





















































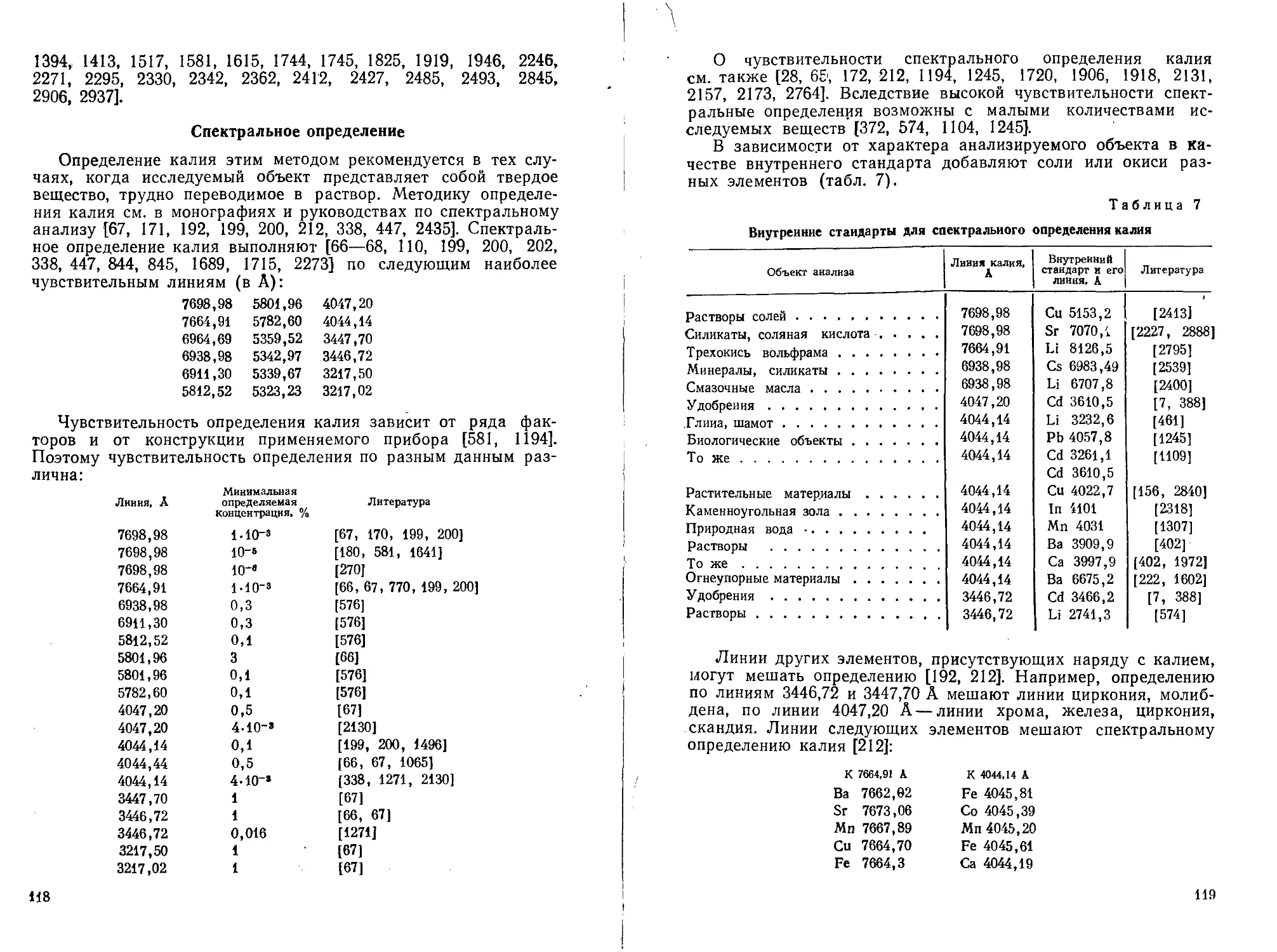






















































Текст
АКАДЕМИЯ НАУК СССР
ИНСТИТУТ ГЕОХИМИИ и аналитической химии им. В. И. ВЕРНАДСКОГО
Серия: «АНАЛИТИЧЕСКАЯ ХИМИЯ ЭЛЕМЕНТОВ»
АНАЛИТИЧЕСКАЯ ХИМИЯ
КАЛИЯ
И. М. К о р е н м а н
ИЗДАТЕЛЬСТВО «НАУКА» Москва 1964
543/545 : 546.32
Серия: «Аналитическая химия элементов»
Главный редактор академик А. П. Виноградов
Редакционная коллегия:
И. П. Алимарин, А. К- Бабко, А. И. Бусев, А. П. Виноградов, А. Н. Ермаков, В. И, Кузнецов, П. Н. Палей, Д. И. Рябчиков, И. В. Тананаев, Ю. А. Чернихов
Редактор тома «Аналитическая химия калия» А. И. Бусев
Адрес редколлегии:
Москва, В-334, Воробьевское шоссе, 47а, Институт геохимии и аналитической химии им. В. И. Вернадского Академии наук СССР
ОТ РЕДКОЛЛЕГИИ
Институт геохимии и аналитической химии им. В. И. Вернадского АН СССР приступил к изданию серии монографий по аналитической химии отдельных элементов. Эта серия — «Аналитическая химия элементов» — составит около пятидесяти томов. Потребность в подобного рода издании давно назрела. Вместе с тем у нас накопился огромный опыт многочисленных лабораторий и теперь стало возможным и необходимым его подытожить. Таким образом, возникло настоящее издание — серия «Аналитическая химия элементов»,— которое осуществляется впервые. Аналитическая химия любого элемента и его различных соединений в настоящее время представляется чрезвычайно разнообразной как вследствие сложности современных объектов исследования и широты диапазона концентраций, которые бывает необходимо определить, так и вследствие разнообразия исшэльзующихся методов.
В связи с этим для монографий был разработан общий план как в смысле содержания, так и последовательности изложения материала.
В монографиях содержатся общие сведения о свойствах элементов и его соединений. Затем излагаются химические реакции, являющиеся основанием для аналитических целей. Методы как физические, так физико-химические и химические излагаются применительно для количественного определения данного химического элемента, начиная с анализа сырья, далее типичных полупродуктов производства, и, наконец, конечной продукции,— металлов или сплавов, окисей, солей и других соединений и материалов. Как правило, приводятся принципы определения и, где это необходимо, дается точное описание всего процесса определения. Необходимое внимание уделяется быстрым методам анализа. Самостоятельное место занимает изложение методов определения так называемых элементов-примесей в чистых материалах.
Обращается внимание на точность и чувствительность методов в связи с общей тенденцией повышения чувствительности методов определения следов элементов-примесей.
3
Монографии содержат обширную литературу, доведенную до последних лет; они рассчитаны на широкий круг химиков, в первую очередь химиков-аналитиков исследовательских институтов и заводских лабораторий различных отраслей хозяйства, а также на химиков-преподавателей и студентов химических высших учебных заведений.
К составлению монографий привлечены наши крупнейшие специалисты, имеющие опыт работы в области аналитической химии того или иного химического элемента.
Отдельные тома серии «Аналитическая химия элементов» будут выходить самостоятельно, по мере их подготовки. Вышли в свет монографии, посвященные торию, таллию, урану, рутению, молибдену и калию, готовятся к печати монографии по аналитической химии плутония и бора.
Мы обращаемся с просьбой ко всем читателям присылать свои замечания и отзывы о монографиях.
От автор а
Калий — один из наиболее распространенных элементов, ему посвящено много исследований, в том числе по вопросам его аналитической химии. В предлагаемой монографии собраны данные почти о 3000 работ по аналитической химии калия и сделана попытка систематизировать и критически их рассмотреть. Понятно, что отдельным реакциям и методам можно было уделить немного места и отметить только главнейшие черты и особенности рассматриваемых методов.
Автор выражает благодарность академику А. П. Виноградову, академику И. В. Тананаеву, профессору В. И. Баранову, профессору А. И. Бусеву, профессору В. И. Кузнецову, доктору химических наук Л. М. Иванцову, доктору химических наук А. В. Карякину, доценту Н. К. Рудневскому за весьма ценные советы, направленные на улучшение рукописи.
И. Коренман
ВВЕДЕНИЕ
Соли калия известны очень давно и столь же давно они широко используются в практике [62, 63, 2837]. Поташ начал применяться приблизительно в 250—380 гг. до нашей эры [149, 153, 1802, 2298]. Поташ был хорошо известен в средние века, и его уже тогда умели отличать от соды [63, 1800—1802, 1879, 2315, 2857, 2927]. Производство поташа и других солей калия в России началось в XV столетии, а может быть и ранее [321]; еще в XVII столетии по производству поташа Россия занимала одно из первых мест в мире [321, 424]. Русские и иностранные ученые выполнили ряд исследований по технологии солей калия [63, 458, 756, 1315а, 1896а] задолго до открытия металлического калия. Последний был получен впервые английским ученым Дэви [1045]. Большинство историков относит это открытие к 1807 г. [63, 72, 153, 353, 2857], хотя о получении щелочных металлов Дэви доложил Королевскому обществу Английской Академии наук еще в октябре 1906 г. [706]. Опыты Дэви были вскоре повторены другими учеными как за границей [1305, 1802], так и в России [396, 457].
Калий представляет собою белый мягкий металл; он очень легко окисляется кислородом воздуха и легко реагирует с водой. Некоторые свойства калия приведены в приложении (стр. 149).
Калий принадлежит к очень распространенным элементам. Пр содержанию в земной коре (в среднем около 2,4% [98, 103]) он уступает только кислороду, кремнию, алюминию, железу, кальцию и натрию. В почвах находится 1—3,6% калия [102, 434, 466, 467]. Калий входит в состав всех растительных и животных организмов [101]. Содержание калия в растениях достигает 1—2% по весу [101]. В золе растений находится до 35—50% поташа [235, 466, 467].
Природный калий содержит главным образом стабильный изотоп Км в количестве 93,08 + 0,09 % [1443, 1822, 2121], по другим данным —93,26% [306, 435, 2321] и 93,3о/о [152, 460, 470, 4711-Второй стабильный изотоп калия К™ содержится в количестве 6,91+0,09% [157, 471, 1443, 1822, 2121]; другие исследователи эту
5
величину считают равной 6,73% [306, 435, 470, 2321] или 6,683% [152, 460, 478]. В природном калии находится радиоактивный изотоп Kia в количестве 0,0119 + 0,0001% [47, 70, 104, 306, 470, 852, 885, 1443, 1822, 2121, 2321, 2349, 2674], по другим данным — 0,011% [152, 435, 441, 460, 478, 951, 1647].
Калий — самый легкий элемент, обладающий естественной радиоактивностью. Радиоактивный изотоп KS образуется по следующим реакциям [460, 2728]:
K^’ + nJ^Ku + r;
Са"4 +
Калий, выделенный из природных солей, пород, метеоритов, из тканей организмов и других материалов, имеет постоянный изотопный состав [851, 1502].
Организмы, усваивая калий, не изменяют его изотопного состава [104, 441, 853, 854, 2504]. На ряд других работ по изотопному составу природного калия мы только сошлемся [672, 720, 849-852, 951, 994, 2193, 2327, 2328, 2821].
В значения атомного веса калия, установленные в различные годы, вносились лишь небольшие изменения:
Годы Атомный вес Литература
1868 39,04 [2628]
1894—1902 39,11 [478]
1903—1908 39,15 [478]
1909—1924 39,10 [478]
1925—1930 39,096 [478]
1931—1933 39,10 [478]
1934—1949 39,096 [478, 649, 1569, 2326, 2889]
1950—1959 39,100 [528, 2890]
1961 39,102 [323а]
Если атомный вес изотопа кислорода О16 принять равным 16,0000, то атомные веса природных изотопов калия имеют следующие значения: К39—38,9761; К40—39,9768; К41—40,9749 [71, 459, 986, 1898].
Радиоактивный изотоп Kie излучает главным образом [3-части-цы и превращается при этом в изотоп кальция Са^ [8, 70, 102, 304, 524, 539, 612, 852, 1202, 1376, 1528, 1556, 1600, 1647, 2870]:
Кы-^Са4’ + [Г.
В результате К-захвата орбитального электрона образуется аргон [8, 70, 102, 304, 852, 1208, 1378, 1455, 1556, 1647, 2175, 2870]:
К“ + е —> Аг“ + Т-
в
Превращение KtS в Аг" сопровождается слабым рентгеновским излучением [122, 2175, 2447, 2669].
На долю’p-излучения приходится 88% общего излучения К", на долю у-излучения — 12% (8,47,86,304], по другим данным — 60 и 40% [152, 1405] или 90 и 10% [195].
Установление относительных количеств К4Й и Аг40 в калиевых минералах позволяет вычислить их геологический возраст [29, 30, 71, 80,*85, 122—126, 278, 287, 373, 478, 596, 603, 612, 1392, 2077, 2125,2414/2545,2708].
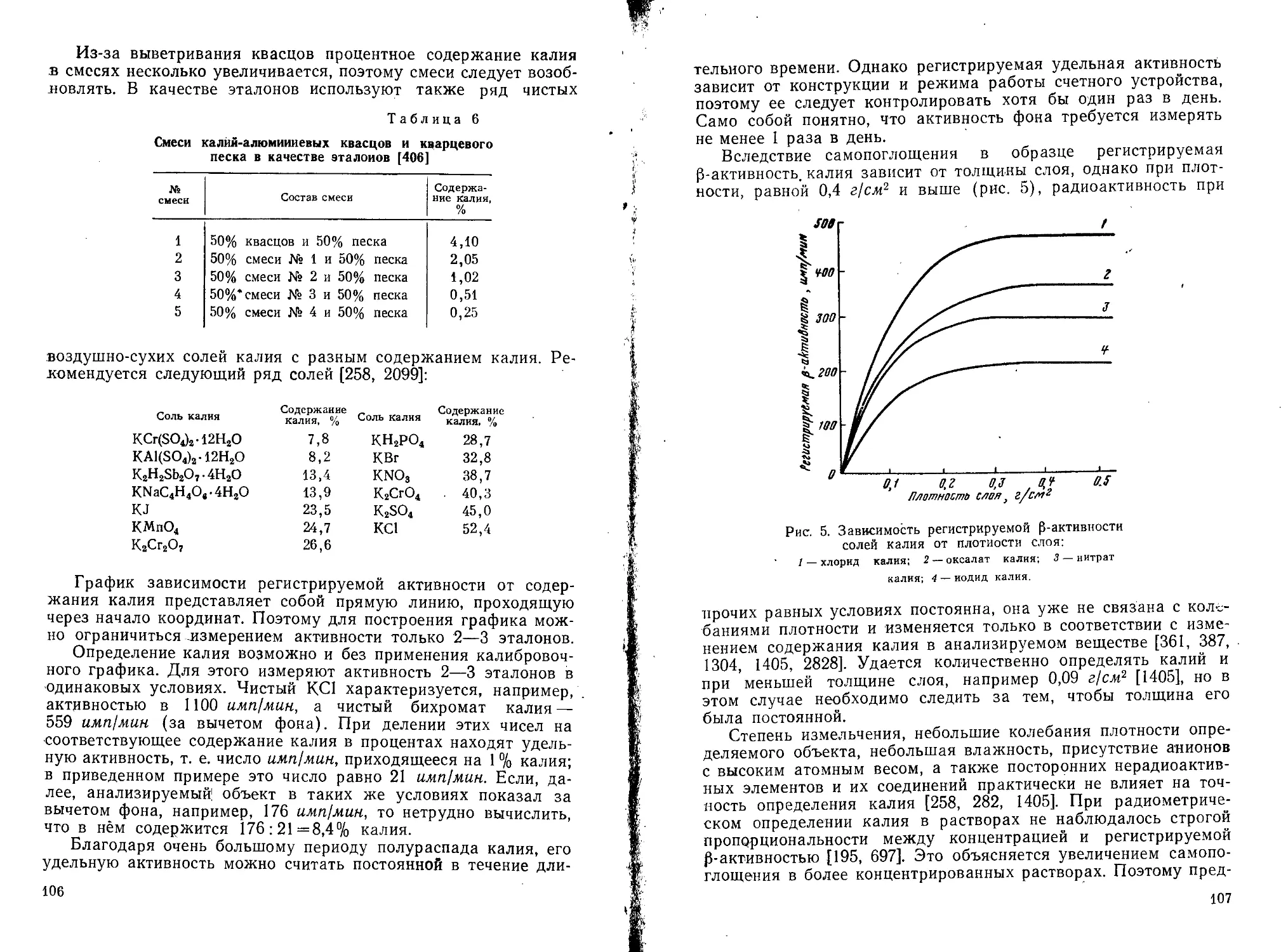
Абсолютная р-активностьжалия, согласно большинству исследований, соответствует 27 —33 [3-распадам на 1 г калия в 1 сек. [835, 885,' 1052, 1186, 1376, 1556, 1557, 2447,2617, 2652, 2674, 2870[; имеются и иные данные: как более низкие — 21—25 [3-распадов [811, 1231, 2079], так и более высокие — 42 — 53 [3-распада на 1 г калия в 1 сек. [785, 1481].
Абсолютная у-активность равна 3,4—3,6 у-кванта [603, 668, 885, 1186, 1348, 2447], по другим данным — 2,96 [2674] или 3,1 у-кванта [1577] на 1 г калия в 1 сек.
Период полураспада калия оценивают различно:
2,4-4,5-108 лет [152, 539, 603, 610, 774, 785, 2669, 2708];
7-8,5-108 лет [306, 435, 612, 942, 1481];
1,2-1,33-10» лет [47, 86, 460, 471, 811, 852, 885, 951, 1556, 1577, 1822, 2321, 2447, 2617, 2652, 2674];
1,42-10» лет [70, 835, 1383, 1443];
1,5-1,8-10» лет [470, 479, 1231, 2079].
Другие данные о периоде полураспада см. [47, 71, 785, 1375, 1390, 1431, 1501, 1528, 1566, 1838, 2162, 2669]. Несмотря на расхождения в оценке периода полураспада К40, порядок этой величины у всех авторов приблизительно одинаков и составляет сотни миллионов лет. Следовательно, относительное содержание радиоактивного изотопа в природном калии в течение ближайших тысячелетий останется на практически постоянном уровне.
Энергия р-частиц (максимальная) 1,3—1,4 Мэв [47, 150, 152, 471, 610, 811, 951, 1195, 1208, 1231, 1577, 1822, 2674, 2870].
Энергия у-квантов (максимальная) 1,46—1,55 Мэв [47, 152, 471, 723, 738, 951, 1249, 1348, 1822, 2260].
P-Частицы характеризуются небольшой проникающей способностью. В воздухе р-частицы, излучаемые калием, могут проходить до десятков сантиметров, но твердое или жидкое вещество толщиною 5 мм способно поглощать их полностью. Напротив, у-лучи калия легко проникают даже через слои жидких и твердых веществ в несколько сантиметров.
7
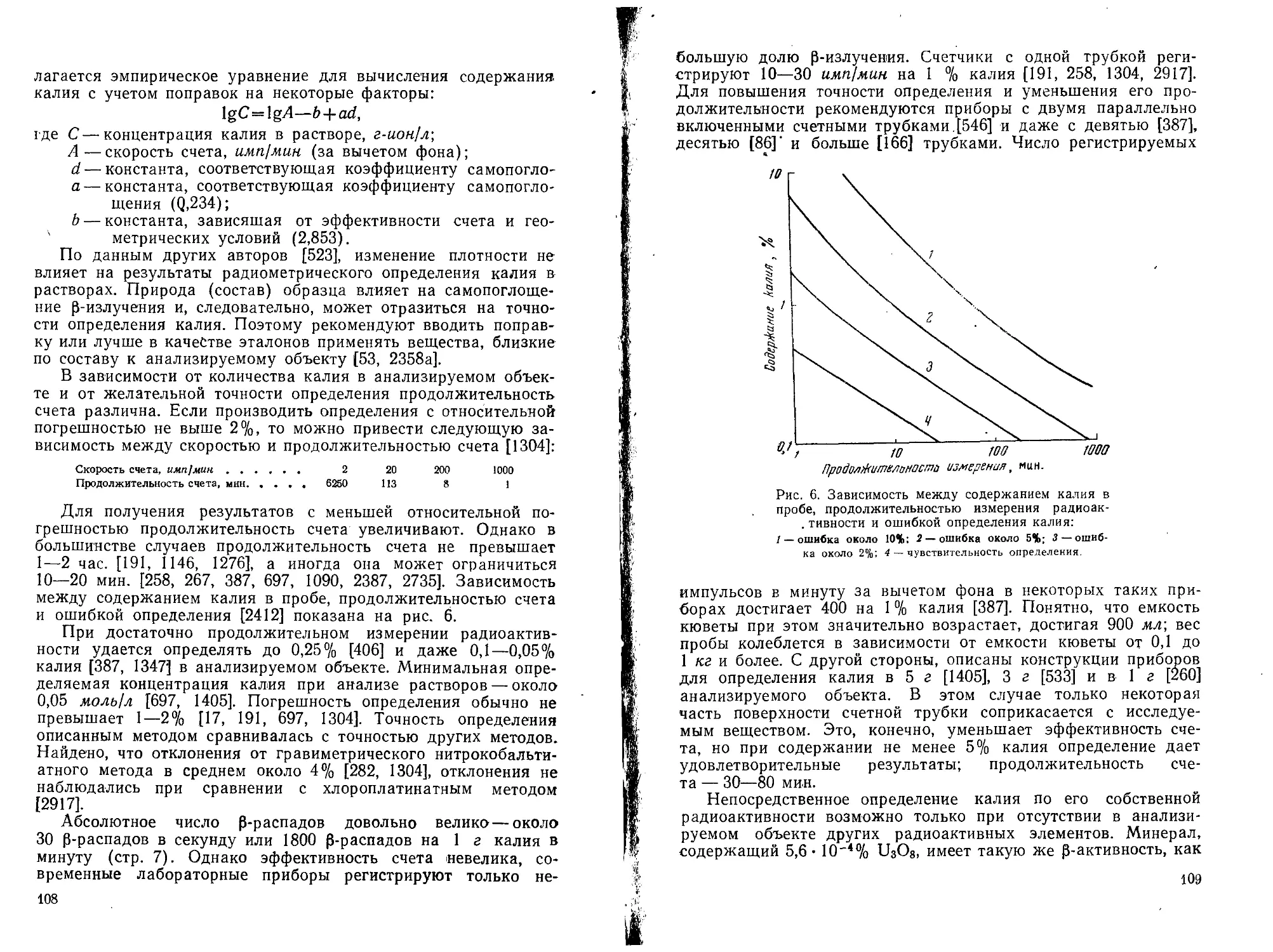
Схема радиоактивного распада Kis представлена на рис. 1 [104, 1348, 1376, 2068, 2175, 2256, 2447, 2669].
Радиоактивность калия была обнаружена Томпсоном в 1905 г. [2711]. Открытие радиоактивности калия иногда приписывают работам Кемпбелла и Вуда [918], которые в 1906 1907 гг. изучали это явление. В течение ряда лет к радиоактивности калия относились
Рис. 1. Схема радиоактивного распада
К?д(~.Ке— захват орбитального
скептически, полагая, что слабая радиоактивность не может быть свойством самого калия и обусловлена примесью других радиоактивных элементов [1923, 2445]. Предполагалось, что радиоактивность зависит от присутствия элемента 87 (франций), невесомая примесь которого изоморфно замещает калий [157]. Отделить калин от предполагаемых радиоактивных примесей не удалось [917, 1510, 1924]. Даже 18—22-кратная перекристаллизация солей калия не приводила к изме-
нению их радиоактивности [1924]. Радиоактивность разных солей калия оказа-
электрона) лась пропорциональной со-
держанию в них калия [1153, 1427, 1924], поэтому было признано, что радиоактивность — собственное свойство калия [1509, 1510]. Путем фракционирования была приготовлена проба калия, обогащенная его тяжелым изотопом, которая характеризовалась большей радиоактивностью, чем обычный калий [1528, 1529, 1892]. Поэтому высказывалось предположение о радиоактивности изотопа К41 или о существовании естественного радиоактивного изотопа К43. Только после открытия изотопа К40 в 1935 г. было показано, что именно этот изотоп сообщает радиоактивность природным солям калия [2587].
В России еще в 1909 г. изучалась радиоактивность калиевых минералов [76—78, 81]. Тогда же было установлено действие солей калия на фотопластинку, которое можно заметить только при экспозиции порядка 5- 6 месяцев [918, 1482, 1868, 2661]. Радиоактивность калия очень мала [77, 78, 117, 1868]. Если ра
диоактивность калия принять за единицу, то радиоактивность тория равна 450, урана—1900 [1185], радия —3- 1010 [2079]. На 8
другие работы, посвященные естественному радиоактивному изотопу калия, мы только сошлемся [117, 151, 655, 773, 787,. 1384, 1483, 1508, 1533, 1835, 2009, 2390, 2706]. Данные об искусственных радиоактивных изотопах калия приведены в приложении (стр. 149).
Калий образует большое число минералов [58а, 393, 529, 756, 1281, 1353, 2321]: сильвин, КС1; сильвинит, КО + NaCl; карналлит, КС1 • MgCl2 • 6Н2О; хлорокальцит, КС1 • СаС12; хло-романганокалит, 4КС1-МпС12; риннеит, ЗКС1 • NaCl • FeCl2; дуглазит, 2КС1 • FeCl2 • 2Н2О; эритросидерит, 2КС1 • FeCl3 • Н2О; селитра," KNO3; лечивит (арканит), .K2SO4; тейлорит, (К, NH4)2SO4; глазерит, 2K2SO4 • Na2SO4; лангбейнит, КгЗО4-•2MgSO4; пикромерит (шенит), K2SO4 • MgSO4 • 6Н2О; мангано-беннит, K2SO4 • 2MnSO4; пальмиерит, (К, Na)2SO4-PbSO4; сингенит, КгЗО4 • CaSO4 • Н2О; леонит, K2SO4 • MgSO4 • 4Н2О; калинит (квасцы), KA1(SO4)2- 12Н2О; алунит, К2О- ЗА12О3- 4SO3- 6Н2О; ярозит, К2О • 3Fe2O3 • 4SO3 • 6Н2О; метавольтин, 5КгО-ЗРе2О3-• 12SO3 • 18Н2О; каинит, КС1 • MgSO4 • ЗН2О; краузит, КгЗО4-• Fe2(SO4)3 • 2Н2О; хлоротионит, Кг5О4-СиС12; полигалит, K2SO4 • MgSO4 • 2CaSO4 • 2Н2О; кругит, K2SO4 • MgSO4 • 4CaSO4 • • 2Н2О; авогадрит, KBF4; калиборит, К2О -4AlgO • 11В2О3- 14Н2О; нефелин, 4(К, Na)2O • 4А12О3 • 9 SiO2; ортоклаз (полевой шпат), К2О • А12О3 • 6SiO2; калиофиллит, КгО • А12О3 • 2SiO2; лейцит, КгО • А12О3 • 4SiO2; мусковит, (К, Н)2О • А12О3 • 2SiO2; биотит, K(Mg, Fe)3 [AlSi3O10](OH, F)2; флогопит, KMg3[AlSi3O]0](OH, F)2; лепидолит, KLi2[AlSi3O6(OH, F)4](OH, F)2; слюда, KH2Al3(SiO4)3.
Калий как примесь содержится во многих минералах. В СССР особенно богатые месторождения калиевых минералов находятся в районе Соликамска [301, 422—424, 474, 559].
Соли калия используются главным образом как удобрения [25, 44, 129, 145, 317, 434, 466, 557, 1281, 2483]; кроме" того, они применяются в стекловарении, медицине, многие из них используются в качестве химических реагентов. Большое значение приобрели сплавы калия [8, 165].
Калию посвящен ряд монографий [8, 129, 466, 571, 756, 1013, 1281].
I. АНАЛИТИЧЕСКАЯ ХАРАКТЕРИСТИКА КАЛИЯ
Для аналитической характеристики калия важно отметить следующие его особенности. Подавляющее большинство солей калия отличаетс'я очень хорошей растворимостью в воде. Даже наименее растворимые соли, в виде которых калий осаждают при качественном и количественном определении, характери--зуются растворимостью порядка 10~2—10~3 моль/л, и только отдельные соли имеют несколько меныпую растворимость. Многие соли калия в отличие от соответствующих солей натрия и лития плохо растворимы в спиртах, ацетоне и некоторых других органических растворителях. Это обстоятельство используется для повышения чувствительности ряда реакций на калий и для его отделения от других элементов.
Значительная часть методов определения калия основана на его предварительном выделении в виде соли, мало растворимой в воде или в смесях воды с органическим растворителем.
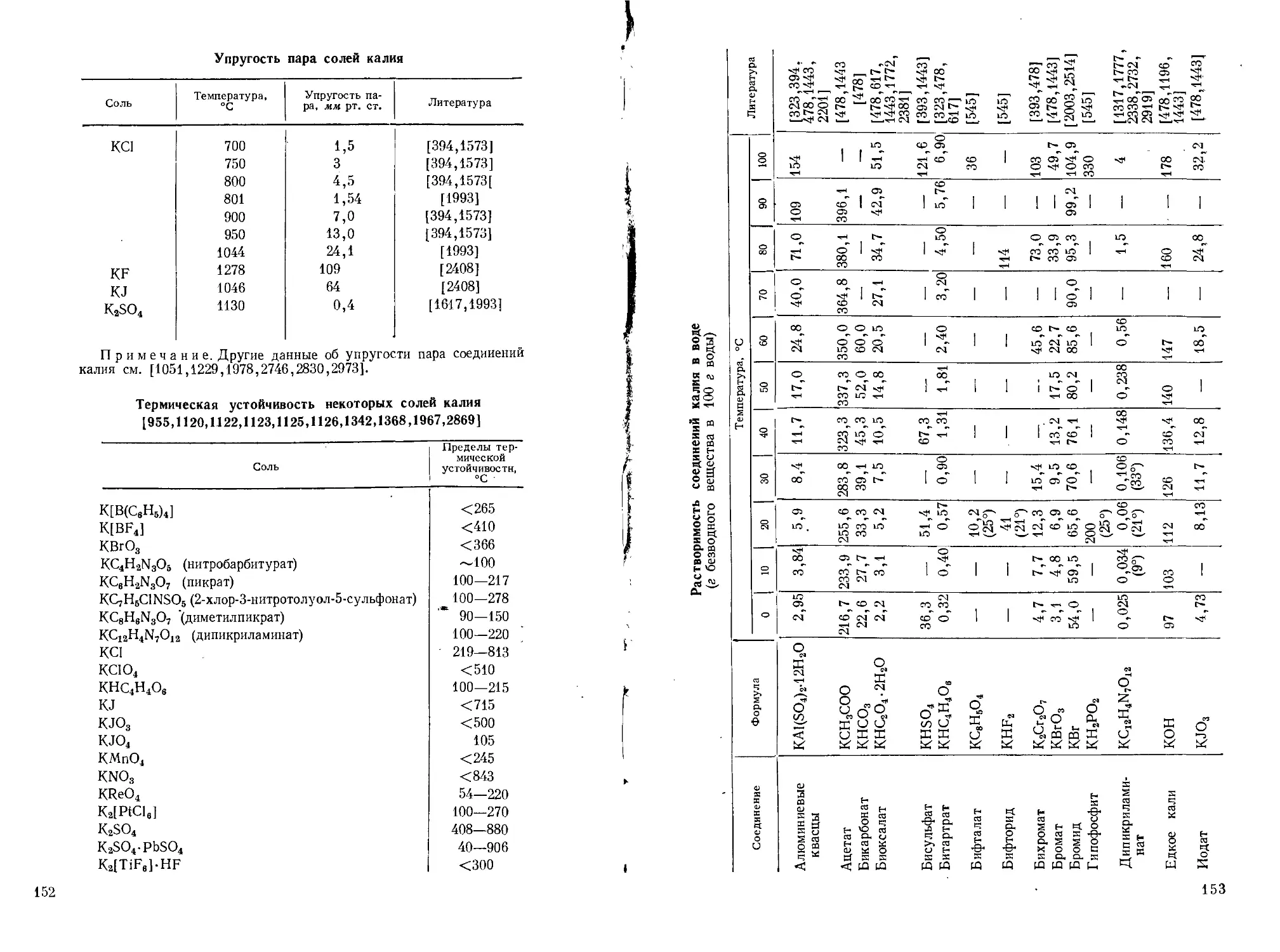
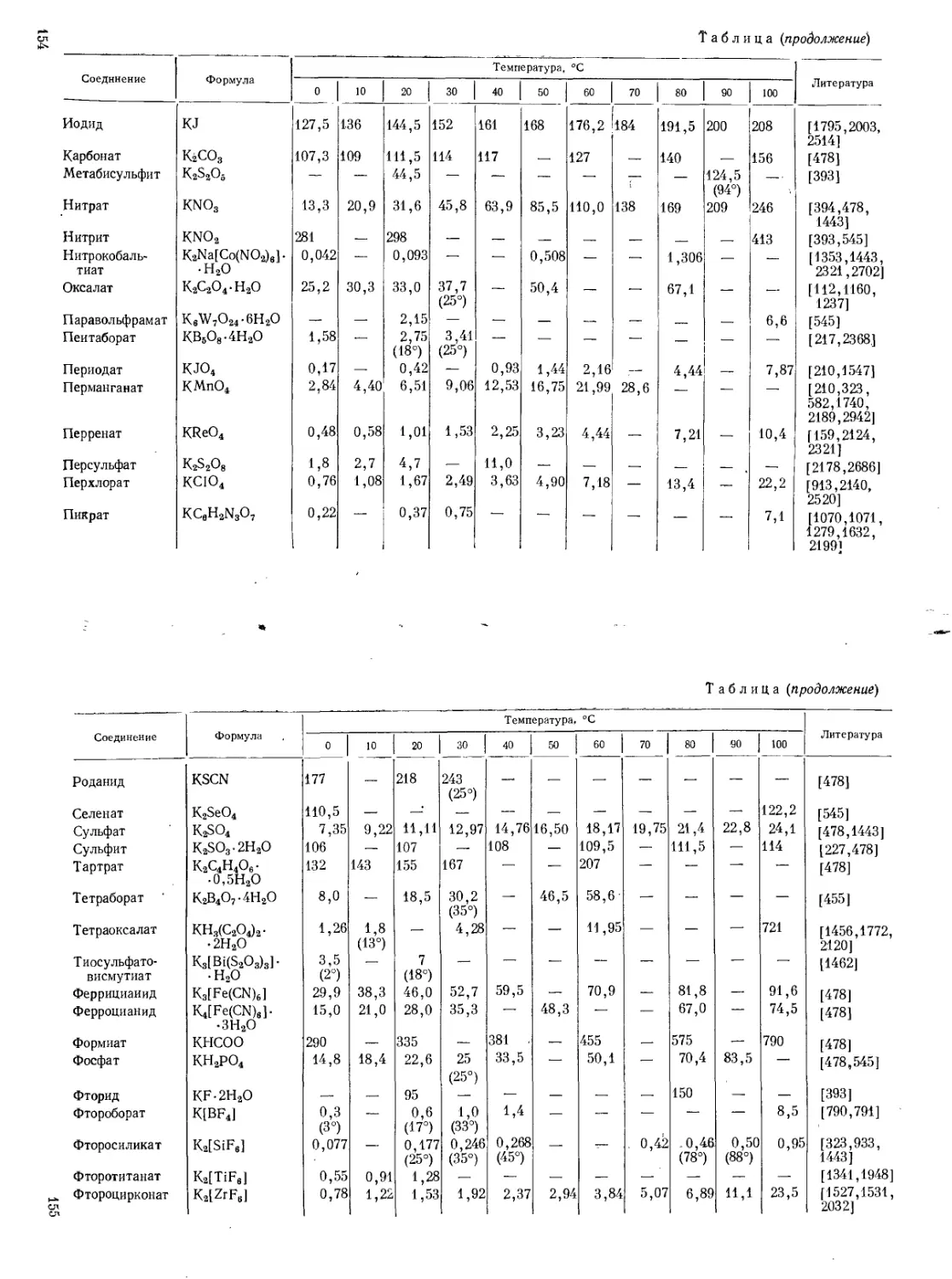
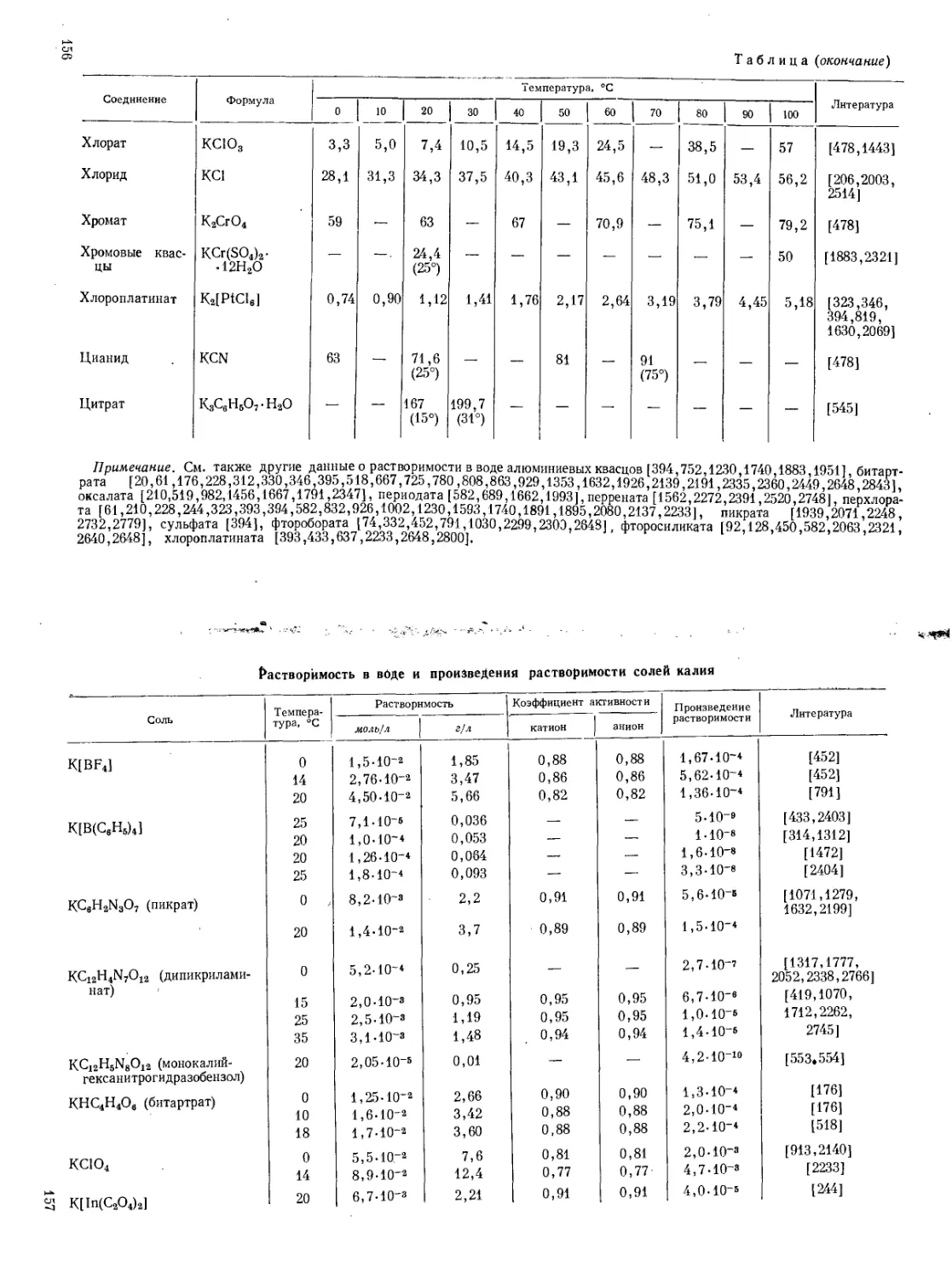
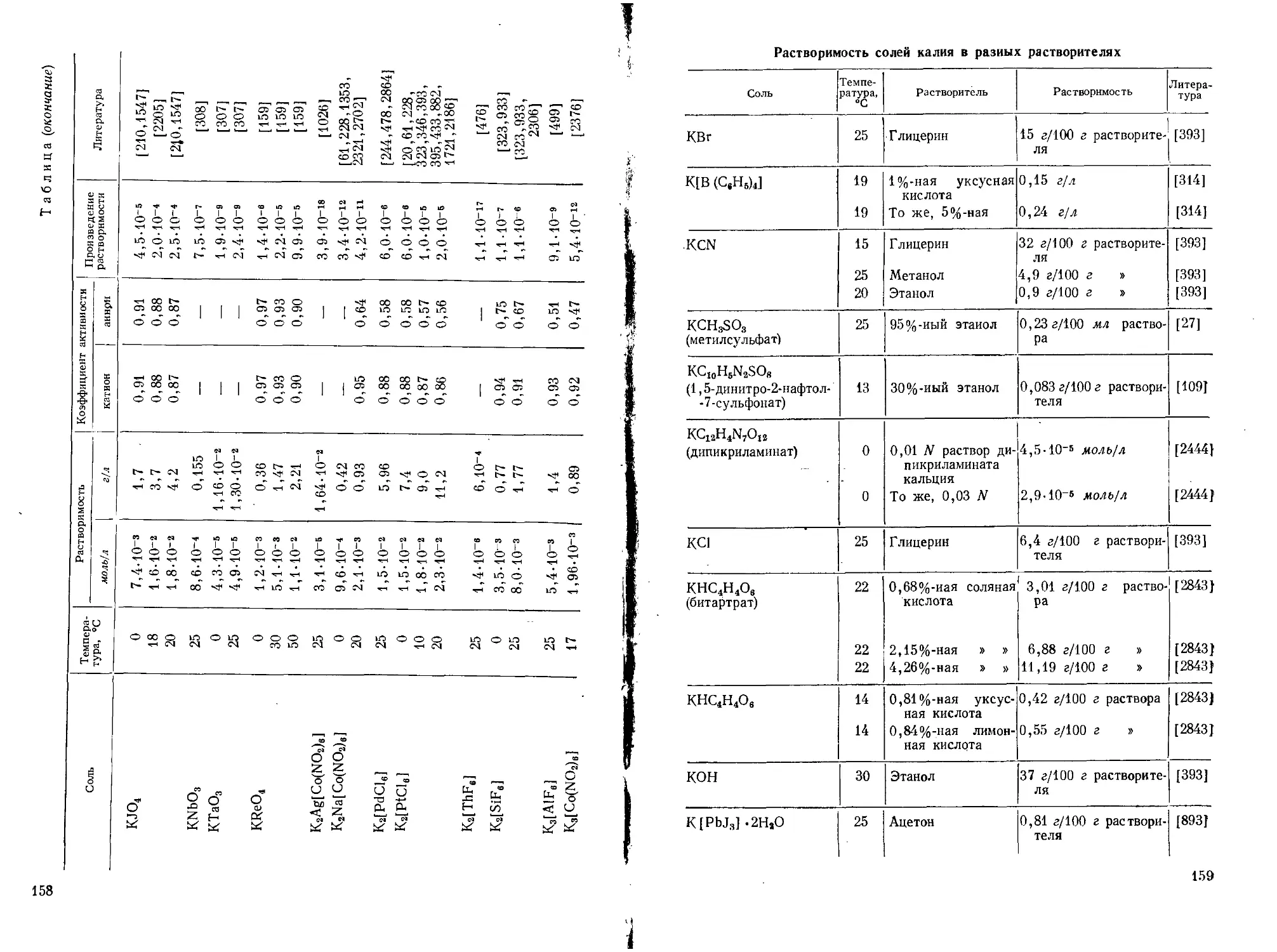
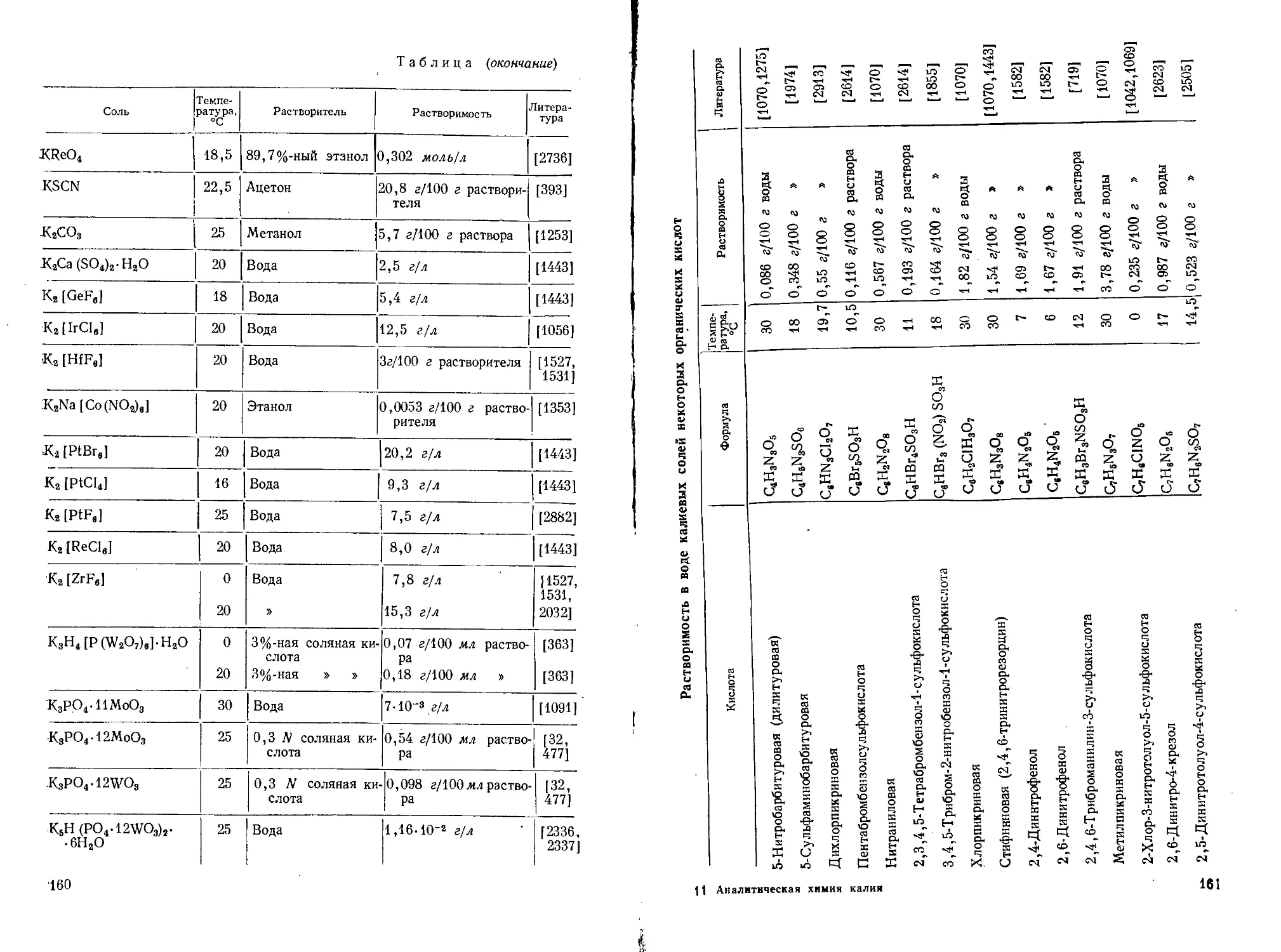
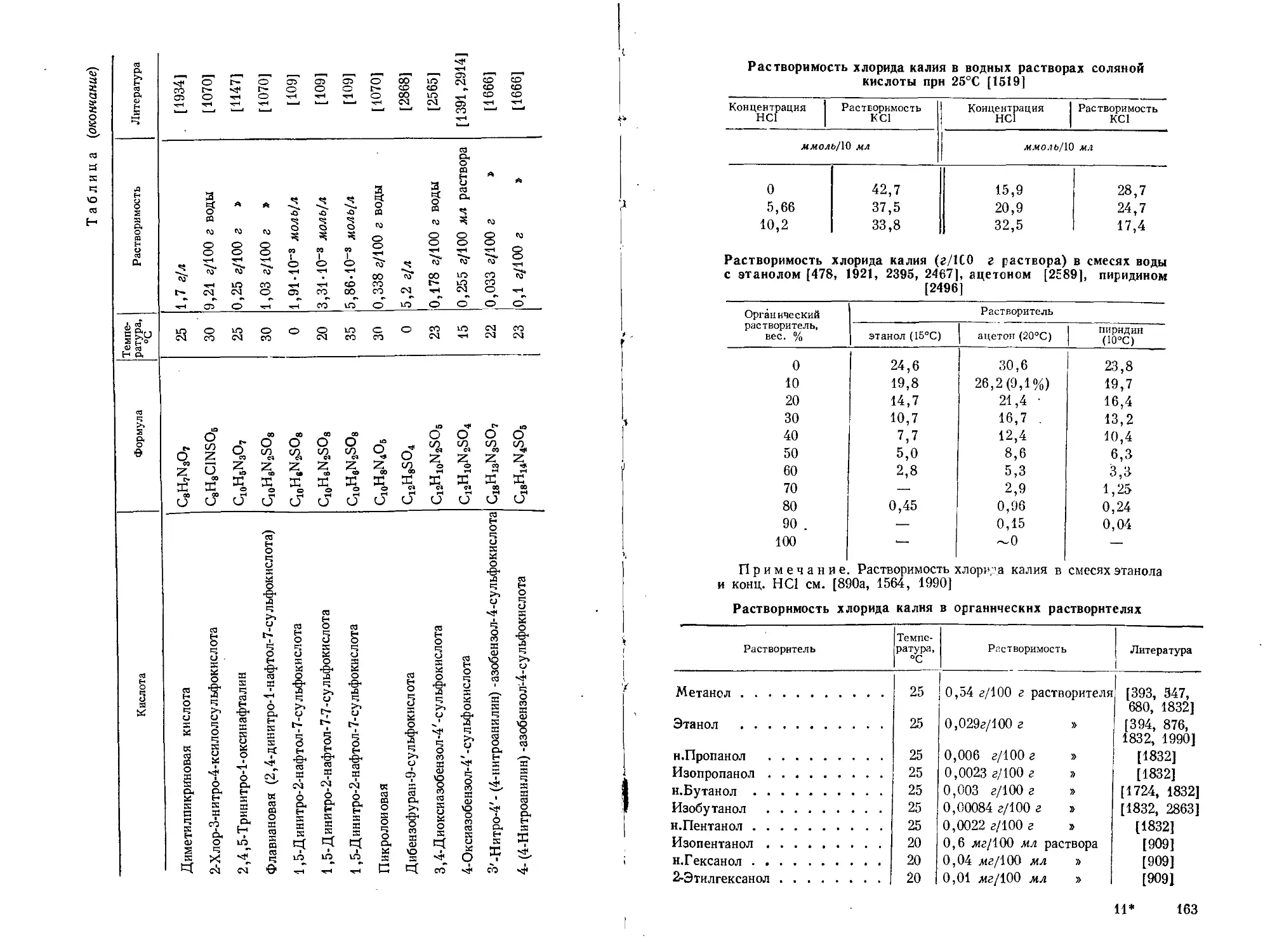
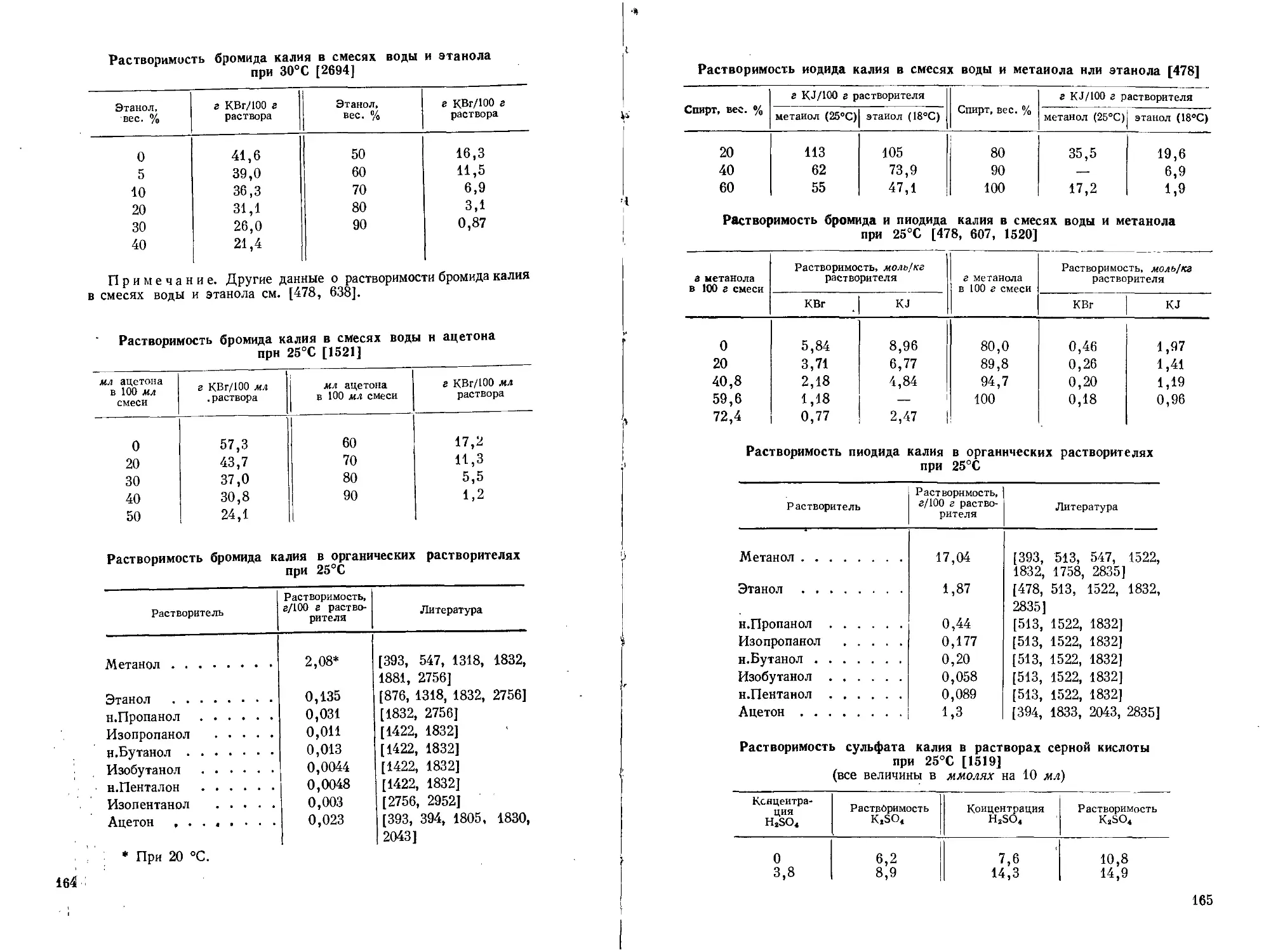
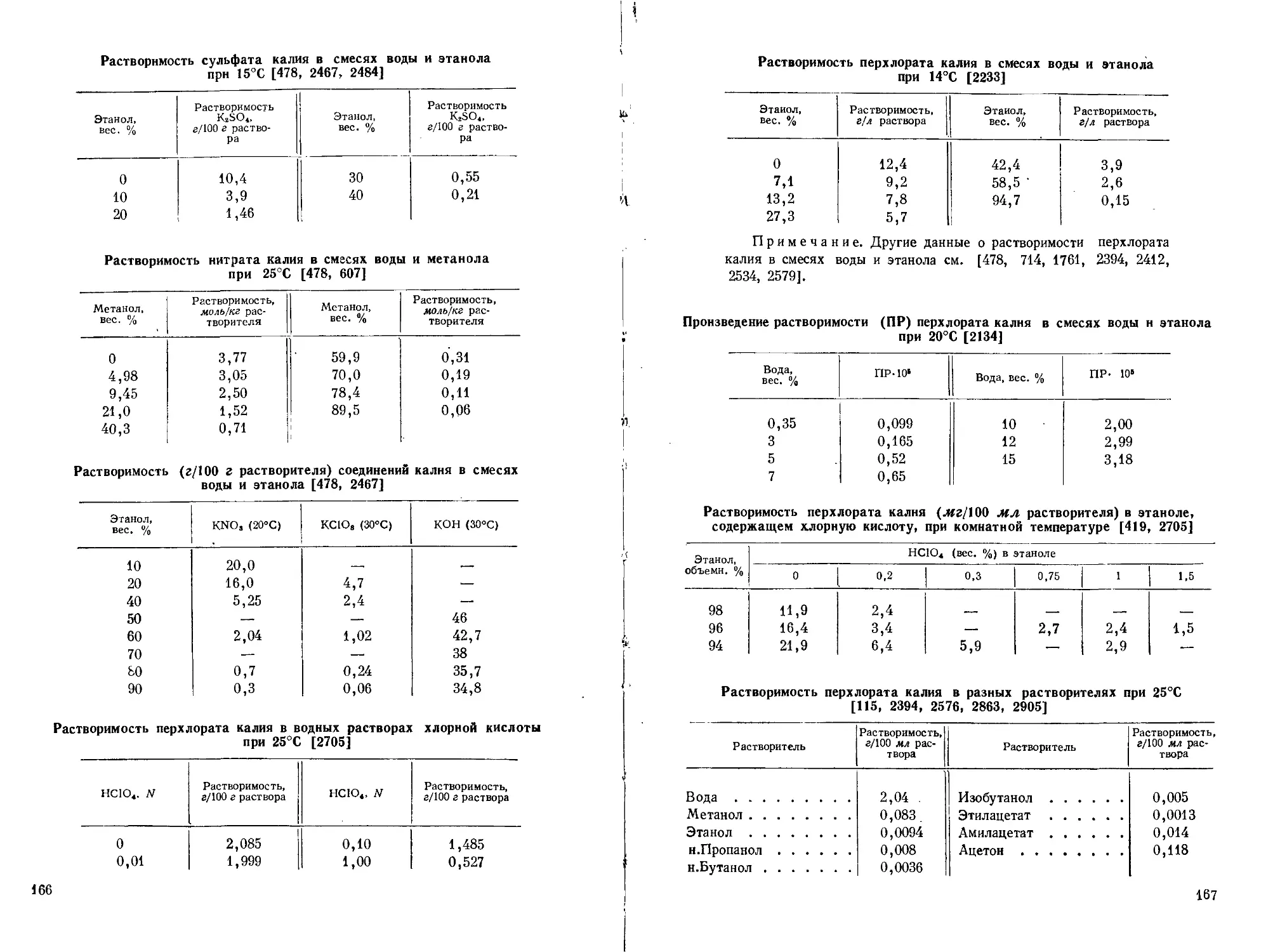
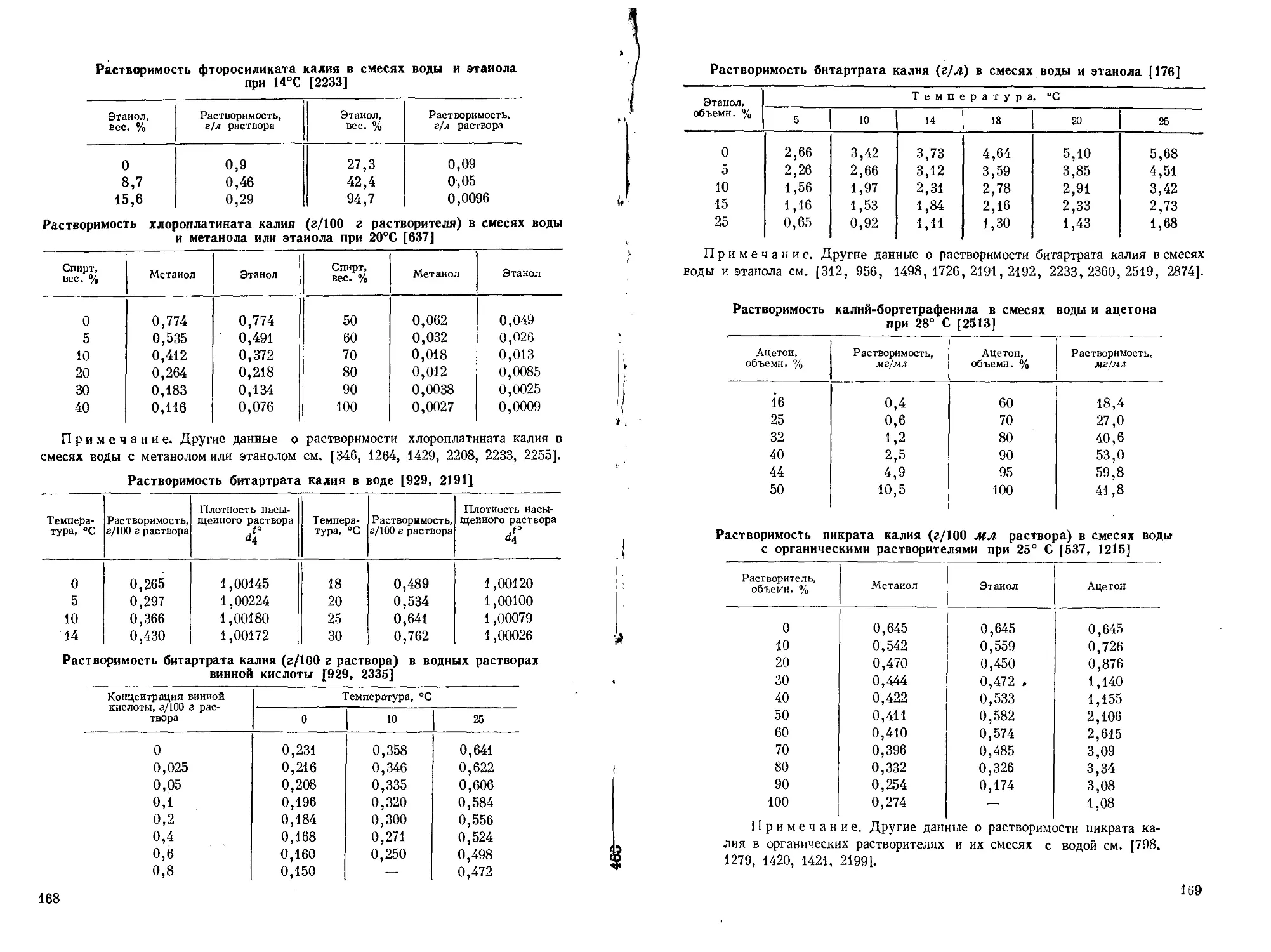
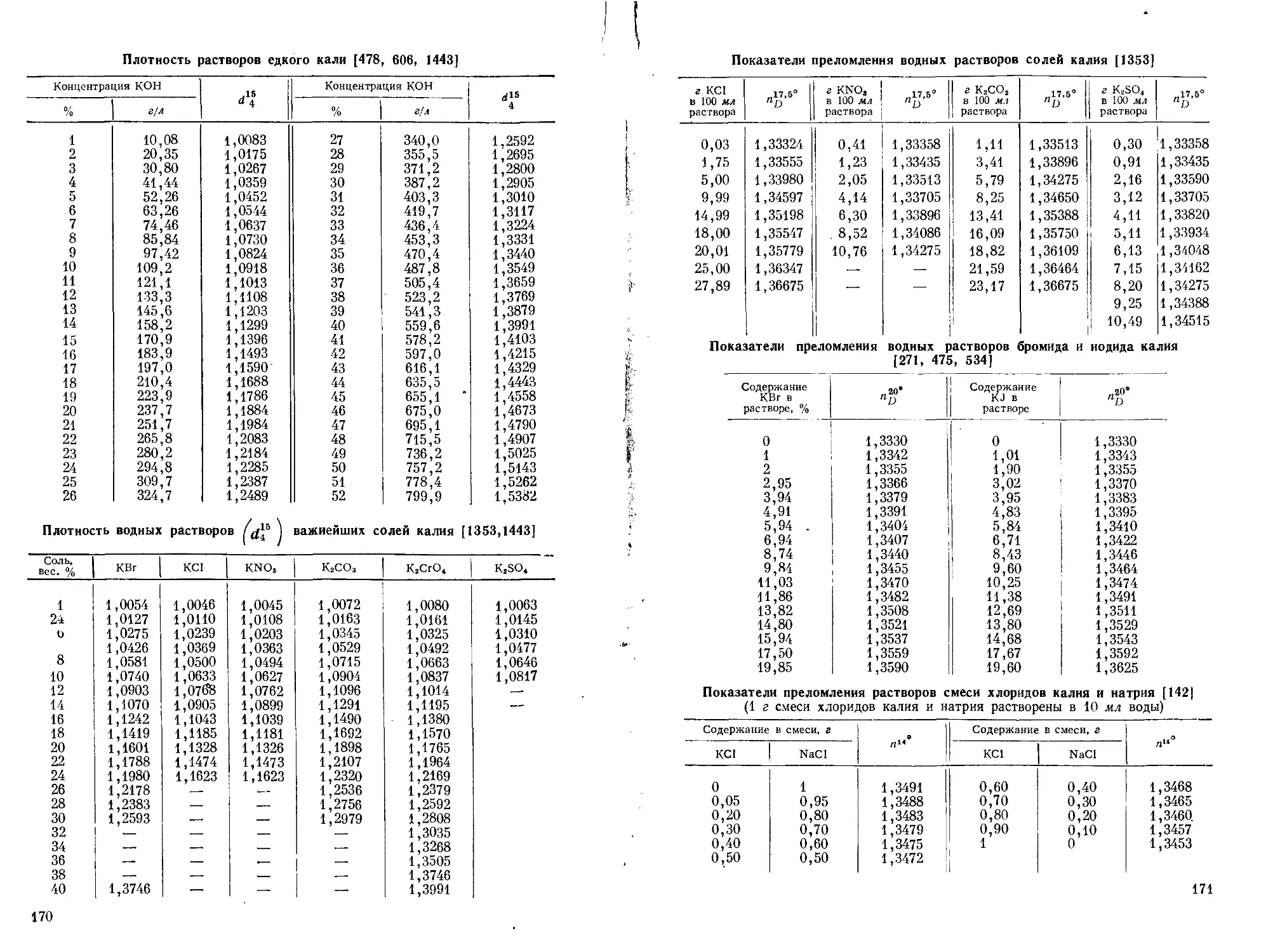
Поэтому в аналитической химии калия очень видное место занимают гравиметрические методы или методы, основанные на предварительном осаждении калия. (Растворимость главнейших солей калия см. стр. 153—169.)
Непостоянство состава малорастворимых соединений калия создает дополнительные трудности при количественных определениях. Состав малорастворимых солей калия нередко зависит от условий осаждения. Неопределенность состава приводит к необходимости пользоваться эмпирическими факторами при вычислении результатов количественного определения калия.
Аналитические свойства ионов калия во многих отношениях близки к свойствам ионов аммония, рубидия, цезия и одновалентного таллия [256]. Вследствие ненадежности количественного отделения калия от.натрия получили распространение косвенные методы определения калия (и натрия), не отличающиеся, однако, высокой точностью.
Ион калия бесцветен, он не дает цветных реакций. Некоторые цветные реакции являются реакциями на анион, его мало-» растворимой соли.
10
В своих соединениях калий только одновалентен, ионы калия не вступают в окислительно-восстановительные реакции. Оксидиметрические методы количественного определения основаны на окислительно-восстановительной способности анионов малорастворимых солей калия.
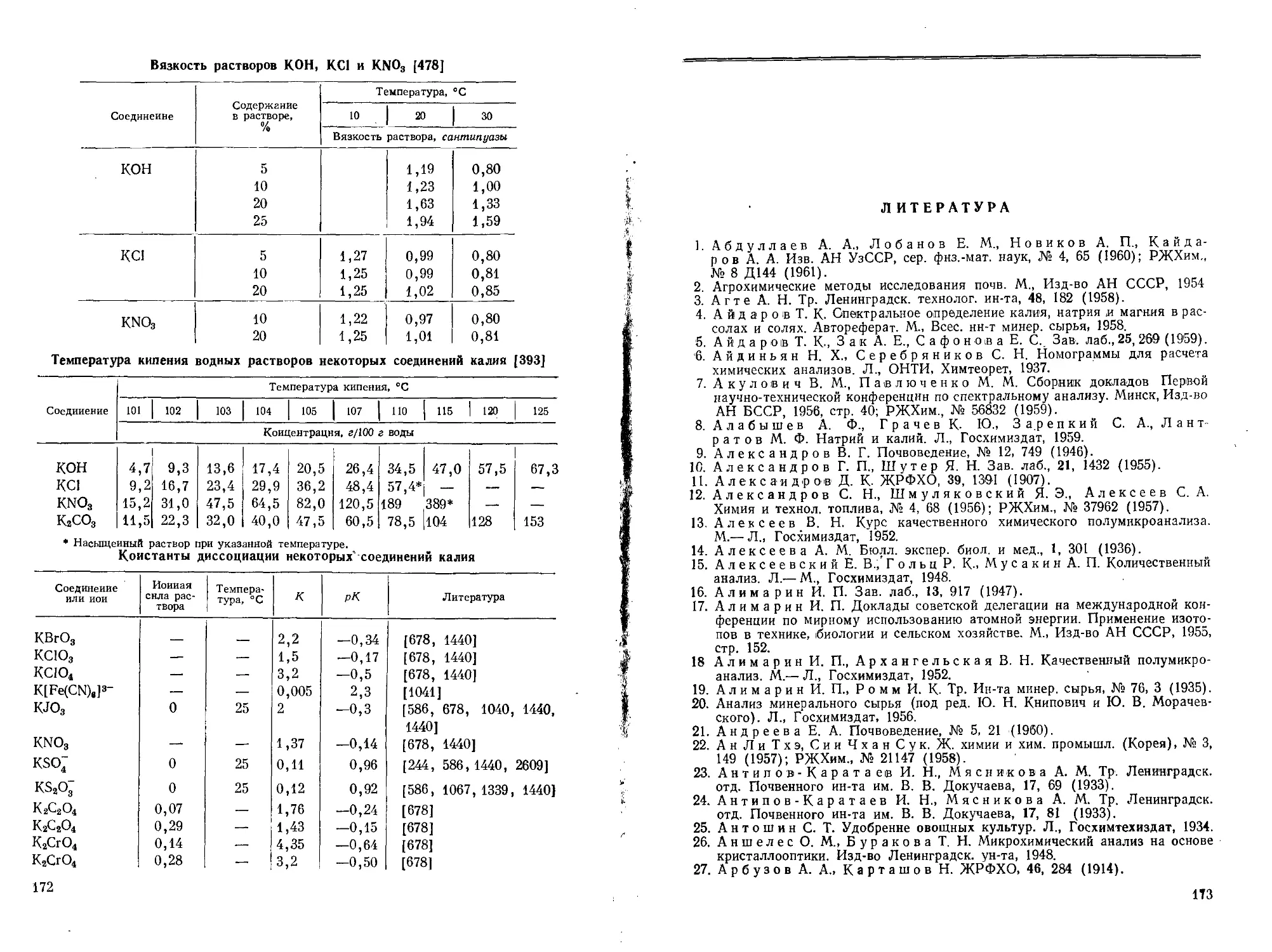
Калий образует очень мало комплексных соединений; они отличаются незначительной стабильностью (см. приложения, стр. 172) и поэтому не находят применения в аналитической химии этого элемента. В отличие от натрия и лития калий не образует комплексного соединения с этилендиаминтетрауксус-ной кислотой [367].
В последнее время для количественного определения калия значительную роль приобрели, физические методы, основанные на использовании естественной радиоактивности и способности окрашивать бесцветное пламя.
В литературе имеется очень много работ, касающихся аналитической химии калия. Здесь мы только сошлемся на обзорные статьи [158, 442, 727, 730, 900, 1117, 1445, 1461, 1501, 1563, 1723, 1841, 1867, 2130, 2337, 2355, 2600, 2627, 2824, 2853]. В некоторых обзорах рассматривается применение отдельных реагентов, например, нитрокобальтиата натрия [280, 2725], натрий-бортетрафенила [544, 584, 694, 903, 1425, 1775, 1831, 2323, 2681, 2698], других органических реагентов [731] или описываются методы определения калия в наиболее важных объектах — биологических материалах [143, 1864, 2142], удобрениях [1089], почвах [858]. Для ускорения вычислений при количественных определениях калия и уменьшения возможности арифметических ошибок'опубликованы специальные таблицы [84, 1142, 1360, 2439] и номограммы [6].
II. ОБНАРУЖЕНИЕ ИОНОВ КАЛИЯ
Для обнаружения ионов калия предложено очень большое число реагентов. Для удобства мы разделим применяемые реагенты на группы.
Образование малорастворимых солей состава KRO4 (R— Cl, Мп, Re)
Наибольшее значение из реакций этой группы имеет образование кристаллического осадка перхлората калия [13, 61, 297, 395, 545, 630, 730, 1000, 1548, 1632, 1849, 2757]. Соли аммония и других катионов (кроме рубидия и цезия) не мешают [61, 297, 1849]. Вследствие заметной растворимости осадка в воде реакция не отличается высокой чувствительностью, удается обнаруживать калий при разбавлении 1 : 1400 [2684]. Рекомендуется микрокристаллоскопическое обнаружение калия в виде КСЮ4 [26, 75, 250, 328, 954, 1311, 1407, 1463, 1670, 2666]; открываемый минимум 0,5 мкг К (1 : 2000) [250] и даже 0,1 мкг К [580]. Небольшие количества перманганата, введенные в раствор до осаждения, окрашивают кристаллы КС1О4 в розово-фиолетовый цвет [346].
Микрореакция образования кристаллического осадка пеп-манганата калия мало чувствительна [229]. Перренат калия KReO4 растворим в воде меньше, чем перхлорат калия (стр. 154), образование кристаллического осадка KReO4 позволяет обнаруживать соли калия.
Образование малорастворимых галоидосодержащих комплексных солей
При добавлении к раствору соли калия 1—2 А раствооа H[BF4] или Na[BF4] образуется кристаллический осадок K[BF4] [2648], что позволяет обнаруживать 2—5 мг К в 5 мл раствооа [1912, 2684]. Чувствительность повышается при добавлении этанола. Об осаждении K[BF4] в микрокристаллоскопии см. [566, 12
699, 1311, 2648]. Соли аммония, рубидия и цезия дают аналогичные реакции.
Несколько большее значение имеет реакция образования K2[SiF6] [58, 1749, 1947, 2649]. Осадителем служит H2[SiF6] либо ее аммонийная или анилиновая соли [250, 1912]. Чувствительность невелика, удается обнаруживать около 5 мг К в 5 мл раствора [1912, 2684]; добавление этанола повышает чувствительность. Соли аммония не мешают [61], соли бария, кальция, натрия дают осадки [56, 1273, 2649]. Эта реакция применяется для микрокристаллоскопического обнаружения калия [26, 56, 75, 250, 346, 580, 699, 724, 807, 1273, 1947, 2649] (10 мкг К в капле). Описан интересный способ обнаружения калия в частицах весом до 10-15 г, находящихся в воздухе, по образованию K^fSiFg] [2516].
Другие малорастворимые калиевые соли комплексных фторсодержащих кислот, например K2[TiF6] [61, 1342], Кг[Т!тР6] [965]„ K^ZrFg] [61], КгГГаР?] [33], не нашли применения для реакций на калий.
В качественном анализе часто пользуются образованием осадка хлороплатината калия KgfPtCls] [58, 228, 518, 1412, 1849, 1928]. Осадителем служит 5—10%-ный раствор H2[PtCls]-Реагент позволяет обнаруживать 1 мг К в 5 мл раствора [58, 1912, 1936, 2684, 2872] и еще меньшие количества калия [228]. Вследствие дороговизны реагента испытание на калий производят на предметном стекле, наблюдая под микроскопом характерные довольно крупные желтые октаэдры [26, 56, 60, 75, 250, 328, 346, 437, 558, 580, 593, 699, 724, 954, 1189, 1356, 1407, 1768* 1856, 1901, 1912, 2223, 2666, 2684, 2775, 2872]. В капле раствора удается заметить 0,01—0,5 мкг К [56, 250, 346, 724]. Добавление этанола повышает чувствительность реакции [228, 2.э0, 346, 580]. Такие же осадки дают ионы аммония, рубидия, цезия, одновалентного таллия. Осаждение хлороплатината применяется для обнаружения калия в гистологических срезах [1620, 2048], биологических жидкостях [751], золе растений [2048], алюминии и магнии [364].
При действии четыреххлористого осмия и этанола на раствор соли калия выпадают темно-красные кристаллы K^OsClg] [2321]. Описано также микрокристаллоскопическое обнаружение калия в виде Кг[РЬС16] — зеленовато-желтые кристаллы [368, 2359, 2865], К[РЬС13] —белые иглы [525, 526, 893, 2361], K2[PdBr6] — черные кубы [1409], K^ReCls] — желто-зеленые октаэдры [2321], К[РЬВг3] — иглы [562, 2361], KatPtJs] — черные кубы и октаэдры [250], K2'[ReJ6] — бурые или черные кристаллы [283, 860], K[PbJ3] • 2Н2О — длинные бледно-желтые иглы [161; 230, 250, 562, 893, 1738, 2100, 2361].
13
Образование малорастворимых комплексных нитритов
Из большого числа реакций осаждения калия в виде комплексных нитритов остановимся на образовании нитрокобаль-тиата, K2Na[Co(NO2)e] • НгО,— одной из наиболее чувствительных реакций на калий [795, 1031, 1996, 2396, 2437, 2463, 2924]. Соединение впервые было синтезировано в середине прошлого столетия Фишером (соль Фишера) [1111, 1211, 1212]. Образование этой соли для качественного анализа было предложено в 1881 г. [1769]. При действии концентрированного раствора нйт-рокобальтиата натрия, Na3[Co(NO2)6], на растворы солей калия выпадает мелкокристаллический желтый осадок, состав которого приближается к приведенной выше формуле (о составе осадка см. стр. 41). На способы приготовления реагента мы только сошлемся [1849, 1914, 2293]. Некоторые рецепты приготовления нитрокобальтиата натрия указаны на стр. 40.
К недостаткам нитрокобальтиата натрия следует отнести его малую устойчивость в растворах, поэтому лучше всего применять растворы реагента не более чем 2—3-дневной давности.
Нитрокобальтиат натрия позволяет обнаруживать калий в, растворах до концентраций порядка 1 : 50 000 [58, 228, 346, 699, 1370, 1686, 1912]. В присутствии этанола удается обнаруживать соли калия при разбавлениях до 1 : 106 {57, 437, 827, 1849, 1936, 2312, 2464, 2872].
Этот реагент применяют в микрокристаллоскопии [26, 75, 250, 328, 484, 580, 2490, 2872], капельном анализе [480], при обнаружении методом растирания [193, 194], электрокапиллярном методе [160] и других. Реагентом пользуются для обнаружения калия в минералах [218, 1282, 1377], стекле [2971], солях натрия [1910, 1911], медикаментах [480], растительных материалах [2048, 2714, 2858], гистологических срезах [132, 1324, 1914, 2048].
Кроме калия, с нитрокобальтиатом взаимодействуют ионы аммония, рубидия, цезия, одновалентного таллия, которые мешают обнаружению калия. Мешают также окислители, свободные щелочи, иодиды [216]. Щелочноземельные металлы, железо, алюминий, цинк и другие катионы не дают осадков [1788, 2379].
Большей чувствительностью отличается осаждение калия в виде K2Ag[Co(NO2)e] [888]. К нейтральному разбавленному раствору соли калия добавляют немного 0,02 N раствора AgNO3 и избыток концентрированного раствора нитрокобальтиата на-; трия; при этом выпадает желтый осадок нитрокобальтиата калия и серебра. В отсутствие калия осадок не образуется. Метод позволяет обнаруживать калий при разбавлениях до 1 : 106 [1271, 1912]. В исследуемом растворе должны отсутствовать хлориды и другие анионы, осаждаемые нитратом серебра. Соли аммония, рубидия, цезия дают такую же реакцию, как и соли
14
каЛия. О применении этой реакции в качественном анализе см, [61, 228, 250, 484, 530, 1199, 1787].
Из других реакций такого типа заслуживает упоминания осаждение K2Pb[Cu(NO2)6], часто применяемое для микрокри-сталлоскопического обнаружения калия [26, 75, ИЗ, 193, 194, 250, 437, 520, 954, 1200, 1311, 1727, 1768, 1902, 1936, 2345, 2872]. Под микроскопом наблюдаются черные блестящие кубически^ кристаллы, открываемый минимум 0,15 мкг К [56, 250, 346, 437, 2684, 2872]. Аналогично взаимодействуют соли аммония, рубидия, цезия и одновалентного таллия. Метод применяется для обнаружения калия в разных объектах [56, 250, 364, 751, 2383].. О приготовлении реагента см. [386].
Описано большое число подобных реакций осаждения калия (рубидия и цезия) в виде следующих комплексных нитритов:
K2Pb[Ni(NO2)6] [248, 250, 1168, 1200, 1727, 1966, 2684]; свинец может быть заменен щелочноземельным элементом [248, 1965, 2076], кадмием, двухвалентной ртутью [1198], а никель — двухвалентным кобальтом [194 , 228, 248, 250, 437, 1032, 1200, 1698, 1966, 2684, 2872], медью [248], железом [1198].
KPb[Co(NO2)6] [160, 250, 341, 507, 1198, 1199].
K4[Ni(NO2)6] [2321], место никеля может занимать двухвалентное железо [1198].
KsCefCufNOsleh, место церия могут занимать катионы других редкоземельных элементов или иттрия, место меди — Со2+, Ni2+, Fe2+ [496, 497, 1033, 1198]. Соединения, содержащие в своем составе Со2+,— темно-зеленого цвета, Ni2+ — красного и Си2+ — черного цвета.
K2[Pb(NO2)4] [250]; KsAg[Pb(NO2)6] • 2Н2О [1625].
Некоторые из указанных соединений еще не нашли применения в аналитической химии.
Образование комплексных (двойных) сульфатов и тиосульфатов
Известны методы обнаружения калия, основанные на осаждении малорастворимых комплексных (двойных) сульфатов или тиосульфатов. Осаждение в виде 3K2SO4 • Bi2(SO4)3 или Кз[В1(5О4)з] позволяет устанавливать до 0,3 мкг К в капле по образованию характерных кристаллов. Мешают соли аммония, рубидия, цезия, щелочноземельных металлов, свинца. Аналогично реагируют соли натрия, которые образуют кристаллы иной формы, что дает возможность одновременно обнаруживать калий и натрий [26, 56, 75, 248, 250, 268, 328, 346, 724, 954, 1287, 1311, 1356, 1499, 1768, 1783, 2223, 2489, 2684].
Предложена микрокристаллоскопическая реакция, основанная на осаждении Кг5О4 • Ce2(SO4)3 • 2Н2О [56, 250]. Описаны также соединения Ks[Ce4(SO4)9] • 8Н2О и K5[Ce(SO4)4] [2321]. Се (IV) образует малорастворимые оранжево-желтые кристаллы K4[Ce(SO4)4] • 2Н2О [2321]. Калий осаждают 10%-ным раствором сульфата цирконила в виде 7г2Оз(5О4К)2'8Н2О [518,
15-
1271 2305, 2378, 2684] или K4[Zr (SO4) 4] • 4H2O [2321]. Реакция позволяет ’обнаруживать калий в присутствии солей аммойия и щелочных металлов.
Осаждение малорастворимых сульфатов типа K2SO4 • PbSO4 [54, 498, 741, 907, 1379, 2216, 2287], в которых место свинца могут занимать щелочноземельные металлы [54, 357, 456, 500, 604, 1378, 2784, 2785], еще не нашло применения в аналитической химии калия.
Подробно изучалось осаждение калия в виде K3[Bi(S20s)3] • • Н2О в присутствии большого количества этанола [9271. Смешивают 1 каплю 0,5 N раствора нитрата висмута, 3 капли 0,5 N раствора тиосульфата натрия, разбавляют 5—10 мл этанола и добавляют небольшой объем исследуемого раствора. При наличии солей калия выпадает желтый осадок [61, 194, 518, 699, 916, 1271, 1370, 1412, 1462, 1554, 2196, 2222]. Предельная концентрация достигает 1 : 125 000 [1912, 2684]. Соли рубидия, цезия, стронция, бария, свинца мешают обнаружению калия. О применении этой реакции в микрокристаллоскопии, см. [250, 483, 484, 580, 1596].
Для приготовления реагента с целью осаждения калия в виде Кб[Со(5гО3)4] [945] к раствору 7 г нитрата кобальта в 100 мл воды добавляют 40 мл метанола; отдельно растворяют 19 г тиосульфата натрия в 50 мл воды. К 10 мл метанола прибавляют по капле этих растворов и несколько капель исследуемого раствора. При наличии калия выпадает синий осадок [945, 1271, 2684, 2924]. Соли аммония, кальция, стронция не дают осадков; соли бария, свинца, серебра, меди, уранила образуют осадки, но не синего цвета [1271].
Образование солей гетерополикислот
Из подкисленного азотной кислотой раствора фосфорномо-либденовая кислота осаждает желтые мелкие кристаллы фос-форомолибдата калия К3РО4 • 12МоОз • «Н2О [1042], предельная концентрация 1 : 10 000 [1598, 2469, 2684]; по другим данным — 1 : 560 [1912]. Об этой реакции см. также [26, 56, 75, 248, 250, 580, 699, 724, 1271, 1297, 1692, 1787, 1878, 2468]. По осаждению белого фосфоровольфрамата калия [2204] удается обнаруживать калий при разбавлении до 1 : 2800 [1912, 2008, 2020, 2684]. См. также [248, 363, 582, 2932, 2943].
Осаждение кремневольфрамата калия K4SiO4 • 12WO3 nH2O применяется редко [250, 580, 1271, 2684]. О составе осадков калиевых солей гетерополикислот см. стр. 47. Гетерополикислоты дают осадки и с растворами солей рубидия, цезия, одновалентного таллия.
16
Образование малорастворимых ферроцианидов
Для осаждения K2Ca[Fe(CN)6] применяют раствор 7 г ферроцианида натрия, 3,5 г хлорида кальция в 95 мл воды; к раствору добавляют 80 мл 96%-ного этанола. Осадок смешанного ферроцианида калия и кальция белого цвета, мало растворим в воде [1295, 1296]. О растворимости этой соли опубликованы противоречивые данные [838, 849, 1271]. Осадки дают также соли аммония, рубидия, цезия [2093, 2174, 2277, 2684, 2924]. В этой реакции соль кальция можно заменить солью магния [2093]. Осадки сложного состава образуются в присутствии уротропина [683]. Для обнаружения калия применяется также раствор ферроцианида лития [2276, 2684] и смесь раствора ферроцианида лития с золем ферроцианида кобальта [495, 2276].
Обнаружение при помощи солей уранила
Ацетат уранила с солями калия образует светло-желтый кристаллический осадок КиО2(СНзСОО)з, используемый для микрокристаллоскопического обнаружения калия [250, 953, 1311, 2384, 2684]. Реакция мало чувствительна. Ацетат уранила дает осадки и с солями натрия, лития и других элементов [250]. Описаны также кристаллические малорастворимые соли КВе(иО2)з(СН3СОО)9 и КиО2(СС13СОО)3 [1076].
Смесь 5%-ных растворов нитрата уранила и хромата натрия с солями калия (рубидия, цезия) дает желтый кристаллический осадок К2СгО4 • 2UO2CrO4 • 6Н2О [1242, 1296, 2894, 2924], • растворимый в кислотах, концентрированных растворах хлорида натрия, в избытке раствора нитрата уранила.
Описаны мало чувствительные микрореакции осаждения калий-магний фосфата, KMgPO4-6H2O [250, 1167, 1732], пентабората калия, KBsO8-4H2O [1415, 2186, 2368, 2965],
K4[Zr(C2O4)4] [2321] и другие [975].
Образование калий-бортетрафенила
Легко растворимый в воде натрий-бортетрафенил, известный также под названием «калигност» и «политест», с растворами солей калия дает малорастворимый белый осадок калий-бортетрафенила, К[В(С6Н5)4] [584, 1192, 1425, 2190, 2404, 2698, 2754, 2928, 2929]. Реагент осаждает также ионы аммония, рубидия, цезия, одновалентного таллия, серебра, органических оснований [2082]. Возможно, что реагентом на калий окажется и натрий-бортетранафтил [436].
2 Аналитическая химия калия 17
Образование осадка битартрата калия
Реакция осаждения белого кристаллического битартрата калия была известна еще в середине XIX столетия [1263, 2240]; ее изучению посвящен ряд исследований [13, 333, 730, 1104, 1259, 2301, 2463]. Реагентом обычно служит раствор битартрата натрия, но применяются и соответствующие соли лития [2922], магния [2684], смесь растворов винной кислоты и ацетата натрия [1849, 2921], раствор винной кислоты, нейтрализованный на 50% едким натром (605], или раствор битартрата анилина [294, 1814]. Реакция позволяет обнаруживать калий при концентрациях не ниже 1 : 1000 [1912, 2684, 2921], по другим данным — 1 : 5000 [228]. При употреблении этанолового раствора битартрата анилина предельная концентрация достигает 1 :25 000 [1814]. Об осаждении в присутствии триэтаноламина см. [1624].
Осаждение битартрата калия часто применяется в микрокристаллоскопии [26, 75, 250, 303, 328, 580, 1311, 1463]. Аналогичные реакции дают соли рубидия и цезия [616, 2921].
Реакцию с солями калия дает правовращающая винная кислота и ее легко растворимые соли. Соответствующие калиевые соли рацемической винной кислоты или недеятельной мезовин-ной кислоты отличаются большей растворимостью в воде [775, 1263, 1479, 2683]. Малорастворима кислая калиевая соль диметилвинной кислоты [816].
Реакции с органическими полинитросоединениями
Эта группа реагентов на калий наиболее многочисленна. Теоретические вопросы, связанные со способностью некоторых полинитросоединений взаимодействовать с солями калия (рубидия, цезия), см. [249, 290, 291, 295].
Наиболее старый реагент этой группы — пикриновая кислота— применяется по крайней мере с 1827 г. [1873]. Насыщенный водный раствор пикриновой кислоты дает с солями калия желтый осадок пикрата калия [1259, 1370, 1511, 1632, 1668, 1840]. Предельная концентрация 1 : 1000—1 : 1250 [1370, 1912, 2684]. Для повышения чувствительности пользуются 10%-ным этаноловым раствором пикриновой кислоты или насыщенным водным раствором пикрата натрия [1912]. Пикрат калия образует крупные желтые иглы и поэтому часто используется в микрокристаллоскопии [26, 75, 250, 379, 580, 954, 1063, 1250, 1258, 1768, 2161, 2188], открываемый минимум 0,2—0,8 мкг К [250, 379]. Аналогичную реакцию дают соли аммония, рубидия, цезия, бария, серебра [2248], одновалентного таллия, ртути, свинца и большие количества натрия [379, 1963].
В настоящее время широко применяется другой реагент этой
18
Таблица 1
Нитросоединения, образующие малорастворимые соли калия (рубидия, цезия)
Реагент Характер осадка соли калия Чувствительность Литература
1,1'-Динитроэтан Желтый осадок — [1983]
1,1'-Динитропропан То же — [1983]
4-Нитрофенол » » — [250,1259, 2381]
2,6-Дибром-2-нитрофенол Темно-красные кристаллы — [2777]
2,4-Динитрофенол Темно-желтые призмы 0,13 мкг* [250,1259, 1582,2381]
2,6-Динитрофенол Красные иглы — [250,1259, 1582,2381]
2, 4, 5-Тринитрофенол Темно-красные иглы — [1511]
Диметилпикриновая кислота Кристаллический осадок 1 : 1000 [1934,2057]
З-Метилпикриновая кислота То же — [295,2057]
З-Этилпикриновая кислота З-Изопропилпикриновая » » — [2057]
кислота » » — [2057]
Метилэтилпикриновая кислота » » — [2057]
Метилпропилпикриновая кислота » » — [2057]
2, 6-Динитро-4-крезол Красные иглы — [2623]
2, 4 ,6-Тринитро-З-крезол Желтые иглы 0,4 мкг* [250,1259, 2381] [250,814,
2, 4, 6-Тринитрорезорцин Оранжевый кристалли- 0,03 мкг*
(стифниновая кислота) ческий осадок 1144,1213, 1511,2381, 2561]
Т ринитрофлороглюцин Желтые и оранжевые кристаллы — [744]
2-Нитро-1-нафтол Красные иглы — [1854]
4, 8-Динитро-1-нафтол Желтый осадок — [1375]
2, 4, 5-Тринитро-1-нафтол Красные иглы — [1147]
2, 4-Динитро-1-нафтол-7- Желто-оранжевый крис- 0,7 мкг[мл [48,250, 263,968, 1104,1258, 1259]
-сульфокислота (нафтоловый желтый S) таллический осадок
7-Нитро-8-оксихинолин-5- -сульфокислота Кристаллический осадок — [2049]
2, 5-Диокси-З, 6-динитро-хинон (нитраниловая кислота) То же Малая [250,1944]
2*
19
Таблица 1 (окончание)
Реагент Характер осадка соли калия | Чувствительность Литература
Пикролоновая кислота Кристаллический осадок 1: 1000 [250,568, 1145,1259, 1841,2819, 2863]
Нитроиндан дион Желтый кристаллический осадок Малая [89]
5-Нитробарбитуровая кислота (дилитуровая кислота) Бесцветный кристаллический осадок 0,02 мг[мл [1214,1257, 1259,1275, 1557,2302, 2472]
Динитробензофуроксан Желтые или оранжевые кристаллы 1-105 [1337,2291]
Т етранитрофенотиазин- — — [2731]
-5,5-диоксид
2,4-Динитро-1,5-бис- — Малая [1606,2732]
-(2,4,6-тринитрофенил-амино)бензол (дигексил)
1 -(2,4,6-Т ринитрофенил-амино)-2,4-динитро- Черный кристаллический осадок —' [1606,2732]
нафталин (а-гексил)
2,4,6,2' ,4' ,6' -гексанитро- Зеленые кристаллы — [553,1845]
гидразобензол
0-Нитро-2-аминофенол- Бурые иглы .— [728]
-4-сульфокислота
1, 5-Динитро-2-нафтол- -7-сульфокислота Желтый кристаллический осадок — [109]
1,1-бис-(2,4-Динитрофе-нил)метан Синий кристаллический осадок 1 : 200 [262]
♦ Микрокристаллоскопическая реакция.
группы — дипикриламин (гексанитродифениламин, 2,2',4,4',6,6'-гексанитродифениламин, ГДПА, ДПА). Это соединение было известно еще в прошлом столетии. Аммонийная соль дипикрил-амина под названием «ауранция» применялась в качестве красителя. Была известна и калиевая соль дипикриламина [11]. Однако только в 1933 г. это соединение было предложено Полуэктовым [407] для обнаружения калия. Реагентом служит раствор натриевой [1191] или магниевой [1827] соли дипикриламина. При добавлении этого реагента к растворам солей калия выпадает кристаллический оранжево-красный осадок ди-пикриламината калия, KC12H4N7O12, что позволяет обнаруживать калий в пробирке при разбавлениях до 1 : 10000—1 :50000 [107, 228, 2684]. Предельная концентрация при капельной реакции 20
Таблица 2
Сульфокислоты, образующие малорастворимые соли калия
Реагент Характер осадка соли калия Чувствительность Литература
Алкилсульфокислоты Бензолсульфокислота З-Бромбензолсу льфокис-лота 4-Бромбензолсульфокис-лота 2,3,4,5-Тетрабромбензол-сульфокислота Пентабромбензолсульфо-кислота 2,4,5-Триброманилин--3-сульфокислота 2-Иодбензол сульфокислота 3, 4, 5-Трииодбензолсуль-фокислота 2-Хлор-З-нитротолуол--5-сульфокислота 2-Бром-З-нитротолуол- -5-сульфокислота 2, 4-Динитротолуо'л--4-сульфокислота 3, 4, 5-Трибром-2-нитро-бензолсу льф окислота Нафталин-1-сульфокис-лота 1-А мино-2-н афтол-6-суль-фокислота (эйконоген) 2, 4-Динитронафтиламино-бензолсульфокислота 5-Сульфаминобарбитуро-вая кислота Ализаринсульфокислый натрий З-Бромазобензолсульфо-кислота 4-Оксиазобензол-4'-суль-фокислота 3,4-Диоксиазобензол-4'-сульфокислота Аморфный осадок Кристаллический осадок То же » » Иглы Иглы Кристаллический осадок Кристаллический осадок То же Кристаллический осадок Аморфный красный осадок 8 мкг/мл 1:2500 1:1000 1:200 1:200 0,1 мг/мл [27,730, 1259,1840а, 2684,2900] [2382] [1639] [1639] [2614] [2614] [719] [671] [830] [730,1042, 1160,1259, 2152,2355, 2385] [295,545, 1069] [2507] [1855] [250,2382] [623,699, 730,1271, 1847,2863] [1135] [1974] [1259,1319, 2684] [2684] [1391,2914] [766,2565]
21
Таблица 2 (окончание)
Реагент Характер осадка соли калия Чувствительность Литература
Метаниловая желтая Кристаллический осадок 0,04 1лг[1лл [1187]
2-Оксинафталин-1 -азонаф-талин-4-сульфокислота — — [730]
4'-(4-Нитроанилин)-азобен-зол-4-сульфокислота Светло-желтые иглы — [1666]
4'-(2, 4-Динитроанилин)- -азобензол-4-сульфокис-лота Темно-красные призмы — [1666]
3'-Нитро-4'-(4-нитроани-лин-азобензол-4-сульфо-кислота) Красные ромбы — [1666]
Дибензофуран-2-сульфо-кислота — [2355,2868]
Фенантрен-З-сульфокис-лота — [2600]
1 : 1000— 1 : 10 000 [13, 296, 407, 437, 530, 563, 1191, 1259, 1778, 1936] и при микрокристаллоскопической— 1 : 10 000— 1 : 500 000 [250, 437, 563, 1768, 1936, 1964, 2781, 2872].
Испытание на присутствие калия следует производить в щелочной или нейтральной средах. В кислых растворах выпадает осадок малорастворимого дипикриламина [560]. Осадки мало-растворимых дипикриламинатов дают также соли рубидия, цезия, одновалентного таллия, свинца, ртути [530, 563, 1778].
Простой способ синтеза дипикриламина [294], возможность обнаружения малых количеств калия и его отделения от натрия, магния, кальция и других катионов способствовали быстрому внедрению этого реагента в практику многих аналитических лабораторий. Дипикриламин применяется для обнаружения калия в минералах [296], крови [296], для гистохимических исследований [132, 575, 924]. Об этой реакции см. также [209, 545, 730, 1082, 1104, 1259, 1713, 2262].
Краткие сведения о других нитросоединениях, способных давать малорастворимые соли калия (рубидия, цезия), приведены в табл. 1 (см. стр. 19—20).
Реакции с сульфокислотами
Многие сульфокислоты способны осаждать соли калия из их не очень разбавленных растворов [255]. В табл. 2 (см. стр. 21—22) приведены краткие данные о некоторых таких сульфокислотах.
22
Известны и другие органические реагенты на калий: фталевая кислота {2684, 2961], кроконовокислый натрий [2253], декаоксициклопентан [2229, 2355], виолуровая кислота [670, 1447, 1448, 2833, 2863], 1-оксинафтойная кислота [2684], 2-нитрозо-1-нафтол-4-сульфокислота [991], 5-оксо-4-оксим-3-фенил-изоксазо-лин [1170], 4-метил-3,5-дициандиоксипиридин [2231], флуоресцеин-комплексон [1773], локановая кислота [327, 1190, 1271, 1412, 1691, 2684, 2863].
Обнаружение солей калия по окраске пламени см. [186, 558, 812а, 881, 884, 894, 922, 1365, 1380, 2257, 2834], об устранении влияния натрия см. [868, 881, 925, 934, 1006, 1524, 1849, 2010, 2362, 2886].
III. КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ КАЛИЯ
ХИМИЧЕСКИЕ МЕТОДЫ
Гравиметрическое определение
Определение в виде сульфата. Известно несколько вариантов определения калия в виде сульфата.
В наиболее простом случае раствор, содержащий только K2SO4, выпаривают, остаток прокаливают, охлаждают и взвешивают. Чтобы уменьшить потери от разбрызгивания, к анализируемому раствору добавляют немного серной кислоты, выпаривают сначала на водяной бане, затем' нагревают на плитке приблизительно при 250° С до полного удаления паров серной кислоты [663, 1221]. При этом образуется бисульфат:
K2SO4 + H2SO4 — 2KHSO4.
При нагревании он переходит в пиросульфат:
2KHSO4— КАО? + Н2О.
Около 600° С пиросульфат начинает разлагаться с образованием сульфата: K2S2O7 —► K2SO4 + SO3.
Однако более быстро пиросульфат калия переходит в сульфат в присутствии карбоната аммония. С этой целью добавляют 0,1—0,2 г чистого карбоната аммония, тигель закрывают и нагревают:
K2S2O7 + (МН4)2СОз ► K2SO4 + (NH4)2SO4 + СО2.
При нагревании улетучивается и сульфат аммония. В остатке находится только K2SO4, который взвешивают после охлаждения [786, 1129, 1761]. Нагревать при более высоких температурах нельзя, выше 800° С заметно улетучивается сам сульфат калия, а при 1500° С и выше он, кроме того, разлагается на окись калия и серный ангидрид [346, 663, 823, 1271, 1699, 2322], остаток от прокаливания оказывается щелочным.
1 вес. ч. K2SO4 соответствует 0,4487 вес. ч. калия.
Если наряду с сульфатом калия в анализируемом растворе присутствует и сульфат натрия или какого-либо другого металла, то способ дает сумму сульфатов. В смеси сульфатов можно определить отдельно калий и натрий косвенными методами (стр. 87). Присутствие солей щелочноземельных металлов или свинца может привести к потере калия вследствие образования малорастворимых соединений, например K2SO4 • CaSO4 • Н2О[357, 456, 604,2784,2785],K2Ca5(SO4)6.H2O [2784, 2785], K2SO4 • PbSO4 [54, 498, 741, 907, 1742, 2216, 2287].
24
В исследуемом объекте или его растворе может содержаться соль летучей кислоты, например карбонат, нитрат, хлорид, бромид, иодид, формиат, ацетат и др. Добавляют немного серной кислоты, выпаривают и прокаливают, как указано выше. В случае бромида и иодида прокаливают в фарфоровом тигле, в присутствии солей других кислот эту операцию производят в платиновом тигле [1271]. Вместо выпаривания с H2SO4 можно выпаривать со смесью сульфата и хлорида аммония [2070].
Калиевую соль органической нелетучей кислоты нагревают при температуре, достаточной для разложения и обугливания. Следует избегать прокаливания при высокой тёмпературе вследствие опасности вспучивания и разбрызгивания пробы. Из обуглившегося остатка извлекают водой образовавшиеся карбонат или окись калия, раствор фильтруют, остаток промывают, фильтрат снова выпаривают и прокаливают. Затем прокаленное вещество обрабатывают водой, раствор фильтруют, фильтрат подкисляют серной кислотой, выпаривают и поступают, как указано выше [1271].
Гравиметрическое определение суммарного количества щелочных металлов в виде сульфатов рекомендуется при анализе ряда объектов.
Определение щелочных металлов в воде. 500 мл исследуемой пробы воды выпаривают досуха, остаток кипятят с раствором Ва(ОН)2 и фильтруют. В фильтрате содержатся щелочные металлы, следы кальция и избыток Ва (ОН)2. Ионы бария и кальция осаждают оксалатом аммония при кипячении, осадок отфильтровывают. Фильтрат выпаривают, остаток прокаливают для удаления солей аммония, обрабатывают серной кислотой, как указано, выше [2831].
Определение щелочных металлов в минералах и горных породах. 10—30 мг анализируемого материала разлагают в платиновом тигле фтористоводородной кислотой и выпаривают. Остаток выпаривают с щавелевой кислотой и прокаливают. Из охлажденного остатка вода извлекает образовавшиеся при прокаливании карбонаты щелочных металлов, а также немного гидроокиси магния и карбонатов щелочноземельных металлов. После осаждения 8-оксихинолином в фильтрате находятся только щелочные металлы (и избыток 8-оксихинолина). Фильтрат обрабатывают серной кислотой и т. д., как указано выше [16]. Можно после разложения фтористоводородной кислотой раствор выпарить досуха и остаток обработать раствором Са(ОН)'2> который осаждает посторонние катионы в виде гидроокисей. Фильтрат, содержащий калий, натрий и избыток гидроокиси кальция, обрабатывают карбонатом аммония для осаждения кальция. В фильтрате определяют суммарное количество калия и натрия в виде сульфата описанным выше способом [35, 311].
Определение щелочных металлов в глине и шамоте. 0,5 г тонкорастер-того анализируемого материала обрабатывают в платиновой чашке 10 мл 40%-ной соляной кислоты и 2 мл серной кислоты (уд. вес 1,84). Выпаривают досуха, прокаливают 10 мин. при 600° С и охлаждают. Остаток смешивают с 30—40 мл горячей воды, к которой прибавлено 4 мл конц. NH4OH, фильтруют и многократно промывают водой. Фильтрат и промывные воды нагревают до кипения, осаждают кальций 3 мл насыщенного раствора оксалата аммония; упаривают до 10—15 мл и для осаждения магния и алюминия прибавляют 2 мл конц. NH4OH и 1,5%-ного раствора 8-оксихинолина до жел-
25-
той окраски раствора. После охлаждения фильтруют,_ фильтрат выпаривают, остаток прокаливают, обрабатывают серной кислотой по описанному выше способу [45}.
Определение щелочных металлов в солях. Из раствора солей удаляют катион в виде соответствующего малорастворимого соединения, напрймер, из раствора нитрата бария осаждают сульфат бария, из раствора нитрата свинца осаждают сульфид свинца и т. п. Фильтрат выпаривают досуха, остаток прокаливают и извлекают водой. К отфильтрованному раствору добавляют несколько капель H2SO4 и т. д. [425, 540]. Можно также удалять катионы меди, кобальта, никеля и других элементов
электролизом, и в оставшемся растворе определять сумму щелочных металлов в виде сульфатов [347].
Относительно определения калия или суммы щелочных металлов в виде сульфатов см. также [517, 761, 841, 1124, 1129, 1156, 1478, 1592, 1807, 2027, 2220, 2242, 2314, 2492, 2774].
Определение в виде хлорида. Для определения калия в растворе, содержащем только КС1, выпаривают определенный объем раствора ла водяной бане. Остаток сушат при 130—150° С и затем прокаливают до постоянного веса при температуре не выше 500° С. После охлаждения взвешивают и вычисляют содержание калия [517, 663, 1271].
1 вес. ч. КС1 соответствует 0,5244 вес. ч. калия.
Хлорид калия часто содержит небольшую примесь MgC^, которая вследствие своей гигроскопичности может быть источником заметных ошибок, поэтому взвешивают в закрытом бю-ксе. Прокаливание при температуре выше 500—600° С приводит к потерям вследствие улетучивания КС1 [346, 722, 930, 1027, 1271, 1332, 1623, 1808, 1874, 2454, 2518, 2580, 2944]. Однако даже прокаливание при температуре около 500° С полностью не освобождает кристаллы КС1 от содержащейся в них воды: при 140° С в кристаллах остается около 0,2% воды, при 550°С — около 0,06% воды [2580].
Если анализу подлежит калиевая соль легко летучей кислоты, то ее нетрудно перевести в хлорид выпариванием с конц. НС1. Карбонат или фторид калия можно перевести в хлорид также и выпариванием с избытком NH4C1 и последующим прокаливанием [1552]. Чтобы получить хлорид из сульфата калия, к раствору последнего прибавляют избыток хлорида бария, и осадок сульфата бария отфильтровывают. Из фильтрата осаждают избыток соли бария карбонатом аммония, фильтрат выпаривают досуха с соляной кислотой и прокаливают [127, 1271]. Другой способ получения хлорида калия из сульфата заключается в добавлении избытка ацетата свинца при нагревании, отфильтровывании и промывании осадка сульфата свинца разбавленным раствором ацетата свинца. В фильтрат пропускают •H2S до насыщения, осадок отфильтровывают и промывают разбавленной уксусной кислотой, насыщенной сероводородом.
26
Фильтрат выпаривают почти досуха, добавляют избыток соляной кислоты, выпаривают, полученный остаток прокаливают при 250° С, охлаждают и взвешивают [127]. Для перевода в хлорид выпаривают сульфат калия 2 раза с хлоргидратом гидразина и прокаливают [2097] или выпаривают 0,25 г сульфата калия с 1,5—2 а смеси, 4 вес. ч. бромида аммония и 1 вес. ч. иодида аммония нагревают около 40 мин. в закрытом тигле. Эту операцию повторяют еще 1—2 раза. Остаток выпаривают 1 раз с хлорной водой и соляной кислотой [2069]. Из нитрата калия довольно трудно получить хлорид выпариванием с соляной кислотой. Поэтому переводят сначала нитрат "в карбонат (выпариванием с щавелевой кислотой и прокаливанием), из которого уже получают хлорид [127, 131]. Аналогичный способ заключается в выпаривании нитрата с 4—6-кратным количеством сахара, остаток прокаливают, растворяют в воде и выпаривают досуха с соляной кислотой [1899].
Бораты могут быть превращены в хлориды выпариванием с метанолом, насыщенным хлористым водородом [127]. Если калий находится в виде фосфата, то для получения хлорида сначала осаждают ионы фосфата в виде основного фосфата железа; хроматы восстанавливают до соли трехвалентного хрома этанолом и соляной кислотой и затем осаждают хром аммиаком [517].
Переведение в хлорид необходимо также и для дальнейшего определения калия хлороплатинатным методом.
Для определения калия в силикатах обрабатывают навеску анализируемого материала фтористоводородной и серной кислотами, извлекают горячей водой, фильтруют. Фильтрат и промывные воды выпаривают до небольшого объема, добавляют несколько граммов карбоната аммония и выпаривают досуха. Из остатка извлекают карбонаты щелочных металлов многократным промыванием водой. Раствор и промывные воды выпаривают досуха. Остаток перемешивают с 20 мл воды, фильтруют; фильтрат подкисляют соляной кислотой, выпаривают; остаток хлоридов высушивают и взвешивают [1750].
Определение калия или суммы щелочных металлов в виде хлоридов применяется при анализе мочи [2258, 2423], золы [2267], металлического натрия [2557]. Определение калия в виде хлорида см. также [1124, 1129, 1478, 1755, 2367, 2734].
Определение по Берцелиусу. Метод определения суммы щелочных металлов в виде хлоридов был описан в 1824 г. [764] и предназначен для анализа силикатов.
Навеску около 1 г измельченного силиката обрабатывают в платиновом тигле 5 мл серной кислоты (1 : 5), перемешивая платиновой проволокой, вводят 5 мл чистой конц. HF и выпаривают до начала испарения серной кислоты. Затем снова добавляют HF в выпаривают. Эту операцию повторяют до полного разложения силиката, после чего удаляют большую часть серной кислоты нагреванием. Для удаления следов HF снова выпаривают с серной кислотой.
При обработке фтористоводородной кислотой главная часть кремнекис-лоты, входящей в состав силиката, удаляется в виде SiF4, катионы щелочноземельных й других металлов превращаются в соответствующие фториды,
27
а щелочные металлы оказываются в виде солей кремнефтористоводородной кислоты. При выпаривании с серной кислотой все эти соли переходят в сульфаты. Далее в полученный раствор вводят небольшой избыток раствора ВаС12 для осаждения сульфатов; не фильтруя, выпаривают досуха. Остаток обрабатывают небольшим количеством горячей воды, добавляют раствор Ва(ОН)2 до щелочной реакции для осаждения гидроокисей магния, алюминия, железа и других катионов; не фильтруя, выпаривают досуха; остаток обрабатывают горячей водой, фильтруют, осадок промывают разбавленным раствором Ва(ОН)2. К нагретому фильтрату добавляют аммиак и карбонат аммония для осаждения ионов бария (нагревают 30 мин.), осадок отфильтровывают и промывают, фильтрат и промывные воды выпаривают досуха в платиновой чашке. При слабом прокаливании удаляют соли аммония. Остаток обрабатывают горячей водой, снова добавляют аммиак и карбонат аммония и т. д. Эту операцию повторяют до тех пор, пока перестанет появляться осадок ВаСОз. Фильтрат выпаривают досуха, остаток смачивают соляной кислотой, выпаривают, прокаливают и после охлаждения взвешивают сумму хлоридов [764].
Определение щелочных металлов этим методом отличается сложностью и длительностью. Описаны более простые варианты метода [31, 37, 505, 599, 634, 1140, 1428, 1897, 2159, 2202, 2440, 2512, 2541, 2597, 2719, 2883].
Определение по Смиту. Метод был предложен в 1853г. [2582—2584].
Навеску (около 0,5 г) тонкоизмельченного силиката тщательно растирают в агатовой ступке с 0,5 г хлорида аммония, добавляют понемногу 4 г чистого СаСОз и продолжают растирать. Для получения хорошо выщелачиваемой в дальнейшем массы добавляют 1 г кварца [1132]. Эту смесь переносят в тигель, на дне которого помещено около 1 г СаСОз. Прокаливают до полного удаления соли аммония, после чего повышают температуру до 1000—1100° С, при которой выдерживают около 1,5 часа. При этом силикат разлагается, Например:
2KAlSi3O8+6CaCO3+2NH4C1^2KCl+6CaSiO3+Al2O +6CO2+2NH3+H2O.
После охлаждения тигель помещают в фарфоровую чашку, наливают в него горячую воду, нагревают и смывают в чашку. Через 15 мин. вводят еще 50 мл воды, нагревают на водяной бане до полного разложения плава. Жидкость декантируют через фильтр, а остаток в чашке снова растирают с. 50 мл воды, дают осесть и декантируют. Такое выщелачивание повторяют несколько раз. Полученный фильтрат, кроме калия и натрия, может содержать магний, кальций, сульфаты и другие элементы. Кальций удаляют осаждением карбонатом аммония и аммиаком при кипячении. Фильтруют, промывают горячей водой. Осадок растворяют в соляной кислоте и снова осаждают карбонат кальция, фильтруют. Все фильтраты выпаривают досуха, прокаливают для удаления соли аммония. Остаток растворяют в 25 мл воды, и для осаждения следов кальция добавляют оксалат аммония и подщелачивают аммиаком. Помещают на водяную баню на 30 мин., фильтруют и промывают осадок 0,1%-ным раствором оксалата аммония. Фильтрат выпаривают досуха в платиновом тигле, прокаливают, остаток смачивают соляной кислотой, снова выпаривают, сушат 30 мин. при 110° С, затем нагревают до начала плавления солей, охлаждают и взвешивают сумму хлоридов щелочных металлов [2584].
Определение щелочных металлов этим способом также отнимает много времени. К недостаткам метода следует отнести возможность улетучивания щелочных металлов при сплавлении, поэтому нагревают при температуре не выше 500° С [127, 2265].
28
Известен ряд вариантов этого метода. При сплавлении заменяют хлорид аммония хлоридом бария; если в пробе присутствуют сульфаты, то их влияние устраняется вследствие образования BaSO4 (1325, 2636]. С этой же целью вместо СаСОз применяют ВаСО3 [902, 1377, 2877]. Вместо смеси хлорида аммония и карбоната кальция иногда берут плавленый и измельченный СаС12, при этом сплавление с анализируемой пробой протекает лучше [1932]; пользуются также смесью окиси и хлорида кальция [1642, 1643, 2487, 2488]. Некоторые авторы разлагают силикаты сплавлением с карбонатом свинца [1634] или окисью свинца [802, 1637], основным нитратом висмута [1507], смесью нитрата и фторида бария [1369], борной кислотой [1635, 1636] и другими реагентами [1633, 1638]. Известны другие модификации этого метода [20, 31, 354, 355, 505, 611, 646, 1093, 1132, 1162, 1290, 1386, 1685, 1757, 1803, 1820, 1945, 2126, 2181, 2634, 2787, 2836, 2902].
Определение в виде иодида. 0,25—0,30 г смеси хлоридов калия и натрия помещают в кварцевую чашку, растворяют в нескольких каплях воды, добавляют конц. HJ и выпаривают досуха на водяной бане. Остаток сушат 1 час при 125° С, охлаждают в эксикаторе. Затем растирают с 5 мл абсолютированного этилового эфира. Фильтруют через фильтр, смоченный изобутанолом. Такое экстрагирование повторяют еще 1—2 раза. Остаток смывают с фильтра смесью 5 мл этанола и 25 мл воды, к раствору добавляют несколько капель HJ, выпаривают, сушат при 125° С, охлаждают и взвешивают.
1 вес. ч. KJ соответствует 0,2355 вес. ч. калия.
Определение в виде калий-бортетрафторид а. К исследуемому раствору добавляют равный объем этанола и затем вводят раствор H[BFJ в избытке. Выпадает осадок K[BF4], который промывают 87%-ным этанолом, сушат при НО—120° С, охлаждают и взвешивают.
1 вес. ч. K[BF4] соответствует 0,3105 вес. ч. калия.
Вследствие заметной растворимости осадка (см. стр. 157) метод позволяет определять только относительно большие количества калия. Для уменьшения потерь от растворимости раствор до фильтрования охлаждают в ледяной бане 1 час и промывают осадок смесью равных объемов метанола и этанола, охлажденной до + 3° С [1938]. При определении 74—460 мг КС1 ошибка составляла около 1% [331, 451]. Большие количества солей лития, магния, кальция, цинка, кадмия, марганца, алюминия, трехвалентного железа, меди, а также хлоридов, нитратов, сульфатов не мешает определению [1938], напротив, соли рубидия и цезия, образующие аналогичные осадки, мешают.
Определение в виде мет^периодата. Гравиметрическое определение калия в виде KJO4 было предложено в 1917 г. [1387].
4—5 мл исследуемого раствора смешивают с раствором 1 г иодной кислоты в 3 мл воды, взбалтывают, добавляют 90 мл смеси равных объемов этанола и этилацетата, охлаждают на льду 30 мин., осадок отфильтровывают через тигель Гуча, промывают безводным этилацетатом, охлажденным до 0° С. Осадок KJO4 сушат 10 мин. при 105° С, охлаждают и взвешивают [736, 2903].
1 вес. ч. KJO4 соответствует 0,1700 вес. ч. калия.
29
Осадок можно промывать также смесью 1 объема изопропанола и 2 объемов этанола, охлажденной до 0° С [1653].
В анализируемом растворе не должны присутствовать хлориды, бромиды и иодиды, восстанавливающие метапериодат.
В их присутствии раствор выпаривают досуха, добавляют 10 мл конц. HNO3 и снова выпаривают досуха. Остаток растворяют в воде, затем добавляют иодную кислоту, как указано выше.
Определению мешают также сульфаты, повышающие вес осадка [2903], и соли рубидия и цезия; не мешают соли лития, магния, кальция, цинка, кобальта, никеля, железа, марганца, алюминия.
Определение в виде перхлората. К исследуемому раствору хлоридов прибавляют 1—2 мл конц. НС1О4 * и выпаривают досуха на плитке. Остаток растворяют в небольшом объеме воды, снова добавляют НСЮ4 и выпаривают. При этой операции хлориды переходят в перхлораты. Остаток обрабатывают 15—20 мл абсолютного этанола, хорошо перемешивают, отфильтровывают через тигель с пористым дном, промывают этанолом. Последний извлекает перхлорат натрия, в тигле остается КС1О4, мало растворимый в этаноле [2371]. Для удаления остатка перхлората натрия растворяют в воде, снова добавляют 1—2 мл хлорной кислоты, выпаривают досуха, извлекают абсолютным этанолом, отфильтровывают через тигель с пористым дном, сушат при 120—130° С, прокаливают при 350° С несколько минут, охлаждают и взвешивают КС1О4 [105, 119, 127, 1271, 1385, 2473].
1 вес. ч. КСЮ4 соответствует 0,2822 вес. ч. калия.
К достоинствам метода следует отнести постоянство состава перхлората калия, поэтому для вычислений пользуются теоретическим фактором.
Заметная растворимость перхлората калия в воде и даже в этаноле (см. стр. 154) приводит к потерям при анализе. Поэтому предложен ряд вариантов этого метода, в которых стараются понизить растворимость перхлората калия и этим уменьшить ошибки определения.
Растворимость может быть понижена применением следующих промывных жидкостей: этанола, к которому добавлено 0,2% хлорной кислоты [127, 442, 656, 762, 1419, 2064, 2525, 2704, 2705, 2827, 2875, 2960], этанола, содержащего 0,2% хлорной кислоты и насыщенного перхлоратом калия [127, 647, 714, 936, 1271, 1363, 2310], либо этанола, насыщенного перхлоратом калия и охлажденного до 0° С [714, 1475].
Однако и в этих жидкостях растворимость перхлората калия оказывается довольно ощутимой (см. стр. 166, 167). Поэтому некоторые авторы вводят поправку на растворимость [1862], величина которой зависит от температуры [1037]. Предложены и другие промывные жидкости.
Хлориды переводят в перхлораты, как указано выше, и смесь перхлоратов растворяют прн нагревании в 3 мл воды и медленно при перемешивании прибавляют 100 мл кипящего абсолютного н.бутанола с примесью 1% хлорной
* Свойства хлорной кислоты и условия обращения с ней см. [630, 1271, 1694, 2460].
30
кислоты. После охлаждения осадок отфильтровывают через тигель с пористым дном, промывают 8—10 раз по 1—2 мл н.бутанола, высушивают, охлаждают и взвешивают.
В н.бутаноле растворимость КСЮ4 очень мала и в большинстве случаев ею можно пренебречь [2575]. Изучалось отделение перхлората натрия от перхлората калия обработкой абсолютным метанолом [2259].
В некоторых вариантах метода осаждают раствором перхлората анилина в абсолютном этаноле [1816], и в качестве растворителя для перхлората натрия пользуются смесью равных объемов безводных н.бутанола и этилацетата [2578]. Промывание ор; паническими растворителями должно быть многократным, обеспечивающим практически полное удаление перхлората натрия [2766].
Относительно температуры высушивания перхлората калия имеются разные указания: 100—105° С [1459, 1853], 120—130° С [119, 519, 656, 661,754, 762, 1271, 1406, 1792, 1862,2280, 2525,2704, 2875] или 130—150° С [1936, 1317, 1618]. Однако при таких температурах в перхлорате калия еще содержится около 0,3% влаги, полное обезвоживание происходит только при 325—350° С [1674,. 2575, 2578, 2764]; при этой температуре перхлорат калия не разлагается.
Соли аммония не мешают определению калия, перхлорат аммония отличается довольно большой растворимостью в воде [1849]. Небольшие количества магния, кальция, стронция, бария, железа, алюминия, марганца, фосфатов не мешают определению [442, 936, 1271].
Определению калия в виде перхлората мешают сульфаты, так как сульфат натрия мало растворим в применяемых органических растворителях и может увеличивать вес остатка перхлората калия [936, 1271, 1476, 1862, 2460, 2488]. Поэтому анализируемый раствор сильно подкисляют соляной кислотой и по каплям прибавляют 10%-ный раствор ВаС12 в небольшом избытке. Калий определяют в фильтрате от осадка BaSO4. Сульфаты удаляют обработкой раствором оксалата бария в соляной кислоте, затем добавляют перекись водорода и аммиак. В осадке находятся сульфат бария, оксалаты щелочноземельных металлов, гидроокиси железа, алюминия, марганца. Фильтрат выпаривают досуха; прокаливают, извлекают водой и осаждают перхлорат калия [2511]. Удаляют сульфат из нейтрального раствора кипячением с ВаСОз, при этом осаждается также и магний [2978].
Для отделения от сульфатов пропускают исследуемый раствор через колонку с катионитом в Н-форме и промывают водой. Катионы, задержанные колонкой, т. е. калий, натрий, литий и другие, вымывают затем 4 N соляной кислотой, фильтрат выпаривают досуха. Остаток растворяют в горячей воде, и в растворе определяют калий перхлоратным методом [2409].
31
Исследуемый раствор пропускают со скоростью 3 мл!мин через колонку с анионитом, предварительно промытым 1 N раствором NaCl. Сульфаты и фосфаты обмениваются на ионы хлора. Колонку промывают затем водой. В фильтрате находится КС1, здесь определяют калий в виде перхлората [1285].
Органические вещества могут вызвать взрыв при нагревании с хлорной кислотой [1271, 1694, 2460], поэтому их предварительно удаляют прокаливанием или многократным выпариванием с царской водкой и затем с соляной кислотой.
При соблюдении всех условий определения дают хорошие результаты, совпадающие с результатами хлороплатинатного метода [1885]. В одном из вариантов метода сначала осаждают калий в виде нитрокобальтиата и таким путем отделяют его от других элементов, а затем уже получают и взвешивают перхлорат.
Осадок нитрокобальтиата калия (стр. 39) растворяют в соляной кислоте при нагревании на водяной бане и выпаривают досуха. Остаток растворяют в малом объеме воды, вводят НС1О«, перемешивают и выпаривают на водяной бане для удаления избытка хлорной кислоты. Такую обработку повторяют еще 1—2 раза, после чего в остатке находятся перхлораты калия, натрия и кобальта. При смешивании остатка с этанолом, содержащим 0,2% хлорной кислоты, в раствор переходят перхлораты натрия и кобальта. Фильтруют через тигель с пористым дном, оставшийся КС1О« промывают этанолом с хлорной кислотой; затем чистым этанолом, выпаривают, высушивают при 120—130° С, охлаждают и взвешив.ают [661, 663, 1459, 1756].
При определении калия перхлоратным методом расходуется большое количество ценных органических растворителей. Возникает вопрос о регенерации их отгонкой из промывных жидкостей. Однако, если в этой жидкости содержится свободная НСЮ4, то образуется этилперхлорат, обладающий взрывчатыми свойствами. Поэтому перед отгонкой кислоту необходимо нейтрализовать взбалтыванием с окисью кальция [1271].
Гравиметрическое определение калия в виде перхлората широко применяется в практике. Мы отметим определение калия в следующих объектах: силикатах [661, 1674, 1968, 2259, 2904], минералах [127, 1037, 1756, 1885, 2460], почвах [119, 1458, 2283, 2459, 2460], удобрениях [754, 936, 1037, 1292, 1328, 1476, 1853, 1862, 2280, 2283, 2459, 2460, 2659, 2827], огнеупорных материалах [1459], цементе [886, 1955, 2126], сточных водах [1292], растительных материалах [762, 2024, 2581], золе [936, 1328, 1406, 1458, 2704], вине [1077, 2310,2525], биологических объектах [2687], мыле [2543, 2653], солях калия [1618, 1739, 1852, 1885, 2031], других материалах 348, 704, 713, 1044, 1294, 1988, 2449а, 2531, 2632, 2742, 2847].
О гравиметрическом перхлоратном методе также см. [1038, 1124, 1762, 1789, 2176, 2501, 2632].
Определение в виде перрената. Исследуемый раствор, не содержащий сульфатов, смешивают с раствором рениевой кислоты и выпари-32
вают досуха. Остаток растворяют в малом количестве воды и снова выпаривают для получения более чистых кристаллов перрената калия, KReO,|. Охлажденный сухой остаток долго перемешивают палочкой с 40 мл 85— 90%-ного этанола, к которому добавляют около 0,5 мл 1%-ного раствора рениевой кислоты. Этанол извлекает соль натрия, этаноловый раствор сливают, остаток омачивают несколькими каплями раствора рениевой кислоты, выпаривают и снова отмывают этанолом. Остаток высушивают при 110° С, охлаждают и взвешивают.
1 вес. ч. KReO< соответствует 0,1351 вес. ч. калия.
Удовлетворительные результаты получают при определении около 20 мг хлорида калия в присутствии 20—100 мг хлорида натрия и 40 мг хлорида магния. К положительным сторонам-метода по сравнении с перхлоратным методом следует отнести довольно большой молекулярный вес KReO4. Из остатков после анализа можно регенерировать рений восстановлением водородом [2736].
Определение в виде хлороплатината. Навеску хлоридов растворяют в воде, помещают в фарфоровую чашку и прибавляют раствор HafPtCle], 10%-ный по платине [20, 127, 240, 505, 517, 1271]. Реагент должен быть в избытке, т. е. в количестве, достаточном для образования хлороплатинатов калия и сопутствующих ему элементов, главным образом натрия. Зная навеску хлоридов и концентрацию платины в применяемом реагенте, можно вычислить необходимый объем раствора HjPtCU]. При вычислениях условно считают, что навеска содержит только NaCl. Табл. 3 позволяет быстро установить необходимое количество реагеита.
Таблица 3
Количество раствора H2[PtCl6] (10% Pt) в зависимостиот взятой иавески хлоридов [20]
Навеска хлоридов, г Объем раствора Н2 [PtCle], мл Навеска хлоридов, г Объем раствора Н2 [PtCle], мл
<0,005 0,2 0,05-0,06 2,0
0,005-0,010 0,3 0,06—0,07 2,5
0,010—0,015 0,5 0,07-0,08 3,0
0,015—0,020 0,7 0,08-0,09 • 3,2
0,020—0,025 0,9 0,09—0,10 3,5
0,025-0,030 1,0 0,10-0,11 4,0
0,030—0,035 1,2. 0,11—0,12 4,2
0,035—0,040 1,4 0,12—0,13 4,5
0,040—0,050 1,7
Смесь нагревают на водяной баие, выпавший вначале осадок KafPtCle] должен раствориться. Если этого не наблюдается, то добавляют воду до тех пор, пока при нагревании весь осадок растворится. Продолжают нагревание и выпаривание жидкости до сиропообразного состояния и охлаждают. Затвердевший при охлаждении остаток обрабатывают 80%-ным этанолом, растворяющим хлороплатинат натрия, но не калия. Фильтруют, чашку и фильтр про
3 Аналитическая химия калия 33
мывают 80%-ным этанолом’ до тех пор, пака вытекающая промывная жидкость станет бесцветной. Осадок на фильтре растворяют в горячей воде, фильтрат собирают во взвешенный платиновый тигель, сюда же переносят осадок из фарфоровой чашки. Выпаривают досуха, нагревают 1 час при 130° С, охлаждают и взвешивают.
Осаждение калия в виде хлороплатината описано в разных вариантах также в других работах [2, 37, 49, 704, 707, 767, 955, 1124, 1265, 1294, 1416, 1465, 1478, 1683, 1916, 2626, 2812].
Теоретически 1 вес. ч. KafPtCle] соответствует 0,1608 вес. ч. калия или 0,3067 вес. ч. хлорида калия. Однако применение при вычислениях теоретических факторов приводит к несколько повышенным результатам. Это объясняется тем, что осадок имеет состав, только приближающийся к формуле К2[РЮб]. Отклонение от теоретического состава вызывается следующими причинами:
а. Гидролизом реагента и продукта реакции [240]. Применяемый реагент вследствие гидролиза содержит примесь H2[Pt(OH)C15] и поэтому наряду с Кг[РЮб] выпадает и некоторое количество К^РЦОН) СЦ. Гидролизу может подвергаться и хлороплатинат калия:
K2[PtCl6]+Н2О MPt (ОН) Cl5] + НС1.
Примесь этой соли вызывает пониженные результаты для калия. По другим данным [442, 517], продукт гидролиза реагента образует с солями калия осадок KH[Pt(OH) CI5], присутствие которого приводит к повышению результатов. Среди продуктов гидролиза может находиться и Н^РЦОН^Си] [2030, 2321] и даже H2[Pt(OH)e] [49]. Для подавления гидролиза и уменьшения влияния этого фактора на точность результатов осаждают калий из раствора, подкисленного соляной кислотой, и обрабатывают хлороплатинат этанолом, подкисленным соляной кислотой [2715].
Утверждают, что замена платинохлористоводородной кислоты ее литиевой солью позволяет получать осадок хлороплатината калия теоретического состава [2579]; хлороплатинат лития получают из H2[PtCl6] и Ы2СО3.
б. Восстановлением хлороплатината калия [240, 369, 1423, 2655]. Этанол, применяемый для отмывания хлороплатината натрия, всегда содержит небольшое количество ацетальдегида, способного восстановить хлороплатинат калия до КдДЧСЦ]. Последний от действия этанола разлагается, при этом получается КО, не растворимый в этаноле. Примесь КО может поэтому содержаться в осадке хлороплатината калия и приводить к пониженным результатам. Полагают [1933], что альдегид восстанавли-
* 80%-ный этанол получают смешиванием 5 объемов 96%-ного этанола С 1 объемом воды.
34
вает хлороплатинат даже до металлической платины, присутствие которой каталитически ускоряет окисление этанола хлороплатинатом. Для устранения этого недостатка применяют этанол, очищенный от примеси альдегида [2655].
В некоторых вариантах метода для удаления хлороплатината натрия промывают остаток метанолом [1114—1116, 2365, 2366, 2703], смесью этанола с ацетоном [2855], чистым ацетоном [1991] или малым количеством воды [2221], но возможность восстановления хлороплатината калия уменьшается мало. Следует учитывать, что хлороплатинат калия в метаноле растворяется больше, чем в этаноле [1114—1116].
в. Неполным удалением воды [240, 2533]. Даже продолжительное высушивание хлороплатината калия не приводит к полному удалению из него воды.
Таким образом, вследствие ряда причин осадок может содержать некоторые примеси, из-за чего применение теоретического фактора пересчета веса осадка на количество калия приводит к ошибкам. Поэтому часто пользуются эмпирическими факторами. Рекомендуется фактор пересчета на КС1, равный 0,3056 [139, 195, 240, 369, 478, 1116, 1262, 1270, 1271, 2823], что соответствует фактору 0,1603 для пересчета на калий. Предлагаются и другие факторы пересчета [49, 1846, 2217, 2252, 2406, 2802, 2825, 2925]. Фактор пересчета подвержен некоторым колебаниям, зависящим от условий выполнения определения и от характера анализируемого объекта. Лучше всего установить фактор путем одновременной обработки эталона с известным содержанием калия. Желательно, чтобы состав эталона приближался к составу анализируемого материала.
Остановимся на возможных источниках ошибок при гравиметрическом определении калия в виде хлороплатината [2386]. В некоторых вариантах этого метода отделяют хлороплатинат калия от соответствующих солей натрия, лития, бария и других элементов отмыванием последних 95%-ным этанолом [1846, 1893, 2000, 2168, 2217, 2255, 2366, 2577, 2724] и даже абсолютным этанолом [1268, 1269, 1270, 1876, 2155]. Однако под влиянием этих растворителей хлороплатинаты могут разлагаться с выделением нерастворимых в этаноле хлоридов натрия и калия [2061, 2867], а также хлорида бария [2345]. Метанол вызывает такой же эффект [2365]. Присутствие хлоридов натрия или бария увеличивает вес осадка и приводит к повышенным результатам определения калия. Поэтому после промывания осадка этанолом необходимо промыть его и водой. Промывание вызывает некоторые, обычно небольшие, потери вследствие растворимости [240, 1583, 1790, 2533] (о растворимости хлороплатината калия в разных растворителях см. стр. 168). Потери уменьшают применением для промывания растворителя, насыщенного хлороплатинатом калия [1177, 1429, 1790].
3*
35
Высушивание хлороплатината калия при температуре выше 270° С приводит к его разложению [142, 442, 955, 1271, 1343, 2769]:
K2[PtCl6]->2 KCl + Pt+2 Cl2.
Однако иногда рекомендуют высушивать этот осадок даже При 350° С [2579]. При высоких температурах хлорид калия улетучивается [346, 722, 2655]. Высушивание на бумажных фильтрах при температуре выше 130° С может вызвать частичное восстановление хлороплатината калия с образованием КС1 и'Р1С12 [369, 517, 1785]. Эти ошибки не имеют места при применении стеклянных фильтров.
Присутствие сульфатов натрия, кальция и других солей, мало растворимых в этаноле, приводит к повышенным результатам для калия [615, 2794]. При одновременном присутствии калия, кальция и сульфата может образоваться K2Ca(SO4)2, мало растворимый в воде и этаноле [2715]; он также понижает точность результатов определения калия [177, 855, 1565, 2761]. Сульфаты необходимо предварительно удалять осаждением раствором ВаС12 [1188, 1661, 1876, 2639, 2761, 2878], избыток последнего осаждают карбонатом натрия.
Присутствие аммиака в воздухе лаборатории приводит к загрязнению осадка хлороплатината калия аналогичной малорастворимой солью аммония, т. е. может быть причиной повышенных результатов определения калия [355, 2654]. Одновременное наличие солей рубидия, цезия, одновалентного таллия, также осаждаемых в виде хлороплатинатов, приводит к повышенным результатам для калия. Цианиды и иодиды препятствуют осаждению хлороплатината калия. Желательно, чтобы исследованию подвергались хлориды. Перевод различных солей калия в хлорид см. стр. 26.
Применяемые при анализе реагенты нередко содержат заметные количества калия. Поэтому полезно параллельно ставить контрольные опыты с соответствующими количествами реагентов. Необходимо, конечно, применять возможно более чистые реагенты [2812].
Другие источники ошибок при этом методе определения калия см. [355, 1268, 1269, 1790, 1791, 2386, 2980]. Для устранения и уменьшения ошибок предложен ряд вариантов хлороплатинат-ного метода. В некоторых модификациях дополнительно растворяют осадок в горячей воде с целью отделения хлороплатината калия от малорастворимых примесей [1240, 2794]. Для устранения влияния посторонних веществ предварительно выделяют калий в виде перхлората [1657, 2577—2579], нитрокобальтиата {57, 73, 1541], битартрата [797] с последующим определением калия в этих осадках хлороплатинатным методом. Возможно хроматографическое выделение калия и натрия с последующим гравиметрическим определением в виде хлороплатината [578].
36
В других вариантах рекомендуются несколько иные условия осаждения, промывания и высушивания хлороплатината калия [1500, 1728, 2006, 2155, 2200, 2252, 2401, 2458, 2761, 2925].
К недостаткам метода следует отнести и дороговизну реагента, который расходуется в довольно больших количествах — на одно определение требуется в среднем около 0,3 а платины. Правда, можно собрать осадки и промывные жидкости и затем регенерировать платину (методы регенерации см. [127, 292, 652, 1430, 1576, 1753, 2255, 2680]). Для уменьшения расхода платины рекомендуются микрохимические способы определения калия [223, 1034, 1459, 1465, 1478]. Если сначала выделить калий в виде перхлората, который затем растворить в воде и осаждать хлороплатинат калия, то реагент расходуется только на осаждение калия, расход реагента на переведение натрия и других элементов в хлороплатинаты отпадает [2579]. Имеются другие способы уменьшения расхода платины [2056, 2112].
Из положительных сторон этого способа можно отметить негигроскопичность осадка [625], сравнительно небольшой фактор пересчета, простоту методики. Несмотря на ряд недостатков, гравиметрический хлороплатинатный метод относится к наиболее точным способам количественного определения калия.
Описан также метод определения калия в виде фтороплатината, K2[PtF6] [2882], или бромоплатината, Kz[PtBr6] [2612]. Вследствие большого молекулярного веса последнего соединения фактор пересчета оказывается значительно меньшим, чем при осаждении хлороплатината.
1 вес. ч. Kz[PtBr6] соответствует 0,09588 вес. ч. калия.
Некоторые' недостатки хлороплатинатного метода устраняются, если осадок KsIPtCle] восстановить и взвешивать металлическую платину [1543, 1545].
Один из вариантов этого метода [127, 879, 1543] заключается в получении осадка хлороплатината калия по описанному выше способу. Промытый осадок растворяют в горячей воде, раствор подкисляют соляной кислотой и вводят магний в виде ленты, предварительно промытой водой [1789]. На каждые 0,2 г калия требуется около 0,5 г магния:
MPtClel+aMg—2KCl+2MgCl2+Pt.
Могут протекать и побочные реакции, например:
K2[PtCl6]+4Mg+4Н2О—2КС1 + 4Mg (ОН) Cl+2Н2+Pt.
Следят за тем, чтобы весь магний растворился. Если выпадают основные соли магния, то снова подкисляют соляной кислотой и кипятят. О полном восстановлении судят по исчезновению желтой окраски раствора (раствор над осадком металлической платины должен стать совершенно бесцветным и прозрачным). Осадок восстановленной платины отфильтровывают, промывают водой; последние промывные воды не должны давать помутнения от добавления раствора AgNOs- Промытый осадок высушивают, прокаливают, охлаждают и взвешивают.
Из приведенного выше уравнения следует, что 1 г-атом платины эквивалентен 2 г-атом калия. Поэтому 1 вес. ч. платины теоретически соответствует
37
0,4005 вес. ч. калия или 0,7638 вес. ч. хлорида калия. Вследствие указанного выше несоответствия между фактическим и теоретическим составом осадка необходимо пользоваться эмпирическим фактором [20, 292, 2112, 2113], устанавливаемым самим экспериментатором в идентичных условиях. Этот способ позволяет определять калий в присутствии сульфатов, боратов, силикатов, щелочноземельных металлов, железа, алюминия и других элементов [1543, 2806]. Можно определять до 0,1 мг калия в 1 г солей натрия [2112].
Описан ряд вариантов метода, основанного на восстановлении хлороплатината калия металлическим магнием [50, 292, 299, 651, 652, 803, 1180, 1204, 1389, 1695, 2112, 2307, 2749].
Восстановление хлороплатината калия другими металлами, например натрием [57], цинком [950, 2806] или железом [1180], не дает удовлетворительных результатов.
Для восстановления раствор хлороплатината нагревают со ртутью в фарфоровой чашке [517, 800, 2601]:
К2[Р t С16]+4Hg— Pt+2КС1 + 2Н g2C 12;
Ki[PtCl6]+2Hg—Pt+2KCl+2HgCl2.
Можно восстанавливать хлороплатинат кипячением с Hg2Cl2 [1995]: K2[PtCl6]+2Hg2Cl2-> Pt + 2KCl + 4HgCl2.
Выпаривают, прокаливают для удаления ртути и ее солей, остаток промывают водой для удаления хлорида калия, слабо прокаливают, охлаждают и взвешивают платину.
Хорошие результаты получают при восстановлении хлороплатината формиатом натрия.
Осадок хлороплатината калия растворяют в горячей воде и добавляют немного формиата натрия [20, 119]:
K2[PtCl6]+2HCOONa-*Pt+2KCl+2NaCl+2HCl+2CO2.
При этом часто получают осадок платины, легко проходящий через фильтр [1646, 2209, 2566, 2806]. Выпаривают досуха для получения платины в легко отфильтровываемом виде, остаток обрабатывают горячей водой, подкисляют соляИой кислотой, прокаливают, охлаждают и взвешивают [20]. Можно также вводить раствор хлороплатината калия небольшими порциями в 10 мл кипящего 20%-ного раствора формиата натрия, кипятить 15—20 мин., сильно подкислить соляной кислотой и снова кипятить. После этого восстановленная платина легко оседает на дно. Раствор над осадком должен быть бесцветным и прозрачным. Платину отфильтровывают, промывают, прокаливают и взвешивают [2938].
Описаны варианты метода, основанного да восстановлении хлороплатината калия формиатом натрия [31, 57, 299, 651, 998, 1007, 1646, 2844] или муравьиной кислотой [1805, 2033, 2680].
Предложены и другие способы восстановления:
а. Восстановление водородом при высокой температуре [517, 875, 1078, 1207, 1834, 2481, 2722, 2755]:
K2[PtCl6]+ 2 H2^Pt + 2 КС1 + 4НС1.
Вместо водорода можно пользоваться светильным газом [1271, 1423, 2112].
38
После прокаливания остаток обрабатывают водой, отфильтровывают, промывают, все растворы выпаривают досуха во взвешенном тигле и взвешивают хлорид калия. Кроме того, прокаливают и взвешивают осадок платины.
б. Восстановление формальдегидом в щелочной среде [1646, 2158]:
K2[PtCl6] + 2HCHO + 6NaOH Pt + 2KCl+4 NaCl + 2 HCOONa +
+4Н2О.
в. Восстановление прокаливанием со щавелевой кислотой 11271} или оксалатом натрия [2047]:
MPtCU]+2Н2С2О4 Pt + 2КС1 + 4НС1 + 4СО2.
В качестве восстановителей в щелочной среде рекомендуют также этанол [119, 797, 950, 1575, 2482], сахар [950], тиоуксусную кислоту [651] и др. [815].
Гравиметрический хлороплатинатный метод в разных модификациях применяется для определения калия в силикатах [37, 803, 883, 1346, 1750, 1751, 1905, 2626, 2802], стекле [31, 139, 2856], минералах [19, 20, 299, 1565, 1737, 1785, 1798, 1805, 1932, 2168, 2370, 2566], почвах [119, 138, 1458, 1866, 2168, 2283, 2386, 2604], природных водах [281, 792, 1377, 1754, 1774, 1791, 2647], удобрениях [615, 650, 910, 1097, 1137, 1139, 1177, 1240, 1254, 1293, 1429, 1555, 1583, 1661, 1665, 1736, 1790, 1791, 2033, 2114, 2187, 2217, 2255, 2386, 2452, 2715, 2721, 2761, 2794], растительных объектах [481, 573, 2187, 2494, 2940], золе растений [ИЗО, 1458, 1574, 1798, 2723], биологических объектах [1188, 1334, 1500, 2158], солях калия и их растворах [41, 131, 348, 797, 1402, 1403, 1790, 2112, 2639, 2761, 2878], других материалах [50, 354, 1444, 1834, 2235, 2543, 2939].
Определение в виде хлорида серебра. При восстановле-нйи хлороплатината калия формиатом образуются хлориды (см. выше). По окончании восстановления раствор'подкисляют азотной кислотой, фильтруют и промывают. К фильтрату и промывным водам добавляют избыток раствора AgNO3. Осадок AgCl отфильтровывают, промывают, высушивают и прокаливают. Зная вес полученного AgCl, вычисляют содержание калия [2033].
1 вес. ч. AgCl соответствует 0,1814 вес. ч. калия.
Опр еделение в виде нитрокобальтиата. Гравиметрическое определение кали'я в виде малорастворимого нитрокобальтиата было предложено еще в конце прошлого столетия [1335], в настоящее время известно много вариантов этого метода.
Один из вариантов заключается в следующем [20, 273]: навеску хлоридов или сульфатов щелочных металлов растворяют в фарфоровой чашке в 5—10 мл воды, подкисляют уксусной кислотой и выпаривают на водяной бане до небольшого объема. К охлажденному раствору добавляют по каплям при перемешивании концентрированный раствор нитрокобальтиата натрия до тех пор, пока раствор над осадком не приобретет интенсивную бурук) окраску. На другой день раствор сливают с осадка в тигель с пористым дном, к осадку прибавляют новую порцию раствора нитрокобальтиата натрия и выпаривают
39
на водяной бане до получения сиропообразной массы. Во время выпаривания раствор дважды подкисляют уксусной кислотой. После охлаждения вводят 5 мл воды для растворения избытка реагента, перемешивают, переносят в тот же тигель с пористым дном, промывают холодной водой до тех пор, пока вытекающая промывная жидкость не станет совершенно бесцветной. После возможно более полного отсасывания воды тигель с осадком сушат 1—2 часа при .110° С, охлаждают и взвешивают.
Большинство исследователей считает, что состав высушенного осадка может быть приблизительно выражен формулой K2Na[Co(N02)6]-H20 [20, 22, 59, 91, 154, 277, 380, 416, 478, 597, 622, 682, 818, 977, 1025, 1271, 1323, 1442, 1610, 2633, 2899]. Поэтому фактор пересчета найденного веса осадка на вес калия принимается равным 0,1721, на вес хлорида калия — 0,3283. Некоторые авторы полагают, что при высушивании соединение полностью теряет кристаллизационную воду, и тогда фактор пересчета на калий увеличивается до 0,1793 [240, 1459].
Нитрокобальтиат натрия выпускается химической промышленностью. Описаны способы получения этого реагента в чистом сухом виде [404, 770, 1028, 1886, 2411]. Известны различные рецепты приготовления раствора нитрокобальтиата натрия из солей кобальта. Приведем несколько вариантов приготовления растворов нитрокобальтиата натрия.
Реагенты из препарата нитрокобальтиата натрия
1. 20—25 г Na^[Co(N02)6] • 0,5Н2О в 100 мл раствора [820, 2182, 2901].
2. 20—30 г нитрокобальтиата натрия и 10—20 г нитрита натрия в 100 мл раствора [941, 2203, 2726].
3. 20%-ный раствор нитрокобальтиата натрия в 4%-ном растворе ацетата натрия [2818]
4. Раствор 45 г нитрокобальтиата натрия, 19 г ацетата натрия и 18 г ледяной уксусной кислоты в 120 мл воды [866, 958, 1697, 2210, 2947].
5. Насыщенный раствор нитрокобальтиата натрия в 0,5—5%-ной уксусной кислоте [247, 2316, 2810].
6. Раствор 12,5 г нитрокобальтиата натрия в 100 мл воды смешивают со 100 мл 95%-ного этанола [1705].
7. Раствор 1,5 г нитрокобальтиата натрия в 30 мл насыщенного раствора хлорида натрия [1629].
Реагенты из солей кобальта
1. Раствор 30 г Co(NO3)2 • 6Н2О в 60 мл воды смешивают с раствором 50 г NaNO2 в 100 мл воды и вводят 10 мл ледяной СНзСООН, через 1—2 дня раствор сливают с осадка или фильтруют [661, 662, 2974].
2. Раствор 113 г Со(СН3СОО)2 • 4Н2О в 300 мл воды смешивают с 100мл конц. СНзСООН и раствором 220 г NaNO2 в 400 мл воды. На другой день раствор фильтруют и разбавляют водой до 1000 мл [416, 857].
3. Раствор 100 г СоС12 6Н2О и 100 мл ледяной СНзСООН в 500 мл воды смешивают с раствором 220 a NaNO2 в 500 мл воды, через раствор пропускают струю воздуха; на другой день фильтруют [2293].
4. Раствор 50 г нитрата кобальта в 150 мл воды смешивают с раствором 100 а нитрита натрия в 150 мл воды и по каплям при взбалтывании вводят 30 мл ледяной СНзСООН [91].
40
Исходные реагенты для приготовления нитрокобальтиата должны быть чистыми, не содержать примеси солей калия и аммония [963]. Нитрит натрия очень часто содержит примесь соли калия. Поэтому в приготовленном растворе нитрокобальтиата натрия появляется желтая муть или небольшой желтый осадок. Последний может получиться и за счет паров аммиака, почти всегда имеющихся в лабораторном воздухе. В таких случаях смесь оставляют на несколько часов или до следующего дня и сливают раствор с выпавшего осадка.
При приготовлении реагента из солей кобальта и нитрита натрия обычно вводится избыток последнего. В раствор, полученный из готового препарата нитрокобальтиата натрия, также рекомендуют прибавлять нитрит натрия. Избыток нитрита способствует более продолжительному сохранению реагента. Аналогично влияет этанол. Напротив, реагенты, содержащие уксусную кислоту, менее стабильны. В кислой среде образуется азотистая кислота, которая постепенно улетучивается. Растворы нитрокобальтиата натрия даже в закрытых сосудах из темного стекла сохраняются сравнительно недолго. При хранении медленно протекает реакция:
2Co3+ + NO; + H20^2Co2+ + NO;+2H+.
Хранение при температуре от 0 до+10° С увеличивает срок годности реагента до 1 месяца [2203, 2974]. Растворы реагента следует готовить в небольших количествах, достаточных для обеспечения работы лаборатории в течение 10—20 дней. Часто рекомендуют готовить отдельно раствор соли кобальта и раствор нитрита натрия и сливать их по мере надобности. Каждый из этих растворов сохраняется длительное время. Приведем рецептуру таких растворов.
1. Раствор 25 г нитрата кобальта в 50 мл воды смешивают с 12,5 мл ледяной СНзСООН. Отдельно растворяют 120 г NaNO2 в 180 мл воды. За день до применения смешивают 1 объем раствора соли кобальта с тремя объемами раствора нитрита [9].
2. Раствор 5 г нитрата кобальта в 26 мл 35%-ной уксусной кислоты и отдельно раствор 50 a NaNO2 в 100 мл воды. Перед применением смешивают равные объемы этих растворов [351, 745].
Главный недостаток нитрокобальтиатного метода определения калия состоит в непостоянстве состава выпадающего осадка. Осадок содержит переменные количества калия, натрия и кристаллизационной воды. Осадку приписывают не только уже указанный выше состав'K2Na[Co(NO2)6]-Н2О. Состав этого соединения выражают также следующими формулами:
K3[Co(N02)e]-nH20 [2297]
K3[Co(N02)6]-2H20 [59, 1118, 1119]
К3[Со(МОг)6]-Н2О [919]
41
K3[Co(NO2)6].1,5H2O
Кз[С°(МО2)6]
K2Na[Co(NO2)6]-6H2O
K2Na[Co(N02),]-3H20
K2Na[Co(N02),J
Ki.oiNal.o»! Co(N02)6 ] • 0,54H2O
Ki,84Nai,ie[Co(NO2)6] -nH2O
Ki,57Nai,13[Co(NO2)e]
Ki,6Nai,6[Co(NO2)6] • nH2O
Ki,36Nai,e5[Co(N02)6]
KNa2[Co(NO2),]-«H2O
[1200]
[490, 594, 1028, 2420]
[1649, 2320]
[817, 2353]
[240, 1459, 2899]
[2491]
[252, 766, 820, 2197, 2210]
[2498]
[252, 417, 600, 1271, 1629, 2293,
2782]
[2354]
[168, 1705]
Вероятно, осадок состоит из смеси двух или трех солей: KNa2[Co(NO2)6], K2Na[Co(N02)6] и Кз[Со(Ы02)6] [266, 1581, 1663, 1769]. В зависимости от условий осаждения и характера анализируемого раствора отношение количеств этих соединений в осадке оказывается различным. Содержание кристаллизационной воды также зависит от ряда факторов, в частности от избытка осадителя [2672], от продолжительности стояния осадка под маточным раствором [2782] и других факторов [268а, 2297, 2657, 2946].
Вследствие неодинакового состава осадков, полученных при различных условиях, фактор пересчета найденного веса осадка на вес калия оказывается непостоянным. Приведем некоторые рекомендуемые факторы пересчета на калий: 0,1437 [2320]; 0,1574 [1649]; 0,1721 (см. выше); 0,1895 [208]. По другим данным [2782], эта величина колеблется в пределах от 0,1555 до 0,1798. Некоторые авторы вообще не считают возможным применять какой-либо один фактор. Полагают, что фактор изменяется с изменением общего количества осадка, полученного данным способом. Например, при определении калия в природных водах рекомендуются следующие факторы [20]:
Вес осадка г нитроко- Фактор Вес осадка нитроко- Фактор
бальтиата пересчета бальтиата пересчета
калия, мг на калий калия, мг на калий
10 0,174 150 0,158
20 0,172 200 0,156
40 0,170 300 0,152
60 0,167 500 0,148
80 0,164 800 0,142
100 0,160 1000 0,138
Непостоянство состава осадка было замечено очень давно
[837, 1176, 2417, 2420, 2660]. Состав осадка нитрокобальтиата
42
калия в зависимости от различных факторов изучался многими учеными [59, 220, 252—254, 266, 268а, 315, 343, 837, 1136, 1610, 1788, 1896, 2417, 2420, 2968]. Состав осадка зависит при прочих равных условиях от содержания калия. Если в общем виде выразить состав осадка формулой Кга№з_п[Со(М02)б] • тН2О, то при изменении содержания калия в растворе от 0,5 до 0,025 мг на 0,5 мл значение п изменяется от 2,04 до 1,58 [266]. Чем боль-[К+1
ше отношение fNT-^ \ v тем больше значение п [253, 254, [Na3Co(NO2)6]
268а, 315], т. е. при увеличении избытка нитрокобальтиата натрия относительное количество калия в осадке уменьшается. С повышением температуры (в пределах 2—31°С), при которой производится осаждение, содержание калия в осадке увеличивается [2238].
Следует указать на опыты по получению осадка практически постоянного состава. Осаждение калия после предварительного введения в раствор большого количества хлорида натрия (почти до насыщения) приводит к получению осадка состава K2Na[Co(NO2)6] • Н2О [22, 280, 941, 1977] и для пересчета на калий можно пользоваться теоретическим фактором 0,1721. Однако, по другим данным, для получения удовлетворительных результатов количество натрия в растворе может превышать количество калия максимум в 15 [818], 100 [211] и в 22—500 раз [804]. Надо также принять во внимание, что в присутствии солей натрия растворимость осадка возрастает [960, 2238, 2660] и осадок вообще может не появиться [211], что, конечно, ведет к потерям калия при анализе.
В качестве.осадителей ионов калия рекомендуются нитро-кобальтиаты лития [919, 1118, 1119], магния [1813, 2762], кобальта [220, 490],. цинка [594, 961, 2762, 2959], применение которых приводит к получению осадка Кз[Со (NO2) б]. Для синтеза таких реагентов пропускают окислы азота через взвесь (или раствор) соответствующей окиси, вследствие чего в растворе образуется нитрит, к которому затем добавляют нитрат кобальта [1813]; для получения нитрокобальтиата цинка окислы азота вводят в раствор ацетатов цинка и кобальта [594]. Эти реагенты образуют с солями калия осадок Кз[Со(Ы02)б] при условии, что в исследуемом растворе не содержатся соли натрия.
Недостатком метода является . также заметная растворимость осадка в воде, при комнатной температуре она равна около 10“3 моль!л (см. стр. 154). Поэтому может наблюдаться неполное осаждение калия и потери при промывании осадка [2238]. Для уменьшения потерь ведут осаждение в присутствии этанола [284, 827, 959, 1681,1705, 2150, 2353, 2498, 2615, 2633, 2635], изопропанола [943, 1059], которые также и ускоряют образование осадка [827, 1681]. Присутствие этанола влияет и на состав осадка [2353]. Потери от растворимости уменьшают
43
осаждением при низких температурах, например при охлаждении льдом [890, 1705, 2289, 2498, 2750, 2818] или в холодильнике [1018, 1686, 2150, 2203, 2206, 2316].
Для уменьшения конечного объема раствора и потерь от растворимости, а также повышения чувствительности определения в исследуемый раствор вводят нитрокобальтиат натрия в сухом виде [83, 279, 309, 690, 943, 2160].
Промывание осадка холодной водой (1686, 2750], водой, насыщенной нитрокобальтиатом калия, или 35%-ным этанолом уменьшает потери от растворения приблизительно в 50 раз [2238, 2498]. Рекомендуется также промывание осадка 0,01 W азотной кислотой, насыщенной нитрокобальтиатом калия, затем 95%-ным этанолом [622, 820, 1459] или 10%-ным этанолом, насыщенным нитрокобальтиатом калия [1559]; 30—70%-ным этанолом [665, 957, 1134, 1158, 1869, 2206, 2320, 2594]; 25%-ным водным раствором ацетона [1579]; водным раствором, содержащим 35% этанола, 1% уксусной кислоты и 0,2% октанола [952, 2947].
Часто на поверхности маточного раствора наблюдается пленка осадка, остающаяся даже после центрифугирования. Для предупреждения этого нежелательного явления к раствору до осаждения добавляют немного 70%-ного этанола, насыщенного камфарой [957].
Методика осаждения и гравиметрического определения калия в виде нитрокобальтиата в разных вариантах описана в ряде работ [22, 154, 208, 365, 662, 742, 801, 1024, 1291, 1559, 1810, 2182, 2281, 2527, 2672, 2826]. Кроме уже отмеченных условий осаждения и промывания, .остановимся еще на некоторых других деталях метода.
Полнота осаждения нитрокобальтиата калия достигается в различных способах через неодинаковые отрезки времени. По некоторым данным, требуется выжидать 2 часа [2182], а при малых количествах калия до 24 час. [820, 1271, 2320].
Осадок высушивают* до постоянного веса при 100—105° С [91, 622, 1649, 2320, 2491], НО—120°С [154, 169, 272, 662, 818, 977, 1335, 1459] или 125—130° С [416, 1025]. Другие условия осаждения нитрокобальтиата калия см. [246, 1024, 2524].
Исследуемый раствор не должен" содержать минеральных кислот, которые разлагают реагент:
[Со (NO2) 6]3~ + 6 Н+ ->Со3+ + 6 HNO2.
По этой же причине нежелательно присутствие солей, подверженных гидролизу, например солей трехвалентного железа, алюминия и других элементов. В таких случаях раствор выпаривают, остаток прокаливают и из полученных окислов извлекают соли калия водой или раствором NaCl [22, 274]. В анализируемом растворе должны отсутствовать едкие щелочи. 44
Оптимальной для осаждения ионов калия поэтому считают слабокислую уксуснокислую среду [2152]. Сильнокйслые анализируемые растворы предварительно подщелачивают едким натром, затем подкисляют уксусной [2498] или азотной кислотой [2182, 2899, 2968], а сильнощелочные растворы подкисляют уксусной кислотой.
Многие авторы осаждают нитрокобальтиат калия из раствора с определенным значением pH, например при pH 4,8 12207, 2936], pH 6 [890, 1622], pH 7 [590].
Исследуемый раствор не должен содержать окислителей или восстановителей, в частности должны отсутствовать иодиды {274]. Соли магния [91, 233, 365, 431, 1846] и кальция [91] н"е мешают определению калия.
Путем,образования нитрокобальтиата калия удается практически полностью выделить калий даже из разбавленного раствора и количественно его определить. Хотя на состав осадка влияет ряд факторов, нитрокобальтиатный метод занимает первое место среди химических способов определения калия. Метод позволяет получать вполне удовлетворительные по точности результаты, если стандартизировать все операции и условия выполнения анализа и применять фактор пересчета, найденный при параллельной обработке объекта с близким и известным содержанием калия [138, 2782]. Ошибки определения калия в микромасштабах достигают только ±3% [1323, 1649]. (О достаточной точности метода см. также [442, 1681].) Некоторые авторы, однако,, считают, что прямое гравиметрическое определение калия в виде нитрокобальтиата не дает удовлетворительных результатов [49, 1335], и осаждение нитрокобальтиата рассматривают только.как удобный и простой способ выделения калия из раствора и отделения его от ряда других катионов. Осадок нитрокобальтиата калия растворяют и в полученном растворе определяют калий каким-нибудь другим способом, например хлороплатинатным [1271, 1335,^1541, 1846], перхлоратным [661, 662, 1271, 1459, 1756, 1806, 1811, 1846], тартратным [1217] и т. д.
Гравиметрическое определение калия в виде нитрокобальтиата рекомендуется при анализе минералов [20, 57, 127, 442], силикатов [1015, 1810, 2817], почв [138, 822, 866, 2108-, 2281, 2304], удобрений [2280], воды [281, 380], огнеупорных материалов [1459], растительных [390, 622, 866, 1875, 2182, 2899] и биологических объектов [143, 642, 662, 1018, 1323, 1649, 2289], пищевых продуктов [1592, 2167, 2939], вина [801], солец натрия [771, 1560] и других материалов [22, 154, 169, 365, 535, 2676].
Нитрокобальтиатному методу определения калия посвящен ряд обзоров [91, 280, 1864, 1865, 2725, 2782].
45
Варианты нитрокобальтиатного метода
а. Осадок Кз[Со(МОг)б]- 2Н2О, полученный осаждением раствором нитрокобальтиата лития, промывают, высушивают и прокаливают при 300° С. Остаток от прокаливания, состоящий из 3KN03 + Co0, взвешивают.
1 вес. ч. остатка соответствует 0,3101 вес. ч. калия [1118].
б. Если нитрокобальтиат натрия подкислить не уксусной, а винной кислотой, то выпадающий осадок имеет постоянный состав, Na (СоО) С4Н4О6 • 7K2Na[Co(NO2)6] • Н2О.
Для приготовления реагента растворяют 90 г нитрата крбальта в 250 мл воды, смешивают с раствором из 180 г нитрита натрия в 250 мл воды и добавляют 50 г винной кислоты. После растворения винной кислоты оставляют на 2 дня, затем раствор фильтруют.
К 5 мл исследуемого раствора добавляют 3—4 г NaCl, нагревают до кипения, вводят избыток реагента и снова кипятят. Осадок отфильтровывают, промывают, сушат при ПО—120° С, охлаждают и взвешивают. Для пересчета на количество калия пользуются теоретическим фактором 0,1598 [2898].
в. Если осаждать нитрокобальтиатом натрия в присутствии AgNOs, то выпадающий осадок содержит серебро, соединению приписывают формулу K2Ag[Co (ЫО2)б] [2730]. Это соединение отличается меньшей растворимостью, чем K2Na[Co(N02)6], и большим молекулярным весом; оба эти фактора благоприятно отражаются на точности количественного определения калия.
Необходимый для осаждения реагент готовят следующим способом [488, 489]. 18 г нитрата кобальта и 6 г нитрата серебра растворяют в 50—60 мл воды, добавляют 60 г нитрита натрия, сильно взбалтывают и вводят еще 50 мл воды для полного растворения солей. К смеси добавляют 25 мл 4 # азотной кислоты, через 3 часа фильтруют, к фильтрату добавляют воду до 195 мл.
Описаны другие способы приготовления реагента [1026].
25 мл исследуемого раствора смешивают при взбалтывании с 5 мл реагента. Через час осадок отфильтровывают, промывают раствором 5 мл конц СНзСООН в I л воды, затем промывают 3—5 мл этанола и 2—3 мл эфира. Сушат при 110° С, охлаждают, взвешивают.
Теоретически 1 вес. ч. K2Ag[Co(N02)6] соответствует 0,1512 вес. ч. калия, однако осадок содержит примесь KAg2[Co(NO2)6], и поэтому эмпирический фактор пересчета оказывается значительно меньшим. Фактор зависит от содержания калия в исследуемом растворе и, следовательно, от веса полу-
ченного осадка [489]:
Вес осадка, мг Фактор пересчета на калий Вес осадка, мя Фактор пересчета на калий
22- 44 0,1124 131—172 0,1155
45- 66 0,1128 173—227 0,1167
67— 87 0,1138 228-280 0,1181
88-130 0,1145 281—332 0,1196
По другим данным, фактор пересчета для колеблется в пределах 0,125—0,166 [2135].
0,05—0,60 г калия
46
Можно не готовить специальный реагент, содержащий серебро. К нейтральному исследуемому раствору добавляют некоторый объем 0,02 N раствора AgNO3 и медленно при взбалтывании вводят избыток 25%-ного раствора нитрокобальтиата натрия [888, 1616]. Осадок имеет переменный состав,
он зависит от ряда факторов [847, 888, 1616]. Средний состав осадка может быть выражен формулой Ki,3s Agi,e5[Co(NO2)6l [252, 2354, 2730], по другим данным — КА^Со(ЫОг)б] [1026]. В большинстве случаев состав осадка приближается к K2Ag[Co(NO2)6] [266].
Описанный метод неприменим, если в исследуемом растворе-присутствуют соли галоидоводородных кислот, а также соли рубидия, цезия, одновалентного таллия. Калий определяют в виде нитрокобальтиата калия и серебра в силикатах [37], биологических [1260, 1453] и других [1012] объектах.
Определение в виде и итро к упро ата свинца и калия. Метод предложен для определения калия в смеси хлоридов калия и натрия.
Навеску растворяют в возможно меньшем объеме воды, добавляют большой избыток реагента (см. ниже) и 1—2 г NaNO2. Смесь оставляют на льду в течение часа, осадок отфильтровывают через стеклянный пористый тигель № 3 или № 4, промывают 95%-ным этанолом, сушат на воздухе, затем 30— 40 мин. при 100° С, охлаждают и взвешивают. Теоретически 1 вес. ч. K2Pb[Cu(NO2)6] соответствует 0,1251 вес. ч. калия. Однако вследствие потерь от заметной растворимости осадка рекомендуется эмпирический фактор 0,1331 [39, 40, 514].
Для осаждения служит раствор 6,6 г ацетата меди и 11,7 г ацетата свинца в 100 мл воды. Перед применением в этой смеси растворяют 13,5 г NaNO2.
Отмечается, что осадок может быть загрязнен хлоридом свинца. Сульфаты мешают определению [40, 514].
Гравиметрическое определение калия возможно также в форме K2Pb[Ni (NO2)6] [286].
Определение в виде солей гетерополикислот. Исследуемый раствор выпаривают досуха, остаток обрабатывают раствором 1 г фос-форномолибденовой кислоты в 10—15 мл воды, хорошо перемешивают, на следующий день осадок отфильтровывают через тигель Гуча [190]. В других вариантах к 1—20 мл анализируемого раствора добавляют 2—3 капли конц. HNO3 и 5 мл насыщенного водного раствора фосфорномолибденовой кислоты. Выпаривают до начала кристаллизации избытка реагента, фильтруют через тигель № 4, промывают 2%-ной азотной кислотой [734, 735]. Осадок высушивают и взвешивают [2294, 2469].
Состав выпадающего желтого осадка зависит от условий осаждения и ряда других факторов [2468, 2605]. Осадку приписывают различный состав:
К3РО4-12МоО3-яН2О [190]
К3[Р(Мо3010)4]-яН20 [1271, 2152]
К3Р04-12Мо03-ЗН20 [26]
К3Р04-11Мо03-6Н20 [1048]
К3Р04-10Мо03.ЗН20 [56, 1768]
47
Содержание воды и отношение К: Мо в осадке подвержено колебаниям. Вследствие этого некоторые авторы считают вообще невозможным количественное определение калия в виде фосфоромолибдата [2605]. При вычислениях пользуются эмпирическим фактором пересчета на калий, который находят путем параллельного анализа стандарта с известным содержанием калия. Положительная сторона метода — возможность определения (или хотя бы выделения) калия в объектах с небольшим содержанием соли этого элемента. Определению мешают соли аммония, рубидия, цезия, одновалентного таллия, органических оснований, дающих малорастворимые соединения с этим реагентом. Гравиметрическое определение калия в виде фосфоромолибдата описано в ряде работ [1388, 2469, 2848].
Осадок фосфоромолибдата калия, полученный как указано выше, растворяют в 10—15 мл 5%-ного раствора аммиака, разбавляют горячей водой приблизительно до 100 мл. Добавляют метиловый оранжевый и нейтрализуют раствором муравьиной кислоты, затем подкисляют этой же кислотой. К раствору добавляют 10 г хлорида аммония, нагревают до кипения и вводят раствор ацетата свинца. Выпадает молибдат свинца РЬМоО4, который отфильтровывают промывают 5%-ным раствором нитрата аммония, сушат, прокаливают при температуре не выше 700° С, охлаждают и взвешивают. Если фосфоромо-либдату калия приписать состав КзРО4 • 12МоО3, то 1 вес. ч. молибдата свинца соответствует 0,02662 вес. ч. калия [734].
Определение в виде фосфоровольфрамата калия имеет такие же недостатки, как и определение в виде фосфоромолибдата. Недостаток заключается в неопределенности состава осадка, которому приписывают следующие формулы: K3PO4-12WO3 [1201, 1327]; КзН4[Р(Ш2О7)6].пН2О [582]; Кз[РО4 • Wi2Oi8(OH)36] [363J; К5Н(РО4-12WO3)2-6H2O [43, 143, 2336, 2337, 2783]. Осадок последнего состава можно рассматривать как смесь эквимолярных количеств КзР04-12\МО3 и К2НРО4 • 12WO3. Однако некоторые авторы считают, что соотношение обоих компонентов в смеси может изменяться. Предполагается, что в осадке содержится 54,5% K3PO4-12WO3 и 45,5% K2HPO4-12WO3 [1232]. Содержание воды в осадке и отношения К:Р и K:W подвержены колебаниям. Поэтому гравиметрическое определение калия в виде фосфоровольфрамата не отличается высокой точностью. Считают, что 1 вес. ч. осадка, высушенного при. 100° С, соответствует 0,0332 вес. ч. калия [2336, 2337]. Необходимо вводить поправку на растворимость осадка [2337].
Определение в виде калий-бортетрафен ил а. 10—100 мл исследуемого раствора, содержащего 2—50 мг калия, подкисляют уксусной кислотой, нагревают до 40" С и по каплям добавляют 3%-иый раствор натрий-бортетрафеиила в небольшом избытке. Выпадающий осадок калий-бортетра-фенила быстро опускается на дно, спустя 10 мин. фильтруют через тигель №4, промывают 1%-ной уксусной кислотой, содержащей немного натрий-бортетра-фенила, затем водой. Осадок сушат около 1 часа при НО—120° С, охлаждают и взвешивают [183, 1312, 2279].
1 вес. ч. К[В(СвН5)4] соответствует 0,1091 вес. ч. калия.
48
Раствор натрий-бортетрафенила, применяемый как осадитель, часто оказывается мутным вследствие разложения [584]:
Na[B (С6Н5) 4]+ЗН2О - С6Н5В (ОН) 2 + ЗС6Н6 + NaOH.
Для удаления примесей взбалтывают раствор со свежеприготовленной и промытой гидроокисью алюминия или добавляют несколько капель раствора соли алюминия. После этого фильтруют, и прозрачный фильтрат применяют в качестве осадителя. Полученный таким образом раствор устойчив в течение длительного времени, особенно если хранить его на льду или в холодильнике [1352, 2803].
Реагент и анализируемый раствор не должны иметь сильно кислой реакции, ускоряющей разложение натрий-бортетрафенила. Оптимум находится при pH 3—6 [183, 433, 948, 979, 1224, 2699], по другим данным,— при pH 4—5 [1312, 1352, 2803]. Некоторые авторы применяют раствор натрий-бортетрафенила в 0,01 W растворе едкого натра [995, 2621] и ведут осаждение калия из раствора, подщелоченного едким натром [1091]. Слабощелочные растворы реагента сохраняются без разложения в течение нескольких недель [995]. Тем не менее, рекомендуются способы осаждения калия при pH 2 [979, 2346] и даже при pH 1 [438, 1759].
Для уменьшения потерь от растворимости калий-бортетра-фенила осаждение ведут при 0°С [314, 805, 2621], и осадок на фильтре промывают охлажденной водой [314] или насыщенным водным раствором калий-бортетрафенила [979, 1218, 1759]. При осаждении на холоду получается очень мелкозернистый, трудно отфильтровываемый осадок; лучше осаждать из теплого или горячего раствора, охладить и затем уже фильтровать. Легко отфильтровываемый осадок получают при применении в качестве реагента свободной кислоты, Н[В(С6Н5)4]. Последнюю получают из раствора ее литиевой соли путем пропускания через катионит в Н-форме. Тетрафенилборная кислота легко разлагается, поэтому ее готовят непосредственно перед применением [1494].
По данным различных авторов, осадок калий-бортетрафенила высушивают при разных температурах: 105—115° С [805, 1470, 1474, 1759, 1818, 2090, 2346, 2699, 2799]; ПО—120°С [183, 753, 948, 979, 1161, 1218, 1826, 2184, 2279, 2621]; 120—130° С [314, 438, 1312, 1512]. Отмечается возможность высушивания осадка в вакуум-эксикаторе при комнатной температуре [2110].
Метод позволяет определять очень малые количества калия [2279]. Можно определять около 1 мг [979] и даже до 0,04 мг калия в пробе [1218]. Концентрации порядка 0,02 мг/мл нельзя определять этим способом [2699]. Ошибки определения 5 мг и больше калия обычно находятся в пределах 1% [314, 584, 2621], при меньших количествах ошибки возрастают, . ’ . . ....
4 Аналитическая химия калия
49
Определение в виде калий-бортетрафенила отличается простотой, хорошей воспроизводимостью и удовлетворительной точностью [1161, 1719]. Определению не мешают сульфаты и фосфаты [753, 2279], соли лития [2279], натрия, магния [438, 805, 2279], кальция [438, 2699], бария, кадмия, хрома, кобальта, никеля, цинка, свинца, олова, уранила [2621]. Если в растворе присутствуют соли трехвалентного железа, то перед осаждением калия вводят NaF [2405].
Натрий-бортетрафенил дает осадки с ионами аммония, рубидия, цезия, серебра, одновалентных ртути и таллия [584], тория [2621], присутствие которых мешает определению калия.
Гравиметрическое определение в виде калий-бортетрафенила применяется при анализе силикатов [1512, 2558, 2799, 2958], цемента [889], стекла [314, 948, 979, 1512, 1826, 2958], огнеупорных материалов [979], удобрений [753, 2506, 2596], золы [733], воды [1470, 2620], пороха [1474, 2184], фармацевтических препаратов [1696, 1734], молока [2486], вина [801, 2310], солей натрия [1696, 1719], солей калия [1818] и других объектов [753, 2087, 2249, 2346, 2616, 2880].
Определению калия при помощи натрий-бортетрафенила посвящен ряд обзорных статей [544, 584, 694, 903, 1351, 1425, 1775, 1831, 2142, 2323, 2681, 2698]. Регенерация реагента из остатков после анализа см. [2313].
Определение в виде битартрата. Раствор хлоридов калия и натрия выпаривают досуха. Остаток растворяют в возможно меньшем количестве воды и осаждают избытком 2%-ного этанолового раствора винной кислоты, насыщенного битартратом калия. После осаждения, которое заканчивается через 2 часа [2922], добавляют 96%-ный этанол, насыщенный битартратом калия, и кипятят для растворения битартрата натрия. Осадок отфильтровывают через тигель Гуча, промывают 96%-ным этанолом, сушат при 80°С, охлаждают и взвешивают [119, 1953, 1954].
1 вес. ч. КНС4Н4Ов соответствует 0,2077 вес, ч. калия.
В других вариантах этого метода осадок высушивают при 100° С [2422]. В качестве осадителя применяют также 0,33 N раствор битартрата натрия [556], раствор 55 г винной кислоты и 14 г едкого натра в 1 л воды [205] или раствор 20 г винной кислоты, 5 г ацетата натрия в 500 мл воды, к раствору добавляют метанол до 1 л [239, 1039]. Не следует в качестве осадителя пользоваться раствором битартрата аммония вследствие его заметного соосаждения с солью калия. В этом случае осадок прокаливают, выпаривают с серной кислотой, прокаливают и взвешивают сульфат калия [1953, 1954].
Рекомендуется осаждать калий раствором битартрата анилина или пиридина [1814].
20 г винной кислоты растворяют в 500 мл воды, добавляют 8 г свежепе-регнаниого анилина или 7 г пиридина и разбавляют метанолом или этанолом до 1 л [185, 374, 1050].
50
Осадителями служат также растворы битартратов магния (1813] или лития * [2922]. Во всех случаях осаждают в присутствии этанола. Применение этих реагентов полезно, если в фильтрате после осаждения калия требуется определить содержание натрия.
Раствор, содержащий около 5 мг!мл калия, смешивают с равным объемом метанола или этанола, добавляют метиловый оранжевый и по каплям при перемешивании вводят раствор битартрата пиридина или анилина до появления желтой окраски индикатора. Через 2 часа осадок отфильтровывают, промывают этанолом, высушивают и взвешивают [185, 374].
\ К достоинствам гравиметрического метода относятся простота и быстрота выполнения определения, соответствие состава осаждаемого соединения приписываемой ему формуле. Однако значительным недостатком является заметная растворимость битартрата калия в воде (см. стр. 153). Для уменьшения потерь от растворимости осаждение ведут в присутствии большого количества этанола, в котором растворимость осадка очень мала [442]. Применение реагентов, предварительно насыщенных битартратом калия, уменьшает потери. Для уменьшения растворимости осадка рекомендуют также промывание его 90— 100%-ным метанолом [73, 143] и введение поправки на растворимость осадка в этом спирте [715].
Исследуемый раствор не должен содержать свободных минеральных кислот или оснований, растворяющих осадок. Наиболее полное осаждение происходит при pH 3,4—3,6 [1262, 1632, 1877]. Присутствие солей лития, натрия, магния, кальция, а также нитратов, хлоридов, сульфатов не мешает определению калия [185, 205, 276, 715]. Из сложных смесей лучше, выделить калий в виде нитрокобальтиата (стр. 39) и затем переосадить в виде битартрата [1’217].
На другие исследования гравиметрического битартратного метода мы только сошлемся [510, 1251, 2029, 2111, 2127, 2154, 2221, 2854].
Определение в виде пикрата. Гравиметрическое определение калия в виде пикрата было предложено в 1881 г. [1420, 1421] для анализа поташа. Метод основан на малой растворимости пикрата калия в 98—99 %-ном этаноле (1:2500) и сравнительно большой растворимости пикрата натрия (1 :80). Исследуемую соль смешивают с 4-кратным количеством пикриновой кислоты, смачивают водой и выпаривают на водяной бане. Остаток осторожно растирают и несколько раз экстрагируют пикрат натрия и избыток пикриновой кислоты 98—99%-ным этанолом. Экстрагирование продолжают до тех пор, пока при выпаривании нескольких капель этанолового экстракта не будет наблюдаться заметный остаток. Полученный таким способом
* 0,5 г карбоната лития и 2 г винной кислоты растворяют в 100 мл воды, добавляют 50 мл 95%-ного этанола и насыщают битартратом калия [2922].
4* 51
пикрат калия сушат при 100° С, охлаждают и взвешивают [1420, 1421].
1 вес. ч. KC6H2N3O7 соответствует 0,1463 вес. ч. калия.
Калий можно осаждать насыщенным водным [2779] или этаноловым [908, 2028] раствором пикриновой кислоты или раствором пикрата натрия [2309], выпавший осадок промывают возможно меньшим объемом холодной воды.
Вследствие заметной растворимости пикрата калия определение дает результаты, заниженные на 2—5% [2309]. В присутствии больших количеств солей натрия могут получаться завышенные результаты для калия. Определению мешает присутствие солей рубидия, цезия, одновалентного таллия, больших количеств аммония, осаждаемых пикриновой кислотой. Не мешают соли магния, кальция, алюминия, железа и других элементов.
Рекомендуется аналогичный способ гравиметрического определения калия в виде диметилпикрата [1934].
Осадителем служит 3%-ный водный раствор диметилпикрата натрия. Осадок диметилпикрата калия промывают холодной водой, сушат при 90—150° С, охлаждают и взвешивают.
1 вес. ч. KCgHsNaO? соответствует 0,1324 вес. ч. калия.
Определение в виде дипикриламината. Исследуемый раствор сначала нейтрализуют в присутствии тимолового синего. Осаждать дипикриламинат калия лучше всего при pH 7—8,8 [657, 658, 1604, 2338]. Осадителем служит раствор дипикриламината магния.
5 г окиси магния и 12 г дипикриламина взбалтывают с 400 мл воды, на другой день фильтруют; таким путем получают 3%-ный раствор дипикриламина [1764].
Нейтральный исследуемый раствор, содержащий хлорид или сульфат калия, выпаривают до возможно меиынего объема, и к горячему раствору добавляют свежеприготовленный горячий раствор дипикриламината магния в избытке. Выпадает оранжево-красный кристаллический осадок дипикрилами-иата калия. На другой день осадок отфильтровывают через стеклянный пористый тигель № 3 и промывают охлажденной водой, насыщенной дипикрилами-натом калия. Осадок высушивают при 100—105° С, охлаждают и взвешивают [20, 392, 1271, 1478].
1 вес. ч. KC12H4N7O12 соответствует 0,08194 вес. ч. калия.
Рекомендуют также высушивание осадка при более высоких температурах—110°С [657, 658, 1271, 1604, 1607, 2441] и даже при 150° С [359]. Для уменьшения ошибок, возникающих вследствие растворимости дипикриламината калия, осаждают при 0°С [1300, 1604, 1607, 1839, 2378] и промывают осадок насыщенным водным раствором дипикриламината калия, также охлажденным до 0° С.
Метод позволяет определять 5—20 мг калия с погрешностью не более 1% [1607, 2262, 2444, 2718]. Микрометодом можно определять десятые доли миллиграмма калия [1128, 2444]. По точ
52 Ч
ности дипикриламинатный способ не уступает хлороплатинат-ному [359] и нитрокобальтиатному [2334] методам определения калия. Сравнительно небольшое содержание калия в осадке относится к числу достоинств метода.
Калий осаждают также раствором дипикриламината натрия.
Последний получают растворением 0,35 г безводного карбоната натрии и 2,7 г дипикриламина в 100 мл воды [2553]. При применении этого реагента желательно переосадить дипикриламинат калия для удаления соосаждениого натрия [2262]. С этой целью отфильтрованный осадок, не промывая, растворяют в 3 мл ацетона, добавляют 5 мл воды и 3 мл раствора дипикриламината натрия, нагревают на водяной бане до удаления ацетона, охлаждают, фильтруют и т. д.
Вследствие соосаждения нельзя определять примесь калия в солях натрия. Тем не менее, дипикриламин оказывается одним из лучших органических нитросоединений, пригодных для отделения и определения калия в присутствии натрия. Это иллюстрирует табл. 4 [1070].
Таблица 4
Растворимость солей калия и натрия (г/100 г воды) некоторых органических нитросоединеиий при 30°С
Нитросоединение Соль натрия Соль калия ^соль Na
^соль К
Метилпикриновая кислота 3,15 3,78 ,0,83
Хлорпикриновая кислота 31,2 1,82 17,15
Стифниновая кислота 8,43 1,54 5,47
Флавиаиовая кислота 9,8 1,03 9,51
Пикриновая кислота 5,6 0,755 7,41
Нитраниловая кислота 0,72 0,567 1,2Т
Пикролоновая кислота 0,28 0,338 0,83
Дипикриламин 11,6 0,146 79,45
Дилитуровая кислота 1,03 0,086 11,97
Соль магния также способна соосаждаться с дипикрил-аминатом калия [1300]. Поэтому рекомендуют осаждать калий раствором дипикриламината лития [627, 1010].
Для получения этого реагента растворяют 0,55 г Li3CO3 в 100 мл воды, добавляют 3 г дипикриламина, нагревают до 50° С. На другой день фильтруют и разбавляют водой до 500 мл [627, 2540].
Присутствие солей лития и кальция не мешает определению калия [2262]. Мешают соли рубидия, цезия, одновалентного таллия, аммония, осаждаемые этим реагентом. Присутствие аммиака в воздухе может вызвать повышенные результаты для калия.
53
Дипикриламин и его соли взрывоопасны. Дипикриламин!] взрывается при 240—250° С, дипикриламинат калия — при^ 325° С [2262]. Взрыв может произойти от удара, трения, искры// [20]. Дипикриламин и его соли вызывают медленно заживаю-'] щие поражения кожи [2262]. Таким образом, необходимо со-’’ блюдать известную осторожность.
Дипикриламинатный способ рекомендуется для анализа силикатов [473], минералов [420], почвы [2480], золы [1330, 2480], воды [1607], вина [657, 658], удобрений [2262, 2480], сплава калия, натрия и свинца [487], стекла [174]. На другие работы, посвященные этому методу, мы только сошлемся [385, 564, 1124, 1558].
Другие способы гравиметрического определения калия
1. Для определения относительно больших количеств калия осаждают и взвешивают его в виде персульфата [2689] (растворимость персульфата калия см. стр. 154); критику этого способа см. [2177].
2. Отмечается возможность гравиметрического определения калия в виде фторосиликата [2374].
3. Описано определение калия (рубидия, цезия) в форме К2О • 2ZrO2 • 2SO3 • nH2O [2953].
4. К раствору сульфата калия прибавляют концентрированный раствор сульфата алюминия и этанол. Выпадает осадок калий-алюминиевых квасцов, который промывают смесью воды и этанола, сушат при 35°С и взвешивают. Если в растворе находятся хлориды или нитраты, то их предварительно переводят в сульфаты выпариванием с серной кислотой.
Метод пригоден для определения сравнительно больших количеств калия [1837, 2195].
5. Определение в виде сульфида висмута. Калий осаждают в виде K5'Na[Bi(S2O3)3]2 (стр. 16), осадок растворяют в воде и добавляют сульфид аммония. Выделившийся осадок сульфида висмута отфильтровывают, промывают, сушат до постоянного веса.
1 вес. ч. Bi2S3 соответствует 0,3802 вес. ч. калия [927].
Метод не давал удовлетворительных результатов [2196].
6. Смешивают 10 мл раствора, содержащего 0,1—0,2 г калия, с 7 мл 10%-ного раствора натриевой соли 2-хлор-З-нитро-толуол-5-сульфокислоты, оставляют на 12 час. при 5° С, осадок отфильтровывают и промывают раствором реагента, затем холодной водой, сушат при 120° С, охлаждают и взвешивают [806, 1042].
1 вес. ч. KC7H5CINSO5 соответствует 0,1349 вес. ч. калия.
Вследствие заметной растворимости осадка (стр. 161) метод позволяет определять только сравнительно большие количест-54
ва калия. Определению мешает присутствие солей аммония, рубидия, цезия, бария.
7. Навеску исследуемого материала растворяют в небольшом объеме воды и добавляют избыток 1%-ного раствора 5-нитробарбитуровой (дилитуровой) кислоты в 50%-ном этаноле. Раствор с выпавшим осадком оставляют на льду до следующего дня, центрифугируют, промывают 50%-ным этанолом, сушат при 100° С, охлаждают и взвешивают.
1 вес. ч. KC4H2N3O7 соответствует 0,1851 вес. ч. калия. Определению мешают соли аммония, рубидия, цезия, бария [1070, 1077, 1374, 2310].
8. Калий осаждают раствором литиевой соли 2,4-динитр’о-нафтиламинобензолсульфокислоты, осадок промывают, высушивают при' 110° С, взвешивают. Определению мешают соли магния [1155].
9. В качестве осадителя для гравиметрического определения калия в присутствии натрия предлагается раствор нафтолового желтого S [1112].
10. Малорастворимая соль калия осаждается при добавлении 1%-ного раствора 3,4-диоксиазобензол-4'-сульфокислоты; осадок содержит 11,9% калия. Определению мешают соли аммония, рубидия, цезия, одновалентного таллия. Соли серебра и золота восстанавливаются реагентом до металла [766].
И. Калий осаждают 1%-ным раствором тетранитрофентиазин-5,5-диоксида в 0,1 М растворе LiOH. Осадок промывают, сушат при 110° С, взвешивают [2731]. »
12. Для определения калия в солях органических кислот навеску сжигают в струе кислорода. Привес трубки после сожжения равен количеству окиси калия, 1 вес. ч. которой соответствует 0,8301 вес. ч. калия [167].
Титриметритеское определение
Метод нейтрализации
Известковый способ. Способ предложен для определения суммы щелочных металлов в силикатах [35, 36].
0,5—1 г размельченного силиката обрабатывают HF в платиновой чашке и выпаривают. При этом большая часть кремния улетучивается в виде SiFi, в остатке находятся фториды магния, кальция, алюминия и других элементов, а также кремнефториды щелочных металлов. Остаток кипятят с избытком раствора Са(ОН)2, что приводит к образованию малорастворимых гидроокисей и фторида кальция, например: .
2FeF3 + 3Ca(OH)2->2Fe(OH)3+3CaF2. '
Кремнефториды щелочных металлов разлагаются с образованием едких щелочей:
KzlS iFe]+4Са (ОН) 2—ЗС aF2 + CaSiO.3+2КОН + ЗН2О.
55
Осадок отфильтровывают, промывают водой. В фильтрат, содержащий едкие щелочи и избыток Са(ОН)2, вводят углекислоту:
Са (ОН) 2+СОг—* СаСОз 4- Н2О;
КОН+СО2—КНСОз.
Снова фильтруют, промывают малым количеством воды, фильтрат кипятят для превращения бикарбонатов в карбонаты и удаления углекислоты:
2КНСОз— К2СО3+С02+Н20
и затем титруют 0,1 N соляной кислотой в присутствии фенолфталеина [38, 94, 505] или метилового оранжевого [35].
Грамм-эквивалент калия при титровании в присутствии фенолфталеина равен 78,2 г, при титровании в присутствии ) метилового оранжевого — 39,1 г.
Баритовый способ. Исследуемый раствор вместе с добавленной серной кислотой выпаривают досуха. Соли щелочных металлов при этом образуют бисульфаты, например:
KC1+H2SO4^KHSO4+HC1. *
Остаток растворяют в воде и добавляют избыток раствора Ва(ОН)2:
KHSO4+Ba(OH)2—*BaSO4+KOH + H2O.
Не фильтруя, пропускают в раствор струю СО2 для удаления избытка Ва(ОН)2, при этом образуется бикарбонат калия (и натрия). При последующем кипячении удаляется избыток СО2, а из бикарбоната получается карбонат. Фильтруют, промывают водой, и карбонат в полученном растворе титруют 0,01—0,1 N соляной кислотой в присутствии фенолфталеина или метилового оранжевого.
1
Значения грамм-эквивалента калия указаны в предыдущем способе [10, 504, 532, 542, 698, 792, 1586, 2317, 2448, 2934].
Для уменьшения ошибки от растворимости ВаСОз следует до фильтрования добавить к раствору половину его объема 95%-ного этанола. (
К фильтрату добавляют отмеренный объем 0,1 N раствора соляной кислоты, кипятят до удаления СО2, и избыток соляной кислоты титруют 0,1 ДО раствором едкой щелочи [2710]. Вместо пропускания струи СО2 можно после реакции с Ва(ОН)2 добавить раствор карбоната аммония и аммиака, затем выпарить на водяной бане для удаления аммиака и карбоната аммония. Остаток растворяют в воде, фильтруют, промывают и титруют [335].
. . кеа латный способ. Исследуемый раствор, ие содержащий сульфатов и фосфатов, смешивают с 5—15 г щавелевой кислоты, выпаривают досуха при постоянном помешивании. При этой операции из солей летучих г кислот образуются оксалаты, например:
2КС1 + Н2С2О4->К2С2О4+2НС1.
От дальнейшего нагревания и прокаливания избыток щавелевой кислоты возгоняется, оксалаты щелочных металлов превращаются в карбонаты [334, 2091а, 503, 506, 508, 2333, 2374, 2511, 2583, 2688а]:
КгС2О4—^КгСОз + СО;
2КяС2О4 + О2^2К2СО3+2СО2.
Остаток охлаж-дают, обрабатывают горячей водой, фильтруют, промывают водой. Полученный раствор титруют 0,1 ДО соляной кислотой в присутствии fi метиловогр оранжевого.
56
Значения грамм-эквивалента калия приведены на стр. 56. Метод применен для анализа буровых вод [334].
Если исследуемый объект содержит сульфаты, то их предварительно осаждают хлоридом бария. Избыток последнего при обработке щавелевой кислотой и прокаливании превращается в ВаСОз, не мешающий дальнейшему определению [2333].
При анализе почвы производят вытяжку 0,2 А раствором щавелевой кислоты, вытяжку затем выпаривают и остаток прокаливают. В прокаленном остатке находятся окислы железа, алюминия, магния, а также карбонаты щелочных металлов.
Этот остаток обрабатывают насыщенным раствором гидроокиси магния, фильтруют, и аликвотную часть фильтрата титруют 0,02 М соляной кислотой в присутствии метилового оранжевого. Отдельно титруют такой же объем насыщенного раствора гидроокиси магния, и эту величину вычитают из общего расхода титрованного раствора кислоты; по разности вычисляют содержание калия или суммы щелочных металлов [561].
Аналогичный метод применен для определения щелочных металлов в силикатах [505].
Навеску силиката обрабатывают избытком HF, затем раствор выпаривают и остаток прокаливают с избытком щавелевой кислоты. Из прокаленного остатка вода извлекает карбонаты щелочных металлов, которые титруют, как указано выше.
Бензидиновый способ. Этот способ позволяет определять содержание калия (или суммы щелочных металлов) при условии, что все соли переведены в сульфаты.
Исследуемый раствор, содержащий около 0,15 мг-экв калия и свободный от фосфатов, солей магния и кальция, смешивают с 0,5 мл конц. HaSO4 и 2 мл конц. HNO3. Выпаривают досуха, остаток прокаливают несколько минут. Пос ле охлаждения извлекают водой и разбавляют до 15 мл. Добавляют 2 мл раствора хлоргидрата бензидина (4 г бензидина обрабатывают 45 мл 1 N соляной кислоты, разбавляют водой до 250 мл и фильтруют):
KjSOi+B -2НС1—2КС1 + В • H2SO4,
где В — молекула бензидина.
Через несколько минут выпавший осадок сульфата бензидина, отфильтровывают. 15 мл фильтрата титруют 0,02 N раствором едкой щелочи в присутствии фенолового красного. Отдельно титруют 2,0 мл раствора хлоргидрата беизндина [1216].
Содержание калия вычисляют по уравнению:
К.™- (V-£v}'N, \ 15 )'
где Кмг.экв —содержание калия (или суммы щелочных металлов), мг-экв-, V — объем раствора едкой щелочи, израсходованный на титрование 2,0 мл раствора хлоргидрата бензидина, мл;
и — объем раствора едкой щелочи, израсходованный на титрование 15 мл фильтрата, мл;
N — нормальность раствора едкой щелочи.'
Этот метод рекомендуется для определения щелочных металлов в биологических объектах [854, 867, 1722].
57
Первоначальный вариант этого способа заключался в титровании промытого осадка сульфата бензидина раствором едкой щелочи [2622]. Однако растворение осадка при промывании вызывает заметные ошибки.
Титрование фторосиликата калия. К анализируемому раствору добавляют избыток раствора H2[SiF6] и равный объем этанола. Выпавший осадок Kz[SiFe] через 30 мин. отфильтровывают, промывают 50%-ным этанолом. Осадок переносят в колбу, взбалтывают с водой, добавляют избыток хлорида кальция и метиловый красный и титруют 0,1 N раствором едкой щелочи до появления желтой окраски [501, 502, 2647]:
[SiF6p- + ЗСа2++4ОН-—>3CaF2+H2SiO3+Н2О.
Вариант метода заключается в добавлении избытка титрованного раствора едкой щелочи, кипячении и титровании избытка щелочи раствором соляной кислоты [2642]. Грамм-эквивалент калия равен 19,550 г.
Предлагается также осаждать калий раствором фторосиликата анилина [1752]. Определению мешают соли аммония и натрия [2642]. Метод рекомендуется для определения калия в фосфатных удобрениях [501], в квасцах [2643].
Титрование продуктов восстановления хлороплатината калия. Осадок хлороплатината калия (стр. 33) взбалтывают с водой, вводят около 1 г платины в порошке (катализатор), добавляют нейтральный этанол и нагревают 1 час:
KilPtCk]+2С2Н5ОН—-4НС1+2КС1 + Pt+2СН3СНО.
После охлаждения добавляют метиловый оранжевый и образовавшуюся кислоту титруют 0,01—0,1 W раствором едкой щелочи [2482]. Теоретический грамм-эквивалент калия равен 19,550 г.
В другом варианте этого способа хлороплатинат восстанавливают в струе водорода при нагревании:
К2[Р tCle]+2Н2—4НС1+2КС1 + Pt.
Выделяющийся хлористый водород поглощают 0,01—0,1 N раствором едкой щелочи, избыток которой титруют затем соляной кислотой в присутствии метилового оранжевого [2655]. Отдельно такой же объем раствора щелочи титруют этим же раствором соляной кислоты. Содержание калия вычисляют по уравнению:
Клг= (Vt-V2)У. 19,550,
где V\ — объем раствора соляной кислоты, израсходованный на титрование раствора едкой щелочи, мл\
У2— объем раствора соляной кислоты, израсходованный на титрование остатка щелочи, мл-,
N — нормальность раствора соляной кислоты.
Метод сложен и вряд ли может иметь практическое применение.
Титрование нитрокобальтиата. Осадок нитрокобальтиата калия (стр. 39) растворяют в соляной кислоте и раствор нейтрализуют раствором едкой щелочи в присутствии метилового оранжевого. К этому раствору прибавляют определенный объем 0,1 N раствора карбоната натрия, вследствие чего выпадает осадок карбоната кобальта. Осадок отфильтровывают, промывают водой, в фильтрате и промывных водах титруют избыток карбоната натрия 0,1 N раствором соляной кислоты в присутствии метилового оранжевого [659]. Одновременно такой же объем первоначального раствора карбоната нат-58
рия титруют соляной кислотой. Если принять, что состав осадка выражается формулой K2Na[Co(NOj)e], то грамм-эквивалент калия равен 19,550 г.
Содержание калия вычисляют по формуле
Кл,г=(У1-У»)ЛМ9,55,
где Vi — объем раствора соляной кислоты, израсходованный на титрование взятого объема раствора карбоната натрия, мл;
V2 — объем раствора соляной кислоты, израсходованный на титрование избытка карбоната натрия, мл;
N — нормальность раствора соляной кислоты.
По другому варианту осадок нитрокобальтиата кипятят с водой, добавляют избыток 0,05—0,1 N раствора соляной кислоты, содержащей мочевину (3 г/л), нагревают на водяной бане:
2NOa- + СО (NH2) 2+2Н+—- 2N2+СО2+ЗНгО.
Охлаждают, добавляют метиловый красный или бромтимоловый синий, и избыток соляной кислоты титруют 0,05—0,1 N раствором едкой щелочи [2211, 2212]. Для определения титра применяемых растворов по калию необходимо одновременно и в таких же условиях осадить калий из его стандартного раствора. При проверке этого метода были получены неточные результаты [960].
Осадок нитрокобальтиата калия растворяют при нагревании в определенном объеме 0,1 N раствора серной кислоты, избыток последней титруют 0,1 N раствором едкой щелочи в присутствии тимолового синего. Этот метод предложен для определения калия в почвах [1066].
Титрование фосфоромолибдата калия. Осадок фосфоромо-либдата калия (стр. 47) отфильтровывают, промывают 2%-иой азотной кислотой, затем насыщенным раствором нитрата калия. Фильтр с осадком взбалтывают в колбе с 50 мл воды и вводят 0,1 N раствор едкой щелочи: ,
К3РО4-12МоО3+23КОН - К2НР04+12К2Мо04+11Н20.
Избыток щелочи титруют 0,1 W раствором соляной кислоты [190, 734, 735].
3-39,1
Теоретически грамм-эквивалент калия равен - = 5,100 г. Однако грамм-
23
эквивалент калия в этом процессе — величина непостоянная, так как состав осадка подвержен колебаниям (см. стр. 47).
Содержание калия вычисляют по уравнению
= (^KOH’^KOH ~ ^HCT^HCl)'5’100’
где VK0H и VHci — объемы растворов щелочи и кислоты, мл;
^КОН и ^НС1 — нормальность растворов щелочи и кислоты.
Метод рекомендуется для определения калия в крови [735]. При определении 5—58 мг калия погрешность составляет 0,1 — 0,7 мг [190].
Предложен также следующий способ определения калия.
Осадок фосфоромолибдата калия после промывания растворяют в аммиаке и прибавляют магнезиальную смесь. Выпавший осадок NH4MgPO4 отфильтровывают, промывают водой и растворяют в определенном объеме 0,01 М раствора серной кислоты:
NH4MgPO4+H2SO4 - MgSO4+NH4H2PO4.
Избыток кислоты титруют 0,01 N раствором щелочи в присутствии метилового оранжевого [2469], грамм-эквивалент калия при этом равен 58,65 г.
Титрование фосфоровольфрамата калия. 0,2 мл раствора, содержащего хлориды калия, натрия и других металлов, смешивают с 0,5 мл~
59
0,8%-ного раствора фосфорновольфрамовой кислоты и выпаривают досуха на водяной бане. Остаток полностью смывают водой в центрифужную пробирку, добавляют воду до 6 ял, центрифугируют, раствор удаляют. В пробирку вводят 10 мл воды и щепотку окиси алюминия (для равномерного кипения), кипятят и прибавляют из бюретки 0,02 N раствор едкой щелочи до растворения осадка и еще избыток до сильно щелочной реакции:
К6Н (PO4.12WOs)2-|-47KOH -» 2K2HPO4+24K2WO4+23H,O.
Избыток щелочи титруют 0,02М серной кислотой в присутствии смешанного индикатора (тимоловый синий и фенолфталеин) [43, 143, 363, 1232, 1683, 2535, 5-39 1
2733, 2783]. Грамм-эквивалент калия равен ——— = 4,159 г. к!
Содержание калия вычисляют по формуле
Кл1г = (^KOH’^KOH ~~ 'ZH1SO4’^H>SoJ,^>1'594-1,75,
где V кон — объем введенного раствора едкой щелочи, мл;
^КОН — нормальность раствора едкой щелочи;
Vh2so — израсходованный объем раствора серной кислоты, мл;
Л,н2ЗО1—нормальность раствора серной кислоты;
1,75—поправка на растворимость осадка в воде, мг.
Недостаток метода заключается в непостоянстве состава осадка (стр. 48), вследствие чего величина грамм-эквивалента калия может меняться. Метод рекомендуется для определения калия в биологических объектах [43, 1232, 2783].
Титрование калий-бортетрафенила. Калий-бортетрафенил легко растворяется в ацетоне, в этом растворе он легко реагирует с солями двухвалентной ртути с выделением хлористого водорода [584, 1538, 2085, 2346а, 2929, 2930]: K[B(C6H5)4] + 4HgCl2 + 3H2O ->3HCl + H3BO3 + 2KCl + 4C6HsHgCl.
Эта реакция позволяет определять калий методом нейтрализации.
Осадок калий-бортетрафенила (стр. 48) растворяют в нейтральном ацетоне [1219, 1224] или* метаноле [2929], добавляют HgCl2, и выделившуюся соляную кислоту титруют 0,01—0,17V раствором едкого натра в присутствии бромкрезол-пурпурного [1223 , 2929]. Грамм-эквивалент калия равен 13,033 г.
Если к нейтрализованному раствору добавить маннит, то можно оттитровать и образовавшуюся в указанной реакции борную кислоту [2929].
Промытый осадок калий-бортетрафенила растворяют в 5 мл нейтрального ацетона и 5 мл насыщенного раствора HgCl2, добавляют 10 мл 0,1 N раствора едкой щелочи, нагревают, вводят 5 мл 10%-ного раствора KJ и титруют 0,1Л? раствором НС1 в присутствии метилового красного до розовой окраски. Затем добавляют еще 1—2 мл раствора НС1, кипятят для удаления СО2 и титруют 0,iN раствором едкой щелочи до появления желтой окраски [1219, 1872, 2957].
Содержание калия вычисляют по формуле
Kju2 = (^NaOH'^NaOH ~ ^НСГ^Нш)’13’033>
60
где Умаон и ^hci ~~ общие объемы введенных растворов щелочи и кислоты, мл;
^NaOH и Уцс! — нормальности растворов щелочи и кислоты.
Для этого титрования предлагается также индикатор, состоящий из смеси 1 объема 0,2%-ного этанолового раствора метилового красного и 3 объемов 0,1%-ного этанолового раствора бромкрезолового зеленого, переход окраски при pH 5,5 от красной (кислая среда) до зеленой (щелочная среда) [1872].
Раствор калий-бортетрафенила в ацетоне разбавляют равным объемом воды и пропускают через колонку с вофатитом в Н-форме, к вытекающему раствору добавляют раствор HgCh и т. д., как описано выше, при этом грамм-эквивалент калия равен 9,775 г [628]. Способ позволяет определять до 0,4 мг калия в пробе.
Водно-ацетоновый раствор калий-бортетрафенила можно пропустить через колонку с катионитом в Н-форме, колонку промывают водой. Фильтрат и промывные воды титруют 0,01 N раствором NaOH в присутствии метилового красного и метиленового голубого до появления устойчивой зеленой окраски. Грамм-эквивалент калия равен 39,102 г [1225].
Предложены варианты этого способа. Учитывая возможность взаимодействия ацетона с солью ртути, рекомендуется не растворять калий-бортетрафенил в ацетоне, а непосредственно обрабатывать его водным раствором HgCU [2054].
Оригинальный вариант определения калия заключается в следующем.
В анализируемый раствор вводят небольшое количество хлорида аммония и добавляют натрий-бортетрафенил. Осаждение аммоний-бортетрафенила способствует осаждению малых количеств калия и уменьшает потери последнего при промывании. Высушенный осадок затем прокаливают [584, 1222, 1224]:
К[В (С6Н5)4]+30О2—>-КВО2-1-24СОа+ IOH2O;
NH4B (С6Н5) 4]+ 30О2— НВО2+24СО3+10Н2О + NH3.
Для определения количества образовавшегося метабората калия к остатку от прокаливания добавляют 1 мл 0,01—0,001 N раствора НС1, нагревают до кипения, и избыток НС1 титруют 0,01—0,001 N раствором NaOH в присутствии смеси метилового красного и метиленового голубого [1224, 2502].
Отметим еще один метод.
5—10 мл анализируемого раствора, содержащего около 20 мг калия, подкисляют соляиой кислотой и добавляют 15 мл 0,05 N раствора натрий-бор-тетрафенила. Через 10 мин. фильтруют и промывают водой. К фильтрату и промывным водам добавляют 20 мл 0,03 N раствора NH4C1. Осадок аммоний-бортетрафенила отфильтровывают, промывают, и в фильтрате определяют избыток соли аммония по методу Крапивина. Для этого фильтрат нейтрализуют в присутствии метилового красного, добавляют 5 мл нейтрализованного раствора формальдегида, и освободившуюся кислоту титруют 0,05 N раствором NaOH в присутствии фенолфталеина [183, 184]. Таким образом, определение сводится к титрованию второго остатка.
: Титрование битартрата калия. Это один из наиболее старых методов определения калия, предложенный более
61
100 лет назад [2045]. Осадок битартрата калия (стр. 50) растворяют в горячей воде (80° С) и титруют 0,01—0,1 N раствором NaOH в присутствии фенолфталеина [73, 143, 185, 239, 369, 374, 442, 715, 718, 971, 1039, 1101, 1217, 1261, 1690, 1920, 2551, 2606, 2677, 2922]:
КНС4Н4О6 4- NaOH ->KN аС4Н4О6+Н2О.
Таким способом можно определить сравнительно большие количества калия, порядка 0,08—0,22 г [971] или даже 0,3—0,6 г [1039] с удовлетворительной точностью. В качестве индикатора при этом определении применялись также лакмус [935] и розо-ловая кислота [2246]. Грамм-эквивалент калия равен 39,102 г. Для повышения точности определения рекомендуется установить титр раствора едкой щелочи по навескам битартрата калия [2922] или лучше по навескам КС1 или его стандартному раствору (5 лгКМл), с которым производят такие же операции и в таких же условиях, как и с исследуемыми растворами [185, 1039, 1050].
Вариант метода заключается в осаждении калия титрованным раствором битартрата натрия и титрования избытка последнего щелочью в присутствии фенолфталеина [205, 442, 516, 556, 794, 810, 1581, 2001, 2002, 2266]. Можно также осадок битартрата растворить в титрованном растворе едкой щелочи, избыток которой титровать раствором HCI в присутствии метилового оранжевого [442]. Описан вариант метода, основанный на растворении битартрата калия в растворе муравьиной кислоты и титровании избытка последней раствором едкой щелочи [1729]. Определение в виде битартрата обычно дает несколько пониженные результаты вследствие заметной растворимости осадка [275, 556, 971]. (О растворимости битартрата калия см. стр. 153).
Следует о,тметить способ определения калия, основанный на его осаждении в виде битартрата в присутствии соли аммония.
Раствор хлоридов, нитратов или ацетатов щелочных металлов слабо подкисляют соляной кислотой, выпаривают до 20 мл, добавляют немного ацетата аммония и осаждают калий винной кислотой. После перемешивания вводят I—1,5 объема 95%-ного этанола, снова перемешивают и оставляют на 1 час. Осадок отфильтровывают, промывают 60%-ным этанолом, высушивают и прокаливают:
2КНС4Н4Ов+5О3—► KjCO3+7СО3+5Н2О;
2NH4HC40s *4 50?—*2NHg 4- 8СО34-6Н3О.
Осадок следует прокаливать осторожно, иначе наблюдается вспучивание и потери. В остатке от прокаливания содержится К3СО3, который растворяют в-воде и титруют 0,05—0,1 М раствором соляной кислоты в присутствии метилового оранжевого. Грамм-эквивалент калия равен 39,102 г [1227].
В этом варианте достигается практически полное осаждение калия и минимальные потери вследствие растворимости осадка.
62
Титрование дипикриламината калия. Осадок дипикрилами-ната калия (стр. 52) растворяют в нейтральном ацетоне, прибавляют 5—10 мл воды, нагревают и добавляют определенный объем 0,1 N раствора НС1:
KC12H4N7O12 4“ НС1 —* Ci2HgN20j2 4~ КС1.
Растворимость дипикриламина значительно меньше растворимости его калиевой соли. Она составляет 8 • 10~6 моль/л при 15°С и 1,2 • 10~5 моль!л при 25° С [1712]. Поэтому от действия НС1 осаждается свободный дипикриламин.
Раствор нагревают на водяной бане' для коагуляции дипикриламииа и удаления ацетона. Охлаждают ледяной водой, фильтруют через стеклянный фильтр, промывают ледяной водой. Фильтрат и промывные воды нагревают до кипения для удаления СО3, и избыток НС1 титруют 0,1 N раствором NaOH в присутствии бромтимолового синего до перехода желтой окраски в синюю. Грамм-эквивалент калия равен 39,102 г.
При определении около 10 мг калия получены удовлетворительные результаты [241, 1764, 2668]. Вариант метода заключается в аналогичном определении избытка дипикриламината натрия или магния в маточном растворе после осаждения калия [2444].
Другие способы определения калия методом нейтрализации мы отметим очень кратко.
1. Навеску хлоридов или нитратов калия, натрия и аммония прокаливают для удаления соли аммония, остаток растворяют в 10—20 мл воды. Весь раствор или его аликвотную часть пропускают через колонку ‘с катионитом (во-фатит KS) в Н-форме и несколько раз промывают водой. В растворе содержится кислота в количестве, эквивалентном суммарному содержанию калия и натрия. Добавляют метиловый красный и метиленовый голубой и титруют 0,01 N раствором NaOH [1222]. 1 мл этого раствора соответствует 0,01 мг-экв солей калия и натрия.
Для определения калия в комплексных цианидах пропускают исследуемый раствор через колонку с анионитом в ОН-форме (ионит обрабатывают раствором аммиака, затем водой). Колонку промывают водой. Полученный раствор титруют 0,1 N раствором НС1 в присутствии метилового оранжевого [1283].
2. Нейтральный раствор КС1 взбалтывают 10 мин. с 1—1,5 г основания Миллона:
[OHg2NH2]OH+КС1—*[OHg2NH3]Cl + КОН.
Фильтрат и промывные воды титруют 0,1 N НС1 в присутствии метилового оранжевого. Грамм-эквивалент калия равен 39,102 г [1326].
Метод неспецифичен, таким же способом можно определять и концентрацию растворов хлоридов других металлов. Необходимый для определения препарат основания Миллона получают взбалтыванием желтой окиси ртути с водным раствором аммиака.
3. Содержание K[BF<] в смеси с нейтральными солями (хлориды, нитраты и другие) может быть определено, исходя из реакции:
K[BF4]+2СаС12+4NaOH—-NaBO2+2CaF3+ КС1+3NaCl+2H3O.
Исследуемый раствор смешивают с 50 мл 4 N раствора СаС13 и 20 мл 1 N раствора NaOH, нагревают иа песчаной бане. Избыток щелочи, а также метаборат натрия титруют 1 N раствором НС1 в присутствии метилового оранжевого [1361]. Грамм-эквивалент калия равен 13,033 г.
Другие способы см. [821, 1009, 2399, 2502].
63
Аргентометрический метод
Титрование хлороплатината калия после его восстановления. Метод основан на предварительном выделении калия в виде хлороплатината. Хлор, входящий в состав этого комплексного соединения, нельзя непосредственно титровать раствором AgNO3. Поэтому хлороплатинат сначала восстанавливают тем или иным способом. Выделившийся хлорид титруют раствором AgNO3. Один из наиболее старых способов предложен Мором [2047].
Осадок хлороплатината калия (стр. 33) сплавляют с двойным количеством оксалата натрия:
Ка[Р tCl6]+2Na3C3O4—-2КС1 + 4NaCI + Pt+4CO2;
2Na3C3O4+O3—*Na3CO3+2CO3.
Плав выщелачивают водой, нейтрализуют уксусной кислотой и титруют хлориды 0,1 А раствором AgNO3 в присутствии К2СгО4. Грамм-эквивалент калия равен 13,033 г.
Более удобно восстанавливать хлороплатинат при помощи формиата [1770].
Осадок хлороплатината калия растворяют в горячей воде, добавляют немного формиата натрия,
Кг[Р tCle]+2HCOONa—< 2КС1 + 2NaCl+2НС1+2СО2+ Pt.
Нагревают до тех пор, пока раствор над осадком восстановленной платины станет совершенно бесцветным, что свидетельствует о полноте восстановления. Раствор взбалтывают с небольшим количеством СаСО3 для нейтрализации образовавшейся НО, фильтруют, промывают осадок на фильтре горячей водой. К фильтрату и промывным водам добавляют К2СгО4 и титруют 0,1 N раствором AgNOj.
Хлороплатинат можно восстанавливать металлическим магнием [879, 1180] или цинком [1074, 1193, 1322, 2940].
Раствор хлороплатината калия обрабатывают на холоду достаточным количеством цииковой пыли:
К2[Р tC 16]+2Zn—2КС1+2ZnCl3+Pt.
По окончании восстановления фильтруют, фильтрат и промывные воды подкисляют азотной кислотой и титруют хлориды по Фольгарду.
В некоторых вариантах этого метода рекомендуется восстанавливать хлороплатинат калия водородом при прокаливании [906]:
K2[PtCl6]+2H2 ->2КС1 +4НС1 + Pt.
По окончании восстановления и охлаждения отмывают хлорид калия из остатка водой, фильтруют, фильтрат подкисляют азотной кислотой и титруют 0,01—0,1 N раствором AgNO3 по Фольгарду. В данно!И случае грамм-эквивалент равен 39,102 г.
Этот вариант менее точен вследствие относительно большого значения грамм-эквивалента и менее удобен по технике вос-
46
становления. Однако положительная сторона восстановления водородом состоит в том, что можно не опасаться влияния солей аммония. Хлороплатинат аммония при обработке водородом превращается в хлорид аммония, который при прокаливании полностью улетучивается и не влияет, следовательно, на результаты титрования хлорида калия.
Описаны и другие модификации титрования образующегося при восстановлении хлорида калия по способу Фольгарда [1102, 1454]. Полученный при восстановлении хлорид можно титровать раствором AgNO3 в присутствии адсорбционного индикатора— дихлорфлуоресцеина [879].
Титрование Нитрокобальтиата калия. Осадок нитрокобальтиата калия и натрия (стр. 39) растворяют в конц. НС1, выпаривают досуха для удаления избытка НС1 и сушат при 125—150° С, затем растворяют в возможно меньшем количестве воды и снова выпаривают досуха. После нескольких таких операций остаток не содержит примеси свободной соляной кислоты. Его растворяют в воде, и образовавшиеся хлориды (2КС1 + NaCl + C0CI2) титруют по Фольгарду. Для этого раствор подкисляют азотной кислотой, вводят избыток 0,02—0,05 N раствора AgNOa, который титруют раствором роданида аммония такой же нормальности в присутствии железо-аммонийных квасцов [208, 1029, 1052, 1435]. Если считать, что осадок имеет состав 2-39,102
K2Na[Co(N02)6], то грамм-эквивалент калия равен--------= 15,64 г. Лучше
5
всего титр раствора AgNOa определить параллельным осаждением стандартного раствора соли калия в виде нитрокобальтиата, который затем растворяют и титруют, как указано выше. «
Осадок нитрокобальтиата калия и серебра (стр. 46) промывают 50%-ным ацетоном, затем 80%-ным ацетоном и растворяют в небольшом объеме HNO3 (1:4) при нагревании на кипящей водяной бане до полного удаления окислов азота. После охлаждения вводят раствор железо-аммонийных квасцов и титруют ионы серебра’ 0,0025—0,01 N раствором роданида аммония до устойчивого розового окрашивания. Титр раствора роданида аммония устанавливают параллельным осаждением и титрованием стандартного раствора K.NO3 [888, 1026, 1616, 2860].
Титрование перхлората калия после его восстановления. Осадок перхлората калия (стр. 30) кипятят около 1 часа с сульфатом трехвалентного титана в присутствии серной кислоты:
С1О4 +8Т1Ш +8Н+—C1- + 8T1IV +4Н2О.
Избыток соли трехвалеитного' титана окисляют раствором перманганата, и ионы хлора титруют по Фольгарду 0,1 N раствором AgNO3. Грамм-эквивалент калия равен 39,102 г [2393, 2624, 2657, 2739].
Восстановление до хлорида производят также сплавлением КСЮ4 с едким натром [2909], с карбонатом натрия [1244, 1735], с нитритом натрия [2720], с карбонатом натрия в присутствии сульфата гидразина [2657] или восстанавливают цинком в сернокислой среде [2532], сульфатом двухвалентного железа в щелочной среде [2567]. Плав растворяют в воде, подкисляют азотной кислотой и титруют ионы хлора по Фольгарду. Применяемые реагенты не должны содержать примеси хлоридов.
5 Аналитическая химия калия
65
Титрование перрената калия. Осадок перрената калия (стр. 32) растворяют в теплой воде, добавляют КгСгО4 и титруют 0,05 А раствором нитрата серебра:
KReO4+AgNO3—»AgReO4+KNO3.
Титрование при взбалтывании продолжают до появления красно-коричневой окраски осадка [1099, 2736]. ‘
Грамм-эквивалент калия равен 39,102 г. При определении около 20 мг калия ошибка достигает 2,5%.
Титрование калий-бортетрафенила. Иои калия осаждают в виде калий-бортетрафенила (стр. 48), промытый осадок растворяют в 5 л.г ацетона [186, 342, 2929], добавляют 10 мл воды 'и 0,1 мл 0,5%-ного раствора КгСгО4 в 50%-ном ацетоне. Полученный раствор титруют 0,01 N раствором AgNO3 в 50%-ном ацетоне:
[В (С6Н5) J-+Ag+—► Ag[B (C6HS) 4].
Когда закончится образование осадка серебряной соли бортетрафенила, тогда небольшая избыточная капля раствора AgNO3 вызывает появление устойчивой буро-красной окраски хромата серебра. Грамм-эквивалент калия равен 39,102 г.
Этот метод рекомендуется для определения калия в удобрениях [755, 1424]. В качестве индикатора при этом титровании предложен ацетат вариамина [1165]. В водно-ацетоновом растворе после подкисления азотной кислотой можно определить бортетрафенил по Фольгарду [2005].
Модификация метода заключается в растворении калин,-бортетрафенила в 10—12 мл ацетона, к раствору добавляют 5 лл 2 У уксусной кислоты, вводят 1 мл 0,1 N раствора КВт и несколько капель 1%-ного раствора эозина и титруют 0,1 А раствором AgNO3 до изменения окраски [1236, 2405, 2981]. Переход окраски без бромида нерезкий. Поэтому в титруемый раствор вводят, как было указано, отмеренное количество бромида калия, который титруется после осаждения бортетрафенила. Отдельным опытом определяют расход раствора AgNO3 на титрование взятого количества бромида, и этот объем вычитают из объема, потраченного на титрование всей пробы [2405].
Таким способом определяют калий в воде [2981], удобрениях [1236] и других объектах [2478].
Растворимость Ag[B(CsH5)4] в ацетоне- довольно заметна, поэтому для растворения осадка калий-бортетрафенила применяют возможно меньшее количество этого растворителя.
Серебро-бортетрафенил в отличие от нитрата серебра растворяется в диэтиловом эфире. Это обстоятельство используется для количественного определения калия.
Осадок калий-бортетрафенила растворяют в 20 мл ацетона, добавляют избыток 0,05 N раствора AgNO3, разбавляют водой до 100 мл, вводят диэтиловый эфир и железо-аммонийные квасцы. При взбалтывании Ag[B(C6H5)4] переходит в эфирный слой, который затем отделяют, а в водном слое титруют избыток нитрата серебра 0,05 N раствором роданида аммония [2405]. См. также [2150а, 1324а].
66
М е р к у р и м е т р и ч е с к и й метод
Хлороплатинат калия (стр. 33) растворяют в горячей воде и кипятят с формиатом натрия:
К2[Р t С16]+2HCOONa—2КС1+2NaCl+2НС1 + 2СО2+ Pt.
Фильтруют, промывают горячей водой, к фильтрату и промывным водам прибавляют 1%-ный раствор дифенилкарбазона в метаноле в качестве индикатора, титруют хлориды 0,01—0,1 раствором Hg(NO3)2, подкисленным азотной кислотой, до устойчивой синей окраски [969]. Грамм-эквивалент калия равен 13,033 г.
Другой меркуриметрический способ заключается в предварительном осаждении калий-бортетрафенила (стр. 48).
Осадок нагревают с избытком 0,1 N раствора ацетата двухвалентной ртути:
К[В(С6Н5)4] + 4Hg(CH3COO)2 + 3H2O^4CH3COOHgC6H5 + СН3СООК+ +ЗСН3СООН+Н3ВО3.
Избыток ацетата ртути титруют 0,1 W раствором роданида аммония в присутствии железо-аммонийных квасцов до устойчивого порозовения [2054]. Грамм-эквивалент калия равен 4,888 г.
К омплек с онометричес кий метод
Осадок перхлората калия восстанавливают до хлорида (стр. 65) и осаждают ионы хлора нитратом серебра. Промытый осадок AgCl обрабатывают раствором K2[Ni(CN)4]:
2AgCl + [Ni(CN)4F-->Ni2+ + 2[Ag(CN)2]- + 2Cl-.
Освободившиеся ионы никеля титруют раствором комплексона III в присутствии мурексида [2608]. Вследствие большого числа операций, каждая из которых может быть причиной потерь, метод не отличается достаточной точностью.
К промытому осадку нитрокобальтиата калия (стр. 39) добавляют 1 мл НС1 (1 : 10), вводят щепотку мочевины и 4 мл воды и кипятят до растворения осадка. В раствор вносят немного ацетата натрия, нагревают, добавляют чскорбиновую кислоту, затем 2 капли смеси равных объемов 0.1 М растворов сульфата меди и комплексона III. В горячий раствор вводят 2 капли 0,05%-ного этанолового раствора а-пиридил-азо-|3-нафтола и титруют 0,01 М раствором комплексона III до перехода фиолетовой окраски в желтую [1221, 1561, 2203].
Значительно проще и, вероятно, точнее следующий вариант.
Осадок нитрокобальтиата растворяют в горячей НС1 (1:20), кипятят до удаления окислов азота, охлаждают, нейтрализуют аммиаком и титруют ионы кобальта 0,01—0,1 W раствором комплексона III в присутствии мурексида [2686]. Рекомендуют также растворить нитрокобальтиат калия в соляной кислоте, как указано выше, нейтрализовать аммиаком, добавить избыток титрованного раствора комплексона III, а также буферный раствор с pH 10. Избыток комплексона Щ титруют раствором сульфата цинка в присутствии эрио-хромчерного Т [2234, 2510]. Вследствие непостоянства состава иитрокобальти-
5*
67
ата калия (стр. 41) необходимо титр раствора комплексона III устанавливать путем осаждения и титрования известных количеств соли калия.
Можно осадить калий в виде K2Na(Co(NO2)6] (стр. 48).
Осадок растворяют при нагревании в небольшом объеме 0,05 N HNO3, охлаждают, добавляют 1—2 г роданида аммония и равный объем ацетона, полученный синий раствор титруют 0,01—0,02 N раствором комплексона III до обесцвечивания [2526].
Осадок калий-бортетрафенила (стр. 48), растворяют в ацетоне, добавляют избыток комплексоната двухвалентной ртути и воду до 200 мл:
[B(CgH5)4]- + 4HgY- + 4Н2О-> 4C6HsHg+ + H3BO3+4HY3 ' + +он-,
где Y — анион комплексона III.
Добавляют 5—10 мл ацетатного буферного раствора с pH 6—6,3, вводят несколько капель метанолового раствора а-пиридил-азо-Р-нафтола, освободившуюся этилендиаминтетрауксусную кислоту титруют 0,1 AI раствором ZnCh до появления оранжевой окраски [1226].
В аналогичном способе осадок калий-бортетрафенила растворяют в N.N'-диметилформамиде, освободившуюся этилендиаминтетрауксусную кислоту титруют 0,1 N раствором соли магния в присутствии эриохромчерного Т [2416].
Через колонку с катионитом пропускают сначала 5%-ный раствор хлорида магния (для перевода катионита в Mg-форму) и промывают водой. Затем через колонку пропускают нейтральный исследуемый раствор со скоростью 5 мл/мин и промывают водой. В фильтрате содержится теперь соль магния в количестве, эквивалентном сумме калия и натрия. Фильтрат нагревают до 80° С, добавляют аммиачный буферный раствор с pH 9, титруют ионы магния раствором комплексона III в присутствии эриохромчерного Т. При определении 0,1—50 мг калия и натрия ошибка не превышает 1% [1286].
Другие комплексонометрические способы определения калия см. Г415, 433, 1773].
Перманганатометрический мет од
Титрование перхлората калия. Перхлорат калия (стр. 30) растворяют в воде (приблизительно 40 мл воды на 0,1 г перхлората) и добавляют 20—60 мл серной кислоты (1 :4). Через колбу с этим раствором пропускают струю СО2 в течение 15 мин. для удаления воздуха и вводят избыток 0,2 N раствора сульфата трехвалентиого титана. Не прекращая пропускания СО2, кипятят 1—1,5 часа. После охлаждения избыток трехвалентного титана титруют 0,5 N раствором перманганата [100, 572, 2619]. Грамм-эквивалент калия равен 4,888 г.
Малую величину грамм-эквивалента можно рассматривать как достоинство метода. Однако определение сложно, продолжительно, титр раствора соли трехвалентного титана легко изменяется при соприкосновении с воздухом.
Титрование хлороплатината калия. Осадок хлороплатината (стр. 33) промывают водой, растворяют в горячей воде и вводят железо в порошке:
K2[PtCl6]+2Fe— 2КС1+2FeCl2 + Pt.
68
Раствор фильтруют, промывают осадок, фильтрат и промывные воды подкисляют серной кислотой; образовавшуюся соль двухвалентного железа титруют 0,01—0,1 N раствором перманганата [1180]. Грамм-эквивалент калия равен 39,102 г.
Этот способ не давал удовлетворительных результатов, что можно объяснить частичным окислением образовавшейся соли двухвалентного железа кислородом воздуха.
Титрование нитрокобальтиата калия. Осаждение калия в виде нитрокобальтиата и последующее перманганатометрическое титрование относится к числу весьма распространенных способов определения калия. Первый вариант этого метода был предложен в 1900 г. [597] и заключается в следующем.
Осадок нитрокобальтиата калия (стр. 39) промывают 10%-ной уксусной кислотой, затем водой и кипятят с разбавленным раствором едкой щелочи:
[Co(N02)6]3- + 30H-^Co(OH)3+6N02 ,
Осадок гидроокиси кобальта можно выделить Также кипячением осадка с раствором бикарбоната натрия [404]. В некоторых модификациях кобальт осаждают в виде фосфата добавлением 1—2 мл 2%-ного раствора Na3PO4 [1579] или Na2HPO4 [957, 1865]. Осадок гидроокиси или фосфата кобальта отфильтровывают и промывают водой, фильтрат подкисляют серной кислотой и титруют 0,1 N раствором перманганата до устойчивого розового окрашивания:
5NO ; + 2МпО ~ + 6Н+^5NO~+2Мп2 + + ЗН2О.
Принимая, что осадок имеет состав K2Na[Co(NO2)6], находим, что грамм-экви-39 1
валент калия равен -1. =6,517 г, и поэтому 1 мл 0,1 N раствора КМпО4
6
соответствует 0,6517 мг калия. В отдельной пробе определяют объем раствора перманганата, уходящий только на подкрашивание титруемого-раствора, и эту величину вычитают щз объема раствора перманганата, израсходованного на титрование образовавшегося нитрита [259].
Аналогичный способ определения калия см. [83, 976].
При любых вариантах перманганатометрического определения осадок можно собрать либо в пористом тигле с асбестовым фильтрующим слоем, либо в стеклянном фильтрующем тигле. При макроопределениях удобно отделять осадок от маточного раствора и промывать его центрифугированием в пробирке.
Значительно более просты модификации метода, в которых не выделяют кобальт в виде малорастворимого соединения и в которых трехвалентный кобальт также участвует в окислении нитрита, поэтому расход перманганата несколько уменьшается [827, 1917, 2036, 2037, 2554, 2778, 2968]. Мы остановимся на одном из распространенных вариантов метода, предложенном для анализа удобрений [1100].
Осадок нитрокобальтиата калия (стр. 39) переносят в стеклянный тирель-с фильтрующим дном и промывают. Тигель вместе с осадком вводят в отмеренный объем 0,1 N раствора перманганата, разбавленного водой приблизи
69
тельно в 10 раз и нагретого почти до кипения. Взбалтывают несколько минут До появления бурого осадка МпО(ОН)г и только тогда подкисляют серной кислотой, добавляют отмеренный объем 0,1 W раствора щавелевой кислоты, в 1 л которого содержится 50 мл конц. H2SO4. При этом раствор обесцвечивается, избыток щавелевой кислоты титруют 0,1 N раствором перманганата. Процесс протекает по следующему суммарному уравнению:
5[Со (NO2) 6]3- +11МпО; +28Н+—5Со2++1 lMn2++30NO;+ 14Н2О.
39,1
Теоретический грамм-эквивалент калия равен —— 5,5
= 7,109 г. Для опреде-
ления малых количеств калия пользуются 0,02—0,005 N растворами перманганата [827, 1917, 1981, 2037, 2778, 2968]. Содержание калия вычисляют по формуле:
Кмг — (^КМпО/^КМпОч- ^Н2С2О4 ЛГН2С2О1),7>109>
где ^кМпО —общий объем введенного раствора перманганата за вычетом объема, уходящего на подкрашивание жидкости, мл;
Vh2c2O —объем раствора щавелевой кислоты, мл;
^КМпО4и ^Н2с2О4 — нормальности растворов перманганата и щавелевой кислоты.
Такой же способ определения калия описан и в ряде других работ [1020, 1579, 1611].
Отметим микромодификацию метода, часто применяемую для анализа биологических объектов [1780, 1782].
В центрифужную пробирку помещают 1 мл исследуемого раствора и 2 мл концентрированного раствора нитрокобальтиата натрия, взбалтывают. Через 45 мин. разбавляют 3 мл воды и центрифугируют. Раствор над осадком осторожно удаляют пипеткой, осадок взбалтывают с 3 мл воды, снова центрифугируют и т. д. до тех пор, пока вода над осадком станет совершенно бесцветной. Обычно достаточно 2—3 промываний. К промытому осадку добавляют из бюретки 2—5 мл 0,01 N раствора перманганата и 1 мл серной кислоты (1 : 5). Пробирку помещают в кипящую водяную баню на 1—2 мин. и перемешивают тонкой стеклянной палочкой. Если введенного объема раствора перманганата оказывается недостаточно и раствор быстро обесцвечивается, то прибавляют еще 1—2 мл раствора перманганата и ?нова нагревают 1—2 мин. в кипящей водяной бане. При этом желтый осадок нитрокобальтиата полностью растворяется, и раствор сохраняет розовую окраску. Теперь вводят 3 мл 0,01 N раствора щавелевой кислоты и обесцветившийся раствор титруют 0,01 W раствором перманганата до устойчивого порозовения. В отдельной пробе определяют объем раствора перманганата, уходящий только на подкрашивание такого же объема жидкости в розовый цвет. Этот объем вычитают из общего объема израсходованного раствора перманганата [259]. Еще лучше титровать параллельно контрольную пробу, т. е. титровать все реагенты, включая щавелевую кислоту, взятые в тех же объемах и в тех же условиях [1579]. В этом случае содержание калия вычисляют по формуле
-7,109, где V—объем раствора перманганата, израсходованный на титрование контрольной пробы, мл;
v — объем раствора перманганата, израсходованный на титрование нитрокобальтиата калия, мл;
N — нормальность раствора перманганата.
Таким способом можно определять 0,01—5 мг калия с удовлетворительной точностью [43, 259, 404, 427, 1542, 1578, 2750]. 70
Перманганатометрический метод позволяет определять до 10~3% калия в магнии и его сплавах [417]. Описан способ определения до 5- 10—4% калия в почве [2818].
Только в отдельных работах отмечаются неудовлетворительные [1049] или пониженные [778, 1055] результаты, получаемые этим методом. В присутствии аммиака или солей аммония определение не может быть точным [2737]. Нежелательно присутствие солей бария, стронция и кальция, которые могут соосаж-даться с солью калия и повышать результаты определения калия [23, 240, 2884].
Перманганатометрически,й метод очень удобен, прост, определения выполняются легко и быстро. Недостаток метода заключается в непостоянстве состава осадка (стр. 41), при вычислениях рекомендуются следующие эмпирические величины:
1 мл 0,01V раство- ра КМпО4 соответ- Литература ствует мг калия: 1 мл 0,01V раствора КМпО4 соответствует мг калия: Литература
0,07109 (теоре- [31, 43, 57, 197, 0,0662 [2524]
тическая вели- 427, 1083, 1100, 0,06515 [1651]
чина> 1669, 1782, 2060, 0,065 [1579, 1686, 1864]
2542, 2899, 2968] 0,0645 [2353]
0,070 [2027] 0,06118 [1917]
0,06901 [208] 0,0567 [2498]
0,06725 [2778]
0,0668 [2037]
По другим данным [404, 1271, 1710, 2023, 2354, 2701], эта величина изменяется в пределах от 0,07109 до 0,049. Фактор пересчета зависит от состава образующегося осадка, который в свою очередь зависит от условий осаждения (стр. 42). Кроме того, эта величина связана и с условиями, при которых производится титрование полученного осадка [281]. Поэтому рекомендуется устанавливать титр раствора перманганата путем одновременного осаждения и титрования калия в определенном объеме стандартного раствора в таких же. условиях. Еще лучше параллельно анализировать раствор или навеску аналогичного объекта с известным содержанием калия.
Определение путем перманганатометрическото титрования осадка нитрокобальтиата калия очень часто применяется при анализе минералов и силикатов [57, 140, 1331], почвы [2, 9, 23, 42, 105, 147, 197, 293, 316, 430, 431, 579, 703, 726, 1686, 1890, 2023, 2281, 2456, 2542, 2610, 2630, 2701, 2727, 2818, 2895], стекла [31], цемента [1417], магния и его сплавов [417], удобрений [1100, 2750], растительных объектов [622, 1669, 2701, 2899], золы растений [789, 957, 2023], пищевых веществ [2044], воды и рассолов [41, 83, 281, 1999, 2296], биологических объектов [43, 143, 259, 590, 778, 834, 1020, 1049, 1061, 1172, 1579, 1706, 1780, 1864,
71
2369, 2615, 2635, 2737, 2751, 2829], других материалов [140, 146, 240, 416, 442, 732, 921, 1118, 1209, 1650, 1652, 1708, 1829, 1977, 2026, 2060, 2065, 2491, 2657, 2709, 2725, 2791].
Принципиально иной способ перманганатометрического титрования заключается в следующем.
Осадок нитрокобальтиата калия растворяют при нагревании в 2 мл 1 А НС1, раствор нейтрализуют аммиаком и осаждают кобальт 4%-ным раствором а-нитрозо-р-нафтола. Осадок отфильтровывают, промывают и прокаливают. Полученную окись кобальта восстанавливают водородом при 600° С до металла, охлаждают, не прекращая пропускания водорода. Кобальт растворяют затем в растворе фосфорномолибденовой кислоты:
Со+МоОз—>СоО+МоО2.
Образовавшаяся двуокись молибдена окрашена в интенсивный синий цвет. После полного растворения кобальта синий раствор титруют 0,01 N раствором перманганата до обесцвечивания:
5МоО2+2МпО ~ +6Н+^5МоО3+2Мп2+ + ЗН2О.
Грамм-эквивалент калия равен 39,102 г.
Авторы метода [1054] определяли 0,24—1,05 мг калия в пробе с ошибкой не более 3%. Следует отметить сложность и продолжительность этого определения.
Титрование нитрокобальтиата калия и серебра. К нейтральному разбавленному раствору соли калия прибавляют 0,02 N раствор AgNO3 и медленно при взбалтывании вводят 25%-ный раствор нитрокобальтиата натрия в избытке. Спустя 30 мин. осадок K2Ag[Co(NO2)6] отфильтровывают через тигель Гуча, промывают его охлажденной водой и вносят в 1 N серную кислоту, добавляют определенный объем 0,1 N раствора перманганата калия, нагревают до растворения, вводят известный объем 0,1 N раствора щавелевой кислоты и титруют 0,1 N раствором перманганата до порозовения [888]. Содержание калия вычисляют по такому же уравнению, как при титровании K2Na[Co(N02)6] (стр. 70).
Титрование нитрокобальтиата калия и свинца. 50 мл раствора, содержащего 20—100 мг калия, смешивают с 5—10 мл реагента (см. ниже) и оставляют на 15—20 мин. К выпавшему осадку КР,Ь[Со(К02)б] после промывания добавляют избыток 0,1 N раствора перманганата, серную кислоту .(1:3), через несколько минут вводят избыток 0,1 N раствора щавелевой кислоты, который титруют 0,1 N раствором перманганата [464, 878].
5[Со (N02) 6]з - + 11 МпО 4- + 28Н +—>5Со2+ +11 Мп2+ + 30N О +14Н2О.
Теоретический грамм-эквивалент калия равен 3,554 г.
Для приготовления реагента растворяют 22 г нитрита натрия, 3 г нитрата кобальта и 5 г нитрата свинца в 80 мл воды и добавляют 5 мл конц. СНзСООН, дают отстояться и раствор сливают с осадка [464].
Вариант метода заключается в предварительном осаждении кобальта раствором едкой щелочи.
Осадок гидроокиси кобальта удаляют, фильтрат и промывные воды подкисляют серной кислотой, прибавляют избыток 0,1 N раствора перманганата и т. д. В этом случае грамм-эквивалент калия равен 3,258 г [464].
Предлагается также осадок КРЬ[Со(ЫОг)б] обрабатывать серной кислотой, раствор выпаривать до удаления серной кислоты и прокаливать.
72
Из остатка извлекают сульфат кобальта водой, раствор фильтруют, выпаривают до небольшого объема, осаждают кобальт добавлением концентрированного раствора щавелевой кислоты и этанола. Выпавший осадок оксалата кобальта растворяют в серной кислоте и титруют 0,1 N раствором перманганата. Грамм-эквивалент калия равен 19,55 г [464].
Этот способ, требующий .выполнения большого числа операций, менее удобен и, вероятно, менее точен.
Состав осадка может изменяться в зависимости от условий осаждения [266], поэтому эмпирический грамм-эквивалент следует определять параллельно по стандартным растворам соли калия.
Титрование нитрокобальтиата калия и свинца. Раствор, содержащий 20—25 мг калия, выпаривают до небольшого объема, добавляют по каплям при помешивании 10—15 мл реагента (см. ниже), взбалтывают 15—20 мин. Выпадает крупнокристаллический темно-зеленый, почти черный осадок К2РЬ[Со(ЬЮ2)б], который промывают декантацией водой, растворяют в горячей воде, добавляют избыток 0,1 N раствора перманганата и серную кислоту (1:3). Затем вводят избыток 0,1 А раствора Н2С2О4 и титруют 0,1 А раствором перманганата:
5[Co(NO2)6]4~ + 12МпО.4“+36Н+->5Со2++ Г2Мп2+ +3ONO3+ 18Н2О*
Теоретический грамм-эквивалент калия равен 6,617 г. Вследствие неточного соответствия состава выпадающего осадка приписываемой ему формуле грамм-эквивалент калия оказывается несколько меньшим — 6,521 г [465, 515], по другим данным,—• 6,517 г [877].
Реагент для осаждения готовят растворением 11,5 г нитрата свинца в малом объеме горячей воды, охлаждают, добавляют 1,5 г нитрита натрия,. 10 г нитрата кобальта и воду до 100 мл. Через некоторое время раствор декантируют с выпавшего осадка. Для повышения чувствительности к полученному раствору добавляют этанол [121].
Титрование малораствори.мых ферроцианидов. Исследуемый раствор, содержащий хлорид кальция, смешивают с избытком1 реагента, состоящего из раствора 7 г ферроцианида натрия и 3,5 г хлорида кальция в 95 мл воды и смешанного с 80 мл 96%-ного этанола. Выпадает осадок К2Са[Ре(СМ)б], который отфильтровывают, промывают смесью воды и этанола, затем водой, растворяют в разбавленной серной кислоте и титруют 0,05— 0,1 А раствором перманганата [493, 1295, 1296, 2174]:
5[Fe(CN) 6]4- +МпО ; + 8Н+—5[Fe(CN) 6]3- +Мп2+ +4Н2О.
Рекомендуется в качестве индикатора вводить раствор метилового фиолетового [493]. Грамм-эквивалент калия равен 78,2 г.
Соли аммония, дающие аналогичный осадок, а также соли тяжелых металлов мешают определению. Не мешают соли натрия и магния. Метод применялся при анализе удобрений [2174].
Описан аналогичный метод, основанный на образовании осадка 4K4[Fe(CN)6] • 5Cd2[Fe(CN)6] или иначе КштСФл • [Fe(CN)6] [237, 494].
К 20 мл 0,1 М раствора сульфата кадмия прибавляют исследуемый раствор КО, вводят 20 мл 0,1 Af раствора ферроцианида лития и разбавляют водой до 149 мл. Взбалтывают 15 мин., добавляют 1 мл 1 М. раствора СаС12 и
73-
фильтруют. 100 мл фильтрата подкисляют серной кислотой и титруют 0,1 N раствором перманганата. Чем больше калия содержалось в анализируемом растворе, тем .меньше расходуется перманганата на титрование. Заранее строят график зависимости между содержанием калия в растворе и расходом перманганата на титрование.
Определению мешают ионы аммония, рубидия, цезия, тяжелых металлов.
Титрование пикрата калия. Промытый осадок пикрата калия (стр. 51) растворяют в воде и разбавляют до 100 мл. Аликвотную часть раствора, содержащую 1—1,5 мг калия, переносят в другую колбу, добавляют 40—'60 мл 0,1 N раствора перманганата, нагревают до кипения, вводят 12 мл серной кислоты (1 : 7) и 4 мл 1%-ного раствора сульфата марганца. Кипятят 15 мин., добавляют 40—60 мл 0,1 N раствора щавелевой кислоты, избыток которой титруют 0,1 N раствором перманганата [569, 1550, 1553]: 5С6Н|2(МО2)зОК + 28КМп04+37Н2804—l15KNO<i+9KiSO4+28Mn&O4+30CO2+ +42Н2О.
Грамм-эквивалент калия равен
39,102
28
= 1,396 г.
Хроматометрический метод
Осадок нитрокобальтиата калия обрабатывают определенным объемом Ю,01—0,1 N раствора бихромата калия в 40%-ной серной кислоте при нагрева-нии на водяной бане до полного растворения:
6[Со (N О2) 6]3- +11 Сг2О2 ~+82Н+—►6Со2+ + 22Cr3+ + 36NO |+41Н2О.
После охлаждения избыток бихромата титруют 0,01—0,1 N раствором соли Мора в присутствии фенантролина [820]. Параллельно в тех же условиях проводят контрольный опыт, титруя такой же объем раствора бихромата без нитрокобальтиата. Содержание калия вычисляют по формуле:
= -Э,
тде V—объем раствора соли Мора, израсходованный на титрование в контрольном опыте, мл;
v — объем1 раствора соли Мора, израсходованный на титрование пробы, мл;
N — нормальность раствора соли Мора; ..
Э — эмпирический грамм-эквивалент калия, определяемый осаждением и» титрованием известного количества соли калия. .
Теоретический грамм-эквивалент калия равен 7,109 г. >
Избыток бихромата можно определять также иодометриче- j ским методом [337, 821]. Другие работы о хроматометрическом i определении калия см. [821, 1605, 1857, 2707].
Периметрический метод !
Осадок нитрокобальтиата калия (стр. 39) промывают 70%-ным этанолом и высушивают при 80° С. К осадку добавляют избыток 0,01—0,02 N раствора 'CefSCUh в 1 N серной кислоте и нагревают на водяной бане до полного оаст-ворения:
[Co(NO2)e]3-+llCe4++6H2O^Co2+ + llCe3+ + 6NO; + 12H+.
74
После охлаждения титруют избыток четырехвалентного церия разными способами.
а. Избыток четырехвалентного церия титруют раствором соли Мора:
Се4+ + Fe2+ ->Се3+ + Fe3+
в присутствии фенантролина до появления устойчивой розовой окраски [866, 1568, 1697]. Соль Мора можно заменить раствором сульфата двухвалентного железа, подкисленным серной кислотой, индикатор — эриохромглауцин [746, 1616].
б. Избыток четырехвалентного церия определяют иодометрически, добавляя к раствору иодид калия:
2Ce4+ + 2J-->2Ce3++J2,
и выделившийся иод титруют 0,01 N раствором тиосульфата в присутствии крахмала [1688, 2288, 2592].
В каждом из вариантов параллельно в таких же условиях титруют такой же объем. раствора Ce(SO4)2 без нитрокобальтиата [1688]. Эмпирический грамм-эквивалент калия равен 6,95 г. Метод дает удовлетворительные результаты.
Известны другие варианты периметрического определения нитрокобальтиата калия [143, 958, 1003, 1159, 1452, 2015, 2027, 2357, 2358, 2716, 2725, 2737].
Описан способ осаждения 0,1—200 мкг калия в виде нитрокобальтиата калия и серебра с последующим периметрическим титрованием [848, 1730]. Погрешность определения около 2%. Этот метод рекомендуется для определения калия в морской воде [2350, 285 Г].
Отметим метод, основанный на окислении калий-бортетрафенила, растворенного в ацетоне, раствором NH4[Ce(NOs)5] в 2 N НС1О4 при 92—97° С, избыток соли четырехвалентного церия затем титруют раствором соли Мора в 2 N серной кислоте в присутствии ферроина [2616].
Титанометрический метод
Исследуемый раствор, содержащий КС1, помещают в центрифужную пробирку и осаждают избытком раствора дипикриламината магния. Выпавший ди-пикриламинат калия промывают насыщенным при 0° С раствором сульфата магния. Осадок затем растворяют в смеси равных объемов ацетона и воды, этим же раствором разбавляют с таким расчетом, чтобы в 1 мл раствора содержалось не более 10 мкг калия. Коническую колбу заполняют СО2, вводят определенный объем титрованного раствора сульфата трехвалентного титана и исследуемый ацетоновый раствор:
2KN[C6H2 (NO2) зЬ+36Ti2 (SO4) з+37H2SO4—2HN[C6H2 (NH2) 3]2 + 72Ti (SO4) 2+ + K2SO4+24H2O.
Через 5 мин. избыток соли трехвалентного титана титруют раствором железо-аммонийных квасцов в присутствии роданида аммония. В таких же усло
75
виях параллельно проводят контрольный опыт, заменяя раствор дипикриламината калия водой [565].
Достоинством метода является очень малый грамм-эквивалент калия, который, как видно из приведенного уравнения, разд {о2
вен д6 =1,086 г. Недостаток метода — способность трехвалентного титана легко окисляться кислородом воздуха, поэтому титрование производят с указанной выше предосторожностью и устанавливают нормальность раствора сульфата трехвалентного 4. титана одновременно с определением калия.
На таком же принципе основано определение в осадке калиевой соли 2-хлор-3-нитротолуол-5-сульфокислоты (стр. 54):
C7H5Cl(NO2)SO3K + 3Ti2(SO4)3+3H2SO4 -> C7H5C1(NH2)SO3K+ + 6Ti(SO4)2 + 2H2O.
Погрешность определения 0,02—0,1 г калия достигает 6% [2896].
Аналогичный метод заключается в восстановлении дипикриламината калия титрованным раствором соли двухвалентного ванадия, избыток которого титруют затем раствором железо-аммонийных квасцов в присутствии сафранина [116, 565]. Другой титанометрический способ основан на осаждении КСЮ4 (стр. 30). 4
Осадок растворяют в 10 мл насыщенного раствора щавелевой кислоты, добавляют 10 мл конц. H2SO4 и 10 мл титрованного раствора Т1С1з-’
сю;+8Ti3+ + 8H+^Cl- + 8Ti4++4H2O.
Нагревают 5 мин., охлаждают, добавляют роданид аммония и избыток трехвалентного титана титруют раствором железоаммонийных квасцов [1743] или
раствором FeCk [2375]. *
Йодометрический метод
I
Титрование йодата калия. Исследуемые соли калия и натрия 'S
переводят в иодиды выпариванием с конц. HJ, раствор выпаривают досуха; иодид натрия извлекают смесью безводных изобутанола и диэтилового эфи- «
ра. Остаток иодида калия растворяют в воде, подкисляют фосфорной кисло- &]
той и добавляют хлорную воду в избытке-. При этом иодид окисляется до йодата: j
2J—4-CI2—►J2+2C1~; I
J2+6C12+6H2O—2HJO3+10HC1.
Кипятят для удаления избытка хлора, охлаждают и вводят иодид калия в нз- ।
бытке: f
JO; +5J"+6H+—-3J2+3H2O. 7
Выделившийся иод титруют 0,01 раствором тиосульфата [1487]. Грамм-эк- :
внвалент калия равен 6,517 г. 1
Титрование метапериодата калия. Осадок KJO4 (стр. 29) S
растворяют в 125 мл буферного раствора с pH 7,5 (5 г борной кислоты и 5 г (
тетрабората натрия в 125 мл воды) и добавляют около 3 г иодида калия:
JO7+2J- +Н2О—►ЛО;+Л2+2ОН_1
76
Выделившийся иод титруют 0,1 N раствором NagAsOg в присутствии крахмала [1035, 1927, 2083, 2426, 2903]. Грамм-эквивалент калия равен 19,550 г. Таким путем можно определять 0,7—80 мг калия с погрешностью около 0,1 мг [2903]. Вариант метода заключается в растворении осадка KJO4 в 10 лл 4 W серной кислоты, добавлении к раствору 25 мл воды и избытка иодяда калия:
JO; +7J" + 8Н+—>4J2+4H2O.
Освободившийся иод титруют 0,1 N раствором тиосульфата в присутствии крахмала [1653, 2021, 2290]. В этом случае грамм-эквивалент калия равен 4,888 г. Таким путем можно определять до 0,1 мг калия [2021].
Следует отметить, что KJO4 непосредственно способен реагировать с тиосульфатом [931]:
jo; +8S20f +8H+->J-+4S40r +4Н2О;
JO; + S2Of+H2O^J~ + 2SO*- + 2H+.
Аналогичная реакция протекает с избытком сернистой кислоты [1819]:
jo;+4sor->j-+4sor. 4 О 4
Метапериодат восстанавливается до иодида амальгамой магния [2109]. Возможность применения этих реакций для определения калия еще не изучалась.
Титрование нитрокобальтиата калия. Йодометрическое определение калия можно рассматривать как вариант описанного на стр. 69 перманганатометрического способа [597].
Ионы калия осаждают в виде нитрокобальтиата, осадок отделяют от маточного раствора центрифугированием и этим же способом промывают его. К промытому оса.дку добавляют 3—5 мл 0,01 N раствора перманганата и 0,25—0,5 мл 25%-ной серной кислоты. Если при стоянии на холоду в течение 30 мин. раствор обесцвечивается, то вводят еще отмеренный объем раствора перманганата и снова выжидают 30 мин. Для определения избытка перманганата вводят 1 мл 10%-иого раствора KJ и выделившийся иод титруют 0,01 N раствором тиосульфата в присутствии крахмала. Одновременно в таких же условиях титруют такой же объем раствора перманганата без ннтро-кобальтиата. Понятно, что на это титрование расходуется больший объем раствора тиосульфата [2972]. Содержание калия вычисляют по уравнению
KMS=(V-v)N-3,
тде V — объем раствора тиосульфата, израсходованный на титрование в контрольном опыте, мл-,
v — объем раствора тиосульфата, израсходованный на титрование анализируемой пробы, лл;
N — нормальность раствора тиосульфата;
Э — эмпирический грамм-эквивалент калия.
Все сказанное о грамм-эквиваленте калия при перманганатометрическом определении (с>р. 71) относится и к этому способу. Величину эмпирического грамм-эквивалента калия лучше всего устанавливать одновременно в тех же условиях путем осаждения калия из определенного объема стандартного раствора КС1 и последующего титрования в описанных выше условиях. Метод
77
находит применение при анализе биологических объектов [834, 1061, 1865]. Варианты этого метода см. [143, 834, 1061, 1393, 1651, 1714, 1865].
Для йодометрического определения можно воспользоваться способностью нитрокобальтиата калия непосредственно реагировать с иодидом калия в солянокислой среде:
2[Со (NO2) б]3- + 14 J" + 24Н+ ->7J2 + 2Со2+ + 12NO + 12Н2О.
Выделившийся иод титруют раствором тиосульфата в присутствии крахмала [340]. Если считать, что осадок имеет состав 39 102 K2Na[Co(NO2)6], то грамм-эквивалент калия равен —-— = 3,5
= 11,71 г, однако лучше устанавливать грамм-эквивалент эмпирически. Метод очень прост, но следует помнить, что образующаяся при реакции окись азота легко окисляется кислородом воздуха до двуокиси, которая снова выделяет иод из иодида калия.
Титрование т р и т и о с у л ь ф а т о в и с м у т и а т а калия. Йодометрическое определение калия по Ks[Bi (520з)з] было предложено в 1878 г. [928].
Исследуемый раствор, содержащий 15—225 мг КО, выпаривают досуха, остаток охлаждают и добавляют ивбыток свежеприготовленного реагента. Последний представляет собой смесь 1 объема 2 М раствора тиосульфата натрия и 2 объемов 0,33 М раствора нитрата висмута, подкисленного азотной кислотой (другой способ приготовления реагента см. [370]). К этому раствору добавляют большой избыток 96%-ного этанола до толучення смесн 80%-ной по этанолу (добавляют 4—5-кратный объем этанола). Спустя 2 часа фильтруют через стеклянный фильтр № 3, промывают 80%-ным этанолом до получения бесцветного промывного раствора и обрабатывают раствором едкой щелочи. Осадок гидроокиси висмута промывают на фильтре водой. Фильтрат нейтрализуют соляной кислотой н титруют 0,1 W раствором иода в присутствии крахмала [51, 52, 375].
Состав осадка подвержен колебаниям и не точно отвечает приведенной выше формуле. Осадок всегда содержит примесь соответствующей соли натрия, что делает метод определения калия мало точным [1815]. В указанных выше условиях выпадает осадок K5Na[Bi(S2O3)3]2 [51, 52, 415]. Титр раствора иода следует устанавливать в параллельном опыте со стандартным раствором КС1. Разработаны другие варианты определения калия в виде тритиосульфатовисмутиата [1008, 1023].
Титрование калий-бортетрафенила. Осадок калий-бортетрафенила (стр. 48) несколько раз промывают водой и растворяют на фильтре в 10 мл 5%-ного горячего раствора ацетата двухвалентной ртути в ледяной уксусной кислоте:
К[В(С6Н5)4] + 4Hg(CH3COO)2 + 3H2O-S4CH3COOHgC6H5 + СН3СООК +
+ЗСН3СООН+Н3ВО3.
Фильтруют, промывают 3—4 раза ледяной уксусной кислотой, прибавляют 2 мл конц. НС1 (выпадает фенилмеркурхлорид) и вводят отмеренный объем 78
бромид-броматной смеси, при этом осадок бромируется и растворяется:
ClHgC6H5+Br2—ClHgCeHjBr+HBr.
Избыток бромата определяют иодометрически. Для этого добавляют 10 мл 10%-ного раствора KJ, разбавляют водой, выделившийся иод титруют 0,1 N раствором тиосульфата. Грамм-эквивалент калия равен 4,888 г [2109].
Калий-бортетрафенил способен реагировать с иодом по уравнению [436]:
К[В (С6Н5) 4] + J2 ->KJ + C6H5J + (С6Н5) Зв.
Эта реакция еще не применялась для количественного определения калия. Другие способы йодометрического определения калия см. [967].
Другие методы
титриметричес к о г о определения калия
Титрование раствором метиленовой синей. 1 мл исследуемого раствора, содержащего хлорид или перхлорат калия, смешивают е 20 мл этанолового раствора пикрата кальция. На'Другой день осадок отфильтровывают, промывают 4%-ным раствором этанола в диэтиловом эфире, последняя промывная жидкость должна быть бесцветной. Промытый осадок пикрата калия растворяют в 5 мл горячей воды, охлаждают, добавляют 5 мл хлороформа и титруют при взбалтывании 0,01 N водным раствором метиленовой синей в воде (3,74 г/л).
Образующийся пикрат метиленовой синей растворяется в хлороформе, окрашивая его в зеленый цвет, а желтый водный раствор бледнеет. В точке эквивалентности водный слой оказывается бесцветным, и одна лишняя капля титрованного раствора окрашивает его в синий цвет. Титр раствора метиленовой синей устанавливают титрованием определенного объема стандартного раствора пикриновой кислоты или пикрата калия [798, 2027]. Вариант этого метода см. [799].
Необходимый для работы раствор пикрата кальция готовят кипячением 24 г пикриновой кислоты и 10 г СаСОз со 100 мл воды. Раствор фильтруют, фильтрат выпаривают досуха, остаток растворяют в 100 мл этанола и фильтруют.
Титрование раствором сульфата цинка. Исследуемый раствор смешивают с 10 мл 0,1 Af раствора нитрата кальция, 20 мл 0,1 М раствора сульфата кадмия, разбавляют водой до 200 мл и прн помешивании вводят 20 мл 0,1 М раствора ферроцианида натрия. Взбалтывают 1 мин., разбавляют водой до 250 мл. Через несколько минут центрифугируют. К 200 мл центрифугата добавляют 10 мл конц. H2SO4, 4 г сульфата аммония, 2—3 капли 1%-ного раствора феррицианида калия, несколько капель раствора дифениламина в серной кислоте и избыток ферроцианида титруют 0,05 Af раствором сульфата цинка до появления сиреневой окраски. Зависимость между содержанием калия в пробе и расходом раствора сульфата цинка устанавливают заранее путем аналогичного титрования нескольких стандартных растворов хлорида калия и построенного на этом основании калибровочного графика.
79
Метод применен для определения калия в сильвинитах [238].
Отметим аналогичный метод, основанный на образовании K2Ca[Fe(CN)e] в 50%-ном этаноле и титровании в присутствии нитрата кобальта (внутренний индикатор) [1075, 1298] или молибдата аммония, подкисленного уксусной кислотой, в качестве внешнего индикатора [1299] для определения избытка ферроцианида.
Титрование раствором тиосульфата натрия. Осадок •хлороплатината калия (стр. 33) растворяют в Г—2 мл горячей воды, добавляют 1 мл 2 N раствора KJ и нагревают до кипения. При этом образуется иодоплатинат, отличающийся интенсивной красной окраской:
[Р t С16]2 - + 6J-—[Pt J6]2- + 6С1-.
Горячий раствор титруют 0,01 N раствором тиосульфата натрия до перехода красной окраски в светло-желтую [2098, 2549]:
[Pt С16]2~ + 2S2Of—[PtJ4 [2“+2.1' + S4Of.
Грамм-эквивалент калия равен 39,102 г.
Титруемый раствор должен быть нейтральным, поэтому растворяют хлороплатинат калия в буферном растворе с pH 7; растворение даже в горячем буферном растворе протекает медленно и иногда требует нескольких часов [2128]. Индикатором служит сам иодоплатинат, следовательно, целесообразно растворять К2[Р1С16] в возможно меньшем объеме воды или буферного раствора. Для уменьшения конечного объема пользуются концентрированным раствором KJ [1457]. Метод отличается достаточной точностью и чувствительностью. Удается определять 10’4— 10-5 мг-экв калия в пробе [992, 1349, 1352, 2128, 2563, 2871]. Описаны микро- и ультрамикромодификации определения калия путем титрования иодоплатината раствором тиосульфата [223, 1349, 2128]. Метод применяется для определения калия в биологических объектах [992, 1432, 1457], воде [1975], почве [2] и удобрениях [2563].
Титрование раствором сульфам и новокислого калия. Осадок нитрокобальтиата калия (стр. 39) смешивают с избытком 0,002 М раствора сульфаминовокислого калия и 1 лл 2 Д' соляной кислоты и нагревают на водяной бане до 80° С:
no; +h2nso;+н+—n2+hso; +Н2О.
Избыток сульфаминовокислого калия титруют раствором NaNO2 до тех пор, пока капля жидкости даст положительную реакцию с KJ-крахмальной бумажкой. Отдельно устанавливают соотношение между растворами сульфаминовокислого калия и нитрита натрия [1355]. .Еще лучше установить титр раствора NaNO2 по стандартному раствору КС1.
Применение внешнего индикатора и малая распространенность одного из реагентов делают этот метод мало пригодным.
Титрование раствором хлорной кислоты. Осадок калий-бортетрафенила (стр. 48) промывают, высушивают и растворяют при нагре-.80
вании в безводном ацетоне. Добавляют несколько капель 0,2%-кого раствора кристаллического фиолетового в безводном ацетоне (индикатор) и титруют раствором HCIO4 до перехода ярко-фиолетовой окраски в синюю. Титрованный раствор готовят смешиванием 0,6 мл 70%-ной хлорной кислоты с 200 мл лед. СНзСООН и 3 мл уксусного ангидрида. Титр раствора устанавливают по навескам калий-бортетрафенила в таких же условиях.
Погрешность при определении 2—8 мг калия не превышает 1% [1220].
Титрование раствором бромида цетил-три-метиламмония. Из исследуемого раствора осаждают калий в виде калий-бортетрафенила. Избыток осадителя титруют в центрифугате или фильтрате раствором бромида цетил-триме-тиламмония в присутствии бромфенолового синего или титанового желтого в ка-честве индикатора [1163, 2451]. Вместо бромида цетил-триметиламмония можно применять растворимые соли других четвертичных аммониевых оснований [1163].
Кондуктометрическое определение
Известен ряд способов кондуктометрического титрования солей калия.
Титрование раствором хлорной кислоты. Метод основан на том, что в этаноловом растворе едкие щелочи ведут себя как слабые основа-
ния. При кондуктометрическом титровании раствором НСЮ4 вследствие образования малорастворимого перхлората в первую очередь титруется едкое кали, затем едкий натр.
К 5 мл раствора, содержащего около 1—5 ммоля хлоридов калия и натрия, добавляют влажную свежеприготовленную окись серебра, взбалтывают 10 мин. и фильтруют в мерную колбу емкостью 250 мл. Осадок промывают 5 мл воды. Затем этанолом и 5 мл насыщенного раствора перхлората калия, разбавляют этанолом до 250 мл. В этом растворе находятся едкие кали и натр, аликвотную часть раствора титруют кондуктометрически 0,1 Л' раствором НСЮ4 в этаноле. На кривой титрования (рис. 2) видны два излома, соответствующие нейтрализации КОН и NaOH [2133, 2134].
I , - I______I_______1_______ь
О 2 * S 8
OJ N раствор Н С10 4 в этаноле. мл
Рис. 2. Кривые кондуктометрического титрования смеси 50 мл 0,005 А' КОН (/) и 50 мл 0,005 N NaOH в этаноле 0,1 N раствором НСЮ4 в этаноле (2)
Большой расход этанола является недостатком метода. Можно также осадить калий из нейтрального раствора избытком титрованного раствора хлорной кислоты в присутствии этанола, 6 Аналитическая химия калия 8!
избыток хлорной кислоты кондуктометрически титруют щелочью [2759].
Титрование раствором перхлората натрия. Исследуемый раствор, охлажденный до +2° С, помещают в сосуд для определения электропроводности и разбавляют водой до 50 мл. Сосуд помещают в толченый лед и выжидают несколько минут до достижения постоянства электропроводности. Раствор титруют из микробюретки 8 W раствором1 NaClOi. После прибавления каждой порции титрованного раствора выжидают 1 мин. и производят отсчет, около точки эквивалентности выжидают 2—3 мин. Титр раствора NaClO.» устанавливают в таких же условиях по стандартному раствору КС1 [57, 1631, 2228]
Метод рекомендуется для определения калия в его солях. Для уменьшения растворимости перхлората калия в титруемый раствор вводят метанол, этанол, ацетон или глицерин [2862].
Титрование раствором кремнефтористоводородной кислоты. Нейтральные растворы солей калия кондуктометрически титруют раствором H2[SiF6] в среде 50%-ного этанола [419, 420]:
2КС1 + H2[S iF6] -*Ki[SiF6] + 2HCl.
Так как соляная кислота сильнее кремнефтористоводородной, то в точке эквивалентности наблюдается перелом кривой титрования. Погрешность определения около 2% [419] обычно в сторону уменьшения [143].
Титрование раствором нитрокобальтиата натрия. Описано кондуктометрическое титрование кадия раствором нитрокобальтиата натрия [2665]. Недостаток метода —малая стабильность титрованного раствора, вследствие чего его приходится готовить и устанавливать каждый день заново по стандартному раствору КС1. Определение затрудняется, если исследуемый раствор содержит большие количества натрия.
Титрование раствором ферроцианида кальция. Нейтральный 0,015—0,06 N раствор соли калия смешивают с таким же объемом 96%-ного этанола и избытком ацетата кальция. Титруют из микробюретк» 0,3—1 М раствором ферроцианида кальция. Конечную точку устанавливают кондуктометрически по возрастанию электропроводности раствора. Раствор ферроцианида кальция стандартизируют по раствору соли калия в таких же условиях. Погрешность определения около ±2% [819].
Титрование раствором пикрата натрия. В сосуд для титрования помещают определенный объем 0,1 Л' раствора пикрата натрия, вводят щепотку сухого пикрата калия и затем прибавляют исследуемый раствор-хлоридов калия и натрия. Избыток пикрата натрия титруют кондуктометрически 0,1 ;V раствором хлорида калия. Раствор пикрата натрия стандартизируют по раствору хлорида калия.
Способ позволяет определять 50—150 мг хлорида калия в присутствии 10—65 мг хлорида натрия, растворенные в возможно меньшем объеме воды [527].
82
Определение сульфатов щелочных металлов и магния
Раствор, содержащий сульфаты калия, натрия и магния, титруют раствором Ва(ОН)2. В первую очередь протекает реакция с сульфатом магния с образованием двух малорастворимых соединений:
MgSO4+Ba(OH)2-Mg(OH)2+BaSO4.
Вследствие удаления ионов Mg2+ и SOJ~ электропроводность раствора уменьшается (рис. 3). При дальнейшем прибавлении Ва(ОН)2 начинают реагировать сульфаты щелочных металлов:
M2SO4+Ba(OH)2-»BaSO4+2MOH.
Теперь электропроводность возрастает, так как вместо сульфат-
ионов в растворе появляются более подвижные гидроксильные ионы. По окончании этого процесса и появлении в растворе избытка Ва(ОН)2 наблюдается новое, но не резко выраженное увеличение электропроводности. График на рис. 3 позволяет легко установить объем раствора Ва(ОН)2, израсходованный на титрование сульфата магния. Объем этого раствора, потраченный на титрование сульфатов щелочных металлов,- установить труднее, здесь наблюдается только слабый перегиб кривой. Поэтому после достижения первой точки перегиба про-
Рис. 3. Кривая кондуктометрического титрования смеси сульфатов магния, калия и натрия раствором гидроокиси бария [389].
должают титрование раство-
ром Ва(ОН)2, чтобы получить часть восходящей кривой и этим определить положение точки А (рис. 4). После этого титруют раствором ацетата бария:
M2SO4+Ba(CH3COO)2->BaSO4+2CH3COOM.
Вместо сульфат-ионов в растворе теперь находятся ацетат-ионы, что приводит к уменьшению электропроводности. По окончании этого процесса и появлении в растворе избытка ацетата бария электропроводность вновь возрастает. По кривой находят расход реагентов на титрование сульфата магния и отдельно на титрование суммарного количества сульфатов калия и натрия.
Для уменьшения погрешности вследствие большой адсорбционной способности выпадающих осадков и медленного измене-
6*
83
Рис. 4. Кривая кондуктометрического титрования смеси сульфатов магния, калия и натрия растворами гидроокиси и ацетата бария [389].
ния электропроводности рекомендуется титровать в водно-этано-ловой среде раствором Ва(ОН)2> к которому добавлено большое количество магнезона II. Этанол и магнезон II уменьшают адсорбцию ионов из исследуемого раствора и реагентов. Нормальность раствора Ва(ОН)2 устанавливают титрованием раствором соляной кислоты; нормальность раствора ацетата бария — кондуктометрическим титрованием навесок сульфатов магния и калия. Метод предложен для анализа воды и рапы [389].
Описаны и другие способы количественного определения, основанные на кондуктометрическом титровании растворов солей калия:
1) титрование раствором H2[PtCl6] в присутствии большого количества этанола [ПОЗ];
2) титрование раствором натрий-бортетрафенила
' [1626, 2618] или лучше ли-
’ тий-бортетрафенила [2279] при pH 5—10 в случае анализа золы, удобрений [95, 1823];
3) титрование дипикриламината калия, растворенного в ацетоне и воде, 0,1 N раствором соляной кислоты или нафталин-1-сульфокислоты [418, 419, 1712, 2476, 2919];
4) титрование растворами литиевой соли 2,4-динитронафтил-аминобензолсульфокислоты [1155];
5) титрование растворами рениевой кислоты [2736];
- 6) титрование растворами винной кислоты [204] и др. [1381,
.2912].
Кондуктометрический метод был применен для определения состава некоторых комплексных солей, например КгРЬХ^, -К[РЬХз], где X — ион галоида [1058].
Потенциометрическое определение
1. Калий осаждают в виде хлороплатината, промытый осадок -растворяют в горячей воде и кипятят с формиатом натрия (стр. 38). Осадок отфильтровывают и промывают, в фильтрате -определяют ионы хлора потенциометрическим титрованием -0,01 М раствором AgNO3[1027, 2564] или Hg(NO3)2 [1027]. Рекомендуется также восстанавливать хлороплатинат магнием в по
84
рошке (стр. 37), а затем титровать ионы хлора потенциометрически 0,01 N раствором AgNO3 в присутствии ацетона [2153].
2. Осаждают калий раствором ферроцианида кальция в виде K2Ca[Fe(CN)6], избыток ферроцианида титруют в фильтрате потенциометрически раствором сульфата цинка. Способ позволяет определять 40—130 мг калия с ошибкой около 0,5 мг [57, 366, 512, 1785, 1836, 2292]. Варианты метода заключаются в титровании в присутствии этанола по образованию 4K4[Fe(CN)6] • 5Cd2[Fe(CN)g] [491] и аналогичной соли никеля [492].
3. Осадок калий-бортетрафенила (стр. 48) растворяют в ацетоне, добавляют равный объем воды и титруют потенциометрически 0,01—0,001 N раствором AgNO3 при потенциале 200 мв. Таким способом можно определить 0,04—0,15 мг калия [1469, 1471] и большие количества [1472, 1766] в водно-ацетоновом растворе. Относительная нормальность раствора AgNO3 зависит от концентрации ацетона в смеси [1679]. Предлагается в титруемый водно-ацетоновый раствор вводить ацетатный буферный раствор с pH 4,8 [1014, 1784]. Вариант этого метода заключается в осаждении калия титрованным раствором натрий-бортетрафенила, избыток которого титруют потенциометрически [1537]. Метод применялся для определения калия в фармацевтических препаратах и в солях калия [1173, 1725].
Описано потенциометрическое титрование дипикриламината калия раствором соли трехвалентного титана [1709], титрование избытка дипикриламината магния 0,1 N раствором соляной кислоты [108, 188]. Другие потенциометрические методы определения калия см. [555, 1956—1959].
Ь- Амперометрическое определение
Титрование раствором натрий-бортетрафенила. К раствору соли калия прибавляют ацетатный буферный раствор с pH 5,6 и титруют при +0,08 в 3%-ным раствором натрий-бортетрафенила, титр которого установлен по стандартному раствору КС1 при тех же условиях [629, 2881].
Метод предложен для Определении калия в силикатах [2573].
Титрование раствором нитрата серебра или одновалентного таллия. Осадок калий-бортетрафенила растворяют в смеси 15 мл ацетонитрила и 5—8 мл воды и титруют амперометрически при —0,1 в водным 0,03 N раствором AgNO3.
1—20 мг калия определяют со средней погрешностью около 1,5% [1206]. Избыток натрий-бортетрафенила титруют также раствором T1NO3 [1701, 2475].
Титрование раствором днникриламнната натрия. Исследуемый раствор смешивают с фосфатным буферным раствором (pH 11,5— 12), раствор охлаждают льдом и при —1,6 в титруют 0,07—0,1 М раствором дипикриламината натрия. Способ позволяет определять 4—120 мг калия в 10 мл раствора с погрешностью не выше 0,4%.
85
Соли лития и натрия не мешают определению [2434]. Метод предложен для определения калия в силикатах. Аналогично определяют в присутствии боратного буферного раствора с pH 9; способ позволяет определять 1 мг калия при наличии 17—400 мг натрия [1645].
Другие титриметрические методы определения калия см. [2095].
Газометрическое определение
Определение по реакции хлороплатината калия с йодноватой кислотой. Осадок хлороплатината калия (не более 0,2 г) растворяют в 10 мл воды, добавляют около 1 г чистой кристаллической йодноватой кислоты и нагревают при взбалтывании до кипения. В момент закипания нагревание прекращают, добавляют 30 мл 95%-ного этанола, взбалтывают и оставляют на 1—2 часа. При этом выпадает трииодат калия, нерастворимый в этаноле:
K2[PtCl6] 4- 6ШОз—2КНД 4О3)з+H2[PtCld.
Осадок отфильтровывают, промывают нейтральным 95%-ным этанолом до нейтральной реакции. Фильтр с осадком отжимают между листами фильтровальной бумаги, помещают в прибор для выделения и измерения газа и вводят 50 мл 2%-ного водного раствора сульфата гидразина:
2KH2(.IOl1)3+9N2H4 • H2SO4—>9N2+K2SO4-f-8H2SO44-i6iHJ+ I8H2O.
По объему выделившегося азота можно вычислить количество калия в пробе [2340]. Измеренный объем 'азота приводят к нормальным условиям (0° С и 760 мм рт. ст.).
1 мл азота при 0° С и 760 мм рт. ст. соответствует 0,38 мг калия.
Азот выделяется (в других количествах) и при непосредственном действии сульфата гидразина на хлороплатинат [128]: 2K2[PtCl6] + N2H4-H2SO4^2K2[PtC14] + N2 + 4HCl + H2SO4.
Определение по реакции нитрокобальтиата с солью двухвалентного железа. Промытый осадок нитрокобальтиата калия (стр. 39) помещают в эвдиометр с концентрированным раствором сульфата двухвалентного железа в 2 N серной кислоте:
[Со (NO2) в]3" + 7Fe2+ + 12Н+—-Со2+ + 7Fe3+ + 6NO+6Н2О.
Объем окис .1 азота, выделившегося при этой реакции, приводят к 0° С и 760 мм рт. ст.
Если осадок имеет состав K2Na[Co(NO2)e], то 1 г-иону калия соответствует 3 г-молекулы окиси азота или, другими словами, 1 мл окиси азота при нормальных условиях соответствует 0,580 мг калия [93, 106, 134, 1629, 2722]. Соотношение между единицей объема окиси азота и количеством калия лучше всего установить параллельной обработкой стандартного раствора соли калия. Продолжительность определения — около 50 мин. Метод предложен для определения калия в почве [134]. Возможно газометрическое определение калия путем аналогичной обработки осадка КгМа[Со(ЫО2)б] [2860].
86 •
Определение по реакции нитрокобальтиата калия с сульфаминовой кислотой. Осадок нитрокобальтиата калия растворяют при нагревании в небольшом объеме 5%-кого раствора Na3PO4 или Na2HPO4. Раствор помещают в нитрометр, где нитрит вступает во взаимодействие с избытком 5%-него раствора сульфаминовой кислоты:
HNO2+H2N • SO3H—N2+H2O+H2SO4.
I мл азота при нормальных условиях соответствует 0,580 мг калия [952]. Определение по реакции нитрокобальтиата калия с мочевиной. Осадок нитрокобальтиата калия обрабатывают щелочью и удаляют осадок гидроокиси кобальта. К полученному раствору, помещенному в аппарат Ван Слайка, добавляют мочевину:
6NO' +3Co(NH2)2—ЗСО|" +6H2O + 6N2.
1 мл азота при нормальных условиях соответствует 0,580 мг калия.
Метод позволяет определять 0,07—0,45 мг калия с погрешностью не более 0,01 мг [1781].
Определение по реакции битартрата калия с бикарбонатом. Осадок битартрата калия (стр. 50) растворяют в воде и добавляют борную кислоту. Образующаяся боровинная кислота при взаимодействии с бикарбонатом натрия выделяет СОг, объем которой измеряют и пересчитывают на калий [2436].
Определение на основе термического разложения перхлората калия. При температуре выше 510° С перхлорат калия разлагается с выделением кислорода £1967]:
КСЮ4->КС1+2О2.
На этом основан газометрический метод определения калия (115]. 1 мл кислорода при 0°С и 760 мм рт. ст. соответствует 0,873 мг калия.
Косвенные методы определения
Выше приводились некоторые методы выделения и одновременного количественного определения калия и натрия без их предварительного разделения. Выделение производится чаще всего в виде хлоридов (стр. 24) или сульфатов (стр. 26). В ряде случаев знание суммарного количества калия и натрия оказывается недостаточным и возникает вопрос о дополнительном раздельном определении калия и натрия. Это можно сделать следующими способами: прямым определением калия (навеску смеси солей растворяют, в полученном растворе определяют калий осаждением в виде перхлората, хлороплатината, нитрокобальтиата и других солей с гравиметрическим, титриметриче-ским, фотометрическим окончанием); косвенными методами, к описанию которых мы переходим.
87
Косвенные методы можно разделить на группы в зависимости от характера исходных солей калия и натрия. Однако во всех случаях задача сводится к решению системы двух уравнений с двумя неизвестными. Эти методы не отличаются достаточной—- точностью. Небольшая влажность исходной навески, присутствие незначительных количеств примесей, небольшие ошибки взвешивания и другие факторы могут заметно отразиться на полученных результатах. Чем выше значения постоянных коэффш циентов в уравнениях для вычисления (см. ниже), тем больше возможная ошибка определения.
Известно суммарное количество хлоридов калияинатрия
а. Гравиметрическое определение хлорида в виде хлорида серебра. Навеску смеси хлоридов растворяют в воде, и содержание хлорида определяют гравиметрическим методом в виде AgCl [15, 323, 349, 663, 1016, 1181, 1203, 1257, 1774, 1915, 1968, 2111, 2916]. Допустим, что навеска хлоридов калия и натрия равна А г, содержание в ней калия в граммах обозначим через х и содержание натрия — через у. Допустим также, что из этой навески после ряда операций получено В г хлорида серебра,- Сказанное щие уравнения:
позволяет составить
следую-
КС1 , NaCl
К Na
AgCl х AgCl
К Na
// = Л;
У=В.
(1)
(2)
Na
Из уравнения (2) находим, что у —---------—
AgCl
Это значение у вводят в уравнение (1):
КС1 к
AgCl
= А,
откуда
= 2.4281Л— 0,9904В.
х = К AgCl
КС1 - NaCl
б. Определение хлорида титрованием раствором нитрата серебра. Это один из наиболее старых косвенных методов определения калия [2046]. Навеску смеси хлоридов калия и натрия растворяют в воде и содержание хлорида
(3)
88
определяют титрованием по Мору [349, 984, 1181, 1266, 1590, 1915, 2007, 2046, 2105, 2916] или по Фольгарду [-349, 2789]. Допустим, что исходная навеска смеси хлоридов калия и натрия равна А г и что на титрование раствора этой навески в воде израсходовано V мл раствора нитрата серебра с нормальностью, равной N. Если содержание калия в навеске равно х, а натрия — У, то
КС1 . NaCl , ,,,
— * + -— У = А\ (I)
К Na
= VN. (2}
10-3К 10-3Na
Решая эти уравнения относительно х, находим:
х = К(Л-10-3№С1-^у) = 2Д281—0,1419WV. (3)
Определение путем перевода в сульфаты. Навеску смеси хлоридов калия и натрия выпаривают с конц. H2SO4 (стр. 25), прокаливают, охлаждают и взвешивают полученную сумму сульфатов калия и натрия [15, 1797, 2713].
Обозначив навеску хлоридов через А, количество полученных сульфатов — через В и содержание в них калия и натрия в граммах соответственно через хну, получаем:
откуда
КС1 , NaCl
•--- х Н-------
К • Na
У. = А,
2SO4 , Na2SO4 _ 2Na У
K(Na2SO4-^ — 2NaCl-5)
Na2SO4-KCl—KaSO4-NaCl
13,7174— 11,288В.
(1}
(2)
(3>
х —
Определение путем перевода в нитраты. Навеску хлоридов калия и натрия несколько раз выпаривают с конц. HNO3, высушивают и взвешивают полученную сумму нитратов [2046, 2274].
Приняв прежние обозначения, получаем:
КС! . NaCl . ...
--- х---------У = А, (1)
К Na v Л
KNO3 v NaNO3
---- х п-------У
К Na
(2)
откуда
х = KCNaNO^-X-NaCl-B) = 7 772Д _5 344В (3)
KCl-NaNOs-KNOs-NaCl ’ ’
Определение путем перевода в хлорид натрия. Навеску А г смеси хлоридов калия и натрия растворяют в воде и раствор про
8ft
пускают через колонку с катионитом в натриевой форме, затем промывают водой, фильтрат и промывные воды выпаривают, остаток высушивают, охлаждают и взвешивают.
Теперь навеска состоит только из хлорида натрия, новый вес равен В г. Количество калия в первоначальной навеске в граммах обозначим через х, количество натрия — через у. На основании сказанного составляют два уравнения [1284]:
х + ^аЯ у = А', К Na
NaCl . NaCl D
--- x 4-----у ~ В.
К Na
(1)
(2)
Вычитаем второе уравнение из первого:
KCI NaCi
Л> ---/к
к к
-откуда х= КИ--В) А~В
КС1 — NaCl 0,4144 ’
Аналогичным способом можно пропустить раствор навески хлоридов калия и натрия через катионит в калиевой форме, в этом случае уравнение (2) приобретает следующий вид:
+ (2а)
К Na '
Из уравнений (1) и (2а) находим, что
х = L&'л - Na2^ = 2,4284 — 1,903В. (За
Известно суммарное количество сульфатов калия и натрия
Гравиметрическое определение сульфата в виде сульфата бария. Навеску смеси сульфатов калия и натрия растворяют в воде, и содержание сульфата определяют гравиметрическим методом в виде BaSO4 [155, 156, 189, 323, 663, 1422, 1751, 1797, 1880, 2007, 2713]. Если вес суммы сульфатов калия и натрия равен А г, полученный вес сульфата бария— В г и если количество калия и натрия (граммы) в навеске обозначить через х и
K2SO4 2К
У, то
Na^ д
1 ОМ. ’ \ /
BaSO4 , BaSO4 - х 4 - у = 2К---------------2Na а
'90
откуда
2К (BaSO4-Д — NaaSO4-B) BaSO4 (K2SO4 — Na2SO4)
= 2,4281A—1,4777В.
(3)
Прочие, косвенные методы
1. Определение, основанное на титровании (стр. 58) навески смеси KjSiF6] и Na2[SiF6] раствором едкого натра [2642].
2. Определение по плотности раствора, полученного растворением определенной навески смеси хлоридов калия и натрия в постоянном количестве воды [987, 1274].
3. Определение по показателю преломления раствора 1 г смеси хлоридов калия и натрия в 10 мл воды [142, 887, 2548, 2571] (см. приложения, стр. 171).
4. Кондуктометрическое титрование раствора навески сульфатов калия и натрия раствором ацетата бария в присутствии равного объема этанола [204].
5. Прокаливание навески нитратов калия и натрия с навеской двуокиси кремния и определение потери веса при этом 11788].
6. Определение по понижению температуры известного объема насыщенного раствора хлорида натрия при растворении в нем известной навески смеси хлоридов калия и натрия [825, 1001, 1794] (см. стр. 123).
7. Определение по электропроводности раствора 5 г смеси хлоридов калия и лития в 1 л воды [2969].
Некоторые косвенные методы определения калия в присутствии натрия см. [162, 1234, 1746, 1754, 2169, 2466, 2627, 2831], в присутствии рубидия или цезия [708, 779, 970, 1525].
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ
Нефелометрическое определение
Определение в виде нитрокобальтиата калия. В несколько одинаковых пробирок помещают по 5 мл анализируемого раствора и отдельно по 5 мл стандартных растворов КС1 или KNO3, содержащие 1—12 мкг калия в'1 мл. Во все пробирки вводят по 0,5 г NaCl и после его растворения добавляют по 0,3 г нитрокобальтиата натрия, взбалтывают до растворения реагента; через 30 мин. помутнение, получившееся в исследуемом растворе, сравнивают с помутнением стандартных растворов [20, 279, 284, 300].
Для ускорения образования помутнения и увеличения чувствительности определения к исследуемым и стандартным растворам добавляют одинаковые количества 85—95% этанола [284, 690, 716, 840, 1354, 1971, 2150], изопропанола [840, 1059, 2160, 2936] или смесь метанола, пропиленгликоля и глицерина [2726].
91
С целью увеличения стабильности помутнения вводят 0,25 %-ный раствор гуммиарабика [2936].
Для определения калия в почвах рекомендуется следующий способ [429].
Навеску исследуемого образца обрабатывают 1 Л' раствором NaCl. Из этого экстракта отбирают в мерные цилиндры два разных объема. К меньшему из них добавляют определенное количество стандартного раствора соли калия (например, 0,2 мг калия) и объем раствора в обоих цилиндрах доводится до одинакового уровня введением 1 W раствора NaCl. К обоим растворам добавляют одинаковые количества раствора нитрокобальтиата натрия, перемешивают; более мутный раствор разбавляют 1 N раствором NaCl, к которому предварительно добавлено такое же количество нитрокобальтиата натрия, до уравнения интенсивности помутнения. В этом случае справедливо ]
следующее отношение: ’
хУг -f- а _ xV2
откуда
У2Г1— VjWi’ I
где х — количество калия в исходном растворе, мг'мл; ' ' I
а — количество добавленного калия, мг;
V, и V2 — начальные объемы анализируемого раствора, мл;
Wi и — конечные объемы при одинаковой мутности, мл.
Более точный метод заключается в измерении интенсивности помутнения на фотоколориметре [890, 2691], фотометре [840] либо, спектрофотометре [943] при 610 [1135], 650 [2936] или 710 ммк [943, 2160].
Для получения воспроизводимых результатов выполняют определение при постоянном значении pH. К исследуемым растворам добавляют буферный раствор или готовят вытяжку из анализируемого объекта (чаще всего из почвы) на буферном ацетатном растворе с pH 4,8 [1059, 2207, 2936], pH 3—5 [1107] или pH 6 [890]. Колебания температуры в пределах 12—20° С [391] и даже 15—35° С [2160] не отражается на результатах нефелометрического определения калия. Небольшие количества солей кальция, магния, сульфата не мешают определению [2726].
При избранных условиях выполняют реакции с несколькими i стандартными растворами соли калия и измеряют оптическую плотность или процент светопоглощения на фотометре при выбранном светофильтре. На основании полученных результатов строят калибровочный график, пользуясь которым находят содержание калия в анализируемом растворе, с которым произве- । дены такие же операции.
Нефелометрическое определение калия в виде нитрокобальтиата рекомендуется для анализа вытяжек из почвы [2, 284, 325, 391, 734, 837, 840, 1059, 1113, 1135, 1354, 1787, 2115, 2203, 2207, 2726, 2816], удобрений [685, 1059], воды [83, 279, 325, 2150], солей i
92
натрия [690, 1719], биологических объектов [1396, 2725, 2726] и других материалов [273, 300, 309, 1107, 1526, 1684, 1971, 2813].
Описаны упрощенные способы количественного определения калия, основанные на получении помутнения от выпадающего нитрокобальтиата калия. Порции исследуемого раствора разбавляют в различных отношениях и находят такое разбавление, при котором добавленный раствор нитрокобальтиата натрия не вызывает появления мути; содержание калия находят по заранее определенным эмпирическим данным [391].
Рекомендуется турбидиметрическое определение калия в удобрениях по образованию K2Ag[Co(NO2)e] [326].
Определение в виде калий-бортетрафенила. При добавлении раствора натрий-бортетрафенила к растворам соли калия образуется малорастворимый калий-бортетрафенил (стр. 48). При малых количествах калия появляется помутнение, интенсивность которого сравнивают с помутнениями серии стандартов [531, 2696]. Более точны варианты этого метода, основанные на измерении интенсивности возникающего помутнения при помощи фотоколориметра [2673] или фотометра [2402]. Метод рекомендуется для определения 4—80 мкг калия в почвах, пищевых продуктах, биологических объектах. Отметим следующий вариант метода.
К 0,2 мл исследуемого раствора добавляют равные объемы 20%-ного раствора трихлоруксусной кислоты, 25%-ного раствора ацетата натрия и стандартного раствора натрий-бортетрафенила (0,5 мг/мл) и центрифугируют. Прозрачный центрифугат помещают в кювету фотометра, избыток осадителя титруют раствором бромида цетил-триметиламмония. При этом образуется муть малорастворимого цетил-триметиламмоний-бортетрафенила. Измерения производят при 400 jujuk. По мере добавления цетил-триметиламмония оптическая плотность раствора возрастает; титруют из микробюретки до практически постоянной оптической плотности.
Метод предложен для определения калия в 0,2 мл сыворотки [1679, 1963].
Возможно нефелометрическое определение калия в форме фосфоромолибдата [2008].
Фотометрическое определение
Среди фотометрических методов определения калия особенно многочисленны и разнообразны способы, основанные на предварительном осаждении калия в виде хлороплатината и нитрокобальтиата.
Определение по окраске раствора хлороплатината калия. Осадок хлороплатината калия (стр. 33) растворяют в горячей воде, охлаждают, разбавляют водой до определенного объема. Раствор окрашен в желтый цвет, интенсивность окраски зависит от количества хлороплатината. Однако интенсивность желтой окраски в разбавленных растворах хлоро
93
платината калия весьма невелика. Значительно большей интенсивностью отличаются растворы этой соли в некоторых органических растворителях. На этом основан способ фотометрического определения калия. Осадок хлороплатината калия растворяют в возможно меньшем объеме смеси бутанола и этилацетата, оптическую плотность полученного раствора измеряют при 264 ммк [1133].
Определение по окраске раствора иодоплатинат а-Осадок хлороплатината калия промывают этанолом. После удаления этанола, выжидают до полного испарения растворителя и растворяют осадок в горячей воде. К охлажденному раствору добавляют каплю конц. НС1 и большой избыток 0,5 М раствора KJ. Для полного протекания реакции между хлороплатинатом и иодидом требуется 3—4 часа. При нагревании максимальная интенсивность окраски наблюдается через несколько минут. Раствор разбавляют водой до 25 или 50 мл, полученный розовый раствор сравнивают в колориметре со стандартным раствором хлороплатината, обработанным таким; же способом [914, 2062].
Оптимальные количества калия, определяемые этим способом,—1 —10 мкг [914], по другим данным — 5—50 мкг [1034,. 1591]. Высокая чувствительность этого метода отмечается многими исследователями [196, 595, 2128]. Погрешность определения: около 6% [914], при очень малых количествах калия — около 10% [1824]. Точность возрастает при измерении оптической плотности при 496 ммк [595, 1794]. Растворы подчиняются закону Ламберта-—Бера. Фотометрическое определение калия в виде иодоплатината применяется при анализе цемента [1235], почвы [914, 1937], биологических объектов [1794].
В присутствии этанола раствор иодоплатината становится желтым. На этом основана следующая модификация метода.
Осадок хлороплатината промывают этанолом и, не ожидая улетучивания остатка этанола, растворяют в горячей воде; к горячему раствору добавляют большой избыток 0,5 N раствора KJ и немного этанола. Вначале появляется розовая окраска, которая через несколько минут становится светло-желтой. Интенсивность желтой окраски сравнивают с окраской стандарта, полученного таким же способом [196, 914, 2018]. Один и тот же раствор можно сначала колориметрировать по розовой окраске, затем по желтой окраске. К. розовому раствору, полученному и исследованному, как указано выше, добавляют немного этанола, нагревают до получения желтой окраски и снова; колориметрируют. Более точные результаты получают колориметрированием, по розовой окраске [1824]. Розовый раствор .после колориметрирования можно еще и оттитровать раствором тиосульфата (стр. 80) [2549]. Кроме того, оттитрованный тиосульфатом раствор подкисляют соляной кислотой, добавляют несколько капель 0,2%-кого раствора перекиси водорода. При этом [PtJJ2-, образовавшийся в результате титрования, снова окисляется до [PtJ6]2- с появлением розовой окраски. Раствор оставляют на 0,5—1 час, разбавляют водой до 25—50 мл и сравнивают со стандартом.
Этот способ применялся для определения калия в биологических о'бъёктах [2421, 2700]. К недостаткам метода следует отнести способность иодоплатината разлагаться с выделением иода 94
под действием света [1794] или большого количества соляной кислоты [914].
Определение по окраске восстановленной платины. Промытый осадок хлороплатината растворяют на фильтре в 30 мл горячей соляной кислоты (1 : 1), промывают фильтр горячей водой и раствор охлаждают. Сюда добавляют 3 мл раствора хлорида олова (75 г SnCl2-2H2O в 400 мл конц. НС1). В зависимости от количества хлороплатината появляется желтая или бурая окраска, вызванная восстановленной коллоидной платиной [2933]. Через 15—20 мин. разбавляют водой до 100 мл и еще через 15 мин. сравнивают со стандартом, полученным одновременно таким же способом [2, 196, 1549, 1824].
Реакция очень чувствительна, она позволяет обнаруживать до 0,1 мкг платины в 1 мл раствора и поэтому удается определять калий даже при концентрациях порядка 1 : 106 [1549]. Этому методу посвящен ряд работ [381, 462, 770, 1372, 2107].
Большое число способов определения калия основано на предварительном осаждении нитрокобальтиата с последующим, получением азокрасителей. В этих способах в качестве стандарта применяется раствор NaNCh, получаемый следующим способом.
В горячей воде растворяют 0,405 г AgNO2, добавляют раствор NaCl в небольшом избытке, затем после охлаждения разбавляют водой до 1000 мл. После выделения хлорида серебра на дно колбы раствор фильтруют. 29,16 мл этого раствора разбавляют водой до 1000 мл. В 1 мл последнего раствора находится количество нитрита, соответствующее 1 мкг калия, если считать, что осадок имеет состав K2Na[Co(NO2)6] [443, 621, 2729].
Можно приготовить стандарт непосредственно из NaNOa, если есть уверенность в его чистоте.
Навеску 1,339 г чистого сухого NaNO2 растворяют в воде и разбавляют до • 1000 мл. Отмеривают 10 мл этого раствора и разбавляют водой до 1000 мл. 10 мл последнего раствора соответствует 25 мкг калия [1705]. Однако правильнее готовить стандарт для сравнения из определенного количества калия в растворе, который осаждают в тех же условиях, как и из исследуемого раствора; полученные осадки нитрокобальтиата калия обрабатывают одновременно и одинаковым способом и затем сравнивают полученные окраски в колориметре. Если в распоряжении имеется фотометр, спектрофотометр или фотоколориметр, то заранее составляют калибровочный график. Данные для построения графика получают обработкой разных известных количеств калия раствором нитрокобальтиата натрия и последующим взаимодействием осадка нитрокобальтиата калия с соответствующими реагентами. Исследуемый раствор анализируют в таких же условиях.
Определение по р_еа_кции Грис с а. Один из старейших методов определения калия заключается во взаимодействии нитрокобальтиата калия с реактивом Грисса [857, 1084].
Для определения используется осадок нитрокобальтиата калия (стр. 39) и такой же осадок, приготовленный в таких же условяих из стандартного раствора соли калия. К каждому осадку добавляют по 5 мл реактива Грисса, нагревают 2 мин. в кипящей водяной бане. Полученные красные растворы переносят в мерные колбы емкостью 100 мл, добавляют еще по 5 мл реактива
95-
Грисса, охлаждают, разбавляют водой до метки, н окраски обоих растворов сравнивают в колориметре [1095].
Отметим другой вариант этого метода.
Осадок нитрокобальтиата калия растворяют в 0,5 мл 2%-кого раствора едкого натра при нагревании на кипящей водяной бане, разбавляют водой до 100 мл в мерной колбе. 5 мл этого раствора смешивают с 5 мл реактива Грис- . са, добавляют воду до 25 мл и оптическую плотность полученного раствора измеряют при зеленом светофильтре [2693, 2947].
Реактив Грисса применяется для определения калия и другими авторами [621, 1118, 1271, 1523, 1652, 1824, 1869, 2725, 2744]. Метод отличается высокой чувствительностью, поэтому для каждого определения требуется очень малое количество анализируемого объекта, например достаточно 0,01—0,1 мл сыворотки [1095, 2947]. Относительная погрешность определения не превышает 3% [1095].
Аналогичное определение предлагается и в случае осаждения калия в виде K2Ag[Co(NO2)e] [704, 847]. Этот способ применен при анализе биологических объектов [2201, 2461, 2692, 2941] и воды [2354].
Отметим метод осаждения калия в виде K2Ca[Ni (МО2)б] с последующим применением реактива Грисса [285, 324].
Необходимый для определения реактив Грисса готовят следующим способом.
1 г а-нафтиламина кипятят со 100 мл воды, добавляют 750 мл 10%-ной уксусной кислоты и смешивают с раствором 2,5 г сульфаниловой кислоты в 750 мл 10%-ной уксусной кислоты.
Определение по реакции с а-нафтиламином. Осадок нитрокобальтиата калия промывают смесью этанола и диэтилового эфира (1 : 1), затем эфиром. Осадок быстро высыхает, к нему добавляют 5 мл 5%-ного раствора МагНРО4, нагревают 5 мин. на водяной бане и центрифугируют для удаления осадка фосфата кобальта. 0,1 мл прозрачного центрифу-гата смешивают с 1 мл 1 %-него этанолового раствора а-нафтиламина, добаз-ляют 5 мл этанола и 2 капли конц. НС1. Появляется фиолетовая окраска, максимальная интенсивность достигается через 30 мин. Оптическую плотность измеряют при желто-зеленом светофильтре. Метод позволяет определять 1 — 100 мкг калия [1705, 2633].
Определение по реакции с а-нафтиламином и хлор-гид ратом новокаина. Осадок нитрокобальтиата калия растворяют в 0,5 мл охлажденной льдом 95%-ной серной кислоты при взбалтывании и затем добавляют полунасыщенный раствор ацетата натрия до 10 мл. 1 мл этого раствора разбавляют водой до 10 мл. 1 мл последнего раствора, в котором -содержится 0,01 часть первоначального количества растворенного нитрокобальтиата, смешивают с 14 мл реагента, через 30—45 мии. измеряют оптическую плотность красного раствора на фотометре. Реагент готовят смешиванием 5 мл 3%-ного раствора хлоргидрата новокаина в 15%-ной уксусной кислоте и 5 мл раствора 0,2 г а-нафтиламина в 30 мл конц. СН3СООН и разбавляют водой до 150 мл.
Этим способом можно определять 1,5—22 мкг калия [1271, 1648, 1652, 1824].
Определение по реакции с а-нафтиламином и /г-нит-ро анилином. Осадок нитрокобальтиата калия растворяют в 0,5 мл 95%-ной серной кислоты, охлажденной льдом, и разбавляют водой до 10 мл.
96
1 мл этого раствора разбавляют ведой до 100 мл, добавляют 0,2 мл реагента и 1 мл серной кислоты (1:1) и через 15 мин. колориметрируют. Реагент готовят растворением 0,5 г и-нитроанилина и 0,7 г а-нафтиламина в 100 мл метанола [2974].
Определение по реакции с сульфаниловой кислотой и фенолом. Промытый осадок нитрокобальтиата калия растворяют при нагревании в 5 мл 0,1 А раствора едкого натра (осадок гидроокиси кобальта не мешает), раствор разбавляют водой до 50 мл, добавляют 2 мл смеси равных объемов 10%-ной соляной кислоты и сульфанилово-фенолового реагента. Через 15 мин. подщелачивают 10%-ным раствором аммиака, разбавляют водой до метки. Желтый раствор сравнивают со стандартом, полученным аналогично из соли калия.
Метод отличается удовлетворительной точностью. Он рекомендуется для определения калия в минералах [339, 443].
Реагент готовят растворением 1 г сульфаниловой кислоты в 100 мл насыщенного при нагревании раствора хлорида аммония и добавлением раствора из 1,5 г фенола в 100 мл воды [1889].
Предлагается вариант метода, в котором калий осаждают в виде КгА§[Со(ЫО2)б] и фенол заменяют (3-нафтолом [2201] или сульфаниловую кислоту заменяют глицином [1882].
Определение по реакции с (3-наф.толом и нафтионатом натрия. Осадок нитрокобальтиата калия нагревают в чашке при 70° С с небольшим объемом воды. Как только осадок растворится, сливают содержимое чашки в мерную колбу емкостью 25—100 мл и раствор разбавляют водой до метки. Аликвотную часть этого раствора помещают в колбу емкостью 100 мл, разбавляют водой приблизительно до 80 мл, вводят 1 мл реагента и 4 капли конц. НС1. Через 30—45 мин. добавляют 1 мл конц,- NH4OH и воду до метки. Полученный красный раствор сравнивают со стандартом.
Метод позволяет определять 1 —1000 мкг калия с ошибкой около 3% [1271, 2339, 2729]. Описаны и другие фотометрические методы определения калия с использованием реакции диазотирования [592, 1891]. Известны способы колориметрического определения калия, основанные на нитрозировании некоторых органических соединений азотистой кислотой, полученной из нитрокобальтиата калия.
Определение по реакции с индолом. Осадок нитрокобальтиата калия растворяют при нагревании на кипящей водяной бане в 5 мл 0,1 N раствора едкого натра. Раствор охлаждают, разбавляют водой до 98 мл, вводят 1,0 мл раствора индола и 1 мл серной кислоты (1:1). Появляется красно-фиолетовая окраска нитрозоиндола, которую сравнивают в колориметре с окраской стандарта [2945, 2946]. Оптическую плотность раствора можно измерять при зеленом светофильтре [2477].
Индоловый способ рекомендуется для определения калия в биологических объектах [2477] и почве [316, 684, 1271, 2945, 2946].
Реагент готовят растворением 0,15 г индола в 10 мл этанола и разбавлением водой до 100 мл.
Метод позволяет определять 10—300 мкг калия с вполне удовлетворительной точностью [1663, 2946]. По некоторым данным,
7 Аналитическая химия калия
97
погрешность определения достигает 10%' [684] и точность' метода не признается достаточной [1523].
Определение по реакции с антипирином. Нитрокобальтиат калия растворяют при нагревании в 1 мл 5%-ного раствора едкого натра, центрифугируют, раствор сливают, осадок промывают водой 3 раза по 1 мл. Центрифугат и промывные воды нейтрализуют серной кислотой и разбавляют водой до 7 мл. Сюда добавляют 3 мл 5%-ного водного раствора антипирина и 1 каплю конц. H2SO4. Образуется нитрозоантипирин, зеленую окраску которого сравнивают с окраской стандарта, полученного таким же способом [2075]. Погрешность определения — около 3%. Метод не отличается высокой чувствительностью.
Определение по реакции с бихроматом. Осадок нитрокобальтиата калия промывают 0,01 N азотной кислотой, к осадку добавляют 5 мл 0,1 N раствора бихромата калия и 2,5 мл конц. H2SO4. Перемешивают н охлаждают до комнатной температуры:
6[Со (NO2) 6]3~ +11 Cr2O7a“+82H+—6Со2+ 4-22Сг3++ 36NO;+41Н2О.
Раствор разбавляют водой до 50 мл и образовавшуюся соль трехвалентного хрома определяют фотометрически [2901] или фотоколориметрически [859] с красным светофильтром. Параллельно такой же опыт производят со стандартным раствором КС1.
Можно также определять избыток бихромата фотометрически при 425 ммк [2841].
/ Недостатком всех способов фотометрического определения ! калия по нитриту является окисление нитрита во время раство-^-рения осадка нитрокобальтиата [549].
Определение путем окисления нитрокобальтиата калия до нитрата. Нитрокобальтиат калия окисляют до нитрата при помощи хлората натрия в дымящей серной кислоте. Полученный нитрат нитрует фенолдисульфокислоту; затем желтую окраску аммиачного раствора образовавшейся нитрофенолдисульфокислоты сравнивают со стандартом [1158]. Этот метод более сложен и не имеет преимуществ перед другими фотометрическими способами определения калия.
Для описанных ниже способов фотометрического определения калия по кобальту, содержащемуся в осадке нитрокобальтиата калия, необходим стандартный раствор соли кобальта.
Сульфат кобальта сушат при 160° С, при этом получается COSO4 • Н2О. Навеску 0,485 г этого препарата растворяют в воде, добавляют 5 мл 1 N серной кислоты и разбавляют водой до 1000 мл. 1 мл этого раствора соответствует 0,20 мг калия [1134]. Растворяют 3,043 г хлорида кобальта, СоСЬ-бНгО, в воде, добавляют 5 мл 5 N соляной кислоты и воду до 1000 мл. 1 мл этого раствора соответствует 1,0 мг калия [2554]. Можно готовить стандартный раствор и из других солей кобальта [1621, 2975].
Определение по реакции с конц. НС1. Осадок нитрокобальтиата калия растворяют в 0,1 мл 27%-ной соляной кислоты, нагревают для удаления окислов азота. После охлаждения разбавляют 27%-ной соляной кислотой до 2 мл. Получают [C0CI4]2- или [C0CI3 • НгО]-, окрашивающие раствор в зеленовато-синий цвет, раствор сравнивают со стандартов. Последний готовят растворением 0,304 г хлорида кобальта в 27%-ной соляной кислоте и разбавлением этой же кислотой до 100 мл. 1 мл полученного раствора соответствует 1.0 мг калия. Для получения растворов меньших концентраций разбавляют 27%-ной соляной кислотой в известное число раз. Таким способом готовят
98
серию стандартов, соответствующую 0,01—2 мг калия. Пробирки запаивают, в таком виде стандарты сохраняются очень долго [1842]. Необходимую здесь 27%-ную соляную кислоту получают смешиванием 700 мл конц. НО уд. веса. 1,19 с 300 мл воды.
Метод позволяет определять 0,03—2 мг калия со средней погрешностью около 10% [1842]. Метод применен для определения калия в крови [1055, 1824, 1842, 2320]. Интенсивность окраски усиливается и чувствительность определения повышается, если кроме соляной кислоты вводить еще и этанол [521].
Определение по реакции с роданидом. Нитрокобальтиат калия растворяют в нескольких каплях конц. HCI, раствор выпаривают досуха, остаток растворяют в 1 мл воды, добавляют 0,5 мл насыщенного раствора роданида аммония, 1 мл 2,5%-ного раствора Na2HPO4 (для маскировки железа) и 10 мл смеси равных объемов ацетона и амилового спирта. Синюю окраску, которая при этом появляется, сравнивают в колориметре со стандартом или же измеряют оптическую плотность на фотоколориметре или фотометре с желтым светофильтром.
Таким способом можно определять 0,1—2 мг калия [1053, 1121, 1622, 1943].
Вариант метода заключается в растворении нитрокобальтиата в воде при кипячении, разбавлении раствора водой до 4 мл и смешивании'с 10 мл реагента. Последний состоит из 100 мл водного раствора роданида аммония (уд. вес 1,1) и 400 мл ацетона [2631, 2975].
Этот способ применялся для определения калия в биологических объектах [1121, 1943, 2012, 2631, 2760], в почве [1622, 1869] и других материалах [219, 549, 1271, 1971, 2740, 2955, 2975]. Такой же способ предложен для определения 0,01 — 1 мг калия в 2 мл раствора после осаждения в виде K2Ag[Co(NO2)6] [939, 962, 1664].
Определение по реакции с тиосульфатом. Осадок нитрокобальтиата калия 'растворяют в 0,1 мл конц. НС1, раствор выпаривают досуха. Остаток растворяют в 1 мл насыщенного раствора тиосульфата натрия и добавляют 0,5 мл этанола, нагревают 30 сек. на водяной бане при 80—90° С. Голубую окраску раствора сравнивают с окраской одинаково приготовленного стандарта из раствора хлорида калия.
Метод позволяет определять калий в 0,05 мл сыворотки [259].
Определение по реакции с сульфидом. Осадок нитрокобальтиата калия, полученный из 0,5 мл сыворотки, растворяют при нагревании в 0,5 мл 2%-ной серной кислоты и раствор разбавляют в мерной колбе до 50 JH4. Помещают 10 мл этого раствора в другую колбу емкостью 50 мл, добавляют 1 мл 0,25 М. раствора NH4C1, 1 мл 0,2%-ного раствора NaOH, нагревают 2 мин. на водяной бане при 70—80° С и вводят 1 мл раствора NaHS. Бурую окраску коллоидного раствора сульфида кобальта сравнивают со стандартом, полученным таким же способом. Необходимый для определения раствор NaHS готовят следующим способом. 10%-ный раствор едкого натра делят на две равные части. Одну часть насыщают сероводородом и смешивают со второй [14].
Аналогично определяют калий после его осаждения в виде K2Ag[Co(NO2)6],
Промытый осадок растворяют при нагревании в 0,2 мл конц. HNOa, раствор переносят в мерную колбу емкостью 25 мл, добавляют 1 мл 1%-ного раствора желатина, 1 мл раствора сульфида аммония иразбавляют водой
7*
99
до метки, взбалтывают. Жидкость приобретает желтую или бурую окраску, вследствие образования сульфидов кобальта и серебра. Интенсивность окраски сравнивают со стандартом. Таким способом можно определять 0,07—0,2 мг калия в 5 мл раствора.
Метод применен для определения калия в биологических объектах [245].
Определение по реакции с бикарбонатом калия и перекисью водорода. Осадок нитрокобальтиата калия растворяют при нагревании в соляной кислоте, выпаривают досуха на водяной бане. Остаток растворяют в воде, добавляют несколько капель 30%-ной перекиси водорода и насыщенный раствор КНСО3. Появляется зеленая окраска основного карбоната трехвалентного кобальта, интенсивность которой сравнивают с окраской стандарта. Вместо перекиси водорода можно применять хлорную воду, вместо бикарбоната калия — бикарбонат натрия.
Метод рекомендуется для определения калия в сыворотке [778], почве [1710] и других объектах [60S, 681, 1271, 1824, 2282].
Определение по1 реакции с комплексоном III. К осадку Нитрокобальтиата калия прибавляют 1 мл 5%-ного раствора комплексона III и 4 мл 3%-ного раствора Н2О2, нагревают в кипящей водяной бане до растворения. При этом образуется комплексонат трехвалентного кобальта, окрашенный в розовый или красный цвет. Оптическую плотность раствора измеряют при 540 ммк, количество калия находят по заранее построенному калибровочному графику [433, 880, 2510].
Определение по реакции с нитрозо-Е-солью. Осадок нитрокобальтиата калия растворяют при нагревании в 5 мл 1 N серной кислоты и разбавляют водой до 100 мл. К 20 мл полученного раствора добавляют 2 мл раствора 544 г/л ацетата натрия и вводят 1 мл раствора 1 г ни-трозо-К-соли в 70 мл ацетона. Появляется красная окраска, интенсивность которой сравнивают со стандартом или измеряют оптическую плотность на фотоколориметре.
Таким способом можно определять 0,5—15 мкг калия в пробе [776, 831, 1118, 1271, 1942, 2206,-2554]. Вместо нитрозо-И-соли можно применять раствор а-нитрозо-Р-нафтола в уксусной кислоте [83], а также нитрозорезорцин [83].
Определение по реакции с цистеином. Промытый осадок нитрокобальтиата калия растворяют в 0,2 мл 6 N соляной кислоты, раствор выпаривают досуха. Остаток растворяют в 1 мл 12,5%-ного раствора пирофосфата калия, добавляют 1 мл свежеприготовленного раствора 3,5 мг хлор-гидрата цистеина и 1,5 мл 0,1%-ной перекиси водорода. Разбавляют водой до 5 мл, и желтую окраску сравнивают с окраской стандарта [1271,2497, 2594].
Определение по реакции с диметилглиоксимом и сульфидом. К раствору нитрокобальтиата калия в азотной кислоте добавляют этаноловый раствор диметилглиоксима и затем сульфид натрия. Колориметрируют по возникающей фиолетовой окраске.
Метод позволяет определять калий в 0,1 мл сыворотки с погрешностью не выше 5% [1271, 1824, 2442, 2962, 2963].
Определение по реакции с холином и ферроцианидом. Осадок нитрокобальтиата калия растворяют при нагревании в 3 мл воды и охлаждают. Добавляют 1 мл 1%-ного водного раствора хлоргидрата холина и 1 мл свежеприготовленного 2 %-нога раствора ферроцианида калия, разбавляют водой до 6 мл. Появляется зеленая окраска, интенсивность которой
100
через 15—20 мин. сравнивают с окраской одновременно полученного стандарта или измеряют оптическую плотность на фотометре при 480 ммк.
При содержании 0,05—0,35 мг калия в пробе наблюдается подчинение растворов закону Ламберта — Бера. Метод применялся для определения калия в почве [1869, 2316, 2805], биологических [43, 700, 1345, 1438, 1621, 1716, 2066] и других [1134] объектах.
Определение по избытку нитрокобальтиата натрия. Осаждают калий точно отмеренным объемом раствора нитробальтиата натрия, 1 мл которого соответствует 10 мг калия. Интенсивность окраски центрифуга-та или фильтрата сравнивают с интенсивностью окраски исходного раствора реагента [1157, 1271, 1824].
Способ пригоден для определения относительно больших количеств калия (0,3—19 мг) в пробе. Следует отметить, трудность приготовления раствора реагента указанной выше концентрации и его малую устойчивость при хранении. Интенсивность окраски реагента может уменьшаться не только вследствие его осаждения в виде соли калия, но и от других причин (например, влияние кислотности раствора и температуры на стабильность реагента) .
Предложены методы, основанные ’ на осаждении нитрокобальтиата калия и реакциях на кобальт с цианатом калия [2039], 8-оксихинолином [665] и другими реагентами [1062, 2214, 2588].
Определение в виде калий-бортетрафенила. Калий-бортетрафенил (стр. 48) растворяют в смеси 75% ацетонитрила и 25% воды. Максимум светопоглощения этого раствора находится при 266 лии/с (молярный коэффициент светопоглощения равен 3225).
Измерение оптической плотности при этой длине волны позволяет определять 2—30 мкгК/мл [2226].
Определение в виде пикрата. Осадок пикрата калия (стр. 51) растворяют в воде, раствор разбавляют до 10—50 мл. Интенсивность желтой окраски раствора сравнивают в колориметре с окраской стандарта [24, 569, 783, 908, 1824]. При содержании 0,8—4,8 мг калия в 50 мл конечного раствора получают удовлетворительные результаты [569]. Можно растворить пикрат калия в воде, добавить карбонат натрия и глюкозу и колориметрировать по красной окраске образовавшейся соли пикраминовой кислоты [1255].
Определение в виде дипикриламината. Определение основано на хорошей растворимости дипикриламината калия в ацетоне [11]. В зависимости от количества дипикриламината получают растворы от желтого до оранжево-красного цвета.
Осадок дипикриламината калия (стр. 52) растворяют в 5 мл ацетона, добавляют смесь равных объемов 0,1 М раствора NaHCOs и 0,05 М Na^COa (pH около 9) и измеряют оптическую плотность в кювете с толщиной слоя 2—5 см при 436 ммк.
101
1
При этих условиях молярный коэффициент светопоглощения равен 2,29±0,05 • 104 [1300], что позволяет определять очень малые количества калия. Возможно определение 1 —10-10 6 моля калия в виде дипикриламината в 1 л конечного раствора. При содержании от 0 до 400 мкг калия в 100 мл конечного раствора соблюдается закон Ламберта — Бера. Окраска сохраняется без изменения 2 часа. Колебания концентрации ацетона в конечном растворе не влияют на оптическую плотность [1300]. Рекомендуется измерять и при 400 ммк [739]. В вариантах этого метода ди-пикриламинат калия растворяют в горячей воде [1446, 1760,1870]. Максимум светопоглощения этих растворов находится при 465— 1
470 ммк [90], поэтому оптическую плотность измеряют с синим или зеленым светофильтром. Водные растворы подчиняются закону Ламберта — Бера в пределах 0,1—0,8 мг калия в 250 мл раствора [90]. Таким способом можно определять 20—200 мкг калия с погрешностью 2%. Описано также определение по интенсивности окраски этаноловых растворов дипикриламината калия [657, 658, 1451], ацетоновых растворов, разбавленных затем подщелоченной водой [462, 983, 2441, 2553]. В ряде работ предлагается измерять оптическую плотность маточного раствора после осаждения калия раствором хорошо растворимой соли дипикрил-амина [627, 1870, 2311, 2540] при 595 ммк [793]. Разные варианты этого метода применяются для определения калия в почве [1839, 2540], удобрениях [1446], латексе [739], растительных тканях [1010], биологических материалах [627, 1010, 1451, 1870, 2311, 2553] и прочих объектах [90].
Известны и другие работы о фотометрическом определении калия в виде дипикриламината [618, 1010, 1179, 1712, 1778, 2338, 2911].
Определение в виде соли гексанитрогидразобензола. В центрифужной пробирке смешивают 1 мл слабокислого исследуемого раствора (pH 4,5—6) с 3 мл насыщенного водного раствора мононатриевой соли гексанитрогидразобензола. Через час центрифугируют, раствор удаляют, а осадок монокалиевой соли гексанитрогидразобензола растворяют в 20 мл этанола, 2 мл этого раствора разбавляют водой до 10 мл, оптическую плотность полученного.красного раствора измеряют при 500 ммк, содержание калия находят по заранее приготовленному калибровочному графику.
Соли лития, магния, кальция, стронция, бария и натрия не мешают определению. Можно определять, например, 10 мкг калия в присутствии 50 Мг натрия [554].
Разработаны фотометрические методы определения калия, основанные на предварительном осаждении растворами соли , висмута и тиосульфата [370, 2210], натриевой солью 2-хлор-З- \ нитротолуол-5-сульфокислоты [2896], 5-нитробарбитуровой кислоты [2180] и реакцией с лимонной кислотой в уксусном ангидриде [1019].
102
Электрохимическое определение
Электролитическое выделение калия. Метод основан на электролизе растворов солей калия (и других щелочных металлов) при 140—160 в и ОД—0,2 а на ртутном катоде [729], по другим данным,— при 3,5 в и 0,25 а [1904]. Электролиз продолжается 0,5—1,5 часа. Выделяющиеся металлы дают со ртутью амальгаму, которая при последующем действии воды разлагается с образованием едкой щелочи. К полученной амальгаме'добавляют 0,1 N соляную кислоту, взбалтывают несколько часов на холоду или несколько минут при кипячении, избыток кислоты титруют 0,1 N раствором едкой щелочи в присутствии ализарина. Погрешность определения находится в пределах около 1% [226, 993, 1340, 1357, 2164, 2638]. О приборах, необходимых для выполнения определения этим способом, см. [1098, 1544]. Таким путем определяют сумму щелочных металлов.
Электролитическое определение. Осадок хлороплатината калия (стр. 33) растворяют в горячей воде и определяют платину электролизом. Выделение платины происходит медленно, процесс заканчивается через' несколько часов. Привес катода, т. е. вес платины, умноженный на 0,4004, дает количество калия в анализируемой пробе. Приведенный фактор — теоретический, вследствие некоторого непостоянства состава осадка (стр. 34) целесообразно экспериментально определять величину фактора [226, 973, 974, 2474, 2505].
Предлагается электролитическое определение и для случая, когда калий осажден в виде нитрокобальтиата [2873]. Промытый осадок (стр. 41) обрабатывают щелочью при нагревании. Выпавший осадок гидроокиси кобальта промывают, растворяют в серной кислоте, затем из этого раствора выделяют кобальт электролизом и взвешивают. По весу кобальта вычисляют содержание калия. Если считать, что осадок имеет состав K2Na[Co(NO2)g], то 1 вес. ч. кобальта соответствует 1,326 вес. ч. калия. Этот фактор лучше всего устанавливать эмпирически по стандартному раствору КС1 в принятых условиях осаждения нитрокобальтиата калия.
Полярографическое определение. Потенциал полуволны калия в водных растворах по разным данным несколько различен:
Фон
Потенциал полуволны, в
Гидроокись тетраметиламмония —2,13
То же —1,88
Гидроокись тетраэтиламмония —1,95
Хлорид тетраметиламмония —2,13
То же —2,15
» » —2,17
Литерат ура [323, 2546, 2811] [1930] [2850] [2811] [2546] [288]
103
В качестве фона для полярографического определения калия, кроме указанных выше электролитов, применяют гидроокиси или хлориды тетрабутил аммония (1434, 2849], фенилтриметиламмония [1277], иодида тетраэтиламмония [164], гидроокись лития [2977].
Потенциал полуволны других щелочных металлов отличается от соответствующей величины калия только на 0,01—0,03 в. Поэтому без предварительного разделения полярографически можно определить только сумму щелочных металлов. Если к части исследуемого раствора добавить сначала небольшое количество хлорида калия, а затем к такому же объему раствора — такое же количество хлорида натрия, то на основании высоты волн без добавки и с добавками соли калия и натрия можно вычислить содержание каждого из этих катионов в первоначальном растворе [1105]. ,
Полярографически определяют калий не только в водной среде, но и в 80%-ном водном растворе диоксана [2213], 50— 80 %-ном водном растворе этанола [472, 2977], в ацетонитриле [2849], морфолине [1411]. Влияние посторонних катионов (алюминий, магний, кальций, цинк, кобальт, никель, кадмий и т. д.) устраняют предварительным добавлением комплексона III [1434] или фосфорной кислоты и гидроокиси тетраметиламмония [1930]. Метод отличается высокой чувствительностью, можно определять калий до концентраций 10-3 моль)л [242] и даже до 3-10~5 моль/л [320]. Погрешность определения около 1—4% [397, 2876]. Полярографический метод рекомендуется для определения калия или суммы щелочных металлов в солях кальция, медц {2850], силикатах [1197, 2770а, 2850, 2876], огнеупорах [164], солях калия [1411], воде [242, 1105, 2264], почве [397,2151], цементе [360], стекле [1277, 2771, 2876], вине [619] и других объектах [320, 1105].
Для определения одного только калия его предварительно выделяют в виде калий-бортетрафенила, который растворяют в Г^М'-диметилформамиде и полярографируют при потенциале — 1,55 в на фоне иодида тетрабутиламмония. Таким способом определяют 0,08— 3 мг калия с удовлетворительной точностью [1206]. Осадок калий-бортетрафенила можно прокалить, и образовавшийся метаборат калия определить полярографически. Рекомендуется осадить калий в виде нитрокобальтиата, осадок растворить, и в растворе определить кобальт полярографически. Если считать, что осадок имеет состав К1,84№а1,!б[Со(ЫО2)б], то 1 мг кобальта соответствует 1,22 мг калия [2197, 2210—2282].
Другой метод основан на осаждении К2РЬ[Си(МОг)б] из 2—5 мл раствора, содержащих 0,5—10 мг калия. Осадок промывают 2—3 раза по 0,5 мл 2,5 М раствора KNO3, растворяют в нескольких каплях конц. HNO3, разбавляют водой до 10 мл, добавляют 10 мл 2,5 М раствора KNO3, 3 капли 1 %-ного раствора бромфенолового синего и получают полярограмму свинца; 1 вес. ч. свинца соответствует 0,3773 вес. ч. калия [236].
104
Можно полярографически определить избыток дипикриламина после осаждения калия при 0,45 в в боратном буферном растворе с pH 8,7—9 [420, 912, 2051, 2052, 2607].
Другие работы по полярографическому определению калия-см. [148, 524а, 548, 591, 932, 1106, 1536, 1539, 1540, 1702, 1888, 1929, 2607]. Об определении калия методом осциллографической, полярографии см. [203, 1675].
РАДИОХИМИЧЕСКИЕ МЕТОДЫ
Определение по естественной радиоактивности калия
Еще в 1908—1910 гг. было установлено, что радиоактивность солей калия пропорциональна содержанию в них калия [1153, 1868]. По собственной радиоактивности калия возможно его количественное определение в различных объектах. Первые
радиохимические определения калия по его у-излучению относятся, вероятно, к 1928—1929 гг. [1763] и тридцатым годам текущего столетия [1382, 1673]. Однако вследствие несовершенства аппаратуры того времени предложенный метод отличался малой точностью [1382] и малой чувствительностью [2457]. Только с появлением современных приборов для измерения радиоактивности стало возможным более точное и сравнительно простое определение калия [255а].
Большинство методов радиометрического определения калия основано на измерении его p-излучения [255а]. Счетную трубку прибора для измерения радиоактивности помещают в кювету с исследуемым р.аствором или с растертым в порошок или гранулированным твердым анализируемым веществом. Вся кювета должна быть заполнена анализируемым объектом, вся поверхность счетной трубки должна быть окружена достаточно плот-
ным слоем анализируемого вещества. В отсутствие калия активность образца (за вычетом фона) равна нулю. При наличии калия в образце измеряемая радиоактив-ность пропорциональна содержанию калия. Предварительно строится калибровочный график зависимости активности от содержания калия.
В качестве эталонов служат смеси хлоридов или нитратов калия и натрия (табл. 5) или других веществ (табл. 6) с известным содержанием калия.
Таблица 5
Смеси хлоридов'калия и натрия в качестве эталонов [2735]
Состав смеси, г
хлорид калия хлорид натрия Содержание калия, %
— 100 0
19,2 80,8 10-
38,4 61,6 20-
57,6 42,4 30-
76,8 23,2 4 О'
100 — 52,4
105
Из-за выветривания квасцов процентное содержание калия в смесях несколько увеличивается, поэтому смеси следует возобновлять. В качестве эталонов используют также ряд чистых
Таблица 6
Смеси калйй-алюмиииевых квасцов и кварцевого песка в качестве эталонов [406]
№ смесн Состав смеси Содержание калия, %
1 50% квасцов и 50% песка 4,10
2 50% смеси № 1 и 50% песка 2,05
3 50% смеси № 2 и 50% песка 1,02
4 50%*смеси № 3 и 50% песка 0,51
5 50% смеси № 4 и 50% песка 0,25
воздушно-сухих солей калия с разным содержанием калия. Рекомендуется следующий ряд солей [258, 2099]:
Соль калия Содержание калия, % Соль калия Содержан: калия,
KCr(SO4)2-12H2O 7,8 КН2РО4 28,7
KA1(SO4)2-12Н2О 8,2 КВг 32,8
K2H2Sb2O7 • 4Н2О 13,4 KNO3 38,7
KNaC4H4O..4H2O 13,0 К2СгО4 . 40,3
KJ 23,5 K2SO4 45,0
КМпО4 24,7 КС1 52,4
К2СГ2О7 26,6
График зависимости регистрируемой активности от содержания калия представляет собой прямую линию, проходящую через начало координат. Поэтому для построения графика можно ограничиться измерением активности только 2—3 эталонов.
Определение калия возможно и без применения калибровочного графика. Для этого измеряют активность 2—3 эталонов в одинаковых условиях. Чистый КС1 характеризуется, например, активностью в 1100 имп/мин, а чистый бихромат калия — 559 имп/мин (за вычетом фона). При делении этих чисел на соответствующее содержание калия в процентах находят удельную активность, т. е. число имп/мин, приходящееся на 1 °/о калия; в приведенном примере это число равно 21 имп/мин. Если, далее, анализируемый! объект в таких же условиях показал за вычетом фона, например, 176 имп/мин, то нетрудно вычислить, что в нём содержится 176:21=8,4% калия.
Благодаря очень большому периоду полураспада калия, его удельную активность можно считать постоянной в течение дли-106
тельного времени. Однако регистрируемая удельная активность зависит от конструкции и режима работы счетного устройства, поэтому ее следует контролировать хотя бы один раз в день. Само собой понятно, что активность фона требуется измерять не менее 1 раза в день.
Вследствие самопоглощения в образце регистрируемая P-активность, калия зависит от толщины слоя, однако при плотности, равной 0,4 г!см2 и выше (рис. 5), радиоактивность при
Рис. 5. Зависимость регистрируемой Р-активности солей калия от плотности слоя:
/ — хлорид калия; 2 — оксалат калия; 3 — нитрат
калия; 4 — иодид калия.
прочих равных условиях постоянна, она уже не связана с коле-баниями плотности и изменяется только в соответствии с изменением содержания калия в анализируемом веществе [361, 387, 1304, 1405, 2828]. Удается количественно определять калий и при меньшей толщине слоя, например 0,09 г/см? [1405], но в этом случае необходимо следить за тем, чтобы толщина его была постоянной.
Степень измельчения, небольшие колебания плотности определяемого объекта, небольшая влажность, присутствие анионов с высоким атомным весом, а также посторонних нерадиоактивных элементов и их соединений практически не влияет на точность определения калия [258, 282, 1405]. При радиометрическом определении калия в растворах не наблюдалось строгой пропррциональности между концентрацией и регистрируемой |3-активностью {195, 697]. Это объясняется увеличением самопоглощения в более концентрированных растворах. Поэтому пред-
107
лагается эмпирическое уравнение для вычисления содержания калия с учетом поправок на некоторые факторы:
lgC=—b + ad,
где С—концентрация калия в растворе, г-ион1л-,
А—скорость счета, имп/мин (за вычетом фона);
d — константа, соответствующая коэффициенту самопогло-а — константа, соответствующая коэффициенту самопогло-щения (Q.234);
b — константа, зависящая от эффективности счета и геометрических условий (2,853).
По данным других авторов [523], изменение плотности не влияет на результаты радиометрического определения калия в растворах. Природа (состав) образца влияет на самопоглоще-ние p-излучения и, следовательно, может отразиться на точности определения калия. Поэтому рекомендуют вводить поправку или лучше в качестве эталонов применять вещества, близкие по составу к анализируемому объекту [53, 2358а].
В зависимости от количества калия в анализируемом объекте и от желательной точности определения продолжительность счета различна. Если производить определения с относительной погрешностью не выше 2%, то можно привести следующую зависимость между скоростью и продолжительностью счета [1304]:
Скорость счета, имп]мин ...... 2 20 200 1000
Продолжительность счета, мнн. ...» 6250 113 8 1
Для получения результатов с меньшей относительной погрешностью продолжительность счета увеличивают. Однако в большинстве случаев продолжительность счета не превышает 1—2 час. [191, 1146, 1276], а иногда она может ограничиться 10—20 мин. [258, 267, 387, 697, 1090, 2387, 2735]. Зависимость между содержанием калия в пробе, продолжительностью счета и ошибкой определения [2412] показана на рис. 6.
При достаточно продолжительном измерении радиоактивности удается определять до 0,25% [406] и даже 0,1—0,05% калия [387, 1347] в анализируемом объекте. Минимальная определяемая концентрация калия при анализе растворов — около 0,05 моль!л [697, 1405]. Погрешность определения обычно не превышает 1—2% [17, 191, 697, 1304]. Точность определения описанным методом сравнивалась с точностью других методов. Найдено, что отклонения от гравиметрического нитрокобальти-атного метода в среднем около 4% [282, 1304], отклонения не наблюдались при сравнении с хлороплатинатным методом [2917].
Абсолютное число p-распадов довольно велико — около 30 p-распадов в секунду или 1800 p-распадов на 1 г калия в минуту (стр. 7). Однако эффективность счета невелика, современные лабораторные приборы регистрируют только не-108
большую долю р-излучения. Счетчики с одной трубкой регистрируют 10—30 ими/мин на 1 % калия [191, 258, 1304, 2917]. Для повышения точности определения и уменьшения его продолжительности рекомендуются приборы с двумя параллельно включенными счетными трубками.[546] и даже с девятью [387], десятью [86]’ и больше [166] трубками. Число регистрируемых
Рис. 6. Зависимость между содержанием калия в пробе, продолжительностью измерения радиоак-. тивности и ошибкой определения калия:
/ — ошибка около 10%: 2 —ошибка около 5%; 3 — ошибка около 2%; 4 — чувствительность определения.
импульсов в минуту за вычетом фона в некоторых таких приборах достигает 400 на 1 °/о калия [387]. Понятно, что емкость кюветы при этом значительно возрастает, достигая 900 мл; вес пробы колеблется в зависимости от емкости кюветы от 0,1 до 1 кг и более. С другой стороны, описаны конструкции приборов для определения калия в 5 г [1405], 3 г [533] и в 1 г [260] анализируемого объекта. В этом случае только некоторая часть поверхности счетной трубки соприкасается с исследуемым веществом. Это, конечно, уменьшает эффективность счета, но при содержании не менее 5% калия определение дает удовлетворительные результаты; продолжительность счета — 30—80 мин.
Непосредственное определение калия по его собственной радиоактивности возможно только при отсутствии в анализируемом объекте других радиоактивных элементов. Минерал, содержащий 5,6- 10-40/о УзОв, имеет такую же ^-активность, как
109
объект с 1% К2О [720, 1185, 1304]. Описаны методы раздельного радиометрического определения калия, тория, урана и радия в одном образце [234, 468, 720, 1090, 1758, 2094]. Кратко опишем способ определения калия в присутствии урана и тория [21]. Сначала измеряют общую • активность образца (N, имп/мин), затем при тех же условиях, но с применением латунного экрана толщиною 0,15 мм снова измеряют общую активность (N3, имп/мин). Предварительными опытами было установлено, что с экраном регистрируют 19,4% от начальной активности калия и 35,8 от активности урана или тория.
Следовательно,
W = «K + nu;
Ws=0,194 nK+0,358 пи ,
где «к и п и — регистрируемая активность калия и урана (тория) без экрана, имп/мин. Решая эти уравнения относительно активности калия, находим:
0,358М — ЛУ nv — —--------имп/мин.
к 0,358 — 0,194
Зная Пк> можно по калибровочному графику найти содержание калия в анализируемом образце. Коэффициенты 0,358 и 0,194 не следует рассматривать как константы. Соответствующие величины измеряют в каждом случае отдельно, они зависят от применяемой счетной установки и режима ее работы, от толщины и материала экрана и т. д. Метод применен для анализа почвы.
Предлагается упрощенный способ определения калия в присутствии урана и тория в осадочных породах [406]. Сначала измеряют p-активность исследуемого объекта и вычисляют количество радиоактивных элементов в процентах калия. Из этой величины вычитают поправку на содержание урана и тория. При этом учитывают, что [3-активность тория приблизительно в 4 раза меньше p-активности урана:
д _ и + °.25Th — 5,6-10-* ’
где А —поправка на содержание урана и тория, выраженная в процентах.калия;
U — содержание урана в анализируемом объекте, %; Th — содержание тория в анализируемом объекте, °/0;
5,6-Ю-4 — содержание урана в процентах, эквивалентное по р-активности 1 % калия.
Определению калия практически не мешает присутствие рубидия, так как его примесь обычно мала и излучение его очень мягкое [697].
110
Преимущество определения калия описанным способом заключается в его простоте. Метод не требует выполнения каких-либо химических операций, при анализе объект сохраняется без изменения, и при необходимости определение может быть повторено. При -содержании 1—2% и больше калия в объекте определение отнимает немного времени. Использование естественной радиоактивности калия дает возможность автоматизировать процесс анализа, позволяет определять содержание калия в потоке и автоматически регулировать некоторые производственные процессы [166].
К недостаткам метода следует отнести поглощение [3-излучения калия твердой или жидкой средой. Для уменьшения этого явления при выполнении определения предусматривается прямой контакт исследуемого объекта со счетной трубкой. Если такой контакт по какой-либо причине невозможен (например, при анализе растворов, действующих на стенки счетной трубки), то определение по p-активности становится неосуществимым.
Правда, для уменьшения влияния природы анализируемого объекта на материал счетной трубки последнюю покрывают тонким слоем лака на основе полихлорвиниловой смолы [166], но это приводит к уменьшению эффективности счета. Этого недостатка лишен метод определения калия по его у-излучению, которое способно проходить даже через несколько сантиметров раствора. Такое свойство позволило определять калий в породах в местах их залегания, в буровых скважинах, пересекающих калиевые горизонты. Счетную у-трубку в герметически закрытой стальной гильзе опускают в скважину на каротажном кабеле. При прохождении через пласты калиевых минералов трубка регистрирует у-лучи. Таким способом можно определять глубину залегания калиевых минералов и содержание в них калия [289, 570]. К недостаткам определения калия по его у-излучению следует отнести значительно меньшую абсолютную у-активность калия по сравнению с его |3-активностью.
Количественное определение калия по его естественной радиоактивности применяется при анализе минералов и руд [289, 406, 468, 749, 1304, 1382, 1587, 1588, 2022, 2828], почвы и песка [749, 1371, 1758, 2358а, 2728, 2892], удобрений [282, 486, 749, 1468, 2892, 2912, 2917], стекла [191, 700а, 1148], золы [2099], солей калия [258, 260, 267, 570, 749, 1090, 1440, 2256, 2457, 2870, 2917], растворов и щелоков [166, 267, 523, 697, 1096, 2828], биологических [1131, 2556] и других объектов [17, 541, 561а, 645, 1185, 1276, 1347, 1431, 1962, 2412, 2976].
Радиометрическое определение калия, несомненно, будет все шире применяться в практике аналитических лабораторий.
111
Определение при помощи искусственных радиоизотопов
Определение при помощи радиоактивного изотопа калия К42- Описанный выше радиометрический метод определения калия пригоден для анализа сравнительно больших количеств исследуемого вещества. Если анализу подлежит очень малая навеска или маленький объем разбавленного раствора, то здесь оказываются пригодными способы, основанные на использовании искусственного радиоактивного изотопа К42. Описан радиометрический метод определения калия в виде хлороплатината с применением К42 в качестве индикатора J1532], Метод изотопного разбавления — осаждение калия в виде перхлората в присутствии того же индикатора [2667] — применен для анализа почвы [686]. На некоторые другие работы о применении К42 в аналитической химии мы только сошлемся [541, 1612].
Недостатком, ограничивающим широкое использование К42 в практике, оказывается малый период полураспада этого изотопа (12,4 часа). Поэтому данной порцией радиоактивного препарата калия или его соли можно пользоваться в течение не более 2—3 дней, после чего следует приобрести свежий препарат.
Описано определение калия и рубидия, одновременно присутствующих в исследуемом объекте, при помощи искусственных радиоактивных изотопов К42 и Rb86 [405]. Исследование потерь при количественном определении калия производилось радиохимическим методом с применением изотопа К42 [1870]. Этот же изотоп используется для изучения распределения калия на бумажных хроматограммах [1278] и ионообменных колонках [980].
О возможности масс-спектрометрического определения калия с применением в качестве индикатора стабильного изотопа К41 см. работу [576а].
Определение при помощи радиоактивного изотопа кобальта Со60. Заслуживают упоминания способы радиометрического определения калия с применением радиоактивных изотопов других элементов и особенно изотопа кобальта Со60 [265—267, 523].
0,5 мл исследуемого раствора, содержащего 0,05—0,5 мг калия, помещают в центрифужную пробирку, подкисляют 2 каплями 2 W уксусной кислоты, добавляют 0,25 мл насыщенного раствора хлорида натрия, затем 0,25 мл 5%-ного раствора нитрата кобальта, меченного Со60, и, наконец, 0,25 мл 50%-ного раствора нитрита натрия. Выпадает желтый осадок K2Na[Co(N02)6], для полного образования которого, особенно при малых количествах калия, выжидают 15—30 мин. и более. Осадок промывают 3 раза смесью из 90 объемов воды, 10 объемов этанола и 1 объема конц. СНзСООН и растворяют в 0,1—0,2 мл 0,5 N соляной кислоты. Полученный раствор количественно переносят на отрезок фильтровальной бумаги (25 X 40 мм), высушивают и изме-112
ряют активность. Отдельно в таких же условиях измеряют удельную активность кобальта. Содержание калия во взятом для анализа объеме раствора вычисляют по формуле.
Кл<е =
Ак-1,326
Ло ’
где Лк—активность осадка, имп/мин-.
Ло — удельная активность, имп/мин на I мг кобальта;
1,326 — коэффициент пересчета количества кобальта на калий.
Для нахождения более точного коэффициента пересчета одновременно производят такие же операции с известным количеством стандартного раствора соли калия [1614].
Погрешность определения составляет в среднем около 5%.' Метод применим для определения калия в шамоте, полевом шпате [265], в металлическом натрии [267], сыворотке [2528], воде [1614], минералах [17] и других объектах [2738].
Аналогично определяют 0,01—0,1 мг калия в 0,5 мл раствора с погрешностью около 4% путем осаждения K2Ag[Co(N02)e] [266]. При анализе сплава калия, натрия и свинца [267] калий осаждают в виде КРЬ[Со(МО2)е], коэффициент пересчета количества кобальта в осадке на калий равен 0,663.
Определение при помощи радиоактивного изотопа серебра Ag110. Исследуемый раствор, содержащий 5—15 мг калия, подкисляют азотной кислотой (pH 4—5), нагревают до 60° С и по каплям вводят избыток 2%-ного раствора натрий-бортетрафенила. По охлаждении осадок калий-бортетрафенила отфильтровывают, промывают насыщенным водным раствором этого соединения, затем промывают водой, растворяют в 30 мл ацетона. К раствору добавляют 3—5 мл 0,1 N раствора AgNO3, меченного радиоактивным изотопом Ag110, перемешивают и разбавляют водой до 100 мл. Осадок серебро-бортетрафенила отфильтровывают и измеряют активность 10 мл фильтрата [1473]. Количество калия вычисляют по формуле
Кл<г= --^---^V-39,1,
А
где v — введенный объем раствора нитрата серебра, мл\
А — активность 1 мл, введенного раствора нитрата серебра, имп/мин,
а—активность 10 мл фильтрата, имп/мин (активности даны за вычетом активности фона).
N — нормальность раствора нитрата серебра.
Можно, конечно, вести определение путем измерения активности осадка Ag[B (СбН5)4]. Погрешность определения — около 0,3%.
Возможно радиометрическое определение калия с применением изотопа свинца, ThB [1141]. Следует отметить определение концентрации растворов иодида калия по интенсивности рассеивания p-излучения радиоактивного изотопа фосфора Р32 [1036].
3 Аналитическая химия калия
ИЗ
Определение методом радиоактивации
Определение калия путем радиоактивации основано на ядер-ной реакции:
Исследуемый объект облучают в реакторе потоком нейтронов с плотностью 5-Ю11—3-1012 нейтронов на см2/сек. [644, 2067]. В зависимости от плотности потока нейтронов, от навески и содержания в ней калия облучение продолжается от 3 дней [2067] до 2 месяцев [833].
Исследуемый объект растворяют, добавляют в качестве носителя небольшое количество соли калия, из раствора выделяют калий либо разделением на колонке с катионитом [904], либо осаждением в виде перхлората калия [644], затем измеряют активность полученного препарата, содержащего К42. Сравнивают с активностью аналогично обработанных эталонных образцов соли калия. Этот метод отличается высокой чувствительностью и позволяет обнаруживать до 10~7 г [644] и даже до 10~8 г калия [596, 1843]. Такой способ применяется для определения до 8-10~5% калия в кремнии [2067], минералах [1,833, 2425, 2917а], крови [2613], сыворотке [1338] и других объектах [195, 426, 828, 861].
ФИЗИЧЕСКИЕ МЕТОДЫ
Фотометрия пламени
Фотометрия пламени очень широко применяется для определения калия. По числу исследований этот метод занимает первое место, оставляя далеко позади остальные способы количественного определения калия. Методика определения калия описана в монографиях и руководствах по фотометрии пламени [409, 410, 1516]. Этот метод пригоден для анализа растворов, содержащих малые количества калия. Последний определяют по интенсивным красным линиям 7698,98 и 7664,91 А. Чувствительность определения калия в зависимости от ряда факторов колеблется в пределах 1-Ю-8—2-Ю-7 г/мл [224, 409, 410, 414, 691, 709, 897, 1046, 1092, 1210, 1871, 1940, 2286, 2529, 2767, 2768, 2885, 2887]. Применением совершенной аппаратуры и чувствительной регистрации интенсивности линий (фотоумножитель) удается понизить определяемое количество калия до 1 • 10-9— 5-Ю-9 г/мл [310, 1178, 2596] и даже до 5-10~10 г/мл [1515]. Вследствие высокой чувствительности для определения достаточен очень малый объем исследуемой жидкости, порядка 2-Ю-2 мл [758] и даже 2-10~4 лтл [1178, 1718, 2286].
К недостаткам определения калия методом фотометрии пламени следует отнести зависимость получаемых результатов от 114
одновременного присутствия других веществ. О влиянии отдельных элементов на определение калия нередко имеются противоречивые данные. Например, некоторые авторы полагают, что соли натрия не влияют или почти не влияют на определение калия [2004, 2271; 2494], по данным других авторов присутствие натрия уменьшает результаты количественного определения калия [1047, 2183] и, наконец, можно встретить много утверждений о повышении результатов определения калия, если в пробе присутствует натрий [413, 444, 842, 1466, 1644, 1909, 1970, 2081, 2879]. Очевидно, характер влияния солей натрия зависит от отношения количеств калия и натрия. В зависимости от конструкции применяемых приборов, характера пламени и используемой линии спектра калия количество натрия может без существенных ошибок превышать содержание калия в 100—100 000 раз [409, 410, 758, 842, 2685]. О влиянии натрия на определение Далия см. также [5, 144, 632, 679, 758, 772, 843, 1280, 1491, 1976, 1984, 2146, 2555, 2664, 2770]. Для уменьшения влияния натрия рекомендуют и в стандартные растворы вводить соответствующие количества хлорида натрия [1909, 2050, 2446] или применять красный светофильтр [1851]. Влияние солей натрия уменьшается, если в исследуемый раствор ввести хлорид бария [1491, 2752].
Для определения очень малой примеси калия в металлическом натрии (или в хлориде натрия) растворяют 1 г натрия в метаноле, раствор осторожно подкисляют соляной кислотой, выпаривают досуха, остаток растворяют в 4—5 мл воды. Полученный раствор подкисляют соляной кислотой до pH 4—5, добавляют в качестве коллектора около 1 мг хлорида аммония, осаждают калий и аммоний добавлением 1 мл 0,1 М раствора натрий-бортетрафенила. Осадок отфильтровывают, промывают 1%-ной уксусной кислотой. Осадок растворяют в 1 капле ацетона, для переосаждения добавляют 5 мл воды и 0,5 мл раствора натрий-бортетрафенила. Осадок растворяют в 4 мл смеси тетрагидрофурана и воды (2:3), в этом растворе определяют калий методом фотометрии пламени по линии 7698,98 А.
Этот способ позволяет определять до 7-10"5% калия в натрии с погрешностью около 5% [2103, 2117]. В некоторых работах изучалось влияние лития [409, 410, 679, 1495, 2271, 2278, 2770], рубидия и цезия [409, 410, 412, 1246, 1597, 2066, 2269—2271] на определение калия. Присутствие лития мало отражается на результатах определения калия; рубидий и цезий повышают результаты определения калия. Тем не менее удается определять до 4-10'4% калия в солях цезия [412].
В ряде работ можно встретить указания об отсутствии влияния магния на определение калия [1495], в других работах утверждается, что не мешают только 10-кратные количества магния [2004], а большие количества этого элемента понижают результаты определения калия [2183, 2494]. О влиянии магния см. также [144, 842, 843, 2185, 2770, 2796]. Большое внимание уделено исследованию влияния солей кальция на результаты определения калия [144, 679, 842, 843, 1466, 1619, 1909, 1960,
8*
115
1976, 1984, 2185, 2555, 2770, 2796, 2879]. Соли кальция почти не влияют на определение калия или влияют очень мало [2004, 2446, 2494, 2879]; иногда они только немного повышают результаты в присутствии 200—100 000-кратных количеств кальция по сравнению с количеством калия [409, 410]. Следует, однако, отметить и указание о снижении определяемых количеств калия, если одновременно присутствуют соли кальция [2183]. Для устранения влияния кальция вводили его соль в эталонные растворы [2050]. Добавление LiCl устраняет влияние солей кальция [144]. Метод фотометрии пламени позволяет определять до 10% калия в СаС1г [588]. Мы ограничимся только ссылками на работы, посвященные влиянию солей аммония [842, 843, 2004, 2183, 2796, 2814], бериллия [2084], стронция [144, 2555, 2770], бария [144, 2183, 2284, 2555], марганца [2183, 2237], алюминия [1495, 2004, 2814], железа [1495, 2183, 2185, 2746], хрома, кобальта, никеля, меди, цинка, молибдена [2185], вольфрама [1485], рения [1992].
Свободные азотная, соляная, серная, фосфорная, борная, щавелевая кислоты уменьшают результаты определения калия [842, 1047, 1284, 1301, 2025, 2185, 2908], однако при концентрации около 0,1 N это влияние уже не ощущается [1909]. Для уменьшения влияния кислот рекомендуется разбавить исследуемый раствор водой в известное число раз [1909]. Анионы несколько уменьшают результаты определения калия [985, 2004, 2091, 2879]. Изучалось влияние хлоридов [842, 985, 2055, 2770, 2879], сульфатов [842, 843, 1970, 2185, 2879], фосфатов [842, 843, 985, 1017, 1280, 1301, 1922, 1960, 1970, 2004, 2237, 2537], карбонатов [2770, 2894] и других анионов [2555, 2568]. Для удаления сульфатов и фосфатов, мешающих определению калия., исследуемый раствор пропускают через колонку с анионитом [1309].
Присутствие органических растворителей, смешивающихся с водой, повышает результаты определения [1301, 1718]. Такой эффект вызывает присутствие в растворе до 20% метанола [2555]. Поэтому для увеличения чувствительности определения к анализируемым растворам специально добавляют метанол [2555], смесь бутанола, ацетона и хлоргидрата лауриламина [2670]. Использование ацетоновых или диоксановых растворов повышает чувствительность определения калия [1718]. О влиянии органических соединений см. также [1092, 1718, 1909, 2861].
На точность определения калия этим методом влияют и другие факторы, например, вязкость растворов [938], состав и давление газо-воздушной смеси [2935] и т. д. Все эти факторы могут быть источником ошибок при определении калия [759, 23771, для устранения которых вводят поправки [759]. Точность определения калия этим методом обсуждается в ряде работ [2979]. Разные исследователи не одинаково оценивают точность этого
116
метода. Наряду со скептической характеристикой точности метода [710] можно встретить и чрезвычайно высокую оценку точности — погрешности составляют только сотые доли процента [1238, 2500]. Па данным большинства исследователей, погрешность определения калия находится в пределах 0,5—5% [1301, 1495, 2081, 2103, 2171, 2295, 2499, 2670, 2908]. При определении очень малых количеств калия (около 24 мкг!л) погрешность достигает 8°/о [2596]. Точность определения методом фотометрии пламени сравнивалась с точностью гравиметрического нитрокобальтиатного [136, 536], хлороплатинатного [1072], периметрического [2015] и других методов [179, 691].
Очень много работ посвящено определению калия методом фотометрии пламени в самых разнообразных объектах: силикатах [310, 589, 836, 905, 1043, 1047, 1228, 1437, 1490, 1567, 1619, 2343, 2446, 2471, 2536, 2570, 2752, 2814, 2979], минералах и рудах [111, 144, 413, 632, 695, 702, 999, 1064, 1480, 1489, 1747, 1821, 1908, 2166, 2343, 2344, 2772, 2908, 2910], почве [2, 136, 137, 179, 182, 352, 371, 639, 654, 869, 895, 923, 937, 939, 947, 999, 1060, 1205, 1288, 1303, 1373, 1437, 1497, 1513, 1518, 1848, 1922, 1984—1987, 2034, 2078, 2096, 2104, 2148, 2218, 2236, 2268, 2324, 2363, 2515,
2586, 2591, 2625, 2679, 2743, 2788, 2801, 2956], цементе [356, 717,
1174, 1238, 1288, 1863, 2198], угле [1079, 2261], стекле [862, 978,
1228, 1480, 1484, 1491, 1495, 2081, 2251, 2291, 2297, 2299, 2392,
2397, 2635, 2763], огнеупорных материалах [769, 1333, 1771, 2055, 2500, 2807], керамических материалах [997, 2308, 2560], удобрениях [660, 842, 1021, 1086, 1239, 1241, 1310, 1314, 1328, 1851, 1960, 2004, 2239, 2450, 2453, 2499, 2547, 2776], золе [550, 630, 824, 1017; 1328, 1619, 1707, 1851, 2237, 2479, 2500], воде [173, 181, 772, 809, 964, 1085, 1786, 1970, 1994, 2050, 2245, 2256, 2268, 2670, 2770, 2796, 2879], нефти и нефтепродуктах [225, 990, 1678], реактивах и медикаментах [412, 482, 896, 1087, 1092, 1359, 1441, 1655, 1700, 1909, 2103, 2419, 2500, 2606, 2859], пищевых продуктах [453, 550, 772, 1308, 1779, 1903], молоке и молочных продуктах [1467, 1693, 2165, 2508, 2773], вине [801, 1077, 2950], биологических объектах [614, 635, 710, 737, 757, 758, 964, 1068, 1088, 1150, 1151, 1178, 1252, 1433, 1436, 1466, 1514, 1546, 1564, 1570,
1580, 1584, 1585, 1660, 1718, 1741, 1809, 1858—1860, 1941, 1943,
1950, 2013, 2025, 2074, 2091, 2147, 2163, 2171, 2219, 2260, 2263,
2268, 2351, 2415, 2517, 2537, 2572, 2598, 2611, 2658, 2805, 2907,
2918, 2923, 2935], растениях [130, 585, 653, 712, 870, 940, 1350, 1513, 1595, 1658, 1922, 1973, 1985, 2078, 2146, 2236, 2268, 2348, 2494, 2495, 2550, 2746а], других объектах [12, 34, 215, 636, 679, 796, 1143, 1404, 1599, 1601, 2038, 2149 , 2529 , 2650, 2662].
На обзорные статьи [408, 1127, 1850, 2446, 2611, 2967] и другие работы, в которых рассматривается определение калия методом фотометрии пламени, мы только сошлемся [5, 224, 401, 411, 449, 696, 760, 777, 826, 989, 1138, . 1247, 1306, 1315, 1316,
117
1394» 1413, 1517, 1581, 1615, 1744, 1745, 1825, 1919, 1946, 2246, 2271, 2295, 2330, 2342, 2362, 2412, 2427, 2485, 2493, 2845, 2906, 2937].
Спектральное определение
Определение калия этим методом рекомендуется в тех случаях, когда исследуемый объект представляет собой твердое вещество, трудно переводимое в раствор. Методику определения калия см. в монографиях и руководствах по спектральному анализу [67, 171, 192, 199, 200, 212, 338, 447, 2435]. Спектральное определение калия выполняют [66—68, 110, 199, 200, 202, 338, 447, 844, 845, 1689, 1715, 2273] по следующим наиболее чувствительным линиям (в А):
7698,98 5801,96 4047,20
7664,91 5782,60 4044,14
6964,69 5359,52 3447,70
6938,98 5342,97 3446,72
6911,30 5339,67 3217,50
5812,52 5323,23 3217,02
Чувствительность определения калия зависит от ряда факторов и от конструкции применяемого прибора [581, 1194]. Поэтому чувствительность определения по разным данным различна:
Линия, Л Минимальная определяемая Концентрация, % Литература
7698,98 1.10-’ [67, 170, 199, 200]
7698,98 10-ь [180, 581, 1641]
7698,98 1о-« [270]
7664,91 МО’3 [66, 67, 770, 199, 200]
6938,98 0,3 [576]
6911,30 0,3 [576]
5812,52 0,1 [576]
5801,96 3 [66]
5801,96 0,1 [576]
5782,60 0,1 [576]
4047,20 0,5 [67]
4047,20 4-Ю-’ [2130]
4044,14 0,1 [199, 200, 1496]
4044,44 0,5 [66, 67, 1065]
4044,14 4-10-» [338, 1271, 2130]
3447,70 1 [67]
3446,72 1 [66, 67]
3446,72 0,016 [1271]
3217,50 1 [67]
3217,02 1 [67]
118
О чувствительности спектрального определения калия см. также (28, 65, 172,212, 1194, 1245, 1720, 1906, 1918, 2131, 2157, 2173, 2764]. Вследствие высокой чувствительности спектральные определения возможны с малыми количествами исследуемых веществ [372, 574, 1104, 1245].
В зависимости от характера анализируемого объекта в качестве внутреннего стандарта добавляют соли или окиси разных элементов (табл. 7).
Таблица 7
Внутренние стандарты для спектрального определения калия
Объект анализа Линия калия, А Внутренний стандарт н его линия, А Литература
Растворы солей 7698,98 Си 5153,2 [2413]
Силикаты, соляная кислота 7698,98 Sr 7070,1 [2227, 2888J
Трехокись вольфрама 7664,91 Li 8126,5 [2795]
Минералы, силикаты 6938,98 Cs 6983,49 [2539]
Смазочные масла 6938,98 Li 6707,8 [2400]
Удобрения 4047,20 Cd 3610,5 [7, 388]
Глииа, шамот 4044,14 Li 3232,6 [461]
Биологические объекты 4044,14 Pb 4057,8 [1245]
То же 4044,14 Cd 3261,1 [1109]
Cd 3610,5
Растительные материалы 4044,14 Си 4022,7 [156, 2840]
Каменноугольная зола 4044,14 In 4101 [2318]
Природная вода 4044,14 Mn 4031 [1307]
Растворы 4044,14 Ba 3909,9 [402]
То же 4044,14 Ca 3997,9 [402, 1972]
Огнеупорные материалы 4044,14 Ba 6675,2 [222, 1602]
Удобрения 3446,72 Cd 3466,2 [7, 388]
Растворы 3446,72 Li 2741,3 [574]
Линии других элементов, присутствующих наряду с калием, могут мешать определению [192, 212]. Например, определению по линиям 3446,72 и 3447,70 А мешают линии циркония, молибдена, по линии 4047,20 А — линии хрома, железа, циркония, скандия. Линии следующих элементов мешают спектральному определению калия [212]:
К 7664,91 A К 4044,14 A
Ba 7662,02 Fe 4045,81
Sr 7673,06 Co 4045,39
Mn 7667,89 Mn 4045,20
Си 7664,70 Fe 4045,61
Fe 7664,3 Ca 4044,19
119
Чтобы уменьшить влияние наложения линий других элементов, рекомендуется [192] определять калий по паре линий в длинноволновой части спектра (7698,98 и 7664,91 А) и одновременно по паре линий в коротковолновой области (4047,20 и 4044,14 А). Точность результатов зависит от характера анализируемого объекта, от способа возбуждения спектра, от особенностей применяемых приборов и других факторов, а также от содержания калия в исследуемом веществе. Ошибка определения калия по разным данным находится в пределах 2—20% [68, 180, 372, 402, 2764, 2795]. Спектральное определение калия дает практически такие же по точности результаты, как и хло-роплатинатный и нитрокобальтиатный методы [372]. ?
Спектральный метод рекомендуется для определения калия в самых разнообразных материалах: минералах и рудах [66, 68, ПО, 400, 439, 440, 445, 448, 624, 740, 1477, 1817, 2539], силикатах, песке и стекле [88, 118, 212, 428, 461, 469, 1488, 2053, 2227, 2398], удобрениях [7, 388], металлах и сплавах [171, 2852], це-менте и огнеупорах [222, 461, 1460, 1503, 1504, 1602, 2058], почве [96, 178, 372, 576, 581, 898, 1152, 1248, 1366, 1497], растительных материалах [156, 372, 576, 626, 1913, 2014, 2059, 2086, 2157, 2840], золе [402, 631, 1329, 1972, 2053, 2106, 2318, 2690], воде [201, 1307], пыли [2362, 2697], солях натрия [232, 399, 677, 2173],
солях редких элементов [69, 141], биологических объектах [763, 829, 981, 1108, 1109, 1245, 1395, 1640, 2130, 2225, 2585], растворах солей [4, 402, 448, 574, 601, 1972, 2273, 2413], других объектах [172, 207, 1184, 2250, 2400, 2795].
Ряд других работ, посвященных спектральному определению калия, см. [224, 440, 446, 964, 1110, 1246, 1256, 1336, 1357, 1506, 1570, 1711, 1733, 1748, 1887, 1900, 1907, 1976, 1982, 2034, 2351, 2538, 2603, 2712. 2717, 2772, 2809].
Мы ограничимся только ссылками на рентгеноспектральный метод определения калия [87, 213, 1274, 1555а, 2435, 2970], который применялся для определения калия в полевых шпатах [1154], почве [911, 1271, 1530], удобрениях [2275], биологических [2102] и других [1408] объектах. '
ПРОЧИЕ МЕТОДЫ ₽
Седиментометрическое определение. Впервые седиментометрическое определение калия при помощи нитрокобальтиата натрия было предложено в 1915 г. [1439]. Осадок нитрокобальтиата калия очень удобен для таких определений, так как он состоит из мелких, приблизительно одинаковых по размерам кристаллов.
2 мл исследуемого раствора помещают в центрифужную пробирку с капиллярным дном. Длина капилляра 15—30 мм, внутренний диаметр — 0,5— 1 мм. К раствору добавляют 0,5 мл 25%-иого раствора нитрокобальтиата натрия в 0,5%-ной уксусной кислоте, взбалтывают и через 15—30 мин. центри-120
1075
\
• фугируют. Осадок входит при этом в капилляр, где в зависимости от количества калия в исследуемом растворе, а значит и от количества выпавшего осадка, образуется столбик большей или меньшей высоты. Центрифугируют при 2000—3000 оборотах в минуту до постоянства высоты столбика осадка. Высота столба осадка и служит критерием для оценки содержания калия. Высота столба ие строго пропорциональна содержанию калия, поэтому требуется заранее выявить эту зависимость и построить калибровочный график.
Можно определять 0,07—0,6 мг калия в пробе с погрешностью 2—10% [143, 246, 580, 643, 1439, 1464, 1581]. Простота этого способа позволяет применять его для массовых анализов. Метод применяется для исследования вытяжек из почвы [2, 915, 2341], биологических объектов [1172, 1652, 2810]. На другие работы, посвященные седиментометрическому определению калия в виде нитрокобальтиата, мы только сошлемся [442, 643, 1344, 2136, 2544].
Вариант метода заключается в том, что осадок нитрокобальтиата калия растворяют в соляной кислоте, подщелачивают аммиаком, подкисляют уксусной кислотой, затем осаждают кобальт раствором а-нитрозо-Р-нафтола в уксусной кислоте. Образуется красный осадок нитрозонафтолата кобальта; центрифугируют и измеряют высоту столбика осадка [83].
Описано седиментометрическое определение калия в виде дипикриламината.
В седиментометрическую пробирку помещают 1—3 мл исследуемого раствора, содержащего около 1 мг калия в 1 мл, вводят при помешивании 1 мл 0,2 N раствора дипикриламината магния и воду до 4,5 мл. Перемешивают не-скопько минут, центрифугируют, измеряют высоту столбика осадка; по заранее составленному калибровочному графику находят содержание калия.
Метод позволяет определять до 8 мг калия в пробе с погрешностью около 2—3% [348, 2042].
Предлагается также седиментометрическое определение ка-лия в виде квасцов [1676], перхлората и нитробарбитурата [2042].
Ареометрическое определение. Плотность раствора зависит при прочих равных условиях от его концентрации. Таким образом, измеряя плотность раствора ареометром или пикнометром, можно судить о его концентрации. Данные о плотности растворов едкого кали и некоторых солей калия см. стр. 170. Ареометрическое определение возможно при наличии в растворе только одного растворенного соединения. Этот метод позволяет анализировать смеси хлоридов калия и натрия. Навеску смеси растворяют в определенном объеме воды, определяют плотность полученного раствора при известной температуре; по заранее составленной таблице или графику находят содержание хлорида калия в смеси [987].
Рефрактометрическое определение. Для быстрого определения концентрации соли калия в растворах можно измерять их показатели преломления. Метод пригоден при условии, что в растворе содержится только одна соль. Такие случаи могут встретиться, например, при анализе растворов
1 24
•бромида или иодида калия, применяемых в качестве лекарств г[271, 475, 534]. Зная показатель преломления раствора, по таблицам (стр. 171) находят содержание соли в растворе. Связь между концентрацией соли калия и показателем преломления раствора дает следующее уравнение (1997]:
п—п0 =р(1—ечс ),
где п — показатель преломления раствора, п0 — показатель преломления воды, n'J;
С — концентрация соли калия в растворе, моль/л-, е — основание натуральных логарифмов [2, 7183], 1g е = 0,4343;
рпц—константы, характеризующие данную соль. Из этого уравнения следует, что
С= 3--------P-J-.
0,4343?
Значения констант р и q для растворов нескольких солей калия приведены в табл. 8 [1997].
Таблица 8
Ковстаиты р и q для растворов солей калия
Соль калия р
Хлорид 0,1691 0,0582
Бромид 0,3206 0,0441
Иодид 1,1292 0,0165
Нитрат 0,0902 0,1077
Сульфат 0,0528 0,4175
Нитрит 0,1325 0,0653
Представляет интерес раздельное рефрактометрическое определение калия и натрия в смесях их хлоридов, основанное на разнице в показателях преломления 20%-ных растворов при 25° С [2548]:
для 20%-ного раствора хлорида калия п25= 1,35992;
для 20%-ного раствора хлорида натрия п25= 1,36829.
Если показатель преломления 20%-ного раствора смеси при 25° С равен п, то содержание хлорида калия равно:
(1,36829—/г)-100
% КС1 = 1,36829—1,35992 •
i
Точность определения зависит от степени чистоты смеси (смесь не должна содержать влаги и посторонних примесей) и измерения показателя преломления точно при 25° С. См. также (142, 887].
Поляриметрическое определение. При добавлении соли калия к раствору, содержащему битартрат натрия и молибдат аммония, угол вращения плоскости поляризации уменьшается, что может служить основанием для определения содержания калия [2949].
122
Термометрическое определение. В смеси хлоридов калия и натрия определяют содержание КС1 по понижению температуры, которое наблюдается при растворении определенной навески смеси в определенном объеме насыщенного раствора NaCl. При этом растворяется только КС1 (988].
В сосуд Дюара помещают 100 мл насыщенного при 20° С раствора NaCl и вводят 10 г смеси хлоридов калия и натрия или 10 г сильвинита; навески должны иметь такую же температуру. Для ускорения растворения перемешивают мешалкой. В раствор погружен термометр, позволяющий определять температуру с точностью ±0,01® С.
В зависимости от содержания КС1 наблюдается более или менее значительное понижение температуры раствора, другими словами, по понижению температуры раствора можно •судить о содержании КС1 в смеси (825, 1001, 1271, 1794]. В качестве растворителя применяется также и вода (825]. На одно определение требуется только 5—10 мин. Аналогичный способ предложен для быстрого анализа смеси сульфатов калия и натрия (784].
Для анализа смеси нитратов калия и натрия используется их разная температура плавления и зависимость этой величины от относительного содержания компонентов (табл. 9) [345].
Таблица 9
Температур а плавления смесей нитратов калия и натрия
Содержание KNO3, % Т. пл., °C Содержание KNOlt % Т.’пл., °C
0 316 54,3 231
10 298 70 242
20 283 80 284
30 268 90 306
40 242 100 339
Этот способ пригоден для определения примеси нитрата калия в нитрате натрия и наоборот. Минимальная температура плавления наблюдается у смеси эквимолярных количеств обоих компонентов.
Содержание калия и натрия в их плаве определяют по температуре затвердевания (плавления) (46, 2838].
Около 40—50 г плава помещают в большую пробирку или узкий стакан и погружают в фарфоровый стакан с вазелиновым маслом, нагретым приблизительно до 70° С. В расплавленный металл опускают термометр иа 100° С, градуированный на 0,1° С, и на металл наливают тонкий слой вазелинового масла. После этого стакан с вазелиновым маслом удаляют и наблюдают за движением столбика ртути в термометре. Момент остановки падения уровня
123
столбика ртути соответствует температуре плавления. Для проверки опыт повторяют еще раз. Отклонения в параллельных опытах находятся в пределах — 0,1 ±0,2° С. При содержании 92% калия в плаве и более вычисляют по уравнению:
К=0,259/+83,5, где К — содержание калия в плаве, %;
/ — температура затвердевания плава, °C.
Хронометрическое определение. Ориентировочные определения содержания калия, основанные на скорости появления помутнения в растворе с добавленным реагентом, относятся к числу очень простых, но только ориентировочных методов. Скорость появления помутнения зависит от многих факторов. Отметим некоторые такие методы определения калия.
* 1. Смешивают 5 мл исследуемого раствора с 5 мл 96%-ного этанола и
Добавляют 1 мл 10%-ного раствора нитрокобальтиата натрия, Если помутнение при комнатной температуре появляется позже чем через 2 мин., то в. растворе содержится менее 0,05% калия [690].
2. В пробирку помещают 4 мл анализируемого раствора, растворяют в них 0,23 г хлорида натрия и добавляют 0,5 мл свежеприготовленного раствора 3 г нитрокобальтиата натрия и 1 г нитрита натрия в 10 мл воды. При содержании 6 мг К/л осадок появляется через 3,5—4 часа, при больших концентрациях образование осадка происходит быстрее, а при 250 мг К/л осадок выпадает тотчас [9411.
3. К 10 мл 2 N раствора NaCl, исследуемого на присутствие калия, добавляют 4 мл 20%-ного раствора нитрокобальтиата натрия. В зависимости от скорости появления помутнения судят о содержании калия [83]:
0,39 мг К..........Осадок выпадает тотчас
' 0,19 мг К.............. » » через 30 мин.
0,03 мг К........., » » через 12 час.
Определение с использованием хроматографии. 1. При прохождении раствора соли калия через колонку с катионитом в Н-форме происходит количественный обмен калия на ионы водорода, которые затем можно оттитровать 0,01—0,1 N раствором щелочи [2428, 2893]. Такой способ предложен для определения нитрата калия в черном порохе. 10 г измельченного пороха несколько раз промывают горячей водой. Фильтрат или его аликвотную часть пропускают через колонку с катионитом в Н-форме и затем промывают колонку водой. Азотную кислоту в фильтрате титруют 0,1—0,05 N раствором едкой щелочи в присутствии метилового красного [2431J. Колонку промывают затем избытком соляной кислоты, полученный раствор выпаривают, остаток хлорида калия высушивают и взвешивают [2431].
2. Анализируемый раствор пропускают через колонку с катионитом в Са-форме. Ионы калия и натрия вытесняют кальций из колонки. Количество кальция в фильтрате и промывных водах, эквивалентное количеству калия и натрия во взятом 124
объеме анализируемого раствора, определяют комплексонометрически [2232]. Аналогичный метод основан на пропускании исследуемого раствора через катионит в Mg-форме [666].
3. Раствор, содержащий хлориды щелочных металлов, пропускают через анионит в ОН-форме; в фильтрате находятся едкие щелочи и аммиак. Фильтрат выпаривают, обрабатывают соляной кислотой, выпаривают, слабо прокаливают, охлаждают и взвешивают сумму хлоридов щелочных металлов [2089].
4. Анионит обрабатывают сначала раствором комплексона III и промывают водой. Через подготовленную таким способом колонку пропускают исследуемый раствор, содержащий соли щелочных металлов, магния и кальция. Фильтрат и промывные воды пропускают затем через другую колонку с анионитом в ОН-форме, промывают водой. В полученном теперь фильтрате титруют образовавшиеся едкие щелочи 0,01—0,1 N раствором соляной кислоты [2514]. Описан аналогичный метод с применением анионита в цитратной форме [454].
5. Хроматографический метод позволяет легко сконцентрировать калий и другие щелочные металлы, что применяют для определения его малых количеств в природных водах. 5 л воды пропускают через катионит в Н-форме и промывают малым объемом соляной кислоты и водой. В этом растворе определяют калий каким-либо известным методом. Удовлетворительные результаты достигаются даже при содержании около 1 мг калия в 1 л воды [2143].
Концентрирование применяется и для качественного анализа. 1—3 мл разбавленного раствора, в котором требуется установить наличие соли калия, взбалтывают несколько минут с 10—20 набухшими зернами катионита. Затем зерна помещают на влажную фильтровальную бумагу, палочкой переносят 3—5 зерен в каплю раствора сульфата висмута или раствора Na2Pb[Cu(NO2)e] и рассматривают под микроскопом. Таким путем удается установить присутствие калия при разбавлениях до 1 :3,5 -105 [231].
6. По площади пятна, полученного при проявлении бумажной хроматограммы (стр. 147), можно судить о содержании калия в капле раствора, взятой для хроматографирования. Работая в таких же условиях со стандартными растворами соли калия, получают ряд окрашенных пятен с разной площадью. Строят график зависимости площади пятна от содержания калия, которым пользуются для установления содержания калия в исследуемом растворе [721, 2523, 2793, 2808].
Рекомендуется следующий вариант этого метода. Одновременно с проведением хроматографического разделения для капли исследуемого раствора в таких же условиях ставят опыты с двумя стандартными растворами соли калия. В капле
125
такого же объема одного из стандартных растворов содержится заведомо большее количество калия, в капле другого — заведомо меньшее количество калия, чем в капле исследуемого раствора. После хроматографирования и проявления1 в одинаковых условиях измеряют площади окрашенных пятен и вычисляют содержание калия в капле анализируемого раствора по уравнению [2522]:
42 — 41
где Кх—содержание калия в капле анализируемого раствора, мкг-,
Р\—контрольное количество калия (меньшее), мкг;
Р2—то же (большее), мкг-,
Qx — площадь пятна с неизвестным количеством калия, см2;
Qi — площадь пятна с меньшим контрольным количеством; калия, см2;
Q2 — площадь пятна с большим контрольным количеством калия, см2.
Применение хроматографии при определениях калия см. также стр. 141.
Биологические методы. Биологические методы определения калия рекомендуются главным образом при анализе почвы.
Выращивают плесень Aspergillus niger в 30 мл полноценной питательной среды, но без калия и в таких же средах с разными известными количествами калия. В такую же среду без калия вводят взвесь, полученную взбалтыванием! 2,5 г почвы с водой. Во все растворы вводят равные количества взвеси спор указанной плесени, колбы закрывают ватой и оставляют при 35° С. Через 5 дней мицелий промывают водой, сушат 8 час. при 60° С и затем 3 часа при 100—105° С, охлаждают в эксикаторе и взвешивают. Вес мицелия пропорционален содержанию калия в пробе, его сравнивают с весом мицелия, выросшего в средах с известным содержанием калия. Разница в весе по сравнению с контрольным опытом заметна еще при 0,2 мг калия в пробе [1164]'. Рекомендуют питательную среду, содержащую 10% сахара, 1 % лимонной кислоты, 0,6% сульфата аммония, 0,1% пептона, 0,075% Р2О5 (в виде NH4H2PO4), 0,01 % магния, 2 - 10—4% марганца, 1,5 • 10~4% меди, по 1 • 10—‘% цинка и железа (все металлы в виде сульфатов) [1164, 2123]. В состав другой питательной среды вводят еще мочевину, экстракт дрожжей, гуминовокислык натрий [1320]. Среду перед применением стерилизуют.
Приводятся другие работы по определению калия при помощи Aspergillus niger [1320, 1321, 1952, 1998, 2122, 2388].
Предложен также метод определения калия с применением Azotobacter chrococcum. В результате его жизнедеятельности питательная среда в зависимости от содержания калия более или менее интенсивно окрашивается (чернеет), интенсивность
126
окраски измеряется фотометром или фотоколориметром. Таким? способом можно определять -до 2,5 мкг калия в пробе.
Опыт ведется при 36° С в течение 14 час. Для этого определения необходима среда, которую получают растворением 0,8 г мононатрийфосфата, 0,075 г хлорида кальция, 0,25 г сульфата магния, 0,5 г хлорида натрия, 0,008 г сульфата марганца и 0,015 г сульфата двухвалентного железа в 200 мл воды. 50 мл этого раствора разбавляют водой до 1000 мл, добавляют 2 г бензоата натрия, вводят раствор едкого натра до pH 7,45 н стерилизуют [2842]. , i \
Мы только отметим биологические методы определения калия с помощью Lactobacillus easel [2364], Lactobacillus helveti-cus [701] и Streptococcus faecalis R [701], применение которых требует сложных питательных сред. В этих случаях о количестве калия судят по изменению pH среды [2364] или по интенсивности ее помутнения [701].
IV. ОТДЕЛЕНИЕ КАЛИЯ ОТ ДРУГИХ ЭЛЕМЕНТОВ
Отделение калия в систематическом ходе •анализа катионов. В систематическом ходе анализа сначала осаждают катионы группы соляной кислоты, групп сероводорода и сульфида аммония, выделяют катионы щелочноземельных металлов, после чего в ’ растворе остаются соли щелочных металлов, аммония и иногда магния. Схема анализа раствора, содержащего эти катионы, представлена в табл. 10.
Таблица 10
Анализ хлоридов щелочных металлов [743, 1936]
Раствор хлоридов щелочных металлов выпаривают досуха, остаток прокаливают для удаления соли аммония. Остаток растворяют в воде, добавляют раствор H2[PtClg], центрифугируют.
Раствор: хлориды «атрия и лития Осадок: хлороплатинаты калия, рубидия, цезия; промывают, высушивают, прокаливают, охлаждают, извлекают водой
Осадок: платина Раствор: хлориды калия, рубидия, цезия; добавляют раствор Н [B1J<], центрифугируют
Осадок: Cs [BiJ4] Раствор: калий, рубидий, следы цезия, висмут; выпаривают с HNO3 для удаления С1_ и J-. Остаток растворяют в воде, добавляют растворы Bi(NO3)3 и NaNO2, центрифугируют
Осадок: Rb2Na[Bi(NO2)], и следы цезия Раствор: калий, немного рубидия; добавляют раствор Co(NO3)2, центрифугируют
Осадок: Rb2Na[Co(NO2)g], немного калия Раствор: калий; добавляют раствор Na3[Co(NO2)g], выпадает KaNafCo^O^g]
Разделение щелочных металлов и магния. Раствор подщелачивают аммиаком до pH 9—11, добавляют NH<C1 н избыток раствора
8-оксихинолина. Осадок окснхинолината магния отфильтровывают, фильтрат подкисляют соляной кислотой, вводят небольшой избыток НС1О4 и охлаждают. Осадок перхлоратов калия, рубидия н цезия отфильтровывают, высушивают и сильно прокаливают. В остатке теперь находятся хлориды калия, рубидия н цезия. Охлаждают и обрабатывают этаноловым раствором хлористого водорода, при этом растворяются хлориды рубидия и цезия, а хлорид калия остается нерастворенным. Остаток промывают этим же растворителем и испытывают на калий [1632, 2915].
Анализ смеси хлоридов или сульфатов щелочных металлов. Раствор содержащий хлориды щелочных металлов, выпаривают досуха, нагревают до 400—500° С для удаления солей аммония. Остаток растворяют в воде, раствор фильтруют, подкисляют соляной кислотой и снова выпаривают досуха. Остаток хлоридов обрабатывают абсолютным этанолом, извлекающим хлориды лития и цезия, в нерастворимой части содержатся хлориды калия, рубидия, натрия [271].
В другом варианте анализируемый раствор выпаривают досуха, остаток прокаливают для удаления солей аммония, охлаждают и растворяют в малом объеме воды. Ионы SO4 осаждают добавлением 1 М раствора нитрата свинца, фильтрат насыщают сероводородом и осадок отфильтровывают. К полученному фильтрату добавляют 5 мл 9 IV HCIO4, выпаривают досуха, остаток извлекают 5 мл этанола. В последнем растворяются перхлораты лития и натрия, в остатке находятся перхлораты калия, рубидия и цезия. Остаток смешивают с 5—10 мл концентрированного раствора нитрокобальта калия; осадок нитрокобальтиатов калия, рубидия н цезия отфильтровывают и смешивают с 1 мл 9 N раствора нитрита натрия, выпаривают досуха и прокаливают. Остаток растворяют в воде, нейтрализуют уксусной кислотой, кипятят и отфильтровывают окись кобальта. К фильтрату, содержащему нитриты калия, рубидия, цезия и натрия, добавляют -1—2 мл насыщенного раствора нитрата висмута в 4 N уксусной кислоте, через 30 мин. осадки нитровисмутиатов рубидия и цезия отфильтровывают. Фильтрат смешивают с 0,3 М раствором нитрата кобальта; при наличии калия образуется осадок K2Na[(Co(NO2)s] [1271, 2138].
Анализ смеси солей калия, натрия, магния и аммония. Отдельную порцию, раствора смеси этих солей сначала испытывают реактивом Несслера на присутствие солей аммония. Если аммоний обнаружен, то раствор выпаривают, остаток прокаливают до удаления солей аммония. Остаток смачивают соляной кислотой, растворяют в воде и делят на две части. В одной части обнаруживают калий при помощи нитрокобальтиата натрия, присутствие натрия и магния не мешает обнаружению калия. Другую часть раствора испытывают на присутствие натрия и магния [61].
Катионы к+, Na+ и Mg2+ разделяют следующим способом [228, 395].
Исследуемый раствор нейтрализуют или слабо подкисляют соляной кислотой, выпаривают и прокаливают. Остаток растворяют в воде, добавляют раствор Ва(ОН)2, кипятят н фильтруют. В осадке находится Mg(OH)2, в растворе— калий, натрий, барий. Добавляют серную кислоту в небольшом избытке, отфильтровывают осадок BaSO4. В растворе находятся калий, натрий; его нейтрализуют едким натром и добавляют раствор нитрокобальтиата натрия в разбавленной СН3СООН. В присутствии калия выпадает желтый осадок K2Na[Co(NO2)6],
Описаны некоторые другие варианты систематического хода анализа щелочных металлов [330, 351, 518, 1081, 1717, 2765].
Упрощенный ход анализа. В ходе анализа катионов группу щелочных металлов обычно исследуют в последнюю оче
9 Аналитическая химия калия
129
редь. Это влечет за собой частичную потерю щелочных металлов вследствие адсорбции на осадках, кроме того, анализируемый раствор после ряда операций может загрязняться щелочными металлами из стекла. Поэтому рекомендуется более короткий путь исследования.
2—3 мл анализируемого раствора сначала подкисляют соляной кислотой, а затем подщелачивают аммиаком. Это делается для того, чтобы в растворе было достаточное количество соли аммония. Для осаждения большинства катионов вводят избыток карбоната аммония и центрифугируют. К центрифу-гату, содержащему наряду со щелочными металлами также и аммиакаты серебра, кобальта, никеля, меди, кадмия, цинка, добавляют несколько капель раствора сульфида аммония и снова центрифугируют. Центрифугат подкисляют соляной кислотой, выпаривают досуха и прокаливают для удаления солен аммония. Остаток растворяют в воде, магний осаждают в виде 8-окси-хинолината. Полученный теперь фильтрат содержит только соли щелочных металлов, этот раствор исследуют, как указано выше [2152].
В другом варианте упрощенного обнаружения щелочных металлов исследуемый раствор обрабатывают аммиаком и сульфидом аммония; осадок гидроокисей и сульфидов многих металлов отфильтровывают. К фильтрату, содержащему соли щелочных и щелочноземельных металлов, добавляют карбонат аммония и снова фильтруют. В фильтрате находятся магний и щелочные металлы [2152].
Можно также смешать исследуемый раствор с конц. HNO3, выпарить досуха и прокалить. Остаток растворяют в воде, отфильтровывают от мета-оловянной и метасурьмяной кислот, обрабатывают избытком NagCOg и фильтруют. Фильтрат подкисляют уксусной кислотой и добавляют нитрокобаль-тиат натрия [620].
Описаны аналогичные методы [946, 2602].
Анализируемый раствор нейтрализуют едким натром, добавляют раствор Na3PO< в небольшом избытке, который устраняют последующим введением раствора соли магния. Фильтрат подкисляют уксусной кислотой и осаждают калий (аммоний, рубидий, цезий) раствором нитрокобальтиата натрия [577, 583].
Другие способы упрощенного обнаружения калия в смеси с другими катионами см. [114, 1769а, 111а].
Ниже рассматриваются способы отделения калия от некоторых катионов или способы его непосредственного обнаружения в присутствии других катионов.
Обнаружение калия в присутствии аммония. Почти всегда, особенно при анализе сложных объектов, когда обнаружению калия предшествует ряд других операций, приходится считаться с наличием солей аммония в растворе даже и в тех случаях, когда они отсутствовали в первоначальном исследуемом веществе. Соли аммония дают почти всегда такие же реакции осаждения, как и соли калия. Поэтому перед обнаружением или количественным определением калия всегда следует удалить соли аммония или перевести их в соединения, не взаимодействующие с реагентами на калий. Таких способов известно несколько.
Наиболее простым и распространенным способом устранения влияния солей аммония является прокаливание, приводя
130
щее к возгонке солеи аммония или к их разложению, например:
NH4NO3-N2O + 2H2O
NH4C1t±NH3 + HC1
3 (NH4) 2SO4-*4NH3 + 3SO2 + N2+6H2O NH4NaHPO4->-NH3 + NaPO3+H2O (NH4) 2C2O4 -*2NH3 + CO + CO2 + H2O
Прокаливают при температуре около 350° С [395], по другим данным,— при 400—500° С [1271]. Соли калия при этих температурах практически не улетучиваются [2035]. Особенно легко разлагается нитрат аммония, поэтому исследуемый объект сначала выпаривают с конц. HNO3, а затем прокаливают [18, 216]. Для удаления остатка солей аммония прокаленные соли растворяют в возможно меньшем количестве воды, выпаривают и снова прокаливают. Новый остаток растворяют в воде и испытывают на присутствие калия. Удаление солей аммония прокаливанием рекомендуется многими авторами [13, 99, 214, 336, 1665, 2035, 2152, 2511, 2644].
Можно удалить соли аммония кипячением со щелочью Щелочи разлагают соли аммония с образованием аммиака, который улетучивается при кипячении:
NH4+ +oh~->nh3 + h2o.
Подщелачивают раствором едкого натра, не содержащего примеси калия [228, 1020, 1289, 1476, 1665, 2152, 2455]. Если в растворе после удаления аммония требуется обнаружить не только калий, но и натрий, то подщелачивают раствором Са(ОН)2 [416, 936] или раствором Ва(ОН)2 [336]. Полноту удаления аммиака устанавливают, например, при помощи красной лакмусовой бумажки, помещенной в выделяющихся парах. По удалении аммиака раствор подкисляют соляной или уксусной кислотой и обнаруживают калий реакцией с нитрокобаль-тиатом натрия. Можно удалять аммоний кипячением предварительно нейтрализованного исследуемого раствора с мелкозернистым металлическим магнием [3]:
2NH+4 +Mg->2NH3 + H2 + Mg2+.
В фильтрате от избытка магния обнаруживают калий при помощи нитрокобальтиата натрия.
Соли аммония удаляют также нагреванием с концентрированным раствором нитрита натрия [99, 417, 583, 2303]:
NH4+ + NO; -*N2 + 2H2O.
Однако полное разложение наблюдается только после длительного кипячения с избытком нитрита. В остатке обнаруживают калий лучше всего при помощи нитрокобальтиата нат-
9» 131
рия. Указанная реакция положена в основу следующего способа обнаружения калия в присутствии аммония.
К исследуемому раствору добавляют нитрокобальтиат натрия, выпавший осадок промывают водой и нагревают 5—10 мин. на водяной бане с 1 мл воды и 0,5 мл насыщенного раствора нитрита натрия. Нерастворимый остаток указывает на присутствие калия. При отсутствии или малом количестве калия осадок растворяется полностью. К этому раствору добавляют новую порцию раствора нитоокобальтиата натрия; выпавший осадок представляет собой К2\а[Со(.\О2)6] [583].
Рекомендуется удалять соли аммония окислением гипохлоритом или гипобромитом:
2NHt +ЗОС1- + 2ОН- ->N2+3C1" + 5H2O.
Проще всего в качестве окислителя пользоваться хлорной известью.
1 мл исследуемого раствора смешивают с 1 г хлорной извести, добавляют 3—5 мл воды, перемешивают, после прекращения выделения азота фильтруют. Фильтрат подкисляют уксусной кислотой, кипятят для удаления хлора и по охлаждении обнаруживают калий [99, 305, 2726].
Можно также окислить аммоний в щелочной [552, 628, 1996, 2088, 2241] или бикарбопатной [313] среде бромом, подщелачивают раствором динатрий-фосфата [552]:
2NHr +3Br2 + 8OH--N2 + 6Br- + 8H2O.
Одновременно в большей или меньшей степени протекает побочная реакция, также приводящая к окислению солей аммония, ио с образованием нитрита [552]:
NU'j +3Br2 + 8OH"->NO2 + 6Br-+6H2O.
В бикарбопатной среде реакция протекает без побочных процессов [313]:
2NH/t +8НСО3“ +3Br2->N2 + 6Br~+ 8СО2 + 8Н2О.
Соли аммония окисляют выпариванием с царской водкой [2370]-
Очень часто для устранения влияния солей аммония пользуются их способностью реагировать с формальдегидом с образованием гексаметилентетрамина (уротропина) [13, 161, 247, 326, 339, 421, 1160, 1505, 1704, 2405, 2816, 2936]:
2NH4 +6HCHO^C6Hi2N4+6H2O+4H+.
Для смещения равновесия вправо освободившуюся кислоту нейтрализуют.
К исследуемому раствору добавляют избыток формальдегида, вводят 1—2 капли раствора фенолфталеина и при взбалтывании добавляют концентрированный раствор Na2CO3 до появления устойчивой розовой окраски. Если при этом выпадает осадок малорастворимых гидроокисей и карбонатов, то его отфильтровывают (или центрифугируют), к фильтрату добавляют 5%-ную уксусную кислоту до обесцвечивания и, наконец, вводят раствор нитрокобальтиата натрия; при наличии калия выпадает светло-желтый осадок. Только 132
еиень большие количества уротропина вызывают образование крупчокристал лического темно-желтого осадка. В щелочном фильтрате можно обнаружит калин также в виде калий-бортетрафенила.
Рекомендуется осаждать соли аммония растворами хлорной ртути в щелочной среде в виде хлорида меркураммония [1459, 1787]:
NH+4 +HgCl2 + 2OH-->Hg(NH2)CH-Cl- + 2H2O.
Реагент для осаждения готовят растворением 120 г NaCi и 10 г HgCb в 180 мл воды, к раствору добавляют взвесь 10 г MgO в 200 мл воды и взбалтывают. Этот реагент осаждает также п соли некоторых металлов (железо, марганец и т. д.). К фильтрату добавляют нитрокобальтиат натрия.
Другие способы удаления солей аммония из анализируемого раствора см. [1233, 1731, 2804].
Отделение калия от натрия. Калий можно обнаруживать непосредственно в присутствии натрия при помощи ряда описанных выше реагентов (стр. 12) и особенно нитрокобальтиата натрия, дипикриламина [418, 2443], хлорной кислоты [1325], платинохлористоводородной кислоты [336, 2035] и др. Для отделения малых количеств калия от больших количеств натрия [127] необходимо сначала удалить большую часть соли натрия.
С этой целью анализируемый раствор концентрируют выпариванием в конической колбе, но не дают солям выделиться, охлаждают ледяной водой И насыщают хлористым водородом. На каждые 100 мл раствора добавляют 2 мл воды и взбалтывают. При этой операции оседает главная масса натрия в ви де хлорида. Его отфильтровывают, промывают водой, насыщенной хлоридом натрия и хлористым водородом. Фильтрат и промывные воды выпаривают до суха в платиновой чашке, растворяют в воде и осаждают калий раствором H2[PtCl6] [127].
Следующий способ основан на .большей растворимости биоксалата калия в воде по сравнению с растворимостью соог ветствующей соли натрия [2118].
Навеску хлоридов калия и натрия сначала многократно выпаривают . конц. HNOs. Полученные нитраты растворяют в малом количестве воды, до бавляют чистую щавелевую кислоту и выпаривают на кипящей водяной бане досуха. Остаток растворяют в воде, добавляют новую порцию щавелевой кис лоты и снова выпаривают. Остаток обрабатывают малым количеством воды, в раствор переходит биоксалат калия и сравнительно небольшие количества биоксалата натрия. Раствор выпаривают, остаток прокаливают, охлаждают, выпаривают с соляной кислотой. Хлориды растворяют в воде и осаждают калий в виде хлороплатината [1894].
Отмечается, что отличие в растворимости биоксалатов калия и натрия не так' велико [2222] и что лучше осаждать натрий насыщенным раствором оксалата аммония, калий при этом не осаждается [2011].
Описаны способы отделения калия от натрия, основанные на разной растворимости в воде их нитратов [2503, 2695], хлоридов, сульфатов [781], пикратов [798], перйодатов (рис. 7) [1547], цинк-уранилацетатов [329] и других солей [1298а, 2424].
13.3
Отделение калия от лития. Соли лития не мешают обнаружению калия в виде нитрокобальтиата, перхлората, хлороплатината и др. Для отделения калия от лития рекомендуется осаждать последний в виде карбоната [2380] или лучше фосфата [1979, 1980, 1986, 2380]. Для понижения растворимости осаждают фосфат лития в присутствии этанола [841].
Рис. 7. Растворимость KJO4 и NaJO4-3H2O в веде: 1 - NaJO,; 2 — KJO4.
Отделение калия от рубидия и цезия. Предложен ряд способов отделения калия от рубидия и цезия. Некоторые из этих методов мы кратко опишем.
1. Используя разную растворимость хлороплатинатов (рис. 8), можно отделять калий от рубидия и цезия путем фракционированного осаждения [97, 2230, 2356, 2802а].
2. Солянокислый раствор четыреххлористого свинца осаждает рубидий и цезий в виде М2[РЬС1б], в растворе остается практически весь калий [82, 873]. Аналогичными осадителями являются треххлористая сурьма [873] и четыреххлористое олово [2646] в солянокислой среде.
3. Растворимость калий-алюминиевых квасцов при 17° С ?
134
воде в 15 и 7,5 раз выше, чем растворимость соответствующих цезиевых и рубидиевых квасцов (рис. 9), что предлагается использовать для разделения калия, рубидия и цезия [1057, 2303].
4. Осаждение рубидия и цезия раствором Na2Ag[Bi(NO2)e] относят к числу лучших способов их отделения от калия [1449, 1609, 1613].
Рис. 8 Растворимость хлороплатинатов в воде [394, 882, 1721, 2186]:
1 — Ks[PtCl«J; 2 - Rb2[PtCl6]; 3 — Cs2[PtCl6],
5. Кремневольфрамовая кислота количественно осаждает цезий в виде Cs4SiO4 • I2WO3 • нН2О, в растворе остаются калий и рубидий [56, 1272, 1841]. Германовольфрамовая кислота осаждает рубидий и цезий, но не калий [874, 2509], аналогично осаждает кремнемолибденовая кислота [996, 1628, 1630, 2204]. 9-Фосфорномолибденовая кислота осаждает рубидий и цезий, калий остается в растворе [228, 1841].
6. Для отделения хлорида калия (и натрия) от хлоридов рубидия и цезия пользуются удовлетворительной растворимостью последних в конц. НС1 (плотность 1,19) при 25° С [1990]:
Хлорид
натрия калия рубидия цезия
Растворимость, моль/л .... 0,023 0,18 1,56 4,72
135
7. При добавлении CdJ2 и затем Н2[Р1С1б] к исследуемому’ раствору выпадает осадок, содержащий кадмий и цезий, но не калий [23891.
8. Для отделения хлоридов калия и цезия предложена возгонка в вакууме (1—3- 10~5 мм рт. ст.) при 430—440° С. В этих
Рис. 9. Растворимость алюминиевых квасцов в воде [3941:
1 — калиевые квасцы; 2— рубидиевые квасцы;
3 — цезиевые квасцы
условиях упругость пара хлорида цезия приблизительно в 25 раз больше, чем хлорида калия. Поэтому возгоняется хлорид цезия, в остатке находится хлорид калия [2745]. Описаны другие способы разделения калия, рубидия и цезия [228, 318, 1313, 2954].
Отделение калия от магния
1. Одним из способов отделения калия от магния может быть осаждение последнего раствором 8-оксихинолина из аммиачного раствора в присутствии солей аммония [55, 747, 1426].
2. Из аммиачного раствора осаждают магний добавлением фосфата аммония, фильтрат выпаривают досуха. Остаток растворяют в воде, удаляют фосфат в виде фосфата олова, затем в фильтрате обнаруживают калий [198, 2170, 2329].
Описан аналогичный способ отделения магния от калия осаждением в виде NH4MgAsO4 [872], 136
3. К раствору солей магния, натрия и калия прибавляют равные объемы этанола и реагента. Последний представляет собою насыщенный раствор карбоната аммония в смеси 18 мл конц. NHaOH, 80 мл воды и 90 мл этанола. При этом магний почти полностью осаждается в виде MgCO3 • (NH4)2CO3, фильтрат содержит калий и натрий.
При больших количествах калия, часть его осаждается с магнием; в таких случаях осадок растворяют и снова осаждают указанным способом [687, 1364].
4. Солянокислый раствор солей калия (натрия) и магния выпаривают почти досуха, добавляют кристаллический оксалат аммония, выпаривают досуха и слабо прокаливают. Охлажденный остаток кипятят с водой и фильтруют. При этом в осадке находится карбонат магния, в раствор переходит карбонат калия (и натрия) [2462].
5. Магний осаждают в виде гидроокиси раствором NaOH или Ва(ОН)2, в фильтрате обнаруживают калий. Если в фильтрате требуется обнаружить также и натрий, то осаждают раствором гидроокиси тетраэтиламмония [1804].
Другие способы отделения калия от магния см. [920, 972, 1421].
Отделение калия от щелочноземельных металлов. Осаждение калия раствором дипикриламината [418] или нитрокобальтиата [1813] приводит к отделению калия от солей щелочноземельных металлов. Можно осадить последние в виде сульфатов или карбонатов и фильтрат испытать на присутствие калия.
Отделение калия от алюминия, железа, цинка, марганца, магния и других элементов. Отделение осуществляется осаждением калия в виде хлороплатината [1841] или осаждением посторонних катионов 8-оксихино-лином [1426] или пиридином [382, 383].
Отделение калия от уранила. К исследуемому раствору добавляют немного NH4C1 и взвесь HgO. При кипячении осаждается гидроокись уранила и основной уранат ртути, в фильтрате находятся катионы щелочных и щелочноземельных металлов и небольшие количества растворившейся HgO. Ртуть удаляют осаждением сероводородом, в фильтрате обнаруживают калий [613]. Уранил отделяют также осаждением 10—20%-ным водным раствором пиридина при кипячении [382, 383], раствором сульфида аммония [2319] или электро лизом [2574].
Отделение калия от сурьмы. Смесь солей нагревают в струе газообразного хлористого водорода, при этом отгоняется SbCl3, в остатке находятся хлориды щелочных металлов [1627].
137
Отделение калия от вольфрама. Исследуемый раствор подкисляют азотной кислотой, нагревают и осаждают вольфрамат избытком нитрата одновалентной ртути. Фильтрат от желтого осадка вольфрамата одновалентной ртути подкисляют соляной кислотой, выпаривают, остаток прокаливают и растворяют в воде; в полученном растворе находятся хлориды калия и других щелочных металлов [1327].
Отделение калия от ванадия. Добавляют раствор ацетата свинца к нейтральному анализируемому раствору для осаждения ванадата [2372]. Можно предварительно восстановить ванадат сернистой кислотой, избыток которой удаляют кипячением, затем осаждают сульфидом аммония, нагревают, фильтруют, промывают. В фильтрате находятся калий и другие щелочные металлы [2129].
Отделение калия от других элементов см. [2593].
Отделение калия при помощи органических растворителей
Разделение катионов щелочных и других элементов довольно успешно производится на основании разной растворимости их солей в органических растворителях.
Разделение хлоридов. Для отделения калия (и натрия) от лития пользуются хорошей растворимостью LiCl в некоторых безводных спиртах и значительно меньшей растворимостью хлоридов калия (и натрия). С этой целью применяют этанол [648], н. пропанол [384], н. бутанол [1724, 2905], изобутанол [2920], бензиловый спирт [2780], а также смеси спиртов с эфирами, например, этанол и диэтиловый эфир [687, 1362, 1656, 2285, 2583], изобутанол и этилацетат [2073], а также этанол [2179] и н. пропанол [403], насыщенные хлористым водородом. Все эти предварительно осушенные растворители из смеси хлоридов извлекают LiCl, в остатке находятся хлориды калия и других щелочных металлов. Особенно часто для этой цели применяют изопентанол [384, 909, 1362, 1423, 1682, 1777, 2092] и другие высшие спирты [909]. Растворимость хлоридов в некоторых высших спиртах см. табл. 11.
Таким образом, наиболее полное отделение хлорида калия (и натрия) от хлорида лития наблюдается при применении 2-этил-гексанола (октанола). Исследуемый раствор выпаривают досуха. Однако при этом возможен гидролиз хлорида лития с образованием нерастворимой в пентаноле гидроокиси' лития. Для предупреждения этого явления к раствору до выпаривания прибавляют несколько капель конц. НО. Остаток обрабатывают горячим пентанолом [1362].
• Для отделения хлорида калия (и натрия) от хлорида лития предложены и другие растворители, способные растворять хло-138
рид лития, но не калия. К числу таких растворителей относятся безводный диоксан [61, 966, 2562], ацетон [61, 871], пиридин [1671] и другие [1243].
Таблица И
Растворимость хлоридов калия, натрия и лития (г/100 г растворителя) в некоторых безводных спиртах при 25° С [909]
Хлорид Растворитель
изопеитанол н. гексаиол 2-этилгексанол
ЛИТИЯ 7,3 5,8 3,0
натрия 0,0016 0,0004 0,0001
калия 0,0006 0,00004 0,00001
SLiCI SKC1 12170 145 000 300 000
Обработка 90—95%-ным этанолом применяется для удаления примеси хлорида натрия к хлориду калия [2395]. Безводные хлориды магния и кальция хорошо растворимы в этаноле и в смеси этанола с диэтиловым эфиром, а также в пентаноле, что используется для их отделения от хлорида калия [1777, 2017, 2254, 2734].
Растворимость CsCl при 25° С в воде (г/л) приблизительно в 4 раза больше, чем растворимость КС1; в смеси 1 объема конц. НС1 и 2 объемов этанола растворимость CsCl в 100 раз больше, чем КС1 (0,31 г КС1 и 31 г CsCl в литре растворителя). Этот растворитель позволяет отделять хлорид цезия от главной массы хлорида калия [587, 1551, 2072, 2656, 2866].
Разделение бромидов. Бромиды щелочных и щелочноземельных элементов имеют разную растворимость в пентаноле (25° С) [2952]:
Бромид Растворимость, мг/мл Бромид Растворимость, мг/мл
ЛИТИЯ Легко магния Легко растворим
растворим кальция То же
натрия 0,85 стронция 305
калия . 0,014 бария 0,13
рубидия 0,05
цезия 0,06
Таким образом, калий, натрий, рубидий, цезий и барий составляют группу мало растворимых в пентаноле бромидов.
Смесь хлоридов или нитратов выпаривают с 8 N НВг, полученные броми ды растворяют в возможно меньшем объеме воды и растирают с Ю’Л1Л пентанола, в котором растворяются бромиды лития, магния, кальция и стронция
139
Остаток промывают изопентанолом, содержащим НВг. и затем растирают i 95%-ным этанолом, в котором растворяются бромиды бария и натрия. Остч ток представляет собою бромиды калия, рубидия и цезия [2951, 2952].
Разделение иодидов. Отделения калия и натрия мож- , но осуществить, если воспользоваться растворимостью NaJ в смеси равных объемов абсолютных изобутанола и диэтилового эфира, в которой KJ почти нерастворим. 10 мл смеси растворяют около 500 мг NaJ и только 2 мг KJ {2682].
0,25—0,3 г смеси хлоридов калия и натрия растворяют в нескольких кап <
лях воды, добавляют избыток конц. HJ (плотность 1,96) и выпаривают в кварцевой чашке досуха па водяной бане. Высушивают 1 час при 125° С, охлаж дают, добавляют 5 мл абсолютного изобутанола и растирают 10 мин. Далее вводят 5 мл абсолютного диэтилового эфира, перемешивают, спустя несколько ]
минут фильтруют через фильтр, смоченный изобутанолом. Такое извлечение ;
повторяют еще 1—2 раза. Остаток на фильтре (K.J) смывают 5 мл этанола !
и 25 мл воды, добавляют несколько капель HJ, выпаривают, высушивают, f
охлаждают и взвешивают KJ [1486, 2682]. I
Разделение перхлоратов. Смесь хлоридов щелочных и щелочно I.
земельных элементов растворяют в возможно меньшем количестве воды, до у
бавляют 5 мл конц. НС1О<, раствор выпаривают досуха. Остаток нагреваю!
до 300—350° С для удаления остатков воды и кислоты. Таким образом пол\ лают смесь перхлоратов [1674].
Растворимость перхлоратов в различных растворителях пл казана в табл. 12 [2905].
Таблица Г12
Растворимость перхлоратов (г/100 лглраствора) в различных растворителях прн 25°С
Перхлорат Растворитель
вода метанол этанол н .бутанол этилацстат ацетон
аммония 21,9 5,3 1,49 0,014 0,029 1,77
лития 47,4 89,4 79,4 49,2 63,4 76,4
натрия 113,9 35,8 11,1 1,5 8,4 36,6
калия 2,04 0,083 0,0094 0,0036 0,0013 0,11.4
рубидия 1,33 0,047 0,0071 0,0016 0,0014 0,071
цезия 1,96 0,073 0,0086 0,0048 Почти нерастворим 0,118
магния 73,4 37,7 19,4 44,6 54,2 —
кальция 112,3 113,7 89,6 68,4 57,4 —
стронция 157,ц 113,9 100,0 71,2 76,7 —
бария 129,0 119,8 78,5 41,7 80,8
Резкие различия в растворимости перхлоратов позволили'
разработать способы отделения калия от натрия при помощи метанола [2259], этанола [328, 336, 1793, 2530], н.бутанола [1674].. пентанола [2017] или смеси н.бутанола и этилацетата [2073].
140
Для уменьшения растворимости КС1О4 рекомендуют в качестве растворителя н.бутанол с добавкой около 0,5% НС1О4 [1674; 2905].
Об отделении калия в виде перхлората см. стр. 30.
Разделение ди пи крилам и патов. К раствору солей щелочных металлов добавляют избыток раствора дипикриламината кальция. При этом выпадают дипикриламинаты калия, рубидия и цезия, т. е. достигается отделение от натрия и лития. Осадок дипикриламинатов экстрагируют смесью 1 объема диоксана, 1 объема пентанола и 2 объемов ксилола; в раствор переходят соли калия и рубидия. Экстракт выпаривают, остаток обрабатывают смесью 1 объема диоксана, 2 объемов пентанола и 2 объемов ксилола; в рас-1вор переходит соль калия [2443].
Кратко отметим другие способы разделения, основанные на применении органических растворителей:
Из смеси битартратов этанол извлекает соль натрия (см. стр. 50).
Отделение калия от натрия можно осуществить на основании разной растворимости их хлорауратов в диэтиловом эфире [1183].
Возможно разделение сульфатов калия, натрия и лития, основанное на их различной растворимости в водных растворах этанола [2438].
Смесь бензола и нитрометана позволяет выделить соль калия из смеси полииодидов щелочных металлов [788].
Другие способы см. [598, 930, 1005, 1410, 2645, 2747].
Хроматографическое отделение калия от других элементов
Для выяснения возможности хроматографического отделения калия от других элементов приведем сорбционный ряд на окиси алюминия [376, 378, 2116, 2152]:
H+>As3+>Sb3+>(Sn2+, Bi3+, Ge4+) >FIg2+> (Th4+, Cr3+, Fe3+, Hg2+) >UO22+>Pb2+> In3+>Cu2+>Nd3+>Ag+>Mg2+>Zn2+>
>(Co2+, Ni2+, Cd2+, Fe2+, Ba2+)>(Mn2+, T1+) >Sr2+>Ca2+>
>(K+, Rb+, Li+)>Cs+>NH4+.
Последовательность катионов в сорбционном ряду несколько изменяется при употреблении других сорбентов, например, для пермутита натрия установлен следующий ряд [376, 2313]:
H+>(Fe3+, Hg2+)>(Ag+, Pb2+) >Tl+>Mg2+>Ba2+> (UO2 f
Cu2+, Zn2+, Cr3+, Cd2+, Sr2+)>Ca2+>(K+, Mn2+, Fe2+, Co2+,
Ni2+),
141
для сейфунита [376]:
As3+>Sb3+>Sn2+>Fe3+>Hg22+ > (Pb2+, Hg2+) >Zn2+>Ag+>
>Co2+>(Cu2+, Cd2+, Ni2+, Mn2+, Fe2+, Ba2+)>(K+, Sr2+)>
>NHt .
Следующий ряд сорбируемости одновалентных катионов имеет место при применении органических ионообменных смол [454, 567, 711, 1796, 1812, 2430]:
Ag+>Tl+>Cs+>Rb+>K+>NHt>Na+>H+>Li+.
Высокой избирательной поглощающей способностью по отношению к ионам калия (рубидия, цезия) обладает синтетическая смола, весьма близкая по строению к известному реагенту на калий — гексанитродифениламнну [298, 2569, 2931]:
—СН2-СН---СН2 СН—СН2—
O2NZ\nO2 OaN/^NOg
I I no2 no2
Для катионообменных смол, содержащих в своем составе фосфатные группы, характерно следующее расположение катионов щелочных металлов в сорбционном ряду: K+>Na+>Li+ [522]. К этой группе относится, например, смола РФ-1, содержа-ОН
I
щая — . Такой же ряд наблюдается и в случае
смол на основе метакриловой кислоты, и при хроматографическом разделении ацетатов или гидроокисей калия, натрия и лития. Однако при работе с галоидными солями этих металлов сорбционная способность иная: Li+>Na+>K+ [578, 846, 1387,2741].
Для ионного обмена при отделении калия особенно часто рекомендуются следующие адсорбенты- вофатиты [666, 1080, 1222, 1654, 1680, 2144, 2797, 2891, 2893], амберлиты [633, 901, 1149, 1404, 1687, 1734, 1884, 1970, 2089, 2141, 2798, 2897, 2970], катиониты КУ-1, КУ-2 [135, 318, 432, 463, 2409], дауэкс [511, 669,
142
702, 765, 980, 1004, 1011, 1094, 1283, 1734, 2132, 2232, 2432, 2433, 2675, 2678, 2846], другие сорбенты [135, 1603, 2966].
В качестве сорбента для разделения смесей щелочных металлов применяют порошок целлюлозы [891], трепел, пропитанный дилитуровой кислотой [1572], сульфат бария, смешанный с виолуровой кислотой [1170, 1171], вольфрамат цирконила [1022].
Для разделения калия, натрия и лития рекомендуется пропустить раствор через колонку с сульфосмолой СДВ-3 с последующим элюированием растворами соляной кислоты разных концентраций; при применении 0,12 N раствора кислоты в 80%-ном метаноле вымывается литий, 0,25 N соляная кислота вымывает натрий, для удаления из колонки калия промывают 0,6 N соляной кислотой [135]. Аналогичный способ последовательного вымывания щелочных металлов из колонки растворами соляной кислоты разных концентраций см. [1654, 2144] или 0,1 N раствором хлорной кислоты [1687]. Можно также вымывать водным 0,7 N раствором соляной кислоты, при этом вначале отмываются литий и натрий, а затем калий [2675, 2678]. Такая же последовательность вымывания катионов щелочных металлов наблюдается и при применении в качестве растворителя смеси фенола, метанола и конц. НС1 [2798], Описан способ элюирования лития и натрия смесью 10%-ного метанола и 0,2 М соляной кислоты с последующим вымыванием калия 0,5 N соляной кислотой [2141].
Мы только сошлемся на статьи, посвященные хроматографическому отделению калия от других щелочных металлов [317, 463, 511, 633, 669, 705, 750, 891, 980, 1004, 1094, 1175, 1182, 1404, 1687, 1884, 2019, 2116, 2678, 2798, 2846, 2891, 2897], магния [705, 1149, 1170, 1680, 2132, 2797], кальция [1149, 2132, 2433, 2797] и других катионов [1149, 2433].
Хроматографические методы позволяют сравнительно легко отделять калий от анионов, мешающих его определению химическими методами. Для отделения калия от сульфатов и фосфатов пропускают исследуемый раствор через колонку с анионитом в хлоридной форме. При этом сульфат- и фосфат-ионы количественно обмениваются на ионы хлора, в фильтрате содержится калий в виде хлорида. После промывания колонки водой в полученном растворе определяют содержание калия гравиметрическим способом в виде перхлората [1285]. Исследуемый раствор пропускают через колонку с катионитом в Н-форме, калий (и натрий) полностью задерживается, а мешающие анализу анионы проходят в фильтрат в виде соответствующих кислот. Колонку промывают затем водой, фильтрат и промывные воды отбрасывают. Калий (и натрий) вытесняют из колонки промыванием соляной кислотой. В фильтрате содержится теперь калий (и натрий) в виде хлорида [2410]. Для отделения калия (и натрия) от анионов-окислителей нельзя пользо-
143
ваться катионитами в Н-форме, в таких случаях применяют катионит в аммонийной форме. На таком катионите можно отделять ионы щелочных металлов от хромата, молибдата, ванадата и др. [2429].
Хроматографические методы применяются для определения калия в силикатах [2678], стекле [578, 2926], воде [1970, 2143], золе [1149], медикаментах [1734] и других объектах [666, 1182, 2431, 2797, 2846].
Отделению калия от других катионов методом хроматографии на бумаге посвящен ряд работ. Предложено много вариантов, отличающихся применяемыми растворителями, способами проявления и другими деталями. Для разделения калия и натрия рекомендуют растворитель, состоящий из смеси 1 объема конц. НС1, 4 объемов 0,5%-ного раствора винной кислоты и 10 объемов воды [1278]. В качестве растворителя предлагается также смесь 4 объемов 96%-ного этанола и 1 объема 2 N уксусной кислоты [2522, 2600]; 8 объемов этанола и 2 объемов 0,1 N соляной кислоты [721], смесь равных объемов метанола и этанола [2790].
Хорошо разделяются калий и натрий [1278]. Для установления значений применяются радиоактивные изотопы К42 и Na24, а также изотоп иода J131. В этих случаях Rf можно определять авторадиографически [1278, 2470]. Хроматограмму проявляют 0,1 N раствором виолуровой или тиовиолуровой кислоты [1169, 1571, 2352, 2521—2523] и наблюдают в ультрафиолете окраски пятен после высушивания. Можно проявлять раствором 8-оксихинолина с последующим наблюдением хроматограммы в ультрафиолете [1148, 2244] или раствором Хта2РЬ[Со(МО2)б]— зеленое пятно [721].
Для разделения хлоридов калия, натрия и лития достаточно 0,05 мл их раствора с содержанием по 0,1 мг каждой соли, растворитель — метанол, проявитель — нитрат серебра и флуоресцеин [899]. В качестве растворителя рекомендуется также смесь 8 объемов этанола или ацетона и 2 объемов воды [949] или 7 объемов метанола и 3 объемов 2-этилгексанола [864]. Разделяют также при помощи растворителя, состоящего из 7 объемов метанола и 3 объемов пентанола [2822].
Хорошее разделение сульфатов этих трех металлов на бумаге достигается при применении смеси 9 объемов метанола и 1 объема воды. При этом R для калия оказывается равным нулю, для натрия — 0,62 и лития —0,72. Бумагу затем смачивают 0,2%-ным раствором хлорида бария в 70%-ном этаноле, высушивают и проявляют 0,05 %-ным раствором родизоната натрия в 50%-ном этаноле,— наблюдаются желтые пятна на красном фоне |[892].
На полоску бумаги помещают 0,01 мл 0,1 N раствора пропионатов калия, натрия и лития, высушивают и разделяют по восходящему способу, применяя 144
в качестве растворителя смесь 100 объемов абсолютного этанола, 10 объемов воды и 1 объема пропионовой кислоты. Через несколько часов проявляют раствором виолуровой кислоты и высушивают сначала при 50° С, затем при 100° С. Значения Rf при этом для калия — 0,17, натрия — 0,31 и для лития — 0,68 [2040]. О других растворителях для этой цели см. [1450, 2243].
Значительный интерес представляет возможность разделения хлоридов калия, рубидия и цезия методом хроматографии на бумаге. Каплю раствора, содержащего по 5 мкг и больше каждого катиона, наносят на полоску бумаги; растворитель состоит из конц. НС1 (плотность 1,19), метанола, н.бутанола и изобутилметилкетона (55:35:5:5), проявитель — раствор Na2Pb[Co(NO2)6] (стр. 15). Приблизительные значения Rf для калия—0,7, рубидия—0,8, цезия—0,9 [163, 2016]. Для этой же цели предлагается другой растворитель, дающий удовлетворительные результаты: смесь конц. НС1, метанола и и.бутанола (55:40:5) [2016].
В качестве растворителя пользуются также смесью равных объемов 20 %-ной соляной кислоты и насыщенного водного раствора фенола. Хроматограмму проявляют 10%-ным раствором нитрокобальтиата натрия, затем промывают водой и дополнительно проявляют 0,1%-ным этаноловым раствором а-нит-розо-|3-нафтола, подщелоченным едким натром. Значения R f при этом для аммония — 0,11, калия — 0,19, рубидия — 0,27 и цезия — 0,33 [2152, 2629]. Состав другого фенолсодержащего растворителя для разделения щелочных металлов на бумаге см. [1925]. В качестве растворителя рекомендуется" также насыщенный водный раствор нитробензола [2041], 87%-ный этанол [551], метанол с добавкой 1—5% концентрированного раствора аммиака [1961], смесь 98% метанола и2% уксусной кислоты [2224], смесь пиридина, этанола и 1,5 N уксусной кислоты (40:40:20) [398]. Проявителем служит 1%-ный ацетоновый раствор пикрата натрия [2041]. Хроматограмму хлоридов щелочных металлов сначала погружают в 0,1 N раствор нитрата серебра, промывают водой и погружают в раствор- сульфида аммония [551] или после обработки раствором нитрата серебра хроматограмму смачивают раствором флуоресцеина в метаноле [2792, 2797].
Хлориды калия, натрия, лития и аммония хорошо разделяются метанолсодержащими растворителями. Для опытов применялось 0,03 мл 1%-ного раствора хлоридов, проявляют 0,2%-ным раствором 2,6-дихлорфенолиндофенола в метаноле, содержащем 3% нитрата серебра; наблюдались пурпурные пятна на голубом фоне. Значения R см. в табл. 13 [693]. .'
Величина R для хлорида калия зависит от состава применяемого растворителя [2792], см. табл. 14. . , ....
Смесь солей калия, натрия и лития в растворе разделяют следующим способом [1367]. Хроматографическую бумагу про-
1Q Аналитическая химия калия
145
Таблица 13
Значения Rf для хлоридов калия, натрия, лития и аммония
Растворитель Значения Rf
К+ Na+ NH* Li
Этанол и метанол (1:1) 0,08 0,23 0,32 0,65
н. Пентанол и метанол (3:7) .... 0,09 0,25 0,33 0,63
и. Бутанол и метанол (3:7) 0,11 0,26 0,34 0,62
Метилэтилкетон и метанол (2:3) 0,14 0,30 0,33 0,62
Ацетон и метанол (2:3) 0,15 0,25 0,28 0,50
мывают сначала смесью воды, пиридина и ледяной уксусной кислоты (80:15:5) и высушивают. Наносят 0,001 мл раствора, содержащего соли указанных металлов, высушивают и обрабатывают смесью изопропанола, пиридина, ледяной уксусной
Таблица 14
Зависимость R? хлорида калия от состава растворителя
Состав растворителя (объемы) Состав растворителя (объемы) *f
пиридин вода ацетон вода
1 0 0 1 0 0
1 0,5 0,60 1 0,2 0,39
1 1 0,84 1 0,33 0,61
1 2 0,99 1 1 0,97
0 1 1,00 0 1 1,00
кислоты и воды (8:8: 1:4). Проявляют 1%-ным этаноловым раствором цинк-уранилацетата, нагревают 2—5 мин. при 120° С и рассматривают в ультрафиолете. При этом на синем фоне видно желтое флуоресцирующее пятно (калий) и бледно-желтое пятно (натрий, литий) [1367].
Изучалось влияние температуры на величину Rf катионов' щелочных металлов при применении растворителя из метанола, конц. НС1 и воды (8:1:1), проявитель — раствор нитрокобальтиата натрия (табл. 15) [2599].
Высота подъема фронта растворителя мало отражается на Rf хлоридов щелочных металлов [2792]. Колебания абсолютных количеств хлорида калия в довольно широких пределах не влияют на величину Rf [2792].
Процесс впитывания растворителя в бумагу отнимает от нескольких часов до суток [551, 864, 2792, 2793]. К положительным 146
Таблица 15
Зависимость Rf от температуры
Температура, °C
Катион 0 10 20 30 37 50
Значения
к+ 0,07 0,09 0,12 0,16 0,21 0,32
Na+ 0,26 0,27 0,31 0,39 0,42 0,50
Li+ 0,69 0,70 0,74 0,75 0,77 0,82
сторонам способа разделения щелочных металлов методом хроматографии на бумаге относится возможность выполнения исследования с очень малыми каплями раствора, цорядка 0,02— 0,001 мл [693, 721, 782, 2418, 2523, 2793, 2808, 2822]. По величине площади проявленного пятна судят о содержании калия в исследуемом растворе [721, 2523, 2793, 2808], (см. стр. 125). Количество хлорида калия на хроматограмме определяют и другими способами [2822]. Например, зону, соответствующую хлориду калия, вырезают, экстрагируют горячей водой, раствор выпаривают досуха, нагревают 30 мин. при 450° С для удаления следов НС1 (если он входил в состав растворителя), растворяют в небольшом объеме воды и титруют 0,001 N раствором нитрата серебра, устанавливая конечную точку кондуктометрически [2418].
Приводятся другие работы, посвященные выделению и определению калия методом хроматографии на бумаге [322, 609, 640, 641, 692, 748, 766а, 812, 944, 1844, 1931, 2145, 2465, 2688].
Описано отделение калия от других щелочных металлов путем электрофореза на бумаге [640, 748, 1401, 2521, 2753].
Соосаждение калия
Соосаждение с сульфатом бария. Способность калия (и других щелочных металлов) соосаждаться с BaSO4 известна давно [538, 651, 2035, 2373, 2641]. Это явление объясняется окклюзией сульфата калия [2331], адсорбцией [674, 865, 1767, 1969, 2119], образованием твердых растворов [64, 490, 2839] или двойной соли [673, 1677, 1861]. Соосаждение калия может быть значительным. Чем больше калия в растворе до осаждения, тем выше его содержание и в осадке BaSO4 [64, 500]. Содержание сульфата калия в-сульфате бария достигает 4,8%. При прочих равных условиях калий соосаждается больше, чем натрий, что находится в соотвётствии с растворимостью сульфатов этих металлов—меньшей растворимостью характеризуется именно сульфат калия [500]. Таким образом, удаление ионов
10*
14
сульфата или же бария из раствора в виде BaSO4 приводит к некоторым потерям калия [1139, 2786].
Много работ посвящено соосаждению перманганата калия с сульфатом бария. Соосаждение объясняется образованием смешанных кристаллов BaSCU и КМпО4 1664, 673, 675, 676, 1399, 1776, 2832]. Осадок BaSO4 приобретает по этой причине красную окраску. Содержание соосажденного перманганата калия в BaSO4 достигает 20% [1677] и даже 40 мол. % [1397, 1398, 1594].
О соосаждении калия с BaSO4 см., кроме того, [240, 251, 252, 674, 1166, 1271, 1659]. Соли калия способны соосаждаться также с сульфатами стронция [1399] и свинца [1703], с селенатами бария и стронция [1397—1399].
Соосаждение с малорастворимыми хроматами. Соли калия соосаждаются с хроматами бария [350], стронция [221], кальция, свинца, серебра [1400], при этом предполагается образование соединений состава КгВа(СгО4)2. K2Sr(CrO4)2 [1400].
Соосаждение с гидроокисями. Соли калия соосаждаются с малорастворимыми гидроокисями железа и алюминия [133, 344, 2715], марганца [1267, 2815, 2948], меди [1989] и других металлов [1458, 2283а].
Наблюдается соосаждение калия с осадками натрий-цинк-уранилацетата [1949], оксалатов кальция [1765] и магния [1535]. 8-оксихинолината магния [1535], сульфида железа [1935], аммо-ний-магнийфосфата [674, 1535], фосфата кальция [1271] и других малорастворимых соединений [1073, 2651].
Соосаждение с аммоний-бортетрафенилом применено для выделения калия из очень разбавленных растворов, порядка 1 : 108 [187, 2103].
Следовательно, соосаждение калия представляет интерес, главным образом, как источник потерь калия при предварительном осаждении соединений других элементов.
Не менее важным источником ошибок может быть и соосаждение других элементов при осаждении малорастворимых соединений калия:
соосаждение аммония с фосфоромолибдатом калия [261]; лития с хлороплатинатом калия [839]; натрия с дипикриламинатом калия [2444]; рубидия, цезия и таллия с нитрокобальтиатом калия [256, 1608, 1672]; цезия с дипикриламинатом калия [264]; цезия с пикратом калия [257]; цезия с перйодатом калия [2590]; щелочных металлов с калий-бортётрафенилом [2279]; таллия с фосфоромолибдатом калия [256, 261]; таллия с битартратом калия [256, 2215]; кальция, бария с .нитрокобальтиатом калия [220].
ПРИЛОЖЕНИЯ
Некоторые свойства калия
Порядковый номер......................19
Атомный вес...............................39, 102
Плотность, г/см3 при 20° С..........................0,85—0,86
при 62,3° С...............................;о,8з
Температура плавления, °C .......... 62,3—63,7
[323а, 649,1569, 2326]
[8, 362, 393, 478, 1414, 1418, 1443, 2247, 2332]
[478]
[8, 269, 302, 362, 485, 688, 768, 856, 1443, 2101, 2325, 2838]
Температура кипения, °C .............. 762—-770 [8, 269, 362, 393,1443,
1534, 2407, 2663]
Твердость (алмаз-10)........................0,5
Теплоемкость, кал]г..................0,17—0,19
Электропроводность (Hg=l) ...................14
Радиус атома, А ...........................2,33
Радиус иона, А ............................1,33
Подвижность иона при 18°С, ом-1-см2/г-экв ...............................64,6
[362, 393]
[362, 393]
[362]
[244, 393, 1353, 2321]
[244, 602, 1353, 1358, ' 2194, 2321, 2637,
2964]
[1401]
Искусственные радиоактивные изотопы калия [70, 152, 306, 435, 459, 460, 471, 543, 813, 861, 885, 1302, 1443, 1589, 1822, 1828, 1898, 2172, 2321, 2552, 559, 2671]
Изотоп Период полураспада Тип превращения | Энергия, Л/эв Ядерные реакции и превращения, образующие данное ядро
Р-частнц | у-лучей
К37 1,3 сек. 3+ 4,57 — К39 (т, 2п); Са40 (р, а)
К38 7,5 мин. Р+, т 2,53 2,15 С136(а, п), К39 (я, 2п) К39 (т, n), Ca40 (d, а)
К42 12,44 часа т 2,07(25%) 3,58(75%) 1,51 К41 (dp); К41(п, у) Ca42 (n, р); Sc4B (п, а) Ar40 (d, рп)
К43 22,4 часа Р~, т 0,24; 0,81 0,4 Аг40 (я, р)
К.43,44 27 мин. | 18 мин. — — Са43.44 (п, р
149
Свойства некоторых соединений калия [269, 393, 478, 485, 1443, 1799, 1993]
Формула Мол. вес Плотность при 20° С, г/см* Т. пл., °C Т. кип., °C
1 2 3 4 5
KAI (SO4)2 258,19 2,75 —
KAI (SO4)2-12H2O 474,38 1,76 (26°) 92 —
к [BF4] 125,92 2,50 529,5 Разлаг.
KBr 119,01 2,75 728 1376
KBrO3 167,01 3,27 (15°) 370 (разлаг.) —
ксн3соо 98,14 1,8 292 —
КС1 74,56 1,99 768 1415
ксю3 122,56 2,32 356 Разлаг., 400
ксю4 138,56 2,54 (11°) 610 (разлаг.) —
KCN 65,12 1,52 (16°) 634,5 —
KCr (SO4)2-12H2O 499,42 1,83 89 —
KF 58,10 2,48 856 1505
KF-2H2O 94,13 2,45 41 —
KFe (SO4)3- 12H2O 503,26 1,83 33 —
khcoo 84,12 1,91 167,5 Разлаг.
KHCO3 100,12 2,17 — —
KHC2O4 128,13 2,0 Разлаг. —
KHC4H4Oe 188,18 1,96 — —
KHSO4 136,17 2,24 210 Разлаг.
KH2AsO4 180,03 2,87 288 —
kh2po4 136,10 2,34 252,6 I—
KJ 166,02 3,13 680 1329
KJO3 214,02 3,89 560 Разлаг.
KJO4 230,02 3,62 582 —
KMnO4 158,03 2,70 Разлаг. >270 —
kno2 86,11 1,92 298 —
KNO3 101,11 2,11 (16°) 334 Разлаг. 400
KOH 56,11 2,04 360 1320
Kpo3 118,08 2,39 (25°) 450 —
KReO4 289,41 4,89 350 —
KSCN 97,18 1,88 — Разлаг. 500
K2CO3 138,21 2,43 (19°) 891 Разлаг.
K2C2O4-H2O 184,24 2,13 — —
150
Таблица (окончание)
1 । 2 1 3 1 1 5
К2Са (SO4)2-H2O 328,42 2,60 1'004 —
К2СгО4 194,21 2,73 968 —
К2Сг2О7 294,22 2,69 398 Разлаг. >500
К2 [1гС16] 484,04 3,54 Разлаг. —
K2Na[Co (NO2)«1 436,18 1,63 135 —
К2 [PdCI4] 326,73 2,67 105 —
К2[PdCIJ 397,64 2,74 — —
К2 [PtBr«] 752,92 4,66 (24°) Разлаг. <400 —
К2 [PtCI4] 415,26 3,30 — —
К2 [PtCI«] 486,17 3,50 250 —
К2 [PtJ«] 1034,95 5,18 — —
К2 [ReCl6] 477,25 3,34 — —
K2S 110,26 2,66 Разлаг. 250 —
K2SO4 174,26 2,66 1068 —
K2S2O7 254,32 2,27 >300 —
270,32 2,48 Разлаг. <100 —
K2 [SiF6] 220,26 2,66 (кубич.) Разлаг. —
3,08 (гексаг.) — —
K2WO4 362,15 3,13 388 —
K2 [ZrF,] 283,42 3,48 —- —
K3[Fe(CN)6] 329,26 1,89 Разлаг. —
k3po4 212,28 2,56 (17°) 1340 — .
K4 [Fe(CN)6]-3H2O 422,41 1,85 (17°) Разлаг. —
K4P2O7-3H2O 384,41 2,33 — —
Упругость пара калия и некоторых его соединений [485]
Калий и его соединения Упругость пара, мм рт. ст.
1 5 1 10 | 20 | 40 | 60 | 100 | 200
Температура, °C
к 341 408 443 483 524 550 586 643
КВг 795 982 — 994 1050 1087 1137 1212
KCI 821 919 968 1020 1078 1115 1164 1239
KF 885 968 1039 1096 1156 1193 1245 1323
КОН 719 814 863 918 976 1013 1064 1142
KJ 745 840 887 938 995 1030 1080 1152
151
152
Упругость пара солей калия
Соль Температура, °C Упругость пара, мм рт. ст. Литература
KCI 700 1,5 [394,1573]
750 3 [394,1573]
800 4,5 [394,1573[
801 1,54 [1993]
900 7,0 [394,1573]
950 13,0 [394,1573]
1044 24,1 [1993]
KF 1278 109 [2408]
KJ 1046 64 [2408]
K2SO4 ИЗО 0,4 [1617,1993]
Примечание. Другие данные об упругости пара соединений калия см. [1051,1229,1978,2746,2830,2973].
Термическая устойчивость некоторых солей калия [955,1120,1122,1123,1125,1126,1342,1368,1967,2869]
Соль Пределы термической устойчивости, °C
К[В(СвН5)4] <265
K[BF4] <410
KBrOg <366
KC4H2N3O5 (нитробарбитурат) ~100
KCeH2N3O7 (пикрат) 100—217
KC7H5C1NSO5 (2-хлор-3-нитротолуол-5-сульфонат) 100—278
KC8HeN3O7 (диметилпикрат) 90—150
KC12H4N7O12 (дипикриламинат) 100—220
KCI 219—813
KCIO4 <510
кнс4н4о6 100—215
KJ <715
KJO3 <500
KJO4 105
KMnO4 <245
KNO3 <843
KReO4 54—220
K3[PtCl6] 100—270
K2SO4 408—880
K2SO4-PbSO4 40—906
K2[TiF,].HF <300
о С© Ci t-. Ci сч
100 154 1 1 L.O 121. 6. С© со 1 103 49, О 330 178 сч. со
-г-1 ci с© СЧ
8 109 396. 42. 1 1.0 1 1 1 1 Ci Ci 1 1 1
О -г-1 г— о ю о Ci СО L.O 00
S о 1 35 1 1 со 00 ю 1 1 О
со
о 00 0 сч о
О 40. 364. I Ь-сч 1 со 1 1 1 1 о Ci ] 1 1 1
в воде юды) О О 3 24,8 350,0 60,0 20,5 2,40 1 1 45,6 сч" сч 85,6 1 ; 0,56 I 147 18,5
калия 1 100 г е С. а С о Ю 17,0 337,3 52,0 14,8 1,81 i 1 i 17,5 80,2 1 0,238 140 1
SS Ш а [° с* СО СО ю со со СЧ 1 00 00
s та ф CQ Е Н о 1 323. 45. 10. с© 1 1 со с© о" 136, сч
S: Я s о со 8,4 283,8 39,1 7,5 i — 0,90 1 1 15,4 9,5 70,6 1 0,106 (33°) 126
г £ Ci С© со сч кр 5 045- 6“" СО Ci С© с© о сТ со
© S S к s о О. ей Ф СП СС О) н ХО ° 1П , 255. со ю со 0 1© 2 g. сч сч с© Ю с© 200 (25 0, (21 112 00
2 00 со 233,9 г* со сч о 1 о 1 1 4,8 59,5 1 0,034 (9°) ,103 1
ю о г— С© СЧ сч со со t-. О 1Л сч со
О сч 216 сч сч сч с© О со 1 1 со Ю 1 сГ S ST
о
Формула X сч СЗ О (Л. & О О о к о & о СЗ X 04 О ° О о X X & <0 о О % со и К X & & о kft X 00 0 к «3 tb К & Q Ч & о й й СЗ о 0, сз X & «3 о Z X «3 о к 2 м о 2
Соединение 0) S 3 н S s со Н £ та е; й) Й р f-« Ef Ь. .П, S3 § S Л. Я s S « о = Scf (. ю 5 ль ci S и ес О & S тао.5 ё S. 5 Р О w s ’Э* аь. ф ь s 2 Е ея У 5.ея и н Q.SSO Е Е О й 2 S шьйВ о' ь хоое м 5 Ч “ И S S Я S Я я ЯО.О.Я S « £ < ММИЩЩЩЩиЧ. Ы S
153
Таблица (продолжение)
Соединение Формула Температура, °C Литература
0 10 20 30 40 50 60 70 80 90 100
Иодид KJ 127,5 136 144,5 152 161 168 176,2 184 191,5 200 208 [1795,2003, 2514]
Карбонат К2СО3 107,3 109 111,5 114 117 — 127 — 140 — 156 [478]
Метабисульфит k2s2o5 — — 44,5 — — — — — 124,5 — [393]
Нитрат 1 (94°)
KNO3 13,3 20,9 31,6 45,8 63,9 85,5 110,0 138 169 209 246 [394,478, 1443] [393,545]
Нитрит kno2 281 — 298 — — — — — 413
Нитрокобаль- K2Na[Co(NO2)e]. 0,042 — 0,093 — — 0,508 — — 1,306 — [1353,1443,
тиат * H2O 2321,2702]
Оксалат k2c2o4-h2o 25,2 30,3 33,0 37,7 — 50,4 — — 67,1 — [112,1160,
(25°) 1237]
Паравольфрамат KeW7O24-6H2O — — 2,15 — — — — — — — 6,6 [545]
Пеитаборат KB5O8-4H2O 1,58 — 2,75 (18°) 3,41 (25°) — — — — — — [217,2368]
Перйодат KJO4 0,17 — 0,42 — 0,93 1,44 2,16 — 4,44 — 7,87 [210,1547]
Перманганат KMnO4 2,84 4,40 6,51 9,06 12,53 16,75 21,99 28,6 — — — [210,323, 582,1740, 2189,2942]
Перренат KReO4 0,48 0,58 1,01 4,7 1,53 2,25 3,23 4,44 — 7,21 — 10,4 [159,2124, 2321]
Персульфат K2S2O8 1,8 2,7 — 11,0 — — — — — — [2178,2686]
Перхлорат KCIO4 kc9h2n3o7 0,76 1,08 1,67 2,49 0,75 3,63 4,90 7,18 — 13,4 — 22,2 [913,2140, 2520]
Пикрат 0,22 — 0,37 — — — — — — 7,1 [1070,1071, 1279,1632,
21991
Т а б л и ц а (продолжение)
Температура, °C
Соединение Формула 0 10 20 30 40 50 60 70 80 90 100 Литература
Роданид KSCN 177 — 218 243 — — — — — — — [478]
(25°)
Селенат K2SeO4 110,5 — — — — — — — — — 122,2 [545]
Сульфат k2so4 7,35 9,22 11,11 12,97 14,76 16,50 18,17 19,75 21,4 22,8 24,1 [478,1443]
Сульфит K2SO3-2H2O 106 — 107 — 108 — 109,5 — 111,5 — 114 [227,478]
Тартрат к2с4н4о6-• 0,5Н2О 132 143 155 167 — — 207 — — — — [478]
Тетраборат К2В4О7-4Н2О 8,0 — 18,5 30,2 (35°) — 46,5 58,6- — — — — [455]
Тетраоксалат КН,(С2О4)2. 1,26 1,8 — 4,28 — — 11,95 — — — 721 [1456,1772,
•2Н2О (13°) 2120]
Тиосульфато- K3[Bi(SaO3)3]- 3,5 — 7 — — — — — — — — [1462]
висмутиат • Н2О (2°) (18°)
Феррицианид K3[Fe(CN)6] 29,9 38,3 46,0 52,7 59,5 — 70,9 — 81,8 — 91,6 [478]
Ферроцианид K4[Fe(CN)e]- •ЗН2О 15,0 21,0 28,0 35,3 — 48,3 — — 67,0 — 74,5 [478]
Формиат кнсоо 290 — 335 — 381 • — 455 — 575 — 790 [478]
Фосфат КН2РО4 14,8 18,4 22,6 25 33,5 — 50,1 — 70,4 83,5 — [478,545]
(25°)
Фторид KF-2H2O — — 95 — — — — — 150 — — [393]
Фтороборат K[BF4] 0,3 — 0,6 1,0 1,4 — — — — — 8,5 [790,791]
(3°) (17°) (33°) . 0,42
Фторосиликат K2[SiFe] 0,077 — 0,177 0,246 0,268 — — -0,46 0,50 (88°) 0,95 [323,933,
(25°) (35°) (45°) (78°) 1443]
Фторотитанат K2[TiFe] 0,55 0,91 1,28 — — — — — — — — [1341,1948]
Фтороцирконат K2[ZrFe] 0,78 1,22 1,53 1,92 2,37 2,94 3,84 5,07 6,89 11,1 23,5 [1527,1531, 2032]
Таблица (окончание)
Соединение Формула температура, °C Литература
0 10 20 30 40 50 60 70 80 90 100
Хлорат ксю3 3,3 5,0 7,4 10,5 14,5 19,3 24,5 — 38,5 — 57 [478,1443]
Хлорид КС1 28,1 31,3 34,3 37,5 40,3 43,1 45,6 48,3 51,0 53,4 56,2 [206,2003, 2514]
Хромат К2СгО4 59 — 63 — 67 — 70,9 — 75,1 — 79,2 [478]
Хромовые квасцы KCr(SO4)2-• 12Н2О — 24,4 (25°) — — — — — 50 [1883,2321]
Хлороплатинат К3[Р1С1в] 0,74 0,90 1,12 1,41 1,76 2,17 2,64 3,19 3,79 4,45 5,18 [323,346, 394,819, 1630,2069]
Цианид KCN 63 — 71,6 (25°) — — 81 — 91 (75°) — — — [478]
Цитрат к3с6н5о7-н2о — — 167 (15°) 199,7 (31°) — — — — — [545]
Примечание. См. также другие данные о растворимости в воде алюминиевых квасцов [394,752,1230,1740.1883.19511 битаот-рата [20,61,176,228,312,330,346,395,518,667,725,780 ,808,863,929,1353,1632,1926,2139,2191,2335,2360,2449,2648 2843], оксалата [210,519,982,1456,1667,1791,2347], перйодата [582,689,1662,1993], перрената [1562,2272,2391,2520,2748], пепхлопа-та [61,210,228,244,323,393,394,582,832,926,1002,1230,1593,1740,1891,1895,2080 2137,2233], пикрата [1939,2071.2248 2732,2779], сульфата [394], фторобората [74,332,452,791,1030,2299,2303,2648 , фторосиликата [92,128,450,582,2063,2321’ 2640,2648], хлороплатината [393,433,637,2233,2648,2800],
Растворимость в воде и произведения растворимости солей калия
Соль Температура, °C Растворимость Коэффициент активности Произведение растворимости Литература
мэль/л | г! л катион | анион
K[BF4] 0 1,5-10-2 1,85 0,88 0,88 1,67-10-“ [452]
14 2,76-10-2 3,47 0,86 0,86 5,62-10-“ [452]
20 4,50-10-2 5,66 0,82 0,82 1,36-10-“ [791]
К[В(С6Н5)4] 25 20 7,1-10-5 1,0-10-* 0,036 0,053 5-10-» 1-Ю-8 [433,2403] [314,1312]
20 1,26-10-“ 0,064 — — 1,6-10-8 [1472]
25 1,8-10-“ 0,093 — — 3,3-10-8 [2404]
КС6Н2Ц3О7 (пикрат) 0 8,2-10-з 2,2 0,91 0,91 5,6-10-5 [1071,1279, 1632,2199]
20 1,4-10-2 3,7 0,89 0,89 1,5-10-“
KCi2H4N7O12 (дипикрилами- 0 5,2-10-“ 0,25 — — 2,7-10-’ [1317,1777, 2052,2338,2766]
нат) 15 2,0-Ю-з 0,95 0,95 0,95 6,7-10-в [419,1070,
25 2,5-10-з 1,19 0,95 0,95 1,0-10-5 1712,2262,
35 3,1-10-з 1,48 0,94 0,94 1,4-10-5 2745]
KC12H5N8O12 (монокалий- 20 2,05-10-5 0,01 — — 4,2-IO”1» [553.554]
гексанитрогидразобензол) КНС4Н4О6 (битартрат) 0 10 1,25-10-2 1,6-10-2 2,66 3,42 0,90 0,88 0,90 0,88 1,3-10-“ 2,0-10"“ [176] [176]
18 1,7-10-2 3,60 0,88 0,88 2,2-10"“ [518]
ксю4 0 5,5-10-2 7,6 0,81 0,81 2,0-10-з [913,2140]
14 8,9-10-2 12,4 0,77 0,77- 4,7-10'3 [2233]
К[1п(С2О4)2] 20 6,7-10-з 2,21 0,91 0,91 4,0-10-5 [244]
Растворимость солей калия в разных растворителях
Соль Температура, °C Растворитель Растворимость Литература
КВг 25 Глицерин 15 г/100 г растворителя [393]
К[В (С.Н6)4] 19 19 1%-ная уксусная кислота То же, 5%-ная 0,15 г/л 0,24 г/л [314] [314]
KCN 15 25 20 Глицерин Метанол Этанол 32 г/100 г растворителя 4,9 г/100 г » 0,9 г/100 г » [393] [393] [393]
KCH3SO3 (метилсульфат) 25 95%-иый этанол 0,23 г/100 мл раствора [27]
KCI0H5N2SOa (1,5-динитро-2-нафтол- -7-сульфонат) 13 30%-иый этанол 0,083 г/100 г растворителя [109J
KC12H4N7O12 (дипикриламинат) 0 0 0,01 # раствор дипикриламината кальция То же, 0,03 N 4,5-Ю-5 моль/л 2,9-10-6 моль/л [2444} [2444}
КС1 25 Глицерин 6,4 г/100 г растворителя [393]
кнс4н4о6 (битартрат) 22 0,68%-иая соляная кислота 3,01 г/100 г раствора [2843}
22 22 2,15%-ная » » 4,26%-ная » » 6,88 г/100 г » И ,19 г/100 г » [2843} [2843}
кнс4н4о6 14 14 0,81%-ная уксусная кислота 0,84%-ная лимонная кислота 0,42 г/100 г раствора 0,55 г/100 г » [2843} [2843}
кон 30 Этанол 37 г/100 г растворителя [393]
К [PbJ3] «2НаО 25 Ацетон 0,81 г/100 г растворителя [893}
159
158
160 11 Аналитическая химия калия
Растворимость в воде калиевых солей некоторых органических кислот
Кислота | 1 Формула 1 *емпе- 1 атура, °C I Растворимость Литература
5-Нитробарбитуровая (дилитуровая) C4H3N3O5 30 0,086 г/100 г воды [1070,1275] [1974]
C4H5N3SO6 18 0,348 г/100 г
5-Сульфаминобарбитуровая [2913]
C,HN3C12O, 19,7 0,55 г/100 г
Дихлорпикриновая [2614]
C,Br6SO3H 10,5 0,116 г/100 г раствора
Пен табромбен зол сульфокислота 0,567 г/100 г [1070]
c,h2n2o8 30 воды
Нитран иловая 0,193 г/100 г [2614]
CeHBr4SO3H И раствора
2,3,4,5-Тетрабромбензол-1-сульфокислота [1855]
3,4,5-Трибром-2-нитробензол-1-сульфокислота CeHBr3 (NO2) so3h 18 0,164 г/100 г
C3H2C1H3O7 30 1,82 г/100 г воды [1070]
Хлорпикриновая Стифннновая (2,4,6-тринитрорезорцин) CeH3N3O8 30 1,54 г/100 г » [1070,1443]
c«h4n«o6 7 1,69 г/100 г [1582]
2,4-Диннтрофенол 1,67 г/100 г [1582]
c,h4n2o6 ' 6 »
2,6-Динитрофенол 1,91 г/100 г [719]
CcH3Br3NSO3H 12 раствора
2,4,6-Триброманилин-З-сульфокислота 3,78 г/100 г [1070]
C7H6N3O, 30 ВОДЫ
Метилпикриновая CjHjCINOj 0 0,235 г/100 г » [1042,1069]
2-Хлор-3-нитротолуол-5-сульфокислота [2623]
C7HeN2O5 17 0,987 г/100 г воды
2,6-Динитро-4-крезол [2505]
2,5- Дин итротолу ол-4-су льфокисл ота |c7h6n2so7 14,5|0,523 г/100 г »
Таблица {окончание}
Формула 1О 00 00 « „ й Л ° ° ° о z Ч °2. z ч 5 z» z z < Я xs х О О О О о о
Кислота Диметилпикриновая кислота 2-Хлор-3-нитро-4-ксилол сульфокислота 2,4,5-Тринитро-1-оксинафталин Флавиановая (2,4-динитро-1-нафтол-7-су льфокислота) 1,5-Динитро-2-нафтол-7-су льфокислота 1,5-Динитро-2-нафтол-7-7-су льфокислота
_00 ю о о о О
о г/Э о ’S' GO СЗ GO со "2.
сз Z Z 00 о Z о Z о Z 03 Z
X X X X X X X
о о 00 00
о о о о о о о
ср
О
ч
ГJ
я
в
о
4+
43 со
Ч
о
CJ Ч
Я
СО СО ч X о
О о о со •е-
ч Ч X ф ч
я Я сЗ хо
X о 43 сЗ О X о е* 43 О ч о со сЗ ч
ч 5 Ч X о X я о со
X о ++ ч Ф
о 43 я \о
ч о 43 ** ч ч & X сЗ о о со СО
о Он
•е* со
СО о X я X
я oq я 05 X ф \о о е; о X я ч я
о Он сЗ СО О сЗ Он со Я X ф \о Vf< X со о
я в я ЕХ § ч о о, о со X ф X о X ЕХ о со со я о я Он я X
vA х я \о я X о X
< X ч СО сэ
4
i
I
Растворимость хлорида калия в водных растворах соляной кислоты прн 25°С (1519]
Концентрация НС1 Растворимость КС1 Концентрация НС1 Растворимость КС1
ллимь/10 мл жлолб/10 мл
0 42,7 15,9 28,7
5,66 37,5 20,9 24,7
10,2 33,8 J 32,5 17,4
Растворимость хлорида калия (г/1СО г раствора) в смесях воды с этанолом [478, 1921, 2395, 2467], ацетоном [2589], пиридином [2496]
Орган ический растворитель, вес. % Растворитель
этанол (15°С) ! ацетон (20°С) 1 пиридин | (Ю°С)
0 24,6 30,6 I 23,8
10 19,8 26,2(9,1%) 19,7
20 14,7 21,4 • 16,4
30 10,7 16,7 . 13,2
40 7,7 12,4 10,4
50 5,0 8,6 6,3
60 2,8 5,3 3,3
70 — 2,9 1,25
80 0,45 0,96 0,24
90 . — 0,15 0,04
100 — ~0 —
Примечание и конц. НС1 см. [8S Растворимость хлорида калия в Юа, 1564, 1990] смесях этанола
Растворимость хлорида калня в органических растворителях
Растворитель Температура, °C Растворимость | Литература i
Метанол 25 0,54 г/100 г растворителя [393, 547, 680, 1832]
Этанол 25 0,029г/100 г » [394, 876, 1832, 1990]
н.Пропанол 25 0,006 г/100 г » [1832]
Изопропанол 25 0,0023 г/100 г [1832]
н.Бутанол 25 0,003 г/100 г » [1724, 1832]
Изобутанол 25 0,00084 г/100 г [1832, 2863]
н.Пентанол 25 0,0022 г/100 г [1832]
Изопентанол 20 0,6 лтг/100 мл раствора [909]
н.Гексанол 20 0,04 мг/100 мл » [909]
2-Этилгексанол I 20 0,01 лтг/ЮО мл » [9091
11*
163
Растворимость бромида калия в смесях воды и этанола при 30°С [2694]
Этанол, вес. % г КВг/100 г раствора Этанол, вес. % г КВг/100 г раствора
0 41,6 50 16,3
5 39,0 60 11,5
10 36,3 70 6,9
20 31,1 80 3,1
30 26,0 90 0,87
40 21,4
Примечание. Другие данные о растворимости бромида калия в смесях воды и этанола см. [478, 638].
Растворимость бромида калия в смесях воды н ацетона прн 25°С [1521]
мл ацетона в 100 мл смеси г КВг/100 мл .раствора 1 мл ацетона 1 в 100 мл смеси г КВг/100 мл раствора
0 57,3 60 17,2
20 43,7 70 11,3
30 37,0 80 5,5
40 30,8 90 1.2
50 24,1
Растворимость бромида калия в органических растворителях при 25°С
Растворитель Растворимость, е/100 г растворителя Литература
Метанол 2,08* [393, 547, 1318, 1832,
1881, 2756]
Этанол 0,135 [876, 1318, 1832, 2756]
н.Пропанол 0,031 [1832, 2756]
Изопропанол 0,011 [1422, 1832]
Н. Бутанол 0,013 [1422, 1832]
Изобутанол 0,0044 [1422, 1832]
н.Пенталон 0,0048 [1422, 1832]
Изопентанол 0,003 [2756, 2952]
Ацетон ........ 0,023 [393, 394, 1805, 1830,
2043]
* При 20 °C.
164
Растворимость иодида калия в смесях воды и метанола или этанола [478]
Спирт, вес. % г KJ/10Q г растворителя Спирт, вес. % г КJ/100 г растворителя
метаиол (25°С) этанол (18°С) метанол (25°С) этанол (18°С)
20 113 105 80 35,5 19,6
40 62 73,9 90 — 6,9
60 55 47,1 100 17,2 1,9
Растворимость бромида и пиодида калия в смесях воды и метанола при 25°С [478, 607, 1520]
а метанола в 100 а смеси Растворимость, молъ/кг растворителя г метанола в 100 г смеси Растворимость, моль/ка растворителя
КВг KJ КВг KJ
0 5,84 8,96 80,0 0,46 1,97
20 3,71 6,77 89,8 0,26 1,41
40,8 2,18 4,84 94,7 0,20 1,19
59,6 1,18 — ' 100 0,18 0,96
72,4 0,77 2,47
Растворимость пиодида калия в органических растворителях при 25°С
Растворитель Растворимость, 2/100 г растворителя Литература
Метанол 17,04 [393, 513, 547, 1522,
1832, 1758, 2835]
Этанол 1,87 [478, 513, 1522, 1832,
2835]
н.Пропанол 0,44 [513, 1522, 1832]
Изопропанол 0,177 [513, 1522, 1832]
н.Бутанол 0,20 [513, 1522, 1832]
Изобутанол 0,058 [513, 1522, 1832]
н.Пентанол 0,089 [513, 1522, 1832]
Ацетон 1,3 [394, 1833, 2043, 2835]
Растворимость сульфата калия в растворах серной кислоты при 25°С [1519] (все величины в ммолях на 10 мл)
Концентрация H8SO4 Растворимость K1SO4 Концентрация HjSO, Растворимость k2so4
0 6,2 7,6 10,8
3,8 8,9 14,3 14,9
165
Растворимость сульфата калия в смесях воды и этанола прн 15°С [478, 2467, 2484]
Этанол, вес. % Растворимость K2SO4, г/100 г раствора Этанол, вес. % Растворимость K2SO4, г/100 г раствора
0 10,4 30 0,55
10 3,9 40 0,21
20 1,46
Растворимость нитрата калия в смесях воды и метанола при 25°С [478, 607]
Метанол, вес. % Растворимость, моль/кг растворителя Метанол, вес. % Растворимость, моль/кг растворителя
0 3,77 59,9 0,31
4,98 3,05 70,0 0,19
9,45 2,50 78,4 0,11
21,0 1,52 89,5 0,06
40,3 0,71 1
Растворимость (г/100 г растворителя) соединений калия в смесях воды и этанола [478, 2467]
Этанол, вес. % KNO, (20°С) КСЮ, (30°С) КОН (30°С)
10 20,0 — —
20 16,0 4,7 —
40 5,25 2,4 —
50 — — 46
60 2,04 1,02 42,7
70 — — 38
80 0,7 0,24 35,7
90 0,3 0,06 34,8
Растворимость перхлората калия в водных растворах хлорной кислоты при 25°С [2705]
НСЮ4. N Растворимость, г/100 г раствора НСЮ„ N Растворимость, г/100 г раствора
0 2,085 0,10 1,485
0,01 1,999 1,00 0,527
166
Растворимость перхлората калия в смесях воды и этанола при 14°С [2233]
Этанол, вес. % Растворимость, г/л раствора Этанол, вес. % Растворимость, г/л раствора
0 12,4 42,4 3,9
7,1 9,2 58,5 ' 2,6
13,2 7,8 94,7 0,15
27,3 5,7
Примечание. Другие данные о растворимости перхлората калия в смесях воды и этанола см. [478, 714, 1761, 2394, 2412, 2534, 2579].
Произведение растворимости (ПР) перхлората калия в смесях воды н этанола при 20°С [2134]
Вода, вес. % ПР-10‘ Вода, вес. % ПР- 10s
0,35 0,099 10 2,00
3 0,165 12 2,99
5 0,52 15 3,18
7 0,65
Растворимость перхлората калия (лгг/100 мл растворителя) в этаноле, содержащем хлорную кислоту, при комнатной температуре [419, 2705]
Этанол, НС1О< (вес. %) в этаноле
объемы. % 0 0,2 0,3 0,75 1 1,5
98 11,9 2,4 —
96 16,4 3,4 — 2,7 2,4 1,5
94 21,9 6,4 5,9 — 2,9 —
Растворимость перхлората калия в разных растворителях при 25°С [115, 2394, 2576, 2863, 2905]
Растворитель Растворимость, 1 г/100 мл раствора Растворитель Растворимость, г/100 мл раствора
Вода 2,04 . Изобутанол 0,005
Метанол 0,083. Этилацетат 0,0013
Этанол 0,0094 Амилацетат 0,014
н.Пропанол 0,008 Ацетон 0,118
н.Бутанол 0,0036
167
Растворимость фторосиликата калия в смесях воды и этанола при 14°С [2233]
Этанол, вес. % Растворимость, г/л раствора Этанол, вес. % Растворимость, г/л раствора
0 0,9 27,3 0,09
8,7 0,46 42,4 0,05
15,6 0,29 94,7 0,0096
Растворимость хлороплатината калия (г/100 г растворителя) в смесях воды и метанола или этанола при 20°С [637]
Спирт, вес. % Метаиол Этанол Спирт, вес. % Метаиол Этанол
0 0,774 0,774 50 0,062 0,049
5 0,535 0,491 60 0,032 0,026
10 0,412 0,372 70 0,018 0,013
20 0,264 0,218 80 0,012 0,0085
30 0,183 0,134 90 0,0038 0,0025
40 0,116 0,076 100 0,0027 0,0009
Примечание. Другие данные о растворимости хлороплатината калия в смесях воды с метанолом или этанолом см. [346, 1264, 1429, 2208, 2233, 2255].
Растворимость битартрата калия в воде [929, 2191]
Температура, °C Растворимость, г/100 г раствора Плотность насыщенного раствора Л* d4 Температура, °C Растворимость, г/100 г раствора Плотность насыщенного раствора Л°
0 0,265 1,00145 18 0,489 1,00120
5 0,297 1,00224 20 0,534 1,00100
10 0,366 1,00180 25 0,641 1,00079
14 0,430 1,00172 30 0,762 1,00026
Растворимость битартрата калия (г/100 г раствора) в водных растворах винной кислоты [929, 2335]
Концентрация виииой кислоты, г/100 г раствора Температура, °C
0 10 25
0 0,231 0,358 0,641
0,025 0,216 0,346 0,622
0,05 0,208 0,335 0,606
ОД 0,196 0,320 0,584
0,2 0,184 0,300 0,556
0,4 0,168 0,271 0,524
0,6 0,160 0,250 0,498
0,8 0,150 — 0,472
168
Растворимость бнтартрата калия (г/л) в смесях воды и этанола [176]
Этанол, объемы. % Температура, ®С
5 10 14 18 20 25
0 2,66 3,42 3,73 4,64 5,10 5,68
5 2,26 2,66 3,12 3,59 3,85 4,51
10 1,56 1,97 2,31 2,78 2,91 3,42
15 1,16 1,53 1,84 2,16 2,33 2,73
25 0,65 0,92 1,11 1,30 1,43 1,68
Примечание. Другие данные о растворимости битартрата калия в смесях воды и этанола см. [312 , 956, 1498, 1726, 2191,2192 , 2233, 2360, 2519 , 2874].
Растворимость калий-бортетрафенила в смесях воды и ацетона при 28° С [2513]
Ацетон, объемы. % Растворимость, мг!м,л Ацетон, объемы. % Растворимость, мг/мл
16 ОД 60 18,4
25 0,6 70 27,0
32 1,2 80 40,6
40 2,5 90 53,0
44 4,9 95 59,8
50 10,5 100 41,8
Растворимость пикрата калия (г/100 мл раствора) в смесях воды с органическими растворителями при 25° С [537, 1215]
Растворитель, объемы. % Метаиол Этанол Ацетон
0 0,645 0,645 0,645
10 0,542 0,559 0,726
20 0,470 0,450 0,876
30 0,444 0,472 . 1,140
40 0,422 0,533 1,155
50 0,411 0,582 2,106
60 0,410 0,574 2,615
70 0,396 0,485 3,09
80 0,332 0,326 3,34
90 0,254 0,174 3,08
100 0,274 — 1,08
Примечание. Другие данные о растворимости пикрата калия в органических растворителях и их смесях с водой см. [798, 1279, 1420, 1421, 21991.
169
Плотность растворов едкого кали [478, 606, 1443]
Концентрация КОН .15 d 4 Концентрация КОН 4
% г/л % | г/л
1 10,08 1,0083 27 340,0 1,2592
2 20,35 1,0175 28 355,5 1,2695
3 30,80 1,0267 29 371,2 1,2800
4 41,44 1,0359 30 387,2 Ц2905
5 52,26 1,0452 31 403,3 1,3010
6 63,26 1,0544 32 419,7 1,3117
7 74,46 1,0637 33 436,4 1,3224
8 85,84 1,0730 34 453,3 1,3331
9 97,42 1,0824 35 470,4 1,3440
10 109,2 1,0918 36 487,8 1,3549
11 121,1 1,1013 37 505,4 1,3659
12 133,3 1,1108 38 523,2 1,3769
13 145,6 1,1203 39 541,3 1,3879
14 158,2 1,1299 40 559,6 1,3991
15 170,9 1,1396 41 578,2 1,4103
16 183,9 1,1493 42 597,0 1,4215
17 197,0 1,1590 43 616,1 1,4329
18 210,4 1,1688 44 635,5 1,4443
19 223,9 1,1786 45 655,1 " 1 4558
20 237,7 1,1884 46 675,0 1,4673
21 251,7 1,1984 47 695,1 1,4790
22 265,8 1,2083 48 715,5 1,4907
23 280,2 1,2184 49 736,2 1,5025
24 294,8 1,2285 50 757,2 1,5143
25 309,7 1,2387 51 778,4 1,5262
26 324,7 1,2489 52 799,9 1,5382
Плотность водных растворов Л/*5 I важнейших солей калия [1353,1443]
Соль, вес. % КВг КС1 KNO3 К2СО3 К2СгО4 K2SO4
1 1,0054 1,0046 1,0045 1,0072 1,0080 1,0063
24 1,0127 1,0110 1,0108 1,0163 1,0161 1,0145
О 1,0275 1,0239 1,0203 1,0345 1,0325 1,0310
1,0426 1,0369 1,0363 1,0529 Ц0492 1,0477
8 1,0581 1,0500 1,0494 1,0715 1,0663 1,0646
10 1,0740 1,0633 1,0627 1,0904 1,0837 1,0817
12 1,0903 1,0708 1,0762 1,1096 1,1014 -
14 1,1070 1,0905 1,0899 1,1291 1,1195 —
16 1,1242 1,1043 1,1039 1,1490 1,1380
18 1,1419 1,1185 1,1181 1,1692 1,1570
20 1,1601 1,1328 1,1326 1,1898 1,1765
22 1,1788 1,1474 1,1473 1,2107 1,1964
24 1,1980 1,1623 1,1623 1,2320 1,2169
26 1,2178 — — 1,2536 1,2379
28 1,2383 — — 1,2756 1,2592
30 1,2593 — 1,2979 1,2808
32 — — — — 1,3035
34 — — .— — 1,3268
36 — — •— —. 1,3505
38 —. — — — 1,3746
40 1,3746 — — — 1,3991
170
Показатели преломления водных растворов солей калия [1353]
г КС! в 100 мл раствора „17.5° nD г KNOa в 100 мл раствора „17.5° nD ! г К2СО2 в 100 мл раствора „17.5° nD a K2SO4 в 100 мл раствора „17.5° nD
0,03 1,33324 0,41 1,33358 1,11 1,33513 0,30 1,33358
1,75 1,33555 1,23 1,33435 3,41 1,33896 0,91 1,33435
5,00 1,33980 2,05 1,33513 5,79 1,34275 2,16 1,33590
9,99 1,34597 4,14 1,33705 8,25 1,34650 3,12 1,33705
14,99 1,35198 6,30 1,33896 13,41 1,35388 4,11 1,33820
18,00 1,35547 . 8,52 1,34086 16,09 1,35750 5,11 1,33934
20,01 1,35779 10,76 1,34275 18,82 1,36109 6,13 1,34048
25,00 1,36347 — — 21,59 1,36464 7,15 1,34162
27,89 1,36675 — 23,17 1,36675 8,20 9,25 10,49 1,34275 1,34388 1,34515
Показатели преломления водных растворов бромида и иодида калия [271, 475, 534]
Содержание КВг в растворе, % 20* nD Содержание KJ в растворе „20-D
0 1,3330 0 1,3330
1 1,3342 1,01 1,3343
2 1,3355 1,90 1,3355
2,95 1,3366 3,02 1,3370
3,94 1,3379 3,95 1,3383
4,91 1,3391 4,83 1,3395
5,94 . 1,3404 5,84 1,3410
6,94 1,3407 6,71 1,3422
8,74 1,3440 8,43 1,3446
9,84 1,3455 9,60 1,3464
11,03 1,3470 10,25 1,3474
11,86 1,3482 11,38 1,3491
13,82 1,3508 12,69 1,3511
14,80 1,3521 13,80 1,3529
15,94 1,3537 14,68 1,3543
17,50 1,3559 17,67 1,3592
19,85 1,3590 19,60 1,3625
Показатели преломления растворов смеси хлоридов калня и натрия [142]
(1 г смеси хлоридов калия и натрия растворены в 10 мл воды)
Содержание в смеси, г Л14’ Содержание в смеси, г
КС1 NaCl КС1 NaCl
0 1 1,3491 0,60 0,40 1,3468
0,05 0,95 1,3488 0,70 0,30 1,3465
0,20 0,80 1,3483 0,80 0,20 1,3460
0,30 0,70 1,3479 0,90 0,10 1,3457
0,40 0,60 1,3475 1 0 1,3453
0,50 0,50 1,3472
171
Вязкость растворов КОН, КС1 и KNO3 [478]
Содержание в растворе, % Температура, °C
Соединение 10 20 30
Вязкость раствора, сантипуазы
кон 5 1,19 0,80
10 1,23 1,00
20 1,63 1,33
25 1,94 1,59
КС! 5 1,27 0,99 0,80
10 1,25 0,99 0,81
20 1,25 1,02 0,85
KNO3 10 20 1,22 1,25 0,97 1,01 0,80 0,81
Температура кипения водных растворов некоторых соединений калия [393]
Температура кипения, °C
Соединение 101 102 103 104 105 107 по 115 120 125
Концентрация, г/100 г воды
кон 4,7 9,3 13,6 17,4 20,5 26,4 34,5 47,0 57,5 67,3
КС! 9,2 16,7 23,4 29,9 36,2 48,4 57,4* — — — —
KNOa 15,2 31,0 47,5 64,5 82,0 120,5 189 389* — —
к2со3 11,5 22,3 32,0 40,0 47,5 60,5 78,5 104 128 153
Насыщенный раствор при указанной температуре.
Коистанты диссоциации некоторых' соединений калия
Соединение или иои Иоииая сила раствора Температура, °C к РК Литература
КВгО3 — — 2,2 —0,34 [678, 1440]
КС1О3 — — 1,5 —0,17 [678, 1440]
КСЮ4 — — 3,2 —0,5 [678, 1440]
K[Fe(CN),]3- — — 0,005 2,3 [1041]
KJO3 0 25 2 —0,3 [586 , 678, 1040, 1440,
1440]
KNO3 — — 1,37 —0,14 [678, 1440]
kso; 0 25 0,11 0,96 [244 , 586, 1440 , 2609]
ks2o; 0 25 0,12 0,92 [586, 1067,1339, 1440]
K2C2O4 0,07 — 1,76 —0,24 [678]
K2C2O4 0,29 —. 1,43 —0,15 [678]
K2CrO4 0,14 — 4,35 —0,64 [678]
К2СГО4 0,28 —• 3,2 —0,50 [678]
172
Л ИТЕРАТУРА
1. Абдуллаев А. А., Лобанов Е. М„ Новиков А. П„ Кайда-ров А А. Изв. АН УзССР, сер. фнз.-мат. наук, № 4, 65 (1960); РЖХим., № 8 Д144 (1961).
2. Агрохимические методы исследования почв. М., Изд-во АН СССР, 1954
3. Агте А. Н. Тр. Ленинградск. технолог, ин-та, 48, 182 (1958).
4. А й д а р о в Т. К. Спектральное определение калия, натрия и магния в рассолах и солях. Автореферат. М., Всес. нн-т минер, сырья, 1958.
5. Айдаров Т. К-, Зак А. Е., Сафонова Е. С. Зав. лаб., 25, 269 (1959).
•6. Айдиньян Н. X., Серебряников С. Н. Номограммы для расчета химических анализов. Л., ОНТИ, Химтеорет, 1937.
7. Акулович В. М., Павлюченко М. М. Сборник докладов Первой научно-технической конференции по спектральному анализу. Минск, Изд-во АН БССР, 1956, стр. 40; РЖХим., № 56832 (1959).
8. Алабышев А. Ф., Грачев К- Ю., За редкий С. А., Л ант ратов М. Ф. Натрий и калий. Л., Госхимиздат, 1959.
9. Александров В. Г. Почвоведение, № 12, 749 (1946).
10. Александров Г. П., Шутер Я. Н. Зав. лаб., 21, 1432 (1955).
11. Александров Д. К. ЖРФХО, 39, 1391 (1907).
12. Александров С. Н., Шмуляковский Я. Э., Алексеев С. А. Химия и технол. топлива, № 4, 68 (1956); РЖХим., № 37962 (1957).
13. Алексеев В. Н. Курс качественного химического полумикроанализа. М.— Л., Госхимиздат, 1952.
14. Алексеева А. М. Бюлл. экспер. биол. и мед., 1, 301 (1936).
15. Алексеевский Е. В., Г о л ь ц Р. К., М у с а к и н А. П. Количественный анализ. Л.— М., Госхимиздат, 1948.
16. Алима рин И. П. Зав. лаб., 13, 917 (1947).
17. Али марин И. П. Доклады советской делегации на международной конференции по мирному использованию атомной энергии. Применение изотопов в технике, биологии и сельском хозяйстве. М., Изд-во АН СССР, 1955, стр. 152.
18 Алимарин И. П., Архангельская В. Н. Качественный полумикроанализ. М.— Л., Госхимиздат, 1952.
19. Алимарин И. П., Ромм И. К. Тр. Ин-та минер, сырья, № 76, 3 (1935).
20. Анализ минерального сырья (под ред. Ю. Н. Книпович и Ю. В. Морачев-ского). Л., Госхимиздат, 1956.
21. Андреева Е. А. Почвоведение, № 5, 21 (1960).
22. Ан Ли Тхэ, Сии Чхан Сук. Ж. химии и хим. промышл. (Корея), № 3, 149 (1957); РЖХим., № 21147 (1958).
23. Антипов- Каратаев И. Н., Мясникова А. М. Тр. Ленинградск. отд. Почвенного ин-та им. В. В. Докучаева, 17, 69 (1933).
24. Антипов-Каратаев И. Н„ Мясникова А. М. Тр. Ленинградск. отд. Почвенного ин-та им. В. В. Докучаева, 17, 81 (1933).
25. Антошин С. Т. Удобрение овощных культур. Л., Госхимтехиздат, 1934.
26. А н ш е л е с О. М., Буракова Т. Н. Микрохимический анализ на основе кристаллооптики. Изд-во Ленинградск. ун-та, 1948.
27. Арбузов А. А., Карташов Н. ЖРФХО, 46, 284 (1914).
173
28. Арнаутов Н. В. Материалы Второй научно-технической конференции молодых ученых ЗСФ АН СССР. Новосибирск, 1957, стр. 77; РЖХим., № 28399 (1959).
29. Афанасьев Г. Д. Тр. Шестой сессии Комиссии по определению абсолютного возраста геологических формаций, 1957. М, Изд-во АН СССР, 1960, стр. 70; РЖХим., № 7 ПО (1961).
30. Афанасьев Г. Д, Кожина Т. К., Старик И. Е. Сб.: Определение абсолютного возраста дочетвертичных геологических формаций. М, Изд-во АН СССР, 1960, стр. 9; РЖХим., № 6 ПО (1961).
31. Афанасьев Л. А., Гол ба Т. Е., Красно веки й О. В., О х о-тин М. В., Полляк В. В., Фиалковская Р. П., Фрезе Н. А. Практикум по контролю производства стекла. М.— Л., Гизлегпром, 1939
32. Бабаев Н. Б. Изучение растворимости некоторых малорастворимых щелочных солей гетерополикислот. Автореферат, Ташкент, 1960.
33. Б а б а е в а А. В., К л я ч к о - Г у р в и ч Л. П. Ж. общ. химии, 5, 220 (1935).
34. Б а б а л о в а А. А., П о п о в а И. Б. Тр. Уральск, научно-исслед. хим. ин-та, 8, 233 (1959).
34а. Бабина Н. М., Гошпаренко Л. П. Доклады межвузовской научной конференции по спектроскопии и спектральному анализу. Томск, ун-т, 1960, стр. 12; РЖХим, № 14 Д36 (1961).
35. Бабко А. К. Сборник трудов Института строительных материалов. Киев, 1930 (цит. по [37]).
36. Бабко А. К. Сборник трудов Института строительных материалов. Киев, 1931, стр. 47.
37. Бабко А. К-, Пятницкий И. В. Количественный анализ. М., Госхим-издат, 1956.
38. Бабко А. К- Романишина Е. В. Зав. лаб, 16, 1416 (1950).
39. Б а г б а н л ы И. Л, С е м и х а н о в И. Р. ДАН Азерб. ССР, № 2 (1950).
40. Б а г б а н л ы И. Л, Семиханов И. Р. Тр. Комиссии по аналит. химии, 5 (8), 167, 173 (1954).
41. Бадеева Т. И. Аналитическая химия природных рассолов. Автореферат. Саратов, 1952.
42. Балабанов Б. Г. Почвоведение, № 9, 98 (1953).
43. Балаховский С. Д, Б а л аховский И. С. Методы химического анализа крови. М, Медгиз, 1953, стр. 202.
44. Балашев Л. Л, Дружинин Д. В. Калийное удобрение. Л, Научное хим.-техн. изд. НТУ ВСХН, 1929.
45. Б а л ю к. С. Т, Г у р в и ч Г. А. Зав. лаб, 17, 364 (1951).
46. Банных 3. С„ Сачко А. П. Тр. Уральск, научно-исслед. хим. ин-та, № 4, 209 (1957).
47. Баранов В. И. Радиометрия. М, Изд-во АН СССР, 1955, стр. 57.
48. Баранов П. А. Химизация соц. земледелия, № 5, 52 (1940).
49. Бассет Г. Теория количественного анализа и ее практическое применение. Л, Гос. научно-техн, изд-во, 1932.
50. Б а х у л и н М. Д. Почвоведение, № 5, 787 (1938).
51. Белоусова Е. М. Объемное определение калия висмутотиосульфатным методом. Автореферат. Одесса, 1951.
52. Белоусова Е. М. Сборник хим. факультета Одесск. гос. ун-та, 3, 81 (1953).
53. Бельский В. Е, Фомин О. К. Зав. лаб, 26, 707 (1960).
54. Беляев И. Н, Нестерова А. К- ДАН СССР, 86, 949 (1952).
55. Б е р г Р. Применение о-оксихинолина в аналитической химии. М, ОНТИ, 1937, стр. 43.
56. Беренс Г, Клей П. Д. Микрохимический анализ. Л, Научное хим,-техн. изд. НТУ ВСНХ, 1928, стр. 26, 38.
57. Б е р л ь - Л у н г е. Химико-технические методы исследования, т. 2, часть 1, вып. 2. Л, ОНТИ, 1937, стр. 422.
58. Бетгер В. Основы качественного анализа. М.— Л, ГОНТИ, 1931, стр. 303.
58а. Бетехтин А. Г. Курс минералогии. М, Госгеолтехиздат, 1961.
174
59. Благовещенская Г. И. Тр. Казанск хим.-технол. ин-та, № 29, 36 (1960); РЖХим., № 12 Д66 (1961).
60. Б л о к Н. И. Пособие к практическим занятиям по качественному микрохимическому анализу. М., Изд. Моск, текстильн. ин-та, 1939.
61. Блок Н. И. Качественный химический анализ. М.— Л., Госхимиздат, 1952, стр. 170.
62. Б л о х М. А. Природа, 13, 114 (1924).
63. Б л о х М. А. Хронология важнейших событий в области химии и смежных дисциплин и библиография по истории химии. Л.— М„ Госхимиздат, 1940.
64. Боброва В. Н. Радиохимия, 2, 243 (1960).
65. Бондаренко А. С. Научн. записки Херсонск. с.-х. ин-та им. А. Д. Цюрупа, № 1, 196 (1939).
66. Боровик С. А. ДАН СССР, 89, 333 (1953).
67. Б о р о в и к - Р о м а н о ва Т. Ф. Спектрально-аналитическое определение щелочных и щелочноземельных элементов. М., Изд-во АН СССР, 1956, стр. 42.
68. Боровик-Романова Т. Ф. Физический сборник (Львовск. ун-т), № 4, 361 (1958).
69. Б о р о в и к - Р о м а н о в а Т. Ф., Фарафонов М. М. Тр. Комиссии по аналит. химии, 12, 322 (1960).
70. Бреслер С. Е. Радиоактивные элементы. М.— Л., Гостехиздат, 1952, стр. 269, 272.
71. Бродский А. И. Химия изотопов. М., Изд-во АН СССР, 1957, стр. 14, 47, 514, 521.
72. Бродский Д. А. Химия в школе, № 3, 33 (1939).
73, Будягин Ф. Лабор. практика, № 6, 7 (1925).
74. Буз Г., Мартин Д. Химия трехфтористого бора и его производных. М., ИЛ, 1955, стр. 99, 104.
75. Буракова Т. Н. Усовершенствование метода качественного микрохимического анализа на основе использования кристаллооптических констант. Автореферат. Л., 1955.
76. Б у р к с е р Е. С. Очерки явлений радиоактивности. Николаев. Издание журнала «Физик — любитель», 1909.
77. Бурксер Е. С. Записки императорского об-ва сельского хозяйства южной России, 84, 35 (1914).
78. Бурксер Е. С. Тр. Радиевой экспедиции Академии наук, № 7, 2 (1915).
79. Б у р к с е р Е. С. См. по [890а].
80. Б у р к с е р Е. С. Как определять возраст горных пород и земли. Киев. Изд-во АН УССР, 1954, стр. 19.
81. БурксерЕ. С., КондогуриВ. В., Капустин Н. П. Калий, 1, № 3, 12 (1932).
82. Бурксер Е. С., Рутковская В. Л., Бауман А. М. Сов. пат. 24393 (1931).
83. Бутырин П. Н. Полевой количественный химический гидроанализ пробирочно-капельным методом. Л., ГОНТИ, 1931, стр. 74, 82.
84. Бутырин П. Н., Лаптев Ф. Ф. Таблицы для пересчета анализов вод в эквивалентную форму. М.— Л., Госгеолразведиздат, 1933, стр. 33.
85. Быковская Е. В., Полевая Н. И., Подгорная Н. С. Советская геология, № 5, 107 (I960).
86. Вагнер И. В., Новосельская А. И., Титаренко Л. П. Автоматизация и приборостроение, № 1, 64 (1959).
87. Вайнштейн Э. Е„ КаханаМ. М. Справочные таблицы по рентгеновской спектроскопии. М., Изд-во АН СССР, 1953, стр. 129.
88. Вайнштейн Э. Е., Королев В. В. Ж. аналит. химии, 11, 627 (1956).
89. В а н а г Г. Я. Нитроиндандион. Рига, Изд-во АН Латв. ССР, 1954.
90. Вань Бан-хэ, Хуан Фу-чэнь. Чжуиго кэсюэюань шию янь-цзюсо, № 1, 35 (1958); РЖХим., № 53109 (1959).
91. Васильев А. А., Матвеев Н. Ж. прикл. химии, 3, 443 (1930).
92. Васильев А. А., Мартьянова Н. Н. Ж. прикл. химии, 9, 152 (1936).
175
93. Васильев А. М. Уч. зап. Казанск. гос. ун-та, 90, 977 (1930).
94. Васильев X. Лека промишленост, 2, 25 (1953); РЖХим, № 37487 (1955).
95. Вейс А. Р„ Иевиньш А. Ф. Уч. зап. Латв. гос. ун-та, 22, 95 (1958).
96. Вернадский В. И. Изв. Академии наук, 1910, 1924.
97. Вернадский В. И. См. по [2802а].
98. Вернадский В. И. Избранные сочинения. М., Изд-во АН СССР, т. 1> 1954, стр. 33; т. 2, 1955, стр. 24.
99. Ветров А. С. Тр. Комиссии по аналит. химии, 3 (6), 342 (1951).
100. Вильямович Е. Т. Зав. лаб., 9, 1330 (1940).
101. Виноградов А. П. Геохимия живого вещества. Л., Изд-во АН СССР, 1932, стр. 20.
102. Виноградов А. П. Геохимия редких и рассеянных химических элементов в почвах. М., Изд-во АН СССР, 1950, стр. 114, 228, 255.
103. Виноградов А. П. Геохимия, № 1, 6 (1956).
104. Виноградов А. П. ДАН СССР, ПО, 375 (1956).
105. Витынь А. Ж. опыта, агрономии, 13, 192 (1912).
106. Владимиров И. С. Ж. хим. промышл., 8, 972 (1931).
107. Вознесенский С. А. Тр. Всес. конференции по аналит. химии, 1, 66 (1939).
108. Войцеховский А. Е., Страшок А. Ф. Тр. Научно-исслед. ин-та основной химии, 11, 334 (1958).
109. Волочнева Е. П. Ж- прикл. химии, 11, 369 (1938).
НО. Воробьев Г. Г. Физический сборник (Львовск. ун-т), № 4, 378 (1958).
111. Ворсин А. Н. Тр. Пятой сессии Комиссии по определению абсолютного возраста геологических формаций, 1956. М., Изд-во АН СССР, 1958, стр. 274.
111а. Вортман Г. Общий ход качественного химического анализа без применения сероводорода. Киев, 1910, стр. 20.
112. Воскресенская Н. Изв. Ин-та физико-химич. анализа, 3, 458 (1926).
113. Вяхирев Д. А., Кулаев Ф. Н. Тр. Комиссии по аналит. химии, 6 (9), 527 (1955).
114. Ган ч ев Н. Изв. Хим. ин-т Българ. АН, 4, 311 (1956); РЖХим., № 51535 (1957).
115. Г а н ч е в Н. Изв. Хим. ин-т Българ. АН, 6, 149 (1958); РЖХим., № 86072 (1959).
116. Гапченко М. В., Шейн цис О. Г. Зав. лаб., 14, 410 (1948).
117. Г ев еш и Г., П а н е т Ф. Учение о радиоактивности. Берлин, Изд-во «Научная мысль», 1924, стр. 179.
118. Гегечкори Н. М. Зав. лаб., 20, 444 (1954).
119. Гедройц К- К. Химический анализ почвы. М.— Л., Сельхозгиз, 1932, стр. 135.
120. Георга И. Г. См. по [1315а].
121. Герке Ф. К- Успехи аналитической химии. М., Изд-во Моск, высшего технич. училища, 1929, стр. 11.
122. Г е р л и н г Э. К., Титов Н. Е. Изв. АН СССР, Отд. хнм. наук, № 2, 128 (1940).
123. Герлинг Э. К.. Титов Н. Е., Ермолин Г. М. ДАН СССР, 68, 553 (1949).
124. ГерлингЭ. К., Павлова Т. Г. ДАН СССР, 77, 85 (1951).
125. Г ер л инг Э. К-, Рик К- Г. Метеоритика, 10, 37 (1952).
126. Г ер л ин г Э. К., Ященко М. Л. ДАН СССР, 83, 901 (1952).
127. Г и л л е б р а н д В. Ф., Л е н д е л ь Г. Э., Брайт Г. А., Гофман Д. И. Практическое руководство по неорганическому анализу. М., Госхимиздат, 1957, стр. 147, 667, 919.
128. Гильденгершель X. И., Шагисултанова Г. А. Ж. прикл. химии, 26, 222 (1953).
129. Гиммельфарб Б. М., Херасков Н. П. Калийные соли. М., Изд-во «Советская Азия», 1933,
176
F
130. Гинзбург К- Е., Щеглова Г. М. Почвоведение, № 5, 100 (I960).
131. ГинзбургС. А. Зав. лаб., 2, 11 (1933).
132. Глик д. Методика гисто- и цитохимии. М., ИЛ, 1950, стр. 20, 144, 252.
133. Головаты й Р. Н. Ж. прикл. химии, 12, 1241 (1939).
134. Гортиков В. М„ Знаменская Л. А. Тр. Ленинградск. отд. Всес. научно-исслед. ин-та удобрений, 17, 57 (1933).
135. Горшков В. И., Кузнецов И. А., Панченков Г. М. Ж- аналит. химии, 14, 417 (1959).
136. Грабаров П. Г., Ксандопуло Г. И., Солодникова Е. А., Войнова Т. Н. Изв. АН Каз. ССР, сер. ботан. и почвовед., № 2, 60 (1959); РЖХим., 26219 (1960).
137. Грабаров П. Г., Солодникова Е. А. Изв. Каз. ССР, сер. ботан. и почвовед., № 3, 44 (1960); РЖХим., № 80750 (1960).
138. Грабаров П. Г., Уварова 3. А. Тр. Казахск. научно-исслед. ин-та земледелия, 1, 95 (1939).
139. Григорьев П. Н. Методы химического анализа в стекольном производстве. М.— Л„ Гизлегпром, 1939, стр. 57, 63, 65.
140. Григорьев П. Н., Король С. С. Ж. хим. промышл., № 1, 68 (1931).
141. Гр ин г а уз Л. И., Лерман Г. М. Зав. лаб., 7, 314 (1938).
142. Грицдель М. Химизация соц. земледелия, № 5, 101 (1935); Калий, 4, 41 (1935).
143. Гриценко Е. Д. Усп. совр. биологии, 33, 33 (1952).
144. Г у р в и ч И. Г., Ханаев Е. И. Изв. АН СССР, сер. геологич., № 6, 101 (1956); № 4, 104 (1957).
145. Гусейнов Д. М.. Изв. Азерб. филиала АН СССР, № 7, 32 (1944).
146. Давыдов А. Т., Морозова Ю. А. Тр. Ин-та химии Харьковск. ун-та, № 10, 183 (1953).
147. Дай Вань-го. Хуасюэ туинбао, № 9, 29 (1959); РЖХим., № 56687 (1960).
148. Д е л и м а р ск и й Ю. К., Городыский А. В. Электродные процессы и методы исследования в полярографии. Киев, Изд-во АН УССР, 1960, стр. 36.
149. Деркачев Н. Д. Поташное производство. Л„ Госхимтехиздат, 1932, стр. 3.
150. Джелепов Б. С., Воробьев Б., Копьева М. ДАН СССР, 52, 121 (1946).
151. Джелепов Б. С., Драницина Г. Ф. Систематика энергий fl-распада. М.— Л., Изд-во АН СССР, 1960, стр. 47.
152. Джелепов Б. С., Петрович С. Усп. физич. наук, 40, 497 (1950).
153. Диогенов Г. Г. История открытия химических элементов. М., Учпедгиз, 1960, стр. 30.
154. Диета нов Г. К- Ж- общ. химии, 7, 681 (1937).
155. Добржанская 3. С., Подъячева И. Б. Цемент, № 1, 29 (1953); РЖХим., № 1988 (1953).
156. Добринская А. А., Гуревич И. М. Зав. лаб., 13, 216 (1947).
157. Добросердов Д. К. Укр. хим. журн., 1, 491 (1925).
158. Драгунов С. С. Тр. Калийной комиссии, вып. II—VII. Л., ГОНТИ, 1931, стр. 281.
1'5'9. Др у це И. Рений. М., ИЛ, 1951, стр. 56, 73.
160. Дьячковский С. И„ Орлен ко А. Ф. Ж. общ. химии, 10, 83 (1940).
161. Евдокимов В. В. Ж. аналит. химии, 2, 242 (1947).
162. Егоров Н. П„ Ковалев И. А. Ж. аналит. химии, 14, 489 (1959).
163. Елисеева Г. Д. Тр. Комиссии по аналит. химии, 6 (9), 438 (1955).
164. Ермолаева Е. В., Коробка Л. А. Бюлл. научно-техн, информации Всес. ин-та огнеупоров, 2, 89 (1957); РЖХим., № 4280 (1958).
165. Жидкометаллические теплоносители (Перевод под ред. Шейдлина А. Е). М„ ИЛ, 1958, стр. 27.
166. Забродин Н. И., Туркин В. П. Хим. наука и промышл., 3, 104 (1958).
12 Аналитическая химия калия 177
167. Забродина А. С., Мирошина В. П. Вестник Моск. гос. уи-та, сер. матем. и хим., 12, 195 (1957).
168. Заварицкий В. Изв. Моск. с.-х. ин-та, 22, 132 (1946).
169. Заваров Г. В. Зав. лаб., 3, 995 (1934).
170. Зайдель А. Н., К а л и т и е в с к и й Н. И., Л и п и с А. В., Ч а ft-ка М. П. Ж. аналит. химии, 12, 17 (1957).
171. Зайдель А. Н., Калитиевский Н. И., Липис А. В., Чайка М. П. Эмиссионный спектральный анализ атомных материалов. М.— Л., Физматгиз, 1960, стр. 333, 338, 374, 391.
172. Зайдель А. Н., Туркин Ю. И. Тр. ГлаЬн. геофиз. обсерватории, № 93, 88 (1959); РЖХим., № 64953 (1960).
173. Зайдман Н., Оречкин Д. Новости нефтяной техники, № 5, 25 (1955); РЖХим., № 54735 (1956).
174. ЗанькоА. А., ЛевинаН Д. Научн. зап. Львовск. политехи, ин-та, № 50, 57 (1958); РЖХим., № 19085 (1959).
175. Заславский И. И. Природа, 24, 22 (1935).
176. Иванов Б. В. Виноделие и виноградарство СССР, 7, 17 (1947).
177. Иванов Г. Ж. опытной агрономии, № 7 (1906).
178. Иванов Д. Н. Изв. АН СССР, сер. физич., 4, 203 (4940).
179. Иванов Д. Н. Почвоведение, № 7, 423 (1949).
180. Иванов Д. Н. Почвоведение, № 1, 61 (1953).
181. Иванов Д. Н. Гидрохимические материалы, 24, 35 (1955).
182. И в а н о в Д. Н. Почвоведение, № 1, 117 (1959); РЖХим., № 56817 (1959)
183. ИевиньшА. Ф., ГудриниецеЭ. Ю. Ж. аналит. химии, 9, 270 (1954).
184. ИевиньшА. Ф., ГудриниецеЭ. Ю. Изв. АН Латв. ССР, № 8, 131 (1954).
185. Иевинып А. Ф., Озол Я. К. Ж. аналит. химии, 8, 53 (1953).
186. Иевиньш А. Ф., Озол Я. К-, Гудриниеце Э. Ю. Изв. АН Латв. ССР, № 7, 135 (1955).
187. Иевиньш А. Ф., Пейнберг М. Я. Изв. АН Латв. ССР, № 5, 85 (1959); РЖХим., № 842 (1960).
188. Измайлов Н. А., Т а р т ы л л о Ю. И. Фармация, № 9, 1 (1939).
189. Иллиминская В. Т. Цемент, № 4, 28 (1953).
190. Ильменев М. И. Зав. лаб., 6, 1018 (1937).
191. Индебом В. Л., Карчмар Ц. А., Юрков Л. Ф., Глухо в-ская Б. М. Зав. лаб., 22, 1293 (1956).
192. Индиченко Л. Н. Расшифровка спектрограмм руд и минералов. М„ Госгеолиздат, 1951, 'стр. 19, 44.
193. Исаков П. М. Научн. бюлл. Ленинградск. гос. ун-та, № 29, 6 (1951).
194. Исаков П. М. Качественный химический анализ руд и минералов методом растирания порошков. М., Госгеолтехиздат, 1955.
195. Использование радиоактивности при химических исследованиях (под ред. А. Валь и Н. Боннер). М„ ИЛ, 1954, стр. 74, 114, 334, 338, 401, 501.
196. й о у Д. Г. Колориметрия. М., ОНТИ, 1935, стр. 210.
197. Кадер Г. М. Тр. Почвенного ин-та им. В. В. Докучаева, 33, 313 (1950).
198. Калининский Г. ЖРФХО, 62, 1355 (1930).
199. Калинин С. К., Мар зув ан о в В. Л., Наймарк Л. Э., И см а г у-лова К. И. Атлас спектральных линий для стеклянного спектрографа. Алма-Ата, Изд-во АН Каз. ССР, 1960, стр. 23.
200. Калинин С. К., Наймарк Л. Э., Марзуванов В. Л., Исмагу-л о в а К. И. Атлас спектральных линий для стеклянного спектрографа. М„ Госгеолтехиздат, 1956, стр. 17.
201. Калинине. К., П о с о х о в Е. В. Вести. АН Каз. ССР, № 4, 100 (1950).
202. Калинин С. К-, Явнель А. А., Алексеева А. И., Марзува-н о в В. Л., Н а й м а р к Л. Э. Атлас спектральных линий для кварцевого спектрографа. М., Госгеолтехиздат, 1959, стр. 17.
203. Кальвода Р. Зав. лаб., 24, 1319 (1958).
178
204. Калье A. К-, Беляева А. Советское здравоохранение Туркмении, № 1, 45 (1940).
205. Кальнина Н. П. Изв. Ростовск. научно-исслед. ин-та прикл. химии, 1, 104 (1935).
206. Капустинский А. Ф., Стаханова М. С., Василев В. А. Изв. АН СССР, Отд. хим. наук, № 12, 2082 (1960).
207. Карклиньш Р. Я., Паэгле А. К., Силаньш Э. А. Физич. сборник Львовск. ун-та, № 4, 521 (1958).
208. Карницкий В. А., Дизертинская Т. С., Медведев В. И. Уч. зап. Ростовск. гос. ун-та, 2, 122 (1939).
209. Каррер П. Курс органической химии. М., ОНТИ, 1938, стр. 502.
210. Карякин Ю. В., Ангелов И. И. Чистые химические реактивы. М., Госхимиздат, 1955, стр. 185, 210, 423.
211. Касаткин Н. А. Тр. Казанск. гос. мед. ин-та, № 1, 285 (1938).
212, -К а т ч е н к о в С. М. Спектральный анализ горных пород. Л., Гостоптех-издат, 1957, стр. 50, 57, 75..
213. Кахана М. М. Зав. лаб., 26, 1359 (1960).
214. Качественный анализ без сероводорода (под ред. И. Е. Орлова). М.— Л., ОНТИ, 1934, стр. 16.
215. Кельцева 3. А. Цемент, № 219 (1958); РЖХим., № 81286 (1958).
216. Кертман Л. Д. Курс качественного анализа. М., ОНТИ, 1937, стр. 271, 376, 439, 581.
217. Кеш ан А. Д. Синтез боратов в водном растворе и их исследование. Рига, Изд-во АН Латв. ССР, 1955, стр. 68.
218. Китай В. А. Изв. АН СССР, сер. геол., № 3, 101 (1960).
219. Киладзе Д. Н. Тр. Тбилисск. хим. ин-та, 2, 79 (1937).
220. Киладзе Д. Н. К вопросу об определении калия при помощи кобальтнитритного метода. Автореферат, Тбилиси, 1952.
221. Киргинцев А. Н., Гвоздев Б. А. Ж. неорган. химии, 5, 2374 (1960).
222. Кириченко А. И. Физич. сборник Львовск. ун-та, № 4, 479 (1960).
223. Кирк П. Количественный ультрамикроанализ. М., ИЛ, 1952, стр. 163, 168.
224. Кисилевский В. В Тр. Всес. ин-та содовой промышл., 9, 120 (1956).
225. Кисилевский В. В., Тютюникова Т. И. Тр. Научно-исслед. ин-та основной химии, 11, 318 (1958).
226. Классен А. Электроанализ М.— Л., Госхимтехиздат, 1934, стр. 121, 126, 306.
227. Клебанов Г. С., Останкевич Н. А. Ж. неорган. химии, 5, 2329 (1960).
228. К л я ч к о Ю. А., Шапиро С. А. Курс химического качественного анализа. М., Госхимиздат, 1960, стр 240.
229. Книга А. Г. Калий, № 7, 32 (1935).
230. Книга А. Г Ж. прикл. химии, 10, 371 (.1937).
231. КоблянскийА. Г. Тр. Комиссии по аналит. химии, 7, 89. (1956).
232. Ковалев И. А. Ж. аналит. химии, 11, 123 (1956).
233. Ковалев Я. А. Тр. Ленинградск. отд. Ин-та удобрений, № 39 (1935).
234. Ковалевский А. Сборник научно-техн, информации Министерства геологии и охраны недр СССР, № 5, 119 (1957).
235. Коваленко П. Н. Уч. зап. Ростовск. гос. ун-та, 25, 99 (1955).
236. Коваленко П. Н., Теньковцев В. В. Зав. лаб., 17, 273 (1951).
237. Козлов А. С. Методы анализа редких и цветных металлов. Моск. гос. ун-т, 1956, стр. 71; РЖХим., № 44841 (1957).
238. Козлов А. С. Зав. лаб., 23, 157 (1957).
239. Козьмин В. В. Научн. зап. Днепропетровск, гос. ун-та, 15, 43 (1940).
240. Кольтгоф И. М„ Сендэл Е. Б. Количественный анализ. М„ Госхимиздат, 1948, стр. 347, 351, 427, 623.
241. Кольтгоф И. М., Стенгер В. А. Объемный анализ, т. 2. М.— Л., Госхимиздат, 1952, стр. 207.
12*
179
242. Комар Н. В. Зав. лаб., 6, 1074 (1937).
243. Комаровский А. С., Горемыкин В. И. См. по [1768а].
244. К о м а р ь Н. П. Основы качественного химического анализа. Изд-во Харьковск. ун-та, 1955, стр. 388, 419, 431.
245. Кометиани П. А., Долидзе III. В. Биохимия, 11, 29 (1946).
246. Кореиман И. М. Количественный микрохимический анализ. Л., ОНТИ, 1936, стр. 85, 123, 149, 186, 211.
247. Коренман И. М. Уч. зап. Горьковск. гос. ун-та, 10, 68 (1941).
248. Коренман И. М. Уч. зап. Горьковск. гос. ун-та, 15, 61 (1949).
249. Кореиман И. М. Уч. зап. Горьковск. гос. ун-та, 17, 105 (1951).
250. Кореиман И. М. Микрокристаллоскопия. М., Госхимиздат, 1955, стр. 118.
251. Коренман И. М. Уч. зап. Горьковск. гос. ун-та, 32, 63 (1958).
252. Коренман И. М. Уч. зап. Горьковск. гос. ун-та, 32, 79 (1958).
253. Коренман И. М. Восьмой Менделеевский съезд по общей и приклад-
ной химии. Секция аналитической химии. Рефераты докладов и сообщений. М., Изд-во АН СССР, 1958, стр. 5.
254. Коренман И М. Труды совещания работников вузов и заводских лабораторий Юго-Востока СССР по физико-химическим методам контроля производства. Изд-во Ростовск. ун-та, 1959, стр. 314.
255. Коренман И. М. Тр. по химии и хим. технол. (Горький), № 2, 318 (1961).
255а. Коренман И. М. Тр. по химии и хим. технол. (Горький), № 3, 498 (1961).
256. Коренман И. М. Аналитическая химия таллия. М., Изд-во АН СССР, 1960.
257. Коренман И. М„ Ганичев И. А., Горшков В. В. Ж- аналит. химии, 10, 327 (1955).
258. Коренман И. М., Зорин Е. И. Зав. лаб., 21, 1419 (1955).
259. Коренман И. М., Малкина Н. И. Научные работы химических лабораторий Горьковского института гигиены труда, № 6. М., Госхимиздат, 1957, стр. 169.
260. Коренман И. М., Николаев Б. А. Тр. по химии и хим. технол. (Горький), № 2, 102 (1959).
261. Коренман И. М., О к с Р. С. Зав. лаб., 17, 1435 (1951).
262. Коренман И. М„ Переплетчикова Е. М., Этлис В. С. Тр. по химии и хим. технол. (Горький), 3, 284 (1960).
263. Коренман И. М., Фрум Ф. С., Иванова Л. В. Уч. зап. Горьковск. гос. ун-та, 23, 83 (1952).
264. Коренман И. М., Шаталина Г. А. Ж- аналит. химии, 13, 299 (1958).
265. Коренман И. М., Шеянова Ф. Р., Глазунова 3. И. Зав. лаб., 21, 774 (1955).
266. Коренман И. М., Шеянова Ф. Р., Глазунова 3. И. Сб.: Применение меченых атомов в аналитической химии. М., Изд-во АН СССР, 1955, стр. 29.
267. Коренман И. М., Шеянова Ф. Р., Туманов А. А., Глазунова 3. И., Демин О. И. Тр. по химии и хим. технол. (Горький), 2, 94 (1959).
268. Коренман И. М., Ягнятинская Г. Я. Ж. прикл. химии, 10, 1496 (1937).
268а. Коренман Я. И. Тр. по химии и хим. технол. (Горький), 3, 96 (1960). 269. Ко рже в П. П. Справочник по химии. М., Учпедгиз, 1949, стр. 157, 170. 270. Королев Ф. А., Жеенбаев Ж. Изв. высших учебных заведений.
Физика, № 5, 134 (1955).
271. Коростышевская Л. Консультативные материалы Укр. гос. ин-та эксперим. фармации, № 6, 171 (1939).
272. Кос май О. М. Ж- прикл. химии, 6, 262 (1933).
273. Котенева Т. В. Тр. Научно-исслед. ин-та геологии Арктики, 98,- 130 (1959); РЖХим., № 73142 (1960).
180
274. Котляров Р. В. Докл. Моск. с.-х. академии им. К. А. Тимирязева, вып. 10, 158 (1949).
275. Котляров Р. В. Докл. Моск. с.-х. академии им. К. А. Тимирязева, вып. И, 204 (1949).
276. Котляров Р. В. Докл. Моск. с.-х. академии им. К. А. Тимирязева, вып. 12, 163 (1950).
277. Котляров Р. В. Изв. Тимирязевской с.-х. академии, № 1, 189 (1954).
278. Красный Л. И., Полевая Н. И. Сб.: Определение абсолютного возраста дочетвертичных геологических формаций. М., Изд-во АН СССР, 1960, стр. 245; РЖХим., № 6 Г16 (1961).
279. Кривенцов М. И. Почвоведейие, № 1, 52 (1947).
280. Кривенцов М. И. Гидрохимические материалы, 15, 3 (1948).
281. Кривенцов М. И. Гидрохимические материалы, 15, 32 (1948).
282. К р и ч м ар С. И., Кайстра Л. Г. Зав. лаб., 24, 925 (1958).
283. К р о н м а н Е. С., Бибикова В. И., Аксенова М. А., Ж. прикл. химии, 7, 47 (1934).
284. Кру минь ш К. А. Изв. АН Латв. ССР, № 2, 95 (1948).
285. Крупкин А. И. Тр. Комиссии по аналит. химии, 8 (11), 204 (1958).
286. Крупкин А. И., Коновалов В. Л. Зав. лаб., 24, 1444 (1958).
287. Крылов А. Я. Сб.: Определение абсолютного возраста дочетвертичных геологических формаций. М., Изд-во АН СССР, 1960, стр. 222; РЖХим., № 6 Г14 (1961).
288. Крюкова Т. А., Синякова С. И., Арефьева Т. В. Полярографический анализ. М., Госхимиздат, 1959, стр. 66, 514, 681.
289. Кудрявцев Ю. И., Новиков П. И. Ж. хим. промышл., № 1, 88 (1959).
290. Кузнецов В. И. Зав. лаб., 7, 1326 (1938).
291. Кузнецов В. И. Тр. Комиссии по аналит. химии, 6 (9), 249 (1955).
292. Кузнецова Р. К., Загорский М. Ф. Удобрение и урожай, № 4, 364 (1931).
293. Кулаковская Т. Н. Тр. Белорусск. научно-исслед. ин-та мелиорации и водн. хозяйства, 9, 253 (1958); РЖХим., № 78252 (1959).
294. Кульберг Л. М. Синтезы органических реактивов. М.— Л., Госхимиздат, 1947, стр. 15, 59.
295. Кульберг Л. М. Органические реактивы в аналитической химии. М., Госхимиздат, 1950, стр. 127.
296. Кульберг Л. М., Альтерзон Г. С., Вельтман Р. П. Капельный анализ. М,—- Л., Госхимиздат, 1951, стр. 301, 487.
297. Кульман А. Г. Калий, № 10, 10 (1937).
298. Куиии Р., Майерс Р. Ионообменные смолы. М., ИЛ, 1952, стр. 63.
299. Курбанов М. Ш. Тр. Дагестанск. с.-х. ин-та, 9, 176 (1956); РЖХим., № 41440 (1957).
300. Куркаев В. Т. Почвоведение, № 9, 114 (1959); РЖХим., № 13135 (1960).
301' Куриаков Н. С. Изв. Геологии, комитета, 37, 323 (1918).
302. Курнаков Н. С., Пу шин Н. А. ЖРФХО, 33, 590 (1901).
303. Куца ков Ф. М. Ж- аналит. химии, 11, 636 (1956).
304. Кюри М. Радиоактивность. М., Физматгиз, 1960, стр. 448.
305. Лапин Л. Н. Тр. Комиссии по аналит. химии, 5 (8), 77 (1954).
306. Лапицкий А. В., Несмеянов Ан. Н. Сб.: Радиохимия (под ред. В. И. Спицина). Изд-во Моск. гос. ун-та, 1952, стр. 250.
307. Л а п и ц к и й А. В., Степанов Б. А., Пчелкина М. А. Ж. общ. химии, 25, 1866 (1955).
308. Лапицкий А. В., Шишкина Л. Н., Пчелкина М. А., Степанов Б. А. Ж. общ. химии, 25, 1862 (1955).
309. Лебедев Б. А. Бюлл. научно-техн, информ. Уральск, научно-исслед. ин-та с.-х., № 4, 39 (1958); РЖХим., № 64096 (1959).
310. Лебедев В. И. Ж. аналит. химии, 14, 283 (1959).
181
311. Лебо в а Р. Г. Сборник научно-техн, информ. Министерства геологии и охраны недр, № 1, 125 (1955); РЖХим., № 5, 4736 (1956).
312. Левензон И. Л. Винная кислота и ее соли. М.— Л., Снабтехиздат, 1934.
313. Л е в и В. И. Ж- хим. промышл., 8, 393 (1931).
314. Левина Н. Д., Пантелеева Л. И.. Зав. лаб., 23, 285 (1957).
315. Левицкий А. Ю., Лесю ко в а А. А. Химизация соц. земледелия, № 2, 57 (1934).
316. Леммерманн О. Методы почвенных исследований. М., Сельхозгиз, 1935.
317. Ле Пешков И. Н. Калийные соли. М.— Л., Изд-во АН СССР, 1946.
318. Лилова О. М., Преображенский Б. К. Радиохимия, 2, 728 (I960).
319. Ловиц Т. Е. Технолог, журн., 1 (часть 4), 145 (1804); Ловиц Т. Е. Избранные труды ' по химии и химической технологии. М, Изд-в> АН СССР, 1955, стр. 203, 229, 253.
320. Лукашев Л. Изв. Хим. ин-т Българ. АН, 5, 17 (1957); РЖХим., № 77201 (1958).
321. Лукьянов П. М. История химических промыслов и химической промышленности России. М.— Л., Изд-во АН СССР, т. 1, 1948, стр. 16; т. 2, 1949, стр. 7, 144.
322. Л у Мин-лянь, Пан Шу-вэй, Лю Да-чунь, Янь И - и, Е Цзай-дэ. РЖХим., № 74643 (1957).
323. Лурье Ю. Ю. Расчетные и справочные таблицы для химиков. М.— Л., Госхимиздат, 1947, стр. 74, 88, 229, 264.
323а. Лурье Ю. Ю. Справочник по аналитической химии. М., Госхимиздат, 1962, стр. 16.
324. Лурье Ю. Ю., Николаева 3. В. Зав. лаб., 16, 1058 (1951).
325. Лю И-фу. Тутан тунбао, № 1, 63 (1959); РЖХим., № 17561 (1960).
326. Лян Цзи-цзин, Ли Цзу-гуан. Хиасюэ шицзе, 14, 304 (1959); РЖХим., № 26218 (1960).
327. Майер Ф. Естественные органические красящие вещества. М., Госхимиздат, 1940, стр. 214.
328. Майергофер А. Фармацевтические препараты и яды. Л., Научное хим.-техн. изд. НТУ ВСНХ, 1929, стр. 32.
329. Макаров С. 3., Букина В. В. Ж общ. химии, 3, 884 (1933).
330. Маковецкий А. ЖРФХО, 38, 669 (1906).
331. Максим ычева 3. Т., Абдусалямов Н. Зав. лаб., 24, 403 (1958).
332. Максим ычева 3. Т., Джиянбаева Р. X. Узб. хим. ж., № 2, 33 (1960).
333. Малиновская Г. Н. Сборник трудов Пензенского с.-х. ин-та, № 2, 459 (1958); РЖХим., № 73689 (1958).
334. Маляров К. Л. ЖРФХО, 60, 829 (1928).
335. Маляров К. Л. Нефтяное хозяйство, 18, 223 (1930).
336. Маляров К. Л. Качественный микрохимический анализ. Изд-во Моск, ун-та, 1951, стр. 161, Г69.
337. Маляров К. Л., Мацкевич В. Б. Зав. лаб., 3, 705 (1934).
338. Мандельштам С. Л. Введение в спектральный анализ. М.— Л., Госхимиздат, 1946, стр. 48, 162, 248.
339. Манзон Е. Д. Химизация соц. земледелия, № 2, 113 (1938).
340. Маслова А. Л. Тр. Всес. ин-та удобрений и агропочвоведения, 5,423 (1934).
341. Медведева Г. А. Тр. Уральск, политехи, ии-та, № 96, 156 (1960); РЖХим., № 12 Д50 (1961).
342. Межарауп Г. П„ Иевиньш А. Ф. Уч. зап. Латв. гос. ун-та, 3, 49 (1956).
343. Мельников. Ф. Тр. Вятек. гос. ветер, ин-та, 1, 82 (1934).
344. Менделеев Д. И. Аналитическая химия. СПб., 1866, стр. 318.
345. Менделеев Д. И. Основы химии, т. 2. М.— Л., Госхимтехиздат, 1934, стр. 342. ,
182
346. Меншуткин Н. А. Аналитическая химия. М.— Л., Госиздат, 1931, стр. 28, 219, 356.
347. Мервель Р. В. Тр. Ин-та чистых реактивов, 16, 118 (1939).
348. Методы анализа рассолов и солей. Тр. Всес. научно-исслед. ин-та галургии, 22, 56 (1950).
349. Мешалкин С. И. Зав. лаб., 5, 422 (1936).
350. Мещерский И. ЖРФХО, 14, 219 (1882).
351. Миндалев Л. А. Химия в школе, 10, 50 (1955).
352. Михельсон X. К. Почвоведение, № 11, 82 (1958).
353. Могилевский Б. Гемфри Дэви. М., Учпедгиз, 1958.
354. Монастырский Д. Н. Примеры технического анализа в металлургическом производстве. Л., «Кубуч», 1932, стр. 5, 16.
355. Морачевский Ю. В.,Тр. Первого совещания химиков главного геологоразведочного управления. М.— Л., Геолиздат, 1931, стр. 88.
356. Москвин В., Рояк Г. Строительные материалы, 4, 33 (1958).
357. Мукимов С. М., Крылова Н. И., Бергман А. Г. Тр. Ин-та химии АН Узб. ССР, № 2, 94 (1949).
358. Муравлев А. П. См. по [2091а].
359. Мурина Г. А., Искандерова А. Д., Спринцсои В. Д. Информ, сборник Всес. научно-исслед. геолог, ин-та, № 3, 134 (1956); РЖХим., № 27140 (1957).
360. Нагерова Е. И. Тр. Гос. ин-та проектирования предприятий цементной промышл., № 1, 32 (1940).
361. Нейман М. Б., Миллер В. Б. Усп. физич. наук, 50, 93 (1953).
362. Некрасов Б. В. Курс общей химии. М., Госхимиздат, 1952, стр. 670, 686.
363. Никитина Е. А. Ж. общ. химии, 5, 1133 (1935).
364. Никитина Е. И. Зав. лаб., 6, 1206 (1937).
365. Никитина Е. И. Полумикрохимические методы количественного и качественного анализа сплавов алюминия и магния. Автореферат. М., 1952.
366. Никольский Б. П., Лавров И. Н. Тр. Ленинградск. отд. Всес. ин-та удобрений и агропочвоведения, 17, 45 (1933).
367. Никольский Б. П., Трофим о,в А. М., В ы с око о с т р о в-ская Н. Б. Ж- неоргаи. химии, 4, 857 (1959).
368. Николюкин Я. ЖРФХО, 17, 207 (1885).
369. Н о в и к - Б о м Е. 3.. Изв. Центр, научно-исслед. ин-та кожев. промышл., № 6-7, 35 (1932).
370. Нонова Д., Керкенякова М. Годишник Софийского ун-та, 51, 61 (1958); РЖХим., № 86071 .(1959).
371. Оболенская Л. И. Почвоведение, № 7, 420 (1951).
372. Оболенская Л. И. Почвоведение, № 3, 278 (1952).
373. Овчинников Л; Н., Гаррис М. А. Сб.: Определение абсолютного возраста дочетвертйчных геологических формаций. М„ Изд-во АН СССР, 1960, стр. 195; РЖХим., № 6, Г17 (1961).
374. Озол Я. К-, Иевиньш А. Изв. АН Латв. ССР, № 11, 1739 (1951).
375. Оленович Н. Л., Белоусова Е. М., Драницкая Р. М. Сборник хим. факульт. Одесск. ун-та, 4, 79 (1954); РЖХим., № 49141 (1955).
376. Ольшанова К. М. Тр. Комиссии по аналит. химии, 6(9), 277 (1955).
377. Ольшанова К. М. Тр. Моск, техйол. ин-та мясной и молочн. промышл., 6, 163 (1956); РЖХим., № 592 (1957).
378. Ольшанова К. М„ Морозова Н. М. Изв. Высш, учебн. завед. Химия и хим. технология, 2, 498 (1959).
379. Орленко А. Ф., Фесенко Н. Г. Ж. прикл. химии, 9, 2116 (1936).
380. Орлов И. Е. Методы анализа рапы, буровых вод и контроль производства иода и брома. М., ГОНТИ, 1939, стр. 67.
381. Орлов Н. А. См. по [2162а].
382. Остроумов Э. А. Зав. лаб., 6, 16 (1937).
383. Остроумов Э. А. Применение органических оснований в аналитической химии. М., Изд-во АН СССР, 1959, стр. 32, 53, 82, 94, 102, ПО.
183
384. Остроушко Ю. И., Б учи хи и П. И., Алексеева В. В., Набойщик о >в а Т. Ф., К о в д а Г. А., Шелкова С. А., Алексеева Р. Н., Маковецкая М. А. Литий, его химия и технология. М., Атомиздат, 1960, стр. 79.
385. О щ а и о в с к и й В. В., 3 а и ь к о А. А. Научн. зап. Львовск. политехи, ин-та, № 22, 103 (1956).
386. Павлинова А. В., К о р о т у н М. В., Проценко А. Е. Ж. аналит. химии, 15, 124 (I960).
387. Павлова Н. И., Оренкова Т. М. Зав. лаб., 26, 1201 (1960).
388. Павлюченко М. М., Акулович В. М. Изв. АН БССР, сер. физ.-технич. наук, № 2, 109 (1957); РЖХим., № 24883 (1958).
388а. Павлюченко М. М„ Акулович В. М. Сборник научи, работ Ин-та общ. и неорг. химии АН БССР, № 1, 35 (I960); РЖХим., № 15 ДНО (1961).
389. Па со в ск а я Г. Б., Удовенко В. В. Тр. Средиеазиатск. ун-та, 27, 81 (1951); Тр. Комиссии по аналит. химии, 4, 196 (1952).
390. Пахомов Е., Колодязная О. Бюлл. Ин-та табака, 81, 103 (1931).
391. Пейве Я- В. Химизация соц. земледелия, № 6, 71 (1935).
392. Пельш Г. К. Уч. зап. Ленииградск. гос. ун-та, № 297, 125 (1960); РЖХим., № 7 ДЗ (1961).
393. Перельман В. И. Краткий справочник химика. М., Госхимиздат, 1954, стр. 24, 68, 92, 342, 404, 473.
394. Перельман Ф. М. Рубидий и цезий. М., Изд-во АН СССР, 1960, стр. 38, 46, 56.
395. ПетрашеньВ И. Качественный химический анализ. М.— Л., Госхимиздат, 1948, стр. 155, 164, 191, 555.
396. Петров В. Тр. Академии наук, ч. 2, 69 (1823).
397. Петров-Спиридонов А. Е. Удобрение и урожай, № 11, 44 (1957); РЖХим., № 28390 (1958).
398. Петров-Спиридонов А. Е. Изв. Тимирязевской с.-х. академии, № 1, 229 (1959); РЖХим., № 4567 (1960).
399. Печерская Ю. И., Певцов Г. А. Тр. Всес. научно-исслед. ии-та реактивов, 22, 56 (1958); РЖХим., № 45601 (1959).
400. Пилипенко П. П., Калинин П. В. Определитель минералов при помощи паяльной трубки. М.— Л., Госгеолиздат, 1947, стр. 24, 28.
401. Писарев В. Д. Тр. Сибирск. физ.-техн, ин-та (Томск), 35, 182 (1956).
402. Писарев В. Д., Иванова Т. А. Зав. лаб., 20, 586 (1954); Труды Сибирок. физ.-техн, ин-та, 35, 196 (1956).
403. Плющев В. Е., Шахи о И. В. Ж. аналит. химии, 8, 293 (1953).
404. Поздюшин Е. В. Изв. Самарск. с.-х. ин-та, 1, 154 (1923).
405. Полевая Н. И., Миркина С. Л. Информ, сборник Всес. научно-исслед. геологии, ин-та, № 1, 123 '(1955).
406. Полевая И. И., Михайлова В. А. Материалы по литологии. Всес. научно-исслед. геологии, ин-т, 1, 130 (1956).
407. Полуэктов Н. С. Калий, 2, № 10, 44 (1933).
408. Полуэктов И. С. Зав. лаб., 21, 1045 (1955).
409. Полуэктов Н. С. Экспрессные методы анализа при помощи фотометрии пламени в цветной металлургии. М., Металлургиздат, 1958, стр. 22, 48, 50.
410. Полуэктов И. С. Методы анализа по фотометрии пламени. М., Госхимиздат, 1959, стр. 148, 155.
411. Полуэктов Н. С. Исследования в области теории пламенно-фотометрического метода. Автореферат. М., 1960.
412. Полуэктов Н. С., Никонова М. П. Зав. лаб., 24, 528 (1958).
413. Полуэктов Н. С., Никонова М. П., Виткун Р. А. Ж. аналит. химии, 13, 48 (1958).
414. П о л у э к т о в Н. С., Овчар Л. А., Кучмент М. М., Н и к 6 л ь-ский М. А. Зав. лаб., 26, 1152 (1960).
415. Поляк Л. Я. Сов. пат. 117662 (1959).
184
416. П о л я к о в Г. И. К вопросу о количественном определении калия. М„ 1928.
417. Попов К. М. Калий, № 5, 39 (1936).
418. Портнов М. А. Сборник статей Государственного института прикладной химии. Госхимиздат, 1919—1939, стр. 100, 140; Хим. реф. журн., 3, 52 (1940).
419. Портнов М. А., Афанасьев С. К. Зав. лаб., 6, 1442 (1937).
420. Портнов М. А., Афанасьев С. К. Зав. лаб., 7, 421 (1938).
421. Починок X. Н. Ж. прикл. химии, 8, 524 (1935).
422. Преображенский П. И. Горный журн., № 7, 389 (1927); Вест. Геологии. комитета, № 1, 9 (1926).
423. Преображенский П. И. Материалы по общ. и прикл. геологии, 126, 1 (1929).
424. Преображенский П. И. Соликамское калийиое месторождение. Л., Госхимтехиздат, 1933.
425. Пржевальский Е. С., Мервель Р. В. Тр. Ин-та чистых хим. реактивов, 15, 58 (1937).
426. Применение метода меченых атомов в химии (Сб. переводов под ред. В. Г. Васильева). М., ИЛ, 1955, стр. 309.
427. Приходько Г. В. Ж. прикл. химии, 5, 99 (1932).
428. Прокопьева А. Н. Физический сборник. Львовский ун-т, № 4, 446 (1958).
429. Проскурякова Г. Ф. Восьмой Менделеевский съезд по общей и прикладной химии, секция агрономической химии. Рефераты докладов и сообщений. М., Изд-во АН СССР, 1958, стр. 46.
430. Протасов П. В. Почвоведение, № 5, 89 (1939).
431. Протасов П. В., Петрова О. Г. Химизация соц. земледелия, № 12, 61 (1939).
432. Прохоров Ф. Г., Янковский К. А. Зав. лаб., 13, 656 (1947).
433. П р ш и б и л Р. Комплексоны в химическом анализе. М., ИЛ, 1960, стр. 148, 188.
434. Прянишников Д. И. Агрохимия. М., Сельхозгиз, 1940, стр. 64, 190, 218, 344.
435. Радиохимия.-Сборник работ под ред. В. И. Спнцина. Изд-во Московского гос. ун-та, 1952.
436. Разуваев Г. А., Брилкина Т. Г. ДАН СССР, 85, 815 (1952); Ж- общ. химии, 24, 1415 (1954).
437. Реакции и реактивы для качественного анализа неорганических соединений. М.— Л., Госхимиздат, 1950, стр. 191.
438. Резников А. А., Нечаев А. А. Информ, сборник Всес. научно-исслед. геол, ин-та, № 4 145 (1956); РЖХим., № 21148 (1958).
439. Решетина Т. С. Изв. АН СССР, сер. физич., 23, 1150 (1959).
440. Ривкина М. А., Русанов А. К. Изв. АН СССР, сер. физич., 12, 467 (1948).
441. Рик Г. Р., Шу ко л юков Ю. А. ДАН СССР, 94, 667 (1954).
442. Розанов С. Н. Калий, 2, 17 (1933).
443. Розанов С. Н., Казаринова В. А. Калий, 2, № 4, 27 (1933).
444. Рубинштейн М. М„ Григорьев И. Г., Узнадзе Э. Д., Гельма н О. Я., Л а ш х и Б. А. Сообщ. АН Груз. ССР, 24, 683 (1960); РЖХим., № 8 Д66 (1961).
445. Русанов А. К. Зав. лаб., 3, 915 (1934).
446. Русанов А. К. Ж. общ. химии, 6, 1057 (1936).
447. Русанов А. К. Спектральный анализ руд и минералов. М.— Л., Гос-геолиздат, 1948, стр. 213.
448. Русанов А. К., Васильев К. Н. Зав. лаб., 8, 832 (1939).
449. Русанов А. К., Гусяцкая Э. В., Ильясова Н. В. Зав. лаб., 16, 447 (1950).
450. Рысс И. Г. Ж- физ. химии, 21, 197 (1947).
185
451. Рысс И. Г., Нилу с Э. Л. Ж. аналит. химии, 12, 64 (1957); Зав. лаб., 24, 1349 (1958).
452. Рысс И. Г., Хорд ас И. С. Ж. неорган. химии, 3, 1410 (1958).
453. Сагакова В. П., Любивая А. И. Консерви. и овощесуш. промышл., № 10, 24 (1960); РЖХим, № 9 НИ (1961).
454. Самуэльсон О. Применение ионного обмена в аналитической химии. М, ИЛ, 1955, стр. 168.
455. Саука Я. Я., Путинь Я. К. Ж. аналит. химии, 11, 668 (1956).
456. Свешникова В. Н. Изв. АН СССР, Отд. хим. наук, № 1, 44 (1952).
457. Свиньин П. Отечественные записки. СПб, 1818, стр; 69.
458. Севергин В. М. Технолог, журн, 1, 166 (1804).
459. Сегре Э. (ред.). Экспериментальная ядерная физика, т. 1. М, ИЛ, 1955, стр. 639.
460. Селинов И. П. Атомные ядра и ядерные превращения. М.— Л, Гос-теоретиздат, 1951, стр. 42.
461. Семкина Н. В, Шаевич А. Б. Зав. лаб, 25, 1465 (1959).
462. С е н д э л Е. Б. Колориметрическое определение следов металлов. М.— Л, Госхимиздат, 1949, стр. 257.
463. С е и я в и н М. М, Колосова Г. М, Пашков А. Б. Тр. Комиссии по аналит. химии, 11, 406, (1960).
464. Сергеенко П. С. Укр. хим. журн, 5, 113 (1930).
465. Сергеенко П. С. Укр. хим. журн, 7, 36 (1932).
466. Сердобольский И. П. Калий. М,—Л, Изд-во АН СССР, 1944, стр. 11, 78.
467. Сердобольский И. П. Химия почвы. М, Изд-во АН СССР, 1953, стр. 35, 124.
468. Сердюкова А. С, Капитанов Ю. Т. Ж. аналит. химии, 13, 88 (1958).
469. С е т к и н а О. Н. Тр. Ленинградск. технолог, ин-та, 35, 186 (1956); РЖХим, № 15699 (1957).
470. Сиборг Г, Перлмаи И. Таблица изотопов. М, ИЛ, 1951, стр. 15.
471. Сиборг Г, Перлман И, Хеллендер Д. Таблица изотопов. М., ИЛ, 1956, стр. 36, 39.
472. Синякова С. И. Тр. Комиссии по аналит. химии, 11, 361 (1960).
473. Соколова М. Н. Огнеупоры, 23, 139 (1958).
474. Соликамские карналлиты. Сборник статей. М.— Л, ОНТИ, НКТП, 1935.
475. Сольц Л. М, Сольц Ф. М. Аптечное дело, № 1, 22 (1953); РЖХим, № 1985 (1953).
476. Спицин В. И. ЖРФХО, 49, 357 (1917).
477. Спицин В. И,- Бабаев Н. Б. Ж. неорган. химии, 5, 580 (1960).
478. Справочник химика. М, Госхимиздат, т. 1, стр. 154, 434, 491, 670; т 2 1952, стр. 8, 68, 76; т. 3, 4952, стр. 77, 194, 362, 413, 521.
479. Старик И. Е. Радиоактивные методы определения геологического времени. М,—Л, ОНТИ, 1937, стр. 15.
480. Старосельская С. А. Фармация, № 12, 11 (1939).
481. Статнов В. И. Химизация соц. земледелия, № 9, 44 (1940).
482. С те пин'Б. Д, Плющев В. Е. Ж- аналит. химии, 15, 556 (1960).
483. Столяров К. П. Качественный микрохимический анализ катионов в ультрафиолетовых лучах. Автореферат. Л, 1953.
484. Столяров К. П. Методы микрохимического анализа. Изд-во Ленинградок. ун-та, 1960, стр. 28, 82, 134, 150.
485. Стэлл Д. Р. Давление пара индивидуальных веществ. М, ИЛ, 1949, ' стр. 60.
486. Сухоиванов В. А. Вести, с.-х. науки, № 2, 128 (1930).
487. С я в ц и л л о С. В., Лускина Б. М. Зав. лаб., 20, 272 (1954).
488. Тананаев И. В. Изв. Укр. ин-та силикатной промышл. (Киев), 10, 13 (1931).
489. Тананаев И. В. Изв. Научно-исслед. ин-та сахарной промьТшл. (Киев), И, 69 (1931).
186
490. Тананаев И. В. Тр. Комиссии по аналит. химии, 1 (4), 5 (1947).
491. Тананаев И. В., Джапаридзе Е. С. Тр. Тбилисок, хим. ин-та, 1,
281 (1935).
492. Тананаев И. В., Джапаридзе Е. С. Тр. Тбилисск. хим. ин-та, 2, 115 (1937).
493. Тананаев И. В., Джапаридзе Е. С. Зав. лаб., 6, 1079 (1937).
494. Тананаев И. В., Козлов А. С. Ж- аналит. химии, 6, 149 (1951).
495. Тананаев И. В., Левина М. И. Зав. лаб., 15, 887 (1949).
496. Тананаев И. В., Лютая М. Д. Ж. неорган. химии, 4, 97 (1959).
497. Та и ан а ев И. В., Лйтая М. Д. Ж- неорган. химии, 4, 103 (1959).
498. Тананаев И. В., Мизецкая И. Б. Ж. аналит. химии, 1, 6 (1946).
499. Тананаев И. В., Талипов Ш. Ж. общ. химии, 9, 1155 (1939).
500. Тананаев И. В., Ульянов А. И. Тр. Комиссии по аналит. химии,
7, 3 (1956).
501. Тананаев И. В., Хрелашвили С., Кахукашвили С., Дар-хиашвили Т. Изв. Грузинск. индустр. ин-та, № 12, 101 (1939); Хим. реф. журн., 4, № 1, 78 (1941).
502. Тананаев И. В., Хрелашвили С., Салуквадзе Е. Изв. Грузинок. индустр. ин-та, № 12, 89 (193'9); Хим. реф. журн., 4, № 1, 82 (1941).
503. Тананаев Н. А. Тр. Пятого Менделеевского съезда по чистой и при-
кладной химии. Казань, 1928, стр. 50.
504. Тананаев Н. А. Зав. лаб., 1, 51 (1932).
505. Тананаев Н. А. Весовой анализ. Свердловск — Москва, ГОНТИ, 1938, стр. 179, 184.
506. Тананаев И. А. Дробный анализ. М.— Л., Госхимиздат, -1950, стр. 41, 43.
507. Тананаев Н. А. Капельный метод. М.— Л., Госхимиздат, 1954, стр. 44.
508. Тананаев Н. А., Бабко А. К. Тр. Пятого Менделеевского съезда по чистой и прикладной химии. Казань, 1928, стр. 51,
509. Тананаев Н. А., Лазаркевич Н. А. См. по [2688а].
510. Тарасенко М. И. Сб. научн. работ Моск. фарм. ин-та, 2, 115 (1959);
РЖХим., № 34443 (I960).
511. Тацудзо О., Томота Н., Итиро Ф. Кето дайгаку, 11, 16 (1957);
РЖХим., №’42364 (1960).
512. Теодорович И. Л., Зазулина Л. М. Зав. лаб., 20, 415 (1954).
513. Тимофеев В. Ф. Исследование над растворимостью веществ в органических растворителях. Диссертация, Харьков, 1894.
514. Тихомиров В. М., Холмогоров С. Н. Зав. лаб., 7, 33 (1938).
515. Товарницкий В. И., Сергеенко П. С. Ж. сахарн. промышл., 2, 228 (1928).
516. Товарницкий В. И., Слезак К. И. Ж- сахарн. промышл., 2, 462 (1928).
517. Тредвелл Ф. П. Количественный анализ. Одесса, 1927, стр. 29, 36, 42.
518. Тредвелл Ф. П., Го л л В. Т. Качественный анализ. М.— Л., Госхимиздат, 1946, стр. 38, 309, 331.
519. Трифонов Н. А. Уч. зап. Саратовск. ун-та, 1, 22 (1923).
520. Т р и ш и н Ф. И. Тр. Одесск. технол. ин-та, 6, 33 (1954); РЖХим., № 19477 (1956).
521. Троицкая М., Заглодина Т. Зав. лаб., 13, 145 (1947).
522. Трос тян с кая Е. Б., Лосев И. П., Тевлина А. С. Успехи химии,
24, 69 (1955); Ж. аналит. химии, 11, 578 (1956). 1
523. Туманов А. А., Хазанов П С. Тр. по химии и хим. технол. (Горький), № 2, 413 (1958).
524. Туницкий Н. Н. Успехи химии, 6, 830 (1937).
524а. Ту рьян Я. И., Романов В. Ф. Хим. промышл., № 1, 68 (1961).
525. У гай Я. А. ДАН СССР, 70, 653 (1950).
526. У гай Я. А., Шатилло В. А. Ж. физ. химии, 23, 744 (1949).
527. Удовенко В. В., Пасовская Г. Б. Ж. аналит. химии, 7, 161 (1952).
187
528. Уичерс Э. Успехи химии, 28, 639 (1959).
529. Уклонений А. С. Минералогия. М.— Л., Гостоптехиздат, 1940.
530. Файгль Ф. Капельный анализ. М., ОНТИ, 1937, стр. 322.
531. ФаньЧун-ян, ГоСю-линь. Хуасюэ шицзе, 14, 596 (1956); РЖХим., № 69129 (1960).
532. Федоренко В. К. Укр. хим. жури., в, 105 (1931).
533. Фиалков Ю. Я., Демченко А. А. Фармацевт, жури., УССР, № 5, 25 (1959).
534. Фиалков Я. А., С о л ь ц Л. М. Фармация, № 2, 16 (1943).
535. Фиалков Я- А., Шапиро М. И. Сов. фармация, № 6, 29 (1933).
536. Фишер А. М., Финкельштейн А. И. Тр. Комиссии по аналит. химии, 8, 272 (1958).
537. Фишер В. М. ЖРФХО, 46, 1250 (1914).
538. Фрезениус Р. Минеральный количественный анализ. Изд-во А. М. Котомина. СПб., 1875, стр. 60, 133, 145, 398.
539. Фридлендер Г., Кеннеди Д. Введение в радиохимию. М., ИЛ, 1952, стр. 20.
540. Фрост А. В. Тр. Ии-та чистых хим. реактивов, № 6, 38 (1927).
541. Хазанов П. С. Бюлл. по обмену опытом. Главазот, № 10, 76 (1957).
542. Хармадарьян М. О., Бродов и ч К. О. Ж. прикл. химии, 3, 951 (1930).
543. Хе веши Г. Радиоактивные индикаторы М., ИЛ, 1950, стр. 31, 137, 170, 195.
544. Хеги -В. Успехи химии, 25, 903 (1956).
545. Химические реактивы и препараты (под ред. В. И. Кузнецова). М.— Л, Госхимиздат, 1953, стр. 76, 597.
546. Ху Вэнь чей, Линь Чжень-ци а нь, Чжан Ши-цзе. РЖХим.. № 80748 (1960).
547. Цейтлин С. М. Укр. хим. журн., 1, 580 (1925).
548. Цзен Юнь-э. Применение иодной кислоты для количественного определения и разделения элементов. Автореферат. М., 1957.
549. Цинцадзе Ш. Р. Тр. Научно-исслед. ин-та удобрений, 83, 6 (1931);
109, 91 (1932).
550. Цируль В. А. Тр. Центр, науч.-исслед ин-та сахарн. промышл., № 7, 74 (1960); РЖХим., № 6 Н389 (1961).
551. Че лап М. Б. Гласник хем. друштва, 21, 225 (1956); РЖХим., № 74660 (1957).
552. Чепелевецкий М. Л., Позднякова С. Н., Файн Р. Д. Ж. хим. промышл., 8, 396 (1931).
553. Черкесов А. И. Тр. Комиссии по аналитич. химии, 11, 137 (1960).
554. Черкесов А. И., Кульберг Л. М. Зав. лаб., 18, 131 (1952).
555. Чернов В. А. Почвоведение, № 6, 294 (1944).
556. Черняева И. Ю., Красновская Р. В. Ж- хим. промышл., № 10, 57 (1933).
557. Чириков Ф. В. Агрохимия калия и фосфора. М., Сельхозгиз, 1956.
558. Чуева М. Н. Минералогический анализ шлихов и рудных концентратов. М., Госгеолиздат, 1950, стр. 41.
559. Шадлун Н. А. Минер, сырье, № 10-11, 774 (1926).
560. Шапиро М. Я. Зав. лаб., 7, 790 (1938).
561. Шатемиров К. Ш. Почвоведение, № 2, 107 (1960).
561а. Шашкин В. Л. Методы анализа естественных радиоактивных элементов. М., Госатомиздат, 1961, стр. 13.
562. Шведов В. П. Ж. общ. химии, 17, 27 (1947).
563. Шейицис О. Г. Зав. лаб., 4, 1047 (1935); 8, 1198 (1939); Ж. прикл. химии, 13, 1098 (1940).
564. Шейнцис О. Г. Зав. лаб., 9, 162 (1940); Ж. хим. промышл., 14, 1373 (1937).
565. Шейнцис О. Г. Ж. прикл. химии, 11, 1012 (1938); Зав. лаб., 10, 151 (1941).
188
566. Шейнцис О. Г. Ж- прикл. химии, 13, 1101 (1940).
567. Шемякин Ф. М., Мицелоиский Э. С., Романов Д. В. Хроматографический анализ. М., Госхимиздат, 1955, стр. 102.
568. Шеянова Ф. Р., Сидорова К. Н. Уч. зап. Горьковск. гос. ун-та. № 32, 97 (1958).
569. Шипов К. Г. Тр. Вологодск. с.-х. ин-та, 3, 3 (1941).
570. Шпак В. А., Новиков П. И. Хим. промышл., № 3, 11 (1954).
571. Шпаке л ер Г. Разработка месторождений калийных солей. Л., ОНТИ, Химтеорет, 1935.
572. Шпитальский Е., Иофа 3. ЖРФХО, 60, 75 (1928).
573. Штатнов В. И. Химизация соц. земледелия, № 9, 44 (1940).
574. Шубина С. Б. Материалы Второго Уральского совещания по спектроскопии, 1958. Свердловск, Металлургиздат, 1959, стр. 188.
575. Шубич М. Г. Бюлл. эксперим. биол. и медицины, 43, 118 (1957).
576. Шугар А. И. Докл. Московск. с.-х. академии им. К. А. Тимирязева, вып. 10, 153 (1949); вып. 12, 183 (1950).
576а. Шуколюков Ю. А., М а т в е е в а И. И. Ж. аналит. химии, 16, 544 (1961).
577. Щи го ль М. Б. Зав. лаб., 9, 791 (1940).
578. Щипакина Н. К-, Немировская Е. М., С е н я в и н М. М. Ж. аналит. химии, 12, 70 (1957).
579. Щукина А., Ваганова Е. Химизация соц. земледелия, № 12, 65, (1939).
580. Эм их Ф. Микрохимический анализ. Л., Госхимтехиздат, 1932, стр. 101, 133, 189.
581. Эфендиев Ф. М. Изв. АН СССР, сер. физич., 11, 313 (1947).
582. Эфраим Ф. Неорганическая химия. Л., Госхимтехиздат, т. 1, 1932, стр. 332, 406; т. 2, 1933, стр. 231.
583. Эфрос С. М., Голованов П. И. Тр. Ленинградск. технол. ин-та им. Ленсовета, № 27, 108 (1953); РЖХим., № 29310 (1954).
584. Янсон Э. Ю., Швиньш А. Ф. Успехи химии, 28, 980 (1959).
585. Ян Юй-ай, Чень Цюань-юн, Ху Чжен-юань. Тужан тунбао, № 1, 60 (1959); РЖХим., № 17563 (1960).
586. Я ц и м и р с к и й К. Б., Васильев В. П. Константы нестойкости комплексных соединений. М., Изд-во АН СССР, 1959, стр. 97, 108, 115.
587. Ященко М’. Л., Овчинников Г. В., Афанасьева Л. И. Тр. Пятой сессии комиссии по определению абсолютного возраста геологических формаций. М., Изд-во АН СССР, 1958, стр. 296; РЖХим., № 45506 (1959).
588. Aasnes Н. Medd. Norsk tarmac. solskop, 19, 53 (1957); РЖХим., № 24884 (1958)
589. Abbey S., Maxwell J. A. Chem. Canada, 12, 37 (1960); РЖХим., № 8 Д70 (4961).
590. Abelin I. Helv. chim. acta, 24, 611 (1941); Chem. Abstr., 35, 6536 (1941).
591. Abresch K. Chem. Fabrik, No39/40, 380 (1935); Калий, 5, №3,43 (1936).
592. Abul-Fadi M. A. Biochem. J., 44, 282 (1949); Chem. Abstr., 43, 9134 (1949).
593. Adams J. I., Benedetti-Pichler A. A., Bryant J. T. Mikroche-mie, 26, 29 (1939); Chem. Abstr., 33, 3288 (1939).
594. Adams J. I., Hall M., Bailey W. F. Ind. Eng. Chem., Analyt Ed., 7, 310 (1935).
595. Adams M. F., John J. L. Ind Eng. Chem., Analyt. Ed., 17, 435 (1945).
596. Adamski L., BouzykJ., Josefowicz K. Nukleonica, 5, 317 (I960); РЖХим., № 6 Д69 (1961).
597. AdieR. H., WoodT.B.J. Chem. Soc., 77, 1076 (1960); Z. analyt. Chem., 48, 53 (1909).
598. Agarwal B. R., Pollard A. G. J. Sci. Food Agr., 3, 152 (1952); Chem. Abstr., 46, 7268 (1952).
599. Aguirreche F. D. Anales soc. espan. fis. quim., 27, 368 (1929); Chem. Abstr., 24, 1173 (1930).
189
600. Aho V. Suomen Kemistlichti, 9, 135 (1937); Chem. Zbl, 108, I, 2437 (1937).
601. Ahrens L. A. J. S. African Chem. Inst., 26,27 (1943); Chem. Abstr, 37, 4323 (1943).
602. Ahrens L. A. Geochim. et cosmochim. acta, 2, 155 (1955).
603. Ahrens L. A., Evans R. D. Phys. Rev., 74, 279 (1948).
604. Aikawa H. A. Chem. Abstr., 35, 1947 (1941).
605. Ajon G. Rivisti ital. essenze profumi, 11, 6 (1929); Chem. Abstr., 23, 1588 (1929).
606. Akerlof G., Bender P. J. Amer. Chem. Soc, 63, 1085 (1941).
607. Akerlof G., Turck H. E. J. Amer. Chem. Soc., 57, 1746 (1935).
608. Albanese A. A., Wagner D. L. J. Lab. Clin. Med., 30, 280 (1945); -Chem. Abstr., 39, 2301 (1945).
609. Alberti G., Grasini G. J. Chromatogr., 4, 423 (1960); РЖХим., № 12 Д41 (1961).
610. Alburger D. E. Phys. Rev., 75, 1442 (1949).
611. Alcock F. Pharm. J., 21 (4), 615 (1905).
612. Aldrich L. T, Nier A. O. Phys. Rev., 74, 876 (1948).
613. Alibegoff G. Lieb. Ann., 233, 143 (1886); Z. analyt. Chem, 26, 632 (1887).
614. A 1 к e m a d e С. T, Smith J, Ver schur e J. C. Chem. WeekbL, 47, 23 (1951); 48, 342 (1952); Biochem. et biophys. acta, 8, 562 (1942); Chem. Abstr, 47, 168 (1953); Z. analyt. Chem, 136, 353 (1952); 137, 366 (1953).
615. Allen H. R. J. Assoc. Offic. Agr. Chem, 21, 134 (1938); 22, 162 (1939); Chem. Abstr., 32, 3073 (1938); 33, 3055 (1939).
616. Allen O. D. J. prakt. Chem, 88, 83 (1863).
617. Alluard. Compt. rend, 59, 503 (1864); Lieb. Ann, 133, 292 (1865).
618. Al-Machmalgy R. Risalatul-kimia, 3, 302 (1955); РЖХим, № 4716 (1957).
619. Almeida H. Anais inst. vinho Porto, 1, 46 (1950); Chem. Abstr, 46, 7705 (1952).
620. Al mk wist G. Z. anorg. allg. Chem, 103, 221 (1918).
621. Al ten F, Weiland H, Kurmies B. Z. Pflanzenernahr, Diing. Bo-denkunde, 32A, 171 (1933); Chem. Zbl, 105, I, 897 (1934).
622. Alva L. C. Anales 4 conv. asoc. peruana technol. azucar, 1955, p. 129; Chem. Abstr, 52, 12 428 (1958).
623. Alvarez E. P. Compt. rend, 140, 1186 (1905); Chem. News, 91, 146 (1905); Chem. Zbl, 76, I, 1337 (1905); Gazz. chim. ital, 35, II, 463 (1906).
624. Alvarez-Gonzalez F, Roca-Adell M, Fernandes-Cellini R. An. Real. Soc. espanola fis. у quim, B54, 278 (1958); РЖХим, № 15099 (1959).
625. Amano J. Ch-'m. Soc. Japan, Pure chem. sec, 75, 499, 502 (1954); РЖХим, № 23327 (1957).
626. Ambrosis R. J. Rev. facultad cienc. quim, Univ. nacl. La Plata, 17, 231 (1942); Chem. Abstr, 38, 4331 (1944).
627. Amdur E. Ind Eng. Chem, Analyt. Ed, 12, 731 (1940).
628. Amin A. A. Chemist-Analyst, 45, 65 (1956); 46, 6 (1957); РЖХим, № 57779 (1957); № 844 (1958).
629. Amos W, Sympson R. F. Analyt. Chem, 31, 133 (1959).
630. Analytical Methods Committee. Analyst, 84, 214 (1959).
631. Anderson С. H, Beatty C. D. Analyt. Chem, 26, 1369 (1954); РЖХим, № 9663 (1955).
632. A n d о A, Omori E, Yamada T. Bull. Geol. Surv. Japan, 10, 659 (1959); РЖХим, № 73035 (1960).
633. Angot J. M.krochim. acta, 1959, 346.
634. Antonian: C, Nicolini M. Giorn. chim. ind. applicata, 15, 57 (1933); Chem. Abstr, 27, 4616 (1933).
635. Antonini F. M. Sperimentale, 103, 305 (1953); Chem. Zbl, 127, 11799 (1956).
90
636. Archer К., Flint D., Jordan J. Fuel, 37, 421 (1958); РЖХим., № 38356 1959).
637. Archibald E. H., Wilcox W. G., Buckley B. G. J. Amer. Chem. Soc, 30; 747 (1908).
638. Armstrong H. E, Eyre J. V, Hussey A. V, Paddison W. P. Proc. Roy. Soc, London, 79, 564 (1907).
639. Ar nd T, Leisen E. Bodenkunde u. Pflanzenernahr, 30, 51 (1942); Chem. Abstr, 38, 1596 (1944).
640. Arnikar H. J. Compt. rend, 244, 2241 (1957); Nature, 182, 1230 (1958); РЖХим, № 10970 (1958); РЖХим, № 38273 (1959).
641. Arnikar H. J, Ch eml a M. Compt. rend, 244, 68 (1957); РЖХим, № 77319 (1957).
642. Aronson J. K. Proc. Iowa Acad. Sci, 59, 402 (1953); Chem. Abstr, 47, 2811 (1953).
643. Arrhenius O, Riehm H. Medd. Vetenskapsakad. Nobel inst, 6, 66 (1926); Chem. Abstr, 20, 2469 (1926).
644. Atchison G. J, Beamer W. H. Analyt. Chem, 24, 1812 (1952).
645. Aten A. Chem. Weekbl, 40, 189 (1943); Chem. Abstr, 38, 6232 (1944).
646. Athy L. C, Taylor R. K. Chemist-Analyst, 29, 76 (1940); Chem. Abstr, 35, 403 (1941).
647. Atkinson H. Analyst, 46, 354 (1921).
648. Ato S, W a d a I. Sci. Papers Inst. Phys. Chem. Res, 4, 263 (1926); Chem. Abstr, 20, 2801 (1926).
649. Atomge.vichtskommission der Internationalen Union fur Chemie. Вег, 69A, 37 (1936).
650. Atterberg A. Chem. Ztg, 20, 131 (1886); Z. analyt. Chem, 36, 314 (1897).
651. Atterberg A. Chem. Ztg, 22, 522 (1898); Z. analyt. Chem, 38, 172 (1899).
652. Atterberg A. Z. analyt. Chem, 51, 483 (1912)'.
653. At toe O. J. Soil Sci. Soc. Amer, Proc. 12, 131 (1947); Chem. Abstr, 43 1079 (1949).
654. At toe O. J, Truog E. Soil Sci. Soc. Amer, Proc. 11, 221 (1946); Chem. Abstr, 42, 2376 (1948).
655. Auger P. Compt. rend, 194, 1346 (1932); Chem. Abstr, 26, 4241 (1932).
656. Aumann C. Z. analyt. Chem, 48, 504 (1909).
657. Austerweil G. V. Chim. et Ind. (Paris), 1955, 952.
658. Austerweil G. V. Z. analyt. Chem, 150, 395 (1956).
659. Austerweil N. Bull. soc. chim, 49, 1541 (1931); Chem. Abstr, 26, 3203 (1932).
660. Austin H. C, Denson W. P, Epps E. A. J. Assoc. Offic. Agric. Chem, 36, 885 (1953); РЖХим, № 19064 (1955).
661. Autenrieth W. Zbl. f. Miner, u. Geol, 513 (1908); Chem. Zbl, 79, II, 1125 (1908).
662. A u t e nr i e t h W, Bernheim R. Z. physiol. Chem, 37, 29 (1902); Chem. Zbl, 74, I, 61 (1903).
663. Autenrieth W, Keller O. Quantitative chemische Analyse, Dresden, 1954, pp. 64, 114, 259.
664. A ver ell P. R, Walden G. H. J. Amer. Chem. Soc, 59, 906 (1937).
665. BaarS. Analyst, 78, N 353 (1953); РЖХим, № 16406 (1954).
666. Babatschen C. N. Z. analyt. Chem, 173, 121 (1960); РЖХим, № 73143 (1960).
667. В a b о A. Z. analyt. Chem, 22, 109 (1893).
668. Backenstoss G, Goebel K. Z. Naturforsch, 10a, 920 (1955); Chem Zbl, 129, 614 (1958).
669. Baddour R. F, Hawthorn R. D. Ind. Eng. Chem, 47, 2517 (1955); Chem. Zbl, 127, 5206 (1956).
670. Baeyer A. Lieb. Ann, 127, 200 (1863).
671. Bahlmann A. Lieb. Ann., 186, 325 (1877).
191
672. Bainbridge К. T. J. Franklin Inst, 212, 317 (1931). .
673. В al arew D. Z. anorg. allg. Cem., 169, 257 (1928); Kolloid Chem.
Beihefte, 32, 304 (1931); Chem. Abstr., 23, 5399 (1929); 25, 4165 (1931).
674. Balarew D. Z. analyt. Chem., 102, 241 (1935); 120, 393 (1940).
675. Balarew D., Janakiewa B. Z. anorg. allg. Chem., 156, 301 (1926);
Chem. Abstr., 21, 517 (1927).
676. Balarew D., Kaischew R. Z. anorg. allg. Chem., 167, 237 (1927);
Chem. Abstr., 22, 2089 (1928).
677. В a 11 a r d S. S„ M i 11 s С. B. Phys. Rev., 61, 108 (1942).
678. Banks W. H., Righell a to E. C., Davies C. W. Trans. Farad.
Soc., 27, 62.1 (1931).
679. Baranska H. Chem. analyt., 2, 138, 229 (1957); РЖХим., № 11471 (1959); № 64171 (1960).
680. В arber H., Ali D. Mikrochem. ver. Mikrochim. acta, 35, 542 (1950).
681. Barbieri G. A. Atti acad. Lincei, 7(6), 747 (1928); Chem. Abstr., 22,
3854 (1928).
682. Barbieri G. A. Z. analyt. Chem., 79, 146 (1929).
683. Barbieri G. A. Gazz. chim. i.tal., 60, 229 (1930); Chem. Abstr.. 24, 3724 (1930).
684. Barbieri G. A. Chim. et Ind., 1932, 836; Chem. Abstr., 26, 3602 (1932).
685. Barbieri G. A. Ann. Agron., 2, 208 (1941); Chem. Abstr., 37, '4185 (1943).
686. В a r b i e r i G. A., Les at nt M., Tendille C., Tyszkiewicz E.
Bull, document. Assoc, intern, fabr. superphosphates, No 22, 33 (19571; '
Chem. Abstr., 52, 5715 (1958). |
687. Bark L. S., Jones W. F. Mikrochim. acta, 1958, 406.
688. Barker H. C. Amer. J. Set, 24, 162 (1907).
689. Barker T. V. Proc. Chem. Soc., 23, 305 (1907); J. Chem. Soc., London, I 93, 15 (1907); Chem. Zbl., 79, I, 795 (1908).
690. Barkovic D., Bican-Fist er T. Farmac. glasnik, 9, 327 (1953); РЖХим., N 2239 (1955).
691. В a r 1 о w J. S., M a n e г у J. F. Can. J. Med. Sci., 31, 326 (1953); Chem.
Abstr., 47, 10609 (1953).
692. Barnabas J. Naturwiss., 42, 153 (1955).
693. Barnabas T., Badve M. G., Barnabas J. Naturwiss., 41, 478 (1954).
694. Barnard A. J. Chemist-Analyst, 44, 104 (1955); 45, 110 (1956); 47, 46
(1958).
695. Barnes R. B., Berry J. W., Hill W. B. Eng. Mining J., 149, N 9, 92 (1948); Chem. Abstr., 42, 8030 (1948).
696. Barnes R. B., Richardson D., Berry J. W-> Hood R. L. Ind.
Eng. Chem., Analyt. Ed., 17, 605 (1945).
697. В a r n e s R. B., S a 11 e у D. J. Ind. Eng. Chem., Analyt. Ed., 15, 4 (1943).
698. Barnett R. S. Ind. Eng. Chem., Analyt. Ed., 7, 183 (1935).
699. Barral E. Precis d’Analyse chimique qualitative, Paris, p. 133, 303, 1923.
700. Barry J. M., Rowland S. J. Biochem. J., 53, 213 (1953); РЖХим., № 464 (1953).
700a. Barth K. Silikattechnik, 11, 554 (1960); РЖХим., № 17 K289 (1961).
701. Barton-Wright E. C. Analyst, 70, 283 (1945).
702. Bartusk a M._ Silikaty, 1, 87 (1957); Chem. Abstr., 52, 13 527 (1958).
703. В a si lei a dos C. Bodenkunde u. Ptlanzenernahr., 13, 243 (1939); Chem.
Abstr., 34, 206 (1940).
704. В a s s e 11 L. G., В у er ley W. Natl. Nuclear Energy Ser., Analyt. Chem. . Manahathan Project, 1950, p. 339; Chem. Abstr., 45, 1908 (1951).
705. В a tide A. M. Compt. rend., 236, 2056 (1953).
706. Bauer A. Osterreich. Chem. Ztg., 1907, 264.
707. Bauer E. Chem. Ztg., 20, 270 (1896); Chem. Zbl., 67, I, 1028 (1896).
708. Bauer M. E. Seifensieder-Ztg., 56, 417 (1929); Chem. Abstr., 24, 1533 (1930).
192
709. Baum E. Ann. Phys., 34, 377 (1939).
710. Baumann R., Herrmann R. Z. ges. exptl. Med., 119, 487 (1952)'-120, 172 (1953); РЖХим. № 8600 (1953).
711. Baumann W. C, Eichhorn J. J. Amer. Chem. Soc., 69, 2830 (1947).
712. Bauserman H. M, Cerney R. R. Analyt. Chem., 25, 1821 (1953); РЖХим., № 7619 (1955).
713. Baxter G. P. J. Amer. Chem. Soc., 42, 2046 (1920).
714. Baxter G. P., Kobayashi M. J. Amer. Chem. Soc., 39, 249 (1917); 42, 735 (1920).
715. Bayer A. Chem. Ztg., 17, 686 (1893); Z. analyt. Chem., 33, 459 (1894).
716. Beater В. E. Proc. 15th Ann. Conf. S. African Sugar Tech. Assoc., 1940, p. 113; Chem. and Ind., 1952, 1150; Chem. Abstr., 36, 1717 (1942); 47, 2413 (1953).
717. Becker F. Zement-Kalk-Gips, 4, 93 (1951); Analyt. Chem., 26, 1578 (1954).
718. Becker Th. Dingl. polytechn. J., 183, 40 (1866); Z. analyt. Chem., 6, 257 (1867).
719. Beckurts H. Lieb. Ann., 181, 214 (1876).
720. Beers R. F., Goodman C. Bull. Geol. Soc. Amer., 55, 1229 (1944).
721. Beerstecher E. Analyt. Chem., 22, 1200 (1950).
722. Behaghel H. Z. analyt. Chem., 12, 390 (1873).
723. Behounek F. Nature, 126, 243 (1930); Chem. Abstr., 24, 5618 (19301.
724. Behrens H. Z. analyt. Chem., 30, 134, 173 (1891).
725. Behrens H., Kley P. D. Organische mikrochemische Analyse. Leipzig, 1922, p. 340.
726. Behrens W. U. Z. Pflanzenernahr, Diing. Bodenkunde, 24A, 289 (1932); Chem. Abstr., 27, 2396 (1933).
727. Behrens W. U. Forschungsdienst, 4, 463 (1937); 9, 237 (1940); Chem. Abstr., 36, 4650 (1942).
728. Beil st ein. Handbuch der organlschen Chemie, 14, 816 (1931).
729. Belak A., Alfoldy Z. Biochem. Z, 214, 110 (1929); Chem. Abstr., 24, 801 (1930).
730. Belcher R. Industrial Chemist, 23, 673 (1947); 24, 213, 473 (1948).
731. Belcher R.,-Gibbons D., Sykes A. Mikrochem. ver. Mikrochim. acta, 40, 76 (1952).
732. Belcher R, Nutten A. J. Analyt. chim. acta, 4, 475 (1950); Chem. Abstr., 45, 3287 (1951).
733. Belcher R., Nutten A. J., Thomas H. Analyt. chim. acta, 11, 120 (1954); РЖХим., № 7068 (1956).
734. Belcher R., Robinson J. W. Analyt. chim. acta, 8, 239 (1953); РЖХим., № 13230 (1954).
735. Belcher R., Robinson J. W. Mikrochim. acta, 1954, 49.
736. Belcher R., Wilson C. L. New Methods in analytical Chemistry. London, 1955, pp. 55, 258.
737. Belke J., Dierkesmann A. Arch, exptl. Phat. Pharmakoi., 205, 629 (1948); Chem. Abstr, 43, 7070 (1949).
738. Bell P. R, Cassidy J. M. Phys. Rev, 77, 409 (1950).
739. Belmas R. Rev. gen. caoutchouc, 26, 341 (1949); Chem. Abstr, 43, 5619 (1949).
740, Belohoubek A. J. prakt. Chem, 99, 235 (1866); Z. analyt. Chem, 6, 120 (1867).
741. Belton F. G. Chem. News, 91, 191 (1905); Chem. Zbl, 76, I, 1552 (1905).
742. Benedetti-Pichler A. A. Z. analyt. Chem, 64, 409 (1924); Chem. Abstr, 19, 620 (1925).
743. Benedetti-Pichler A. A, Bryant J. T. Ind. Eng. Chem, Analyt. Ed, 10, 107 (1938).
744. Benedikt R. Ber, 11, 1376 (1878).
745. Bennett A. H. Analyst, 41, 165 (1916).
13 Аналитическая химия калия
193
746. Bennett H., Hartwood H. F. Analyst, 60, 677 (1935).
747. Berg R. Z. analyt. Chem., 71, 32 (1927).
748. В er gam ini C., Rapi G. Ann. chimica, 50, 1314 (1960); РЖХим., № 12 Д65-(1961).
749. Bergh HJ Kgk Norske Videnskab. Selskab. Forh., 23, 16, 21 (1950); Chem. Abstr., 46, 2733 (1952).
750. Bergsma F. Chem. Weekbl., 48, 361 (1952); Chem. Abstr., 46, 7841 (1952).
751. Berisso B., Bruno A. Publ. inst. invest, microquim., Argentina, 14, 109 (1950); Chem. Abstr., 46, 7161 (1952).
752. Berkeley E. Phil, trans. Roy. Soc., London, 203A, 189 (1904).
753. Berkhout H. W. Chem. Weekbl., 48,, 909 (1952); Z. analyt. Chem, 140, 61 (1953).
754. Berkhout H. W. Z. analyt. Chem., 152, 248 (1956); РЖХим., № 48279 (1957).
755. Berkhout H. W., Jon gen G. H. Chem. Weekbl., 51, 607 (1955); Z. analyt. Chem., 150, 216 (1956); РЖХим., № 71945 (1956).
756. Berliner J. F. Potash Bibliography to 1928, Bull. Bureau of Mines, . No 327. Washington, 1930.
757. Bernstein R. E. S. African J. Med. Sci., 14, 163 (1949); 17, 101 (1952); Chem. Abstr., 44, 5418 (1950); 47, 7014 (1953).
758. Bernstein R. E. Nature 165, 649 (1950); Amer. J. Clin. Pathol., 23, 933 (1953); Chem. Abstr., 44, 7917 (1960); 47, 12488 (1953).
759. Bernstein R. E. Biochim. et biophysica acta, 9, 576 (1952); Chem. Zbl., 127, 13 520 (1956).
760. Berry J. W., Chappell D. G., Barnes R. Analyt. Chem., 18, 19 (1946).
761. Berthelot M. Ann. de chim. et de phys., 14(6), 86 (1888); Z. analyt. Chem., 29, 106 (1890).
762. Bertrand G. Ann. Sci. agron., 46, 1 (1929); Chem. Abstr., 23, 4493 (1929).
763. Bertrand G., Bertrand D. Mikrochemie, 36/37, 1004 (1951).
764. Berzelius J. J. Pogg. Ann., 1, 169 (1824).
765. Beukenkamp J., Riem an W. Analyt Chem., 22, 582 (1950); Chem. Abstr., 44, 5760 (1950).
766. В evil lard P. Bull. soc. chim., 19 (5), 1053 (1952).
766a. Bhatnagar M. L., Mathur N. K. J. Sci. Ind., BC19, No 12, 501 (1960); РЖХим., № 16 Д38 (1961).
767. Bible С. M. J. Assoc. Agr. Chemists, 8, 420 (1925); Ind. Eng. Chem., Analyt. Ed., 4, 234 (1932); Chem. Abstr., 19, 2612 (1925); 26, 3061 (1932).
768. Bidwell С. C. Phys. Rev., 23, 367 (1924).
769. В if fen F. M. Analyt. Chem., 22, 1014 (1950).
770. Biilmann E. Z. analyt. Chem., 39, 284 (1900).
771. Biilmann E., Thaulow R. Compt. rend. 6e conference intern chim. (Bucarest), 1925, p. 307; Chem. Absrt., 20, 3406 (1926).
772. Bills С. E., McDonald F. G., Niedermeier W., Schwartz M. C. Amer. Dietetic Assoc., 25, 304 (1949); Analyt. Chem., 21, 1076 (1949); Chem. Abstr., 43, 4780, 8943 (1949).
773. Biltz W., Marcus E. Z. anorg. Chem., 81, 369 (1913).
774. Birch F. Phys. Rev., 72, 1128 (1947); Chem. Abstr., 42, 1814 (1948).
775. Bischof f C. A., Walden P. Ber., 22, 1812 (1889).
776. Blackie W. J., Biggs A. I. Agr. J. Fiji, 13, 83 (1942); Chem. Abstr., 37, 1546 (1943).
777. Blackwell T., Ye ger C., Kraus M. J. Assoc. Offic. Agr. Chemists, 36, 898 (1953); РЖХим., № 47264 (1956).
778. Blanchetiere A., Pirlot J. M. Compt. rend, biol., 101, 858 (1929); Chem. Abstr., 25, 3265 (1931).
779. Blank E. W., Rotolo B. J. Amer. Oil Chemists Soc., 26, 102 (1949); Chem. Abstr., 43, 3636 (1949).
780. Blarez N. Compt. rend., 112, 434 (1891).
781. В las dale W. J. Ind. Eng. Chem., 10, 347 (1918).
782. Blasius E. G 6 111 i n g W. Z. analyt. Chem., 162, 423 (1958); РЖХим., № 4284 (1959).
783. Blaszkowska Z., Szperl A. Przemysl Chem., 6, 125 (1950); Chem. Abstr., 45, 10129 (1951).
784. Blaszkowska Z., Tatur H. Przemysl Chem., 6, 297 (1950).
785. BleulerE., Gabriel M. Helv. phys. acta, 20, 67 (1947); Chem. Abstr., 41, 5013 (1947).
786. Biome J. Pharmaz. Zentralhalle, 88, 202 (1949).
787. BocciarelliD. Atti accad. Lincei, 15, 686 (1932); 17, 830 (1933); Chem. Abstr., 26, 5833 (1932); 27, 5243 (1933).
788. Bock R., Hoppe T. Analyt. chim. acta, 16, 406 (1957).
789. Bodnar J., Barta L. Biochem. Z., 227, 429 (1930); Chem. Abstr., 25, 557 (1931).
790. В о e r J. H. Verslag. Akad. Wetenschappen Amsterdam, 36, 161 (1927).
791. В о e r J. H., L i e m p t J. A. Rec. tr.av. chim., 46, 124 (1927).
792: Bohling E. Z. analyt. Chem., 38, 431 (1899).
793. Bois E., Jean M. Analyt. Chem., 26, 727 (1954); РЖХим., № 7620 (1955).
794. Bokemuller H. Kali, 12, 233, 271 (1918); Chem. Zbl., 89, II, 764 (1918).
795. Bokorny Th. Allgem. Brauer- u. Hopfen-Ztg., 52, 113 (1912); Chem. Abstr., 6, 2044 (1912).
796. Bolle-Jones E. W., Ma llikarjuneswara V. R., Ratnasin-gam K. J. Rubber Research Inst. Malaya, 15, 86 (1957); Chem. Abstr., 52, 7401 (1958).
797. Bolley N. J. prakt. Chem., 103, 495 (1868); Z. analyt. Chem., 8, 505 (1869).
798. В ol liger A. Z. analyt. Chem., 94, 403 (1933); J. biol. Chem., 107, 229 (1934); Chem. Abstr., 28, 71 (1934); 29, 1031 (1935).
799. Bolliger A., Day E. J. exp. biol. Med. Sci., 6, 91 (1929); Chem. Zbl., 100, II, 2803-(1929).
800. Bo Im F. Z. analyt. Chem., 38, 348 (1899).
801. Bonastre J. Ann. Falsificat. Fraudes, 48, 347 (1955); Chem. Zbl., 129, 11971 (1958).
802. Bong G. Bull. soc. chim., 29, (2), 50 (1878); Z. analyt. Chem., 18, 270 (1879).
803. B onj ean E. Chem. News, 80, 240 (1899); Z. analyt. Chem., 40, 59 (1901).
804. Bonneau L. Bull. soc. chim., 45, 798 (1929); Chem. Abstr., 24, 800 (1930).
805. Booth E., Parker A. Rept. Atomic Energy Res. Establ. АМН, 1959, p. 2; РЖХим., № 86070 (1959).
806. Borgi oli N., I acozzil 1 i R. Chimica (Milan), 3, 359 (1948); Chem. Abstr., 43, 4178 (1949).
807. В or icky E. Z. analyt. Chem., 18, 95 (1879).
808. Borntrager A. Z. analyt. Chem., 25, 334 (1879).
809. Borowicz A. Chem. analit., 4, 481 (1959); РЖХим., № 22115 (1960).
810. Borsche E. Kali, 14, 275, 303, 358, 374 (1920); Chem. Abstr., 15, 280'7 (1921).
811. Borst L. B., Floyd J. J. Phys. Rev., 74, 989 (1948).
812. Bosian G., Miinz Th., Weinberg u. Keller, 3, 28 (1956); Chem Zbl., 128, 9536 (1957).
812a. В oss net R. Bull. soc. chim., 51 (4), 681 (1932); Compt. rend., 196, 469 (1933).
813. Bothe W. Z. Naturforsch., 1, 179 (1946).
814. Bott ger R., Will H. Lieb. Ann., 58, 273 (1846).
13*
195
815. Bott ger R. J. prakt. Chem., 91, 251 (1864); Z. analyt. Chem., 3, 362 (1864).
816. В ottinger C. Lieb. Ann., 188, 319 (1877).
817. Bouat N. Ann. Ec. Nat. Agric. Montpellier, 23, 197 (1935).
818. Boucher L. An. Soc. espan. fis. quim., 23, 540 (1925); Chem. Abstr., 20, 1366 (1926).
819 В о u 1 a d J. H. J. Soc. Chem. Ind., 52, 270 (1933); Chem. Abstr., 27, 5025 (1933).
820. Bourdon D. Chim. analyt., 31, 154 (1949); 32, 273 (1950); Z. analyt. Chem., 132, 438 (1951).
821. Bourdon D., Cotte J., Gielfrich M. L. Chim. analyt., 33, 170 (1951); Chem. Abstr., 45, 6125 (1951).
822. Bourdon D., Gielfrich M. L. Bull. soc. sci. Bretagne, 23, 117 (1948); Chem. Abstr., 44, 5260 (1950).
823. Boussingault J. prakt. Chem., 102, 94 (1867).
824. Bo vay E. Z. analyt. Chem., 106, 284 (1956); Chem. Zbl., 128, 9179 (1957).
825. Bovis P. Chim. et Ind., 19, 944 (1928).
826. Bowman R. L., Berliner R. W. Federation Proc., 8, 14 (1949).
827. Bowser L. T. Ind. Eng. Chem., 1, 791 (1909); J. Amer. Chem. Soc., 32, 78 (1910); 33, 1752 (1911).
828. Boyd G. Analyt. Chem., 21, 335 (1949).
829. Boyle A. J., Whitehead T., Bird E. J., Batchelor T. M., I s e-r i L. T., J а с о b s о n S. D., Myers G. B. J. Lab. Clin. Med., 34, 625 (1949); Chem. Abstr., 43, 7071 (1949).
830. Boyle R. J. Chem. Soc., 95. 1711 (1909).
831. Boynton D., P e e c h M. J. Amer. Soc. Agron., 37, 404 (1945).
832. Bozorth R. M. J. Amer. Chem. Soc., 45, 2653 (1923).
833. Bradley J. E., Bradley O. Mineralog. Mag. J. Mineralog. Soc., 31, 164 (1956); Chem. Zbl., 129, 815 (1958).
834. Bramante P. Policlinico, Sez. prat., 57, 1371 (1950); Chem. Abstr., 45, 6682 (1951).
835. Bramley A., Brower A. K. Phys. Rev., 53, 502 (1938); Chem. Abstr., 32, 4070 (1938).
836. Brannock W. W., Berthold S. M. Geol. Surv. Bull., No 992, 1 (1953); РЖХим., № 48526 (1954).
837. Braun C. D. Z. analyt. Chem., 7, 313 (1868).
838. Braun J. S. J. Chem. Soc., 91, 1826 (1907).
839. Brauner B. Coll. Czech. Chem. Comm., 2, 442 (1930).
840. Bray R. H. J. Amer. Soc. Agronomy, 24, 312 (1932); Chem. Abstr., 26, 3058 (1932).
841. Bray W. C. J. Amer. Chem. Soc., 31, 611, 621 (1909); Chem. Zbl., 80, II, 1275 (1909).
842. Brea ley L. Analyst, 76, 340 (1951).
843. В re a ley L., Ross R. E. Analyst, 76, 334 (1951).
844. Breckpot R. Ann. Soc. Sci. Bruxelles, 53B, 219 (1933).
845. Breckpot R., Mevis A. Ann. Soc. Sci. Bruxelles, 54B, 99 (1934); 55B, 16 (1935).
846. Bregman J., Murata Y. J. Amer. Chem. Soc., 74, 1867 (1952).
847. В reh F., Gaebler О. H. J. biol. Chem., 87, 81 (1930); Chem. Abstr., 24, 3526 (1930).
848. Brennecke E., Faja ns R., Furman N., Lang R., Stamm H. Neuere massanalytische Methoden. Stuttgart, 1951.
849. Brewer A. K. Phys. Rev., 46, 894 (1934).
850. Brewer A. K. Phys. Rev., 48, 640 (1935).
851. Brewer A. K. J. Amer. Chem. Soc., 58, 365, 370 (1936).
852. Brewer A. K. Ind. Eng. Chem., 30, 893 (1938); Chem. Abstr., 32, 7340 (1938).
853. Brewer A. K. J. Amer. Chem. Soc., 59, 1578 (1957); 61, 1597 (1939).
196
854. Brewer A. К., Madorsky S. L., Westhaver J. W. Science, 104, 156 (1946).
855. Breyer T., Schweitzer H. Chem. Ztg., 16, 1720 (1892); 17, 101, 244 (1893); Z. analyt. Chem., 35, 687, 689 (1896).
856. Bridgman P. W. Phys. Rev., 3, 152 (1914).
857. Bri ggs A. P. J. Biol. Chem., 57, 351 (1923).
858. BrindW. D. Soils a. Fertilizers, 14, 347 (1951).
859. Briscoe H. V., H о 11 P. F. Inorganic microanalysis. London, 1950, pp. 67, 126, 147.
860. Briscoe H. V., Robinson P. L., Rudge A. J. J. Chem. Soc., 3218 (1931); Chem. Abstr., 26, 1207 (1932).
861. В rod a E. Berg- u. Hiittenmannische Monatschr., 101, 228 (1956).
862. Broderick E. J., Zack P. G. Anal. Chem., 23, 1455 (1951).
863. Bronsted J. N. Danske Selsk. Skr., 12, 254 (1914—1916).
864. Broomhead J. A., Gibson N. A. Chem. and Ind., 1956, 1474; РЖХим., № 41 420 (1957).
865. Brouckere L. J. chim. phys., 27, 543 (1930); Chem. Abstr., 25, 3216 (1931).
866. Brown D. S., Robinson R. R., Browning G. M. Ind. Eng. Chem., Analyt. Ed., 10, 652 (1938); Chem. Abstr., 33, 78 (1939).
867. Brown H. B., Shohl A. T. J. biol. Chem., 91, 745 (1931); Chem. Abstr., 25, 3929 (1931).
868. Brown J. A. J. Chem. Educ., 30, 363 (1953).
869. Brown J. G., Lille land O. Proc. Amer. Soc. Hort. Sci., 48, 341 (1946); Chem. Abstr., 41, 7453 (1947).
870. В г о w n J. G., L i 11 e 1 a n d O., Jackson R. K. Proc. Amer. Soc. Hort. Sci., 56, 12 (1950); Chem. Abstr., 46, 1391 (1952L
871. Brown M. H., Reedy J. H. Ind. Eng. Chem., Analyt. Ed., 2, 304 (1930).
872. Browning P. E, D r u s h e 1 W. A. Amer. J. Science, 23 (4), 293 (1907); Z. analyt. Chem., 47, 64 (1908).
873. Browning P. E., Spencer M. Amer. J. Science, 42 (4), 279 (1916).
874. Brukl A. Monatsh. f. Chem., 56, 179 (1930).
875. Brunner C. Pogg. Ann., 122, 155 (1864); Z. analyt. Chem., 4, 415 (1865).
876. Bruyn L. Z. phys. Chem., 10, 787 (1892).
877. Bulli M. N, Fernandes L. Ann. chim. appl., 13, 46 (1923); Chem. Zb!., 94, IV, 562 (1923).
878. Bulli M. N, Fernandes L., Foa N. L’Agricoltura coloniale, 18, 417 (1924); Chem. Abstr., 19, 1610 (1925).
879. Bullock B., Kirk P. L. Ind. Eng. Chem., Analyt. Ed., 7, 178 (1935).
880. Bultasova H., Konopasek E. Chem. listy, 49, 769 (1955).
881. Bunsen R. Lieb. Ann., Ill, 265 (1859); Pogg. Ann., 155, 230, 366 (1875); Z. analyt. Chem., 15, 89 (1876).
882. Bunsen R. Pogg. Ann., 113, 367 (1861).
883. Bunsen R. Lieb. Ann., 122, 348 (1862); Z. analyt. Chem., 26, 355 (1887).
884. Bunsen R., Kirchhoff G. Pogg. Ann., 110, 161 (1860).
885. Burch P. R. Nature, 172, 361 (1953).
886. Burchartz H. Zement, 20, 987 (1931); Chem. Abstr., 26, 2295 (1932).
887. Burger G. Monatsh. f. Chem, 53/54, 985 (1929).
888. Burgess L. L, Kamm O. J. Amer. Chem. Soc, 34, 652 (1912); Univ. Illinois Bull, 10, No 36 (1913); Chem. Zbl, 83, II, 868 (1912); 86, I, 398 (1915).
889. Burglen L, Longuet P. Rev. mater, constr. et trav. publics, No 530, 257 (1959); РЖХим, № 53 623 (1960).
890. Burkhart L. Plant Physiol, 16, 411 (1941); Chem. Abstr, 37, 324 (1943). 890a. Burkser E. S. Z. analyt. Chem, 80, 270 (1930).
891. Burma D. P. Analyst, 77, 382 (1952); Chem. Zbl, 127, 10 774 (1956).
892. Burma D. P. Analyt. chim. acta, 9, 513 (1953); РЖХим, № 46814 (1954)
893. Burrage L. J. Chem. News, 134, 85 (1927); Chem. Abstr, 21, 1602 (1927).
197
894. Burriel-Marti F., Ramirez-Munoz J. Flame photometry. Amsterdam, 1957.
895. Burrie! F, Ramirez-Munoz J., Be n ito Potou s A. Anales edafol. у fisiol. vegetal (Madrid), 16, 167 (1957); Chem. Abstr., 52, 1529 (1958).
896. Burriel F., Jimenez S., Alvarez H. C. Ann. edafol. veget., 14, 213 (1955); An. Real soc. espanola fis. у quim., B50, 663 (1954); РЖХим., № 1085, № 36193 (1956).
897. Burriel-Marti F., Rexach M, Ramirez-Munoz J. Rev. Univ, industr. Santander, 1, 37 (1959); РЖХим., № 8. Д62 (1961).
898. Burriel-Marti F, Lizarduy M. L, Ramirez-Munoz J. Rev. Univ. Ind. Santander, Colombia, 1, 37 (1959); Analyt. Abstr., 7, 114 (I960).
899. Burstall F. H., Davies G. R., Linstead R. P., Wells R. A. J. Chem. Soc., 1950, 516.
900. Burton F. Metallurgia, 34, 49, 105 (1946).
901. В user W. Helv. chim. acta, 34, 1635 (1951).
902. Buttner H. Keram. Rundschau, 47, 101 (1939); Chem. Abstr., 33, 4158 (1939)
903. Buz as L. Magyar Kem. Lapja, 14, 251 (1959); Analyt. Abstr., 7, 851 (1960).
904. Cabell M. J., Thomas A. The Determination of rubidium and caesium in sodium-potassium alloys and related materials by radioactivation. Harwell, 1955.
905. Cadiou P., Montagne P. 19e Congr. G. A. M. S. 1956. Paris, 1958, p. 245; РЖХим., № 69127 (1960).
906. Cahen A. Bull. soc. chim., 3 (5), 640 (1936).
907. Calcagni G., Marotta D. Gazz. chim. ital, 42, 11, 674 (1912).
908. Caley E. R. J. Amer. Chem. Soc., 52, 953 (1930); 53,-539 (1931).
909. С a 1 e у E. R, A x i I г о d H. D. Ind. Eng. Chem., Analyt. Ed., 14, 242 (1942).
910. Caluwe P. Le dosage de la potasse dans les engrais. Bruxelles, 1888; Z. analyt. Chem., 35, 687 (1896).
911. Calvert J. T. Trans. Farad. Soc., 26, 509 (1930); Chem. Abstr., 24, 5912 (1930).
912. Calzolari C., Ciana A. 1st. chim. univ. studi Trieste, Fac. sei., No 2, 1 (1954); РЖХим., № 49140 (1955).
913. Calzolari F. Gazz. chim. ital., 42, II, 85 (1912).
914. Cameron F. K-, Fa i Iyer C. F. J. Amer. Chem. Soc., 25, 1603 (1903}.
915. Campanile S. Ann. sper. agrar. (Rome), 4, 919 (1950); Chem. Abstr., 45, 6781 (1951).
916. Campari G. Arch. Pharm., 221, 67 (1883); Z. analyt. Chem., 23, 60 (1884).
917. Campbell N. R. Proc. Cambridge Philos. Soc., 14, 211, 557 (1907/1908);
15, 11 (1909); Chem. Zbl., 78, II, 1047 (1907); 80, I, 984, 1385 (1909); ЖРФХО, 42, 2102 (1910).
918. Campbell N. R., Wood A. Proc. Cambridge Philos. Soc., 13, 282 (1906); Chem. Zbl., 77, II, 4 (1906); 78, II, 1047 (1907); ЖРФХО 40, 2, 153, 211 (1908).
919. Campo A., Boissier V., Hoyos A. Rev. Real acad. cience exact, fis., Madrid, 34, 352 (1940); Chem. Abstr., 45, 4167 (1951).
920. Candea C., Saucine L. I. Bull. soc. chim., Romania, 14, 76 (1932).
921. Cantani R. A. Anais assoc, quim., Brazil, 3, 131 (1944); Chem. Abstr., 39, 2037 (1945).
922. Cap pel N. Pogg. Ann., 139, 628 (1870).
923. Cardus D., Llaurado J. C. Med. clin. Barcelona, 24, 193 (1955); Chem. Zbl, 127, 14131 (1956).
924. Carere-Comes O. Z. wiss. Mikroskopie, 55, 1 (1938).
925. Carfmell R. Phil. Mag, 16 (4), 328 (1858).
498
926. Carlson В Klason’s Tidsskrift, 1910, 247.
927. Carnot A. Ber. Deutsch, chem. Ges., 9, 1434 (1876); Compt. rend., 83, 338 (1876); 84, 390, 1504 (1877).
928. С a r n о t A. Compt. rend., 86, 478 (1878).
929. Carpenter D. C., Mack G. L. J. Amer. Chem. Soc., 56, 311 (1934).
930. Carpenter F. B., Power R. O. American Fertilizer, No 79, 8 (1933); Ind. Eng. Chem., Analyt. Ed., 6, 62 (934); Калий, 3, № 6, 42 (1934).
931. Carriere E., Da Ip la M. Compt. rend., 205, 1157 (1937); Chem. Abstr., 32 1202 (1938)
932. Carruthers C. Ind. Eng. Chem., Analyt. Ed., 15, 70 (1943).
933. Carter R. H. Ind. Eng. Chem., 22, 886 (1930).
934. Cartmell R. Phil. Mag., 16, (4), 328 (1858).
935. Casamajor P. Chem. News, 34, 231, 242 (1876).
936. Caspari R. Z. angew. Chem., 6, 68 (1893); Z. analyt. Chem., 36, 709 (1897).
937. Catani R. A., Pai va-Neto J. D. Bragantia, 9, 175 (1949); Chem. Abstr., 45, 9786 (1951).
938. Caton R. D., Bremner R. W. Analyt. Chem., 26, 805 (-1954).
939. C a veil A. J. Analyst, 77, 537 (1952).
940. C a veil A. J. J. Sci. Food Agr., 5, 195 (1954).
941. Ceausescu D. Studii si cercetari sciint. Acad. RPR, 4, 103 (1957); РЖХим., № 23017 (1959).
942. С e с c a r e 11 i M., Quareni G., Rostagni A. Phys. Rev., 80, 909 (I960); Nuovo cimento, 8, 132 (1951); Chem. Abstr., 45, 4572 (1951).
943. Cecconi S., Pol esell a A. Ricerca sclent., 23, 2030 (1953); РЖХим., № 32757 (1956).
944. Celap M. B. Glasnik Khem. Drustva, Belgrad, 21, 225 (1956); Chem. Abstr., 52, 15332 (1958).
945. Cel si S. A. An. farm, bioquim., 4, 55 (1933); Z. analyt. Chem., 98, 368 (1934).
946. Cel si S. A. Bol. Soc. quim. Peru, 25, 73 (1959); РЖХим., № 56683 (1960).
947. Cencelj J. Vest. Slov. Kem. Drusltva, 4, 31 (1957); РЖХим., № 38359 (1959).
948. Cenley H. J. Analyst, 80, 354 (1955); J. Soc. Glass Technol., 43, T62 (1959); РЖХим., № 78293 (1959).
949. Chakrabarti S., Burma D. P. Science and Culture, 16 , 485 (1951); Chem. Abstr., 46, 380 (1952).
950. Chalmers J., Tatlock R. R. Chem. News, 17, 199 (1868); Z. analyt. Chem., 8, 88 (1869).
951. Chalmers T. A. Nature, 168, 870 (1951).
952. Chambon M. Bull. soc. chim., 8, 700 (1941); Chem. Abstr., 36, 6436, 6437 (1942); Ann. biol. clin. (Paris), 8, 513 (1950); Chem. Abstr., 45, 3895 (1951).
953. Ch a mot E. M., Bed Sent H. A. Mikrochemie, 6, 13 (1928); Chem. Abstr., 22, 3602 (1928).
954. Ch amot E. M., Mason C. W. Handbook of Chemical Microscopy, vol. 2. N. Y., 1931, pp. 61, 66, 72, 184.
955. Champ P., Fanconnier P., Duval C. Analyt. chim. acta, 10, 443 (1954); РЖХим., № 14 166 (1955).
956. Chancel G. Compt rend., 60, 409 (1865).
957. Chapman G. W. J. Agr. Sci., 37, 29 (1947); Chem. Abstr., 45, 4172 (1951).
958. Chariot G., Bezier D. Methodes modernes d’analyse quantitative minerale. Paris, 1949, pp. 556, 560.
959. C h a t u r v e d i G. K-, Bhattacharya A. K. Z. analyt. Chem., 175, 401 (1960); РЖХим., № 1 126 (1961).
960. Chen S. C., Wilson S. D. J. Chinese Chem. Soc., 16, 35 (1949); Chem. Abstr., 43, 9126 (1949).
199
961. Chen Y. C., Shen S. S. J. Chinese Chem. Soc., 8, 12 (1941); Chem. Abstr., 37, 51 (1943).
962. Chenery E. M. Analyst, 77, 102 (1952); 80, 569 (1955).
963. Chiba H. J. Sci. Soil Manure, Japan, 23, 207 (1953); Chem. Abstr., 47, 12134 (1953).
964. Cholak J., Hubbard D. M. Ind. Eng. Chem., Analyt. Ed., 16, 728 (1944).
965. C h у d e n i u s J. J. Z. analyt. Chem., 2, 365 (1863).
966. Cimermann C., Rzymowska C. J. Mikrochemie, 29, 129 (1936).
967. C iochin a J. Z. analyt. Chem., 71, 45 (1927).
968. С 1 a г к A. W., W i 11 e t s С. O. Ind. Eng. Chem. Analyt. Ed., 8, 209 (1936).
969. C 1 a г к W. G, L e v i t a n N. I., G 1 e a s о n D. F., Greenberg G.
J. biol. Chem., 145, 85 (1942).
970. Clarke C. Amer. J. Sci., 32 (3), 356 (1886).
971. Clarke L., Davidson J. M. Ind. Eng. Chem., Analyt. Ed., 3, 324 (1931); Chem. Abstr., 25, 4486 (1931).
972. Classen A. Z. analyt. Chem., 18, 373 (1879).
973. Classen A. Ber, 17, 2477 (1884).
974. Classen A., Danneel H. Quantitative Analyse durch Elektrolyse. Stuttgart, 1927, pp. 135, 342.
975. Claudatus I., Ghimicescu G. Ann. sci. univ. Jassy, 24, 357 (1938); Chem. Abstr., 32, 8302 (1938).
976. Clausen S. W. J. biol. Chem., 36, 479 (1918).
977. Clerfeyt E. Bull. soc. chim. Belg., 31, 417 (1922); Z. analyt. Chem., 78, 82 (1929).
978. CloseP, Watson M. T. J. Amer, ceram. Soc., 37, 235 (1954); РЖХим., № 29131 (1955).
979. Cluley H. J. Analyst, 80, 354 (1955); Z. analyt. Chem., 149, 142 (1956).
980. Cohn W., Kohn H. J. Amer. Chem. Soc., 70, 1986 (1948).
981. Cohn W. M. J. applied phys., 10, 200 (1939); Chem. Abstr., 33, 3410 (1939).
982. Colani A. Bull. Soc. Chim., 19, (4), 407 (1916); Compt. rend., 163, 123 (1916).
983. Colin J. Bull. soc. chim. biol., 33, 394 (1951); Chem. Abstr., 46, 161 (1952).
984. Collier P. Amer. J. Sci., 37 (2), 344 (1864); Z. analyt. Chem., 4, 413 (1865).
985. С о 11 i n s G. C., Polkinhorne H. Analyst, 77, 430 (1952); Chem. Zbl., 127, 5371 (1956).
986. С о 1 1 i n s T. L., Nier A. O., JohnsonW. H. Phys. Rev., 84, 717 (1951); Chem. Abstr., 46, 2929 (1952).
987. Cornelia G. Ann. chim. appl, 15, 123 (1925); Chem. Abstr., 19, 3441 (1925).
988. Comite consultatif de la Direction des Poudres et Salpetres, Ann. de Chi-mie, 12 (2), 41 (1819).
989. Connors J. J. J. Amer. Water Works Assoc., 42, 33 (1950); Chem. Abstr., 44, 2148 (1950).
990. Conrad A. L, Johnson W. C. Analyt. Chem., 22, 153 (1950).
991. Conrad N. Lieb. Ann., 273, 117 (1893).
992. Consol az io W. V., Talbott J. H. J. Biol. Chem., 126, 55 (1938); Chem. Abstr., 33, 667 (1939).
993. С о n s о 1 a z i о W. V., T a 1 b о 11 J. H. J. Biol. Chem., 132, 753 (1940).
994. Cook K. L. Phys. Rev, 64, 278 (1943).
995. Cooper S. S. Analyt. Chem, 29, 446 (1957); Chemist-Analyst, 46, 62 (1957); РЖХим, № 74633 (1957); № 64144 (1958).
996. Cop aux H. Bull. Soc. franj. mineral, 30, 292 (1907); Chem. Zbl, 79, I, 711 (1908).
997. Coraducci P. Ceramica, 10, 37 (1955); РЖХим, № 46137 (1955).
200
I
Я 998. Corenwinder W.. Contamine G. Compt. rend., 89, 907 (1879);
f Z. analyt. Chem., 35, 72 (1896).
! 999. Corey R. B., Jackson M. L. Analyt. Chem., 25, 624 (1953).
1000. Cornec E., Dickeley J. Compt. rend., 184, 1555 (1927); Bull. soc. chim., 41, 1017 (1927).
1001. Cornec E., Krombach H. Compt. rend., 194, 784 (1932); Bull. soc. chim., 51, 672 (1932).
1002. Cornec E., Neumeister A. Revista Ciliche (Chile), 10, 492 (1929).
1003. Comes J. J. Analyst, 69, 237 (1944); 70, 131 (1945); Chem. Abstr., 38, 5750 (1944); 39, 2855 (1945).
- 1004. Cornet C., Coursier J., Hure J. Analyt. chim. acta, 19, 259 (1958);
РЖХим., № 4259 (1959).
1005. Cornfield A. H., P о 11 a r d A. G. J. Sci. Food Agr., 3, 151 (1952); Chem. Abstr., 46, 7268 (1952).
1006. Cornwall H. B. American Chemist, 2, 366 (1872); Z. analyt. Chem., 11, 307 (1872).
1007. Corradi R. Boll. chim. farm., 41, 419, 675 (1902); Chem. Zbl., 73, II, 1340 (1902).
1008. Costeanu N. D. Bui. Facultat. Stiinti Cernanti, 11, 135 (1937); Chem. Abstr., 33, 794 (1939).
1009. Cotte J., Ducet G. Ann. agron., 16, 225 (1946); Chem. Abstr., 41, 5051 (1947).
1010. Cotton R. H. Ind. Eng. Chem., Analyt. Ed., 17, 734 (1945).
1011. Coursier J., Hure J. Analyt. chim. acta, 18, 272 (1958); РЖХим., № 64147 (1958).
1012. Courtois F. Canad. J. Med. Technol., 13, 15 (1951); Chem. Abstr., 46, 3452 (1952).
1013. Cowie G. A. Potash. N. Y., 1956.
1014. Crane F. E. Analyt. Chem., 28, 1794 (1956).
1 1015. Crasu V., Manol » V. Bull. soc. Romania,- 15, 129 (1933); Chem. Zbl.,
105, II, 476 (1934).
1016. Crawford C. Arch. d. Pharm., 88 (2), 296 (1856).
1017. Crismon J. M. Federation Proc., 7, 74 (1948).
1018. Cristal P., Caron M. Bull. soc. chim. biol., 34, 228 (1952); Chem. Abstr., 47, 660 (1953).
1019. Cr itchfield F. E., Johnson J. B. Taianta, 5, 58 (1960); РЖХим., № 6, Д67 (1961).
1020. Cronheim G. Biochem. Z., 171, 7 (1926); Chem. Abstr., 20, 3407 ' ' (1926).
1021. Crooks R. C. J. Assoc. Offic. Agric. Chemists, 36, 891 (1953); РЖХим., ; № 48529 (1954).
’ 1022. Crouch E. A., Corbett J. A., Willis H. H. The Fast Separation
of the Alkali Metals. Harwell, 1957.
1023. Cuisiner V. Bull. soc. chim., 31, (4), 1064 (1922).
1024. Cullen G. E., Wilkins W. E. J. biol. Chem., 102, 403 (1933).
• 1025. Cumbers C. F., Coppock J. B. J. Soc. Chem. Ind., 56, 405 (1937);
t Chem. Abstr., 32, 877 (1938).
5 1026. Cumings J. N. Biochem. J., 33, 642 (1939); Chem. Abstr., 33, 8229 (1939).
? 1027. Cunningham B., Kirk P. L., Brooks S. C. J. biol. Chem., 139, 21
i (1941).
; 1028. Cunningham M., Perkin F. M. J. Chem. Soc., London, 95, 1562
(1909); Chem. Zbl., 80, II, 1967 (1909).
1029. Cun у L. J. pharm. Chim., 16, 55 (1932); Chem. Abstr., 26, 5510 (1932).
1 1030. Curtius G. H. Iron Age, 155, 54 (1945).
! 1031. Curtmann S. O. Ber., 14, 1951 (1881).
' 1032. Cuttica V. Gazz. chim. ital., 53, 185 (1923).
1033. Cuttica V., Gallo T. Gazz. chim. ital., 53, 374 (1923).
; 1034. Cuvelier В. V. Naturw. Tijdschr., 14, No 3—6, 107 (1932); Chem.
Abstr., 26, 4007 (1932).
201
t
1035. Da hr E. Acta physiol. Scand., 12, 229 (1946); Chem. Abstr., 41, 1271 (1947).
1036. Danguy L. Internat. J. Appl. Radiat. a. Isotopes, 2, 90 (19571; РЖХим., № 57130 (1958).
1037. D’Ans J. Z. angew. Chem., 47, 583 (1934); Калий, 3, № 10 35 (1934).
1038. D’Ans J. Kali, 35, 198 (1941); Chem. Abstr., 37, 1670 (1943),
1039. Daubner W. Angew. Chem., 49, 830 (1936).
1040. Davies C. W. Trans. Farad. Soc., 26, 592 (1930).
1041. Davies C. W. J. Amer. Chem. Soc., 59, 1760 (1937).
1042. Davies H, Davies W. J. Chem. Soc., 107, 1678 (1915); 123, 2976 (1923).
1043. Davis С. E. Ssi. Chem. Ind. Victoria, 51, 1032 (1952); Chem. Zbl., 127, 2552 (1956).
1044. Davis W. A. J. Agric. Science, 5, 52 (1912); Z. anal. Chem., 57, 480 (1918).
1045. Davy H. Phil. Trans., 98, 1 (1808); 99, 39 (1809); 100, 16 (1810).
1046. Deal S. B. Analyt. Chem., 26, 598 (1954).
1047. Debras G. J., Voinovitch I. A. Bull. soc. franc, ceram., No 38, 77 (1958); Compt. rend., 248, 3421 (1959); РЖХим., № 77200 (1958); № 34483 (1960).
1047a. Debras G. J., Voinovitch I. A. Chem. analit. (Polska), 5, 193 (1960); РЖХим., № 7Д37 (1961).
1048. Debray H. Bull. soc. chim., 5 (2), 404 (1866); Z. analyt. Chem., 5, 380 (1866); Compt. rend., 66, 702 (1868).
1049. Dehn O. Z. physiol. Chem., 144, 178 (1925); Chem. Abstr., 19, 2681 (1925).
1050. Deibner L, Benard P. Ann. Falsifikat. Fraudes, 48, 165, 217 (1955); Z. analyt. Chem., 149, 370 (1956).
1051. Deitz V. J. Chem. Phys., 4, 578 (1936).
1052. Delaney C. F. Phys. Rev., 81, 158 (1951); Sci. Proc. Roy. Dublin Soc., 25, 251 (1951); Chem. Abstr., 46, 10928 (1952).
1053. Delaville G., Desbordes J., Schuster G. Ann. pharm. franf, 10, 488 (195'2); Chem. Abstr., 47, 2813 (1953).
1054. Delaville M., Carlier P. Compt. rend., 182, 701 (1926); Compt. rend, biol., 94, 1364 (1926).
1055. Delaville M, Carlier P. Bull. soc. chim. biol., 8, 481 (1926); Compt. rend, biol, 101, 1082 (1929).
1056. Del epine M. Bull. soc. chim., 3 (4), 904 (1908).
1057. Del epine M. Ann. chim. (Paris), 7, 401 (1952); Chem. Abstr., 47, 4233 (1953).
1058. Del ger у I. Compt. rend., 224, 915 (1947).
1059. DemoIon A. Compt. rend. acad. agr. France, 27, 504 (1941); Chem. Abstr., 38, 3405 (1944).
1060. Demortier G., Van Hoeck G. Bull. inst. agron. et stas. recherches Gembloux, 20, 57 (1952).
1061. Denes S. Mikrochemie, 26, 277 (1939); Chem. Abstr., 33, 4618 (1939).
1062. Deniges G. Compt. rend, biol., 84, 63 (1921).
1063. Deniges G. Bull. soc. pharm. Bordeaux, 71, 191 (1933); Chem. Abstr., 27, 5677 (1933).
1064. D e n n e n W. H., Ahrens L. H., Fairbairn H. W. U. S. Geol. Survey Bull., No 980, 25 (1951); Chem. Abstr, 46, 11009 (1952).
1065. Dennen W. H, Fowler W. C. Bull. Geol. Soc. Amer, 66, 655 (1955); РЖХим, № 13166 (1956).
1066. Dennet J. H. Malayan Agr. J, 17, 341 (1929); Ghem. Abstr, 24, 1314 (1930).
1067. D e n n e у T. О, M о n к С. B. Trans. Farad. Soc, 47, 992 (1951).
1068. Denson J. J. biol. Chem, 209, 233 (1954).
1069. Dermer H, Dermer О. C. J. Amer. Chem. Soc, 60, 1 (1938); Chem. Abstr, 32, 1602 (1938).
202
1070. Dermer О. C., Dermer V. Н. J. Amer. Chem. Soc., 61, 3302 (193У).
1071 Designoll.e N. Dingl. polyt. J., 192, 71 (1871).
1072. Despaul J. E., Weissman H. B., Barsky M. H. J. Assoc. Agr. Chem., 36, 1083 (1953).
1073. Devaux H, Aubel E. Compt. rend., 184, 601 (1927); Chem. Abstr., 21, 1909 (1927).
1074. Di am ant J. Z. analyt. Chem., 38, 182 (1899).
1075. Dias de Rada F. Anal. Soc. Esp. fis. quim., 27, 390 (1929); Калий, 2, № 7, 17 (1933).
1076. Di eke G, Duncan A. B. Spectroscopic properties of uranium compounds. Me Graw-Hill Book Company, 1949, p. 290; Chem. Abstr., 44, 432 (1950).
1077. Diemair W., Gundermann C. Z. Lebensmittel-Untersuch, 109, 469;
111, 120 (1959); РЖХим., № 11149, 75134 (1960).
1078. Dittmar W., McArtur J. J. Soc. Chem. Ind., 6, 799 (1887).
1079. Dixon K. Analyst, 83, 362 (1958); РЖХим., № 991 (1959).
1080. Djurfeldt R, Samuelson O. Acta Chem. Scand., 4, 165 (1950).
1081. Dobbins J. T, Markham E. C., Edwards H. L. J. chem. Educ., 16, 94 (1939).
1082. Dobbins J. T, Southern J. A. J. chem. Educ., 19, 479 (1942); Chen!. Abstr, 37, 47 (1943).
1083. Doden W, Rodeck H. Biochem. Z, 320, 413 (1950); Chem. Abstr, 45, 3447 (1951).
1084. Doisy F, Bell G. J. biol. Chem., 57, 353 (1923).
1085. Dojlido J, К oz io rowski B. Gaz, woda i techn. sanit, 34, 140 (1960); РЖХим, № 92022 (1960).
1086. D о 1 a r a A. L, Ortega M, C a n t u r r i F. G. An. Real Acad. Farmac, 23, 225 (1957); Chem. Zbl, 129, 11977 (1958).
1087. Domange L, Longuevalle S. Ann. pharm. franf, 9, 647 (1951); Chem. Abstr, 46, 4170 (1952).
1088. Domingo W. R, К 1 у n e W. Biochem. J, 45, 400 (1949); Chem. Abstr, 44, 3067 (1950).
1089. Donald R. Fertiliser Soc. (Engl.), Proc, No 16, 20 (1952); Chem. Abstr, 46, 7270 (1952).
1090. Donath M, Mathes H. Bergbauwiss, 7, No 4, 86 (1960); РЖХим, № 73037 (1960).
1091. Donk M. G. Bureau C)iem. U. S. Dept. Agr, No 90, 1905, p. 3.
1092, Dorche J, Ro Het M, Costet C. Ann. pharmac. franf, 13, 288 (1955); Chem. Zbl, 129, 3395 (1958).
1098. Dor in g Th. Z. analyt. Chem, 49, 158 (1910).
1094. Drake B. Ark. Kemi, 8, 171 (1955); Chem. Zbl, 128, 502 (1957).
1095. D r e_g_U-S-s-M. Biochem. Z, 233, 375 (1931); Chem. Abstr, 25, 3681 (1931),
1096. Dresia H, Beckmann R. Z. analyt. Chem, 159, 1 (1957).
1097. Dreyfus E. Bull. soc. chim, 38, 162 (1882); Z. analyt. Chem, 22, 62 (1883).
1098. Drossbach P. Z. anorg. allg. Chem, 165, 149 (1927); Chem. Abstr, 21, 3577 (1927).
1099. Druce J. G. Chem. News, 145, 186 (1932); Chem. Abstr, 27, 44 (1933).
1100. Drushel W. A. Amer. J. Sci, 24 (4), 433 (1907); 26 (4), 329, 555 (1908);
Z. anorg. Chem, 56, 223 (1908); 59, 97 (1908); 61, 137 (1909); Z. analyt. Chem, 48, 53 (1909).
1101. Dubaquie J. Ann. fals, 25, 280 (1932); Chem. Abstr, 26, 4907 (1932).
1102. Dubernard. Z. analyt. Chem, 25, 551 (1886).
1103. D u b о u x M. Compt. rend, 159, 320 (1914).
1104. Dubsky J. V, Novakovd M. Chem. Obzor, 15, 136 (1940); Chem. Abstr, 37, 49 (1943).
1105. Duca A. Studii si cercetari stiint, 5, 99 (1954); Studii si cercetari de chim, 4, 131 (1956); РЖХим, № 19512 (1956); Ns 846 (1958).
203
1106. Duca A., Buruleanu N. Rev. chim., 7, 430 (1956); РЖХим., № 19520 (1957).
1107. Ducet G., Bats J., Dupony-Bras M. Ann. inst. natl. recherche agron, 2, 367 (1951); Chem. Abstr., 47, 995 (1953).
1108. Duff end ack O. S, Thomson К. B., Lee W. С., К op pi us O. G. J. biol. Chem., 126, 1 (1938); Chem. Abstr, 33, 667 (1939).
1109. Duff end ack O. S, Wiley F. H, Owens J. S. Ind. Eng Chem, Analyt. Ed, 7, 410 (1935).
1110. D u f f e n d а с к O. S„ W о 1 f 1 a n d R. A, S m i t h R. W. Ind. Eng. Chem, Analyt. Ed, 5, 226 (1933).
1111. D u f 1 о s A, Fischer N. W. Pogg. Ann, 72, 477 (1847).
1112. Du ggan R. E. J. Assoc. Offic. Agr. Chem, 25, 895 (1942); Chem. Abstr, 37, 727 (1943).
1113. Dunkle E. C, Merkle F. G, Anthony R. D. J. Amer. Soc. Agron, 31, 438 (1939); Chem. Abstr, 33, 5110 (1939).
1114. DupreF. T. Dissertation. Halle, 1893; Калий, 2, № 7, 17 (1933).
1115. Dupre F. T. Z. analyt. Chem, 36, 319 (1897).
1116. Dupre F. T. Chem. Ztg, 20, 305 (1896); Chem. Zbl, 67, I, 1144 (1896).
1117. D u p u i s T. Bull. soc. chim, 327 (1949).
1118. Dupuis T. Compt. rend, 237, 256 (1953); Chem. Zbl, 127, 1982 (1956).
1119. Dupuis T. Z. analyt. Chem, 142, 298 (1954).
1120. Dupuis T, Duval C. Analyt. chim. acta, 4, 55 (1949); Chem. Abstr, 44, 6339 (1950).
1121. Durupt A, Schlesinger A. Bull. soc. chim. biol, 13, 700 (1931); Chem. Abstr, 26, 1951 (1932).
1122. Duval C. Analyt. chim. acta, 1, 105 (1947); Compt. rend, 226, 1276 (1948); Chem. Abstr, 42, 5791, 8696 (1948).
1123. DuvalC. Analyt. chim. acta, 4, 159 (1950); Chem. Abstr, 44, 9860 (1950).
1124. Duval C. Chim. analyt, 34, 209 (1952); Chem. Abstr, 47, 68 (1953).
1125. Duval C. Analyt. chim. acta, 15, 223 (1956); 16, 545 (1957); 23, 257 (1960).
1126. Duval T, Duval C. Analyt. chim. acta, 2, 57, 97, 103, 110, 205 (1948).
1127. Dvorak J, Rezac Z. Chem. listy, 53, 588, 1113 (1959).
1128. Dworzak R, Ballczo H. Mikrochem. ver. Mikrochim. acta, 26, 322 (1939); Chem. Abstr, 33, 4903 (1939).
1129. Dworzak R, Friedrich-Liebenberg A. Mikrochim. acta, 1, 168 (1937).
ИЗО. E a s t E. M. J. Amer. chem. Soc, 26, 297 (1904); Z. analyt. Chem, 47, 439 (1908).
1131. Ebersole E. R, Fl у gar e J. K. Proc. Health Phys. Soc, 1, 72 (1956); Chem. Abstr, 52, 12975 (1958).
1132. Echdahl W. R. Chemist-Analyst, 28, 47 (1939); Chem. Abstr, 33, 6753 (1939).
1133. Eckel R. E. J. biol. Chem, 195, 191 (1952).
1134. Eden A. Analyst, 68, 167 (1943); Chem. Abstr, 37, 4984 (1943).
1135. Edson S. N. Better Crops, 37, 25 (1953); РЖХим, № 14195 (1955).
1136. Edson S. N, Smith F. B. Quart. J. Florida Acad. Sci, 14, 266 (1951); Chem. Abstr, 46, 3693 (1952).
1137. Edwards V. Chem. News, 70, 140 (1894); Z. analyt. Chem, 35, 634 (1896).
1138. Eggertsen F. T, W у 1 d G, L у к к e n L. Amer. Soc. Testing Materials, Symposium on Flame Photometric, No 116, 52 (1951); Chem. Abstr, 47, 7367 (1953).
1139. E g g e r t z C. G, Nilson L. F. Z. analyt. Chem, 38, 172 (1899).
1140. Egner H. Kgl. Landtbruks-Akad. Handl. Tid, 64, 416 (1925); Chem. Abstr, 19, 3137 (1925).
1141. Ehrenberg R. Mikrochemie Pregl. Festschr, 1929, 61; Biochem. Z, 197, 467 (1928); Chem. Abstr, 24, 2395 (1930).
204
1142. Ehrhardt R. Tabellen zur Berechnung von Kalianalysen. Halle, 1933.
1143. Ehrlin-Tamm G. Acta Chem. Scand., 4, 1317 (1950); Chem. Abstr., 46, 6992 (1952).
1144. Einbeck H., Jablonski K. Ber., 54, 1084 (1921).
1145. E i s e n b e r g W. V., К e e n a n G. L. J. Assoc. Offic. Agr. Chem., 27, 177 (1944); Chem. Abstr., 38, 2583 , 6237 (1944).
1146. Eklund S. Arkiv Mat., Astron. Fysik, B32, No 3, 6 (1945); Chem. Abstr., 41, 2543 (1947).
1147. Ekstrand A. G. Ber., 11, 163 (1878).
1148. E 1 b e i h I. 1., Me О m i e J. F., Pollard F. H. Trans. Farad. Soc., 7, 183 (1949).
1149. Ellington E., Stanley L. Analyst, 80, 313 (1955).
1150. Elliot F. H. Canad. J. Technol., 29, 111 (1951); Z. analyt. Chem., 135, 37 (1952).
1151. Elliot H. C., Holley H. L. Amer. J. Clin. Pathol;, 21, 831 (1951); J. Alabama Acad. Sci., 23—24, 39 (1953); Chem. Abstr., 45, 10345 (1951); 47, 9401 (1953).
1152. Ells V. R., Marshal С. E. Soil Sci. Soc. Amer. Proc., 4, 131 (1939)-, Chem. Abstr., 35, 247 (1941).
1153. Elster J., Geitel H. Physik. Z„ 11, 275 (1910).
1154. Emerson D. O. Amer. Mineralogist, 44, 661 (1959); РЖХим., № 8834 (1960).
1155. E m i К., T о e i К., В a b a J., I n о n e T. J. Chem. Soc. Japan, Pure Chem. Sec., 79, 598 (1958); РЖХим., № 19083 (1959).
1156. Emich F. Monatsh. f. Chem., 36, 433 (1915).
1157. E m m e r t E. M. J. Assoc. Offic. Agr. Chem., 14, 573 (1931); Chem. Abstr., 26, 937 (1932).
1158. Emmert E. M. Proc. Amer. Soc. Hort. Sci., 44, 381 (1944); 45, 311 (1945); Chem. Abstr., 39, 4388, 5200 (1945).
1159. Endredy E. Mezogazclasagi Kutatasok, 14, 203 (1941); Chem. Abstr.. 36, 2662 (1942).
1160. Engel R. Bull. soc. chim. France, 45 (2), 318 (1886).
1161. Engelbrecht R. M., Me Coy F. A. Analyt. Chem., 28, 1772 (1956).
1162. E p p e r s о n A. W., Rudy R. B. Ind. Eng. Chem., 17, 35 (1925); Chem. Abstr., 19, 795 (1925).
1163. Epps E. M., Burden J. C. Analyt. Chem., 30, 1882 (1958).
1164. Erdenbr echer A. H. Mikrokosmos, 25, 105 (1931).
1165. Erdey L., Buzas I., Vigh K. Taianta, 1, 377 (1958); РЖХим., № 22068 (1960).
1166. Erdey L., Paulik F. Acta chim. Acad. Sci. Hung., 4, 97 (1954); РЖХим., № 16509 (1955).
1167. Erdmann H., Kothner P. Lieb. Ann., 294, 72 (1897).
1168. Erdmann O. L. Z. analyt. Chem., 3, 161 (1864); J. prakt. Chem., 97, 397 (1866).
1169. Erlenmeyer H., Hahn H., Sorkin E. Helv. chim. acta, 34, 1419 (1951).
1170. Erlenmeyer H., Schmid 1 in J. Helv. chim. acta, 24, 1213 (1941); Chem. Abstr., 36, 3451 (1942).
1171. Erlenmeyer H., Schoenauer W. Helv. chim. acta, 24, 878 (1941); Chem. Abstr., 36, 2221 (1942).
1172. Ernst E., Barasits J. Biochem. Z., 209, 438 (1929).
1173. Espersen T. Dansk tisdskr. farmaci, 33, 113 (1959); РЖХим., № 74489 (1960).
1174. E u b a n к W. R., R о g u e R. H. J. Research Natl. Bur. Standards, 43, 173 (1949); Z. analyt. Chem., 133, 202 (1951).
1175. Evans С. H„ Strain H. H„ Analyt. Chem., 28, 1560 (1956).
1176. Evre S. Lieb. Ann., 80, 274 (1851).
1177. Ewan M. A., Ford O. W., Schall E. D. Analyt. Chem., 20, 192 (1948).
205
1178. Exley D. Photoelectr. Spectrum. Group. Bull., No 12, 322 (1959); РЖХим., № 77010 (1960).
1179. Faber R„ Dirkse T. P. Analyt. Chem., 25, 808 (1953).
1180. Fabre C. Compt. rend., 122, 1331 (1896); Chem. Zbl., 67, II, 206 (1896).
1181. Fairbourne A. Analyst, 48, 263 (1923).
1182. Farnworth A. J. Australian J. appl. Sci., 7, 233 (1956); Chem. Zbl., 129, 6424 (1958).
1183. Fasbender N. Nederl. Tijdschr. Pharm., 6, 1 (1894); Chem. Zbl., 65, I, 409 (1894).
1184. Fast E. Analyt. Chem., 22, 320 (1950).
1185. Faul H., Sullivan G. R. Nucleonics, 4, 53 (1949).
1186. Faust W. R. Phys. Rev., 78, 624 (1950).
1187. Fazio С. C. Pubis. Inst, investig. microquim., 23, 55 (1957); РЖХим., № 61085 (1960).
1188. Federer F. Z. physiol. Chem., 89, 232 (1914); Z. analyt. Chem., 54, 486 (1915).
1189. Feichtinger F. Lieb. Ann., 102, 353 (1857).
1190. Feigl F. Mikrochemie, 8, 356 (1930).
1191. Feigl F., Barbosa P. E. Rev. soc. brasil. quim., 10, 137 (1941); Ministerio agr., Dept. nacl. producao mineral., Lab. producao mineral. (Brazil), Bol. No 5, 131 (1942); Chem. Abstr., 36, 4438 (1942); 38, 1976 (1944).
1192. Feigl F., Caldas A. Miikrochim. acta, 1310 (1956).
1193. Feit W., Bokemiiller H. Posts chemisch-technische Analyse, Braunschweig, 1, 919, 1908.
1194. Feldman C. Analyt. Chem., 21, 1041 (1949).
1195. Feldman L„ Wu C. S. Phys. Rev., 81, 298 (1950); 87, 1091 (1952); Chem. Abstr., 46, 10925 (1952).
1196. Ferchland P. Z. anorg. Chem., 30, 130 (1902); Chem. Zbl., 73, 1, 846 (1902).
1197. Ferenzy Z., Almasy A. Epitoahyag, 6, 301 (1954); Chem. Zbl., 127, 1689 (1956).
1198. Ferrari A., Cavalca L., Nardelli M. Gazz. chim. ital., 81, 964, 982 (1951); Chem. Abstr., 46, 9000, 10034 (1952).
1199. Ferrari A., Coghi L., Piovosi N. Gazz. chim. ital., 70, 812 (1940); Chem. Abstr., 35, 7865 (1941).
1200. Ferrari A., Colla C. Atti accad. Lincei, 11, 755 (1930); Chem. Abstr., 24, 5559 (1930).
1201. Ferrari A., Nanni O. Gazz. chim. ital., 69, 301 (1939).
1202. Festa C., Santangelo M. Ann. geofis. (Rome), 3, 95 (1950); Chem. Abstr., 44, 6736 (1950).
1203. Fick er s B. A. J. Chem. Educ., 33, 575 (1956); Chem. Zbl., 128, 12354 (1957).
1204. Fiechter A. Z. analyt. Chem., 50, 629 (1911).
1205. Fieldes M., King P. J., Richardson J. P., S win dale L. D. Soil Sci., 72, 219 (1951); Chem. Abstr., 47, 10162 (1953).
1206. Findeis A. F., Vries T. Analyt. Chem., 28, 209, 1899 (1956).
1207. Fin ken er R. Pogg. Ann., 129, 637 (1866); Z. analyt. Chem., 6, 213 (1867); 36, 321 (1897).
1208. Fireman E. L. Phys. Rev., 75, 1447 (1949).
1209. Fischer H. Landwirtschaftliche Versuchsstationen, 85, 139 (1914).
1210. Fischer J., Doiwa A. Mikrochim. acta, 1956, 353.
1211. Fischer N. W. Pogg. Ann., 72, 477 (1847).
1212. Fischer N. W. Pogg. Ann., 74, 115 (1848).
1213. Fischer R. Mikrochemie, 27, 67 (1939).
1214. Fischer R., Langhammer T. Mikrochem. ver. Mikrochim. acta,. 34, 208 (1949).
1215. Fischer W. M. Z. physik. Chem., 92, 581 (1916/17)
206
1216. Fiske С. H. J. biol. Chem., 51, 55 (1922).
1217. Fiske С. H., Litarczek G. J. biol. Chem., 67, 17 (1926).
1218. Flaschka H. Z. analyt. Chem., 136, 99 (1952).
1219. Flaschka H. Z. analyt. Chem., 144, 415 (1955).
1220. Flaschka H. Chemist-Analyst, 44, 60 (1955); Z. analvt. Chem., 151, 289 (1956).
1221. Flaschka H. Z. physiol. Chem., 308, 183 (1957); Chem. Abstr., 52, 2999 (1958).
1222. Flaschka H, Amin A. M. Chemist-Analyst, 42, 78 (1953); РЖХим., № 603 (1955).
1223. Flaschka H., Amin A. M, Holasek A. Z. analyt. Chem., 138, 241 (1953).
1224. Flaschka H., Holasek A., Amin A. M. Z. analyt. Chem., 138, 161 (1953).
1225. Flaschka H., Sadek F. Chemist-Analyst, 45, 20 (1956); РЖХим., № 71946 (1956).
1226. Flaschka H., Sadek F. Chemist-Analyst, 47, 30 (1958); РЖХим., № 81858 (1959).
1227. Fleischer E. Z. analyt. Chem., 9, 331 (1870).
1228. F 1 e i t c h e r W. W. J. Soc. Glass Technol., 43, 86 (1959); РЖХим., № 86073 (1959).
1229. Flock E., Rod ebush W. J. Amer. Chem. Soc., 48. 2522 (1926).
1230. Flottmann F. Z. analyt. Chem., 73, 1 (1928). 4
1231. Floyd J. J., Borst L. B. Phys. Rev., 75, 1106 (1949).
1232. Fol ch J., Lauren M. J. biol. Chem., 169, 539 (1947).
1233. Folin O., Bell R. D. J. biol. Chem., 29, 329 (1917).
1234. Folling A. Scand. Arch. Physiol., 61, 27 (1931); Cherh. Abstr., 25, 1860 (1931).
1235. Fongi N. Ann. chim. (Rome), 41, 163 (1951); Chem. Abstr., 45, 8394 (1951).
1236. Fontana P., Zanetti B. Publ. univ. cattolica S. Cuore, 60, 201 (1956); Chem. Abstr., 52, 7601 (1958).
1237. Foote H. W, Andrew I. A. Amer. Chem. J., 34, 155 (1905).
1238. Ford C. L.-J. Assoc. Offic. Arg. Chem., 33, 268 (1950); 35, 674 (1952); 36, 649 (1953); 37, 363 (1954).
1239. Ford C. L. Analyt. Chem., 26, 1578 (1954); ASTM Bull., No 233, 57 (1959); РЖХим., № 17557 (1960).
1240. Ford О. W. J. Assoc. Offic. Agr. Chem, 22, 280 (1939); 23, 264 (1940); 24, 285 (1941); 26, 59 (1943); 31, 242 (1948); Chem. Abstr, 33, 7019 (1939); 34, 5235 (1940); 35, 5626 (1941); 37, 2871 (1943); 42, 8397 (1948).
1241. Ford O. W. J. Assoc. Offic. Agr. Chem, 34, 660 (1951); Chem. Abstr, 45, 10460 (1951).
1242. Form a nek J. Lieb. Ann, 257, 102 (1890); Z. analyt. Chem, 39, 409 (1900).
1243. Forster C. F. Analyst, 79, 943 (1954).
1244. Forster O. Chem. Ztg, 22, 357 (1898).
1245. Foster J. S, Langs troth G. O, Me Rae D. R. Proc. Roy. Soc, London, 165A, 465 (1938); Chem. Abstr, 32 , 7952 (1938).
1246. Foster W, Hume D. Analyt. Chem, 31, 2033 (1959).
1247. Fox C. L. Analyt. Chem, 23, 137 (1951).
1248. F о x G. W, Goodwin R. A. Iowa State Coll. J. Sci, 15, 119 (1941); Chem. Asbtr, 35, 5232 (1941).
1249. Franchetti S, Giovanozzi M. Phys. Rev, 74, 102 (1948)'.
1250. Frangopol C. Bull. chim. pura appl. Soc. rom. Stiinti, 37, 259 (1934); Chem. Zbl, 106, II, 2250 (1935).
1251. Frank A. Z. analyt. Chem, 6, 257 (1867).
1252. Frankenberg B, Hospadaruk V, Neufeld A. H. Canad. Med. Assoc. J, 65, 388 (1951); Chem. Abstr, 46, 2609 (1952).
1253. Frank for ter G. B, Frary F. C. J. Phys. Chem, 17, 402 (1913).
207
1254. Fraps G. S. J. Assoc. Offic. Agr. Chem., 9, 192 (1926); Chem. Abstr., 20, 2221 (1926).
1255. Frebault A„ Al о у J. J. pharm. chim., 20 (6), 245 (1904); Chem. Zbl., 75, II, 1385 (1904).
1256. Fredenhagen C. Ann. der Physik, 20 (4), 173 (1906); Chem. Zbl., 77, II, 158 (1906).
1257. Fredholm H. Z. analyt. Chem., 104, 400 (1936).
1258. Frediani H. A. Ind. Eng. Chem., Analyt. Ed., 10, 447 (1938).
1259. Frediani H. A., Gamble L. Mikrochem. ver. Mikrochim. acta, 29, 22 (1941).
1260. Freeman S., Burril M. W. J. Biol. Chem., 157, 287 (1945); Chem. Abstr., 39, 1657 (1945).
1261. Frehden O. Rev. chim., 5, 559 (1954); РЖХим., № 1098 (1956).
1262. Fresenius H., Printon P. H. Z. analyt. Chem., 50, 21 (1911).
1263. Fresenius R. Lieb. Ann., 41, 6 (1842).
1264. Fresenius R. Lieb. Ann., 59, 117 (1846).
1265. Fresenius R. Z. analyt. Chem., 1, 59 (1862).
1266. Fresenius R. Z. analyt. Chem., 4, 413 (1865).
1267. Fresenius R. Z. analyt. Chem., 11, 290, 413 (1872).
1268. Fresenius R. Z. analyt. Chem., 15, 221 (1876).
1269. Fresenius R, Z. analyt. Chem., 16, 63 (1877).
1270. Fresenius.R. Z. analyt. Chem., 21, 234 (1882).
1271. Fresenius W., Yander G. Handbuch der analytischen Chemie. Berlin, Bd. la, 1944, S. 91, 105; Bd. 9, 1956, S. 183.
1272. Freundler P., Menager Y. Compt. rend., 182, 1158 (1926); Chem. Abstr., 20, 2472 (1926).
1273. Frey A. Pharm. Centralhalle, 13, 266 (1872); Z. analyt. Chem., 32, 204 (1893).
1274. Friedenthal A. Ber., 44, 1444 (1911).
1275. Friedholm H. Z. analyt. Chem., 104, 400 (1936).
1276. Friedmann H. Eng. Mining J., 152, 91, 119 (1951); Chem. Abstr., 45, 10136 (1951).
1277. Friedrich M. Chem. listy, 49, 1241 (1955); Sklar a. Keramik, 5, 74 (1955); Chem. Zbl., 127, 8171 (1956); РЖХим., № 1094 (1956).
1278. Frierson W. J., Jones J. W. Analyt. Chem., 23, 1447 (1951).
1279. Frisch K. J. prakt. Chem., 100 (1), 229 (1867).
1280. Fukushima S. Mikrochem. acta, 1957, 35; 1960, 332.
1281. Fulda E. Das Kali. Stuttgart, 1928.
1282. Gabriel A., Cox E. Amer. Mineralogist, 14, 290 (1929).
1283. Gabrielson G. Analyst. 80, 479 (1955); РЖХим., № 7067 (1956). .
1284. Gabrielson G. Analyt. chim. acta, 20, 146 (1959); РЖХим., № 53108 (1959).
1-285. Gabrielson G., Samuelson O. Svensk Kem. Tid., 62, 221 (1950); Chem. Abstr., 45, 4169 (1951).
1286. Gagliardi E., Reimers H. Z. analyt. Chem., 160, 1 (1958); РЖХим., № 57144 (1958).
1287. Gaglioti V., Stolfi A. Atti R. Accad. Lincei, 5, (6), 396 (1917).
1288. Gammon N. Soil Sci., 71, 211 (1951); Chem. Abstr., 46, 675 (1952).
1289. G a n c h ev N. Ann. univ. Sofia, 15, 377 (1937); Chem. Abstr., 33, 76 (1939).
1290. Garmick L. G. Public Roads, 21, 79 (1940); Chem. Abstr., 34, 6035 (1940).
1291. Garola С. V„ Braun V. Ann. falsific. et fraudes, 10, 572 (1917); Z. analyt. Chem., 64, 430 (1924).
1292. Garrick P. Nature, 160, 434 (1947).
1293. Garrignes W. E. J. Amer. Chem. Soc., 17, 47 (1895); Chem. Zbl., 66.
I, 509 (1895).
1294. Gartner E. Monatsh. f. Chem., 41, 489 (1920).
208
1295. Gaspar у ArnalT. An. soc. espan. fis. quim., 24, 99 (1926)- Chem. Abstr., 20, 2129 (1926).
1296. Gaspar у ArnalT. An. soc. espan. fis. quim., 26, 184 (1928); Chem. Abstr., 23, 52 (1929); Z. analyt. Chem., 79, 210 (1930).
1297. Gaspar у Arnal T. Ann. chim. analyt. appl, 11, 97 (1929); Chem. Abstr., 23, 3186 (1929).
1298. Gaspary Arnal T. Chim. analyt., 41, 152 (1959); РЖХим., № 60463 (1959).
1298a. Gaspar yArnalT. Chim. et Ind., 84, 523 (1960); РЖХим., № 9Д39 (1961).
1299. Gaspar у Arnal T., Vega-Bragado J. Chim. analyt., 38, 343 (1956); РЖХим., № 19474 (1954).
1300. Gastinger E. Z. analyt. Chem, 140, 335 (1953); РЖХим, № 2236 (1955).
1301. Gata E., Gat a G. Dariseama sedint. Com. geol. RPR, 42, 273 (1959); РЖХим, № 47017 (I960).
1302. Gatrousis C, Crouthamel C. J. Inorg. Nucl. Chem, 13, 13 (1960); РЖХим., № 15301 (1961).
1303. Gattorta G, Servello V. Ann. Staz. Chim. Agr. Roma, No 142, 15 (1958); Analyt. Abstr, 7, No 785 (1960).
1304. Gau din A. M, Pannell J. H. Analyt. Chem, 20, 1154 (1948).
1305. Gay-Lussac L, Thenard P. Lieb. Ann, 65, 33 (1809).
1306. Gazzi V. Ann. chim. appl., 29, 500 (1939); Chem. Abstr, 34, 3613 (1940).
1307. Gazzi V. Chim. et Ind, 36, 249 (1954); РЖХим, № 51086 (1956).
1308. Gee A, Domingues L. P, Deitz V. R. Analyt. Chem, 26, 1487 (1954).
1309. Gehrke C. W, A f f s p r u n g H. E, W о о d E. L. J. Agric. Food Chem, 3, 48 (1955); РЖХим, № 40353 (1955).
1310. Gehrke C. W, Wood E. L. Missouri Univ, Agr. Expt. Sta, Research Bull, No 635, 60 (1957); Chem. Abstr, 52, 2319 (1958).
1311. G e i 1 m a n n W. Bilder zur qualitativen Mikroanalyse anorganischer Stoffe, Leipzig, 1934.
1312. Geilmann W, Gebauhr W. Z. analyt.Chem, 139, 161 (1953).
1313. Geilmann W, Gebauhr W. Z. analyt. Chem, 142, 241 (1954).
1314. Gelli P. Ann. sper. agrar. (Rome), 6, 359 (1952); Chem. Abstr, 46, 8309 (1952).
1315. Gentini A. An. assoc, quim. farm. Uruguay, 44, 21 (1941); Chem. Abstr, 36, 3113 (1942).
1315a. Georg a J. G. Nova Acta Acad. Sci, 3, 250 (1788).
1316. Gergely G, Varadi F. P. Magyar. Kemiai Folyoirat, 61, 182 (1955); Z. analyt. Chem, 150, 194 (1956).
1317. Gericke S. Analytische Chemie der Dungemittel. Stuttgart, 1949, S, 104, 160.
1318. Germuth F. G. J. Franklin Inst, 212, 346 (1931).
1319. Germuth F. G, Mitchell C. Amer. J. Pharmac, 10, 46 (1929).
1320. Gerretsen F. C. Analyt. chim. acta, 2, 782 (1948); Rept. Proc. 4th Intern. Congr. Microbiol. 1947, 1949, p. 487; Chem. Abstr, 43, 8083 (1949); 44, 8036 (1950).
1321. Gerretsen F. C, Blumendal N. Verslag. Landb. Onderzock, No 46A, 219 (1940); Chem. Agstr, 35, 1563 (1941).
1322. Gerri tz H. W. J. Assoc. Offic. Agr. Chem, 25, 232, 433 (1942); Chem. Abstr, 36, 2339, 4618 (1942).
1323. Ger-schman R, Marenzi A. D. Anales farm, bioquim, 2, 194 (1931); Chem. Abstr, 26, 2479 (1932).
1324. Gersh W. Anat. Record, 70, 311 (1938).
1324a. Geyer R, Chojnacki K- Z. analyt. Chem, 179, 409 (1961).
1325. Ghitti L. Boll. sci. facolta chim. ind. Bologna, 1941, 22; Chem. Abstr, 38, 1978 (1944).
14 Аналитическая химия калия
209
1326. Ghosh N. N. Science and Culture, 21, 165 (1955); Chem. Zbl., 128, 1005 (1957).
1327. Gibbs W. Amer. Chem. J., 1, 219 (1879); 2, 228 (1880); Z. analyt. Chem., 21, 565 (1882).
1328. Giesecke F., Rathje W. Z. Pflanzenernahr. Diing. Bodenkunde, 9-10, 776 (1938); Z. analyt. Chem., 124, 145 (1942).
1329. Giesecke F., Rathje W., Krummel W. Z. Pflanzenernahr. Dflng. Bodenkunde, 7, 173 (1938); Chem. Abstr., 32, 8660 (1938).
1330. Giesecke F., Schulte L. Bodenkunde u. Pflanzenernahr., 6, 139 (1937); Chem. Abstr., 32, 5142 (1938).
1331. G i e s e к i n g J. E., Snider H. J. Ind. Eng. Chem., Analyt. Ed., 9, 232 (1937).
1332. Gieseking J. E., Snider H. J., Getz C. A. Ind. Eng. Chem., Analyt. Ed., 7, 185 (1935).
1333. Giesen K-, Kamp a P. Tonindustrie-Ztg., 77, 383 (1953); РЖХим., № 36399 (1954).
1334. Gilbert В. E. Plant Physiol., 1, 191 (1926); Chem. Abstr., 21, 3652 (1927).
1335. Gilbert K. Die Bestimmung des Kaliums nach quantitativer Absche-dung desselben als Kaliumnatriumkobaltinitrits. Dissertation, Tiibingen, 1898; Z. analyt. Chem., 38, 184 (1899).
1336. Gillis J., Eeckout J. Chem. Weekbl., 43, 387 (1947); Chem. Abstr., 41, 6169 (1947).
1337. Gillis J., Hoste J. Analyt. chim. acta, 1, 326 (1947); Z. analyt. Chem., 130, 250 (1950).
1338. Gillis J., Hoste J. J. pharmac. Belg., 13, 326 (1958); РЖХим., № 67622 (1959).
1339. Gimblett F. G., Monk С. B. Trans. Farad. Soc., 51, 793 (1955).
1340. Ginsberg H. Z. anorg. allg. Chem., 167, 183 (1927); Chem. Abstr., 22, 558 (1928).
1341. Ginsberg H. Z. anorg. allg. Chem., 204, 225 (1932).
1342. Ginsberg H., Holder G. Z. anorg. allg. Chem., 190, 407 (1930); Chem. Abstr., 24, 4725 (1930).
1343. Gire G. Ann. chim., 4, 183, 370 (1925); Chem. Abstr., 20, 694 (1926).
1344. Gisiger L. Landwirtschaftliche Versuchsstationen, 123, 209 (1935).
1344a. Gita G„ Gita E; Rev. chim. (RPR), 11, 528 (1960); РЖХим., № 17 Д12 (1961).
1345. Giuliani V., Zazo S. Rass. med. sper., 2, 240 (1955); Chem Abstr., 52, 3003 (1958).
1346. Glaze F. W. Rock Products, 44, 42, 94 (1941); Chem. Abstr., 35, 6412 (1941).
1347. Gleditsch E., Graf T. Arch. Math. Naturvidenskab., 44, 63 (1941).
1348. Gleditsch E., Graf T. Phys. Rev., 72, 640 (1947); Chem. Abstr., 41, 7246 (1947).
1349. Glick D. J. chem. Educ., 16, 68 (1939).
1350. Glick D., S w i g a r t R. H., N а у у a r S. N., Stecklein H. R., J. Histochem. Cytochem., 3, 6 (1955); Z. analyt. Chem., 151, 151 (1956).
1351. Gloss G. H. Analyst, 42, 50 (1953); Chem. Abstr., 47, 10 398 (1953).
1352. Gloss G. H., Olson B. Analyst, 43, 70 (1954); РЖХим., № 37, 492 (1955).
1353. Gmelins Handbuch der anorganischen Chemie, System-Nummer 22, Kalium. Berlin, 1936—1938.
1354. G о d b о u t G., S с о 11 A. Sci. Agr., 23, 461 (1943); Chem. Abstr., 37, 4176 (1943).
1355. Goehring M., Schlaich J. Z. analyt. Chem., 129, 319 (1949); Chem. Abstr., 44, 1847 (1950).
1356. Goke G. Lab.-Prax., 11, 59 (1959); РЖХим., № 78290 (1959).
1357. Goldbaum J. S., Smith E. F. J. Amer. Chem. Soc., 80, 1705 (1908); Chem. Zbl., 80, I, 581 (1909).
210
1358- Goldschmidt V. M. Geochemische Verteilungsgesetze. Oslo, 1926.
1359. Goldstein S. W, Sanders D. P. Drugs Standards, 22, 137 (1954); РЖХим., № 40357 (1955).
1360. Goltschke Z. analyt. Chem., 37, 1 (1898).
1361. Gonard A. Bull. soc. chim., 9, 917 (1942); Chem. Abstr., 38, 2899 (1944).
1362. Gooch F A. Proc. Amer. Acad. Arts and Sol., 22, 177 (1886); Chem. News, 55, 18, 29, 40, 56, 78 (1878); Amer. Chem. J., 9, 33 (1887); Z. analyt. Chem., 26, 354 (1887).
1363. Gooch F. A, Blake G. R. Amer. J. Sci., 44, 381 (1917); Chem. Abstr., 12, 29 (1918).
1364. Gooch F. A., Eddy E. A. Z. anorg. Chem, 58, 427 (1908); Z. analyt. Chem, 48, 55 (1909).
1365. Gooch F. A, Hart T. S. Amer. J. Sci, 42, (3), 448 (1891); Z. analyt. Chem, 36, 389 (1897).
1366. Goodwin R. A, Iowa State Coll. J. Sci, 14, 35 (1939); Chem. Abstr, 34, 5984 (1940).
1367. Gordon H. T, Hewel C. A. Analyt. Chem, 27, 1471 (1955).
1368. Gordon S, Campbell C. Analyt. Chem, 27, 1102 (1955).
1369. Gore C. J.. Chem. Soc, 15, 105 (1862); Z. analyt. Chem, 2, 429 (1863).
1370. Gorski M. Z“. anorg. Chem, 81, 344 (1913).
1371. Gorski M, Moskal S. Roczniki Nauk Rolniczych, 76, 405 (1957); Chem. Abstr, 52, 12293 (1958).
1372. Gow P. L. Hawaiian Planters Record, 35, 401 (1931); Chem. Abstr, 26, 3751 (.1932).
1373. G о у S. Angew. Chem, 50, 301 (1937); Bodenkunde u. Pflanzenernahr., 3, 308 (1937).
1374. Graaff C. L, Noyons E. C. Chem. Weekbl, 43, 300 (1947); Chem. Abstr, 41, 7303 (1947).
1375. Grabe C, Oeser A. Ber, 35, 145 (1904).
1376. Graf T. Phys. Rev, 74, 831, 1199 (1948).
1377. Grager G. Z. analyt. Chem, 8, 499 (1869).
1378. Graham Ё. R. Analyst, 44, 37 (1955); Z. analyt. Chem, 151, 45 (1956).
1379. Grahmann W. Z. anorg. Chem., 81, 257 (1913).
1380. Grammont A. Compt. rend, 122, 1326, 1411, 1534 (1896).
1381. Grant J. Metal Ind. (London), 53, 538 (1938); Chem. Abstr, 33, 1233 (1939).
1382. Graven H. Berichte Wien. Akad, 139, 181 (1930); Chem. Abstr, 25, 2661 (1931).
1383. Greathouse L. H. Dissertation, University of Michigan, 1917.
1384. Green D, Richardson J. R. Phys. Rev, 83, 891 (1951).
1385. Green M. M. Ind. Eng. Chem., 15, 163 (1923).
1386. Green T. C. Analyst, 16, 16 (1927); Chem. Abstr., 22, 38 (1928).
1387. Gregor H. P, H a m i 11 о n M. J, О z a R. J, В e r n s t e i n F. J. Phys. Chem, 60, 263 (1956); Chem. Zbl, 129, 942 (1958).
1388. Grete A. Chem. Ztg, 33, 1128 (1909).
1389. Grete A. Chem. Ztg, 34, 1040 (1910).
1390. G reuli ng E. Phys. Rev, 61, 568 (1942); Chem. Abstr, 36, 4022 (1942).
1391. Griess P. Ber, 11, 2153, 2196 (1878).
1392. Griffin С. E, Kulp J. L. Bull. Geol. Soc. Amer, 71, 219 (1960); РЖХим, № 91858 (1960).
1393. Griffon H, Bernard A. J. pharm. chim, 12, (8), 118 (1930); Chem. Abstr. 25, 1309 (1931).
1394. Griggs M. A. Science, 89, 134 (1939); Chem. Abstr, 33, 8842 (1939).
1395. Griggs M. A, J о h n s t i n R, Elledge В. E. Ind. Eng. Chem, Analyt. Ed, 13, 99 (1941).
1396. Grill F, Wohlmuth N. Mitt, hoher. Bundeslehr, 2, 244 (1952); Chem. Abstr, 47, 9537 (1953).
1397. Grimm H. G. Z. Elektrochem, 30, 467 (1924).
14*
211
1398. Grimm H. G. Naturwiss, 15, 561 (1927).
1399. Grimm H. G, Wagner G. Z. phys. Chem., 132, 131 (1928); Chem. Abstr., 22, 1880 (1928).
1400. Groger M. Z. anorg. Chem., 54, 185 (1907); Chem. Zbl., 78, II, 516 (1907).
1401. Gross D. Nature, 180, 596 (1957).
1402. Grueber N. Z. angew. Chem., 8, 504 (1895).
1403. Grueber N. Z. analyt. Chem., 36, 311 (1897).
1404. Gualandi C, Mazzei I., Burana G. Ann. chimica, 49, 1941 (1959); РЖХим., № 80724 (1960).
1405. G libel i O., Slammbach K. Helv. chim. acta, 34, 1245, 1253 (1951).
1406. Guimaraes G„ Santini P. Anais assoc, quim. Brasil, 6, 234 (1947); Chem. Abstr., 42, 7655 (1948).
1407. Gulbransen E. A., Phelps R. T, Langer A. Ind. Eng. Chem., Analyt. Ed., 17, 646 (1945).
1408. Gullbransen L. B. Analyt. Chem., 28, 1632 (1956).
1409. Gutbier A., Kreil A. Ber., 38, 2385 (1905); Chem. Zbl., 76, II, 452 (1905).
1410. Gut er G. A., Hammond G. S. J. Amer. Chem. Soc., 78, 5166 (1956); Chem. Zbl., 128, 5950 (1957).
1411. Gutmann V., Nedbalek E. Monatsch. f. Chem., 88, 320 (1957); РЖХим., № 4269 (1958).
1412. Gutzeit G. Helv. chim. acta, 12, 713 (1929); Chem. Abstr., 23, 4644 (1929).
1413. Gyorgy V. Chim. et Ind., 56, 413 (1946); Chem. Abstr, 41, 5244 (1947).
1414. Hackspill L. Compt. rend, 152, 261 (1911).
1415. H а с к s p i 11 L, Claude D, Andres L. Chem. Abstr, 26, 2273 (1932).
1416. Haefcke H. Chem. Ztg, 20, 88 (1896); Chem. Zbl, 67, I, 726 (1896).
1417. Haff R. C, Schwartz E. H. Ind. Eng. Chem, 9, 785 (1917).
1418. Hagen E. W. Wied. Ann, 19, 444 (1883).
1419. Hager G, Kern J. Landw. Vers. Stat, 87, 365 (1915).
1420. Hager'H. Pharm. Zentralhalle, 22, 224 (1881).
1421. Hager H. Z. analyt. Chem, 21, 408, 561 (1882).
1422. Hahn F. L. Ber, 22, 3434 (1922).
1423. Hahn F. L. Leitfaden der quantitativen Analyse. Dresden u. Leipzig, 1922, S. 65, 68.
1424. Hahn F. L. Z. analyt. Chem, 145, 97 (1955); РЖХим, № 37 493 (1955).
1425. Hahn F. L. Ciencia, 14, 249 (1954); РЖХим, № 22 645 (1956).
1426. Hahn F. L, View eg K. Z. analyt. Chem, 71, 122 (1927).
1427. Hahn O, Rothenbach M. Physik. Z, 20, 194 (1919).
1428. HahneH. Z. Pflanzenernahr, Diing, Bodenkunde, 6A, 238 (1926); Chem. Abstr, 20, 3205 (1926).
1429. Haigh L. D. Assoc. Offic. Agr. Chem, 17, 255 (1934); Калий, № 2, 26 (1935).
1430. Haigh L. D, Hall A. R. J. Assoc. Offic. Agr. Chem, 16, 147 (1953); Chem. Abstr, 27, 2109 (1933).
1431. Ha 1 aunbrenner J. Roczniki Chem, 28, 569 (1954). .
1432. H a 1 d .P. M. J. biol. Chem, 103, 471 (1933).
1433. Hald P. M. J. biol. Chem, 167, 499 (1947); Chem. Abstr, 41, 3150 (1947).
1434. Hale H, H a 1 e M. H. Analyt. Chem, 24, 2015 (1952).
1435. H alii к О. Keemia Teated, 2, 7 (1934); Chem. Abstr, 32, 1379 (1938).
1436. Hallman N, Leppanen V. Suomen Kemistilehti, 22B, 55 (1949);
Chem. Abstr, 44, 10031 (1950).
1437. Halstead W. J, Chaitken B. Public Roads, 26, 99 (1950); Chem. Abstr, 45, 796 (1951).
1438. H a m a r N. Arb. physiol, u. angew. Entomol, Berlin-Dahlem 14, 45 (1949); Chem. Abstr, 46, 10260 (1952).
1439. Hamburger H. J. Biochem. Z, 71, 415 (1915).
212
1440. Hamer W. J. The structure of electrolytic solutions. N Y — London, 1959, p. 26.
1441. Hamernik R. Hutn. listy, 14, 142 (1959); РЖХим., № 4587 (1960).
1442. Hamid M. A. Analyst, 51, 450 (1926); Chem. Abstr., 21, 34 (1927).
1443. Handbook of Chemistry and Physics, Editor Hodgman C. D., Cleveland, 1955—1956, pp. 377, 564, 570, 596, 1598, 1878.
1444. Hanna W. C, Bryant L. N, Hicks T. A. Amer. Soc. Testing Materials Bull., No 114, 41 (1942); Chem. Abstr., 37, 245 (1943).
1445. Hanou L. Chemie en technick, 15, 90 (1960); РЖХим., № 80747 (1960).
1446. Hanson W. C. Fertiliser Soc. (Engl.) Proc., No 16, 33 (1952); Chem. Abstr., 46, 7270 (1952).
1447. Hantzsch A. Ber., 42, 986 (1909).
1448. Hantzsch A., Issaias B. Ber., 42, 1000 (1909).
1449. Hara T. Bull. Inst. Chem. Res. Kyoto Univ., 37, 112 (1959); РЖХим., № 30431 (I960).
1450. Harasawa S, Sakamoto T. J. Chem. Soc. japan, 73, 614 (1952); Chem. Abjtr, 47, 3752 (1953).
1451. Harington C. R. Biochem. J., 35, 545 (1941); Chem. Abstr., 35, 6895 (1941).
1452. Harris H. C. Soil Sci, 40, 301 (1935); Chem. Zbl, 107, II, 1051 (1936).
1453. Harris J. E. J. biol. Chem, 136, 619 (1940); Chem. Abstr, 35, 765 (1941).
1454. Harrison H. E, Darrow D. C. J. biol. Chem, 121, 631 (1937); Chem. Abstr, 32, 613 (1938).
1455. Harteck P, Suess H. Naturwiss, 34, 214 (1947); Chem. Abstr, 43, 5712 (1949).
1456. Hartley H, Drugman J, Vlieland C. A, Bourdillon R. J. Chem. Soc, 103, 1747 (1913).
1457. Hartzler E. R. J. biol. Chem, 122, 19 (1937/38); Chem. Abstr, 32, 965 (1938)..
1458. Hasenbaumer J. Chem. Ztg, 28, 210 (1904); Chem. Zbl, 75, I, 1170 • (1904).
1459. Haslam J, Beeley J. Analyst, 66, 185 (1941); Chem. Abstr., 35, 5409 (1941).
1460. Hasler M. F, Harvey С. E, Barley F. W. Amer. Soc. Testing Materials, Proc, 48, 944 (1948); Chem. Abstr, 44, 3691 (1950),
1461. Haurowitz F. Mikrochemie, 2, 185 (1930); Chem. Abstr, 24, 4234 (1930).
1462. Hauser O. Z. anorg. Chem, 35, 4 (1903).
1463. Haushofer K. Mikroskopische Reaktionen. Braunschweig, 1885, S. 37.
1464. Hausler G. F. Z. analyt. Chem, 124, 327 (1942); Chem. Abstr, 37,
4841 (1943).
1465. Hausler H. Z. analyt. Chem, 64, 372 (1924).
1466. Haussler A, Hajdn P. Mitt, deutsch. pharm. Ges, 29, 73 (1959); РЖХим, № 8966 (1960).
1467. Have A. J, Mulder H. Nederl. Melken Zuiveltijdschr, 11, 128 (1957); Chem. Zbl, 129, 6699 (1958).
1468. Havelka S, Rakovic M. Chem. prtimysl, 9, 509 (1959); Analyt. Abstr, 7, 169 (1960).
1469. Havir J. Collect, 25, 595 (1960); РЖХим, № 5, Д58 (1961).
1470. Havir J. Voda, 35, 402 (1956); Chem. Zbl, 129, 11926 (1958).
1471. Havir J. Collect, 24, 1954 (1959); 25, 595 (1960).
1472. Havir J. Chem. listy, 52, 1274 (1958); РЖХим, № 49218 (1959).
1473. Havir J, Krivanek M. Collect, 24, 3183 (1959); РЖХим, № 69128 (1960).
1474. Havir J, Vrestal J. Chem. zvesti, 11, 35 (1957); РЖХим, № 4278 (1958).
1475. Hawkins F. S, Partington J. R. J. Chem. Soc, 1927, 1397; Chem. Abstr, 21, 2858 (1927).
213
1476. Hayashi Y., Kobayashi J. J. sci. Soil Manure, Japan, 12, 29 (1938); Chem. Abstr., 32, 3537 (1938).
1477. Haycock M. H, Russell L. R. Trans. Canad. Inst. Mining Met., 51, 281 (1948); Chem. Abstr., 42, 5375 (1948).
1478. Hecht F, Donau J. Anorganische Mikrogewichtsanalyse. Wien, 1940,
cc 997 930 234 268
1479. Heckel e F. Os’terr. 'chem. Ztg., 31, 29 (1928).
1480 Hedgecock G. A. Soc. Glass Technol, 43, 94 (1959); РЖХим., № 87074 (1959).
1481. Неё A., Coche A., Veller P, Jarovoy M, Wack M. Ann. geophys, 10, 19 (1954); Chem. Zbl., 127, 8798 (1956).
1482. Hee A., Jarovoy M. Ann. geophys., 9, 153 (1953); Chem. Abstr., 47, 9166 (1953).
1483. Hee A., Wack M., Jarovoy M. Ann. geophys., 8, 323 (1952); Chem. Abstr., 47, 3151 (1953).
1484. Hegediis A. J., Dvorszky M. Mikrochim. acta, 1959, 160.
1485. Hegediis A. J., Neugebauer J., Dvorszky M. Mikrochim. acta, 1959 282
I486. Hegediis M. Z. analyt. Chem., 107, 166 (1936).
1487. Hegediis M., Juvancz I, Mikrochem. ver. Mikrochim. acta, 28, 113 (1940).
1488. Hegemann F. Glastechn. Ber., 26, 168 (1953); РЖХим., № 25750 (1954).
1489. Hegemann F, Hert W. Ber. deutsch. keram. Ges., 35, 258 (1958); РЖХим., № 19152 (1959).
1490. Hegemann F, Koster H. M, Neubauer G. Ber. deutsch. keram. Ges., 37, 483 (1960); РЖХим., № И K227 (1961).
1491. Hegemann F., Kostyra H, Pfab B. Glastechn. Ber., 30, 14 (19571; РЖХим., № 54678 (1957).
1492. Hegemann F„ Os ferried O. Glastechn. Ber., 33, 285 (1960) , РЖХим., № 11 Д55 (1961).
1493. Hegemann F., Pfab B. Glastechn. Ber., 28, 437 (1955); РЖХим. № 32755 (1956).
1494. Hegemann F, Pfab B. Glastechn. Ber., 28, 242 (1955); РЖХим № 51100 (1956).
1495. Hegemann F, Schmidt W., Hert W. Glastechn. Ber., 32, lb (1959); РЖХим., № 69132 (1960).
1496. Hegemann F.. Zoellner H. Glas-Email-Keramo-Techn., 3, 367 (1952); Chem. Abstr., 47, 5084 (1953).
1497. Heidel R. H. Proc. Iowa Acad. Sci., 53, 211 (1946); Chem. Abstr., 42,
4298 (1948).
1498. Heidenhain H. Z. analyt. Chem., 27, 689 (1888).
1499. Heintz W. Pogg. Ann., 63, 82 (1844).
1500. Heintz W. Pogg. Ann., 66, 133 (1845); Z. analyt. Chem., 38, 407 (1899).
1501. Heller H., Haurowitz F., Stary Z. Mikrochemie, 8, 182 (1930).
1502. Heller R., Wagner C. L. Z. anorg. allg. Chem., 200, 105 (1931); 206, 152 (1932).
1503. Helz A. W. J. Research Natl. Bur. Standards, 34, 129 (1945); Chem. Abstr., 39, 2631 (1945).
1504. Helz A. W., Scribner B. F. J. Research Nat. Bur. Standards, 38, 439 (1947); Chem. Abstr., 41, 5698 (1947).
1505. Hemmeler A., Stordi F. N. Ann. chim. appl., 29, 536 (1939); Chem.
Abstr., 34, 3612 (1940).
1506. Hempel W., Klemperer R. L. Angew. Chem., 23, 1756 (1910).
1507. Hempel W„ Koch R. F. Z. analyt. Chem, 20, 496 (1881).
1508. Henderson W. J. Phys. Rev, 55, 238 (1939).
1509. Henriot E. Compt rend, 148, 910 (1909); 150, 1750 (1910); 152, 851 (1911); Ann. chim. et phys, 26, 71 (1912).
1510. Henriot E, Vavon G. Compt. rend, 149, 30 (1909).
214
1511. Henriques R. Lieb. Ann., 215, 321 (1882).
1512. Henry H. F. Analyst, 48, 38 (1959); РЖХим, № 30430 (1960).
1513. Herrmann R. Forschungsdienst, 16, 239 (1943).
1514. Herrmann R. Z. ges. exptl. Med., 118, 187 (1952); 122, 84 (1953); Chem. Abstr., 47, 1223 (1953); Analyt. Abstr, 1, 533 (1954).
1515. Herrmann R. Optik, 12, 189 (1955); Chem. Zbl, 127, 3663 (1956).
1516. Herrmann R. Flammenphotometrie. Berlin, 1956, S. 220.
1517. Herrmann R, Baumann R. Z. ges. exptl. Med, 119, 487 (1953); Chem. Abstr, 47, 7020 (1953).
1518. Herrmann R, Lederle P. Bodenkunde u. Pflanzenernahr, 30, 189
(1942); 34, 1 (1944); Chem. Abstr, 38, 1399 (1944); 41, 6651 (1947).
1519. Herz W. Z. anorg. Chem, 73, 274 (1911); Chem. Zbl, 83, I, 638 (1912).
1520. Herz W, Anders G. Z. anorg.. Chem, 52, 164, 271 (1907); Chem. Zbl, 78, I, 617; II, 1294 (1907).
1521. Herz W, Knoch M. Z. anorg. Chem, 45, 263 (1905).
1522. Herz W, Kuhn F. Z. anorg. Chem, 60, 152 (1908).
1523. Herzner R. A. Biochem. Z, 237, 129 (1931).
1524. Herzog A. Chem. Ztg, 42, 145 (1918); Z. analyt. Chem, 57, 373 (1918).
1525. Hess W, Fahey J. Amer. Mineral, 17, 173 (1932).
1526. Hetterschij С. M. Chem. Weekbl, 39, 448 (1942); Chem. Abstr, 38, 4738 (1944).
1527. H eves у G. Chem. and Ind, 42, 929 (1923).
1528. H eves у G. Nature, 120, 838 (1927); Naturwiss, 23, 583 (1935).
1529. H eves у G. Z. anorg. allg. Chem, 171, 1 (1928).
1530. Hevesy G, Calvert J. C. Naturwiss, 18, 529 (1930); Chem. Abstr, 24, 4573 (1930).
1531. Hevesy G, Christiansen J. A, Berglund V. Z. anorg. allg. Chem, 144, 69 (1925); Chem. Abstr, 19, 2155 (1925).
1532. Hevesy G, Hahn L. Kgl. Danske Videnskab. Selskab. Biol, medd, 16, 1 (1941).
1533. Hevesy G, Seith W, Pahl M. Z. phys. Chem, Bodenstein-Festband, 1931, 309; Chem. Abstr, 25, 5618 (1931).
1534. Hey cock С. T, Lamp lough F. E. Proc. Chem. Soc, 28, 4 (1912).
1535. Heyn A. H, Finston H. L. Analyt. Chem, 32, 328 (1960).
1536. Heyrovsk’y A. Chem. listy, 50, 69 (1956); Сб. чехослов. хим. работ, 21, 1150 (1956).
1537. Heyrovsky A. Chem. listy, 52, 40 (1958); РЖХим, № 81251 (1958).
1538. Heyrovsky A. Analyt. chim. acta, 32, 405 (1960).
1539. Heyrovsky J, Bures M. Collection, 8, 446 (1936).
1540. Heyrovsky J, I 1 ко vid D. Collection, 7, 198 (1935).
1541. Hibbard P. L. Ind. Eng. Chem, 9, 504 (1917).
1542. Hibbard P. L, Stout P. R. J. Assoc. Offic. Agr. Chem, 16, 137 (1933); Chem. Abstr, 27, 2109 (1933).
1543. Hicks W. B. Ind. Eng. Chem, 5, 650 (1913).
1544. Hildenbrand J. H. J. Amer. Chem. Soc, 29,. 447 (1907).
1545. Hilgard E. W. Z. analyt. Chem, 32, 184 (1893).
1546. Hilgers A. Z. physiol. Chem, 304, 193 (1956); Chem. Zbl, 128, 229
(1957).
1547. Hill A. E. J. Amer. Chem. Soc, 50, 2678 (1928).
1548. Hill D. U. Amer. J. Sci, 40, 75 (1915); Chem. Abstr, 9, 2363 (1915).
1549. Hill L. A. J. Amer. Chem. Soc, 25, 990 (1903).
1550. Hill L. M, Tompkins F. C. Trans. Soc. S. Africa, 30, 201 (1944); Chem. Abstr, 39, 2688 (1945).
1551. Hillyer J. C. Ind. Eng. Chem, Analyt. Ed, 9, 236 (1937).
1552. Hinden F. Z. analyt. Chem, 45, 332 (1906).
1553. Hinshelwood A. J. Chem. Soc, 115, 1183 (1919).
1554. Hintz E. Z. analyt. Chem, 23, 60 (1884).
1555. Hintz E. Z. analyt. Chem, 35, 685 (1896).
215
1555a. Hirano S., Sasucha H. J. Chem. Soc. Japan, 64, 77 (1961); РЖХим., № 17 Д34 (1961).
1566. Hirzel O., Waffler H. Helv. phys. acta, 19, 216 (1946); Phys. Rev., 74, 1553 (1948); Chem. Abstr., 41, 5013 (1947); 43, 1260 (1949).
1557. Hochstetter K. Anal. quim. farm. (Chile), 1940, 32; Chem. Abstr., 35, 7314 (1941).
1558. Hoffmann O. Bodenkunde u. Pflanzenernahr., 6, 133 (1937); Chem. Abstr., 32, 5142 (1938).
1559. Hoffman W. S. J. Biol. Chem., 120, 57 (1937).
1560. Hol P. J., Leendertse G. C. Chem. Weekbl., 49, 114 (1953); Z. analyt. Chem., 140, 62 (1953).
1561. Holasek A., Lieb H., Pec ar M. Mikrochim. acta, 1960, 750.
1562. Holemann H., Kleese W. Z. anorg. allg. Chem., 237, 172 (1938); Chem. Abstr., 32, 6928 (1938).
1563. Hol gen H. J. Chem. Weekbl., 14, 578 (1917).
1564. Holiday E., Preedy J. Biochem. J., 55, 214 (1953).
1565. H о 11 e m a n A. F. Chem. Ztg., 16, 1920 (1892); Z. analyt. Chem., 35, 688 (1896).
1566. Holmes A., Lawson R. W. Phil. Mag., 2(7), 1224 (1926); Nature, 117, 620 (1926).
1567. Holstead W. J., Chaitken B. Public Roads, 26, 99 (1950).
1568. Honaker W. L. Concrete, 50, 153 (1942); Chem. Abstr., 36, 3923 (1942).
1569. Honigschmid O., Sachtleben R. Z. anorg. allg. Chem., 213, 365 (1933).
1570. Hon ma M., O’Connor J. D. Appl. Spectroscopy, 7, 131 (1953); РЖХим., № 41302 (1954).
1571. Hopf P. P. J. Chem. Soc., 1946, 785.
1572. Hopf P. P., Gardens S. W. J. Chem. Soc., 1946, 785.
1573. Horiba S., В a b a H. Bull. Chem. Soc. Japan, 3, 11 (1928); Chem. Abstr., 23, 1509 (1928).
1574. Hornberger R. Z. analyt. Chem., 18, 361 (1879).
1575. Horsch H. Compt. rend., 168, 167 (1919).
1576. Hough G. J. Ind. Eng. Chem., Analyt. Ed., 1, 162 (1929); Chem. Abstr., 23, 3870 (1929).
1577. Houtermans F. G., Haxel O., Heintze J. Z. physik., 128, 657 (1950).
1578. Hubbard R. S. J. biol. Chem., 100, 557 (1933); Chem. Abstr., 27, 3496 (1933).
1579. Hubbard R. S., Garbutt H. R. Amer. J. Clin. Path., Tech. Suppl., 3, 119 (1939); Chem. Abstr., 33, 5430 (1939).
1580. Hiibener H. J., Kreuziger H., Heintz R., Koch M. Z. ges. exptl. Med., 119, 523 (1952).
1581. Hubert P. Bull. Soc. Ind. Mulhouse, 88, 500 (1922).
1582. Hiibner H., Schneider W. Lieb. Ann., 167, 89 (1873).
1583. Hughes C. W., Ford O. W. Ind. Eng. Chem., Analyt. Ed., 13, 233 (1941).
1584. Hu let W. H. Amer. J. Med. Sci., 229, 81 (1955); Chem. Zbl., 127, 7326 (1956).
1585. Hunter F. R., В erend a G. D. J. Biol. Chem., 192, 701 (1951)
1586. Hurka W. Z. physiol. Chem., 271, 214 (1941); Biochem. Z., 313, 416 (1943); Chem. Abstr., 37, 5993, 6292 (1943).
1587. Hurley P. M. Bull. Geol. Soc. Amer., 67, 405 (1956); Chem. Zbl., 128, 8702 (1957).
1588. Hurley P. M., Fairbairn H. W., Pinson W. H., Faure G., Geo-chim. et cosmochim. acta, 18, 247 (1960); РЖХим., № 91866 (1960).
1589. Hurst D. G„ Walke H. Phys. Rev., 51, 1033 (1937).
1590. Hurtley W. H„ Orton K. J. J. of Physiol., 30, 10 (1903); Chem. Zbl., 74, II, 1474 (1903).
216
1591. Hurwitz C., Batchelor H. W. Soil Sci., 63, 351 (1947); Chem. Abstr., 41, 7602 (1947).
1592. Husband A. D., Godden W. Analyst, 52, 72 (1927); Chem. Abstr., 21, 1852 (1927).
1593. Hut stein J. Brandes Archiv, 65, 159 (1851).
1594. Huttig G. F., Menzel E. Z. analyt. Chem., 68, 343 (1926); Chem.
* Abstr., 21, 29 (1927).
1595. Hutton R. G„ Nye P. H. J. Sci. Food Agr., 9, 7 (1958); Chem. Abstr., 52, 6059 (1958).
1596. Huysse A. C. Z. analyt. Chem., 39, 9 (1900).
1597. Ikeda S. Sci. Repts Res. Insts Tohoku Univ., A9, 1 (1957); РЖХим., № 81281 (1958).
1598. Illingworth J. W., Santos J. A. Nature, 134, 971 (1934).
1599. Industr. Group. U. K. Atomic Energie, Autor. No 174, 9 (1959); РЖХим., s № 47018 (1960).
* 1600. Inghram M. G., Brown H., Patterson C„ Hess D. C. Phys. Rev., 80, 916 (1950).
1601. Inman W. R., Rogers R. A., Fournier J. A. Analyt. Chem., 23, 482 (1951).
1602. Innes D. D. Trans. Brit. Ceram. Soc., 57, 29 (1958); Chem. Zbl., 129, 11931 (1958).
1603. lonescu S., Constantinescu O., Gainar I., Mihalcu M., Top or D. Studii si cercetari chim., 6, 313 (1958); РЖХим., № 38275 (1959).
1604. Iqbal S., В hat G. D. J. Scient. a. Industr. Res., BC17, 13307 (1958); РЖХим., № 30973 (1959).
1605 Iritani N. J. Pharm. Soc. Japan, 68, 63 (1948); Chem. Abstr., 44, 478 (1950).
1606. Ishibashi M. J. Chem. Soc. Japan, Pure Chem. Sec., 76, 104 (1955); РЖХим., № 39823 (1956).
1607. Ishibashi M., Hara T. Bull. Inst. Chem. Res. Kyoto Univ., 37, 167 (1959); РЖХим., № 51648 (1960).
1608. Ishibashi M., Hara T. Bull. Inst. Chem. Res. Kyoto Univ., 37, 172 (1959); РЖХим., № 51649 (1960).
1609. Ishibashi M., Hara T. Bull. Inst. Chem. Res. Kyoto Univ., 37, 185 (1959); РЖХим., № 51651 (1960).
1610. Ishibashi M., Kagi K. J. Chem. Soc. Japan, 59, 954 (1938); Chem. Abstr., 32, 8298 (1938).
1611. Ishibashi M., Kagi K. J. Chem. Soc. Japan, 63, 1414 (1942); Chem. Abstr., 41, 3012 (1947).
1612. Ishibashi M„ Kishi H. J. Chem. Soc. Japan, 57, 1031, 1039 (1936); Chem. Abstr., 31, 625 (1937).
1613. Ishibashi M., Yamamoto T., Hara T. Bull. Inst. Chem. Res. Kyoto Univ., 37, 159 (1959); РЖХим., № 51647 (1960).
1614. Ishimori T., Takashima Y. Bull. Chem. Soc. Japan, 26, 481 (1953); РЖХим., № 2238 (1955).
1615. Isida N. J. Chem. Soc. Japan, Pure Chem. Sec., 76, 56 (1955); РЖХим., № 4698 (1957).
1616. Ismail A. M., Harwood H. F. Analyst, 62, 443 (1937).
1617. Jackson A. Ind. Eng. Chem., 13, 116 (1921).
1618. Jackson C. Analyst., 78, 599 (1953).
1619. Jackson P..J., Smith A. C. J. Appl. Chem., 6, 547 (1956); РЖХим., № 74655 (1957).
1620. Jacobi W., Keuscher W. Arch. Psych. Nerv., 79, 323 (1927); Chem. Abstr., 22, 3898 (1928).
1621. Jacobs H. R., Hoffman W. S. J. Biol. Chem., 93, 685 (1931); Chem. Abstr., 26, 752 (1932).
1622. Jacquet J., Le Nir Y. Ann. Inst. Nat. Rech. Agr., A6, 499 (1955); Z. analyt. Chem., 151, 58 (1956); РЖХим., № 22686 (1956).
217
1623. Jaeger F. M. Z. anorg. Chem., 101, 185 (1917).
1624. Jaffe E. Ann. chim. (Rome), 41, 796 (1951); Chem. Abstr., 46, 6035 (1952).
1625. Jamieson G. S. Amer. Chem. J., 38, 614 (1907); Chem. Zbl., 79, I, 338 (1908).
1626. Jan der G., A n к e A. Z. analyt. Chem., 154, 8 (1957); РЖХим., № 69121 (1957).
1627. Jander G, Bru 11 W. Lieb. Ann., 453, 332 (1927); Chem. Abstr., 21, 2857 (1927).
1628. Jander G, Busch H. Z. anorg. allg. Chem., 187, 165 (1930); 194, 38 (1930).
1629. Jander G., Faber H. Z. anorg. allg. Chem., 173, 225 (1928); 181, 189 (1929).
1630. Jander G., Faber H. Z. anorg. allg. Chem., 179, 321 (1929).
1631. Jander G, Pfundt O. Z. analyt. Chem., 71, 417 (1927); Chem. Fab- -rik, 1, 435, 446 (1928); Chem. Abstr., 21, 3858 (1927); 23, 1838 (1929).
1632. Jander G., Wendt H. Lehrbuch der analytischen und praparativen anorganischen Chemie. Leipzig, 1958, SS. 65, 72, 76, 89.
1633. Jannasch P. Ber., 24, 273 (1891); Z. anorg. Chem, 6, 72 (1894).
1634. Jannasch P. Z. anorg. Chem, 8, 364 (1895).
1635. Jannasch P. Ber, 28, 2822 (1895).
1636. Jannasch P, Heidenreich O. Z. anorg. Chem, 12, 208 (1896).
1637. Jannasch P, Locke J. Z. anorg. Chem, 6, 168, 321 (1894).
1638. Jannasch P, Vogtherr N. Ber, 24, 3206 (1891).
1639. Janowsky I. V. Monatsh. f. Chem, 8, 49 (1887).
1640. Jansen W. H, Heves J. Z. physiol. Chem, 211, 75 (1932); Chem. Abstr, 26, 5867 (1932)’.
1641. Jansen W. H„ Heyes J, Richter C. Z. phys. Chem, 171, 268 (1934).
1642. Japhe D. Sklarske Rozhl, 14, 123 (1937); Chem. Abstr, 32, 7370 (1938).
1643. Japhe D. Chim. et Ind, 39, 915 (1938).
1644. Jaroszewicz K. Chem. analyt, 2, 97 (1957).
1645. Jasumori Y. Bull. Chem. Soc. Japan, 24, 107 (1951); Chem. Abstr., 46, 11028 (1952).
1646. Jean F, Tri 11 at I. A. Bull. soc. chim, 7(3), 228 (1892); Z. analyt. Chem, 38, 181 (1899).
1647. Jeffreys H. Ann. Geophys, 6, 10 (1950); Chem. Abstr, 44, 7667 (1950).
1648. Jendrassik L, Falcsik-Szabo E. Biochem. Z, 261, 110 (1933).
1649. J e n d r a s s i к L, Petras P. Biochem. Z, 226, 381 (1930); Chem.
Abstr, 25, 310 (1931).
1650. Jendrassik L, Polgar A. Z. analyt. Chem, 107, 417 (1936).
1651. Jendrassik L, Szel J. Biochem. Z, 267, 124 (1933).
1652. Jendrassik L, Takacs F. Biochem. Z, 274, 194 (1934).
1653. Jentoft R. E, Robinson R. J. Analyt. Chem, 28, 2011 (1956); 29, 855 (1957).
1654. Jentsch D., Frotscher I. Z. analyt. Chem, 144, 17 (1955); РЖХим, № 34646 (1955).
1655. Jentsch D, Jacob G. Chem. Technik, 7, 93 (1955); РЖХим, № 40355 (1955).
1656. Jenzsch G. Pogg. Ann, 104, 105 (1858).
1657. John J. L, Mid ley M. C. Ind. Eng. Chem, Analyt. Ed, 14, 301 (1942).
1658. Johnston B. R, Duncan C. W, Lawton'K, В e n n e E. J.
J. Assoc. Offic. Agr. Chem, 35, 813 (1952); Chem Abstr, 47, 5301 (1953).
1659. Johnston J, Adams L. J. Amer. Chem. Soc, 33, 529 (1911).
1660. Jones E. S, Collings B. J. Med. Lab. Technol, 13, 68 (1955); РЖХим, № 36123 (1956).
1661. Jones E. W. Chem. News, 70, 172 (1894); Z. analyt. Chem, 35, 634 (1896).
1662. J о n e s J. H, H e с к m a n H. J. Amer. Chem. Soc, 69, 536 (1947).
218
*
1663. Jordan P. Helv. chim. acta, 31, 1483 (1948).
1664. Joukovsky N. I., Lowenthal S. Bull. Soc. Chim. Biol., 31, 1190 (1949); Intern. Congr. Biochem., Abstr. of Communs. 1st Congr. Cambridge,.1949, p. 286; Chem. Abstr., 44, 10027 (1950); 47, 666 (1953).
1665. Joy A. B. Ind. Eng. Chem., Analyt. Ed., 16, 383 (1944).
1666. J nil lard A. Bull. soc. chim., 33, 977, 988 (1880).
1667. Jungfleisch E., Landrieu P. Compt. rend., 158, 1309 (1914).
1668. Justin-Miller'E. J. pharm. chim., 28, 15 (1923); Chem. Abstr., 17, 3658 (1923).
1669. Ju у T. Тужан сю=>бао, 2, 100 (1953); РЖХим., № 29311 (1954).
1670. Kahane E. Bull. soc. chim., 53, 547 (1933).
*1671. Kahlenberg L., Krauskopf F. C. J. Amer. Chem. Soc., 30, 1104 (1908).
1672. Kahn B„ Smith D. K., Straub С. P. Analyt. Chem., 29, 1210 (1957).
1673. Kali-Forschungs-Anstalt. Chem. Abstr., 32, 8846 (1938).
1674. Kallmann S. Ind. Eng. Chem., Analyt. Ed., 16, 712 (1944); 20, 678 (1948).
1675. Kalvoda R. Analyt. chim. acta, 18, 132 (1958); РЖХим., № 77151 (1958).
1676. Kaplan D., Schnerb J. Bull. Council Israel, 3, 448 (1954); 4, 397 (1955); РЖХим., № 18965 (1955); № 58432 (1956).
1677. Karaoglanow Z. Z. analyt. Chem., 105, 36 (1936); Osterr. chem. Ztg., 44, 149 (1941).
1678. К a r c h m e r J. H., Gunn E. L. Analyt. Chem., 24, 1733 (1952).
1679. К a r r m a n K. J., В 1 a d h E., G e d d a P. Mikrocim. acta, 1959, 775, 779.
1680. К assn er B. J. prakt. Chem., 4(4), 306 (1957); Chem. Zbl., 128, II, 793 (1957).
1681. Katakousinos S., Paradimitriou A. Z. Pflanzenernahr., Diing., Bodenkunde, 26A, 166 (1932).
1682. Kato T. J. Elektrochem. Assoc. Japan, 3, 276 (1935); Chem. Abstr., 30, 2517 (1936).
1683. Katz I. Pflung. Arch., 63, 1 (1896).
1684. Kaufmann H. L. Chemist-Analyst, 19, 11 (1930).
1685. Kavina J. Chem. listy, 23, 267, 289 (1929); Chern. Zbl., 100, 1592, 3874 (1929).
1686. К a we A. Z. Pflanzenernahr., Diing., Bodenkunde, 43, 69 (1936); Z. analyt. Chem., 115, 385 (1938/39); Deutsch, med. Wochschr., 71, 266 (1946).
1687. Kayas G. Compt. rend., 228, 1002 (1949); J. Chem. Phys., 47, 408 (1950); Chem. Abstr., 43, 5698 (1949); 45, 61 (1951).
1688. Kaye I. A. Ind. Eng. Chem., Analyt. Ed., 12, 310 (1940).
1689. Kayser H. Runge C. Z. analyt. Chem., 32, 573 (1893).
1690. Kayser R. Z. analyt. Chemie, 21, 427 (1882).
1691. Kayser R. Ber., 18, 3417 (1885).
1692. Kehrmann F. Z. anorg. allg. Chem., 7, 406, 425 (1894).
1693. Keirs R. J., Speck S. J. J. Dairy Sci., 33, 413 (1950); Chem. Abstr., 44, 8012 (1950).
1694. Keitt T. E. Ind. Eng. Chem., 12, 276 (1920).
1695. Keitt T. E„ Shiver H. Ind. Eng. Chem., 10, 219, 994 (1918); II, 1049 (1919).
1696. Keller W„ Weiss F. Pharmazie, 12, 19 (1957); Chem. Zbl., 128, 11954 (1957).
1697. Kelley O. J., Hunter A. S., St er ges A. J. Ind. Eng. Chem., Analyt. Ed., 18, 319 (1946).
1698. Keim E. F., Wilkinson J. A. Proc. Iowa Acad. Sci., 43, 169 (1936); Chem. Abstr., 32, 4100 (1938).
1699. Kemb N. Z. analyt. Chem., 93, Heft 9 (1953); Калий, 3, № 5, 45 (1934).
1700. К emu la W., Brachaczek W., Dancewitz D., Hulanicki A. Chem. analyt. (Varsovie), 3, 729 (1958); РЖХим., № 86234 (1959).
219
1701. Kemula W., Kornacki J. Roczn. chem., 28, 635 (1954); РЖХим., № 43215 (1955).
1702. Kemula W., Michalski M. Atti X° congr. intern, chim., 3, 419, (1939); Chem. Abstr., 34, 1044 (1940).
1703. Kenjiro H., Yasumitsu U. J. Chem. Soc. Japan, Pure Chem. Sec., 81, 906 (1960); РЖХим., № 3, 135 (1961).
1704. Kenny W. R, Hester J. B. J. Amer. Soc. Agron, 28, 682 (1936);
Chem. Zbl., 107, II, 4030 (1936).
1705. Kenyon C., Oventson T. C, Parker C. A. Analyst, 75, 269 (1950).
1706. Kerr S. E. J. biol. Chem., 67, 689 (1926).
1707. Kertscher F. Bodenkunde u. Pflanzenernahr, 9—10, 758 (1938);
Z. analyt. Chem., 124, 146 (1942). '»
1708. Khoi T. T. Ann. chim. analyt. appl., 15, 340 (1933); Chem. Abstr., 27, 5026 (1933).
1709. Ki ba T. J. Chem. Soc. Japan, 60, 1073 (1939); Chem. Abstr., 34, 1591 (1940).
1710. Kibe M. M., Basu J. K. Poona Agr. Coll. Mag., 35, 4 (1943); Chem. Abstr., 38, 3568 (1944).
1711. Kick H. Z. Pflanzenernahr., Diing, Bodenkunde, 60, 163 (1953); РЖХим., № 594 (1955).
1712. Kielland J. J. Amer. Chem. Soc., 59, 1675 (1937); Ber., 71, 220 (1938); Chem. Abstr., 32, 2865 (1938).
1713. Kielland J. Chem. Abstr., 45, 2161 (1951).
1714. Kimura J., Chiba H. J. Sci. Soil Manure, 10, 429 (1936); Chem. Zbl., 108, I, 2415 (1937).
1715. Kimura M., Nakamura G. Sci. Papers Inst. Phys. Chem. Res., Japan, 3, 51 (1925); Chem. Abstr., 19, 2913 (1925).
1716. King E. J., Haslewood G. A., Delory G. E., Beall D. Lancet,
1, 207 (1942); Chem. Abstr., 36, 4536 (1942).
1717. King H. S. Proc. Trans. Nova Scotian Inst. Sci., 16, 30 (1928); Chem. Abstr., 22, 2900 (1928).
1718. Kingsley G. R, Schaffert R. R. Science, 116, 359 (1952); Analyt. Chem., 25, 1738 (1953); J. biol. Chem., 206, 807 (1954).
1719. Kingsley W. K., Wolf G. E., Wolfram W. E. Analyt. Chem., 29, 939 (1957).
1720. Kirchhoff G., Bunsen R. Pogg. Ann., 110, 173 (1860); Z. analyt. Chem., 1, 1 (1862).
1721. Kirchhoff G., Bunsen R. Pogg. Ann., 112, 373 (1861); Z. analvt. Chem., 1, 62 (1862).
1722. Kirjan W. M. Biochem. Z, 237, 73 (1931); Chem. Abstr., 25, 5185 (1931).
1723. Kirk P. Ann. Rev. biochem., 6, 76 (1937); 9, 953 (1940).
1724. Kirn E. R, Dunlap H. L. J. Amer. Chem. Soc., 53, 391 (1931).
1725. Kirsten W. J., Berggren A., Nilsson K. Analyt. Chem., 30, 237 (1958).
1726. Kissel E. Z. analyt. Chem., 8, 410 (1869).
1727. Kisser J. Pharm. Presse, 28, 2 (1923); Chem. Abstr., 17, 1812 (1923).
1728. Kiss ling N. Chem. Ztg., 7, 1139 (1883).
1729. Klapproth W. K. Kali, 17, 343 (1923); Chem. Abstr, 19, 1113 (1925).
1730. Klein B, Jacobi M. Ind. Eng. Chem, Analyt. Ed, 12, 687 (1940); Chem. Abstr, 35, 48 (1941).
1731. Klein G, Taubock K. Biochem. Z, 241, 413 (1931).
1732. Klein J, Strebinger R. Fortschritte der Mikrochemie. Wien, 1928.
1733. Klemperer R. L. Dissertation, Dresden, 1910; Z. analyt. Chem, 53, 278 (1914).
1734. К levstrand R. Med. norsk. farmac. Selsk, 17, 190 (1955); Chem. Zbl, 129, 9571 (1958).
1735. Kling A, Lassieur A. Ann. Falsific, 7, 410 (1914).
1736. Kling M, Engels O. Z. analyt. Chem, 45, 315 (1906).
220
1737. Klinkerfues F. Chem. Ztg., 29, 77, 1085 (1905); Chem. Zbl., 76, I, 693; II, 1465 (19*05).
1738. Klooster H. S., Stearns E. I. J. Amer. Chem. Soc., 55, 4121 (1933); Chem. Abstr., 27, 5624 (1933).
1739. Klopping W. Kali, 35, 105 (1941); Chem. Abstr., 39, 4301 (1945).
1740. Klottmann F. Z. analyt. Chem., 73, I (1928); Chem. Abstr., 22, 1514 (1928).
1741. Klyne W. Spectrochim. acta, 4, 64 (1950).
1742. Knakkergaard-Moller C. Acta Chem. Scand., 8, 81 (1954); РЖХим., № 42669 (1954).
1743. Knecht E. Hibbert E., New Reduction Methods in Volumetric Analysis. London, 1925, pp. 25, 84.
1744. Knemmel D. F., Karl H. L. Analyt. Chem., 26, 386 (1954).
1745. Knichmann E. Z. Pflanzenernahr., Diing., Bodenkunde, 54, 117 (1951); Chem. Abstr., 46, 8565 (1952).
1746. Knigge G. Allgem. 01- u. Fettztg, 27, 223 (1930); Chem. Abstr., 25, 2016 (1931).
1747. Knight S. B., Mathis W. C., Graham J. R. Analyt. Chem. 23, 1704 (1951).
1748. Knop W. J. Chem. Listy, 51, 182 (1957); Сб. чехослов. хим. работ, 22, 1709 (1957).
1749. Knop W. Z. analyt. Chem., 1, 470 (1862).
1750. Knop W. Z. analyt. Chem., 18, 462 (1879).
1751. Knop W. Z. analyt. Chem., 22, 421 (1883).
1752. Knop W., Wolf W. Z. analyt. Chem., 1, 470 (1862).
1753. Knosel Th. Ber., 6, 1159 (1873); Z. analyt. Chem., 13, 51 (1874).
1754. Koch E. Z. angew. Chem., 22, 1442 (1909);. Chem. Zbl., 80, II, 861 (1909).
1755. Koenig E. W. Ind. Eng. Chem., Analyt. Ed., 7, 314 (1935).
1756. Koenig E. W. J. Amer. Ceram. Soc., 22, 164 (1939); Chem. Abstr., 33, 5319 (1939).
1757. Koenigsberger J. Chem. Ztg., 24, 690 (1900).
1758. Kohl J., Na th er R. E., Guinn V. P. Nucleonics, 14, 50 (1956); РЖХим, №.17607 (1958).
1759. Kohler M. Z. analyt. Chem, 138, 9 (1953).
1760. Kohn W. Kali, 35, 108 (1941); Z. analyt. Chem, 128, 1 (1947); Chem. Abstr, 39, 4301 (1945); 42, 481 (1948).
1761. Kolarov N. Z. analyt. Chem, 122, 399 (1941); Chem. Abstr, 36, 4052 (1942).
1762. Kolbe H. J. prakt. Chem, 5, 93 (1872); Z. analyt» Chem, 11, 195 (1872).
1763. Kolhorster W. Naturwiss, 16, 28 (1928); Chem. Abstr, 24, 3729 (1930).
1764. Kolthoff I. M, Bendix G. H. Ind. Eng. Chem, Analvt. Ed, 11, 94 (1939).
1765. Kolthoff I. M, San dell E. B. J. Phys. Chem, 37, 443 (1933); Chem. Abstr, 27, 5017 (1933).
1766. Kolthoff I. M, Verzijl E. Z. anorg. allg. Chem, 132, 318 (1924).
1767. Koluschewa A, Sewrugowa P. Koll. Z, 60, 141 (1932); Chem. Abstr, 26, 5472 (1932).
1768 Komar ek K. Mikrochemie, Mikroanalysa kvalitativni. Praha, 1948, SS. 38, 148.
1768a. Komarovski A. S, Goremikin V. 1. Z. analyt. Chem, 87, 339 (1932)
1769. Koninck L. L. Z. analyt. Chem, 20, 390 (1881); 49, 53 (1910).
1770. Koninck L. L. Chem, News, 44, 144 (1881); Z. analyt. Chem, 21, 406 (1882); 35, 72 (1896); Chem. Ztg, 19, 901 (1895).
1771. Konopicky K, Schmidt W. Ber. deutsch. keram. Ges, 37, 368 (I960); РЖХим, № 6 Д70 (1961).
1772. Koppel J, Cahn M. Z. anorg. Chem, 60, 53 (1908).
221
1773. KorblJ., VydraF., P F i b i 1 R. Taianta, 1, 281 (1958); РЖХим., № 22060 (1960).
1774. Korn E. Z. angew. Chem., 22, 1442 (1909); Z. analyt. Chem., 49, 60 (1910).
1775. Kornacki J. Wiadom. chem., 8, 538 (1954).
1776. Kortum G, Schottler H. Z. Elektrochem, 57, 353 (1953); Chem. Abstr., 47, 11971 (1953).
1777. Koszegi D. Acta Sci. Regiae Univ. Hung., 11, 214 (1947); Chem. Abstr., 23, 791 (1929).
1778. Kourim V., К r t i 1 J., Konecny C. Chem. listy, 52, 262 (1958): Collect., 24, 1474 (1959); РЖХим., № 77180 (1958); № 8832 (1960).
1779. Kovar C. Sbornik Ceskoslov. akad. zemedelskych ved., 2, 805 (1957); Chem. Abstr., 52, 7567 ,(1958).
1780. Kramer B. J. biol. Chem., 41, 263 (1920).
1781. Kramer B., Gittleman J. Proc. Soc. Exptl. Biol. Med., 24, 241 (1926); Chem. Abstr., 21, 2709 (1927); Z. analyt. Chem., 74, 160 (1928). .
1782. Kramer B., Tisdall F. J. biol. Chem., 46, 339 (1921).
1783, Kramer J. Z. analyt. Chem., 109, 16 (1937).
1784. Krane F. E. Analyt. chim. acta, 16, 370 (1957); РЖХим., № 808 (1958).
1785. Krause G. Arch. d. Pharmac., 2, 407 (1874); Z. analyt. Chem., 14, 184 (1875).
1786. Krause R. Vom Wasser, 23, 279 (1956); Chem. Zbl., 129, 6065 (1958).
1787. К-rauss J. Z. Bodenkunde u. Pflanzenern., 48, 310 (1936); Калий, 6, № 5—6, 45 (1937).
1788. Kraut K. Z. analyt. Chem., 26, 604 (1887).
1789. Kraut K, Orrman L., Kusel W. Z. analyt. Chem., 14, 152 (1875).
1790. Kray bi 11 H. R. J. Assoc. Offic. Agr. Chem., 18, 237 (1935); Калий, 4, 39 (1935).
1791. Kray bi 11 H. R., Thornton S. E. Amer. Fertilizer, 17, 24 (1934);
18, 260 (1935); Калий, 4, № 2, 27; № 8, 38 (1935).
1792. Kreider D. A. Z. anorg. Chem, 9, 342 (1895); 10, 277 (1895); Amer. 3. Sci., 49, 433 (1895); 50, 287 (1895); Z. analyt. Chem., 36, 711 (1897).
1793. Kreider D. A, Breckenridge J. E. Amer. J. Science, 2, 263 (1896), Chem. Zbl., 67, II, 928 (1896).
1794. Kreisky F. Mikrochim. acta, 1959, 243.
1795. Kremann R, Kerschbaum F. Z. anorg. Chem, 56, 218 (1907); Chem. Zbl, 79, I, 212 (1908).
1796. Kressman T. R„ Kitchner J. A. J. Chem. Soc, 1949, 1190, 1201, 1208, 1211.
1797. Kretschy M.,Z. analyt. Chem, 15, 37 (1876).
1798. Kretschmar M. Chem. Ztg, 11, 418 (1887); Z. analyt Chem, 28, 95 (1889).
1799. Krickmeyer R.- Z. phys. Chem, 21, 53 (1896); Chem. Zbl, 67, II, 993 (1896).
1800. Krische P. Kali, 2, 425, 441, 461 (1908).
1801. Krische P. Die Entwicklungsgeschichte der Chemie des Kaliums. Halle, 1908.
1802. Krische P. Das Kali. Stuttgart, Bd. I, 1923; Bd. 2, 1928.
1803. Krishnayya H. V. Chem. News, 107, 100 (1913); Z. analyt. Chem, 52, 777 (1913).
1804. Krokovski T.‘Z. analyt. Chem, 112, 183 (1938).
1805. Krug W. H, Me Elroy К. P. J. analyt. Chem, 6, 184 (1892).
1806. Kriigel C, Retter A. Das Superphosphat, 9, 85 (1933); Z. analyt. Chem, 96, 314 (1934); Калий 4, 40 (1935).
1807. Kriiss G, Nilson L. F. Ber, 20, 1682 (1887).
1808. К ii h 1 H. Pharm. Zentralhalle, 50, 127 (1909); Chem. Zbl, 80, 1041 (1909).
1809. Ktihns К, M ii 11 e r G. Z. phvsiol. Chem, 294, 86 (1954); РЖХим, № 49797 (1954).
222
1810. Kuisel H. F. Helv. chim. acta, 18, 896 (1935).
1811. Kiikel A. Das Superphosphat, No 4 (1933); Калий, 3, № 2, 36 (1934).
1812. Kunin R. Analyt. Chem., 21, 87 (1949).
1813. Kunz J. Helv. chim. acta, 16, 3 (1933).
1814. Kunz J. Helv. chim. acta, 16, 259 (1933).
1815. Kiister F. W., Griiters M. Z. anorg. Chem., 36, 325 (1903); Chem.
Zbl., 74, II, 908 (1903).
1816. Kuzirian S. B. Proc. Iowa Acad. Sci., 24, 547 (1917); Chem. Abstr., 14, 1273 (1920).
1817. Kvalheim A. J. Optic Soc. Amer., 37, 585 (1947).
1818. Kvapil J. Kvasny prumysl, 4, 87 (1958); РЖХим.; № 81921 (1959).
1819. Lal К., Lal К. R. Res. Bull. Panjab Univ., 10, 257 (1960); РЖХим, № 11, B68 (1961).
1820. Lamar M. O., Hazel W, M., O’Leary W. J. Ind. Eng. Chem., Analyt. Ed., 7, 429 (1935).
1821. Landergren S., Mu Id W. Mikrochim. acta, 1955, 245.
1822. Landolt H., Bernstein R. Zahlenwerte und Funktionen. Bd. I, Teil 5. Berlin, 1952, SS. 80, 206, 246.
1823. Lane E. S. Analyst, 82, 406 (1957).
1824. Lange B. Kolorimetrische Analyse. Verlag Chemie, Weinheim, 1952, SS. 46, 68, 73.
1825. Lange J. Silikattechnik, 7, 54 (1956); РЖХим., № 66376 (1957).
1826. Lange J. Sprechsaal Keramik, Gias, Email, 92, 305 (1959); РЖХим., № 838 (I960).
1827. Langhans N. Nitrocellulose, 10, 104, 130 (1939); Chem. Abstr., 33,.
8181 (1939).
1828. Langmuir R. V. Phys. Rev., 74, 1559 (1948).
1829. Lanik J. Chem. Obzor, 21, 111 (1946); Ind. Eng. Chem., Analyt. Ed., 21, 165 (1949).
1830. Lannung A. Z. phys. Chem., 161 A, 255 (1932).
1831. Lappert M. F. Chem. Rev., 56, 1036 (1956).
1832. Larson R, G., Hunt H. J. phys. Chem., 43, 417 (1939).
1833. Lasczynski S. Ber., 27, 2285 (1894).
1834. Laspeyres H. J. prakt. Chem., 94, 193 (1865); Z. analyt. Chem., 4, 415 (1865).
1835. Latour A. La Nature, 35, 23 (1907).
1836. Laur A. Acta Comm. Univ. Tartuensis, 16A, 66 (1929); Chem. Abstr., 23,
5430 (1929).
1837. Lavoye M. J. pharm. Belg., 13, 160 (1931).
1838. Layvson R. W. Nature, 117, 620 (1926).
1839. Lawton K. Soil Sci. Soc. Amer., Proc., 10, 126 (1945); Chem. Abstr., 41,
2192 (1947).
1840. Lea A. Amer. J. Science, 32 (2), 183 (1861); Chem. News, 3, 180 (1861). 1840a. Lea per F. M. Textile Colorist, 55, 833 (1933).
1841. Leary W. J., P apish J. Ind. Eng. Chem., Analyt. Ed., 6, 107 (1934).
1842. Lebermann F. Biochem. Z., 150, 548 (1924); Klin. Wochenschr., 3, 1632 (1924); Chem. Abstr., 19, 87 (1925).
1843. Leddi cotte G. W„ Reynolds S. A. ASTM Bull., N 188, 29 (1953).
1844. Lederer M. Mikrochim. acta, 1956, 43.
1845. Leemann H., Grandmougin E. Ber., 41, 1296 (1908); Chem. Zbl., 79, I, 2094 (1908).
1846. Lee nt F. H. Z. analyt. Chem., 40, 569 (1901).
1847. Leffmann H. J. Franklin Inst., 162, 371 (1906); Chem. Zbl., 78, I, 372 (1907).
1848. Legg J. O., A x 1 e у J. H. Soil. Sci. Soc. Amer. Proc., 22, 287 (1958); РЖХим., № 23016 (1959).
1849. Lehmann F. Analytische Chemie, Bd. 3. Leipzig, 1957, SS. 119, 415.
1850. Lehmann H., Pra low W. Tonind.-Ztg., 76, 33 (1952); Chem. Abstr., 46, 4424 (1952).
223
1851. Lehmann W. Bodenkunde u. Pflanzenernahr., 9—10, 766 (1938); Angew. Chem., 51, 595 (1938); Chem. Abstr., 33, 78, 2436 (1939); Z. analyt. Chem., 124. 197 (1942).
1852. Leimbach G. Caliche, 8, 254 (1926); Chem. Abstr., 21, 2631 (1927).
1853. Lejeune G. Compt. rend., 227, 434 (1948).
1854. Lellmann E., Remy A. Ber., 19, 802 (1886).
1855. Lenz N. Lieb. Ann., 181, 40 (1876).
1856. Lenz W. Z. analyt. Chem., 54, 27 (1915).
1857. Leontovitsch N, Benard J. Bull. soc. chim., 157 (1946); Chem. Abstr., 50, 5660 (1946).
1858. Leppanen V., Forsander O. Scand. J. Clin. Lab. Invest., 3, 33 (1951); Chem. Abstr., 46, 161 (1952).
1859. Leppanen V., Hallman N. Scand. J. Clin. Lab. Invest., 5, 250 (1953); РЖХим., № 49795 (1954).
1860. Leppanen V., Krusius F. E., Mettinen T. Ann. Med. Exptl. Biol. Fenniae, 30, 305 (1952); Chem. Abstr., 47, 4947 (1953).
1861. Lepper W. Z. analyt. Chem., 108, No 1, 1 (1937); Калий, 6, № 5—6, 45 (1937).
1862. Lepper W. Z. analyt. Chem., 122, 243 (1941); 124, 27 (1942); Bodenkunde u. Pflanzenernahr., 17, 112 (1940); Chem. Abstr., 35, 262 (1941); 36, 4958 (1942).
1863. Les ar D. S. Afric. Industr. Chemist, 11, 236 (1957); РЖХим., Я» 50034 (1958).
1864. Leuler A. Bull. soc. chim. biol., 15, 158 (1933).
1865. Leuler A., Velluz L., Gruffon H. Bull. soc. chim. biol., 10, 891 (1928); Chem. Abstr., 23, 627 (1929).
1866. Levi A. Staz. sperim. agrar. ital., 37, 595 (1904); Chem. Zbl., 75, II, 1255(1904).
1867. Levi G. R. Arquiv. biol. (Sao Paulo), 34, 95 (1950); Chimica et indust-ria (Milan), 33, 9 (1951); Chem. Abstr., 45, 62, 5566 (1951).
1868. Lewin M, Ruer R. Physik Z, 9, 248 (1908); 10, 576 (1909).
1869. Lewis A. H., Marmoy F. B. J. Soc. Chem. Ind., 52, 177 (1933); Chem. Abstr., 27, 4189 (1933).
1870. Lewis P. R. Analyst, 80, 768 (1955).
1871. Leyton L. Analyst, 76, 723 (1951).
1872. Lieber W. Cement-Kalk-Gips, 10, 61 (1957); РЖХим., № 60829 (1957).
1873. Liebig J. Pogg. Ann., 13, 201 (1827).
1874. Linderstrom-Lang K. Compt. rend. trav. lab. Carlsberg, Ser. chim., 21, 111 (1936).
1875. Lindner R. C. Plant Physiol., 19, 76 (1944); Chem. Abstr., 39 2712 (1945).
1876. Lindo-Gladding N. Chem. News, 44, 77, 86, 97, 129 (1881); 53, 202, 296 (1886); Z. analyt. Chem., 21, 406 (1882).
1877. Ling an e J. J. Analyt. Chem., 19, 810 (1947).
3878. Linz A. Ind. Eng. Chem., Analyt. Ed., 15, 459 (1943).
1879. Lippmann E. O. Chem. Ztg., 32, 977 (1908); 34, 1217, 1226, 1235 (1910); Chem. Zbl., 79, II, 1409 (1908); 82, I, 289 (1911).
1880. List E. Lieb. Ann., 80, 117 (1851); Z. analyt. Chem, 15, 38 (1876).
1881. Lloyd E, Brown С. B, Glynwyn D, Bonnel R, Jones W. J J. Chem. Soc., 1928, 658.
1882. Lochhead H. B, Purcell M. K. Amer. J. Clin. Path, 21, 877 (1951); Chem. Abstr, 45, 10288 (1951).
1883. Locke J. Amer. Chem. J, 26, 174 (1901).
1884. Logie D, Rayner J. E. J. Inst. Fuel, 26, 146 (1953); РЖХим, , № 25749 (1954).
1885. Lohr P. Z. offentl. Chem, 10, 421, 439 (1904); Chem. Zbl, 76, I, 296 (1905).
1886. Lohse H. W. Ind. Eng. Chem, Analyt. Ed, 7, 272 (1935).
1887. Lohse H. W. Canad. J. Res, 12, 519 (1935).
•224
1888. Lohuis D., Meloche F., Jud а у C. Trans. Wise. Acad. Sci., 31,265 (1938).
1889. Lombard M. Bull. soc. chim., 13(4), 304 (1913); Z. analyt. Chem., 53, 135 (1914).
1890. Lons t ein I. J. S. African Chem. Inst., 10, 27 (1927); Chem. Abstr., 23, 4292 (1929).
1891. Looney J. M, Dyer C. G. J. Lab. Clin. Med., 28, 355 (1942); Chem. Abstr., 37, 6688 (1943).
1892. Loring H. F., Druce J. G. Chem. News, 140, 34 (1930).
1893. Losche P. Chem. Ztg., 20, 38 (1896); Z. analyt. Chem., 36, 320 (1897).
1894. Loughridge R. H. Amer. Chemist, 3, 369 (1875); Z. analyt. Chem., 14, 341 (1875).
1895. Louguinine W. Lieb. Ann., 121, 123 (1862).
1896. Louise H. W. Ind. Eng. Chem., Analyt. Ed., 7, 272 (1935).
1896a. Loviz T. E. Nova Acta Acad. Sci., 15, 131 (1806).
1897. Low A. H. Chem. News, 67, 185 (1893).
1898. Low W., Townes С. H. Phys. Rev., 80, 608 (1950).
1899. Lucanus B. Z. analyt. Chem., 3, 403 (1864).
1900. Lucas H. Physik. Z, 31, 803 (1930); Z. anorg. allg. Chem., 195, 321 (1931); Chem. Abstr., 24, 5663 (1930); 25, 1732 (1931).
1901. Ludwig E. Bull. soc. chim. Romania, 2, 28 (1920); Chim. et Ind., 7, 60 (1922); Chem. Abstr., 14, 3614 (1920); 16, 1543 (1922).
1902. Ludwig E., Spirescu H. Bull. soc. chim. Romania, 2, 78 (1920); Chem. Zbl., 92, II, 1008 (1921).
1903. Luh B. S„ Nikatic G. Food Res., 24, 305 (1959); РЖХим., № 49796 (1960).
1904. Lukens H. S„ Smit E. F. J. Amer. Chem. Soc, 29, 1455 (1907).
1905. Lund L. J. Geol, 59, 490 (1948); Chem. Abstr, 43, 520 (1949).
1906. Lundegarth H. Arkiv Kemi Mineral Geol, 10, 26 (1928); Chem. Abstr, 23, 791 (1929).
1907. Lundegarth H, Boratynski K. Svensk Kern. Tid, 50, 135 (1938); Chem. Abstr, 32, 6972 (1938).
1908. Lundegarth P. H. Geol. Foren. i. Stockholm Forh, 72, 151 (1950); Chem. Abstr, 44, 10597 (1950).
1909. Lundgren-P. J. Pharmacy a. Pharmacol, 5, 511 (1953); РЖХим, № 4130 (1956).
1910. L fl n i n g О, H a u t о g H. Z. Nahr. Genussm, 49, 1 (1925); Chem. Abstr, 19, 1916 (1925).
1911. L u p a n t - A n d г ё F. J. pharm. Belg, 13, 63 (1958); Chem. Abstr, 52, 14080 (1958).
1912. Lutz O. Z. analyt. Chem, 59, 145 (1920).
1913. Luzanski N. Тек. Ukeblad, 86, 311 (1939); Chem. Abstr, 34, 4205 (1940).
1914. M a c a 11 u m A. B. J. of Physiol, 32, 95 (1905); Ergebnisse d. Physiol, 7, 600 (1908); Arch. Neerland. Physiol, 7, 304 (1922); Chem. Abstr, 17, 781 (1923).
1915. McCrudden F, Sargent C. J. biol. Chem, 33, 235 (1918).
1916. McCurdy M. Proc. Trans. Nova Scotian Inst. Sci, 16, 142 (1928); Chem. Abstr, 22, 3109 (1928).
1917. McDougall F. H. J. Amer. Chem. Soc, 34, 1684 (1912).
1918. McDuffie B. J. Chem. Educ, 30, 454 (1953).
1919 MeGo van G. K, Smart J. Biochem. J, 49, 31 (1951).
1920. MacheleidtR. Kali, 16, 33 3(1922); Chem. Abstr, 16, 3044 (1922); 17, 2092 (1923).
1921. Me Intosh D. J. Phys. Chem, 7, 350 (1903).
1922 Me Kay D. C, D e L о n g W. A. Canad. J. Agric. Sci, 34, 451 (1954); РЖХим, № 4116 (1956).
1923. Me Lennan J. C. Nature, 78, 29 (1908); Le Radium, 5, 142 (1908); Physical. Z, 9, 510 (1908); Chem. Zbl, 79, II, 485, 757 (1908).
15 Аналитическая химия калия
225
1924. McLennan J. C., Kennedy W. T. Phil. Mag., 16, 377 (1908); Chem. Zbl., 79, II, 1332 (1908).
1925. Magee R. J., Headridge J. B. Analyst, 82, 95 (1957); РЖХим., № 54653 (1957).
1926. Magnanini G. Gazz. chim. ital., 31, 542 (1901).
1927. Magnier P., Khrounoff A., Martin M., Daudel P., Dan-del R. Bull. soc. chim., 1947, 626; Chem. Abstr., 42, 2535 (1948).
1928. Magnus G. Ann. d. Physik, 14, 241 (1827).
1929. Mainzhausen L. Bodenkunde u. Pflanzenernahr., 14, 290 (1939)-Chem. Abstr., 34, 565 (1940).
1930. Maj er V. Z. analyt. Chem., 92, 321 (1933).
1931. Majumdar А. К., В i j о 1 i К- P. Z. analyt. Chem., 174, 429 (1960).
1932. Makinen E. Z. anorg. Chem., 74, 74 (1912).
1933. Makris C. G. J. pharm. chim., 13(8), 569 (1931); Chem. Abstr., 26, 2394 (1932).
1934. Malatesta L. Gazz. chim. ital., 77, 147 (1947); Chem. Abstr., 41, 7299 (1947).
1935. Malfatti H. Z. analyt. Chem., 47, 133 (1908); Chem. Zbl., 79, I, 1029 (1908).
1936. Malissa H., Benedetti-Pichler A. A. Anorganische qualitative Mikroanalyse. Wien, 1958, SS. 115, 291.
1937 Malychin F. Chem. listy, 36, 2 (1942); Chem. Abstr., 37, 4848 (1943).
1938. Manasevit H. M. Analyt. Chem., 27, 81 (1955),
1939. Marchand R. E. J. prakt. Chem., 44, 91 (1848).
1940. Marek J. Sbirka praci vizkumn. ust., No 17—21, 37 (1956); РЖХим., № 24733 (1958).
1941. Marenzi A. D. Compt. rend, biol., 129, 1241 (1938); Chem. Abstr., 33, 2929 (1939).
1942. Marenzi A. D., Card ini С. E. Anales farm, bioquim., 12, 32, 45 (1941); Chem. Abstr., 35, 5526, 7994 (1941).
1943. Maronzi A. D., Gerschmann R. Compt. rend, biol., 110, 145 (1932); Anales farm, bioquim., 3, 107 (1932); 9, 51, 85 (1938); Chem. Abstr., 26, 4352 (1932); 27, 3232 (1933); 33, 1357 (1939).
1944. Marenzi A. D.1 V i 1 a 11 о n g a F. Anales farm, bioquim., 11, 105 (1940); Chem. Abstr., 35, 2442 (1941).
1945. Margosches B. Chem. Ztg., 29, 385 (1905).
1946. Margoshes M., Vallee B. L. Analyt. Chem., 28, 180 (1956).
1947. Marignac C. Ann. Mines, 12(5), 19 (1857).
1948. Marignac C. Ann. chim. phys., 8(4), 65 (1866).
1949. Marin F. R. Anales asoc. quim. argentina, 30, 49 (1942); Chem. Abstr., 36, 6108 (1942).
1950. Marinis T. P., M u i r h e a d E. E., Jones F„ Hill J. M. J. Lab. Clin. Med., 32, 1208 (1947); Chem. Abstr., 42, 635 (1948).
1951. Marino L.-Gazz. chim. ital., 35, II, 351 (1905).
1952. Marroquin A. S., Tamayo R. Anales escuela nacl. cienc. biol. (Mex.), 4, 199 (1946); Chem. Abstr., 41, 2192 (1947).
1953. Marschall F. Chem. Ztg., 38, 585, 615 (1914).
1954, Marschall F. Z. analyt. Chem., 54, 167 (1915).
1955. Marschner W. Zement, 20, 466 (1931); Chem. Abstr., 25, 5536 (1931).
1956. Marshall С. E. J. Phys. Chem., 43, 1155 (1939).
1957. Marshall С. E. J. Amer. Chem. Soc., 63, 1911 (1941).
1958. Marshall С. E. J. Phys. Chem., 46, 52, 325 (1942).
1959. Marchall С. E. J. Amer. Chem. Soc., 64, 1814 (1942).
1960. Martens P. H. Chim. analyt., 39, 361 (1957); РЖХим., № 24737 (1958).
1961. Martin E. C. Analyt. chim. acta, 22, 142 (1960).
1962. Martinek K., Gottfried J. Chem. prumysl, 10, 529 (I960); РЖХим., № 6 Д72 (1961).
1963. Martinez F. B., Bori M. G. Chemist-Analyst, 49, 69 (1960) ; РЖХим.» № 7 Д65 (1961).
226
1964. Martini A. Pub. inst. investig. microquim., Univ. nacl. litoral, Argentina, 2, 69 (1939); Chem. Abstr., 33, 7237 (1939).
1965. Martini A. Pub. inst. investig. microquim., Univ. nacl. litoral, Argentina, 4, 63 (1942) ; Chem. Abstr., 36, 1264 (1942).
1966. Martini A, Rizza L. S. Anales asoc. quim. Argentina, 31, 83 (1943); Chem. Abstr., 38, 528 (1944).
1967. Marvin G. G., Wool a ver L. B. Ind. Eng. Chem., Analyt. Ed., 17, 474 (1945).
1968. Marvin G. G., Wool a ver L. B. Ind. Eng. Chem., Analyt. Ed., 17, 554 (1945).
1969. Masaji M. J. Sci. Hiroshima Univ., A17, 161 (1953); РЖХим., № 580 (1950).
1970. Mashiko Y. J. pharm. soc. Japan, 76, 689, 1272 (1956); РЖХим., № 15734, № 48280 (1957).
1971. Mason A. C. Analyst, 76, 176 (1951).
1972. Mathis W. T. Analyt. Chem., 25, 943 (1953).
1973. Mathis W. T. J. Assoc. Offic. Agr. Chem., 39, 419 (1956); Chem. Zbl., 128, 12268 (1957).
1974. Matigton C. Ann. Chim. et de Phys., 28 (6), 309 (1893).
1975. Matsui H. J. Chem. Soc. Japan, 64, 645 (1943); Chem. Abstr., 41, 3396 (1947).
1976. Mavrodineanu R., Boiteux H. L’analyse spectrale quantative par la flamme, t. 8. Paris, 1954, p. 101.
1977. Maw С. B., Miller K. R. Proc. Utah Acad. Sci., 8, 61 (1931); Chem. Abstr., 26, 4007 (1932).
1978. Mayer J. E., H 6 1 d e r - W i n t n e r I. J. Chem. Phys., 6, 301 (1938).
1979. Mayer W. Lieb. Ann., 98, 193 (1856).
1980. Mayer W. Z. analyt. Chem., 26, 354 (1887).
1981. Mazza F. P., Rossi A. Arch. >ci. biol. (Italy), 13, 466 (1920); Chem. Abstr., 23, 5208 (1929).
1982. Mazza L. Atti X° congr. intern, chim., 3, 438 (1939); Chem. Abstr., 33, 9181 (1939).
1983. Meer E. Ber., 8, 1080 (1875).
1984. Mehlich A. J. Assoc. Offic. Agr. Chem., 39, 330 (1956); Chem. Zbl., 128, 14110 (1957).
1985. Mehlich A., H ar ward M. E. J. Assoc. Offic. Agr. Chem., 36, 227 (1953); Chem. Abstr., 47, 12114 (1953).
1986. Mehlich A., Monroe R. J. J. Assoc. Offic. Agr. Chem., 35, 588 (1952);
Chem. Abstr., 47, 2624 (1953).
1987. Mehlich A., Reitmeier R. F., Mason D. D. J. Assoc. Offic. Agr. Chem., 34, 589 (1951); Chem. Abstr., 45, 10454 (1951).
1988. Mehlich J. P. J. Chem. Educ., 4, 1537 (1927); Chem. Abstr., 22, 926 (1928).
1989. Mehrotra M. R., Dhar N. R. J. Phys. Chem., 33, 216 (1929); Chem. Abstr., 23, 1550 (1929).
1990. Meier D, Treadwell W. D. Helv. chim. acta, 34, 805 (1951).
1991. Meillere G. J. Pharm. Chim, 7(7), 281 (1913); Chem. Zbl, 84, I, 1725 (1913).
1992. Mel a ven A. D, Chadwell A. J. Analyt. Chem, 25, 1934 (1953).
1993. Mellor J. W. A Comprehensive Treatise on Inorganic and Theoretical Chemistry. London, 1938, pp. 473, 534, 663, 813.
1994. Mengoli M, Boari A. Boll. lab. chim. provinc, 9, 341 (1958); РЖХим, № 49220 (1959).
1995. Mercier A. Bull. soc. chim. Belg, 10, 403 (1897).
1996. Merlan R, Romain P. Bull. Soc. Pharm. Bordeaux, 93, 25 (1955); РЖХим, № 75255 (1956).
1997. Me ria nd A. Compt. rend, 227, 189 (1948); Chem. Abstr, 42, 4605 (1948).
15*
227
1998. Merlich A., Trug E., Fred E. B. Soil Science, 35, 259 (1933); Chem. Abstr., 27, 3277 (1933).
1999. Mestayer H. Eau, 36, 174 (1949); Chem. Abstr., 44, 3636 (1950).
2000. Methoden zur Untersuchung der Kunstdiingemittel. Verein Deutscher Dfln-gen-Fabrikanten, 1925.
2001. Meurice R. Ann. chim. analyt. appl., 7, 161 (1925); Chem Abstr., 19, 2613 (1925).
2002. Meurice R. Ann. chim. analyt. appl, 8, 129 (1926); Chem. Abstr., 20, 2800 (1926).
2003. Meusser A. Z. anorg. Chem., 44, 79 (1905); Chem. Zbl., 76 I, 854 (1905).
2004. Mevel N., Angot J., Vanoverberghe L. Chim. analyL, 42, 15 (1960); РЖХим., № 73039 (1960).
2005. Mevel N., Lacruche B. Mikrochim. acta, 1958, 241.
2006. Meyer A. Z. analyt. Chem., 36, 159 (1897).
2007. Meyer A. Chem. Ztg., 51, 778 (1927); Chem. Abstr., 22, 39 (1928).
2008. Meyer G. C. Chem. Ztg., 31, 158 (1907); Chem. Zbl., 78, I, 909 (1907).
2009. Meyer H. A., SchwachheimG., Souza Santos M. D. Phys. Rev., 71, 908 (1947).
2010. Meyer J. Helv. chim. acta, 8, 146 (1925); Chem. Abstr., 19, 1984 (1925).
2011. Meyerfeld J. Z. analyt. Chem., 67, 150 (1925); Chem. Abstr., 20, 1189 (1926).
2012. Miethke M., Finzenhagen H. Milch. Forsch., 16, 426 (1934).
2013. Milani C. Biochim. appl. 4, 401 (1957); Chem. Abstr. 52, 8260 (1958).
2014. Mil bourn M. J. soc. Chem. Ind., 56, 205 (1937).
2015. Miles I. E. J. Assoc. Offic. Agr. Chem., 32, 370 (1949); Chem. Abstr., 43, 7172 (1949).
2016. Miller С. C., Magee R. J. J. Chem. Soc., 1951, 3188; Chem. Abstr., 46, 3453 (1952).
2017. M i 11 e г С. C„ T r a v e s F. J. Chem. Soc., 139, 1390 (1936).
2018. Miller C. F. Chemist-Analyst, 20, 8 (1931); Chem. Abstr., 25, 5364
(1931).
2019. Miller H. C„ Kline G. E. J. Amer. Chem. Soc., 73, 2741 (1951).
2020. Miller S. P. Proc. Iowa Acad. Sci., 43, 179 (1936); Chem. Abstr., 32,
4100 (1938).
2021. Miller W. J., Andrews J. T. J. Amer. Oil Chemists Soc., 26, 309 (1949); Chem. Abstr., 43, 5976 (1949).
2022. Milmoe J. O. Quart. Colorado School Mines, 51, 1 (1956); Chem. Zbl., 128, 4008 (1957).
2023. Milne G. J. Agr. Sci., 19, 541 (1929); Chem. Abstr., 23, 5535 (1929).
2024. Milner R. T. J. Assoc. Offic. Agr. Chem., 21, 356 (1938); Chem. Abstr., 32, 8301 (1938).
2025. M i 11 о n R. F., Duffield W. D. Chem. and Ind., 11, 280 (1955); Chem.
Zbl., 127, 1395 (1956).
2026. M i 11 о n R. F., Hoskins J. L., J а с к m a n W. H. Analyst, 69, 299 (1944).
2027. M i 11 о n R. F., Waters W. A. Methods of quantitative microanalysis. London, 1949, pp. 40, 98, 170, 193.
2028. Minovici S., Jonescu A. Bull. Soc. chim. Romania, 3, 25 (1921); Chem. Abstr., 15, 3045 (1921).
2029. Minovici S., Kollo C. Bull. Soc. chim. Romania, 3, 17 (1921); Chem.
Abstr., 15, 3044 (1921).
2030. Miolati A. Z. anorg. Chem., 22, 445 (1900).
2031. Mir J. Afinidad, 19, 365 (1942); Chem. Abstr., 37, 3698 (1943).
2032. Missenden J. Chem. News, 124, 326 (1922).
2033. Mitchell H. L., Ford O. W. Ind. Eng. Chem., Analyt. Ed., 15, 56 (1943).
2034. Mitchell R. L. Spectrochim. acta, 4, 62 (1950).
2035. Mitscherlich A. J. Prakt. Chem., 83, 460, 485 (1861); Z. analyt.
Chem., 1, 58, 63 (1862).
228
2036. Mitscherlich E. A., Celichowski K., Fischer E A Z analyt. Chem., 51, 898 (1912).
2037. Mitscherlich E. A., Fischer H. Die landwirtschaftliche Versuchs-stationen, 78, 75 (1912); Z. analyt. Chem., 52, 587 (1913).
2038. Miyamoto S., Anan K., Matsumura Y., Osaki R., S u g a i A J. Japan. Biochem. Soc., 27, 632 (1956); Chem. Zbl., 129, 2238 (1958).
2039. Modreanu F. Acad rep. populare Romine, 6, 273 (1955); Chem. Abstr., 52, 13530 (1958).
2040. Modreanu F. J. Chromatogr., 1, 554 (1958); РЖХим., № 38272 (1959).
2041. Modreanu F., Fisel S., Carpov A. Studii si cercetari stiint. Acad. RPR, 7, 25 (1956); РЖХим., № 64170 (1958); Nature, 181, 1618 (1958). .
2042. Modreanu F., lorga N. An. stiint. Univ. Iasi, No 2, 143 (1958); РЖХим., № 30429 (1960).
2043. Moelwyn-Hughes E. A. Trans. Farad. Soc., 45, 167 (1949).
2044. Mohler H„ Hammerle W., Hartnagel J. Mitt. Lebensmitt. Hyg., 32, 96 (1941); Chem. Abstr., 37, 6361 (1943).
2045. Mohr F. Lieb. Ann., 119, 123 (1861); Z. analyt. Chem., 1, 59 (1862).
2046. Mohr F. Z. analyt. Chem., 7, 173 (1868).
2047. Mohr F. Z. analyt. Chem., 12, 137 (1873).
2048. Molisch H. Mikrochemie der Pflanze. Jena, 1923, S. 61.
2049. Mol land J. Tids. Kjemi Bergvesen, 19, 119 (1939); Chem. Abstr., 34, 1932 (1940).
2050. Molt E. L„ Tio Liory Hien. Chem. Weekbl., 52, 313 (1956); РЖХим., № 75256 (1956).
2051. Monnier D., Besso Z. Analyt. chim. acta, 7, 380 (1952); Chem. Abstrt., 47, 4789 (1953).
2052. Monnier D., Besso Z„ Wenger P. E. Helv. chim. acta, 34, 433 (1951); Chem. Abstr., 45, 7186 (1951).
2053. Monnot G. A. Chim. analyt., 35, 274 (1953); РЖХим., № 25742 (1954).
2054. M о n t e g u i R., Doadrio A., Serrano C. An. Real. sci. espanola fis. у quim., B53, 447 (1957); B54, 29 (1958); РЖХим., № 14149, № 7968 (1958).
2055. Monvoisin J., Mavrodineanu R. Spectrochim. acta, 4, 152 (1950); Chem. Abstr., 44, 10487 (1950).
2056. Moore С. C. Z. angew. Chem., 11, 559 (1898).
2057. Moore С. E., L a 11 у M. M., A n d e г s о n R. L., В r a d у J. L., M c Lafferty J. J. Analyt. chim. acta, 15, 1 (1956); РЖХим., № 54650 (1957).
2058. Morand M., Ki ehl M. Bull. soc. fran?. ceram., No 33, 19 (1956); РЖХим., № 44843 (1957).
2059. Morel A., Policard A. Bull. soc. chim., 51, 1125 (1932).
2060. Morgulis S., Perley A. J. biol. Chem., 77, 647 (1928); Chem. Abstr.„ 22, 2763 (1928).
2061. Morozewicz J. Bull, de 1’Acad. de Sciences de Cracovie, 1906, 796; Z. analyt. Chem., 47, 422 (1908).
2062. Morrell T. T. J. Amer. Chem. Soc., 2, 145 (1880).
2063. Morris D„ Sutherland B., Wright C. Canad. Chem. a. Metallurgy 21,271 (1937).
2064. Morris R. L. Analyst, 45, 349 (1920).
2065. M о г г i s R. L. Analyst, 48, 250 (1923).
2066 Morris V. H., Ger del R. W. Plant. Physiol., 8, 315 (1933); Chem. Abstr., 27, 3733 (1933).
2067. Morrison G. H., Cos grave J. F. Analyt. Chem., 27, 810 (1955).
2068. Morrison P. Phys. Rev., 82, 209 (1951).
2069. Moser L., Marian S. Ber., 59, 1335 (1926).
2070. Moser L., Maxymowicz W. Ber., 60, 646 (1927).
2071. Moser L., Ritschel E. Monatschr. f. Chem., 46, 9 (1925).
2072. Moser L., Ritschel E. Z. analyt. Chem., 70, 184 (1927).
2073. Moser L„ Schutt K. Monatsh. f. Chem., 51, 23 (1929).
229
2074. Mosher R. E., Boyle A. J., Bird E. J., Jacobson S. D., Batch-elor T. M„ Iseri L. T„ Myers G. B. Amer. J. Clin. Path., 19, 461 (1949); Chem. Abstr., 43, 5441 (1949).
2075. Mousserson M. Biochem. Z., 233, 375 (1931); Bull, soc chim biol, 13, 831 (1931); Chem. Abstr., 26, 5273 (1932).
2076. Mo u s se r s о n M„ Cariteau R. J. pharm. chim., 16, 382 (1932); Chem. Abstr., 27, 1839 (1933).
2077. MousufA. K. Phys. Rev., 88, 150 (1952).
2078. Mueller R. T. Analyt. Chem., 27, 1848 (1955).
2079. Miihlhoff W. Ann. d. Physik, 7, 205 (1930); Chem. Abstr., 25, 3238 (1931).
2080. Muir M. M. Chem. News, 33, 15 (1876).
2081. Mukai K-, Ogihara M. Repts Tokyo'Metropol. Industr. Res. Inst., № 8, 59 (1959); РЖХим., № 26217 (1960).
2082. M u к h e г j i A. K., Sant B. R. Mikrochim. acta, 1959, 370.
2083. Miiller E., Friedberger O. Ber., 35, 2652 (1902).
2084. Mullin H. R., Sheehy T. P. Analyt. Chem., 25, 529 (1953).
2085. Munro D. S., Renschler H., Wilson G. M. J. Physiol., 128, 68 (1955); Chem. Zbl., 128, 2574 (1957).
2086. Muntz J. H., Melsted S. W. Analyt. Chem., 27, 751 (1955).
2087. Mur аса R. F. Chemist-Analyst, 43, 69 (1954); Chem. Zbl., 127, 9537 (1956).
2088. Mu г аса R. F., Collier H. E., В о n s а с к J. B., Jacobs E. S. Chemist-Analyst, 43, 102 (1954).
2089. Mu race J. Chem. Soc. Japan, Pure chem. sec., 75, 227 (1954); РЖХим.. № 1213 (1954).
2090. Muraire M. Chim. analyt., 39, 184 (1957); РЖХим., № 845 (1958).
2091. Mu ran is T. P., Murihead E. E., Jones F., Hill J. M. Lab. Clin. Med., 32, 1208 (1947).
2091a. Muraviev A. N. Z. analyt. Chem., 72, 15 (1927).
2092. Murmann E. Z. analyt. Chem., 50, 171 (1911).
2093. Murmann E. Oesterr. chem. Ztg., 28, 42 (1925); Chem. Abstr., 19, 1827 (1925).
2094. M u г г а у E. G., A d a m s J. A. Geochim. et cosmochim. acta, 13, 260 (1958); Chem. Abstr., 52, 11675 (1958).
2095. Murray K- A. S. African Ind. Chemist, 6, 270 (1952); Chem. Abstr., 47, 6074 (1953).
2096. Myers A. T., Dy al R. S., Borland J. B. Soil Sci. Soc. Amer. Proc., 12, 127 (1947); Chem. Abstr., 43, 1130 (1949).
2097. Mylius W. Sprechsaal, 63, 972 (1930); Chem. Zbl., 102, I, 836 (1931).
2098. N.agami S., Tashiro T. J. Chem. Soc. Japan, 53, 1147 (1932); Chem. Abstr., 27, 928 (1933).
2099. N a g a у a m a S., Tsumori K., Shimizu S. Bull. Inst. Chem. Res., Kyoto Univ., 31, 207 (1953); Chem. Abstr., 47, 11072 (1953).
2100. Na r dell i M., Cavalca L., Braibant A. Ricerca sci., 1958, 1886; Chem. Zbl., 128, 9308 (1957).
2101. Narthrup E. F. Trans. Elektrochem. Soc., 20, 200 (1911).
2102. Na telson S., Stephen L. Microchem. J., 3, 19 (1959); РЖХим., № 71210 (1959).
2103. Neeb К. H., Gebauhr W. Z. analyt. Chem., 162, 167 (1958).
2104. Nehring K. Bodenkunde u. Pflanzenernahr., 30, 36 (1942); Chem. Abstr., 38, 1061 (1944).
2105. Neidig R. E„ Bollen W. B. Ind. Eng. Chem., 19, 154 (1927); Chem. Abstr., 21, 975 (1927).
2106. Nelson R. C., Hamm P. C., Tsiang Y. S. Plant Physiol., 18, 699 (1943); Chem. Abstr., 38, 529 (1944).
2107. Ntmec A. Biochem. Z„ 189, 50 (1927); Chem. Abstr., 22, 658 (1928).
2108. Nemec A. Zemedelske Archivy, 27, 506 (1936); Chem. Abstr., 33, 1075 (1939).
230
2109. Neogi P., Bhattacharyya R. C. J. Indian. Chem. Soc, 6, 333 (1929); Chem. Abstr., 24, 798 (1930).
2110. Neu R. Z. analyt. Chem., 143, 254 (1954).
2111. Neubauer C. Z. analyt. Chem., 11, 474 (1872).
2112. Neubauer H. Z. analyt. Chem., 39, 481 (1900).
2113. Neubauer H. Z. analyt. Chem., 43, 14 (1904).
2114. Neubauer H. Z. analyt. Chem., 46, 311 (1907).
2115. Neugebauer V., Tanasijevic G. Arkh. Ministerstva Pol’ opriv-rede, 6, 27 (1939); Chem. Abstr., 36, 5302 (1942).
2116. Neugebauer W„ Schafer H. Z. anorg. allg. Chem., 274, 281 (1953); РЖХим., № 29107 (1955).
2117. Neumann В. Casop. lekaru Ceskych, 93, 1223 (1954); РЖХим., № 34671. (1955).
2118. Nichols N. Chem. News, 22, 244 (1870).
2119. Nichols M. J. phys. Chem., 45, 411 (1941).
2120. Nichols W. R. Z. anorg. Chem., 60, 106 (1908).
2121. Nier A. O. Phys. Rev., 48, 283 (1935); 50, 1041 (1936); 77, 789 (1950).
2122. Niklas H., P о s c h e n r i e d e r H. Ernahr. Pflanze, 28, 86 (1932); Chem Abstr., 26, 5371 (1932).
2123. Niklas H., Toursei O. Bodenkunde u. Pflanzenernahr., 18, 79 (1940).
2124. Nod dak I., Nod dak W. Z. angew. Chem., 44, 215 (1931).
2125. Nod dak I., Ze i tie г G.. Z. Elektrochem., 58, 643 (1954).
2126. Nogueira J. J., Maffei F. J. Anais assoc, quim. Brasil, 3, 8 (1944); Chem. Abstr., 38, 6509 (1944).
2127. N 5 11 n e г C. J. prakt. Chem., 103, 495 (1868).
2128. Norberg B. Mikrochim. acta, 1, 212 (1937); Compr. rend. trav. lab. Carlsberg, Ser. chim., 21, 233 (1938); Chem. Abstr., 32, 5865 (1938).
2129. N о г b 1 a d J. A. Z. analyt. Chem., 14, 344 (1875) .
2130. Norinder E. Biochem. Z., 299, 168 (1938); Upsala Lakareforen. Forh., 45, 419 (1940); Chem. Abstr., 33, 2163 (1939); 34, 4784 (1940).
2131. Norman D. P., Johnson W. W. Ind. Eng. Chem., Analyt. Ed., 15, 152 (1943).
2132. Norman N., Beck J. C., Browne J. S. J. Lab. Clin. Med., 50, 308 (1957); Chem. Abstr., 52, 489 (1958).
2133. Novak V.'Sb. vedec. praci Visokeskoly chem.-technol. Pardubice. Praha, 1959, p. 121; 1960, p. 129; РЖХим., № 77014 (1960); № 11 Д56 (1961).
2134. Novak V., Krajina A. Collec. czechoslov. chem. communs., 24, 1808 (1959); Chem. listy, 52, 1506 (1950); РЖХим., № 49219 (1959); № 8830 (1960).
2135. Novakova M., D у к у i J. Z. Zuckerind. Bohmen Mahren, 66, 229 (1943); Chem. Abstr., 39, 473 (1945).
2136. Nowak H. Przemysl Chem., 18, 509 (1934).
2137. Noyes A. A., Boggs C. R, Farrell F. 8., Stewart M. A. J. Amer. Chem. Soc.,.33, 1650 (1911); Chem. Zbl., 83, 540 (1912).
2138. Noyes A. A., Bray W. C. A System of Qualitative Analysis for the Rare Elements. N. Y., 1927.
2139. Noyes A. A., Clement A. A. Z. phys. Chem., 13, 412 (1894).
2140. Noyes A. A., Sammet G. V. Z. phys. Chem., 43, 530 (1903).
2141. Nozaki T., Manabe K. Japan Analyst, 9, 316 (I960); РЖХим., № 84399 (1960).
2142. Nutten A. J. Industrial Chemist, 30, 29 (1954); РЖХим., № 5781 (1955).
2143. Nydahl F. Proc. Intern. Assoc, of Theor. Appl. Limnology, 11, 276 (1951).
2144. Oehlmann F., Heimer M. Chem. Techn., 10, 296 (1958); РЖХим., № 19065 (1959).
2145. Oehme F. Z. analyt. Chem., 150, 93 (1956).
2146. OelschlagerW. Z. analyt. Chem., 149, 190 (1956).
2147. Oer A., Hofert H. J. Arch. expl. Path. Pharmakoi., 214, 109 (1951); Z. analyt. Chem., 137, 366 (1953); Chem. Abstr., 47, 2240 (1953).
231
2148. Oertel A. C„ Stace H. C. J. Soc. Chem. Ind., 65, 350 (1946); Chem. Abstr., 41, 1788 (1947).
2149. О h a s h i K., Y a s u i E„ Suzuki H. J. Chem. Soc. Japan, 57, 16 (1954). 2150. Ohle W. Vom Wasser, 14, 158 (1939/40); Chem. Abstr., 35, 3370 (1941). 2150a. Oka S., M u t a G., V a d a J. Bull. Soc. Salt Sci. Japan, 14, 179 (I960)’ РЖХим., № 17 Д36 (1961).
2151. Okac A. Bodenkunde u. Pflanzenernahr., 36, 37 (1945); Chem Abstr 41, 5244 (1947).
2152. Okac A. Qualitative analytische Chemie. Leipzig, 1960, SS. 94 181 213 536 557 ’
2153. Okac A., Те th al T. Chem. Listy, 46, 610 (1952); Z. analyt. Chem., 140, 370 (1953).
2154. Okada K. Mem. Coll. Sci., Kyoto, 1, 89 (1914); Chem. Abstr., 9, 2492 (1915).
2155. Okinaka H. Bunseki to Shiyaku, 3, 14 (1949); Chem. Abstr., 46, 10042 (1952).
2156. Olivari L., Benassi R. Boll. lab. chim. provinc., 11, 230 (1960); РЖХим., № 6 И257 (1961).
2157. Oliver J., Galbis J. A. Boll. inst. nacl. invest., agron. (Madrid), 16, 255 (1956); Chem. Abstr., 52, 4112 (1958).
2158. Olmer D., Payan L., BerthierJ. Compt. rend, biol., 87, 865 (1922); Chem. Zbl., 94, II, 510 (1923).
2159. Olsen S. R., Shaw В. T. J. Amer. Soc. Agron., 35, 1 (1943).
2160. Olson R. V. Soil Sci. Soc. Amer., Proc., 17, 20 (1953); РЖХим., № 14194 (1955).
2161. Oluf sen D. Mikrokosmos, 22, 15 (1928); Chem. Abstr., 23, 55 (1929).
2162. Orban A. Akad. Wiss. Wien. Ber., 140, 121 (1931).
2162a. Orlov N. A. Farmaz. J., 42, 1737 (1903); Analyst, 29, 201 (1904).
2163. Orskov S. L. Acta Physiol. Scand., 27, 321 (1953); Chem. Abstr., 47, 5476 (1953).
2164. Orskov S. L., Ratjen E. Acta Physiol. Scand., 13, 238 (1947); Chem. Abstr., 41, 4529 (1947).
2165. Ortega-Mata M. Anales bromatol. (Madrid), 6, 423 (1954).
2166. Osborn G. H., Johns H. Analyst, 76, 410 (1951).
2167. Osborn R. A. J. Assoc. Offic. Agr. Chem., 25, 410 (1942); 26, 324 (1943); Chem. Abstr., 36, 4617 (1942); 37, 6048 (1943).
2168. Osthaus В. B. J. Amer. Ceram. Soc., 33, 377 (1950); Chem. Abstr., 45, 1462 (1951).
2169. Ott W. Seifensieder-Ztg., 54, 584 (1927); Chem.' Abstr., 21, 2759 (1927).
2170. Otto C. J. Amer. Chem. Soc., 48, 3016 (1926); Chem. Abstr., 21, 717 (1927).
2171. Overman R. R., Davis A. K. J. biol. Chem., 168, 641 (1947); Chem. Abstr., 41, 6595 (1947).
2172. Overstreet R., Jacobson L., Stout P. R. Phys. Rev., 75, 231 (1949).
2173. Owens J. S. Ind. Eng. Chem., Analyt. Ed., 10, 64 (1938).
2174. Pacheco J. R., Lopez-Rubio F. B. Inform, quim. analyt., 1, 121 (1947); Chem. Abstr., 43, 342 (1949).
2175. Paganelli M., Quareni G. Phys. Rev., 86, 423 (1952).
2176. Page H. J. J. Agric. Sci., 14, 133 (1924).
2177. Pajetta R. Gazz. chim. ital., 36, II, 150 (1906); 37, II, 82 (1907); Chem. Zbl., 77, II, 1086 (1906); 78, II, 1016 (1907).
2178. Pajetta R. Gazz. chim. ital., 36, II, 298 (1906); Chem. Zbl., 77, II, 1523 (1906).
2179. Palkin S. J. Amer. Chem. Soc., 38, 3226 (1916); Chem. Abstr., 10, 3043 (1916).
2180. Palous R., Pavelka V., Mara M. Naturwiss., 45, 627 (1958); Collect. Czechoslov. chem. Communs., 24, 3910 (1959); РЖХим., № 45508 (1959); № 1, 126 (1961).
232
2181. P a p e W. Sprechsaal, 60, 163 (1927); Chem. Abstr., 21, 3327 (1927)
2182. Parks R. Q„ Hood S. L„ Hurwitz C., Ellis G. H. Ind. Eng Chem Analyt. Ed., 15, 527 (1943).
2183. Parks T. D., Johnson H. O., Lykken L. Analyt. Chem., 20, 822 (1948).
2184. Parpaillen M. Mem. poudres, 39, 417 (1957): РЖХим., № 77206 (1958).
2185. Parra J. Cenicafe, 8, 28 (1957); РЖХим., № 24731 (1958).
2186. Pascal P. Traite de chimie minerale, t. 6. Paris, 1934, pp. 5, 74, 93, 368, 724.
2187. Passon M. Z. angew. Chem., 15, 1263 (1902); Chem. Zbl., 74, I, 250 (1903).
2188. Patschovsky N. Ber. deutsch. botan. Ges., 43, 489 (1925); Chem. Abstr., 20, 3716 (1926).
2189. Patterson A. M. J. Amer. Chem. Soc., 28, 1734 (1906); Chem. Zbl., 78, I, 450 (1907).
2190. Paul A. D„ Gibson J. A. J. Chem. Educ., 36, 380 (1959); РЖХим., № 16383 (1960).
2191. Paul Th. Z. Elektrochem., 23, 65 (1917); Arb. Reichsgesundh. Amt., 57, 94 (1926).
2192. Paul Th., Dietzel R., Sonneborn W. Arb. Reichsgesundh. Amt., 57, 99, 102 (1926).
2193. Paul W. Z. physik., 124, 244 (1948).
2194. Pauling L. J. Amer. Chem. Soc., 49, 765 (1927).
2195. Pau He D. Bull. soc. chim., 34, 190 (1923).
2196. Pauly C. Pharm. Centralhalle, 8, 187 (1887); Z. analyt. Chem., 36, 512 (1897).
2197. Pavlova M., Jordanov N. Докл. Волг. акад, наук, 10, 33 (1957).
2198. Pawlowska Н., Zlotowska Z. Mater, budowl., 9, 196, 277 (1954); РЖХим., № 9664, № 34670 (1955).
2199. Payen A. Dingl. polytechn. J., 192, 67 (1869).
2200. Payne G. F. Chem. News, 66, 251 (1892); Z. analyt. Chem., 35, 690 (1896).
2201. Payza N. Turk. Ijien Tecriibi Biol. Degrisi, 11, 417 (1951); Chem. Abstr., 47, 1762 (1953).
2202. Pazler J. Listy Cukrovar, 48, 17 (1929); Chem. Abstr., 24, 190 (1930).
2203. Pec ar M. Mikrochim. acta, 1960, 567.
2204. Pechard E. Compt. rend., 110, 754 (1890); Ann. chim. phys., 22 (6), 187 (1891).
2205. Pedersen K. J. Kgl Danske Videnskab. Selskab. Math.-fys. Medd., 18, 26 (1941); Chem. Abstr., 35, 6500 (1941).
2206. Peech M. Ind. Eng. Chem., Analyt. Ed., 13, 436 (1941); Soil Sci., 59, 25 (1945).
2207. Peech M. English L„ Soil Sci., 57, 167 (1944); Chem. Abstr., 38, 4362 (1944).
2208. Peligot M. Z. analyt. Chem., 36, 322 (1897).
2209. Pellet H. Ann. chim. analyt., 22, 146, 179 (1917).
2210 Pence ft N. P. Rev. chim. (RPR), 10, 231 (1959); РЖХим., № 841 (1960).
2211. Peng C. Trans. Sci. Soc. China, 8, No 2 (1934).
2212. Peng G. Science (China), 17, 542 (1933); Chem. Abstr., 27, 3419 (1933).
2213. Peracchio E. S., Mel о ch e V. M. J. Amer. Chem. Soc., 60, 1770 (1938).
2214. Pereira R. S. J. biol. chem., 160, 617 (1945).
2215. Perey M. Theses, Univ, de Paris, 1946; цит. no [172].
2216 Perrier C., Bellanca A. Periodico mineral., 11, 163 (1940); Chem. Abstr., 37, 303 (1943).
2217. Perrin С. H. Analyt. Chem., 21, 984 (1949).
233
2218. Peter H., Kreissing G. Z. Pflanzenernahr., Diing., Bodenkunde, 42,
41 (1948); Chem. Abstr., 43, 6871 (1949).
2219. Peters H. J. Weekbl., 48, 710 (1952).
2220. Pettit J. H. J. Amer. Chem. Soc., 25, 496 (1903); Chem. Zbl.. 74, II 144 (1903). ’ }
2221. Pfaff С. H. Handbuch der analytischen Chemie. Altona, Bd 1 1821
S. 97- Bd. 2 1822 S. 22.
2222. Pfeiffer E. Z. analyt. Chem., 24, 410 (1885).
2223. Pfeil E. Chem. Labor, u. Betrieb, 5, 418 (1954); РЖХим., № 31806 (1955).
2224. Pfeil E„ Friedrich A., Wachsmann T. Z. analyt. Chem., 158, i
429 (1957).
2225. Pfeilsticker K. Spectrochim. acta, 4, 100 (1950); Chem. Abstr., 44, 10776 (1950).
2226. Pflaum R, No wick L. Analyt. Chem., 28, 1542 (1956).
2227. Pfrunder V. R., Schwander H. Zement-Kalk-Gips, 11, 291 (1958);
РЖХим., № 11473 (1959).
2228. Pfundt O. Z. analyt. Chem., 71, 417 (1927); 73, 439 (1928); Chem. Abstr., 22, 2899 (1928).
2229. Philpot J. S., Graham-Horgan V. J., Watson J. Research (London), 1, 273 (1948). '?
2230. Piccard J. J. prakt. Chem., 86, 449 (1862); Z. analyt. Chem., 1, 470 (1862).
2231. Piccinini G. Rend. soc. chim. ital., 1907, 6.
2232. Pichler M. E. Magyar Kern, lapja, 14, 127 (1959); РЖХим, № 26216 (1960).
2233. Pierrot M. Compt. rend, 172, 1041 (1921). ' ?
2234. Pilleri R. Z. analyt. Chem, 157, 1 (1957).
2235. Pillitz W. Z. analyt. Chem, 18, 66 (1879).
2236. Pinta M. Ann. Inst. Nat. Rech. Agronom, Ser. A, 6, 189 (1955); Z. analyt. Chem, 151, 57 (1956); Chim. analyt, 36, 126 (1954); РЖХим, № 2221 (1955).
2237. Pinta M, Bove C. Mikrochim. acta, 1956, 1788.
2238. Piper C. S. J. Soc. Chem. Ind, 53, 392 (1934); 54, 157 (1935).
2239. Piper E, Hagedorn H. Arch. Eisenhiittenw, 22, 299 (1951); Chem.
Abstr, 46, 2223 (1952).
2240. Plunkett W. Chem. Gaz, 1858, 217; Z. analyt. Chem, 5, 381 (1866). 1
2241. Polcin J. Chem. zvesti, 13, 446 (1959); РЖХим, № 51690 (1960).
2242. P о 1 i c a r d A, P i 11 e t D. Bull, histol. appl. physiol, et techn. mikro-scop, 3, 230 (1926).
2243. Pollard F. H, Me О m i e J. F, Banister A. J. Chem. and Ind,
74, 1598 (1955); Chem. Zbl, 128, 2926 (1957).
2244. Pollard F. H, McOmieJ. F, Elbeih J. J. J. Chem. Soc, 1951, •
466.
2245. Pompowski T, Wiewiorowski E. Chem. analit, 4, 487 (1959); РЖХим, № 17562 (1960).
2246. Porter P, Wyld G. Analyt. Chem, 27, 733 (1955).
2247. Posnjak E. J. Phys. Chem, 32, 357 (1928).
2248. Post J, Mehrtens H. Ber, 8, 1549 (1875).
2249. Postis J. Chem. Rev, 223, 1006 (1946).
2250. Potgieter J. E. Union S. Africa Dept. Agr, Sci. Bull, No 312, 10 (1950); Chem, Abstr, 45, 8937 (1951).
2251. Powell R. J, Todd J. J. Soc. Glass Technol, 43, 173 (1959); РЖХим, № 8963 (1960).
2252. Prager A. Chem. Ztg, 20, 260 (1896); Z. analyt. Chem, 36, 318 (1897).
2253. Prebendowski S. Roczn. chem, 31, 329 (1957);. РЖХим, № 24735 (1958).
2254. Precht H. Z. analyt. Chem, 18, 438 (1879).
234
2255. Precht H. Z. analyt. Chem., 18, 509 (1879); Chem. Ztg., 20, 209 (1896);
36, 315 (1897).
2256. Prehn A. Bergbautechnik, 6, 601 (1956); Chem. Zbl., 129, 10757 (1958).
2257. Preus E. Mikrochim. acta, 1956, 382.
2258. Pribram R, Gregor G. Z. analyt. Chem., 38, 401 (1899).
2259. Primbsch E. O. Emailwaren-Ind, 17, 97 (1940); Chem. Abstr., 35, 1955 (1941).
2260. Pringle R. W, Standil S., Roulston К. I. Phys. Rev., 77, 841 (1950).
2261. Pringle W. J. Fuel, 36, 257 (1957); Chem. Zbl., 129, 8801 (1958).
2262. Prodinger W. Organische Fallungsmittel in der quantitativen Analyse. Stuttgart, 1954, S. 8.
2263. Proehl E. C., Nelson W. P. Amer. J. Clin. Path., 20, 806 (1950); Chem. Abstr., 44, 10779 (1950).
2264. Proszt J., Gyorbiro K- Chem. zvesti, 11, 198 (1957); РЖХим., № 969 (1958).
2265. Priissing C. Zement, 20, 360 (1931); Chem. Abstr., 25, 5365 (1931).
2266. Przibilla C. Kali, 2, 401 (1908); 6, 473 (1912); Chem. Abstr., 7, 2024 (1913).
2267. Pucci F., Santini P., Barzaghi L. Anais assoc, quim. Brasil., 3, 188 (1944); Chem. Abstr., 39, 5200 (1945).
2268. P u f f e 1 e s M., N e s s i m N. E. Analyst, 82, 467 (1957).
2269. Pun go r E. Magyar tud, Akad. Kem. tud. oszt Kozl, 12, 225 (1959); РЖХим., № 6 Д31 (1961).
2270. Pungor E., Hegediis A. J., Konkoly-Thege I., Zapp E. E. Mikrochim. acta, 1956, 1247.
2271. Pungor E., Zapp E. E. Acta chim. Acad. Sci. Hung., 7, 185 (1955);
Magyar Kem. folyoirat. 61, 117 (1955) РЖХим., № 22685, № 32754 (1956).
2271a. Pungor E., Zapp E. Magyar Kem. folyoirat, 66, 523 (1960); Analyt.
Abstr,, 8, No 2791 (1961).
2272. P u s h i n N. А., К о v a c h D. Z. anorg. allg. Chem., 199,.369 (1931); Chem. Abstr., 25, 5822 (1931); 26, 1165 (1932).
2273. Putzmann H. Spectrochim. acta, 1, 408 (1940); Chem. Abstr., 35, 6212 (1941).
2274. Quantaroli A. Gazz. chim. ital., 50, II, 64 (1920).
2275. Rabillon R., Griffoul R. Rev. univer. mines, 15, 536 (1959); РЖХим., № 4549 (1960).
2276. Rada F. D. Anales soc. espan fis. quim., 24, 442 (1926); 27, 390 (1929);
Chem. Abstr., 21, 368 (1927); 23, 4161 (1929).
2277. Rada F. D., Gaspar у ArnalT. Anales soc. espan. fis. quim., 24, 150 (1926); Chem. Abstr., 20, 2297 (1926).
2278. R a d d i n g S. B., Phillips R. С., H i e s t e r N. K. Analyt. chim. acta, 11, 538 (1954).
2279. Raff P„ Brotz W. Z. analyt. Chem., 133, 241 (1951).
2280. Rafols Rovira J. M. Inform, quim. analit, 13, 14 (1959); РЖХим, № 51 646 (1960).
2281. Rajagopalan T. Indian J. Agr. Sci., 9, 745 (1939); Chem. Abstr., 34, 2113 (1940).
2282. Ralea R., Modreanu F. Studii si cercetari stiint. Acad. RPR, 8, 231 (1957); РЖХим., № 23010 (1959).
2283. Rama swami M. R., Ayyar M. R. Madras Agr. Dept. Yearbook, 1926, 1 (1927); Chem. Abstr., 22, 3722 (1928).
2283a. Rameshwar P., Arun D. J. Indian. Chem. Soc., 37, 747 (1960);
РЖХим., № 15 До (1961).
2284. Ramirez-Munoz J., Burriel-Marti F. An Real. Soc. espan. fis. у quim., B52, 169 (1956); РЖХим, № 15 702 (1957).
2285. Rammelsberg C. Pogg. Ann, 66, 85 (1845); Z. analyt. Chem, 26, 355 (1887).
235
2286. R am s a у J. A., Brown R. H, F a 11 о о n S. W. J. Exptl biol., 30, 1 (1953); РЖХим., № 5113 (1953).
2287. Randall M, Shaw J. J. Amer. Chem. Soc., 57, 427 (1935).
2288. Rappaport F. Klin. Wochenschr., 12, 1774 (1933).
2289. Rappaport F„ Eichhorn F. Acta Med. Orient, 5, 199 (1946): Chem. Abstr., 43, 5065 (1949).
2290. Rappaport F., Reiter I., Weinmann H. Mikrochim. acta 1 290 (1937).
2291. Rathsburg H., Scheuerer A. Die Chemie, 56, 123 (1943); Chem. Abstr.. 38, 5468 (1944).
2292. Rauch A. Z. anorg. allg. Chem., 160, 77 (1927); Chem. Abstr., 21, 1605 (1927).
2293. Rauch A. Z. analyt. Chem., 98, 385 (1934).
2294. Raulin J. Compt. rend., 110, 289 (1890); Z. analyt. Chem., 31, 359 (1892).
2295. Rauterberg R., Knippenberg E. Bodenkunde u. Pflanzenernahr., 20, 364 (1940); Ang. Chem., 53, 477 (1940); Ernahr. Pflanze. 37, 73 (1941); Chem. Abstr., 35, 706 (1941); 36, 4052, 5108 (1942).
2296. RavaglioJ. A Retorta, 1, 73 (1945/46); Chem. Abstr., 40, 5356 (1946).
2297. Ray P. J. Chem. Soc., 89, 551 (1906).
2298. R а у P. Quart. J. Indian. Chem. Soc., 1, 230 (1925).
2299. Ray R. C, Chatterji K. J. Chem. Soc., 384 (1932).
2300. Ray R. C„ Mitra H. C. Trans. Farad. Soc., 30, 1161 (1934); 31, 1312 (1935).
2301. Reckleben H. Z. angew. Chem., 26, 375 (1913); Chem. Abstr., 7, 3292 (1913).
2302. Redemann С. E., Niemann C. J. Amer. Chem. Soc., 62, 590 (1940).
2303. Radtenbacher N. J. prakt. Chem., 94, 442 (1865); Z. analyt. Chem., 4, 97 (1865).
2304. Reed J. F, Mehlich A., P i 1 a n d J. R. Proc. Soil Sci. Soc. Amer., 9, 56 (1944).
2305. R e e d R. D., W i t h г о w J. R. J. Amer. Chem. Soc., 50, 1515, 2985 (1928);
51, 1062, 3238 (1929); 52, 2666 (1930); Chem. Abstr., 22, 2899 (1928); 23, 791, 2390 (1929); 24, 567, 4237 (1930).
2306. Rees A. G., Huddleston L. J. J. Chem. Soc., 1931, 1648.
2307. Re gel K. Chem. Ztg., 30, 684 (1906); Chem. Zbl., 77, II, 558 (1906).
2308. Reich H, Grabbe F. Tonind.-Ztg., 79, 127 (1955); РЖХим., № 55317 (1955).
2309. Reichard C. Z. analyt. Chem., 40, 377 (1901); Chem. Ztg., 25, 1151 (1901).
2310. Reichard O. Z. analyt. Chem., 140, 188 (1953).
2311. Reimann W. Z. physiol. Chem., 287, 210 (1951); Chem. Abstr., 47, 8807 (1953).
2312. Reimers F., Gottlieb K. R. Contrib. Danish Pharm. Comm., 1, 12 (1946); Chem. Abstr., 41, 2856 (1947).
2313. Reimers H. Chem. Ztg., 81, 357 (1957); РЖХим, № 7534 (1958).
2314. Reinhardt C. Stahl u. Eisen, 16, 448 (1896); Z. analyt. Chem, 40, 59 (1901).
2315. Reisenegger H. Chem. Ztg, 32, 1071 (1908); Chem. Zbl, 79, II, 1713 (1908).
2316. Reitemeier R. F. Ind. Eng. Chem, Analyt, Ed, 15, 393 (1943).
2317. R e i t h J. F, Looyen A. Pharm. Weekbl, 75, 690 (1938); Chem. Abstr, 32, 6783 (1938).
2318. Rekus A. Appl. spectroscopy, 12, 141 (1958); РЖХим, № 64095 (1959).
2319. Rem el e A. J. de Pharm. et de Chim, 677 (1885); Z. analyt. Chem, 26, 631 (1887).
2320. Remy E. Arch. d. Pharm, 269, 678 (1931); Chem. Abstr, 26, 1211 (1932).
3221. Remy H. Lehrbuch der anorganischen Chemie. Leipzig, 1957, Bd. 1, SS. 201, 221, 239, 432. 1957; Bd. 2, 271, 282, 432, 593, 770.
236
2322. Remy H., Siegmund R. Z. analyt. Chem., 93, 327 (1933).
2323. Renault J. Mises point chimie analyt. pure appl. Paris, 1958, p. 109.
2324. Ren dig V. V. Soil Sci., 12, 449 (1947); Chem. Abstr., 43, 2721 (1949).
2325. Rengade E. Compt. rend., 156, 1898 (1913).
2326. Report of Committee an Atomic Weights. J. Amer. Chem. Soc., 70, 3531 (1948); 71, 1141 (1949).
2327. Reutersward C. Arkiv Fysik, 4, 203 (1951); 11, 1 (1956); Chem. Zbl., 129, 9972 (1958).
2328. Reutersward C. On the Isotopic Constitution of Potassium. Upsala, 1956.
2329. Reynoso A. Compt. rend., 56, 873 (1863); Z. analyt. Chem., 2, 364 (1663).
2330. Rich С. I. Agronomy J., 48, 430 (1956); Chem. Zbl., 128, 13761 (1957).
2331. Richards T. Z. anorg. Chem., 23, 383 (1900).
2332. Richards T. W., Brink F. N. J. Amer. Chem. Soc., 29, 123 (1907).
2333. Richardson C. Amer. Chem. J., 3, 422 (1881); Z. analyt. Chem., 23, 409 (1884).
2334. Richer A. C. Better Crops with Plant Food, 31, 26, 42 (1947); Chem. Abstr., 41, 3173 (1947).
2335. Richert P. H. J. Amer. Chem. Soc., 52, 2241 (1930).
2336. Rieben W. K, Van Slyke D. D. J. biol. Chem., 156, 765 (1944); Chem. Abstr., 39, 1189 (1945).
2337. Rieben W. K. Uber die Kalibestimmung in biologischer Substanz. Basel, 1947.
2338. Riedler K., Schreiner L. Chem. Techn., 11, 593 (1959); РЖХим., № 51645 (1960).
2339. Riegler E. Z. analyt. Chem., 36, 377 (1897).
2340. Riegler E. Z. analyt. Chem., 43, 211 (1904).
2341. Riehm H. Z. Pflanzenernahr., 39, 28 (1934); Калий, 5, № 1, 42 (1936).
2342. Riehm H. Bodenkunde u. Pflanzenernahr., 36, 109 (1945); Z. analyt.
Chem., 128, 249 (1948); Chem. Abstr., 41, 4600 (1947); 42, 5795'(1948).
2343. Riley J. P. Analyt. chim. acta, 19, 413 (1958); РЖХим, № 42122 (1959).
2344. Riley J. P, Williams H. P. Mikrochim. acta, 1959, 804.
2345. Rischbieth R. Z. phys. chem. Unterricht, 39, 67 (1926); Chem. Abstr, 22, 2298 (1928).
2346. Riva B. Ann. chimica, 48, 50 (1958); РЖХим, № 67269 (1958).
2346a. Riva B. Ann. chimica, 51, 52 (1961); РЖХим, № 19 Д30 (1961).
2347. Rivett A. C, O’Connor T. A. J. Chem. Soc, 115, 1348 (1919).
2348. Roach W. A. J. Soc. Chem. Ind, 65, 33 (1946); Chem. Abstr, 40, 3693 (194b).
2349. Roberts T. R, Nier A. O. Phys. Rev, 79, 198 (1950).
2350. Rodertson J. D, Webb B. A. J. Exptl. biol. 1,6, 155 (1939); Chem. Abstr, 33, 6374 (1939).
2351. R о b i n s о n A. R, Newman K. J., S c h о e b E. J. Analyt. Chem, 22, 1026 (1950).
2352. Robinson G. Mettallurgia, 37, 45, 107 (1947); Chem: Abstr, 42, 2204 (1948).
2353. Robinson R. J, Hanschildt J. D. Ind. Eng. Chem, Analyt. Ed, 12, 676 (1940).
2354. Robinson R. J, Putnam G. L. Ind. Eng. Chem, Analyt. Ed, 8, 211 (1936).
2355. Robinson W. Chem. Age, 66, 447, 507, 573 (1952).
2356. Robinson W. O. Ind. Eng. Chem, 10, 50 (1918).
2357. Rodeck H. Deutsch. Z. Nervenheilkunde, 166, 268 (1951); Arztl. Wochen-schr, 7, 182 (1952); Chem. Abstr, 46, 7152 (1952); Chem. Zbl, 127, 12366 (1956).
2358. Rodeck H, Doden W. Klin. Wochenschr, 28, 515 (1950); Chem. Abstr, 45, 3898 (1951).
237
2358a. Rodriguez J., Mattingly G., J. Sci. Food. a. Agric., 11, 717 (1960); РЖХим., № 13 Д70 (1961).
2359. Rodziewicz W., Sz у ch bin ski J. Roczn. chem., 28, 657 (1954); РЖХим., № 40352 (1955).
2360. Roelofsen J. A. Amer. Chem. J., 16, 467 (1894).
2361. Roger L. Ann. chim., 19, 362 (1944); Chem. Abstr., 41, 26 (1947).
2362. Rogers D. P. Chem. and Metallurg. Eng., 25, 161 (1921).
2363. Rogers L. Soil Sci., 12, 124 (1947); Chem. Abstr., 43, 1130 (1949).
2364. Rogosa M. J. biol. Chem., 154, 307 (1944); Chem. Abstr., 38, 5238 (1944).
2365. Roh land P. Z. anorg. Chem., 15, 412 (1897); Z. analyt. Chem., 38, 175 (1899).
2366. Rohl and P. Z. analyt. Chem., 49, 358 (1910).
2367. Rokosz A., Dyrek M. Chim. analit., 4, 705 (1959); РЖХим., № 65063 (1960).
2368. Rollet A. P., Andres L. Compt. rend., 191, 375, 567 (1930); Bull. soc.
chim., 49, 1065, 1086 (1931); Chem. Abstr., 25, 654 (1931); 26, 21 (1932).
2369. Rona P., Haurowitz F., Petow H. Biochem. Z., 149, 392 (1924).
2370. Roode R. J. Amer. Chem. Soc., 17, 85 (1895); Z. analyt. Chem., 36, 321 (1897).
2371. Roscoe H. E. Lieb. Ann., 121, 246 (1861); J. Chem. Soc., 16, 82 (1863).
2372. Roscoe H. E. Phil. Trans., 160, II, 317 (1870); Lieb. Ann., Suppl., 8, 95 (1872).
2373. Rose H. Pogg. Ann., 113, 627 (1861).
2374. Rose H., Finkener H. Handbuch der analytischen Chemie. Leipzig, 1871.
2375. Rosenberg A. Z. analyt. Chem., 90, 103 (1932).
2376. Rosenbl att Th. Ber., 19, 2515 (1886).
2377. Rosendahl A. Tonind-Ztg., 80, 223 (1956).
2378. Rosenheim A., Frank P. Ber., 38, 812 (1905).
2379. Rosenheim A., Koppel I. Z. anorg. Chem., 17, 35 (1898).
2380. Rosenheim A., ReglinA. Z. anorg. Chem., 120, 109 (1922).
2381. Rosenthaler L. Der Nachweis organischer Werbindungen. Stuttgart.
1923, SS. 326, 479, 483.
2382. Rosenthaler L. Mikrochemie, 2, 29 (1924).
2383. Rosenthaler L. Amer. J. Pharm., 101, 784 (1929); Chem. Abstr., 24, 465 (1930).
2384. Rosenthaler L. Mikrochemie, 13, 83 (1933); Chem. Abstr., 27, 2902 (1933).
2385. Rosenthaler L. Mikrochemie, 14, 363 (1934); Chem. Abstr., 28, 6385 (1934).
2386. RossW. H. Amer. Fertilizer, No 5, 24 (1934); J. Assoc. Offic. Agr. Chem., 18, 327 (1935); Калий, 4, № 2, 27; № 8, 42 (1935).
2387. Rosseler M. Kernenenergie, 3, 388 (1960); РЖХим., № 84403 (1960).
2388. RosseletF. South African J. Sci., 49, 344 (1953); 51, 331 (1955); Chem.
Abstr., 47, 11623 (1953); Chem. Zbl., 128, 6874 (1957).
2389. Rossi L., Dy г os si J. L., Chem. Abstr., 39, 1379 (1945).
2390. Rostagni A. Geofis. pura e appl. (Milan), 18, 128 (1950); Chem. Abstr., 46, 833 (1952).
2391. Roth W. A. Becker G. Z. phys. Chem., 159 A, 39 (1932).
2392. Ro t her me 1 D. L„ Nordberg M. E. Amer. Ceram. Soc. Bull., 31, 324 (1952); 46, 10564 (1952).
2393. Rothmund V, Z. anorg. Chem., 62, 108 (1909).
2394. Rothmund V. Z. phys. Chem., 69, 539 (1909).
2395. Rotter F„ Precht H. Ber., 18, 2076 (1885).
2396. Rotter G. Z. analyt. Chem., 63, 163 (1923).
2397. Roy N. Analyt. Chem., 28, 34 (1956).
2398. Rozsa J. T. J. Amer. Ceram. Soc., 31, 280 (1948); Chem. Abstr., 43, 829 (1949).
238
2399. Rozsa P. Acta pharm. hung., 38 (1953); Chem. Zbl., 127 5371 H9561
2400. Rozza J. T„ Zeeb L. E. Petroleum Proces., 8, 1708 (19531- Chem Zbl., 127, 2911 (1956). ’’
24011. Rube C. J. prakt. Chem., 94, 117 (1865); Z. analyt. Chem., 4, 415 (1865)
2402. Rubia P. J., Blasco L. F. Chemist Analyst, 44, 58 (1955); Inform' quim. analit, 9, 21 (1955); РЖХим., № 1096; № 51101 (1956).
2403. Rfldorff W., Zannier H. Angew. Chem., 64, 613 (1952).
2404. Rudorff W., Zannier H. Z. Naturforsch., 8b, 611, (1953).
2405. Rudorff W., Zannier H. Z. analyt. Chem., 137, 1 (1952); 140, 241 (1953); Angew. Chem., 66, 638 (1954).
2406. Ruer R. Chem. Ztg., 20, 270 (1896); Z. analyt. Chem., 36, 319 (1897).
2407. Ruff 0., Johansen O. Ber., 38, 3603 (1905).
2408. Ruff 0. Z. anorg. Chem., 117, 147 (1921); 123, 87 (1922).
2409. Runeberg G. Svensk Kem. Tid., 57, 114 (1945).
2410. Runeberg G., Samuelson O. Svensk Kem. Tid., 57, 91 (1945).
2411. Rupp E., Poggendorf A. Apoth. Ztg., 47, 282 (1932); Chem. Abstr., 26, 3200 (1932).
2412. Russel O. J. Brit. J. appl. Physics, 3, 47 (1952).
2413. Russell R. D. Analyt. Chem., 22, 904 (1950).
2414. R u s s e 11 R. D., S h i 11 i b e e r H. A., F a г q u h a г R, M., M о u s u f A. K. Phys. Rev., 91, 1223 (1953).
2415. Ryssing E. Scand. J. Clin. Lab. Investig., 3, 17 (1951); Z. analyt. Chem., 135, 37 (1952).
2416. Sadek F. S., Reilley C. N. Analyt. Chem., 31, 494 (1959).
2417. S ad tier S. P. Z. analyt. Chem., 10, 101 (1871).
2418. Saeki H., Fujii K. Chem. Engng., 2, 814 (1957); Soil and Plant Food, 2, 190 (1957); РЖХим., № 77202; № 81283 (1958).
2419. Safir К. Chemie (Praha), 8, 230 (1952); Chem. Abstr., 47, 10905 (1953).
2420. Saint-Evre E. J. prakt. Chem., 54, 84 (1891); Compt. rend., 33, 166 (1851).
2421. S a 1 i t P. W. J. biol. Chem., 136, 191 (1940); Chem. Abstr., 34, 7954 (1940).
2422. Salkowski E. Arch. d. Physiol., 6, 209 (1872); Z. analyt. Chem., 11, 474 (1872).
2423. Salkowski E., Munk J. Virchow’s Arch, pathol. Anat., 53, 210 (1899); Z. analyt. Chem., 38, 406 (1899).
2424. Salmon-Legagneur F. Compt. rend., 243, 1325 (1956); РЖХим., № 41393 (1957).
2425. Salmon L. The Determination of Sodium and Potassium in a Sample of Dunite by Radioactivation Analyse. Harwell, 1957; Repts. Atomic Energy Res. Establ. NC/M323, 6 (1957); РЖХим., № 57146 (1958).
2426. S amsel E. P., Bush S. H., Warren R. L., Gordon A. F. Analyt. Chem., 20, 142 (1948).
2427. Samuel T. Bull. Centre beige etude et docum. eaux, No 74, 66 (1957); РЖХим., № 54679 (1957).
2428. Samuelson O. Z. analyt. Chem., 116, 328 (1939).
2429. Samuelson O. Svensk. Kem. Tid., 51, 195 (1939); Chem. Abstr., 34, 1271 (1940).
2430. Samuelson O. Dissertation, Stockholm, 1944.
2431. Samuelson O. Svensk Kem. Tid., 57, 158 (1945).
2432. Samuelson O., Sj ostrom E. Analyt. Chem., 26, 1908 (1954).
2433. Samuelson O., Sjostrom E., Tor sb 10 m S. Z. analyt. Chem., 144, 323 (1955).
2434. Sandberg B. Svensk. Kem. Tid., 58, 147 (1946); Sbornik mezinarod. polarograf. Sjezdu Praze, 1, 227 (1951); Chem. Abstr.; 41, 349 (1947); 46, 6993 (1952).
2435. S a n u e r H. Die К-Emissions- und Absorptionsspektren der Elements Kalium bis Kupfer. Uppsala, 1941.
2436. Saporta D. J. Pharm. Chim., 18 (6), 63 (1903); Chem. Zbl.. 74, II, 461 (1903).
239
2437. Sa re do Y. F. Ph., II (4), No 1, 43 (1939); Chem. Abstr., 34, 6540 (1940).
2438. SaredoY. F. Anales asoc. quim. farm. Uruguay, 47, 100 (1945).
2439. Sartori A. Tabellen zur Berechnung quantitativen chemischer Analysen, Z. analyt. Chem., 40, 201 (1901).
2440. Sartori M. Atti III congresso naz chim. pura appl., 1930, p. 547; Chem. Abstr. 25, 1018 (1931).
2441. Sato M. Bunseki to Shiyaku, 3, 55 (1949); J. Chem. Soc. Japan, 72, 182, 249 (1951); 73, 2 (1952); Chem. Abstr., 46, 852, 3452, 10047 (1952); Chem. Zbl., 127, 811 (1956).
2442. Sato M„ Murat a K- Bull. Agric. Chem. Soc. Japan, 13, 34 (1937); Chem. Zbl., 108, II, 886 (1937).
2443. Sato S. Journ. Chem. Soc. Japan, 72, 420 (1951); Chem. Abstr., 46, 1919 (1952).
2444. Sato S„ Ujino Z. J. Chem. Soc. Japan, 72, 450 492 (1951); 73, 2 (1952); Chem. Abstr., 46, 3452, 8569 (1952).
2445. Satterly J. Proc. Cambridge Phil. Soc., 16, 67 (1912).
2446. Sauman Z. Chem. zvesti, 11, 168 (1957); РЖХим., № 4279; № 35883 (1958); Stavivo, 35, 483 (1957).
2447. Sawyer G. A., Wiedenbeck M. L. Phys. Rev., 76, 1535 (1949); 79, 490 (1950).
2448. S a z E. Ann. chim. analyt. appl., 11, 289 (1929); Chem. Abstr., 24, 36 (1930).
2449. Sea a chi A. Atti Accad. Napoli, 3, 13 (1866—68).
2449a. Schall C. J. Amer. Chem. Soc., 36, 2085 (1914).
2450. Schall E. D. Assoc. Offic. Agr. Chem., 36, 902 (1953); РЖХим., № 48 527 (1954).
2451. Schall E. D. Analyt. Chem., 29, 1044 (1957).
2452. Schall E. D„ Ford O. W. J. Assoc. Offic. Agr. Chem., 31, 397 (1948); Chem. Abstr., 42, 8396 (1948).
2453. Schall E. D., H a g e 1 b e r g R. R. J. Assoc, offic. Agr. Chem., 35, 757 (1952); Z. analyt. Chem., 140, 42 (1953).
2454. Scharrer K-, Munk H. Agrochimica, 1, 44 (1956); Chem. Abstr., 52, 6695 (1958).
2455. Scharrer K., Schorstein H. Landw. Versuchsstationen, 123, 227 (1935).
2456. Schedd О. M. Ind. Eng. Chem., 1, 302 (1909); 2, 379 (1910).
2457. Scheel К. C. Angew. Chem., 66, 102 (1954).
2458. Scheer er Th. Lieb. Ann., 112, 177 (1859); Z. analyt. Chem., 4, 415 (1865).
2459. Schenke V. Landw. Versuchsstationen, 68, 61 (1908); Chem. Zbl., 79, I, 1423 (1908).
2460. Schenke V., Kruger P. Landw. Versuchsstationen, 67, 145 (1907); Chem. Zbl., 78, II, 1759 (1907); Z. analyt. Chem., 48, 505 (1909).
2461. Schenker A. C. Canad. J. Med. Techn., 5, 108 (1943); Chem. Abstr., 38, 992 (1944).
2462. Scherer Th. J. prakt. Chem., 3, 476 (1871); Z. analyt. Chem., 11, 197 (1872).
2463. Scheringa K. Chem. Weekbl., 30, 598 (1933); Chem. Abstr., 28, 4333 (1934).
2464. Scheuler I. E., Thomas R. P. Ind. Eng. Chem., 5, 163 (1933).
2465. Schier O. Angew. Chem., 68, 63 (1956); РЖХим., № 4709 (1957).
2466. Schiff H. Lieb. Ann., 103, 219 (1857); Z. analyt. Chem., 15, 38 (1876).
2467. Schiff H. Lieb. Ann., 118, 365 (1861); Z. analyt. Chem., 1, 65 (1862).
2468. Schlicht A. Chem. Ztg., 30, 1299 (1906).
2469. Schlicht A. Chem. Ztg., 32, 1125, 1138 (1908); 33, 1128 (1909).
2470. Schiller P., Tolgyessy J. Chem. zvesti, 11, 508 (1957); РЖХим., № 28397 (1958).
2471. Schinkmann A. Silikat-Technik, 2, 163 (1951).
240
2472. Sc hl ieper S. Lieb. Ann., 56, 24 (1847).
2473. S ch 16 si ng Th. Compt. rend., 73, 1269 (1872); Z. analyt Chem., 11, 193 (1872).
2474. Schlumm 0. Z. analyt. Chem., 40, 385 (1901).
2475. Schmidt H. J. Z. analyt. Chem., 157, 321 (1957).
2476. Schmidt L. Forschungsdienst, 5, 144 (1938); Chem. Abstr., 32, 5136 (1938).
2477. Schmidt V. Scand. J. Clin. Lab. Invest., 3, 352 (1951); Chem. Abstr., 46, 3106 (1952).
2478. Schmied W., Stegmiiller L. Ber., 34, 135 (1957); Chem. Zbl., 128, 11691 (1957).
2479. Schmitt L., Breitweiser W. Bodenkunde u. Pflanzenernahr., 9—10, 750 (1938); Chem. Abstr., 33, 2436 (1939).
2480. Schmitt L., Ott M. Bodenkunde u. Pflanzenernahr., 6, 127 (1937); Chem. Abstr., 32, 5142 (1938).
2481. Schmitz B. Chem. Ztg., 33, 1127 (1909).
2482. Schmitz B. Z. analyt. Chem., 52, 589 (1913).
2483. Schneidemind W. Die Kalidungung. Berlin, 1922.
2484. Schneider H. Z. analyt. Chem., 135, 191 (1952).
2485. Schnell J. Spectrochim. acta, 2, 71 (1941); Chem. Abstr., 36, 5729 (1942).
2486. Schober R, Fricker A. Z. Lebensm. Untersuch. u. Forsch., 95, 107 (1952); 97, 177 (1953); Chem. Abstr., 46, 11479 (1952); РЖХим, № 43089 (1954).
2487. Scholes S. R, Wessels V. E. Chemist-Analyst, 25, 38 (1936).
2488. Scholes S. R, Wessels V. E. Chem. Abstr, 33, 71 (1939).
2489. Schoorl N. Chem. Weekbl, 4, 687 (1907); 5, 107 (1908); Z. analyt. Chem, 47, 734 (1908); 48, 593, 601 (1909); Chem. Zbl, 79, 1, 1086 (1908).
2490. Schoorl N. Beitrage zur mikrochemischen Analyse. 1909.
2491. Schoorl N. Rec. trav. chim. Pays-Bas, 61, 91 (1942); Chem. Abstr, 37, 3694 (1943).
2492. Schoorl N, Kolthoff I. M. Chem. Weekbl, 17, 425 (1920); Chem. Zbl, 91, IV, 496 (1920).
2493. Schrenk W. G. Analyt. Chem, 22, 1202 (1950).
2494. S c h r e n к W. G, Glendening B. L. Analyt. Chem, 27, 1031 (1955).
2495. Schrenk W. G, Smith F. M. Analyt. Chem, 22, 1023 (1950).
2496. Schroeder J. J. Prakt. Chem, 77 (2), 267 (1908); Chem. Zbl, 79, I, 1368 (1908).
2497. Schubert M. P. J. Amer. Chem, Soc, 53, 3851 (1931).
2498. Schueler J. E, Thomas R. P. Ind. Eng. Chem, Analyt. Ed, 5, 163 (1933).
2499. Schuhknecht W. Angew. Chem, 50, 299 (1937); Optik, 10, 245 (1953); РЖХим, № 22136 (1954); Z. analyt. Chem, 124, 147 (1942).
2500. Schuhknecht W, Schinkel H. Z. analyt. Chem, 143, 321 (1954); Brennstoff-Chemie, 38, 275 (1957); РЖХим, № 34669 (1955); № 21145 (1958).
2501. Schulek E, Koros E. Acta chim. Acad. Sci. Hung, 3, 281 (1963); РЖХим, № 25748 (1954).
2502. Schulek E, Koros E. Ann. Univ, scient. Budapest, Sec. chim, 2, 561 (1960); РЖХим, № 10 Д39 (1961).
2503. Schultz C. Z. analyt. Chem, 9, 278 (1870).
2504. Schumb W, Evans R, Leaders W. J. Amer. Chem. Soc, 63, 1203 (1941).
2505. Schumm O. Z. analyt. Chem, 40, 385 (1901).
2506. Schwaibold J, Kohler M. Landwirtsch. Jahrb. f. Bayern, 30, 1 (1953).
2507. Schwanert A. Lieb. Ann, 186, 353 (1877).
2508. Schwarz G, Krausz B. Kiel. Milchwirt. Forschungsber, 4, 579 (1952); Chem. Abstr, 47, 10144 (1953).
16 Аналитическая химия калия
241
2509. Schwarz M. С. Ber., 63, 2428 (1930).
2510. S с h w а г z е n b а с h G. Helv. chim. acta. 29, 1338 (1946).
2511. Schweitzer H., Lungwitz E. Chem. Ztg., 18, 1320 (1894); Z. analyt. Chem., 36, 191 (1897).
2512. Schwerdt K. Bodenkunde u. Pflanzenernahr., 27, 221 (1942); Chem. Abstr., 37, 4183 (1943).
2513 Scott A. D„ Hunziker H. H., Reed M. G. Chemist-Analyst, 48, 11 (1959); РЖХим., № 46977 (1960).
2514. Scott A. F., Franzier W. R. J. phys. Chem., 31, 459 (1909).
2515 Sedlacek B., Calicek J. Sbornik Ceskoslov. Akad. Zemedelske, 21, 136 (1948); Chem. Abstr., 44, 7000 (1950).
2516. Seely В. K. Analyt. Chem., 27, 93 (1955).
2517. S e g er A. J., Va n Zo о n E. J., L i к i n s M. R. Amer. J. Med. Techno]., 18, 281 (1952); Chem. Abstr., 47, 5479 (1953).
2518. Seibert R. A., Huggins R. A., Bryan A. R. J. Lab. Clin. Med., 38, 640 (1951); Chem. Abstr., 46, 6688 (1952).
2519. Seidell A. Treasury Depart, publ. Health U. S. hygien. Labor. Bl., No 67, 83 (1910).
2520. Seidell A. Solubilities, Vol. 1. N. Y., 1953, pp. 856, 1175.
2521. Seiler H., Artz K., Erlenmeyer H. Helv. chim. acta, 39, 783 (1956); РЖХим., № 1212 (1957).
2522. Seiler H., Sorkin E., Erlenmeyer H. Helv. chim. acta, 35, 120 (1952).
2523. Seiler H., Sorkin E., Erlenmeyer H. Helv. chim. acta, 35, 2483 (1952).
2524. Selke W. Landwirtsch. Versuchsstationen, 114, 321 (1933); Chem. Abstr., 27, 4499 (1933).
2525. Semichon L., Flanzy M. Ann. fals., 23, 517 (1930); Chem. Abstr., 25, 1028 (1931).
2526. Sen A. Z. analyt. Chem., 157, 2 (1957); Analyt. chim. acta, 19, 320 (1958); РЖХим., № 17536 (1958); № 11471 (1959).
2527. Sen A., Bose A. C. Science and Culture, 17, 434 (1952); Chem. Abstr., 46,7467 (1952).
2528. Serano E., Santos J. L. Bol. radiotividad, 24, 49 (1951); Chem. Abstr., 47, 7011 (1953).
2529. Serfass E. J., Levine W. S„ Oliver J. E. Plating, 35, 260, 297 (1948); Chem. Abstr., 42, 4086 (1948).
2530. Serullas M. Ann. chim. phys., 46, 294, 297 (1831).
2531. Serullas M. Z. analyt. Chem., 11. 193 (1872).
2532. Sestini F. Z. analyt. Chem., 1, 409 (1862).
2533. Seubert C. Lieb. Ann., 207, 1 (1881); Z. analyt. Chem., 36, 317 (1897).
2534. Seward R. P., Schumb W. C. J. Amer. Chem. Soc., 52, 3962 (1930).
2535. Shank R. E., Hoagland C. L. Science, 98, 592 (1943).
2536. Shapiro L., Brannock W. U. S. Geol. Survey Circ., No 165, 17 (1952); Chem. Abstr., 46, 7933 (1952).
2537. Shapiro S., Hoagland H. Amer. J. Physiol., 153, 428 (1948); Chem.
Abstr., 43, 266 (1949).
2538. Shaver W. W. Trans. Roy. Soc. Canada, 18, Sect. Ill, 23 (1924); Chem. Abstr., 19, 1096 (1925).
2539. Shaw D. M., Wickremasinghe О. C., Weber J. N. Analyt. chim. acta, 22, 398 (1960).
2540. Shawarbi M. Y. Mikrochemie, 36/37, 366 (1951).
2541. She ad A. C., Smith G. F. J. Amer. Chem. Soc., 53, 483 (1931).
2542. Shedd О. M. J. Ind. Eng. Chem., 1, 302 (1909); Chem. Zbl., 80, II, 1774 (1909).
2543. Sheely M. L. Oila, Soap, 15, 17 (1938); Chem. Abstr., 32, 1967 (1938).
2544. Sherrill E. Ind. Eng. Chem., 13, 227 (1921); Z. analyt Chem., 78, 82 (1929); Chem. Abstr., 26, 2673 (1932).
2545. Shillibeer H. A., Watson K. Science, 121, 33 (1955).
242
2546. Shinanawa M. J. Sci. Hiroshima Univ., 16A, 145 (1952): Chem Zb!
127, 1685 (1956).
2547. Shioroya S. Japan Analyst, 5, 384 (1956); РЖХим., № 19523 (1957).
2548. S hippy B., Burrows G. H. J. Amer. Chem. Soc., 40, 185 (1918); Z. analyt. Chem., 57, 372 (1918).
2549. Shohl A. T„ Bennett H. B. J. biol. Chem., 78, 643 (1928).
2550. Shoji K-, Ma kata S. Proc. Amer. Soc. Hort. Sci., 64, 299 (1954).
2551. Shukla В. K., Mehta D. J. Z. analyt.. Chem., 176, 355 (1960).
2552. Shull F. B., Feenberg E. Phys. Rev., 75, 1768 (1949).
2553. Sica V., Gigante D. Diagnostica tec. lab. (Napoli), Riv. mensile, 12, 27 (1941); Chem. Abstr., 41, 3155 (1947).
2554. Si der is С. P. Ind. Eng. Chem., Analyt. Ed., 9, 145 (1937); 14, 821 (1942).
2555. Siebert H., Rapoport S. Biochem. Z., 326, 413 (1955); Z. analyt. Chem., 151, 81 (1956); РЖХим., № 68639, № 71947 (1956).
2556. Sievert R. M. Proc. int. Conf, peaceful Uses Atomic Energy, 13, 187 (1956); Chem. Zbl., 129, 5564 (1958).
2557. Silverman L., Trego K. Analyst, 78, 717 (1953); РЖХим., № 29132 J1954).
2558. Simek M. Collect. Czechoslov. cliem. Communs., 20, 502 (1955); Chem Zbl., 128, 8304 (1957).
2559. Sinclair W. K-, Holloway A. F. Nature, 167, 365 (1951).
256Q. Singer S. S. Brit. Clayworker, 64, 262 (1955); Chem. Zbl., 127, 8190 (1956).
2561. Singh S., Singh K. Chem. and Ind. (London), 1957, 1297; Chem. Abstr., 52, 1831 (1958).
2562. Sink a A. Z. analyt. Chem., 80, 430 (1930).
2563. Sioiri M., Y one da S. Imp. Agr. Expt. Sta., Spec. Rept, 1940, 1; Chem. Abstr., 35, 4902 (1941).
2564. Sisco R. C., Cunningham B., Kirk P. L. J. Biol. Chem., 139, 21 (1941); Chem. Abstr., 35, 4405 (1941).
2565. Sisley P. Bull. soc. chim., 25 (3), 869 (1892).
2566. Sj oil ema B. Chem. Ztg., 21, 739 (1897); 26, 1014 (1902); Chem. Zbl., 73, II, 1391- (1902).
2567. Sjollema B. Z. anorg. Chem., 42, 127 (1904).
2568. Skarzynski J., Panusz H., Greger J. Chem. analit., 3, 553 (1958)j РЖХим., № 8967 (1960).
2569. Skogseid A. Dissertation. Oslo, 1948.
2570. Skupinski S., Idkiewicz A., Smoluchowska B. Prace Inst. Meeh., 3, 3 (1954).
2571. Slawson С. B. Amer. Mineral., 14, 293 (1929); Chem. Abstr, 24, 1599 (1930).
2572. Smit J., Alkemade С. T., Verschure J. C. Biochem. biophys. acta, 6, 508 (1951); Chem. Weekbl., 47, 23 (1951); Chem. Abstr., 46, 11302 (1952).
2573. Smith D. L., Jamieson D. R., E1 v i n g P. J. Analyt. Chem., 32. 1253 (1960).
2574. Smith E. F. Ber., 21, 260 (1882).
2575. Smith G. F. J. Amer. Chem. Soc., 45, 2072 (1923).
2576. Smith G. F. J. Amer. Chem. Soc., 47, 762 (1925); Chem. Abstr., 19, 1548 (1925).
2577. Smith G. F„ Gring J. L. J. Amer. Chem. Soc., 55, 3957 (1933); Chem. Abstr., 27, 5674 (1933).
2578. Smith G. F„ Ross J. F. J. Amer. Chem. Soc., 47, 774, 1020 (1925); Chem. Abstr., 19, 1548 (1925).
2579. Smith G. F„ Shead A. J. Amer. Chem. Soc., 53, 947 (1931); 54, 1772 (1932).
2580. Smith G. F„ Stubblefield F. M„ Mid die ton E. B. Ind. Eng. Chem., Analyt. Ed., 6, 314 (1934).
16*
243
2581. Smith I. Tids. Kjemi, Bergvesen Met, 2, 89 (1942); Chem. Abstr., 38, 3924 (1944).
2582. Smith J. L. Amer. J. Sci., 15, 234 (1853); J. prakt. Chem., 59, 159 (1853);
60, 244 (1853).
2583. Smith J. L. Amer. J. Sci., 16, 56 (1853); Z. analyt. Chem., 26, 355, 373 (1887).
2584. Smith J. L. Chem. News, 23, 222, 234 (1871); 27, 316 (1873); Amer. J. Sci., 50, 269 (1871); Lieb. Ann., 159, 82 (1871); Z. analyt. Chem., 11, 85 (1872); Amer. Chemist, 3, 241 (1873).
2585. S m i th R. G„ C r a i g P„ В i r d E. J., Boy 1 e A. J., I s er i L. T„ Jacobson E. D., Myers G. B. Amer. J. Clin. Path., 20, 263 (1950); Chem. Abstr., 44, 5421 (1950).
2586. Smitt C., Breitweiser W. Bodenkunde u. Pflanzenernahr., 15, 268 (1939); Chem. Abstr., 34, 2117 (1940).
2587. Smythe W. R., Hemmendinger A. Phys. Rev., 51, 178 (1937).
2588. Snell F., Snell C. Colorimetric Methods of Analysis. N. Y., 1936, p. 432.
2589. Snell J. F. J. prakt. Chem., 2, 474 (1898).
2590. Snyder E., Robinson R. J. Analyt. Chem., 31, 474 (1959).
2591. Snyder J. Q. Proc. Okla. Acad. Sci., 31, 134 (1950); Chem. Abstr., 46, 6550 (1952).
2592. Sobel A. E. Ind. Eng. Chem., Analyt. Ed., 17, 242 (1945).
2593. Sobel A. E., Hanok A., Kramer B. J. biol. Chem., 144, 363 (1942); Chem. Abstr., 36, 6565 (1942).
2594. Sobel A. E., Kramer B. J. biol. Chem., 100, 561 (1933); Chem. Abstr., 27, 3496 (1933).
2595. Sol ari J. Chim. analyt., 38, 91 (1936); Ind. Agric. Aliment, 73, 25 (1956).
2596. Solomon A. K-, Caton D. C. Analyt. Chem., 27, 1849 (1955).
2597. Solon K. Deutsch. Zuckerind., 57, 739, 759 (1932); Chem. Abstr., 27, 5133 (1933).
2598. Sommer A. J. Amer. J. Med. Technol., 17, 276 (1951); Chem. Abstr., 46, 8181 (1952).
2599. Sommer G. Z. analyt. Chem., 151, 336 (1956).
2600. Sondquist N. Lieb. Ann., 369, 105 (1909).
2601. Sonstadt E. Z. analyt. Chem., 36, 501 (1897); 38, 186 (1899).
2602. Soos P., V i r f L., В 1 a z s e к A. Studii si cercetarii chim. Acad. RPR,
8, 231 (1957); РЖХим., № 22984 (1959).
2603. S or e t J. L. Z. analyt. Chem., 18, 94 (1879).
2604. Sornay P. Bull, de 1’Assoc. chim. Suer, et Dist., 26, 976 (1909); Chem. Zbl., 80, II, 1008 (1909).
2605. Sornay P. Bull, de 1’Assoc. chim. Suer, et Dist., 26, 978 (1909); Chem. Zbl., 80, II, 1009 (1909).
2606. Sosin Z., Zaremba J. Chem. analyt., 3, 883 (1958); РЖХим., № 64167 (1958).
2607. Sou ch a у P. Analyt. chim. acta, 2, 17 (1948); Chem. Abstr., 43, 64 (1949).
2608. Sousa A. Analyt. chim. acta, 22, 522 (1960); РЖХим., № 84402 (I960). •
2609. S p e d d i n g F. H., J a f f e S. J. Amer. Chem. Soc., 76, 882 (1954).
2610. Spek J., Dokker M. Verslag. Landb. Onderzoek, No 44, 613 (1938); Chem. Abstr., 33, 3508 (1939).
2611. Spencer A. G. Lancet, 259, 623 (1950); Chem. Abstr., 45, 3017 (1951).
2612. Spencer E., Sen К. B. Analyst, 54, 224 (1929); Chem. Abstr., 23, 2904 (1929).
2613. Spencer R. P., Mitchell T. G., King E. R. Amer. J. Roentgenol. Radium Therapy Nuclear Med., 79, 1053 (1958); Chem. Abstr., 52, 13837 (1958).
2614. Spiegelberg N. Lieb. Ann., 197, 274, 292 (1879).
2615. Spier H. W. Z. physiol. Chem., 285, 146 (1950); Chem. Abstr., 45, 3449 (1951).
244
2616. Spier H. W. Biochem. Z„ 322, 467 (1952); Chem. Abstr., 46, 11028 (1952).
2617. Spiers F. W. Nature, 165, 356 (1950).
2618. Spillner F. Chem. Eng. Techn., 29, 24 (1957); РЖХим., № 51567 (1957).
2619. Spitalsky E., Jo fa S. Z. anorg. Chem., 169, 309 (1928).
2620. Sporek K. F. Analyst, 81, 540 (1956); РЖХим., № 23391 (1957).
2621. Sporek K-, Williams A. F. Analyst, 80, 347 (1955); РЖХим., № 1097 (1956).
2622. Stadie W. C., Ross E. C. J. biol. Chem., 65, 735 (1925).
2623. Staedel W. Lieb. Ann., 217, 169 (1883).
2624. Stabler A. Chem. Ztg., 33, 759 (1909).
2625. Stanford G., English L. Agron. J., 41, 446 (1949); Chem. Abstr., 43, 9318 (1949).
2626. Starck G. Z. analyt. Chem., 48, 415 (1909).
2627. Stary Z. Mikrochemie, 2, 299 (1930); Chem. Abstr., 24, 4234 (1930).
2628. Stas J. S. Z. analyt. Chem., 7, 160 (1868).
2629. Steel A. E. Nature, 173, 315 (1954); РЖХим., № 18963 (1955).
2630. Steenkamp J. L. J. South African Chem. Inst., 13, 64 (1930); Chem. Abstr., 25, 3754 (1931).
2631. Steffen F. Schweiz, med. Wochenschr., 62, 13 (1932); Chem. Abstr, 26, 4368 (1932).
2632. Stein A. Z. angew. Chem., 42, 179 (1929).
2633. Steiner Z. Studii si cercetari stiint. Acad. RPR, 2, 165 (1955); РЖХим. № 19521 (1957).
2634. Steilen R. L. Chem. Ztg., 29, 364 (1905); Z. analyt. Chem., 46, 529 (1907).
2635. Stephenson N. R. J. Lab. Clin. Med., 30, 80 (1945); Chem. Abstr.. 39. 4022 (1945).
2636. Stevens R. E. Ind. Eng. Chem., Analyt. Ed., 12, 413 (1940):
2637. Stocker K. Helv. chim. acta, 33, 1409 (1950).
2638. Stoddard J. L. J. biol. Chem., 74, 677 (1927); Chem. Abstr., 21, 3639 (1927).
2639. Stohmahn F. Z. analyt. Chem., 5, 306 (1866).
2640. St ol ba F. J. prakt. Chem., 90, 196 (1863).
2641. Stolba F. Dingl. polytechn. J., 168, 43 (1863).
2642. Stolba F. Z. analyt Chem., 2, 397 (1863); 3, 298 (1864); J. prakt Chem., 89, 129 (1863); 94, 24 (1865).
2643. Stolba F. Dingl. polytechn. J., 176, 38 (1865); Z. analyt. Chem., 4, 218 (1865).
2644. Stolba F. Z. analyt. Chem., 9, 96 (1870).
2645. Stolba F. Z. analyt. Chem., 9, 100 (1870).
2646. Stolba F. Dingl. polytechn. J., 198, 225 (1873); Z. analyt. Chem., 12, 440 (1873).
2647. Stolba F. Z. analyt. Chem., 13, 79 (1874).
2648. Stolba F. Z. analyt. Chem., 14, 339 (1875).
2649. S t о 1 b a F. Z. analyt. Chem., 21, 601 (1882).
2650. Stone I. Gray P. P„ Kenigsberg M. Wallerstein Labs. Communs, 14, 297 (1951).
2651. Stortenbeker W. Rec. trav. chim. Pays-Bas, 24, 53 (1904); Chem.
Zbl., 76, I, 1212 (1905).
2652. Stout R. W. Phys. Rev., 75, 1107 (1949).
2653. Strauss R. Seifensieder-Ztg., 65, 429 (1938); Chem. Abstr., 33, 1980 (1939).
2654: Strebinger R. Oester. Chem. Ztg., 21, 74 (1918).
2655. Strebinger R., Holzer H. Z. analyt. Chem., 90, 81 (1932).
2656. Strecker W., Diaz F. O. Z. analyt. Chem., 67, 321 (1925/26).
2657. Strecker W„ Junge к A. Z. analyt. Chem., 63, 161 (1923).
245
2658. Strengers T., Klinkenbergh E. A. Chem. Weekbl., 48, 705 (1952); Chem. Abstr., 47, 2251 (1953).
2659. S tri gel A., Do d t J. Landwirtsch. Versuchsstationen, 33, 1752 (1913); Z. analyt. Chem., 52, 588 (1913).
2660. Stromeyer A. Lieb. Ann., 96, 218 (1855).
2661. Strong W. W. Amer. Chem. J., 42, 147 (1909); Phys. Rev., 29, 170 (1909); Chem. Zbl., 80, II, 1623 (1909).
2662. Stuart W. A., Simpson'M., Hardwick W. H. Analyst, 82, 200 (1957); РЖХим., № 69124 (1957).
2663. Stull D. R. Ind. Eng. Chem., 39, 517 (1947).
2664. S t u m p f К. E., G о n s i о r T. Proc. Colloquium Spectroscopicum Internat. VI, London, 35 (1957).
2665. Such tel en F. H., Itano A. J. Amer. Chem. Soc., 36, 1793 (1914).
2666. Sudo T. J. Japan. Assoc. Mineral Petrol. Econ. Geol., 31, 13 (1944); Chem. Abstr., 42, 481 (1948).
2667. Siie P. Nature, 157, 622 (1946); Bull. soc. chim., 405 (1947).
2668. Sueda H., Kaneko M. Bull. Chem. Soc. Japan, 16, 137 (1941); Chem. Abstr., 35, 6210 (1941).
2669. Suess H. E. Phys. Rev., 73, 1209 (1948).
2670. Sugawara R., Koyama T., Kawasaki N. Bull. Chem. Soc. Japan, 29, 679 (1956); РЖХим., № 27121 (1957).
2671. Sun C. R., Wright В. T. Bull. Amer. Phys. Soc., 1, 253 (1956); Phys.
Rev., 109, 109 (1958).
2672. Sunawala S. D., Krishnaswami K. R. J. Indian Inst. Sci., 17A, 105 (1934); Chem. Zbl., 106, I, 1276 (1935).
2673. Sunderman F. W. Amer. J. Clin. Pathol., 29, 95 (1958); Chem. Abstr.,
52, 10273 (1958).
2674. Suttle A. D„ Libby W. F. Analyt. Chem., 27, 921 (1955).
2675. Sutton W. J., Al my E. F. J. Dairly Sci., 36, 1248 (1953); РЖХим.,
№ 36401 (1954).
2676. Suzuki K-, Ken jo M. Rept. Covt. Expt. Sta. Taiwan, 1936, 103; Chem. Abstr., 32, 3181 (1938).
2677. Suzuki N. Bull. Fac. Fisherits Hokkaido Univ., 3, 58 (1952); Chem. Abstr., 47, 12118 (1953).
2678. Sweet R. C., Riem an W., Beukenkamp J. Analyt. Chem., 24, 952 (1952).
2679. Swindal L., Fieldes M. Soil Sci., 74, 287 (1952).
2680. Swisher M., Hummel F. Ind. Eng. Chem., Analyt. Ed., 11, 162 (1939).
2681. Sykes A. Industr. Chemist, 31, 245, 305 (1955); 32, 164, 233 (1956).
2682. Szebelledy L., Schick K. Z. analyt. Chem., 97, 106 (1934).
2683. Szebelledy L., Jonas J. Z. analyt. Chem., 107, 114 (1936).
2684. Tables of Reagents for Inorganic Analysis. Leipzig, 1938, S. 261.
2685. Takashima S., Hagino Y. Rec. Oceanogr. Works Japan, 2, 45 (1955); РЖХим., № 23389 (1957).
2686. Taketatsu T. J. Chem. Soc. Japan, 78, 640 (1957); РЖХим., № 57147 (1958).
2687. T a 1 a p a t r a S. K-, Ray S. C., Sen К- C. Indian J. Vet. Sci., 10, 243 (1940); Chem. Abstr., 35, 2616 (1941).
2688. Tamura Z. Japan Analyst, 1, 117 (1952); Chem. Abstr., 47, 4792 (1953).
2688a. Tananaev N. A., Lasarkevich N. A.«Z. analyt. Chem., 81, 117 (1930).
2689. Tarugi N. Gazz. chim. ital., 34, I, 329, 366 (1904); Chem. Zbl., 75, II, 366 (1904); 78, II, 559 (1907).
2690. Tate F. G„ Whalley H. K. Analyst, 65, 587 (1940); Chem. Abstr., 35, 849 (1941).
2691. Taycock G. H. J. Sci. Instrum., 35, 171 (1958); Chem. Abstr., 52, 13 327 (1958).
246
2692. Taylor F. A. Amer. J. Clin. Pathol., 22, 501 (1952); Chem. Abstr., 46, 6545 (1952).
2693. Taylor F. H. J. biol. Chem., 87, 27 (1930); Chem. Abstr., 24, 3526 (1930).
2694. Taylor S. F. J. phys. Chem., 1, 301, 468 (1897).
2695. Taylor S. F. J. phys. Chem., 1, 723 (1897).
2695a. Taylor S. R. Geochim. et cosmochim. acta, 20, 85 (1960); РЖХим., № 15 Г15 (1961).
2696. T e e r i A. E., Sesin P. G. Amer. J. Clin. Pathol., 29, 86 (1958); Chem. Abstr., 52, 8262 (1958).
2697. Те i chert F. Z. Meteorol., 9, 302 (1955); РЖХим., № 61813 (1956).
2698. Teijgeler C. A. Chem. en pharmac. techn., 13, 48, 57, 75, 90 (1957); РЖХим., № 32155 (1958).
2699. Tendille C. Ann. Inst. nat. rech. agron., A6, 1055 (1955); РЖХим., № 78420 (1956).
2700. Tenery R. M„ Anderson С. E. J. Biol. Chem., 135, 659 (1940).
2701. Ten Have J. Chem. Weekbl., 44, 484 (1948); Chem. Abstr., 42, 9027 (1948).
2702. Terlikowski F., Sozanski S. Roczn. Nauk rolniczych i lesnych, 37, 1 (1936); Chem. Zbl., 107, II, 2431 (1936).
2703. Teschemacher F. T„ Smith J. D. Chem. News, 17, 244 (1868); Z. analyt. Chem., 8, 89 (1869).
2704. Teshinia S. J. Sci. Soil Manure Nippon, 15, 515 (1941); Chem. Abstr., 44, 1852 (1950).
2705. Thin R. H., Cumming A. G. J. Soc. Chem., 107, 361 (1915); Chem. Abstr., 9, 1442 (1915).
2706. Thirring H. Phys. Z., 14, 406 (1913).
2707. Thivolle L., Sonntag G. Trav. membres soc. chim. biol., 23, 1296 (1941); Chem. Abstr., 36, 7046 (1942).
2708. Thompson F. C., Rowlands S. Nature, 152, 103 (1943).
2709. Thompson N. Ind. Eng. Chem., 3, 398 (1911).
2710. Thomson J. E. J. Amer. Chem. Soc., 30, 420 (1908); Chem. Zbl., 79, I, 1575 (1908).
2711. Thomson!. J. Phil. Mag., 10 (6), 584 (1905).
2712. Thomson К. B., Duff end ack O. S. J. optic. Soc. Amer., 23, 101 (1933).
2713. Thomson T. Lieb. Ann., 20, 205 (1836); Z. analyt. Chem., 15, 38 (1876).
2714. Thornton S. F. J. Amer. Soc. Agron., 25, 473 (1933); Chem. Abstr., 27, 4336 (1933).
2715. Thornton S. F., Kraybill H. R. J. Assoc. Offic. Agr. Chem., 18, 281 (1935); Amer. Fertilizer, 85, 9 (1936); Калий, 4, № 5, 40 (1935); 6, № 8, 43 (1937).
2716. Thrun W. E. Ind. Eng. Chem., Analyt. Ed., 5, 79 (1933).
2717. Thruston M. N. J. Soc. Chem. Ind., 61, 144 (1942); Chem. Abstr., 37, 573 (1943).
2718. Thun R., Wenzel O. Bodenkunde u. Pflanzenernahr., 6, 136 (1937); Chem. Abstr., 32, 5142 (1938).
2719. Thiirmer A. Chem. Ztg., 52, 974 (1928); Chem. Abstr., 23, 1077 (1929).
2720. Tian A. Bull. soc. chim., 47, 698 (1930).
2721. Tietjens L. Chem. Ztg., 20, 202 (1896); Z. analyt. Chem., 36, 315 (1897).
2722. Tietjens L„ Roemer H. Laboratoriumsbuch ffir die Kaliindustrie. Halle, 1910, S. 69.
2723. Tilden D. H. J. Assoc. Offic. Agr. Chem., 11, 445 (1928); Chem. Abstr., 23, 217 (1929).
2724. Tilden D. H. J. Assoc. Offic. Agr. Chem., 12, 362 (1929); Chem. Abstr., 24, 438 (1930).
2725. Tinsley J. Analyst, 73, 86 (1948).
2726. Tinsley J. Analyst, 74, 167 (1949).
247
2727. Tinsley J., Pizer N. H. J. Soc. Chim. Ind., 59, 206, 265 (1940); Chem. Abstr., 35, 555, 3019 (1941).
2728. Tiratsoo E. N. Petroleum, 12, 117, 166 (1949); Chem. Abstr., 43, 7331 (1949).
2729. Ti scher J. Biochem. Z., 238, 148 (1931); Mikrochemie Molisch-Festschr., 1936, 418; Chem. Abstr., 25, 5641, 5868 (1931); 31, 4229 (1937).
2730. Tischer J. Z. analyt. Chem., 109, 119 (1937).
2731. Toei K. J. Chem. Soc. Japan, 77, 1270 (1956); РЖХим., № 23390 (1957).
2732. Toei K. J. Chem. Soc. Japan, 78, 1379 (1957); Chem. Zbl., 129, 12351 (1958).
2733. Toennies G., Elliott M. J. biol. Chem., Ill, 61 (1935).
2734. Tokarski J. Bull, intern, acad. polon. sci., Ser. A, 107 (1948); Chem. Abstr., 43, 7860 (1949)._
2735. Tolgyessy J., Sarsunova M., M a j e r J. Pharmazie, 14, 524 (1959); РЖХим., № 70598 (1960).
2736. Tollert H. Naturwiss., 18, 849 (1930); Z. anorg. Chem., 204, 140 (1932); Chem. Abstr., 25, 892 (1931); 26, 2138 (1932).
2737. Tomasson H. Biochem. Z., 195, 475 (1928); Chem. Abstr., 22, 4606 (1928).
2738. Tomitaro J., Yoshimasa T. Bull. Soc. Chem. Japan, 26, 481 (1953).
2739. Tomula E. S. Ann. Acad. Sci. Fenn., 29A, N 21 (1927).
2740. Tomula E. S. Z. analyt Chem., 83, 6 (1931).
2741. Tooper E. B., D’Amico E. S., Bregman J. I. Drug. a. Allied Inds, 39, 9 (1953); РЖХим., № 3921 (1955).
2742. Torstensen A. Dansk Tids. Farm., 2, 258 (1928); Chem. Abstr., 23, 1839 (1929).
2743. Toth S. J., Prince A. L. Soil Sci., 66, 459 (1948); 67, 439 (1949); Chem. Abstr., 43, 3314, 7620 (1949).
2744. Traszkowski R., Zwemer R. L. Biochem. J., 30, 1345 (1936); 31, 229 (1937).
2745. Treadwell W. D., Hepenstrick H. Helv. chim. acta, 32, 1903 (1949).
2746. Treadwell W. D., Werner W. Helv. chim. acta, 36, 1438, 1445 (1953).
2746a. Trendelenburg F. J. Sci. Ind. Res., India, 19, 542 (1960); Analyt. Abstr., 8, No 2745 (1961).
2747. T r e n e 1 M., Trey H. I. Z. Pflanzenernahr. Diing. Bodenkunde, 24A, 293 (1932); Chem. Abstr., 27, 2238 (1933).
2748. Trib al at S. Rhenium et technetium. Paris, 1957, p. 51.
2749. Trnka R. Z. analyt. Chem., 51, 103 (1912).
2750. Truog E., Volk G. W., Pong C., Volk N. J. Amer. Fertilizer, 85, 6 (1936); Калий, 6, № 1, 43 (1937).
2751. Tschopp E. Biochem. Z., 203, 267 (1928); Chem. Abstr., 23, 1531 (1929).
2752. Tsubaki I., Moriya Y. Bull. Osaka Industr. Res. Inst., 9, 109 (1958); РЖХим., № 53107 (1959).
2753. Tuckerman M. M., Strain H. H. Analyt. Chem., 32, 695 (1960).
2754. Tufts B. J. Analyt. Chem., 31, 242 (1959).
2755. Turk us B. Ann. chim. analyt., 22, 101 (1917); Chem. Zbl., 88. II, 646 (1917).
2756. Turner W. E„ Bissett С. C. J. Chem. Soc., 103, 1904 (1913).
2757. Tut ton A. E. Proc. Roy. Soc. London, 111A, 462 (1926); Chem. Abstr., 20, 3105 (1926).
2758. Tyrer D. J. Chem. Soc., 97, 621, 1778 (1910).
2759. Uhl A. F. Bodenkunde u. Pflanzenernahr., 19, 360 (1940); Chem. Abstr., 36, 5940 (1942).
2760. Uhl A. F. Z. analyt. Chem., 123, 322 (1942).
2761. Ulex Z. analyt. Chem., 17, 175 (1878).
248
2762. Underwood G., Bailley W. F. Trans. Illinois State Acad. Sci., 39, 74 (1946); Chem. Abstr., 41, 3393 (1947).
2763. Unger I, Unger L. Glastechn. Ber., 29, 15 (1956); РЖХим., № 58435 (1956)
2764. Urbain P., Wada M. Compt. rend., 199, 1199 (1934); Bull. soc. chim., 3, 163 (1936).
2765. Valentin F., Sucharowa M. Chem. Zvesti, 4, 68 (1950); Chem. Abstr., 44, 9303 (1950).
2766. Valkenburg H. B., McDaniels W. B. J. Colo-Wyo. Acad. Sci., 1, 44 (1930); Chem. Abstr., 26, 3454 (1932).
2767. Vallee B. L. Nature, 174, 1050 (1954); Chem. Zbl., 127, 10775 (1956).
2768. Vallee B. L.,-Margoshes M. Analyt. Chem., 28, 175 (1956).
2769. Vai let P. Compt. rend., 195, 1074 (1932); Chem. Abstr., 27, 1290 (1933).
2770. Valori P, Sovoini F. Ricerca sceint., 27, 1204 (1957); РЖХим., № 17619 (1958).
2770a. Vance a M, Berindeanu G. Ind. usoara, 7, 399 (1960); РЖХим., № 16 Д 45 (1961).
2771. Vandenbosch V. Analyt. chim. acta, 2, 566 (1948); Chem. Abstr., 43, 5558 (1949).
2772. Van den Hen de A., Cottenie A. H., Jonghe P. Ind. chim. beige, 17, 35 (1952); Chem. Abstr, 46, 5743 (1952).
2773. Van der Have A. J., Mulder H. Neth. Milk. Dairy Journ., 11, 128 (1957); Chem. Abstr., 52, 3188 (1958).
2774. Van Du in C. F. Chem. Weekbl., 17, 283 (1920).
277'5. Van Duijn C. Mikrokosmos, 36, 16 (1942); Chem. Abstr., 38, 3178 (1944).
2776. Vanek J., Jur cik F. Sbor. Vysoke skoly zemed. Lesn. fok. Brne, No 4, 261 (1956); РЖХим., № 24736 (1958).
2777. Van Erp A. Rec. trav. chim., 29, 206 (1910). 1
2778. Van Horn J. L. Chem. Weekbl., 10, 182 (1913); Chem. Zbl., 84, I, 1362 (1913).
2779. Van Itallie L. Pharm. Weekbl., 42, 190 (1905); Chem/Zbl, 76, 1, 1117 (1905).
2780. Van Nieuwenburg C. J., Nitenbrock G. Analyt. chim. acta, 3, 572 (1949); Z. analyt. Chem., 131, 364 (1950).
2781. Van Nieuwenburg C. J., Van der Hoek T. Mikrochemie, 18, 175 (1935); Chem. Abstr., 30, 43 (1936).
2782. Van Rysselberge P. J. Ind. Eng. Chem., Analyt. Ed., 3, 3 (1931).
2783. Van Slyke D. D„ Rieben W. K. J. biol. Chem., 156, 743 (1944); Chem. Abstr., 39, 1189 (1945).
2784. V a n’t Hoff J. H. Sitzungsber. Kgl. Akad. Wiss. Berlin, 1904, S. 935; Z. anorg. Chem., 47, 244 (1905); Chem. Zbl., 75, II, 469 (1904); 76, II, 1689 (1905).
2785. V a n’t Hoff J. H., V о e r m a n G. L., В 1 a s d a 1 e W. C. Sitzungsber. Kgl. Akad. Wiss. Berlin, 1905, S. 305; Chem. Zbl., 76, I, 1184 (1905).
2786. Van’t Kruijs M. J. Chem. Weekbl., 6, 735 (1909); Chem. Zbl., 80, II. 1495 (1909).
2787. Van Tongeren W. Chem. Weekbl., 32, 224 (1935).
2788. Varallyav G. Mezogazdasagi Kutatasok, 17, 95 (1944); Chem. Abstr., 41, 7602 (1947).
2789. Varga B. Magyar Kemikosok Lapia, 10, 122 (1955); РЖХим., № 46139 (1955).
2790. Vasudeva A. R., Narayan V. A. J. Indian Inst. Sci., 42, No 3, 51 (1960); РЖХим, № 12 Д40 (1961).
2791. Vaubel W. Z. offentl. Chem, 20, 426 (1914); 21, 1 (1915).
2792. V a v r u c h I, Hejtmanek M. Chem. listy, 49, 200 (1955); Сб. че-хослов. хим. работ, 21, 1363 (1956).
2793. Vavruch 1, .Hejtmanek M, Bouzkova J. Chem. listy, 49, 1782 (1955); Сб. чехослов. хим. работ, 21, 1376 (1956).
249
2794. Veitch F. P. J. Amer. Chem. Soc., 27, 56 (1905); Chem. Zbl., 76, 1, 693 (1905).
2795. V elek er T. J., Dyck R. Analyt. Chem., 31, 387 (1959).
2796. Vendange J. Bull. Direct, mines et geol. Gouv. Gen., No 8, 151 (1951); РЖХим., № 64174 (1958).
2797. Vender M. Chem. Listy, 40, 771 (1955); РЖХим., № 19515 (1956).
2798. Venturello G., Gualandi M, Mazzei I. Ann. chimica, 49, 149 (1959); РЖХим., № 86040 (1959).
2799. Venturini A. Boll. lab. chim. provinc., 9, 425 (1958); РЖХим., № 67637 (1959).
2800. Ver man de V. Pharm. Weekbl, 55, 1131 (1918).
2801. Vermeulen F. H. T. N. O. nieuws, 11, 524 (1956); РЖХим., № 37964 (1957).
2802. Vermey A. Chem. Weekbl., 6, 452 (1909); Z. analyt. Chem., 48, 760 (1909); Chem. Zbl., 80, II, 751 (1909).
2802a. Vernadski V. I. Bull. Soc. France mineral., 36, 258 (1913).
2803. Vicaire P. Compt. rend. Maroc, 24, 185 (1958); РЖХим., Кг 47 019 (1960).
2804. Vickery H. В., Pucher G. W. J. biol. Chem., 83, 1 (1929).
2805. Viergiver E. Bull. Ayer. Clin. Lab. Penna. Hosp., 4, 71 (1951); Chem. Abstr., 45, 5798 (1951).
2806. V i 11 i e r s А., В о r g F. Comptr. rend., 116, 1526 (1893); Z. analyt. Chem., 36, 322 (1897).
2807. Vilnat J., Lio dec N, Voinovitch I. A. Bull. soc. franc, ceram. No 47, 107 (1960); РЖХим., № 9 Д144 (1961).
2808. Viswanathan R., Pillai V. K- J- Scient. Industr Res., B13, 770 (1954); РЖХим., № 46 108 (1955).
2809. Viterbi E. Ann. chim. analyt. appl., 19, 329 (1929); Chem. Zbl., 101, I, 411 (1930).
2810. Vladesco R. Compt. rend., 212, 394 (1941); Chem. Abstr., 36, 506 (1942).
2811. Vlcek A. A. Chem. listy, 48, 1485 (1954); Collect. Czechoslov. chem. Communs., 20, 413 (1955).
2812. Vogel H, Haefcke H. Landw. Vers. Stat., 47, 97 (1896); Z. analyt. Chem., 38, 174 (1899).
2813. Vogelenzang E. H. Pharm. Weekbl., 90, 138 (1955); РЖХим., № 43197 (1955).
2814. Voinovitch I, Debras J. Bull. Soc. fran?. ceram, No 32, 29 (1956), РЖХим, № 60828 (1957).
2815. Volhard J. Lieb. Ann, 198, 318 (1879).
2816. Volk G. M. Soil Sci. Soc. Florida, Proc, 3, 99 (1941); Chem. Abstr, 39, 772 (1945).
2817. Volk N. J„ Truog E. J. Amer. Soc. Agron, 26, 537 (1934).
2818. Volk N. J. J. Amer. Soc. Agron, 33, 684 (1941); Chem. Abstr, 35, 7619 (1941).
2819. Vol mar Y, Leber M. J. pharm. chim, 17, 366, 427 (1933); Ann. chim. analyt. appl, 16, 70, 113 (1934).
2820. Vortman G. См. no [111а].
2821. V о s h a g e H, Hintenberger H. Z. Naturforsch, 14a, 216 (1959); Z. analyt. Chem, 170, 366 (1959); РЖХим, № 81920 (1959); Кг 38392 (1960).
2822. Vries G, Van Dalen E. Rec. trav. chim, 73, 1028 (1954); РЖХим., № 7042 (1956).
2823. Vries H. J. Chem. Weekbl, 4, 231, 333, 455 (1907); 5, 176, 261 (1908); Chem. Zbl, 79, I, 1647 (1908).
2824. Viirtheim A. Chem. Weekbl, 15, 827 (1918).
2825. Viirtheim A. Chem. Weekbl, 17, 637 (1920); Z. analyt. Chem, 67, 457 (1925/26).
2826. Viirtheim A. Rec. trav. chim, 40, 593 (1921); Калий, 2, № 7, 17 (1933).
250
2827. Viirtheim A. Chem. Weekbl., 22, 138 (1925); Chem. Abstr., 19, 2462 (1925).
2828. Wack M. Ann. Geophysique, 8, 337 (1952); Chem. Zbl., 127, 11801 (1956).
2829. Wacker L. Biochem. Z.,245, 149 (1932); Chem. Abstr., 26, 3273 (1932).
2830. Waetenberg A. Z. Elektrochem., 27, 162 (1921).
2831. Wagenaar M. Pharm. Weekbl., 53, 232 (1916); Chem. Zbl., 87, II, 423 (1916).
2832. WagnerG. Z. phys. Chem., 2 B, 27 (1929); Chem. Abstr., 23, 5076 (1929).
2833. W.agner J. Z. phys. Chem., 12, 314 (1893).
2834. Waibel F. Wiss. Veroffentl. Simens-Konzern, 14, 32 (1935).
2835. Walden P. Z. phys. Chem., 55, 712 (1906).
2836. Walker O. J. J. Chem. Soc., 123, 2336 (1923).
2837. Wall er ins J. G. Mineralogy. Stockholm, 1749.
2838. Walters S. L., Miller R. R. Ind. Eng. Chem., Analyt. Ed., 18, 468 (1946).
2839. Walton A. J. Amer. Chem. Soc., 68, 1742 (1946).
2840. Wander I. W., Brode W. R. J. Optical Soc. Amer., 31, 402 (1941).
2841. Wander I. W. Ind. Eng. Chem., Analyt. Ed., 14, 471 (1942).
2842. Wang T. L. Plant a. Soil, 3, 41 (1951); Chem. Abstr., 45, 8178 (1951).
2843. Warington R. J. Chem. Soc., 28, 946 (1875).
2844. Warren H. N. Chein. News, 75, 256 (1897); Z. analyt. Chem., 38, 181 (1899).
2845. Warren R. L. J. Sci. Instrum., 29, 284 (1952).
2846. Watanabe S., Yamagata N. Bunseki Kagaku, 6, 97 (1957); Chem. Abstr., 52, 9332 (1958).
2847. Watson S. I. Analyst, 47, 285 (1922).
2848. Wavelet. Ann. chim. analyt., 5, 289 (1900); Chem. Zbl., 71, II, 689 (1900).
2849. Wawzonek S., Runner M. E. J. Electrochem. Soc., 99, 457 (1952); Chem. Abstr., 47, 51 (1953).
2850. Weaver J. R., Lykken L. Analyt. Chem., 19, 372 (1947).
2851. Webb A. J. Exptl. Biol., 16, 178 (1939); Chem. Abstr., 33, 5714 (1939).
2852. Webb R. J. Repts. Atomic Energy Res. Establ., No C/R 2116, 7 (1958); РЖХим., № 67322 (1958).
2853. Weber H. Z. analyt. Chem., 36, 311 (1897); 38, 171 (1899).
2854. Weber H. Z. analyt. Chem., 54, 167 (1915).
2855. Webster P. A., Crane R. M. Ind. Eng. Chem., Analyt. Ed., 15, 36 (1943).
2856. Webster P. A., Lyle A. K. J. Amer. Ceram. Soc., 23, 235 (1940).
2857. Weeks M. E. J. Chem. Educ., 9, 1035 (1932).
2858. Weavers T. Recueil trav. bot. Neerlandais, 8, 202, 289 (1911).
2859. Wehner G., Bunge W. Chem. Technik, 5, 251 (1953).
2860. Weichselbaum T. E., Somogyi M., Rusk H. A. J. biol. Chem., 132, 343 (1940).
2861. Weichselbaum T. E., Varney P. L. Proc. Soc. Exptl. Biol. Med., 71, 570 (1949).
2862. Weiner R„ Koller L. Z. analyt. Chem., 136, 246 (1952).
2863. Welcher E. J. Organic analytical Reagents, N. Y., vol. 1, pp. 108, 234, 398; vol. 3, p. 287; vol. 4, pp. 40, 305, 1948.
2864. Wellmann H. B. J. Amer. Chem. Soc., 52, 985 (1930).
2865. Wells H. L. Z. anorg. Chem., 4, 335 (1893); Z. analyt. Chem., 34, 76 (1895).
2866. Wells R. C., Stevens R. E. Ind. Eng. Chem., Analyt. Ed., 6, 439 (1934).
2867. Wells R. C., Bailey R. K-, Fairchild J. G. Ind. Eng. Chem., 16, 935 (1924).
2868. W e n d 1 a n d R., Smith С. H., M u r a c a R. J. Amer. Chem. Soc., 71, 1593 (1949); Proc. North Dakota Acad. Sci., 2, 40 (1949); Chem. Abstr., 43, 5021, 6203 (1949).
251
2869. Wendlandt W. W. Analyt. Chem., 28, 1001 (1956); Chemist-Analyst, 46, 38 (1957).
2870. Wendt I. Geol. Jahrb., 70, 385 (1955); РЖХим., № 4717 (1957).
2871. Wenger P., Cimerman C., Rzymowska C. J. Arch. Sci. phys. nat. Geneve, 17 (5), Supl. 89, 140 (1935); Brit. Chem. Abstr., 1935, 1472, 1519.
2872. Wenger P., Duckert R. Reagents for qualitativen inorganic analysis. London, 1948, p. 231.
2873. Wenger P., Hemen C. Ann. chim. analyt. appl., 2, 198 (1920).
2874. Wenger W. H. Amer. Chem. J., 14, 624 (1892).
2875. Wense W. Z. angew. Chem., 4, 691 (1891); 5, 233 (1892); Z. analyt. Chem., 36, 707 (1897).
2876. Werner C. D. Fortschr. Mineralog., 33, 147 (1955); Chem. Zbl., 128, 6255 (1957).
2877. Werther G. J. prakt. Chem., 91, 321 (1864); Bull. soc. chim., 2, 48 (1864).
2878. West A. Z. analyt. Chem., 20, 357 (1881).
2879. West P. W., Folse P., Montgomery D. Analyt. Chem., 22, 667 (1950).
2880. West T. S. Metallurgia, 47, 97 (1953); Chem. Abstr., 47, 4791 (1953).
2881. West T. S. Chem. Age, 81, 245 (1959); РЖХим., № 45503 (1959).
2882. Wheeler T. E., P e г г о s T. P., N a e s e r C. R. J. Amer. Chem. Soc., 77, 3488 (1955).
2883. Wheeting L. C. Soil Sci., 29, 1 (1930); Chem. Abstr., 24, 1452 (1930).
2884. Whelan M. Amer. J. Physiol., 76, 233 (1926); Chem. Abstr., 21, 3576 (1927).
2885. Whisman M., Eccleston В. H. Analyt. Chem., 27, 1861 (1955).
2886. White С. E. J. Chem. Educ., 21, 501 (1944).
2887. White J. Analyt. Chem., 24, 394 (1952).
2888. Whitehead T., Williams E. V. Ind. Eng. Chem., 17, 490 (1945)'.
2889. Wi chers E. J. Amer. Chem. Soc., 72, 1431 (1950).
2890. Wi chers E. J. Amer. Chem. Soc., 74, 2447 (1952); 76, 2033 (1954); 78, 3235 (1956).
2891. Wickbold R. Z. analyt. Chem., 132, 241, 321, 401 (1951).
2892. W ierzbicka Z. Przem. chem., 10, 34 (1954); РЖХим., № 50201 (1954).
2893. Wiesenberger E. Mikrochem. ver. Mikrochim. acta, 30, 176 (1942).
2894. Wiesner A. Jahresber. f. Chem., 332 (1882); Lieb. Ann., 257, 102 (1890).
2895. Wiessmann H., Bollmann A., Muller F. W. Z. Pflanzenernahr. Diing. Bodenkunde, 11, 441 (1932).
2896. Wiggins W. R., Wood С. E. J. Inst. Petrol. Technol., 21, 200 (1935).
2897. W i к 1 a n d e r L., Nilsson E. Acta Agr. Scand., 2, 197 (1952); Chem. Abstr., 47, 6215 (1953).
2898. Wikul M. Z. anorg. allg. Chem., 151, 338 (1926); 181, 121 (1929); Z. analyt. Chem., 72, 345 (1927).
2899. Wilcox L. V. Ind. Eng. Chem., Analyt. Ed., 9, 136 (1937).
2900. Wildman E. A. Proc. Indana Acad. Sci., 44, 121 (1934).
2901. Wilkanowicz M. Szklo i ceram., 5, 1 (1954); РЖХим., № 29130 (1955).
2902. Wilke E. Z. analyt. Chem., 51, 755 (1912).
2903. Willard H. H., Boyle A. J. Ind. Eng. Chem., Analyt. Ed., 13, 137 (1941).
2904. Willard H. H., Liggett L. M., Diehl H. Ind. Eng. Chem., Analyt. Ed., 14, 234 (1942).
2905. Willard H. H., Smith G. F. J. Amer. Chem. Soc., 44, 2816 (1922);
45, 286 (1923).
2906. Willard H .H., Van Lente K. A., Van Atta R. E. J. Chem. Educ., 34, 192 (1957).
252
2907. Willebrands A. F. Chem. Weekbl., 45, 344 (1949); 48, 708 (1952); Nederland. Tijdschr. Geneesktinde, 93, III, 2351 (1949); Rec trav chim «9, 799 (1950); Chem. Abstr., 43, 9137 (1949); 44, 185, 8984 (1950); 47' 2251 (1953)
2908. Wil 1 gal fis A. Z. analyt. Chem, 157, 249 (1957).
2909. W i 11 i a m s J. G. Chem. News, 119, 8 (1919).
2910. Williams J. P, Adams P. B. J. Amer. Ceram. Soc, 37, 306 (1954); РЖХим, № 40356 (1955).
2911. Williams W. O. Proc. Amer. Soc. Hort. Sci, 39, 47 (1941); Chem. Abstr, 36, 4774 (1942).
2912. W i 11 i g e n A. H, Groot P. W. Landbouwkund Tijdschr, 59, 227 (1947).
2913. Wil 1st after R. Ber, 51, 787 (1918).
2914. Wil sing N. Lieb. Ann, 215, 232 (1882).
2915. Wilson C. L, Wilson D. W. Comprehensive Analytical Chemistry, vol. 1A, Amsterdam, 1959, p. 348.
2916. Wilson H. N. Chem. News, 118, 3 (1919).
2917. Wilson H. N, Lees D. S, Broomfield W. Analyst, 76, 355 (1951). 2917a. Winchester J. W. Analyt. Chem, 33, 1007 (1961).
2918. W i n e r A. D, Ernst K. F. U. S. Armed Forces med. J, 5, 823 (lb4 Chem. Zbl, 127, 13813 (1956).
2919. Winkel A, Maas H. Angew. Chem, 49, 611, 827 (1936); Chem. Absti, 31, 627 (1937).
2920. Winkler L. W. Z. analyt. Chem., 52, 628 (1913).
2921. Winkler L. W. Z. angew. Chem, 26, 208 (1913); Chem. Abstr, 7, 2174 (1913).
2922. Winkler L. W. Pharm. J., 94, 741 (1915); Chem. Abstr, 9, 2042 (1915).
2923. Winter K. Z. ges. inn. Med. Grenzgebiete, 11, 324 (1956); Chem. Zbl, 128, 229 (1957).
2924. Winteron R. J. Analyt. chim. acta, 8, 1 (1953); Chem. Abstr, 47, 4789 (1953).
2925. Winton A. L. J. Amer. Chem. Soc, 17, 459 (1895); Z. analyt. Chem, 36, 315 (1897).
2926. Witkowsk H. Chem. analit, Warszawa, 3, 1049 (1958); Analyt Abstr, 7, N 368 (1560).
2927. Witt O. N. Chem. Ztg, 32, 1029 (1908); Chem. Zbl, 79, II, 1713 (1908).
2928. Wittig G. Angew. Chem, 62A, 231 (1950).
2929. Wittig G, Keich er G, Riickert A, Raff P. Lieb. Ann, 563, 110 ... (1949).
2930. W i 11 i g G, R a f f P. Lieb. Ann, 573, 195 (1951).
2931. Woermann D, Bonhoeffer K- F, Helfferich F. Z. phys. Chem. (Neue Folge), 8, 265 (1956).
2932. Woerner E. Ber, 10, 4 (1899).
2933. Wohler L, S pen gel A. Z. Chem. Ind. Kolloide, 7, 243 (1910); Chem. Zbl, 81, II, 1870 (1910).
2934. W о h 1 к A. Dansk Tids Farm, 2, 315 (1928); Chem. Abstr, 23, 1839 (1929).
2935. Woldring M. G. Pharm .Weekbl, 87, 725 (1952); Analyt. chim. acta, 8, 150 (1953); Chem. Abstr, 47, 1768 (1953).
2936. Wolf B. Ind. Eng. Chem, Analyt. Ed, 15, 248 (1943); 16, 121 (1944).
2937. Wolfram W, Stanczuk T. Przemysl Chem, 6, 383 (1950).
2938. Wonssen N. J. Chem. Soc, 54, 89 (1888); Z. analyt. Chem, 28, 342 (1889).
2939. Wood C. A. J. Assoc. Offic. Agr. Chem, 24, 391, 454 (1941); 25, 429 (1942); Chem. Abstr, 35, 5584, 5585 (1941); 36, 4618 (1942).
2940. Wood C. A. J. Assoc. Offic. Agr. Chem, 26, 472 (1943); Chem. Abstr, 37, 6049 (1943).
2941. Wood E. H. J. lab. clin. Med, 27, 960 (1942); Chem. Abstr, 36, 3208 (1942).
2942. Worden E. C. J. Soc. Chem. Ind, 26, 452 (1907); Chem. Zbl, 78, II, 778 (1907).
253
2943. Wo гпе г E. Ber. deutsch. pharm. Ges., 10, 4 (1899); Chem. Zbl., 71, I, 517 (1900).
2944. Woy R. Z. offentl. Chem., 8, 389 (1902); Chem. Zbl., 73, II, 1429 (1902).
2945. Wrangel M. Z. analyt. Chem., 82, 224 (1930).
2946. Wrangel M., Beute Ispracher H. Z. analyt. Chem., 90, 401 (1932).
2947. Wretling K- A. Acta Physiol. Scand., 1, 43 (1940); Chem. Abstr., 35, 1432 (1941).
2948. Wright A., Menke A. E. J. Chem. Soc., 37, 22 (1880).
2949. Wrobel A. Rocznici Chem., 4, 287 (1924); Chem. Abstr., 19, 2463 (1925).
2950. Wurziger J. Deutsch. Lebensmittel-Rundschau, 51, 124 (1955); Z. analyt. Chem., 149, 371 (1956).
2951. Yagoda H. J. Amer. Chem. Soc., 52, 3068 (1930).
2952. Yagoda H. J. Amer. Chem. Soc., 54, 984 (1932).
2953. Yajnik N. A., Tan don G. L. J. Indian Chem. Soc., 7, 287 (1930); Chem. Abstr., 24, 4236 (1930).
2954. Ya ma gat a N., Taj i ma E. Bull. Chem. Soc. Japan, 36, 674 (1957); РЖХим., № 35885 (1958).
2955. Yamane Y. Kagaku no Ryoiki,3,417 (1949); Chem. Abstr.,46,2116 (1952).
2956. Yamasaki T., Kusano S. Bull. Divis. Plant Breeding Cultivat, No 3.
1 (1956); Chem. Zbl., 128, 14110 (1957).
2957. Yamazaki M. Bull. Soc. Salt Sci., Japan, 12, 47 (1958); РЖХим., № 81285 (1958).
2958. Yankov H. F. Chemist-Analyst, 48, 38 (1959); Analyt. Abstr., 7, No 877 (1960).
2959. Yieng Chung Chen, Shing Sung Chen. J. Chinese Chem. Soc., 8, 12 (1941); Chem. Abstr., 37, 51 (1943).
2960. Yoe J. H. Ann. chim. analyt. appl., 7, 193 (1925); 8, 1 (1926); Z. analyt Chem., 69, 61 (1926).
2961. Yorks K. P-, Willard M. L. Mikrochemie, 19, 227 (1936).
2962. Yoshimatsu S. Tohoku J. Exptl. Med., 8, 174 (1926); Chem. Abstr., 21, 1133 (1927).
2963. Yoshimatsu S„ Uga Y. Tohoku J. Exptl. Med., 19, 156 (1932); Chem. Abstr., 26, 5592 (1932).
2964. Zach aria sen W. H. Z. Kristallogr., 80, 137 (1931).
2965. Zach ariasen W. H. Z. Kristallogr., 98, 266 (1937).
2966.- Z a g г о d z к i S., Zaorski H. Rozn. chem., 29, 902 (1955); РЖХим., № 13190 (1956).
2967. Zak B., Mosher R. E., Boyle A. J. Amer. J. Clin. Pathol., 23, 60 (1953); РЖХим., № 449 (1953).
2968. Zaleski L. Landw. Versuchsstationen, 83, 221 (1913); Z. analyt. Chem., 55, 306 (1916).
2969. Zatloukal J. Chem. prumysl, 9, 131 (1959); РЖХим., № 71227 (1959).
2970. Z ema ny P. D„ W e 1 bon W. W., G a i nes G. L. Analyt. Chem., 30, 299 (1958).
2971. Zerf oss S., Hess R. L. J. Amer. Ceram. Soc., 28, 16 (1945); Chem. Abstr., 39, 1029 (1945).
2972. Z e u 1 i e г A., V e 11 u z L., G r i f f о n H. Bull. Soc. chim. biol., 10, 891 (1928).
2973. Zimm В. H., Mayer J. E. J. Chem. Phys., 12, 362 (1944).
2974. Zimmer H. Z. analyt. Chem., 155, 337 (1957).
2975. Zin z adz eC. Chim. et Ind., 27, 841 (1932); Chem. Abstr., 26,3751 (1932).
2976. Zlotowski I. Przemysl Chem., 31, 439 (1952); Chem. Abstr., 47, 10404 (1953).
2977. Zlotowski I., Koi th off I. M. J. Amer. Chem. Soc., 64, 1297 (1942); Ind. Eng. Chem., Analyt. Ed., 14, 473 (1942).
2978. Zocheddu E. Industria chimica, 4, 1116 (1929); Chem. Abstr., 24, 2081 (1930).
2979. Zoellner H. Glass-Email-Keramo-Techn., 3, 290, 378 (1951); Chem. Abstr., 46, 5, 3446 (1952).
2980. Zuckschwerdt N. Z. analyt. Chem., 20, 185 (1881).
2981. Zymny E. Prakt. Chem., 6, 327 (1955); Chem. Zbl., 127, 7633 (1956).
содержание
От редколлегии........................................................ 3
От автора............................................................. 4
Введение........................................................ . 5
I. Аналитическая характеристика калия.............................10
II. Обнаружение ионов калия.......................................12
Образование малорастворимых солей состава KRO4 (R— С1, Мп, Re)...................................................12
Образование малорастворимых галоидосодержащих комплексных солей.................................................... 12
Образование малорастворимых комплексных нитритов ... 14
Образование комплексных (двойных) сульфатов и тиосульфатов 15
Образование солей гетерополикислот....................16
Образование малорастворимых ферроцианидов .................. 17
Обнаружение при помощи солей уранила..................17
Образование калий-бортетрафенила ........ 17
Образование осадка битартрата калия ........................ 18
Реакции с органическими полинитросоединениями 18
Реакции с сульфокислотами.............................22
III. Количественное определение калия...............................24
Химические методы................................................Й
Гравиметрическое определение................................. 24
Титриметрическое определение ..................................55
Газометрическое определение .................................. 86
Косвенные методы определения...................................87
Физико-химические методы....................................... 91
Нефелометрическое определение..................................91
Фотометрическое определение....................................93
Электрохимическое определение.................................103
Радиохимические методы . ......................................105
Определение по естественной радиоактивности калия .... 105
Определение при помощи искусственных радиоизотопов . . . 112
Определение методом радиоактивации .......................... 114
Физические методы..............................................114
Фотометрия пламени............................................114
Спектральное определение .................................... 118
Прочие методы..................................................120
IV. Отделение калия от других элементов..........................128
Хроматографическое отделение калия от других элементов . . 141
Соосаждение калия...........................................147
Приложения.....................................................149
Литература.....................................................173
Израиль Миронович Кореям.
Аналитическая химия кал>
Утверждено к печати
Институтом геохимии и аналитичь кой химии им. В. И. Вернадского Академии наук СССР
Редактор Е. К.. Корчемная
Технические редакторы П. С. Кашина, А. П. Гусева
РИСО АН СССР № 18-В. Сдано в набор 2/IX 1963 г.
Подписано к печати 18/XII 1963 г.
Формат 60 X 90716. Печ. л. 16. Уч.-изд. л. 20. Тираж 3500 экз.
Т. 13652. Изд. № 1970. Тип. зак. № 5858
Цена 1 р. 60 к.
Издательство «Наука»
Москва, К-62, Подсосенский пер., 21
2-я типография Издательства «Наука»
Москва, Г-99, Шубинский пер., 10
ИСПРАВЛЕНИЯ И ОПЕЧАТКИ
Страница Строка Напечатано Должно быть
20 табл. 1 1 10- 1 : 105
31 17 сн. 2488 2449а
50 15 св. 2596 2595
69 13 сн. макроопределениях микроопределениях
104 8 сн. 2210—2282 2210, 2282
108 6 св. ' d — константа... d — плотность раствора
123 14 св. 1271, 1794 1271
129 23 св. калия натрия
136 14 сн. 2745 2746
145 4 и 5 сн. R 1551
163 13 сн. 1564
165 7 и 17 св. пиодида ' иодида
167 14. сн. 419 1419
Г4 3 св. 1959 1958
174 6 св. 70 107
179 Ссылка 215 № 219 № 2, 19
224 » 1857 50 40
227 » 1969 1950 1955
Аналитическая химия калия