз).
Весьма распространенным типом гомогенного катализа явля-
ется кислотный катализ. В качестве примера рас-
смотрим кислотный катализ дегидратации спиртов:
СНз—СН—СНз -> СНз—СН=СН2 + Н2О
I
он
Дегидратация этого спирта при обычной температуре происходит
очень медленно; процесс ускоряется при повышении темпера-
туры. Энергия активации реакции относительно высока, так как
дегидратация соответствует, в частности, разрыву связи угле-
род — кислород спирта (С—ОН), энергия разрыва которого при-
близительно равна 380 кДж/моль.
Если добавить в реакционную смесь немного кислоты, напри-
мер H2SO4, которая служит источником протонов Н+, то реакция
пойдет быстрее.
Когда протон присоединяется к молекуле спирта, атом
кислорода заряжается положительно и притягивает электроны
связи углерод — кислород; легко осуществляется разрыв этой
связи:
н н
+Н+ | I
сн3—СН—СН3 н3с—с—сн3 *=* сн3—с—сн3 + нон
I -н+ I +
он |
о
143
Карбкатион быстро нейтрализуется, образуя соответствующий
олефин:
сн3—с—сн2 сн2—сн = сн2 + Н30+
н
Л
Протон Н+, играющий роль катализатора, в конце реакции
регенерируется в виде иона гидроксония НзО+:
н3о+ Н+ + Н2О
Автокатализ. Это процесс каталитического ускорения хими-
ческой реакции одним из ее продуктов. Выше было показано,
как протоны могут ускорить, например, реакцию гидролиза
сложного эфира; при автокатализе эти протоны могут возникнуть
в системе, например, в результате диссоциации образующегося
продукта. Так, при гидролизе этилацетата
СН3СООС2Н5 + Н2О СНзСООН + С2Н5ОН
продукт реакции — уксусная кислота диссоциирует с образова-
нием протонов (вернее, ионов гидроксония, что в общем одно
и то же):
СНзСООН + Н2о СНзСОО- + Н3О+
Н3О+^Н+ + Н2О
которые ускоряют реакцию гидролиза.
Характерная особенность автокаталитической реакции состо-
ит в том, что она идет при переменной возрастающей концен-
трации катализатора. Поэтому скорость автокаталитической
реакции в начальный период возрастает, и лишь на последующих
стадиях в результате убыли концентрации реагентов рост ско-
рости сменяется падением. Начальный участок кинетической кри-
вой продукта реакции обращен выпуклостью вниз (рис. 58).
Кинетическая кривая продукта автокаталитической реакции
Рис. 58. Кинетическая кри-
вая автокаталитической
реакции
имеет S-образный вид.
Гетерогенный катализ. В гетерогенном
катализе катализатор и превращаемые
вещества находятся в различных агрегат-
ных состояниях. Катализатор в основном
представляет собой твердое вещество, а
реагирующие вещества являются газами
или жидкостями. Большинство катализа-
торов, применяющихся в химической
промышленности, состоит из оксидов,
сульфидов, металлов и солей, т. е. прак-
тически всех элементов периодической
системы Менделеева. Действие гетероген-
144
ного катализатора связано с взаимодействием реагирующих мо-
лекул с его поверхностью —адсорбцией (см. гл. XI).
Так как каталитический эффект, вызываемый гетерогенным
катализатором, определяется прежде всего поверхностью ката-
лизатора, обычно стремятся по возможности ее увеличить. Для
этого применяют только измельченные активные вещества. Но
порошкообразный катализатор легко уносится струей газа, а
взятый в больших массах создает уже значительное сопротив-
ление потоку газа. Поэтому обычно активное вещество осаждают
на пористых инертных подкладках — носителях (силикагеле,
алюмогеле, солях и т. п.) — или прессуют, придавая катализа-
тору форму таблеток, шариков или цилиндров.
Применение катализаторов особенно важно для проведения
медленных реакций, сопровождающихся выделением теплоты.
Хотя при этом константа равновесия обеспечивает высокий
выход продукта при умеренной температуре, скорость реакции
оказывается настолько низкой, что реакцию нельзя использовать
в практических целях. При повышении температуры скорость
реакции возрастает, но при этом равновесие для экзотерми-
ческих реакций сдвигается в сторону уменьшения выхода про-
дукта, как это непосредственно следует из принципа Ле Шателье.
Например, синтез такого ценного вещества, как аммиак, из
азота и водорода практически идет лишь при очень высоких
температурах, так как энергия активации процесса соединения
этих газов велика. Но чем выше температура, тем меньше полу-
чится аммиака в равновесной смеси, так как равновесие
сдвигается с ростом температуры в сторону образования реаген-
тов и, следовательно, нагревание невыгодно. Введение катализа-
тора дает возможность быстро достигнуть равновесия при уме-
ренных температурах, т. е. более выгодных для практики
условий.
Для проведения реакции при температурах, когда константа
равновесия обеспечивает достаточный выход продукта, исполь-
зуются специальные железные катализаторы. Чтобы сделать
катализатор более эффективным и предохранить его от слишком
быстрой дезактивации прд действием загрязняющих газ при-
месей, в него добавляют ванадий и другие металлы. Вещества,
повышающие активность катализатора, называются промото-
рами, а вещества, снижающие каталитическую активность, —
ядами. Поскольку только некоторая доля поверхности ката-
лизатора принимает участие в реакции, легко понять, почему
даже относительно малое содержание промоторов и ядов может
оказывать значительное действие на активность.
В процессе каталитической реакции на поверхности катали-
затора адсорбируются (см. гл. XI), по крайней мере некоторые
из реагирующих молекул, в результате чего их реакционная
способность повышается. Это может быть обусловлено ориента-
цией молекул, благоприятной для последующей реакции, и
ослаблением некоторых внутримолекулярных связей, которые
145
далее разрываются с боль-
шей легкостью, что приводит
к снижению энергии актива-
ции стадии, определяющей
скорость реакции.
Изменение прочности связи при
адсорбции можно иногда непосред-
ственно показать с помощью ин-
фракрасных спектров адсорбиро-
ванных молекул.
На рис. 59 показаны инфра-
красные спектры поглощения окси-
да углерода, адсорбированного на
четырех различных металлах. Мо-
лекула газообразного оксида угле-
рода почти неполярна и имеет
лишь слабое поглощение при
2143 см-1. Кетоны поглощают из-
лучение в области 1900... 1600 см-1.
Как видно из рис. 59, при адсорб-
ции оксида углерода на меди часто-
та колебаний связи изменяется не-
значительно, а при адсорбции на
палладии частота становится почти
такой же, как частота колебаний
карбонильной группы в кетонах,
молекула оксида углерода адсорби-
Частота-, см'1
Рис. 59. ИК-спектры оксида углерода, ад-
сорбированного на различных металлах
Полученные данные свидетельствуют, чтс
руатся на атоме меди или платины в виде М—С=О, а с никелем или палладием
О
II
образуются мостики между двумя атомами металла М—С—М, которые явля-
ются более реакционноспособными частицами. Поэтому никель и палладий — хо-
рошие катализаторы гидрирования оксида углерода в метан, тогда как медь и
платина — неактивные катализаторы этой реакции.
Пусть в отсутствие катализатора протекает бимолекулярная
реакция
А 4- В -* АВ* Продукты
а в присутствии катализатора “скорость ее возрастает, но про-
дукты реакции остаются теми же. Если считать, что активное
адсорбционное состояние аналогично активированному комплек-
су АВ* некаталитической реакции, то весь процесс можно
изобразить следующим образом.
1. Адсорбция исходных веществ на поверхность катализа-
тора:
А + В + Кт — АВКт
Этот процесс, как правило, активированный и экзотермический.
2. Перевод адсорбированного состояния в активное:
АВКт — АВКт*
Этот процесс требует затраты определенной энергии, называемой
истинной энергией активации гетерогенной катали-
тической реакции.
146
3. Реакция в адсорбированном состоянии с образованием
адсорбированных конечных продуктов:
АВКт* — Продукты Кт
4. Десорбция продуктов реакции, приводящая к регенерации
катализатора:
Продукты Кт — Кт + Продукты
Таким образом, и в гетерогенном катализе ускоряющее дей-
ствие катализатора, так же как и в гомогенном катализе, связано
с тем, что реагирующие вещества образуют промежуточные
соединения, что приводит к снижению энергии активации.
Современные представления о механизме гетерогенного ката-
лиза свидетельствуют о сложности и многостадийности процесса,
и в настоящее время нет единой теории гетерогенного катализа.
В 1925 г. английский ученый Г. Тейлор предложил теорию
активных центров катализатора. Согласно этой теории
активные центры возникают в тех же местах поверхности
катализатора, где атомы слабее всего связаны с кристалличе-
ской решеткой, т. е. там, где силовое поле наименее насыщено
(выступы, «пики» поверхности, грани кристаллов). Таких актив-
ных центров, или «пиков», очень мало. Они составляют всего
лишь 0,1% от всей поверхности катализатора. Установлено, что
адсорбция веществ наблюдается на поверхности многих катали-
заторов, однако только некоторые из них способны вызвать
данную химическую реакцию и провести ее в определенном
направлении.
Для кристаллических катализаторов, характеризующихся
правильным пространственным расположением частиц, были
установлены закономерности, связывающие расстояния между
атомами в превращаемой молекуле с расстоянием и геометри-
ческим расположением частиц катализатора. Эти закономерности
составляют содержание теории мультиплетов А. А. Ба-
ландина, в которой впервые рассматривается состав активного
центра гетерогенного катализатора.
Основные положения этой теории, впервые высказанные
в 1929 г., следующие.
1. Активный центр катализатора представляет собой сово-
купность определенного числа адсорбированных центров, распо-
ложенных на поверхности в геометрическом соответствии со
строением молекулы, претерпевающей превращение (принцип
геометрического или структурного соответствия).
2. При адсорбции реагирующих молекул на активном центре
образуется мультиплетный комплекс, в результате чего про-
исходит перераспределение связей, приводящее к образованию
продуктов реакции.
Мультиплетная теория ставит геометрическое расположение
адсорбционных центров в активном центре в прямую зависи-
мость от геометрического строения катализируемых молекул.
Эту теорию можно было бы назвать теорией геометрического
147
подобия активного центра и реагирующей молекулы. Для различ-
ных реакций число адсорбционных центров (каждый из которых
отождествляется с одним атомом металла) в активном центре
принимается равным 2, 3, 4, 6 и т. д. Подобные активные центры
были названы дублетами, триплетами, квадру-
плетами, секстетами, а в общем случае мульти-
плетами.
Например, дегидрирование этилового спирта согласно теории
мультиплетов происходит на дублете, причем к одному атому
дублета притягиваются водородные атомы групп СН2 и ОН, а
к другому — атом кислорода и углеродный атом группы СН2.
В результате происходит разрыв связей С—Н и О—Н и возник-
новение связей Н—Н и С=О с образованием молекул уксус-
ного альдегида и водорода (атомы дублета на поверхности ката-
лизатора изображены кружками):
I °l । °
н н Н н—н
Если расстояния между атомами катализатора в дублете будут
иными, то возможен другой процесс.
Схематическое изображение частиц на мультиплете платины
при дегидрировании циклогексана приведено на рис. 60. Для
простоты на рисунке приведены лишь атомы углерода циклогек-
сана (а—f) и связи между ними. Атомы секстета 1, 3 и 5 оттяги-
вают атомы водорода от атомов углерода циклогексана, в ре-
зультате чего происходит перераспределение связей и на них
образуются молекулы водорода. В то же время под воздей-
ствием атомов катализатора 2, 4 и 6 образуют двойные связи
между углеродными атомами а и Ь, с и d, е и f.
Конечное состояние отвечает возникновению молекулы бен-
зола и трех молекул водорода.
Развитием мультиплетной теории является принцип энергетического соот-
ветствия (А. А. Баландин, 1935), согласно которому энергия активации гете-
рогенной реакции является сложной величиной. Она в первом приближении
состоит из двух слагаемых, одно из которых зависит только от энергии связи
между составными частями реагирующих молекул, а другое — от энергии
взаимодействия между катализатором и составными частями мультиплетного
комплекса.
В отличие от мультиплетной теории теория каталитически ак-
тивных ансамблей Н. И. Кобозева предусматривает возможность
существования активных центров из атомов, не входящих в кристаллическую
решетку. Из этой теории следует, что лишь сочетание определенного (обычно
небольшого) числа частиц катализатора (ансамбль) способно проявлять ка-
талитическую активность. Так, для реакции соединения азота и водорода не-
обходимо три атома катализатора (железа), сгруппированных в активный
ансамбль. Для реакции присоединения водорода к органическим соединениям,
ускоряемой палладием, необходимо два атома палладия и т. д. Отдельные
ансамбли на поверхности, твердого катализатора не могут соединяться друг с
другом, потому что поверхность катализатора очень неоднородна, и частицы
148
Рис. 60. Схема дегидрирования циклогексана на платиновом сек-
стете
вещества не могут беспрепятственно двигаться по всей поверхности. Их дви-
жение ограничено лишь небольшой частью поверхности.
Согласно теории каталитически активных ансамблей, атомы активных
центров образуют «аморфную» фазу на поверхности носителя или на поверх-
ности кристаллических граней самого металла. Последний при этом выполняет
роль носителя.
4. ФЕРМЕНТАТИВНЫЙ КАТАЛИЗ
Ферменты (от лат. fermentum — закваска) — биологические
катализаторы белковой породы, присутствующие во всех живых
клетках. Практически все биохимические реакции, протекающие
в любом организме и обеспечивающие в совокупности обмен
веществ, совершаются при участии ферментов.
Все известные ферменты представляют собой длинные цепи из а-амино-
кислот (относительная молекулярная масса порядка 0,5 млн), свернутые в ком-
пактную форму, в которых имеется несколько реакционноспособных участков.
Изучение природы ферментов показало, что, помимо белка, многие из них содер-
жат и другие соединения. Так, например, в составе окислительных ферментов
были обнаружены органические соединения железа. Эти соединения у различ-
ных окислительных ферментов оказались одинаковыми по составу. Кроме того,
было выяснено, что такие же соединения железа входят и в гемоглобин крови,
переносящий кислород в организме человека и животных. Комплексное
соединение железа (гем) можно отделить от белка. Однако после этого ни бе-
лок, ни гем не проявляют ферментативных свойств. Отсюда следует, что высо-
кая активность и специфичность свойственны только сложной системе, состоя-
щей из белка и гема. В состав различных ферментов входят и комплексные
соединения других металлов. В некоторых ферментах обнаружены медь, цинк,
марганец, хром и другие элементы. Для некоторых ферментов уже известна
первичная структура, т. е. последовательность аминокислот в длинной цепи.
Вторичная структура — общий характер спирали, образуемый цепью, прибли-
женно установлена для нескольких ферментов. О третичной структуре, т. е.
природе реакционноспособных поверхностных участков молекулы, известно
очень мало.
Ферменты обладают двумя характерными особенностями.
Во-первых, они катализируют химические реакции, заставляя их
протекать с огромными скоростями, недостижимыми при ис-
пользовании любых искусственных катализаторов.
149
Так, константа скорости разложения пероксида водорода,
ионами Fe2+ равна 56,0 (если измерять концентрацию в кмоль/м3
а время — в секундах). Константа скорости той же реакции, про-
водимой каталазой, составляет 3,5- 107. Иными словами, реак-
ция под действием фермента протекает в миллион раз быстрее.
Константа скорости гидролиза мочевины под действием кислоты
равна 7,4- 10-7, а фермент уреаза позволяет проводить эту
реакцию со скоростью, константа которой на тринадцать поряд-
ков больше — 5,0-106.
Такие ускорения реакции связаны с тем, что ферменты резко
снижают энергетические барьеры на реакционном пути. Напри-
мер, энергия активации для реакции распада пероксида водоро-
да под действием иона железа (II) и молекулы каталазы соот-
ветственно равны 42 и 7,1 кДж/моль; для гидролиза мочевины
кислотой и уреазой — соответственно 103 и 28 кДж/моль.
Во-вторых, еще более удивительная способность действия
ферментов состоит в том, что ферменты весьма специфичны. Так,
амилаза, содержащаяся в слюне, легко и быстро расщепляет
крахмал, молекула которого состоит из огромного числа одина-
ковых глюкозных звеньев. Но она не катализирует процесс
распада сахарозы.
Рассмотрим реагенты, которые составляют субстрат S биоло-
гической реакции, катализируемой ферментом Е с образованием
продуктов Р. Начальные растворы ферментов и субстратов раз-
бавлены; исследуем влияние изменения концентрации субстрата
на скорость реакции.
Экспериментально показано, что если повысить концентрацию
субстрата, сохраняя постоянной концентрацию фермента, то ско-
рость возрастает пропорционально концентрации субстрата. При
дальнейшем увеличении концентрации субстрата наступает такой
момент, когда скорость перестает меняться с ростом концентра-
ции субстрата. Это происходит потому, что, когда все активные
центры фермента заняты, дальнейшее повышение концентрации S
в растворе не приводит к увеличению скорости реакции.
Согласно обычным представлениям о механизме катализа
считают, что субстрат S реагирует с ферментом Е, образуя сна-
чала ферментсубстратный комплекс ES. Эта реакция обратима,
комплекс ES может диссоциировать на субстрат S и фермент Е,
но равным образом приводит к продуктам Р реакции:
S + E^ES-^P -И Е
Ферментсубстратный комплекс образуется очень быстро и
всегда находится в равновесии с субстратом и ферментом, а
расщепление комплекса происходит сравнительно медленно и
практически не влияет на концентрацию ферментсубстратного
комплекса. Тогда скорость реакции v связана с концетрацией
субстрата уравнением
_ ^макс
= 1 + Km/S
150
Здесь г>Макс — макси-
мальная скорость реак-
ции, достигаемая при
больших значениях S, т. е.
тогда, когда знаменатель
правой части уравнения
близок к единице, что со-
ответствует почти плоско-
му, горизонтальному уча-
стку кривой на рис. 61;
Кт' (называется констан-
той Михаэлиса) — кон-
Рис. 61. Скорость реакции, катализируемой
ферментом
станта
акции
лекса
страт,
имеет
диссоциации в ре-
образования комп-
фермент — суб-
Как и все константы диссоциации, константа Михаэлиса
размерность концентрации и при v = ’/гамаке величина
S = Кт. Таким образом, Кт равна концентрации субстрата, при
которой скорость реакции составляет половину максимальной.
Константа Михаэлиса имеет большое значение при изучении
ферментативных реакций, так как она служит мерой сродства
между ферментом и субстратом: чем больше их способность к
образованию комплекса ES, тем меньше Кт, и наоборот. Зна-
чение константы Михаэлиса зависит от природы исследуемого
фермента, а в тех случаях, когда он действует на несколько
субстратов, — и от природы субстрата, на который он действует.
Одна из наиболее характерных особенностей действия фер-
ментов состоит в чрезвычайно сильной зависимости фермента-
тивной активности от pH среды, в которой происходит реакция.
Ферменты активны лишь в узком интервале значений pH, и для
них характерно наличие в этом интервале определенной мак-
симальной активности, резко уменьшающейся по обе стороны от
оптимального значения pH.
Большинство ферментов обладает максимальной активно-
стью цри значениях pH, близких к 7, так как pH вне- и внутри-
клеточных жидкостей организма, в которых действуют ферменты,
также близки к этому значению. Однако известны и некоторые
весьма широко распространенные ферменты, которые активны в
сильнокислом или сильнощелочном растворе.
Изменение pH может действовать на ферменты по-разному:
оно может влиять на соединение фермента и субстрата с обра-
зованием комплекса ES, а также замедлять или ускорять разло-
жение этого комплекса с высвобождением продуктов реакции.
При значительном изменении pH ферменты становятся неустой-
чивыми, разрушение фермента может происходить по одну или
по обе стороны от оптимального значения pH.
Влияние pH на ферментативную активность, как и на всякий
химический процесс, обусловлено изменением ионизации одного
или нескольких веществ (в том числе самого фермента), прини-
151
мающих участие в реакции. Молекула фермента имеет различ-
ные химические группы, степень диссоциации которых изменя-
ется в зависимости от кислотности или щелочности среды. Сле-
довательно, данный фермент может существовать в нескольких
различных ионных формах, переходящих друг в друга при изме-
нениях pH. Молекула фермента существует преимущественно в
какой-то одной ионной форме в весьма узком интервале pH, и
соответственно небольшая протяженность интервала, в котором
большинство ферментов проявляет активность, указывает на
то, что каталитической активностью обладает лишь одна из этих
возможных форм.
Контрольные вопросы
1. Известно, что использование катализатора снижает энергию активации.
А как влияет катализатор на равновесие?
2. Почему уменьшение энергии активации на 20...50% вызывает увеличение
скорости реакций в 102...105 раз?
3. Почему при гидролизе этилацетата скорость реакции резко увеличива-
ется после довольно медленной начальной стадии?
4. Зачем применяют катализатор в промышленном синтезе аммиака?
5. Почему очень незначительное содержание ядов может резко снизить актив-
ность катализатора?
6. Сопоставьте действия синтетических катализаторов и ферментов.
7. Как действует изменение pH на ферментативную активность? Укажите
причину.
Глава VI
ФОТОХИМИЧЕСКИЕ РЕАКЦИИ
1. ВЗАИМОДЕЙСТВИЕ ИЗЛУЧЕНИЯ С ВЕЩЕСТВОМ
При рассмотрении кинетики химических реакций было пока-
зано, что для совершения элементарного акта химического про-
цесса молекулы должны обладать запасом энергии, достаточным
для преодоления активационного барьера, разделяющего реаген-
ты и продукты. Следовательно, чтобы при определенных усло-
виях увеличить число превращающихся молекул, нужно пере-
дать исходным молекулам некоторое дополнительное количество
энергии, т. е. активировать их.
Большей частью активацию молекул осуществляют за счет
передачи энергии в форме теплоты; эти реакции иногда назы-
вают темновыми.
Передача энергии может быть осуществлена также с по-
мощью облучения реагентов. Реакции, происходящие под дей-
ствием света, называются фотохимическими, а раздел
химии, изучающий эти реакции — фотохимией. Реакции,
протекающие под действием излучений высоких энергий (у-излу-
чение, рентгеновское излучение, потоки электронов, протонов,
нейтронов, а-частиц и т. д.), называются радиационно-хи-'
мическими; изучением их занимается радиационная химия.
Еще в 1817 г. Ф. И. Гротгус заметил, что на химическую
реакцию оказывает влияние свет, причем вызывают реакцию
только то излучение, которое поглощается данной средой. Излу-
чением фотохимических реакций занимался К. А. Тимирязев.
Он пришел к выводу, что содержание образующегося при фото-
химической реакции продукта пропорционально количеству по-
глощенной световой энергии.
Весьма важным принципом фотохимии является закон
Штарка — Эйнштейна, который говорит, что каждый поглощен-
ный фотон активирует только одну молекулу. А. Эйнштейн посту-
лировал, что вся энергия кванта сообщается при поглощении
света одному-единственному электрону, вследствие чего этот
электрон поднимается на более высокий энергетический уровень.
Чтобы лучше понять процесс поглощения света, необходимо
рассмотреть роль электронов в акте поглощения.
С классической точки зрения электрон представляет собой
заряженную частицу, которая движется по некоторой орбитали
вокруг ядра атома. Энергия электрона зависит, как от положения
153
орбитали в пространстве, так и от того, насколько быстро дви-
жется электрон по орбитали. Увеличение энергии электрона
в результате поглощения фотона может быть использовано для
переноса этого электрона на орбиталь, которая обладает более
высокой энергией, чем исходная орбиталь, либо для увеличения
скорости движения электрона вокруг ядра по сравнению с той
скоростью, с какой он двигался до поглощения света. Положе-
ние возможных электронных орбиталей и скорости движения
по ним электронов взаимно ограничены некоторыми определен-
ными дискретными или разрешенными величинами, установлен-
ными квантовой механикой. Это означает, что электрон может
поглощать только определенные порции энергии и, следователь-
но, общая энергия электронов атома или молекулы соответ-
ствует дискретным условиям. Только свет определенной длины
волны обладает подходящей энергией для того, чтобы вызвать
переход электрона с одного уровня энергии на другой. Поэтому,
чтобы происходило поглощение света, энергия фотона, опреде-
ляемая уравнением E — hv, должна равняться разности энергий
некоторого разрешенного возбужденного состояния атома или
молекулы и начального состояния, которым обычно является
основное состояние, или состояние с наименьшей энергией.
С квантово-механических позиций избирательное отношение
веществ к излучению связано с различием уровней энергии и
атомов и молекул. В атомах энергия излучения поглощается
только электронами. В зависимости от значения кванта энергии hv
электрон, поглотивший этот квант, переходит на более высо-
кий энергетический уровень или отрывается от ядра. В молекулах
происходит возбуждение электронных, колебательных и враща-
тельных уровней либо ионизация и диссоциация, т. е. отрыв
электрона от молекулы и распад молекулы на атомы.
В конечном результате взаимодействие света с веществом
проявляется в следующих трех направлениях.
1. Возбуждение энергетических уровней атомов и молекул.
Этот процесс можно выразить уравнениями
А + /ту — А*
АВ + Ь->АВ*
где А — атом; АВ — молекула; hv — поглощенный квант света;
А* и АВ* — атом или молекула в возбужденном состоянии.
2. Ионизация атомов и молекул, происходящая с отрывом
электронов и образованием простых А+ или молекулярных АВ+
ионов:
А + /zv—кА+ + е~
АВ + hv~,АВ+ + е~
3. Диссоциация молекулы, приводящая к отрыву одного из
атомов в сложной молекуле или распаду простой молекулы на
атомы. При диссоциации молекул образуются активные час-
154
тицы, имеющие неспаренные электроны: атомы или свободные
радикалы. Уравнение диссоциации в общем виде
АВ + hv А • + В •
где А* и В* — атомы или радикалы.
Химические процессы ионизации и диссоциации, непосред-
ственно идущие за поглощением энергии излучения, называются
первичными в отличие от вторичных процессов дальнейшего
превращения ионов, атомов и радикалов.
Энергию поглощающих квантов можно рассчитать по извест-
ному соотношению Планка, которое в пересчете на моль квантов
(постоянная Авогадро) запишется так:
Е = N^hv.
Если вместо частоты использовать длину волны в наномет-
рах, то после подстановки численных значений Ад и h получим
следующее выражение для расчета энергии квантов в джоулях:
Е = 0,119- 109/Х.
Например, энергия красного света (Х = 600нм) равна
О 11 0.1 о9
= 105 дж~ 200 кДж.
6- 102
Этой энергии достаточно, чтобы вызвать распад молекул брома
и иода, энергии диссоциации которых соответственно равны
180 и 148 кДж/моль.
2. ЗАКОН ФОТОХИМИЧЕСКОЙ ЭКВИВАЛЕНТНОСТИ ЭЙНШТЕЙНА
И КВАНТОВЫЙ ВЫХОД
Если излучение осуществляет химическое превращение, то один поглощен-
ный квант энергии вызывает единственный первичный химический процесс.
Этот закон фотохимической эквивалентности А. Эйнтшейна
справедлив только для световых квантов, и его применение
ограничивается лишь первичными процессами взаимодействия
фотона с молекулой. На практике же фотохимическая реакция
включает также последующие вторичные процессы, и для описа-
ния всей реакции вводится такая характеристика, как кванто-
вый выход, который отражает эффективность реакции. Он удо-
бен для описания экспериментальных фактов и полезен, когда
нужно сделать заключение о механизме реакции.
Квантовым выходом у называется отношение числа N частиц, претерпев-
ших химическое превращение, к числу поглощенных квантов:
N
7— Q/hv ’
где Q — энергия поглощенного монохроматического излучения.
В первичном процессе один квант света поглощается одной
155
молекулой, молекула активируется и тем самым переходит в
более высокое энергетическое состояние:
Образовавшаяся в результате поглощения излучения активи-
рованная молекула может вести себя по-разному, вызывая про-
текание следующих процессов:
1) протекание простой стехиометрической реакции с образо-
ванием целого числа или доли молей продукта реакции;
2) осуществление цепной реакции; при этом одна активиро-
ванная молекула распадается на активные частицы, которые
вызывают многократное повторение реакции;
3) потеря энергии активированной молекулы (например, в
процессе люминесценции), при этом активированная молекула
исчезает, не давая жизнь химическому акту;
4) рекомбинация активированной молекулы, приводящая к
гибели активной частицы.
В зависимости от характеристики активированной молекулы
(или активных частиц, на которые она распадается) квантовый
выход может принимать различные значения, например от ты-
сячных долей до миллиона. Характеристика же активированных
молекул или активных частиц зависит в основном от времени их
жизни, что, в свою очередь, связано с их стабильностью.
Чем дольше существует активированная молекула или активная
частица, образовавшаяся при ее диссоциации, тем с большим
квантовым выходом осуществляется фотохимическая реакция.
В табл. 19 и 20 приведены некоторые типичные фотохимиче-
ские реакции в газовой фазе и в растворах. Согласно Я. И. Гера-
симову, все фотохимические реакции в зависимости от значения
квантового выхода можно подразделить на четыре группы:
1) реакции, в которых квантовый выход у= 1 (например, обра-
зование бромциклогексана, пероксида водорода, нитрозометана,
брома в результате реакции хлора с трихлорбромметаном,
разложение сероводорода в бензольном растворе и др.); 2) реак-
ции, в которых квантовый выход у<1 (например, разложение
Таблица 19. Параметры некоторых фотохимических реакций
в газовой фазе
Реакции Поглощаю- щее свет вещество Длина вол- ны, нм Квантовый выход
С12 + Н2 2НС1 С12 500 106
С!з “I- О2 —О2С12 С12 420 1
Вг2-|- CeHi2 —*- CgHuBr Ч- НВг Вг2 470 1
2НВг Н2 + Вг2 НВг 207...253 2
2NO2 -> 2NO + О2 о2 313...405 2
СНз! -* (СН4, С2Н6> 12) СНз! 202...210 МО"2
СНзВг (СН4, Вг2) СНз.Вг 253 4•10~3
Г56
Таблица 20. Параметры некоторых фотохимических реакций
в растворах
Реакция Раство- ритель Погло- щающее свет ве- щество Длина “волны, нм Квантовый выход
С12 + 2СС1зВг -> 2CCU + Вг2 СС14 С12 470 1
2НС1О 2НС1 + О2 н2о нею 366...436 2
СвНб + ЗВг2—»- СвНбВгб СбНб Вг2 300...550 0,4...0,9
2H2O22НгО + Оз н2о Н2О2 275...366 20...500
H2S н2 + S- Н2о H2S 207...253 0,2...0,3
аммиака, ацетона, уксусной кислоты, образование гексабром-
бензола); 3) реакции, в которых квантовый выход у>1 (напри-
мер, образование бромоводорода, озона, разложение диоксида
азота, азометана, хлорноватистой кислоты и др.); 4) реакции,
в которых квантовый выход (например, реакции взаимо-
действия хлора с водородом и с оксидом углерода и др.).
Рассмотрим некоторые примеры.
С квантовым выходом у<1 протекают многие реакции фото-
химического разложения органических соединений. Так, разло-
жение ацетона с квантовым выходом 0,2 вызвано нестабильно-
стью активных частиц, образующихся на первой стадии фото-
химической реакции. Механизм таких реакций изучен слабо.
С квантовым выходом у — 1 протекает большее число реак-
ций. Как правило, это обычные химические процессы, где
молекула реагента получила дополнительную энергию не в виде
теплоты (как в темновых процессах), а в виде излучения.
С квантовым выходом у>1 протекают многие важные реак-
ции. Первичной ступенью реакции образования озона является
поглощение кванта лучистой энергии молекулой кислорода:
Ог т hv == о?
Поглотив один квант энергии активирования, молекула кис-
лорода вступает в реакцию с другими молекулами кислорода:
+ О2 = Оз -|- О
Ог + О = Оз
Записав реакцию одним уравнением, получим
ЗО2 И- hv = 20з
Квантовый выход реакции равен 3, так как на один поглощен-
ный квант превращаются три молекулы кислорода.
Фотохимические реакции с квантовым выходом у^>1 обычно
протекают по механизму цепных процессов, где кванты света обра-
зуют первичную возбужденную частицу, которая далее создает
цепь (вторичная реакция). Так, с квантовым выходом ~50 осу-
157
ществляется фотохимический распад пероксида водорода. Урав-
нение первичного процесса
Н2о2+ /iv —2ОН*
Вторичный процесс цепной, включающий следующие стадии:
Н2О2 + • ОН — НО2*+ Н2О + 116,2 кДж |
Н2О2 + НО2*-> Н2О + О2 + • ОН + 83,7 кДж / (ЦеПЬ)
Н°2‘+ °2 + Н£ + 30516 КДЖ J (обрыв цепи)
ОН + Стенка-»-Продукт )
Известны фотохимические реакции с очень большим кван-
товым выходом. Так, рассмотренный ранее цепной процесс полу-
чения хлороводорода из хлора и водорода протекает с кванто-
вым выходом, достигающим 106. Уравнение первичного процесса
может быть записано так:
С12 +/iv —> 2С1 *
Вторичный цепной процесс включает следующие стадии:
С1-4-Н2^НС14-Н’1 }
Н* + С12^НС1 + С1Ч
2С1 С12 1 / к х
ои. 2 (обрыв цепи)
2г1 —► г12 )
Квантовый выход реакций, протекающих в растворах, очень
часто оказывается меньше единицы. Это происходит вследствие
дезактивации возбужденных молекул, образующихся в резуль-
тате передачи энергии молекулам растворителя или в резуль-
тате рекомбинации возникших при фотодиссоциации атомов и
молекул, причем рекомбинация облегчается молекулами раство-
рителя, играющими роль трех частиц. Такое уничтожение
реакционноспособных частиц получило название эффекта
ячейки (клетки) Франка — Рабиновича.
Таким образом, в соответствии с законом фотохимической
эквивалентности квантовый выход первичных процессов всегда
равен единице. Однако в подавляющем большинстве случаев
протекают вторичные процессы, благодаря которым квантовый
выход может стать больше или меньше единицы. При этом
справедливо следующее правило. Если в результате первичного
процесса образуются стабильные радикалы, то квантовый выход
реакции больше единицы; если при первичном распаде образу-
ются первичные радикалы, которые далее разлагаются или ре-
комбинируют, то квантовый выход равен или меньше единицы.
3. СКОРОСТЬ ФОТОХИМИЧЕСКИХ РЕАКЦИЙ
Скорость фотохимических реакций так же, как и темновых
реакций, зависит от концентрации превращаемых веществ и тем-
пературы. Кроме того, она пропорциональна количеству погло-
158
щенной энергии излучения. Согласно закону Ламберта — Бера
между интенсивностью /о падающего монохроматического излу-
чения и интенсивностью излучения /, прошедшего через слой
вещества, существует зависимость
/ = (6.1)
где С — концентрация вещества, кмоль/м3; I — толщина слоя
вещества; е — молярный коэффициент поглощения.
Количество поглощенной энергии пропорционально разности
/о— I — M. Из уравнения (6.1) следует, что
I = /о - /ов-еС/ = /0(7 - е-сС/). (6.2)
Если предположить, что вся поглощающая энергия превра-
щается в химическую, то, разделив обе части равенства на
Sdl = dV, можно получить концентрацию превращающихся час-
тиц в элементе объема dV. Для площади 5 = 1 м2 концентрация
реагирующих частиц равна еС/.
Поскольку скорость химической реакции определяется изме-
нением концентрации вещества в единицу времени в единице
объема, скорость первичного химического процесса равна
= knzCl,
где kn — константа скорости первичного акта распада.
Обычно фотохимические превращения не ограничиваются
первичным процессом; протекают вторичные реакции, при кото-
рых первичные частицы взаимодействуют с другими веществами
или рекомбинируют. Предположим, что первичные частицы
взаимодействуют с веществом В, концентрация которого С'.
Скорость вторичных реакций пропорциональна концентрациям
активных частиц вещества А и В: еС7 и С', т. е.
vB = kBCC'l,
где — константа скорости вторичной реакции.
Сенсибилизированные реакции. Первичным фотохимическим
реакциям близки так называемые реакции фотохимической сен-
сибилизации. Сущность этого явления состоит в том, что иногда
вещество непосредственно не поглощает свет с данной длиной
волны, но может при столкновении принять энергию от другого
атома, возбужденного облучением. Вещества, поглощающие
энергию излучения и передающие ее при столкновении превра-
щающимся молекулам, называются сенсибилизатор а-
м и. Примером сенсибилизированной фотохимической реакции
может служить диссоциация молекул водорода на атомы. Для
распада молекулы Н2 на атомы требуется 431,219 кДж/моль.
Легко подсчитать, что такое количество энергии сообщает излу-
чение с длиной волны 275,9 нм. Однако диссоциации молекул оно
не вызывает, так как свет с такой длиной волны не поглощается
молекулой водорода. Атомы же ртути поглощают излучение с
длиной волны 253,75 нм, переходят в возбужденное состояние
159
и при столкновении с молекулами водорода вызывают их дис-
социацию. Уравнение этой сенсибилизированной фотохимической
реакции запишется так:
Hg + hv Hg*
Hg*-J-Н2Hg + 2Н
' Кроме атомов ртути сенсибилизаторами в реакции диссо-
циации молекулярного водорода могут быть атомы кадмия, цин-
ка, поглощающие излучение с длиной волны соответственно
228,87 и 213,93 нм, и другие атомы и молекулы.
Многие вещества поглощают свет в труднодоступной дале-
кой ультрафиолетовой области спектра, что усложняет проведе-
ние фотохимических реакций с этими веществами. Тогда к этим
веществам примешивают сенсибилизатор, который поглощает
свет в более доступной области спектра и переходит в возбуж-
денное состояние. Этот прием используется в фотографии. Вве-
дение в эмульсию подходящего сенсибилизатора делает ее све-
точувствительной в тех областях спектра, в которых без них
чувствительность отсутствует.
В качестве сенсибилизаторов кроме названных веществ
часто применяют и галогениды, что объясняется легкостью их
активации. Спектр поглощения галогенидов расположен в види-
мой или близкой ультрафиолетовой области.
Важную роль играет сенсибилизация в фотосинтезе, где в
качестве сенсибилизатора выступает хлорофилл.
4. ФОТОСИНТЕЗ
Развитие жизни на Земле связано с реакциями, которые тер-
модинамически не способны протекать самопроизвольно. Эти
реакции сильно эндотермические и сопровождаются возраста-
нием изобарно-изотермического потенциала.
Важнейшими фотохимическими реакциями такого рода
являются реакции фотосинтеза, протекающие в растениях.
К. А. Тимирязев установил, что синтез углеводов из диоксида
углерода и воды осуществляется растениями за счет энергии
солнечного света, поглощаемого ими, и что к этому процессу
полностью применим закон сохранения энергии. Работы К. А. Ти-
мирязева нанесли решительный удар идеалистическим теориям,
согласно которым такой синтез происходит под действием особой
«жизненной силы».
Практически вся энергия, созидающая жизнь, по своему пер-
воначальному происхождению является энергией электромаг-
нитного излучения Солнца. Поэтому рассмотрим некоторые
характеристики интенсивности солнечного света, получаемого
Землей.
Согласно П. Нобелу, общее дневное количество лучистой
энергии Солнца, достигающей поверхности Земли, составляет в
безоблачный день около 360 Дж/см2. Около 41% всех квантов,
V60
попадающих на поверхность
Земли, приходится на ин-
фракрасную область, 7% —
на ультрафиолетовое излу-
чение, а остальные — на ви-
димый свет.
На рис. 62 показано от-
носительное количество фо-
тонов солнечного света, по-
лучаемое земной атмосферой
и поверхностью Земли в за-
висимости от длины волны.
Большая часть ультрафио-
Рис. 62. Распределение по длинам волн
интенсивности потоков фотонов солнечного
света, падающих на земную атмосферу 3
и поверхность Земли 2. Показано влияние
озона 1 и газов атмосферы 4
[ поглощает часть видимого излу-
)0 нм) и эффективно экранирует
летового компонента солнеч-
ного излучения, попадающе-
го в атмосферу, не достигает
поверхности Земли из-за
присутствующего в верхних
слоях атмосферы озона Оз. <
чения (особенно в области
коротковолновое излучение, сильно поглощая в области ниже
300 нм. Большая часть инфракрасного излучения Солнца взаимо-
действует с водяными парами атмосферы. Вода сильно погло-
щает в области 900...1100 нм и выше 1200 нм, имея основную
полосу поглощения в инфракрасной области при 1400 нм.
Распределение фотонов, достигающих поверхности Земли, по
длинам волн оказывает глубокое воздействие на жизнь. Напри-
мер, сильное поглощение ультрафиолетового излучения озоном
уменьшает потенциальную угрозу мутагенных эффектов, вызы-
ваемых коротковолновым излучением. Широкий максимум в об-
ласти 680 нм для фотонов, достигающих поверхности Земли
(рис. 62), совпадает с полосой поглощения хлорофилла.
Фотосинтез является непременным условием жизни растений
и животных, будучи фактически самым крупномасштабным син-
тетическим процессом на Земле. Как считает П. Нобел, за год
фотосинтезирующими организмами фиксируется и переводится
в форму органических соединений около 5-1016 г (50 млрд, т)
углерода, причем большая часть его фиксируется фитопланкто-
ном, живущим вблизи поверхности океанов. Это количество
соответствует параллелепипеду, сложенному из фотосинтетиче-
ских продуктов, с основанием 1 км2 и высотой несколько более
100 км. Источником углерода для фотосинтеза служит атмос-
ферный СОг (содержание в атмосфере составляет 0,03%), а
также СО2 и НСОз^ растворенные в воде озер и океанов. Из
продуктов фотосинтеза, кроме органических соединений, очень
важное значение имеет кислород, необходимый для всех орга-
низмов, обладающих дыханием. Весь кислород, содержащийся
в атмосфере, был образован путем фотосинтеза за несколько
тысячелетий.
161
6—696
Фотосинтез состоит из большого числа весьма сложных
реакций, однако суммарный его результат выражается следую-
щей простой схемой:
6СО2 + 6Н2О-^ C6Hi2O6 + 6О2, bG° = 2861,9 кДж/моль
или в более общем виде
СО2 + Н2О-^> (СН2О)4-О2, Дб° = 477,0 кДж/моль
Таким образом, суммарный результат фотосинтеза состоит в
связывании диоксида углерода, окислении воды до молеку-
лярного кислорода и синтеза углеводов. Образование кислорода
как побочного продукта фотосинтеза не является универсальным
свойством фотосинтезирующих организмов. Например, у некото-
рых бактерий фотосинтеза процесс выражается схемой
2H2S + со2^ СН2О + Н2О + 2S
где вместо кислорода образуется свободная сера. Более общая
схема суммарного процесса такова:
2Н2Х + СО2 сн2о 4- Н2О + 2Х
где X — элемент, окисляющийся в ходе фотосинтеза.
Фотосинтез состоит из двух основных стадий. Первая из них,
называемая световой стадией, связана с поглощением
фотонов. Этот процесс осуществляется с огромной скоростью,
возможно, за время 10“12... 10“8 с. Затем следует ряд химических
превращений, которые в совокупности образуют темновую
стадию. Этот процесс осуществляется в отсутствие света.
Темновые реакции лимитируют весь процесс в целом.
Молекулы хлорофилла представляют собой хромофоры, с помощью которых
поглощается свет. В фотосинтезирующих организмах могут содержаться два и
более типов молекул хлорофилла. В зеленых растениях содержатся хлоро-
филлы а и Ь, структура которых показана на рис. 63. Поглощение света в
видимой области спектра обусловлено наличием сильно сопряженной порфири-
новой системы. Как видно из рис. 64, хлорофилл поглощает свет наиболее
интенсивно в синей и красной областях спектра, но отражает зеленый, желтый
и оранжевый свет. Этим определяется характерный зеленый цвет растений.
Хлорофилл b непосредственно не участвует в фотосинтетических превра-
щениях. Его роль состоит в том, что он улавливает и собирает свет. После фото-
возбуждения хлорофилл b быстро передает избыточную энергию молекуле хло-
рофилла а, которая затем принимает непосредственное участие в превращениях
фотосинтеза.
В растениях хлорофилл связан с липопротеиновыми мембранами, нахо-
дящимися в специальных органеллах клетки — хлоропластах. Типичная расти-
тельная клетка содержит от 50 до 200 хлоропластов. Каждый хлоропласт имеет
длину около 1000 нм. Кроме двух наружных мембран хлоропласты содержат
систему внутренних мембран, образующих многослойные структуры, упакован-
ные в пачки. Это так называемые. граны. Внутренние мембраны ограничи-
вают замкнутые объемы, отделенные от остальной части хлоропласта. Хлоро-
филл и другие пигменты находятся в ламеллах гран, ламеллах стромы, и именно
в этих частях хлоропласта начинается процесс фотосинтеза.
После поглощения фотона возбужденная молекула хлорофилла может уча-
ствовать в процессах двух типов. Она может флуоресцировать или использо-
вать избыточную электронную энергию для проведения каких-то энергетически
невыгодных реакций.
162
Рис. 63. Фотосинтетические пигменты
J3-каротин
Поглощение
Длина волны}нм
Рис. 64. Спектры поглощения фотосинтетических пигментов
6*
Так как суммарный процесс фотосинтеза состоит в окислении воды до
кислорода и восстановлении диоксида углерода до углеводов, можно следующим
образом оценить энергетические параметры процесса. Окислительно-восстано-
вительный потенциал пары (см. гл. X, разд. 6) Н2О/О2 равен +0,81 В, а пары
углевод/СОг равен —0,42 В. Таким образом, перенос одного электрона от воды
на диоксид углерода требует затраты 0,82 — (—0,42) = 1,24 В, или
119,6 кДж/моль. В реакции
СО2 -> СН2О
необходимо перенести 4 электрона (или 4 атома Н), что соответствует
478,4 кДж/моль. Эта энергия получается в фотохимическом процессе следующим
образом. В каждом реакционном центре молекула хлорофилла (Chi) тесно
связана с донором электронов X и акцептором электронов А. После фотовозбуж-
дения происходят реакции переноса заряда
XChlA^L XChlA*—.XCh 1+А_—►Х+СЫА_
Молекула А принимает электрон от возбужденного хлорофилла (ХСЫА) и
превращается в А-. Электрон, удаленный из молекулы пигмента, может быть
затем заменен на электрон, пришедший от молекулы донора X. Поскольку X и
А в достаточной степени отдалены друг от друга, они не взаимодействуют
между собой непосредственно, и возникшие в этом процессе частицы Х+ и А-
могут далее принимать участие в других окислительно-восстановительных реак-
циях.
Контрольные вопросы
1. При каких условиях взаимодействия квантов света с веществом произой-
дет фотохимическая реакция?
2. Известно, что согласно закону Эйнштейна один поглощенный квант энер-
гии вызывает единственный химический процесс. В то же самое время квантовый
выход может быть равен 106. Как объяснить это кажущееся противоречие?
3. Почему квантовый выход фотохимических реакций в жидкой среде может
быть очень мал? Каковы причины малого квантового выхода некоторых реак-
ций, протекающих в газовой среде?
4. Почему некоторые фотохимические реакции требуют применения сенси-
билизатора?
5. Какую роль играет хлорофилл в фотосинтезе?
6. Является ли образование кислорода как побочного (но очень важного)
продукта фотосинтеза универсальным свойством фотосинтеза?
7. Какие реакции лимитируют весь процесс фотосинтеза?
Глава VII
РАСТВОРЫ НЕЭЛЕКТРОЛИТОВ
1. ОБРАЗОВАНИЕ РАСТВОРОВ. РАСТВОРИМОСТЬ
Исследование растворов — весьма важная область физиче-
ской химии, поскольку большая часть процессов, представляю-
щих интерес для химии, биохимии и биологии, протекает в жид-
кой фазе.
Раствором называется однородная смесь, состоящая из двух или большего
числа веществ, состав которой в некоторых пределах может непрерывно изме-
няться без скачкообразных изменений свойств.
Раствор может иметь любое агрегатное состояние — твердое,
жидкое или газообразное, т. е. различают газовые, жидкие и
твердые растворы. Для газообразной системы более принято
говорить о газообразной смеси, а не о газовом растворе. Твер-
дые растворы менее обычны, чем жидкие, но с термодинамиче-
ской точки зрения они аналогичны последним. Жидкие растворы
делят на две группы: растворы неэлектролитов и
растворы электролитов.
Наибольшее практическое значение имеют жидкие растворы.
Именно в них создаются наиболее благоприятные условия как
для перемещения молекул, так и для их сближения, необходи-
мого для химического взаимодействия.
Простейшие составные части раствора, которые могут быть
выделены в чистом виде и смешением которых можно получить
растворы любого возможного состава, называются компо-
нентами раствора.
С термодинамической точки зрения все компоненты раствора
равноценны, поэтому деление их на растворитель и растворенное
вещество носит условный характер. Обычно растворите-
лем называют компонент, присутствующий в растворе в значи-
тельно большем количестве по сравнению с другими компонен-
тами.
Если одним из веществ, которые составляют раствор, явля-
ется жидкость, а другими — газы или твердые вещества, то рас-
творителем обычно считают жидкость.
Процесс растворения — это не только процесс простого рас-
пределения молекул или ионов одного вещества среди молекул
или ионов другого. Он большей частью связан с различными
взаимодействиями химического и физического характера между
165
ними. Поэтому свойства растворов определяются взаимодейст-
вием компонентов, которое зависит не только от вида сил, дейст-
вующих между частицами, но и от формы и размеров частиц.
Образование растворов является процессом самопроизволь-
ным, идущим с увеличением беспорядка в системе, т. е. с повы-
шением энтропии. Лучше всего это иллюстрируется частным
случаем образования раствора, когда частицы растворяемого
вещества переходят из более упорядоченного (а в предельном
случае растворения кристалла, полностью упорядоченного) со-
стояния в менее упорядоченное. При этом с увеличением энтро-
пии (А5>0) уменьшается свободная энергия системы (AG<0).
Если речь идет об образовании раствора из двух жидкостей,
то движущаяся сила процесса растворения обусловлена стремле-
нием компонентов раствора к выравниванию концентраций, что
также приводит к увеличению энтропии, т. е. AS>0, a AG<0.
Процесс образования растворов является процессом динами-
ческим. Часть молекул растворяемого вещества непрерывно пе-
реходит в раствор, а часть молекул растворенного вещества из
раствора возвращается обратно к растворяемому веществу.
Последний процесс усиливается по мере увеличения концентра-
ции растворенного вещества в растворе. В итоге устанавлива-
ется некоторая предельная для данной температуры концентра-
ция растворенного вещества, при которой число молекул, посту-
пающих в раствор и уходящих из раствора в единицу времени,
становится равным. Устанавливается истинное динамическое
равновесие AG = 0; образовавшийся раствор называют насы-
щенным, а достигнутую предельную концентрацию насыщен-
ного раствора — растворимостью. Ее часто выражают не
только в единицах молярной концентрации, но и в массовых
единицах, т. е. числом килограммов растворенного вещества в
единице объема раствора или на единицу массы (кг) раство-
рителя.
Если получен пересыщенный раствор, т. е. такой, концентра-
ция которого выше концентрации насыщенного раствора, систе-
ма находится не в истинном, а кажущемся, неустойчивом равно-
весии (AG>>0). Эта система очень легко самопроизвольно или
под слабым внешним воздействием (встряхивание, внесение
кристаллов и т. д.) переходит в состояние истинного равновесия
(AG = 0), характерное для насыщенного раствора.
Очень часто для очистки вещества применяют способ пере-
кристаллизации, позволяющий удалить из данного вещества при-
меси, которые обладают такой же растворимостью, что и очищае-
мое вещество. Допустим, что вещество содержит 90% выделяе-
мого вещества и 10% какой-нибудь примеси, имеющей такую же
растворимость. Смесь обрабатывают таким количеством раство-
рителя, чтобы растворить и выделяемое вещество, и примесь.
Если предположить, что разделяемые вещества не влияют на
взаимную растворимость, то раствор будет насыщен по отноше-
нию к выделяемому веществу, но находиться ниже точки насы-
166
щения примесью. При удалении части растворителя, например
выпариванием, раствор делают пересыщенным по отношению к
выделяемому веществу, так что часть последнего выпадает в
осадок. Но раствор все же остается ненасыщенным по отноше-
нию к примеси, и она не выпадает в осадок. Поэтому можно про-
должать удалять растворитель и таким образом получать кри-
сталлы чистого вещества, пока не будет достигнуто насыщение
раствора примесью. После этого операцию следует остановить,
ибо далее чистое вещество и примесь стали бы осаждаться в
равных отношениях. Этим путем получают большую часть чисто-
го вещества с потерей Vio части, которая сильно загрязнена при-
месью.
Идеальные растворы. При смешении жидкостей, молекулы
которых неполярны и сходны между собой по структуре, хими-
ческой связи и величине молекул, тепловые и объемные измене-
ния очень малы. Например, такое наблюдается для процесса сме-
шения толуола с бензолом. Если при смешении двух жидкостей
происходит лишь хаотическое распределение частиц без измене-
ния межчастичного взаимодействия, то теплота смешения равна
нулю (АЯР=О), изменения объема системы не происходит
(объем смешения тоже равен нулю АУР=О), энтропия растет
лишь' в результате выравнивания концентрации за счет диф-
фузии.
Смеси, в процессе образования которых отсутствуют тепло-
вые и объемные эффекты, т. е. АЯР=О и AVP=O, называются
идеальными растворами. Иногда для выражения этой
особенности идеальных растворов говорят, что для них энталь-
пия и объем аддитивны. Идеальный раствор есть воображаемое
состояние; реальные растворы могут быть только более или менее
близки к идеальным.
Свойства идеальных растворов, подобно свойствам смесей
идеальных газов, не зависят от природы растворенного веще-
ства, а определяются лишь концентрацией. Следовательно, един-
ственной причиной их образования (как и образования газооб-
разных смесей) является возрастание энтропии при смешении.
Таким образом, даже для идеального раствора АЗР=#О (так как
растворение связано с изменением термодинамической вероятно-
сти для любого раствора). Однако ASP в этом случае не будет
зависеть от природы компонентов, а однозначно определится
их соотношением (молярными долями).
По некоторым свойствам к идеальным растворам близки
так называемые бесконечно разбавленные рас-
творы; в них можно пренебречь взаимодействием между ча-
стицами растворенного вещества и взаимодействием между
частицами растворенного вещества и растворителем.
По мере возрастания концентрации растворенного вещества
простые закономерности, характерные для предельно разбавлен-
ных растворов, начинают искажаться, так как становятся зна-
чительными межчастичные взаимодействия.
Реальные растворы. Образование реальных растворов сопро-
вождается тепловыми и объемными эффектами (АЯР^=О, АУР^=О).
Эти эффекты могут быть значительными; например, при раство-
рении спирта в воде происходит заметное увеличение температу-
ры и уменьшение объема на 3,5%.
Растворимость при заданной температуре зависит от природы
растворенного вещества и растворителя, их агрегатных состоя-
ний и внешних условий. До настоящего времени нет теории, с
помощью которой можно было бы предсказать и вычислить рас-
творимость. Это обусловлено сложностью взаимодействия частиц
в растворе, а также отсутствием общей теории жидкого состоя-
ния. Тем не менее накопленный богатый экспериментальный
материал позволяет объяснить качественно процессы раство-
рения.
В смеси двух жидкостей А и В, состоящих из молекул с ко-
валентными связями, энергия взаимодействия частиц А и В не
отличается существенно от энергии взаимодействия между части-
цами А и А или частицами В и В. Поэтому различные жидкости
с ковалентной связью в молекулах обычно неограниченно
растворяются друг в друге (например, растворимость
толуола в бензоле не ограничена).
Указанные закономерности были замечены еще алхимиками,
сделавшими вывод: подобное растворяется в подобном.
Другие соотношения характеризуют образование раствора
при смешении полярных и неполярных (или малополярных)
веществ. Энергия взаимодействия молекул А и А или В и В боль-
ше, чем А и В, и одинаковые молекулы одного и того же компо-
нента предпочтительно будут связываться между собой, а не с
молекулами другого компонента. В результате этого раствори-
мость сильно понизится, чем, например, и можно объяснить
Рис. 65. Растворимость
анилина в воде
анилин > % ^Gc)
ограниченную раствори-
мость полярного хлороводорода в
неполярном бензоле и небольшую раст-
воримость неполярных и малополяр-
ных веществ в полярном растворителе,
например в воде. Молекулы НгО в
жидкой воде связаны между собой
сильными водородными связями. По-
этому притяжение неполярных молекул
неэлектролита к молекулам воды будет
меньше, чем притяжение молекул воды
друг к другу.
Ограниченная растворимость жид-
костей наблюдается, например, при
смешении воды и анилина. Кривая
(рис. 65) разделяет области существо-
вания гомогенных и гетерогенных си-
стем. Заштрихованная площадь — это
область расслаивания жидкостей, а в
168
любой точке на кривой (например, в точках с и d) имеются две
фазы, образованные взаимно насыщенными растворами.
Температура, соответствующая точке k, — критическая
температура растворения. Это та температура, начи-
ная с которой происходит неограниченная взаимная смешивае-
мость обоих компонентов. Рост взаимной растворимости с повы-
шением температуры обусловлен эндотермичностью процесса
растворения.
Таким образом, можно утверждать, что когда растворяемое
вещество проявляет химическое сродство к растворителю, оно
более или менее растворимо в этом растворителе. Органические
соединения, содержащие гидроксигруппу, очень часто раствори-
мы в воде, и чем больше относительное содержание в их моле-
куле гидроксигрупп, 'тем\они более растворимы. Так, высшие
одноатомные спирты ряда СПН2«+1ОН, например СбН1зОН, мало
растворимы в воде, так как многоатомные спирты, такие, как
маннит СбНв(ОН)б, легко растворимы. Углеводороды очень труд-
но растворимы в простых соединениях, содержащих гидрокси-
группу, например в воде, спирте; вещества же, содержащие от-
носительно мной) гидроксигрупп, очень слабо растворимы в
углеводородах.
Если в двухфазную систему, состоящую из двух практически
нерастворимых жидкостей, внести какое-либо вещество, то оно
распределяется между обеими фазами. Рассмотрим этот процесс
на примере растворения иода в воде и тетрахлориде углерода.
При перемешивании иод будет растворяться (распределяться)
как в одной, так и в другой жидкости, причем в тетрахлориде
углерода растворимость иода во много раз больше, чем в воде.
Титрованием можно определить содержание иода как в той,
таки в другой жидкости (Ci и С2).
Концентрация иода в двух слоях по мере введения дополни-
тельной массы иода (в пределах, ограниченных растворимостью
данного вещества в указанных растворителях) будет изменяться,
однако отношение концентраций С1/С2 практически остается
неизменным. Этот факт, экспериментально подтвержденный при
исследовании различных систем, позволил сформулировать
закон распределения:
отношение концентраций вещества, распределяющегося между двумя несме-
шивающимися жидкостями, является для каждой температуры величиной по-
стоянной, не зависящей от абсолютных и относительных количеств каждого
из растворителей и распределяемого вещества:
Сх/С2 = К.
Постоянная величина К, характерная для данного вещества
и данной пары растворителей, является константой равновесия
и называется коэффициентом распределения, ко-
торый зависит от температуры.
Чтобы извлечь из раствора вещество, растворимое в данном
растворителе, нужно добавить к нему другой растворитель, не
169
смешивающийся с первым. Этот процесс называется экстрак-
цией. Ее применяют, когда необходимо разделение веществ,
кипящих в узком интервале температур или обладающих малой
летучестью паров и высокой температурой кипения.
Влияние на растворимость внешних условий. Так как при
получении раствора устанавливается истинное равновесие
(AG=0), то для определения влияния температуры и давления
на растворимость пользуются принципом Ле Шателье. Это зна-
чит, что надо учитывать знак теплового эффекта образования
раствора или, иначе говоря, энтальпию смешения АЯР и знак
изменения объема АГР. Знак энтальпии смешения будет опреде-
лять характер действия температуры, а знак изменения объема —
характер действия давления. Величина влияния воздействия
определится абсолютным значением АЯР и АУР.
Рассмотрим влияние давления и температуры на примере
растворения газообразных веществ в жидкости. При растворении
смеси газов растворимость каждого из них пропорциональна
его парциальному давлению. Так, например, в воде растворя-
ется столько же кислорода воздуха, сколько бы его растворялось
при соприкосновении воды с чистым кислородом, находящимся
под давлением 21 кПа (парциальное давление кислорода в воз-
духе) .
Растворение газов почти всегда сопровождается выделением
теплоты, так как происходит сольватация их молекул. Поэтому,
согласно принципу Ле Шателье, повышение температуры пони-
жает растворимость газов. Так как при растворении газообраз-
ных веществ в жидкости АУр<0, то давление способствует росту
растворимости газов. Малорастворимые газы подчиняются
закону Генри (1802):
растворимость газа при постоянной температуре пропорциональна его
давлению:
C = kp,
где С — концентрация газа в жидкости; k — коэффициент про-
порциональности (константа Генри); р — давление газа над рас-
твором. В табл. 21 приведены значения констант Генри для
растворимости некоторых газов в воде при постоянной темпера-
туре.
Таблица 21. Константы Генри для растворимости некоторых газов
Газ ky моль «мПа 1 Газ k, моль-мПа 1
н2 140 о2 82
Не 280 со2 3,4
Аг 70 H2S 1,1
N2 170
170
Проявление закона Генри иллюстрируется образованием
обильной пены прц откупоривании бутылки газированной воды;
здесь происходит резкое уменьшение растворимости газа (в ос-
новном диоксида углерода) при понижении его парциального
давления. Этот же закон объясняет возникновение кесонной
болезни. На глубине около 40 м ниже уровня моря общее дав-
ление составляет около 600 кПа и растворимость азота в плазме
крови на этой глубине в 9 раз больше, чем на поверхности моря.
При быстром подъеме водолаза с глубины растворенный азот
выделяется в кровь пузырьками, что может привести к тяжелым
последствиям и даже к смерти.
Закон Генри выполняется, во-первых, только для разбавлен-
ных растворов и неприменим к растворам более высокой кон-
центрации. Во-вторых, если газ реагирует с водой, то может
наблюдаться аномально высокая растворимость. Например, вы-
сокая растворимость диоксида углерода определяется реакцией
СО2 4- Н2О Н2СО3 Н+ 4- НСОГ 2Н+ 4- СО1~
Эта реакция представляет значительный интерес для биологиче-
ских процессов. Другими примерами газов, реагирующих при
растворении, могут служить H2S, НС1, SO2 и NH3. Поведение
кислорода в крови характеризует еще одно отклонение от закона
Генри. Обычно кислород лишь незначительно растворим в воде,
однако его растворимость резко повышается в присутствии гемо-
глобина или миоглобина.
На растворимость жидкостей в жидкостях давление почти не
влияет, поскольку при растворении объемнее эффекты невелики.
Также практически не сказывается оно и на растворимости твер-
дых веществ, так как АУр=0; его влияние становится ощутимым
лишь при очень высоких давлениях (108...109 Па).
Так как растворение газа является экзотермическим про-
цессом, то, согласно принципу Ле Шателье, растворимость газов
уменьшается с нагреванием и увеличивается при охлаждении.
Растворимость некоторых газов в воде при постоянном давлении
(0,1 МПа) и разных температурах приведена в табл. 22.
Таблица 22. Растворимость некоторых газов в воде
при различных температурах
Темпера- тура, к Растворимость (1 м3 газа/1 м3 воды) газов
n2 о2 н2 СО2 H2S SO2
273 0,0239 0,0489 0,0215 1,710 4,67 79,8
293 '/Ч 0,0164 0,0310 0,0182 0,878 2,58 39,4
з1з .г ' 0,0118 0,0230 0,0164 0,530 1,66 18,8
171
Растворимость данного вещества обычно уменьшается при
добавлении других веществ. Так, растворимость газов в воде
часто сильно уменьшается при внесении в воду солей или иных
растворимых в ней веществ. Например, в 1 г воды при 293 К рас-
творяется около 3*10~3м3 хлора, а в 1 г насыщенного раствора
NaCl растворяется в 10 раз меньше.
Подобно газам растворимость многих жидкостей понижается
в присутствии солей. Например, растворимость фенола в чистой
воде больше, чем в солевом растворе.
Понижение растворимости в присутствии солей называется
высаливанием. Одной из причин высаливания может быть
сольватация солей, ведущая к уменьшению числа свободных мо-
лекул растворителя, а с ним и к понижению растворяющей спо-
собности жидкости.
2. СОСТАВ РАСТВОРОВ. ПАРЦИАЛЬНАЯ МОЛЯРНАЯ ВЕЛИЧИНА.
КОНЦЕНТРАЦИЯ
Вещество, переходя в раствор, становится компонентом раство-
ра и в какой-то степени теряет свою индивидуальность. Свойства
раствора могут быть охарактеризованы термодинамическими
величинами: V, Н, U, F, G и т. д. Так как между молекулами
компонентов раствора происходит взаимодействие, термодинами-
ческие характеристики приходится относить к раствору как к
целому, а не только к составляющим его веществам. Так, напри-
мер, легко измерить общий объем раствора, однако нельзя опре-
делить, какие объемы занимают в нем отдельные компоненты.
Любой неидеальный раствор характеризуется не только соб-
ственной энергией молекул компонентов, но и энергией их взаи-
модействия. Это взаимодействие не может быть отнесено к ка-
кому-либо из компонентов. Однако важно знать, какую долю
вносит данный компонент в то или иное свойство раствора.
Такая характеристика называется парциальной молярной вели-
чиной.
Г. Льюис назвал парциальной молярной величиной компонента частную
производную от какой-либо экстенсивной величины g по количеству вещества
(молей) nt этого компонента раствора при постоянных давлении, температуре и
количеству вещества (молей) всех компонентов, кроме данного:
г. = f-M
\ дп[
Соблюдение условий, требуемых этим определением, может быть достигнуто,
если, например, к весьма большому объему раствора данной концентрации при
постоянных давлении и температуре добавить 1 моль какого-либо компонента.
Тогда концентрация раствора практически не изменится, и соответствующее
изменение свойства раствора будет парциальной молярной величиной добав-
ленного компонента.
Например, парциальный молярный объем определяется уравнением
X о п -t /р> Т,
172
Если раствор образуется из компонентов без изменения объема (AVP = O), то
очевидно, что_ парциальный молярный объем компонента равен его молярному
объему, т. е. V?, где индекс нуль означает, что вещество находится в чистом
состоянии. В отличие от молярного объема парциальный молярный объем
может быть отрицательной величиной. Последнее имеет место, если вследствие
молекулярного взаимодействия при добавлении компонента к раствору происхо-
дит уменьшение объема.
Важнейшей характеристикой раствора является его состав
или концентрация компонентов. Наиболее удобно выражать кон-
центрацию раствора в молярных долях. Молярной долей XL
/-го компонента называется отношение количества вещества
(молей) nt к сумме всех компонентов раствора (молей)
Сумма молярных долей компонентов раствора равна единице.
Иногда концентрацию выражают числом киломолей Ct рас-
творенного вещества в 1 м3 раствора (молярная концентрация).
Это, однако, не всегда удобно, так как концентрация раствора
вследствие термического расширения зависит от температуры.
В связи с этим часто также пользуются моляльностью miy т. е.
количеством растворенного вещества (молей) в 1 кг раствори-
теля (воды), величина которой не зависит от температуры. Меж-
ду молярной долей и моляльностью в водных растворах сущест-
вует соотношение
3. ПОНИЖЕНИЕ ДАВЛЕНИЯ НАСЫЩЕННОГО ПАРА
РАЗБАВЛЕННЫХ РАСТВОРОВ
Представим себе, что в равновесную систему жидкость — пар
введено нелетучее вещество, переход которого в паровую фазу
исключен. При образовании раствора концентрация раствори-
теля уменьшается, его молярная доля становится меньше 1;
это вызывает нарушение равновесия жидкость — пар. В соответ-
ствии с принципом Ле Шателье начинает протекать процесс,
стремящийся ослабить влияние воздействия, т. е. конденсация.
Благодаря этому процессу давление пара над раствором стано-
вится меньше, чем над чистым растворителем.
Давление насыщенного пара растворителя над раствором всегда меньше,
чем над чистым растворителем. При этом понижение давления пара будет тем
большим, чем больше концентрация растворенного вещества в растворе.
Для разбавленных растворов, близких к идеальным, когда
растворитель и растворенное вещество близки между собой по
составу и строению молекул, давление насыщенного пара рд
данного компонента А над раствором прямо пропорционально
относительному содержанию его молекул в растворе, т. е. его
молярной доле Хд:
рА = £ХА.
173
При Хд=1 давление рд представляет собой давление насы-
щенного пара данного компонента в свободном состоянии рд.
Следовательно, й=рд. Тогда
Ра — ^аРа-
Введем в растворитель растворенное вещество В. Концент-
рация растворенного вещества в растворе при выражении ее
в молярных долях вещества Х& связана с Хк простым соотно-
шением: Хд + Хв=1. Тогда
Ра=(1-Хв)рХ.
Отсюда следует, что
Разность Ра —рд называется понижением давления насыщенного
п
Ра — Ра
пара, а отношение — ---относительным понижением давле-
РА
ния насыщенного пара.
Относительное понижение давления насыщенного пара растворителя над
раствором равно молярной доле растворенного вещества в растворе (первый
закон Рауля).
Следует еще раз подчеркнуть, что закон Рауля выполняется
только для идеальных и реальных разбавленных растворов, близ-
ких к идеальным (АЯР^О, ДУР^О).
4. ТЕМПЕРАТУРА КРИСТАЛЛИЗАЦИИ РАЗБАВЛЕННЫХ РАСТВОРОВ
Раствор в отличие от чистой жидкости не отвердевает цели-
ком при постоянной температуре. Кристаллы начинают выделять-
ся при какой-то одной температуре, но по мере понижения тем-
пературы число их растет до полного отвердения раствора.
Температурой начала кристаллизации рас-
твора называют температуру, при которой в результате
охлаждения раствора (в условиях, исключающих возможность
образования пересыщенных растворов) происходит образование
кристаллов.
В условиях, исключающих образование твердых растворов,
кристаллы растворителя находятся в равновесии с раствором
данного состава. Если температура начала кристаллизации не
выше комнатной, ее называют температурой замерза-
ния раствора.
Еще М. В. Ломоносов заметил, что разбавленный раствор
замерзает при температуре более низкой, чем чистый раствори-
тель. Так, морская вода замерзает не при 273 К, а при несколько
более низкой температуре. Многочисленные эксперименты пока-
зали, что такое изменение температуры замерзания раствора
можно рассматривать как общее правило.
174
Температуры замерзания растворов
характеризуются величиной понижения
температуры замерзания ДГ3, которая
равна разности между температурами
замерзания чистого растворителя Г? и
раствора Г3:
АГ3=Г3°-Г3.
Рассмотрим р—Г-диаграмму со-
стояния воды (растворитель) и раство-
ров, полученных добавлением в воду
различных количеств растворенного ве-
щества (рис. 66). Кривая ОА представ-
ляет собой зависимость давления на-
Рис. 66. Понижение давления
насыщенного пара раствори-
теля над раствором
сыщенного пара чистой воды над водой от температуры, а кри-
вые ВС, DE и т. д. — давления насыщенного пара воды над раст-
ворами с различными концентрациями растворенного вещества.
Они должны расположиться, очевидно, ниже кривой ОА, так как
раствор, в соответствии с законом Рауля, обладает меньшим дав-
лением насыщенного пара. Кривая Off выражает температурную
зависимость давления насыщенного пара воды над льдом. Кри-
сталлы растворителя будут находиться в равновесии с раствором
только тогда, когда давление насыщенного пара растворителя
над кристаллами и над раствором одинаково, т. е. когда кривая
Off пересечется с кривой давления насыщенного пара над раст-
вором данной концентрации. Температура, отвечающая этому
условию, должна быть более низкой, чем температура замерза-
ния чистого растворителя. Рассматривая бесконечно разбавлен-
ные растворы, считают отвечающие им бесконечно малые участ-
ки OB, ВВ', OD, DD', OF, FF' кривых НО, ВС, DE и FG прямо-
линейными. Из подобия треугольников BOB', DOD', FOF' сле-
дует, что понижение температуры замерзания пропорционально
понижению давления пара и, следовательно, понижение темпера-
туры замерзания пропорционально концентрации растворенного
вещества в растворе:
\Т3 = Кт,
(7-1)
где т — моляльная концентрация растворенного вещества, вы-
ражаемая в молях на 1000 г растворителя. Это уравнение иногда
называют вторым законом Рауля.
Для каждого данного растворителя коэффициент пропорцио-
нальности К является величиной постоянной. Он называется
моляльным понижением температуры замер-
зания или криоскопической постоянной. Крио-
скопические константы наиболее важных растворителей приве-
дены в табл. 23.
Уравнение (7.1) используют для определения концентрации
растворенного вещества в разбавленном растворе. Оно может
175
Таблица 23. Криоскопические и эбуллиоскопические константы
некоторых растворителей
Вещество К,град/моль Е, град/моль Вещество К, град/моль Е, град/моль
Уксусная
кислота 3,90 2,93 Этанол — 1,23
Бензол 5,12 2,64 Нафталин 6,8 5,65
Камфора 40,0 — Толуол — 3,37
Вода 1,86 0,514
быть применено также для определения молекулярной массы
растворенного вещества.
Если в т\ г растворителя было растворено п моль веще-
ства, то
1000
т\
т = п
m2 дпгЮОО
п=~ИТ’ т = —П---->
М Мт\
где m2 — навеска вещества; М — его молярная масса.
Поэтому, подставляя это выражение т в уравнение (7.1),
получаем
1000
/П1 ’
откуда
Рис. 67. Прибор для крио-
скопического определения
молекулярной массы:
1 — термометр; 2 — «рубаш-
ка»; 3, 6, 8 — пробирки;
4 — термометр Бекмана;
5, 9 — мешалки; 7 — боко-
вой отросток; 10 — крыш-
ка; 11 — стакан
.. К/п21000
М =-----Г-=
miAT
(7.2)
Эту формулу используют для вычисле-
ния молекулярной массы методом крио-
скопии.
Молекулярную массу криоскопическим
методом определяют с помощью прибора,
изображенного на рис. 67. В пробирку 6
вводят взвешенную массу растворителя.
В широкий толстостенный стакан 11, снаб-
женный мешалкой 9, помещают охлади-
тельную смесь из снега и поваренной соли.
В эту смесь опускают широкую пробир-
ку 3, служащую воздушной «рубашкой» и
обеспечивающую более медленное и рав-
номерное понижение температуры в про-
бирке 6, измеряемое термометром 4.
Для точного определения температуры
замерзания применяют термометр Бек-
мана, с помощью которого возможен от-
счет изменения температуры с точностью
до 0,005°. Термометр Бекмана с мешал-
176
кой 5 помещают в пробирку 3 и, перемешивая жидкость в пробирке
с помощью мешалки, следят по термометру за понижением темпе-
ратуры и отмечают точку замерзания То чистого растворителя.
После этого (по расплавлении кристаллов льда, образовавшихся
при замерзании растворителя) через боковой отросток 7 в пробир-
ку 3 вносят навеску исследуемого вещества и растворяют, поме-
шивая раствор. Опыт повторяют с раствором и определяют темпе-
ратуру замерзания раствора Т3. Молекулярную массу растворен-
ного вещества вычисляют по уравнению (7.2).
5. ТЕМПЕРАТУРА КИПЕНИЯ РАЗБАВЛЕННЫХ РАСТВОРОВ
Температура кипения растворов нелетучего вещества в лету-
чих растворителях всегда выше температуры кипения чистого
растворителя при том же давлении. Рассмотрим р-Т-диаграмму
для воды (растворитель) и разбавленных водных растворов,
содержащих различную массу растворенного вещества (рис. 68).
Любая жидкость — растворитель или раствор — кипит при
той же температуре, при которой давление насыщенного пара
становится равным внешнему давлению. Соответственно темпе-
ратуры, при которых изобара, равная атмосферному давлению
(101,3 кПа), пересечет кривые О А, ВС и т. д., будут температу-
рами кипения соответствующих жидкостей при этом давлении.
Для растворов эти температуры являются более высокими,
чем для чистого растворителя, и разность между ними
АТ -- Т __ ТО
кип— 1 кип 1 кип
будет тем большей, чем выше концентрация раствора. Для бес-
конечно разбавленных растворов бесконечно малые участки кри-
вых СС', ЕЕ' и т. д. можно считать прямолинейными. Тогда
из подобия треугольников ACC', АЕЕ' и т. д. вытекает, что
повышение температуры кипения
пропорционально понижению давления
насыщенного пара и, следовательно,
повышение температуры кипения про-
порционально концентрации раствора:
A7KHn = E/n. (7«3)
Для каждого данного рас-
творителя коэффициент про-
порциональности Е является
величиной постоянной. Он на-
зывается моляльным повыше-
нием температуры кипения или
эбуллиоскопической
постоянной. Значения
Рис. 68. Повышение температуры ки-
пения разбавленных растворов
177
эбуллиоскопической постоянной констант для некоторых раство-
рителей приведены в табл. 23.
6. ОСМОТИЧЕСКОЕ ДАВЛЕНИЕ
В 1748 г. А. Нолле впервые наблюдал, как растворитель про-
ходит через мембрану из разбавленного раствора в более кон-
центрированный. Если к более концентрированному раствору
приложить давление, то в зависимости от его величин течение
растворителя может быть замедлено или остановлено. Это явле-
ние было названо осмосом.
Осмотическим давлением раствора называется то наименьшее давление,
которое помимо давления самого растворителя необходимо приложить к раство-
ру, чтобы предотвратить перемещение растворителя к раствору через мембрану,
разделяющую раствор и растворитель, причем мембрана непроницаема для
молекул растворенного вещества.
Иллюстрацией осмотического давления может служить
рис. 69. В левом отделении прибора находится чистый раствори-
тель, в правом — раствор. Эти две жидкости отделены друг от
друга полупроницаемой мембраной (например, мембра-
ной из целлофана). Поры в мембране достаточно велики, чтобы
через них свободно проходили молекулы растворителя, но мемб-
рана препятствует проникновению молекул растворенного веще-
ства из правой части сосуда в левую. Скорость перехода рас-
творителя из левой части сосуда в правую в начальный момент
больше, чем скорость его перемещения в обратном направлении.
Поэтому высота столба жидкости в правой части сосуда увели-
чивается до тех пор, пока не будет достигнуто равновесие, при
котором разность уровней жидкости в левой и правой частях (Л)
отвечает осмотическому давлению.
Рис. 69. Прибор для демонстрации осмо-
тического давления:
кружки — молекулы растворителя; квадрати-
ки — молекулы растворенного вещества
В 1877 г. В. Пфеффер из-
мерил осмотическое давление
л нескольких растворов, при-
готовленных путем растворе-
ния одной и той же массы
вещества в разных объемах
растворителя. При этом он
показал, что если поддержи-
вать температуру постоян-
ной, то произведение л V всег-
да будет одним и тем же. Для
данного раствора с повыше-
нием температуры осмотиче-
ское давление увеличивается,
причем отношение л/Г сохра-
няется постоянным. Я. Вант-
Гофф обобщил эти резуль-
таты и предложил эмпириче-
ское уравнение для описания
178
осмотического давления растворов:
(7.4)
где л — осмотическое давление; С — молярная концентрация
раствора; Т — температура.
Линейная зависимость осмотического давления от концентра-
ции раствора и от температуры соблюдается только для идеаль-
ных растворов. Поэтому уравнение (7.4) можно применять толь-
ко для разбавленных растворов. Если растворенное вещество
диссоциирует и имеет степень диссоциации а (см. гл. VIII,
разд. 2.1), то в простейшем случае диссоциации одной частицы
на две имеем
АВ^А+4-В“
Число недиссоциированных частиц, получившихся из 1 моль,
равно (1—а) моль, число продуктов диссоциации равно 2а моль,
а всего молей будет
1 — а 4- 2а = 1 + а.
Сумму 1 +а обозначают буквой L Это так называемый изото-
нический коэффициент Вант-Гоффа (см. гл. VIII).
Тогда уравнение осмотического давления (7.3) принимает
вид
Осмотическое давление можно опреде-
лять двумя основными методами: статиче-
ским и динамическим.
Статический метод основан на
том, что осмотическое давление раствора
уравновешивается давлением столба жид-
кости, возникающем в результате проник-
новения растворителя в раствор.
Осмометр (рис. 70) состоит из камеры 2
вместимостью ~ 10 мл из стекла или хроми-
рованной латуни. Камера присоединяется с
помощью винтов к пластинке, в середине
которой имеются отверстия (диаметром
1 мм). Нижняя сторона камеры плотно при-
жимает мембрану к пластинке — сетке, тол-
щина которой должна быть не больше
0,5 мм. Часть осмометра, в которой находит-
ся раствор, называется осмотической ячей-
кой. Раствор наливается в ячейку через
верхнее отверстие /, куда для отсчета дав-
ления вставляется пришлифованный градуи-
рованный капилляр диаметром 1 мм и дли-
ной 50 см. Нижняя пришлифованная часть
капилляра входит в камеру на 0,5... 1 мм для
предохранения камеры от пузырьков воздуха
при заполнении.
Рис. 70. Осмометр для
определения осмотиче-
ского давления стати-
ческим методом
179
Рис. 71. Осмометр для определения осмо-
тического давления динамическим мето-
дом
Камера с раствором
вставляется в сосуд 5, напол-
ненный чистым растворите-
лем. Сосуд закрывается при-
шлифованной крышкой во
избежание испарения рас-
творителя. При измерении
осмотического давления ос-
мометр помещают в термо-
стат.
Динамический ме-
тод основан на том, что
осмотическое давление ком-
пенсируется наложенным на
раствор переменным проти-
водавлением. Осмотическое
давление вычисляется на основании измерения скорости проник-
новения растворителя через мембрану. Преимущество динами-
ческого метода заключается в быстроте измерений.
На рис. 71 представлен компенсационный осмометр А. В. Ду-
манского. Осмотическая ячейка 6 соединяется с мембраной 7,
аспиратором 1 и манометром 3. Ячейку и внешний сосуд поме-
щают в термостат 5. Поднятием верхней части аспиратора 2
регулируется внешнее давление таким образом, чтобы оно было
больше или меньше осмотического давления. При избыточном
внешнем давлении мениск в капилляре 4 опускается вниз со ско-
ростью с/1, пропорциональной избыточному давлению р\— л. При
пониженном внешнем давлении мениск поднимается со скоро-
стью ^2, пропорциональной разности л—рг, тогда отношение ско-
ростей перемещения растворителя по капилляру v\/vz будет
равно отношению разностей давлений, обусловливающих подня-
тие или опускание мениска в капилляре, т. е.
V\ __ pi — л
VI л — Р2
отсюда
___ У 1р2 4" У2р1
Я ^14-^2
Осмотическое давление данного раствора может быть экспе-
риментально определено по понижению температуры замерза-
ния или по повышению температуры кипения раствора.
Предполагая равенство моляльной и молярной концентрации
(т^С) для разбавленных растворов, можно при сопоставлении
уравнений
_\Т=Кт, n = CRT
получить значение осмотического давления, связанного с величи-
ной АГ, которое определяют экспериментально:
180
л
Аналогично можно установить связь осмотического давления
с повышением температуры кипения и эбуллиоскопической кон-
стантой.
Осмотическое давление играет большую роль в жизни клеток. Каждая рас-
тительная клетка покрыта прочной целлюлозной оболочкой, к которой плотно
прилегает протоплазма клетки. Поверхностный слой этой протоплазмы обладает
свойствами полупроницаемой оболочки и, следовательно, свободно пропускает
воду и не пропускает или почти не пропускает многие растворенные в воде
вещества. Целлюлозная оболочка свойствами полупроницаемости не обладает
и поэтому легко проницаема для всех растворенных веществ.
Если растительную клетку перенести в концентрированный раствор
какого-нибудь вещества (например сахара или хлорида натрия), молярная кон-
центрация которого будет выше, чем концентрация растворенных веществ в
клетке, то наблюдается осмотическое высасывание воды из клетки в окружающий
ее внешний раствор (экзосмос). Протоплазма уменьшается в объме и отстает от
стенок целлюлозной оболочки. Объем протоплазмы делается тем меньше, чем
большей концентрации был раствор, в который погружена клетка. При соответ-
ствующих условиях протоплазма принимает шарообразную форму, уменьшаясь
в несколько раз. Это явление называется плазмолизом. Если плазмоли-
зированную клетку поместить снова в раствор обычной для нее концентрации
или в дистиллированную воду, клетка, благодаря осмотическому всасыванию
•растворителя, увеличивает свой объем, возвращаясь в свое исходное положение.
Таким образом, плазмолиз является обратимым процессом.
Если нормальную растительную клетку поместить в раствор, концентрация
которого будет ниже, чем концентрация растворенных веществ в самой клетке,
или просто в дистиллированную воду, то под влиянием более высокого осмоти-
ческого давления содержимого клетки происходит осмотическое всасывание воды
в клетку (эндосмос). Объем клетки при этом увеличивается, растягивая стенки
целлюлозной оболочки: клетка при этих условиях находится в состоянии напря-
жения. Состояние осмотического напряжения клетки, обусловленное повышенным
осмотическим давлением, называется тургором. Он поддерживает в напря-
жении ткани и органы у растений. Увядание растений связано с уменьшением
тургора.
Естественно, что клетки окажутся неизменными, если их поместить в среду,
осмотическое давление которой одинаково с внутриклеточным осмотическим дав-
лением. Все упомянутые осмотические процессы присущи также животным
тканям и клеткам.
Растворы, осмотическое давление которых одинаково с осмотическим давле-
нием клеток и тканей, называются изоосмотическими или изото-
ническими. Растворы, молярная концентрация которых, а стало быть и осмо-
тическое давление, выше, чем внутри клеток и тканей, называются гипер-
тоническими. Растворы, молярная концентрация которых, а следователь-
но, и осмотическое давление, ниже, чем в клетках и тканях, называются гипо-
тоническими.
Итак, если изотонические растворы не вызывают изменений в клетках, то
растворы гипертонические обусловливают явление плазмолиза, а растворы
гипотонические — явление тургора.
Нормальные растительные клетки всегда тургоризированы. Иначе говоря,
концентрация растворенных веществ клетки выше, чем концентрация растворен-
ных веществ в окружающей среде. Тургор растительных клеток является необ-
ходимым условием для роста и нормальной жизнедеятельности.
В растениях наблюдается значительное осмотическое давление, достигаю-
щее 0,5...2,0 МПа. Некоторые растения пустынь и засоленных почв, которым
приходится особенно упорно бороться за влагу, имеют осмотическое давление,
достигающее 5 МПа и даже 17 МПа.
Особенно высоко осмотическое давление в точках роста. Так, у клеток стеб-
181
левых узлов злаков оно достигает 5 МПа. Корни всегда имеют более высокое
осмотическое давление, чем почвенный раствор, откуда они всасывают воду и
питательные вещества.
7. СОСТАВ ПАРА ИДЕАЛЬНЫХ И РЕАЛЬНЫХ РАСТВОРОВ
Если компоненты раствора в чистом состоянии летучи, то пар
будет содержать эти компоненты. Однако относительное содер-
жание компонентов в парах, как правило, будет отличаться от
относительного содержания их в жидкости. Пар относительно
богаче более летучим компонентом смеси.
Расссмотрим смесь двух жидкостей с неограниченной взаим-
ной растворимостью. Пусть даны вещества А и В. Обозначим
молярные доли их Ха и Хв. По закону Рауля имеем
Ра = Ра^а, Рв = Рв^в»
где р° — давление насыщенного пара чистого вещества.
Рис. 72. Зависимость парциальных и общих давлений пара реального
(сплошные линии) и идеального (пунктир) бинарных растворов от
состава при положительных (а) и отрицательных (6) отклонениях от
закона Рауля
Эти уравнения графически выражаются прямыми, если на
осях отложены давления пара и молярные доли (рис. 72). Общее
давление пара раствора равно
Р = %аРа + ^вРв
или, так как
Ха= 1-Хв,
то
Р = Ра + *в(р°в — Ра) •
Эти соотношения являются линейными относительно Хв.
Таким образом, для идеальных растворов (АЯР =0, AVp = O)
зависимость общего и парциальных давлений пара от состава
раствора (при повышении его в молярных долях) оказывается
линейной (пунктирные линии на рис. 72). Такими являются,
182
например, системы бензол — толуол, //-гексан — я-гептан, смеси
изомерных углеводородов и др.
Для реальных растворов (A//n#:0, AVp=/=0) простая линейная
зависимость рд = РаХа и рв = рвХв осложняется и фактическая
зависимость изображается уже кривой, а не прямой линией.
Если молекулы данного компонента взаимодействуют друг с
другом сильнее, чем с молекулами другого компонента, то истин-
ные парциальные давления пара над смесью будут больше
вычисленных по закону Рауля (положительные отклонения).
Если же частицы разных компонентов притягиваются друг к
другу сильнее, чем частицы одного и того же компонента, то
парциальные давления паров компонентов будут меньше вы-
численных (отрицательные отклонения).
Реальные растворы, характеризую щиесй положительными отклонениями
давления пара, образуются из чистых компонентов большей частью с поглоще-
нием теплоты.
Поглощение теплоты при образовании раствора приводит к
уменьшению количества теплоты, необходимого для превращения
жидкости в пар, что при прочих равных условиях облегчает про-
цесс испарения. Поэтому давление пара каждого данного ком-
понента над раствором становится большим, чем для идеальных
растворов.
Реальные растворы, характеризующиеся отрицательными отклонениями дав-
ления пара, образуются из чистых компонентов обычно с выделением теплоты.
Вследствие этого теплота испарения компонентов из такого
раствора оказывается большей, чем из чистых растворителей.
Поэтому давление насыщенного пара оказывается меньшим,
чем соответствующих идеальных растворов.
8. ПОНЯТИЕ АКТИВНОСТИ
Если концентрация растворенного вещества не превышает
0,1 кмоль/м3, раствор неэлектролита обычно считают разбавлен-
ным. В таких растворах существуют взаимодействия между
молекулами растворителя, а также между молекулами раство-
ренного вещества и растворителя, однако первые существенно
преобладают и поэтому вторыми можно пренебречь. В концент-
рированных растворах взаимодействием между молекулами раст-
воренного вещества и растворителем уже нульзя пренебречь.
В настоящее время имеющиеся данные о межмолекулярных
силах недостаточны для теоретического учета отклонений от
идеальности, потому что приходится ограничить описание систе-
мы эмпирическими методами. Для этого вводится величина,
называемая активностью а, которая заменяет концентра-
цию С и связана с ней соотношением
где f — коэффициент активности. Коэффициент
183
активности не является постоянной величиной, он зависит от
концентрации С.
Таким образом, активность можно рассматривать как «эффек-
тивную концентрацию». В предельном случае бесконечного раз-
бавления
lim — = limf— 1.
С-0 С с^о/
В зависимости от природы раствора возможны положитель-
ные и отрицательные отклонения от единичного значения коэф-
фициента активности.
В табл. 24 приведены данные по коэффициентам активности
некоторых электролитов.
Таблица 24. Коэффициенты активности некоторых электролитов
при 298 К
Электролит f для молярной концен- трации, кмоль/м3 Электролит f для молярной концен- трации, кмоль/м3
0,01 0,1 1,0 0,01 0,1 1,0
AgNO3 0,90 0,72 0,40 кон 0,90 0,80 0,76
ВаС12 0,72 0,49 0,39 MgSO4 0,40 0,18 0,06
Ca(NO3)2 0,71 0,48 0,35 NH4C1 0,88 0,74 0,57
CdCl2 0,53 0,22 0,06 NaCl 0,92 0,80 0,65
C11SO4 0,40 0,16 0,05 Pb(NO3)2 0,69 0,37 0,11
HNO3 0,91 0,79 0,73 ZnCl2 0,71 0,50 0,33
H2SO4 0,54 0,27 0,13 ZnSO4 0,39 0,15 0,05
КС1 0,90 0,78 0,61
Контрольные вопросы
1. Чем отличаются идеальные растворы от реальных?
2. Используя принцип Ле Шателье, рассмотрите влияние внешних условий
на процесс растворения газов в жидкости.
3. Используя принцип Ле Шателье, рассмотрите процесс понижения давления
насыщенного пара разбавленных растворов.
4. При образовании насыщенного раствора ДС = 0, а каково ее значение для
перенасыщенного раствора?
5. Как можно доказать, что раствор спирта в воде является не идеальным
раствором?
6. Рассмотрите р — Г-диаграмму для растворителя и разбавленных растворов
в разных концентрациях с описанием каждой кривой и каждой точки пересечения.
7. Какие факторы определяют неограниченную и ограниченную растворимость
двух жидкостей?-
Глава VIII
РАСТВОРЫ ЭЛЕКТРОЛИТОВ
I. ОБРАЗОВАНИЕ ИОНОВ В ВОДНЫХ РАСТВОРАХ
Электролитами называются соединения, которые существуют в растворе
в виде ионов независимо от прохождения или непрохождения через раствор
электрического тока/
Причиной, вызывающей распад растворенного вещества на
ионы, является интенсивное взаимодействие ионов с молеку-
лами растворителя, т. е. сольватация ионов. Частный
случай сольватации ионов есть гидратация, т. е. взаимо-
действие их с водой.
Впервые представление о гидратации ионов было введено
Д. И. Менделеевым в его химической теории растворов. Даль-
нейшие исследования И. А. Каблукова и В. А. Кистяковского, а
также теория электролитической диссоциации С. Аррениуса
оказались плодотворными для объяснения свойств растворов
электролитов.
На рис. 73 приведена схема, иллюстрирующая образование
гидратов при взаимодействии ионов с молекулами воды. Если
образуются стойкие гидраты, то их удается выделить при охлаж-
дении или выпаривании раствора. Так, из растворов гидрокар-
боната натрия можно получить кристаллы, содержащие десять
молекул воды на молекулу №гСОз. Если же гидраты нестойки,
то они разлагаются при выделении, и их наличие удается дока-
зать при помощи спектрального исследования растворов.
Учитывая сказанное, процесс растворения можно предста-
вить в виде уравнения
р>М+(г) +Х"(г)
МХ(т) — 1 (8Л)
‘L^M+(ag) 4- X\aq)
Изменение энтальпии для стадии а срответствует энергии, необ-
ходимой для удаления ионов из кристаллической решетки на
бесконечное расстояние, что эквивалентно процессу сублимации
по схеме а. Эту энергию называют энергией решетки Uq.
Чтобы разрушить решетку, необходимо затратить значительную
работу, так как между ионами имеется сильное электростатиче-
ское притяжение. Так, для преодоления притяжения между раз-
ноименными ионами, имеющими заряд, равный заряду одного
185
Рис. 73. Сольватация ионов в водном растворе
электрона, и находящимися на расстоянии 0,2 нм, нужно затра-
тить энергию около 600 кДж/моль. Изменение энтальпии для
стадии в представляет собой теплоту растворения АЯР или теп-
лоту, которая поглощается или выделяется при растворении
твердого вещества MX в воде. Стадия б характеризует собой
теплоту гидратации ионного газа.
По определению, теплота гидратации АЯГ равна количеству
теплоты, которая выделяется при переводе 1 моль ионов из ва-
куума в водный раствор. Величина А/7Г может быть найдена из
экспериментальных данных; имеются также методы теоретиче-
ского расчета.
Для рассмотренной схемы (8.1) теплота гидратации, соглас-
но закону Гесса, определяется по уравнению
ЬНГ = ЬНР- Uo, (8.2)
где А//р — теплота растворения.
Величину АЯР можно измерить экспериментально. Uo можно
оценить, если известна структура 'решетки или если эта величи-
на непосредственно измерена. Для NaCl С/0 = 765 кДж/моль
по расчетным данным и (70 = 778 кДж/моль по данным экспе-
римента; А//р = 3,8 кДж/моль, так что АЯГ=3,8 —778 =
= —774,2 кДж/моль. Таким образом, при гидратации ионов Na+
и С1“ освобождается значительное количество теплоты.
Обобщение большого числа подобных примеров приводит к
выводу, что с энергетической точки зрения образование ионов
в растворе значительно облегчается по сравнению с газовой
фазой, поскольку затрата энергии на преодоление электростати-
ческого взаимодействия разноименно заряженных ионов в значи-
тельной степени компенсируется энергией взаимодействия обра-
зовавшихся ионов с их сольватными оболочками.
При расчете Д{7Г по уравнению (8.2) получают сумму теплот гидратации
ионов обоих видов, образующих соль, — катионов и анионов. Для нахождения
теплот гидратации отдельных ионов эту величину нужно разделить на состав-
ляющие для катионов и анионов; выбор правильного метода разделения
186
представляет довольно трудную задачу. В 1977 г. С. И. Дракиным было уста-
новлено, что теплоту гидратации отдельных ионов можно довольно точно опре-
делить как сумму
ДЯг = А^г(1) + ДЯГ(2) + Ас, (8.3)
где Д/7Г(1) — тепловой эффект взаимодействия иона с ближайшими молекулами
воды; ДлГ(2) — тепловой эффект взаимодействия иона с остальными моле-
кулами воды; Ас — работа разрыва водородных связей при образовании ионов
в растворе.
Для первого слагаемого уравнения (8.3) известны экспериментальные зна-
чения (определены тепловые эффекты взаимодействия ионов с молекулами воды
в газовой фазе), два других слагаемых можно рассчитать с точностью от 5 до
9 кДж/моль. Для иона Na+ было получено ДЯГ=—473 кДж/моль. Зная
Д//г иона одного вида и суммарные ДЯГ для солей, можно найти ДЯГ других
ионов (табл. 25).
Таблица 25. Теплоты гидратации некоторых ионов
Ион дяг, кДж/моль Ион ДЯп кДж/моль Ион дяг, кДж/моль
Li+ —587 сг -312 Sr2+ -1579
Na+ -473 Вг~ -281 Ва2+ -1441
К+ -389 Г —241 А13+ —4877
Rb+ -364 Ве2+ -2619 Sc3+ -4130
Cs+ -340 Mg2+ -2058 -3788
F+ —452 Са2+ -1712 La3+ —3473
Естественно, поставить вопрос о роли типа растворителя в
процессе образования раствора электролита. Растворение солей
в воде происходит вследствие ослабления связей между ионами.
По закону Кулона сила взаимного притяжения между ионами
пропорциональна произведению зарядов ионов и обратно пропор-
циональна квадрату расстояния между ионами:
(8.4)
8/2 ’
где f — сила притяжения между ионами; е\ и е% — заряды ионов;
I — расстояние между ионами; е — диэлектрическая постоянная.
Диэлектрическая постоянная для абсолютной пустоты равна
единице. Для других сред она больше единицы и показывает, во
сколько раз сила взаимодействия между двумя наэлектризован-
ными телами в данной среде меньше, чем в пустоте. Диэлек-
трическая постоянная воды равна ~80. Это означает, что в воде
сила притяжения между ионами в 80 раз меньше, чем в пустоте.
Согласно уравнению (8.4) можно сделать вывод, что увеличе-
ние диэлектрической постоянной приводит к уменьшению сил
взаимодействия между ионами. Поэтому сольватирующая спо-
собность растворителя растет с увеличением его диэлектрической
постоянной. Следовательно, диэлектрическая постоянная может
служить количественной характеристикой ионизирующей способ-
ности растворителя. В табл. 26 приведены значения диэлектриче-
187
Таблица 26. Диэлектрическая постоянная для ряда жидкостей
Растворитель Е Растворитель Е
Ацетон 21,5 Серная кислота 84,0
Бензол 2,23 Сероуглерод 2,65
Вода 81,0 Толуол 2,29
к-Гексан 1,89 Уксусная кислота 6,4
Глицерин 56,2 (288 К) Уксусный ангидрид 20,5
Диоксан 2,0 Хлорбензол 10,3
Метиловый спирт 33,7 Хлороформ 5,1
Муравьиная кислота 58,5 Циклогексан 2,05
Нитробензол 36,4 Тетрахлорид углерода 2,23
Пиридин 12,5 Этиловый спирт 25,8
ской постоянной ряда жидкостей, чаще всего используемых в
химии в качестве растворителей. Как и следовало ожидать, легче
всего ионы образуются в растворителях с большими значениями
диэлектрической постоянной. Наоборот, в таких неполярных
растворителях, как бензол, гексан, диоксан, толуол, тетрахлорид
углерода, ионы образовываться в заметной мере не могут и в
таких растворителях процессы ионообразования практически не
осуществляются.
Если рассматривать процесс образования раствора с пози-
ции растворяемого вещества, то возникновение ионов может
идти двумя основными путями. Если электролитами являются
ионные соединения, то положительные и отрицательные ионы
существуют в кристалле еще до его растворения. При этом
растворители с большой диэлектрической постоянной, в частно-
сти вода (е = 81), разрушая кристаллическую решетку, создают
в растворе ионы противоположных зарядов. Эти ионы сольва-
тируются молекулами растворителя
Cl Na+ (кристалл)— Cl“(aq) + Na+(aq)
Соединения с ковалентными связями до попадания в раствор
не содержат ионов, и тогда процесс растворения представляет
собой обычную химическую реакцию между ковалентным соеди-
нением и растворителем, выступающим в роли донора электро-
нов.
Так, например, связь Н—С1 соляной кислоты подвергается ге-
теролитическому разрыву под действием диэтилового эфира
(СгНбЬО; при этом образуются ионы (С2Н5)2ОН+ и С1~.
Ион(С2Н5)2ОН+ возникает при переносе протона кислоты на мо-
лекулу эфира, с которой он связывается при помощи одной из
свободных электронных пар атома кислорода. При этом осво-
бождается анион С1“. Реакция записывается в виде
С2Н5'
С2Н5'
Н—С1
С2Н5ч
+—н] ci-
[
188
Значение диэлектрической постоянной эфира мало (е = 4). Поло-
жительные и отрицательные ионы, возникающие в процессе иони-
зации, остаются по соседству друг с другом, образуя ионную
пару. Поэтому растворы, содержащие ионные пары, не проводят
ток. Вода же может сначала ионизировать ковалентные соеди-
нения (например, кислоты), а затем диссоциировать образовав-
шиеся ионные пары на свободные ионы, сольватированные мо-
лекулами воды, и тогда раствор будет проводить ток. Реакция
воды с соляной кислотой запишется так:
Н\ Гнх
>0 4- и— С1 >о+—Н + С1-
hz Lhz
Таким образом, образование ионов в растворе электролитов
представляет собой сложный физико-химический процесс, приво-
дящий к системам более сложным, чем растворы неэлектроли-
тов. В растворах электролитов находятся разноименные частицы,
взаимодействующие как между собой, так и с молекулами
растворителя. Образующиеся в растворе ионы резко отличаются
от молекул по физическим, химическим и физиологическим па-
раметрам.
2. СЛАБЫЕ И СИЛЬНЫЕ ЭЛЕКТРОЛИТЫ
По способности образовывать ионы в растворе электролиты
делят на две группы: слабые и сильные.
К сильным электролитам принадлежат сильные кислоты, силь-
ные основания и большая часть солей. К ним относятся все соли,
образованные сильным основанием или сильной кислотой, и
большая часть солей, образованных слабым основанием и сла-
бой кислотой. Многие сильные электролиты кристаллизуются в
кристаллах с ионной решеткой; часто сильными электролитами
называют только такие вещества.
К слабым электролитам относятся ковалентные соединения,
подвергающиеся в воде частичной диссоциации. Это прежде
всего слабые кислоты и слабые основания, а также некоторые
соли. К ним принадлежат большинство органических кислот,
фенолы, амины. Диссоциация связана с поляризацией ковалент-
ной связи, степень которой определяет число образующихся
ионов. Хорошей иллюстрацией этого могут служить хлоруксус-
ные кислоты. Если последовательно присоединять к а-углеро-
ду уксусной кислоты один, два и, наконец, три атома хлора, то
смещение электронов благодаря влиянию атомов хлора будет
приводить к усилению поляризации связи О—Н карбоксильной
группы. В трихлоруксусной кислоте электронная пара уже на-
столько сильно смещена в сторону кислорода, что эта кислота в
воде будет в сильной степени диссоциирована. Сравнение кон-
189
стант диссоциации (см. гл. VIII, разд. 2.1) показывает, что ве-
личина А для трихлоруксусной кислоты равна 2-10“1 кмоль/м3,
а для уксусной кислоты — всего лишь 1,86-10-5 кмоль/м3.
2.1. Слабые электролиты. Степень диссоциации
Процесс диссоциации слабых электролитов является обратимым.
Молекулы распадаются на ионы, а образующиеся ионы проти-
воположного знака, встречаясь в растворе, могут вновь соеди-
няться в молекулы. Для электролита вида АВ процесс диссоциа-
ции можно записать так:
Ав^ а+ + В-
Как и во всяком обратимом процессе, здесь устанавливается
некоторое динамическое равновесие. Так как слабые электролиты
подчиняются, закону действующих масс, это равновесие может
количественно характеризоваться константой равновесия, назы-
ваемой в данном случае константой диссоциации.
Для рассматриваемого электролита, распадающегося на два
иона (так называемый бинарный электролит), эта константа вы-
разится соотношением
Следует подчеркнуть, что эта константа может достаточно
корректно характеризовать лишь разбавленные растворы слабых
электролитов.
Количественная характеристика равновесного состояния дис-
социации слабого электролита может быть получена также с
использованием степени диссоциации а, которая показывает, ка-
кая часть молекул (Электролита в растворе распалась на ионы:
__ Число распавшихся молекул
Общее число растворенных молекул
Значение а всегда меньше единицы. Умножая а на 100, полу-
чают долю ионизированных молекул в процентах.
Степень диссоциации связана с константой диссоциации. Так,
для бинарного электролита АВ концентрации ионов [А+] и [В+]
связываются с общей концентрацией электролита С соотноше-
нием
[А+] = [В+] = аС.
Степень диссоциации зависит от природы электролита, при-
роды растворителя, температуры раствора и степени его разбав-
ления. Как уже было рассмотрено, чем больше диэлектрическая
постоянная растворителя, тем значительнее при прочих равных
условиях электролитическая диссоциация растворенного в нем
вещества. Диссоциация молекул — процесс эндотермический.
Следовательно, по принципу Ле Шателье с повышением темпе-
190
ратуры степень диссоциации должна возрастать. Однако при
повышении температуры уменьшается диэлектрическая постоян-
ная растворителя, что благоприятствует образованию молекул
из ионов. У большинства электролитов степень диссоциации
по мере повышения температуры увеличивается, а у некоторых
(например, NH4OH, СНзСООН) достигает максимума, а затем
уменьшается.
С разбавлением раствора электролита вероятность взаимо-
действия ионов в растворе уменьшается, степень электролити-
ческой диссоциации увеличивается. При этом константа диссоци-
ации, вычисленная для данного слабого электролита, в сильно
разбавленном растворе имеет постоянное значение (табл. 27).
Экспериментальные данные свидетельствуют, что растворы элек-
тролитов обнаруживают всегда большие отклонения от свойств
чистого растворителя, чем растворы неэлектролитов. Отклонения оп-
ределяются большим числом (концентрацией) частиц растворенного
вещества в растворе электролита вследствие диссоциации его
молекул на ионы. В результате диссоциации число частиц ока-
зывается. больше расчетного. Следовательно, в уравнениях, ко-
торые выражают зависимость свойств раствора от его концентра-
ции для неэлектролитов, требуется ввести поправочный множи-
тель, отражающий это явление (табл. 28).
Оценка отклонения позволяет определять степень диссоциа-
ции. Поэтому значение степени диссоциации может быть опреде-
лено по величине осмотического давления, понижению темпера-
Таблица 27. Значение а и К для уксусной кислоты
при различных разбавлениях
Разбавление, м3/кмоль а-102 К-105
13,57. 1,570 1,845
27,14 2,216 1,851
108,56 4,380 1,849
868,4 11,90 1,850
3474,0 22,36 1,855
Таблица 28. Сопоставление некоторых уравнений
для растворов неэлектролитов и электролитов
Для растворов неэлектролитов Для растворов электролитов
Ар «1 р0 П14-П2 АТз = Кт АТКИП = Ет n^CRT Ар Zrti ро /П|Ч-Л2 АГ3 = 1Кт ДТКИП =i iEm л = iCRT
191
туры замерзания раствора и данным электрической проводи-
мости.
Если в растворе до диссоциации находилось У молекул и
если степень диссоциации их при данных условиях равна а, то
число диссоциированных молекул будет
ДО—Мх = У(1 - а).
Когда каждая молекула способна распадаться на п ионов,
число всех частиц (молекул и ионов) равно
N(1 — a) + пДОа = ДО(1 — а -|- па).
Осмотическое давление пропорционально числу частиц, следо-
вательно, наблюдаемое осмотическое давление лопр пропорцио-
нально общему числу частиц (молекул и ионов) после диссоциа-
ции, т. е. числу N(1 — a + na). Вычисляемое же осмотическое
давление лВыч пропорционально числу частиц (молекул) до дис-
социации, т. е. Af. Отсюда
i = = Ml — « + ««) + a(n _ Q (8.5)
Л-выч **
ИЛИ
Уравнение (8.5) позволяет понять физический смысл изотони-
ческого коэффициента.
Изотонический коэффициент показывает, во сколько раз суммарная эф-
фективная концентрация недиссоциированных молекул и ионов больше, чем
концентрация молекул до диссоциации.
В частном случае для бинарного электролита (п — 2)
i — 1 + a, a = i — 1.
Таким образом, определив осмотическое давление лопр экспе-
риментально и вычислив Лвыч из формулы л = ICRT, можно полу-
чить степень диссоциации слабого электролита.
Другим методом экспериментального определения степени
диссоциации является криоскопический метод, основанный на
определении кажущейся молекулярной массы М\, которая пред-
ставляет собой среднее значение масс ионов и недиссоциирован-
ных молекул:
где М — истинная молекулярная масса, откуда
А--1
п — 1
а для бинарного электролита
М - Mi
a =---—---.
192
2.2. Сильные электролиты. Активность
С. Аррениус, создавая теорию электролитической диссоциа-
ции, ошибочно предполагал, что в растворе сильного электролита
существует такое же динамическое равновесие между молекула-
ми и ионами, как и у слабых электролитов. Он,также ошибочно
считал, что, как и слабые электролиты, сильные электролиты
лишь частично диссоциированы в растворах любых концентраций
и разница между сильными и слабыми электролитами заключа-
ется лишь в разной степени диссоциации. Отсюда следовал не-
правильный вывод о применимости закона действующих масс к
сильным электролитам.
Экспериментальные исследования показали, что закон дейст-
вующих масс неприменим к диссоциации сильных электролитов.
Для слабых электролитов константа диссоциации с разбавлением
раствора остается величиной постоянной, а для сильных электро-
литов она изменяется с изменением концентрации.
Исследование растворов сильных электролитов с помощью
спектральных методов показало, что в них не существует недис-
социированных молекул, которые этими методами всегда обнару-
живаются в растворах слабых электролитов. Это позволило сде-
лать вывод, что сильные электролиты в отличие от слабых
электролитов не только в разбавленных, но и в растворах значи-
тельной концентрации содержатся только в виде ионов, иначе
говоря, все молекулы сильного электролита в растворе распада-
ются на ионы.
Из сказанного следует, что закономерности, выведенные для
слабых электролитов, не могут применяться без соответствую-
щих поправок к сильным электролитам.
Теория сильных электролитов, разработанная П. Дебаем и
Г. Хюккелем (1923), позволила объяснить противоречивые, на пер-
вый взгляд, данные о поведении сильных электролитов, а также
создала основу для количественных
сильных электролитов.
П. Дебай и Г. Хюккель предполо-
жили, что основной причиной резко-
го различия в поведении сильных и
слабых электролитов является нали-
чие в растворах сильных электроли-
тов межионных взаимодействий. Ио-
ны, находящиеся в растворе, хотя и
отделены друг от друга молекулами
растворителя, все же испытывают
слабое притяжение. Чем выше кон-
центрация, тем ближе ионы располо-
жены друг к другу. В результате
этого каждый ион оказывается окру-
женным «атмосферой» ионов проти-
воположного знака, что несколько
расчетов характеристик
© © ©
Рис. 74. Схематическое изобра-
жение ионной атмосферы Дебая
и Хюккеля
7—696
193
ограничивает свободу его движения (рис. 74). Благодаря ограни-
чению в движении ионов растворенного вещества ослабляется
их .влияние на процесс испарения растворителя. Поэтому дав-
ление пара растворителя понижается уже не строго пропорцио-
нально числу присутствующих ионов, а в несколько меньшей
степени.
Усиление взаимного притяжения ионов, вызываемое увеличе-
нием концентрации, приводит к изменению свойств в том же на-
правлении, как действовало бы частичное соединение ионов в
молекулы, т. е. уменьшение степени диссоциации. Поэтому повы-
шение концентрации раствора даже при полной диссоциации
электролита влияет на свойства раствора в известной степени
аналогично тому, как если бы при этом уменьшалась степень
диссоциации электролита.
Следовательно, определяя степень диссоциации для не. очень
разбавленных растворов сильных электролитов, находят не ис-
тинную, а так называемую кажущуюся степень дис-
социации, т. е. величину с поправками на межионные силы
взаимодействия. Чем выше концентрация раствора, тем сильнее
будет взаимодействие разноименно заряженных ионов между со-
бой, тем меньше будет и кажущаяся степень диссоциации, хотя
на самом деле молекулы сильных электролитов диссоциированы
полностью.
Количественные расчеты характеристик растворов сильных
электролитов осуществляются с введением понятий об актив-
ности электролита аэ и активности его ионов:
а+ — активность катионов, а_ — активность анионов. Актив-
ность аэ выражает эффективную концентрацию диссоциирован-
ной части электролита в растворе с учетом межионных взаимо-
действий, т. е. взаимного притяжения разноименных ионов.
Тогда активность равна произведению коэффициента активности
на концентрацию:
a+ = f+C+, a_ = f_C_,
где и f_ — коэффициенты активности катионов и анионов со-
ответственно; и С- — аналитические концентрации катионов
и анионов.
Практически определяют средний коэффициент активности
для катионов и анионов, так как в любом растворе одновременно
содержатся те или другие ионы. Для простоты средний коэффи-
циент активности f± обозначают через f.
Для однозарядного электролита средняя активность аэ дается
уравнением
аэ == у/а+а_.
Аналогичным образом коэффициент активности выражают
как средний коэффициент активности ионов электролита:
194
Для очень разбавленных растворов, когда межионные силы
равны нулю, = С+, а_ = С_ и, следовательно, f = 1. В более
концентрированных растворах каждый ион притягивает сосед-
ние противоположно заряженные ионы и связывает молекулы
растворителя, вследствие чего уменьшается его активность,
т. е. f < 1.
П. Дебаем и Г. Хюккелем был разработан метод расчета f.
Для бинарного электролита в предельной форме, относящейся к
разбавленным водным растворам при 298 К, уравнение имеет вид
lg/ — — 0,51z2/F,
где z — заряд иона (в единицах заряда электрона), для которого
рассчитывается коэффициент активности f; I — ионная сила
раствора, взятая с учетом всех веществ, присутствующих в
растворе и подверженных диссоциации.
Ионная сила представляет собой удобный параметр, который
выражает концентрацию всех растворенных веществ и одновре-
менно учитывает их как молекулярную концентрацию (или мо-
ляльность), так и их заряд. Так как поведение иона обусловлено
влиянием ионной атмосферы, рассматриваются все ионы, присут-
ствующие в растворе. Ионная сила / рассчитывается как полу-
сумма концентраций всех ионов, умноженных на квадрат их
заряда:
/ = 0,5£Gz?.
В качестве примера рассчитаем ионную силу 0,1 М КН2РО4 и 0,05 М К2НРО4
растворов.
Сначала необходимо выписать все виды ионов, присутствующих в растворе:
К4, НгРОг и НРОГ . Данные соли образуют растворы, значения pH которых
лежат в интервале от 4 до 10, так что присутствием ионов НзО+ и ОН", образую-
щихся в результате собственной диссоциации воды, можно пренебречь. Сум-
марная концентрация каждого из ионов будет равна: [К+] = 0,1 + (2• 0,05),
[Н2РО4"1 =0,1, [НРОГ] =0,05.
Подставив значения концентраций и зарядов в выражение для ионной силы,
получим
\ Z = 0,5([К+] • I2 4- [Н2РО4"]-12+ [НРОГ] -22) =
= 0,5(0,2 + 0,1 + 0,05-4) = 0,25.
Теория П. Дебая и Г. Хюккеля обладает применимостью
только при концентрациях, не превышающих 0,01...0,05 кмоль/м3.
При более высоких концентрациях эта теория оказывается недо-
статочно полной, чтобы описать возникшие сложные системы.
Так, например, имеются данные, что при более высоких концен-
трациях может происходить ассоциация ионов, когда противо-
положно заряженные ионы сближаются и взаимодействуют меж-
ду собой, хотя не так сильно, как при типичном образовании
молекул.
7*
195
3. РАЗВИТИЕ ПОНЯТИЯ КИСЛОТЫ И ОСНОВАНИЯ
В 1887 г. С. Аррениус впервые установил критерии кислот-
ности и основности. По Аррениусу, кислота — это соединение с
характерным кислым вкусом, действующее на окрашенные инди-
каторы. Эти свойства определяются присутствием в водном
растворе ионов Н+, возникающих при диссоциации кислоты.
Основание также воздействует на окрашенные индикаторы и
кислоты, что связано с наличием в водном растворе ионов ОН-.
В дальнейшем было показано, что некоторые вещества, мо-
лекулы которых не содержат гидроксигруппы, обладают свой-
ствами оснований (например, жидкий или газообразный ам-
миак). Кроме того, теория С. Аррениуса не объясняла роли воды
при диссоциации кислоты и предполагала существование сво-
бодного протона Н+ в водном растворе, а не иона гидроксо-
ния Н3О+.
В 1923 г. появились две новые более общие теории кислот и
оснований: теории Дж. Бренстеда и Г. Льюиса.
Согласно электронной теории, разработанной Г. Льюисом, ос-
нование — это соединение, поставляющее электронные пары для
образования химической связи, т. е. донор электронных пар.
Кислота — вещество, принимающее электронные пары, т. е% ак-
цептор электронных пар. Кислотно-основное взаимодействие,
согласно электронной теории, заключается в образовании донор-
но-акцепторной связи. В результате взаимодействия кислоты с
основанием образуются солеподобные вещества, называемые
аддуктами. Часто (но не всегда) их удаемся выделить как инди-
видуальные соединения.
Ниже приведено несколько примеров реакций, которые, со-
гласно электронной теории, представляют кислотно-основное вза
имодействие:
Н Н ।
H:N + H:C1: ^[h:N:h]+ :С1:~ \
н н
основание кислота
(CH3)3N + BCI3-* (СНз)з1Ч:ВС1з
основание кислота
(Н:6:Г4-н+ — Н:О:Н
•• ••
основание кислота
:О: :О:
:O:S + :О:Н-+н2[
:О: н :0: 1
кислота основание
Таким образом, электронная теория Г. Льюиса рассматри-
вает нейтрализацию в водных растворах, взаимодействие аминов
с галогенидами бора, комплексообразование, реакции ангидри-
196
дов с водой как сходные процессы. Вещества, являющиеся до-
норами электронных пар, называют основаниями Лью-
иса, а акцепторы электронных пар — кислотами Лью-
иса.
Согласно теории Бренстеда кислота — донор протона, а основание — ак-
цептор протона.
Таким образом, при реакции воды с соляной кислотой
НС1 + Н2о НзО+ 4- Cl-
молекула НС1 отдает протон молекуле воды, следовательно, она
кислота. Молекула Н2О принимает протон, образуя ион гидро-
ксония Н'зО+, значит, она основание. Хотя соляная кислота имеет
сильную тенденцию отдавать протон молекуле воды, можно пред-
ставить себе обратную реакцию, в которой НзО+, отдавая про-
тон, будет играть роль кислоты, а С1-, принимая протон, —
роль основания:
Н3О+ 4- сг Н2О 4- НС1
Ион гидроксония НзО+ называется кислотой, сопряженной с ос-
нованием Н2О, а ион С1“ — основанием, сопряженным с кисло-
той НС1.
Поэтому можно рассматривать реакцию воды с соляной кис-
лотой в равновесии в виде
НС1 4- Н2О НзО+ 4- Cl-
кислота основа- сопряжен- сопря-
ние ная кис- женное
лота основа-
ние
или
НС1 4- Н2о НзО+ 4- сг
кисло- основа- кислота 2 основа-
та 1 ние 2 ние 1
Таким образом, все реакции, в которых принимают участие
кислоты и основания, образуют кислотно-основные пары, соответ-
ствующие равновесиям, более или менее смещенным в ту или
другую сторону, согласно соответствующим силам сопряженных
кислот и оснований.
Рассмотрим еще несколько примеров кислотно-основных пар:
Кислота 1 4- Основание 2 Кислота 2 4-Основание 1
НС1 4- Н2О H3O+ + cr (8.6)
HNO3 4- Н2О Н3О+ + NO3" (8.7)
СНзСООН 4- Н2О H3O+ + CH3COO- (8.8)
Н2О 4- NH3 NHt + OH- (8.9)
Н2О 4- CN“ HCN + OH- (8.10)
Н2О 4- RO- ROFJ ,0—H + OH" (8.П)
H2SO4 + СНзСООН СН3СГ + SO4H- (8.12)
ЮН
197
В уравнениях (8.6) — (8.8) вода — основание по отношению к
соляной, азотной и уксусной кислотам: она принимает протон.
В уравнениях (8.9) — (8.11) вода — кислота по отношению к
аммиаку, цианид- и алкоголят-иону: она отдает протон. Уксусная
кислота СНзСООН является кислотой по отношению к воде (8.8)
и, наоборот, основанием по отношению к серной кислоте (8.12),
поскольку она принимает протон от этой кислоты.
Таким образом, можно сделать вывод об относительности
понятий кислоты и основания. Одно соединение может быть и
кислотой и основанием по отношению к реагенту, с которым оно
вступает в реакцию. Например, если СНзСООН принимает про-
тон от H2SO4 (8.12), то это значит, что уксусная кислота явля-
ется более слабой кислотой, чем серная; напротив, в реак-
ции (8.8) СНзСООН более сильная кислота по сравнению с во-
дой.
Естественно, для проведения как теоретических, так и при-
кладных исследований проведенное выше чисто качественное со-
поставление недостаточно. Необходимо осуществить количествен-
ную оценку силы кислот и оснований, для чего следует опреде-
лить критерий оценки этой силы и создать шкалу количественных
измерений.
4. СИЛА КИСЛОТ И ОСНОВАНИЙ
Критерий установления силы кислотуи оснований очень прост.
Сила кислот и оснований обусловлена той легкостью, с которой они отдают
или принимают протон.
Кислота тем сильнее, чем легче она отдает протон. Основание
тем сильнее, чем легче оно принимает протон.
Рассмотрим химическое равновесие кислоты с водой:
HA + HoOi А“+Н2О+
Чем сильнее кислота, тем больше реагента превращается в про-
дукт. Для сильной кислоты равновесие резко смещено в направ-
лении 1 и наблюдается лишь слабая тенденция к протеканию
обратной реакции (направление 2), когда основание может при-
нять протон от иона НзО+. Поэтому сопряженное основание, со-
ответствующее сильной кислоте, должно быть слабым основа-
нием.
Так, при равновесии соляной кислоты с водой
НС1 + н2о i н3о+ + СГ
2
концентрация ионов НзО+ и С1~ велика, а концентрация Моле-
кул НС1 очень мала. Поскольку кислота НС1 сильно диссоцииро-
вана, она является сильной кислотой и очень легко отдает протон
воде; сопряженное ей основание, наоборот, очень слабое, так как
оно очень трудно принимает протон от иона гидроксония НзО+.
Аналогично можно показать, что сильному основанию соот-
198
ветствует слабая сопряженная кислота: основание очень легко
принимает протон, а сопряженная ему кислота с трудом отдает
его.
Таким образом, основание, сопряженное кислоте, тем слабее,
чем сильнее кислота; кислота, сопряженная основанию, тем сла-
бее, чем сильнее основание.
Сравним теперь между собой силы двух кислотно-основных
пар на примере реакции соляной кислоты с водой:
НС1 + н2о i н3о+ + • сг
кислота 1 основание 2 2 кислота 2 основа-
(более (более (более ние 1 (бо-
сильная) сильное) слабая) лее сла-
бое)
В рассмотренной реакции равновесие сильно смещено в на-
правлении 1, НС1 отдает протон Н2О легче, чем НзО+ иону С1“.
Следовательно, НС1 — более сильная кислота, чем Н3О+,
а Н2О — более сильное основание, чем С1-.
Рассмотрим реакцию уксусной кислоты с водой:
СНзСООН + Н2О i СНзСОО- + НзО+
2
При равновесии концентрация молекул СНзСООН велика, а кон-
центрация ионов СНзСОО- и Н3О+ относительно мала. В этом
случае Н3О+ отдает протон основанию СНзСОО- легче, чем
СНзСООН основанию Ц2О. Основание СНзСОО- принимает
протон от НзО+ легче, чем вода от уксусной кислоты. Следова-
тельно, СНзСООН — более слабая кислота, чем НзО+, а
Н2О — более слабое основание, чем СНзСОО-; это можно
изобразить схемой, где равновесие смещено в направлении 2:
СНзСООН
кислота 1
(более сла-
бая)
+ Н2О
основа-
ние 2
(более
слабое)
i Н3О+ + СНзСОО-
2 кислота 2 основание 1
(более (более силь-
сильная) ное)
Соляная кислота, растворенная в воде, является более сильной
кислотой, чем уксусная.
Теперь следует установить шкалу для количественного сопо-
ставления кислот и оснований. Для этого нужно выбрать веще-
ство, способное фиксировать протон в присутствии наибольшего
числа реагентов. Таким веществом является вода.
Шкала кислотности по отношению к воде установлена для
сравнения силы, с которой кислоты НА переносят протон на
молекулу воды, играющую роль основания, согласно уравнению
реакции (направление 1)
НА + Н2О i А" + Н3О+
2
(8.13)
И наоборот, сила основания А- обусловлена легкостью, с кото-
рой оно принимает протон от иона гидроксония (направление 2.)
Будем пользоваться шкалой кислотности, установленной для
199
сравнения силы, с которой основание А- принимает протон от
воды (последняя здесь играет роль кислоты): в этой реакции
освобождается ион ОН-:
А” + Н2О i АН + ОН“. (8.14)
2
В одном случае равновесие (8.13) определяет шкалу относительно
пары НгО/НзО^, в другом равновесие (8.14) характеризует шка-
лу относительно пары Н2О/ОН-.
5. ИОННОЕ ПРОИЗВЕДЕНИЕ ВОДЫ. ПОНЯТИЕ pH
При количественном определении кислотности водных
растворов рассматриваются только разбавленные водные раство-
ры, т. е. такие растворы, в которых количество вещества (молей)
кислоты или основания, растворенных в литре воды, гораздо
меньше количества вещества (молей) воды (приблизительно
55,5 кмоль/м3). Тогда можно считать, что количество вещества
(молей) воды, например, в 1 л остается практически постоянным,
несмотря на химические реакции, которые могут протекать в
растворе, приводя к распаду или возникновению молекул воды.
Рассмотрим диссоциацию самой воды. Равновесие диссоциации
воды выражается уравнением
Н2О + Н2О НзО+ + ОН-
Диссоциация воды очень мала, поскольку при 298 К константа
равновесия
[ii2OJ
Концентрацию воды Н2О в разбавленном растворе можно
считать постоянной. Тогда
ги ™ 1000 сс с /3
[Н2О] = ——— = 55,5 кмоль/м3.
1 о
Концентрации ионов НзО+ и ОН- при этом равны, так как
раствор электронейтрален. Тогда,
[Н3О+] [ОН-] = 55,52-3,24* 10~18 (кмоль/м3)2 = Ю-14 (кмоль/м3)2 — Ку,
[Н3О+] = [ОН'] = 10“7 кмоль/м3,
где Kw — ионное произведение воды.
Так как Kw 0, не может быть водного раствора, в котором
концентрация ионов НзО+ или ОН- равнялась бы нулю. Следо-
вательно, в любом водном растворе всегда присутствуют совмест-
но ионы Н3О+ и ОН-.
Следует также отметить, что диссоциация воды является эн-
дотермическим процессом, а ее образование из ионов — экзотер-
мическим. Отсюда в соответствии с принципом Ле Шателье тем-
пература будет оказывать значительное влияние на Kw
(табл. 29).
200
Таблица 29. Зависимость Kw и pH от температуры
т, к Kw pH
273 1,139- 10-,е 7,97
291 5,702-10-15 7,11
298 1,008-10~14 6,99
323 5,474 • 10-14 6,63
373 5,90-10‘3 6,12
Раствор называется кислым, если концентрация ионов
НзО+ выше 10-7 кмоль/м3, а концентрация ионов ОН- ниже
этого значения. И наоборот, щелочной раствор соответствует
концентрации ионов ОН- выше 10-7 кмоль/м3 при концентра-
ции ионов НзО+ ниже этого значения. Если концентрации
ионов НзО+ и ОН- равны 10-7 кмоль/м3, то раствор называется
нейтральным.
Разбавленные растворы кислот всегда соответствуют очень
низким концентрациям ионов НзО+, обычно выраженным отри-
цательной степенью 10. Для краткого выражения концентрации
Зёренсен ввел в 1920 г. новое понятие, известное под названи-
ем pH.
Величина pH раствора определяется как отрицательный десятичный лога-
рифм числа, выражающего концентрацию ионов Н3О+ в этом растворе.
Итак,
pH = - 1g [НзО+] = 1g.
Но концентрации ионов НзО+ и ОН- водного раствора всегда
должны удовлетворять равновесию диссоциации воды:
[НзО+] [ОН-] = Kw = Ю-14 (кмоль/м3)2.
Отсюда вытекает следующая связь между концентрациями
ионов НзО+ и ОН- водного раствора:
1g [Н3О+] +lg [ОН-] =-14.
Тогда, воспользовавшись аналогичным понятием рОН =
= —1g [ОН-], можно записать
pH 4- рОН = 14.
На самом деле значением рОН не пользуются, а вычисляют pH
щелочного раствора из соотношения
pH = 14 - рОН.
pH кислых и щелочных водных растворов рассчитывают по-
разному, исходя из силы кислоты или основания по отношению
к воде. Поэтому необходимо рассмотреть классификацию кислот
и оснований по отношению к воде.
201
6. КЛАССИФИКАЦИЯ КИСЛОТ И ОСНОВАНИЙ ПО ОТНОШЕНИЮ
К ВОДЕ
Понятие рКа. Как было уже рассмотрено, для сильной кисло-
ты равновесие НА + H2oi А- + Н3О+ (8.15) кисло- 2 сопря- та женное
основа-
ние
смещено в направлении 1, так как сопряженное основание пред-
ставляет собой слабое основание. Константа кислотности Ка
определяется выражением
[Н3О+] [А-]
Ла [АН]
Теоретически термин сильная кислота обозначает
кислоту, полностью диссоциированную в водном растворе, что
отвечает бесконечному значению Ка. Практически при Ка > 2
или 3 кислоту считают полностью диссоциированной.
Силу кислот часто характеризуют величиной ее рКа, которая равна отри-
цательному логарифму
pAa=-lgAa-
Полностью диссоциированная кислота (Ка = оо) соответству-
ет величине рКа, равной —оо. ,Для сильных кислот величи-
ны рКа отрицательны (табл. 30). Чем слабее кислбта, тем больше
значение рКа этой кислоты. Необходимо отметить также, что
кислота сильнее воды, если ее рКа < 7.
Как известно, можно сравнивать силы оснований по их спо-
собности принимать протон от иона НзО+ по схеме
А- + НзО+^НА + Н2О 4 (8.16)
Равновесие (8.16) обратно равновесию (8.15), а константа рав-
новесия, характеризующая тенденцию А- принимать протон от
НзО+, равна
1 _ [НА]
Ка [А-] [Нзб^Т '
Таблица 30. Значения Ка и рКв для сильных
и слабых кислот
Сильные кис- лоты Ка ?Ка Слабые кис- лоты Ка рКа
НС1О4 ю8 -8 hno2 4-10-“ +3,4
НС1 103 -3 СНзСООН 1,8-10~5 +4,75
HNO3 102 —2 HCN 4-10-10 +9,4
202
В этом случае основность, как и для кислот, относится к паре
НзО+ДЬО. Следовательно, можно дать единую классификацию
кислотно-основных пар относительно пары НзО+/Н2О, поскольку
кислоты легче, чем НзО+, отдают протон воде, а основания легче,
чем НгО, принимают протон от Н3О+.
Можно охарактеризовать силу основания, рассматривая тен-
денцию основания принимать протон не от иона НзО+, а от моле-
кулы Н2О, согласно следующей реакции:
А- + Н2О^АН + ОН~ (8.17)
Здесь основность рассматривается относительно пары НгО / ОН-,
и чем больше значение константы равновесия (8.17), тем больше
сила основания. Константа равновесия (8.17) определяется со-
отношением
[АН] [ОН-]
Kb = [А=] '
Можно заметить, что константа основности Кь связана с констан^
той кислотности Ка сопряженной ему кислоты; действительно,
_ [АН] [ОН-] _ [АН] [ОН-] [Н3О+] _ Kw
[А-] [АД [Н3О+1 Аа *
Следовательно, для оснований нет необходимости устанавливать
шкалу основности по константам равновесия Кь или рКь. Достаточ-
но знать константы кислотности Ка сопряженных кислот относи-
тельно кислотноосновной пары НзО+/Н2О.
6.1. Расчет pH кислых и щелочных растворов
Для определения pH необходимо с помощью эксперимента
определить концентрацию ионов НзО+. Рассмотрим сильные кис-
лоты и основания, значение рКа которых таково, что их можно
считать практически полностью диссоциированными в воде.
Сильные кислоты и сильные основания. Ионы Н3О+ возни-
кают, с одной стороны, при диссоциации кислоты по реакции
АН4-Н2О-+A--hH3O+
и, с другой стороны, при диссоциации воды. Последняя, как
известно, — очень слабая кислота, кроме того, нужно отметить,
что в присутствии кислоты, т. е. избытка ионов НзО+, равно-
весие диссоциации воды
Н2О 4- Н2О^НзО+ + ОН-
2
еще больше смещено в направлении 2, т. е. вода очень слабо
диссоциирует. Поэтому можно пренебречь ионами Н3О+, возни-
кающими при диссоциации воды, и учитывать только те ионы
НзО+, которые образуются при диссоциации кислоты. При этом
предполагается, что кислота диссоциирована полностью.
203
Определим, например, pH раствора соляной кислоты, началь-
ная концентрация которого равна С кмоль/м3. В рассматривае-
мых условиях можно записать
[Н3о+] « [НС1] «С кмоль/м3,
откуда
рН= —lg[H3O+] = — IgC.
Для сильных оснований А-, которые предполагаются пол-
ностью диссоциированными, равновесие целиком смещено в ука-
занном направлении:
А- -р Н2О-^АН + ОН-
Так, например, для алкоголята натрия, предполагая полную
диссоциацию, реакцию можно записать в виде
С2Н5О- + Н2О-^С2Н5ОН + ОН-
Тогда можно считать, что ионы ОН- — возникают при реакции
алкоголята, вода достаточно слабо диссоциирует и числом ио-
нов ОН-, образующимися при диссоциации, можно пренебречь.
Следовательно, чем больше молекул алкоголята в растворе, тем
больше число ионов ОН-. Для таких оснований, как гидроксид
натрия, весь растворенный NaOH диссоциирован на Na+ и ОН-.
Поэтому, исходя из принятых приближений для концентрации
раствора, С кмоль/м3 можно записать
[ОН-] ^[Сильное основание]^ С кмоль/м3, откуда
POH = -lg[OH-] = - IgC
и
pH = 14 - рОН.
Слабые кислоты и слабые основания. В качестве слабой
кислоты рассмотрим уксусную кислоту:
СНзСООН + Н2О^СН3СОО~ 4- НзО+ (8.18)
Как и для сильной кислоты, расчет pH раствора заключается в
определении концентрации ионов НзО+ раствора. Здесь еще
можно пренебречь в первом приближении небольшим числом
ионов НзО+, возникающих при диссоциации воды. Таким обра-
зом, будут рассматриваться только ионы НзО+, образующиеся
при диссоциации кислоты. Тогда выполняется равенство
[СНзСОО-] = [Н3О+] (8.19)
Константа кислотности определяет отношение между концентра-
циями ионов гидроксония НзО+, ацетат-ионов СНзСОО- и
недиссоциированных молекул уксусной кислоты СНзСООН при
равновесии в растворе:
„ [СНзСОО-] [НзО+]
~ [СНзСООН]
204
Из этого уравнения можно определить концентрацию ионов
НзО+ в растворе. Для этого надо выразить равновесные кон-
центрации СНзСОО- и СНзСООН через концентрацию един-
ственного неизвестного НзО+. Введем параметр а — степень дис-
социации кислоты, которая в рассматриваемом примере пред-
ставляет собой долю диссоциированных молекул кислоты, отне-
сенную к одному киломолю уксусной кислоты в растворе. Следо-
вательно, учитывая соотношение (8.19), можно записать
СНзСООН 4- Н2О СНзСОО' 4- Н3О+
1 — а а а
С(1 — а) Са Са
принимая соответственно начальную концентрацию СНзСООН
равной 1 кмоль/м3 либо С кмоль/м3.
При равновесии
[СНзСОО-] равн [НзО+] равн _ [НзО+] равн
[СНзСООН] равн [СНзСООН] равн •
Константа кислотности, выраженная через начальные кон-
центрации, равна
Г2 2 С 2
Если кислота достаточно слабо диссоциирована, то можно
использовать приближение
[СНзСООН] равн — [СНзСООН] нач- [СНзСООН] дисс« [СНзСООН] нач.
Это значит, что степень диссоциации а настолько незначительна,
что ею можно пренебречь по сравнению с единицей, т. е. 1^>а.
Тогда
С(1- а)« С
и соотношение (8.20) преобразуется к виду
к . [НзО+lU ияи к . CW 2 (8 21)
Ка [СНзСООН] нач К“ С с (8'2 )
Отсюда следует
[НзО+] = Са [СНзСООН] на; или [Н3О+] =
Это приближение считают правильным до тех пор, пока концен-
трация ионов НзО+, вычисленная таким способом, не превысит
1/10 от начальной концентрации кислоты С.
Из соотношения (8.21) можно вывести выражение
а=
Это выражение показывает, что
степень диссоциации увеличивается с разбавлением (закон Оствальда).
205
Если нельзя пользоваться приближением С(1 — а) «С, со-
отношение (8.20) приводит к уравнению второй степени по а:
а2С = /Саа — Ка = 0,
откуда
а =
2С
и
[НзО+] = аС
2
Расчет pH для слабых оснований рассмотрим на примере
реакции NH3 с водой:
NH3 + H2O^NH4+ + ОН-
Равновесие описывается константой
[NHfl (ОН-]
[NH3 ’
Если воспользоваться теми же понятиями и теми же приближе-
ниями, что были предложены при расчете слабых кислот, можно
получить
If eV г 2 __, / ^ь
Кь ~ С(1 - а) ~ Са ’ а — У ’
где а — степень превращения аммиака.
Приближенный расчет дает
[ОН-] « С а &
Как уже отмечалось, константа Кь, соответствующая основанию
NH3, связана с константой кислотности Ка сопряженной кис-
лоты NH^" соотношением
Kb = Kw/Ka.
Таким образом, в рамках принятых приближений
[ОН-] = VKwCfKa,
откуда
(НзО+1 = — 1/
13 J [он-] V с
Если приближение не выполняется ([ОН-] >0,1 С), то решение
уравнения второй степени по а, аналогичное предыдущему, дает
точное значение а.
При проведении точного расчета pH раствора слабой кислоты или слабого
основания нужно учитывать ионы НзО+, возникающие при диссоциации воды.
Тогда, например, для слабой кислоты следует рассматривать два равновесия:
НА + Н2О^Н3О+ + А'
206
и
Н20 4- Н2О^Н3О+ 4- 0Н“
(1)
В равновесиях участвуют четыре компонента, концентрации которых нужно
рассчитать: АН, А", ОН- и НзО+.
Для этого необходимо составить четыре уравнения. Два уравнения можно
вывести, используя значения констант двух равновесий:
г _ [А-] [Н3О+1
[АН)
^-^[НзО+1 [ОН"]. (2)
Третье уравнение получают, исходя из электронейтральности, т. е. записы-
вая для рассматриваемого раствора равенство концентраций положительных
ионов НзО+ и суммы концентраций отрицательных ионов А и ОН-:
[Н3О+] = [A-J + [ОН-] (3)
Четвертое уравнение получают из начальной концентрации кислоты, которая
частично существует в недиссоциированной форме, а частично — в форме
ионов А- Следовательно, полагая начальную концентрацию С известной,
запишем
С = [АН] + [А-] (1)
Решение этой системы из четырех уравнений приводит к выражению
[н.о+1 {№0*1-2^}
1\а-----------------J7------ .
С-[Н3О+] + [Нз0+]
Это уравнение третьей степени по НзО+.
6.2. Вычисление pH водных растворов солей
Реакция воды с солями называется гидролизом со-
лей. Она относится к кислотно-основному типу.
Гидролиз соли сильной кислоты и сильного основания. Эту
реакцию рассмотрим на примере взаимодействия NaCl с водой.
Хлорид натрия в водном растворе диссоциирован на свободные
сольватированные ионы Na+(aq) и Cl '(aq), но эти ионы факти-
чески не реагируют с водой. Таким образом, в рассматриваемом
примере гидролиз не происходит. Значение pH раствора остается,
следовательно, таким же, как pH абсолютно чистой воды, т. е.
pH =7 (при 298 К).
Гидролиз соли сильной кислоты и слабого основания. Кри-
сталлический хлорид Аммония, растворенной в воде, диссоцииру-
ет по схеме
NH4C1 (т) ~>Cl~(aq) + NH4+(aq)
Кислота NH^- находится в присутствии двух оснований: воды и
иона С1_. Вода — более сильное основание, чем С1~; она прини-
мает протон от иона NH^ образуя сопряженную ему кислоту
НзО4" и NH3, что мЬжно представить в виде равновесия:
СГ + NHt+ H2oiNH3 + НзО+ + сг
207
При протекании обратной реакции (направление 2) более силь-
ное основание NH3 принимает протон от НзО+. Хлорид-анион
практически не участвует в реакции, которую можно записать в
виде
nh4+ 4- h2o^>nh3 4- н3о+
Опять имеет место равновесие диссоциации слабой кислоты, с
которой переносится протон на молекулу воды. Это равновесие
типа (8.18). Поэтому расчет pH водного раствора NH4C1 прово-
дится так же, как расчет pH раствора слабой кислоты:
„ [NH3] [НзО+]
Ка [NHt]
Если использовать те же приближения, что и при расчете pH
раствора слабой кислоты, то
[Н3о+] = )П^С.
Для раствора соли степень диссоциации кислоты часто называ-
ется степенью гидролиза. Запишем ее в виде
а = /Ка/С или а = yTKw/KbC ,
так как
Kw/Kb.
Из этого следует, что чем слабее основание, тем большую
роль играет гидролиз соли сильной кислоты и слабого основа-
ния и, следовательно, тем выше концентрация ионов НзО+ в
растворе.
Гидролиз соли слабой кислоты и сильного основания. Рас-
смотрим эту реакцию на примере гидролиза ацетата натрия
CHsCOONa, который в водном растворе диссоциирован по схеме
СНзСОО” + Na+ + H20J-СНзСООН + ОН- + Na+
2
В противоположность Na+ основание СНзСОО- (ацетат-ион)
может присоединять протон воды, образуя уксусную кислоту и
освобождая ионы гидроксила. При обратной реакции (направ-
ление 2) уксусная уислота отдает протон основанию ОН-, при
этом образуются ацетат-ион и вода. Следовательно, ионы Na+
не принимают участия в реакции гидролиза, которую можно
представить в виде схемы
СНзСОО- 4- Н2О^СНзСООН 4- ОН~
Итак, в водном растворе ацетат натрия ведет себя как слабое
основание. Его pH вычисляют так же, как pH раствора слабо
диссоциированного основания. Выражение для константы гидро-
лиза будет выглядеть следующим образом:
[СНзСООН] [ОН-] _ _ Kw
[СНзСОО-] ка ’
208
где Кь — константа основности СНзСОО-, а Ка — константа
кислотности сопряженной ему кислоты СНзСООН.
Степень гидролиза равна
а = ^Kb/& ~ VKw/KaC,
откуда
[ОН-] = Са = = 1/-J4 с
’ Ла
И
[н3о+] = у/кмс.
Чем меньше константа диссоциации кислоты, из которой
образована соль, тем большую роль играет гидролиз и, следова-
тельно, тем выше концентрация ионов ОН- в растворе.
Гидролиз соли) слабой кислоты и слабого основания. Эта реакция рассмот-
рена на примере гидролиза монохлорацетата аммония. Схемы реакций гидро-
лиза ионов СН2С1СОО и NHt выглядят следующим образом:
СН2С1СОСГ + Н2О^СН2С1СООН + он-
NH^+ H2O^NH3 4- Н3О+
Они характеризуются соответственно константами равновесия К' и /С":
[СН2С1СООН] [ОН-] [NH3] [Н3О+]
“ [СН2С1СОО~] ’ [NHfl
Эти константы можно выразить в зависимости от констант К\ и Х2 двух кис-
лотно-основных пар. Известно, что
Ki (СН2С1СООН/СН2С1СОО-) = 1,6-ю-4,
K2(NHt/NH3) = 5,5‘1О-10.
Имеем
К' = J^-=0fi-10-'°, К"= К.2 = 5,5- Ю-10.
Л1
Если значения констант К' и К" довольно близки (как в этом примере),
то концентрация ионов ОН- и Н3О+ в растворе, так же как концентрации
СН2С1СООН и NH3, практически будут равны. В этих условиях суммарную
реакцию гидролиза можно представить в виде
NH4+ 4- СН2С1СОСГ 4- 2Н2О СН2С1СООН 4- МН3 4- Н3О+ 4- ОН~
Экспериментально установлено, что
[СН2С1СОО-] « [NHt]
[СН2С1СООН] « [NH3]
а константа рассматриваемого равновесия равйа
[СН2С1СООН] [NH3] Kw _ K2KW _ [NH3]2^
K~ [CHzCICOO-] [NHi] (H2OJ2- Я, [НгО]2 [NHt]2 [H2O]2 ’
0ТКУда K2 [NH3]2
Ki [NH^]2 '
Кроме того, известно, что
[NH3]2 _ Kl
[NHt]2 [H3O+]2 ’
209
следовательно,
[Н3О+] = /Л7КГ.
Отсюда можно рассчитать pH раствора монохлорацетата аммония, который
равен 6,52.
Таким образом, pH при гидролизе соли слабой кислоты и слабого основа-
ния зависит только от значения констант и А'г, относящихся к двум кислотно-
основным парам, а не от начальной концентрации соли. Если произведение
констант больше 10 '14, то раствор будет кислым, в противоположном случае —
щелочным.
Роль концентрации водородных ионов в биологических процессах. Разви-
тие внутренней среды организмов в большой степени связано с концентрацией
водородных ионов. Специфика процессов, протекающих в тканях, и активность
этих процессов (особенно ферментативных) в большой степени зависят от pH.
Изменение интервала pH может изменить направление ферментативного про-
цесса. Так, например, тканевые катепсины в нейтральной среде катализируют
синтез белка, а при кислой реакции его расщепляют.
Концентрация водородных ионов во многом обусловливает протекание бак-
териальных процессов и использование растениями питательных элементов из
почвы (табл. 31).
Таблица 31. Оптимальные значения pH для развития растений
(по Прянишникову)
Растения Значения pH Растения Значения pH
оптимальные допустимые оптимальные допустимые
Люпин 4...5 4...6 Пшеница 6...7 5...8
Картофель 5 4...8 Горох 6...7 5...8
Овес 5...6 4...8 Клевер 6...6,5 5...8
Рожь 5...6 4...7 Свекла 7 6...8
Лен 5...6 4...7 Кукуруза 7 6...8
7. БУФЕРНЫЕ СИСТЕМЫ
Буферными называются растворы, представляющие собой смесь слабой
кислоты и одной из ее солей с сильным основанием или слабого основания и
одной из его солей с сильной кислотой.
В качестве смеси слабой кислоты и соли слабой кислоты и
сильного основания рассмотрим раствор, концентрация уксусной
кислоты которого равна С\ кмоль/м3, а концентрация ацетата
натрия С2 кмоль/м .
Равновесие диссоциации уксусной кислоты в водном растворе
СНзСООН 4- Н2О^ СНзСОО- + НзО+ (8.22)
соответствует слабой диссоциации; оно смещено в направлении
2. В-растворе имеется также ацетат натрий, который реагирует
с водой: /
СНзСОО—ННзО^СНзСООН+ОН- (8.23)
2
210
Равновесие (8.22) характеризуется константой кислотно-
сти Ка-
[СНзСОО-] {Н3о+]
а [СНзСООН]
Следовательно, концентрация ионов Н3О+ в растворе равна
[СНзСООН]
[ИзО ] [СНзСОО-] К°'
Итак, для расчета pH необходимо знать концентрации уксус-
ной кислоты и ацетат-ионов в растворе при равновесии.
Рассмотрим влияние друг на друга равновесных систем
(8.22), (8.23). Введение в раствор ионов СН3СОО_ в форме соли
вызывает смещение равновесия (8.22) в направлении 2; в присут-
ствии избытка ацетат-ионов ионизация уксусной кислоты стано-
вится еще слабее, так как добавление общего иона приводит к
уменьшению степени диссоциации. Поэтому степень диссоциации
а достаточно мала, что позволяет пренебречь концентрацией
диссоциированной уксусной кислоты по сравнению с начальной
концентрацией кислоты. Тогда можно записать
[СН3СООН]=С1(1 - а)«С1.
Гидролиз ацетата приводит к возрастанию концентрации
уксусной кислоты в данном растворе. Однако присутствие уксус-
ной кислоты подавляет гидролиз, и равновесие (8.23) смещается
в направлении 2. Поэтому количеством уксусной кислоты, обра-
зовавшейся при гидролизе, можно также пренебречь по сравне-
нию с количеством кислоты, введенной первоначально в раствор.
Отсюда следует, что
[СНзСООН] «С1. (8.25)
Концентрация ацетата-ионов будет определяться следующими
факторами:
1) ацетат натрия полностью диссоциирован в воде и концен-
трация ацетат-ионов, первоначально введенных в раствор в
виде соли, равна С2;
2) уменьшение концентрации ацетат-ионов, вызванное гидро-
лизом соли, достаточно мало при избытке кислоты и им можно
пренебречь; ;
3) уксусная кислота при избытке ацетат-ионов диссоциирует
довольно слабо, чтобы можно было пренебречь концентрацией
ацетат-ионов, возникающих при диссоциации кислоты, по срав-
нению с начальной концентрацией ацетат-ионов С2. Тогда можно
считать, что
[СНзСОО-] » G . (8.26)
и с учетом (8.25) и (8.26) выражение (8.24) для концентрации
ионов Н3О+ примет вид
[Н3О+]=^ ~
С2
211
Таким образом, pH легко определить, зная начальные концен-
трации кислоты и соли в растворе:
рН=рКа + 1g
т. е. в общем случае
pH=pKfl+lg
[соль]
[кислота]
При рассмотрении смеси слабого основания и соли слабого
основания и сильной кислоты ход рассуждений такой же, как и
для раствора, содержащего слабое основание и одну из солей
сильной кислоты. Если взять в качестве примера водный раствор
аммиака и хлорида аммония, то реакция между основанием NH3
и водор представлена равновесием
NH3 + н2о nh4+ + он~
а реакция гидролиза соли—равновесием
NH4+ + Н2О NH3 + Н3О+
Концентрация ионов НзО+ и pH раствора определяется из
соотношения
(но+1_к №1
1Нз° 1 ПмъГ’
где Ка— константа кислотности пары МН^/МНз-
Воспользовавшись для определения равновесных концентра-
ций теми же приближениями, что и в предыдущем примере,
можно получить
[NH3] = С,
И
[NHfl = С2,
где Ci и С2 — начальные концентрации NH3 и NH4C1 соответ-
ственно. Имеем
[Н3о+]=ха
С1
Если концентрации основания и соли равны, то
[НзО+] = Ка, рН = рКа.
В общем случае
о „ . , Ci тт „ . [основание]
pH = рКа + 1g—-,т. е. pH = рКа + 1g-5—------------
С2 [соль]
Свойства буферных растворов. В противоположность раство-
рам слабой кислоты и слабого основания pH этих растворов не
212
зависит от концентрации, если не учитывать коэффициенты
активности компонентов.
Действительно, как уже было показано, для двух типов
буферных растворов pH выражается соотношениями
[НзО+] = [кисл2?а1 , [Н3О+] = Ки . [С0ЛЬ] -j- .
1 [соль] [основание]
При разбавлении раствора меняется концентрация кислоты и
соли или основания и соли, но их отношения остаются постоян-
ными.
Важнейшее свойство буферных растворов заключается в том,
что их pH мало изменяется при добавлении умеренных количеств
сильной, кислоты или сильного основания.
Рассмотрим буферный раствор, состоящий из смеси уксусной
кислоты и ацетата натрия, и рассчитаем изменение pH, вызван-
ное прибавлением 10“* кмоль соляной кислоты к 1 м(з раствора,
содержащего 1 кмоль уксусной кислоты и 1 кмоль ацетата нат-
рия. Соляная кислота, полностью ионизированная на ионы Н3О+
и С1”, реагирует с ацетатом натрия:
СНзСОО-+ H3oi СНзСООН + Н2О
Это равновесие сильно смещено в направлении 1; соответствую-
щая константа равновесия равна
„[СНзСООН] =_1_ = 55.104
[СНзСОО-] [НзО+] Ка
Следовательно,‘Практически образуется 10-2моль уксусной кис-
лоты и исчезает 10-2моль ацетата. В этих условиях концентрация
ионов НзО+ раствора равна
= Ка_ [кислота] = 1+0£1
[соль] 1 — 0,01
т. е. изменение составляет только 2% кислоты и соли. Для pH
получаем
pH = рКа -> lg-^- = 4-75 - 0,13 = 4,62.
Это значение сравнимо со значением, соответствующим исход-
ному раствору, содержащему 1 кмоль/м3 уксусной кислоты и
ацетата натрия (рН= p/G = 4,75).
Таким образом, изменение pH незначительно. Необходимо
отметить, что прибавление 10-2кмоль соляной кислоты в 1 м3 чис-
той воды изменит pH раствора с 7 до 2, т. е. на 5 порядков.
Прибавление сильного основания к буферному раствору так-
же лишь слегка изменит количество образовавшейся соли и
уменьшит количество кислоты, присутствующей в растворе, взяв
только очень небольшое изменение pH. Можно показать, что
сильнее всего буферное свойство проявляется для раствора
213
эквимолекулярных количеств кислоты и соли или основания и
соли.
Чем меньше изменение pH при добавлении в буферный рас-
твор кислоты или основания, тем сильнее выражено буферное
свойство раствора.
Интервал, в котором проявляется буферное свойство, называется буферной
емкостью.
Она определяется количеством сильной кислоты или основа-
ния (в кмолях), которое необходимо добавить в 1 м3 буферного
раствора, чтобы сместить pH на единицу:
» СУ
(pHi —рН2) Убуф ’
где В — буферная емкость; С — концентрация сильной кислоты
или основания, кмоль/м3; V — объем добавленной кислоты или
основания, м3; Убуф — объем буферного раствора, м3; pHi и
рН2 — водородные показатели соответственно до и после добав-
ления сильной кислоты или основания.
Биологическое значение буферных растворов^. Буферные системы почв.
Буферные растворы особенно важны для биологических сред, в которых pH
должен .оставаться неизменным. Так, например, pH крови должен быть постоян-
ным и близким 7,4. Однако питание и обмен веществ могут вызвать приток
кислых или щелочных соединений. Для поддержания pH крови постоянным в
биологических средах существуют системы эффективных регуляторов, состоящие
из буферных растворов, например смесей NaHCOg — Na2COg или NaH2PO4 —
— Na2HPO4. Наряду с этими химическими простыми буферными системами
крови следует отметить так называемый гемоглобиновый буфер, играющий важ-
ную роль в поддержании постоянства pH крови, так как он обеспечивает около
75% буферной емкости крови. Гемоглобиновый буфер представляет собой
смесь калиевой соли гемоглобина и свободного гемоглобина, являющегося
слабой органической кислотой.
В живых клетках постоянство pH обеспечивается так называемым белко-
вым буфером, который состоит из протеина и его натриевой соли.
Почва и почвенные растворы также обладают определенной буферной
характеристикой, которая обусловливается наличием коллоидов и поглощенных
катионов. Большое значение имеют энергия поглощения водородных ионов
коллоидами и степень диссоциации коллоидов. Органическое вещество почвы
преимущественно состоит из слабых кислот, поэтому оно увеличивает буфер-
ность почвы.
Почвенный раствор обладает буферным свойством по отношению к кислотам, =
если в нем присутствуют соли сильных оснований и слабой кислоты. К сильным
основаниям относятся натрий, калий, к менее сильным — кальций и магний.
Слабые кислоты в почве представлены гуминовыми кислотами, фульвокисло-
тами, щавелевой, угольной и др. Сильные — серной, азотной, соляной. Послед-
ние частично попадают в почву с удобрениями либо освобождаются при погло-
щении растениями питательных веществ из физиологически кислых удобрений,
например аммония из сульфата аммония.
Буферную характеристику почв необходимо учитывать в сельскохозяйствен-'
ном производстве. Так, в слабобуферных почвах реакция может резко изме-
ниться от внесения кислых удобрений, что влияет на развитие растений.
Хорошие буферные свойства по отношению к кислотам имеют почвы, богатые
органическими коллоидами (черноземы, луговые, некоторые торфяные), мине-
ральными коллоидами (суглинистые, глинистые), содержащие много погло-
щенных оснований. Почвы, бедные органическими и минеральными коллоидами
(щебнистые, песчаные), содержащие много поглощенного водорода и мало каль-
214
ция и магния, обладают плохой буферной характеристикой. Кислые почвы,
например подзолистые, имеют хорошую буферную характеристику по отноше-
нию к щелочам и плохую по отношению к кислотам.
Улучшение буферной характеристики почв осуществляют внесением орга-
нических или минеральных коллоидов и ила; в кислых почвах это достигается
внесением извести. Кислые минеральные удобрения в почвы, не обладающие,
буферными свойствами, вносят небольшими дозами или одновременно с известью.
Особенно важна эта осторожность в начальный период развития растений.
Контрольные вопросы
1. Чтобы растереть в порошок куски поваренной соли, нужно приложить
значительные усилия, а растворить в воде гораздо легче, почему?
2. Почему один и тот же электролит в разных растворителях образует рас-
творы либо проводящие, либо не проводящие тока?
3. Почему нельзя использовать принцип Ле Шателье для описания изме-
нения степени диссоциации при изменении температуры?
4. Почему основное уравнение, характеризующее растворы электролитов,
не может быть без соответствующих поправок использовано для растворов
электролитов? Покажите, какие вносятся поправки.
5. Почему закон действующих масс неприменим к сильным электролитам?
6. Почему при определении pH необходимо поддержание постоянной тем-
пературы?
7. Зачем нужны буферные растворы в биологических средах?
Глава IX
ЭЛЕКТРИЧЕСКАЯ ПРОВОДИМОСТЬ РАСТВОРОВ
ЭЛЕКТРОЛИТОВ
1. СКОРОСТЬ ДВИЖЕНИЯ ИОНОВ. ЧИСЛА ПЕРЕНОСА
Для всех агрегатных состояний электрическую проводимость
можно подразделить на четыре типа.
1. Металлическая проводимость, обусловленная подвижно-
стью электронов, являющихся носителями заряда. При увеличе-
нии температуры проводимость металлических проводников
ухудшается, поскольку движение электронов через решетку кри-
сталла затруднено вследствие более активного теплового движе-
ния атомов в решетке. Вещества, характеризующиеся металли-
ческой проводимостью, называются проводниками I рода.
2. Электролитическая проводимость жидкостей, вызванная
подвижностью ионов; носителями заряда являются катионы и
анионы. При увеличении температуры проводимость электриче-
ских проводников улучшается, поскольку при более высоких
температурах ионы движутся с большей скоростью за счет
понижения вязкости и уменьшения сольватации ионов. Вещества,
характеризующиеся электролитической проводимостью, называ-
ются проводниками Ирода. К проводникам II рода
относятся растворы электролитов (кислоты, соли, основания).
При наложении внешнего электрического поля анионы движутся
к положительно заряженному электроду — аноду, катионы —
к отрицательно заряженному электроду — катоду. Поскольку
скорости движения ионов в растворе значительно меньше, чем
скорости движения электронов в металлах, электрическая про-
водимость металлов, например меди и серебра, примерно в
миллион раз больше, чем для растворов электролитов.
3. Полупроводимость твердых тел, возникающая вследствие
заселенности электронами незаполненной зоны, отделенной от
полностью заполненной зоны энергетическим барьером. Прово-
димость полупроводников экспоненциально возрастает с темпе-
ратурой.
4. Электрическая проводимость газов, возникающая за счет
ионов газа и электронов.
Нами будет рассмотрена электролитическая проводимость,
связанная с прохождением электрического тока через объем
раствора электролита, помещенного в специальный сосуд, назы-
ваемый электролитической ячейкой. В отсутствие
внешнего электрического поля катионы и анионы электролита
216
передвигаются в растворе беспорядочно, причем любое направ-
ление движения является равновероятным. При наложении
электрического поля на раствор электролита одно из направле-
ний движения, а именно движение к электродам, становится
основным, причем преимущество этого направления будет тем
больше, чем больше градиент потенциала, т. е. чем больше паде-
ние напряжения на единицу расстояния между электродами.
Скоростью движения иона называется скорость его передвижения в направле-
нии одного из электродов, выраженная в м/с.
Скорости движения ионов различны и зависят как от при-
роды иона, так и от градиента потенциала. Поэтому для сравне-
ния скоростей движения различных ионов пользуются понятием
абсолютной скорости движения ионов, которая
равна отношению скорости движения иона в электрическом поле
к напряженности электрического поля Е.
Напряженность электрического поля по определению равна
отношению силы, действующей на точечный заряд, к величине
заряда. В качестве единичного электрического поля принимается
такое поле Е, в котором на точечный заряд в один кулон дейст-
вует сила в один ньютон. Следовательно, Е имеет размерность
Н/Кл. Электрический потенциал, который измеряется в вольтах,
представляет собой энергию, приходящуюся на единицу заряда.
Таким образом, 1 В= 1 Дж/Кл=1 Н-м/Кл, откуда следует, что
1 В/м=1 Н/Кл. Так как напряженность электрического поля
имеет размерность В/м, абсолютная скорость движения ионов
имеет размерность (м/с)/(В/м) =м2/(В-с).
Абсолютные скорости движения ионов в растворах невелики
(табл. 32); они существенно меньше, чем скорости движения мо-
лекул в газах. Например, ион водорода движется в водной среде
приблизительно в 108 раз медленнее, чем молекула водорода
в газовой среде. Это объясняется тем, что в водной среде гидра-
тированные ионы испытывают при передвижении большое сопро-
тивление со стороны среды.
Как видно из таблицы, скорость движения ионов НзО+ и ОН-
аномально высока, несмотря на то, что их гидратированные ра-
диусы сопоставимы с радиусами, остальных ионов. Такая подвиж-
Таблица 32. Абсолютные скорости движения ионов
при 298 К [м2/(В-с)]
Ион U-IO'® Ион и-10-® Ион у.10-’
Н3О+ 36,3 Ва2+ 6,60 г 7,96
к+ 7,62 Са2+ 6,16 сг 7,90
nh4+ 7,60 Sr2+ 6,16 NO3“ 7,40
Ag+ 6,42 Mg2+ 5,50 НСОГ 4,61
Na+ 5,20 ОН“ 20,5 СНзСОО- 4,24
Li+ 4,01 Вг- 8,12 5ОГ 8,27
217
ность этих ионов проявляется только в водных растворах и в
растворах простейших спиртов, что связано с особым механиз-
мом перемещения этих ионов.
Средняя продолжительность жизни иона гидроксония состав-
ляет 10“11 с. В цепочке, построенной из молекул воды, которые
связаны между собой водородными связями, заряд переходит от
одного конца цепочки к другому в результате сравнительно
небольшого (а значит, и быстрого) перемещения, показанного
на следующей схеме:
Н—О...Н—О...Н—О...Н—О...Н^О...Н-О...Н—б—н
I I I I I I I
—н -<—н
+ I
Н—О—Н... О—Н...О—Н... О—Н...О—Н... О—Н...О—
н н н н н н н
Аналогично передвигается и гидроксил-ион:
О...Н—О...Н—О...Н—О...Н-—О...Н—О
I I I I I I I
-----гп—н *—гм— гм----------------н
О—Н... О—Н...О—Н... О—Н... О—Н... О—Н...О
н н н н н н н
Как видно из приведенной схемы, протоны проходят не весь
путь до катода, а только расстояние между молекулами воды.
Они как бы передаются по эстафете от одной молекулы воды до
другой. Поэтому механизм их передвижения был назван эста-
фетным. Большая скорость движения ОН“-ионов объясня-
ется таким же механизмом, однако при этом протон передается
от молекулы воды к гидроксил-ионам. В результате этот процесс
выглядит как перемещение ОН”-ионов к аноду.
В растворах электролитов носителями заряда являются как
катионы, так и анионы. Поэтому общее количество перенесенного
электричества равно сумме количества электричества, перене-
сенного катионами, и части количества электричества, перенесен-
ного анионами, и зависит от концентрации ионов и скорости их
движения. Если рассматривать бинарный электролит, то концент-
рации катионов и анионов одинаковы и, следовательно, их уча-
стие в переносе электричества определяется только скоростью
их движения. Для оценки участия данного вида ионов в переносе
электричества пользуются понятием числа переноса
ионов.
Числом переноса иона называется отношение количества электричества,
перенесенного ионами данного вида, к общему количеству электричества, про-
шедшего через электролит, т. е. числа перекоса показывают долю электриче-
ства, переносимую данным видом монов.
218
Для бинарного электролита числа переноса для катиона и
аниона выражаются следующими уравнениями:
к
t - -
а ^к+^а’
(9.1)
(9.2)
где UK и Ua — абсолютные скорости движения ионов.
Очевидно, что для вещества, распадающегося на ионы двух
видов, сумма чисел переноса (т. е. сумма долей) всегда будет
равна единице. Из уравнений (9.1) и (9.2) следует, что
число переноса представляет собой отношение скорости движения данного
иона к сумме скоростей движения катиона и аниона.
Как и скорости движения ионов, число переноса зависит от
концентрации и температуры, но в значительно меньшей степени.
Очень часто числа переноса с повышением температуры при-
ближаются к 0,5, т. е. скорости движения катиона и аниона ста-
новятся приблизительно равны (табл. 33).
Таблица 33. Числа переноса катионов для 0,01 М
растворов при 298 К
Электролит к Электролит
НС! 0,8251 AgNO3 0,4648
LiCl 0,3289 CH3COONa 0,5537
NaCl 0,3918 CaCb 0,4264
КС1 0,4902 K2SO4 0,4829
KBr 0,4833 LaCl3 0,4625
KNO3 0,5084 K3[Fe(CN)6] 0,4315
2. УДЕЛЬНАЯ ЭЛЕКТРИЧЕСКАЯ ПРОВОДИМОСТЬ ЭЛЕКТРОЛИТОВ
Электрическое сопротивление R проводника определяется из
уравнения
где I — длина проводника; S — площадь поперечного сечения;
q и н — коэффициенты пропорциональности, называемые соот-
ветственно удельным сопротивлением и удель-
ной электрической проводимостью.
Таким образом, удельная электрическая проводимость явля-
ется величиной, обратной удельному сопротивлению:
1 1 I
н~ q""R S'
Единицей электрического сопротивления является Ом. Следо-
вательно, как видно из уравнения (9.3), в СИ удельная электри-
219
ческая проводимость х имеет размерность См/м (См — сименс).
По физическому смыслу удельная электрическая проводи-
мость раствора электролита — это проводимость раствора объе-
мом в 1 м3 при расстоянии между электродами в 1 м.
Мерой электрической проводимости L растворов электроли-
тов является количество электричества, выраженное в кулонах,
которое проходит через раствор за единицу времени:
I=LE,
где I— сила тока; Е — электродвижущая сила.
При £=1 I=L. Тогда
I=E/R. (9.4)
Используя уравнения (9.3) и {9.4), можно связать электриче-
скую проводимость раствора с удельной электрической проводи-
мостью:
s
L = xy. (9.5)
В табл. 34 приведены опытные значения удельной электриче-
ской проводимости проводников I и II рода.
Согласно этим данным растворы электролитов имеют прово-
димость, в десятки и сотни тысяч раз меньшую, чем металлы,
но на много порядков больше, чем типичные диэлектрики.
Удельная электрическая проводимость растворов электроли-
тов зависит от нескольких факторов: концентрации раствора,
скорости движения ионов, заряда ионов, температуры.
Удельная электрическая проводимость растворов электроли-
тов зависит от концентрации переносчиков заряда, т. е. от числа
ионов, содержащихся в единице объема раствора.
Графически эта зависимость представлена на рис. 75. Как
видно из рисунка, с увеличением концентрации электролита
электрическая проводимость х увеличивается, достигая опреде-
ленного максимального значения. При дальнейшем увеличении
концентрации удельная электрическая проводимость начинает
уменьшаться. Такая зависимость очень четко выражена для
сильных электролитов и значительно хуже для слабых, напри-
Таблица 34. Электрическая проводимость различных веществ
при 291 К
Проводник I рода Проводник II рода Диэлектрик
Вещество х, См/м Вещество х, См/м Вещество х, См/м
Серебро Алюминий Платина Ртуть 6-Ю3 4-Ю3 9-102 1 • 102 H2SO4 (30%-ная) КС1 (0,01 М) С2Н5ОН Н2О о СО Ю 00 — 1111 о о оо ео 4 Сера Кварц Парафин — СП ND ООО 1 1 1 ЬЭ — 00 О СО
220
мер для уксусной кислоты. Наличие
максимумов на кривых объясняется
тем, что в разбавленных растворах
сильных электролитов скорость движе-
ния ионов почти не зависит от концент-
рации и х растет почти прямо пропор-
ционально числу ионов, которое, в
свою очередь, увеличивается с кон-
центрацией. При достижении опреде-
ленной концентрации в растворах силь-
ных электролитов скорость движения
ионов существенно уменьшается из-за
наличия ионной атмосферы, в резуль-
тате чего х также уменьшается.
Для слабых электролитов при до-
стижении определенной концентрации
начинает уменьшаться степень диссо-
циации. В результате число ионов в
растворе возрастает в меньшей степе-
х,См/м
Рис. 75. Зависимость удель-
ной электрической проводи-
мости некоторых электроли-
тов от концентрации
ни, чем аналитическая концентрация раствора, что приводит к
уменьшению удельной электрической проводимости.
Удельная электрическая проводимость растворов электро-
литов зависит также и от индивидуальных свойств ионов, в част-
ности, от скорости их движения и заряда. Естественно, что чем
больше скорость движения ионов, тем большее количество элект-
ричества перенесет данный ион через единицу объема раствора
за единицу времени. Согласно данным табл. 32 и 33 больше
всего переносить электричество будут ионы гидроксония и гид-
роксила, которые характеризуются наибольшей скоростью дви-
жения. Точно так же чем выше заряд иона, тем большее коли-
чество электричества переносит данный ион соответствующему
электроду. Например, двухзарядный ион при. всех прочих равных
условиях (температура, вязкость среды, концентрация и т. д.)
перенесет в два раза большее количество электричества, чем
однозарядный.
Зависимость удельной электрической проводимости растворов электролитов
от температуры является довольно сложной, поскольку температура влияет на
несколько характеристик такой системы, как раствор электролита (вязкость,
степень диссоциации, гидратация ионов, скорость движения и т. д.). Однако
в общем случае х растворов всегда увеличивается с повышением температуры
(табл. 35).
Таблица 35. Удельная электрическая проводимость 0,1 М
КС! при различных температурах
Температура, К %. 10 5, См/м Температура,К к-10 5, См/м
273 7,1295 293 10,6593
283 9,3158 303 14,0996
221
При нагревании раствора на один градус электрическая проводимость
возрастает примерно на 2%, что, в частности, приводит к необходимости при-
менения термостатов при точных измерениях. Зависимость удельной электриче-
ской проводимости от температуры для разбавленных водных растворов элект-
ролитов описывается уравнением, в котором температура принята равной 291 К:
х( = х29, [1 + а'(Т-291)4- р'(Т-291)2].
Температурный коэффициент а' для сильных кислот равен 0,0164, для силь-
ных оснований 0,0190 и для солей 0,0220. Отсюда следует, что наибольшим тем-
пературным коэффициентом характеризуются ионы с относительно небольшой
скоростью движения. Положительное влияние температуры на электрическую
проводимость растворов электролитов объясняется уменьшением вязкости при
увеличении температуры. Для большинства ионов температурный коэффициент
скорости движения в водных растворах равен 2,3...2,5%.
Для слабых электролитов температурная зависимость электрической прово-
димости может иметь максимум. В этом случае, несмотря на то, что скорость
движения ионов с температурой увеличивается, степень диссоциации может
уменьшаться, в результате чего уменьшается общее число ионов в растворе.
Уменьшение степени диссоциации при увеличении температуры происходит
вследствие уменьшения диэлектрической проницаемости раствора, в результате
чего силы взаимодействия между ионами увеличиваются.
Таким образом, удельная электрическая проводимость раство-
ров электролитов зависит от многих факторов. Поэтому для
более общего учета влияния на электрическую проводимость
растворов электролитов их концентрации и взаимодействия
между ионами введено понятие молярной электриче-
ской проводимости.
3. МОЛЯРНАЯ ЭЛЕКТРИЧЕСКАЯ ПРОВОДИМОСТЬ ЭЛЕКТРОЛИТОВ
Молярной электрической проводимостью называется электрическая про-
водимость объема раствора, содержащего 1 кмоль растворенного вещества и по-
мещенного между электродами, находящимися на расстоянии 1 м друг от друга.
Физический смысл этой величины можно пояснить следую-
щим образом. Допустим, что имеется какой-то объем V электро-
лита, содержащий 1 кмоль растворенного вещества и находя-
щийся между электродами, отстоящими друг от друга на рас-
стояние 1 м. Электрическая проводимость 1 м3 данного раствора
будет являться удельной. Суммарная, т. е. в данном случае
молярная, электрическая проводимость будет равна произведе-
нию удельной электрической проводимости на число кубических
метров раствора V, в котором содержится 1 кмоль электролита:
Концентрация вещества С связана с объемом раствора V
соотношением
V== 1/С.
Величина V=l/C называется разведением или раз-
бавлением раствора и показывает объем раствора (в м3)
данной концентрации, в котором содержится 1 кмоль растворен-
ного вещества. Например, для 0,1 М раствора разведение равно
222
V = 1/0,1 = 10 м3, т. е. именно в 10 м3 0,1 М раствора содержится
1 кмоль растворенного вещества.
С учетом концентрации уравнение для молярной электриче-
ской проводимости запишется в виде
ку = к/С.
Таким образом, молярная электрическая проводимость рас-
твора электролита равна произведению удельной электрической
проводимости на разведение (в м3 на 1 кмоль электролита).
В результате единицей измерения молярной электрической про-
водимости будет См-м2/кмоль. Последний множитель иногда
опускается и молярную электрическую проводимость выражают
в этом случае как См-м2, имея в виду 1 кмоль растворенного
вещества.
Молярная электрическая проводимость как сильных, так и
слабых электролитов увеличивается с уменьшением концентра-
ции, т. е. с увеличением разведения раствора, и достигает неко-
торого предельного значения, называемого молярной элек-
трической проводимостью при бесконечном
разбавлении као. Это иллюстрируется рис. 76 и 77, на
которых показана зависимость молярной электрической проводи-
мости от разведения и от концентрации, а также эксперименталь-
ными данными, приведенными в табл. 36.
Как видно из табл. 36 и рис. 76 и 77, молярная электрическая
проводимость сильных электролитов отличается от электрической
проводимости слабых электролитов не только по значению, но
и по характеру ее зависимости от концентрации или разведения.
Для слабого электролита различие в молярных электрических
проводимостях при разных разведениях обусловлено, в основном,
только различием в числе ионов, образующихся при соответст-
вующем разведении, т. е. степенью диссоциации электролита.
Поэтому можно принять, что отношение молярной электрической
проводимости при каком-то конкретном разведении Ли к моляр-
ной электрической проводимости при бесконечном разведении Лоо
равно соответствующей степени диссоциации:
^у/^оо Cty/ctoo.
Таблица 36. Молярная электрическая проводимость некоторых
электролитов в водных растворах при 298 К
Концентрация, кмоль/м3 Ху• 10 5, См«м2, для веществ
КС1 AgNO3 НС1 кон СНзСООН NH4OH
1,0 98,3 67,8 301,0 184,0 1,32 0,89
о,1 112,0 94,3 351,0 213,0 4,60 3,3
0,01 122,4 107,8 370,0 228,0 14,3 9,6
0,001 127,3 113,2 377,0 234,0 41,0 28,0
0,0001 129,1 115,0 — — 107,0 66,0
223
Л,См-м2/кмоль
Рис. 76. Зависимость молярной элек-
трической проводимости некоторых
электролитов от концентрации
Рис. 77. Зависимость моляр-
ной электрической проводи-
мости некоторых электроли-
тов от разведения
Поскольку doo 1 ,
Ху == CtyX<
(9.6)
Следует, однако, отметить, что у слабых электролитов при-
ближение Ху к своему пределу Хоо происходит очень медленно и
значение предела в большинстве случаев экспериментально
установить нельзя.
Так как сильные электролиты полностью диссоциируют в
растворах различных концентраций, то, казалось бы, можно ожи-
дать, что Ху для таких растворов должна быть равна молярной
электрической проводимости при бесконечном разведении Х«.
Однако по мере увеличения концентрации ионов в растворе силь-
ного электролита, т. е. по мере уменьшения разведения, средние
расстояния между ними уменьшаются. Это приводит к усилению
Рис. 78. Зависимость
молярной 'электриче-
ской проводимости
разбавленных раство-
ров электролитов от
л/С
взаимодействия между ионами и, как след-
ствие, к уменьшению скорости их движения.
При увеличении разведения происходит об-
ратное явление, т. е. вследствие уменьшения
взаимодействия между ионами увеличивает-
ся скорость их движения, и молярная элект-
рическая проводимость увеличивается, до-
стигая своего предельного значения при
бесконечном разведении.
Определенный интерес представляет зависимость
молярной электрической проводимости от -у) С (рис. 78).
Для сильных электролитов наблюдается медленное
линейное уменьшение Ху с увеличением -у/С, которое
для разбавленных растворов хорошо описывается
эмпирической формулой Кольрауша
Ху = Хто — а~\/ С,
где а — эмпирическая константа, зависящая от при-
роды растворителя, температуры и заряда ионов. Вто-
рой член этого уравнения учитывает уменьшение
224
электрической проводимости, которое обусловлено уменьшением скорости движения
ионов и наличием ионных атмосфер. Следует отметить, что для растворов слабых
электролитов указанная зависимость в области небольших концентраций не
соблюдается (рис. 78).
Молярная электрическая проводимость, так же как и удельная, зависит от
температуры и обычно с увеличением температуры на один градус она увеличи-
вается на 2...2,5%. Это опять-таки объясняется увеличением скорости движения
ионов. Зависимость молярной электрической проводимости от температуры выра-
жается уравнением
ХГ=Х298[1 +а(Т-298)+ р(7’-298)2],
где кт и Я298 — молярные электрические проводимости при температурах Г и
298 К; а и р — температурные коэффициенты, зависящие от природы ионов и
растворителя. Обычно при расчетах используют только коэффициент а, так
как р очень мал.
4. СВЯЗЬ МОЛЯРНОЙ ЭЛЕКТРИЧЕСКОЙ ПРОВОДИМОСТИ
СО СКОРОСТЯМИ ДВИЖЕНИЯ ионов
Вычисление электрической проводимости раствора осуще-
ствляется при подсчете числа ионов, проходящих через любое
поперечное сечение электролитической ячейки в единицу времени
при стандартных условиях, т. е. при градиенте потенциала 1 В/м.
Допустим, что имеется раствор бинарного электролита с моляр-
ной концентрацией (кмоль/м3) растворенного вещества, скорости
движения катиона и аниона которого равны соответственно vK
и va, а степень электролитической диссоциации равна а. Раствор
помещен в электролитическую ячейку, площадь поперечного
сечения которой S, а расстояние между электродами I (рис. 79).
Под действием приложенного к ячей-
ке напряжения Е катионы и анионы бу-
дут двигаться к электродам со скоро-
стями
Е Е
vK=UK-j- и va=U.d-p
и Ua — абсолютные скорости дви-
катиона и аниона.
где U к
жения
Подсчитаем число катионов и анио-
нов, прошедших через поперечное сечение
ячейки S за 1с. За это время в одну сто-
рону пройдут все катионы, отстоящие в
начальный момент от выбранного сечения
на расстояние, численно равное ик, т. е.
число катионов, находящихся в объеме,
равно vKS. Поскольку в 1 м3 данного рас-
твора содержится а С ионов, то число
катионов пк, прошедших через выбранную
площадь сечения, равно
Рис. 79. Схема, поясняю-
щая связь молярной элек-
трической проводимости
со скоростями движения
ИОНОВ '
nK = SvKaC.
8—696
225
Тогда число анионов па, прошедших через ту же площадь сече-
ния, равно
na = SvaaC.
Отсюда общее число ионов лОбщ, прошедших за 1 с через выбран-
ную площадь поперечного сечения, равно
яобщ = Як + Яа = SvKaC + SvaaC = SaC(vK + va).
Заменив наблюдаемые скорости движения ионов на абсолютные,
получим выражение
е
Яобщ — ЗаС —((7 к + ^а)-
Следовательно, общим числом ионов, прошедших через выбран-
ную площадь поперечного сечения, будет перенесено количество
электричества, равное
е
FSvC—(U*+Uay==Q,
где F—постоянная Фарадея (F=96 500 Кл).
Поскольку сила тока в амперах определяется количеством
электричества в кулонах, прошедшего через поперечное сечение
проводника за 1 с, в рассматриваемой электролитической ячейке
возникает электрический ток, равный
F
,/ = FSaCT((/K+t7a). (9.7)
Согласно уравнениям (9.4) и (9.5) имеем
Приравняв уравнения (9.7) и (9.8), получим уравнение
K<sF F
( 2f££_=FSaC^-(GK+l/a),
сократив которое, получим
x=CaF(t/K+t/a). (9.9)
После некоторого преобразования уравнение (9.9) примет вид
-g-=a£(t/a+t/a). (9Л0)
Поскольку выражение х/С представляет собой молярную элект-
рическую проводимость, уравнение (9.10) принимает окончатель-
ный вид (уравнение Аррениуса):
Xv = aF(i7K4- Ua)-
Таким образом, уравнение Аррениуса показывает, что моляр-
ная электрическая проводимость растворов электролитов зависит
от абсолютных скоростей движения катиона и аниона.
226
5. ЗАКОН НЕЗАВИСИМОСТИ ДВИЖЕНИЯ ИОНОВ
Ф. Кольрауш в своих исследованиях экспериментально пока-
зал, что в растворах электролитов, доведенных до бесконечного
разведения, каждый из ионов вносит свой независимый вклад
в молярную электрическую проводимость. Иначе говоря, моляр-
ная электрическая проводимость является аддитивным свойством
электролита, т. е. суммой двух независимых переменных
Хоо^к + Ьа, (9.11)
которые представляют собой молярную электрическую проводи-
мость катиона и аниона при бесконечном разведении (Хк и Ха
соответственно) и называются электролитическими
подвижностями. Таким образом, открытый Ф. Кольрау-
шем закон независимости движения ионов может быть сформу-
лирован следующим образом:
молярная электрическая проводимость при бесконечном разведении Х^ равна
сумме электролитических подвижностей катиона и аниона данного электролита.
Молярная электрическая проводимость при конкретном разве-
дении описывается также уравнением Аррениуса
Xv = aF(t/K+t/a),
в которое входит степень диссоциации электролита. Если разве-
дение раствора стремится к бесконечному, степень диссоциации а
стремится к 1, а молярная электрическая проводимость — к Хоо.
В этом случае уравнение Аррениуса можно переписать в виде
Хоо=^к+^а)
или
Хоо =FUK + FUa. (9А2)
Сопоставив уравнение (9.11) и (9.12), можно получить, что
XK=(/KF и Ka = FUa.
Электролитическая подвижность ионов есть не что иное, как произведение
абсолютной скорости движения ионов на постоянную Фарадея.
Электролитическая подвижность является важнейшей харак-
теристикой ионов, отражающей их специфическое участие в
электрической проводимости растворов электролитов. С помощью
закона Кольрауша и значений электролитических подвижностей
(табл. 37) можно легко вычислить молярную электрическую про-
водимость при бесконечном разведении соответствующих раство-
ров. Для растворов солей молярная электрическая проводимость
при бесконечном разведении выражается значениями порядка
1,0...1,3 м2-См/кмоль; растворы кислот, в силу большой скорости
движения иона гидроксония, характеризуются значениями Х^ ,
в 3—4 раза большими; растворы оснований характеризуются
промежуточными значениями Х«, .
8*
227
Электролитическая подвижность зависит от скорости движе-
ния ионов, которая, в свою очередь, обратно пропорциональна
их радиусам. Если рассмотреть щелочные металлы (Li,+, Na+,
К+), то в указанном ряду истинные радиусы ионов увеличива-
ются и, следовательно, в такой же последовательности должна
уменьшаться их подвижность. Однако, как видно из табл. 37,
подвижность ионов при переходе от Li+ к К+ увеличивается
почти в два раза. Таким образом, ионы в растворе и в кристалличе-
ской решетке имеют разные радиусы, причем чем меньше истинный
(кристаллохимический) радиус иона, тем больше его эффектив-
ный радиус в растворе электролита. Это объясняется тем, что в
растворе все ионы гидратируются (в общем случае сольватиру-
ются) и, следовательно, эффективный радиус движущегося иона
определяется степенью его гидратации, т. е. числом .связанных
с ионом молекул воды. Сила взаимодействия, а также число свя-
занных ионом молекул воды будет зависеть от плотности заряда
на поверхности ионов. Плотность заряда на поверхности иона ли-
тия значительно больше, чем на поверхности иона калия, потому
что поверхность первого меньше поверхности второго, а радиус,
т. е. расстояние диполей воды от эффективного точечного заряда
в центре иона, наоборот, меньше. В результате степень гидрата-
ции иона лития больше степени гидратации иона калия, радиус
гидратированного иона лития больше, чем у калия, скорость дви-
жения, а следовательно, и электролитическая подвижность гид-
ратированного иона калия больше, чем у лития (табл. 37 и 38).
Скорость движения многозарядных ионов мало отличается от
скорости движения однозарядных, что обусловлено большей
степенью гидратации первых вследствие большей плотности
заряда на их поверхности.
Электролитическая подвижность ионов зависит, кроме того,
от температуры (табл. 39) и вида растворителя.
Как уже отмечалось, зависимость подвижностей ионов от температуры
обусловлена уменьшением вязкости среды и степени сольватации ионов, в связи
с чем возрастает скорость движения ионов, однако произведение электролити-
Таблица 37. Электролитические подвижности ионов в водных
растворах электролитов при 298 К (К См*м2/кмоль)
Катион V 1СГ1 Катион V 101 Анион >.„•10 ' Анион la-10—1
Н3О+ 34,98 Mg2+ 5,30 OH- 19,76 SO?“ , 8,0
Li+ 3,86 Ca2+ 5,95 F" 5,54 C2O?- 7,41
Na+ 5,01 Sr2+ 5,95 ci- 7,64 соГ 6,93
к+ 7,35 Ba2+ 6,36 Br~ 7,81 Fe(CN)|- 9,91
Rb+ 7,78 Cu2+ 5,66 1“ 7,68 Fe(CN)64- 11,10
Cs+ 7,72 Zn2+ 5,28 NOT 7,14
Ag+ 6,19 Al3+ 6,30 СНзСООН 4,09
NHt 7,35 Fe2+ 5,35
Fe3+ 6,80
228
Таблица 38. Числа гидратации ионов
Ион vio-2, См-м2/кмоль rгидр '10 1, НМ Гист' Ю *, НМ Число молекул Н2О, связанных с ионом
Na+ 50,1 1,83 0,97 5
Li+ 38,68 2,37 0,60 7
Mg2+ 53,05 3,46 0,65 12
Са2+ 59,50 3,09 0,99 10
Sr2+ 59,45 3,09 1,13 10
Ва2+ 63,63 2,88 1,35 9...10
Zn2+ 53,0 3,46 0,74 12
La3+ 69,75 3,95 1,15 13...14
Таблица 39. Электролитическая подвижность ионов
в водных растворах при различных температурах
Ион X-10 2 (См*м2/кмоль) при
273 К 291 К 298 К 373 К
Н3О+ 225 315 349,8 650,0
Li+ 19,4 32,8 38,6 115,0
Na+ 26,4 42,8 50,1 145,0
к+ 40,7 63,9 73,5 195,0
Ag+ 33,1 53,5 61,9 175,0
NHt 40,2 63,9 73,5 180,0
Mg2+ 28,9 44,9 53,0 165,0
Ca2+ 31,2 50,7. 59,5 180,0
Ba2+ 34,0 54,6 63,6 195,0
OH~ 105,0 171,0 197,6 450,0
Cl- 41,0 66,0 76,4 212,0
sor 41,0 68,4 80,0 260,0
ческой подвижности иона на вязкость среды т|о практически не изменяется в ши-
роком диапазоне температур. Например, для ацетатного иона в водном растворе
произведение X0-qo остается практически постоянным:
Т, К .
^СН3СОО-по
273 291 298 373
0,366 0,368 0,366 0,368
Можно предположить, что указанная зависимость, т. е. постоянство произ-
ведения подвижности иона и, следовательно, молярной электрической проводи-
мости на вязкость среды, сохраняется одинаковой и для неводных растворителей
(так называемое правило Вальдена — Писаржевского):
—const.
Однако это правило очень часто непригодно для объяснения влияния раство-
рителя на электрическую проводимость растворов, потому что оно предусматри-
вает равенство радиусов сольватированных ионов в различных растворителях,
что в действительности не соблюдается. Невыполнимость правила Вальдена
как раз подтверждает тот факт, что в различных растворителях ионы сольва-
тируются в разной степени.
229
Закон Кольрауша подтверждается следующими экспериментальными дан-
ными. Если сравнивать молярные электрические проводимости при бесконечном
разведении различных солей с одинаковыми катионами, но различными анио-
нами, то всегда получается одинаковая разность. Например, для сульфатов
калия и натрия имеем
А = Хоо (K2SO4)—^00 (Na2SO<) =0,21 1, .
для фторидов
А = Х00 (KF) — Хоо (NaF) =0,21 1.
Молярные электрические проводимости при бесконечном разбавлении хло-
рида и нитрата натрия соответственно равны
Х^ (NaCJ) == 1,090 И Хоо (NaNOj) = 1,052.
Разность между ними равна 0,038. Точно такая же разность получится, если
рассматривать молярные электрические проводимости Х^ различных солей,
имеющих разные катионы, но те же самые анионы. Именно на основании подоб-
ных экспериментальных данных Ф. Кольрауш пришел к выводу, что в разбавлен-
ных растворах каждый из ионов вносит свой независимый вклад в молярную
электрическую проводимость.
6. ЗАВИСИМОСТЬ ЭЛЕКТРИЧЕСКОЙ ПРОВОДИМОСТИ
ОТ ПРИРОДЫ ЭЛЕКТРОЛИТА
Уменьшение молярной электрической проводимости при уве-
личении концентрации С. Аррениус объяснял не уменьшением
скорости движения ионов, а уменьшением степени диссоциации
электролита. Однако с дальнейшим развитием теории растворов
электролитов было показано, что эти представления не верны.
Для сильных электролитов его молекулы в растворе диссоцииро-
ваны полностью при любой концентрации. Каждый ион окружен
ионной атмосферой, состоящей из ионов противоположного зна-
ка, при этом плотность ионной атмосферы увеличивается с по-
вышением концентрации электролита. П. Дебай и Г. Хюккель
объясняли уменьшение молярной электрической проводимости с
увеличением концентрации именно наличием ионной атмосферы.
В результате при движении иона возникает два тормозящих
эффекта: электрофоретический (катафоретический) и
эффект релаксации (асимметрии).
Электрофоретический эффект торможения обусловлен тем,
что при наложении электрического поля ион начинает двигаться
в одну сторону, а его ионная атмосфера — в другую. Движение
разноименно заряженных ионов в противоположные стороны
создает дополнительное торможение движению рассматривае-
мого иона. С увеличением концентрации плотность ионной
атмосферы увеличивается, следовательно, увеличивается тормо-
зящий электрофоретический эффект.
При движении под действием электрического поля ион выхо-
дит из своей ионной атмосферы, которая вновь воссоздается
в новом положении иона. Скорость восстановления ионной
атмосферы в другом элементе объема называется временем
релаксации. Для бинарного электролита в разбавленных
230
растворах оно обратно пропорционально концентрации раствора
и заряду ионов. Например, для 0,1 М раствора одно-однозаряд -
ного электролита время релаксации равно 0,6-10-9 с, а для
0,001 М — 0,6-10“' с, т. е. атмосфера восстанавливается в опре-
деленный интервал времени. Это приводит к тому, что в каждый
момент времени концентрация ионов в ионной атмосфере движу-
щегося иона сзади всегда несколько выше, чем впереди него,
т. е. наблюдается некоторая несимметричность ионной атмосфе-
ры. Наличие такой асимметрии ионной атмосферы создает допол-
нительное тормозящее действие. Следует отметить, что электро-
форетический эффект примерно в два раза сильнее эффекта
релаксации.
Для описания зависимости молярной электрической проводимости растворов
сильных электролитов от концентрации Л. Онзагер вывел следующее теорети-
ческое уравнение:
^ = ^00 -И+В%оо /С).
где А и В — постоянные, зависящие только от температуры и вита раство-
рителя;
. 82,4 „ 8,20-105
(87-)|/2г) И В~ (еГ),/2 ’
где е — диэлектрическая постоянная среды; т] — вязкость среды.
Уравнение Онзагера справедливо для одно-однозарядных электролитов
небольшой концентрации. Это уравнение хорошо согласуется с эмпирической
формулой Кольрауша в интервале средних концентраций:
Ху=Хоо — д
Именно в силу указанных причин молярная электрическая проводимость элект-
ролитов достигает своего предельного значения Хоо только при достаточно
большом разведении, несмотря на то, что они полностью диссоциируют при
любых концентрациях.
Для учета влияния сил межионного взаимодействия в раство-
рах сильных электролитов используется коэффициент электриче-
ской проводимости Д:
Л —Ху/Хоо,
который показывает, во сколько раз действительное значение
молярной электрической проводимости меньше теоретической
проводимости при бесконечном разведении для данной концент-
рации электролита. Коэффициент зависит от лрироды и концент-
рации электролита. В 0,1 М растворе бинарного электролита
f%=0,8, для одно-двухзарядного электролита Д=0,75, для одно-
трехзарядного электролита той же концентрации Д=0,4. По мере
разведения раствора коэффициент Д увеличивается, и при дости-
жении бесконечного разведения для всех типов электролитов
он становится равным 1, а молярная электрическая проводи-
мость достигает своего предельного значения.
Для слабых электролитов уменьшение молярной электриче-
ской проводимости при увеличении концентрации объясняется
несколько иначе. Слабые электролиты характеризуются тем, что
не все молекулы электролита распадаются на ионы, а лишь неко-
231
торая часть, которая учитывается степенью диссоциации. Сте-
пень диссоциации, как известно, уменьшается с увеличением
концентрации, т. е. уменьшается число ионов в растворе. В соот-
ветствии с этим уменьшается и молярная электрическая прово-
димость растворов. Наоборот, с увеличением разведения степень
диссоциации увеличивается, достигая единицы при бесконечном
разведении, а молярная электрическая проводимость достигает
своего максимального значения tao. Необходимо отметить, что
константа электролитической диссоциации в отличие от степени
диссоциации не зависит от концентрации раствора.
Для бинарного слабого электролита его способность к диссо-
циации характеризуется константой электролитической диссоциа-
ции, которая связана со степенью диссоциации согласно урав-
нению
а2
(9.13)
Поскольку из уравнения (9.6) a=Zv/Xoo, подставив значе-
ние а в уравнение для константы диссоциации, получим
Это уравнение позволяет непосредственно оценить константу
электролитической диссоциации, если известна молярная элект-
рическая проводимость раствора.
7. ПРАКТИЧЕСКОЕ ПРИМЕНЕНИЕ ЭЛЕКТРИЧЕСКОЙ
ПРОВОДИМОСТИ
Измерение электрической проводимости растворов является
основой кондуктометрических методов анализа. Эти методы
просты, практически очень удобны, достаточно точны и позво-
ляют решить ряд важных научно-исследовательских и производ-
ственных задач, не поддающихся решению, другими аналитиче-
скими методами. Измеряя электролитическую проводимость рас-
творов, можно определить основность органических кислот,
растворимость и произведение растворимости малорастворимых
соединений, влажность различных объектов, степень минерали-
зации вод, почв и грунтов. Большое значение имеет также опре-
деление кислотности различных растворов методом кондуктомет-
рического титрования.
Определение растворимости малорастворимых соединений.
Рассмотрим этот вопрос на примере хлорида серебра. Так как
AgCl очень мало растворим, то степень его диссоциации в вод-
ном растворе должна быть близка к единице, т. е. электрическая
проводимость насыщенного раствора этой соли практически не
отличается от таковой при бесконечном разведении. Допустим,
что х воды, в которой растворен AgCl, равна 1,60-10-8 См/м
при 298 К, а х насыщенного раствора хлорида серебра —
232
3,14-Ю8 См/м. Увеличение электрической проводимости при
растворении в воде хлорида серебра обусловлено появлением
в растворе ионов Ag+ и С1“. Следовательно, хлорид серебра
обеспечивает электрическую проводимость, которая равна
XAgC[ = Храствора — ЛН2о = 3,41 -108 — 1,60-108 = 1,81 -10“8 См-м2/кмоль.
Молярную электрическую проводимость при бесконечном разве-
дении находим по закону Кольрауша, пользуясь табличными
значениями электролитических подвижностей ионов Ag+ и С1”:
Xoo=XAg+ + X!cr = 0,6192 + 0,7634 = 1,3826 См-м2/кмоль.
Поскольку для данного случая а=1, Zi/ = tao. Таким образом,
можно записать, что
Х^ =х/С,
откуда
Подставляя в последнее уравнение численные значения отдель-
ных величин, получим растворимость хлорида серебра:
181 1О8
С=- -= 1.31 • Ю-8 кмоль/м3.
I ,oozo
Так как для хлорида серебра, который является бинарным элект-
ролитом [Ag+] = [C1~], то, зная концентрацию ионов, можно
определить и произведение растворимости данного соединения:
[ПР] =[Ag+) [СГ] = 1,31 • 10~8-1,31 • 10-8 = 1,72- 1(Г16.
Аналогичным образом определяют растворимость и произведение
растворимости других труднорастворимых соединений.
Определение солесодержания. Измерение электрической про-
водимости используется при контроле процесса очистки вод, в
частности при получении дистиллированной воды. Теоретическая
удельная электрическая проводимость совершенно чистой воды
при 291 К, рассчитанная на основе ионного произведения воды
и подвижностей водородных и гидроксильных ионов при беско-
нечном разведении, равна 3,8- 10“t0 См/м. Удельная электриче-
ская проводимость полученной дистиллированной воды, пере-
гнанной один раз в вакууме при 291 К, равна 4,41 • 1О^10 См/м.
Для обычных лабораторных целей используют дистиллирован-
ную воду с х» 1 • 10~8 См/м. Таким образом, измерение электри-
ческой проводимости воды позволяет судить о ее качестве, в
частности о наличии в воде (минеральной, речной, морской
и т. д.) водорастворимых солей.
Количественное содержание солей в воде может быть вычислено по изме-
рению электрической проводимости с использованием коэффициента пересчета ky
который зависит от состава солей и заданных условий анализа:
C = nk.
Средний состав присутствующих примесей обычно известен. Небольшое
изменение состава примесей несущественно изменяет коэффициент пересчета,
233
так как подвижности катионов и анионов, за. исключением Н3О+ и ОН-,, в общем
мало различаются. Для нахождения коэффициента пересчета измеряют электри-
ческую проводимость воды с известным содержанием примесей.
Очень важной задачей является определение солесодержания в почвах.
Во-первых, степень засоленности почвы очень часто является решающим факто-
ром при освоении новых земель и выборе наиболее оптимальных методов
орошения. Во-вторых, неумелое использование поливной воды без учета физико-
химических и водно-физических свойств орошаемой почвы может привести к их
вторичному засолению. Поэтому на орошаемых полях необходим контроль за
динамикой солей в почве, чтобы предотвратить вторичное засоление.
Путем непосредственного измерения электрической проводимости почвы опре-
делить содержание солей не удается, поскольку электрическая проводимость
почвы зависит также от очень многих других факторов. Поэтому определение
солесодержания в почвах в настоящее время проводят по значению электриче-
ской проводимости почвенной вытяжки с помощью так называемых солемеров.
Определение влажности. Для определения влажности самых
различных объектов (органические растворители, газы, твердые
соли, текстильные материалы, бумага, зерно, почвы и т. д.) при-
меняют прямую кондуктометрию. Принцип измерения основан на
проводимости исследуемых объектов. За последние годы в прак-
тике сельского хозяйства подобные приборы получили широкое
применение для определения влажности зерна. Некоторый объем
зерна помещается в измерительную ячейку между электродами и
измеряется сопротивление этой пробы. Чем большей влажностью
характеризуется зерно, тем меньшим сопротивлением оно обла-
дает. Обычно приборы градуируются в процентах (мае.) влаж-
ности для каждого вида зерна. Кондуктометрический метод
определения влажности зерна отличается быстротой и достаточно
высокой точностью.
В настоящее время такое же широкое распространение в сельском хозяй-
стве получил кондуктометрический метод определения влажности почвы при
помощи датчиков с промежуточной средой из гипса (так называемый метод
гипсовых датчиков). В почву на необходимую глубину помещают на несколько
лет датчики, которые представляют собой гипсовые блоки с залитыми в них
некорродирующими электродами. Сопротивление такого датчика зависит от
влажности окружающей почвы: чем выше влажность почвы, тем меньшим со-
противлением оно обладает. Метод измерения влажности при помощи гипсовых
датчиков свободен от целого ряда недостатков, свойственных измерению влаж-
ности методами прямого определения электрической проводимости почвы.
Кондуктометрическое титрование. Сущность кондуктометриче-
ского титрования заключается в измерении электрической прово-
димости раствора, меняющейся в процессе химической реакции
после добавления очередной порции титранта. По полученным
данным строят графическую зависимость электрической прово-
димости раствора от количества вещества добавленного реа-
гента. Подобные графические зависимости называются кривыми
кондуктометрического титрования. Для кондуктометрического
определения вещества, как правило, пригодны такие реакции,
на кондуктометрических кривых титрования которых имеется
четкий излом в точке эквивалентности. Если титруется смесь
веществ, то число изломов на кривой титрования будет равно
числу компонентов титруемой смеси.
234
Кривые кондуктометрического титрования характеризуются
многообразием форм. Поэтому точки эквивалентности на этих
кривых устанавливаются графическим методом. Следует отме-
тить, что графическое установление точек эквивалентности позво-
ляет использовать в какой-то мере обратимые химические реак-
ции, что является существенным преимуществом кондуктометри-
ческого метода титрования. Например, возможно кондуктометри-
ческое определение в водных растворах кислот с р/(а=10, что
нельзя осуществить индикаторным или потенциометрическим ме-
тодами анализа.
Преимуществом кондуктометрического титрования является
также то, что иногда представляется возможным использовать
реакции, при которых вещества реагируют не в стехиометриче-
ских отношениях. Однако обязательным условием при этом явля-
ется постоянство состава образующихся продуктов реакции.
В отличие от ряда других методов анализа кондуктометрическое
определение возможно в окрашенных и мутных растворах, а
также в присутствии окислителей и восстановителей.
Следует также отметить, что кондуктометрический метод
титрования обладает достаточно большой чувствительностью и
точностью. Концентрации титруемых растворов могут достигать
0,0001 кмоль/м3 и менее.
Имеется классификация методов кондуктометрического титро-
вания, которая основана на типах реакций, используемых для
определения содержания того или иного соединения. В этих
Методах используются следующие реакции: 1) кислотно-основ-
ного взаимодействия; 2) комплексообразования; 3) осаждения;
4) окисления — восстановления.
Наибольшее распространение получили методы кондуктомет-
рического титрования, основанные на использовании кислотно-
основных реакций. Так как при кислотно-основных взаимодей-
ствиях в растворах изменяются концентрации высокоподвижных
водородных и гидроксильных ионов, то кривые титрования в
данном случае имеют резко выраженные изломы, что сущест-
венно облегчает нахождение точек эквивалентности.
Рассмотрим в качестве примера кривые титрования сильных
и слабых кислот сильным основанием. Если титруется сильная
кислота, например, НС1, сильной щелочью, например NaOH, то
в результате кислотно-основного взаимодействия высокоподвиж-
ные ионы Н+ и ОН- образуют слабодиссоциирующее соединение
НгО, а в растворе накопятся менее подвижные ионы Na+ и С1“.
Электрическая проводимость уменьшится и достигнет минимума
в точке эквивалентности, когда в растворе будут находиться
только ионы Na+ и С1_. При дальнейшем прибавлении NaOH
электрическая проводимость снова возрастает, поскольку в тит-
руемый раствор вводятся высокоподвижные ионы гидроксила
(рис. 80, кривая /).
При титровании слабой кислоты изменение электрической
проводимости титруемого раствора происходит несколько иначе.
235
Рис. 80. Кривые кондукто-
метрического титрования:
/ — сильной кислоты; 2 — сла-
бой кислоты; 3 — смеси силь-
ной и слабой кислот (т. э. —
точк£ эквивалентности)
До прибавления щелочи в растворе
находится кислота в молекулярной
форме, а также незначительное число
ионов Н+ и анионов кислоты. При до-
бавлении щелочи образуется малодис-
социирующее вещество — вода, но
электрическая проводимость раствора
медленно увеличивается вплоть до
точки эквивалентности за счет даль-
нейшей диссоциации новых порций
кислоты, а также за счет увеличения
числа катионов основания и анионов
кислоты. После достижения точки
эквивалентности электрическая прово-
димость титруемого раствора увеличи-
вается более резко за счет введения
в раствор высокоподвижных ионов гид-
роксила (рис. 80, кривая 2).
При титровании смеси сильной и слабой кислот на кривой
титрования будет две точки эквивалентности. Поскольку сильная
кислота подавляет диссоциацию слабой кислоты, последняя
практически не влияет на электрическую проводимость титруе-
мого раствора. Из этого следует, что до первой точки эквивалент-
ности титруется сильная кислота и электрическая проводимость
раствора уменьшается. После первой точки эквивалентности нач-
нет титроваться слабая кислота, при этом электрическая прово-
димость раствора медленно увеличивается до второй точки экви-
валентности. После второй точки эквивалентности электрическая
проводимость титруемого раствора увеличивается за счет введе-
ния высокоподвижных ионов гидроксила (рис. 80, кривая 3).
Методы анализа, основанные на использовании реакций
комплексообразования, очень разнообразны. Развитию этих ме-
тодов способствовало применение кондуктометрии для изучения
комплексных соединений, а также в связи с использованием
комплексонов. Также получила широкое распространение группа
методов, основанная на реакциях осаждения. В отличие от гра-
виметрического химического анализа кондуктометрическое титро-
вание не требует много времени и имеет более высокую чувст-
вительность.
Группа методов, основанная на реакциях окисления—вос-
становления, распространена в меньшей степени, поскольку про-
ведение таких реакций в кислых или щелочных растворах за-
трудняет использование кондуктометрического титрования.
Линейное изменение электрической проводимости раствора
на ветвях кондуктометрических кривых титрования наблюдается
только в некоторых случаях (титрование сильных кислот или
сильных оснований, титрование солей слабых кислот или слабых
оснований, не подвергающихся заметному гидролизу, титрование,
сопровождающееся образованием осадков, и некоторые другие).
236
Нелинейность многих кривых кондуктометрического титрования
может быть вызвана рядом причин. Основными из них являются
смещение ионных равновесий в процессе титрования и обрати-
мость используемых для определения реакций.
Хронокондуктометрическое титрование. Особое преимущество по сравнению
с классическими методами имеет хронокондуктометрический метод титрования,
отличающийся автоматической записью кривых титрования, объективностью
измерений, экспрессностью и большой точностью. Этот метод титрования осно-
ван на определении содержания вещества по продолжительности титрования.
Содержание вещества находят с помощью измерений удельной электрической
проводимости раствора в процессе химических реакций. Если в классическом
методе кондуктометрического титрования устанавливают объем титранта, всту-
пившего в реакцию, то в хронокондуктометрическом титровании находят про-
должительность титрования в секундах. Характерной особенностью хронокон-
дуктометрического титрования является постоянная скорость подачи титранта
в анализируемый раствор. В хронокондуктометрическом титровании условия
титрования легко воспроизводимы, что позволяет использовать реакции, проте-
кающие с малой скоростью, реакции, в которых вещества взаимодействуют
в нестехиометрических отношениях, а также реакции, сопровождающиеся
явлениями адсорбции на осадках.
Контрольные вопросы
1. Какие данные, характеризующие раствор, можно получить с помощью
измерения электрической проводимости слабых и сильных электролитов?
2. Как можно измерить подвижность ионов?
3. Почему абсолютные скорости движения ионов гидроксония и гидроксил-
иона выше скоростей других ионов?
4. Какие факторы влияют на удельную электрическую проводимость?
5. Сопоставьте молярную электрическую проводимость слабых и сильных
электролитов.
6. Электролитическая подвижность не коррелируется со значениями истин-
ных радиусов ионов из-за гидратации. Почему?
7. Как можно измерить константу электролитической диссоциации слабых
кислот и оснований?
Глава X
ЭЛЕКТРОХИМИЧЕСКИЕ ПРОЦЕССЫ
1. ЭЛЕКТРОДНЫЕ ПОТЕНЦИАЛЫ. ЭЛЕКТРОХИМИЧЕСКИЕ
ЭЛЕМЕНТЫ И ЭЛЕКТРОДВИЖУЩИЕ СИЛЫ
Электрохимическим (гальваническим) элементом называется устройство
(прибор) для получения электрического тока за счет химических реакций, про-
текающих на электродах.
Простейший электрохимический элемент состоит из двух полу-
элементов — металлических пластин (проводников I рода), кото-
рые помещены в растворы электролитов (проводники II рода).
Полуэлементы пространственно отделены друг от друга и соеди-
нены между собой проводником I рода. Растворы электролитов
соединяются между собой проводником II рода.
Важнейшей количественной характеристикой электрохимиче-
ского элемента является электродвижущая сила
(ЭДС, обозначаемая в дальнейшем буквой Е), которая равна
равновесной разности потенциалов между двумя полуэлемен-
тами.
Если при прохождении электрического тока в разных направ-
лениях на поверхности электрода протекает одна и та же хими-
ческая реакция, но в противоположных направлениях, то такие
электроды, а также элементы, составленные из них, называют
обратимыми. ЭДС обратимых элементов является их термо-
динамической характеристикой, т. е. зависит только от темпера-
туры, давления, природы веществ, составляющих электроды и
растворы элемента, и концентрации этих растворов.
Как обратимые, так и необратимые элементы могут работать
в различных термодинамических условиях, которые определяют
обратимость или необратимость процесса. Иначе говоря, с одной
стороны, обратимость (или необратимость) элемента — это ха-
рактеристика самой цепи; она не связана с условиями проведе-
ния процесса, а определяется химическими процессами, проте-
кающими в элементе. С другой стороны, термодинамическая
обратимость (или необратимость) процесса — это характеристи-
ка условий, в которых осуществляется процесс, присущий данной
цепи.
Термодинамические условия обратимости применительно к ра-
боте электрохимических элементов заключаются в следующем.
Электрохимический элемент работает обратимо, если, во-первых,
его ЭДС лишь на бесконечно малое значение превышает прило-
женную к нему извне и противоположно направленную ЭДС
238
(так называемая обратимость условий рабо-
ты). Во-вторых, если реакция в элементе мо-
жет быть полностью обращена в противопо-
ложном направлении при подключении к нему
извне противоположно направленной ЭДС, ко-
торая лишь на бесконечно малое значение пре-
вышает ЭДС данного элемента (обратимость
самой цепи, т. е. химических реакций, проте-
кающих на электродах).
Таким образом, нужно четко различать не-
обратимость работы данного элемента, вызван-
ную лишь условиями работы элемента при
принципиальной обратимости самой цепи, и
необратимость самой цепи. Примером обрати-
мого элемента может служить элемент Даниэля — Якоби, кото-
рый, согласно международной конвенции, схематически изобра-
жается следующим образом (рис. 81):
(-) Zn|Zn2+||Cu2+!Cu (+)
При работе элемента идут следующие реакции на электродах:
Zn = Zn2+ + 2e- и 2е~-}-Cu2+ = Cu
При пропускании от внешнего источника через элемент тока бес-
конечно малой силы на электродах протекают обратные реакции
Zn2+ + 2^ = Zn; Cu=Cu2+4-2^
Рис. 81. Схема галь-
ванического эле-
мента Даниэля —
Якоби
и такому элементу соответствует схематическая запись:
Cu)Cu2+|JZn2+|Zn
В качестве примера необратимого элемента рассмотрим элемент
Вольта
Zn|H2SO4|Cu
При работе этого элемента протекают электродные реакции
Zn = Zn2+-|-2е~ и 2Н+Ч-2е-=Н2
При пропускании от внешнего источника тока бесконечно
малой силы электродными реакциями будут
2Н+-{-2е_ = Н2 и Cu = Cu2+ + 2e-
т. е. восстановление ионов цинка в данном случае не происходит.
Электрохимические реакции на электродах, вызывающие про-
хождение тока в проводнике, протекают только при замкнутой
цепи и прекращаются при ее размыкании. В необратимом эле-
менте обычно возможно протекание химической реакции и при
разомкнутой внешней цепи (например, реакция Zn-|-H2SO4
в элементе Вольта). Но и обратимая, с точки зрения химизма
процесса, цепь в общем далека от термодинамического равнове-
сия. Если такую цепь замкнуть на конечное сопротивление и пре-
доставить самой себе, то во внешней цепи возникнет электриче-
231'
ский ток измеримой силы, т. е. цепь совершает работу, необра-
тимо приближаясь к состоянию термодинамического равновесия.
Основные электрохимические явления — это процессы, проте-
кающие на границах раздела различных фаз. Работа электро-
химического элемента и его ЭДС — это лишь суммарное прояв-
ление совокупности процессов, совершающихся на этих границах
раздела.
Принцип работы электрохимического элемента. Как уже ука-
зывалось, ток в электрохимическом элементе возникает в резуль-
тате химической реакции. Такая реакция состоит из двух полу-
реакций: окисления, при которой восстановленная форма отдает
электроны, и восстановления, при которой окисленная форма
принимает электроны. Например, в элементе Даниэля — Якоби
суммарная реакция следующая:
Cu2+ + Zn = Си + Zn2+
Таким образом, электрохимический элемент можно охаракте-
ризовать как прибор для пространственно разделенного проте-
кания окислительной и восстановительной полуреакций, что поз-
воляет использовать возникший ток для получения полезной
работы.
ЭДС элемента зависит от константы равновесия реакции,
происходящей в электрохимическом элементе. Чем больше кон-
станта равновесия, тем больше ЭДС элемента. Итак, на основе
измерения ЭДС можно вычислить некоторые термодинамические
параметры химической реакции, протекающей в элементе.
Если при работе электрохимического элемента переносится
1 Ф электричества, то в элементе произойдут следующие про-
цессы:
1) */г кмоль Zn расходуется на образование 1 кмоль ионов
Zn2+;
2) 1 Ф электричества (96 500 Кл) соответствует числу элект-
ронов, равному постоянной Авогадро, которое переходит по
внешней цепи к медному электроду;
3) 1 кмоль ионов Си2+ превращается в !/2 кмоль Си, осаж-
дающейся на медном электроде.
Химическая реакция в электрохимическом элементе будет
идти самопроизвольно до тех пор, пока не будет достигнуто
состояние равновесия. При достижении состояния равновесия
ЭДС элемента будет равно нулю.
ЭДС электрохимического элемента является величиной поло-
жительной, так как она всегда соответствует самопроизвольно
протекающему процессу, дающему положительную работу.
Обратному процессу, который не может протекать самопроиз-
вольно, соответствовала бы отрицательная ЭДС.
Стандартная ЭДС элемента £° есть разность стандартных
электродных потенциалов, т. е. потенциалов, возникающих на
электродах при активности электродного электролита, равной 1.
Чтобы вычислить стандартную ЭДС электрохимического эле-
240
мента для реакции этого элемента и решить, является ли реакция
самопроизвольной, используют следующие три правила.
1. Стандартная электродвижущая сила элемента равна стан-
дартному электродному потенциалу правого электрода Минус
стандартный электродный потенциал левого электрода.
Электрод слева должен быть записан в последовательности
металл—ион. Электрод справа записывается в последовательно-
сти ион — металл. Например, для элемента
Zn | Zn2+ || Cu2+ | Си (I)
E° = 0,337 — (—0,763) = 1,100В при 298 К.
Для элемента
Си I Cu2+HZn2+IZn (II)
стандартная ЭДС будет равна
£° = —0,763 —0,337 = — 1,100 В при 298 К (П элемент).
2. Реакция, протекающая на левом электроде, записывается
как окислительная, а реакция на правом электроде записывается
как восстановительная. Реакция гальванического элемента равна
сумме этих двух реакций.
Реакция для I элемента:
(левый электрод) Zn = Zn2+ + 2е~ (окисление)
(правый электрод) Cu2+4-2e”=Cu (восстановление)
Zn + Cu2+ = Zn2+ + Си (суммарная реакция)
Реакция для II элемента:
(правый электрод) Zn2“f’+ 2e“ = Zn (восстановление)
(левый электрод) Cu = Cu2+4-2e_ (окисление)
Си + Zn2+= Си2+ + Zn (суммарная реакция)
3. Если EQ элемента, вычисленная по правилу 1, является
положительной, то реакция элемента, записанная по правилу 2,
будет самопроизвольной, когда активности исходных веществ и
конечных продуктов реакции равны единице.
Таким образом, реакция в I элементе, для которой £°= 1,100 В,
является термодинамически самопроизвольной, а реакция во
II элементе, для которой £° =— 1,100 В, — несамопроизвольной.
Это правило вытекает из того факта, что самопроизвольность
реакции определяется отрицательным значением AG°, a AG° =
= —nFEy где п — число электронов, £ — постоянная Фарадея:
А6° =—2’96 500-1,100=—212,3 кДж/моль (для I элемента).
Таким образом, критерием самопроизвольности работы эле-
мента является отрицательное значение AG0 электрохимической
реакции.
Поскольку
ДО° = -ЯПпЛ.
можно рассчитать константу этой реакции. Так как
ЯТ1пК==п£Е°,
241
то
nF
а константа химической реакции равна
nFEQ
К
л = е
или в логарифмической форме
Для элемента Даниэля — Якоби £° = 1,100 В. Отсюда
Иными словами, реакция
Cu2+ + Zn = Zn2+ + Си
практически полностью сдвинута вправо, т. е. протекает до
конца.
2. ИЗМЕРЕНИЕ ЭДС. НОРМАЛЬНЫЙ ЭЛЕМЕНТ ВЕСТОНА
Измерение ЭДС. Для измерения равновесной (обратимой)
ЭДС электрохимического элемента необходимо, чтобы процесс
совершался бесконечно медленно, т. е. чтобы элемент работал
при бесконечно малой силе тока. Это условие выполняется в
компенсационном методе измерения, который основан на том, что
элемент включается против внешней разности потенциалов, и
последняя подбирается так, чтобы ток в цепи отсутствовал.
В этом случае внешняя разность потенциалов равна ЭДС изу-
чаемого элемента. Пользуясь компенсационным методом (мето-
дом Поггендорфа), можно непосредственно измерить значение
равновесной ЭДС элемента.
В лабораторной практике предпочитают чаще сравнивать
ЭДС изучаемого элемента с ЭДС
так называемых стандартных (нор-
мальных) элементов, которая при их
кратковременной работе практиче-
ски постоянна и мало меняется с
изменением температуры. Для отно-
сительного измерения ЭДС элемен-
та используют, таким образом, срав-
нительный метод, при котором схема
измерения также является компен-
сационной (рис. 82). Реохорд АВ —
Рис. 82. Схема для измерения
ЭДС
однородная по толщине проволока
со значительным сопротивлением —
242
натянута на линейку или на барабан со шкалой. Концы реохорда
соединены с источником постоянного тока С, ЭДС которого
равна Е. Скользящий контакт на реохорде соединен с одним из
концов реохорда А через гальванометр G и ключ К, к которому
поочередно подсоединяется изучаемый элемент Ех или нормаль-
ный элемент Ен. Как источник постоянного тока С, так и элемен-
ты Ех и Ев подсоединены к левому концу реохорда одноименны-
ми полюсами. Передвигая скользящий контакт (положение точ-
ки D на реохорде АВ), находят положение, при котором стрелка
гальванометра стоит на нуле, т. е. в цепи ток отсутствует. Эту
операцию производят дважды: сначала для изучаемого элемента
Ех, затем для нормального £н. Допустим, для элемента Ех на
реохорде найдена точка D, когда ток в цепи АСВЕХ отсутствует
(точка компенсации). Тогда разности потенциалов между точка-
ми Л и D, создаваемые двумя противоположно направленными
источниками тока С и Ех, одинаковы и равны ЭДС элемента Ех.
Это значение составляет часть ЭДС аккумулятора Е, пропор-
циональную отношению AD/AB, т. е.
Е
AD
АВ
(10.1)
Для нормального элемента Ев с известной ЭДС, равной Eq,
положение скользящего контакта, при котором ток в цепи отсут-
ствует, найдено при D'. Отсюда
А ТУ
Е^-^-Е. (10.2)
Из выражений (10.1) и (10.2) получают уравнение для расчета
ЭДС изучаемого элемента Ех,
rj AD „ ах с
AD ац
где AD соответствует компенсации на реохорде для изучаемого
элемента (ах), AD' — компенсации на реохорде для стандартно-
го (нормального) элемента
мента. Для определения
ЭДС элемента описанным
методом нет необходимос-
ти измерять силу тока и
напряжения, а также знать
ЭДС внешнего источни-
ка тока (аккумулятора).
Нужно лишь иметь нор-
мальный элемент и такой
реохорд, сопротивление
любого отрезка которого
было бы строго пропорцио-
нально длине этого отрезка.
Нормальный элемент
Вестона. Основным нор-
(ан), Eq — ЭДС стандартного эле-
243
мальным элементом, используемым при измерении ЭДС компен-
сационным методом, является насыщенный элемент Вестона
(рис. 83), который схематически записывается следующим об-
разом:
(Hg + Cd) I CdSO4-8/3H2O (т) I CdSO4, aq | CdSO4-8/3H2O (t) | HgSO4 (t) | Hg
На электродах элемента Вестона протекают следующие
электродные реакции:
на левом (отрицательном) — окисление
Cd + SOl-aq + 8/3Н2О (ж) = CdSO4’8/3H2O (т) + ге-
на правом (положительном) — восстановление
Hg2SO4 4- 2е“ = 2Hg^) + SO?',' aq
Таким образом, суммарная химическая реакция, идущая в
элементе Вестона, записывается следующим образом:
Cd + Hg2SO4 (т) + 7зН2О = 2Hg (ж) + CdSO4 -8/3Н2О (т)
ЭДС элемента Вестона определена путем тщательных измере-
ний при различных температурах. В частности, при 293 К это зна-
чение равно 1,0180 В. Температурный коэффициент элемента мал
и равен в среднем 0,000005 'В/К-
3. ТЕРМОДИНАМИКА ЭЛЕКТРОХИМИЧЕСКИХ ЭЛЕМЕНТОВ
Электрохимические элементы часто применяют для того, чтобы
определить изменение изобарного потенциала химической реакции.
Электрическая энергия, вырабатываемая элементом, работающим
обратимо, равна полезной работе суммарного процесса, протекаю-
щего в элементе, который рассматривается как термодинамическая
система. Как известно, полезная работа обратимого процесса
является максимальной и равна изменению изобарного потенциала
системы AG. Это изменение изобарного потенциала вызвано со-
вокупностью электрохимических реакций на электродах, т. е. сум-
марной химической реакцией или другими физико-химическими
процессами (растворение, выравнивание концентраций, фазовое
превращение и т. д.), протекающими обратимо. Если процесс яв-
ляется обратимым, можно заставить элемент работать в условиях
почти полной компенсации ЭДС элемента подключением внеш-
ней разности потенциалов. При этом можно провести процесс в
электрохимическом элементе бесконечно медленно, приближаясь
бесконечно близко к состоянию равновесия. Такому процессу и
соответствует измеренная величина Е, зная которую можно вы-
числить изменение изобарного потенциала системы AG.
При проведении электрохимического процесса в таких близких
к обратимым условиях суммарное значение AG элемента и внеш-
него источника тока равно нулю (осуществляется состояние рав-
новесия) :
АбСумм — АбЭл Т Абист.тока = 0.
244
Количество электричества, соответствующее числу молей, ука-
занному в химическом уравнении электрохимического элемента,
равно nF. Если это количество электричества перенести при разно-
сти потенциалов Е, то общая работа электрического тока электро-
химического элемента, работающего обратимо, будет равна
A3J}=~nFE. (10.3)
Отсюда следует, что для элемента должно соблюдаться равен-
ство
А(?эл == —ЛОист.тока == —tlFE. (10.4)
ЭДС элемента не зависит от стехиометрических коэффициентов
в равновесной химической реакции, но изменение изобарного по-
тенциала AG зависит от п, которое, в свою очередь, зависит от
того, как записано стехиометрическое уравнение.
Работа электрического тока, вычисленная по уравнению (10.3),
выражается в джоулях. Используя температурный коэффициент
ЭДС, можно рассчитать изменение энтропии для реакции, проте-
кающей в электрохимическом элементе, поскольку
(~) (105)
\ дТ Jp
Сопоставляя уравнение (10.4) с уравнением (10.5), находим, что
nF(-^) =AS-
\ дТ
Таким образом, измерив ЭДС элемента и ее температурный
коэффициент, можно легко найти значения AG и ДЗ для суммар-
ного процесса, протекающего в электрохимическом элементе. По-
скольку этот процесс является самопроизвольным, ДG с 0.
Еще одну важную термодинамическую характеристику этого
процесса — изменение энтальпии — вычисляют по уравнению
Гиббса — Гельмгольца
откуда получается значение энтальпии
v' г(^), —"«*+
Пример. В электрохимическом элементе
(4-)Pt(Cl2)|HCl(aq), AgCl[Ag(—)
протекают следующие электрохимические реакции:
(окисление) А^(т) + Cl- = AgCl(т) + е“
(восстановление) ’/2С12 + е~ — С1 “
Суммарная электрохимическая реакция представляет собой химическую реак-
цию образования кристаллического AgCl из серебра и газообразного хлора:
Ag(r) + ’/2С12 = АИС1(т)
ЭДС элемента при 298 К равна 1,132 В, температурный коэффициент
245
дЕ \
= —0,000477 В/К. Подставив эти значения в приведенные формулы,
получим
AG — —nFE = —1 -96500-1,132 == —109 238 Дж/моль,
AS = = 1.96 500-0,000477 = —46,031 Дж/(моль-К),
FAS = —298-46,031 = —13717,24 Дж/моль,
AW = AG + TAS = - 109 238 — 13717,24 = —122955,24 Дж/моль.
Из полученных значений изменения термодинамических параметров можно
непосредственно получить величины теплот образования AgCl для двух процес-
сов (оба при р ~ const):
1) обратимого процесса, протекающего в электрохимическом элементе
при бесконечно малой силе тока (бесконечно медленный процесс; AG = 0)
Qo6p = TAS = —13717,24 Дж/моль;
2) предельно необратимого процесса, протекающего при постоянном давле-
нии и полном отсутствии полезной работы АПол (непосредственное взаимодей-
ствие серебра и хлора)
Фнеобр = А//= —122955,24 Дж/моль.
Значение Q06p равно теплоте, выделяемой работающим электрохимическим
элементом. Если элемент поместить в калориметр, то, в принципе, можно изме-
рить Qo6p как поглощенную теплоту. Но значение это будет не совсем точным,
так как нельзя соблюсти условие бесконечной медленности процесса. Однако,
зная ЭДС при разных температурах, нетрудно вычислить Q06p = TAS, как это
сделано в разобранном примере. Величина фнеобр — это стандартная (табличная)
теплота образования AgCl, ее можно либо измерить экспериментально, либо
вычислить с помощью закона Гесса по результатам калориметрических изме-
рений.
Результаты определения величины QHeo6p = АЯ электрохимическим или кало-
риметрическим методом, естественно, совпадают, если измерения в обоих случаях
достаточно точны.
Таким образом, измерение ЭДС электрохимических элементов
является простым^и точным методом вычисления термодинамиче-
ских характеристик химических реакций и некоторых важных
физико-химических процессов в растворах. Методом ЭДС для
измерения и расчета указанных характеристик можно пользо-
ваться только тогда, когда процесс можно осуществить обратимо
в электрохимическом элементе, разбив этот процесс на два, про-
текающих обратимо на двух электродах.
4. ЭЛЕКТРИЧЕСКИЕ ПОТЕНЦИАЛЫ НА ФАЗОВЫХ ГРАНИЦАХ
При соприкосновении проводника I рода с электролитом на
границе электрод — раствор возникает двойной электри-
ческий слой. В качестве примера рассмотрим медный
электрод, погруженный в раствор медного купороса.
В концентрированных растворах при погружении металла в
раствор часть ионов Си2+ из раствора перейдет на металл, соз-
дав на нем положительный заряд. Возникший положительный за-
ряд будет препятствовать дальнейшему переходу ионов Си2+
246
из раствора на металл
и приведет к образова-
нию двойного электри-
ческого слоя вблизи
поверхности металла
за счет притянутых, к
нему вследствие элект-
ростатического взаимо-
действия анионов SO2"
(рис. 84, а). Этот про-
цесс будет идти до тех
пор, пока не установит-
ся так называемое
электрохимическое рав-
новесие:
Рис. 84. Схема двойного электрического слоя
переход ионов с электрода в раствор =<=► переход ионов из
раствора на электрод
При погружении металла в разбавленный раствор будет на-
блюдаться обратный процесс перехода ионов меди из электри-
чески нейтральной кристаллической решетки металла в раствор и
электрод окажется заряженным отрицательно за счет избытка
электронов. Этот заряд будет препятствовать дальнейшему пере-
ходу ионов Си24" в раствор, в результате чего установится новое
электрохимическое равновесие (рис. 84, б).
Можно подобрать концентрацию раствора электролита такую,
что на поверхности электрода не возникнет двойного электриче-
ского слоя. Растворы такой концентрации называют нулевы-
ми растворами. Согласно Нернсту, единственным источ-
ником ЭДС электрохимического элемента является двойной
электрический слой на поверхности электродов, т. е.
Е ------ Е1 ------1>2,
где 81 и 82 — скачки потенциала в ионных двойных слоях. Таким
образом, потенциал металла в нулевом растворе Нернста опреде-
ляется как абсолютный нуль потенциалов. Однако на основании
экспериментальных данных было установлено, что ЭДС электро-
химических элементов, составленных из двух различных электро-
дов, погруженных в свои нулевые растворы, весьма значительно
отличаются от нуля. Следовательно, потенциал электрода в ну-
левом растворе, называемый потенциалом нулевого
заряда, нельзя рассматривать как абсолютный нуль потен-
циалов.
Контактным потенциалом называется разность потенциалов
между двумя точками в вакууме (или в диэлектрике), находя-
щимися вблизи поверхностей двух металлов (на расстоянии
10-7...10“6 м от поверхности каждого металла). А. Н. Фрумки-
ным было установлено и экспериментально проверено, что изме-
ряемая разность потенциалов между двумя различными метал-
лами, помещенными в свои нулевые растворы, т. е. разность
247
между потенциалами их нулевых зарядов, приблизительно равна
контактной разности потенциалов этих металлов в вакууме.
Если металлы погружены не в'нулевые растворы, то на грани-
цах электродов с раствором возникают, кроме того, ионные двой-
ные электрические слои. Таким образом, измеряемая ЭДС галь-
ванического элемента, состоящего из двух электродов, суммиру-
ется из контактного потенциала, металлов в воде как изоляторе
ек и разности потенциалов в ионных двойных слоях 81 и 82, воз-
никающих в результате обмена ионами между металлами и
раствором:
Е = ei — 82 + ек.
При этом ионные двойные слои на электродах всегда таковы, что
ЭДС электрохимического элемента соответствует суммарному
процессу, протекающему в этом элементе.
При изучении термодинамики электрохимических процессов
достаточно знать, что изменение энергии электрохимического
элемента полностью определяется химическими реакциями на
электродах.
Величина и знак электродного потенциала. Скачок потен-
циала на границе металл — раствор, как и разность потенциалов
между двумя точками, находящимися в различных фазах, экспе-
риментально измерить невозможно. Можно измерить лишь ЭДС
электрохимического элемента. Иначе говоря, опытным путем
можно определить только относительные величины так называе-
мых электродных потенциалов, т. е. ЭДС цепи, со-
ставленной из данного электрода и некоторого стандартного
электрода, потенциал которого условно принимается равным ну-
лю. Таким стандартным электродом, или электродом
сравнения, является обратимый водородный электрод, в ко-
тором газообразный водород находится при давлении 101,3 кПа
Рис. 85. Водородный электрод
и насыщает платиновую плас-
тинку. Раствор, в который
погружен водородный электрод,
содержит ионы водорода, при-
чем активность ионов Н+ рав-
на единице (рис. 85):
Pt, Н2 1н+А~
(рн2 = 101,3 кПа) aH+ — 1
Так как адсорбированный пла-
тиной водород находится в
равновесии с газообразным во-
дородом, можно рассматривать
электрохимическое равновесие
в следующем виде:
8
Н2(г) + 2Н2О 2НзО+ + 2е-
248
Электродный потенциал нестандартного водородного электро-
да, не равный нулю, зависит от давления водорода и активности
ионов водорода в растворе
с
Pt, Н2 Н+А~
(рн2^1) (ан+^1)
и определяется по уравнению Нернста
0 . RT . RT .
8 = 8°+ -у- 1пан+ — 1прн2.
Так как по условию е° — 0, можно записать уравнение для
электродного потенциала нестандартного водородного электрода
RT RT
8 = —р~ 1пан+ 1п^н2-
Электродным потенциалом электрода называется ЭДС элемента, состав-
ленного из данного электрода (справа) и стандартного водородного электрода
(слева).
Например, для нахождения электродного потенциала цинка
составляют элемент
Pt, H2|2H+||Zn2+|Zn
Стандартный водородный электрод всегда записывается слева.
При активности ионов цинка, равной 1, ЭДС элемента равна
—0,763 В. Это и есть стандартный электродный потенциал цинко-
вого электрода, поскольку потенциал стандартного водородного
электрода принят равным 0. Чтобы найти электродный потенциал
меди, составляют элемент
Pt, H2|2H+(|Cu2+|Cu
Определенная в данном случае ЭДС и, следовательно, стандарт-
ный электродный потенциал медного электрода равны -j-0,337 В.
В схеме элемента целесообразно записывать сочетание ме-
талл— раствор иона в том порядке, который имеется в записи
элемента, составленного из стандартного водородного электрода
и испытуемого электрода. Именно для записанных таким обра-
зом электродов приведены электродные потенциалы с соответст-
вующим знаком (табл. 40). Знак плюс соответствует, как уже
отмечалось, движению ионов слева направо (от раствора к
электроду в элементе, в котором рассматриваемый электрод сое-
динен с водородным).
Стандартные электродные потенциалы. В табл. 40 приведены
стандартные потенциалы е° некоторых электродов в водных раст-
ворах. Так как они измеряются относительно стандартного водо-
родного электрода, знак величины 8° указывает на знак заряда
этого электрода и знак ЭДС всего элемента, составленного из
данного электрода и стандартного водородного электрода:
Pt, Н2|2Н+||Мл+|М
249
Таблица 40. Стандартные электродные потенциалы
в водных растворах
Электрод e", В Реакция в полуэлементе
K+IK -2,925 К+ + е- = К
Na+|Na -2,714 Na+ + е~ = Na
Mg2+|Mg -2,370 '/2Mg2+ + е~ = '/2Mg
А13+|А1 -1,660 7зА13+ + е~ = '/зА!
Zn2+|Zn -0,763 72Zn2+ + e- = 72Zn
Fe2+|Fe -0,440 72Fe2+ + е~ = 72Fe
Cd2+|Cd -0,403 72Cd2+ + е~ = 72Cd
Pb2+|Pb -0,126 72Pb2+ + е~ = 72РЬ
2H+|(H2)Pt 0,000 Н+ +е- = 7гН2
Cu2+|Cu +0,337 7гСи2+ + е~ = 7аСи
Pb4+Pb +0,70 7«РЬ4+ + е~= 7<РЬ
Hgi+lHg +0,789 72Hgi+ + е- = Hg
Ag+|Ag +0,799 Ag+ + е~ = Ag
Hg2+|Hg +0,854 72Hg2+ + е~ = 72Hg
Au3+|Au + 1,29 7зАи3+ + е~ = 7зАи
Стандартные электродные потенциалы всех электродов, изме-
ренные относительно нормального водородного электрода, со-
ставляют ряд напряжений. Следует помнить, что таблич-
ные значения 8° относятся к растворам с ам+ = 1. Электроды в
таблице записаны в последовательности ион — металл, а все
электродные реакции — в виде реакций присоединения электронов.
Значение стандартного электродного потенциала есть мера того,
что реакция на электроде будет протекать в направлении восста-
новления, т. е. принятия электронов. Чем ниже реакция располо-
жена в таблице, тем больше стремление окисленной формы при-
нять электроны и восстановиться. Чем выше реакция располо-
жена в таблице, тем больше стремление восстановленной формы
отдать электроны и окислиться. Например, активные металлы
натрий и калий имеют очень большие отрицательные стандарт-
ные электродные потенциалы и большую склонность к отдаче
электронов.
Восстановленная форма любого элемента или иона при актив-
ности, равной единице, будет восстанавливать окисленную форму
такого элемента или иона (при активности равной единице),
стандартный электродный потенциал которого имеет менее отри-
цательное, т. е. более положительное значение. Помещенные в
табл. 40 стандартные электродные потенциалы могут быть ис-
пользованы для вычисления ЭДС электрохимических элементов
и констант равновесия соответствующих химических реакций.
250
5. КЛАССИФИКАЦИЯ ЭЛЕКТРОДОВ.
ЭЛЕКТРОДЫ ПЕРВОГО И ВТОРОГО РОДА
Все электроды по типу электродной реакции можно разделить
на две группы.
Электроды первого рода. К ним относятся электроды, состоя-
щие из металлической пластинки, погруженной в раствор соли
этого же металла, например Zn|Zn2+, Cu|Cu2+, и водородный
электрод. В зависимости от знака ЭДС элемента, в который
включен электрод, на металлической пластинке идет процесс
перехода катиона из металла в раствор или из раствора в ме-
талл, т. е. данные электроды обратимы по катиону. Потенциал
электрбдов первого рода связан с активностыб катиона в раст-
воре уравнением Нернста
6=80+1пам+’ (,о-6)
где е — электродный потенциал; 8° — нормальный (стандарт-
ный) электродный потенциал, измеряемый в стандартных усло-
виях (Г = 298 К, р= 101,3 кПа при активности электродного
электролита 1 кмоль/м3); ам+ — активность ионов металла в
растворе.
Электроды первого рода используют, как правило, в качестве
индикаторных электродов. Индикаторные электроды
позволяют определять активность ионов металлов путем измере-
ния их потенциалов.
Электроды второго рода. К ним относятся электроды, в кото-
рых металл покрыт слоем малорастворимой соли этого металла и
находится в растворе, насыщенном этой солью и содержащем
другую легкорастворимую соль с тем же анионом. Таковы, на-
пример, каломельный и хлорсеребряный электроды. Электроды
этого типа обратимы как относительно катиона, так и относитель-
но аниона, но регулировать можно только концентрацию аниона,
и только таким образом можно влиять на их электродный по-
тенциал. Электроды второго рода используют в качестве элек-
тродов сравнения, так как при соответствующем приго-
товлении они имеют постоянные значения потенциала. Электро-
дами сравнения могут быть как электроды первого рода (водо-
родный), так и электроды второго рода (каломельный, хлорсе-
ребряный) .
В отдельную группу выделяются окислительно-восстанови-
тельные электроды.
Каломельный электрод. Работа с водородным электродом в
качестве электрода сравнения требует соблюдения ряда пре-
досторожностей. Гораздо проще работать с каломельным элект-
родом, который может быть легко приготовлен и потенциал кото-
рого относительно водородного электрода точно известен (рис. 86).
Каломельным электродом называется ртутный электрод, помещен-
251
ный в раствор КС1 определен-
ной концентрации и насыщен-
ный Hg2Ch (каломель)
Hg|Hg2Cl2, КС1
Потенциал ртутного электрода
связан с концентрацией соли
ртути в растворе по уравнению
Нернста
EHg = SHg+-^-lnatHg§+]. (1о7)
В насыщенном растворе Hg2Ch
произведение растворимости
ПР = fl[Hgi+]aci- есть величина
Рис. 86. Каломельный электрод постоянная. Концентрация
ионов хлора задана концентра-
цией КО и может быть изменена. Выразив из произведения рас-
творимости активность ионов [Hg2+] и подставив ее в уравнение
(10.7), получим
о I RT 1 ГТП RT | 2
eHg = 8^g + -^7-In ПР — — In а£г.
(10.8)
Первые две величины в правой части уравнения (10.8) являются
постоянными и могут быть объединены в одну еХ. В этом случае
уравнение Нернста для каломельного электрода запишется в виде
с ______Ро
ЕКЛМ 8клм
RT .
-^lnaCi->
т.е. потенциал этого электрода зависит от
активности ионов С1“. Полученное уравнение
справедливо для всех электродов второго рода.
В общем виде оно запишется так:
RT
е — е°--^rlnaaH. (10.9)
Каломельный электрод при заданной кон-
центрации ионов хлора (т. е. концентрации
КС1) имеет совершенно определенный потен-
циал и может служить электродом сравнения.
В последнее время в качестве основного
электрода сравнения, применяемого на прак-
тике, используется хлорсерефяный электрод
(рис. 87)
Рис. 87. Хлорсереб-
ряный электрод:
/ — серебряная про
волока; 2 — слой
AgCl; 3— раствор
НС1 или КС1; 4 —
шлиф
AglAgCl, КО
потенциал которого связан с концентрацией
ионов хлора по уравнению Нернста для элект-
родов второго рода (10.9).
252
Стеклянный электрод. По прин-
ципу работы стеклянный электрод
относится к так называемым ион-
селективным (мембранным) элект-
родам. В основе работы таких
электродов лежат ионообменные ре-
акции, протекающие на границах
мембран с растворами электролитов,
т. е. в электродных реакциях элект-
роны участия не принимают. Ионсе-
лективные электроды могут быть
обратимы как по катиону, так и по
аниону в зависимости от свойств
используемой мембраны.
Принцип действия мембранных
электродов заключается в следую-
щем. Мембрана, являющаяся осно-
вой электрохимической ячейки,
должна быть селективна по отноше-
Рис. 88. Схема мембранного
электрода:
/ — внутренний электродный раст-
вор; 2 — внутренний электрод срав-
нения; 3 — гальванометр; 4 —
внешний электрод сравнения; 5 —
исследуемый раствор; 6 — мембра-
на
нию к данному иону. Она разделяет
два раствора с различной активностью этого иона. Разность по-
тенциалов, установившаяся между двумя сторонами мембраны,
измеряется с помощью двух электродов (рис. 88). При соответст-
вующем составе и строении мембраны ее потенциал зависит толь-
ко от активности данного иона по обе стороны мембраны. Ника-
кой другой процесс, протекающий в мембране, не влияет на
мембранный потенциал.
Теория стеклянного электрода была разработана Б. П. Ни-
кольским и основана на представлении о существовании обмена
ионами между стеклом и раствором. Таким образом, электродная
реакция сводится к обмену ионами Н+ между двумя фазами —
стеклом и раствором:
^нс+.
Потенциал стеклянного электрода с учетом подобного обмена мо-
жет быть выражен уравнением
8еТ = е0 +4^1п-ф-. (10.10)
ан+
В ионный процесс кроме водорода вовлекаются также ионы
щелочных металлов, которые входят в состав стекла. В результате
этого процесса они часто заменяются на ионы водорода, а между
поверхностью стекла и раствором устанавливается следующее рав-
новесие:
н£ + М^ « Нр+ 4; Met
где М+ — ион щелочного металла, входящего 9 состав стекла.
Установившееся равновесие описывается соответствующей
константой обмена:
р ст
flH+aM+
ст р
аН+аМ+
Аобм
(10.11)
253
Значение константы обмена зависит от состава электродного стекла и от •
температуры. В настоящее время изготавливают стекла с константой обмена от
10-4 до 10-15. Стеклянные электроды, используемые для измерения pH, характе-
ризуются константами обмена 10“и...10~15.
Для данного стекла число обменных мест, занятых ионами водорода и иона-
ми металл^, остается величиной постоянной, т. е.
„СТ I „ст „
ан + । ам+ — а‘
С учетом этого можно записать уравнение (10.11) для константы обмена следую-
щим образом:
1Z _ ан+(а—ан+)
Лобм--------------•
После преобразования этого уравнения получим
°н+_ Он+^обмОм'1' (10.12)
СТ ’ ,
dpi + CL
Подставив полученное значение afp /ан+ (Ю. 12) в уравнение электродного по-
тенциала стеклянного электрода (10.10), имеем
eCT = e°-^lna + -^ln(afI+ + (10.13)
Первые два члена уравнения (10.13) в правой части являются величинами
постоянными для данного сорта стекла и могут быть объединены в одну eJT. Тогда
уравнение запишется так
^ = 8?. +-£L|n(a?! +Кобм<4ь). (10.14)
Из уравнения (10.14) следует, что потенциал стеклянного
электрода зависит от двух величин: активности ионов водорода
и активности ионов щелочного металла в растворе.
Если Кобм будут величиной порядка 10~4...10~15, то ан+^>
/СобмЯм+ и поэтому величиной ЛобмЯм+ в уравнении (10.14) мож-
но пренебречь. Отсюда потенциал стеклянного электрода вы-
разится уравнением
есг = е?т + -^-1п^+. (10.15)
т. е. целиком обусловлен активностью ионов водорода в растворе.
Такие электроды называются электродами с водород-
ной функцией, и именно они используются для потенциомет-
рического определения pH растворов.
Если /Собм имеет большое значение (порядка 10-1...10~3), то
произведением /<Обмам+ пренебречь нельзя, поскольку а^+ может
быть существенно меньше этого произведения, т. е. а^+ <С КОбм«м+-
Тогда уравнение (10.14) для потенциала стеклянного электрода
примет вид
0 1 1 гл 1 RT t р
8СТ Ест t in Лобм "1 р 1П
Поскольку Кобм является величиной постоянной для данного
сорта стекла, то, объединив ее с е?т в одну постоянную, получим
уравнение для стеклянного электрода с металличе-
ской функцией в виде
254
ем — е° + In а^+.
Такие электроды используют в качестве индикаторных для опре-
деления активности катионов соответствующего щелочного ме-
талла.
Если раствор содержит смесь двух катионов (например, смесь солей калия
и натрия), то при высоких значениях pH, т. е. незначительной активности ионов
водорода, уравнение стеклянного электрода примет вид
RT
6Ст==Ест-] (aNa+ + Ак NaflK+)*
где Лк, Na — константа ионного обмена.
Для электродов с нерезко выраженной специфичностью по ионам одного
вида (например, для ионов натрия), а также при средних значениях констант
обмена и при наличии ионов нескольких щелочных , металлов уравнение для
потенциала стеклянного электрода существенно усложняется:
RT
6ст = £?т + ~ р 1п(дн+ + Ан, Na^Na+ + Ан, КаК+)-
Таким образом, в зависимости от сорта стекла и, следовательно, от константы
обмена стеклянный электрод может характеризоваться либо водородной функ-
цией, либо металлической.
Стеклянный электрод с водородной функцией схематически
записывается следующим образом:
AglAgCl, НС1|стекло|Н^ (раствор)
где —неизвестная концентрация ионов водорода в исследуе-
мом растворе.
В качестве внутреннего электрода сравнения в стеклянном
электроде используется хлорсеребряный электрод. Наиболее ча-
сто стеклянный электрод выполняется в виде стеклянной трубки
с напаянной на конце мембраной в виде шарика (рис. 89).
Шарик заполняется раствором НС1 с определенной концентра-
цией ионов Н+, в который погружается внутренний электрод
сравнения.
Водородная функция стеклянного электрода связана
с составом стекла, его гигроскопичностью, химической
устойчивостью и толщиной мембраны. При подготовке
стеклянного электрода к работе происходят гидратация и
набухание поверхностного слоя мембраны. Гидратация
мембраны оказывает заметное влияние на водородную
функцию электрода: чем больше гидратация мембраны,
тем в большей степени водородная функция приближается
к идеальной.
На стабильность водородной функции электрода за-
метное влияние оказывает так называемый потенциал
асимметрии. Если по обе стороны стеклянной мембраны
помещен один и тот же раствор, в который опущены
одинаковые вспомогательные электроды, то в такой ячейке
будет возникать небольшая разность потенциалов. По-
скольку потенциалы двух вспомогательных электродов
равны по значению и противоположны по знаку, эта
разность потенциалов должна быть обусловлена неодина-
ковыми свойствами поверхностных слоев мембраны, т. е.
ее асимметрией.
Рис. 89. Стеклян-
ный электрод:
/ — стеклянная мем-
брана; 2 — внутрен-
ний электродный
раствор сравнения;
3 — стеклянная труб-
ка; 4 — внутренний
электрод сравнения
255
Потенциал асимметрии меняется со временем и поэтому влияет на водород-
ную функцию стеклянного электрода, однако большим и внезапным изменениям
он не подвержен. В принципе, он может рассматриваться как некоторая кон-
станта измерительного прибора. Именно поэтому стеклянный электрод перед
измерением pH исследуемого раствора предварительно калибруют по стандарт-
ным буферным растворам, pH которых известен. В силу особенностей стеклян-
ного электрода, а точнее мембраны, он требует определенного хранения и ухода.
Для получения наиболее точных результатов новые или оставшиеся сухими
электроды перед употреблением следует вымачивать в течение 1...2ч или даже
оставлять в растворе на всю ночь. При этом электроды, предназначенные для
измерения в растворах, где pH меньше 9, могут быть вымочены в воде или
фосфатном буферном растворе (рН6,81). Электроды, которые употребляются
исключительно в щелочных растворах, необходимо вымачивать в буферных рас-
творах с большим значением pH. При работе необходимо следить, чтобы рН-чув-
ствительный конец электрода не подвергался сильным механическим воздейст-
виям. Стеклянные электроды нельзя погружать в хромовокислые растворы или
в растворы других дегидратирующих агентов.
6. ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ЭЛЕКТРОДЫ
И ИХ ПОТЕНЦИАЛЫ
В отличие от описанных электрохимических процессов полу-
чение и отдача электронов на окислительно-восстановительных
электродах осуществляется без изменения заряда ионов в рас-
творе.
Окислительно-восстановительным электродом называется система, состоящая
из инертного металла, опущенного в раствор двух солей, который содержит
одноименные катионы или анионы переменной степени окисления.
Если опустить платиновый электрод в раствор, содержащий
двух- и трехзарядные ионы железа, то ионы Fe3+ могут восста-
новиться до Fe2+ за счет электронов, полученных от платины:
Fe3+ -J- е~ —>Fe2+
Атомы самой платины не участвуют в электродном процессе, т. е.
она служит лишь переносчиком электронов. В результате этого
процесса электрод получает положительный заряд и притягивает
анионы из раствора. На поверхности электрода образуется двой-
ной электрический слой с определенным скачком потенциала.
Этот электродный потенциал зависит от концентрации ионов
Fe2+ и Fe3+. Знак потенциала и его значение определяются отно-
сительно стандартного водородного электрода в элементе:
Pt, H2|2H+||Fe3+, Fe2+|Pt
В написанном элементе протекает реакция
72H2 + Fe3+ = H+ + Fe2+
ЭДС этого элемента, равная потенциалу левого электрода (по-
скольку потенциал стандартного водородного электрода равен 0),
находится по уравнению
£ = £°_-^1|п .
aFe3+0H;
256
По условию рн2=1 (стандартные условия), ань=1. Тогда ЭДС
составленного элемента будет равна
E = EQ--^\n-?^±, (10.16)
F aFe3+
где Е'0=8ок-Вос — стандартный потенциал электрода Fe3+, Fe2+|Pt
при равных активностях ионов Fe2+ и Fe3+. При 298 К £Fe3+,Fe2+iPt =
= +0,771 В (табл. 41).
Таблица 41. Стандартные (нормальные) окислительно-
восстановительные потенциалы
Электрод Электродный процесс e”, В
Cr2+, Cr3+|Pt Cr3+ + e- : = Cr2+ -0,41
Sn2+, Sn4+|Pt Sn4+ + 2e+ = Sn2+ +0,153
Cu+, Cu2+|Pt Cu2+ + e~ = Cu+ +0,167
Fe2+, Fe3+|Pt Fe3+ + e~ = Fe2+ +0,771
Mn2+, Mn3+|Pt Mn3+ + e~ = Mn2+ +0,51
Co2+, Co3+|Pt Co3+ + e~ : = Co2+ + 1,840
Очень часто уравнение (10.16) для электродного потенциала
окислительно-восстановительного электрода записывают в виде
е = е0 + -^1п-2^ ‘ (10.17)
F aFe2+
или в общем виде
_ __ о । ЯГ . [Ox] fin
®ОК-ВОС ^ОК-ВОС Ч ГП-’ (10.18)
nF [Red]
где [Ох] и [Red] — концентрации окисленной и восстановленной
формы соответственно.
При составлении электрохимических элементов с участием
окислительно-восстановительных электродов электродная реак-
ция на них может быть либо восстановительной, либо окисли-
тельной.
Если составить электрохимический элемент из электрода
Pt|Fe3+, Fe2+ и второго электрода, имеющего более положитель-
ный электродный потенциал, то при работе этого элемента на
окислительно-восстановительном электроде будет протекать реак-
ция окисления с отдачей электронов электроду:
Fe2+= Fe3+ + е~~
Если второй электрод будет иметь электродный потенциал
меньше, чем потенциал электрода Pt|Fe3+, Fe2+, то на окисли-
тельно-восстановительном электроде будет протекать реакция
9—696
257
восстановления с присоединением электронов к ионам Fe3+ на
платиновом электроде:
Fe3++e~ —Fe2+
Уравнения для окислительно-восстановительного потенциала были выведены
для простейших реакций, когда кроме окислителя и восстановителя в системе не
присутствуют другие компоненты. В более сложных системах необходимо учи-
тывать равновесные концентрации всех участвующих в реакции соединений,
т. е. под окисленной формой следует понимать все соединения в правой стороне
уравнения реакции
Red = Ох + е
а под восстановительной — все соединения слева от знака равенства.
Например, реакции окисления — восстановления органических соединений
довольно часто идут с участием водородных ионов. При этом возможны три
случая:
1) водородные ионы не участвуют в1 окислительно-восстановительной
реакции; 4
2) водородные ионы непосредственно участвуют в окислительно-восстано-
вительных реакциях;
3) водородные ионы влияют на окислительно-восстановительную реакцию
косвенным образом.
В первом случае ионы водорода не принимают никакого участия в окисли-
тельно-восстановительных реакциях. В основном это реакции окислеция — вос-
становления простых ионов металлов и ряда анионов типа
Fe3+ + e" = Fe2+
Cu2+ + e“=Cu+
Fe(CN)j- + e- = Fe(CN)^-
Во всех подобных реакциях 80К.В0с не зависит от р.Н, и окислительно-восстано-
вительный потенциал рассчитывается по уравнению (10.17).
Во втором случае, например в реакциях окисления — восстановления орга-
нических соединений, 8ок-вос будет зависеть от pH системы. Примером подобных
реакций могут служить реакции типа
H2O2 + 2e~ + 2H+ = 2H2O
NO? + 8e~ + 10H+ = NHt + 3H2O
или в общей форме
(а)
(б)
Ох + ле 4-mH+ = Red
Уравнение для еок.вос такой системы с учетом ионов водорода будет запи-
сываться в виде '
е с° I щ [°х1 »Н+]
Вок вое—е -F nF in [Red}
откуда
_ П, RT , [Ox] , mRT , [u+1
еок-вое-8 4-_ln-[Red] +——InlH ].
(10.19) '
Из уравнения (10.19) следует, что по мере подкисления раствора еок.вос будет
увеличиваться. Величина сдвига 80К.в0С зависит от числа электронов п и ионов
водорода т, участвующих в реакции, т. е. от отношения т/п.
В третьем случае ионы водорода оказывают на 80КВ0С косвенное влияние
из-за изменения растворимости компонентов окислительно-восстановительной
системы при изменении pH. В качестве примера рассмотрим равновесие в си-
стеме Fe3+^+Fe2+. Если коэффициенты активности близки к единице, то еок.вос
вычисляется по уравнению (10.18). При pH, равном, например, 6,0, число ионов
258
железа в растворе можно вычислить из произведений растворимости соответ-
ствующих гидроксидов:
[Fe2+] [ОН~] 2= 4.8-10-16; [Fe3+] [ОН~]3 =4,5-10~4°,
откуда
3+1 4,5- 1О-40 4,5- 10—40 . _ ,._1в ,3
[Feэ+] = [0Н-]3 =——=4,5-10 кмоль/м3,
2+1 4,8-10~16 4,8-10-'6 ,о ,3
[Fe2+] = -- _р-=——=4,8 кмоль/м3.
Концентрация ОН" была найдена из ионного произведения воды и для
pH 6,0 [ОН~] = 10~8 кмоль/м3.
Из полученных данных видно, что при pH 6,0 ионы Fe2+ находятся в рас-
творе, а ионы Fe3+ практически отсутствуют. Следовательно, при данном pH
Сок-вос такой системы целиком определяется концентрацией Fe2+ при условии, что
в растворе отсутствуют органические вещества, способствующие переводу ионов
Fe3+ в раствор. Если в уравнении для концентрации ионов Fe3+ заменить ОН"
на Н+, используя ионное произведение воды, то получим зависимость концент-
рации Fe3+ от pH среды
[Fe-]=AL2p!|iE.==4,5.102[H+]3,
откуда после логарифмирования получим
lg [Fe3+] =2,65 + 31g [Н+]
или
lg [Fe3+] =2,65 —ЗрН.
Следовательно, концентрация ионов Fe3+ в растворе контролируется исклю-
чительно значением pH (табл. 42).
Таблица 42. Активность ионов Fe3+ в зависимости от значения pH
pH 1g аРез+ aFe3+ , кмоль/м3 pH lgaFe3+ ; aFe3+, кмоль/м3
1,0 -0,35 4,47-10-1 6,0 -15,35 4,47-10-16
2,0 -3,35 4,47- IO"4 7,0 — 18,35 4,47-10“19
3,0 -6,35 4,47-IO-7 8,0 -21,35 4,47-10"22
4,0 -9,35 4,47-IO'10 9,0 -24,35 4,47-10~25
5,0 -12,35 4,47-IO-13 10,0 -27,35 4,47-10"28
Таким образом, влияние pH на еок-вос подобных систем обусловлено, как уже
говорилось, изменением растворимости компонентов, в данном случае гидроксида
железа (III), что оказывает существенное влияние на окислительно-восстано-
вительные свойства системы.
Окислительно-восстановительные реакции и потенциалы в почвах. Почва
представляет собой сложную естественную окислительно-восстановительную сис-
тему. Поэтому окислительно-восстановительные реакции играют важную роль
в процессах почвообразования. Кроме того, нормальный рост и развитие растений
возможны при определенном окислительно-восстановительном состоянии почвы.
Окислительно-восстановительные реакции, протекающие в почве, чаще всего
являются необратимыми. Обратимые реакции свойственны только некоторым
почвенным окислительно-восстановительным системам, например окисление и вос-
становление железа (Fe3+ Fe2+), марганца (Мп4+Mn2 t ), азота (N5+ +_>
^*N3+). Важнейшим окислительным агентом в почвенных системах является
молекулярный кислород почвенного воздуха и почвенного раствора. Поэтому
направление и протекание окислительно-восстановительных процессов в почве
9*
259
зависит от ее аэрации и, следовательно, от таких свойств почвы, как структура,
плотность, механический состав, влажность. Любые причины, вызывающие ухуд-
шение аэрации почвы, приводят к уменьшению ее окислительно-восстановитель-
ного потенциала. Как правило, в нормально аэрируемых почвах окислительно-
восстановительный потенциал колеблется в пределах 300...650 мВ. Развитие таких
почвообразовательных процессов, как заболачивание и оглеение, снижают его
до 200 мВ и менее.
Окислительно-восстановительные реакции в почве влияют на подвижность и,
следовательно, доступность растениям таких элементов питания, как железо,
марганец, азот, сера и др. Например, при разложении органических соединений
в условиях высоких значений окислительно-восстановительного потенциала сера
переходит преимущественно в сульфаты, а при низких значениях, т. е. в анаэроб-
ных условиях, — образуются сульфиды.
Изменение окислительно-восстановительного потенциала в зависимости от
внешних условий в разных почвах происходит неодинаково. Иногда изменение
Еок-вос происходит резко, а иногда — медленно. Это объясняется тем, что окис-
лительно-восстановительные системы почв характеризуются различной емкостью
и, следовательно, податливостью к изменениям. Максимальная стабильность
окислительно-восстановительной системы наблюдается тогда, когда окисленная
и восстановленная формы данной системы Находятся примерно в одинаковой
концентрации. При этом абсолютная концентрация всех компонентов данной
системы должна быть больше всех остальных окислительно-восстановительных
систем, находящихся в растворе. Однако в почвенных окислительно-восстано-
вительных системах отношение окисленной формы к восстановленной редко
бывает равным единице, а концентрация окислительно-восстановительных систем
в целом достаточно низкая. Этим и объясняется тот факт, что £ок-вос почвы
подвержен значительным изменениям под влиянием внешних условий, например
влажности почвы.
Для измерения еок-вос почвы или других объектов исследования использу-
ются платиновые электроды самых различных систем. В качестве электрода
сравнения используется хлорсеребряный.
7. КОНЦЕНТРАЦИОННЫЕ ЭЛЕМЕНТЫ
И ДИФФУЗИОННЫЙ ПОТЕНЦИАЛ
Концентрационным элементом называется элемент, в котором
работа электрического тока получается при замыкании внешней
цепи в процессе переноса вещества при самопроизвольном вы-
равнивании концентрации между двумя электролитами — раст-
ворами одного и того же вещества.
Обычный концентрационный элемент состоит из одинаковых
металлических электродов, опущенных в электролиты различной
концентрации (активности), содержащие какую-либо соль метал-
ла, из которого изготовлены электроды. Примером может слу-
жить элемент
Ag | AgNO3(a') || AgNO3(a") | Ag
где а' > а". При работе концентрационного элемента оба элек-
трода в совокупности не испытывают термодинамического изме-
нения. В правом электролите активность Ag+ увеличивается,
а в левом — уменьшается. Таким образом, единственным ре-
зультатом суммарного процесса
Ag+(a') — Ag+(a")
является перенос растворенного вещества AgNO3 из левого раст-
вора в правый, т. е. из более концентрированного в более раз-
260
бавленный. Этот процесс является самопроизвольным и поэтому
сопровождается уменьшением изобарного потенциала. Посредст-
вом диффузии он может протекать необратимо без совершения
работы; в элементе же он протекает обратимо, в результате чего
получается работа электрического тока.
Если не учитывать диффузионный потенциал, возникающий
на границе двух растворов, то подсчет максимальной работы
может быть осуществлен следующим образом:
&G = — nFE.
ЭДС концентрационного элемента рассчитывается по формуле
Е = ei — е2
с использованием уравнения электродного потенциала Нернста
(10.6).
Для правого электрода
Е2 = £2 + Ina",
г
для левого
о , RT 1 ,
В1 = е¥ -|-1па .
г
Отсюда ЭДС концентрационного элемента будет равна
Z7 0,^1, 0 RT 1 „
Е = 82 Я-Ina' — ei-— Ina".
F F
Поскольку электроды состоят из одного и того же металла, то
е? = Е2. В результате получаем
Р %т I , RT 1 „ RT , а'
Е = —р- Ina'-— Ina" = In — .
F F Fa"
В общем случае
E
RT
In
a'
a‘
(10.20)
T
Так как a' > а", ЭДС рассматриваемого концентрационного
элемента, вычисленная по уравнению (10.20), положительна
и элемент работает самопроизвольно. Таким образом, работа
электрического тока в концентрационных элементах — это рабо-
та диффузионного процесса, который проводится обратимо
путем разделения его на несколько различных по направлению
обратимых электродных процессов. Каждый из этих процессов
связан с определенной максимальной работой (убылью G)
и лишь разность этих величин равна работе переноса растворен-
ной соли.
Примером концентрационного элемента иного типа является
элемент с двумя водородными электродами, находящимися под
261
разными давлениями (pi >£2), помещенными в растворы с оди-
наковыми активностями (^1 = ^2):
Pt,H2 1 HCl(ar) 1ГНС1(а2) | Н2, Pt
Pi Ръ
ЭДС такого элемента с учетом уравнения Нернста для водород-
ного электрода будет равна
£ = ~ 4к1прГ‘ (10.21)
Концентрационные водородные элементы рассмотренного типа
можно использовать для определения парциального, давления
водорода в смесях с благородными газами (азот, аргон и т. д.),
т. е. для анализа газовых смесей.
Возникновение диффузионного потенциала связано с диффу-
зией ионов электролита в растворе против градиента концент-
рации. Так как ионы, обладающие большей подвижностью,
диффундируют в более разбавленный раствор с большей ско-
ростью, на границе соприкосновения двух растворов создается
двойной электрический слой с соответствующим скачком потен-
циала. Возникающая разность потенциалов будет ускорять дви-
жение менее подвижного иона и замедлять движение более
подвижного, пока не наступит стационарное состояние, при кото-
ром скорости диффундирующих ионов сравняются и растворен-
ное вещество начнет диффундировать как единое целое. Таким
образом, дальнейшее взаимное перемещение ионов прекра-
щается. Равновесная разность потенциалов, установившаяся
в пограничном слое между двумя растворами, носит название
диффузионного потенциала.
Значение диффузионного потенциала как функцию концентра-
ции можно определить в простейшем случае при соприкосновении
двух растворов одной и той же соли различной концентрации.
В качестве примера рассмотрим концентрационный элемент
Ag | AgNO3(a') || AgNO3(a") | Ag
при условии, что а' > а". При прохождении через элемент F кулонов электри-
чества в правом полуэлементе происходят следующие изменения: 1) растворяет-
ся 1 кмоль серебра, 2) переходит налево tK кмоль Ag+; 3) поступит слева /а кмоль
NO? (где ta и /к — числа переноса аниона и катиона, выраженные через электро-
литические подвижности соответственно, равные ta = . . \а и tK — -у——)•
Т Лк Ла । Л-к
В сумме в правом полуэлементе появляется 1 — tK = ta кмоль Ag+ и ta кмоль
NOr. За это время в левом полуэлементе исчезает /а кмоль обоих ионов.
Таким образом, суммарный процесс состоит в переносе ta кмоль обоих ионов
от раствора с активностью а' к раствору с активностью а". Изменение изобарного
потенциала этого процесса равно
AG = taRЛп + <аД Лп = 2гаЯЛп = - FE,
“к Яа Яка
откуда
Е = 2/а ДД In = 2 (-г-4тг)
г Яка \ Аа Т^к / г ака
262
Если бы диффузионного потенциала не было, т. е. скорости ионов разного знака
были бы равны, то /а = 0,5 и 2/а = 1, что дает для ЭДС концентрационного
элемента
р __ RT 1П gKa
£«--^-10-^.
Представив ЭДС концентрационного элемента как сумму собственно концентра-
ционной ЭДС £к и диффузионного потенциала £д:
£ = £к + £д,
получим
ЕД = Е-ЕК = 2 ( . V-r)
\ А’к “г / г ака г пка
или, подставив числа переноса,
Ед = (2/а — 1) -Щ- 1П Л*-. (10.22)
Г ика
Если Ха > Хк, т. е. подвижность аниона больше подвижности катиона, то
£д>0,.и тогда диффузионный потенциал, вычисленный по уравнению (10.22),
прибавляется к разности электродных потенциалов, увеличивая ЭДС элемента.
Если же Ха <С Хк, то диффузионный потенциал имеет обратный знак относи-
тельно £к и вычитается из нее, уменьшая таким образом ЭДС элемента.
Величина £д невелика: для растворов, отношение концентраций которых
равно 10, значение £д находится в пределах ±30 мВ при ta — 0,20...0,70.
Для КС1, числа переносов ионов которого близки к 0,5, £д при соотношении
концентраций 10:1 составляет всего 1 мВ.
Второй частный случай связан с наличием двух растворов одинаковой кон-
центрации разных солей с общим ионом. При этом диффузионный потенциал
возникает вследствие различия в подвижностях разных ионов в двух растворах.
Диффузионный потенциал на такой границе выражается уравнением
£д=4^~|п V'tv - (Ю.23)
к г Л-a “г
Если привести в соприкосновение два электролита, в которых растворителями
являются разные жидкости, то различия в свойствах этих жидкостей вызывают
появление диффузионного потенциала, величина которого не подчиняется приве-
денным выше уравнениям (10.22) и (10.23).
Как уже отмечалось, электрохимические элементы с диффу-
зионными потенциалами, на ЭДС которых влияют числа перено-
са, называются элементами с переносом. При измерениях невы-
сокой точности можно существенно уменьшить значение Ед,
включив между двумя растворами так называемый солевой
мостик, т. е. концентрированный раствор электролита, например
КС1 или NH4NO3. Резкое уменьшение Ед связано с тем, что ионы
концентрированного раствора проводят практически весь ток
в зонах соприкосновения, а числа переноса указанных солей
близки к 0,5. Кроме того, один диффузионный потенциал на пер-
воначальной границе при введении солевого мостика заменяется
двумя потенциалами меньшего значения. К тому же эти два по-
тенциала часто оказываются противоположными по знаку.
8. ПОТЕНЦИОМЕТРИЯ
Под потенциометрией понимается ряд методов анализа и оп-
ределения физико-химических характеристик электролитов и
химических реакций, основанных на измерении электродных по-
тенциалов и электродвижущих сил гальванических элементов.
Потенциометрические измерения являются наиболее надежными
при изучении констант равновесия электродных реакций, термо-
динамических характеристик реакций, протекающих в растворах,
определении растворимости солей, коэффициентов активности
ионов, pH растворов. Особенно обширное применение нашли
потенциометрические измерения именно при определении pH,
которое является важнейшей характеристикой жидких систем.
Для этого используют электрохимическую цепь, составленную
из электрода сравнения и индикаторного электрода, потенциал
которого зависит от концентрации (активности) ионов Н+ (так
называемые электроды с водородной функцией). К таким элек-
тродам относятся, например, рассмотренные ранее водородный
и стеклянный электроды.
Для определения pH применяют два типа потенциометри-
ческих измерений:
1) прямую потенциометрию, основанную на определении ак-
тивности и концентрации по измеренным значениям электродного
потенциала и электродвижущей силы;
2) потенциометрическое титрование, основанное на исполь-
зовании измерений ЭДС для нахождения точки эквивалентности
в реакциях нейтрализации, осаждения, окисления — восстанов-
ления, комплексообразования.
Потенциомерические методы анализа имеют ряд преимуществ.
Они очень чувствительны, позволяют проводить измерения в мут-
ных и окрашенных растворах, вязких пастах. Поскольку равно-
весное значение потенциала устанавливается быстро, то потен-
циометрические измерения не требуют значительных затрат
времени. Существуют модификации потенциометрического опре-
деления, позволяющие проводить анализ в пробах объемом до
десятых долей миллиметра, что особенно важно в биологических
исследованиях.
Определение pH растворов. К числу наиболее распространен-
ных приложений прямой потенциометрии относятся задачи по
определению pH раствора. Основным индикаторным электродом
при этом является стеклянный электрод с водородной функцией.
Потенциал этого электрода описывается уравнением (10.15),
которое можно записать в виде
с _ t.o 2,303ЯГ „
Ест — Ьет.--р--pH.
В качестве электрода сравнения применяется хлорсеребряный
электрод. Таким образом, используемая для потенциометриче-
ского определения pH стеклянно-хлорсеребряная цепь схемати-
чески может быть записана следующим образом:
264
Ag I AgCl, HC1 |стеклянная мембрана I | KC1, AgCl | Ag
ЭДС этой цепи будет равна
р р ро I 2,3037?/’ гт
£ Ехс бет “I-~р--- pH-
Поскольку 8ст — величина постоянная для данного электрода, то
.о _ F , П „ X
Данным методом можно определять pH растворов в присутствии
окислителей-восстановителей, каталитических ядов, многих со-
лей.
Потенциометрическое титрование. Это титрование кислот и
оснований основано на фиксировании точки эквивалентности по
резкому изменению потенциала индикаторного электрода в про-
цессе реакции нейтрализации. Для кислотно-основного титрова-
ния применяется стеклянно-хлорсеребряная цепь, так же как и
при потенциометрическом определении pH.
Потенциометрическое титрование проводят следующим обра-
зом. В раствор титруемой кислоты помещают стеклянный электрод
(индикаторный) и электрод сравнения (хлорсеребряный). Затем
в раствор анализируемой кислоты из бюретки определенными
дозами добавляется основание, которое уменьшает активности
ионов Н+ в растворе кислоты, вследствие чего изменяется значе-
ние измеряемой ЭДС. Результаты титрования представляют в ви-
де графической зависимости изменения ЭДС (pH) в зависимости
от объема прибавленного основания. Такая зависимость назы-
вается кривой потенциометрического титро-
вания.
Кривая потенциометрического тит-
рования может быть разделена на три
участка, которые отличаются темпом
изменения pH на единицу объема до-
бавленного титранта (рис. 90). На-
чальный участок характеризуется мед-
ленным изменением pH, вблизи точки
эквивалентности незначительное при-
бавление основания вызывает резкое
изменение pH. При дальнейшем до-
бавлении основания после точки экви-
валентности темп изменения pH замед-
ляется. Значение наибольшего измене-
ния pH вблизи точки эквивалентности
называется скачком титрова-
ния.
Точка эквивалентности на кривой
потенциометрического титрования на-
ходится графическим методом, как по-
казано на рис. 90. Зная объем основа-
Рис. 90. Кривая потенциомет-
рического титрования слабой
кислоты. Графическое опреде-
ление точки эквивалентности
(т. э.) и константы диссоциа-
ции
265
ния в точке эквивалентности, находят концентрацию титруемой
кислоты, используя для этого закон эквивалентов.
Метод потенциометрического титрования позволяет наряду
с определением концентрации получить характеристику тит-
руемой кислоты. По числу скачков титрования устанавливают
основность кислоты, постольку каждый скачок соответствует
одной ступени ее диссоциации. По кривой потенциометрического
титрования определяют также силу кислоты. Если скачок тит-
рования равен 7... 10 единиц pH и выражен четко и резко, то
титруемая кислота является сильной. Для слабых кислот харак-
терен довольно плавный скачок титрования, а его значение
равно 3...5 единицам pH. Кроме того, для слабых кислот метод
потенциометрического титрования позволяет оценить константу
диссоциации, которая является мерой силы слабых кислот и ос-
нований. При этом используется уравнение буферных растворов
pH — pKaH-lg [соль] — 1g [кислота].
применение которого обусловлено тем, что в процессе титрования
слабой кислоты (основания) сильным основанием (кислотой)
в титруемом растворе образуется смесь слабой кислоты (осно-
вания) и ее соли с сильным основанием (кислотой), т. е. буфер-
ный раствор. Согласно уравнению буферных растворов рК будет
равно pH в тот момент, когда концентрация образовавшейся в
процессе титрования соли станет равной концентрации еще неот-
титрованной кислоты. В данный момент объем титранта будет
равен половине объема титранта в точке эквивалентности, и на
кривой потенциометрического титрования эта точка находится
графическим методом.
Контрольные вопросы
1. Способность посылать ионы в раствор различна у различных металлов.
Каковы причины этого различия?
2. Как изготовить концентрационный элемент и какие данные можно полу-
чить, измеряя ЭДС этого элемента?
3. Как возникает разность потенциалов на границе между двумя жидкостя-
ми? Как влияет она на измеряемую ЭДС элемента? Каким образом можно ее уста-
новить?
4. Какими свойствами должны обладать электроды, используемые для изме-
рения pH потенциометрическим методом?
5. Как с помощью потенциометрического метода определить константу элек-
тролитической диссоциации слабой кислоты и слабого основания?
6. Как рассчитать константы равновесия химической реакции, происходящей
в элементе, используя значение нормальных потенциалов электродов, составляю-
щих гальванический элемент?
7. Каким образом можно охарактеризовать окислительно-восстановительное
состояние почвенных систем? Какие электроды при этом надо использовать?
Глава XI
ПОВЕРХНОСТНЫЕ ЯВЛЕНИЯ
1. АДСОРБЦИЯ. АДСОРБЦИЯ НА ГРАНИЦЕ
ТВЕРДОЕ ТЕЛО — ГАЗ
Любая гетерогенная система, т. е. система, обладающая по-
верхностью раздела фаз, характеризуется определенным запасом
поверхностной энергии. Эта энергия тем больше, чем больше
поверхность раздела фаз. Так как разные поверхности обладают
различной поверхностной энергией, то для их сравнения исполь-
зуют понятие удельной поверхностной энергии —
энергии, приходящейся на единицу площади поверхности.
В изотермических условиях и при постоянном объеме изме-
нение энергии поверхности равно изменению изохорно-изотерми-
ческого потенциала:
F = aS,
где F — изохорно-изотермический потенциал, отвечающий общей
энергии поверхности; S — площадь поверхности; о — удельная
(приходящаяся на 1 м2) поверхностная энергия.
Согласно второму началу термодинамики процессы, идущие
с уменьшением потенциала F, протекают самопроизвольно. В ге-
терогенных системах изохорно-изотермический потенциал может
уменьшиться либо при сокращении поверхности S, либо при
уменьшении поверхностной энергии о. Поверхность твердых тел
самопроизвольно не уменьшается, она постоянна. Тогда само-
произвольное уменьшение поверхностной энергии может быть
только результатом снижения поверхностной энергии о при
поглощении молекул газа или растворенного вещества поверх-
ностью твердого тела.
Поглощение каким-либо веществом других веществ называет-
ся сорбцией.
Если процесс сорбции идет только на поверхности, то его называют адсорб-
цией, которая представляет собой увеличение концентрации вещества на границе
раздела фаз. Если поглощаемое вещество диффундирует в Глубь поглотителя
и распределяется по объему, то это явление называется абсорбцией.
То вещество, на поверхности которого идет адсорбция, при-
нято называть адсорбентом, а вещество, которое адсорби-
руется, — адсорбатом. Адсорбцию Г обычно выражают
соотношением адсорбата х, приходящимся на единицу площади
поверхности адсорбента S, в кмоль/м2:
г = x/S,
267
Если адсорбентом является твердое пористое тело, общую
поверхность которого определить невозможно, то адсорбцию Г
относят к единице массы адсорбента в кмоль/кг:
Г = х/т.
Адсорбция может идти на поверхности раздела следующих
фаз: газ — твердое тело, раствор — твердое тело, газ — раствор.
Адсорбцию газа углем наблюдал К. Шееле еще в XVIII в.
На явление же адсорбции веществ из раствора впервые обратил
внимание русский акад. Т. Е. Ловиц (1785). Н. Соссюр (1814)
определил, что все пористые тела, т. е. тела с большой удельной
поверхностью, способны адсорбировать газы и что при этом
обычно выделяется теплота. Отсюда следует, что процесс адсорб-
ции является экзотермическим процессом.
Описание взаимодействия молекул адсорбата и молекул ад-
сорбента представляет собой весьма сложную и до сих пор нере-
шенную задачу. Силы взаимодействия адсорбента и адсорбата,
определяющие адсорбцию, различны, и обычно рассматривают
два крайних случая, когда адсорбция характеризуется физиче-
скими либо химическими взаимодействиями: так называемая
физическая или химическая адсорбция.
Физическая адсобция возникает за счет ван-дер-
ваальсовых взаимодействий. Она характеризуется хорошей обра-
тимостью, отсутствием стехиометрических соотношений, уменьше-
нием адсорбции при повышении температуры, близостью эффектов
адсорбции к теплотам снижения или испарения (обычно 10...
80 кДж/моль). Такова, например, адсорбция благородных газов
на угле.
Химическая адсорбция (хемосорбция) осуществляется
только путем химического взаимодействия. Здесь адсорбция
почти необратима, тепловой эффект близок к энергии образо-
вания химических соединений.
Так как хемосорбция является химическим процессом, требу-
ющим энергии активации порядка 40... 120 кДж/моль, повышение
температуры способствует хемосорбции в отличие от физической ад-
сорбции. Примером такой адсорбции является адсорбция кислорода
на вольфраме или кислорода на серебре при повышенных темпе-
ратурах.
При химической адсорбции молекулы адсорбата, связанные
с адсорбентом прочными химическими силами, не могут переме-
щаться по поверхности последнего (локализованная адсорбция):
В отличие от хемосорбции при физической адсорбции могут
иметь место как нелокализованная адсорбция, когда
молекулы адсорбата способны передвигаться по поверхности
адсорбента, так и локализованная адсорбция.
С повышением температуры локализованная физическая ад-
сорбция может переходить в нелокализованную.
Как уже указывалось, физическая адсорбция протекает само-
произвольно, и этот процесс является динамическим: наряду
с адсорбцией идет обратный процесс — десорбция, которая ха-
268
рактеризуется удалением адсорбированных молекул с поверх-
ности адсорбента. Скорость адсорбции с течением времени
уменьшается, а скорость десорбции увеличивается. Эти измене-
ния происходят до тех пор, пока обе скорости не становятся
одинаковыми, т. е. не наступает адсорбционное равновесие:
Адсорбция «=* Десорбция
Для каждой температуры существует свое состояние равно-
весия. Чем выше концентрация адсорбата, тем больше адсорб-
ция, а чем выше температура, тем меньше физическая адсорб-
ция. Влияние температуры на физическую адсорбцию отвечает
принципу Ле Шателье, поскольку десорбция как процесс, обрат-
ный адсорбции, сопровождается поглощением теплоты.
Так как химическая адсорбция обусловлена образованием
связей, близких к химическим, десорбция протекает с большим
трудом. При этом энергия активации возрастает с повышением
степени покрытия поверхности хемосорбированными молекулами,
что можно объяснить лишь существованием активных центров
с различными энергиями активации.
Следует подчеркнуть, что явления физической и химической
адсорбции четко различаются лишь в крайних случаях. Обычно
осуществляются промежуточные варианты, когда основная мас-
са адсорбированного вещества связывается сравнительно слабо
(физическая адсорбция) и лишь небольшая часть связана проч-
но и может быть удалена длительным прогреванием и вакууми-
рованием (химическая адсорбция). Например, кислород на ме-
таллах или водород на никеле адсорбируются при низких темпе-
ратурах по законам физической адсорбции, но при повышении
температуры начинает протекать адсорбция с заметной энергией
активации. В определенном интервале повышения температур
прирост химической адсорбции перекрывает падение физической
адсорбции, и на кривой температурной зависимости адсорбции
возникает промежуточный максимум (рис. 91).
Адсорбционное равновесие является гетерогенным химиче-
ским равновесием. Поэтому каждому состоянию газа или пара
с определенным давлением или температурой соответствует неко-
торое количество адсорбирован-
ного вещества, которое изменяет-
ся в соответствии с изменением
параметров, определяющих со-
стояние газа.
Уравнением адсорбции назы-
вают функциональную зависи-
мость вида
/(Г, С, Г) = 0,
где Г — количество адсорбиро-
ванного вещества (в кмолях),
рассчитанное на 1 м2 поверхности
Рис. 91. Зависимость адсорбции от
температуры при адсорбции Н2 на
никеле
269
адсорбента (или на 1 кг адсорбента) ; С — равновесная концент-
рация газа; Т — температура.
Это уравнение может быть представлено в виде
г = f(C, Т),
определяющем зависимость количества адсорбированного газа
от концентрации и температуры.
Если температура постоянная, то адсорбированное количество
вещества есть функция исключительно равновесной концентра-
ции газа:
Г = f(C); Т = const.
Это уравнение называется изотермой адсорбции.
Часто изотермой адсорбции называют также графическую за-
висимость адсорбции от равновесного давления при постоянной
температуре.
2. ТЕОРИИ АДСОРБЦИИ
В настоящее время нет общей теории, которая достаточно
корректно описывала бы все виды адсорбции на различных
адсорбентах ♦ и разных поверхностях раздела фаз. Поэтому рас-
смотрим наиболее распространенные теории, которые (несмотря
на большое число допущений) позволяют на качественном уров-
не получить представление о таком сложнейшем процессе, как
адсорбция.
Теория молекулярной адсорбции Ленгмюра. В основе этой
теории лежат следующие положения.
1. Адсорбция является локализованной й вызывается силами,
близкими к химическим.
2. Адсорбция молекул адсорбата происходит на активных
центрах, всегда существующих на поверхности адсорбента.
Такими центрами являются пики и возвышения, имеющиеся на
любой поверхности. Так, например, на поверхности кристалла
известкового шпата есть выступы высотой в 10”7... 10“12 м, тонко
отполированные зеркала имеют на поверхности выступы до
3-10“1бм.
также, что активными центрами
являются ребра и углы кри-
Ряд исследователей считают
Рис. 92. Схематическое изображение по-
верхности восстановленного никеля
сталлов и границы зерен в
микронеоднородном адсор-
бенте.
3. Активные центры ха-
рактеризуются большой не-
насыщенностью силового по-
ля, благодаря чему на цент-
рах удерживаются налетаю-
щие молекулы газа. Приме-
ром поверхности с центрами
270
\ различной активности может служить поверхность восстанов-
ленного никеля, для которой предложена следующая схема
строения (рис. 92). Чем больше свободных связей у атомов
адсорбента или, иначе говоря, чем меньше данный атом связан
с другими атомами, образующими поверхность адсорбента, тем
сильнее осуществляется на них адсорбция.
4. Каждый активный центр обладает малым радиусом дей-
ствия и способен насыщаться. Поэтому активный центр может
провзаимодействовать лишь с одной молекулой адсорбата.
В результате этого на поверхности адсорбента может образо-
ваться только один (мономолекулярный) слой адсорбата (моно-
молекулярная адсорбция).
5. Адсорбированные молекулы удерживаются данным актив-
ный центром только в течение определенного промежутка време-
ни. Через некоторое время молекулы отрываются от активного
центра и переходят в газовую фазу. Взамен этих молекул актив-
ные центры могут адсорбировать новые молекулы, которые,
в свою очередь, десорбируются, и т. д. Время пребывания моле-
кулы в адсорбированном состоянии сильно зависит от температу-
ры. При низких температурах это время может быть сколь угодно
большим. При высоких температурах порядка 1300...2300 К вре-
мя пребывания молекулы в адсорбированном состоянии может
равняться всего 10 “6 с.
. 6. Силы взаимодействия между адсорбированными молеку-
лами не учитываются. Поэтому время пребывания молекул газа
на активных центрах не зависит от того, заняты молекулами
газа соседние активные центры или нет.
На основании приведенных положений было выведено урав-
нение изотермы локализованной адсорбции,
пригодное как для описания адсорбции газов, так и растворен-
ных веществ.
Представим себе однородную поверхность адсорбента, нахо-
дящуюся при постоянной температуре в контакте с газом. Равно-
весие устанавливается тогда, когда число молекул адсорбата,
остающихся на поверхности в единицу времени, становится рав-
но числу молекул, покидающих поверхность за то же время.
Число молекул, остающихся на адсорбенте, пропорционально
концентрации С и доле поверхности, которая остается свобод-
" ной. Если вся рассматриваемая поверхность имеет площадь,
равную единице, и занята ее доля, равная 0, то доля (1 —0)
свободная. Следовательно, скорость адсорбции, т. е. скорость
связывания молекул поверхностью,
4т- = Л.С(1 -0).
Скорость десорбции пропорциональна занятой поверхности:
- -ar = fe2e-
При равновесии скорость адсорбции равна скорости десорб-
ции. Поэтому, приравняв обе скорости, получим
271
0 = П> klCb Г ИЛИ 0 = ?17
(11.1)
где
а = ki/k[.
Так как величина 0 равна отношению адсорбированного
количества вещества Г' к максимальной адсорбции Гмакс при
полном заполнении поверхности,
А _ Г с
^макс С -р Д
Отсюда
г =^77^-, (11.2)
где Г — адсорбция; С — равновесная концентрация газа; а —
отношение скоростей десорбции и адсорбции.
Уравнение (11.2) называется уравнением изотер-
мы адсорбции Ленгмюра. Оно показывает, что при малых
концентрациях (при С <С а) адсорбция пропорциональна кон-
центрации:
г = Гма«с- г = кг
а при высоких концентрациях (С^>а) адсорбция стремится
к предельному значению Гмакс (рис. 93). При адсорбции газов С
может быть заменено пропорциональной величиной давления
газа. При адсорбции из раствора, для которой это уравнение,
в принципе, такжё остается справедливым, С означает концент-
рацию растворенного вещества.
Уравнение Фрейндлиха. Представления, развитые И. Ленгмю-
ром, в значительной степени идеализируют и упрощают дейст-
вительную картину адсорбции. На самом деле поверхность боль-
шинства адсорбентов неоднородна, между адсорбированными
частицами имеет место взаимодействие, и адсорбция часто не
ограничивается образованием мономолекулярного слоя. В этом
случае уравнение изотермы адсорбции усложняется. Г. Фрейнд-
лих предположил, что масса адсорбированного газа или раство-
Рис. 93. Изотерма адсорбции
Ленгмюра
ренного вещества, приходящаяся на
единицу массы адсорбента, должна
быть пропорциональна равновесно-
му давлению (для газа) или равно-
весной концентрации (для твердого
вещества, адсорбируемого из рас-
твора), возведенной в какую-то
дробную степень. Другими словами,
чем выше давление и чем больше
концентрация растворенного веще-
ства, тем больше вещества будет
адсорбироваться на поверхности,
однако пропорциональность должна
272
носить не прямой, а экспоненциальный характер. Это положение
выражается эмпирическим уравнением
X = Кр1/п или х = КС1/л, (11.3)
где х — масса адсорбированного вещества, приходящаяся на
1 г адсорбирующего материала, г; р — равновесное давление;
С — равновесная концентрация; /Сип — константы.
Переписывая уравнение в логарифмической форме, получаем
Igx = 4-(lgC) + IgK. (11.4)
Зависимость Igx от IgC выражается прямой линией, тангенс
угла наклона которой равен 1/п, отсекающей на оси ординат
отрезок, равный lg/C
Предполагая экспоненциальное распределение активных цент-
ров по энергиям адсорбции, Я. Б. Зельдович теоретически полу-
чил уравнение, которое ранее эмпирически было установлено
Г. Фрейндлихом.
На рис. 94 приведены изотермы адсорбции уксусной и бен-
зойной кислот на древесном угле при 298 К, подчиняющиеся
уравнению Фрейндлиха.
Трория полимолекулярной адсорбции. Эксперименты показа-
ли, что на практике, особенно при адсорбции паров, встречаются
изотермы, правая часть которых круто поднимается кверху
(рис. 95), что свидетельствует о взаимодействии адсорбирован-
ных слоев молекул с адсорбатом, когда адсорбированные моле-
кулы наслаиваются друг на друга. Для объяснения этого явления
и описания так называемых S-образных изотерм адсорбции
М. Поляни (1915) предложил теорию полимолекулярной адсорб-
ции. В основе этой теории лежали следующие положения.
1. Адсорбция создается чисто физическими силами.
2. На поверхности адсорбента нет активных центров, а ад-
Рис. 94. Графики Фрейндлиха, построенные с использованием уравнений
(11.3) (а) и (11.4) (б):
1 — уксусная кислота в воде; 2 — бензойная кислота в бензоле
/1 огари ср и концентрации
д')
273
Рис. 95. Изотерма полимолекулярной
адсорбции
в адсорбционном объеме других
сорбционные силы действуют
вблизи от поверхности адсор-
бента, образуя непрерывное
силовое поле.
3. Силовое поле, обусловли-
вающее адсорбцию,' действует
на расстояния, которые боль-
ше, чем размеры отдельных
молекул адсорбата. Иначе го-
воря, у поверхности адсорбента
существует так называемый
адсорбционный объем, который
заполняется при адсорбции
молекулами адсорбата.
4. Притяжение молекулы
адсорбата поверхностью ад-
сорбента не зависит от наличия
молекул, вследствие чего воз-
можна полимолекулярная адсорбция.
5. Адсорбционные силы не зависят от температуры и, следо-
вательно, с изменением температуры адсорбционный объем не
изменяется.
Обе рассмотренные теории адсорбции — теория мономолеку-
лярной адсорбции Ленгмюра и теория полимолекулярной адсорб-
ции не могут описать с достаточной полнотой сложного процесса
адсорбций.
Обобщенная теория Брунауэра, Эммета и Теллера. Делались
попытки обобщить представления И. Ленгмюра и М. Поляни
и описать изотермы различной формы с помощью одного урав-
нения. Такая обобщенная теория была развита С. Брунауэром,
П. Эмметом и Е. Теллером (1935...1940) применительно к адсорб-
ции паров. Она получила название БЭТ по первым буквам фа-
милий авторов. Основные положения этой теории следующие.
1. На поверхности адсорбента имеется определенное число
равноценных в энергетическом отношении активных центров.
2. Взаимодействия соседних адсорбированных молекул в пер-
вом и последнем слоях отсутствуют.
3. Каждая молекула предыдущего слоя представляет собой
возможный активный центр для адсорбции следующего адсор-
бционного слоя.
4. Предполагается, что все молекулы во втором и более дале-
ких слоях ведут себя подобно молекулам жидкости.
Таким образом, адсорбированная фаза может быть представ-
лена как совокупность адсорбционных комплексов — цепей моле-
кул, первая из которых связана с поверхностью адсорбента. Все
эти цепи энергетически не взаимодействуют друг с другом. Схема
строения адсорбционного слоя по теории БЭТ показана на
рис. 96.
С. Брунауэр, П. Эммет и Е. Теллер вывели уравнение
274
Рис. 96. Схема строения адсорбционного
слоя по теории БЭТ
Рис. 97. Изотерма сорбции
при капиллярной конден-
сации
Р _ 1 . С-1 р
V(po - Р) VMC VMC ро ’
(И.5)
где ро — давление насыщенного пара; V — объем адсорбирован-
ного газа при данном давлении газа р; VM — объем адсорбирован-
ного газа в монослое; С = e^/RT (Ае — разность между теплотой
адсорбции газа в первом слое и теплотой сжижения газа).
Находя зависимость левой части уравнения (11.5) от р/ро,
получают на графике прямую линию, наклон которой равен
С — 1/(VMC), а отрезок на оси ординат 1/(VMC). Отсюда можно
определить величины 14 и С.
Из значения VM (соответствующего также точке В на рис. 95)
может быть определена полная адсорбирующая поверхность
адсорбента 3, что имеет важное практическое значение, а из зна-
чения С вычисляется теплота адсорбции газа в монослое (обыч-
но около 8... 10 кДж/моль). Например, адсорбирующая поверх-
ность 1 кг силикагеля составляет 5-1O5 м2/кг, а активированного
угля — до 1-Ю6 м2/кг.
Капиллярная конденсация. При сорбции паров на твердых
пористых адсорбентах адсорбционный процесс может перейти в
так называемую капиллярную конденсацию. Сначала пар адсор-
бируется на стенках пор (капилляров) сорбента, а затем конден-
сируется в жидкость. Далее слои этой жидкости соединяются,
заполняя частично самые тонкие капилляры жидкостью, обра-
зующей вогнутый мениск. Давление насыщенного пара над во-
гнутым мениском всегда меньше давления пара над плоской
поверхностью жидкости (см. гл. XI, раздел 4) и поэтому пар
начинает конденсироваться над вогнутым мениском и капилляры
полностью заполняются жидкостью. С увеличением равновесного
давления капиллярная конденсация охватывает более крупные
капилляры по сравнению с первичными капиллярами.
Изотерма сорблии—тфи наличии капиллярной конденсации
имеет S-образную форму (рис. 97). При малых давлениях кривая
представляет собой обычную изотерму адсорбции, при приближе-
нии к пределу адсорбции она резко поднимается вверх. Этот
участок соответствует массе сорбированного вещества в резуль-
тате капиллярной конденсации.
275
3. АДСОРБЦИЯ НА ГРАНИЦЕ ТВЕРДОЕ ТЕЛО — РАСТВОР
3.1. Молекулярная адсорбция из растворов
Первые исследования по адсорбции из раствора принадле-
жат русскому ученому Т. Е. Ловицу (1785). Он впервые предло-
жил применять уголь для очистки спирта от сивушных масел и
для удаления из воды неприятно пахнущих веществ.
Изотермы адсорбции растворенных веществ из раствора в
общем по своему виду аналогичны адсорбционным изотермам
для газов, и, как было показано, для разбавленных растворов
эти изотермы достаточно хорошо подчиняются уравнению Фрейн-
длиха или уравнению Ленгмюра, если в них подставить равно-
весную концентрацию С растворенного вещества в растворе.
Однако адсорбция из растворов по сравнению с газовой адсорб-
цией оказывается значительно более сложным явлением прежде
всего потому, что наряду с адсорбцией растворенного вещества
(адсорбата) на поверхности адсорбента может происходить и
адсорбция самого растворителя. В результате этого между
адсорбатом и растворителем происходит конкуренция за «обла-
дание адсорбентом» и чем хуже адсорбируется растворитель,
тем лучше адсорбируется растворенное вещество из раствора
(адсорбат). Поэтому различного рода искажения обычной изо-
термы адсорбции происходят довольно часто.
/Адсорбция вещества из раствора идет медленнее адсорбции
газа, так как уменьшение концентрации в граничном слое может
восполняться только путем диффузии растворенного вещества
в поры адсорбента, что осуществляется довольно медленно.
Для ускорения установления адсорбционного равновесия часто
применяют перемешивание системы.
Да адсорбцию влияет также способность растворителя раство-
рять адсорбент. Чем лучше растворитель растворяет адсор-
бент, тем хуже идет адсорбция из раствор^} С повышением тем-
пературы адсорбция обычно уменьшается, .однако не так сильно,
как адсорбция газов, но иногда, например, когда температурный
коэффициент растворимости вещества отрицательный, адсорбция
увеличивается.
Массу а вещества, молекулярно-адсорбированного 1 кг адсор-
бента из раствора, обычно вычисляют по уравнению
а = У(Ср - Ci)
т
где Со и Ci — начальная и равновесная концентрация адсорбата
соответственно, кмоль/м3; V — объем раствора, из которого про-
исходит адсорбция, м3; т — масса адсорбента, кг. Если есть воз-
можность определить площадь адсорбции, величину адсорбции
относят к единице поверхности (m2)J
Зависимость адсорбции от строения молекул адсорбата очень
сложна. Многие молекулы органических веществ (кислот, спир-
276
тов, аминов) состоят из двух частей: полярной группы и непо-
лярного углеводородного радикала. Такие молекулы называются
дифильными. к полярным группам относятся группы
—СООН, —ОН, —NH2, —SH, —CN, —NO2, —СНО, —SO2OH.
Эти группы при взаимодействии с водой хорошо гидратируются,
т. е. являются гидрофильными. В отличие от них углеводород-
ные радикалы гидрофобны. Они не взаимодействуют с водой, но
сольватируются молекулами неполярных растворителей. Схема-
тически дифильные молекулы изображают в виде символа, в ко-
тором прямой чертой обозначен углеводородный (неполярный)
радикал, а кружком — полярная группа (рис. 98).
При адсорбции на твердом адсорбенте дифильные молекулы
растворенного вещества ориентируются на его поверхности так,
чтобы полярная часть молекулы была обращена к полярной
Рис. 98. Ориентация дифильных молекул на поверхности адсорбента
фазе, а неполярная — к неполярной. Так, при адсорбции органи-
ческой кислоты из водного раствора на неполярном адсорбен-
те — угле молекулы ее будут ориентироваться «хвостами» к ад-
сорбенту (рис. 98, а). При адсорбции этой же кислоты из раство-
ра в бензоле (неполярный растворитель) на полярном адсор-
бенте— силикагеле ориентация будет другой (рис. 98,6).
^Следовательно, гидрофобные адсорбенты (уголь, тальк) луч-
ше адсорбируют органические (дифильные) вещества из водных
растворов, а гидрофильные адсорбенты (силикагель, глины) луч-
ше адсорбируют их из неполярных и слабополярных жидкостей?
3.2. Адсорбция электролитов
Адсорбция из водных растворов, как правило, происходит та-
ким образом, что на твердом адсорбенте из раствора адсорби-
руются преимущественно ионы одного вида.
Преимущественная адсорбция из раствора или катиона, или
аниона зависит от природы адсорбента или от природы ионов —
их заряда, радиуса и степени гидратации. Чем больше заряд
277
иона, тем лучше он адсорбируется. Из ионов с одинаковым за-
рядом лучше адсорбируется ион, имеющий наибольший радиус,
так как он имеет наименьшую степень гидратации. Гидратная
оболочка препятствует адсорбции ионов, и чем она меньше, т. е.
чем меньше степень гидратации, тем ион адсорбируется лучше.
Рассмотрим адсорбцию ионов раствора KI на поверхности
кристалла Agl (рис. 99, а). На поверхности кристаллов иодида
серебра в определенном порядке расположены ионы Ag+ и I-.
Иодид-ионы, которые могут образовывать с ионами серебра, на-
ходящимися в кристаллической решетке, малорастворимое соеди-
нение, адсорбируются на поверхности, создавая на ней избыток
отрицательных зарядов. Ионы калия не адсорбируются, так как
они не образуют с ионами иода нерастворимое соединение, но
под действием электростатического притяжения они располага-
Рис. 99. Схемы адсорбции иодид-ионов (а) и ионов серебра (б)
на поверхности кристалла иодида серебра:
А — двойной электрический слой
ются вблизи поверхности. Иодид-ионы, сорбированные поверх-
ностью, и ионы калия, находящиеся в жидкой фазе, образуют
двойной электрический слой.
Если кристаллы Agl находятся в контакте с раствором
AgNCh, то на поверхности кристаллов будут адсорбироваться
ионы Ag+, ионы NO3" остаются в жидкой фазе (рис. 99,6).
Таким образом, на поверхности кристаллического твердого
тела из раствора адсорбируется тот из ионов, который входит
в состав кристаллической решетки или может образовывать
с одним из ионов решетки малорастворимое соединение (правило
Фаянса — Пескова).
3.3. Обменная адсорбция
Обменная адсорбция представляет собой процесс обмена
ионов между раствором и твердой фазой — адсорбентом, точнее,
обмен ионов между двойным электрическим слоем адсорбента
и средой. При этом твердая фаза поглощает из раствора ионы
4 одного знака (катионы или анионы) и вместо них выделяет в
раствор эквивалентное число других ионов того же знака.
278
Обменная адсорбция иуеет ряд особенностей.
1. Обменная адсорбция специфична, т. е. для данного адсор-
бента к обмену способны только определенные ионы. Различают
кислотные и основные адсорбенты. Кислотные адсорбенты, подоб-
но кислотам, способны обменивать с растворами катионы; основ-
ные адсорбенты ведут себя как основания и обменивают анионы.
Существуют также амфотерные адсорбенты, которые в одних
условиях обменивают катионы, а в других — анионы.
2. Обменная адсорбция не всегда обратима.
3. Обменная адсорбция протекает более медленно, чем моле-
кулярная адсорбция. Это особенно заметно тогда, когда осущест-
вляется обмен ионов, находящихся в глубине адсорбента. Время
обмена лимитируется скоростью диффузии ионов из раствора
в глубь адсорбента. Вытесненные из адсорбента ионы затем пе-
реходят в раствор.
4. При обменной адсорбции может изменяться pH среды.
Это наблюдается тогда, когда обмениваемый адсорбентом ион
является водородным или гидроксильным ионом. Если адсорбент
заменяют на какой-нибудь водородный ион, то последний, посту-
пая в раствор, уменьшает pH среды. Если адсорбент меняет на
какой-нибудь анион гидроксильный ион, то pH раствора, наобо-
рот, увеличивается. Схематически обмен ионов можно предста-
вить следующими реакциями:
Адсорбент ~Н+ 4- Na+ 4- Cl- -> Адсорбент ~Na+ -|- Н+ 4- С1~
Адсорбент +ОН- + Na+ -|- С1“ Адсорбент +С1- 4- Na+ 4- ОН-
Б. П. Никольский (1939) предложил уравнение, количествен-
но характеризующее обмен ионов на твердой поверхности:
гт1/2' Л1/2’
= (Н.6)
62 а2
где gb g2 — содержание обменивающихся ионов в адсорбенте,
моль/г; И1, аг — активности обменивающихся ионов в растворе;
Zi, Z2 — заряд ионов; К — константа.
При zi = 22 = 1 уравнение (11.6) упрощается:
gl/g2 = K\al/a2). (11.7)
При небольших концентрациях электролита вместо актив-
ности а можно использовать концентрации. Тогда уравне-
ние (11.7) принимает вид
£1/£2=Г(С1/С2). (1L8)
Обменная адсорбция имеет большое значение для земледелия,
так как от природы поглощенных почвой катионов зависит ее
плодородие. Исследуя обмен ионов в почве, К. К. Гейдройц
(1933) установил, что процесс происходит в строго эквивалент-
ных соотношениях, т. е. поглощение почвой какого-либо иона
сопровождается эквивалентным выделением из нее в раствор
другого иона. Например, почва способна поглощать и удерживать
279
катионы К+ и NH^, содержащиеся в удобрениях и необходимые
для питания растений. Взамен этих катионов почва выделяет
эквивалентные количества других катионов, например Са2+
и Mg2+. Анионы, как, например, Cl~, NO3", SO4 , почти не погло-
щаются почвой. Согласно К. К. Гейдройцу, поглощать катионы
способен так называемый поглощающий комплекс — высокодис-
персная смесь нерастворимых алюмосиликатов и органомине-
ральных соединений. От природы поглощенных ионов в значи-
тельной мере зависят физические и агротехнические свойства
почвы.
К обмену ионов способны не только почвы, но и ряд природ-
ных и синтетических силикатов. В последние десятилетия был
осуществлен промышленный синтез ионообменных смол — иони-
тов. В зависимости от того, какой вид ионов участвует в обмене,
иониты делят на катиониты и аниониты. Катиониты спо-
собны обменивать катионы, в том числе и ион водорода, анио-
ниты обменивают анионы, в том числе гидроксил-ион.
Иониты широко применяют при получении в производствен-
ных условиях деминерализованной воды, т. е. воды, не содержа-
щей растворенных солей. Для полного обессоливания воды ее
последовательно пропускают через катионитовый и анионитовый
фильтры. Катионит должен содержать способный к обмену Н+
(Н-форма катионита), анионит — ОН- (ОН-форма анионита).
Рассмотрим в качестве примера удаление NaCl из воды. Катионит, взаимо-
действуя с хлоридом натрия, обменивает ион натрия на ион водорода, который
поступает в раствор:
RH (т) + NaCI (р) -> RNa (т) + НС1 (р)
Далее воду обрабатывают анионитом в ОН-форме, при этом поглощаются
хлорид-ионы:
R'OH (т) + НС1 (р) R'Cl (т) + Н2О
где RH и R'OH представляют собой иониты, способные к обмену катионов
или анионов соответственно.
После использования иониты могут быть восстановлены — регенерированы —
обработкой раствором серной или хлороводородной кислот (катионит) или
растворами гидроксида натрия или гидрокарбоната натрия (анионит).
Большое значение имеют иониты при охране окружающей среды. Так, на-
пример, в сточных водах многих производств содержатся ионы тяжелых метал-
лов, которые очень вредны для живых организмов. Так как концентрация тя-
желых металлов в сточных водах очень мала, применение обычных методов
удаления (например, осаждение) неэффективно и дорого. Сточные воды обра-
батывают катионитом, причем можно использовать катионит, содержащий не
ион водорода, а, например, ион натрия. Ионы тяжелых металлов, обмениваясь
на ион натрия, поглощаются катионитом, из которого их можно легко извлечь
и использовать в народном хозяйстве.
4. ПОВЕРХНОСТНОЕ НАТЯЖЕНИЕ. СМАЧИВАНИЕ
Гетерогенная система, представляющая собой чистую жид-
кость, которая соприкасается с другой средой, например с ее соб-
ственным паром, характеризуется тем, что жидкость на поверх-
280
ности находится в особых услови-
ях по сравнению с остальной мас-
сой жидкости (рис. 100).
Каждая молекула во внутрен-
них слоях жидкости подвергается
одинаковому воздействию других
молекул, окружающих ее со всех
сторон. Поэтому силы молекуляр-
ного воздействия здесь уравнове-
шиваются. Если же молекула
жидкости находится в поверхно-
стном слое на границе с газом,
то взаимодействие этой молекулы
Рис. 100. Схема действия межмоле-
кулярных сил внутри жидкости и на
ее поверхности:
/ — молекулы; 2 — сфера действия сил
с молекулами газа намного
меньше, чем с молекулами жидкости. В результате этого моле-
кулы на границе с газом стремятся втянуться внутрь жидкости,
а это значит, что поверхность раздела жидкость — газ стремится
к уменьшений). Этот процесс обусловлен известным термодина-
мическим стремлением свободной энергии к уменьшению. Наи-
более ярким проявлением этого процесса может служить шаро-
образная форма свободной капли жидкости; известно, что шар из
всех геометрических фигур данного объема обладает минималь-
ной поверхностью.
Итак, если известно, что молекулы из поверхности раздела
самопроизвольно стремятся внутрь, то ясно, что обратный про-
цесс — перемещение молекулы из объема на поверхность — по-
требует затраты энергии. Увеличение поверхности жидкости тре-
бует затраты энергии F, равной работе at, необходимой для пере-
мещения одной молекулы из объема на поверхность, умноженной
на число перемещенных молекул и:
F = по 1.
Если работа проводится в равновесных изотермических усло-
виях, то величина F, входящая в это уравнение, называется
свободной поверхностной энергией или поверх-
ностной энергией, которая, отнесенная к единице поверхности,
называется поверхностным натяжением:
a = F/S,
где в — поверхностное натяжение, Дж/м2; F— поверхностная
энергия; S — площадь поверхности.
Поверхностное натяжение для различных жидкостей неоди-
наково и особенно велико у .воды. В табл. 43 приведены значе-
ния поверхностного натяжения для некоторых веществ.
Поверхностное натяжение жидкости о — это сила, приложен-
ная по касательной к плоскости поверхности (приходящаяся
на единицу длины), которая препятствует увеличению поверх-
ности. Для иллюстрации этого определения на рис. 101 изоб-
ражай идеализированный эксперимент, в котором подвижная
проволока под действием силы F растягивает жидкую пленку
281
Рис. 101. Схема, иллюст-
рирующая существование
у жидкости поверхностно-
го натяжения
подобно тому, как это происходит с мыль-
ной пленкой на проволочной рамке. По-
верхностное натяжение можно вычислить
по уравнению
о = F/2Z,
где I — длина проволоки; множитель 2
учитывает наличие у жидкой пленки двух
поверхностей — передней и задней.
Поверхностное натяжение чаще вы-
числяют, зная массу капли жидкости в
момент отрыва ее от капилляра, по урав-
нению
а == mg/(2jir),
где г — радиус капилляра; о — поверхностное натяжение; g~
ускорение свободного падения.
Массу одной капли находят по уравнению
т ~ Vq/п,
где п — число капель в объеме V жидкости, вытекающей из
капилляра; р — плотность жидкости.
Отсюда
О = VQg/(2nrn).
Если две жидкости, занимающие одинаковые объемы, пооче-
редно пропускать через один и тот же капилляр, то
€>1 _ Qltt2
02 $2П}
Таблица 43. Поверхностное натяжение некоторых жидкостей
при различной температуре и газовой атмосфере
(П — пар данного вещества)
Вещество Газовая атмосфера г, К о, Дж/м2
Бром Воздух . 286 44,1
Сера » 411 58,3
Цинк Вакуум 700 772,2
Ртуть » 293 471,6
Вода Воздух 4- П 373 58,80
Ацетон Воздух 293 23,7
Метанол » 288 22,99
Масляная кислота П 288 27,32
Октан Воздух 293 31,11
Бензол » 293 28,88
Толуол » 293 27,4
Нафталин 353 32,06
Фенол П 328 36,5
Анилин » 292,5 43,3
282
откуда находим
Для определения поверхностного натяжения через установи
ленный вертикально капилляр пропускают определенный объем
исследуемой жидкости и считают капли, отрывающиеся от ка-
пилляра. Затем пропускают через капилляр такой же объем
жидкости с известным поверхностным натяжением, обычно воду.
Зная число капель п\ и п,2 в объеме V цля двух жидкостей и их
плотность^ вычисляют поверхностное натяжение исследуемой
жидкости.
Поверхностное натяжение на границе двух несмешивающихся
жидкостей называется межфазным поверхностным натяжением.
Чем меньше поверхностное натяжение на границе двух жид-
костей (оа,Д тем выше взаимная растворимость жидкостей и при
критической температуре полного смешения обоих жидкостей
(например, для системы вода — фенол при 339 К) оа,б = 0.
По правилу Антонова межфазное поверхностное натяжение
двух жидкостей, находящихся во взаимном равновесии, связано
с поверхностным натяжением каждой из них соотношением
Gab == (Ja (Jft,
где Gab — поверхностное натяжение на поверхности раздела двух
жидкостей; оа и вь — соответственно поверхностные натяжения
жидкостей а и b на поверхности раздела жидкость/пар.
Растекание одной жидкости по поверхности другой. Гаркинс и
его сотрудники подробно рассмотрели термодинамику образова-
ния новой поверхности, возникающей при соприкосновении двух
жидкостей. Если жидкость В растекается изотермически и обра-
тимо на поверхности жидкости А, то поверхность а исчезает и
вместо нее появляется поверхность 6; кроме этого, образуется
поверхность раздела ab. Уменьшение свободной энергии AG,
сопровождающее растекание, дается выражением
* Дб = Ga — Gab — Gb',
так как
Дд = Ga — Gab “Н Gb, Д{( = 2(7fe,
TO
Дб = Да — Дк,
где Аа — работа прилипания (адгезии); Ак— работа сцепления
(когезии) жидкости.
Рассмотрим подробнее, что из себя представляют адгезия и
когезия. Если имеется система, состоящая из молекул однород-
ной жидкости (например, вода), то для создания новых поверх-
ностей раздела (например, вода — воздух) потребуется затрата
работы. Эта работа, затраченная на преодоление сил сцепления
283
©между молекулами однородной жидкости,
называется когезией.
Предположим, что имеется система, со-
[yj стоящая из двух неоднородных жидкостей
с некоторой поверхностью раздела между
ними (например, масло — вода). Если те-
перь создать новые поверхности раздела
(например, масло — воздух и вода — воз-
дух), то работа, связанная с «разрезанием»
по имеющейся ранее поверхности раздела
(масло — вода), будет затрачена на пре-
одоление сил сцепления между разными
молекулами. Такая работа называется
адгезией.
Сказанное можно проиллюстрировать
схемой, приведенной на рис. 102. Предполо-
жим, что в цилиндрическом объеме 1а нахо-
дится вода а. «Разрежем» этот цилиндр
плоскостью, перпендикулярной поверхно-
сти цилиндра по направлению стрелки 1. Об-
стрирующая - понятия разуются две поверхности раздела вода —
«адгезия» и «когезия» В03дуХ (они заштрихованы на рис. 102, Па).
Работа когезии Лк будет равна
А'к = 2оа,
оа равна поверхностному натяжению на границе воздух—вода.
Рассмотрим такой же цилиндр /б, содержащий теперь не во-
ду, а масло Ь. Проведем ту же операцию «разрезания» плоско-
стью в направлении стрелки 2. При этом образуются две поверх-
ности раздела масло — воздух. Работа когезии Л£ в этом случае
отлична от и равна
А" = 2оь,
где <5ь равно поверхностному натяжению на границе масло —
вода.
Рассмотрим теперь цилиндр, содержащий две жидкости: а и Ь.
Здесь уже имеется поверхность раздела вода — масло. Осущест-
вим разрез (по стрелке 3) так, чтобы «разрезающая» плоскость
проходила через уже имеющуюся поверхность раздела между
неоднородными жидкостями. Тогда работа адгезии равна
Лд = ва 4- Оь — &ab-
Согласно правилу, установленному Гаркинсом, растекание
происходит, если прилипание между двумя жидкостями больше,
чем сцепление растекающейся жидкости. Следовательно, при
Ла — Лк > 0 происходит растекание, если же ЛА — Лк <С О,
растекание не имеет места.
Следует заметить, что если жидкость а растекается на по-
верхности жидкости Ь, то это не значит, что жидкость b будет
обязательно растекаться по поверхности жидкости а. Например,
284
почти все органические жидкости растекаются по поверхности
воды, но вода растекается по поверхности очень немногих орга-
нических веществ. В табл. 44 приведены характеристики расте-
кания некоторых органических жидкостей (при 293 К). Данные
взяты из работы Гаркинса, содержащей сведения о 71 органиче-
ской жидкости.
Наличие полярных групп не обязательно, чтобы происходило
растекание. Однако полярные группы повышают величину Ла
по отношению к воде значительно больше, чем Лк, так что раз-
ность Лд — Лк увеличивается. Отсутствие растекания, как пра-
вило, вызывается высоким значением Лк для данной жидкости.
Поверхностное натяжение на границе трех фаз. На границе
раздела трех фаз наблюдаются более сложные соотношения
между межфазными поверхностными натяжениями. Если на
твердую поверхность 3 (рис. 103) нанесена капля воды 1 и обе
поверхности граничат с газом 2, то капля образует с твердой
поверхностью краевой угол смачивания 6 (измеряемый в водной
фазе). По уравнению Лапласа величина cosO при равновесии
связана с межфазными поверхностными натяжениями следую-
щим соотношением:
аз,2 — аз,1
cos 0 = —:--— ,
а 1,2
где индексы о указывают границы раздела фаз.
В зависимости от значений равновесного краевого угла раз-
личают три основных вида смачивания:
1) несмачивание (плохое смачивание) — краевой угол тупой:
180° > 0 > 90°. Пример: вода на парафине или тефлоне;
2) смачивание (ограниченное смачивание) — краевой угол
острый: 90° > 0 > 0°. Например, вода на металле, покрытом
оксидной пленкой;
3) полное смачивание. Равновесный краевой угол не уста-
навливается, капля растекается в тонкую пленку, например ртуть
на поверхности свинца, очищенной от оксидной пленки.
Таблица 44. Значение (Аа — Ак) для растекающихся
и нерастекающихся по воде жидкостей (по Гаркинсу)
Вещество Вещество
Растекающиеся жидкости
Этиловый спирт 50.4 Фенетол 10,66
Муравьиная кислота 35,5 Трихлорэтилен 5,09
Анилин 24,45 Гексан 2,31
Хлороформ 13,04 Октан 0,22
Нерастекающж еся жидкости
Дибромэтилен -3,19 Бромоформ -9,58
Сероуглерод -6,94 Ди подметан -26,46
285
Рис. 103. Смачивание на границе
раздела трех фаз (по Ребиндеру):
1 — вода; 2 — среда; 3 — твердая по-
верх ность
Значение равновесного крае-
вого угла определяется соотноше-
нием сил притяжения жидкости к
твердому телу (или к жидкой под-
ложке) и сил взаимного притяже-
ния между частицами (молеку-
лами) самой жидкости. Эту зави-
симость можно продемонстриро-
вать с помощью очень простых
и наглядных опытов.
Рассмотрим смачивание при контакте воды с тщательно обез-
жиренным стеклом. Если стеклянную пластину погрузить в воду,
а затем снова вытащить на воздух, на стекле остается тонкий
слой воды' Это означает, что сйлы притяжения жидкости к твер-
дому телу преобладают над взаимным' притяжением. молекул,
жидкости. Напротив, при отсутствии смачивания (например, при
контакте ртути со стеклом) после аналогичного опыта на твердой
поверхности не остается следов жидкости^
Связь между молекулярным взаимодействием жидкости и
твердого тела и видом смачивания выявляется особенно отчет-
ливо при избирательном смачивании, когда с твердым телом
одновременно контактируют две жидкости, различные по своей
молекулярной природе (полярная и неполярная). По виду изби-
рательного смачивания все твердые тела подразделяют на три
основные группы:
1) гидрофильные (или олеофобные) материалы, которые луч-
ше смачиваются водой: кальцит, кварц, большинство силикатов
и окисленных минералов, галогениды щелочных металлов;
2) гидрофобные (олеофильные) материалы, которые лучше
смачиваются неполярной жидкостью (мылом)? графит, уголь,
сера;
3) абсолютно гидрофобные тела; в эту группу входят пара-
фин, тефлон, битумы.
Помимо краевого угла смачивания, другой мерой гидрофиль-
ности поверхности является теплота смачивания, так как гидро-
фильные поверхности смачиваются водой с положительным теп-
ловым эффектом. По П. А. Ребиндеру, гидрофильность поверх-
ности следует характеризовать по отношению теплот ее смачива-
ния водой q\ и бензолом Для гидрофильной поверхности
> 1 (для агара 35),
?2
для гидрофобной поверхности
< 1 (для угля 0,34).
q2
Капиллярное давление. В различных процессах, связанных
со смачиванием, важную роль играет капиллярное давление, ко-
торое возникает из-за искривления поверхности жидкости.
286
Для выпуклой поверхности капил-
лярное давление положительно,
для вогнутой оно отрицательно.
Если поверхность раздела жид-
кости выпуклая (рис. 104), то мо-
лекулы из поверхности жидкости
(например, молекулы А) втягива-
ются внутрь жидкости меньшим
числом молекул, т. е. слабее, чем
из плоской поверхности (SAS'),
а из вогнутой поверхности
(САС')— большим числом моле-
Рис. 104. Выпуклая (ВАВ'), пло-
ская (S4S') и вогнутая (САС')
кул жидкости, т. е. сильнее, чем поверхности жидкости
из плоской поверхности. Поэтому / ? • v
между искривленной и плоской поверхностью раздела существует
разность молекулярных давлений, которая называется капилляр-
ным давлением р. Искривление поверхности характеризуют ра-
диусом кривизны г, направленным внутрь жидкости при выпук-
лой поверхности (в этом случае г считается положительным) и
наружу при вогнутой поверхности (г отрицателен). Для плоской
поверхности г = оо. По уравнению Лапласа
р — 2(5/г,
откуда видно, что для плоской поверхности р = 0, для выпуклой
поверхности р> 0и для вогнутой поверхности р<0; эти давле-
ния р суммируются с внешним давлением, оказываемым на
жидкость.
При опускании тонкого стеклянного капилляра в воду
(рис. 105, а) краевой угол смачивания близок к нулю и поэтому
мениск вогнутый. Давление р при этом ниже, чем давление при
Рис. 105. Капиллярное поднятие (а) и
опускание (б) жидкости
плоской поверхности. В ре-
зультате мениск поднимается
на высоту /г, при которой вес
поднятого столба жидкости
уравновешивает разность
давлений между обеими по-
верхностями. При погруже-
нии капилляра в несмачива-
емую жидкость (рис. 105, б),
напротив, происходит опуска-
ние уровня жидкости в ка-
пилляре.
Из рис. 104 также следу-
ет, что да выпуклой поверх-
ности молекулы легче могут
перейти в пар, чем на плоской
или на вогнутой поверхно-
стях. Давление пара жидкос-
ти на выпуклой поверхности
287
больше, а на вогнутой поверхности — меньше, чем нормальное
давление пара на плоской поверхности жидкости. Эта законо-
мерность выражается уравнением Томсона
где Др/р— относительное изменение давления насыщенного
пара по сравнению с нормальным; М — молекулярная масса;
р — плотность жидкости; г — радиус капилляра или капли жид-
кости.
Из уравнения (11.9) следует, например, что для капель воды
с г ~ 10-8 упругость пара на 10% выше, чем для воды с плоской
поверхностью; поэтому если в замкнутой системе содержатся
одновременно крупные и мелкие капли, последние перегоняются
к крупным каплям. Уравнение (11.9) объясняет также более вы-
сокую растворимость мелких твердых частиц по сравнению с
крупными и ряд других явлений.
5. АДСОРБЦИЯ НА ГРАНИЦЕ РАЗДЕЛА РАСТВОР—ГАЗ
Свободная поверхностная энергия F равна произведению по-
верхности S на поверхностное натяжение о:
Поэтому свободная поверхностная энергия может быть уменьше-
на не только за счет уменьшения поверхности S, но и за счет
понижения поверхностного натяжения ст; соответственно F может
быть увеличена за счет увеличения о, что достигается в раство-
рах изменением их состава.
Если в растворе содержится вещество, перенос которого в
поверхностный слой требует меньше энергии, чем перенос в по-
верхностный слой молекул растворителя, то будет происходить
накопление этого вещества в поверхностном слое. Иначе говоря,
будет происходить ^адсорбция и растворенного вещества в по-
верхностном слое станет больше, чем в слое такой же толщины
в объеме раствора.
Если в растворе содержится вещество, перенос которого в
поверхностный слой требует больше энергии, чем перенос моле-
кул растворителя, то будет происходить уменьшение этого ве-
щества в поверхностном слое и растворенного вещества в по-
верхностном слое станет меньше, чем в слое такой же толщины
в объеме раствора.
Количественное соотношение между адсорбцией Г раство-
ренного вещества (в кмоль на 1 м2 поверхности) и изменением
поверхностного натяжения с концентрацией раствора опреде-
ляется уравнением Гиббса (1878):
Г =------— _ (11.10)
RT de ’ v '
288
Из уравнения (11.10) следует, что
направление процесса — концентриро- g I -—~~ 2
вание вещества в поверхностном слое у
или, наоборот, переход его в объем жид- § \
кой фазы — определяется знаком про- \
изводной do/dC. Значение do/dC<0 | \
соответствует накоплению вещества в § \
поверхностном слое (Г>0), a do/dC^C §
<0 — уменьшению вещества в поверх- <&
ностном слое (Г<0). Величину do/dC _______
называют поверхностной а к- Концентрация
т и в н о с т ь ю. ' >
На рис. 106 представлены Три ВОЗ- Рис. 106. Зависимость по-
МОЖНЫХ случая изменения поверхност- верхностного натяжения от
ного натяжения с ростом концентрации. воров н к
Все три графика начинаются в одной
точке, которая соответствует поверхностному натяжению чистого
растворителя (в данном случае воды). Вещества типа 1 снижа-
ют поверхностное натяжение растворителя. Вещества типа' 2 не
влияют на поверхностное натяжение, а вещества типа 3 его слегка
повышают.
Вещества, добавление которых к растворителю снижает по-
верхностное натяжение, принято называть поверхностно-
активными веществами; соединения типа 2 и 3 называют
поверхностно-неактивными. Адсорбция поверхност-
но-активных веществ положительна.
К поверхностно-инактивным веществам относятся неоргани-
ческие электролиты — кислоты, щелочи и соли. Поверхностно-ак-
тивные вещества представлены некоторыми органическими соеди-
нениями, содержащими полярные группы, — спиртами, жирными
кислотами, аминами, мылами, детергентами.
При очень малых концентрациях, когда значение С прибли-
жается к АС, уравнение Гиббса (11.10) при замене do/dC
на Ао/АС переходит в уравнение
Ла = Go — а — р == RTF.
В этом уравнении р аналогично давлению в поверхностном слое,
а Г = 1 /S, где S — площадь, занимаемая адсорбированным
веществом в поверхностном слое; тогда
pS = RT.
(H.ll)
Уравнение (11.11) является двухмерным аналогом уравнения
состояния идеальных газов. Поэтому можно предположить, что
при очень малых концентрациях молекулы адсорбированного
вещества находятся в поверхностном слое в псевдогазовом со-
стоянии.
А. Фрумкиным (1925) были введены поправки, учитывающие
собственную площадь адсорбированных молекул So и силы взаим-
10—696 289
ного притяжения между ними а. Полученное уравнение Фрум-
кина
(11.12)
является двухмерным аналогом уравнения Ван-дер-Ваальса.
С его помощью удается определить зависимость поверхностного
натяжения растворов от концентрации растворов в широком ин-
тервале концентраций. По аналогии с уравнением Ван-дер-Ва-
альса уравнение (11.12) называется уравнением состо-
яния адсорбционного слоя.
Исследуя поверхностное натяжение водных растворов органи-
ческих веществ, Траубе (1891) установил правило:
в любом гомологическом ряду при малых концентрациях удлинение угле-
родной цепи на СН2-группу увеличивает поверхностную активность в 3—3,5 раза.
Это означает, что для каждого последующего гомолога кон-
центрация, вызывающая одинаковое понижение поверхностного
натяжения, уменьшается примерно втрое. И. Ленгмюр определил,
что правило Траубе справедливо лишь при свободном располо-
жении адсорбированных молекул в поверхностном слое парал-
лельно поверхности.
Исследование водных растворов жирных кислот показало, что
для них зависимость поверхностного натяжения от концентрации
выражается эмпирическим уравнением Шишковского (1909):
Да = 6 In + 1
(11.13)
где Ь и К — эмпирические постоянные. При этом значение b оди-
наково для всего гомологического ряда, а К увеличивается в
3...3,5 раза для каждого последующего члена ряда в соответст-
вии с правилом Траубе.
Ориентация молекул в поверхностном слое. Ранее было по-
казано, что когда концентрация раствора мала, адсорбция тоже
невелика, и состояние системы описывается уравнением (11.11).
Применимость этого уравнения (которое является аналогом
уравнения, описывающего состояние идеального газа) свидетель-
ствует о псевдогазовом состоянии адсорбированных веществ.
Увеличение концентрации раствора приводит к увеличению
адсорбции. В этом случае (как и при переходе от идеальных
газов к реальным) необходимо учитывать собственную площадь
и силы взаимодействия адсорбированных молекул (11.12).
При дальнейшем увеличении адсорбции поверхностного веще-
ства Г достигает своего максимального значения ГмаКс, при ко-
тором площадь S = 1 /Гмакс, занимаемая молекулой в адсорб-
ционном слое, также приобретает свое предельное значение.
При сопоставлении уравнений (11.10) и (11.13) было показа-
но, что при высоких концентрациях 6=/?7ТмаКс. Это значит, что
независимо от длины цепи в гомологическом ряду каждая мо-
290
лекула занимает одну и ту
же площадь в поверхност-
ном слое. Так, для различ-
ных жирных кислот эта
величина равна 2,1 нм2.
Если эта молекула «ле-
жит» на поверхности, то
из-за различия в длине
углеводородного радикала
Рис. 107. Предельная ориентация молекулы
в адсорбционном слое
они должны занимать различную
площадь.
Молекулы многих поверхностно-активных веществ обладают
дифильным строением. Поэтому расположение таких молекул в
поверхностном слое энергетически наиболее выгодно при условии
погружения полярных групп в воду, а углеводородных цепей —
в воздух или в неполярную фазу. Было подсчитано, что вероят-
ность погружения гидроксигруппы в воду уже для молекулы ме-
тилового спирта в сотни тысяч раз больше вероятности обратной
ориентации.
При малой концентрации адсорбированных молекул в по-
верхностном слое тепловое движение нарушает их ориентацию и
молекулы в основном расположены в поверхностном слое гори-
зонтально — псевдогазовые пленки. При повышении концентра-
ции усиливается взаимодействие углеводородных цепей между
собой, что благоприятствует вертикальной ориентации молекул,
и при насыщении адсорбционного слоя Гмакс создается возмож-
ность образования «молекулярного чистокола» из вертикально
расположенных молекул (рис. 107). В этом состоянии площадь
So =1/Гмакс, занимаемая молекулой, определяется лишь пло-
щадью полярной группы, постоянной для всех членов гомоло-
гического ряда, что и объясняет постоянство константы b в урав-
нении Шишковского. Длина углеводородных цепей и энергия их
взаимодействия изменяются при каждом удлинении цепи на
группу СН2. Это объясняет изменение второй константы К в
уравнении Шишковского.
Контрольные вопросы
1. Смешиваются равное число молекул воды и спирта. Будет ли поверхност-
ное натяжение полученного раствора равно полусумме поверхностных натяжений
компонентов раствора?
2. Какой фактор является определяющим в переносе частиц ПАВ в растворе
к границе раздела фаз?
3. Процесс физической адсорбции обратим. Для каких видов адсорбции про-
цесс может быть необратимым?
4. Почему при создании новых поверхностей раздела в системе требуется
произвести работу?
5. Почему адсорбция частиц адсорбата происходит на активных центрах?
6. Почему отношение смачивания водой и бензолом определяет гидрофиль-
ность и гидрофобность поверхности?
7. Каков по знаку тепловой эффект адсорбции и как влияет (в случае нали-
чия равновесия) на адсорбцию увеличение и уменьшение температуры?
10*
Глава XII
КОЛЛОИДНЫЕ СИСТЕМЫ
И МЕТОДЫ ПОЛУЧЕНИЯ
ЛИОФОБНЫХ КОЛЛОИДОВ
1. ОБЩАЯ ХАРАКТЕРИСТИКА КОЛЛОИДНЫХ СИСТЕМ
Возникновение коллоидной химии как самостоятельной науки
связывают обычно с именем английского ученого Т. Грэма, кото-
рый в 1861 г. разработал ряд методов приготовления и очистки
коллоидных растворов. Т. Грэм предложил все вещества класси-
фицировать на две группы:
1) кристаллоиды — сравнительно быстро диффунди-
рующие вещества, образующие истинные растворы, из которых
вещество при определенных условиях может быть выделено в
виде кристаллов;
2) коллоиды — вещества, обладающие малой скоростью
диффузии, низким осмотическим давлением и образующие кол-
лоидные растворы, студни или клееподобные аморфные осадки.
Т. Грэм ошибочно считал, что коллоиды и кристаллоиды
представляют собой совершенно обособленные классы веществ,
между которыми нет никакого сходства. На основе теоретиче-
ских соображений и тщательного экспериментального исследова-
ния русские ученые Н. Г. Борщев (1869) и П. П. Веймарн
(1904...1916) сделали правильный вывод о том, что любое веще-
ство можно перевести в коллоидное состояние, создавая соответ-
ствующие условия. /
И <
Если коллоидные частицы находятся в жидкой среде, то такие системы
обычно называют коллоидными растворами или золями, у
В течение XIX и в начале XX вв. был выполнен ряд фунда-
ментальных исследований, сыгравших большую роль в развитии
коллоидной химии. На основе этих исследований было установ-
лено, что в коллоидных растворах частицы находятся в высокой
степени раздробления или диспергирования, но они гораздо
больше молекул.
Таким образом, коллоидные системы относятся к диспер-
сным системам, т. е. к системам, где одно вещество в виде
частиц различной величины распределено в другом веществе.
Степень раздробленности частиц называется степенью
дисперсност иД Среда, в которой находится раздробленное
(диспергированное) вещество, называется дисперсионной
средой, а раздробленное вещество в виде частиц разных раз-
меров — дисперсной фазой.
292
Дисперсные системы чрезвычайно многообразны и можно ска-
зать, что они представляют основу всего биологического мира.
Распространенность их в технике также очень велика, так как
различие химического состава отдельных компонентов дисперс-
ных систем, величины частиц и разнообразие дисперсионных
сред обеспечивают получение и использование колоссального
числа продуктов.
Дисперсионные системы прежде всего классифицируют по
размеру частиц дисперсионной фазы или, иначе говоря, по степе-
ни дисперсности (табл. 45). Кроме того, их разделяют на группы,
отличающиеся по природе и агрегатному состоянию дисперсной
фазы и дисперсионной среды.
Таблица 45. Дисперсные системы
Системы
Диаметр частицы, м
Грубодисперсные системы
Коллоидные системы
Истинные растворы
нт7...кг5
ю-9...1(г7
<1(Г9
Грубодисперсные системы. Если дисперсная фаза состоит из
твердых частиц, то система называется взвесью или сус-
пензией. В качестве примера можно привести взмученную
глину в воде. Если дисперсная фаза представлена капельками
жидкости, то ее называют эмульсией; пример эмульсии —
капли масла в воде. Эти системы неустойчивы.
Коллоидные системы. Частицы коллоидных систем значитель-
но больше молекул (ионов), из которых состоит дисперсионная
среда, что приводит к наличию поверхности раздела между час-
тицами и средой. Если суспензии можно наблюдать с помощью
микроскопа, то коллоидные частицы таким способом не обнару-
живаются.
Коллоидные системы относительно устойчивы, но со временем
они разрушаются. При получении коллоидных систем затрачи-
вается внешняя энергия.
Истинные растворы. Их иначе называют молекулярно-дис-
персными или ионно-дисперсными системами. Эти растворы
устойчивы, не разрушаются и получаются самопроизвольно.
Исследования коллоидных систем, особенно изучение зависи-
мости их устойчивости от наличия и концентрации электролитов
в растворе, показали, что одна лишь дисперсность не может оха-
рактеризовать коллоидные системы и объяснить их свойства.
Н. П. Песков (1917) установил, что свойства коллоидных
систем зависят не только от размеров частиц, но и в гораздо
большей мере от наличия поверхностей раздела со значительной
свободной поверхностной энергией. Был сделан вывод, что кроме
кинетической устойчивости, зависящей от размера частиц, име-
293
ется устойчивость частиц к взаимному слипанию (агрегативная
устойчивость).
Коллоидным системам свойственна агрегативная неустойчи-
вость, преодолеваемая лишь путем адсорбции ионов или моле-
кул на частицах дисперсной фазы. Таким образом, агрегативно-
устойчивая коллоидная система, в принципе, должна состоять
из трех компонентов: диспергированных частиц, среды и стабили-
затора.
Глубокое изучение процессов и характера взаимодействия дис-
персной фазы и дисперсионной среды привело к тому, что колло-
идные системы не могут рассматриваться как единое целое, а
имеются две группы, резко отличные по взаимодействиям между
частицами дисперсной фазы и дисперсионной среды: лиофоб-
ные коллоиды и ра створы высокомолекуляр-
ных соединений (растворы ВМС), которые ранее называ-
лись лиофильными коллоидами.
К лиофобным коллоидам относятся системы, в которой части-
цы дисперсной фазы не взаимодействуют или слабо взаимодейст-
вуют с дисперсионной средой. Эти растворы получают с затратой
энергии, и они устойчивы лишь в присутствии стабилизаторов.
К растворам ВМС относятся системы, образованные самопро-
извольно, благодаря сильному взаимодействию дисперсной фазы
и дисперсионной среды.
Работами В. А. Каргина и его школы было показано, что
растворы ВМС являются термодинамически обратимыми моле-
кулярными гомогенными (однофазными) системами, способными
сохранять агрегативную устойчивость без стабилизатора в двух-
компонентном растворе.
Лиофобные коллоиды и растворы ВМС отличаются также и
по структуре частиц, составляющих дисперсную фазу. Для лио-
фобных частиц характерной единицей структуры является м и-
целла, представляющая собой сложный многокомпонентный
агрегат с переменным числом адсорбированных ионов или моле-
кул. Растворы ВМС представляют собой истинные растворы.
2. МЕТОДЫ ПОЛУЧЕНИЯ ЛИОФОБНЫХ КОЛЛОИДОВ
Коллоидные растворы занимают по степени дисперсности
промежуточное положение между истинными растворами или,
иначе говоря, молекулярно- и ионно-дисперсными системами и
грубодисперсными системами. Поэтому они могут быть получены
либо путем ассоциации (конденсации) молекул и ионов истинных
растворов, либо раздроблением частиц грубодисперсных систем.
Методы получения коллоидных растворов представлены двумя
группами: методы конденсации и дисперсионные методы. В от-
дельную группу может быть выделен метод получения коллоид-
ных растворов с помощью пептизации.
Коллоидные растворы, полученные любыми методами, обычно
294
содержат ряд примесей (исходные вещества или побочные про-
дукты) . Все эти вещества изменяют свойства коллоидных систем
и поэтому должны быть удалены (см. гл. XIII, разд. 5).
2.1. Методы конденсации
Рис. 108. Схема прибора
С. 3. Рогинского и
А. И. Шальникова:
1,3 — отростки для бензола и
металлического натрия; 2 — от-
росток для сбора коллоидного
раствора; 4 — пробирки с жид-
ким воздухом
Методы физической конденсации. Один из методов конден-
сационного получения золей был предложен С. 3. Рогинским
и А. И. Шальниковым. Этот метод основан на конденсации паров
в вакууме на поверхности сосуда, охлажденной жидким возду-
хом (рис. 108). Для этого в отростках / и 3 прибора подверга-
ются испарению одновременно диспергируемое вещество (напри-
мер, натрий) и дисперсионная среда (например, бензол) при
температуре 673 К. Пары этих веществ конденсируются на по-
верхности сосуда 4, охлаждаемого жидким воздухом до 193 К;
при этом охлажденный твердый бензол, намерзающий на стен-
ках, содержит затвердевший натрий. После удаления из сосуда 4
жидкого воздуха температура постепенно повышается, оттаяв-
шая смесь бензола с натрием попадает в отросток 2, образуя
коллоидный раствор натрия в бензоле. Этот метод используют
при получении золей щелочных металлов в органических жид-
костях (бензоле, толуоле, гексане и др
При замене одного растворителя
другим можно перевести растворенное
вещество, находящееся в молекулярно-
дисперсном раздроблении, в коллоидное
раздробление. Для этого необходимо
иметь два смешивающихся между собой
растворителя, один из которых хорошо
растворяет вещество, а другой не рас-
творяет. В качестве примера можно
привести образование гидрозоля масти-
ки. Мастика хорошо растворима в спир-
те и почти нерастворима в воде. При
добавлении спиртового раствора масти-
ки в воде происходит резкое понижение
растворимости мастики, в результате
чего молекулы соединяются в коллоид-
ные частицы и образуется коллоидный
раствор. Также получают коллоидные
растворы серы или канифоли, которые
растворяются в этиловом спирте, обра-
зуя истинный раствор. В воде сера и
канифоль практически нерастворимы,
поэтому при добавлении воды к их спир-
товому раствору молекулы конденсиру-
ются в более крупные агрегаты и
образуется коллоидный раствор.
295
Методы химической конденсации. К этой группе относятся ме-
тоды, основанные на проведении в растворе химических реак-
ций, сопровождающихся образованием нерастворимых или труд-
норастворимых веществ. Используются различные типы реак-
ций: восстановления, окисления, разложения, гидролиза и др.
Так, красный золь золота получают восстановлением соли
золота формальдегидом до металлического золота с образова-
нием золя золота по схеме
2NaAuO2 + ЗНСОН + Na2CO3 = 2Au + 3HCOONa + NaHCO3 + H2O
На образующихся частичках (кристалликах золота) адсорбиру-
ются ионы АиОг. Строение частиц можно представить схемой
(см. гл. XII, разд. 7.1)
{[Au]m-nAuOr •(« ~x)Na+}x-xNa+
Аналогичным образом получают из очень разбавленных раство-
ров нитрата серебра.желто-коричневый золь серебра.
Для получения золей других элементов применяют окислитель-
ные реакции. Например, золи серы и селена получают действием
кислорода на растворы сероводорода и селеноводорода:
2H2S + 0'2 -> 2Н2О + 2S
2H2Se + 02 -> 2Н2О + 2Se
Золь серы получается также при действии на сероводород окси-
да серы (IV):
2H2S + S02 3S + 2Н2О
Здесь строение золя серы можно представить схемой
{[S]m'nHS~-(n — х)Н+}х~хН+
Примером применения реакции разложения может служить
получение золя серы разложением тиосульфатов и полисуль-
фидов:
H2S2O3 Н20 + so2 + S
(NH4)2S2 + H2SO4 -> (NH4)2SO4 + h2s + s
При действии на каменную соль NaCl радиоактивного излу-
чения выделяется металлический натрий, который, распределяясь
в кристаллической среде, обусловливает голубое окрашивание
кристаллов.
р?оли образуются также в результате химической реакции
двойного обмена между двумя растворимыми веществами, в ре-
зультате которых образуется нерастворимое вещество, которое
при соответствующих условиях превращается в коллоидный ра-
створ. Этим методом, например, получают золь хлорида серебра:
AgNO3 + NaCl -> AgCl + NaNO3
При избытке нитрата серебра коллоидные частицы схематиче-
ски можно представить как
296
([AgCl]m-nAg-(n - x)NO3-}x+xNO3-
Золь сульфида мышьяка получают пропусканием в разбав-
ленный водный раствор оксида мышьяка (III) сероводорода:
As2O3 4" 3H2S —> ЗН2О 4~ As2S3
Коллоидная частица имеет строение
([As2S3]m-/:HS_ •(« - х)Н+}х-хН+
Процесс гидролиза различных растворимых соединений
часто сопровождается образованием нерастворимых веществ,
которые превращаются в коллоидные растворы. Так, например,
при гидролизе хлорида железа (III) образуется нерастворимый
гидроксид железа (III), большое число молекул которого при
соответствующих условиях образует коллоидные частицы:
FeCl3 4- ЗН2О -> Fe(OH)3 4- ЗНС1
nFe (ОН) з 4- nHCl nFeOCl 4- «Н2О
nFeOCl FeO+ 4- яС1“
nFe(OH)3 4- nFeO+ 4- яС1“ ->
([Fe(OH)3]m-nFeO+ *(п — х)С1-)х+хС1_
Равновесие гидролиза зависит от концентрации и темпера-
туры. С увеличением разбавления и повышением температуры
степень гидролиза возрастает, что используют для получения
золя гидроксида железа (III). Раствор хлорида железа (III)
приливают к кипящей воде, при этом получают красно-корич-
невый золь гидроксида железа (III). С помощью гидролиза
могут быть также получены золи кремниевой, вольфрамовой,
титановой и других кислот, нерастворимых в воде.
2.2. Дисперсионные методы
В основе этих методов лежит раздробление твердых тел до
частиц коллоидного размера и образование таким образом
коллоидных растворов. Процесс раздробления можно осущест-
вить следующими методами: механическим дроблением, электри-
ческим раздроблением, действием ультразвука.
Получение коллоидных растворов механическим раздробле-
нием твердых тел. Для получения коллоидных растворов этим
методом производится растирание и дробление твердых тел в
специальных машинах — коллоидных мельницах.
Первая коллоидная мельница (рис. 109) была сконструиро-
вана русским инженером К. Плауссоном (1920). Она представ-
ляет собой геометрически закрытый, быстро вращающийся ме-
ханизм ударного действия. Насаженные на валу лопасти Ь и не-
подвижные выступы а расположены очень близко друг от друга,
но в разных положениях. Частицы вещества, подлежащего дис-
пергированию, в предварительно измельченном виде смешивают-
ся с соответствующей жидкостью, содержащей стабилизатор,
и в виде взвеси подаются через загрузочное отверстие с. При
297
помощи быстро вращающихся
лопастей взвесь приводится в бы-
строе вращение, в результате чего
частицы вещества приобретают
скорость и, ударяясь о неподвиж-
ные выступы, разбиваются на мел-
кие частицы. Готовый тонкодис-
персный продукт удаляется через
отверстие d.
Получение коллоидных раство-
ров методом электрического рас-
пыления. Этот метод состоит в том,
что через какую-либо дисперсион-
ную среду, например воду, пропус-
кают электрический ток между
электродами, изготовленными из
ницы109 Схема коллоиднои мель“ материала, коллоидный раствор
которого хотят получить. Так, для
приготовления коллоидного раствора платины создают под водой
электрическую дугу между платиновыми электродами. При этом
один электрод распыляется. Вначале при пропускании электри-
ческого тока осуществляется молекулярное раздробление, но
затем молекулы конденсируются в коллоидные частицы, образуя
коллоидный раствор.
Метод электрического распыления применяют для получе-
ния коллоидных растворов золота, серебра, платины и других
металлов.
Получение коллоидных растворов при помощи ультразвука.
Колебания воздуха с большой частотой (1О5...1(г Гц) назы-
ваются ультразвуковыми волнами, которые образуются благодаря
применению так называемых пьезоэлектрических осцилляторов. >
При этом взвесь грубодисперсного вещества, подлежащего
раздроблению, под действием ультразвуковых волн размель-
чается до коллоидного состояния. С помощью ультразвуковых
волн можно получить коллоидные растворы смол, гипса, графита,
металлов, красителей, крахмала и многих других веществ.
2.3. Метод пептизации
Многие^ осадки, практически нерастворимые в воде, перехо-
дят в коллоидный раствор при действии на них некоторых ве-
ществ. Этот широко известный способ получения золей был
предложен впервые биохимиками.
В отличие от других методов образования коллоидных
растворов при пептизации не происходит изменения степени
дисперсности частиц. Пептизаторы, вернее ионы пептизатора,
хорошо адсорбируются на поверхности коллоидных частиц осад-
ка и обусловливают их переход в золь. .
Пептизация по механизму действия осуществляется двумя
298
путями. В соответствии с этим рассматривают непосред-
ственную и посредственную пептизации. Если на
поверхности частиц перед их разделением адсорбируется не-
посредственно добавленный пептизатор, то такую пептизацию
называют непосредственной. При посредственной пептизации
на поверхности частиц адсорбируется продукт взаимодействия
пептизатора с веществом дисперсной фазы (точнее, ионы вновь
полученного пептизатора).
Примером непосредственной пептизации может служить дис-
пергирование студенистого осадка гидроксида железа (III) при
действии на него раствора хлорида железа (III). При этом
ионы железа, адсорбируясь на поверхности частиц, сообщают
им положительный заряд. Одновременно заряженные частицы
благодаря взаимному отталкиванию переходят из осадка в ра-
створ. Процесс пептизации схематически можно представить
следующим образом:
/nFe(OH)3 + nFeCl3 -> ([Fe(OH)3]„t-rtFe3+-3(л - х)С1~}3х+ЗхС\~
Примером посредственной пептизации может служить получе-
ние того же золя гидроксида железа (III) при действии раз-
бавленной соляной кислоты на осадок гидроксида железа (III).
При этом часть молекул гидроксида железа (III) взаимодей-
ствует с соляной кислотой с образованием хлороксида железа
FeOCl. Вновь образованное соединение FeOCl (точнее ионы
FeO+), адсорбируясь на поверхности частиц осадка гидроксида
железа (III), переводят его в коллоидное состояние:
Fe(OH)3 + НС1 -> FeOHCl + 2Н2О
nFeOCl nFeO+ 4~ nCl”
Fe(OH)3 + nFeO+ 4- nCl“ ->
->( [Fe (OH) з]^ • nFeO+;(n - х)СГ)х+ • xC 1 "
Контрольные вопросы
1. Возможен ли переход любого вещества в коллоидное состояние?
2. Какими двумя основными факторами определяются свойства коллоидных
систем?
3. Чем отличаются друг от друга по структуре лиофобные коллоиды и раство-
ры ВМС?
4. Чем обусловлена возможность получения лиофобных коллоидов двумя
основными способами: методом конденсации и дисперсионным методом?
5. Чем отличается метод получения лиофобных коллоидов с помощью пепти-
зации от двух основных методов?
Глава XIII
МОЛЕКУЛЯРНО-КИНЕТИЧЕСКИЕ, ОПТИЧЕСКИЕ
И ЭЛЕКТРИЧЕСКИЕ СВОЙСТВА ЛИОФОБНЫХ
КОЛЛОИДОВ
1. БРОУНОВСКОЕ ДВИЖЕНИЕ
Коллоидные системы по молекулярно-кинетическим свойствам
принципиально не отличаются от истинных растворов. Взвешен-
ные в растворе частицы находятся в постоянном беспорядоч-
ном тепловом движении. При столкновении частиц происходит
обмен количеством движения и в результате этого устанавли-
вается средняя кинетическая энергия, одинаковая для всех
частиц. Однако наблюдается большая разница в скоростях
молекул и коллоидных частиц: если для молекул газов средние
скорости движения измеряются сотнями метров в секунду, то
для частиц размером в 3...5 мкм они составляют доли миллимет-
ра в секунду, что обусловлено гигантскими (по сравнению с мо-
лекулами) размерами коллоидных частиц.
В 1827 г. английский ботаник Р. Броун заметил, что микро-
скопические частицы пыльцы и спор растений непрерывно и
хаотически передвигаются в воде. Это движение, названное
броуновским (рис. 110), вначале связывалось с жизненны-
ми процессами, но затем было установлено, что оно свойствен-
но любым мельчайшим частицам как органического, так и неор-
ганического происхождения и проявляется тем интенсивнее, чем
выше температура и вязкость среды.
Дальнейшие исследования показали, что характер броунов-
ского движения изменяется, а интенсивность его возрастает с
повышением степени дисперсности. Так, ультрамикроскопические
исследования показали, что в отличие от колебательных дви-
жений более крупных (по сравнению с истинно коллоидными)
частиц суспензий и эмульсий наблюдалось весьма быстрое
зигзагообразное поступательное передвижение коллоидных час-
тиц не только в плоскости поля зрения, но и в его объеме.
Это явление долгое время объясняли внешними причинами:
например, вибрацией аппаратуры или конвективными потоками
в жидкости.
Во второй половине XIX в. тщательными исследованиями
ряда ученых было установлено, что, несмотря на любые меры
по соблюдению точного механического и термического равнове-
сия, броуновское движение продолжало иметь место. Позже
было обнаружено и вращательное броуновское движение, пред-
ставляющее собой вращение частиц вокруг собственной оси.
300
Весь этот экспериментальный ма-
териал привел в 80-х годах к за-
ключению, что источником бро-
уновского движения являются не
внешние причины, а внутренние,
присущие системе. Был сделан
вывод, что броуновское движение
есть результат вечно существу-
ющего молекулярного движения.
Оно отражает взаимодействие мо-
лекул дисперсионной среды со
сравнительно крупными части-
цами дисперсной фазы.
Ж. Перрен сравнивал броунов-
ское движение с качанием лодки
Рис. ПО. Схема броуновского дви-
жения частицы
на поверхности волнующегося моря. Наблюдатель издали не
замечает волн, но может их обнаружить по качаниям лодки; так
же и здесь — движения молекул жидкости не видны, но результаты
их сказываются на более крупных взвешенных в жидкости час-
тицах, которые видны в микроскопе. Если частица очень велика,
то в каждый момент она испытывает много соударений со всех
сторон, причем в среднем число их с каждой стороны одинаково.
Равнодействующая соударений близка к нулю и во всяком слу-
чае недостаточно велика, чтобы передвинуть взвешенную частицу
на достаточно большое расстояние, при котором такое передви-
жение стало бы заметным. Поэтому частицы с диаметром больше
4 мкм уже не испытывают броуновского движения. Небольшие
частицы испытывают одновременно лишь малое число соударе-
ний, и их распределение не будет уже равномерным. В резуль-
тате легкая частица получает поступательное движение тем более
быстрое, чем она меньше.
Элементарные исследования броуновского движения проводи-
лись Р. Зигмонди, Ж. Перреном, Т. Сведбергом, а теория
этого движения была развита А. Эйнштейном и М. Смолухов-
ским (1905). В результате этих работ удалось установить основ-
ные закономерности броуновского движения и показать, что оно
в действительности является строгим выражением полной беспо-
рядочности («идеальный беспорядок»), вытекающей из закона
больших чисел. Поэтому статистическая теория броуновского
движения, разработанная А. Эйнштейном, в качестве основного
постулата исходит из предположения о завершенной хаотичности
движения частиц.
Истинный путь частицы при, броуновском движении, как и
при молекулярном, проследить в ультрамикроскоп невозможно,
так как коллоидная частица за 1 с успевает претерпеть десятки
и сотни миллионов ударов молекул среды, и путь частицы изме-
няется по направлению до 1О20 раз в 1 с. Глаз человека способен
улавливать не более 10 изменений направлений в 1 с и притом
лишь в крупном масштабе. Поэтому визуальное наблюдение за
301
броуновским движением в ультрамикроскопе передает заметные
сдвиги лишь как суммарный результат миллионов отдельных,
не заметных для глаза сдвигов, происшедших за время рас-
смотрения системы.
В теорию броуновского движения вместо средней квадратич-
ной скорости для газовых молекул было введено понятие сред-
ний сдвиг (смещение) ±Ах, представляющий собой проек-
цию расстояния между двумя положениями частиц А и В за вре-
мя t двух последовательных наблюдений (см. рис. НО). Хаоти-
ческое движение частицы охватывает определенный объем про-
странства, увеличивающийся во времени. В горизонтальной
плоскости он соответствует возрастающей площади, пропорцио-
нальной квадрату среднего сдвига Ах2. Как показал А. Эйн-
штейн, среднее значение квадрата смещения частицы Ах2, вы-
численное из большого числа измерений смещения Ах за про-
межутки времени /, можно найти из уравнения
Ах2 RT
t ЗлЛ^А'Пг
(13.1)
(13.2)
где R — универсальная газовая постоянная; Т — абсолютная
температура; N\ — постоянная Авогадро; т] — вязкость среды;
г — радиус взвешенных частиц. '
Уже указывалось, что помимо поступательного движения
частицы обладают также вращательным броуновским движе-
нием, которое характеризуется средним квадратом угла враще-
ния Асе2 за время t. Энергия вращения частицы близка энергии
поступательного движения. По Эйнштейну,
Да2 RT
t 4л#АЛг3
Теория А. Эйнштейна, на основании которой были выведены
уравнения (13.1) и (13.2), получила многочисленные и неоспо-
римые экспериментальные доказательства. Блестящим подтверж-
дением теории явились работы Ж. Перрена, который в своих
опытах использовал сферические частицы мастики с точно из-
вестным радиусом, равным ~ 1 мкм. Измеряя на этом золе
поступательное и вращательное смещения частиц при извест-
ных значениях Г и т), Ж. Перрен вычислил по формулам (13.1)
и (13.2) постоянную Авогадро Na, которая оказалась равной
6,5-1023, что хорошо согласовалось с другими измерениями
этой постоянной. Таким образом было доказано, что закономер-
“ ности молекулярно-кинетического движения коллоидных частиц и
движения молекул в растворе одинаковые. '
А. Эйнштейн, изучая броуновское движение, установил связь
коэффициента диффузии D со средним сдвигом
Ах2 = 2Dt,
что дало возможность экспериментально определять D по методу
сдвига с наилучшими результатами для тонких суспензий и для
302
не слишком высокодисперсных золей, в которых особенно удобно
вести наблюдение за средними сдвигами в ультрамикроскопе.
Рассматривая силу трения, действующую со стороны раство-
рителя на диффузию молекул растворенного вещества, А. Эйн-
штейн предложил следующее соотношение:
п = кГ/Л (13.3)
где к — постоянная Больцмана; f—коэффициент трения для
растворенных молекул. В единицах СИ f измеряется в Н*с/м.
Частица со стороны растворителя испытывает силу трения, или
сопротивление, которая равна произведению f на скорость моле-
кулы. Дж, Стокс показал, что для сферической частицы
f = 6ЛЦГ,
где т] — вязкость растворителя; г — радиус частицы.
Сопоставляя уравнения (13.3) и (13.4), получим
(13.4)
(13.5)
смысл
Уравнения (13.3) и (13.5) выражают физический
коэффициента диффузии. Величина кТ — мера тепловой, или ки-
нетической, энергии молекулы; f или ц — мера вязкостного
сопротивления диффузии. Отношение двух противоположных
величин кинетической энергии и вязкости определяет, насколько
легко частица растворенного вещества диффундирует в растворе.
Оценим, например, коэффициент диффузии сферической частицы радиусом
1,5* 10“9
Т = 300
м в воде при 300 К. Запишем исходные данные: к= 1,38*10 Дж/К,
К, ц = 1.Ю“3 Н’С/м2, г— 1,5* 10-9 м. Тогда по уравнению (13.5)
_ (1,38* 10~23 Дж/К) (300 К) 10
D 6л(0,001 Н-с/м2) (1,5-10-9 м) 1>46-10 Дж-м/(Н-с).
1 Дж = 1 Н*м. то D — 1.46* 10“10 м2/с.
Так как
Пользуясь уравнением (13.5), можно оценить радиус частицы, если из-
вестны Риц.
2. фСМОТИЧЕСКОЕ ДАВЛЕНИЕ
Уравнение Вант-Гоффа применимо лишь к идеальным или
разбавленным растворам. Однако это уравнение с некоторыми
оговорками может быть использовано и при рассмотрении кол-
лоидных растворов.
Прежде всего следует отметить, что значение осмотического
давления пропорционально числу частиц растворенного вещества
в растворе. Поэтому при переходе от истинных растворов к
коллоидным, где размер частиц намного больше (а значит,
число частиц при той же концентрации намного меньше), прои-
зойдет резкое изменение давления. Так, можно показать, что
если в 1 М истинном растворе осмотическое давление равно
22,5 кПа, то для 0,1%-ного золя золота с размером частиц
ДО”8 м осмотическое давление будет приблизительно равно
303
22,5 ♦ 10-7 кПа, т. е. в 107 раз меньше. Столь малое значение не
только нельзя использовать для проверки применимости теории
к коллоидным растворам, но ее почти невозможно измерить с
необходимой точностю, учитывая погрешности осмотических экс-
периментов.
Таким образом, в процессе осмоса не обнаруживается ни-
каких принципиальных качественных различий между коллоид-
ными и молекулярными растворами (основные закономерности
едины), но большие количественные различия в значениях
концентрации приводят к слабому проявлению осмоса в коллоид-
ных системах.
Размер частиц в коллоидных растворах может колебаться
в довольно широких пределах, поэтому для различных коллоид-
ных растворов значения осмотического давления будут резко
отличаться, что связано с разной дисперсностью коллоидных
систем. Простой расчет позволяет установить количественную
зависимость между осмотическим давлением, числом частиц п
в единице объема раствора, средним радиусом г частиц, а зна-
чит, и степенью дисперсности z.
Так, массу диспергированного вещества в единице объема
раствора можно выразить через (где d — плотность
раствора). Тогда для двух дисперсных систем с однакэъым
растворителем и при одинаковых температурных условиях можно
записать
4/3nr?<irti = 4/зЛГ2^/12
и
rbl =
Таким образом,
_ Pi __ d _ Zi
Пч р2 Z2 '
т. е. осмотическое давление обратно пропорционально кубу ра-
диуса частиц и, следовательно, прямо пропорционально кубу
степени дисперсности.
Это* значит, что даже самое небольшое изменение в разме-
рах частиц дисперсных систем должно повлечь за собой весьма
значительные изменения в осмотическом давлении. Например, >
при увеличении размера частиц в 2 раза осмотическое давле-
ние должно уменьшиться в 8 раз при той же концентрации ра-
створа.
Растворы лиофобных коллоидов характеризуются непостоян-
ным размером частиц. При увеличении концентрации, напри-
мер, возможно увеличение размеров этих частиц благодаря
процессу агрегации (слипания), а при уменьшении концентра-
ции— процессу дезагрегации (разукрупнения). Эти процессы
приводят к тому, что с изменением концентрации осмотическое
давление изменяется не прямо пропорционально последней, как
304
следует из уравнения Вант-Гоффа, а значительно больше или
меньше.
Так как на процессы укрупнения и разукрупнения частиц
определенное влияние оказывает и температура, осмотическое
давление с ростом температуры изменяется для ряда коллоидных
систем аномально: с повышением температуры осмотическое дав-
ление растет не по прямой в соответствии с уравнением Вант-
Гоффа, а по быстро восходящей кривой и с понижением оно
аномально падает и притом по иной, нисходящей, кривой.
3. СЕДИМЕНТАЦИЯ
Процесс оседания коллоидных частиц в растворе под действием силы тя-
жести называется седиментацией.
Из курса физики известно, что для шарообразных частиц
с радиусом г при их падении сила трения F в жидкости с вяз-
костью т] равна
F — 6лх]ги,
где U — скорость оседания частиц.
Эффективный вес G этих частиц равен:
б = 7злг3(^ — p)g,
где d — плотность частиц; р — плотность среды; g — ускорение
силы тяжести.
Постоянная скорость оседания достигается при равенстве
F = G, тогда
6лГТ]Г == 4/3ЛГ3(б/ — Q)g,
а скорость оседания, выведенная на основании данного равен-
ству, равна
U = i r^~Q)g (13.6)
У Г]
Из уравнения (13.6) следует прежде всего то, что если части-
цы легче жидкости (например, эмульсия масла в воде), то раз-
ность (d — р) < 0 и тогда U < 0. Иначе говоря, вместо оседа-
ния произойдет всплывание частиц. Если (d — р) > 0, то кол-
лоидные частицы будут оседать, причем скорость оседания в
данной жидкости будет очень сильно зависеть от радиуса час-
тиц, так как в уравнение входит величина г2. В результате
можно показать, что частицы с диаметром 200 мкм проходят
1 см за 0,05 с, при диаметре 2 мкм — за 500 с, а при диаметре
20 нм — за 58 дней.
Таким образом, если исходить только из уравнения (13.6),
все коллоидные частицы независимо от размера за достаточ-
ный промежуток времени осядут на дно сосуда. На самом деле
это не так, ибо процессу седиментации, приводящему к концент-
рированию частиц на дне сосуда, противодействует другой про-
305
цесс — диффузия частиц, который приводит к равномерному рас-
пределению коллоидных частиц по всему объему раствора. Про-
цесс диффузии, осуществляемый под влиянием броуновского
движения, проявляется тем сильнее, чем меньше радиус коллоид-
ной частицы. Отсюда следует, что если процесс агрегации (сли-
пания) частиц не происходит, то только их размер будет опре-
делять, произойдет вообще седиментация или нет (табл. 46).
Из табл. 46 следует, что вероятность седиментации частиц
с диаметром 100 нм мала, однако она очень велика для частиц
с диаметром 10 000 нм. Процесс диффузии сопоставим с процес-
сом седиментации для частиц с диаметром 1000 нм и является
превалирующим для частиц с диаметром 100 нм.
Таблица 46. Соотношение расстояний, проходящих частицами Ag
разного размера под воздействием диффузии
и седиментации (по Бертону)
Диаметр частиц, нм Расстояние, проходимое частицей за 1 с (в нм)
Диффузия Седиментация
100 10000 67,6
1000 3162 6760
10000 1000 676000
Если статическое равновесие между действием сил тяготе-
ния и броуновского движения таково, что основная масса
частиц за сравнительно короткое время оказывается на дне со-
суда, то система называется кинетически неустойчи-
вой, если же частицы в основном остаются распределенными
в объеме раствора, то система называется кинетически
устойчивой. Лиофобные золи обычно имеют такие размеры
и плотности частиц, что обладают достаточно высокой кине-'
тической устойчивостью в системах, где не происходят процес-
сы, изменяющие степень дисперсности.
Заметное оседание частиц в системе, обладающей высо-
кой кинетической устойчивостью, можно вызвать, если использо-
вать значительные по величине центробежные силы. Впервые это
сделал А. В. Думанский (1913), применивший центрифугу для
осаждения коллоидных частиц. В 1923 г. Т. Сведбер! разработал
специальную центрифугу с большим числом оборотов, называе-
мую ультрацентрифугой (рис. 111).,Для центрифугирования тре-
буются приборы, которые позволяют работать при точно извест-
ных скоростях с малыми отклонениями без температурных коле-
баний. Современные ультрацентрифуги работают при больших
ускорениях до 420 000 ± 100g с контролем температуры в пре-
делах десятой градуса. Существует два типа приборов — анали-
тические и препаративные. Аналитические центрифуги снабже-
306
ны оптической системой, позволяющей опре-
делять распределение концентраций в любой
момент времени в течение всего измерения,
в то время как при использовании препа-
ративных центрифуг осуществляют фрак-
ционирование содержимого центрифужной
ячейки. В ультрацентрифуге фракциони-
руют не только мельчайшие частицы гид-
рофобных коллоидов, но и молекулы бел-
ков и высокомолекулярных веществ.
В поле земного тяготения ускорение,
действующее на частицу, остается постоян-
ным, а в ультрацентрифуге оно увеличи-
вается по мере оседания частицы. Поэтому
скорость оседания U выражают в виде
dx/d/, где х — расстояние от частицы до
оси вращения. Значение этой скорости, от-
несенное к единице действующей силы, на-
зывается константой седимента-
ции X частицы:
Рис. 111. Схема ульт-
рацентрифуги:
/ — ротор; 2 — кюветы;
3 — источник света; 4 —
корпус; 5 — регистри-
рующее оптическое
устройство
„ dx/dt .
------2— >
0) X
скорость вращения ротора,
является характеристиче-
растворенного вещества. За
единицу измерения
где co — угловая
Величина X
ской константой r г _
константы седиментации принят 1 сведберг= 10~13 с. Константа
седиментации колеблется от 2 (при относительной молекулярной
массе около 104) до 200 (при относительной молекулярной массе
около 107).
Относительная молекулярная масса связана с величиной X
уравнением
м =-------
где D — коэффициент диффузии; <р — парциальный удельный
объем; q — плотность растворителя.
Полидисперсность и кривые распределения. Методы седимен-
тации и ультрацентрифугирования применяются для изучения
так называемой полидисперсности коллоидных систем.
Большинство коллоидных систем характеризуется наличием час-
тиц или молекул различных размеров, что сильно отражается
на их свойствах. Изучение полидисперсности для установления
количественного распределения частиц или молекул по размерам
(так называемых кривых распределения) производит-
ся разными методами для различных дисперсных систем. Си-
стемы с очень грубыми частицами (свыше 0,2 мм) исследуются
простым ситовым анализом. Суспензии же и эмульсии с размера-
ми частиц в интервале 1 ...200 мкм изучаются методами седимен-
307
Рис. 112. Седиментометр Фигу-
ровского
тации, вследствие чего анализ поли-
дисперсности в этом случае назы-
вается седиментационным
анализом.
Седиментационный анализ поли-
дисперсных систем осуществляется
по измерению кинетики накопления
слоя «осадка» в суммарном про-
цессе седиментации, или, иначе, по
возрастанию эффективного веса та-
кого слоя со временем. Такие изме-
рения осуществляются наиболее
простым и достаточным способом
с помощью так называемого седи-
ментометра — гидростатических упругих микровесов Фигуров-
ского.
В седиментометре Фигуровского (рис. 112) к упругому стек-
лянному или кварцевому стержню 1 прикреплена на стеклян-
ной нити 2 с крючком чашечка 3, на которой накапливается
осадок суспензии. Прогиб плеча измеряется по шкале при помо-
щи микроскопа. По мере оседания частиц дисперсной фазы
прогиб увеличивается вначале быстро вследствие преимущест-
венного выпадения более тяжелых частиц, а затем все медлен-
нее, почти до полного окончания оседания. Седиментационную
кривую накопления р = f(f) строят, откладывая по оси абсцисс
время седиментации t от 0 до tm, а по оси ординат — относи-
тельное накопление «осадка» (в %) р (от 0 до 100). Если высота
столба суспензии равна / м, то при времени оседания t скорость
оседания составит U = l/t. По уравнению (13.6) можно рассчи-
тать критический радиус гкр частиц, обладающих этой скоростью
соединения.
Любая ордината кривой накопления р = /(/), определяющая
(в %) относительную массу всех частиц, осевших за данное вре-
мя t из исследуемого столба, характеризует две фракции:
308
1) вполне определенную, це-
ликом выделившуюся фрак-
цию to частиц, радиус кото-
рых равен или больше гра- /|ж\
ничного гкр, и 2) неопреде- / Ц \
ленную фракцию всех более / [Ц \
мелких частиц т с радиуса- / Ш
ми г С Гкр; при этом р = J
= w-\-m. Определение w и »
т производится по кривой 6
седиментации графическим Рис. 114. Кривая распределения
способом, так как w равно
отрезку ординаты, отсекаемому касательной к кривой седимен-
тации, а т равно разности (p—w) для каждого значения t. На-
пример, кривая на рис. 113 показывает, что за интервал времени
до 5 мин полностью оседает около 22% суспензии, за интервал
времени от 5 до 10 мин оседает 60—22=38% суспензии, за
интервал времени от 10 до 15 мин оседает 71—60=11% и т. д.
Таким образом, для каждого интервала размеров частиц, т. е.
для каждого интервала времени оседания, может быть опреде-
лена доля выпавшей суспензии.
Проводя анализ кривой седиментации на рис. 113, можно
рассчитать кривую распределения для данной суспензии
(рис. 114), которая характеризует относительное содержание в
суспензии частиц различного размера. Обычно кривые распре-
деления содержат один максимум, который определяет гв — наи-
более вероятный радиус частиц.
4. ВЯЗКОСТЬ
Добавление коллоидных частиц к растворителю с вязкостью
т]о приводит к увеличению вязкости раствора т]. Повышение
вязкости есть результат увеличения трения между прилегающи-.
ми мономолекулярными слоями жидкости, вызванного тем, что
частицы крупнее молекул растворителя. Изменение вязкости
обычно выражают как отношение ц/т]о, называемое относи-
тельной вязкостью цотн. Если коллоидные частицы име-
ют сферическую форму и не взаимодействуют между собой
(разбавленные растворы), то, как было установлено А. Эйн-
штейном,
ЛоТН -------
1 -|- 0,5<р
(1-ф)2 ’
где ф — доля объема суспендированных частиц в единицу объема
суспензии.
Это уравнение может быть переписано в виде ряда членов
с возрастающими степенями ф. Для разбавленных растворов
можно использовать уравнение, содержащее два члена:
Лотн == л/ло = 1 + 2,5ср.
(13.7)
309
При введении еще одного члена
Чотн = 1 4- 2,5ф 4- 14,1 ср2
(13.8)
Рис. .115. Схема диализатора
область использования уравнения расширяется и оно в ряде слу-
чаев может быть применено для 30%-ных растворов.
Если частицы не сферические, но симметричные, осуществ-
ляют эмпирический подбор коэффициентов. Так, для частиц
эллипсоидальной формы с отношением полуосей 6/а=10
уравнение примет вид
Чотн = 1 4- 5<р 4- 48ф2.
Зависимость вязкости коллоидных растворов от температуры
имеет более сложный характер, чем для истинных растворов.
Линейная зависимость т] от 1/Г обычно нарушается из-за агре-
гации и дезагрегации частиц при колебаниях температуры.
5. ОЧИСТКА КОЛЛОИДНЫХ РАСТВОРОВ
Некоторые молекулярно-кинетические свойства коллоидных
систем используют для осуществления очистки коллоидных раство-
ров от электролитов и молекулярно растворенных веществ^
которые часто образуются одновременно с веществами, имею-
щими коллоидную степень раздробления. Естественно, наличие
указанных примесей в коллоидных растворах искажает резуль-
таты исследований, и их следует удалить.
Наиболее распространенными методами очистки коллоидных
растворов от примеси кристаллоидов являются диализ и ультра-
фильтрация.
Диализ. Это процесс очистки (отделения) коллоидных раство-
ров, который основан на свойстве полупроницаемой мембраны
пропускать примеси ионов и молекул малых размеров и задер-
живать коллоидные частицы.
Прибор для очистки коллоидов называется диализатором.
Простейший диализатор представляет собой сосуд /, у которого
нижнее отверстие затянуто полупроницаемой мембраной 2
(рис. 115). В сосуд 1 наливают коллоидный раствор, после
чего он погружается в сосуд 5, наполненный чистой водой.
Обычно применяют диализатор, в котором вода в наружном
сосуде меняется периодически, либо проточный диализатор с
непрерывной сменой воды.
Молекулы и ионы, способные
проходить через мембрану, диффун-
]/5=з — дируют в растворитель, при постоян-
ритель ной смене которого можно очистить
— коллоидный раствор от примесей.
Для проведения диализа в простейшем
диализаторе требуется очень много времени
(недели, месяцы), поэтому в современных
диализаторах увеличивают поверхность мем-
браны или создают высокий градиент кон-
310
центрации по обе стороны мембраны, или же, наконец, повышают температуру.
Все это существенно сокращает время участия.
Необходимо помнить, что степень очистки ограничивается устойчивостью
коллоидных частиц; слишком продолжительный диализ может привести к уда-
лению частиц, стабилизирующих золь, а это значит, что исследуемая коллоид-
ная система будет разрушена.
Для более быстрой и полной очистки коллоидов от электроли-
тов в производстве применяют так называемый электро-
диализ. Прибор, с помощью которого осуществляется элект-
родиализ, называется электродиализатором.
Электродиализатор (рис. 116) состоит из трех частей. Сред-
няя часть электродиализатора 2 заполняется коллоидным раство-
ром; она отделена от двух примыкающих к ней частей мембра-
нами, сделанными из полупроницаемого вещества (коллодия,
пергамента, целлофана и т. п.). Части 1 и 3 прибора имеют
специальные отверстия: 5 — для подачи воды, 4 — для ее выхода
и 6 — для ввода электродов, примыкающих к внутренним по-
Рис. 116. Схема электродиализатора
верхностям мембран. При электродиализе обычно используют
невысокую плотность тока, так как в противном случае наблю-
дается сильное разогревание, существенно меняющее свойства
золя. Обычно, если золь содержит большой объем электролита,
его предварительно подвергают обычному диализу, удаляющему
основную часть электролитов, и только после такой предвари-
тельной очистки применяют электродиализ. Преимущество элект-
родиализа по сравнению с обычным диализом заключается в
возможности удаления даже следов электролитов, что обыкно-
венным диализом не достигается.
Ультрафильтрация. Это процесс отделения дисперсной фазы
от дисперсионной среды путем фильтрования коллоидных раство-
ров через полупроницаемые мембраны. При ультрафильтрации
коллоидные частицы остаются на фильтре (мембране), а фильт-
рат, содержащий электролиты, переходит в растворитель.
Для ускорения ультрафильтрацию проводят под давлением.
Разность давлений получают либо создавая разряжение под
фильтром (ультрафильтрация в вакууме), либо производя дав-
ление на фильтрующийся раствор (ультрафильтрация под давле-
нием). Для ультрафильтрации в ваку(уме применяют специаль-
ную воронку (рис. 117), которую на пробке вставляют в сосуд,
311
Рис. 117. Схема во-
ронки с ультра-
фильтром:
1 — мембрана; 2 —
пористая пластинка;
3 — воронка
соединенный с вакуумным насосом. В воронку,
на дне которой находятся обыкновенный
фильтр (пористая пластинка) и ультрафильтр
(мембрана), наливают коллоидный раствор.
Создается вакуум, благодаря которому ультра-
фильтрат проходит через поры фильтра, и
дисперсная фаза остается в воронке. Если
нужно провести фракционирование частиц
коллоидного раствора, то применяют ультра-
фильтры с различной пористостью. Зная раз-
мер пор ультрафильтрата, можно провести
приблизительную оценку размера частиц в от-
дельных фракциях.
Для очистки применяют также метод
электроультрафильтрации’, пред-
ставляющий собой ультрафильтрацию в элект-
рическом поле.
В табл. 47 сопоставлены относительные скорости очистки
наиболее распространенными методами.
Таблица 47. Относительные скорости
очистки коллоидных растворов
Метод
Относительные скорости
удаляемого вещества
NaCl сахар
Диализ
Электродиализ
Ультрафильтрация
Электроультрафильтрация
Г
163
14
182
0,3
2
.14
14
6. ОПТИЧЕСКИЕ СВОЙСТВА
Светорассеяние. В 1869 г. Дж. Тиндаль заметил, что если
пропустить пучок сходящихся лучей через коллоидный раствор,
то наблюдается образование светящегося конуса. Это явление
в честь автора названо явлением Тиндаля. Первона-
чально? предполагали, что этим свойством обладают только
коллоидные системы. Но впоследствии было найдено, что явле-
ние Тиндаля наблюдается у всех дисперсных систем, степень
дисперсности которых равна или больше степени дисперсности
коллоидной системы.
Если пучок параллельных лучей попадает на поверхность
какой-нибудь частицы, линейные размеры которой велики по
сравнению с длиной волны падающего излучения, то наблюдает-
ся отражение по законам геометрической оптики. Когда длина
волны падающего света велика (примерно в 10 раз больше)
312
сравнительно с линейными размерами светорассеивающей части-
цы, то в этом случае наблюдается дифракция световой волны,
вызывающая светорассеяние. Такое светорассеяние и является
причиной эффекта Тиндаля.
Дж. Релей создал теорию светорассеяния коллоидных раство-
ров и предложил уравнение, характеризующее этот процесс:
24ул2У2А2/ - nj
\ Л1 И- 2«2
(13.9)
где I — общее количество световой энергии, рассеянной едини-
цей объема; v — число частиц в единице объема; V — объем
частицы; X — длина волны падающего излучения; А — амплиту-
да колебания излучений; п2 и П\ — показатели преломления
дисперсионной среды и дисперсной фазы соответственно.
Из уравнения 13.9 видно, что чем меньше длина волны па-
дающего излучения, тем больше будет рассеяние. Следовательно,
если на частицу падает белый свет, то наибольшее рассеивание
будут испытывать синие и фиолетовые компоненты. Поэтому
если пропускать через коллоидную систему белый луч, то в
проходящем свете раствор будет окрашен в красноватый цвет,
а в боковом, отраженном, — в голубой. Это хорошо видно на зо-
лях мастики, серы.
Обозначив
уравнение (13.9) можно записать следующим образом:
vV2
р
,Так как число частиц v пропорционально концентрации С,
а объем частицы, если принять ее шарообразной, V = 4/злг3, то
v = С/( Vq),
где q — плотность вещества.
Подставляя это выражение в уравнение (13.10), получим
vqV 3 qV
Обозначив все постоянные величины через К', получаем для
данной длины волны
/ = К'Сг\
(13.11)
Количество рассеянной энергии пропорционально выражению
/ ft? — nj \2
\ П? 4- «2 / ’
являющемуся сомножителем в уравнении (13.9). Отсюда следует,
что когда п\ = п2, этот сомножитель превращается в нуль и,
31$
Рис. 118. Схема ультрамикроскопа
следовательно, /=0, т. е.
светорассеяние (явление
Тиндаля) не происходит.
Ультрамикроскопические
исследования. Эффект Тин-
даля был использован в
1903 г. Р. Зигмонди для
устройства прибора, назван-
ного им ультрамикроскопом.
Коллоидные частицы с
радиусом меньше 200 нм не
видны при использовании
современных микроскопов
из-за ограничений, связанных с разрешающей силой. С помощью
же ультрамикроскопа можно наблюдать частицы с радиусом
3... 150 нм. При использовании ультрамикроскопа коллоидную
систему наблюдают не в проходящем свете (как это делается
в обычном микроскопе), а в боковом, отраженном. Таким обра-
зом, рассматривают не сами частицы, а дифрагированные пучки
света.
Схема ультрамикроскопа приведена на рис. 118. Источник
света /, в качестве которого применяют вольтову дугу, образует
пучок света, проходящий через отверстие экрана 2. Концентри-
рование этого света в небольшом объеме кюветы 5, содержащей
коллоидный раствор, достигается применением соответствующих
линз 7 и 6 и диафрагмой 3. Наблюдение за частицами произ-
водится с помощью микроскопа 4.
Ввиду того что рассматриваются дифрагированные пучки света, ультра-
микроскоп не дает возможности видеть размер и форму частиц, а удается
только обнаружить присутствие частиц и наблюдать их движение. Однако,
пользуясь некоторыми приемами, можно приближенно определить и размер
частиц. Так, выделяют определенный оптический объем Уо, в котором подсчи-
тывают число частиц п. Зная массовую концентрацию частиц С и число их
в единице объема v, находят объем одной частицы V:
V = С/(yd) = CVQ/(nd\
где d — плотность дисперсной фазы.
Для кубической и сферической форм частиц можно вычислить радиус г
по формулам
г — /Vиг = 3|/37/4л .
Другой описанный ранее способ определения г заключается в измерении
среднего сдвига иастицы Ах по серии снимков, полученных с помощью фото-
насадки.
В поточном ультрамикроскопе аэрозоль или гидрозоль протекает через
специальную кювету в направлении оси микроскопа при боковом освещении.
Соотношение числа отблесков, видимых на темном фоне, и объемной скорости
потока определяет число частиц в единице объема, а следовательно, V и г.
В этом приборе можно регулировать яркость освещения, что позволяет по-
строить кривую определения частиц по размерам путем подсчета числа частиц
при различных степенях яркости.
Применение ультрамикроскопа позволяет наблюдать частицы с размерами
до 3 нм, т. е. отодвигает границу видимости почти на два порядка, охваты-
314
вая практически всю коллоидную область. Однако в последнее время большую
распространенность начинает приобретать метод электронной микроскопии,
обладающей гораздо большей разрешающей способностью.
Нефелометрические измерения. Коллоидные системы можно
исследовать другим методом — методом нефелометрии, который
основан на сравнении интенсивности светорассеяния.
Пусть имеются два золя Agl одинаковой концентрации, но
различной дисперсности. Согласно уравнению (13.11) размеры
частиц обоих растворов будут равны
Г1 у к,с , Г2 у ,
откуда
Если для одного золя, являющегося эталоном, радиус час-
тиц известен, то можно определить степень дисперсности дру-
гого золя.
В другом случае, когда золи отличаются между собой
концентрацией, но содержат частицы одинакового размера, со-
гласно уравнению (13.11) справедливо соотношение
/1 Ci
/2 Сг
- тхе., сравнивая интенсивности светорассеяния, можно опреде-
лить концентрацию исследуемого золя. Такие измерения прово-
дят в приборах, называемых нефелометрами.
Схема нефелометра показана на рис. 119. Пучок лучей от
источника света /, пройдя через линзу 2, освещает испытуемый
раствор в сосуде 7. Фотоэлемент 3, питаемый от электрической
батареи 6, под влиянием света, рассеянного раствором, при-
водит в действие через сопротивление 4 зеркальный гальва-
нометр 5, чувствительность которого достигает 10-9 А. Сравни-
вая отклонения стрелки для стандартного и исследуемого раство-
ров, находят концентрацию последнего или размер частиц в нем.
Поглощение света. При
прохождении света через
молекулярные растворы и
другие оптические однород-
ные среды интенсивность
прошедшего света I всегда
меньше интенсивности па-
дающего света /о вследствие
некоторого поглощения све-
та в этой среде. Согласно
закону Ламберта — Бера, в
данном слое однородной сре-
ды отношение 1/1о определя-
2
Рис. 119. Схема нефелометра
315
ется только числом поглощающих частиц или молекул:
— е >
где I — толщина слоя; С — молярная концентрация растворен-
ного вещества; 8 — молярный коэффициент поглощения вещест-
ва. Величина 8 не зависит от концентрации, но изменяется с
длиной волны, температурой й природой растворителя. При
выражении закона Ламберта — Бера через десятичные логариф-
мы величина 8' называется коэффициентом экстинкции, или
поглощения (е' = 0,434s).
Однако закон Ламберта — Бера не всегда может быть
применен при описании поглощения света коллоидными раство-
рами. В зависимости от природы коллоидных частиц одни кол-
лоидные растворы подчиняются этому закону, а другие — нет.
По-другому проявляется связь между строением, составом
и окраской коллоидных растворов по сравнению с молекуляр-
ными растворами. Если для молекулярных растворов окраска не
связана с размерами частиц, то для коллоидных растворов
такая связь окраски с размерами частиц имеет место и поэтому
в большинстве случаев окраска коллоидных золей зависит от
степени их дисперсности.
7. ЭЛЕКТРИЧЕСКИЕ СВОЙСТВА
7.1. Строение мицеллы
Строение мицеллы может быть рассмотрено лишь в первом
приближении, так как она не имеет определенного состава.
В настоящее время нет возможности учесть влияние на строе-
ние мицеллы всех процессов, обусловленных сложными ионно-
молекулярными взаимодействиями на поверхности раздела двух
фаз в растворе.
Если исключить влияние растворителя, в котором образует-
ся коллоидная система, то упрощенные схемы мицеллы золя
иодида серебра, полученные при сливании сильно разбавлен-
ных растворов AgNO3 и KI, можно представить следующим
образом (рис. 120, 121).
Элементарная коллоидная частица — мицелла — содержит
нерастворимое в данной дисперсной среде ядро (рис. 120).
Ядро состоит из диспергированного вещества и представляет
собой микрокристалл или агрегат из многих микрокристаллов*.
Работами В. А. Каргина и его школы было показано, что иногда
сначала образуется аморфное ядро, которое затем может крис-
таллизоваться. Так, при получении золя сульфида мышьяка с
помощью электронного микроскопа было установлено, что сна-
чала образуются шарообразные или бесформенные частицы,
* Ряд авторов называют твердофазную часть мицеллы агрегатом (согласно
терминологии Пескова), а агрегат с адсорбированными ионами — ядром.
316
имеющие аморфную
структуру. В дальней-
шем же внутри аморф-
ных частиц возникают
кристаллы. Благодаря
возникновению кри-
сталлических образова-
ний внутри аморфных
частиц создаются на-
пряжения и частицы
распадаются на боль-
шое число мелких кри-
сталлических образова-
ний. Процесс кристал-
лизации протекает во
времени. Так, для золя
Рис. 120. Мицелла золя иодида серебра, полу-
ченного по реакции AgNCh + KI при избытке
KI
золота процесс кри-
сталлизации при ком-
натной температуре на-
чинается через 5 мин;
для золя оксида ванадия (V) — через 1 ч; для оксида титана
(IV) —через 1...2ч; для гидроксида алюминия — через сутки,
а для кремниевой кислоты требуется примерно два года. В зави-
симости от природы вещества дисперсной фазы форма и размер
микрокристаллов могут быть различными.
Поверхность ядра обладает запасом свободной энергии и
избирательно адсорбирует ионы из окружающей среды. Экспе-
риментально было показано, что на поверхности ядра преиму-
щественно адсорбируются ионы, одноименные с составом ядра.
На основании обширного экспериментального материала было
сформулировано правило: на поверхности твердого вещества
предпочтительно адсорбируются ионы, способные достраивать
его кристаллическую решетку или образующие с ионами, входя-
щими в состав кристаллической решетки, наиболее трудно раст-
воримые соединения.
Таким образом, в рассматриваемом примере ядро мицеллы,
состоящее из кристалликов Agl, может адсорбировать из раство-
ра ионы Ag+ или I-. Определяется это избытком тех или иных
ионов в растворе. Если реакция AgNO3 и KI осуществляется
при избытке последнего, то ядро будет адсорбировать ионы I-
(рис. 120); при избытке AgNO3 ядро адсорбирует ионы Ag+
(рис. 121).
В результате этого процесса ядро с адсорбированными ио-
нами приобретает отрицательный либо положительный заряд.
Ионы, создающие этот заряд, называются потенциалоп-
ределяющими. Естественно, после возникновения заряда
агрегат начинает притягивать из раствора ионы с противо-
положными знаком (противоионы); образуется двойной
электрический слой. Некоторая часть противоионов очень прочно
317
Потенииалопределяютие
Рис. 121. Мицелла золя иодида серебра, получение- .
го по реакции AgNO3 Ч- KI при избытке AgNO3
притягивается к агрегату, образуя адсорбцйонный слой проти-
воионов. Агрегат вместе с адсорбированным слоем называют
частицей или гранулой. Таким образом, частица (гра-
нула) характеризуется двойным электрическим слоем, который
образуется из потенциалопределяющих ионов и противоионов.
Противоионы, которые не входят в двойной электрический
слой и находятся на более далеком расстоянии от ядра, об-
разуют так называемый диффузный слой противоионов. Этот
слой обычно называют просто диффузным ело е м.
Состав мицеллы может быть изображен схематически сле-
дующим образом. При избытке KI золь иодида серебра может
быть представлен формулой
гранула
1---------------
{[Agl)m- ЛГ (л -х) К+} ’хК+
ядро
1______;_____________I
мицелла
Потенциалопределяюшие ионы п\~ придают грануле отрицатель-
ный заряд, так как п > (п — х).
При избытке AgNO3 гранулы имеют положительный заряд,
поскольку потенциалопределяющими ионами являются Ag+. Про-
тивоионы nNOr располагаются в адсорбционном слое в коли-
честве п — х, а в диффузном — х ионов. Так как п>(п — х), гра-
нула золя будет иметь положительный заряд
гранула
I-------------------’7П
{lAgI]m • rtAg+(k-x)NoJ xNO;
l_____I
ядро
I__________________________I
мицелла
318
Необходимо отметить, что в целом мицелла электронейтраль-
на, и поэтому число ионов, входящих в диффузный слой (х),
определяется из условия электронейтральности мицеллы. Иначе
говоря, противоионы диффузного слоя нейтрализуют свободный
(избыточный) заряд гранулы.
7.2. Двойной электрический слой
и электрокинетические явления
При рассмотрении строения мицеллы было показано, что
при взаимодействии лиофобных коллоидов с электролитами на
поверхности ядра адсорбируются определенные ионы из раство-
ра. Ядро с адсорбированными на нем ионами того или иного
знака взаимодействует с окружающим раствором. При этом
благодаря электростатическому притяжению ионы, обладающие
знаком, противоположным по отнощению к потенциалопределяю-
щим ионам, стремятся расположиться к ним как можно ближе.
В результате этого образуются два близко расположенных
слоя ионов: один на поверхности (потенциалобразующие ионы)
и другой в растворе (противоионы). Такая система называет-
ся двойным электрическим слоем Гельмгольца
(рис. 122). Следует помнить, что в целом эта система электроней-
тральна. В представлении Гельмгольца двойной электрический $
слой подобен плоскому конденсатору, внутренняя обкладка
которого находится в твердой фазе, а внешняя — расположе-
на в жидкости параллельно твердой поверхности ядра на рас-
стоянии молекулярного порядка. Общий термодинамический по-
Рис. 122. Схема'двойного электрического слоя:
а—по Гельмгольцу; б — по Гун; в — по Штерну; вверху — располо-
жение ионов; внизу — кривые падения потенциала; ср — термодинами-
ческий потенциал; g — электрокинетический потенциал
319
тенциал ф уменьшается линейно от поверхности в соответствии
с теорией плоского конденсатора.
Однако картина распределения ионов в растворе более
сложна, чем в описанном ранее случае. Часть ионов, имеющих
знак, обратный потенциалопределяющим ионам (противоионы),
образует, благодаря тепловому движению около твердой поверх-
ности ядра, диффузную ионную атмосферу. В результате часть
противоионов удаляется от твердой поверхности на расстоя-
ние, превышающее молекулярное (рис. 122,6). Такая структура
двойного слоя (с изменяющейся толщиной от 1 до 10 мкм) была
предложена Гуи. Здесь изменение термодинамического потенциа-
ла происходит по более сложному закону.
Предложенная Штерном структура двойного электрического
слоя является промежуточной между двумя крайними случаями,
описанными Гельмгольцем и Гуи. Согласно теории Штерна,
часть противоионов находится на молекулярном расстоянии от
поверхности ядра (слой Гельмгольца), а другая часть образует
диффузный двойной слой по Гуи.
Разность потенциалов между подвижной (диффузной) и неподвижной
(адсорбированной) частями двойного электрического слоя называется электро-
кинетическим потенциалом или иначе ^-потенциалом (дзета-потенциалом).
Если коллоидную частицу поместить в постоянное электри-
ческое поле, то в ней, как и в растворах простых электролитов,
происходит движение зарядов к противоположно заряженным
электродам: коллоидная частица движется в одну сторону, про-
тивоионы — в другую. Если бы все ионы могли передвигаться
независимо от частицы, то процесс отвечал бы обычному пере-
носу ионов. Однако коллоидная частица движется не только с
адсорбированными на ней ионами, но и с той частью противо-
ионов, которые непосредственно к ней прилегают (рис. 123),
что соответствует гельмгольцевской части двойного электриче-
ского слоя (см. рис. 122, а). Поэтому граница смещения ион-
ных слоев коллоидных частиц в электрическом поле не совпа-
дает с границей поверхности частиц, а несколько смещена в
сторону раствора (пунктирная линия на рис. 122).
Скорость передвижения коллоидных частиц в электрическом
поле пропорциональна ее потенциалу. Если бы коллоидная
частица передвигалась без части противоионов, то ее измеряе-
мый потенциал соответствовал бы полной разности потенциалов
между поверхностью частицы и глубиной раствора, т. е. обычно
определяемому в электрохимии полному, или термодинамическо-
му, потенциалу <р (см. рис. 122). Однако на самом деле часть
противоионов увлекается вместе с коллоидной частицей, поэтому
определяемый по передвижению в электрическом поле дзета-
потенциал (см. рис. 122) составляет лишь часть потенциала <р.
Наличие у частиц дисперсных систем электрического заряда
было открыто еще в 1808 г. проф. Московского университета
Ф. Ф. Рейссом. В опыте были использованы две стеклянные
320
Рис. 123. Схема движения коллоидной частицы
в электрическом поле
Рис. 124. Схема опыта Рейс-
са по электрофорезу
трубки, погруженные в сырую глину и заполненные водой.
При наложении электрических потенциалов (рис. 124) жидкость
в трубке с положительным полюсом мутнела, а в трубке с отри-
цательным полюсом вода оставалась прозрачной, т. е. частицы
глины переносились в электрическом поле к положительному
полюсу. Более поздними исследованиями было установлено, что
частицы движутся в электрическом поле с постоянной скоростью.
Эта скорость тем больше, чем выше приложенная разность
потенциалов и диэлектрическая постоянная среды, и тем меньше,
чем больше вязкость среды. Перенос частиц в электрическом
поле получил название электрофореза.
В других опытах Ф. Ф. Рейсс показал, что при пропуска-
нии постоянного тока через прибор, состоявший из U-образной,
наполненной водой трубки, средняя часть которой была запол-
нена мелким кварцевым песком (рис. 125), вода в колене с от-
рицательным электродом (катодом) поднималась, а в колене4с
положительным электродом опускалась, что свидетельствовало
о положительном заряде воды.
Подобно электрофорезу, этот процесс идет с постоянной ско-
ростью, и объем перенесенной жидкости прямо пропорционален
приложенной разности потенциалов и диэлектрической постоян-
ной и обратно пропорционален вязкости среды. Было установлено,
что объем жидкости, прошедший через капилляры пористой диаф-
рагмы, пропорционален силе тока и
при постоянной силе тока не зависит
от площади сечения или толщины
диафрагмы. Это явление было названо
электроосмосом.
Последующими исследованиями бы-
ли обнаружены два явления, как бы
обратимые к электрофорезу и электро-
осмосу. Г. Дорн (1878) обнаружил,
что при оседании каких-либо частиц
в жидкости, например песка в воде,
возникает электродвижущая сила меж-
ду двумя электродами, введенными в
Рис. 425. Схема опыта по
электроосмосу
11—696
321
разные места столба жидкости. Это явление получило название
эффекта Дорна или потенциала седиментации.
При продавливании жидкости через пористую перегородку, по
обеим сторонам которой находятся электроды, также было обна-
ружено возникновение разности потенциалов. Явление это, откры-
тое Квинке (1859) и обратное электроосмосу, было названо по-
тенциалом протекания или потенциалом те-'
ч е н и я.
Все четыре указанных явления, поскольку в них происходит
передвижение частиц или жидкости при приложении разности
потенциалов или, наоборот, возникает разность потенциалов при
передвижении частиц или жидкости, получили ,общее название
электрокинетических явлений.
Электрокинетические явления классифицируют следующим
образом.
1. Электрокинетические явления первого рода — относитель-
ное перемещение фаз под действием приложенного напряжения;
к ним относятся: а) электрофорез — движения частиц дисперс-
ной фазы в неподвижной дисперсионной среде; б) электро-
осмос — движение жидкости относительно неподвижной твердой
поверхности пористых мембран.
2. Электрокинетические явления второго рода — возникнове-
ние разности потенциалов вследствие вынужденного относитель-
ного движения фаз; различают: а) потенциал оседания (эффект
Дорна) — возникновение разности потенциалов при движении
частиц в неподвижной жидкости; б) потенциал протекания
(эффект Квинке) — возникновение разности потенциалов при
движении жидкости относительно неподвижной твердой фазы.
Для вычисления скорости электрофореза и дзета-потенциала
золей по макроскопическому методу применяют уравнение
Гельмгольца — Смолуховского:
и=-^т- (13.12)
Из уравнения (13.12) получают значение дзета-потенциала
S гЁ~'
Величина г| (для дисперсионной среды) может быть получена
из справочных таблиц, а 8 и £ легко измеримы; поэтому опре-
деление дзета-потенциала сводится к возможно точному измере-
нию U путем наблюдения за перемещением границы раздела фаз.
Скорость электроосмоса, т. е. скорость передвижения жид-
кости в капиллярах или в пористой диафрагме (порошке) под
влиянием приложенной разности потенциалов, измеряют объемом
(или массой) жидкой фазы, прошедшим через диафрагму в еди-
ницу времени. Эта скорость, как и в случае электрофореза,
находится в прямой зависимости от д, £, /, е, а также от площа-
ди сечения всех капилляров S и в обратной — от г| и /; поэтому
322
уравнение электроосмоса имеет почти такой же вид, как и урав-
нение электрофореза
U_^S_ g/s
4лт)/ 4лт)х
(13.13)
где I — сила тока; х— удельная электрическая проводимость
жидкости.
Уравнение (13.13) дает возможность определить дзета-потен-
циал
___ 4лт]х£/
е /е
Контрольные вопросы
1. Почему нельзя визуально наблюдать истинный путь коллоидной частицы?
Как определяется средний сдвиг (смещение)?
2. Почему практически нельзя измерить осмотическое давление большинства
лиофобных коллоидов?
3. Как, используя оптические свойства, легко и быстро отличить раствор
лиофобных коллоидов от истинного раствора?
4. Почему мицелла нейтральна?
5. Почему на поверхности ядра адсорбируются ионы из раствора с опреде-
ленным зарядом?
6. Как экспериментально определяют электрокинетический потенциал?
11*
Глава XIV
УСТОЙЧИВОСТЬ И КОАГУЛЯЦИЯ
ЛИОФОБНЫХ КОЛЛОИДОВ
1. КИНЕТИЧЕСКАЯ И АГРЕГАТИВНАЯ УСТОЙЧИВОСТЬ ,
Частицы дисперсной системы, с одной стороны, испытывают
действие земного притяжения; с другой стороны, они подвер-
жены диффузии, стремящейся выравнять концентрацию во всех
точках системы. Когда между этими двумя силами наступает
равновесие, частицы дисперсной фазы определенным образом
распределяются относительно поверхности Земли.
Если бы устойчивость лиофобных коллоидов определялась
только равновесием, то повышение температуры и связанное
с ним усиление броуновского движения дисперсных частиц
должно было бы всегда вести к увеличению устойчивости систе-
мы. Однако в золях, суспензиях и эмульсиях повышение темпе-
ратуры ведет, наоборот, к понижению устойчивости. Это проти-
воречие было полностью устранено Н. П. Песковым, когда он в
1920 г. предложил различать два типа устойчивости — кине-
тическую и агрегативную устойчивость.
Под кинетической устойчивостью Н. П. Песков понимал
свойство дисперсных частиц удерживаться во взвешенном сос-
тоянии, не седиментируясь, а распределяясь по высоте в соответ-
ствии с гипсометрической формулой Лапласа — Перрена:
NaV
tl2
где п\/пъ — относительное изменение числа частиц в единице
объема в двух слоях жидкости, отстоящих на расстояние h
(слой 2 выше слоя 1); Na — постоянная Авогадро; V — объем
отдельной частицы; d — плотность частицы; Q — плотность среды.
Н. П. Песков использовал в качестве критерия кинетической устойчивости
понятие о высоте столба (слоя) в седиментационном равновесии. Высота стол-
ба — это высота, на которой частичная концентрация данной дисперсной
системы становится предельно малой. Н. П. Песков предложил в качестве
такого предела принять убыль частичной концентрации П\1п<2. в 10б раз по
сравнению с концентрацией у основания столба, обозначив такую высоту че-
рез Яо.
По другому предложению Н. П. Пескова мерой высоты столба можно счи-
тать высоту, на которой частичная концентрация ni/n2 убывает в 2 раза
324
(n1/n2 = 2), что в практическом отношении гораздо удобнее; обозначим ее
через hQ. Тогда из уравнения
ПО1 о IW(d-Q)ghQ
можно рассчитать /z0.
Значение высот Но и ho определяется результатом действия двух противо-
положных сил — силы тяжести, увлекающей частицы на дно сосуда, и броунов-
ского движения, ведущего к равномерному распределению частиц по всей высоте
сосуда. Отсюда следует, что определяющим фактором в кинетической устой-
чивости системы является степень дисперсности дисперсной фазы' Чем она выше,
т. е. чем меньше размеры и масса частиц и, следовательно, чем больше энергия
их броуновского движения, тем выше кинетическая устойчивость. Повышение
температуры, ведущее к усилению броуновского движения, увеличивает кине-
тическую устойчивость.
Однако не только степень дисперсности определяет общую
устойчивость лиофобных коллоидов. Опыт показывает, что раз-
мер частиц со временем, и особенно при повышении темпера-
туры, становится больше. Происходит как бы слипание частиц
или< иначе говоря, их агрегация. Более крупные агрегаты кол-
лоидных частиц продолжают сталкиваться друг с другом, сли-
паясь при столкновении, так что вскоре размер агрегатов стано-
вится заметным для человеческого глаза. Происходит помутнение
коллоидного раствора и, наконец, дисперсная фаза выпадает
в осадок.
Этот тип устойчивости коллоидных систем Н. П. Песков на-
звал агрегативной устойчивостью.
Таким образом, агрегативная устойчивость коллоидных сис-
тем есть способность частиц дисперсной фазы оказывать со-
противление их слипанию и тем удерживать определенную
степень дисперсности этой фазы в целом.
Представление Н. П. Пескова о двух типах устойчивости
легко объясняет указанное противоречивое влияние темпера-
туры на устойчивость золей. Повышение температуры, а значит,
и энергии броуновского движения препятствует оседанию частиц
и тем способствует повышению кинетической устойчивости.
Повышение кинетической энергии частиц способствует более час-
тым и эффективным столкновениям, преодолению сил отталки-
вания и, следовательно, слипанию частиц, т. е. ведет к пониже-
нию агрегативной устойчивости. Одновременно с понижением
агрегативной устойчивости уменьшается и кинетическая устой-
чивость, и в целом общая устойчивость коллоидной системы.
Исходя из сказанного можно было бы сделать неправильный
вывод о невозможности существования коллоидных растворов
в течение долгого времени. Ведь броуновское движение непре-
рывно и вечно, и оно приводит к очень частым столкновениям
коллоидных частиц. Опыт опровергает такой вывод. На самом
деле большинство лиофобных золей обладает заметной агрега-
тивной устойчивостью, которая обеспечивает время их сущест-
вования, измеряемое десятками лет. Следовательно, несмотря
на непрерывное столкновение, коллоидные частицы не слипаются,
325
не образуют более крупных агрегатов. Что же стабилизирует
коллоидные частицы, что мешает их столкновению?
В общем можно предположить, что двойной электрический
слой и диффузные ионы обеспечивают защитную оболочку
вокруг частицы и препятствуют их слипанию при столкновении.
Если это так, то частицы должны тем больше отталкиваться друг
от друга, чем выше дзета-потенциал. Тогда, повышая дзета-
потенциал (например, введением электролита-стабилизатора),
можно было бы повысить стабильность коллоидных растворов.
Однако ряд наблюдений привел к выводу, что иногда добав-
ка электролита-стабилизатора хотя и ведет к увеличению заряда
частицы и к повышению дзета-потенциала, но устойчивость
золя при этом не повышается, а понижается. В других случаях,
наоборот, с падением дзета-потенциала устойчивость золя все же
не снижается. Описаны такие явления, когда агрегативная
устойчивость золя сохраняется даже, когда дзета-потенциал
равен нулю.
Эти факты, а также некоторые теоретические соображения
приводят к выводу, что для полного объяснения устойчивости
дисперсных систем одного фактора — величины дзета-потенциа-
ла — совершенно недостаточно.
До настоящего времени вопрос б механизме стабилизации
окончательно не решен, особенно если рассматривать количе-
Рис. 126. Схематическое изображение сольватирован-
ной мицеллы:
1 — ядро; 2 — потенциалопределяющие ионы; 3 — сольват-
ная оболочка потенциалопределяющих ионов; 4 — сольват-
ная оболочка противоионов; 5 — сольватная оболочка диф-
фузных ионов
326
ственную сторону явления. Однако, по всей видимости, основная
причина устойчивости связана с процессом гидратации (сольва-
тации) .
Само по себе ядро мицеллы нерастворимо в данной среде и,
следовательно, не сольватировано. Ионы, адсорбированные на
поверхности ядра, и противоионы двойного электрического слоя
сольватированы (рис. 126). Благодаря этому вокруг ядра созда-
ется ионно-сольватная оболочка. Толщина ее зависит от распре-
деления ионов двойного слоя: чем больше противоионов нахо-
дится в диффузном слое, тем больше и толщина сольватной
оболочки. Сжатие двойного слоя уменьшает степень сольватации
ионов. В изоэлектрическом состоянии (дзета-потенциал равен
нулю) сольватная оболочка, вокруг ядра предельно тонка (по-
рядка 1О~10 м). Такие тонкие слои не защищают мицеллы от
слипания при столкновении, в результате начинается агрегация
частиц. Толщина сольватных слоев в устойчивых золях значи-
тельно больше и достигает 10“8 м.
Такими образом, оболочка из гидратированных противоионов
диффузного слоя вокруг коллоидной частицы является средством
защиты частиц от слипания и причиной агрегативной устой-
чивости гидрозолей в целом. Чем толще диффузный слой (т. е.
чем больше в нем гидратированных противоионов), тем плотнее
общая гидратная оболочка и тем стабильнее соответствующий
гидрозоль.
Следовательно, устанавливается прямая зависимость между
толщиной (плотностью) сольватных (гидратных) оболочек или,
иначе, степенью гидратации противоионов и значением дзета-
потенциала и агрегативной устойчивостью коллоидной системы.
2. КОАГУЛЯЦИЯ
Лиофобные коллоиды являются термодинамически неустойчи-
выми системами, существующими благодаря стабилизации за
счет возникновения защитных ионных или молекулярных слоев.
Следовательно, изменение состояния этих слоев может привести
к потере устойчивости и затем к выпадению дисперсной фазы.
Процесс слипания коллоидных частиц с образованием более крупных агре-
гатов (потеря агрегативной устойчивости) с последующей потерей кинетической
устойчивости называется коагуляцией.
Коагуляция коллоидных растворов может быть вызвана
различными факторами: 1) прибавлением электролитов; 2) на-
греванием или замораживанием; 3) механическим воздействием;
4) высокочастотными колебаниями; 5) ультрацентрифугирова-
нием.
Наиболее важным и изученным фактором коагуляции являет-
ся действие электролитов.
327
2.1. Действие электролитов
Установлен ряд эмпирических закономерностей воздействия
электролитов, которые известны под названием правил коа-
гуляции.
1. Любые электролиты могут вызвать коагуляцию, однако за-
метное воздействие они оказывают при достижении определенной
концентрации. Минимальная концентрация электролита, которая
вызывает коагуляцию, получила название порога коагу-
ляции; он обычно обозначается буквой у и выражается в
моль/м3. Порог коагуляции определяется по началу помутнения
раствора, по изменению окраски раствора или по началу .
выделения дисперсной фазы в осадок.
2. Коагулирующим действием обладает лишь тот ион элект-
ролита, заряд которого противоположен заряду потенциалопре-
деляющих ионов мицеллы, причем его коагулирующая способ-
ность выражается тем сильнее, чем выше заряд; эта законо-
мерность называется правилом значности или правилом
Шульце — Г арди. Если принять коагулирующую способ-
ность однозарядного иона за единицу, то коагулирующая спо-
собность двухзарядных ионов будет на порядок больше, а трех-
зарядного — на два порядка.
В табл. 48 в качестве примера приведены значения порогов
коагуляции для двух золей — сульфида мишьяка и гидроксида
железа.
Правило Шульца — Гарди имеет приближенный характер и
описывает действие ионов лишь неорганических соединений. Не-
которые органические однозарядные ионы (катионы морфина
и др.) обладают более сильным коагулирующим действием,
чем двухзарядные ионы. Это объясняется тем, что объемные
органические ионы обладают более сильной специфической
адсорбируемостью и легче входят во внутреннюю часть двойного
электрического слоя коллоидных частиц.
3. В ряду органических ионов коагулирующее действие воз-
растает с повышением адсорбционной способности.
Таблица 48. Значения порога коагуляции различными
электролитами для двух золей
Золь AS2S3 Золь Fe(OH)2
Электролит Коагулирую- щий ион (катион) у, моль/м3 Электролит Коагулирую- щий ион (анион) у, моль/м3
NaCl Na+ 51 КС1 сг 9,0
КС1 К+ 49,5 КВг Вг~ 12,5
MgCl2 Mg2+ 0,72 KI Г 10,0
СаС12 Са2+ 0,65 KNO3 NOT 12,0
ВаС12 Ва2+ 0,69 K2SO4 SO42- 0,205
А1С1з А13+ 0,092 КгСггО? Сг2О?“ 0,195
328
4. В ряду неорганических ионов одинаковой зарядности их
коагулирующая активность возрастает с уменьшением гидрата-
ции; например, в ряду однозарядных катионов и анионов коагу-
лирующая активность и гидратация изменяются следующим
образом:
возрастание коагулирующей активности
Li+ Na+ К+ Rb+ ~
возрастание степени гидратации
возрастание коагулирующей активности
С1~ Вг~ I" CNS~
•возрастание степени гидратации
называются лиотропными рядами или рядами Гоф-
Эти ряды
мейстера.
5. Очень часто началу коагуляции соответствует снижение
дзета-потенциала до критического значения (около 0,03 В).
6. В осадках, получаемых при коагуляции электролитами,
всегда присутствуют ионы, вызывающие ее; например, при коагу-
ляции золя сульфита мышьяка, частицы которого имеют отри-
цательный заряд, хлоридом бария в осадке содержится некоторое
число ионов Ва2+.
Совместное действие электролитов при коагуляции. При коа-
гуляции золей смесями электролитов они очень редко действуют
независимо, иначе говоря, аддитивного действия не наблюдается:
чаще происходит как бы противодействие их друг другу (антаго-
низм) либо усиление коагулирующего действия
На рис. 127 отображены три предельных
случая коагулирующего действия электроли-
тов. Для построения графика в качестве
координатных осей используют концентра-
ции коагулирующих электролитов. Каждая
точка на графике отвечает порогу коагу-
ляции, соответствующему отложенным по
осям концентрациям электролитов. Точки
Yi и у2 представляют собой пороговые
концентрации для каждого из двух электро-
литов в чистом виде.
Аддитивное действие электролитов. На
рис. 127 оно характеризуется прямой /,
соединяющей значения порогов коагуляции
yi и 72 каждым электролитом. Электролиты
действуют как бы независимо и их коагули-
рующее действие в смеси определяют по пра-
вилу простого сложения. Такое явление на-
блюдается, когда ионы-коагуляторы обла-
дают одинаковой зарядностью и близкой
степенью гидратации (смеси КС1 и KNO3
или NaCl и КС1).
(синергизм).
Рис,
действие электролитов
при коагуляции:
/— прямая, характери-
зующая аддитивное дей-
ствие электролитов; 2 —
кривая, характеризую-
щая антагонизм; 3 —
кривая, характеризую-
щая синергизм
л
Г, —в»
127. Совместное
329
Антагонизм электролитов. Иногда антагонизм проявляется в
такой мере, что в коагулирующей смеси содержание каждого
электролита может значительно превышать его собственную
пороговую концентрацию. Г. Фрейндлих считал, что причиной
антагонизма является способность одного иона понижать адсорб-
ционную способность, а следовательно, и коагулирующую силу
другого иона. Антагонизм наблюдается при коагуляции золей
Agl смесями А1(МОз)з и K2SO4, Т1(МОз)4 и Na2SO4.
Синергизм электролитов. Электролиты как бы усиливают
действие каждого из них, и для коагуляции золя их требуется
меньше, чем по правилу аддитивности. Это явление встречается
реже, чем антагонизм. Примером может служить смесь LiGl и
СаС1г в их действии на гидрозоль H2S.
Взаимная коагуляция золей. Еще в прошлом веке было
обнаружено, что при взаимодействии золей с разноименными
знаками зарядов частиц происходит коагуляция. Этот факт
объясняли действием противоположных знаков зарядов частиц
дисперсной фазы обоих золей. Таким образом, взаимную коагу-
цию рассматривали как частный случай коагуляции электроли-
тами, если один из золей принят в качестве электролита
с большим ионом-коагулятором. Так, например, положительно
заряженную коллоидную частицу золя Ее(ОН)з рассматривали
по отношению к отрицательной частице золя AS2S3 как ион-
коагулятор, прибавление которого снижает дзета-потенциал
частиц золя AS2S3. Естественно, что при этом золи должны быть
взяты в таком количественном соотношении, которое обеспечи-
вало бы более или менее полную нейтрализацию заряда
частиц.
В дальнейшем С. И. Соколовым, С. С. Воюцким и Б. В. Деря-
гиным было доказано, что возможен и другой механизм взаимной
коагуляции. Экспериментами было установлено, что взаимная
коагуляция коллоидных систем может наблюдаться и тогда,
когда частицы обоих золей несут электрический заряд одного
и того же знака. В этом случае причиной потери устойчивости
данной дисперсной системы в присутствии другой является
адсорбция стабилизатора данной системы поверхностью другой
системы и снижение вследствие этого концентрации стабилиза-
тора в золе данной системы.
Взаимная коагуляция золей широко распространена в приро-
де и играет важную роль в ряде технологических процессов.
Так, образование илистых отложений в дельтах рек связано
со смешением морской и речной воды. Ионы солей морской
воды адсорбируются на заряженных частицах коллоидов речной
воды, в результате чего происходит их коагуляция. Это процесс
приводит к скапливанию ила на дне. Большую роль играет
процесс взаимной коагуляции в образовании почвенных кол-
лоидов.
330
2.2. Коагуляция и дзета-потенциал
Различают два вида коагуляции коллоидных растворов
электролитами — концентрационную и нейтрали-
зационную.
Концентрационная коагуляция связана с увеличением кон-
центрации электролита, не вступающего в химическое взаимо-
действие с компонентами коллоидного раствора. Такие электро-
литы называют индифферентными; они не имеют ионов, способ-
ных достраивать ядро мицеллы и вступать в реакцию с потен-
циалопределяющими ионами. При увеличении концентрации
индифферентного электролита диффузный слой противоионов
мицеллы сжимается, переходя в адсорбционный слой. В резуль-
тате уменьшается электрокинетический потенциал и он может
стать равным нулю. Такое состояние коллоидной системы
называется изо эл е к т р и ч е с к и м. С уменьшением электро-
кинетического потенциала агрегативная устойчивость коллоидно-
го раствора снижается и при критическом значении дзета-потен-
циала начинается коагуляция. Термодинамический потенциал
при этом не изменяется.
При нейтрализационной коагуляции ионы прибавляемого
электролита нейтрализуют потенциалопределяющие ионы, умень-
шается термодинамический потенциал и соответственно умень-
шается и дзета-потенциал.
Когда в коллоидные системы вводят порциями электролиты,
содержащие многозарядные ионы с зарядом, противоположным
заряду частицы, золь сначала остается устойчивым, затем
в определенном интервале концентраций происходит коагуляция,
далее золь снова становится устойчивым и, наконец, при высоком
содержании электролита опять наступает коагуляция, уже окон-
чательная. Подобное явление могут вызывать и объемные орга-
нические ионы красителей или алкалоидов.
Рассмотрим механизм этого явления (рис. 128). Вначале со-
держание введенного электролита многозарядных ионов недоста-
точно, чтобы скоагулировать золь; при этом дзета-потенциал
частиц выше критического его значения. Дальнейшее добавле-
ние электролита приводит к тому,
что его ионы начинают проявлять
коагулирующее действие. Указан-
ный интервал концентраций отве-
чает значениям электролитиче-
ского потенциала частиц от крити-
ческого дзета-потенциала gKp одно-
го знака до £кр противоположного
знака. При последующем добав-
лении многозарядных ионов начи-
нается перезарядка коллоидных
частиц и золь становится опять
устойчивым. В этой зоне дзета-
Рис. 128. Чередование зон. устойчи-
вости (заштрихованы) при введе-
нии в золь электролита
331
потенциал снова выше критического значения, но его знак обра-
тен знаку дзета-потенциала частиц исходного золя. Если продол-
жать добавлять электролит, то многозарядные ионы снова и окон-
чательно коагулируют золь.
Влияние различных концентраций многозарядных ионов было
отмечено давно, и оно получило название явления неправильных
рядов. В то время перезарядка частиц золей была мало иссле-
дована и только потом выяснилось, что чередование зон устой-
чивости, и неустойчивости при добавлении многозарядных ионов
вполне закономерно и поэтому термин неправильные
ряды должен рассматриваться как условный.
2.3. Теория коагуляции электролитами
Выяснение механизма коагуляции тесно связано с установ-
лением причины устойчивости самого золя и его существования.
Поэтому разрабатывались и были предложены различные теории
коагуляции электролитами, большинство из которых, рассматри-
вающие один из факторов коагуляции, постепенно утратили
свое значение. В настоящее время общепринятой является
физическая теория коагуляции электролитами, которая основана
на общих принципах статистической физики, теории молекуляр-
ных сил и теории растворов.
Все факторы коагуляции, рассмотренные в ранних теориях,
приводят к перестройке двойного электрического слоя, при кото-
ром происходит его сжатие. Физическая теория коагуляции
объясняет, почему сокращение толщины двойного слоя вызывает
коагуляцию.
Для коагуляции коллоидные частицы должны сблизиться
на такое расстояние, при котором энергия их взаимного моле-
кулярного притяжения, обусловленного ван-дер-ваальсовыми
силами, была бы больше энергии теплового (броуновского) дви-
жения. Выполнение этого условия для сферических частиц воз-
можно, если наименьшее расстояние между поверхностями час-
тиц станет малым по сравнению с радиусами частиц.
Сближению коллоидных частиц на достаточно малые рас-
стояния препятствует электростатическое отталкивание между
их двойными электрическими слоями. Б. В. Дерягин установил,
что эти силы электростатического отталкивания возникают лишь
при перекрытии ионных атмосфер коллоидных частиц и А2
(рис. 129). Внешняя оболочка двойного электрического слоя
в значительной мере экранирует заряд коллоидных частиц,
и до тех пор, пока диффузные слои коллоидных частиц практиче-
ски не перекроются, электростатическое отталкивание не имеет
существенного значения. Когда происходит сближение частиц
в зоне перекрытия ионных атмосфер Ао (рис. 129), происходит
перераспределение ионов с местным изменением их концентра-
ции, вследствие чего появляются дополнительные электроста-
тические силы, приводящие к результативному отталкиванию
332
коллоидных частиц. Если же тол-
щина двойного слоя уменьшается,
то коллоидные частицы получают
возможность более тесного сбли-
жения без возникновения силы от-
талкивания. При достаточно ма-
лых расстояниях силы межчастич-
ного притяжения преобладают над
броуновским движением частиц, и
в этот момент происходит слипа-
ние, или коагуляция, частиц.
Суммарные энергии взаимо-
действия W двух коллоидных ча-
стиц в зависимости от расстояния
между их поверхностями изме-
няются по кривой 1 (рис. 130),
где положительные значения W
соответствуют отталкиванию, а
отрицательные — притяжению
частиц. Такие кривые, получае-
мые суммированием рассчитан-
ных сил межчастичного притяже-
ния и электростатического оттал-
кивания, называются потен-
циальными кривыми.
На рис. 130 виден ‘барьер от-
талкивания S на расстояние го,
который препятствует сближению
частиц. При достаточно высокой
концентрации электролитов барь-
ер S устраняется и начинают пре-
Рис. 129. Перекрытие ионных атмо-
сфер двух коллоидных частиц (по
Дерягину)
Рис. 130. Энергия взаимодействия
частиц в зависимости от расстояния
между ними:
1 — устойчивые системы; 2 — неустой-
чивые системы
обладать силы притяжения. Частицы сближаются при коагуля-
ции до расстояния Гг- При этом вся потенциальная кривая ле-
жит в области отрицательных значений W (кривая 2).
Б. В. Дерягин и Л. Д. Ландау установили, что порог коагу-
ляции определяется следующим уравнением:
ceW
У Я'еУ
где С — константа; 8— диэлектрическая постоянная; А — кон-
станта ван-дер-ваальсового притяжения; е — заряд электрона;
z — зарядность коагулирующего иона; к — константа Больц-
мана.
Из этого уравнения видно, что значение порога коагуляции
обратно пропорционально z6.
Тиксотропия. Рассмотрение энергии взаимодействия двух
коллоидных частиц (рис. 130, кривая /) показывает, что на
больших расстояниях Г\ между частицами имеется неглубокий
ззз
минимум. Этот минимум, незначительный для обычных гидро-
фобных золей, становится весьма заметным для крупных асим-
метричных частиц, имеющих форму палочек или пластинок.
Для таких частиц энергия взаимодействия в точке М может быть
в несколько раз больше энергии броуновского движения, что
приводит к взаимному притяжению этих частиц на больших
расстояниях.
Таким образом, создается возможность возникновения слабо-
связанных структур с гелеобразным состоянием. Так как энергия
взаимного притяжения здесь довольно слаба, то эти структуры
легко разрушаются при простом встряхивании, однако через
некоторое время покоя образующийся золь снова превращается
в гель.
Это явление изотермического обратимого перехода золь
«=* гель получило название тиксотропии.
Тиксотропные структуры возникают лишь при определенных
концентрациях коллоидных частиц и электролитов, причем пе-
риод их застудневания — величина постоянная для каждой
данной системы.
Явление тиксотропии распространено; оно наблюдается в золях V2O5, WO3,
Ре2Оз, в различных суспензиях бентонита, в растворе вируса табачной мозаики,
а также в протоплазме клеток при их делении.
Тиксотропия играет отрицательную роль в земледелии, так как тиксотропные
почвы плохо проницаемы для воды и воздуха, поэтому в них часто развиваются
восстановительные процессы и оглеение. Улучшения физических свойств таких
почв можно достичь' высушиванием, внесением коагуляторов с минеральными
удобрениями: кальция, магния.
Тиксотропия резко выражена в почвах тундровой зоны, а также иногда
в плывунах. Поверхность тиксотропной почвы ничем не отличается от почвы,
не обладающей тиксотропными свойствами. При сотрясении или механической
обработке выступает вода и почва становится текучей, однако через некоторое
время вязкость и затвердевание увеличиваются.
2.4. Кинетика коагуляции
Процесс коагуляции всегда совершается в измеримый отрезок
времени и поэтому, естественно, был поставлен вопрос и о коли-
чественном измерении скорости коагуляции, которая зависит не
только от концентрации самого золя, но и от концентрации
электролита-коагулятора.
График, характеризующий изменение скорости коагуляции
в зависимости от концентрации электролита, представлен на
рис. 131. На кривой Оабв отрезок Оа отвечает периоду скрытой
коагуляции. В точке а, которой отвечает значение критического
дзета-потенциала, начинается явная коагуляция. На отрезке аб
скорость коагуляции сильно зависит от концентрации электро-
лита, причем скорость сначала быстро увеличивается, а затем
(начиная с точки б) становится постоянной и, значит, незави-
симой от концентрации электролита; в точке б значение дзета-
потенциала равно нулю и отрезок бв соответствует быстрой
коагуляции.
334
Теория быстрой коагуля-
ции была разработана
М. Смолуховским. Она осно-
вана на предположении, что
в области быстрой коагуля-
ции любое столкновение ча-
стиц приводит к их слипа-
нию и что все мицеллы золя
до коагуляции имеют одина-
ковый размер и сфериче-
скую форму. Число столкно-
вений в единицу времени оп-
ределяется интенсивностью
броуновского движения ча-
Рис. 131. Влияние концентрации электро-
лита на скорость коагуляции
СТИЦ.
М. Смолуховский считал, что вокруг частиц существует
силовое поле, которое действует в пределах с радиусом (7?-|-а),
где 7? — радиус шарообразной частицы, а — расстояние, на кото-
ром от поверхности действуют силы притяжения. По М. Смолу-
ховскому, для коагуляции достаточно, чтобы силовые поля двух
частиц перекрывались. Для количественного описания коагуля-
ции М. Смолуховский использовал уравнения формальной хими-
ческой кинетики. При этом были использованы следующие допу-
щения:
1) при столкновении двух первичных частиц, из которых со-
стоял золь до начала коагуляции, появляются более крупные
частицы, называемые вторичными;
2) при столкновении вторичных частиц с первичными появ-
ляются третичные частицы;
3) частицы более высокого, четвертого, порядка образуются
либо в результате слипания двух вторичных частиц, либо третич-
ной частицы с первичной; подобные процессы приводят к обра-
зованию частиц большей кратности.
Если обозначить через п,\, nz, n%, ..., nz-— соответственно кон-
центрации первичных, вторичных, третичных, z-й кратности ча-
стиц, то общая концентрация частиц всех порядков (кратности)
в системе будет равна
/I — S tit П1 -|- П2 “р ^3 “Н • • • “И ^1-
Так как при любом столкновении частиц уменьшается ча-
стичная концентрация, в системе, уравнение скорости этого про-
цесса можно рассматривать как реакцию второго порядка:
(14.1)
где k — константа скорости.
М. Смолуховский установил, что на константу k влияют
интенсивность броуновского движения, мерой которого служит
коэффициент диффузии Z), и радиус силового поля R-\-a. Зави-
335
132.
Коагуляционные
Рис. 1
кривые
симость между ними можно предста-
вить следующим образом:
k — &siD(R + a)-
М. Смолуховский вывел также
уравнение, позволяющее оценить в
процессе коагуляции изменение отно-
сительно числа частиц любой кратно-
сти. Если кратность ту то число ча-
стиц с этой кратностью
На рис. 132 представлены кривые,
характеризующие изменение относи-
тельно числа частиц во времени от на-
чала коагуляции. Как следует из рисунка, общее число частиц
непрерывно уменьшается, причем число первичных частиц убы- s
вает с наибольшей скоростью. Число вторичных частиц проходит
через максимум; величина максимума падает с повышением
кратности частиц.
М. Смолуховский развил теорию медленной коагуляции ча-
стиц, сохраняющих некоторую стабильность, предполагая, что
не каждое столкновение частиц приводит к их слипанию. Если
эффективной является лишь часть столкновений а, то константа
скорости реакции уравнения (14.1) равна
k = 4n,DRa.
2.5. Старение золей и пептизация /
Лиофобные коллоиды, характеризующиеся слабым взаимо-
действием дисперсной фазы и дисперсионной среды, характеризу-
ются неустойчивостью и склонностью к уменьшению дисперсно-
сти со временем. Избыток свободной поверхностной энергии,
полученной частицами при их образовании, является, согласно
второму началу термодинамики, основной причиной перехода в
более устойчивое состояние, которое определяется укрупнением
частиц.
Самопроизвольный процесс укрупнения частиц (уменьшения степени дис-
персности) в лиофобных золях называется старением или автокоагуляцией.
Скорость автокоагуляции гораздо медленнее, чем коагуляция
под воздействием электролитов. Известны, например,'золи золота,
сохраняющиеся без видимых изменений десятилетия. Причиной
старения является медленно совершающийся процесс перекри-
сталлизации вещества ядра и изменение дзета-потенциала при
некоторых столкновениях частиц.
Пептизацией (дезагрегацией) называется расщепление
336
коагулята (коагеля) на первичные частицы с образованием
золя. Она противоположна коагуляции, поэтому этот процесс
называют также декоагуляцией. Термин пептизация был введен
на основании внешнего сходства этого процесса с переводом
белка в раствор в результате расщепления его на пептоны под
действием фермента пепсина. Термин этот не вполне удачен и
поэтому в современной литературе все чаще применяют термин
дезагрегация.
Пептизация возможна лишь тогда, когда структура частиц
в коагуляте не<изменена по сравнению с первоначальной и они
не сращиваются друг с другом. Поэтому лучше всего пептизи-
руются свежеосажденные рыхлые осадки, содержащие воду, на-
пример Ре(ОН)з, А1(ОН)з. Чтобы пептизировать коагулят, его
необходимо отмыть от коагулирующего электролита и ввести в
среду стабилизатор. В качестве стабилизаторов наиболее употре-
бительны электролиты, содержащие ионы, которые выступают в
качестве потенциалопределяющих ионов на поверхности частиц.
Их называют пептизирующими электролитами или пептизато-
рами. Так, например, для коагулята гидроксида железа (III)
пептизаторами являются FeCU, НО.
Аналогично коагуляции при пептизации не наблюдается сте-
хиометрических соотношений между содержанием пептизатора и
пептизированного осадка. Для пептизации осадка не требуется,
чтобы вся поверхность частиц была покрыта слоем адсорбиро-
ванного пептизатора; часто достаточно, чтобы пептизатором
было покрыто 10...25% площади пептизируемого осадка.
Скорость пептизации (при данном содержании пептизатора)
обычно возрастает с увеличением температуры и при переме-
шивании. Процесс пептизации вначале идет с большой скоро-
стью, а потом постепенно замедляется.
Была установлена эмпирическая закономерность (называе-
мая иногда правилом осадка), заключающаяся в том,
что при постоянном содержании пептизатора увеличение массы
осадка, взятого для растворения, приводит к увеличению, а за-
тем к уменьшению массы осадка, перешедшей в раствор. Эта
закономерность, иллюстрируемая графиком (рис. 133), может
быть объяснена следующим образом,
одной частицы осадка требуется неко-
торое минимальное содержание пепти-
затора, при введении первых порций
осадка пептизатор оказывается в из-
бытке и осадок легко переходит в золь.
По мере добавления осадка относи-
тельное содержание пептизатора ста-
новится все меньше и меньше, что при-
водит к снижению содержания декоагу-
лята. Затем, когда пептизируемого ве-
Так как для пептизации
Рис. 133. График, иллюстри-
рующий правило осадка
щества в системе становится много,
пептизатора не хватает; осадок уже
12—696
337
не растворяется и даже часть уже растворенного осадка может
снова коагулировать.
Пептизация играет значительную роль в почвенных процессах. Так, в черно-
земных почвах коллоидные частицы содержат в диффузном слое преимущественно
ионы Са2+ и Mg2+, что обусловливает небольшое значение дзета-потенциала,
и почвенные коллоиды находятся в скоагулированном состоянии. Обработка
почвы раствором NaCI приводит к замещению ионов Са2+ и Mg2+ в диффузном..
слое на Na+. В результате происходит пептизация с образованием золя, кото-
рый легко вымывается из верхних в нижние горизонты. При этом почва теряет
ценные агрономические свойства. В солонцовых почвах, которые содержат зна-
чительное число ионов натрия, этот процесс, идущий спонтанно, резко ухудшает
структуру почвы.
Процесс пептизации с помощью NaCI и последующей промывкой почв
водой используется в качестве метода выделения почвенных коллоидов.
2.6. Защитное действие молекулярных
адсорбирующих слоев
В лиофобных золях поверхность коллоидных частиц является
резкой границей раздела двух фаз и обладает свободной поверх-
ностной энергией, определяющей возникновение адсорбционных
слоев на поверхности. Эти слои могут быть образованы молеку-
лами поверхностно-активных веществ на поверхности коллоид-
ных частиц. Но при этом большое значение имеет характер
ориентации поверхностно-активных молекул в адсорбционном
слое. Адсорбционные слои могут покрывать не всю поверхность
частиц. Часто стабилизация достигается при покрытии моно-
слоем всего 40...60% поверхности коллоидных частиц, когда .
защитный слой имеет прерывный (в форме островков) характер. -
Однако максимальная устойчивость коллоидных систем до-
стигается именно при образовании полного мономолекулярного
слоя. Например, при добавлении желатины и некоторых других
белков к золям или суспензиям кварца устойчивость системы
обеспечивалась при толщине адсорбционного слоя около 1 нм,
что соответствует сплошному мономолекулярному слою. В таком
состоянии молекулы желатины образуются двухмерные гелеоб-
разные структуры с большим числом гидратированных полярных
групп. Подобные адсорбционные слои способны наиболее эффек-
тивно защищать коллоидные частицы от возможности слипания.
На рис. 134 схематически изображен процесс осаждения золя
золота под действием электролита и защитное коллоидное дейст-
вие желатины на золь. Частица золя золота в результате адсорб-
ции анионов приобретает заряд. В отсутствие защитного коллои-
да эта частица не обладает защитной оболочкой из молекул
растворителя и поэтому при добавлении в раствор хлорида нат-
рия легко осаждается. Ионы натрия нейтрализуют заряд золя,
частицы золя уже не отталкивают друг друга и агрегируют.
Если к золю добавить желатину, белок прикрепляется к
частицам золя и образует вокруг них защитную пленку. Ионная
поверхность желатины сообщает частицам дополнительный за-
338
Частица
золя
Агрегатная
форма
+Ма
Желатина
\
Пленка
желатины
Ncfcr
Na
'Водная
пленка
Частица не
^осаждается
Рис. 134. Защитное действие
желатины на золь золота
ряд, как положительный, так и отрицательный. Поверхность
белка адсорбирует молекулы воды, что обеспечивает частицам
золя золота добавочную защиту. Добавление хлорида натрия
уменьшает заряд золя, но не столь эффективно, как это наблю-
далось в отсутствие защитного коллоида. В результате частица
не осаждается.
Р. Зигмонди предложил количественное определение защит-
ного действия того или иного защитного коллоида — так назы-
ваемое золотое число. Известно, что коллоидное золото,
весьма чувствительное к прибавкам электролитов, в высокодис-
персном состоянии имеет красный цвет. При уменьшении степени
дисперсности золь золота приобретает голубой цвет.
Золотое число выражается количеством миллиграммов защитного коллоида,
которое достаточно, чтобы предотвратить изменение цвета 10 мл красного золя
золота в голубой при прибавлении 1 мл 10%-ного раствора хлорида натрия.
Чем меньше золотое число, тем больше защитное действие
коллоида. Различные защитные коллоиды обладают отличаю-
щимся защитным действием и, следовательно, характеризуются
различным золотым числом.
Было определено также защитное действие в отношении
золей серебра (серебряное число), конго рубинового (рубиновое
число), серы и т. д. (табл. 49).
Явление защиты играет важную роль в ряде физиологических процессов.
Так,, например, защитные вещества белкового характера удерживают в мелко-
дисперсном состоянии находящиеся в крови труднорастворимые фосфат и кар-
бонат кальция. При некоторых заболеваниях содержание защитных веществ
в крови понижается, что приводит к выпадению указанных солей в осадок (обра-
зование камней в почках, в печени, отложение солей в суставах). Многие лекар-
ственные вещества являются защитными золями (калларгон, протаргол и др.).
В фотографии используют светочувствительные коллоидные препараты бромида
серебра, защищенные желатиной. Широко применяется желатина как защитный
коллоид в пищевой промышленности.
В почвах содержатся высокомолекулярные органические соединения, назы-
12* 339
Таблица 49. Защитное действие различных коллоидов
Защитный коллоид Число
золотое серебряное рубиновое серное
Желатина 0,01 0,035 2,5 0,00012
Казеинат натрия 0,01 — 0,4 —-
Гемоглобин 0,03...0,07 — 0,8 —
Яичный альбумин 2,5 1,5 2,0 0,025
Гуммиарабик 0,5 1,25 — 0,024
Декстрин 20 100 — 0,125
Картофельный крахмал 20 — 20 —
Сапонин 115 35 — 0,015
ваемые гумусовыми веществами (гумус). Как полагают, гумус, находящийся
в растворенном состоянии, оказывает защитное действие по отношению к поч-
венным коллоидам. Такие содержащиеся в почве коллоиды, как гидроксид
железа (III), гидроксид алюминия,и др., не будучи защищены гумусом, коагу-
лируют в присутствии даже небольшого содержания электролитов. В почвах,
содержащих высокодисперсный (так называемый кислый) гумус, коллоиды,
защищенные гумусом, не коагулируют и, находясь в состоянии золей, вымыва-
ются из почвы, что лишает ее наиболее ценной составной части.
2.7. Роль процессов коагуляции в образовании почв
Почвы образуются при разрушении горных пород в результате выветривания,
выщелачивания и гидролиза. Эти процессы приводят к образованию нераствори-
мых оксидов типа SiO2, А12О3, Ге20з (точнее — гидроксидов) и растворимых
типа МО, М2О (где М — металл). В дальнейшем в процессе взаимной коагуля-
ции образуются структурированные коагуляты, близкие по свойствам к гелям,
называемые коагелями. Эти коллоидно-химические процессы определяют
все многообразие существующих типов почв.
Например, подзолистые почвы, типичные для средних районов нашей страны,
образуются в условиях малого содержания органического вещества и большой
влажности, которая способствует вымыванию оксидов основного характера (МО
и МгО). Частицы оксидов типа М2О3, защищенные гуминовыми кислотами, так-
же вымываются в этих условиях. Остающиеся коагели характеризуются высоким
содержанием SiO2 и малым содержанием питательных веществ, необходимых
для растений.
Черноземные почвы средней полосы РСФСР образуются в условиях малой
влажности. В этих условиях ионы Са2+ и Mg2+ не вымываются и, взаимодейст-
вуя с гуминовыми кислотами, образуют нерастворимые высокомолекулярные
коллоидные частицы — гуматы кальция и магния. В процессе взаимной коагу-
ляции положительно заряженных частиц М2О3 с отрицательно заряженными
гуматами и SiO2 возникают структурированные коагели, обусловливающие обра-
зование плодородной почвы.
Возникновение иловых почв в дельтах рек также связано с коагуляционными
процессами. Так, дельта Нила образуется в результате слияния двух рек —
Белого и Голубого Нила. Воды Белого Нила, вытекающие из болот, несут
много органических веществ, частично защищающих минеральные частицы.
Благодаря защитному действию гуматов высокодисперсная система весьма устой-
чива, и воды Белого Нила на всем его протяжении характеризуются значительной
мутностью. Голубой Нил, стекая с горных хребтов Эфиопии, содержит много
минеральных солей, вызывающих коагуляцию и осаждение гидрофобных мине-
ральных частиц, поэтому воды Голубого Нила совершенно прозрачны. После
слияния двух рек вода Нила продолжает оставаться мутной, так как концентра-
340
ция солей в воде Голубого Нила не достигает порога коагуляции, соответст-
вующего сильно гидрофилизированным частицам, содержащимся в воде Белого
Нила. Коагуляция наступает лишь в устье, где речная вода встречается с соле-
ными водами Средиземного моря и остановка течения способствует седимента-
ции коагулированных агрегатов, приводящей к образованию плодородной
дельты.
Рассмотрев лиофобные коллоиды, можно определить их об-
щие характеристики.
1. Всякий лиофобный зол^ образуется с затратой работы.
В результате этого возникающая система обладает избытком
свободной энергии в виде свободной поверхностной энергии.
Отсюда. следует термодинамическая неустойчивость лиофобных
коллоидов.
2. Лиофобные золи имеют мицеллярное строение, подразуме-
вающее наличие кристаллического ядра в дисперсионной среде.
Таким образом, гетерогенность состава является отличительной
характеристикой лиофобных коллоидов.
3. Лиофобные золи всегда находятся в неустойчивом равно-
весии и, являясь системами, агрегативно неустойчивыми, в прин-
ципе не могут без стабилизатора сохранять постоянство мицел-
лярной концентрации. Процесс слипания частиц является само-
произвольным процессом.
4. Так как лиофобные коллоиды являются псевдорастворами,
то для них наблюдается необратимость процессов, связанных
с изменением температуры, давления и состава.
5. Все частицы лиофобных золей несут электрический заряд,
обусловленный присутствием стабилизатора.
6. Так как лиофобные золи могут существовать без защиты
только в ничтожно малых концентрациях, они способны прояв-
лять в очень слабой степени диффузионные и осмотические
свойства, а значения вязкости мало отличаются от вязкости чис-
той дисперсионной среды.
Контрольные вопросы
1. Какие процессы протекают при нагревании большинства растворов лио-
фобных коллоидов? Как эти процессы влияют на устойчивость?
2. Почему правило Шульце — Гарда выполняется не во всех случаях?
3. В чем причина автокоагуляции и какова скорость этого процесса в срав-
нении с коагуляцией?
4. Если имеются два одинаковых по составу, но разных «по возрасту» ко-
агулянта, то какой из них будет легче пептизировать?
5. Какова роль пептизатора в почвенных процессах?
6. Какова роль защитного действия коллоидов в почвенных процессах?
7. Сольватировано ли само ядро мицеллы?
Глава XV
МИКРОГЕТЕРОГЕННЫЕ СИСТЕМЫ
1. СУСПЕНЗИИ
Суспензии — очень распространенные микрогетерогенные си-
стемы с размером частиц 10~7...10“5. К ним относятся глинистые,
цементные и известковые «растворы», масляные краски. Суспен-
зии играют важную роль в производстве инсектицидов и фунги-
цидов. Велико значение их в почвенной агротехнике.
Отличаясь от лиофобных коллоидов в основном только более
низкой степенью дисперсности, суспензии, в принципе, могут быть
получены как конденсационными, так и дисперсными методами.
Однако практически их получают путем диспергирования нерас-
творимых твердых веществ в жидкой среде или взмучиванием
в этой среде предварительно полученного порошка. 7
Благодаря низкой степени дисперсности в суспензиях слабо
проявляется или отсутствует такое молекулярно-кинетическое
свойство, как броуновское движение, а значит, и диффузия.
Осмотическое давление, весьма слабо выраженное в лиофобных
коллоидах, в суспензиях практически не обнаруживается, так
как частичная концентрация в них еще меньше, чем в лиофобных
коллоидах. Вязкость разбавленных суспензий мало отличается
от вязкости дисперсионной среды. Высококонцентрированные
суспензии (пасты) имеют свойства структурированных систем
и характеризуются высокой вязкостью.
Суспензии не проявляют светорассеяния, и к ним неприменим
закон Релея, так как размер частиц в суспензиях исключает
возможность дифракции.
Вследствие низкой степени дисперсности суспензии являются
кинетически неустойчивыми системами (они легко седименти-
руются), а для достижения агрегативной устойчивости необхо-
димо выполнение по крайней мере одного из двух условий:
1) смачиваемость поверхности частиц дисперсной фазы диспер-
сионной средой, 2) наличие стабилизатора. Добавляемый ста-
билизатор вводят либо в виде ионов, заряжающих и стабилизи-
рующих частицы суспензии, либо в виде поверхностно-активных
веществ, либо в виде защитного высокомолекулярного соеди-
нения. Если стабилизатор отсутствует, но частицы суспензии
хорошо смачиваются дисперсионной средой, то на их поверхно-
сти образуется сольватная оболочка, обладающая упругими
342
свойствами и препятствующая со-
единению частиц в крупные агре-
гаты.
Примером агрегативно-устойчи-
вых суспензий без стабилизатора с
сольватационным механизмом устой-
чивости являются суспензии^кварца
в воде и сажи в бензоле. Так как
кварц хорошо смачивается водой,
а сажа — бензолом, эти суспензии
агрегативно-устойчивы без третьего
компонента — стабилизатора. Если
Рис. 135. Стабилизация суспен-
зии сажи в воде поверхностно-
активным веществом
заменить дисперсионную среду, ис-
ключив тем самым смачивание (например, размешать порошок
сажи в воде), то получается агрегативно-неустойчивая система.
При этом частицы сажи с водой не смачиваются, гидратная обо-
лочка на поверхности частиц не образуется и незащищенные
частички легко соединяются друг с другом. Такой суспензии
можно придать устойчивость, вводя в нее третий компонент —
стабилизатор — поверхностно-активное вещество, растворимое
в дисперсионной среде. Молекулы поверхностно-активного веще-
ства, адсорбируясь на частицах сажи неполярными группами,
сообщают поверхности частиц свойство смачиваться водой, т. е.
гидрофилизируют ее (рис. 135). В результате смачивания обра-
зуется гидратная оболочка, которая обеспечивает агрегативную
устойчивость суспензии. Неустойчивую суспензию тоже можно
сделать устойчивой, если добавить в дисперсионную среду рас-
творимое в ней поверхностно-активное вещество (стеариновую
или олеиновую кислоту). Таким образом, механизм стабилиза-
ций носит адсорбционный характер.
Если в качестве стабилизаторов в суспензию добавляют высо-
комолекулярные соединения, механизм их действия аналогичен
механизму защиты лиофобных золей. При этом частицы суспен-
зии окружаются длинными цепочкообразными макромолекулами,
которые образуют защитный слой.
Высокомолекулярные соединения применяют также в каче-
стве стабилизаторов концентрированных суспензий. При этом
в суспензиях образуются структурные сетки, захватывающие
большие объемы дисперсионной жидкости, что приводит к резкому
увеличению вязкости системы. Стабилизатор — защитный поли-
мер — образует на поверхности частиц суспензии механически
прочные студнеобразные пленки.
2. ЭМУЛЬСИИ
Эмульсия — система, состоящая из двух жидких фаз, одна из
которых диспергирована в виде капелек в другой. Жидкость,
раздробленная на капельки, называется дисперсной фазой, а
жидкость, заполняющая объем между капельками, — диспер-
сионной средой. Для существования устойчивой эмульсии необ-
343
ходимо, чтобы жидкости, образующие эмульсию, были практиче-
ски взаимно нерастворимы или обладали достаточно малой
растворимостью. Размер частиц дисперсной фазы эмульсий ко-
леблется в пределах от 10-7 до 10“^ м, и поэтому их можно
отнести к микрогетерогенным системам.
Характерной чертой эмульсии является то, что в зависимости
от условий возникновения любая из двух жидкостей, образую-
щих дисперсионную систему, может оказаться как дисперсной
фазой, так и дисперсионной средой. Наиболее частый случай —
эмульсия воды (В) и нерастворимой в ней органической жидко-
сти (например, масло, бензол, хлороформ), которую условно
называют маслом (М). Возможны два типа таких эмульсий:
эмульсии, в которых дисперсной фазой является масло, и эмуль-
сии с водной дисперсной фазой. Первый тип эмульсии называют
эмульсиями масла в воде (сокращенно М/В) или эмуль-
сиями первого рода (прямые). Второй тип — эмульсии
воды в масле (В/М) или эмульсии второго рода
(обратные).
Чтобы выяснить, какая из двух4 жидкостей является дисперс-
ной фазой эмульсии, чаще всего применяют кондуктометриче-
ский метод. Известно, что удельная электрическая проводимость
воды и ее растворов значительно больше (в 1014 раз) удельной
электрической проводимости нерастворимых в ней органических
жидкостей. Электрическая проводимость дисперсной системы по
значению близка к электрической проводимости дисперсионной
среды. Поэтому, если электрическая проводимость эмульсии
достаточно высока, это означает, что присутствует эмульсия
типа М/В, а в случае низкой электрической проводимости —
В/М. Для установления типа эмульсий используют также метод
флуоресценции. Эмульсии В/М под действием ультрафиолетового
излучения приобретают видимую в темной камере окраску; это
отличает их от эмульсий М/В, которые обычно не флуоресци-
руют.
Эмульсии получают методом механического диспергирования,
хотя, в принципе, возможно использование и методов конденса-
ции. Для диспергирования применяют различные мешалки, сме-
сители, гомогенизаторы, коллоидные мельницы. Высокодиспер-
сные эмульсии часто получают способом ультразвукового дис-
пергирования.
В зависимости от концентрации дисперсной фазы различают
эмульсии разбавленные, концентрированные и высококонцентри-
рованные. К разбавленным эмульсиям относят эмульсии, содер-
жащие не больше 0,1% (об.) дисперсной фазы. Концентриро-
ванными считают эмульсии с содержанием дисперсной фазы не
более 74% (об.), в которых сохраняется сферическая форма
частиц. Эмульсии с содержанием дисперсной фазы больше 74%
(об.) называют высококонцентрированными.
Геометрическая форма частиц в высококонцентрированных
эмульсиях может значительно. отличаться от шарообразной.
344
Рис. 136. Микрофотография кон-
центрированной эмульсии
На рис. 136 показана микрофото-
графия концентрированной эмуль-
сии воды в каучуке, которая ха-
рактеризуется сильной деформа-
цией шариков воды с образова-
нием плоских участков поверхно-
сти. Такие плоские поверхности
соприкосновения между соседни-
ми частицами наблюдаются и для
других высококонцентрированных
эмульсий. Частицы имеют при
этом форму различных непра-
вильных многогранников и разде-
ляются тонкой пленкой диспер-
сионной среды.
Эмульсии являются седимен-
тационно неустойчивыми систе-
мами. Если дисперсная фаза и дисперсионная среда отличаются
по плотности, то возможна седиментация (или всплывание) ка-
пелек дисперсной фазы, т. е. нарушение однородности концентра-
ции. Агрегативная неустойчивость эмульсий проявляется в само-
произвольном слиянии капелек в дисперсной фазе — коалес-
ценция. Этот процесс может привести к разрушению эмуль-
сии и разделению ее на два жидких слоя.
Эмульсии, как и все микрогетерогенные системы, обладают
большой поверхностью раздела фаз. Образование поверхности
раздела всегда требует затраты работы, и работа эта тем боль-
ше, чем выше поверхностное натяжение на этой поверхности.
Поэтому легкость образования эмульсии и повышение ее устой-
чивости обеспечиваются введением веществ, крторые, адсорби-
руясь на поверхности раздела фаз, уменьшает поверхностное
натяжение на “
рами.
Бода
ней. Такие вещества называются эмульгато-
Наряду с понижением поверхностного
натяжения эмульгаторы могут стабилизиро-
вать эмульсию также и тем, что на поверх-
ности раздела образуется компактная плен-
ка из эмульгатора, обладающая известной
механической прочностью. Такие пленки за-
щищают частицы эмульсии от коалесцен-
ции. В отличие от суспензий этот фактор
может быть более важным, чем создание на
поверхности капелек электрических зарядов.
Рассмотрим подробнее механизм эмуль-
гирования на примере действия гидрофиль-
ного эмульгатора в эмульсии М/В. Поляр-
ные (дифильные) молекулы эмульгатора
(рис. 137) адсорбируются на поверхности
капельки масла, растворяясь неполярными
Масло
Рис. 137. Схематиче-
ское изображение
взаимодействия эмуль-
гатора и капли эмуль-
сии
345
углеводородными радикалами в масле, а полярными группами
в воде. В результате адсорбции эмульгатора поверхностное натя-
жение капли масла понижается, значит, значение поверхност-
ной энергии уменьшается. В результате система становится
устойчивее. Кроме того, образующаяся гидратная клетка прочно
свяжет капельки масла с дисперсионной средой (в данном слу-
чае с водой), что будет препятствовать коалесценции.
Если в качестве эмульгатора используют молекулы, способные
к диссоциации на ионы (например, мыло, представляющее собой
соль жирных кислот), то капелька масла зарядится отрицательно,
что приведет к еще большей стабильности эмульсии.
В качестве эмульгаторов могут быть использованы и твердые
вещества, применяемые в виде порошка. В этом случае механизм
эмульгирования связан со смачиваемостью порошка жидкостью,
входящей в состав эмульсии, и с образованием вокруг капелек,
прочных твердых оболочек (рис. 138, а, б). Гидрофильные эмуль-
гаторы, такие, как глина, мел, гипс, стабилизируют эмульсии типа
М/В, а гидрофобные (порошок сажи) — эмульсии типа В/М.
Специфическим свойством большинства эмульсий является
взаимное превращение эмульсий двух типов:
М/ВВ/М
Этот процесс, получивший название обращение фаз, при-'
водит к тому, что дисперсная фаза данной эмульсии становится
дисперсионной средой вновь образованной системы, а дисперсион-
ная среда данной эмульсии — дисперсной фазой вновь образован-
ной эмульсии. Осуществляется это введением поверхностно-ак-
тивного вещества, которое стабилизирует обратный тип эмульсии.
Например, эмульсию типа М/В, стабилизированную олеатом нат-
рия, переводят в эмульсию В/М введением избытка олеата каль-
ция. Эмульсию бензола в воде, стабилизированную мылом щелоч-
ного металла, превращают в эмульсию воды в бензоле прибавле-
нием к ней при встряхивании небольшой массы хлорида кальция.
Образующаяся при этом кальциевая соль мыла, хорошо раствори-
мая в бензоле, стабилизирует эмульсию воды в бензоле.
Зода
а)
Рис. 138. Схема действия твердого гидро-
фильного эмульгатора
Обращение эмульсии
иногда может быть вызвано
длительным механическим
воздействием. Так, сбивание
сливок (эмульсия типа М/В)
приводит к получению масла
(эмульсия типа В/М с ма-
лым содержанием воды в
виде дисперсной фазы).
Биологическое значение
эмульсий очень велико. На-
пример, молоко и яичный
белок представляют собой
эмульсии типа М/В. Усвое-
346
ние жиров в организме осуществляется через их эмульгирование
под влиянием желчи. Млечный сок каучуконосных растений
(латекс) также представляет собой эмульсию. Эмульсии находят
широкое применение в промышленности: битумные эмульсии для
асфальтирования, краски, «режущие эмульсии», используемые
при обработке металлов.
В сельскохозяйственном производстве эмульсии играют важ-
ную роль при изготовлении и применении средств опрыскивания.
Например, основным фактором, определяющим действие контакт-
ных инсектицидов, являются поверхностное натяжение, поверх-
ностная активность и смачивающая способность. Водные растворы
должны не только обладать токсичным действием, но и хорошо рас-
текаться по обрабатываемой поверхности.
Очень часто возникает необходимость разрушить эмульсию и,
выделить ее составные части. Для разбавленных эмульсий, стаби-
лизатором которых является электролит и устойчивость опреде-
ляется двойным ионным слоем, коалесценция может быть вызвана
теми же методами, что и коагуляция коллоидных растворов. Го-
раздо сложнее разрушить эмульсию, стабилизированную неионо-
генным эмульгатором.
Разрушение эмульсий (деэмульгирование) осуществляется
следующими способами:
1) разрушением защитных пленок сильными реагентами (на-
пример, кислотами);
2) вытеснением эмульгатора веществом, которое лучше адсор-
бируется, чем эмульгатор, но не является эмульгатором;
3) использованием механического воздействия: сбивание,
центрифугирование, фильтрование;
4) применением полей высокого напряжения;
5) нагреванием, ведущим к десорбции эмульгатора.
3. ПЕНЫ
Пенами называются высококонцентрированные грубодисперс-
ные системы, в которых дисперсионная среда — жидкость, а дис-
персная фаза — газ.
Пузырьки газов в пенах имеют размеры порядка миллимет-
ров (и даже сантиметров); они разделены тонкими жидкими плен-
ками, которые обладают размером коллоидных частиц. Газовые
пузырьки взаимно сдавливают друг друга, теряют правильную
сферическую форму, а сама пена приобретает ячеистую сотообраз-
ную структуру (рис. 139).
Как правило, пены получают с помощью диспергационных ме-
тодов: интенсивное встряхивание или перемешивание жидкости,
продавливание газа в жидкость через пористые фильтры, Реже
используют конденсационные методы.
Пены характеризуются следующими показателями: 1) пенисто-
стью, т. е. отношением объема пены к объему жидкости в виде
пленок; 2) дисперсностью (средним размером пузырьков и средней
347
толщиной жидкостных пле-
нок) ; устойчивостью (опре-
деляется по времени суще-
ствования пен).
В настоящее время нет
единой теории устойчивости
пен. Мера устойчивости пе-
ны определяется временем
ее жизни, т. е. временем от
момента ее образования до
самопроизвольного разруше-
ния. Устойчивую пену мож-
но получить только в при-
сутствии стабилизатора —
пенообразователя, от природы и концентрации которого в основ-
ном зависит время существования пены. В качестве пенообразо-
вателей используют поверхностно-активные вещества с доста-
точно длинными углеводородными радикалами. Пенообразова-
тель с длинной молекулярной цепью, адсорбируясь на границе
вода — воздух, образует высоковязкую структурированную плен-
ку, препятствующую стеканию жидкости. При этом толщина слоя
жидкости между пузырьками газа уменьшается медленно, и пена
может существовать длительное время.
С повышением температуры время жизни пены уменьшается,
так как при этом снижается адсорбция пенообразователя на гра-
нице фаз и уменьшается вязкость жидкости.
С увеличением вязкости жидкости устойчивость пены возраста-
ет. Увеличение испарения (если пенообразователь летучий) умень-
шает время жизни пены, так как концентрация его в поверхност-
ном слое определяет прочность пленки. Введение электролитов,
как правило, снижает время жизни пены.
Пена и пенообразование имеют важное практическое значение.
Всем известно положительное действие пены при удалении за-
грязнений с любых поверхностей. Стойкие пены используют при
тушении пожаров и во флотационных процессах. Пены обеспечи-
вают оптимальный технологический процесс в виноделии и в кон-
дитерском производстве.
Иногда образование пены приводит к нежелательным послед-
ствиям. Так, например, она мешает перемешиванию и выпарива-
нию жидкостей. Большой вред наносят пены, образующиеся в
сточных водах, которые содержат пенообразователи. Эти пены
покрывают слоем поверхность водоемов и, прекращая доступ кис-
лорода в воду, делают невозможным развитие в ней лтрбых орга-
низмов.
4. АЭРОЗОЛИ
Аэрозолями называются дисперсные системы, дисперсионной
средой которых является газ (воздух), а дисперсной фазой могут
быть твердые частицы или капельки жидкости.
348
Обычно аэрозоли классифицируют по агрегатному состоянию
дисперсной фазы. Аэрозоли с жидкой дисперсной фазой называют
туманами, с твердыми частицами — дымами. Аэрозоли с
твердой дисперсионной фазой, размеры частиц которых больше,
чем у дымов, называют обычно пылью.
Размер частиц аэрозолей лежит в пределах 10”7...10“4 м.
В табл. 50 приведены размеры частиц некоторых типичных аэро-
золей.
Таблица 50. Размеры частиц некоторых аэрозолей
Аэрозоль Размеры частиц, м Аэрозоль Размеры частиц, м
Туман Слоистые облака Дождевые облака 5-Ю-7 1 • 10~6...1 • 10~6 1-10~5...1-1(Г4 ZnO (дым) Табачный дым Топочный дым 5-10~8 1 • 10~7. 1 • 10~6 ЫО-Л.-ЫО-’
Кривая полидисперсности аэрозоля зависит от его происхожде-
ния и вторичных процессов, происходящих после его получения.
Форма частиц аэрозолей зависит от агрегатного состояния
вещества дисперсной фазы. В туманах капельки жидкости шаро-
образны, в дымах форма частиц может быть самой разнообразной:
игольчатой, пластинчатой, звездообразной.
Как и любые дисперсионные системы, аэрозоли образуются
двумя методами — конденсационным и диспергационным. К кон-
денсационному методу относится возникновение тумана при ох-
лаждении насыщенного пара.
При диспергационных методах получения аэрозолей твердые
или жидкие тела размельчаются обычно механическим путем, а за-
тем твердые частицы или жидкие капельки распределяются в газе.
Например, пневматическое распыление жидкостей осуществляется
с помощью так называемых аэрозольных баллончиков при полу-
чении парфюмерно-косметических аэрозолей, аэрозолей инсекти-
цидов, эмалей.
По оптическим свойствам аэрозоли очень близки к растворам
лиофобных коллоидов. В частности, для них также характерно
светорассеяние. Однако вследствие большой разницы в показате-
лях преломления газовой дисперсионной среды и жидкой или
твердой дисперсной фазы светорассеяние у аэрозолей более интен-
сивно, и они не пропускают свет. На этом свойстве аэрозолей осно-
вано применение маскировочных дымовых завес. Благодаря силь-
ному светорассеянию аэрозоли, находящиеся в верхних слоях ат-
мосферы, уменьшают интенсивность солнечной радиации и влияют
на климатические условия.
Для аэрозолей характерны специфические процессы, связан-
ные с их кинетическими свойствами: термофорез, фотофорез и
термопреципитация. Явление термофореза заключается в
движении частиц аэрозоля в направление снижения температуры.
349
Это объясняется тем, что на более нагретую сторону частицы моле-
кулы газа налетают с большей скоростью, чем на менее нагретую.
Фотофорез заключается в передвижении частиц при односто-
роннем их освещении. Термофорез и фотофорез имеют большое
значение в движении атмосферных аэрозолей, например при обра-
зовании облаков.
Термопреципитация представляет собой осаждение
частиц аэрозоля на холодных поверхностях. Это происходит пото-
му, что при взаимодействии с такими поверхностями частицы те-
ряют кинетическую энергию.
У частиц аэрозолей нет двойного электрического слоя, но в
определенных условиях они приобретают электрический заряд. За-
ряд частиц аэрозолей может появиться в результате трения при
их распылении или вследствие адсорбции на поверхности частиц
ионов газов, образующихся под действием космического излуче-
ния. В отличие от обычных коллоидных растворов, где заряд
частиц обусловлен адсорбцией ионов электролита и определяется
равновесием между частицей и окружающей средой, у аэрозолей
заряд частицы большей частью случаен. В общем все-таки наблю-
дается закономерность между дисперсностью и величиной заряда:
заряд частицы аэрозоля тем больше, чем больше ее размеры.
Электрические заряды отдельных частиц аэрозолей невелики и
поэтому они не могут определять агрегативную устойчивость
аэрозолей. При высокой дисперсности и кинетической устойчиво-
сти аэрозоли агрегативно неустойчивы. Для них характерна
быстрая коагуляция.
Практическое значение аэрозолей достаточно велико. Очень
широко применяют аэрозоли ядохимикатов в сельском хозяйстве
при опылении лесов и полей для борьбы с вредными насекомыми.
С помощью аэрозолей (дым костров) защищают фруктовые сады
от внезапных заморозков.
Контрольные вопросы
1. Сопоставьте кинетическую и агрегативную устойчивость суспензии с соот-
ветствующими характеристиками лиофобных коллоидов.
2. При рассмотрении эмульсии В/М и М/В трудно определить, какая из
двух жидкостей является дисперсной фазой. Какими методами можно это сде-
лать?
3. Как осуществить обращение фаз эмульсии?
4. Какова роль эмульсии в сельскохозяйственном производстве?
5. Какова роль аэрозолей в сельскохозяйственном производстве?
6. Какой процесс ответствен за оседание пыли на холодных поверхно-
стях?
7. Чем отличается процесс светорассеяния у аэрозоли по сравнению с лио-
фобными коллоидами?
Г л а в a XVI
РАСТВОРЫ ВЫСОКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИЙ
(РАСТВОРЫ ВМС)
h ОБЩИЕ ХАРАКТЕРИСТИКИ РАСТВОРОВ ВМС
Рассмотренные ранее (см. гл. XII—XV) гетерогенные системы
представляют собой одну из двух больших групп коллоидов. Раст-
воры высокомолекулярных соединений (растворы ВМС) представ-
ляют вторую группу коллоидных систем.
На первых этапах развития коллоидной химии растворы ВМС
были отнесены к золям или, иначе, к псевдорастворам, имеющим
мицеллярное строение. Близость некоторых свойств лиофобных
коллоидов и растворов ВМС привела к тому, что они были неудач-
но названы лиофильными золями.
Исследования В. А. Каргина и его школы опровергли мицелляр-
ную теорию строения растворов ВМС, а последующие исследова-
ния показали, что не только по строению, но и по образованию и
поведению растворы ВМС никак не могут считаться золями (псев-
дорастворами).
Растворы ВМС характеризуются следующими особенностями.
1. Растворы ВМС представляют собой гомогенные (а не гете-
рогенные, как лиофобные коллоиды) системы, являясь истинными
растворами, где взвешенные частицы не содержат ядер, а пред-
ставлены макромолекулами — молекулами гигантских размеров.
2. Растворение ВМС осуществляется с образованием менее
упорядоченной системы из более ^упорядоченной и, значит, этот
процесс протекает с увеличением энтропии (AS > 0). Растворение
ВМС — самопроизвольный процесс, осуществляемый с уменьше-
нием свободной энергии: AG = А//— TAS <0.
Изменение энтальпии (знак АЯ) при растворении может быть
положительным (эндотермическая реакция), отрицательным (эк-
зотермическая реакция) или равным нулю.
Так как растворы ВМС образуются самопроизвольно с умень-<
шением свободной энергии, они представляют собой термодинами-
чески устойчивые системы, способные существовать без стабилиза-
тора неограниченное время в весьма больших массовых и значи-
тельных молярных концентрациях.
3. В отличие от лиофобных коллоидов растворы ВМС представ-
ляют собой равновесные системы, к которым применимо правило
фаз.
4. Растворы ВМС, подобно растворам низкомолекулярных сое-
динений, могут быть и молекулярными, и ионными, причем в по-
351
следнем случае природа зарядов связана с наличием функцио-
нальных групп. В образовании зарядов стабилизатор не принимает
участия.
5. ВМС способны образовывать не только истинные растворы,
но и типичные лиофобные золи, если в качестве дисперсионной
среды использовать такую жидкость, по отношению к которой дан-
ное высокомолекулярное вещество является лиофобным, т. е. не
способным растворяться в нем. Дополнительная энергия, необхо-
димая на диспергирование вещества, и добавка стабилизатора
приводит это вещество во взвешенное состояние и обусловливает
все характерные свойства лиофобного золя.
6. Являясь истинными растворами, растворы ВМС все же отли-
чаются от растворов низкомолекулярных соединений. Прежде
всего огромные размеры молекул являются ответственными за
большинство физических свойств растворов ВМС, отличающихся
от низкомолекулярных соединений. На поведение растворов ВМС
сильное влияние оказывают форма и отдельные фрагменты строе-
ния микромолекул.
Поведение молекул ВМС в растворе сходно с поведением свернутых в клубок
длинных нитей, причем пространственное положение этих нитей в растворе непре-
рывно меняется в результате теплового движения. Таким образом, если частицы
лиофобных коллоидов под влиянием теплоты испытывают броуновское движение,
гигантские молекулы ВМС изменяют свою конформацию. Однако форма клубка
нитей ВМС всегда остается близкой к форме вытянутого эллипсоида вращения.
Макромолекулы в растворе находятся в сольватированном состоянии. При
растворении ВМС изменение энтальпии системы (АЯ) может быть равно нулю или
иметь разный знак. Экспериментами установлено, что при растворении ВМС с
полярными группами в полярных растворах (ацетон, вода) происходит экзотер-
мическая реакция (АЯ<0) и тепловой эффект равен 40... 120 кДж/моль. При этом
изменение АЯ обусловлено сильным взаимодействием молекул ВМС и растворите-
ля; происходит сольватация (гидратация) ВМС. Растворение неполярных ВМС
(например, полиизобутилен в изооктане) осуществляется при слабом взаимодей-
ствии ВМС с растворителем, и тепловой эффект растворения очень мал (АЯ«0)
или даже может быть отрицательным (АЯ>0). Здесь большую роль играют фак-
торы растворения ВМС, связанные с энтропийным членом в уравнении AG =
— АЯ—TAS.
При сольватации активность растворителя вблизи от макромолекулы ВМС
понижается по сравнению с активностью чистого растворителя. Например, при
гидратации полярные группы ВМС (например, функциональные группы белковой
молекулы) взаимодействуют с диполями воды с образованием водородных связей.
В то же время молекулы воды не взаимодействуют с остальной частью ВМС, кото-
рая не содержит функциональных групп. Гидратный слой поэтому располагается
по макромолекуле «островками».
2. ЭЛЕКТРИЧЕСКИЕ, МОЛЕКУЛЯРНО-КИНЕТИЧЕСКИЕ
И ОПТИЧЕСКИЕ СВОЙСТВА РАСТВОРОВ ВМС
Заряд частицы ВМС. Изоэлектрическая точка (ИТ). Одной из
важных проблем, возникающих при изучении ВМС, является проб-
лема появления на поверхности молекул заряда, который возни-
кает по разным причинам.
Прежде всего поверхность ВМС может иметь собственный за-
ряд, возникающий благодаря расположенным на ней анионным и
катионным группам. Так, белки содержат карбоксильные анионы,
352
несущие отрицательный заряд, и протонированные основные груп-
пы, которые сообщают молекуле положительный заряд. Нуклеино-
вые кислоты заряжены отрицательно, что обусловлено диссоциа-
цией фосфатных групп, которые в структуре остова молекулы че-
редуются с молекулами пентозы. Кроме того, если даже молекуляр-
ная структура не содержит ионизируемых групп (что, в частно-
сти, характерно для амилозы и целлюлозы), то поверхность моле-
кулы связывает ионы растворителя или другие имеющиеся в раст-
воре ионы за счет поляризации вблизи гидроксигрупп или атомов
кислорода, входящих в состав ВМС (в данном частном случае вхо-
дящих в кольцо рибозы).
Даже тогда, когда макромолекулы совсем не содержат поляр-
ных групп, в них возникает заряд, благодаря неравномерному рас-
пределению электронов. Так, на инертной, на первый взгляд, по-
верхности парафинового масла возникает индуцированная поля-
ризация, которая приводит к связыванию ионов растворителя.
Таким образом, заряженная поверхность является одной из
особенностей крупных частиц, отличающей их от обычных низко-
молекулярных растворенных веществ.
Доказательством наличия заряда у частицы ВМС является ее
поведение при электрофорезе: заряженная частица, присутствую-
щая в растворе, в частности микромблекула, под действием элект-
рических сил движется к электроду противоположного знака.
При исследовании белка экспериментально определяют ско-
рость движения макромолекул в электрическом поле. Поскольку
отдельные макромолекулы увидеть нельзя, измеряют скорость
перемещения фронта окрашенного белка в растворителе. Положе-
ние этого фронта, назы-
ваемого границей, ре-
гистрируется оптическими
методами. Первый элект-
рофоретический аппарат
с оптическим устройст-
вом, предназначенным для
точного измерения скоро-
сти движения границ,
сконструировал А. Тизели-
ус; за эту работу он был
удостоен Нобелевской пре-
мии.
На рис. 140 схемати-
чески показана граница
(полоса) чистого белка, а
также границы (полосы),
полученные в результате
разделения компонентов
белковой смеси в электро-
форетической кювете. В ле-
вой кювете видна един-
Рис. 140. Разделение компонентов белковой
смеси при электрофорезе:
/ — буфер; 2 — единственная полоса; 3 — не-
сколько полос
353
ственная полоса белка, а в пра-
вой кювете, где разделялась
смесь белков, образовалось не-
сколько полос. Электрофорети-
ческий прибор может быть ис-
пользован для самых разных
целей. Его применяют для раз-
деления смесей макромолекул,
установления чистоты белка, а
также для определения изо-
электрической точки белка в
данном буферном растворе. ,
Если внести в электрофо-
ретическую кювету раствор, со-
приложить электрическое напря-
Рис. 141. Вид графика при эксперимен-
тальном определении изоэлектрической
точки
держащий различные белки, и
жение, то скорость движения каждого белка будет зависеть от
плотности заряда, которая определяется как суммарный заряд
частицы на единицу площади поверхности. В идеальном случае
каждый индивидуальный белок будет двигаться со скоростью, от-
личной от скорости движения остальных белков. Поэтому при до-
статочных размерах кюветы и за достаточно большое время смесь
белков разделится на индивидуальные компоненты. Секции кю-
веты можно отделить друг от друга так, чтобы в каждую попал
индивидуальный белок. Таким образом, электрофоретическая
техника может быть использована для получения чистых белков.
Для белков между зарядом молекул и электрофоретической
подвижностью существует прямая пропорциональная зависимость
в широком интервале рЦ. Если при некотором значении pH число
всех положительных зарядов на белковой молекуле равно общему
числу отрицательных зарядов, то при этом pH молекула не пере-
двигается в электрическом поле.
Значение pH, при котором электрофоретическая подвижность белка равна
нулю, называется изоэлектрической точкой.
Изоэлектрическая точка может быть определена эксперимен-
тально измерением электрофоретической подвижности белка в не-
котором интервале значений pH, внутри которого наблюдается
изменение знака заряда. При pH меньшем, чем pH изоэлектриче-
ской точки, белок заряжен положительно, а большем — отрица-
тельно. Значение pH, при котором график пересекает линию нуле-
вой подвижности, соответствует изоэлектрической точке
(рис. 141).
Аминокислоты и белки в водных растворах находятся преиму-
щественно в форме биполярных ионов:
NH2— R—COOH
II
NH* —R—СОО
NH*—R—COOH NH2—R— COO'
354
В кислой среде, когда в
результате избытка водород-
ных ионов подавлена иони-
зация карбоксильных групп,
молекула белка ведет себя
как основание, приобретает
положительный заряд и при
электрофорезе движется к
катоду. В щелочной среде,
наоборот, подавлена иониза-
ция аминогрупп, и молекула
белка ведет себя как кислота
и при электрофорезе передви-
рН<ИТ " pH-ИТ рН>ИТ
Рис. 142. Схематическое изображение
формы отдельных участков молекулы бел-
ка при различных pH
отрицательный заряд всех находя-
гается к аноду. В изоэлектри-
ческой точке суммарный за-
ряд частицы равен нулю, т. е.
щихся на частице анионных групп точно скомпенсирован поло-
жительным зарядом катионных групп.
В изоэлектрическом состоянии свойства растворов белков рез-
ко меняются: при этом они имеют, например, наименьшую вяз-
кость, плохую растворимость, что связано с изменением формы
микромолекул (рис. 142). При значении pH, близком к изоэлект-
рической точке, разноименно заряженные группы —NHj^ и
СОО~ притягиваются друг к другу и нить закручивается в спи-
раль. При смещении pH среды от изоэлектрической точки одно-
именно заряженные группы отталкиваются и цепь выпрямляется.
Молекулы ВМС в развернутом состоянии придают растворам
более высокую вязкость, чем молекулы ВМС, свернутые в спи-
раль или клубок.
В табл. 51 в качестве примера приведены изоэлектрические
точки различных белков.
Т а б лл ц а 51. Изоэлектрические точки некоторых белков
Кислые белки ИТ Основные белки ИТ
Казеин 4,6 Гемоглобин 6,8
Желатина 4,7 Глобулин 5,4
Альбумин яйца - 4,8 Глиадин пшеницы 9,8
Растворам белков свойственна, как и лиофобным золям, спо-
собность менять не только значение, но и знак заряда частиц
(способность перезаряжаться). Однако в растворах белков изме-
нение значения и знака заряда происходит от изменения концент-
рации водородных ионов, а у мицелл лиофобных золей переза-
рядка обусловлена специфической адсорбцией ионов.
Мембранное равновесие. Присутствие в организмах солей
белков, отделенных клеточной мембраной от растворов электро-
355
литов, приводит к перераспределению электролитов и соответст-
венно влияет на осмотическое давление по обе стороны мембра-
ны. Перераспределение электролитов подчиняется выведенному
Донанном уравнению мембранного равновесия.
Рассмотрим систему, в которой белок может быть представлен
в виде R“Na+. Весь белок находится в растворе с внутренней
стороны мембраны и не может проходить через нее. Пусть по
другую сторону мембраны находится раствор NaCl, т. е. Na+ и
С1“ находятся с внешней стороны мембраны. Схематическое опи-,
сание системы дано ниже:
Начальное состояние системы
Внутри
IR_ 'внутр
[Na+]BHyn,
^внутр = [Na ] ВНутр
С внешней стороны
[С1 1 внешн
[Na ]внешн
[CI кнешн= [Na ]внешн
Мембрана
Квадратные скобки означают концентрации соответствующих
ионов, причем индексы «внутр» и «внешн» относятся к началь-
ным концентрациям соответственно с внутренней и внешней сто-
роны мембраны.
Мембрана проницаема для малых ионов, и поэтому ионы хло-
ра будут двигаться справа налево, так как ионы стремятся рав-
номерно распределиться по всему объему. Каждый ион хлора,
проходящий через мембрану, должен сопровождаться ионом
натрия, иначе с внешней стороны мембраны накапливались бы
положительные заряды. Пусть х означает концентрацию, кото-
рой достигли ионы хлора за время установления равновесия.
Белок не может диффундировать через мембрану и остается с ее
внутренней стороны. В результате перераспределения при равно-
весии система будет находиться в следующем состоянии:
Конечное состояние системы (равновесие)
Внутри
[R- 1 внутр
[Na JjjHyip + X
[СГ] = х
С внешней стороны
1 внешн- х
[Na 1 внешн- х
[С1 ] внешн х ~ [Na ]внешн х
Мембрана
При достижении равновесия соблюдается принцип электро-
нейтральности, тогда
[Na + ] внутр [С1 ] внутр = [Na+] внешн. [С1 внешн]
356
и, следовательно, ([Na+] внутр + х)х=( [Ма+]внешн — х)2. Решая
это уравнение относительно х, имеем
______ [Na+]2BHeiiiH __ ЛАП
[Na+]BHyTp -|- 2[Na+]BHeiIIH
Полученное основное уравнение (16.1) позволяет рассчитать
равновесные концентрации независимо от того, какими они были
вначале. Например, если начальная концентрация натриевой
соли белка равна 0,1 кмоль/м3 и начальная концентрация хло-
рида натрия, находящегося с внешней стороны мембраны, равна
0,1, хр х = 0,033 кмоль/м3, т. е. через мембрану прошла % хло-
рида на;трия. Это перераспределение приведет к заметному по-
вышению осмотического давления раствора, находящегося с
внутренней стороны мембраны.
Уравнение Доннана разобрано для случая, когда снаружи
имеется соль, а внутри клетки — только один из ее ионов. Одна-
ко положение не изменяется, если у белка и электролита вне
клетки нет общего иона.
Таким образом, при соприкосновении клетки с раствором
электролита некоторая часть электролита всегда перейдет в
клетку, поэтому осмотическое давление, зависящее от концент-
рации ионов электролита плюс концентрации белка, всегда будет
выше, чем в окружающем растворе. Этот факт способствует
поддержанию тургора клеток даже в изотонических растворах.
В то же время в гипертонических растворах происходит не
только потеря клеткой воды, но и переход некоторой части соли
внутрь нее.
Равенство произведений концентраций разнозаряженных ио-
нов по обе стороны мембраны совпадает с равенством сумм кон-
центраций (т. е. сумм числа ионов) только при отсутствии в клет-
ке белка. Если .же в клетке содержится белок, то суммы концент-
раций ионов по обе стороны мембраны будут неодинаковы, что
обусловит возникновение разности потенциала (мембранного
потенциала).
Эффект Доннана (т. е. неравномерное распределение Электро-
литов между клеткой и омывающей их жидкостью) оказывает
большое влияние на жизнедеятельность клеток, в частности на
величину биопотенциалов.
Вязкость. Вязкость растворов, содержащих макромолекулы,
обычно выше вязкости растворов низкомолекулярных соединений
и коллоидных растворов тех же концентраций. Например, у раст-
воров каучука аномально высокая вязкость наблюдается уже
при массовой доле порядка 0,05%. Только очень разбавленные
растворы ВМС можно считать подчиняющимися законам Ньюто-
на и Пуазейля. Вязкость растворов ВМС не подчиняется также
закону. Эйнштейна и возрастает с увеличением концентраций.
Графически эта зависимость изображается кривой, обращенной
выпуклостью к оси концентраций (рис. 143).
Долгое время высокую вязкость растворов ВМС объясняли
357
большой сольватацией макромолекул.
А. В. Думанский поэтому ввел в урав-
нение Эйнштейна поправку, учитываю-
щую сольватацию:
Чотн= —= 1+2,5(ч> 4- V),
По
где ф — доля объема суспендирован^
ных частиц в единице объема суспен-
зии; V — сольватный объем частиц
дисперсной фазы.
В дальнейшем было показано, что
сольватация не играет столь важной
роли при образовании растворов ВМС.
Основной причиной отклонения вязко-
сти растворов ВМС от законов, кото-
Рис. 143. Зависимость вязко-
сти раствора от концентра-
ции:
1 — лиофобные коллоиды; 2 —
растворы ВМС
рым подчиняются растворы низкомолекулярных веществ,’^явля-
ется взаимодействие вытянутых и гибких макромолекул, часто
образующих структурированные системы (ассоциаты^ Эти ас-
социаты, естественно, сильно увеличивают вязкость раствора^ по
сравнению с раствором лиофобных коллоидов, где взаимодейст-
вием частиц можно пренебречь. При низкой концентрации рас-
творов ВМС вероятность структурирования не так велика, и
поэтому для сильно разбавленных растворов может быть исполь-
зовано уравнение Эйнштейна. При высокой концентрации эти
взаимодействия очень велики. Кроме того, так как макромоле-
кулы в растворе находятся в виде клубков, включающих боль-
шой объем растворителя, то объем этого растворителя, простран-
ственно связанного с полимером, также следует отнести к объему
дисперсной фазы.
Часто значительное структурирование происходит при стоя-
нии раствора ВМС. В этом случае, как это, например, показано
для раствора желатины при 398 К, возможно значительное изме-
нение вязкости полученного раствора (рис. 144).
С повышением температуры вяз-
кость растворов ВМС может изменять-
ся по-разному. Если раствор образо-
ван сильно разветвленными молеку-
лами, то вязкость раствора понижает-
ся с увеличением температуры за счет
уменьшения структурирования. Вяз-
кость растворов, содержащих длинные
неразветвленные молекулярные цепи,
с повышением температуры может по-
вышаться из-за увеличения интенсив-
ности движения фрагментов макромо-
лекулы, что препятствует ориентации
линейной макромолекулы в потоке.
Таким образом, вязкость растворов
Рис. 144. Увеличение относи-
тельной вязкости раствора
желатины при 298 К со вре-
менем
358
в ВМС сложным образом связана с формой макромолекул и
характером межмолекулярных взаимодействий. С одной стороны,
это затрудняет надежное определение значения вязкости, а с
другой — позволяет использовать определение вязкости для опи-
сания сложных процессов, происходящих в растворах ВМС.
При исследовании растворов ВМС часто используют значе-
ние так называемой характеристической вязко-
сти, обозначаемой через [ц]. Она представляет собой значе-
ние цуд/С, экстраполированной к нулевой концентрации, т. е.
(X fa] = Игп (т|уд/С)с^о-
Удёльная вязкость т|уд связана с относительной вязкостью
выражением
Луд = Лотн — 1 •
Она пропорциональна массовой концентрации высокомолекуляр-
ного соединения в растворе.
Г. Штаудингер показал, что характеристическая вязкость
количественно связана с молекулярной массой полимера. Уравне-
нение Штаудингера содержит эмпирические константы, которые
приходится оценивать для каждого конкретного вещества, раст-
воренного в данном растворителе. Общая форма модифицирован-
ного уравнения Штаудингера имеет следующий вид:
[л] = W, (16.2)
где К и а — эмпирические константы; М—молекулярная масса.
Константа а связана с формой молекул; для гибких нитевидных
молекул, свернутых в клубок, она составляет от 0,5 до 1,0, а для
палочкообразных молекул достигает 1,8. Так, например, для
растворов амилозы в 1 М растворе гидроксида калия были полу-
чены значения К = 0,85-10~\ а — 0,76. В дальнейшем эти кон-
станты использовались для расчета относительной молекулярной
массы по уравнению (16.2) кристаллических препаратов амило-
зы, выделенных из рисового крахмала. Данные для амилозы и
других макромолекул приведены в табл. 52.
Таблица 52. Значение относительной молекулярной массы М
и характеристической вязкости для различных биополимеров
Биополимер м fa], м3/кг Растворитель
Амилоза из риса сорта Рексоро 325000 209 1 м кон
сорта Сенчури Патна 100000 84 То же
Рибонуклеаза 13683 3,3 Водный солевой
Сывороточный альбумин 65000 3,7 раствор То же
Тропомиозин 93000 52 »
Коллаген 345000 1150 »
ДНК 6000000 5000 »
Сопоставление относительных
молекулярных масс, вычисленных
из данных по характеристической
вязкости, седиментации и диффузии,
позволяет судить о гибкости макро-
молекул, а также об их способно-
сти к гидратации.
Осмотическое давление раство- v
ров ВМС. Согласно уравнению
Вант-Гоффа осмотическое давление
растворов увеличивается прямо про-
порционально концентрации. Для
растворов ВМС эксперимент пока-
зывает, что осмотическое давление
значительно выше, чем это требует-
ся по закону Вант-Гоффа, и в своей
Вант-Гоффа непреложимо для таких
Рис. 145. Зависимость осмотиче-
ского давления от концентрации
раствора ВМС:
/ — высокомолекулярное вещество;
2 — низкомолекулярное вещество
(сахар); 3 — слабый электролит
уравнение
схематически сопоставлены изменения осмотиче-
с концентрацией для растворов низкомолекуляр-
(кривая 2), слабого электролита (кривая 3) и
обычной форме
растворов.
На рис. 145
скоро давления
ного вещества
высокомолекулярного соединения (кривая /). Как видно из ри-
сунка, осмотическое давление раствора низкомолекулярного ве-
щества возрастает прямо пропорционально концентрации. Для
слабого электролита осмотическое давление раствора обусловле-
но не только числом молекул, но и ионов, а так как степень иони-
зации уменьшается с повышением концентрации, осмотическое
давление возрастает более медленно, чем концентрация. Об этом
свидетельствует выпуклость кривой, которая обращена в сторону
ординаты. Наконец, осмотическое давление раствора высокомо-
лекулярного вещества возрастает быстрее, чем увеличивается
концентрация. Это происходит из-за того, что макромолекула
благодаря большим размерам и гибкости ведет себя в растворе
как несколько более коротких молекул. Поэтому роль кинетиче-
ского элемента играет уже не вся макромолекула, а Соответст-
вующие ее фрагменты. Чем более гибка молекула, тем при про-
чих равных условиях осмотическое давление выше и тем больше
оно отклоняется от значения, вычисленного по уравнению Вант-
Гоффа. На основании таких представлений для описания зависи-
мости осмотического давления от концентрации полимеров было
предложено уравнение
CRT -4~ яг2
~м~+вс
или
л
RT I RC
~с—М~ + ВС’
(16.3)
где С — концентрация; М — относительная молекулярная масса
360
полимера; В — некоторый коэффициент, характеризующий откло-
нение от уравнения Вант-Гоффа.
В таком виде уравнение (16.3) представляет собой уравнение
прямой, по которой графически легко найти относительную мо-
лекулярную массу полимера. Для этого определяют осмотическое
давление при разных концентрациях, строят график в координа-
тах л/С—С и экстраполируют прямую до пересечения с осью ор-
динат. Отрезок, отсеченный прямой на оси ординат, равен RT/M,
а В определится как тангенс угла, образуемого прямой и осью
абсцисс.
Таким способом с применением современных осмометров,
снабженных оптическим детектором и сервоуправляемой систе-
мой давления, удается автоматически определить относительные
молекулярные массы ВМС в области от 20 тыс. до 1 млн.
Светорассеяние и поглощение света. Цепные молекулы поли-
меров нельзя обнаружить в растворах при ультрамикроскопиче-
ских наблюдениях. Это объясняется тем, что растворы полиме-
ров гомогенны и линейные макромолекулы приближаются к кол-
лоидным частицам только по длине, а в двух других направле-
ниях соответствуют размерам обычных молекул. Кроме того,
коэффициент преломления полимеров, как правило, сравнитель-
но близок к коэффициенту преломления среды.
Растворы ВМС так же, как и лиофобные коллоиды, характе-
ризуются светорассеянием, хотя величина рассеяния для них не
так велика, как для лиофобных систем. Изменение величины рас-
сеяния света используют в методе определения относительной
массы полимеров. Метод основан на измерении мутности разбав-
ленных растворов ВМС. При этом экспериментально измеряется
коэффициент ослабления света в результате светорассеяния при
прохождении его через слой раствора.
Светорассеяние концентрированных растворов полимеров
обусловлено их неоднородностью, которая возникает из-за не-
прерывных небольших отклонений концентраций. Последние вы-
зывают в свою очередь отклонения (флуктуации) показателя
преломления от его среднего значения.
Окраска растворов макромолекул и белков зависит прежде
всего от истинного поглощения. Максимумы этого поглощения,
как правило, расположены вне видимой области спектра. Поэто-
му растворы, бесцветные в видимых лучах, могут обладать силь-
ным поглощением в ультрафиолетовой и инфракрасной областях
спектра.
Ультрафиолетовые спектры белков отличаются сильным по-
глощением, характеристическим для ароматических фрагментов
аминокислот, входящих в их состав: фенилаланин, тирозин, трип-
тофан. Эти спектры поглощения используют для аналитического
определения остатков указанных аминокислот. Резкий максимум
поглощения, характерный для нуклеиновых кислот и нуклеопро-
теидов, позволяет определить их содержание в отдельных клет-
ках.
361
Инфракрасные спектры поглощения ВМС очень сложны и их
редко используют при исследовании растворов ВМС, но они иг-
рают важную роль в современных исследованиях твердых поли-
меров.
Полимеры, образованные из оптически активных мономеров, с
преобладанием одного из антиподов (например, белки построены
исключительно из /-аминокислот, полисахариды — из d-углево-
дов), изменяют направление плоскости поляризации при прохож-
дении через них поляризованного света. Вращение плоскости
поляризации измеряется специальными приборами — поляримет-
рами.
Если поляризованный падающий луч при прохождении через
слой раствора / с концентрацией С растворенного вещества, рав-
ной 1 кг на 1 кг раствора, и плотностью раствора q поворачи-
вается на угол а, то удельное вращение [а] растворенного ве-
щества равно
=-Йг-
Для белков удельное вращение всегда отрицательно и колеблется
для различных белков от —30 до —60°. В растворах желатины
удельное вращение изменяется в процессе застудневания; это
явление называется мутаротацией. Величина оптического
(Вращения в значительной степени зависит от pH, состава и кон-
фигурации полипептидной цепи, и в настоящее время измерения-
ми удельного вращения широко пользуются для изучения процес-
са денатурации в полипептидах и белках.
Чувствительным методом исследования конфигурации белков и полипептидов
в нативном и денатурированном состоянии является метод, основанный на изме-
нении зависимости величины удельного вращения от длины волны света — дис-
персии оптического вращения.
Как известно, все аминокислоты, за исключением глицина, имеют асиммет-
рический атом углерода в a-положении. Все они относятся к /-аминокислотам
и обладают одними и теми же заместителями у а-углерода: группами —NH2
и —СООН и боковой цепью, характерной для каждой аминокислоты. Долгое
время полагали, что оптическое вращение полипептидов и белков является ад-
дитивным свойством и зависит исключительно от доли, вносимой каждым ами-
нокислотным остатком в отдельности. Однако значительный рост левого вращения
белков при денатурации (от —50 до —100°) и при застудневании желатины при-
водит к выводу, что эти изменения связаны с конформационными изменениями
полипептидной цепи. При исследовании эмпирическую величину удельного
оптического вращения [а] заменяют на величину эффективного вращения
цепи [т]:
г 1 3 А4о г I
W + Т ТосГ [“1-
где вместо концентрации в килограммах на 1 кг раствора вводится молярная
концентрация звена с относительной молекулярной массой MQ. Для белков
MQ = 125.
По измерению оптической активности рассчитывают степень спиральности
(упорядоченности) молекулы. У пепсина, например, она равна 28%, а у миогло-
бина —_70%.
362
3. НАБУХАНИЕ И РАСТВОРЕНИЕ ВМС
Наличие больших макромолекул приводит к своеобразию про-
цесса растворения ВМС, который для них сопровождается явле-
нием набухания.
Набухание представляет собой самопроизвольный процесс
поглощения, ВМС больших объемов низкомолекулярной жидко-
сти, сопровождающийся значительным увеличением объема ВМС.
В отличие от процесса растворения низкомолекулярных веществ,
где осуществляется диффузия растворяемого вещества в раство-
ритель, в процессе растворения ВМС происходит главным обра-
зом диффузия молекул растворителя в высокомолекулярное ве-
щество. Это обусловлено двумя факторами: 1) большей подвиж-
ностью маленьких по сравнению с макромолекулами ВМС моле-
кул растворителя; 2) неплотной упаковкой макромолекул ВМС.
В результате теплового движения этих макромолекул возникают
свободные межмолекулярные объемы, куда могут проникать ма-
ленькие молекулы растворителя.
Процесс проникновения молекул растворителя в макромоле-
кулы ВМС приводит к тому, что при набухании объем полимера
всегда увеличивается, а объем всей системы (полимер + раство-
ритель) обычно уменьшается. Это особенно заметно при набуха-
нии полярных полимеров в полярных растворителях. Уменьшение
объема системы при набухании, называемое контракцией,
в большинстве случаев хорошо описывается следующим эмпири-
ческим уравнением с двумя константами:
V = ат/(р + т),
где V — контракция; m — масса жидкости, поглощенной при на-
бухании 1 кг полимера (степень оводнения); аир — константы.
Контракция системы при набухании полимера объясняется
ориентацией молекул растворителя в результате их «Поглоще-
ния» макромолекулами, что способствует увеличению плотности
системы. Частично контракция происходит за счет того, что при
набухании относительно небольшие молекулы растворителя прони-
кают в пространства между громоздкими макромолекулами,
вследствие чего компактность упаковки молекул возрастает.
Таким образом, на первой стадии взаимодействия ВМС и
низкомолекулярной жидкости образуется гетерогенная система,
состоящая из ВМС и свободной низкомолекулярной жидкости.
Эта система при дальнейшем поглощении низкомолекулярной
жидкости превращается в однофазную гомогенную систему.
В обычных условиях набухания (при Т = const и р = const) процесс на-
бухания характеризуется уменьшением свободной энергии системы: ДО =
= ДЯ — TAS < 0.
В первой стадии происходит специфическое взаимодействие молекул раст-
ворителя и ВМС. Процесс этот экзотермичен (ДЯ <0), а Д5 в этом процессе
близко к нулю, а иногда даже ДЗ < 0 в тех случаях, когда сольватация приводит
к увеличению жесткости цепи. Однако |ДЯ| > IFASI и ДО <0.
Во второй стадии набухания теплота взаимодействия молекул растворителя
и ВМС почти или совсем не выделяется (ДЯ 0), но зато возрастает энтропия.
363
Последнее происходит потому, что разрыхление сетки ВМС приводит к частич-
ному освобождению макромолекул и переходу их в раствор. А это отвечает пере-
ходу системы из более упорядоченного состояния в менее упорядоченное. Отсюда
следует, что вторая стадия характеризуется следующими уравнениями: TAS > 0;
&G ж —TAS < 0.
Суммарный тепловой эффект при набухании ВМС обычно по-
ложительный. Различают интегральную теплоту набухания t/инт,
т. е. общее количество теплоты, выделившейся при набухании
1 кг сухого полимера, и дифференциальную теплоту набухания
<7диф, представляющую собой количество теплоты, выделившейся
при поглощении 1 кг жидкости сухим или же набухшим высоко-
молекулярным веществом.
Итак, набухание представляет собой специфическую стадию
процесса растворения ВМС, а весь процесс растворения можно
разделить на четыре стадии.
1. Исходная стадия. Система гетерогенна, двухфазна: чистая
низкомолекулярная жидкость и чистый полимер Ж1 + Жг
(рис. 146, а).
Рис. 146. Схематическое изображение стадии набухания
2. Стадия набухания. Система расслаивается на две жидкие
фазы: одна фаза — раствор низкомолекулярного компонента в
компоненте ВМС Ж1 ->Жг, где Жг — набухший ВМС, а Ж1 — чис-
тая низкомолекулярная жидкость (рис. 146, б). Вторая фаза
представлена чистой низкомолекулярной жидкостью.
3. Стадия образования второго раствора Жг->Ж1— Система,
также расслоена на две фазы: одна фаза прежняя — набухший
полимер Ж1->Жг; Другая фаза — раствор полимера в низкомо-
лекулярной жидкости Жг->Ж1 (рис. 146, в).
4. Стадия полного растворения — превращение гетерогенной
(двухфазной) системы в гомогенную Ж f ->Жг (рис. 146, г).
Различают два вида набухания — неограниченное и ограни-
ченное.
Неограниченное представляет собою набухание, последова-
тельно переходящее через все четыре стадии в полное растворение,
т. е. с образованием однофазной системы. Так набухают каучуки в
364
бензоле, нитроцеллюлоза в ацетоне, белок в воде, целлюлоза в
медно-аммиачном растворе.
Ограниченное — набухание, не переходящее в полное растворе-
ние, а останавливающееся на второй или третьей его стадии. Так
набухают цри комнатной температуре желатина и целлюлоза в
воде. Процесс, доходящий до третьей стадии, аналогичен сме-
шению двухх низкомолекулярных жидкостей с ограниченной
взаиморастворимостью (например, фенола и воды). Повышение
температуры ведет к усилению растворимости ВМС и даже к
полному его растворению. Таким свойством обладает, например,
желатина: при комнатной температуре она набухает ограниченно,
а при повышенных температурах — неограниченно.
Однако во многих случаях ограниченное набухание останав-
ливается на второй стадии (рис. 146, б) и не переходит в раство-
рение даже при повышении температуры. Такой вид ограничен-
ного набухания происходит тогда, когда цепи полимеров связаны
между собой мостиковыми связями. Так ведет себя, например,
вулканизированный каучук, который набухает в бензоле, но не
растворяется в нем. При этом молекулы низкомолекулярной жид-
кости проникают в свободное пространство между макромолеку-
лами, раздвигают их, но не способны порвать мостики.
Степень набухания и скорость набухания. Степень набухания а
определяется массой жидкости (в кг), которая поглощается на
данной стадии набухания и при данной температуре 1 кг высоко-
полимера:
Ш2 —
а =--------
т\
Скорость набухания (имеется в виду ограниченное набуха-
ние) обычно выражают в объемных единицах, поскольку в непре-
рывном процессе набухания удобнее вести наблюдение за измене-
нием объема (в особых приборах — дилатометрах).
Уравнение кинетики набухания в простейшем виде может быть
дано известным уравнением реакции первого порядка
_ЁН —(у _ ул
dt I и°° °’
показывающем, что скорость набухания прямо пропорциональна
разности между предельным объемом набухшего ВМС К» и
объемом Vt в момент времени t и обратно пропорциональна пер-
воначальной толщине / слоя набухающего полимера. Константа
скорости набухания k зависит от природы полимера и раство-
рителя.
Уравнение кинетики набухания выражается графически в
виде кривых набухания (рис. 147). Эти кривые показывают, что
скорость набухания наибольшее значение имеет в первые его
моменты, а затем постепенно уменьшается и при предельном
набухании становится равной нулю. При прочих равных условиях
степень набухания и его скорость различным образом зависят от
365
природы полимера и растворителя. Так, из рис. 147 видно, что
полимер 1 набухает быстрее, но с меньшей степенью предельного
набухания, чем полимер 2.
Факторы набухания. На степень и скорость набухания одного
и того же ВМС в одном и том же растворителе влияют такие
факторы, как температура, давление, pH среды, присутствие
посторонних веществ, в особенности электролитов, степень из-
мельченности полимера, возраст (свежесть) полимера.
При повышении температуры скорость набухания увеличива-
ется, а степень предельного набухания уменьшается (рис. 148).
Примером резкого снижения степени набухания с повышением
температуры является набухание целлюлозы в растворах щелочи.
Увеличение давления повышает и степень набухания. Названные
два фактора следуют принципу Ле Шателье,
Рис. 149. Влияние pH на на-
бухание
Рис. J47. Схемати- ^Рис. 148. Влияние тем-
ческий вид кривых пературы на скорость
набухания набухания
Влияние pH среды хорошо изучено для белков и целлюлозы.
Минимум набухания лежит в области изоэлектрической точки
(для желатины, например, при pH 4,7), по ту и другую сторону
от этой точки степень набухания возрастает и, достигнув мак-
симумов (из них больший в кислой зоне при pH 3,2), вновь
уменьшается (рис. 149). Такое влияние pH на набухание связа-
но с тем, что в изоэлектрической точке заряд макромолекул бел-
ков минимален, а вместе с этим минимальна степень гидратации
белковых ионов.
Влияние электролитов (нейтральных солей) своеобразно
прежде всего тем, что главным образом на набухание влияют
анионы и лишь в значительной степени катионы. Причем одни
анионы усиливают набухание, а другие ослабляют. По влиянию
анионов на набухание последние можно расположить в лио-
тропный ряд. Анионы, расположенные до хлорид-иона, усиливают
набухание в нисходящем порядке. Хлорид занимает близкое к
нейтральному положение, а последующие анионы не только не
усиливают набухание, а, наоборот, все больше его задерживают:
CNS~>I->Br->NOI>Cr>CH3COO->SOr
366
При установлении ряда необходимо, чтобы растворы солей
были достаточно концентрированными, а среда в целом нейтраль-
ной или слабощелочной. В достаточно кислой среде все анионы
уменьшают набухание.
Влияние концентрации водородных ионов и солей на набуха-
ние нашло большое практическое применение, например, при
дублении кож, в варке целлюлозы, в производстве дубителей
из древесной коры.
На скорость набухания влияет повышение степени измель-
ченное™. Это связано с увеличением общей поверхности веще-
ства, благодаря чему ускоряется проникновение молекул низко-
молекулярной жидкости внутрь ВМС.
Влияние возраста всегда однозначно: чем свежее (моложе)
ВМС, тем больше степень набухания и скорость набухания.
Давление набухания. Одним из ярких проявлений процесса
набухания является увеличение объема набухающего тела. Если
создать препятствие увеличению объема набухающего тела, то
при этом развивается давление, называемое давлением набуха-
ния рн. При помощи особых маномётров удалось эксперимен-
тально определить, что давление набухания рн выражается урав-
нением, аналогичным эмпирическому уравнению адсорбции:
Рн = КСя,
где К — константа, зависящая от природы полимера и раствори-
теля; п — константа, почти не зависящая от природы последних
и в среднем приблизительно равная 3; С — концентрация, выра-
женная в килограммах сухого ВМС в 1 м3 образовавшейся сис-
темы.
Давление набухания может быть очень большим, достигая
иногда до 10 МПа. Так, известны случаи трагической гибели
морских судов от набухания зерна в их трюмах вследствие по-
падания воды при пробоинах.
Набухание играет большую роль в физиологии животных и растений.
Так, первой фазой прорастания зерна является его набухание, т. е. увеличение
его объема после смачивания. Лишь после набухания зерна возможны реак-
ции, сопровождающие рост и развитие, не идущие при сухом состояния геля.
В молодых растущих организмах все процессы обмена совершаются особенно
энергетично, и содержание воды в них, степень набухания их коллоидов тем
больше, чем моложе организм. В период интенсивного роста, усиленного деле-
ния клеток в начале утробной жизни младенца степень набухания коллоидов
столь велика, что вода составляет 95% массы плода. Содержание воды у ново-
рожденного составляет уже 70...75%, у взрослого — 59...60%. Постепенное ста-
рение коллоидов сопровождается уменьшением способности к набуханию и в
живом организме к старости замедляются процессы обмена, замечается бук-
вально высыхание человека, уменьшение его объема: морщины, являющиеся
характерным признаком старости, связаны с потерей коллоидами способности
к набуханию.
4. НАРУШЕНИЕ УСТОЙЧИВОСТИ РАСТВОРОВ ВМС
Растворы ВМС, образующиеся с понижением свободной энер-
гии и находящиеся в равновесном состоянии, агрегативно устой-
чивы, как и истинные растворы. Их устойчивость главным обра-
зом определяется растворимостью данного ВМС в растворителе,
а другие факторы, которые играют основную роль в устойчиво-
сти лиофобных коллоидов, например заряд частицы и сольвати-
рующая способность, практически не влияют на устойчивость.
Так, известно, что белки устойчивы в изоэлектрической точке,
где дзета-потенциал равен нулю. Поэтому теории, которые объ-
ясняют агрегативную устойчивость растворов ВМС действием
электрического заряда либо сольватацией, в настоящее время
надо признать устаревшими. Заряд и сольватация, конечно, игра-
ют роль, но только в той степени, в которой они влияют на
растворимость ВМС. Так, растворимость белков зависит от pH;
она минимальна в изоэлектрической точке. При смещении от
изоэлектрической точки увеличение заряда и гидратация молекул
белка повышают растворимость его в воде, и поэтому увеличива-
ется устойчивость раствора ВМС.
ИВсе процессы нарушения устойчивости растворов ВМС свя-
заны с переходом от полного растворения ВМС к ограниченному
растворению или к нерастворимости. Изменение растворимости
ВМС может быть вызвано либо понижением температуры, либо
изменением состава раствора путем добавления жидкости, в ко-
торой ВМС не растворяется. Чаще всего нарушение устойчиво^
сти растворов ВМС связывают с введением в раствор электро-
литов, при введении которых наблюдается превращение гомо-
генной системы раствора ВМС в гетерогенную, т. е. происходит
выделение ВМС из раствора.
Нарушение устойчивости растворов ВМС при введении элек-
тролитов нельзя отождествлять с коагуляцией лиофобных кол-
лоидов. Коагуляция золей происходит при введении малых кон-
центраций электролита и представляет собой обычно необрати-
мое явление. Выделение из раствора ВМС происходит при до-
бавлении относительно больших объемов электролита, на 3...5 по-
рядков превышающих порог коагуляции и не подчиняющихся
правилу Шульце—Гарди. Процесс является обратимым, и после
удаления из осадка электролита ВМС снова способно к раство-
рению.
Механизм коагуляции лиофобных коллоидов и нарушения
устойчивости ВМС различны. Коагуляция золей происходит обыч-
но в результате сжатия двойного электрического слоя и умень-
шения или полного исчезновения электрического заряда на по-
верхности частицы, являющегося в этом случае основным фак-
тором устойчивости. Выделение из раствора ВМС при добавле-
нии электролита объясняется уменьшением растворимости ВМС
в концентрированном растворе электролита. Поэтому по анало-
гии с подобными явлениями в растворах низкомолекулярных
368
веществ такое выделение ВМС из раствора называют выса-
ливанием.
Можно сказать, что при добавлении электролитов между
макромолекулами и вводимыми ионами электролита происходит
борьба за обладание молекулами воды. При этом немаловаж-
ную роль играют величина и распределение заряда в ионе.
Основная роль в высаливании, как и в набухании, принадле-
жит анионам, и по высаливающему действию они могут быть
расположены в следующий ряд:
C2Or>SO42->CH3COO->Cl->NO3->Br->I->CNS
В аналогичный ряд, но с меньшим воздействием на высалива-
ние располагаются и катионы:
Li+>Na+>K+>Pb+>Cs+>Mg2+>Ca2+>Sr2+>Ba2+
Эти ряды называют лиотропными рядами Гофмейстера.
Лиотропное действие связано с гидратацией ионов: чем больше
требуется воды для гидратации иона, тем меньше воды остается
на растворимость ВМС и тем легче происходит высаливание.
Первые члены лиотропных рядов наиболее сильно гидратируются
и оказывают повышенное высаливающее действие. Гидратация
иона зависит от плотности заряда, приходящегося на единицу
поверхности иона. Чем больше заряд, тем выше степень гидра-
тации. Так, Li+ и Cs+ — однозарядные ионы, но радиус у цезия
почти в два раза больше, чем у лития, и, следовательно, при
одном и том же заряде плотность заряда на единицу поверхно-
сти будет больше у лития. Вследствие этого литий сильнее
притягивает диполи воды и сильнее проявляет дегидратирующие
свойства.
Высаливание белков концентрированными растворами солей
является одним из основных методов фракционирования белко-
вых смесей на альбумины и глобулины. Понижения раствори-
мости белков можно достичь также добавлением спирта и дей-
ствием низкой температуры. На сочетании действия спирта, со-
лей и охлаждения основаны способы детального фракциониро-
вания белковых смесей по Кону; например, из сыворотки крови
этим путем выделено свыше 12 белков.
Под влиянием высокой и низкой температуры, при действии
высококонцентрированных кислот и щелочей, дубильных ве-
ществ, лучистой энергии, ультразвука, механического воздей-
ствия, высокого давления происходит специфическое необрати-
мое осаждение белков, которое называется денатурацией.
Типичным примером может служить денатурация куриного
белка (альбумина) при варке: из прозрачного растворимого в
воде вещества он превращается в более или менее твердую,
непрозрачную, нерастворимую в воде массу. Исследования по-
казали, что денатурация или свертывание белка при нагревании
большей частью складывается из двух различных процессов:
13—696
369
1) понижения растворимости белка (собственно денатурация);
2) коагуляции золей денатурированного белка.
Обе стадии невозможно отделить одну от другой. Сущность
тепловой денатурации можно рассмотреть на примере глобуляр-
ных белков. Основная молекула глобулярного белка, как извест-
но, состоит из одной или нескольких полипептидных цепей,
сложенных складками и образующих клубки. Такая структура
стабилизируется непрочными связями, среди которых большую
роль играют водородные связи, образующие поперечные мос-
тики между параллельными пептидными цепями или их склад-
ками. При нагревании белков происходит усиленное движение
полипептидных цепей или их складок, что вызывает разрыв
непрочных связей между ними. В результате этого наблюдается
развертывание и перегруппировка складок, сопровождаемые
перераспределением полярных и неполярных радикалов, причем
неполярные радикалы концентрируются на поверхности глобул,
понижая их гидрофильность, а следовательно, и растворимость.
Для денатурации белка необходимо присутствие некоторого
минимального содержания воды. Безводный белок при нагрева-
нии не подвергается денатурации. Так, сухой альбумин выдер-
жйвает нагревание в струе сухого воздуха при температуре
393 К без заметного изменения его растворимости.
Своеобразной формой коагуляции растворов некоторых ВМС
и, в частности, белка, совершающейся под действием солей и
сопровождающейся, как и высаливание, образованием гетероген-
ной системы, является процесс, получивший название к о а-
цервации (от лат. acervus — рой). От высаливания коа-
цервация отличается тем, что белок не выделяется сразу в
виде хлопьев, которые уплотняются в осадок, а собирается сна-
чала в невидимые невооруженным глазом жидкие капли, кото-
рые сливаются далее в видимые глазом капли и лишь затем
происходит расслоение.
Контрольные вопросы
1. Для лиофобных коллоидов характерно наличие мицелл. А каково строение
частиц растворов ВМС?
2. Как доказать, что в большинстве случаев у частиц ВМС имеется заряд?
Чем отличается этот заряд от заряда частицы лиофобных коллоидов?
3. Почему возникает биопотенциал?
4. Каковы движущие силы (термодинамические) процесса набухания? Осу-
ществляются ли аналогичные процессы в лиофобных коллоидах?
5. Какова роль набухания в физиологии животных и растениях?
6. Сопоставьте механизмы коагуляции лиофобных коллоидов и нарушение
устойчивости растворов ВМС.
7. Какие факторы вызывают денатурацию белка? Каков механизм денату-
рации?
Глава XVII
ГЕЛИ. СТУДНИ. ПОЛУКОЛЛОИДЫ
1. Гели. Студни*
Гели представляют собой пространственные сетки, образован-
ные либо твердыми коллоидными частицами, либо гибкими мак-
ромолекулами, в промежуточных объемах которых находится
растворитель. Если гели образуются твердыми коллоидными час-
тицами типа SiC>2, ЕезОз,V2O5, то они называются хрупкими
гелями. Если пространственная сетка образована макромо-
лекулами ВМС, гели называются эластичными гелями
или студнями. Хрупкие гели имеют двухфазную гетероген-
ную структуру, а эластичные гели (студни) представляют собой
гомогенную систему.
Гели играют важную роль в практической деятельности че-
ловека и в биологических процессах. В частности, значение
гелей велико в процессах почвообразования и жизни почвы, так
как в. почве коллоиды находятся преимущественно в состоянии
геля.- К гелям относятся различные пористые и ионообменные
адсорбенты, ультрафильтры, искусственные мембраны, волокна
мышечных тканей, хрящи, клеточные оболочки, оболочки эритро-
цитов и различные Мембраны в организмах. Основным содержа-
нием любой живой клетки является протоплазма, которую
можно рассматривать как весьма подвижный студень, построен-
ный в основном из молекул белка.
Хрупкие гели. Структура хрупких гелей характеризуется
большим числом капилляров диаметром 2...4 нм, в которых проч-
но адсорбируется вода при обводнении высушенного геля. Так
как жесткие частицы создают прочный каркас, хрупкие гели
при обводнении и высушивании практически не изменяют своего
объема, за что они получили название ненабухающих
гелей.
Поглощение жидкости благодаря капиллярной структуре геля
происходит по законам капиллярной конденсации, т. е. по физи-
ческим законам. Отсюда следует, что хрупкие гели могут погло-
щать любую смачивающую их жидкость. Этот процесс сопро-
вождается своеобразным явлением, получившим название гис-
терезиса оводнения и обезвоживания. Явление
гистерезиса впервые было изучено на гелях S1O2 и РегОз и за-
ключается оно в том, что в гелях при одинаковых условиях
опыта процессы оводнения и обезвоживания осуществляются не
13*
371
Рис. 150. Кривые оводнения и
обезвоживания геля
по одной и той же кривой, как это
свойственно обратимым процессам,
а по разным. Явление гистерезиса
легко понять при рассмотрении кри-
вой давления пара при оводнении и
обезвоживании кремниевой кислоты
(рис. 150). Поглощение и обезво-
живание воды гелем характеризует-
ся циклическим процессом, однако
кривые оводнения и обезвоживания
не совпадают. Причина гистерезиса
заключается либо в необратимых изменениях, происходящих в
системе во время прямого процесса, либо в очень большой дли-
тельности срока установления в системе равновесного состояния.
Так, при обезвоживании узкие капилляры способны быстро
уменьшать свой объем (высыхать) и гораздо медленнее восста-
навливать его при оводнении.
Студни. Они представляют собой гомогенную систему, сос-
тоящую из ВМС и растворителя. Сплошная пространственная
сетка имеет в сечении молекулярные размеры и образована не
ван-дер-ваальсовыми, а химическими и водородными связями.
С одной стороны, студень можно рассматривать как раствор
ВМС, который образуется в том случае, если процесс растворе;
ния останавливается на второй стадии набухания, а с другой
стороны, как раствор ВМС, который под воздействием внешних
факторов потерял свою текучесть. Такие определения обуслов-
лены двумя возможными способами, получения студня. Студень
образуется из раствора полимера при его охлаждении, выпари-
вании или при добавлении в небольших объемах электролита;
по другому способу студень получают при ограниченном набу-
хании полимера в низкомолекулярной жидкости:
охлаждение
ограниченное
-- набухание
Раствор ВМС-------------—------------►- Студень-*-----------— ВМС
концентрирование,
добавление электролита
Процесс образования студня из раствора ВМС называется
застудневанием. Этот процесс состоит в возникновении
новых прочных связей между макромолекулами ВМС, которые
ранее существовали в растворе в качестве самостоятельных
кинетических единиц. Как говорилось ранее, в растворе ВМС
происходит возникновение и распад ассоциатов макромолекул.
Если связи между макромолекулами в ассоциатах становятся
настолько прочными, что ассоциат не распадается, то происходит
образование пространственной сетки. Причины возникновения
таких прочных связей могут быть разными. Так, например, если
полимер содержит ионогенные группы, то взаимодействие этих
групп, несущих противоположные по знаку заряды, является
одной из причин образования межмолекулярных связей. Поляр-
372
< . >
ные группы макромолекул также могут взаимодействовать
друг с другом. Иногда возможно образование и водородных
связей, и обычных химических связей.
Повышение концентрации растворов ВМС всегда увеличивает
вероятность застудневания, так как при этом возрастает веро-
ятность столкновения макромолекул или их фрагментов. Увели-
чение числа столкновений повышает возможность образования
межмолекулярных связей. Обычно в этом же направлении дей-
ствует и понижение температуры, хотя для отдельных систем
иногда может наблюдаться и обратная картина. Это бывает
лишь тогда, когда наблюдается отрицательный температурный
коэффициент растворимости ВМС в данном растворителе. Пере-
ход раствора в студень совершается при охлаждении непрерывно
и не характеризуется какой-либо определенной температурой.
Большое влияние на процесс застудневания в водных раство-
рах амфотерных ВМС имеет pH раствора. Процесс лучше всего
осуществляется при pH, близких к изоэлектрической точке.
В изоэлектрическом состоянии по всей длине макромолекулярной
цепи распределены противоположно заряженные группы, которые
взаимодействуют с такими же группами других молекул, спо-
собствуют установлению межмолекулярных связей.
Свойства студней. Наиболее характерной особенностью студ-
ней является их эластичность. Растворы ВМС, обладающие сла-
быми межмолекулярными связями, характеризуются большой
текучестью. Студень способен противостоять течению, и вплоть
до определенного значения приложенного сдвига ведет себя как
эластичное твердое тело. Степень эластичности связана с концен-
трацией: студни, содержащие в единице объема малое число
межмолекулярных связей, весьма эластичны, в то время как уве-
личение числа межмолекулярных связей увеличивает жесткость
образованного студня. Так, например, увеличение концентрации
раствора в 8 раз (от 0,05 до 0,4%) приводит к повышению кри-
тического напряжения сдвига более чем в 15 тысяч раз.
Некоторые студни обладают тиксотропными свойствами. Ме-
ханическое воздействие нарушает связь между макромолекула-
ми, и вся система становится текучей. Через определенное время
эти связи восстанавливаются и снова образуется студень.
Скорость диффузии низкомолекулярных веществ в студне
определяется сложностью пространственной сетки. В студнях
невысоких концентраций диффузия низкомолекулярных веществ
происходит практически с такой же скоростью, что и в чистом
растворителе. Это обусловлено наличием достаточно больших
промежутков между макромолекулами, соединенными друг с дру-
гом в трехмерную структуру. С увеличением концентрации студ-
ня, а также с возрастанием размера диффундирующих частиц
скорость диффузии уменьшается. Если размеры частиц диффун-
дирующего вещества так велики, что частицы не могут пройти
через макромолекулярную сетку, то диффузии вообще не будет.
На этом основано применение полупроницаемых мембран, обыч-
373
но являющихся типичными студнями. Так, при диализе малень-
кие молекулы или ионы легко диффундируют через мембрану, а
крупные коллоидные частицы или макромолекулы задерживаются.
При образовании студня равновесное состояние часто дости-
гается лишь после завершения процесса застудневания. Связи
между молекулами ВМС продолжают упрочняться в уже обра-
зованном студне, что приводит к специфическому явлению, кото-
рый называется синерезисом. При синерезисе объем
студня уменьшается, и жидкость (растворитель), выделяемая из
студня, образует новую макрофазу. При этом другая студнеоб-
разная макрофаза продолжает сохранять форму сосуда, в кото-
ром находился первоначальный студень.
Синерезис может также явиться следствием достижения рав-
новесия в студне при его охлаждении. Иногда синерезис проте-
кает благодаря осуществлению в студне химических процессов,
например разложения ВМС.
Уменьшение объема студня связано с концентрацией раство-
рителя в исходном студне: чем выше концентрация раствори-
теля, тем больше синерезис. При этом не существует определен-
ной зависимости скорости синерезиса от концентрации исходного
студня.
Для студней амфотерных белков наибольший синерезис осу-
ществляется в изоэлектрической точке. С отклонением pH среды
в ту или другую сторону от изоэлектрической точки синерезис
уменьшается, так как фрагменты макромолекулы приобретают
одноименный заряд, что приводит к взаимному отталкиванию
цепочек макромолекул друг от друга. Это в свою очередь вызы-
вает увеличение объема студня, а следовательно, и уменьшение
синерезиса. Влияние низкомолекулярных электролитов на синере-
зис различно, но, как правило, электролиты, способствующие
набуханию, уменьшают синерезис.
2. ПОЛУКОЛЛОИДЫ
Системы, характеризующиеся равновесными переходами:
Молекулярный раствор^ЗольГель
называются полуколлоидами или семиколлоид а-
м и. Полуколлоидные системы образуются при растворении по-
верхностно-активных веществ, а также некоторых красителей
мыла и таннинов. Из указанных систем наиболее хорошо изу-
чены растворы поверхностно-активных соединений.
В настоящее время коллоидные поверхностно-активные ве-
щества (ПАВ) используют в различных отраслях промышлен-
ности: для стирки и обработки тканей, в качестве диспергаторов
твердых веществ, эмульгаторов в производстве фармацевтиче-
ских и косметических препаратов. В последнее время они начи-
нают находить возрастающее применение в биологических
• 374
исследованиях, например для деструкции биологических мем-
бран.
Для полуколлоидов характерно образование в растворах ми-
целл. Этот факт для коллоидных ПАВ получил подтверждение
при изучении рассеяния света, осмотического давления и давле-
ния пара растворителя.
В настоящее время нет единого мнения относительно строе-
ния мицелл в растворах коллоидных ПАВ. В растворах • кол-
лоидных электролитов, согласно Мак-Бэну, содержатся плас-
тинчатые мицеллы (рис. 151); их сочетание определяет форму
кривой электрической проводимости. Существование пластинча-
тых мицелл было доказано рентгенографическими исследова-
ниями В. Филиппова. Пластинчатые мицеллы с уточнениями,
предложенными В. Филипповым, схематически изображены на
рис. 152. Одинаковые концы молекул, составляющих мицеллы,
обращены друг к другу, причем неполярные части молекул
образуют своего рода углеводородную фазу.
Г. Гартли предположил, что своеобразная углеводородная
фаза в мицеллах менее упорядочена, чем полагали Мак-Бэн
и В. Филиппов. Согласно Г. Гартли, мицеллы имеют сферическую
форму (рис. 153). Гидрофильные группы располагаются на по-
верхности мицелл, а неполярные звенья молекул обращены на
ее объем. Внутренняя часть таких мицелл близка по свойствам
и к жидким углеводородам. П. Дебай на основании данных по
Рис. 151. Структура пла-
стинчатых мицелл по
Мак-Бэну
Рис. 152. Пластичная ми-
целла по Филиппову
рассеянию света пришел к выводу, что мицеллы могут иметь
палочкообразную форму. Мицелла, по П. Дебаю, состоит из
большого числа плоских слоев, в каждом из которых полярные
группы располагаются по окружности и обращены к воде, а угле-
водородные «хвосты» направлены друг к другу (рис. 154).
Важнейшей характеристикой полуколлоидной системы служит
критическая концентрация мицеллообразования (ККМ). Так
называют минимальную концентрацию растворенного вещества,
при которой можно экспериментально обнаружить коллоидно-
дисперсную фазу. ККМ выражается в кмоль/м3 или в % раство-
ренного вещества. Критические концентрации мицеллообразова-
ния некоторых соединений приведены в табл. 53.
Особенности строения мицелл ПАВ обусловливают специфи-
ческое растворение в воде различных органических соединений,
обычно нерастворимых в воде без добавок ПАВ. Этот процесс
называется солюбилизацией. Механизм солюбилизации
различен для разного типа растворяемых соединений. Он обыч-
но осуществляется по одному из трех способов (рис. 155). Солю-
билизация неполярных соединений, например бензола, гексана,
объясняется их внедрением в углеводородную часть мицеллы.
Соединения, содержащие полярную группу, при солюбилизации
располагаются в мицелле так, чтобы их углеводородный «хвост»
находился внутри мицеллы, а полярная
группа была обращена наружу. Для со-
единений, содержащих несколько поляр-
ных групп, наиболее вероятна адсорбция
на поверхности мицеллы.
Содержание солюбилизированного ве-
щества возрастает с увеличением кон-
центрации раствора ПАВ. Данной темпе-
ратуре и концентрации ПАВ соответ-
ствует вполне определенное насыщение
раствора органическим веществом. В ре-
зультате солюбилизации получают устой-
чивые дисперсные системы.
Рис. 153. Сферическая ми-
целла по Гартлу
Рис. 154. Мицелла
по Дебаю
376
f I'D
Поверхностно- 1 3
активное вещество Солюбилизат
Рис. 155. Три возможных способа солюбилизации:
/ — соединения с .полярной группой и гидрофобной частью; 2—соединения без
гидрофобной части; 3 — неполярные соединения
Таблица 53. Критические концентрации мицеллообразования
некоторых соединений
Соединение
Критическая
концентрация
(кмоль/м3) • 104
Соединение
Критическая
концентрация
(кмоль/м3) • 104
Стеарат калия
Олеат калия
Пальмиат калия
Додецилсульфат
натрия
323
298
323
298
12
22
Додециламмо-
нийхлорид
Холат натрия
Дезоксихолат
натрия
Моностеарат са-
харозы
303
298
298
293
140
130
21
0,046
8
Экспериментально установлено, что солюбилизация углево-
дородов падает с ростом длины цепи, а солюбилизирующая
способность ПАВ в пределах одного гомологического ряда воз-
растает по мере увеличения числа углеродных атомов. Очень
высока солюбилизирующая активность биологически активных
коллоидных электролитов — холата и дезоксихолата натрия.
Солюбилизация — один из первых актов усвоения животными
жиров из пищи.
Наиболее важное свойство растворов коллоидных ПАВ —
это их моющее действие. Как правило, частицы загрязняющих
веществ являются гидрофобными, т. е. не смачиваются водой.
Поэтому даже при высокой температуре моющее действие одной
воды очень мало. Удаление загрязнений с любой поверхности
значительно облегчается, если для этого применяют раствор кол-
лоидных ПАВ.
При контакте раствора ПАВ — мыла или любого моющего
377
средства — с загрязненной поверхностью молекулы ПАВ адсор-
бируются на частицах грязи и на очищаемой поверхности.
Молекулы ПАВ постепенно проникают между загрязняющими
частицами и очищаемой поверхностью, создавая так называе-
мое расклинивающее действие, которое отрывает частицу грязи
от поверхности. Отдельные частицы загрязняющих веществ об-
разуют в жидкой фазе эмульсию или суспензию, которые стаби-
лизируются молекулами ПАВ. Кроме того, коллоидные ПАВ
являются хорошими пенообразователями, и частицы грязи вмес-
те с пеной легко удаляются водой. Таким образом, моющее
действие включает в себя ряд коллоидно-химических процессов:
адсорбцию, эмульгирование, стабилизацию суспензии и пенооб-
разование.
Отрицательной стороной синтетических ПАВ как моющих
средств в быту и промышленности является их стойкость к
биологическому разложению. Если моющие ПАВ являются хими-
чески и биологически устойчивыми веществами, то, попадая в
воду или почву, они загрязняют окружающую среду.
Контрольные вопросы
1. Как влияет на процесс застудневания концентрация растворов ВМС и pH?
2. Как влияет синерезис на достижение равновесия в студне?
3. Сопоставьте строение мицелл в лиофобных коллоидах и в растворе ПАВ.
4. Какова роль солюбилизации в биологических процессах?
5. Каково значение гелей в процессах почвообразования и жизни почв?
ЗАКЛЮЧЕНИЕ
Основы физической и коллоидной химии позволяют заложить
фундамент развития качественных и количественных представ-
лений об окружающем мире. Эти знания необходимы для даль-
нейшего изучения таких специальных дисциплин, как агрохимия,
почвоведение, агрономия, физиология растений и животных
и др. Современное состояние науки характеризуется рассмотре-
нием основных физико-химических процессов на атомно-молеку-
лярном уровне. Здесь главенствующую роль играют термодина-
мические и кинетические аспекты сложных физико-химических
взаимодействий, определяющих в конечном счете направление
химических превращений. Выявление закономерностей протека-
ния химических реакций в свою очередь подводит к возможности
управления этими реакциями при решении как научных, так и
технологических задач. Роль каталитических (ферментативных)
и фотохимических процессов в развитии и жизни растений и
организмов чрезвычайно велика. Большинство технологических
процессов также осуществляется с применением катализа. Поэ-
тому изучение основ катализа и фотохимии необходимо для
последующего правильного подхода к процессам, происходящим
в природе, и четкого определения движущих сил этих процессов
и влияния на них внешних факторов. Перенос энергии часто
осуществляется с возникновением, передачей и изменением
значений заряда частиц. Для понимания этой стороны сложных
превращений необходимо знание электрохимических процессов.
Зарождение жизни на Земле и ее развитие невозможно без
участия растворов, представляющих собой ту необходимую сре-
ду, где облегчается переход от простого к сложному и создаются
благоприятные условия для осуществления реакций, особенно
успешно протекающих на разделе двух фаз.
Коллоидная химия связана прежде всего с изучением двух
крупных классов систем с высокоразвитой поверхностью —
лиофобных коллоидов и растворов высокомолекулярных соеди-
нений. Роль и значение этих систем не требует доказательств.
Достаточно упомянуть, что к ним относятся такие соединения,
как белки. Решение основных задач по интенсивной технологии
в сельскохозяйственном производстве диктует необходимость
получения будущими специалистами фундаментальных знаний, в
частности, по физической и коллоидной химии.
ЛИТЕРАТУРА
Балезин С. А., Ерофеев Б. В., Подобаев Н. Н. Основы физической и коллоид-
ной химии. — М.: Просвещение, 1975.
Болдырев А. И. Физическая и коллоидная химия. — М.: Высшая школа, 1983.
Воюцкий С. С. Курс коллоидной химии. — М.: Химия, 1976.
Глазов В. М. Основы физической химии. — М.: Высшая школа, 1981.
Киреев В. А. КурС физической химии. — М.: Химия, 1978.
Кнорре Д. Г., Крылова Л. Ф., Музыкантов В. С. Физическая химия. — М.:
Высшая школа, 1981. J
Николаев Л. А. Физическая химия. — М.: Высшая школа, 1979.
Нобел П. Физиология растительной клетки (физико-химический подход). —
М.: Мир, 1973. ;
Фичини ж., Лаи(брозо-Бадер Н., Депезе Ж. К. Основы физической хи-
мии. — М.: Мир, 1972)
ПРИЛОЖЕНИЕ
1. Атомные массы
Элемент Символ Атомная масса Элемент Символ Атомная масса
Азот N 14,0067 Лоуренсий, Lw [257]
Актиний Ас [227] Лютеций Lu 174,97
Алюминий А1 26,9815 Магний Mg 24,312
Америций Ат [243] Марганец Мп 54,9380
Аргон Аг 39,948 Медь Си 63,54
Астат At [210] Менделевий Md [256]
Барий Ва 137,34 Молибден Mo 95,94
Бериллий Be 9,0122 Мышьяк As 74,9216
Берклий Bk [247] Натрий Na 22,9898
Бор В 10,811 Неодим Nd 144,24
Бром Вг 79,909 Неон He 20,183
Ванадий У 50,942 Нептуний Np [237]
Висмут Bi 208,980 Никель Ni 58,71
Водород Н 1,00797 Ниобий Nb 92,906
Вольфрам W 183,85 Нобелий No [254]
Гадолиний Gd 157,25 Олово Sn 118,69
Галлий Ga 69,72 Осмий Os 190,2
Гафний Hf 178,49 Палладий Pd 106,4
Гелий Не 4,0026 Платина Pt 195,09
Германий Ge 72,59 Плутоний Pu [242]
Гольмий Но 164,930 Полоний Po [210]
Диспрозий Dy 162,50 Празеодим Pr 140,907
Европий Eu 151,96 Прометий Pm [147
Железо Fe 55,847 Протактиний Pa [231
Золото Au 196,967 Радий Ra [226
Индий Id 114,82 Радон Rn [222
Иод I 126,9044 Рений Re 186,2
Иридий Ir 192,2 Родий Rh ’ 102,905
Иттербий Yb 173,04 Ртуть Hg 200,59
Иттрий Y 88,905 Рубидий Rb 85,47
Кадмий Cd 112,40 Рутений Ru 101,07
Калий К 39,102 Самарий Sm 150,35
Калифорний Cf [247] Свинец Pb 207,19
Кальций Ca 40,08 Селен Se 78,96
Кислород 0 15,9994 Сера S 32,064
Кобальт Co 58,9332 Серебро Ag 107,870
Кремний Si 28,086 Скандий Sc 44,956
Криптон Kr 83,80 Стронций Sr 87,62
Ксенон Xe 131,30 Сурьма Sb 121,75
Кюрий Cm [247] Таллий T1 204,37
Лантан La 138,91 Тантал Ta 180,948
Литий Li 6,939 Теллур Те 127,60
381
Элемент Символ Атомная масса Элемент Символ Атомная масса
Тербий ТЬ 158,924 Фтор F 18,9984
Технеций Тс 97 Хлор С1 35,453
Титан Ti 47,90 Хром Сг 51,996
Торий Th 232,038 Цезий Cs 132,905
Тулий Tm 168,934 Церий Се 140,12
Углерод С 12,01115 Цинк Zn 65,37
Уран и 238,03 Цирконий Zr 91,22
Фермий Fm [253] Эйнштений Es [254]
Фосфор Р 30,9738 Эрбий Er 167,26
Франций Fr [223]
2. Соотношения между некоторыми единицами СИ
и единицами других систем
(отношение единицы СИ к единице указанной системы)
Величина Обозна- чения единиц СИ Число единиц других систем, содержащихся в единице СИ
обозна- чение соотноше- ние обозна- чение соотно- шение обозна- чение соотно- шение
Длина м СМ 102 см ю2 м 1
Масса кг г 103 г 10’ т.е.м. 0,102
Время с с 1 с 1 с 1
Сила Н дин 105 дин ю5 кГ 0,102
Энергия Дж эрг 107 эрг ю7 кГм 0,102
Мощность Вт эрг/с ’ 107 эрг/с ю7 кГм/с 0,102
Давление Па дин/см2 10 дин/см2 10 кГ/м2 0,102
Сила тока А — 3-109 — 10-' — —
Количество элек- тричества Кл — 3-109 — 10-' — —
Напряжение В — 1/300 — ю8— —
Электрическая индукция Кл — 4л-3-105 — 4л-10-5 — —
Поток электри- ческой индукции Кл/м2 — 4л-3-109 — 4л-10-1 — —
Напряженность электрического В/м — 4--ю-4 — 106 — —
поля Электрическая емкость Ф см 3 9-10" 10-9
Электрическое сопротивление Ом — — 109 — —
Напряженность
магнитного поля А/м — 4л-3-107 э 4л-1О-3 — —
Магнитная ин- дукция Тл — ю-6 Гс / 104 — —
Диэлектрическая 4л-9-109 4л-10-"
проницаемость Ф/м — — — —
382
3. Внесистемные единицы
Наименование величины Единица
Наименование Обозначение Размер единицы в СИ
Длина микрон мк 10“6 м
миллимикрон ммк . 10“9 м
ангстрем А 10-10м
Масса тонна т 103 кг
центнер ц 102 кг
Время час ч 3600 с
минута мин 60 с
Плоский угол градус О W рад
минута / w 10-2 рад
секунда ft \ \ 10-3 рад
Площадь ар а 100 м2
гектар га Ю4 м2
Объем литр л ~ 10-3 м3
Мощность лошадиная сила л.с. ~ 735,5 Вт
Работа и энергия ватт-час Вт-ч 3,6-103 Дж
киловатт-час кВт-ч 3,6-106 Дж
электрон-вольт эВ ~ 1,602-10-” Дж
мегаэлектрон-вольт МэВ ~ 1,602-10-13 Дж
калория кал ~ 4,19 Дж
Давление бар миллиметр ртутно- бар 105 Н/м2
го столба мм рт.ст. 133,322
техническая атмос-
фера ат 9,81-104
4. Некоторые важнейшие физические константы
Скорость света в вакууме
Нормальное ускорение силы тяжести
Постоянная Авогадро
Универсальная газовая постоянная
Постоянная Больцмана
Постоянная Планка
Заряд электрона
Масса покоя электрона
протона
нейтрона
Удельный заряд электрона
Электрическая постоянная
Магнитная постоянная
с — 2,998-108 м/с
g = 9,81 м/с2
Nk = 6,023-1026 кмоль'1
R = 8314 Дж/ (кмоль - К)
k = 1,38-10“23 Дж/К
Л = 6,62-10~34 Дж-с
е — 1,6-10~19 Кл
те = 9,1 • 1О.“31 кг
тр ~ 1,672-10~27 кг
тп = 1,675-10~27 кг
е/те = 1,756- 10й к/кг
80 = 8,85-10~12 Ф/м
Ио= 1,26-10~6 Гн/м
383
5. Множители и приставки для образования десятичных кратных
и дольных единиц
Множи- тель Приставки Множи- тель Приставки
Наимено- вание Обозначение Наимено- вание Обозначение
русское между- народное русское между- народное
ю18 экса Э Е 10~' деци Д d
1015 пета П Р 10~2 санти с с
ю12 тера Т Т 10-’ милли м m
ю9 гига Г G 1(Г6 микро мк И
106 мега м М 10~9 нано н п
103 кило к к 10-12 ПИКО п Р
102 (гекто) г h 10~15 фемто ф f
10* (дека) да da 10-18 атто а а
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Абсорбция 267
Абсолютный нуль 87
Абсолютно гидрофобные тела 286
Автокатализ 144
Агрегат мицеллы 318
Агрегативная устойчивость 294, 321
Адгезия 284
Адсорбат 267, 268
Адсорбент 267, 268
Адсорбция 145, 146
— газов 267
— изотерма 269
— из растворов 276
— локализованная 268
— молекулярная 270
— нелокализованная 268
— обменная 278
— полимолекулярная 274
— теории 270
— физическая 268,269
— химическая 268, 269
~ электролитов 277
Активированный комплекс 131
Активность 183, 194, 195
— фермента 150, 151
Активный комплекс 139, 141, 143, 146
— центр 123, 142, 147, 150, 273
Акцептор 196
Аллотропия 28
Аминокислоты 362
Аморфное состояние 27, 28
Аниониты 280
Антагонизм электролитов 330
Ассоциаты 358
Аэрозоли 348
Барьер энергетический 132, 133, 150
Белки 353, 354, 355, 361, 362, 369, 370
Биоаэрозоли 350
Биополимеры 359
Биопотенциалы 357
Биотехнология 3
Биполярные ионы 354
Броуновское движение 87, 300, 301
Буферная емкость 214
Буферные растворы 210, 211
Буферные системы почв 214
Вероятность,
— взаимной ориентации 135
— математическая 85, 86, 135
— термодинамическая 84, 135
Взаимодействие
— дипольное 25,26
— индукционное 25, 26
— кислотно-основное 196
— межионное 193
— межмолекулярное 25, 26
— межчастичное 167, 168
— ориентационное 25, 26
Внешняя среда 50
Вода 44, 45
— диаграмма состояния 106, 107
— диссоциация 200, 201
— ионное произведение 200
— полярность молекул 46
— структура 45, 46
Второе начало термодинамики 79, 80
— формулировка Клаузиуса 81
— формулировка Кельвина и Планка
81
Вязкость 39—42
— лиофобных коллоидов 309
— относительная 42, 358
— растворов ВМС 357
— характеристическая 359
385
Газы 9, 10
— адсорбция 267
идеальные 9, 10, 85
— кинетическая теория 15
— основные законы 10, 11
— парциальное давление 14
— реальные 21
— уравнение состояния
Гальванический элемент 239, 240
Гели 371, 372
Гидратация 185, 186
Гидраты 185, 186, 211
Гидролиз 207, 208, 213, 297
Гидрофильные материалы 286
Гидрофобные материалы 286
Гистерезис оводнения 371
Гранула 318, 319
Давление 52, 57
— капиллярное 286
— критическое 24
— набухания 367
— насыщенного пара 43
— осмотическое 179
— парциальное 14
Двойной электрический слой
Двусторонние химические реакции 121
Денатурация 369
Десублимация 8
Дзета-потенциал 320, 321
Диаграммы
— фазовые 106
— энергий 131, 132, 139
Диализ 310
Дипольный момент 37
Дисперсионная среда 293
Дисперсная фаза 593
Дисперсность 293, 304, 342
Диссоциация 184
Диффузионный потенциал 262, 263
Диффузия 306
Диффузный слой
Диэлектрическая постоянная 187, 188
Длина цепи 124
Донор 196
Дым 349
Жидкости
— ассоциация 48, 358
— взаимная растворимость 168 .
— вязкость 39
— вязкость ньютоновская 40
— несмешивающиеся 169
— плотность 36
Желатина 338 _>
Закон(ы)
— Авогадро 15
— Бойля — Мариотта 10
— Генри 170
— Гей-Люссака 11
— Гесса 71, 72, 73, 186
— Грэма 17
— Дальтона 14
— . Джоуля 65
— действующих масс 100, 101
— Кирхгофа 69
— Кольрауша 227
— ; Кулона 187
— Лавуазье — Лапласа 74
— Ламберта — Бера 159
— Максвелла — Больцмана 19
— Освальда 205
— распределения 169
— Рауля 174, 175
— Фика 17
— фотохимической эквивалентности
155
— Шарля 11
— Штарка — Эйнштейна 153
Зарождение цепи 123, 124
Заряд частицы ВМС 354
Защитное действие растворов ВМС 338
Золи 296, 297
Изоморфизм 28
Изотерма адсорбции 269
Изотропность 27
Изоэлектрическая точка 352
Интервал размягчения 27
Инфракрасные спектры поглощения
112, 146, 362
Иониты 270
Ионндя пара 189
Ионная сила растворов 195
Ионное произведение воды 200
Ионов
— абсолютная скорость 217
386
— взаимодействие 186, 187
— заряды 186, 187, 221
— образование 185, 186
— скорость движения 216
Калориметр 69
Капиллярная конденсация 275
Катализ 139, 140
— гетерогенный 142, 144, 145
— гомогенный 142, 143
— кислотно-основной 141, 14?
— кислотный 143
— окислительно-восстановительный
141, 142
— ферментативный 149
Катализатора
— активность 145, 147
— носители 145
— поверхность 145
— промоторы 145
— специфичность 143, 150
— яды 145
Катиониты 280
Квантовый выход 155, 156
Кванты света 153, 154, 155
Кинетическая устойчивость 293, 306,
321, 322
Кислотно-основные пары 197, 198
Кислоты Льюиса 197
Коагуляция 327, 328
— быстрая 335
— в почвенных процессах 340
— взаимная 330
— кинетика 334, 335
- — концентрационная 331
— механизм 331
— нейтрализационная 331
— правила 327
— скрытая 334
— теория 332, 335
— электролитами 327, 328
Коалесценция 345
Коацервация 370
Когезия 284
Коллоидная мельница 297
Кондуктометрическое титрование 234
Константа
— Больцмана 15
— Ван-дер-Ваальса 22
— Генри 170
— Михаэлиса 151
— кислотности 202
— криоскопическая 170
— критическая 214
— основности 203
— равновесия 96
— седиментация 307
— скорости реакции 116, 117
— эбуллиоскопическая 177
Контракция 363
Коэффициент
— активности 183
— вириальный 23
— вязкости 39
— диффузии 302, 335
— изотонический 179, 192
— полезного действия 80, 81
— распределения 169
— сжимаемости 21
— трения 303
Краевой угол 285
Кривые
— давления пара 106, 107, 182
— кинетическая 113, 114
— плавления 106, 107
— распределения 307
— расслоения 168'
— сублимации 106, 107
Криоскопия 175
Кристаллоиды 292
Кристаллы
— жидкие 48, 49
— лиотропные 48, 49
— нематические 49
— смектические 48
— термотропные 49
—' холестерические 49
Критическое давление 24, 25
Критический объем 24, 25
Критическая температура 24, 25
Критическая точка 24
Лед 44, 45, 106, 107
Лиотропные ряды 369
Лиофобные коллоиды-
— — коагуляция 327, 328
— — очистка 311
— — поглощение света
387
— — светорассеяние 312
— — устойчивость 321
Макромолекулы 352, 363, 374
Масс-спектрометрия 112
Мембраны 49, 178, 322
Механизм реакции 111, 140
— — каталитической 141, 145, 146
Мицелла 316, 317
Мицеллообразование 375
Молекулы
— активные 131, 136
— возбужденные 154, 159
— дифильные 277, 291
— неполярные 168
— ориентированные 135, 145
— полярные 168
— устойчивые 71
Молекулярный «чистокол» 291
Моляльность 173, 175
Молярная доля 173
Молярная концентрация 173
Мультиплеты 147, 148
Мутаротация 362
Набухание 313, 314
— давление 367
— кинетика 364
— неограниченное 364
— ограниченное 364
— скорость 364
— степень 364
— факторы 365
Направление процесса реакции 140,
141
Насыщенный пар 173
Нефелометрия 315
Обратимые реакции 121
Обрыв цепи 123, 124
Объем 52
— критический 24, 25
— молярный 32, 36
— парциальный молярный 172, 173
— соответственный 25
— стандартный молярный 12
Определение
— влажности 234
— солесодержания 233
388
— энергии активации 133, 134
Оптическая активность 362
Осмометр 179, 180
Осмотическое давление 178, 179, 304
— — лиофобных коллоидов 303
— — растворов ВМС 360
Основания Льюиса 197
Параметры 105
— зависимые 53
— интенсивные 53
— макроскопические 84
— микроскопические 84
— независимые 52
— термодинамические 52
— фотохимических реакций 156, 157
— экстенсивные 53
Парообразование 43
Парциальное давление 14
Парциальный объем 172
Пенообразователи 348
Пены 347
Пептизация 298, 337
— посредственная 299
— непосредственная 299
Первое начало термодинамики 60
Перезарядка золей 331
Переход 105
— фазовый 105
— электрона 154
Плазма 8
Плазмолиз 181
Плотность 36
Поверхностно-активные вещества 289,
290, 374
Поверхностно-инактивные вещества
289, 290
Поверхностное натяжение 57, 280.
Поглощение энергии излучения 153,
155
Подвижность ионов 227
Полидисперсность 307
Полиморфизм 28
Полуколлоиды 374
Поляризуемость 39
Полярность
— молекул 37—39
Порог коагуляции 327
Постоянная
— Авогадро 15
- - Генри 170
— диэлектрическая 38
— Фарадея 227
Потенциал
— гравитационный 57
— диффузионный 260, 261
— изобарно-изотермический 89
— изохорно-изотермический 90
— контактный 247
— мембранный 357
— окислительно-восстановительный
256, 257
— протекания 322
— седиментации 322
— химический 93, 94
— электродный 248
— электродный стандартный 249
— электрокинетический 320
Потенциалопределяющие ионы 317, 318
Потенциометрия 264
Потенциометрическое титрование 265
Правило
— Антонова 283
— Гаркинса 284
— осадка 337
— Траубе 289
— фаз 105, 351
— Фаянса — Пескова 278
— Шульце — Гарди 327
Превращение
— необратимое 52, 82, 83
— обратимое 54, 82, 83
— термомёханическое 51, 92
Предэкспоненциальный множитель 131,
135, 136, 140
Принцип
— Ле Шателье 97, 98, 170, 173, 190,
200, 366
— энергетического соответствия 148
Проводимость
— металлическая 216
— твердых тел 216
— электрическая 216
— электрическая газов 216
— электролитическая 216
Продукты 63, 112, 113, 131, 132
Произведение растворимости 252
Промежуточный продукт 120, 136
Противоионы 317, 318
Процесс
— биологический 130, 171, 181, 200,
214, 259, 367
— изобарный И
— изотермический 10
— изохорный 11
— каталитический 139, 140
— необратимый 54, 82, 83, 93
— обратимый 54, 82, 83, 93
— обратный 95, 97, 121
— почвенный 259, 279, 334
— прямой 95, 97, 121
— самопроизвольный (см. необрати-
мый)
— экзотермический 268
— элементарный 111
Пыль 349
Работа 51, 55, 56
— гравитационная 57
— изменения поверхности 56, 57
— механическая 55, 56; 57, 60
— расширения газа 56, 57
— электрическая 56, 57
Равновесие 53, 54, 86, 95, 96, 122, 140
— адсорбционно-десорбционное 269
— влияние внешних условий 97, 98,
102, 103
— гетерогенное 269
— гидролиза 297
— динамическое 95, 122
— диссоциации 210
— истинное 140
— константа 96, 108, 140, 145
— мембранное 356
— устойчивое 95
— фазовое 105, 106
— химическое 95
— электрохимическое 247
Радикал 120, 123, 142
— рекомбинация 120, 156, 158, 159
— свободный 158
— стабильный 120
Развитие цепи 123
Расплав 34, 36
Распределение по скоростям 19, 131
Растворение 85, 86, 165, 363
Растворимость
389
— влияние внешних условий 170
— газа 170
— малорастворимых соединений 232
— неограниченное 168
— ограниченное 168
Растворитель 165, 188
Растворов
— высаливание 172
— замерзание 174
— компоненты 165, 172, 185
— кристаллизации 175
— образование 165, 166, 185, 186
— перекристаллизация 166
— разбавление 222, 223 .
— свойства 167, 172, 191
— состав 167, 173, 182
Растворы
— бесконечно разбавленные 167
— буферные 210
— водные 185, 186
— высокомолекулярных соединений
294, 351, 352
— газовые 165
— гипертонические 181
— гипотонические 181
— жидкие 165
— реальные 167, 168
— изотонические 181
— истинные 293, 295, 303
— кислые 201, 203, 204-
— коллоидные
— насыщенные 166
— нейтральные 201
— неэлектролитов 165, 166
— нулевые 247
— пересыщенные 166
— разбавленные 173, 174
— ^реальные 168, 182
— твердые 165
— щелочные 201, 203, 204
— электролитов 185, 186
Растекание 283
Рассеяние свеТа 312
Реагенты 63, 112, 113, 131, 132, 136,
139
Реакции
— бимолекулярные 117
— второго порядка 128, 129
— гетерогенные 112, 122
— гомогенные 112
— двусторонние 121
— каталитические 139, 140
— константа скорбсти 114, 116
— лимитирующие 122
— медленные 118
— молекулярность 117, 118
— мономолекулярные 117
— нулевого порядка 129
— обратные 121, 122
— обратимые 121
— окислительно-восстановительные
256, 259
— первого порядка 125, 126
— порядок 114, 115, Г17
— последовательные 119
— промежуточные 112
— прямые 121, 122
— радиационно-химические 153
— радиоактивного распада 127
— сенсибилизированные 159
— скорость 113, 114
— сложные 118, 119, 129, 130
— сопряженные 120
— стадия диффузионная 122
— стадия кинетическая 122
— темновые 153
— тримолекулярные 117
— ферментативные 130
— фотохимические 153, 154
Решетка
— ионная 30, 31
— ковалентная 30, 31, 32
— кристаллическая 30, 33
— металлическая 30, 33
— молекулярная 30, 32
Ряд
— лиотропный 328, 369
— напряженный 250
Световая стадия фотосинтеза 162 *
Светорассеяние лиофобных коллоид-
ных растворов ВМС 361
Свободный пробег молекул 18
Связь
— внутримолекулярная 145
— водородная 168
— ионная 31
— ковалентная 168
390
Седиментация 305
Седиментационный анализ 308
Седиментометр 308
Сенсибилизатор 159
Синерезис гелей 374
Система 50, 51
— буферная 210
— двухфазная 106, 107
— дивариантная 106, 107
— дисперсионная 293
— дисперсная 293
— закрытая 50, 63, 89
— изолированная 50, 81, 82, 83, 85
— инвариантная 106, 107
— коллоидная 292
— моновариантная 106, 107
— однофазная 106, 107, 294
— открытая 51, 137
— химический анализ 112
— химический состав 85
Скорость молекул 19, 20, 85
— — наиболее вероятная 19, 20
— — среднеарифметическая 19, 20
— — средняя квадратичная 19, 20
Скорость реакции
— — влияние температуры 129
— — зависимость от концентрации
125
— — каталитической 140
— — константа 114
— — мгновенная 114
— — средняя 113
— — фотохимической 158
Слипание частиц 325
Смачивание 280, 281
Смещение частиц 302
Солесодержание 233
Солюбилизация 376
Сольватация 170, 185
— мицеллы 326
Сопряженные кислоты и основания 199
Сорбция 267
Состояние
— агрегатное 8
— аморфное 27, 28
— газообразное 9
— жидкое 34, 35, 36
— кристаллическое 27, 28
— критическое 23, 24
— макроскопическое 85, 86, 87
— мезоморфное 48
— микроскопическое 85, 86
— паракристаллическое 48
— переходное 136
— равновесное 53
— соответственное 25
— стандартное 64
— фазовое 142
— энергетическое 155
Специфичность катализатора 141, 142
Старение золей 336
Стандартный молекулярный объем 12
Стандартная ЭДС 240
Степень
— гидролиза 208, 209
— дисперсности 292
— диссоциации 190, 191
Студни 371
Сублимация 185
Субстрат 150
Суспензии 342
Темновая стадия фотосинтеза 162
Температура
— абсолютная 28, 52
— замерзания раствора 174
— кипения 43, 44
— кипения раствора 177
— критическая 24
— критическая растворения 169
— плавления 27
Температура инактивации ферментов
130
Температурный оптимум растений 130
Теория
— адсорбции 270
— активных центров 147
— ансамблей 148 '
— Бренстеда 197
— Брунауэра, Эмпета и Теллера 274
— газов кинетическая 15
— Гедройца 279
— Гельмгольца 319
— Гуи 320
— коагуляции 332
— Льюиса 196
— мультиплетов 147
— переходного состояния 134
391
— сильных электролитов 193, 194
— столкновений 134
— Штещш,Л18
— электролитической диссоциации
‘ 185, 186
Теплоемкость 66, 67
— изобарическая 66, 68
— изохорическая 66, 68
— молярная 66
— продуктов 68, 69
— реагентов 68, 69
— средняя 66
— удельная 66
Теплота 52, 56, 79, 80
— гидратации 186, 187
— испарения 183
— растворения 185, 186
— реакции 63, 66, 73
— сгорания 75
— скрытая 70
— смачивания 286
Термометр Бекмана 176
Термопреципитация 350
Термофорез 349
Течение
— ламинарное 40
— турбулентное 40
Тиксотропия 333
Титрование
— • кондуктометрическое 234
— потенциометрическое 265
Тиксотропия 333
Третье начало термодинамики 87, 88
Тройная точка 107
Туманы 349
Тургор 181, 357
Удельная
— поверхностная энергия 267
— электрическая проводимость 220
Ультразвук 298
Ультрамикроскопия 314
Ультрафильтрация 311
Ультрацентрифугирование 307
Универсальная газовая постоянная 12,
13
Уравнение
— адсорбции 269
— Аррениуса 131
— Ван-дер-Ваальса 2U 22,.
— Вант-Гоффа 103
— вариальное 24
— Гиббса 288
— Гиббса — Гельмгольца 245
— Карно 80
— кинетическое 115, 116, 119
— Клапейрона 108, 109
— Лапласа 283, 287
— Ленгмюра 272
— Нернста 249
— Никольского 279
— Релея 313
— состояния адсорбционного слоя 290
— состояния идеального газа 13
— Томсона 288
— Фрейндлиха 272
— Фрумкина 290
— Шишкове кого 290
Устойчивость золей
— — агрегативная 294, 321
— — кинетическая 293, 321
Устойчивость_растворов
— — лиофебных 324
.---ВМС 368
Фаза 105, 112
Ферментативная активность 151
Ферментов структуры 149
Ферментсубстратный комплекс 150, 151
Ферменты 49, 130
Физическая конденсация 295
Флуоресценция 344
Фотосинтез 160, 161
Фотосинтетические пигменты 163
Фотохимические реакции 154, 155 .
— — в газовой фазе 156
— — в растворе 156
Фотохимическое
— возбуждение 154, 155, 162
— диссоциация 154, 155
— ионизация 154, 155
Фотофорез 349
Функция состояния 57, 58, 73, 74
Хемосорбция 268
Химическая индукция 120
Химическая конденсация 296
Хлоропласты 49, 162
392
Хлорофилл 160, 162, 163
Хронокондуктометрическое титрование
237
Частота столкновений 20, 21
Частица промежуточная 119
Число
— золотое 309
— компонентов 105, 106
— переноса 218
— плоскостей 107
— рубиновое 310
— серебряное 310
— серное 310
— степеней свободы 105, 106
— фаз 105, 106
Цепные реакции
— — инициированные 124
— — простые 124
— — разветвленные 124
— — сплошь разветвленные 125
Эбуллиоскопия 177
Экстракция 170
Электрическая проводимость
— — молярная 222, 223
— — удельная 220, 221
Электрическое распыление 298
Электрод
— водородный 248
— второго рода 251
— индикаторный 251
— каломельный 251
— окислительно-восстановительный
256
— первого рода 250
— с водородной функцией 254
— с металлической функцией 254
— сравнения 248
— стеклянный 253, 255
— хлорсеребряный 252
Электродвижущая сила 220, 238, 239,
261
Электродиализ 311
Электрокинетические давления 319
Электролиты 185
— бинарные 190
— сильные 193, 194, 223
— слабые 190, 191, 221, 223
Электронные орбитали 154
Электроосмос 321
Электроультрафильтрация 312
-Электрофорез 321, 353, 355
Элемент,
— Вестона 243
— Даниэля — Якоби 239
— концентрационный 260
— необратимый 238, 239
— обратимый 238, 239
Эмульгаторы 345, 346
Эмульсии 343
— второго рода 344
— высококонцентрированные 344
— концентрированные 344
— первого рода 344
— разбавленные 344
— разрушенные 347
Энергия
— активации 20, 130, 131, 269
— вращательная 55
— внутренняя 55, 60, 61, 63, 64, 91
— внутримолекулярная 55
— внутриядерная 55
— гравитационная 51
— излучения 153, 154
— кинетическая 55
— колебательная 55
— лучистая 55
— механическая 80
— молекул 131
— поверхностная 267
— поглощенных квантов 155
— поступательного движения 55
— потенциальная 55, 57, 81
— решетки 135
— свободная Гельмгольца 90, 91, 94,
140
— свободная Гиббса 89, 90, 94, 100
и 101, 363
— свободная образования 91
— свободная поверхностная 281
— связанная 81
— связи 75, 76, 131
— удельная поверхностная 267
— электрона 153, 154
Энтальпия
— образования стандартная 71
393
— продуктов 64
— реагентов 64
— растворения 185
— смешения 165, 166, .170
Энтропийный фактор 137
Энтропия 82, 83
— абсолютная 88
— активация 135, 139
— продуктов 89
— растворения 166
— реагентов 89
— статистическая интерпретация 84
Эстафетный механизм 218
^Эффект
— Доннана 357
— Тиндаля 312
— Франка — Рабиновича 158
Эффективные соударения 131, 132, 133
Эффузия 17
ИМЕННОЙ УКАЗАТЕЛЬ
Авогадро 15, 16, 41, 155, 240, 302, 324
Антонов 283
Аррениус 131, 133, 135, 137, 185, 193,
196, 226, 227
Баландин 147, 148
Бачинский 40
Бекман 1,76
Бер 315, 316
Бойль 10, 12, 23
Больцман 15, 19, 84, 85, 86, 136, 303
Борщев 292
Бренстед 196, 197
Броун 300
Брунауэр 274
Вааге 102
Ван-дер-Ваальс 21, 22, 25, 36, 290
Вант-Гофф 103, 178, 179, 303, 304, 305,
360, 361
Веймарн 292
Вестон 242, 243, 244
Вин 47
Воюцкий 330
Гарди 327, 368
Гаркинс 284, 285
Гартли 374
Гедройц 279, 280
Гей-Люссак 11, 12
Гельмгольц 90, 245, 319, 320, 322
Генри 170, 171
Герасимов 156
Гесс 72, 73, 74
Гиббс 89, 105, 245, 288, 289
Урбтгус 153
Грэм 16, 17, 292
Гузман де 41
Гуи 320
Гульдберг 102
Дальтон 14
Даниэль 240, 242
Дебай 38, 193, 195, 230, 374, 376
Дерягин 330, 332, 333
Джоуль 65
Доннан 357
Дорн 321, 322
Дракин 187
Думанский 306
Зельдович 273
Зёренсен 201
Зигмонди 301, 314, 339
Каргин 294, 316
Квинке 322
Кельвин 81
Кирхгоф 69
Кистяковский 185
Клапейрон 108, 109
Клаузиус 60, 81, 82
Кобозев 148
Кольрауш 227, 230
Кон 369
Крист 96
Кулон 187
Ламберт 315, 316
Ландау 333
Лаплас 285, 287, 324
Ленгмюр 270, 272, 274, 290
Ле Шателье 97, 103, 173, 269
Ловиц 268
Ломоносов 5, 174
Лондон 26
Льюис 196, 197
Мак-Бэн 374
Максвелл
Мариотт 10, 12, 23
Менделеев 185
Михаэлис 151
Нернст 247, 249, 251, 252, 262
395
Никольский 253, 279
Нобел 161
Нолле 178
Ньютон 39, 357
Онзагер 231
Перрен 301, 302, 324
Песков 278, 293, 321, 322
Планк 55, 81, 136, 155
Плауссон 297
Поггендорф 242
Поляни 273, 274
Пуазейль 41, 357
Пфеффер 178
Рабинович 158
Рауль 174, 175, 182
Ребиндер 286
Рейсс 320, 321
Релей 312
Рентген 46
Рогинский 295
Свердберг 301, 306
Семенов 123
Смолуховский 301, 322, 335, 336
Соколов 330
Соссюр 268
Тейлор 96, 147
Теллер 274
Тизелиус 353
Тимирязев 7, 153, 160
Тиндаль 312
Траубе 290
Фарадей 226, 227, 241
Фаянс 278
Фигуровский 308
Филиппов 374
Франк 47, 158
Фрейндлих 272, 273, 276, 330
Фрумкин 247, 289, 290
Хиншельвуд 123
Хюккель 193, 195, 230
[Пальников 295
Шарль 10, 11, 12
Шеелс 268
Шерага 217
Шишковский 291
Штарк 153
Штаудингер 159
Штерн 320
Шульце 327, 368
Эйринг 48
Эммет 274
Эйнштейн 153, 301, 302, 357, 358
Якоби 240, 242
ОГЛАВЛЕНИЕ
Предисловие............................................... . . 3
Введение ............................................ . . . 5
Глава I. Агрегатные состояния вещества . . ... 8
1. Классификация состояний.................. ... 8
2. Газообразное состояние....................................... 9
2.1. Законы идеальных газов..................................... 10
2.1.1. Закон Бойля—Мариотта ....... .......... 10
2.1.2. Закон Шарля.............................................. 10
2.1.3. Закон Гей-Люссака........................................ 11
2.1.4. Уравнение состояния идеального газа ... ............. 12
2.2. Парциальное давление. Закон Дальтона................... . . 14
2.3. Кинетическая интерпретация абсолютной температуры . . . 15
2.4. Закон Авогадро......................................... . . 15
2.5. Скорость молекул. Диффузия. Эффузия.......................... 16
2.6. Реальные газы............................................ . 21
2.6.1. Уравнение Ван-дер-Ваальса.............................. . 21
2.6.2. Конденсация газов и критическое состояние .... 23
2.6.3. Соответственные состояния.............................. . 25
2.6.4. Межмолекулярные взаимодействия......................... . 25
3. Твердое состояние......................................... . 27
3.1. Аморфное и кристаллическое состояния. Полиморфизм . 27
3.2. Ионные решетки............................................... 30
3.3. Ковалентные решетки......................... . 31
3.4. Молекулярные решетки ....:. . 32
3.5. Металлические решетки.................. . . 33
4. Жидкое состояние ..... .... 34
4.1. Плотность и молярный объем . .... 36
4.2. Электрический дипольный момент ..... 37
4.3. Вязкость .................- . . . . . . 39
4.4. Давление насыщенного пара жидкости . . 42
4.5. Вода и лед............ .... . 44
4.6. Жидкие кристаллы ....... 48
Глава II. Химическая термодинамика . . 50
1. Классическая и статистическая термодинамика .... . 50
2. Система и внешняя среда............................. . 50
3......................, Термодинамические параметры состояния системы................. 52
4. Состояние равновесия. Обратимые и необратимые превращения 53
5. Энергия. Работа. Теплота .... 55
6. Функция состояния............................................. 57
7. Первое начало термодинамики................................... 60
8. Приложение первого начала термодинамики к химическим процес-
сам .............................................................. 63
8.1. Теплота реакции (тепловой эффект)............................ 63
8.2. Теплоемкость. Зависимость теплоты реакции от температуры 66
8.3. Скрытая теплота изменения состояния ... ............. 70
397
8.4. Энтальпия образования . ... .... 71
Закон Гесса ... ............ .... 72
8*6? Энергия связи........................................ .... 75
9. Второе начало термодинамики . . . . ........................... 79
9.1. Энтропия. Математическая формулировка второго начала термо-
динамики ....................._............... . .... 82
9.2. Статистическая интерпретация энтропии....................... 84
10. Третье начало термодинамики.................................. 87
11. Свободная энергия............................................. 89
12. Свободная энергия и направление химических реакций . 90
13. Понятие о химических потенциалах ...... 93
Глава III. Химические и фазовые равновесия . 95
1. Химические равновесия................................ . . 95
1.1. Влияние изменения внешних условий на равновесие ....;. 97
1.2. Закон действующих масс . . ................................. 100
1.3. Количественная оценка смещения равновесия с изменением тем-
пературы и давления...................... . . ... 102
2. Равновесие между фазами и диаграмма состояния................ 105
2.1. Правило фаз............................................. 105
2.2. Уравнение Клапейрона . ... ..... 108
Глава IV. Химическая кинетика........................................ 111
1. Кинетика и механизм реакций. Реагенты и продукты .... 111
2. Скорость химической реакции................................... ИЗ
3. Порядок и константа скорости реакции......................... 114
4. Элементарные (простые) реакции. Молекулярность реакции . . . 117
5. Сложные реакции............................................. 118
6. Количественные соотношения между скоростью некоторых реакций
и концентрациями реагентов.............................. . . . . 125
7. Влияние температуры на скорость реакции. Энергия активации 129
8. Теории столкновений и переходного состояния.................. 134
9. Понятие о кинетике реакций в открытых системах .... 137
Глава V. Катализ . . ........... 139
1. Каталитические процессы и катализаторы................. .... 139
2. Кислотно-основной и окислительно-восстановительный катализ . . 141
3. Гомогенный и гетерогенный катализ . ..................... 142
4. Ферментативный катализ ... . . ... 149
Глава VI. Фотохимические реакции ... 153
1. Взаимодействие излучения с веществом................... .... 153
2. Закон фотохимической эквивалентности Эйнштейна и квантовый
выход .................................................... .... 155
3. Скорость фотохимических реакций . . . . ... 158
4. Фотосинтез..................... ... . . 160
Глава VII. Растворы неэлектролитов ... 165
1. Образование растворов. Растворимость. . . .................... 165
2. Состав растворов. Парциальная молярная величина. Концентрация 172
3. Понижение давления насыщенного пара разбавленных растворов 173
4. Температура кристаллизации разбавленных растворов............ 174
5. Температура кипения разбавленных растворов.................... 177
6. Осмотическое давление........................................ 178
7. Состав пара идеальных и реальных растворов .... 182
8. Понятие активности....................................... . 183
Глава VIII. Растворы электролитов .... . Qt-
1 оО
1. Образование ионов в водных растворах . . 185
2. Слабые и сильные электролиты................................. 189
2.1. Слабые электролиты. Степень диссоциации . 190
398
2.2. Сильные электролиты. Активность . . . >..................... 193
3. Развитие понятия кислоты и основания . . . ..... 196
4. Сила кислот и оснований.................. . . . 198
5. Ионное произведение воды. Понятие pH . ............ . . 200
6. Классификация кислот и оснований по отношению к воде .... 202
6.1. Расчет pH кислых и щелочных растворов.................. ... 203
6.2. Вычисление pH водных растворов солей ... . , 207
7. Буферные системы ... ........................ 210
Глава IX. Электрическая проводимость растворов электролитов . . 216
1. Скорость движения ионов. Числа переноса....................... 216
2. Удельная электрическая проводимость электролитов ... . . 219
3. Молярная электрическая проводимость электролитов . . 222
4. Связь молярной электрической проводимости со скоростями дви-
жения ионов...................................................... 225
5. Закон независимости движения ионов............................ 227
6. Зависимость электрической проводимости от природы электролита 230
7. Практическое применение электрической проводимости . . 232
Глава X. Электрохимические процессы................................... 238
1. Электродные потенциалы. Электрохимические элементы и электро-
движущие силы................................................... 238
2. Измерение ЭДС. Нормальный элемент Вестона..................... 242
3. Термодинамика электрохимических элементов..................... 244
4. Электрические потенциалы на фазовых границах .....' 246
5. Классификация электродов. Электроды первого и второго рода . 251
6. Окислительно-восстановительные электроды и их потенциалы . 256
7. Концентрационные элементы и диффузионный потенциал . . 260
8. Потенциометрия.............................................. 264
Глава XI. Поверхностные явления ... ... 267
1. Адсорбция. Адсорбция на границе твердое тело — газ . 267
2. Теории адсорбции.............................. . 270
3. Адсорбция на границе твердое тело — раствор .... 276
3.1. Молекулярная адсорбция из растворов .... 276
3.2. Адсорбция элекролитов....................................... 277
3.3. Обменная адсорбция..................... . 278
4. Поверхностное натяжение. Смачивание .... 280
5. Адсорбция на границе раздела раствор — газ.................... 288
Глава XII. Коллоидные системы и методы получения лиофобных коллои-
дов ............................................................ 292
1. Общая характеристика коллоидных систем . . . ... 292
2. Методы получения лиофобных коллоидов . ................ 294
2.1. Методы конденсации....................................... . 295
2.2. Дисперсионные методы................... . 297
2.3. Метод пептизации. . . . -............................. .... 298
Глава XIII. Молекулярно-кинетические, оптические и электрические свой-
ства лиофобных коллоидов....................... ... ... 300
1. Броуновское движение....................... ... ... 300
2. Осмотическое давление....................... ... .... 303
3. Седиментация........................ ... .... 305
4. Вязкость..................................... ... . 309
5. Очистка коллоидных растворов . . .............. ... 310
6. Оптические свойства.................................... ... 312
7. Электрические свойства...................................... 316
7.1. Строение мицеллы.......................................... 316
7.2. Двойной электрический слой и электрокинетические явления . 319
Глава XIV. Устойчивость и коагуляция лиофобных коллоидов . 324
ч/ 1. Кинетическая и агрегативная устойчивость . . т 324
399
2. Коагуляция ...................... ... . 327
2.1. Действие электролитов .... . . 328
2.2. Коагуляция и дзета-потенциал . ................. 331
2.3. Теория коагуляции электролитами . ....... 332
2.4. Кинетика коагуляции...................................... 334
2.5. Старение золей и пептизация................................ 336
2.6. Защитное действие молекулярных адсорбирующих слоев . . 338
2.7. Роль процессов коагуляции в образовании почв ... . 340
Глава XV. Микрогетерогенные системы . . 342
1. Суспензии . ... . . ... . . 342
2. Эмульсии . . ... . . ... . ... 343
3. Пены...................................... ... 347
4. Аэрозоли . . ... ... ... . ... 348
Глава XVI. Растворы высокомолекулярных соединений (растворы ВМС) 351
1. Общие характеристики растворов ВМС........................... 351
2. Электрические, молекул яр но-кинетические и оптические свойства
растворов ВМС................................................... 352
3. Набухание и растворение ВМС *...... ...... 363
4. Нарушение устойчивости растворов ВМС ... . ... 368
Глава XVII. Гели. Студни. Полуколлоиды . 371
L ГелХи. Студни ...... 371
2. Полуколлоиды . . . . 374
Заключение ... 379
Литература ......................................................... 380
Приложение ......................................................... 381
Предметный указатель . 385
Именной указатель . . 395
Учебное издание
Рюрик Аркадьевич Хмельницкий
ФИЗИЧЕСКАЯ И КОЛЛОИДНАЯ ХИМИЯ
Зав. редакцией С. Ф. Кондрашкова. Редактор В. Н. Бораненкова. Мл.
редакторы С. М. Ерохина, Л. С. Макаркина. Художник В. В. Гарбузов.
Художественный редактор Е. К. Косырева. Технический редактор
Н. В. Яшукова. Корректор С. К. Завьялова.
ИБ № 6816
Изд. № ХИМ—784. Сдано в набор 12.06.87. Подп. в печать 03.03.88.
Формат 60 X OO’/ie- Бум. офс. № 2. Гарнитура литературная. Печать офсет-
ная. Объем 25 усл. п. л. +0,25 усл. п. л. форз. 50,5 усл. кр.-отт. 25,68
уч.-изд. л.+ 0,48 уч.-изд. л. форз. Тираж 37 000 экз. Заказ № 696.
Цена 1 р. 20 к.
Издательство «Высшая школа», 101430, ГСП-4, Москва, Неглинная ул.,
д. 29/14.
Ярославский полиграфкомбинат Союзполиграфпрома при Государственном
комитете СССР по делам издательств, полиграфии и книжной торговли.
150014, Ярославль, ул. Свободы, 97.